Embed Size (px)
Citation preview

Uniwersytet WrocławskiInstytut Genetyki i Mikrobiologii
Zakład Genetyki
Jacek Skała
ĆWICZENIA Z GENETYKI MOLEKULARNEJ
Wrocław 2008

2
Spis treściSpis treści ................................................................................................................................................2
Organizacja ćwiczeń .......................................................................................................................................4
KRYTERIA OCENY ........................................................................................................................................6L ITERATURA POMOCNICZA .........................................................................................................................6
PODSTAWOWE INFORMACJE O PLAZMIDACH I SZCZEPACH..... ................................................8
PLAZMIDY ....................................................................................................................................................8SZCZEPY .....................................................................................................................................................10
KLASYCZNA METODA IZOLACJI DNA PLAZMIDOWEGO Z ESCHERICHIA COLI ..................11
SZYBKA METODA IZOLACJA DNA PLAZMIDOWEGO Z ESCHERICHIA COLI ........................17
ULTRA SZYBKA METODA IZOLACJA DNA PLAZMIDOWEGO Z ESCHERICHIA COLI .........19
WYBRANE METODY IZOLACJI, OCZYSZCZANIA I ZAG ĘSZCZANIA KWASÓWNUKLEINOWYCH......................................................................................................................................21
IZOLACJA I OCZYSZCZANIE DNA I RNA NA KOLUMNACH ZE ZŁO śEM KRZEMIONKOWYM ..................21OCZYSZCZANIE KWASÓW NUKLEINOWYCH FENOLEM ............................................................................25ZAGĘSZCZANIE ROZTWORÓW KWASÓW NUKLEINOWYCH PRZEZ WYTR ĄCANIE ....................................28M IKRODIALIZA ..........................................................................................................................................30
SPEKTROFOTOMETRYCZNE OZNACZANIE ST ĘśENIA I CZYSTO ŚCI KWASÓWNUKLEINOWYCH......................................................................................................................................31
TRAWIENIE DNA ENZYMAMI RESTRYKCYJNYMI.............. ...........................................................32
K ILKA RAD PRAKTYCZNYCH O PRACY Z ENZYMAMI RESTRYKCYJ NYMI ................................................33K IEDY ENZYM RESTRYKCYJNY NIE CHCE CI ĄĆ........................................................................................34ENZYM RESTRYKCYJNY PRZY PRACY .......................................................................................................35
ELEKTROFOREZA DNA W śELACH AGAROZOWYCH ..................................................................38
WPŁYW ILO ŚCI DNA I CZASU TRWANIA ELEKTROFOREZY NA ROZDZIAŁ FRAGMENTÓ W FAGA λλλλ W
śELU AGAROZOWYM .................................................................................................................................41ELEKTROFOREZA DNA W AGAROZIE O NISKIM PUNKCIE TOPNIENIA ...................................................42OGÓLNE REGUŁY POSTĘPOWANIA ............................................................................................................42ROZDZIAŁ I ODZYSKANIE DNA Z AGAROZY LMP ...................................................................................43
DEFOSFORYLACJA WEKTORA.............................................................................................................46
LIGACJA.......................................................................................................................................................49
ELEKTROTRANSFORMACJA.................................................................................................................50
PRZYGOTOWANIE DNA DO ELEKTROTRANSFORMACJI ...........................................................................50PRZYGOTOWANIE ELEKTROKOMPETENTNYCH KOMÓREK ESCHERICHIA COLI .....................................51
Elektrotransformacja Escherichia coli..................................................................................................53PRZYGOTOWANIE ELEKTROKOMPETENTNYCH KOMÓREK SACCHAROMYCES CEREVISIAE....................56
Elektrotransformacja Saccharomyces cerevisiae..................................................................................58
CHEMICZNA TRANSFORMACJA SACCHAROMYCES CEREVISIAE..............................................60
PRZYGOTOWANIE KOMÓREK DRO śDśOWYCH DO TRANSFORMACJI CHEMICZNEJ ................................60TRANSFORMACJA SACCHAROMYCES CEREVISIAE.....................................................................................61
PODŁOśE SELEKCYJNE Z X-GAL I IPTG.............................................................................................62
SELEKCJA TRANSFORMANTÓW ESCHERICHIA COLI Z WEKTOREM ZAWIERAJ ĄCYMDROśDśOWY GEN HIS3 ..........................................................................................................................64

3
BEZPOŚREDNIE WYKAZANIE OBECNO ŚCI GENU HIS3 W TRANSFORMANTACHDROśDśOWYCH ........................................................................................................................................69
PCR ................................................................................................................................................................70
OGÓLNE ZASADY PROJEKTOWANIA STARTERÓW DO PCR......................................................................73Startery do wykrywania droŜdŜowego genu HIS3...............................................................................74
PREPARACJA DNA GENOMOWEGO Z DRO śDśY SACCHAROMYCES CEREVISIAE..................76
ZESTAWIENIE REAKCJI PCR Z DNA DROśDśOWYM ...............................................................................78PROGRAM PCR DO WYKRYWANIA DRO śDśOWEGO GENU HIS3 STARTERAMI HIS -P I HIS-K ....................80

4
Organizacja ćwiczeń
Grupa studencka dzieli się na dwuosobowe zespoły. KaŜdy zespół
otrzymuje dwa szczepy Escherichia coli JM109. Jeden z nich zawiera pusty
wektor wahadłowy pFL44S, natomiast w drugim znajduje się plazmid pD11
złoŜony z części bakteryjnej oraz 1764 bp fragmentu genomowego DNA
droŜdŜowego z genem HIS3 (Rys. 1). Zadaniem zespołu jest:
a. przeniesienie genu HIS3 do wektora wahadłowego,
b. transformacja nowo wytworzonym plazmidem zawierającym gen HIS3
szczepu Saccharomyces cerevisiae FY73,
c. wyselekcjonowanie transformantów droŜdŜowych zdolnych do wzrostu
na podłoŜu minimalnym bez histydyny oraz określenie wydajności trans-
formacji wytworzonym plazmidem,
d. bezpośrednie wykazanie obecności genu HIS3 w transformantach droŜ-
dŜowych metodą PCR,
e. opracowanie mapy restrykcyjnej wytworzonego plazmidu
Realizacja postawionego zadania zajmuje pięć tygodni i odbywa się
według poniŜszego planu:
I tydzień
• Izolacja plazmidów pFL44S i pD11 ze szczepów bakteryjnych.
• Trawienie plazmidów pFL44S i pD11 enzymem BamHI.
• Elektroforeza kontrolna w Ŝelu agarozowym.
II tydzień
• Defosforylacja końców rozciętego plazmidu pFL44S.

5
• Rozdział fragmentów plazmidu pD11 w agarozowym Ŝelu preparatywnym
o niskim punkcie topnienia (LMP).
• Odzyskanie z agarozy fragmentu plazmidu pD11 zawierającego gen HIS3.
• Ligacja plazmidu pFL44S (wektora) z wyizolowanym fragmentem DNA
droŜdŜowego zawierającym gen HIS3.
III tydzie ń
• Przygotowanie elektrokompetentnych komórek E. coli JM109.
• Przygotowanie elektrokompetentnych komórek S. cerevisiae FY73.
• Przygotowanie podłoŜa selekcyjnego LB z ampicyliną, X-gal i IPTG.
• Dializa roztworu ligacyjnego
• Elektrotransformacja E. coli JM109 ligowanym DNA i wysiew na podłoŜe
selekcyjne LB z ampicyliną, X-gal i IPTG.
IV tydzień
• Obserwacja wzrostu i róŜnicowania transformantów bakteryjnych na pod-
łoŜu selekcyjnym z X-gal i IPTG.
• ZałoŜenie hodowli płynnych wybranych białych kolonii transformantów.
• Preparacja DNA z hodowli wybranych transformantów bakteryjnych.
• Trawienie restrykcyjne wyizolowanego DNA plazmidowego, elektrofore-
za kontrolna oraz opracowanie mapy restrykcyjnej.
• Transformacja komórek droŜdŜowych wytworzonym plazmidem waha-
dłowym z genem HIS3.
V tydzień
• Obserwacja wzrostu transformantów droŜdŜowych na podłoŜu selekcyj-
nym bez histydyny.

6
• ZałoŜenie hodowli płynnych wybranych transformantów droŜdŜowych.
• Izolacja DNA genomowego z transformantów droŜdŜowych.
• Wykazanie obecności genu HIS3 w transformantach droŜdŜowych przy
uŜyciu techniki PCR
• Końcowe omówienie wyników i zaliczenie ćwiczeń.
Kryteria oceny
Do zaliczenia ćwiczeń wymagane jest uzyskanie pozytywnych ocen za:
a) aktywność intelektualną na zajęciach
b) działalność praktyczną na zajęciach
c) uzyskane wyniki
d) kolokwium zaliczeniowe
Literatura pomocnicza
1. Birnboim H. C., Doly J. (1979). A rapid alkaline extraction procedure for
screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513 -
2. Dale J. W., von Schantz M. (2007). From genes to genomes. Concepts and
applications of DNA technology. Second edition. John Wiley, England.
3. PCR primer. A laboratory manual. (1995). Dieffenbach C.W., Dveksler G.
S. [ed.], Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
4. Sambrook J., Russell D. (2001). Molecular cloning. A laboratory manual.
Third edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
7
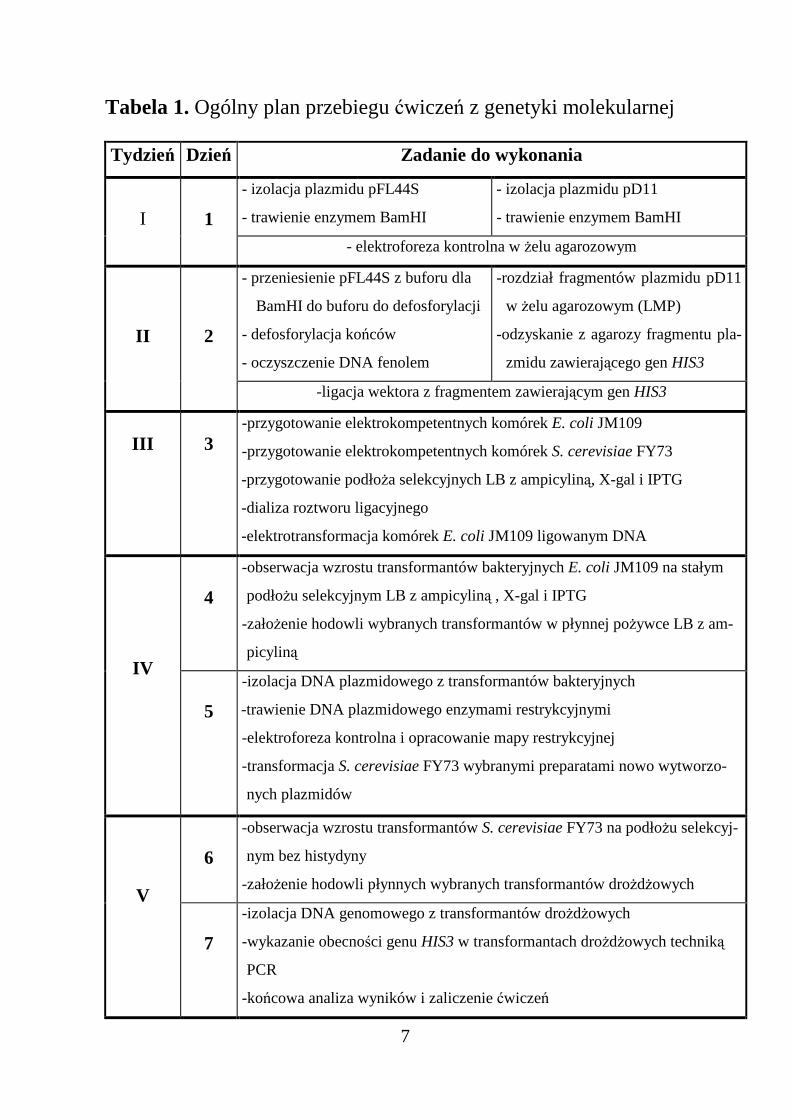
Tabela 1. Ogólny plan przebiegu ćwiczeń z genetyki molekularnej
Tydzień Dzień Zadanie do wykonania
- izolacja plazmidu pFL44S
- trawienie enzymem BamHI
- izolacja plazmidu pD11
- trawienie enzymem BamHII 1- elektroforeza kontrolna w Ŝelu agarozowym
- przeniesienie pFL44S z buforu dla
BamHI do buforu do defosforylacji
- defosforylacja końców
- oczyszczenie DNA fenolem
-rozdział fragmentów plazmidu pD11
w Ŝelu agarozowym (LMP)
-odzyskanie z agarozy fragmentu pla-
zmidu zawierającego gen HIS3
II 2
-ligacja wektora z fragmentem zawierającym gen HIS3
III 3-przygotowanie elektrokompetentnych komórek E. coli JM109
-przygotowanie elektrokompetentnych komórek S. cerevisiae FY73
-przygotowanie podłoŜa selekcyjnych LB z ampicyliną, X-gal i IPTG
-dializa roztworu ligacyjnego
-elektrotransformacja komórek E. coli JM109 ligowanym DNA
4
-obserwacja wzrostu transformantów bakteryjnych E. coli JM109 na stałym
podłoŜu selekcyjnym LB z ampicyliną , X-gal i IPTG
-załoŜenie hodowli wybranych transformantów w płynnej poŜywce LB z am-
picylinąIV
5
-izolacja DNA plazmidowego z transformantów bakteryjnych
-trawienie DNA plazmidowego enzymami restrykcyjnymi
-elektroforeza kontrolna i opracowanie mapy restrykcyjnej
-transformacja S. cerevisiae FY73 wybranymi preparatami nowo wytworzo-
nych plazmidów
6
-obserwacja wzrostu transformantów S. cerevisiae FY73 na podłoŜu selekcyj-
nym bez histydyny
-załoŜenie hodowli płynnych wybranych transformantów droŜdŜowychV
7
-izolacja DNA genomowego z transformantów droŜdŜowych
-wykazanie obecności genu HIS3 w transformantach droŜdŜowych techniką
PCR
-końcowa analiza wyników i zaliczenie ćwiczeń

8
Podstawowe informacje o plazmidach i szczepach
Plazmidy
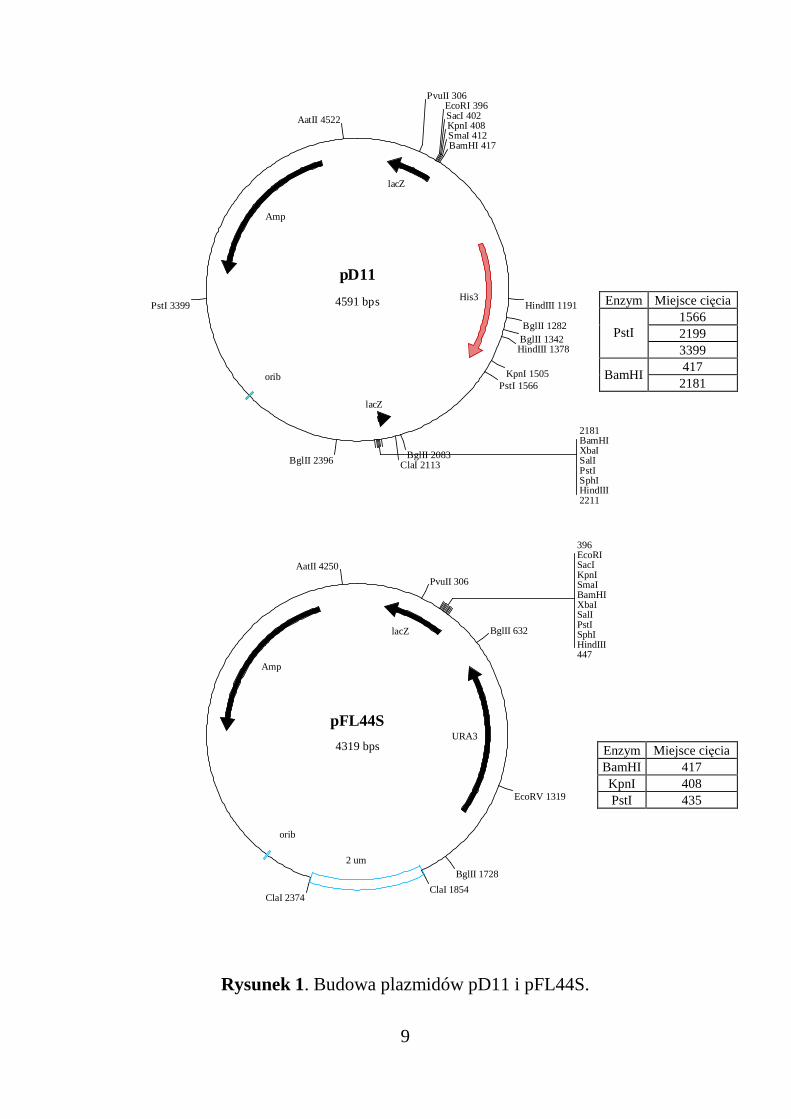
pFL44S - wielkość 4319 bp, składa się następujących elementów (Rys. 1):
• bakteryjny gen Amp kodujący β−laktamazę warunkującą oporność na
ampicylinę
• droŜdŜowy gen URA3
• bakteryjny region startu replikacji orib pochodzący z plazmidu ColE1
• promotor operonu lac wraz z częścią genu lacZ kodującą N-końcową
część β−galaktozydazy (α-peptyd) o długości 145 aminokwasów
• polilinker wbudowany do fragmentem genu lacZ kodującego α-peptyd
pD11 - wielkość 4591 bp, składa się z następujących elementów (Rys. 1):
• gen Amp kodujący β−laktamazę warunkującą oporność na ampicylinę
• Fragment BamHI (1764 bp) z XV chromosomu Saccharomyces cerevi-
siae zawierający gen HIS3. Gen ten koduje enzym ze szlaku biosyntezy
histydyny - IGP dehydratazę. Uszkodzenie geneu HIS3 objawia się u
droŜdŜy brakiem wzrostu na poŜywce minimalnej bez histydyny
• bakteryjny region startu replikacji orib pochodzący z plazmidu ColE1
9
Rysunek 1. Budowa plazmidów pD11 i pFL44S.
Enzym Miejsce cięcia15662199PstI3399417
BamHI2181
Enzym Miejsce cięciaBamHI 417KpnI 408PstI 435
pFL44S
4319 bps
PvuII 306
396EcoRISacIKpnISmaIBamHIXbaISalIPstISphIHindIII447
BglII 632
EcoRV 1319
BglII 1728
ClaI 1854ClaI 2374
AatII 4250
lacZ
URA3
2 um
orib
Amp
pD11
4591 bps
PvuII 306EcoRI 396SacI 402KpnI 408SmaI 412BamHI 417
HindIII 1191
BglII 1282BglII 1342HindIII 1378
KpnI 1505PstI 1566
BglII 2083ClaI 2113
2181BamHIXbaISalIPstISphIHindIII2211
BglII 2396
PstI 3399
AatII 4522
lacZ
His3
lacZ
orib
Amp

10
Szczepy
Saccharomyces cerevisiae FY73 (MATα, ura3-52, his3-∆200, GAL2)
MATαααα - droŜdŜowy typ płciowy „α”; ura3-52 - mutacja w genie metaboli-
zmu uracylu, uniemoŜliwia wzrost na podłoŜu minimalnym bez tego związ-
ku; his3-∆∆∆∆200 - delecja o długości 1036 bp, obejmująca całą sekwencję ko-
dującą genu HIS3 wraz z regionem promotorowym (Rysunek 4), fenotypowo
objawia się brakiem wzrostu na podłoŜu minimalnym bez histydyny; GAL2 -
zdolność do wykorzystywania galaktozy jako jedynego źródła węgla
Escherichia coli JM109 [F’, traD36, proA+B+, lacIq, lacZ∆Μ15] recA, rK-,endA1, gyrA96, thi1, supE44, ∆(lac-proAB)
F’ – obecność plazmidu F’ warunkującego wytwarzanie pili płciowych;
traD36 - mutacja blokująca przekazywanie plazmidu F’ podczas koniugacji;
proAB - mutacje w genach metabolizmu proliny powodujące brak wzrostu
na podłoŜu minimalnym bez tego aminokwasu; lacIq - mutacja powodująca
nadprodukcję represora operonu lac; lacZ∆∆∆∆M15 - delecja fragmentu genu
lacZ kodującego N-końcową część β-galaktozydazy (α-peptyd), pozostawio-
na jest natomiast część kodująca C-koniec (ω-peptyd); recA - mutacja w ge-
nie warunkującym rekombinację i naprawę DNA, uniemoŜliwia rekombina-
cję obcego DNA z genomem gospodarza; rK- - brak systemu restrykcji; en-
dA1 - mutacja w genie endonukleazy I, zwiększa wydajność i poprawia ja-
kość izolowanego DNA plazmidowego; gyrA96 - mutacja w podjednostce A
gyrazy, wywołuje oporność na kwas nalidiksowy; thi1 - mutacja powodująca
brak wzrostu na podłoŜu minimalnym bez tiaminy; supE44 - supresor muta-
cji amber (UAG); ∆∆∆∆(lac-proAB) - delecja operonu laktozowego i genów
proAB

11
Klasyczna metoda izolacja DNA plazmidowego z Escherichia coli
Obecnie do izolacji DNA plazmidowego z komórek bakteryjnych sto-sowane są metody róŜniące się niekiedy dość znacznie w poszczególnychetapach preparacji. Wszystkie je jednak cechuje zastosowanie lizy komórekSDS-em w warunkach alkalicznych. Podstawy tej metody opracowali Birn-boim i Doly w roku 1979. Podany wówczas sposób postępowania doczekałsię licznych, zazwyczaj drobnych modyfikacji skracających czas preparacji, aczasem równieŜ poprawiających czystość uzyskiwanych plazmidów. Najbar-dziej znacząca modyfikacja polegała na zastosowaniu gotowych kolumn zezłoŜem krzemionkowym co zostało opisane w rozdziale „Izolacja i oczysz-czanie kwasów nukleinowych na kolumnach”.
PoniŜej podano opis klasycznej metody izolacji DNA plazmidowego zbakterii z uŜyciem lizy alkalicznej i oczyszczania fenolem jak podali Birn-boim i Doly (1979). Do protokołu postępowania wprowadzono jedynie mo-dyfikacje dostosowujące go do potrzeb zająć ćwiczeniowych i posiadanegowyposaŜenia laboratoryjnego. Metoda ta daje dobre wyniki nawet w rękachosób początkujących. Przy jej uŜyciu moŜna izolować plazmidy o wielkoścido 15 kbp, a po nabyciu wprawy nawet do około 30 kbp. Wyizolowane pla-zmidy nie są całkowicie wolne od zanieczyszczeń, ale stopień ich czystościjest wystarczający do analizy restrykcyjnej, reakcji PCR i ligacji, a po nabra-niu wprawy równieŜ i do sekwencjonownia.
Metoda ta działa bardzo dobrze równieŜ po zmianie skali preparacji.MoŜna więc izolować DNA plazmidowe z hodowli bakteryjnych o innej ob-jętości niŜ podane 3 ml. W tym celu objętości poszczególnych roztworówroboczych naleŜy zmienić proporcjonalnie do zmiany objętości hodowli.
1. 3,5 ml poŜywki LB (wyciąg droŜdŜowy 1 %, trypton 1 % , NaCl 0,5 %) zampicyliną (100 µg/ml) zaszczepić szczepem E. coli zawierającym od-powiedni plazmid.
2. Hodowlę inkubować przez 18 od 20 godzin w temp. 37oC z wytrząsa-niem (150 - 160 cykli na minutę).
3. Do probówki Eppendorfa przenieść 1,5 ml hodowli bakteryjnej po czymwirować przez 3 min.
JeŜeli podano jedynie czas to znaczy, Ŝe wirowanie naleŜy wykonać wwirówce typu Eppendorf, w temperaturze pokojowej. Wirówki tej klasynajczęściej nie mają chłodzenia oraz moŜliwości regulacji obrotów ipracują ze stałą prędkością około 14000 obr/min.

12
W przypadkach gdy obok czasu podano równieŜ temperaturę i pręd-kość obrotową naleŜy uŜyć wirówki z moŜliwością regulowania tych pa-rametrów.
4. Usunąć supernatant przy pomocy pompki próŜniowej zachowując osadbakteryjny. Do probówki przenieść kolejne 1,5 ml hodowli i ponowniewirować przez 3 min.
5. Supernatant usunąć przy pomocy pompki próŜniowej zbierając równieŜkropelki pozostające na ściance probówki.
6. Osad bakteryjny zawiesić przy pomocy pipety w 200 µl buforu TGE (50mM Tris-HCl, 50 mM glukoza, 10 mM EDTA, pH 8,0) tak, aby powstałajednolita, pozbawiona grudek zawiesina, co jest warunkiem prawidłowe-go przebiegu lizy.
7. Do zawiesiny bakterii dodać 300 µl świeŜo przygotowanego roztworu dolizy (1% SDS, 0,2 N NaOH), po czym całość dokładnie wymieszać przezkilkukrotne odwrócenie probówki do góry dnem. NaleŜy unikać gwał-townego potrząsania probówką. Liza komórek następuje natychmiast pododaniu roztworu SDS z NaOH i objawia się wzrostem lepkości orazprzejrzystości zawiesiny przy równoczesnym zaniku barwy beŜowej.
8. Probówkę z lizatem umieścić na 3 min. w lodzie.
9. Do lizatu dodać 240 µl 3M octanu sodu (pH 5,2) i wymieszać przez kil-kukrotne odwracanie probówki do góry dnem. W lizacie powinny poja-wić się białawe "serowate" kłaczki wytrąconych białek.
10. Lizat z wytrąconymi białkami inkubować przez 10 min. w temp. -20oC.
11. Probówkę z lizatem wirować przez 15 min. (14 000 obr/min., 4oC).
W wyniku wirowania lizat powinien się rozdzielić na jasno beŜowyosad i lekko śluzowaty, przejrzysty supernatant.
− jeŜeli rozdział nie jest dobry to wirowanie (15 min, 14 000 obr/min.,4oC) naleŜy powtórzyć po ewentualnym rozcieńczeniu lizatu buforemTGE
− w przypadku dobrego rozdziału supernatant naleŜy ostroŜnie zebraćpipetą znad osadu i przenieść do czystej probówki Eppendorfa

13
12. Do supernatantu zebranego znad osadu dodać jedną objętość fenolu i ca-łość energicznie wytrząsać do wytworzenia jednorodnej emulsji.
Patrz równieŜ rozdział „Oczyszczanie kwasów nukleinowych fenolem”.
13. Probówkę z emulsją wodno-fenolową wirować przez 5 min.
Zawartość probówki powinna rozdzielić się na dwie wyraźne odgra-niczone fazy: fenolową (dolna) i wodną (górna). Zanieczyszczenia biał-kowe gromadzą się w fazie fenolowej i na granicy faz.
14. Pipetą zebrać fazę wodną i przenieść do czystej probówki. Przy zbieraniuszczególną uwagę naleŜy zwrócić na to, by nie pobrać zanieczyszczeńznajdujących się na granicy faz.
15. Do fazy wodnej dodać około dwie objętości eteru. Probówkę dokładniezamknąć i energicznie wytrząsać do wytworzenia jednorodnej emulsji.
16. Probówkę z emulsją wodno-eterową wirować przez 5 min.
RównieŜ i tym razem zawartość probówki powinna rozdzielić sięna dwie fazy: wodną (dolna) i eterową (górna). Zanieczyszczenia groma-dzą się w fazie eterowej i na granicy faz.
17. Posługując się pompką wodną odessać fazę eterową wraz z interfazą. Dlazminimalizowania strat naleŜy unikać zasysania fazy wodnej.
18. Powtórzyć postępowanie opisane w punktach od 15 do 17 po czymprzejść do punktu 19.
19. Do fazy wodnej pozostałej po dwukrotnej ekstrakcji eterem dodać jednąobjętość zimnego (-20oC) izopropanolu i całość wymieszać przez kilku-krotne odwracanie probówki do góry dnem. JeŜeli stęŜenie DNA w roz-tworze jest wysokie to po dodaniu izopropanolu pojawi się strąt w posta-ci pływającego kłaczka. Przy małym stęŜeniu DNA strąt moŜe być nie-widoczny.
20. Probówkę z zawartością inkubować w temp. -20oC przez 10 do 15 min.Inkubacja w niskiej temperaturze przyspiesza wytrącanie kwasów nu-kleinowych i poprawia wydajność.

14
21. Po zakończeniu inkubacji probówkę z zawartością wirować przez 10 min(14 000 obr/min, 4oC). Osad kwasów nukleinowych (DNA i RNA) zbie-ra się na ściance bocznej i dnie probówki i najczęściej jest słabo widocz-ny.
22. OstroŜnie, tak aby nie naruszyć osadu kwasów nukleinowych, usunąćsupernatant. Następnie do probówki dodać 1 ml 70 % etanolu.
23. Wirować przez 1 min.
24. Supernatant usunąć znad osadu przy pomocy pompki próŜniowej. ZebraćmoŜliwie wszystkie kropelki osadzone na ściance probówki.
25. Osad wysuszyć w suszarce próŜniowej z wirującym rotorem (speedvac).
26. Wysuszony osad kwasów nukleinowych rozpuścić w 31 µl wody miliQ.
27. Po rozpuszczeniu osadu pobrać 1 µl roztworu do nowej probówkiEppendorfa i zachować do kontroli.
28. Do pozostałych 30 µl roztworu kwasów nukleinowych dodać 1 µl roz-tworu RNazy o stęŜeniu 10 mg/ml.
29. Część preparatu plazmidowego pociąć enzymem restrykcyjnym BamHI.W tym celu zestawić roztwory reakcyjne przenosząc poszczególneskładniki do probówek Eppendorfa jak podano w Tabeli 2. Następniekrótko zwirować (10 do 15 sekund) co zapewnia zebranie wszystkichskładników reakcyjnych na dnie probówki i ułatwia ich wymieszanie. Powymieszaniu zawartość probówki naleŜy ponownie krótko zwirować.
Tabela 2. Trawienie plazmidów pFL44S i pD11 enzymem BamHI
Numer probówki 1 2DNA 10,0 µl pFL44S 10,0 µl pD11Bufor „Bam” 2,0 µl 2,0 µlWoda 6,0 µl 6,0 µlEnzym BamHI (10 U/µl) 2,0 µl 2,0 µl
30. Roztwory reakcyjne inkubować przez 2 godz. w cieplarce w temp. 37oC.
31. Po zakończeniu inkubacji przygotować próbki do elektroforezy kontrol-nej. W tym celu wskazane w Tabeli 3 objętości poszczególnych roztwo-

15
rów przenieść do probówek Eppendorfa, krótko zwirować, wymieszać iponownie zwirować.
Pozostałą część roztworów restrykcyjnych oraz preparaty plazmi-dów zamrozić w temp. -20oC.
Tabela 3. Przygotowanie próbek do elektroforezy kontrolnej.
Probówka A B C D E F
DNA1 µµµµl
pFL44Sbez RNazy
1 µµµµlpFL44S
2 µµµµlpFL44S/BamHI
1 µµµµlpD11 bez
RNazy
1 µµµµlpD11
2 µµµµlpD11/BamHI
Woda 7 µµµµl 7 µµµµl 6 µµµµl 7 µµµµl 7 µµµµl 6 µµµµlBarwnik donanoszenia 2 µµµµl 2 µµµµl 2 µµµµl 2 µµµµl 2 µµµµl 2 µµµµl
33. Przygotowane próbki nanieść na 0,9 % Ŝel agarozowy w buforze TBE zbromkiem etydyny (0,25 µg/ml). Na Ŝel nanieść równieŜ marker masy oznanym stęŜeniu i przeprowadzić elektroforezę pod napięciem od 150 Vdo 190 V w zaleŜności od wielkości Ŝelu.
Patrz równieŜ rozdział „Elektroforeza DNA w Ŝelach agarozowych”
34. Wykonać zdjęcie Ŝelu w przechodzącym świetle ultrafioletowym (mak-simum emisji lampy przy 312 nm).
35. Na podstawie zdjęcia ocenić jakość wyizolowanych plazmidów pD11 ipFL44S, sporządzić krzywą kalibracyjną i wyznaczyć wielkość frag-mentów liniowych obu plazmidów oraz oszacować błąd metody.
36. Na podstawie porównania intensywności świecenia prąŜków plazmido-wych z intensywnością świecenia markera masy oszacować ilość uzy-skanego plazmidowego DNA.
37. Posługując się spektrofotometrem do pomiarów kwasów nukleinowychw małych objętościach zmierzyć absorbancję uzyskanych preparatówplazmidowych przy długości fali 230 nm, 260 nm i 280 nm. Na tej pod-stawie określić ich stęŜenie i czystość.
Patrz równieŜ rozdział „Spektrofotometryczne oznaczanie stęŜenia i czy-stości kwasów nukleinowych.”

16
Materiały− poŜywka LB (wyciąg droŜdŜowy 1 %, trypton 1 % , NaCl 0,5 %)− ampicylina (10 mg/ml)− bufor TGE (50 mM Tris-HCl, 50 mM glukoza, 10 mM EDTA, pH 8,0)− roztwór do lizy (SDS 1%, NaOH 0,2 M)
Roztwór do lizy przygotowuje się bezpośrednio przed uŜyciemmieszając ze sobą równe objętości 2 % SDS i 0,4 M NaOH.
− octan sodu (3 M, pH 5,2)− fenol (nasycony roztwór fenolu w 0,1 M Tris-HCl (pH 8,0) z 0,1 % dodat-
kiem hydroksychinoliny)− eter− izopropanol− 70 % etanol− woda destylowana jałowa (miliQ)− RNaza (10 mg/ml)− enzym restrykcyjny BamHI (Fermentas, nr kat. ER0051)− bufor „Bam” zalecany przez producenta do trawienia enzymem BamHI
(10 mM Tris-HCl (pH 8,0), 5 mM MgCl2, 100 mM KCl, 0,02 % merkapto-etanol, 0,1 mg/ml BSA)
− agaroza LE (Prona)− marker masy - DNA faga λ trawiony enzymami EcoRI i HindIII− barwnik do nanoszenia na Ŝel 5 x stęŜony (Ficoll 400 15 %, błękit bromo-
fenolowy 0,15 %)− bufor TBE 10 x stęŜony (Tris 100,89 g, kwas borny 59 g, EDTA 9,35 g,
woda do 1000 ml, pH 8,0)− bromek etydyny (10 mg/ml)
Literatura
1. Birnboim H. C. i Doly J. (1979). A rapid alkaline extraction procedure forscreening recombinant plasmid DNA. Nucleic Acids Res. 7:1513
2. Sambrook J., Fritsch E.F. i Maniatis T. (1989). Molecular cloning. A labo-ratory manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor.

17
Szybka metoda izolacja DNA plazmidowego z Escherichia coli
Klasyczną metodę izolacji DNA plazmidowego z Escherichia colimoŜna znacznie uprościć usuwając z niej etapy związane z oczyszczaniemfenolem. Skrócona wersja doskonale sprawdza się w sytuacjach gdy wyse-lekcjonowanie poŜądanej konstrukcji plazmidowej wymaga przeszukaniaduŜej liczby transformantów. Ponadto, podobnie jak jej rozszerzona wersja,daje dobre wyniki równieŜ w rękach osób o małym doświadczeniu. Uzyska-ne tą drogą plazmidy nadają się do analizy restrykcyjnej i jako matryca doreakcji PCR, ale zazwyczaj nie są wystarczająco czyste do innych zastoso-wań.
1. W probówce Eppendorfa odwirować (3 min) 1,5 ml całonocnej hodowli(poŜywka LB z odpowiednim antybiotykiem) szczepu E.coli zawierają-cego badany plazmid. Supernatant usunąć przy pomocy pompki próŜ-niowej zachowując osad bakteryjny.
2. Osad bakteryjny dokładnie zawiesić w 200 µl buforu TGE (50 mMTris-HCl, 50 mM glukoza, 10 mM EDTA, pH 8,0).
3. Do zawiesiny bakterii dodać 300 µl świeŜo przygotowanego roztworu dolizy (1% SDS, 0,2 N NaOH), po czym całość dokładnie wymieszać przezkilkukrotne odwrócenie probówki do góry dnem.
4. Do lizatu dodać 240 µl 3M octanu sodu (pH 5,2) i wymieszać przez kil-kukrotne odwracanie probówki do góry dnem.
5. Lizat z wytrąconymi białkami inkubować przez 10 min. w temp. -20oC.
6. Probówkę z lizatem wirować przez 15 min. (14 000 obr/min., 4oC).
7. Supernatant przenieść do nowej probówki i dodać jedną objętość zimne-go (-20oC) izopropanolu. Całość wymieszać przez kilkukrotne odwraca-nie probówki.
8. Probówkę inkubować w temp. -20o C przez 10 min.
9. Po inkubacji probówkę z zawartością wirować przez 10 minut (14 000obr/min, 4oC).

18
10. OstroŜnie, nie naruszając osadu, usunąć supernatant. Następnie do pro-bówki dodać 1 ml 70 % etanolu.
11. Wirować przez 1 min.
12. Supernatant usunąć znad osadu przy pomocy pompki próŜniowej. ZebraćmoŜliwie wszystkie kropelki osadzone na ściance probówki.
13. Osad wysuszyć w suszarce typu speedvac.
14. Wysuszony osad rozpuścić w 29 µl wody miliQ po czym dodać 1 µlroztworu RNazy o stęŜeniu 10 mg/ml.
15. Uzyskany preparat wykorzystać w dalszych etapach prowadzonego eks-perymentu lub zamrozić w temp. –20oC.
Materiały− poŜywka LB (wyciąg droŜdŜowy 1 %, trypton 1 % , NaCl 0,5 %)− bufor TGE (50 mM Tris-HCl, 50 mM glukoza, 10 mM EDTA, pH 8,0)− roztwór do lizy (SDS 1%, NaOH 0,2 M)
Roztwór do lizy przygotowuje się bezpośrednio przed uŜyciemmieszając ze sobą równe objętości 2 % SDS i 0,4 M NaOH.
− octan sodu (3 M, pH 5,2)− izopropanol− 70 % etanol− woda destylowana jałowa (miliQ)− RNaza (10 mg/ml)

19
Ultra szybka metoda izolacja DNA plazmidowego z Escherichia coli
PoniŜej przedstawiono kolejną metodę izolacji DNA plazmidowego wwarunkach lizy alkalicznej. Tym razem jako główny cel postawiono sobiemaksymalne skrócenie czasu trwania preparacji, nawet kosztem znacznegozmniejszenia wydajności i pogorszenia jakości izolowanego DNA. Metoda tanadaje się wyłącznie do selekcji transformantów i, co najbardziej chyba za-skakujące, bardzo często zawodzi w rękach osób z małym doświadczeniemlaboratoryjnym. Podany poniŜej protokół opracowali Cormack i Somssich(1997).
1. Do probówki zawierającej 1 ml poŜywki SB (wyciąg droŜdŜowy 2 %,pepton 3,2 %, NaCl 0,5 %) z ampicyliną (100 µg/ml) przenieść jedną,dobrze wyrośniętą kolonię bakteryjną.
2. Hodowlę inkubować przez 18 godzin w temp. 37oC z energicznym wy-trząsaniem (150 -160 cykli na minutę).
3. Do probówki Eppendorfa przenieść 250 µl hodowli i dodać do niej 250µl świeŜo przygotowanego roztworu do lizy (1% SDS, 0,2 N NaOH), poczym całość dokładnie wymieszać przez kilkakrotne odwrócenie pro-bówki do góry dnem.
4. Do lizatu dodać 250 µl 3M octanu sodu (pH 5,2) i wymieszać przez od-wracanie probówki do góry dnem.
5. Probówkę z lizatem wirować przez 6 min.
6. Pipetą ostroŜnie zebrać supernatant i przenieść do czystej probówkiEppendorfa.
7. Do zebranego supernatantu dodać 750 µl zimnego (-20oC) izopropanolui wymieszać przez odwracanie probówki.
8. Wytrącone DNA odwirować (4 min.)
9. Przy pomocy pompki próŜniowej usunąć supernatant nie naruszając osa-du.

20
10. Probówkę krótko wirować i dokładnie usunąć pozostałe resztki superna-tantu.
11. Osad rozpuścić w 20 µl jałowej wody destylowanej i dodać 1 µl roztwo-ru RNazy o stęŜeniu 10 mg/ml.
16. Uzyskany preparat wykorzystać bezpośrednio w dalszych etapach pro-wadzonego eksperymentu lub zamrozić w temp. –20oC.
Materiały
− poŜywka SB (wyciąg droŜdŜowy 2 %, pepton 3,2 %, NaCl 0,5 %)− ampicylina (10 mg/ml)− roztwór do lizy (SDS 1%, NaOH 0,2 M)
Roztwór do lizy przygotowuje się bezpośrednio przed uŜyciem mie-szając ze sobą równe objętości 2 % SDS i 0,4 M NaOH.
− 3 M octan sodu, pH 5,2− izopropanol− jałowa woda miliQ− RNaza (10 mg/ml)
LiteraturaCormack R. S., Somssich I. E. (1997). Ultra-fast alkaline lysis plasmidextraction (UFX). Technical Tips Online, 1, 25:T01210.

21
Wybrane metody izolacji, oczyszczania i zagęszcza-nia kwasów nukleinowych
Izolacja i oczyszczanie DNA i RNA na kolumnach ze złoŜem krze-mionkowym
W roku 1979 Vogelstein i Gillespie zaobserwowali, Ŝe w obecnościjodku sodu liniowe, krótkie fragmenty dwuniciowego DNA adsorbują się napowierzchni sproszkowanego szkła typu flint. W kolejnych latach wykazano,Ŝe zjawisko to obejmuje równieŜ i pozostałe rodzaje kwasów nukleinowych,to jest dsDNA, ssDNA i RNA.
Flint to szkło krzemowo-ołowiowe o wysokim współczynniku załama-nia światła zawierające od 4 do 60 % tlenku ołowiu. Obecnie, ze względu natoksyczne właściwości ołowiu, do produkcji flintu uŜywany jest dwutlenektytanu lub cyrkonu.
Sole chaotropowe (jodek sodu, chlorowodorek guanidyny, izotiocyja-nian guanidyny) są to związki, które w roztworach wodnych wprowadzajązaburzenia w sieci wiązań wodorowych w cząsteczkach wody. PowodująrównieŜ rozpad błon komórkowych i denaturację białek, co sprzyja izolacjikwasów nukleinowych.
W obecności wysokich stęŜeń soli chaotropowych tylko kwasy nukle-inowe wiąŜą się ze złoŜem krzemionkowym podczas gdy wszystkie innezwiązki pozostają w roztworze. Wiązanie jest odwracalne i po zmianie pHoraz obniŜeniu siły jonowej kwasy nukleinowe odłączają się od złoŜa krze-mionkowego. Wiele firm biotechnologicznych, wykorzystując to zjawisko,opracowało zestawy zawierające kolumny z uformowanymi złoŜami krze-mionkowymi oraz odpowiednie bufory przeznaczone do izolacji DNA pla-zmidowego i genomowego oraz RNA z takich źródeł jak bakterie, droŜdŜe,tkanki roślinne i zwierzęce, a takŜe wydzieliny i płyny ustrojowe. Opraco-wano równieŜ zestawy do odzyskiwania kwasów nukleinowych z Ŝeli agaro-zowych oraz do oczyszczania po reakcjach enzymatycznych takich jak zna-kowanie, cięcie restrykcyjne czy PCR.
Aktualnie najbardziej rozpowszechnione są złoŜa wykonane w postacimembran z niemodyfikownej chemicznie krzemionki. Kolumny z tego typuzłoŜami są względnie tanie i pozwalają na uzyskanie kwasów nukleinowycho czystości wystarczającej do analizy restrykcyjnej, ligacji, znakowania, am-plifikcji (PCR i RTPCR) oraz sekwencjonownia.
ZłoŜa najnowszej generacji wykonywane są z mikrokuleczek krze-mionkowych do których na trwale przyłączono na przykład dietyloaminoeta-nol (DEAE). Działanie tego typu złoŜa opiera się na interakcji między ujem-nie naładowanymi grupami fosforanowymi kwasu nukleinowego, a dodatnio

22
naładowanymi grupami DEAE. Dzięki przyłączeniu cząsteczek DEAE dokulek krzemionkowych uzyskuje się duŜe zwiększenie gęstości dodatniegoładunku powierzchniowego, co powoduje mocniejsze wiązanie oczyszczane-go kwasu nukleinowego ze złoŜem. Dzięki temu moŜna zastosować bardziejenergiczne odpłukiwanie zanieczyszczeń, przekładające się w konsekwencjina lepsze oczyszczenie izolowanego materiału. Stopień czystości uzyskanychtą drogą kwasów nukleinowych odpowiada czystości uzyskiwanej w wynikudwukrotnego wirowania w gradiencie chlorku cezu. Jest to czystość wyma-gana w tak krytycznych zastosowaniach jak transfekcja, mikroinjekcja i tera-pie genowe.
Wszystkie firmy dostarczają produkowane przez siebie zestawy wrazze szczegółowymi instrukcjami dotyczącymi ich przeznaczenia i sposobuuŜycia. Zazwyczaj instrukcje te nie podają składu oferowanych buforów orazzawierają jedynie podstawową informację o rodzaju uŜytego złoŜa. Mimodość duŜej róŜnorodności wszystkie oferowane zestawy wiąŜe kilka cechwspólnych:
1. Przygotowanie materiału do naniesienia na kolumnę− lizat komórek bakteryjnych poddaje się wirowaniu w celu odrzucenia
kompleksów białko-SDS oraz DNA chromosomalnego− tkanki zwierzęce trawi się proteinazą K i następnie dodaje bufor o wy-
sokim stęŜeniu soli chaotropowych− tkanki roślinne rozbija się mechaniczne (młynek Retcha lub rozciera-
nie ręczne w moździerzu w obecności płynnego azotu) i przenosi dobufor o wysokim stęŜeniu soli chaotropowych
− agarozę z której ma być odzyskany DNA roztapia się w buforze o wy-sokim stęŜeniu soli chaotropowych w temp. 50 do 60oC
− do roztworów po reakcjach enzymatycznych dodaje się bufor z solamichaotropowymi
− w przypadku stosowania kolumn NUCLEOBOND (produkowanychprzez firmę Macherey i Nagel) wstępnie przygotowany materiał, przednaniesieniem na kolumnę, oczyszcza się na dodatkowym sączku
2. Przepuszczenie wstępnie przygotowanego materiału przez kolumnęW zaleŜności od skali preparacji stosowane są kolumny o przepływie
grawitacyjnym (np. NUCLEOBOND ) lub o przepływie wymuszonymprzez wirowanie (np. QIAGEN oraz A&A Biotechnology).
Kwasy nukleinowe zawarte w przepływającym przez kolumnę mate-riale zostają selektywnie związane ze złoŜem silikonowym. Wszystkieinne składniki przechodzą do odbieralnika kolumny.

23
3. Płukanie złoŜa ze związanymi kwasami nukleinowymi w celu usunięciazanieczyszczeń oraz osuszenie kolumny.
4. Odmycie kwasów nukleinowych ze złoŜa.W zaleŜności od zaleceń producenta bezpośrednio do złoŜa dodaje się
bufor TE (10 mM Tris-HCl, 1 mM EDTA, pH 8,0), 10 mM Tris (pH 8,0)lub wodę. W wyniku zmiany siły jonowej i pH złoŜa, kwasy nukleinowezostają uwolnione i przechodzą do roztworu.
Dokonując wyboru środka wymywającego naleŜy ściśle przestrzegaćzaleceń producenta, gdyŜ w przypadku niektórych kolumn (np. QIA-GEN-owskich) wydajność odmywania zaleŜy od pH uŜytego eluentu. Wtakich przypadkach uŜycie wody, zamiast buforu, powoduje drastycznyspadek wydajności elucji. Kolumny A&A Biotechnology są pod tymwzględem bardziej tolerancyjne.
5. Większość kolumn przeznaczonych do oczyszczania DNA po reakcjachenzymatycznych pozwala na pracę z fragmentami o wielkości do 7 kb, aniekiedy do 10 kb. PowyŜej tych wielkości wydajność odzyskiwaniafragmentów z kolumny drastycznie spada.
Fotgrafia 1. Kolumna Nucleobond AX100 (Macherey-Nagel) o swobodnymprzepływie grawitacyjnym (1). Jej złoŜe (ZN) jest w stanie związać 100 µgkwasów nukleinowych. (2) kolumna produkowana przez polską firmę A&ABiotechnology z Gdyni umieszczona w odbieralniku (O). Jest to kolumna oprzepływie wymuszanym przez wirowanie. Jej złoŜe (ZA), widoczne w po-staci białego paska tuŜ pod niebieskim pierścieniem mocującym moŜe zwią-zać 20 µg DNA, 2A – ta sama kolumna po wyjęciu z odbieralnika.
1
ZN
2
2A
ZA
O
O

24
Literatura pomocnicza
Vogelstein B., Gillespie D. (1979). Preparative and analytical purification ofDNA from agarose. Proc. Natl. Acad. Sci. USA, 76, 615-619.
Materiały informacyjne firmy Macherey-Nagel:Nucleobond AX. Properties and application (1999)Plasmid DNA purification (2002)www.macherey-nagel.de
Materiały informacyjne firmy Qiagen, www.qiagen .comMateriały informacyjne firmy A&A Biotechnology, www.aabiot.com

25
Oczyszczanie kwasów nukleinowych fenolem
Powszechnie kiedyś uŜywanym sposobem odbiałczania kwasów nu-kleinowych była ekstrakcja ich roztworów fenolem. W jej wyniku zdenatu-rowane białka przechodzą z fazy wodnej do fazy fenolowej i są wraz z niąodrzucane. Rozwój metod opartych na wykorzystaniu kolumn ze złoŜemkrzemionkowym usunął metodę fenolową w cień. Ale ciągle w niektórychzastosowaniach, na przykład przy izolacji wysoko cząsteczkowego DNA ge-nomowego, ekstrakcja fenolem ciągle jest niezastąpiona.
Ekstrakcja fenolem moŜe być jednym z etapów izolacji DNA geno-mowego czy plazmidowego (patrz rozdział „Klasyczna metoda izolacji DNAplazmidowego z Escherichia coli”). Stosuje się ją równieŜ wtedy, gdy w ko-lejnych etapach eksperymentu stosowane są enzymy wiąŜące się z kwasaminukleinowymi lub działające antagonistycznie względem siebie, a oczysz-czanie na kolumnach zawodzi.
Ekstrakcje fenolem wykonywane są w dwóch wariantach:- z uŜyciem wyłącznie nasyconego roztworu fenolu,- z uŜyciem nasyconego roztworu fenolu z dodatkiem chloroformu i alko-holu izoamylowego
Sam fenol nie eliminuje całkowicie aktywności RNazy i działa jak roz-puszczalnik dla cząsteczek RNA zawierających długie fragmenty poli(A).Właściwości tych pozbawiony jest nasycony roztwór fenolu z chloroformemi alkoholem izoamylowym, sporządzony w stosunku 25:24:1.
Podany poniŜej protokół stosowany jest do oczyszczania roztworówDNA. W pracach z RNA powinien być stosowany fenol z chloroformem ialkoholem izoamylowym.
Pracując z fenolem trzeba pamiętać, Ŝe POWODUJE ON BARDZOBOLESNE I TRUDNO GOJĄCE SIĘ RANY. Miejsce przypadkowegokontaktu skóry z fenolem naleŜy spłukać duŜą ilością bieŜącej wody, a na-stępnie wodą z mydłem. W razie rozległych oparzeń konieczna jest inter-wencja lekarska.
1. Do probówki Eppendorfa zawierającej wodny roztwór DNA dodać rów-ną objętość fenolu (dokładne objaśnienie terminu „fenol” podane jestponiŜej w części „Materiały”). Probówkę dokładnie zamknąć.
2. Probówkę wytrząsać energicznie do wytworzenia jednorodnej emulsji.
3. Probówkę z zawartością wirować 5 min. Podczas wirowania zawartośćprobówki rozdziela się na fazę górną (wodną) i dolną (fenolową). Za-

26
nieczyszczenia białkowe gromadzą się w fazie fenolowej oraz na granicyfaz.
4. Zebrać fazę wodną i przenieść do czystej probówki. Dla zminimalizowa-nia strat zebrać trzeba jak najwięcej fazy wodnej nie naruszając przy tyminterfazy. Jeśli interfaza jest wyjątkowo duŜa lub została naruszona topostępowanie opisane w punktach od 1 do 4 naleŜy powtórzyć.
5. Do probówki zawierającej fazę wodną dodać dwie objętości eteru. Pro-bówkę szczelnie zamknąć.
Pracując z eterem naleŜy unikać jego wdychania, a jeśli jest to moŜli-we naleŜy pracować pod działającym wyciągiem chemicznym.
6. Zawartość probówki wytrząsać energicznie do wytworzenia jednorodnejemulsji.
7. Wirować 3 min. (14 000 obr/min., temp. pokojowa). W trakcie wirowa-nia zawartość probówki rozdziela się na fazę górną (eterową) i dolną(wodną). Zanieczyszczenia gromadzą się w fazie eterowej oraz na grani-cy faz.
8. Posługując się pompką wodną odessać fazę eterową wraz z interfazą. Dlazminimalizowania strat naleŜy unikać zasysania fazy wodnej.
9. Powtórzyć postępowanie opisane w punktach od 5 do 8.
10. W zastosowaniach wraŜliwych nawet na śladowe ilości fenolu preparo-wane kwasy nukleinowe moŜna dodatkowo oczyścić przez wytrącanieetanolem lub izopropanolem w obecności octanu sodu.
Patrz równieŜ rozdział „Zagęszczanie roztworów kwasów nukleino-wych przez wytrącanie”
Materiały
W powyŜszym protokole "fenol" zawsze oznacza zbuforowany (Tris-HCl, pH>7,8), nasycony wodny roztwór fenolu z 0,1 % dodatkiem 8-hydroksychinoliny. pH fenolu decyduje o podziale DNA między fazę wodnąi fenolową. Przy pH kwaśnym DNA w znacznej części przechodzi do fazyfenolowej.

27
Zwykły fenol handlowy, w zaleŜności od stopnia zanieczyszczenia, makolor od jasno róŜowego do ciemno fioletowego i przed uŜyciem musi byćoczyszczony. Destyluje się go w szklanym aparacie z termostatowaną, prostąchłodnicą płaszczową. Temperatura wrzenia fenolu wynosi 182oC, a krzep-nięcia 41oC.
Spływający z chłodnicy destylarki fenol zbiera się do szklanej butelkiz szerokim otworem, zawierającej 0,5 M Tris-HCl (pH 8) w ilości okołodwukrotne większej od przewidywanej objętości fenolu. Po przedestylowa-niu zakładanej ilości fenolu do butelki naleŜy dodać 8-hydroksychinolinę wilości 0,1 % względem fenolu. Hydroksychinolina przechodzi do fazy feno-lowej i zabarwia ją na Ŝółty kolor.
Następnie, szczelnie zakręconą, butelkę wytrząsa się energicznie dowytworzenia jednorodnej emulsji i pozostawia do rozdzielenia faz. Po czymnaleŜy usunąć fazę wodną (górną), a do pozostałej w butelce fazy fenolowejdodać jedną objętość 0,1 M Tris-HCl (pH 8) i powtórzyć wyŜej opisaneczynności aŜ pH buforu nad fenolem osiągnie wartość 7,8 lub więcej (czystyfenol ma kwaśne pH). Zwykle do tego celu wystarczają 2 - 3 zmiany buforu.
Obok względnie brudnego fenolu wymagającego destylacji przed uŜy-ciem, wyspecjalizowane firmy oferują równieŜ fenol destylowany. Jest ondostarczany w opakowaniach wypełnionych gazem szlachetnym w celuochrony przed dostępem powietrza. Oferowane są równieŜ gotowe do uŜyciazbuforowane roztwory fenolu. Trzeba jednak pamiętać, Ŝe po otwarciu ory-ginalnych opakowań materiały te mają krótki okres trwałości (zwykle około1 miesiąca), a ponadto są znacznie droŜsze od zwykłego fenolu handlowego.

28
Zagęszczanie roztworów kwasów nukleinowych przez wytrąca-nie
W przypadkach gdy zachodzi konieczność uzyskania wyŜszego stęŜe-nia DNA lub RNA w roztworze stosuje się wytrącanie.
Do roztworu o niskim stęŜeniu kwasu nukleinowego dodaje się octansodu oraz izopropanol powodujące wytrącenie znajdującego się tam DNAlub RNA. Następnie roztwór wiruje dla zebrania wytrąconego kwasu nukle-inowego w osadzie. Osad przemywa się 70 % etanolem i po wysuszeniu roz-puszcza w wodzie lub buforze o poŜądanej objętości. Wytrącanie stosuje sięrównieŜ wtedy gdy zachodzi konieczność zmiany buforu w którym rozpusz-czony jest kwas nukleinowy. PoniŜej podano szczegółowy opis postępowa-nia przy zagęszczaniu roztworów kwasów nukleinowych przez wytrącanie.
1. Do probówki zawierającej roztwór z którego mają być wytrącone kwasy
nukleinowe dodać 0,1 objętości 3 M octanu sodu (pH 5,2) i 1 objętość
izopropanolu lub 2 objętości 96 % etanolu. Wybór alkoholu zaleŜy od
preferencji eksperymentatora przy czym naleŜy pamiętać, Ŝe izopropanol
jest znacznie tańszy. Oba alkohole powinny być przed uŜyciem schłodzo-
ne do -20oC.
2. JeŜeli stęŜenie kwasów nukleinowych jest bardzo niskie (około 20 ng/ml)
roztwór moŜna inkubować w temp. -20oC przez 10 do 20 min. JeŜeli stę-
Ŝenie jest wyŜsze to inkubacja w niskiej temperaturze nie jest konieczna.
3. Wytrącone DNA odwirować (15 min., 14 000 obr., 4oC).
4. OstroŜnie, tak aby nie naruszyć osadu, usunąć supernatant. Następnie do-
dać do probówki 1 ml 70 % etanolu.
5. Wirować przez 1 min.
6. Przy pomocy pompki próŜniowej usunąć supernatant znad osadu. Zebrać
moŜliwie wszystkie kropelki osadzone na ściance probówki.
7. Osadzone DNA wysuszyć w suszarce próŜniowej z wirującym rotorem
(speed vac).

29
8. Osad DNA rozpuścić w odpowiedniej ilość jałowej wody destylowanej
lub w odpowiednim buforze. Najczęściej uŜywa się do tego celu 10 mM
Tris-HCl lub bufor TE (10 mM Tris-HCl, 1 mM EDTA, pH 8,0).

30
Mikrodializa
Wraz z pojawieniem się membran z porami o średnicy 0,025 µm (Mil-lipore, nr kat. VSWP01300) powstała moŜliwość dializowania roztworów obardzo małych objętościach. Stosując dializę moŜna szybko usunąć związkiniskocząsteczkowe z roztworu kwasu nukleinowego. Jest to metoda bardzoprzydatna na przykład do odsalania roztworów ligacyjnych przed elektropor-cją. PoniŜej podano szczegółowy sposób postępowania.
1. Do jałowej szalki Petriego nalać około 25 ml jałowej wody destylowanej.
2. Na powierzchnię wody połoŜyć krąŜek wykonany z membrany półprze-puszczalnej (np. Millipore, nr kat. VSWP01300) tak aby pomiędzy mem-braną i wodą nie utworzyły się pęcherze powietrza (Fot. 2).
MoŜna uŜyć membrany dowolnego producenta pod warunkiem, Ŝeśrednia wielkość jej por nie przekracza 0,025 µm. Nakładanie na po-wierzchnię wody najlepiej wykonać przy pomocy pęsety o łopatkowatozakończonych końcach. Membrany nie wolno dotykać palcami, bo powo-duje to zatykanie por. Membrana musi pływać na powierzchni wody, niemoŜe być zanurzona (Fot. 2).
3. NaleŜy odczekać około 1 do 2 minut aŜ membrana wysyci się równomier-nie wodą, co moŜna poznać po jej delikatnym ściemnieniu.
4. Na powierzchnię membrany nanieść dializowany roztwór. NaleŜy to robićbardzo ostroŜnie aby nie zatopić membrany. Na membranę o średnicy 13mm moŜna nanieść do 50 µl roztworu.
5. Po upływie 10 do 20 min (w zaleŜności o objętości dializowanego roz-tworu) naleŜy pipetą odessać z membrany naniesiony tam uprzednioroztwór, po czym jest on gotowy do dalszego uŜycia.
Fotografia 2. Dializowanie roztworu DNA na krąŜkach z membrany pół-przepuszczalnej.

31
Spektrofotometryczne oznaczanie stęŜenia i czystościkwasów nukleinowych
StęŜenie kwasów nukleinowych w roztworach moŜna wyznaczyć
spektrofotometryczne kierując się następującymi zaleŜnościami:
1. Dla długości fali 260 nm i drogi optycznej 1 cm, absorbancja (OD260) wy-
nosi 1 dla czystych roztworów kwasów nukleinowych o następujących
stęŜeniach:
• dwuniciowy DNA 50 µg/ml
• jednoniciowy DNA i RNA 40 µg/ml
• jednoniciowy oligonukleotyd 20 µg/ml
2. Stosunek absorbancji OD260/OD280 pozwala na oszacowanie czystości
roztworu kwasu nukleinowego
• dla czystego roztworu RNA stosunek absorbancji OD260/OD280 wynosi 2,0
• dla czystego roztworu DNA stosunek absorbancji OD260/OD280 wynosi 1,8
• dla czystego roztworu DNA stosunek absorbancji OD260/OD230 jest więk-
szy niŜ 2,2
• zanieczyszczenie roztworu DNA cukrami powoduje spadek stosunku
OD260/OD230 poniŜej wartości 2,0
• zanieczyszczenie roztworu DNA białkami i/lub fenolem powoduje spadek
stosunku OD260/OD280 poniŜej wartości 1,8
• zanieczyszczenie roztworu DNA kwasem rybonukleinowym powoduje
podniesienie stosunku OD260/OD280 powyŜej wartości 1,8
3. JeŜeli stosunek OD260/OD280 znacznie odbiega od wartości 1,8 dokładne
oznaczenie stęŜenia DNA nie jest moŜliwe.

32
Trawienie DNA enzymami restrykcyjnymi
Endonukleazy restrykcyjne klasy II (zwane krócej enzymami restryk-cyjnymi lub restryktazami) są enzymami, które po rozpoznaniu i przyłącze-niu się do specyficznej sekwencji DNA rozcinają wiązania fosfodwuestrowew jej obrębie. Rozpoznawane sekwencje są najczęściej jednoznacznie zdefi-niowanymi 4, 5, 6 lub 8 nukleotydowymi palindromami (Tabela 4). Znane sąrównieŜ enzymy rozpoznające jednoznacznie określone sekwencje niepalin-dromowe o długości nawet do 40 nt oraz enzymy rozpoznające i rozcinającemiejsca o zmiennej sekwencji nukleotydowej (Tabela 4).
Tabela 4. Przykładowe sekwencje nukleotydowe rozpoznawane i rozcinaneprzez enzymy restrykcyjne.
Enzym Długość roz-poznawanejsekwencji
Rozpoznawana sekwencja z zaznaczonymi miej-scami cięcia
Rodzaj genero-wanych końców
HhaI 4 5’...GCG|C...3’
3’...C|GCG...5’lepkie 3’
Sau3AI 4 5’...|GATC ...3’
3’... CTAG|...5’lepkie 5’
HinfI 5 5’...G|ANTC...3’
3’...CTNA|G...5’lepkie 5’
FokI 5 5’...GGATG(N) 9|...3’
3’...CCTAC(N) 13|...5’lepkie 5’
SmaI 6 5’...CCC|GGG...3’
3’...GGG|CCC...5’tępe
BamHI 6 5’...G|GATCC...3’
3’...CCTAG|G...5’lepkie 5’
KpnI 6 5’...GGTAC|C...3’
3’...C|CATGG...5’lepkie 3’
NotI 8 5’...GC|GGCCGC...3’
3’...CGCCGG|CG...5’lepkie 5’
TstI 12 5’...| 8(N)CAC(N) 6TCC(N) 12|...3’
3’...| 13(N)GTG(N) 6AGG(N)7 |...5’lepkie 3’
I-SceI 18 5’...TAGGGATAA|CAGGGTAAT...3’
3’...ATCCC|TATTGTCCCATTA...5’lepkie 3’

33
Większość enzymów restrykcyjnych przecina obie nici cząsteczkiDNA w obrębie rozpoznawanej sekwencji lub bezpośrednio na jej końcach(Sau3AI). Enzymy z podklasy IIS, na przykład FokI i TstI, przecinają czą-steczkę w miejscu leŜącym w stałej odległości od rozpoznawanej sekwencjibez względu na to jaki jest tam układ nukleotydów. Przy czym enzym TstItnący po obu stronach rozpoznawanej sekwencji powoduje jej usunięcie ztrawionego DNA.
Enzymy naleŜące do klasy II i IIS wymagają do swego działania obec-ności jonów magnezu.
PoniewaŜ rozpoznawane są jedynie ściśle określone kombinacje nu-kleotydów, to dowolna cząsteczka DNA rozcinana jest przez dany enzym naograniczoną liczbę fragmentów. Wykorzystanie tej właściwości enzymówrestrykcyjnych umoŜliwiło powstanie i rozwój inŜynierii genetycznej.
Analizę nieznanego DNA rozpoczyna się zwykle od sporządzenia jegomapy restrykcyjnej. Dobór enzymów zaleŜny jest od wielkości badanegoDNA. Przy cząsteczkach znacznie dłuŜszych niŜ 5000 bp, analizę warto roz-począć od enzymów rozpoznających 6 nukleotydów. Sekwencje szóstkowemają szansę wystąpienia średnio raz na 4096 nukleotydów (46 = 4096). Poustaleniu kilku pierwszych punktów orientacyjnych moŜna przystąpić domapowania z uŜyciem częściej tnących enzymów, pamiętając, Ŝe sekwencjepiątkowe występują raz na 1024 (45), a czwórkowe raz na 256 (44) nukleoty-dów.
Kilka rad praktycznych o pracy z enzymami restrykcyjnymi
A. Enzymu restrykcyjne są dość drogie i wymagają umiejętnego obchodzeniasię z nimi.
B. Enzymy restrykcyjne dostarczane są przez producenta w 50 % glicerolu.Transportuje się je w suchym lodzie i przechowuje w temp. -20oC.
C. Przed pobraniem enzymu z probówki dobrze jest ją krótko zwirować. En-zymu czasami jest tylko kilkanaście µl i probówka moŜe początkowo wy-dawać się pusta.
D. Enzymy restrykcyjne dostarczane są zwykle w stęŜeniu 10 U/µl.
E. Jedna jednostka aktywności jest to taka ilość enzymu, która w warunkachoptymalnych dla danego enzymu, w ciągu 1 godziny trawi całkowicie 1 µgodpowiedniego substratu.

34
Warunki trawienia to temperatura, najczęściej 37oC, ale są i wyjątkinp. dla enzymu AccIII 65oC, skład i pH buforu do inkubacji. Substratemjest najczęściej DNA faga λ. Dane te są zawsze podawane w opisie kon-kretnego enzymu. Producenci wraz z enzymem dostarczają równieŜ opty-malny dla niego bufor.
F. 10 U/µl to duŜa aktywność, enzym naleŜy więc przed uŜyciem odpowied-nio rozcieńczyć. Nadmiar enzymu nie tylko niepotrzebnie zwiększa kosz-ty, ale często pogarsza wyniki trawienia, a w skrajnych przypadkach tnieniespecyficznie.
G. PoniewaŜ rozcieńczone enzymy bardzo szybko tracą swoje właściwości, tonaleŜy rozcieńczać tylko tyle enzymu ile się od razu zuŜyje.
Kiedy enzym restrykcyjny nie chce ciąć
Zdarzają się sytuacje, iŜ mimo poprawnego doboru enzymu restrykcyj-nego, buforu i temperatury, trawienie DNA nie zachodzi. MoŜe to być spo-wodowane zanieczyszczeniem DNA lub jego metylacją. DNA moŜna oczy-ścić fenolem lub na kolumnach, co pomaga prawie we wszystkich przypad-kach. Druga moŜliwość wymaga nieco szerszego omówienia.
Sekwencje nukleotydowe rozpoznawane przez niektóre enzymy re-strykcyjne mogą być równieŜ identyfikowane przez dam i dcm metylazy zEscherichia coli. Metylaza dam przenosi grupę metylową na adeninę w se-kwencji GATC przekształcając ją w GmATC. Metylaza dcm metyluje drugącytozynę w sekwencji CCWGG (gdzie W = A lub T) przekształcając ją wCmCWGG. DNA z dodatkowymi grupami metylowymi przestaje być sub-stratem rozpoznawanym przez enzym restrykcyjny. Przykładowo: enzymBclI rozpoznaje i rozcina sekwencję TGATCA, ale nie TGmATCA, zaś Eco-RII rozpoznaje i tnie sekwencję CCWGG, ale nie CmCWGG.
JeŜeli plazmid ma być cięty enzymem restrykcyjnym wraŜliwym nametylację rozcinanej przez niego sekwencji nukleotydowej to naleŜy gonamnaŜać w szczepie E. coli JM110 mającym mutacje w genach dam i dcm.Mutacja dam powoduje inaktywację metylazy odpowiedzialnej za metylacjęadeniny w obrębie sekwencji 5’-GATC-3’. Natomiast mutacja genu dcmuniemoŜliwia metylację wewnętrznej cytozyny w obrębie sekwencji 5’-CCAGG-3’ oraz 5’-CCTGG-3’. Wadą szczepu E. coli JM110 jest jegomniejsza Ŝywotność w porównaniu z innymi standartowymi szczepami tegogatunku.

35
Enzym restrykcyjny przy pracy
Trawienie DNA przeprowadza się w jak najmniejszej objętości roz-tworu reakcyjnego, w skład którego wchodzą: DNA, enzym, bufor i woda.Reguły rządzące ilościowym doborem poszczególnych składników są nastę-pujące:
1. Ilość DNA brana do trawienia powinna w przybliŜeniu zawierać się wprzedziale od 0,1 do 1 µg. Przy mniejszej ilości prąŜki nie będą widocznew Ŝelu, zaś przy większej mogą się nie rozdzielać i są nieostre. Wraz zewzrostem liczby fragmentów na jakie cięte jest dane DNA, naleŜy zwięk-szać jego ilość braną do trawienia.
2. Ilość enzymu naleŜy dobrać do ilości trawionego DNA oraz ilości miejsccięcia w jego obrębie. Trzeba pamiętać, Ŝe aktywność enzymu podanaprzez producenta spada w miarę jego przechowywania. Właściwie enzy-mom bardziej szkodzą wielokrotne zmiany temperatury spowodowanewyjmowaniem z zamraŜarki niŜ sam czas jaki upłynął od daty ich wytwo-rzenia. Dlatego teŜ enzym po wyjęciu z zamraŜarki powinien być natych-miast umieszczany w lodzie lub przenośnym zasobniku zimna. DuŜe por-cje enzymów powinny być dzielone na mniejsze części natychmiast ponadejściu przesyłki od producenta.
3. Enzymy dostarczane są przez producentów w buforach o składzie opty-malnym dla kaŜdego z nich. Trzeba przy tym pamiętać, Ŝe we wszystkichtych buforach znajduje się 50 % glicerolu stosowanego jako krioprotek-tor. Dla umoŜliwienia prawidłowego działania enzymu restrykcyjnegostęŜenie glicerolu w roztworze reakcyjnym nie moŜe przekraczać 5 %objętości końcowej. W praktyce oznacza to, Ŝe enzym nie moŜe stanowićwięcej niŜ jedną dziesiątą objętości końcowej roztworu reakcyjnego.
4. Wraz z enzymem producenci dostarczają optymalny bufor w postacidziesięciokrotnie stęŜonego roztworu. W nielicznych przypadkach moŜnasię teŜ spotkać z buforami pięciokrotnie stęŜonymi. Niektóre enzymy (naprzykład EcoRII i NdeII) wymagają dodatkowo obecności w roztworzereakcyjnym DTT albo albuminy. Producenci często dostarczają te skład-niki w postaci oddzielnych roztworów o ile nie włączyli ich w skład bufo-ru optymalnego dla danego enzymu.
5. Wielu producentów wraz z buforem optymalnym dla danego enzymu do-starcza równieŜ bufor uniwersalny, który moŜe być uŜywany do jednocze-snego trawienia DNA dwoma enzymami o róŜnych wymaganiach wzglę-

36
dem buforu. Bufory uniwersalne na ogół znacznie ułatwiają pracę. Jednakzdarzają się przypadki, Ŝe ich uŜycie nie przynosi zadawalających rezul-tatów. W takich sytuacjach trzeba wykonywać trawienia kolejno po sobiestosując bufory optymalne dla uŜytych enzymów.
6. Woda stanowi dopełnienie do wymaganej objętości końcowej. Powinnabyć o najwyŜszym moŜliwym stopniu czystości.
7. Po połączeniu wszystkich składników, roztwór reakcyjny inkubuje się wtemp. 37oC przez około 2 godziny lub przez całą noc.
8. JeŜeli trawione DNA ma być uŜyte następnie do ligacji to lepiej jest niestosować długich czasów trawienia ze względu na moŜliwość degradacjikońców lub całego DNA. Niektóre partie enzymów, przy przedłuŜonymczasie inkubacji, wykazują nadmierną niespecyficzną aktywność nukle-olityczną degradującą trawione DNA. Tego rodzaju przypadłości zdarzająsię czasem enzymom pochodzącym od najbardziej nawet renomowanychproducentów.
9. Ze względu na skraplanie się pary wodnej na zimnej pokrywce probówki(co zaburza skład roztworu), wszystkie reakcje restrykcyjne powinny byćinkubowane w cieplarce, a nie w łaźni wodnej.
10. Niektóre enzymy mają inną optymalną temperaturę działania niŜ 37oC. Naprzykład dla SmaI wynosi ona 50oC. W razie wątpliwości naleŜy odszu-kać odpowiednie dane w katalogu.
11. JeŜeli po restrykcji, w następnym etapie doświadczenia, uŜyta ma być li-gaza lub polimeraza, to konieczne jest całkowite zinaktywowanie enzymurestrykcyjnego. Najczęściej stosuje się w tym celu ogrzewanie w temp.60oC przez 10 min. Niektóre enzymy (np. BamHI, TaqI i HindIII) wyma-gają wyŜszej temperatury i dłuŜszego czasu inaktywacji. W razie wątpli-wości naleŜy odszukać odpowiednie dane w katalogu producenta enzymu.
12. JeŜeli produkty trawienia mają być analizowane w elektroforezie, to pozakończeniu inkubacji do roztworu reakcyjnego dodaje się jedną piątąobjętości barwnika obciąŜającego, całość dokładnie miesza i nanosi naŜel.
Barwnik obciąŜający (5 x stęŜony): 15,00 % ficoll 400 0,15 % błękit bromofenolowy 0,15 % ksylenecyjanol

37
Zadaniem ficollu 400 jest obciąŜenie próbki tak, aby przy nanoszeniuna Ŝel opadała na dno gniazda. Barwniki umoŜliwiają wizualne śledzeniepostępów elektroforezy. Błekit bromofenolowy migruje w Ŝelu agarozo-wym z buforem TBE jak dwuniciowy, liniowy DNA o długości około 300bp. W tych samych warunkach ksylenecyjanol migruje jak dwuniciowy,liniowy DNA o długości około 4 000 bp. ZaleŜności te moŜna uznać zastałe w Ŝelach agarozowych o stęŜeniu od 0,5 % do 1,5 %.

38
Elektroforeza DNA w Ŝelach agarozowych
Droga migracji DNA w Ŝelu, w pewnym przedziale, jest w przybliŜe-niu wprost proporcjonalna do wielkości rozdzielanych fragmentów. Górnagranica tego przedziału zaleŜy od zastosowanej techniki elektroforetycznejoraz rodzaju i stęŜenia Ŝelu. Prawidłowe dobranie tych parametrów pozwalana uzyskanie dobrego rozdziału cząsteczek DNA o wielkości od kilku, doponad miliona nukleotydów, co odpowiada całym chromosomom organi-zmów eukariotycznych.
W Ŝelu poliakrylamidowym łatwo rozróŜnialne są fragmenty DNAróŜniące się wielkością o jeden nukleotyd. śel poliakrylamidowy uŜywanyjest do rozdziału cząsteczek o wielkości do około 500 nukleotydów. PowyŜejtej wielkości najczęściej stosuje się Ŝel agarozowy. ZaleŜność między jegostęŜeniem a wielkością cząsteczek które mogą być rozdzielane podano w Ta-beli 5. Praktyczne operowanie Ŝelem agarozowym o stęŜeniu poniŜej 0,5 %jest juŜ bardzo kłopotliwe, ze względu na jego małą wytrzymałość mecha-niczną.
Tabela 5. Wpływ stęŜenia agarozy na rozdzielczość Ŝelu
StęŜenie agaro-zy (%)
Wielkość rozdziela-nych cząsteczek (kb)
0,3 5,0 - 600,6 1,0 - 200,7 0,8 - 100,9 0,5 - 71,2 0,4 - 61,5 0,2 - 32,0 0,1 - 2
W latach osiemdziesiątch rozwinięto kilka technik elektroforezy pul-sacyjnej (FIGE, OFAGE i CHEF) pozwalających na rozdział w Ŝelach agaro-zowych, cząsteczek o wielkości ponad 1000 kbp. Stosując te techniki roz-dzielono i wypreparowano, między innymi, wszystkie chromosomy Saccha-romyces cerevisiae o wielkościach od 223 do 1640 kbp.
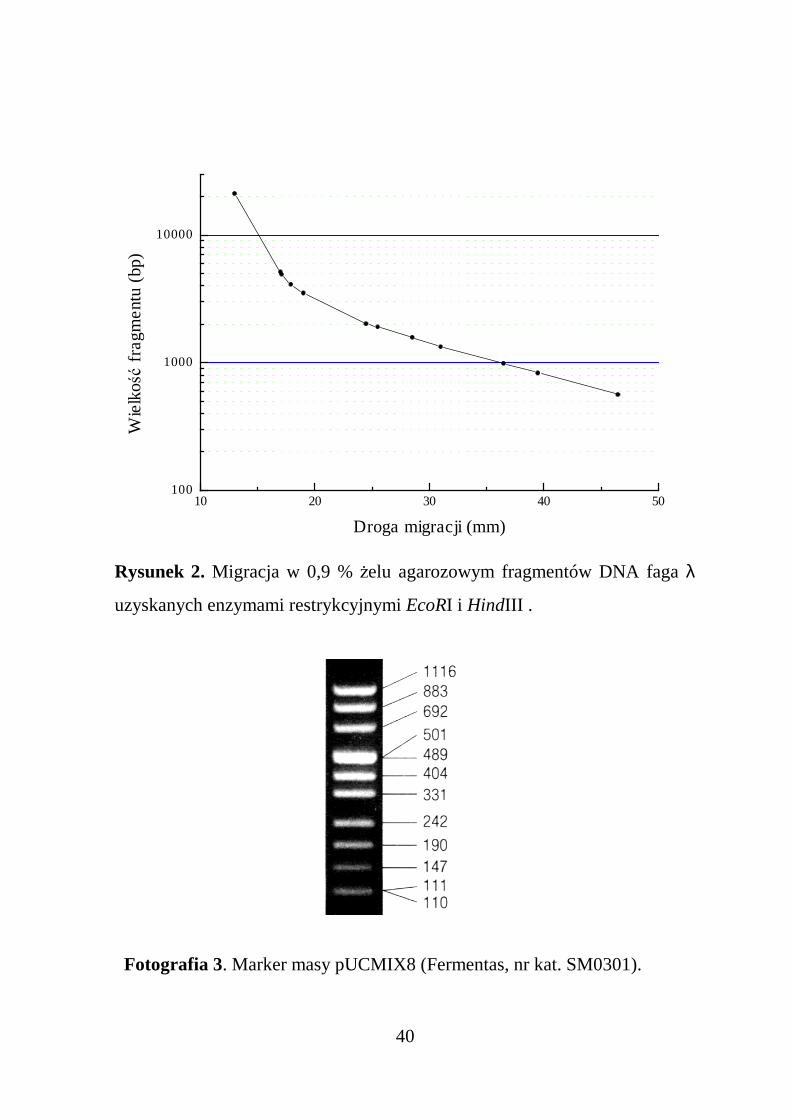
Masę fragmentów DNA wyznacza się migrując badaną próbkę wobecności wzorca, którym najczęściej jest DNA faga λ pocięty odpowiedniodobranymi enzymami restrykcyjnymi (Tabela 6). Dla wzorca sporządza siękrzywą kalibracyjną zaleŜności drogi migracji od logarytmu masy (Rysu-nek 2). W przedziale do około 3 kb jest ona linią prostą. Z wykresu, po zmie-rzeniu drogi migracji badanego fragmentu, odczytać moŜna jego masę.
39
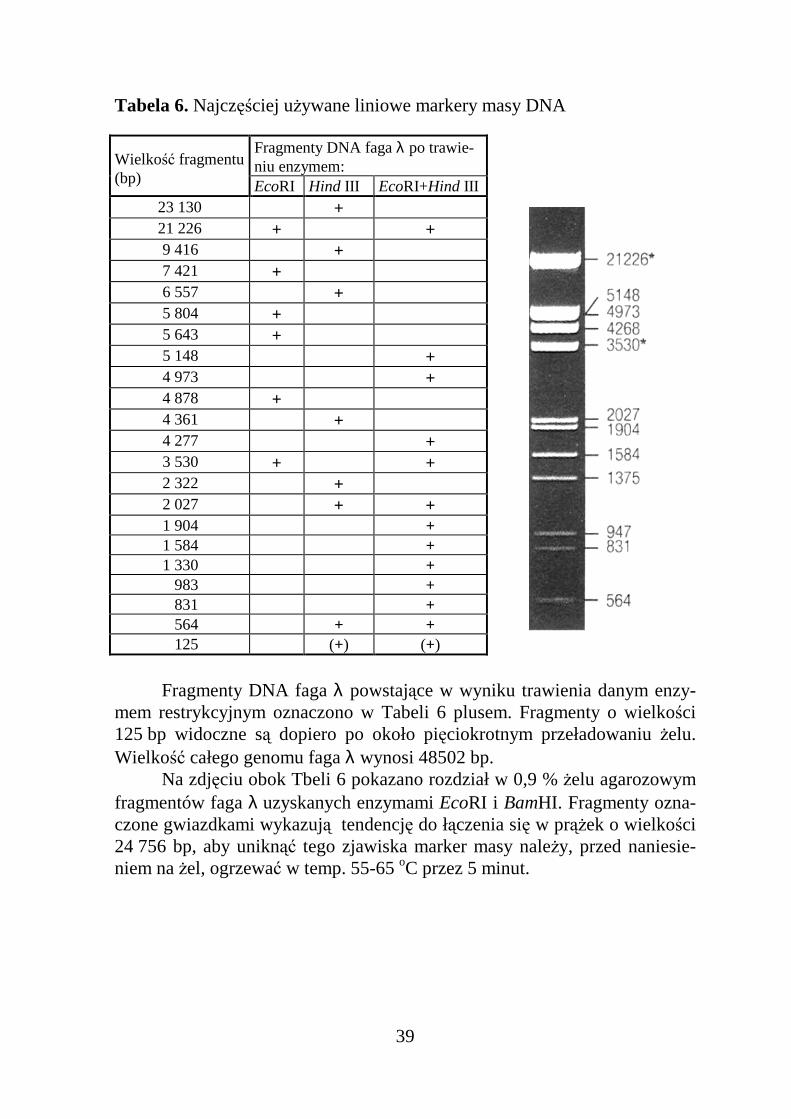
Tabela 6. Najczęściej uŜywane liniowe markery masy DNA
Fragmenty DNA faga λ po trawie-niu enzymem:Wielkość fragmentu
(bp) EcoRI Hind III EcoRI+Hind III23 130 +21 226 + +
9 416 + 7 421 + 6 557 + 5 804 + 5 643 + 5 148 + 4 973 + 4 878 + 4 361 + 4 277 + 3 530 + + 2 322 + 2 027 + + 1 904 + 1 584 + 1 330 + 983 + 831 + 564 + + 125 (+) (+)
Fragmenty DNA faga λ powstające w wyniku trawienia danym enzy-mem restrykcyjnym oznaczono w Tabeli 6 plusem. Fragmenty o wielkości125 bp widoczne są dopiero po około pięciokrotnym przeładowaniu Ŝelu.Wielkość całego genomu faga λ wynosi 48502 bp.
Na zdjęciu obok Tbeli 6 pokazano rozdział w 0,9 % Ŝelu agarozowymfragmentów faga λ uzyskanych enzymami EcoRI i BamHI. Fragmenty ozna-czone gwiazdkami wykazują tendencję do łączenia się w prąŜek o wielkości24 756 bp, aby uniknąć tego zjawiska marker masy naleŜy, przed naniesie-niem na Ŝel, ogrzewać w temp. 55-65 oC przez 5 minut.
40
10 20 30 40 50100
1000
10000
Wie
lkość
fra
gme
ntu
(bp)
Droga migracji (mm)
Rysunek 2. Migracja w 0,9 % Ŝelu agarozowym fragmentów DNA faga λ
uzyskanych enzymami restrykcyjnymi EcoRI i HindIII .
Fotografia 3. Marker masy pUCMIX8 (Fermentas, nr kat. SM0301).

41
Wpływ ilo ści DNA i czasu trwania elektroforezy na rozdział frag-mentów faga λλλλ w Ŝelu agarozowym
Fotografia 4. Wpływ ilości DNA na rozdział fragmentów w Ŝelu agarozo-wym. Fragmenty uzyskane w wyniku trawienia DNA faga λ enzymami Eco-RI i HindIII rozdzielono w 0,9 % Ŝelu agarozowym w buforze TBE. Czastrwania elektroforezy 2 godz. przy napięciu 100 V. Na poszczególne ścieŜkinaniesiono po: 0,5 µg, 1 µg, 2 µg i 4 µg DNA. Kolejno od góry widoczne sąfragmenty o następujących masach wyraŜonych w parach nukleotydów (bp):21226; 5148 i 4973 (te fragmenty w omawianych warunkach nie rozdzielająsię); 4277; 3530; 2027; 1904; 1548; 1330; 983; 831; 564 i 125 (fragment tennie jest widoczny).
Fotografia 5. Wpływ czasu trwania elektroforezy na rozdział fragmentówDNA w Ŝelu agarozowym. Ten sam Ŝel co na Fot. 1 ale po 4 godzinach elek-troforezy, pozostałe parametry nie zostały zmienione. Fragmenty 5148 i 4973nadal zlewają się ze sobą, podczas gdy prąŜki 2027 i 1904 zostały dobrzerozdzielone. Fragmenty 564 i 125 wypłynęły juŜ z Ŝelu.

42
Elektroforeza DNA w agarozie o niskim punkcie topnienia
Ogólne reguły postępowania
• Elektroforezę naleŜy wykonać bez bromku etydyny, który działa niszczą-
co na DNA migrujące w Ŝelu. Po zakończeniu elektroforezy Ŝel barwi się
przez 3 do 5 min. w roztworze bromku etydyny o stęŜeniu 0,5 µg/ml.
• Czas naświetlania Ŝelu promieniowaniem UV powinien być tak krótki jak
tylko to moŜliwe. W zasadzie nie powinien przekraczać 1 min. O ile to
moŜliwe naleŜy pracować przy obniŜonej mocy lampy, przy długości fali
300 nm lub więcej. Zbyt długa ekspozycja i krótki UV powodują pękanie i
(lub) dimeryzację DNA co obniŜa wydajność reakcji enzymatycznych z
jego udziałem.
• NaleŜy unikać przeładowania Ŝelu powodującego rozmycie prąŜka DNA.
Rozmyty prąŜek zajmuje duŜą objętość agarozy co jest niekorzystne w
dalszych etapach pracy. Przy wycinaniu prąŜka z Ŝelu naleŜy maksymalnie
pozbyć się agarozy nie zawierającej DNA.
• Przy topieniu agarozy zawierającej DNA nie wolno przekroczyć temp.
65oC. WyŜsza temperatura moŜe spowodować denaturację DNA i obniŜyć
wydajność ligacji.

43
Rozdział i odzyskanie DNA z agarozy LMP
1. Nastawić trawienie badanego DNA wybranym enzymem restrykcyjnym.
2. W trakcie trawienia przygotować 0,7 % Ŝel z agarozy o niskim punkcie
topnienia (agaroza SeaPlaque GTG) w buforze TAE.
Do Ŝelu i do buforu do elektroforezy nie dodawać bromku etydyny !
3. Strawione DNA nanieść na Ŝel i przeprowadzić elektroforezę.
4. śele sporządzone na buforze TAE łatwo przegrzewają się, a w skrajnych
przypadkach nawet topią. Aby tego uniknąć nie naleŜy przekraczać na-
pięcia 100 V, a aparat do elektroforezy najlepiej umieścić w lodówce.
5. Po zakończeniu elektroforezy przenieść Ŝel do buforu TAE z dodatkiem
bromku etydyny w ilości 0,5 µg/ml. Po upływie około 5 min. obejrzeć
Ŝel pod lampą UV. JeŜeli prąŜki będą juŜ widoczne, natychmiast przystą-
pić do ich wycinania, w przeciwnym wypadku Ŝel ponownie umieścić w
roztworze bromku etydyny na około 3 min.
6. Wycięty fragment Ŝelu pokroić na drobne kawałeczki i przenieść do czy-
stej, sterylnej i uprzednio zwaŜonej probówki.
7. ZwaŜyć probówkę wraz z zawartością. Obliczyć objętość agarozy przyj-
mując, Ŝe 1g agarozy zajmuje objętość 1 ml.
Wycięty fragment agarozy moŜe być przechowywany przez dłuŜszy czas
w temp. 4oC bez dostępu światła.

44
8. Do probówki z Ŝelem agarozowym dodać około 5 objętości buforu TE
(10 mM Tris-HCl, 1 mM EDTA, pH 8,0) i umieścić w łaźni wodnej o
temp. 65oC.
9. Po pięciu minutach inkubacji sprawdzić czy Ŝel uległ całkowitemu sto-
pieniu. Jeśli tak, to przystąpić do następnego punktu, w przeciwnym
przypadku przedłuŜyć inkubację.
10. Schłodzić zawartość probówki do temperatury pokojowej.
11. Do probówki dodać jedną objętość fenolu i całość energicznie wymie-
szać.
12. Probówkę z emulsją wodno-fenolową wirować przez 5 min. Agarozazbiera się na granicy faz w postaci białawego „koŜucha”.
13. Pipetą zebrać fazę wodną i przenieść do czystej probówki. Przy zbieraniuszczególną uwagę naleŜy zwrócić na to, by nie pobrać agarozy znajdują-cej się na granicy faz. W razie wystąpienia duŜej ilości agarozy w inter-fazie powtórzyć punkty 11 - 13 w przeciwnym przypadku przejść od razudo punktu 14.
14. Do fazy wodnej dodać dwie objętości eteru. Probówkę dokładnie za-mknąć i energicznie wytrząsać do wytworzenia jednorodnej emulsji.
15. Probówkę z emulsją wodno-eterową wirować przez 5 min.
16. Posługując się pompką wodną odessać fazę eterową wraz z interfazą. Dlazminimalizowania strat naleŜy unikać zasysania fazy wodnej.
17. Powtórzyć postępowanie opisane w punktach od 14 do 16 po czymprzejść do punktu 18.
18. Do fazy wodnej pozostałej po dwukrotnej ekstrakcji eterem dodać 0,1objętości 3 M octanu sodu (pH 5,2) i 2 objętości 96 % zimnego (-20oC)etanolu. Całość wymieszać przez odwracanie probówki.

45
19. Inkubować w temp. -20oC przez 10 do 20 min.
20. Odwirować wytrącone DNA (10 min, 14 000 obr/min, 4oC).
21. OstroŜnie, tak aby nie naruszyć osadu, usunąć supernatant. Następnie doprobówki dodać 1 ml 70 % etanolu.
22. Wirować przez 1 min.
23. Supernatant usunąć znad osadu przy pomocy pompki próŜniowej. ZebraćmoŜliwie wszystkie kropelki osadzone na ściance probówki.
24. Osad wysuszyć w suszarce próŜniowej z wirującym rotorem (speed vac)
i rozpuścić w 4 µl wody.
Materiały:
− agaroza SeaPlaque GTG (FMC BioProducts)
− bufor TAE 10 x stęŜony (Tris 60,57 g, octan sodu bezwodny 16,4 g,
EDTA 7,44 g, woda do objętości 1000 ml, pH 8,0)
− bromek etydyny 10 mg/ml
− bufor TE (10 mM Tris-HCl, 1 mM EDTA, pH 7,5)
− fenol (nasycony roztwór fenolu w 0,1 M Tris-HCl (pH 8,0) z 0,1 % dodat-
kiem 8-hydroksychinoliny)
− 3 M octan sodu, pH 5,2
− eter etylowy
− 70 % etanol
− 96 % etanol
− jałowa woda miliQ
− barwnik do nanoszenia na Ŝel 5 x stęŜony (ficoll 400 15 %, błękit bromo-
fenolowy 0,15 %)

46
Defosforylacja wektora
W obecności ligazy liniowa forma wektora, uzyskana działaniem en-
zymu restrykcyjnego, ma większą zdolność do odtworzenia swojej pierwot-
nej postaci niŜ do połączenia się z inną cząsteczką DNA. Aby zapobiec temu
zjawisku na rozcięty wektor działa się alkaliczną fosfatazą usuwającą grupy
fosforanowe z 5’ końca. Dla zapewnienia optymalnych warunków w miejsce
buforu dla enzymu restrykcyjnego wprowadza się bufor dla fosfatazy. Po de-
fosforylacji konieczne jest ponowne oczyszczenie DNA w celu usunięcia
fosfatazy i jej buforu.
1. Do probówki z DNA strawionym enzymem restrykcyjnym dodać
0,1 objętości 3 M octanu sodu (pH 5,2) i 2 objętości 96 % zimnego
(-20oC) etanolu.
2. Inkubować w temp. -20oC przez 10 do 20 min
3. Wytrącone DNA odwirować (15 min., 14 000 obr., 4oC).
4. OstroŜnie, tak aby nie naruszyć osadu, usunąć supernatant. Następnie
dodać do probówki 1 ml 70 % etanolu.
5. Wirować przez 1 min.
6. Przy pomocy pompki próŜniowej usunąć supernatant znad osadu. Zebrać
moŜliwie wszystkie kropelki osadzone na ściance probówki.
7. Osad wysuszyć w suszarce próŜniowej z wirującym rotorem (speed vac).
8. Osad zawiesić w 49 µl buforu do defosforylacji

47
9. Dodać 1 U alkalicznej fosfatazy z jelit cielęcia (CIAP)
10. Inkubować w temp. 37oC przez 30 min.
11. Po zakończeniu inkubacji do probówki dodać jedną objętość fenolu icałość energicznie wymieszać.
12. Probówkę z emulsją wodno-fenolową wirować przez 5 min.
13. Pipetą zebrać fazę wodną i przenieść do czystej probówki. Przy zbieraniuszczególną uwagę naleŜy zwrócić na to, by nie pobrać zanieczyszczeńznajdujących się na granicy faz.
14. Do fazy wodnej dodać dwie objętości eteru. Probówkę dokładnie za-mknąć i energicznie wytrząsać do wytworzenia jednorodnej emulsji.
15. Probówkę z emulsją wodno-eterową wirować przez 5 min.
16. Posługując się pompką wodną odessać fazę eterową wraz z interfazą. Dlazminimalizowania strat naleŜy unikać zasysania fazy wodnej.
17. Powtórzyć postępowanie opisane w punktach od 14 do 16 i przejść dopunktu 18.
18. Do fazy wodnej pozostałej po dwukrotnej ekstrakcji eterem dodać 0,1objętości 3 M octanu sodu (pH 5,2) i 2 objętości 96 % zimnego (-20oC)etanolu. Całość wymieszać przez odwracanie probówki.
19. Inkubować w temp. -20oC przez 10 do 20 min
20. Wytrącone DNA odwirować (10 min, 14 000 obr/min, 4oC).
21. OstroŜnie, tak aby nie naruszyć osadu, usunąć supernatant. Następnie doprobówki dodać 1 ml 70 % etanolu.
22. Wirować przez 1 min.
23. Supernatant usunąć znad osadu przy pomocy pompki próŜniowej. ZebraćmoŜliwie wszystkie kropelki osadzone na ściance probówki.

48
24. Osad wysuszyć w suszarce próŜniowej z wirującym rotorem (speed vac)
i zawiesić w 4 µl wody.
Materiały:
− 3 M octanu sodu, pH 5,2
− 96 % etanol
− 70 % etanol
− bufor do defosforylacji 10 x stęŜony (PROMEGA): 500 mM Tris-HCl (pH
9,3), 10 mM MgCl2, 10 mM ZnCl2, 10 mM spermidyna
Dla porównania skład 10 x stęŜonego buforu E (Promega): 6 mM Tris-HCl (pH 7,5), 6 mM MgCl2, 100 mM NaCl
− alkaliczna fosfataza (CIAP, PROMEGA)
− nasycony roztwór fenolu w 0,1 M Tris-HCl (pH 8,0) z 0,1 % dodatkiem
8-hydroksychinoliny
− eter
− jałowa woda destylowana

49
Ligacja
1. Przygotować roztwór do ligacji przenosząc do jednej probówki Eppen-
dorfa następujące składniki:
DNA wektora 4 µl
DNA wstawki 4 µl
bufor do ligacji 10 x stęŜ. 1 µl
T4 DNA ligaza 1 µl (1 U, Weiss)
Całkowita objętość: 10 µl
2. Wszystkie składniki dokładnie wymieszać. Probówkę inkubować przez
noc w temperaturze 15oC. Po tym czasie przystąpić do transformacji lub
zamrozić w -20oC.
Materiały:
- bufor do ligacji 10 x stęŜ. (660 mM Tris-HCl (pH 7,5), 50 mM MgCl2, 10
mM DTE, 10 mM ATP)

50
Elektrotransformacja
Przygotowanie DNA do elektrotransformacji
Roztwór DNA, który ma być uŜyty do elektrotransformacji powinien w
minimalnym stopniu zaburzać przewodność elektryczną transformowanej za-
wiesiny komórek. Bufory do ligacji i trawień restrykcyjnych nie spełniają tego
warunku, dlatego teŜ, wraz z zawartym w nich DNA, wymagają około 5-cio
krotnego rozcieńczenia przed elektroporacją. Sposób ten, aczkolwiek bardzo
szybki i prosty, eliminuje około 80 % materiału. Aby uniknąć tej straty, moŜna
wytrącić DNA etanolem z roztworu reakcyjnego (patrz rozdział „Zagęszczanie
roztworów kwasów nukleinowych przez wytrącanie”) i po zawieszeniu w wo-
dzie wykorzystać w całości.
Jeśli jednak dysponuje się membranami o wielkości por 0,025 µm to
najlepszą metodą z wyboru jest mikrodializa (patrz rozdział „Mikrodializa”).
Przygotowane tą drogą DNA doskonale nadaje się do elektroporacji.

51
Przygotowanie elektrokompetentnych komórek Escherichia coli
1. Dwa mililitry poŜywki LB (bez ampicyliny) zaszczepić szczepem E. coli
JM109 pobranym bezpośrednio z kolekcji utrzymywanej w temp. -70oC.
2. Inkubować przez noc w temp. 37oC z wytrząsaniem
3. Do 100 ml poŜywki LB o temp. 37oC dodać 1 ml prekultury
4. Inkubować w temp. 37oC z wytrząsaniem do osiągnięcia gęstości
optycznej OD600 = 0,6 (dla szczepu JM109 trwa to zwykle około dwie i pół
godziny)
5. Hodowlę schłodzić przez 5 min. w lodzie, a następnie przenieść do dwóch
probówek typu „FALCON”
6. Hodowlę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
7. Pierwsze przemycie
− do osadu komórkowego w obu probówkach dodać po około 5 ml
zimnej, jałowej wody miliQ, a następnie posługując się
miniwytrząsarką doprowadzić do zawieszenia komórek
− obie probówki dopełnić wodą do objętości około 50 ml i zawartość
dokładnie wymieszać
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
8. Drugie przemycie

52
− do osadów komórkowy w obu probówkach dodać po około 5 ml
zimnej, jałowej wody miliQ
− osady zawiesić w dodanej wodzie, a następnie przenieść do jednej
probówki FALCON i dopełnić wodą do objętości około 50 ml
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
9. Trzecie przemycie
− osad zawiesić w około 5 ml zimnej, jałowej wody miliQ po czym
uzupełnić wodą do objętości 50 ml
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
10. Czwarte przemycie
− komórki zawiesić w około 5 ml zimnego, jałowego 10 % glicerolu po
czym dopełnić probówkę glicerolem do objętości 50 ml
− zawiesinę komórek odwirować w temp. 4oC (7000 rpm, 10 min.),
supernatant odrzucić
11. Osad bakteryjny zawiesić w około 250 µl oziębionego w lodzie, jałowego
10 % glicerolu
Uzyskane komórki elektrokompetentne naleŜy moŜliwie szybko uŜyć do
transformacji lub, po podzieleniu na małe porcje, zamrozić w temp. -70oC.
Komórki mogą być w tym stanie przechowywane nawet przez kilka lat bez
utraty swoich właściwości. NaleŜy jedynie przestrzegać zasady, Ŝe porcja
komórek raz rozmroŜona nie moŜe być ponownie zamraŜana.

53
Elektrotransformacja Escherichia coli
1. Probówkę z 10 µl roztworu DNA umieścić w lodzie i przenieść do niej 40
µl elektrokompetentnych komórek E. coli. Całość dokładnie wymieszać i
inkubować w lodzie przez 1 min.
Jako kontrolę przygotować probówkę zawierającą 40 µµµµl komórek
elektrokompetentnych i 10 µµµµl wody. Z próbą kontrolną postępować w
dalszym ciągu doświadczenia jak z próbą zawierającą DNA.
2. Zawiesinę komórek przenieść do schłodzonej kuwety do elektroporacji.
Zawiesina musi się stykać z oboma ściankami metalowymi kuwety i nie
moŜe zawierać pęcherzyków powietrza.
3. Napełnione kuwety chłodzić przez 1 min w lodzie
4. Ustawić następujące parametry elektroporacji:
− napięcie 1250 V (dla kuwety o odległości ścianek 0,1 cm daje to
natęŜenie pola 12,5 kV/cm)
− pojemność kondensatora 25 µF
− opór zewnętrzny 200 omów
Dla tych parametrów stała czasowa odczytana po wyzwoleniu impulsu
powinna mieścić się w przedziale od 3 do 5 msec.
5. Umieścić kuwetę w komorze do elektroporacji i po zamknięciu pokrywy
wyzwolić impuls prądowy naciskając jednocześnie oba przyciski PULSE
do momentu usłyszenia sygnału dźwiękowego.

54
6. Po usłyszeniu sygnału zwolnić przyciski. Wyjąć kuwetę z komory i
natychmiast dodać do niej 950 µl poŜywki SOC w taki sposób aby
zawiesina komórek bakteryjnych została dobrze wymieszana.
7. Zanotować rzeczywiste parametry elektroporacji (napięcie, opór próbki i
stałą czasowa).
8. Zawiesinę komórek przenieść do probówki Eppendorfa i inkubować przez
40 min. w temp. 37oC z wytrząsaniem.
9. Wysiać po 100 µl zawiesiny na dwie szalki Petriego z podłoŜem
selekcyjnym (poŜywka LB z ampicyliną, X-gal i IPTG - patrz instrukcja
„PodłoŜe selekcyjne z X-gal i IPTG”).
10. Pozostałe 800 µl zawiesiny wirować przez 3 min. Odrzucić 600 µl
supernatantu, osad bakteryjny zawiesić w pozostałych 200 µl i wysiać po
100 µl na dwie kolejne szalki Petriego z podłoŜem selekcyjnym (LB z
ampicyliną, X-gal i IPTG).
11. Probówkę z zawiesiną kontrolną wirować przez 3 min. Odrzucić 600
µµµµl supernatantu, osad zawiesić w pozostałych 400 µµµµl. Wysiać po 100 µµµµl
na dwie szalki Petriego z podłoŜem selekcyjnym (LB z ampicyliną, X-
gal i IPTG) oraz na dwie z takim samym podłoŜem ale bez ampicyliny.
12. Szalki inkubować w temp. 37oC do uzyskania wyraŜnego wzrostu. UnikaćprzedłuŜonej inkubacji.
Materiały
− poŜywka LB (wyciąg droŜdŜowy 1 %, trypton 1 %, NaCl 0,5 %)
− poŜywka SOC (poŜywka SOB z 20 mM dodatkiem glukozy)

55
− poŜywka SOB (trypton 2,0 %, wyciąg droŜdŜowy 0,5 %, NaCl 0,5 %,
KCl 0,2 %, pH 6,8 do 7,0).
Składniki poŜywki SOB jałowi się w autoklawie i po ostudzeniu
dodaje sterylnie 1M MgCl2 i 1 M MgSO4 w takiej ilości aby
stęŜenie końcowe tych soli wynosiło po 10 mM.
− 10 % jałowy glicerol
Przygotowuje się przez rozcieńczenie handlowego glicerolu jałową
wodą miliQ. Glicerol naleŜy wyjałowić przez sączenie przez sączek
bakteriologiczny (0,22 µm) lub w autoklawie.
− jałowa woda miliQ

56
Przygotowanie elektrokompetentnych komórek Saccharomyces cerevisiae
1. Dwa mililitry poŜywki YPG zaszczepić szczepem S. cerevisiae FY73
pobranym bezpośrednio z kolekcji utrzymywanej w temp. -70oC.
2. Inkubować przez noc w temp. 28oC z wytrząsaniem
3. Do 50 ml poŜywki YPG o temp. 28oC dodać 0,5 ml prekultury
4. Inkubować w temp. 28oC z wytrząsaniem do osiągnięcia gęstości
optycznej OD600 = 0,6 - 1 (dla szczepu FY73 trwa to zwykle około 20
godzin)
5. Hodowlę inkubować przez 5 min. w lodzie
6. Hodowlę odwirować w probówce typu „FALCON” w temp. 4oC (7000
rpm, 10 min.), supernatant odrzucić
7. Pierwsze przemycie
− komórki zawiesić w około 5 ml zimnej, jałowej wody miliQ, a
następnie probówkę dopełnić wodą do objętości około 50 ml
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
8. Drugie przemycie
− komórki zawiesić w około 5 ml zimnej, jałowej wody miliQ, a
następnie probówkę dopełnić wodą do objętości około 50 ml
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić

57
9. Trzecie przemycie
− komórki zawiesić w około 5 ml zimnej, jałowej wody miliQ, a
następnie probówkę dopełnić wodą do objętości około 50 ml
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
10. Czwarte przemycie
− komórki zawiesić w około 5 ml zimnego, jałowego 1 M sorbitolu, a
następnie probówkę dopełnić sorbitolem do objętości około 50 ml
− zawiesinę odwirować w temp. 4oC (7000 rpm, 10 min.), supernatant
odrzucić
11. Osad droŜdŜowy zawiesić w około 250 µl oziębionego w lodzie, jałowego
1 M sorbitolu
Uzyskane komórki elektrokompetentne naleŜy moŜliwie szybko uŜyć do
transformacji lub, po podzieleniu na małe porcje, zamrozić w temp. -70oC.
Komórki mogą być w tym stanie przechowywane przez kilka lat bez utraty
swoich właściwości. NaleŜy jedynie przestrzegać zasady, Ŝe porcja komórek
raz rozmroŜona nie moŜe być ponownie zamraŜana.

58
Elektrotransformacja Saccharomyces cerevisiae
1. Probówkę z 10 µl roztworu DNA umieścić w lodzie i przenieść do niej
40 µl elektrokompetentnych komórek S. cerevisiae. Całość dokładnie wy-
mieszać i inkubować w lodzie przez 1 min.
Jako kontrolę przygotować probówkę zawierającą 40 µµµµl komórek
elektrokompetentnych i 10 µµµµl wody. Z próbą kontrolną postępować w
dalszym ciągu doświadczenia jak z próbą zawierającą DNA.
2. Zawiesinę komórek przenieść do schłodzonych kuwet do elektroporacji.
Zawiesina musi się stykać z oboma ściankami metalowymi kuwety i nie
moŜe zawierać pęcherzy powietrza.
3. Napełnione kuwety chłodzić przez 1 min w lodzie
4. Ustawić następujące parametry elektroporacji:
− napięcie 600 V (dla kuwety o odległości ścianek 0,1 cm daje to
natęŜenie pola 6 kV/cm)
− pojemność kondensatora 25 µF
− opornik zewnętrzny 200
Dla tych parametrów stała czasowa odczytana po wyzwoleniu impulsu
powinna mieścić się w przedziale od 3 do 5 msec.
5. Umieścić kuwetę w komorze do elektroporacji i po zamknięciu pokrywy
wyzwolić impuls prądowy naciskając jednocześnie oba przyciski PULSE
do momentu usłyszenia sygnału dźwiękowego.

59
6. Po usłyszeniu sygnału zwolnić przyciski. Wyjąć kuwetę z komory i
natychmiast dodać do niej 950 µl 1 M sorbitolu w taki sposób aby za-
wiesina komórek droŜdŜowych została dobrze wymieszana.
7. Zanotować rzeczywiste parametry elektroporacji (napięcie, opór próbki i
stałą czasową).
8. Zawiesinę komórek przenieść do 1,5 ml probówki Eppendorfa.
9. Zawiesinę odwirować przez 1 min. Odrzucić 800 µl supernatantu, osad
droŜdŜowy zawiesić w pozostałych 200 µl. Wysiać po 100 µl zawiesiny
na dwie szalki Petriego z podłoŜem selekcyjnym (G0 z uracylem).
10. Probówkę z zawiesiną kontrolną wirować przez 1 min. Odrzucić 600
µµµµl supernatantu, osad zawiesić w pozostałych 400 µµµµl. Wysiać po 100 µµµµl
na dwie szalki Petriego z podłoŜem selekcyjnym (G0 z uracylem) oraz
na dwie z takim samym podłoŜem, ale z dodatkiem histydyny.
11. Szalki inkubować w temp. 28oC do uzyskania wyraźnego wzrostu, co
zwykle trwa 3 do 7 dni.
Materiały
− poŜywka YPG (wyciąg droŜdŜowy 1 %, bactopepton 1 %, glukoza 2 %)
− poŜywka G0 (yeast nitrogen base 0,67 %, glukoza 2 %, agar 2%). PoŜyw-
kę, po wyjałowieniu w autoklawie, uzupełnia się stosownie do potrzeb od-
dzielnie przygotowanymi roztworami aminokwasów, tak aby ich końcowe
stęŜenie wynosiło po 20 do 50 µg/ml.− 1 M sorbitol
− jałowa woda miliQ

60
Chemiczna transformacja Saccharomyces cerevisiae
Przygotowanie komórek droŜdŜowych do transformacji chemicznej
1. Dwa mililitry poŜywki YPG zaszczepić szczepem S. cerevisiae FY73
pobranym bezpośrednio z kolekcji utrzymywanej w temp. -70oC.
2. Inkubować przez noc w temp. 28oC z wytrząsaniem.
3. Do 50 ml poŜywki YPG o temp. 28oC dodać 0,5 ml prekultury.
4. Inkubować w temp. 28oC z wytrząsaniem do osiągnięcia gęstości optycznej
OD600 = 0,6 - 1 (dla szczepu FY73 trwa to zwykle około 20 godzin).
5. Dwa razy po 1,5 ml hodowli przenieść do dwóch probówek Eppendorfa
(ilość na jedną transformację) i wirować przez 1 min. Przygotować
równieŜ dodatkową porcję komórek do transformacji kontrolnej.
6. Supernatant z obu probówek odrzucić. Osady komórkowe zawiesić w
500 µl jałowej wody destylowanej i przenieść do jednej probówki.
7. Zawiesinę wirować przez 1 min., supernatant odrzucić.
8. Komórki zawiesić w 1000 µl 100 mM LiAc i inkubować przez 10 min. w
temp. 30oC.
9. Po zakończeniu inkubacji zawiesinę wirować przez 2 min., supernatant
odrzucić. Uzyskane w ten sposób droŜdŜowe komórki uŜyć do trans-
formacji.

61
Transformacja Saccharomyces cerevisiae
1. DroŜdŜowe komórki kompetentne (punkt 9 powyŜszej instrukcji) zawie-sić w 100 µl świeŜo przygotowanego roztworu do transformacji.
2. Do zawiesiny komórek droŜdŜowych w roztworze do transformacji dodaćokoło 10 µl wybranego roztworu DNA.
Jako kontrolę negatywną przygotować dodatkową probówkęzawierającą 100 µµµµl komórek kompetentnych i 10 µµµµl wody. Z próbąkontroln ą postępować w dalszym ciągu doświadczenia jak z próbązawierającą DNA.
3. Zawiesinę inkubować 30 min. w temp. 45oC.
4. Do zawiesiny komórek dodać 900 µl jałowej wody destylowanej, całośćdokładnie wymieszać.
5. Zawiesinę wirować (5000 obr., 2 min.), usunąć około 800 µl supernatan-tu.
6. Komórki zawiesić w pozostałej objętości supernatantu.
7. Zawiesinę komórek rozgłaskać na szalce Petriego z odpowiednim podło-Ŝem selekcyjnym.
8. Płytki inkubować przez 3 do 7 dni w temp. 30oC.
Materiały
− poŜywka YPG (wyciąg droŜdŜowy 1 %, bactopepton 1 %, glukoza 2 %)− 100 mM octan litu (LiAc)− Roztwór do transformacji
2 M octan litu 100 µl50 % PEG 4000 800 µl1 M DTT 100 µl
− poŜywka G0 (yeast nitrogen base 0,67 %, glukoza 2 %, agar 2 %). PoŜyw-kę, po wyjałowieniu w autoklawie, uzupełnia się stosownie do potrzeb od-dzielnie przygotowanymi roztworami aminokwasów, tak aby ich końcowestęŜenie wynosiło po 20 do 50 µg/ml.
− jałowa woda miliQ

62
PodłoŜe selekcyjne z X-gal i IPTG
Na szalkę Petriego zawierającą poŜywkę LB z ampicyliną (70 µg/ml)
nanieść po 50 µl roztworów X-gal i IPTG. Roztwory natychmiast
rozprowadzić jałowym głaszczkiem po całej powierzchni poŜywki. Otwarte
szalki suszyć w komorze laminarnej przez 1 godzinę.
PoŜywka z X-gal i IPTG, ze względu na bardzo wysoką cenę tych
odczynników, powinna być przygotowywana bezpośrednio przed uŜyciem w
ilości dokładnie potrzebnej w danym doświadczeniu.
PoŜywka LB
wyciąg droŜdŜowy 1 %
trypton 1 %
chlorek sodu 0,5 %
agar 2 %
PoŜywkę jałowi się w autoklawie przez 20 min. w temp. 121oC (1 atn),
następnie schładza do temp. około 45-50oC i po dodaniu ampicyliny do
końcowego stęŜenia 70 µg/ml, wylewa na szalki Petriego. JeŜeli płytki mają
być natychmiast wykorzystane to suszy się je w komorze laminarnej przez 1
godzinę. Płytki z poŜywką LB i ampicyliną moŜna przechowywać w temp. 4oC
przez okres do jednego miesiąca, pod warunkiem, Ŝe są dobrze zabezpieczone
przed wysychaniem.
Ampicylina
− 1 % roztwór ampicyliny w wodzie destylowanej
Roztwór jałowi się sącząc przez 0,22 µm filtr, dzieli na małe porcje i
przechowuje w -20oC. Ampicylinę stosuje się najczęściej w stęŜeniach: 70
µg/ml do poŜywek stałych i 100 µg/ml do poŜywek płynnych.

63
X-gal
− 1 % roztwór X-gal w dwumetyloformamidzie
Roztworu nie trzeba jałowić. Przechowywać w -20oC po uprzednim
podzieleniu na małe porcje, chronić przed światłem.
IPTG
− 1 % roztwór IPTG w wodzie destylowanej
Roztwór wyjałowić przesączając przez 0,22 µm filtr, podzielić na małe
porcje, przechowywać w -20oC.

64
Selekcja transformantów Escherichia coli z wektorem zawiera-jącym droŜdŜowy gen HIS3
1. Do probówki zawierającej 2 ml poŜywki LB z ampicyliną (100 µg/ml)przenieść jedną, dobrze wyrośniętą, białą kolonię bakteryjną.
2. Hodowlę inkubować przez około 20 godzin w temp. 37oC z energicznymwytrząsaniem (150 - 160 cykli na minutę).
3. Do probówki Eppendorfa przenieść 1,5 ml hodowli i wirować przez 3min. Supernatant usunąć przy pomocy pompki próŜniowej zachowującosad bakteryjny.
4. Osad bakteryjny wymieszać ze 100 µl buforu TGE (50 mM Tris-HCl, 50mM glukoza, 10 mM EDTA, pH 8,0) tak, aby powstała jednolita, po-zbawiona grudek zawiesina, co jest warunkiem prawidłowego przebiegulizy.
5. Do zawiesiny bakterii dodać 150 µl świeŜo przygotowanego roztworu dolizy (1% SDS, 0,2 N NaOH), po czym całość dokładnie wymieszać przezkilkakrotne odwrócenie probówki do góry dnem. NaleŜy unikać gwał-townego potrząsania probówką. Liza komórek następuje natychmiast pododaniu roztworu SDS z NaOH i objawia się wzrostem lepkości orazprzejrzystości zawiesiny przy równoczesnym zaniku barwy beŜowej.
6. Probówkę z lizatem umieścić na 3 min. w lodzie.
7. Do lizatu dodać 120 µl 3M octanu sodu (pH 5,2) i wymieszać przez od-wracanie probówki do góry dnem. W lizacie powinny pojawić się biała-we "serowate" kłaczki wytrąconych białek.
8. Lizat z wytrąconymi białkami inkubować przez 10 min. w temp. -20oC.
9. Probówkę z lizatem wirować przez 15 min. (14 000 obr/min., 4oC). Wwyniku wirowania lizat powinien się rozdzielić na jasno beŜowy osad iśluzowaty, przejrzysty supernatant.
− jeŜeli rozdział nie jest dobry to wirowanie naleŜy powtórzyć (14 000obr/min., 4oC, 15 min.)

65
− w przypadku dobrego rozdziału supernatant naleŜy ostroŜnie zebraćpipetą znad osadu i przenieść do czystej probówki Eppendorfa.
10. Do zebranego supernatantu dodać jedną objętość fenolu i całość ener-gicznie wytrząsać do wytworzenia jednorodnej emulsji.
11. Probówkę z emulsją wodno-fenolową wirować przez 5 min. Zawartośćprobówki powinna rozdzielić się na dwie wyraźne fazy: fenolową (dol-na) i wodną (górna). Zanieczyszczenia białkowe gromadzą się w faziefenolowej i na granicy faz.
12. Pipetą zebrać fazę wodną i przenieść do czystej probówki. Przy zbieraniuszczególną uwagę naleŜy zwrócić na to, by nie pobrać zanieczyszczeńznajdujących się na granicy faz.
13. Do fazy wodnej dodać dwie objętości eteru. Probówkę dokładnie za-mknąć i energicznie wytrząsać do wytworzenia jednorodnej emulsji.
14. Probówkę z emulsją wodno-eterową wirować przez 5 min. Zawartośćprobówki powinna rozdzielić się na dwie wyraźne fazy: wodną (dolna) ieterową (górna). Zanieczyszczenia gromadzą się w fazie eterowej i nagranicy faz.
15. Posługując się pompką wodną odessać fazę eterową wraz z interfazą. Dlazminimalizowania strat naleŜy unikać zasysania fazy wodnej.
16. Powtórzyć postępowanie opisane w punktach od 13 do 15 i przejść dopunktu 17.
17. Do fazy wodnej pozostałej po dwukrotnej ekstrakcji eterem dodać jednąobjętość zimnego (-20oC) izopropanolu i całość wymieszać przez odwra-canie probówki. JeŜeli stęŜenie DNA w roztworze jest wysokie to po do-daniu izopropanolu pojawi się strąt w postaci pływającego kłaczka. Przymałym stęŜeniu DNA strąt moŜe być niewidoczny. Inkubacja w niskiejtemperaturze (-20oC) przyspiesza wytrącanie i poprawia jego wydajność.
18. Probówkę z zawartością inkubować w temp. -20oC przez 10 do 20 min.
19. Wytrącone DNA odwirować (10 min, 14 000 obr/min, 4oC).

66
20. OstroŜnie, tak aby nie naruszyć osadu, usunąć supernatant. Następnie doprobówki dodać 1 ml 70 % etanolu.
21. Wirować przez 1 min.
22. Supernatant usunąć znad osadu przy pomocy pompki próŜniowej. ZebraćmoŜliwie wszystkie kropelki osadzone na ściance probówki.
23. Osad wysuszyć w suszarce próŜniowej z wirującym rotorem (speedvac).
24. Wysuszony osad rozpuścić w 10 µl wody miliQ i dodać 1 µl roztworuRNazy o stęŜeniu 10 mg/ml.
25. Uzyskane preparaty plazmidowe oraz DNA kontrolne pociąć enzymamirestrykcyjnymi BamHI i PstI. W tym celu przygotować reakcje jak poda-no w Tabeli 7 i Tabeli 8.
Tabela 7. Trawienie plazmidów enzymem BamHI
Probówka A B C D DNA 2 µl pFL44S* 2 µl pD11** 2 µl transf. 1 2 µl transf. 2 bufor BamHI 1 µl 1 µl 1 µl 1 µl woda 6,2 µl 6,2 µl 6,2 µl 6,2 µl enzym BamHI 0,8 µl 0,8 µl 0,8 µl 0,8 µl
* plazmid pFL44S wyizolowany na pierwszych zajęciach** plazmid pD11 wyizolowany na pierwszych zajęciach
Tabela 8. Trawienie plazmidów enzymem PstI
Probówka E F G H DNA 2 µl pFL44S* 2 µl pD11** 2 µl transf. 1 2 µl transf. 2 bufor O 1 µl 1 µl 1 µl 1 µl woda 6,2 µl 6,2 µl 6,2 µl 6,2 µl enzym PstI 0,8 µl 0,8 µl 0,8 µl 0,8 µl
* plazmid pFL44S wyizolowany na pierwszych zajęciach** plazmid pD11 wyizolowany na pierwszych zajęciach
26. Dodatkowo przygotować kontrole nietrawionych plazmidów jak podanow Tabeli 9.

67
Tabela 9. Kontrole nietrawionych plazmidów
Probówka I J DNA 2 µl pFL44S* 2 µl pD11** bufor O 1 µl 1 µl woda 7 µl 7 µl enzym PstI - -
* plazmid pFL44S wyizolowany na pierwszych zajęciach** plazmid pD11 wyizolowany na pierwszych zajęciach
27. Wszystkie roztwory inkubować przez 2 godz. w temp. 37oC.
28. Po zakończeniu inkubacji do wszystkich probówek (od A do J) dodać po5 µl barwnika do nanoszenia na Ŝel.
29. Przygotowane próbki nanieść na 0,9 % Ŝel agarozowy i przeprowadzićelektroforezę w obecności markera o znanej masie cząsteczkowej.
30. Wykonać zdjęcie Ŝelu w przechodzącym świetle ultrafioletowym.
31. Na podstawie zdjęcia wybrać preparaty do transformacji droŜdŜy.
Materiały
− poŜywka LB (wyciąg droŜdŜowy 1 %, trypton 1 % , NaCl 0,5 %)− bufor TGE (50 mM Tris-HCl, 50 mM glukoza, 10 mM EDTA, pH 8,0)− roztwór do lizy (SDS 1%, NaOH 0,2 M)
Roztwór do lizy przygotowuje się bezpośrednio przed uŜyciemmieszając ze sobą równe objętości 2 % SDS i 0,4 M NaOH.
− 3 M octan sodu, pH 5,2− fenol (nasycony roztwór fenolu w 0,1 M Tris-HCl (pH 8,0) z 0,1 % dodat-
kiem 8-hydroksychinoliny)− eter− izopropanol− 70 % etanol− jałowa woda miliQ− RNaza 10 mg/ml− enzym restrykcyjny BamHI wraz z buforem Bam (Fermentas)

68
− enzym restrykcyjny PstI wraz z buforem O (Fermentas)− agaroza− marker masy - DNA faga λ trawiony enzymami EcoRI i HindIII− barwnik do nanoszenia na Ŝel, 5 x stęŜony (ficoll 400 15%, błękit bromo-
fenolowy 0,15 %)− bufor TBE, 10 x stęŜony (Tris 100,89 g, kwas borny 59 g, EDTA 9,35 g,
woda do objętości 1000 ml, pH 8,0)− bromek etydyny 10 mg/ml

69
Bezpośrednie wykazanie obecności genu HIS3 w
transformantach droŜdŜowych
Po transformacji fragmentami plazmidu skonstruowanego na ćwicze-
niach, szczep Sacharomyces cerevisiae FY73 (MATa, ura3-52, his3-∆200)
tworzy kolonie na podłoŜu minimalnym z uracylem (y.n.b. 0,67 %, glukoza 2
%, agar 2%, uracyl 50 µg/ml). Na tej podstawie moŜna wnioskować, Ŝe jeden z
fragmentów DNA uŜyty do transformacji zawiera prawidłową kopię genu hi-
stydynowego znoszącą efekt delecji his3-∆200. Hipotezę tą moŜna zweryfi-
kować przy pomocy primerów specyficznych dla genu HIS3 w reakcji PCR
z DNA genomowym transformantów uŜytym jako matryca. Z praktycznego
punktu widzenia całą procedurę podzielić moŜna na pięć etapów obejmują-
cych:
− zaprojektowanie primerów
− izolację DNA genomowego z uŜytego do transformacji szczepu S. cerevi-
siae FY73, transformantów oraz kontrolnego szczepu S. cerevisiae WY91
zawierającego dziką kopię genu HIS3
− reakcję PCR
− elektroforezę produktów uzyskanych w reakcji PCR
− interpretację wyniku elektroforezy i wnioski końcowe

70
PCR
Powielania DNA in vitro w łańcuchowej reakcji polimerazy znane
jest lepiej pod skrótem PCR, utworzonym z pierwszych trzech liter angiel-
skiej nazwy Polymerase Chain Reaction. Metoda PCR została opracowana
przez Kary Mullis’a (Specific synthesis of DNA in vitro via polymerase-
catalyzed chain reaction. Methods in Enzymology 155, 335-350, 1987) w
amerykańskiej firmie biotechnologicznej CETUS i początkowo była uŜywa-
na wyłącznie w badaniach podstawowych w dziedzinie genetyki molekular-
nej. Obecnie, po wprowadzeniu automatycznych termocyklerów, metoda ta
znalazła tak wiele zastosowań, Ŝe trudno jest znaleźć dziedzinę badań biolo-
gicznych w której by nie była stosowana.
Zasada metody PCR
Reakcje PCR przeprowadza się zwykle w 25 do 35 kolejno następują-
cych po sobie jednakowych cyklach. W kaŜdym cyklu wyodrębnić moŜna
trzy fazy, a mianowicie: denaturację, annealing oraz wydłuŜanie (Rysunek
4).
• Denaturacja polega na ogrzaniu badanego DNA do temperatury około
95oC powodującej rozdzielenie dwuniciowych cząsteczek na pojedyncze
nici. Następnie roztwór reakcyjny jest szybko chłodzony co umoŜliwia
przejście do fazy annealingu.
• Annealing jest to przyłączenie primerów do miejsc homologicznych w
badanym DNA zachodzące w temperaturze około 55 do 80oC. Dokładną

71
temperaturę annealingu ustala się na podstawie składu nukleotydowego
primerów. Na etapie annealingu PCR kierowany jest na jedną z dwóch
nawzajem wykluczających się dróg, a mianowicie:
− primer wykrywa w badanym materiale sekwencję homologiczną i łączy
się z nią tworząc dwuniciowy fragment DNA. Utworzona struktura jest
sygnałem dla polimerazy DNA do powielenia tego regionu. Powielony
w wielu cyklach DNA jest łatwy do zidentyfikowania w Ŝelu agarozo-
wym.
− primer nie wykrywa w badanym materiale sekwencji homologicznej, nie
moŜe więc utworzyć struktury dwuniciowej koniecznej dla zapoczątko-
wania aktywności polimerazy. Tym samym badany materiał nie zostanie
powielony, co równieŜ łatwo wykazać w elektroforezie kontrolnej.
• WydłuŜanie to faza PCR w której temperatura roztworu reakcyjnego pod-
noszona jest do 74oC, co umoŜliwia polimerazie rozpoznanie dwunicio-
wego kompleksu DNA docelowego oraz primera i wydajną syntezę no-
wych fragmentów DNA.
PCR jest metodą bardzo czułą. Teoretycznie moŜliwe jest wykazanie
obecności nawet jednej cząsteczki DNA. W praktyce przyjmuje się jednak,
Ŝe dolna granica czułości to kilkanaście cząsteczek w jednym mililitrze ba-
danego roztworu.

72
5’ 3’ Dwuniciowe DNA docelowe (poszukiwne) 3’ 5’
Denaturacja. Ogrzanie DNA w temp. 940C powoduje jego roz-dział na dwie nici.
5’ 3’ Cząsteczka DNA docelowego rozdzielona na
pojedyncze nici
3’ 5’
Annealing. Primery “wykrywają” sekwencje homologiczne wDNA docelowym i przyłączają się do nich. W przypadku brakuhomologii primery nie mają się do czego przyłączyć i dalszy prze-bieg procesu zostaje zatrzymany.
5’ 3’ 3’ 5’ Jednoniciowy DNA docelowy z primerami przyłączonymi do miejsc homologicznych. 5’ 3’ 3’ 5’
WydłuŜanie. Termostabilna polimeraza rozpoznaje dwuniciowefragmenty złoŜone z DNA docelowego oraz primerów i rozpoczy-na od nich syntezę nowych nici DNA.
5’ 3’ 3’ 5’ W pierwszym cyklu DNA docelowe zostaje podwojone, a kaŜda jego cząsteczka składa 5’ 3’ się z jednej “starej” i jednej “nowej” nici. 3’ 5’
Rysunek 4. Powielenie DNA docelowego w pierwszym cyklu PCR
Legenda
pojedyncza nić DNA docelowego primer nowo zsyntetyzowana nić DNA

73
Ogólne zasady projektowania starterów do PCR
Startery do PCR naleŜy tak projektować, aby w maksymalnym stopniu
spełniały podane poniŜej reguły.
• Długość startera: 18 - 30 nukleotydów
• Udział nukleotydów G i C: 40 - 60 %
• Temperatura denaturacji (topnienia) Tm:
− temperaturę denaturacji startera oblicza się według wzoru:
Tm = 2oC x (A+T) + 4oC x (G + C)
− wartości Tm pary starterów powinny być bardzo zbliŜone a najlepiej
jednakowe
− rzeczywista temperatura annealingu moŜe odbiegać od wyliczonej
wartości Tm zaleca się jednak aby była o 5oC niŜsza i mieściła się w
przedziale od 55 do 80oC
• Sekwencja nukleotydowa:
− naleŜy unikać komplementarności ostatnich 2 - 3 nukleotydów na 3’
końcach pary starterów
− naleŜy unikać ciągu trzech lub więcej nukleotydów G lub C na 3’
końcu
− naleŜy unikać nukleotydu T na 3’ końcu
− naleŜy unikać sekwencji komplementarnych wewnątrz startera i mię-
dzy parą starterów
• StęŜenie starterów:
− stęŜenie starterów w reakcji PCR moŜe wahać się w granicach od 0,1
do 0,5 µM (najczęściej uŜywa się po 0,2 µM kaŜdego startera)
− startery jednej pary zawsze stosuje się w jednakowych stęŜeniach

74
Startery do wykrywania droŜdŜowego genu HIS3
Startery his-p i his-k zostały zaprojektowane do wykrywania części
kodującej droŜdŜowego genu HIS3. PołoŜenie starterów względem tego genu
pokazano na Rysunku 5.
Starter his-p
Lokalizacja: 5’ koniec nici kodującej genu HIS3
Sekwencja:
5’ - TGA CAG AGC AGA AAG CCC TAG TAA AGC G - 3’
Udział nukleotydów GC: 48,3 %
Temperatura denaturacji (Tm): 86 oC
Starter his-k
Lokalizacja: 5’ koniec nici niekodującej genu HIS3
Sekwencja:
5’ - AAC ACC TTT GGT GGA GGG AAC ATC G - 3’
Udział nukleotydów GC: 52 %
Temperatura denaturacji (Tm): 76 oC

75
10 20 30 40 50 BamHI | | | | | 5’ - 1 GGATCCGCTG CACGGTCCTG TTCCCTAGCA TGTACGTGAG CGTATTTCCT 51 TTTAAACCAC GACGCTTTGT CTTCATTCAA CGTTTCCCAT TGTTTTTTTC 101 TACTATTGCT TTGCTGTGGG AAAAACTTAT CGAAAGATGA CGACTTTTTC 151 TTAATTCTCG TTTTAAGAGC TTGGTGAGCG CTAGGAGTCA CTGCCAGGTA 201 TCGTTTGAAC ACGGCATTAG TCAGGGAAGT CATAACACAG TCCTTTCCCG 251 CAATTTTCTT TTTCTATTAC TCTTGGCCTC CTCTAGTACA CTCTATATTT 301 TTTTATGCCT CGGTAATGAT TTTCATTTTT TTTTTTCCAC CTAGCGGATG TATA box
351 ACTCTTTTTT TTTCTTAGCG ATTGGCATTA TCACATAATG AAT TATACAT 401 TATATAAAGT AATGTGATTT CTTCGAAGAA TATACTAAAA AATGAGCAGG starter his-p 5’ - tgacagagc agaaagccct agtaaagcg 451 CAAGATAAAC GAAGGCAAAG ATGACAGAGC AGAAAGCCCT AGTAAAGCGT start HIS3
501 ATTACAAATG AAACCAAGAT TCAGATTGCG ATCTCTTTAA AGGGTGGTCC 551 CCTAGCGATA GAGCACTCGA TCTTCCCAGA AAAAGAGGCA GAAGCAGTAG 601 CAGAACAGGC CACACAATCG CAAGTGATTA ACGTCCACAC AGGTATAGGG 651 TTTCTGGACC ATATGATACA TGCTCTGGCC AAGCATTCCG GCTGGTCGCT 701 AATCGTTGAG TGCATTGGTG ACTTACACAT AGACGACCAT CACACCACTG 751 AAGACTGCGG GATTGCTCTC GGTCAAGCTT TTAAAGAGGC CCTACTGGCG 801 CGTGGAGTAA AAAGGTTTGG ATCAGGATTT GCGCCTTTGG ATGAGGCACT 851 TTCCAGAGCG GTGGTAGATC TTTCGAACAG GCCGTACGCA GTTGTCGAAC 901 TTGGTTTGCA AAGGGAGAAA GTAGGAGATC TCTCTTGCGA GATGATCCCG 951 CATTTTCTTG AAAGCTTTGC AGAGGCTAGC AGAATTACCC TCCACGTTGA 1001 TTGTCTGCGA GGCAAGAATG ATCATCACCG TAGTGAGAGT GCGTTCAAGG 1051 CTCTTGCGGT TGCCATAAGA GAAGCCACCT CGCCCAATGG TACCAACGAT starter his-k 3'-gcta 1101 GTTCCCTCCA CCAAAGGTGT TCTTATGTAG TGACACCGAT TATTTAAAGC caagggaggt ggtttccaca a-5' stop HIS3
1151 TGCAGCATAC GATATATATA CATGTGTATA TATGTATACC TATGAATGTC 1201 AGTAAGTATG TATACGAACA GTATGATACT GAAGATGACA AGGTAATGCA 1251 TCATTCTATA CGTGTCATTC TGAACGAGGC GCGCTTTCCT TTTTTCTTTT 1301 TGCTTTTTCT TTTTTTTTCT CTTGAACTCG AGAAAAAAAA TATAAAAGAG 1351 ATGGAGGAAC GGGAAAAAGT TAGTTGTGGT GATAGGTGGC AAGTGGTATT 1401 CCGTAAGAAC AACAAGAAAA GCATTTCATA TTATGGCTGA ACTGAGCGAA 1451 CAAGTGCAAA ATTTAAGCAT CAACGACAAC AACGAGAATG GTTATGTTCC 1501 TCCTCACTTA AGAGGAAAAC CAAGAAGTGC CAGAAATAAC ATGAGCAACT 1551 ACAATAACAA CAACGGCGGC TACAACGGTG GCCGTGGCGG TGGCAGCTTC 1601 TTTAGCAACA ACCGTCGTGG TGGTTACGGC AACGGTGGTT TCTTCGGTGG 1651 AAACAACGGT GGCAGCAGAT CTAACGGCCG TTCTGGTGGT AGATGGATCG 1701 ATGGCAAACA TGTCCCAGCT CCAAGAAACG AAAAGGCCGA GATCGCCATA 1751 TTTGGTGTCC CCGA GGATCC – 3’ BamHI
Rysunek 5. Sekwencja nukleotydowa nici kodującej genu HIS3 (kolor niebieski) wraz z 5’ i3’ regionami flankującymi. BamHI – miejsca rozpoznawane przez enzym restrykcyjny.TATA box oraz kodony start i stop translacji podkreślono i odpowiednio opisano. StarterywyróŜniono małymi literami i kolorem czerwonym. Delecja his3-∆200 w szczepieSacharomyces cerevisiae FY73 rozciąga się od pozycji 95 do 1130.

76
Preparacja DNA genomowego z droŜdŜy Saccharomyces
cerevisiae
1. 2 ml poŜywki YPG zaszczepić odpowiednim szczepem droŜdŜowym.
2. Inkubować przez noc w temp. 28oC z energicznym wytrząsaniem.
3. 1500 µl hodowli przenieść do probówki Eppendorfa i wirować przez 1
min.
4. Supernatant odrzucić, komórki zawiesić w 1 ml jałowej wody
destylowanej.
5. Zawiesinę komórek wirować przez 1 min.
6. Supernatant odrzucić, komórki zawiesić w 200 µl świeŜo przygotowanego
roztworu zymolazy w buforze do trawienia ściany komórkowej.
7. Zawiesinę inkubować przez 20 min. w temp. 37oC wytrząsając od czasu
do czasu.
8. Do zawiesiny dodać 200 µl świeŜo przygotowanego roztworu do lizy (1 %
SDS, 0,2 M NaOH). Wymieszać dokładnie przez odwracanie probówki.
Liza komórek objawia się wzrostem lepkości oraz przejrzystości zawiesiny
ale oba zjawiska są mniej wyraźne niŜ w przypadku lizy komórek bakte-
ryjnych.
9. Lizat inkubować przez 10 min. w temp. 65oC.
10. Do lizatu dodać 200 µl 3 M octanu sodu i dokładnie wymieszać.
11. Inkubować przez 10 min. w temp. -20oC.
12. Lizat odwirować (15 min, 14 000 obr/min., 4oC).
13. OstroŜnie zebrać supernatant znad osadu i przenieść do nowej probówki.
14. Do supernatantu dodać 1 objętość zimnego (- 20oC) izopropanolu i całość
wymieszać przez odwracanie probówki. Zaobserwować wytrącanie kwa-
sów nukleinowych.

77
15. Wytrącone DNA odwirować (5min, 14 000 obr/min., 4oC), supernatant
odrzucić.
16. Osad przemyć dwukrotnie 1 ml porcjami 70 % etanolu.
17. Osad wysuszyć w suszarce próŜniowej z rotorem (speedvac) i zawiesić w
100 µl wody.
18. Do roztworu DNA dodać 3 µl RNazy o stęŜeniu 10 mg/ml.
Materiały
− poŜywka YPG (pepton 1 %, wyciąg droŜdŜowy 1 %, glukoza 2 %)
− jałowa woda destylowana
− bufor do trawienia ściany komórkowej (50 mM Tris-HCl (pH 7,5), 25 mM
EDTA, 5 mg/ml zymolazy (20 000 U/mg), 1 % β-merkaptoetanol)
β-merkaptoetanol i zymolazę dodaje się do pozostałych
składników buforu bezpośrednio przed uŜyciem.
− roztwór do lizy (1% SDS, 0,2 M NaOH)
Przygotowuje się bezpośrednio przed uŜyciem mieszając ze sobą
równe objętości 2% SDS i 0,4 M NaOH.
− 3 M octan sodu (pH 5,2)
− izopropanol
− 70 % etanol
− RNaza 10 mg/ml
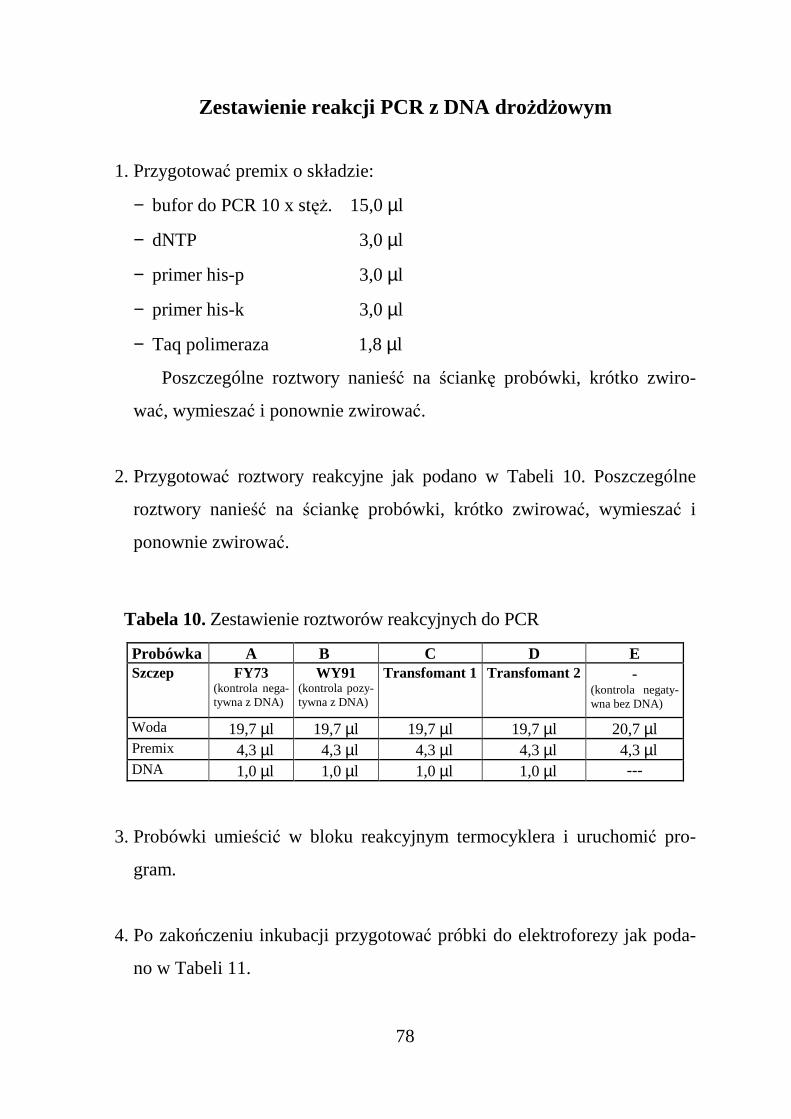
78
Zestawienie reakcji PCR z DNA droŜdŜowym
1. Przygotować premix o składzie:
− bufor do PCR 10 x stęŜ. 15,0 µl
− dNTP 3,0 µl
− primer his-p 3,0 µl
− primer his-k 3,0 µl
− Taq polimeraza 1,8 µl
Poszczególne roztwory nanieść na ściankę probówki, krótko zwiro-
wać, wymieszać i ponownie zwirować.
2. Przygotować roztwory reakcyjne jak podano w Tabeli 10. Poszczególne
roztwory nanieść na ściankę probówki, krótko zwirować, wymieszać i
ponownie zwirować.
Tabela 10. Zestawienie roztworów reakcyjnych do PCR
Probówka A B C D ESzczep FY73
(kontrola nega-tywna z DNA)
WY91 (kontrola pozy-tywna z DNA)
Transfomant 1 Transfomant 2 -(kontrola negaty-wna bez DNA)
Woda 19,7 µl 19,7 µl 19,7 µl 19,7 µl 20,7 µlPremix 4,3 µl 4,3 µl 4,3 µl 4,3 µl 4,3 µlDNA 1,0 µl 1,0 µl 1,0 µl 1,0 µl ---
3. Probówki umieścić w bloku reakcyjnym termocyklera i uruchomić pro-
gram.
4. Po zakończeniu inkubacji przygotować próbki do elektroforezy jak poda-
no w Tabeli 11.

79
Tabela 11. Przygotowanie próbek do elektroforezy
Nr probówki 1 2 3 4 5Produkt PCR 10 µl z A 10 µl z B 10 µl z C 10 µl z D 10 µl z EBarwnik do na-noszenia na Ŝel
2 µl 2 µl 2 µl 2 µl 2 µl
5. Przygotowane próbki nanieść na 1 % Ŝel agarozowy i przeprowadzićelektroforezę w obecności markera o znanej masie cząsteczkowej.
6. Wykonać zdjęcie Ŝelu w przechodzącym świetle ultrafioletowym.

80
Program PCR do wykrywania droŜdŜowego genu HIS3
starterami his-p i his-k
Control method: CALCULATED
1 = 94oC for 2:00
2 = 94oC for 0:30
3 = 65oC for 0:45
4 = 72 oC for 1:30
5 = Go to 2, 24 times
6 = 72 oC for 2:00
7 = 4 oC for ever
8 = End
81
Tabela 12. Przedrostki do tworzenia wielokrotności i podwielokrotnościjednostek miar w systemie metrycznym.
Skrót Przedrostek MnoŜnik
Y yotta 1024
Z zetta 1021
E exa 1018
P peta 1015
T tera 1012
G giga 109
M mega 106
k kilo 103
h hekto 102
deka da 101
decy d 10-1
c centy 10-2
m mili 10-3
µ mikro 10-6
n nano 10-9
p pico 10-12
f femto 10-15
a atto 10-18
z zepto 10-21
y yocto 10-24






















