Embed Size (px)
Citation preview

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
1
LABORATORIO IV: ELECTROFORESIS DE PROTEÍNAS
La gran mayoría de los polímeros biológicos presentan carga eléctrica. Las proteínas pueden ser consideradas como polianfolitos débiles. Las cargas derivan de los aminoácidos con grupos laterales ionizables. Éstos son: los residuos básicos de asparagina, lisina e histidina y los residuos ácidos del ácido glutámico y aspártico. Las cadenas laterales de tirosina también contribuyen a la carga total de la proteína. Debido a que estos grupos presentan diferentes grados de ionización la carga neta de las proteínas es muy dependiente del pH del medio. Las modificaciones post-traduccionales como fosforilación y glicosilación también le aportan carga a las proteínas.
Al someter moléculas cargadas a un campo eléctrico, producido por el pasaje de corriente eléctrica a través de la solución, éstas se desplazan. Dicho fenómeno se conoce como electroforesis. La velocidad de migración depende de la carga, tamaño y forma de las partículas (Cuadro 1). Esto permite separar componentes de una mezcla compleja, siempre y cuando, presenten velocidades de migración suficientemente diferentes. Cuadro1. Migración electroforética. Para poder entender la teoría de electroforesis se analizará el desplazamiento de una partícula cargada aislada en un campo eléctrico. En el momento que se aplica el campo eléctrico, E (volts/cm), la acción de la fuerza eléctrica (Fe) hará que la partícula se acelere. Esta fuerza depende de la fuerza del campo y de la carga (q) de la partícula: Fe=qE Tan pronto como la partícula empieza a moverse por la acción de la fuerza eléctrica, se manifiesta una fuerza que se opone al movimiento, Fv, generada por la fricción del medio. Fv= -fv f= coeficiente de fricción; v= velocidad de la partícula. Si se asume que la partícula es esférica se puede aplicar la Ley de Stokes y entonces el coeficiente de fricción se define como: f = 6πηr siendo r el radio de la partícula y η la viscosidad del medio.
Entonces: Fv= 6πηrv Debido a que la fuerza de fricción o fuerza viscosa es proporcional a la velocidad de migración de la partícula ésta se acelerará hasta que la fuerza viscosa se iguale con la fuerza eléctrica. A partir de ese momento la fuerza neta sobre la partícula será nula y se alcanzará un estado estacionario en el que la partícula moverá a velocidad constante. 6πηrv = qE Rearreglando esta ecuación se puede definir la movilidad electroforética (μ) como: v/E = q / 6πηr = μ Dado que la viscosidad está dada por el medio, la movilidad electroforética depende de la relación q/r que presente cada partícula. Partículas con diferente relación q/r podrán moverse con diferente velocidad durante la corrida electroforética y, si esta diferencia es suficiente, se separarán.
En la realidad, la separación de macromoléculas ocurre en soluciones acuosas que contienen, además de la macromolécula de interés, los iones constituyentes de los buffers y sales. En este caso no se estudia la macromolécula sola sino en presencia de otras especies cargadas que influenciarán el campo local además de interaccionar con la macromolécula generando una capa de contraiones y dificultando su análisis. En consecuencia, la definición de movilidad electroforética, deducida en el cuadro 1 para el caso ideal de una partícula cargada en una solución diluida de un solvente no conductor, es sólo una aproximación al comportamiento de una macromolécula en condiciones reales. A pesar de los esfuerzos realizados para introducir correcciones a esta ecuación sólo se han logrado aproximaciones.

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
2
Las técnicas electroforéticas son utilizadas para separaciones efectivas y resolución de mezclas de proteínas para su posterior análisis y caracterización así como para controles de pureza. La electroforesis fue descrita como método bioquímico de separación por Tiselius a mediados de 1930. Estos primeros experimentos se realizaron en “solución libre” (sin la presencia de soporte) pero la inestabilidad del aparato y la difusión de la muestra, además del efecto de calentamiento producido por el campo eléctrico comprometían la resolución de la separación. La introducción del “medio soporte” como los geles de almidón y derivados de papel superó en parte esos problemas. El propósito del “medio soporte” era contener el electrolito o buffer de corrida y generar algún impedimento al movimiento libre de los componentes de la muestra tal que se minimizara la difusión al azar. Electroforesis en soporte Se han usado muchos medios soportes que se pueden clasificar en dos tipos:
• los soportes que se utilizan principalmente para minimizar la convección como: papel,
acetato de celulosa y los materiales con que se hace cromatografía en capa fina. La separación se basa en la densidad de carga (relación carga/masa) de las proteínas al pH de trabajo de la misma forma que ocurre en la electroforesis libre.
• los soportes que además de evitar la convección y minimizar la difusión tienen una
estructura porosa (geles de almidón, agarosa y poliacrilamida), tal que si el tamaño de las moléculas a separar es del mismo orden que el tamaño del poro, causan un efecto de tamiz molecular. Las moléculas con menor tamaño pasarán por los poros del gel más fácilmente que aquellas con tamaño mayor. La separación de las moléculas dependerá, entonces, de la densidad de carga y del tamaño.
De esta forma dos proteínas con igual densidad de carga pero distinto tamaño probablemente no podrán ser separadas mediante electroforesis en papel, mientras que, si la diferencia de tamaño es suficiente, podrían ser separadas por electroforesis en geles de poliacrilamida. Electroforesis en gel Geles de almidón Los geles de almidón son una red tridimensional formada por almidón parcialmente hidrolizado. No se pude controlar demasiado el tamaño de poro. Actualmente son poco usados. Geles de agarosa La agarosa es un polímero de poligalactosa. El tamaño de poro es muy grande y se utiliza para separar moléculas de tamaño mayor a 200 KDa. Variando la concentración de agarosa se puede controlar el tamaño de poro. Se aplica principalmente para la separación de ácidos nucleicos o proteínas muy grandes. Geles de poliacrilamida Los geles de poliacrilamida son ampliamente utilizados para la separación de proteínas y fragmentos pequeños de ácidos nucleicos. El gel se obtiene mediante la polimerización de monómeros de acrilamida generando cadenas largas y el entrecruzamiento de éstas por compuestos bifuncionales como la N,N’-metilen bisacrilamida que reaccionan con los grupos funcionales libres en los extremos de las cadenas (figura 1). La reacción de polimerización de acrilamida es iniciada con persulfato de amonio y calor
Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
3
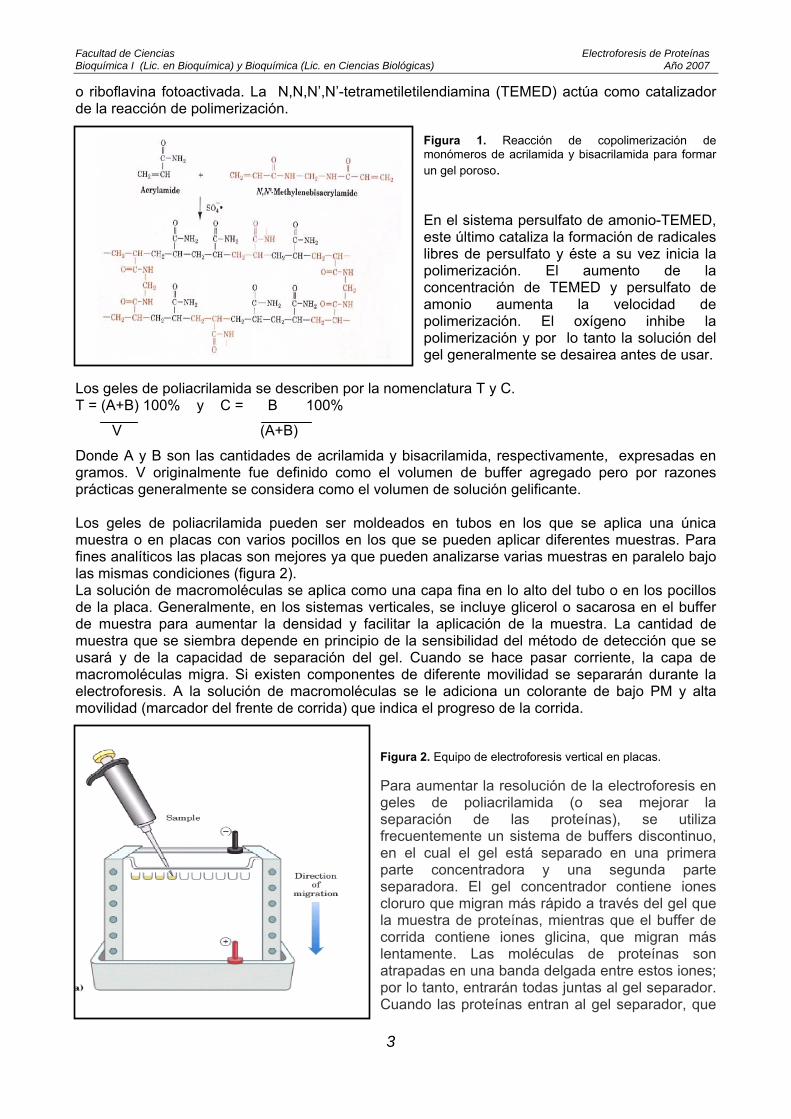
o riboflavina fotoactivada. La N,N,N’,N’-tetrametiletilendiamina (TEMED) actúa como catalizador de la reacción de polimerización.
Figura 1. Reacción de copolimerización de monómeros de acrilamida y bisacrilamida para formar un gel poroso. En el sistema persulfato de amonio-TEMED, este último cataliza la formación de radicales libres de persulfato y éste a su vez inicia la polimerización. El aumento de la concentración de TEMED y persulfato de amonio aumenta la velocidad de polimerización. El oxígeno inhibe la polimerización y por lo tanto la solución del gel generalmente se desairea antes de usar.
Los geles de poliacrilamida se describen por la nomenclatura T y C. T = (A+B) 100% y C = B 100%
V (A+B)
Donde A y B son las cantidades de acrilamida y bisacrilamida, respectivamente, expresadas en gramos. V originalmente fue definido como el volumen de buffer agregado pero por razones prácticas generalmente se considera como el volumen de solución gelificante. Los geles de poliacrilamida pueden ser moldeados en tubos en los que se aplica una única muestra o en placas con varios pocillos en los que se pueden aplicar diferentes muestras. Para fines analíticos las placas son mejores ya que pueden analizarse varias muestras en paralelo bajo las mismas condiciones (figura 2). La solución de macromoléculas se aplica como una capa fina en lo alto del tubo o en los pocillos de la placa. Generalmente, en los sistemas verticales, se incluye glicerol o sacarosa en el buffer de muestra para aumentar la densidad y facilitar la aplicación de la muestra. La cantidad de muestra que se siembra depende en principio de la sensibilidad del método de detección que se usará y de la capacidad de separación del gel. Cuando se hace pasar corriente, la capa de macromoléculas migra. Si existen componentes de diferente movilidad se separarán durante la electroforesis. A la solución de macromoléculas se le adiciona un colorante de bajo PM y alta movilidad (marcador del frente de corrida) que indica el progreso de la corrida.
Figura 2. Equipo de electroforesis vertical en placas.
Para aumentar la resolución de la electroforesis en geles de poliacrilamida (o sea mejorar la separación de las proteínas), se utiliza frecuentemente un sistema de buffers discontinuo, en el cual el gel está separado en una primera parte concentradora y una segunda parte separadora. El gel concentrador contiene iones cloruro que migran más rápido a través del gel que la muestra de proteínas, mientras que el buffer de corrida contiene iones glicina, que migran más lentamente. Las moléculas de proteínas son atrapadas en una banda delgada entre estos iones; por lo tanto, entrarán todas juntas al gel separador. Cuando las proteínas entran al gel separador, que

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
4
tiene un tamaño de poro menor, un mayor pH y una alta concentración salina respecto al concentrador, la glicina se ioniza y el gradiente de pH se disipa y las proteínas son separadas según su tamaño (figura 3).
Figura 3. Esquema de una electroforesis en geles de poliacrilamida con un sistema de buffers discontinuo.
Tipos de electroforesis Electroforesis no desnaturalizante o nativa Una proteína en su estructura nativa tendrá una determinada relación q/r y migrará en un campo eléctrico a una velocidad proporcional a esa relación. Si el medio en que se realiza la electroforesis es un gel con tamaño de poro adecuado además de la relación q/r deberá tenerse en cuenta el efecto tamiz aportado por el gel. Las proteínas poseen una carga neta a valores de pH diferentes de su punto isoeléctrico (PI), en el cual la carga neta de la molécula es nula. La forma de la proteína dependerá de cómo se pliegue la cadena polipeptídica (estructuras secundaria y terciaria) y de su peso molecular. Dos proteínas con igual peso molecular pueden tener diferente forma. Una proteína globular (con forma semejante a una esfera) y una proteína fibrosa (con forma de bastón) con el mismo peso molecular, debido a que tienen formas diferentes, tendrán radios diferentes. Bajo condiciones experimentales seleccionadas (pH, fuerza iónica, características del soporte, voltaje, temperatura, tiempo de corrida) se podrán separar moléculas con diferente relación q/r. Este tipo de electroforesis, al no desnaturalizar la muestra, permite realizar ensayos biológicos posteriores a la separación. La separación electroforética puede realizarse a cualquier pH entre 3 y 11. En caso de emplearse poliacrilamida como soporte, se utiliza la abreviatura PAGE (del inglés polyacrylamide gel electrophoresis). En el cuadro 2 se describen algunos parámetros del sistema que pueden ser variados para optimizar la separación.
A Al inicio de la corrida los aniones son ion cloruro (verde) en los geles concentrador y separador y glicina (naranja) en las compartimentos de electrodos. Las proteínas se encuentran en los pocillos (azul).
B Al establecerse la banda en el gel concentrador, las proteínas se concentran en una capa delgada entre los “iones líderes” cloruro y las moléculas de glicina los “iones atrasados”.
CCuando los componentes alcanzan el gel separador su migración será de acuerdo a sus movilidades individuales como en cualquier sistema homogéneo.

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
5
Cuadro 2. Parámetros a variar para optimizar la separación electroforética. 1. Dependientes del campo eléctrico a. Voltaje El gradiente de potencial es V/d si la separación de los electrodos es d (cm) y la diferencia de potencial entre ellos es V (volts). La fuerza sobre un ión que tiene una carga q es por tanto Vq/d y por tanto la velocidad de migración es proporcional a Vq/d. Un aumento en el gradiente de potencial aumenta proporcionalmente la velocidad de migración. b. Corriente Cuando se aplica una diferencia de potencial entre los electrodos, se genera una corriente (medida en Coulomb/segundo o Ampere). La magnitud de esta corriente está determinada por la resistencia del medio y es proporcional al voltaje. La corriente es conducida en la solución entre los electrodos, principalmente por los iones del buffer y en una pequeña proporción por los iones de la muestra. El aumento del voltaje aumentará la carga total/ segundo que converge al electrodo.
La distancia migrada por los iones será proporcional a la corriente aplicada y al tiempo. 2. Dependientes de la muestra Carga La velocidad de migración aumenta al aumentar la carga neta. La magnitud de la carga es en general dependiente del pH y por tanto dependiente del sistema de buffers utilizado. 3. Dependientes del soporte Tamiz molecular Este efecto se presenta en geles de almidón, agarosa y poliacrilamida. El movimiento de las moléculas es frenado al tener que pasar a través de los poros. Dependiendo del radio de las proteínas a separar se debe seleccionar el tipo de gel y tamaño de poro adecuado para que exista un efecto tamiz del soporte.
Electroforesis desnaturalizante Las proteínas pueden ser desnaturalizadas por una variedad de agentes tales como urea e hidrocloruro de guanidina así como detergentes iónicos fuertemente positivos o negativos. Dentro de éstos, el detergente aniónico dodecil sulfato, normalmente como su sal sódica (SDS del inglés, Sodium Dodecyl Sulfate; fig. 4), ha sido extremadamente útil combinado con separaciones electroforéticas en geles de poliacrilamida (SDS-PAGE). El SDS en la concentración adecuada se une a la mayoría de las proteínas en una relación peso / peso constante (1,4 g de SDS/ g de proteína).
Figura 4. Estructura química del SDS. Habrá un número definido de cargas negativas del SDS por residuo de aminoácido y por lo tanto la carga aportada por este detergente será proporcional al PM de la proteína y todos los complejos SDS-proteína tendrán la misma densidad de carga. Además, el SDS al unirse a las proteínas mediante interacciones hidrofóbicas, las desnaturaliza (elimina sus estructuras secundaria, terciaria y cuaternaria).
Preparación de la muestra El buffer de muestra se compone de SDS, glicerol, Tris, EDTA, un agente reductor como el ditiotreitol (DTT) y/o 2-mercaptoetanol y azul de bromofenol que sirve como tinta marcadora. El EDTA es un agente preservante que actúa como agente quelante de iones divalentes, lo que reduce la actividad de enzimas proteolíticas que requieren iones Ca2+ y Mg2+ como cofactores. El Tris actúa como buffer que es muy importante en los sistemas de electroforesis discontinua que requieren un pH específico. El glicerol hace a la muestra más densa y permite que ésta permanezca en el fondo del pocillo y no se disperse. La tinta permite al investigador seguir y controlar el proceso de electroforesis. El agregado de agentes reductores como 2-mercaptoetanol o ditiotreitol (DTT) y un tratamiento térmico a ebullición durante unos minutos permite lograr la

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
6
completa desnaturalización de las proteínas. Estos agentes reducen los puentes disulfuro intra y/o intercatenarios de las proteínas. Se ha comprobado que todos los complejos SDS-proteína adoptan una conformación extendida o de bastón, eliminándose la influencia de la forma en la movilidad electroforética. Debido a que todos los complejos SDS-proteína tendrán la misma densidad de carga y conformación, en una electroforesis en solución libre (sin la presencia de soporte poroso) migrarán con la misma velocidad y por lo tanto no se separarán. Cuando la electroforesis se realiza en geles de poliacrilamida, con la porosidad adecuada para generar el efecto de tamiz molecular, la separación ocurre exclusivamente en función del peso molecular. Los complejos con mayor tamaño molecular presentarán más resistencia a pasar por los poros del gel y migrarán con menor velocidad que los de menor tamaño. Para cada tipo particular y concentración de gel, existe una relación aproximadamente lineal entre el log del PM de los complejos SDS-proteína y su Rf. El Rf se define como la relación entre la distancia migrada por la proteína y la migrada por el marcador de frente de corrida. Para determinar el PM aparente de las proteínas contenidas en una muestra se corre en el mismo gel un estándar de PM constituido por proteínas con relativa pureza y PM conocido (figura 5). Luego de terminada la electroforesis, teñido y desteñido del gel se determinan los Rf para cada una de las proteínas del estándar y de las proteínas de interés. Al graficar el log PM de las proteínas estándar en función de su Rf generalmente se obtiene una relación lineal. Los PM aparentes de las proteínas de la muestra pueden determinarse combinando su valor de Rf con el punto adecuado en la curva de calibración. Para calcular el Rf debe tenerse en cuenta la variación en las dimensiones del gel antes y después del proceso de teñido. Este tipo de electroforesis es particularmente aplicable cuando se quiere analizar proteínas relativamente insolubles en los buffers comunes ya que en presencia de SDS éstas se solubilizan. Es una técnica relativamente rápida, económica y simple para determinar el PM de proteínas.
Figura 5. Determinación del PM de una proteína desconocida mediante SDS-PAGE. (a) En el carril 1 se corrió un estándar de PM y en el carril 2 la proteína desconocida. (b) Se grafica el log PM de las proteínas del estándar en función de sus Rf. Mediante el valor de Rf de la proteína desconocida se obtiene su PM. Algunas proteínas no tienen un comportamiento normal en SDS-PAGE. Las glicoproteínas no unen la cantidad SDS promedio y las estimaciones del PM aparente deben hacerse con mucho cuidado. Los pesos moleculares pueden ser sobrestimados hasta en un 30%. En el caso de las proteínas extremadamente cargadas positivas como las histonas, una cantidad significativa de las cargas pertenecientes al SDS unido está balanceada por las cargas propias de la proteína, lo que hace difícil la estimación de su PM aparente. Las proteínas muy ácidas como la pepsina, papaina y glucosa oxidasa unen cantidades mucho menores de SDS.

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
7
Debe tenerse en cuenta que este método de separación, al desnaturalizar las proteínas, hace que pierdan su actividad biológica. Existen algunas proteínas con estabilidad poco común que mantienen su actividad en presencia de SDS y en otros casos ciertas proteínas pueden recuperar su actividad al cambiar este detergente fuerte por otro más débil.
Cuadro 3. Masa molecular y peso molecular La masa molecular (M) se expresa en Daltons (Da). Un Da se define como 1/12 la masa del 12C. El peso molecular no es lo mismo que la masa molecular. También se conoce como masa molecular relativa (Mr). El peso molecular se define como la proporción de la masa de una macromolécula al 1/12 de la masa del átomo 12C. Es una cantidad adimensionada. Es incorrecto expresar un peso molecular o Mr en Da, pero de todas maneras se encuentra en la literatura el peso molecular expresado en kiloDaltons. Isoelectroenfoque (IEF) Es una técnica electroforética en la cual son fraccionados compuestos anfóteros de acuerdo a su PI a lo largo de un gradiente de pH continuo. Las proteínas (polianfolitos) migrarán hacia el ánodo o cátodo de acuerdo a su carga neta hasta alcanzar la región del gradiente de pH que coincida con su PI. A pH igual a su PI tendrán carga neta cero, no podrán migrar y se concentrarán o enfocarán en zonas estrechas. El gradiente de pH en el gel puede lograrse mediante un sistema buffer con anfolitos transportadores o mediante un sistema de gradiente de pH inmovilizado. El IEF es una técnica de equilibro y el resultado no depende (dentro de límites razonables) del modo de aplicación de la muestra, la carga total de proteína o el tiempo de corrida. Permite determinar un parámetro fisicoquímico intrínseco de las proteínas: su PI. Se alcanzan excelentes resoluciones de proteínas cuyos PI difieren sólo en 0,02 unidades de pH; las bandas de proteína son muy nítidas debido al efecto de enfocado. Luego de una corrida de IEF el gradiente de pH en el gel puede ser medido tanto utilizando un electrodo con membrana para superficies como cortando el gel en pequeños intervalos, eluyendo las muestras de cada trozo y midiendo su pH. Otra alternativa es usar una mezcla de proteínas estándar de PI conocidos para calibrar el gradiente de pH alcanzado durante el IEF. Electroforesis bidimensional Los métodos analizados anteriormente separan mezclas complejas de proteínas y bajo condiciones óptimas pueden resolver hasta 100 zonas diferentes. En los años recientes, se ha vuelto necesario analizar muestras de mayor complejidad como aquellas procedentes de células enteras o tejidos. Este nivel de complejidad no puede analizarse por ninguno de los procedimientos electroforéticos en una dimensión. Además, en cualquiera de estos procedimientos la separación se basa en una única propiedad fisicoquímica (tamaño, carga). En consecuencia, por ejemplo, si se realizó una SDS-PAGE, las bandas obtenidas luego de la separación contendrán todas las proteínas de la muestra con igual PM, que no necesariamente serán una misma proteína. La electroforesis bidimensional resulta de la combinación de dos técnicas electroforéticas en una dimensión en un procedimiento en dos dimensiones. La combinación más usada es realizar un IEF en la primera dimensión y a continuación un SDS-PAGE en la segunda dimensión (figura 6), resultando en una separación basada en dos propiedades fisicoquímicas independientes (carga y tamaño). Esta metodología se ha adaptado a un amplio rango de muestras. Electroforesis capilar Es una técnica surgida recientemente, más rápida y sensible que la electroforesis en gel. La separación electroforética ocurre en capilares de 50-100μm de diámetro y 20-60cm de largo. Debido a que la resistencia es grande dada la pequeña sección de los capilares, es posible aplicar

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
8
gradientes de voltaje bastante altos sin que la corriente se incremente demasiado. Esto hace que la separación sea rápida. Debido a que la capilaridad inhibe la mezcla por convección no es necesario utilizar soporte, aunque a veces se incorpora un soporte poroso para generar el efecto tamiz molecular. La detección se realiza por absorbancia UV o fluorescencia a medida que los componentes pasan por un punto determinado en el capilar. La sensibilidad es de fentomoles (10-
15) con detección UV y zeptomoles (10 –21) con detección fluorescente.
Figura 6. Esquema de un experimento de electroforesis bidimensional en la que se combina IEF en la primera dimensión y SDS-PAGE en la segunda. Métodos de detección de proteínas en el gel Luego de realizada la separación electroforética es necesario visualizar y, en algunos casos, cuantificar las bandas proteicas en el gel. A continuación se describen brevemente algunas de las técnicas más utilizadas. Tinción con colorantes orgánicos El gel es embebido en una solución de colorante que se une a las proteínas. Para la visualización de las bandas proteicas es necesario desteñir el gel con el solvente utilizado para disolver el colorante. Azul de Coomasie (Coomasie Brillant Blue, CBB): Es el método más común de tinción de proteínas. El CBB es un colorante orgánico que en medio ácido se une a las proteínas mediante interacciones electrostáticas con los grupos amino. Luego de teñido y desteñido el gel las

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
9
proteínas se visualizan como bandas azules. La tinción con CBB es simple pero presenta baja sensibilidad; se pueden detectar hasta 0,2 – 0,5 μg de proteína por banda. Amido Black: Es un colorante orgánico que permite teñir proteínas con una sensibilidad 3 veces menor que la tinción con CBB. Tinción con sales de plata Los procedimientos de tinción con plata se basan en la reducción de la plata y se pueden agrupar en dos categorías. Los métodos alcalinos se basan en el uso de plata amoniacal o como solución diamina. Los iones de plata complejados con las proteínas son reducidos por formaldehído en medio ácido. En el segundo grupo de métodos, se usa una solución débilmente ácida de nitrato de plata para embeber el gel. El revelado se logra por la reducción selectiva de los iones plata a plata metálica por formaldehído en medio alcalino. La tinción con plata es monocromática, produciendo bandas de color marrón oscuro. La sensibilidad es su gran ventaja siendo 20-200 veces mayor que con CBB y pudiéndose detectar hasta 0,1 ng de proteína por banda. Entre las desventajas se encuentra el alto costo, el hecho de ser un método laborioso, lento y poco reproducible y que no todas las proteínas se tiñen. Marcado con colorantes fluorescentes Se han desarrollado métodos en los que las proteínas son marcadas con colorantes fluorescentes antes de la corrida electroforética y las bandas proteicas son detectadas mediante escaneo. El colorante más usado es la fluorescamina. Las mayores ventajas de este reactivo son su alta sensibilidad (se pueden detectar aproximadamente 5 ng de proteína unida a fluorescamina) y la facilidad de poder monitorear el avance de la separación electroforética exponiendo el gel a luz UV. Otros marcadores fluorescentes usados son MDPF (2-metoxi-2, 4-difenil-3(2H)-furanona), que en geles de poliacrilamida mantienen la fluorescencia por algunos meses y el o-ftaldialdehido (OPA) que presenta una sensibilidad 10 veces mayor que la fluorescamina. Las bandas proteicas también pueden ser detectadas mediante unión a colorantes fluorescentes luego de realizada la electroforesis. Las moléculas usadas son ANS (1-anilina-8-sulfonato de naftaleno) y OPA. Marcado con moléculas radioactivas Se han desarrollado procedimientos por los que se realiza el marcado de las muestras con isótopos radiactivos tanto antes de la separación electroforética como luego de la misma mediante marcado in situ de las bandas proteicas separadas en la matriz. En comparación con los métodos de teñido con colorantes, el uso de marcado radiactivo ofrece algunas ventajas como mayor sensibilidad y en condiciones que pueden ser no desnaturalizantes de forma que los componentes separados pueden ser extraídos manteniendo su actividad biológica. El revelado generalmente se realiza poniendo en contacto el gel con una película de rayos-X durante un tiempo apropiado (autorradiografía). Revelado específico de enzimas Esta técnica consiste en ensayar la actividad enzimática directamente en el gel o recortar y eluir las bandas proteicas separadas y entonces ensayar la actividad enzimática en solución. En el primer caso, luego de la separación electroforética el gel se pone en contacto con una solución apropiada del sustrato (coloreado o fluorescente) o un soporte impregnado con el mismo. Transcurrido el tiempo de reacción adecuado se detecta el producto de la reacción enzimática. Mediante este tipo de revelado es posible identificar las bandas que presentan actividad enzimática del total de bandas proteicas obtenidas en la separación. Western blotting Este procedimiento consiste en transferir las proteínas separadas electroforéticamente desde el gel a una matriz, generalmente nitrocelulosa o membranas de difluoruro de polivinilideno (PVDF).

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
10
Estas matrices son más manejables y permiten el revelado mediante cualquier método general para proteínas. Es posible, además, localizar proteínas de interés mediante anticuerpos específicos para ellas. Debido a que la unión antígeno-anticuerpo es específica se logra de esta forma identificar la proteína buscada. Aplicaciones de la electroforesis de proteínas La electroforesis de proteínas en todas sus variables, tiene numerosas aplicaciones que incluyen: - determinación del peso molecular aparente de las proteínas - identificación de proteínas - control de pureza de una muestra - identificación de puentes disulfuro - cuantificación de proteínas - determinación de pI - estudio de isoformas - estudio de multisubunidades La electroforesis es frecuentemente usada en el diagnóstico clínico. En el suero humano se encuentran las proteínas albúmina, alfa-, beta- y gama- globulinas. La variación en la cantidad relativa de estas proteínas se asocia a algunas patologías. Mediante electroforesis en acetato de celulosa, estas proteínas pueden separarse y cuantificarse para diagnosticar ciertas enfermedades. Además, es empleada como herramienta en estudios evolutivos, taxonómicos, genéticos y de biología de la conservación. La electroforesis de proteínas ha permitido ampliar el conocimiento existente sobre las relaciones de parentesco entre las especies, así como conocer la diversidad y variabilidad genética de las poblaciones y la manera en que se relacionan filogenéticamente. Esto se ha realizado estudiando las aloenzimas (formas alternativas de una enzima codificada por diferentes alelos del mismo locus), las cuales presentan movilidad diferencial en una electroforesis. Algunos ejemplos: - Los patrones de proteínas presentes en la pared celular de levaduras obtenidos por SDS-PAGE han sido utilizados para la identificación de especies. - Las gliadinas son un grupo heterogéneo de proteínas que se encuentran en plantas de trigo. La electroforesis en gel ha sido utilizada para examinar la composición de estas proteínas en diferentes genotipos de trigo, ya que están relacionadas a la taxonomía y evolución de esta especie, así como a su calidad para la elaboración de pan. Bibliografía Andrews, A. Electrophoresis. Theory, Techniques an Biochemical and Clinical Applications (2a edición).
Editado por Clarendon Press, Oxford, 1986. Dunn, MJ. Gel Electrophoresis: proteins. BIOS Scientific Publishers Limited, 1993, Oxford. Hames,BD & Rickwood D, editores. Gel Electrophoresis of Proteins. A Practical Approach (2a edición). IRL
Press, Oxford & New York, 1990. Jakoby. “Enzyme Purification and Related Techniques” En: Methods in Enzymology, vol 104, Part C.
Academic Pess, Inc, 1984. Laas, Torgny. “Electrophoresis in gel” En: Protein Purification, Hight-Resolution Methods, and Applications,
pp 463-493 (Segunda edición). Editado por Jan-Christer Janson & Lars Rydén. Wiley-VCH Inc., New York, 1998.

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
11
Análisis de proteínas por electroforesis en geles de poliacrilamida con SDS ( SDS_PAGE)
Objetivos: En esta práctica se evaluará el proceso de purificación de lisozima mediante el análisis por SDS-PAGE de las fracciones obtenidas en la práctica anterior. En particular analizaremos -la composición proteica de cada una de las fracciones de la cromatografía de intercambio iónico -el grado de pureza de la fracción F2 obtenida -el peso molecular aparente de la proteína purificada. Reactivos: - Solución Acrilamida: Acrilamida 30%, N,N’ metilrnbisacrilamida 0.8%. - Buffer I (4X): Tris. HCl 1.5 M, SDS 0.4 %, pH 8.8 - Buffer II (4X): Tris HCl 0.5 M, SDS 0.4 %, pH 6.8 - TEMED: N,N,N’,N’, tetrametilendiamina. - Buffer de corrida (10X): Tris 50 mM, glicina 384 mM, SDS 10% - Buffer de muestra (4X): 1.52g Tris base, 2g SDS, 2mL 2-mercaptoetanol, 20mL glicerol, 1mg Azul de bromofenol, Ajustar pH 6.8, H2O csp 50mL. - Solución colorante de Coomasie: 2g Coomasie Brillant Blue R-250, 300mL etanol, 70mL ácido acético glacial y agua csp 1,0 L. Preparación del gel: 1- Se arman las placas molde del gel de forma vertical. 2- Para un gel separador al 12% de 10 cm X 8 cm X 1 mm, mezclar:
3 mL Solución acrilamida 1,875 mL Buffer I 2,6mL agua 0.5 mL glicerol 25 μL APS 7 μL TEMED
3- Se transfiere esta solución, con ayuda de una pipeta al molde, se dejan 2 cm aprox. del molde sin llenar. Se deja polimerizar (15-30min). 4- Una vez polimerizado el gel separador, se prepara el gel concentrador. Se mezclan:
0.35 mL Solución acrilamida 0.65 mL Buffer II 1,5 mL agua 10 μL APS 5 μL TEMED
5- Se coloca el peine en los 2 cm del molde sin gel y se transfiere la nueva solución a molde del gel. Se deja polimerizar (15-30min). Preparación de la muestra: 1- Mezclar un volumen de Buffer de muestra con 3 volúmenes de muestra 2- Calentar en baño de agua a ebullición durante 5 minutos Corrida electroforética: 1- Una vez polimerizado, colocar el gel en la cuba de electroforesis. 2- Quitar los peines. 3- Agregar el buffer de corrida hasta cubrir totalmente los pocillos y en la cuba inferior 4- Cargar 10 μL de muestra por pocillo. Anotar el orden en que se cargaron las muestras.

Facultad de Ciencias Electroforesis de Proteínas Bioquímica I (Lic. en Bioquímica) y Bioquímica (Lic. en Ciencias Biológicas) Año 2007
12
5- Encender la fuente de poder y realizar la corrida a 120V (70 min aprox) 6- Finalizada la corrida, desconectar el equipo, quitar el buffer en la cuba superior y sacar las placas de las cubas. 7- Desmoldar los geles de las placas cuidadosamente, marcar la esquina derecha de cada gel cortando una punta con la espátula 8- Sumergir el gel en la solución colorante durante 5-10 minutos. 9- Retirar la solución colorante (no descartarla), y agregar al recipiente con el gel agua caliente agitando hasta que se satura con colorante. Cambiar el agua y repetir hasta que se observen bandas proteicas azules en un fondo transparente.
Preguntas 1. Indique en qué se basa la separación en las siguientes técnicas electroforéticas:
a. Electroforesis nativa b. SDS-PAGE c. IEF
2. Se obtienen los siguientes resultados de un SDS-PAGE de una muestra que contiene una proteína aislada:
a) La muestra previamente tratada con 2-mercaptoetanol muestra un patrón
con dos bandas que migraron 10,0 y 10,6 cm respectivamente. b) Una serie de proteínas estándar migraron según se muestra: c) El azul de bromofenol migró una distancia de 12 cm. Teniendo en cuenta estos resultados ¿qué podría decir sobre la estructura cuaternaria de la proteína de la muestra?.
PM distancia (cm)seralbumina bovina 67000 1.1 ovalbumina 43000 3.9 anhidrasa carbonica 30000 6.6 inh. de tripsina 20100 9.3 alfa-lactalbumina 14400 11.7

























![Electroforesis [Bioseparaciones Mecánicas]](https://img.pdfslide.tips/doc/110x75/556de82ed8b42a524e8b5110/electroforesis-bioseparaciones-mecanicas.jpg)


![Informe Electroforesis[1]](https://img.pdfslide.tips/doc/110x75/5571f81549795991698c9940/informe-electroforesis1.jpg)