Embed Size (px)
Citation preview

Gutartige Tumoren und tumorähnliche Läsionendes KnochensPer-Ulf Tunn1, Hans Roland Dürr2
1Klinik für Chirurgie und Chirurgische Onkologie, Universitätsmedizin Berlin, Charité Campus Buch,Helios Kliniken Berlin2Schwerpunkt Tumororthopädie, Orthopädische Klinik, Klinikum der LMU München, Standort Groß-hadern
SchlüsselwörterGutartige Knochentumoren, tumorähnliche Läsionen, Diag-nose, Therapie
ZusammenfassungBenigne Knochentumoren und tumorähnliche Läsionensind bei weitem häufiger als primär maligne Knochentumo-ren. Ein Großteil der Läsionen kann aus dem Röntgenbildund der klinischen Symptomatik diagnostiziert werden undbedarf keiner weiteren Therapie. Trotzdem können benigneTumoren wie z. B. der Riesenzelltumor auch Metastasensetzen und nichttumoröse Läsionen, wie z. B. aneurysmaleKnochenzysten oder die fibröse Dysplasie therapeutisch er-heblich herausfordern. Über die Jahre hat sich das Wissenum Genese und Therapie der Läsionen gewandelt, das Os-teoidosteom wird so heute nur noch in den seltensten Fäl-len offen reseziert werden müssen. Multiple kartilaginäreExostosen sind im langfristigen Verlauf kontrollpflichtig, ei-ne umfassende Beratung der Patienten und Eltern ist not-wendig. Chondrome und Enchondrome bereiten diagnos-tisch aufgrund fehlender klarer Differenzierung zu hochdif-ferenzierten Chondrosarkomen manchmal Probleme, sindjedoch zumeist nicht therapiepflichtige Zufallsbefunde. DasChondroblastom wie auch das Chondromyxoidfibrom sindso selten, dass nur wenige Kollegen je einen Patienten se-hen werden. Die Therapie der zystischen Läsionen ist viel-fältig, wesentlich ist natürlich die Differenzierung in eherbenigne verlaufende juvenile und eher aggressiv verlaufen-de aneurysmale Knochenzysten. Letztere müssen auch vomteleangiektatischen Osteosarkom abgegrenzt werden. Beider fibrösen Dysplasie wurden in den letzten Jahren einigeArbeiten zur Bisphosphonattherapie publiziert, die den Ein-satz der Präparate zumindest beim Erwachsenen erwä-genswert machen.
KeywordsBenigne bone tumours, tumor-like lesions, diagnosis, ther-apy
SummaryBenign bone tumours and tumour-like lesions of the boneare much more common than primary malignant bone tu-mours. The majority of the lesions can be diagnosed fromthe radiographs and the clinical symptoms and require nofurther therapy. Nevertheless benign tumours as forexample the giant cell tumour are capable of distant meta-stasis, and some tumour-like lesions such as the aneurys-mal bone cysts or fibrous dysplasia may cause substantialtherapeutical challenges. Over the years the knowledgeabout genesis and therapy of the lesions has changed, sonowadays osteoidosteomas will be treated only seldomlyby open resection. Multiple cartilaginous exostoses dorequire a life-long control, comprehensive advice to the pa-tient and, in childhood, the parents is necessary. Chondro-mas and enchondromas sometimes provide diagnosticproblems due to a missing distinct differentiation fromhighly differentiated chondrosarcomas. However most ofthem are coincidental findings requiring no therapy. Os-teoblastomas, chondroblastomas as also the chondromy-xoidfibroma are so exceedingly rare that only few col-leagues will see a patient. The therapy of cystic lesionsoffers a lot of options, most important is the differentiationinto rather benign juvenile and rather aggressive aneurys-mal bone cysts. The latter must be distinguished also fromteleangiectatic osteosarcoma. In fibrous dysplasia studiesregarding bisphosphonats have been published in the lastyears, which consider the therapy of at least adult patientsworthwhile.
Benign bone tumours and tumour-like lesions of thebone
arthritis + rheuma 2007; 27: 129–140
E twa 40 bis 50 Prozent aller Kno-chentumoren sind als benigne zubeurteilen; berücksichtigt man
auch die die tumorähnlichen Läsionen, so
überwiegen benigne Läsionen bei weitem.Die prinzipielle Einteilung erfolgt dabeinach der historisch gewachsenen und fort-laufend ergänzten Klassifikation der WHO,
die als wesentliches Kriterium eine eventu-ell vorhandene Matrixproduktion der Läsi-on oder sonstige histologische, klinischeund radiologische Kriterien verwendet(Tab. 1) (1, 2). Oft ist die Differenzierungder Läsion aufgrund allein histologischerKriterien nicht eindeutig möglich. Radiolo-gie und Klinik sind deshalb von besondererBedeutung. Umgekehrt lassen sich diemeisten der benignen Knochenläsionen al-lein schon aufgrund ihrer typischen Mor-phologie im Röntgenbild und des klinischenVerlaufs ohne histologische Sicherung di-agnostisch zuordnen.
Insbesondere die sogenannten tumor-ähnlichen, also nicht neoplastischen Läsio-nen, sind in vielen Fällen radiologische Zu-fallsbefunde, die keiner weiteren Diagnos-tik und Therapie bedürfen. Die Grenzenzwischen „echten“ Tumoren und tumor-ähnlichen Läsionen sind dabei eher his-torisch gewachsen als definitiv gesichertzu verstehen. So sind die typischen solitä-ren kartilaginären Exostosen vermutlicheher Resultate einer pathologischen Ent-wicklung von Teilen der Wachstumsfugeals originäre Tumoren, Hämangiome sindeher Hamartome als Neoplasien währenddie aneurysmale Knochenzyste umgekehrtaufgrund neuerer Untersuchungen eherden Tumoren zugerechnet werden muss.Viele Läsionen, wie die pigmentierte villo-noduläre Synovitis, zeigen auch Kriterienbeider Gruppen. Auch finden sich inner-halb der benignen Tumoren Läsionen, wieder Riesenzelltumor, die inklusive einerMetastasierung vergleichsweise maligneverlaufen können.
Eine Stadieneinteilung der benignenKnochentumoren nach Enneking (3) oderbeim Riesenzelltumor auch nach Campa-nacci (4) ist möglich, erfolgt jedoch auf-
Tumororthopädie 129
© 2007 Schattauer GmbH
arthritis + rheuma 3/2007

arthritis + rheuma 3/2007
130
Tunn, Dürr
grund fehlender klinischer Relevanz eherselten.
DiagnostikUnverzichtbar und in vielen Fällen völligausreichend ist nach wie vor eine Röntgen-aufnahme der Läsion in zwei Ebenen. ZurFrage der Binnenstruktur, zum Nachweisossifizierter Matrix, zur Klärung der Rand-begrenzung wie auch in der Abgrenzung zutumorsimulierenden Läsionen, wie Ermü-dungsfrakturen, ist die Computertomogra-fie (CT), vor allem in Dünnschichttechnik,eine sinnvolle Ergänzung des konventionel-len Röntgens. Die Magnetresonanztomo-grafie (MRT) erlaubt die präzise Beschrei-bung des nicht ossären Läsionsinhalts wieauch die vergleichsweise exakte Abgren-zung der Läsion hin zu den Weichteilen.
Auch die Frage der Kontrastmittelaufnahmekann in einigen Läsionen (z.B. Enchon-drom) wichtige Zusatzinformationen ge-ben. In der Planung einer Biopsie erlaubt siedie Lokalisation der sinnvollsten Zielregi-on. Der Skelettszintigrafie wie auch der Po-sitronenemissionstomografie (PET oderPET-CT) kommt insbesondere im Scree-ning auf weitere Herde, wie auch in der Fra-ge der biologischen Aktivität einzelner Lä-sionen, Bedeutung zu (5).
Knochenbildende TumorenOsteomOsteome sind in der Regel Zufallsbefundbei asymptomatischen Patienten (Abb. 1).Sie existieren sowohl als intramedulläre(Enostose, Knocheninsel) wie auch als jux-
Tab. 1Einteilung und Häufigkeitprimär benigner Tumorendes Knochens
primär benigne Tumoren des Knochens Häufigkeit (%)
knochenbildende Tumoren● Osteom● Osteoidosteom● Osteoblastom
1003
knorpelbildende Tumoren● kartilaginäre Exostosen● Chrondrome● Chrondroblastom● Chrondromyxoidfibrom
48230502
Riesenzelltumor 10
vaskuläre Tumoren● Hämangiom● Glomustumor
04<1
intraossäre Weichgewegstumoren● Desmoidtumor (Fibromatose)● Lipom● benignes fibröses Histiozytom
<1<102
intraossäre neurogene Tumoren● Neurinom● Neurilemmom
<1<1
tumorähnliche Läsionen● juvenile Knochenzysten● aneurysmale Knochenzyste● fibröse Dysplasie● steofibröse Dysplasie● Langerhans-Zell-Histiozytose● fibröser metaphysärer Defekt● pigmentierte villonoduläre Synovitis● Morbus Paget● .......
arthritis + rheuma 3/2007
132
Tunn, Dürr
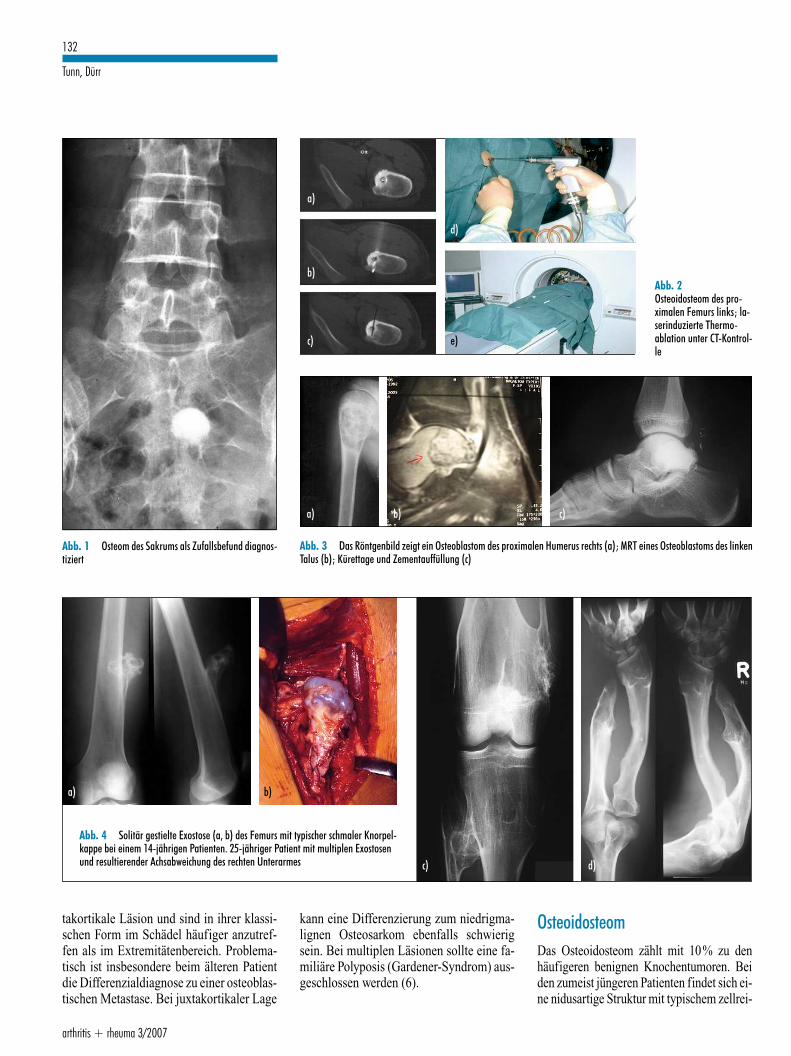
Abb. 1 Osteom des Sakrums als Zufallsbefund diagnos-tiziert
Abb. 2Osteoidosteom des pro-ximalen Femurs links; la-serinduzierte Thermo-ablation unter CT-Kontrol-le
b)
c)
d)
e)
a)
Abb. 3 Das Röntgenbild zeigt ein Osteoblastom des proximalen Humerus rechts (a); MRT eines Osteoblastoms des linkenTalus (b); Kürettage und Zementauffüllung (c)
a) b) c)
Abb. 4 Solitär gestielte Exostose (a, b) des Femurs mit typischer schmaler Knorpel-kappe bei einem 14-jährigen Patienten. 25-jähriger Patient mit multiplen Exostosenund resultierender Achsabweichung des rechten Unterarmes
a)
c)
b)
d)
takortikale Läsion und sind in ihrer klassi-schen Form im Schädel häufiger anzutref-fen als im Extremitätenbereich. Problema-tisch ist insbesondere beim älteren Patientdie Differenzialdiagnose zu einer osteoblas-tischen Metastase. Bei juxtakortikaler Lage
kann eine Differenzierung zum niedrigma-lignen Osteosarkom ebenfalls schwierigsein. Bei multiplen Läsionen sollte eine fa-miliäre Polyposis (Gardener-Syndrom) aus-geschlossen werden (6).
OsteoidosteomDas Osteoidosteom zählt mit 10% zu denhäufigeren benignen Knochentumoren. Beiden zumeist jüngeren Patienten findet sich ei-ne nidusartige Struktur mit typischem zellrei-

Abb. 6 Beispiel der malignen Transformation eines Enchondroms des distalen Femurs; auffällig ist hier das Verschwindender ehemals vorhandenen Kalzifikation im distalen Läsionsbereich sowie die neu entwickelte kortikale Destruktion
a) b) c)
arthritis + rheuma 3/2007
133
Benigne Tumoren und tumorähnliche Läsionen des Knochens
chen, stark vaskularisiertem Gewebe, meistnicht größer als ein Zentimeter, umgeben voneiner oft ausgedehnten Knochensklerose. DieLäsion ist schmerzhaft, da der Tumor Prosta-glandine produziert. In vielen Fällen führtdeshalb die Gabe eines nichtsteroidalen An-tirheumatikums („Aspirin-Test“) zur Be-schwerdelinderung. Ob es sich bei der Läsionum einen tatsächlichen Tumor handelt, bleibtsuspekt, da eine Ausheilung nach zum Teilmehrjährigem Verlauf spontan erfolgen kann.Die klassische Therapie ist die Resektion desmanchmal schwierig zu findenden Nidus.Abgelöst wurde diese durch mittlerweile alsStandard eingesetzte minimalinvasive Ver-fahren, bei denen typischerweise CT-gesteu-ert eine Thermoablation (Laser- oder Radio-frequenzablation) oder eine Ausbohrung mit/ohne Ethanolapplikation erfolgt (Abb. 2). DieRezidivraten liegen bei entsprechender Tech-nik unter zehn Prozent (siehe auch Beitragvon Trumm et al. „Moderne interventionelleVerfahren bei primären Knochentumoren undKnochenmetastasen“ auf Seite 169 ff. in die-sem Heft). Problematisch ist in einigen Fällendie Diagnose der Läsion, da bei gelenknaherLage ausgedehnte reaktive Synovitiden deneigentlichen Herd sowohl klinisch wie auchradiologisch maskieren können.
OsteoblastomDas Osteoblastom findet sich vergleichs-weise selten. Beschrieben wird es in etwa
3% aller benignen Knochentumoren. His-tologisch ist es vom Osteoidosteom nicht zudifferenzieren. Klinisch findet sich dieMehrzahl der Patienten in der zweiten Le-
Abb. 5 (a) Multiple Enchondrome des Handskeletts bei jugendlichem Patienten; typische umschriebene Osteolysen mit partiellen Kalzifikationen; (b) periostales Chondrom des Fingersbei 16-jährigem Patienten; (c) typischer Zufallsbefund eines Enchondroms des proximalen Humerus eines Erwachsenen; deutlich verstärkte Kalzifikation der Läsion
a) b) c) d)
e) f) g)
arthritis + rheuma 3/2007
134
Tunn, Dürr
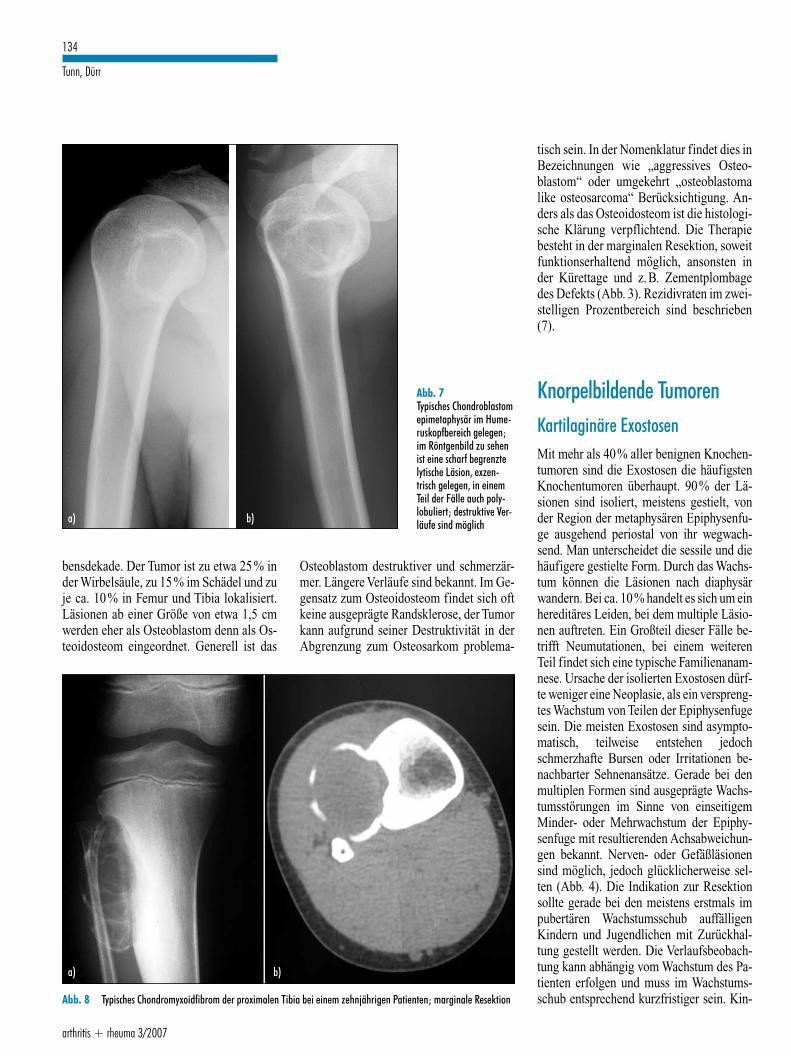
bensdekade. Der Tumor ist zu etwa 25% inder Wirbelsäule, zu 15% im Schädel und zuje ca. 10% in Femur und Tibia lokalisiert.Läsionen ab einer Größe von etwa 1,5 cmwerden eher als Osteoblastom denn als Os-teoidosteom eingeordnet. Generell ist das
Osteoblastom destruktiver und schmerzär-mer. Längere Verläufe sind bekannt. Im Ge-gensatz zum Osteoidosteom findet sich oftkeine ausgeprägte Randsklerose, der Tumorkann aufgrund seiner Destruktivität in derAbgrenzung zum Osteosarkom problema-
tisch sein. In der Nomenklatur findet dies inBezeichnungen wie „aggressives Osteo-blastom“ oder umgekehrt „osteoblastomalike osteosarcoma“ Berücksichtigung. An-ders als das Osteoidosteom ist die histologi-sche Klärung verpflichtend. Die Therapiebesteht in der marginalen Resektion, soweitfunktionserhaltend möglich, ansonsten inder Kürettage und z.B. Zementplombagedes Defekts (Abb. 3). Rezidivraten im zwei-stelligen Prozentbereich sind beschrieben(7).
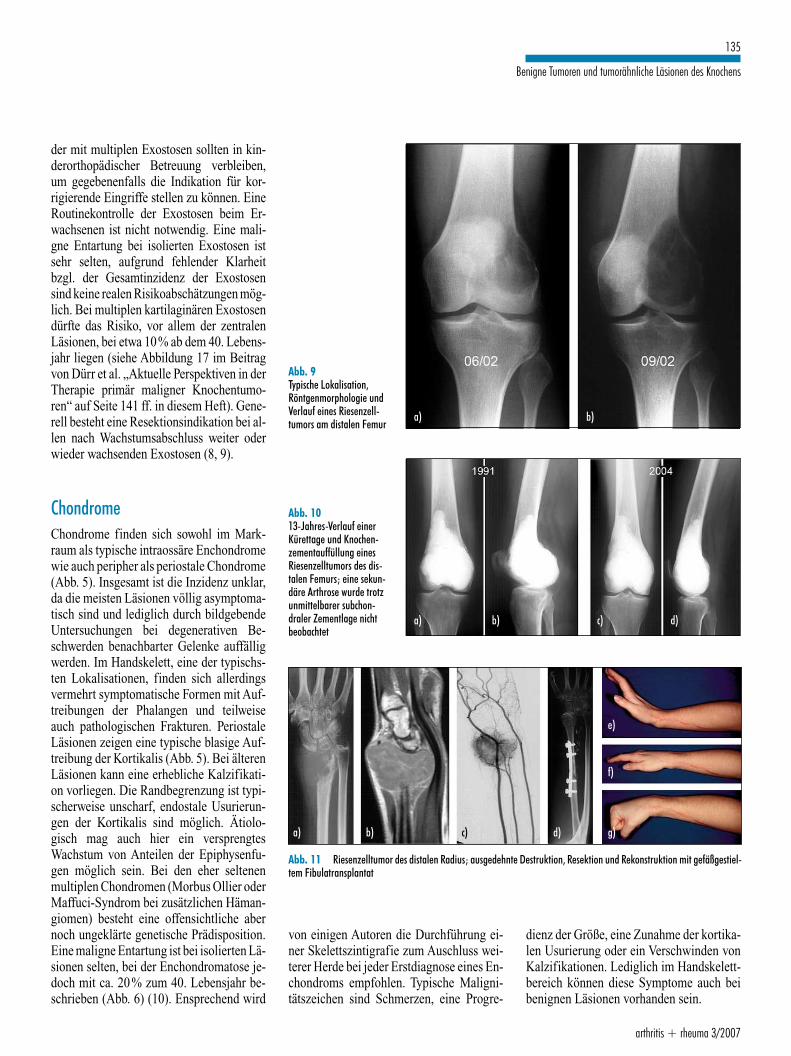
Knorpelbildende TumorenKartilaginäre ExostosenMit mehr als 40% aller benignen Knochen-tumoren sind die Exostosen die häufigstenKnochentumoren überhaupt. 90% der Lä-sionen sind isoliert, meistens gestielt, vonder Region der metaphysären Epiphysenfu-ge ausgehend periostal von ihr wegwach-send. Man unterscheidet die sessile und diehäufigere gestielte Form. Durch das Wachs-tum können die Läsionen nach diaphysärwandern. Bei ca. 10% handelt es sich um einhereditäres Leiden, bei dem multiple Läsio-nen auftreten. Ein Großteil dieser Fälle be-trifft Neumutationen, bei einem weiterenTeil findet sich eine typische Familienanam-nese. Ursache der isolierten Exostosen dürf-te weniger eine Neoplasie, als ein verspreng-tes Wachstum von Teilen der Epiphysenfugesein. Die meisten Exostosen sind asympto-matisch, teilweise entstehen jedochschmerzhafte Bursen oder Irritationen be-nachbarter Sehnenansätze. Gerade bei denmultiplen Formen sind ausgeprägte Wachs-tumsstörungen im Sinne von einseitigemMinder- oder Mehrwachstum der Epiphy-senfuge mit resultierenden Achsabweichun-gen bekannt. Nerven- oder Gefäßläsionensind möglich, jedoch glücklicherweise sel-ten (Abb. 4). Die Indikation zur Resektionsollte gerade bei den meistens erstmals impubertären Wachstumsschub auffälligenKindern und Jugendlichen mit Zurückhal-tung gestellt werden. Die Verlaufsbeobach-tung kann abhängig vom Wachstum des Pa-tienten erfolgen und muss im Wachstums-schub entsprechend kurzfristiger sein. Kin-
Abb. 7Typisches Chondroblastomepimetaphysär im Hume-ruskopfbereich gelegen;im Röntgenbild zu sehenist eine scharf begrenztelytische Läsion, exzen-trisch gelegen, in einemTeil der Fälle auch poly-lobuliert; destruktive Ver-läufe sind möglicha) b)
Abb. 8 Typisches Chondromyxoidfibrom der proximalen Tibia bei einem zehnjährigen Patienten; marginale Resektion
a) b)
arthritis + rheuma 3/2007
135
Benigne Tumoren und tumorähnliche Läsionen des Knochens
dienz der Größe, eine Zunahme der kortika-len Usurierung oder ein Verschwinden vonKalzifikationen. Lediglich im Handskelett-bereich können diese Symptome auch beibenignen Läsionen vorhanden sein.
a) b)
Abb. 9Typische Lokalisation,Röntgenmorphologie undVerlauf eines Riesenzell-tumors am distalen Femur
a) d)
Abb. 1013-Jahres-Verlauf einerKürettage und Knochen-zementauffüllung einesRiesenzelltumors des dis-talen Femurs; eine sekun-däre Arthrose wurde trotzunmittelbarer subchon-draler Zementlage nichtbeobachtet
b) c)
a)
Abb. 11 Riesenzelltumor des distalen Radius; ausgedehnte Destruktion, Resektion und Rekonstruktion mit gefäßgestiel-tem Fibulatransplantat
b) c) d)
e)
f)
g)
der mit multiplen Exostosen sollten in kin-derorthopädischer Betreuung verbleiben,um gegebenenfalls die Indikation für kor-rigierende Eingriffe stellen zu können. EineRoutinekontrolle der Exostosen beim Er-wachsenen ist nicht notwendig. Eine mali-gne Entartung bei isolierten Exostosen istsehr selten, aufgrund fehlender Klarheitbzgl. der Gesamtinzidenz der Exostosensind keine realen Risikoabschätzungen mög-lich. Bei multiplen kartilaginären Exostosendürfte das Risiko, vor allem der zentralenLäsionen, bei etwa 10% ab dem 40. Lebens-jahr liegen (siehe Abbildung 17 im Beitragvon Dürr et al. „Aktuelle Perspektiven in derTherapie primär maligner Knochentumo-ren“ auf Seite 141 ff. in diesem Heft). Gene-rell besteht eine Resektionsindikation bei al-len nach Wachstumsabschluss weiter oderwieder wachsenden Exostosen (8, 9).
ChondromeChondrome finden sich sowohl im Mark-raum als typische intraossäre Enchondromewie auch peripher als periostale Chondrome(Abb. 5). Insgesamt ist die Inzidenz unklar,da die meisten Läsionen völlig asymptoma-tisch sind und lediglich durch bildgebendeUntersuchungen bei degenerativen Be-schwerden benachbarter Gelenke auffälligwerden. Im Handskelett, eine der typischs-ten Lokalisationen, finden sich allerdingsvermehrt symptomatische Formen mit Auf-treibungen der Phalangen und teilweiseauch pathologischen Frakturen. PeriostaleLäsionen zeigen eine typische blasige Auf-treibung der Kortikalis (Abb. 5). Bei älterenLäsionen kann eine erhebliche Kalzifikati-on vorliegen. Die Randbegrenzung ist typi-scherweise unscharf, endostale Usurierun-gen der Kortikalis sind möglich. Ätiolo-gisch mag auch hier ein versprengtesWachstum von Anteilen der Epiphysenfu-gen möglich sein. Bei den eher seltenenmultiplen Chondromen (Morbus Ollier oderMaffuci-Syndrom bei zusätzlichen Häman-giomen) besteht eine offensichtliche abernoch ungeklärte genetische Prädisposition.Eine maligne Entartung ist bei isolierten Lä-sionen selten, bei der Enchondromatose je-doch mit ca. 20% zum 40. Lebensjahr be-schrieben (Abb. 6) (10). Ensprechend wird
von einigen Autoren die Durchführung ei-ner Skelettszintigrafie zum Auschluss wei-terer Herde bei jeder Erstdiagnose eines En-chondroms empfohlen. Typische Maligni-tätszeichen sind Schmerzen, eine Progre-

arthritis + rheuma 3/2007
136
Tunn, Dürr
rechtfertigen, bei Hinweisen auf Malignitätdie Resektion im Gesunden. Da Enchondro-me ein äußerst aktives histologisches Bild mitvielen Mitosen und zum Teil Kernpolymor-phie aufweisen und damit eine Abgrenzungzum Chondrosarkom Grad 1 sehr schwierigbis unmöglich ist, ist die Biopsie radiologischtypischer Befunde wenig hilfreich (11, 12).
ChondroblastomDas Chondroblastom gehört zur seltenenGruppe jener Knochenläsionen, die eine epi-theliale Differenzierung aufweisen. Auf-grund seiner typischen Zellmorphologie vonpolygonalen relativ gleichförmigen chondro-blastenähnlichen Zellen wird er den Knor-peltumoren zugerechnet. Er bildet eine knor-pelige Matrix mit Kalzifikationen. Weltweitsind einige hundert Fälle beschrieben. Typi-scherweise finden sich 80% der Läsionen imzweiten Lebensjahrzehnt, klassischerweisein der Epiphyse der langen Röhrenknochenlokalisiert (Abb. 7). Führende Symptomatikist der Schmerz, die Diagnose wird durch ei-ne Biopsie gestellt. Therapeutisch wird wiebei vielen benignen im Verlauf jedoch ag-gressiven Läsionen die Kürettage und Auf-füllung, gegebenenfalls mit Knochenzementempfohlen. Lokale Adjuvantien sind mög-lich und empfohlen, die Wirksamkeit vonAl-kohol oder Phenol ist jedoch umstritten. DieRezidivrate wird dabei mit 10-20% angege-ben. Diskutiert wird zusätzlich der Begriffdes „malignen Chondroblastoms“, da sich ineinigen Fällen trotz typischer Histologie einmaligner Verlauf mit Fernmetastasen (typi-scherweise pulmonal) finden kann (13, 14).
ChondromyxoidfibromNoch seltener als das Chondroblastom istder metaphysäre Tumor typischerweise beiKindern zwischen 5 und 15 Jahren anzutref-fen. Er zeigt namensgebend sowohl chon-droide wie auch myxoide und fibroide An-teile. Klassischerweise findet man eine ova-läre Osteolyse, exzentrisch gelegen, manch-mal lobuliert mit oder ohne ausgeprägtenSklerosesaum. Matrixossifikationen sindmöglich aber selten (Abb. 8). Die Therapiebesteht, soweit machbar, in der marginalen
Generell wird man bei typischen Befun-den ein zuwartendesVerhalten mit regelmäßi-gen Kontrollen in größeren Abständen (jähr-lich oder alle zwei Jahre) anraten. Eine Aus-
nahme stellen die prognostisch schwierigenperiostalen Läsionen dar, die operativ ver-sorgt werden sollten. Im Zweifelsfall ist dieKürettage und Auffüllung der Läsion zu
Abb. 12Juvenile Knochenzyste desHumerus; Verlauf nachInjektionstherapie mitMethylprednisolonazetat
a) b)
Abb. 13 Aneurysmale Knochenzyste des proximalen Radius (a) bei einem neunjährigen Jungen; typischer Flüssigkeits-spiegel der zellulären Blutbestandteile in der MRT (b); Ausheilungszustand vier Jahre nach Kürettage unter Belassung desPeriosts und allogener Spongiosaplastik
Abb. 14Typischer Aspekt einerAKZ des distalen Femurs(a) bei einem 13-jährigenJungen; exzentrische me-taphysäre Läsion, denKnochen auftreibend mitmultiplen Septen im MRT(b)
a) b) c)
a) b)

Wichtig ist zudem in jedem Fall die Bestim-mung des Parathormons, da histologisch ei-ne Differenzierung zum „braunen Tumor“bei Hyperparathyreoidismus nicht möglichist. Die klassische Therapie besteht in derintraläsionalen Kürettage unter Anwendungzusätzlicher mechanischer (Hochgeschwin-digkeitsfräse) und physikalisch/chemischerAdjuvantien (Knochenzement, Alkohol,Phenol, Kryochirurgie, Kauterisierung)(17).
Das Rezidivrisiko nach marginaler Re-sektion, soweit diese machbar ist, liegt beinahezu 0%, nach optimalen intraläsionalenVorgehen bei etwa 10%. Entscheidend isthier die ausgedehnte Freilegung des Tumorsmit kompletter Entdachung der Läsion undsorgfältigstem chirurgischen Vorgehen. Imeigenen Krankengut bewährt hat sich hierdie Methymetacrylatauffüllung des De-fekts. Die Knochenzementplombe bietet ei-nen guten thermoablativen Effekt und ge-währleistet die sofortige und dauerhafteStabilität. Die Rezidivdiagnostik wird er-heblich vereinfacht, eine nachteilige Wir-kung auf das Gelenk auch bei unmittelbarsubchondraler Implantation wurde nicht be-obachtet (Abb. 10). Da die Tumoren jedochtypischerweise epimetaphysär vorliegen istder Gelenkerhalt nicht immer möglich(Abb. 11). Aufgrund der Neigung der Rie-senzelltumoren zu Spätrezidiven sollte dieNachsorge über längere Zeiträume (z.B.zehn Jahre) erfolgen.
Zystische Läsionen des KnochensKnochenzysten zählen zu den häufigstentumorähnlichen Läsionen des Knochens.Man unterscheidet die „einfache“ juvenileKnochenzyste, eine mit seröser Flüssigkeitgefüllte Läsion, die in nahezu 80% der Fäl-le im ersten und zweiten Lebensjahrzehntdiagnostiziert und nur bei Frakturen symp-tomatisch wird und meist als Zufallsbefundvorliegt von der aneurysmalen Knochen-zyste. Diese ist, wie wir heute wissen, eineNeoplasie mit chromosomaler Translokati-on (t[16;17][q22;p13]).
Juvenile Knochenzysten sind in ihrerÄtiologie völlig ungeklärt und zeigen ins-besondere in Phasen des allgemeinen Kör-perwachstums eine deutliche Progredienz.Sie finden sich bevorzugt im Bereich derlangen Röhrenknöchen, meist an der pro-ximalen Meta- und Diaphyse, im Verlaufzur Diaphyse hin wachsend (Abb. 12). Istdie Diagnose gesichert, ist eine Verlaufs-beobachtung möglich, bei Progredienz exis-tieren eine Reihe konkurrierender Therapie-verfahren. So kann eine Spontanheilungnach Fraktur eintreten – wird oft überschätzt–, eine Instillation von Methylprednisolonoder Knochenmarkaspirat erfolgen, kanü-lierte Schrauben können eingedreht, flexi-ble Nägel zur zusätzlichen mechanischenSchienung und Eröffnung der Zyste ver-wandt werden. Eine Auffüllung mit Kno-chen oder eine größere Osteosynthese wirdnur selten notwendig. Die Läsion ist prinzi-piell mit mehr oder weniger großem Restde-fekt selbstheilend, man sieht jedoch hin undwieder auch im Erwachsenenalter Frakturenan kritischen Lokalisationen (z.B. proxima-ler Femur) (18).
Aneurysmale Knochenzysten (AKZ)sind prinzipiell in zwei Formen vorliegend:der primären, tatsächlich einer primären Lä-sion entsprechenden Form – nur diese zeigtdie Translokation – und der sekundären, alsreaktive Begleitläsion bei einem sonstigenneoplastischen oder nicht neoplastischenProzess des Knochens. Die Unterscheidungist wesentlich, da die Primärläsion (z.B. einRiesenzelltumor) einer sekundären AKZdurchaus maligne sein könnte oder eine an-dere Therapie erfordern würde. Differenzi-aldiagnostisch muss vor allem ein telean-giektatisches Osteosarkom ausgeschlossen
Resektion oder der intraläsionalen Küretta-ge. Rezidive sind ebenfalls in 10-20% be-schrieben, eine Metastasierung kann mög-lich sein (15).
RiesenzelltumorNamensgebend für den Riesenzelltumor(Osteoklastom) ist das histologische Bild,das durch das multiple Auftreten osteoklas-tischer Riesenzellen geprägt ist. Diese sindjedoch nur als die reaktive Komponente an-zusehen, als eigentlicher Tumor muss die fi-brozytäre Gerüstsubstanz betrachtet wer-den. Der Riesenzelltumor findet sich imepimetaphysären Bereich, meist exzen-trisch gelegen, vergleichsweise gut um-schrieben, ausschließlich osteolytisch ohneRandreaktion (Abb. 9). Das typische Er-krankungsalter liegt bei zehn bis 30 Jahren,in einer eigenen Untersuchung an 87 Patien-ten wurde ein durchschnittliches Erkran-kungsalter von 28 Jahren festgestellt (16).40% der Läsionen lagen im distalen Femurund proximalenTibiabereich, in der Häufig-keit gefolgt vom distalen Radius. In dreiFällen fanden sich zusätzlich pulmonaleMetastasen, ausschließlich bei jenen Patien-ten, bei denen es zum Lokalrezidiv kam. Inder Literatur werden diese bis zu 10% be-schrieben, eine entsprechende Unter-suchung vor und im Verlauf der Therapieund Nachsorge wird empfohlen. Der Spon-tanverlauf ist nicht absehbar, histologischePrognosefaktoren existieren ebenfalls nicht.
Abb. 15Typische polyostotischeForm einer fibrösen Dys-plasie mit Ausbildung ei-ner Coxa vara („Hirten-stab“) bei einem 19-jäh-rigen Patienten; die meis-ten Läsionen sind hemi-mel verteilt und zeigen ei-ne milchglasartige Trans-parenz
a) b)
arthritis + rheuma 3/2007
138
Tunn, Dürr

werden. Histologisch zeigt sich dieAKZ mitunterschiedlichen blutgefüllten Hohlräu-men gefüllt, unterteilt durch Bindegewebs-septen, welche Bälkchen aus Osteoid undosteoklastären Riesenzellen enthalten. Miteiner Inzidenz von etwa drei bis vier Prozentaller registrierten Knochentumoren ist sieselten, sie findet sich vor allem im zweitenLebensjahrzehnt. Die Symptomatik ist un-spezifisch, die Therapie besteht in der Kü-rettage gegebenenfalls mit Adjuvantien undAuffüllung der Läsion. Marginale Resektio-nen sind selten möglich. Röntgenologischzeigt sich die Läsion metaphysär, im An-fangsstadium als exzentrische Osteolyse,im späteren Verlauf mit Auftreibung desKnochens und schalenartiger Neokortikalis(Abb. 13, 14) (19). Prognostisch zeigen ins-besondere Kinder unter zehn Jahren erhebli-che Neigungen zu Lokalrezidiven. Die in-traläsionale Applikation von entzündungs-auslösenden Reagenzien oder die vaskuläreEmbolisation ist möglich und wird unter-schiedlich diskutiert. Letztere kann ins-besondere auch vor Kürettagen an schwieri-gen Lokalisationen (z.B. Becken) die intra-operative Übersicht und den Blutverlust er-heblich optimieren. In Ausnahmefällenkann die niedrig dosierte Strahlentherapiediskutiert werden (20).
Fibröse DysplasieDie fibröse Dysplasie ist eine vergleichswei-se häufige Läsion des Knochens, bei der ein(monostotisch) oder mehrere (polyosto-tisch) Knochen betroffen sind. Es zeigt sichdabei ein fibröser Defekt des Knochens, be-reits in früher Kindheit beginnend, bei demhistologisch Knochen durch Bindegewebeersetzt ist, welches im Sinne einer DysplasieOsteoid bildet. Das Osteoid ist dabei feinverteilt, wirkt radiologisch milchglasartig,und zeigt sich gesäumt von Fibroblasten an-stelle der normalerweise vorliegenden Os-teozyten. Ätiologisch fand sich die Ursachein der aktivierenden Mutation eines Gens fürdie Alpha-Untereinheit des stimulatorischenG-Proteins (G[s]alpha lokalisiert an20q13.2–13.3). Die meisten Läsionen sindmonostotisch und asymptom. Syndrom-erkrankungen sind bekannt. PolyostotischeFormen oder ausgedehntere Läsionen gera-
Fazit für die PraxisBenigne Tumoren und tumorähnlicheLäsionen des Knochens erfordern in derMehrzahl der Fälle lediglich die beruhi-gende Beratung des Patienten. Bei denhäufigen Exostosen ist die Wachstums-beeinflussung oder die seltene Kompres-sion von Nerven und Gefäßen von Be-deutung. Bei multiplen Vorkommenmuss auf eine maligne Entartung im Ver-lauf geachtet werden. Enchondrome sindähnlich zu beurteilen, gerade große Lä-sionen können hier jedoch differenzial-diagnostisch problematisch sein. Riesen-zelltumoren, Chondroblastome oder Os-teoblastome erfordern die sorgfältigeoperative Therapie, Adjuvantien werdenempfohlen. Da die Läsionen oft sehr de-struktiv sind und zu Rezidiven neigen, isteine Behandlung im Zentrum sinvoll. Ei-ne pulmonale Metastasierung ist mög-lich und muss beachtet werden. JuvenileKnochenzysten sind nur bei Fraktur oderFrakturgefahr von Bedeutung, bei aneu-rysmalen Knochenzysten ist ein eventu-ell zugrundeliegender Tumor oder ein te-leangieektatisches Osteosarkom aus-zuschließen. Bei der fibrösen Dysplasiesind neuere Therapieansätze mitBisphosphonaten vielsprechend.
Literatur1. Fletcher CDM, Unni KK, Mertens F, Hrsg. World
Health Organization Classification of Tumours:Pathology and Genetics of Tumours of Soft Tissueand Bone. Lyon: IARC Press 2002.
2. Freyschmidt J, Ostertag H, Jundt G. Knochentu-moren. Klinik, Radiologie, Pathologie. Berlin,Heidelberg, NewYork: Springer 2003; 9 und 679.
3. Enneking WF, Spanier SS, Goodman MA. A sys-tem for the surgical staging of musculoskeletalsarcoma. Clin Orthop Rel Res 1980; 198 (153):106–120.
4. Campanacci M, Baldini N, Boriani S et al. Giant-cell tumor of bone. J Bone Joint SurgAm 1987; 69(1): 106–114.
5. Freyschmidt J. Standards und diagnostische Stra-tegien bei der Diagnostik von Knochen-geschwülsten und geschwulstähnlichen Läsionen.Radiologe 1998; 38: 287–300.
6. Peyser AB, Makley JT, Callewart CC et al. Osteo-ma of the long bones and the spine. A study of ele-ven patients and a review of the literature. J BoneJoint Surg Am 1996; 78: 1172–1180.
7. Papagelopoulos PJ, Galanis EC, Sim FH et al. Cli-nicopathologic features, diagnosis, and treatmentof osteoblastoma. Orthopedics 1999; 22:244–247.
8. Stieber JR, Dormans JP. Manifestations of heredi-tary multiple exostoses. J Am Acad Orthop Surg2005; 13: 110–120.
9. Saglik Y, Altay M, Unal VS et al. Manifestationsand management of osteochondromas: a retro-spective analysis of 382 patients. Acta OrthopBelg 2006; 72: 748–755.
10. Müller PE, Dürr HR, Nerlich A et al. Malignanttransformation of a benign enchondroma of thehand to secondary chondrosarcoma with isolatedpulmonary metastasis. Acta Chir Belg 2004; 104:341–344.
11. Müller PE, Dürr HR, Wegener B et al. Solitary en-chondromas: is radiographic follow-up sufficientin patients with asymptomatic lesions? Acta Or-thop Belg 2003; 69: 112–118.
12. Delling G, Jobke B, Burisch S, Werner M. Knor-pelbildende Tumoren Klassifikation, Vorausset-zungen für die Biopsie und histologische Charak-teristika. Orthopäde 2005; 34: 1267–1281.
13. Kirchhoff C, Buhmann S, Mussack T et al. Ag-gressive scapular chondroblastoma with seconda-ry metastasis-a case report and review of literatu-re. Eur J Med Res 2006; 11: 128–134.
14. Lin PP,ThenappanA, Deavers MT et al.Treatmentand prognosis of chondroblastoma. Clin OrthopRelat Res 2005; 438: 103–109.
arthritis + rheuma 3/2007
139
Benigne Tumoren und tumorähnliche Läsionen des Knochens
de an lasttragenden Bereichen können je-doch ein operatives Vorgehen notwendigmachen (Abb. 15) (21). Entscheidet mansich für eine Operation, sollten aufgrund derhohen Rezidivquote bei der Verwendung au-tologer Spongiosa besser kortikale Trans-plantate (z.B. Fibula) verwandt werden.Frakturen durch Läsion der fibrösen Dyspla-sie zeigen auch bei konservativer Therapieeine gute Ausheilung. Auch der Einsatz vonBisphosphonaten ist möglich, wengleich ge-rade bei Kindern die relativ lang wirkendenPräparate noch wenig untersucht sind (22).
Sonstige LäsionenWeitere häufige nicht tumoröse Läsionensind metaphysäre Knochendefekte, meist
ohne jegliche Relevanz, intraossäre Gan-glien oder seitens der benignen TumorenHämangiome, die jedoch eher den Fehlbil-dungen zuzurechnen sind. Alle anderennicht tumorösen Läsionen oder benignenTumoren stellen zwar zum Teil sehr interes-sante, aber seltene Befunde dar.

arthritis + rheuma 3/2007
140
Tunn, Dürr
Korrespondenzadresse:Dr. med. Per-Ulf TunnKlinik für Chirurgie und Chirurgische OnkologieUniversitätsmedizin BerlinCharité Campus BuchLindenberger Weg 8013125 BerlinE-Mail: [email protected]
15. Dürr HR, Lienemann A, Nerlich A et al. Chondro-myxoid fibroma of bone. Arch Orthop TraumaSurg 2000; 120: 42–47.
16. Tunn PU, Schlag PM. Der Riesenzelltumor desKnochens. Eine Analyse von 87 Patienten. Z Or-thop Ihre Grenzgeb 2003; 141: 690–698.
17. Dürr HR, Maier M, Jansson V et al. Phenol as anadjuvant for local control in the treatment of giantcell tumour of the bone. Eur J Surg Oncol 1999;25: 610–618.
18. Wilkins RM. Unicameral bone cysts. J Am AcadOrthop Surg 2000; 8: 217–224.
19. Freyschmidt J, Ostertag H, Jundt G. Knochentu-moren. Klinik, Radiologie, Pathologie. Berlin,Heidelberg, New York: Springer 2003; 754–776.
20. MendenhallWM, Zlotecki RA, Gibbs CP et al.Aneu-rysmalbonecyst.AmJClinOncol2006;29:311–315.
21. DiCaprio MR, Enneking WF. Fibrous dysplasia.Pathophysiology, evaluation, and treatment. J Bo-ne Joint Surg Am 2005; 87: 1848–1864.
22. Egner-Hobarth S, Welkerling H, Windhager R.Bisphosphonate in der Therapie der fibrösen Dys-plasie : Relevante Daten und praktische Aspekte.Orthopäde 2007; 36: 124–130





























