Embed Size (px)
Citation preview

IMMUNOLOGY
Host DNases prevent vascularocclusion by neutrophilextracellular trapsMiguel Jiménez-Alcázar,1* Chandini Rangaswamy,1* Rachita Panda,1
Josephine Bitterling,1 Yashin J. Simsek,1 Andy T. Long,1 Rostyslav Bilyy,2 Veit Krenn,3
Christoph Renné,4 Thomas Renné,1,5 Stefan Kluge,6 Ulf Panzer,7 Ryushin Mizuta,8
Hans Georg Mannherz,9 Daisuke Kitamura,8 Martin Herrmann,10
Markus Napirei,9 Tobias A. Fuchs1,5†
Platelet and fibrin clots occlude blood vessels in hemostasis and thrombosis. Here we reporta noncanonical mechanism for vascular occlusion based on neutrophil extracellular traps(NETs), DNA fibers released by neutrophils during inflammation.We investigated which hostfactors control NETs in vivo and found that two deoxyribonucleases (DNases), DNase1and DNase1-like 3, degraded NETs in circulation during sterile neutrophilia and septicemia. Inthe absence of both DNases, intravascular NETs formed clots that obstructed blood vesselsand caused organ damage.Vascular occlusions in patients with severe bacterial infections wereassociated with a defect to degrade NETs ex vivo and the formation of intravascular NETclots. DNase1 and DNase1-like 3 are independently expressed and thus provide dual hostprotection against deleterious effects of intravascular NETs.
Inflammation is an essential host responsefor the control of invading microbes and heal-ing of damaged tissues (1). Uncontrolled andpersistent inflammation causes tissue injuryin a plethora of inflammatory disorders. Neu-
trophils are the predominant leukocytes presentduring acute inflammation. During infections,neutrophils generate extracellular traps (NETs),lattices of DNA filaments decorated with toxichistones and enzymes that immobilize and neu-tralize bacteria (2). Extracellular deoxyribonucle-ases (DNases) serve as virulence factors in several
pathogenic bacteria, demonstrating the relevanceof NETs in host defense (3, 4). However, inap-propriately releasedNETsmay harmhost cells asa result of their cytotoxic, proinflammatory, andprothrombotic activity (5–7). Indeed, NETs arefrequently associated with inflammatory orischemic organ damage, and the therapeutic in-fusion of DNases limits host injury in variousanimal models (8, 9).How the host degrades NETs in vivo to limit
tissue damage during episodes of inflammationis poorly understood. Earlier work has shown
that DNase1 in serum digests the DNA backboneof NETs in vitro (10). We analyzed serum fromwild-type mice by zymography and detected twoenzymatically active DNases, DNase1 and DNase1-like 3 (DNase1L3) (Fig. 1A). Both enzymes aremembers of the DNase1 protein family, but differin their origin and substrate affinity. DNase1is expressed by nonhematopoietic tissues andpreferentially cleaves protein-free DNA (11, 12).DNase1L3, also knownasDNasegamma, is secretedby immunecells and targetsDNA-proteincomplexes,such as nucleosomes (11, 13). We generated micethat lackedDNA-degrading activity in serumdueto a combined deficiency of DNase1 and DNase1L3(Fig. 1A). In vitro–generated NETs remained intactafter exposure toDnase1–/–Dnase1l3–/– sera,whereassera from wild-type, Dnase1–/–, and Dnase1l3 –/–
mice degraded NETs (Fig. 1, B and C). We thenstably expressedDnase1 orDnase1l3 cDNA in thelivers of Dnase1–/– Dnase1l3 –/– mice. Given thatboth enzymes contain a secretory protein signalsequence (11), this approach restored the activityof DNase1 or DNase1L3 in circulation (Fig. 1D)and the capacity of sera fromDnase1–/–Dnase1l3–/–
mice to degrade NETs (Fig. 1, E and F). Thus, twoindependently expressed host enzymes, DNase1and DNase1L3, degrade NETs in vitro.To test the requirementofDNase1 andDNase1L3
for NET degradation in vivo, we chronically stim-ulated wild-type, Dnase1–/–, Dnase1l3–/–, andDnase1–/– Dnase1l3–/– mice with the granulocytecolony-stimulating factor (G-CSF), which triggersneutrophilia—a hallmark of acute inflammation—and stimulates a subpopulation of neutrophils tospontaneously release NETs ex vivo (14). Hepaticexpression of Csf3 cDNA, which encodes G-CSF,resulted in chronically elevated concentrationsof G-CSF in plasma (fig. S1A). Consequently, theneutrophil blood count steadily increased, andspontaneously formed NETs were detected in
RESEARCH
Jiménez-Alcázar et al., Science 358, 1202–1206 (2017) 1 December 2017 1 of 5
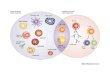
Fig. 1. DNase1 and DNase1L3 in circulationdegrade NETs in vitro. Characterization ofDNA-degrading activity of sera from wild-type(WT), Dnase1–/– (D1–/–), Dnase1l3–/– (D1l3–/–),and Dnase1–/– Dnase1l3–/– (D1−/− D1l3−/−)mice. (A) Detection of DNase1 (D1), DNase1L3(D1L3), and total DNase activity by thezymographic assays denaturing polyacrylamidegel electrophoresis zymography (DPZ) and singleradial enzyme diffusion (SRED). (B) Images and(C) quantification of DNA staining of NETs gen-erated in vitro after incubation with sera fromindicated genotypes (N = 6). Scale bar, 50 mm.(D) DPZ and SRED analysis of sera from D1–/–
D1l3–/– mice stably expressing a plasmidwith D1 or D1l3 or a control plasmid (Ctrl) for7 days. (E) Images and (F) quantification of DNAstaining of NETs generated in vitro afterincubation with buffer or sera from D1–/– D1l3–/–
mice expressing D1, D1l3, or Ctrl (N = 5). Scalebar, 50 mm. Images are representative of two ormore independent experiments. (C) and (F) one-wayanalysis of variance (ANOVA) followed byBonferroni’s multiple comparisons post hoc test;***P < 0.001 versus all other groups.
WT D1-/- D1l3-/- D1-/-D1l3-/-
A
WT
D1-/-
D1l3-/-
D1-/- D1l3
-/-0
20
40
60
80
100
NE
T-D
NA
(%
of b
uffe
r)
***C
WW W
B
D1-/-D1l3-/- D1l3-/-
D1-/-
W: loading well
D1-/-D1l3-/-
+D1l3D1-/-D1l3-/-
+D1D1-/-D1l3-/-
+Ctrl
E
In v
itro
NE
T-d
eg
rad
atio
n
Buffer
D1-/-D1l3-/- + D1l3D1-/-D1l3-/- + D1
D1-/-D1l3-/- + Ctrl
W
WWW
W: loading well
D1
D1L3
TotalDNaseactivity
D
WT
Ctrl D1D1l3
0
20
40
60
80
100
NE
T-D
NA
(%
of b
uffe
r)
***
D1-/-D1l3-/-
F
DP
ZS
RE
D
In v
itro
NE
T-d
eg
rad
atio
n
D1
D1L3
TotalDNaseactivity
DP
ZS
RE
D
on March 28, 2020
http://science.sciencem
ag.org/D
ownloaded from
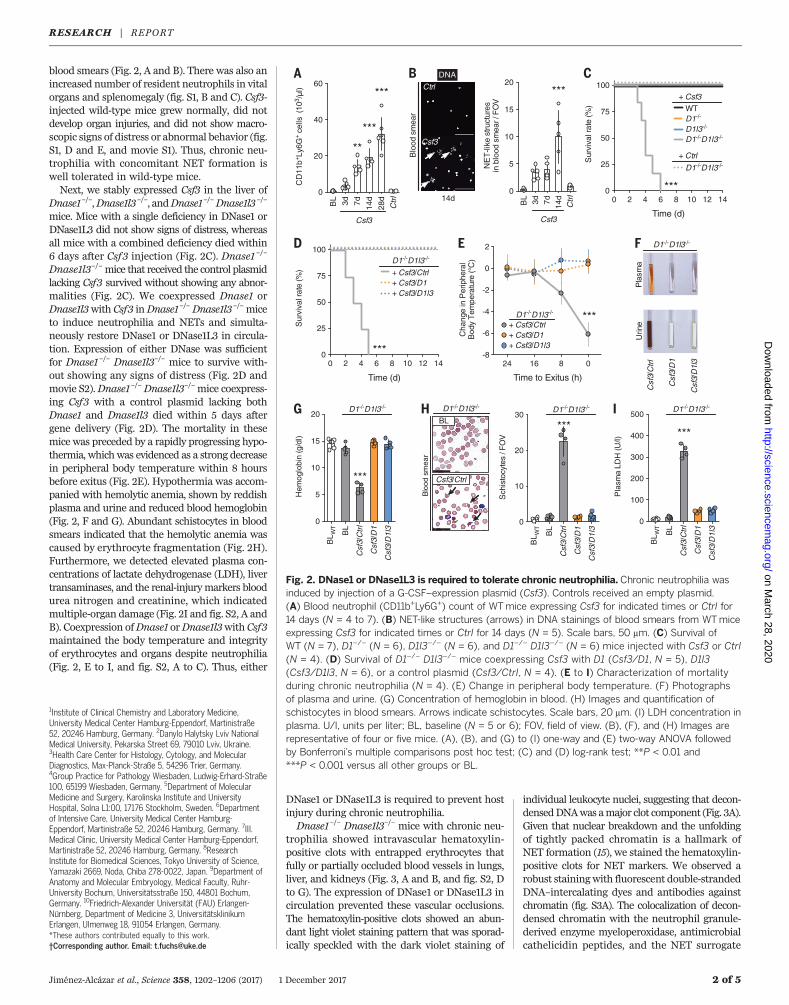
blood smears (Fig. 2, A and B). There was also anincreased number of resident neutrophils in vitalorgans and splenomegaly (fig. S1, B and C). Csf3-injected wild-type mice grew normally, did notdevelop organ injuries, and did not show macro-scopic signs of distress or abnormal behavior (fig.S1, D and E, and movie S1). Thus, chronic neu-trophilia with concomitant NET formation iswell tolerated in wild-type mice.Next, we stably expressed Csf3 in the liver of
Dnase1–/–,Dnase1l3–/–, andDnase1–/–Dnase1l3–/–
mice. Mice with a single deficiency in DNase1 orDNase1L3 did not show signs of distress, whereasall mice with a combined deficiency died within6 days after Csf3 injection (Fig. 2C). Dnase1 –/–
Dnase1l3–/–mice that received the control plasmidlacking Csf3 survived without showing any abnor-malities (Fig. 2C). We coexpressed Dnase1 orDnase1l3with Csf3 inDnase1–/–Dnase1l3–/–miceto induce neutrophilia and NETs and simulta-neously restore DNase1 or DNase1L3 in circula-tion. Expression of either DNase was sufficientfor Dnase1–/– Dnase1l3–/– mice to survive with-out showing any signs of distress (Fig. 2D andmovie S2).Dnase1–/–Dnase1l3–/–mice coexpress-ing Csf3 with a control plasmid lacking bothDnase1 and Dnase1l3 died within 5 days aftergene delivery (Fig. 2D). The mortality in thesemice was preceded by a rapidly progressing hypo-thermia, whichwas evidenced as a strong decreasein peripheral body temperature within 8 hoursbefore exitus (Fig. 2E). Hypothermia was accom-panied with hemolytic anemia, shown by reddishplasma and urine and reduced blood hemoglobin(Fig. 2, F and G). Abundant schistocytes in bloodsmears indicated that the hemolytic anemia wascaused by erythrocyte fragmentation (Fig. 2H).Furthermore, we detected elevated plasma con-centrations of lactate dehydrogenase (LDH), livertransaminases, and the renal-injurymarkers bloodurea nitrogen and creatinine, which indicatedmultiple-organ damage (Fig. 2I and fig. S2, A andB). Coexpression ofDnase1 orDnase1l3with Csf3maintained the body temperature and integrityof erythrocytes and organs despite neutrophilia(Fig. 2, E to I, and fig. S2, A to C). Thus, either
DNase1 or DNase1L3 is required to prevent hostinjury during chronic neutrophilia.Dnase1–/– Dnase1l3–/– mice with chronic neu-
trophilia showed intravascular hematoxylin-positive clots with entrapped erythrocytes thatfully or partially occluded blood vessels in lungs,liver, and kidneys (Fig. 3, A and B, and fig. S2, Dto G). The expression of DNase1 or DNase1L3 incirculation prevented these vascular occlusions.The hematoxylin-positive clots showed an abun-dant light violet staining pattern that was sporad-ically speckled with the dark violet staining of
individual leukocyte nuclei, suggesting that decon-densedDNAwas amajor clot component (Fig. 3A).Given that nuclear breakdown and the unfoldingof tightly packed chromatin is a hallmark ofNET formation (15), we stained the hematoxylin-positive clots for NET markers. We observed arobust stainingwith fluorescent double-strandedDNA–intercalating dyes and antibodies againstchromatin (fig. S3A). The colocalization of decon-densed chromatin with the neutrophil granule-derived enzyme myeloperoxidase, antimicrobialcathelicidin peptides, and the NET surrogate
Jiménez-Alcázar et al., Science 358, 1202–1206 (2017) 1 December 2017 2 of 5
1Institute of Clinical Chemistry and Laboratory Medicine,University Medical Center Hamburg-Eppendorf, Martinistraße52, 20246 Hamburg, Germany. 2Danylo Halytsky Lviv NationalMedical University, Pekarska Street 69, 79010 Lviv, Ukraine.3Health Care Center for Histology, Cytology, and MolecularDiagnostics, Max-Planck-Straße 5, 54296 Trier, Germany.4Group Practice for Pathology Wiesbaden, Ludwig-Erhard-Straße100, 65199 Wiesbaden, Germany. 5Department of MolecularMedicine and Surgery, Karolinska Institute and UniversityHospital, Solna L1:00, 17176 Stockholm, Sweden. 6Departmentof Intensive Care, University Medical Center Hamburg-Eppendorf, Martinistraße 52, 20246 Hamburg, Germany. 7III.Medical Clinic, University Medical Center Hamburg-Eppendorf,Martinistraße 52, 20246 Hamburg, Germany. 8ResearchInstitute for Biomedical Sciences, Tokyo University of Science,Yamazaki 2669, Noda, Chiba 278-0022, Japan. 9Department ofAnatomy and Molecular Embryology, Medical Faculty, Ruhr-University Bochum, Universitätsstraße 150, 44801 Bochum,Germany. 10Friedrich-Alexander Universität (FAU) Erlangen-Nürnberg, Department of Medicine 3, UniversitätsklinikumErlangen, Ulmenweg 18, 91054 Erlangen, Germany.*These authors contributed equally to this work.†Corresponding author. Email: [email protected]
BL W
T
BL
Csf
3/C
trl
Csf
3/D
1
Csf
3/D
1l3
0
5
10
15
20
Hem
oglo
bin
(g/d
l)
***
D1-/-D1l3-/-
0 2 4 6 8 10 12 14
100
75
50
25
0
Time (d)
Sur
viva
l rat
e (%
)
+ Csf3
***
WT-
D1-/-
D1l3-/-
D1-/-D1l3-/-
D1-/-D1l3-/-
+ Ctrl
24 16 8 0-8
-6
-4
-2
0
2
Time to Exitus (h)
Cha
nge
in P
erip
hera
l B
ody
Tem
pera
ture
(°C
)
D1-/-D1l3-/-
+ Csf3/Ctrl-
+ Csf3/D1-/-
+ Csf3/D1l3-
***B
L 3d 7d 14d
28d
Ctr
l0
20
40
60
CD
11b+ L
y6G
+ ce
lls (
103 /
l)
***
***
**
Csf3
A
+ Csf3/Ctrlz
+ Csf3/D1-/-
+ Csf3/D1l3-/-
0 2 4 6 8 10 12 14
100
75
50
25
0
Time (d)
Sur
viva
l rat
e (%
)
D1-/-D1l3-/-
***
BL W
T
BL
Csf
3/C
trl
Csf
3/D
1
Csf
3/D
1l3
0
100
200
300
400
500
Pla
sma
LDH
(U/l)
***
D1-/-D1l3-/-I
C
E F
G
Pla
sma
D1-/-D1l3-/-
BL 3d 7d 14d
Ctr
l0
5
10
15
20
NE
T-li
ke s
truct
ures
in
blo
od s
mea
r / F
OV
***
Csf3
DNA
Blo
od s
mea
r
CSF3
14d
Csf3
Ctrl
B
D
Urin
e
Csf
3/D
1l3
Csf
3/C
trl
Csf
3/D
1
BL W
T
BL
Csf
3/C
trl
Csf
3/D
1
Csf
3/D
1l3
0
10
20
30
Sch
isto
cyte
s / F
OV
D1-/-D1l3-/-
***H
Blo
od s
mea
r
D1-/-D1l3-/-
BL
Csf3/Ctrl
Fig. 2. DNase1 or DNase1L3 is required to tolerate chronic neutrophilia. Chronic neutrophilia wasinduced by injection of a G-CSF–expression plasmid (Csf3). Controls received an empty plasmid.(A) Blood neutrophil (CD11b+Ly6G+) count of WTmice expressing Csf3 for indicated times or Ctrl for14 days (N = 4 to 7). (B) NET-like structures (arrows) in DNA stainings of blood smears from WTmiceexpressing Csf3 for indicated times or Ctrl for 14 days (N = 5). Scale bars, 50 mm. (C) Survival ofWT (N = 7), D1–/– (N = 6), D1l3–/– (N = 6), and D1–/– D1l3–/– (N = 6) mice injected with Csf3 or Ctrl(N = 4). (D) Survival of D1–/– D1l3–/– mice coexpressing Csf3 with D1 (Csf3/D1, N = 5), D1l3(Csf3/D1l3, N = 6), or a control plasmid (Csf3/Ctrl, N = 4). (E to I) Characterization of mortalityduring chronic neutrophilia (N = 4). (E) Change in peripheral body temperature. (F) Photographsof plasma and urine. (G) Concentration of hemoglobin in blood. (H) Images and quantification ofschistocytes in blood smears. Arrows indicate schistocytes. Scale bars, 20 mm. (I) LDH concentration inplasma. U/l, units per liter; BL, baseline (N = 5 or 6); FOV, field of view. (B), (F), and (H) Images arerepresentative of four or five mice. (A), (B), and (G) to (I) one-way and (E) two-way ANOVA followedby Bonferroni’s multiple comparisons post hoc test; (C) and (D) log-rank test; **P < 0.01 and***P < 0.001 versus all other groups or BL.
RESEARCH | REPORTon M
arch 28, 2020
http://science.sciencemag.org/
Dow
nloaded from
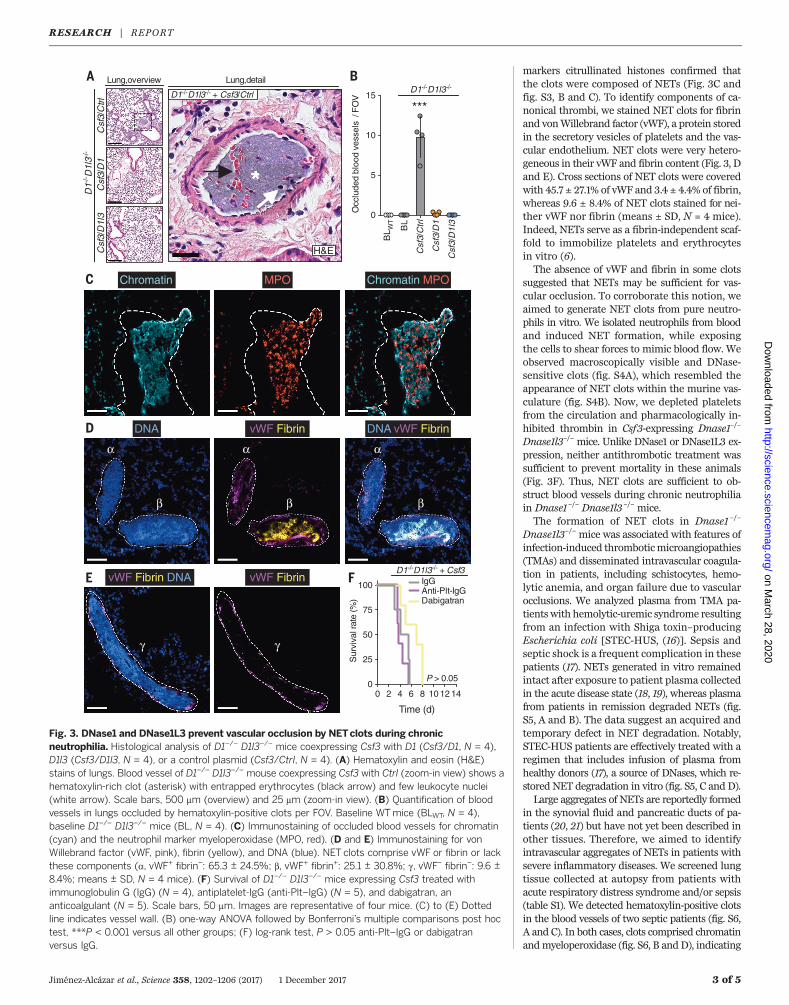
markers citrullinated histones confirmed thatthe clots were composed of NETs (Fig. 3C andfig. S3, B and C). To identify components of ca-nonical thrombi, we stained NET clots for fibrinand vonWillebrand factor (vWF), a protein storedin the secretory vesicles of platelets and the vas-cular endothelium. NET clots were very hetero-geneous in their vWF and fibrin content (Fig. 3, Dand E). Cross sections of NET clots were coveredwith 45.7 ± 27.1% of vWF and 3.4 ± 4.4% of fibrin,whereas 9.6 ± 8.4% of NET clots stained for nei-ther vWF nor fibrin (means ± SD, N = 4 mice).Indeed, NETs serve as a fibrin-independent scaf-fold to immobilize platelets and erythrocytesin vitro (6).The absence of vWF and fibrin in some clots
suggested that NETs may be sufficient for vas-cular occlusion. To corroborate this notion, weaimed to generate NET clots from pure neutro-phils in vitro. We isolated neutrophils from bloodand induced NET formation, while exposingthe cells to shear forces to mimic blood flow. Weobserved macroscopically visible and DNase-sensitive clots (fig. S4A), which resembled theappearance of NET clots within the murine vas-culature (fig. S4B). Now, we depleted plateletsfrom the circulation and pharmacologically in-hibited thrombin in Csf3-expressing Dnase1–/–
Dnase1l3–/– mice. Unlike DNase1 or DNase1L3 ex-pression, neither antithrombotic treatment wassufficient to prevent mortality in these animals(Fig. 3F). Thus, NET clots are sufficient to ob-struct blood vessels during chronic neutrophiliain Dnase1–/– Dnase1l3 –/– mice.The formation of NET clots in Dnase1 –/–
Dnase1l3–/– mice was associated with features ofinfection-induced thromboticmicroangiopathies(TMAs) and disseminated intravascular coagula-tion in patients, including schistocytes, hemo-lytic anemia, and organ failure due to vascularocclusions. We analyzed plasma from TMA pa-tients with hemolytic-uremic syndrome resultingfrom an infection with Shiga toxin–producingEscherichia coli [STEC-HUS, (16)]. Sepsis andseptic shock is a frequent complication in thesepatients (17). NETs generated in vitro remainedintact after exposure to patient plasma collectedin the acute disease state (18, 19), whereas plasmafrom patients in remission degraded NETs (fig.S5, A and B). The data suggest an acquired andtemporary defect in NET degradation. Notably,STEC-HUS patients are effectively treated with aregimen that includes infusion of plasma fromhealthy donors (17), a source of DNases, which re-stored NET degradation in vitro (fig. S5, C and D).Large aggregates of NETs are reportedly formed
in the synovial fluid and pancreatic ducts of pa-tients (20, 21) but have not yet been described inother tissues. Therefore, we aimed to identifyintravascular aggregates of NETs in patients withsevere inflammatory diseases. We screened lungtissue collected at autopsy from patients withacute respiratory distress syndrome and/or sepsis(table S1). We detected hematoxylin-positive clotsin the blood vessels of two septic patients (fig. S6,A and C). In both cases, clots comprised chromatinandmyeloperoxidase (fig. S6, B andD), indicating
Jiménez-Alcázar et al., Science 358, 1202–1206 (2017) 1 December 2017 3 of 5
0 2 4 6 8 10 12 14
100
75
50
25
0
Time (d)
Sur
viva
l rat
e (%
)
D1-/-D1l3-/- + Csf3IgGAnti-Plt-IgGDabigatran
P > 0.05
A
Csf
3/C
trlC
sf3/
D1
Csf
3/D
1l3
BL W
T
BL
Csf
3/C
trl
Csf
3/D
1
Csf
3/D
1l3
0
5
10
15
Occ
lude
d bl
ood
vess
els
/ F
OV
***D1-/-D1l3-/-3
D1-/-
D1l
3-/-
BLung,overview
D1-/-D1l3-/- + Csf3/Ctrl
H&E
Lung,detail
Chromatin/MPOChromatin MPOC
D DNA vWF/FibrinDNA vWF/Fibrin
FE vWF/Fibrin/DNA
β
α
γ
β
α
β
α
vWF/Fibrin
γ
*
Fig. 3. DNase1 and DNase1L3 prevent vascular occlusion by NETclots during chronicneutrophilia. Histological analysis of D1–/– D1l3–/– mice coexpressing Csf3 with D1 (Csf3/D1, N = 4),D1l3 (Csf3/D1l3, N = 4), or a control plasmid (Csf3/Ctrl, N = 4). (A) Hematoxylin and eosin (H&E)stains of lungs. Blood vessel of D1–/– D1l3–/– mouse coexpressing Csf3 with Ctrl (zoom-in view) shows ahematoxylin-rich clot (asterisk) with entrapped erythrocytes (black arrow) and few leukocyte nuclei(white arrow). Scale bars, 500 mm (overview) and 25 mm (zoom-in view). (B) Quantification of bloodvessels in lungs occluded by hematoxylin-positive clots per FOV. Baseline WTmice (BLWT, N = 4),baseline D1–/– D1l3–/– mice (BL, N = 4). (C) Immunostaining of occluded blood vessels for chromatin(cyan) and the neutrophil marker myeloperoxidase (MPO, red). (D and E) Immunostaining for vonWillebrand factor (vWF, pink), fibrin (yellow), and DNA (blue). NET clots comprise vWF or fibrin or lackthese components (a, vWF+ fibrin–: 65.3 ± 24.5%; b, vWF+ fibrin+: 25.1 ± 30.8%; g, vWF– fibrin–: 9.6 ±8.4%; means ± SD, N = 4 mice). (F) Survival of D1–/– D1l3–/– mice expressing Csf3 treated withimmunoglobulin G (IgG) (N = 4), antiplatelet-IgG (anti-Plt–IgG) (N = 5), and dabigatran, ananticoalgulant (N = 5). Scale bars, 50 mm. Images are representative of four mice. (C) to (E) Dottedline indicates vessel wall. (B) one-way ANOVA followed by Bonferroni’s multiple comparisons post hoctest, ***P < 0.001 versus all other groups; (F) log-rank test, P > 0.05 anti-Plt–IgG or dabigatranversus IgG.
RESEARCH | REPORTon M
arch 28, 2020
http://science.sciencemag.org/
Dow
nloaded from
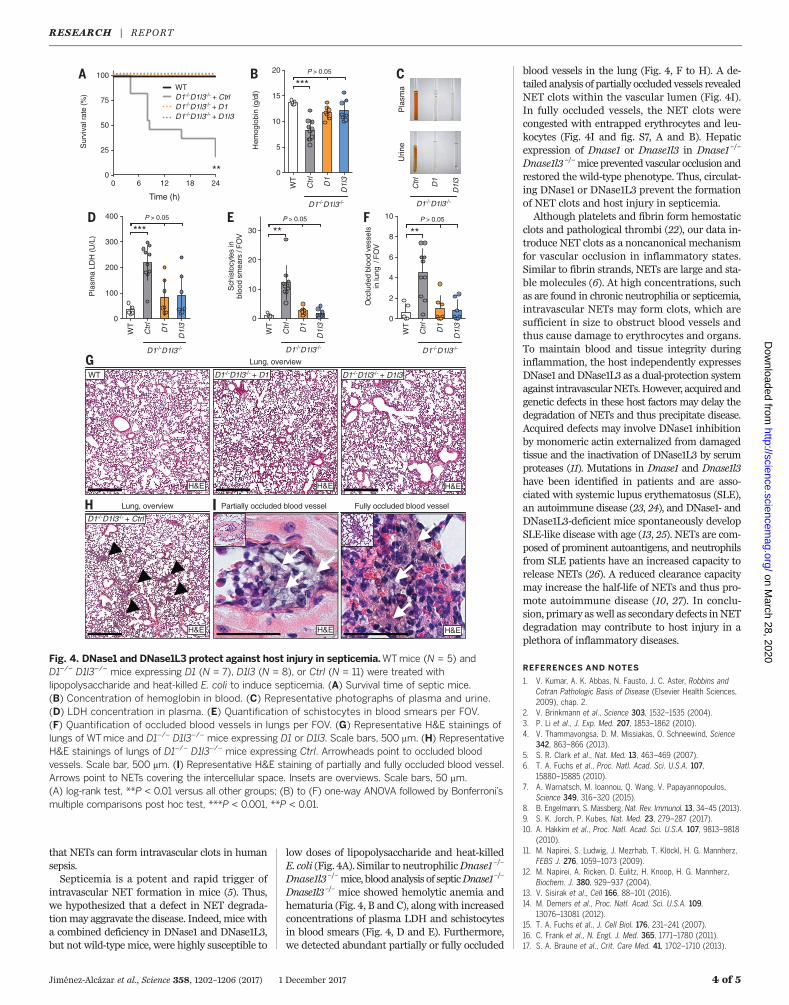
that NETs can form intravascular clots in humansepsis.Septicemia is a potent and rapid trigger of
intravascular NET formation in mice (5). Thus,we hypothesized that a defect in NET degrada-tionmay aggravate the disease. Indeed, mice witha combined deficiency in DNase1 and DNase1L3,but not wild-type mice, were highly susceptible to
low doses of lipopolysaccharide and heat-killedE. coli (Fig. 4A). Similar to neutrophilicDnase1–/–
Dnase1l3–/–mice, bloodanalysis of septicDnase1–/–
Dnase1l3–/– mice showed hemolytic anemia andhematuria (Fig. 4, B and C), along with increasedconcentrations of plasma LDH and schistocytesin blood smears (Fig. 4, D and E). Furthermore,we detected abundant partially or fully occluded
blood vessels in the lung (Fig. 4, F to H). A de-tailed analysis of partially occluded vessels revealedNET clots within the vascular lumen (Fig. 4I).In fully occluded vessels, the NET clots werecongested with entrapped erythrocytes and leu-kocytes (Fig. 4I and fig. S7, A and B). Hepaticexpression of Dnase1 or Dnase1l3 in Dnase1–/–
Dnase1l3–/–mice prevented vascular occlusion andrestored the wild-type phenotype. Thus, circulat-ing DNase1 or DNase1L3 prevent the formationof NET clots and host injury in septicemia.Although platelets and fibrin form hemostatic
clots and pathological thrombi (22), our data in-troduce NET clots as a noncanonical mechanismfor vascular occlusion in inflammatory states.Similar to fibrin strands, NETs are large and sta-ble molecules (6). At high concentrations, suchas are found in chronic neutrophilia or septicemia,intravascular NETs may form clots, which aresufficient in size to obstruct blood vessels andthus cause damage to erythrocytes and organs.To maintain blood and tissue integrity duringinflammation, the host independently expressesDNase1 and DNase1L3 as a dual-protection systemagainst intravascularNETs.However, acquired andgenetic defects in these host factors may delay thedegradation of NETs and thus precipitate disease.Acquired defects may involve DNase1 inhibitionby monomeric actin externalized from damagedtissue and the inactivation of DNase1L3 by serumproteases (11). Mutations in Dnase1 and Dnase1l3have been identified in patients and are asso-ciated with systemic lupus erythematosus (SLE),an autoimmune disease (23, 24), and DNase1- andDNase1L3-deficient mice spontaneously developSLE-like disease with age (13, 25). NETs are com-posed of prominent autoantigens, and neutrophilsfrom SLE patients have an increased capacity torelease NETs (26). A reduced clearance capacitymay increase the half-life of NETs and thus pro-mote autoimmune disease (10, 27). In conclu-sion, primary as well as secondary defects inNETdegradation may contribute to host injury in aplethora of inflammatory diseases.
REFERENCES AND NOTES
1. V. Kumar, A. K. Abbas, N. Fausto, J. C. Aster, Robbins andCotran Pathologic Basis of Disease (Elsevier Health Sciences,2009), chap. 2.
2. V. Brinkmann et al., Science 303, 1532–1535 (2004).3. P. Li et al., J. Exp. Med. 207, 1853–1862 (2010).4. V. Thammavongsa, D. M. Missiakas, O. Schneewind, Science
342, 863–866 (2013).5. S. R. Clark et al., Nat. Med. 13, 463–469 (2007).6. T. A. Fuchs et al., Proc. Natl. Acad. Sci. U.S.A. 107,
15880–15885 (2010).7. A. Warnatsch, M. Ioannou, Q. Wang, V. Papayannopoulos,
Science 349, 316–320 (2015).8. B. Engelmann, S. Massberg, Nat. Rev. Immunol. 13, 34–45 (2013).9. S. K. Jorch, P. Kubes, Nat. Med. 23, 279–287 (2017).10. A. Hakkim et al., Proc. Natl. Acad. Sci. U.S.A. 107, 9813–9818
(2010).11. M. Napirei, S. Ludwig, J. Mezrhab, T. Klöckl, H. G. Mannherz,
FEBS J. 276, 1059–1073 (2009).12. M. Napirei, A. Ricken, D. Eulitz, H. Knoop, H. G. Mannherz,
Biochem. J. 380, 929–937 (2004).13. V. Sisirak et al., Cell 166, 88–101 (2016).14. M. Demers et al., Proc. Natl. Acad. Sci. U.S.A. 109,
13076–13081 (2012).15. T. A. Fuchs et al., J. Cell Biol. 176, 231–241 (2007).16. C. Frank et al., N. Engl. J. Med. 365, 1771–1780 (2011).17. S. A. Braune et al., Crit. Care Med. 41, 1702–1710 (2013).
Jiménez-Alcázar et al., Science 358, 1202–1206 (2017) 1 December 2017 4 of 5
WT
Ctr
l
D1
D1l
3
0
5
10
15
20
Hem
oglo
bin
(g/d
l)
***P > 0.05
D1-/-D1l3-/-
WT
Ctr
l
D1
D1l
3
0
2
4
6
8
10
Occ
lude
d bl
ood
vess
els
in lu
ng /
FO
V
**P > 0.05
D1-/-D1l3-/-
D1-/-D1l3-/- + D1 D1-/-D1l3-/- + D1l3WT
D1-/-D1l3-/- + Ctrl
Partially occluded blood vessel
C
Pla
sma
D1-/-D1l3-/-
D
Urin
e
D1l
3
Ctr
l
D1
BWTD1-/-D1l3-/- + Ctrl D1-/-D1l3-/- + D1D1-/-D1l3-/- + D1l3
0 6 12 18 24
100
75
50
25
0
Time (h)
Sur
viva
l rat
e (%
)
**
A
G
WT
Ctr
l
D1
D1l
30
10
20
30S
chis
tocy
tes
in
bloo
d sm
ears
/ F
OV **
P > 0.05
D1-/-D1l3-/-
E
H
WT
Ctr
l
D1
D1l
3
0
100
200
300
400
Pla
sma
LDH
(U/L
)
***P > 0.05
D1-/-D1l3-/-
F
Fully occluded blood vessel
Lung, overview
Lung, overview
H&EH&E H&E
H&EH&E H&E
I
Fig. 4. DNase1 and DNase1L3 protect against host injury in septicemia.WTmice (N = 5) andD1–/– D1l3–/– mice expressing D1 (N = 7), D1l3 (N = 8), or Ctrl (N = 11) were treated withlipopolysaccharide and heat-killed E. coli to induce septicemia. (A) Survival time of septic mice.(B) Concentration of hemoglobin in blood. (C) Representative photographs of plasma and urine.(D) LDH concentration in plasma. (E) Quantification of schistocytes in blood smears per FOV.(F) Quantification of occluded blood vessels in lungs per FOV. (G) Representative H&E stainings oflungs of WTmice and D1–/– D1l3–/– mice expressing D1 or D1l3. Scale bars, 500 mm. (H) RepresentativeH&E stainings of lungs of D1–/– D1l3–/– mice expressing Ctrl. Arrowheads point to occluded bloodvessels. Scale bar, 500 mm. (I) Representative H&E staining of partially and fully occluded blood vessel.Arrows point to NETs covering the intercellular space. Insets are overviews. Scale bars, 50 mm.(A) log-rank test, **P < 0.01 versus all other groups; (B) to (F) one-way ANOVA followed by Bonferroni’smultiple comparisons post hoc test, ***P < 0.001, **P < 0.01.
RESEARCH | REPORTon M
arch 28, 2020
http://science.sciencemag.org/
Dow
nloaded from

18. M. Jiménez-Alcázar et al., J. Thromb. Haemost. 13, 732–742(2015).
19. J. Leffler et al., J. Innate Immun. 9, 12–21 (2017).20. M. Leppkes et al., Nat. Commun. 7, 10973 (2016).21. C. Schauer et al., Nat. Med. 20, 511–517 (2014).22. V. Kumar, A. K. Abbas, N. Fausto, J. C. Aster, Robbins and
Cotran Pathologic Basis of Disease (Elsevier Health Sciences,2009), chap. 4.
23. S. M. Al-Mayouf et al., Nat. Genet. 43, 1186–1188 (2011).24. K. Yasutomo et al., Nat. Genet. 28, 313–314 (2001).25. M. Napirei et al., Nat. Genet. 25, 177–181 (2000).26. S. Gupta, M. J. Kaplan, Nat. Rev. Nephrol. 12, 402–413
(2016).27. R. Mizuta et al., PLOS ONE 8, e80223 (2013).
ACKNOWLEDGMENTS
We thank A.V. Failla of the Microscope Core Facility,K. Hartmann of the Mouse Pathology Core Facility of the UniversityMedical Center Hamburg-Eppendorf, and V. Vovk of the LvivNational Medical University for their support. This study wassupported in part by the German Research Society (KFO 306, FU742/4-1; SFB 841, INST 152/621-1; SFB 877, INST 257/433-1; SFB1192, INST 152/692-1, INST 152/686-1), the Stiftung fürPathobiochemie und Molekulare Diagnostik of the German Societyfor Clinical Chemistry and Laboratory Medicine, the FoRUM-program of the Ruhr-University Bochum (F505-2006),Hjärt Lungfonden (20110500), Vetenskapsrådet (K2013-65X-21462-04-5), and the European Research Council (ERC-StG-2012-311575_F-12, PIIF-GA-2013-628264). M.H. acknowledges generous
support by Ardea Biosciences, Inc. The data are contained inthe manuscript and the supplementary materials.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/358/6367/1202/suppl/DC1Materials and MethodsSupplementary TextFigs. S1 to S9Table S1References (28–30)Movies S1 and S2
20 March 2017; resubmitted 10 July 2017Accepted 11 October 201710.1126/science.aam8897
Jiménez-Alcázar et al., Science 358, 1202–1206 (2017) 1 December 2017 5 of 5
RESEARCH | REPORTon M
arch 28, 2020
http://science.sciencemag.org/
Dow
nloaded from

Host DNases prevent vascular occlusion by neutrophil extracellular traps
Mannherz, Daisuke Kitamura, Martin Herrmann, Markus Napirei and Tobias A. FuchsRostyslav Bilyy, Veit Krenn, Christoph Renné, Thomas Renné, Stefan Kluge, Ulf Panzer, Ryushin Mizuta, Hans Georg Miguel Jiménez-Alcázar, Chandini Rangaswamy, Rachita Panda, Josephine Bitterling, Yashin J. Simsek, Andy T. Long,
DOI: 10.1126/science.aam8897 (6367), 1202-1206.358Science
, this issue p. 1202; see also p. 1126Sciencewhen these DNases were absent.vessels were occluded by NET clots. Furthermore, the damage unleashed by clots during septicemia was enhanced Gunzer). Knockout mice lacking these enzymes were unable to tolerate chronic neutrophilia, quickly dying after bloodDNASE1 and DNASE1L3, have partially redundant roles in degrading NETs in the circulation (see the Perspective by
report that two deoxyribonucleases (DNases),et al.good by promoting inflammation and thrombosis. Jiménez-Alcázar cytoplasmic proteins that trap and neutralize microbes. However, their inappropriate release may do more harm than
Neutrophil extracellular traps (NETs) are lattices of processed chromatin decorated with select secreted andBlood DNases hack the NET
ARTICLE TOOLS http://science.sciencemag.org/content/358/6367/1202
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2017/12/01/358.6367.1202.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/4/157/157ra141.fullhttp://stm.sciencemag.org/content/scitransmed/5/178/178ra40.fullhttp://stm.sciencemag.org/content/scitransmed/8/361/361ra138.fullhttp://stm.sciencemag.org/content/scitransmed/8/369/369ra176.fullhttp://science.sciencemag.org/content/sci/358/6367/1126.full
REFERENCES
http://science.sciencemag.org/content/358/6367/1202#BIBLThis article cites 28 articles, 10 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.ScienceScience, 1200 New York Avenue NW, Washington, DC 20005. The title (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
Science. No claim to original U.S. Government WorksCopyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of
on March 28, 2020
http://science.sciencem
ag.org/D
ownloaded from