Embed Size (px)
Citation preview

(2007) 301–311www.elsevier.com/locate/cellsig
Cellular Signalling 19
Induction of high-molecular-weight (HMW) tumor necrosis factor(TNF)alpha by hepatitis C virus (HCV) non-structural protein 3 (NS3)
in liver cells is AP-1 and NF-κB-dependent activation
Mohamed Hassan a,⁎, Denis Selimovic b, Hanan Ghozlan c, Ola Abdel-Kader d
a Department of Dermatology, Faculty of Medicine, University of Duesseldorf, Mooren Str. 5, 40225 Duesseldorf, Germanyb Department of Operative Dentistry and Periodontology, University of Homburg an der Saar, Kirrberger Str.1, 66424 Homburg/Saar, Germany
c Department of Microbiology, Faculty of Science, University of Alexandria, Alexandria, Egyptd Department of Microbiology, Medical Research Institute, University of Alexandria, Alexandria, Egypt
Received 23 June 2006; accepted 5 July 2006Available online 17 August 2006
Abstract
Chronic infection of hepatitis C virus (HCV)-infected patients is associated with the production of serum and interhepatic inflammatory cytokinesincluding tumor necrosis factor α (TNF-α). In this study, we delineated part of the mechanism whereby HCV induces the synthesis of TNF-α inhuman liver cell lines HepG2 and Huh7. HepG2 transiently transfected with the full-length HCV cDNA expressed high-molecular-weight (HMW)TNF-αmRNAs, which were absent in the control cells. In addition tightly regulated expression of HCVNS3 in both HepG2 and Huh7 was found toinduce the expression of HMWmRNAs and subsequently the production of biologically active TNF-α. Interestingly, the expression of NS3 proteinin HepG2-NS3 or in Huh7-NS3 resulted in the activation of kinase (IKK-α) of NF-αB inhibitor (IαB) and in the enhancement of the DNA-bindingactivity of the nuclear transcription factor NF-κB. The inhibition of the transcription of TNF-α mRNAs and subsequently TNF-α productionfollowing the treatment of HepG2-NS3 or Huh7-NS3 transfectants with the inhibitor of NF-κB, Bay 11-7082, suggesting the importance of NF-κBfor the regulation of NS3-mediated TNF-α expression in HepG2 and HeLa cells. Interestingly, data obtained from luciferase assays, in liver and innon-liver cells showed the contribution of NS3 protein in the regulation of TNF-α promoter through the activation of AP-1 and NF-κB. Our dataindicate that the intrahepatic TNF-α production induced by HCV is transcriptionally up-regulated by HCV NS3. Therefore, HCV NS3 may have apotential role in the induction of intrahepatic inflammatory processes that occur during acute and chronic hepatitis C.© 2006 Elsevier Inc. All rights reserved.
Keywords: HCV NS3; JNK; NF-kB; TNF-α
1. Introduction
Hepatitis C Virus (HCV) is a major cause of chronic liverdiseaseworldwide. Persistent infection often progresses to chronichepatitis, cirrhosis, and hepatocellular carcinoma. Neither a vac-cine nor an effective therapy is currently available for HCVotherthan interferon alpha combined with ribavirine [1].
HCV genome has a long open reading frame, flanked with 5′and 3′ untranslated region, which encodes a polyprotein precursorof about 3010 to 3033 amino acids (aa) residues [2]. This poly-
⁎ Corresponding author. Tel.: +49 211 811 16173; fax: +49 211 811 8840.E-mail address: [email protected] (M. Hassan).
0898-6568/$ - see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.cellsig.2006.07.002
protein is cleaved by both host and viral proteases to generate fourstructural proteins (C, E1; E2 and P7) and six non-structuralproteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B) [2]. Virusmultiplication is dependent upon viral proteins expressed fromHCV genome, including nonstructural proteins that are expectedto participate in genome transcription and replication [3].
In particular, the non-structural protein 3 (NS3) that possessesserine protease activity, which is essential for viral protein pro-cessing [2] and nucleotide triphosphatase-RNA helicase activitythat is essential for virus replication [4,5]. Thus, based on func-tions related directly to viral multiplication, the HCVNS3 proteinis considered an important target for anti-HCV therapies. Besidesits pivotal role in viral protein processing and virus replication, the

Fig. 1. Northern blot analysis of HCV RNA and TNF-αmRNA levels in HepG2cells and HCV-transiently transfected HepG2 cells. Approximately 20 μg oftotal cellular RNA of three independent samples from HepG2-mock cells andHCV-transiently transfected HepG2 cells was electrophoresed in agarose gelsand transferred onto nylon membrane. Blots were hybridized with an HCV-derived DNA probe spanning from nucleotides 350 to 3500 of the HCV genomegenotype 4 A (A) and with human TNF-α cDNA probe (B). The equal loadingof mRNAwas judged by GAPDH. The position of the TNF-αmRNAs and HCVgenomic transcript is indicated.
302 M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
HCV NS3 protein was reported to be involved in the modulationof many cellular functions [6,7], including regulation of pro- andanti-inflammatory cytokines [8].
An increase in serum levels of pro-inflammatory cytokineshas been noted in chronic liver diseases [9], including chronichepatitis C [10]. However, the most important pro-inflammatorycytokines, including TNF-α and IL-1β are expressed in the livermostly by resident macrophages (Kupffer cells) and T-cells [11].
Although, hepatocytes have been reported to augment theproduction of pro-inflammatory cytokines in response to liverinjury and infection, including viral hepatitis [12,13], the triggerof the hepatocyte-derived cytokines in chronic hepatitis C remainsunknown. However, an increased TNF-α mRNA levels in liverand mononuclear cells compared to healthy controls have beendemonstrated in untreated patients with chronic hepatitis C [14].
Although various cell types are capable to produce TNF-α[15,16], the regulatory mechanisms controlling TNF-α expres-sion seem to depend on the type and the differentiation state ofthe cells.
In the present study, we addressed the question of whetherTNF-α transcription is regulated in HCV-infected hepatocytes,and if so, what is the role of HCVNS3 protein in this context. Wesought to use an in vitro HCV- and HCV NS3 expression systemand to investigate whether HCV NS3 protein is the trigger of theintrahepatic TNF-α and to set out the mechanism whereby theHCV NS3 protein regulates TNF-α in hepatocytes. Here, wepresent evidence that the HCV NS3 protein-induced TNF-αproduction, in the hepatocytes, is regulated through the activationof both IKKα/NF-κB and JNK/AP-1 signaling pathways.
2. Material and methods
2.1. Cell lines
Human hepatoma cells (HepG2 and Huh7), and human cervical carcinomacells (HeLa) (ATCC, Rockville, MD, USA). HeLa Tet-Off cells, whichconstitutively express the tetracycline-controlled transactivator and the Retro-Pack™ pT67 cells, were purchased from Clontech (California, USA). Cells weregrown in Dulbecco's modified Eagle's medium (Sigma, Daisenhofen) sup-plemented with 10% fetal bovine serum.
2.2. Extraction of RNA from sera, cDNA synthesis and plasmidconstruction
Patients with high HCVRNA titers (105–108 copies/ml) were selected in orderto obtain sufficient amount of RNA. All patients had detectable HCV RNA of thegenotype 4A (Genbank Acc. Nr.: Y11604) when we analyzed the genotyping asdescribed previously [17–19]. Nucleic acids were extracted from 100 μl of serumusingQIAamp viral RNA extraction kit (Qiagen, Hilden, Germany). The completeHCV cDNA was synthesized with Genscript (Genecraft, Munster, Germany) asdescribed previously [17,18,20] using random hexamers. The HCV cDNA (nt280–9306) was amplified from the complete HCV cDNA by conventional PCRusing the following primers: 5′-GAC CGT GCA CCATGA GCA CG-3′ (sense)and 5′-CTGCCTACCGAGCAGGCAGCA-3′. The PCR product wasmodifiedto generate blunt ends and then cloned into the EcoR V site of pcDNA3.1(+)(Invitrogen, Groningen, The Netherlands) to generate pcDNA3.1-HCV. The HCVNS3 protein-encoding region (nt 3340–5254) was amplified from the pcDNA3.1-HCV plasmid comprising the complete HCV cDNA using the following primershaving a start codon (bold) and a Cla I restriction site (underlined) 5′-CCATCGATATGAAGGGG TGGAGACTC CTT GC-3′ (sense) and 5′-CCATCG ATTGTC ACTACC TCG AGATCA GC-3′ (anti-sense). The amplified product was
digested with Cla I and then cloned into the Cla I site of pRevTRE (Clontech,California, USA) to generate pRevTRE-NS3 plasmid.
2.3. Generation of viruses
The packaging cell line RetroPack™ pT67 (CLONTECH) was grown inDMEM with 10% FCS, 2 mM L-glutamine (all from Sigma, Daisenhofen,Germany) at 37 °C and in 5% CO2. The cells were transfected with the appro-priate retroviral construct e.g. pRev Tet-Off, pRevTRE-luciferase, or pRevTRE-NS3 by nucleafector™ Kit (AMAXA BIOSYSTEMS). 48 h post-transfection,the supernatant was collected, filtered through a 0.45-μm syringe filter, and spunat 50.000 ×g for 1.5 h. Pelleted virus was resuspended in a 0.1 or 0.05 the originalvolume of medium at 4 °C for 4 h.
2.4. Infection of target cells
The development of HepG2, Huh7 and HeLa cells inducibly expressing NS3protein was performed as described [17,18,21]. G418 and Hygromycin resistantclones, termed,HepG2-NS3,Huh7-NS3 andHeLa-NS3 transfectantswere screenedfor expression ofHCVNS3protein byRT-PCR. Positive clones,with high inductionefficiency, were expanded and re-screened by RT-PCR and immunoblotting usinganti-HCV NS3 antibody for the expression of HCV NS3 protein.
2.5. Luciferase assay
The luciferase assay was performed as described [21]. The relative lightunits (RLU) were measured in illuminometer (EG and G Berthold, Berthold,Germany).

303M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
2.6. Immunoblot
Immunoblot analysis was performed according to the standard procedures.The following antibodies were used at the indicated dilution: anti-HCV NS3protein (Research Diagnostic, Inc, USA), 1:1,000; anti-IKKα (SC-7218), 1:2,000;anti-IκB-α (Sc-203), 1:2.000; anti-Actin (SC-1615), 1:5.000 (Santa CruzBiotechnology, California, USA).
2.7. Northern blot, EMSA and kinase assay
Total RNA extraction, agrose/formaldehyde gels, Northern blot, electropho-retic mobility shift assays, gel shift assays, preparation of nuclear extracts andradiolabeled probes, protein purification and kinase reactions were performed asrecently described [17,18,21].
2.8. Transfection of HepG2 cells with the complete HCV cDNA
The HepG2 cells were plated into a 10-mm3 dish (Nunc) and cultured incomplete medium. 48 h later, the cells (1×106) were co-transfected with 5 μg ofpcDNA3.1-HCV plasmid and 1 μg of pβGal (CLONTECH) by nucleofactor kit(AMAXA BIOSYSTEM). 48 h later, the cells were harvested and the total RNAextraction was carried out using RNeasy Mini Kit (Qiagen, Hilden, Germany).The RNA concentration was measured and normalized to the transfection effi-ciency, which was determined by direct visualization of the β-Gal-expressing cellsas described previously [22].
2.9. Construction of the reporter plasmids
The reporter plasmid with full-length human TNF-α promoter (pTNF 1051;from − 1051 bases to +26 bases relative to the transcription initiation site) wasamplified by PCR and inserted into the pGEM-T-easy vector (Promega,Madison, USA). Kpn I-Hind III of the resulting plasmid was subcloned into aluciferase reporter vector, pGL2-basic (PROMEGA).
2.10. Cell transfection and luciferase assay
The HepG2-NS3, Huh7-NS3 and HeLa-NS3 transfectants (1×106 each)were plated into a 10-mm3 dish (Nunc) and cultured in medium with (+Tc) 4 μg/ml tetracycline. 48 h later, the cells (1×106) were co-transfected with 2 μg of thereporter plasmid and 1 μg of pβGal (CLONTECH) by nucleofactor kit
Fig. 2. Detection of HCVNS3 protein expression in both HepG2-NS3, Huh7-NS3 andNS3 transfectants were cultured in medium with (+Tc) or without (−Tc) 4 μg/ml tewhole cell lysate (50 μg); or B) in the whole cell lysate (100 μg), in the cytoplasmantibody. Arrow indicates expressed HCV NS3 protein.
(AMAXA BIOSYSTEM). After the transfection, the cells were plated into a10-mm3 dish (Nunc) and cultured in medium with (+Tc) 4 μg/ml tetracycline.24 h later, the cells were allowed to grow in mediumwith (+Tc) or without (−Tc)and then pretreated or not treated with the specific inhibitors of JNK and/or NF-κB. 48 h later, the luciferase activity was measured and normalized to thetransfection efficiency as described for HepG2 transfection with pcDNA3.1-HCV.
2.11. Detection of TNF-α by enzyme-linked immunosorbent assay anddetermination of the biological activity
TNF-α production either in cell culture supernatants or cell extracts weredetermined by enzyme-linked immunosorbent assay (ELISA), using HBThuman TNF-α DECA kit (Hycult biotechnolgy, Uden, Netherlands). TNF-αbioactivity was determined as described previously [23] with minor modifica-tions that cytotoxicity was also assessed by the tetrazolium salt dye method [24].
3. Results
3.1. Analysis of TNF-α mRNA expression in HepG2 cellstransiently transfected with the complete HCV cDNA
The HCV RNA was analyzed in HepG2 cells transientlytransfected with the complete HCV cDNA by Northern blot. Asshown in Fig. 1A, the HCV-transfected HepG2 cells expressedas expected, the HCV transcript of about 9.0 kb compared tocontrol cells. We further studied the transcriptional status ofTNF-α gene in HCV-transfected HepG2 cells. Northern blotanalysis (Fig. 1B) revealed that HCV-transfected HepG2 cells,but not HepG2 control cells expressed two major TNF-αtranscripts of 3.6 kb and 4.3 kb in addition to the typical 1.7 kbTNF-α mRNA. An HMW TNF-α mRNA species of 3.6 kb hasbeen previously reported and characterized. These TNF-αmRNA species have been demonstrated to be polyadenylatedand free of intronic and intergenic elements [25]. We have alsoconfirmed the absence of the intronic sequences in TNF-αtranscripts isolated from HCV-transfected HepG2 cells (data not
HeLa-NS3 transfectants byWestern blotting. HepG2-NS3, Huh7-NS3 or HeLa-tracycline. 48 h later, the expression of HCV NS3 protein were analyzed; A) in(50 μg) or in nuclear extracts (50 μg) by immunoblotting using anti-HCV NS3

Fig. 3. Effect of HCV NS3 protein on IKKα, IκBα. HepG2-NS3 and Huh7-NS3 transfectants were cultured in medium with (+Tc) or without (−Tc) 4 μg/mltetracycline. 48 h later, the whole cell lysate or cytoplasmic lysate was prepared. Kinase assay: equal amounts of whole cell lysate (100 μg) were immunoprecipitatedusing anti-IKKα antibody (upper panel). Subsequently, the immunocomplex was subjected to an in vitro kinase assay in the presence of GST-IκBα as a substrate forIKKα. The protein kinase complexes were resolved by 15% SDS-PAGE followed by autoradiography. Western blot: equal amounts of whole cell lysate (100 μg) weresubjected to immunoblot analysis using anti-IKKα antibody (middle panel). Western blot: equal amounts of cytoplasmic protein (100 μg) were subjected toimmunoblot anti-IκBα antibody (lower panel). The same blots were re-probed with an anti-actin antibody to compare loading and transfer. In vitro kinase assays andWestern blots are representative of four independent experiments performed with identical results.
304 M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
shown). These results suggested that an HCV-related proteincould be involved in the transcriptional up-regulation of TNF-αgene in hepatocytes.
3.2. Establishment of cell lines regulated expression of HCVNS3 protein
To examine the potential role of HCV NS3 protein of thegenotype 4A HCV in the transcriptional up-regulation of theTNF-α gene in hepatocytes, we amplified the cDNA comprisingNS3 encoding region from the entire nucleotide sequence of thegenotype 4A HCV genome isolated from patients with chronichepatitis C. Furthermore, we established liver and non-liver cellmodels for the controlled expression of HCV NS3 protein. Thedelivery of the regulatory and the responsive cassettes of thetetracycline-regulated expression system into HepG2, Huh7 orHeLa cells was made as described [17,18,21] using retroviralgene transfer system (CLONTECH). We screened all HepG2-Tet-Off clones by transient infection with recombinant pRev-TRE-luc virus comprising the luciferase reporter gene. HepG2and Huh7 Tet-Off clones with high induction level and lowbackground (data not shown), and HeLa Tet-Off clones wereused for the infection with recombinant pRevTRE-NS3. Stabletransfectants with high induction capacity and low background
Fig. 4. Effect of HCV NS3 protein on NF-κB. A) Activation of NF-κB in HepG2 andcultured in medium with (+Tc) or without (−Tc) 4 μg/ml tetracycline. 48 h lateB) Inhibition of HCVNS3 protein-induced activation of NF-κB by Bay11-7082. Hep4 μg/ml tetracycline. 1 h prior to the withdrawal of tetracycline from the culture meextracts were prepared and EMSAwas performed. C) Supershift analysis and specifHepG2-NS3 and Huh7-NS3 transfectants cultured in medium with (+Tc) or withouantibodies to p50 (SC-114), p65 (SC-372) or c-Rel (SC-272) (Santa CRUZ Biotechnreaction was monitored by EMSA with a labeled NF-κB probe. The Oct-1 probe wEMSAs are representative of three independent experiments performed with identic
(data not shown) as assessed by RT-PCR, were further expandedand examined for the expression of HCV NS3 protein. Theexpression of HCV NS3 protein was detected in HepG2-NS3,Huh7-NS3 and HeLa-NS3 transfectants in the absence oftetracycline, while the 70 kDa band of the HCV NS3 proteinwas turned off by the addition of tetracycline (4 μg/ml) to theculture medium for 48 h (Fig. 2A). The detection of HCV NS3protein in the cytoplasmic and the nuclear extracts obtained fromHepG2-NS3 and Huh7-NS3 transfectants-expressing HCV NS3protein by immunoblotting (Fig. 1B) confirmed earlier findingsreported in HCV NS3 transfected cells [26].
3.3. Activation of NF-κB pathway by HCV NS3 protein
Based on its role in the regulation of pro-inflammatory cyto-kine including TNF-α, we investigated whether the expression ofHCV NS3 proteins triggers the NF-κB signalling pathway inhuman hepatoma cell lines HepG2 and Huh7. Results obtainedfromWestern blot and in vitro kinase assay (Fig. 3) demonstratedthat the expression of HCV NS3 protein does not alter theexpression of IKKα kinase, but enhances its basal activity inHepG2 and Huh7 cells. We further examined the effect of theexpression of HCV NS3 protein on IκB-α, the physiological
Huh7-NS3 cells-expressing HCV NS3 protein. HepG2-NS3 transfectants werer, nuclear extracts were prepared and assayed for NF-κB activity by EMSA.G2-NS3 and Huh7-NS3 transfectants were seeded for 24 h in medium with (+Tc)dium, the cells were pretreated with Bay 11-7082 (5 μg/ml). 48 h later, nuclearicity of HCV NS3 protein-induced NF-κB activation. The nuclear extracts fromt (−Tc), in the presence or in the absence of Bay11-7082, were incubated withology) and a 50-fold excess of an unlabeled competitor for 30 min at 37 °C. Theas used to monitor the concentration of nuclear extracts in the different lanes.al results.

305M. Hassan et al. / Cellular Signalling 19 (2007) 301–311

Fig. 5. Effect of HCV NS3 protein on the expression and the activation of JNKin Huh7 cells. In vitro kinase assay: equal amounts of whole cell lysate (100 μg)were immunoprecipitated using anti-JNK antibody. Subsequently the immuno-complex was subjected to an in vitro kinase assay in the presence of GST-c-Junfusion protein as a substrate for JNK. The protein kinase complex was resolvedby 15% SDS-PAGE followed by autoradiography. Western blot: equal amountsof whole cell lysate (50 μg) were subjected to immunoblot analysis using anti-JNK antibody. The same blot was re-probed with an anti-actin antibody tocompare loading and transfer. These results are representative of three inde-pendent experiments.
306 M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
substrate of IKKα. Results from Western blot analysis (Fig. 3)revealed that the expression of HCV NS3 protein enhances theproteolytic breakdown of IκB-α. These results suggest that theactivation of IKKα kinase may be an important intermediatewhereby HCVNS3 protein induces NF-κB signalling pathway inliver cells.
Fig. 6. Effect of HCV NS3 protein on AP-1. A) Inhibition of HCV NS3 protein-inducseeded for 24 h in medium with (+Tc) 4 μg/ml tetracycline. 1 h prior to the withdSP600125 (20 μM/ml). 48 h later, nuclear extracts were prepared and EMSAwas perfactivation. The nuclear extracts from Huh7-NS3 transfectants cultured in medium wincubated with antibodies to c-Fos (SC-253G) or c-Jun (SC-1694) (Santa Cruz Biolabeled AP-1 probe. The Oct 1 probe was used to monitor the concentration of thindependent experiments.
3.4. HCV NS3 protein enhances NF-κB activity
To examine whether HCV NS3 protein-induced activation ofIKKα, in HepG2 and Huh7 cells, is associated with the enhance-ment of DNA-binding activity of NF-κB. EMSAwas performedusing the nuclear extracts prepared from HepG2-NS3 and Huh7-NS3 transfectants cultured in a medium with (+Tc) or without(−Tc) tetracycline for 48 h. Results from EMSA (Fig. 4A)demonstrated that the expression of HCV NS3 protein enhancesthe activity of NF-κB in both HepG2-NS3 and Huh7-NS3 cellsinduced to express HCV NS3 protein. We ascertained the speci-ficity of the NF-κB band following incubation of nuclear extractswith antibody to p50 subunit by EMSA. Antibody to the p50subunit shifted the band to a higher molecular weight (Fig. 4C) inboth HepG2-NS3 and Huh7-NS3 cells-expressing HCV NS3protein. These results indicate that the activation of NF-κB in bothHepG2-NS3 and Huh7-NS3 cells-expressing HCV NS3 proteinmay result from HCV NS3 protein-induced activation of IKKα.
3.5. HCV NS3 protein induces the activation of the MAP kinasesignalling pathway JNK in Huh7 cells
The ability of HCV NS3 protein to induce the activation of theMAP kinase signalling pathway JNKwas recently reported in ourlaboratory [17]. To determine whether results obtained withHepG2 cells are applicable to the human hepatoma cell line
ed activation of AP-1 by JNK inhibitor, SP600125. Huh7-NS3 transfectants wererawal of tetracycline from the culture medium, the cells were pretreated withormed. B) Supershift analysis and specificity of HCV NS3 protein-induced AP-1ith (+Tc) or without (−Tc) in the presence or in the absence of SP600125 weretechnology) for 30 min at 37 °C. The reaction was monitored by EMSAwith ae nuclear extracts in the different lanes. Similar results were obtained in three
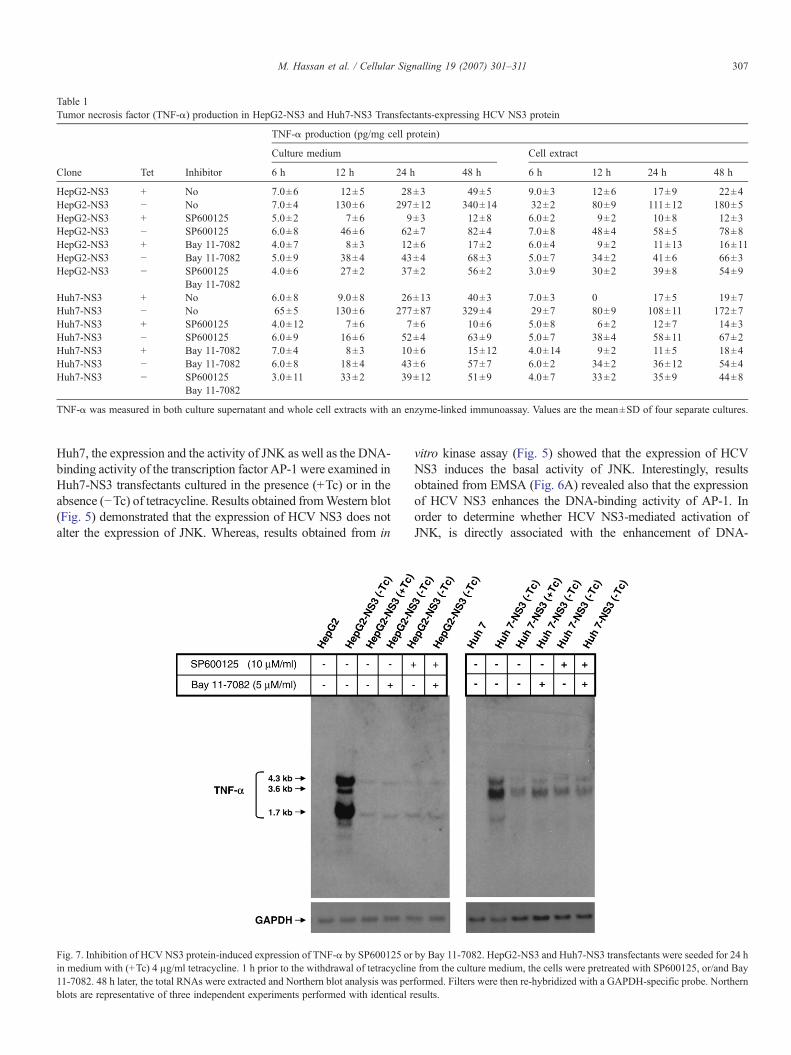
Table 1Tumor necrosis factor (TNF-α) production in HepG2-NS3 and Huh7-NS3 Transfectants-expressing HCV NS3 protein
TNF-α production (pg/mg cell protein)
Culture medium Cell extract
Clone Tet Inhibitor 6 h 12 h 24 h 48 h 6 h 12 h 24 h 48 h
HepG2-NS3 + No 7.0±6 12±5 28±3 49±5 9.0±3 12±6 17±9 22±4HepG2-NS3 − No 7.0±4 130±6 297±12 340±14 32±2 80±9 111±12 180±5HepG2-NS3 + SP600125 5.0±2 7±6 9±3 12±8 6.0±2 9±2 10±8 12±3HepG2-NS3 − SP600125 6.0±8 46±6 62±7 82±4 7.0±8 48±4 58±5 78±8HepG2-NS3 + Bay 11-7082 4.0±7 8±3 12±6 17±2 6.0±4 9±2 11±13 16±11HepG2-NS3 − Bay 11-7082 5.0±9 38±4 43±4 68±3 5.0±7 34±2 41±6 66±3HepG2-NS3 − SP600125 4.0±6 27±2 37±2 56±2 3.0±9 30±2 39±8 54±9
Bay 11-7082Huh7-NS3 + No 6.0±8 9.0±8 26±13 40±3 7.0±3 0 17±5 19±7Huh7-NS3 − No 65±5 130±6 277±87 329±4 29±7 80±9 108±11 172±7Huh7-NS3 + SP600125 4.0±12 7±6 7±6 10±6 5.0±8 6±2 12±7 14±3Huh7-NS3 − SP600125 6.0±9 16±6 52±4 63±9 5.0±7 38±4 58±11 67±2Huh7-NS3 + Bay 11-7082 7.0±4 8±3 10±6 15±12 4.0±14 9±2 11±5 18±4Huh7-NS3 − Bay 11-7082 6.0±8 18±4 43±6 57±7 6.0±2 34±2 36±12 54±4Huh7-NS3 − SP600125 3.0±11 33±2 39±12 51±9 4.0±7 33±2 35±9 44±8
Bay 11-7082
TNF-α was measured in both culture supernatant and whole cell extracts with an enzyme-linked immunoassay. Values are the mean±SD of four separate cultures.
307M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
Huh7, the expression and the activity of JNK as well as the DNA-binding activity of the transcription factor AP-1 were examined inHuh7-NS3 transfectants cultured in the presence (+Tc) or in theabsence (−Tc) of tetracycline. Results obtained fromWestern blot(Fig. 5) demonstrated that the expression of HCV NS3 does notalter the expression of JNK. Whereas, results obtained from in
Fig. 7. Inhibition of HCV NS3 protein-induced expression of TNF-α by SP600125 orin medium with (+Tc) 4 μg/ml tetracycline. 1 h prior to the withdrawal of tetracyclin11-7082. 48 h later, the total RNAs were extracted and Northern blot analysis was perblots are representative of three independent experiments performed with identical r
vitro kinase assay (Fig. 5) showed that the expression of HCVNS3 induces the basal activity of JNK. Interestingly, resultsobtained from EMSA (Fig. 6A) revealed also that the expressionof HCV NS3 enhances the DNA-binding activity of AP-1. Inorder to determine whether HCV NS3-mediated activation ofJNK, is directly associated with the enhancement of DNA-
by Bay 11-7082. HepG2-NS3 and Huh7-NS3 transfectants were seeded for 24 he from the culture medium, the cells were pretreated with SP600125, or/and Bayformed. Filters were then re-hybridized with a GAPDH-specific probe. Northernesults.

308 M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
binding activity of AP-1, we sought to block JNK activity usingthe JNK inhibitor, SP600125 and to investigate the effect of HCVNS3 expression on the extent of the DNA-binding activity of AP-1. Results from EMSA (Fig. 6A) demonstrated that the SP600125inhibits HCV NS3-induced activation of AP-1 in Huh7 cells.These data suggest that the HCV NS3-induced activation of JNKis involved in the enhancement of DNA-binding activity of AP-1.
3.6. Both IKKα/NF-κB and JNK/AP-1 pathways are involvedin the modulation of HCV NS3 protein-induced TNF-αsynthesis in HepG2 and Huh7 cells
To examine whether HCV NS3 protein-induced activation ofIKKα/NF-κB and/or JNK/AP-1 pathways are involved in theregulation of TNF-α expression in hepatocytes. We sought toblock the activation of both IKKα/NF-κB and JNK/AP-1 path-ways using Bay 11-7082 [27] and SP600125 [18], respectively.HepG2-NS3 and Huh7-NS3 transfectants were cultured in amedium with (+Tc) or without (-Tc) for 48 h in the presence andin the absence of Bay 11-7082 or/and Sp600125. We collectedthe culture medium, prepared the total cell extracts, nuclear
Fig. 8. HCV NS3 protein trans-activates the TNF-α promoter in HepG2, Huh7-NS3 atransfected with either reporter plasmids, pTNF1151-Luc, pPro-Luc (negative control)pβGal control plasmid was included in each transfection point. Luciferase activities areHepG2-NS3 and HeLa-NS3 cells over the value of HepG2-NS3, Huh7-NS3 and HeLa-and the data from a representative experiments are shown. B) Transiently transfectiontransfectants, which were pretreated with either SP600125 or/and Bay 11-7082 and indinduction over the values obtained from HepG2, Huh7 and HeLa-mock transfectants.
extracts and the total RNA. Results from EMSA revealed thatpre-treatment of HepG2-NS3 and Huh7-NS3 cells-expressingHCV NS3 with Bay 11-7082 inhibited HCV NS3 protein-induced activation of NF-κB (Fig. 4B). Based on the role ofHCV virus in the triggering of TNF-α production [8], weinvestigated whether the HCV-related protein NS3 effects theexpression of TNF-α in hepatocytes. We examined the ex-pression of TNF-α at mRNA level in both HepG2-NS3 andHuh7-NS3 cells induced to express HCV NS3 protein. Resultsfrom Northern blot analysis (Fig. 6) demonstrated that theexpression of HCV NS3 protein, either in HepG2 or in Huh7cells, induces the expression of HMW TNF-α mRNA species(4.3 and 3.6 kb) in addition to the typical 1.7 kb TNF-αmRNA,which were absent in the parent HepG2 and Huh7 cells. Inter-estingly, when we blocked the expression of HCV NS3 protein,the 3.6 kb TNF-α transcript becomes undetectable, in HepG2-NS3, and weakly detectable in Huh7-NS3 cells, whereas, thetypical 1.7 kb TNF-α transcript becomes undetectable in Huh7-NS3 and weakely detectable in HepG2-NS3 and becomes un-detectable in Huh7-NS3 cells. While the blockade of HCV NS3expression by re-addition of tetracycline resulted in theinhibition of the HMW 4.3 kb transcript in both HepG2 and
nd HeLa cells. A) HepG2-NS3, Huh7-NS3 and HeLa-NS3 cells were transiently, or pPA1Pro-Luc (positive control). To normalize the transfection efficiency, therepresented as fold induction in reporter pTNF1151-Luc in transiently transfectedNS3 cells-transfected with pPro-Luc. Each transfection was performed three times,of the reporter plasmid pTNF1151-Luc in HepG2-NS3, Huh7-NS3 or HeLa-NS3-uced to express HCV NS3 protein. The luciferase activities are represented as foldSimilar results were obtained in four independent experiments.

309M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
Huh7 cells. Interestingly, the same observations were noted alsoin HepG2-NS3 and in Huh7-NS3 transfectants induced toexpress HCV NS3 protein following the treatment with NF-κBand/or JNK inhibitors. To demonstrate whether HCV NS3protein-induced TNF-α mRNA can be translated into protein,the TNF-α was measured by ELISA in the culture medium aswell as in the soluble cell lysates (Table 1). Indeed, the markedincrease in TNF-α mRNA observed in both HepG2-NS3 andHuh7-NS3 cells induced to express HCV NS3 protein was fol-lowed by TNF-α protein secretion. The biologically active TNF-αbecomes weakly detectable in the supernatants of both HepG2-NS3 and Huh7-NS3 cultures 6 h after the withdrawal of tetra-cycline, and increased thereafter as shown in Table 1. The biolo-gical activity of TNF-α secreted into the culture supernatant byHepG2 and Huh7 cells-expressing HCVNS3 protein was roughlyequivalent to 100 pg/ml of recombinant TNF-α as determinedwithL–M cells assay. Taken together these results demonstrate that theHCV-related protein NS3 plays a potential role in the regulation ofHCV-induced production of TNF-α in hepatocytes. Results fromNorthern blot (Fig. 7) and ELISA (Table 1) demonstrated that thepre-treatment of HepG2-NS3 and Huh7-NS3 cells induced toexpress HCV NS3 protein with Bay 11-7082 and/or SP600125inhibited the transcription of themRNA species 3.6 kb, 4.3 kb and1.7 kb (Fig. 7). These results confirm the importance of HCVNS3protein-induced activation of IKKα/NF-κB and JNK/AP-1 path-ways in the transcriptional regulation of TNF-α. However, theability ofBay 11-7082 andSP600125 to inhibitHCVNS3 protein-induced expression of TNF-α indicating that the IKKα/NF-κBand JNK pathways are involved in the modulation of HCV NS3protein-mediated up-regulation of the pro-inflammatory cytokine,TNF-α in hepatocytes.
3.7. Comparative functional analysis of TNF-α promoteractivity in liver and non liver cells-expressing HCV NS3 protein
To determine whether the up-regulation of TNF-α gene ismediated by changes in the activity of TNF-α 5′-upstreamregulatory sequences. The HepG2-NS3, Huh7-NS3 and HeLa-NS3 transfectants were transiently transfected with the TNF-αpromoter-derived construct, pTNF1051-Luc, and the promoteractivity was analyzed in HepG2-NS3, Huh7-NS3 and HeLa-NS3transfectants cultured in medium with (+Tc) or without (−Tc)4 μg/ml tetracycline. In addition, the reporter plasmids, pAP1Pro-Luc, and pOro-Luc, which contained the minimal rat prolactinpromoter either with or without AP-1 binding sites were used ascontrol. The plasmid, pβGal, was included in each transfection toevaluate the transfection efficiency,whichwas quantified by directvisualization of the β-Gal expressing cells. HepG2-NS3, Huh7-NS3 and HeLa-NS3 displayed a higher normalized luciferaseactivity than those cultured in the presence of tetracycline whentransfected with pTNF1051-Luc (Fig. 7). In addition, the plasmid,pAP1Pro-Luc, was also stimulated in HepG2-NS3, Huh7-NS3and HeLa-NS3, where as the plasmid pPro-Luc, which lackedinducible elements, displayed similar promoter activity in all cells.The pre-treatment of eitherHepG2-NS3,Huh7-NS3 orHeLa-NS3clones, when transfected, with the inhibitor of JNKor/andwith theinhibitor of NF-κB blocked the HCV NS3-stimulated activity of
TNF-α promoter. These results confirmed that the HCV NS3-induced activation of JNK/AP-1 and IKKα/NF-κB pathways areessential for the transcriptional regulation of TNF-α gene (Fig. 8).
4. Discussion
The results presented herein provide an insight into the mecha-nism of the intrahepatic TNF-α production in chronic infection ofhepatitis C virus (HCV)-infected patients. In addition, our resultsdemonstrate that the production of TNF-α, in vivo, by hepatocytesin chronic hepatitis may be a consequence of the intracellularpresence of viral compounds and is not caused, at least exclusively,by immune cell-derived inflammatory mediators.
In this work, we have used an in vitro expression system toprove the authenticity of the enhanced intrahepatic TNF-α byHCV-infected hepatocytes as well as the mechanism responsi-ble for its up-regulation. Our results demonstrated that the pro-inflammatory pathways, including TNF-α production aretriggered by HCV NS3 protein in hepatocytes. Induction ofTNF-α gene expression by viruses has been shown to take placein a variety of in vitro models [28,29].
Induction of the inflammation-related genes, including TNF-αis regulated by the activation ofNF-κB site [30] or by the activationof AP-1 site [31,32] in their promoter region, which are commonlyinduced by pathogens, including certain viruses [33–35]. Wefound that the expression of HCV NS3 protein resulted in theactivation of IKKα that was coincident with a rapid decrease incytoplasmic I-κBα protein levels, which is an initial step in theactivation of the NF-κB intracellular signaling pathway inhepatocytes [30]. Upon hepatocyte activation, I-κBα is phosphor-ylated, then degraded via proteosom pathway resulting in NF-κBnuclear translocation and subsequent activation of NF-κB-driveninflammatory genes [30]. In HCV NS3 protein-expressing hepa-tocyte system, NF-κB activation is likely to occur as a result ofsignaling events between the endogenously expressed HCV NS3protein and the intracellular portion of the NF-κB signalingpathway. Indeed, the expression of pro-inflammatory cytokinesincluding TNF-α requires NF-κB activation.
The transcription factor, AP-1, is a major target of the signaltransduction pathways mediated by MAP kinases. Therefore, it islikely that AP-1 might participate in the up-regulation of theTNF-α gene either by its single binding to a specific DNA recog-nition sites within the proximal region of the TNF-α promoter, orin cooperation with heterologous transcription factors, throughtargeting composite response elements. Recently,we demonstratedthe activation of JNK/AP-1 signalling pathway in cells-expressingHCV NS3 protein [17] and we could confirm this finding also inHuh7 cells-expressing HCV NS3 protein. Therefore, the role ofHCV NS3 protein-induced activation of JNK/AP-1 in theregulation of TNF-α in hepatocytes was considered. Althoughthe treatment of HepG2 and Huh7 cells-expressing HCV NS3protein, with the inhibitor of JNK, abrogated completely the actionofHCVNS3 protein onAP-1, however, the inhibition ofAP-1wasnot able to block completely the expression of TNF-α gene. Itshould be noted that the inhibition of TNF-α-induced productionby HCV NS3 protein, through the blockage of JNK/AP-1 and/orIKKα/NF-κBpathways, is not necessarily complete because of the

310 M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
existance of multiple transcription factors. Hence, the inhibition ofone transcription factor causes a reduction in the production ofTNF-α rather than suppression. Although the importance of HCVNS3 protein-mediated activation of JNK and NF-κB pathways inthe regulation of TNF-α gene expression in hepatocytes, thecontribution of further signaling pathways is considered. In ourexperiments, HCV NS3 protein was found to induce the ex-pression of TNF-α, since the blockage of HCV NS3 proteinexpression resulted directly in the repression of TNF-α production.TNF-α has been reported to be involved in angiogenesis processes[36]. In this regard, the observed neo-angiogenesis in the liver ofHCV-infected patients [37], suggesting a potential role for TNF-αin the development of viral hepatitis-associated liver tumors,including HCC.
In the present work, we identified for the first time HMWtranscripts in HCV-transfected hepatocytes. The expression ofthese transcripts are specific for the expression of HCV-relatedprotein NS3 in hepatocytes. Our data indicated that HCV-mediated induction of TNF-α transcripts, in hepatocytes, isregulated at the transcriptional level. First, the HMW TNF-αtranscripts (4.3 and 3.6 kb) and the typical TNF-αmRNA 1.7 kb,which were noted in HCV-transfected HepG2 cells or in HepG2-NS3 and Huh7-NS3 transfectants-expressing HCV NS3 protein,were absent in the parent HepG2 cells. Second, pre-treatment ofHepG2 and Huh7 cells-expressing HCV NS3 protein with theinhibitors of JNK and/or NF-κB resulted in the inhibition of TNF-α transcripts (4.3 and 1.7 kb) HMW TNF-α 3.6 transcript,suggesting a clear correlation between the expression of HCVNS3 protein and TNF-α secretion.
Our results, in agreement with previously reported studies[25,38], demonstrated a marked accumulation of TNF-αmRNA,includingHMW transcripts (4.3 and 3.6 kb) and protein secretion.However, the induction of TNF-αmRNA expression and TNF-αprotein secretion, by the expression of HCV NS3 protein or afterthe treatment of HepG2 and Huh7 cells with IL-1β or phorbolester [39], indicating that TNF-α gene up-regulation can betriggered in hepatocytes with several stimuli, that is likely tohappen by acting through common signal transduction pathways.
Our finding that the TNF-α secreted by HepG2 or Huh7cells-expressing HCV NS3 protein, is biologically active,suggesting that the non-secreted form of TNF-α might have arole in the progression of hepatitis C. Since TNF-α has beenshown to contribute to the liver injury that accompanies experi-mentally induced hepatitis in mice [40].
The proximal region of TNF-α promoter contains variousrecognition sequences for several transcription factors includingNF-κB andAP-1 [41]. In the present work, we demonstrated thatHCV NS3 protein induced up-regulation of TNF-α promoter, inliver and non-liver cells, are mediated mainly through theactivation of AP-1 and NF-κB pathways. Our finding is furthersupported by the observed enhancement of the basal activity ofIKKα in HepG2 and Huh7 cells-expressing HCV NS3, theactivation of JNK in Huh7 cells-expressing HCVNS3 protein aswell as by our recent finding demonstrating the enhancement ofJNK activation by the expression of HCV NS3 in liver and non-liver cell, independent from the co-expression of its cofactorNS4A protein [17]. Therefore, the effect of HCVNS3 protein on
TNF-α secretion may result from the enhanced phosphorylationof c-Jun NH2-terminal activation domain as well as from thephosphorylation of the kinase of NF-κB inhibitor. NF-κB andAP-1 are likely to mediate the induction of cytokines includingTNF-α, leading to inflammatory responses [42]. The HCV NS3protein has been previously detected in the cytoplasm as well asin the nucleus of HCV NS3 transfected cells [26], and in thiswork, we obtained similar results by immunoblotting. Ourfindings, therefore, suggest that HCVNS3 protein might possessa dual function, acting at both the nuclear and the cytoplasmiccompartments. The nuclear function of HCV NS3 protein maybe mediated by the physical association of HCV NS3 proteinwith the proteins of the transcriptional machinery, where as, thecytoplasmic function of HCV NS3 protein involves the acti-vation of JNK/AP-1 and IKKα/NF-κB signaling pathways,which in turn are implicated in the activation of the trans-criptional factors AP-1 and NF-κB.
In conclusion, we identified HMW TNF-α transcripts, whichare specific to the expression ofHCVNS3 protein inHepatocytes,and present an explanation for the possible mechanism of theintrahepatic TNF-α production in vivo. Therefore, the analysis ofother HCV related proteins may provide more information tounderstand themechanisms associatedwith the induction of TNF-α production following HCV infection of liver cells in theinflammatory process in vivo. Therefore, the development of atherapeutic approach based on the modulation of JNK/AP-1 and/or IKKα/NF-kB pathwaysmay become an attractive approach forthe treatment of chronic hepatitis in the future.
Acknowledgements
We thank Professor Dr. Stefan Zeuzem (Internal Medicine II,Saarland University Hospital) for the helpful comments on themanuscript.
References
[1] G.L. Davis, R. Esteban-Mur, V. Rustgi, J. Hoefs, S.C. Gordon, C. Trepo,M.L. Shiffman, S. Zeuzem, A. Craxi, M.H. Ling, J. Albrecht, N. Engl. J.Med. 339 (1998) 1493.
[2] A. Takamizawa, C. Mori, I. Fuke, S. Manabe, S. Murakami, J. Fujita, E.Onishi, T. Andoh, I. Yoshida, H. Okayama, J. Virol. 65 (1991) 1105.
[3] P. Neddermann, L. Tomei, C. Steinkuhler, P. Gallinari, A. Tramontano, R.De Francesco, Biol. Chem. 378 (1997) 469.
[4] Z. Hong, E. Ferrari, J. Wright-Minogue, R. Chase, C. Risano, G. Seelig,C.G. Lee, A.D. Kwong, J. Virol. 70 (1996) 4261.
[5] D.W Kim, Y. Gwack, J.H. Han, J. Choe, Biochem. Biophys. Res.Commun. 215 (1995) 160.
[6] D. Sakamuro, T. Furukawa, T. Takegami, J. Virol. 69 (1995) 3893.[7] T. Fujita, S. Ishido, S. Muramatsu, M. Itoh, H. Hotta, Biochem. Biophys.
Res. Commun. 229 (1996) 825.[8] A. Dolganiuc, K. Kodys, A. Kopasz, C. Marshall, T. Do, L. Romics Jr., P.
Mandrekar, M. Zapp, G. Szabo, J. Immunol. 170 (2003) 5615.[9] H. Tilg, A. Wilmer, W. Vogel, M. Herold, B. Nolchen, G. Judmaier, C.
Huber, Gastroenterology 103 (1992) 264.[10] A. Kasprzak, M. Zabel, W. Biczysko, J. Wysocki, A. Adamek, R.
Spachacz, J. Surdyk-Zasada, J. Histochem. Cytochem. 52 (2004) 29.[11] P.J. Winwood, M.J. Arthur, Semin. Liver Dis. 13 (1993) 50.[12] R. Gonzalez-Amaro, C. Garcia-Monzon, L. Garcia-Buey, R. Moreno-
Otero, J.L. Alonso, E. Yague, J.P. Pivel, M. Lopez-Cabrera, E. Fernandez-Ruiz, F. Sanchez-Madrid, J. Exp. Med. 179 (1994) 841.

311M. Hassan et al. / Cellular Signalling 19 (2007) 301–311
[13] Y. Oyanagi, T. Takahashi, S. Matsui, S. Takahashi, S. Boku, K. Takahashi,K. Furukawa, F. Arai, H. Asakura, Liver 19 (1999) 464.
[14] P.H. McGuinness, D. Painter, S. Davies, G.W. McCaughan, Gut 46 (2000)260.
[15] K.L. MacNaul, N.I. Hutchinson, J.N. Parsons, E.K. Bayne, M.J. Tocci,J. Immunol. 145 (1990) 4154.
[16] P. Vassalli, Annu. Rev. Immunol. 10 (1992) 411.[17] M. Hassan, H. Ghozlan, O. Abdel-Kader, Virology 333 (2005) 324.[18] M. Hassan, H. Ghozlan, O. Abdel-Kader, Cell Signal 16 (2004) 1375.[19] M. Furione, L. Simoncini,M.Gatti, F. Baldanti,M.GraziaRevello, G. Gerna,
J. Clin. Virol. (1999) 121.[20] R.W. Chamberlain, N. Adams, A.A. Saeed, P. Simmonds, R.M. Elliott,
J. Gen. Virol. 78 (1997) 1341.[21] M. Hassan, Interference of hepatitis C virus (HCV) core protein with
intracellular signal transduction processes in liver cells, First ed., ShakerVerlag, Aachen, 2001.
[22] J. Alam, J.L. Cook, Anal. Biochem. 188 (1990) 245.[23] S.M. Kramer, M.E. Carver, J. Immunol. Methods 93 (1986) 201.[24] M. Wolff, W. Jelkmann, Ann. Hematol. 66 (1993) 27.[25] E. Lara-Pezzi, P.L. Majano, M. Gomez-Gonzalo, C. Garcia-Monzon, R.
Moreno-Otero, M. Levrero, M. Lopez-Cabrera, Hepatology 28 (1998) 1013.[26] J.E. Kim, W.K. Song, K.M. Chung, S.H. Back, S.K. Jang, Arch. Virol. 144
(1999) 329.[27] Y. Dai, M. Rahmani, S. Grant, Oncogene 22 (2003) 7108.[28] J.H. Gong, H. Sprenger, F. Hinder, A. Bender, A. Schmidt, S. Horch, M.
Nain, D. Gemsa, J. Immunol. 147 (1991) 3507.[29] J. Gosselin, L. Flamand, M. D'Addario, J. Hiscott, J. Menezes, J. Clin.
Invest. 89 (1992) 1849.
[30] Q. Li, I.M. Verma, Nat. Rev., Immunol. 2 (2002) 725.[31] Y. Fu, K.K. Ishii, Y. Munakata, T. Saitoh, M. Kaku, T. Sasaki, J. Virol. 76
(2002) 5395.[32] K.L. Rhoades, S.H. Golub, J.S. Economou, Virus Res. 40 (1996) 65.[33] H.A. Haeberle, R. Takizawa, A. Casola, A.R. Brasier, H.J. Dieterich, N.
Van Rooijen, Z. Gatalica, R.P. Garofalo, J. Infect. Dis. 186 (2002) 1199.[34] A.C. Erlandsson, L.G. Bladh, P. Stierna, T. Yucel-Lindberg, O.
Hammarsten, T. Modeer, J. Harmenberg, A.C. Wikstrom, J. Endocrinol.175 (2002) 165.
[35] H.J. Kwon, K.W. Lee, S.H. Yu, J.H. Han, D.S. Kim, Biochem. Biophys.Res. Commun. 311 (2003) 129.
[36] S. Yoshida, M.Ono, T. Shono, H. Izumi, T. Ishibashi, H. Suzuki, M.Kuwano, Mol. Cell. Biol. 17 (1997) 4015.
[37] R. Mazzanti, L. Messerini, L. Monsacchi, G. Buzzelli, A.L. Zignego, M.Foschi, M. Monti, G. Laffi, L. Morbidelli, O. Fantappie, F. Bartoloni SaintOmer, M. Ziche, Hepatology 25 (1997) 229.
[38] J. Oquendo, S. Karray, P. Galanaud, M.A. Petit, Res. Immunol. 148 (1997)399.
[39] S. Frede, J. Fandrey, W. Jelkmann, Pflugers Arch. 431 (1996) 923.[40] C.C. Solorzano, R. Ksontini, J.H. Pruitt, P.J. Hess, P.D. Edwards, A.
Kaibara, A. Abouhamze, T. Auffenberg, R.E. Galardy, J.N. Vauthey, E.M.Copeland, C.K. Edwards, G.Y. Lauwers, M. Clare-Salzler, S.L. MacKay,L.L. Moldawer, D.D. Lazarus, J. Immunol. 158 (1997) 414.
[41] E.Y. Tsai, J. Yie, D. Thanos, A.E. Goldfeld, Mol. Cell. Biol. 16 (1996) 5232.[42] Z.G. Liu, H. Hsu, D.V. Goeddel, M. Karin, Cell 87 (1996) 565.

![ax - Anna Nagar Ayyappa · PDF fileHmw KW]Xtb \ax Hmw Khmw ]Xtb \ax SRI AYYAPPA TEMPLE, ANNANAGAR, CHENNAI - 600 040. 3 Hmw AlÀ¸Xtb \ax 70 Hmw PSm[cmb \ax Hmw Pe\n`mb \ax](https://img.pdfslide.tips/doc/110x75/5a78a70c7f8b9a07028c24b6/ax-anna-nagar-ayyappa-kwxtb-ax-hmw-khmw-xtb-ax-sri-ayyappa-temple-annanagar.jpg)





![POLİETİLEN (PE) [HMW-PE500] - Hakan Metal · 2019. 9. 1. · POLİETİLEN (PE) [HMW-PE500] Standart Renkler: Siyah / Beyaz Mekanik Özellikler g/cm³ 0.95 MPa 28 % 300 MPa 850 MPa](https://img.pdfslide.tips/doc/110x75/6069edddb1195a708f57fd36/poletlen-pe-hmw-pe500-hakan-metal-2019-9-1-poletlen-pe-hmw-pe500.jpg)





















