Embed Size (px)
Citation preview

Analysis of Human Tyrosyl-DNA Phosphodiesterase ICatalytic Residues
Amy C. Raymond1,2, Marc C. Rideout2, Bart Staker2, Kathryn Hjerrild2
and Alex B. Burgin Jr1,2*
1Biology Department, SanDiego State University, 5500Campanile Drive, San DiegoCA 98182-4614, USA
2deCODE geneticsBioStructures Group, 7869 NEDay Road West, BainbridgeIsland, WA 98110, USA
Tyrosyl-DNA phosphodiesterase I (Tdp1) is involved in the repair ofDNA lesions created by topoisomerase I in vivo. Tdp1 is a member of thephospholipase D (PLD) superfamily of enzymes and hydrolyzes 30-phos-photyrosyl bonds to generate 30-phosphate DNA and free tyrosine invitro. Here, we use synthetic 30-(4-nitro)phenyl, 30-(4-methyl)phenyl, and30-tyrosine phosphate oligonucleotides to study human Tdp1. Kineticanalysis of human Tdp1 (hTdp1) shows that the enzyme has nanomolaraffinity for all three substrates and the overall in vitro reaction is diffusion-limited. Analysis of active-site mutants using these modified substratesdemonstrates that hTdp1 uses an acid/base catalytic mechanism. Theresults show that histidine 493 serves as the general acid during the initialtransesterification, in agreement with hypotheses based on previouscrystal structure models. The results also argue that lysine 495 andasparagine 516 participate in the general acid reaction, and the analysisof crystal structures suggests that these residues may function in a protonrelay. Together with previous crystal structure data, the new functionaldata provide a mechanistic understanding of the conserved histidine,lysine and asparagine residues found among all PLD family members.
q 2004 Elsevier Ltd. All rights reserved.
Keywords: tyrosyl-DNA phosphodiesterase I; phospholipase Dsuperfamily; topoisomerase I; DNA repair; proton relay*Corresponding author
Introduction
DNA topoisomerases are ubiquitous enzymesthat catalyze changes in DNA topology by alteringthe linkage of DNA strands.1 Eukaryotic topo-isomerase I (TopoI) uses an active-site tyrosineresidue to cleave one strand of DNA forming a 30-phosphotyrosine intermediate. This opening of theDNA backbone is necessary to allow the removalof superhelical tension that is generated duringreplication and transcription. The phosphodiesterDNA backbone is restored when the 50-hydroxyl,generated during cleavage, attacks the 30-phospho-tyrosyl phosphodiester.2 Because the rate of
re-ligation is normally much faster than the rate ofcleavage, the steady-state concentration of topo-isomerase–DNA adducts is extremely low. This isimportant to maintain the integrity of the genome;however, TopoI–DNA adducts can accumulate inthe presence of naturally occurring DNA damagesuch as nicks,3 abasic sites,4 modified bases,5 modi-fied sugar molecules6 or as a result of exposure to avariety of chemotherapeutic drugs.7,8 Tyrosyl-DNAphosphodiesterase I (Tdp1) has been shown to actas a specific repair enzyme for TopoI lesionsin vivo,9 and catalyzes the hydrolysis of the phos-phodiester bond between the 30 end of DNA and asingle tyrosine residue in vitro.10 Tdp1 is thereforean important DNA repair enzyme and a potentialmolecular target for new anti-cancer drugs.11
Tdp1 is a member of the phospholipase D (PLD)superfamily and is conserved from yeast to man.12
The PLD superfamily represents an extremelydiverse family of enzymes, including phospholipidhydrolases, cardiolipin synthases, phosphatidylserine synthases, poxvirus envelope proteins,endonucleases, and a Yersinia murine toxin.13,14
0022-2836/$ - see front matter q 2004 Elsevier Ltd. All rights reserved.
E-mail address of the corresponding author:[email protected]
Abbreviations used: Tdp1, tyrosyl-DNAphosphodiesterase I; hTdp1, human tyrosyl-DNAphosphodiesterase I; TopoI, topoisomerase I; PLD,phospholipase D; yTdp1, yeast Tdp1; 4-nitro, 30-(4-nitro)phenyl phosphate DNA; 4-methyl, 30-(4-methyl)phenyl phosphate DNA.
doi:10.1016/j.jmb.2004.03.013 J. Mol. Biol. (2004) 338, 895–906

Despite this diverse array of substrates and bio-logical functions, it had been shown that allmembers of this family shared a signatureHxKx4D motif.15 Tdp1 has two such motifs,marked by histidine 263/lysine 265 (H263/K265)and histidine 493/lysine 495 (H493/K495). Sur-prisingly, discovery of the Tdp1 subfamily showedthat the aspartic acid (D) is not conserved and thatTdp1 represents a unique subclass within the PLDsuperfamily.12 Characterization of the Tdp1orthologs also showed that two asparagine resi-dues are more highly conserved (N283 and N516).
Previous mechanistic and structural studies ofother phospholipase D superfamily membersargue that Tdp1 uses a two-step general acid/basereaction mechanism to cleave 30-phosphotyrosylbonds.12,16,17 On the basis of crystal structuremodels, it has been proposed that in the first stepof the reaction H263 acts as a nucleophile forminga 30-phosphohistidine Tdp1 covalent intermediate,and that H493 protonates the phenoxy anion ofthe tyrosine leaving group.18,19 Free Tdp1 and 30-phosphate DNA are presumably generated byhydrolysis of the 30-phosphohistidine intermediatein a second step. This two-step general acid/basereaction is summarized in Figure 1. It has alsobeen proposed that K265, N283, K495 and N516are all involved in substrate binding and transitionstate stabilization.18
Here, we use a new series of Tdp1 substrateanalogs to show that Tdp1, and presumably othermembers of the PLD superfamily, use a generalacid/base catalytic mechanism. This functionaldata validates the proposed roles of H263 andH493. Surprisingly, the results show that K495 andN516 participate directly or indirectly in protonat-ing the tyrosine leaving group during the firsttransesterification reaction. This result suggeststhat the conserved lysine and asparagine residuesmay have multiple functions during the course ofthe reaction. A comparison of hTdp1 crystal struc-
tures before and after substrate binding, suggestsa mechanism for how these residues participate inthe general acid reaction.
Results
Although Tdp1 has been shown to be involvedin the repair of topoisomerase I–DNA covalentadducts, the natural in vivo substrate is notknown. In vitro, Tdp1 hydrolyzes 30-phospho-tyrosyl DNA, but does not cleave 50-phospho-tyrosyl DNA or 30-phosphoseryl DNA.10 Single ordouble-stranded oligonucleotides containing a 30-tyrosine residue or a small topoIB peptide frag-ment (four to eight amino acid residues) are alsoefficient substrates in vitro.12,20 Oligonucleotidescontaining larger peptide fragments (.11 aminoacid residues) are hydrolyzed very slowly and it ispresumed that in vivo the topoisomerase is proteo-lyzed to one (i.e. tyrosine residue) or a few aminoacid residues before it is ultimately acted upon byTdp1.10 Tdp1 can cleave 30-tyrosine or 30-(4-nitro)-phenyl mononucleotides,10,21 but these substratesare relatively poor. The preference for oligonucleo-tide substrates versus mononucleotide substrates isexpected, since three conserved Tdp1 residuesmake specific hydrogen bond contacts to two phos-phodiester bonds upstream of the cleavage site.Structural data also suggest that there might be aconserved stacking interaction (F259) with anupstream base.18 Unlike contacts between Tdp1and the DNA portion of the substrate, bindinginteractions with the leaving group are less welldefined. For example, one crystal structure showsthat a topoI fragment of eight amino acid residuesmakes three hydrogen bond contacts to Tdp1;18
however, these residues are not conserved and theconformation of the peptide is significantlydifferent from the conformation of the correspond-ing region found in the crystal structures oftopoisomerase I.22 In addition, yeast Tdp1 (yTdp1)yielded identical kinetics (KM and kcat) witholigonucleotide substrates containing four aminoacid residues or a single tyrosine residue(kcat=KM ¼ 1 £ 106 M21 s21).20 Taken together, theseresults show that the only feature of a leavinggroup that is shared by all efficiently cleaved sub-strates is a single phenol moiety.
This observation prompted us to synthesize andcompare two oligonucleotide substrates thatwould result in minimal leaving groups whencleaved by Tdp1: 4-nitro-phenol and 4-methyl-phenol. These two leaving groups, diagrammed inFigure 2a, were chosen because they wouldoccupy very similar space within the enzymeactive site, but the quality of the two leavinggroups is significantly different. The presence of astrong electron withdrawing group (–NO2) lowersthe pKa of the phenoxy anion, creating a betterleaving group, whereas a methyl group at the 4position increases the pKa of the phenoxy anionsimilar to the b-methylene carbon atom of tyrosine.
Figure 1. Two-step reaction mechanism for Tdp1. Inthe first transesterification step, the Tdp1 nucleophileH263 attacks the substrate 30-phosphotyrosyl bond. Thetyrosine phenoxide is protonated by the Tdp1 generalacid H493. In the second step, the 30-phosphohistidineintermediate is hydrolyzed to produce 30-phosphateDNA and free Tdp1.
896 Functional Analysis of Tdp1 Catalytic Residues

To test the efficiency of these new substrates wecompared them to identical oligonucleotidescontaining a 30-phospho-tyrosine residue. Sub-strates were 50-end labeled and incubated withhTdp1 D1-148. All crystal structures of hTdp1have been obtained using this N-terminal deletionand we have independently confirmed previousstudies12,23 demonstrating that full-length hTdp1and hTdp1 D1-148 have indistinguishable specificactivities (data not shown). The N terminus ofTdp1 is extremely variable in size and the amino
acid identity among Tdp family members, whichargues that this region is not important foractivity.12 hTdp1 D1-148 was used for all of thekinetic studies described below so that the com-parison to the structural data would be moredirect. After incubation with the enzyme, the reac-tion products were resolved on a denaturingsequencing gel. As expected, all the three sub-strates are converted to 30-phosphate DNAproducts that co-migrate in the gel (Figure 2b).The 30-tyrosyl phosphate DNA substrate migratesmore slowly in the gel than the 30-(4-nitro)phenylphosphate or 30-(4-methyl)phenyl phosphate DNA,abbreviated 4-nitro and 4-methyl, due to the slightincrease in molecular mass. The identities of allthree starting materials and the Tdp1-mediatedcleavage products (30-phosphate DNA) were con-firmed by mass spectrophotometric analysis ofunlabelled reactions (data not shown).
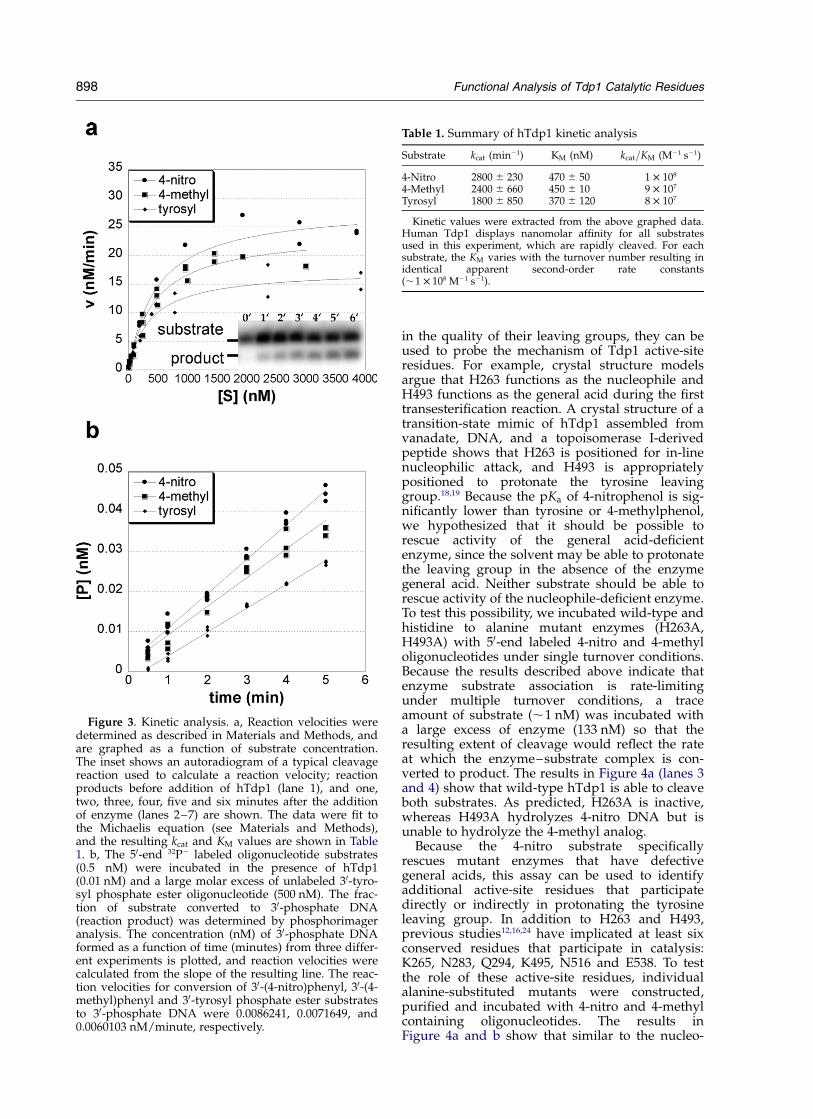
To determine how well hTdp1 recognizes andcleaves these three substrates, reaction velocitieswere measured as a function of substrate concen-tration (Figure 3a). The results in Table 1 showthat all three substrates are cleaved rapidly withkcat values of 1800, 2400, 2800 min21 for the tyrosyl,4-methyl and 4-nitro substrates, respectively. Theapparent KM values also varied with the samemagnitude: 370, 450, and 470 nM for the tyrosyl,4-methyl and 4-nitro substrates, respectively. As aresult, the calculated kcat=KM values are statisticallyindistinguishable between the three substrates,,1 £ 108 M21 s21. This value is important becauseit represents the apparent second-order rateconstant for the overall reaction, and provides ameasure of how rapidly the enzyme can work atlow substrate concentrations, presumablyobserved in vivo. In this case, the observed kcat=KM
value is close to the expected rate of diffusion-controlled encounter of enzyme and substrate(108–109 M21 s21).
The observation that all three substrates displayvery similar kcat=KM values predicts that hTdp1cannot distinguish between the three substrates ina competition experiment. To test this prediction,a trace amount (0.5 nM) of each 50-end labeled sub-strate was incubated in the presence of a largeexcess of 30-tyrosyl DNA (500 nM) in three separatereactions. If Tdp1 cleaves the tyrosyl substratemore efficiently than the 4-nitro or 4-methyl sub-strates, then the observed velocities should bedecreased relative to the tyrosyl substrate. Theresults in Figure 3b show that the reactionvelocities are essentially identical for all three sub-strates; in fact, the observed velocities differ asexpected from the statistically insignificantdifferences in the calculated kcat=KM values(Table 1). Taken together these results show thathTdp1 cannot distinguish the 4-methyl and 4-nitrosubstrates from a standard tyrosine substrate, andall three derivatized oligonucleotides are valid invitro substrates for hTdp1.
Because 4-nitro and 4-methyl substrates havesimilar molecular mass but differ significantly
Figure 2. The 30-derivatized oligonucleotide substrates.a, The 30-derivatized oligonucleotide Tdp1 substrates arediagrammed on the left. The 30-(4-nitro)phenyl phos-phate ester DNA is shown at the top and is abbreviated4-nitro: 30-(4-methyl)phenyl phosphate ester DNA and30-phosphotyrosine DNA, abbreviated 4-methyl andtyrosyl, respectively, are shown below. The secondcolumn shows the products of Tdp1 cleavage, and thethird column shows the pKa of the leaving group foreach substrate. b, The 50-end-labeled 30-tyrosyl, 30-(4-methyl)phenyl and 30-(4-nitro)phenyl phosphate esterDNA substrates were incubated in the presence (þ) orabsence (2 ) of hTdp1. The reaction products wereresolved on a 20% polyacrylamide gel containing 8 Murea, and the resulting autoradiogram is shown. Thepositions of substrates and 30-phosphate DNA productswithin the gel are indicated on the right.
Functional Analysis of Tdp1 Catalytic Residues 897
in the quality of their leaving groups, they can beused to probe the mechanism of Tdp1 active-siteresidues. For example, crystal structure modelsargue that H263 functions as the nucleophile andH493 functions as the general acid during the firsttransesterification reaction. A crystal structure of atransition-state mimic of hTdp1 assembled fromvanadate, DNA, and a topoisomerase I-derivedpeptide shows that H263 is positioned for in-linenucleophilic attack, and H493 is appropriatelypositioned to protonate the tyrosine leavinggroup.18,19 Because the pKa of 4-nitrophenol is sig-nificantly lower than tyrosine or 4-methylphenol,we hypothesized that it should be possible torescue activity of the general acid-deficientenzyme, since the solvent may be able to protonatethe leaving group in the absence of the enzymegeneral acid. Neither substrate should be able torescue activity of the nucleophile-deficient enzyme.To test this possibility, we incubated wild-type andhistidine to alanine mutant enzymes (H263A,H493A) with 50-end labeled 4-nitro and 4-methyloligonucleotides under single turnover conditions.Because the results described above indicate thatenzyme substrate association is rate-limitingunder multiple turnover conditions, a traceamount of substrate (,1 nM) was incubated witha large excess of enzyme (133 nM) so that theresulting extent of cleavage would reflect the rateat which the enzyme–substrate complex is con-verted to product. The results in Figure 4a (lanes 3and 4) show that wild-type hTdp1 is able to cleaveboth substrates. As predicted, H263A is inactive,whereas H493A hydrolyzes 4-nitro DNA but isunable to hydrolyze the 4-methyl analog.
Because the 4-nitro substrate specificallyrescues mutant enzymes that have defectivegeneral acids, this assay can be used to identifyadditional active-site residues that participatedirectly or indirectly in protonating the tyrosineleaving group. In addition to H263 and H493,previous studies12,16,24 have implicated at least sixconserved residues that participate in catalysis:K265, N283, Q294, K495, N516 and E538. To testthe role of these active-site residues, individualalanine-substituted mutants were constructed,purified and incubated with 4-nitro and 4-methylcontaining oligonucleotides. The results inFigure 4a and b show that similar to the nucleo-
Figure 3. Kinetic analysis. a, Reaction velocities weredetermined as described in Materials and Methods, andare graphed as a function of substrate concentration.The inset shows an autoradiogram of a typical cleavagereaction used to calculate a reaction velocity; reactionproducts before addition of hTdp1 (lane 1), and one,two, three, four, five and six minutes after the additionof enzyme (lanes 2–7) are shown. The data were fit tothe Michaelis equation (see Materials and Methods),and the resulting kcat and KM values are shown in Table1. b, The 50-end 32P2 labeled oligonucleotide substrates(0.5 nM) were incubated in the presence of hTdp1(0.01 nM) and a large molar excess of unlabeled 30-tyro-syl phosphate ester oligonucleotide (500 nM). The frac-tion of substrate converted to 30-phosphate DNA(reaction product) was determined by phosphorimageranalysis. The concentration (nM) of 30-phosphate DNAformed as a function of time (minutes) from three differ-ent experiments is plotted, and reaction velocities werecalculated from the slope of the resulting line. The reac-tion velocities for conversion of 30-(4-nitro)phenyl, 30-(4-methyl)phenyl and 30-tyrosyl phosphate ester substratesto 30-phosphate DNA were 0.0086241, 0.0071649, and0.0060103 nM/minute, respectively.
Table 1. Summary of hTdp1 kinetic analysis
Substrate kcat (min21) KM (nM) kcat=KM (M21 s21)
4-Nitro 2800 ^ 230 470 ^ 50 1 £ 108
4-Methyl 2400 ^ 660 450 ^ 10 9 £ 107
Tyrosyl 1800 ^ 850 370 ^ 120 8 £ 107
Kinetic values were extracted from the above graphed data.Human Tdp1 displays nanomolar affinity for all substratesused in this experiment, which are rapidly cleaved. For eachsubstrate, the KM varies with the turnover number resulting inidentical apparent second-order rate constants(,1 £ 108 M21 s21).
898 Functional Analysis of Tdp1 Catalytic Residues
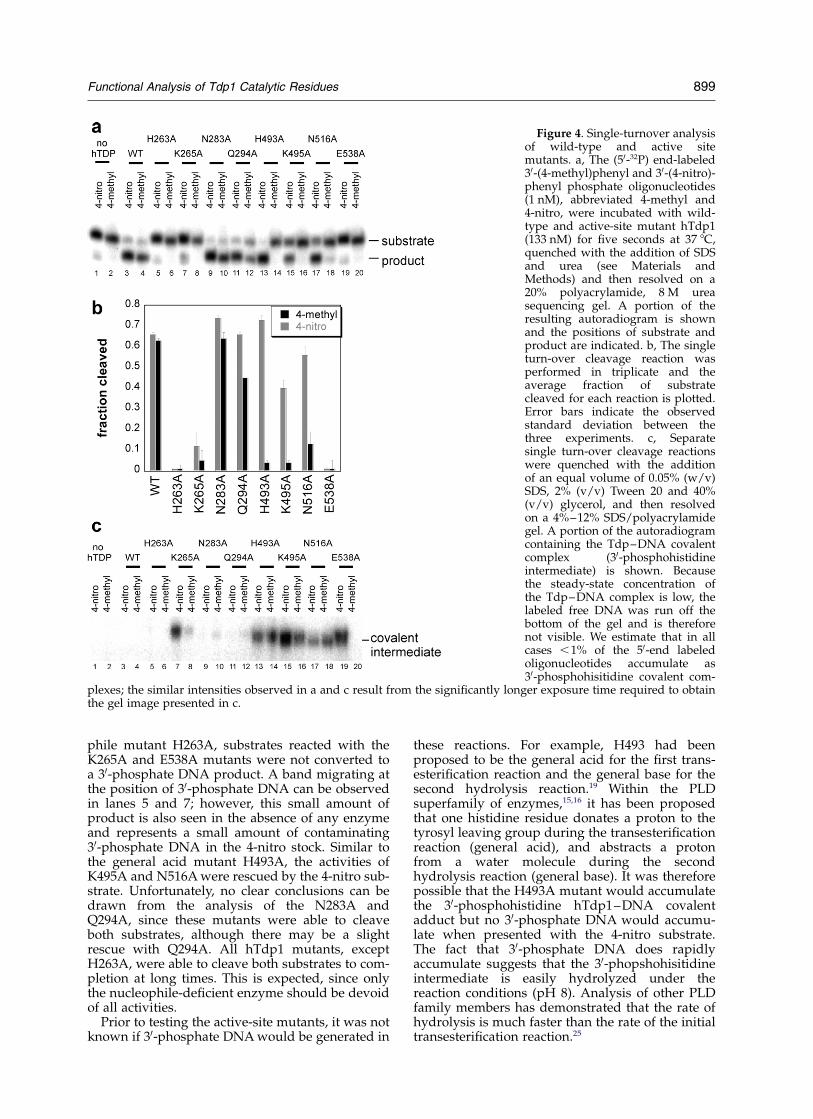
phile mutant H263A, substrates reacted with theK265A and E538A mutants were not converted toa 30-phosphate DNA product. A band migrating atthe position of 30-phosphate DNA can be observedin lanes 5 and 7; however, this small amount ofproduct is also seen in the absence of any enzymeand represents a small amount of contaminating30-phosphate DNA in the 4-nitro stock. Similar tothe general acid mutant H493A, the activities ofK495A and N516A were rescued by the 4-nitro sub-strate. Unfortunately, no clear conclusions can bedrawn from the analysis of the N283A andQ294A, since these mutants were able to cleaveboth substrates, although there may be a slightrescue with Q294A. All hTdp1 mutants, exceptH263A, were able to cleave both substrates to com-pletion at long times. This is expected, since onlythe nucleophile-deficient enzyme should be devoidof all activities.
Prior to testing the active-site mutants, it was notknown if 30-phosphate DNA would be generated in
these reactions. For example, H493 had beenproposed to be the general acid for the first trans-esterification reaction and the general base for thesecond hydrolysis reaction.19 Within the PLDsuperfamily of enzymes,15,16 it has been proposedthat one histidine residue donates a proton to thetyrosyl leaving group during the transesterificationreaction (general acid), and abstracts a protonfrom a water molecule during the secondhydrolysis reaction (general base). It was thereforepossible that the H493A mutant would accumulatethe 30-phosphohistidine hTdp1–DNA covalentadduct but no 30-phosphate DNA would accumu-late when presented with the 4-nitro substrate.The fact that 30-phosphate DNA does rapidlyaccumulate suggests that the 30-phopshohisitidineintermediate is easily hydrolyzed under thereaction conditions (pH 8). Analysis of other PLDfamily members has demonstrated that the rate ofhydrolysis is much faster than the rate of the initialtransesterification reaction.25
Figure 4. Single-turnover analysisof wild-type and active sitemutants. a, The (50-32P) end-labeled30-(4-methyl)phenyl and 30-(4-nitro)-phenyl phosphate oligonucleotides(1 nM), abbreviated 4-methyl and4-nitro, were incubated with wild-type and active-site mutant hTdp1(133 nM) for five seconds at 37 8C,quenched with the addition of SDSand urea (see Materials andMethods) and then resolved on a20% polyacrylamide, 8 M ureasequencing gel. A portion of theresulting autoradiogram is shownand the positions of substrate andproduct are indicated. b, The singleturn-over cleavage reaction wasperformed in triplicate and theaverage fraction of substratecleaved for each reaction is plotted.Error bars indicate the observedstandard deviation between thethree experiments. c, Separatesingle turn-over cleavage reactionswere quenched with the additionof an equal volume of 0.05% (w/v)SDS, 2% (v/v) Tween 20 and 40%(v/v) glycerol, and then resolvedon a 4%–12% SDS/polyacrylamidegel. A portion of the autoradiogramcontaining the Tdp–DNA covalentcomplex (30-phosphohistidineintermediate) is shown. Becausethe steady-state concentration ofthe Tdp–DNA complex is low, thelabeled free DNA was run off thebottom of the gel and is thereforenot visible. We estimate that in allcases ,1% of the 50-end labeledoligonucleotides accumulate as30-phosphohisitidine covalent com-
plexes; the similar intensities observed in a and c result from the significantly longer exposure time required to obtainthe gel image presented in c.
Functional Analysis of Tdp1 Catalytic Residues 899

However, to see if any of the active-site mutantswere partially defective for the second hydrolysisstep, the same single-turnover reactions wererepeated and the products resolved by SDS-PAGE.Because the oligonucleotide substrates were 50-endlabeled, the 30-phosphohistidine hTdp1–DNAintermediate should be detectable as a 32P-labeledprotein product. The autoradiogram of theresulting gel shows that the three residues thatwere implicated to be involved in protonating thetyrosine leaving group (H493A, K495A andN516A) also resulted in a steady-state accumu-lation of the 30-phoshophohistidine covalentadduct (Figure 4c). This result is consistent withthese residues being involved in the general basereaction for the second hydrolysis reaction. Phos-phorimager analysis of the SDS gel (Figure 4c)and the denaturing DNA gel (Figure 4a) showedthat only a very small fraction (approximately 1%)of the total DNA is present within the covalentcomplex band. Surprisingly, K265A and E538Amutants also resulted in an accumulation of theTpd1–DNA intermediate, but only whenincubated with the 4-nitro substrate. It is importantto emphasize that this assay alone cannot be usedto directly address the importance of activeresidues during hydrolysis, since the observedsteady-state accumulation depends upon both therate of transesterification (formation of inter-mediate) and hydrolysis (destruction of inter-mediate to form 30-phosphate DNA). We arecurrently developing assays to isolate the hydroly-sis reaction in the absence of the initial trans-esterification reaction to determine the roles ofactive residues during Tdp1-catalyzed hydrolysis.
Crystal structures of hTdp1 have been describedin the presence of a transition-state mimic.18,19
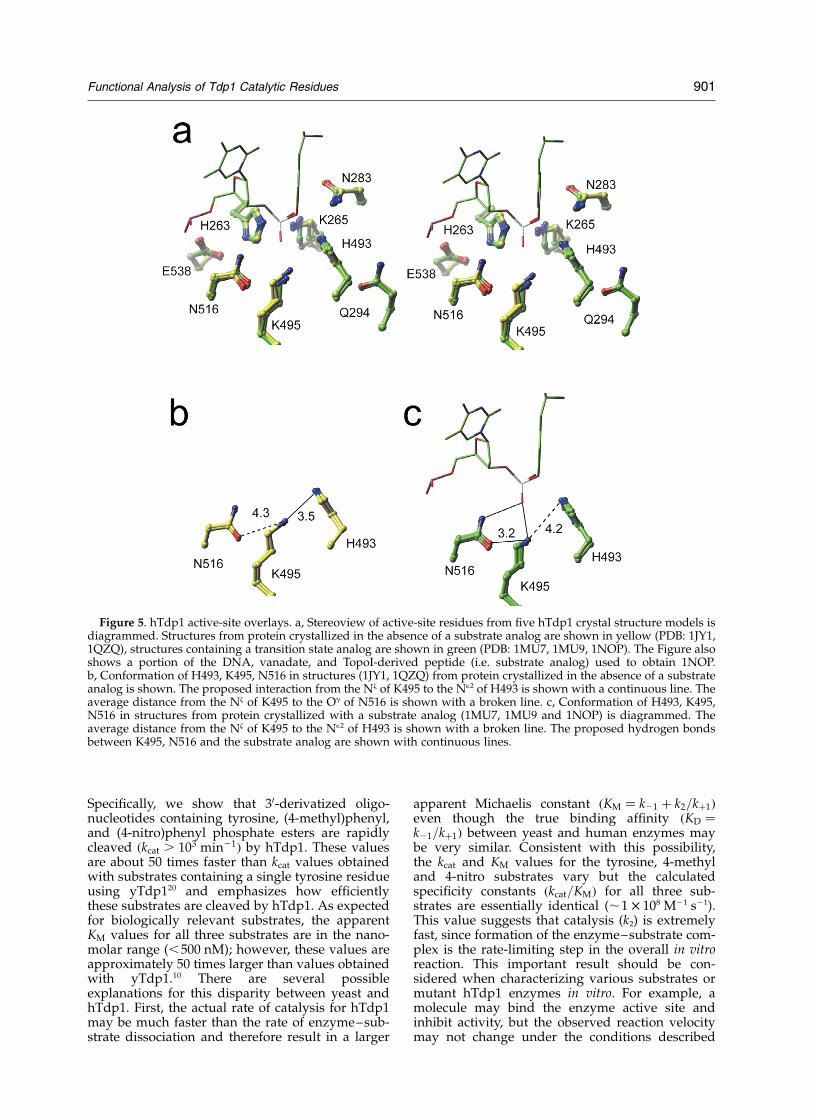
These structures have suggested that K495 andN516 are important for substrate binding and tran-sition-state stabilization;18 however, our resultssuggest that K495 and N516 participate in thegeneral reaction (i.e. protonating the tyrosineleaving group). The structural and functional datacan be reconciled by proposing that these residueshave multiple roles before and after substrate bind-ing. To examine this possibility, we solved anadditional X-ray crystal structure of hTdp1(Table 2) in the absence of a transition-state mimic.The active site of this new crystal structure is over-laid with the four other published hTdp1 struc-tures in Figure 5a. It is important to note that themajority of the active-site residues overlay remark-ably well from all five models; however, K265 andK495 are observed in two distinct conformations,depending upon the presence or absence of a sub-strate analog. As expected, the crystal structureshows that K495 and N516 are in position to inter-act with each other and could participate in thegeneral acid reaction during the first trans-esterification reaction. In two structures(Figure 5b), obtained in the absence of a tran-sition-state mimic, the Nz of K495 is 3.5 A from theN12 of H493 (3.51 A and 3.39 A in two models)
and 4.3 A from the Od1 of N516 (4.27 A and4.29 A). Whereas in three structures obtained inthe presence of different transition-state mimics(Figure 5c), the Nz of K495 is 4.2 A from the N12 ofH493 (4.25, 4.32 and 3.89 A) and 3.2 A from theOd1 of N516 (3.20, 3.12 and 3.13 A).
Discussion
yTdp1 has been shown to be involved in therepair of topoisomerase I DNA lesions in vivo9 andpresumably hydrolyzes the topoisomerase fromthe 30-end of the DNA during double-strand breakrepair.26,27 Human Tdp1 (hTdp1) is also believedto be involved in the repair of topoisomerase I-induced lesions and may function during single-strand break repair.28,29 For both hTdp1 andyTdp1, it is presumed that the topoisomerase isfirst proteolyzed before being acted upon byTdp1. Only substrates containing one to fouramino acid residues are efficient yTdp1 substratesin vitro,10,20,26 and native topoisomerase I linked tothe 30-end of oligonucleotide DNA is a very poorsubstrate for hTdp1, but a trypsin-resistantremnant peptide can be hydrolyzed.12 It is import-ant to note that Tdp1 may have other roles in thecell. For example, it has been shown that humanand yTdp1 are able to cleave double-strand 30-phosphoglycolate DNA to 30-phosphate DNAproducts;30 however, this in vitro activity is rela-tively inefficient and no genetic data is availableto address the importance of this activity in vivo.
In order to better understand the in vitro sub-strate requirements of hTdp1, we have performeda kinetic analysis of hTdp1 using oligonucleotidesubstrates containing a single tyrosine residue.
Table 2. hTdp1 crystallographic refinement statistics
Resolution (A) 50.0–2.4 (2.55–2.4)No. of reflections 40,794 (6326)Rsym
a 9.7 (40.3)Completeness (%) 99.4 (99.8)Redundancy 6.1 (5.8)I/sI 18.5 (3.6)Space group P212121
Unit cell dimensionsa (A) 50.0b (A) 105.1c (A) 194.1
Reflections used in Rfree (%) 2067 (5)Molecules per asymmetric unit 2No. of solvent atoms 243R-factor 18.5 (22.6)Rfree 23.4 (30.5)r.m.s. deviations from ideal stereochemistryBond lengths (A) 0.009Bond angles (deg.) 1.5Impropers (deg.) 0.92Dihedral angles (deg.) 24.6
Mean B-factor for all atoms (A3) 30.8
Numbers in parentheses represent the final shell of data.a Rsym ¼
PlIi 2 Iml
PIm where Ii is the intensity of the
measured reflection and Im is the mean intensity of all sym-metry-related reflections.
900 Functional Analysis of Tdp1 Catalytic Residues
Specifically, we show that 30-derivatized oligo-nucleotides containing tyrosine, (4-methyl)phenyl,and (4-nitro)phenyl phosphate esters are rapidlycleaved ðkcat . 103 min21Þ by hTdp1. These valuesare about 50 times faster than kcat values obtainedwith substrates containing a single tyrosine residueusing yTdp120 and emphasizes how efficientlythese substrates are cleaved by hTdp1. As expectedfor biologically relevant substrates, the apparentKM values for all three substrates are in the nano-molar range (,500 nM); however, these values areapproximately 50 times larger than values obtainedwith yTdp1.10 There are several possibleexplanations for this disparity between yeast andhTdp1. First, the actual rate of catalysis for hTdp1may be much faster than the rate of enzyme–sub-strate dissociation and therefore result in a larger
apparent Michaelis constant ðKM ¼ k21 þ k2=kþ1Þeven though the true binding affinity ðKD ¼k21=kþ1Þ between yeast and human enzymes maybe very similar. Consistent with this possibility,the kcat and KM values for the tyrosine, 4-methyland 4-nitro substrates vary but the calculatedspecificity constants ðkcat=KMÞ for all three sub-strates are essentially identical (,1 £ 108 M21 s21).This value suggests that catalysis (k2) is extremelyfast, since formation of the enzyme–substrate com-plex is the rate-limiting step in the overall in vitroreaction. This important result should be con-sidered when characterizing various substrates ormutant hTdp1 enzymes in vitro. For example, amolecule may bind the enzyme active site andinhibit activity, but the observed reaction velocitymay not change under the conditions described
Figure 5. hTdp1 active-site overlays. a, Stereoview of active-site residues from five hTdp1 crystal structure models isdiagrammed. Structures from protein crystallized in the absence of a substrate analog are shown in yellow (PDB: 1JY1,1QZQ), structures containing a transition state analog are shown in green (PDB: 1MU7, 1MU9, 1NOP). The Figure alsoshows a portion of the DNA, vanadate, and TopoI-derived peptide (i.e. substrate analog) used to obtain 1NOP.b, Conformation of H493, K495, N516 in structures (1JY1, 1QZQ) from protein crystallized in the absence of a substrateanalog is shown. The proposed interaction from the Nz of K495 to the N12 of H493 is shown with a continuous line. Theaverage distance from the Nz of K495 to the Og of N516 is shown with a broken line. c, Conformation of H493, K495,N516 in structures from protein crystallized with a substrate analog (1MU7, 1MU9 and 1NOP) is diagrammed. Theaverage distance from the Nz of K495 to the N12 of H493 is shown with a broken line. The proposed hydrogen bondsbetween K495, N516 and the substrate analog are shown with continuous lines.
Functional Analysis of Tdp1 Catalytic Residues 901

because the enzyme–substrate complex formation,rather than enzymatic chemistry, is limiting.Another explanation for the discrepancy betweenyeast and hTdp1 is that hTdp1 is one componentof a multi-enzyme complex and additional proteinsmay be involved in recognizing 30-phoshotyrosylDNA in vivo; hTdp1 has been shown to associatewith XRCC1.31 The kinetic data cannot directlyaddress this possibility; however, the results doshow that additional proteins are not required forefficient hTdp1-mediated cleavage in vitro.
As mentioned above, the precise in vivo substrateof hTdp1 is not known. The substrate-bindingpocket of hTdp1 can accommodate a fairly largetopoisomerase I peptide fragment,18 and hTdp1can cleave a 12 amino acid residue trypsin-resistantfragment of topoisomerase I linked to the 30-end ofDNA.12 It is therefore attractive to speculate thathTdp1 makes additional contacts to the topo-isomerase I-derived peptide that are necessary forrecognition or cleavage by hTdp1. However, theonly feature that is shared by the three efficientlycleaved substrates in this study is a single phenylring, demonstrating that a single phenyl ring issufficient for Tdp1-mediated recognition andcleavage in vitro. Although a detailed comparisonof these substrates with larger substrates isnecessary to draw any conclusions, the efficiencywith which the 4-methyl and 4-nitro substrates arecleaved by hTdp1 in vitro raises the possibilitythat the topoisomerase I is degraded to a singletyrosine residue before cleavage by hTdp1 in vivo.Another possibility that could be entertained isthat hTdp1 may have a general role in the repairof many different small residues linked to the30-end of DNA. For example, it has been shownthat yTdp1 can cleave 30-phosphoglycolate linkagesin vitro.30
Previous crystal structure models have shownthat histidine 263 (H263) is positioned for in-lineattack of the 30-phosphotyrosyl bond and histidine493 (H493) is appropriately positioned to protonatethe tyrosine leaving group.18,19 It was thereforeproposed that H263 is the nucleophile and H493was the general acid; however, structural dataalone cannot assign these roles, since the modelsrepresent the most stable conformation in thecrystallization condition and may not be thecatalytically active conformation. We have shownthat when histidine 263 is mutated to alanine(H263A), the enzyme is catalytically inactive onany substrate,18,19 whereas the H493A mutant canconvert 4-nitro DNA to 30-phosphate product, butthe 4-methyl DNA remains intact. While only the4-nitro DNA was converted to product, the H493Amutant formed covalent intermediate with bothsubstrates. The H493A covalent intermediateformed with both substrates is the result of thetransesterification step in the reaction, which pro-ceeds prior to the general acid activity required inthe hydrolysis step. H493A is able to convert the4-nitro DNA to product because the lower pKa ofthe phenoxide oxygen atom relieves the mutant
enzyme of the responsibility for protonating thesubstrate leaving group and the reaction proceedseven in the absence of a general acid. The functionaldata are therefore consistent with H263 serving asthe nucleophile and demonstrates that H493 is thegeneral acid. More importantly, the data provide thefirst direct evidence that members of the phosho-lipase D (PLD) superfamily of enzymes use a generalacid/base catalytic mechanism.
These results also allowed us to identifyadditional active-site residues that are involved inprotonating the tyrosine leaving group. Weexamined six conserved residues that have beenproposed to participate in catalysis: K265, N283,Q294, K495, N516, and E413. No conclusions canbe drawn from the analysis of K265A and E538Abecause both mutants failed to cleave the 4-methylor the 4-nitro substrates. This is expected, since ithas been proposed that E538 coordinates the H263(nucleophile), and K265 coordinates the non-bridging oxygen atom of the phosphotyrosylbond.18,19 In addition, no conclusions can bedrawn from the analysis of N283A and Q294A,because these mutants were able to cleave bothsubstrates during the course of the reaction. Struc-tural studies show that N283 is one of several resi-dues (N516, K495 and K265) that can coordinatenon-bridging oxygen atoms of the scissile phos-phodiester. It is therefore possible that these otherresidues supply enough redundant function thatstabilization by N283 is not necessary for the firsttransesterification reaction. The observation thatQ294A is able to cleave both substrates is more sur-prising, because it has been proposed that Q294coordinates the H493 general acid;18,19 if H493requires coordination, the Q294A mutant shouldcleave the 4-nitro substrate, but should not cleavethe 4-methyl substrate. Although there may be amodest reduction of activity using with the4-methyl substrate (Figure 4), the simplestexplanation of this data is that the coordination ofH493 by Q294 alone is not critical for hTdp1activity in vitro.
The most definitive conclusions come from theanalysis of K495A and N516A, since these mutantsare able to cleave the 4-nitro substrate, but areunable to cleave the 4-methyl substrate. Theseresults demonstrate that K495 and N516 aredirectly or indirectly involved in protonating the50-leaving group during the wild-type reaction. Anoverlay of five different crystal structures ofhTdp1 shows that K495 lies adjacent to both N516and the general acid H493. In two structures,obtained in the absence of a transition state mimic,the Nz of K495 is close to the N12 of H493 (3.5 A)and is not in position to make a hydrogen bondcontact to the Od1 of N516. Whereas in three struc-tures obtained in the presence of different tran-sition-state mimics, the Nz of K495 has movedaway from the N12 of H493 and is now in positionto make a hydrogen bond contact to the Od1 ofN516 (3.2 A). It is important to note that althoughthe two conformations of K495 only differ by
902 Functional Analysis of Tdp1 Catalytic Residues

1–2 A and the resolution of five crystal structuresvaries between 2.0 A and 2.4 A, the five structureswere refined independently and represent threedifferent crystal forms obtained in two differentlaboratories.
One mechanism that explains the functional andstructural data is that K495 participates in a“proton relay” and transfers a proton to thegeneral acid H493 prior to substrate binding.Upon substrate binding, the lysine residue movesaway from the general acid and makes a hydrogenbond contact to the Od1 of N516. According to thismodel, K495 participates directly in the generalacid reaction by protonating H493, which in turntransfers a proton to the tyrosine leaving group.N516 participates indirectly in the general acidreaction by contacting and stabilizing K495; thehydrogen bond contact between N516 and K495made upon substrate binding, and presumably thehydrogen bond contacts to the scissile phospho-diester, allows H493 to protonate the tyrosineleaving group.
Another possible explanation for the roles ofK495 and N516 is that K495 is sterically importantfor positioning the H493 general acid. When K495is mutated, or when K495 cannot be appropriatelypositioned through a hydrogen bond contact toN516, H493 is destabilized and cannot act as ageneral acid. We disfavor this explanation, sinceother residues that have been proposed to helpposition the general acid do not significantlyinhibit the single turn-over reaction; for example,Q294A mutants are able to cleave both 4-methyland 4-nitro substrates (Figure 4).
K265 and K493 are the only active-site residuesthat are observed in two distinct conformations,depending upon the presence or absence of a tran-sition-state mimic, and lysine has been shown toact as a general acid during phosphoryl transferreactions catalyzed by the type 1B topoisomerase/tyrosine recombinase superfamily of enzymes.32,33
It is therefore interesting to speculate that theindividual lysine residues within each of the twoconserved HKD motifs (K265 and K493) have simi-lar roles during Tdp1 catalysis. For example, ifK493 transfers a proton to H493 during the firsttransesterification reaction as we propose above, itis possible that K265 transfers a proton to H263during the second hydrolysis reaction. In otherwords, K265 would function as the general acidfor the second hydrolysis step. This may explainwhy an increased steady-state concentration of theTdp1–DNA covalent adduct (30-phosphohistidine)was observed in the single-turnover reactionswhen K265A was used. Specifically, if K265A wasable to cleave a small portion of the more reactive4-nitro substrate, but unable to promote thegeneral acid reaction allowing hydrolysis of the30-phosphohistidine intermediate, very little30-phosphate product would be observed(Figure 4a, lane 7) and the hTdp1–DNA covalentcomplex would accumulate (Figure 4c, lane 7). IfE538 is important for coordinating H263 during
the hydrolysis reaction, then this would alsoexplain the accumulation of the 30-phospho-histidine intermediate when E538A was used inthe single turnover reactions (Figure 4a, lane 19;Figure 4c, lane 19). Clearly, developing an assay toisolate the hydrolysis reaction that does notdepend upon the initial transesterification reactionis necessary to address these possibilities.
In conclusion, we have shown that single-strandDNA oligonucleotide substrates containing asingle 30-phospho-phenyl ring are efficient sub-strates for hTdp1 in vitro, and the rate of sub-strate–enzyme association, rather than catalysis, israte limiting. The efficiency of these substratessuggests that a single phenyl ring is sufficient forcatalysis and potential contacts between hTdp1and topoisomerase I peptide fragments are notnecessary for efficient cleavage. The ability ofH493A to efficiently cleave 4-nitro substrates pro-vides evidence that hTdp1 uses a general acid/base catalytic mechanism. Because all PLD familyshare similar structures,23,34,35 it suggests that allPLD members share this same general acid/basecatalytic mechanism. Finally, the ability of the4-nitro substrate to relieve the need for additionalactive site residues (K495 and N516) shows thatthese residues participate directly or indirectly inprotonating the tyrosine leaving group. On thebasis of the functional and structural data, we pro-pose that N516 participates in the general acidreaction by coordinating K495, and that K495transfers a proton to H493, which in turn transfersa proton to the tyrosine leaving group.
Materials and Methods
Synthesis of 30-derivatized oligonucleotidesubstrates
The 30-(4-nitro)phenyl phosphate oligonucleotideswere prepared as described.36 The 30-phosphotyrosineoligonucleotides were purchased from Midland CertifiedReagent Company (Midland, TX). The 30-(4-methyl)-phenyl oligonucleotides were prepared by post-syntheticcondensation. Oligonucleotides (6 £ 1 mmol) were syn-thesized using 30-PO4 CPG (Glen Research, Sterling, VA)and standard DNA phosphoramidites on an ABI 392automated DNA synthesizer. The resin was incubatedin concentrated ammonium hydroxide at 65 8C for 12hours, and the 50-OH containing DNA was ethanol-precipitated. The resulting DNA pellet was resuspendedin 1 ml of 2 mM MgCl2, 100 mM Mes (pH 5.5), andaliquots of 1 ml 99% para-cresol (Aldrich, Milwaukee,WI) and 0.048 g of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC) (Aldrich) wereadded sequentially. The resulting two immiscible layerswere mixed vigorously at 65 8C to form an emulsion.After 20–24 hours, the aqueous layer was extractedthree times with 2 ml each of ethyl acetate and the DNAwas recovered from the aqueous phase by ethanol-precipitation. This EDC-derivatization protocol wasrepeated a total of three times. Typically, ,5–10%of the 30-PO4 (starting material) was converted to30-cresol DNA, which was purified by anion-exchange
Functional Analysis of Tdp1 Catalytic Residues 903

chromatography on a Hitachi analytical HPLC instru-ment using a DNAPac PA-100 4 mm £ 250 mm column(Dionex, Sunnyvale, CA) at 1 ml/minute, 5% to 60%(w/v) 2 M sodium chloride gradient (buffered with20 mM sodium phosphate, pH 7.0) over 15 minutes.Peak fractions (typically 0.2 minute longer retentiontime than underivatized DNA) were pooled, precipi-tated, resuspended in water and stored at 220 8C. Thesequence of all oligonucleotides was 50-CGTTGAAGCCTGCTTTY, where Y is tyrosine or the tyrosine analog.
Preparation of hTdp1
The hTdp1 D 1-148 cDNA was PCR-amplified fromIMAGE clones 3900062 and 740522 (ResGen InvitrogenCorp. Carlsbad CA). The cDNA was amplified in twohalves: DNA encoding G149 through L379 was amplifiedfrom IMAGE clone 3900062, and DNA encoding K380through S608 was amplified from IMAGE clone 740522.These two halves were ligated via a naturally occurringHindIII site embedded in the sequence coding for L379and K380. The full-length gene and the N-terminaldeletion (D 1-148) were cloned into a modified pBAD(Invitrogen) expression vector. The final expressed pro-tein contains an N-terminal 6 £ His tag and a sevenamino acid epitope tag (EEYMPTE). Mutant and wild-type hTdp1 D 1-148 constructs were sequence verifiedand transfected into TOP10 cells (Invitrogen), andgrown at 30 8C. Protein expression was induced by add-ing arabinose to 0.2% (w/v) at mid-log phase, and cellswere harvested after four hours, concentrated and thenlysed in 100 mM NaCl2, 20 mM Tris–HCl (pH 9), 2 mMBME, and 0.1 mg/ml of lysozyme by nitrogen cavitation.Lysate was clarified by centrifugation and filtration, andloaded onto HiTRAP nickel chelating columns(Amersham, Piscataway, NJ). Protein was purified at1 ml/minute in 100 mM NaCl2, 20 mM Tris–HCl (pH 8)and eluted in a gradient of 24% to 100% (w/v) 500 mMimidazole. Peak fractions were pooled and loaded ontoa 20 ml affinity column (monoclonal antibody whichrecognizes the EEYMPTE epitope tag) equilibrated in500 mM NaCl2, 20 mM Tris–HCl (pH 8). The columnwas then loaded with 20 ml of 5 mg/ml of peptide (EEYMPTE) in the same equilibration buffer and incubatedfor 30 minutes at 4 8C. hTdp1 was eluted from thecolumn with 1 mg/ml of peptide and fractions were ana-lyzed by SDS-PAGE. Pure hTdp1 was dialyzed exhaus-tively in 250 mM NaCl2, 15 mM Tris–HCl (pH 8.2),3 mM DTT at 4 8C. Following purification, all versionsof hTdp1 D 1-148 migrated as a single band of the pre-dicted molecular mass on SDS-PAGE, and the identityof wild-type and mutant hTdp1 was confirmed byMALDI TOF mass spectroscopy. All residue numberingcorresponds to full-length protein.
Kinetic determinations
hTdp1 activity assays were performed in 50 mM Tris–HCl (pH 8.0), 5 mM MgCl2, 80 mM KCl, 2 mM EDTA,1 mM DTT, and 40 mg/ml of BSA. Enzyme stocks werediluted in 50 mM Tris–HCl (pH 8.0), 100 mM NaCl,5 mM DTT, 10% (v/v) glycerol, 500 mg/ml of BSA anddiluted tenfold for each reaction. Cleavage substrateswere 50-radiolabeled with [a-32P]ATP (Perkin Elmer,Boston, MA) using phage T4 polynucleotide kinase(New England Biolabs, Beverly, MA) at 37 8C for15 minutes, followed by ten minutes at 90 8C to denaturethe kinase.
Unless otherwise noted, all reactions were performedat 37 8C in 96 well V-bottom reaction plates andquenched by the addition of an equal volume of 8 Murea, 0.05% (w/v) SDS, 30% glycerol, 0.25% (v/v)bromophenol blue, and resolved on 20% (w/v) poly-acrylamide sequencing gels. The concentration of 30-phosphate DNA was determined by measuring the frac-tion of substrate converted to product by densitometryanalysis of the gel image. Initial velocities were deter-mined by plotting the concentration of 30-phosphateDNA as a function of time; only concentrations repre-senting less than 20% of initial substrate concentrationswere used, all lines extrapolated to zero product at thestart of the reaction, and at least five time-points wereused to determine the slope of the line (velocity).Finally, all velocity measurements were performed intriplicate on three different days. Apparent KM and Vmax
values were determined by fitting the initial velocityversus substrate concentration to the Michaelis equation,v ¼ ðVmax½S�Þ=ðKM þ ½S�Þ: Very similar values wereobtained from linear regression analysis of Eadie–Hofstee plots of the same data (data not shown).
hTdp1 crystallization
Pure protein was concentrated to 6.5 mg/ml andscreened at 4 8C against 10–20% (w/v) PEG 8000,100 mM Ches (pH 9.4–10), 8 mM spermine. Crystalswere observed in 16%, 18% and 20% PEG 8000 for allChes pH ranges within one week, with fully formedcrystals observed after one month. Crystals were cryo-protected by soaking in 30% glycerol, 20% PEG 8000,100 mM Ches (pH 10), 8 mM spermine for 30 secondsand flash-cooled in liquid nitrogen prior to diffraction.Data were collected at 100 K at COM-CAT, Sector 32,Advanced Photon Source, Argonne National Laboratory.The structure was solved by molecular replacementwith AmoRe37 by using the published D1-148 hTdp1structure (PDB: 1JY1).23 The structure was rebuilt andrefined by CNX with simulated annealing38 and iterativemodel adjustments by XTALVIEW (see Table 2).39
Protein Data Bank accession numbers
Coordinates have been deposited in the Protein DataBank with accession number 1QZQ.
Acknowledgements
We thank Zachary Halloran for technicalassistance in growing Tdp1-expressing cells andS. Lovell at COM-CAT (Sector 32) AdvancedPhoton Source, Argonne National Laboratory forassistance in data collection. We thank Anca Segall,Lance Stewart and Mark Gurney for criticalreading of the manuscript. ACR was supportedby Predoctoral Fellowship 1F31 GM66372-01 fromthe National Institutes of Health. This researchwas funded in part by the Cooperative Planningof the SDSU/UCSD Cancer Partnership (grantCCA92079-01A2) to ACR.
904 Functional Analysis of Tdp1 Catalytic Residues

References
1. Wang, J. C. (2002). Cellular roles of DNA topo-isomerases: a molecular perspective. Nature Rev.Mol. Cell. Biol. 3, 430–440.
2. Champoux, J. J. (2001). DNA topoisomerases: struc-ture, function, and mechanism. Annu. Rev. Biochem.70, 369–413.
3. Pourquier, P., Pilon, A. A., Kohlhagen, G.,Mazumder, A., Sharma, A. & Pommier, Y. (1997).Trapping of mammalian topoisomerase I andrecombinations induced by damaged DNA contain-ing nicks or gaps. Importance of DNA end phos-phorylation and camptothecin effects. J. Biol. Chem.272, 26441–26447.
4. Pourquier, P., Ueng, L. M., Kohlhagen, G.,Mazumder, A., Gupta, M., Kohn, K. W. & Pommier,Y. (1997). Effects of uracil incorporation, DNA mis-matches, and abasic sites on cleavage and religationactivities of mammalian topoisomerase I. J. Biol.Chem. 272, 7792–7796.
5. Lesher, D. T., Pommier, Y., Stewart, L. & Redinbo,M. R. (2002). 8-Oxoguanine rearranges the activesite of human topoisomerase I. Proc. Natl Acad. Sci.USA, 99, 12102–12107.
6. Chrencik, J. E., Burgin, A. B., Pommier, Y., Stewart, L.& Redinbo, M. R. (2003). Structural impact of theleukemia drug 1-beta-D-arabinofuranosylcytosine(Ara-C) on the covalent human topoisomeraseI-DNA complex. J. Biol. Chem. 278, 12461–12466.
7. Meng, L. H., Liao, Z. Y. & Pommier, Y. (2003). Non-camptothecin DNA topoisomerase I inhibitors incancer therapy. Curr. Top. Med. Chem. 3, 305–320.
8. Pizzolato, J. F. & Saltz, L. B. (2003). The campto-thecins. Lancet, 361, 2235–2242.
9. Pouliot, J. J., Yao, K. C., Robertson, C. A. & Nash,H. A. (1999). Yeast gene for a Tyr-DNA phospho-diesterase that repairs topoisomerase I complexes.Science, 286, 552–555.
10. Yang, S. W., Burgin, A. B., Jr, Huizenga, B. N.,Robertson, C. A., Yao, K. C. & Nash, H. A. (1996).A eukaryotic enzyme that can disjoin dead-endcovalent complexes between DNA and type Itopoisomerases. Proc. Natl Acad. Sci. USA, 93,11534–11539.
11. Connelly, J. C. & Leach, D. R. (2004). Repair of DNAcovalently linked to protein. Mol. Cell, 13, 307–316.
12. Interthal, H., Pouliot, J. J. & Champoux, J. J. (2001).The tyrosyl-DNA phosphodiesterase Tdp1 is a mem-ber of the phospholipase D superfamily. Proc. NatlAcad. Sci. USA, 98, 12009–12014.
13. Koonin, E. V. (1996). A duplicated catalytic motif in anew superfamily of phosphohydrolases and phos-pholipid synthases that includes poxvirus envelopeproteins. Trends Biochem. Sci. 21, 242–243.
14. Ponting, C. P. & Kerr, I. D. (1996). A novel family ofphospholipase D homologues that includes phos-pholipid synthases and putative endonucleases:identification of duplicated repeats and potentialactive site residues. Protein Sci. 5, 914–922.
15. Waite, M. (1999). The PLD superfamily: insights intocatalysis. Biochim. Biophys. Acta, 1439, 187–197.
16. Gottlin, E. B., Rudolph, A. E., Zhao, Y., Matthews,H. R. & Dixon, J. E. (1998). Catalytic mechanism ofthe phospholipase D superfamily proceeds via acovalent phosphohistidine intermediate. Proc. NatlAcad. Sci. USA, 95, 9202–9207.
17. Iwasaki, Y., Horiike, S., Matsushima, K. & Yamane, T.(1999). Location of the catalytic nucleophile of
phospholipase D of Streptomyces antibioticus in theC-terminal half domain. Eur. J. Biochem. 264,577–581.
18. Davies, D. R., Interthal, H., Champoux, J. J. & Hol,W. G. (2003). Crystal structure of a transition statemimic for tdp1 assembled from vanadate, DNA,and a topoisomerase I-derived peptide. Chem. Biol.10, 139–147.
19. Davies, D. R., Interthal, H., Champoux, J. J. & Hol,W. G. (2002). Insights into substrate binding andcatalytic mechanism of human tyrosyl-DNA phos-phodiesterase (Tdp1) from vanadate and tungstate-inhibited structures. J. Mol. Biol. 324, 917–932.
20. Debethune, L., Kohlhagen, G., Grandas, A. &Pommier, Y. (2002). Processing of nucleopeptidesmimicking the topoisomerase I-DNA covalentcomplex by tyrosyl-DNA phosphodiesterase. Nucl.Acids Res. 30, 1198–1204.
21. Cheng, T. J., Rey, P. G., Poon, T. & Kan, C. C. (2002).Kinetic studies of human tyrosyl-DNA phospho-diesterase, an enzyme in the topoisomerase I DNArepair pathway. Eur. J. Biochem. 269, 3697–3704.
22. Redinbo, M. R., Champoux, J. J. & Hol, W. G. (2000).Novel insights into catalytic mechanism from acrystal structure of human topoisomerase I incomplex with DNA. Biochemistry, 39, 6832–6840.
23. Davies, D. R., Interthal, H., Champoux, J. J. & Hol,W. G. (2002). The crystal structure of human tyrosyl-DNA phosphodiesterase, Tdp1. Structure (Camb), 10,237–248.
24. Sung, T. C., Roper, R. L., Zhang, Y., Rudge, S. A.,Temel, R., Hammond, S. M. et al. (1997). Mutagenesisof phospholipase D defines a superfamily includinga trans-Golgi viral protein required for poxviruspathogenicity. EMBO J. 16, 4519–4530.
25. Yang, H. & Roberts, M. F. (2003). Phosphohydrolaseand transphosphatidylation reactions of twoStreptomyces phospholipase D enzymes: covalentversus noncovalent catalysis. Protein Sci. 12,2087–2098.
26. Pouliot, J. J., Robertson, C. A. & Nash, H. A. (2001).Pathways for repair of topoisomerase I covalentcomplexes in Saccharomyces cerevisiae. Genes Cells, 6,677–687.
27. Vance, J. R. & Wilson, T. E. (2002). Yeast Tdp1 andRad1-Rad10 function as redundant pathways forrepairing Top1 replicative damage. Proc. Natl Acad.Sci. USA, 99, 13669–13674.
28. Caldecott, K. W. (2003). DNA single-strand breakrepair and spinocerebellar ataxia. Cell, 112, 7–10.
29. Takashima, H., Boerkoel, C. F., John, J., Saifi, G. M.,Salih, M. A., Armstrong, D. et al. (2002). Mutation ofTDP1, encoding a topoisomerase I-dependent DNAdamage repair enzyme, in spinocerebellar ataxiawith axonal neuropathy. Nature Genet. 32, 267–272.
30. Inamdar, K. V., Pouliot, J. J., Zhou, T., Lees-Miller,S. P., Rasouli-Nia, A. & Povirk, L. F. (2002). Conver-sion of phosphoglycolate to phosphate termini on 30
overhangs of DNA double strand breaks by thehuman tyrosyl-DNA phosphodiesterase hTdp1.J. Biol. Chem. 277, 27162–27168.
31. Plo, I., Liao, Z. Y., Barcelo, J. M., Kohlhagen, G.,Caldecott, K. W., Weinfeld, M. & Pommier, Y. (2003).Association of XRCC1 and tyrosyl DNA phospho-diesterase (Tdp1) for the repair of topoisomeraseI-mediated DNA lesions. DNA Repair (Amst), 2,1087–1100.
32. Krogh, B. O. & Shuman, S. (2000). Catalytic
Functional Analysis of Tdp1 Catalytic Residues 905

mechanism of DNA topoisomerase IB. Mol. Cell, 5,1035–1041.
33. Krogh, B. O. & Shuman, S. (2002). Proton relaymechanism of general acid catalysis by DNA topo-isomerase IB. J. Biol. Chem. 277, 5711–5714.
34. Leiros, I., Secundo, F., Zambonelli, C., Servi, S. &Hough, E. (2000). The first crystal structure of aphospholipase D. Struct. Fold. Des. 8, 655–667.
35. Stuckey, J. A. & Dixon, J. E. (1999). Crystal structureof a phospholipase D family member. Nature Struct.Biol. 6, 278–284.
36. Woodfield, G., Cheng, C., Shuman, S. & Burgin, A. B.(2000). Vaccinia topoisomerase and Cre recombinasecatalyze direct ligation of activated DNA substrates
containing a 30-para-nitrophenyl phosphate ester.Nucl. Acids Res. 28, 3323–3331.
37. Navaza, J. (2001). Implementation of molecularreplacement in AMoRe. Acta Crystallog. sect. D, 57,1367–1372.
38. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography & NMR system: a new softwaresuite for macromolecular structure determination.Acta Crystallog. sect. D, 54, 905–921.
39. McRee, D. E. (1999). XtalView/Xfit–a versatileprogram for manipulating atomic coordinates andelectron density. J. Struct. Biol. 125, 156–165.
Edited by D. E. Draper
(Received 19 January 2004; received in revised form 28 February 2004; accepted 2 March 2004)
906 Functional Analysis of Tdp1 Catalytic Residues
























![[ITDG] BIOL](https://img.pdfslide.tips/doc/110x75/5571fcdd49795991699815b6/itdg-biol.jpg)




