Embed Size (px)
Citation preview
Genetyka człowieka
Podstawy
1

Wyjątkowość człowieka?
2
} Człowiek jest typowym organizmem diploidalnym, pod względem mechanizmów genetycznych nie odróżnia się od innych, ale
} Jako obiekt badań genetycznych jest szczególny: } istotne są różnice między osobnikami (w przeciwieństwie do
jednorodnych genetycznie organizmów modelowych) } istotne są różnice środowiska (w przeciwieństwie do
kontrolowanych warunków laboratoryjnych) } istnieją ograniczenia etyczne i praktyczne dopuszczalnych
metod badawczych

Fenotypy badane w genetyce człowieka
3
} W większości choroby } jednogenowe (Mendlowskie) } wieloczynnikowe } nowotwory (mutacje somatyczne) } zaburzenia chromosomowe
} Inne cechy (elementy zmienności prawidłowej) } jednogenowe (rzadko) } markery molekularne } wieloczynnikowe
} np. inteligencja, cechy behawioralne
} Badania obserwacyjne, nie eksperymentalne } zasada największej wiarygodności
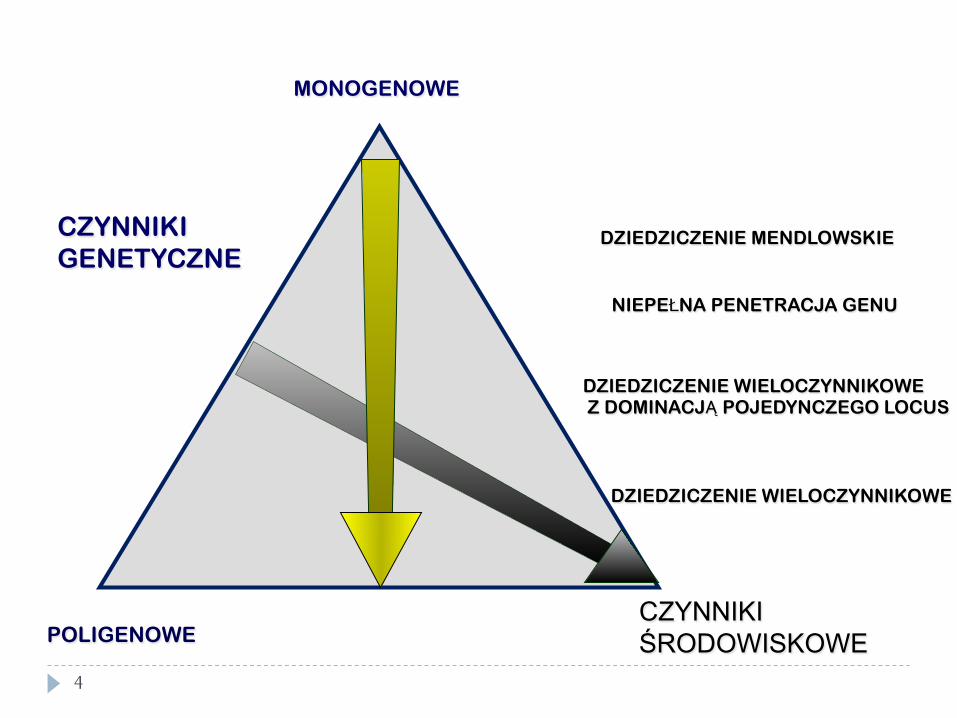
CZYNNIKI GENETYCZNE
CZYNNIKI ŚRODOWISKOWE POLIGENOWE
DZIEDZICZENIE MENDLOWSKIE
NIEPEŁNA PENETRACJA GENU
DZIEDZICZENIE WIELOCZYNNIKOWE Z DOMINACJĄ POJEDYNCZEGO LOCUS
DZIEDZICZENIE WIELOCZYNNIKOWE
MONOGENOWE
4
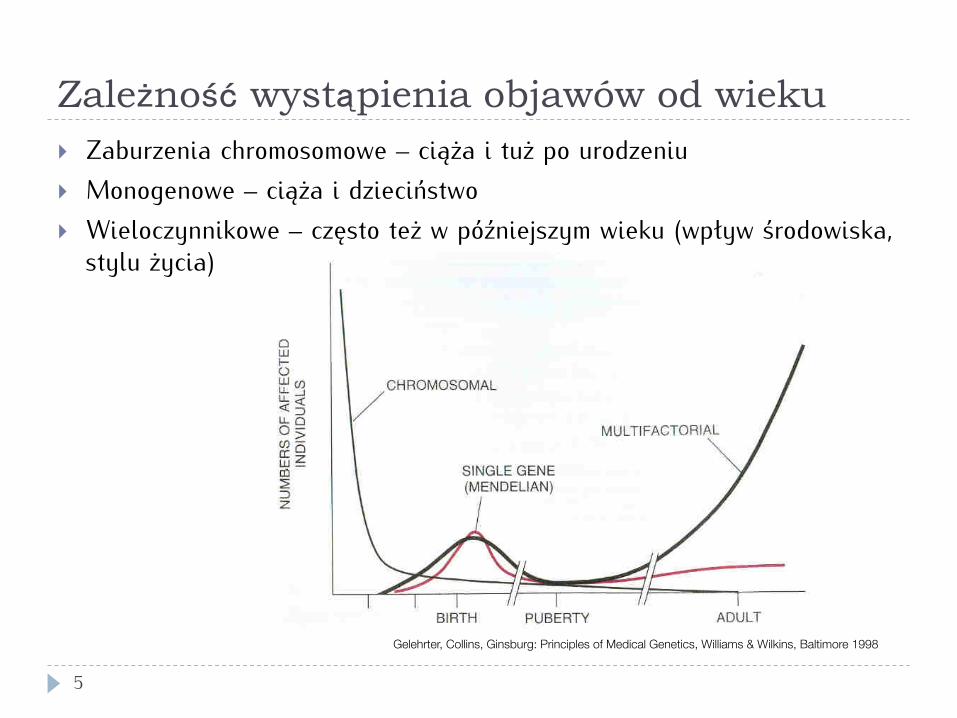
Zależność wystąpienia objawów od wieku
5
} Zaburzenia chromosomowe – ciąża i tuż po urodzeniu } Monogenowe – ciąża i dzieciństwo } Wieloczynnikowe – często też w późniejszym wieku (wpływ środowiska,
stylu życia)
Gelehrter, Collins, Ginsburg: Principles of Medical Genetics, Williams & Wilkins, Baltimore 1998

Odziedziczalność
6
} Za każdy fenotyp odpowiada interakcja genotypu ze środowiskiem
} Odziedziczalność: proporcja zmienności fenotypowej wyjaśnianej zmiennością genetyczną w populacji } badania bliźniąt
} Monozygotyczne (MZ) vs. dizygotyczne (DZ) } badania adopcji } agregacja rodzinna
} częstość objawów u krewnych I stopnia przewyższa obserwowaną u dalszych krewnych i osób niespokrewnionych

Badania bliźniąt - statystyka
7
} Model ACE: trzy składowe: } A (additive genetics) – suma czynników genetycznych } C (common environment) – wspólne środowisko } E (unique environment) – różnice środowiska
} Bliźnięta monozygotyczne: wspólne geny (A) i środowisko (C) – korelacja (rMZ) szacuje A+C; E = 1 - rMZ
} Bliźnięta dizygotyczne: wspólne 50% genów i środowisko – korelacja (rMZ) szacuje ½A + C
} Stąd:
€
rMZ = A +C
rDZ =12A +C
A = 2(rMZ − rDZ )
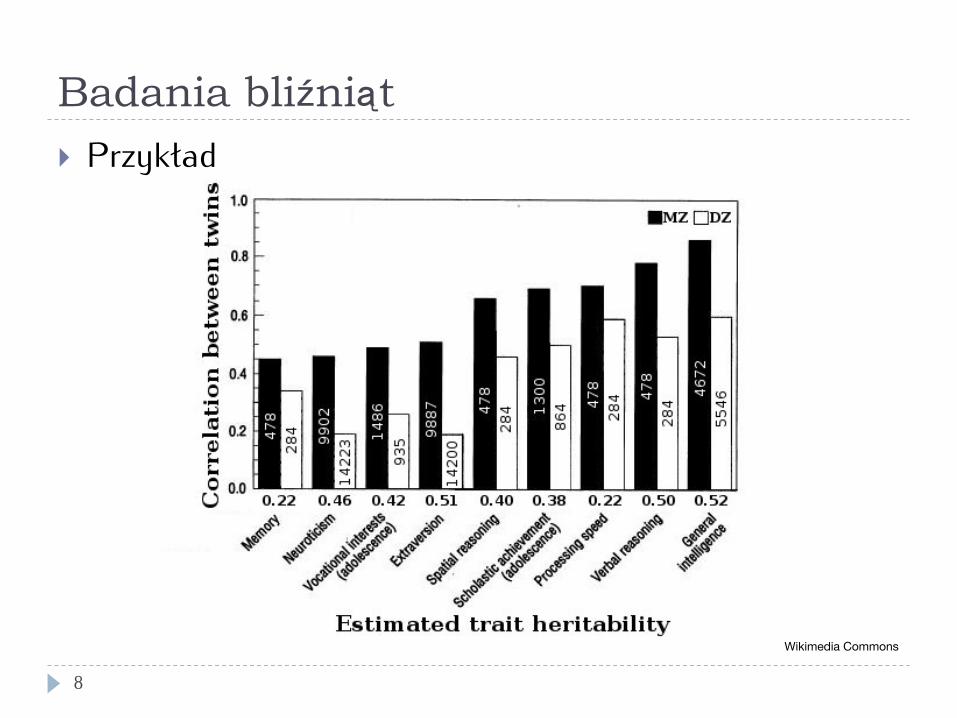
Badania bliźniąt
8
} Przykład
Wikimedia Commons

Agregacja rodzinna
9
} Choroby afektywne (ostre cykliczne zaburzenia nastroju) } jednobiegunowa (epizody depresyjne) } dwubiegunowa (epizody maniakalne i depresyjne)
} Choroba jednobiegunowa } ryzyko w populacji ~3 % (mężczyźni) ~5-9% (kobiety) } krewni I stopnia osoby chorej – ryzyko ~10% } stosunkowo mała odziedziczalność
} Choroba dwubiegunowa } ryzyko w populacji ~1% } krewni I stopnia osoby chorej – ryzyko ~ 20% } istotna odziedziczalność

Cechy jednogenowe
10
} Prosty wzór dziedziczenia } Bardzo wysoka odziedziczalność (często ~100%) } Oprócz chorób
} nieliczne cechy o charakterze anegdotycznym } zdolność do zwijania języka w trąbkę, odstający płatek uszny,
smak PTC, itp. } markery immunologiczne (grupy krwi)
} układ AB0, czynnik Rh } markery molekularne

Choroby jednogenowe
11
} Znanych jest bardzo wiele chorób jednogenowych } ~4500, z tego dla ~1700 nieznany gen } Większość jest bardzo rzadka (najczęstsze ~1/1000 - 1/2000
urodzeń) } problem “chorób sierocych” (orphan diseases)
} W sumie ~4/1000 żywych urodzeń } Autosomalne
} ~4100 chorób, z tego ~1600 nieznany gen } Sprzężone z X
} ~ 360 chorób, z tego ~140 nieznany gen } Sprzężone z Y
} 9 chorób, z tego 5 nieznany gen } Mitochondrialne
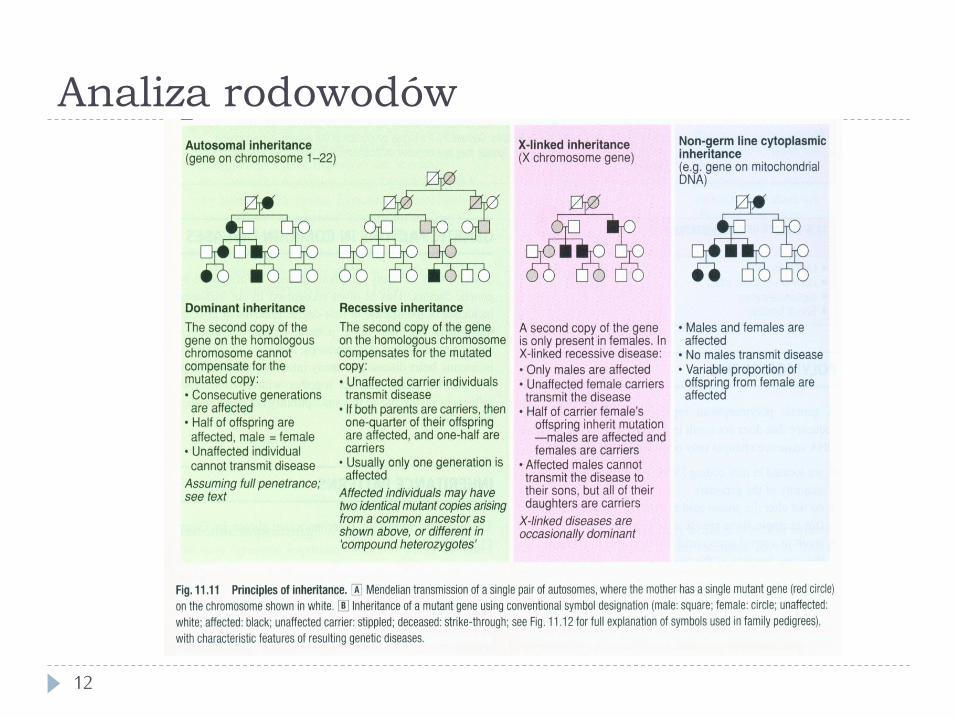
Analiza rodowodów
12
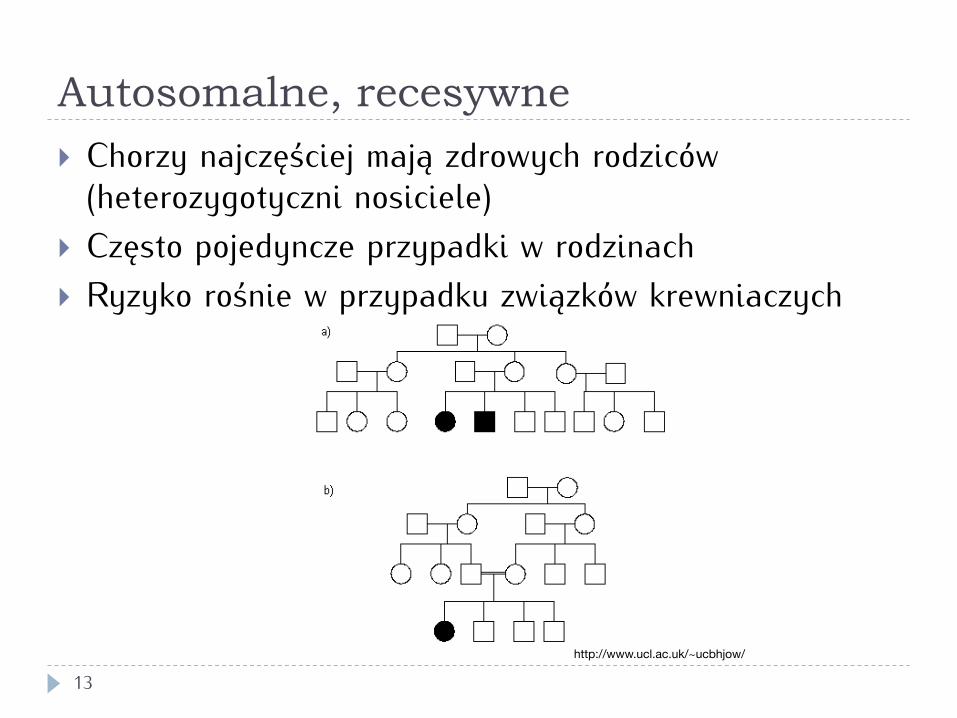
Autosomalne, recesywne
13
} Chorzy najczęściej mają zdrowych rodziców (heterozygotyczni nosiciele)
} Często pojedyncze przypadki w rodzinach } Ryzyko rośnie w przypadku związków krewniaczych
http://www.ucl.ac.uk/~ucbhjow/

Jednogenowe autosomalne choroby krwi
14
} Talasemie } mutacje null i hipomorficzne w genach globin (np. defekty
splicingu) } najczęstsze choroby jednogenowe
} występują głównie w populacjach śródziemnomorskich (do 10% w niektórych populacjach) i u Arabów, także w Azji
} Anemia sierpowata } 1/500 urodzeń u Afroamerykanów
} Wysoka częstość w populacjach z obszarów malarycznych – oporność na malarię heterozygot
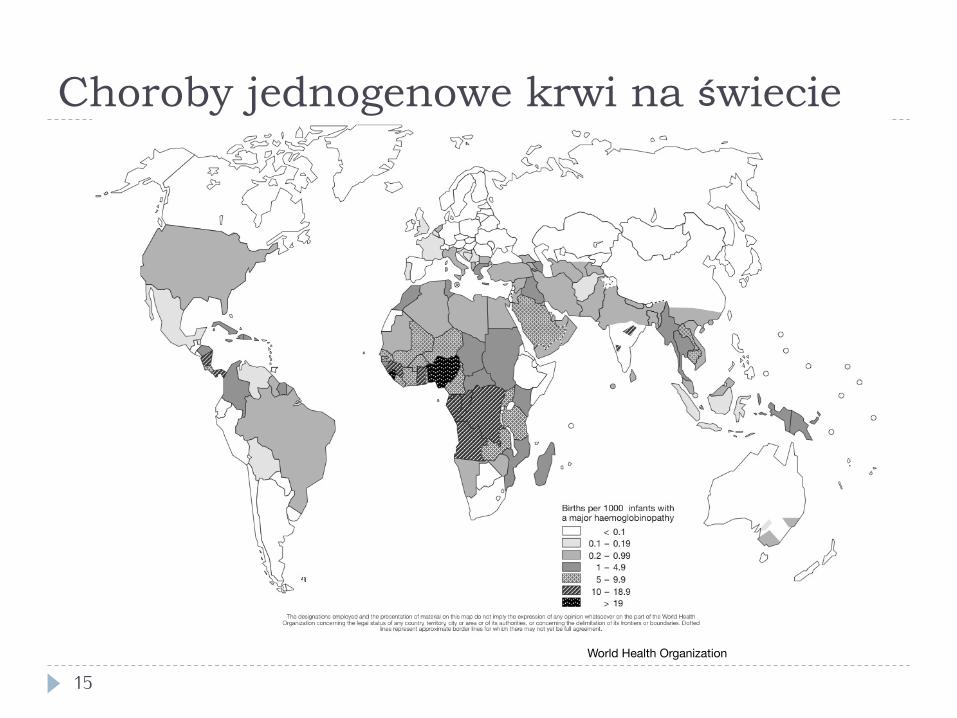
Choroby jednogenowe krwi na świecie
15 World Health Organization
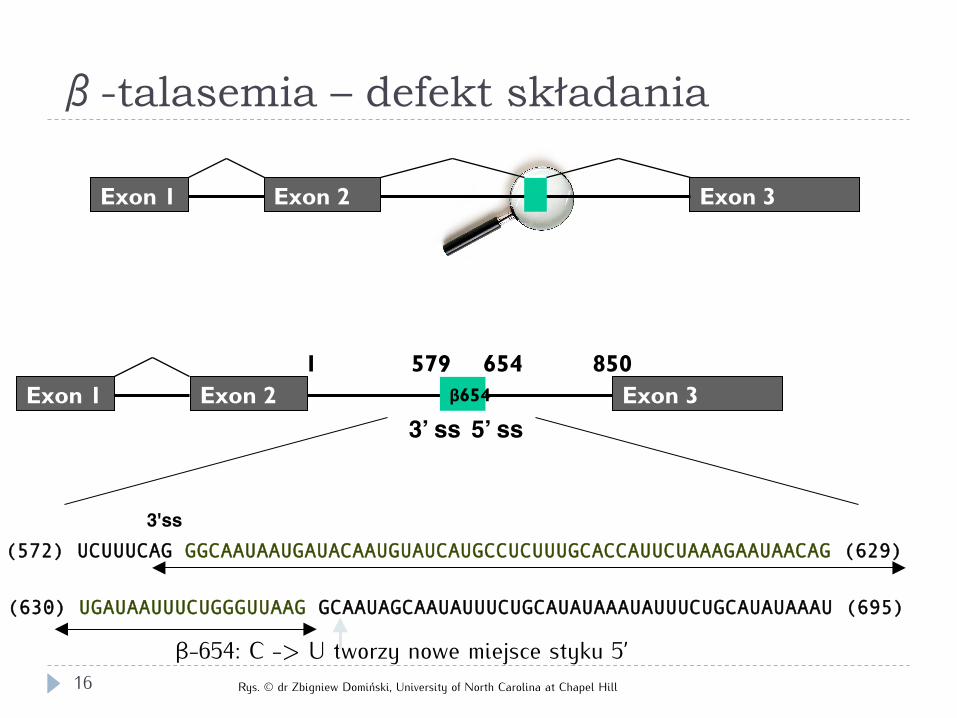
(572) UCUUUCAG GGCAAUAAUGAUACAAUGUAUCAUGCCUCUUUGCACCAUUCUAAAGAAUAACAG (629)
(630) UGAUAAUUUCUGGGUUAAG GCAAUAGCAAUAUUUCUGCAUAUAAAUAUUUCUGCAUAUAAAU (695)
3'ss
β-654: C -> U tworzy nowe miejsce styku 5’
579 654 850 1
3’ ss 5’ ss Exon 2 Exon 3 β654 Exon 1
Exon 1 Exon 2 Exon 3
β-talasemia – defekt składania
Rys. © dr Zbigniew Domiński, University of North Carolina at Chapel Hill 16
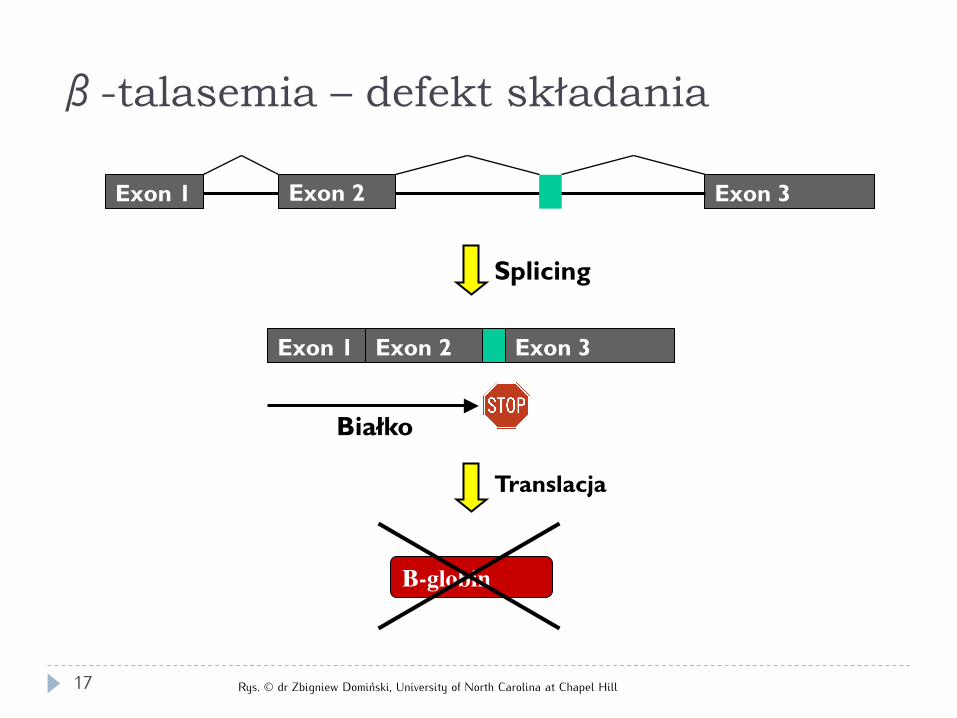
Exon 1 Exon 2 Exon 3
Exon 1 Exon 2 Exon 3
Splicing
Translacja
Białko
Β-globin
β-talasemia – defekt składania
Rys. © dr Zbigniew Domiński, University of North Carolina at Chapel Hill 17

Terapia składania?
18

Mukowiscydoza
19
} Najczęstsza choroba } Najczęstsza choroba autosomalna Europy recesywna w
populacjach Europy północnej } ~1/2000 urodzeń
} Znana od średniowiecza: } “Nieszczęsne dziecko, które pocałowane w czoło zostawia na ustach słony smak. Jest nawiedzone przez złe duchy
i

Mukowiscydoza
20
} Mutacje utraty funkcji genu CFTR } 70% delecja jednego aminokwasu, zaburzenia transportu białka do błony } 15% - mutacje częste (kilkanaście) } 15% - mutacje rzadkie
} CFTR koduje białko transportujące jony Cl- przez błonę
Wikimedia Commons

Choroba Tay-Sachsa
21
} Silna zależność populacyjna } u Żydów aszkenazyjskich 1 osoba na 30 jest nosicielem, w innych grupach 1
na 300 } podobnie wysoka częstość u Kanadyjczyków pochodzenia francuskiego (inna
mutacja niż u Aszkenazyjczyków) i Cajunów w USA } Defekt enzymu (heksozaminidaza A)
} akumulacja gangliozydów (związki lipidowe) w neuronach } neurodegeneracja } śmierć ok. 4 r. ż.

Przyczyny wysokiej częstości allelu recesywnego w populacji
22
} Efekt założyciela } Izolowane populacje wywodzące się z małych grup
} Przewaga heterozygot } Talasemie, anemia sierpowata - malaria } Mukowiscydoza: większa oporność na enterotoksyny
bakteryjne (np. cholera) } Choroba Tay-Sachsa: wyższa inteligencja heterozygot?? –
kontrowersyjna teoria } Kompensacja rodzicielska
} Rodzice, którzy utracili dziecko chętniej decydowali się na kolejne
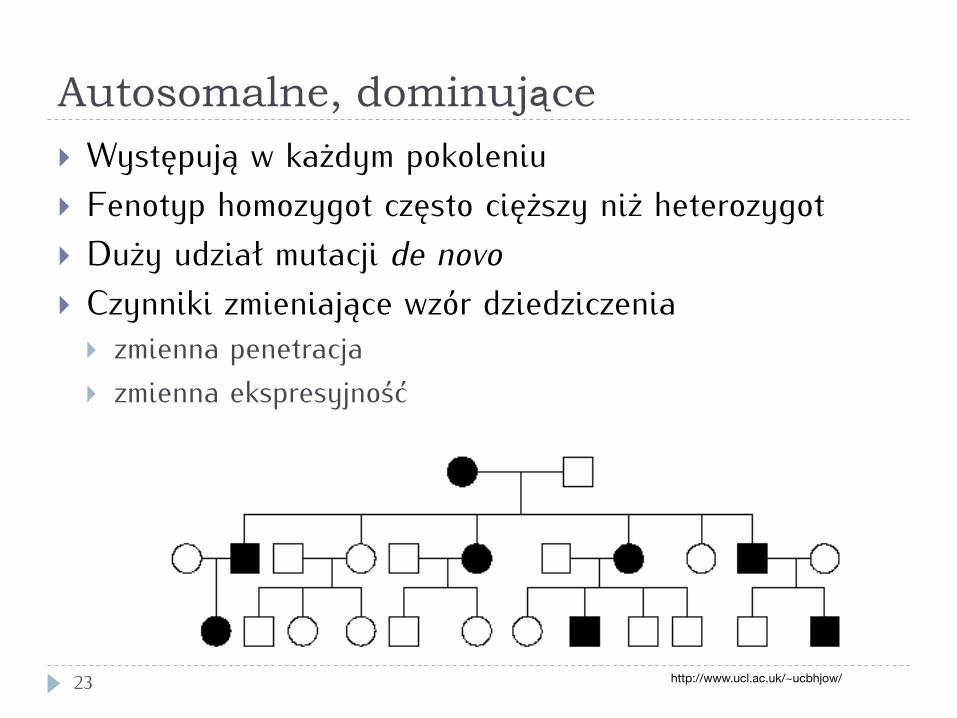
Autosomalne, dominujące
23
} Występują w każdym pokoleniu } Fenotyp homozygot często cięższy niż heterozygot } Duży udział mutacji de novo } Czynniki zmieniające wzór dziedziczenia
} zmienna penetracja } zmienna ekspresyjność
http://www.ucl.ac.uk/~ucbhjow/

Rodzinna hipercholesterolemia
24
} Dominacja przez haploinsuficjencję } Mutacje w genach LDLR (receptor LDL – low density
lipoprotein) i ApoB (apolipoproteina B – część kompleksu LDL odpowiedzialna za oddziaływanie z receptorem)
} Heterozygoty: podwyższony poziom LDL we krwi, miażdżyca, choroby serca ok. 40 r. życia } leczenie: statyny, dieta
} Homozygoty: ciężkie schorzenia serca i naczyń już w dzieciństwie } leczenie: trudne, wysokie dawki statyn, przeszczep wątroby
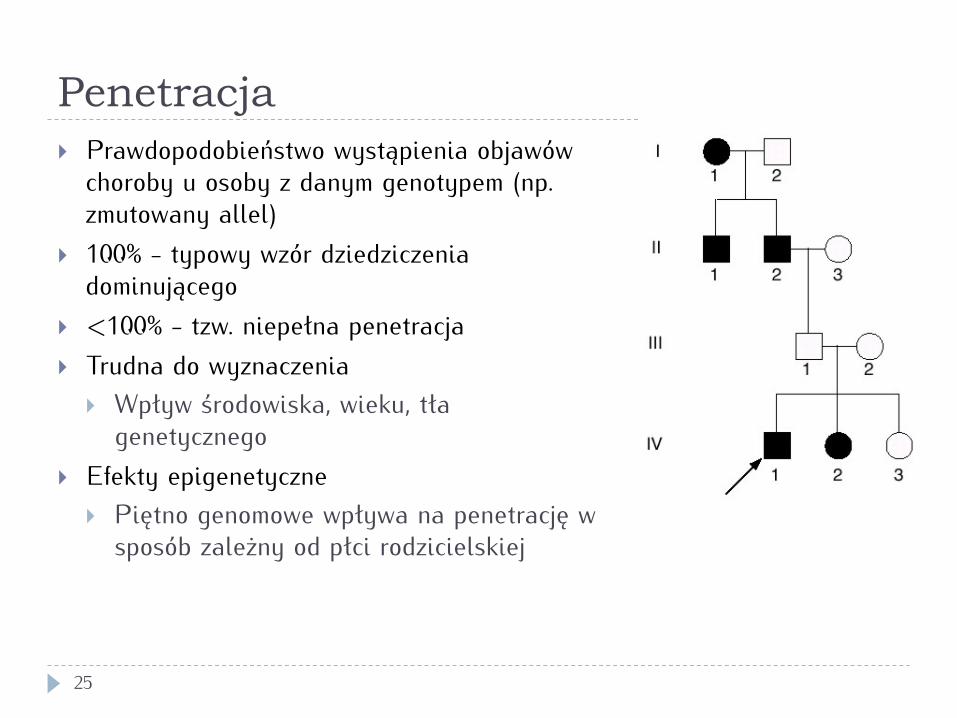
Penetracja
25
} Prawdopodobieństwo wystąpienia objawów choroby u osoby z danym genotypem (np. zmutowany allel)
} 100% - typowy wzór dziedziczenia dominującego
} <100% - tzw. niepełna penetracja } Trudna do wyznaczenia
} Wpływ środowiska, wieku, tła genetycznego
} Efekty epigenetyczne } Piętno genomowe wpływa na penetrację w
sposób zależny od płci rodzicielskiej

Ekspresyjność
26
} Ten sam odziedziczony allel może dawać różne efekty fenotypowe u różnych osób } Penetracja – czy będzie jakikolwiek fenotyp } Ekspresyjność – jaki będzie fenotyp
} Podłoże } Wpływ środowiska, wieku, tła genetycznego
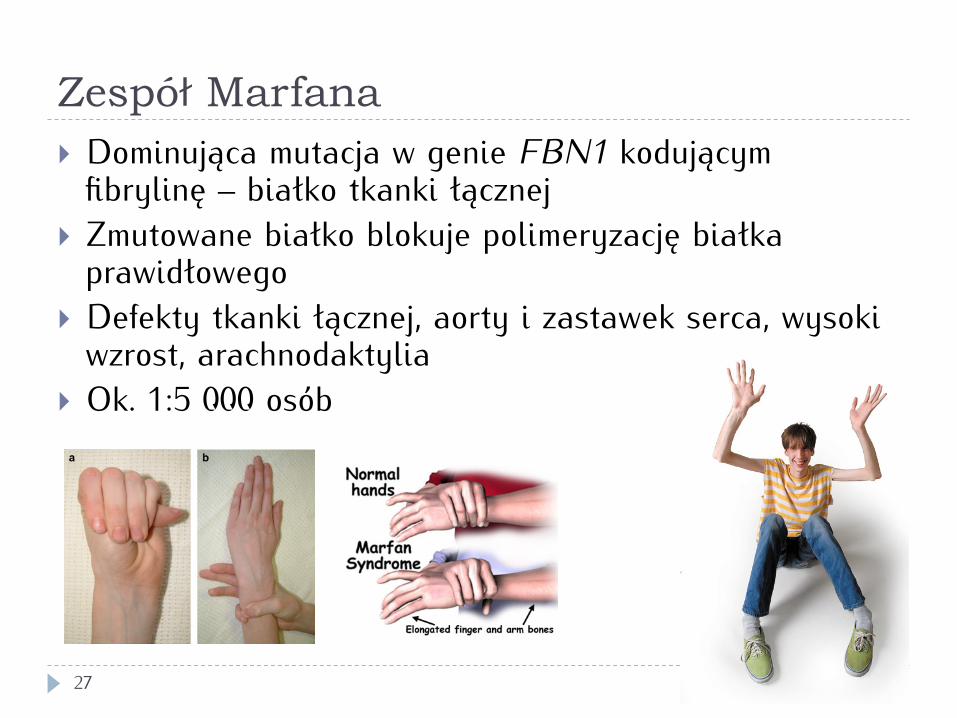
Zespół Marfana
27
} Dominująca mutacja w genie FBN1 kodującym fibrylinę – białko tkanki łącznej
} Zmutowane białko blokuje polimeryzację białka prawidłowego
} Defekty tkanki łącznej, aorty i zastawek serca, wysoki wzrost, arachnodaktylia
} Ok. 1:5 000 osób
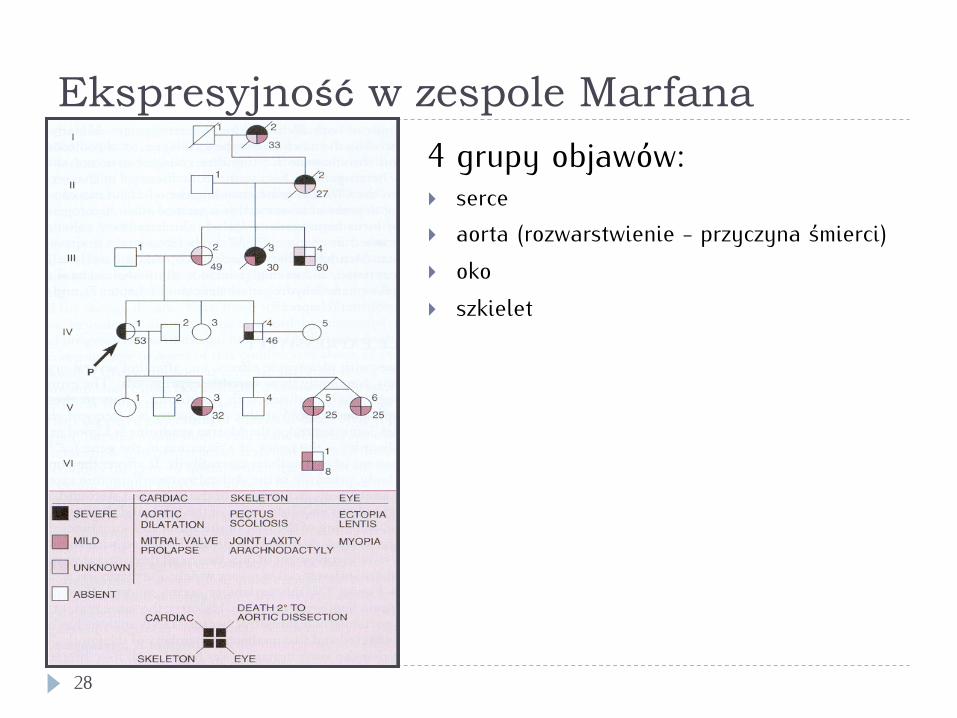
Ekspresyjność w zespole Marfana
28
4 grupy objawów: } serce } aorta (rozwarstwienie - przyczyna śmierci) } oko } szkielet
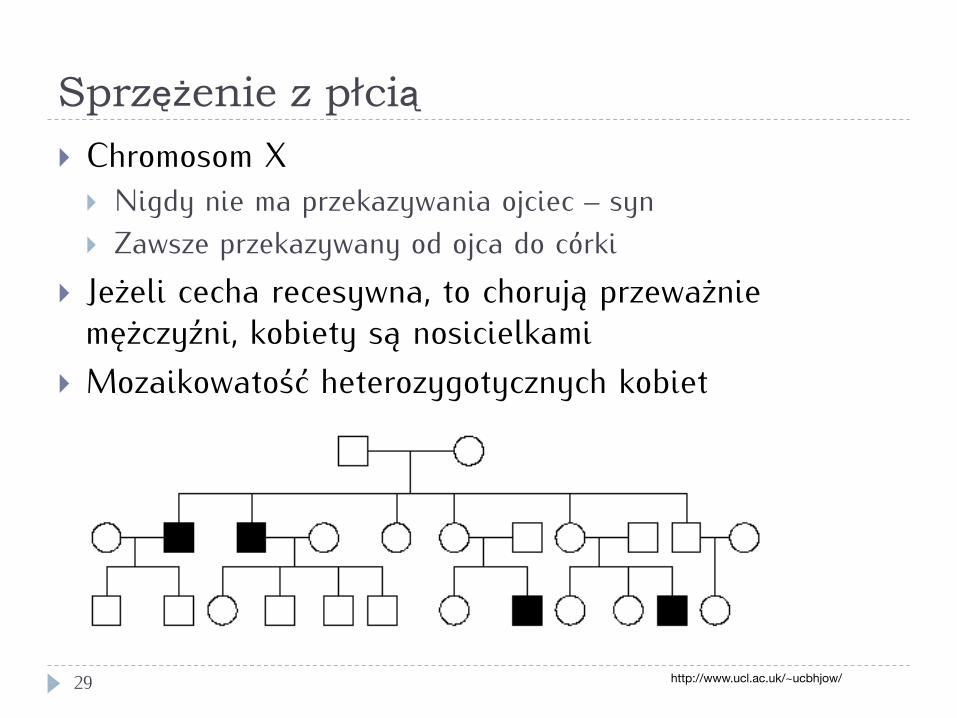
Sprzężenie z płcią
29
} Chromosom X } Nigdy nie ma przekazywania ojciec – syn } Zawsze przekazywany od ojca do córki
} Jeżeli cecha recesywna, to chorują przeważnie mężczyźni, kobiety są nosicielkami
} Mozaikowatość heterozygotycznych kobiet
http://www.ucl.ac.uk/~ucbhjow/

Mozaikowatość inaktywacji X
30
} Mutacja genu odpowiadającego za barwę futra (czarne lub rude) na chromosomie X
} U samic łaty czarne i rude zależnie od inaktywacji
} Samce albo czarne, albo rude
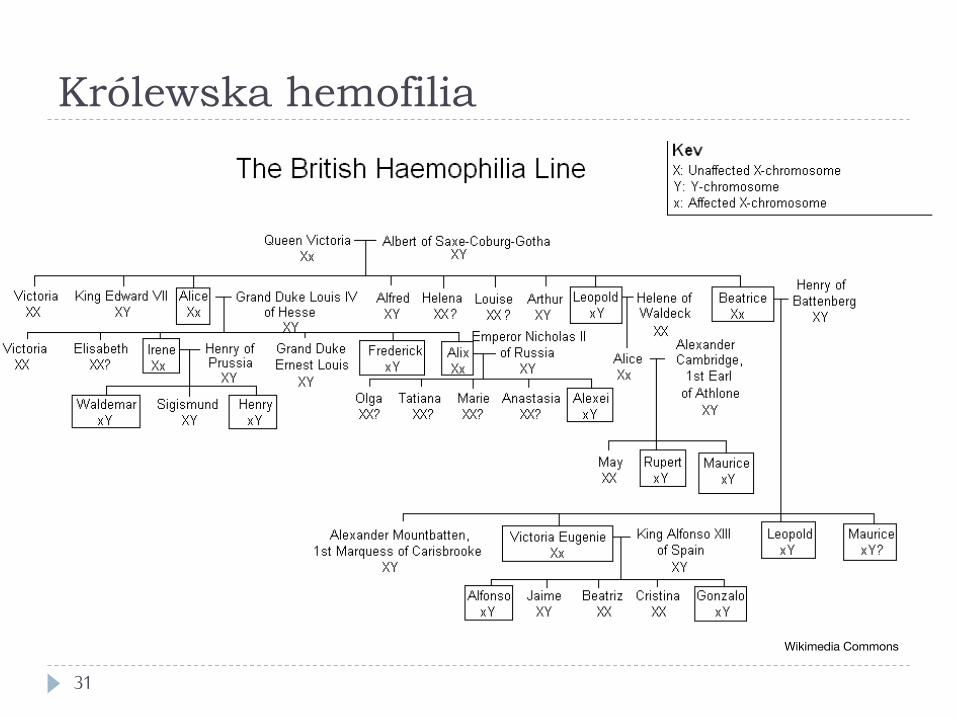
Królewska hemofilia
31
Wikimedia Commons
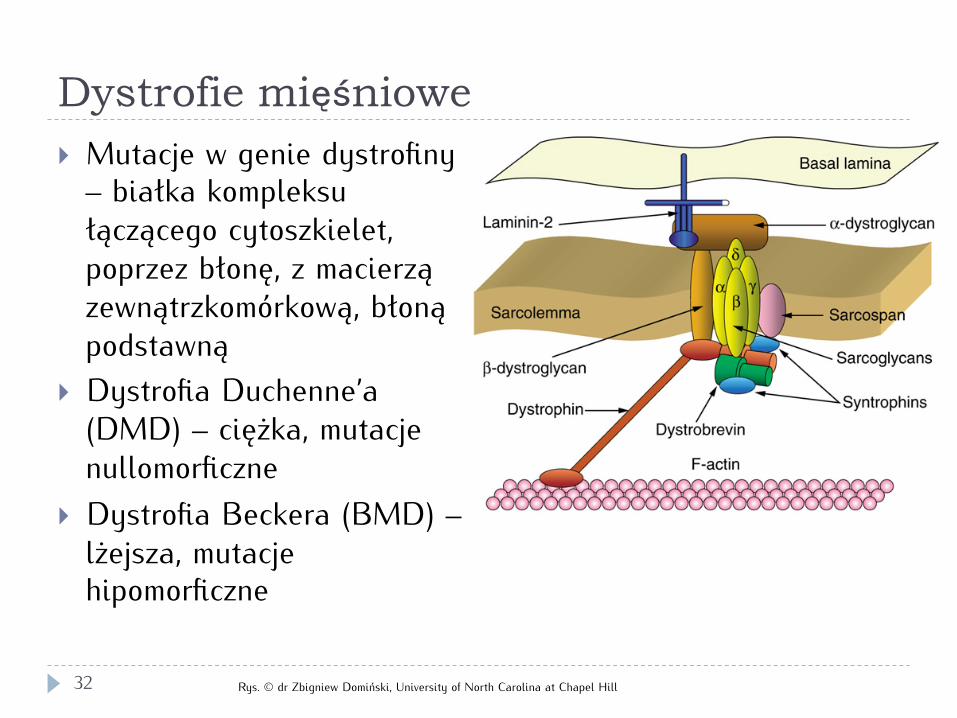
Dystrofie mięśniowe
32
} Mutacje w genie dystrofiny – białka kompleksu łączącego cytoszkielet, poprzez błonę, z macierzą zewnątrzkomórkową, błoną podstawną
} Dystrofia Duchenne’a (DMD) – ciężka, mutacje nullomorficzne
} Dystrofia Beckera (BMD) – lżejsza, mutacje hipomorficzne
Rys. © dr Zbigniew Domiński, University of North Carolina at Chapel Hill
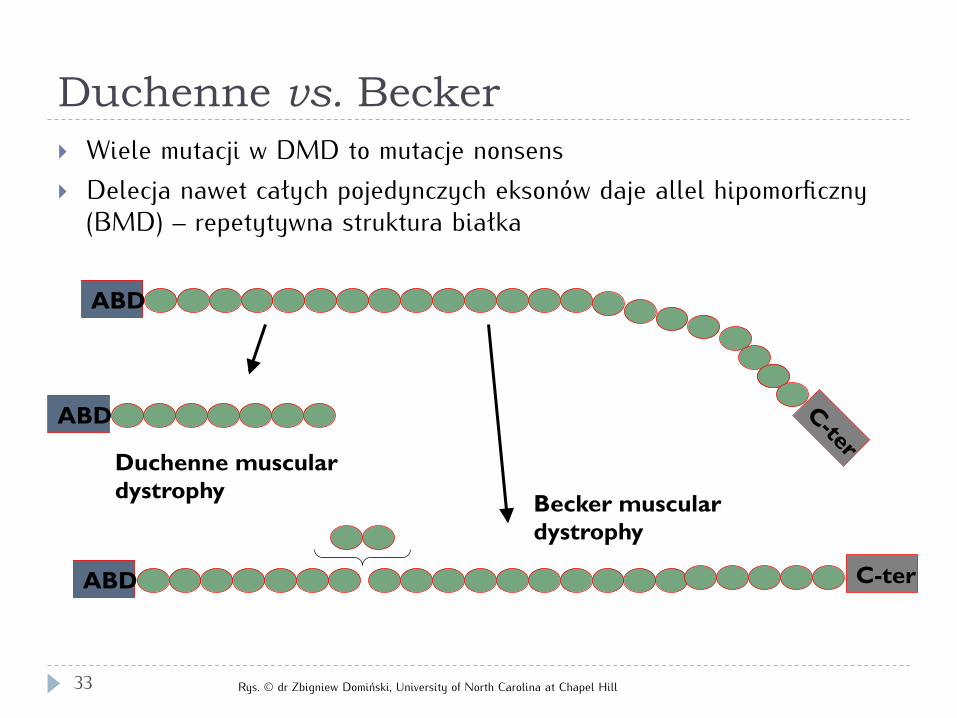
Duchenne vs. Becker
33
} Wiele mutacji w DMD to mutacje nonsens } Delecja nawet całych pojedynczych eksonów daje allel hipomorficzny
(BMD) – repetytywna struktura białka
ABD
Duchenne muscular dystrophy
ABD
Becker muscular dystrophy
ABD C-ter
Rys. © dr Zbigniew Domiński, University of North Carolina at Chapel Hill
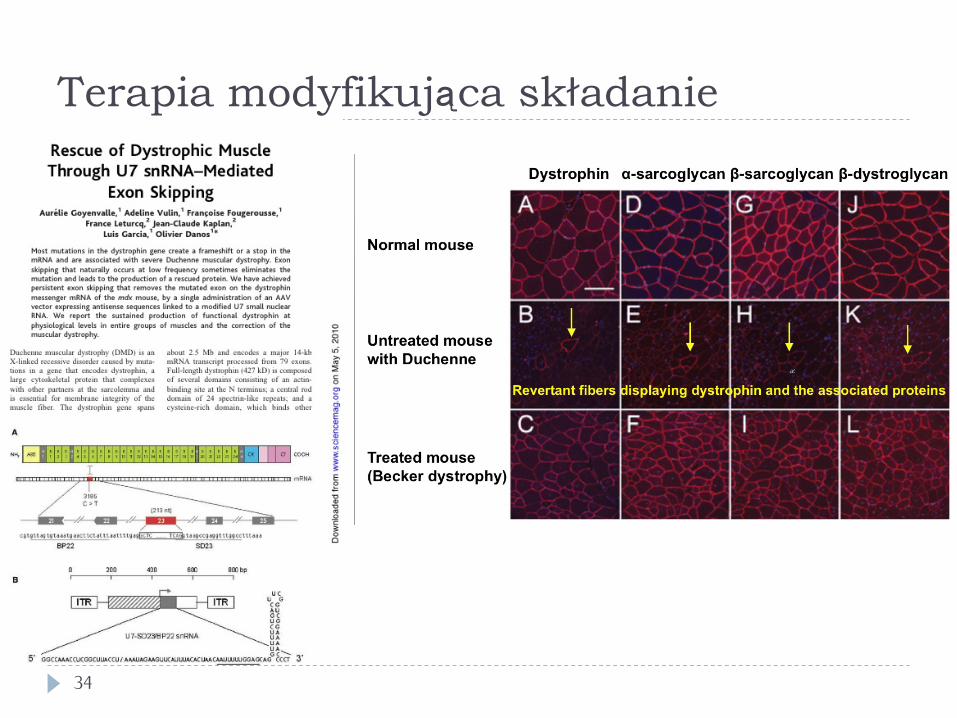
Terapia modyfikująca składanie
34

Faza I/IIa – wstępne badania kliniczne
35

Dziedziczenie mitochondrialne
36
} Charakterystyczny wzór rodowodu (dziedziczenie matczyne) } Najczęściej neuropatologie (w tym zaburzenia wzroku i
słuchu), miopatologie, zaburzenia metaboliczne (kwasica mleczanowa, cukrzyca)
} Mutacje punktowe, rozległe delecje lub deplecja mtDNA } Przy ciężkich mutacjach najczęściej heteroplazmia, stąd
duże zróżnicowanie objawów u różnych osób w rodzinie (efekty progowe)
} Złożone relacje genotyp/fenotyp – dana mutacja może powodować różne objawy, a podobne objawy mogą być wywołane różnymi mutacjami

mtDNA człowieka } 16 568 par zasad } koduje - 13 białek łańcucha
oddechowego (z 84), 2 rRNA (16 S i 12 S), 22 tRNA
} Zwarty - brak intronów, brak lub niewielkie obszary niekodujące między genami, czasem brak pełnego kodonu STOP (wprowadzany poprzez poliadenylację)
} Dziedziczenie od matki (mitochondria plemnika niszczone przez mechanizm zależny od ubikwitynacji
37
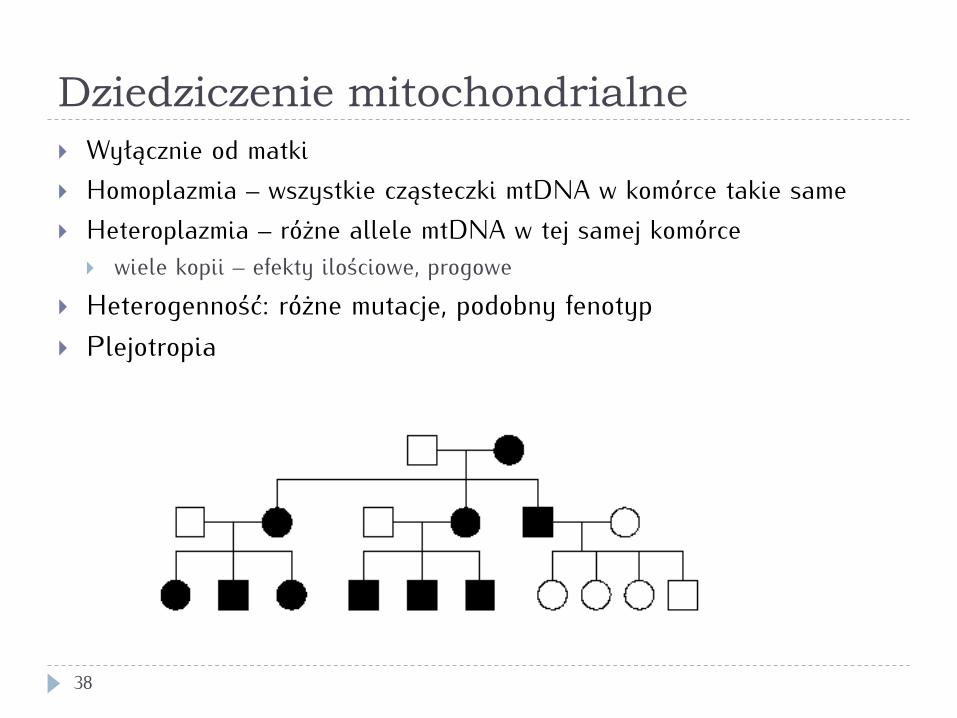
Dziedziczenie mitochondrialne
38
} Wyłącznie od matki } Homoplazmia – wszystkie cząsteczki mtDNA w komórce takie same } Heteroplazmia – różne allele mtDNA w tej samej komórce
} wiele kopii – efekty ilościowe, progowe } Heterogenność: różne mutacje, podobny fenotyp } Plejotropia

Zaburzenia chromosomowe
39
} W przypadku autosomów ciężki i plejotropowy fenotyp } przeważnie letalny – tylko 3 wyjątki
} Główna przyczyna spontanicznych poronień (często nie dochodzi do zagnieżdżenia zarodka)
} W przypadku chromosomów płci (X,Y) fenotyp może być łagodniejszy

Zaburzenia autosomów
40
} Trisomia 21 – zespół Downa } ~1/800 urodzeń, zależnie od wieku matki } Częste poronienia samoistne (75%) } Zaburzenia rozwojowe, opóźnienie umysłowe } Choroby serca, otyłość, cukrzyca, Alzheimer
} Trisomia 13 – zespół Patau } ~1/8000 – 1/120000 urodzeń } Wady rozwojowe (przepukliny, serce, czaszka i mózg) } Średnia przeżywalność: 2 dni, mniej niż 6 miesięcy, bardzo rzadko dłużej
} Trisomia 18 – zespół Edwardsa } 1/3000 – 1/8000 urodzeń } Liczne wady rozwojowe, niedorozwój, mikrocefalia, wady serca } 5% szans na przeżycie 1 r. ż.
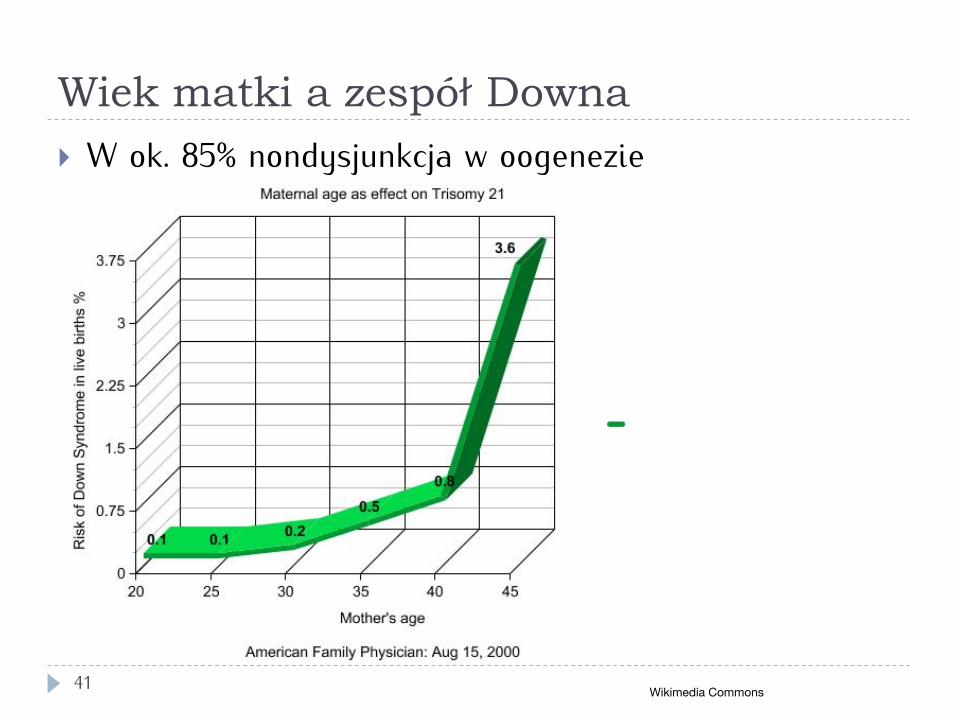
Wiek matki a zespół Downa
41
} W ok. 85% nondysjunkcja w oogenezie
Wikimedia Commons
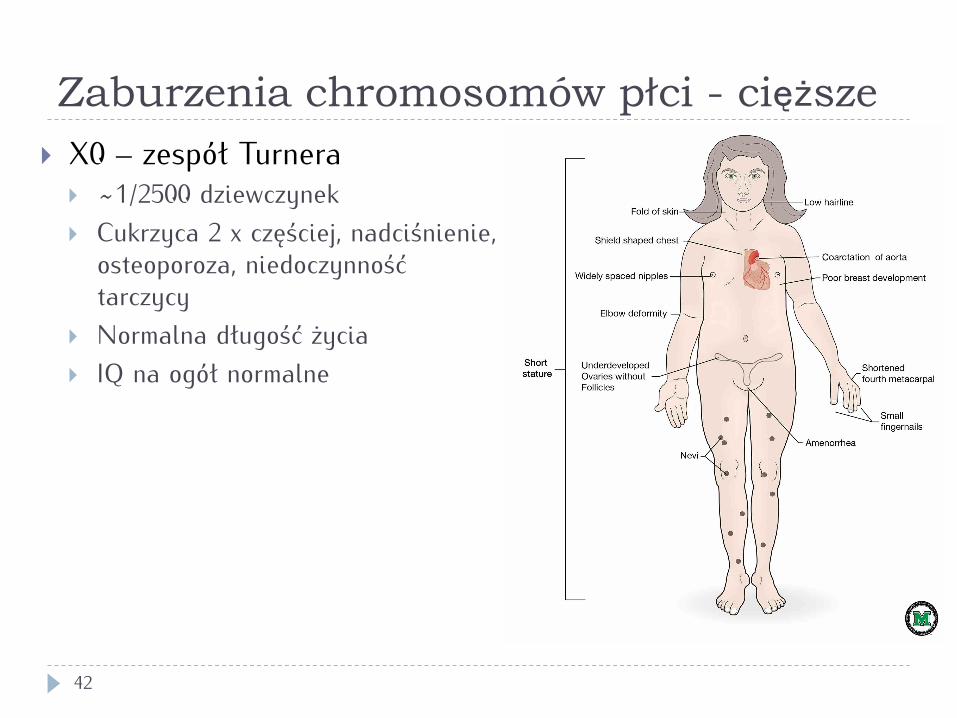
Zaburzenia chromosomów płci - cięższe
42
} X0 – zespół Turnera } ~1/2500 dziewczynek } Cukrzyca 2 x częściej, nadciśnienie,
osteoporoza, niedoczynność tarczycy
} Normalna długość życia } IQ na ogół normalne
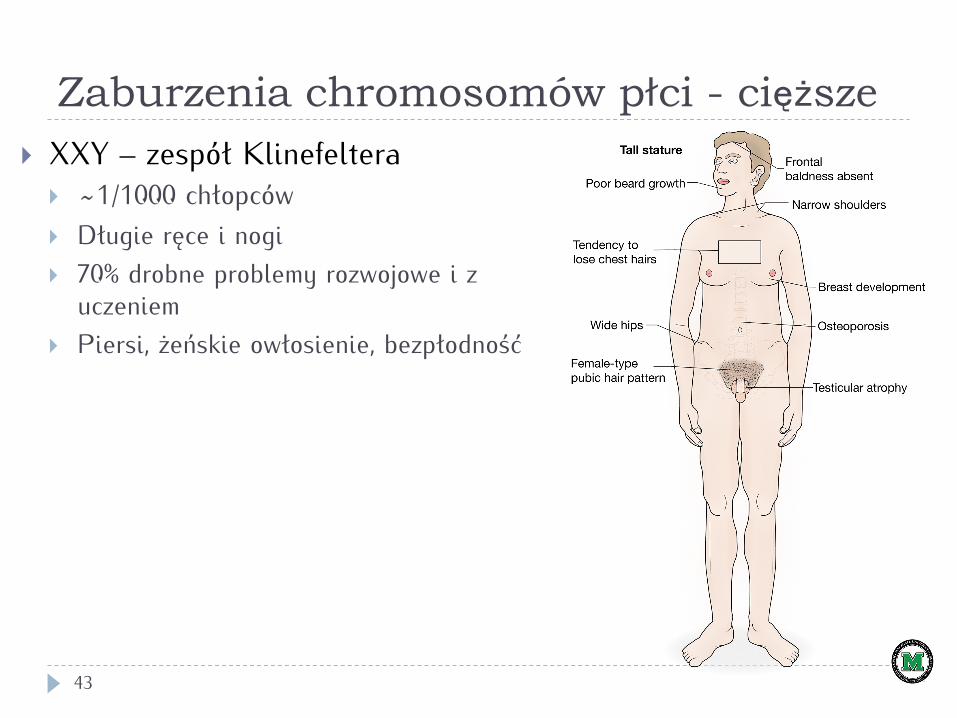
Zaburzenia chromosomów płci - cięższe
43
} XXY – zespół Klinefeltera } ~1/1000 chłopców } Długie ręce i nogi } 70% drobne problemy rozwojowe i z
uczeniem } Piersi, żeńskie owłosienie, bezpłodność

Zaburzenia chromosomów płci - lekkie
44
} XXX } Lekkie objawy ze względu na inaktywację X, zwykle brak ewidentnych
zewnętrznych objawów } Niekiedy zaburzenia cyklu menstrualnego i umiarkowane problemy rozwoju
intelektualnego, wysoki wzrost } ~1/1000 dziewczynek } bardzo rzadko XXXX i XXXXX (kilkaset przypadków w historii, cięższe
objawy) } XYY
} Zwykle brak ewidentnych zewnętrznych objawów, >90% nie wie, że ma ten kariotyp
} ~1/1000 chłopców } Nieco zwiększone ryzyko opóźnień w nauce, wysoki wzrost, normalny poziom
testosteronu } Wcześniejsze doniesienia o korelacji z zachowaniem agresywnym - fałszywe


















![Genetyka Zwierząt - 2004 - Krystyna M. Charon [PL]](https://img.pdfslide.tips/doc/110x75/5571f26e49795947648c8e67/genetyka-zwierzat-2004-krystyna-m-charon-pl.jpg)