Embed Size (px)
Citation preview

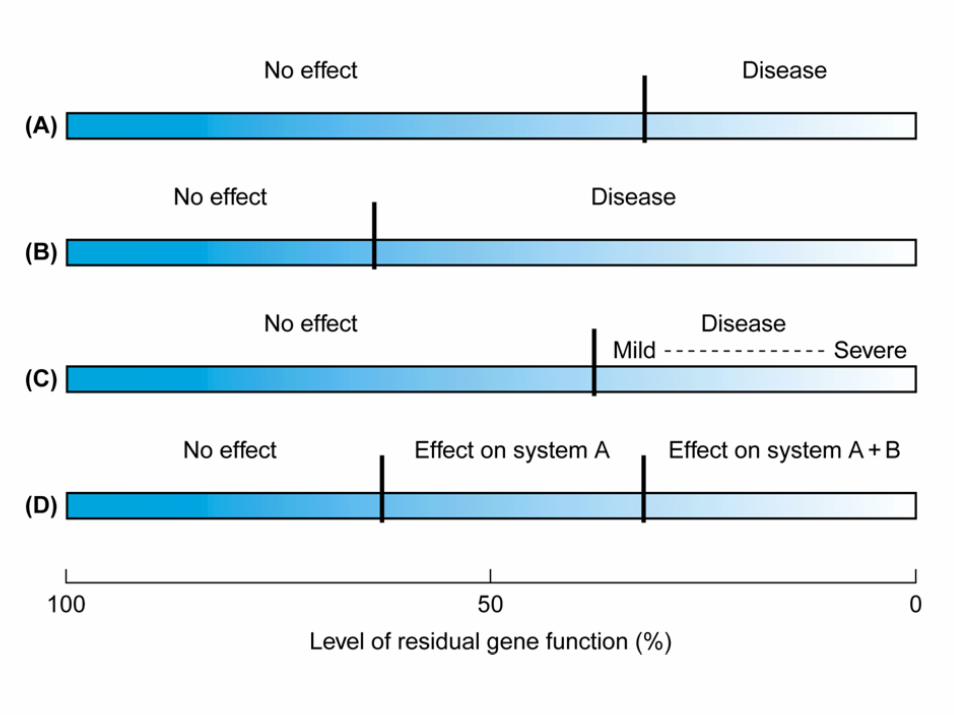
malattie genetiche da mutazione in 1 allele
Le mutazioni monoalleliche possono causare disordini a trasmissione dominante o recessiva legata all’X negli uomini
• Se la malattia a trasmissione dominante è grave in età fertile e pertanto limita o annulla la capacità riproduttiva (bassa fitness), le mutazioni monoalleliche sono nuove e spesso distribuite in modo casuale
• Se la malattia dominante non è grave in età fertile e non limita in alcun modo la capacità riproduttiva (normale fitness), le mutazioni monoalleliche sono ereditate da un genitore e spesso si tramandano da molte generazioni
• Se la malattia è recessiva legata all’X ed è letale ha una vita media di tre generazioni, perché le donne trasmettono gli alleli mutati in eterozigosi e gli uomini li eliminano

eredità autosomica dominante a penetranza completa(malattia che non modifica la fitness riproduttiva)

Cos’è una mutazione causativa?
Una variazione della sequenza del DNA ….
• ..che è trovata solo negli individui affetti
• ..che non è mai ritrovata in quelli non affetti
• ..che spiega il processo patologico
• ..che, quando corretta per tempo, fa recuperare un fenotipo normale

….che è trovata solo negli individui affetti..che non è mai ritrovata in quelli non affetti
penetranza incompleta
che è ritrovata più frequentemente negli individui affetti rispetto ai non affetti…

malattie genetiche da mutazione in 2 alleli
Le mutazioni bialleliche possono causare disordini a
trasmissione autosomica recessiva
• Se la malattia a trasmissione recessiva è grave in età fertile e limita o annulla la capacità riproduttiva (bassa fitness), le mutazioni non si estinguono comunque perché i portatori sani sono 10-10.000 volte più numerosi degli affetti
• Le mutazioni in genere si trasmettono da 100-1000 generazioni, mentre le nuove mutazioni sono rare
• Solo se la malattia è biallelica le mutazioni hanno una firma etnicache caratterizza una località di origine e un fondatore comuneeterozigote sano

Malattie genetiche da 2 alleli
• L’alto numero di portatori è un fattore di rischio per l’eterozigosi composta (due mutazioni differente nei due alleli). Questo potrebbe essere causato da una fitness migliore degli eterozigoti nei confronti di un fattore negativo vedi A
• La consanguineità è un fattore di rischio per l’omozigosità (due alleli identici) anche se la mutazione è rarissima vedi B

Imprinting

Imprinting
• Nelle cellule germinali primordiali l’imprinting viene cancellato del tutto e il DNA è demetilato
• Successivamente nella linea germinale maschile si determina un pattern di imprinting che in alcuni loci ècomplementare a quello della linea germinale femminile
• I cromosomi su cui avviene l’imprinting (7, 11, 15) manterranno questo pattern e lo riprodurranno ad ogni mitosi
• Si potranno sempre distinguere l’espressione genica del cromosoma materno e paterno

Disomia uniparentale
• Due copie dello stesso cromosoma sono ereditate dallo stesso genitore
• Spesso questo avviene attraverso un fenomeno transitorio di trisomia, seguito dalla perdita del cromosoma singolo e mantenimento del cromosoma doppio

Angelman
• 70% dei casi delezione della regione cromosomica 15q11-q13, che è soggetta al fenomeno dell'imprinting del cromosoma paterno
• Il gene materno (l'unico espresso) può essere alterato con 4 meccanismi noti:– delezione– disomia uniparentale paterna– difetti nell'imprinting– mutazioni a carico del gene UBE3A (ubiquitin ligasi)
• La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi


• "happy puppet syndrome" si può identificare in Cucciolo (Dopey) "addormentato", il più giovane dei nani che non ha mai imparato a parlare
• ritardo mentale con assenza del linguaggio, difficoltànell'equilibrio, eccessivo buon umore
Angelman

• L'incidenza è 1/20.000 nati• crisi epilettiche e comunque
alterazioni dell'EEG e microcefalia relativa
Angelman

Prader-Willi
• iperfagia>obesità• eccessiva assunzione di liquidi• reazioni abnormi ai sedativi• acromicria, criptorchidismo• insensibilità al dolore, lesioni cutanee• sbalzi di umore

Prader-Willi
1/15.000

Mutazioni dinamiche

Circa il 2% della popolazione ha un IQ<70 (ritardo mentale)
il 15-20% di tutti I ritardi mentali sono attribuibili a geni del cromosoma X
Il ritardo mentale legato al cromosoma X (XLMR) è geneticamente eterogeneo con 202 loci responsabili di forme che si sovrappongono clinicamente
46 geni sono stati a tutt’oggi identificati
il locus che contribuisce alla frazione maggiore causa la sindrome di Martin-Bell, oggi nota come
sindrome dell’X fragile

X fragile
ritardo mentale: IQ tra 20 e 70
•deficit di memoria a breve termine di informazioni complesse•ritardo nel linguaggio•ridotte abilità visuo-spaziali•ipersensibilità agli stimoli•iperattività con deficit di attenzione•comportamento autistico
•Macrocefalia con fronte, mento e orecchie sporgenti•Macroorchidismo (<30ml) dopo la pubertà•Anomalie connettivali: prolasso della mitrale, lassità articolare, piede piatto•Disfunzioni ipotalamiche?


Nel 1969 Lubs osservò una costrizione (marker X) sul braccio lungo del cromosoma X in quattro maschi affetti e tre carriers obbligate della stessa famiglia

Il sito fragile a Xq27.3
rottura o costrizione dei cromosomi in metafase che insorge quando le cellule sono esposte ad una perturbazione della replicazione del DNA
siti fragili sono su tutti i cromosomi e prendono il nome della banda cromosomica, es fra(X)(q27.3)
la nomenclatura HUGO chiama questo sito FRAXA, cioè il primo sito fragile identificato sul cromosoma X

1
1 3 4
1
2
1 2 3 4 5
I
II
IV
III
Segregazione, paradosso di Sherman
perché non è affetto?
perché è affetta?
Il 20% dei maschi che portano l’allele mutato sono normali (NTM)
Il 30% delle carrier presenta ritardo mentale

Fragile X syndrome


X fragile al microscopio a forza atomica

Il gene FMR

controlli premutazioni
200
150
100
50
0
Zhong et al. Am J Hum Genet 1995

Eag I
allele normale:stabile nella famiglia e nell’individuo,instabile nella popolazione(polimorfismo)
premutazione:instabile nella famiglia,stabile nell’individuo
mutazione piena:instabile nell’individuo(mutazioni somatiche)reprime la trascrizionedi FMR1
CpG island/5 ’UTRFMR1 geneprobe
(CGG) ~ 6 to 50
(CGG) 59 to ~ 200
(CGG) > 250Methylation
EcoRI EcoRI
2.4kb 2.8kb

diagnosi di X Fragile : analisi mediante Southern blot di espansione e metilazione
Rousseau et al. NEJM 1991

Premutazioni e mutazioni
• Le premutazioni si espandono quando sono trasmesse dalla madre
• La donna con premutazioni ha un maggiore rischio di menopausa precoce POF (premature ovarian failure)
• Il più corto allele descritto che in una sola generazione èdiventato mutazione piena è di 59 triplette
Espansione stabile (CGG)9-AGG-(CGG)9-AGG-(CGG)9Ha almeno 2 A che interrompono la serie di 9 triplette
Espansione instabile (CGG)9-(CGG)9-(CGG)9- (CGG)9……NON ha A che interrompono la serie

Il gene FMR1 (fragile X mental retardation 1) è all’interno di deserto genico: quindi il fenotipo NON è da geni contigui
Mutazioni puntiformi o delezioni di FMR1 causano un fenotipo identico alle espansioni e questo dimostra che il ruolo del gene non è importante nelle prime fasi dello sviluppo, quando le triplette non sono ancora metilate

Trasmissione X fragile

cosa fa FMR1?
• FMR1 codifica per una RNA-binding protein selettiva associata con i poliribosomi ed espressa nei neuroni
• nelle spine dendritiche regola la traduzione degli mRNA, funzione cruciale per la plasticità sinaptica e la maturazione neuronale
• interagisce con gli mRNA e con il pathway dei miRNA
• Nell’X fragile le spine dendritiche sono immature e lunghe

Spine dendritiche nel neocortex lunghe ed immature anche nel topo KO

Malattie da triplette ripetute
Disease Gene Locus/Protein Repeat Location
Fragile X syndromeFragile XE syndromeFriedreich ataxiaMyotonic dystrophy 1
Myotonic dystrophy 2Spinobulbar muscularatrophy
Huntington diseaseDentatorubralpallidoluysianatrophy
SCA type 1SCA type 2SCA type 3(Machado-Joseph disease)SCA type 6
SCA type 7SCA type 8SCA type 12
Xq27.3/FMR-1 proteinXq28/FMR-2 protein9q13-9q21.1/frataxin19q13/myotonic dystrophyprotein kinase
3q21Xq13-Xq21/androgen receptor
4p16.3/huntington12p13.31/atrophin-1
6p23/ataxin-112q24/ataxin-214q32.1/ataxin-3
19p13/α-1A (voltage-ependentcalcium channel subunit)
3p12-3p13/ataxin-713q12/none identified5q31-5q33
CGGGCCGAACTG
CCTGCAG
CAGCAG
CAGCAGCAG
CAG
CAGCTGCAG
NoncodingNoncodingNoncodingNoncoding
NoncodingCoding
CodingCoding
CodingCodingCoding
Coding
Coding?
Noncoding

Malattie da triplette ripetute non codificanti
CGGCGG
CGGCGGCGGCGGCGGCGGCGGCGG
5' UTR 3' UTR
mutazionecompleta
normale
222222000000000000------ >>>>>>222222000000000000000000
60-200
6-52 7-22
60-80
5-37
Sindrome delX fragile
Atassia diFriedreich
Distrofiamiotonica
pre-mutazione
CTGCTGCTGCTGCTGCTGCTGCTGCTGCTG
esoneintrone introneesone esone
222222000000000000------ >>>>>>999999000000000000 222222000000000000------ >>>>>>222222000000000000000000GAAGAAGAAGAAGAAGAAGAAGAAGAAGAA

anticipazione nella distrofia miotonica

distrofia miotonica DM1
• fenomeno “miotonico”, difficoltà al rilasciamento muscolare dopo una contrazione
• ipotonia al volto, non debolezza importante• cataratta precoce• alterazioni ritmo cardiaco• disfunzione tiroidea• trasmissione autosomica dominante (1/8000)• forma congenita con grave ipotonia neonatale

distrofia miotonica DM1
• La distrofia miotonica di Steinert è la più comune distrofia muscolare dell’adulto
• è causata da un’espansione CTG nel 3’UTR del gene DMPK (nell’RNA CUG) a 19q13.3
• presenta eredità autosomica dominante con anticipazione
• sono state identificate RNA binding proteins che interagiscono con l’espansione CUG

distrofia miotonica DM2
• cromosoma 3p21• un’espansione simile nell’ introne 1 di un repeat CCUG
nel gene ZNF9 (zinc finger protein 9) causa la distrofia miotonica 2
• la DM2 è detta anche distrofia miotonica prossimale

Malattie da triplette ripetute di poliglutammina
ORF
CAGCAGCAGCAGCAGCAGCAGCAGCAGCAG
CAGCAGCAGCAGCAGCAGCAGCAGCAG
CAGCAGCAGCAGCAGCAGCAGCAGCAG
CAGCAGCAGCAGCAGCAGCAGCAGCAG
CAGCAGCAGCAGCAGCAGCAGCAGCAG
CAGCAGCAGCAGCAGCAGCAGCAGCAG
CAGCAGCAGCAGCAGCAGCAG
5' UTR 3' UTRnormale
ammalato
normale
ammalato
121
36346
41396
81
49257
88
683613
79
2935
59
153440
62
111621
27
4
Corea diHuntington
Atassiaspinocerebellare
di tipo 1
Atassiaspinocerebellare
di tipo 2
Atassiaspinocerebellare
di tipo 6
Malattia diMachado-Joseph
Atrofiadentatorubrale-pallidoluysiana
Atrofiamuscolare
spinobulbare

Còrea di Huntington
• descritta da George Huntington nel 1872, è detta anche còrea che in greco indica la danza
• alla base vi è una degenerazione programmata geneticamente dei neuroni dei gangli basali (nuclei caudato e pallido) e della corteccia
• prevalenza di 1/10,000 e presenta il fenomeno dell'anticipazione
• si trasmette nel 97% dei casi come carattere autosomico dominante associato al gene huntingtinasul cromosoma 4p16.3
• solo il 3% dei casi è dovuto a nuove mutazioni• un'espansione dinamica della tripletta CAG che
codifica per la glutammina

quante glutammine?
• fino a 28 = numero max di CAG per un soggetto non a rischio
• 29 - 39 CAG la malattia si potrebbe presentare alla generazione successiva (premutazione)
• oltre 39 CAG il soggetto è considerato affetto anche se la patologia non si è ancora manifestata
• il test è in grado di prevedere che la patologia si manifesterà in futuro
• l'esecuzione in soggetti sani solleva problemi di natura etica

Caratteri complessi
Vincenzo Nigro
Dipartimento di Patologia GeneraleSeconda Università degli Studi di Napoli
Telethon Institute of Genetics and Medicine (TIGEM)

Caratteri non mendeliani(non dipendenti dal genotipo in un singolo locus )
•• continui (quantitativi)continui (quantitativi)•• discontinui (dicotomi)discontinui (dicotomi)

~25,000 gene e altri elementi di ~25,000 gene e altri elementi di regolazioneregolazione
Tratti genetici o malattieTratti genetici o malattie
Rapporto genotipo-fenotipo


L’ereditabilità delle malattie comuni è dovuta a geni multipli ciascuno con un piccolo effetto



un carattere non Mendeliano ha una componente genetica?
• i genitori trasmettono– i geni– l’ambiente (questo vale specialmente per caratteri quali IQ e i
disordini psichiatrici)
• anche le abitudini alimentari, il clima, ecc.
• Occorre provare il ruolo dei geni al di là della ricorrenza familiare

• gemelli monozigoti• fratelli, genitori-figli• fratellastri, zii-nipoti• cugini I grado• cugini II grado
• 1/1 consanguineità• 1/2 consanguineità• 1/4 consanguineità• 1/8 consanguineità• 1/32 consanguineità
geni condivisi

gemelli monozigoti MZ
• hanno lo stesso sesso• hanno gli stessi alleli• hanno gli stessi polimorfismi• se femmine, hanno un differente pattern di inattivazione
dell’X• hanno un differente repertorio di immunoglobuline• hanno un differente TCR• spesso hanno un ambiente più simile

gemelli dizigoti DZ
• hanno lo stesso sesso nel 50% dei casi• hanno il 50% degli alleli in comune• hanno il 50% dei polimorfismi in comune
Occorre considerare il rapporto MZ/DZ che può essere inferiore a 2 o superiore a 2 in funzione del numero dei geni coinvolti


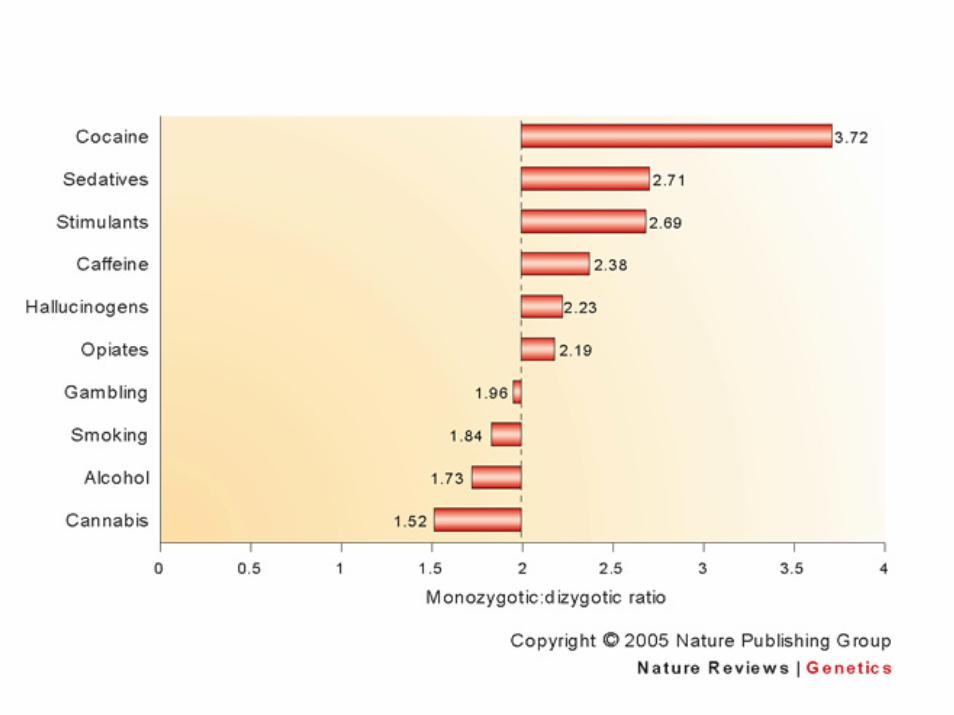

Errori sistematici di accertamento
• quanti figli affetti ha un coppia di portatori di un tratto recessivo?
• ¼?• se hanno due figli• 8/14 (sarebbero 8/32, ma in 9 famiglie non ci sono
affetti)
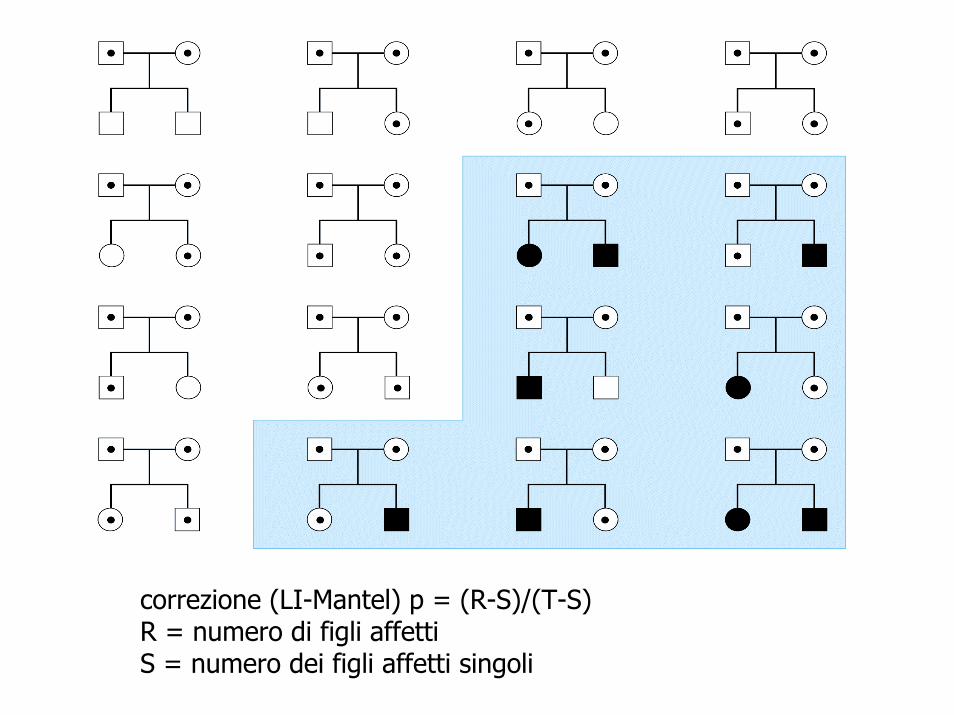
correzione (LI-Mantel) p = (R-S)/(T-S)R = numero di figli affetti S = numero dei figli affetti singoli

Analisi parametricarichiede un preciso modello di trasmissione
una piccola parte degli affetti può avere condizioni Mendeliane indistinguibili dalla maggioranza non Mendeliana e l’identificazione della causa Mendeliana potrebbe on essere di aiuto
Breast Cancer (11 loci), Alzheimer (AD1-AD15) ALS (1-8)Per caso molti fattori di suscettibilità sono presenti nella
maggior parte delle persone studiate e quindi si considera un gene che fa spostare l’equilibrio (Hirschsprung)

Analisi nonparametrica
Senza un modelloNon si sa a priori se la trasmissione è dominante o
recessiva o mista o poligenica, eccSi valuta quanta parte si condivide dei segmenti di DNA
nelle famiglie o nelle popolazioni

identity by state IBS
significa che gli alleli sono uguali apparentemente, ma non è conosciuta la loro origine
identity by descent IBD
significa che gli alleli sono uguali perché hanno una comune origine

con una segregazione casuale una coppia di fratelli (sib-pair) condivide 2 alleli AC, 1 allele AD e BC, oppure o alleli BD per ogni segmento di DNA
affected sib pairs ASP

Se la ipotetica condizione è dominante, 1 allele deve essere in comune
affected sib pairs ASP

necessariamente i fratelli devono avere in comune 2 alleli

‘Non-parametric’ linkageMisura se gli affetti condividono dei marcatori ereditati da un comune progenitore IBD più frequentemente rispetto al caso
Marker unlinkedto susceptibility gene sib-pairs share alleles according to chance
sib-pairs share alleles more often than expected by chance
25% 50% 25%
40% 55% 5%Marker linked to susceptibility gene
A B C D A B C D A B C D
2 alleli 1 allele 0 alleliA C A C A C A D A C B D
* * *
* * * * **

Problemi con il linkage non parametrico
Meno sensibile e quindi sono richiesti più sib-pairsPiù imprecisoPiù difficilmente si può arrivare al geneSoglia di significatività più elevataLOD score significativo tra 3.6 e 5.3LOD score altamente significativo >5.3Difficoltà a replicare gli studi in una differente
popolazione

Studi di associazione
• Si testa se la presenza di una specifica variante genica aumenta il rischio di ammalarsi
• Confrontano la frequenza dei markers tra casi e controlli
Case-control
Test for different frequency of an allele incases vs controls
Parental alleles:Trasmitted -> A ANon trasmitted -> B B
A B A B
A A
Family based association tests(TDT)
Parental alleles not trasmitted to probandsare used as “internal controls”

TdT
• Transmission disequilibrium test• (a-b)2/(a+b)• a è il numero delle volte in cui un allele è
trasmesso • b è il numero delle volte in cui l’altro allele
è trasmesso

Linkage disequilibrium mapping
M=allele of a marker on the same haplotype as the *
mutation
Se le variazioni nella stessa regione non sono indipendenti sono in linkage disequilibrium

GWA (genomic wide association) studies
• GWAs servono a comprendere la base genetica dei tratti complessi
• Per alcuni tratti ci sono molti loci: diabete, cancro della prostata e della mammella, malattia infiammatoria intestinale, ecc
• Per altri tratti i loci sono pochi: asma, malattia coronarica, fibrillazione atriale
• Sono diventati il metodo migliore per identificare fattori di rischio per malattie complesse
• Più potente del linkage se le varianti che conferiscono suscettibilità sono comuni, mentre è meno potente se le varianti sono rare
• È necessario procedere con una mappa che comprende molti marcatori a breve distanza l’uno dall’altro

usare SNPs comuniAssociazione indiretta
Gli SNPs sono genotipizzati sulla base del linkage disequilibrium (LD)
Minor allele frequency = MAF• MAF > 0.05 alleli comuni usati per GWA• MAF >0.01 e <0.05 polimorfismi non comuni• MAF<0.01 varianti rare innocue o mutazioni
recessive

Infinium, IlluminaInfinium HD
BeadChipMarkers
(per sample)
Median Marker Spacing
Human1M-Duo
>1.1 million 1.5kb
Human660W-Quad
>658,000 2.5kb
HumanCytoSNP-12
~300,000 10kb
Human510S-Duo* >510,000 3.2kb
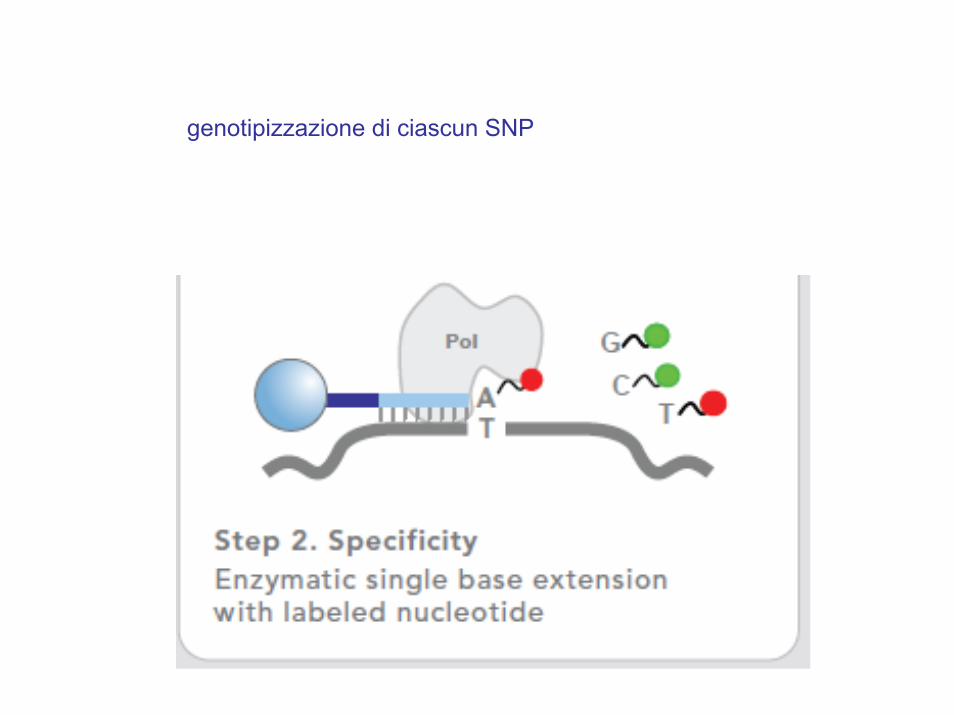
genotipizzazione di ciascun SNP

aploblocchi nel genoma
all common variants>0.05; 6 000 K
gene nr 24680
100 kb

Gli aploblocchi sono dovuti alla mancanza di ricombinazioneGli aploblocchi sono dovuti alla mancanza di ricombinazione

punti critici dei GWAs• Selezione dei casi
– minimizzare l’eterogeneità fenotipica?– focalizzazione su casi familiari?– scegliere casi ad insorgenza precoce?
• Selezione dei controlli– Controlli comuni a differenti tratti?– 3000 controlli della Wellcome Trust Case Control
Consortium• Significatività statistica
– GWS genome wide significance = p < 5 x 10–8– Replica dello studio su una differente popolazione

Tremore• 02/25/09 Stefansson
February 01, 2009 Nat GenetVariant in the sequence of the LINGO1 gene confers risk of essential tremor Essential tremor
• 452 cases• 14,378 controls• 15q24.3• LINGO1• 1 x 10-9 • Illumina [305,624]

obesità• 01/15/09Thorleifsson December 14, 200Nat Genet
Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity
• Body mass index• Studio iniziale: 80,969 individuals• Replica: 11,036 individuals• 16q12.2 FTO 1 x 10-47 • 2p25.3 TMEM18 4 x 10-17 • 16q12.2 18q21.32 19q13.11 1p31.1 3q27.2 16p11.2 11p14.1 11p14.1 1q25.2
12q13.13 1p21.3 11p14.1• FTO MC4R KCTD15, CHST8 NEGR1 SFRS10, ETV5, DGKG SH2B1, ATP2A1
BDNF BDNF SEC16B, RASAL2 BCDIN3D, FAIM2 NR BDN• 4 x 10-13 1 x 10-12 7 x 10-12 1 x 10-11 7 x 10-11 3 x 10-10 5 x 10-10 9 x 10-10 6 x
10-8 1 x 10-7 4 x 10-6 8 x 10-6 8.04 [6.96-9.12] % SD 6.12 [4.69-7.55] % SD 5.25 [3.82-6.68] % SD 4.38 [3.16-5.60] % SD 4.18 [2.98-5.38] % SD 3.77 [2.67-4.87] % SD 4.42 [3.09-5.75] % SD 3.63 [2.49-4.77] % SD 4.58 [3.07-6.09] % SD 3.85 [2.62-5.08] % SD 3.36 [2.14-4.58] % SD 3.28 [2.06-4.50] % SD 2.6 [1.50-3.70] % SD 3.15 [1.78-4.52] % SD
• Illumina [305,846]

Aneurismi intracranici• Bilguvar November 09, 2008 Nat Genet
Susceptibility loci for intracranial aneurysm in European and Japanese populations
• 1,580 European cases, 6,276 European controls• 495 Japanese cases, 676 Japanese controls• 8q11.23 SOX17 1 x 10-10 • 9q21.3 CDKN2A CDKN2B 1 x 10-10 • Illumina [289,271]

Manhattan plot
GWA Diabete di tipo 2Crom 10 TCF7L2 transcription factor 7-like 2Crom 16 FTO fat mass and obesity associatedCrom 6 CDKAL1 = CDK5 regulatory subunit associated protein 1-like 1
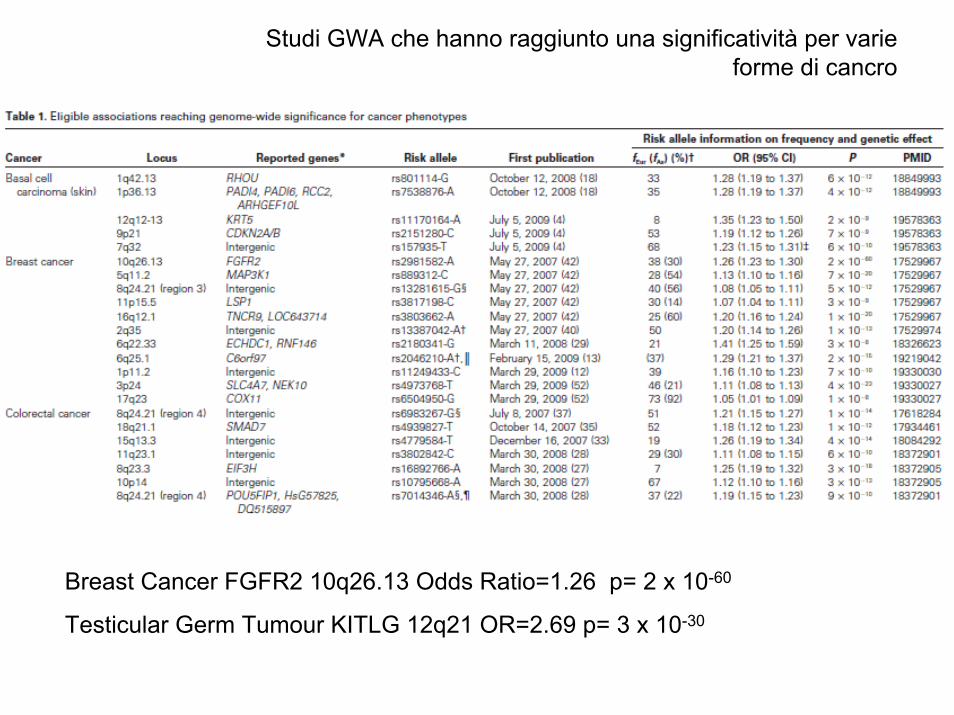
Studi GWA che hanno raggiunto una significatività per varie forme di cancro
Breast Cancer FGFR2 10q26.13 Odds Ratio=1.26 p= 2 x 10-60
Testicular Germ Tumour KITLG 12q21 OR=2.69 p= 3 x 10-30

Effetto combinato delle varianti in quattro forme di cancro

ruolo delle delezioni nella predisposizione genetica a una malattia

Associazione sinteticaIl blocco di LD non èabbastanza grande da includere le varianti più rare associate alla malattia e si potrebbe pensare che la variante comune abbia una funzione regolatoria








Materia oscuraDark matter


Concetti generali di terapia genica
• Inibizione mirata dell’espressione genica per bloccare un processo patologico
• Distruzione mirata geneticamente di specifiche cellule• Supplementazione: fornire una copia funzionante del
gene difettoso• Sostituzione: sostituire il gene mutante con una copia
funzionante in situ




Supplementazione: fornire una copia funzionante del gene difettoso
• ex vivo: trasferire i geni clonati in cellule in coltura, selezionare le cellule, espanderle in vitro e poi immetterle nel paziente
• in vivo: trasferire direttamente i geni nel tessuto bersaglio o in circolo, facendo in modo che poi arrivi al tessuto bersaglio

ex vivo

Vettori non virali per la terapia genica
• Liposomi: vescicole sintetiche che si formano quando alcuni lipidi sono in soluzione acquosa. Possono essere anionici e circondano il DNA o cationici e legano il DNA all’esterno
• iniezione diretta di plasmidi o bombardamento del tessuto del DNA attaccato a pellets di metallo (biolistico). Il trasferimento è molto basso e il DNA non è integrato.

Liposomi

Virus per la terapia genica
• Retrovirus: sono ad RNA e sintetizzano cDNA che integrano casualmente nel genoma dell’ospite quando si dissolve la membrana nucleare (divisione cellulare)
• Adenovirus: sono a DNA e capaci di trasdurre ad alti titoli tutte le cellule, ma in forma episomale. E’ molto forte la reazione immunitaria. Morte di Jesse Gelsinger nel 1999
• Adeno-associati (AAV) hanno DNA a singola elica e si potrebbero integrare sul cromosoma 19q13 grazie al gene rep. Ma il 96% del genoma è deleto. Possono ospitare fino a 4.5kb
• Lentivirus: sono retrovirus specializzati che infettano anche le cellule non in divisione. Sono più complessi dei retrovirus e sono capaci di un’espressione a lungo termine.

Vettori virali per la terapia genica
Genoma Stato Capacità Target
AAV Dna IntegratoEpisomal
4.7 kb D/ND
Adeno Dna Episomal < 36 kb D/ND
Retro Rna Integrato 8 kb D
Lenti Rna Integrato 8 kb D/ND
HSV Dna Episomal >50kb D/ND

AAV2
Virus Adeno-associato
Famiglia: ParvovirideGenere : DependovirusGenoma : DNAss 4.7kbBersaglio: Cellule in divisione e cellule non in divisioneStato : episomale/integrazione sito specifica su 19q13.3 Sierotipi : 10 AAV1-10
ITR
Rep Cap
ITR
Rep78
Rep68
Rep52
Rep40
VP1
VP2
VP3
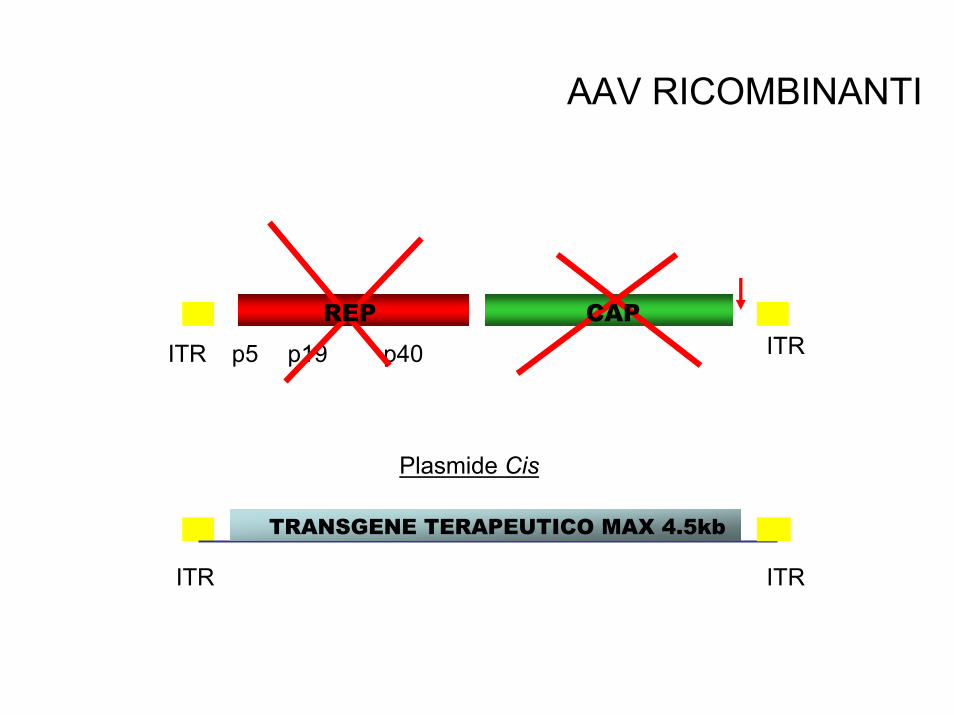
AAV RICOMBINANTI
Plasmide Cis
ITR ITR
TRANSGENE
p5 p19 p40ITR ITRREP CAP
TRANSGENE TERAPEUTICO MAX 4.5kb

Tripla trasfezione
HEK 293 cells(E1)
Transrep2 cappro
E2A E4 VAI
Adeno Helper
TransgeneITR2 ITR2
Cis
Produzione di vettori rAAV
rAAV
Pro pA

AAV2/1 (6)
AAV2/2 Rep 2 Cap 2ITR 2ITR 2
AAV2/5 Rep 2 Cap 5ITR 2ITR 2
AAV2/3 Rep 2 Cap 3ITR 2ITR 2
AAV2/7 Rep 2 Cap 7ITR 2ITR 2
AAV2/4 Rep 2 Cap 4ITR 2ITR 2
AAV2/8 Rep 2 Cap 8ITR 2ITR 2
Cap 1ITR 2ITR 2
Rep 2gene terapeutico
rAAV Pseudotipizzati

DIFFERENZE TRA SIEROTIPI AAV 1-8
PROTEINE CAPSIDE
RECETTORIRECETTORI
AAV2 AAV2 FGFR1,EPARINAFGFR1,EPARINA
AAV4AAV4
Ac.SIALICOAc.SIALICO
AAV5 AAV5
PDGFR, Ac.SIALICOPDGFR, Ac.SIALICO
TROPISMOTROPISMO
AAV1AAV1 Muscolo,retinaMuscolo,retina
AAV2 AAV2 Muscolo,fegatoMuscolo,fegato
AAV4AAV4 CervelloCervello
AAV5AAV5 Cervello,polm.Cervello,polm.
AAV6 AAV6 Muscolo,retinaMuscolo,retina
AAV7AAV7 Muscolo,fegatoMuscolo,fegato
AAV8AAV8 FegatoFegato
ANTIGENICITA’

•Infezione di cellule in divisione e non in divisione
Vantaggi
Svantaggi•Produzione di anticorpi anti-AAV AAV2
No risomministrazione del vettore virale
•Dimensioni dell’inserto non superiori alle 4.5kb
•Non patogeni•Espressione genica efficiente e a lungo termine
•Pochi effetti immunologici•Ampia varietà di cellule ospiti






























