Embed Size (px)
Citation preview

30/10/2009
1
INMUNODEFICIENCIAS
Dr. Agustín Sansosti Servicio de Alergología
Dr.Sansosti Octubre 2009
g gHospital Universitario “Virgen de la Arrixaca”
Murcia – EspañaOctubre 2009
www.alergomurcia.com
INTRODUCCIÓN
Desde que Bruton describió la agammaglobulenia en 1952, más de 150 sindromes de imuno-deficiencias han sido descritos.Estos trastornos pueden afectar uno o varios componentes del sistema inmune, incluyendolinfocitos T,B.NK, celulas fagociticas y complemento.
Paradojicamente, algunas inmunodeficiencias pueden acompañarse de producción excesiva de IgE, o de autoanticuerpos.Por el uso rutinario de antibióticos, las enfermedades alérgicas o autoinmunes pueden ser la primera manifestación de estos cuadros.
También existe un riesgo aumentado de desarrollar tumores, especialmente en aquellos condefectos en celulas B y T. Esto puede deberse a una alteración para eliminar celulas tumoraleso bien a la infección por agentes asociados a malignidad.
Las inmunodeficiencias primarias son diagnosticadas fundamentalmente en la infancia donde
Dr.Sansosti Octubre 2009
Las inmunodeficiencias primarias son diagnosticadas fundamentalmente en la infancia, dondeocurren con mayor frecuencia en niños, mientras que en la edad adulta se dan preferentemente en mujeres más que en hombres.
www.alergomurcia.com

30/10/2009
2
PRIMARIAS
INMUNODEFICIENCIAS
SECUNDARIAS
Dr.Sansosti Octubre 2009
www.alergomurcia.com
Defectos ligados al cromosoma X
Defectos autosomicos cuya base molecular
Inmunodeficiencias primarias
es conocida
Defectos autosomicos cuya base moleculares desconocida
Dr.Sansosti Octubre 2009
Deficits del sistema de complementowww.alergomurcia.com

30/10/2009
3
DEFECTOS LIGADOS AL CROMOSOMA X
• Agammaglobulinemia de Bruton
• Inmunodeficiencia con hiper IgM
fi i d f l d l d i l ( O)• Deficit de factor nuclear modulador esencial (NEMO)
• Sindrome de Wiskott Aldrich
• Enfermedad linfoproliferativa (Enfermedad de Duncan)
• Sindrome IPEX
Dr.Sansosti Octubre 2009
• Sindrome IPEX
• Enfermedad granulomatosa cronica
• Inmunodeficiencia combinada severa ligada a cromosoma Xwww.alergomurcia.com
Agammaglobulinemia de Bruton ( XLA)
Descrita en 1952 por Ogden Bruton, estos pacientes padecen de infeciones recurrentes por patogenos encapsulados ( neumococo, haemophilus)
Gracias a la IgG materna los primeros meses de vida transcurren sin problemas excepto porGracias a la IgG materna, los primeros meses de vida transcurren sin problemas, excepto porinfecciones mucosas debidas al deficit de IgA.
Los pacientes padecen un déficit de todo tipo de inmunoglobulinas, por tanto, las infeccionespueden ser sistemicas (sepsis, meningitis), o bien afectando superficies mucosas (sinusitis,otitis, neumonia, infecciones digestivas), articulaciones o piel.
Las infecciones micoticas y virales en general no son un problema y se manejan con normalidad.
Dr.Sansosti Octubre 2009
y g p y j
La concentración de todas las inmunoglobulinas son extremadamente bajas, al igual que los linfocitos B. En cambio, la concentración y funcion de celulas T y NK son normales.
www.alergomurcia.com

30/10/2009
4
El trastorno básico en esta enfermedad, reside en un deficit de una tirosin kinasa intracelular,llamada Btk (Bruton tirosin kinasa), la cual se expresa con altos niveles en celulas B. De ahí que estos pacientes carezcan practicamente de linfocitos B, y por ende de anticuerpos.
En general, el pronóstico es razonablemente bueno si se instituye precozmente la terapia coninmunoglobulinas vía endovenosa.Las infecciones sistemicas se pueden prevenir con la administracion de las mismas a una dosisde 400 mg/kg cada 3-4 semanas. Además, es necesario la terapia antibiotica cronica para el manejo efectivo de pansinusitis y bronquiectasias.
Dr.Sansosti Octubre 2009
Más allá de las infecciones, el pronostico se ensombrece por el desarrollo de neoplasias linfoproliferativas, con una frecuencia de un 6%.
www.alergomurcia.com
Inmunodeficiencia con hiper IgM
Estos pacientes se asemejan a los de la agammaglobulinemia de Bruton por la tendencia a infecciones por germenes encapsulados.
Las concentraciones de IgE IgA IgG son muy bajas mientras que la de IgM es normal oLas concentraciones de IgE, IgA, IgG son muy bajas, mientras que la de IgM es normal oelevada. Es muy frecuente la formación de autoanticuerpos, con anemia hemolítica, trombopeniay neutropenia.
Se creia que el defecto residia en las celulas B, pero se ha visto que el número de estas es normal, y que cuando se cultivan con diferentes celulas T, responden produciendo Igs.Por tanto, parece que el defecto reside en esta última linea celular.
Dr.Sansosti Octubre 2009
Dado que el pronóstico no es bueno, el tratamiento de elección es el transplante de médula osea.El resto del tratamiento es similar al de la enfermedad de Bruton.
www.alergomurcia.com

30/10/2009
5
Deficit de factor nuclear modulador esencial (NEMO)
Este sindrome se caracteriza clinicamente por la presencia de displasia ectodermica anhidroticaasociada a inmunodeficiencia, debido al deficit de una proteina reguladora denominada NEMO.
La displasia ectodérmica puede afectar, en diferentes grados, a todas las estructuras asociadascon el desarrollo de la piel. La ausencia o disminución de la sudoración en personas condisplasia ectodérmica anhidrótica es causada por la carencia de glándulas sudoríparas.Los niños afectados tienen dificultad para controlar fiebres, y las enfermedades leves pueden producir fiebres extremadamente altas a causa de la falta de regulación de la temperatura a través de la sudoración. Los adultos afectados son incapaces de tolerar un ambiente cálido y requieren medidas especiales para mantener una temperatura corporal normal.
Dr.Sansosti Octubre 2009
NEMO es una proteina reguladora relacionada con la produccion de TNF-alfa e IL-12. Por tanto, estos pacientes son incapaces de sintetizar de manera adecuada estos mediadores.
www.alergomurcia.com
Di if t i d l di l i t d i hid ti
Dr.Sansosti Octubre 2009
Diversas manifestaciones de la displasia ectodermica anhidrotica
www.alergomurcia.com

30/10/2009
6
Sindrome de Wiskott Aldrich
Este sindrome se caracteriza por la presencia de :
• ECCEMA
• PURPURA TROMBOCITOPENICA
• SUCEPTIBILIDAD A INFECCIONES
Los pacientes suelen presentar sangrado prolongado en la zona de circunsición, diarrea san-
Dr.Sansosti Octubre 2009
guinolenta, además de las infecciones recurrentes.Son frecuentes también la dermatitis atopica, asma bronquial alergico, como tambien otroscuadros alergicos.
www.alergomurcia.com
Las infecciones son por germenes encapsulados, como neumococo, produciendo neumoniaotitis media, sepsis y meningitis.Posteriormente las infecciones por germenes oportunistas,como Pneumocystis jiroveci einfecciones virales se tornan mas problematicas.
De todas maneras, la supervivencia es rara después de la adolescencia, siendo las causas demuerte las infecciones, las hemorragias y las neoplasias linfoproliferativas.
El trastorno en estos pacientes se ha localizado en una proteina denominada WASP, lo cualaltera la funcion plaquetaria y de celulas B y T.
Se observa una tasa acelerada tanto de sintesis como de catabolismo de albumina e Igs, porlo que la concentración de estas ultimas resulta sumamente variable.Por lo general existen valores bajos de IgM altos de IgA e IgE y normales o ligeramente
Dr.Sansosti Octubre 2009
Por lo general, existen valores bajos de IgM, altos de IgA e IgE, y normales o ligeramentedisminuidos de IgG.
www.alergomurcia.com

30/10/2009
7
El tratamiento de elección para este trastorno es el transplante de médula osea.
Actualmente, la principal causa de muerte en estos pacientes son las neoplasias linfoproliferativasinducidas por el virus Epstein-Bar.
Dr.Sansosti Octubre 2009
Obtenido de pagina web de Universidad de Valencia
www.alergomurcia.com
Dr.Sansosti Octubre 2009
Costras sanguinolentas en una dermatitis atopica en un paciente con Sindrome de Wiskott-Aldrich
De www.iqb.es
www.alergomurcia.com

30/10/2009
8
Enfermedad linfoproliferativa (Enfermedad de Duncan)
Se caracteriza por una respuesta anormal a la infeccion por el virus de Epstein Bar.
Los pacientes afectados son aparentemente normales hasta que experimentan mononucleosis infecciosa.
Se distinguen 3 fenotipos: mononucleosis infecciosa fulminante (50%), linfomas (20%) ehipogamaglobulinemia (30%).La mononucleosis es mortal debido a la extensa necrosis hepatica.
La edad media de presentación es por debajo de los 5 años. Los pacientes que sobrevivendesarrollan linfomas o hipogamaglobulinemia.
Dr.Sansosti Octubre 2009
El trastorno en esta enfermedad radica en una proteina denominada SH2D1A.
La funcion de esta proteina se relaciona con chequear y controlar la proliferación de lascelulas T en respuesta a la infeccion por EBV, y posiblemente otras infecciones .
www.alergomurcia.com
Sindrome IPEX
cd4+ cd25+ foxp3+
Dr.Sansosti Octubre 2009
CELULA T REGULADORA
www.alergomurcia.com

30/10/2009
9
Imune disregulation
PolyendocrinopathyPolyendocrinopathy
Enteropathy
X linked
Dr.Sansosti Octubre 2009
X-linked
www.alergomurcia.com
Este cuadro se caracteriza por la presencia de autoinmunidad sistémica, generalmente bajo laforma de Diabetes tipo 1, enteropatia severa con diarrea acuosa sanguinolienta, dermatitiseccematosa, y la presencia de infecciones recurrentes causadas por Enterococcus y Staphylococcus.
El trastorno se debe a la mutación en el gen que codifica para la proteina FOXP3.
Foxp3 es una proteina que se expresa en las celulas T reguladoras. El numero de celulas T enestos pacientes es normal, pero no poseen dicha proteína, por lo que su función esta alterada.
Dr.Sansosti Octubre 2009
Esto demuestra el papel de FOXP3 en el mantenimiento de la propia tolerancia, y la prevenciónde enfermedades alergicas y autoinmunitarias.
www.alergomurcia.com

30/10/2009
10
Enfermedad granulomatosa crónica
Este trastorno se caracteriza por una destrucción intracelular defectuosa de bacterias y hongos.Se calcula que tiene una frecuencia de 1 cada 500000 recien nacidos. Si bien se presenta en lainfancia, se han descrito casos en edades mas tardías, entre los 13 y 30 años.
Se caracteriza por infecciones por organismos catalasa positivos, como Staphylococcus, Serratia,Pseudomonas, Salmonella; pero no por catalasa negativos, como neumococo y haemophilus.
La osteomielitis por Serratia, y posteriormente por hongos (Aspergillus) es caracteristica de estaforma de inmunodeficiencia.
Dr.Sansosti Octubre 2009
www.alergomurcia.com
El trastorno radica en la incapacidad de los fagocitos para destruir los microorganismos ingeridos, debido a un fracaso en el “estallido respiratorio“, en el que el NADP es transformadoen NADPH.
Debido a esta incapacidad, y como consecuencia de una respuesta compensadora por parte delas celulas linfoides, se forman granulomas, particularmente en higado, via urinaria y tubodigestivo.
El tratamiento se basa en el manejo agresivo de las infecciones con antibióticos, juntamente con la terapia crónica con antifungicos, como Itraconazol.
Dr.Sansosti Octubre 2009
con la terapia crónica con antifungicos, como Itraconazol.
www.alergomurcia.com

30/10/2009
11
Inmunodeficiencia combinada severa ligada a cromosoma X
Se trata de un grave síndrome caracterizado por un profundo déficit en la funcion de celulas T y B, producido por diversas causas geneticas, siendo la forma mas frecuente la ligada alcromosoma X.
Los afectados presentan en los primeros meses de vida, episodios de diarrea, neumonia, otitis e infecciones cutaneas y sepsis.Además, infecciones por germenes oportunistas como Candida, Pneumocystis Jiroveci, etc.
A l d d d 6 7 d l í d l i t di ti d t
Dr.Sansosti Octubre 2009
A la edad de 6-7 meses, cuando la mayoría de los pacientes son diagnosticados, presentan LINFOPENIA, además las concentraciones séricas de inmunoglobulinas son nulas, y luego de la vacunación no existe formación de anticuerpos.
www.alergomurcia.com
En el año 1976, John Travolta protagonizó una película para la televisión (El chico de la burbuja de plástico)
Dr.Sansosti Octubre 2009
e a o 9 6, Jo a o ta p otago ó u a pe cu a pa a a te e s ó ( c co de a bu buja de p ást co)que contaba la historia de un niño que tenía un sistema inmunológico que no funcionaba correctamente. El contacto con el aire sin filtrar podía acabar con su vida y esto le obligó a permanecer en un ambiente esterilizado, aislado del mundo exterior. Este niño, en el que se inspiraba la película, existió realmente y sellamó David Vetter.David Vetter nació con inmunodeficiencia severa combinada (SCID)
www.alergomurcia.com

30/10/2009
12
El gen alterado codifica para una cadena proteica común a diversos receptores de citocinas, entre ellos el de IL-2, IL-4, IL-7, IL-9, IL-15 e IL-21.
La SCID constituye una emergencia pediátrica. A no ser que se efectúe el transplante de médula osea, la muerte suele ocurrir antes del primer año de vida, y casi invariablemente antes del segundo.
Por otro lado, el transplante de médula osea en los primeros meses de vida proporciona una probabilidad de supervivencia de más del 96%.
Por tanto el diagnóstico precoz es esencial.
Dr.Sansosti Octubre 2009
www.alergomurcia.com
Defectos autosomicos cuya base molecular es conocidaDefectos autosomicos cuya base molecular es conocida
Dr.Sansosti Octubre 2009
www.alergomurcia.com

30/10/2009
13
Déficit de adenosin desaminasa
Estos pacientes tienen la misma susceptibilidad a infecciones que los pacientes con SCID-X.Además, presentan algunas anomalías óseas características, similares al rosario raquítico.
Generalmente, existe una linfopenia más profunda que en otros tipos de inmunodeficiencia, dado que el déficit de ADA afecta primariamente a las células T.
El déficit enzimático causa la acumulación de adenosina, deoxyadenosina y metiladenosina, los
Dr.Sansosti Octubre 2009
y ycuales conducen directa o indirectamente a la apoptosis de timocitos y linfocitos circulantes,lo que causa la inmunodeficiencia.
www.alergomurcia.com
El tratamiento consiste en el transplante de médula ósea.
Cabe decir que en esta enfermedad fue la primera en la que se intentó la terapia genica empleando virus como vectores, para reemplazar el gen defectuoso.
Dr.Sansosti Octubre 2009
De www.udp.cl
www.alergomurcia.com

30/10/2009
14
Inmunodeficiencia combinadaEste termino se emplea para diferenciar a los pacientes con una baja, pero no ausente, función de células T, de aquellos con SCID.
Entre ellas se encuentran:
- DEFICIT DE PURIN-NUCLEOSIDO FOSFORILASA
- HIPOPLASIA PELO-CARTILAGINOSA
Dr.Sansosti Octubre 2009
- ATAXIA TELANGIECTASIA
www.alergomurcia.com
Ataxia telangiectasia
Dr.Sansosti Octubre 2009
de www.nature.com
www.alergomurcia.com

30/10/2009
15
La ataxia telangiectasia es un trastorno que se caracteriza clinicamente por :
Ataxia cerebelosa progresiva, telangiectasias oculocutaneas, enfermedad sinopulmonar crónica, inmunodeficiencia humoral y celular y una alta incidencia de tumores malignos (linfomas y adenocarcinomas).
No se ha encontrado todavía un tratamiento satisfactorio. El pronóstico es muy malo, a pesar deque algunos pacientes pueden alcanzar la edad adulta.
Las causas más comunes de muerte son las neoplasias linfoides y la enfermedad neurologicaprogresiva.
Dr.Sansosti Octubre 2009
www.alergomurcia.com
Sindrome de Di George
Resulta de una dismorfogenesis del 3° y 4° arco branquial, lo que lleva a una aplasia o hipoplasia del timo y glandulas paratiroides.Aparte suelen encontrarse otras alteraciones, como úvula bífida y malformaciones cardíacas.
Lo que sí se ve es que es mucho mas frecuente la hipoplasia que la aplasia total, por lo que muchos pacientes no tienen grandes problemas infecciosos y crecen normalmente.
En general, el diagnóstico se realiza en el período neonatal debido a las convulsiones porhipocalcemia.
Dr.Sansosti Octubre 2009
www.alergomurcia.com

30/10/2009
16
Sindrome linfoproliferativo autoinmune
Se trata de un trastorno donde fracasa la apoptosis de los linfocitos,lo que lleva a su proliferación, con hiperesplenismo y citopenias autoinmunes.
La causa genética más común consiste en la mutación del gen que codifica para el receptor CD 95 ( bi ll d FAS) l l di d d l iCD 95 (tambien llamado FAS) , el cual es un mediador de la apoptosis.
Las factores de morbimortalidad lo constituyen la severidad de la enfermedad autoinmune, el hiperesplenismo y los linfomas.
Esto pone de manifiesto la importancia de los receptores de membrana en eliminar los linfocitos con potencial oncogénico y autoinmune.
Dr.Sansosti Octubre 2009
www.alergomurcia.com
Sindrome de Chediak Higashi
Se caracteriza por la presencia de albinismo oculocutáneo y susceptibilidad a infecciones respiratorias recurrentes.
Existen lisosomas gigantes, lo cuales carecen de la capacidad de fusionarse con los fagosomas,l l b t i i id d d t idpor lo que las bacterias ingeridas no pueden ser destruidas.
El 85% de los pacientes sufren una fase acelerada de la enfermedad, con hepatoesplenomegalia,ictericia, pancitopenia y cambios neurológicos.Una vez que ocurre la fase acelerada, la muerte sobreviene, por término medio, a los 30 meses;a no ser que se efectúe transplante de médula ósea.
Dr.Sansosti Octubre 2009
www.alergomurcia.com

30/10/2009
17
Inmunodeficiencia común variable (CVID)
También llamada hipogammaglobulinemia adquirida, se parece en muchos aspectos a la XLA.De todas maneras en estos pacientes las infecciones suelen ser menos severas y presentarse mástardiamente.Este cuadro se ha asociado de manera variable a linfoma intestinal, colitis, atrofia gástrica, timoma y otros trastornos.y
Si bien en estos pacientes el número de linfocitos B es normal, no se diferencian en célulasproductoras de inmunoglobulinas.
Además, existe tendencia a la formación de autoanticuerpos, por ello existen casos de CVID quese transforman en LES. Llama la atención que en muchos pacientes el cuadro se resuelve de manera temporal o
Dr.Sansosti Octubre 2009
Llama la atención que en muchos pacientes el cuadro se resuelve de manera temporal o permanente al adquirir la infección por VIH.
www.alergomurcia.com
Dado que este trastorno ocurre en familiares de 1° de pacientes con déficit de IgA, y que muchospacientes con este déficit, con el tiempo se tornan panhipogammaglobulinemicos, se sospecha que ambos trastornos pueden tener un origen genético común.
El tratamiento es similar al de la XLA, con inmunoglobulinas IV, y tratamiento antibióticocrónico.
El pronóstico es razonablemente bueno, salvo que se desarrollen trastornos autoinmunes o neoplasias linfoides.
Dr.Sansosti Octubre 2009
neoplasias linfoides.
www.alergomurcia.com

30/10/2009
18
Déficit selectivo de IgA
Se cree que es la inmunodeficiencia más frecuente, con una frecuencia de 1 cada 300-400 dadores de sangre.
Como cabe esperar, las infecciones se dan preferentemente a nivel de mucosas (respiratoria,digestiva, urinaria).g , )De manera similar a la CVID, existe un mayor riesgo de enfermedad autoinmune y neoplasias linfoides.
Salvo la IgA, la concentración del resto de inmunoglobulinas es normal.
De gran trascendencia clínica es el hecho de que un porcentaje alto de estos pacientes (40-50%), presentan anticuerpos anti IgA (particularmente IgE anti IgA).
Dr.Sansosti Octubre 2009
Por ello, pueden sufrir reacciones anafilácticas al recibir derivados sanguíneos que contengan IgA.De ahí que deben pre-tratarse los productos antes de transfundirlos,o bien emplear aquellos de otras personas con deficit de IgA.
www.alergomurcia.com
El defecto que causa este déficit de momento se desconoce. Se sospecha que puede tener unacausa similar a la CVID.
No hay un tratamiento específico, excepto el tratamiento intensivo de las infecciones con ATBadecuados.En algunos niños, se ha visto que el trastorno puede ser transitorio y desaparecer con el tiempo,mientras que en adultos suele ser más bien persistente, y en algunos casos convertirse en unaCVID.
Dr.Sansosti Octubre 2009
www.alergomurcia.com
30/10/2009
19
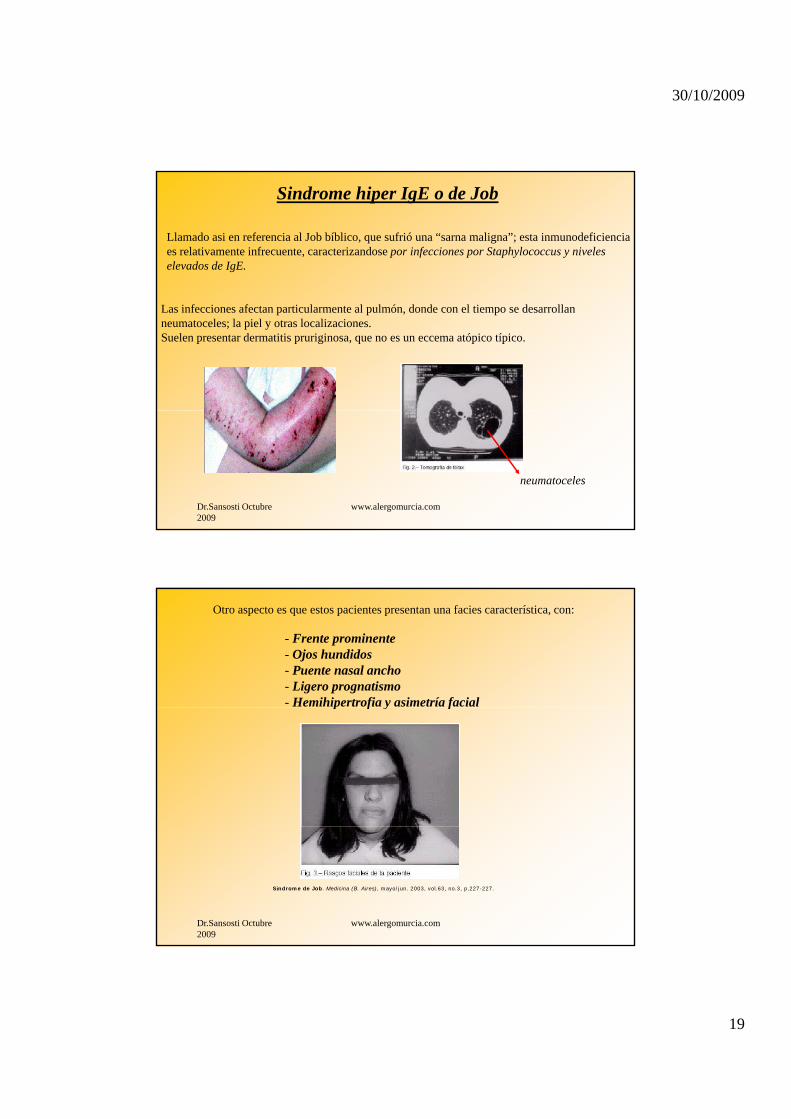
Sindrome hiper IgE o de Job
Llamado asi en referencia al Job bíblico, que sufrió una “sarna maligna”; esta inmunodeficienciaes relativamente infrecuente, caracterizandose por infecciones por Staphylococcus y niveleselevados de IgE.
Las infecciones afectan particularmente al pulmón, donde con el tiempo se desarrollanneumatoceles; la piel y otras localizaciones.Suelen presentar dermatitis pruriginosa, que no es un eccema atópico típico.
Dr.Sansosti Octubre 2009
neumatoceles
www.alergomurcia.com
Otro aspecto es que estos pacientes presentan una facies característica, con:
- Frente prominente- Ojos hundidos- Puente nasal ancho- Ligero prognatismo- Hemihipertrofia y asimetría facialp f y f
Dr.Sansosti Octubre 2009
Síndrome de Job. Medicina (B. Aires), mayo/jun. 2003, vol.63, no.3, p.227-227.
www.alergomurcia.com

30/10/2009
20
También existen alteraciones en la dentición, y alteraciones óseas, tales como fracturas patológicas, y escoliosis.
A nivel analítico, destaca la elevación de IgE (2000 a 20000 o más), y la elevada eosinofilia ensangre y esputo.
El trastorno se debe a una mutación en el gen STAT3. El STAT3 es una proteína que esta implicada en la transducción de la señal de muchas citocinas, entre ellas IL-6, IL-10 e IL-21.implicada en la transducción de la señal de muchas citocinas, entre ellas IL 6, IL 10 e IL 21.
El fenotipo clínico es, ya sea por inflamación insuficiente o excesiva. Las infecciones del pulmón que llevan a neumatoceles son un ejemplo de inflamación excesiva; mientras que losabscesos cutaneos (“fríos”) son un ejemplo de inflamación insuficiente.Esta dicotomía es esperada por citocinas proinflamatorias (IL-6) y antiinflamatorias (IL-10),ambas mediadas por el STAT3.
El tratamiento consiste en el manejo adecuado de las infecciones ,teniendo en cuenta que en sumayor parte son producidas por staphylococcus.
Dr.Sansosti Octubre 2009
A. Freeman, S. Holland. Pediatr Res 65:32R-37R,2009
mayor parte son producidas por staphylococcus.
www.alergomurcia.com
Déficits en componentes del sistema de complemento
Se han descrito déficits en casi todos los componentes del sistema de complemento.
Dr.Sansosti Octubre 2009
www.alergomurcia.com

30/10/2009
21
Los pacientes con déficits en la vía clásica o componentes tardíos no suelen tener grandes problemas, salvo cuando sufren sepsis o infección por gérmenes patógenos de alto grado.
En cambio, aquellos con déficits en la vía alternativa suelen tener infecciones más frecuentesy más severas, especialmente en la infancia, debido a que la vía alternativa es de gran valordurante la primera exposición a un agente infeccioso.
Por otro lado, se ha visto en los pacientes con estos déficits, una alta incidencia de trastornosautoinmunes
C1q, C1r, C1s, C2 y C4
Dr.Sansosti Octubre 2009
Déficits en sistemade complemento C3
Déficits de componentes terminales
www.alergomurcia.com
DÉFICIT DE C1q, C1r, C1s, C2 y C4
En estos casos las infecciones son más leves y menos frecuentes que aquellos con déficit enla vía alternativa.
El trastorno más frecuente es el deficit de C2, con una frecuencia de 1 cada 10.000 sujetos. En estos casos pueden ser normales o padecer ,con carácter recurrente, sepsis, meningitis yosteomielitis.
Los pacientes con estos déficits pueden tener síntomas que asemejan a lupus discoide o LES
Dr.Sansosti Octubre 2009
Los pacientes con estos déficits pueden tener síntomas que asemejan a lupus discoide o LES.En estos casos, suelen presentar fotosensibilidad y compromiso de la funcion renal a edadestempranas; los títulos de anticuerpos antinucleares pueden ser negativos.
www.alergomurcia.com

30/10/2009
22
Déficit de C3
Los déficits en la vía alternativa por lo general conducen a infecciones recurrentes por patógenosencapsulados, provocando neumonia, otitis media y sinusitis.
El déficit de C3 puede ser debido a una disminución en su síntesis o a un catabolismo aumentado.
La consecuencia de esto es una pérdida del control de la vía alternativa
Dr.Sansosti Octubre 2009
www.alergomurcia.com
Déficits de componentes terminales (C5 C6 C7 C8)
Estos déficits pueden verse en individuos sanos, o asociarse a una mayor incidencia de infeccióno trastorno autoinmune.
Pacientes con deficit de C5 pueden verse afectados de LES, fenómeno de Raynaud, nefritis,neumonia recurrente por neumococo, e infección gonocócica diseminada.
Los pacientes con deficit de C6, C7 o C8 pueden sufrir meningitis recurrente por meningococoe infección gonocócica diseminada.
Por último, tener en cuenta que no hay un tratamiento específico para los déficits del sistemade complemento
Dr.Sansosti Octubre 2009
de complemento.El mismo se basa en el tratamiento adecuado de las infecciones con ATB, e incluso puederealizarse la antibioticoterapia crónica de manera profiláctica.
www.alergomurcia.com

30/10/2009
23
F I N
Dr.Sansosti Octubre 2009
www.alergomurcia.com