Embed Size (px)
Citation preview

CASE REPORT
Idiopathic Giant Cell Granulomatous Hypophysitis withHypopituitarism, Right Abducens Nerve Paresis
and Masked Diabetes InsipidusTohru Fujiwara, Kozo Ota, Noriko Kakudo, Shozo Rikimaru, Tomohiro Sugawara, Katsura Yamada*,Tatsuyuki Satoh, Mitsushi Yano, Eiichi Tamate, Masaetsu Miura, Hidetoshi Ikeda** and Tokihisa Kimura
Abstract
A 38-year-old man presented with headache, fever, anddouble vision associated with right abducens nerve pare-sis. He had neither nuchal rigidity nor visual field defect.Laboratory data revealed elevated erythrocyte sedimenta-tion rate (ESR), eosinophilia, and lymphocytic pleocytosisin the cerebrospinal fluid (CSF). Provocation tests of pitu-itary hormones showed partial hypopituitarism. Magneticresonance imaging (MRI) revealed swelling of the hypo-physis and a mass lesion expanding into the right cavern-ous sinus. The supplement dose of dexamethasone for hy-pothalamic hypocortisolism manifested diabetes insipidus.Biopsy, carried out through the transsphenoidal approach,revealed giant cell granuloma. Systemic granulomatousdiseases were ruled out, and the lesion was considered tobe idiopathic giant cell granulomatous hypophysitis. Rightabducens nerve paresis, diabetes insipidus and dysfunctionof the anterior lobe were amendedby the treatment withprednisolone for 4 months, and findings of the pituitarygland and stalk were normalized. The present case showsthat glucocorticoid has an effect on amendmentof idiopathicgiant cell granulomatous hypophysitis.(Internal Medicine 40: 915-919, 2001)
Key words: magnetic resonance imaging, transsphenoidal bi-opsy, prednisolone, hypothalamic hypocortisolism
Introduction
Idiopathic giant cell granulomatous hypophysitis is a raredisease of the pituitary gland, presenting with hypopituitarismas well as symptomsdue to expanding mass lesions (1-5). His-tological characteristics are the infiltration of multinucle-
ated giant cells, plasma cells and lymphocytes. Granuloma-tous lesions of the pituitary gland are generally associated withsystemic granulomatous diseases such as sarcoidosis, tubercu-losis, syphilis, mycosis, and Langerhans cell histiocytosis.Therefore, granulomatous systemic disorders should be ex-cluded in order to diagnose idiopathic giant cell granuloma-tous hypophysitis.We, herein, describe a 38-year-old man, who developedhypopituitarism, right abducens nerve paresis and masked dia-betes insipidus due to idiopathic giant cell granulomatoushypophysitis; he was successfully treated with oral cortico-steroids.
Case ReportA 38-year-old man was admitted to Furukawa City Hospi-tal for the evaluation of abducens nerve paresis of the right eyein April 1999. He had been suffreing from headache and low-grade fever for 40 days before admission. Brain computed to-mography (CT) revealed no abnormal findings, but he beganto complain of gradual development of diplopia over the sub-sequent 20 days, and was diagnosed as right abducens nerveparesis. Magnetic resonance imaging (MRI) showed no abnor-mal findings in the right orbit. His family and past historieswereunremarkable.On admission, he had a mild fever (37.5°C) and right ab-ducens nerve paresis. Mydriasis and ptosis were not noticed inhis right eye. The pupillary response to light, visual fields andvisual acuity were normal, and ocular fundus was clear. He didnot complain of any pain in the orbital regions, and there wereno pathological findings in his skin. Laboratory data showedmild anemia, eosinohphilia, a mild elevation of transaminases,and elevated erythrocyte sedimentation rate (ESR) and an in-creased C-reactive protein (CRP) level (Table 1). Chest X-rayand CTwere not remarkable and no mediastinal adenopathywas noticed. Endocrinological studies showed secondary hy-
From the Department of Internal Medicine and *Department of Ophthalmology, Furukawa City Hospital, Furukawa and **Department of Neurosurgery,Tohoku University School of Medicine, Sendai
Received for publication October 26, 2000; Accepted for publication April 19, 200 1Reprint requests should be addressed to Dr. Tohru Fujiwara, the Department of Clinical Pharmacology and Therapeutics, Tohoku University School of Medi-
cine, 1-1 Seiryo-cho, Aoba-ku, Sendai 980-8574
Internal Medicine Vol. 40, No. 9 (September 2001) 915

Fujiwara et al
Table 1. Labolatory DataC o m p l e t e b l o o d c o u n t I m mu n i ty t e st s
Wh i te bl o od c el l 7 , 60 0/ mm 3 A n t i - n u c l e a r A b ( - )N e u tro p h il 3 6 % A n ti -p i tu it a ry g l an d A bL y m p h o c y te 4 2 % A n te rio r lo b e (- )M o n o c y te 10 % P o ste rio r lo b e (- )E o sin o p h il 1 1 % R F (- )B a so p h il 1 % T h y r o i d t e s t ( - )
R e d b l oo d c e l l 3 72 x l O 4/ m m3 M i c r o s o m e t e s t ( - )H e m o g lo b in 10 .2 g/d l Co a gu l at i on te s tP la te le t 22 .3 x l O 4 /m m3 E S R 5 5 m m ( l h )
B l o o d c he m i s t r y 9 2 m m ( 2 h )T ot al p ro te in 6.2 g/ dl U rin ary sisA lb u m in 6 0 . 8 % G ra v ity 1 .0 3 0
a , - gl obu lin 3 . 1 % P r o t e i n ( - )
cc 2 -globul in 1 0 . 7 % G lu c o s e (- )B -g lo b u lin 9 . 4 % U r i n e v o l u m e 1 , 9 8 0 m l / d a yY -g lo b u lin 1 6 . 0 % O s m o l a l i t y 3 7 1 m O s m / k g
A S T 3 7 I U/ 1 C e re b ro sp in a l flu idA L T 7 2 I U/ / C o lo r C o lo rle ssL D H 21 0 IU / / C h r o l i d e 1 2 6 m E q / /A L P 27 0 I U / Z G l u c o s e 4 9 m g / d lT o ta l c h o le ste ro l 1 6 2 mg / d l T o t a l p r o t e i n 6 7 m g / d lT rig ly c e rid e 1 0 6 m g / g l C e l l c o u n t s 6 9 / 3 m m 3
Ur e a n i t r og e n 7 . 7 m g /d l C e ll ty p e M N L : 8 7 %C re a tin in e l .O mg / dl P N L : 1 3 %S o d iu m 1 3 7 m E q / /P o ta ss iu m 4 . 3 m E q / lC h lo rid e 9 9 m E q / /C a lc iu m 9 .5 m g/ d lP h o sp h a te 5 . 9 m g /d lG lu c o se 9 1 m g / d l
C -r e ac ti v e pr ot e in 1. 2 m g /d lFerriti n 4 0 0 n g/ m lA C E 10 . 8 ug / m l
L y so z y m e 2 0 . 7 U
AST:asparate aminotransferase, ALT:alanine aminotransferase, LDH:lactate dehydrogenase,ALP: alkaline phosphatase, ACE:angiotensin converting enzyme, RF: rheumatoid factor, ESR:erythrocyte sedimentation rate, MNL:mononuclear lymphocyte, PNL: polynuclear lympho-cyte.
pogonadism, hypothyrotropinism without apparent secondaryhypothyroidism, hypottialamic hypocortisolism and hyperpro-lactinemia, as well as partial central diabetes insipidus (Tables2, 3, 4). Skull X-ray revealed no abnormal findings. Brain MRIshowed swelling of the pituitary gland and stalk with invasioninto the right cavernous sinus (Fig. 1A). The mass wasisointense on the Tl-weighted image with hyperintensity onthe T2-weighted image, and it showed homogeneous enhance-ment due to gadolinium- diethylenetriaminepenta-acetic acid(Gd-DTPA).
Following the diagnosis of secondary hypocortisolism, dexa-methasone (0.5 mg/day) was orally administered. Immediatelyafter the treatment, the urine volumewas markedly increasedfrom about 2 to 5 //day. The presence of masked diabetesinsipidus was suspected, and the treatment with DDAVPwas
initiated, thereby leading to normalization of urine volume.Transsphenoidal biopsy was carried out for a precise diag-nosis. The sella and dura mater showed the fibrous mass. His-tological examination showedextensive well-formed non-caseating granulomas without cholesterol crystals and necro-sis. Granulomas were composed of multinucleated giant cellscoexisting with accumulation of plasma cells and lymphocytes(Fig. 2). There were several areas where the normal glandularstructure of the hypophysis was destroyed due to granuloma-tous inflammation. Since no systemic granulomatous diseaseswere noted, he was diagnosed as idiopathic giant cell granulo-matous hypophysitis.After the diagnosis, oral prednisolone (20 mg/day) was ini-tiated. Serological findings showed an improvement of ESRand CRP. Urinary concentrating ability was normalized with-
916 Internal Medicine Vol. 40, No. 9 (September 2001)
Granulomatous Hypophysitis and Hypopituitarism
Table 2. Hormonal Provocative TestsH o rm o n e B a s a l v a lu e R e s po n s i v e va l u e s
(N o rm a l ra n g e )1 5 m i n 3 0 m in 6 0 m in 1 2 0 m in
T R H t e s t T S H ( L i U / m l ) 0 . 01 (0 . 2 3- 4 ) 0 .0 1 0 . 0 2 0 . 0 2 0 . 0 2P R L ( n g / m l ) 1 9. 9 (1 .5 -9 . 7) 2 4 . 6 2 2 . 9 2 1 . 0 2 5 . 1
G R F t e s t G H ( n g / m l ) 2 . 95 (< 1 .4 6 ) 2 6 .1 8 2 5 . 5 4 1 6 . 3 4 9 . 1 2C R H t e s t A C T H ( p g / m l ) 1 8 (6 - 3 6 ) 2 5 7 3 7 7 2 7 7 1 0 5
C o rtiso l (ju g /d l) 1.5 (3 - 1 5 .2 ) 3.4 6 . 7 9 . 7 9 . 9L H R H t e s t F S H ( m l U / m l ) 0 .9 ( 1 .8 -1 3 .6 ) 1.2 1 . 7 2 . 5 3 . 2
L H ( m l U / m l ) <0 . 5 ( 1. 1- 1 .8 ) < 0 .5 1.3 1.3 1. 3A C T H t e s t C o rtiso l (ji g /d l) < 1. 0 ( 3 - 15 . 2 ) 7 . 8 1 0 . 7 1 2 . 8 1 2 . 3
CRH:corticotropind-releasing hormone, GH:growth hormone, ACTH:adrenocorticotropic hor-mone, GRF: growth hormone-releasing hormone, TSH: thyroid-stimulating hormone, LH: lutenizinghormone, TRH: thyroid-stimulating hormone-releasing hormone, FSH: follicle-stimulating hor-mone, PRL: prolactin, LH-RH: lutenizing hormone-releasing hormone.
Table 3. Endocrinological DataH o rm o n e B a sa l v a lu e
(N o rm al ra n g e )
S e ru mT 3 (n g /m l) 2. 4 (0 . 8- 1. 8)T 4 ( u g /d l ) l l. 3 (4 .5 - l l .7 )A D H ( p g / m l ) 1. 0 ( 0. 8- 6.3 )O s m o l a l i t y ( m O s m / k g ) 2 9 3 (2 7 0 - 2 9 3 )
Ur i n e
F ree c ort is ol (j ig/ day ) 2 0 ( 3 4- 1 6 0 )1 7 - O H C S ( m g / d a y ) 3. 2 (3 .1 -8 .7 )1 7 -K S (n g /d a y ) 1 .4 ( 4 .2 -1 2. 4)O sm o lality (m O s m /k g ) 3 10
ADH: antidiuretic hormone, 1 7-OHCS: 1 7-Hydroxycor-ticosteroid, 17-KS: 17-Ketosteroid.
Table 4. Dehydration-Pitressin Tests
Responsive valuesDehydrarion %AUosm
value 30 min 60 min
Urinary osmolality (Uosm)(mOsm/kg) 305 330 349 1 5.A
Serum osmolality (Posm)(mO sm/kg) 29 3
Responsive values are measured by an intravenous injection of aqueous pitressin( 100 mU), after 1 0 hours of dehydration in the presence of dexamathasone. %Aosmindicates the ratio of a percent increase in urinary osmolality after pitressin.
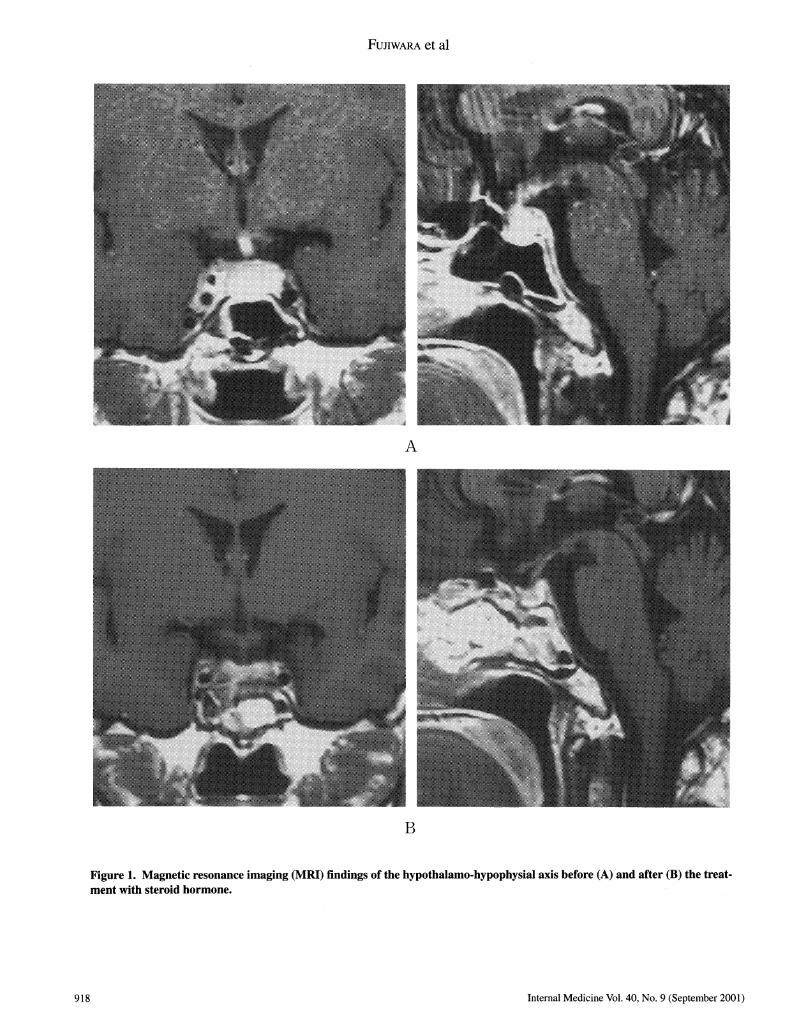
out DDAVPtreatment. MRIshowed the disappearance of theswelling of the pituitary gland and stalk 4 weeks after pred-nisolone (Fig. IB). 4 months after treatment, basal levels ofgonadotropin, prolactin and thyrotropin were normalized andurinary concentrating ability recovered without DDAVPtreat-ment. The dose of prednisolone was gradually reduced with-out recurrence.
Discussion
Giant cell-containing granulomatous hypophysitis of thepituitary gland (granulomatous hypophysitis) was first de-scribed in 19 17, and is considered a clinico-pathological entitiywith the isolated involvement of the pituitary gland ( 1 , 2). Clini-cal evaluation must exclude specific granulomas. Serologicaland histological examination are helpful in distinguishing id-iopathic giant cell granulomatous hypophysitis from othergranulomatous diseases, such as sarcoidosis, tuberculosis andsyphilis. Unlike giant cell granulomatous hypophysitis, sar-coidosis is characterized by the presence of various systemic
Internal Medicine Vol. 40, No. 9 (September 2001) 917
Fujiwara et al
Figure 1. Magnetic resonance imaging (MRI) findings of the hypothalamo-hypophysial axis before (A) and after (B) the treat-ment with steroid hormone.
918 Internal Medicine Vol. 40, No. 9 (September 2001)

Granulomatous Hypophysitis and Hypopituitarism
Figure 2. Photomicrograph of the surgical specimen showing agranulomatous area with multinucleated giant cells, plasma cellsand lymphocytes (HE stain, x400).
lesions in the lung, skin, and eye. In addition, increased levelsof angiotensin-converting enzyme (ACE) in the serum or CSFare indicative of sarcoidosis. In the present case, there wasneither characteristic lesions in any organs, nor increased ACE.Moreover,the Mantouxreaction was normal.TPHAwasnega-tive. Therefore, specific granulomatous diseases were not con-sidered.Lymphocytic hypophysitis is another type of idiopathic in-flammatory disease of the pituitary gland. Pathologically, dif-fuse infiltration of lymphocytes and plasma cells is character-istic. This entity has commonlybeen reported in womendur-ing pregnancy and after delivery (6, 7). But lymphocytichypophysitis, not associated with pregnancy, has been reportedto occur more frequently than previously considered (8). Thepathophysiology of this disorder is thought to be due to au-toimmune disorders and to similar backgrounds as giant cellgranulomatous hypophysitis (4). Howeverin the present case,no serological data indicative of autoimmuneanomalies werepresent.Headacheis a characteristic sign in inflammatory lesions ofthe pituitary gland and stalk, and is frequently observed ingranulomatous hypophysitis (2). Signs and symptoms such asfever, headache, nausea, vomiting, and abnormal CSFcellcounts are present in not only acute meningitis, but also in id-iopathic giant cell granulomatous hypophysitis (4, 9). In thepresent case, a slight increase in lymphocytes in the CSF wasobserved, but no typical signs and symptomsof acute menin-gitis were noticed.
In contrast to lymphocytic hypophysitis (6, 7), visual dis-turbance is rarely associated with giant cell granulomatoushypophysitis. To our knowledge, only one autopsy and twosurgical cases have been reported showing symptoms ofchiasmal involvement (1, 4). Three cases of the 13 reportedgiant cell granulomas were associated with oculomotor nerveparesis, but not with visual disturbance (3-5). Therefore, giant
cell granulomatous hypophysitis should be considered, whenthere is paresis of the oculomotor nerves without visual fielddefect.
It has been reported that the incidence of diabetes insipidusdue to giant cell granulomatous hypophysitis is low, comparedwith that due to pituitary sarcoidosis and Langerhans cell his-tiocytosis (1, 10). In the present case, however, there was par-tial diabetes insipidus. Granulomatous hypophysitis was re-ported to mainly affect the anterior pituitary lobe, but to partlyextend to the posterior lobe, pituitary stalk, and hypothalamus(1). Indeed, co-existence of diabetes insipidus, hyperprolac-tinemia and hypothalamic hypocortisolism were indicative ofextension of lesion into the neurohypophysis as well as hypo-thalamus.The treatment for giant cell granulomatous hypophysitis hasnot yet been established. The mass is surgically removed bythe transsphenoidal approach in most cases, but corticoste-roids might be useful for specific granulomatous disorders ofunknownetiology such as sarcoidosis and Tolosa-Hunt syn-drome. Indeed, in the present case, cranial nerve paresis wasimproved due to treatment with glucocorticoid hormone.Honegger et al (8) reported that treatment with oral cortico-steroids was effective in decreasing the size of pituitary mass.Onthe other hand, surgical resection of the granulomatous massis recommended, if visual disturbance is not amendeddue toconservative treatment with steroid hormones.
Acknowledegment:This study was supported in part by the Health Sci-ence Research Grants for Research on Specific Diseases, the Ministry of Healthand Welfare, Japan.
References
1) Rickards AG, Harvey PW. Giant cell granuloma and the other pituitarygranulomata. Quart J Med 23: 425-439, 1954.
2) Scanarini M, d'Avella D, RotilioA, Kitromilis N, Mingrino S. Giant-cellgranulomatous hypophysitis: a distinct clinicopathological entity. J
Neurosurg 71: 681-686, 1989.3) Taylon C, Duff TA. Giant cell granuloma involving the pituitary gland.
Case report. J Neurosurg 52: 584-587, 1980.4) Yoshioka M, Yamakawa N, Saito H, et al. Granulomatous hypophysitis
with meningitis and hypopituitarism. Intern Med 31: 1147-1150, 1992.5) Inoue T, Kaneko Y, Mannoji H, Fukui M. Giant cell granulomatoushypophysitis manifesting as an intrasellar masswith unilateral ophthal-moplegia. Neurol Med Chir (Tokyo) 37: 766-770, 1997.
6) Asa SL, Bilbao JM, Kovacs K, Josse RG, Kreines K. Lymphocytichypophysitis of pregnancy resulting in hypopituitarism: A distinct clinico-pathological entity. Ann Intern Med 95: 166-171, 1981.
7) Baskin DS, Townsend JJ, Wilson CB. Lymphocytic adenohypophysitisof pregnancy simulating a pituitary adenoma: A distinct pathological en-tity. J Neurosurg 56: 148-153, 1982.
8) Honegger J, Fahlbusch R, Bornemann A, et al. Lymphocytic and granu-lomatous hypophysitis: Experience with nine cases. Neurosurgery 40:713-723, 1997 (see comments).
9) Cooper R, Belilos E, Drexler S, Efton A, Ferrara E, Tollin SR. Idiopathicgiant-cell granulomatous hypophysitis mimicking acute meningitis. AmJ Med Sci 318: 339-342, 1999.
10) Nishio S, Mizuno J, Barrow DL, Takei Y, Tindall GT. Isolated histiocyto-sis X of the pituitary gland: case report. Neurosurgery 21: 718-721, 1987.
Internal Medicine Vol. 40, No. 9 (September 2001) 919








![Prezentacja programu PowerPoint - OncoArendi Therapeutics · 2019-07-17 · CCL4 in BAL fluid [pg/ml] l ) 0 2 4 6 8 10 n s n) Organized granulomatous structures **** Granulomatous](https://img.pdfslide.tips/doc/110x75/5f5b3733c1c4190b59080a36/prezentacja-programu-powerpoint-oncoarendi-therapeutics-2019-07-17-ccl4-in-bal.jpg)




















