Embed Size (px)
Citation preview


Imprime MICHELENA ARTES GRÁFICAS S.L. Astigarraga (Gipuzkoa) http://www.michag.es

ORGANIZADOR Sociedad Española de Química Analítica (SEQA) COMITÉ LOCAL Grupo de Química Analítica Departamento de Química Aplicada Facultad de Química de San Sebastián Universidad del País Vasco / Euskal Herriko Unibertsitatea Carlos Ubide (Presidente) Miren Ostra Esmeralda Millán (Secretaria) Juan Zuriarrain Gloria del Campo Ainara Barriola Iñaki Berregi Maider Vidal Rosa Garcia Ane Bordagaray COMITÉ CIENTÍFICO Carmen Cámara (Presidenta) (UCM) José M. Vadillo (UMA) Yolanda Madrid (Secretaria) (UCM) Arántzazu Narváez (Tragsa) Manuel Silva (UCO) M. Teresa Galcerán (UB) Darío Prada (UDC) Enrique Barrado (UVA) Vicente Ferreira (UNIZAR) Carlos Ubide (UPV/EHU) Alfredo Sanz-Medel (UNIOVI) PATROCINADORES Universidad del País Vasco / Euskal Herriko Unibertsitatea (UPV/EHU) Donostia-Gipuzkoa Kutxa / Caja Gipuzkoa-San Sebastián Uda Ikastaroak / Cursos de Verano (UPV/EHU) Eusko Jaurlaritza / Gobierno Vasco Ministerio de Ciencia e Innovación (Gobierno de España) EMPRESAS COLABORADORAS Agilent Technologies Air Liquide Camo A/S Fisher Scientific-Afora Ocean Optics LQC s.l. Perkin Elmer

PROGRAMA DOMINGO 19 DE JULIO (PALACIO DE MIRAMAR) 18:00-20:00 Entrega de documentación
20:00 Recepción de bienvenida LUNES 20 DE JULIO (PALACIO DE MIRAMAR) 8:30-9:00 Entrega de documentación
9:00-9:30 Apertura oficial Sesión: Química Analítica en Ciencias de la Vida y Alimentación
Moderadores: Santiago Maspoch (UAB) y Jorge Ruiz Encinar (UNIOVI)
9:30-10:15 Conferencia Plenaria (CP-1)
Bio-chemometrics: multivariate data modelling in systems biology
HARALD MARTENS, Achim Kohler
Nofima, Centre for Biospectroscopy and Data Modelling, and CIGENE, IMT, Norwegian U. of Life Sciences, Aas Norway.
Moderadores: Javier Galbán (UNIZAR) y Olatz Zuloaga (UPV/EHU)
10:15-10:45 Conferencia específica (CE-1)
Alimentos funcionales. Lípidos. Retos analíticos MANUELA JUÁREZ, M.A. de la Fuente
Instituto del Frío, CSIC
10:45-11:15 Café y carteles
Moderadores: Antonio Molina (UJAEN) y Riansares Muñoz (UCM)
11:15-11:45 Conferencia específica (CE-2)
Retos analíticos en la seguridad alimentaria ANA Mª TRONCOSO
Agencia Española de Seguridad Alimentaria y Nutrición
Moderadores: Alfredo Sanz-Medel (UNIOVI) y Vicente Ferreira (UNIZAR)
11:45-12:45 Comunicaciones flash (CF-2 a CF-7, CFD-15 y CFD-16)
12:45-14:15 Sesión de carteles 1
14:15-16:00 Comida Sesión: Docencia y Sociedad
Moderadores: Senén Durand (UNED) y María Dolores Marazuela (UCM)
16:00-16:30 Conferencia específica (CE-3)
La Química Analítica en los estudios de Grado de Química BARTOLOMÉ SIMONET, Miguel Valcárcel, M. Silva
Universidad de Córdoba

16:30-17:00 Conferencia específica (CE-4) El título de Máster en la nueva organización de las enseñanzas
universitarias MANUEL SILVA
Universidad de Córdoba
17:00-17:30 Conferencia específica (CE-5)
La Química Analítica española a la luz de la bibliometría. ¿Zidanes o Pavones?
JOSÉ M. VADILLO
Universidad de Málaga
17:30-18:30 Café y carteles
18:30-20:00 Asamblea de la SEQA
MARTES 21 DE JULIO (PALACIO DE MIRAMAR) Sesión: Medio ambiente, cambio climático y energías renovables
Moderadores: Mercedes Gallego (UCO) y Antonio Moreda (USC)
9:30-10:15 Conferencia plenaria (CP-2)
Los usos de la energía y el cambio climático a lo largo de la historia humana
JOAN GRIMALT
CSIC
Moderadores: José Juan Santana (ULPGC) y Ruth María Fernández (US)
10:15-10:45 Conferencia específica (CE-6)
Presencia de contaminantes minoritarios y emergentes en aguas superficiales y subterráneas
LUCILA CANDELA
Universidad Politécnica de Cataluña 10:45-11:15 Café y carteles
Moderadores: Marisol Larrechi (URV) y Ana Sayago (UHU)
11:15-11:45 Conferencia específica (CE-7)
Disruptores endocrinos. Su presencia en el medio y sus efectos NICOLÁS OLEA
Hospital Universitario, Universidad de Granada 11:45-12:15 Conferencia específica (CE-8)
Chemical imaging. From RGB to hyperspectral. Features, benefits and drawbacks
JOSÉ MANUEL AMIGO
Universidad de Copenhague
Moderadores: Darío Prada (UDC) y Maite Galcerán (UB)
12:15-13:15 Comunicaciones flash (CF-8 a CF-14, CF-1)
13:15-14:15 Sesión de carteles 2 (alternativa a la siguiente)
13:15-14:15 Reunión del Grupo de Especiación (alternativa a la anterior)
14:15-16:30 Comida (incluyendo Sesión de clausura)
16:30 Visita a Chillida Leku (Autobuses desde el Hotel Costa Vasca)
21:00 Cena de la reunión (Cofradía Vasca de Gastronomía)


ÍNDICE CONFERENCIAS PLENARIAS .............................................................9
CP-1 y CP-2
CONFERENCIAS ESPECÍFICAS ........................................................13
CE-1 a CE-8
COMUNICACIONES FLASH .............................................................23
CF-1 a CF-14
CFD-15 y CFD-16
COMUNICACIONES EN CARTEL.......................................................41
C1-1 a C1-74
C1-166
C2-75 a C2-79
C2-82
C2-84 a C2-150
COMUNICACIONES DE DOCENCIA EN CARTEL ............................... 189
CD1-151 a CD1-158
CD2-159 a CD2-161
CD2-163 a CD2-165
ÍNDICE DE AUTORES .................................................................. 205
LISTA DE ASISTENTES ................................................................ 213


CONFERENCIAS PLENARIAS

CP-1 CONFERENCIAS PLENARIAS
10
BIO-CHEMOMETRICS: MULTIVARIATE DATA MODELLING IN SYSTEMS
BIOLOGY
Harald Martens; Achim Kohler Nofima, Centre for Biospectroscopy and Data Modelling, Ås, Norway and CIGENE,
IMT, Norwegian University of Life Sciences, Ås, Norway E-mail: [email protected], [email protected]
Combining “-omics” and “-metrics”: The rapid developments in systems biology
and in the different “-omics” fields reflect that bio-scientist want overview of real-life complexity, not only studies of individual mechanisms in model systems. Thus, modern biology creates lots of data big tables - some of them of the traditional “long-thin” type (n >>p) and others of the “short-fat” type (p>>n). How can the various data tables be interpreted [1] - and by whom [2]? Some novel applications of bio-chemometrics in systems biology and functional genomics will here be outlined.
(1) From bio-spectroscopy via genetics to genomics: Low-cost high-throughput
FTIR bio-spectroscopy, calibrated for fatty acid composition by PLSR, was applied to millions of milk samples from individual animals at various times (Kohler et al 2008, in prep). Preliminary analysis shows that when the predictions were linked to pedigree etc by BLUP, clear heritabilities were found. When daughter-averages for about 2000 breeding bulls (sires) were related to 20 000 SNPs (Affymetrix) by QTL analysis, several fatty-acid related regions of the DNA genome were indicated (Lien et al. 2008, in prep).
(2) A difficult mathematical model explored statistically: A high-dimensional
non-linear dynamic model concerning pattern formation during cell differentiation was studied by super-computer simulations according to fractional factorial designs. The obtained patterns were described by computerized image analysis and human sensory analysis, and the results explored by chemometrics, giving overview as well as detailed interpretation. Several unexpected patterning systems were discovered (Martens H. et al 2008, in prep).
(3) From time series data via soft multivariate regression to mechanistic non-
linear differential models: A new approach is outlined for the tentative identification of potential mechanisms that could have caused the dynamics of state variables observed in a system. The approach combines inductive/explorative multivariate techniques from chemometrics and psychometrics with deductive/confirmative planning-and validation techniques from computational statistics plus mechanistic non-linear dynamics from systems biology [2]. [1] Faergestad E M, Sæbø S, Langsrud Ø, Høy M, Kohler A, Liland K H, Hollung K,
Almergren J, Anderssen E, Martens H, (2008) Analysis of megavariate data in functional genomics, in: Comprehensive Chemometrics, Walczak B, Tauler R, Ferré J, Brown S. (eds), Elsevier, in press.
[2] Martens, H. and Kohler, A. (2008) Mathematics and measurements for high-throughput quantitative biology. Submitted to Biological Theory, Special issue on Quantifying Biology.
Key words and phrases: Chemometrics, multivariate calibration, PLS, rank-reduced regression, nominal-level, non-linear dynamic modeling, high-dimensional time series analysis, FTIR, bio-spectroscopy, sensory analysis of non-linear dynamic model behaviour.

CONFERENCIAS PLENARIAS CP-2
11
LOS CAMBIOS CLIMÁTICOS ABRUPTOS, UNA INCÓGNITA MÁS DE LA
EVOLUCIÓN DE NUESTRO PLANETA
Joan O. Grimalt
Consejo Superior de Investigaciones Científicas. Barcelona [email protected]
Los cambios abruptos son transiciones climáticas que han dado lugar a
modificaciones muy importantes de las condiciones climáticas en zonas muy amplias de nuestro planeta, como por ejemplo todo el hemisferio norte.
Los cambios climáticos regulares han dado lugar a los periodos glaciares e interglaciares del pasado. Son debidos a las variaciones de insolación que experimenta la Tierra debido a cambios pequeños de trayectoria o de inclinación del eje de rotación por influencia de otros astros presentes en el sistema planetario, fundamentalmente Júpiter y la Luna. Como es conocido, la trayectoria orbital de la Tierra es elíptica. La excentricidad puede ser más o menos acusada, aproximándose a una circunferencia en el segundo caso. Como consecuencia de este carácter elíptico nuestro planeta a veces se encuentra más cerca o más lejos del sol, siendo el perihelo y el afelio los momentos que corresponden a mayor proximidad o lejanía, respectivamente. Los cambios de excentricidad son cíclicos y siguen periodos de 100,000 y 400,000 años. Otra característica orbital de la tierra es la oblicuidad. Como sabemos, la Tierra gira sobre si misma respecto a un eje que está inclinado con referencia al plano de traslación. La inclinación es variable, entre 21.5º y 24.5º, en la actualidad 23.5º. Los cambios de inclinación dan lugar a variaciones estacionales más o menos marcadas, y varían siguiendo unos periodos de 41,000 años. Otro proceso orbital importante es la precesión, que resulta de la combinación de la excentricidad y los cambios estacionales. El perihelio y el afelio, actualmente 147 y 152 millones de kilómetros, respectivamente, tienen una relevancia diferente si ocurren en invierno o en verano. Los cambios de precesión siguen unos periodos de unos 19,000, 22,000 y 24,000 años.
Los cambios abruptos son aquellos que ocurren en intervalos menores a estos ciclos, usualmente comprenden periodos de 1,000 a 5,000 años. Dichos cambios suponen intervalos de tiempo menores que los orbitales pero aún así se trata de periodos climáticos muy largos si se contemplan a escala humana. Hay que destacar que su mayor brevedad relativa no implica que su intensidad sea menor en comparación con las variaciones climáticas asociadas a las transiciones orbitales. Los cambios abruptos pueden ser de menor, semejante o mayor intensidad que los cambios entre épocas glaciares e interglaciares.
Dichos cambios abruptos fueron algo muy frecuente en el último período glaciar, entre los últimos 20,000 y 70,000 años. Existieron unos seis episodios de fuertes fríos y duración del orden de 5,000 años en los que se produjo una fusión muy importante de hielos provenientes de icebergs en toda una franja entre 40 y 55ºN del Océano Atlántico. Durante ellos las aguas del Atlántico alcanzaron las temperaturas más bajas características de la época glaciar. Otro tipo de fenómenos abruptos que también se produjeron en esta época glaciar tuvieron una duración más corta (en el orden de 1,000-2,000 años, a veces 250 años) y no fueron acompañados de procesos masivos de fusión de hielo. A pesar de su duración menor, las caídas de temperatura que los caracterizaron son a menudo de la misma intensidad que los episodios descritos anteriormente.
En esta presentación se describirán estos cambios y se mostrará como la química analítica ha contribuido a su estudio.


CONFERENCIAS ESPECÍFICAS

CE-1 CONFERENCIAS ESPECÍFICAS
14
ALIMENTOS FUNCIONALES. LÍPIDOS. RETOS ANALÍTICOS
M. Juárez y M.A. de la Fuente
Dpto. Productos Lácteos. Instituto del Frío (CSIC), Madrid.
E-mail: [email protected]
Desde hace décadas se reconocen constituyentes de los alimentos como ingredientes de interés para la salud: componentes derivados de las proteínas, lípidos, oligosacáridos, minerales, vitaminas y antioxidantes. Dentro de los lípidos funcionales, destacan por su interés y desarrollo en productos comercializados los esteroles vegetales, los ácidos grasos omega-3 y en menor medida el ácido linoleico conjugado (CLA).
En cuanto a los alimentos que llevan incorporado β-sitosterol los avances en el análisis de la fracción esterólica han estado sobre todo dirigidos a simplificar las etapas para lograr el aislamiento de esa fracción previo al análisis por cromatogra-fía de gases (GC), incluida la combinación con cromatografía de líquidos (HPLC).
La trazabilidad de los ácidos grasos omega-3 en los alimentos funcionales comercializados, puede resolverse por GC, utilizando la columna adecuada y de suficiente longitud, aunque los tiempos de análisis son largos.
Respecto al estudio del CLA en alimentos, por GC es posible cuantificar los isómeros mayoritarios. Sin embargo, la determinación del perfil de hasta los 24 isómeros posicionales y geométricos potencialmente presentes necesariamente requiere la utilización de dos técnicas cromatográficas (GC y HPLC) en combinación con espectrometría de masas (MS). Actualmente se comercializan derivados lácteos que incluyen una mezcla de los isómeros cis-9 trans-11 y trans-10 cis-12 C18:2, en proporción comparable, obtenida a partir de aceite de cártamo. La detección y cuantificación de estos dos isómeros es viable tanto por GC, como por HPLC.
La grasa de leche tiene ácidos grasos trans-monoinsaturados, cuyo perfil isomérico es diferente del que presentan las grasas hidrogenadas, con el isómero trans-11 C18:1 (ácido vacénico, VA) -precursor fisiológico del CLA- como mayoritario. La suplementación de la dieta del ganado con semillas o aceites vegetales ricos en ácidos grasos poliinsaturados ha dado lugar a incrementos en los niveles de CLA y VA, además de cambios en el perfil de ácidos grasos hacia una composición más beneficiosa para la salud humana. Esta práctica es una alternativa para incrementar la funcionalidad de la grasa de la leche y ya se comercializan productos con la composición en ácidos grasos modificada en esa línea.
La determinación del perfil de CLA, así como el de los ácidos trans-monoinsaturados es imprescindible ya que los distintos isómeros pueden tener distintas funciones biológicas. La separación de los trans- C18:1 es factible utilizando previo a la GC, otra técnica de fraccionamiento como la cromatografía en capa fina, la HPLC o la extracción en fase sólida con ión plata [1].
La GC y GC-MS de los ésteres metílicos, permiten cuantificar e identificar los isómeros mayoritarios del CLA. Un análisis de la posición de los dobles enlaces se puede realizar parcialmente usando GC-MS de otros derivados de los ácidos grasos como dimetiloxazolina. La GC-FTIR puede también contribuir a confirmar la geometría (cis o trans) de los dobles enlaces, aunque no permite discriminar entre configuración cis-trans y trans-cis. La espectrometría de masas en tandem, con ionización química de acetonitrilo, ha logrado avances destacados en la identificación de la estructura molecular de isómeros CLA.
La Ag+-HPLC con columnas en serie y detector UV juega un papel destacado en la separación de los isómeros individuales de CLA, principalmente los trans-trans [2].
[1] P. Luna, V. Rodríguez-Pino, M.A. de la Fuente. Food Chem. (en prensa, 2009) [2] P. Luna, J. Fontecha, M. Juárez, J. de la Fuente. Dairy Res. 72, 415-424 (2005)

CONFERENCIAS ESPECÍFICAS CE-2
15
RETOS ANALÍTICOS EN LA SEGURIDAD ALIMENTARIA
Ana Mª Troncoso
Agencia Española de Seguridad Alimentaria y Nutrición
E-mail: [email protected]
La Seguridad Alimentaria está diseñada partiendo de los estudios toxicológicos de los componentes o contaminantes presentes o potencialmente presentes en los alimentos, y es exigida mediante las correspondientes legislaciones. No obstante, existe un condicionante sustancial para una aplicación adecuada de la Seguridad Alimentaria: Disponer de métodos de análisis adecuados para poder constatar el cumplimiento de las legislaciones.
Los métodos analíticos de control tienen un reto esencial: poder determinar inequívocamente dentro del intervalo del 95% de seguridad, la sustancia o substancias que se investigan a los límites que los estudios toxicológicos exigen. Desgraciadamente, no siempre las exigencias toxicológicas están en concordancia con la sensibilidad analítica. El gran campo de actividad de la química analítica es poder proporcionar métodos fiables con la sensibilidad requerida para las sustancias con mayor riesgo toxicológico, ya que precisamente son las que tienen unos niveles permitidos menores. Los aspectos mas significativos donde se puede incidir en esta actividad para mejorar la Seguridad Alimentaria, como se ha puesto de manifiesto las investigaciones de los últimos años, son:
• Componentes de alimentos con significación toxicológica, en concentraciones muy variables: Acrilamida y Furano en alimentos procesados.
• Impurezas de aditivos alimentarios con riesgo toxicológico • Componentes de materiales procedentes de los procesos de fabricación o contaminantes de materiales reciclados
• Residuos de medicamentos veterinarios • Toxinas
El campo de actuación es muy amplio, ya que gracias a sensibilidad y selectividad de las nuevas técnicas analíticas, ha sido posible poner de manifiesto nuevos componentes, unas veces por su búsqueda directa y otras por la elucidación sustancias interferentes en procesos rutinarios de análisis de control. Como ejemplos mas recientes de manifiesta necesidad de métodos analíticos en el campo de la seguridad alimentaria se exponen los siguientes:
• Impurezas de aditivos alimentarios en los alimentos o en los propios aditivos en los procesos de la comprobación de la Normas de Identidad y Pureza
• Nuevos Componentes minoritarios de las tintas de imprimir los envases ya que estas tienen una compleja formulación no sólo de colorantes, sino de barnices de aplicación plastificantes
• Productos minoritarios de los alimentos originados en sus procesos culinarios • Productos de reacciones secundarias de la obtención de polímeros plásticos, barnices y pinturas, siliconas, papel y cartón, etc.
• Integrantes de alimentos “exóticos” (dietas orientales) con riesgo toxicológico • Diferenciación química de plantas con “actividad funcional” por los riesgos toxicológicos implícitos
• Principios “activos funcionales” de plantas utilizadas como complementos alimenticios
• Aspectos contemplados en la página de Seguridad Alimentaria de la UE, http://ec.europa.eu/food/food/chemicalsafety.

CE-3 CONFERENCIAS ESPECÍFICAS
16
LA QUÍMICA ANALÍTICA EN LOS PLANES DE ESTUDIO
DEL GRADO DE QUÍMICA
M. Valcárcel, B.M. Simonet, M. Silva
Departamento de Química Analítica. Edificio Marie Curie Anexo. Campus de Rabanales. Universidad de Córdoba. 14071 Córdoba.
E-mail: [email protected]
Después de una década de preparación del sistema universitario español para su Convergencia con Europa en el marco de la Declaración de Bolonia, la sensación de fracaso y desánimo es bastante común. Como botones de muestra, pueden mencionarse el reparto de créditos entre áreas/departamentos como criterio fundamental de las comisiones de Planes de Estudio, así como el planteamiento burocrático y superficial de las competencias estudiantiles por parte de las mismas. Es curioso indicar que se las autoridades políticas han mezclado institucionalmente el cumplimiento de los hitos de la Declaración de Bolonia de 1999 (objetivo básico) con la renovación de las metodologías docentes (objetivo devenido).
En esta comunicación se pretende resumir las actividades de la Red de
Docencia de la SEQA en los últimos años, y fundamentalmente los logros alcanzados. En este contexto, cabe indicar que desde la red de docencia se han elaborado los siguientes documentos:
- Situación actual de la implementación de las nuevas Titulaciones de Grado en Química;
- Resultados previstos en el Grado: indicadores; - Justificación del Título: referencias externas; - Unidad de Garantía de Calidad de las Titulaciones; - Evaluación de las competencias estudiantiles del Grado de Química; y - Posibles materias y contenidos mínimos de Química Analítica para impartir en el
Grado de Química.
Por otra parte, se analizará la situación de la Química Analítica en los Planes de Estudio de las universidades españolas que ya han obtenido (o están en vías de hacerlo) el informe positivo de verificación en al menos tres aspectos:
1) % de créditos asignados a la Química Analítica; 2) Distribución modular y por materias/asignaturas del contenido de la Química
Analítica; y 3) Comparación de las competencias específicas que tendrán que adquirir los
estudiantes en las materias de Química Analítica.
Necesariamente se trata de una visión parcial, ya que muchos de los Títulos de Grado en Química están en proceso de verificación o todavía no han sido remitidos al Consejo de Universidades. Se trata de un adelanto en estos momentos de transición. La Red de Docencia de la SEQA publicará los resultados globalizados una vez que hayan finalizado todos los procesos de verificación pendientes.

CONFERENCIAS ESPECÍFICAS CE-4
17
EL TÍTULO DE MÁSTER EN LA NUEVA ORGANIZACIÓN DE LAS ENSEÑANZAS
UNIVERSITARIAS
Manuel Silva
Departamento de Química Analítica, Edificio Marie-Curie (Anexo), Campus de
Rabanales. Universidad de Córdoba. E-mail: [email protected] En el ámbito de la construcción del Espacio Europeo de Educación Superior
(EEES) se introduce en el año 2005 el título de Master Universitario en la nueva organización de las enseñanzas universitarias en España mediante el Real Decreto 56/2005 en el que se regulan los estudios universitarios oficiales de Posgrado. Sin embargo, este marco jurídico para el desarrollo de estas enseñanzas fue verdaderamente efímero ya que en la Ley Orgánica de Universidades 4/2007 se fijó una nueva estructuración de las enseñanzas y títulos universitarios oficiales suprimiendo el concepto de Posgrado aunque manteniendo los tres títulos oficiales de Grado, Master y Doctorado, cuya ordenación se establece en el Real Decreto 1393/2007. A pesar de estos avatares, las finalidades de las enseñanzas de Master se han mantenido intacta: adquisición por el estudiante de una formación avanzada, de carácter especializado o multidisciplinar, orientada a la especialización académica o profesional, o bien a promover la iniciación en las tareas investigadoras. Con objeto de armonizar la elaboración y propuestas de Títulos de Master, el Real Decreto 1393/2007 estableció un proceso de verificación de los mismos por el Consejo de Universidades, antes de su implantación, conforme a un protocolo elaborado por ANECA. En la actualidad se está desarrollando esta etapa de verificación no sólo de los títulos presentados conforme a este Real Decreto, sino además de aquellos previamente autorizados según lo dispuesto en el Real Decreto de Posgrado, en este caso, a través de un procedimiento abreviado.
En esta comunicación se pretende reflexionar sobre diversos aspectos
relacionados con el devenir del titulo de Master en estos escasos años de existencia en las enseñanzas universitarias en España. Su introducción en un espacio temporal inadecuado (antes de la propuesta y desarrollo de los estudios de grado) ha generado una serie de malas prácticas que quizá sean difíciles de erradicar. En este sentido, las finalidades recogidas en el Real Decreto para estos estudios de Master se han restringido en la gran mayoría de los casos a “promover la iniciación en las tareas investigadoras”. Así, un número muy significativo de los títulos de Master propuestos tanto en el marco de los estudios de Posgrado como al amparo del Real Decreto 1393/2007, proceden de la transformación de Programas de Doctorado, en su mayoría con mención de calidad. Este proceso ha mermado considerablemente el presumible potencial y prestigio de estos estudios, ya que hoy por hoy en la Universidad española todavía no se ha tomado conciencia de lo que realmente representa un título de Master tal como se considera en otros países del entorno del EEES.
Finalmente, se analizará estadísticamente la implantación de los diferentes
títulos en cada una de las ramas de conocimiento, y en especial en la Rama de Ciencias en la que se encuadran los posibles títulos de Master con una participación del área de conocimiento de Química Analítica.

CE-5 CONFERENCIAS ESPECÍFICAS
18
LA QUÍMICA ANALÍTICA ESPAÑOLA A LA LUZ DE LA BIBLIOMETRÍA:
¿ZIDANES O PAVONES?
José Miguel Vadillo
Departamento de Química Analítica, Universidad de Málaga [email protected]
El factor de impacto, el índice de inmediatez, el número H o el número de citas medias son algunos de los indicadores bibliométricos con los que se intenta cuantificar la calidad del investigador y de su investigación. La polémica está servida desde su origen, no sólo por el intrínseco sesgo que conlleva cualquier sistema de medición, sino por las implicaciones asociadas en un mundo competitivo como el de la publicación científica. Este espíritu competitivo, se encuentra retroalimentado en alguna medida por la lícita competencia entre las grandes multinacionales de la publicación. Abusando de la demagogia, se podría argüir que a grandes científicos nunca se les asoció un indicativo de su excelencia, y que los vaivenes de los indicadores de calidad tampoco auguran que hayamos encontrado el mecanismo correcto de medición de la calidad científica. Sin embargo las odiosas comparaciones se establecen, basadas en frías estadísticas que proyectan una imagen distorsionada de lo que somos y de lo que hacemos … ¿o no?
La comunicación que se presenta intenta mostrar la situación actual de la
química analítica española en varios niveles distintos: el panorama nacional; en su contexto internacional; y referida a otras disciplinas químicas. El estudio se ha realizado en base a las publicaciones realizadas en el periodo 2000-2007, aunque se mostrarán algunos datos comparados con el periodo 1993-2000 que pueden resultar ilustrativos.

CONFERENCIAS ESPECÍFICAS CE-6
19
PRESENCIA DE CONTAMINANTES MINORITARIOS Y EMERGENTES EN
AGUAS SUPERFICIALES Y SUBTERRÁNEAS.
Lucila Candela
Universidad Politécnica de Cataluña Jordi Girona 1-3, Campus Nord, Edificio D2, 08034 Barcelona
Desde los años 70, los impactos de la contaminación en las aguas se han focalizado exclusivamente en los denominados contaminantes tradicionales, especialmente en los plaguicidas tóxicos/carcinogénicos y los derivados de la industria que presentaban una notable persistencia en el medio ambiente. Recientemente, un grupo de compuestos de naturaleza muy variada, potencialmente contaminantes y que se le ha denominado emergentes ha empezado ser objeto de atención.
El término contaminante emergente hace referencia a toda una serie de
compuestos químicos entre los que se encuentran los productos farmacéuticos y de higiene personal, surfactantes y agentes fluorescentes blanqueantes, otros productos químicos de uso doméstico y nanomateriales. Emergente, no necesariamente significa que sean nuevos, dado que estos compuestos han sido vertidos al medio ambiente desde su comercialización y su presencia en las aguas ya fue detectada desde principios de los años 80. Generalmente se introducen en el medio acuático en forma continua y mediante mezclas complejas, a través de aguas y lodos procedentes de depuradora, escorrentía superficial, o de vertidos incontrolados. Al contrario que los contaminantes convencionales persistentes, los emergentes no necesitan presentar esta característica al ser introducidos en el medio ambiente de forma constante y continuada, y aunque de forma individual se presenten en bajas concentraciones, la presencia de numerosos compuestos con el mismo tipo de acción puede presentar efectos adversos. La continua presencia en suelos y masas de agua es preocupante dado su efecto acumulativo y en muchos casos, el desconocimiento de sus adversos efectos sobre los organismos.
Aunque los procesos bióticos y abióticos determinan su presencia en el medio
acuático, la bibliografía muestra que numerosos compuestos sobreviven a la biodegradación, muchos de sus metabolitos muestran persistencia, generalmente se desconoce su toxicidad ambiental o y dada su continua introducción en el medio ambiente pueden mostrar alta persistencia. Mayoritariamente se han detectado en aguas procedentes de plantas depuradoras, y algunos de ellos en aguas de abastecimiento especialmente cuando su origen es subterráneo. El problema derivado de su presencia en el medio reside en que pueden llegar a producir procesos neurovegetativos, alteraciones inmunológicas y endocrinas con efecto acumulativo en humanos. Además hay que tener en cuenta que estos efectos pueden ser mucho más graves sobre la biota, dado que el umbral de daños es inferior al requerido por los humanos.
A la posible amenaza medioambiental (especialmente si consideramos el
creciente uso de las aguas procedentes de las EDAR) en relación con su comportamiento en el suelo y medio acuático, hay que añadir la problemática que encierra su identificación analítica; desconocimiento de tipos de compuestos y cantidades utilizadas, eficiencia en la eliminación de los compuestos y las posibles interacciones entre ellos u otros compuestos químicos.

CE-7 CONFERENCIAS ESPECÍFICAS
20
DISRUPTORES ENDOCRINOS. SU PRESENCIA EN EL MEDIO Y EFECTOS
Nicolás Olea
Laboratorio de Investigaciones Médicas. CIBERESP
Hospital Clínico. Universidad de Granada 18071 Granada; [email protected]
Algunos efectos adversos sobre la salud humana observados recientemente podrían deberse a la exposición a sustancias químicas con capacidad de alterar el equilibrio hormonal. La evidencia de esta asociación, demostrada en poblaciones animales, ha generado el contexto doctrinal en el que se basa la hipótesis de la disrupción endocrina (DE) en humanos. DE es un problema emergente de salud medioambiental que ha cuestionado algunos de los paradigmas en que se fundamenta el control y la regulación de uso de los compuestos químicos, ya que anticipa el impacto sobre la salud humana del efecto combinado (aditivo, sinérgico o antagónico) de los compuestos químicos –rara vez explorados de forma combinada. La confirmación de la exposición humana a disruptores endocrinos, que ocurre de forme inadvertida en muchos de los casos, junto a la demostración de efecto en algunas poblaciones con una especial sensibilidad, como son población infantil y mujeres en edad fértil y durante el embarazo-lactancia, obliga a reconsiderar los riesgos ambientales de los individuos no profesionalmente expuestos y exige actuar con medidas preventivas. Algunos ejemplos de asociación exposición-efecto deberían ser investigados con más detalle y ayudarían al sostenimiento de la hipótesis de disrupción endocrina si en los diferentes foros científicos se popularizara la hipótesis de trabajo. La exposición infantil a bisfenoles provenientes de resinas epoxy de los pegamentos o el policarbonato de los biberones, a benzofenonas de las cremas solares con filtros UV, a ftalatos de los ablandadores y aditivos de los plásticos empleados en tetinas y biberones, o de tributilestaño en las pinturas de empleos múltiples, no son más que unos pocos ejemplos para los cuales la comunidad científica debería estar convenientemente informada. Para abordar este problema emergente bajo la perspectiva del “principio de precaución” habría que establecer un control mas estricto sobre las sustancias químicas presentes en el medioambiente, alimentos y en bienes de consumo, mejorar los sistemas de evaluación de la toxicidad incluyendo los estudios de múltiples compuestos químicos y establecer un sistema de biomonitorización de la exposición humana que pudieran ser utilizados para implementar medidas preventivas y evaluar su efectividad.

CONFERENCIAS ESPECÍFICAS CE-8
21
CHEMICAL IMAGING. FROM RGB TO HYPERSPECTRAL.
FEATURES, BENEFITS AND DRAWBACKS
José M. Amigo
Dpt. of Food Science Quality and Technology, Faculty of Life Sciences University of Copenhagen, Rolighedsvej 30, DK-1958 Frederiksberg C, Denmark
In the last years, Chemical Imaging techniques have appeared as surface analysis techniques with the aim not only to cover weak points in conventional spectroscopic techniques but also to extend the knowledge onto the measured surface. Imaging techniques result especially attractive because of the possibility of providing huge spectral and spatial information of one sample (thousands of spectra per sample) in a short time analysis [1].
Merging spectroscopic imaging and chemometric methods enhances the
outcomes of instrumental technology and data analysis. Knowing in detail how the chemical compounds are distributed and how physical artifacts are affecting the image gives valuable information about essential issues as correct distribution assessment of compounds in pharmaceutical tablets, morphological information in medicine, growing patterns of contaminants in cultural heritage, etc. The power to detect and locate minor compounds can also be enhanced, providing information about, for example, contaminants or impurities in the scanned surface.
The Image Analysis has been divided into different aspects, considering the
different Chemometric tools applied so far: a) Exploratory purposes, the so-called Multivariate Image Analysis (MIA) [2], b) Image segmentation [3], c) Quantification of images, Multivariate Image Regression (MIR) [4] and, finally, d) Image resolution, which are specifically focused on the recovery of the real distribution maps and pure spectra of the image constituents [5].
The target of this conference is three-fold: a) To offer a perspective of the
different meanings of “Chemical Image”, b) To extent the knowledge of the most common Chemometric methods applied to each particular aspect of Image Analysis and, finally, c) the main part of the presentation will be devoted to illustrate the benefits and drawbacks of Image analysis in different real applications (Environmental Analysis, Food industry, Pharmaceutical research, etc…). [1] E.N. Lewis, L.H. Kidder, E. Lee, K.S. Haber, Spectrochemical Analysis Using
Infrared Multichannel Detectors, Blackwell Publishing Ltd., Oxford, 2005. [2] J.M. Amigo, J. Cruz, M. Bautista, S. Maspoch, J. Coello, M. Blanco, Trac-Trends
in Analytical Chemistry, 27 (2008) 696. [3] T.N. Tran, R. Wehrens, L.M.C. Buydens, Chemometrics and Intelligent
Laboratory Systems, 77 (2005) 3. [4] H. Grahn, P. Geladi, Techniques and applications of hyperspectral image
analysis, Wiley & Sons, 2007. [5] A. de Juan, R. Tauler, R. Dyson, C. Marcolli, M. Rault, M. Maeder, Trac-Trends
in Analytical Chemistry, 23 (2004) 70.


COMUNICACIONES FLASH

CF-1 COMUNICACIONES FLASH
24
CHEMOMETRIC ASSISTED SOLID-PHASE MICROEXTRACTION FOR THE
DETERMINATION OF ANTI-INFLAMMATORY AND ANTIEPILEPTIC DRUGS IN RIVER WATER BY LIQUID CHROMATOGRAPHY DIODE ARRAY DETECTION
M. Martínez Galeraa, L. Vera Candiotib, M.D. Gil Garcíaa, H.C. Goicoecheab
aDpto. Hidrogeología y Química Analítica, Univ.Almería, Almería, España. E-mail:
[email protected] bLab. Desarrollo Analítico y Quimiometría, Cátedra de Química Analítica, Univ.
Nacional del Litoral, Santa Fe, Argentina
Environmental sample matrices such as ground superficial and waste-waters are complex samples, often containing compounds which can interfere with the compounds of interest, so that direct analysis may not be possible and, moreover, pharmaceuticals are generally found in these matrices at trace concentration levels. Therefore, it is necessary to perform an initial sample preparation step, including purification and concentration of the analytes. Sample preparation may be achieved by a wide range of techniques but all of them shows the two above mentioned goals, in addition to provide a robust and reproducible method which is independent of variations in the sample matrix [1]. Even though traditional sample-preparation methods are still in use (e.g. solid phase extraction, SPE), trends in recent years are focused towards smaller initial simple sizes, small volumes or no organic solvents, greater specificity or greater selectivity in extraction and increased potential for automation. Solid phase microextraction (SPME) is a modern sampling preparation technique for isolating and preconcentrating organic compounds, which is in compliance with all these requisites and, additionally, is highly sensitive and can be used for polar and no polar analytes.
In the present work, an analytical method for the simultaneous determination
of seven non steroidal anti-inflammatory drugs (naproxen, ketoprofen, diclofenac, piroxicam, indomethacin, sulindac and diflunisal) and the anticonvulsant carbamazepine is reported. The method involves preconcentration and clean-up by SPME using polydimethylsiloxane/divinylbenzene (PDMS/DVB) fibers, followed by liquid chromatography with diode array detection analysis (LC-DAD). Parameters that affect the efficiency of SPME step such a soaking solvent, soaking period, desorption period, stirring rate, extraction time, sample pH, ionic strength, organic solvent and temperature were investigated using a Plackett-Burman screening design. Then, the factors presenting significant positive effects on the analytical response (soaking period, stirring rate, stirring time) were considered in a further central composite design (CCD) to optimize the operational conditions for the SPME procedure. Additionally, multiple response simultaneous (MRS) optimization function was used to find the optimum experimental conditions for the on-line SPME procedure. The best results were obtained using a soaking period of 5 min, stirring rate of 1400 rpm and stirring time of 44 min. The use of SPME avoided matrix effect and allowed to quantify the analytes in river water samples by using Milli-Q based calibration graphs. Recoveries ranging from 71.6 to 122.8 % for all pharmaceuticals proved the accuracy of the proposed method in river water samples. Method detection limits were in the range of 0.5 to 3.0 µg L-1 and limits of quantitation were between 1.0 and 4.0 µg L-1 for pharmaceutical compounds in river water samples. The expanded uncertainty associated to the measurement of the concentration ranged between 8.5% and 29.0% for 20 µg L-1 of each analyte and between 9.0% and 29.5% for the average of different concentration levels.
[1] R.M. Smith, J. Chromatogr. A 1000 (2003) 3-27.

COMUNICACIONES FLASH CF-2
25
STUDY OF THE SIZE-BASED BIOAVAILABILITY OF METALS ASSOCIATED TO
NATURAL ORGANIC MATTER BY STABLE ISOTOPE EXCHANGE AND QUADRUPOLE ICP-MS COUPLED TO As-FlFFF
F. Laborda, S. Ruiz-Beguería, E. Bolea, J.R. Castillo
Analytical Spectroscopy and SensorsGroup (GEAS).
Institute of Environmental Sciences (IUCA). University of Zaragoza. Pedro Cerbuna, 12. 50009 Zaragoza.Spain
E-mail: [email protected] The potential bioavailability of a metal in a solid environmental sample (soil,
sediment, sludge, compost…) can be determined by isotopic exchange. Although this methodology was developed for radiactive isotopes, enriched stable isotopes can also be used [1]. Basically, a suspension of the sample is spiked with a small amount of the enriched isotope, which reequilibrates with the exchangeable fraction ot the metal in the solid. At equilibrium, the enriched isotope should be distributed equally in the exchangeable fraction of the solid (considered potentially bioavailable) and in the solution (assumed fully exchangeable). The isotope ratio is measured in the solution and the bioavalibility calculated as:
samplemeasured
measuredspikebsample
bspikesample
sample IR IR
IR IR
A M
m M
m1
E−
−=
where E is the bioavailability (mg kg-1); msample, the mass of sample (kg); Msample, the atomic weight of the element in sample; Mb: the atomic weight of isotope b; Ab
sample, the abundance of isotope b in the sample; mspike, the mass of the isotope spiked in the sample; IRmeasured, the isotope ratio (a/b) in the spiked sample; IRspike, the isotope ratio (a/b) in the spike; IRsample, the isotope ratio (a/b) in the sample.
Metals can be associated to different forms of organic matter and colloids in the
aqueous extract, which can show different bioavailabilities. When isotope ratios are measured directly in the aqueous extracts, an averaged bioavailability is obtained. In order to get detailed information about the metal bioavalibility with respect to molecular mass and colloid size, the isotopic exchangeability has been investigated by on line coupling of a size-based separation techniques (assymetric flow field flow fractionation, As-FlFFF) to quadrupole ICP-MS. Acquisiton parameters for isotope ration measurements were selected as a compromise between sampling frequency and attainable precision. Results for copper and lead in organic amendments will be presented.
[1] A.L. Nolan et al., Anal. Bioanal. Chem. 380 (2004) 789.
This work has been sponsored by the Spanish Ministry of Science (BQU 2006-00894).

CF-3 COMUNICACIONES FLASH
26
COMPLEMENTARY MOLECULAR AND ELEMENTAL MASS SPECTROMETRY FOR THE IDENTIFICATION OF ARSENIC COMPOUNDS IN HUMAN METABOLISM
AFTER FEEDING WITH SEAFOOD
M. Contreras-Acuña, T. García-Barrera, J.L. Gómez-Ariza
Dpto. de Química y CC.MM. Facultad de Ciencias Experimentales. Universidad de Huelva. Campus de El Carmen.21007 Huelva. (SPAIN)
E-mail: [email protected]
Seafood is a major dietary source of arsenic which includes at least 28 different arsenicals. Marine algae and shellfish are the more frequent seafood exposure sources with exhibits the greatest diversity of arsenicals. Among these arsenic species, the potential for biotransformation upon ingestion varies considerably. For example, it is generally established that ingested arsenobetaine (AsB) is excreted unchanged in urine, although recently Jenkins has found the biotransformation of arsenobetaine by microorganisms from the human gastrointestinal tract. However, inorganic arsenic produce mono- and dimethylarsenate (MMA and DMA) as major urinary metabolites. Arsenosugars, typically associated with seaweed ingestion, are also biotransformed to produce primarily urinary DMA (10–12). Although DMA is the major urinary metabolite detected, the fact that one conversion involves arsenic–carbon bond formation and the other arsenic–carbon bond cleavage denotes very different metabolic pathways. The lack in arsenic metabolism is especially marked for arsenosugars (oxo-arsenosugars) which can partially converted in thio-arsenosugars that multiplies the number of species involved in the metabolism of this element, and promote the occurrence of sulphur-containing arsenic species.
The purpose of this study is the combined use of HPLC-ICP-MS and HPLC-ESI-MS to identify the presence of arsenic and thio-arsenic species in clams and anemone (Anamonia sulcata) from the southwest Spain. In addition, the metabolism of arsenic in human feed with these typical seafood products has been studied applying combined mass techniques on food and urine.

COMUNICACIONES FLASH CF-4
27
ENZYME-MODIFIED NANOPARTICLES USING BIOMIMETICALLY
SYNTHESIZED SILICA FOR SENSING APPLICATIONS
Patricia Zamora, Arántzazu Narváez, Elena Domínguez Department of Analytical Chemistry, University of Alcalá, Alcalá de Henares, Madrid.
E-mail: [email protected]
The entrapment of enzymes within biomimetic silica nanoparticles offers unique and simple immobilization protocols that merge the stability of proteins confined in solid phases with the high loading and reduced diffusion limitations inherent to nano-sized structures. Herein, we report on the biomimetic silica entrapment of chemically derivatized horseradish peroxidase for amperometric sensing applications. The so-called "biomimetic silification reaction" is inspired on the biomineralisation process of the diatoms. This provides a biocompatible, nano-sized, and a simple method for enzyme immobilisation. Scanning electron microscopy evidences the formation of enzyme-modified nanospheres using poly(ethylenimine) as template for silicic acid condensation.
The adsorption of these enzyme-silica nanoparticles on graphite electrodes
resulted in cathodic currents upon addition of H2O2 at -100 mV vs. Ag|AgClsat, showing direct electron transfer between the electrode surface and the modified horseradish peroxidase. The entrapment of the negatively charged peroxidase within silica nanoparticles at gold surfaces with a self-assembled monolayer of poly(ethylenimine) is also demonstrated by microgravimetric measurements and SEM images. These in-situ biomimetically synthesized peroxidase nanospheres are catalytically active enabling direct bioelectrocatalysis at 0 mV vs. Ag|AgClsat with long-term stability. This methodology also enables the co-inmmobilization of hydrogen peroxide producing oxidases and peroxidases enlarging the spectrum of analytes and samples. More generally, the possibility to customize the shape and size of these nanoparticles extends the range of applications and properties to be fully explored and exploited in the field of nanobiotechnology.
Financial support from the EU FP6 RTD Project “Synthesis and nanoapplication of tethered silicates” (NMP4-CT-2006-033254) is fully acknowledged.

CF-5 COMUNICACIONES FLASH
28
MULTIVARIATE CALIBRATION PROCEDURES APPLIED TO HIGH-
PERFORMANCE LIQUID CHROMATOGRAPHY COUPLED TO FAST-SCANNING FLUORESCENCE DETECTION FOR THE DETERMINATION OF
FLUOROQUINOLONES IN DIFFERENT MATRICES
A. Espinosa Mansilla,1 A. Muñoz de la Peña, 1 J. A. Arancibia,2
F. Cañada Cañada,1 G. M. Escandar,2 G. A. Ibañez,2 A. C. Olivieri2
1Departamento de Química Analítica, Facultad de Ciencias Universidad de Extremadura, 06071 Badajoz, España
[email protected] 2Instituto de Química Rosario (CONICET-UNR), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, 2000, Rosario,
Argentina.
Different second-order multivariate calibration algorithms, namely parallel factor analysis (PARAFAC), N-dimensional partial least-squares (N-PLS) and multivariate curve resolution-alternating least-squares (MCR-ALS) have been compared for the analysis of eight fluoroquinolones in aqueous solutions, including some human urine samples. The assayed analytes were: pipemidic acid (PIPE), belonging to the first generation of quinolone antibiotics; marbofloxacin (MARBO), ofloxacin (OFLO), norfloxacin (NOR), ciprofloxacin (CIPRO), enrofloxacin (ENRO) and lomefloxacin (LOME) belonging to the second generation; and danofloxacin (DANO) belonging to the third generation. These analytes were chosen due to their importance in human medicine (second-generation fluoroquinolones) and also in veterinary medicine (MARBO, ENRO, and DANO).
Data were measured in a short time with a chromatographic system operating
in the isocratic mode. The detection system consisted of a fast-scanning spectrofluorimeter, which allows one to obtain second-order data matrices containing the fluorescence intensity as a function of retention time and emission wavelength. The developed approach enabled us to determine eight analytes with overlapped profiles, without the necessity of applying an elution gradient, and thus significantly reducing both the experimental time and complexity. The study was employed for the discussion of the scopes of the applied second-order chemometric tools.
The quality of the proposed technique coupled to each of the evaluated
algorithms was assessed on the basis of the figures of merit for the determination of fluoroquinolones in the analyzed water and urine samples. Univariate calibration of four analytes led to limits of detection in the range 20–40 ng mL–1 and root mean square errors for the validation samples in the range 30–60 ng mL–1 (corresponding to relative prediction errors of 3–8%). The ranges for second-order multivariate calibration (using PARAFAC and N-PLS) of the remaining four analytes were: limit of detection, 2–8 ng mL–1, root mean square errors, 3–50 ng mL–1 and relative prediction errors, 1–5%.
Acknowledgements: Financing from the Junta de Extremadura (Project
GRU09092) and the Ministerio de Ciencia e Innovación of Spain are acknowledged (Project CTQ2008-06657-C02-01).

COMUNICACIONES FLASH CF-6
29
DETERMINATION OF PCBS FROM EXTREMELY SMALL BIOLOGICAL SAMPLES USING ULTRASONIC PROBE: APPLICATION TO BIOACUMULATION IN
ZEBRA FISH EMBRYOS. M. Pena-Abaurrea1, S. El-Amrani2, J. Sanz-Landaluze2, L. Ramos1 and
C. Cámara2
1Department of Instrumental Analysis and Environmental Chemistry, IQO G
(CSIC), Juan de la Cierva 3. 28006 Madrid, Spain. 2Department of Analytical Chemistry, Faculty of Chemistry Sciences, Complutense University of Madrid (UCM), 28040 Madrid, Spain. E-mail: [email protected]
Increased interest in the fate, transport and toxicity of polychlorinated biphenyls (PCBs) over the past few years has led to a variety of studies reporting different methodologies for analysis of these persistent organic pollutants. The analytical methods commonly used for the determination of PCBs in complex biological matrices with high fat content are tedious procedures, involving a number of steps for extraction, purification and final isolation of the environmental relevant congeners (i.e. priority and planar toxic congeners) from the bulk of the PCBs and from other co-extracted material. These sample treatment procedures are expensive in terms of solvent, sorbent and time consumption, and are prone to losses and contamination because of the constant manipulation of the extracts. Most separation-plus-detection instruments nowadays available allow to achieve extremely low detection limits, which results in the effective injection of only a small fraction of the final extract into the chromatograph.
In this study, the feasibility of ultrasonic extraction, using a 0.6 mm ultrasonic
probe, has been investigated for the fast and exhaustive extraction of priority and toxic PCB congeners from a variety of biological samples. The latter were previously investigated within the frame of interlaboratory exercises. Once optimised, the method allowed quantitative recoveries of the endogenous PCBs in only 40 s. Accurate determination was possible using only 20 mg of sample and 150 µL of n-hexane as extraction solvent. The obtained fatty extracts were further purified by direct elution through a 1 mL column containing silica modified with sulphuric acid. Gas chromatography equipped with an electron capture microdetector was used for final determination of the target compounds. The efficiency of the method has been demonstrated by comparison with more conventional sample preparation procedures for this type of analysis.
The optimised method was applied to evaluate bioaccumulation of PCB#104 in
Zebra fish embryos as part of an alternative protocol to OCDE 305. Actually, this OCDE method is the reference for evaluation of bioaccumulation and forms part of the requirements of the EU REACH regulation for better and earlier identification of the intrinsic properties of chemical substances The protocol has a cost higher than 100.000 € for each evaluated compound, as 108 adult fishes are needed.
Acknowledgements: Authors acknowledge the Spanish Ministry for Environment
for financial support (046/PC08/2-14.4). S. El-Amrani also thanks AECI for their predoctoral grant.

CF-7 COMUNICACIONES FLASH
30
0
0,025
0, 05
0,075
0,1
GG -7 GG-10 GG - 3 GG- 2 GG -6 GG -5 GG-8 GG -11 GG -4 GG - 9 GG - 1 GG -14 GG - 13 GG- 12
Inhibitory Concentration 50 (IC50)
0
0,025
0, 05
0,075
0,1
GG -7 GG-10 GG - 3 GG- 2 GG -6 GG -5 GG-8 GG -11 GG -4 GG - 9 GG - 1 GG -14 GG - 13 GG- 12
Inhibitory Concentration 50 (IC50)
Fuente vegetal Extracción de polifenoles
Caracterización con LC/MS Valoración de bioactividad
Procesamiento de datosCorrelación entre datos de MS y bioactividad
Elección de candidato/s y caracterización por RMN
2 Compuestos polifenólicosO H
C OOH
OH
CO OH
OHO H
OH
O
O
OH
O H
OHOH
OH
O
O
OH
OH
OHOH OOH
O H
O HO H
O H
O
O
OH
OHOMe
OMe
3
1
4
-1 0 1 2 3 4 5
x 1 06
0
10
20
30
40
50
60
70
80
90
1 00
Ac P in 4 15 r =- 0. 83 31 7 3 R2= 69 .4 % p= 0. 00 0 21 50 5 4 a = -1 .4 92 88 e -00 5 b = 66 .3 09 7
int en si da d
activ
idad
rel
ativa
(%
)
0 5 00 10 0 0 15 00-0 .4
-0 .2
0
0 .2
0 .4
0 .6
0 .8
1
5
OH
OH
OH
O+
OH
OH
Hibiscus sabdariffa: CARACTERIZACIÓN ANALÍTICA MEDIANTE HPLC-DAD- ESI-TOF/IT, CAPACIDAD ANTIOXIDANTE Y BÚSQUEDA DE COMPUESTOS
BIOACTIVOS MEDIANTE TÉCNICAS DE DESREPLICACIÓN. 1S. Fernández-Arroyo, 1I. C. Rodríguez-Medina, 2S. Mota, 2A. de la Torre,
1A. Segura-Carretero, 1A. Fernández-Gutierrez 1Departamento de Química Analítica, Facultad de Ciencias, Universidad de Granada.
2Dpto. Teoría de la Señal, Telemática y Comunicaciones, E.T.S. Ingeniería Informática y de Telecomunicación, Universidad de Granada
E-mail: [email protected]; [email protected]
Hibiscus sabdariffa, una planta de la familia Malvaceae es conocida desde hace bastante tiempo por sus propiedades medicinales beneficiosas para la salud. Contiene gran cantidad de productos fenólicos que se conocen por sus propiedades antioxidantes y que podrían reducir el riesgo de enfermedades cardiovasculares y contra el cáncer. La fracción fenólica y otros compuestos polares del extracto acuoso de Hibiscus sabdariffa fue separada y caracterizada por HPLC con detección diodo array (DAD) acoplada a tiempo de vuelo (TOF) y trampa de iones (IT) para análisis de MS/MS. Entre los compuestos identificados destacan el ácido hidroxicítrico, su lactona, el ácido hibíscico, ácido clorogénico e isómeros, quercetina 3-rutinosa y antocianinas como delfinidina 3-sambubiosa y cianidina 3-sambubiosa.
La capacidad antioxidante del extracto acuoso de Hibiscus sabdariffa fue determinada mediante diferentes métodos, como son el poder antioxidante para reducir el ión férrico (FRAP), la capacidad de absorber radicales de oxígeno (ORAC), la capacidad antioxidante en equivalentes de Trolox (TEAC) y la capacidad para inhibir la peroxidación lipídica (TBARS).
Por último, se ha desarrollado una técnica rápida de “desreplicación” basándose en datos fuente de espectrometría de masas generados mediante LC/MS(TOF/IT) a partir del extracto crudo de Hibiscus sabdariffa y datos de bioactividad en diferentes líneas celulares (cáncer de mama, adipocitos y condrocitos). Esta técnica permite, tanto a nivel de filtrado de señales como a nivel estadístico, establecer la correlación entre bioactividad y la composición en polifenoles de las muestras vegetales. Esto permitirá la localización del/os componente/s bioactivo/s del extracto crudo en su perfil cromatográfico-MS con un número pequeño de ensayos y el desarrollo de modelos de predicción para futuros extractos crudos potencialmente bioactivos (Figura).
Esquema de la técnica de desreplicación en la búsqueda de compuestos bioactivos

COMUNICACIONES FLASH CF-8
31
ESTUARINE SEDIMENT QUALITY ASSESSMENT BY FOURIER TRANSFORM
INFRARED SPECTROSCOPY Javier Moros 1, Ricardo J. Cassella 2, María C. Barciela-Alonso 3, Antonio Moreda-Piñeiro 3, Paloma Herbello-Hermelo 3, Pilar Bermejo-Barrera 3,
Salvador Garrigues 1 and Miguel de la Guardia 1
(1) Department of Analytical Chemistry, University of Valencia, 50th Dr. Moliner Street,46100 Burjassot, Valencia, Spain
(2) Department of Analytical Chemistry, Universidade Federal Fluminense, Outeiro de São João Batista s/n, Niterói RJ 24020-141, Brazil
(3) Department of Analytical Chemistry, Nutrition and Bromatology, University of Santiago de Compostela,
Avenida das Ciencias s/n, Santiago de Compostela E-15782, Spain
Partial least squares Fourier-transform infrared (PLS-FTIR) models were developed for the quality assessment of estuarine sediments through the evaluation of several physicochemical parameters.
Models were based on the chemometric treatment of attenuated total reflection
(ATR) spectra directly obtained from samples previously lyophilized and sieved through a lower than 63 µm grid.
Spectra were scanned from 3997 to 523 cm-1, averaging 36 scans per spectrum
with a nominal resolution of 8 cm-1. Models were built using reference data obtained for sediment samples collected from Ria de Arousa estuary. Hierarchical cluster classification of sediment ATR spectra was employed for the establishment of the independent calibration and validation sample sets.
Several parameters, like trace metal content (Sn, Pb, Cd, As, Sb and total Cr
and also acid soluble, reducible and oxidable Cr fractions), elemental composition (carbon (C), nitrogen (N) and hydrogen (H) content), pH, redox potential (Eh), together with the amount of fulvic and humic acids, were evaluated.
Residual predictive deviation (RPD) values ranging from 1.79 to 2.28 were
obtained for all trace metals with the exception of Pb, which exhibited a RPD value of 1.5, similar than those obtained for the different extractable fractions of Cr. Regarding to the elemental analysis, excellent RPD values were also obtained for C and N, but for H only adequate values for screening purposes were achieved. Prediction errors for pH and Eh were 0.13 units and 30 mV respectively, thus indicating the good prediction capabilities of the method. For humic substances, excellent results were obtained for humic acids when compared with fulvic ones.
In short, direct ART-FTIR measurements on solid sediments allow us a rapid
screening of many components of sediments and quantitative data of the main physico-chemical parameters.
The authors acknowledge the financial support of Ministerio de Educación y Ciencia (Project AGL2007-64567 and CTQ2008-05719).

CF-9 COMUNICACIONES FLASH
32
MULTIELEMENT DETERMINATION OF TRACE METALS IN SEAWATER BY ICP-MS WITH ON-LINE PRECONCENTRATION/SEPARATION BY A CHELATING
RESIN-PAKED MINICOLUMN
I. Sánchez Trujillo, E. Vereda Alonso, M.T. Siles Cordero, A. García de Torres and J.M. Cano Pavón
Department of Analytical Chemistry, Faculty of Sciences, University of Málaga.
E-mail: [email protected]
The analysis of trace metals in seawater is difficult due to both the low concentrations and the seawater matrix. Nowadays, inductively coupled plasma mass spectrometry (ICP-MS) has become one of the most powerful analytical techniques for trace element analysis with high sensitivity; however, weak tolerance to dissolved salts and polyatomic interferences is its principal disadvantage. Although the sample matrix effects can be minimised by on-line dilution of the sample, this solution is not the most adequate when the analytes concentration are very low. In these cases, it is better the on-line separation and preconcentration of the analytes coupled with ICP-MS.
In this work the multielement determination of trace metals in sea water was
carried out by ICP-MS coupled with an on-line preconcentration/separation system. In this system a minicolumn packed with silica gel chelating resin functionalised with 1-(di-2-pyridyl)methylene thiocarbonohydrazide was placed in the injection valve of a simple flow manifold. The system was applied to the preconcentration/separation and determination of cobalt, chromium and nickel in sea water. According to the scheme showed in figure 1, sample solutions (adjusted to pH 8.6) were passed through the column. After washing the column with water, the adsorbed metals were subsequently eluted into the plasma with 2% nitric acid. Detection limits of the trace metals (180 s sample loading time) were 0.006 ng ml-1 for Co, 0.53 ng ml-1 for Cr and 0.13 ng ml-1 for Ni, with enrichment factors of 3,52; 7,18 y 2,26, respectively. The precision, expressed as relative standard deviation, was 1.38% for 0.04 ng ml−1 of Co, 1.28% for 1.0 ng ml-1 of Cr and 1,34% for 0.2 ng ml-1 of Ni. Results from the determination of these metals in certified reference materials of seawaters (SLEW 3 and NASS-5) were in agreement with the certified values.
Figure 1

COMUNICACIONES FLASH CF-10
33
MICROFLUIDIC CHIPS WITH ELECTROCHEMICAL DETECTORS BASED ON
MULTIWALLED CARBON NANOTUBES AS FLOW INJECTION AND SEPARATION ELECTROKINETIC DRIVEN SYSTEMS
A. G. Crevillén, M. Pumera, M.C. González and A. Escarpa
Dpto. Química Analítica e Ing. Química, Univ. Alcalá, Alcalá de Henares.
E-mail: [email protected] Detection is one of the main challenges for microfluidic chips, since very
sensitive techniques are needed as a consequence of the ultra-small sample volumes used (pL-nL). Electrochemical detection (ED) is one of the most commonly routes used because apart from its high sensitivity and inherent miniaturization, another added functionality is the opened opportunity to modify these surfaces suitably with nanomaterials. An excellent example of this is carbon nanotubes (CNTs). CNTs are a group of nanomaterials which offers notably favorable possibilities involving the large active surface at electrodes of small dimensions and the enhancement of electronic transfer. These properties have clear influence on their analytical sensitivity which is enhanced by the use of these nanomaterials. There are two main types of carbon nanotubes that can have high structural perfection: single-walled nanotubes (SWCNT) which consist of a single graphite sheet seamlessly wrapped into a cylindrical tube, and multi-walled nanotubes (MWCNT) which comprise an array of such nanotubes that are concentrically nested like rings of a tree trunk.
In this work, the analytical potency of microfluidic chips with multiwalled
carbon nanotubes, in two analytical formats-flow injection and separation electrokinetic driven systems- using real samples is explored. Accordingly, two applications of high significance have been chosen to demonstrate the suitability of the electrokinetic’s platform integrating nanoelectrochemical detectors (See Figure 1): the determination of total isoflavones with integrated calibration and the fast detection of the major natural antioxidants where microfluidic chips were used as flow injection and separation formats, respectively.
Figure 1. Dual format of electrokinetic microfluidic chips with carbon nanotubes detectors for analytical domains. Flow injection format for total analyte detection (total isoflavones, with sequential calibration: RB1, C1 and S1) and separation format for individual analyte detection (antioxidant detection, RB2, and S2). (RB1-2 running buffers, C calibration/control reservoir, S1-2, sample reservoirs)
A.G. Crevillén, M. Pumera, M.C. González, A. Escarpa Lab Chip 9 (2009) 346-353

CF-11 COMUNICACIONES FLASH
34
ELECTROANALYTICAL SCHEMES FOR PARAQUAT DETECTION AT CARBON NANOTUBE-MODIFIED ELECTRODES AND BISMUTH-BASED ELECTRODES
Alberto Sánchez Arribas1, Mónica Moreno1, José Antonio Pérez López1,
Esperanza Bermejo1, Antonio Zapardiel2, Manuel Chicharro1
1Dpto. Química Analítica y Análisis Instrumental, Universidad Autónoma de Madrid. C/ Francisco Tomás y Valiente, 7, 28049 Madrid. E-mail: [email protected]
2Dpto. Ciencias Analíticas. Universidad Nacional de Educación a Distancia. Pº Senda del Rey, 9, 28040 Madrid.
Paraquat (1,1’-Dimethyl-4,4’-bipyridinium dichloride, also known as
methylviologen) is commonly used as a nonselective contact herbicide due to its extreme toxicity linked to its redox potential. The uncontrolled use of paraquat (PQ) and its high persistence in the environment, where it is only slightly absorbed by soil but is a potential contaminant of waters owing to its high solubility, lead to different health risks since it could enter to the food chain. Many analytical schemes have been employed in PQ detection but most of them are either suffer from the instability of the reagent and the instrumental system or require extensive sample pretreatment. On the other hand PQ electrochemical properties have attracted considerable attention over the past years resulting in many possible electrochemical applications which make voltammetry a suitable technique for the determination of this compound.
In this work, the electroanalytical possibilities of carbon nanotube-modified
electrodes and bismuth-based electrodes were examined for PQ detection. These mercury-free electrodes showed promising characteristics for cathodic electrochemical detection of this pesticide, even in the presence of dissolved oxygen. The presence of a carbon nanotubes layer onto a glassy carbon electrode promote the electrochemical reduction of oxygen close to -0.2 V (vs Ag/AgCl/KCl 3M reference electrode) while PQ was reduction was observed at -0.55 V. Interesting positive potential shifts due to adsorptive processes were noticed when using carbon nanotubes compared to signals obtained at bare glassy carbon. Moreover, bismuth-based electrodes efficiently supressed oxygen voltammetric signals and improved the electrode kinetics of PQ reduction. Direct voltammetric detection using differential pulse voltammetry leaded to LODs close to 5 µM at the studied electrodes in 0.10 M Na2SO4. These LODs were lowered more than 200 fold by modifying the electrodes with Nafion polymer in connection with a 6 minute accumulation step, taking advantage of the polymer cation-exchange properties.

COMUNICACIONES FLASH CF-12
35
ELECTROTHERMAL ATOMIC ABSORPTION SPECTROMETRIC
DETERMINATION OF CADMIUM AND LEAD AFTER MICROEXTRACTION BASED ON SOLIDIFICATION OF A FLOATING ORGANIC DROP
Ricardo E. Rivas, Ignacio López García, Manuel Hernández Córdoba
Department of Analytical Chemistry, Faculty of Chemistry, University of Murcia,
30071-Murcia, Spain. E-mail: [email protected]
Following the success of solid-phase microextraction (SPME) methodology, a number of approaches have been developed in the last years for the purpose of miniaturizing the stage of separation/preconcentration of the analytes [1-2]. Most of these procedures have as a common denominator the use of a very low amount of organic solvent, or none, so that they can be considered, in practice, as solventless extraction procedures. This is the case of liquid-phase microextraction (LLME) which can be carried out in a variety of ways. One of such ways, termed dispersive liquid–liquid microextraction (DLLME) uses a small drop of extracting solvent together with a disperser solvent. When the mixture of sample and solvents is shaken, the large contact areas between extraction solvent and aqueous sample results in a very fast and efficient extraction process. Recently [3], a new mode of liquid-phase microextraction based on solidification of a floating organic droplet (LLME-SFO) has been developed. In this technique, a microdrop of the organic solvent is floated on the surface of an aqueous sample which is being stirred by means of a magnetic stirrer. Under suitable conditions, the floated microdrop can remain in the top-center position of the aqueous sample. After the completion of the extraction, the sample vial is transferred into a cold water bath for a short time in order the low melting point organic solvent to be solidified. More recently [4], a new method (DLLME-SFO) combining the advantages of DLLME and LLME-SFO has been introduced. Most the miniaturized approaches for sample preconcentration have been devised to deal with organic analytes but it is clear they also offer interesting advantages for inorganic compounds. In the case of the determination of very low amounts of metallic elements, the combination of electrothermal atomic absorption spectrometry (ETAAS) and the mentioned methodologies is of a particular interest since ETAAS uses only a small volume of sample. Thus, and due to the preconcentration stage, a very high sensitivity is achieved and the resulting procedure represents an alternative choice to mass-based methods.
In this communication, the combination of LLME-SFO or DLLME-SFO with
ETAAS for the purpose of determining trace elements is studied. The applicability of the approach is demonstrated for the determination of cadmium and lead in water samples. Factors affecting the extraction efficiency, such as solution pH, concentration of organic reagent, extraction time, sample volume, and ionic strength are optimized. Concentrations at the sub-ppb level for both analytes can be measured in a reliable way.
[1] C. Nerín, J. Salafranca, M. Aznar, R. Batlle, Anal. Bioanal. Chem. 393 (2009)
809-833. [2] F. Pena-Pereira, I. Lavilla, C. Bendicho, Spectrochim. Acta 64B (2009) 1-15 [3] M.R.K. Zanjani, Y. Yamini, S. Shariati, J.A. Jönsson, Anal. Chim. Acta 585
(2007) 286-293 [4] H. Xu, Z. Ding, L. Lv, D. Song, Y.Q. Feng, Anal. Chim. Acta 636 (2009) 28-33

CF-13 COMUNICACIONES FLASH
36
DESARROLLO DE UN SISTEMA DE CUANTIFICACIÓN ABSOLUTA DE COMPUESTOS ORGÁNICOS MEDIANTE GC-COMBUSTIÓN-(EI)MS Y DILUCIÓN ISOTÓPICA POST-COLUMNA EMPLEANDO 13CO2 COMO
TRAZADOR
S. Cueto, J. Ruiz, A. Sanz-Medel, J. I. García
Dpto. Quimica Física y Analítica, Univ. Oviedo, Oviedo. E-mail: [email protected]
La cromatografía de gases con detección por espectrometría de masas (GC-MS)
se ha convertido en una técnica analítica imprescindible tanto en laboratorios de investigación como de rutina, debido a su gran potencial en términos de sensibilidad y selectividad así como a la capacidad de identificación de compuestos orgánicos en base a su espectro de masas. Sin embargo, las fuentes de ionización moleculares utilizadas habitualmente en GC-MS, como impacto electrónico, presentan distinta sensibilidad en función de las propiedades químicas o estructurales de las moléculas a analizar y, por tanto, para obtener información cuantitativa, su respuesta debe ser calibrada para cada analito mediante patrones específicos. Desafortunadamente, estas estrategias de calibración repercuten negativamente en el tiempo y coste por análisis. Además, el número de compuestos de interés analítico crece continuamente y en muchos casos los estándares puros no están disponibles comercialmente.
En este trabajo [1] se presenta el desarrollo de un sistema de cuantificación de
compuestos orgánicos basado en GC-MS y dilución isotópica post-columna, capaz de proporcionar de manera exacta y precisa la cantidad de compuesto presente en una muestra dada sin necesidad de realizar calibración externa ni recurrir a patrones específicos. La idea se basa en la transformación en línea de los compuestos separados previamente en el cromatógrafo en CO2, mediante una reacción en fase sólida en un tubo de combustión relleno de hilos de cobre y platino y calentado a 850 ºC. Posteriormente se mezcla este flujo con otro que contiene el trazador isotópico (13CO2 disuelto en He) y que se usará como patrón genérico. La medida en continuo de la relación isotópica 44/45 permite la obtención de un cromatograma de flujo másico de carbono en el cual, la integración de los picos proporciona de manera directa la cantidad de este elemento (en masa) presente en cada pico.
Para aplicar esta técnica absoluta de cuantificación, se modificó un GC-MS
comercial, situando la interfase de combustión desarrollada entre la columna y la fuente de ionización. Para la adición post-columna del 13CO2 disuelto en He, se diseñó un sistema consistente en un cilindro de acero y un controlador de flujo másico a la salida para lograr un control preciso de este flujo. Además, se incluyó una válvula de seis vías en el interior del horno cromatográfico. Esta válvula, además de evitar que el disolvente llegue al tubo de combustión, acortando su vida útil, permite transferir los compuestos que eluyen de la columna directamente a la fuente de ionización para obtener información estructural. El sistema fue validado mediante el análisis de distintas disoluciones patrón de compuestos orgánicos, alifáticos, aromáticos y funcionalizados. Se demostró la aplicabilidad a muestras reales complejas mediante el análisis de un gasóleo. Los resultados obtenidos fueron excelentes en términos de exactitud (±6%) y precisión (<5%). [1] S. Cueto, J. Ruiz, A. Sanz-Medel, J. I. García, Angew. Chem. Int. Ed. 48 (2009) D2561-D2564.

COMUNICACIONES FLASH CF-14
37
HIGH EFFICIENCY AND HIGHLY RETENTIVE MONOLITHIC SILICA
CAPILLARY COLUMNS COATED WITH POLY(OCTADECYL METHACRYLATE) FOR REVERSED-PHASE µ-HPLC SEPARATIONS
O. Núñez1, M.T. Galceran1, T. Ikegami2, N. Tanaka2
1Dpt. Analytical Chemistry, University of Barcelona, Av. Diagonal, 647. 08028
Barcelona. E-mail: [email protected] 2Dpt. Biomolecular Engineering, Kyoto Institute of Technology, Goisho-Kaido-cho,
Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan Monolithic silica columns have proven to be a good alternative for high
efficiency separations in HPLC. Because of their small-sized skeletons and wide through-pores much higher separation efficiency can be achieved than with particle-packed columns at a similar pressure drop [1]. Silica gel-based monolithic columns can be prepared using sol-gel technology to create a continuous network by the gelation of a sol solution within the column. Once the monolith is formed, it can be chemically functionalized in order to obtain the desired stationary phase [2]. Recently, polimerization procedures have been proposed for the chemical modification of silica monoliths. In these cases an anchor is first bonded to the silica before the chemical modification is performed. The objective of this study is to develop high efficiency and highly retentive monolithic silica capillary columns for reversed-phase (RP) chromatography. Then, columns with high peak capacity (high resolution) could be obtained for future 2D-HPLC applications such as the analysis of complex mixtures in life sciences or clinical applications.
In the present work, hybrid type monolithic silica capillary columns (25 cm x 200 µm I.D.) based on silica prepared from a mixture of tetramethoxysilane and methyltrimethoxysilane were used [3]. The columns were modified for RP-HPLC using octadecyl methacrylate via free radical polymerization for 3 h at 80 oC (ODM columns) [4]. The effects of monomer and radical initiator (α,α´-azobis-isobutyronitrile, AIBN) concentrations in the reaction mixture were examined. Hybrid monolithic silica capillary columns modified with octadecylsilil-(N,N-diethylamino)silane (ODS columns) were obtained for comparison. The performance of these columns was tested using different test analytes (such as alkylbenzenes) in terms of chromatographic efficiency and retention behaviour. It was observed that highly retentive and high efficiency ODM monolithic silica columns can be obtained by increasing monomer and initiator concentrations.
The performance of ODM columns for the RP-HPLC separations of some polar and non-polar compounds was also studied, and compared to that of ODS columns. Benzene and naphthalene derivatives, polycyclic aromatic hydrocarbons, steroids, alkyl phthalates, and tocopherol homologues were used as test samples [5]. In general, the polymer coated monolithic silica stationary phase ODM seems to have the greater preference (higher retention) for compounds with aromatic character, rigid and planar structures, and smaller length-to-breadth ratios (more compact structures).
[2] G. Guiochon, J. Chromatogr. A 1168 (2007) 101-168. [3] O. Núñez, K. Nakanishi, N. Tanaka, J. Chromatogr. A 1191 (2008) 231-252. [4] M. Motokawa, H. Kobayashi, N. Ishizuka, H. Minakuchi, K. Nakanishi, H. Jinnai,
K. Hosoya, T. Ikegami, N. Tanaka, J. Chromatogr. A 961 (2002) 53-63. [5] O. Núñez, T. Ikegami, W. Kajiwara, K. Miyamoto, K. Horie, N. Tanaka, J.
Chromatogr. A 1156 (2007) 35-44. [6] O. Núñez, T. Ikegami, K. Miyamoto, N. Tanaka, J. Chromatogr. A 1175 (2007)
7-15.

CFD-15 COMUNICACIONES FLASH
38
ESTRATEGIAS DE INNOVACIÓN DOCENTE EN LA ASIGNATURA TÉCNICAS
ANALÍTICAS DE SEPARACIÓN DE LA LICENCIATURA QUÍMICA: APRENDIZAJE BASADO EN LA INVESTIGACIÓN
M.J. Lobo Castañón, M. Teresa Fernández Abedul, M. Rosario Pereiro García, M. Dolores Gutiérrez Álvarez, Domingo Blanco Gomis.
Departamento de Química Física y Analítica, Universidad de Oviedo, Asturias.
E-mail: [email protected] Ante la continua demanda de cambios y mejoras en el proceso de enseñanza-
aprendizaje que permita incrementar la calidad docente, el equipo de profesores que impartió la asignatura Técnicas Analíticas de Separación durante el curso académico 2008-2009 en la Facultad de Química de la Universidad de Oviedo, nos planteamos implantar nuevas estrategias didácticas que fomenten el aprendizaje activo del estudiante. Esta es una asignatura troncal de 4º curso de Licenciatura en Química, en la que se combinan clases teóricas y prácticas de laboratorio para dar una introducción general a la teoría y práctica de un conjunto de técnicas imprescindibles en el análisis actual. Siguiendo la idea básica de aprendizaje basado en el descubrimiento guiado más que en la mera transmisión de información, se han planteado las siguientes estrategias de aprendizaje con las que se pretende fomentar que el estudiante elabore su propio conocimiento:
1. Diseño de una Webquest, actividad de investigación guiada utilizando recursos principalmente de Internet, titulada “¿Qué hay detrás del aceite de orujo de oliva? Análisis de benzopirenos en aceites comestibles”. En esta actividad se planteó a los estudiantes investigar el problema social que surgió tras la retirada del mercado en 2001 de aceites de orujo de oliva con elevados contenidos en benzopirenos y qué metodologías analíticas se desarrollaron para controlar los niveles de estos compuestos en aceites de orujo de uso alimentario. El resultado de esta actividad grupal se presentó como informe escrito y en un debate, tratando de trabajar competencias de comunicación tanto escrita como oral.
2. Diseño de un ejercicio de laboratorio que demuestre las posibilidades y limitaciones que los algoritmos quimiométricos ofrecen y cómo estos pueden ser usados en el tratamiento de resultados cromatográficos. Concretamente, se propone a los estudiantes el análisis quimiométrico de datos cromatográficos obtenidos en la caracterización de diversas variedades de manzanas de sidra asturiana, amparadas por la denominación de origen protegida “Sidra de Asturias”, utilizando para ello los resultados [1] obtenidos por SPME-HSCG durante la ejecución de un proyecto de investigación que tenía como objetivo el análisis cualitativo y cuantitativo de los compuestos volátiles que forman el aroma de las manzanas.
En esta comunicación se presentarán los resultados de estas experiencias que
se han combinado con una mejora de los materiales electrónicos utilizados durante el curso y el acceso permanente a los mismos a través de la plataforma del Campus Virtual que ofrece la Universidad de Oviedo.
Agradecimientos: Al Área de Innovación del Vicerrectorado de Informática y
Comunicaciones de la Universidad de Oviedo por su financiación a través del proyecto de innovación docente PB-08-025.
[1] P. Arias Abrodo, D. Díaz Llorente, S. Junco Corujedo, E. Dapera de la Fuente, M.D. Gutiérrez
Álvarez, D. Blanco Gomis. Food Chemistry. Manuscrito enviado para publicación, 2009.

COMUNICACIONES FLASH CFD-16
39
EL CONGRESO VIRTUAL COMO HERRAMIENTA DE APRENDIZAJE EN
QUÍMICA ANALÍTICA
M. A. Herrador, M.T. Morales
Dpto. Química Analítica, Fac. Farmacia, Univ. Sevilla, Sevilla. E-mail: [email protected]
Los nuevos retos planteados por el EEES han exigido cambios en la
metodología docente y la implementación de procesos de innovación [1]. En este sentido se ha desarrollado un proyecto de innovación docente en el que se ha utilizado un Congreso Virtual como nueva herramienta para el aprendizaje de la Química Analítica.
Los objetivos que pretendían alcanzarse con este trabajo fueron: i) Conseguir
que el alumno se implicara en el proceso de enseñanza-aprendizaje tomando un papel activo en la resolución de casos relacionados con la asignatura; ii) Fomentar la adquisición de competencias específicas de la asignatura haciendo comprender al alumno la importancia de cada una de las etapas del proceso analítico general; iii) Fomentar la adquisición de competencias trasversales; iv) Fomentar la adquisición de competencias profesionales relacionadas con la Química Analítica.
El proyecto se ha llevado a cabo en el segundo cuatrimestre del curso 2008/09.
Durante su ejecución se ha mantenido contacto permanente con los alumnos a través de la plataforma WebCT, que se ha utilizado como soporte para el desarrollo de la parte virtual de la actividad. El proyecto se ha desarrollado en 7 fases según el cronograma que se muestra en la siguiente Tabla:
Febrero Marzo Abril Mayo Junio
Fase 1. Formación grupos de trabajo. Asignación problema analítico.
Fase 2. Búsqueda bibliográfica y elaboración borrador de trabajo.
Fase 3. Seguimiento tutorizado y corrección.
Fase 4. Realización resumen en forma de cartel virtual en Power Point.
Fase 5. Colocación material creado en plataforma virtual WebCT.
Fase 6. Defensa oral trabajos. Encuesta opinión/satisfacción.
Fase 7. Evaluación.
La utilización de esta nueva herramienta de aprendizaje ha permitido fomentar el trabajo autónomo, la capacidad de análisis y síntesis, la identificación y resolución de problemas, la creatividad, la responsabilidad y el entrenamiento de habilidades profesionales. Por otra parte, la difusión de estos trabajos en forma de cartel a través de la plataforma WebCT ha permitido a los alumnos, además de comunicar sus conocimientos y conocer los ajenos, valorar la importancia de las TICs y sus posibilidades en el desarrollo de nuevos modos de aprendizaje y, a los profesores, evaluar los resultados de este aprendizaje expresados en términos de competencias.
[1] A.G. Asuero, G. Galán, M.A. Herrador, A.M. Jiménez, M.T. Montaña, M.T. Morales, M.J. Navas, Trends in Pharmacy Education, European Association of Faculties of Pharmacy, Annual Conference, 56-57.


COMUNICACIONES EN CARTEL

C1-1 COMUNICACIONES EN CARTEL
42
CARACTERIZACIÓN DE LAS ESPECIES COMERCIALES DE CAFÉ (ARÁBICA Y ROBUSTA) SEGÚN SU CONTENIDO EN CERAS
M.J. Martín, F. Pablos, M. León-Camacho, A. Alcázar, J.M. Jurado
Dpto. Química Analítica, Univ. Sevilla, Sevilla. E-mail: [email protected]
Una de las bebidas más consumidas en todo el mundo es el café, obtenido a partir de las semillas tostadas de un arbusto que pertenece al género Coffea. En su elaboración, se emplean fundamentalmente dos especies Coffea arabica y Coffea canephora, denominadas comúnmente arabica y robusta. Los cafés comerciales se preparan a partir de granos de las variedades arabica y robusta o mezclas de los dos. Ambas especies son bastante diferentes desde el punto de vista organoléptico: la especie arabica tiene mejor sabor y un aroma más suave y por ello es más apreciada por los consumidores, alcanzando un mayor precio en el mercado.
En cuanto a su composición química, el grano de café es bastante complejo
siendo fundamental el tipo de tueste al que es sometido, así como su duración. Durante esta etapa de su elaboración, se origina un gran número de compuestos que cambian las características organolépticas de la bebida final. Agua, carbohidratos, sustancias nitrogenadas, contenido mineral y compuestos volátiles, entre otros son los principales constituyentes de este producto.
Mención especial debe realizarse a la fracción grasa del café. El aceite extraído
de esta semilla está compuesto por ácidos grasos, tocoferoles, triglicéridos, esteroles, alcoholes, entre otros. Varios han sido los estudios realizados analizando el contenido de estos compuestos en ambas especies de café [1-3]. Otro de los compuestos que forman parte del aceite del café son las ceras, ésteres de ácidos grasos y alcoholes alifáticos de cadena larga.
En esta comunicación se realiza un estudio del contenido de ceras en granos de
café arabica y robusta mediante cromatografía de gases con detector de ionización de llama. Posteriormente, se realiza un tratamiento matemático de los datos obtenidos aplicando, en primer lugar, tests no paramétricos que confirman diferencias significativas en el contenido de diversas ceras en una especie con respecto a la otra y, en segundo lugar, técnicas de reconocimiento de patrones como análisis discriminante lineal y análisis cluster empleando el contenido de ceras analizado como descriptores químicos para establecer una diferenciación y caracterización de ambas especies.
[1] M.S. Valdenebro, M. León-Camacho, F. Pablos, A.G. González, M.J. Martín, The
analyst 124 (1999) 999-1002. [2] M.J. Martín, F. Pablos, A.G. González, M.S. Valdenebro, M. León-Camacho,
Talanta 54 (2001) 291-297. [3] A.G. González, F. Pablos, M.J. Martín, M. León-Camacho, M.S. Valdenebro,
Food Chem. 73 (2001) 93-101.

COMUNICACIONES EN CARTEL C1-2
43
ANALYSIS OF METAL RESIDUES IN EDIBLE OILS AND VEGETABLES
FROM SPAIN AND MOROCCO
K. Bakkali,1 N. Ramos,1 B. Souhail,2 E. Ballesteros3
1Departamento de Química Física y Analítica, Facultad de Ciencias, Universidad de Jaén, E-23071, Jaén, Spain. E-mail: [email protected]
2Départment de Chimie, Faculté des Sciences, Université Abdelmalek Essaadi, Tétouan, Maroc
3Departamento de Química Física y Analítica, E.P.S. de Linares, Universidad de Jaén, E-23700 Linares, Spain. E-mail: [email protected]
The metals and metalloids present in the earth’s crust, soils, water, the
atmosphere and biospheres, and hence those in foods, are stable, environmentally persistent species having a slow rate of elimination, accumulating in tissues, and undergoing some transformations that increase their toxicity. Specially worrisome among the metals and metalloids present in foods are lead, cadmium, arsenic and mercury on account of their toxicological potential. As such, they have elicited a response in most countries in the form of legal restrictions on their presence in foods [1]. Other metals including cooper, iron, tin, zinc, antimony, aluminium, magnesium, manganese, molybdenum and chromium, which are present in trace amounts in the human diet, may also be the source of toxic hazards to humans under specific conditions [2].
A number of methods for improving quality assurance in analytical methods for
the determination of metals in food samples have been developed most of which include a sample digestion step. Wet and dry ashing are the two most widely used methodologies for digesting these samples; however, they have some drawbacks including long sample processing times, and high costs and hazards. In recent years, microwave ovens have been broadly employed for the acid digestion of a variety of food samples as an effective alternative to classical digestion procedures. Also, atomic spectrometric techniques continue to be the preferred choices for determining metals in food samples [3].
In the present communication, a simple, expeditious method for the
determination of trace metals (cadmium, chromium, copper, manganese and lead) is proposed. The metals are extracted from their matrix by using nitric acid and hydrogen peroxide in a closed-vessel microwave digestion system and subsequently detected by graphite furnace atomic absorption spectrometry (GFAAS) or inductively coupled plasma mass spectrometry (ICP-MS). A comparative study of both detection methods in terms of sensitivity, selectivity, accuracy and precision was conducted. Accuracy was assessed by using two certified reference materials (NCS ZC85006 Tomato and Used Oil HU-1). Also, both methods were applied to analysis of vegetables (tomato, pepper, onion) and edible oils (olive, sunflower, sunflower, maize and pomace olive). The metal contents in the studied foods were found to depend on food type and country of origin.
[1] EEC, Economic European Communities. Setting maximum levels for certain
contaminants in foodstuffs. Official Journal of the European Communities, L 364/5-L 364/24, Directive No. 1881/2006, Brussels.
[2] F. E. Ahmed. Trace metal contaminants in food. In C. F. Moffat, K. J. Whittle (Eds.), Environmental Contaminants in Food (pp 146–214). Sheffield: CRC Press. 1999.
[3] J. R. Bacon, K. L. Linge, R. R. Parrish, L. van Vaeck. J. Anal. At. Spectrom. 23 (2008) 1130–1162.

C1-3 COMUNICACIONES EN CARTEL
44
QUANTITATIVE DETERMINATION OF CAFFEINE, FORMIC ACID,
TRIGONELLINE AND 5-(HYDROXYMETHYL)FURFURAL IN SOLUBLE COFFEES BY 1H NMR SPECTROMETRY
G. del Campo, I. Berregi, J. Zuriarrain, R. Caracena
Department of Applied Chemistry, University of the Basque Country
Donostia-San Sebastián. E-mail: [email protected] A quantitative method for the determination of caffeine, formic acid,
trigonelline and 5-(hydroxymethyl)furfural (5-HMF) in soluble coffees by applying the proton nuclear magnetic resonance technique (1H NMR) is proposed. Each of these compounds records a singlet signal at the 7.6-9.5 ppm interval of the spectrum, and its area is used to determine the concentration. 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid (TSP) is added in an exact known concentration as a reference for δ = 0.00 ppm and as an internal standard.
Figure 1 shows the low field 1H NMR spectrum of a commercial soluble coffee.
Working signals of caffeine, formic acid, trigonelline and 5-HMF are indicated. The signal of TSP, not shown here, is another singlet at 0 ppm.
Figure 1. Low field 1H NMR spectrum of Baqué soluble coffee. The calibration graphs are obtained by plotting the ratio between the peak
areas of each analyte (A) and the internal standard TSP (ATSP) against analyte concentration (C, mg/L). The general equation is A/ATSP = a × C(mg/L) + b. The number of experimental points is 9 in all cases. Results are summarized in table 1.
Table 1. Calibration data for the general equation A/ATSP = a × C(mg/L) + b
Analyte a ± Sa b ± Sb R Sy/x LOD mg/L
LOD mg/g
CV %
Caffeine (4.832±0.054)×10–3 (1.446±4.658)×10–2 0.9996 8.5655×10–2 53 1.32 4.2
Formic acid (1.530±0.027)×10–2 (3.968±5.653)×10–2 0.9989 9.3334×10–2 18 0.45 2.6
Trigonelline (4.295±0.053)×10–3 (5.811±18.16)×10–3 0.9995 3.3403×10–2 23 0.58 2.4
5-HMF (4.854±0.062)×10–3 (3.351±11.01)×10–3 0.9994 1.8674×10–2 12 0.30 7.3
Sy/x: standard error for regression line LOD: limit of detection CV: coefficient of variation
The method is applied to commercial soluble coffees and satisfactorily
compared with results obtained by HPLC and enzymatic techniques. It is a direct method and no previous derivatization is needed.
Caffeine
Formic acid
Trigonelline 5-HMF
ppm8.008.509.009.50

COMUNICACIONES EN CARTEL C1-4
45
STUDY OF SELENIUM BIOACCESSIBILITY IN SOILS
V. Funes-Collado, J.Fermín López-Sánchez, Roser Rubio
Departament de Química Analítica. Facultat de Química. Universitat de Barcelona.
E-mail: [email protected]
The effects of soil pollutants upon living organisms depend on the degree to which the pollutants can be adsorbed or metabolised by the receptors. Bioaccessibility, through in vitro tests using simulated gastrointestinal digestions, allows to establish a correlation with in vivo bioavailability.
The main source to incorporate selenium into the organism is through
vegetables, then is first necessary to need the concentration of selenium that contain the soil where has been cultivated. Some studies have been reported on bioaccessibility of metals from contaminated soils for estimating their bioavailability through oral exposure. None of the studies reported deal on selenium and moreover all of the studies consider only the measurement of total metal concentration in the digestion extracts from the in vitro tests applied. One of the most accepted scheme for these studies is the method of Ruby et al, (1993), called Physiologically Based Extraction Test (PBET). This method establishes conditions that simulate the gastric and intestinal phases of the process of human digestion, establishing values of pH, temperature and time of residence for each of the phases.
In the present work, studies of bioaccessibility have been started as a
preliminary step to estimate the bioavailability of selenium in the human organism. Two soil samples and two certificated reference materials, with very different concentration levels of selenium, have been selected for the study. The determination of the total Se in the extracts, as well as in soils, is reported by HG-AFS and ICPMS. Moreover, for the first time, the analytical speciation of Se is carried out in the digestion extracts. For this, the coupling LC-UV-HG-AFS and LC-ICPMS are used. Moreover, the results from the PBET method has been compared with those from a phosphoric extraction, usually applied for assessing extractable of metals in soils. Finally, quality parameters, as recoveries, repeatability, reproducibility and detection and quantification limits when applying the PBET test from both, total Se and Se speciation, have been established.

C1-5 COMUNICACIONES EN CARTEL
46
APLICACIÓN DE LA MICROEXTRACCIÓN LÍQUIDO-LÍQUIDO (LPME) A LA
DETERMINACIÓN CROMATOGRÁFICA (HPLC) DE DICLOFENACO, IBUPROFENO Y ÁCIDO SALICÍLICO EN AGUAS RESIDUALES
M.D. Ramos-Payán, M.A. Bello-López, R. Fernández-Torres,
M. Callejón-Mochón, M. Villar-Navarro
Dpto de Química Analítica. Univ. de Sevilla. E-mail:[email protected]
Los productos farmacéuticos son un amplio y diverso grupo de productos químicos elaborados y utilizados para producir efectos biológicos en la salud humana y animal.
El creciente consumo de productos farmacéuticos en todo el mundo está
generando una serie de residuos o microcontaminantes que hace tan solo unos años no existían o sus niveles no eran, en absoluto, preocupantes. La posibilidad de que se encuentren en el medio ambiente se ha convertido en una cuestión importante en los últimos años ya que por su propia naturaleza su presencia en el medio ambiente es problemática debido a su intensa actividad biológica inherente. Además, la introducción en el mercado de nuevos fármacos aumenta de forma exponencial, apareciendo un enorme espectro de componentes activos cuya actividad biológica es, hoy día, en parte desconocida. Tras su ingesta, estas sustancias son transformadas a través de los procesos metabólicos y, una vez liberadas, pueden permanecer como tal o bien transformarse en nuevos productos que en muchos casos poseen una actividad biológica incluso mayor que los productos de partida.
Numerosas evidencias indican que los fármacos que se consumen
habitualmente alcanzan el suelo, las aguas subterráneas y superficiales. La concentración presente en el medio ambiente de estas sustancias depende de su consumo y de la tasa de excreción a través de las heces y la orina. Estos productos terminan alcanzando los sistemas de alcantarillado y llegando a las estaciones depuradoras de aguas residuales, donde sus niveles pueden, o no, descender; los efluentes de salida se incorporan las aguas naturales, que se pueden ver, de esta forma, contaminadas.
En este trabajo se propone una novedosa metodología analítica para la
extracción y determinación simultánea de tres antiinflamatorios en aguas residuales. Los principios activos farmacológicos estudiados han sido el ácido salicílico (principal producto de degradación del ácido acetilsalicílico), ibuprofeno y diclofenaco. El método se basa en la puesta a punto de un procedimiento de microextracción en fase líquida (LMPE) empleando fibras huecas lo que permite aumentar la sensibilidad y selectividad del método propuesto, y posterior determinación por cromatografía líquida de alta resolución con detectores en serie de fila de diodos y fluorescencia. Dicho procedimiento se ha aplicado satisfactoriamente a la determinación simultánea de ácido salicílico, ibuprofeno y diclofenaco en aguas residuales.

COMUNICACIONES EN CARTEL C1-6
47
OPTIMIZATION OF A SPE-HPLC-ESI-MS/MS METHOD FOR THE
DETERMINATION OF PHARMACEUTICAL RESIDUES AND ITS APLICATION TO GROUNDWATERS
S. Montesdeoca1, E. Estévez 2, M.C. Cabrera2, Z. Sosa 1, J.J. Santana 1
1Department of Chemistry 2Department of Physics.
Faculty of Marine Sciences, University Las Palmas de G.C., 35017 Las Palmas de Gran Canaria. E-mail: [email protected]
A liquid chromatography-electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) method was developed for the determination of different pharmaceutical compounds: atenolol (B-bloker), metamizol (analgesic), fluoxetine (antidepressant), caffeine (stimulant), and its metabolite para-xanthine, as well as nicotine and permethrin, in environmental water matrices.
These analytes can reach to the sewage water treatment plants, which are
unable to remove them completely. Thus, urban wastewaters typically have a strongly contaminating effect on the natural aquatic systems [1].However, their environmental concentrations are very low, for that these analytes need to be extracted and preconcentrated prior to environmental analysis. In this way, a solid-phase extraction step prior to their determination has been very efficient [2].
In this work we optimize the MS/MS condition for each compound by direct
infusion of pure standard. Parameters that affect the SPE process were also optimized and the analytical characteristics of the method (linearity, reproducibility and validation) were established.
Finally, the proposed method was applied to the determination of these target
analytes in different groundwater samples taken in different wells in Gran Canaria island. [1] M.J. Martínez, A. Agüera, M.J. Gómez, M.D. Hernando, J.F. García-Reyes, A.R.
Fernández Alba, Anal Chem. 79 (2007) 9372 [2] Z. Moldovan, Chemosphere 64 (2006) 1808

C1-7 COMUNICACIONES EN CARTEL
48
DEVELOPMENT OF A NEAR INFRARED SPECTROSCOPY FINGERPRINT OF
PANAX GINSENG BY PATTERN RECOGNITION APROACHES
J. R. Lucio-Gutiérrez, J. Coello, S. Maspoch
Unitat de Química Analítica, Departament de Química, Universitat Autònoma de Barcelona E-08193-Bellaterra (Spain). Phone: +34 935812122;
Fax: +34 935812357; e-mail: [email protected]
Ginseng, the root of Panax ginseng C.A. Meyer, is one of the most famous herbal medicines used as dietary supplement, due to its wide spectrum of pharmacological properties and physiological activities. These include cancer-related, antiinflamatory, antiallergic, immunomodulatory, anti-diabetic and adaptogenic activities. The ginseng herb has traditionally been authenticated by morphological and histological means. However, this approach is hardly reliable nowadays because ginseng species are not only morphological similar, but commercial products are also prepared in various ways which make it harder to identify by smell, taste and appearance. Furthermore, as ginseng is expensive, adulteration with cheaper products occurs and adulterators are continuously trying to develop ways to make their product's chemical profile similar to the authentic ginseng. This raises many concerns with regards to its quality control, which is still a common problem today. Currently, there is a common practice among natural products analysts to select one or more compounds as "markers" for purposes of identification and quality assessment. As many herbs and related species could contain the same compounds and only few compounds are used, such approach can fail. To cope this problem, a pattern approach can be useful to develop the fingerprint since it considers the whole spectrum as a feature, in this way the whole herbal medicine is regarded as one active ingredient.
In this work, the NIR spectral fingerprints of the ginsengs (1100-2498 nm)
were used for the analysis since the difference in chemical composition of the ginsengs would be reflected in the combination and overtone bands of their near infrared (NIR) spectra. Samples of Panax ginseng and herbs consider "ginsengs" were obtained from several international suppliers and herb stores of metropolitan area of Barcelona. A minimum treatment of the sample was needed: the dried roots were ground with a blender and screened through a 100 mesh sieve. The spectra were collected using a NirSystems 5000 with a module for Rapid Content Analyzer from Foss. Soft independent modeling of class analogy (SIMCA), partial least squares discriminant analysis (PLS-DA) and linear discriminant analysis (LDA) were performed using the software packages Unscrambler 9.8 (CAMO) and SPSS 15.0. Standard normal variate (SNV) was selected like spectra pretreatment. According to the results obtained with the different algorithms, proper classification of Panax ginseng samples and mixtures detection can be achieved, being SIMCA the algorithm which exhibit the more reliable and readily results using a significance level of 5%.
“Supported by the Programme Alβan, the European Union Programme of High
Level Scholarships for Latin America, scholarship No. E07D401919MX”

COMUNICACIONES EN CARTEL C1-8
49
FOCUSED ULTRASOUND ASSISTED ENZYMATIC HYDROLYSIS OF NONYL-
AND OCTYL PHENOL GLUCURONIDES. APPLICATION TO THE DETERMINATION OF NONYL- AND OCTYLPHENOLS IN FISH BILE
A. Vallejo, A. Usobiaga, I. Martinez-Arkarazo, L.A. Fernández, O. Zuloaga
Kimika Analitikoa Saila, Euskal Herriko Unibertsitatea, PK 644, 48080 Bilbao.
E-mail: [email protected] Nonyl- and octylphenols (NPs and OPs) are some of the priority organic
pollutants considered by the Water Framework Directive (WFD) due to their endocrine disruptive properties [1]. Since analytes such as NPs and OPs are not completely removed in Waste Water Treatment Plants (WWTPs), their concentrations are often measured in wastewater effluents [2-3]. However, no conclusive association has been proven yet between environmentally relevant concentrations and estrogenic effects. In this sense, the measurement of those analytes in in-vivo experiments is necessary to understand their estrogenic activity. NPs and OPs tend to accumulate in the bile of fishes as glucuronide derivative but, however, very little is known about their concentration in such tissues [4].
Since NPs and OPs are found as glucuronide derivatives in fish bile, their
determination requires of an enzymatic hydrolysis previous to the preconcentration and analysis steps. Classical enzymatic hydrolysis procedures are usually time-consuming (16-18 hours) [4]. In this work, ultrasound energy was used in order to reduce this step by means of titanium ultrasound probes. Variables such as the hydrolysis time (1-20 min), ultrasound power (5-25%) and the number of cycles (1-7) were studied by means of an experimental design approach. Since NP and Op glucuronides are not commercially available, β-D-nitrophenol glucuronide, 4-nitrophenol sulphate and and salicin glucose were used as substrates. The concentration of the enzymes glucuronidase, sulfatase and glucosidase were kept constant during the whole optimisation. Under optimum conditions (1 cycle, 20 min and 10% power), repeatability of the method was studied. After solid-phase extraction (SPE) clean-up of the extracts, the compounds were analysed with and without derivatisation in a gas chromatograph coupled to a mass spectrometer (GC-MS). Linear ranges were within 16-240 ng for the derivatised analytes. The optimised method was applied to the determination of NPs and OPs from fish biles from the estuary of Urdaibai (Basque Country).
AKNOWLEDGEMENTS: This project has been financially supported by the
University of the Basque Country through the UNESCO07/09 project and the Spanish Ministry of Science and Innovation through the CTQ2008-02775/BQU project project.
[1] M. Coquery, A. Morin, A. Bécue, B. Lepot, Trends Anal. Chem. 24 (2005) 117-127.
[2] B.L.L. Tan, D.W. Hawker, J.F. Müller, L.A. Tremblay, H.F. Chapman, Water Res. 42 (2008) 404-412.
[3] D. Barceló, M. Petrovic, Trends Anal. Chem. 26 (2007) 647-649. [4] R. Gibson, C.R. Tyler, E.M. Hill, J. Chromatogr A 1066 (2005) 33-40.

C1-9 COMUNICACIONES EN CARTEL
50
ESPECIACIÓN DE ARSÉNICO EN MUESTRAS DE ALGAS MARINAS
MEDIANTE HPLC-ICP/MS
T.Llorente*, M.J.Ruiz-Chancho*, M.Barbero**, R.Rubio*, J.F. López-Sánchez*
*Dpto. Química Analítica, Universidad de Barcelona, Barcelona.
E-mail: [email protected] **Dpto. de Biología Vegetal (Botánica), Universidad de Barcelona, Barcelona
El objetivo de este trabajo es el estudio de la especiación de arsénico en una población natural de algas de una zona del litoral de la costa catalana (Mar Mediterráneo). En particular el estudio se centró en los comúnmente llamados arsenoazúcares, los compuestos de arsénico que se acostumbran a encontrar mayoritariamente en algas.
Se tomaron muestras de diversas especies de algas en las zonas rocosas de la
playa de Santa Cristina de Lloret de Mar (Costa Brava, Catalunya), durante la marea baja, en Noviembre de 2008. Algunas algas tenían gran cantidad de epífitos depositados en su superficie que fueron difíciles de eliminar. Así pués, se procedió a analizar estas muestras sin epífitos y con epífitos.
El contenido total de arsénico de las muestras se determinó por ICP-MS
después de una digestión ácida utilizando un equipo de microondas. Para llevar a cabo la especiación de arsénico se aplicó una extracción con agua mediante agitación mecánica. Para la separación de las especies de As presentes en los extractos acuosos se utilizaron dos tipos de separaciones cromatográficas y se utilizó el acoplamiento HPLC-ICP/MS. Uno de los métodos utilizó una columna de intercambio aniónico con una columna Hamilton PRP-X100 y una fase móvil de dihidrogeno fosfato de amonio. El otro método utilizado fue de intercambio catiónico con una columna Zorbax 300-SCX y una fase móvil de piridina. En ambas separaciones la salida de la columna se conectó directamente al nebulizador del instrumento ICP/MS utilizando un tubo de PEEK.
Los resultados obtenidos mostraron un intervalo de concentración de As total
entre 2.0 y 39.0 mg As kg-1. Se obtuvieron concentraciones más elevadas en algas que contenían epifitos, siempre que se compararon algas de la misma especie. En general, dentro de las algas que se encontraban sumergidas a la misma profundidad, las algas que pertenecen a la división Chlorophyta contienen mayor concentración de As total que las que pertenecen a las divisiones Rhodophyta y Heterokontophyta (Cl. Phaeophyceae). Los rendimientos de extracción de las especies de arsénico dependieron mucho del tipo de alga y variaron entre el 43% y el 89 %. Se identificaron y cuantificaron una gran proporción de arsenoazúcares en la mayoría de las algas, no obstante, algunas presentaban altos porcentajes de arsénico inorgánico en forma de As (V). También se determinaron otras especies minoritarias como MMA, DMA y As (III). Se detectaron pequeñas cantidades de Arsenobetaina en muchas de las algas estudiadas, que probablemente se podrían atribuir a la presencia de los epífitos adheridos a la superficie de las muestras.

COMUNICACIONES EN CARTEL C1-10
51
DETERMINACIÓN DE COMPUESTOS DE As EN ALGAS COMESTIBLES
MEDIANTE HPLC-ICP/MS
T.Llorente*, M.J.Ruiz-Chancho*, M.Barbero**, R.Rubio*, J.F. López-Sánchez*
*Dpto. Química Analítica, Universidad de Barcelona, Barcelona.
E-mail: [email protected] **Dpto. de Biología Vegetal (Botánica), Universidad de Barcelona, Barcelona
La especiación de arsénico es de especial interés para conocer los posibles compuestos de este elemento que se pueden encontrar en alimentos, muestras medioambientales, entre otros, con el fin de poder estimar su posible toxicidad. En el caso de las algas estos estudios son importantes ya que hay diversas variedades que se destinan al consumo humano. Las algas son muy populares en la cocina china y japonesa, forman parte de su dieta habitual y por lo tanto su consumo es bastante elevado. Hoy en día, su uso también se ha extendido a países occidentales a causa del su alto contenido mineral y sus propiedades terapéuticas reconocidas. La gran mayoría de las algas estudiadas contienen casi exclusivamente arsenoazúcares. El metabolismo de estos compuestos derivados de los carbohidratos es casi desconocido, pero podrían tener una toxicidad potencial. La información sobre la presencia de las especies de arsénico en las algas comestibles ayudará a evaluar el riesgo asociado con la entrada de arsénico en la cadena alimentaria humana.
En el presente trabajo se realizó un estudio de especiación de arsénico en algas
comerciales, destinadas al consumo humano, (procedentes de la Costa Gallega). En particular el estudio se centró en los arsenoazúcares, los compuestos de arsénico que se acostumbran a encontrar mayoritariamente en algas y también en las especies inorgánicas As(III) y As(V) por su elevada toxicidad. Para ello se llevó a cabo una extracción con agua y agitación mecánica y se realizó la determinación de las especies de arsénico mediante el acoplamiento HPLC-ICP/MS. Para obtener la mayor información sobre las especies de arsénico se utilizaron dos sistemas cromatográficos, utilizando intercambio aniónico y catiónico respectivamente. El contenido total de arsénico de las muestras se determinó por ICP-MS después de la digestión ácida de las algas utilizando un equipo de microondas.
Se encontraron niveles relativamente elevados de As en las algas estudiadas,
en un intervalo desde 5.8 a 56.8 mg As kg-1. Los rendimientos de extracción de las especies fueron en general elevados. Se identificaron y cuantificaron mayoritariamente arsenoazúcares en las especies Porphyra purpurea, Chondrus crispus, Ulva rigida, Laminaria ochroleuca, Laminaria saccharina y Undaria pinnatifida.

C1-11 COMUNICACIONES EN CARTEL
52
PREDICTION OF THE GENETIC VARIETY OF EXTRA VIRGIN OLIVE OILS PRODUCED AT LA COMUNITAT VALENCIANA, SPAIN, BY FOURIER-
TRANSFORM INFRARED SPECTROSCOPY
M.J. Lerma-García1, V. Concha-Herrera2, J.M. Herrero-Martínez1, E.F. Simó-Alfonso1
1Dto. Química Analítica, Univ. Valencia, Burjassot, Valencia. Email: [email protected]
2Unidad Académica de Ciencias Químicas, Univ. Autónoma de Zacatecas, Zacatecas, México
Fourier-transform infrared spectroscopy (FTIR), followed by multivariate treatment of the spectral data, was used to classify extra virgin olive oils (EVOO) from La Comunitat Valenciana according to their genetic variety. EVOO samples corresponding to 7 different genetic varieties (Arbequina, Borriolenca, Canetera, Farga, Hojiblanca, Picual and Serrana) were analyzed. The wavelength scale of the FTIR spectra of the oils was divided in 20 regions. The normalized absorbance peak areas within these regions were used as predictor variables. Classification of the EVOO samples according to their genetic variety was achieved by linear discriminant analysis (LDA). An excellent resolution among all categories was achieved using an LDA model constructed with only 9 predictor variables. With this LDA model, an 88 % of the EVOOs were correctly classified with assignment probabilities higher than 95 %.
Project CTQ2007-61445 (MEC of Spain and FEDER funds). V. Concha-Herrera
thanks the University of Valencia for a contract. M.J. Lerma-García also thanks the Generalitat Valenciana for an FPI grant for PhD studies.

COMUNICACIONES EN CARTEL C1-12
53
DETERMINATION OF FOODSTUFFS ADULTERATION WITH NMR SPECTROMETRIC TECHNIQUE AND CHEMOMETRIC APPROACH
C. Di Anibal, M.P. Callao, I. Ruisánchez
Departament de Química Analítica i Química Orgànica. Universitat Rovira i Virgili.
Campus Sescelades, C/. Marcel�lí Domingo, s/n 43007 Tarragona, SPAIN [email protected]; [email protected];
Sudan dyes I, II, III, IV are an azo-family of red synthetic dyes categorized as Class 3 carcinogens by the International Agency for Research on Cancer (IARC) [1]. Among its different uses in industry as colorant, Sudan dyes have been discovered in spices like chilli powder and food products containing chilli powder, curry, turmeric, mild and hot paprika, among others. Therefore, Sudan dyes (I to IV) are illegal as additives in foodstuffs destined to human consumption according to both the FSA [2] and the European Union.
In this work, we propose the detection of Sudan dyes in spices through NMR
spectrometric measurements and a multivariate classification technique as Partial Least Squares-Discriminant Analysis (PLS-DA) [3,4]. Nuclear Magnetic Resonance has progressed to become one powerful analytical tool [5,6] and it could proportionate a rapid screening method for detecting Sudan dyes in spices destined to human consumption.
A NMR spectrum have several thousands measurement points containing only a
few of them (hundred of features) relevant information, in our case to discriminate among a predefined classes. Because of this, variable selection has to be done in order to facilitate data visualization and data understanding, to reduce time computation and to select the most relevant variables to improve the classification results. The main goal is to select those NMR zones that are indicative of the presence of any Sudan dye using a univariate variable selection procedure, in which the statistic are calculated at each NMR frequency (data point) using the non-contaminated samples spectra. These selected variables are subjected to further PLS-DA analysis in order to obtain the classification results.
We work with 27 commercial spices (curry, turmeric and mild and hot paprika),
distributed as 5 classes: natural samples not contaminated and samples spiked independently with Sudan I, II, III or IV, giving a total of 135 samples. Before the NMR analysis, each commercial sample was analysed by HLPC-DAD to confirm that they were free of Sudan dyes. Very good classification results have been obtained and which is more important no false negative results are obtained, samples contaminated with any of the four Sudan dyes are not classified as non- contaminated. [1] IARC (1975) Monographs on the evaluation of the carcinogenic risk of chemical
to man: some aromatic azo compounds (Vol. 8). Lyon, France, International Agency for Research on Cancer, pp. 224-231.
[2] http://www.food.gov.uk/foodindustry/guidancenotes/foodguid/sudanguidance [3] M. Barker, W. Rayens, Journal of Chemometrics 17 (2003) 166-173. [4] K. Hovde Liland, U. Geir Nidal, Journal of Chemometrics 23 (2009) 7-18. [5] A. M. Weljie, J. Newton, P. Mercier, E. Carlson, C. M. Slupsky, Analytical
Chemistry 78 (2006) 4430-4442. [6] M. Lolli, D. Bertelli, M. Plessi, A. G. Sabationi, C. Restani. Journal of Agricultural
and Food Chemistry 56 (2008) 1298-1304.
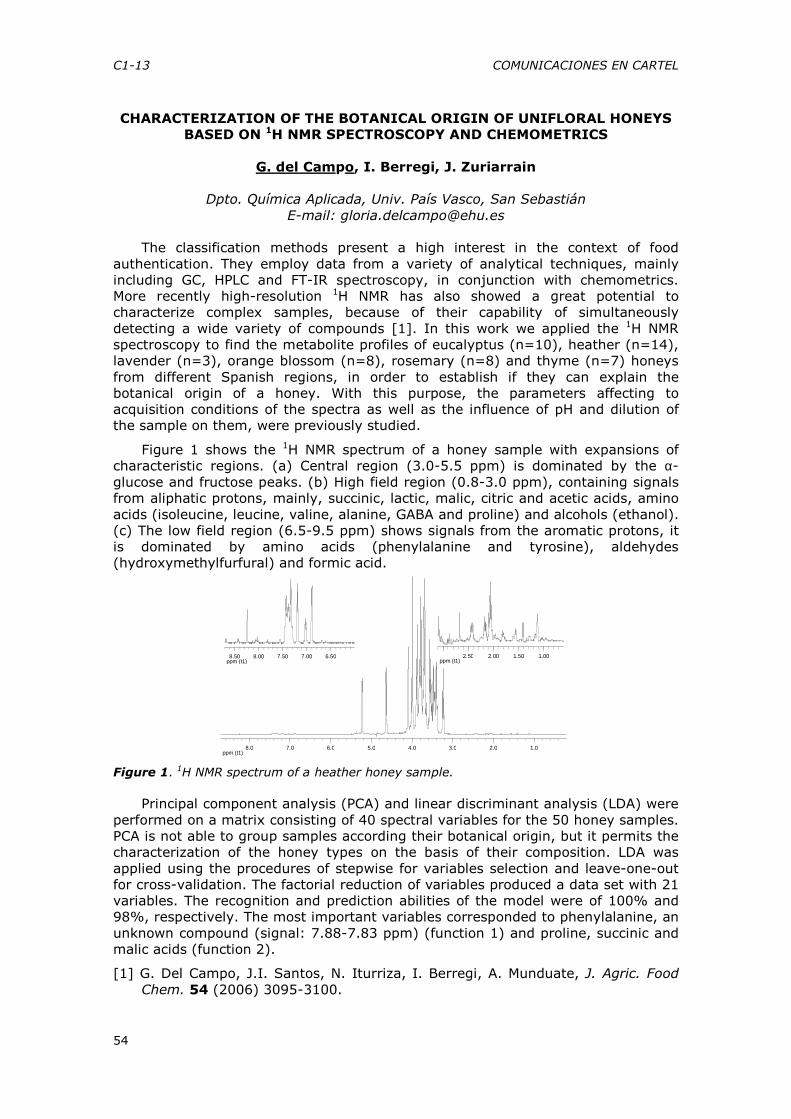
C1-13 COMUNICACIONES EN CARTEL
54
ppm (t1)1.02.03.04.05.06.07.08.0
ppm (t1)6.507.007.508.008.50
ppm (t1)1.001.502.002.50
CHARACTERIZATION OF THE BOTANICAL ORIGIN OF UNIFLORAL HONEYS
BASED ON 1H NMR SPECTROSCOPY AND CHEMOMETRICS
G. del Campo, I. Berregi, J. Zuriarrain
Dpto. Química Aplicada, Univ. País Vasco, San Sebastián E-mail: [email protected]
The classification methods present a high interest in the context of food
authentication. They employ data from a variety of analytical techniques, mainly including GC, HPLC and FT-IR spectroscopy, in conjunction with chemometrics. More recently high-resolution 1H NMR has also showed a great potential to characterize complex samples, because of their capability of simultaneously detecting a wide variety of compounds [1]. In this work we applied the 1H NMR spectroscopy to find the metabolite profiles of eucalyptus (n=10), heather (n=14), lavender (n=3), orange blossom (n=8), rosemary (n=8) and thyme (n=7) honeys from different Spanish regions, in order to establish if they can explain the botanical origin of a honey. With this purpose, the parameters affecting to acquisition conditions of the spectra as well as the influence of pH and dilution of the sample on them, were previously studied.
Figure 1 shows the 1H NMR spectrum of a honey sample with expansions of characteristic regions. (a) Central region (3.0-5.5 ppm) is dominated by the α-glucose and fructose peaks. (b) High field region (0.8-3.0 ppm), containing signals from aliphatic protons, mainly, succinic, lactic, malic, citric and acetic acids, amino acids (isoleucine, leucine, valine, alanine, GABA and proline) and alcohols (ethanol). (c) The low field region (6.5-9.5 ppm) shows signals from the aromatic protons, it is dominated by amino acids (phenylalanine and tyrosine), aldehydes (hydroxymethylfurfural) and formic acid.
Figure 1. 1H NMR spectrum of a heather honey sample.
Principal component analysis (PCA) and linear discriminant analysis (LDA) were performed on a matrix consisting of 40 spectral variables for the 50 honey samples. PCA is not able to group samples according their botanical origin, but it permits the characterization of the honey types on the basis of their composition. LDA was applied using the procedures of stepwise for variables selection and leave-one-out for cross-validation. The factorial reduction of variables produced a data set with 21 variables. The recognition and prediction abilities of the model were of 100% and 98%, respectively. The most important variables corresponded to phenylalanine, an unknown compound (signal: 7.88-7.83 ppm) (function 1) and proline, succinic and malic acids (function 2).
[1] G. Del Campo, J.I. Santos, N. Iturriza, I. Berregi, A. Munduate, J. Agric. Food Chem. 54 (2006) 3095-3100.

COMUNICACIONES EN CARTEL C1-14
55
AUTOMATED PARAFAC MODELING OF FLUORESCENCE EEM DATA
R. Broa, M. Vidalb*
aDepartment of Food Science, Quality and Technology, Faculty of Life Sciences, University of Copenhagen, Rolighedsvej 30, DK-1958 Frederiksberg C, Denmark
bDpto. Química Aplicada, Facultad de Química, Universidad del País Vasco, 20080–San Sebastián, Spain *E-mail: [email protected]
PARAFAC is a superior technique for modelling multivariate fluorescence EEM
data. By mathematical chromatography, it allows to estimate pure spectra and concentrations from mixture measurements under mild conditions. In practice, however, widespread use of PARAFAC is hindered by the problems of finding the right PARAFAC model. It is crucial, in particular for PARAFAC, that the right number of components is used and that non-consistent parts of the data are removed, but this has not been traditionally an easy task.
The proposal of this communication is to show an automated method that can
ease the model building dramatically. This has been achieved through a new algorithm, the EEMizer function, with automatically runs a series of PARAFAC models for different variables introduced.
The algorithm is simply called as follows (MATLAB notation):
result=EEMizer(X,Factors) where X is the original data array, and Factors refers to the span of number of components tested.
Despite the simple call, the algorithm, will test
• different number of components, • different approaches to handling Rayleigh scatter • possible outliers • low excitation wavelengths that are disturbing the model. The output of the function is an overall quality measure between 0 and 1 with 1
indicating a perfect model. Among all the calculated models, the best ones are chosen based on this quality measure. The measure is reflecting the explained variance, the core consistency and the results of split-half validation. A value close to 0 indicates that the model is not fitting the data suitably because the data are not enough trilinear or, on the contrary, because a wrong choice in the number of components etc. has been made. However, the higher the value, the better the model is.
The function has been tested on very different fluorescence datasets and it is
found that it greatly simplifies modelling when the data are of a sufficient quality (enough samples and adequately spanning of the fluorophores). It allows, therefore, to obtain good PARAFAC model without time-consuming use of skilled personnel.
ACKNOWLEDGEMENT. M.V. acknowledges financial support from GV in the form
of a pre-doctoral fellowship, MICINN (Project CTQ2008-06751-C02-02/BQU) and UPV/EHU (Project GIU07/58).

C1-15 COMUNICACIONES EN CARTEL
56
FORENSIC ANALYSIS OF DRUGS OF ABUSE AND OTHER STIMULANTS IN HAIR BY RAPID RESOLUTION LIQUID CHROMATOGRAPHY TIME-OF-FLIGHT
MASS SPECTROMETRY Juan C. Domínguez-Romero, Juan F. García-Reyes and Antonio Molina-Díaz
Analytical Chemistry Research Group, Department of Physical and Analytical
Chemistry, University of Jaén, Campus Las Lagunillas, Ed. B-3, 23071 Jaén, Spain. [email protected]
The analysis of drugs in hair samples has become very popular in recent years, since it has potential applications in forensic and clinical toxicology. It has become an alternative and a complementary approach to drug abuse detection and offers some advantages over urine assays. The collection of hair specimens is less embarrassing and intrusive, drugs and metabolites incorporated into hair persist longer than in urine, hair can be stored at room temperature and do not need to be analysed shortly after collection. In order to increase the ability of drug testing, development of a rapid and simultaneous method for the analysis of multiple drugs is required.
In this study we have developed a rapid method for simultaneous quantitative
determination of 30 multiclass drugs of abuse (including cannabinoids, amphetamines and opiates) in hair samples using liquid chromatography time-of-flight mass spectrometry (LC-TOFMS). The proposed methodology comprises a sample treatment step consisting of three different extraction protocols (methanolic extraction in ultrasonic bath, acidic methanol extraction in ultrasonic bath and alkaline digestion). The separation was carried out using a high-performance liquid chromatography (HPLC) system (Agilent series 1200, Agilent Technologies) equipped with a reversed-phase XDB-C18 analytical column of 4.6mm x 50mm and 1.8µm particle size (Agilent Technologies). 20µL of the sample extract were injected. Mobile phases A and B were water with 0.1% formic acid and acetonitrile. The chromatographic method held the initial mobile phase composition (10%B) constant for 3 min, followed by a linear gradient to 100% B up to 15 min and kept for 5 min at 100% B. The flow rate used was 0.5mL min-1. The HPLC system was connected to a time-of-flight mass spectrometer (Agilent 6220 accurate mass TOF, Agilent Technologies) equipped with an electrospray interface operating in positive ion mode, using the following operating parameters: capillary voltage, 4000V; nebulizer pressure, 40psig; drying gas flow rate, 9.0 L min-1; gas temperature, 325ºC; skimmer voltage, 65V; octapole 1 rf, 250V; fragmentor voltage 190V. LC-MS accurate mass spectra were recorded across the range of 50-1000 m/z. The identification of the analytes was performed by accurate mass measurements of the protonated ion [M+H]+ of targeted species and characteristic fragments if any (mass error <3ppm). The method was applied to spiked and real samples with satisfactory results, for instance, the method is able to detect as low as 0.4 pg (absolute) of cocaine. It was applied to the detection of cocaine in hair samples and in bank notes

COMUNICACIONES EN CARTEL C1-16
57
SENSITIZED FLUORIMETRIC DETERMINATION OF PICEID IN WINE BASED
ON ITS IMMOBILIZATION ON NYLON MEMBRANES
Diego Airado-Rodríguez, Mª del Pilar Godoy Caballero, Isabel Durán-Merás, Teresa Galeano-Díaz
Department of Analytical Chemistry, University of Extremadura, Avda. de Elvas s/n,
06071, Badajoz, Spain. E-mail: [email protected]
Piceid (3,4’,5-trihydroxystilbene-3-β-mono-D-glucoside) is a phenolic compound found in many families of plants. Piceid is physiologically as important as its aglycone, decreasing the risk of coronary heart disease and preventing cancer and other degenerative disorders such as Alzheimer’s disease or dementia. Piceid can be found in grape products, e.g. wines, with the concentration of the glycoside usually being significantly higher than the those of its aglycone (3,4’,5-trihydroxystilbene) [1].
This work presents a novel method for the sensitive determination of piceid.
The approach combines the sensitivity of the luminescence methodologies and the ability of nylon membranes to retain piceid on its surface. Nylon has proved to be an adequate solid support for detecting fluorescence of certain organic compounds [2]. Piceid is slightly fluorescent in solution; however its quantum fluorescence yield is greatly enhanced as a result of its immobilization of piceid in a nylon solid surface, as a consequence on the increase of the medium rigidity.
Variables involved in the deposition and measurement processes were
optimized, and the analytical figures of merit were obtained under optimal conditions.
The method has been satisfactorily applied to wine samples, prior extraction of
piceid into ethyl acetate, which is directly deposited in the nylon membrane and dried with N2 stream. The emission spectra were recorded between 350-500 nm and under these conditions a linear relationship between fluorescence signal and the concentration range of 22-700 ppb for piceid was observed.
The required instrumental involved is completely non-sophisticated, so the
presented methodology could be easily implemented in routine laboratories.
[1] R. M. Lamuela Raventos, A. L. Waterhouse, Methods Enzymol. 299 (1999) 184 [2] S. A. Bartolato, J. A. Arancibia, G. M. Escandar, Anal. Chim. Acta 613 (2008)
218

C1-17 COMUNICACIONES EN CARTEL
58
SINTESIS Y CARACTERIZACION DE POLIMEROS DE IMPRESIÓN MOLECULAR PARA LA DETERMINACION DE ACIDO CLOFIBRICO
N. Molina, E.E. Paniagua, C. Cacho, C. Pérez-Conde
Dpto. Química Analítica, Univ. Complutense de Madrid, Madrid.
E-mail: [email protected] El ácido clofíbrico es un medicamento ampliamente utilizado para la
disminución del colesterol sanguíneo. Su empleo masivo en los últimos años ha propiciado su clasificación dentro del grupo de los contaminantes emergentes al haber aparecido recientemente en el medio ambiente. Actualmente, los métodos empleados en su determinación resultan largos y complicados, ya que requieren de numerosas etapas de limpieza de las muestras. La tecnología de impresión molecular permite la preparación de materiales altamente selectivos, capaces de simplificar considerablemente el análisis al facilitar el tratamiento de las muestras.
En el presente trabajo, se han sintetizado y caracterizado polímeros impresos
capaces de interaccionar selectivamente con el ácido clofíbrico. Para ello, se han preparado dos polímeros impresos empleando ácido clofíbrico como analito plantilla, ácido metacrílico y 4-vinilpiridina como monómeros funcionales, etilenglicoldimetacrilato como entrecruzante, azo-bis-isobutironitrilo como iniciador y acetonitrilo como porogen. Una vez sintetizados los polímeros, el ácido clofíbrico se ha eliminado mediante extracción Soxhlet con una disolución de metanol:ácido acético (1:1, v/v) durante 10 horas.
Se ha estudiado la selectividad de la interacción de ambos polímeros impresos
con ácido clofíbrico, comparando su funcionamiento con el de los correspondientes polímeros no impresos, sintetizados en ausencia del analito plantilla. Los resultados obtenidos de este estudio demostraron que tan sólo el polímero sintetizado empleando 4-VP como monómero funcional fue capaz de reconocer selectivamente el ácido clofíbrico, por lo que dicho polímero se seleccionó para los estudios posteriores.
Una vez optimizadas las distintas etapas del proceso de extracción en fase
sólida empleando el polímero de 4-VP como adsorbente selectivo, se ha caracterizado la interacción entre el ácido clofíbrico y dicho polímero, para lo cual se ha evaluado la recuperación obtenida al cargar concentraciones crecientes del analito sobre el MIP. Los resultados obtenidos de este estudio se han ajustando al modelo matemático de la isoterma de Langmuir-Freundlich, ampliamente empleada en la caracterización de materiales tanto homogéneos como heterogéneos.
Donde B representa la cantidad de ácido clofíbrico unida selectivamente al
polímero impreso, F la cantidad de ácido clofíbrico que permanece en disolución, q la capacidad del polímero impreso, a un parámetro relacionado con la afinidad del MIP hacia el ácido clofíbrico y m la homogeneidad de los puntos de unión. AGRADECIMIENTOS: Los autores quieren agradecer al MICINN (CTQ2008-01031), a la CAM (S-0505-AMB-0312) y a la UCM (CCG08-UCM/PPQ) por la financiación de este trabajo.
B =q K F m
1 + K F mB =
q K F m
1 + K F m
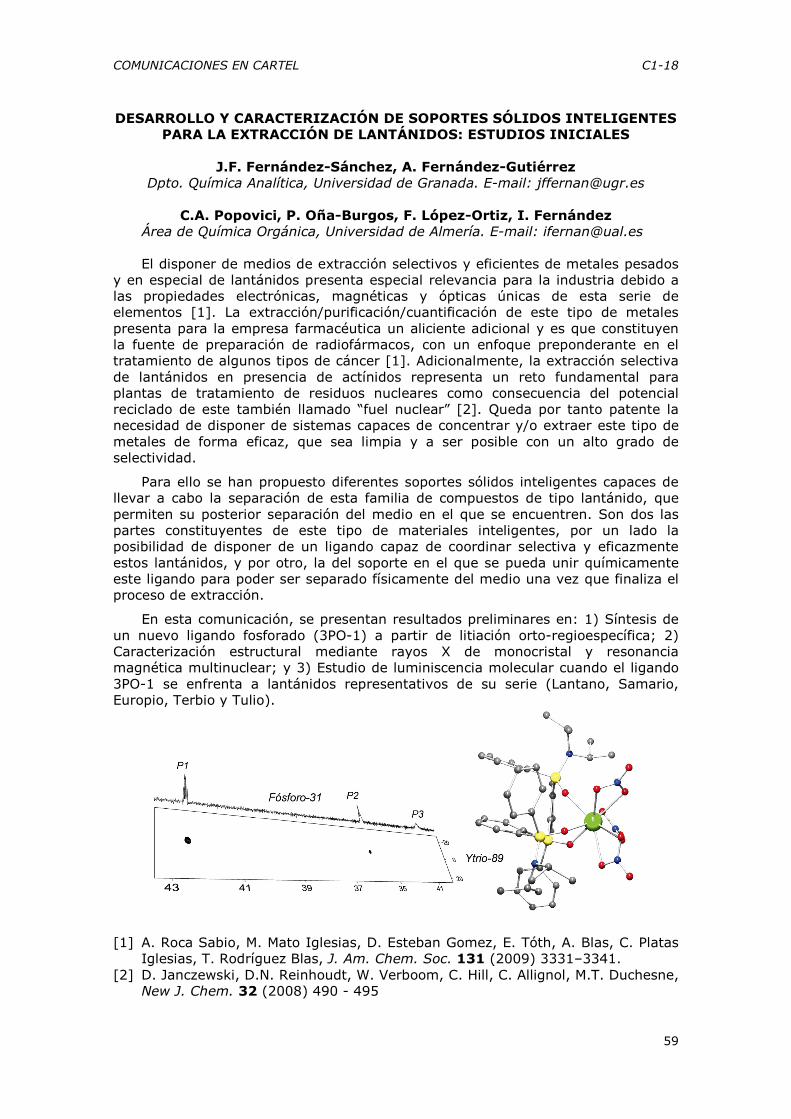
COMUNICACIONES EN CARTEL C1-18
59
DESARROLLO Y CARACTERIZACIÓN DE SOPORTES SÓLIDOS INTELIGENTES
PARA LA EXTRACCIÓN DE LANTÁNIDOS: ESTUDIOS INICIALES
J.F. Fernández-Sánchez, A. Fernández-Gutiérrez Dpto. Química Analítica, Universidad de Granada. E-mail: [email protected]
C.A. Popovici, P. Oña-Burgos, F. López-Ortiz, I. Fernández Área de Química Orgánica, Universidad de Almería. E-mail: [email protected] El disponer de medios de extracción selectivos y eficientes de metales pesados
y en especial de lantánidos presenta especial relevancia para la industria debido a las propiedades electrónicas, magnéticas y ópticas únicas de esta serie de elementos [1]. La extracción/purificación/cuantificación de este tipo de metales presenta para la empresa farmacéutica un aliciente adicional y es que constituyen la fuente de preparación de radiofármacos, con un enfoque preponderante en el tratamiento de algunos tipos de cáncer [1]. Adicionalmente, la extracción selectiva de lantánidos en presencia de actínidos representa un reto fundamental para plantas de tratamiento de residuos nucleares como consecuencia del potencial reciclado de este también llamado “fuel nuclear” [2]. Queda por tanto patente la necesidad de disponer de sistemas capaces de concentrar y/o extraer este tipo de metales de forma eficaz, que sea limpia y a ser posible con un alto grado de selectividad.
Para ello se han propuesto diferentes soportes sólidos inteligentes capaces de llevar a cabo la separación de esta familia de compuestos de tipo lantánido, que permiten su posterior separación del medio en el que se encuentren. Son dos las partes constituyentes de este tipo de materiales inteligentes, por un lado la posibilidad de disponer de un ligando capaz de coordinar selectiva y eficazmente estos lantánidos, y por otro, la del soporte en el que se pueda unir químicamente este ligando para poder ser separado físicamente del medio una vez que finaliza el proceso de extracción.
En esta comunicación, se presentan resultados preliminares en: 1) Síntesis de un nuevo ligando fosforado (3PO-1) a partir de litiación orto-regioespecífica; 2) Caracterización estructural mediante rayos X de monocristal y resonancia magnética multinuclear; y 3) Estudio de luminiscencia molecular cuando el ligando 3PO-1 se enfrenta a lantánidos representativos de su serie (Lantano, Samario, Europio, Terbio y Tulio).
[1] A. Roca Sabio, M. Mato Iglesias, D. Esteban Gomez, E. Tóth, A. Blas, C. Platas
Iglesias, T. Rodríguez Blas, J. Am. Chem. Soc. 131 (2009) 3331–3341. [2] D. Janczewski, D.N. Reinhoudt, W. Verboom, C. Hill, C. Allignol, M.T. Duchesne,
New J. Chem. 32 (2008) 490 - 495

C1-19 COMUNICACIONES EN CARTEL
60
COPs FORMATION HPLC MONITORING IN ELECTRON-BEAM IRRADIATED
FOODS
J.J. Lozada-Castro, M. Gil-Díaz, M.J. Santos-Delgado, S. Rubio-Barroso, L.M. Polo-Díez
Department of Analytical Chemistry, Faculty of Chemistry, Complutense University
of Madrid, 28040 Madrid, Spain. E-mail: [email protected] Nowadays, changes in dietary habits have produced a high increase in the
consumption of ready-to-eat (RTE) foods. Preparation of RTEs requires different treatments to minimize microbial contamination and to increase shelf-life. Electron-beam (E-beam) irradiation may be a useful anti-microbial technique, easy to apply in mass production and environmentally friendly, but food security also involves chemical control of RTEs [1,2]. During this treatment cholesterol could be oxidized, forming a group of oxides (COP) potentially more harmful than cholesterol itself. These oxides have received considerable attention in recent years because they have been associated with human diseases, such as inhibition of cholesterol biosynthesis, atherosclerosis, changes in membrane properties and carcinogenesis.
In the present work, COPs have been determined by HPLC with UV detection in
two different RTE foods, “Soft cheese” and “Serrano ham”, after treatment with E-beam. Firstly, these samples were prepared as thin slices and they were irradiated at different doses, 1, 2, 4, 6 and 8 kGy; the evolution of three COPs (25- hydroxycholesterol (25-OH), 7-ketocholesterol (7-Keto) and 6-ketocholestanol (6-Keto)) often found in processed foods was monitored. The analytical method was based on solvent extraction and clean-up by SPE. Recovery studies at three concentration levels were carried out and they were between 67 and 95%; relative standard deviation levels were, in general, lower than 10%.
COPs were detected after E-beam irradiation; the concentration levels of 25-OH
and 7-Keto increased with the irradiation dose in the two kinds of RTE foods, above to 6 kGy dose. However, 6-Keto was not found in any of the samples analyzed.
[1] M. Gil-Díaz, M.J. Santos-Delgado, S. Rubio-Barroso, L.M. Polo-Díez. Meat Science, 82, (2009) 24-29
[2] N. Rosales-Conrado, M.E. León-González, L.V. Pérez-Arribas, L.M. Polo-Díez. Analytical and Bioanalytical Chemistry 391 (2008) 1433-1442

COMUNICACIONES EN CARTEL C1-20
61
CARACTERIZACIÓN DE LA FRACCIÓN FENÓLICA EN TÉ ROOIBOS MEDIANTE HPLC-DAD ACOPLADA A ELECTROESPRAY-TIEMPO DE VUELO Y TRAMPA DE
IONES (HPLC-DAD-ESI-TOF-MS/IT-MS)
I. Iswaldi, I. C. Rodríguez-Medina, A. Segura Carretero, A. Fernández-Gutiérrez
Departamento de Química Analítica, Facultad de Ciencias, Universidad de Granada,
C/Fuentenueva s/n, 18071, Granada. E-mail: [email protected]; [email protected]
Es indudable que la salud es la principal preocupación de todos los seres humanos y la alimentación es parte inseparable de una buena salud. En los últimos años los productos naturales con un alto contenido en compuestos polifenólicos han despertado un creciente interés debido precisamente a estas propiedades saludables que han sido ampliamente estudiadas y que podrían reducir el riesgo de contraer enfermedades cardiovasculares y cáncer.
En este sentido destacan las distintas variedades de té, en concreto el té
Rooibos en el que se centra este trabajo. Este té no tiene cafeína y sólo la mitad de taninos que el té común. Además de ser una bebida refrescante, tiene la ventaja de contener minerales y poderosos antioxidantes entre ellos los polifenoles.
Experimental Para la caracterización del té Rooibos se emplearon dos sistemas de detección
distintos acoplados a HPLC: espectroscopía UV visible con DAD y espectrometría de masas con analizadores de tiempo de vuelo (TOF) y trampa de iones (IT). La espectroscopía UV/visible proporciona información acerca de las familias a las que pertenecen los distintos compuestos, lo que permitió acotar el campo de búsqueda. El acoplamiento HPLC-MS utilizando el analizador TOF proporciona unos valores de masa molecular muy exactos en un amplio rango, lo cual unido al uso de la distribución isotópica (True Isotopic Pattern) permite generar la fórmula molecular de cada compuesto. El método cromatográfico fue optimizado y en 35 minutos se obtuvieron los perfiles de los distintos polifenoles. El método de MS fue optimizado igualmente y se analizó el té en condiciones de modo negativo de ionización obteniéndose entre otros aspalatina, vitexina, quercetina 3- rutinosa, quercetina, luteolina 7-glucosa, luteolina. Posteriormente se confirmaron los compuestos en base a los patrones de fragmentación obtenidos por MS/MS.

C1-21 COMUNICACIONES EN CARTEL
62
EVALUACIÓN DE LA BIODISPONIBILIDAD DE ARSÉNICO EN ALGAS COMESTIBLES MEDIANTE EL ESTUDIO DE LA FRACCIÓN DIALIZABLE
C. García Sartal, M.C. Barciela-Alonso, V. Romarís-Hortas,
A. Moreda-Piñeiro, P. Bermejo-Barrera
Dpto. Química Analítica, Nutrición y Bromatología. Universidad de Santiago de Compostela. Avenida das Ciencias s/n 15782 Santiago de Compostela.
E-mail: [email protected]
El objetivo del presente trabajo es la evaluación de la biodisponibilidad de arsénico en diferentes tipos de algas comestibles cultivadas en las costas gallegas (Noroeste de España). En el estudio se incluyen dos tipos de algas rojas (Palmaria Palmate (Dulse) y Porphyra umbilicales (Nori)), tres algas pardas (Laminaria ochroleuca (Kombu), Undaria pinnatifida (Wakame) y Himanthalia elongate (Espagueti de mar)), una alga verde (Ulva rigida (Lechuga de mar)) y una alga microscópica (Spirulina plantesis). También se incluyen en el estudio una muestra de alga cocida que consiste en un preparado comercial compuesto por dos tipos de algas, Himanthalia elongata y Saccorhiza polyschides y un producto derivado (agar-agar) del alga roja Gelidium sesquipedale. En cada una de las muestras de alga se determinó el arsénico total así como la concentración de arsénico biodisponible utilizando para ello un método de digestión in vitro. El estudio in vitro reproduce las condiciones gastrointestinales de un adulto en dos etapas. En la primera etapa se reproduce la digestión gástrica (pH 5, solución de pepsina) y en la segunda la digestión intestinal (pH 7, sales biliares-pancreatina), usando en esta etapa una membrana de diálisis rellena de solución PIPES para simular la absorción intestinal. La determinación del arsénico total en la muestra de alga y en la fracción no dializada fue realizada por Generación de Hidruros-Espectroscopía de Fluorescencia Atómica (HG-AFS). La determinación de arsénico en la fracción dializada se realizó mediante Plasma Acoplado Inductivamente-Espectrometría de Masas (ICP-MS). Para la evaluación de la exactitud del método para la determinación del arsénico total se utilizó el material de referencia NIES-09. La exactitud del método in Vitro fue evaluada realizando un balance de masas de todo el proceso para un alga tipo Kombu, obteniendo una buena exactitud para todo el proceso (intervalo de confianza del 95%). Los resultados obtenidos muestran que las concentraciones más altas de arsénico se encuentran en las algas tipo Kombu, espagueti de mar y wakame (168.4±11.7, 38.4±1.8 y 29.7±1.9 µg g-1 respectivamente), mientras que los mayores porcentajes de dializabilidad se encuentran en las algas tipo Dulse y Nori (102.9±12.1 y 76.2±6.9% respectivamente).

COMUNICACIONES EN CARTEL C1-22
63
NANOPARTÍCULAS COMO BIOMARCADORES. SÍNTESIS, CARACTERIZACIÓN
Y FUNCIONALIZACIÓN DE QUANTUM DOTS EN MEDIO ACUOSO.
M. J. Almendral Parra, A. Alonso Mateos, Sara Sánchez Paradinas y Juan Francisco Boyero Benito
Departamento de Química Analítica, Nutrición y Bromatología. Universidad de
Salamanca. E-mail:[email protected].
Uno de los principales avances que permite aumentar considerablemente las aplicaciones prácticas de los Quantum Dots (QDs) en bioquímica y medicina incluye la síntesis de QDs de alta calidad en grandes cantidades y la optimización de los procesos de modificación de su superficie por bioconjugación con moléculas funcionales apropiadas [1]. Las numerosas aplicaciones posibles existentes demandan QDs de alta calidad, siendo en este sentido un factor de especial relevancia el rendimiento cuántico, que está ampliamente influenciado por el estado de la superficie del QD. La modificación de esta superficie puede también aumentar la estabilidad de los nanocristales y prevenir la formación de agregados entre ellos y hace a los QDs disponibles para interacciones con analitos marcados, de crucial interés para su aplicación como sensores químicos o biosensores [2,3].
Los trabajos dirigidos a la mejora en los procedimientos de síntesis y
recubrimiento de QDs son numerosos en la actualidad y pueden considerarse como un área pujante que implica diversas ramas de conocimiento [4]. Hasta la actualidad, la gran mayoría de los métodos descritos utilizan medios orgánicos en los procesos de síntesis. Sin embargo, la síntesis en medio acuoso resulta considerablemente menos laboriosa y permite ampliar el campo de aplicación en el mundo biológico.
El trabajo que se presenta refiere la optimización de las condiciones de síntesis
en medio acuoso de los QDs CdS y CdSe, con especial hincapié en el control cinético de la obtención de las nanopartículas. Se muestran los resultados obtenidos en estudios de estabilidad, rendimiento cuántico y caracterización, obteniéndose QDs de elevada eficacia cuántica (alta fluorescencia) y estabilidad superior a 3 meses. Se describen diversos procedimientos de modificación de su superficie, con vistas a su biofuncionalización y se muestran los resultados obtenidos empleando polietilenimina y otros modificadores.
Referencias: [1] R. E. Bailey, A. M. Smith, S. Nie. Physica E, 25 (2004)1-12. [2] P. Sharma, S. Brown, G. Walter. Advances in Colloid and Interface Science
123-126 (2006) 471-485. [3] M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos. Science 281
(1998) 2013-2015. [4] A. M, Smith, S. Nie. Analyst 129 (2004) 672.

C1-23 COMUNICACIONES EN CARTEL
64
BIOACUMULACIÓN DE METALES Y ESPECIACIÓN DE SELENIO Y MERCURIO
EN CLARIAS GARIEPINUS
I. López- Heras1, Z. Pedrero-Zayas1, S. Murillo1, C. Cámara1, Y. Madrid1, L. Lima-Cazorla2, S. Olivares-Rieumonut2 y D. de la Rosa-Medero2
1Dpto. Química Analítica, Facultad de Ciencias Químicas. Univ. Complutense de
Madrid, 28040 Madrid. Spain. E-mail:[email protected] 2Instituto Superior de Tecnologías y Ciencias Aplicadas. Ave. Salvador Allende
Luaces. La Habana. A.P. 6163. Cuba
El Clarias gariepinus (pez gato) se introdujo en Cuba en 1999 y rápidamente se extendió por muchos ríos de Cuba, incluyendo el río Sagua la Grande. Estos pescados constituyen una de las principales fuentes de alimentación de la mayoría de los habitantes de la zona. Por otra parte, muy cerca de Sagua la Grande se encuentra la mayor planta de producción de cloro-álcali de Cuba que utiliza el mercurio en células electroliticas [1]. La combinación de estos dos factores hace a estos pescados interesantes indicadores del nivel de contaminación y permitirá establecer el potencial riesgo que su consumo produce a la población. En el presente estudio se llevó a cabo la determinación de As, Se y Hg en el músculo del pescado (parte comestible) y la especiación de mercurio y selenio
Para el estudio de las concentraciones de los metales seleccionados se tomaron
muestras de pescado de distinto tamaño, peso y sexo en tres puntos diferentes, puente (a 7 Km de la ciudad Sagua la Grande), dique (próximo a la planta de producción) y en un canal de irrigación que utiliza agua del río Sagua la Grande. La determinación se llevó a cabo mediante ICP-MS previa mineralización de la muestra con horno de microondas. Los niveles de Hg y As en el músculo fueron, entre 0.3 y 0.8 µg/g y 0.2-0.4 µg/g, respectivamente. Es interesante destacar las elevadas concentraciones de Se encontradas en el músculo del pescado, oscilando entre 7 y 15 mg/g, sin que se tenga hasta ahora una idea clara del origen de estas elevadas concentraciones. No se observaron diferencias significativas entre pescados de distinto sexo, peso y tamaño, respecto a las concentraciones totales de cada elemento.
La especiación de selenio y mercurio se realizó mediante HPLC-ICP-MS. En el caso del mercurio, la especie mayoritaria fue metilmercurio y en el caso del selenio la especie mayoritaria encontrada fue la selenometionina y una pequeña cantidad selenocisteína, identificada previa carboximetilación con iodoacetamida. Se discuten los resultados obtenidos para la especiación de selenio con distintos tipos de tratamiento de muestra y la relación entre las concentraciones de las especies de selenio y mercurio para los distintos pescados procedentes de los diferentes lugares de muestreo.
[1] D. De la Rosa, L. Lima, S. Olivares-Rieumont, D. W. Graham, I. Enriquez, O.
Diaz, J. M. Bastías, O. Muñoz, Bull Environ Contam Toxicol 82 (2009) 101-105.

COMUNICACIONES EN CARTEL C1-24
65
TRATAMIENTO DE MUESTRA PARA LA IDENTIFICACIÓN DE COMPUESTOS FENÓLICOS Y SUS METABOLITOS EN SUERO Y PLASMA MEDIANTE
HPLC/nanoLC-ESI-TOF (MS) R. Quirantes-Piné1, V. Verardo2, D. Arraez-Román1, A. Segura-Carretero1,
A. Fernández-Gutiérrez1
1Dpto. Quimica Analítica, Univ. Granada, Granada. E-mail: [email protected]
2Dipartimento di Scienze degli Alimenti, Università di Bologna, Cesena (FC), Italy En los últimos años los compuestos fenólicos han despertado un creciente
interés debido a los numerosos estudios in vitro e in vivo que han demostrado su bioactividad como antioxidantes, antialergénicos, antiarterogénicos, antiinflamatorios, antimicrobianos, cardioprotectores y vasodilatadores. Para dilucidar su mecanismo de acción a nivel tanto tisular como celular es necesario establecer las rutas metabólicas de los distintos compuestos fenólicos llevando a cabo determinaciones de ellos y sus metabolitos en fluidos biológicos como suero o plasma empleando diferentes metodologías analíticas.
Experimental La gran complejidad de estas muestras biológicas hace que el tratamiento
previo de la muestra sea un paso clave del proceso de análisis. Por ello, en este trabajo se ha llevado a cabo la comparación de diversos protocolos para el tratamiento de muestras de suero y plasma incluyendo etapas de precipitación de proteínas, limpieza mediante extracción sólido-líquido con diferentes fases sólidas e hidrólisis enzimática de proteínas. Para realizar esta comparativa se ha empleado suero bovino fetal fortificado con patrones de compuestos fenólicos representativos de cada familia, como son naringenina, luteolina, apigenina, rutina, ácido p-coumárico, ácido siríngico y catequina. El análisis de estas muestras se ha llevado a cabo mediante cromatografía líquida de alta resolución acoplada a espectrometría de masas con analizador de tiempo de vuelo (HPLC-ESI-TOF).
El tratamiento de muestra elegido como óptimo se ha aplicado al estudio del
metabolismo de ratas sobrealimentadas con un extracto de lippia citriodora rico en fenilpropanoides, flavonoides e iridoides que ha demostrado tener actividad antioxidante y antiinflamatoria. Para realizar este estudio se han analizado muestras de plasma de 9 ratas Wistar, 3 de ellas controles que no habían tomado el extracto y 6 que habían ingerido 1440 mg/Kg de extracto 20 minutos antes de la extracción de la sangre. Estas muestras fueron analizadas mediante HPLC/nanoLC-ESI-TOF. Hasta el momento ésta es la primera aplicación de nanoLC en el ámbito de la metabolómica resultando de gran utilidad debido al incremento en la sensibilidad que supone respecto a los sistemas de cromatografía líquida convencionales y a la posibilidad de analizar pequeñas cantidades de muestra con ninguna o muy poca dilución. Empleando esta metodología se han llegado a detectar en el plasma hasta 11 compuestos fenólicos presentes en el extracto y algunos posibles metabolitos.

C1-25 COMUNICACIONES EN CARTEL
66
INTERPRETATION OF BCR-SM&T CHEMICAL FRACTIONATION RESULTS BY
TWO-WAY AND THREE-WAY PRINCIPAL COMPONENT ANALYSIS
R. Pardo, M. Vega, M.I. Sánchez, L. Debán
Departamento de Química Analítica, Facultad de Ciencias, Universidad de Valladolid, Prado de la Magdalena s/n, 47005 Valladolid (Spain)
E-mail: [email protected]
Heavy and trace metals are a hazardous group of pollutants because of their toxic and accumulative characteristics. Polluted soils can act as temporary reservoirs from which these elements could be released back, so a full evaluation of their environmental risk must take into account their mobility/availability, which can be evaluated by chemical fractionation. The BCR-SM&T sequential extraction procedure is becoming one of ‘de facto’ standard approaches for chemical fractionation of metals in soils and sediments.
Since BCR-SM&T scheme uses three reagents of increasing reactivity (acetic acid, hydroxylamine hydrochloride and hydrogen peroxide), three fractions of decreasing mobility/availability corresponding or related to metals bound to (i) exchangeable and carbonate phases, (ii) iron/manganese oxide and hydroxide phases, and (iii) organic and sulphide phases, are found. A fourth fraction, labelled ‘Residual’ and calculated by difference from the ‘pseudo-total’ contents or by digestion of the last residue, is routinely added. Therefore, the resulting dataset is a tri-dimensional matrix X, whose dimensions are: (no. of samples x no. of metals x no. of fractions).
Whereas X is usually described and discussed by using univariate statistical procedures, a better understanding of its information can be accomplished by using multivariate chemometric tools such as Principal Components Analysis. The classic PCA is designed for bi-dimensional matrixes, but can be also applied to tri-dimensional ones, by unfolding X into a bi-dimensional matrix and applying the so-called matrix augmentation-PCA (MA-PCA) approach. The method allows a full recovering of information from variables, but that from samples and fractions becomes mixed. On the contrary, the use of true n-dimensional PCA methods (3-PCA in this case), such as PARAFAC of Tucker3, allow to fully separate the information contained in X and corresponding to samples, metals and fractions.
In this communication we have applied the BCR-SM&T scheme to 13 soil samples collected in Medina del Campo (Valladolid. Spain), an area with environmental problems derived from the presence of a lead smelter. The elements studied were Cd, Co, Cu, Cr, Ni, Pb and Zn, which were determined in the four BCR-SM&T fractions by ETAAS or ICP-MS. The resulting X dataset (13x7x4) has been studied by MA-PCA, after a previous column-wise unfolding, and by 3-PCA methods after j-scaling.
MA-PCA and PARAFAC models (Tucker3 did not yield good results) originated similar two-factor models explaining, respectively, the 74.3% and the 63.8% of the information, or variance, contained in X. In the case of 3-PCA, the second PARAFAC factor is biased towards the presence of cadmium and lead in the BCR-SM&T fraction 2. Because of the relative availability of this fraction and the toxic nature of these two elements, the second PARAFAC factor could be taken as a measurement of the ‘environmental risk’, allowing its use to locate the more hazardous points in the studied zone.
Acknowledgments. Authors wish to thank Spanish MEC (CTM2006/02249) and Junta de Castilla y León (VA015A06) for financial support.
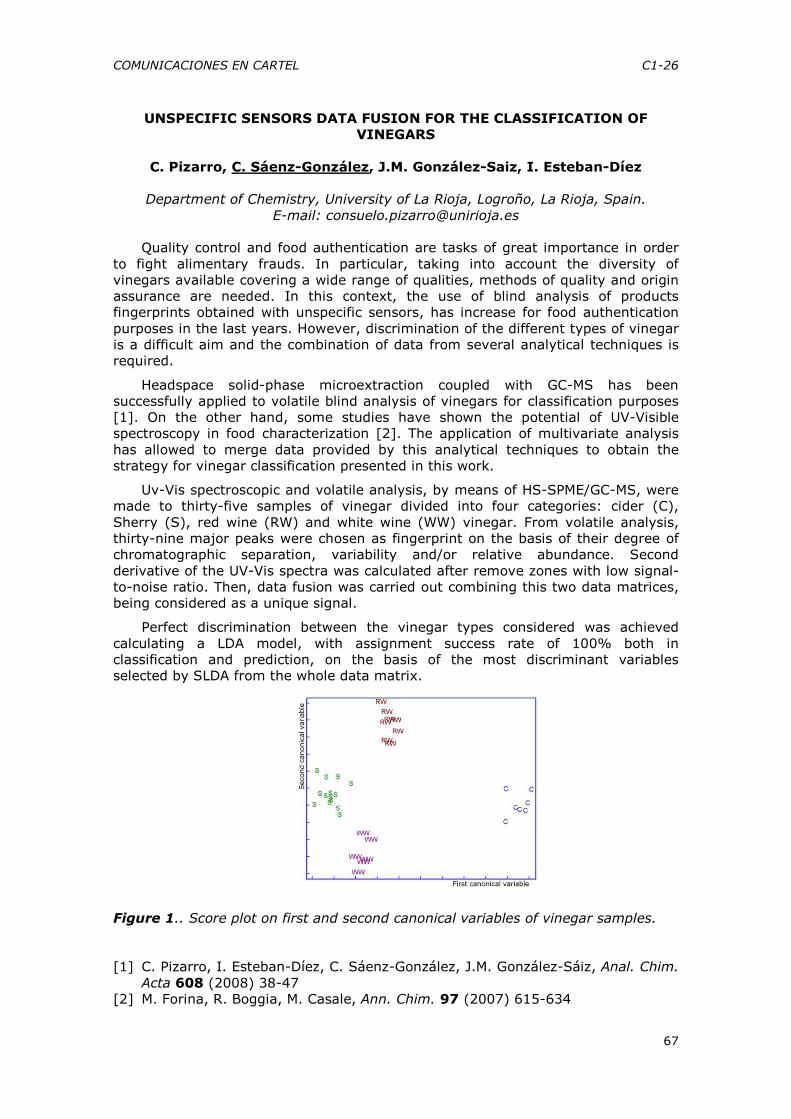
COMUNICACIONES EN CARTEL C1-26
67
UNSPECIFIC SENSORS DATA FUSION FOR THE CLASSIFICATION OF
VINEGARS
C. Pizarro, C. Sáenz-González, J.M. González-Saiz, I. Esteban-Díez
Department of Chemistry, University of La Rioja, Logroño, La Rioja, Spain. E-mail: [email protected]
Quality control and food authentication are tasks of great importance in order
to fight alimentary frauds. In particular, taking into account the diversity of vinegars available covering a wide range of qualities, methods of quality and origin assurance are needed. In this context, the use of blind analysis of products fingerprints obtained with unspecific sensors, has increase for food authentication purposes in the last years. However, discrimination of the different types of vinegar is a difficult aim and the combination of data from several analytical techniques is required.
Headspace solid-phase microextraction coupled with GC-MS has been successfully applied to volatile blind analysis of vinegars for classification purposes [1]. On the other hand, some studies have shown the potential of UV-Visible spectroscopy in food characterization [2]. The application of multivariate analysis has allowed to merge data provided by this analytical techniques to obtain the strategy for vinegar classification presented in this work.
Uv-Vis spectroscopic and volatile analysis, by means of HS-SPME/GC-MS, were made to thirty-five samples of vinegar divided into four categories: cider (C), Sherry (S), red wine (RW) and white wine (WW) vinegar. From volatile analysis, thirty-nine major peaks were chosen as fingerprint on the basis of their degree of chromatographic separation, variability and/or relative abundance. Second derivative of the UV-Vis spectra was calculated after remove zones with low signal-to-noise ratio. Then, data fusion was carried out combining this two data matrices, being considered as a unique signal.
Perfect discrimination between the vinegar types considered was achieved calculating a LDA model, with assignment success rate of 100% both in classification and prediction, on the basis of the most discriminant variables selected by SLDA from the whole data matrix.
Figure 1.. Score plot on first and second canonical variables of vinegar samples. [1] C. Pizarro, I. Esteban-Díez, C. Sáenz-González, J.M. González-Sáiz, Anal. Chim.
Acta 608 (2008) 38-47 [2] M. Forina, R. Boggia, M. Casale, Ann. Chim. 97 (2007) 615-634

C1-27 COMUNICACIONES EN CARTEL
68
SPECIATION OF MERCURY AND SELENIUM TO STUDY THEIR METABOLISM
IN MICROALGAE FEED WITH THE ELEMENTS
F. Moreno, T. García-Barrera, J.L. Gómez-Ariza
Dpto. de Química y CC.MM. Facultad de Ciencias Experimentales. Universidad de Huelva. Campus de El Carmen.21007 Huelva (Spain).
E-mail: [email protected] As previously stated in the literature selenium prevents against some
neurotoxic effects of mercury and the protective effect is related to the ability of the different selenospecies to form HgSe with Hg2+. For this reason, new analytical approaches for the simultaneous speciation of mercury and selenium are claimed and have a great concern.
In a previous study, a column switching method for the simultaneous speciation
of mercury and selenium including chiral species was developed allowing the resolution of several species in less than 25 minutes. In addition, several efficient sample treatment procedures for the extraction of all the species from biological samples was optimized and compared by using four certified reference materials, namely: SELM-1 (selenium enriched yeast), DORM-2 (dogfish), TORT-2 (lobster hepatopancreas), IAEA-407 (fish homogenate).
The proposed analytical method simplifies the extraction strategy since the
extract can be directly injected in the HPLC–ICP-MS, and memory effect was overcome by using 2-mercaptoethanol in the mobile phase. This work presents an outline about the best procedure for each type of sample that allows the extraction of the most important mercury and selenium species. In addition several toxicologically experiments have been carried out in microalgae supplemented with methylmercury, seleno-dl-methionine, L-selenomethionine and D-selenomethionine in order to determine which selenospecie increases the formation of HgSe and to test the protective effect of each selenospecie.

COMUNICACIONES EN CARTEL C1-28
69
CHARACTERIZATION OF COPPER CONTAINING PROTEINS IN THE BRAIN
OF THE GENETICALLY SEQUENCED Mus musculus QqTOF
M. Gónzalez-Fernándeza, T. García-Barreraa, J.López-Bareab, C. Pueyob and J.L. Gómez-Arizaa
aDpto. de Química y CC.MM. Facultad de Ciencias Experimentales. Universidad de
Huelva. Campus de El Carmen.21007 Huelva (Spain). E-mail.: [email protected]
bDpto. de Bioquímica y Biología Molecular. Campus Universitario de Rabanales. Edificio Severo Ochoa (C-6), 2ª planta. Ctra Madrid-Cádiz Km 396a, 14071 Córdoba (Spain).
The atomic specific detector ICP-MS copes for most of elements with biological
significance and is usually employed in analytical methodologies for metallomics in combination with size exclusion chromatography (SEC). For unknown metallobiomolecules the identification step is covered by the use of organic mass spectrometry techniques, namely MALDI-TOF and Qq-TOF, this later for MS/MS analysis.
However, to get a more global vision about the behavior of metal in their
interactions with the cells compartments and metabolic pathways, the combination of metallomics results with those from proteomics can be very useful. In addition, the relation of both with genetic information can close the circle of the cell or biological tissue behavior.
In the present work, Mus musculus from which the genome is well known has
been studied using a Metallomics approach. The organs of several mice were pooled, frozen and the cytosolic fraction extracted. The extracts were analyzed by SEC-UV-ICP-MS and the metal containing fraction studied. The main result is that a Cu-containing fraction in the brain extract of about 7 kDa to 10 kDa is not present in any other organ of Mus musculus. This fraction was collected lyophilized and analyzed by anion exchange chromatography coupled to ICP-MS that gives two cooper containing peaks. Finally, unknown metallobiomolecules were characterized by using Qq-TOF.
A traditional proteomics approach was applied in parallel on the basis of 2D-
PAGE separation, tryptic digestion of the spots and protein characterization and sequencing by MALDI-TOF and Qq-TOF. The results obtained from both studies were compared and correlated with the Mus musculus genome data base.

C1-29 COMUNICACIONES EN CARTEL
70
OPTIMIZATION OF AN EXTRACTION METHOD FOR POPs IN FOOD SAMPLES
BY MEMBRANE EXTRACTION AND GC-ECD/MS
J.M. Manso-Sayago, T. García-Barrera, J.L. Gómez-Ariza
Dpto. de Química y CC.MM. Facultad de Ciencias Experimentales. Universidad de Huelva. Campus de El Carmen.21007 Huelva (Spain).
E-mail: [email protected] Membrane technology has become the method of choice for a wide variety of
analytes and samples through the many types of synthetic membranes produced nowadays. Due to its multidisciplinary character, this technology, employing flat sheet (FS) and hollow fibre (HF) synthetic organic membranes, is becoming widely used in medicine, biology, chemistry, biotechnology and water treatment. This technique allows analyte extraction, enrichment and clean-up from broad spectrum of matrices, such as air, water, human urine and plasma, soil and sediments, in addition to many others. In comparison with most reliable techniques such as solid phase microextraction (SPME) membrane extraction has low cost and it is disposable which completely eliminates carry-over problems.
Persistent organic pollutants (POPs) are a group of chemicals targeted for
reduction, or already banned, because of their persistence, potential for long-range transport and their ability to bioaccumulate in food-webs. In the present work a method based in HF-LPME (Hollow fibre liquid-phase microextraction) has been optimised for the extraction of forty POPs, which includes PCBs, PBDEs and pesticides.
In HF-LPME the extraction, a syringe needle tip is inserted into the top of the
few-centimetre long HF and the assembly is then immersed in the organic solvent to impregnate the pores of the fibre wall. Then, a microlitre-volume of organic extraction solvent is withdrawn into a microsyringe. After that, the HF is taken and put into the aqueous sample. The best organic solvent, time of the extraction and stirring was optimised for all the compounds in order to obtain maximum reproducibility and recoveries. The procedure was satisfactory applied to fruit juices fortified with analytes. The detection was carried out by GC with two dimensional detection based in ECD (electron capture detector) and MS (mass spectrometry) that allows high sensitivity and unequivocal identification. The performance of the method was successful for all the compounds and sample matrix effects were not present. Further studies will be carried out in biological high complex matrices such us blood and hair.

COMUNICACIONES EN CARTEL C1-30
71
ANALYSIS OF CYSTEINE AND GLUTATHION-DERIVED FLAVOUR PRE-
CURSORS FROM GRAPE JUICE BY SPE AND QUANTIFICATION BY HPLC-MS. A. Peña-Gallego *, P. Hernández-Orte, P. Herrero, V. Ferreira and J. Cacho.
Dpto. Química analítica. Facultad de Ciencias. Univ. Zaragoza. Plaza San Francisco
s/n. 50009. Zaragoza. España. Tfno: 976762503. Fax: 976761292. E-mail:[email protected].
Some non polar amino-acids have a relevant biological significance. Among
them there are some derivatives of cysteine ((4-S-4-methylpentan-2-ona)-L-Cysteine (Cys-MP), 3-S-(hexan-1-ol)-L-Cysteine (Cys-MH) or glutathione ((4-S-4-methylpentan-2-ona)-L-Glutathione (Glu-MP), 3-S-(hexan-1-ol)-L-Glutathione (Glu-MH), which are precursors of some of the most powerful aroma compounds existing in nature. There is an obvious interest, in developing methods for their selective isolation and further determination.
The direct analysis of those compounds using some of the many derivatization techniques proposed in the literature is not possible because of the low concentration which they are found and because of the interferences caused by the rest of amino-acids present in the matrix. Therefore, a selective isolation of these compounds is compulsory. The major goal of the present research is to develop a strategy for the selective isolation of cysteine-derivatives.
The studies have been carried out with verdejo must and verdejo must soaked with skins at the natural pH 3.5 spiked with the two cystein-derivatives Cys-MP, Cys-MH and two glutathione-derivatives Glu-MP and Glu-MH. A method with hydrophobic retention on polymeric sorbent to the extraction has been used. The maximum loading volumes, nature and volumes of rinsing solutions, eluting (minimum eluted volume) and the effect of pH have been determined.
The retention of these compounds is made in Bond Elut-ENV sorbents. 200 mg cartridge is used and breakthrough volumes not higher than 10 ml must be loaded and not more than 2 ml of eluted solution are needed to remove the aroma precursors.
Different chromatographic strategies have been studied (HPLC with fluorescence detection and HPLC with Mass detection have been used to analyze the isolated compounds). The first method needs a derivatization stage to improve the sensitivity and put the molecules in the most appropriate conditions to their subsequent analysis. When AQC [1] (6-aminoquinolyl-N-Hydroxysuccinimidyl Carbamate) cystein and glutathione-derivatives are formed, sensibility obtained to cystein and glutathione-conjugates derivatized with AQC were not enough to analyze the precursors in the order of magnitude there appear in the must. The best results have been obtained when HPLC-MS are used, without derivatization, a simple chromatographic method and Electrospray Ionisation positive (ESI+) [2] with Single Ion Monitoring (SIM) are used as the best technique to analyzed our precursors.
Keywords: Cys-MP, Cys-MH, Glu-MP, Glu-MH, AQC, ESI+, SIM Acknowledgements: This work has been funded by the Spanish MICYT
AGL2007-65139/ALI. [1] P. Hernández-Orte, M.J. Ibarz, J. Cacho, V. Ferreira, Chromatographia, 58
(2003), D29-D35. [2] B. Fredrizzi, K. H. Pardon, M. A. Sefton, G. M. Elsey, and D. W. Jeffery, J. Agric.
Food Chem., 57 (2009) D991-D995.

C1-31 COMUNICACIONES EN CARTEL
72
ARSENIC SPECIATION IN SEAWEED SAMPLES BY MICROWAVE-ASSISTED EXTRACTION AND
DETERMINATION BY HPLC-HG-ICP-AES AND HPLC-(UV)-HG-AFS
S. García Salgado, M.A. Quijano Nieto, M.M. Bonilla Simón Dpto. Ingeniería Civil: Tecnología Hidráulica y Energética, E.U.I.T. Obras Públicas,
Univ. Politécnica de Madrid, Madrid. E-mail: [email protected]
Arsenic is a toxic element widely distributed in the environment, and the estimation of its toxicity requires quantification of the individual arsenic species in biological material. Therefore, efforts have been focused on the development of methods for the separation and detection of the arsenic compounds. For the application of these methods to natural samples, an extraction step that makes the arsenic compounds available for analysis is necessary. Arsenic compounds should be extracted quantitatively without decomposition or chemical conversion. It is known that marine organisms can accumulate high arsenic concentrations, up to µg g-1 level, which may be harmful to human beings. Although the main arsenic species found in seaweed are arsenoribosides (arsenosugars), which are considered to be non-toxic to living organisms, potentially toxic inorganic species can be found in high percentages [1].
In the present work an analytical procedure was developed for arsenic speciation in seaweed. The samples studied were: Sargassum fulvellum (NIES No. 9), Hizikia fusiformis (Hijiki) and Laminaria digitata (slimming capsules). An optimised microwave-assisted extraction method, with deionised water as extractant, was used to extract the arsenic species present in the samples [2]. The method allowed us to achieve an arsenic extracted recovery between 78 and 98%, depending on the kind of alga analysed. Hyphenated techniques, HPLC-HG-ICP-AES and HPLC-(UV)-HG-AFS, were used for arsenic species determination. The separation was performed on a Hamilton PRP-X100 anion exchange column. When HPLC-HG-ICP-AES was employed, a 17 mmol L-1 phosphate buffer at pH 5.5 was used as mobile phase, and the arsines were generated with 0.5% (w/v) NaBH4 and 4.0 M HCl. Under these conditions, only toxic arsenic species (As(III), As(V), MMA and DMA) were detected. However, toxic and non-toxic arsenic species were detected when HPLC-(UV)-HG-AFS was used. In this case, the separation was performed with phosphate buffer at pH 9.0 as mobile phase, with a concentration gradient from 5 to 10 mmol L-1. The chromatographic system was coupled to the atomic fluorescence spectrometer via hydride generation, with a photo-oxidation step, which employs a UV lamp and a solution of potassium peroxodisulfate (2% (w/v) K2S2O8 / 2% (w/v) NaOH). Hydrides were generated with 1.4% (w/v) NaBH4 and 8.0 M HCl. This method led us to the separation of AsB, As(III), DMA, MMA and As(V). The chromatographic analysis of the samples allowed us to identify and quantify DMA species in all algae analysed, while As(V) species was only found in Hijiki and Sargasso material. Furthermore, three non-identified arsenic species, probably arsenosugars, were detected only by HPLC-(UV)-HG-AFS in all the samples analysed.
Acknowledgements: The authors would like to thank Universidad Politécnica
de Madrid (project 188/Q065815-146) and Ministerio de Educación y Ciencia of Spain (project CTM2007-66432) for financial support. [1] P. Bhattacharya, A.H. Welch, K.G. Stollenwerk, M.J. McLaughlin, J. Bundschuh,
G. Panaullah, Sci. Total Environ. 379 (2007) 109-120. [2] S. García Salgado, M.A. Quijano Nieto, M.M. Bonilla Simón, J. Chromatogr., A
1129 (2006) 54-60.

COMUNICACIONES EN CARTEL C1-32
73
MSFIA-SPECTROPHOTOMETIC DETERMINATION OF SULFUR DIOXIDE IN WINES USING A POLYSULFONE IMMERSION PRECIPITATION MEMBRANE
J. A. Rodriguez1, I. Enciso1, M.E. Paez-Hernandez1, E. Barrado2
1Centro de Investigaciones Químicas, Universidad Autónoma del Estado de Hidalgo.
Pachuca, Hgo, Mexico. E-mail: [email protected] 2QUIANE/ Departamento de Química Analítica, Universidad de Valladolid. Valladolid.
Spain. E-mail: [email protected] Sulfur dioxide (SO2) is an important compound in winemaking; it serves as
antibiotic and antioxidant, protecting wine from spoilage by bacteria and oxidation. The upper limit of SO2 allowed in wine is 350 mg l-1 in US, whilst in EU are 160 and 210 mg l-1 for red and white wines respectively. Different methods for quantitative determination of SO2 have been reported however the spectrophotometric determination with the reaction among SO2, formaldehyde and pararosaniline has been commonly used in flow analysis.
A requirement to generate a robust system for analysis of red and white wines
is the incorporation of a gas diffusion unit with polyvinylidene fluoride (PVDF) commercial porous membranes. Porous membranes with reproducible porous size can be obtained by the immersion precipitation process. In this work, a Multisyringe Flow Injection Analysis (MSFIA) with spectrophotometric detection using a polysulfone porous membrane (Figure 1) is proposed to determine SO2 in wines.
Figure 1. Proposed system used to determine SO2.
The membrane was formed using polysulfone, dimethylformamide and water as
polymer, solvent and non-solvent respectively. The analytical cycle begins with the aspiration of the sample into the injection loop (145 µl) then is injected into the carrier solution (HCl 1.0 mol·l-1). The SO2 diffuses through the membrane and is mixed with the chromogenic reagent (formaldehyde and pararosaniline). The mixture is transported at a flow rate of 2 ml min-1, and the signal is recorded at 580 nm. The influence of several parameters such as coiler reactor length, flow rate, carrier solution concentration and sample injection volume are evaluated using a Box-Behnken design. Linear calibration curve is obtained in the concentration curve between 100 and 500 mg l-1. The lowest limit of detection found under optimal conditions is around 40 mg l-1. Finally the procedure was applied to red and white samples.
Acknowledgements The authors wish to thank the CONACyT (project 61310), and the Consejería de Educación y Cultura de la Junta de Castilla y León (project VA029A07) for financial support.

C1-33 COMUNICACIONES EN CARTEL
74
NANOSTRUCTURED AMPEROMETRIC IMMUNOSENSORES FOR OCHRATOXIN A ( OTA) BASED ON SCREEN PRINTED CARBON ELECTRODES (SPCEs)
FUNCTIONALIZED WITH AuNPs-OTA-BSA
P. Duato, J.C. Vidal, L. Bonel and J.R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS), Institute of Environmental Sciences (IUCA), University of Zaragoza, C/ Pedro Cerbuna 12. 50009 ZARAGOZA.
E-mail: [email protected] Ochratoxin A (OTA) is a mycotoxin with nephrotoxic, teratogenic, carcinogenic
and immunotoxic activity in human and animals. OTA occurs in several foodstuffs such as cereals, coffee beans, nuts and cocoa. There are very strict mycotoxin regulations all over the world.
Chromatographic analytical methods available for the analysis of mycotoxins in food, beverages,and foodstuffs are usually validated by AOAC International Official Methods of Analysis. The direct voltammetric analysis of OTA is possible, but the oxidation of this molecule needs a very high potential (+1.5 V). Besides, sensitivity is low, and then the direct amperometric determination of OTA is not suitable at the level of low ppb required. For these reasons the use of immobilized antibodies is more convenient when amperometric transducer are used.
We are developing a rapid and sensible OTA immunosensors using advantages of the immunochemical assay and the nanostructured screen-printed technology. We have studied the influence of gold nanoparticles (AuNP, 20 nm. diameter) directly immobilized on screen printed graphite electrodes (SPCEs) made in our lab in order to increase the analytical signal obtaining higher voltammetric currents in the detection step. The Quartz Crystal Microbalance (QCM) was used to demonstrated that the nanofunctionalization process with AuNP increases the real sensoring surface of the quartz crystal device. The use of AuNP as versatile and efficient substrates for the immobilization of antibody or antigen showed not only enhance the amount of antibodies or antigens immobilized on the electrode sensing surface, also preserve the activity of the immobilized biomolecules. Nanometer-size (20nm) AuNPs exhibit excellent catalytic activity and these AuNPs have a relative high surface area-to-volume ratio.
We used indirect immunoassays in a competitive way by immobilizing AuNPs-OTA-BSA (ochratoxin A and serum albumin bovine with gold colloidal nanoparticles) conjugate on the SPCEs using ExtrAvidin-horse-radish peroxidise as transduction enzymes to generate the amperometric signal by the union to a biotinilated monoclonal antibody against OTA. Also we used casein 1% for blocking and avoiding unspecific adsorption. We also are working in a direct competitive immunosensor by immobilizing AuNPs-ST (gold colloidal nanoparticles with streptavidin) conjugate on the SPCEs and using the same antibody. In this case the amperometric signal is generated by the competition between OTA and OTA-HRP conjugate. We use biotin 10 mM for blocking and avoiding unspecific adsorption.
The electrochemical substrate for generating the amperometric signal was hydroquinone/H2O2 in both cases. The enzymatic product was detected by differential pulse voltammetry (DPV). Besides, the analytical results were validated with official methods based on HPLC with fluorescence detection. And the SPCEs with AuNPs were observed by Scanning Electron Microscopy (SEM) in order to characterize their nanostructured surface.
In conclusion, SPCEs inmunosensors for OTA allow the quick and specific determination of this mycotoxin at the ppb levels. The formation of a layer of nanostructured particles (AuNPs) increased the sensitivity of the transduction process in this new OTA amperometric immunosensors.
This work has been financed by the Aragon Government (Science, Technology and University Depart.) Project PM 027/2007. Patricia Duato thanks ACP S.A. a research grant.

COMUNICACIONES EN CARTEL C1-34
75
OPTIMIZATION OF A VALIDATION METHOD TO DETERMINE
OCHRATOXINE A IN FOODS BY HPLC WITH FLUORESCENCE AND UV-VIS DETECTION
E. Irisarri, A. López Molinero, J.R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS)
Institute of Environmental Sciences (IUCA) University of Zaragoza, C/Pedro Cerbuna 12, 50009 ZARAGOZA. Spain
Email: [email protected]
Ochratoxin A (OTA) is a naturally occuring mycotoxin produced primarily by Aspergillus ochraceus and Penicillium verrucosum usually present in a variety of foods. It is mainly found as a contaminant of cereals, cereal products and coffee beans.
Our research group works in the development of immunoelectrochemical
sensors and aptasensors to stablish rapid and sensitive methods for monitoring and quantifying OTA in contaminated food. The analytical use of these sensors need well stablished methods, with solid, seguros, and contratado procedures, useful to validate the results. Several standard methods for OTA determination have been stablished by HPLC – RP using fluorimetric detection, with a detection limit in the order of 0.1 ug/L.
In our laboratory a study has been accomplished in order to evaluate between
different analytical methods and asses the most useful in control and validation of analytical results in cereals, grapes, must and wines. Currently, the key step is found in the clean-up process. Different immunoaffinity columns, IAC’s, can be used, and a critical comparison will be performed.
We checked HPLC separation with RP C-18 and alternatively fluorimetric and
UV-VIS detection are evaluated. Best results were obtained by fluorimetric detection, with linear range between: 20-400 ug/l, and a detection limit of 7 ng/mL. The chromatographic conditions: mobile phase, eluyentes mixtures, isocratic and gradient procedure have been evaluated. Typical chromatograms, in optimum conditions, are obtained in 15 min.
The optimized method has been applied to the analysis of certified reference
material of OTA in wheat samples , and certified reference material BCR 471, blank sample. Results are in accordance with values certified. The method is used in our laboratory to validate the analytical results obtained with our new ochratoxine sensors.
We thank the financial support of the Aragon Gouverment Project PM 27-2007
and the P.Duato´s grant to the ACP SA

C1-35 COMUNICACIONES EN CARTEL
76
NANOSTRUCTURED ELECTROCHEMICAL APTASENSORS FOR
OCHRATOXIN A (OTA) DETERMINATION
L. Bonel, P. Duato, J.C. Vidal, J.R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS), Institute of Environmental Sciences (IUCA), University of Zaragoza, C/ Pedro Cerbuna 12. 50009 ZARAGOZA.
E-mail: [email protected]
Ochratoxin A is a naturally occuring mycotoxin produced primarily by Aspergillus ochraceus and Penicillium verrucosum usually present in a variety of foods. It is mainly found as a contaminant of cereals, cereal products and coffee beans. Development of immunochemical analysis has led to many rapid and sensitive methods for monitoring and quantifying OTA in contaminated food. Several groups have stablished immunoassays for OTA.
In the literature has been reported the combination of electrochemical immunosensor using gold nanoparticles (AuNPs) or carbon nanotubes (CNTs). Nanostructured materials have proven as one of the most powerful tool in new technologies and research, due to their absolutely peculiar properties at nanometer size scale. AuNPs serve as label in “sandwich” manner stripping analysis as well as effective electrocatalysys for nanostructuring of electrode surface. Colloidal gold nanoparticles strongly adsorb some proteins or antibodies.
The interesting approach of this work is related to the using of a selective aptamer to OTA. Aptamers are nucleic acids (DNA or RNA) that selectively bind to low molecular weight organic or inorganic substrates or to macromolecules such as proteins. The affinity constant of aptamers towards their substrates is in the micromolar to nanomolar range, comparable to the binding constants of antibodies to antigens. Anyway, this work is the first step in the realization of an assay for OTA based on the use of the specific aptamer explointing the known advantages of these biomimetic receptors. In literature only two papers report the development of an assay for OTA detection using specific aptamer.
A disposable electrochemical assay involving AuNPs and carbon-based screen-printed electrodes (SPCEs) was developed for the detection of OTA. The assay was based on a direct competitive format in which a DNA aptamer was used as biorecognition element, and horse-radish-peroxidase (HRP) was used as enzymatic label.
The assay developed for OTA detection was based on a direct format. All steps of the assay were carried out onto AuNPs; only the electrochemical detection was performed transferring the functionalized nanoparticles onto the working electrode of a screen-printed electrode. The immobilisation of the biotinylated aptamer was based on streptavidin-biotin interaction using streptavidin coated AuNPs.
The performance of the assay in terms of sensitivity, reproducibility and selectivity were studied. The calibration curve carried out shows a LOD and LOQ 0.2 and 6 µg/l respectively and the average coefficient of variation (ACV) resulted 8 %.
Finally, this approach will be applied to the analysis of some OTA samples to determine the concentration of OTA and predict the risk of a possible contamination with OTA.
This work has been financed by the Aragon Government (Science, Technology
and University Department) with Project PM 027/2007. Patricia Duato thanks a research grant from ACP S.A.

COMUNICACIONES EN CARTEL C1-36
77
STUDY OF THE INFLUENCE OF THE ELECTROPHORETIC PROCEDURE ON THE DETECTION OF METALS BOUND TO PROTEINS BY SUBSEQUENT LA-ICP-MS
L. Rodríguez, María S. Jiménez, María T. Gómez and Juan R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS)
Institute of Environmental Sciences. University of Zaragoza Pedro Cerbuna, 12. ZARAGOZA-50009 (SPAIN)
E-mail: [email protected] Laser Ablation (LA) Inductively Coupled Plasma (ICP) Mass Spectrometry (MS)
has been increasingly used as a system to detect metals bound to proteins after separation by Polyacrylamide Gel Electrophoresis (PAGE). Because of this, it is of great importance to keep the integrity of the metal-protein bindings during the procedure. When heteroatom-containing proteins are been investigated, the interactions metal-protein are strongly enough to remain intact during electrophoresis. Nevertheless when the object of the study are metal-binding proteins with weaker interactions it can occur that the metals are lost during the process and they cannot be detected bound to the proteins subsequently by LA-ICP-MS. This fact makes necessary the development of new procedures to separate proteins preventing the loss of metals bound to these proteins.
Starting from a previous paper [1], we have carried out some experiences to
determine the influence of some factors concerning the electrophoretic procedure in the detection of metals by La-ICP-MS of two proteins ,superoxide dismutase (SOD), containing Zn and Cu, and alcohol dehydrogenase (ADH), containing Zn. Not only the influence of the nature of the electrophoretic method has been studied, but also the effect of other aspects such as intensity applied, trailing ion chosen and post-separation gel treatment. For a denaturing PAGE of SOD, we recommended tricine as trailing ion . While non denaturing PAGE based on tricine is better for ADH in order to maintain the integrity of metal-protein binding.
Regarding to the intensity applied, as higher it is the possibilities of metal-
protein binding losses are higher. All of our results have demonstrated that it is preferred to avoid staining steps because they can alter the stability of the metal-binding proteins and prevent detecting metals bound to them.
[1] María S. Jiménez, María T. Gómez, L. Rodríguez, L. Martínez, Juan R. Castillo, Anal. Bioanal. Chem., 2009, 393, 699-707. The authors acknowledge the financial support from the Spanish Ministry of
Science and Technology (project no. CTQ 2006-00894).

C1-37 COMUNICACIONES EN CARTEL
78
CARACTERIZACIÓN DE VINOS TINTOS DE LA DENOMINACIÓN DE ORIGEN “VINOS DE MADRID” MEDIANTE EL ESTUDIO DE SU CONTENIDO METÁLICO
A.A. Sánchez, A. Gallego, R.M. Garcinuño, P. Fernández, J.S. Durand
Dpto. Ciencias Analítica, Facultad de Ciencias, UNED
E-mail: [email protected] La identificación y cuantificación de metales en vinos se considera de gran
interés ya que permite la definición de lo que se llama “huella dactilar” de cada uno de ellos, verificándose en cada caso su Denominación de Origen. Esta “huella” muestra que la composición del vino está fuertemente relacionada con factores propios del área específica de producción (variedad de uva, suelo y climatología, levaduras y prácticas de cultivo y vinificación), por lo que siempre habrá metales característicos para cada zona de cultivo. Además, el análisis de metales en el vino es de gran importancia para el control de la calidad y autenticidad del mismo.
La investigación llevada a cabo se centra en la determinación analítica de 11
elementos metálicos, mayoritarios y minoritarios tales como Ca, Cd, Cu, Fe, Mg, Mn, Ni, Zn, Li, Na y K, en muestras de vino tinto de la Denominación de Origen “Vinos de Madrid”, procedentes de las tres subzonas geográficas que la integran (Navalcarnero, Arganda y San Martín de Valdeiglesias). Los vinos analizados son tintos jóvenes del año 2004, procedentes de diferentes bodegas de estas tres subzonas de producción de la Comunidad de Madrid.
La determinación analítica de estos elementos se ha llevado a cabo empleando
la técnica de espectrometría de absorción y emisión atómica con llama. Por otro lado, se ha llevado a cabo la correlación de los resultados obtenidos
del contenido de dichos metales con el origen geográfico de los vinos, mediante un tratamiento estadístico adecuado. Para ello, se han realizado unos diagramas de cajas y bigotes con cada uno de los analitos de forma independiente. Finalmente, se ha aplicado a la matriz de datos distintos métodos estadísticos multivariantes, tales como el análisis de componentes principales y el análisis de clúster.
Tras estos estudios estadísticos se deduce que las subzonas de Navalcarnero y
San Martín de Valdeiglesias pueden diferenciarse en función de las variables (metales) estudiados. La subzona de Arganda, en general, posee valores muy dispersos no diferenciables, posiblemente debido a valores de caliza activa y materia orgánica diferentes entre parcelas de esta subzona.
Las muestras de vino fueron proporcionadas desinteresadamente por Consejo
Regulador de la Denominación de Origen de la Comunidad de Madrid. Los resultados obtenidos han proporcionado datos interesantes tanto para la Comunidad de Madrid como para llevar a cabo futuros estudios de investigación relacionados con este tipo de muestras.
[1] Anuario de estadística agroalimentaria, Ministerio de Agricultura, Pesca y
Alimentación, 2006 [2] Pérez, C., Técnicas de análisis multivariantes de datos. Aplicaciones con SPSS.
Pearson Educ., 2004

COMUNICACIONES EN CARTEL C1-38
79
CONTROL DE MADURACIÓN IN-SITU ACEITUNAS DESTINADAS A LA PRODUCCIÓN DE ACEITE DE OLIVA MEDIANTE ESPECTROSCOPÍA
INFRARROJA CERCANA (NIRS)
M. Ybarra-Lalor.*; R. Bouza-Deaño*, J. Espejo-Maqueda; A.J. Fernandez-Espinosa*, M. Ternero-Rodríguez*, J.C. Mohino-Guerrero**
*Departamento de Química Analítica. Facultad de Química. Universidad de Sevilla.
c/Profesor García Gonzalez, 1. 41012 Sevilla, España. (1) [email protected] **Soluciones Integrales de Laboratorio, S.L. (TECNILAB). c/Valencia, 978.
19170 – El Casar (Guadalajara). España La elección del momento de recolección de la aceituna destinada a la
producción de aceite de oliva es algo decisivo para los agricultores, ya que influye en la calidad del aceite obtenido finalmente. Resulta por tanto muy interesante para el agricultor el disponer de técnicas analíticas in-situ que le ofrezcan la información objetiva necesaria para llevar a cabo una correcta elección del momento idóneo para la recolección.
Los valores del porcentaje de humedad y grasa contenidos en el fruto, así como el índice de acidez del aceite son los parámetros utilizados por los agricultores para realizar esta decisión, realizándose estos siempre en el laboratorio mediante métodos normalizados. La posibilidad de realizar estos análisis de una forma totalmente automatizada y en campo, directamente sobre la fruta intacta, sin incluso desprenderla del árbol resulta lo suficientemente tentadora como para estudiar diversas técnicas analíticas que permitan la consecución de tal objetivo.
En las últimas décadas se han desarrollado numerosas aplicaciones similares utilizando la espectroscopia de infrarrojo cercano (Near InfraRed Spectroscopy, NIRS) para la predicción de parámetros de calidad interna de frutas intactas [1]. Referente a la aplicación de esta técnica en olivas y productos derivados, cabe destacar que la determinación del contenido de aceite y humedad en pasta de aceituna está siendo utilizado ya de forma rutinaria en numerosos laboratorios [2], con eficacias comparables a las obtenidas por técnicas aceptadas como la resonancia magnética nuclear (NMR) o la extracción por Soxhlet.
En la presente comunicación se presentan los resultados obtenidos para la predicción de parámetros de calidad de aceitunas (humedad, grasa y acidez), mediante análisis directos en el fruto por medio de espectrometría de infrarrojo cercano (NIRS), evaluándose simultáneamente la utilidad de un espectrómetro NIRS portátil para la utilización sobre la oliva en el árbol. Se han desarrollado respectivamente modelos predictivos y calibraciones utilizando como análisis de referencia análisis de laboratorio según métodos oficiales. Los parámetros analizados fueron: humedad del fruto, contenido en aceite del mismo referido a peso fresco y acidez libre del aceite. Los resultados indican un gran potencial de predicción mediante ambos métodos y alientan al perfeccionamiento de los modelos obtenidos mediante la ampliación de las calibraciones. [1] Tsuchikawa S, Sakai E, Inoue K. 2003. Application of Time-of-flight Near-
infrared Spectroscopy to detect sugar and acid content in Satsuma mandarin. J.Amer. Soc. Hort. Sci. 128, 391-396.
[2] García A., Ramos N, Ballesteros E. 2005. Estudio comparativo de distintas técnicas analíticas (espectroscopía de NIR y RMN y extracción mediante Soxhlet) para la determinación del contenido graso y de humedad en aceitunas y orujo de Jaén. Grasas y Aceites 56, 220-227.

C1-39 COMUNICACIONES EN CARTEL
80
ANALYSIS OF ALKYLMETHOXYPYRAZINES IN WINE BY MIXED-MODE SPE
AND GC-MS
E. Gracia-Moreno, R. López, J. Cacho, V. Ferreira
Laboratorio de Análisis del Aroma y Enología-I3A, Dpto. Química Analítica, Facultad de Ciencias, Univ. Zaragoza, Zaragoza. E-mail: [email protected] Alkylmethoxypyrazines are a family of aromatic compounds with an
endogenous presence in grapes [1]. Their concentration depends on the variety of grapes and the different techniques of viticulture [2]. It is also been reported their occurrence due to the presence of Harmonia axyridis lady beetle [3,4]. These compounds are considered responsible of important aromas in wines, particularly in those made from Sauvignon grapes. Their importance is related to their extremely low sensory thresholds [5-7]. Determination of these compounds in wine is a highly difficult task because of their low concentration levels. In this communication it is shown the development and validation of a new analytical method to quantify these analytes in their natural range of occurrence in wine.
Highly selective methods are needed to analyse compounds that are in the
ultra-trace levels, as in this case. For this reason, the proposed method is based in solid phase extraction with mixed mode sorbents coupled to gas chromatography-mass spectrometry. In the development of the method, several sorbents were tested to ascertain which one could provide the optimum breakthrough volume for those compounds. Usually, the most important problem when the analytes are in the ultra-trace levels is the presence of interferences. For this reason, once the loading conditions of the sample were optimized, several washing steps were tested. Finally, a comparison of eluents and the recoveries provided by them was carried out.
The method proposed in this communication allows the determination of 3-
isobutyl-2-methoxypyrazine, 3-isopropyl-2-methoxypyrazine and 3-sec-butyl-2-methoxypyrazine using 3-isopropyl-2-ethoxypyrazine as internal standard.
Linear ranges between 0.5 and 25 ng/L and detection limits lower than 0.5
ng/L have been achieved for these analytes thanks to the high selectivity of the sample processing method. The quantitation limits are enough to ascertain the sensorial importance of alkylmethoxypyrazines in wine.
[1] D. Roujou de Boubee, C. Van Leeuwen, D. Dubourdieu, J. Agric. Food Chem.
48 (2000) 4830-4834. [2] C. Sala, O. Busto, J. Guasch, F. Zamora, Journal of the Science of Food and
Agriculture 85 (2005) 1131-1136. [3] G.J. Pickering, J. Lin, A. Reynolds, G. Soleas, R. Riesen, International Journal of
Food Science and Technology 41 (2006) 77. [4] G.J. Pickering, J. Lin, R. Riesen, A. Reynolds, I.D. Brindle, G. Soleas, American
Journal of Enology and Viticulture 55 (2004) 153. [5] M.S. Allen, M.J. Lacey, R.L.N. Harris, W.V. Brown, American Journal of Enology
and Viticulture 42 (1991) 109-112. [6] S. Boutou, P. Chatonnet, Journal of Chromatography A 1141 (2007) 1-9. [7] Y. Kotsteridis, A. Anocibar Beloqui, A. Bertrand, J.P. Doazan, American Journal
of Enology and Viticulture 49 (1998) 44-48.

COMUNICACIONES EN CARTEL C1-40
81
PARÁMETROS DE CALIDAD NUTRICIONAL Y FUNCIONAL DE HORTALIZAS Y FRUTAS DE ALTO CONSUMO. ACTUALIZACIÓN Y MEJORA DE LAS TABLAS
DE COMPOSICIÓN DE ALIMENTOS
M. Gómez-Romero; E. Hurtado-Fernández; A. Carrasco-Pancorbo; A. Fernández-Gutiérrez
Dpto. Química Analítica, Facultad de Ciencias, Univ. Granada, Granada, España
E-mail: [email protected] Las tablas de composición de alimentos son importantes porque proporcionan
información detallada acerca de las concentraciones de nutrientes y componentes nutricionalmente importantes en los alimentos. Entre otras cosas, se emplean para conocer el valor nutricional de los alimentos en relación con la salud, en programas de educación e intervención nutricional en centros docentes y en colectivos, y en estudios de epidemiología nutricional.
Las tablas de composición de alimentos más completas existentes en España,
han sido elaboradas en la Universidad de Granada por miembros del Instituto de Nutrición y Tecnología de los Alimentos de dicha Universidad (Mataix J. y col. Tablas de Composición de Alimentos. Editorial Universidad de Granada, 2003), siendo utilizadas muy extensamente desde su publicación. Las citadas tablas tienen una fracción importante de datos basados en análisis propios, pero la mayor parte son datos bibliográficos, obtenidos de distintos países, fundamentalmente Estados Unidos e Inglaterra, que pueden no coincidir en muchos casos con los contenidos existentes en nuestro país con una climatología y condiciones ambientales bastante diferentes a las de aquellos.
Tanto en esas tablas como en otras publicadas en España no se recoge un
parámetro que toma cada vez más importancia, como es el contenido en antioxidantes. La importancia de estas sustancias radica principalmente en su capacidad para contrarrestar el daño celular causado por especies reactivas de oxígeno, dentro de las cuales se encuentran los radicales libres y los compuestos oxidantes. Los alimentos que los contienen se denominan alimentos funcionales, ya que, además de cualidades nutricionales, tienen un valor añadido por presentar propiedades saludables, bien con acción preventiva o terapéutica frente a distintas enfermedades crónicas como cáncer, enfermedades cardiovasculares, etc.
Con el fin de aportar datos referentes a la concentración de estos componentes
en nuestra dieta se someterán a estudio frutas y verduras producidas en la cuenca Mediterránea, que son muy ricas en dos familias de antioxidantes: los compuestos fenólicos (ácidos fenólicos y flavonoides) y los carotenoides (licopeno, luteína, β-caroteno y β-criptoxantina). Serán utilizadas 26 frutas y hortalizas, seleccionando distintas variedades, entre las que deben contemplarse la más consumida, la de mayor valor nutricional, alguna variedad ecológica, etc. Una vez extraídos los antioxidantes de la matriz alimentaria se procederá a su determinación mediante técnicas separativas, como son la electroforesis capilar, la cromatografía líquida y la cromatografía de gases acopladas a distintos detectores (fundamentalmente espectrometría de masas utilizando distintas interfases y analizadores).

C1-41 COMUNICACIONES EN CARTEL
82
CARACTERIZACIÓN DE VARIEDADES DE FRESAS Y OTRAS BERRIES A
PARTIR DE LA COMPOSICIÓN DE SU FRACCIÓN VOLÁTIL
Mª Carmen Robles y Ángeles F. Recamales
Área Química Analítica. Universidad de Huelva. E-mail: [email protected]
El estudio de los compuestos volátiles presenta un gran interés ya que constituyen una parte importante de los compuestos responsables del aroma de los alimentos y, por lo tanto, de la aceptación por parte de los consumidores. Asimismo, el aroma es esencial para la evaluación de la calidad de los productos frescos y procesados. El aroma típico de la fresa es el resultado de un equilibrio entre diferentes tipos de moléculas volátiles incluyendo ésteres, alcoholes, aldehídos, cetonas, y furanonas, siendo los compuestos que más contribuyen el hexanoato de etilo, butanoato de etilo, furaneol, butanoato de metilo y hexanoato de metilo. Muchos otros compuestos volátiles, aunque presentes en bajas concentraciones, son importantes en determinadas variedades.
En este trabajo se ha determinado la composición de la fracción volátil de tres
variedades de fresas (Camarosa, Candonga y Festival) cultivadas en la provincia de Huelva, en sistemas sin suelo utilizando cinco sustratos (geotextil, geotextil en saco, fibra de coco, lana de vidrio y perlita). También se han analizado dos variedades de mora y una de arándano y frambuesa. Las determinaciones se han realizado mediante un procedimiento de microextracción con disolventes asistida por ultrasonidos. En las muestras analizadas se han determinado ácidos, ésteres, furanonas, aldehídos y cetonas, lactonas, fenoles volátiles y terpenos.
A partir de los resultados obtenidos y mediante técnicas de análisis
multivariante se ha podido caracterizar las muestras analizadas. El análisis de la varianza aplicado a las muestras de fresa pone de manifiesto que existen diferencias significativas en la composición volátil de las variedades estudiadas, mientras que el tipo de sustrato prácticamente no tiene efecto significativo sobre las variables consideradas. El análisis discriminante permite asimismo diferenciar las tres variedades de fresas en base a su composición volátil. La función canónica 1 permite diferenciar la variedad Camarosa de la Candonga y Festival que a su vez se diferencian a través de la función canónica 2. Geraniol, furaneol, t-nerolidol, benzaldehído y ácido 2-metilbutírico son las variables con mayor poder discriminante. El LDA también permite diferenciar entre fresa y los otros tres tipos de frutas del bosque (mora, arándano y frambuesa) siendo en este caso las variables con mayor poder de discriminación el t-2-hexen-1-ol, etil-2-metilbutirato, t-2-hexenilacetato y hexanoato de metilo.

COMUNICACIONES EN CARTEL C1-42
83
GATHERING INFORMATION FROM NEAR INFRARED AND UV-VISIBLE SPECTROSCOPY FOR THE CLASSIFICATION OF SPANISH EXTRA VIRGIN
OLIVE OILS
C. Pizarro, S. Rodríguez-Tecedor, N. Pérez-del-Notario, J.M. González-Saiz, I. Esteban-Díez
Department of Chemistry, University of La Rioja, Logroño, La Rioja, Spain
E-mail: [email protected] Virgin olive oil is the juice obtained from the fruit of the olive tree (Olea
Europaea L.) by purely mechanical process, without solvent extraction. This quality category is the only one that can be consumed directly. The protected designation of origin (PDO) was created in order to guarantee and promote high quality virgin olive oils, that show particular quality properties due to their origin region.
Sample characterization and quality control processes are generally aimed to
knowing whether a given sample really comes from its declared origin or has the quality claimed. The European Union (EU) and the International Olive Oil Council (IOOC) have established official methods in order to guarantee the quality of the different olive oils. Since these methods, focused on the determination of some physical-chemical parameters, are laborious and time-consuming, other faster and simpler procedures are required.
Near Infrared and UV-Visible Spectroscopy, based on unspecific physical
quantities, have been applied in recent food authentication researches [1,2]. This spectroscopic profiles represent a “fingerprint” of the sample and can be considered as a continuous and unspecific signal without identifying any peak or compound.
In the present work, extra virgin olive oils from different Spanish origins,
including POD “Aceite de La Rioja”, were analyzed by Near Infrared and UV-Visible Spectroscopy. In order to classify the samples on the basis of their geographical origin, an strategy based on the application of Linear Discriminant Analysis (LDA) was applied. LDA was performed after pretreatment of signals and selection of the most discriminant wavelengths. The study was carried out in two steps. Firstly, data from NIR and UV-Vis were processed separately, and then joined to evaluate the effect of synergy of the information obtained from both techniques.
The clear separation achieved between the studied categories (with high
percentages of correct classifications in both training and prediction), confirms the feasibility of applying the proposed strategy for extra virgin olive oil quality assurance purposes.
[1] M. Casale, C. Armanino, C. Casolino, M. Forina, Anal. Chim. Acta. 589 (2007),
89-95. [2] C. Pizarro, I. Esteban-Díez, C. Sáenz-González, J.M. González-Sáiz, Anal. Chim.
Acta 608 (2008), 38-47.

C1-43 COMUNICACIONES EN CARTEL
84
EMPLEO DE QUANTUM DOTS DE CdSe COMO “MARCAS” ELEMENTALES PARA LA CUANTIFICACIÓN DE PROTEÍNAS MEDIANTE ESPECTROMETRÍA
DE MASAS ELEMENTAL
Antonio R. Montoro Bustos, Jorge Ruiz Encinar, María T. Fernández-Argüelles, José M. Costa-Fernández y Alfredo Sanz-Medel
Dpto. Química Física y Analítica, Universidad de Oviedo, Oviedo.
E-mail: [email protected] Los “Quantum Dots” (QDs) son nanopartículas semiconductoras, típicamente
esféricas con un diámetro comprendido entre 1 y 10 nm, compuestos por pocos átomos (200-10000 átomos). Presentan una serie de propiedades optoelectrónicas excepcionales que los convierten en un sistema de transducción especialmente atractivo para el desarrollo de nuevos (bio)sensores y “marcas” fluorescentes. Sin embargo, la propiedad más interesante y explotada de los QDs es la posibilidad de modificar su longitud de onda de emisión fotoluminiscente simplemente variando su tamaño y su composición elemental. Por esta razón el control exhaustivo de los procesos de síntesis de éstas nanopartículas es hoy crítico. Actualmente las aplicaciones bioanalíticas más recientes y prometedoras de los QDs están dirigidas a la detección de biomoléculas a las que han de bioconjugarse. Por tanto, el potencial analítico del empleo de QDs como “marcas” elementales para el análisis cuantitativo depende en último término de la determinación de la estequiometría QD-biomolécula, siendo aquí donde el ICPMS puede jugar un papel fundamental debido a su capacidad de cuantificación.
En un trabajo recientemente desarrollado [1] se ha empleado la utilización de
la Espectrometría de Masas con fuente de Plasma de Acoplamiento Inductivo (ICPMS) para la caracterización cuantitativa a nivel elemental de QDs de CdSe de manera precisa, logrando determinar de manera fiable el número de átomos (Cd y Se) existentes por QD sintetizado.
Una vez que los QDs han sido caracterizados desde el punto de vista elemental,
se propone su empleo como “marcas” elementales para la cuantificación de proteínas. Para poder traducir la señal elemental cuantitativa proporcionada por el QD en cantidad de proteína es necesario realizar un estudio en profundidad de la estequiometría proteína/QD, la cual se concreta en la medida de la relación S/Cd, entre el contenido de S de la proteína y el Cd aportado por el QD haciendo uso de un equipo ICPMS de alta resolución.
Como sistema modelo se ha escogido la proteína Avidina (Av) a la que se
bioconjugarán los QDs de CdSe empleando 3-(dimetilaminopropil)carbodiimida (EDC) como agente catalizador.
[1] A. R. Montoro Bustos, J. Ruiz Encinar, M. T. Fernández-Argüelles, J. M. Costa-Fernández, A. Sanz-Medel, Chem. Commun. DOI: 10.1039/b901493d (2009).

COMUNICACIONES EN CARTEL C1-44
85
ESTUDIOS CINÉTICOS DE ALTA RESOLUCIÓN DEL GRADO DE
FOSFORILACIÓN DE PÉPTIDOS/PROTEÍNAS MEDIANTE µHPLC-ICP-MS
Jorge Ruiz Encinar, Alejandra Simón Mañogil, Ana Pereira Navaza, Alfredo Sanz-Medel
Departamento de Química Física y Analítica, Universidad de Oviedo, Oviedo. Las células están continuamente recibiendo estímulos del exterior a los cuales
deben responder. Es bien sabido hoy en día que la fosforilación juega un papel esencial en dichas repuestas por lo que es estrictamente necesario cuantificar la dinámica de fosforilación de cada proteína para descubrir la relación entre las reacciones de señalización y las respuestas biológicas observadas.
La espectrometría de masas molecular se ha establecido actualmente como la
herramienta más poderosa para el estudio de cambios temporales en la fosforilación debido a su versatilidad y sensibilidad. Las estrategias basadas en la espectrometría de masas para estudiar dinámicas temporales de la señalización celular están basadas, principalmente, es el uso de estrategias de marcaje con isótopos estables. Todas estas estrategias de marcaje proporcionan únicamente la cuantificación relativa de la fosforilación de la proteína. Además, la resolución en el tiempo que se consigue está siempre limitada tanto por el número de marcas isotópicas disponibles como por la relativamente alta variabilidad en los resultados cuantitativos (10-20% RSD). Recientemente, han sido reportados métodos de cuantificación (AQUA, QCAT) pero requieren la síntesis de péptidos/proteínas homólogos marcados con isótopos pesados para ser usados como patrones internos de cada péptido/proteína en estudio.
La respuesta por espectrometría de masas elemental (ICPMS) es independiente
de la especie y de la matriz permitiendo una cuantificación absoluta y específica de los fosfopéptidos generados después de su digestión tríptica y que corresponden a los distintos sitios de fosforilación de la proteína (conocidos de antemano o no). Los resultados cuantitativos obtenidos en este trabajo por µHPLC-ICPMS también muestran una exactitud y precisión excepcional (<5%RSD) [1]. En este trabajo se muestra el extraordinario potencial de esta estrategia para discriminar entre cambios temporales muy pequeños en los niveles de fosforilación de las proteínas [2]. La alta precisión conseguida en la cuantificación, la cual permite discriminar entre grados de fosforilación muy próximos y la no limitación por el uso de isótopos estables permite realizar estudios cinéticos de fosforilación/defosforilación de proteínas con una resolución temporal (hasta 15 puntos) no alcanzada anteriormente. Estos estudios temporales de alta resolución podrían servir en el futuro para conocer en detalle el papel de las proteínas estudiadas en el proceso de señalización celular en el que están involucradas. [1] A. Pereira Navaza, J. Ruiz Encinar, M. Carrascal, J. Abian, A. Sanz-Medel,
Analytical Chemistry, 2008, 80, (5),1777-1787 [2] A. Pereira Navaza, J. Ruiz Encinar, A. Sanz-Medel Chemical Communications,
2008, 6230–6232

C1-45 COMUNICACIONES EN CARTEL
86
DETERMINACIÓN DE AMINAS VOLATILES EN PRINCIPIOS ACTIVOS
FARMACEUTICOS POR SPME-GC-MS-NCI
J. Zapata, V. Ferreira, J. Cacho
Laboratorio de Análisis del Aroma y Enología, Dpto. de Química Analítica, Facultad de Ciencias, Universidad de Zaragoza, 50009 Zaragoza, España.
E-mail: [email protected]
Algunas aminas volátiles son empleadas en la síntesis de principios activos farmacéuticos y se encuentran presentes en ellos de forma residual, otras se generan por degradación de los principios farmacéuticos activos o material residual. Muchas de estas aminas presentan alto riesgo por su toxicidad, como en el caso de la 2-Cloroetilamina que es usado en la síntesis de principios activos farmacéuticos, la cantidad residual de esta puede reaccionar, vía ciclación, para producir la aziridina, un potente agente carcinogénico [1]. Por eso como requisito para la fabricación y desarrollo de drogas, se hace necesario desarrollar métodos analíticos adecuados para la determinación de estos compuestos a niveles de mg/Kg.
La técnica idónea para el análisis de compuestos volátiles es la cromatografía de gases, pero el análisis de las aminas volátiles por esta técnica es particularmente difícil debido a sus propiedades fisicoquímicas; alta volatilidad, alta polaridad y carácter básico, capacidad de formar puentes de hidrogeno y alta solubilidad en agua. Por ello las estrategias para su determinación requieren emplear técnicas de derivatización que normalmente demandan múltiples etapas y consumen demasiado tiempo para su análisis [2]. El objetivo de esta comunicación es presentar un método analítico rápido y completamente automatizado, mediante la técnica de HS-SPME basado en la derivatización-extracción de las aminas volátiles sobre una fibra de PDMS/DVB y su posterior análisis por GC-MS-NCI, empleando como agente derivatizante el 2,3,4,5,6-pentafluorobenzoil-cloruro. Para el desarrollo del método se emplearon dos aminoácidos, glicina y fenilalanina, como modelos de principios activos farmacéuticos, estos fueron puestos en viales estándar de SPME con agua basificada para el análisis directo de las respectivas Pentaflourobencil-amidas formadas.
Inicialmente se realizo un estudio de la cinética de fijación del PFBCl en la fibra y la cinética total de la extracción-reacción de las aminas para la optimización del método y posteriormente se realizo la validación analítica. Los resultados de la validación mostraron que el método presenta como mayor fortaleza una alta sensibilidad, con límites de detección del orden de los µg/Kg, las reproducibilidades encontradas están en el rango de 3-20 %RSD, la linealidad fue satisfactoria, en todos los casos el coeficiente R2>0,99, las recuperaciones del método son satisfactorias, si bien el método debe ser recalibrado al cambiar la fibra de SPME.
El método presentado puede ser una alternativa útil, rápida y económica para el análisis de aminas volátiles residuales en principios activos farmacéuticos.
Agradecimientos
Este trabajo fue financiado por MEC-DGI, proyecto AGL 2007-65139. Referencias:
[1] 1. P. E. de Haan, D. d. J. J. H. M. v. d. B. C. G. K., Journal of High Resolution Chromatography 1989, 12, (9), 604-607.
[2] 2. Ábalos, M.; Mas, R.; Suc, V.; Bayona, J.,. Chromatographia 2001, 54, (1), 109-113.

COMUNICACIONES EN CARTEL C1-46
87
UHPLC-MS/MS DETERMINATION OF MYCOTOXINS IN DIFFERENT
FOOD COMMODITIES
E. Beltrán, J.V. Sancho, M. Ibáñez, F. Hernández
Research Institute for Pesticides and Water, University Jaume I, Castellón, Spain. E-mail: [email protected]
Mycotoxins are secondary metabolites produced by molds, without biochemical
significance in fungal growth and development. Mycotoxins have adverse effects on humans and animals and cause economic losses on crops due to their contamination.
Mycotoxins are molecules with various chemical structures and, therefore various biological effects. There are known over 300 compounds that grow under a wide range of climatic conditions on agricultural commodities. Not all secondary metabolites from molds are toxic, so it is necessary to regulate those mycotoxins that can be dangerous for human health
Due to the serious effects on humans and animals, the European Commission (EC) has established the maximum permitted levels for several mycotoxins. The main mycotoxins regulated are: Aflatoxins, Ochratoxin A, Patulin, Deoxynivalenol, Zearalenone, Fumonisins, and HT-2 and T-2 Toxin. It is therefore necessary to apply sensitive and reliable analytical methods able to reach the regulatory levels established by the EC.
A rapid multianalyte-multiclass method with little sample manipulation has
been developed for the simultaneous determination of 11 mycotoxins in different food commodities by using ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry (UHPLC-MS/MS) [1]. Toxins were extracted from the samples with ACN:H2O (80:20, v/v) 0.1% HCOOH and, after a two-fold dilution with water, directly injected into the system. Thanks to the fast high-resolution separation of UHPLC, the eleven mycotoxins were separated by gradient elution in only 4 minutes. The method has been validated in three food matrices (maize kernels, dry pasta (wheat), and 8-multicereal babyfood (wheat, maize, rice, oat, barley, rye, sorghum, millet)) at four different concentration levels. Satisfactory recoveries were obtained (70-110%) and precision (expressed as relative standard deviation) was typically below 15% with very few exceptions. Quantification of samples was carried out with matrix-matched standards calibration. The lowest concentration successfully validated in sample was as low as 0.5 µg/Kg for aflatoxins and ochratoxin A in babyfood, and 20 µg/Kg for the rest of selected mycotoxins in all matrices tested. Deoxynivalenol could be only validated at 200 µg/Kg, due the poor sensitivity for this mycotoxin analysis. With only two exceptions (HT-2 and deoxynivalenol), the LODs, estimated for a signal-to-noise of 3 from the chromatograms of samples spiked at the lowest level validated, varied between 0.1 and 1 µg/Kg in the three food matrices tested. The method was applied to the analysis of different kinds of samples. Positive findings were confirmed by acquiring two transitions (Q quantification, q confirmation) and evaluating the Q/q ratio.
Present research is focussed on Aflatoxin B1 and its metabolite Aflatoxin M1 in order to reach the sensitivity required for babyfood analysis, accordingly to the very low concentration levels admitted by the European Commission [1] E. Beltran, M. Ibáñez, J.V. Sancho, F. Hernández, Rapid Commun. Mass
Spectrom. 23 (2009) 1-9

C1-47 COMUNICACIONES EN CARTEL
88
APLICACIÓN DE MCR-ALS A DATOS DE HPLC-MS PARA EL ESTUDIO
COMPARATIVO DEL COMPORTAMIENTO DE LOS METABOLITOS ENDÓGENOS PRESENTES EN DOS VARIEDADES DE TOMATE TRATADAS CON
CARBOFURANO
M. Martínez Galeraa, I. Sánchez Péreza, M.D. Gil Garcíaa, H.C. Goicoecheab
aDpto. Hidrogeología y Química Analítica, Univ.Almería, Almería, España
E-mail: [email protected] bLab. Desarrollo Analítico y Quimiometría, Cátedra de Química Analítica, Univ.
Nacional del Litoral, Santa Fe, Argentina
Se ha llevado a cabo un estudio metabonómico basado en la aplicación de resolución multivariada de curvas y mínimos cuadrados alternantes (MCR-ALS) a datos tridimensionales obtenidos mediante HPLC-MS de muestras de dos variedades tomate de tratadas carbofurano. Así, ha sido posible la detección de diferentes comportamientos en el tiempo para determinados metabolitos endógenos presentes simultáneamente en variedades de tomate Rambo y Raf, tras su tratamiento con dicho plaguicida.
En primer lugar, se llevó a cabo la extracción (método QuEChERS) y el análisis
mediante HPLC-MS (en modo SCAN) de diferentes muestras recolectadas durante un periodo de 21 días tras el tratamiento con carbofurano. En segundo lugar, los datos obtenidos de cada matriz (muestras tratadas y no tratadas) fueron analizados con MCR-ALS mediante la metodología de matrices ampliadas por columnas, a fin de obtener información acerca de la evolución en el tiempo de los diferentes componentes (componentes principales estimados mediante descomposición en valores singulares, SVD). MCR-ALS también proporcionó los perfiles espectrales de masas para dichos componentes, los cuales se utilizaron para identificar componentes comunes presentes en las muestras tratadas y muestras no tratadas con carbofurano, en ambas matrices (Rambo y Raf). Tras este estudio se encontró que la presencia de plaguicida afectaba al comportamiento de diversos componentes de la matriz del tomate alterando su perfil de evolución en el tiempo, tanto en tomates de variedad Rambo como en tomates Raf.
Además, un estudio comparativo (mediante el uso de una matriz de
correlación) de los perfiles espectrales obtenidos en los análisis por MCR-ALS de las muestras de ambas variedades mostró que determinados componentes, presentes en ambas variedades, se comportaban de forma diferente en el tiempo tras el tratamiento con el plaguicida. Por otra parte, se encontró que otros componentes, presentaban perfiles de evolución en el tiempo idénticos tanto en Rambo como en Raf.
La comparación de dichos perfiles con los de las muestras no tratadas con
carbofurano puso de manifiesto que la presencia del plaguicida provoca en las plantas una situación de estrés fisiológico que afecta de igual o diferente manera a un grupo de metabolitos (componentes principales) presentes simultáneamente en ambas variedades de tomate.

COMUNICACIONES EN CARTEL C1-48
89
ON-LINE PRECONCENTRATION SYSTEM USING A SHORT-LIQUID
CHROMATOGRAPHY COLUMN COUPLED WITH LIQUID CHROMATOGRAPHY DIODE ARRAY DETECTION FOR THE SOLID-PHASE EXTRACTION AND CLEAN-UP OF BASIC DRUGS IN ENVIROMENTAL WATER SAMPLES
M. Martínez Galeraa, M.D. Gil Garcíaa, M.J. Culzonib, H.C. Goicoecheab
aDpto. Hidrogeología y Química Analítica, Univ.Almería, Almería, España
E-mail: [email protected] bLab. Desarrollo Analítico y Quimiometría, Cátedra de Química Analítica, Univ.
Nacional del Litoral, Santa Fe, Argentina
Human and veterinary drugs are used in large quantities and significant fractions of the original substances are excreted (as un-metabolized form or active metabolites) via urine or faeces. Moreover, some of them are not degraded in waste treatment plants and enter into hydrologic systems in their original form, with hazardous effects on human health and aquatic ecosystems. In the last decades, different studies carried out in some countries showed that most of these pharmaceuticals can be found in wastewater, river water and even in drinking water at levels of up to a few µg L−1. Therefore, several methods have been developed for the determination of pharmaceuticals in water in the lower ng L-1 range using normally off-line solid phase extraction (SPE) techniques combined with LC or GC. These extraction techniques involve the manipulation of samples, and therefore, they may lead to the partial loss of some of the compounds, as well as an increase of labour, time and cost.
The main goal of the present work is the optimization of a simple and
automated method using a first LC short column for the preconcentration of several basic drugs in river water samples coupled with a second conventional LC column for separation and diode array detection. Eleven pharmaceuticals compounds, among the more commonly used in human, have been selected in this study.
The use of a short LC column (C-1) coupled to LC analytical column (C-2)
allows the preconcentration, clean up and analysis of large volumes of water samples and the determination of drugs at ng L-1 range. In order to obtain the lower quantitation limits, the optimized parameters for peconcentration step were percentage of organic modifier in the water sample, flow and time of preconcentration in the first column. The best results were found with a flow rate of 1.5 mL min-1 during 20 min (corresponding to a load of water sample of 30 mL) and 0.4 % of methanol as organic modifier in the aqueous sample. Transference and separation of analytes in C-2 column were performed using a ternary gradient mobile phase composed of 0.25 mol L-1 KH2PO4 adjusted at pH 3.0, methanol and acetonitrile. Several mobile phases were evaluated in order to allow good peak resolutions for the eleven analytes although non resolution of chromatographic peaks was found for some analytes even for long time of analysis. In addition, when real river water samples were analyzed interferent peaks and matrix effect were found for some analytes. In order to solve these problems, standard addition methodology combined with Multivariate Curve Resolution (MCR) has been proposed as a complementary tool to traditional chromatographic approaches. The proposed methodology was validated using spiked blank river water samples. Quantitation limits were ranged between 0.1 and 0.5 µg L-1 with recovery percentages of basic drugs in the river water near to 100 %.

C1-49 COMUNICACIONES EN CARTEL
90
QUASI-REAGENTLESS ELECTROCHEMICAL INMUNOSENSORS FOR THE
DETECTION OF QUINOLONES
Javier Jiménez, Arántzazu Narváez, Elena Domínguez
Department of Analytical Chemistry, University of Alcalá, Alcalá de Henares, Madrid. E-mail: [email protected]
The increasing demand of food products of animal origin for human
consumption has led to the use of certain compounds for the prevention of animal farm diseases. Quinolones, extremely active substances at low doses, are broad-spectrum antibiotics frequently used in the treatment of animal infections and, sometimes, as growth promoters fraudulently [1]. This massive use provokes the presence of quinolones residues in food chain. This fact causes concerns for consumers’ health, so that the UE has introduced in its legislation (Regulation 2377/90/CEE) maximum tolerable concentrations of residues for different target tissues.
This work describes a novel quasi-reagentless amperometric immunosensors
based on multilayered heterofunctional assemblies [2,3] for the detection of quinolones. The rationale of our approach relays on the completion of the affinity reaction within the assemblies using enzyme labelled immunoreagents. Detection is then based on the displacement of the labelled immunoreagent by the presence of the sample rendering a concomitant signal decrease. The analysis only requires the addition of a sample drop to the immunochips and incubation for 30 minutes. Amperometric responses are then obtained at +50 mV vs. Ag/AgClsat by the addition of the substrate. The work covers the analytical description of the immunochips as well as the preparation and characterisation of the layered nanostructures for the integration of the affinity and transduction events. The obtained results show a limit of detection in buffer of 0.05 ppb and a EC50 of 0.8 ppb for ciprofloxacin.
Financial support from the Spanish Ministry of Education and Science (AGL2008-05578-C05-02/ALI) is acknowledged.
[1] A. Anadon, M.R. Martinez-Larranaga, Livestock Prod. Sci. 59 (1999) 183. [2] A. Narváez, G. Suárez, I.C. Popescu, I. Katakis & E. Domínguez. Biosens.
Bioelectron. 15 (2000) 43. [3] E. Domínguez, O. Rincón, A. Narvaez. Anal. Chem. 76 (2004) 3132.

COMUNICACIONES EN CARTEL C1-50
91
COMPARATIVE STUDY OF DIFFERENT CLONES OF GRAPE USED IN THE VINIFICATION OF TXAKOLI RED WINE OF BIZKAIA THROUGH THE
ANTHOCYANIN PROFILE
D.M. López-Márquez, M.B. Sánchez-Ilárduya, M. Romera-Fernández, Luis A. Berrueta, B. Gallo, F. Vicente
Departamento de Química Analítica, Facultad de Ciencia y Tecnología, UPV/EHU,
Apdo. 644, E-48080, Bilbao, E-mail: [email protected] Bizkaia (Basque Country) has a rich and varied heritage vineyard, composed by
local and foreign varieties. The red grape is the main source of anthocyanin compounds which are the predominant pigments responsible for the colour of red wine. These compounds, which are stored in the skin of grape berries, change during the maturation and their biosynthesis is affected by factors such as climatic conditions, ripening, winemaking practices, grape variety, etc...
To carry out this work, different clones of the local variety Hondarrabi Beltza and French varieties Cabernet Franc and Cabernet Sauvignon have been supplied by the Experimental Winery of the Diputación Foral de Bizkaia, with which a clonal and a comparative study to differentiate among varieties according with anthocyanin composition have been developed.
The samples analyzed were collected indistinctively in 2004, 2005 and 2006 vintages: 2 clones of the variety H. Beltza (clon A and B), 3 clones of the variety C. Franc (clon 396, 622 and plot 1) and 5 clones of the variety C. Sauvignon (clon 15, 171, 191,216 and 685). Each grape sample was pressed in the laboratory using a traditional wooden press, obtaining on one side the skins, and in the other side the monovarietal musts. The skins were quickly frozen with liquid nitrogen to prevent oxidation. Subsequently the skins were freeze-dried in order to remove all the water and finally were crushed to obtain a fine and dried violet powder. On the other hand, the monovarietal musts were centrifuged and aliquots were taken to carry out the analysis of the anthocyanin content.
The determination of the anthocyanin profile of each sample was performed by HPLC-DAD analysis after a prior solid-liquid extraction for the skins and by direct injection in the case of musts. Quantification of the anthocyanins was performed at 530 nm, while the UV-visible absorption spectra, collected between 250-600 nm, were used to confirm which anthocyanin family each compound belongs to.
The results of this study permitted to define exactly the anthocyanin profile of the grape varieties harvested in Bizkaia. This study shows that the anthocyanin profiles were similar among the different grapes studied. However the mean content of anthocyanin was higher for the H. Beltza grapes than for the foreign varieties C. Franc and C. Sauvignon. Besides, each type of grape had a distinctive anthocyanin pattern. So, C. Franc and C. Sauvignon showed the major content in malvidin-3-glucoside, while H. Beltza can be differentiated based on the higher content in peonidin-3-glucoside. In the same way, PCA analysis of anthocyanin profiles show differents clusters depending on grape varieties.
From the results obtained we can concluded that the local grapes, H. Beltza, are an interesting plant material to obtain high colored txakolis red wines with a suitable anthocyanin composition.
Acknowledgements: The authors thank to Departamento de Agricultura y Pesca del Gobierno Vasco/ Eusko Jaurlaritza (Ph. D. grant of D.M. López-Márquez) for financial support and express their gratitude to the Diputación Foral de Bizkaia.

C1-51 COMUNICACIONES EN CARTEL
92
INFLUENCE OF DIFFERENT ANTHOCYANIN DERIVED PIGMENTS ON THE
COLOUR OF RED WINES FROM RIOJA
M. B. Sánchez-Ilárduya, S. Garmón-Lobato, M. Viloria-Bernal, L. A. Berrueta, B. Gallo & F. Vicente
Departamento de Química Analítica, Facultad de Ciencia y Tecnología, UPV/EHU, Apdo. 644, E-48080 Bilbao, España. E-mail: [email protected]
Reactions between anthocyanins and other substances present in wine lead to
the formation of new pigments that provide different hues to aged wine. Some of these new pigments, called pyranoanthocyanins, for example adducts with pyruvic acid, acetaldehyde, acetone, vinylphenol, vinylcatechol, vinylguaiacol, vinylsyringol and vinylflavanols, provide orange hues to red wine because of their visible absorption maxima present hypsochromical shifts with respect to simple anthocyanins. Other kind of compounds, which are formed by direct or by an ethyl brigde condensations between anthocyanins and flavanols, provide bluish hues to red wine due to batochromical shifts in their visible absorption maxima.
The absorption λmax of different malvidin derived pigments are shown in the following table.
Table 1. Absorption λmax of different malvidin derived pigments.
Pigment Absorption λmax (nm)
Mv-3-glc 526
Mv-3-glc-pyruvic acid 510
Mv-3-glc-acetaldehyde 502
Mv-3-glc-acetone 499
Mv-3-glc-vinylphenol 503
Mv-3-glc-vinylcatechol 510
Mv-3-glc-vinylguaiacol 511
Mv-3-glc-vinylsyringol 517
Mv-3-glc-vinyl(epi)catechin 506
Mv-3-glc-vinyldi(epi)catechin 511
Mv-3-glc-vinyl(epi)galocatechin 506
(Epi)catechin-Mv-3-glc 532
Di(epi)catechin-Mv-3-glc 536
Mv-3-glc-8-ethyl-(epi)catechin 541
The colour provided by the anthocyanins and anthocyanin derived pigments to wine could be modified towards bluish or orange hues in accordance with the proportion of each type of pigments. These shifts can be observed in the absorption spectra of two wines (Figure 1). Wine 1 contains higher concentrations of pyranoanthocyanins and wine 2 contains higher concentrations of direct condensations of anthocyanins with flavanols.
Normalized absorption spectra
35
45
55
65
75
85
95
105
115
125
350 400 450 500 550 600 650
Wavelength (nm)
No
rmal
ized
ab
sorb
ance
Wine 1
Wine 2
Figure 1. Comparison between the absorption spectra of two different wines.
Acknowledgements: Authors thank to Departamento de Industria, Comercio y Turismo (Projects S-PE06UN14 and S-PE07UN34) of Gobierno Vasco/Eusko Jaurlaritza for financial support. M. B. Sánchez-Ilárduya thanks to UPV/EHU for her Ph.D Grant.

COMUNICACIONES EN CARTEL C1-52
93
RAPID DETERMINATION OF THE ANTHOCYANINS CONTENT IN THE GRAPE
MUSTS OF RIOJA WINES USING FT-IR
M. Romera-Fernández, C. Sánchez-Fernández, M. Viloria-Bernal, L. A. Berrueta, Blanca Gallo, Francisca Vicente
Departamento de Química Analítica. Facultad de Ciencia y Tecnología, UPV/EHU,
Apdo. 644, E-48080 Bilbao. E-mail: [email protected]
The knowledge of the anthocyanins content in the grape must is very important in order to obtain great quality wines; it is necessary to control the technologic, aromatic and phenolic maturation, since it helps to determine the grape ripeness and deciding the optimal time of vintage. Analysis of anthocyanins content in grapes during maturation is used to monitor the development of these molecules and classify the vineyards and even individual parcels according to their phenolic content. Taking into account that under similar conditions in the winemaking practices, the grapes with a higher anthocyanic content will generate more wine color, determining the time when the anthocyanins content in grapes reaches a maximum is a rather important aspect that should be controlled. At optimum phenolic ripeness, the grapes have both a high power of pigmentation and a good ability to release these substances to the wine.
A chromatographic analysis of 78 samples of grape musts of the 2007 harvest has been carried out to obtain the concentrations of individual anthocyanins in musts (reference values). These musts belong to five wineries of Rioja (39 musts have been extracted at pH 1.0 and 39 at pH 3.2). At pH 1, all anthocyanins are soluble and removable, while extraction at pH 3.2 is comparable to the procces that occurs in the winemaking process. The percentage of anthocyanic content extracted at pH 3.2 is higher for the non-acylated in comparison to acetylated and coumaroylated anthocyanins, and among the acylated anthocyanins extraction is higher for acetylated. The FT-IR spectra of the same 78 grape musts were also obtained.
A principal component analysis of the reference values at both pHs has been performed. Two clearly different groups have been observed depending on the pH of the extraction. Therefore, two PLS regression treatments for the FT-IR instruments of the wineries were built, one includes samples of musts at pH 1.0 and the other those at pH 3.2.
Cross validation errors in these PLS regressions show no significant differences for most of anthocyanins predictions, however they are slightly better for the calibrate generated using the samples extracted at pH 1.0 (the concentrations of anthocyanins were higher, and therefore the methodology will have less difficulty to find areas of spectrum where these polyphenolic compounds are better defined); these improvements ranged from 0.5 to 18.4 percentage points when compared with the calibration from samples at pH 3.2. In both calibrates, eight of anthocyanins, including the Malvidin-3-glucoside and the sum of non-acylated anthocyanins, are predicted with an error less than 15%.
At pH 3.2, the percentage of extraction of anthocyanins is quite similar to that in the winemaking process and the differences in the cross-validation errors are not so significant between the two calibrations, at least for major anthocyanins. Therefore, final calibration implemented in the wineries for determining anthocyanins in grape musts is that one built with the must samples at pH 3.2. Acknowledgements: This study was supported by Departamento de Agricultura, Pesca y Alimentación del Gobierno Vasco. Miriam Romera thanks UPV/EHU for her Ph.D. Grant.
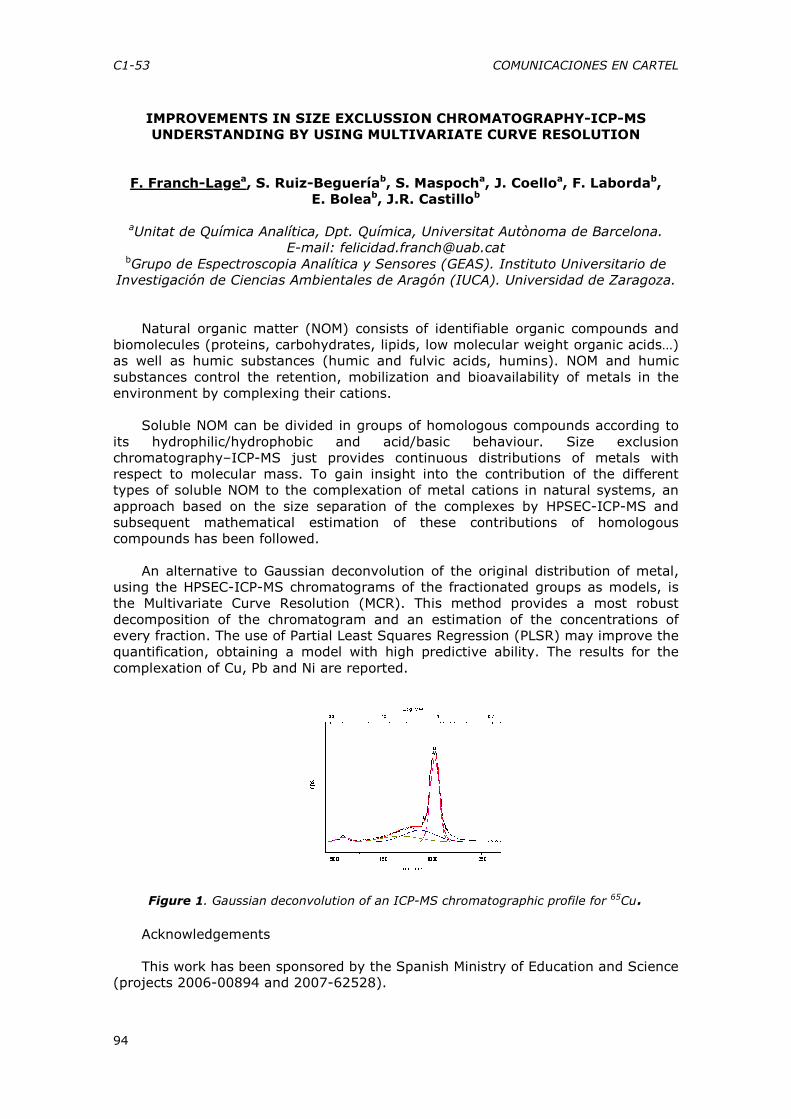
C1-53 COMUNICACIONES EN CARTEL
94
IMPROVEMENTS IN SIZE EXCLUSSION CHROMATOGRAPHY-ICP-MS UNDERSTANDING BY USING MULTIVARIATE CURVE RESOLUTION
F. Franch-Lagea, S. Ruiz-Begueríab, S. Maspocha, J. Coelloa, F. Labordab,
E. Boleab, J.R. Castillob
aUnitat de Química Analítica, Dpt. Química, Universitat Autònoma de Barcelona. E-mail: [email protected]
bGrupo de Espectroscopia Analítica y Sensores (GEAS). Instituto Universitario de Investigación de Ciencias Ambientales de Aragón (IUCA). Universidad de Zaragoza.
Natural organic matter (NOM) consists of identifiable organic compounds and
biomolecules (proteins, carbohydrates, lipids, low molecular weight organic acids…) as well as humic substances (humic and fulvic acids, humins). NOM and humic substances control the retention, mobilization and bioavailability of metals in the environment by complexing their cations.
Soluble NOM can be divided in groups of homologous compounds according to
its hydrophilic/hydrophobic and acid/basic behaviour. Size exclusion chromatography–ICP-MS just provides continuous distributions of metals with respect to molecular mass. To gain insight into the contribution of the different types of soluble NOM to the complexation of metal cations in natural systems, an approach based on the size separation of the complexes by HPSEC-ICP-MS and subsequent mathematical estimation of these contributions of homologous compounds has been followed.
An alternative to Gaussian deconvolution of the original distribution of metal,
using the HPSEC-ICP-MS chromatograms of the fractionated groups as models, is the Multivariate Curve Resolution (MCR). This method provides a most robust decomposition of the chromatogram and an estimation of the concentrations of every fraction. The use of Partial Least Squares Regression (PLSR) may improve the quantification, obtaining a model with high predictive ability. The results for the complexation of Cu, Pb and Ni are reported.
Figure 1. Gaussian deconvolution of an ICP-MS chromatographic profile for 65Cu.
Acknowledgements This work has been sponsored by the Spanish Ministry of Education and Science
(projects 2006-00894 and 2007-62528).

COMUNICACIONES EN CARTEL C1-54
95
ACETYLSALICYLIC ACID DISTRIBUTION IN COMMERCIAL TABLETS BY
USING NIR-CHEMICAL IMAGING.
Jordi Cruz1*, Manel Bautista1, José Manuel Amigo2, Marcel Blanco1
1Unidad de Química Analítica. Departamento de Química. Facultad de Ciencias. Universitat Autónoma de Barcelona. 08193 Bellaterra, Barcelona. España.
*[email protected] 2Department of Food Science Quality & Technology. University of Copenhagen.
Rolighedsvej 30, DK-1958 Frederiksberg-C. Denmark
Near Infrared Chemical Imaging (NIR-CI) is demonstrating an increasing interest in pharmaceutical field meeting the challenging analytical needs of pharmaceutical quality in a non destructive and non invasive, manner, representing an important complement to conventional NIR spectroscopy.
Hyperspectral Imaging techniques provide a NIR spectrum in each pixel of the
image, generating a huge amount of information, which is difficult to be treated. To treat this amount of information, easy to use chemometric methods are continuously demanded. Some algorithms, like Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) has been already used to quantify each pixel from the image and to obtain a concentration map without doing a previous calibration.
In this work, MCR-ALS has been applied to commercial acetylsalicylic acid
tablets to quantify the Active Principal Ingredient (API) concentration. API concentration maps and histograms have been calculated for each tablet, studding in this way the distribution of the API in each tablet. A wide range of API concentration, between 82% and 12%, was covered for the studied commercial tablets, which present different compositions depending on the manufacturer.
The results showed a good API distribution despite the different origins of the
tablets. Good prediction values of the API content from the tablets were obtained. No significant differences regarding the nominal concentration values, defined as the ratio between the quantity of API by the tablet weight, could be observed.
Moreover, the average differences between the calculated concentrations with
the MCR-ALS algorithm and the nominal concentrations were comprised between 2-3%.

C1-55 COMUNICACIONES EN CARTEL
96
RP-HPLC-ESI-MS PROFILING OF TRANSGENIC AND NON-TRANSGENIC
MAIZE CULTIVARS
M.C. García1, V. García-Cañas2, M.L. Marina1
1Dpto. Química Analítica, Facultad de Química, Universidad de Alcalá, 28871 Alcalá de Henares (Madrid). E-mail: [email protected]
2Dpto. Análisis de Alimentos, Instituto de Fermentaciones Industriales, CSIC, C/ Juan de la Cierva, 3, 28006 Madrid.
Maize (Zea mays L.) is one of the most important cereal crops worldwide
constituting a cheap source of food and feed. The huge genetic diversity of this crop, the differences regarding quality and characteristics of every maize variety, and the development of maize improvement programs are only some of the pursued goals by plant breeders and scientific programs. Moreover, advances in genetics have enabled the introduction of DNA fragments into the maize genome to improve its resistance to certain plagues such as the European corn borer (Ostrinia nubilalis) and the tolerance to certain herbicides. Nevertheless, the development of these new cultivars is surrounded by a great controversy concerning its safety, environmental and ethical impact, potential negative effect on human health, etc.
The growing interest in evaluating and genetically improving the quality of
maize has originated the need for the development of methodologies for maize characterization. They are usually based on plant morphological and agronomical characters, immunochemical assays or DNA markers. Another strategy has been the analysis of proteins. Maize contains around 10 % proteins classified according to their solubility in albumins, globulins, glutelins, and the most abundant called zeins or prolamins. Zeins analysis by MALDI-TOF-MS showed that this technique could be a suitable analytical tool for genotype identification [1].
In this work, an analytical methodology using RP-HPLC-ESI-MS (ion trap) is
proposed for the first time for the profiling of transgenic and non-transgenic maize cultivars. To optimize chromatographic conditions the following parameters were studied: column, gradient, and ion-pairing reagent. Moreover, the influence on the MS signal of the variation of the capillary voltage and the accumulated ions in the trap were also investigated. The developed method was applied to the profiling of different maize samples, namely, maize cultivars from different geographical origins (USA, Canada, France, and Spain), transgenic maize samples with certified GMO content, and three transgenic Bt maize cultivars with their isogenic non-transgenic counterparts (Aristis Bt vs. Aristis, PR33P67 vs. PR33P66, and DKC6575 vs. Tietar). Also, different protein fractions (albumin, globulin, prolamin, and glutelin) isolated from the Bt transgenic and non-transgenic maize cultivars were profiled by this method.
Mass spectra obtained for certain peaks in the maize cultivars studied resulted,
in some occasions, useful for cultivar characterization showing the potential of RP-HPLC-ESI-MS analysis for the profiling of maize cultivars. The comparison of UV and MS profiles and mass spectra corresponding to the protein fractions isolated from different transgenic and non-transgenic maize cultivars enabled their assignment in the whole seed profile.
[1] W.R. Adams, S. Huang, A. L. Kriz, M. H. Luethy, J. Agric. Food Chem., 52
(2004) 1842-1849.

COMUNICACIONES EN CARTEL C1-56
97
APLICACIÓN DE BAJAS TEMPERATURAS COMO TÉCNICA DE VINIFICACIÓN ECOLÓGICA EN LA ELABORACIÓN DE NUEVOS VINOS TINTOS ANDALUCES:
ESTUDIO DEL PERFIL FENÓLICO.
V. Gallo1, F.J. Forero1, F.J. Heredia2, M.L. González-Miret2 y D. Hernanz1
1Área de Química Analítica, Facultad de Ciencias Experimentales, Univ. de Huelva
E-mail:[email protected] 2 Laboratorio de Color y Calidad de Alimentos, Área de Nutrición y Bromatología,
Facultad de Farmacia, Univ. de Sevilla
Dentro de las técnicas enológicas encaminadas a la mejora de la calidad del color de vinos tintos se encuentra la maceración prefermentativa de los mostos con los hollejos a temperatura controlada, para la extracción de compuestos antociánicos y fenólicos en general. Además, el mantenimiento de bajas temperaturas a lo largo de la fermentación, estabilización y conservación del vino produce una importante disminución de la necesidad de adicionar conservantes tales como el anhídrido sulfuroso, producto hasta ahora imprescindible en una vinificación industrial estándar.
En zonas cálidas, como es el suroeste de España, las condiciones climatológicas
hacen que las cualidades de la uva, y por tanto del vino elaborado, tengan características diferentes a las de otras zonas vitivinícolas. En este trabajo se investiga la extracción de materia colorante en la criomaceración prefermentativa de vinos ecológicos elaborados con la variedad de uva Tempranillo. Se ha realizado el estudio comparativo de 16 ensayos de vinificación: 8 ensayos de maceración prefermentativa a 10-15 °C de temperatura durante 7 días, y 8 ensayos de vinificación tradicional. Se definen las diferentes fases de la vinificación y se determina en ellas el perfil polifenólico. Como resultado se podrá evaluar la posibilidad de la elaboración ecológica de vinos tintos en climas cálidos, ya que uno de los principales problemas que presentan estos vinos es la “caída de color”, sobre todo cuando son sometidos a crianza.
Las diferencias entre mostos y vinos obtenidos con diferentes procesos de
elaboración fue establecida mediante análisis estadísticos multivariantes tales como análisis discriminate y análisis en componentes principales. Se encontraron que los compuestos flavonoides tales la catequina, epicatequina, rutina y derivados de la quercetina junto a los ácidos hidroxicinámicos, como el ácido cafeico, ácido p-cumárico y sus derivados, fueron los que mejor discriminaban entre vinos ecológicos con y sin criomaceración.

C1-57 COMUNICACIONES EN CARTEL
98
CARACTERIZACIÓN DE VINOS ARGENTINOS EN FUNCIÓN DE SUS CUALIDADES NUTRICIONALES COMO PROTECTORES DEL ESTRÉS
OXIDATIVO
V. Gallo1, R. Di Paola2, D. Wunderlin2, F.J. Heredia3 y D. Hernanz1
1 Área de Química Analítica, Faculta de Ciencias Experimentales, Univ. Huelva E-mail: [email protected]
2 Departamento de Bioquímica Clínica-CIBICI/CONICET, Facultad de Ciencias Químicas, Univ. Nacional de Córdoba (Argentina)
3 Laboratorio de Color y Calidad de Alimentos, Área de Nutrición y Bromatología, Facultad de Farmacia, Univ. de Sevilla
Estudios epidemiológicos, químicos y biológicos realizados en los últimos años han aportado considerables evidencias sobre el rol beneficioso de los nutrientes con capacidad antioxidante (CA) en la salud y en la protección en ciertas patologías frecuentes y graves como las oncológicas o las cardiovasculares. En vinos, los principales compuestos fenólicos son el ácido cafeico, epicatequina, catequina, ácido gálico, rutina, miricetina, quercetina y resveratrol, entre otros. Estos fenoles, además de contribuir a las características organolépticas del vino, poseen en mayor o en menor grado propiedades antioxidantes. Es importante mencionar que la CA y la composición fenólica de los vinos, no sólo van a estar influenciadas por la variedad de uva, sino también por el proceso de elaboración, tiempo de almacenamiento y el envejecimiento del mismo.
El objetivo planteado es evaluar la CA de los vinos mediante la determinación
de su capacidad de inhibición de estrés oxidativo por acción de compuestos antioxidantes naturales. Así, se ha determinado y comparado la CA y fenoles totales en muestras de vinos tintos monovarietales de Merlot, Cabernet Sauvignon, Frambua, Malbec y Sangiovese. Las muestras fueron obtenidas de bodegas del Municipio de Colonia Caroya (Córdoba, Rep. Argentina).
Los fenoles totales se determinaron espectrofotométricamente por el método
de Folin-Ciocalteau y los resultados se expresaron en mg/L de ácido gálico. La capacidad antioxidante de los vinos fue evaluada por dos métodos in vitro, el método TEAC (Capacidad Antioxidante Equivalente al Trolox) y el método FRAP (Poder Antioxidante de Reducción del Ión Férrico). El primero se basa en la capacidad de un compuesto para captar el radical coloreado ABTS•+, en un medio acuoso, convirtiendo el mismo en un producto decolorado [1]. El método FRAP se basa en la capacidad que presentan los compuestos antioxidantes presentes en la muestra para donar electrones y reducir el ión férrico a ferroso en medio acuoso ácido [2]. En ambos métodos los resultados se expresaron como miliequivalentes de Trolox, utilizado como estándar.
Se realizó, como estudio preliminar, un análisis estadístico de correlaciones,
encontrándose una correlación significativa (p<0,05) entre el contenido en fenoles totales y la actividad antioxidante de los vinos medida por el método FRAP (r=0.75). [1] R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang, C. Rice-Evans. Free
Radical Biology and Medicine. 26 (1999) 1231-1237. [2] F.F. Benzie, J.J. Strain, Analytical Biochemistry. 239 (1996) 70-76.
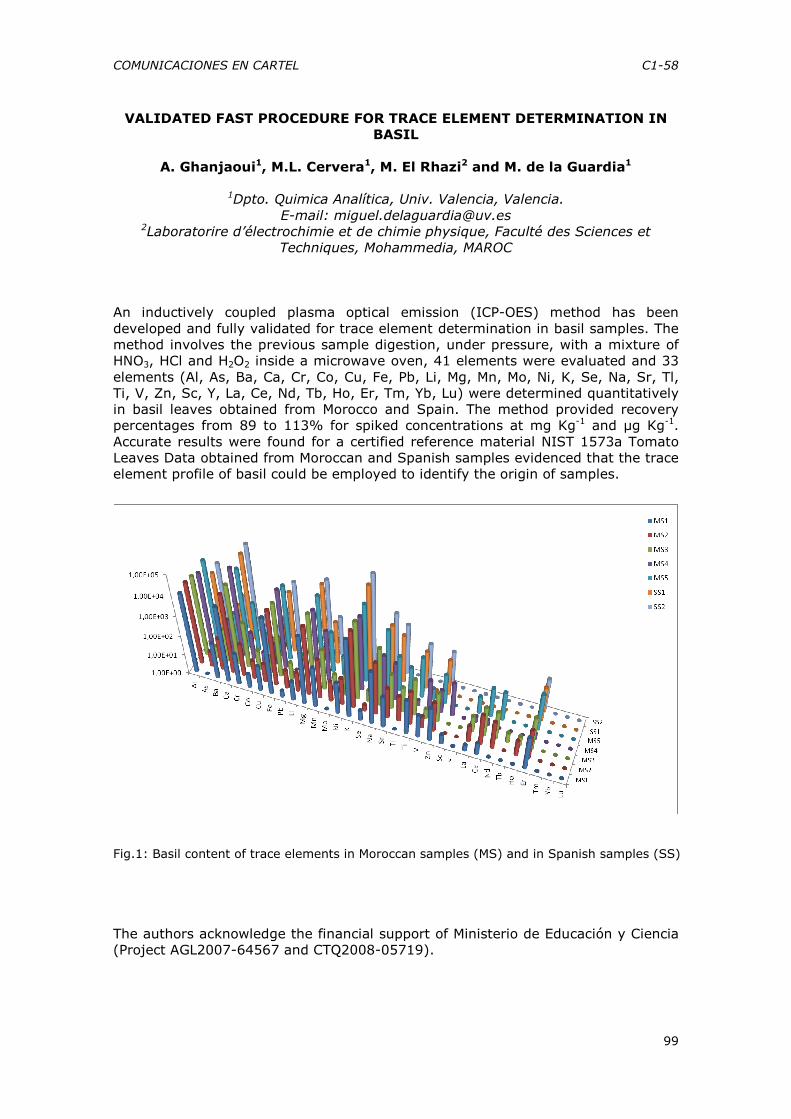
COMUNICACIONES EN CARTEL C1-58
99
VALIDATED FAST PROCEDURE FOR TRACE ELEMENT DETERMINATION IN
BASIL
A. Ghanjaoui1, M.L. Cervera1, M. El Rhazi2 and M. de la Guardia1
1Dpto. Quimica Analítica, Univ. Valencia, Valencia. E-mail: [email protected]
2Laboratorire d’électrochimie et de chimie physique, Faculté des Sciences et Techniques, Mohammedia, MAROC
An inductively coupled plasma optical emission (ICP-OES) method has been developed and fully validated for trace element determination in basil samples. The method involves the previous sample digestion, under pressure, with a mixture of HNO3, HCl and H2O2 inside a microwave oven, 41 elements were evaluated and 33 elements (Al, As, Ba, Ca, Cr, Co, Cu, Fe, Pb, Li, Mg, Mn, Mo, Ni, K, Se, Na, Sr, Tl, Ti, V, Zn, Sc, Y, La, Ce, Nd, Tb, Ho, Er, Tm, Yb, Lu) were determined quantitatively in basil leaves obtained from Morocco and Spain. The method provided recovery percentages from 89 to 113% for spiked concentrations at mg Kg-1 and µg Kg-1. Accurate results were found for a certified reference material NIST 1573a Tomato Leaves Data obtained from Moroccan and Spanish samples evidenced that the trace element profile of basil could be employed to identify the origin of samples.
Fig.1: Basil content of trace elements in Moroccan samples (MS) and in Spanish samples (SS) The authors acknowledge the financial support of Ministerio de Educación y Ciencia (Project AGL2007-64567 and CTQ2008-05719).

C1-59 COMUNICACIONES EN CARTEL
100
ESPECIACION DE ANTIMONIO Y TELURO EN CEREALES POR
ESPECTROMETRIA DE FLUORESCENCIA ATOMICA CON GENERACION DE HIDRUROS
M.N. Matos-Reyes, M.D. González, M.L. Cervera, M. de la Guardia
Dpto. Quimica Analítica, Univ. Valencia, Valencia. E-mail: [email protected]
Los metales se consideran contaminantes potenciales en los alimentos. Suelos
y aguas contaminadas, herbicidas, fungicidas e insecticidas son fuentes importantes de metales. Por lo tanto, es necesario el control del contenido de estos elementos en los alimentos con el fin de garantizar una buena salud y calidad de vida. El arroz es un cereal que es la base de la dieta de muchos países alrededor del mundo pues se trata de un alimento económico, energético y digerible. En la literatura existen algunos datos del contenido de As(III), As(V) y algunas especies metiladas en arroz, sémola. Sin embargo la información acerca de las especies de antimonio y teluro es escasa.
Se ha desarrollado un método no cromatográfico rápido, sensible y fácil para la
determinación de Sb(III), Sb(V), Te(IV), y Te(VI) en arroz. El método consiste en la extracción asistida por ultrasonidos y posterior determinación por espectrometría de fluorescencia atómica con generación de hidruros empleando para especiar diferentes condiciones de generación de los hidruros. Se realizaron estudios preliminares para obtener la mejor eficiencia de extracción a partir de la muestra sólida utilizando H3PO4
1 mol L-1, HNO3 3 mol L-1, agua regia, H2SO4 1 mol L-1, y HCl 6 mol L-1. La extracción con H3PO4, HNO3, y HCl fueron inviables, con agua regia se presentó una clara interconversión de las especies, oxidación al estado superior, durante el proceso; siendo el mejor extractante el H2SO4 1 mol L-1, con una eficiencia de extracción mayor al 90% con respecto al contenido total de antimonio y teluro determinados por otro método. De los resultados obtenidos se puede observar que las especies mayoritarias son el Sb(V) y Te(VI). Los estudios de recuperación adicionando las cuatro especies proporcionaron porcentajes de recuperación en todos los casos superiores al 90%, poniendo de manifiesto la no-interconversión de especies.
El método desarrollado se ha aplicado a diferentes muestras adquiridas en el
comercio local. The authors acknowledge the financial support of Ministerio de Educación y Ciencia (Project AGL2007-64567 and CTQ2008-05719).
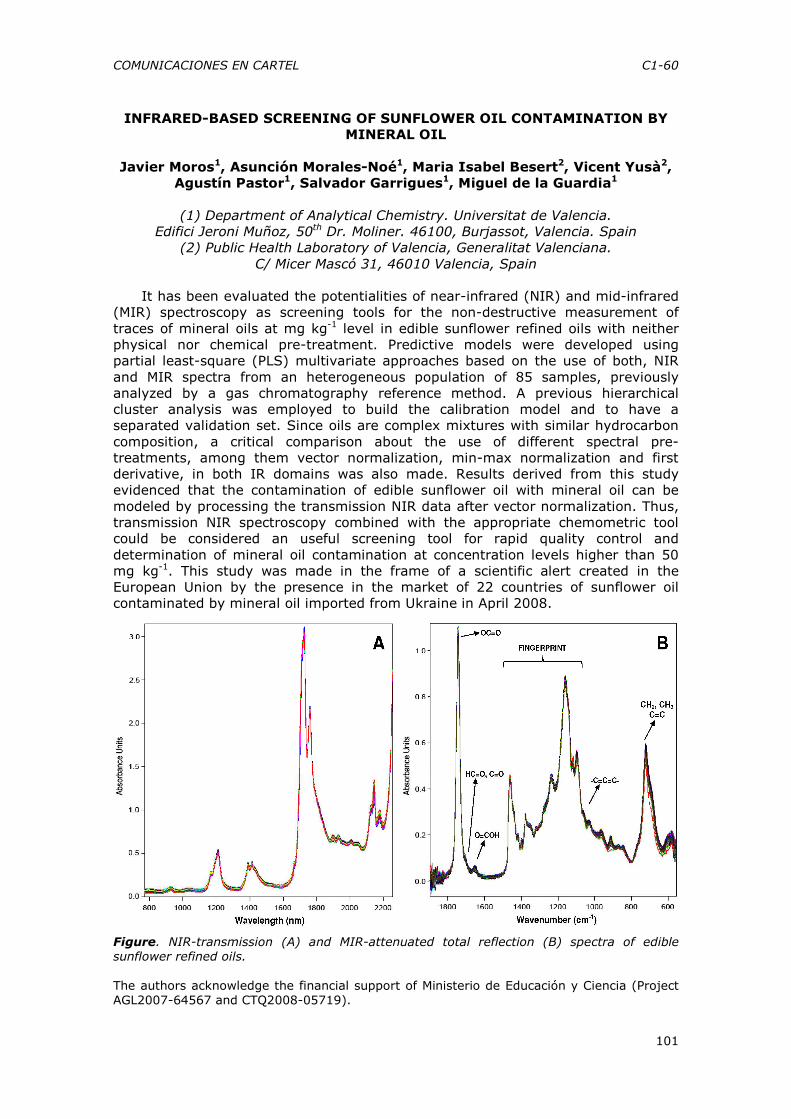
COMUNICACIONES EN CARTEL C1-60
101
INFRARED-BASED SCREENING OF SUNFLOWER OIL CONTAMINATION BY
MINERAL OIL
Javier Moros1, Asunción Morales-Noé1, Maria Isabel Besert2, Vicent Yusà2, Agustín Pastor1, Salvador Garrigues1, Miguel de la Guardia1
(1) Department of Analytical Chemistry. Universitat de Valencia.
Edifici Jeroni Muñoz, 50th Dr. Moliner. 46100, Burjassot, Valencia. Spain (2) Public Health Laboratory of Valencia, Generalitat Valenciana.
C/ Micer Mascó 31, 46010 Valencia, Spain It has been evaluated the potentialities of near-infrared (NIR) and mid-infrared
(MIR) spectroscopy as screening tools for the non-destructive measurement of traces of mineral oils at mg kg-1 level in edible sunflower refined oils with neither physical nor chemical pre-treatment. Predictive models were developed using partial least-square (PLS) multivariate approaches based on the use of both, NIR and MIR spectra from an heterogeneous population of 85 samples, previously analyzed by a gas chromatography reference method. A previous hierarchical cluster analysis was employed to build the calibration model and to have a separated validation set. Since oils are complex mixtures with similar hydrocarbon composition, a critical comparison about the use of different spectral pre-treatments, among them vector normalization, min-max normalization and first derivative, in both IR domains was also made. Results derived from this study evidenced that the contamination of edible sunflower oil with mineral oil can be modeled by processing the transmission NIR data after vector normalization. Thus, transmission NIR spectroscopy combined with the appropriate chemometric tool could be considered an useful screening tool for rapid quality control and determination of mineral oil contamination at concentration levels higher than 50 mg kg-1. This study was made in the frame of a scientific alert created in the European Union by the presence in the market of 22 countries of sunflower oil contaminated by mineral oil imported from Ukraine in April 2008. Figure. NIR-transmission (A) and MIR-attenuated total reflection (B) spectra of edible sunflower refined oils. The authors acknowledge the financial support of Ministerio de Educación y Ciencia (Project AGL2007-64567 and CTQ2008-05719).

C1-61 COMUNICACIONES EN CARTEL
102
DETERMINACIÓN DE FOSFATIDILCOLINA BASADA EN LA FLUORESCENCIA
DE LA ENZIMA COLINA OXIDASA COMBINADA CON HIDRÓLISIS ENZIMÁTICA
A. Domínguez, S. de Marcos, J. Galbán, I. Sanz-Vicente
Grupo de Biosensores Analíticos, Dpto. Química Analítica, Facultad de Ciencias.
Instituto de Nanociencia de Aragón. Universidad de Zaragoza. E-mail: [email protected]
El uso de las propiedades ópticas de las enzimas como base de métodos
analíticos para la determinación de sus correspondientes sustratos es una alternativa robusta para el desarrollo de métodos analíticos en general y biosensores ópticos reversibles (biosensores autoindicadores) en particular. En este contexto, la colina se puede determinar mediante su conocida reacción con la colina oxidasa (ChOx, reacción (2)) usando como señal el cambio de fluorescencia intrínseca (o el de determinados fluoróforos ligados a la ChOx) observado durante la reacción [1].
Tomando esta metodología como base, estamos desarrollando métodos
analíticos y biosensores para la determinación de algunos fosfolípidos, en particular para los dos fosfolípidos mayoritarios en el plasma humano: la esfingomielina (SM, 20% del total) y fosfatidilcolina (PC, en torno al 70% del total). Desde un punto de vista clínico es importante conocer la concentración de SM y la relación SM/PC en plasma; ambos parámetros, junto con el colesterol, dan una visión muy completa del riesgo de sufrir problemas cardiovasculares. Dado que ambos fosfolípidos tienen un resto colina, la metodología que se está siguiendo se basa en la acción hidrolítica de una fosfolipasa, seguida de la reacción enzimática con la ChOx. En el caso particular de la PC la secuencia de reacciones que se está ensayando es la combinación de la Fosfolipasa D (PLD), que libera directamente colina, con la ChOx:
PC + H2O == (PLD) ==> Acido Fosfatídico + Colina (1) Colina + O2 == (ChOx) ==> Aldehido betaina + H2O2 (2)
Hasta la fecha se ha trabajado usando 1,2-dioctanoil-sn-glicero-3-fosfocolina (PC-C8), un emulador de la PC que es soluble en agua. Debido a que cada enzima tiene distinto rango de pH óptimo, el trabajo ha seguido dos variantes:
1) Realizar las dos reacciones por separado, a pH 5 para la PLD y a pH 9 para
la ChOx. En este caso, el rendimiento en la formación de colina depende del tiempo de incubación de la concentración de lipasa. Por el momento se han llegado a detectar concentraciones de PC-C8 de 1,5 x 10-3 M pero sería posible bajar aún más esta concentración puesto que el límite inferior del rango lineal de la ChOx llega hasta 5 x 10-4 mM.
2) Llevar a cabo las dos reacciones simultáneamente. Tras diversos estudios se ha encontrado un pH óptimo en 7, en el que ambas enzimas muestran un comportamiento aceptable. Por el momento y en las condiciones de trabajo usadas (140 U/ml de PLD como máximo), la hidrólisis de PC-C8 está en torno al 10% en 25 minutos. A partir de los resultados obtenidos a este pH se ha determinado la constante de velocidad del proceso de hidrólisis de la PC-C8 (en torno a 0,8 M-1s-1). Agradecimientos. Este trabajo se está desarrollando bajo la financiación otorgada por el Ministerio de Ciencia e Innovación, proyecto CTQ 2008-06751-C02-01/BQU. [1] J. Galbán, O. Sánchez-Monreal, Y. Andreu, S. de Marcos, J. R. Castillo. Anal.
Biochem. 334 (2004) 207-215.

COMUNICACIONES EN CARTEL C1-62
103
INFLUENCE OF PHYSICAL PARAMETERS ON: NIR SPECTRA AND
CONSTRUCTION OF CALIBRATION MODELS
M. Blanco y A. Peguero
Departament de Química. Edifici Cn. Universitat Autònoma de Barcelona 08193-Bellaterra (Barcelona). Spain. [email protected]
The manufacturing process of pharmaceutical tablets entails several steps (blending, granulation, compression and coating), where the physical properties and chemical composition changes during the process. NIR spectroscopy is a suitable technique for process control, as physical and chemical information is contained in the NIR spectrum. The multivariate calibration models for the determination of active principle ingredient (API), needs the use of similar laboratory samples than those from the process, matching the physical characteristics and chemical composition. Also, the reference values required during the method development are obtained by an external method, such as HPLC.
In this presentation the predictive ability of calibration models calculated using
only laboratory samples has been evaluated, during the different steps of a real manufacturing process. These laboratory samples were prepared by mixing appropriate amounts of API and excipients, previously weighted with an analytical balance. In this case, no reference method is needed. Different variables that affect both manufacturing process and NIR spectra have been assessed, such as particle size, shape, compaction pressure, coating thickness and chemical composition. The dissimilarity grade between laboratory and production samples has been evaluated to establish suitable laboratory sample sets to construct models. The authors have used not supervised pattern recognition methods, such as hierarchical cluster analysis and principal component analysis. The spectral differences between laboratory and product samples have been corrected or reduced using chemometrical tools. This strategy allowed the obtaining of suitable partial least squares calibration models to quantify API content.
In this work it is demonstrated the obtained calibration models, calculated
using only laboratory samples, allows an accurate and rugged control of API content during the complete manufacturing process of pharmaceutical tablets. This strategy demonstrates the ability of the NIR technique for the intended purpose, without the need of a reference method.

C1-63 COMUNICACIONES EN CARTEL
104
DETERMINACIÓN DE GLUTATION OXIDADO Y REDUCIDO MEDIANTE CROMATOGRAFÍA LÍQUIDA PREVIA FORMACIÓN DE ADUCTO
FLUORESCENTE.
J.J. Berzas, R.C. Rodríguez, F.J. Guzmán, C. Rodríguez
Dpto. Química Analítica y Tecnología de Alimentos. Univ. Castilla-La Mancha. Toledo. E-mail: [email protected]
El glutatión (GSH) es un tripéptido antioxidante que está implicado en varias
funciones biológicas entre las cuales se encuentra su papel detoxificador frente a xenobióticos y a especies reactivas de oxígeno, con un electrón desapareado, que se generan en el metabolismo de éstos. El mecanismo protector del GSH tiene como resultado la aparición de su forma oxidada, el glutatión disulfuro (GSSG). Así pues, una forma de evaluar el estrés oxidativo es determinando la relación entre GSH/GSSG y por lo tanto es necesario contar con las metodologías analíticas adecuadas para la determinación de ambas especies.
En bibliografía se han encontrado fundamentalmente determinaciones por cromatografía líquida y detección espectroscópica y electroquímica, pero también se ha utilizado como técnica separativa la electroforesis capilar y como detección la espectrometría de masas. En el caso de la detección espectroscópica es necesario derivatizar los analitos porque no presentan absorción ni fluorescencia nativas. En el caso de utilizar el derivado fluorescente se mejora significativamente la selectividad y la sensibilidad de la determinación.
El objetivo de este trabajo es desarrollar y validar un método para la determinación de GSH y GSSG utilizando cromatografía líquida y detección espectrofluorimétrica previa derivatización, con el fin de poder aplicarlo a tejidos de organismos sometidos a estrés oxidativo por mercurio.
El agente derivatizante fue el orto-ftalaldehído, que forma un aducto fluorescente con GSH pero no con GSSG. Para la determinación de GSSG es necesario reducirlo a GSH, en este caso con ditioeritritol (DTT) y posteriormente proceder a la derivatización y medida. Así pues, son necesarias dos determinaciones: en la primera se determina el GSH y en la segunda se determina la suma de GSH y GSSG. Por diferencia se calcularía la concentración de GSSG.
El desarrollo, optimización y validación del método se realizó con un cromatógrafo de líquidos Agilent 1200 equipado con una bomba binaria y un inyector manual con un bucle de 5 µL. Se utilizó una columna Eclipse XDB-C18 RRHT (5 cm × 4,6 mm × 1,8 µm). El resultado de la optimización fue utilizar como fase móvil tampón fosfato 20 mM (pH = 7,0):acetonitrilo (90:10, v:v) a un caudal de 2 mL min-1 y termostatizar la columna a 25 ºC. Las longitudes de onda de excitación y emisión fueron 340 nm y 420 nm, respectivamente.
En estas condiciones, se obtuvieron límites de detección y cuantificación instrumentales de 10,43 nmol L-1 y 34,78 nmol L-1, respectivamente, e intervalos de linealidad desde el límite de cuantificación hasta 65 µmol L-1. La precisión obtenida fue del 2,44 % en términos de desviación estándar relativa de las áreas de pico (n = 20). Para evaluar la exactitud se prepararon disoluciones de concentración conocida de ambas especies, pero de proporciones de GSH:GSSG que variaban desde 85:15 hasta 10:90, encontrándose concordancias próximas al 100 % con los patrones en todos los casos.
Se pretende aplicar esta metodología al análisis de lombrices expuestas a Hg para evaluar cómo varía la relación GSH/GSSG en presencia de este metal. [1] R.T. Williams, Detoxication Mechanisms, Chapman and Hall. London, 1959 [2] E. Camera, M. Picardo. J. Chromatogr. B, 781 (2002) 181-206

COMUNICACIONES EN CARTEL C1-64
105
DESIGN SPACE DETERMINATION IN PHARMACEUTICAL GEL FABRICATION
BY NEAR INFRARED SPECTROSCOPY
J. Rosas, M. Alcalà, M. Blanco
Departamento de Química, Grupo de Quimiometría Aplicada, Universitat Autònoma de Barcelona, 08193 Bellaterra E-mail: [email protected], [email protected]
The near infrared spectroscopy (NIR) technique is gaining a greater interest as
tool for the determination of active principles in pharmaceutical formulations. This importance is because it is a fast technique, low cost and non-destructive. Near infrared spectroscopy can be used for the determination of both chemical composition and physical characterization. The aim of this work consists in: a) the determination of chemical composition of a pharmaceutical gel by transmittance NIR spectroscopy, and b) the study of the evolution of viscosity and pH during the manufacturing process, with the scope of defining a design space.
For the preparation of gel formulations, a full factorial experimental design was
used, with three factors at two levels each (ethanol, water and Carbopol content), including five central level experiments. NIR spectra were acquired and viscosity and pH were determined from the fifteen experiments. Partial least squares calibration models were calculated for the determination of viscosity, pH, ethanol and water content. Ten spectra per experiment were selected for inclusion in the calibration set. The remaining five were used as validation set. The analysis of the results showed that the most influential factors of the viscosity are the water and ethanol content. The obtained calibration models showed good predictive ability, presenting relative errors below 5% in all cases.
It is demonstrated that the NIR technique is an alternative tool for the
monitoring of viscosity, pH and chemical composition during the manufacturing of a pharmaceutical gel. The obtained methods provided enough information to assure the quality of the final product with the specifications established.

C1-65 COMUNICACIONES EN CARTEL
106
APLICACIÓN DEL DISEÑO EXPERIMENTAL A LA OPTIMIZACIÓN DE LA
MICROEXTRACCIÓN DE FENOLES EN ACEITE DE OLIVA
M. Beltrán, M.L. Pizarro, R. Beltrán, A. Sayago.
Área de Química Analítica. Facultad de Ciencias Experimentales. Universidad de Huelva. Avd. Tres de Marzo S/N. 21071. Huelva
E-mail: [email protected]
Los beneficios que el consumo de aceite de oliva virgen tiene para la salud se explican con dos series de compuestos, los ácidos grasos y los compuestos fenólicos. Aunque los primeros contribuyen por igual en todos los aceites, no ocurre lo mismo con la composición en fenoles, cuya concentración varía sustancialmente fruto de la interacción de varios factores como las condiciones climatológicas, las características de la tierra de cultivo, la variedad de la aceituna, su estado de maduración, tipo de almacenamiento y la tecnología utilizada en su transporte y procesamiento [1, 2]. La importancia de los fenoles en la salud está llevando a las multinacionales a incluir la concentración de fenoles en la etiqueta como reclamo de las ventajas para la salud de sus aceites. Los compuestos fenólicos, por tanto, representan un importante indicador de calidad, ya que afectan no sólo a la estabilidad del aceite y a sus propiedades organolépticas sino a sus funciones nutricionales y terapéuticas, basadas principalmente en el carácter antioxidante de los mismos.
El objetivo principal del empleo de un diseño experimental consiste en extraer
la máxima cantidad de información imparcial en relación con los factores que afectan a un proceso realizando el menor número de observaciones posibles.
En el presente trabajo se ha puesto a punto un método de extracción líquido-
líquido para el análisis de compuestos fenólicos en el aceite de oliva. La optimización se ha llevado a cabo mediante la aplicación de dos diseños experimentales. Inicialmente se realizó un screening para determinar de las variables propuestas (peso de la muestra, volumen de extractante, tiempo de agitación, tipo de agitación y tiempo de centrifuga), aquellas que ejercían un mayor efecto sobre el porcentaje de extracción de cada uno de los fenoles estudiados. Posteriormente con las variables seleccionadas se realizó un segundo diseño para afianzar los resultados del primero y estudiar las posibles interacciones entre los distintos factores. Una vez optimizada la técnica, se estudió su repetibilidad y su reproducibilidad. La aplicabilidad del método se comprobó en aceites de oliva virgen extra. [1] T. Gutfinger JAOCS, 58 (1981) 966-968 [2] E. Gimeno, A.I. Castellote, R. Lamuela-Raventós, M.C. de la Torre-Boronat,
M.C. López-Sabater. Food Chemistry 78 (2002) 207-211

COMUNICACIONES EN CARTEL C1-66
107
DESARROLLO DE UNA METODOLOGÍA BASADA EN LA ELECTROFORESIS CAPILAR PARA LA DETERMINACIÓN DE POLIFENOLES EN EL ACEITE DE
OLIVA VIRGEN EXTRA.
M.L. Pizarro, M. Beltrán, J.C. Santos, A. Sayago, R. Beltrán.
Área de Química Analítica. Facultad de Ciencias Experimentales. Universidad de Huelva. Avd. Tres de Marzo S/N. 21071. Huelva
E-mail: [email protected]
La metodología basada en la electroforesis capilar como técnica separativa para la determinación de compuestos fenólicos en diversas matrices, ha adquirido una gran importancia en los últimos años, debido a las ventajas que presenta frente a otras técnicas de separación como la cromatografía líquida de alta resolución que suele ser la técnica de elección para la determinación de este tipo de compuestos. Entre estas ventajas podrían señalarse una disminución en el requerimiento de pequeñas cantidades de muestra y de reactivos, pero sobre todo del tiempo de análisis [1].
Los compuestos fenólicos, generalmente denominados como polifenoles,
constituyen un elemento básico de la dieta humana, debido a sus propiedades antioxidantes y al papel positivo que juegan en la prevención de determinadas enfermedades crónicas [2,3]. El aceite de oliva virgen extra obtenido por extracción en frío a partir de los frutos del olivo (Olea Europea) se considera como una fuente rica en este tipo de compuestos.
En el presente trabajo se ha llevado a cabo una optimización de los diferentes
parámetros que afectan a la técnica de separación basada en la electroforesis capilar aplicada a los compuestos de la fracción fenólica del aceite de oliva, tales como: tirosol, hidroxitirosol, ácido vainíllico, ácido m-cumárico, ácido p-cumárico, vainillina, luteolina, apigenina, oleuropeína y ácido caféico. Los parámetros estudiados fueron la composición de la disolución reguladora, así como su concentración y pH, el voltaje y la temperatura del capilar y el tiempo de inyección. Las medidas para la optimización de la separación de los compuestos fenólicos, se han realizado con un equipo de electroforesis capilar P/ACETM MDQ (Beckman Instruments, Palo Alto, CA, USA), equipado con un detector de diodo array (190-600 nm) cuya longitud de onda fue fijada a 200 nm. [1] A. Gomez Caravaca, A. Carrasco Pancorbo, B. Cañabate Diaz, A. Segura
Carretero, A. Fernández Gutiérrez, Electrophoresis, 26 (2005), 3538–3551 [2] D. Boskou, Trends Food Sci. Technol. 17 (2003) 505–512. [3] A. Bendini, L. Cerretani, A. Carrasco-Pancorbo, A.-M. Gomez-Caravaca, A.,
Segura-Carretero, A. Fernandez-Gutierrez, G. Lerker, Molecules 12 (2007)1679–1719

C1-67 COMUNICACIONES EN CARTEL
108
ESPECIACIÓN DE YODO EN LA FRACCIÓN BIODISPONIBLE DE ALGAS
COMESTIBLES
V. Romarís Hortas, A. Moreda Piñeiro, P. Bermejo Barrera
Dpto. Química Analítica Nutrición y Bromatología, Univ. de Santiago de Compostela, Santiago de Compostela. E-mail: [email protected]
El creciente uso de las algas como alimento para consumo humano hace
necesario no sólo un buen cocimiento de la composición química, sino también de la fracción que es asimilable por el organismo. Aunque hay gran cantidad de trabajos sobre biodisponibilidad de elementos traza, pocos tratan sobre el yodo y menos todavía sobre las especies presentes en la fracción biodisponible.
En este trabajo se presentan los resultados del análisis de diferentes tipos de
algas pertenecientes a los tres grandes grupos: pardas, rojas y verdes. Se ha determinado la concentración de yodo total por espectrometría de masas atómica con plasma acoplado por inducción (ICP-MS), tras una extracción alcalina. Posteriormente se ha estudiado la fracción biodisponible por medio de un método in vitro con membranas de diálisis, realizándose la determinación de yodo por ICP-MS. Finalmente se ha llevado a cabo un estudio de especiación en la fracción biodisponible por cromatografía de intercambio iónico acoplada a ICP-MS (IC-HPLC-ICP-MS).
Los resultados muestran que el comportamiento de cada alga es realmente
diferente, tanto en relación a la concentración de yodo total, como de porcentaje de biodisponibilidad, como de las especies presentes en la fracción biodisponible. El alga Kombu es, con diferencia, la que mayor cantidad de yodo contiene (alrededor de 6ooo µg�g-1, referido a masa seca) y la que mayor porcentaje de biodisponibilidad presenta (18 %). En cuanto a las especies que aparecen en la fracción biodisponible, el yoduro es la especie mayoritaria en todas las muestras analizadas. Algunas algas presentan picos al mismo tiempo de retención que la diyodotirosina (DIT), la monoyodotirosina (MIT) y el yodato, aunque su presencia no ha sido confirmada. Además, algunos extractos de dializado presentan picos no identificados, siendo el caso del alga Kombu el que mayor número de especies presenta en los cromatogramas.
El método de determinación de yodo total ha sido validado mediante el análisis
del material de referencia NIES-09, mientras que la exactitud del método de biodisponibilidad se ha evaluado mediante un balance de masas, obteniéndose en ambos casos resultados positivos. El límite de detección (LOD) correspondiente al estudio de la biodisponibilidad es de 1,5 µg�g-1 (referido a masa seca). Los LOD correspondientes a las especies presentes en la fracción biodisponible son 4,8 ng�g-1 para yodato, 1,1 ng�g-1 para MIT, 4,3 ng�g-1 para DIT y 26,9 ng�g-1 para yoduro (referido a masa seca).

COMUNICACIONES EN CARTEL C1-68
109
DIRECT FLUORESCENCE SCREENING OF URINE SAMPLES FOR NON-POLAR HETEROCYCLIC AMINES AND ALTERNATIVE CONFIRMATION METHOD BASED ON CAPILLARY LIQUID CHROMATOGRAPHY WITH EVAPORATIVE
LIGHT SCATTERING DETECTION
Fernando de Andrés, Mohammed Zougagh, Gregorio Castañeda, Ángel Ríos
Department of Analytical Chemistry and Food Technology, Faculty of Chemistry, University of Castilla-La Mancha, Av. Camilo José Cela Nº10, E-13004 Ciudad Real,
Spain. E-mail: [email protected]
Heterocyclic amines (HCAs) are formed in the surface layer of meat during cooking and are carcinogenic in animal models, but the effect in human beings remains to be elucidated. It has been observed that the rising cancer incidences in humans in the developed countries, especially diseases of colon, large intestine, prostate, liver and kidneys are related to the greater consumption of meat as well as preserved meat [1]. Thus, methods have been developed to determine these compounds in biological samples (mainly urine samples) to reflect recent HCAs exposure.
A rapid and simple method for direct screening and confirmation of six non
polar heterocyclic amines (harman, norharman, 2-amino-3-methyl-9H-pyrido[2,3-b]indole, 2-amino-9H-pyrido[2,3-b]indole, 3-amino-1-methyl-5H-pyrido[4,3-b]indole and 3-amino-1,4-dimethyl-5H-pyrido[4,3-b]indole) in human urine samples is proposed. This method comprises solid phase extraction and capillary liquid chromatography with fluorescence detection for the screening and light scattering detection for the confirmation of positive samples. Analytes were extracted and pre-concentrated from the sample using methanol/acetonitrile on Strata X reversed phase cartridges. For screening purposes, the urine extract was directly introduced in the fluorescence detector and the total non-polar heterocyclic amines content of the sample determined. For confirmatory analysis, another aliquot of the extracted sample was directed to the capillary chromatographic column and evaporating light scattering detector (ELSD), which provided the individuals non-polar heterocyclic amines profiles. The use of a mobile phase of acetonitrile and ammonium acetate 35mM at pH 4.5 through a gradient of composition is proposed. Conditions for fluorescence and light scattering detection were both optimized. Good agreements were obtained in the analysis of the spiked urine samples assayed following the screening and confirmatory methods.
Acknowledgments: The Spanish Ministry of Education and Science and JJCC
Castilla-La Mancha are gratefully acknowledged for funding this work with Grants CTQ2007-61830 and PCC08-0015-0722, respectively. [1] F.U Shah, T. Barri, J.A. Jönson, K. Skog, J. Chromatogr. B. 870 (2008) 203-208

C1-69 COMUNICACIONES EN CARTEL
110
DIRECT SCREENING OF AGROCHEMICALS IN FOODSTUFFS BY AMBIENT
DESORPTION/IONIZATION MASS SPECTROMETRY
Juan F. García-Reyes, Ayanna U. Jackson, Joshua S. Wiley, Jason D. Harper, Nicholas A. Charipar, Antonio Molina-Díaz and R. Graham Cooks
Analytical Chemistry Research Group (FQM 323), Department of Physical and
Analytical Chemistry, University of Jaén, Campus Las Lagunillas, Ed. B-3, E-23071 Jaén, Spain.
Aston Labs of Mass Spectrometry, Department of Chemistry, Purdue University, West Lafayette, IN, USA. E-mail:[email protected]
Detection of analytes in complex matrices is of increasing importance in analytical chemistry. The targeted applications demand rapid, sensitive and selective analytical techniques. Reducing or removing sample preparation steps prior to analysis is a key to moving the analysis of complex samples out of the lab and towards automated, in situ protocols. Some of the most promising techniques for direct, real time analysis are based on new mass spectrometry methods in which samples are ionized in the ambient environment. This innovation in mass spectrometry, so-called ambient ionization mass spectrometry [1,2], allows the acquisition of mass spectra on ordinary samples, in their native environment, without sample preparation or preseparation by creating ions outside the instrument. Of these methods, Desorption Electrospray Ionization Mass Spectrometry (DESI-MS) [3] (introduced in 2004) has proven to be a widely applicable ionization technique which allows rapid in situ analysis with minimal or without any sample pretreatment, while retaining high molecular specificity and broad applicability. In DESI, a charged, high-velocity spray of microdroplets is directed toward a surface of interest, and the ions generated from the compound are transferred through air to the atmospheric pressure interface of a mass spectrometer. Due to its speed and ease of use, DESI is becoming a useful tool in a wide variety of applications [1].
Recently, a new ambient plasma-based ionization technique has been
introduced, so called low temperature plasma (LTP) [4] that permits the direct analysis of trace compounds in solid surfaces and complex matrices, without needing solvents or reagents besides the carrier/discharge gas. The Low Temperature Plasma (LTP) ionization is an ambient ionization method permitting the direct mass analysis of samples in their native atmospheric environment with minimal sample preparation. LTP utilizes dielectric barrier discharges (DBD) to create a plasma, guided by a gas flow, to interact with the analyte. In this work, the potential of DESI-MS and LTP-MS for the rapid, in situ, direct qualitative and quantitative (ultra)trace analysis of agrochemicals in foodstuffs has been evaluated and compared in detail. [1] R.G Cooks, Z. Ouyang, Z. Takats, J.M. Wiseman. Science. 311 (2006) 1566-
1570. [2] A. Venter, M. Nefliu, R.G Cooks. Trends Anal. Chem. 27 (2008) 284-290. [3] Z. Takats, J.M. Wiseman, B. Gologan, R.G Cooks. Science. 306 (2004) 471-
473. [4] J.D. Harper, N.A. Charipar, C.C. Mulligan, X.R. Zhang, R.G Cooks, Z. Ouyang.
Anal. Chem. 80 (2008) 9097-9104.

COMUNICACIONES EN CARTEL C1-70
111
DESARROLLO Y CARACTERIZACIÓN DE UN APTASENSOR PARA
TOBRAMICINA MEDIANTE MEDIDAS DE SPR
E. González-Fernández, N. de-los-Santos-Álvarez, M.J. Lobo-Castañón, A.J. Miranda-Ordieres y P. Tuñón-Blanco
Dpto. Química Física y Analítica, Universidad de Oviedo 33006 Oviedo, España
E-mail:[email protected] La tobramicina es un antibiótico de amplio espectro perteneciente al grupo de
los aminoglicósidos, que muestran un rango terapéutico estrecho debido a los efectos secundarios de ototoxicidad y nefrotoxicidad [1] observados, por lo que se requiere un control muy estricto de su dosis.
Una característica de este grupo de compuestos es su carencia de propiedades
electroquímicas y espectroscópicas útiles, lo que hace muy necesario el desarrollo de métodos de análisis sensibles que no requieran derivatización o reactivos marcados.
Para resolver este problema, se propone el uso de un aptámero anti-
tobramicina como elemento de reconocimiento molecular acoplado a medidas mediante Espectroscopía de Resonancia de Plasmón Superficial (SPR), empleando un esquema general de diseño previamente desarrollado en nuestro grupo [2].
Se han descrito varios aptámeros capaces de reconocer la tobramicina [3],
pero todos ellos son oligonucleótidos de ARN, lo cual los hace especialmente vulnerables a la acción de las endonucleasas y limita su aplicabilidad en muestras biológicas. Por ello para el desarrollo de la fases sensora se ha modificado el aptámero de ARN en la posición 2´-OH de todas las ribosas excepto en una, puesto que dicho grupo hidroxilo parece estar directamente implicado en el enlace con la tobramicina [4].
En este punto es de suma importancia conocer si dicha modificación altera la
afinidad entre el aptámero y el aminoglicósido. Por ello se llevó a cabo la caracterización de la interacción aptámero-tobramicina mediante SPR empleando el aptámero natural sin modificar y el aptámero modificado. La afinidad de ambos aptámeros ha sido comparada en términos de las constantes cinéticas y de afinidad obtenidas.
[1] Megoulas, N. C.; Koupparis, M. A. Analytical and Bioanalytical Chemistry 2005, 382, 290-296.
[2] de-los-Santos-Alvarez, N.; Lobo-Castañon, M. J.; Miranda-Ordieres, A. J.; Tuñon-Blanco, P. Journal of the American Chemical Society 2007, 129, 3808-3809.
[3] Wang, Y.; Rando, R. R. Chemistry & Biology 1995, 2, 281-290. [4] Jiang, L. C.; Patel, D. J. Nature Structural Biology 1998, 5, 769-774.

C1-71 COMUNICACIONES EN CARTEL
112
NEAR INFRARED SPECTROSCOPY TO HOMOGENEITY MONITORING OF
COMPLEX POWDER BLENDS IN PHARMACEUTICAL PROCESS
M. Blanco, R. Cueva-Mestanza
Dpto. Quimica, Facultad de Ciencias, Univ. Autónoma de Barcelona E-08193 Bellaterra, Barcelona, Spain. E-mail: [email protected].
Las formas farmacéuticas prescritas más frecuentemente son las formulaciones
farmacéuticas sólidas. El proceso de mezclado constituye uno de las etapas más importantes durante el proceso de fabricación de formas farmacéuticas sólidas en la industria farmacéutica y es el determinante de la calidad del producto final. Asegurar que cada unidad individual del fármaco presente una correcta dosificación, solamente se puede conseguir mediante una perfecta distribución de todos los componentes del preparado. La verificación de la homogeneidad de la mezcla se realiza mediante análisis de uniformidad de contenido en unidades de dosis del producto final; un incorrecto proceso de mezcla producirá resultados analíticos con amplio rango de valores y un riesgo importante de no cumplir con las especificaciones que establecen las farmacopeas que conduce a la no comercialización del lote de producción. Disponer de un método que permita realizar el control del proceso de mezcla durante su realización tiene indudables ventajas no sólo por la reducción del tiempo del proceso sino también por su efecto económico. Este control de homogeneidad es especialmente importante en las formulaciones con una baja concentración de principio activo (API).
En este trabajo se ha estudiado un método basado en Espectroscopia de
Infrarrojo Cercano (NIRS) como una herramienta que permita determinar el tiempo necesario para alcanzar la homogeneidad en el producto final y realizar un seguimiento del proceso de mezclado sin necesidad de extraer muestra. Se ha estudiado una formulación que contiene 7 componentes en el cual el principio activo corresponde al 3.33% w/w del total de la mezcla. Los registros de los espectros fueron realizados on-line mediante una sonda de fibra óptica y los espectros sucesivos registrados a lo largo del tiempo se han comparado, usando diferentes algoritmos, para evaluar su evolución temporal. Las pequeñas diferencias que presentan los espectros en situaciones próximas a la homogeneidad exige la aplicación de técnicas quimiométricas sensibles y a la vez aplicables en continuo para asegurar la consecución de la mezcla homogénea. El espectro NIR de una muestra en polvo contiene información tanto de características físicas como químicas de la muestra, lo que dificulta la selección del tratamiento matemático que indique que la repetitividad de los espectros es indicativa de la homogeneidad. En este trabajo se investigan los posibles pre-tratamientos, junto con varias formas en el establecimiento de homogeneidad de mezcla, que incluye la aplicación de bloques móviles desviación estándar, estudio de similaridad (cluster) y análisis de componentes principales (PCA).

COMUNICACIONES EN CARTEL C1-72
113
BIOTRANSFORMACIÓN DE SELENIO EN LA FABRICACIÓN DE CERVEZA
E.G.P da Silva2, T. Pérez-Corona1, S.L.C. Ferreira2, C. Cámara1, Y. Madrid1
1Dpto. Quimica Analítica, Univ. Complutense de Madrid, España E-mail: [email protected]
2Dpto. Quimica Analitica, Univ. Federal da Bahia, Salvador-Ba, Brasil E-mail: [email protected]
El selenio es un elemento esencial al que se le asignan tanto propiedades
antioxidantes como propiedades de agente quimiopreventivo frente a algunos tipos de cáncer (cáncer de próstata). Sus efectos en la salud humana están directamente relacionados con su forma química y concentración. La principal fuente de selenio es la dieta, sin embargo existen regiones en el mundo en las que su dieta tiene una carencia importante de este elemento, llegando a ser insuficiente [1]. Como consecuencia de esto ha cobrado interés el desarrollo de alimentos enriquecidos en selenio. Uno de los aspectos más estudiados en este tema es la capacidad de ciertas levaduras utilizadas en procesos de fermentación para biotransformar el selenio. La cerveza es un producto, que además de ser muy popular, es consumida a niveles importantes en todo el mundo y conlleva un proceso de fermentación, lo que la convierte en una matriz ideal para el estudio de estos procesos de biotransformación de selenio. El objetivo de este trabajo es estudiar la acumulación y transformación de Se(IV) añadido en el proceso de fermentación implicado en la fabricación de cerveza, tanto de tipo “ale” como “lager”, como paso inicial para el desarrollo de cervezas enriquecidas en este elemento.
El procedimiento seguido ha supuesto la reproducción en el laboratorio del
proceso de elaboración de cervezas. Para ello se ha realizado la adición de selenio, en forma Se(IV) y en distintas concentraciones (250-1000 µg), al conjunto del mosto procedente de la malta pre-fermentada y la levadura, todo ello en distintas condiciones de fermentación: Saccharomyces cerevisiae para fermentación “ale” (18-25°C) y levadura Saccharomyces uvarum para “lager” (14-18°C). Transcurridos 12 días del proceso se separó la cerveza de la levadura, y en ambas fracciones se determinó tanto el contenido total de selenio como el de las especies. La determinación del contenido total de selenio se llevó a cabo mediante ICP-MS, midiendo 82Se previa digestión por horno microondas. Los resultados obtenidos indicaron que sólo una pequeña fracción de selenio adicionado se había incorporado a la cerveza (10%) quedando la mayor parte de selenio acumulada en la levadura (>70%). La determinación de las especies se llevó a cabo mediante el acoplamiento HPLC-ICP-MS utilizando dos mecanismos de separación: fase inversa (C18 Waters Symmetryshield) y de intercambio aniónico (Hamilton PRP X-100), con el fin de realizar una mejor confirmación de las especies. Se utilizaron diversos tratamientos de muestra: hidrólisis enzimática mediante sonda de ultrasonidos y baño, así como introducción directa (sólo en el caso de la cerveza). Tanto en la levadura como en la cerveza obtenida por ambos tipos de fermentación (“ale” y “lager”) la especie mayoritaria encontrada fue selenometionina. Sin embargo, esta selenometionina aparece ligada a proteínas en el caso de las levaduras y en el caso de las cervezas aparece libre. En conclusión, en el proceso de elaboración de la cerveza el selenio es transformado por parte de las levaduras a selenometionina y ésta es incorporada a la cerveza en forma libre. [1] Z. Pedrero, Y. Madrid, Anal. Chim. Acta, 634 (2009) 135-152.

C1-73 COMUNICACIONES EN CARTEL
114
EXTRACCIÓN CON DISOLVENTES PRESURIZADOS (PLE) APLICADO AL ANÁLISIS DE RESIDUOS DE FLUOROQUINOLONAS EN ALIMENTOS
INFANTILES
E. Rodríguez, F. Navarro, M.D. Marazuela y M.C. Moreno-Bondi
Dpto. Química Analítica. Facultad de CC. Químicas. Univ. Complutense de Madrid. Madrid. E-mail: [email protected]
Las fluoroquinolonas (FQs) pertenecen a un grupo de agentes antibacterianos
ampliamente utilizados en clínica y veterinaria. Inhiben la ADN-girasa, por lo que la síntesis del ADN bacteriano, y en consecuencia la multiplicación bacteriana, se bloquean, mostrando una fuerte actividad contra microorganismos Gram(-) y algunos Gram(+) [1]. Estas sustancias se emplean con fines profilácticos y terapéuticos para el tratamiento de animales de granja, lo que puede conducir a la presencia de residuos en productos como la leche, carne, huevos. Esto suscita gran preocupación en salud pública, debido a la aparición de importantes fenómenos de resistencias bacterianas en humanos. La ruta principal por la que pueden transmitirse las bacterias resistentes desde los animales hacia el hombre es mediante el contacto directo con los animales que están siendo tratados. Alternativamente, sería el consumo o manipulación de productos de origen animal, que contengan flora resistente a los antimicrobianos. Con el fin de asegurar un nivel adecuado de protección sanitaria y facilitar los intercambios comerciales de alimentos de origen animal, la Unión Europea (UE) ha establecido límites máximos de residuos (LMRs) para diferentes drogas veterinarias, entre las que se encuentran las FQs, en algunos alimentos de origen animal (principalmente, tejidos animales y leche). Sin embargo, no se ha establecido ningún valor de LMRs para FQs en alimentos infantiles, por lo que el principio de “tolerancia cero” [2] es el que se debe aplicar a este tipo de alimentos. De ahí, que sea necesario disponer de métodos analíticos sensibles y fiables que faciliten el control oficial de residuos de estas sustancias presentes en este tipo de alimentos.
En el presente trabajo se ha desarrollado un nuevo método de análisis basado
en el empleo de PLE y posterior determinación por LC con detección fluorescente para 6 FQs en diferentes alimentos infantiles (leches de fórmula y potitos infantiles). Las principales variables que afectan a la extracción PLE de estos analitos, tales como composición del disolvente de extracción, temperatura, presión y número de ciclos, se han optimizado aplicando un diseño experimental con objeto de obtener la máxima recuperación de las FQs. La validación de la metodología propuesta, se desarrolló con base en la Directiva Europea 657/2002/EC [3] y finalmente el método se ha aplicado con éxito al análisis de muestras de alimentos infantiles, procedentes del mercado español, detectándose la presencia de residuos de enrofloxacina en una muestra de potitos analizada.
Agradecimientos: Este trabajo ha sido financiado por el MEC (ref. CTQ2006-
15610-CO2) y la CAM (ref. S-0505/AMB/0374). E.R. agradece al programa Albán de la UE la concesión de una beca doctoral (No. E06D101058CO) y a la Universidad de Cartagena (Colombia).
[1] A.A.M. Stolker, U.A.Th. Brinkman, J. Chromatogr. A 1067 (2005) 15. [2] Th. Hereber, M. Lahrssen, H. Schafft, K. Abraham, H. Pzyrembel, K.J. Henning,
M. Schauzu, J. Braeuning, M. Goetz, L. Niemann, U. Gundert, A. Luch, B. Appel, U. Banasiak, G. Fleur Böl, A. Lampen, R. Wittkowski, A. Hensel, Toxicol. Letters 175 (2007) 118.
[3] Commission Decision 2002/657/EC of 12th August. Off. J. L 221 2002.

COMUNICACIONES EN CARTEL C1-74
115
MULTI-CLASS ANALYSIS OF ANTIBIOTIC RESIDUES IN HONEY BY ULTRA-HIGH-PRESSURE LIQUID CHROMATOGRAPHY-TANDEM
MASS SPECTROMETRY
J.L. Martínez Vidal, A. Garrido Frenich, M.M. Aguilera-Luiz, R. Romero-González
Dpto. Química Analítica, Univ. Almería, Almería. E-mail: [email protected]
Honey is a complex product that has always been considered as a natural and healthy food, free of impurities. However, in apiculture, antibiotics are mainly used for the treatment of bacterial brood diseases, such as American foulbrood (Bacillus larvae) and European foulbrood (Streptococcus pluton). In general, there is a zero-tolerance policy to drug residues in honey, so it is necessary to develop sensitive analytical methods with low limits of detection in order to detect traces of antibiotics in honey. Furthermore, some surveys show that positive samples can be contaminated by more than one class of antibiotics, so new sensitive multi-class methods are necessary for the simultaneous determination of antibiotics at trace levels in this matrix.
A method has been developed and validated for the simultaneous analysis of
different veterinary drug residues (macrolides, tetracyclines, quinolones and sulfonamides) in honey. Honey samples were dissolved with Na2EDTA and veterinary residues were extracted from supernatant by solid-phase extraction (SPE), using OASIS HLB cartridges. The separation and determination was carried out by ultra high pressure liquid chromatography coupled to tandem mass spectrometry (UHPLC-MS/MS), using an electrospay ionization source (ESI) in positive mode. Data acquisition under MS/MS was achieved by applying multiple reaction monitoring (MRM) of two fragment ion transitions per compound to provide a high degree of sensitivity and specificity. The method was validated and mean recoveries were evaluated at three concentration levels (10, 50 and 100 µg/kg), ranging from 70 to 120 % except for doxycycline, erythromycin and tylmicosin with recovery higher than 50 % at the three levels assayed. Relative standard deviations (RSDs) of the recoveries were less than 20% within the intraday precision, and less than 25% within the interday precision. The limits of detection (LODs) ranged from 0.1 µg/kg to 1.0 µg/kg, whereas limits of quantification (LOQs) were always lower than 4 µg/kg. The developed procedure was applied to sixteen honey samples, and erythromycin, sarafloxacin, and tylosin were found in a few samples.
ACKNOWLEDGMENTS The authors gratefully acknowledge Spanish Ministry of Science and Innovation
(AGL2007-63569) for financial support.

C1-166 COMUNICACIONES EN CARTEL
116
ANALYTICAL METHOD FOR SCREENING VOLATILE COMPOUNDS IN PLASTIC
MATERIALS FROM WASTE LANDFILL
J.S. Félix, C. Domeño, C. Nerín
Dpto. Química Analítica, Univ. Zaragoza, Zaragoza. E-mail: [email protected]
The consumption of plastics worldwide increases daily. Particularly Spain,
according to the Spanish Association of Manufacturers of Plastics (ANAIP), occupies the seventh position in global consumption and ninth position in production of plastics, which means that there are approximately consumption of 115 kilos/year [1]. However, in parallel with this growth, the problem of plastic waste and all solid waste generated has been increasing. At present, it promotes the recycling of all materials in order to recover the waste, minimizing their production simultaneously.
The development of recycled materials requires safety certification in
accordance with the intended use. Therefore, it is necessary to ensure that the new recycled packaging materials are safe. Thus, it is essential to develop efficient analytical techniques, simple, rapid and, if possible, automatic.
The aim of this work was to develop the analytical method for screening volatile
compounds from PE and PE/EVA materials from waste landfill. So, solid-phase microextraction (SPME) in headspace mode (HS) coupled to gas chromatography-mass spectrometry (GC-MS) was used. Among the many factors that can affect the SPME, kind of fiber, time and temperature of extraction and stirring were evaluated during the experimental design. The MODDE software was used to design the experiment and to perform the optimization study.
The divinylbenzene/carboxen/polydimethylsiloxane fiber (DVB/CAR/PDMS -
50/30 µm) provided the highest number of compounds, with the highest peak area. The best conditions of time, temperature and stirring to extract the studied compounds were 10 minutes, 100 °C and 500 rpm, respectively. The optimized procedure can be considered an efficient and fast technique for screening volatile compounds in plastic materials from waste.
[1] Asociación Española de Industriales de Plásticos. http://www.anaip.es/
This project has been financed by Profit-2008-2010.

COMUNICACIONES EN CARTEL C2-75
117
NOVEL STRATEGIES FOR MERCURY VAPOUR GENERATION IN ATOMIC SPECTROMETRY: COMBINED USE OF ULTRAVIOLET AND ULTRASOUND
IRRADIATION IN THE PRESENCE OR FORMIC ACID E. Staniszb, H. Matusiewiczb, S. Gila, F. Pena-Pereiraa, M. Costasa, A. López-
Roucoa, I. Lavillaa and C. Bendichoa
aDepartamento de Química Analítica y Alimentaria, Área de Química Analítica, Universidad de Vigo, Facultad de Química, As Lagoas - Marcosende s/n, 36310 Vigo
(Spain). Email: [email protected] bPolitechnika Poznanska, Department of Analytical Chemistry, 60-965 Poznan,
Poland
The combined use of ultraviolet and ultrasound radiation in a sequential mode has proved very efficient for the generation of Hg vapour in a miniaturized batch reactor coupled to an atomic absorption spectrometer. Fast reduction of ionic Hg to Hg(0) occurs under ultraviolet irradiation, whereas fast mass transfer rate from the sample solution to the gas phase is achieved by ultrasound irradiation. The method yields comparable sensitivity and detection limit in regard to previous UV reduction methods for total Hg determination and fast atomic absorption signals. The use of formic acid fulfils two functions in this method, i.e. as a precursor of species needed for Hg2+ and CH3Hg
+ reduction and for solubilization of biological tissues. The latter operation is dramatically speeded up by ultrasound thus yielding a remarkable simplification of the overall method for determination of total Hg in biotissues. UV reduction times of 10 s and 30 s were needed for Hg(II) and CH3Hg
+, respectively. A 5 s ultrasound irradiation time was enough to yield an atomic absorption signal integrable within 20 s. The method was applied to the determination of total Hg in four certified reference materials corresponding to marine biological tissues following fast ultrasound-assisted solubilization with formic acid. LODs of Hg in the biological tissues ranged from 0.025 to 0.06 µg/g. Within-batch precision was around 4 %, whilst between-batch precision was in the range 6-11%.
Acknowledgements
Financial support from the Spanish Education and Science Ministry (project CTQ2006-04111/BQU) is gratefully acknowledged. This work has been undertaken as part of the EU sponsored COST Programme (Action D32, working group D32/005/04).

C2-76 COMUNICACIONES EN CARTEL
118
DETERMINATION OF METHYLCYCLOPENTADIENYL-MANGANESE TRICARBONYL IN WATERS BY GAS CHROMATOGRAPHY-MASS
SPECTROMETRY AFTER HEADSPACE SINGLE-DROP MICROEXTRACTION
Elefteria Psillakisb, Nicolas Kalogerakisb, Francisco Pena-Pereiraa, Marta Costasa, Sandra Gila, Isela Lavillaa and Carlos Bendichoa
aDepartamento de Química Analítica y Alimentaria, Facultad de Ciencias de Vigo,
Universidad de Vigo As Lagoas-Marcosende, E-36200 Vigo, Spain, Email: [email protected]
bDepartment of Environmental Engineering, Technical University of Crete, Polytechneioupolis, GR-73100 Chania, Greece
Headspace single-drop microextraction coupled to gas chromatography–mass spectrometry has been applied as a simple, fast and virtually solventless analytical protocol for analysis of aqueous samples contaminated with methylcyclopen-tadienylmanganese tricarbonyl (MMT). Parameters influencing the microextraction process were controlled and optimised and the optimum conditions found were: 2.5 µL octane microdrop exposed for 20 min to the headspace of a 10 mL aqueous sample (15 ml vial) containing 20% (w/v) NaCl and stirred at 1250 rpm. The calculated calibration curves gave a high level of linearity for MMT with regression coefficients greater than 0.9995 after conducting a three-day study. The limit of detection was calculated to be 0.21 µg/L. The proposed method achieved an enrichment factor of the order of 2100 and a 53 % recovery after extracting the spiked aqueous solution for 20 minutes under the optimised experimental conditions. The repeatability and intra-day reproducibility of the proposed method, expressed as relative standard deviation were 8.4 and 6.4 % respectively. Analysis of spiked tap and wastewater samples revealed that matrix had little effect upon extraction.
Acknowledgements:
Financial support from the Spanish Education and Science Ministry (project CTQ2006-04111/BQU) is gratefully acknowledged. This work has been undertaken as part of the EU sponsored COST Programme (Action D32, working group D32/005/04).

COMUNICACIONES EN CARTEL C2-77
119
IONIC LIQUID-BASED SINGLE-DROP MICROEXTRACTION COMBINED WITH HIGH PERFORMANCE LIQUID CHROMATOGRAPHY-PHOTODIODE ARRAY
DETECTION FOR SPECIATION OF MERCURY Isela Lavillab, Francisco Pena-Pereirab, Carlos Bendichob, Lorena Vidala and
Antonio Canalsa
aDepartamento de Química Analítica, Nutrición y Bromatología, Universidad de Alicante, P.O. Box 99, E-03080 Alicante, Spain. Email: [email protected]
bDepartamento de Química Analítica y Alimentaria, Area de Química Analítica, Facultad de Química, Universidad de Vigo, Campus As Lagoas-Marcosende s/n,
36310 Vigo, Spain.
Matrix separation and preconcentration of mercury species (MeHg+, EtHg+, PhHg+ and Hg2+) following formation of the corresponding dithizonates and microextraction of these neutral chelates onto a microdrop of an ionic liquid was developed. The separation and determination were subsequently carried out by high performance liquid chromatography with a photodiode array detector. Variables affecting the formation and extraction of mercury dithizonates were optimized. The optimum conditions found were: microextraction time, 20 min; stirring rate, 900 rpm; pH, 11; ionic liquid type, 1-hexyl-3-methylimidazolium hexafluorophosphate ([C6MIM][PF6]); drop volume, 4 µL; and no sodium chloride addition. Limits of detection were between 1.0 and 22.8 µg/Lfor the four species of mercury, while the repeatability of the method, expressed as relative standard deviation, was between 3.7 and 11.6% (n = 8). The method was finally applied to the determination of mercury species in different water samples.
This study demonstrates the suitability of ionic liquids for mercury speciation by
combination of SDME with high-performance liquid chromatography. The proposed method allows the determination of inorganic and organomercury species in water samples, being easy to use, fast, economical and virtually extractant-free.
Acknowledgments
F. P.-P. thanks Xunta de Galicia for financial support as a researcher of the Maria Barbeito. F. P.-P also thanks Universidade de Vigo for a grant. The authors gratefully acknowledge the financial support of the Spanish Government (projects n. PET2006-706-00, CTQ2005-09079-C03-01/BQU and CTQ2006-04111/BQU), the Regional Governments of Comunidad Valenciana and Galicia (projects n. ACOMP07/053, ARVIV/2007/062 and PGIDIT05PXIB31401PR) and the EU-COST Program (Action D32, working group WG/D32/005/04: “Microwave and ultrasound activation in chemical analysis”).

C2-78 COMUNICACIONES EN CARTEL
120
TRAPPING OF STIBINE ONTO A PALLADIUM-CONTAINING AQUEOUS DROP FOR ANTIMONY SPECIATION BY ELECTROTHERMAL-ATOMIC ABSORPTION
SPECTROMETRY
Francisco Pena-Pereira, Inmaculada de La Calle, Noelia Cabaleiro, Marta Costas, Sandra Gil, Isela Lavilla and Carlos Bendicho
Departamento de Química Analítica y Alimentaria, Area de Química Analítica,
Facultad de Química, Universidad de Vigo, Campus As Lagoas-Marcosende s/n, ESP 36310Vigo, Spain. Email: [email protected]
The coupling of headspace-single drop microextraction with electrothermal atomic absorption spectrometry (ETAAS) has been applied to the determination of antimony (III) and total antimony following stibine generation and trapping of the hydride onto a Pd(II)-containing aqueous drop. Pd(II) acts as an efficient trapping agent of the stibine generated in situ and as matrix modifier in the graphite furnace. The system has been fully optimized studying the following variables: hydrochloric acid and sodium tetrahydroborate concentrations, sample volume, palladium (II) concentration in the acceptor phase and microextraction time were fully optimized. A 26-2IV factorial fractional design was initially used for screening the effect of the variables, followed by an univariate approach. The method proved a great tolerance to potential interferences caused by hydride-forming elements and transition metals. The detection limit of Sb(III) was 25 pg/mL. A preconcentration factor of 176 is achieved in 3 minutes. The repeatability, expressed as relative standard deviation, was 4.7 %. The method was successfully validated against two certified reference materials (NWRI-TM 27.2 and NIST 2711) and applied to the determination of Sb(III) and total Sb in waters.
Acknowledgements:
Financial support from the Spanish Education and Science Ministry (project CTQ2006-04111/BQU) is gratefully acknowledged. This work has been undertaken as part of the EU sponsored COST Programme (Action D32, working group D32/005/04).

COMUNICACIONES EN CARTEL C2-79
121
LARGE-SCALE SCREENING OF PESTICIDES IN OLIVE OIL BY RAPID
RESOLUTION LIQUID CHROMATOGRAPHY ELECTROSPRAY TIME-OF-FLIGHT MASS SPECTROMETRY
Bienvenida Gilbert-López1, Juan F. García-Reyes1,
Amadeo R. Fernández-Alba2 and Antonio Molina-Díaz1
1Analytical Chemistry Research Group, Department of Physical and Analytical Chemistry, University of Jaén, Campus Las Lagunillas, Edificio B-3, 23071 Jaén,
Spain. E-mail: [email protected] 2Pesticide Residue Research Group, Department of Hydrogeology and Analytical
Chemistry, University of Almería, 04120 La Cañada de San Urbano, Almería, Spain.
The most extensively applied agrochemicals in olive plantations of Mediterranean countries (Greece, Spain and Italy) are by far herbicides and insecticides. Consequently, both the European Union and the Codex Alimentarius Commission of the Food and Agriculture Organization of the United Nations (FAO) have established maximum pesticide residue limits in olives and olive oil. In this study, we have developed a large-scale multiresidue method for the analysis of 119 multiclass pesticides in olive oil samples using rapid resolution liquid chromatography time-of-flight mass spectrometry (RRLC-TOFMS). The proposed methodology comprises a sample treatment step based either on QuEChERS methodology (liquid liquid partitioning followed by dispersive solid-phase extraction clean-up) or on Matrix Solid-Phase Dispersion (MSPD) using aminopropyl as sorbent material and a final clean-up performed in the elution step using Florisil. Recoveries in the range 70-120 % were obtained in most cases using both sample treatment protocols. The identification and quantitation of the selected agrochemicals was accomplished with the accurate mass measurements of the protonated molecule ([M+H]+) and those of the main fragment ions observed. The linearity, precision, accuracy, matrix effects and limits of detection obtained (in the range 1-10 ng g-1), compared well with the values generally achievable with triple quadrupole instruments, thus illustrating the potential of RRLC-TOFMS for the rapid screening of agrochemicals in fatty vegetable samples.

C2-82 COMUNICACIONES EN CARTEL
122
DETERMINATION OF ALIPHATIC AMINES IN LIQUID MEDIA USING DERIVATIZATION AND HEADSPACE SOLID-PHASE MICROEXTRACTION
FOLLOWED BY GC-FID
N. Longás, A. Bordagaray, R. Garcia Arrona, E. Millán
Dpto. Quimica Aplicada, UPV/EHU, Donostia-San Sebastián. E-mail: [email protected]
Aliphatic smaller amines are used as raw materials or as intermediate in
manufacturing of several industrial chemicals. Also are common components of biological systems as degradation products of organic matter such us amino acids, proteins and other nitrogen-containing organic compounds. Besides hygienic problems due to the unpleasant smell, these compounds may be hazardous to health; i.e., irritants to skin, mucous membranes, throat and lungs. In addition, they can react with nitrosating agents, leading to formation of potentially carcinogenic N-nitrosamines [1].
Analysis of aliphatic amines in aqueous media is very difficult due to the high
polarity of these compounds. The determination is often achieved using a derivatization procedure followed by an extraction/concentration step, prior to chromatographic analysis. The most common reactions for gas chromatography (GC) analysis of amines are acylation, silylation and formation of Schiff bases and carbamates [1]. Among Schiff base type reactions pentafluorobenzaldehyde (PFBAY) has been used for short chain aliphatic amines [2]. Solid phase microextraction (SPME) is a convenient alternative for extraction, since SPME does not require solvents, and can be carried out directly from the liquid phase or from the headspace (HS) over the samples. The analytical procedure between SPME and derivatization can be arranged in different modes: derivatization in the sample matrix, derivatization on the SPME fiber after sampling, simultaneous sampling and on-fiber derivatization, and derivatization in the injection port [3]. In this work, after different tries with derivatizating chemicals and arranged extraction types, derivatization was done with PFBAY in liquid matrix and the derivatives were extracted by poly(acrylate)(PA)-coated fiber in HS mode in the same vial.
There are several variables affecting the derivatization and HS-SPME overall
procedure. Some of the experimental parameters were fixed according to practical experience (80 oC reaction temperature, 10 mL of sample at pH 9.3, 30 mL of headspace volume).The effect of derivatization time, sodium chloride addition, extraction time and extraction temperature was studied. A 24-1 fractional factorial design for obtaining the best experimental conditions was used. Those were the chosen conditions: derivatization time, 15 min; sodium chloride, 300 g L-1; extraction temperature, 80 ºC; and extraction time, 40 min. Working with those conditions and GC-FID, the method was applied to the determination of methylamine, dimethylamine, ethylamine, propylamine and butylamine. Linear range and precision were examined. Calibration curves showed good linearity, and RSD mostly was between 12-17%. The detection limits were between 0.07 and 0.23 µg mL-1. The method was used with a urine sample where the studied amines were detected and quantified. The developed method is useful for industrial waste water and biological fluids with medium-high levels of aliphatic amines. [1] H. Kataoka, J. Chromatogr. A 733 (1996) 19-34 [2] C. Deng, N. Li, L. Wang, X. Zhang, J. Chromatogr. A 1131 (2006) 45-50 [3] E.E. Stashenko, J.R. Martínez, TrAC 23 (2004) 553-561

COMUNICACIONES EN CARTEL C2-84
123
EXPLORATORY AND QUANTITATIVE ANALYSIS OF A MODEL CURING REACTION OF EPOXY RESINS FROM RENEWABLE RESOURCES BY
CHEMOMETRIC ANALYSIS OF NIR DATA
Vanessa del Río, M.Pilar Callao, M. Soledad Larrechi
Chemometrics, Qualimetrics and Nanosensors Group Analytical Chemistry and Organic Chemistry Department
Rovira i Virgili University E-mail: [email protected]
In recent years, the demand for green raw materials is increasing because of the escalating prices of petrochemicals and high rate of depletion of the natural mineral resources. In this way, natural oils have attracted renewed attention as alternatives to conventional petrochemicals for the manufacture of organic chemical and polymeric materials. Among such materials, epoxy resins from vegetable oils are extremely promising as environmentally friendly polymers for industrial applications because they share many of the characteristics of conventional petro-chemical based epoxy resins.
In order to optimise the end-use properties of these new green resins, it is
important to evaluate the extension of the reaction and to know the pathway in which the reaction undergoes. This works presents an exploratory study of a model polymerization reaction to obtain epoxy resins from vegetable oils, between an epoxidized derivative oleic oil (EMO) and aniline as curing reagent, and gives an interpretation of the effects of the experimental conditions in the kinetic pathway of the reaction. Due to the chemical structure of the initial derivative oil reagent, competitive reactions can be done (Scheme 1). Therefore, determining whether these reactions are taking place is of interest from the point of view of the physical and chemical characteristics of the polymers that are obtained and it is a challenging from the chemometric treatment point of view.
CH3O
O O
7 7
+ BF3·OEt2CH3O
O
7
OH
7
NH
+
(A) (B) (C)
(C) (B) (D)
(a)
(b)
CH3O
O
7
OH
7
NH
NH2
NH2
O
7
OH
7
NH
HN
Scheme 1. Reaction between EMO and aniline at 95 ºC: (a) epoxy resin formation, (b) amide formation. (A) EMO, (B) aniline, (C) secondary amine, (D) amide.

C2-85 COMUNICACIONES EN CARTEL
124
SIMULTANEOUS DETERMINATION OF NITROIMIDAZOLES IN WATER
SAMPLES USING HPLC-UV DETECTION WITH SOLID PHASE MICROEXTRACTION AND MICELLAR DESORPTION
Z. Sosa Ferrera, M.E. Torres Padrón, C. Mahugo Santana,
J.J. Santana Rodríguez
Departamento de Química, Universidad de Las Palmas de Gran Canaria. 35017 Las Palmas de Gran Canaria. E-mail: [email protected]
Nitroimidazoles are a class of veterinary drugs with antibiotic and anticoccidial
activities in poultry and swine. They have also been used as feed additives for growth promotion and improvement of feed efficiency. However, they have been banned from use in food producing animals in many countries [1] because of their potentially harmful effects on human health. One of the ways to administer these drugs to animals could be via drinking water or via oral. Thus, it is possible to find residues of these compounds in water samples from fish farms located in the sea.
Standard and official methods to extract and determine organic compounds in
liquid environmental samples involve usually the liquid-liquid extraction (LLE). Therefore, there has been a need for developing sensitive and reliable analytical methods to analyze the residues of these compounds in water samples.
In this work, we propose the application of the solid-phase microextraction
(SPME) and an external desorption using a micellar medium as desorbing agent (MD) to extract seven nitroimidazoles in water samples. The main property of these organized structures is their good capacity to solubilize different types of solutes. Moreover, the most commonly used surfactants are commercially available and they are compatible with the mobile phase used in HPLC analysis. SPME-MD have been applied to the determination of phenolic compounds [2] and pharmaceutical residues in environmental liquid samples [3,4].
The proposed method have been used to optimize the extraction and
preconcentration of different nitroimidazoles (etanidazole, ronidazole, metronidazole, dimetridazole and ornidazole) and some of their metabolites (metronidazole-OH and ipronidazole-OH) in liquid environmental samples and their following determination by high performance liquid chromatography with diode array detection.
[1] Establishment of Maximum Residue Levels of Veterinary Medical Products in
foodstuffs of animal origin, Council Regulation (EEC) Nº 2377/90, Off. J.Eur. Commum. L224, 1990.
[2] M.E. Torres Padrón, C. Mahugo Santana, Z. Sosa Ferrera, J.J. Santana Rodríguez J. Chromatogr. Sci. 46 (2008) 325.
[3] S. Montesdeoca Esponda, M.E. Torres Padrón, Z. Sosa Ferrera, J.J. Santana Rodríguez, Anal. Bioanal. Chem. (2009), DOI 10.1007/s00216-009-2629-8
[4] M.E. Torres Padrón, Z. Sosa Ferrera, J.J. Santana Rodríguez, Biomed. Chromatogr. (2009), DOI 10.1007/s00216-009-2629-8

COMUNICACIONES EN CARTEL C2-86
125
NUEVO MÉTODO DE IDENTIFICACIÓN DE MUESTRAS USANDO UN ÚNICO
PULSO DE ABLACIÓN LÁSER D. Marcos-Martínez, F.J. Ortega-Nogales, M. del Valle and J.O. Cáceres
Departamento de Química Analítica. Facultad de Ciencias Químicas
Universidad Complutense de Madrid Av. Complutense s/n 28040 Madrid, Spain [email protected]
Un único pulso de ablación láser (LIBS) ha sido utilizado para la identificación
instantánea y la clasificación automática de diferentes muestras sin ningún tratamiento previo. La identificación instantánea de las muestras es obtenida utilizando una librería espectral, la cual fue obtenida antes del análisis utilizando un único pulso láser de muestras representativas y almacenadas en un ordenador. Los resultados obtenidos, alcanzan valores prácticamente ideales de 100% de selectividad y 100 % especificidad. Las muestras utilizadas fueron divididas en dos grupos y seleccionadas con matrices y composición química muy similar tales como ciertos estándares y productos alimentarios. El objetivo de esta comunicación es presentar las ventajas de este método simple y directo, desarrollado en nuestro grupo de investigación. Los resultados más significativos así como su potencial aplicación para el análisis y su uso en la industria serán presentados y discutidos.

C2-87 COMUNICACIONES EN CARTEL
126
HIGHLY SELECTIVE FLUORIMETRIC CHEMOSENSOR FOR MERCURY (II)
BASED IN A RHODAMINE 6G DERIVATIVE
A. Muñoz de la Peña1, M.I. Rodríguez-Cáceres1, M.C. Hurtado1, D. Bohoyo1 and R. Babiano2
1Department of Analytical Chemistry, University of Extremadura, Badajoz, Spain 2Department of Organic and Inorganic Chemistry, University of Extremadura,
Badajoz, Spain E-mail: [email protected]
Sensors based on metal ion inducing changes in fluorescence are particularly atractive, as useful tools for detection of environmentally relevant metal ions. Due to the high toxicity of mercury, considerable attention has been devoted to the development of new fluorescent chemosensors for the detection of mercury with sufficient selectivity. Recently, Rhodamine derivatives have been utilized as fluorescent chemosensors for Hg2+ [1].
In this communication, the analytical properties of a synthesized Rhodamine 6G derivative are presented. The reagent shows a highly selective reaction with Hg2+, giving rise to strong fluorescence emission (λex = 525 nm, λem = 550 nm), at neutral pH.
The chemosensor is bearing a thiospirolactone moiety and is not presenting native fluorescence. The reaction with Hg2+ specifically produces the ring-opening process of spirolactone to give the ring open amide binding to the metal ion in a 2:1 reagent:Hg2+ stoichiometric ratio, by the coordination of two sulfur atoms to Hg2+, and an “off-on” type fluorescence.
The procedure will be implemented in a portatil instrument (Figure 1) composed of a 515 nm LED as excitation source, two optical fibers and a CCD camera as detector, connected to a portable computer for data acquisition and analysis, intended for “in situ” determination of mercury in water effluents.
Figure 1.- Portable fiber-optic spectrometer for fluorimetric mercury determination
Acknowledgements: Financiation from the Junta de Extremadura (Project GRU09092) and the Ministerio de Ciencia e Innovación of Spain are acknowledged (Project CTQ2008-06657-C02-01). [1] X. Chen, S.W. Nam, M.J. Jou, Y. Kim, S.J. Kim, S. Park and J. Yoon, Organic
Letters, 10 (2008) 5235-5238.
Prizmatic-LED515 AvaSpec
FC-UV600-2 FC-UV600-2

COMUNICACIONES EN CARTEL C2-88
127
SCREENING AND MODELING OF THE AZO DYES SIMULTANEOUS
PHOTODEGRADATION
C. Fernández, M.S. Larrechi, M.P. Callao
Department of Analytical and Organic Chemistry, Rovira I Virgili University, Marcel�lí Domingo s/n Campus Sescelades, E-43007 Tarragona, Spain.
E-mail: [email protected] The simultaneous photodegradation of three azo dyes used in textile industry
(Acid Red 97, Acid Orange 61 and Acid Brown 425) has been studied. The spectra UV-vis of the dye mixture was registered during the photodegradation process and a curve resolution method (MCR-ALS) was applied to the spectra obtained in order to determine the concentration of each dye in each moment of the degradation.
There are many efforts dedicated to find the efficient conditions to carry out the photodegradation processes because the ability on the removal of dyes will vary depending on the synergistic or antagonistic action among the different factors. For this reason in the last years experimental designs are used [1-2].
The aim of our study is apply multivariate techniques to design the conditions that enable the quantitative description of parameters affecting the degradation of the three studied dyes. In a first stage the effect of multiple variables was studied (kind of catalyst, oxidant concentration, pH, catalyst concentration and stirring). In a second stage we use the obtained information to reduce the number of variables and find a suitable approximating function to predict the behaviour of the degradation of Acid Brown, which is the most persistent dye, versus the selected factors (H2O2, Acid Red concentration and Acid Orange concentration). Fig.1 shows a scheme of the procedure followed in order to obtain the response surface for the photodegradation process.
Fig.1 Scheme of the experimental design procedure The response surfaces obtained could be useful for the tanning industry
because for a fixed concentration of dyes is possible to determine the optimum concentration of H2O2 for the degradation. With this methodology could be reduced the cost of time and money for the waste water treatment.
[1] B. K. Körbahti, M. A. Rauf, Chemical Engineering Journal 138 (2008) 166 [2] A. F. Caliman, C. Cojocaru, A. Antoniadis, I. Poulios 144 (2007) 265
Screening design
Central composite design
Full factorial design
Yes
No
Not validated
Influential factors
Optimal experimental domain?
First-order response surface
Second-order response surface

C2-89 COMUNICACIONES EN CARTEL
128
ION-MOBILITY SPECTROMETRY: A FAST TECHNIQUE
FOR LOW CONTENT DETERMINATION
Manel Alcalà, Marcel Blanco, David Zamora Grup de Quimiometria Aplicada, Departament de Química (U. Analítica), Facultat de
Ciències, Universitat Autònoma de Barcelona, 01893 Bellaterra, Spain. Tel.: +34 935811712 Fax: +34 935812379 E-mail address: [email protected]
Quick, efficient, and low-cost analytical methods for quality monitoring are constantly demanded. Cleaning verification is related to the screening of certain analytes on manufacturing equipment, which could contaminate future products and impurity determination in products for human use. Routinely used techniques for control such total organic carbon (TOC) and high-performance liquid chromatography (HPLC), suffer the shortcomings of low speed and limited sensitivity and accuracy, among others. IMS has been applied to diverse fields, such as detection of trace explosives, screening of chemical-warfare agents, environmental monitoring and screening of illicit drugs. Nowadays, its use in the chemical/pharmaceutical industry is increasing. [1,2]
IMS is based on the measurement of ion mobility upon sample ionization.
Direct analysis of formulations and wastes with a minimum sample preparation is possible, obtaining both high sensitivity and selectivity. Low cost and maintenance of worker health and safety can help save money and increase overall efficiency of production. Two IMS applications are presented: I) determination of low drug content for cleaning verification purposes and II) impurity determination in a complex sample from a manufacturing process.
In summary, both methods have been developed between the concentration
range of 500-4000ppb (R2>0.999), obtaining a LOD/LOQ for the cleaning verification and for the impurity testing of 110/380ppb and 60/200 ppb, respectively. In all cases, the maximum RSD% has been lower than 4%. The total time for each method development is less than two hours and each determination can be performed in one minute. It is demonstrated that IMS gives the opportunity to develop fast, accurate and rugged analytical methods for the intended purposes.
[1] G. Eiceman. Trends in Analytical Chemistry 21(4) (2002), 259-275. [2] R. O-Donnell, X. Sun, P. Harrington. Trends in Analytical Chemistry 27(1)
(2008), 44-53.

COMUNICACIONES EN CARTEL C2-90
129
OPTIMISATION OF PTV-LV INJECTION FOR DETERMINATION OF
PYRETHROID, ORGANOPHOSPHORUS AND CARBAMATE PESTICIDES IN EDIBLE SEAWEED SAMPLES USING GAS CHROMATOGRAPHY–MASS
SPECTROMETRY
R.A. Lorenzo, D. García-Rodríguez, A.M. Carro, R. Cela
Dpto. de Química Analítica, Nutrición y Bromatología, Facultad de Química, Instituto de Investigaciones y Análisis Alimentarios (IIAA). Universidad de Santiago de Compostela, Avda. de las Ciencias, s/n 15782-Santiago de Compostela, Spain.
E-mail: [email protected]
The developement of seaweed industry is based on the extensive use of this plant for various purposes such as food, feed or fodder [1] as well as seaweed are considered in EU feed legislation as feed materials not requiring any registration as feed additives [2].
The use of Pyrethroid (Pyrs), organophosphorus (OPPs) and carbamate (CAR) pesticides by the aquaculture industry as veterinary chemotherapeutants may lead in discharges of these pollutants directly to the marine environment [3]. Due to their relatively high hydrophobic, these compounds tend to associate to particulate matter such as seaweed producing a bioaccumulative effect through the food chain with the consequent risk for human health [4,5].
A rapid and simultaneous method has been developed for residue identification and quantification of three pyrethroid insecticides, permethrin, a-cypermethrin and deltamethrin; two organophosphorus pesticides, chlorpiryfos and chlorpiryifos-methyl; and a carbamate insecticide, carbaryl in edible seaweed. The procedure is based on a pressurized liquid extraction (PLE) with in-cell clean-up [6]. Gas chromatography coupled to tandem mass spectrometry and large-volume injections with a programmed-temperature vaporizer (PTV-LV) were used to increase sensitivity and selectivity of the pesticide determination.
The PTV technique is used to introduce large sample volumes into capillary GC columns at a temperature slightly below the boiling point of solvent. LVI using PTV can improve GC system detection limits by one to two orders of magnitude over conventional split/splitless inlet [7]. Due to the complicated PTV injection process, many factors can affect the performance and efficiency of this system, such as liner type, injection volume, initial inlet temperature and temperature ramp rate. The optimization of the PTV-LVI parameters was studied in detail. Besides, the inlet vent flow and splitless time are factors which will affect the system sensitivity. These last factors were investigated using a Doehlert response surface design.The optimized method gives recoveries between 92 and 108 % with R.S.D ranged from 2 to 10%. The limit of detection ranged between 0.5 and 12,5 pg/g when 0,2 g of seaweed were analysed.
[1] V.K. Dhargalkar, X.N. Verlecar Aquaculture 287 (2009) 229–242 [2] Noël Dierick, Anneke Ovyn, Stefaan De Smet J Sci Food Agric 89 (2009) 584–594 [3] D. García-Rodríguez, A. M. Carro, R. A. Lorenzo, F. Fernández, R. Cela, J. Sep. Sci. 31
(2008) 2882-2890 [4] D. V. Moreno, Z. S. Ferrera, J. J. S. Rodríguez, Microchemical J. 87 (2007) 139-146 [5] B. Gowland, L. Webster, R. Fryer, I. Davies, C. Moffat, R. Stagg, Environ. Poll. 120
(2002) 805-811 [6] D. García-Rodríguez, A. M. Carro, R. A. Lorenzo, R. Cela, 12as Jornadas de Análisis
Instrumental (JAI) PO-MA-3 Barcelona 21-23 octubre 2008 [7] Darinka Štajnbaher, Lucija Zupančič-Kralj J. Chromatography A, 1190 (2008) 316–326

C2-91 COMUNICACIONES EN CARTEL
130
STIR BAR SORPTIVE EXTRACTION AND IN-TUBE DERIVATIZATION
THERMAL-DESORPTION-GAS CHROMATOGRAPHY-MASS SPECTROMETRY FOR THE DETERMINATION OF PRIORITY AND EMERGING POLLUTANTS
O. Basauri, P. Navarro, A. Prieto, O. Zuloaga, A. Usobiaga, N. Etxebarria,
L.A. Fernández
Kimika Analitikoa Saila, Euskal Herriko Unibertsitatea, PK 644, 48080 Bilbao. E-mail: [email protected]
Pharmaceuticals, hormones and endocrine disruptor compounds (EDCs) have
become important emerging contaminants due to their presence in environmental waters, the threat they suppose to drinking water sources and the concern about their possible estrogenic and other kind of effects [1]. The inclusion of such compounds in legislations such as the Water Framework directive requires the development of robust analytical procedures that guarantee the reliability of the results that will emerge from the determination of such compounds in different environmental compartments.
Such contaminants are found at trace levels in water samples and require,
therefore, extraction and preconcentration steps. Recently and due to the importance that miniaturisation has reached in the analytical chemistry field, extraction techniques such as Stir Bar Sorptive Extraction (SBSE) have attained great importance in the preconcentration of analytes from water samples [2] since they minimise the use of organic solvents (green chemistry) and analysis time and improve the sensitivity of the global analytical method [3]. SBSE can be coupled to thermal desorption (TD) when coupled to gas chromatography (TD-GC) and, in this case, the use of pollutant organic solvents is null. However, compounds such as pharmaceuticals, hormones or EDCs are highly polar and are not best chromatographied by GC. In this case, a derivatization step is necessary and, in some cases, this derivatization can occur in-situ in the TD unit [4].
In the present work SBSE and in-situ derivatization TD-GC-MS was optimized
for the determination of a large variety of polar organic pollutants (octylphenols, nonylphenols, bisphenol A, diethylstilbestrol, cis-androsterone, estrone, equilin, 17-β-estradiol, testosterone, equilenin, mestranol, 19-norethisterone, 17-α-ethynyl estradiol, progesterone, estriol, coprostanol, cholesterol, stigmastanol). First of all, the best derivatization reagent [2,2,2-trifluoro-N-trimethylsilyl-1-trimethylsilyloxy-ethanimine (BSTFA)] was studied. Then, chemical variables (pH, NaCl and methanol addition) affecting the extraction step were analyzed by means of an experimental design approach. Once the optimum extraction conditions were fitted (360 min extraction time, 20% NaCl, pH=7.5 and without the addition of methanol), in-situ derivatization thermal desorption conditions were optimised including cryo-focusing temperature, derivatisation reagent volume, flow, time and desorption temperature. Preliminary results are shown in the present communication.
ACKNOWLEDGEMENTS: This project has been financially supported by the
Spanish Ministry of Science and Innovation through the CTQ2008-02775/BQU project. [1] S.D. Richardson, Anal. Chem. 79 (2007) 4295-4324. [2] P. Popp, L. Montero, M. Rueckert, J. Chromatogr. A 1071 (2005) 155-162. [3] F. David, B. Tienpont, P. Sandra, LC-GC Europe July (2003) 2-7. [4] M. Kawaguchi, R. Ito, H. Honda, N. Endo, N. Okanouchi, K. Saito, Y. Seto, H.
Nakazawa, J. Chromatogr. A 1200 (2008) 260-263.

COMUNICACIONES EN CARTEL C2-92
131
ELECTROCHEMICAL TECHNIQUES IN PYROCHEMICAL PROCESSES.
I.- CHEMICAL AND ELECTROCHEMICAL BEHAVIOUR OF Yb(III) IN MOLTEN CHLORIDES.
Y. Castrillejo(a), P. Fernández(a), J. Medina(b), M. Vega(a) and E. Barrado(a)
Dpto (a)Química Analítica.(b) F. Mat. Condensada Cristalografía y Mineralogía. Universidad de Valladolid. Prado de la Magdalena s/n. 47005 Valladolid Spain.
E-mail: [email protected] (presenting author)
Partitioning and transmutation (P&T) is one of the powerful strategies for reducing the radiotoxicity and volume of nuclear wastes. In this strategy, the separation (partitioning) of long-lived radionucleides, i.e. minor actinides (MAs), from other fission products especially lanthanides (Lns), is required before transmuting them into other stable or lower-level nuclides, otherwise the Lns absorb neutrons effectively and hence prevent neutron capture by the transmutable actinides. However, the separation of MAs from Lns is not simple due to the amount of Lns in the fission products and the similarity of their chemical properties.
This work is concerned with the electrochemistry of ytterbium, a heavy
lanthanide present in the fission products. The study has been carried out in the eutectic LiCl-KCl mixture, using GC, W and Al electrodes.
On GC and W electrodes, the electro-reduction of Yb(III) ions proceeds in one
electrochemical step, Yb(III)/Yb(II). Yb metal reacts with the melt to displace lithium, according to the reaction:
Yb(0) +2Li(I) ↔ Yb(II) + 2Li(0)
Therefore, electrodeposition of metal cannot be observed within the electrochemical window.
The electrochemical system Yb(III)/Yb(II) was found to be quasi-reversible and
the values of the E1/2 and the kinetic parameters, k0 and α, have been calculated by simulation of the cyclic voltammograms. Mass transport towards the electrode was demonstrated to be a simple diffusion process, and the diffusion coefficient of Yb(III) ions has been calculated.
Cyclic voltammetry, open-circuit chronopotentiometry and potentiostatic
electrolysis pointed out that Yb(III) can be reduced on an aluminium electrode to form Yb-Al alloys. Therefore, the use of a reactive cathode made of aluminium could be a pertinent route for the extraction of ytterbium from chloride media.
Potentiometric titrations of Yb(III) solutions with oxide donors, using a yttria
stabilized zirconia membrane electrode “YSZE” as a pO2- indicator electrode, have shown the formation of YbOCl and Yb2O3 compounds, and their solubility products have been determined at 723K. E-pO2- predominance diagram of Yb in the studied melt has been constructed. Acknowledgements.- The authors thank the Consejería de Educación y Cultura de la Junta de Castilla y León (project VA029A07) for the financing of this work.
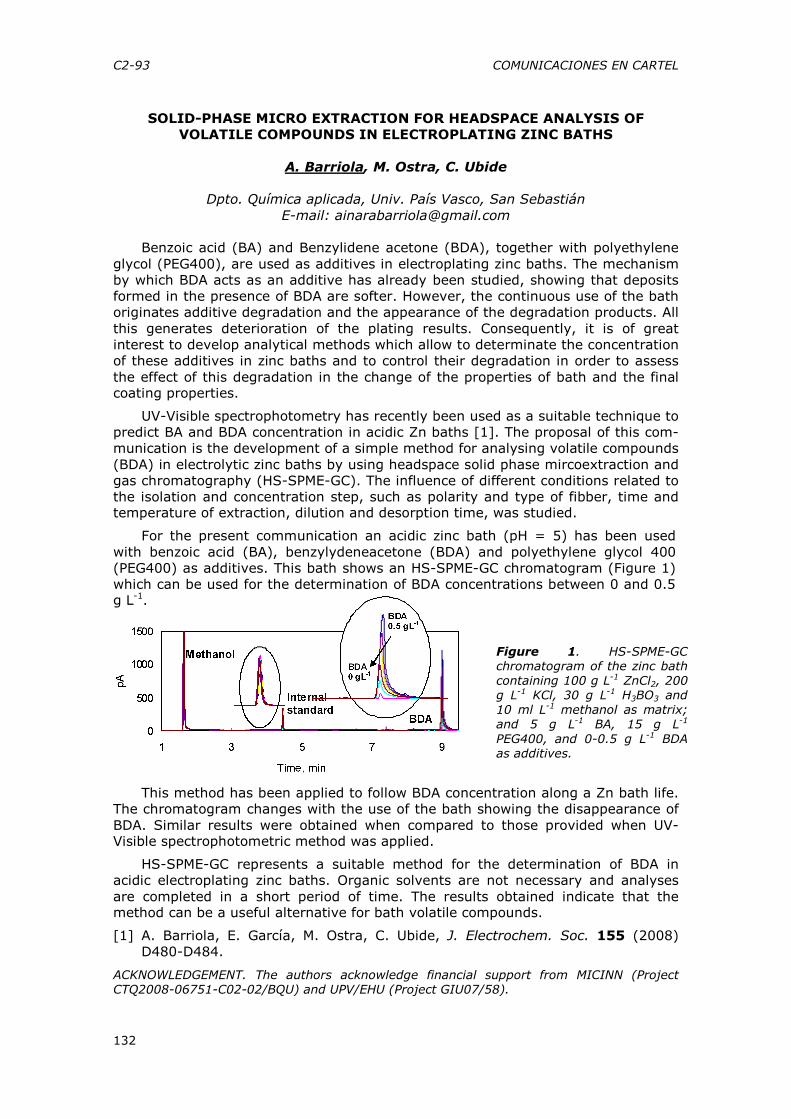
C2-93 COMUNICACIONES EN CARTEL
132
SOLID-PHASE MICRO EXTRACTION FOR HEADSPACE ANALYSIS OF
VOLATILE COMPOUNDS IN ELECTROPLATING ZINC BATHS
A. Barriola, M. Ostra, C. Ubide
Dpto. Química aplicada, Univ. País Vasco, San Sebastián E-mail: [email protected]
Benzoic acid (BA) and Benzylidene acetone (BDA), together with polyethylene
glycol (PEG400), are used as additives in electroplating zinc baths. The mechanism by which BDA acts as an additive has already been studied, showing that deposits formed in the presence of BDA are softer. However, the continuous use of the bath originates additive degradation and the appearance of the degradation products. All this generates deterioration of the plating results. Consequently, it is of great interest to develop analytical methods which allow to determinate the concentration of these additives in zinc baths and to control their degradation in order to assess the effect of this degradation in the change of the properties of bath and the final coating properties.
UV-Visible spectrophotometry has recently been used as a suitable technique to predict BA and BDA concentration in acidic Zn baths [1]. The proposal of this com-munication is the development of a simple method for analysing volatile compounds (BDA) in electrolytic zinc baths by using headspace solid phase mircoextraction and gas chromatography (HS-SPME-GC). The influence of different conditions related to the isolation and concentration step, such as polarity and type of fibber, time and temperature of extraction, dilution and desorption time, was studied.
For the present communication an acidic zinc bath (pH = 5) has been used with benzoic acid (BA), benzylydeneacetone (BDA) and polyethylene glycol 400 (PEG400) as additives. This bath shows an HS-SPME-GC chromatogram (Figure 1) which can be used for the determination of BDA concentrations between 0 and 0.5 g L-1.
Figure 1. HS-SPME-GC chromatogram of the zinc bath containing 100 g L-1 ZnCl2, 200 g L-1 KCl, 30 g L-1 H3BO3 and 10 ml L-1 methanol as matrix; and 5 g L-1 BA, 15 g L-1 PEG400, and 0-0.5 g L-1 BDA as additives.
This method has been applied to follow BDA concentration along a Zn bath life. The chromatogram changes with the use of the bath showing the disappearance of BDA. Similar results were obtained when compared to those provided when UV-Visible spectrophotometric method was applied.
HS-SPME-GC represents a suitable method for the determination of BDA in acidic electroplating zinc baths. Organic solvents are not necessary and analyses are completed in a short period of time. The results obtained indicate that the method can be a useful alternative for bath volatile compounds.
[1] A. Barriola, E. García, M. Ostra, C. Ubide, J. Electrochem. Soc. 155 (2008) D480-D484.
ACKNOWLEDGEMENT. The authors acknowledge financial support from MICINN (Project CTQ2008-06751-C02-02/BQU) and UPV/EHU (Project GIU07/58).
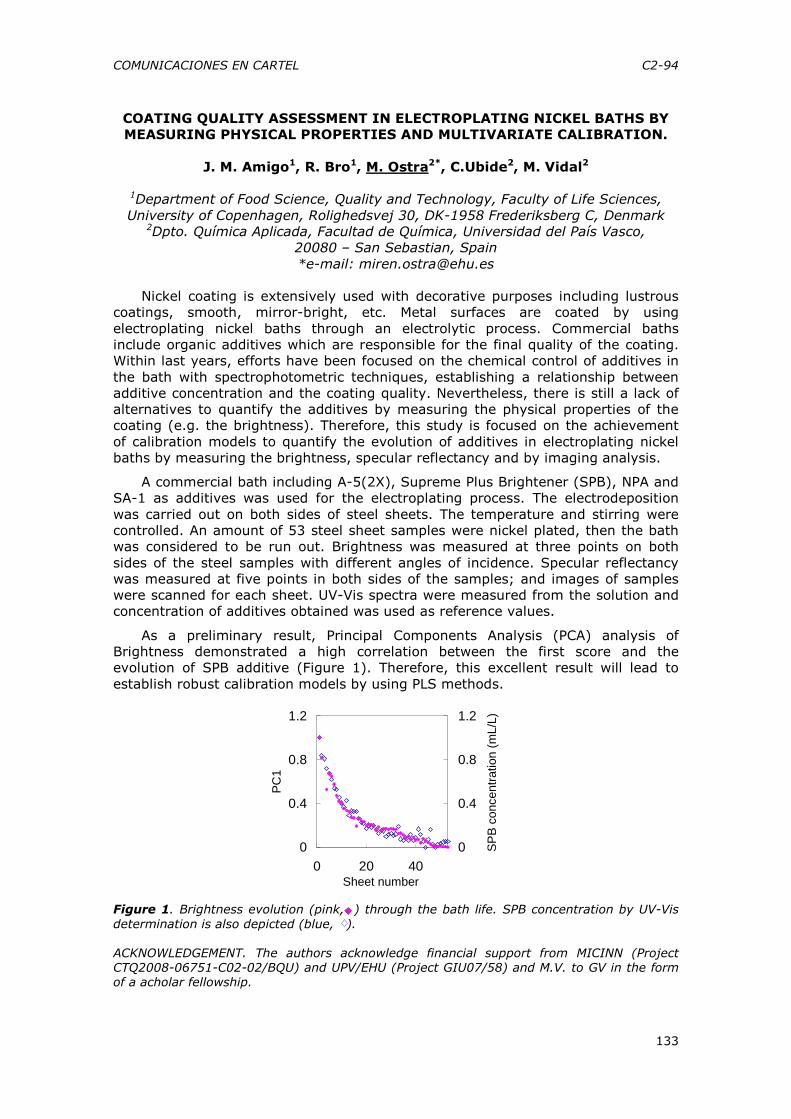
COMUNICACIONES EN CARTEL C2-94
133
0
0.4
0.8
1.2
0 20 40Sheet number
PC
1
0
0.4
0.8
1.2
SP
B c
once
ntra
tion
(mL/
L)
COATING QUALITY ASSESSMENT IN ELECTROPLATING NICKEL BATHS BY MEASURING PHYSICAL PROPERTIES AND MULTIVARIATE CALIBRATION.
J. M. Amigo1, R. Bro1, M. Ostra2*, C.Ubide2, M. Vidal2
1Department of Food Science, Quality and Technology, Faculty of Life Sciences, University of Copenhagen, Rolighedsvej 30, DK-1958 Frederiksberg C, Denmark
2Dpto. Química Aplicada, Facultad de Química, Universidad del País Vasco, 20080 – San Sebastian, Spain *e-mail: [email protected]
Nickel coating is extensively used with decorative purposes including lustrous
coatings, smooth, mirror-bright, etc. Metal surfaces are coated by using electroplating nickel baths through an electrolytic process. Commercial baths include organic additives which are responsible for the final quality of the coating. Within last years, efforts have been focused on the chemical control of additives in the bath with spectrophotometric techniques, establishing a relationship between additive concentration and the coating quality. Nevertheless, there is still a lack of alternatives to quantify the additives by measuring the physical properties of the coating (e.g. the brightness). Therefore, this study is focused on the achievement of calibration models to quantify the evolution of additives in electroplating nickel baths by measuring the brightness, specular reflectancy and by imaging analysis.
A commercial bath including A-5(2X), Supreme Plus Brightener (SPB), NPA and SA-1 as additives was used for the electroplating process. The electrodeposition was carried out on both sides of steel sheets. The temperature and stirring were controlled. An amount of 53 steel sheet samples were nickel plated, then the bath was considered to be run out. Brightness was measured at three points on both sides of the steel samples with different angles of incidence. Specular reflectancy was measured at five points in both sides of the samples; and images of samples were scanned for each sheet. UV-Vis spectra were measured from the solution and concentration of additives obtained was used as reference values.
As a preliminary result, Principal Components Analysis (PCA) analysis of Brightness demonstrated a high correlation between the first score and the evolution of SPB additive (Figure 1). Therefore, this excellent result will lead to establish robust calibration models by using PLS methods. Figure 1. Brightness evolution (pink, ) through the bath life. SPB concentration by UV-Vis determination is also depicted (blue, ). ACKNOWLEDGEMENT. The authors acknowledge financial support from MICINN (Project CTQ2008-06751-C02-02/BQU) and UPV/EHU (Project GIU07/58) and M.V. to GV in the form of a acholar fellowship.

C2-95 COMUNICACIONES EN CARTEL
134
MULTICLASS DETERMINATION OF 60 ORGANIC POLLUTANTS IN
WASTEWATER SAMPLES BY GAS CHROMATOGRAPHY TRIPLE QUADRUPOLE MASS SPECTROMETRY
J. Robles-Molina, B. Gilbert-López, J.F. Garcia-Reyes, A. Molina-Díaz
Analytical Chemistry Research Group, Department of Physical and Analytical
Chemistry, University of Jaen, Jaen. E-mail: [email protected] In Spain, although more than 50% of urban wastewaters are currently being
treated, only half of them are subjected to biological treatments and only 3% undergo advanced treatment technologies. Consequently, the application of more exhaustive wastewater treatment protocols, including the use of new and improved technologies, the application of wider and integrated quality control strategies and the development of water reuse strategies are priority tasks to tackle with. In this project, we have used as a reference, the European Water Framework Directive (WFD; Directive 2000/60/CE), which establishes a framework for Community action in the field of water policy, setting a list of priority compounds to be monitored in water in order to evaluate their levels. The aim of the present study is to evaluate the presence of such compounds in different wastewater treatment plants. For this purpose, in this work, we have developed and validated a multi-residue method for the analysis of 60 multi-class organic contaminants in wastewater samples using gas chromatography coupled to triple quadrupole mass spectrometry.
The proposed method is based on a sample treatment using liquid-liquid
extraction with n-hexane followed by identification, confirmation and quantitation with gas chromatography tandem mass spectrometry using a triple quadrupole analyzer in the selected reaction monitoring mode. Three MS/MS transitions were selected for unambiguous confirmation of the target chemicals. The method was validated at different concentration levels obtaining recovery rates in the range 70-110 % in most cases. The limits of quantitation obtained for most of the compounds tested were in the low nanogram per litre range. Samples of different origin (industrial, coastal and urban) provided by several wastewater treatment plants (WWTPs) located throughout Spain were tested. Results so far showed that most of the samples assayed did not contain large amount of these contaminants. Hexachlorobenzene was found to be the more frequently detected contaminant in the studied samples, but at levels below 5µg L-1.

COMUNICACIONES EN CARTEL C2-96
135
DETERMINACION DE CROMO(VI) EN AGUA MEDIANTE
ELECTROQUIMIOLUMINISCENCIA DE COMPLEJOS DE RUTENIO
A. Chiappino, L. Sanz, N. Pérez, C. Cacho, S. Rubio, A.M. Gutiérrez, C. Pérez-Conde
Dpto. Química Analítica, Univ. Complutense de Madrid, Madrid.
E-mail: [email protected] La electroquimioluminiscencia (ECL) es una técnica analítica basada en la
medida de la radiación emitida durante el transcurso de una reacción electroquímica. Esta técnica presenta las ventajas de elevada selectividad, amplio intervalo lineal, bajo coste y la posibilidad de adaptarla al desarrollo de sensores. En los últimos años, la ECL de los complejos de rutenio y, en especial, la del tris(2,2’-bipiridina)rutenio(II) (Ru(bpy)3
2+), se ha empleado ampliamente para la determinación de numerosos compuestos orgánicos, tales como ácidos carboxílicos, aminas y antibióticos tetraciclínicos, pero su aplicación a la determinación de metales es considerablemente más escasa.
El principal objetivo del presente trabajo es la puesta a punto de un método
para la determinación de cromo(VI), basado en el efecto ejercido por éste sobre la señal ECL del Ru(bpy)3
2+. Para ello, se ha diseñado una celda electroquímica que permite la oxidación del Ru(bpy)3
2+ en continuo empleando Pt como electrodos de trabajo y auxiliar y Ag/AgCl como electrodo de referencia. El Ru(bpy)3
3+ generado electroquímicamente se reduce posteriormente con oxalato a Ru(bpy)3
2+*, cuya emisión se mide en el detector de quimioluminiscencia. En la siguiente figura se muestra un esquema del diseño FIA empleado.
Figura 1. Diseño FIA empleado en la determinación de Cr(VI) en muestras de agua de grifo y pozo.
Se han optimizado las distintas variables que influyen en el sistema como concentración de reductor y Ru(bpy)3
2+, fuerza iónica, caudal, potencial y tiempo de oxidación. Se ha demostrado que el Cr(VI) aumenta considerablemente la señal ECL del Ru(bpy)3
2+, mientras que el Cr(III) no la varía, siendo posible determinar el Cr(VI) en presencia de grandes cantidades de Cr(III), permitiendo la especiación de este elemento.
El método desarrollado se ha aplicado a la determinación de Cr(VI) en
muestras de agua de grifo y pozo enriquecidas, obteniendo recuperaciones superiores al 90% en todos los casos, siendo la RSD inferior al 8%. Los límites de detección obtenidos (5-15 µg/L) permiten la determinación del Cr(VI) a niveles de concentración inferiores a los valores máximos permitidos por la OMS en aguas potables (50 µg/L). AGRADECIMIENTOS:
Los autores quieren agradecer al MICINN (CTQ2006-15610-C02-02/BQU), a la CAM (S-0505-AMB-000374), al Fondo Social Europeo, al Fondo Europeo de Desarrollo Regional y a la Universidad Complutense de Madrid por la financiación de este trabajo.
Detector dequimioluminiscencia
Válvula inyección
Celdaelectroquímica
BO
MB
AP
ER
IST
ALT
ICA
Ru(bpy)32+ 1 mM
Fosfato 100 mM, pH 3
Oxalato 10 mMFosfato 100 mM, pH 3
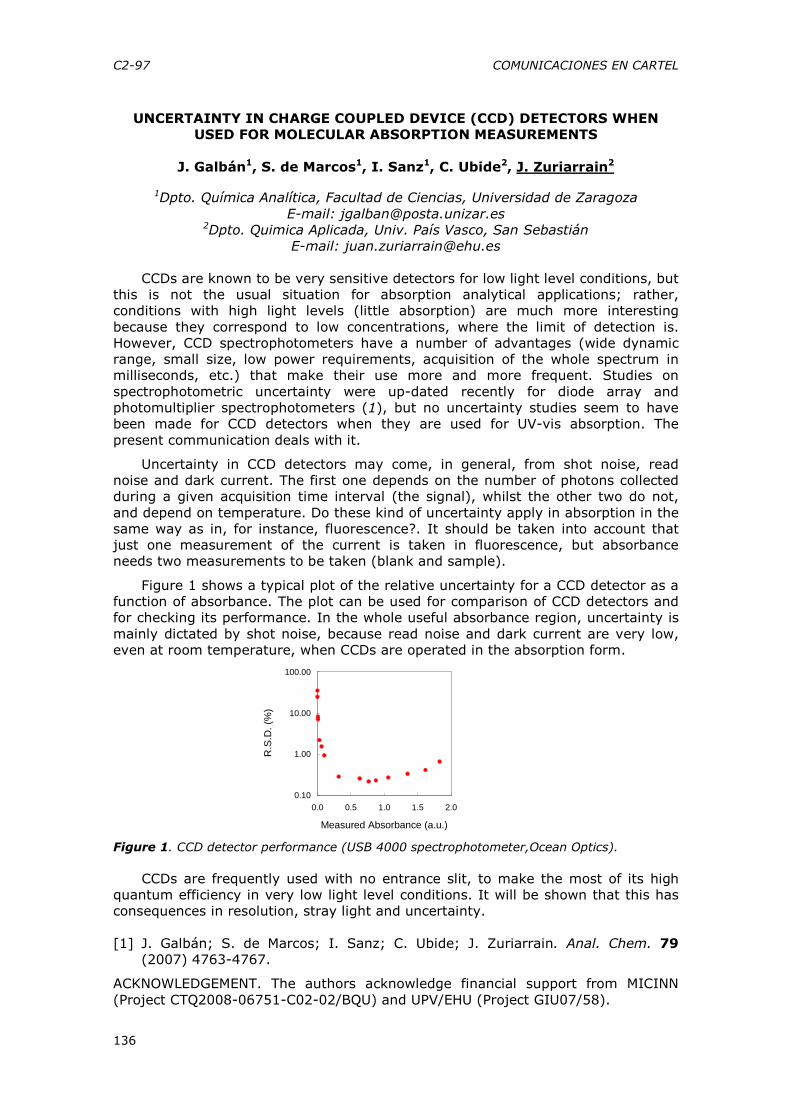
C2-97 COMUNICACIONES EN CARTEL
136
0.10
1.00
10.00
100.00
0.0 0.5 1.0 1.5 2.0
Measured Absorbance (a.u.)
R.S
.D. (
%)
UNCERTAINTY IN CHARGE COUPLED DEVICE (CCD) DETECTORS WHEN
USED FOR MOLECULAR ABSORPTION MEASUREMENTS
J. Galbán1, S. de Marcos1, I. Sanz1, C. Ubide2, J. Zuriarrain2
1Dpto. Química Analítica, Facultad de Ciencias, Universidad de Zaragoza E-mail: [email protected]
2Dpto. Quimica Aplicada, Univ. País Vasco, San Sebastián E-mail: [email protected]
CCDs are known to be very sensitive detectors for low light level conditions, but
this is not the usual situation for absorption analytical applications; rather, conditions with high light levels (little absorption) are much more interesting because they correspond to low concentrations, where the limit of detection is. However, CCD spectrophotometers have a number of advantages (wide dynamic range, small size, low power requirements, acquisition of the whole spectrum in milliseconds, etc.) that make their use more and more frequent. Studies on spectrophotometric uncertainty were up-dated recently for diode array and photomultiplier spectrophotometers (1), but no uncertainty studies seem to have been made for CCD detectors when they are used for UV-vis absorption. The present communication deals with it.
Uncertainty in CCD detectors may come, in general, from shot noise, read noise and dark current. The first one depends on the number of photons collected during a given acquisition time interval (the signal), whilst the other two do not, and depend on temperature. Do these kind of uncertainty apply in absorption in the same way as in, for instance, fluorescence?. It should be taken into account that just one measurement of the current is taken in fluorescence, but absorbance needs two measurements to be taken (blank and sample).
Figure 1 shows a typical plot of the relative uncertainty for a CCD detector as a function of absorbance. The plot can be used for comparison of CCD detectors and for checking its performance. In the whole useful absorbance region, uncertainty is mainly dictated by shot noise, because read noise and dark current are very low, even at room temperature, when CCDs are operated in the absorption form.
Figure 1. CCD detector performance (USB 4000 spectrophotometer,Ocean Optics).
CCDs are frequently used with no entrance slit, to make the most of its high
quantum efficiency in very low light level conditions. It will be shown that this has consequences in resolution, stray light and uncertainty.
[1] J. Galbán; S. de Marcos; I. Sanz; C. Ubide; J. Zuriarrain. Anal. Chem. 79
(2007) 4763-4767.
ACKNOWLEDGEMENT. The authors acknowledge financial support from MICINN (Project CTQ2008-06751-C02-02/BQU) and UPV/EHU (Project GIU07/58).

COMUNICACIONES EN CARTEL C2-98
137
SIMULTANEOUS LIQUID–LIQUID MICROEXTRACTION/METHYLATION FOR THE DETERMINATION OF HALOACETIC ACIDS IN DRINKING WATERS BY
HEADSPACE GAS CHROMATOGRAPHY
M.J. Cardador, A. Serrano, M. Gallego
Department of Analytical Chemistry, Campus of Rabanales, University of Córdoba, E-14071 Córdoba, Spain. E-mail: [email protected]
Extensive research has focused on the formation of disinfection byproducts (DBPs), with emphasis on trihalomethanes and, more recently, haloacetic acids (HAAs), the two most prominent kinds of DBPs. HAAs are formed by the interaction of hypochlorite with natural organic matter and bromide, if present. Although there are nine haloacetic acid congeners that contain chlorine or bromine, the EPA has established a maximum contaminant level (MCL) of 60 µg L-1 for the sum of only five HAAs: monochloroacetic acid, dichloroacetic acid, trichloroacetic acid, monobromoacetic acid, and dibromoacetic acid. After reviewing the methods developed to date for the determination of HAAs in waters, two serious drawbacks have been found: the time required for the sample treatment (40–120 min) and the degradation of the most unstable species. From that premise, the aims of this work were: (i) to reduce the time of sample treatment (derivatisation/extraction), (ii) to cut down on organic solvents (extraction), and (iii) to minimise the degradation of the HAAs during the whole procedure.
A novel analytical method that combines simultaneous liquid–liquid
microextraction/methylation and headspace gas chromatography–mass spectrometry for the determination of nine haloacetic acids (HAAs) in water was reported. A mechanistic model on the basis of mass transfer with chemical reaction in which methylation of HAAs was accomplished in n-pentane–water (150 µL–10 mL) two-phase system with a tetrabutylammonium salt as phase transfer catalyst was proposed. Derivatisation with dimethylsulphate was completed in 3 min by shaking at room temperature. The methyl ester derivatives and the organic phase were completely volatilised by static headspace technique, being the gaseous phase analysed. Parameters related to the extraction/methylation and headspace generation of HAAs were studied and the results were compared with methyl haloacetate standards to establish the yield of each step. The thermal instability of HAAs, by degradation to their respective halogenated hydrocarbon by decarboxylation, and the possible hydrolysation of the methyl esters were rigorously controlled in the whole process. The proposed method is simpler and faster than those described in the literature since the sample treatment has been shortened using softer conditions, which also minimised the degradation of HAAs, obtaining a reliable and robust method. In addition, the sensitivity (LODs from 0.02 to 0.4 µg L−1) and precision (RSD 7.5%) were similar to the most sensitive method developed to date [1].
Finally, the method was validated with the US Environmental Protection Agency
(EPA) method 552.2 for the determination of HAAs in drinking and swimming pool water samples. [1] M.N. Sarrion, F.J. Santos, M.T. Galcerán, Anal. Chem. 72 (2002) 4865-4873.

C2-99 COMUNICACIONES EN CARTEL
138
ANALYSIS OF LOW-MOLECULAR MASS ALDEHYDES IN DRINKING WATERS
THROUGH CAPILLARY ELECTROPHORESIS WITH LASER INDUCED FLUORESCENCE DETECTION
Clara Eugenia Baños and Manuel Silva
Department of Analytical Chemistry, Campus of Rabanales. University of Cordoba
E-14071, Cordoba, Spain. E-mail: [email protected] Low-molecular mass aldehydes are acknowledged to be harmful organic
pollutants that play an important role in tropospheric chemistry, in ground waters as their atmospheric water-soluble fraction and also in potable waters as the disinfection by-products produced primarily from the organic matter present in the water from ozone treatment [1,2]. These volatile, polar and reactive organic compounds produce adverse health effects and various aldehydes are also recognized to be irritants and allergens. So, the quality control of water samples demands the need for sensitive and reliable methods of analysis to measure concentrations of low aliphatic aldehydes at µg/L levels in this kind of samples. In recent years, liquid chromatography (LC) is increasingly being viewed as an interesting alternative to gas chromatography for the determination of low aliphatic aldehydes in water samples; however, the potential of capillary electroforesis (CE) in this field as a possible option to LC methods is on its way to develop [5].
In this communication, a simple sensitive method was developed for the simultaneous determination of the seven major aldehydes found in ozonated drinking waters such as formaldehyde, acetaldehyde, propionaldehyde, butyraldehyde, valeraldehyde, glyoxal and methylglyoxal by CE with laser induced fluorescence (LIF) detection using an air-cooled argon ion laser. The optimum procedure includes a derivatization step of the low aliphatic aldehydes with fluorescein-5-thiosemicarbazide (FTSC) at pH 6.5 (60°C for 2 hours in the dark) and direct injection for CE analysis, which is conducted within about 25 min using a running buffer consisted of 10 µM Triton X-100 and 60 mM sodium borate at pH 10. The lowest detectable analyte concentration ranged from 80-450 ng/L with a precision of 5.0-8.3%. The method was applied to the determination of low-molecular mass aldehydes in mineral and bottled (treated with ozone) water samples with average recoveries ranging from 99.5 to 104.8%. These results indicate that the proposed method is a straightforward and sensitive tool for the determination of these aldehydes in drinking water samples. To our knowledge, this contribution is the first reporting the analysis of these aldehydes by CE-LIF in water samples. [1] S.D. Richardson, J. Environ. Monit. 4 (2002) 1. [2] S.D. Richardson, T.V. Caughran, T. Poiger, Y. Guo, F.G. Crumley, Ozone Sci.
Eng. 22 (2000) 653. [3] C. Zwiener, F.H. Frimmel, Anal. Bioanal. Chem. 372 (2002) 615. [4] C.E. Baños, M. Silva, Talanta 77 (2009) 1597. [5] N. Dossi, S. Susmel, R. Toniolo, A. Pizzariello, G. Bontempelli, J. Chromatogr. A 1207 (2008) 169.
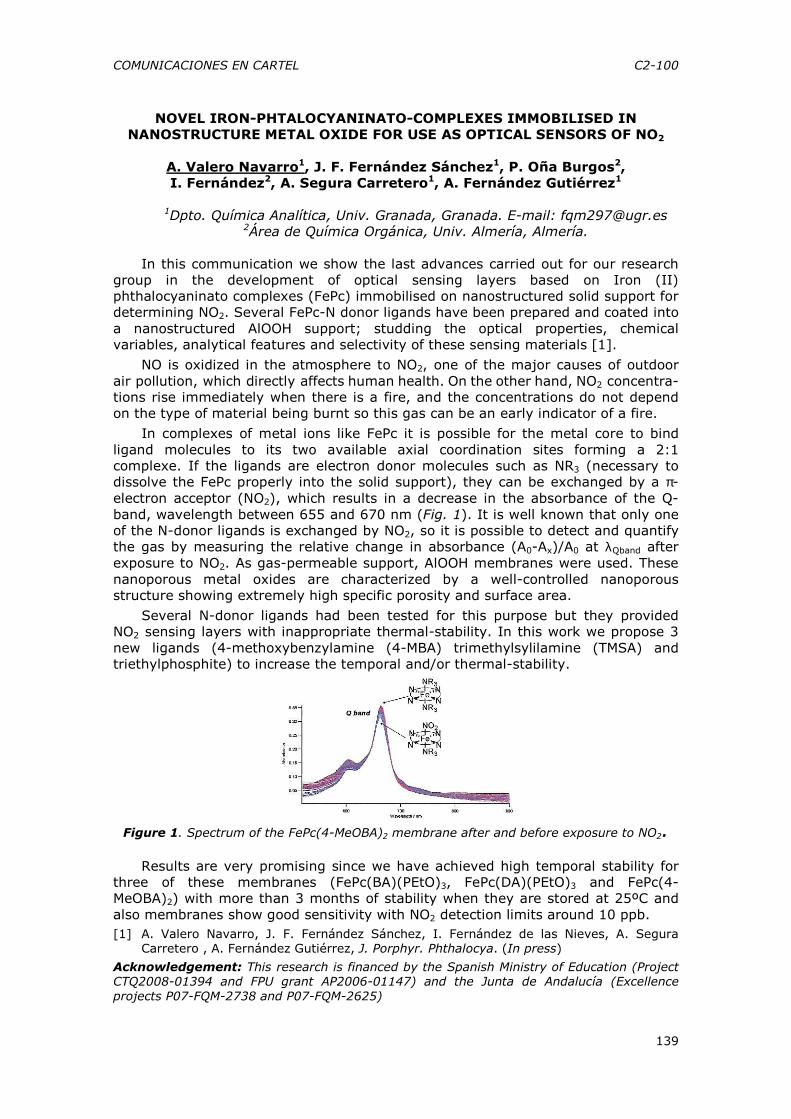
COMUNICACIONES EN CARTEL C2-100
139
NOVEL IRON-PHTALOCYANINATO-COMPLEXES IMMOBILISED IN
NANOSTRUCTURE METAL OXIDE FOR USE AS OPTICAL SENSORS OF NO2
A. Valero Navarro1, J. F. Fernández Sánchez1, P. Oña Burgos2, I. Fernández2, A. Segura Carretero1, A. Fernández Gutiérrez1
1Dpto. Química Analítica, Univ. Granada, Granada. E-mail: [email protected]
2Área de Química Orgánica, Univ. Almería, Almería. In this communication we show the last advances carried out for our research
group in the development of optical sensing layers based on Iron (II) phthalocyaninato complexes (FePc) immobilised on nanostructured solid support for determining NO2. Several FePc-N donor ligands have been prepared and coated into a nanostructured AlOOH support; studding the optical properties, chemical variables, analytical features and selectivity of these sensing materials [1].
NO is oxidized in the atmosphere to NO2, one of the major causes of outdoor air pollution, which directly affects human health. On the other hand, NO2 concentra-tions rise immediately when there is a fire, and the concentrations do not depend on the type of material being burnt so this gas can be an early indicator of a fire.
In complexes of metal ions like FePc it is possible for the metal core to bind ligand molecules to its two available axial coordination sites forming a 2:1 complexe. If the ligands are electron donor molecules such as NR3 (necessary to dissolve the FePc properly into the solid support), they can be exchanged by a π-electron acceptor (NO2), which results in a decrease in the absorbance of the Q-band, wavelength between 655 and 670 nm (Fig. 1). It is well known that only one of the N-donor ligands is exchanged by NO2, so it is possible to detect and quantify the gas by measuring the relative change in absorbance (A0-Ax)/A0 at λQband after exposure to NO2. As gas-permeable support, AlOOH membranes were used. These nanoporous metal oxides are characterized by a well-controlled nanoporous structure showing extremely high specific porosity and surface area.
Several N-donor ligands had been tested for this purpose but they provided NO2 sensing layers with inappropriate thermal-stability. In this work we propose 3 new ligands (4-methoxybenzylamine (4-MBA) trimethylsylilamine (TMSA) and triethylphosphite) to increase the temporal and/or thermal-stability.
Figure 1. Spectrum of the FePc(4-MeOBA)2 membrane after and before exposure to NO2. Results are very promising since we have achieved high temporal stability for
three of these membranes (FePc(BA)(PEtO)3, FePc(DA)(PEtO)3 and FePc(4-MeOBA)2) with more than 3 months of stability when they are stored at 25ºC and also membranes show good sensitivity with NO2 detection limits around 10 ppb. [1] A. Valero Navarro, J. F. Fernández Sánchez, I. Fernández de las Nieves, A. Segura
Carretero , A. Fernández Gutiérrez, J. Porphyr. Phthalocya. (In press)
Acknowledgement: This research is financed by the Spanish Ministry of Education (Project CTQ2008-01394 and FPU grant AP2006-01147) and the Junta de Andalucía (Excellence projects P07-FQM-2738 and P07-FQM-2625)

C2-101 COMUNICACIONES EN CARTEL
140
CARACTERIZACIÓN DE COMPLEJOS DE IRIDIO PARA LA DETERMINACIÓN
DE OXÍGENO MEDIANTE ATENUACIÓN DE LA FLUORESCENCIA
M. Marín Suárez del Toro, J.F. Fernández Sánchez, A. Fernández Gutiérrez
Dpto. Química Analítica, Universidad de Granada, Granada. E-mail: [email protected] El oxígeno molecular juega un papel muy importante en la vida, estando
presente en multitud de reacciones químicas, tanto como reactivo como producto, lo cual hace que la medida de este gas tome especial interés en numerosas aplicaciones en la industria, medicina, medioambiente… etc. Aunque la detección del oxígeno ha sido ampliamente estudiada, hoy en día toma especial importancia el desarrollo de sensores basados en la atenuación de la luminiscencia. El uso de este tipo de sensor permite obtener un tiempo de respuesta más corto, con menor consumo de energía, y además hace posible su miniaturización y consiguiente utilización “in-situ”.
Para cuantificar el oxígeno de esta forma, existen tres métodos básicos:
medida de la intensidad, medida de tiempo de vida y medidas relativas. Las dos primeras presentan una serie de desventajas, ya que la medida directa de intensidad puede verse afectada por cambios en el instrumento o degradación del material fosforescente, mientras que las medidas de tiempo de vida requieren un instrumento de medida muy complejo. Por otro lado, las medidas relativas de intensidad, al presentar una referencia interna, dan lugar a sistemas de medida más robustos y menos afectados por lo cambios de intensidad no relacionados con la presencia de oxígeno.
En esta comunicación se presenta la caracterización de cuatro complejos
fosforescentes de iridio, bis(2,4-difluorofenilpiridina)-4,4’-tert-butil-2,2’bipiridina iridio(III) (N-969), 4,7-difenil-1,10-fenantrolina-bis(2fenilpiridina) iridio(III) (N-1001), [Ir(2,4-difluorofenilpiridina)2(4,7-difenil-1,10-fenantrolina)] (N-1008) y [Ir(2-fenilpiridine)-2-(4-bromo-2,2’-bipiridina)] (N-1010), para el desarrollo de fases sensores para la determinación de oxígeno molecular. Estos complejos, fueron inmovilizados tanto en poliestireno (con y sin el plastificante o-cianometil octil éter) como en un material nanoestructurado de óxido de aluminio.
Para su caracterización se estudiaron sus características espectroscópicas
(longitud de onda de excitación, longitud de onda de emisión y tiempo de vida) así como sus parámetros analíticos (constante de Stern-Volmer), obteniéndose que, en función del soporte y la longitud de onda de excitación, se pueden llevar a cabo medidas directas y/o relativas para la determinación de O2.
La utilización de plastificante en la formación de las membranas poliméricas de
poliestireno produce una atenuación de la fosforescencia, disminuyendo su sensibilidad. Por otro lado, la inmovilización de estos complejos en un material nanoestructurado de un óxido metálico muestra un incremento de la sensibilidad en comparación con estas membranas clásicas.
Por último, destacar que el complejo 1001 inmovilizado sobre óxido de aluminio
nanoestructurado se atenúa totalmente a una concentración en volumen del 1% de O2, por tanto es un complejo muy útil para controlar la presencia de oxígeno en muy baja cantidad (envases al vació, atmósferas inertes, etc.) Agradecimientos: Esta investigación está financiada por la Junta de Andalucía (Proy. Excelencia P07-FQM-2738 y P07-FQM-2625).

COMUNICACIONES EN CARTEL C2-103
141
ROBUSTNESS TEST OF A MICROWAVE-ASSISTED EXTRACTION METHOD FOR THE DETERMINATION OF HALOANISOLES AND HALOPHENOLS IN
CORK STOPPERS USING EXPERIMENTAL DESIGN
C. Pizarro, N. Pérez-del-Notario, J.M. González-Saiz
Department of Chemistry, University of La Rioja, Logroño, La Rioja, Spain. E-mail: [email protected]
Nowadays, in the field of enology, researches are focusing their work on the
development of analytical methods that can be widespread used in their own industry. However, one of the most important steps of any method validation, i.e., robustness evaluation, has often been ignored. Robustness can be described as the ability to reproduce an analytical method in different laboratories or under different circumstances without the occurrence of unexpected differences in the obtained results; and a robustness test is an experimental set-up to evaluate the robustness of a given method.
Before performing the robustness test on an analytical procedure, the parameters that influence the process must be optimised. In this case, the evaluated method is based on microwave-assisted extraction (MAE) method optimised for the analysis of four haloanisoles (2,4,6-trichloro-, 2,3,4,6-tetrachloro, pentachloroanisol and 2,4,6-tribromoanisol), as well as their precursors halophenols (2,4,6-trichloro-, 2,3,4,6-tetrachloro-, pentachlorophenol and tribromophenol), involved in the presence of cork taint in wine. The origin of these haloanisoles, which are the direct responsible of this off-flavour, is related to the microbial degradation of the corresponding halophenols used as insecticides in different wood materials present in the wineries and the microflora and hypochlorite used to bleach wine cork. These compounds can cause this organoleptic defect in wine when they are present at the ng/l level.
The final aim of this work was testing the robustness of this MAE analytical method, and, consequently, ensuring its correct implementation in the enological industry. With that objective in mind, some factors, which represent potential sources of variability (solvent volume, extraction time and temperature), were selected and examined in an interval around the nominal level using experimental design. The robustness test included in this research was developed according to the experimental matrix of a 3-factor hybrid design. The experimental ranges selected were representative of the variations which can be expected when the method is transferred. From the statistical analysis of the results obtained it was possible to conclude that the proposed procedure based on MAE can be considered robust for haloanisoles and halophenols extraction because the changes of the three factors considered do not affect significantly to the measured responses for these analytes. [1] W.J. Youden, E.H. Steiner, Statistical Manual of the Association of Official
Analytical Chemistry, The Association of Official Analytical Chemists ed., Arlington, 1975, p. 33-36, 70-71, 82-83.
[2] J.A. Van Leeuwen, L.M.C. Buydens, B.G.M. Vandeginste, G. Kateman, P.J. Schoenmakers, M. Mulholland, Chemom. Intell. Lab. Syst. 10 (1991) 337-347.
[3] G.T. Wernimont, Use of Statistics to develop and evaluate Analytical Methods, W. Spendley ed., Association of Official Analytical Chemists, Arlington, 1985, 78-82.
[4] B. Campisi, D. Chicco, D. Vojnovic, R. Phan-Tan-Luu, J. Pharm. Biomed. Anal., 18 (1998) 57-65.

C2-104 COMUNICACIONES EN CARTEL
142
DETERMINACIÓN VOLTAMPEROMÉTRICA DE CINC, CADMIO Y PLOMO SOBRE ELECTRODOS IMPRESOS MODIFICADOS POR ADSORCIÓN DE
NANOPARTÍCULAS DE BISMUTO
M.A. Granado, M. Olivares, E. Pinilla
Dpto. Química Analítica, Univ. Extremadura Campus universitario, 06071 Badajoz
E-mail: [email protected] La monitorización de los niveles de metales pesados tales como el plomo, el
cadmio y el cinc en aguas naturales y en efluentes líquidos industriales requiere la aplicación de sensores específicos que puedan proporcionar medidas exactas con buena resolución temporal y espacial en condiciones de campo. En este trabajo nos centramos en la aplicación de un tipo concreto de sensor electroquímico, electrodos impresos de carbono (SCPE), modificando su superficie con bismuto para mejorar su respuesta a distintos metales pesados. En bibliografía se encuentran descritos distintos métodos de modificación entre ellos formación de películas [1, 2] y de síntesis de nanopartículas [3].
Se propone un sencillo procedimiento de síntesis química de nanopartículas de
bismuto y su adsorción sobre un electrodo impreso de carbono para la determinación de niveles traza de cinc, cadmio y plomo en muestras de agua mediante voltamperometría de redisolución anódica con barrido de onda cuadrada. El método de síntesis está basado en un método publicado por Zhao et al [4]. Se comparan dos configuraciones hidrodinámicas diferentes (celda convectiva y celda de flujo). Se optimizan distintas variables experimentales que afectan a las señales de redisolución de los metales de interés, como son el tiempo de deposición (en celda convectiva y de flujo) y la velocidad de agitación (en celda convectiva) y caudal de muestra (en celda de flujo). Con ambas configuraciones se obtienen para los tres metales pesados límites de detección del orden de las ppb. Se investiga su aplicabilidad a muestras de aguas naturales y residuales. Agradecimientos
Proyecto financiado por la Junta de Extremadura (PDT07A016). Mara Olivares agradece una beca de A.G. Siderurgia Balboa. [5] A. Economou. Trends in Anal. Chemistry. 4 (2005) 334 [6] J. Wang. Electroanalysis. 17 (2005) 1341 [7] N. A. Malakhova, N.Y. Stojko, K.Z. Brainina, Electrochem. Commun. 9 (2007)
223 [4] Y. Zhao, Z. Zhang, H. Dang, Mater. Lett. 58 (2004) 790

COMUNICACIONES EN CARTEL C2-105
143
DETERMINATION OF POLYBROMINATED DIPHENYL ETHERS IN INDOOR DUST BY PRESSURISED LIQUID EXTRACTION WITH IN CELL CLEAN-UP
AND GAS CHROMATOGRAPHY-MASS SPECTROMETRY M.P. Martínez, A.R.Hernández, A. Garrido-López, J.D. Carrillo and M.T. Tena
Department of Chemistry, University of La Rioja
C/ Madre de Dios 51. 26006 Logroño (La Rioja). Spain E-mail: [email protected]
Polybrominated diphenyl ethers (PBDEs) are polymer additives commonly used because of their flame-retardant properties. Since they are lipophylic compounds structurally similar to polychlorinated biphenyl ethers, they are suspected of being potentially toxic for humans because they can bioaccumulate and cause adverse effects on endocrine system. Their use as additives implies they are not part of the polymer structure and thus they can easily migrate from this to the environment. Indoor dust can result in an important PBDE contamination source from common domestic goods that contain PBDEs because it acts as repository of PBDEs and other semivolatile organic compounds, and it is one of the main sources of human exposition to PBDEs.
A selective pressurised liquid extraction (PLE) and gas chromatography-mass
spectrometry/mass spectrometry (GC-MS/MS) method is proposed for the determination of polybrominated diphenyl ethers in indoor dust. Selective PLE is based on the use of Florisil mixed with the sample to perform an in-cell clean-up. This approach provided cleaner and almost colourless extracts ready to be injected in the gas chromatograph. The PLE conditions were studied by experimental design, first a 4×3×2 multifactor categorical design to screen sorbent, solvent and temperature, and then a central composite design to optimise sorbent mass, temperature and time. Finally, the number of extraction cycles was studied.
The method was characterised in terms of limit of detection, reproducibility and
recovery. Limits of detection were between 0.06 ng g-1 and 1.64 ng g-1 for BDE-47 and BDE-138, respectively. The relative standard deviations (calculated under reproducibility conditions) were lower than 10%. Recovery values ranged from 82% to 101%, for BDE-3 and BDE-100, respectively. Finally, the accuracy of the method was checked by the analysis of a standard reference material. Moreover, it is advantageous over other methods in terms of simplicity, automation, analysis time and solvent consumption.

C2-106 COMUNICACIONES EN CARTEL
144
DETERMINATION OF ENDOCRINE DISRUPTING COMPOUNDS IN SEWAGE
SLUDGE USING PRESSURISED LIQUID EXTRACTION AND GAS CHROMATOGRAPHY-MASS SPECTROMETRY
M.P. Martínez and M.T. Tena
Department of Chemistry, University of La Rioja
C/ Madre de Dios 51. 26006 Logroño (La Rioja). Spain E-mail: [email protected]
Alkylphenols (APs), and bisphenol A (BPA) have been shown to exhibit endocrine disrupting properties in wildlife and laboratory animals. Trace levels of these compounds can potentially cause adverse effects in humans.
The analysis of sewage sludges is important because they are commonly used
as agricultural manure by their properties (relatively high content of organic matter and essential plant nutrients) and they can contain certain pollutants (proceeding from pharmaceutics and personal care products) that can affect crops and soil microorganisms and even can contaminate water resources.
In this communication, a method to determine endocrine disrupting compounds
(EDCs) in sewage sludge using pressurised liquid extraction, derivatization with BSTFA and gas chromatography-mass spectrometry is proposed. The analytes include butylphenol, t-butylphenol, pentylphenol, hexylphenol, heptylphenol, octylphenol, t-octylphenol, nonylphenol and bisphenol-A. Deuterated bisphenol-A (BPA-d16) was used as internal standard.
Pressurised liquid extracts of sewage sludge were very dirty and needed a
clean-up before their chromatographic analysis. The aim of the developed method is to avoid the extract clean-up step using on-line combination of pressurized liquid extraction (PLE), matrix solid-phase dispersion (MSPD) and solid-phase extraction (SPE) under high pressure and temperature conditions. The development of the method consisted of a multifactor study of variables (temperature, solvent, sorbent, time, cycles) and the method was finally characterised in terms of sensitivity, accuracy and precision.

COMUNICACIONES EN CARTEL C2-107
145
DETERMINATION OF Na, K, Ca, Mg AND P BY ICP-AES IN BIODIESEL SAMPLES PRODUCED BY HOMOGENEOUS AND HETEROGENEOUS
CATALYSIS C. Molina Mayoa, S. García Salgadoc, D. García Casillasc, A. Brito Alayóna, M.T. García Rodrígueza, F. Jiménez Morenob, M.A. Quijano Nietoc,
M.M. Bonilla Simónc
aDpto. Ingeniería Química y Tecnología Farmacéutica, Facultad de Química, Univ. de La Laguna; bDpto. Química Analítica, Nutrición y Bromatología, Facultad de Química, Univ. de La Laguna; cDpto. Ingeniería Civil: Tecnología Hidráulica y
Energética, E.U.I.T. Obras Públicas, Univ. Politécnica de Madrid. E-mail: [email protected]
Biodiesel is a biodegradable, renewable, eco-friendly and non-toxic alternative to diesel fuel, which is produced by the transesterification of vegetable oils or animal fats with an alcohol and often employing NaOH or KOH added as a catalyst. This reaction produces alkyl esters from long chain fatty acids and glycerin, as a subproduct. Biodiesel contributes to the reduction of greenhouse gases and other harmful emissions, and produces quite less sulfur dioxide and about 60% less net carbon dioxide emissions than petroleum-based diesel. Furthermore, it reduces black smoke normally associated with diesel vehicles and other particulate matter emissions that cause respiratory tract damage [1].
The determination of metals in biodiesel is important, since they can promote
the decomposition of the fuel or corrosion of motor parts. Monitoring Na and K is especially important, since their hydroxides are employed as catalysts in the biodiesel production process. These elements may be present as abrasive solids or soluble soaps and can contribute to engine parts wear (injector, fuel pump, piston, ring, etc.) and/or to deposits on the parts. Similarly, the determination of other elements, such as P, Ca and Mg is also important [2].
The aim of the present work consisted of determining the content of Na, K, Ca,
Mg and P, by inductively coupled plasma atomic emission spectrometry (ICP-AES), according to the European standards UNE-EN-14538:2006 [3] and UNE-EN-14107:2003 [4], in biodiesel samples obtained by homogeneous and heterogeneous catalysis from not used sunflower and corn oils, as well as from fried sunflower oil and a mixture of fried oils. In this way, these same elements were determined in the source oils, in order to compare these elements contents both in the raw materials used and in the main product of the transesterification reaction. [1] M.G. Andrade Korn, D.S. Sodré dos Santos, B. Welz, M.G. Rodrigues Vale,
A. Paixão Teixeira, D. Castro Lima, S.L. Costa Ferreira, Talanta 73 (2007) 1–11 [2] E.S. Chaves, T.D. Saint’Pierre, E.J. Santos, L. Tormen, V.L.A.F. Bascuñana, A.J.
Curtiusa, J. Braz. Chem. Soc. 19 (2008) 856-861. [3] UNE-EN-14538:2006. Productos derivados de grasas y aceites. Esteres
metílicos de ácidos grasos (FAME). Determinación del contenido de Ca, K, Mg y Na por análisis espectral de emisión óptica con plasma acoplado inductivamente (ICP OES).
[4] UNE-EN-14107:2003 Productos derivados de grasas y aceites. Esteres metílicos de ácidos grasos (FAME). Determinación del contenido de fósforo mediante espectrometría de emisión con plasma acoplado inductivo (ICP).

C2-108 COMUNICACIONES EN CARTEL
146
IONIC LIQUID BASED DISPERSIVE LIQUID-LIQUID MICROEXTRACTION OF
POLYCYCLIC AROMATIC HYDROCARBONS IN WATER SAMPLES
T. Pena, C. Casais, C. Mejuto, R. Cela
Departamento de Química Analítica, Nutrición y Bromatología. Facultad de Química. Instituto de Investigación y Análisis Alimentario. Universidad de Santiago de
Compostela. 15782 Santiago de Compostela. Spain. E-mail: [email protected]
Coastal and inland waters usually act as receptors for sewage effluents,
industrial effluents and urban and rural run-off. As streams and rivers, lakes and ponds are frequently used for potable water supply, contamination of water resources is particularly undesirable [1].
PAHs are important pollutants that extensively exist in environmental waters. Because of their high toxicity and potential carcinogenicity, the study of these compounds in water samples is an important issue. Most of the problems associated with the determination of PAHs in the aqueous environment are derived from their low concentrations owing to their extremely low solubility in water. Therefore, there is an increasing demand for developing sensitive, simple and rapid methods for the determination of PAHs in water samples.
Recent research trends involve miniaturisation of the traditional methodologies and reduction of organic solvent consumption by using microextraction techniques. One of the emerging techniques in this area is dispersive liquid-liquid microextraction (DLLME), where a mixture of a high-density solvent (extractant) and a water miscible polar solvent (disperser) is employed. DLLME offers several important advantages over conventional solvent extraction methods such as faster operation, easier manipulation, no needs to large amounts of organic extraction solvent and low time and cost.
On the other hand, one of the principal driving forces for the broad investigations in the area of new materials is the need to find replacements for environmentally hazardous volatile organic solvents. In this sense, ionic liquids (Ils), have attracted much attention taking into account their special features like low-vapour pressure, high viscosity and good thermal stability [2]. Thus, use of IL can replace use of highly toxic chlorinated solvents, usually employed as extractants in DLLME, and allows facile injection into reversed-phase system.
In the present work, the use of an ionic liquid in DLLME is proposed for the isolation of 18 PAHs from water samples. For the first time ever, ionic liquid based dispersive liquid liquid microextraction (IL-DLLME) was applied for the extraction of PAHs from water samples. The influence of several variables (e.g. type and volume of dispersant and extraction solvents, ionic strength, etc.) on the performance of the sample preparation step was carefully evaluated. Analytical determinations were carried out by high performance liquid chromatography (HPLC) with fluorescence detection. The developed procedure exhibits good recoveries as well as high enrichment factors for the studied compounds. Quantification limits obtained for all of these compounds were between 0.1 and 7 ng L-1. [1] E. Manoli, C. Samara, Trends Anal. Chem. 18 (1999) 417. [2] M. Koel, Crit. Rev. Anal. Chem. 35 (2005) 177. Acknowledgements:
This research was financially supported by the Spanish Ministry of Education and Science (project CTQ2006-03334/BQU), E.U. FEDER funding and the Xunta de Galicia (project PGIDIT06PXIB237039PR). T. Pena gratefully acknowledges her doctoral FPI grant from the Spanish Ministry of Education and Science.

COMUNICACIONES EN CARTEL C2-109
147
ELECTROCHEMICAL TECHNIQUES IN PYROCHEMICAL PROCESSES.
I.-CHEMICAL AND ELECTROCHEMICAL EXTRACTION OF THULIUM FROM MOLTEN CHLORIDES.
P. Fernández(a), Y. Castrillejo(a), J. Medina(b), M. Vega(a) and E. Barrado(a)
Dpto (a)Q. Analítica.(b) F. Mat. Condensada Cristalografía y Mineralogía.
Universidad de Valladolid. Prado de la Magdalena s/n. 47005 Valladolid Spain. E-mail: [email protected] (presenting author)
Partitioning and transmutation (P&T) of Minor Actinides (MAs) and Long-Lived
Fission Products (LLFP) arising out of the back-end of the fuel cycle would be one of the key-steps in any future sustainable nuclear fuel cycle. Pyrochemical separation methods would form a critical stage of P&T by recovering long-lived elements and thus reducing the environmental impact by the back-end of the fuel cycle. The reduction of the radiotoxicity and volume of the nuclear waste will be only possible if the most radiotoxic elements present in spent nuclear fuels, i.e. MAs, can be efficiently separated from the fission products (FPs), especially from lanthanides (Ln), and destroyed by transmutation.
This work is concerned with the electrochemistry of thulium a heavy lanthanide, as part of the program to look into the Ln series in chloride melts which has impacts for fuel reprocessing. The study has been carried out with a solution of TmCl3 using inert (W) and reactive (Al) electrodes.
On an inert electrode, Tm(III) ions are reduced to metallic thulium through two consecutive steps Tm(III)/Tm(II), Tm(II)/Tm(0).Consequently, corrosion of Tm metal in the molten chloride media is expected in the presence of Tm(III) according to the reaction: 2Tm(III) + Tm(0) ↔ 3Tm(II)
This corrosion reaction could be responsible for a low current yield in the electrolysis and a low stability of the deposits.
When and Al wire was used as working electrode, the electrochemical signals of Tm(III) were observed at more positive potentials values than those at the inert electrode. This potential shift was due to a lowering of the activity of Tm in the metal phase due to the formation of intermetallic compounds. The stabilization of Tm metal in the Al phase by alloy formation avoids its corrosion in the presence of Tm(III). Therefore, for the extraction of thulium from molten chlorides, the use of a reactive electrode made of aluminium leading to different Al-Tm alloys seems to be a pertinent route.
Tm-Al alloy films were obtained using potentiostatic electrolysis, and the samples were characterized by X-ray diffraction analysis and scanning electron microscopy.
On the other hand, using an YSZME, it was possible to follow the pO2- during the potentiometric titration of Tm(III) solutions by O2- ions. The experimental curves pointed out the formation of solid oxychloride TmOCl and oxide Tm2O3, which were identified by XRD analysis of the obtained solids. The corresponding solubility products, pks(TmOCl) = 8.0 ± 0.3 and pks(Tm2O3) = 18.8± 0.7, were determined by mathematical modelling of the experimental curves.
Experimental solubilization tests of solid Tm2O3 and TmOCl carried out by bubbling gaseous HCl through the melt were carried out, showing efficiencies close to 100%.
Acknowledgements.- The authors thank the Consejería de Educación y Cultura de la Junta de Castilla y León (project VA029A07) for the financing of this work.

C2-110 COMUNICACIONES EN CARTEL
148
ASSESSMENT OF CONTAMINATION BY ARSENIC AND OTHER HEAVY METALS IN SOILS FROM AN ABANDONED MINE AND ESTIMATION OF METAL BIOAVAILABILITY BY SEQUENTIAL EXTRACTION METHODS
D. García Casillas, S. García Salgado, M.A. Quijano Nieto,
M.M. Bonilla Simón Dpto. Ingeniería Civil: Tecnología Hidráulica y Energética, E.U.I.T. Obras Públicas,
Univ. Politécnica de Madrid, Madrid. E-mail: [email protected]
Mining activities are one of the most important sources of toxic element contamination in the surface environment, and their effects can persist in abandoned mines. Soil organisms and dynamic processes are affected by many pollutants, particularly toxic and persistent elements such as heavy metals. However, the ecotoxicity and mobility of these metals in the environment depend strongly on their specific chemical forms or method of binding. For this reason, partitioning methods are necessary to assess the mobility rate of heavy metals in the soil-plant system, for a better evaluation of the environmental impact of a contamination event [1].
Abandoned metal mines in Sierra de Guadarrama, Madrid (Spain), are often located in areas of high ecological value. The area studied corresponds to the “Mina Mónica” surroundings, located at the Northeastern part of Bustarviejo (Madrid). This is one of the mines that exist in the North part of Madrid, which was exploited between 17th and 20th centuries, for arsenic (arsenopyrite, FeAsS) and silver (matildite, AgBiS2) extraction.
This work focused on the assessment of contamination by arsenic and other heavy metals in soils from the mining site “Mónica”. This study is a part of a bigger one, whose aim is the study of the relationship between the plant biodiversity and the bioavailability of arsenic and other heavy metals in an abandoned mine environment [2]. The trace and major elements selected were: Al, As, Ca, Cd, Cr, Cu, Fe, Mg, Mn, Ni, Pb and Zn. Total determinations were carried out by ICP-AES, using rhodium as internal standard. The efficiency of different digestion methods, including alkaline melting and microwave acid digestion with nitric acid or mixtures of nitric and hydrochloric acids, was evaluated on three soil certified reference materials (NCS DC 73319; 73323 and 73324). The results showed that microwave digestion with aqua regia was suitable to carry out the solubilisation of most elements studied, so this method was applied to soil samples. On the other hand, the modified BCR sequential extraction procedure was used to determine the relative distribution, in the soils samples, of metals in different chemical forms and their correlations with the total content in mining areas [3]. This method allowed us to assess the metals partitioning along the fractions: exchangeable (soluble species, associated with carbonates), reducible (Fe and Mn oxyhydroxides), oxidizable (organic matter and sulphides) and residual. This study was complemented with particle size, pH and organic matter analysis. Acknowledgements: The authors would like to thank Universidad Politécnica de Madrid (project 188/Q065815-146) and Ministerio de Educación y Ciencia of Spain (project CTM2007-66432) for financial support. [1] D.C. Adriano. Trace elements in terrestrial environments (2nd ed.). Springer, Berlin
Heidelberg New York, 2001, p. 867. [2] J. Pastor, S. García Salgado, A.J. Hernández, M.A. Quijano, M.M. Bonilla, Pastos, clave
en la gestión de los territorios: Integrando disciplinas (2008) 91-97. [3] C.R.M. Rao, A. Sahuquillo, J.F. Lopez Sanchez, Water, Air, Soil Pollut. 189 (2008) 291-
333.

COMUNICACIONES EN CARTEL C2-111
149
ANTHROPOGENIC POLLUTION ASSESMENT IN THE GUADALQUIVIR RIVER ESTUARY USING INORGANIC NUTRIENTS AND CARBON SPECIES AS
INDICATORS
J.A. López-López, M. García-Vargas, C. Moreno
Dpto. Química Analítica, Univ. de Cádiz, Puerto Real (Cádiz) E-mail: [email protected]
Pollution control of natural water resources has become a necessity since this
precious good presents economical, environmental and health implications. Besides, since water pollution levels are related with human land uses (industrial, urban, and agricultural activities) have to be taken into account.
The Guadalquivir River supposes the biggest source of freshwater in the
Spanish Atlantic basin. In particular, alongside its estuary -115 Km long- different human activities have been established. The upper estuary is characterized by the city of Seville (700,000 inhabitants) and its outskirts as source of urban pollutants. Secondly, an agricultural area with cereals as main crop can be found. Finally, until its mouth, the river flows beside the Doñana National Park and is controlled by the tide effect.
Inorganic nutrients (NO3
-, NH4+, NO2
-, and PO43-), total dissolved nitrogen
(TDN), dissolved carbon species (total (TDC), inorganic (DIC), and organic carbon (DOC)), pH, dissolved oxygen (DO), and total (TSS) as well as volatile suspended solids (VSS) concentrations were determined along the Guadalquivir estuary. In order to study their spatial-temporal behavior, samples of surface water were collected for a two-year period (2007-2008) during both wet and dry seasons. Spatial distribution of the different variables offers information of their characteristic area of influence. However, better correlations may be obtained employing chemometric tools. With this in mind, the use of two-way ANOVA was useful to determine significant differences among the different sampling campaigns, especially in the case of total suspended solids (TSS). In addition, Principal Components (PCA) and Cluster Analyses (CA) were performed to establish relationships among the sampling station and variables.
Summarizing, on the one hand, some pollutants can be clearly classified within
three groups: the first one is constituted by nitrogen species and volatile suspended matter, as urban pollution; the second one, related to the agricultural area is formed by total suspended solids and DOC; while the latter corresponds with the mouth of the river as well as variables such as pH, DO, TDC, or DIC. On the other hand, PO4
3- and TSS for 2008 sampling campaigns show an intermediate behavior as a result of urban and agricultural origin.
This work has been supported by the Andalusian Consejería de Innovación,
Ciencia y Empresa (PAI05-RNM-0117.18INPR0437) and the Spanish Ministerio de Educación y Ciencia (CTM2007-64678/TECNO).

C2-112 COMUNICACIONES EN CARTEL
150
SELECTIVE SEPARATION OF ORGANIC AND INORGANIC SPECIES OF
COPPER IN SEAWATER BY SOLVENT EXTRACTION
C. Vergel Rodríguez, C. Mendiguchía, M. García-Vargas, C. Moreno
Dpto. Química Analítica, Univ. de Cádiz, Puerto Real (Cádiz). E-mail: [email protected]
Speciation studies has became of major importance in environmental sciences.
In aquatic ecosystems, dissolved heavy metals may exist in different chemical forms. Thus, in addition to free hydrated ions, heavy metals may form complexes with both organic and inorganic ligands. Each species plays a different role in the ecosystem and may present different toxicity, which should to be related to the different biological availability of the species.
To quantify the concentration of the different species of any heavy metal in
natural waters several analytical procedures may be applied, mostly involving the use of an electroanalytical device or an advanced separation method followed by another analytical technique. Among these separation methods, liquid-liquid extraction may be used to perform the extraction and, if needed, back-extraction processes.
When using liquid-liquid extraction, we need to ensure the inalterability of the
sample during the extraction process and it is especially important to work at natural pH values. This may be achieved by using two types of extractants: acidic and solvating.
In the present work, we have studied different extraction systems to perform
the selective separation of the different fractions of copper present in seawater at natural pH. They were based on the use of a solvating reagent, tri n-butyl phosphate (TBP) and an acidic reagent, 1,2-cyclohexanedione bis-benzoylhydrazone (1,2-CHBBH), which required the addition of a buffer solution to the samples.
As a part of the optimization process, we studied the influence of different
variables, such as the type of organic solvent, the addition of modifiers to the organic solvent, reagent concentrations, sample pH and extraction time.
Under selected conditions, we also studied, on the one hand, the influence of
both organic and inorganic ligands present in seawater on the efficiency of the extraction process. Thus, we studied the effect of the major anions species present in seawater (Cl-, HCO3
-, SO42-, Br-, …) and the addition of humic substances.
On the other hand, we have studied the chemical conditions allowing the back-
extraction of copper species previously extracted from seawater.
This work has been supported by the Spanish Ministerio de Educación y Ciencia (CTM2007-64678/TECNO).

COMUNICACIONES EN CARTEL C2-113
151
FUNCTIONAL SPECIATION OF METAL ASSOCIATIONS TO ENVIRONMENTAL
MICROPARTICLES, NANOCOLLOIDS AND MACROMOLECULES BY ASYMMETRICAL-FIFFF COUPLED TO ICP-MS.
E. Bolea, F. Laborda, J.R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS)
Institute of Environmental Sciences (IUCA) University of Zaragoza
C/Pedro Cerbuna 12, 50009 ZARAGOZA. Spain. E-mail: [email protected]
A methodological approach for the characterization of environmental
microparticles, nanocolloids and macromolecules by Asymmetrical Flow Field Flow Fractionation (AsFlFFF) is presented. The procedure is optimized to minimize the possible alteration of the original size distribution and metal associations of the species characterized. Prior to separation by AsFlFFF, samples are subjected to two pre-fractionation procedures: gravitational settling of the solid suspension, which removes particles larger than 50 µm, and a centrifugation of the settled sample to remove particles larger than 1 µm.
In the settled sample, those microparticles larger than 1 µm are eluted in steric
mode in the AsFlFFF, whereas colloids and macromolecules coeluted in normal mode. Both pre-fractionation procedures are used to provide information on microparticles. The centrifuged sample allows the proper characterization of both colloids and macromolecules applying different operational conditions in the AsFlFFF system. The methodology proposed has been applied to compost leachates.
The coupling of As-FlFFF with ICP-MS ensures a detection capability in
accordance to the concentrations expected. Compost leachates show associations of heavy metals with macromolecules (in the range of a few kDa), colloids (20-60 nm) and microparticles (5-20 µm).
Finally, an on-line isotopic dilution (ID) technique coupled with AsFlFFF-ICP-MS
has been applied for the determination of these metallic associations for some elements (copper and lead).
This work has been sponsored by the Spanish Ministry of Science (project
2006-00894).

C2-114 COMUNICACIONES EN CARTEL
152
SOLID PHASE MICROEXTRACTION COUPLED TO REVERSE PHASE-HPLC IN
THE CHARACTERIZATION OF NATURAL ORGANIC MATTER (NOM)
Elena Peña, A. López Molinero, E. Irisarri, I. Tello, J. Jiménez, M.A. Gómez, J.R.Castillo
Analytical Spectroscopy and Sensors Group (GEAS)
Institute of Environmental Sciences (IUCA) University of Zaragoza, C/Pedro Cerbuna 12, 50009 ZARAGOZA. Spain
Email: [email protected]
Common characterization of the main component of Natural Organic Matter (Humic Substances) are carried out by Size Exclusion-HPLC techniques, depending on the size of the compounds. However studies using Reversed Phase separation, depending on the polarity of the compounds could give a useful complementary information.
We will present the results of a study developped by the technique of Solid
Phase Microextraction (SPME), as a rapid method for extraction,and preconcentration coupled with HPLC separation using Reverse Phase and UV-VIS Diode Array detection.
The application of SPME requires a previous and significant optimization
process of the large number of variables affecting the extraction. It was implemented by a Placket-Burman design for 7 factors at 2 levels, where its influence is considered in two stages: i) extraction: fiber type, time, Temperature, stirring time , and salting-out effect. And ii) re-extraction: solvent and time.
The separation of the extracts was carried out by HPLC with Reverse Phase C-
18 and mobile phase Acetonitrile / water gradient. The total development is achieved within 22 min. Cleaning and conditioning steps are necessary. The response to the extraction of samples is evaluated based on two chromatographic criteria: number of peaks and concentration recovery.
The results show the highest influence of fiber type and the solvent desorption.
So that the best conditions were obtained with PDMS fiber and Acetonitrile/water/80/20%v/v.The working range was 0.01-100 mg / l of Humic Substances
The method has been applied to the characterization of Humic Substances in
samples of different origins: leonardite and natural waters, and to characterize the Humic Substances decomposition by using different oxidation processes.
We thank the financial support of the Spanish Department of Science. Project
CTQ 2006-00894 BQU

COMUNICACIONES EN CARTEL C2-115
153
ELECTROCHEMICAL SPECIATION OF ARSENIC IN NATURAL WATERS USING
HEMATITE MODIFIED CARBON PASTE ELECTRODES
S. Hamida, G. Cepriá, F. Laborda, J.R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS) Institute of Environmental Sciences(IUCA)
University of Zaragoza, C/ Pedro Cerbuna 12, 50009 Zaragoza.Spain E-mail: [email protected]
Arsenic is a target analyte in all environmental samples due to its high toxicity
and ubiquitous character. The total content of As in a sample does not provide much information since the toxicity and mobility of As(V) and(III) is very different. The former is not very mobile and stays sorbed on organic mater and on iron oxides and it is not as toxic as As(III) which is a mobile physicochemical form. This implies that the methods developed to analyze the As content must be able to distinguish between these forms. It has to be taken into account that the ratio As(V):As(III) might change depending on the conditions of the environment, namely pH redox potential and presence of oxides of iron or manganese.
From this point of view, electroanalytical techniques have offered valuable solutions. The main drawback of these methods is the requirement of a previous reducing agent of As(V) to As(III) because As(V) cannot be reduced directly on the electrode unless the pH is strongly acidic and the reduction potential extremely negative and the need of gold electrodes which can be fouled by the organic mater present in the environmental.
The procedure developed in our group is based in the reduction of As(V) and As(III) to Asº on the surface of a carbon paste electrode modified with hematite. As(V) is reduced on this electrode by the Fe(II) produced on its surface and subsequently deposited on the electrode as Asº. The selective determination of As(V) and As(III) is possible selecting adequately the pH [1]. The procedure exhibits a broad dynamic linear range, up to 50 ppb with a limit of detention below the requirements of the WHO.
The influence of several inorganic interferences was checked. It turned out that Cu was a decisive interference at 200 ppb but Se, Bi, Sb and Hg did not interfere. Humic acid did not cause any problem if the pH is set to 0 (HCl 1 M) for the deter-mination of total arsenic and to 2 (HCl 0.01 M) for the determination of As(III).
A reference material, TMDA-54.4 lot1205 was used to validate the method. The certified value of total was 43.6±3.5 (mean ± standard deviation) ppb and the obtained value in our lab was 49.24±2.3 ppb (mean of three different determinations ± standard deviation). The concentration of As(III) was 50.0 ±2.2 ppb (mean of three different determinations ± standard deviation). This value was expected to be similar to the total content of As since the natural water was spiked with As(III) to yield the certified value.
This method was applied to the tap water of the city of Zaragoza, and the obtained values are below the limits recommended by the WHO: Total As: 1.5 ±0.5 ppb and As(III):0.4±0.1 ppb (mean of two separate determinations ± standard deviation) [1] G. Cepriá, S. Hamida, F. Laborda, J.R. Castillo, J. Appl. Electrochem. 37 (2007)
1171-1176. We thank the financial support of the project CTQ 2006-00894BQU of the Spanish Science Department

C2-116 COMUNICACIONES EN CARTEL
154
ESTUDIO DE INTERRELACIÓN ENTRE CONTAMINANTES INORGÁNICOS TOTALES Y SOLUBLES EN PARTÍCULAS ATMOSFÉRICAS EN SUSPENSIÓN
Y EN EL AGUA DE LLUVIA
R. Montoya, A.J. Fernández, D. Trigo, M. Ternero
Departamento de Química Analítica. Facultad de Química. Universidad de Sevilla. C/ Prof. García Glez 1. Campus de Reina Mercedes. 41012 –Sevilla.
Email: [email protected]
El estudio de la composición química de la materia particulada atmosférica, en particular del contenido total y soluble de metales pesados, es de gran interés por su efecto nocivo e impacto sobre los sistemas biológicos y medio ambiente, en general. A su vez, la composición química del agua de lluvia presenta una alta variabilidad debido a que es el resultado de un proceso complejo donde intervienen factores naturales y antropogénicos y procesos de transformación química y de transporte y deposición de partículas atmosféricas. Dentro de este contexto, como continuación de trabajos realizados con anterioridad y de acuerdo con la bibliografía existente [1,2] se ha desarrollado, junto con el Dpto de Química Analítica de la Universidad de Cádiz, un Proyecto de Investigación subvencionado por la Consejería de Innovación, Ciencia y Empresa que ha contemplado entre sus objetivos “Evaluar cualitativa y cuantitativamente la entrada (o “inputs”) de contaminantes en el ecosistema fluvial a través de la atmósfera.
En la presente comunicación se presentan los resultados de un año de muestreo, tanto de filtros de partículas en suspensión totales (TSP), como de agua de lluvia. El muestreo de agua de lluvia se ha llevado a cabo con muestreadores de precipitaciones atmosféricas (“wet only”), y el de partículas atmosféricas se ha realizado con muestreadores de alto volumen de partículas en suspensión totales.
Los filtros de TSP han sido procesados en el laboratorio de manera que se han analizado tanto los contenidos totales en metales (digestión ácida), como la parte soluble de estas partículas (lixiviación en agua), analizando en esta última fracción los metales traza solubles, así como los compuestos iónicos mayoritarios. En el agua de lluvia han sido determinados los mismos elementos y compuestos que en dicha fracción soluble.
Para la determinación de metales pesados ha sido utilizada la espectrometría de emisión por plasma con acoplamiento inductivo con detección de espectrometría de masas (ICP-MS), y la determinación de compuestos iónicos se ha realizado mediante cromatografía iónica con una columna de alta resolución, detector de conductividad y supresión química.
Se ha evaluado, por un lado, el grado de eliminación de contaminantes a través de la deposición húmeda (agua de lluvia), estudiando las diferencias entre las concentraciones de analito en los filtros de TSP recogidos justo antes y después de cada uno de los episodios de lluvia estudiados; por otro lado, se ha estudiado la interrelación existente entre estos niveles atmosféricos y los niveles de las correspondientes muestras de agua de lluvia recogidas en dichos episodios de precipitación.
[1] D. Trigo, M. Ternero, A.J. Fernández y R. Bouza. Estudio de los aportes de metales
pesados, nutrientes y otros aniones en las precipitaciones de la ciudad de Sevilla como punto de entrada de contaminantes al ciclo integral del agua. VI Simposio sobre el agua en Andalucía. 2006. Pág 1143-1150. ISBN: 84-7840-579-8.
[2] Vázquez, A., Costoya, M., Peña, R.M., García, S., Herrero, C., 2003. A rainwater quality monitoring network: a preliminary study of the composition of rainwater in Galicia (NW Spain). Chemosphere 51, 375-386

COMUNICACIONES EN CARTEL C2-117
155
HERRAMIENTAS DE ASEGURAMIENTO DE CALIDAD DE DATOS ANALÍTICOS PARA LA MONITORIZACIÓN DEL PERFIL DE ELEMENTOS EN AEROSOLES
ATMOSFÉRICOS MEDIANTE ICP-MS
M.R. Palomo, C. Marín, L. Calvo, E. Pinilla
Dpto. Química Analítica, Univ. Extremadura Campus universitario, 06071 Badajoz
E-mail: [email protected] El perfil de elementos constituye una valiosa fuente de información para la
asignación de origen de los aerosoles atmosféricos. Por otra parte, la evaluación de algunos elementos tóxicos como Pb, Cd, Cd, As o Ni en aerosoles, es una exigencia legal como parte de los programas rutinarios de evaluación de la calidad del aire (por ejemplo, en España están recogidas en los RR.DD. 1073/2002 y 812/2007).
La evaluación del perfil de elementos mayoritarios y elementos traza en
aerosoles ambientales mediante ICP-MS es un procedimiento analítico multietapa complejo que comprende la toma de muestras por aspiración del aire sobre filtros de fibra de cuarzo, seguida de análisis gravimétrico, digestión ácida, detección y determinación de un número variable de analitos que puede alcanzar los 65 elementos, en amplios intervalos de concentración. Para obtener información exacta y representativa en la aplicación de estas metodologías es esencial el aseguramiento de la calidad de los resultados, especialmente en aquéllas situaciones en que los niveles de concentración de diversos elementos traza se encuentran próximos a los límites de detección del método. En este trabajo se evalúan críticamente las herramientas de control de resultados que se emplean rutinariamente en la red de vigilancia atmosférica de Extremadura para el control del método de análisis de aerosoles, que incluyen evaluación de blancos de filtros, recuperación de materiales de referencia certificados, cálculo de incertidumbres y ensayos interlaboratorios. Se presentan los resultados obtenidos durante los años 2007 y 2008. Agradecimientos
Proyecto financiado por la red de vigilancia atmosférica de Extremadura (Junta de Extremadura, Consejería de Industria, Energía y Medio Ambiente)

C2-118 COMUNICACIONES EN CARTEL
156
SEASONAL VARIATION IN PARTICLE SIZE DISTRIBUTION AND METAL
CONTENT IN THE AEROSOL OF AN URBAN ENVIRONMENT
M. González, M.A. Barrero, L. Cantón
Grupo de Ingeniería Química, Dpto. Quimica Aplicada, Univ. País Vasco, San Sebastián.E-mail: [email protected]
The solid phase of atmospheric aerosol is essentially composed of suspended
particles. This particulate matter (PM) shows a wide range of particle size, morphology, composition, etc. Moreover, these properties are determined by its origin as well as transformation processes in the atmosphere. Therefore, changes in emission sources and atmospheric conditions cause variations over time. The study of certain key characteristics of PM reveals its seasonal variation and provides strong clues towards the identification of possible sources of particles in the studied area.
In urban environments, the analysis of particle size distribution is influenced by
traffic. Fine particles are usually related with engine combustion processes with coarser fractions of PM arising from soil erosion and coagulation of smaller particles from tailpipes. Many environmental pollution studies [1] have identified various toxic metals that can be used as tracers of pollution sources, so their analysis on PM allows the possible attribution of some particle origins. In addition, certain morphologic characteristics of PM are useful for source identification. For example, spherical particles are normally associated with combustion processes.
The main objective of this work has been the description of the seasonal
variation of particulate matter in an urban environment of the Basque Country (Hernani) in terms of particle size distribution, metal composition and morphology.
For this purpose, 24-hour PM10 samples were collected by every week for one
year. In addition, a cascade impactor was used for determining the particle size distribution. The metal composition in the collected PM samples was analysed by atomic absorption spectroscopy and five metals were determined: Cu, Fe, Mn, Pb and Zn. The morphology of PM was examined by scanning electron microscopy.
0%
20%
40%
60%
80%
100%
Summer Winter
>10 µm
4,9-10 µm
2,7-4,9 µm
1,3-2,7 µm
0,61-1,3 µm
<0,61 µm
Figure 1. Particle size distribution determined in summer and winter seasons in an urban atmosphere.
Seasonal variations were detected both in particle size distribution (Figure 1)
and in trace metal content. In summer, coarse particles composed about 60% of PM with the fine fraction making up 21%, while in winter the amount of coarse particles decreased to 36% and the amount of fine particles increased to 43%.
[1] Amato et al., 2009; Querol et al., 2007.

COMUNICACIONES EN CARTEL C2-119
157
GEOCHEMICAL BACKGROUND OF ANTIMONY AND THALLIUM IN THE
REGION OF MURCIA (SE, SPAIN) AND POSSIBLE RELATIONSHIP BETWEEN MINERALOGY AND SOIL PROPERTIES.
C. Pérez-Sirvent, M.J. Martínez-Sánchez, J. Molina and M.L. García-Lorenzo
Dpto. Química Agrícola, Geología y Edafología, Universidad de Murcia, Murcia
E-mail : [email protected].
Background values for trace elements in soils can act as a true reference level
for estimating the extent of soil pollution with these elements. For some samples, the soils have been influenced by the erosion of surrounding mountains, which belong to different geologic units and, as a consequence, background and baseline value determination is more difficult. The background value is highly dependent on the mineralogical composition of the parent material and on the weathering processes that have led to the formation of the soil clay, as well as is affected by the organic matter content. The socioeconomic relevance of the subject explains why, in recent years, many governments have promoted research to define geochemical background and baseline levels to serve as a basis for legislation to diagnose, prevent and reduce soil contamination. Moreover, the purpose of this paper was to study the possible relationship between soil properties, mineralogical composition and metal content in soil samples.
This communication seeks to establish the geochemical background for thallium
and antimony in the Region of Murcia [1]. The possible relationship between soil properties and target metals concentration has been studied. In the present study, background concentrations were established by analysing a large number of soil samples considered unaffected, or at least minimally affected, by human activities. Samples were analysed for antimony content by atomic fluorescence spectrometry (AFS) and by inductively coupled plasma spectrometry (ICP-MS) for thallium content. Other soil characteristics such as electrical conductivity (EC), organic matter (OM), pH, soluble salts, granulometry and calcium carbonate content were also measured to determine their influence on trace element content. Four different areas were established according to geological composition, and the results suggested that thallium and antimony were positively correlated with calcium carbonate content and pH. Moreover, metal concentration was positively correlated with fine soil fraction. Four different groups were established based on significant relationship with the mineralogical composition in soil samples, using discriminant analysis. The results showed that the concentration of antimony and thallium is correlated with the mineralogical composition. Thus, the metal level was found to be positively correlated with the phylosilicate content. On the other hand, it was found that both metals were positively correlated. Background levels for thallium and antimony, showed as the median, were 0.35 mg kg−1 and 1.15 mg kg−1, respectively. [1] M.J. Martínez-Sánchez, C. Pérez-Sirvent (2008) Niveles de fondo y niveles
genéricos de referencia de metales pesados en suelos de la Región de Murcia. Consejería de Desarrollo Sostenible y Ordenación del Territorio, Comunidad Autónoma de la Región de Murcia

C2-120 COMUNICACIONES EN CARTEL
158
ESPECIACIÓN DE ARSÉNICO EN MATERIAL PARTICULADO ATMOSFÉRICO (PM10 Y PM2.5) EN FONDO URBANO CON INFLUENCIA INDUSTRIAL
Y. González a , A. Sánchez de la Campaa , J. de la Rosa a ,
V. Oliveira b, D. Sánchez-Rodas b
aDepartamento de Geología. Facultad de Ciencias Experimentales. Universidad de Huelva. Campus de El Carmen. 21071-Huelva bDepartamento de Química y Ciencia de los Materiales. Facultad de Ciencias Experimentales. Campus de El Carmen. 21071-Huelva. E-mail: [email protected]
El material particulado atmosférico representa una amenaza para la salud humana a través de la respiración. El tamaño de particular afecta a la capacidad de penetración en el organismo. La bioacumulación de metales pesados puede ejercer una influencia negativa en los procesos bioquímicos de los seres vivos. La presencia antropogénica de arsénico en material particulado se debe a actividad industrial relacionada con la fundición y refino de metales no ferrosos y emisiones de vehículos. El valor propuesto por la Comisión Europea para arsénico en partículas con diámetro inferior a 10 µm (PM10) es de 6 ng m-3 (EC Directiva 2004).
Se seleccionaron para el estudio de especiación 34 muestras (17 de PM10 y 17
de PM2.5) tomadas en muestreos sincrónico y con mayor contenido en arsénico de un total de 76 muestras repartidas a lo largo del año 2006, utilizando la estación de medida situada en el Campus de El Carmen (ciudad de Huelva), perteneciente a la Red de Vigilancia y Control de la Contaminación Atmosférica de la Junta de Andalucía. Cada muestra corresponde a un muestro de 24 h. El interés de la estación del Campus de El Carmen reside en representar un fondo urbano influenciado por la actividad industrial del Polo Químico de Huelva.
Se caracterizó la composición química referida a la concentración de hasta 49
elementos mediante ICP-MS, seleccionándose aquellos relacionados con la actividad industrial (As, Se, Ti, V, Ni, Cu, Zn, Cd, La, Pb, Bi). Para los estudios de especiación de arsénico en PM10 y PM2.5, se procedió a la extracción de las especies de los filtros empleando un procedimiento basado en el empleo de disoluciones de clorhidrato de hidroxilamina y radiación de microondas. Los extractos fueron analizados mediante acoplamiento instrumental HPLC-HG-AFS, según metodología analítica previamente puesta a punto para este tipo de muestras [1].
Los resultados mostraron la presencia de las especies de As(V) y en menor
medida de As(III), concentradas preferentemente en la fracción más fina (PM2.5 frente a PM10), estando la media de la concentración de As(V) por encima del nivel guía de 6 ng m-3.Se observó también la correlación entre niveles elevados de As y vientos con componentes adecuadas para el transporte del material particulado desde el Polo Químico al a ciudad de Huelva. [1] A. Sánchez de la Campa, J. de la Rosa, D. Sánchez-Rodas, V. Oliveira, X.
Querol, A. Alastuey, J.L Gómez-Ariza, Atmospheric Environment 42 (2008) 6487-6495

COMUNICACIONES EN CARTEL C2-121
159
CARACTERIZACION ANALITICA DEL IMPACTO AMBIENTAL DE LA
APLICACIÓN DE LOS NEUMÁTICOS FUERA DE USO (NFUS) EN OBRA CIVIL
I. Seijo, A.J. Fernández, M. Ternero
Departamento de Química Analítica. Facultad de Química. Universidad de Sevilla. c/Profesor García González, s/n. 41012 Sevilla, España. Email: [email protected]
La masiva fabricación de neumáticos y las dificultades para hacerlos desaparecer una vez usados, constituye uno de los más graves problemas medioambientales de los últimos años en todo el mundo. Un neumático necesita grandes cantidades de energía para ser fabricado y también provoca, si no es convenientemente reciclado, contaminación ambiental al formar parte, generalmente, de vertederos incontrolados. Las posibilidades de reciclaje de los NFUs, es decir el aprovechamiento de sus componentes materiales para otros usos distintos de la valorización energética, han experimentado en los últimos tiempos un importante aumento.
Los objetivos del Proyecto se centran en la caracterización analítica de los NFUs con miras a evaluar el impacto de su posible utilización como relleno de construcción en obra civil [2]. Para dicha evaluación es necesario medir las propiedades físicas de los NFUs troceados y las propiedades químicas del lixiviado que se produzca en su utilización al verse afectado por las condiciones climáticas.
Las muestras de NFUs serán obtenidas a través de plantas troceadoras, diseñadas para la trituración de este tipo de residuo, que se encuentran ubicadas generalmente en vertederos. En base a los antecedentes bibliográficos [1], se evaluará el impacto medioambiental de los NFUs a través del análisis de lixiviados que se obtendrán según un procedimiento basado en la norma europea EN 12457.
Desde el punto de vista medioambiental, se evaluarán los contenidos de elementos que previsiblemente pueden formar parte de los lixiviados. Así se determinarán los siguientes elementos:
• Aniones y cationes mayoritarios por cromatografía iónica • Metales pesados: Al, As, Ba, Cd, Cr, cu, Fe, Pd, Mn, Hg, Ni, Se, Ag, Zn, etc.
por espectrometría de emisión por plasma ICP-OES [2]. • Fenoles: 4-tert-Octylphenol, 4-n-Nonylphenol, iso-nonylphenol, Bisphenol-F,
Bisphenol-A, entre otros, que se determinaran por cromatografía de gases con detección por espectrometría de masas. Asimismo se determinara el contenido en fenoles totales, por métodos de espectrofotometría de absorción molecular UV-VIS.
• Hidrocarburos aromáticos poli-cíclicos, por cromatografía de gases con detección por espectrometría de masas.
Los resultados analíticos permiten evaluar que impacto medioambiental tendría la reutilización de estos residuos utilizados como relleno de construcción y si los niveles medidos en os lixiviados serian admisibles de acuerdo con las distintas normativas aplicables
[1] A Mohammad, F. Azizian, Peter O. Nelson, Pugazhendhi Thayumanavan, Kenneth J. Williamson, Environmental impact of highway construction and repair materials on surface and ground waters: Case study: crumb rubber asphalt concrete. Waste Management 23 (2003), 719–728.
[2] Yoon, S., Prezzi, M., Siddiki, N. Z., & Kim, B. Construction of a test embankment using a sand–tire shred mixture as fill material. Waste Management, 26(9) (2006), 1033-1044.

C2-122 COMUNICACIONES EN CARTEL
160
USE OF THE HSV COLOR SPACE AS A QUANTITATIVE PARAMETER FOR
BITONAL OPTICAL SENSORS
I. de Orbe-Payá1, M.M. Erenas1, K. Cantrell2, L.F. Capitán-Vallvey1
1ECsens, Department of Analytical Chemistry, Campus Fuentenueva, University of Granada, E-18071 Granada, Spain. 2Department of Chemistry, The University of
Portland, Portland, OR 97203, USA
We are interested in the use of color for chemical analysis. Colors are qualities that are present or represented in visual experiences [1] and could be objectify using a number of methods or color spaces that specify color in terms of three primary colors, similarly to human color perception. Signals coming from different imaging devices have been used for quantitative purposes; thus, different types of scanners [2], CCD arrays, video cameras [3], digital photographic cameras [4] and digital color analyzers. The most used color space by far is the RGB and if the measurement is selective enough the calibration function is obtained by means the most sensitive color coordinate, red(R), green(G) and blue(B), through a curvilinear or linearized function.
The hue or H component of the HSV color space has been studied as a quantitative analytical parameter for bitonal optical sensors. The main feature of this color space is the representation of the cognitive color information in a single parameter, the hue component H. The robust nature of this parameter affords it superior precision for the measurement of sensor materials which change colors with the speciation of some indicator molecule. This parameter has been compared to RGB intensity and RGB absorbance along with differences and ratios of both intensity and absorbance, and has been demonstrated to be 2 to 3 times superior. The H value maintains this superior precision with variations in indicator concentration, membrane thickness, detector spectral responsivity, and illumination. Because this parameter is stable, simple to calculate, easily obtained from commercial devices such as digital cameras and scanners, continuous over the entire color gamut, and bound between values of 0 and 1 it shows great promise for use in a variety of sensing applications including imaging, automated analysis, pharmaceutical sensing, lab-on-a-chip devices, and quality control applications.
We have studied a one-shot sensor for K(I) [5] based in ionophore-chromoionophore chemistry using a scanner as imaging technique. In order to check the independence of this parameter to changes in path length and chomoionphore concentration we prepare membranes of different thickness and with different concentration. The coefficient of variation obtained was about 0.5% in both cases.
We also performed a precision study, intra and intermembrane, and the results coefficient of variation obtained were above 0.3%. Finally we calibrated the sensor (see figure) with 11 potassium concentration obtaining five values from each solution. The calibration is similar in shape as obtained with other optical technique but the precision obtained is much higher.
[1] Maund, Barry. Color. 2006. Metaphysics Research Lab, CSLI, Stanford University .
Stanford Encyclopedie of Philosophy. [2] Lapresta-Fernandez, A.; Capitan-Vallvey, Luis Fermin Sens.Actuators B 2008, B134,
694-701. [3] Tohda, Koji; Gratzl, Miklos Anal.Sci. 2006, 22, 383-88. [4] Apyari, V. V.; Dmitrienko, S. G. J.Anal.Chem. 2008, 63, 530-37. [5] Capitan-Vallvey, Luis Fermin; Fernandez Ramos, M. D.; Al Natsheh, Muneer
Sens.Actuators B 2003, 88, 217-22.
0.5
0.6
0.7
0.8
-8 -6 -4 -2 0Log[K]
H

COMUNICACIONES EN CARTEL C2-123
161
MÉTODOS DE ANÁLISIS RÁPIDOS BASADOS EN TECNOLOGÍAS DE
INFRARROJO CERCANO (NIRS) EN EL CONTROL ANALÍTICO DE PROCESOS DE DEPURACIÓN DE AGUAS RESIDUALES URBANAS
V. Fernández*, R. Bouza*, A.J. Fernández*, M. Ternero*, J.C. Mohino**
*Departamento de Química Analítica. Facultad de Química. Universidad de Sevilla.
C/Profesor García González, 1. 41012 Sevilla, España. E-mail: [email protected] **Soluciones Integrales de Laboratorio, S.L. (TECNILAB). C/Valencia, 978. 19170 –
El Casar (Guadalajara). España La Directiva de Aguas Residuales Urbanas y la Directiva Marco de Aguas son el
referente para un adecuado control analítico de la calidad de las aguas y una mejora de las instalaciones de saneamiento en toda la Unión Europea. Uno de los principales problemas a los que se enfrentan los responsables de las Estaciones Depuradoras de Aguas Residuales Urbanas (EDARs) es la demora en la obtención de los resultados analíticos de las distintas muestras tomadas en sus plantas de tratamiento de forma rutinaria, que va desde algunas horas en los mejores casos hasta cinco días en el caso de la demanda biológica de oxígeno. Para el óptimo control de una EDAR sería necesario disponer de método de análisis in situ, rápidos y económicos. Si bien esto es posible actualmente para determinados parámetros que aunque aportan información del estado de una estación de tratamiento de aguas tienen un elevado coste de inversión y un complejo mantenimiento, requiriendo personal cualificado para el mismo.
Las ondas electromagnéticas cuya longitud de onda se encuentra entre 700 y
2500 nm son las que pertenecen la denominada región del infrarrojo cercano NIR, siendo esta una región del espectro donde la materia orgánica y otros compuestos de interés desde el punto de vista de la contaminación de las aguas absorben una importante cantidad de luz debido principalmente a la absorción de energía vibración al de los enlaces, especialmente de hidrógeno con otros átomos. (OH, CH, NH, SH, PH).
Aunque han sido numerosas las aplicaciones industriales que con éxito se han
desarrollado mediante esta técnica, son muy pocas las investigaciones que se han realizado para su aplicación al control de procesos de aguas residuales [1,2], por lo que la actual línea de investigación presenta un indudable interés práctico ya que no sólo pretende la monitorización del sistema, sino la cuantificación analítica del mayor número de parámetros de control posible, permitiendo que con un solo equipo analítico, bajo coste y prácticamente nulo mantenimiento sea posible conocer los parámetros analíticos que requiere la gestión de una EDAR de forma instantánea.
Los objetivos del proyecto se centran por tanto en el desarrollo y validación de
metodologías analíticas, preferiblemente in situ, que permitan la obtención de los principales parámetros analíticos para el control de procesos de depuración de aguas residuales urbanas. [1] Dias, A.M.; Moita, I.; Alves, M.M.; Ferreira, E.C.; Páscoa, R.; Lopes, J.A.
Activated sludge process monitoring through in situ near-infrared spectral analysis. Water Sci. Technol. 2008; 57(10):1643-50.
[2] Stephens, A.B.; Walter, P.N. Near-infrared spectroscopy as a tool for real-time determination of BOD5 for single-source samples. Transactions of the ASAE, 2002, vol. 45, nº2, pp. 451-458.

C2-124 COMUNICACIONES EN CARTEL
162
STUDY OF NATIVE FLUORESCENCE OF PHENOLIC COMPOUNDS FOR MONITORING HPLC SEPARATIONS AND THEIR CHARACTERIZATION
S. Garmón-Lobato, D.M. López-Márquez, C. Sánchez-Fernández,
L. A. Berrueta, B. Gallo, F. Vicente, A. Arranz
Departamento de Química Analítica. Facultad de Farmacia, UPV/EHU, Paseo de la Universidad, 7. 01006 Vitoria. E-mail: [email protected]
UV-Vis detection has been traditionally the most applied technique for phenolics
characterization; however some phenolic compounds exhibit native fluorescence and almost everyone presents fluorescence under certain conditions.
Fluorimetric determination could report many benefits over UV-Vis detection, for instance, their sensibility can be greater in certain conditions; and even more interesting, the selectivity can hugely enhanced, because of the uniqueness of fluorescence process and optimizing specific λexc and λem.
In order to analyse the suitability of fluorimetric detection, several groups of phenolic standards (80 compounds), have been analysed employing an multipurpose chromatographic method, successfully applied before with UV-Vis detection. Two detectors in line have been used, the first one is a DAD which records spectra and chromatograms at 280 nm, 320 nm and 370 nm, and the second one is a spectrofluorimeter which in four successive runs, has registered four fluorescence chromatograms and excitation (at λem= 340 nm and 420 nm) and emission (at λexc=250 nm and 280 nm) on-line spectra for each compound. These wavelengths have been selected to comprise the fluorescence of all families studied, thus they are not the optimal ones for all of them.
Hidroxybenzoic acids show moderate native fluorescence at λem= 360 nm, with an instrumental sensitivity rather similar to the UV-Vis one but with presumably much better selectivity. Gentisic acid has a shifted fluorescent emission band at 450 nm. Excitation spectra correlate well with absorption bands as expected.
By other side, hidroxycinnamic acids show an emission band at 400-450 nm depending on their substitution. For example, the fluorimetric sensitivity of ferulic acid is about 50 times lower than using absorption at 320 nm. The only studied coumarin, the scopoletin has very strong emission at 450 nm, with an enhanced sensitivity in comparison with UV detection. The hydroquinone arbutin is fluorescent (λexc=280 nm, λem= 320 nm), being its sensitivity two times greater than UV-Vis.
As it has been reported, flavonoids have very little native fluorescence, HPLC polar mobile phases inhibit it although acetic acid use as modifier can enhance it as well as complexation with metal ions. Free hydroxylation at C3 of the chromane ring is very important for their native fluorescence. Thus, only flavanols and flavonols have important native fluorescent. Flavanols and procyanidins show an intense emission band at 323-328 nm which makes them excellent candidates for fluorimetric detection with an improved sensitivity. Among flavonols, only free quercetin aglycone has shown little fluorescence, its emission spectrum has two characteristic bands, being emission at 528 nm caused by the tautomer (ESPT).
On the other hand, acylated flavonoids like epigalocatechin-gallate and kaempferol-3-p-coumaroylglucoside show only emission bands corresponding to the acyl moiety. Interestingly, only free aglycone flavones have exhibited a little native fluorescence, polymetoxylated flavones have an emission band at ~440 nm.
To sum up, native fluorescence would allow a more selective and confident detection way for flavanols, coumarins, hydroquinones and phenolic acids, being a good alternative to UV-Vis absorption. Fluorescence for the other flavonoids is very low and in order to be suitable, actions for enhancing it must be taken like metal complexation. Acknowledgements: Authors thank to Universidad del País Vasco/Euskal Herriko Unibertsitatea for financial support (project nº 15978/2004) and S. Garmón thanks Gobierno Vasco/ Eusko Jaurlaritza for his Ph.D. Grant.

COMUNICACIONES EN CARTEL C2-125
163
DETERMINACIÓN DE METALES PESADOS EN CULTIVOS ORGANOPÓNICOS URBANOS Y PERIURBANOS PROCEDENTES DE CIUDAD DE LA HABANA
M. Melaniuk, K. Januszenicz, M. Liva, S.A. Veitia,
R. Muñoz-Olivas, C. Cámara
Dpto. Química Analítica, Fac. CC. Químicas, Univ. Complutense de Madrid, Madrid. E-mail: [email protected]
El término de cultivo organopónico hace referencia a aquellos cultivos basados en la producción de especies vegetales de ciclo corto - entre 3 y 4 meses - sobre tierras que pueden ser en principio aptas o no aptas, en cuyo caso se preparan los suelos por medios naturales. Por tanto, se emplean sustratos orgánicos en descomposición, así como biofertilizantes. Estos cultivos han tomado gran relevancia en determinadas zonas de Cuba, especialmente desde que se creó el Programa Nacional de Agricultura Urbana en Cuba en 1994 durante el llamado “Período Especial”.
La procedencia de los fertilizantes no aporta contaminación de metales a estos
cultivos; sin embargo, la proximidad a las ciudades con tráfico rodado, fábricas y otras contaminaciones relacionadas con el medio urbano, hacen necesario un control de algunos elementos para conocer la aptitud para su consumo humano. Por ello, se ha llevado a cabo el análisis de algunos elementos tóxicos y/o esenciales como son As, Cd, Cu, Hg, Mn, Pb, Sn, Zn para evaluar sus contenidos totales y la movilidad y disponibilidad de dichos elementos junto con su potencial toxicidad. Para ello, se han desarrollado diversos tratamientos de muestra teniendo como objetivos la rapidez de los mismos así como la obtención de elevados rendimientos de extracción del máximo número de elementos posible.
Con estos objetivos, se han estudiado métodos rápidos de digestión
(microondas) y de extracción (baño y sonda de ultrasonidos) en muestras de zanahorias y espinacas por ser dos de los vegetales más representativos y consumidos por la población cubana. Los análisis se han llevado a cabo por FAAS para el Mn, Zn y Cu y por ICP/MS para el As, Hg, Sn, Pb y Cd y han sido comparados con los resultados obtenidos en un ejercicio de inter-comparación realizado en Cuba quedando demostrada la validez de los métodos de tratamiento de muestra desarrollados y descritos en este trabajo. Asimismo, la validación de los métodos se llevó a cabo mediante el análisis de materiales de referencia certificados.

C2-126 COMUNICACIONES EN CARTEL
164
CARACTERIZACION Y EVALUACION DE POLIMEROS IMPRESOS
SINTETIZADOS CON OBJETO DE ESPECIAR EL TBT Y SUS PRINCIPALES PRODUCTOS DE DEGRADACION
M. Liva, R. Muñoz-Olivas, C. Cámara
Dpto. Química Analítica, Univ. Complutense de Madrid, Madrid.
E-mail: [email protected]
Las posibilidades analíticas que brindan los polímeros impresos tanto para el tratamiento de muestra como para la separación de compuestos son ampliamente conocidas. En el caso de las especies de estaño se han realizado ya varios trabajos que han probado la selectividad de los materiales sintetizados para esas especies.
En este trabajo se presenta el empleo de unos polímeros sintetizados mediante
la técnica de emulsión tomando como compuesto plantilla el tributilestaño (TBT), así como una mezcla del TBT con sus compuestos más importantes de degradación: dibutilestaño (DBT) y monobutilestaño (MBT). Se presenta un estudio de caracterización de estos materiales así como las posibilidades de separación de dichas especies empleando este polímero como fase estacionaria en cromatografía líquida. Como técnica de detección se ha utilizado el ICP-MS acoplado al HPLC. Para la caracterización superficial de estos polímeros se han empleado las técnicas SEM y BET, calculando los diámetros de las partículas obtenidas, así como la porosidad y otras propiedades características del análisis superficial. Se han evaluado las isotermas de adsorción con el objetivo de poder explicar las interacciones de estos materiales impresos con las diferentes especies de estaño. Por último, se presentan y discuten también los resultados obtenidos en su empleo como relleno de columnas cromatográficas.

COMUNICACIONES EN CARTEL C2-127
165
DETERMINACIÓN SIMULTANEA DE SULFONATOS DE ALQUILBENCENO LINEALES, NONILFENOLES Y DI(2-ETILHEXIL)FTALATO EN SUELOS
TRATADOS CON LODOS DE DEPURADORAS
M.M González, J.L. Santos, I. Aparicio, E. Alonso
Dpto. de Química Analítica, Escuela Politécnica, Universidad de Sevilla, C/ Virgen de África, 7, 41011 Sevilla. E-mail: [email protected]
La aplicación de lodos de depuradora al suelo constituye uno de los principales destinos de los residuos generados durante el tratamiento de aguas residuales. Sin embargo, los lodos originados como subproducto del proceso de depuración tienden a concentrar los contaminantes presentes en las aguas residuales, pudiendo causar diversos efectos en los ecosistemas tras su aplicación al suelo [1].
La Unión Europea publicó en el año 2000 el tercer borrador de una futura
Directiva que regulará la aplicación de lodos de depuradora al suelo y en la que se establecen límites de concentración para algunas de las familias de compuestos orgánicos presentes en los lodos [2]. De éstas, las que aparecen a mayores concentraciones son sulfonatos de alquilbenceno lineales (LAS), nonilfenoles (NPEs; suma de nonilfenol y nonilfenoles mono- y dietoxilados) y el compuesto di(2-etilhexil)ftalato (DEHP) [3] para las que, la futura Directiva Europea, establece unas concentraciones límite de 2600, 50 y 100 mg kg-1, respectivamente.
En la actualidad existen diversos métodos en la bibliografía para la
determinación individual de estas familias de compuestos en suelos tratados con lodos de depuradora, sin embargo no se han encontrado métodos analíticos que permitan su determinación de manera simultánea.
En este trabajo, se presenta un método de rutina para la determinación
simultánea de LAS, NPEs y DEHP en suelos a los que se les ha aplicado lodo. El método se basa en la extracción de los contaminantes en baño de ultrasonidos, limpieza por extracción en fase sólida y determinación por cromatografía líquida de alta resolución con detectores ultravioleta de fila de diodos y fluorescencia. Las recuperaciones obtenidas estuvieron en todos los casos entre 63% y 99%. Los límites de detección se situaron entre 0.3 y 1.1 mg kg-1 para la detección ultravioleta y entre 0.2 y 0.6 mg kg-1 para la detección por fluorescencia. Los límites de cuantificación se situaron entre 0.6 y 3.4 mg kg-1 empleando el detector ultravioleta de fila de diodos y entre 0.3 y 1.5 mg kg-1 empleando el detector de fluorescencia.
Este trabajo ha sido financiado por la Secretaría General para la Prevención de
la Contaminación y el Cambio Climático del Ministerio de Medio Ambiente y Medio Rural y Marino (Proyecto nº 269/PC08/1-04.1) y por la Agencia Andaluza del Agua (con cofinanciación FEDER), de la Consejería de Medio Ambiente de la Junta de Andalucía (Proyecto nº 2007/1481). [1] I. Angelidaki, A.S. Mogensen, B.K. Ahring. Biodegradation 11 (2000) 377-383. [2] Working Document on Sludge, Third Draft, European Union, Brussels, Belgium
(2000). [3] J.L. Santos, M.M. González, I. Aparicio, E. Alonso, Int. J. Environ. Anal. Chem.
87 (2007) 1033-1042.

C2-128 COMUNICACIONES EN CARTEL
166
A COMPARISON BETWEEN ON-LINE AND OFF-LINE SOLID PHASE
EXTRACTION METHODOLOGY FOR THE MULTIELEMENTAL PRECONCENTRATION OF TRACE METALS IN SEAWATER
C. Mendiguchía(1), C. Moreno(1), H. Garraud(2), O.F.X. Donard(3)
(1)Dpto. Química Analítica, Univ. Cádiz, Puerto Real.
E-mail: [email protected] (2) UT2A, Pau (France)
(3) IPREM, CNRS UMR 5254, Pau (France)
In the last years analytical techniques have improved their detection capabilities achieving very low detection limits, as in the case of inductively coupled plasma mass-spectrometry (ICP-MS). Nevertheless, in the case of very complex matrices, as seawater, the problem is not yet solved. The high content in salts of seawater can block the interface of ICP-MS or generate poliatomic interferences making difficult obtaining accurate results. Although some attempts had been made for direct analysis by ICP-MS using collision cell [1,2], it was necessary a previous dilution of the samples decreasing still more the natural low concentration of trace metals in seawater, and even a standard addition calibration was required [2]. For these reasons usually a separation/preconcentration step prior to the analytical determination is preferable and, in the case of saline samples, it is always needed. This step often implies a risk of sample contamination and a longer time of analysis, and then many efforts had been focused on developing automatized methods in order to reduce sample manipulation. Currently, there are several methods available to carry out this previous step, but SPE is the most extensively applied due its accesability, simplicity, high concentration factors and it can be easily automatized.
In this work, a comparison between an on-line and an off-line methodology to
preconcentrate metals from seawater prior to its determination by ICP-MS was performed. In both cases the preconcentration was carried out by means of commercial SPE cartridges with aminodiacetate groups following a four-step procedure. This process comprises: (1) sorbent conditioning, (2) sample loading, (3) removal of interferences, and (4) elution of the analyte of interest into a receiving vessel. Both systems were previously optimized using experimental design with the objectives of reduce interferences and increase the analites elution. For the on-line methodology a preconcentration module, previously used to multielemental speciation of trace elements in estuarine waters [3], was selected, while for the off-line procedure the SPE cartridges were coupled to a vacuum system manifold to perform the extraction. All experiments were carried out in a clean room in order to avoid sample contamination. Analysis of reference seawater was performed under the optimized values of the variables to obtain the recoveries for V, Mn, Co, Ni, Cu, Mo, Cd, Pb and U. Although the results obtained by both methodologies exhibited good reproducibility, only the on-line procedure achieved recoveries close to 100 % for all of the elements. Moreover, lead contamination of samples was observed for off-line procedure, showing that on-line methodology is preferable for very low metal concentrations. [1] H. Louie, M. Wu, P. Di, P. Snitch, G. Chapple, J. Anal. At. Spectrom. 17 (2002) 587. [2] P. Leonhard, R. Pepelnik, A. Prange, N. Yamada, T. Yamada, J. Anal. At. Spectrom. 17
(2002) 189. [3] D. Point, G. Bareille, H. Pinaly, C. Belin, O.F.X. Donard, Talanta 72 (2007) 1207.

COMUNICACIONES EN CARTEL C2-129
167
ANALIZADOR PORTATIL PARA LA DETERMINACIÓN DE PEROXIDOS EN
AGUAS BASADO EN UN BIOSENSOR ÓPTICO AUTOINDICADOR.
E. Mateos, S. de Marcos, V. Sanz and J. Galbán
Grupo de Biosensores Analíticos, Dpto. Quimica Analítica, Facultad de Ciencias, Instituto de Nanociencia de Aragón, Universidad de Zaragoza.
E-mail: [email protected] Nuestro grupo de investigación está desarrollando métodos analíticos
enzimáticos basados en la utilización de las propiedades espectroscópicas (absorción o fluorescencia), tanto intrínsecas como extrínsecas (después de la modificación química), de las proteínas con la finalidad de diseñar biosensores autoindicadores. Este trabajo utiliza las propiedades de Absorción Molecular de la peroxidasa (HRP) como base de un biosensor para la determinación de peróxido de hidrógeno en aguas procedentes de depuración.
En un primer trabajo [1] se desarrolló un biosensor óptico autoindicador de laboratorio, consistente en una célula de flujo dotada de una lámina sensora (de 0.3 mm de paso óptico) formada por HRP inmovilizada en poliacrilamida (HRP-PAA), por la que se hace pasar una disolución portadora/regeneradora de tirosina, sobre la que inyectan las alícuotas del analito. El biosensor estaba basado en que durante la reacción el HRP se oxida a un intermedio, con propiedades de absorción UV-visible diferentes de la enzima original, produciéndose cambios de absorbancia proporcionales a la concentración de peróxido; la tirosina regenera la HRP. El desarrollo va acompañado de un estudio teórico que modela el comportamiento del biosensor.
Para transformar el biosensor de laboratorio en un analizador portátil autónomo se han tenido que:
1) Incorporar los elementos necesarios para la hidrodinámica del proceso (una bomba peristáltica miniaturizada y dos válvulas de tres vías)
2) Desarrollar la óptica. Las fuentes de radiación son dos LEDs, uno para la referencia (r) y otro para la medida (m); tras un meticuloso estudio se han elegido finalmente dos LEDs cuyas λ están centradas en 650nm (r) y 420nm (m).
3) Diseñar la electrónica y el sistema de control y procesamiento de señal. El analizador mide los valores de área y altura de la señal transitoria y los transforma en concentración.
4) Rediseñar los componentes del biosensor original, tomando como base el modelo matemático desarrollado. En particular se ha incrementado el paso óptico de la lámina sensora hasta los 6 mm, se ha rediseñado la célula de flujo y se ha optimizado el tipo y concentración de regenerador.
El resultado ha sido un analizador que mide concentraciones de peróxido comprendidas entre 1 y 20 mg/l, con una reproducibilidad razonable (5%) , que permite realizar en torno a 100 medidas consecutivas sin necesidad de cambiar la lámina sensora y que está optimizado para la determinación del analito en aguas procedentes de depuración. Actualmente se sigue trabajando en la química del sistema para incrementar el tiempo de vida de las láminas.
Agradecimientos. Este trabajo ha sido financiado con el proyecto PROFIT CIT-320100-2007-30 del DGCYT y el proyecto BQU2008. También agradecemos su ayuda y soporte científico al Servicio de Instrumentación Científica de la Universidad de Zaragoza. [1] V. Sanz, S. de Marcos, J. Galbán, Anal.Chim.Acta 583, 332-339 (2007).

C2-130 COMUNICACIONES EN CARTEL
168
DISEÑO DE UN SENSOR ÓPTICO DE VIDA LARGA PARA LA MEDIDA DE pH
BASADO EN ROJO CONGO INMOVILIZADO.
A. Delgado-Camón2, S. de Marcos1, J. Galbán1, E. Romero2, V. Cebolla2
1Grupo de Biosensores Analíticos, Dpto. Química Analítica, Facultad de Ciencias.
Instituto de Nanociencia de Aragón, Universidad de Zaragoza. 2Instituto de Carboquímica (CSIS)-Zaragoza. E-mail: [email protected]
La investigación sobre sensores ópticos para pH basados en la inmovilización de indicadores ácido-base sobre soportes sólidos, sigue siendo un campo de interés, a pesar de todo el trabajo de investigación que se ha publicado. Una de las razones que lo impulsa es conseguir un dispositivo que realmente tenga una vida útil larga, ya que el principal problema de estos dispositivos es la lixiviación del indicador o de los reactivos que se añaden para mejorar las respuestas. El objetivo de este estudio es ensayar procedimientos alternativos a los habitualmente utilizados que permitan conseguir sensores de pH de vida larga.
La idea general es utilizar indicadores ácido-base con grupos amino terminales
para su unión covalente al soporte. Se han ensayado Rojo Congo, Rojo Neutro, Azul Nilo y Ácido Fucsia, como reactivos de absorción UV-visible, y aminofluoresceina y bromuro de etidio, como reactivos de fluorescencia. En cuanto a los tipos de soportes se han ensayado fundamentalmente:
a) Vidrio químicamente activado con isotiocianato. No se han conseguido sensores duraderos.
b) Membranas comerciales que están ya químicamente modificadas con grupos adecuados para la unión covalente de los indicadores, como las APEG-membranes™ (polipropileno de celulosa con polietilenglicol como espaciador) o ULTRABIND™ (polietersulfonato modificado). Ambas láminas han dado muy buenos resultados, cualitativamente hablando, desde un punto de unión rápida y duradera, empleando cualquiera de los indicadores anteriormente mencionados. Estas láminas solo se pueden usar para hacer medidas por reflexión (absorción o fluorescencia).
c) Polianilina, para conseguir un fenómeno mixto de entrampamiento y polimerización. Se ha conseguido la fabricación de la lámina sensora pero la sensibilidad, por el momento, es baja.
d) Poliacrilamida (PAA), usando glutaraldehido (GA) para unir químicamente el indicador a la red. Los resultados obtenidos con Rojo Congo (RC) han sido muy satisfactorios. Se pueden usar para medir absorción de luz por reflexión o transmisión.
A la vista de estos resultados se está fabricando un sensor óptico basado en
medidas de absorción por transmisión para la medida de pH en torno a 4, para lo que se está utilizando la inmovilización en PAA y el RC como indicador. El RC se hace reaccionar con el GA y posteriormente con el monómero de la PAA (acrilamida), antes de formar la lámina. La lámina de PAA-GA-RC obtenida da un cambio de color bastante rápido, el pK del RC no se altera, la lámina es estable durante varios meses, se puede trabajar a 495 o 595 nm y se puede medir pH en el intervalo de 3 a 5. La lámina de PAA-GA-RC se ha acoplado a la terminación de una fibra óptica para la realización de medidas en continuo con resultados satisfactorios.
Agradecimientos. Este trabajo se está desarrollando bajo la financiación otorgada por el Ministerio de Ciencia e Innovación, proyecto CTQ 2008-06751-C02-01/BQU.

COMUNICACIONES EN CARTEL C2-131
169
MONITORING OF PRIORITY AND EMERGING CONTAMINANTS IN EFFLUENT WASTEWATERS FROM SPANISH WASTEWATER TREATMENT PLANTS
J. Robles-Molina, B. Gilbert-López, J.C. Domínguez-Romero, J.F. García-Reyes, M.J. Gómez-Ramos, A. Agüera, A.R. Fernández-Alba, A. Molina-Díaz
Analytical Chemistry Research Group, Department of Physical and Analytical Chemistry, University of Jaén, Campus Las Lagunillas, Ed. B-3, 23071 Jaén, Spain.
E-mail: [email protected]
Urban wastewaters, which include domestic and some industrial waters, among others, typically have a strongly contaminating effect on the natural aquatic systems. Despite wastewaters are submitted to treatment, many studies report on the presence of multiple organic compounds, such as pharmaceuticals, personal care products, hormones, and other disrupting compounds, some of them are becoming ubiquitous in the environment. Identification, analysis, and characterization of the risks posed by the presence of these compounds in the environment are important issues of concern within the scientific community. Consequently, the application of more exhaustive wastewater treatment protocols, including the use of new and improved technologies, the application of wider and integrated quality control strategies, comprising chemical, microbiological, and toxicological analysis, and the study and development of wastewater reuse strategies are tasks that are necessary undertake. With this aim, an ambitious program (TRAGUA) financed by the Spanish Government, which involves the participation of a multidisciplinary team formed by 24 research groups, has been initiated. The program attempts to tackle the different aspects involved in the reuse of wastewater coming from the sewage treatment plants (STPs). As a part of this program, the present work has been focused on the development of suitable analytical methodology, which allows an exhaustive characterization of wastewater effluents. This work describes the analytical methods used for the simultaneous screening and identification of over 180 multi-class priority and emerging pollutants in effluent wastewaters. The ultimate goal of the present study is to evaluate the presence of such compounds in different wastewater treatment plants and assess the effectiveness of the current wastewater treatment technologies to eliminate their presence.
Samples of different origin (industrial, coastal and urban) are provided by several wastewater treatment plants (WWTPs) located throughout Spain. Several mass spectrometric techniques are being used to perform this study, according to compounds’ chemical characteristics: Inductively Coupled Plasma Mass Spectrometry (ICP-MS) is used for the direct analysis of heavy metals (Hg, Cd, Pb and Ni); Gas Chromatography coupled to mass spectrometry (GC-MS/MS) was used for the analysis of organic pollutants and, liquid chromatography coupled to time-of-flight mass spectrometry (LC-TOFMS) was used for polar and/or non-volatile analytes (i.e. selected priority pesticides, pharmaceuticals, surfactants and plasticizers).
The GC-MS/MS (triple quadrupole) multi-residue method covered the analysis of 60 multi-class organic contaminants in wastewater samples. It was based on a sample treatment using liquid-liquid extraction with n-hexane followed by identification, confirmation and quantitation with gas chromatography tandem mass spectrometry using a triple quadrupole analyzer in the selected reaction monitoring mode. Three MS/MS transitions were selected for unambiguous confirmation of the target chemicals.
The authors acknowledge funding from the Spanish Ministerio de Ciencia y Tecnología (CONSOLIDER 2010, CSD 2006-00044, TRAGUA network).

C2-132 COMUNICACIONES EN CARTEL
170
EMPLEO DE LA DISPERSIÓN DE MATRIZ EN FASE SÓLIDA EN LA
ACELERACIÓN DEL PROCEDIMIENTO BCR PARA SEDIMENTOS MARINOS
Marta Martínez Fernández, Mª Carmen Barciela Alonso, Antonio Moreda Piñeiro, Pilar Bermejo Barrera
Departamento de Química Analítica, Nutrición y Bromatología, Universidad de
Santiago de Compostela, Facultad de Química, Avda. das Ciencias s/n Campus Sur, 15782, Santiago de Compostela, Spain. E-mail: [email protected]
El Protocolo BCR es un método de extracción secuencial de metales en muestras medioambientales sólidas. Este tipo de procedimiento da información sobre la química de los metales pesados en su interacción con otros componentes de la muestra como arcillas, materia orgánica y solución del suelo; siendo también válidos para la evaluación de la movilidad, retención y biodisponibilidad de los mismos para las plantas.
En el año 1992, la Community Bureau of Referente (BCR) de la Comisión de las Comunidades Europeas, propuso este Protocolo, que comprende tres etapas de trabajo, en las cuales se extraen secuencialmente cuatro fracciones del sedimento: metal en forma de iones intercambiables y carbonatos (f1), metal asociado a óxidos de hierro y manganeso (f2), metal ligado a materia orgánica (f3) y fase residual o litogénica (f4).
El principal inconveniente de este método de extracción, está en que cada una
de las etapas comprende una fase de agitación de 16 horas, lo cual aumenta enormemente el tiempo de trabajo.
Se han desarrollado diversos métodos para acelerar el procedimiento BCR,
tales como agitación por ultrasonidos o calentamiento mediante microondas. En este trabajo, se ha propuesto utilizar la dispersión de matriz en fase sólida
(MSPD), como técnica sustitutiva a la agitación mecánica de la muestra. La MSPD es una técnica en la cual la muestra se mezcla con un agente dispersante en un mortero, provocando una ruptura de la arquitectura de la misma y dando lugar a una nueva estructura de la que es más fácil liberar los analitos de interés.
El método ha sido optimizado para las dos primeras etapas del Protocolo BCR
dando resultados satisfactorios para muestras de sedimentos marinos. Se ha estudiado el agente dispersante, la relación del mismo y la muestra y el volumen de extractante a utilizar. Se ha comprobado que los mejores resultados se obtienen usando arena como agente dispersante en una relación 1:2 con la muestra; es decir, 0.5 gramos de muestra y 1 gramo de arena. Para la primera etapa, el volumen óptimo de extractante son 25 mL de ácido acético 0.11 M y en la segunda, 20 mL de hidrocloruro de hidroxilamina 0.1 M. Los eluyentes utilizados en la extracción son los propuestos por el Protocolo BCR.

COMUNICACIONES EN CARTEL C2-133
171
DETERMINACIÓN DE Ni EN AGUA DE MAR POR ICP-OES EMPLEANDO UNA PRECONCENTRACIÓN EN LÍNEA BASADA EN POLÍMEROS DE IMPRESIÓN
IÓNICA
Natalia García-Otero, Jacobo Otero-Romaní, Elena Peña-Vázquez, Antonio Moreda-Piñeiro, Pilar Bermejo-Barrera
Dpto. de Química Analítica, Nutrición y Bromatología. Facultad de Química.
Universidad de Santiago de Compostela. Avenida de las Ciencias, s/n 15782 – Santiago de Compostela. España. E-mail: [email protected]
Se ha desarrollado un método de preconcentración on line empleando un polímero de impresión iónica (IIP) como soporte de extracción en fase sólida. El polímero de impresión iónica de Ni se sintetizó por la técnica de polimerización por precipitación, utilizando como analito plantilla el cloruro de Ni (NiCl2), monómero el 2-(dietilamino) etil metacrilato (DEM), agente complejante la 8-Hidroxiquinolina
(8-HQ), entrecruzante el divinilbenceno (DVB), iniciador el 2,2´-
azobisisobutyronitrilo (AIBN) y porogén acetonitrilo-tolueno (3:1). El método se aplicó a la preconcentración/determinación de Níquel en agua de mar por espectrometría de emisión óptica por plasma acoplado inductivamente.
En la pre-concentración (carga), la muestra de agua de mar se pasa a través
del cartucho que contiene el IIP mediante un flujo de 2.25 mL min-1 durante dos minutos. Mientras tanto la disolución de elución (ácido nítrico 3M) es bombeada por la bomba 2 y la bomba 1 hacia el detector (Fig. 1 Load). En la elución, la disolución de ácido nítrico pasa a través del cartucho arrastrando los analitos y se introduce directamente en el detector (inyección).
Antes de la etapa de carga se pasa a través del cartucho una disolución tampón
0.1M/0.1M NH3/NH4Cl a pH 9.0 mediante un flujo de 2.25 mL min-1 durante 60s para acondicionar el IIP. Esta misma disolución tampón se pasa a través del cartucho justo antes de la elución para eliminar compuestos interferentes que se hayan quedado retenidos de forma inespecífica.
El límite de detección y el limite de cuantificación obtenidos fueron 0.33 y 1.10
µg L-1 respectivamente. El factor de preconcentración obtenido fue de 15. La precisión del método se evaluó mediante el coeficiente de variación de los resultados obtenidos de preconcentrar 11 veces una muestra de agua de mar; el valor del RSD fue del 8%. La exactitud del método fue evaluada mediante el análisis de dos materiales de referencia el TM-24 y el SLEW-3.
Con la incorporación de un cartucho con el IIP a una vávula de seis vías de
inyección se pueden analizar hasta 12 muestras por hora. Los tiempos de análisis obtenidos son mucho menores que empleando el procedimiento convencional de extracción en fase sólida en modo off-line.

C2-134 COMUNICACIONES EN CARTEL
172
MONITORING OF PESTICIDE RESIDUES IN FRUIT-BASED SOFT DRINKS
FROM THE SPANISH MARKET
M. Villar-Pulido, B. Gilbert-López, L. Jaén-Martos, J.F. García-Reyes, N. Ramos-Martos, A. Molina Díaz
Analytical Chemistry Research Group (FQM 323), Department of Physical and
Analytical Chemistry, University of Jaén, E-23071 Jaén, Spain. E-mail: [email protected]
There are regulations for pesticides in fruits, vegetables, baby food or drinking
water. However, scarce attention has been paid to derivate products, which may contain these commodities as an ingredient. Unlike other kinds of commodities (fruit, vegetables, water), the soft drink industry that comprises large companies, remains unregulated in terms of pesticide regulation worldwide. In a recent work [1], we measured over one hundred fruit-based soft drink samples, purchased from 15 different countries from brands of companies distributed worldwide (i.e. Coca Cola Co., PepsiCo, etc) and reported on the presence of relatively large concentration levels of pesticides in fruit-based or juice-based soft drinks. Some of the detected pesticides are those applied to crops at final stages of production (post harvest treatment) and might have hazardous effects to infants, the main target of these products.
This work reports on the follow-up study and monitoring of pesticides in fruit
based soft drink samples from the Spanish Market. Over 40 samples have been collected and analyzed from different regions of Spain during 2009. The analytical methodology used in this monitoring study [2] consists of a sample treatment protocol based on solid-phase extraction using hydrophilic-lipophilic balanced polymer-based reverse-phase HLB Oasis cartridges followed with identification and quantitation of the targeted species by rapid resolution liquid chromatography-mass spectrometry using electrospray ionization in the positive ionization mode and a short C18 column (4.6 x 50 mm) with 1.8 µm particle size. The unambiguous confirmation of the compounds was based on accurate mass measurements of the protonated molecules ([M+H]+) and their main fragment ions. The proposed method was successfully implemented to conduct the survey, revealing the presence of selected pesticides in a high percentage of the studied samples in the µg L-1 range
[1] J.F. García-Reyes, B. Gilbert-López, A. Molina-Díaz, A.R. Fernández-Alba, Anal. Chem. 80 (2008) 8966-8974.
[2] B. Gilbert-López, J.F. García-Reyes, M. Mezcua, A.R. Fernández-Alba, A. Molina Díaz, submitted for publication.

COMUNICACIONES EN CARTEL C2-135
173
DESARROLLO DE UNA METODOLOGÍA ANALÍTICA PARA LA
MONITORIZACIÓN DE PRINCIPIOS ACTIVOS FARMACOLÓGICOS EN AGUAS RESIDUALES Y SUPERFICIALES
D. Camacho-Muñoz, J. Martín, J.L. Santos, I. Aparicio, E. Alonso
Departamento de Química Analítica, Escuela Politécnica, Universidad de Sevilla,
C/ Virgen de África 7, 41011 Sevilla. E-mail: [email protected]
Cada año se consumen cientos de toneladas de medicamentos en todo el mundo. Como consecuencia de ello, los principios activos farmacológicos contenidos en los mismos son descargados a las estaciones depuradoras de aguas residuales (EDARs) a través de las redes de alcantarillado. Estas EDARs no están diseñadas específicamente para la eliminación de tales contaminantes y, en consecuencia, se produce una descarga continua de estos contaminantes al medio acuático [1].
En los últimos años, la presencia de principios activos farmacológicos en aguas
residuales y superficiales ha sido ampliamente descrita en la mayoría de países desarrollados [2], sin embargo, la información acerca de la presencia de estos compuestos en las aguas residuales y superficiales procedente de otras localizaciones geográficas es escasa. Este hecho podría estar motivado porque, hasta el momento, la mayoría de métodos analíticos descritos para la determinación de principios activos farmacológicos están basados en métodos cromatográficos acoplados a la espectrometría de masas. Estos métodos, aunque adecuados en términos de exactitud, sensibilidad y selectividad, son costosos y no se encuentran disponibles en la mayoría de los laboratorios. Esto hace que el uso de técnicas de cromatografía líquida con detectores de fila de diodos y fluorescencia constituya una interesante alternativa para la determinación de estos contaminantes en muestras medioambientales.
En este trabajo se propone un método analítico para la determinación
simultánea de diecisiete principios activos farmacológicos en aguas residuales y superficiales. El método se basa en la extracción y preconcentración simultánea mediante extracción en fase sólida y posterior determinación por cromatografía líquida de alta resolución con detectores ultravioleta de fila de diodos y fluorescencia. Los principios activos farmacológicos seleccionados fueron seis antiinflamatorios (ácido salicílico, diclofenaco, ibuprofeno, ketoprofeno, naproxeno y paracetamol), un psicoestimulante (cafeína), dos antibióticos (sulfametoxazol y trimetoprim), dos reguladores lipídicos (ácido clofíbrico y gemfibrozilo), un antiepiléptico (carbamacepina), un β-bloqueante (propanolol) y cuatro hormonas (17α-etinilestradiol, 17β-estradiol, estriol y estrona). Las recuperaciones obtenidas se encontraron en el rango comprendido entre el 61% y el 116 %. Los límites de detección se situaron entre 0.003-0.30 µg L-1 para el detector de ultravioleta y entre 0.001-0.137 µg L-1 para el detector de fluorescencia. El método fue aplicado a la determinación de principios activos farmacológicos en aguas residuales y superficiales de la provincia de Sevilla.
Este trabajo ha sido financiado por el Ministerio de Educación y Ciencia
(Proyecto nº CGL2007-62281/HID). [1] J.L. Santos, I. Aparicio, E. Alonso, Environ. Int. 33 (2007) 596-601. [2] K. Fent, A.A. Weston, D. Caminada, Aquat. Toxicol. 76 (2006) 122–159.

C2-136 COMUNICACIONES EN CARTEL
174
FAST ANALYSIS USING LASER INDUCED PLASMA SPECTROSCOPY
J. Anzano, R.J. Lasheras, J. Casas-González1, B. Bonilla, B. Montull-Ibor and
C. Bello
Laser Laboratory & Environment, Department of Analytical Chemistry, Univ. Zaragoza, Zaragoza. E-mail: [email protected]
www.unizar.es/janzano 1Department of Condensed Matter Physics. Faculty of Sciences, University of
Zaragoza Pedro Cerbuna #12, 50009-Zaragoza, SPAIN Laser-induced plasma (LIPs) has adquired great interest in recent years as
spectroscopy sources. LIPs gave excellent results in respect of reproducibility, sensitivity, non-destructiveness, rapid elemental analysis, and depth-profile analysis. In our lab three LIPS research fields are carried out: (i) optimization of LIPS parameters, (ii) identification of materials and (iii) quantitative analysis. (i) Optimization of LIBS parameters
LIBS instrumental parameters have evaluated: laser pulse energy, delay time and integration time gate, with regard to their influences on signal-to-noise-ratio (SNR) of seven elements in soil sample. (ii) Identification of materials
It was demonstrated the application of parametric correlation methods for identification of materials such as polymers,[1] kidney stones[2] and analgesic pills[3]. Other method studied consisted in the measurement of ratios as C, H, N, O and C2 (Swan bands). Others identification methods have studied using LIPS: Principal Component Analysis (PCA), Euclidean Distance, Euclidean Distance Squares, Pearson’s Correlation Coefficient and Blocks. (iii) Quantitative analysis
Quantitative analysis of iron and the simultaneous determination of calcium and magnesium were carried out in samples of pottery manufactured in ancient settlements of early Roman.
[1] J.Anzano, B. Bonilla, B. Montull-Ibor, R.J. Lasheras and J. Casas-González, J.Poly.Eng. (2009) In press.
[2] J.Anzano and R.J. Lasheras, Talanta (2009) DOI:10.1016/j.talanta.2009.03.065. [3] J. Anzano, B. Bonilla, B.Montull-Ibor and J. Casas-González, Med. Chem.Res.
(2009) DOI:10.1007/s00044-008-9157-5.

COMUNICACIONES EN CARTEL C2-137
175
SIMS DEPTH PROFILING ANALYSIS OF NANOLAYERED STRUCTURES FOR
MICROELECTRONIC APPLICATIONS
H. Téllez, J.M. Vadillo, J.J. Laserna
Dpto. Quimica Analítica, Univ.Málaga, Campus Teatinos, E-mail: [email protected] A wide array of nanolayered structures and materials is being employed in
today´s microelectronic industry where the questions to be solved by analytical chemistry refer to extremely low concentrations and spatial dimensions. Examples of such are the dopant/impurity diffusion in multi-nanostructured III-V semiconductors or the degradation processes in nano-metallized film capacitors. These processes give rise to critical failures or efficiency losses during service life of such microelectronic devices. Thus, this sort of nanolayered structures represents a great challenge in the field of surface chemical analysis. Additionally, analytical techniques capable of determining the possible atomic/molecular composition variations along the nanometric thicknesses are required for quality assurance in microelectronics.
Dynamic SIMS (d-SIMS) has been traditionally used in this field as it provides
layer-by-layer analysis of the multilayered samples with subnanometric resolution and complete atomic/molecular information generated by the analysis of the fragments emitted during the sputtering process. In combination with kinetic energy distribution filtering, d-SIMS represents a powerful approach to solve a variety of microelectronic problems at the nanometric scale. Examples related to the revealing of corrosion phenomena in nano-metallized film capacitors or the analysis of new generation of Ge-based concentration photovoltaic devices will be presented and discussed.
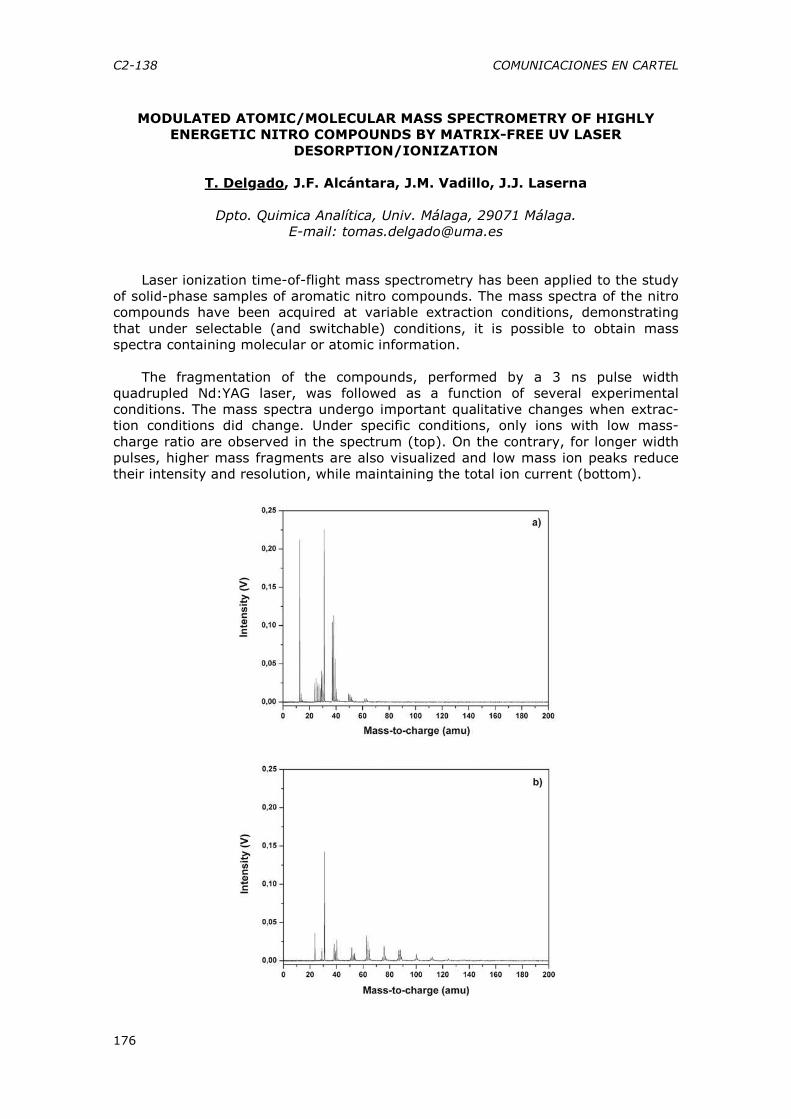
C2-138 COMUNICACIONES EN CARTEL
176
MODULATED ATOMIC/MOLECULAR MASS SPECTROMETRY OF HIGHLY
ENERGETIC NITRO COMPOUNDS BY MATRIX-FREE UV LASER DESORPTION/IONIZATION
T. Delgado, J.F. Alcántara, J.M. Vadillo, J.J. Laserna
Dpto. Quimica Analítica, Univ. Málaga, 29071 Málaga.
E-mail: [email protected] Laser ionization time-of-flight mass spectrometry has been applied to the study
of solid-phase samples of aromatic nitro compounds. The mass spectra of the nitro compounds have been acquired at variable extraction conditions, demonstrating that under selectable (and switchable) conditions, it is possible to obtain mass spectra containing molecular or atomic information.
The fragmentation of the compounds, performed by a 3 ns pulse width
quadrupled Nd:YAG laser, was followed as a function of several experimental conditions. The mass spectra undergo important qualitative changes when extrac-tion conditions did change. Under specific conditions, only ions with low mass-charge ratio are observed in the spectrum (top). On the contrary, for longer width pulses, higher mass fragments are also visualized and low mass ion peaks reduce their intensity and resolution, while maintaining the total ion current (bottom).

COMUNICACIONES EN CARTEL C2-139
177
QUANTITATIVE DETERMINATIONS OF ZrC IN NEW CERAMIC MATERIALS BY
FOURIER TRANSFORM INFRARED SPECTROSCOPY.
M.M. López Guerrero, E. Vereda Alonso, A. García de Torres, M.T. Siles Cordero, J.M. Cano Pavón
Department of Analytical Chemistry, Faculty of Sciences, University of Málaga.
E-mail: [email protected] The ceramic materials, called biomorphic ceramics, show at low density,
excellent mechanical propierties such as high-strength, stiffness, toughness, good oxidation and good thermal shock resistence [1]. ZrC show interesting physical and mechanical properties which are suitable for applications as structural components, as refractory material, in electronic devices or as substitute for SiC coatings in the nuclear industry [2].
Zirconium carbide-based biomorphic ceramics have been fabricated by the
pyrolysis and infiltration of natural wood with zirconia-sol. This infiltration offers a versatile method to manufacture novel ceramics. The results of the process of synthesis have been studied in this and other biomorphic ceramics using thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS) analysis and scanning electron microscopy (SEM) and X-ray diffraction (XRD). For evaluating the yield of the synthesis, a new method by Fourier transform infrared spectrometry (FTIR) has been developed for the direct determination of ZrC by absorbance measurements in KBr pellets. The procedure was based on the use of the ratio between the absorbance of the characteristic band of zirconium carbide and those of an oxalate internal standard added to samples. A multivariate calibration strategy based on inverse least squares and the standard addition approach were employed for quantification. The results obtained for all biomorphic ceramics studied and synthetic samples prepared by mixing pyrolyzed wood with pure ZrC were satisfactory.
Figure 1. SEM micrographs of zirconium carbide biomorphic ceramic.
[1] E. Vereda Alonso, A. García de Torres, M.T, Siles Cordero, J.M. Cano Pavón. Talanta. 75 (2008) 424-431.
[2] Storms EK. The refractory carbides. London: UK Academic Press; 1997.

C2-140 COMUNICACIONES EN CARTEL
178
OPTIMIZATION OF THE BCR SPECIATION OF METAL IONS IN SOILS AND
SEDIMENTS ASSISTED BY MICROWAVE OVEN
M. Alonso Castillo, E. Vereda Alonso, M.T. Siles Cordero, A. García de Torres and J.M. Cano Pavón
Department of Analytical Chemistry, Faculty of Sciences, University of Málaga.
E-mail: [email protected]; [email protected]
The sequential speciation of trace metals in sediments permits to obtain information about the origin, biological and physic-chemistry availability and transport of the trace metals through of the environment. The most accepted method for the sequential speciation of metals is the BCR, but this method is tedious and slow. In this work a fast microwave assisted extraction procedure was developed and optimized for their eventual exploitation in three-stage sequential extraction procedure proposed by modified BCR protocol. In order to optimize the method was development a design of experiments by the software Statgraphics. The microwave oven used was an Anton Paar, model Multiwave 3000. The parameters optimized were the microwave power, ramp and hold time. Extractable metal obtained were measured by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) with a Dynamic Reaction Cell (DRC) with methane.
Following a factorial design 22+star (central composite design), the conditions
optimized for stage 1 were the same for Ni, Cd, Cr and Pb. However, for Cu and Zn the conditions had to be stronger. The conditions optimized for stages 2 and 3 were the same for the six elements.
The material used for the optimization of the procedure was a sample of soil
collected from Guadalhorce River in Malaga (Spain) which was previously analyzed by modified BCR protocol. The results obtained in the three stages by microwave treatment on the extraction of Cd, Cr, Cu, Ni, Pb and Zn were compared with the certified data of a certified reference material (BCR 701 Lake sediment) with good agreement.

COMUNICACIONES EN CARTEL C2-141
179
OCCURRENCE AND FRACTIONATION OF ARSENIC IN SOILS OF AN ARSENIC AFFECTED AQUIFER AND ITS RELATION WITH SOIL GEOCHEMISTRY
M. Vega, M.C. Carretero, M.I. Sánchez, R. Pardo, Y. Castrillejo
Dept. Química Analítica, Facultad de Ciencias, Universidad de Valladolid,
47005 Valladolid, Spain. E-mail: [email protected] Arsenic is relatively scarce in the environment, but it is widely distributed. It
can be found in detectable amounts in most rocks and soils. Arsenic concentrations in sedimentary rocks are usually above the terrestrial average content (5 mg/kg). The major minerals binding arsenic are metal oxides, particularly those of Fe and Mn, and clays. Although the dominant source of arsenic in soils is geological, additional inputs may be derived locally from anthropogenic sources.
The recent finding that groundwater from Tierra de Pinares (a sedimentary region located in the Duero basin amongst the provinces of Ávila, Segovia and Valladolid) is heavily enriched with arsenic, has prompted the need to investigate its distribution in soils and rocks from the area to understand the mobilization pathways. A relatively simple and well-adopted method to assess the distribution and to predict the mobilization of metals in soils is the fractionation by sequential extraction with reagents of increasing dissolution strength. A number of fractionation schemes have been devised which attempt to allocate elements to particular mineralogical phases in soils, but few of them can be applied to arsenic due to its anionic character.
In this work, the fractionation scheme described by Wenzel el al. [1] has been applied to soil and rock samples collected from different geological strata in a confined aquifer affected by arsenic contamination. The scheme extracts sequentially: (1) non-specifically adsorbed As, with (NH4)2SO4; (2) specifically adsorbed As, with NH4H2PO4; (3) As bound to amorphous Fe, Mn and Al oxides, with ammonium oxalate buffer (pH 3.25); (4) As bound to crystalline Fe, Mn and Al oxides, with ammonium oxalate buffer + ascorbic acid (pH 3.25 and 90ºC); and (5) residual As, by microwave digestion with HNO3/H2O2. The geochemistry of the aquifer has been also established in an attempt to correlate the arsenic fractionation with the mineralogy of the soil samples.
Arsenic fractionation results showed that, in general, arsenic is mainly bound in fraction 4. Total arsenic concentration in the soils varied widely with the geological stratum, and hence with the mineralogical composition of the soil. Values below 10 mg/kg were found in deeper rocks (Facies “Toro”, constituted mainly by silt and clay) and upper heights (Facies “Transición al Páramo”, limestones). Arsenic concentrations higher than 20 mg/kg were detected in midway sediments: Facies “Cuestas” (clay, marls, gypsum), Facies “Dueñas” (marls, limestone) and Facies “Villalba de Adaja” (clay, sand, gravel), Facies “Tierra de Campos” (clay, gypsum).
The significant correlation of arsenic with contents of clay, iron, and some other heavy elements seems to indicate that arsenic and other metals are bonded to iron-rich clay materials by adsorption processes. In some geological strata arsenic was not related to clay content, thus pointing to the occurrence of other arsenic-bearing phases (sulphides, arsenates…).
[1] Wenzel, W.W., Kirchbaumer, N., Prohaska, T., Stingeder, G., Lombi, E.,
Adriano, D.C., Anal. Chim. Acta, 436 (2001) 309. Acknowledgements. Thanks to Junta de Castilla y León, Consejería de Educación (VA077A05) and to Spanish MEC (CTM2006/02249) for financial support.

C2-142 COMUNICACIONES EN CARTEL
180
OPTIMIZATION OF SOLID-PHASE EXTRACTION PROCEDURE FOR THE
DETERMINATION OF 1,3-DICHLORO-2-PROPANOL AND 3-MONOCHLORO-1,2-PROPANDIOL IN WATER BY BSTFA DERIVATIZATION AND GC-MS
A.M. Carro*; P. González; R.A. Lorenzo and R. Cela
Dpto. de Química Analítica, Nutrición y Bromatología, Facultad de Química, Instituto
de Investigaciones y Análisis Alimentarios (IIAA). Universidad de Santiago de Compostela, Avda. de las Ciencias, s/n 15782-Santiago de Compostela, Spain.
*E-mail address of corresponding author: [email protected]
The resins formed by polyamidoamine-epichlorhydrin (PAAE) are used in the paper industry [1]. During the conversion of polyamidoamine, chloropropanols compounds could be formed by reaction of epichlorhidrin with chloride ions. Moreover, the presence of dichloropropanols in water is a consequence of polyamine flocculants used in the treatment of drinking water [2].
Previous investigations have developed various types of extraction methods for
chloropropanols in environmental samples, including Liquid–liquid extraction (LLE) and purification by solid-phase extraction (SPE) using an Extrelut column [3].
This study proposes a sensitive method for the determination of 1,3 dichloro-2-
propanol (1,3-DCP) and 3-monochloro-1,2-propandiol (3-MCPD) in water samples using bis-(trimethylsilyl)trifluoracetamide (BSTFA) as derivatizant agent and gas chromatography-mass spectrometry. The method uses 1,3-DCP-d5 and 3-MCPD-d5 as internal standard. The sorbent Oasis HLB was used in a SPE process to extract chloropropanols in water samples, such as tap water, mineral water, river water, influent and effluent wastewater from a paper industry. The parameters that may affect the recovery of the chloropropanols, such as water properties: pH value, salinity were investigated by a response surface Doehlert design [5]. NEMROD©W software was used for the generation and evaluation of the experimental design [4]. It was observed that better SPE efficiency is achieved for high levels of pH and high amounts of NaCl for 1,3-DCP. However, 3-MCPD shows a different behaviour and better responses were acquired at high amounts of NaCl but any pH value is valid. In order to find the best simultaneous conditions, multicriteria decision-making strategies using desirability function optimization were applied without additional experimentation [5]. Final optimal conditions of SPE procedure are: 1,5 g NaCl added to the samples and pH value of 6.
Under optimal conditions, the analytical performance of the SPE-derivatization-
GC-MS was evaluated showed good linearity, precision, accuracy and detection limits. References
[1] Bodén L, Lundgren M, Stensiö K, Gorzynski M (1997) J Chromatogr, A 788: 195-203.
[2] Schuhmacher R, Nurmi-Legat J, Oberhauser A, Kainz M, Krska R (2005) Anal. Bioanal. Chem. 382:366-371
[3] Wenzl T, Lachenmeier DW, Gökmen V (2007) Anal. Bioanal. Chem. 389:119-137
[4] Mathieu D, Nony J, Phan-Tan-Luu R, NemrodW, version 2001, LPRAI, Marseille [5] Lewis G A, Mathieu D, Phan-Tan-Luu R, Pharmaceutical Experimental Design,
Marcel Dekker, Inc., New York, 1999

COMUNICACIONES EN CARTEL C2-143
181
DETERMINATION OF PESTICIDES IN SOILS BY PRESSURIZED LIQUID
EXTRACTION AND GC-MS/MS AND LC-MS/MS ANALYSIS
A. Garrido Frenich, J.L. Martínez Vidal, P. Plaza-Bolaños, J.A. Padilla-Sánchez, M.N. Barco-Bonilla
Dpto. Química Analítica, Univ. Almería, Almería. E-mail: [email protected]
Pesticides are widely used in intensive agriculture in order to preserve optimal crop yields. However they present estrogenic and carcinogenic effects and pesticide residues are pollutants that can be found in environment samples such as water and soil. Soils are considered as a reservoir of this kind of pollutants that could be transported to further growing plants.
Due to the large range of pesticides that can be used in agriculture, the
development of quick and multiresidue methods that evaluate polar and non polar pesticides is demanded. The efforts are also focused on the reduction of analysis time and sampling process. In this sense, pressurized liquid extraction is a good alternative to conventional extraction procedures such as Soxhlet and it was used for the simultaneous extraction of a wide range of polar and non polar pesticides. Different extraction conditions such as type of extractant solvent, preheat time and extraction temperature were tested. After simultaneous extraction, the analysis of non polar pesticides was performed by gas chromatography (GC), and polar pesticides were analyzed by ultra performance liquid chromatography (UPLC). In both cases, GC and UPLC were coupled to a triple quadrupole mass spectrometer (MS/MS).
The optimized extraction procedure was validated in terms of linearity,
accuracy, precision (intra and interday precision), detection and quantification limits. The average extraction recoveries ranged from 70 to 110 %, with precision values lower than 20 %. The matrix effect was also evaluated, and matrix-matched calibration was selected in order to compensate the matrix effect.
In order to assess the applicability of the proposed method, 15 soil samples
were extracted with the proposed PLE method and analyzed by GC-MS/MS and UPLC-MS/MS. ACKNOWLEDGMENTS
The authors are grateful to Andalusian Regional Government (Regional Ministry of Innovation, Science and Enterprise) for financial support (Project Ref. P07-AGR-02922).
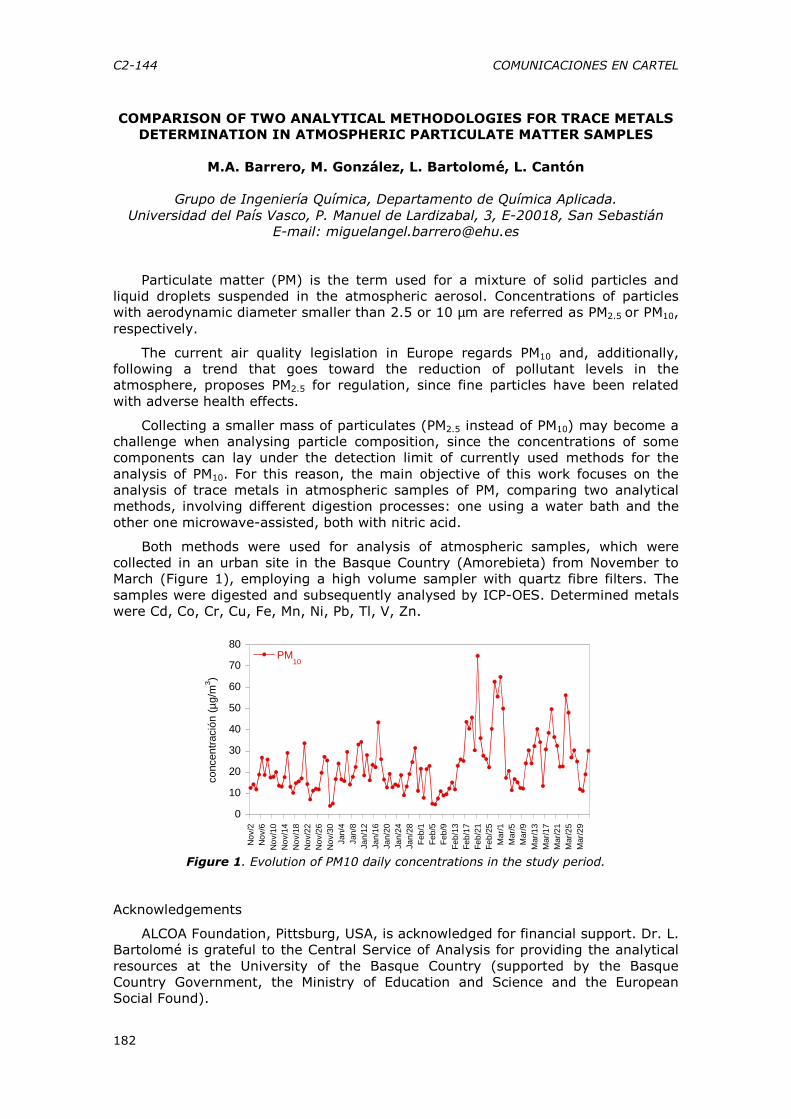
C2-144 COMUNICACIONES EN CARTEL
182
COMPARISON OF TWO ANALYTICAL METHODOLOGIES FOR TRACE METALS DETERMINATION IN ATMOSPHERIC PARTICULATE MATTER SAMPLES
M.A. Barrero, M. González, L. Bartolomé, L. Cantón
Grupo de Ingeniería Química, Departamento de Química Aplicada.
Universidad del País Vasco, P. Manuel de Lardizabal, 3, E-20018, San Sebastián E-mail: [email protected]
Particulate matter (PM) is the term used for a mixture of solid particles and
liquid droplets suspended in the atmospheric aerosol. Concentrations of particles with aerodynamic diameter smaller than 2.5 or 10 µm are referred as PM2.5 or PM10, respectively.
The current air quality legislation in Europe regards PM10 and, additionally, following a trend that goes toward the reduction of pollutant levels in the atmosphere, proposes PM2.5 for regulation, since fine particles have been related with adverse health effects.
Collecting a smaller mass of particulates (PM2.5 instead of PM10) may become a challenge when analysing particle composition, since the concentrations of some components can lay under the detection limit of currently used methods for the analysis of PM10. For this reason, the main objective of this work focuses on the analysis of trace metals in atmospheric samples of PM, comparing two analytical methods, involving different digestion processes: one using a water bath and the other one microwave-assisted, both with nitric acid.
Both methods were used for analysis of atmospheric samples, which were collected in an urban site in the Basque Country (Amorebieta) from November to March (Figure 1), employing a high volume sampler with quartz fibre filters. The samples were digested and subsequently analysed by ICP-OES. Determined metals were Cd, Co, Cr, Cu, Fe, Mn, Ni, Pb, Tl, V, Zn.
0
10
20
30
40
50
60
70
80
Nov
/2N
ov/6
Nov
/10
Nov
/14
Nov
/18
Nov
/22
Nov
/26
Nov
/30
Jan/
4Ja
n/8
Jan/
12Ja
n/16
Jan/
20Ja
n/24
Jan/
28F
eb/1
Feb
/5F
eb/9
Feb
/13
Feb
/17
Feb
/21
Feb
/25
Mar
/1M
ar/5
Mar
/9M
ar/1
3M
ar/1
7M
ar/2
1M
ar/2
5M
ar/2
9
PM10
conc
entr
ació
n (µ
g/m
3)
Figure 1. Evolution of PM10 daily concentrations in the study period.
Acknowledgements
ALCOA Foundation, Pittsburg, USA, is acknowledged for financial support. Dr. L. Bartolomé is grateful to the Central Service of Analysis for providing the analytical resources at the University of the Basque Country (supported by the Basque Country Government, the Ministry of Education and Science and the European Social Found).
COMUNICACIONES EN CARTEL C2-145
183
STRATEGIES FOR LIQUID RESIDUES OF ORGANOPHOPHOROUS
SUBSTANCES DETERMINATION IN A SOIL MATRIX USING LASER-INDUCED BREAKDOWN SPECTROSCOPY
A. Fernández-Bravoa, P. Lucenaa, A. Doñab, J.J. Lasernaa
aDpto. Quimica Analítica, Univ. Málaga, Málaga. E-mail: [email protected]
bIndra sistemas, León, Spain This work describes the capabilities of single shot-dual pulse laser-induced
breakdown spectroscopy (LIBS) to identify and characterize five organic substances, simulant of chemical warfare agents (CWA) in this case. CWA´s are organic compounds which presents a heteroatom, such as phosphorus, sulphur, arsenic, fluorine and chlorine in their structure and they are used as chemical weapons. On the other hand, we have to differentiate between simulants or precursors of CWA, in our study we used a group of liquid substances, that contain a heteroatom in their formula for this purpose. Identification of these substances usually was carried out mainly by the P, S, As, F and Cl detection. We used organophosphorous compounds in this work like liquid substances and certificate soil sample like solid matrix. Self-absorption of soil emission lines was evaluated. Preliminary studies such as timing parameters and the fluence of the laser pulses was done. Finally, calibration curves and estimation of the detection limits (LOD), for five different organophosphorous substances, were also estimated.
Figure 1. Experimental set-up for LIBS experiment.
Figure 2. Evaluation of self-absorption of soil emission lines.

C2-146 COMUNICACIONES EN CARTEL
184
ACUMULACIÓN Y TRANSFORMACIÓN DE MERCURIO EN PLANTAS
TRANSGÉNICAS DE Medicago truncatula
S. Cuello , M. Palomo, T. Pérez-Corona, Y. Madrid
Dpto. Química Analítica, Univ. Complutense, 28040 (Madrid). E-mail: [email protected]
C.R. Ochoa-Hueso; E. Manrique, J.J. Pueyo
Instituto de Recursos Naturales, Centro de Ciencias Medioambientales, C.S.I.C. Madrid.
M.E. Pérez-Corona
Dpto. Ecología. Univ. Complutense, 28040 (Madrid) El mercurio es un elemento que se libera al medio ambiente a través de
procesos naturales o artificiales. Este elemento puede depositarse en el suelo, dando lugar a suelos con una elevada concentración de mercurio. La contaminación de mercurio de los suelos es producida mayoritariamente por mercurio inorgánico (Hg(II)). Algunos componentes del suelo pueden inmovilizar el mercurio pero éste también puede ser absorbido tanto por microorganismos como por plantas.
La tolerancia y la elevada acumulación metálica que caracteriza a determinadas especies vegetales frente a los metales ha sido utilizada como estrategia para recuperar espacios contaminados de una forma limpia, económica y respetuosa con el medio. Dicha tecnología se conoce como fitorremediación.
El desarrollo de organismos mejor adaptados a condiciones de estrés puede incrementar la eficiencia del uso de las especies vegetales para restauración de zonas contaminadas.
El objetivo de este trabajo es evaluar el comportamiento de Medicago truncatula en presencia de mercurio tanto a través de los posibles procesos de bioacumulación en la planta de dicha especie, como de la biotransformación del metilmercurio en otras especies. Por otro lado se estudia el efecto de la presencia de mercurio en el medio en la producción de biomasa.
El procedimiento seguido implicó el cultivo de tres líneas de Medicago truncatula (una salvaje y dos modificadas genéticamente para acumular prolina, con una (P18) o dos (P2) copias adicionales del gen p5cs, respectivamente), en un medio hidropónico empleando perlita como sustrato inerte. Las plantas germinadas (72) fueron regadas durante 4 semanas con solución nutritiva de Hoagland. Transcurrido ese periodo las plantas fueron tratadas durante 20 días con la misma solución nutritiva suplementada con 1mg L-1 de mercurio. Las plantas se cosecharon y dividieron en raíces, tallos y hojas y se determinó el peso seco. Además se llevó a cabo el análisis de especies de mercurio. Para ello se empleó una columna cromatográfica de fase inversa acoplada al ICP-MS. Las condiciones cromatográficas utilizadas fueron las siguientes: flujo de 1mL min-1 y fase móvil 0,1% HFBA, 0,1% ácido fórmico, 2 % de MeOH, 1 mM cisteína.
Los resultados obtenidos pusieron de manifiesto que la especie mayoritaria en el caso de los tratamientos con CH3Hg
+, era el CH3Hg+, y que hay una mayor
acumulación en las raíces. En cuanto a la producción de biomasa, la comparación entre las variedades revela que en el tratamiento control y Hg (II) no hay diferencias mientras que con metil mercurio el transgénico P2 presentan mayor biomasa que las otras dos variedades (p<0,05; Anova, Tukey).

COMUNICACIONES EN CARTEL C2-147
185
ANALYSIS OF ORGANOCHLORINE PESTICIDES FROM SEDIMENTS USING PRESSURIZED HOT WATER EXTRACTION COUPLED TO SOLID- PHASE MICROEXTRACTION- GAS CHROMATOGRAPHY- MASS SPECTROMETRY
E. Concha-Graña1, V. Fernández-González1,2, S. Muniategui-Lorenzo1*;
P. López-Mahía1,2, D. Prada-Rodríguez1,2
1Department of Analytical Chemistry. University of A Coruña. Campus da Zapateira. E-15071 A Coruña, Spain..
2University Institute of Environment, Pazo de Lóngora, Liáns, Oleiros E-15179, A Coruña, Spain. E-mail: [email protected]
A fully automated, environmentally friendly, simple, and sensitive method was
developed for the analysis of 28 organochlorine pesticides in sediment samples. The procedure is based on pressurized hot water extraction (PHWE) followed by solid phase microextraction (SPME) and determination by gas chromatography-mass spectrometry (Figure 1).
For PHWE, parameters such as organic modifier, percentage of organic
modifier, temperature, and static extraction time were studied. For SPME, extraction temperature and time, desorption temperature and time, and effect of an organic modifier were studied.
AUTOMATIC SPME EXTRACTIONPHWE EXTRACTION-ASE-200 extractor
-0.5 g of sediment sample
-No dispersing agent
-Pressure: 1500 psi; Temperature: 150ºC
-Organic modifier: methanol 5%
-Static time: 5 minutes
-Fiber: 65 µm PDMS/DVB
-Extraction T: 60ºC;Extraction time:45 min
- Desorption T: 270ºC; Desorption time: 5 minutes
- Splitless time: 2 minutes
18 ml
SPME EXTRACTION
GC-MS INJECTION
PHWE EXTRACTION
AUTOMATIC SPME EXTRACTIONPHWE EXTRACTION-ASE-200 extractor
-0.5 g of sediment sample
-No dispersing agent
-Pressure: 1500 psi; Temperature: 150ºC
-Organic modifier: methanol 5%
-Static time: 5 minutes
-Fiber: 65 µm PDMS/DVB
-Extraction T: 60ºC;Extraction time:45 min
- Desorption T: 270ºC; Desorption time: 5 minutes
- Splitless time: 2 minutes
18 ml
SPME EXTRACTION
GC-MS INJECTION
PHWE EXTRACTION
Figure 1. Schematic view of the developed PHWE-SPME-GC-MS method. When these parameters were selected, the figures of merit were calculated.
The detection and quantification limits were between 0.11 - 16 µg kg-1 and between 0.24 - 22 µg kg-1, respectively. The linearity of the method was assessed spiking sediment samples at six levels of concentration ranged between LODs and 500 µg kg-1 for most of the studied pesticides. The method was validated by analysis of a reference marine sediment material (SRM 1944). The obtained results are in very good agreement with the certificate material. Another sediment reference material (SRM 1941b), with a very low concentration of pesticides was also analysed with good results. The developed method seems to be suitable for the analysis of organochlorine pesticides at ultratrace levels in environmental matrices as sediment samples.
Acknowledgements: This work was supported by “Xunta de Galicia” (Project
PGIDIT07PXIB103193PR). The authors would like to thank to Spanish Oceanographic Institute (IEO) for supply the samples.

C2-148 COMUNICACIONES EN CARTEL
186
ESTUDIO DE LA DEGRADACIÓN DE LOS HIDROCARBUROS ALIFÁTICOS Y AROMÁTICOS DE CRUDOS DE PETRÓLEO Y FUELES PESADOS BAJO
CONDICIONES SIMULADAS DE ENVEJECIMIENTO
R. Fernández-Varela, J.M. Andrade, S. Muniategui, D. Prada
Dpto. Química Analítica, Univ. da Coruña, Campus da Zapateira, s/n, E-15071, A Coruña. E-mail: [email protected]
Las características físicas y químicas del petróleo empiezan a modificarse casi
en el mismo instante en el que se produce su derrame en el medio marino. Esto se produce mediante fenómenos de extensión, evaporación, disolución, dispersión en la columna de agua, formación de emulsiones agua-hidrocarburo, oxidación fotoquímica, degradación microbiana, adsorción a material particulado en suspensión y sedimentación. Todos estos procesos interaccionan unos con los otros y se denominan colectivamente “envejecimiento del petróleo” y se manifiestan a través de cambios temporales en la composición química del vertido.
De cara a la identificación y seguimiento de los hidrocarburos vertidos al medio
marino, resulta importante realizar un estudio del envejecimiento de los mismos porque las muestras que arriban a las playas no siempre son inmediatas al momento del accidente o descarga ilegal. De hecho, el producto puede haber estado en el medio natural días o semanas, con la correspondiente degradación y, por tanto, la identificación con el origen del vertido no es en absoluto trivial. Para llevar a cabo estudios de envejecimiento de productos vertidos al medio marino y envejecidos en condiciones naturales se vertieron muestras representativas en recipientes especialmente diseñados con objeto de simular un derrame en el mar y su evolución posterior (Figura 1). El estudio de su comportamiento en el mar, y de los efectos del envejecimiento, puede ayudar a la identificación de la potencial fuente de la contaminación. Además, el grado de envejecimiento puede dar una indicación aproximada del tiempo que ha pasado desde que el producto ha sido vertido.
57
56
29
13
6
21
2
Aire
Agua
Agua
57
56
29
13
6
21
2
Aire
Agua
Agua
Figura 1. Esquema del recipiente para los vertidos simulados.
Para este estudio se han seleccionado seis productos de características
diferentes: cuatro crudos (Ashtart (medio-pesado), Brent (ligero), Maya (pesado) y Sahara Blend (muy ligero)) y dos fueles (el fuel que transportaba el buque Prestige y un fuel marino intermedio que proporcionó el Ministerio de Fomento por sus características similares al Prestige, que denominaremos IFO Marino). Agradecimientos Se agradece el apoyo económico de los gobiernos español y gallego (PROFIT-310200-2005-37, 07MDS031103PR), con la participación de los fondos FEDER.

COMUNICACIONES EN CARTEL C2-149
187
DETERMINATION OF Cu, Fe, Ni AND Zn BY FLAME AND ELECTROTHERMAL ATOMIC ABSORPTION SPECTROMETRY IN BIODIESEL SAMPLES PRODUCED
BY HOMOGENEOUS CATALYSIS
P. Gil Hernándeza, C. Molina Mayob , A. Brito Alayónb, M.T. García Rodríguezb, J.J. Arias Leóna, F. Jiménez Morenoa
aDpto. Química Analítica, Nutrición y Bromatología, Facultad de Química. Univ. de La Laguna; bDpto. Ingeniería Química y Tecnología Farmacéutica, Facultad de
Química, Univ. de La Laguna. San Cristóbal de La Laguna, Tenerife. E-mail: [email protected]
The eminent fossil fuel scarcity has motivated the search for alternative sources of energy, such as biodiesel, which is obtained from transesterification of vegetable oils or animal fats with short chain alcohol (ethanol or methanol), in presence of sodium or potassium hydroxide as a catalyst [1]. Raw materials (in general, the seeds that are the source of the oils used to produce biodiesel) can absorb/uptake some metals from water and soil during its grown. Moreover, metals can be integrated in the biodiesel composition during its production, transport and/or storage. It is known that the presence of some metals in biodiesel can lead to several problems, such as fuel decomposition or corrosion of engine parts [2,3]. Therefore, the assessment of metals concentration in biodiesel composition is of paramount importance in order to implement it as a diesel fuel.
The elements of most concern are catalyst residues, mainly sodium and potassium, as they can promote ash build-up in the engine. Catalyst poison elements, such us lead and vanadium, also need to be monitored. These are elements that are usually present at low levels in raw materials. However, they can be found in the final product (biodiesel) due to contamination in oil processing during fatty acids methyl esters production. It is also necessary to carry out the determination of copper, iron and nickel contents in biodiesel composition, because of their strong catalytic effect on the oxidation process of the fuel [4].
The goal of the this work was the development of a method for determination of Cu, Fe, Ni and Zn contents, by flame and electrothermal atomic absorption spectrometry, in biodiesel samples obtained by homogeneous catalyst from not used oils, and mixture of fried oils. The results obtained using both conventional (wet digestion) and microwave assisted digestion procedures were compared for all the samples analyzed. [1] M.G. Andrade Korn, D.S. Sodré dos Santos, B. Welz, M.G. Rodrigues Vale,
A. Paixão Teixeira, D. Castro Lima, S.L. Costa Ferreira, Talanta 73 (2007) 1–11.
[2] A.P. de Oliveira, R.D. Villa, K.C. Pinheiro, A. de Magalhães, E. Castro, Fuel 88 (2009) 764–766.
[3] A. de Jesus, M.M. Silva, M.G. Vale, Talanta 74 (2008) 1378–1384. [4] G. Knothe, R.O. Dunn, Journal of American Oil Chemists Society 80 (2003)
1021-1026.

C2-150 COMUNICACIONES EN CARTEL
188
SENSOR LUMINISCENTE SOBRE FIBRA ÓPTICA PARA LA MONITORIZACIÓN
DE TEMPERATURA EN VERTEDEROS
Silvia Mateos,1 María C. Moreno-Bondi,2 Guillermo Orellana1
Grupo de Sensores Químicos Ópticos y Fotoquímica Aplicada (GSOLFA) Dpmts. de Química Orgánica1 y Química Analítica2
Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid Tel.: +34-913944220; fax: +34-913944103; E-mail: [email protected];
Los vertederos son emplazamientos esenciales en la sociedad moderna como lugares donde depositar residuos sólidos urbanos. El residuo sólido urbano acumulado en un vertedero constituye un bioreactor que produce de forma aeróbica y luego anaeróbica, metano (CH4) y dióxido de carbono (CO2), así como un lixiviado contaminante. Aunque estos vertederos presentan una serie de capas de aislamiento con la finalidad de evitar tanto infiltraciones en las aguas subterráneas como escapes de gases a la atmósfera, existe siempre un riesgo de contaminación medioambiental. Por esta razón, la supervisión de determinados parámetros químicos y físicos, debe ser esencial con la finalidad de prevenir un posible impacto medioambiental.
La temperatura es un parámetro de gran relevancia en multitud de áreas distintas, entre las que se incluye la monitorización del estatus de un vertedero.[1] La medida de la temperatura puede constituir un buen sistema indicativo de las diferentes etapas por las que evoluciona un vertedero, jugando un papel muy importante en el análisis de la estabilidad y el comportamiento a largo plazo del mismo. Además, la práctica totalidad de los sensores químicos conocidos, entre los que se encuentran los sensores luminiscentes basados en complejos poliazaheterocíclicos de Ru(II), son dependientes de la temperatura, requiriendo una medida paralela de dicho parámetro que permita corregir la medida del analito de interés.[2]
Nuestro principal objetivo es el desarrollo y caracterización de un robusto sensor químico luminiscente sobre fibra óptica de plástico (POF) para la monitorización de la temperatura, a través de su integración en geotextiles en formato cuerda para su implantación en el interior del vertedero en un conjunto de sensores químicos sobre POF. Dicho sensor utiliza un indicador luminiscente sensible a la temperatura, Ru[(phen)2(4-Clp)]
2+, inmovilizado en un soporte polimérico impermeable al oxígeno. La interrogación de la película fina sensora se ha llevado a cabo tanto en el extremo de la monofibra óptica como en el campo evanescente de la luz que es guiada a través de la fibra óptica, mediante un sistema optoelectrónico multicanal “a medida” (Interlab IE OPTOSEN®). del equipo emplea LEDs azules de excitación, cuya intensidad se modula sinusoidalmente, y detección de la emisión roja sensible a la fase. El intervalo de operación estudiado del sensor está comprendido entre 5 y 65 ºC, con una resolución de 0,1 ºC, gran reproducibilidad y repetibilidad.
El sensor desarrollado puede servir como sistema de corrección en la monitorización de otros parámetros químicos de gran importancia en vertederos (O2, humedad, CO2, etc.).
Agradecimientos: Este proyecto (“Polyfunctional Technical Textiles Against Natural Hazards”, POLYTECT) ha sido financiado por la EU (NMP2-CT-2006-026789) y por la UCM-B. Santander (Ayudas a Grupos de Investigación 2008).
[1] S.L. Bartelt-Hunt, M.A. Barlaz, D.R.U. Knappe, P. Kjeldsen, Environ. Sci. Technol. 2006, 40, 4219–4225.
[2] G. Orellana y D. García-Fresnadillo, en Optical Sensors: Industrial, Environmental and Diagnostic Applications, R. Narayanaswamy y O. S. Wolfbeis (Eds.), Springer Series on Chemical Sensors and Biosensors Vol. 1, Springer, Berlin-Heidelberg, 2004; pp. 309–357.

COMUNICACIONES DE DOCENCIA EN CARTEL

CD1-151 COMUNICACIONES DE DOCENCIA EN CARTEL
190
TRIVIAL DE QUÍMICA ANALÍTICA (QUIMITRIVIAL)
Cristina Nerín, Ana Moreno, Blanca Garro, Jorge Ruiz, Eduardo Andrés,
Ana Mª Espasa
Dpto. Química Analítica, Centro Politécnico Superior (CPS), Universidad de Zaragoza Mª de Luna 3, 50018, Zaragoza. [email protected]
La asignatura de Química Analítica que se imparte en el 4º semestre de
Ingenieros Químicos del C. P. S. resulta una materia difícil para los alumnos. Contiene muchos conceptos que deben asimilar y relacionar entre sí, lo cual no es fácil y además requiere tiempo, que es muy escaso en un cuatrimestre. Esta asignatura tiene un gran contenido químico y por tanto requiere que los alumnos tengan una buena base de química, incluyendo el conocimiento del sistema periódico, valencias y estados de oxidación, conceptos básicos de enlaces, pH, equilibrio, nomenclatura, etc.
Con la idea de estimular el aprendizaje y hacerlo además de forma amena y eficaz, se ha elaborado con un grupo de 5 alumnos, un TRIVIAL de QUÍMICA ANALÍTICA. El mecanismo ha consistido en diseñar el juego, con el modelo del TRIVIAL convencional, pero adaptado a Química ANALÍTICA, y realizar todas las preguntas. Se han establecido 6 categorías con un número total de preguntas que se acerca a 1.000, distribuidas en:
a) EQUILIBRIOS; b) COMPLEJOS; c) ACIDO-BASE; d) PRECIPITACIÓN Y REDOX; e) INTERCAMBIO IÓNICO; f) MISCELÁNEA
Se han elaborado las tarjetas de distinto color en cada categoría y un tablero
hexagonal, con capacidad máxima para 6 jugadores. El diseño del tablero corresponde a un anillo bencénico, en el que los dobles enlaces forman a su vez parte del recorrido. Como fichas se han diseñado y fabricado micro-erlenmeyer de vidrio, pintados interiormente con los colores elegidos para el juego.
En lugar de “quesitos” que forman parte del juego tradicional, se ganan “viales”, que contienen una disolución coloreada, obtenida a partir de colorantes seleccionados en el laboratorio. Cada jugador dispone además de una gradilla de polipropileno, para colocar los viales ganados hasta completar los 6 viales que lo clasificarán como ganador de la partida.
Una vez completado el TRIVIAL, se han organizado varias sesiones de juego, fuera del horario de clase, premiando al ganador de cada tablero. El juego completo está disponible en la Hemeroteca del Campus de Ingeniería para que lo usen los alumnos como ayuda al estudio. Sin duda este TRIVIAL ha servido de estímulo para el aprendizaje, de manera divertida y amena pero muy eficaz. Permite además que los alumnos trabajen juntos, interaccionen entre ellos y comprueben el grado de asimilación y comprensión de la materia de una forma diferente. El éxito conseguido ha permitido además que la asignatura se vea de otra forma. Se presenta un juego completo a esta Sección de Innovación Docente puesto que creemos que la mejor manera de apreciarlo es ver el resultado del esfuerzo y trabajo realizado.
Si el nuevo sistema de enseñanza siguiendo las directrices de Bolonia impulsa el proceso de aprendizaje del alumno y su participación activa, este es un buen ejemplo de la nueva metodología.

COMUNICACIONES DE DOCENCIA EN CARTEL CD1-152
191
ESTRATEGIAS INNOVADORAS EN UN ENTORNO VIRTUAL PARA EL ESTUDIO
DE LA ‘TOXICOLOGÍA ANALÍTICA’. APRENDIZAJE COLABORATIVO Y CONVERGENCIA DE MEDIOS
R.M. Garcinuño, P. Fernández, A. Gallego, J.S. Durand
Dpto. Ciencias Analíticas, Facultad de Ciencias, UNED
E-mail: [email protected]
La Toxicología Analítica es uno de los campos fundamentales de la Toxicología, pues en cualquiera de las ramas de ésta, se deben utilizar los métodos de análisis químicos, entre otros. La asignatura Toxicología Analítica queda englobada en el Máster en Ciencia y Tecnología Química de la UNED, y se imparte en el Departamento de Ciencias Analíticas de la Facultad de Ciencias desde el curso académico 2008/2009, con 21 estudiantes matriculados. Esta asignatura forma parte del Módulo I. Química Analítica del citado Máster. Tiene un carácter teórico-práctico, con 12 créditos ECTS (anual), repartidos en cuatro bloques temáticos (con 14 unidades) y un último bloque con un programa práctico. Las competencias (tanto específicas como transversales) y destrezas que se espera que el estudiante adquiera y desarrolle al finalizar esta asignatura, están relacionadas con el Conocimiento, Habilidades y Actitudes.
Para seguir el curso se utiliza
la metodología propia de la UNED, empleando, fundamentalmente, la plataforma educativa aLF a través de Internet. Esta asignatura, por lo tanto, no tiene clases presenciales, por lo que los contenidos teóricos se impartirán a distancia. Al inicio de cada Unidad se propone al estudiante matriculado, un plan de trabajo personalizado así como un conjunto de tareas que deberá realizar al finalizar el estudio de cada Unidad y de cada Bloque Temático. Todas estas actividades
junto con los exámenes que se realizan en la plataforma educativa, forman parte de la evaluación continua del estudiante. Para los que no superen esta evaluación, se programa un examen presencial en su Centro Asociado en la convocatoria extraordinaria del mes de septiembre.
Todo el material desarrollado (Curso Virtual, documentos interactivos, Blog,
material audiovisual, Guías de Estudio, etc.) se comprueba que funcione con cualquier sistema operativo (Windows, Mac OS X, Linux, dando versiones para su funcionamiento en ‘iPhone’ , ‘iPod classic’, ‘iPod touch’) antes de ponerlo en la red. Se personaliza el seguimiento de cada estudiante mediante la utilización conjunta de una base de datos personal para organizar toda la información generada y el ePortfolio correspondiente. Todo el material se ha desarrollado por el Equipo Docente, utilizando máquinas Mac provistas del sistema operativo ‘Leopard’, tarjetas de grabación de sonido externas ‘M-Audio’ y microfonos ‘Rode’ de alta sensibilidad. Todos los programas informáticos utilizados para el desarrollo del material funcionan bajo el sistema operativo de ‘Apple’, y los ficheros generados, por lo general, tienen formato .pdf, .mov, y .m4a.

CD1-153 COMUNICACIONES DE DOCENCIA EN CARTEL
192
PROGRAMACIÓN EN ECTS DE UNA ASIGNATURA PRÁCTICA UTILIZANDO EL
“CONTRATO DE APRENDIZAJE”
E. Barrado, Y. Castrillejo
Dpto. Quimica Analítica. Facultad de Ciencias. Univ.Valladolid, Valladolid E-mail: [email protected]
En esta comunicación se presentan los resultados obtenidos al utilizar el
denominado "Contrato docente" o "Contrato de aprendizaje", como forma de planificación/programación en ECTS y su desarrollo-aplicación a la asignatura práctica “Experimentación en Química”, correspondiente al segundo curso de la titulación de “Ingeniero Químico”.
La metodología consiste en elaborar un “Contrato de Aprendizaje”, por acuerdo
entre el profesor y los alumnos, que debe respetar las limitaciones reales impuestas al proceso (organización docente, calendarios y horarios, disponibilidad de espacios e instalaciones, etc.), y que convierte en explícitos los "supuestos" acuerdos implícitos, normalmente establecidos entre el que enseña y el que aprende [1].
Mediante el contrato, la vinculación entre las dos partes contratantes, profesor
y alumno, adopta una forma concreta y con estipulaciones en cuanto a contenidos, espacios, tiempos, plazos, obligaciones, derechos y modos de evaluación y consecución de objetivos [2,3]. Consta de una primera parte general en la que se especifican una serie de pactos, distribuidos en tres títulos: I.-Estructura y Objetivos; II.-Obligaciones del profesorado; III.-Obligaciones del alumno. A continuación, y como anexos se incluyen las especificaciones para cada uno de los apartados del proyecto. En este caso, al tratarse de una asignatura desarrollada en el laboratorio, para cada práctica (realizada en una sesión diaria de 4 horas presenciales), se especifican: 1.-Objetivos: general y específicos. 2.- Compromisos del alumno, que incluyen la revisión previa de los conocimientos teóricos requeridos para el desarrollo de la práctica (necesarios para superar la evaluación diaria sobre los mismos); la realización de la parte práctica (presencial) y los cálculos para la obtención de un resultado (generalmente cuantitativo), así como la cumplimentación de las hojas de control y la elaboración de un informe según un modelo de "PNT" (Procedimiento Normalizado de Trabajo), para introducir al alumno en los conceptos de calidad (Quality Assurance) y buenas prácticas de laboratorio (Good Laboratory Practices). 3.-Compromisos del profesorado en cada supuesto y 4.-Criterios de evaluación, incluyendo la revisión diaria antes citada, que exime de un examen escrito.
Se presentan los resultados de la evaluación y se comparan con los que obtuvieron los alumnos en la primera parte de la asignatura, que se desarrolló de forma convencional, así como los resultados de la encuesta contestada por los alumnos al final del proceso.
[1] Knowles, M. “Using learning contracts: practical approaches to individualising
and structuring learning”. Jossey Bass, San Francisco. 1986. [2] García Castelló, E. et al. “El Contrato de aprendizaje”. 10ª Experiencia de
Innovación Educativa. Servei de Formació Permanent. Ed. Valencia. 2007. [3] Franquet Sugrañes, T. et al. “El contrato de aprendizaje en la enseñanza
universitaria”. Grup d’Innovació Docent de la Facultat de Ciències Jurídiques de la Universitat Rovira i Virgili. 2007.

COMUNICACIONES DE DOCENCIA EN CARTEL CD1-154
193
ELABORACIÓN DE PROTOCOLOS NORMALIZADOS DE TRABAJO (PNTs)
COMO SOPORTE A LA DOCENCIA EN LAS ASIGNATURAS PRÁCTICAS DE LOS LABORATORIOS DE CIENCIAS EXPERIMENTALES
D. Hernanz, V. Gallo, R. Beltrán, A. Sayago
Área de Química Analítica, Facultad de Ciencias Experimentales, Univ. Huelva.
E-mail:[email protected]
Desde la Convergencia Europea se nos propone un cambio de la conceptualización del modelo de Universidad. Esta innovación afecta al estilo docente del profesorado universitario que debe adaptarse a las nuevas exigencias. Con este proyecto de innovación docente se intenta trasladar a los laboratorios de prácticas el concepto de Sistema de Gestión de la Calidad bajo los principios de Buenas Prácticas de Laboratorio (BPL), mediante una mayor implicación del alumnado que refuerce y ayude a la comprensión de determinados contenidos del temario. Por otro lado, se pretende que el alumno además de adquirir los conceptos se familiarice con el uso de recursos y herramientas imprescindibles en los futuros egresados, permitiendo una mejora en la adquisición de habilidades y competencias.
La calidad es hoy día un recurso estratégico para la competitividad, con una gran incidencia en numerosos ámbitos profesionales, productivos y sociales. Los laboratorios analíticos no son una excepción, y si bien el concepto de calidad de la medida ha estado siempre implícito en la química analítica, ha sido en los últimos años cuando se ha producido un planteamiento explícito y sistemático de la calidad en los laboratorios analíticos, derivados del concepto de “garantía de calidad”. En las buenas prácticas de laboratorio se define un procedimiento normalizado de trabajo como aquellos documentos que describen como realizar ensayos o actividades normalmente no detalladas en los protocolos o guías. La norma UNE-EN-ISO 9000 (UNE 2000a) define un procedimiento como una forma específica de llevar a cabo una actividad o un proceso.
Los objetivos de este proyecto son implicar directamente al alumnado en el aprendizaje teórico-práctico de la asignatura Métodos Analíticos de Control de Calidad, concretamente en la elaboración de material didáctico que podrá ser utilizado además de para su propia formación, para la formación de cualquier alumno que realice asignaturas de carácter práctico. Todo ello de acuerdo con la filosofía de trabajo propuesto por los nuevos planes de estudio según el Espacio Europeo de Enseñanza Superior (EEES). Estimular la participación de los alumnos en el desarrollo de la asignatura, y así conseguir una mayor motivación del alumnado, que sirva como refuerzo positivo para el estudio práctico de la asignatura.
En un primer paso el equipo docente participante del proyecto se reunió para seleccionar los equipos de laboratorio en los que se gestionarán los PNTs. El trabajo se realizó en grupos reducidos de alumnos, a cada uno de los cuales se le asigno uno de los equipos básicos de uso más frecuente dentro del laboratorio. Este trabajo en grupo constituye una buena ocasión para completar mutuamente la formación del profesorado y alumnado, supone además, un acercamiento al conocimiento y manejo de referencias bibliográficas analíticas y de calidad, que les serán de extraordinaria ayuda a la finalización de su formación académica. Los materiales multimedia elaborados posibilitan la realización de los objetivos y son, además un soporte a la docencia en las asignaturas prácticas de los laboratorios de Ciencias Experimentales.

CD1-155 COMUNICACIONES DE DOCENCIA EN CARTEL
194
ADAPTACIÓN DE LAS PRÁCTICAS DE ANÁLISIS INSTRUMENTAL AL ESPACIO DE CONVERGENCIA EUROPEA. ELABORACIÓN DE MATERIAL
DIDÁCTICO
A. Sayago, D. Hernanz, V. Gallo, R. Beltrán.
Área de Química Analítica. Facultad de Ciencias Experimentales. Universidad de Huelva. Avd. Tres de Marzo S/N. 21071. Huelva
E-mail: [email protected]
En la actualidad, la metodología didáctica más extendida entre el profesorado universitario en la impartición de asignaturas de carácter práctico es fundamentalmente unidireccional, en la que el alumno se limita básicamente a seguir las pautas indicadas en los protocolos de prácticas previamente elaborados por el profesor, ya que los contenidos de los mismos no se encuentran recogidos en uno o unos pocos textos de consulta, sino que aparecen dispersos en multitud de documentos de diversa índole. El aprendizaje autónomo dentro del marco de este tipo de actividades, parece una competencia consolidada si se tiene en cuenta que el alumno participa activamente en el desarrollo de las mismas. No obstante, el conocimiento del fundamento teórico mediante la búsqueda de información a partir de los recursos disponibles, favorece la motivación del alumnado que no se limitaría sólo a seguir los protocolos.
El trabajo desempeñado durante el desarrollo de este proyecto de innovación
docente ha permitido trasladar a los laboratorios un concepto más amplio de aprendizaje autónomo fomentando una mayor implicación de los alumnos (a la hora de buscar, adquirir y elaborar los conocimientos) que ha reforzado y ayudado a la compresión de determinados contenidos propuestos en el temario. Por otro lado, se pretendía que el alumno además de adquirir los conceptos se familiarizara con el uso de recursos y herramientas imprescindibles en su futuro profesional. Este trabajo en grupo constituye una buena ocasión para completar mutuamente la formación del profesor y alumnos, supone además, un acercamiento al conocimiento y manejo de referencias bibliográficas analíticas e instrumentales, que les serán de extraordinaria ayuda a la finalización de sus estudios.
Los cuadernos de prácticas elaborados como resultado de este trabajo serán
editados para la consulta y aplicación de todos aquellos alumnos, personal docente y técnicos de laboratorio implicados en esta asignatura.

COMUNICACIONES DE DOCENCIA EN CARTEL CD1-156
195
DISEÑO DE UNA ACTIVIDAD DE APRENDIZAJE PARA LA ASIGNATURA
“ESPECTROSCOPÍA”
M. Teresa Gómez, M. Sierra Jiménez y Juan R. Castillo
Analytical Spectroscopy and Sensors Group (GEAS) Institute of Environmental Sciences. University of Zaragoza
Pedro Cerbuna, 12. ZARAGOZA-50009 (SPAIN) E-mail: [email protected]
En este estudio presentamos una actividad de aprendizaje basada en fomentar
el trabajo en grupo entre alumnos de la asignatura “Espectroscopía” del II Ciclo de la Licenciatura de Químicas de la Universidad de Zaragoza. Se trata de una actividad que los profesores participantes están realizando en cursos anteriores pero que, en esta adaptación progresiva hacia el EEES, consideramos que se debe canalizar y estructurar de forma que las actividades llevadas a cabo por los alumnos sean consideradas como parte de la enseñanza no presencial y encuadrada dentro de la evaluación continuada del estudiante. El trabajo invertido por los alumnos en “Espectroscopía”, trabajo no presencial y de carácter grupal, debe ser evaluado y valorado y éste es el objetivo primordial del estudio.
Se intenta fomentar competencias transversales como:
- capacidad de trabajo en equipo - búsqueda de información y de documentación - capacidad de discriminación de información relevante - valoración del trabajo personal, tanto del propio como del resto de los alumnos - exposición oral de los informes y trabajos realizados
La experiencia se divide en dos etapas:
- preparación del material por parte del profesorado. Comprende la aportación de documentación sobre aplicaciones de técnicas instrumentales atómicas, el diseño de un diario personal basado en la elaboración de fichas sobre las búsquedas bibliográficas, la preparación de cuestionarios de evaluación y autoevaluación que tienen en cuenta la redacción, presentación audiovisual o exposición oral del trabajo realizado entre otros aspectos.
- implementación de la actividad en la clase formando grupos reducidos de alumnos que desarrollan un trabajo basado en:
o realización de un trabajo escrito sobre la documentación proporcionada por el profesor y relacionada con los conocimientos teóricos de la asignatura
o búsqueda de información en bases de datos relacionada con el tema concreto del trabajo
o exposición oral del trabajo realizado mediante presentaciones power point por parte de un miembro de cada grupo elegido de forma aleatoria momentos antes del inicio de la sesión
o realización de cuestionarios de evaluación y de autoevaluación
Estas actividades se realizan bajo la supervisión y la asistencia tutorizada de los profesores según un calendario y una organización de tiempos planteada por los mismos.
Como conclusión podemos destacar que la experiencia es muy eficaz en el caso de los alumnos y resulta enriquecedora para los profesores. Además, el hecho de que el trabajo sea considerado dentro de la actividad continuada de la asignatura y valorado como parte de la calificación global hace que el esfuerzo no presencial del alumno adquiera relevancia y complemente la importancia del examen final de la asignatura.

CD1-157 COMUNICACIONES DE DOCENCIA EN CARTEL
196
INFLUENCIA DEL INSTRUMENTO EN LA CALIDAD DEL MÉTODO ANALÍTICO Y SU APLICACIÓN EN ESPECTROFOTOMETRÍA DE ABSORCIÓN MOLECULAR
UV-VIS (EAM UV-VIS).
J. Galban1, S. de Marcos1, I. Sanz-Vicente1 , C. Ubide2 y J. Zuriarrain2.
1Dpto. Química Analítica, Facultad de Ciencias, Universidad de Zaragoza E-mail: [email protected]
2Dpto. Quimica Aplicada, Univ. País Vasco, San Sebastián E-mail: [email protected]
¿Qué criterios analíticos se deben usar a la hora de elegir un instrumento? La
presente comunicación incide en este aspecto tan fundamental de un método analítico, usando la EAM UV-vis como técnica modelo. Se propone:
1) una forma de mostrar al alumno cómo el instrumento induce errores sistemáticos y aleatorios en la absorbancia medida, en qué medida afectan a las características analíticas de un método y de que parámetros de fabricación del instrumento dependen.
2) cómo evaluar esos parámetros a partir de datos extraídos de las especificaciones técnicas del equipo, dando al estudiante un criterio de elección.
Este enfoque supone replantear la forma de abordar y estructurar la Instrumentación durante la presentación docente de una técnica instrumental.
Cuando se usa un espectrofotómetro de absorción molecular UV-visible el parámetro de calidad más importante inherente al propio instrumento es el intervalo de cumplimiento de la ley de Beer, que condiciona el rango de respuesta lineal del método. Ese intervalo depende, por un lado, del ruido instrumental (error aleatorio) y, por otro, de la luz parásita (error sistemático). En las figuras dentro de la tabla siguiente se muestra cómo el primero afecta especialmente a valores de absorbancia bajos mientras que el segundo afecta a los altos; se recuerdan también las ecuaciones para su cálculo.
Zona de absorbancia
Efecto (tipo de error)
Ecuación que lo rige Representación
Altas/bajas Ruido (aleatorio)
43 4100 1 10AbsAbss ´
DERAbs Abs
= = +k
0
1
2
0 0,5 1
DER, %
Abs
k=0,001
Altas Luz parásita
(sistemático)
100 110 Abs
Abs%error
log p−
= + −
0
1
2
0 1 2 3
%
error
Abs
p=0,0001 (0.01%)
En estas ecuaciones k y p dependen de cada instrumento y pueden evaluarse a partir de las especificaciones técnicas del fabricante; así, las que se refieren al Ruido Fotométrico o a la Precisión Fotométrica permiten calcular k (se dan como incertidumbres para valores de absorbancia concretos), mientras que la que habla de Luz Parásita ofrece el valor de p máximo del equipo en forma %T. Conocidos los valores de k y p, es posible comparar “sobre el papel” la calidad de distintos instrumentos o comprobar el correcto funcionamiento de un equipo en particular. Por ejemplo para los valores de k y p dados en las figuras, si no se quiere que la incertidumbre en la absorbancia sea mayor del 1% se deberían usar valores de absorbancia comprendidos entre 0´050 y 2´800 aproximadamente.
Agradecimientos. Este trabajo ha sido financiado con el proyecto coordinado CTQ-2008-06751-C02/BQU del MICINN y por UPV/EHU (Project GIU07/58).

COMUNICACIONES DE DOCENCIA EN CARTEL CD1-158
197
DESARROLLO Y EVALUACIÓN DE COMPETENCIAS GENÉRICAS EN LA
ASIGNATURA DE QUIMIOMETRÍA
M.I. Sánchez-Báscones
Dpto. Quimica Analítica Escuela Universitaria Politécnica, Univ. Valladolid, Valladolid. E-mail: [email protected]
La adaptación de la Universidad al Espacio Europeo de Educación Superior
(EEES) implica una serie de cambios sustanciales en diversos ámbitos, como los planes de estudio, las metodologías docentes, las actividades a realizar o los métodos de evaluación que actualmente se emplean en nuestras Universidades
Desde hace algunos años, la Universidad de Valladolid (UVa), dentro del proceso de reforma para su adaptación al Espacio Europeo de Educación Superior (EEES), está llevando a cabo una serie de acciones especiales, orientadas a la modificación del modelo de enseñanza, más centrado en el aprendizaje de los estudiantes y dirigido a la adquisición y desarrollo de competencias.
La incorporación de las competencias genéricas a los objetivos de las asignaturas en el proceso de adaptación al nuevo EEES, obliga a renovar las modalidades organizativas y los métodos de enseñanza [1], lo que nos lleva a la realización, de una serie de actividades que fomenten el desarrollo de estas competencias tanto en clase como de forma no presencial.
En este trabajo se presenta la metodología de aprendizaje y los procedimientos de evaluación llevados a cabo en la asignatura, Quimiometría, impartida en tercer curso la Titulación de Ingeniero Técnico Industrial en Química Industrial, en la Escuela Universitaria Politécnica de la Universidad de Valladolid. La competencia genérica a desarrollar en este caso ha sido el “Trabajo en equipo”
La experiencia diseñada ha consistido en el estudio de uno de los módulos de la asignatura denominado Calibración, mediante el análisis de la bibliografía suministrada y su aplicación a la resolución de problemas. Se han formado equipos de cuatro estudiantes y se les ha asignado la resolución de un número de problemas de una colección, además se les ha suministrado bibliografía y material para facilitar la comprensión del tema. Todos los grupos deben asistir a una tutoría obligatoria donde el profesor valora en primer término el grado de consecución de los objetivos, entregar los problemas resueltos con una introducción teórica previa a cada problema y realizar la exposición de uno de los problemas en clase.
La evaluación que se ha realizado ha tenido en cuenta por un lado los conocimientos adquiridos y por otro la competencia genérica considerada asignando un peso en la calificación a cada aspecto. La evaluación de Trabajo en equipo se ha realizado por coevaluación y por evaluación directa [2], para ello se ha elaborado una rúbrica [3]cuyo formato se presenta en esta comunicación y que cumple con una doble función, formativa y de evaluación
El trabajo recoge además la opinión de los estudiantes sobre los aspectos recogidos en las rúbricas y su utilización
[1] M. de Miguel Díaz, “Modalidades de Enseñanza Centradas en el Desarrollo de
Competencias”, Oviedo 2006. [2] R. Piqué, J. Domingo. “Taller sobre Evaluación de Competencias Genéricas”.
2007. [3] R. Patiño, E. Alarcia, B. Martínez, C. Pérez, M.I. Sánchez .” INECE 08 Madrid

CD2-159 COMUNICACIONES DE DOCENCIA EN CARTEL
198
DECONSTRUCTING LABORATORY PRACTICES: PROJECT BASED LEARNING
FOR ANALYTICAL EXPERIMENTATION
A. Gustavo González, Raquel Fuentes, Rocío Franco, Antonio Díaz, Andrés Escudero, Antonio Delgado, Amo Sánchez
Departamento de Química Analítica, Universidad Hispalense, Sevilla.
E-mail: [email protected] European Higher Education Area (EHEA) involves a student centred educational
paradigm with the focus on learning outcomes and acquired competencies (capacities, attitudes, abilities, skills). Teach to learn involves items such as multidisciplinarity, projects and cases, directed learning, working teams, autoevaluation, peer-to-peer evaluation and so forth.
In this way, Project Based Learning (PBL) is a didactical strategy in which the students (organized in groups) develop projects. The aims of PBL are the integration of knowledge and competencies of different areas, the developing of intellectual abilities and to promote independent learning, team-working and autoevaluation. These projects can be carried out for laboratory practices with sufficient number of credits to enable the students to design their own protocols and properly elaborate reports involving documentation and multimedia presentations. Portable documentation (pdf, djvu) written in English together commented videos of experiences exhibit the transversality (offimatics, language, multimedia skills). In our case we have performed such a task in the subject “Experimentación Química” (Fifth course of the present-day degree in Chemistry). I have worked with six students.
The general topic was the study of oscillating chemical reactions. Each student spends three days to search for the topic. Aside from textbooks and monographies, internet research is fully recommended. Tools such as SCOPUS, Scirus, SciFinder (Web engine), Sciencedirect were used. After these three days, students make an appointment with me and in the meeting, they are split in working couples and a subtopic is selected by them. In our case the topics were: The Briggs-Rauscher reaction, and the Belousov-Zhabotinskii reaction in time and in space.
Each couple has a week to prepare a protocol for developing a chemical demonstration and written in English. The protocol includes Introduction, Outline of the experiment, Material and Reagents, Apparatus, Procedures, Hazards and Disposals and Bibliography. This stage is performed by continuous supervision of the teacher. The final revisited document is submitted to me as a pdf file (from a Word file by using some free software such as PrimoPDF). Once the protocol is suitable to be push ahead, the couples have to carry out the experiments within the next week.
Then each couple records a video of the chemical demonstration (the cam can be brought by the students or loaned by the Faculty). Students are trained to compress the video recorded using a MPEG4 codec such DivX or Xvid with the help of free software such as VirtualDubMod. According to the duration of the video, students perform a speech in English introducing and explaining the experiment. Then they record it (generally with a microphone attached to a PC by using a free software audio editor like Audacity) and prepare a final audio file compressed in mp3 containing the audio of the speech combined with a background music audio suitably selected. Finally video and audio are multiplexed for instance with Windows Movie Maker in order to have a final AVI container with a MPEG4 compliant video stream (DivX, Xvid) and a mp3 audio stream (44.1 MHz, 129 kbps, stereo) with a maximum size of 1 GB and a maximal duration of 10 minutes.
The students will upload the corresponding videos to Youtube to disseminate their work.

COMUNICACIONES DE DOCENCIA EN CARTEL CD2-160
199
DEVELOPMENT OF SPOKEN/WRITTEN FLUENCY IN SCIENTIFIC ENGLISH
APPLIED TO ANALYTICAL CHEMISTRY
D. Bellido, R. Castro, L. Cubillana, E. Durán, E. Espada, M.D. Galindo, M.V. García, M.D. Granado, D. Guillén, C. Mendiguchía, M. Milla,
I. Naranjo, R. Natera, J.J. Pinto, M.C. Rodríguez
Dpto. Química Analítica, Univ. Cádiz, Polígono Río San Pedro, s/n, C.P.11510 Puerto Real, (Cádiz)
E-mail: [email protected] One of the main objectives of the Governing team of the University of Cádiz is
focused on favouring the development and innovation of its Teaching and Researching Staff. With this in mind, the Vice-Chancellor for Information Technologies and Teaching Innovation has recently set up a Development and Innovation Plan designed for all the University staff. Inside this Plan, the first stage is the preparation of Self-Reports of Development of Competences, carried out individually, by the University Teaching and Researching Staff. Using these reports, a final Development Report for each department is made by the Vice-Chancellor’s office. The final Development Report of the Analytical Chemistry Department revealed the need to improve one of the studied abilities: the spoken/written fluency in a second language.
Furthermore, the Vice-Chancellor, in its Program of European Convergence for
improvement and innovation in teaching for staff, provides the possibility of support for different proposals related to the Development and Innovation Plan mentioned above. Using this opportunity, the Department of Analytical Chemistry from the University of Cádiz, requested a project focused on the improvement of the spoken/written level of scientific English of the participants.
The learning activities were proposed by the group and supervised by the
English teacher. The total length of the project was fifty hours and it was composed of three kinds of activities: lectures with contact hours, individual activities and group work. A total amount of thirteen sessions of two hours each of contact time was applied with an English teacher. Different types of oral presentations were carried out by the participating staff during the sessions such as notional learning classes; description of laboratory work; the use of multimedia interactive exercises; and explanation of scientific articles. The individual activities were sixteen hours long and four sessions were employed to discuss the work and activities program and the learning outcomes of the project. All the activities were carried out using the English language as the communication tool both for speaking and writing.
With this proposal, the Teaching and Researching Staff of the Analytical
Chemistry Department of the University of Cádiz have improved their skills in scientific English according to the requirements of the new Common European Space in Higher Education.
Acknowledgements: This work has been supported by Vice-Chancellor for Information Technologies and Teaching Innovation from the University of Cádiz: European Project, Department Development Plan-CIA05, 2008-2009. The authors gratefully acknowledge the kindness and teaching of English professor Antonio Manuel Brenes Castaño.

CD2-161 COMUNICACIONES DE DOCENCIA EN CARTEL
200
DESARROLLO DE UN PROGRAMA EDUCATIVO INFORMÁTICO PARA EL
TRATAMIENTO DE DATOS CINÉTICOS
Teresa Galeano-Díaz, Anunciación Espinosa, Arsenio Muñoz de la Peña, Isabel Durán-Merás e Isabel Acedo
Department of Analytical Chemistry, University of Extremadura, Avda. de Elvas s/n,
06071, Badajoz, Spain. E-mail: [email protected]
Se plantea el desarrollo de un programa educativo informático para el tratamiento de señales cinéticas, que cubra las necesidades de los alumnos en los trabajos experimentales de laboratorio del área de Química Analítica. Dicho programa facilitará al alumno la comprensión del tratamiento de datos cinéticos y su conveniencia para llevar a cabo determinaciones cuantitativas en análisis de rutina. Se pretende la divulgación de dicho programa en formato CD, acompañado con las instrucciones de aplicación del mismo, para ser empleado en las prácticas de los estudiantes de asignaturas del área de Química Analítica, que incluyen contenidos de análisis instrumental, mediante medidas cinéticas. El Programa se desarrollará en el entorno de programación MATLAB, e incluirá rutinas relacionadas con el cálculo de velocidades de reacción sobre curvas cinéticas experimentales. El programa está basado en un trabajo educativo desarrollado previamente en el Departamento de Química Analítica, por varios miembros del grupo (1). Este trabajo de desarrolla y financia como Proyecto de Innovación Educativa en la modalidad de “Elaboración de Materiales para Tutorías y Apoyo a la Enseñanza Presencial”, dentro de la Convocatoria de Plan de Actuación de la UEX al Espacio Europeo de Educación Superior correspondiente al curso 2008-2009.
Se pretende así disponer de una herramienta metodológica que posibilitará una
mayor implicación del alumno en el análisis de sus datos cinéticos experimentales de laboratorio, que incidirá en una mejor comprensión de los mismos y eliminará, previsiblemente, la falta de confianza en las medidas cinéticas que el alumno a veces tiene en base a la dificultad del tratamiento de los datos. Los aspectos teóricos y prácticos relacionados con las medidas cinéticas son descriptores en varias asignaturas troncales u obligatorias de la Licenciatura de Química actual, como son Análisis Instrumental (3º curso de Químicas, obligatoria), Química Analítica Avanzada (4º curso de la Licenciatura en Química, troncal) y Experimentación en Química Analítica (5º curso de Químicas, troncal) y de la Licenciatura en Ciencias Ambientales, en Introducción a las Técnicas Instrumentales y Separativas. Además, en el proyecto del nuevo Plan del Grado en Química, aún en revisión, también se desarrollarán estos contenidos en asignaturas tales como Análisis Instrumental y Química Analítica Avanzada, entre otras, tanto desde el punto de vista teórico como experimental.
[1] A. Espinosa Mansilla, A. Muñoz de la Peña, F. Cañada-Cañada, D. Bohoyo Gil, D. González Gómez, “Experimentos computacionales en Química Analítica. Estudio cinético educativo usando programación en Matlab”, Chemical Educator, 12 (2007) 190-194.

COMUNICACIONES DE DOCENCIA EN CARTEL CD2-163
201
ACCIONES DE INNOVACIÓN DOCENTE EN LA ASIGNATURA “ANÁLISIS DE
ALIMENTOS”
M.A. Bello-López, R. Fernández-Torres, M. Callejón-Mochón, M.D. Ramos-Payán, M. Villar-Navarro, J.L. Pérez Bernal
Dpto. de Química Analítica. Univ. de Sevilla. E-mail:[email protected]
Ante el inminente proceso de adaptación denominado “Espacio Europeo de
Educación Superior (EEES)” por el que, a diferencia del modelo de enseñanza tradicional (en el que el alumno recibía una formación pasiva), el alumno es parte activa y protagonista en su formación, el docente se enfrenta al reto de conjugar una formación clásica con nuevas metodologías de enseñanza.
El nuevo sistema de créditos ahora se basa, fundamentalmente en el trabajo
personal del estudiante para la consecución de las competencias marcadas en cada asignatura con el fin de mejorar e implementar su propia formación. En este sentido juega un papel muy importante el educador, que debe involucrarse activamente en el desarrollo y aplicación de nuevas metodologías enseñanza-aprendizaje, lo cual conlleva un nuevo diseño estructural de las asignaturas.
El docente debe cambiar su papel de líder dentro del grupo para convertirse en asesor y conseguir así guiar a los alumnos hasta la consecución de los objetivos marcados en cuanto a competencias se refiere. Este planteamiento implica la completa integración y uso de nuevas tecnologías de información y comunicación (TICs) como herramienta docente.
La interacción alumno-docente se extiende no solo al aula sino al espacio
virtual que componen los nuevos recursos educativos disponibles creándose de este modo una comunicación fluida y continua que le permite afrontar con mayores posibilidades de éxito su etapa formativa.
En este sentido se vienen desarrollando desde el año 2007 una serie de
acciones de innovación para adaptarse al nuevo modelo de enseñanza, que a continuación se detallan:
Uso de la plataforma WebCT, que supone un contacto directo y continuo entre
educador y alumno, siendo una vía rápida de transmisión bidireccional de comunicación y conocimientos. Además, se fomenta la formación integral del alumnado implicándolo activamente en el uso de nuevas tecnologías en el proceso formativo.
Creación de foros de debate, herramienta que permite un contacto permanente
entre profesor-alumno y alumno-alumno. La tutorización de los mismos se puede realizar de forma activa lo que asegura un seguimiento de la evolución del aprendizaje del alumnado.
Establecimiento de tareas, mediante las cuales se pueden ampliar los contenidos de la asignatura entre las que se encuentra la preparación de temas, evaluables, para su exposición al resto del alumnado.
Elaboración de preguntas de auto-evaluación, herramienta con la que el
alumno puede realizar un seguimiento real de las competencias y destrezas que ha adquirido.

CD2-164 COMUNICACIONES DE DOCENCIA EN CARTEL
202
APRENDIZAJE BASADO EN PROBLEMAS (ABP) COMO HERRAMIENTA DE INNOVACIÓN DOCENTE EN LA ASIGNATURA ANÁLISIS QUÍMICO
G. Galán y M.T. Montaña
Dpto. Química Analítica, Fac. Farmacia, Univ. Sevilla.
E-mail: [email protected]
La técnica de PBL o ABP (aprendizaje basado en problemas) es una estrategia didáctica muy utilizada para el aprendizaje activo del alumno en el entorno del Espacio Europeo de Educación Superior (EEES), ya que permite al estudiante conectar la teoría con la aplicación, relacionar conocimientos diversos y conseguir una formación más globalizada. Es una actividad grupal en la que el estudiante toma responsabilidades y decisiones que son básicas para su proceso formativo [1]. En el presente trabajo se plantea la incorporación de la metodología ABP en la enseñanza de la asignatura Análisis Químico de la Licenciatura de Farmacia.
La metodología seguida se ha desarrollado en distintas etapas:
1ª Etapa: Planteamiento del problema a los alumnos. Se realizó en el aula, al comienzo del segundo cuatrimestre, para poder aplicar los conocimientos teóricos impartidos en el primer cuatrimestre referente al Proceso Analítico General.
En esta sesión los profesores responsables facilitaron al alumno el título del problema, una pequeña introducción y el enunciado del mismo. Así mismo, se incluyeron materiales de apoyo como los objetivos a conseguir, los diferentes puntos de control establecidos para el seguimiento de su desarrollo, los distintos recursos de que dispone el alumno (se promoverá que el alumno obtenga información de distintas fuentes: libros, revistas especializadas, prensa, internet, etc..), tutorías presenciales y on-line haciendo uso de las herramientas de la plataforma virtual de la Universidad (webCT), tiempo del que disponen para la resolución del problema y criterios de evaluación. 2ª Etapa: Formación en el Aula de grupos de aproximadamente 5 alumnos para el estudio y resolución del problema. Estos grupos han permanecido a lo largo de todo el cuatrimestre. Cada grupo se ha organizado internamente, asignando un rol a cada uno de los miembros. 3ª Etapa: Estudio individual del alumno de todos los conocimientos teóricos previos necesarios para poder resolver el problema planteado. 4ª Etapa: Los estudiantes tienen una primera discusión para decidir qué conceptos ya conocen, qué más necesitan saber y como repartirse el trabajo .Así mismo establecen reuniones periódicas entre ellos mismos para comprobar la evolución del trabajo individual de cada uno y elaborar el informe final. 5ª Etapa: Previa petición de una tutoría por parte de cada grupo, los profesores han revisado el trabajo realizado y le han aportado los datos necesarios para la correcta resolución final del problema. 6ª Etapa: El Informe final de cada grupo ha sido enviado por escrito y en CD a los profesores responsables a través de la plataforma webCT y deberá ser defendido y razonado ante el resto de los grupos de trabajo y del resto de la clase.
La evaluación este sistema de ABP presenta una triple vertiente: Evaluación de
la participación de los compañeros de grupo, valoración del trabajo en grupo, evaluación de esta herramienta de aprendizaje por parte de los alumnos. [1] A.G. Asuero, G. Galán, M.A. Herrador, A.M. Jiménez, M.T. Montaña, M.T.
Morales, M.J. Navas, Trends in Pharmacy Education, European Association of Faculties of Pharmacy, Annual Conference (2007) pp. 56-57.

COMUNICACIONES DE DOCENCIA EN CARTEL CD2-165
203
THE TECHNIQUE OF PROBLEM-BASED LEARNING (PBL) APPLIED TO THE TEACHING-LEARNING PROCESS OF ANALYTICAL CHEMISTRY, DEGREE
PROJECT
R.A. Lorenzoa, A.M. Carroa, P. Fernándezb
aDpto. de Química Analítica, Nutrición y Bromatología, Facultad de Química. Avda. de las Ciencias, s/n
bDpto. de Anatomía Patológica y Ciencias Forenses. Servicio de Toxicología Forense. Facultad de Medicina y Odontología. R/ de San Francisco, s/n
Universidad de Santiago de Compostela, 15782-Santiago de Compostela, Spain. E-mail: [email protected]
The subject Degree Project (18 ECTS credits, 18x25 = 450 hours of student work to distribute during the course) belongs to the block training experimental basis and is providing the future formation of a chemical introduction to research in the laboratory through a proposed work and protected by teachers of the University. It is appropriate to the methodology of Problem-Based Learning (PBL).
The Problem-Based Learning appears like an innovating educative proposal that
is characterized so that the learning is student centred, promoting significant learning as well as the development of a number of important skills and abilities in the present professional surroundings. The professor becomes a facilitator of the learning process. Today PBL may be one of the methods appropriate for new models of Higher Education based on learning.
In the development of this project of educational innovation, a virtual class in
the WebCT platform was created. Students will train in the use of tools and technologies necessary to perform the experimental work in laboratory Analytical Chemistry having to take their own learning in analytical aspects using the analytical equipment and instrumentation needed for this. Being this is the essence of the method of PBL, the work is directed to:
1. Definition of the problem to develop. 2. Bibliographic Search. 3. Performance and validation of the analytical method through the analytical
properties. 4. Data acquisition and processing. 5. Application of analytical methodology for analysis of real samples. 6. Development of a report of the experimental work performed in the laboratory
in which it describes in detail each of the stages. The results obtained are discussed with the teacher's guide, for making decisions about the initial problem.
7. Development of an oral presentation of experimental work and the ability to answer the questions and comments of the court before which presents the work.
References
• P. Morales Bueno, V. Landa Fitzgerald, Theoria, Vol. 13: 145-157, 2004 • B. Duch, S. Gron, and D. Allen, Ed. The Power of Problem-Based Learning,
Stylus Publishing, Virginia (2001) • http://www.udel.edu/chem/white/teaching/BiochEd/articles.html


ÍNDICE DE AUTORES

ÍNDICE DE AUTORES
206
A Acedo, I. ................................... 200 Agüera, A. ................................. 169 Aguilera-Luiz, M.M. ..................... 115 Airado-Rodríguez, D......................57 Alcalà, M. ...........................105, 128 Alcántara, J.F. ............................ 176 Alcázar, A. ...................................42 Almendral Parra, M.J.....................63 Alonso, E. ...........................165, 173 Alonso Castillo, M. ...................... 178 Alonso Mateos, A. .........................63 Amigo, J.M. .................... 21, 95, 133 Andrade, J.M.............................. 186 Andrés, E. ................................. 190 Anzano, J. ................................. 174 Aparicio, I...........................165, 173 Arancibia, J.A...............................28 Arias León, J.J............................ 187 Arraez-Román, D..........................65 Arranz, A................................... 162 B Babiano, R................................. 126 Bakkali, K. ...................................43 Ballesteros, E. ..............................43 Baños, C.E................................. 138 Barbero, M............................. 50, 51 Barciela Alonso, M.C. ....... 31, 62, 170 Barco-Bonilla, M.N. ..................... 181 Barrado, E. ............73, 131, 147, 192 Barrero, M.A .......................156, 182 Barriola, A. ................................ 132 Bartolomé, L. ............................. 182 Basauri, O. ................................ 130 Bautista, M. .................................95 Blanco, M. .......95,103, 105, 112, 128 Blanco Gomis, D...........................38 Bellido, D. ................................. 199 Bello, C. .................................... 174 Bello-López, M.A................... 46, 201 Beltrán, E. ...................................87 Beltrán, M...........................106, 107 Beltrán, R. ........... 106, 107, 193, 194 Bendicho, C. ......... 117,118, 119, 120 Bermejo, E. .................................34 Bermejo Barrera, P. ......................... ...................... 31, 62, 108, 170, 171 Berregi, I............................... 44, 54 Berrueta, L.A. ........... 91, 92, 93, 162 Berzas, J.J. ................................ 104 Besert, M.I. ............................... 101 Bohoyo, D. ................................ 126 Bolea, E. ........................ 25, 94, 151 Bonel, L................................. 74, 76
Bonilla, B................................... 174 Bonilla Simón, M.M. ....... 72, 145, 148 Bordagaray, A. ........................... 122 Bouza-Deaño, R. .................. 79, 161 Boyero Benito, J.F. .......................63 Brito Alayón, A. ...................145, 187 Bro, R. ................................ 55, 133 C Cabaleiro, N............................... 120 Cabrera, M.C................................47 Cáceres, J.O. ............................. 125 Cacho, C. ............................ 58, 135 Cacho, J. ..........................71, 80, 86 Callao, M.P. .................. 53, 123, 127 Callejón-Mochón, M. ............. 46, 201 Calvo, L..................................... 155 Camacho-Muñoz, D..................... 173 Cámara, C. ...... 29, 64, 113, 163, 164 Canals, A................................... 119 Candela, L. ..................................19 Cano Pavón, J.M. ........... 32, 177, 178 Cantón, L. ..........................156, 182 Cantrell, K. ................................ 160 Cañada-Cañada, F. .......................28 Capitán-Vallvey, L.F. ................... 160 Caracena, R. ................................44 Cardador, M.J............................. 137 Carrasco-Pancorbo, A....................81 Carretero, M.C. .......................... 179 Carrillo, J.D................................ 143 Carro, A.M. ................. 129, 180, 203 Casais, C. .................................. 146 Casas-González, J....................... 174 Cassella, R.J. ...............................31 Castañeda, G. ............................ 109 Castillo, J.R...... 25, 74, 75, 76, 77, 94 .......................... 151, 152, 153, 195 Castrillejo, Y. ....... 131, 147, 179, 192 Castro, R. .................................. 199 Cebolla, V.................................. 168 Cela, R. ...................... 129, 146, 180 Cepriá, G................................... 153 Cervera, M.L. ....................... 99, 100 Charipar, N.A. ............................ 110 Chiappino, A. ............................. 135 Chicharro, M. ...............................34 Coello, J. ............................... 48, 94 Concha-Graña, E. ....................... 185 Concha-Herrera, V. .......................52 Contreras-Acuña, M. .....................26 Cooks, R.G. ............................... 110 Costa-Fernández, J.M....................84 Costas, M. .................. 117, 118, 120 Crevillén, A.G...............................33 Cruz, J. .......................................95

ÍNDICE DE AUTORES
207
Cubillana, L................................ 199 Cuello, S. .................................. 184 Cueto, S. .....................................36 Cueva-Mestanza, R. .................... 112 Culzoni, M.J. ................................89 D da Silva, E.G.P. .......................... 113 de Andrés, F. ............................. 109 de la Calle, I. ............................. 120 de la Fuente, M.A. ........................14 de la Guardia, M. ......31, 99, 100, 101 de la Rosa, J. ............................. 158 de la Rosa-Medero, D....................64 de la Torre, A...............................30 de-los-Santos-Álvarez, N. ............ 111 de Marcos, S. .................................. ...................102, 136, 167, 168, 196 de Orbe-Payá, I.......................... 160 Debán, L. ....................................66 del Campo, G. ........................ 44, 54 del Río, V. ................................. 123 del Valle, M................................ 125 Delgado, A................................. 198 Delgado, T................................. 176 Delgado-Camón, A...................... 168 Di Anibal, C. ................................53 Di Paola, R...................................98 Díaz, A. ..................................... 198 Domeño, C. ............................... 116 Domínguez, A. ........................... 102 Domínguez, E. ....................... 27, 90 Domínguez-Romero, J.C. ....... 56, 169 Donard, O.F.X. ........................... 166 Doña, A..................................... 183 Duato, P. ............................... 74, 76 Durán, E.................................... 199 Durán-Merás, I..................... 57, 200 Durand, J.S. ........................ 78, 191 E El-Amrani. S. ...............................29 El Rhazi M. ..................................99 Enciso, I. .....................................73 Erenas, M.M............................... 160 Escandar, G.M..............................28 Escarpa, A. ..................................33 Escudero, A. .............................. 198 Espada, E. ................................. 199 Espasa, A.M. .............................. 190 Espejo-Maqueda, J........................79 Espinosa Mansilla, A.............. 28, 200 Esteban-Díez, I. ..................... 67, 83 Estévez, E. ..................................47 Etxebarria, N. ............................ 130
F Félix, J.S. .................................. 116 Fernández, C.............................. 127 Fernández, I. ....................... 59, 139 Fernández, L.A. .................... 49, 130 Fernández, P................................... .....................78, 131, 147, 191, 203 Fernández, V.............................. 161 Fernández Abedul, M.T. .................38 Fernández-Alba, A.R. ...........121, 169 Fernández-Argüelles, M.T. .............84 Fernández-Arroyo, S. ....................30 Fernández-Bravo, A. ................... 183 Fernández-Espinosa, A.J................... ............................79, 154, 159, 161 Fernández-González, V................ 185 Fernández-Gutiérrez, A. ................... ............. 30, 59, 61, 65, 81, 139, 140 Fernández Sánchez, J.F. . 59, 139, 140 Fernández-Torres, R. ............ 46, 201 Fernández-Varela, R. .................. 186 Ferreira, S.L.C............................ 113 Ferreira, V. .......................71, 80, 86 Forero, F.J. ..................................97 Franch-Lage, F. ............................94 Franco, R................................... 198 Fuentes, R. ................................ 198 Funes-Collado, V. .........................45 G Galán, G.................................... 202 Galbán, J. .....102, 136, 167, 168, 196 Galcerán, M.T...............................37 Galeano-Díaz, T.................... 57, 200 Galindo, M.D. ............................. 199 Gallego, A............................ 78, 191 Gallego, M. ................................ 137 Gallo, B. ................... 91, 92, 93, 162 Gallo, V. ..................97, 98, 193, 194 García, J.I....................................36 García, M.C..................................96 García, M.V................................ 199 Garcia Arrona, R......................... 122 García-Barrera, T......... 26, 68, 69, 70 García-Cañas, V. ..........................96 García Casillas, D. ...............145, 148 García de Torres, A. ....... 32, 177, 178 García-Lorenzo, M.L. ................... 157 García-Otero, N. ......................... 171 García-Reyes, J.F............................. ............. 56, 110, 121, 134, 169, 172 García-Rodríguez, D.................... 129 García-Rodríguez, M.T..........145, 187 García-Salgado, S.......... 72, 145, 148 García Sartal, C............................62

ÍNDICE DE AUTORES
208
García-Vargas, M.................149, 150 Garcinuño, R.M. ................... 78, 191 Garmón-Lobato, S. ............... 92, 162 Garraud, H. ............................... 166 Garrido Frenich, A. ..............115, 181 Garrido-López, A. ....................... 143 Garrigues, S. ....................... 31, 101 Garro, B. ................................... 190 Ghanjaoui, A................................99 Gil, S. ......................... 117,118, 120 Gil-Díaz, M...................................60 Gil García, M.D. .................24, 88, 89 Gil Hernández, P......................... 187 Gilbert-López, B. .. 121, 134, 169, 172 Godoy Caballero. M.P. ...................57 Goicoechea, H.C. ...............24, 88, 89 Gómez, M.A. .............................. 152 Gómez, M.T. ........................ 77, 195 Gómez-Ariza, J.L. ........ 26, 68, 69, 70 Gómez-Ramos, M.J. .................... 169 Gómez-Romero, M........................81 González, A.G. ........................... 198 González, M. .......................156, 182 González, M.C. .............................33 González, M.D............................ 100 González, M.M............................ 165 González, P................................ 180 González, Y................................ 158 González-Fernández, E................ 111 González-Fernández, M. ................69 González-Miret, M.L. .....................97 González-Saiz, J.M........... 67, 83, 141 Gracia-Moreno, E..........................80 Granado, M.A............................. 142 Granado, M.D............................. 199 Grimalt, J.O. ................................11 Guillén, D. ................................. 199 Gutiérrez, A.M............................ 135 Gutiérrez Álvarez, M.D. .................38 Guzmán, F.J............................... 104 H Hamida, S. ................................ 153 Harper, J.D. ............................... 110 Herbello-Hermelo, P......................31 Heredia, F.J. .......................... 97, 98 Hernández, A.R. ......................... 143 Hernández, F. ..............................87 Hernández Córdoba, M. .................35 Hernández-Orte, P. .......................71 Hernanz, D. .............97, 98, 193, 194 Herrador, M.A. .............................39 Herrero, P....................................71 Herrero-Martínez, J.M. ..................52 Hurtado, M.C. ............................ 126 Hurtado-Fernández, E. ..................81
I Ibáñez, G.A. ................................28 Ibáñez, M. ...................................87 Ikegami, T. ..................................37 Irisarri, E............................. 75, 152 Iswaldi, I. ....................................61 J Jackson, A.U. ............................. 110 Jaén-Martos, L. .......................... 172 Januszenicz, K............................ 163 Jiménez, J. .......................... 90, 152 Jiménez, M.S. ...................... 77, 195 Jiménez Moreno, F...............145, 187 Juárez, M.....................................14 Jurado, J.M. .................................42 K Kalogerakis, N............................ 118 Kohler, A. ....................................10 L Laborda, F. ..............25, 94, 151, 153 Larrechi, M.S. .....................123, 127 Laserna, J.J. ............... 175, 176, 183 Lasheras, R.J. ............................ 174 Lavilla, I. ............. 117, 118, 119, 120 León-Camacho, M.........................42 Lerma-García, M.J. .......................52 Lima-Cazorla, L. ...........................64 Liva, M. ..............................163, 164 Llorente, T. ............................ 50, 51 Lobo Castañón, M.J............... 38, 111 Longás, N. ................................. 122 López, R. .....................................80 López-Barea, J. ............................69 López García, I. ............................35 López Guerrero, M.M................... 177 López-Heras, I. ............................64 López-López, J.A. ....................... 149 López-Mahía, P........................... 185 López-Márquez, D.M. ............ 91, 162 López-Molinero, A. ................ 75, 152 López-Ortiz, F. .............................59 López-Rouco, A. ......................... 117 López-Sánchez, J.F. ...........45, 50, 51 Lorenzo, R.A. .............. 129, 180, 203 Lozada-Castro, J.J. .......................60 Lucena, P. ................................. 183 Lucio-Gutiérrez, J.R. .....................48

ÍNDICE DE AUTORES
209
M Madrid, Y...................... 64, 113, 184 Mahugo Santana, C..................... 124 Manrique, E. .............................. 184 Manso-Sayago, J.M.......................70 Marazuela, M.D. ......................... 114 Marcos-Martínez, D..................... 125 Marín, C. ................................... 155 Marín Suárez del Toro, M. ............ 140 Marina, M.L..................................96 Martens, H...................................10 Martín, J.................................... 173 Martín, M.J. .................................42 Martínez, M.P. .....................143, 144 Martínez-Arkarazo, I. ....................49 Martínez Fernández, M. ............... 170 Martínez Galera, M.............24, 88, 89 Martínez-Sánchez, M.J. ............... 157 Martínez Vidal, J.L. ..............115, 181 Maspoch, S. ........................... 48, 94 Mateos, E. ................................. 167 Mateos, S. ................................. 188 Matos-Reyes, M.N....................... 100 Matusiewicz, H. .......................... 117 Medina, J............................131, 147 Mejuto, C. ................................. 146 Melaniuk, M. .............................. 163 Mendiguchía, C............ 150, 166, 199 Milla, M. .................................... 199 Millán, E. ................................... 122 Miranda-Ordieres, A.J.................. 111 Mohino-Guerrero, J.C. ........... 79, 161 Molina, J.................................... 157 Molina, N.....................................58 Molina Díaz, A. ................................ ............. 56, 110, 121, 134, 169, 172 Molina Mayo, C....................145, 187 Montaña, M.T. ............................ 202 Montesdeoca, S............................47 Montoro Bustos, A.R. ....................84 Montoya, R. ............................... 154 Montull-Ibor, B........................... 174 Morales, M.T. ...............................39 Morales-Noé, A........................... 101 Moreda Piñeiro, A. ........................... ...................... 31, 62, 108, 170, 171 Moreno, A.................................. 190 Moreno, C................... 149, 150, 166 Moreno, F. ...................................68 Moreno, M. ..................................34 Moreno-Bondi, M.C. .............114, 188 Moros, J. ............................. 31, 101 Mota, S. ......................................30 Muniategui-Lorenzo, S. ........185, 186 Muñoz de la Peña, A. ..... 28, 126, 200 Muñoz-Olivas, R. .................163, 164
Murillo, S.....................................64 N Naranjo, I.................................. 199 Narváez, A............................. 27, 90 Natera, R................................... 199 Navarro, F. ................................ 114 Navarro, P. ................................ 130 Nerín, C..............................116, 190 Núñez, O. ....................................37 O Ochoa-Hueso, C.R. ..................... 184 Olea, N........................................20 Olivares, M. ............................... 142 Olivares-Rieumonut, S. .................64 Oliveira, V. ................................ 158 Olivieri, A.C. ................................28 Oña Burgos, P. ..................... 59, 139 Orellana, G. ............................... 188 Ortega-Morales, F.J..................... 125 Ostra, M. ............................132, 133 Otero-Romaní, J. ........................ 171 P Pablos, F. ....................................42 Padilla-Sánchez, J.A.................... 181 Páez-Hernández, M.E. ...................73 Palomo, M. ................................ 184 Palomo, M.R. ............................. 155 Paniagua, E.E...............................58 Pardo, R. ............................. 66, 179 Pastor, A. .................................. 101 Pedrero-Zayas, Z..........................64 Peguero, A................................. 103 Pena, T. .................................... 146 Pena-Abaurrea, M.........................29 Pena-Pereira, F..... 117, 118, 119, 120 Peña, E. .................................... 152 Peña-Gallego, A............................71 Peña-Vázquez, E......................... 171 Pereira Navaza, A. ........................85 Pereiro García, M.R. ......................38 Pérez, N. ................................... 135 Pérez Bernal, J.L......................... 201 Pérez-Conde, C. ................... 58, 135 Pérez-Corona, M.E. ..................... 184 Pérez-Corona, T. .................113, 184 Pérez-del-Notario, N. ............ 83, 141 Pérez López, J.A. ..........................34 Pérez-Sirvent, C. ........................ 157 Pinilla, E. ............................142, 155 Pinto, J.J. .................................. 199 Pizarro, C. ...................... 67, 83, 141

ÍNDICE DE AUTORES
210
Pizarro, M.L. .......................106, 107 Plaza-Bolaños, P......................... 181 Polo-Díez, L.M. .............................60 Popovici, C.A................................59 Prada-Rodríguez, D..............185, 186 Prieto, A. ................................... 130 Psillakis, E. ................................ 118 Pueyo, C......................................69 Pueyo, J.J. ................................. 184 Pumera, M. ..................................33 Q Quijano Nieto, M.A......... 72, 145, 148 Quirantes-Piné, R. ........................65 R Ramos, L. ....................................29 Ramos-Martos, N.................. 43, 172 Ramos-Payán. M.D. .............. 46, 201 Recamales, A.F. ...........................82 Ríos, A. ..................................... 109 Rivas, R.E....................................35 Robles, M.C. ................................82 Robles-Molina, J. .................134, 169 Rodríguez, C. ............................. 104 Rodríguez, E. ............................. 114 Rodríguez, J.A..............................73 Rodríguez, L. ...............................77 Rodríguez, M.C........................... 199 Rodríguez, R.C. .......................... 104 Rodríguez-Cáceres, M.I. .............. 126 Rodríguez-Medina, I.C............. 30, 61 Rodríguez-Tecedor, S....................83 Romarís Hortas, V. ............... 62, 108 Romera-Fernández, M. ............ 91, 93 Romero, E. ................................ 168 Romero-González, R. .................. 115 Rosas, J. ................................... 105 Rubio, R. ..........................45, 50, 51 Rubio-Barroso, S. ................. 60, 135 Ruisánchez, I. ..............................53 Ruiz, J................................. 36, 190 Ruiz-Beguería, S. ................... 25, 94 Ruiz-Chancho, M.J. ................. 50, 51 Ruiz Encinar, J. ...................... 84, 85 S Sáenz-González, C........................67 Sánchez, A. ............................... 198 Sánchez, A.A. ..............................78 Sánchez Arribas, A. ......................34 Sánchez-Báscones, M.I. . 66, 179, 197 Sánchez de la Campa, A. ............. 158 Sánchez-Fernández, C........... 93, 162
Sánchez-Ilárduya, M.B. ........... 91, 92 Sánchez Paradinas, S....................63 Sánchez Pérez, I. .........................88 Sánchez-Rodas, D. ..................... 158 Sánchez Trujillo, I.........................32 Sancho, J.V..................................87 Santana Rodríguez, J.J. ......... 47, 124 Santos, J.C. ............................... 107 Santos, J.L..........................165, 173 Santos-Delgado, J.M. ....................60 Sanz, L...................................... 135 Sanz, V. .................................... 167 Sanz-Landaluze, J.........................29 Sanz-Medel, A...................36, 84, 85 Sanz-Vicente, I. .......... 102, 136, 196 Sayago, A............ 106, 107, 193, 194 Segura-Carretero, A... 30, 61, 65, 139 Seijo, I...................................... 159 Serrano, A. ................................ 137 Siles Cordero, M.T. ........ 32, 177, 178 Silva, M.......................... 16, 17, 138 Simó-Alfonso, E.F. ........................52 Simón Mañogil, A..........................85 Simonet, B.M. ..............................16 Sosa Ferrera, Z. ................... 47, 124 Souhail, B....................................43 Stanisz, E. ................................. 117 T Tanaka, N....................................37 Téllez, H.................................... 175 Tello, I. ..................................... 152 Tena, M.T. ..........................143, 144 Ternero-Rodríguez, M....................... ............................79, 154, 159, 161 Torres Padrón, M.E. .................... 124 Trigo, D..................................... 154 Troncoso, A.M. .............................15 Tuñón-Blanco, P. ........................ 111 U Ubide, C. ............. 132, 133, 136, 196 Usobiaga, A. ........................ 49, 130 V Vadillo, J.M. ................... 18,175, 176 Valcárcel, M. ................................16 Valero Navarro, A. ...................... 139 Vallejo, A.....................................49 Vega, M.................66, 131, 147, 179 Veitia, S.A. ................................ 163 Vera Candioti, L............................24 Verardo, V. ..................................65 Vereda Alonso, E. ........... 32,177, 178

ÍNDICE DE AUTORES
211
Vergel Rodríguez, C. ................... 150 Vicente, F. ................ 91, 92, 93, 162 Vidal, J.C............................... 74, 76 Vidal, L...................................... 119 Vidal, M............................... 55, 133 Villar-Navarro, M. ................. 46, 201 Villar-Pulido, M. .......................... 172 Viloria-Bernal, M..................... 92, 93 W Wiley, J.S. ................................. 110 Wunderlin, D................................98 Y Ybarra-Lalor, M. ...........................79 Yusà, V. .................................... 101 Z Zamora, D. ................................ 128 Zamora, P. ..................................27 Zapardiel, A. ................................34 Zapata, J. ....................................86 Zougagh, M. .............................. 109 Zuloaga, O........................... 49, 130 Zuriarrain, J.............44, 54, 136, 196


LISTA DE ASISTENTES

LISTA DE ASISTENTES
214
A Alcántara Leiva, José Francisco Almendral Parra, Mª Jesús Alonso Álvarez, Esteban Alonso Castillo, Manuel Amigo Rubio, José Manuel Anzano Lacarte, Jesús Aparicio Gómez, Irene Arráez Román, David Arranz García, Adela B Bakkali, Karima Barciela Alonso, Mª del Carmen Barrado Esteban, Enrique Barrero, Miguel Angel Barriola Goicoechea, Ainara Bartolomé, Luis Beltrán Lucena, Rafael Bendicho Hernández, Carlos Bermejo Barrera, Pilar Bermejo Benito, Esperanza Berregi Abalde, Iñaki Berrueta Simal, Luis Angel Berzas Nevado, Juan José Blanco Romia, Marcelo Bolea Morales, Eduardo Bonel Sanmartín, Laura Bonilla Simón, Mª Milagros Bordagaray Eizaguirre, Ane C Caceci, Marco Cáceres, Jorge Omar Callao Lasmarias, Mª Pilar Calvo Blázquez, Lorenzo Cámara Rica, Carmen Candela Lledó, Lucila Castillo Suárez, Juan R. Castrillejo Hernández, Mª Yolanda Cepriá Pamplona, Gemma Coello Bonilla, Jordi Coïc, Daniela Contreras Acuña, Manuel Costas Rodríguez, Marta Cruz Sánchez, Jordi Cuello Núñez, Susana Cueto Díaz, Sergio Cueva Mestanza, Rubén Eduardo D de Andrés Segura, Fernando de la Guardia Cirugeda, Miguel
de las Heras Álvarez, Concepción de Marcos Ruiz, Susana de Orbe Payá, Ignacio del Cacho Vicente, Carmen del Campo Martínez, Gloria del Río Sánchez, Vanessa Delgado Camón, Mª Arantzazu Delgado Pérez, Tomás Di Anibal, Carolina Vanesa Domeño Recalde, Celia Domínguez Cañas, Elena Domínguez Romero, Juan Carlos Duato Calvo, Patricia Durán Guerrero, Enrique Durán Martín-Merás, Isabel Durand Alegría, Jesús Senén E Escarpa Miguel, Jesús Alberto F Falcao, María Fernández Arroyo, Salvador Fernández Cegrí, Victoria Fernández Gutiérrez, Alberto Fernández Recamales, Mª Ángeles Fernández Requejo, Patricia Fernández Sánchez, Jorge Fernando Fernández Torres, Rut Fernández-Bravo del Olmo, Ángel Ferreira González, Vicente Ferrer Blanco, Isabel Florido, José Manuel Franch Lage, Felicidad Funes Collado, Virginia G Galán Alfonso, Guillermina Galbán Bernal, Javier Galcerán Huguet, Mª Teresa Gallego Fernández, Mercedes Gallo Hermosa, Blanca Gallo Mazzuchino, Mª Valeria García Alonso, José Ignacio Garcia Arrona, Rosa García Barrera, Tamara García Casillas, David García Otero, Natalia García Reyes, Juan Fco. García Salgado, Sara Garrido Frenich, Antonia Garrigues Mateo, Salvador Gil García, Mª Dolores Gil Hernández, Patricia

LISTA DE ASISTENTES
215
Gilbert López, Bienvenida Gómez Ariza, José Luis Gómez Cotín, Mª Teresa Gómez González, Miguel Ángel Gómez Romero, María González Aguirre, Marta González González, A. Gustavo González Martín, Mª Cristina Gracia Moreno, Elisa Granado Rico, Mª Ángeles Grimalt Obrador, Joan Guitart de Juan, Ana H Hernández Córdoba, Manuel Hernanz Vila, Dolores Herrero Piedrafita, Paula Irisarri Jiménez, Enrique I Iswaldi, Ihsan J Jiménez Centelles, Javier Jiménez García-Alcalá, Mª Sierra Jiménez Gómez, Yolanda Jiménez Lamana, Javier Jiménez Palacios, Francisco Joaquín Juárez Iglesias, Manuela L Laborda García, Francisco Larrechi García, Mª Soledad Lavilla Beltrán, Isela Lerma García, María Jesús Liva Garrido, Mª Beatriz Llorente Mirandes, Toni Lobo Castañón, Mª Jesús López Guerrero, Mª del Mar López Heras, Mª Isabel López López, José Antonio López Oyagüe, Alaia López Sánchez, José Fermín Lorenzo Ferreira, Rosa Antonia Lozada Castro, Juan José Lucio Gutiérrez, Juan Ricardo M Madrid, Yolanda Manso Sayago, José Manuel Mantecón, Jesús
Marazuela Lamata, Mª Dolores Marcos Martínez, Daniel Marina Alegre, Mª Luisa Martens, Harald Martínez Fernández, Marta Martínez Galera, María Martínez Moral, Mª Pilar Martínez Ortíz del Río, Javier Martínez Vidal, José Luis Maspoch Andrés, Santiago Mateos García, Silvia Mejuto Martí, Mª del Carmen Mendiguchía Martínez, Carolina Millán Martín, Esmeralda Mir Marín, José Maria Molina Díaz, Antonio Molina Mayo, Carlos Montaña González, Mª Teresa Montoro Bustos, Antonio Rafael Montoya Mayor, Rocío Morales Millán, Mª Teresa Moreda Piñeiro, Antonio Moreno Aguilar, Carlos Moreno Barambio, Mónica Moreno Bondi, Mª Cruz Moreno Cordero, Bernardo Muniategui Lorenzo, Soledad Muñoz de la Peña Castrillo, Arsenio Muñoz Olivas, Riansares N Narváez, Arantzazu Nerín de la Puerta, Cristina Núñez Burcio, Oscar O Olea Serrano, Nicolás Ostra Beldarrain, Miren P Pablos Pons, Fernando Palacios Corvillo, Mª Antonia Palomo Marín, Mª del Rosario Pardo Almudí, Rafael Peguero Gutiérrez, Anna Pena Pereira, Francisco Peña Margelí, Elena Pérez Conde, Concepción Pérez Corona, Mª Teresa Pérez del Notario García, Nuria Pérez Fernández, Juan Manuel Pérez Outeiral, Jessica Pérez Pavón, José Luis Pérez Sirvent, Carmen

LISTA DE ASISTENTES
216
Pinilla Gil, Eduardo Pizarro Millán, Consuelo Prada Rodríguez, Darío R Ramos Martos, Natividad Ríos Castro, Ángel Robles Molina, José Rodríguez Álvarez, Carolina Rodríguez Cavallo, Erika Rodríguez Castañedas, José Luis Rodríguez Martín-Doimeadiós, Rosa Carmen Rodríguez Sanz, Laura Rodríguez Tecedor, Sofía Romarís Hortas, Vanessa Rosas Portugal, Juan Guillermo Rosat Rodrigo, Carlos Rubio Rovira, Roser Ruisánchez Capelástegui, Itziar Ruiz Encinar, Jorge S Sáenz González, Cristina Salvadó Martín, Victoria Sánchez Arribas, Alberto Sánchez Báscones, Mª Isabel Sánchez Pérez, Isidro Sánchez-Rodas Navarro, Daniel Alejandro Sancho Llopis, Juan Vicente Santana Rodríguez, José Juan Santos Morcillo, Juan Luis Sanz Landaluze, Jon Sanz-Medel, Alfredo Sayago Gómez, Ana Seijo Delgado, Inmaculada Siano, Gabriel Germán Siles Cordero, Mª Teresa Silva Félix, Juliana Silva Rodríguez, Manuel Simonet Suau, Bartolomé Sosa Ferrera, Zoraida T Téllez Lozano, Helena Tena Vázquez de la Torre, Mª Teresa Troncoso González, Ana Mª U Ubide Sebastián, Carlos Usobiaga Epelde, Aresatz
V Vadillo Pérez, José Miguel Valcárcel Cases, Miguel Valencia Mirón, Carmen Valero Navarro, Ángel Vega Alegre, Marisol Vereda Alonso, Elisa Isabel Vicente Estévez, Francisca Vidal Postigo, Maider Villar Navarro, Mercedes Villar Pulido, Marina Y Ybarra Lalor, Miguel Z Zamora Bonachela, Patricia Zamora Zamora, David Zapardiel Palenzuela, Antonio Zapata Ochoa, Julián Andrés Zougagh, Mohammed Zuloaga Zubieta, Olatz Zuriarrain Ocio, Andoni Zuriarrain Ocio, Juan
















![[1993]City Hunter CD2](https://img.pdfslide.tips/doc/110x75/56d6be561a28ab301691ae4d/1993city-hunter-cd2.jpg)





![[1993]City Hunter CD1](https://img.pdfslide.tips/doc/110x75/56d6be561a28ab301691ad90/1993city-hunter-cd1.jpg)







