Embed Size (px)
Citation preview

Guide d’aide à la mise en place de la Biovigilance
dans un Etablissement de Santé
Version SEPTEMBRE 2006
Document élaboré par le groupe de travail multidisciplinaire sur la biovigilance
143/147, bd Anatole France – F-93285 Saint-Denis cedex – Tél. +33 (0) 1 55 87 30 00 – www.afssaps.sante.fr
REPUBLIQUE FRANCAISE

2
SOMMAIRE
Le correspondant local de biov igilance dans un établissement de santé ___________________3
En pratique… _________________________________________________________________5
Annexe 1 : La problématique ORGANES ____________________________________________7 1) Identification de mes interlocuteurs___________________________________________________________ 7
2) Moy ens et outils _________________________________________________________________________ 7
3) Le signalement et la déclaration de biovigilance (Figure 1) ________________________________________ 8
4) L’urgence en biov igilance (Figure 2)__________________________________________________________ 9
5) L’enquête de biov igilance (Figure 3)_________________________________________________________ 10
6) Et si des tissus ont été prélev és lors du prelev ement multi-organes (PMO)?__________________________ 11
Annexe 2 : La problématique TISSUS______________________________________________12 1) Identification de mes interlocuteurs__________________________________________________________ 12
2) Moy ens et outils ________________________________________________________________________ 12
3) Le signalement et la déclaration de biovigilance (Figure 1bis)_____________________________________ 13
4) L’urgence en biov igilance _________________________________________________________________ 14
5) L’enquête de biov igilance (Figure 3bis)_______________________________________________________ 14
6) Cas particulier des tissus prélevés lors d’un PMO______________________________________________ 15
7) Cas particulier des résidus opératoires_______________________________________________________ 15
Annexe 3 : La problématique des préparations de thérapie cellulaire _____________________16 1) Identification de mes interlocuteurs__________________________________________________________ 16
2) Moy ens et outils ________________________________________________________________________ 16
3) Le signalement et la déclaration de biovigilance (Figure 1ter) _____________________________________ 17
4) L’urgence en biovigilance_________________________________________________________________ 18
5) L’enquête de biov igilance (Figure 3ter)_______________________________________________________ 18
6) Cas particulier de la moelle osseuse non transf ormée___________________________________________ 19
7) Cas particulier des greffons de CSH allogéniques non apparentés _________________________________ 19
8) Cas particulier des résidus opératoires_______________________________________________________ 20
Annexe 4 : La problématique PTA en AMP__________________________________________21 1) L’AMP v igilance_________________________________________________________________________ 21
2) Identification de mes interlocuteurs__________________________________________________________ 21
3) Le signalement et la déclaration de biovigilance (Figure 1) _______________________________________ 21
Annexe 5 : Les SRA et le RFGM de l’ABM __________________________________________23
Que déclarer en biov igilance ? 10 grands principes___________________________________24
Annexe 6 : Que déclarer avec les organes ? ________________________________________25
Annexe 7 : Que déclarer avec les Tissus ?__________________________________________27
Annexe 8 : Que déclarer avec les preparations ______________________________________29
de thérapie cellulaire ?_________________________________________________________29
Abrév ia tions _______________________________________________________________31
Glossaire ___________________________________________________________________32
Règlementation applicable______________________________________________________35

3
LE CORRESPONDANT LOCAL DE BIOVIGILANCE DANS UN ETABLISSEMENT DE SANTE
La biovigilance, mise en place par le décret n° 2003-1206 du 12 décembre 2003, a pour objet la surveillance des incidents* et des risques d’incident relatifs aux éléments et produits du corps humain utilisés à des fins thérapeutiques, et aux produits, autres que les médicaments, qui en dérivent, aux dispositifs médicaux les incorporant et aux produits thérapeutiques annexes, ainsi que des effets indésirables* résultant de leur util isation.
L’objectif in fine étant d’améliorer la sécurité sanitaire de ces mêmes éléments et produits.
Produits de santé relevant de la biovigilance Produits de santé n e relevant pas de la biovigilance
! Organes et tissus issus du corps humain et utilisés à des fins thérapeutiques chez l'homme, ainsi que leurs déri vés ! Préparati ons de thérapie cellulaire ! Dispositifs médicaux incorporant des éléments et produits issus du corps humai n
! Tous les pr oduits thérapeutiques annexes* (PTA) y compris ceux utilisés en assistance médicale à la procréation (AMP)
! Gamètes (Agence de la Biomédecine - ABM)
! Produits de thérapies cellulaire et génique soumis à une autorisation de mise sur le marché AMM (pharmacovigilance)
! Préparati ons de thérapie génique et préparations de thérapie cellulaire xénogénique (pharmacovigilance)
! Tout autre dispositif médical ( matériovigilance)
! Produits sanguins labiles (hémovigilance)
! Médicaments biologiques : médicaments déri vés du sang, protéines d'extraction… (pharmacovigilance)
! Eléments ou produits d'origine animale utilisés à des fins thérapeutiques chez l’homme.
! Dispositifs médicaux de di agnostics in vitro (réactovigilance)
! produits cosmétiques (cosmétovigilance)
! produits de tatouages à visée esthétique (cosmétovigilance)
Le correspondant local de biovigilance (CLB) est un acteur central du réseau national de biovigilance. Ce réseau étant dépourv u d’échelon régional, le CLB se trouve directement à l ’interface entre les professionnels de santé et l ’Agence française de sécurité sanitaire des produits de santé (Afssaps) et l ’Agence de la biomédecine (ancien Etablissement français des greffes), le cas échéant.
RAPPEL : Toute nomination officielle d’un correspondant local de biovigilance doit être communiquée, par le responsable de la structure concernée, à l’Afssaps (Cellule de biovigilance fax : 01 55 87 34 92) et à l’Agence de la biomédecine (Pôle sécurité – qualité fax : 01 55 93 69 36). L’Afssaps tient à jour l ’annuaire national des correspondants locaux de biovigilance et diffuse, de manière régulière, cet annuaire au réseau national des CLB.
Les missions du correspondant local de biovigilance exerçant dans un établissement de santé sont précisées aux articles R. 1211-42 et 43 du code de la santé publique.

4
L’article R. 1211-42 précise que le CLB est chargé de :
! recueillir l’ensemble des informations portées à sa connaissance et relatives aux incidents et effets indésirables
! déclarer immédiatement à l’Afssaps tout incident ou tout effet indésirable grave
! informer sans délai le CLB de l’ABM de tout incident ou effet indésirable survenu dans le cadre des activités de prélèvement ou de greffe d’organes ou de tissus.
! informer, dès lors qu’i ls sont concernés, les autres CLB ainsi que les correspondants des autres vigilances des produits de santé
! procéder aux investigations et examens appropriés dans le ou les établissements ou la ou les structures où il exerce ses fonctions de CLB et, le cas échéant, dans l’unité ayant signalé l’effet indésirable ou l’incident et en tant que de besoin, informer de leurs résultats les CLB des autres établissements ou structures susceptibles de poursuivre ces investigations et enquêtes dans le ou les établissements ou la ou les structures auxquels ils sont rattachés, et en avise l’Afssaps.
! signaler à l’Afssaps toute difficulté susceptible de compromettre le bon fonctionnement du dispositif de biovigilance.
! informer les autres intervenants du système national, à la demande de l’Afssaps.
L’article R. 1211-43 précise que : Lorsqu’il exerce ses fonctions au sein d’un établissement de santé, d’un établissement de transfusion sanguine ou de toute autre structure publique ou privée qui prélève, collecte, administre ou greffe des produits relevant de la biovigilance, le CLB est chargé de s’assurer de la mise en place, par les services concernés par ces activités, des circuits ou des procédures visant au recueil : ! de tout document utile à la traçabilité* des produits d’origine humaine utilisés à des fins
thérapeutiques de façon à permettre d’établir un lien entre donneur et receveur
! de tout document uti le à la traçabilité des produits thérapeutiques annexes (PTA), depuis leur cession par le fabricant jusqu’à leur utilisation, de façon à établir un lien entre le lot de fabrication du PTA utilisé et le produit d’origine humaine avec lequel il a été en contact
! des résultats des analyses biologiques et des tests de dépistage pratiqués sur le donneur ainsi que des contrôles pratiqués sur les éléments prélevés.
! pour les services de greffe, des résultats des tests de dépistage et examens biologiques pratiqués chez le receveur préalablement, ou, le cas échéant, postérieurement à la greffe d’organes ou de tissu ou à l’administration de cellules ou de produits de thérapie cellulaire
Le CLB est chargé de :
! collaborer, dans le cadre de ses missions, avec les équipes de prélèvement ou de greffe de l’ES ou de la structure à laquelle il est rattaché, ainsi qu’avec la structure de coordination hospitalière du prélèvement.
Les cabinets d’exercice libéral ayant des activités d’administration ou de greffe de ces produits ne sont pas concernés par cette disposition. Remarques sur les éléments et produits d’origine animale Concernant les cellules d’origine animale, que ce soient des spécialités pharmaceutiques ou des préparations de thérapie cellulaire xénogéniques mises sur le marché, le décret relatif à la pharmacovigilance leur est applicable. L’article R.5121-150 du Code de la santé publique qui fixe le champ d’application de la pharmacovigilance précise que "la pharmacovigilance a pour objet la surveillance du risque d'effet indésirable résultant de l'utilisation des médicaments et produits à usage humain mentionnés aux articles L. 5111-2 et L. 5121-1 (...)". Les préparations de thérapie cellulaire xénogéniques définies au 13° de l 'article L. 5121-1 sont soumises au décret relatif à la pharmacovigilance et aux bonnes pratiques de pharmacovigilance (arrêté du 28/04/2005). Les organes et les tissu s d'origine animale ne peuvent être utilisés à des fins thérapeutiques que dans le cadre d'essais cliniques (Art. L. 1125-2 du Code de la santé publique). * Renvoi Glossaire

5
EN PRATIQUE… J’ai été désigné correspondant local de biovigilance dans mon établissement de santé. Pour mener à bien mes missions de biovigilant, la première étape est d’identifier de manière exhaustiv e, au sein de mon établissement, les éléments et produits issus du corps humain, ainsi que les activités qui relèvent du champ de la biovigilance. Pour cela, il conviendra que je me pose notamment les questions suivantes.
Au sein de mon établissement : # Y a-t-i l des activités de prélèvement ou de greffe d’organes ? Si oui, quels sont les organes
concernés ? # Y a-t-il des activités de prélèvement ou de greffe de tissu s* ? Si oui, quels sont les tissu s
concernés ? # Y a-t-il des activités de prélèvement ou de greffe de cellules, d’administration de préparations de
thérapie cellulaire* ? Si oui, quels sont les produits concernés ? # Y a-t-il des activités de greffe de produits biologiques dérivant d’organes, de tissu s ou de cellules
d’origine humaine ? De la poudre d’os obtenue à partir d’une tête fémorale, une suspension de kératinocytes obtenue à partir d’une biopsie de peau par exemple ?
# Y a-t-il des activités d’assistance médicale à la procréation* (AMP)?
# Existe-t-il une ou des structures chargées de la préparation, de la transformation, de la conservation, du transport, de la distribution, de la cession, de l’importation, de l’exportation, de la répartition ou de l’attribution des produits entrant dans le champ de la biovigilance ? Cette structure peut être une unité de thérapie cellulaire, une banque de tissu s par exemple.
# Quels sont les produits thérapeutiques annexes (PTA)* utilisés ou stockés ? Cet inventaire des PTA, très spécifique, ne sera possible qu’auprès des professionnels concernés et interviendra par conséquent dans un deuxième temps.
A l’issue de cette première étape, primordiale, je dev rais être en mesure de dire quelle(s) est (sont) la(les) problématique(s) rencontrée(s) dans mon établissement de santé.
En matière de biovigilance, la problématique AMP méritera d’être abordée de manière différente par rapport aux 3 problématiques précédentes. En effet, pour les tissus et les préparations de thérapie cellulaire, l ’Afssaps e st compétente d’une part pour autoriser les procédés et la mise sur le marché des PTA et d’autre part pour gérer leur vigilance (y compris celle des organes). En revanche, pour l ’AMP, elle ne sera compétente que pour autoriser les PTA utilisés dans ce cadre et pour la vigilance des PTA. Les autorisations d’activités cliniques et biologiques relatives à l’assistance médicale à la procréation sont délivrées par l ’Agence de la biomédecine. Pour l ’AMP, un décret précisant le champ et les modalités de mise en œuvre de l’AMP vigilance, sous la responsabilité de l’ABM, est en cours de préparation. Ainsi, en AMP, seuls les incidents, risques d’incidents et effets indésirables susceptibles d’être liés à un PTA dev ront être déclarés en biovigilance.
La problématique ORGANES
Annexe 1
La problématique TISSUS Annexe 2
La problématique Préparations de TC
Annexe 3

6
Un même établissement de santé pourra bien entendu cumuler plusieurs de ces problématiques. Ce sera le cas notamment des structures telles que les CHU. Néanmoins, ces 4 problématiques mériteront d’être abordées indiv iduellement. Pour chacune d’elle, il vous est conseillé :
- d’identifier vos interlocuteurs potentiels : certains acteurs pourront être communs aux 4 problématiques (laboratoires d’analyses biologiques, pharmacie, autres vigilances,…) alors que d’autres seront exclusifs d’une problématique (l ’unité de thérapie cellulaire, la banque de tissu s, le laboratoire de procréation médicale assistée…)
- de préciser, par le biais de procédures, le rôle de chacun en cas i) de déclaration, i i) de situation d’urgence et i ii) d’enquête de biovigilance .
* Renvoi Glossaire
La problématique PTA en AMP
Annexe 4

7
ANNEXE 1 : LA PROBLEMATIQUE ORGANES 1) IDENTIFICATION DE MES INTERLOCUTEURS
# En interne Quels sont les professionnels de santé exerçant au sein de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des organes ?
- les équipes médico-chirurgicales de prélèvement ou de greffe ; - les équipes médicales de suivi des receveurs et des donneurs vivants ; - l ’équipe de la coordination des prélèvements ; - les laboratoires d’analyses : qualification biologique du don, contrôles microbiologiques des
PTA, suivi sérologique des receveurs, laboratoires d’immuno-hématologie, laboratoires HLA , laboratoire d’anatomie-pathologie…
- la pharmacie, si les PTA tels que les milieux de conservation et de transport des organes y sont conservés
- la cellule de gestion des risques et notamment le centre de coordination et de lutte contre les infections nosocomiales (CLIN), les acteurs des autres vigilances ; - la cellule d’hygiène, le cas échéant ; - le responsable de la banque de tissu s, le cas échéant ; - autres…
# En externe Quels sont les professionnels de santé exerçant en dehors de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des organes ?
- le service de régulation et d’appui (SRA) de l’Agence de la biomédecine (Annexe 5) ; - les CLB d’autres établissements de santé (cf. Annuaire national des CLB) ;
J’ai eu un problème de biovigilance avec un organe greffé ou devant être greffé dans mon établissement : quels sont les CLB des établissements de santé où ont été greffés les autres organes et les tissus issus du mê me prélèvement multi-organes (PMO), le cas échéant?
- les laboratoires d’analyses extérieurs avec lesquels travaille mon établissement de santé (convention souhaitable entre le laboratoire et l ’établissement de santé pour définir les responsabilités de chaque partie)
- les CLB des fabricants de PTA (cf. Annuaire national des CLB) ; - le responsable et le CLB de la banque de tissu s, le cas échéant ; - la cellule de biovigilance de l’Afssap s - autres…
2) MOYENS ET OUTILS
Quels sont les outils mis à ma disposition, ou dont je peux disposer, et qui me permettront de communiquer de façon optimale avec mes interlocuteurs ? - répondeur téléphonique spécifique, ordinateur, boîte mail spécifique, fax spécifique, téléphone ; - existe-t-il des moyens déjà développés pour les autres vigilances notamment et dont je pourrai
bénéficier ? Fiche unique de signalement, guichet unique des vigilances, logiciel de déclaration, modalités d’archivage de ces informations,… ;
- quelles sont les réunions organisées, internes à mon établissement, auxquelles je peux d’ores et déjà participer pour d’une part mieux me faire connaître et mieux faire connaître la biovigilance aux acteurs concernés, et d’autre part stimuler les échanges d’informations avec les équipes de prélèvement et de greffe notamment.

8
Quels sont les outils de travail que je devrai élaborer, en collaboration avec mes interlocuteurs, pour assurer la mise en place et le bon fonctionnement de la biovigilance ? - procédures pour préciser les modalités de signalement des incidents et des effets indésirables
par les professionnels de santé concernés - procédures relatives notamment i) au signalement et à la déclaration (c’est-à-dire en dehors
de toute situation d’urgence), ii) à la gestion de l’urgence et à i ii) l’enquête de biovigilance. Ces procédures doivent avant tout préciser le rôle de chacun des acteurs concernés et permettre de s’a ssurer que le CLB est destinataire de l’ensemble des informations qui le concerne au sein de son établissement de santé
- mise en place d’une structure spécifique chargée de la biovigilance (comité de biovigilance ou équivalent) qui pourrait se réunir 1 à 2 fois par an et dont les missions pourraient être de valider et d’actualiser l ’ensemble des procédures relatives à la biovigilance afin d’optimiser le système de biovigilance dans son ensemble. A défaut d’un comité spécialement dédié à la biovigilance, il peut être envisagé de prévoir une cession biovigilance au sein de réunions déjà existantes telles que le comité technique des greffes, le comité technique des banques de tissus, la cellule de gestion des risques, le comité de coordination des vigilances….
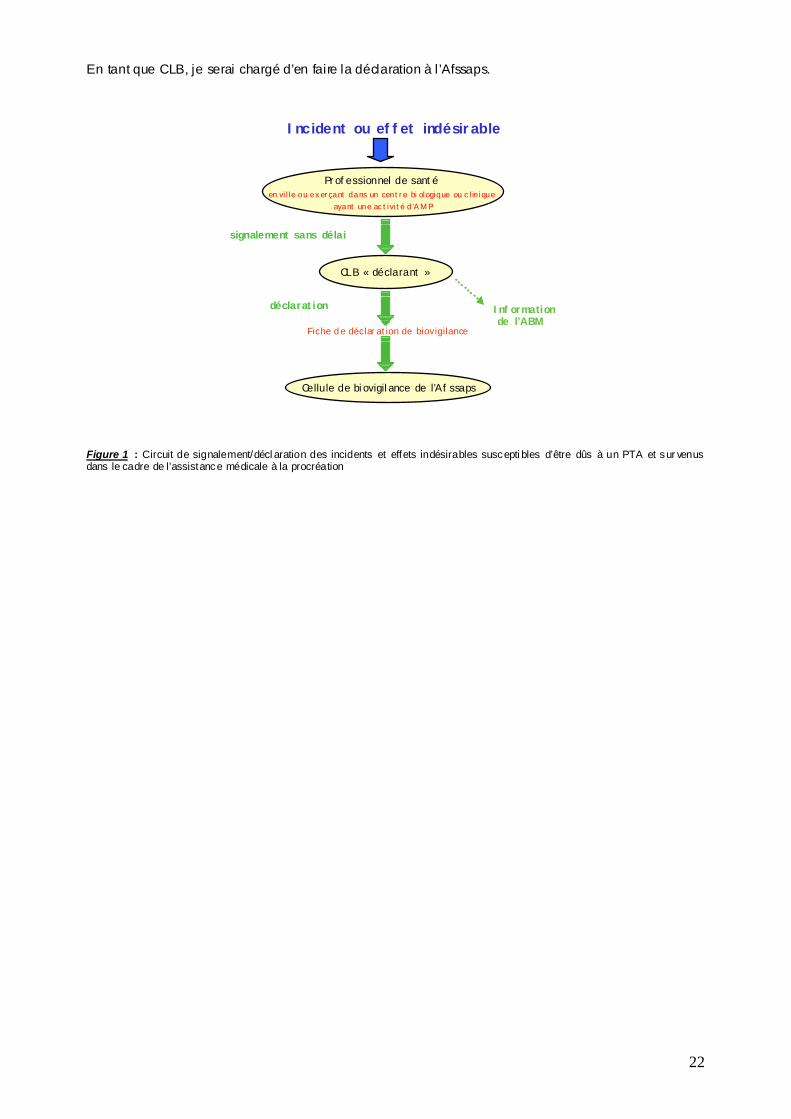
3) LE SIGNALEMENT ET LA DECLAR ATION DE BIOVIGILANCE (Figure 1)
Tout professionnel de santé de mon établissement devra me signaler, sans délai dès lors qu’i l en aura eu connaissance, tout incident ou effet indésirable selon les procédures que nous aurons conjointement rédigées.
En tant que CLB, je serai chargé d’en faire la déclaration à l’Afssaps.
- Quels sont les incidents et effets indésirables que je dois déclarer en biovigilance ? (Annexe 6) - Quels sont les délais dont je dispose pour faire ma déclaration ? (Annexe 6) - Comment je déclare ? Fiche de déclaration + guide de remplissage disponibles sur le site Internet
de l’Afssaps (www.afssaps.sante.fr)
Figure 1 : Circuit de signalement/déclarati on des i ncidents et effets indésirables de biovigilance en rapport avec des organes * Dans l e cadre de la problématique Organes , le professionnel de santé sera notamment le chirurgien pr éleveur ou greffeur ou bien l’équipe de la coordi nati on des prélèvements L’agence de la biomédecine doit être systématiquement informée de toute déclaration de biovigilance en rapport avec des organes, quelles que soient leurs modalités de prélèvement (PMO, PPM, donneurs vivants). En pratique, parallèlement à la déclaration à la cellule de biovigilance, une copie de la fiche de déclaration de biovigilance sera transmise au SRA du centre donneur. Le CLB de l’agence de la biomédecine sera informé selon les procédures internes mises en place au sein de l’ABM.
Incident ou effet indésirable
signalement sans délai
Cellule de biovigilance de l’Afssaps
CLB « déclarant »
Professionnel de santé*exerçant dans un établissement ou
une structure disposant d’un CLB
Fiche de déclaration de biovigilance
déclarationInformation du CLB de l’ABM (SRA du centre donneur)

9
Remarque : le fonctionnement interne de l’Agence de la biomédecine est tel que, pour tout incident ou effet indésirable survenu avec un organe, ce sera le SRA du centre donneur qui devra être prioritairement informé. En effet, c’est ce SRA qui est à même d’identifier les sites receveurs des autres organes prélevés à partir d’un même donneur.
4) L’URGENCE EN BIOVIGILANCE (FIGURE 2)
En biovigilance, l ’urgence peut être définie dès lors qu’un incident grave ou un effet indésirable grave, su sceptible de mettre en jeu la sécurité immédiate d’autres donneurs ou d’autres receveurs, survient. Elle consistera par conséquent à gérer, dans les meilleurs délais, une situation à risque pour un autre patient, le plus souvent receveur. La gestion de l’urgence, en matière d’organes, est assurée à tout moment, 24h sur 24, 7 jours sur 7 par les SRA de l’ABM. Les services de régulation et d’appui (SRA) de l’Agence de la biomédecine, de par les missions qui leur sont confiées, sont compétents pour gérer ces situations d’urgence. Ces structures disposent notamment des données relatives à la traçabilité de tous les organes et des tissus prélevés sur donneur décédé ainsi que les coordonnées des autres équipes de greffe à informer, le cas échéant. La gestion de l’urgence fait donc intervenir prioritairement le professionnel de santé qui constate l’incident ou l’effet indésirable et le SRA du centre donneur qui informe les autres équipes de greffe et l ’équipe de prélèvement, le cas échéant. Aussi, comme cela est précisé à l’article R.1211-46 du code de la santé publique, le CLB peut ne pas être, d’emblée, impliqué dans la gestion d’une urgence de biovigilance et ce d’autant plus si une telle situation survient la nuit ou le week-end. Figure 2 : Gesti on de l’urgence en biovigilance en rapport avec des organes Dans un second temps, dès que la gestion de l’urgence aura été assurée par le SRA, le professionnel de santé qui a constaté l’incident ou l’effet indésirable le signalera, dans les meilleurs délais, au CLB de son établissement qui se chargera d’en faire la déclaration à la cellule de biovigilance de l’Afssaps. Selon les procédures élaborées entre les différents acteurs, la déclaration de biovigilance à l’Afssaps pourra être réalisée par le CLB de l’ABM ou bien le CLB de l’établissement de santé. Dans ce 2nd cas, une copie de la déclaration devra être transmise à l’ABM.
Quel que soit le circuit de déclaration adopté, il est indispensable, en matière d’organes, que le SRA du centre donneur et le CLB soient conjointement informés de tout incident ou effet indésirable, chacun ayant son rôle à jouer dans l’enquête de biov igilance.
Incident ou effet indésirable
Alerte du SRA
Professionnel de santéexerçant dans un établissement ouune structure disposant d’un CLB
Information du SRA du centre donneur (ABM)
Alerte des autres équipes de greffe
Autres équipes de greffe
Signalement dans un second temps
CLB

10
5) L’ENQUETE DE BIOVIGILANCE (FIGURE 3)
Les objectifs de l’enquête de biovigilance seront notamment d’identifier : - les causes de l’incident ou de l’effet indésirable ; - si des dysfonctionnements ont eu lieu et à quel(s) niveau(x) ; - les autres partenaires du réseau pouvant être concernés : autres vigilances, autres équipes de
greffe ou équipe de prélèvement via les SRA, CLIN ; - si les PTA ont été utilisés dans des conditions conformes à leur autorisation de mise sur le
marché. Comme on l’a v u précédemment, une même déclaration de biov igilance peut impliquer plusieurs établissements de santé. Seule l’agence de la biomédecine, par l ’intermédiaire de ses SRA, sait quels sont ces autres établissements.
L’enquête de biov igilance doit être coordonnée par une seule et même personne, le correspondant local de biov igilance « déclarant » qui, à ce titre, est l’interlocuteur priv ilégié de la cellule de biov igilance de l’Afssaps.
Chaque CLB impliqué dans une enquête de biovigilance a pour mission d’une part de récupérer, au sein de l’établissement dans lequel il a été désigné, toutes les informations utiles à la conclusion de l’enquête (par exemple les résultats des examens bactériologiques réalisés chez le donneur ou les receveurs, sur les milieux de conservation des organes, les résultats des contrôles sérologiques) et d’autre part de transmettre au CLB « déclarant » ces mêmes conclusions.
Si vous avez fait la déclaration de biovigilance à l’Afssaps, vous serez à ce titre chargé de centraliser l ’ensemble des informations obtenues au sein de votre établissement ainsi que celles obtenues par les autres CLB impliqués.
Figure 3 : Interacti ons et échanges d’infor mati ons lors d’une enquête de biovigilance entre les différents acteurs concernés dans l’hypothèse où l’incident ou l’effet indésirable a lieu dans un établissement greffeur
Le CLB « déclarant » dev ra communiquer les conclusions de l’enquête à la cellule de biovigilance de l’Afssaps.
CLB « déclarant »
Autres vig ilances
CLB des autrescentres greffeurs
CLB de l’ABM (SRA)
Autres CL B : fabricant PTA, labo d’analyses,…
Professionnels de santé
Cellule de biovigilance de l’Afssaps
Éta
bliss
emen
t ou
stru
ctur
edé
clar
ante
Aut
re é
tabl
issem
ent
ou s
truc
ture
impl
iqué
Con
clus
ions
d’en
quêt
es
CLB ducentre préleveur

11
6) ET SI DES TISSUS ONT ETE PRELEVES LORS DU PRELEVEMENT MULTI-ORGANES (PMO)?
Certains tissus peuvent être prélevés dans le cadre d’un prélèvement multi-organes et notamment la cornée, l ’os cortical, la peau, les valves et artères (arrêté du 2 août 2005). En biovigilance, 2 situations pourront se présenter :
- le point de départ est un incident ou un effet indésirable survenu avec un organe ; - le point de départ est un incident ou un effet indésirable survenu avec un tissu
Dans la 1ère situation, au même titre que l’information des autres équipes de greffes, seul le SRA du centre donneur sera en mesure de savoir si des tissu s ont été prélevés lors du PMO et dans quelle(s) banque(s) de tissus ils ont été adressé s. L’information de(s) la banque(s) sera réalisée par le SRA du centre donneur et les coordonnées de cette (ces) dernière(s) seront communiquées au CLB de l’établissement où a eu lieu l’incident ou l’effet indésirable. Le CLB de la banque de tissus sera chargé de mener l ’enquête au sein de sa structure et d’en communiquer les conclusions au CLB « déclarant ».
Les procédures précédentes décrites aux 3), 4) et 5) devront être adaptées en conséquence. Pour la seconde situation, se reporter à l ’annexe 2 : la problématique TISSUS.

12
ANNEXE 2 : LA PROBLEMATIQUE TISSUS 1) IDENTIFICATION DE MES INTERLOCUTEURS
# En interne Quels sont les professionnels de santé exerçant au sein de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des tissu s ?
- les équipes chirurgicales de prélèvement ou de greffe ; - l ’équipe de la coordination des prélèvements si des tissu s sont prélevés sur donneurs
décédés ; - les laboratoires d’analyses : qualification biologique du don, contrôles microbiologiques des
PTA, suivi sérologique des receveurs, laboratoires HLA, laboratoires d’immuno-hématologie, laboratoire d’anatomie-pathologie …
- le responsable de la banque de tissu s ; - la pharmacie si les milieux de conservation des tissus y sont conservés ; - la cellule de gestion des risques et notamment le CLIN, les acteurs des autres vigilances ; - la cellule d’hygiène, le cas échéant ; - autres…
# En externe Quels sont les professionnels de santé exerçant en dehors de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des tissus ?
- le responsable et le CLB de la (les) banque(s) de tissu s avec laquelle mon établissement a établi une convention (cf. Annuaire national des CLB) ;
- le service de régulation et d’appui (SRA) de l’Agence de la biomédecine (Annexe 5) si des tissu s sont prélevés sur donneurs décédés ;
- les CLB d’autres établissements de santé (cf. Annuaire national des CLB) ; J’ai eu un problème de biovigilance avec un tissu greffé ou devant être greffé dans mon établissement, quels sont les CLB des établissements de santé où ont été envoyés les éventuels autres tissus ou organes issus du mê me prélèvement multi-organes ?
- les laboratoires d’analyses biologiques avec lesquels travaille mon établissement de santé (convention souhaitable entre le laboratoire et l ’établissement de santé pour définir les responsabilités de chaque partie) ;
- les CLB des fabricants de PTA (cf. Annuaire national des CLB) ; - la cellule de biovigilance de l’Afssap s ; - autres…
2) MOYENS ET OUTILS
Quels sont les outils mis à ma disposition, ou dont je peux disposer, et qui me permettront de communiquer de façon optimale avec mes interlocuteurs ? - répondeur téléphonique spécifique, ordinateur, boîte mail spécifique, fax spécifique, téléphone ; - existe-t-il des moyens déjà développés pour les autres vigilances notamment et dont je pourrai
bénéficier ? Fiche unique de signalement, guichet unique des vigilances, logiciel de déclaration, modalités d’archivage de ces informations,… ;
- quelles sont les réunions organisées, internes à mon établissement, auxquelles je peux d’ores et déjà participer pour d’une part mieux me faire connaître et mieux faire connaître la biovigilance aux acteurs concernés, et d’autre part stimuler les échanges d’informations avec les équipes de prélèvement et de greffe notamment.

13
Quels sont les outils de travail que je devrai élaborer, en collaboration avec mes interlocuteurs, pour assurer la mise en place et le bon fonctionnement de la biovigilance ?
- procédures pour préciser les modalités de signalement des incidents et des effets indésirables par les professionnels de santé concernés ;
- procédures relatives notamment i) au signalement et à la déclaration (c’est-à-dire en dehors de toute situation d’urgence), ii) à la gestion de l’urgence et à i ii) l’enquête de biovigilance. Ces procédures doivent avant tout préciser le rôle de chacun des acteurs concernés et permettre de s’a ssurer que le CLB est destinataire de l’ensemble des informations qui le concerne au sein de son établissement de santé
- mise en place d’une structure spécifique chargée de la biovigilance (comité de biovigilance ou équivalent) qui pourrait se réunir 1 à 2 fois par an et dont les missions pourraient être de valider et d’actualiser l ’ensemble des procédures relatives à la biovigilance afin d’optimiser le système de biovigilance dans son ensemble. A défaut d’un comité spécialement dédié à la biovigilance, il peut être envisagé de prévoir une cession biovigilance au sein de réunions déjà existantes telles que le comité technique des greffes, le comité technique des banques de tissus, la cellule de gestion des risques, le comité de coordination des vigilances….
3) LE SIGNALEMENT ET LA DECLAR ATION DE BIOVIGILANCE (FIGURE 1BIS)
Tout professionnel de santé de mon établissement devra me signaler, sans délai dès lors qu’i l en aura eu connaissance ou fait la constatation, tout incident ou effet indésirable selon les procédures internes que nous aurons conjointement rédigées.
En tant que CLB, je serai chargé d’en faire la déclaration aux autorités compétentes.
- Quels sont les incidents et effets indésirables que je dois déclarer en biovigilance ? (Annexe 7) - Quels sont les délais dont je dispose pour faire ma déclaration ? (Annexe 7) - Comment je déclare ? Fiche de déclaration + guide de remplissage disponibles sur le site Internet
de l’Afssaps http://afssaps.sante.fr Figure 1bis : Circuit de signalement/décl aration des incidents et effets i ndésirables de biovigilance en rapport avec des tissus
Un incident ou effet indésirable survenu avec un tissu prélevé, greffé ou devant être greffé peut avoir des conséquences chez les receveurs des éventuels autres tissu s ou organes issus du même donneur.
La banque de tissus qui a libéré le greffon, de par les missions qui lui sont confiées, est la seule structure qui est en mesure de confirmer si oui ou non d’autres patients sont impliqués. Si le tissu a été prélevé sur un donneur décédé, elle devra de surcroît se rapprocher du SRA du centre donneur pour avoir une vision exhaustive des autres patients impliqués et en particulier des receveurs d’organes.
Incident ou effet indésirable
signalement sans délai
Cellule de biovigilance de l’Afssaps
CLB « déclarant »
Professionnel de santéexerça nt da ns un établissement ouune structure disposant d’un CLB
Fiche de déclaration de biovigilance
déclarationInformation du
CLB de la banque de tissus
Information du CLB de l’ABM (SRA du centre donneur)

14
Selon les procédures élaborées entre les différents acteurs, la déclaration de biovigilance à l’Afssaps pourra être réalisée par le CLB de la banque de tissu s ou le CLB de l’établissement de santé. Dans tous les cas, une copie de la fiche de déclaration devra être transmise au CLB de l’ABM pour information.
Quel que soit le circuit de déclaration adopté, il est indispensable, en matière de tissus, que les CLB de l’établissement de santé et de la banque de tissus soient conjointement et systématiquement informés de tout incident ou effet indésirable, chacun ayant son rôle à jouer dans l’enquête de biov igilance.
4) L’URGENCE EN BIOVIGILANCE
En biovigilance, l ’urgence peut être définie dès lors qu’un incident grave ou un effet indésirable grave, su sceptible de mettre en jeu la sécurité immédiate d’autres donneurs ou d’autres receveurs, survient. Elle consistera par conséquent à gérer, dans les meilleurs délais, une situation à risque pour un autre patient, le plus souvent receveur. La gestion de l’urgence telle qu’elle se rencontre avec les organes (Annexe 1), c’est-à-dire réalisable 24h sur 24, 7 jours sur 7, n’existe pas avec les tissu s. Les tissu s prélevés à partir de donneurs vivants ou décédés sont le plus souvent greffés lors d’opérations programmées pendant les heures ouvrables. De surcroît, le délai notamment de quarantaine entre le prélèvement et la greffe du tissu laisse souvent plus de temps à la gestion des problèmes que ce que l’on observe en greffe d’organes. 5) L’ENQUETE DE BIOVIGILANCE (FIGURE 3BIS)
Les objectifs de l’enquête de biovigilance seront notamment d’identifier : - les causes de l’incident ou de l’effet indésirable - si des dysfonctionnements ont eu lieu et à quel(s) niveau(x) - les autres partenaires du réseau pouvant être concernés : autres vigilances, autres équipes de
greffe via la banque de tissus, CLIN - si les PTA ont été utilisés dans des conditions optimales.
Comme on l’a v u précédemment, une même déclaration de biov igilance peut impliquer plusieurs établissements de santé. Seule la banque de tissus sait quels sont ces autres établissements. Aussi, dès lors qu’i l est informé de l’incident ou de l’effet indésirable, le CLB de la banque de tissu s communique au CLB « déclarant » de l’établissement de santé où a eu lieu l’incident ou l’effet indésirable la liste exhaustive de ces autres établissements. Les premières informations qu’il aura pu récupérer lors de la gestion de l’alerte devront également être communiquées au CLB « déclarant » chargé de coordonner l ’enquête.
L’enquête de biov igilance doit être coordonnée par une seule et même personne, le correspondant local de biov igilance « déclarant » qui, à ce titre, est l’interlocuteur priv ilégié de la cellule de biov igilance de l’Afssaps.
Chaque CLB impliqué dans une enquête de biovigilance a pour mission d’une part de récupérer au sein de l’établissement dans lequel i l a été désigné toutes les informations utiles à la conclusion de l’enquête (par exemple les résultats des examens bactériologiques réalisés chez le donneur ou les receveurs, sur les milieux de conservation des organes, les résultats des contrôles sérologiques) et d’autre part de transmettre au CLB « déclarant » ces mêmes conclusions.
Si vous avez fait la déclaration de biovigilance à l’Afssaps, vous serez à ce titre chargé de centraliser l ’ensemble des informations obtenues au sein de votre établissement ainsi que celles obtenues par les autres CLB impliqués.

15
Le CLB « déclarant » dev ra communiquer les conclusions de l’enquête à la cellule de biovigilance de l’Afssaps. Figure 3bis : L’ enquête de biovigilance : interactions et échanges d’informations entre l es dif férents ac teurs concernés 6) CAS PARTICULIER DES TISSUS PRELEVES LORS D’UN PMO
Certains tissus peuvent être prélevés dans le cadre d’un prélèvement multi-organes et notamment la cornée, l ’os cortical, la peau, les valves et artères (arrêté du 2 août 2005). En biovigilance, 2 situations pourront se présenter :
- le point de départ est un incident ou un effet indésirable survenu avec un organe ; - le point de départ est un incident ou un effet indésirable survenu avec un tissu
Pour le première situation, se reporter à l ’annexe 1 : la problématique ORGANES Dans la seconde situation, seul le CLB de la banque de tissus sera en mesure de savoir si le tissu provient d’un PMO. Si tel est le cas, il devra informer le SRA du centre donneur de l’incident ou de l’effet indésirable qui se chargera d’informer les autres équipes de greffes et de prélèvement, le cas échéant.
Les procédures précédentes décrites aux 3), 4) et 5) devront être adaptées en conséquence. 7) CAS PARTICULIER DES RESIDUS OPERATOIRES
Les résidus opératoires* sont recueillis à l ’occasion d’une intervention chirurgicale et en premier lieu pour le bénéfice direct du patient opéré (membranes amniotiques, têtes fémorales,…).
Tout résidu opératoire prélevé à des fins thérapeutiques doit nécessairement être adressé à une banque de tissus autorisée.
L’Agence de la biomédecine, au même titre que ses SRA, n’est pas du tout impliquée dans la traçabilité des ré sidus opératoires. Aussi, tout incident ou effet indésirable en rapport av ec des résidus opératoires n’impliquera à aucun moment ni l’ABM ni ses SRA.
Les procédures précédentes décrites aux 3), 4) et 5) devront être adaptées en conséquence. * Renvoi Glossaire
CLB « déclarant »
Autres vig ilances
CLB des autrescentres greffeurs
CLB de l’ABM (SRA)
Autres CLB : fabricant PTA par exemple
Professionnels de santé
Cellule de biovigilance de l’Afssaps
Éta
blis
seme
nt o
u st
ruct
ure
décl
aran
teA
utre
éta
bliss
emen
tou
str
uctu
re im
pliq
ué
Con
clus
ions
d’en
quêt
es
CLB de la banque de tissus

16
ANNEXE 3 : LA PROBLEMATIQUE DES PREPARATIONS DE THERAPIE CELLULAIRE
1) IDENTIFICATION DE MES INTERLOCUTEURS
# En interne Quels sont les professionnels de santé exerçant au sein de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des préparations de thérapie cellulaire ?
- les équipes de prélèvement ou de greffe ; - les laboratoires d’analyses : qualification biologique du don, contrôles microbiologiques des
PTA, suivi sérologique des receveurs, laboratoires HLA, laboratoires d’immuno-hématologie,… ; - le responsable de l’unité de thérapie cellulaire ; - la pharmacie si les PTA util isés avec les préparations de thérapie cellulaire y sont
conservés, comme l’albumine par exemple ; - la cellule de gestion des risques et notamment le CLIN, les acteurs des autres vigilances ; - la cellule d’hygiène, le cas échéant ; - autres…
# En externe Quels sont les professionnels de santé exerçant en dehors de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des préparations de thérapie cellulaire ?
- le responsable et le CLB de l’unité de thérapie cellulaire qui m’a délivré le greffon si cette unité ne fait pas partie de mon établissement de santé (cf. Annuaire national des CLB) ;
- le CLB de l’Agence de la biomédecine (cf annexe 5) ; - les CLB d’autres établissements de santé (cf. Annuaire national des CLB) ;
- les laboratoires d’analyses biologiques avec lesquels travaille mon établissement de santé (convention souhaitable entre le laboratoire et l ’établissement de santé pour définir les responsabilités de chaque partie) ;
- les CLB des fabricants de PTA (cf. Annuaire national des CLB) ;
- la cellule de biovigilance de l’Afssap s ; - autres…
2) MOYENS ET OUTILS
Quels sont les outils mis à ma disposition ou dont je peux disposer et qui me permettront de communiquer de façon optimale avec mes interlocuteurs ? - répondeur téléphonique spécifique, ordinateur, boîte mail spécifique, fax spécifique, téléphone ; - existe-t-il des moyens déjà développés pour les autres vigilances notamment et dont je pourrai
bénéficier ? Fiche unique de signalement, guichet unique des vigilances, logiciel de déclaration, modalités d’archivage de ces informations,… ;
- quelles sont les réunions organisées, internes à mon établissement, auxquelles je peux d’ores et déjà participer pour d’une part mieux me faire connaître et mieux faire connaître la biovigilance aux acteurs concernés, et d’autre part stimuler les échanges d’informations avec les équipes de prélèvement et de greffe notamment.
Quels sont les outils de travail que je devrai élaborer, en collaboration avec mes interlocuteurs, pour assurer la mise en place et le bon fonctionnement de la biovigilance ?

17
- procédures pour préciser les modalités de signalement des incidents et des effets indésirables par les professionnels de santé concernés
- procédures relatives notamment i) au signalement et à la déclaration (c’est-à-dire en dehors de toute situation d’urgence), ii) à la gestion de l’urgence et à i ii) l’enquête de biovigilance. Ces procédures doivent avant tout préciser le rôle de chacun des acteurs concernés et permettre de s’a ssurer que le CLB est destinataire de l’ensemble des informations qui le concerne au sein de son établissement de santé
- mise en place d’une structure spécifique chargée de la biovigilance (comité de biovigilance ou équivalent) qui pourrait se réunir 1 à 2 fois par an et dont les missions pourraient être de valider et d’actualiser l ’ensemble des procédures relatives à la biovigilance afin d’optimiser le système de biovigilance dans son ensemble. A défaut d’un comité spécialement dédié à la biovigilance, il peut être envisagé de prévoir une cession biovigilance au sein de réunions déjà existantes telles que le comité technique des greffes, le comité technique des banques de tissus, la cellule de gestion des risques, le comité de coordination des vigilances….
3) LE SIGNALEMENT ET LA DECLAR ATION DE BIOVIGILANCE (FIGURE 1TER)
Tout professionnel de santé de mon établissement devra me signaler, sans délai dès lors qu’i l en aura eu connaissance ou fait la constatation, tout incident ou effet indésirable selon les procédures internes que nous aurons conjointement rédigées.
En tant que CLB, je serai chargé d’en faire la déclaration aux autorités compétentes.
- Quels sont les incidents et effets indésirables que je dois déclarer en biovigilance ? (Annexe 8) - Quels sont les délais dont je dispose pour faire ma déclaration ? (Annexe 8) - Comment je déclare ? Fiche de déclaration + guide de remplissage disponibles sur le site Internet
de l’Afssaps http://afssaps.sante.fr Figure 1ter : Circuit de signalement/déclarati on des incidents et effets indésirables de bi ovigilance en rapport avec des prépar ations de thérapie cellul aire Un incident ou effet indésirable survenu avec une préparation de thérapie cellulaire prélevée, administrée ou devant être administrée peut avoir des conséquences chez d’autres patients y compris dans le cas d’un produit autologue (si problème en relation avec un PTA notamment).
L’unité de thérapie cellulaire qui a libéré le produit, de par les missions qui lui sont confiées, est la seule structure qui est en mesure de confirmer si oui ou non d’autres patients sont impliqués. Selon les procédures élaborées entre les différents acteurs, la déclaration de biovigilance à l’Afssaps pourra être réalisée par le CLB de l’unité de thérapie cellulaire ou le CLB de l’établissement de santé. Dans tous les cas, une copie de la fiche de déclaration devra être transmise à l’ABM.
Incident ou effet indésirable
signalement sans délai
Cellule de biovigilance de l’Afssaps
CLB « déclarant »
Professionnel de santéexerç ant da ns un éta blis sement ouune struc ture disposa nt d’u n CLB
Fiche de décl aration de biovigilance
déclarationInformation du CLB
de l’unité de thérapie cellulaire
Information de l’ABM

18
Quel que soit le circuit de déclaration adopté, il est indispensable, en matière de préparations de thérapie cellulaire, que les CLB de l’établissement de santé et de l’unité de thérapie cellulaire soient conjointement et systématiquement informés de tout incident ou effet indésirable, chacun ayant son rôle à jouer dans l’enquête de biov igilance.
4) L’URGENCE EN BIOVIGILANCE
En biovigilance, l ’urgence peut être définie dès lors qu’un incident grave ou un effet indésirable grave, su sceptible de mettre en jeu la sécurité immédiate d’autres donneurs ou d’autres receveurs, survient. Elle consistera par conséquent à gérer, dans les meilleurs délais, une situation à risque pour un autre patient, le plus souvent receveur. La gestion de l’urgence telle qu’elle se rencontre avec les organes (Annexe 1), c’est-à-dire réalisable 24h sur 24, 7 jours sur 7, n’existe pas avec les préparations de thérapie cellulaire. 5) L’ENQUETE DE BIOVIGILANCE (FIGURE 3TER)
Les objectifs de l’enquête de biovigilance seront notamment d’identifier : - les causes de l’incident ou de l’effet indésirable ; - si des dysfonctionnements ont eu lieu et à quel(s) niveau(x) ; - les autres partenaires du réseau pouvant être concernés : autres vigilances, autres équipes de
greffe via l ’unité de thérapie cellulaire, CLIN ; - si les PTA ont été utilisés dans des conditions optimales.
Comme on l’a v u précédemment, une même déclaration de biov igilance peut impliquer plusieurs établissements de santé. Seule l’unité de thérapie cellulaire sait quels sont ces autres établissements. Aussi, dès lors qu’i l est informé de l’incident ou de l’effet indésirable, le CLB de l’unité de thérapie cellulaire communique au CLB « déclarant » la l iste exhaustive de ces autres établissements. Les premières informations qu’i l aura pu récupérer lors de la gestion de l’alerte devront également être communiquées au CLB « déclarant » chargé de coordonner l ’enquête.
L’enquête de biov igilance doit être coordonnée par une seule et même personne, le correspondant local de biov igilance « déclarant » qui, à ce titre, est l’interlocuteur priv ilégié de la cellule de biov igilance de l’Afssaps.
Chaque CLB impliqué dans une enquête de biovigilance a pour mission d’une part de récupérer au sein de l’établissement dans lequel i l a été désigné toutes les informations utiles à la conclusion de l’enquête (par exemple les résultats des examens bactériologiques réalisés chez le donneur ou les receveurs, sur les milieux de conservation des organes, les résultats des contrôles sérologiques) et d’autre part de transmettre au CLB « déclarant » ces mêmes conclusions.
Si vous avez fait la déclaration de biovigilance à l’Afssaps, vous serez à ce titre chargé de centraliser l ’ensemble des informations obtenues au sein de votre établissement ainsi que celles obtenues par les autres CLB impliqués.
Le CLB « déclarant » dev ra communiquer les conclusions de l’enquête à la cellule de biovigilance de l’Afssaps.

19
Figure 3ter : L’enquête de biovigilance : interactions et échanges d’informations entre les différents acteurs concer nés
6) CAS PARTICULIER DE LA MOELLE OSSEUSE NON TRANSFORMEE
En accord avec la loi n°2004-800 du 6 août 2004 relative à la bioéthique, la moelle osseuse est désormais considérée comme de la cellule et non plus comme un organe. Toutefois, elle conserve une spécificité par rapport aux autres cellules pour ce qui concerne l’importation et l ’exportation. En effet, les établissements de santé autorisés à prélever et à greffer des cellules hématopoïétiques issues de la moelle osseuse peuvent exporter et importer des cellules de la moelle osseuse non transformées, respectivement, sans qu’elles « transitent » par une unité de thérapie cellulaire (Art.L.1245-5 du CSP). Si un incident ou un effet indésirable survient avec un greffon de cellules hématopoïétiques issues de moelle osseuse non transformée et n’ayant pas « transité » par une unité de thérapie cellulaire, aucun correspondant local de biovigilance d’unité de thérapie cellulaire ne sera impliqué, ni dans la déclaration ni dans l’enquête de biovigilance. Par contre, les CLB des établissements de santé concernés seront impliqués. Les procédures précédentes décrites au 3), 4) et 5) devront être adaptées en conséquence.
7) CAS PARTICULIER DES GREFFONS DE CSH ALLOGENIQUES NON APPARENTES
Depuis 1987, France Greffe de Moelle (FGM), association qui gère le registre national de donneurs de cellules hématopoïétiques, est l ’association de référence pour les greffes de cellules hématopoïétiques (origine médullaire, origine sang périphérique, origine placentaire) chez les patients n’ayant pas de donneur familial. Le décret n° 2005-1391 du 8 novembre 2005 a organisé le transfert du fichier des donneurs tenu par FGM à l’Agence de la biomédecine. Aussi, pour tout incident ou effet indésirable survenu avec un greffon de CSH allogéniques non apparentés importé, le professionnel de santé devra systématiquement informer le référent biovigilance du registre FGM (Annexe 5), via le CLB de l’ABM, Pour tout incident ou effet indésirable survenu avec un greffon de CSH allogéniques non apparentés exporté, le référent biovigilance du registre FGM, informé directement par le Registre International
CLB « déclarant »
Autres vig ilances
CLB des autrescentres greffeurs
CLB de l’unité de thérapie cellulaire
Autres CL B : fabricant PTA, labo d’analyses,…
Professionnels de santé
Cellule de biovigilance de l’Afssaps
Éta
bliss
emen
t ou
stru
ctur
edé
clar
ante
Aut
re é
tabl
issem
ent
ou s
truc
ture
impl
iqué
Con
clus
ions
d’en
quêt
es

20
concerné, le signale au CLB de l’ABM qui fait la déclaration de biovigilance à l’Afssaps. L’unité de thérapie cellulaire du centre préleveur devra être informée pour participer, le cas échant, à l ’enquête. Les procédures précédentes décrites au 3), 4) et 5) devront être adaptées en conséquence.
8) CAS PARTICULIER DES RESIDUS OPERATOIRES
Les résidus opératoires* sont recueillis à l ’occasion d’une intervention chirurgicale et en premier lieu pour le bénéfice direct du patient opéré (membranes amniotiques, têtes fémorales,…).
Tout résidu opératoire prélevé à des fins thérapeutiques doit nécessairement être adressé à une banque de tissus autorisée.
L’Agence de la biomédecine, au même titre que ses SRA, n’est pas du tout impliquée dans la traçabilité des ré sidus opératoires. Aussi, tout incident ou effet indésirable en rapport av ec des résidus opératoires n’impliquera à aucun moment ni l’ABM ni ses SRA.
Les procédures précédentes décrites aux 3), 4) et 5) devront être adaptées en conséquence. * Renvoi Glossaire

21
ANNEXE 4 : LA PROBLEMATIQUE PTA EN AMP
1) L’AMP VIGILANCE
En matière de biovigilance, la problématique AMP méritera d’être abordée de manière différente par rapport aux 3 problématiques précédentes. En effet, pour les tissus et les préparations de thérapie cellulaire, l ’Afssaps est compétente d’une part pour autoriser les procédés et la mise sur le marché des PTA et d’autre part pour gérer leur vigilance (y compris celle des organes). En revanche, pour l ’AMP, elle ne sera compétente que pour autoriser les PTA utilisés dans ce cadre et pour la vigilance des PTA. Les autorisations d’activités cliniques et biologiques relatives à l’assistance médicale à la procréation sont délivrées par l ’Agence de la biomédecine. Le dispositif de l’assistance médicale à la procréation, créé par la loi du 6 Août 2004, est actuellement en cours de mise en œuv re par l’ABM et sera précisé ultérieurement. Dans l’attente, la présente annexe se veut volontairement très générale.
2) IDENTIFICATION DE MES INTERLOCUTEURS
# En interne En tant que CLB, quels sont les professionnels de santé exerçant au sein de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des PTA utilisés en AMP ?
- Le responsable de la vigilance liée à l’AMP mise en place par l ’ABM ; - les équipes médico-chirurgicales ; - le centre clinique et biologique effectuant des activités d’AMP ; - les laboratoires d’analyses : contrôles microbiologiques des PTA, laboratoires d’immuno-
hématologie, … - la pharmacie si les PTA utilisés en AMP y sont conservés ; - la cellule de gestion des risques et notamment le CLIN, les acteurs des autres vigilances ; - l ’équipe opérationnelle d’hygiène, le cas échéant ; - autres…
# En externe Quels sont les professionnels de santé exerçant en dehors de mon établissement et avec lesquels je serai amené, le cas échéant, à collaborer dans le cadre de la biovigilance des PTA utilisés en AMP?
- l ’Agence de la biomédecine ; - les CLB d’autres établissements de santé ; - les CLB des fabricants / fournisseurs de PTA ; - la cellule de biovigilance de l’Afssap s ; - autres…
Les serv ices de régulation et d’appui (SRA) de l’ABM, très impliqués dans la problématique organes, ne sont pas impliqués dans la problématique AMP.
3) LE SIGNALEMENT ET LA DECLAR ATION DE BIOVIGILANCE (FIGURE 1)
Tout professionnel de santé ayant une activité d’assistance médicale à la procréation devra me signaler, sans délai dès lors qu’il en aura eu connaissance, tout incident ou effet indésirable lié ou susceptible d’être lié aux PTA utilisés, selon les procédures internes que nous aurons conjointement rédigées.
22
En tant que CLB, je serai chargé d’en faire la déclaration à l’Afssaps.
Figure 1 : Circuit de signalement/décl aration des incidents et effets indésirables suscepti bles d’être dûs à un PTA et sur venus dans le cadre de l’assistance médicale à la procréation
Inc ident ou effet indésirable
Professionnel de santéen ville ou exerça nt da ns un centre bi ologiq ue ou c lin ique
aya nt une ac tivité d’AMP
signalement sans délai
Cellule de biovigilance de l’Afssaps
CLB « déclarant »
Fiche de déclaration de biovigilance
déclaration Information de l’ABM

23
ANNEXE 5 : LES SRA ET LE RFGM DE L’ABM
Inter-région Adresse Tel / fax SRA I - Nord
Agence de la biomédecine Hôpital Calmette - Pavillon Breton 59037 LILLE CEDEX
T : 03.20.44.59.14 F : 03.20.44.46.84
SRA II - Est
Agence de la biomédecine Faculté de Médecine 9 avenue de la Forêt de Haye Bâtiment E – 1er étage 54519 VANDOEUVRE LES NANCY
T : 03.83.68.38.10 F : 03.83.68.38.19
SRA III - Centre-Est/La Réunion
Agence de la biomédecine Home Lacassagne 162 avenue Lacassagne 69424 LYON CEDEX 03
T : 04.72.11.02.77 F : 04.72.11.52.22
SRA IX - Sud
Agence de la biomédecine Hôtel Dieu - 6 place Daviel 13224 MARSEILLE CEDEX 02
T : 04.91.56.52.18 F : 04.91.56.52.07
SRA VI - Ouest
Agence de la biomédecine CHRU Pontchaillou Rue Henri Le Guilloux – Bât. B2 35033 RENNES CEDEX 9
T : 02.99.28.41.23 F : 02.99.54.53.00
SRA VII - Ile-de-France – Centre – Les Antilles
Agence de la biomédecine CHU Bicêtre - Bâtiment Paul Langevin 78 rue du Gal Leclerc 94276 LE KREMLIN BICETRE
T : 01.58.46.15.40 F : 01.58.46.15.59
Siège de l’Agence de la biomédecine (ABM)
Agence de la biomédecine Unité de biovigilance 1, avenue du stade de France 93212 SAINT-DENIS LA PLAINE cedex
T : 01 55 93 64 53
F : 01 55 93 69 36
Registre France Greffe de Moelle
Agence de la biomédecine 1, avenue du stade de France 93212 SAINT-DENIS LA PLAINE cedex
T : 01 55 93 65 32
F : 01 49 98 37 14

24
QUE DECLARER EN BIOVIGILANCE ? 10 GRANDS PRINCIPES ! Déclarer immédiatement les incidents et effets indésirables graves de biovigilance susceptibles de mettre en jeu la sécurité d’autres patients, donneurs ou receveurs
! Les incidents et effets indésirables inattendus doivent être déclarés en biovigilance
! Les effets indésirables survenus chez les receveurs et liés ou susceptibles d’être liés à un défaut de qualité du greffon ou d’un PTA doivent être déclarés en biovigilance
! Les non conformités de qualifications biologique et/ou clinique du donneur ne seront à déclarer en biovigilance que si le greffon a effectiv ement été libéré par le « producteur »
! D’une manière plus générale par rapport au point précédent, les incidents gérés dans le cadre d’un système de management de la qualité et ayant abouti à la non libération du greffon ne sont pas à déclarer en biovigilance ! Les greffons libérés en dehors des spécifications * (nature du greffon, nombre,…) pourront être précisés dans le rapport annuel de biovigilance rédigé par le CLB, et dont le format est fixé par Décision du Directeur général de l’Afssaps (Art.R.1211-45 du CSP) ! Les effets indésirables survenus chez des receveurs de greffons libérés en dehors des spécifications devront être déclarés immédiatement s’i ls sont considérés comme graves sinon au plus tard dans les 15 jours
! Les incidents survenus au cours de la fabrication des produits thérapeutiques annexes ne seront à déclarer en biovigilance que si les lots de PTA ont effectiv ement été libérés et sont par conséquent susceptibles d’entraîner des incidents ou des effets indésirables ! Les incidents mettant en cause des dispositifs médicaux utilisés pour le conditionnement ou le transport des greffons ne seront à déclarer en biovigilance que s’i l y a une incidence sur le greffon lui-même et potentiellement chez le receveur. Par contre, ils devront systématiquement faire l ’objet d’une déclaration de matériovigilance.
! Indépendamment des précédents points, les notions de fréquence de survenue et d’effet-site devront être prises en compte et appréciées selon les conséquences possibles sur le donneur, le greffon ou le receveur
* Renvoi Glossaire
25
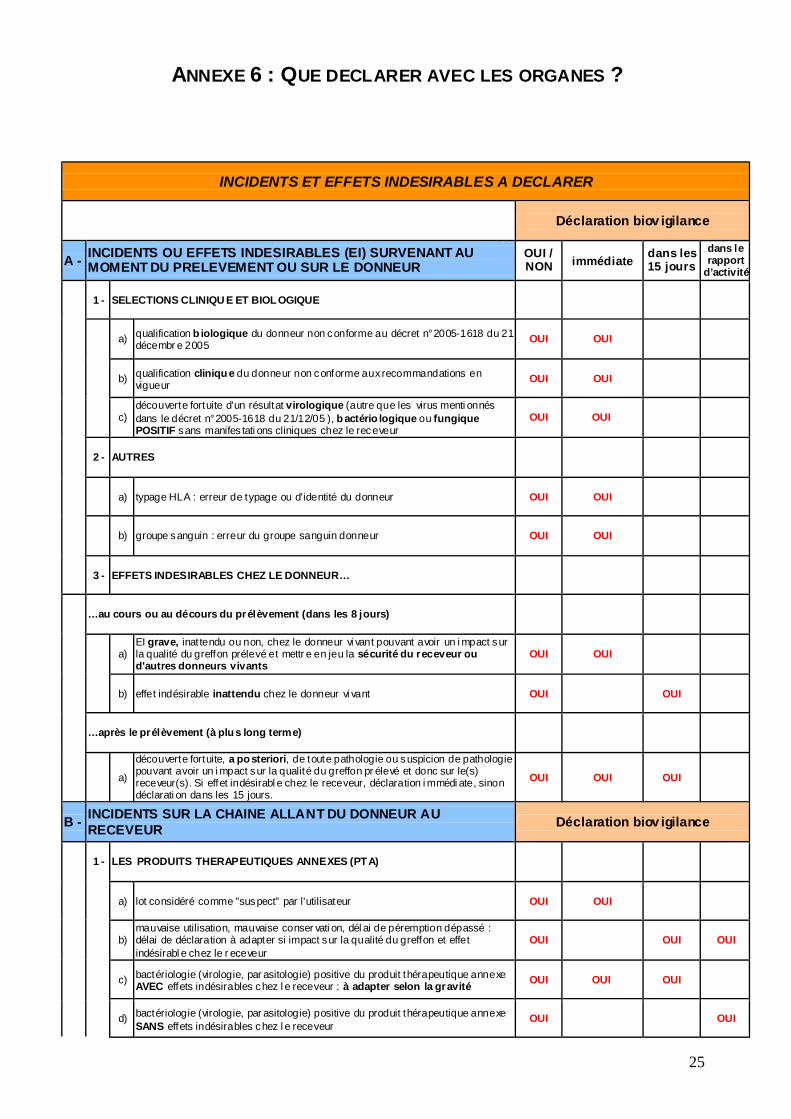
ANNEXE 6 : QUE DECLARER AVEC LES ORGANES ?
INCIDENTS ET EFFETS INDESIRABLES A DECLARER
Déclaration biov igilance
A - INCIDENTS OU EFFETS INDESIRABLES (EI) SURVENANT AU MOMENT DU PRELEVEMENT OU SUR LE DONNEUR
OUI / NON immédiate dans les
15 jours dans le rapport
d’activité
1 - SELECTIONS CLINIQU E ET BIOL OGIQUE
a) qualification b iologique du donneur non conforme au décret n°2005-1618 du 21 décembr e 2005 OUI OUI
b) qualification cliniqu e du donneur non conforme aux recommandations en vigueur OUI OUI
c) découverte fortuite d'un résultat virologique (autre que les virus menti onnés dans le décret n°2005-1618 du 21/12/05 ), b actério logique ou fungique POSITIF sans manifes tati ons cliniques chez le receveur
OUI OUI
2 - AUTRES
a) typage HLA : erreur de typage ou d'identité du donneur OUI OUI
b) groupe sanguin : erreur du groupe sanguin donneur OUI OUI
3 - EFFETS INDESIRABLES CHEZ LE DONNEUR…
…au cours ou au décours du prélèvement (dans les 8 jours)
a) EI grave, inattendu ou non, chez le donneur vi vant pouvant avoir un i mpact sur la qualité du greffon prélevé et mettr e en jeu la sécurité du receveur ou d'autres donneurs vivants
OUI OUI
b) effet indésirable inattendu chez le donneur vi vant OUI OUI
…après le prélèvement (à plu s long terme)
a)
découverte fortuite, a po steriori, de toute pathologie ou suspicion de pathologie pouvant avoir un i mpact sur la qualité du greffon pr élevé et donc sur le(s) receveur(s). Si effet indésirabl e chez le receveur, déclaration i mmédi ate, sinon déclarati on dans les 15 jours.
OUI OUI OUI
B - INCIDENTS SUR LA CHAINE ALLANT DU DONNEUR AU RECEVEUR Déclaration biov igilance
1 - LES PRODUITS THERAPEUTIQUES ANNEXES (PT A)
a) lot considéré comme "suspect" par l'utilisateur OUI OUI
b) mauvaise utilisation, mauvaise conser vati on, dél ai de péremption dépassé : délai de déclaration à adapter si impact sur la qualité du greffon et effet indésirabl e chez le r eceveur
OUI
OUI OUI
c) bactériologie (virologie, par asitologie) positive du produit thérapeutique annexe AVEC effets indésirables chez l e receveur : à adapter selon la gravité OUI OUI OUI
d) bactériologie (virologie, par asitologie) positive du produit thérapeutique annexe SANS effets indésirables chez l e receveur
OUI
OUI

26
e) traçabilité PTA défectueuse OUI
OUI
2 - LE CONDITIONNEMENT
a) non conformité du conditionnement aux recommandations du f abricant OUI OUI
b) conditi onnement (dispositif médical) défectueu x OUI OUI
C - INCIDENTS OU EFFETS INDESIRABLES (EI) SURVENANT AU MOMENT DE LA GREFFE OU SUR LE RECEVEUR Déclaration biov igilance
1 - DECES EN PER-OPER ATOIRE OU PERI-OPERATOIRE OUI OUI
2 - ERREUR DE GROUPE SANGUIN, HLA…
a) … entraînant l e décès du receveur, ou l a perte du greffon OUI OUI
b) ...sans conséquence pour le receveur OUI OUI
3 - EFFETS INDESIRABLES CHEZ LE RECEVEUR
… en per-opératoire ou p éri-opératoire
a) EI grave, inattendu ou non, pouvant mettre en j eu la sécurité d'autres receveurs OUI OUI
b) EI inattendus OUI OUI
…après la greffe (à plu s long terme)
a) symptomatologie post-greffe grave, inattendue ou non OUI OUI
27
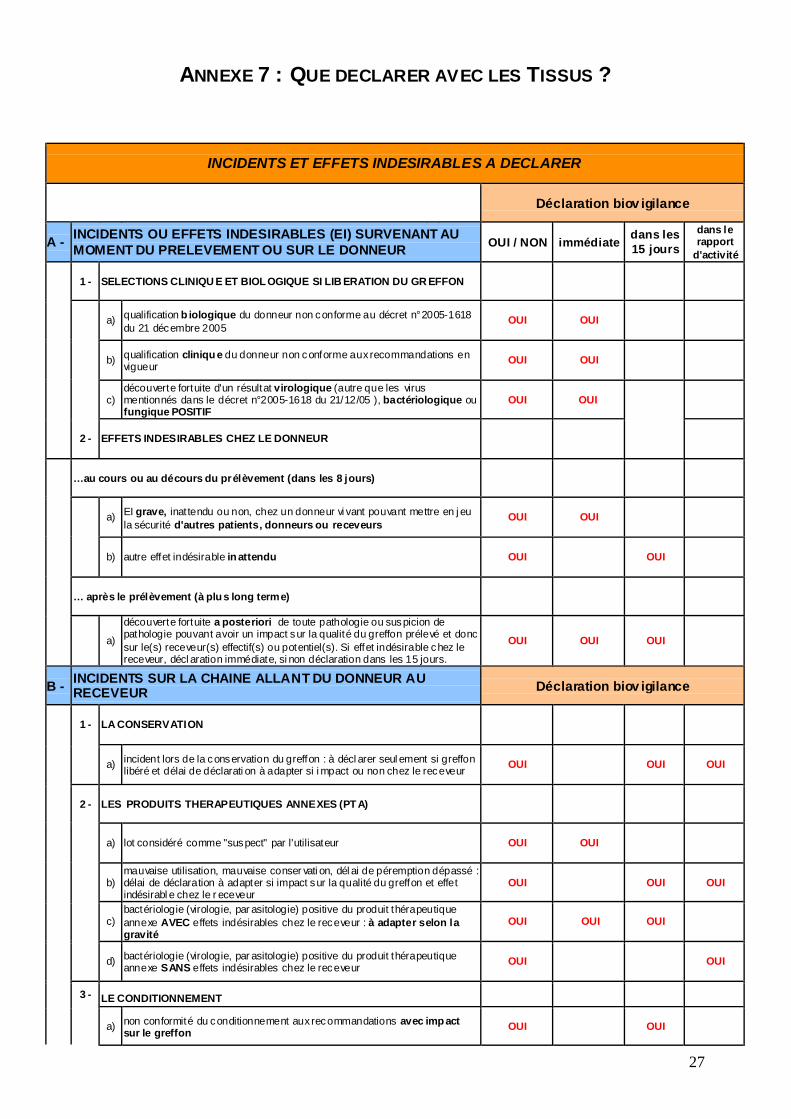
ANNEXE 7 : QUE DECLARER AVEC LES TISSUS ?
INCIDENTS ET EFFETS INDESIRABLES A DECLARER
Déclaration biov igilance
A - INCIDENTS OU EFFETS INDESIRABLES (EI) SURVENANT AU MOMENT DU PRELEVEMENT OU SUR LE DONNEUR OUI / NON immédiate dans les
15 jours dans le rapport
d'activité
1 - SELECTIONS CLINIQU E ET BIOL OGIQUE SI LIB ERATION DU GR EFFON
a) qualification b iologique du donneur non conforme au décret n°2005-1618 du 21 décembre 2005
OUI OUI
b) qualification cliniqu e du donneur non conforme aux recommandations en vigueur OUI OUI
c) découverte fortuite d'un résultat virologique (autre que les virus mentionnés dans le décret n°2005-1618 du 21/12/05 ), bactériologique ou fungique POSITIF
OUI OUI
2 - EFFETS INDESIRABLES CHEZ LE DONNEUR
…au cours ou au décours du prélèvement (dans les 8 jours)
a) EI grave, inattendu ou non, chez un donneur vi vant pouvant mettre en j eu la sécurité d'autres patients, donneurs ou receveurs
OUI OUI
b) autre effet indésirable in attendu OUI OUI
… après le prélèvement (à plu s long terme)
a)
découverte fortuite a posteriori de toute pathologie ou suspicion de pathologie pouvant avoir un impact sur la qualité du greffon prélevé et donc sur le(s) receveur(s) effectif(s) ou potentiel(s). Si effet indésirable chez le receveur, décl aration immédiate, si non déclaration dans les 15 jours.
OUI OUI OUI
B - INCIDENTS SUR LA CHAINE ALLANT DU DONNEUR AU RECEVEUR Déclaration biov igilance
1 - LA CONSERVATION
a) incident lors de la conservation du greffon : à décl arer seul ement si greffon libéré et délai de déclarati on à adapter si i mpact ou non chez le receveur OUI OUI OUI
2 - LES PRODUITS THERAPEUTIQUES ANNEXES (PT A)
a) lot considéré comme "suspect" par l'utilisateur OUI OUI
b) mauvaise utilisation, mauvaise conser vati on, dél ai de péremption dépassé : délai de déclaration à adapter si impact sur la qualité du greffon et effet indésirabl e chez le r eceveur
OUI
OUI OUI
c) bactériologie (virologie, par asitologie) positive du produit thérapeutique annexe AVEC effets indésirables chez le receveur : à adapter selon la gravité
OUI OUI OUI
d) bactériologie (virologie, par asitologie) positive du produit thérapeutique annexe SANS effets indésirables chez le receveur OUI
OUI
3 - LE CONDITIONNEMENT
a) non conformité du conditionnement aux recommandations avec imp act sur le greffon OUI OUI

28
b) conditi onnement (dispositif médical) défectueu x avec imp act sur le greffon OUI OUI
4 - LE TRANSPORT
a) incident lors du transport : délai de décl aration à adapter si impact ou non chez le receveur OUI OUI OUI
5 - LA QU ALITE DU GREFFON
a) greffon libéré en dehors des spécifications : dél ai de déclaration à adapter si impac t ou non chez le receveur OUI OUI OUI
6 - LA TRAC ABILITE
a) traçabilité du greffon défec tueuse : délai de déclaration à adapter si impact possible sur d’ autres patients OUI OUI OUI
b) traçabilité PTA défectueuse OUI
OUI
C - INCIDENTS OU EFFETS INDESIRABLES (EI) SURVENANT AU MOMENT DE LA GREFFE OU SUR LE RECEVEUR Déclaration biov igilance
1 - PERTE DU GREFFON
a) prélevé et libéré par le " producteur" OUI
OUI
2 - LA GREFFE
a) erreur d'attribution OUI OUI
3 - EFFETS INDESIRABLES CHEZ LE RECEVEUR
… au cours ou au d écours de la greffe (dans les 8 jours)
a) EI grave, inattendu ou non, pouvant mettre en j eu la sécurité d'autres receveurs
OUI OUI
b) EI inattendus OUI OUI
…après la greffe (à plu s long terme)
a) symptomatologie post-greffe grave, inattendue ou non OUI OUI
b) mauvaise ou non prise de greffe OUI
OUI
29
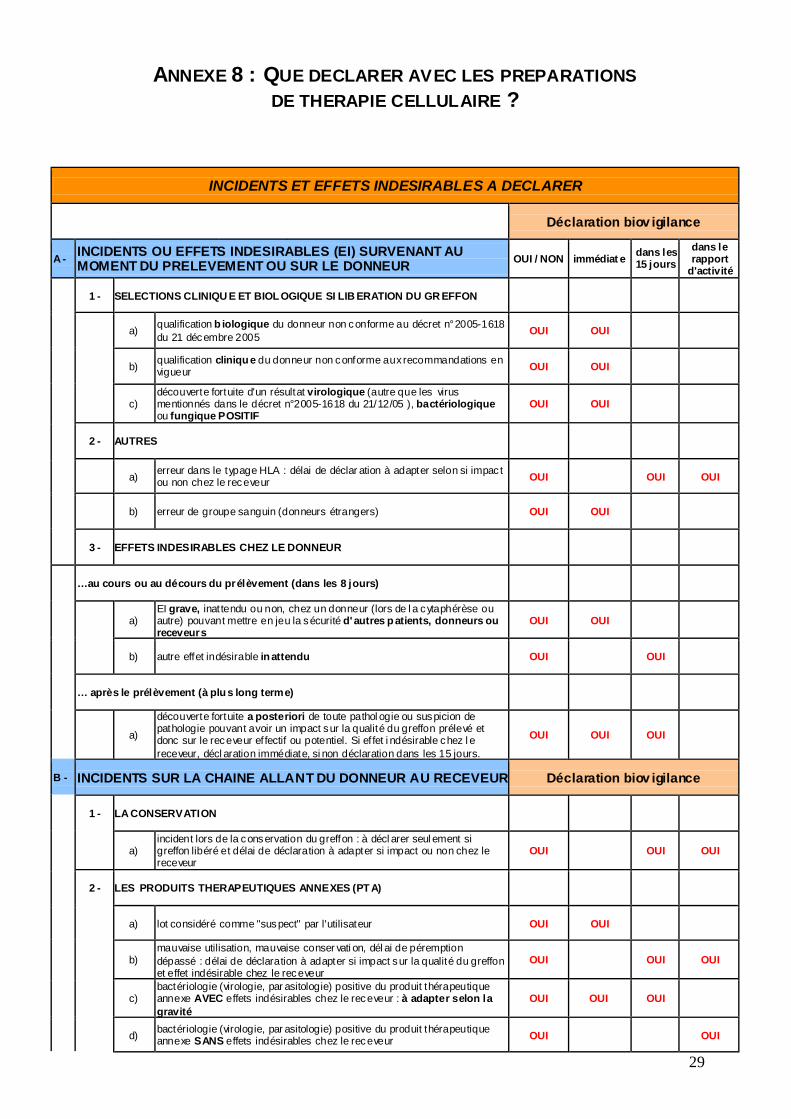
ANNEXE 8 : QUE DECLARER AVEC LES PREPARATIONS DE THERAPIE CELLULAIRE ?
INCIDENTS ET EFFETS INDESIRABLES A DECLARER
Déclaration biov igilance
A - INCIDENTS OU EFFETS INDESIRABLES (EI) SURVENANT AU MOMENT DU PRELEVEMENT OU SUR LE DONNEUR OUI / NON immédiat e dans les
15 jours dans le rapport
d'activité
1 - SELECTIONS CLINIQU E ET BIOL OGIQUE SI LIB ERATION DU GR EFFON
a) qualification b iologique du donneur non conforme au décret n°2005-1618 du 21 décembre 2005 OUI OUI
b) qualification cliniqu e du donneur non conforme aux recommandations en vigueur OUI OUI
c) découverte fortuite d'un résultat virologique (autre que les virus mentionnés dans le décret n°2005-1618 du 21/12/05 ), bactériologique ou fungique POSITIF
OUI OUI
2 - AUTRES
a) erreur dans le typage HLA : délai de déclar ation à adapter selon si impac t ou non chez le receveur OUI OUI OUI
b) erreur de groupe sanguin (donneurs étrangers) OUI OUI
3 - EFFETS INDESIRABLES CHEZ LE DONNEUR
…au cours ou au décours du prélèvement (dans les 8 jours)
a) EI grave, inattendu ou non, chez un donneur (lors de l a cytaphérèse ou autre) pouvant mettre en jeu la sécurité d'autres p atients, donneurs ou receveurs
OUI OUI
b) autre effet indésirable in attendu OUI OUI
… après le prélèvement (à plu s long terme)
a)
découverte fortuite a posteriori de toute pathol ogie ou suspicion de pathologie pouvant avoir un impact sur la qualité du greffon prélevé et donc sur le receveur ef fectif ou potentiel. Si ef fet i ndésirable chez l e receveur, décl aration immédiate, si non déclaration dans les 15 jours.
OUI OUI OUI
B - INCIDENTS SUR LA CHAINE ALLANT DU DONNEUR AU RECEVEUR Déclaration biov igilance
1 - LA CONSERVATION
a) incident lors de la conservation du greffon : à décl arer seul ement si greffon libéré et délai de déclaration à adapter si impact ou non chez le receveur
OUI
OUI OUI
2 - LES PRODUITS THERAPEUTIQUES ANNEXES (PT A)
a) lot considéré comme "suspect" par l'utilisateur OUI OUI
b) mauvaise utilisation, mauvaise conser vati on, dél ai de péremption dépassé : délai de déclaration à adapter si impact sur la qualité du greffon et effet indésirable chez le receveur
OUI
OUI OUI
c) bactériologie (virologie, par asitologie) positive du produit thérapeutique annexe AVEC effets indésirables chez le receveur : à adapter selon la gravité
OUI OUI OUI
d) bactériologie (virologie, par asitologie) positive du produit thérapeutique annexe SANS effets indésirables chez le receveur OUI
OUI

30
3 - LE CONDITIONNEMENT
a) non conformité du conditionnement aux recommandations avec imp act sur le greffon OUI
OUI
b) conditi onnement (dispositif médical) défectueu x avec imp act sur le greffon
OUI
OUI
4 - LE TRANSPORT
a) incident lors du transport : délai de décl aration à adapter si impact ou non chez le receveur OUI
OUI OUI
5 - LA QU ALITE DU GREFFON
a) greffon libéré en dehors des spécifications : dél ai de déclaration à adapter si impac t ou non chez le receveur OUI OUI OUI
6 - LA TRAC ABILITE
a) traçabilité du greffon défec tueuse : délai de déclaration à adapter si impac t possible sur d’autres patients OUI OUI OUI
b) traçabilité PTA défectueuse OUI
OUI
C - INCIDENTS OU EFFETS INDESIRABLES (EI) SURVENANT AU MOMENT DE LA GREFFE OU SUR LE RECEVEUR Déclaration biov igilance
1 - PERTE DU GREFFON
a) prélevé et libéré par le " producteur" OUI
OUI
b) autre : OUI OUI
2 - LA GREFFE
a) erreur d'attribution OUI OUI
3 - EFFETS INDESIRABLES CHEZ LE RECEVEUR
… au cours ou au d écours de la greffe (dans les 8 jours)
a) EI grave, inattendu ou non, pouvant mettre en j eu la sécurité d'autres receveurs OUI OUI
b) EI inattendus OUI OUI
…après la greffe (à plu s long terme)
a) symptomatologie post-greffe grave, inattendue ou non OUI OUI
c) mauvaise ou non prise de greffe OUI
OUI

31
ABREVIATIONS
ABM Agence de la Biomédecine
AMM Autorisation de Mise sur le Marché
AMP Assistance Médicale à la Procréation
CHU Centre Hospitalo-Universitaire
CLB Correspondant Local de Biovigilance
CLIN Comité de Lutte contre les Infections Nosocomiales
CSH Cellules Souches Hématopoïétiques
CSP Code de la santé publique
ES Etablissement de Santé
FGM France Greffe de Moelle
HLA Human Leucocyte Antigen (typage d’antigènes leucocytaires)
PMO Prélèvement Multi-Organes
PPM Prélèvement Post-Mortem
PTA Produit Thérapeutique Annexe
RFGM Registre France Greffe de Moelle
SRA Service de Régulation et d’Appui (de l’Agence de la biomédecine)

32
GLOSSAIRE
AGENCE FRANÇAISE DE SECURITE SANITAIRE DES PRODUITS DE SANTE (AFSSAPS)
Etablissement public de l’Etat à caractère administratif, placé sous tutelle du ministre chargé de la santé. Son champ de compétence s’étend à l’ensemble des produits à finalité sanitaire destinés à l’homme, y compris les produits à finalité cosmétique. Pour ces produits, l ’Afssaps a ssure cinq missions principales :
- l ’évaluation, - le contrôle, - l ’inspection, - la vigilance, - l ’information des professionnels de santé et du public.
Elle dispose d’un pouvoir de police sanitaire (ensemble des moyens juridiques et matériels ayant pour but d’assurer la sécurité sanitaire). Elle organise et coordonne la mise en œuvre des systèmes de vigilance relatifs aux produits de santé, en particulier par le recueil et l ’évaluation des informations concernant les effets indésirables liés à l ’utilisation de ces produits et assure la fonction d’alerte sanitaire en cas de risque pour la santé publique. ASSISTANCE MEDICALE A LA PROCREATION (AMP)
L’assi stance médicale à la procréation s’entend des pratiques cliniques et biologiques permettant la conception in vitro, le transfert d’embryons et l ’insémination artificielle, ainsi que toute technique d’effet équivalent permettant la procréation en dehors du processu s naturel, dont la l iste est fixée par arrêté du ministre chargé de la santé, après avis de l’agence de la biomédecine (Art L.2141-1 du Code de la santé publique). Elle est destinée à répondre à la demande parentale d’un couple. Elle a pour objet de remédier à l ’infertili té dont le caractère pathologique a été médicalement diagnostiqué ou d’éviter la transmission à l’enfant ou à un membre du couple d’une maladie d’une particulière gravité (Art. L. 2141-2 du Code de la santé publique). LIBERATION EN DEHORS DES SPECIFICATIONS
Un greffon peut être libéré par le responsable de la banque même s’i l ne répond pas aux spécifications précisées dans les procédés/produits autorisés par l ’Afssaps. Cette l ibération sera notamment laissée au choix du clinicien sur la base de l’évaluation du rapport bénéfice/risque. EFFET INDESIRABLE
Manifestation nocive et non recherchée, survenant chez un patient, donneur vivant ou receveur, attribuée à un produit ou une activité relevant du champ de la biovigilance.
Exemples : allergie à un PTA, un excipient entraînant fièvre, frissons, ou douleur, érythème au site d'injection, kératite après greffe de cornée suite à une contamination du milieu de conservation Est considéré comme grav e l 'effet indésirable :
- pouvant entraîner la mort, - su sceptible de mettre en jeu le pronostic vital du patient, - su sceptible de mettre en jeu la sécurité d'un ou plusieurs donneurs vivants et/ou d'un ou
plusieurs receveurs.
Exemples : transmission d'une maladie infectieuse (paludisme), séroconversion receveur, choc anaphylactique, décès…

33
INCIDENT
Défaillance ou altération d'un élément isolé, d'un processu s ou d'un système, liée aux activités entrant dans la champ de compétence de la biovigilance et susceptible d'entraîner un effet indésirable chez le patient, le donneur vivant ou le receveur. Exemples : lot défectueux de PTA libéré par la fabricant, nécessitant l 'uti lisation d'un nouveau lot et le rappel de toutes les unités de ce lot Est considéré comme grav e :
- l 'incident susceptible de se répéter et pouvant mettre en jeu la sécurité d'un ou plusieurs patients, donneurs vivants ou receveurs
- tout incident pouvant entraîner un effet indésirable grave.
Exemples : contamination microbiologique ou parasitologique d'un PTA ou d'un greffon avant sa libération par la banque, défail lance du système de traçabilité, défaillance au cours d'un protocole d'inactivation virale, défaut d’alimentation en azote d’une cuve contenant des greffons de cellules souches hématopoïétiques PRODUITS CELLULAIRES A FINALITE THERAPEUTIQUE (Art.L.1243-1)
A l'exception des produits sanguins labiles, sont des produits cellulaires à finalité thérapeutique les cellules humaines utilisées à des fins thérapeutiques autologues ou allogéniques, quel que soit leur niveau de transformation, y compris leurs dérivés. Lorsque ces produits cellulaires à finalité thérapeutique sont des spécialités pharmaceutiques ou d'autres médicaments fabriqués industriellement, ils sont régis par les dispositions du titre II du livre Ier de la cinquième partie. Ils entrent dans le champ de la pharmacovigilance. Dans les autres cas, ce sont des préparations de thérapie cellulaire, y compris lorsque les cellules humaines servent à transférer du matériel génétique."
Seule cette dernière catégorie est soumise à la biovigilance. Exemples : - thérapie substitutive : cellules souches hématopoïétiques, cellules nerveuses… - immunothérapie adoptive : cellules dendritiques, macrophages… - thérapeutiques de reconstruction : kératinocytes, chondrocytes, myoblastes… PRODUITS THERAPEUTIQUES ANNEXES
Les produits thérapeutiques annexes sont définis comme tout produit, à l ’exception de dispositifs médicaux, entrant en contact avec des organes, tissu s, cellules ou produits issu s du corps humain ou d’origine animale au cours de leur conservation, de leur préparation, de leur transformation, de leur conditionnement ou de leur transport avant leur uti lisation thérapeutique chez l’homme, ainsi que tout produit entrant en contact avec des embryons dans le cadre d’une activité d’assistance médicale à la procréation. Selon les cas, ce s produits peuvent avoir reçu une autorisation de PTA, ce sera le cas notamment des milieux de conservation des organes, des cornées mais également des milieux utilisés en assistance médicale à la procréation. Ils peuvent également posséder une autorisation de mise sur le marché (AMM) tels que l'albumine, les facteurs de croissance, les antibiotiques présents dans les milieux de culture des cellules ou avoir le statut de dispositif médical ou de dispositif médical de diagnostic in vitro… RESIDUS OPERATOIRES
Ils désignent les tissu s, cellules et produits du corps humain recueillis à l ’occasion d’une intervention médicale lorsqu’ils sont conservés en vue d’une util isation ultérieure (arrêté du 1er avril 1997).
Ils sont prélevés en premier lieu pour le bénéfice direct du patient opéré et peuvent être utilisés à des fins scientifiques ou thérapeutiques. Dans ce dernier cas, ils doivent obligatoirement être adressés à une banque de tissus autorisée (exemples : membranes amniotiques, têtes fémorales).

34
Les règles de sécurité sanitaire applicables en cas de prélèvement de résidus opératoires sont identiques à celles effectuées lors des prélèvements de tissu s sur une personne décédée. TISSUS
Les tissus sont définis comme toute partie constitutive du corps humain constitué de cellules (directive 2004/23/CE).
Exemples : os, peau, valves cardiaques, cornées, tendons, artères, veines, dure-mères ainsi que les tissu s fœtaux recueillis lors d'avortement, le placenta et le cordon ombilical
En 1997, les bonnes pratiques de prélèvement donnent comme définition aux tissu s « les éléments prélevés sur le corps humain que sont notamment les os, les éléments de l 'appareil locomoteur, les valves cardiaques, les vaisseaux (artères et veines), la peau, les tissu s endocriniens selon la réglementation applicable. Cette réglementation ne correspond en fait qu'à des textes interdisant, pour des raisons de sécurité, le prélèvement de certains tissu s tels que les dures-mères ou les tympans." TRAÇABILITE
On entend par traçabilité l 'ensemble des informations et des mesures permettant de suivre et de retrouver rapidement chacune des étapes allant de l’examen clinique du donneur à l 'utilisation thérapeutique de cet élément ou produit du corps humain en passant par le prélèvement, la transformation, la conservation, le transport, la distribution et la dispensation à un patient. La traçabilité, établie à partir d'une codification préservant l'anonymat des personnes, permet d’établir un lien entre le donneur et le ou les receveur(s).

35
REGLEMENTATION APPLICABLE Textes généraux
Décret n° 2006-477 du 26 avril 2006 modifiant le chapitre 1er du titre II du livre 1er de la première partie du code de la santé publique relatif aux recherches biomédicales (dispositions réglementaires) Directive 2006/17/CE de la commission du 8 février 2006 portant application de la directive 2004/23/CE du Parlement européen et du Conseil concernant certaines exigences techniques relatives au don, à l ’obtention et au contrôle de tissus et de cellules d’origine humaine Décret n°2005-1618 du 21 décembre 2005 relatif aux règles de sécurité sanitaire portant sur le prélèvement et l ’uti lisation des éléments et produits du corps humain et modifiant le code de la santé publique (partie réglementaire) Arrêté du 21 décembre 2005 pris en application des articles R.1211-14, R.1211-15, R.1211-16 et R.1211-21 du code de la santé publique Décret n°2005-1391 du 8 novembre 2005 relatif au transfert à l ’Agence de la biomédecine du fichier des donneurs tenu par l ’association France greffe de moelle Décret n° 2004-829 du 19 août 2004 relatif aux conditions d’autorisation de mise sur le marché des produits thérapeutiques annexes et modifiant le code de la santé publique (partie réglementaire) Loi n° 2004-800 du 6 août 2004 relative à la bioéthique Directive 2004/23/CE du 31 mars 2004 du Parlement européen et du Conseil relative à l 'établissement de normes de qualité et de sécurité pour le don, l 'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissu s et cellules humains Décret n° 2003-1206 du 12 décembre 2003 portant organisation de la biovigilance et modifiant le code de la santé publique (partie réglementaire) Loi n° 98-535 du 1er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l’homme Arrêté du 9 octobre 1995 fixant les modalités de transmission des informations nécessaires au suivi et à la traçabilité des éléments et produits du corps humain (organes, tissus et cellules ou leurs dérivés) utilisés chez l’homme à de fins thérapeutiques Textes en rapport avec les organes Arrêté du 2 août 2005 fixant la liste des organes pour lesquels le prélèvement sur une personne décédée présentant un arrêt cardiaque et respiratoire persistant est autorisé Arrêté du 27 février 1998 portant homologation des règles de bonnes pratiques relatives au prélèvement d’organes à finalité thérapeutique sur personne décédée Textes en rapport avec les tissus Arrêté du 2 août 2005 fixant la liste des tissu s et des cellules pour lesquels le prélèvement sur une personne décédée présentant un arrêt cardiaque et respiratoire persistant est autorisé Décret n° 2002-1125 du 2 septembre 2002 relatif aux conditions d’autorisation des procédés de préparation, de conservation et de transformation de tissus du corps humain et de leurs dérivés mis en œuvre en vue d’un usage thérapeutique et modifiant le code de la santé publique

36
Arrêté du 29 décembre 1998 portant homologation des règles de bonnes pratiques relatives à la conservation, à la transformation et au transport des tissu s d’origine humaine utilisés à de fins thérapeutiques Arrêté du 1er avril 1997 portant homologation des règles de bonnes pratiques relatives au prélèvement des tissus et au recueil des résidus opératoires issu s du corps humain utilisés à de fins thérapeutiques Textes en rapport avec les produits de thérapie cellulaire Décret n° 2001-909 du 1er octobre 2001 relatif aux cellules et aux produits de thérapies génique et cellulaire fixant les conditions d’autorisation des établissements, organismes, procédés, produits et protocoles d’essais cliniques et modifiant le code de la santé publique Arrêté du 16 décembre 1998 portant homologation des règles de bonnes pratiques relatives au prélèvement, au transport, à la transformation, y compris la conservation, de cellules souches hématopoïétiques issues du corps humain et des cellules mononucléées sanguines à des fins thérapeutiques





























