Embed Size (px)
Citation preview

Metallfreie KatalyseDOI: 10.1002/ange.200901826
Heteroatome steigern die Selektivit�t der oxidativen Dehydrierung anNanokohlenstoff**Benjamin Frank, Jian Zhang, Raoul Blume, Robert Schl�gl und Dang Sheng Su*
Die Oberfl�chen von Kohlenstoffmaterialien weisen �bli-cherweise eine Vielzahl sauerstoffhaltiger funktionellerGruppen auf,[1, 2] was die Aufmerksamkeit vieler Forschungs-bereiche auf sich zieht.[3] Unter diesen Oberfl�chenspeziessind Keto- und Chinongruppen elektronenreich und damitpotenziell in der Lage, eine Redoxreaktion zu koordinieren(Schema 1). Lewis-basische Zentren k�nnen Wasserstoffato-
me aus Alkanmolek�len abstrahieren, wobei das entspre-chende Alken gebildet wird. Sauerstoff aus der Gasphasewiederum regeneriert das aktive Zentrum durch Reaktionmit dem abstrahierten Wasserstoff unter Bildung von Wasserals Nebenprodukt.[4, 5]
Ein faszinierender Bereich in der Kohlenstoffchemie istder Einsatz von Nanostrukturen, insbesondere von Nano-r�hren (CNTs), als metallfreie Katalysatoren. �ber beacht-liche katalytische Eigenschaften von Nanokohlenstoff in deroxidativen Dehydrierung (ODH) von Kohlenwasserstoffenwurde in letzter Zeit berichtet.[4,6] Dieser Fortschritt er�ffneteinen weiten Horizont f�r die Anwendung von CNTs undbietet neue M�glichkeiten zur detaillierten Erforschung der
ODH. Anders als tr�gerfixierte �bergangsmetalloxide istNanokohlenstoff als einfache Plattform den Bedingungen f�rkinetische und mechanistische Untersuchungen dieser kom-plexen Redoxreaktion gut anzupassen. K�rzlich konnten wirbereits �hnlichkeiten zum Mechanismus der ODH an Va-nadiumoxidkatalysatoren aufzeigen.[4,7] Zudem kann es unterODH-Bedingungen durchaus zur Abscheidung von Kohlen-stoff auf der Oberfl�che von Metalloxiden kommen,[8] was einneues Licht auf die katalytische Rolle von Kohlenstoff in derODH wirft. Ethylbenzol ist bislang das �berwiegend ver-wendete Substrat der kohlenstoffkatalysierten ODH, da dieAusbeute wegen des stabilen konjugierten Elektronensytemsdes Produkts Styrol hoch ist. Kurzkettige Alkane sind jedochweit weniger reaktiv als die entsprechenden Alkene.[9] Diegeringe Selektivit�t zu und Ausbeute an Alkenen ist bislangdas Haupthindernis f�r eine industrielle Anwendung derODH-Technik – trotz zahlreicher Vorteile des oxidativenReaktionspfades gegen�ber nichtoxidativen Prozessen derAlkenproduktion.[10]
Nucleophiler Sauerstoff wie O2� in den aktiven Zentrenwurde als Voraussetzung f�r eine hohe Alkenselektivit�t er-kannt. Leider sind jedoch die wahrscheinlichsten Interme-diate der Sauerstoffaktivierung, O2
2�, O2� und O� , allesamt
elektrophil und bewirken daher unweigerlich die Totaloxi-dation, insbesondere des gebildeten Alkens mit seiner elek-tronenreichen p-Bindung.[11] Elektronenziehende Dotierun-gen wie Bor oder Boroxid unterdr�cken die Bildung hoch-reaktiver Formen von Sauerstoff[12] und sollten daher dieSelektivit�t zu Propen erh�hen. Im Folgenden berichten wir,dass Bor-modifizierte CNTs bemerkenswert selektive Kata-lysatoren der ODH von Propan sind. Der Austausch vonSauerstoffisotopen und Transientenexperimente zeigen dieOptimierung der Sauerstoffaktivierung, was in einer Pro-penselektivit�t �hnlich der gering beladener Vanadiumoxid-katalysatoren resultiert. Wir zeigen eine nachhaltige Strategief�r die Propensynthese auf, die frei von der Umweltbelastungdurch toxische �bergangsmetalle ist.
F�r die Studie der katalytischen Eigenschaften in derODH von Propan wurden kommerzielle CNTs (CNTs), oxi-dierte (oCNTs) und Boroxid-modifizierte CNTs (x = 0.003–5 Gew.-% B2O3, gekennzeichnet als xB-oCNTs) herangezo-gen. Eine Probe mit 5 Gew.-% P2O5 (5P-oCNTs) wurdeanalog pr�pariert.[4] Einzige Reaktionsprodukte warenPropen, CO, CO2 und H2O. Reaktionen in der Gasphasekonnten durch Leerrohr-Experimente ausgeschlossenwerden. S�mtliche Proben zeigten nach einigen Stunden sta-bile katalytische Eigenschaften und wurden im Detail unter-sucht: Str�mungsgeschwindigkeit und Reaktionstemperaturwurden variiert, Propan und Sauerstoff wurden abwechselndeingespeist, und auf dynamischen Sauerstoffaustausch wurde
Schema 1. Reaktionsschema der oxidativen Dehydrierung von Alkanenan funktionalisierten CNTs.
[*] Dr. B. Frank, Dr. J. Zhang, Dr. R. Blume, Prof. Dr. R. Schl�gl,Dr. D. S. SuAbteilung Anorganische Chemie, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4–6, 14195 Berlin (Deutschland)Fax: (+ 49)30-8413-4401E-Mail : [email protected]
[**] Diese Arbeit wurde vom EnerChem-Projekt (Max-Planck-Gesell-schaft) gef�rdert. Die Autoren danken Prof. R. Schom�cker (Tech-nische Universit�t Berlin), E. Kitzelmann, G. Weinberg, Dr. D. Ro-senthal und Dr. Bo Zhu f�r experimentelle Unterst�tzung und hilf-reiche Diskussionen.
Hintergrundinformationen zu diesem Beitrag sind im WWW unterhttp://dx.doi.org/10.1002/ange.200901826 zu finden.
Zuschriften
7046 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2009, 121, 7046 –7051

getestet. Wie Abbildung 1a zu entnehmen ist, weisen dieProben mit Bor- und Phosphoroxid eine h�here Selektivit�tzu Propen auf als die nichtmodifizierten Proben CNT undoCNT. Dieser Anstieg geht einher mit einer verbesserten
chemischen Stabilit�t nach der Modifizierung. Durch tem-peraturprogrammierte Oxidation (TPO) ließ sich eine an-steigende Widerstandsf�higkeit gegen Oxidation in der Rei-henfolge CNTs< oCNTs ! 2B-oCNTs� 5B-oCNTs< 5P-oCNTs (Abbildung S1) belegen. Mit steigendem Boroxidge-halt verschieben sich Starttemperatur und Lage des Maxi-mums zu h�heren Temperaturen. In der Tat wird angenom-men, dass das Modifizieren mit Borat oder Phosphat dieVerbrennungszentren, die als Angriffspunkt f�r O2 dienen,blockiert, was die Verbrennung des Kohlenstoffger�sts beierh�hten Temperaturen merklich unterdr�ckt.[12, 13] Vom ki-netischen Standpunkt aus spiegelt sich die Schutzwirkunggegen die Aktivierung von O2 in einer um etwa 40 kJmol�1
erh�hten Aktivierungsenergie f�r den Sauerstoffverbrauchwider (Abbildung 1a). Die Aktivierung von Propan wirddurch die Modifizierung nicht beeinflusst, und die Aktivie-rungsenergie bleibt bei (130� 5) kJmol�1.
Die modifizierten CNTs sind weitaus selektiver als zahl-reiche auf unterschiedlichen Tr�gern fixierte Vanadiumoxid-katalysatoren (Abbildung S2).[14] Bei 673 K folgt die Pro-penselektivit�t dem Trend VOx-Al2O3> 2B/5B-oCNTs�VOx-TiO2> 5P-oCNTs>VOx-CeO2>VOx-SiO2>VOx-ZrO2> (o)CNTs. Der Selektivit�tsverlust bei steigendemUmsatz ist bedingt durch die schwache C-H-Bindung inPropen und die resultierende Folgeoxidation.[9] Die direkteTotaloxidation von Propan dagegen wird durch die B- und P-Modifizierung unterdr�ckt, wie nahezu 100 % Propenselek-tivit�t bei differentiellem Umsatz zeigt. Nur Kohlenstoff insp2-Konfiguration erlaubt eine stabile und selektive ODH-Katalyse.[15] In dieser Studie fiel die Wahl auf CNTs, da sieeinen Kompromiss zwischen hoher Reaktivit�t (sp3, Aktiv-kohle) und hoher Stabilit�t (sp2, Graphit) darstellen. EineReferenzprobe, Aktivkohle beladen mit 5 Gew.-% B2O3,erwies sich in der Tat als unbrauchbar (Abbildung S3).
Struktur- und Oberfl�chenbeschaffenheit der Katalysa-toren vor und nach der Reaktion wurden mit mikroskopi-schen, spektroskopischen und thermoanalytischen Methodenuntersucht. Energiegefilterte Transmissionselektronenmi-kroskopie (EF-TEM) belegt die gute Verteilung von B und Oinnerhalb der CNT-Agglomerate (Abbildung S4). Das Kom-posit widersteht den Reaktionsbedingungen, und s�mtlichesB2O3 (1.7 Atom-% B in 5B-oCNT) konnte nach der Ver-wendung in der ODH �ber Elektronenenergieverlustspek-troskopie (EELS) nachgewiesen werden (Abbildung S5). DasCNT-Ger�st wird weder durch die Modifizierung noch durchdie oxidierende Gasatmosph�re unter Reaktionsbedingungenbeeintr�chtigt, wie Raman-Spektroskopie, Raster- (SEM)und Transmissionselektronenmikroskopie (TEM) sowie N2-Physisorption belegen (Abbildung S1 und S5). Das Intensi-t�tsverh�ltnis von D- zu G-Bande, ein Indikator f�r die De-fektdichte von Kohlenstoff, steigt leicht an und bewegt sich imBereich von 2.0–2.1 (Tabelle S1). Nach den SEM- und TEM-Aufnahmen ist die Morphologie vor und nach der Reaktionweitgehend unver�ndert. Auch die Lage des TPO-Maximumsist nahezu unver�ndert, jedoch verschwindet ein Tieftempe-ratursignal bei etwa 550 K, wohl bedingt durch die Beseiti-gung instabiler Kohlenstoffspezies im oxidierenden Reak-tantenstrom. Die qualitative und quantitative Ver�nderungder sauerstoffhaltigen Oberfl�chenspezies wurde mittelstemperaturprogrammierter Desorption (TPD) und thermo-gravimetrischer Analyse (TGA) bestimmt (Abbildung S6).
Die anf�ngliche Entwicklung der katalytischen Eigen-schaften einiger CNTs ist in Abbildung S7 dargestellt. Mitzunehmender Reaktionszeit steigt die Aktivit�t kommerzi-eller und oxidierter CNTs. Ursache ist die In-situ-Bildungaktiver funktioneller Gruppen sowie der Abbrand instabilerGruppen und amorpher Kohlenstoffverunreinigungen in deroxidierenden Reaktantengasmischung,[16] was durch TPO-und TPD-Ergebnisse best�tigt wird. Eine geringere Defekt-dichte und der Verlust von oberfl�chennahem Sauerstoffwerden durch Raman-Spektroskopie, Ex-situ-R�ntgenpho-toelektronenspektroskopie (XPS) und TPD belegt (Tabel-le S1). Im Allgemeinen zeigen die TPD-Profile einen Verlustlabiler Carboxy-, Anhydrid- und Lactongruppen durch Frei-setzen von CO2 im Temperaturbereich zwischen 400–800 Ksowie die Bildung von Keto- und Chinonfunktionalit�ten, die
Abbildung 1. Katalytische Eigenschaften von CNT-Katalysatoren in derODH von Propan, VOx-Al2O3 als Referenz.[14] a) Scheinbare Aktivie-rungsenergie EA f�r den Propanverbrauch, den Sauerstoffverbrauchund die Propenbildung (Balken) sowie normierte Aktivit�t rnorm undPropenselektivit�t S (in %) bei 5% Propanumsatz (Symbole) undb) Propenselektivit�t bei 5% Propanumsatz (&) und Reaktionsge-schwindigkeit r (*) als Funktion der B2O3-Beladung wB2O3
. Reaktions-bedingungen f�r die CNT-Katalysatoren: 1 g, C3H8/O2/N2 = 1:1:4, 10–140 mLn min�1, 673 K.
AngewandteChemie
7047Angew. Chem. 2009, 121, 7046 –7051 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

zwischen 800 und 1200 K CO freisetzen (Abbildung S6).[2]
Letztere dominieren die Oberfl�che zusammen mit hoch-temperaturstabilen Lacton- und Anhydridgruppen. DieoCNTs zeigen eine h�here Anfangsaktivit�t als die kom-merziellen CNTs. Jedoch verschwindet der Unterschied nach5 h sowohl strukturell als auch katalytisch. Diese Aktivie-rungsperiode kann durch scharfe Oxidation der oCNTs (1 h,773 K, Luft) simuliert werden. Die derart vorbehandeltenoCNTs zeigen von Beginn an eine stabilere Leistung. Eingenereller Anstieg der Oberfl�che insbesondere im Bereichder weniger als 5 nm messenden Poren spricht f�r einenstarken Einfluss der �ffnung verschlossener CNTs durch dieReaktion mit O2. Die hier erzeugten aktiven Zentren sindallerdings unselektiv und beg�nstigen die Bildung von CO2,wie der drastische Abfall der Propenselektivit�t innerhalb derAnfangsperiode zeigt (Abbildung S7).
Die B2O3-modifizierten CNTs dagegen werden zu Beginnin geringem Maße desaktiviert, was mit der Zersetzung undthermischen Spreitung der B2O3-Vorstufe zusammenh�ngenmag. Die Balance zwischen Aktivit�tsverlust und Selektivi-t�tsgewinn kann �ber die Menge an B2O3 eingestellt werden(Abbildung 1b). Beide Kenngr�ßen h�ngen stark von derBeladung ab. Eine weitere Ursache f�r die gesteigerte Pro-penselektivit�t auf den modifizierten CNTs k�nnte neben derunterdr�ckten Totaloxidation von Propan auch eine gerin-gere Sauerstoffkapazit�t der aktiven Zentren sein. Boroxidals elektronenarmer Promotor erschwert die dissoziativeAdsorption von O2, und somit �berwiegen auf der Katalysa-toroberfl�che die selektiven Sauerstoffspezies.[17] Die Wir-kung von B2O3 kann bereits bei einer Beladung von nur0.003 Gew.-% beobachtet werden (Abbildung 1b), was zu-gleich verdeutlicht, dass nur ein sehr geringer Teil der de-tektierten Oberfl�chenfunktionalit�ten tats�chlich kataly-tisch aktiv ist. Die Propenselektivit�t steigt hier von 17 auf28%, wohingegen die Reaktionsgeschwindigkeit von 4.3 auf3.2 mmolg�1 h�1 abf�llt. Insgesamt nimmt jedoch die Pro-duktion von Propen um 18% zu. Die maximale Propenbil-dung wurde bei einer B2O3-Beladung von 0.01% gemessen;man beachte hierbei allerdings, dass die Propenausbeute beigleichem Umsatz mit der Temperatur ansteigt, wohingegendie thermische Stabilit�t mit der B2O3-Beladung zunimmt.Die kombinierte Optimierung der Reaktionsbedingungenund der Beladung bezogen auf die Propenausbeute ist dahersehr komplex.
Die F�higkeit zum Sauerstoffaustausch der CNTs mit undohne B2O3-Modifizierung wurde im Wechsel von 16O2 zuisotopenmarkiertem 18O2 untersucht. Wie in Abbildung 2agezeigt, findet auf den oCNTs ein reger Austausch von Sau-erstoffatomen statt, und die Bildung von partiell markiertem18O16O wird sogar in Abwesenheit von Propan beobachtet.Dagegen sinkt die Menge an 18O16O mit steigender B2O3-Beladung und verschwindet g�nzlich auf den hochbeladenenProben (Abbildung 2b), �hnlich wie beim gering beladenenVOx-Al2O3-Katalysator, der hier als Referenzsystem mitguten Katalyseeigenschaften in der ODH von Propandient.[14] Elektrophile (intermedi�re) Sauerstoffspezies wiePeroxide oder Superoxide auf nichtmodifizierten CNTs be-wirken sehr wahrscheinlich eine hohe Aktivit�t, aber einegeringe Propenselektivit�t.
Katalyse und Charakterisierung lassen eine einheitlicheStruktur der aktiven Zentren vermuten. Im station�ren Zu-stand sind kommerzielle wie oxidierte CNTs von �hnlicherAktivit�t und Selektivit�t, und auch die scheinbaren Akti-vierungsenergien f�r Propanverbrauch, Sauerstoffverbrauchund Propenbildung sind �hnlich (Abbildung 1a). Gleiches giltf�r die mit jeweils 5 Gew.-% Bor- und Phosphoroxid modi-fizierten CNTs. Man kann daraus schließen, dass weder Bnoch P direkt in die aktive Dom�ne eingebunden sind, an derdie ODH-Umsetzung abl�uft, und dass diese Heteroatomedas aktive Zentrum auch nicht beeinflussen. Die Experi-mente belegen, dass Bor- und Phosphoroxid lediglich unse-lektive Zentren blockieren und solche, an denen die Ver-brennung des Katalysatores stattfindet. Dies stimmt mit denTEM/EELS- und XPS-Ergebnissen (Abbildung S5 und S8)�berein. Bedingt durch die Pr�parationsmethode ist B2O3
�berwiegend im Innern und an den Enden der CNTs zufinden.[18] Nur ein geringer Teil der �ußeren Oberfl�che ist miteiner amorphen Schicht (� 1–2 nm) bedeckt, wohingegeneine hochdispergierte Submonolage von BOx-Spezies allge-genw�rtig zu sein scheint, da die geringere BET-Oberfl�cheauch mit dem Auff�llen/Gl�tten von oberfl�chlichen De-fektstrukturen erkl�rt werden k�nnte. Die hohe thermischeStabilit�t der dotierten Proben spricht f�r eine kovalente
Abbildung 2. Sauerstoffaustausch an a) oCNTs und b) 5B-oCNTs nacheinem Wechsel der Gasatmosph�re von 2% 16O2/Ne auf 2% 18O2/2%Ar/96% Ne, 673 K, 100 mg, 10 mLn min�1 als Ionenstrom I aufgetra-gen. Schwarze durchgezogene Linie: 16O2, schwarze gestrichelte Linie:18O2, graue durchgezogene Linie: 16O18O (300fach gegen�ber den an-deren Signalen verst�rkt).
Zuschriften
7048 www.angewandte.de � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2009, 121, 7046 –7051
Bindung an das CNT-Ger�st, wohingegen die B2O3/P2O5-F�llung der CNTs als Schutz gegen die Verbrennung fungiert.
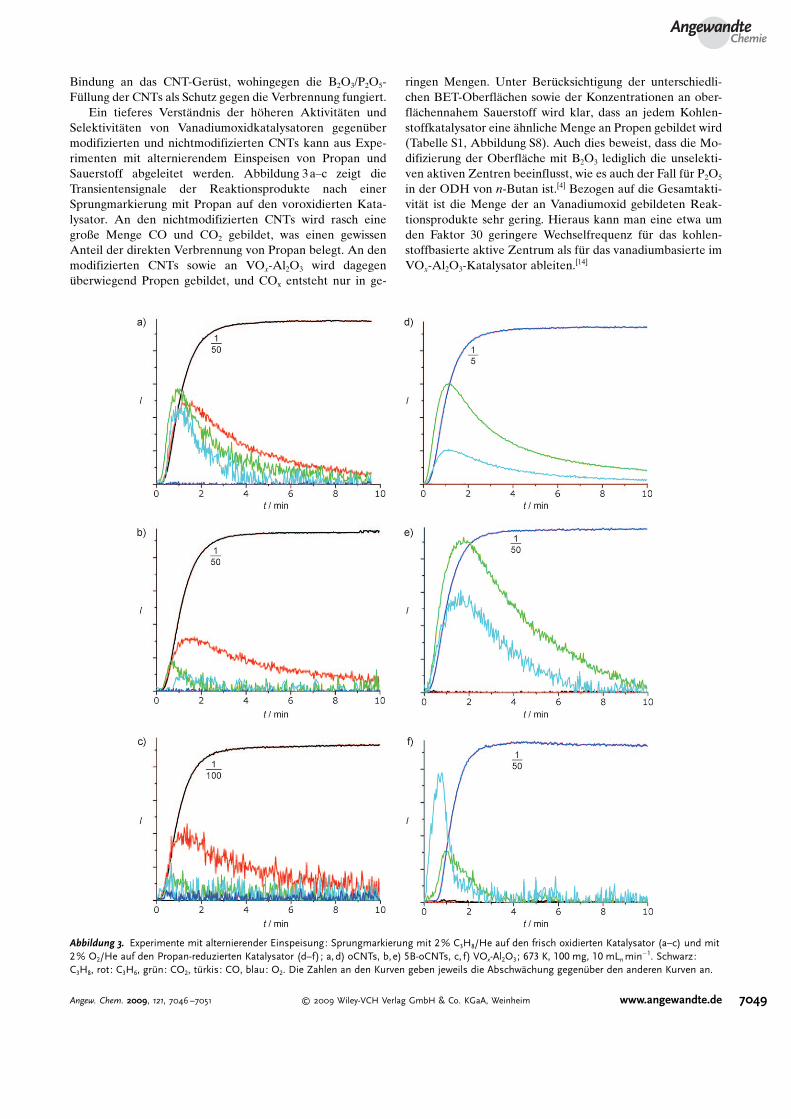
Ein tieferes Verst�ndnis der h�heren Aktivit�ten undSelektivit�ten von Vanadiumoxidkatalysatoren gegen�bermodifizierten und nichtmodifizierten CNTs kann aus Expe-rimenten mit alternierendem Einspeisen von Propan undSauerstoff abgeleitet werden. Abbildung 3a–c zeigt dieTransientensignale der Reaktionsprodukte nach einerSprungmarkierung mit Propan auf den voroxidierten Kata-lysator. An den nichtmodifizierten CNTs wird rasch einegroße Menge CO und CO2 gebildet, was einen gewissenAnteil der direkten Verbrennung von Propan belegt. An denmodifizierten CNTs sowie an VOx-Al2O3 wird dagegen�berwiegend Propen gebildet, und COx entsteht nur in ge-
ringen Mengen. Unter Ber�cksichtigung der unterschiedli-chen BET-Oberfl�chen sowie der Konzentrationen an ober-fl�chennahem Sauerstoff wird klar, dass an jedem Kohlen-stoffkatalysator eine �hnliche Menge an Propen gebildet wird(Tabelle S1, Abbildung S8). Auch dies beweist, dass die Mo-difizierung der Oberfl�che mit B2O3 lediglich die unselekti-ven aktiven Zentren beeinflusst, wie es auch der Fall f�r P2O5
in der ODH von n-Butan ist.[4] Bezogen auf die Gesamtakti-vit�t ist die Menge der an Vanadiumoxid gebildeten Reak-tionsprodukte sehr gering. Hieraus kann man eine etwa umden Faktor 30 geringere Wechselfrequenz f�r das kohlen-stoffbasierte aktive Zentrum als f�r das vanadiumbasierte imVOx-Al2O3-Katalysator ableiten.[14]
Abbildung 3. Experimente mit alternierender Einspeisung: Sprungmarkierung mit 2% C3H8/He auf den frisch oxidierten Katalysator (a–c) und mit2% O2/He auf den Propan-reduzierten Katalysator (d–f); a, d) oCNTs, b,e) 5B-oCNTs, c, f) VOx-Al2O3; 673 K, 100 mg, 10 mLn min�1. Schwarz:C3H8, rot: C3H6, gr�n: CO2, t�rkis: CO, blau: O2. Die Zahlen an den Kurven geben jeweils die Abschw�chung gegen�ber den anderen Kurven an.
AngewandteChemie
7049Angew. Chem. 2009, 121, 7046 –7051 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Eine Ursache daf�r l�sst sich aus der anschließendenReoxidation der mit Propan reduzierten Katalysatoren ab-leiten (Abbildung 3 d–f). Am Vanadiumoxidkatalysator ad-sorbiert nur sehr wenig Propan, und dessen Beseitigung durchVerbrennung geschieht viel schneller als an den CNT-Kata-lysatoren. Da an den Kohlenstoffkatalysatoren eine deutlichgr�ßere Menge an Propan adsorbiert, das auf den aktivenZentren haftet, wird zun�chst eine großen Menge COx ge-bildet, bevor das aktive Zentrum reoxidiert werden kann.Folgerichtig weisen die CNTs �berlegene katalytische Ei-genschaften unter sauerstoffreichen Reaktionsbedingungenauf (Abbildung S9). Vanadiumoxidkatalysatoren als selek-tivste Metalloxidkatalysatoren arbeiten hingegen gut beiniedrigen Sauerstoffpartialdr�cken, bei denen die Folgeoxi-dation des gebildeten Propens reduziert ist. Der kritischeAspekt der Selbstoxidation der Kohlenstoffkatalysatorenwurde in Langzeitstudien an ausgew�hlten Systemen unter-sucht (Abbildung S10). Bei einem Sauerstoff/Kohlenwasser-stoff-Verh�ltnis von 1:1 zeigen VOx-Al2O3, 5B-oCNTs undsogar nichtmodifizierte oCNTs eine hohe Stabilit�t (bis zu200 h). Im verd�nnten Reaktantenstrom weisen 5B-oCNTseine �hnliche Propenselektivit�t auf wie VOx-Al2O3.
[12] Einstetiger Anstieg der Aktivit�t von 5B-oCNTs, bei gleichblei-bend hoher Selektivit�t, spricht noch f�r ein gewisses Poten-zial zur Herstellung weiter optimierter Katalysatoren.
Nanokohlenstoff ist eine attraktive Alternative zu Me-talloxidkatalysatoren f�r ODH-Reaktionen nicht nur vonEthylbenzol sondern auch von kurzkettigen Alkanen. Propanwird von sauerstoffhaltigen funktionellen Gruppen aktiviert,und Propen wird mit beachtlicher Selektivit�t gebildet.Propan adsorbiert allerdings auch auf der reduzierten Kata-lysatoroberfl�che, was der Alkenselektivit�t abtr�glich ist.Effektive Abhilfe schafft hier die Modifizierung mit geringenMengen Boroxid, die zugleich die Bildung unselektiver Sau-erstoffspezies unterdr�ckt. Die bislang sehr geringe Ge-samtaktivit�t der Kohlenstoffkatalysatoren kann durch dieGenerierung einer h�heren Konzentration selektiver aktiverZentren gesteigert werden.
Experimentelles und MethodenKatalysatorpr�paration und -charakterisierung: Die kommerziellenCNTs wurden von Nanocyl bezogen (NC3100; CNTs). Zur Herstel-lung der oCNTs wurden die CNTs in konz. Salpeters�ure(100 mLgCNT
�1) 2 h unter R�ckfluss gekocht, anschließend mitDeionat auf pH 6–7 gewaschen und �ber Nacht bei 393 K an Luftgetrocknet. Die B- und P-modifizierten Proben (xB-oCNTs, 5P-oCNTs) wurden �ber Nassimpr�gnierung der oCNTs (6 mLg�1) mitw�ssrigen L�sungen von (NH4)2B10O16·8H2O bzw. (NH4)2HPO4
(Sigma Aldrich, > 99 %) hergestellt; die Beladungen x betrugen f�rB2O3 zwischen 0.003 und 5 Gew.-% und f�r P2O5 5 Gew.-%. Dieimpr�gnierten Proben wurden �ber Nacht bei 393 K an Luft ge-trocknet. Spezifische Oberfl�chen (BET) und Porenradienverteilun-gen (BJH) der CNTs wurden �ber die Adsorption von N2 bei 77 K miteinem Micromeritics-2375-BET-Ger�t ermittelt. Das Verh�ltnis vonamorphem (D) zu graphitischem Kohlenstoff (G) wurde mittelsLaser-Raman-Spektroskopie mit einem ISA-LabRam-Instrumentausgestattet mit einem Olympus-BX40-Mikroskop ermittelt. DieAnregungswellenl�nge betrug 632.8 nm bei einer spektralen Aufl�-sung von 0.9 cm�1. Die TEM-Analyse geschah an einem Philips-CM200-FEG-Transmissionselektronenmikroskop (Beschleunigungs-
spannung 200 kV) mit hochaufl�sender Aufnahme, EELS und EDX(energiedispersive R�ntgenspektroskopie). Elementspezifische Auf-nahmen wurden im STEM-Modus �ber den EDX-Detektor gemacht.F�r die SEM-Analyse wurde ein HITACHI-S-4800-Instrument (Be-schleunigungsspannung 2–15 kV) genutzt. Die Elementaranalysewurde mit dem EDAX-Genesis-4000-System mit Saphir-Detektordurchgef�hrt. Die Oxidationsbest�ndigkeit der Kohlenstoffspezieswurde mittels TPO bestimmt. 20-mg-Proben wurden in einemQuarzrohr unter 13.3 Vol.-% O2 in He (15 mLn min�1) linear von 323auf 1100 K aufgeheizt. Der Produktstrom wurde massenspektrome-trisch analysiert (Pfeiffer-QME-200), die Massenzahlen 4, 17, 18, 28,32 und 44 wurden f�r die Detektion von He, NH3, H2O, CO, O2 bzw.CO2 mit einer zeitlichen Aufl�sung von 4 s aufgezeichnet. DieOberfl�chenanalytik mittels XPS geschah an der ISISS-Strahllinievon BESSY II, Berlin. Die XP-Spektren wurden mit der Strah-lungsintensit�t korrigiert und ein Shirley-Hintergrund subtrahiert.Die Peakfl�chen wurden mit den theoretischen Wirkungsquer-schnitten normalisiert, um die relative Elementzusammensetzung ander Oberfl�che zu ermitteln.
Katalyseversuche: Standardprozeduren wurden in einem bereitsvorgestellten Versuchsaufbau durchgef�hrt.[14] Jeweils 1 g granulierteCNT-Probe (100–450 mm) wurde bei 673 K in einem Fluss von C3H8/O2/N2 = 1:1:4 gehalten (Str�mungsgeschwindigkeit: 60 mLn min�1),bis ein konstanter Umsatz zu verzeichnen war. Anschließend wurdedie Geschwindigkeit zwischen 10 und 120 mLn min�1 variiert. Diescheinbare Aktivierungsenergie wurde bei 60 mLn min�1 durch stu-fenweises Abk�hlen (10-K-Schritte) bis auf 623 K ermittelt. Isoto-penversuche (SSITKA) und Experimente mit alternierendem Ein-speisen wurden mit 100-mg-Proben und einer Str�mungsgeschwin-digkeit von 10 mLn min�1 nach einer Alterungszeit von 5 h in C3H8/O2/N2 = 1:1:4 durchgef�hrt. Die folgenden Gasmischungen wurdenverwendet: 2% 16O2/He, 2% C3H8/He, 2% 16O2/98% Ne und 2%18O2/2 % Ar/96% Ne.
Eingegangen am 5. April 2009,ver�nderte Fassung am 2. Juli 2009Online ver�ffentlicht am 7. August 2009
.Stichw�rter: C-H-Aktivierung · Heterogene Katalyse ·Nanomaterialien · Oxidationen · Selektivit�t
[1] H. P. Boehm, Carbon 1994, 32, 759.[2] J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas, J. J. M.
�rf¼o, Carbon 1999, 37, 1379, zit. Lit.[3] a) K. P. de Jong, J. W. Geus, Catal. Rev. Sci. Eng. 2000, 42, 481;
b) P. Serp, M. Corrias, P. Kalck, Appl. Catal. A 2003, 253, 337;c) A. Rochefort, P. Avouris, J. Phys. Chem. A 2000, 104, 9807;d) Y. Maniwa, Y. Kumazawa, Y. Saito, H. Tou, H. Kataura, H.Ishii, S. Suzuki, Y. Achiba, A. Fujiwara, H. Suematsu, Mol. Cryst.Liq. Cryst. 2000, 340, 671; e) J. L. Figueiredo, M. F. R. Pereira,Catal. Today 2009, DOI: 10.1016/j.cattod.2009.04.010.
[4] J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schl�gl, D. S. Su,Science 2008, 322, 73.
[5] J. A. Maci-Agull, D. Cazorla-Amors, A. Linares-Solano, U.Wild, D. S. Su, R. Schl�gl, Catal. Today 2005, 102–103, 248.
[6] a) D. S. Su, N. Maksimova, J. J. Delgado, N. Keller, G. Mestl,M. J. Ledoux, R. Schl�gl, Catal. Today 2005, 102–103, 110; b) X.Liu, D. S. Su, R. Schl�gl, Carbon 2008, 46, 547; c) M. F. R.Pereira, J. L. Figueiredo, J. J. M. �rf¼o, P. Serp, P. Kalck, Y. Kihn,Carbon 2004, 42, 2807; d) D. E. Resasco, Nat. Nanotechnol. 2008,3, 708; e) J. Sui, J. H. Zhou, Y. C. Dai, W. K. Yuan, Catal. Today2005, 106, 90.
[7] a) K. Chen, A. Khodakov, J. Yang, A. T. Bell, E. Iglesia, J. Catal.1999, 186, 325; b) P. Mars, D. W. van Krevelen, Chem. Eng. Sci.1954, 3, 41.
Zuschriften
7050 www.angewandte.de � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2009, 121, 7046 –7051

[8] a) G. Mul, M. A. Ba�ares, G. Garcia Cort�z, B. van der Linden,S. J. Khatib, J. A. Moulijna, Phys. Chem. Chem. Phys. 2003, 5,4378; b) A. T. Bell in Catalyst Deactivation (Hrsg.: E. E. Peter-sen, A. T. Bell), Marcel Dekker, New York, 1987, S. 235 – 260.
[9] B. K. Hodnett in Supported Catalysts and Their Applications(Hrsg.: D. C. Sherrington, A. P. Kybett), Royal Society of Che-mistry, London, 2000, S. 1 – 8.
[10] a) E. A. Mamedov, V. Cort�s Corbern, Appl. Catal. A 1995,127, 1; b) I. E. Wachs, Catal. Today 2005, 100, 79; c) F. Cavani, N.Ballarini, A. Cericola, Catal. Today 2007, 127, 113.
[11] a) H. W. Zanthoff, S. A. Buchholz, A. Pantazidis, C. Mirodatos,Chem. Eng. Sci. 1999, 54, 4397; b) B. Grzybowska-Swierkosz,Appl. Catal. A 1997, 157, 409.
[12] L. E. Jones, P. A. Thrower, J. Chim. Phys. 1987, 84, 1431.[13] a) D. W. McKee, Chem. Phys. Carbon 1991, 23, 173; b) Y. J. Lee,
L. R. Radovic, Carbon 2003, 41, 1987.
[14] a) B. Frank, A. Dinse, O. Ovsitser, E. V. Kondratenko, R.Schom�cker, Appl. Catal. A 2007, 323, 66; b) A. Dinse, B. Frank,C. Hess, D. Habel, R. Schom�cker, J. Mol. Catal. A 2008, 289, 28.
[15] a) D. S. Su, N. Maksimova, J. J. Delgado, N. Keller, G. Mestl,M. J. Ledoux, R. Schl�gl, Catal. Today 2005, 102–103, 110; b) J.Zhang, D. S. Su, A. H. Zhang, D. Wang, R. Schl�gl, C. H�bert,Angew. Chem. 2007, 119, 7460; Angew. Chem. Int. Ed. 2007, 46,7319.
[16] K. Behler, S. Osswald, H. Ye, S. Dimovski, Y. Gogotsi, J. Na-nopart. Res. 2006, 8, 615.
[17] a) E. V. Kondratenko, M. Cherian, M. Baerns, Catal. Today 2006,112, 60; b) O. V. Buyevskaya, M. Baerns, Catal. Today 1998, 42,315.
[18] J. Zhang, Y. S. Hu, J. P. Tessonnier, G. Weinberg, J. Maier, R.Schl�gl, D. S. Su, Adv. Mater. 2008, 20, 1450.
AngewandteChemie
7051Angew. Chem. 2009, 121, 7046 –7051 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de





























