Embed Size (px)
DESCRIPTION
Hematologija
Citation preview

1
HEMATOLOGIJA
Sadražaj: *Hematopoeza_____________________________________________________________ 2 I. Eritrociti_________________________________________________________________2 1. Anemije………………………………………………………..…….............................................3 2. Aplastiĉne anemije…………………………………................................………...……..4 3. Refraktarna anemija (Mijelodisplazni sy)…………..…..............................…….……...5 4. Megaloblastna anemija…………………………...............................………….….….…6 4.1.Perniciozna anemija……………………...............................………….…...…..6 5. Hiposideremijska anemija…………………………...............................……….…..……7 6. Talasemija……………………………………………...................................……….…...8 7. Hemolizne anemije………………………………………................................………….9 7.1.Korpukularne hemolizne anemije…….............................……………..……..10 7.2.Extrakorpuskularne hemolizne anemije…...........................………….……..11 8. Prava policitemija……….……………….……………………...............................….…12 9. Akutna agranulocitoza…..……………….…………….............................………….…12 10.Hroniĉna idiopatska agranulocitoza…….…………………............................……….13 11.Infektivna mononukleoza……………….…………………….........................................…….13 II.Leukociti________________________________________________________________14 1. Leukemije…………………………................................…….……………………….....14 2. Acc mijeloidna leukemija………...............................…….………………………….....15 3. Acc limfoidna leukemija……………................................…………………………..….17 4. Chr mijeloidna leukemija………….…………………………..…..................................17 5. Mijelofibroza…………………….…………………………...................................……..18 6. Chr limfoidna leukemija………………………….………….................................….…19 7. Hodgkin limfom………………………………….……………..…..................................20 8. Non-Hodgkin limfom…………………………….……………..….................................21 9. Multipli mijelom – plazmocitom………………….…………….….................................22 10.Macroglobulinemija (Waldenstrom)……………..…………….…...............................22 III.Hemoragijski sindromi_____________________________________________________23 1.Vaskularni hemoragijski sindromi……………….……………….…..............................26 2.Nasledna hemoragijska telangiektazija………………………………...........................26 3.Steĉene vaskularne purpure – Henoch-Schonlein……………………........................26 4.Mb von Wilerbrandt…………………………………………………................................27 IV.Hemofilije______________________________________________________________27 V.DIK____________________________________________________________________28 VI.Bolesti slezine___________________________________________________________29 1.Benigni i maligni tumori slezine………………………………..….................................29 2.Splenomegalije……………………………………………….............................…....…..29 3.Hipersplenizam…………………………………………..............................…………….29 VII.Trombocitopenije i trombocitopatije__________________________________________30 1.Trombocitopenija………………..…….………............................………………….…...30 3.Trombocitopatije……………………………….............................………………………31 VII.Tromboza_____________________________________________________________31
www.belimantil.info

2
*HEMATOPOEZA je proces sinteze tj obnavljanja krvnih ćelija -U hematopoezi se odigravaju 3 meĊusobno zavisna procesa: 1.PROLIFERACIJA ćelijski rast, tj deoba 2.DIFERENCIJACIJA usmeravanje u pojedinaĉnu ćelijsku lozu 3.MATURACIJA sazrevanje do funkionalno aktivnih ćelija -Organi hematopoeze su: *Koštana srž: najvećim delom pjosnate kosti (karlica, kiĉma, sternum, rebra, proximalni okrajci femura i humerusa) *Timus *Limfne žlezde *Slezina -Hematopoezno tkivo ima veliki rezervni kapacitet; u uslovima povećane potrebe, masna kosna srž prelazi u aktivnu srvenu kosnu srž. I.ERITROCITI *FUNKCIJA: -transportuju Hb, a ovaj nosi O2 iz pluća u tkiva -sadrže karboanhidrazu koja ubrzava reakciju izmeĊu CO2 i H2O (to omogućava da krv primi velike koliĉine CO2 i da ga transportuje kao HCO3) -Hb deluje kao pufer *Eritrociti se posle roĊenja sintetišu u kosnoj sržI: Pluripotentna unipotentna proeritroblasti retikulociti eritrociti matiĉna ćelija matiĉna ćelija *REGULACIJA PROIZVODNJE ERITROCITA:
- Transport O2 u tkiva (anemija, velike nadmorske visine, insuf.srca)proizvodnja Er -Glavni faktor koji stimuliše proizvodnju Er je ERITROPOETIN iz bubrega -Potrebni su i VITAMIN B12 (za stvaranje DNK koja je neophodna za diferencijaciju ćelija i njihovo sazrevanje). Kada nedostaje B12 razvijaju se megaloblasti, a posle makrociti (dobro transportuju O2, ali su slabe graĊe, pa brzo propadaju. -Potrebna je i FOLNA KISELINA, takoĊe za stvaranje DNK *METABOLIZAM ER:-ER u krvotoku obiĉno ostane 120 dana, a onda se raspada u slezini -Iz raspadnutog ER makrofagi fagocituju Hb i razgraĊuju ga, naroĉito u jetri -Feritin odlazi u kosnu srž (za proizvodnju novih ćelija), a porfirin se pretvori u bilirubin, koji se otpusti u krv, a odavde ga jetra izluĉI u žuĉ. ZA NORMALNU ERITROPOEZU POTRENBI SU: 1)Metali: Fe, Mn, Co 2)Vitamini: B12, C, E, B6 (piridoxin), riboflavin, tiamin, pantotenska kiselina 3)Aminokiseline 4)Hormoni: faktori matiĉnih ćelija, Il-3, GM-CSF, eritropoertin, androgeni i tiroxin *Anemija je smanjenja ER u krvi 1.Hipohromna 2.Aplastiĉna 3.Megaloblastna 4.Hemolitiĉka
www.belimantil.info

3
1.ANEMIJE
-Anemije su bolesti koje odlikuje mase ER, vrednosti Hb, hematokrita baz obzira na etiologiju. -Anemije nastaju kao posledica poremećaja odnosa izmeĊu sinteze i razgradnje eritrocita. -DG anemije se postavlja kada je koncentracija Hb manja od: *muškarci 140g/l *Eritrociti: muškarci=4,2-5,8 x 10 na 12/l *žene 120g/l žene=3,7-5,2 x 10 na 12/l *PATOFIZIOLOŠKA PODELA ANEMIJA: 1)ANEMIJE ZBOG NEDOVOLJNOG STVARANJE ER: A)Nedostatak faktora potrebnih za eritropoezu: 1.Megaloblastne anemije: nedostatak B12, folata, pirimidina, purina 2.Hipohromne anemije: nedostatk Fe 3.Talasemije, Hemoglobinopatije: poremećaj sinteze globina B)Insuficijencija kosne sržI (por.na nivou pluripotentnih ćelija): 1.Aplastiĉne anemije 2.Refraktarne anemije
2)ANEMIJE ZBOG RAZGRADNJE ER (HEMOLIZNE): A)Hemolizne (nasledne) hemolitiĉke anemije: (faktor hemolize je u ER) 1.Poremećaj opne ER: -Sferocitoza -Eliptocitoza -Akantocitoza 2.Poremećaj enzima ER: -Deficit glukoza-6-fosfat-dehidrogenaze -Deficit piruvat kinaze 3.Poremećaj sinteze globina: -talasemija -hemoglobinopatije -porfirije -paroksizmalna noćna hemoglobinurija B)Extrakorpuskularne (steĉene) hemolizne anemije: (faktor hemolize je u plazmi) 1.Imune: Izoimune – posttransfuziona *Fetalna eristoblastoza Autoimune – idiopatska autoimuna *Topla at *Hladna at *virusi, lekovi, hem.supstance *poremećaj imunog odgovora 2.Neimune: *Hemijske - toxiĉne hemolizne anemije *Fiziĉke – mehabiĉke i anemije usled opekotina *Mikroorganizmi – hemolizna anemija u infekcijama
www.belimantil.info

4
3)ANEMIJE ZBOG GUBITKA KRVI: A)Akutna posthemoragijska anemija Nagli gubitak krvi B)Hroniĉna posthemoragijska anemija Postepen gubitak Fe *MORFOLOŠKA PODELA ANEMIJA: Zasniva se na: *veliĉini Er (normo, makro i mikrocitne) i *koliĉini Hb (normo i hipohromne) 1)MAKROCITNA MCV>95fl -Megaloblastna: perniciozna, nutritivna -Nemegaloblastna: hemolizna, aplastiĉne, alkohol, bolesti jetre 2)MIKROCITNA MCV<80fl; MCH<27pg -Hipohromna -Poremećaj sinteze globina -deficit Fe, talasemije… -Poremećaj sinteze porfirina, hema 3)NORMOHROMNA, NORMOCITNA MCV je 85-95fl; MCH>26pg -Akutno krvarenje -Hemoliza -Aplastiĉna anemija -Leukemije, plazmocitom, insuf.bubrega i jetre, hipopituarizam, mixedem *OPŠTA SIMPTOMATOLOGIJA ANEMIJA: 1)KLINIĈKI: -Pojava simptoma ne mora biti vezana zastepen anemije -Za pojavu opštih simptoma važni su: 1*uzroĉni poremećaji koji su doveli do anemije 2*promene ukupnog volumena krvi i 3*smanjena oxigenacija krvi 2)SUBJEKTIVNE MANIFESTACIJE: -umor, slabost, malaxalost, lako zamaranje -tahikardija i lupanje srca, srĉana insuficijencija
-glavobolja, nesvestice, vrtoglavice, zujanje u ušima, koncentracije, nesanica -gubitak apetita, muka, gaĊenje -poremećaj menstrualnog ciklusa
-t, metabolizam 3)OBJEKTIVNE MANIFESTACIJE: -bledilo kože i vidljivih sluzokoža -trofiĉke promene adnexa kože: lomljivi nokti, kosa bez sjaja, opada -stomatitis
-laka proteinurija uz koncentrovanje urina -lako uvećana jetra i slezina (kod teških oblika) -sporije zarastanje rana 2.APLASTIČNE ANEMIJE -Normocitne, normohromne -zbog nedivoljnog stvaranja Er
www.belimantil.info

5
-Poremećaj na nivou kosne sržI, tj pluripotentnih ćelija (Er, Tr i granulociti su ) *ETIOLOGIJA:
PRIMARNA: -Fankonijeva hipoplastiĉna anemija – kongenitalna -lekovi: citostatici, hloramfenikol – steĉena (soli Au, DDT, zraĉenje, tolbutamid)
SEKUNDARNA: -hemijski agensi: lekovi, jonizujuće zraĉenje -virusi: Epstein-Barr, hepatitisi -metaboliĉka: pankreatitis, trudnoća, imunološke *PATOGENEZA: -Poremećaj u proliferaciji -Defekt pluripotentne matiĉne ćelije (poremećaj hematopoetskog proliferativnog kapaciteta kosne sržI) ĉiji uzrok nije jasan -Odgovor matiĉne ćelije na hematopoetske faktore je smanjen ili potpuno odsutan -Uspešno leĉenje imunosupresivnom terapijom je dokaz postojanja imunoloških ĉinilaca u njjenoj patogenezi -Preosetljivost na lekove i direktno toxiĉno dejstvo leka *KLINIĈKI: -Brzo zamaranje, lupanje srca, slabost, gubitak apetita, mršavljenje, -Bledilo, tahikardija, glavobolja -Težina infekcije i krvarenja zavisi od stepena smanjenja broja trombocita i granulocita –
petehije po kožI i sluznicama, ekhimoze i krvarenja na sluznicama nosa, desni, retine, t, infekcije *LABORATORIJA: -Normocitna, normohromna anemija -MCV (srednja vrednost zapremine Er) = normalna -MCHC (srednja vrednost koc.Hb u Er) = normalna
-Fe u serumu = Fe je u Er normalno, ali je Fe u serumu (jer se ne iskoristi svo) -punkcija kosne sržI = masno, tj neaktivno hematopoetsko tkivo *KOMPLIKACIJE: Teška krvarenja, sepsa, gljiviĉne infekcije *TERAPIJA: -Androgeni (u cilju stimulacije eritropoeze) -glukokortikoidi -faktori rasta matiĉnih ćelija -imunosupresivi (ciklosporin A) -transplantacija kosne sržI 3.REFRAKTARNA ANEMIJA (Sy myelodyspalsticum) -Zbog nedovoljnog stvaranja Er (insuf. kosne sržI, tj. por. na nivou pluripotentnih ćelija) -Hroniĉna anemija ili MIJELODISPLAZNI SINDROMI su grupa oboljenja koji nastaju malignom transformacijom na nivou pluripotentnih ćelija hematopoeze -Poremećaj u diferenciranju je osnovni poremećaj -Podeljen je u 5 grupa: 1.RA refrakratna anemija sa manje od 5% blasta 2.RARS RA sa ringrd sideroblastima u kosnoj sržI; više od 15% 3.RAEB RA sa viškom blasta u kosnoj sržI (5-20%) 4.CMML chr mijelomonocitna leukemija sa preko 1x10 na 9/l monocita
www.belimantil.info

6
5.RAEB-t RA sa blastima u transform+blasti u krvi=5% i kosnoj sržI 20-30% *ETIOPATOGENEZA:
-To je maligno oboljenje kosne sržI sa leuko i trombocitopenijom, serumskog Fe i velikim procentom blasta u kosnoj sržI, nepoznate etiologije -Može nastati kao primarno ili sekundarno oboljenje, posle primene citostatskih lekova, kod zraĉenja, primene hemijskih supstanci i mogućim sledstvenim oštećenjem kariotipa *KLINIĈKI: -Bolest se javlja kod osoba starijih od 50 god, ĉešće muškaraca -Tok može da bude postepen i da se Dg postavi sluĉajno -Simptomi i znaci su uslovljeni postojanjem anemije, granulocitopenije i trombocitopenije -zamaranje, dispnea, malaxalost -Infekcija respiratornih puteva, kože, urinarnog trakta; moguća septikemija -Hemoragijski sindromi: krvarenje po kožI, ekhimoze, petehije -U manjeg broja pacijenata uvećana je slezina -Kod CMML postoji hepatosplenomegalija i limfadenopatija *LABORATORIJA:
-PANCITOPENIJA Er makrocitni, br retikulocita, granulociti, trombociti -RUNGED-sideroblasti *DIJAGNOZA: -Na MDS treba misliti kada postoji citopenija i displazne osobine (gore navedene) -Aspirat kosne sržI = hipercelularan *TOK zavisi od tipa MDS. Najbolja je prognoza za RA i RARS (70 meseci), a najlošija je za CMML (5 meseci) -Najteža komplikacija je prelazak u akutnu leukemiju *TERAPIJA: -konzervativna sa transfuzijama Er i Tr -antibiotici -citostatici (ali oni mogu i da pogoršaju stanje) -transplantacija kosne srži 4.MEGALOBLASTNE ANEMIJE -Nastaju zbog: 1)Nedostatka folata (nema neuroloških simptoma) 2)Nedostatka B12 (obavezna je neuropatologija) -Nastaju zbog nedovoljnog stvaranje Er ili zbog nedostatka faktora potrebnih za eritropoezu (B12, folna kiselina, purini, pirimidini) -Megaloblastne anemije nastaju zbog smanjene sinteze DNK. Deoba ćelija je usporena, a razvoj citoplazme se normalno odvija, tako da ćelije postaju uvećene -U megaloblastne anemije ubrajaju se kao najĉešće: 1.perniciozna anemija 2.anemija zbog gastrektomije (kod totalne 100%, kod parcijalne ne mora=posle 3-4god) 3.anemije izazvane bakter. I crevnim parazitima (jer troše B12; Skandinavija=riblja pantljiĉara) 4.anemije zbog malapsorpcije (spru, TBC, resekcije ileuma=tu dolazi do resorpcije B12) 5.anemije zbog nedostatka folne kiseline 4.1.Perniciozna anemija - najĉešća megaloblastna anemija -Nastaje zbog nedostatka vitamina B12 u organizmu, zbog nedostatka intrizing faktora u želudaĉnom soku, koji je neophodan za resorpciju vitamina B12. *ETIOPATOGENEZA: -Genski uslovljena -Autoimuna (auto-At na parijetalne ćelije)
www.belimantil.info

7
-Deficit vitamina B12 nastaje zbog: deficita u ishrani, loše aposorpcije, naslednih poremećaja i povećanih potreba za B12. -Bolesnici imaju atrofiju želudaĉne sluznice i parijetalnih ćelija i gubitak unutrašnjeg faktora
*Smanjenje folata zbog unosa: alkoholiĉari, narkomani, ljudi na dijalizi
*Smanjenje folata zbog potreba: trudnice, anemije (kod hemolizne anemije folati su
, zbog hematopoeze) *KLINIĈKI:-Bolest je podmukla, razvija se postepeno i sporo napreduje -osoben nalaz je atrofiĉni gastritis; sve ostalo je njegova posledica -Poĉinje malaxalošću, opštom slabošću, zamaranjem -Lupanje srca, bledo žuta boja lica (kao slama); turgor kože je slab -Jezik gladak, sjajan, atrofiĉan, nema papila = Hunter glositis -depresije, por. pamćenja, parestezije, otežan hod (funikularna mijeloza),poremećaj dubljeg senzibiliteta poremećaji NS lokalizovani su uglavnom u zadnjim rogovima kiĉmene moždine(demijelinizacija perifernih živaca koja se može proširiti i na produženu moždinu), a posledica su, verovatno, poremećaja sinteze DNK -muka, gaĊenje, povraćanje, nadimanje
*LABORATORIJA:-u odmaklom stadijumu razvoja bolesti zapaža se jako broja Er,
nesrazmerno sa Hb; lako su leukociti i trombociti -kosna srž je hipercelularna sa puno megaloblasta (Er) i džinovskih metamijelocita (leu)
-u serumu je vit. B12, Fe
-je želudaĉna sekrecija, prisutna su antiparijetalna At u serumu *DIJAGNOZA:-Anamneza i objektivni pregled; gastroskopija -želudaĉna ahlorhidrija posle histamina-jer mena parijetalnih ćelija -u razmazu veliki broj eritriblasta, megaloblasta i džinovskih metamijelocita -+ Šilingov test: test apsorpcije vit.B12 sa radioaktivnim Co; posle 1-3 dana ispituje se radioaktivnost mokraće (18% normalno; u pernicioznoj anemiji 0-1% *TERAPIJA: vitamin B12=parenteralno. Folana kiselina je kontraindikovana, jer može da pogorša nervne poremećaje. -Ako se ne leĉI, tok bolesti je progresivan do smrti!!! Pern.anemiĉari=5x većI rizik za Ca želuca 5.HIPOSIDEREMIJSKE ANEMIJE -hipohromne -mikrocitna -Nastaju zbog nediviljnog stvaranja Er, tj nedostaju supstance koje stimulišu eritropoezu -Najĉešća su grupa anemija i nastaju zbog deficita Fe u organizmu *ETIOPATOGENEZA: 1)Nedovoljan unos Fe hranom 2)Loša resorpcija Fe u org. za varenje (celijaĉna bolest, ahlorhidrija, gasterktomija…)
3)gubitak Fe iz organizma (krvarenja, kamen u bubregu, ulkus, varixi jednjaka, hernija, karcinom)
4)potrebe organizma za Fe (pubertet, trudnoća, dojenje…) *DEFICIT Fe: -Prolazi kroz tri faze: *PRELATENTNI PERIOD: pražnjenje rezerve Fe, još uvek nema biohemijskih i hematoloških znakova *LATENTNI PERIOD: smanjenje rezerve Fe u serumu, još nema anemije
www.belimantil.info

8
*PERIOD DEFICITA SA ANEMIJOM: Fe u kosnoj sržI je sada nedovoljan za sintezu Hb *KLINIĈKI: -U većine pacijenata tegobe poĉinju da se javljaju u naporu, jer je tada povećana potreba za O2. Tegobe su uslovljene hipoksijom tkiva. -otežano disanje, lupanje srca, zamaranje, slabost, razdražljivost, glavobolja -promene na jeziku: helitis, angulitis -disfagija, gastritis -bleda koža i vidljive sluznice, sedefast sjaj sklera -nokti krti (koilonihija), kosa krta, sedi -neuralgija extremiteta *LABORATORIJA: -U perifernoj krvi anemija sa mikrocitozom i hipohromijom Er
-Hb, Fe,Ht, Er
-u kosnoj sržI hiperplazija sa brojem Er -anizocitoza = razliĉiti oblici Er *TERAPIJA: -Preparati Fe: oralno (Ferosulfat, Feroglukonat) ili parenteralno (Fedex) -Vitamin C *DIJAGNOZA: -Kod dijagnostifikovanja ove vrste anemije, važno je otkriti etiološki faktor, jer je ova anemija pretežno sekundarna, te je ĉesto znak nekog hroniĉnog ili malignog oboljenja. 6.TALASEMIJE
Mediteranska (Cooleyeva) anemija - talasemija (major) -Hemoglobinopatija -poremećaj u sintezi globulina -? zbog nedovoljnog stvaranja Er tj faktora za neophodnih za eritropoezu ? -Ovo su nasledne bolesti u kojim ja poremećena sinteza jedne ili više subjedinica hemoglobina, gde su sintetisani lanci Hb normalne strukture, ali se stvaraju u neadekvatnoj koliĉini. *PODELA:
-Dele se prema tome koji je lanac globina stvoren u manjku:
talasemija delecija globulinskog gena
talasemija MINOR – lakšI oblik MAJOR – težI oblik
TALASEMIJA: lanac kodiraju 4 gena; bolest će se smrtno završiti samo ako su sva 4
defektna; u suprotnom predominiraju ne- lanci; ova forma je blaža od -talasemije, jer ovde
predominantni ne- lanci nisu tako toxiĉni kao lanci (koji predominiraju u -talasemiji),
takoĊe, ne- lanci su rastvorljiviji u vodi
TALASEMIJA: Er su ovde hipohromni (jer je redukovana sinteza HbA); prisutna je hemolitiĉka komponenta
(ne znog nedostatka -lanaca, već zbog viška -lanaca ĉija je sinteza normalna); -lanci predominiraju i zato stvaraju agregate koji precipitiraju unutar Er; takvi Er su fragilni i meta su za ćelije RES-a; to je destrukcija Er u kosnoj sržI=infektivna eritropoeza, koja ima za posledicu porast resorpcije per os unetog Fehemosideroza *PATOGENEZA:
www.belimantil.info

9
-Zamena peptidnog (reĊe ) lanca u globulinu lancem kao posledica genskog poremećaja dovodi do talasemije.
-Autozomo – dominantni poremećaj hromozoma 11() i 16() -Minor talasemija: većina bolesnika nema tegobe, 20% ima. Th nije potrebna. -Major talasemija:sreće se kod naroda naseljenih oko Sredozemnog mora. Major talasemija postaje vidna kada se fetalni Hb treba zameniti sa HbA. Retardi od roĊenja. Karakteriše se prisustvom LEPTOCITA (tanki Er), mikrocita, hipohromnih Er “target cells”; smrta u 17.godini *KLINIĈKI: -Simptomi su posledica anemije, hemolize i prekomernog deponovanja Fe.
-Produkcija lanaca premašuje produkciju ; on se nakuplja u višku i precipitiraju prekursorima Er. Ovi precipitati Hb menjaju propustljivost opne Er i liziraju ga. -Postoji kompenzatorna hiperplazija u kosnoj srži. -ikterus + anemija + deponovanje melanina = osobena boja lica
-t, zaostajanje u rastu, hepatosplenomegalija, promene na kostima; defor.kosti lica *LABORATORIJA: -Hipohromna, mikrocitna anemija *DIJAGNOZA: -leptociti, target cells, eritroblasti u krvi
-Fe u serumu, bilirubin *TERAPIJA: -U težim oblicima daju se transfuzije da poprave hipoxiju tkiva -Deferoxamin infuzije (za odstranjenje viška Fe) -splenektomija, transplantacija kosne sržI 7.HEMOLIZNE ANEMIJE -Mogu biti: 1)Intravaskularne 2)Extravaskularne (slezina) -Nastaju zbog povećane destrukcije Er u cirkulaciji; Er su ovde skraćenog veka i do 6x (normalno je 120 dana, a u anemiji je 10-15 dana)
-Hemolizna anemija je uvek praćena konc.bilirubina i pojavom ikterusa, je i konc.urobilinogena u urinu i stolici i u žuĉI – predispozicija za holelitijazu
-Kompenzatorni odgovor organizma na hemolizu je eritropoeza koja se ogleda u hiperplaziji kosne sržI – crvene loze i otpuštanju povećanog broja retikulocita na periferiju (retikulocitoza) ! *1*HRONIĈNI OBLIK: -Uglavnom nasledni; kod korpuskularnih anemija -anemija, splenomegalija (splen uklanja oštećene Er), ulceracije na potkolenicama -ispupĉenja kostiju lobanje (zbog hiperplazije kosne sržI) -hemolizne krize – tokom napora i infekcija (bol u trbuhu, groznica) -reĊe su aplastiĉne krize, koje znaĉe prestanak funkcije kosne srži *2*AKUTNI OBLIK: -Kod steĉenih, extrakorpuskularnih anemija -groznica, bolovi, šok, ikterus, anemija, bubrežna insuficijencija *LABORATORIJA: 0skraćenje veka Er
-hiperbilirubinemija - žuĉnih boja u urinu i stolici -hemoglobinemija, hemoglobinurija -retikulocitoza, eritroblastoza, posebni oblici Er
-granulocitne i trombocitne loze
www.belimantil.info

10
*OSNOVNO U HEMOLIZNIM ANEMIJAMA: 1)Pojaĉana razgradnja ER (u jetri=više oštećeni Er; u slezini=manje oštećeni Er) 2)Pojaĉana eritropoeza u kosnoj sržI *Posledica pojaĉane rezgradnje ERporast konjugovanog i nekonjugovanog bilirubinaurobilirunemija i hiperholiĉne stolicehemoglobinurijaoligurija, anurija, acc bubrežna insuficijencija 7.1.Korpuskularne hemolizne anemije Sve su uroĊene i posledica su štetnog dejstva intrakorpuskularnih faktora A)MEMBRANOPATIJE – poremećaj graće opne Er 1.Nasledna sferocitoza; nasleĊuje se autosomo-recesivno -Odlikuje se povećanim brojem sferocita tj eritrocita, koji zbog poremećene strukture membrane pokazuju sniženu osmotsku rezistenciju i pojaĉanu razgradnju u slezini
-Ovde postoji deficit lanaca spektrina, što u opni Er dovodi do propustljivosti za jon Na
-Zato je potreba za aktivnošću pumpe za Na
-To se postiže glikolizom u Er
-Kada Er stignu u slezinu se glikoliza, pa se smanjuje rad pumpe, Na i voda ulaze u Er, koji dobijaju loptast (sferiĉan) izgled, što smanjuje njihovu elastiĉnosti i povećava mogućnost oštećenja pri prolasku kroz slezinu, gde se i razrgaĊuju *Kliniĉka slika: -uz znake anemije (bledilo, malaxalost, vrtoglavica, tinitus, tahikardija) -na hemolizu ukazuje i žutica i splenomegalija (zbog zadržavanja sferocita u slezini umnožavaju se makrofagne ćelije slezine); kalkuloza žuĉne kese od detinjstva
*Dijagnoza: -broj retikulocita, nalaz sferocita u razmazu periferne krvi -volumen sferocita je obiĉno manji i govori u prilog MIKROCITOZI *Terapija je splenektomija 2.Hereditarna akantocitoza -Stanje koje karakterišu Er sa nepravilnim produžecima koji im daju makazast izgled u razmazu periferne krvi
-To je uroĊeni nedostatak -lipoproteina, kod tih pacijenata se viĊa kao i u nekim bolestima jetre (ciroza) i bubrega 3.Hereditarna eliptocitoza -UroĊeni poremećaj opne Er (aut-domin), koji dovodi do pojave elipsoidnih oblika u periferiji. -Er su kobasiĉastog izgleda -Bez simptoma je; otkriva se sluĉajno u razmazu krvi, obiĉno sa sferocitozom B)ENZIMOPATIJE – poremećaj enzimskih sistema u Er 1.EHA zbog deficita piruvat kinaze -deficit enzima potrebnih za Embden-Meyerov put (anaerobna glikoliza; obezbeĊuje 90% energije za Er), što uzrokuje nedovoljno stvaranje energije u Er i nedovoljno obnavljanje ATP-a, što skaraćuje vek Er. 2.EHA zbog deficita gly-6-dehidrogenaze -deficit enzima potrebnog za hexozo-monofosfatni put
(aerobni put; daje 10% energije za Er; stvaranje metHb.) (ovaj put se stvara ATP, kojeg u ovom sluĉaju nema, a on,redukuje met Hb u Hb) -denaturacija Hb i taloženje Heinzovih telašaca (precipitati denaturisanog Hb) smanjuju savitljivost Er, što dovodi do hemolize u slezini); Th nije potrebna C)HEMOGLOBINOPATIJE – poremećaj sinteze globulina
www.belimantil.info

11
1.Paroxizmalna noćna hemoglobinurija je steĉena bolest matiĉnih ćelija hematopoeze koja se karakteriše stvaranjem krvnih ćelija preosetljivih na komplement, kada stradaju sve ćelije, a naroĉito Er. *Kliniĉka slika: Poĉinje slikom hroniĉne hemolizne anemije, a javlja se i noćna hemoglobinurija koja se ispoljava tamnom do crvenom jutarnjom mokraćom (zbog velike koliĉine Hb) *Dijagnoza: postojanje Er osetljivih na komplement *Terapija:-kod težih oblika=transfuzija opranih Er; deficit Fe=nadoknada Fe preparatima -u sluĉaju tromboze (v.hepatis) = antikoagulansi -transplantacija kosne sržI 2.Hemoglobinopatije vezane su za poremećaj aminokiselinske strukture Hb. To su: a)Drepanocitoza (HbS i srpasti Ermikroinfarkti i hemoliza u slezini; crnci) b)Nestabilni hemoglobini (sklonost Hb da precipitira=Heizova telašca) c)Hb sa patološkim afinitetom za O2 (povećan ili smanjen afinitet ) d)Methemoglobinemije 3.Talasemije 4.Porfirije 7.2.Extrakorpuskularne hemolizne anemije su steĉene i posledica su dejstva extrakorpuskularnih faktora *1.IMUNE EHA - IZOIMUNE a)Izoimuna bolest novoroĊenĉeta Javlja se u fetalnom ili poroĊajnom periodu i uslovljena je Rh (D) ili ABO nepodnošljivošću krvnih grupa majke i ploda. -Velika razgradnja Er u periferiji dovodi do extramedularne hematopoeze, naroĉito u jetri, sa prelaskom velikog broja eritroblasta na periferiju *Kliniĉka slika: -anemija, ikterus, hepatosplenomegalija -encefalopatija i hidrops fetusa -kernikterus, ispoljava se pospanošću, hipotonijom i areflexijom (sisanje) *Terapija i profilaxa: Zaštita od primarne imunizacije Rh- porodilje, sprovodi se davanjem anti Rh(D) antitela u toku prvih 72h od roĊenja Rh+(D) deteta = ROGAM (anti-D-globulin) -posle roĊenja fototerapija, jer dejstvom svetla bilirubin ide u dipirole koji se lako izluĉe b)Posttransfuziona reakcija je anafilaktiĉka r-ja na RhD ili ABO nepodudarnost -Uneti Er sadrže Ag koje organizam primaoca nema, pa se na njih stvaraju At koja dovode do razgradnje Er. -OslobaĊa se valika koliĉina Hb, praćena hemoglobinurijom, anurijom i acc bubrežnom insuficijencijom. Moguća je i DIK. *)Autoimune -Odlikuju se prisustvom autoAt u krvnoj plazmi, na sopstvene Er Moguće je da neki ĉinioci (virusi, lekovi..) nemaju Ag strukturu opne Er protiv koje sopstveni sistem stvara At. Ili je možda reĉ o grešci u samom imunom sistemu. -Postoje oblici sa: 1)Hladnim At
IgM se vežu se na manje od 30 C (mikoplazma infekcije; mononukleoza…) 2)Toplim At
IgG auto-At; se vežu na više od 37C(idiopatske anemije; SLE; lekovi; limfomi i leukemije) *Kliniĉka sl: hepato i splenomegalija, bledilo, žutica i taman urin zbog hemoglobinurije *Terapija: -veće doze KS (1-2mg/kg/dan) uz transfuziju većih doza opranih Er
www.belimantil.info

12
-Splenektomija: kod onih koji ne reaguju na pomenutu Th *2.NEIMUNE EHA 1)Mehaniĉke: veštaĉke valvule, mikroangiopatska 2)Infekcije: Closrtidium Welchi, malarija 3)Lekovi, hemijska jedinjenja 4)Diseritrocitopoeza 8.PRAVA POLICITEMIJA je nepoznatog porekla; to je klonska proliferacija svih loza hematopoeze sa dominantnim uvećanjem mase Er. -Karakteriše se lividno-crvenom bojom lica i vidljivih sluzokoža, splenomegalijom,
povećanjem broja Er, konc.Hb, visokim hematokritom, broja granulocita, naroĉito trombocita *KLINIĈKA SLIKA: -glavobolje, slabost, malaxalost, lako zamaranje, tinitus, zagasito rumena boja lica -vidljive sluzokože lividne, a konjuktive crvene -pruritus, tromboze, epistaxe, splenomegalija ĉesto *DIJAGNOZA:
-broja Er, Hb *eritropoetin je smanjen (zbog porasta Hb) -Er su normohromni, normocitni
-bazofilija, alkalna fosfataza
-leu, trombocitoza *KOMPLIKACIJE: -Izražena viskoznost krvi, zbog povećanja mase Er, praćena usporenom cirkulacijom, naroĉito kroz manje krvne sudove i
-broj trombocita krvarenja i tromboza (venske tromboze sa embolijama i tromboflebitisi) *TOK: Bolest je hroniĉnog toka, može da, može da traje i do 15 godina. *TERAPIJA: -hemioterapijska mijelosupresija (bisulfan, hidroxiurea) -venepunkcijau poĉetku 500-2000ml krvi nedeljno, a kasnije 500ml na 2-3 meseca -radioaktivni P, RTG zraĉenje 9.ACC AGRANULOCITOZA -uzrok nastanka = uzimanje lekova(indometacin, salicilati, diazepam, hlorpromazin,ibuprofen, tiouracilpenicilamin, kinin, DDT, soli zlata, antitireoidni lekovi, ranitidin…) -to je sindrom izazvan nižim vrednostima neutrofila (<0,5x10 na 9/l) ili potpunim odsustvom -karakteriše se teškom opštom infekcijom, zbog Išĉezavanja granulocita iz krvi *ETIOLOGIJA: najvećI broj granulocita nastaje zbog preosetljivosti izazvane nekim lekom (analgetici, antitireoidni lekovi, antibiotici, fenotiazini, antihistaminici)
*KLINIĈKA SLIKA:-bolest poĉinje naglo (t, malaxalost na tonzilama i nepĉanim lukovima javlja se angina sa prljavo sivim naslagama)+gangrenozni ulceri vagine, rektuma i anusa -otežano gutanje i govor zbog pratećeg otoka -Ly žlezde vrata su oteĉene -bledilo, sluznice normalno prokrvljene -opšte stanje je vrlo teško -može doćI do upale pluća, bubrega, creva, sepse, smrti
*LABORATORIJA: -broj granulocita (>200-300mm3)
www.belimantil.info

13
-u kosnoj sržI se ne nalaze ćelije granulocitne loze
-SE i fibrinogen *TERAPIJA: -uklanjanje uzroĉnika (lekovi, hem.agensi) +hidratacija -antibiotici +parenteralna ishrana -transfuzija neutrofila u teškim sluĉajevima Simptomi obiĉno traju oko 2 nedelje; mortalitet je 20% bez obzira na primenjenu Th 10.CHR IDIOPATSKA AGRANULOCITOZA Bolest je hroniĉnog toka, nepoznatok uzroka, a karakteriše se ĉestim infekcijama zbog niskog broja granulocita u krvi.
*Kliniĉka slika: infekcije razliĉitih organa ili sistema, praćene visokom t *Dijagnoza: -anamneza,
-broj granulocita u krvi -odsustvo mijelocita i metamijelocita u kosnoj sržI *Terapija: antibiotici i antimikotici 11.INFEKTIVNA MONONUKLEOZA -Prouzrokovana je Epsten-Barr virusom koji pripada grupi herpes virusa. -Akutnog je toka, obiĉno benigna i ĉešće se javlja kod mladih (15-25 god) -Širi se kapljiĉnim putem (kissing disease), aerogeneo, transfuzijom krvi *Kliniĉka slika: Oboljenje može pokazati vrlo širok spektar simptoma -prodromalni stadijum od nekoliko dana ide pod slikom malaxalosti, zamora, glavobolje, grozniĉavosti -u punom zamahu javlja se temperetura, cervikalna adenopatija i grebanje u grlu sa hiperemijom sluznice -od drugih znakova ĉešća je splenomegalija od hepatomegalije -na kožI se može pojaviti raš, nekad i blažI ikterus -bolest obiĉno traje 2 nedelje, oporavak u 3.nedelji -kod teškog oblika mogu se javiti nervne komplikacije, prljavo-sive skrame u grlu, generalizovano uvećanje limfnih žlezda i krvarenje *Dijagnoza:
-leu, ly, monociti i nalaz atipiĉnih limfocita (virociti) + Paul-Bunell-Davidsonova reakcija *Diferencijalna dijagnoza: -oboljenja izazvana citomegalovirusom -difteriĉne naslage (difterija) -acc.leukemija *Komplikacije- prolazna hemolizna anemija -miokarditis -ruptura slezine (retko) -oštećenja CNS *Terapija: -simptomatska -ispiranje guše -analgetici, antipiretici -glukokortikosteroidi 10-15mg u izraženim oboljenjima
www.belimantil.info

14
II.LEUKOCITI -Stvaraju se delom u kosnoj sržI (granulociti, monociti i deo ly) i delom u limfatiĉnom tkivu (ly i plazma ćelije) -Oni uĉestvuju u odbrani organizma: a)uništavanjem agenasa fagocitozom b)stvaranjem At i senzibilisanih ly *Belu krvnu lozu ĉine: 1)NEUTROFILI: 55-60% leu; vrše fagocitozu agenasa i njihovu razgradnju (pomoću lizozoma); u krvotoku kod zapaljenja, oni odmah dospevaju na mesto procesa 2)EOZINOFILI: 2-3% leu; imaju sposobnost hemotaxe; slabije fagocituju; puno ih je kod infekcija parazitima; u alergijskim reakcijama inaktiviraju heparin i histamin otpuĉtene iz neutrofila, bazofila ili mastocita 3)BAZOFILI:0-1% leu; otpuštaju heparin (spreĉava grušanje krvi), ubrzavaju odstranjivanje masti iz krvotoka, imaju ulogu u alergijama (IgE se vezuje za bazofile) 4)MONOCITI: 6-8% leu; tek kada dospeju u tkiva poĉnu da bubre, pa postaju makrofagi koji vrše fagocitozu, zatim taj fagocitovani materijal predaju limfocitima koji stvaraju At 5)LIMFOCITI: 20-40% leu; B-ly = humuralni imunitet (proizvodnja At-Ig) T-ly = celularni imunitet (citotoxiĉne ćelje – direktno ubija Ag; helper ćelije – aktiviraju makrofage, b-ly, T-ly; supresorne T-ćelije – spreĉavaju da prejako reaguju) *Granulociti, nakon otpuštanja iz kosne sržI žive 4-8h u krvi i još 4-5 dana u tkivu. *Ukupan broj leukocita je 4x10 na 9 *Ukupan broj trombocita je 150-400x 10 na 9 1.LEUKEMIJE su maligna oboljenja hematopoeznog tkiva, koja se karakterišu prekidom sazrevanja pojedinih vrsta ćelija i njihovim nagomilavanjem u kosnoj sržI, slezini, jetri, ly žlezdama, i intersticijumu, tj mezenhimu organa. Ovi nenormalni ćelijski elementi se pojavljuju i u cirkulišućoj krvi. 1)U pitanju je metaplazija lokalnih retikulo-histocitnih elemenata u leukemij. ćeliji 2)Leukemij. ćelije se brzo ubacuju u krv ĉim se stvore u kosnoj sržI, analogno emisiji normalnih zrelih krvnih ćelija -U chr leukemijama, u poĉetku je oĉuvano sazrevanje i diferencijacija ćelija Etiologija leukemija: jonizujuće zraĉenje, hemijski kancerogeni (benzen i derivati), neki lekovi (citostatici), leukemogeni virusi (HTLV-1), endogeni faktori (porodiĉna slkonost) *Supstrat leukemijskog procesa može da predstavlja bilo koji ćelijski elemenat hematop tkiva. *Najĉešće su: -MIJELOIDNA LEUKEMIJAumnoženi granulociti ili prethodnici -LIMFOIDNA LEUKEMIJAumnožavanje ly i prethodnika -monocitna leu, plazmocitna, eritroleukemija *Etiologija leukemija: je nepoznata, najverovatniji uzroĉnici su jonizujuće zraĉenje, genetska predispozicija, virusi *Sa kliniĉkog aspekta svaki oblik leukemije može se javiti u acc i chr obliku
RES:1)pokretni makrofagi
2)fixirani tkivni makrofagi
Fixirani tkivni makrofagi su: Kupferove ćelije u jetri,
makrofagi u ly čvorovima, slezini I kosnoj sržI,
alveolarni makrofagi, tkivni histociti, mikroglija u
mozgu
www.belimantil.info

15
2.ACC MIJELOIDNA LEUKEMIJA -Je maligna klonalna bolest hematopoezno tkiva *Karakteriše se: 1.proliferacijom malignog tkiva mijeloidne loze 2.progresivnom infiltracijom 3. I zamenom normalnih ćelija kosne sržI patološkim ćelijama (blastima)
*U toku bolesti se broj malignih leu, a br Er i Tr, što dovodi do infekcije anemija, tromboze i krvarenja *Etiologija: smatra se da u pojavi bolesti ulogu igraju unutrašnji, nasledni ĉinioci i spoljašnji ĉinioci. SPOLJAŠNJI FAKTORI izazivaju promene u genomu neke od hematopoeznih ćelija, što vodi ka malignom preobražaju te ćelije (ONKOGENI) VIRUSNA TEORIJApostoji, a ona kaže da se virus inkorporiše u genom krvnih ćelija, a pod dejstvom odreĊenih spoljnih i unutrašnjih ĉinilaca dolazi do aktiviranja virusa i stvaranja patoloških ćelija. *Spoljni faktori su: jonizujuće zraĉenje, neka hemijska jedinjenja (derivati benzena, citostatici, hloramfenikol...) *Pojavi leukemije mogu doprineti neka uroĊena ili steĉena stanja imunodeficijencije, genska i rasna konstitucija, genska predispozicija *Patogeneza: najviše je pogoĊena unipotentna ćelija -U acc leukemiji postoji infektivna hemocitopoeza koja naroĉito pogaĊa unipotentnu (matiĉnu) ćeliju uz prisustvo smanjene ili abnormalne koliĉine mijelocitopoetskih faktora -Pretpostavlja se da postoji odsustvo regulatora u toku razmnožavanja unipotentne ćelije -Smatra se da leukemijska ćelija duže živi od normalne, zbog poremećaja mijelocitopoeze, te je njihovo brojno povećanje više posledica nagomilavanja, nego proliferacije -Zloćudni preobražaj nastaje u jednoj od matiĉnih hematopoetskih ćelija. Ako imunski sistem uništi ovu ćeliju, nastaje matiĉna ćelija leukemijskog klona. Neprekidnim deljenjem i razmnožavanjem ovog klona, nagomilava se masa leukemijskih ćelija koje potiskuju normalno hematopoezno tkivo. *Kliniĉka slika: 1)Znaci insuficijencije kosne sržIzbog nagomilavanja leuk.ćelija 2)Sy i znaci nastali usled usled infiltacije tkiva i organa (koža,testisi) 3)Sistemski poremećaji -Da bi se ispoljili simptomi potrebno je 10 na 12 ćelija -Poĉetak bolesti može biti vrlo raznolik. Od slabo izraženih pojava slabosti, malaxalosti, bledila, do naglih krvarenja, bolova u kostima, temperature -Anemijau razvijenoj bolesti zapaža se bledilo, malaxalost i mršavljenje u ½ bolesnika -Trombocitopenijakrvarenje u kožu, vidljive sluznice, iz desni, nosa, bolovi u kostima, trbuhu -Splenomegalija, reĊe hepatomegalija, uvećanje jedne limfne žlezde -Ĉeste su infekcije respiratornog i urinarnog trakta -Infiltracija meningea koja se opisuje kao MENINGEALNA LEUKEMIJA, zapaža se kod manjeg broja pacijenata i praćena je jakom glavoboljom, poremećajem vida, kranijalnih nerava i povraćanjem *Komplikacije su posledica hematoloških, infiltrativnih, metaboliĉkih i nutritivnih promena:
www.belimantil.info

16
1)Hematološki poremećaji: granulocitopenija praćena infekcijama, trombocitopenija sa krvarenjem i anemija 2)Metaboliĉki poremećaji: hiperurikemija, DIK, hipo Ili hiperkalijemija, i hepatiĉka insuficijencija 3)Nutritivni poremećaji: gubitak telesne težine *Dijagnoza: pregled periferne krvi i kosne sržI: unipolarnostpatoloških blast ćelija meĊu kojima se teško pronalaze normalne ćelije kosne sržI
br.leu izmeĊu 10-50000mm3/može i do 100000 u leukocitnoj formuli dominiraju patološki blasti broj trombocita je nizak, ispod 50000
faktori koagulacije su što doprinosi krvarenju
mokraćna kiselinaacc bubrežna insuficijencija (naroĉit posle hemoterapije); *Podela: MO: nediferencirana M1: mijeloblasti bez sazrevanja M2: sa granulocitnim sazrevanjem M3: acc promijelocitna M4: monocitno i granulocitno sazrevanje (mijelomonocitna) M5: monocitna ili monoblastna M6: eritroleukemija M7: megakarioblastna *Terapija: se sprovodi u 4 faze: 1)Indukciona faza=cilj je postizanje remisije 2)Faza konsolidacije 3)Intenzifikaciona faza 4)Faza održavanja +Suportativna Th (davanje Er, Tr, plazme, krioprecipitata, antibiotika, antimikotika, faktora rasta) -Daje se kombinacija 2 ili više citostatika, koji deluju u razliĉitim fazama ćelijskog ciklusa, sve do postizanja remisije (odsustvo blasta u krvi i manje od 5% u kosnoj sržI), a kada se postigne remisija, nastavlja se sa Th održavanja (Merkaptopurin) Th indukciona: Citozinrabinozid + Daunorubicin = postiže se remisija u 60% sluĉajeva (ako se ovim ne postigne remisija, prognoza je loša) Th konsolidacije: najbolji je rezultat posle 2-3 ciklusa konsolidacione Th; remisija traje oko 10-28 meseci; 10-30% bolesnika preživi 5 godina -Posle remisije nastaje relaps bolesti – obiĉno u toku 2 godine. Tada je bolest manje osetljiva na Th, a preživljavanje je 3-6 meseci Transplantacija kosne sržI posle postizanja remisije *Podela leukemija: 1)Akutne: -Mijeloidna (mijeloblastna) -Limfoblastna 2)Hroniĉne: -Mijeloidna (granulocitna) -Limfocitna -Ostale: leukemija vlasastih ćelija, polimorfocitne leukemije, limfom/leukemija sindrom
www.belimantil.info

17
3.ACC LIMFOIDNA LEUKEMIJA -ovde su u osnovi limfoblasti; umnoženi su i prethodnici -Acc limfoidna leukemija je maligna bolest nastala klonskom proliferacijom i nagomilavanjem limfoblasta. -Leukemijski klon može da ispolji osobine B i T limfocita -Javlja se pretežno u prve tri decenije života
-Ćelijske promene su sliĉne onim u acc mijeloidnoj leukemiji i sastoje se u tome što postoji broj leukocita, udruženo sa visokom proporcijom ćel. tipa limfoblasta !!! -Sazrevanje leukemijskih ćelija prestaje na nivou limfoblasta !!! *Kliniĉka slika: -Poĉetak bolesti je nagao, postoje: anemija, granulocitopenija i trombocitopenija -slabost i malaxalostzbog anemije -bolovi u kostima i zglobovima kod dece (zbog osteoliznih promena), leuk. CNS -infekcije kože, orofarinxa, pluća, krvarenjatrombocitopenija (hematomi, petehije) -80% bolesnika ima uvećanu slezinu, jetru, ly žlezde (ovde je to obavezno !!!) *Dijagnoza: 1)u perifernoj krvi: anemija, granulocitopenija, trombocitopenija
2)leu mogu biti ili normalni 3)kosna srž je hipercelularna uglavnom limfoblasti *Podela: L1: blast mali; visok nukleo-citoplazmatski odnos L2: blast krupniji; heterogeni L3:vakuolizovani blast, bazofilna citoplazma *Prognoza: -U dece izmeĊu 3-10 godina postiže se izleĉenje hemioTh i transplantacijom -Kod odraslih se postižu remisije koje u 80% traju 18 meseci *Terapija: Indukciona Th: za postizanje remisije (u 75-85% u toku 4 nedelje)
Vincristin + Prednison + Adriblastin + asparginaza profilaxa CNS leukemije = Metotrexat intratektalno i zraĉenje CNSa Th konsolidacije: posle uspostavljanja remisije 6-tioganin, Merkaptopurin, Metotrexat, Dexorubicin, Cikolfosfamid Th održavanja: 6-merkaptopurin i Metotrexat 2-3 godine Transplantacija kosne sržI posle postizanja remisije 4.CHR MIJELOIDNA LEUKEMIJA Je izazvana širenjem klona i subklonova malignih ćelija koji akumulitaju nove mutacije dovodećI do acc ili subacc (ubrzane) faze oboljenja --ima odlike acc leukemije rezistentne na dosadašnju Th *Etiologija: je nepoznata; nema herediteta -U 90% pacijenata postoji “FILADELFIJA HROMOZOM”=translokacija dugog kraka 22 na 9 hromozom -Bolest se odvija u 3 faze: 1)Chr fazatraje 3-4 godine 2)Faza pogoršanjatraje 3-4 meseca
www.belimantil.info

18
3)Faza akutizacije Blastna transformacija *Kliniĉka slika: -Ĉešća je u muškaraca srednje životne dobi -U 80% poĉinje postepeno: opšta slabost, zamor, noćno znojenje, mršavljenje, bol pod levim rebarnim lukom, limfne žlezde su neznatno uvećane ili normalne veliĉine -20% je asimptomatiĉno pri postavljanju dijagnoze skoro svi imaju splenomegaliju pri postavljanju Dg. Ponekad hepatomegalija -Retke su anemije, trombocitopenije i infekcije *Dijagnoza: Izražena leukocitoza (do 500000mm3), sa predominacijom elemenata granulocitne loze u svim fazama sazrevanja. Mijelociti i metamijelociti su nesrazmerno zastupljeni. Bazofilija upadljiva. u kosnoj sržI izražena granulocitopoeza
aktivnost alkalne fosfataze hiperurikemija, naroĉito posle Th splenomegalija Filadelfija hromozom *Terapija:
-interferon = prolazno dovodi do etiološke eradikacije malignog klona -hidroxiurea -autologa transplantacija *Prognoza: -Preživljavanje do 33 meseca, sa interferonom do 50 meseci -Ako se u toku evolucije i leĉenja chr mijeloidne leukemije razvije MIJELOFIBROZA, to je loš prognostiĉki znak MIJELOPROLIFERATIVNE BOLESTI: (pluripotentna matiĉna ćelija) 1.Chr mijeloidna leukemija(granulocitna) leukemija 2.Plicitemija rubra vera 3.Mijelofibroza 4.Esencijalna trombocitopenija 5.MIJELOFIBROZA Je chr mijeloproliferativno oboljenje kosne sržI, koje se karakeriše progresivnom fibrozom kosne sržI (stimuliše se umnožavanje fibrocita od strane malignih ćelija hematopoeze) sa extramedularnom hematopoezom Obzirom da postoji fibroza krvnih sudova, hematopoeza se odigrava u drugim tkivima-jetri i slezini *Etiologija: Hem. I jon. zraĉenje, metastaze u kosnoj sržI, TBC -Sekrecija ĉinioca stimulacije fibrocita dovodi do umnožavanja fibroznog tkiva kosne sržI i progresivne splenomegalije ali zbog naseljavanja tkiva slezine matiĉnim ćelijama hematopoeze, koje su umnožene u cirkulaciji, a ne zbog insuficijencije hematopoeze u kosnoj srži *Kliniĉka slika: -Poĉinje postepeno, sa razvojem anemije i uvećanjem slezine i jetre kod 50% to daje osećaj nelagodnosti i punoće pod levim rebarnim lukom -Znaci anemije: zamaranje, slabost, bledilo, tahikardija -sklonost krvarenju, modrice, noćna znojenja
www.belimantil.info

19
-može se javiti i portna hipertenzija zbog intrahepatiĉne tromboze -ponekad se javlja krvarenje u kožI i sluznicama GITa (zbog trombocitopenije i smanjenja faktora protrombinskog komplementa) *Dijagnoza: -u parijetalnoj krvi prisutni mijelociti i eritroblasti (“Er u obliku suze”) -LAB: varijacije krvne slike i svih krvnih loza, anemija u 50% -Patohistološki: histologija kosne sržIeritroblastemija, leukocitoza u 50% -umnoženost retikulinskih vlakana; moguća do osteoskleroze *Terapija: -transfuzije Er u stanju teške anemije -androgeni preparati=zbog insuf,eritropoeze -hidroxiurea, hemioTh kod velike splenomegalije -splenektomija indikovana kod ometanja rada trbušnih organa *Tok: -Bolest traje dugo, 5-6 godina, može i do 20 godina -Ako jako dugo traje, može prećI u acc granulocitnu leukemiju 6.CHR LIMFOIDNA LEUKEMIJA Je maligno limfoproliferativno oboljenje koje nastaje zbog maligne klonske proliferacije malih, dobro diferentovanih limfocita (obiĉno B tipa) i njihovom akumulacijom u kosnoj sržI, perifernoj krvi, slezini, ly žlezdama, jetri i drugim organima. -Javlja se u 25% bolesnika sa leukemijama, najĉešće u osoba preko 50 godina. -Ima dugotrajnu evoluciju i priliĉno povoljnu prognozu *Etiologija: -Moguća je uloga hromozomskih anomalija, onkogena i retrovirusa *Kliniĉka slika: -Može poĉeti asimptomatski, (25% se otkriva sluĉajno) -Kod ostalih, simptomi nastaju zbog tkivne infiltracije limfocitima, citopenije u perifernoj krvi i imunosupresije -Ĉesto postoji pojava autoAt prema Er i trombocitima. Može se javiti hemoragiĉni sindrom, mada u odmaklim stadijumima -Javljaju se simptomi anemije i generalizovane limfadenopatije. Uvećane limfne žlezde nisu bolne, meke su, pojedinaĉne (vrat, axila, ingvinum)
-t zbog infekcija; H.zpster infekcije -kožni infiltrati, hepatosplenomegalija *Dijagnoza: -perzistentna limfocitoza preko 5000/ml (pretežno B-ly; uglavnom zrele ćelije) -infiltracija kosne sržI limfocitima, limfadenopatija -anemija- delimiĉno hemolizna, kasnije trombocitopenija
- serumski globulin *Prognoza: -Preživljavanje je 5-10 godina; stepenovanje na osnovu kliniĉke slike 0 = limfocitoza * preživljavanje je 12 godina 1 = limfocitoza + limfadenopatija * preživljavalje je 8 godina
2 = “ + ” + splenomegalija
3 = ” + “ + ” + anemija (Hb<100g/l)
4 = ” + “ + ” + “ + trombocitopenija (Tr< 100x10 na 9/l) *Terapija:
www.belimantil.info

20
-Nije potrebna ako je stadijum O. -Th j potrebna ako je 2, pa uvećane žlezde komprimuju okolno tkivo; potrebna je i u 3 -Kortikostroidi = kod insuficijencije kosne sržI -Citostatici = radioTh -splenektomija = kod hemolize Er i trombocitopenije koje ne reaguju na Th 7.HODGKIN LIMFOM Je maligno oboljenje limfoidnog tkiva koje se karakteriše bujanje karakteristiĉnih Reed-Sternbergovih ćelija i bezbolnim progresivnim uvećanjem limfnog tkiva (najĉešće žlezda) *Etiologija: je nepoznata; imaju uĉešća infekcije (infektivna mononukleoza), zraĉenje, onkogeni virusi, Epstein-Barr virus; Javlja se ĉešće kod muškaraca u trećoj i petoj deceniji -Ovde je poremećena funkcija imunološkog sistema, naroĉito ćelijsog, kao posledica smanjenja i insuficijencije T=ly, dok je humuralni imunitet (koji odžava funkciju B=ly) u poĉetku bolesti oĉuvan, sve do terminalne faze. *Kliniĉka slika: Bolest poĉinje neprimetno, bezbolnim uvećanje ly žlezda na vratu i u supraklavikularnoj regiji, reĊe axilama i preponama -žlezde su ĉvrste i vremenom rast. širi se zahvatajućI organe RESa.
-Kod nekih se javi porast t, noćno znojenje, svrab, ikterus, mršavljenje -Najĉešće zahvaćeni organi su: pluća, kosna srž i jetra
-porast tje tipa Pel-Epstajn: naizmeniĉno , normalna i subfebrilna *Kliniĉka klasifikacija: 1.STADIJUMzahvaćeni ly ĉvorovi jednog regiona 2.STADIJUM2 ili više regiona sa iste strane dijafragme 3.STADIJUMly ĉvorovi sa obe str dijafragme i slezine ili 1 extra ly organ 4.STADIJUMdifuzna zahvaćenost više extralimfoidnih organa *Histološka klasifikacija: 1)Limfocitna predominacija: obilje malih ly, uz prisustvo histocita; retke R-S ćelije 2)Nodularna skleroza: kolagena vlakna oiviĉavaju tkivo i stvaraju noduluse RS su zastupljene, kao i ly, eozinofili i histociti 3)Mešovita celularnost:ly i histocit retki,R-S brojne, puno eozinof, plazmoci i fibroblasta 4)Limfocitna deplecija: obilje R-S, ly nema, retki eozinofili i plazmociti *Laboratorija: -u 50-70% dominira leukocitoza, a u 30% eozinofilija -normocitna anemija,(ponekad hemolizna), limfocitopenija, ubrzana SE
- globulina, alkalne fosfataze (znak zahvatanja jetre i kostiju) *Dijagnoza:-Kliniĉki: uvećanje ly žlezda (supraklavikularno, axilarno, prepone) -biopsija najstarije zahvaćenog ly ĉvora -limfografija, CT, laparotomija sa extripacijom retroperitonealnih ly žlezda -limfopenija -Po uspostavljanju Dg, bitno je odrediti kliniĉki stadijum bolesti *Prognoza: Zavisi od kliniĉkog stadijuma i patohistološkog tipa. Znatno bolju prognozu imaju bolesnici sa ranim kliniĉkim stadijumima (1,2), osobe ženskog pola, osobe mlaĊe od 30 godina i bolesnici koji nemaju veliku tumorsku masu. Moguće izleĉenje u 60% uz savremenu Th. *Terapija: zavisi od kliniĉkog stadijuma bolesti -kombinacija radio i hemioTh -kod primarne lokalizacije hemioTh *LIMFOPROLIFERATIVNE BOLESTI:*
www.belimantil.info

21
1)Limfomi = nastaju iskljuĉivo u limfnim žlezdama 2)Acc limfoblastna leukemija 3)Chr limfocitna leukemija 8.NON-HODGKIN LIMFOM Su heterogena grupa tumora koji proistiĉu iz neoplastiĉnog klona B i T-ly u toku njihovog diferentovanja. To su promarni maligni tumori imunog sistema. -Mogu da nastanu u bilo kom delu tela, ali su najĉešćI u organima sa velikom koliĉinom limfocita ly ĉvorovi, slezina, timus, tkivo nazofarinxa, želudaĉno-crevnog trakta. NHL mogu nastati i u nelimfatiĉnim organima, a ukoliko od poĉetka zahvataju i kosnu srž, teško ih je razlikovati od leukemija. -Sazrevanje ly može nastati u raznim fazama njihovog diferentovanja. Pošto NHL nastaje samo iz ćelije (1.klona), smatra se da neoplastiĉni limfociti predstavljaju klonalnu expanziju normalnih ly. ZnaĉI, premda je sazrevanje, NHL nije i proliferacija ly, jer podrazumeva samo njihovo povećanje. -NHL skoro uvek sadržI samo ćelije jedne vrste i stog stepena zrelosti, dok se kod HL vidi izrazit pleomorfizam u sastavu neoplastiĉnog tkiva *Etiologija je nepoznata -uticaj virusa (HTLV 1,2,3), genskih ĉinilaca, uroĊenog i steĉnenog deficita imuniteta (AIDS), jonizujućeg zraĉenja, autoimunih bolesti, onkogenog dejstva nekih lekova i hemijskih jedinjenja -uticaj EBV se posebno izdvaja, jer neprekidni podsticaj ovim virusom dovodi do malignog preobražaja ly *Klasifikacija: -Nodularni -Difuzni *Histološka klasifikacija: -limfocitni limfom -mešoviti limfom -histocitni limfom -nediferentovani limfom -imunoblastni limfom -limfoblastni limfom -mycosis fungoides limfom *Kliniĉka klasifikacija: A)NISKOG STEPENA MALIGNOSTI: 1.malih limfocita 2.folikulski, malih ćelija 3.folikulski mešoviti, malih i velikh ćelija B)SREDNJI STEPEN MALIGNOSTI: 4.folikulski velikih ćelija 5.difuzni malih ćelija 6.difuzni mešoviti, malih i velikh ćelija C)VISOKOG STEPENA MALIGNOSTI: 7.difuzni velikih ćelija, imunoblasta 8.limfoblastni 9.difuzni, Buritov limfom *Kliniĉka slika: -Dugo ne postoje nikakvi simptomi
www.belimantil.info

22
-Poĉinje limfadenopatijom, prvo na vratu, a potom u axilama i preponama ĉvorovi su bezbolni, pojedinaĉni ili meĊusobno sliveni -u 15% postoji intratorakalna lokalizacija ly ĉvorovi medijastinuma, pluća, pleure. Bilo kao posledica neposrednog prelaska limfoma sa susednih ĉvorova, bilo hematogenom diseminacijom -Lokalizacija NHL u abdomenu je ĉesta, a zahvaćeni su retroperitonealni i mezenterijalni ly ĉvorovi -Poĉetna lokalizacija NHL na koštanom sistemu je retka i znak je 4.stadijuma bolesti, recidiva ili progresije bolesti -U pojedinih postoji anemija zbog potiskivanja crvene loze limfoblastnim tkivom u kosnoj sržI, trombocitopenija, leukocitopenija -Mogu se javiti febrilnost, mršavljenje, znojenje, malaxalost, koje sve zajedno predstavljaju sistemske manifestacije NHL *Dijagnoza: Kliniĉka slika i biopsija limfnog ĉvora ili drugog zahvaćenog organa. *Prognoza: Zavisi od histološkog tipa, veliĉine tumora, stepena diseminacije i lokalizacije tumora. -Nepovoljnim ĉiniocima se smatraju: lokalizacija u CNS, testisima, medijastinumu, GITu, kliniĉki stadijumi 3. I 4. (stadijumi su kao kod Hodgkin limfoma), tumorska masa u abdomenu preko 10cm, sistemske pojave *Terapija: zavisi od histološkog tipa (ne kao kod HL, od kliniĉkog stadijuma) -radioTh = palijativna -hemioTh = osnovni vid Th -hiruška Th kod primarne lokalizacije -transpalnatcija kosne srži 9.MULTIPLI MIJELOM-PLAZMOCITOM Je maligna klonska proliferacija B-ly, potpuno diferentovanih do krajnjeg funkcionalnog stadijuma plazma ćelija. Ovo bujanje se dešava u kosnoj sržI i drugim tkivima, a retko prelaze u perifernu krv. Izmenjeni plazmociti stvaraju IgA, D, E -povećan je nivo imunoglobulina u serumu i urinu *Kliniĉka slika: -Malaxalost, kostobolja, patološke frakture (zbog osteoliznih promena na kostima) -smanjenje telesne visine; kompresija k.moždine sbog kolapsa pršljenova -infekcije (zbog granulocitopenije), trombocitopenije -proteinurija, hiperkalcemija, hiperkalemija
-SE, viskoznost krvi -anemija je veoma ĉesta, zbog potiskivanja hematopeznog tkiva plazmocitima *Dijagnoza: -sternalni punktat -elektroforeza proteina (M komponenta) -RTG = osteolizne promene kostiju -anemija, pancitopenija, porast plazma ćelija *Terapija:-citostatici (Melfalan) i prednison=4 dana, protokol ponavljati na 4-6 nedelja -lokalna radioTh (destrukcija kosti) -interferon -analgetici, antibiotici, rehidratacija *Proseĉno preživljavanje je 40 meseci
www.belimantil.info

23
10.MACROGLOBULINEMIJA (Waldenstrom) Je retko oboljenje sa prliferacijom B ćelija (limfoplazmocitnih) koje produkuju IgM; ĉešća je kod muškaraca starijih od 50 godina -Karakteriše se uvećanim ly žlezdama, slezinom, ĉestim krvarenjima iz sluznica nosa i digestivnog trakta; infiltracija k.sržI; anemija, trombocitopenija, granulocitopenija -hiperviskoznos, anemija, Reynov sy (krioglobulinemija) -hemoragiĉna dijeteza (dejstvo makroglobulina na faktore koagulacije=hemoragijski sy) *Terapija: -Hlorambucil i drugi alkilansi (citostatici) -kombinacija citostatici + Prednison -Preživljavanje je 10 godina IMUNOPROLIFERATIVNE BOLESTI: (bolesti plazmocitne loze) bujenje jednog klona B-ly ili limfocita ili limfoplazmocitaĉija je posledicanastanak M belanĉevine, koja je zapravo imunoglobulin III.HEMORAGIJSKI SINDROMI -Poremećaji hemostaze koji dovode do sklonosti ka krvarenju mogu biti uroĊeni i steĉeni. -Nasledni hemoragijski sindromi su skoro uvek izazvani poremećajem samo jednog od ĉinilaca hemostaze -Steĉeni hemoragijski sindromi su najĉešće posledica deficita većeg broja ĉinilaca -Narušavanje integriteta vaskularnog endotela može da remeti hemostazu, bilo u smeru hemoragiskog sindroma, bilo prema trombozi -U mehanizmu hemostaze uloga krvnih sudova je višestruka: 1.vazokonstrikcijom spreĉavaju izlivanje krvi 2.omogućavaju normalnu adheziju (sy von-Wilerbrandovog faktora) 3.stimulišu agregaciju i spolj. put koagulacije (oslobaĊaju tkivni tromboplastin) 4.imaju antikoagulantnu aktivnost 5.utiĉu na fibrinolitiĉku aktivnost
*Hemostaza zaustavljanje krvarenja 1.grĉ krvnog suda 2.stvaranje trombocitnog ĉepa 3.zgrušavanje krvi 4.urastanje veziva u ugrušak **Obajšnjenje za stavku 2: Kada trombociti doĊu u kontakt sa povreĊenim krvnim sudom, oni menjaju svoje karakteristike, bubre i lepe se na kolagene niti i luĉe ADP. Njihovi enzimi stvaraju TROMBOXAN A, koji sa ADP-om aktivira susedne trombocite, koji se lepe na ove prethodne. Tako se stvara TROMBOCITNI ĈEP, u koji se kasnije upliću fibrinske niti.
www.belimantil.info
24
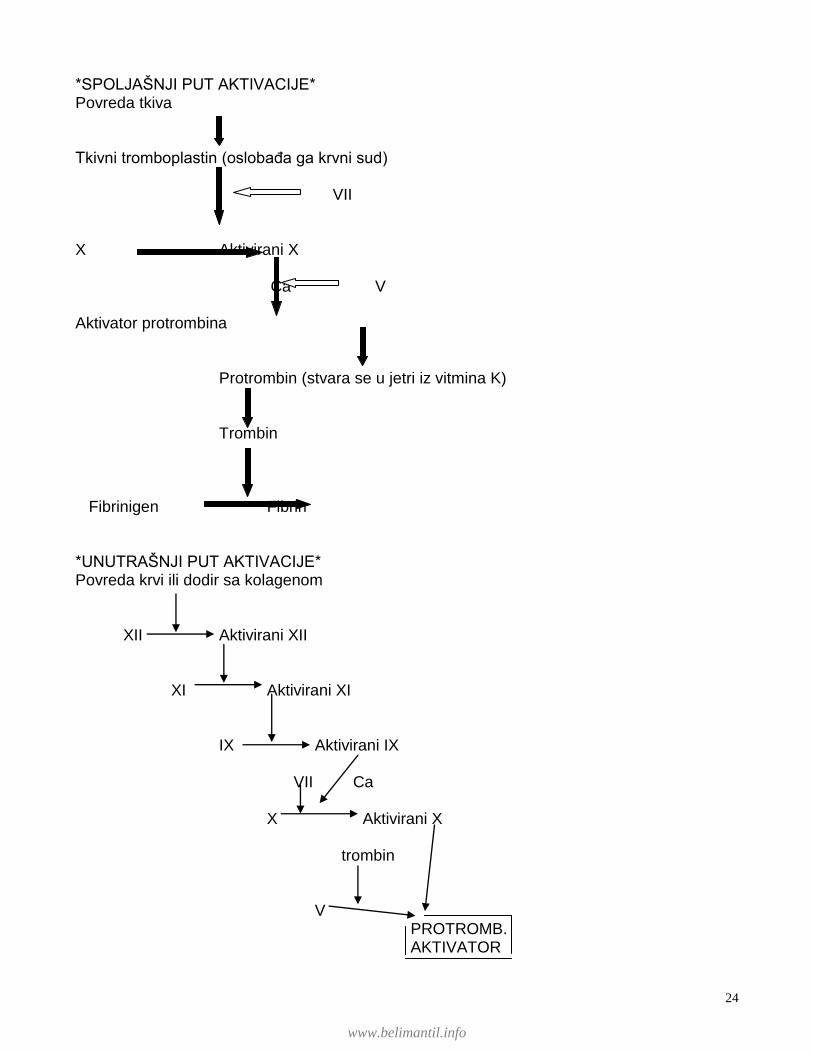
*SPOLJAŠNJI PUT AKTIVACIJE* Povreda tkiva Tkivni tromboplastin (oslobaĊa ga krvni sud) VII X Aktivirani X Ca V Aktivator protrombina Protrombin (stvara se u jetri iz vitmina K) Trombin Fibrinigen Fibrin *UNUTRAŠNJI PUT AKTIVACIJE* Povreda krvi ili dodir sa kolagenom XII Aktivirani XII XI Aktivirani XI IX Aktivirani IX VII Ca X Aktivirani X trombin V PROTROMB. AKTIVATOR
www.belimantil.info

25
-Joni Ca su neophodni za zgrušavanje krvi -Vitamin K je neophodan za stvaranje protrombina, faktora 7, 9 i 10 *Vreme koagulacije po Lee-White: normalno iznosi 6-10min produženo je kod deficita ĉinilaca koji uĉestvuju u stvaranju tkivnog tromboplastina (hemofilija) *Trombocitno vreme – Quick: je vreme koagulacije plazme posle davanja tkivnog tromboplastina normalno iznosi 11-15sec produženo je kod smanjenja faktora 5, 7, 10 *Vreme krvarenja: normalno je 1-8 Min produženo je u imunol.trombocitopeniji *PODELA HEMORAGIJSKIH SINDROMA: 1)Poremećaji krvnih sudova A)NASLEDNE VASKULARNE PURPURE: -Nasledna hemoragijska telangiektazija = Mb.Osler B)STEĈENE VASKULARNE PURPURE: -Alergijska purpure = Henoch – Schonlein -Nealergijska purpura:skorbut,dijabet,anemija, ortostatska, senilna purpura 2)Poremećaj trombocita karakteriše se petehijalnim krvarenjima u kožu, sluzokože, serozne opne, parenhimatozne organe A)KVANTITATIVNI POREMEĆAJI: (poremećaji u broju)
Trombocitopenije zbog stvaranja trombocita
Trombocitopenije zbog razgradnje trombocita 1.uroĊene neimune trombocitopenije 2.steĉene neimune trombocitopenije 3.uroĊene imune trombocitopenije 4.steĉene imune trombocitopenije 5.autoimune trombocitopenije
Idiopatska trombocitopenijska purpura: 1.acc oblik 2.chr oblik 3.sekundarne autoimune trombocitopenije 4.imune trombocitopenije izazvane lekovima
Trombocitoza B)KVALITATIVNI POREMEĆAJI Su Trombocitopatije 3)Poremećaj koagulacije krvi Karakterišu se površinskim krvarenjima u kožu, krvarenjem u zglobove i telesne duplje, kao i hematomima mekih tkiva A)NASLEDNI POREMEĆAJI KOAGULACIJE KRVI: 1.Hemofilije=por.ĉinioca 1.faze koagulacije (deficit 8 i 9) 2.Deficit 2.faktora 3.Deficit 3.faktora=nasledni poremećaj fibrinogena 4.Deficit 7.faktora
www.belimantil.info

26
5.Von-Wilerbrandtova bolest B)STEĈENI POREMEĆAJ KOAGULACIJE KRVI : 1.deficit koag.ĉinilaca koji zavise od K vitamina (2, 7, 9 i 10) 2.por.koag.krvi zbog inhibicije koagulacije 3.sindrom defibrinacije 4.patološka fibrinoliza 5.DIK (diseminovana intravaskularna koagulacija) 1.VASKULARNI HEMORAGIJSKI SINDROMI Nastaju zbog naslednih ili steĉenih poremećaja funkcije kapilara, venula ili arteriola 2.NASLEDNA HEMORAGIJSKA TELANGIEKTAZIJA (Mb Osler) Bolest se karakteriše uĉestalim pojavama krvarenja iz ograniĉeno proširenih, savijenih sitnih vena i kapilara (telangiektazije) -Osnovni poremećaj bolesti je istanjenost zidova venula i kapilara, koji se na pojedinim mestima sastoje samo od endotelskog sloja (zato izostaje kontrakcija zida pri povredi) -Obiĉno se manifestuje izmeĊu 2. I 3. decenije života, krvarenjima iz nosa. Nešto reĊe se javlja melena, hematurija, hemoptizije. -Zbog ponavljanih krvarenja razvija se anemija *Telangiektazija se vidi kao jasno crvena ili lako cijanotiĉna promena na kožI i sluznicama, razliĉite veliĉine, od jedva vidljivih, do preĉnika od ½ cm. -Najĉešće se otkrivaju na kožI lica, dlanova, stopala, sluznice nosa, usne duplje, usana i jezika *Dijagnoza: -anamneza o ponovljenim krvarenjima -pregled *Laboratorija : hipohromna, hiposideremijska anemija *Terapija: -specijalo leĉenje ne postoji --daje se simptomatska Th: preparati Fe transfuzija krvi krvarenja na pristipaĉnim mestima zaustavljaju se pritiskom, primenom fibrinske pene, trombina, laserom, teĉnim N2 hiruška intervencija 3.STEČENE VASKULARNE PURPURE Henoch – Schonlein Ovde je pojaĉana sinteza IgA -U osnovi bolesti ležI imuno-alergijski mehanizam koji dovodi do oštećenja krvnih sudova kože, creva i bubrega -Usled toga dolazi do povećane propustljivosti zidova krvnih sudova i izlaska krvne plazme i Er u okolno tkivo. -Kao alergeni mogu se javiti: bakterije, alergeni hrane (jaja, mleko), ujed insekta, lekovi -Bolest se,uglavnom javlja u dece i mlaĊih osoba. Skoro uvek poĉinje naglo.
*Kliniĉka slika:-Malaxalost, glavobolja, t, gubitak apetita -kožne promene (purpure), svrab, parestezije -bolovi i otok zglobova
www.belimantil.info

27
-mogu se javiti i abdominalni bolovi (crna stolica ili krvav proliv= “purfura”) -bubrežne promene se ispoljavaju hematurijom, proteinurijom, bubrežnom insuficijencijom 30-50%
*Laboratorija:-SE, Leu + neutrofilija
-kod izraženih krvarenja anemija *Terapija:-plazmaferezom ukloniti imune komplexe -ako je moguće, ukloniti alergen -odmor u postelji + AB + antihistaminici + kortikosteroidi 4.Mb von Wilerbrandt -je najĉešćI uroĊeni hemoragijski sindrom. Nastaje sbog smanjenja ili prestanka sinteze glikoproteina plazme = von Wilerbrandtovog faktora *Funkcija vW faktora: -stabilizacija faktora koagulacije 8 -pomoć u adheziji Tr na zid povreĊenog krvnog suda *Kliniĉka slika: najpre samo produženo krvarenje pri hiruškim zahvatima, a kasnije spontane epistaxe, krvarenja iz modrica, kod žena menometroragija i postpartalna krvarenja; u težim sluĉajevima i hemartroze praćene bolovima *Dijagnoza: -produženo vreme krvarenja -snižen nivo faktora koagulacije 8. *Tok bolesti: bolest se stišava sa starenjem *Terapija: blagi oblici tretiraju se deamino-D-arginin-vazopresinom; tokom operacija i u težim sluĉajevima daje se krioprecipitat i koncentrovani preparat faktora koagulacije 8. IV.HEMOFILIJE Poremećaj hemostaze koji se odlikuje doživotnom sklonošću ka krvarenju -uroĊeni poremećaj koagulacije krvi -Predstavljaju vrlo ozbiljan poremećaj hemostaze koji se nasleĊuje recesivnim genom na X hromozomu, zato su žene prenosioci, a oboljevaju samo muškarci -Ovde postoje kvalitativne ili kvantitativne promene 8.faktor (hemofilija A), ili faktora 9.koagulacije (hemofilija B)prestanak ili smanjenje sinteze 8. I 9.faktora -Deficit 8. Ili 9.faktora koagulacije u hemofiliĉara dovodi do krvarenja zbog nedovoljnog
stvaranja protrombinaze, što povlaĉI pretveranje protrobmina u trombin i sledstveno
pretvranje fibrinogena u fibrin -Do <1% faktora8 teška hemofilija sa obilnim spontanim krvarenjima 1-5% faktora 8 umerena hemofilija 6-25% faktora 8 retka krvarenja 25% faktora 8 otkrije se kod teže povrede ili operacije *Kliniĉka slika: -sklonost krvarenju postoji tokom celog života -uĉestalost i težina krvar.srazmerne su stepenu nedostatka faktora 8 i 9 -javljaju se krvarenja u zglobovima, naroĉito kolenima i skoĉnom zglobu, lakat, rame, ruĉje -ova ponovljena krvarenja izazivaju osteoartritis, ankilozu zgloba i atrofiju mišIća -krvarenja iz organa (GIT, mokraćnih i polnih) -krvarenja u mozgu = ĉesto smrtonosna veće povrede (vaĊenje zuba) izazivaju krvarenja koja mogu trajati satima, danima i nedeljama. Ovakvo krvarenje se zaustavlja zahvaljujućI primarnoj hemostazi, ali se posle nekoliko dana nastavlja u vidu malog, ali dugotrajnog krvarenja
www.belimantil.info

28
*Laboratorija: Vreme koagulacije, test utroška protrombina -Taĉna Dg se uspostavlja odreĊivanjem koncentracije 8 i 9 faktora koagulacije u plazmi *Terapija: -Supstituciona Th ima za cilj da se poveća koncentracija 8. I 9. faktora iznad 0,3 od normalne vrednosti, i da se održI dok se ne uspostavi hemostaza, odnosno, izvede hiruška ihtervencija. U tom cilju daje se ANTIHEMOFILNA PLAZMA, koncentrovani preparati 8 i 9 faktora ili sveža krv. -Uz navedenu Th dati i antifibrinozne lekove, jer se time smanjuje liza fibrina, tako da koagulum posle extrakcije postaje ĉvršćI i spreĉava naknadno krverenje -Male posekotine mogu se leĉiti lokalnom primenom trombina ili fibrinske mreže *Sinteza faktora koagulacije: 8 = u jetri 9 = u jetri; potreban je vitamin K Nedostatak 9 faktora = hemofilija C Steĉeni hemoragijski sy: ĉešćI su i komplikovaniji V.DIK Je steĉeni hemoragiski sindrom u mnogim bolestima i odlikuje se brzom intravaskularnom portošnjom većeg broja faktora koagulacije i trombocita, zbog stvaranja mnogih koaguluma, naroĉito u mikrocirkulaciji -Kliniĉki se ispoljava brojnim blagim ili teškim krvarenjima -Kao komplikacija javlja se u septiĉkim infekcijama izazvanim G- bakterijama, patološkoj trudnoćI, malignim tumorima, promijelocitnoj leukemiji, teškoj hipoksiji *Patofiziologija: -DIK je poremećaj koagulacije u koji su ukljuĉeni: zid krvnih sudova, trombociti, faktori koagulacije, fibrinolitiĉki sistem, inhibitori i komplement -Prodiranjem u krvotok tromboplastinskih materija iz oštećenih tkiva (pluća, prostata, mozak, posteljica) ili oštećenog endotela krvnih sudova, pokreće se spoljni i unutrašnji mehanizam aktivacije koagulacionog sistema. -Dolazi do diseminovanog stvaranja koaguluma u mukrocirkulaciji, usled toga se smanjuje koncentracija fibrinogena, protrombina, 5, 7 faktora koagulacije i trombocita koji su se utrošili u procesu koagulacije -Smanjenje koliĉine ovih faktora dovodi do pojave hemoragijskog sindroma -Kliniĉki se može ispoljiti kao acc ili chr *Kliniĉka slika: -Acc oblik se odlikuje vrlo izraženim spontanim krvarenjima u kožI, sluznicama, organima, iz operativne rane -Skoro uvek je prisutan šok i znaci insuf.bubrega -Kod chr oblika samo postoje laboratorijski poremećaji, bez kliniĉkih znakova *Laboratorija:
-br.Tr (jer se od njih stvaraju trombi), konc.fibrinogena, protrombina, 5 i 7 faktora -liza krvnog ugruška je spora
-proizvoda razgradnje fibrin/fibrinogen *Terapija: -i.v. heparin
www.belimantil.info

29
-antifibrinolizni lekovi (u sluĉaju izražene fibrinolize) -davanje konc.trombocita u sluĉaju izražene trombocitopenije VI.BOLESTI SLEZINE -Slezina uĉestvuje u hematopoezi do 5.meseca intaruterinog života, ali u njoj ostaje sposobnost za hematopoezu tokom celog života -Slezina je najvećI limfocitni organ i sadržI T i B ly -U njoj se normalno, posle roĊenja stvaraju plazmociti i monociti -Slezina ima sposobnost da fagocituje ostarele i oštećene Er i Tr, i predstavlja rezervorar krvnih ćelija, naroĉito limfocita, monocita i trombocita *Podela bolesti slezine: -primarne (tumori i primarni hipersplenizam) -sekundarne -Mnogo ĉešće se radi o promenama slezine tokom drugih bolesti krvi ili nekih drugih organa 1)KONGESTIVNA SPLENOMEGALIJA usled pasivne venske staze, uslovljene povećanim Pa u veni porti (ciroza jetre, tromboza vene porte ili vena slezine) 2)TROPSKA SPLENOMEGALIJA izazivaju je parazitarne tropske bolesti , posebno kala-azar 1.BENIGNI i MALIGNI TUMORI SLEZINE 1)HEMANGIOM primarni maligni tumori slezine su retki (sarkom) metastaze kasnijih tumora sreću se znatno ĉešće (Ca kože, dojke,grlića, pluća) 2)CISTIĈNE TVOREVINE 2.SPLENOMEGALIJA Sreće se u: 1.virusnim infekcijama (infektivna mononukleoza) 2.bakterijskim inf. (sepsa, subacc bakt.endokarditis, TBC, lues) 3.parazitskim bolestima 4.cistiĉnim bolestima vezivnog tkiva -Uvećanje slezine javlja se u ovim bolestima zbog imunog odgovora limfocitnog tkiva i hiperplazije mononukleofagocitnih ćelija -Izražena splenomegalija prati CHR.HEMOLIZNE ANEMIJE -Uvek postoji u LIMFOPROLIFERATIVNIM BOLESTIMA (chr LL, maligni limfom) -TakoĊe i u MIJELOPROLIFERATIVNIM BOLESTIMA -I u EXTRAMEDULARNOJ HEMATOPOEZI (Talasemija) 3.HIPERSPLENIZAM Je kliniĉki sindrom koji se karakteriše splenomegalijom, pancitopenijom ili monocitotopenijom u perifernoj krvi i hiperplazijom prethodnika ovih krvnih elemenata u kosnoj sržI, koji su u perifernoj krvi smanjeni -Smanjenje broja ćelija periferne krvi, posledica je povećanog rada slezine, odnosno povećane aktivnosti jedne ili više njenih funkcija *PRIMARNI HIPERSPLENIZAM: -Javlja se u autoimunim bolestima krvi, kao što su: hemolizna anemija nepoznate etiologije, trombocitopenijska purpura *SEKUNDARNI HIPERSPLENIZAM: -javlja se kao pratećI sindrom u patološkim stanjima koja dovode do splenomegalije *Kliniĉka slika:
www.belimantil.info

30
-anemija, leukopenija, trombocitopenija, ili smanjenje broja jedne ili dve krvne loze -prisutni su i znaci osnovnog oboljenja koje je dovelo do hipersplenizma *Terapija: -kortikosteroidi -imunosupresivi -suptituciona Th (transfuzije krvi ili pojedinaĉnih elemenata Er, Tr, Leu) -leĉenje osnovnog oboljenja VII.TROMBOCITOPENIJE i TROMBOCITOPATIJE 1.TROMBOCITOPENIJE je stanje sa smanjenim brojem Tr (manje od 150x10 na 9/l Tr)
-Pošto postoje fiziološke varijacije u broju Tr (dnevne, zavisne od t, menstrualnog ciklusa), vrednosti ispod 100x10 na 9/l smatraju se pouzdanim znakom trombocitopenje -Sklonost krvarenju se obiĉno manifestuje pri vrednosti Tr ispod 50x10 na 9/l -Spontana krvarenja nastaju pri 10-20x10 na 9/l Tr -Krvarenja su najĉešće u vidu petehija, ekhimoza po kožI, iz sluzokože, u vidu epistaxe i menoragije -Salicilati i antibiotici potenciraju krvarenje kod trombocitopenije -Najopanije je krvarenje u CNS *Podela trombocitopenija prema mehanizmu nastanka:
1)Trombocitopenije uzrokovene proizvodnje Tr = AMEGAKARIOCITNE -Smanjena proizvodnja nastaje zbog hipoplazije kosne sržI, potiskivanja sržI malignim tkivom (mijeloprolifertivna i limfoproliferativna oboljenja, sideropenijske i megaloblastne anemije, u toku virusnih infekcija, HIV, radioterapija) -Dg: na osnovu broja elemenata u krvi, razmaza krvi, ispitivanja kosne srži
2)Trombocitopenije uzrokovane potrošnjom i razaranjem Tr = MEGAKARIOCITNE -Pojaĉana razgradnja Tr je posledica imunoloških bolesti, senzibilizacije prema lekovima, autoimune hemolizne anemije, SLE, leukoza, u toku DIK-a, posle ugradnje veštaĉke valvule) 3)Trombocitopenije zbog poremećene raspodele Tr u cirkulaciji: -zbog akumulacije cirkulišućih Tr u velikoj slezini *Kliniĉka slika trombocitopenija: -osnovno je produženo vreme krvarenja i slaba retrakcija koaguluma, -smenjenje Tr ispod 10000 manifestuje se izraženim krvarenjem i pojavom petehija, ekhimoza, krvarenja iz sluznica. Opasno je !!! A)IMUNE TROMBOCITOPENIJE: a)Idiopatska trombocitopenijska purpura je autoimuno oboljenje u kome su Tr obloženi velikom koliĉinom IgG, pa podležu pojaĉanoj razgradnji u makrofazima slezine -Etiopatogeneza: pokretaĉI su virusi, lekovi, nutritivni alergeni -Kliniĉka slika: krvarenje, petehije, ekhimoze, iz nosa, GIT-a -Terapija:kortikosteroidi (leĉenje traje 4-6 nedelja) b)Sekundarne imune trombocitopenijeske purpure: -Nasataju zbog stvaranja At na virusne Ag, koji se adsorbuju na porvšini Tr -Javlja se kod: SLE, leukemija, limfoma c)Izoimunme trombocitopenijske purpure -Posttransfuzijska i neonatalna d)Trombocitopenije izazvane lekovima -Mijelosupresivni lekovi, tiazidi, etanol, estrogeni, antibiotici, sedativi, aspirin, soli zlata
www.belimantil.info

31
B)NEIMUNE TROMBOCITOPENIJE: a)Trombolitiĉna-trombocitopenijska purpura = Moskoviĉev sy b)Hemolitiĉno – uremijski sy 3.TROMBOCITOPATIJE -Obuhvataju grupu oboljenja kod kojih je poremećena funkcija Tr -Funkcionalno insuficijentni Tr uzrok su krvarenja i pored normalnog broja Tr *Kliniĉka slika: spontana krvarenja u kožI i sluzokožama vreme krverenja je produženo broj Tr je normalan A)URO\ENE TROMBOCITOPATIJE: a)Bernard-Solierov syporemećaj adhezije Tr b)Glanzmanova trombastenijaporemećaj agregacije Tr
c)Nedostatak i granulaporemećaj sekrecije B)STEĈENE TROMBOCITOPATIJE: -Kod bolesnika sa bubrežnom insuficijencijom, cirozom jetre (deficit faktora koagulacije i smanjenje agregacije Tr), mijeloproliferativne bolesti, paraproteinemije VIII. TROMBOZA -Prekomerna aktivacija hemostaznog sistema, na pogrešnom mestu u krvnom sudu može dovesti do stvaranja koaguluma, odnosno tromba -Postoje venske i arterijske tromboze -Arterijske tromboze smanjuju dotok krvi O2 u tkivo tj u organ -Venske tromboze su ĉešće i one se obiĉno komplikuju stvaranjem tromboembolija -Tromboza se javlja u okviru drugih bolesti, ali može biti i posledica uroĊenog ili steĉenog poremećaja hemostaze (stanje povećane aktivnosti hemostaznog sistema, bez znakova njegove aktivacije, naziva se trombofilija) *URO\ENA PROTROMBINOZNA STANJA: nedostatak: antitrombina III, proteina C, proteina S, heparinskog kofaktora II, 7.ĉinioca koagulacije, plazminogena, aktivatora plazminogena, inhibitora tkivnog faktora; nedovoljno oslobaĊanje aktivatora plazminogena… *BOLESTI i STANJA UDRUžENA SA TROMBOZOM: chr srĉana insuficijencija veštaĉke valvule, ateroskleroza, obimne traume, hiruški zahvati i postoperativna stanja, gojaznost, imobilizacija, starost, trudnoća, postpartalni period, nefrotski sy, malignitet i metastaze, mijeloproliferativne bolesti, paroxizmlna noćna hemoglobinurija, hiperviskozni sy, primena oralnih kontraceptiva i preparata estrogena… *PATOGENEZA: *Oštećenje endotela i poremećaj protokazadržavanje aktiviranih faktora koagulacijeTR adheriraju za subednotelne strukturesekretuju ADP koji dovodi do agregacije Tr i stvaranja trombocitnog ĉepa *Istovremeno iz oštećenog endotela oslobaĊa se tkivni faktor koji sa aktiviranim 7 ĉiniocem aktivira 10.ĉinilac on u prisutstvu 5.ĉinioca, jona Ca i fosfolipida pretvara protrombin u trombin koji pretvara fibrinogen u fibrin; koji se stabilizuje i postaje nerastvorljiv koagulum, odnosno tromb *KLINIĈKA SLIKA: -Trobmoza se ispoljava razliĉitim znacima, zavisno od lokalizacije -Razlikuju se slike venskih i arterijskih tromboza *ARTERIJSKE TROMBOZE: uzrokuju smanjen ili prekinut dotok krvi u tkivo koje data aretrija vaskularizuje
www.belimantil.info

32
-koronarne arterije=angina pektoris ili infarkt -cerebralne arterije=pareze i paralize -a.centralis retinae=gubitak vide -bubrežna arterija=slabinski bol, hematurija, proteinurija i hipertenzija -mezenterijalne arterije=bol u trbuhu, muka, gaĊenje, povraćanje, proliv, gubitak apetita, melena -raĉve aorte i aa.extremiteta=nagli bol,parestezije,paraliza,hladna,bleda koža,bez pulsa, gangrena *VENSKE TROMBOZE: uzrokuju poremećaj oticanja krvi iz odreĊenog podruĉja. Ispoljavaju se bolom, cijanozom i edemom zahvaćenog podruĉja -Ako se ne dogodi tromboliza, već se tromb organizuje, nastaje posttrombozni sy, najĉešće na donjim ekstremitetima (trajni otok i cijanoza) -Zasto u oticanju krvi daje trofiĉke promene=hiperpigmentacija, fibroza i pojava grizlica -hepatiĉne vene=hepatomegalija, bol u desnom hipohondrijumu, acsites; žutica i splenomegalija kod 1/3 bolesnika -v.porta=ascites, hematemeza i melena, zbog krvarenja iz proširenih vena jednjaka ili želuca i uvećanja slezine -Tromboembolije se javljaju kao posledica venske tromboze. Najĉešće se ispoljavaju u plućima i ĉesto ugrožavaju život bolesnika. *DIJAGNOZA: postavlja se na osnovu simptoma, zavisno od vrste i lokalizacije tromboze -kliniĉka slika, scintigrfaija, angiografija, laboratorija *TERAPIJA: -Osnovni princip zasniva se na ranoj dijagnostici i brzoj primeni antitrombozne Th, koja obuhvata TROMBOLITIĈNU, ANTIKOAGULANTNU i ANTIAGREGACIONU TERAPIJU *TROMBOLITIĈNA TERAPIJA: cilj je rastvaranje trimba i rekanalizacija krvnog suda. -Indikovana je u 4 osnovna sluĉaja: duboke venske trobmoze; plućne embolije; acc okluzije perifernih arterija i acc infarkt miokarda -Lekovi koji se primenjuju: streptokinaza, urokinaza, tkivni aktivator plazminogena i komplex streptokinaze sa plazminogenom (Th acc infrakta miokarda) -Ova vrsta Th je manje prihvaćena za duboke venske tromboze i plućnu emboliju, zbog rizika od krvarenja. Ovde se daju heparin i antagonisti K vitamina per oralno *ANTIKOAGULANTNA Th HEPARINOM: heparin se veže za antitrombin 3 i inaktivira ga; to je snažan antokoagulans koji smanjuje stvaranje trombina i fibrina -Heparin se primenjuje u Th arterijskih i venskih tromboza ili embolija; daje se parenteralno tako da se parcijalno tromboplastinsko vreme poveća 1,5-2x u odnosu na kontrolno (najĉešće se postiže sa 1000 jedinica heparina na sat) u kontinuiranoj infuziji; kada se protrombinsko vreme uvede u terapijski opseg, heparin se obustavlja, a nastavlja se sa peroralnim antikoagulansima -Dugotrajno kontinuirano parenteralno davanje (infuzijom) heparina primenjuje se kod bolesnika sa rekurentnim trombozama koji ne reaguju na per os antikoagulantnu Th, kod trudnica sa tromboembolijama i u DIK-u -Manje doze heparina daju se kao prevencija venskih tromboza -Komplikacija heparinske Th je najĉešće na mestima heparinske imtramuskulrne injekcije ili hiruških intervencija. Istovremena primena aspirina doprinosi nastanku krvarenja. -Snažan antidot heparina je protamin sulfat, ali se on ne daje za obustavu krvarenja, jer se isto postiže samo smanjenjem doze heparina; Moguća je pojava trombocitopenije u 10% bolesnika
www.belimantil.info

33
*PERORALNA ANTOKOAGULANTNA TERAPIJA: kumarinski antikoagulansi podrazumevaju varfarin i dikumarol. Oni spreĉavaju metabolizam K vitamina na nivou mikrozama jetre i tako izazivaju stanje sliĉno nedostatku K vitamina. -Koriste se kod prevencije: rekurentnih tromboza, plućnih embolija, cerebralnih arterijskih embolija u postojanju srĉanog tromba, kod arterioskleroze i delimiĉno kod stenoze karotida ili vertebralnih arterija -Leĉenje varafrinom zapoĉinje se tokom Th heparinom u dozi 5-10mg, takoĊe da se protrombinsko vreme produžI 1,5-2 puta u odnosu na kontrolnu vrednost; tada se obustavi heparin i nastavlja se samo sa peroralnim antikoagulansom -Antikoagulantno leĉenje traje 3-6 meseci kod prve nekomplikovane tromboembolije; u težim sluĉajevima, Th je doživotna. Doživotno se daje, uprkos opasnosti od krverenja, i bolesnicima sa veštaĉkom valvulom, teškom mitralnom stenozom, fibrilacijom pretkomora, chr kongestivnom srĉanom insuficijencijom… *ANTIAGREGACIONA TERAPIJA: koriste se lekovi koji smanjuju hemostaznu funkciju Tr= aspirin (inhibiranjem ciklooxigenaze spreĉava stavaranje tromboxana A2 koji bi izazvao agregaciju Tr) -Aspirin se daje u dnevnoj dozi 150mg ili svaki drugi dan po 300mg, bolesnicima sa arterijskim vaskularnim bolestima i tromboembolijama
-Još se mogu koristiti i tiklopidin, dipiridamol, NSAIL, -blokarori… INDIKACIJE ZA PRIMENU ANTITROMBOZNE TERAPIJE
Indikacije za fibrinolitiĉku terapiju
Indikacije za antikoagulantnu terapiju
Indikacije za antiagregacionu terapiju
Acc infarkt miokarda Profilaxa u hirurgiji Tranzitorni ishemiĉni atak
Acc okluzija periferne arterije
Profilaxa u kongestivnoj srĉanoj insuficijenciji
Sekundarana prevencija cerebrovaskularnih inzulta
Masivna embolija pluća Acc infarkt miokrada Veštaĉka valvula
Tromboza axilarne vene Venska tromboembolija Nestabilana angina
Masivna tromboza aleofemoralne vene
Akutna venska ili arterijska tromboza
Primarna i sekundarana prevencija acc infarkta miokrada
Okluzija A-V šanta Profilaxa u trudnoćI Koronarni by-pass
Arterijska ili venska kanila DIK Održavanje A-V kanile
www.belimantil.info





























