Embed Size (px)
Citation preview

1
1
Phylogenie IIPhylogenie II
WS 2006/2007
Genomforschung und Sequenzanalyse
Einführung in Methoden der Bioinformatik
Bernhard Lieb &Tom Hankeln
MolekulareMolekulare
2
Themen
Grundlagen und Begriffe der molekularen Phylogenie
Evolutionsmodelle
BerechnungPrüfung
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen
2
3
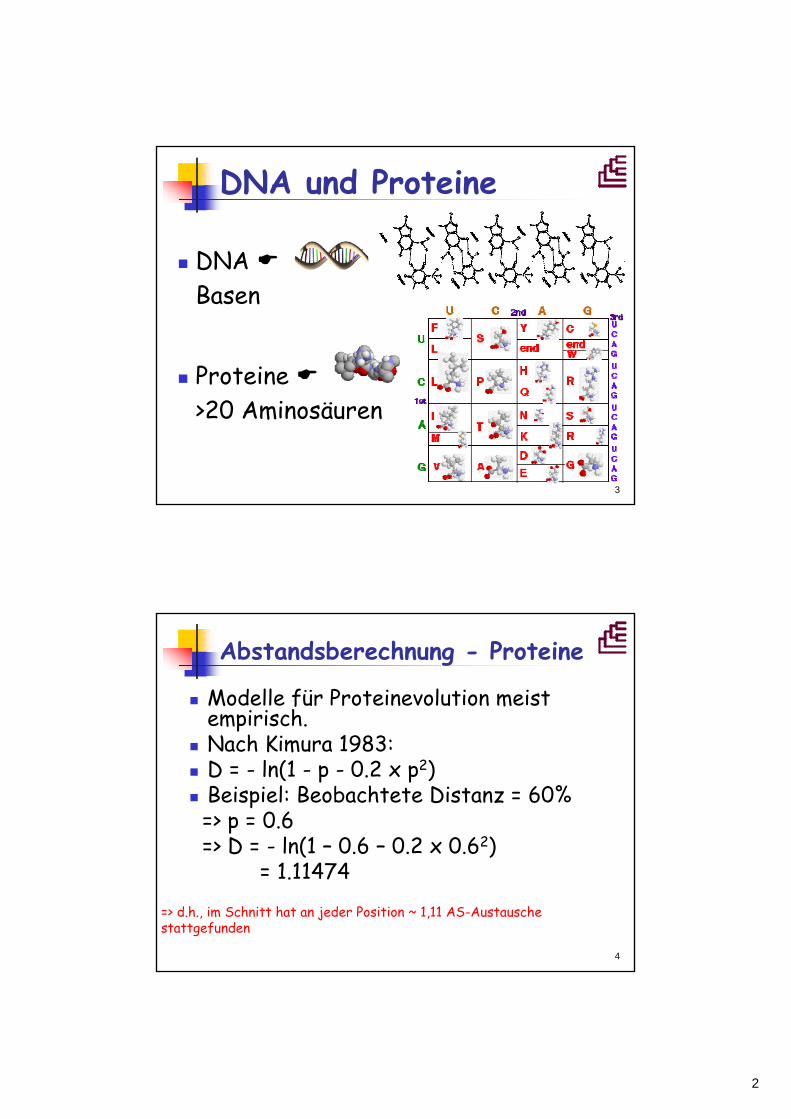
DNA und Proteine
DNA Basen
Proteine >20 Aminosäuren
4
Abstandsberechnung - Proteine
Modelle für Proteinevolution meist empirisch.Nach Kimura 1983:D = - ln(1 - p - 0.2 x p2)Beispiel: Beobachtete Distanz = 60%=> p = 0.6=> D = - ln(1 – 0.6 – 0.2 x 0.62)
= 1.11474
=> d.h., im Schnitt hat an jeder Position ~ 1,11 AS-Austausche stattgefunden

3
5
Aber…
…das Modell ist zu einfach!
Jeder Aminosäureaustausch wird gleich bewertet.
Aber:
6
AminosäurenNicht jeder Aminosäureaustausch ist gleich wahrscheinlich!
Aminosäure-Eigenschaften:
CP
GGAVIL
MF
YW H
KR
E Q
DNS
TCSH
S+S
Sehr klein
klein
geladen
positivpolararomatisch
aliphatisch
hydrophob
4
7
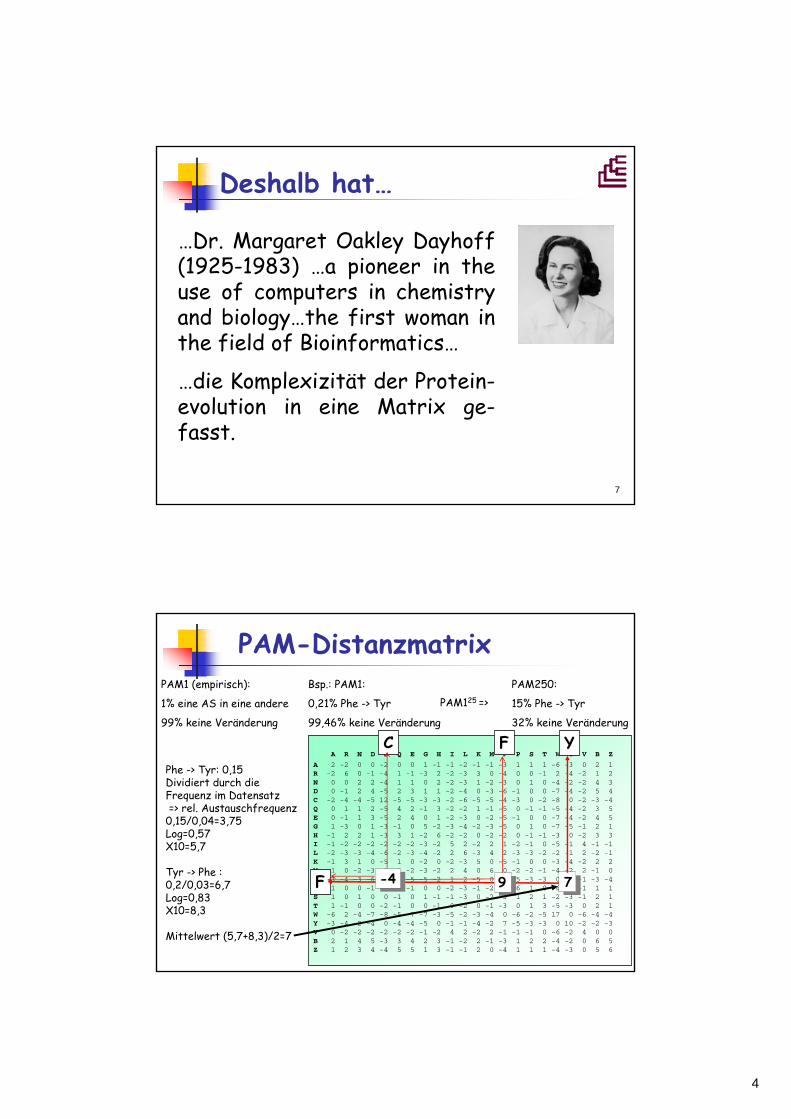
Deshalb hat…
…Dr. Margaret Oakley Dayhoff(1925-1983) …a pioneer in theuse of computers in chemistryand biology…the first woman in the field of Bioinformatics…
…die Komplexizität der Protein-evolution in eine Matrix ge-fasst.
A R N D C Q E G H I L K M F P S T W Y V B Z
A 2 -2 0 0 -2 0 0 1 -1 -1 -2 -1 -1 -3 1 1 1 -6 -3 0 2 1 R -2 6 0 -1 -4 1 -1 -3 2 -2 -3 3 0 -4 0 0 -1 2 -4 -2 1 2 N 0 0 2 2 -4 1 1 0 2 -2 -3 1 -2 -3 0 1 0 -4 -2 -2 4 3 D 0 -1 2 4 -5 2 3 1 1 -2 -4 0 -3 -6 -1 0 0 -7 -4 -2 5 4 C -2 -4 -4 -5 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8 0 -2 -3 -4 Q 0 1 1 2 -5 4 2 -1 3 -2 -2 1 -1 -5 0 -1 -1 -5 -4 -2 3 5 E 0 -1 1 3 -5 2 4 0 1 -2 -3 0 -2 -5 -1 0 0 -7 -4 -2 4 5 G 1 -3 0 1 -3 -1 0 5 -2 -3 -4 -2 -3 -5 0 1 0 -7 -5 -1 2 1 H -1 2 2 1 -3 3 1 -2 6 -2 -2 0 -2 -2 0 -1 -1 -3 0 -2 3 3 I -1 -2 -2 -2 -2 -2 -2 -3 -2 5 2 -2 2 1 -2 -1 0 -5 -1 4 -1 -1 L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 -3 4 2 -3 -3 -2 -2 -1 2 -2 -1 K -1 3 1 0 -5 1 0 -2 0 -2 -3 5 0 -5 -1 0 0 -3 -4 -2 2 2 M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 0 -2 -2 -1 -4 -2 2 -1 0 F -3 -4 -3 -6 -4 -5 -5 -5 -2 1 2 -5 0 9 -5 -3 -3 0 7 -1 -3 -4 P 1 0 0 -1 -3 0 -1 0 0 -2 -3 -1 -2 -5 6 1 0 -6 -5 -1 1 1 S 1 0 1 0 0 -1 0 1 -1 -1 -3 0 -2 -3 1 2 1 -2 -3 -1 2 1 T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 3 -5 -3 0 2 1 W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -6 -2 -5 17 0 -6 -4 -4 Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 -2 -2 -3 V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4 0 0 B 2 1 4 5 -3 3 4 2 3 -1 -2 2 -1 -3 1 2 2 -4 -2 0 6 5 Z 1 2 3 4 -4 5 5 1 3 -1 -1 2 0 -4 1 1 1 -4 -3 0 5 6
F
PAM-DistanzmatrixPAM1 (empirisch):
1% eine AS in eine andere
99% keine Veränderung
7
YF
9
PAM250:
15% Phe -> Tyr
32% keine Veränderung
PAM125 =>Bsp.: PAM1:
0,21% Phe -> Tyr
99,46% keine Veränderung
Phe -> Tyr: 0,15Dividiert durch dieFrequenz im Datensatz=> rel. Austauschfrequenz
0,15/0,04=3,75Log=0,57X10=5,7
Tyr -> Phe :0,2/0,03=6,7Log=0,83X10=8,3
Mittelwert (5,7+8,3)/2=7
C
-4
5
9
Transmembran-Proteine
Evolutionsmodelle
Globuläre Proteine
K
R N
K
10
Sequenzmatrizes
BLOSUM => aus BlöckenPAM => empirischGonnet => "moderne" PAMJTT => "moderne" PAMVT => WahrscheinlichkeitsmodellWAG => Wahrscheinlichkeitsmodell
Stammbaumberechnung
Multiples Alignment
Auswahl der Methode
Algorithmus
Ergebnisüberprüfung
Sequenzen

6
11
Stammbaumerstellung1.Matrix-orientierte Methoden• UPGMA (Unweighted Pair-Group Method with Arithmetric Means)• Neighbor-joining• Minimal Evolution (least squares)
=> Sequenzen werden in Distanzmatrix konvertiert(„1 Information“ pro Sequenz)
2. Charakter-orientierte Methoden• Parsimony• Maximum Likelihood etc.
=> jede Position wird als informative Einheit betrachtet
12
Datentypen
Sequenz 1 0.000 0.236 0.621 0.702 1.510Sequenz 2 0.000 0.599 0.672 1.482Sequenz 3 0.000 0.112 1.561Sequenz 4 0.000 1.425Sequenz 5 0.000
Sequenz 1 0.000 0.236 0.621 0.702 1.510Sequenz 2 0.000 0.599 0.672 1.482Sequenz 3 0.000 0.112 1.561Sequenz 4 0.000 1.425Sequenz 5 0.000
Sequenz 1 TATAAGCATGACTAGTAAGCTTAGCAATSequenz 2 TAT---CATGACTGGTAACCTCAACAATSequenz 3 TAT---CATGACTAGCAGGCTTAACATT Sequenz 4 TGTTGCCACGATTAGCTACCATAGCGAT Sequenz 5 CGTAGCTATGACCAACGGGCACAGCGAT
Sequenz 1 TATAAGCATGACTAGTAAGCTTAGCAATSequenz 2 TAT---CATGACTGGTAACCTCAACAATSequenz 3 TAT---CATGACTAGCAGGCTTAACATT Sequenz 4 TGTTGCCACGATTAGCTACCATAGCGAT Sequenz 5 CGTAGCTATGACCAACGGGCACAGCGAT
Charaktere
Distanzen

7
13
Matrix-orientierte Methoden
Zwei Schritte: 1.Berechnen der korrigierten
paarweisen Abstände zwischen den Sequenzen=> Evolutionsmodelle!
DNADNA: JC, K2P ...ProteinProtein: PAM, BLOSUM...
2. Erstellen eines Stammbaums anhand dieser Abstandsdaten
14
Distanzmatrix
Sequenz 1 0,000 0,236 0,621 0,702 1,510Sequenz 2 0,000 0,599 0,672 1,482Sequenz 3 0,000 0,112 1,561Sequenz 4 0,000 1,425Sequenz 5 0,000
Sequenz 1 0,000 0,236 0,621 0,702 1,510Sequenz 2 0,000 0,599 0,672 1,482Sequenz 3 0,000 0,112 1,561Sequenz 4 0,000 1,425Sequenz 5 0,000
Ausgedrückt i.d.R. als Mutationen pro PositionAbstand kann > 1 werden!Bsp. Jukes-Cantor: p = 0.6 => K = 1.21
Berechnen des paarweisen Abstands
⎟⎠⎞
⎜⎝⎛ −−= pK
341ln
43
8
15
Stammbaumerstellung
Wie kommen wir von einer Distanzmatrix zu einem Stammbaum?=> Algorithmus berechnet aus den Distanzen den "besten" Stammbaum.Sequenzen selbst werden nicht mehr berücksichtigt.
16
A B C D OTU A 0 6 10 18 OTU B 0 12 20OTU C 0 19OTU D 0
A B C D OTU A 0 6 10 18 OTU B 0 12 20OTU C 0 19OTU D 0
A/B C D OTU A/B 0 11 19OTU C 0 19OTU D 0
A/B C D OTU A/B 0 11 19OTU C 0 19OTU D 0
3 A
3 B
6
A/B
C
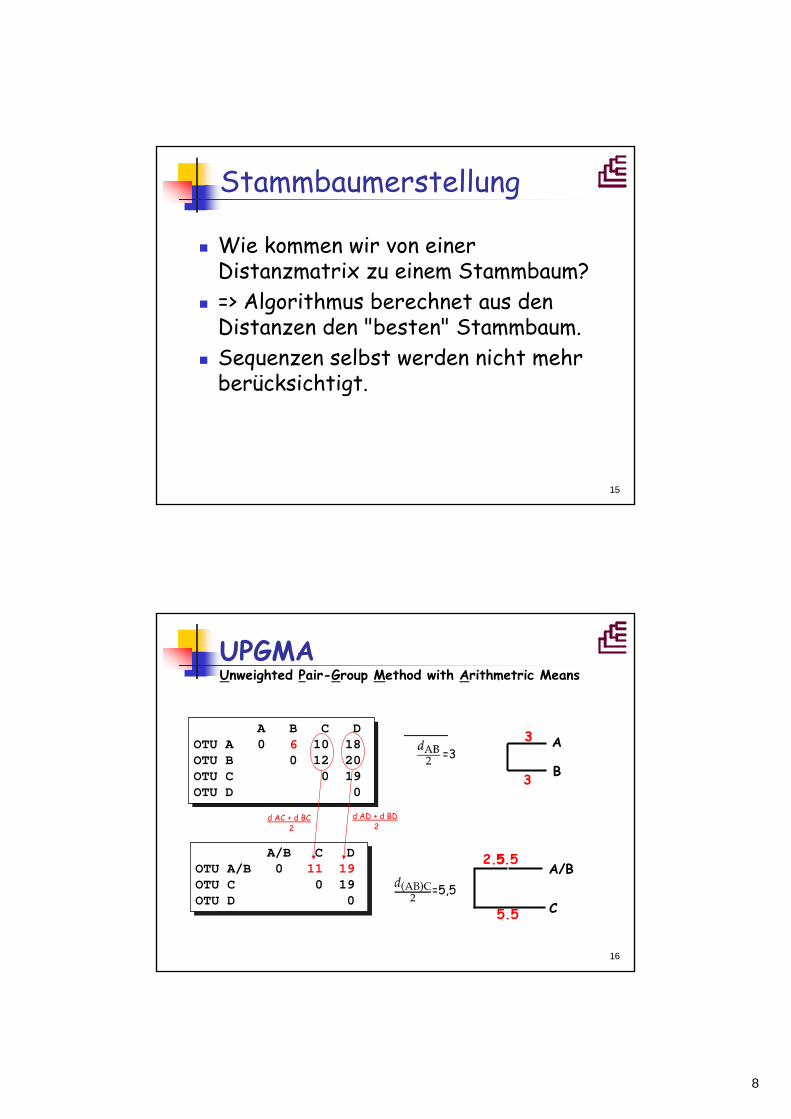
5.5
5.5
2.5
=3
d AD + d BD2
=5,5
UPGMA Unweighted Pair-Group Method with Arithmetric Means
d AC + d BC2
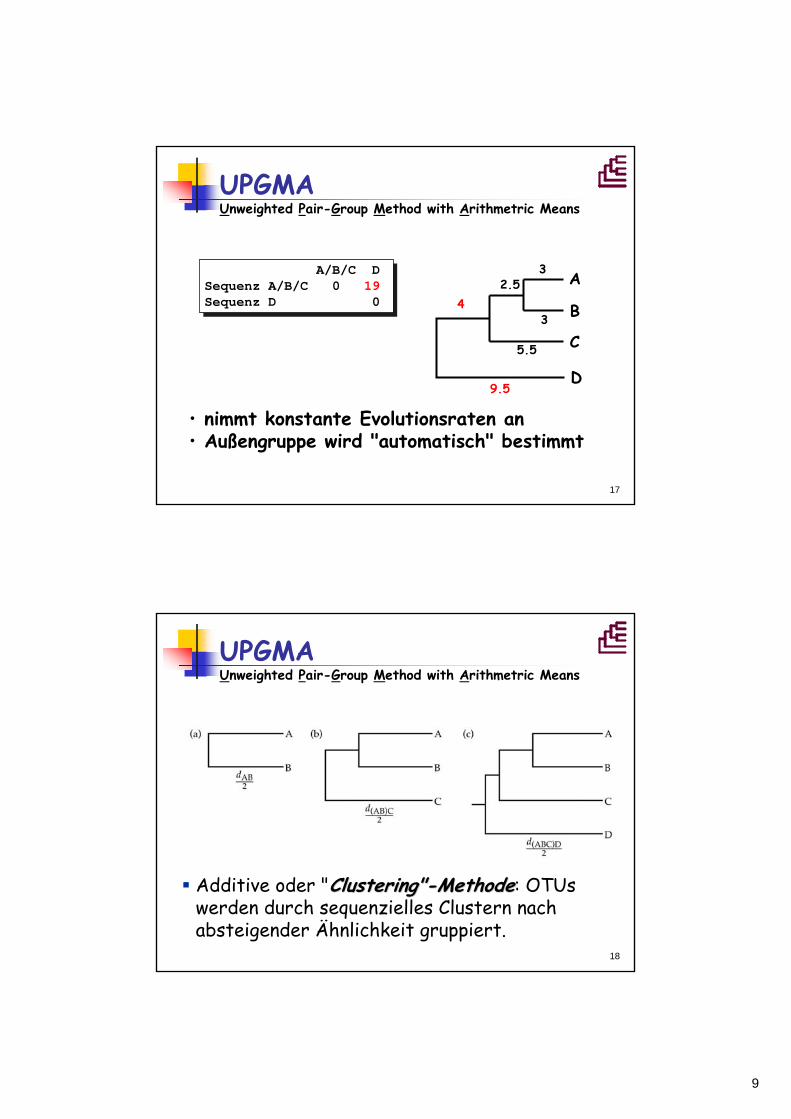
9
17
A/B/C D Sequenz A/B/C 0 19Sequenz D 0
A/B/C D Sequenz A/B/C 0 19Sequenz D 0
A
3 B
2.5
5.5 C
D
4
9.5
3
• nimmt konstante Evolutionsraten an• Außengruppe wird "automatisch" bestimmt
UPGMA Unweighted Pair-Group Method with Arithmetric Means
18
Additive oder "Clustering"Clustering"--MethodeMethode: OTUswerden durch sequenzielles Clustern nach absteigender Ähnlichkeit gruppiert.
UPGMA Unweighted Pair-Group Method with Arithmetric Means
10
19
A B C D OTU A 0 6 10 18 OTU B 0 12 20OTU C 0 19OTU D 0
A B C D OTU A 0 6 10 18 OTU B 0 12 20OTU C 0 19OTU D 0
A B C D OTU A 0 6 11 19 OTU B 0 11 19OTU C 0 19OTU D 0
A B C D OTU A 0 6 11 19 OTU B 0 11 19OTU C 0 19OTU D 0
A
3 B
2.5
5.5 C
D
4
9.5
3
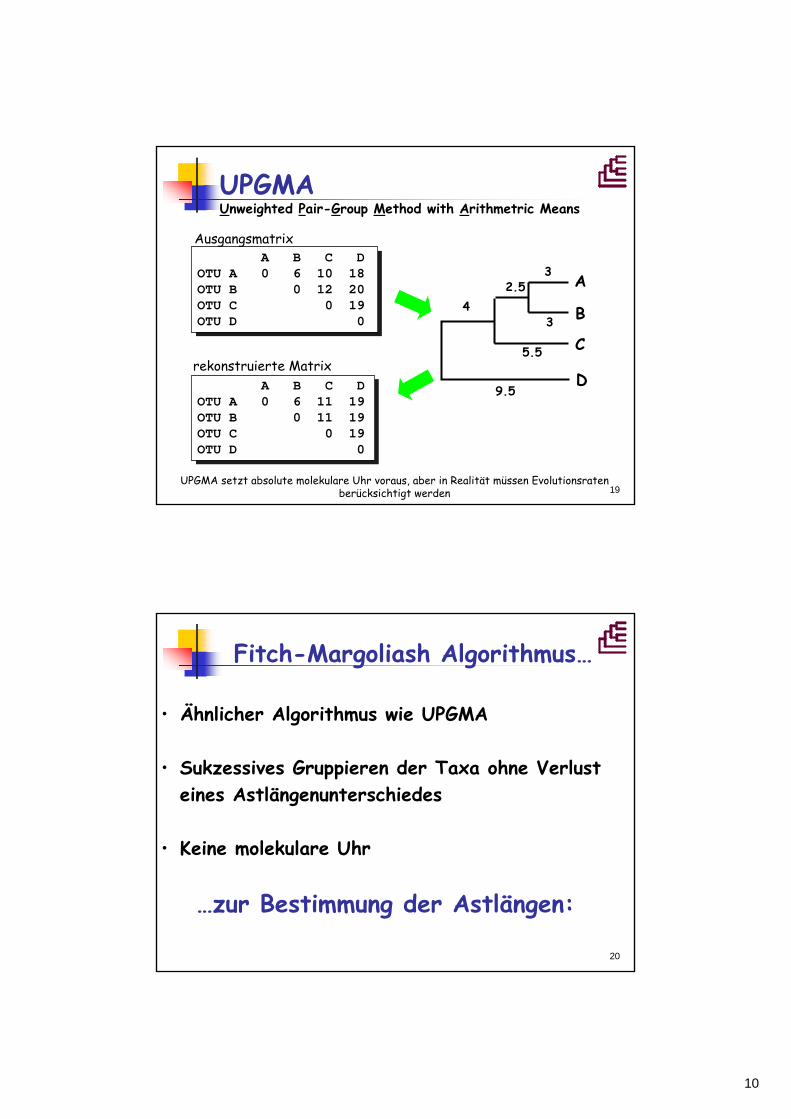
Ausgangsmatrix
rekonstruierte Matrix
UPGMA Unweighted Pair-Group Method with Arithmetric Means
UPGMA setzt absolute molekulare Uhr voraus, aber in Realität müssen Evolutionsraten berücksichtigt werden
20
Fitch-Margoliash Algorithmus…
…zur Bestimmung der Astlängen:
• Ähnlicher Algorithmus wie UPGMA
• Sukzessives Gruppieren der Taxa ohne Verlust eines Astlängenunterschiedes
• Keine molekulare Uhr
11
21
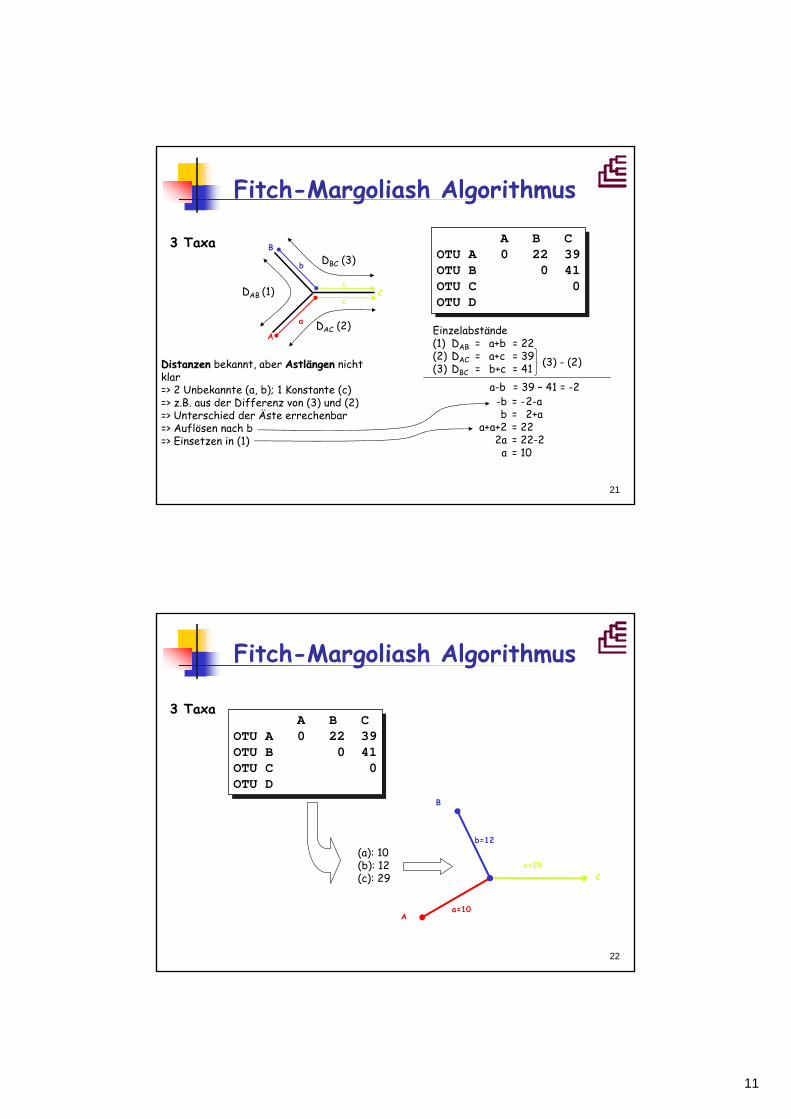
Fitch-Margoliash Algorithmus
3 Taxa B
C
A
c
A B COTU A 0 22 39OTU B 0 41OTU C 0OTU D
A B COTU A 0 22 39OTU B 0 41OTU C 0OTU D
a
c
b
DAB (1)
DAC (2)
DBC (3)
Einzelabstände(1) DAB = a+b = 22(2) DAC = a+c = 39(3) DBC = b+c = 41DistanzenDistanzen bekannt, aber AstlAstläängenngen nicht
klar=> 2 Unbekannte (a, b); 1 Konstante (c)=> z.B. aus der Differenz von (3) und (2)=> Unterschied der Äste errechenbar=> Auflösen nach b=> Einsetzen in (1)
(3) - (2)
a-b = 39 – 41 = -2-b = -2-ab = 2+a
a+a+2 = 222a = 22-2
a = 10
22
Fitch-Margoliash Algorithmus
3 TaxaA B C
OTU A 0 22 39OTU B 0 41OTU C 0OTU D
A B COTU A 0 22 39OTU B 0 41OTU C 0OTU D
B
C
A
c=29
a=10
b=12(a): 10(b): 12(c): 29

12
23
Fitch-Margoliash Algorithmus
5 Taxa A B C D EOTU A 0 22 39 39 41OTU B 0 41 41 43OTU C 0 18 20OTU D 10OTU E 0
A B C D EOTU A 0 22 39 39 41OTU B 0 41 41 43OTU C 0 18 20OTU D 10OTU E 0
B C
D
E
A
bc
de
af
D und E geringste Distanz
Durschnittswert aus ABC
ABC bilden 3.Taxon
Berechung der Astlängen D und E
Neue Matrix
g
A B C D/EOTU A 0 22 39 40OTU B 0 41 42OTU C 0 19OTU D/E 0
A B C D/EOTU A 0 22 39 40OTU B 0 41 42OTU C 0 19OTU D/E 0
:2
usw…
24
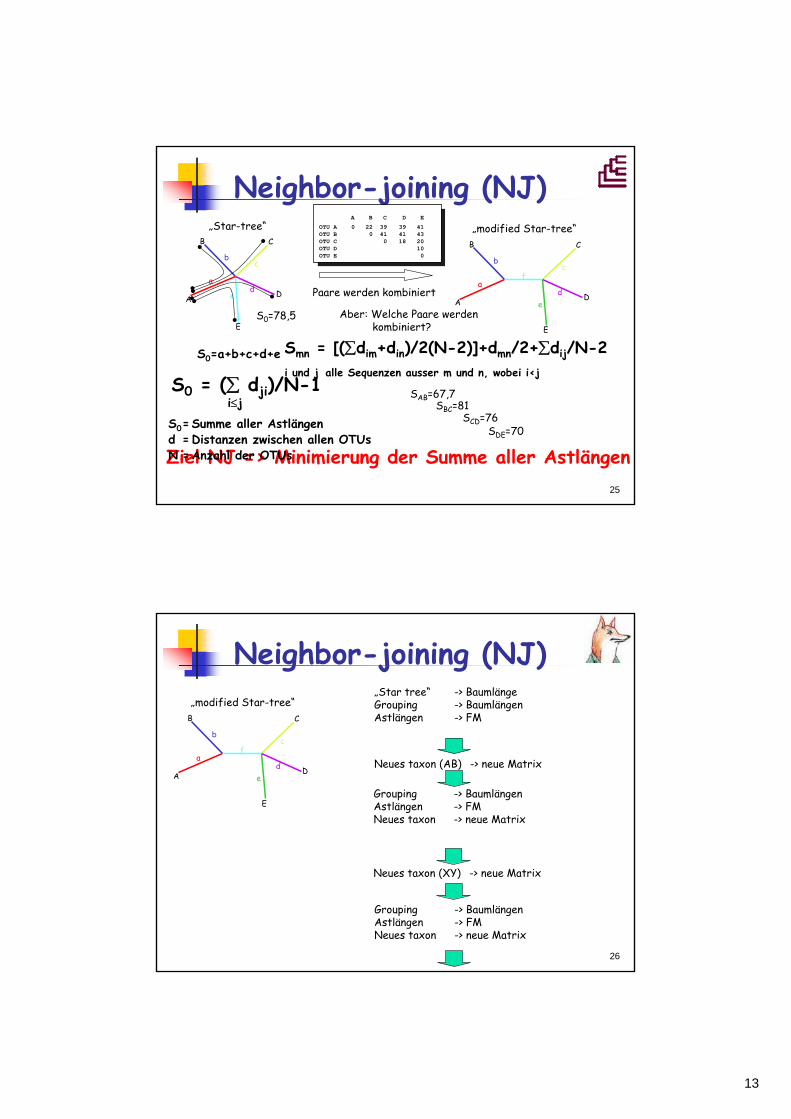
Neighbor-joining (NJ)Saitou und Nai, 1987
• Ähnlicher Algorithmus wie UPGMA bzw. FM• Sukzessives Gruppieren der Taxa ohne Verlust eines
Astlängenunterschiedes• „unrooted tree“ oder ungewurzelter Baum (radial)• Keine molekulare Uhr• Besonders sinnvoll, wenn Evoltuionsraten der
verschiedenen Linien unterschiedlich sind
Minimierung der Gesamt-Astlängen des Baums=> Stammbaum wird aufgelöst
13
25
Neighbor-joining (NJ)
Ziel NJ => Minimierung der Summe aller Astlängen
B C
D
E
A
bc
de
a
S0 = (∑ dji)/N-1i≤j
S0=Summe aller Astlängend =Distanzen zwischen allen OTUsN =Anzahl der OTUs
S0=a+b+c+d+e
„Star-tree“B C
D
E
A
bc
de
af
„modified Star-tree“
Paare werden kombiniert
Smn = [(∑dim+din)/2(N-2)]+dmn/2+∑dij/N-2i und j alle Sequenzen ausser m und n, wobei i<j
A B C D E
OTU A 0 22 39 39 41OTU B 0 41 41 43OTU C 0 18 20OTU D 10OTU E 0
A B C D E
OTU A 0 22 39 39 41OTU B 0 41 41 43OTU C 0 18 20OTU D 10OTU E 0
SAB=67,7
S0=78,5
SBC=81SCD=76
SDE=70
Aber: Welche Paare werden kombiniert?
26
Grouping -> BaumlängenAstlängen -> FMNeues taxon -> neue Matrix
B C
D
E
A
bc
de
af
„modified Star-tree“„Star tree“ -> BaumlängeGrouping -> BaumlängenAstlängen -> FM
Neighbor-joining (NJ)
Neues taxon (AB) -> neue Matrix
Grouping -> BaumlängenAstlängen -> FMNeues taxon -> neue Matrix
Neues taxon (XY) -> neue Matrix
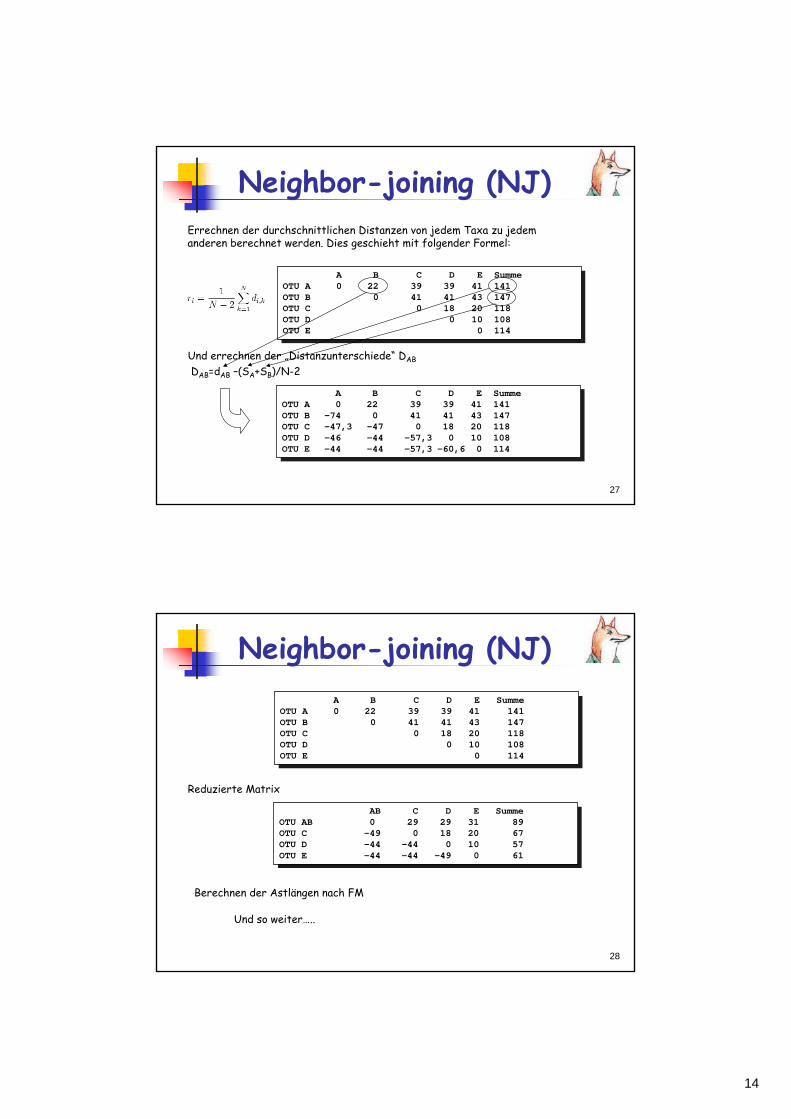
14
27
Neighbor-joining (NJ)
A B C D E SummeOTU A 0 22 39 39 41 141OTU B -74 0 41 41 43 147OTU C -47,3 -47 0 18 20 118OTU D -46 -44 -57,3 0 10 108OTU E -44 -44 -57,3 -60,6 0 114
A B C D E SummeOTU A 0 22 39 39 41 141OTU B -74 0 41 41 43 147OTU C -47,3 -47 0 18 20 118OTU D -46 -44 -57,3 0 10 108OTU E -44 -44 -57,3 -60,6 0 114
Errechnen der durchschnittlichen Distanzen von jedem Taxa zu jedem anderen berechnet werden. Dies geschieht mit folgender Formel:
A B C D E SummeOTU A 0 22 39 39 41 141OTU B 0 41 41 43 147OTU C 0 18 20 118OTU D 0 10 108OTU E 0 114
A B C D E SummeOTU A 0 22 39 39 41 141OTU B 0 41 41 43 147OTU C 0 18 20 118OTU D 0 10 108OTU E 0 114
Und errechnen der „Distanzunterschiede“ DAB
DAB=dAB –(SA+SB)/N-2
28
Neighbor-joining (NJ)
AB C D E SummeOTU AB 0 29 29 31 89OTU C -49 0 18 20 67OTU D -44 -44 0 10 57OTU E -44 -44 -49 0 61
AB C D E SummeOTU AB 0 29 29 31 89OTU C -49 0 18 20 67OTU D -44 -44 0 10 57OTU E -44 -44 -49 0 61
A B C D E SummeOTU A 0 22 39 39 41 141OTU B 0 41 41 43 147OTU C 0 18 20 118OTU D 0 10 108OTU E 0 114
A B C D E SummeOTU A 0 22 39 39 41 141OTU B 0 41 41 43 147OTU C 0 18 20 118OTU D 0 10 108OTU E 0 114
Reduzierte Matrix
Berechnen der Astlängen nach FM
Und so weiter…..
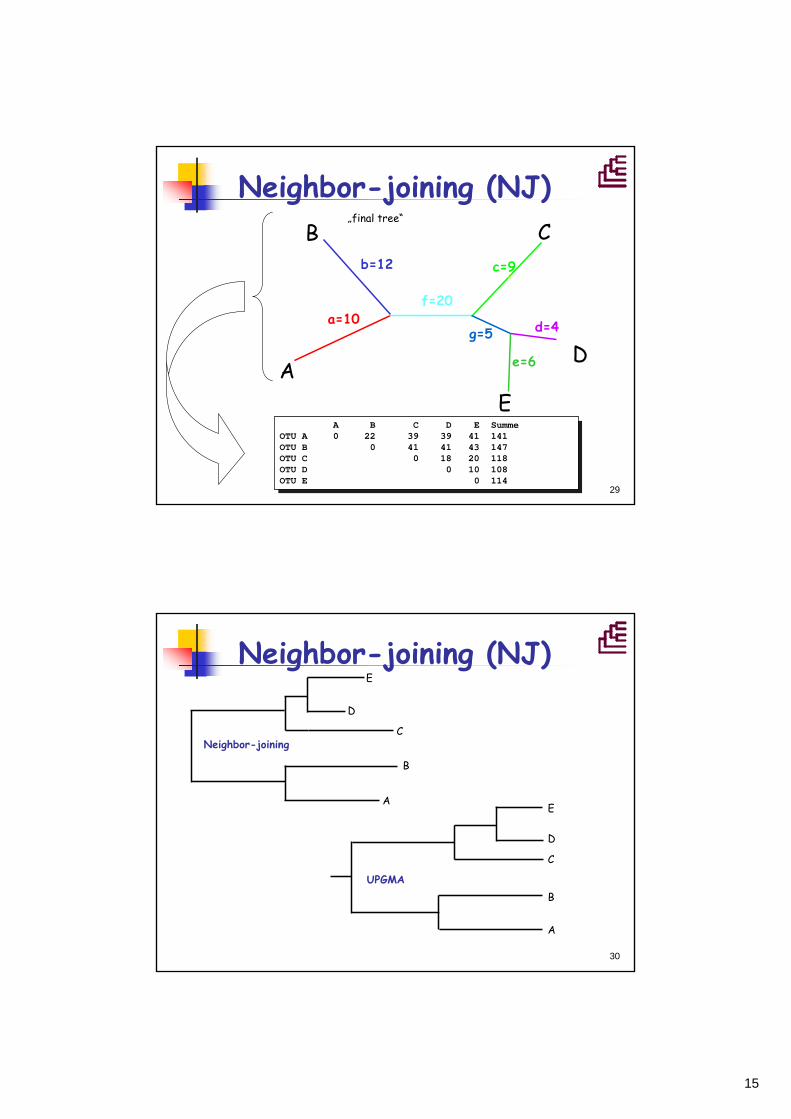
15
29
Neighbor-joining (NJ)B C
D
EA
b=12c
d=4
e=6
a=10f=20
„final tree“
g=5
c=9
A B C D E SummeOTU A 0 22 39 39 41 141OTU B 0 41 41 43 147OTU C 0 18 20 118OTU D 0 10 108OTU E 0 114
A B C D E SummeOTU A 0 22 39 39 41 141OTU B 0 41 41 43 147OTU C 0 18 20 118OTU D 0 10 108OTU E 0 114
30
D
E
C
B
A
D
E
C
B
A
Neighbor-joining
UPGMA
Neighbor-joining (NJ)
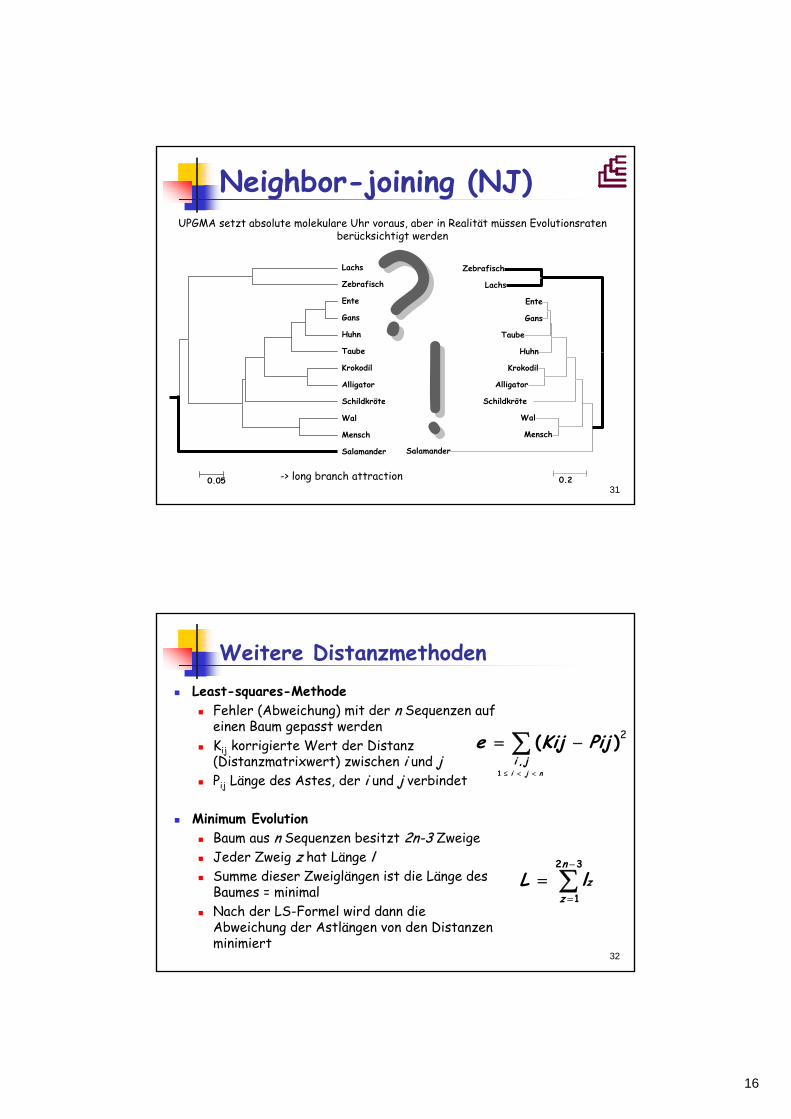
16
31
Neighbor-joining (NJ)UPGMA setzt absolute molekulare Uhr voraus, aber in Realität müssen Evolutionsraten
berücksichtigt werden
Lachs
Zebrafisch
Ente
Gans
Huhn
Taube
Krokodil
Alligator
Schildkröte
Wal
Mensch
Salamander
0.05
Zebrafisch
Lachs
Ente
Gans
Taube
Huhn
Krokodil
Alligator
Schildkröte
Wal
Mensch
Salamander
0.2
??-> long branch attraction
!!
32
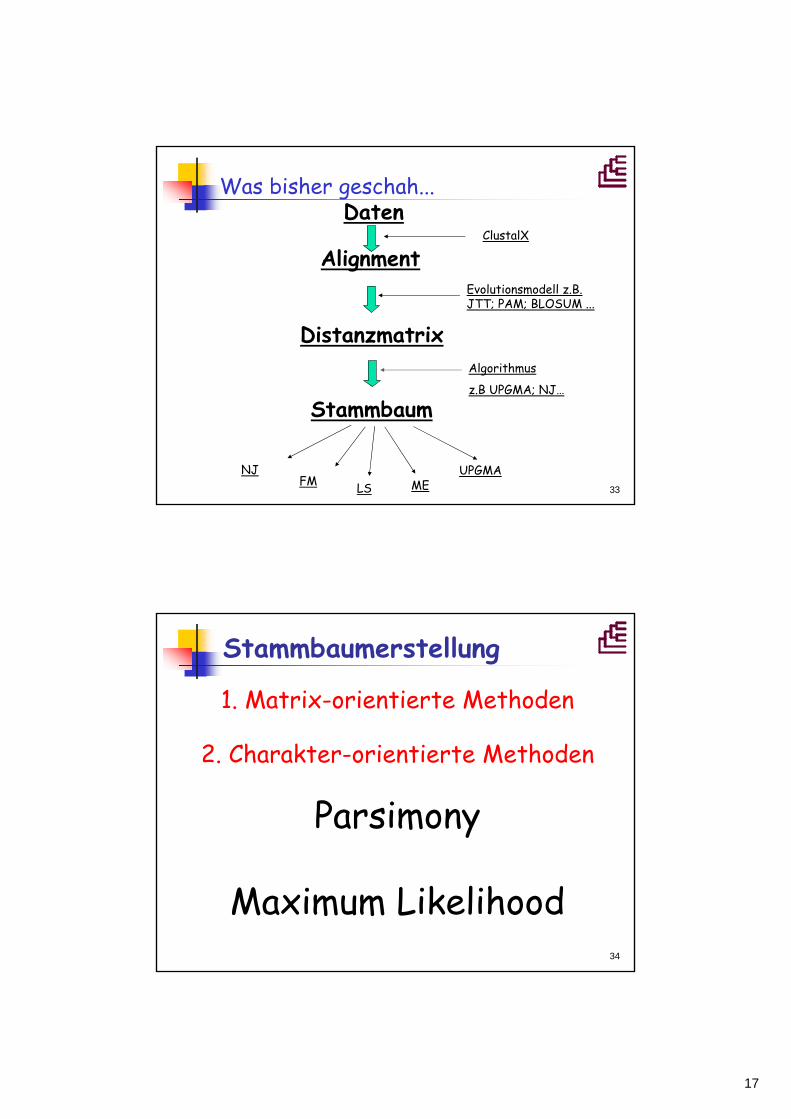
Weitere DistanzmethodenLeast-squares-Methode
Fehler (Abweichung) mit der n n Sequenzen auf einen Baum gepasst werdenKij korrigierte Wert der Distanz (Distanzmatrixwert) zwischen ii und jjPij Länge des Astes, der i i und j j verbindet
Minimum EvolutionBaum aus nn Sequenzen besitzt 2n2n--3 3 ZweigeJeder Zweig zz hat Länge llSumme dieser Zweiglängen ist die Länge des Baumes = minimalNach der LS-Formel wird dann die Abweichung der Astlängen von den Distanzen minimiert
∑−
=
=32
1
n
zzlL
∑ −=ji
PijKije,
)( 2
nji <<≤1
17
33
Was bisher geschah...
Alignment
Distanzmatrix
Evolutionsmodell z.B. JTT; PAM; BLOSUM ...
Stammbaum
Algorithmus
z.B UPGMA; NJ…
DatenClustalX
UPGMANJFM LS ME
34
Stammbaumerstellung
1. Matrix-orientierte Methoden
2. Charakter-orientierte Methoden
Parsimony
Maximum Likelihood

18
35
Charakter-orientierte Methoden
1. Maximum Parsimony (MP)2. Maximum Likelihood (ML)
Arbeiten direkt mit dem AlignmentExtrahieren mehr Information als Matrix-orientierte Methoden
36
Was sind Charaktere?
• kontinuierliche oder diskontinuierliche Eigenschaften.
• Nukleotide und Aminosäuren können als diskrete, diskontinuierliche Charaktere behandelt werden.
• Der phylogenetische Stammbaum wird anhand des Musters der Änderungen der Charaktere berechnet
1,2,3,4.... = kontinuierliche CharaktereA,T,G,C = diskontinuierliche Charaktere

19
37
Maximum Parsimony (MP)
WilliWilli HennigHennig19131913--19761976
• Methode des "maximalen Geizes" bzw. der "maximalen Sparsamkeit"
• Entwickelt für morphologische Charaktere
1950 „Grundzüge einer Theorie der phylogenetischen Systematik“,
38
Maximum Parsimony
• Annahme: Evolution ging den kürzesten Weg• “Ockham's razor” : "Pluralitas non est ponenda
sine neccesitate" ("Ohne Notwendigkeit sollkeine Vielfältigkeit hinzugefügt werden")
• =>minimalistische Ökonomieprinzipien• kürzester Stammbaum wird berechnet, d.h. der
die wenigsten evolutiven Schritten benötigt• "Schritte" = Änderungen von Charakteren
William of Ockham (1285-1349)
20
39
• Erklärung mit morphologischen Charakteren
• Gleiche Prinzipien sind für Sequenzen (Basenpaare, Aminosäuren) gültig
Maximum Parsimony
40
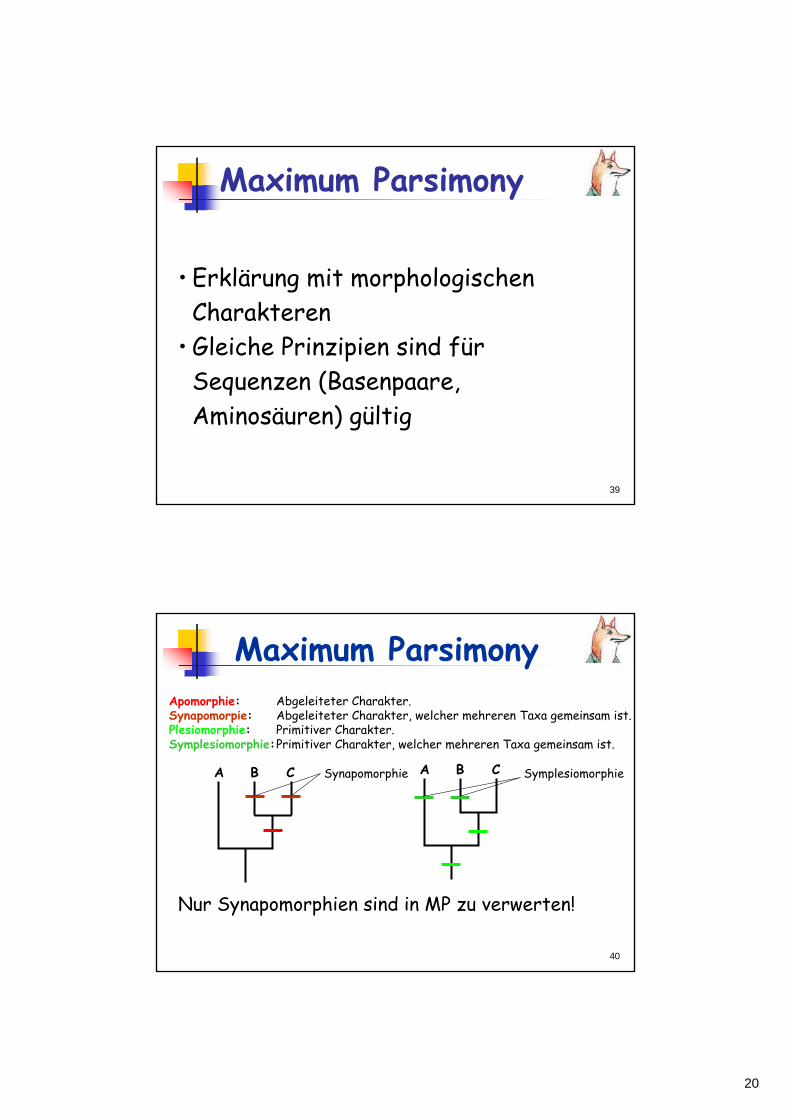
Apomorphie: Abgeleiteter Charakter. Synapomorpie: Abgeleiteter Charakter, welcher mehreren Taxa gemeinsam ist.Plesiomorphie: Primitiver Charakter. Symplesiomorphie:Primitiver Charakter, welcher mehreren Taxa gemeinsam ist.
Nur Synapomorphien sind in MP zu verwerten!
A B C Synapomorphie A B C Symplesiomorphie
Maximum Parsimony
21
41
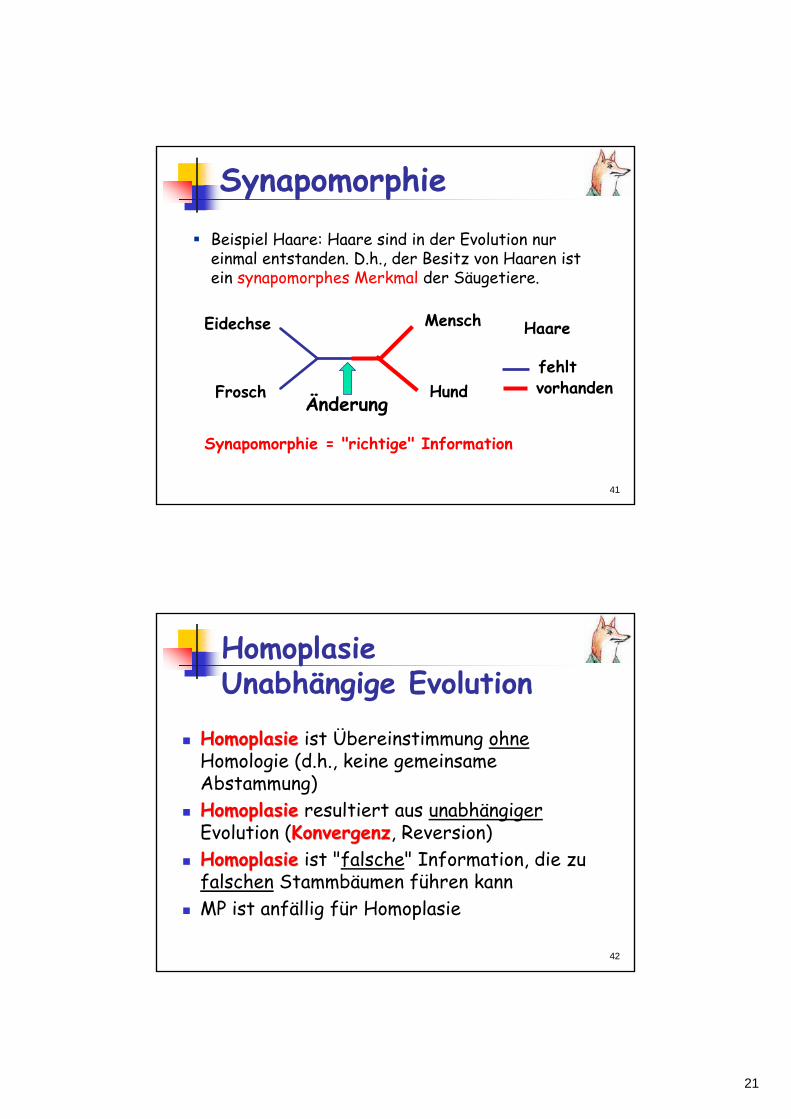
SynapomorphieBeispiel Haare: Haare sind in der Evolution nureinmal entstanden. D.h., der Besitz von Haaren istein synapomorphes Merkmal der Säugetiere.
Eidechse
Frosch
Mensch
Hund
Haare
fehltvorhanden
Änderung
Synapomorphie = "richtige" Information
42
HomoplasieUnabhängige Evolution
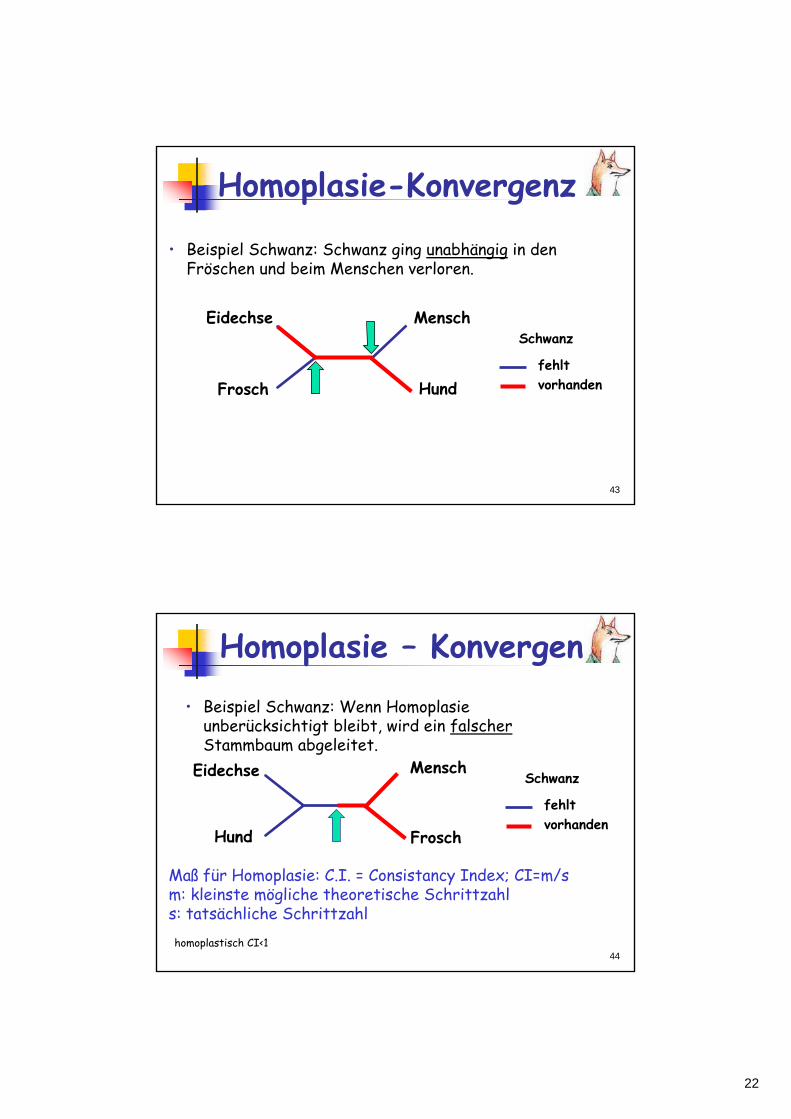
HomoplasieHomoplasie ist Übereinstimmung ohneHomologie (d.h., keine gemeinsame Abstammung)HomoplasieHomoplasie resultiert aus unabhängigerEvolution (KonvergenzKonvergenz, Reversion)HomoplasieHomoplasie ist "falsche" Information, die zu falschen Stammbäumen führen kannMP ist anfällig für Homoplasie
22
43
Homoplasie-Konvergenz
Schwanz
fehltvorhanden
• Beispiel Schwanz: Schwanz ging unabhängig in den Fröschen und beim Menschen verloren.
Eidechse
Frosch
Mensch
Hund
44
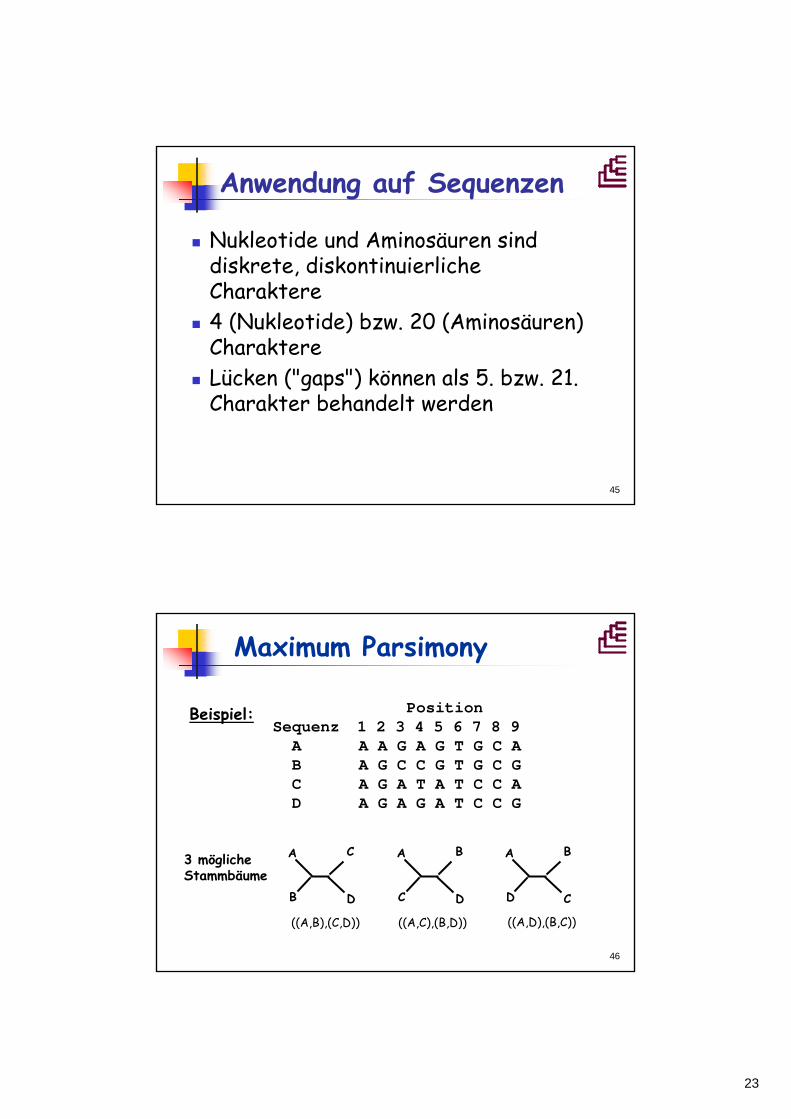
Homoplasie – Konvergenz
Schwanz
fehltvorhanden
• Beispiel Schwanz: Wenn Homoplasieunberücksichtigt bleibt, wird ein falscherStammbaum abgeleitet.
Eidechse
Frosch
Mensch
Hund
Maß für Homoplasie: C.I. = Consistancy Index; CI=m/sm: kleinste mögliche theoretische Schrittzahls: tatsächliche Schrittzahlhomoplastisch CI<1
23
45
Anwendung auf Sequenzen
Nukleotide und Aminosäuren sind diskrete, diskontinuierliche Charaktere4 (Nukleotide) bzw. 20 (Aminosäuren) CharaktereLücken ("gaps") können als 5. bzw. 21. Charakter behandelt werden
46
Maximum Parsimony
Position Sequenz 1 2 3 4 5 6 7 8 9 A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
Beispiel:
A
B
C
D
A
C
B
D
A
D
B
C
3 mögliche Stammbäume
((A,B),(C,D)) ((A,C),(B,D)) ((A,D),(B,C))
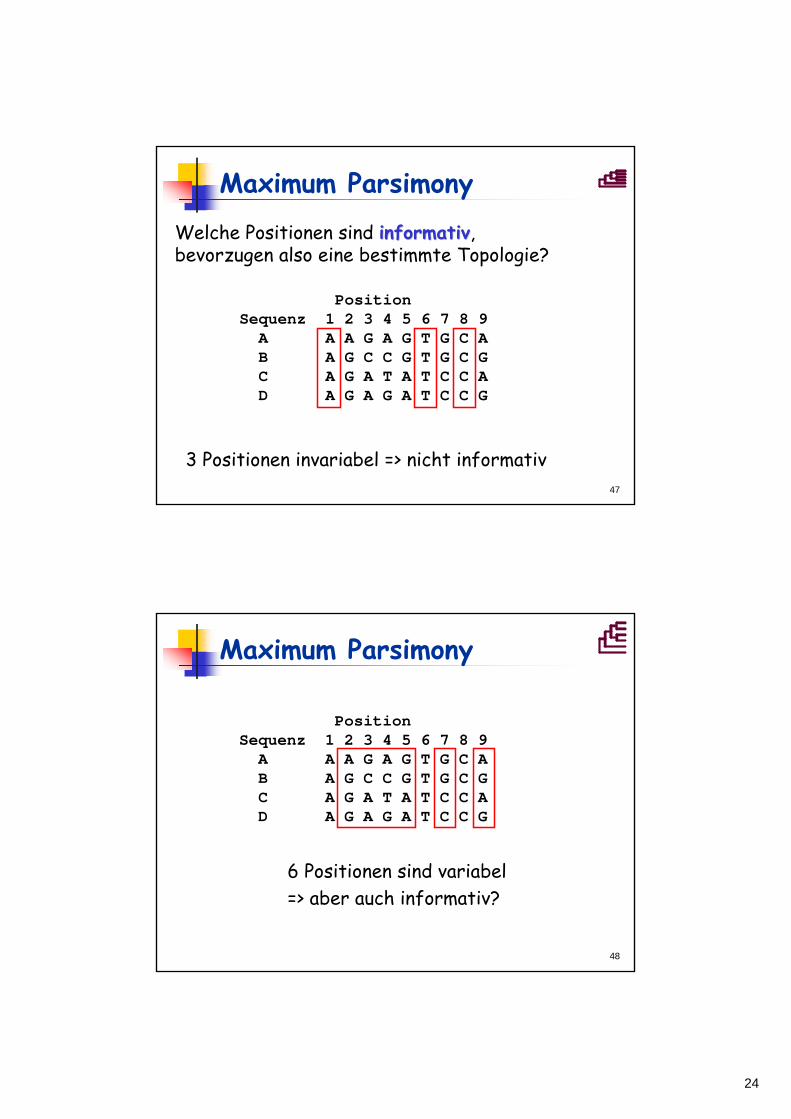
24
47
Position Sequenz 1 2 3 4 5 6 7 8 9 A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
3 Positionen invariabel => nicht informativ
Welche Positionen sind informativinformativ, bevorzugen also eine bestimmte Topologie?
Maximum Parsimony
48
Position Sequenz 1 2 3 4 5 6 7 8 9 A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
6 Positionen sind variabel=> aber auch informativ?
Maximum Parsimony

25
49
Position Sequenz 1 2 3 4 5 6 7 8 9 A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
3 Positionen sind zwar variabel, aber nicht informativ
Maximum Parsimony
50
Position Sequenz 1 2 3 4 5 6 7 8 9 A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
* * *
Welche Positionen sind aber nun informativ?
=> nur 3 von 9 Positionen sind informativ, d.h., favorisieren eine best. Topologie.
10 11- A - GC GC G*
=> Indels sind Charaktere!
Maximum Parsimony
26
51
Position 3:
((A,B),(C,D)) ((A,C),(B,D)) ((A,D),(B,C))
G
C
A
A
G
A
A
C
G
A
A
C
•• •
••
•A AAAAA
G
G
A
A
G
A
A
G
G
A
A
G
••
••
•G AAAAA
A
G
A
G
A
A
G
G
A
G
A
G•• ••
•A AAGAA
Position 5:
Position 9:
?
MaximumParsimony
Position Sequenz 1 2 3 4 5 6 7 8 9
A A A G A G T G C AB A G C C G T G C GC A G A T A T C C AD A G A G A T C C G
- + +
52
A
B
C
D
A
C
B
D
A
D
B
C
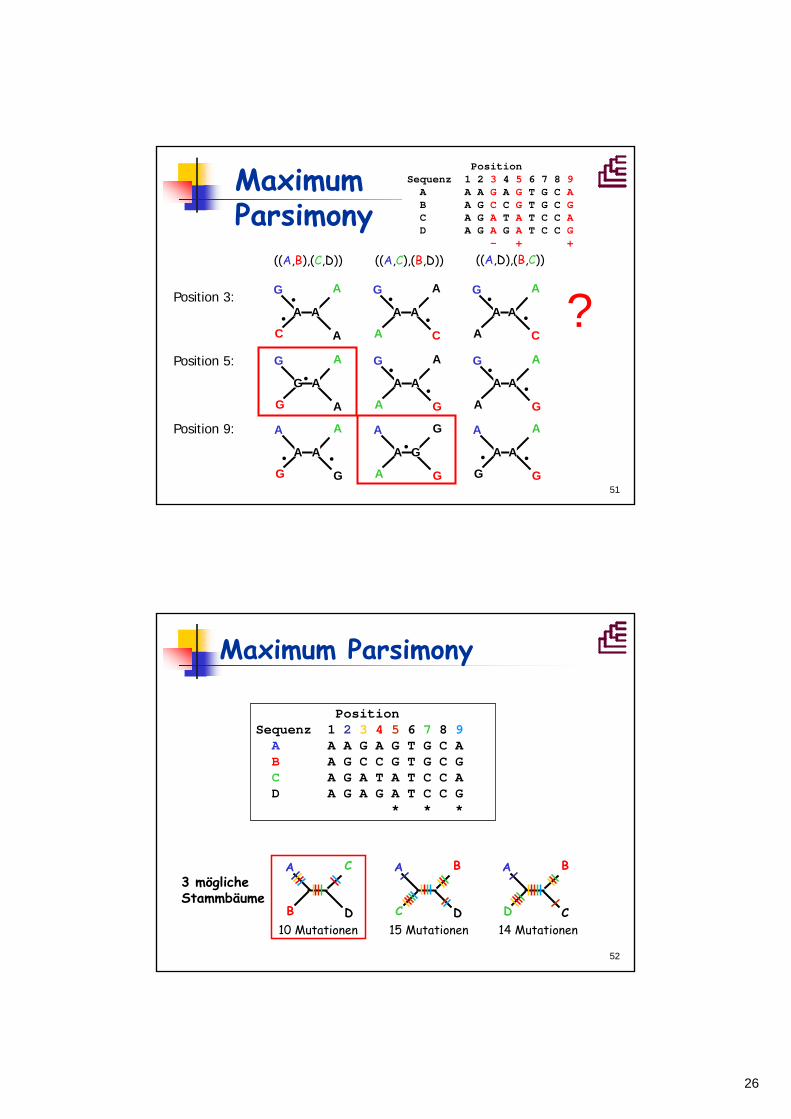
3 mögliche Stammbäume
10 Mutationen 15 Mutationen 14 Mutationen
Position Sequenz 1 2 3 4 5 6 7 8 9A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
* * *
Maximum Parsimony
27
53
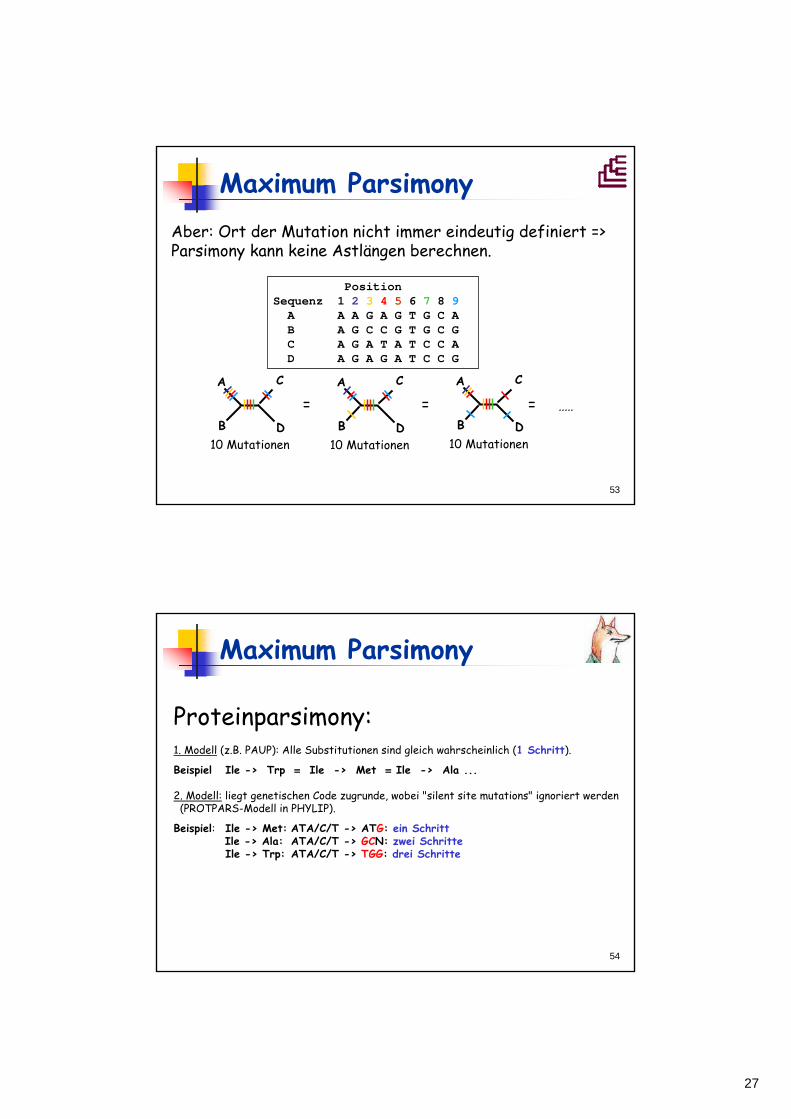
Position Sequenz 1 2 3 4 5 6 7 8 9A A A G A G T G C A B A G C C G T G C G C A G A T A T C C A D A G A G A T C C G
Aber: Ort der Mutation nicht immer eindeutig definiert => Parsimony kann keine Astlängen berechnen.
A
B
C
D10 Mutationen
A
B
C
D10 Mutationen
A
B
C
D10 Mutationen
= = = .....
Maximum Parsimony
54
Proteinparsimony:1. Modell (z.B. PAUP): Alle Substitutionen sind gleich wahrscheinlich (1 Schritt).
Beispiel Ile -> Trp ≡ Ile -> Met ≡ Ile -> Ala ...
2. Modell: liegt genetischen Code zugrunde, wobei "silent site mutations" ignoriert werden (PROTPARS-Modell in PHYLIP).
Beispiel: Ile -> Met: ATA/C/T -> ATG: ein SchrittIle -> Ala: ATA/C/T -> GCN: zwei SchritteIle -> Trp: ATA/C/T -> TGG: drei Schritte
Maximum Parsimony

28
55
Maximum Parsimony
Exhaustive = Alle Stammbäume werdenuntersucht, der beste Stammbaum wirderhalten (garantiert).
Branch-and-Bound = Einige Stammbäumewerden berechnet, bester Stammbaumgarantiert.
Heuristic = Einige Stammbäume werdenberechnet, bester Stammbaum nichtgarantiert.
56
Maximum Parsimony:Exhaustive Search
A
B C(1)Start: 3 beliebige Taxa
(2a)
A
B DC
A
BD C
A
B CD
(2b) (2c)
+ 4. Taxon (D) in jeder möglichen Position -> 3 Bäume
+ 5. Taxon (E) in jeder der fünf möglichen Positionen=> 15 Stammbäume etc.
E
E
EE
E
"Branch addition“

29
57
Problem: Anzahl der möglichen Stammbäume
=> bei > ~10 Sequenzenausführliche Suche allerStammbäume de factounmöglich
Maximum Parsimony:Exhaustive Search
Numberof OTUs
Number ofrooted trees
Number ofunrooted trees
2 1 13 3 14 15 35 105 156 954 1057 10395 9548 135135 103959 2027025 135135
10 34459425 2027025
58
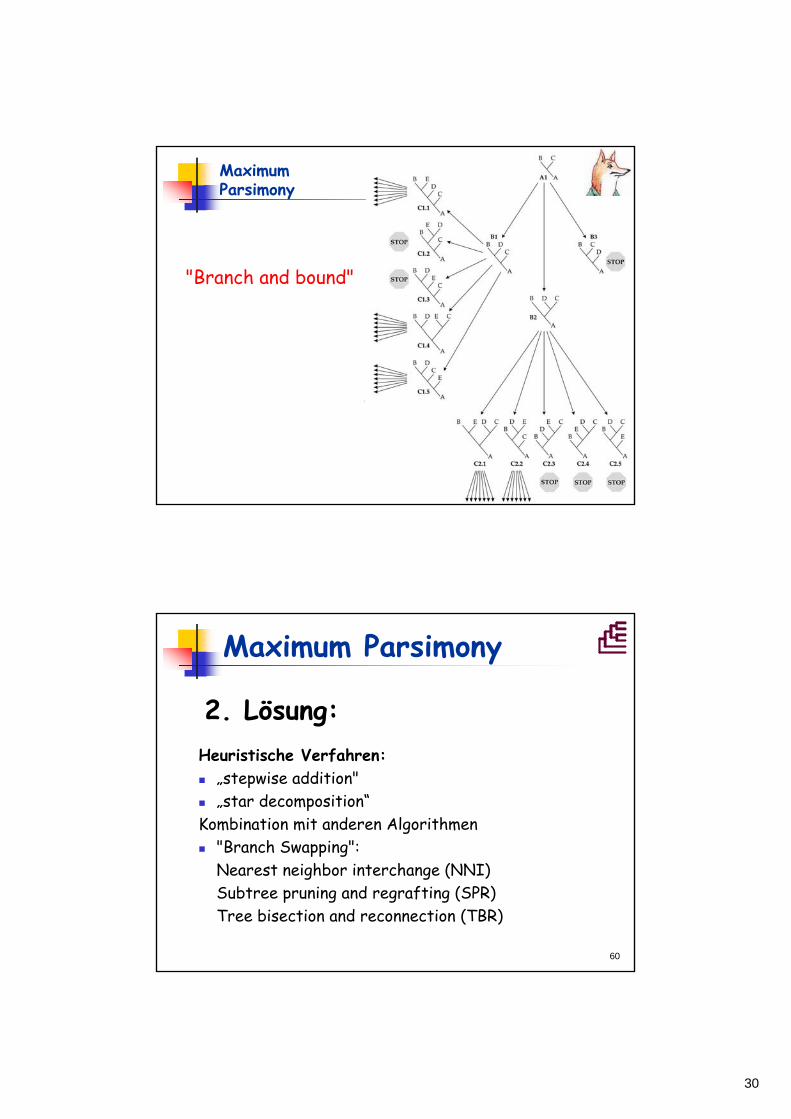
Maximum Parsimony
1. Lösung "Branch and bound":• Stammbaum wird mit schneller Methode (z.B. NJ) berechnet, die Anzahl der notwendigen Schritte (L) wird berechnet.
• => verwirft Gruppen von Bäumen, die nicht kürzer werden können als L.
• Kann für Problemlösungen mit < ~ 20 Taxa verwendet werden.
30
59
MaximumParsimony
"Branch and bound"
60
Maximum Parsimony
2. Lösung:Heuristische Verfahren:
„stepwise addition" „star decomposition“
Kombination mit anderen Algorithmen"Branch Swapping":
Nearest neighbor interchange (NNI) Subtree pruning and regrafting (SPR) Tree bisection and reconnection (TBR)
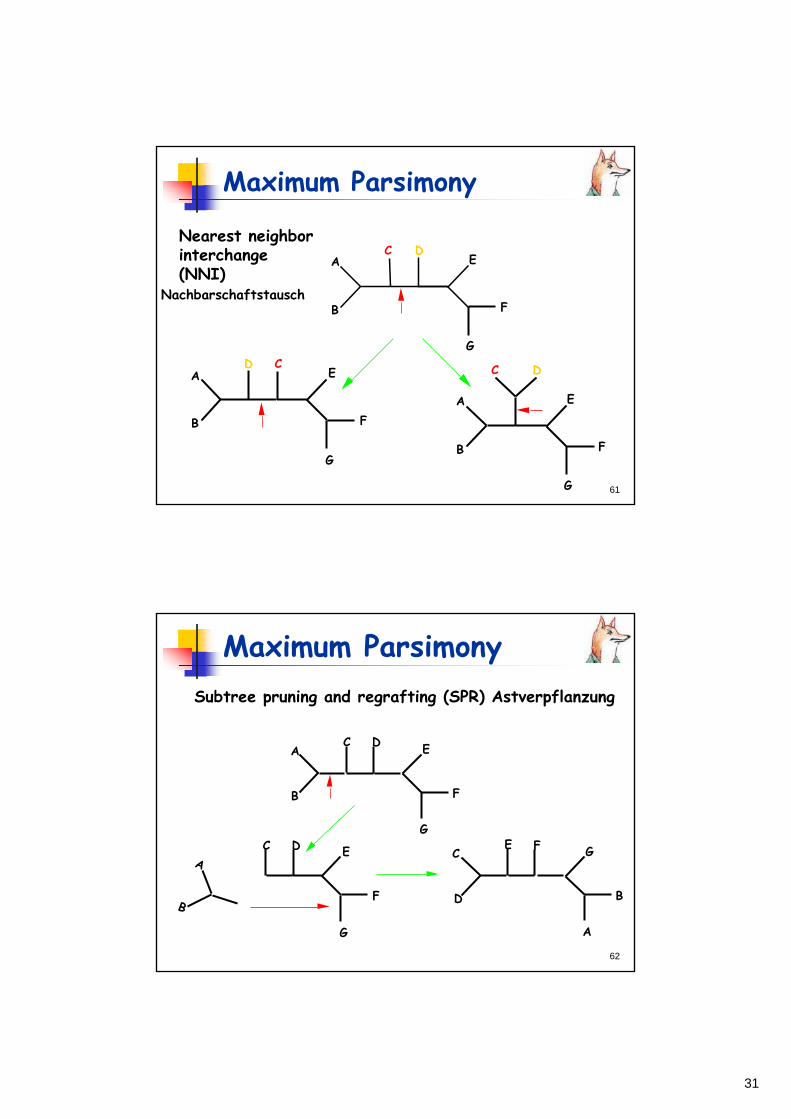
31
61
Maximum Parsimony
Nearest neighborinterchange(NNI)
Nachbarschaftstausch
A
B
C DE
F
G
A
B
D CE
F
G
A
B
C D
E
F
G
62
Maximum ParsimonySubtree pruning and regrafting (SPR) Astverpflanzung
A
B
C D E
F
G
A
B
C D E
F
G
C
D
G
B
A
E F
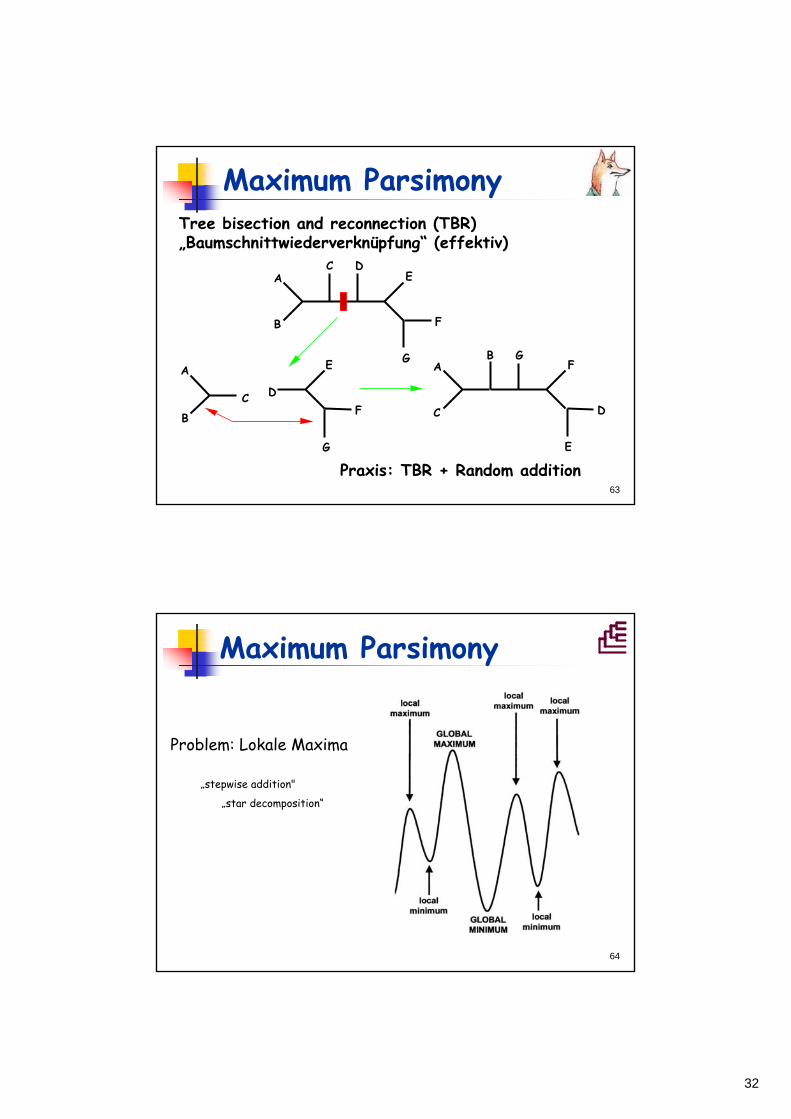
32
63
Maximum ParsimonyTree bisection and reconnection (TBR) „Baumschnittwiederverknüpfung“ (effektiv)
A
B
C DE
F
GA
B
C D
E
F
G
A
C
F
D
E
B G
Praxis: TBR + Random addition
64
Maximum Parsimony
Problem: Lokale Maxima
„stepwise addition"
„star decomposition“

33
65
Maximum Parsimony
einfachohne konkretes EvolutionsmodellErrechnung ancestraler Positionenfunktioniert gut mit konsistenten Datensätzen
Vorteile:
empfindlich gegen Homoplasien (Konvergenz)empfindlich gegen "Long Branch Attraction"Astlängen werden unterschätztkein Evolutionsmodell möglich!
Nachteile:
66
Charakter-orientierte Methoden
1. Maximum Parsimony (MP)
2. Maximum Likelihood (ML)

34
67
Charakter-orientierte Methoden
Maximum Likelihood (ML)
Eigenschaften:Annäherung an wahre Parameter bei
zunehmender Datenmenge(KonsistenzKonsistenz) und minimalerStreuung (VarianzVarianz) um den tatsächlichen Wert (EffizienzEffizienz)
68
Maximum LikelihoodL = P(data|hypothesis)
• Wahrscheinlichkeit der beobachteten Daten(Sequenzen!) im Lichte der Hypothese(Stammbaum).
• d.h, es wird der Stammbaum errechnet, der die beobachteten Daten (also die aligniertenSequenzen) am besten (unter der Annahme des Modells) erklärt.
• Likelihood veranschaulicht das Verhältniss von verschiedenen Hypothesen

35
69
Maximum Likelihood
Probability (P) = Wahrscheinlichkeit
Wahrscheinlichkeiten summieren sich stets auf 1 auf:Wie wahrscheinlich ist es, dass ich eine 6 würfele? Antwort: 1/6. Wie wahrscheinlich ist es, dass ich keine 6 würfele? Antwort 5/6. => 1/6 + 5/6 =1.
Likelihood (L) ≠ Wahrscheinlichkeit (P)
Für "Likelihood"-Werte summieren sich nicht auf 1 auf:=> Wie wahrscheinlich ist meine Hypothese unter dem gegebenen Randbedingungen?
70
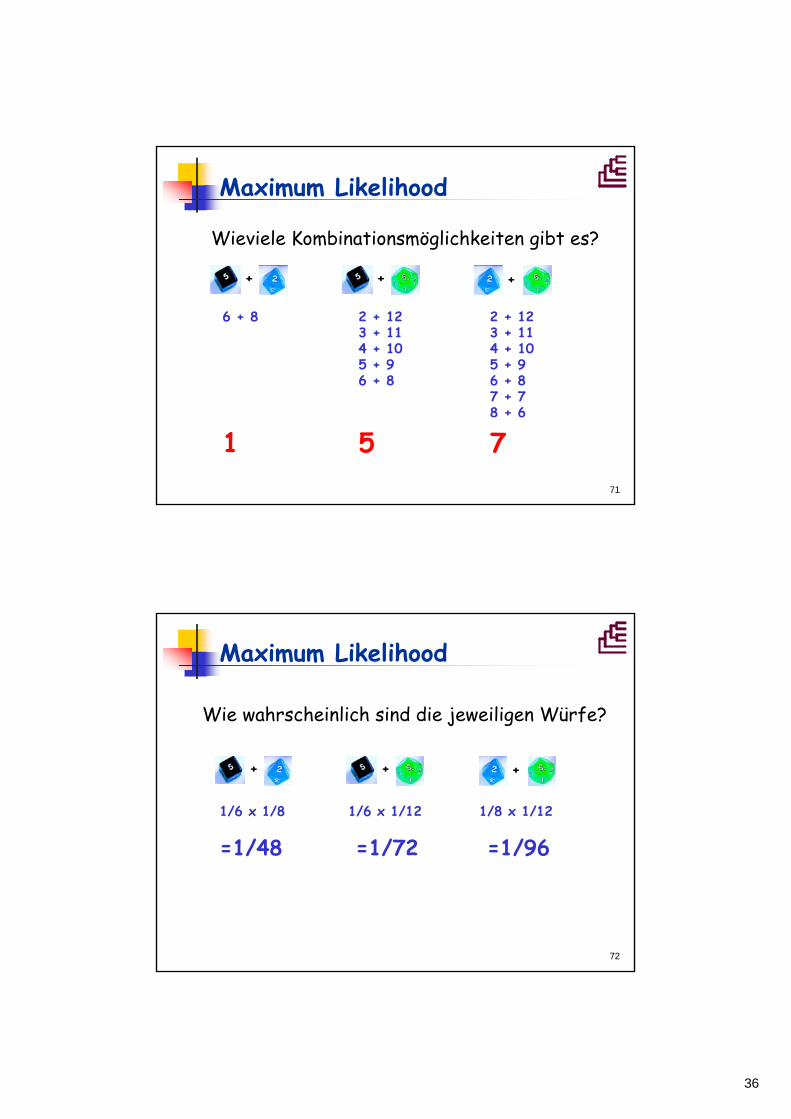
Maximum Likelihood
Beispiel: 3 Würfel
Würfel mit 6 Seiten
Würfel mit 12 Seiten
Würfel mit 8 Seiten
Gegeben: Eine Person würfelt mit zwei Würfeln die Summe "14".
Frage: Welches Würfelpaar ist mit der größten Wahrscheinlichkeit für dieses Ergebnis verantwortlich?
Equivalent: Welcher Stammbaum hat die gegebenen Sequenzen verantwortlich?
36
71
Maximum Likelihood
+ + +
6 + 8 2 + 12 2 + 123 + 11 3 + 114 + 10 4 + 105 + 9 5 + 96 + 8 6 + 8
7 + 78 + 6
1 5 7
Wieviele Kombinationsmöglichkeiten gibt es?
72
Maximum Likelihood
+ + +
1/6 x 1/8 1/6 x 1/12 1/8 x 1/12
=1/48 =1/72 =1/96
Wie wahrscheinlich sind die jeweiligen Würfe?

37
73
Maximum Likelihood
+ + +
1 x 1/48 5 x 1/72 7 x 1/96
Multiplikation der Einzelwahrscheinlichkeiten
0.0208 0.0694 0.0729maximum likelihood
74
Maximum Likelihood
• benötigt ein explizites Evolutionsmodell• Parameter werden aus Daten + Modell
errechnet.• Explizite Verbindung Daten + Modell +
Stammbaum.• aber: schlechtes Modell => schlechter
Stammbaum• Alternative Stammbäume lassen sich testen
=> keine Methode extrahiert mehr Information aus den Daten; aber: sehr rechenintensiv
38
75
Maximum Likelihood
•Evolutionsmodell:
Für DNA-Sequenzen:=> JC, K2P, F81, HKY, REVFür Protein-Sequenzen:=> PAM, BLOSUM, JTT, WAG ...
76
Maximum Likelihood
Seq1 CGAGACSeq2 AGCGACSeq3 AGATTASeq4 GGATAG
1
2
3
4Frage: Wie hoch ist die Wahrscheinlichkeit, daßder Stammbaum A für die Daten (Sequenzen) unter dem gegebenen Modell verantwortlich ist?
A
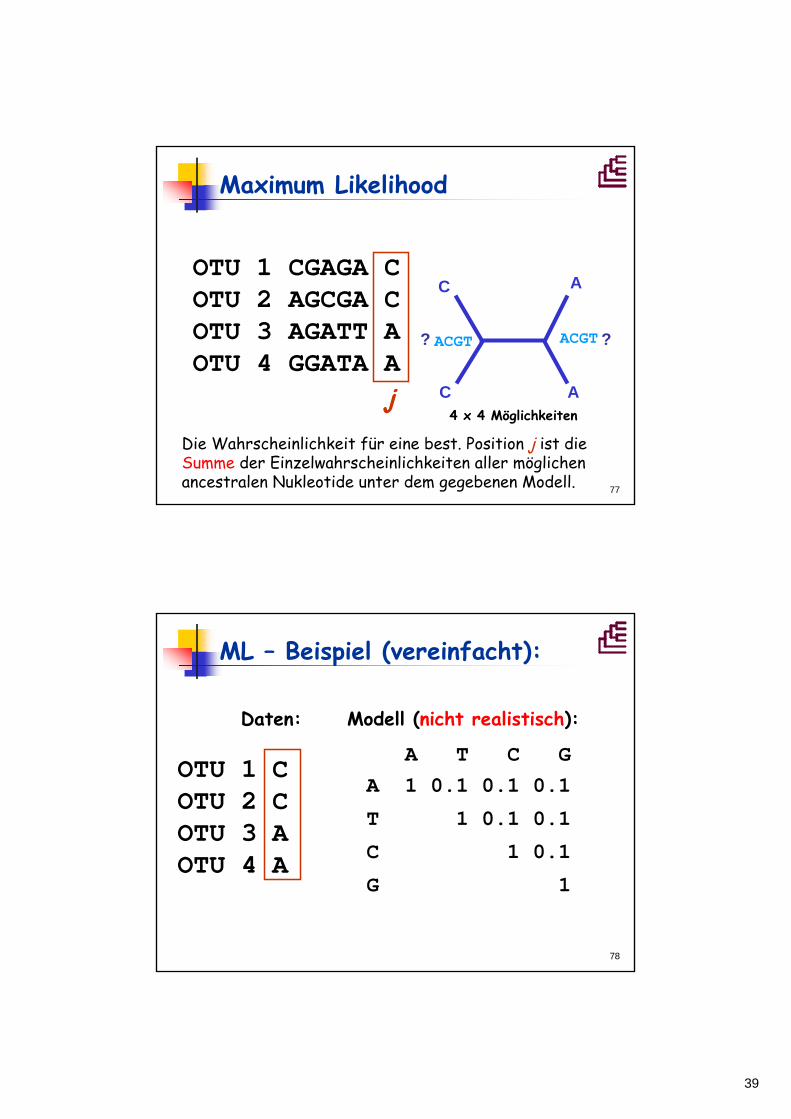
39
77
Maximum Likelihood
OTU 1 CGAGA COTU 2 AGCGA COTU 3 AGATT AOTU 4 GGATA A
j
ACGT ??
C
C
A
A
ACGT
4 x 4 Möglichkeiten
Die Wahrscheinlichkeit für eine best. Position j ist die Summe der Einzelwahrscheinlichkeiten aller möglichen ancestralen Nukleotide unter dem gegebenen Modell.
78
ML – Beispiel (vereinfacht):
CCAA
Daten: Modell (nicht realistisch):
A T C GA 1 0.1 0.1 0.1
T 1 0.1 0.1
C 1 0.1
G 1
OTU 1 OTU 2OTU 3OTU 4
40
79
ML - Beispiel:
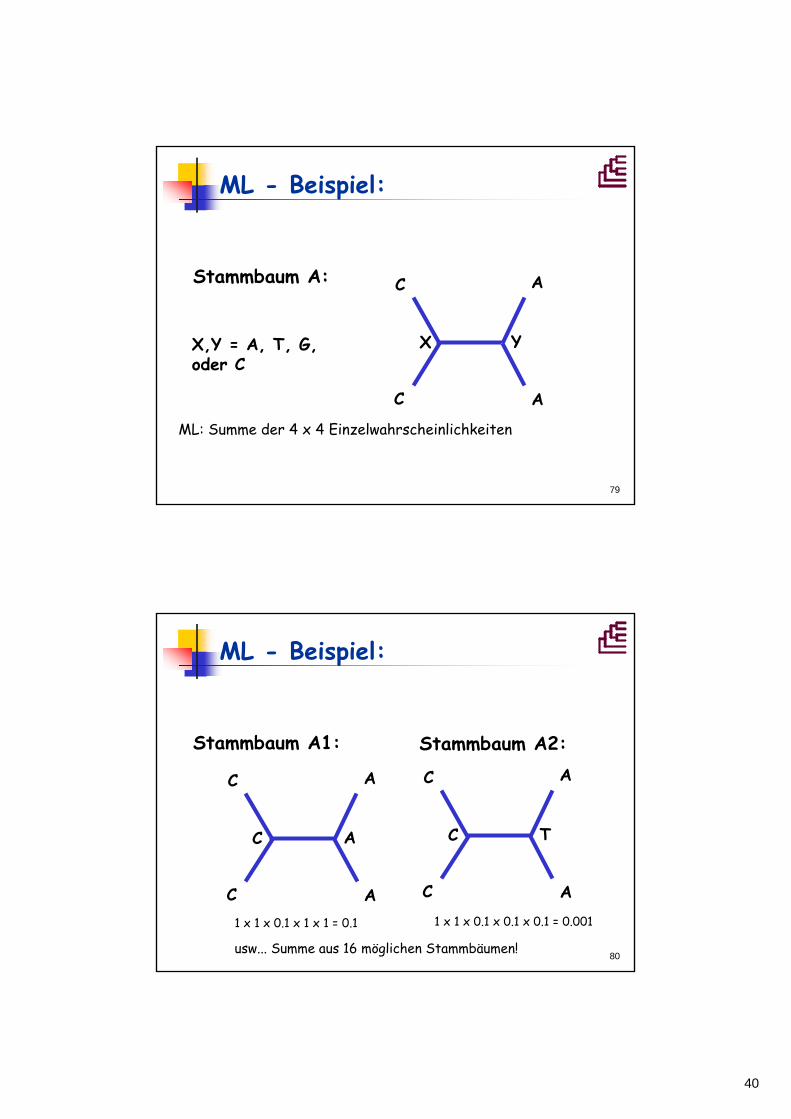
C
C
A
A
Stammbaum A:
X YX,Y = A, T, G, oder C
ML: Summe der 4 x 4 Einzelwahrscheinlichkeiten
80
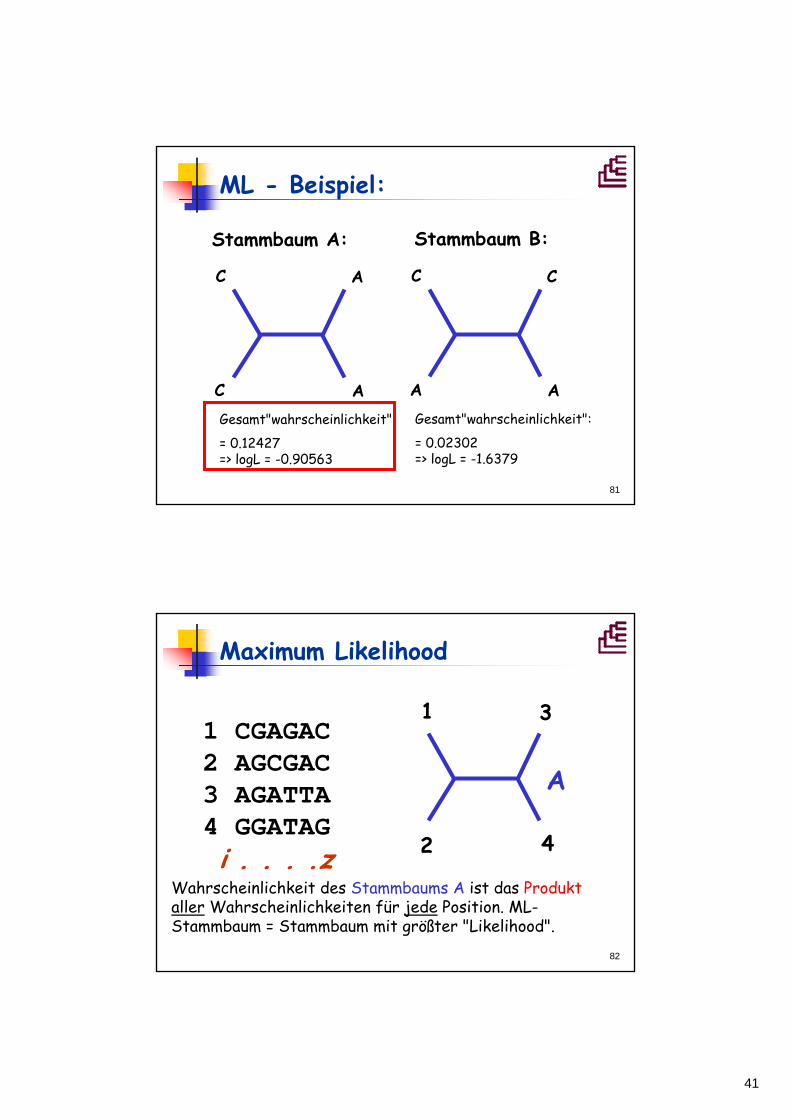
ML - Beispiel:
Stammbaum A1:
C
C
A
A
C T
C
C
A
A
C A
1 x 1 x 0.1 x 1 x 1 = 0.1 1 x 1 x 0.1 x 0.1 x 0.1 = 0.001
usw... Summe aus 16 möglichen Stammbäumen!
Stammbaum A2:
41
81
ML - Beispiel:
Stammbaum A:
C
C
A
AGesamt"wahrscheinlichkeit":
= 0.12427=> logL = -0.90563
C
A
C
AGesamt"wahrscheinlichkeit":
= 0.02302=> logL = -1.6379
Stammbaum B:
82
Wahrscheinlichkeit des Stammbaums A ist das Produktaller Wahrscheinlichkeiten für jede Position. ML-Stammbaum = Stammbaum mit größter "Likelihood".
Maximum Likelihood
1 CGAGAC2 AGCGAC3 AGATTA4 GGATAGi . . . .z
1
2
3
4
A

42
83
X,Y = A, T, G, oder CA T C G
A 1,0 0,1 0,2 0,4
T 1,0 0,3 0,6
C 1,0 0,1
G 1,0
Sequence 1 CGAGAASequence 2 AGCGAASequence 3 AGATTTSequence 4 GGATAT
1x1x1x0,1x0,1=0,01
Sequence 1 CGAGAASequence 2 AGCGAASequence 3 AGATTTSequence 4 GGATAT
CGAGAAAGCGAAAGATTTGGATAT
Likelihood einer vorgegebenen Topologie ist das Produkt aller
Wahrscheinlichkeiten jederPosition
Berechnen aller Möglichkeiten für eine Topologie und eine Position
Maximum Likelihood
84
Maximum Likelihood - Vorteile
Mathematisch gut definiertFunktioniert gut in Simulationsexperimenten Erlaubt explizite Verbindung von Evolutionsmodell und Daten (Sequenzen) "Realistische" Annahmen zur EvolutionVerschiedene Modelle und Stammbäume lassen sich testen

43
85
Maximum Likelihood - Nachteile
Maximum likelihood ist nur konsistent (ergibt einen "wahren" Stammbaum) wenn die Evolution nach den gegebenen Modell ablief: Wie gut stimmt mein Modell mit den Daten überein? Computertechnisch nicht zu lösen wenn zu viele Taxa oder Parameter berücksichtigt werden müssen.
86
Maximum Likelihood
Bei vielen Taxa sind computertechnisch nicht alle möglichen Stammbäume berechenbar Lösung: "Intelligente Algorithmen"- Quartet puzzling- Bayessche Methode + MCMCMC

44
87





























