Embed Size (px)
Citation preview

/ OBTENCION ELECTROQUIMICA DE DEPOSITOS DE ESPECIES DE
SUURIO, An€RICIO Y PLUTONIO.
CALCüLO DE LAS PROBABILIDADES DE MISION ALFA DE
Ai-241, Ri-239 Y Pu-240
ARdBECERRIL VILCHIS
PARA OBTMER EL GRADO DE:
L)oclwt EN CIENCIAS
UNIVERSIDAD AunmollA CtETRopOLITANA
DIVISION DE CIEWCAS BASICAS E INGENIERIA
EWER0 DE 1994

1 4 4 1 5 9
4
Estetrrrbqjo de tesis fue dirigido por e l k . Yunny Meas Voug y en eiia se encuentra tambh la colaborsclbn de: Alberto Rojas, John Bland,
- Jean " d f y , Christine Upy, IRsIcIpi Tavera, I[nnacio Gondez y n en la
discusi6n y revisión: Sivia Buibuiian, Elsa Arce, Laura Gaiicia y Ariel Tejera.
.. algunas personas mgs. De la mhna manera parmplw
$

Se contd con el apoyo económico de ias sigui- iastitucioneS:
CONACyT, México SEP, México O m , Austria I", México UAM-I, M e 0 CEA, FRancia Univemd de w, Canadá.
a
,

INDICE
Resinen
I. Introducción
11. Generalidades
11.1. Revisión bibliográfica
11.2. Estados de oxidación
I I. 2.1. Lantánidos
11.2.2. Actínidos
111. Fisicoquímica de l a disolución
111.1. Teoría
111.2. Resultados
I I I. 2.1. Sistema Sm/H2S04/H20/e-
I1 I. 2.2. Sistema Am/H2C04/H20/e-
I I I. 2.3. Sistema WH2S04/H20/e-
IV. Modelo de Hansen
IV. 1. Teoría
IV. 2. Resultados
V. Técnicas experimentales
V. 1. Equipos
V.l.l; Electrodos y celdas electroquímicas
V.1.2. Motor
V. 1.3. Caja de Faraday
Página 1
4
8
8
10
10
12
16
18
31
31
37
45
53
54
60
63
63
63
67
67 !

V. 1.4. Potenciostato
V. 1.5 . Sistema de medidas electroquímicas
V. 1.6 . Disco rotatorio
V. 1.7 . Medidor de pH
V. 1.8. Balanzas
V. 1.9. Caja de Guantes
V. 1.10. Espectrómetro de f,uorescencia de Rx
V. 1.11. Espectrómetro gamma
V. 1.12. Espectrómetro alfa
V. 1.13. Analizador de imágenes
V. 2. Reactivos
V. 3. Procedimientos
V. 3.1. Estudio electroquímico
V. 3.1.1. Voltamperornetria cíclica
V. 3.1.2. Voltamperornetria en régimen de
difusión estacionario
V. 3.1.3. Impedancia electroquímica
V. 3.2. Optimización del método de depósito
V.3.2.1. pH y pS0; del electrolito soporte
V. 3.2.2. Densidad del corriente
V.3.2.3. Velocidad de rotación del cátodo
V. 3.2.4. Tiempo de depósito
V. 3.3. Espectrometría
V. 3.3.1. Fluorescencia de rayos X
V. 3.3.2. Espectrometría gamma
68
60
68
68
69
69
69
70
70
71
71
72
72
72
73
74
75
76
77
78
78
79
79
79

V. 3.3.3. Espectrometría alfa
V. 3.4. Caracterización física de los depósitos
V. 3.4.1. Alfagrafía
V. 3.4.2. Perf ilometría
VI. Estudio electroquímico
VI.l. Voltamperometría en régimen de
difusión estacionario
VI. 2. Voltamperometría de barrido triangular
VI.3. Análisis armónico de la impedancia
electroquímica
VII. Estudio de parámetros experimentales
VI I. 1. Resultados VI I I. Espectrome tr í a
VIII.l. Métodos de ajuste
VI I I. 1 . 1 . Adquisición de la medida
VIII. 1.2. Función de ajuste
VIII. 1.2.1. Modelo matemático
VIII. 1.3. Criterio de ajuste .- ___
VIII. 1.3.1. Criterio de mínimos cuadrados
VIII. 1.3.2. Criterio de x” VIII. 1.3.3. Criterio de Poisson
VIII. 1.4. Algoritmo de ajuste
VIII. 1.5. Evaluación de la incertidumbre de los
parámetros de ajuste VI I I. 2. Resultados VIII.2.1. Espectrometría alfa
80
81
81
82
83
84
87
89
92
97
112
112
114
114
115
119
119
120
121
121
122
124
125

VIII.2.1.1. Americio-241
VIII.2.1.2. Plutonio-239 y 240
VIII.2.2. Espectrometría X y gamma
VIII. 2.2.1. Espectrometría X
VIII. 2.2.2. Espectrometría gamma
IX. Conclusiones
Anexos
Referencias
126
133
136
137
137
139
143
173

La generación de desechos emisores alfa como el plutonio y
el americio provenientes principalmente de las centrales nucleoeléctricas y de la industria nuclear y su posterior disposición en formaciones geológicas así corno el empleo de combustible mixto U/Pu en los nuevos reactores generadores de electricidad y el empleo de los mismos radisótopos en investigación básica, plantean la necesidad de contar con datos nucleares de dichos elementos. Por ejemplo, el estudio de la estructura fina de emisión alfa por espectrometría, depende de un espectro con alta resolución y de un valor alto de la relación máximo del pico/fondo espectral. La calidad de los espectros con las características descritas depende básicamente de la calidad de la fuente radiactiva origen del espectro alfa y de la resolución intrínseca del sistema de detección utilizado. A su
vez, la calidad de la fuente radiactiva depende de la calidad de la película de material radiactivo depositada.
Por otro lado, la precisión del cálculo de las probabilidades de emisión alfa depende de que el algoritmo de convolución de los picos del espectro tome en cuenta los fenómenos que concurren en la detección de las partículas alfa. _ _ _
El método electroquímico de depósito, de uso generalizado, reportado en la literatura 11-33] ha sido estudiado de manera semiempírica, proporcionando aún asi depósitos uniformes y
delgados. Sin embargo, el método electroquímico puede proporcionar depósitos de mejor calidad a condición de que se realicen estudios sistemáticos y con pleno conocimiento de los fundamentos. La calidad de los depósitos depende de las características fisicoquímicas de la solución electrolítica y del tipo de celda electroquímica empleada.
A través del estudio fisicoquinico es posible predecir la
1

naturaleza química de las especies presentes en la solución y
también las condiciones para la formación de nuevas fases sólidas (depósitos). Utilizando el modelo de Hansen [15,161 ylos datos proporcionados por el estudio fisicoquímico, es posible calcular las mejores condiciones de pH de la solución y de densidad de corriente para la obtención de depósitos delgados y uniformes.
Por otro lado, aplicando la teoría del electrodo disco
rotatorio es posible determinar los criterios de diseño de la celda electroquímica que se va a emplear con el fin de optimizar los depósitos. La presencia de una capa límite de difusión (en regimen estacionario) en la superficie del cátodo y de un flujo laminar de solución hacia la superficie del mismo permiten controlar la calidad del depósito en espesor y uniformidad.
El diseño y análisis estadistico de los experimentos de optimización del proceso de depósito permi ten anal izar de manera sistemática y consistente la participación de los parámetros experimentales (pH y pS0 ' de la solución, densidad de corriente (i), velocidad de rotación del cátodo (u) y tiempo de depósito (t)). En cuanto al algoritmo de convolución de los espectros alfa se refiere, es preciso el empleo de una función de ajuste que tome en cuenta las diferentes contribuciones a la forma de los picos alfa del espectro, como por ejemplo (asumiendo que se emplea un
I
detector de estado sólido tipo PIPS): ~ _ _
1 . el "Straggling" de la pérdida de energía de los electrones en la capa muerta del detector,
2. las variaciones del espesor de la capa muerta del detector, 3. la ionización y excitación electrónica (en el detector) por
interacción con las partículas alfa,
4. la estadística con la que se crean los pares ión-agujero en el detector,
5. la resolución del sistema de amplificación.
2

Se calcularon las probabilidades de emisión alfa del Americio 241 y del Plutonio 239 y 240, así como la relación isotópica de un muestra problema de Pu239/240. La línea a3 del
Am-241. cuyo último estudio data de los años 60 [los], fue indentificada y calculada su intensidad relativa de emisión. El resto de las líneas estudiadas coinciden con los valores reportados [63,106,1071 aunque se ganó en la precisión del cálculo. Igualmente se determinaron las intensidades de emisión del Pu-239 y 240 difiriendo de manera notable con la literatura [lo71 los valores de intensidades de emisión de las líneas a3 y a4 del Pu-239.
Los resultados del estudio electroquímico del depósito, llevado a cabo con el samario como elemento representativo del Am-241 y del Pu-239 en el sistema electroquímico. pudieron hacerse extensivos para el estudio de optimización del depósito de Am-241 y Pu-239.
El estudio fisicoquímico de la solución proporcionó criterios más precisos para el uso del modelo de Hansen lo cual condujo a una mayor correlación entre las previsiones teóricas y
los resultados experimentales. El diseño de la celda electroquímica basado en criterios
hidrodinámicos y los resultados del análisis estadísticos de la ~
optimización del proceso de depósito permitió obtener depósitos delgados (< 50 1 1 y uniformes (210%) con altos rendimientos de--- depósito (97.5+2.5%).
3
i

I. INTRODUCCION
Existe un gran interés en la fisicoquímica de los lantánidoc y de los actínidos. Su uso y aplicaciones en la física y la tecnología
nucleares dependen de los estudios fundamentales sobre dichos
elementos. Algunos aspectos que denotan la relevancia de dichos estudios son la presencia de actínidos como el plutonio en el ambiente y el problema de la disposición definitiva de los desechos radiactivos. El uso de reactores nucleares de tercera generación para la producción de energía eléctrica requiere de elementos combustibles mixtos a base de uranio y de plutonio, de
ahí la necesidad de métodos de control de pureza isotópica, y de datos nucleares como las probabilidades de emisión alfa que son utilizados en investigación fundamental y aplicada. En todos estos aspectos encontramos tres elementos comunes y de gran importancia: la fisicoquímica de los elementos de la serie f, el método de separación y el método de análisis como la espectrometria alfa. Cabe señalar que la investigación con actínidos involucra algunas limitantes relacionadas con su naturaleza radiotóxica y con lo limitado de las cantidades disponibles. Los experimentos se deben realizar en campanas aspiradas con filtros absolutos y/o en cajas de guantes hermeticas y provistas con filtros absolutos. Es de primera importancia la contención del elemento radiactivo y el diseño y tipo de experimentos. Por otra parte, la gran mayoría de los trabajos publicados tienen que ver más con el trabajo experimental semiempírico que con el estudio sistemático retroalimentado con elementos teóricos. Se hace referencia al método de depósito electroquímico [l-321 en medio acuoso para la separación y recuperación de actínidos. En particular este método presenta ventajas tales como la abundancia de datos electroquímicos y fisicoquímicos en la literatura, y la
~-
-~
4

f
fácil realización experimental (el equipamiento es sencillo, de bajo costo y los experimentos no son sofisticados). Además, los resultados que proporciona el método son de calidad comparable con la de otros métodos más sofisticados y onerosos. El método electroquímico Hansen [15,161 presenta un desarrollo experimental basado en consideraciones teóricas, aunque con limitaciones en lo que se refiere a la hidrodinámica y transporte de materia, y a la fisicoquímica de las disoluciones entre otras cosas.
Para tener resultados reproducibles en la obtención de películas delgadas y uniformesse requiere del estudio del comportamiento de
las especies químicas en solución y de las condiciones de formación de nuevas fases, así como de las condiciones
hidrodinámicas del dispositivo electroquímico. También se requiere encontrar las mejores condiciones de obtención de
películas analizando de manera sistemática y consistente los parámetros experimentales que intervienen en el proceso. El trabajo abordará el estudio fisicoquímico de la solución electrolitica, el análisis de las condiciones hidrodinámícas del dispositivo electroquimico y el diseño y análisis estadístico de
los experimentos de optirnización del proceso de depósito. Por otro lado, el cálculo preciso de las probabilidades de emisión alfa requiere de un algoritmo capaz de convolucionar picos alfa considerando los fenómenos que se presentan durante la detección de las partículas alfa y que determinan la forma de los picos y de los espectros. El objetivo de este trabajo es medir y
calcular las probabilidades de emisión alfa del Am-241 del Pu-239 y del Pu-240 así como la relación isotópica de una mezcla de
Pu-2391240.
Entonces en el presente trabajo se contemplan los siguientes aspectos:
- Un estudio f isicoquímico de la disolución electrolítica
(electrolito soporte + sal del elemento + agua) para l a
predicción de la distribución y predominio de las diferentes
i

especies químicas presentes al inicio del proceso, y para la
predicción de las condiciones de aparición de nuevas fases (depósito de un sólido). Todo esto en función de la concentración del electrolito soporte, de la acidez de la disolución y de la concentración del elemento. La predicción de las especies presentes y las condiciones de aparición de nuevas fases pueden contribuir a la comprensión del estudio electroquímico (cap. 111). La predicción de las condiciones de aparición de nuevas fases se puede complementar con la teoría de Hansen para definir condiciones Óptimas de depósito (cap. IV).
- Un estudio electroquimico del proceso de depósito de actínidos simulando su presencia por medio del samario que tiene propiedades fisicoquímicas similares a las de los actínidos trivalentes. El samario presenta la ventaja de no ser radiactivo y de fácil disponibilidad. Métodos como la voltamperornetria cíclica, la voltamperornetria en régimen de difusión estacionario y la espectroscopía de impedancia electroquímica, proporcionan información sobre la influencia del medio químico sobre el proceso y sobre los mecanismos de reacción del proceso (cap. VI.
- Un estudio hidrodinámico para diseñar la celda electroquímica. Una celda con un cátodo rotatorio diseñados para satisfacer las condiciones hidrodinámicas contempladas en la teoría del electrodo disco rotatorio (RDE), permiten emplear la teoría de Hansen basada en condiciones estacionarias de depósito conservando un flujo laminar normal a la superficie del cátodo (anexo 3) .
- Un diseño experimental para la optimízación del método de depósito. El diseño estadístico de experimentos permite optimizar el método de depósito con un mínimo de experimentos. Los resultados tienen en ese momento significancia estadística como
6

consecuencia de un estudio sistemático apoyado en métodos estadísticos. En el estudio se conoce la influencia de cada parámetro y se obtiene la función analítica que la describe. También se puede analizar la influencia de parámetros
interrelacíonados (como es el caso de la densidad de corriente y
el pH de la disolución) y a su vez obtener la función analitica que la describa donde se incluye el término cruzado (cap. VII).
- Un método de análisis de espectros alfa de alta resolución que permita análizar el espectro, medir y calcular las probabilidades de emisión alfa tomando en cuenta la estructura fina de emisión.
Además, medir y calcular las fracciones o relaciones isotópicas de una fuente radiactiva. La convolución de espectros alfa empleando un método iterativo para ajustar una función a un espectro experimental aplicando un criterio de miniiización como el SIMPLEX permite analizar la bondad del ajuste y calcular los errores asociados a los valores calculados (cap. VIII).
7

11. GEWERUIDADES
11.1. Revisión bibliográfica
Las fuentes radiactivas se obtienen por diferentes métodos de los cuales cuatro son los mas importantes [l, 21 : El primero es la electropulverización que consiste en pulverizar y evaporar una disolución del material radiactivo, que se va a depositar, bajo el efecto de un campo eléctrico muy intenso (del orden de 10000 VI. La sal del material se deposita en finos cristales sobre el cátodo con un rendimiento del orden del 70%.
la calidad de los depósitos es regular y la realización complicada. El segundo es la sublimación al vacío (de a lo-' Pa), este consiste en sublimar el óxido del material radiactivo por efecto joule, por bombardeo electrónico o por aplicación de un laser. Los depósitos son de buena calidad pero con un rendimiento del orden de 10%. la pérdida del material radiactivo tiene como
consecuencia la contaminación del equipo y un proceso costoso. El tercero es la pulverhación catódica o "sputtering" donde el material radiactivo se deposita sobre un disco metálico por
efecto de un haz de iones argón focalizado sobre el recipiente del material (el haz eyecta el material del recipiente proyectándolo sobre el disco metálico). El rendimiento es del orden del 20% y la realización es difícil y costosa. La ventaja de este Último método es la calidad de los depósitos que proporcionan espectros con valores de resolución de 10f0.1 KeV. Los métodos descritos anteriormente presentan rendimientos bajos, necesitan de una instrumentación costosa e involucran procedimientos complicados y largos.
8

El cuarto es el método electroquímico, el cual es sencillo en su
realización y de bajo costo. Los métodos de depósito electroquímico de actinidos en medios orgánicos 13-51, en medios mixtos 16-91 y en sales fundidas [ l o ]
son empíricos debido a que se conoce mal el mecanismo de depósito y la influencia de los parámetros, además de que es difícil controlar las condiciones experimentales. Esto se explica en parte por la falta de información necesaria para llevar a cabo dichos estudios. Por el contrario el método en medio acuoso ha sido más estudiado 111-321 y en la parte teórica sobresalen los trabajos de Hansen [15,161. Se reportan rendimientos de hasta 100% con depósitos adherentes y poco uniformes. Tiene además el inconveniente de que se requieren tiempos largos de depósito, del orden de 100
mi nut os.
El depósito electroquímico de lantánidos y de actínidos es diferente del depósito metálico, esto se debe a dos características presentes en ambas familias de elementos: - todos los potenciales redox de los lantánidos y de los actínidos son mucho más negativos que el potencial de reducción del agua en medio ácido [331. Esto hace que en medio acuoso ácido la reducción del agua se lleve a cabo antes que la de los lantánidos y actínidos y que el estado metálico de los mismos sea ines tab 1 e,
- los lantánidos y los actínidos se hidrolizan bajo ciertas
-
___
condiciones de pH. La ecuación simplificada de la hidrólisis es:
-5 nM"+ + JOH- *
donde es el ión del lantánido o del actínido y OH el ión hidroxilo, con Ks= Ihf+In IOH-IJ como la constante de solubilidad. Además, como se verá en la sección correspondiente para cada
9

actínido y lantánido y para cada estado de oxidación existen numerosas reacciones incluidas las de formación de especies polimérícas como es el caso del plutonio y que es necesario también considerar. Las características mencionadas arriba se combinan para indicar que el mecanismo de depósito que involucra al menos una reacción electroquímica y una química. De manera general y esquemática el proceso se divide en dos etapas: - una reacción electroquímica de reducción del agua en medio ácido:
-t-- H ~ O + H+ + 2 e H T + OH- 2
que como se observa involucra la formación de hidrógeno gaseoso. Además en la interfase cátodo-disolución se forma un gradiente de pH por la formación de iones hidroxilo en la superficie del cátodo generando un'auento en el pH de la interfase, - una reacción química donde los lantánidos o los actínidos precipitan en forma de hidróxidos.La precipitación del hidróxido se inicia cuando el pH aumenta hasta el dominio de las especies hidroxiladas insolubles.
11.2. Estados de oxidación
11.2.1. Lantánidos El estado de oxidación más estable de los lantánidos iLn) es el trivalente. La razón para la estabilidad del estado trivalente no se debe a la configuración electrónica sino más bien a las condiciones termodinámicas de las especies hidratadas. Las energías de ionización ( del orden de 1500 KJ mol-') y las de hidratación (del orden de 3000 KJ mol-') son de la magnitud y signo apropiado para que el estado trivalente sea el más estable
10

[341. Como se observa en la tabla 11.1, también existen los
estados 2 y 4 de algunos lantánidos: Eu(II1, Yb(II1, Sm(II), Ce(IV), Tb(IV) y Pr(1V). La presencia de estas especies se debe
en parte a la configuración electrónica de los iones. Las especies son más estables cuando las capas 4f se encuentran vacias (4f0), semiiienas (4f7) o llenas (4fi4). Por ejemplo, el Ce(1V) es el lantánido tetravalente más estable pues tiene una configuración 4f0, el Eu(I1) es el divalente más estable pues tiene una configuración 4f’. En la tabla 11.2 aparecen las configuraciones de los átomos y de los iones.
Tabla 11.1 Estados de oxidación de los lantánidos
Símbolo
Ce Pr Nd h Sm Eu Gd
Tb
DY Ho Er , Tm Yb
Lu
Estado dea oxidac i ón
3 - a. Las especies entre paréntesis sólo se
de carbonatos y las subrayadas son las más conocen en medio es tab1 es.
11

Tabla 11.2. Configuración electrónica de los lantánidos
Símbolc
Ce
Pr Nd Pm Sm Eu Gd
Tb
DY Ho Er Tm Yb
Lu -
Atomo
4f '5d16s2
4f 36s2
4f '6s2 4f56s2 4f 66s2 4f 76s2
4f '5d16s2 4f 96s2
4f1 06s2
4f 116s2
4f1 26s2 4f1 36s2 4f1 46s2
4f ''5d16s2
4f
4f
4f ' 4f 4f7
4f 75d1
4f
4f1°
4f'l
4f12 4f13 4f"
i ón h ( I I I )
4fl
4f2
4f3
4f4 4f5 4f6
4f7
4f8
4f9
4f1°
4f" 4fi2 4fi3
4f"
4f0
4f'
4f
4f8
a.Ln(I1). El estado divalente es conocido para el Sm, Ey e Yb [34], aunque el Sm(I1) y el Yb(I1) son inestables en agua y aire oxidandose [351.
b.Ln(II1). Es el estado de oxidación más estable para todos los lantánidos.
c.Ln(iV). El estado tetravalente de Ce, Pr y Tb ha sido reportado l34.361. En solución acuosa el Ce(1V) es un poderoso oxidante. El Tb(1V) y el Pr(1V) oxidan el agua por lo que se requiere de
agentes complejantes para estabilizarlos.
11.2.2. Actínidos Como se puede observar en la tabla 11.3 los actínidos (An) .
presentan un mayor núaaero de estados de oxidación que los
12

lantánidos. A partir del americio se encuentra el grupo con un comportamiento similar al de los lantánidos trivalentes y el estado de oxidación más estables es el I11 (excepto para el No).
En contraste el estado de oxidación más estable para el U es el
VI, el cual tiene una configuración Sf'. La configuración de los actínidos se enlista en la tabla 11.4.
Tabla 11.3 Estados de oxidación de los Actínidos
Símbo 1 o
Th Pa U
NP Pu Am
Cm Bk Cf
Es
Fm Md
No Lr
Estado dea oxidación
4
4 s
3 4 5 6 - 3 4 5 6 6 - 3 4 5 6 7 -
-
(2) 3 4 5 E (2) 3 4
3 4 - (2) 3 2 3 2 3 2 3
2 3 - 3
-
- a.Las especies entre paréntesis fueron producidas por radiólisis pulsada y las subrayadas son las más estables.
13
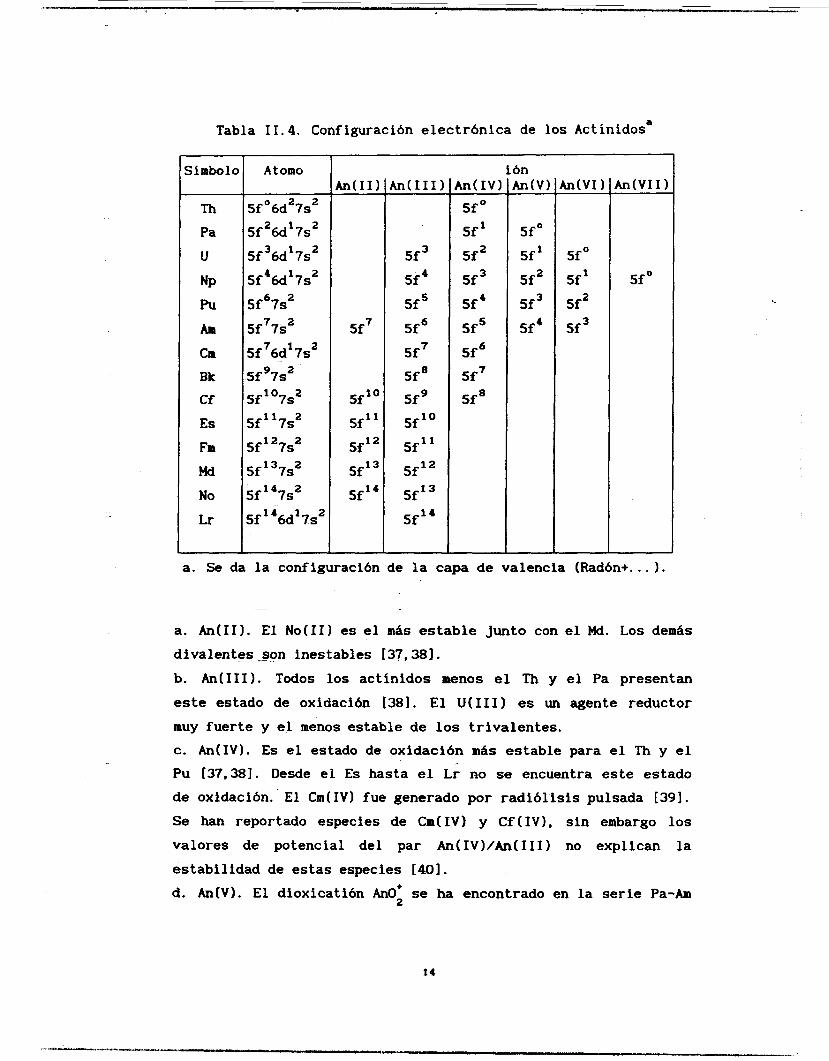
Tabla I I. 4, Configuración electrónica de los Act ínidos'
Síabole
Th Pa U
NP Pu Am Ca Bk Cf Es Fm
Md
No Lr
At omo
5f 06d27s2 5f 26d' 7s2 5f 36d17s2 5f ',6d17S2 5f 67s2
5f 77s2
5f 97s2
5f l O7S2
5f l '7s2 5f 27s2
5f l 37s2
5f 47s2
5f 76d1 7s2
5f1 '6d' 7s2
h(II1
5f7
5fla
5f" 5f12 5f13 5f"
An(II1)
5f
5f 5f5 5f6 5f7 5f 5f Sf lo 5f1' 5f12 5f13 5fl'
4n( IVI 5f0
5fl
5f3 5f4 Sf5 5f6 5f7 5f8
5f
i ón
5f0
5f2 5f
5f 5f
4n(VI)
5f o
5f l 5f2 5f
An(V1 I)
5f O
a. Se da la configuración de la capa de Valencia (Ftadón+ ... 1.
_ _ _
a. An(I1). El No(I1) es el más estable junto con el Md. Los demás divalentes son inestables 137,381.
b. An(II1). Todos los actínidos menos el Th y el Pa presentan este estado de oxidación [381. El U(II1) es un agente reductor muy fuerte y el menos estable de los trivalentes. c. An(IV1. Es el estado de oxidación mas estable para el Th y el Pu [37,381. Desde el Es hasta el Lr no se encuentra este estado de oxidación. El Cm(IV) fue generado por radiólisis pulsada [391.
Se han reportado especies de Crn(IV) y Cf(IV1, sin embargo los valores de potencial del par An(IV)/An(IIII no explican la estabilidad de estas especies [401.
d. An(V1. El dioxicatión Ano; se ha encontrado en la serie Pa-Am
14
.

E 1381. El estado pentavalente es el más estable para el Pa y el
Np. El U(V1, el Pu(V) y el h(V) tienden a dismutar pero pueden ser estabilizados bajo ciertas condiciones [381.
e. An(V1). El estado hexavalente de U,Np,Pu y Am existe como Amor [381. El U(V1) es el estado de oxidación más estable de este elemento. f. An(VI1). El Np(VI1) y el’ Pu(VI1) pueden existir en medio
(3-n) + fuertemente alcalino probablemente bajo 1371. El Np(VI1) tiene la configuración 5f0. El Am(VI1) ha sido
reportado [411 pero otros investigadores no han podido reproducirlo [371.
la forma AnO2(0HIn
15

111. FISIUXJUfl4ICA DE LA DlSOLWIdH
La mayoría de las investigaciones en química nuclear y en radioquímica incluyen la preparación de fuentes radíactivas delgadas y uniformes. Por ejemplo, un número grande de actínidos y lantánidos han sido depositados electroquímicamente [ 1,321,
sobre sustratos metálicos, a partir de reacciones adecuadas de precipitación [15,161. Este tipo de fuentes es muy Útil en estudios de espectroscopía alfa, de probabklidades de emisión alfa y de relaciones ísotópicas de interés en la tecnología nuclear como la elaboración de combustibles mixtos de uranio/plutonio para reactores. En la literatura existen muchas publicaciones sobre diferentes métodos de depósito electroquímico 11-32]. En estos se describen los procedimientos de elaboración y de tratamiento semiempírico de los correspondientes parámetros experimentales. Esto podría ser explicado si se toma en cuenta que el depósito electroquímico de una fase sólida es un proceso multiparamétríco, algunos de estos parámetros son:
~~
- comportamiento fisicoquímico de la solucibn electrolítica, - perfil hidrodinámico de la celda electroquímica, - naturaleza de los electrodos, - potencial/corriente de depósito,
_-
- tiempo de depósito ....
El comportamiento fisicoquímico de la solución electrolítica y la composición del electrolito soporte son tratados de manera empírica y en ciertas ocasiones no son objeto de estudio. Generalmente se estudia la influencia del electrolito soporte sobre el rendimiento de depósito tomando en cuenta la composición y la concentración del mismo.
16

A ú n cuando métodos termodinámicos de estudio como los diagramas de zonas de predominio L48.491 y los diagramas tipo Pourbaix [751
presentan limitaciones como:
- desviaciones cinéticas al comportamiento termodinámico del sistema,
- desviaciones al comportamiento ideal del mismo sistema,
éstos han contribuido al entendimiento de la relación que existe entre la fisicoquímica y la composición de la solucióri electrolítica y el proceso de depósito electrolítico. A partir de 1986 Rojas et al publicaron una serie de artículos 142-471 sobre un nuevo método para predecir el comportamiento de las diferentes especies quimicas presentes en una disolución dada y para predecir el comportamiento de la misma solución en un sistema electroquímico. Por extensión este método puede ser utilizado para predecir las condiciones iniciales de la solución electrolítica y las condiciones de depósito, en la interfase electrodo-solución, cuando se aplica una densidad de corriente constante a un electrodo de disco rotatorio. A partir de cierto momento se alcanza un "equilibrio dinámico" donde el gradiente de
- _-
pH en la interfase presenta un perfil casi constante como el espesor de la capa límite de difusión. El método de Rojas et al se basa en los conceptos de especies y equilibrios generalizados para representar la distribución de las especies químicas en solución y en fase condensada. En ambas fases se toman encuenta todas las especies complejadas y polinucleares del lantánido o del actinido al interaccionar con el electrolito soporte y con las especies generadas durante la reducción del agua en medio ácido. A partir de constantes termodinámicas publicadas se construyen diagramas tipo PZD, PED y PTD. Es preciso señalar que tanto las
!
17

constantes de equilibrio como los potenciales redox deben ser seleccionados cuidadosamente por lo que es conveniente contar con datos provenientes de al menos dos autores diferentes para seleccionar los valores que se consideren mas confiables y
precisos. Los sistemas estudiados son los siguientes:
1. Samar io/H2S04/H20/e-
2. her icio/H2S04/H20/e-
3. P lu tonio/H2S04/H20/e-
111.1. Teoría
El método de las especies y equilibrios generalizados de Rojas-Hernández et al [42-471 para el estudio de la distribución de las especies químicas en solución acuosa se basa en la generalización de los conceptos de Ringboi 1481 y Charlot [491. A
partir de especies y equilibrios generalizados es posible estudiar sistemas multicomponentes y multirreaccionantes considerando especies complejadas y/o polinucleares en solución, en equilibrio con fases condensadas. El tratamiento matemático de dichos equilibrios proporciona valores o ecuaciones que definen constantes condicionales, las cuales gráficamente se encuentran expresadas por lineas rectas o curvas que aparecen como fronteras entre las zonas de predominio de las diferentes especies en solución y en fases condensadas (diagramas de zonas de predominio
(PZD) y diagramas de existencia-predominio (PED)). Es conveniente señalar que al imponerse una fuerza iónica, es posible expresar las constantes de equilibrio en función de las concentraciones molares de las especies. Esto es conveniente puesto que las zonas de predominio que aparecen en los diferentes diagramas pueden interpretarse como ZOMS de predominio de
18

concentración. Ahora bien, si a los diagramas de existencia-predominio (PED) y a los de zonas de predominio (PZD) les aplicamos restricciones como la concentración del ión de interés y la concentración del electrolito soporte (en un sistema electroquímico) se generan intervalos unidimensionales de predominio de especies y fases en su caso. Si a dichas especies las involucramos en reacciones de transferencia de electrones entonces podemos obtener las ecuaciones de potencial en función del pH y a partir de ellas obtener los diagramas tipo Pourbaix (PTD). En los PTD aparecen todos los estados de oxidación del elemento de interés. Cabe aclarar que cada estado de oxidación del elemento de interés requiere de un diagrama PZD y / o de un PED. La generalización del método ha sido demostrada por Rojas et al [42-471 por lo que a continuación se expone la teoría que se aplica a un sistema electroquímico de cuatro componentes, M/L/X/e . En el desarrollo se omiten, por simplicidad, las cargas eléctricas de las especies. En la primera parte se expone el fundamento sobre la construcción de los diagramas, familiarizando al lector con los conceptos y
nomenclatura utilizada en el desarrollo del método. Y en la segunda parte se presentan los algoritmos empleados para construir los diferentes diagramas partiendo del caso de especies solubles mononuclares (complejos mixtos), luego el de especies solubles (complejos mixtos y polinucleares), después el de especies solubles en equilibrio con fases condensadas (complejos mixtos y polinucleares presentes en ambas fases). En los casos anteriores solo se analiza un sólo estado de oxidación. Finalmente el caso de especies solubles (complejos mixtos y
polinucleares) en equilibrio con fases condensadas (complejos
mixtos y polinucleares) interviniendo en equilibrios redox. En el método, L y X representan las particulas que se intercambian y en el primer caso intervienen en el siguiente equilibrio de formación:
19

(111.11
A la sustancia ML se le conoce como donador y a la M como receptor conjugado, [MLI, [MI y [Ll representan las concentraciones molares de las especies correspondientes. Km es
la constante de equilibrio de concentraciones, la cual sólo es constante cuando los coeficientes de actividad de todas las especies permanecen constantes. Si L, M y ML pueden reaccionar con X, en el sistema también se
L
presentan los siguientes equilibrios:
Ox KOx = K O x = 1. liX0 llLX0
Por definición LXo=L, Mo=M, HW(o=ML y KLxo
Otros equilibrios en el sistema son:
-__
l a ecuación 111.4 puede ser expresada como un producto de las anteriores:
KLX L #LXk K - - Km [ KJX KIX 1'
iíXJ U 1
(111.61

Se señala que estableciendo el balance de materia de cada
equilibrio y teniendo las constantes de equilibrio asociadas, es posible calcular las fracciones iolares de cada especie complejada o no de M.
Como se puede observar la presencia de X implica un gran número de equilibrios que aumentan la complejidad del análisis. Ahora
bien, para facilitar su estudio se impone como condición la
concentración de X [condición de amortiguamiento) ya que de ese
modo quedan determinadas cuales de las especies de mi, MXJ y MLxir predominan en el sistema [421. Todo esto permite de manera aproximada, reducir el conjunto de reacciones probables representadas en la ecuación 111.4, a un equilibrio predominante formado sólo por las especies predominantes. A dicho equilibrio
se le llama equilibrio ~ representativo.
Por otro lado, de la constante de equilibrio de la ecuación
111.4, bajo condiciones de amortiguamiento en X, se puede definir una nueva constante:
(111.71
. - .-
Como se puede ver Kijk depende de [XI impuesta y se le denomina
constante condicional.
Si ahora se consideran diferentes valores de [XI impuestos, el
equilibrio de la ecuación 111.4 es diferente para cada uno de estos valores debido al cambio de las especies predominantes
representadas por LXi, MXJ y Mwút. %on la finalidad de estudiar el cambio en el conjunto de las especies presentes en los equilibrios representativos con su
respectiva constante condicional, se definen las especies
generalizadas L', M' y ML' como las especies predominantes de
LXi, MXJ y Mwúr respectivamente para un valor dado de [XI . De esta forma el conjunto de equilibrios representativos (ec.111.41,
21

en su zona de predominio respectiva, queda definido por un equilibrio generalizado de la forma:
(111.8)
Aplicando la mecánica utilizada arriba, a un sistema del tipo
L/M/ML/ML2/X se pueden definir los siguientes equilibrios de
formación sucesivos:
(111.9)
(111.10)
el equilibrio de formación global:
(111.11) - + - - KE2= p2= [ML2 1 M+2L-+nLz [M1[L12 ’
y el equilibro de dismutaci6n: ~ ~-
(111.12)
Como los equilibrios I I I .7 ,8,9,10 son interdependientes pueden
relacionarse de la siguiente manera:
Y
2 L L L @2= Km2= Km.Km, (111.13)
(111.141
22

Igualmente que en el caso anterior, la relación de la ecuación de balance de materia para cada equilibrio y su correspondiente
constante de equilibrio, permiten definir las fracciones molares de las diferentes especies de M. Suponiendo que L,M.ML y ML2 reaccionan con X entonces se pueden
presentar los siguientes equilibrios:
i E {~,í..,a), (111.1s) c-- ix
[LI [X I l KLXi= L + ix __j Lxl
[ia2xi1 1~ {O,l,. . ,d). (111.18) t ML2 + iX ___) M12Xi KLl=
[ E 2 1 [XI -
Por definición W(o=L, Mx04 , MLXo=ML, ML2Xo=ML2 y
Kox ox ox ox =K =K =K =1, Otros equilibrios presentes pueden ser: LXO M(0 W O 1Iw(o
Por medio de l a ley de Hess se puede demostrar que las constantes
de equilibrio KiJk.
equilibrios 111.9 a 111.18. Como se vi6 anteriormente, bajo condiciones de amortiguamiento en
Kikl, KiJ,. y KJki son dependientes de los
23

1
.I i
[ X I , los equilibrios 111.19 a 111.22 quedan definidos por las siguientes constantes condicionales:
(111.23)
( I I I. 24 1
(111.25) (1-J-21 I s [ML2XlI K; J1= KiJ1 [XI [MxJl [Lxi l2 '
Kikl= KJkl [XI (l+J-21), - IML2X11 [MXJI ( I I I. 26 1 [Mw(k 1
Si consideramos la posibilidad de diferentes valores de [XI
impuestos, probablemente los equilibrios 111.19 a 1 1 1 . 2 2 serán
diferentes por el d i o de las especies predominantes de u(&,
MXj, MLXk y W i , por lo que para considerar el conJunto de
posibilidades de cambio es necesario definir las especies
generalizadas y Sus respectivos equilibrios generalizados de la
siguiente manera:
Se toman L' ,M' , ML' y "2 como las especies predominantes de U&, MXJ, Mu(k y MLzxi respectivamente, con los siguientes
equi 1 ibrios:
( I I I. 27)
[ML; 1 (111.28) e-
ML' + L' ML; &,= [ML'][L'] 9
2 [ML; 1
( I I I. 29 1 t- 2L' M' + L' -+ ML; Km, = 2 [M'1[L'12 '
[MLiI [M' 1 . (111.30) e- 2HL' 2 ML' - MLi + M' Km' =
2 [ML' l 2
En diagramas bidimensionales donde por definición pX= -log[Xl,
pL= -log [Ll y pM= -log[Ml, las constantes condicionales previamente definidas se traducen en líneas que al intersectarse
I .
1;
24

entre si definen planos donde predomina alguna de las especies
resultantes del estudio. En la vecindad de dichas líneas
coexisten las especies involucradas en los dos planos adyacentes. A continuación se presentan los algoritmos resultado de la generalización del método aplicado a sistemas de interés para los
casos estudiados.
- Sistema WYX, con especies solubles mononucleares, con
amortiguamiento en [XI y un sólo estado de oxidación. Para
sistemas que incluyen especies generalizadas como L’ y ML’ éstas
pueden ser definidas como sigue: J ’
a
[L’ I= [LXhl= [L1aLtx,
(111.31) h=O I
donde j E (O, 1.. ,nl, y son el máximo número de partículas
son los capaces de complejar L o MLJ. aLtX)
coeficientes de complejación de L y MLJ respectivamente y
dependientes de [XI . Dichos coeficientes obedecen a las siguientes relaciones:
y a i a J ( X )
a = i + K ’ ~ ~ [ X I + . . . . . . . . + KEl 1x1’ L ( X ) 1. (111.32)
Estas especies estan relacionadas entre si a través de los siguientes equilibrios generalizados de formación y de dismutación, y de sus correspondientes constantes condicionales:

. (111.33) 1 Combinando las ecuaciones 111.31 y 111.33 es posible expresar las constantes condicionales en función de los coeficientes de comp 1 e jac i 6x1:
~
Para la obtención de diagrams de zonas de predominio (PP)) en el
espacio pL'/pX se utiliza la siguiente ecuación:
- Sistemas M/wx con especies insolubles polinucleares, amortiguamiento en pL' y pX, y un sólo estado de oxidación. En este caso se inicia por definir las especies generalizadas a segundo orden:
26

..
donde :
a es el coeficiente de complejación a segundo orden n ( L ' , X ) 1
de Hi dependiente de L' y X ,
es el coeficiente de complejación a segundo orden de
M' dependiente de L',
n; (L' 1
n; (XI
a
1
a es el coeficiente de complejación a primer orden de
M1 dependiente de X con id1.2,. . ,n}.
Las especies generalizadas definidas en la ecuación 11.36
intervienen en los siguientes equilibrios:
-
- formación:
- polinucleación: ~ __
- dismutación:
i0{1,2 ,.., ( j - 1 ) ) J42.3 ,.., (r-1)) kr((j+i),(j+z) ,.., ni) (111.39)
De acuerdo con Rojas et al 142-471, existen diferentes maneras de obtener el algoritmo para la construcción de los diagrams (PZD y PED). Una de las maneras es obtener directamente del equilibrio
27

de formación el algoritmo que se va a utilizar:
- Sistemas WYX con especies polinucleares, con amortiguamiento
en pL' y pX y un sólo estado de oxidación en condiciones de saturación. En sistemas donde hay todo tipo de especies en solución, incluidas las polinucleares, se puede llegar a la saturación. Tratándose de 3 componentes puede haber una o mas fases coexistiendo en la solución. En este nuevo caso también es posible definir las especies generalizadas para las fases condensadas, de tal manera que:
donde son las deltas de Kroeneker para . las especies insolubles tales que:
6' = 1 para las especies insolubles 6 = O para el resto de las especies
11
lk=i J
A partir de esta definición puede construirse \in diagrama de fases condensadas en el plano pL'/pX de l a solución saturada, ____
mediante los equilibrios generalizados de interconversión a primer orden:
(111.42)
Por otra parte el diagrama de zonas de predominio define la especie generalizada total de segundo orden:
}. (111.43) t [ M"I= [M " l + 2[M"]+ 2 ...+ m[M"]
20

con t se consideran todos los polimeros de M en la misma fase.
Ahora bien, tomando en cuenta todos los polimeros posibles obtenemos una especie generalizada global insoluble, tal que:
'HI1 ~ allMll + a I I M I I +...+ ¿Pn" (111.441 ( f ) 1 ( f ) 2 2 ( f ) h h í f )
Las especies generalizadas de las ecuaciones 111.43 y III.44
permiten definir el equilibrio generalizado total a segundo orden:
de la ecuaciones 111.44 y 111.45 tenemos que:
Además de la ecuación 11.45 tenemos directamente:
(111.4'7)
A partir de la ecuación 11.45 podemos obtener el diagrama correspondiente en el plano p M'/pH. - Sistemas W e - con polinucleación, con amortiguamiento en
t
I
pL' y pX y considerando fases condensadas en un sistema cerrado. Para construir diagramas donde hay reacciones de transferencia de
carga, es necesario partir de los diagramas p M'/pL'/pX, cada uno de ellos definiendo una especie generalizada global, tal que:
t
29

con J definiendo el estado de oxidación de M tal que fe{. . ,-2. - l , O , 1,2,. . 1. De aquí que se pueden obtener los equilibrios generalizados globales redox a segundo orden:
siendo E, el potencial bicondicional del par redox de
la ecuación 111.49. Igualmente son posibles equilibrios generalizados globales de dismutación a segundo orden:
n~~ (k) ( J )
La existencia de los equilibrios 11.49 y 11.50 debe tomar en cuenta la regla de las fases. Las ecuaciones descritas arriba permiten construir diagramas tipo
Pourbaix con un sistema multicomponente (EYp M"/pL'/pX). El método descrito presenta la ventaja de llevar, con relativa facilidad, el modelo generalizado a un sistema en particular. Además, es posible obtener tantos diagramas como sea necesario para un mismo sistema. Por otro lado, las limitaciones del método derivan de un estudio, en el equilibrio, de un sistema ideal donde no se hacen consideraciones de tipo cinético.
*

144159
I I I. 2. Resu1 tados
. -
I I I. 2.1. Sistema W á c i d o sulfúrico/agua/e-
Las especies generalizadas globales Sm" ( I I I) y Sm" ( I I 1 estan * *
definidas como sigue:
* - Sm"(II1)
con respecto a condiciones de saturación:
con respecto a SO::
Sm" ( i I I )= Sm' ( ii i )+S~;+Sm(SO,); (I I1 1
Sm"(III)(s,= Sm' (III)(s,
con respecto a OH-:
* - cr"(11)
Wo'existen datos reportados de Sm(I1) A partir de las especies generalizadas se obtienen los siguientes equilibrios generalizados (E. G. 1 :
31

E. G.
E. G.
E. G.
32

E. G.
E. G.
E. G.
E. G.
- __
Las constantes termodinámicas utilizadas para la construcción de
los diagramas son las publicadas por Hogfeldt en 1982 [761 y por Kragten en 1978 [771 y se encuentran resumidas en la tabla 111.1.
33

Tabla 1II.l.Constantes de equilibrio de los complejos de Sm(II1)
con los ligandos SO' y OH-. 4
ión
H+
Sm ( I I I 1
equi 1 i brio log K
1 . 9
3.52
1.67
7.5
7.7
-17.5
19.5
ref
76
76
76
77
77
77
77
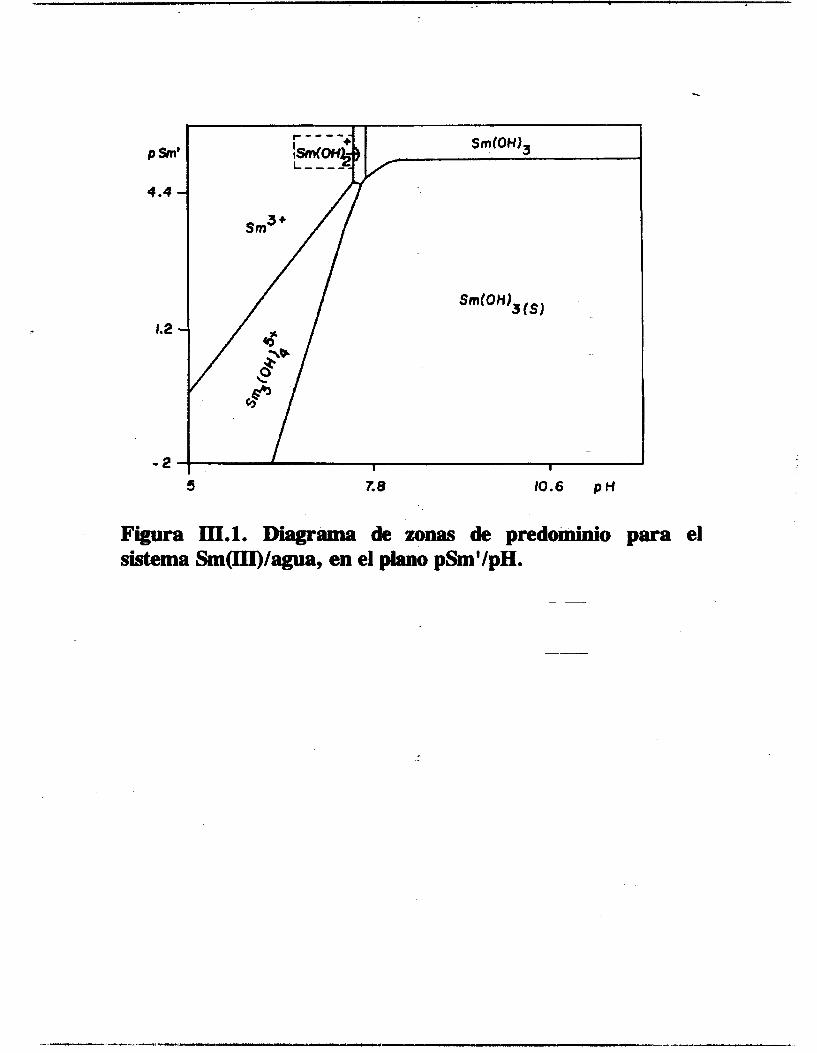
El diagrama PED para el sistema Sm(III)/SO~/H20, figura 111.1,
muestra las zonas de predomhh-de las diferentes especies de
samario en solución y la aparición de una fase sólida en el
espacio bidimensional pCi'/pH.
34
P *'
4.4 -
1.2 8
- 2 - 5 7.8 10.6 p H
Figura III.1. Diagrama de zonas de predominio para el sistema Sm(rn)/agua, en el piano pSmVpH.

Cuando se realiza un corte del plano en pSm'=3 y se traza la
línea recta correspondiente paralela al eje del pH, la
intersección de esta línea con las líneas que delimitan las zonas
de predominio de las diferentes especies y fases define una serie de intervalos de pH asociados a estas especies y fases. Esta operación da origen a un diagrama PED unidimensional del tipo:
sm3+ Sm3(OH):+ Sm (OH 1 (s) I I I
I PH 6.' 7 7:2
De la misma manera si la condición es una concentración global de samario tal que pSm'=6, el corte del plano de la figura 111.1 proporciona un PZD unidimensional de la forma:
sr3+ Sin (OH 1 Sm(OHI3 I I I PH
7.5 7.7
A partir de los dos diagramas unidimensionales se construyen el
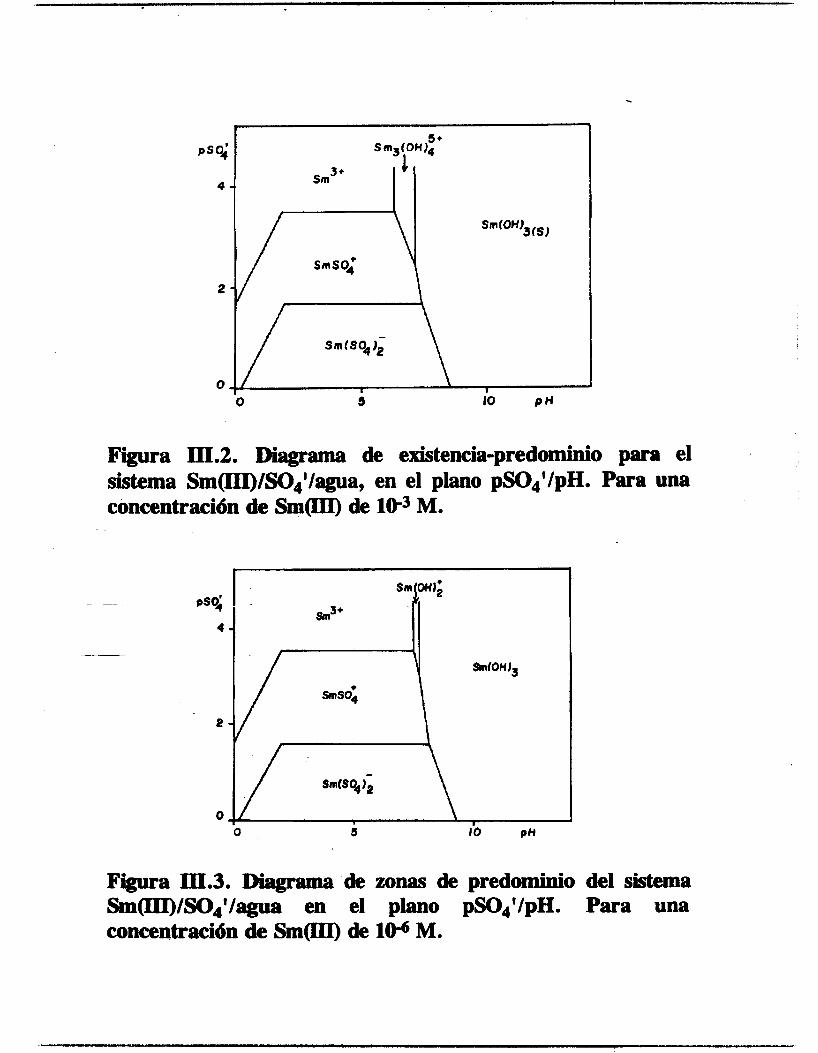
diagrama tipo PED para el primer caso y el tipo PZD para el segundo caso. El PED se muestra en la figura 111.2 y el PZD en la figura 111.3. Ambos diagramas se construyen en el espacio--
bidimensional pSOf/pH. En el primer diagrama se observa que
existen 4 diferentes especies solubles y dependiendo del valor de pSm' aparece una nueva fase correspondiente a un hidróxido
insoluble de samario. La fase sólida se inicia en el intervalo de pH de 7.1 a 8.5. Además se observa que en el intervalo de
concentraciones usuales de electrolito soporte y de pH inicial de
la solución aparece una especie aniónica de sdario, Sm(S041~. En el segundo diagrama hecho a partir de un pSm'=6 sólo se observan especies solubles de samario, donde también se observa que hay
una ZOM de pSO;/pH donde aparece una especie aniónica de samario, la misma que en el diagrama anterior.
Partiendo del diagrama 111.2 (pSd-3 y pSOf4.5231 se construye
35
.
Figura III.2. Diagrama de existencia-predominio para el sistema Srn(III)/SO4'/agua, en el plano pS0JpH. Para una concentración de S m o de le3 M.
PSO;
4
2
Figura III.3. IBiagrama de zonas de predominio del sistema Sm(III)/S04'/agua en el plano pS04'/pH. Para una concentración de S m o de lo6 M.

el diagrama unidimensional PED a partir del corte con pSO~=O.S23 y las intersecciones de las diferentes zonas de lac especies presentes, obteniendo:
Por otro lado se obtienen los equilibrios generalizados utilizados para 'la construcción del PTD:
* -t* Srn"(iii) + e Sm"(I1)
-t- 2+
- - 2-
3H' + Srn(OH)3(s, + e Sm + H20
srn(W4)S + e - sm2+ + 290~
snico + e -t sm2+ + SO, 4 -
-c-- sm2+ + 2e - smo En una segunda etapa, utilizando los valores de potencial de la tabla 111.2, se construye el PTD en función del potencial, E, y
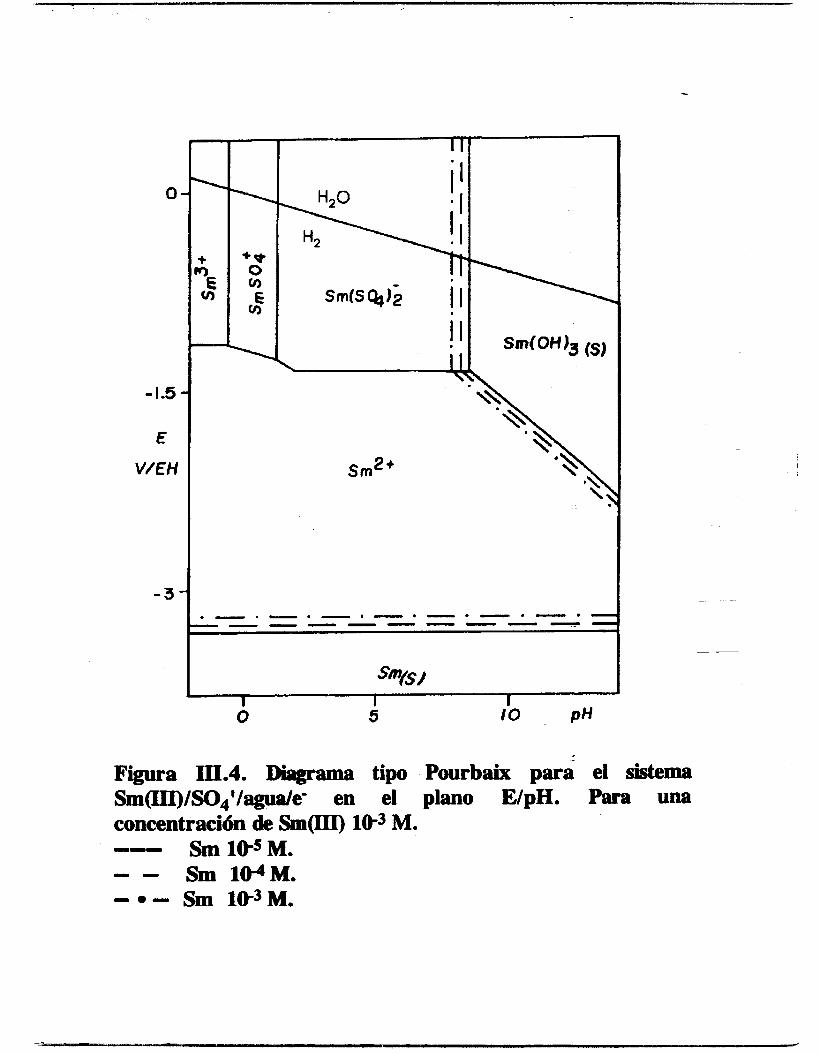
del pH (figura 111.4) .
36
O
E
V/Eh
-3
-
\
+P O v> E
v)
\
I if
1 1 I o 5 10 PH
Figura III.4. Diagrama tipo Pourbaix para el sistema Sm(III)/S04'/agua/e- en el plano E/pH. Para una concentración de Sm(III) lW3 M. --- Sm lWM. - - Sm 1WM. - e - Sm W M .

Tabla III.2.Potenciales estándares del samario para los
diferentes estados de oxidación 176,331
par redox
sm3+/~Epo
2smo/sm203
smso;/smo
sm3 +/SR' +
sm2 +/smo
Sm (OH ) 3/Smo
Eo,V
-2.41
-2. o
-2.48
-1.15
-3.12
-2.83
En el PTD se observan todas las especies solubles y la presencia de una fase sólida del hidróxido de samario y otra del samario metálico. La línea que cruza el diagrama indica la evolución del potencial de reducción del agua sefíalando la imposibilidad de llegar a reducir al samario al estado metálico en un medio acuoso. En los límites de la zona de predominio de la fase sólida del hidróxio de samario se marca con una serie de líneas la evolución de esa frontera en función de la concentración de samario, en todos los casos este límite oscila alrededor del pH=8. También1 esta presente la especie aniónica de sulfato de samario, sm(so4);.
111.2.2. Sistema Wácido sulfÚrico/agua/e-
Siguiendo la misma mecánica de los casos anteriores, definimos las especies globales generalizadas:

* - Am"(II1):
con respecto a condiciones de saturación:
con respecto a SO;:
Am" ( I I I 1 = Ani' ( I I I 1 +Amso; ( I I I 1 +Am (so4 1; ( I I I )
Am"(III)(S)= Am' (III)(s,
ni' (III)= A~~++A~(oH)~+
con respecto a OH-:
Aiaso;(III)= Amso;
Am' (III)(s)= Am(OH)3(s)
* - Am"(1V):
con respecto a condiciones de saturación:
con respecto a SO; y OH-:
A~YIV)= ~ m ' ( I V ) = A ~ ~ +
Am"( IV) (s)= Am' (IV) (S)= Amo2
38

con respecto a condiciones de saturación:
con respecto a So; y OH-:
Am"(V)= Am' (v,=Amo;
* - Am"(V1):
con respecto a condiciones de saturación:
con fespecto a SO; y OH-:-
Ani" (VI )= An' (V)=Am02
Dichas especies generalizadas intervienen en los siguientes equilibrios generalizados:
39

E. G.
E. G.
E. G.
E. G.
E. G.
Las constantes termodinámicas utilizadas para construir los
diagrams PZD y PED son las publicadas por Bard [331 y Martell
1781 y se encuentran resumidas en la tabla 111.3.
40
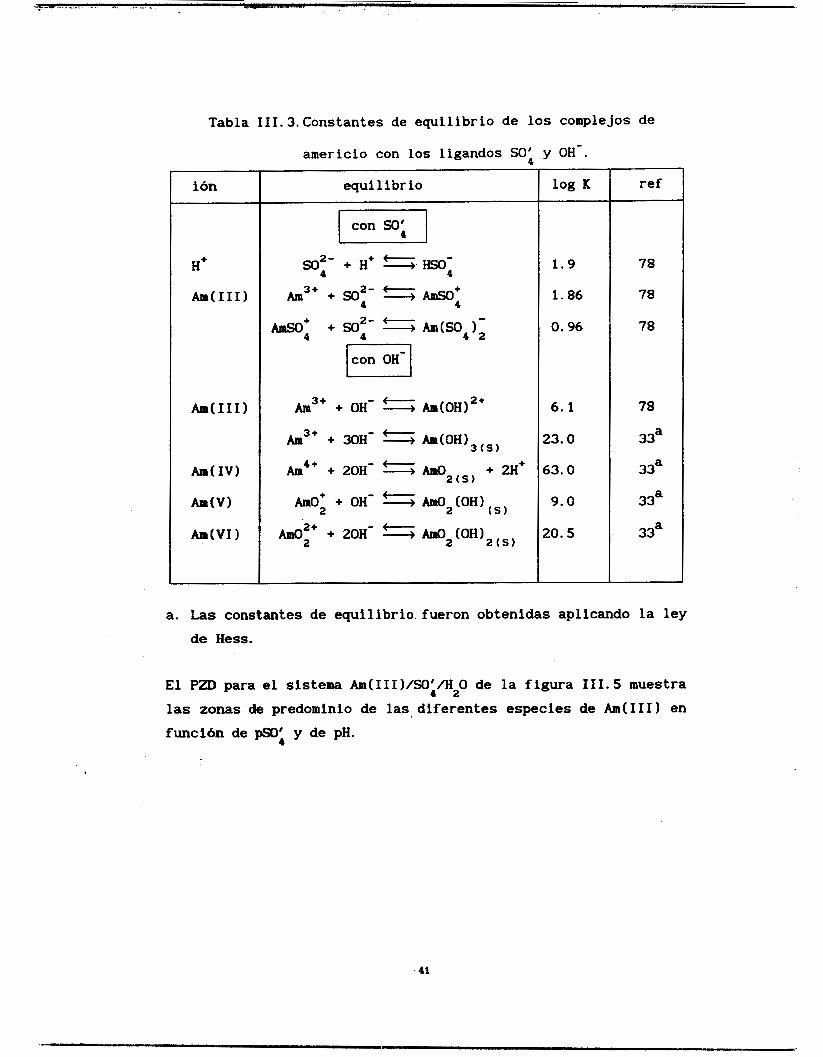
Tabla III.3.Constantes de equilibrio de los complejos de
americio con los ligandos So: y OH-.
ión
H+
Am(II1)
Am(III1
log K
1.9
1.86
O. 96
6 .1
23. O
53. o
9. o
20.5
ref
78
78
78
78
33a
33a
33a
33a
a. Las constantes de equilibrio.fueron obtenidas aplicando la ley de Hess.
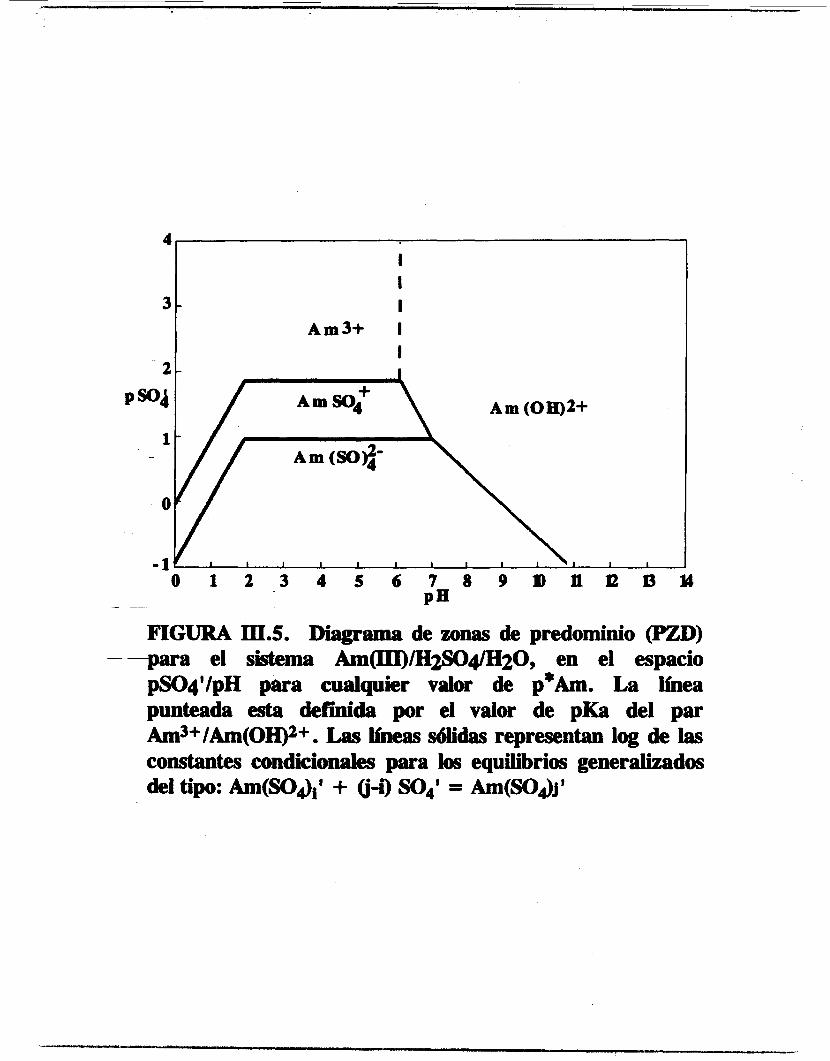
El PZD para el sistema Am(II11/SO;M20 de la figura 111.5 muestra las zonas de predominio de las diferentes especies de Am(III1 en función de pS0; y de pH.
41
0 1 2 3 4 5 6 7 8 9 D í l i 2 i 3 M - __ PH
FIGURA III.5. Diagrama de zonas de predominio (PZD) - -para el sistema Am(III)/H2SO4/H20, en el espacio
pSOq'/pH para cualquier vahw de p*Am. La íínea punteada esta defdda por el valor de pKa del par Am3+/Am(OH)2+. Las beas sólidas representan log de las constantes cmdicionaies para los equilibrios generalizados del tipo: Am(SO&' + Q-i) SO4' = Am(SO,&'

Para una concentración generalizada de sulfatos pSOi=O.523, una linea paralela a la escala de pH al intersectarse con las líneas de las zonas de predominio define una serie de intervalos de pH. De esta manera se obtienen las especies de americio que son predominantes en cada intervalo de pH. El PZD unidimensional resultante es el siguiente:
h(s0,) ; Am (OH 1 2+ I PH
Am3+ I =; I
O. 56 1.46 7.87
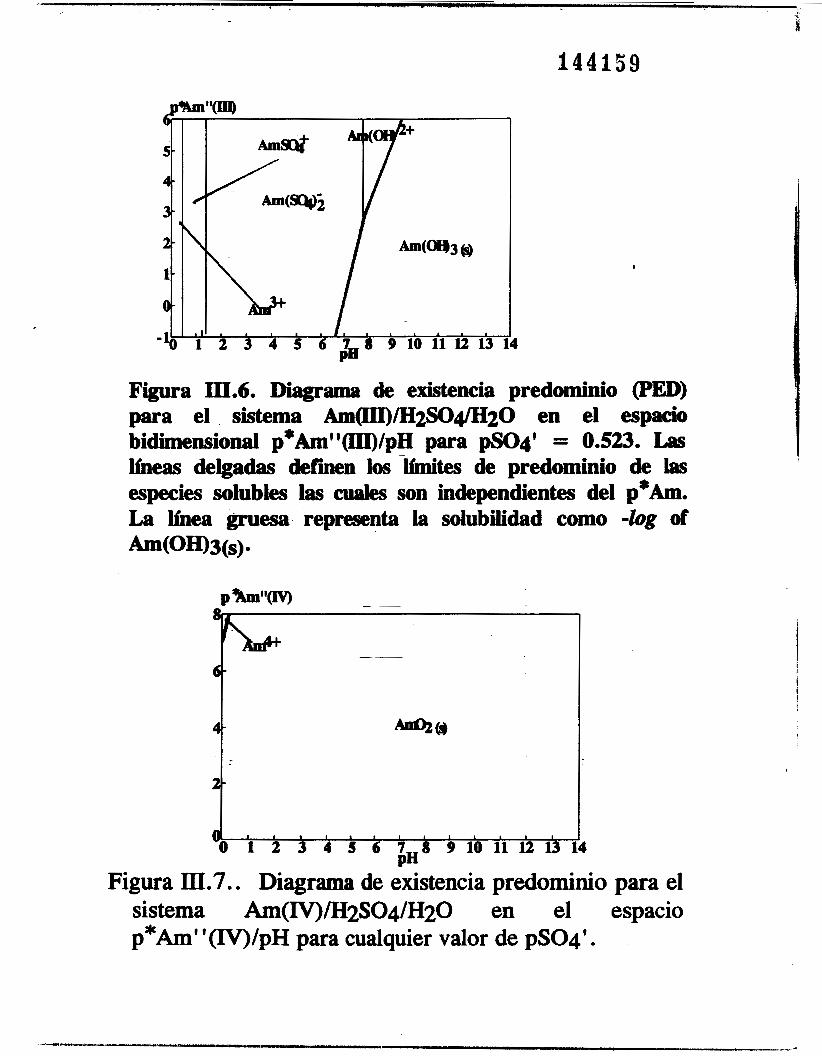
Basándose en este diagrama y tonando en cuenta las especies insolubles de americio, se construye el PED, figura 111.6, para el sistema ~ ( I I I ) / A m ( I I I ) ~ ~ , / s O ~ ~ ~ O en función de p Am" y pH. En la figura se observan las diferentes especies de americio. Puesto que no hay constantes de equilibrio para complejos de sulfato del americio V y VI solo se construyen los PED en función de p Am" y pH. Las figuras 111.7, 111.8 y 111.9 son los PED para los sistemas Am(IV)/SO;/H20, Am(VI/CO;/H20 y Am(VII/SO;/H2O respectivamente. En cada diagrama, aparece solo una especie soluble, es decir, Am4* para el h( IV ) , Amo; para el Am(V1) y Amo: para el Am(V1). También aparece una sola fase sólida en
para el Am(IV1, Am02(0H)(s, para cada diagrama, es decir, el Am(V) y Amo2(OH)2(s, para el Am(V1). En cada diagrama la fase sólida esta presente en una gran parte del intervalo de pH. Por otro lado el PTD para el sistema Am/SO;/H20 se construye utilizando el PED de cada estado de oxidación de americio e imponiendo p Am"=6 y pcO;=O.523. Como primer paso, se obtiene el PED unidimensional con los valores impuestos para
ill
Am02 (€3)
--
*
* Am" y So; :
Am3* ASO; Am(CO,); Am(0HI2+ AmII(OH)3(c) de la figura 111.6: I I I I I PH
0.56 1.46 7.87 9.5
42
h
> I l , , , , I I . I I I a I 1
1 2 3 4 5 6 7 8 9 1 0 1 1 1 2 1 3 m
144159
I
I
Figura III.6. Diagrama de existencia predominio (PED) para el sistema Am(III)/H2SO4/H20 en el espacio bidimensional p*Am"(III)/pH para pS04' = 0.523. Las líneas delgadas defmn lw-Minites de predominio de las especies solubles las cuales son independientes del p * h . La línea gruesa representa la solubilidad como -log of Am(OH)3(S).
1 # I I 1 1 1 1 1 1 1 I
1 2 3 4 5 6 7 8 9 1 0 1 1 1 2 1 3 : PH
4
Figura III.7.. Diagrama de existencia predominio para el sistema Am(IV)íH2SO4/H2O en el espacio p*Am' '(IV)/pH para cualquier valor de pSO4'.
4
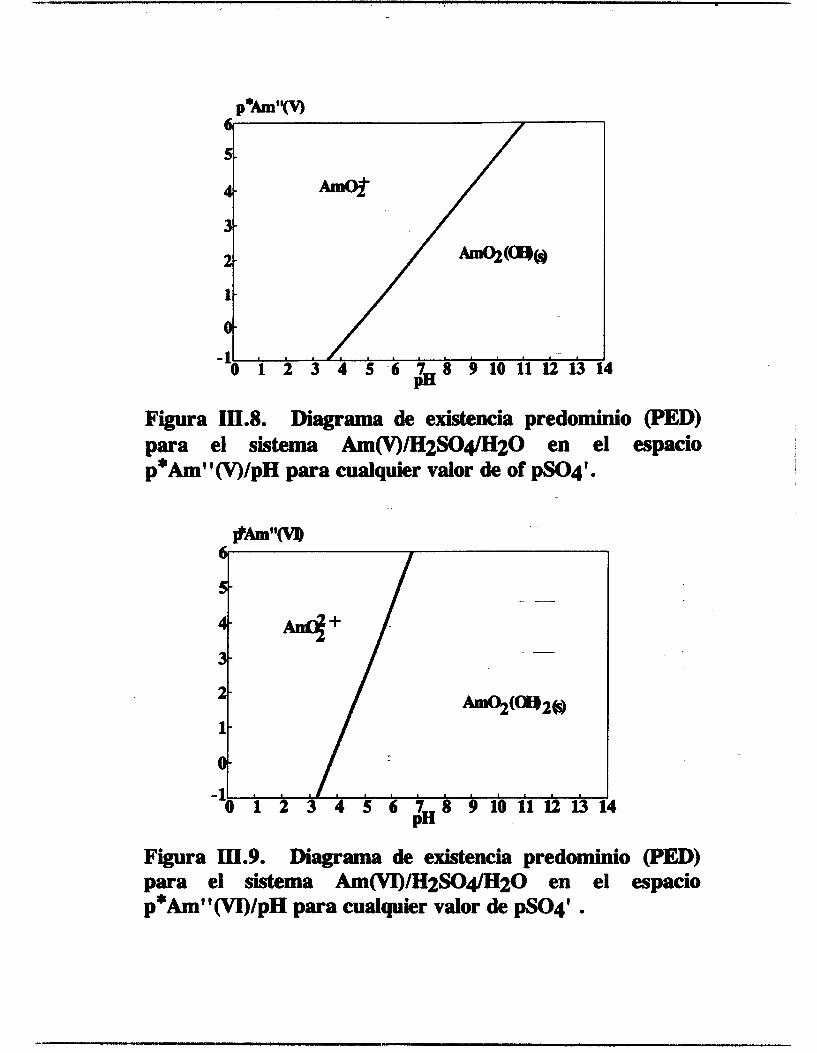
Figura III.8. Diagrama de existencia predominio (PED) para el sistema AmcV)/H2SO4/H~O en el espacio p*Am"(V)/pH para cualquier valor de of pSO4'.
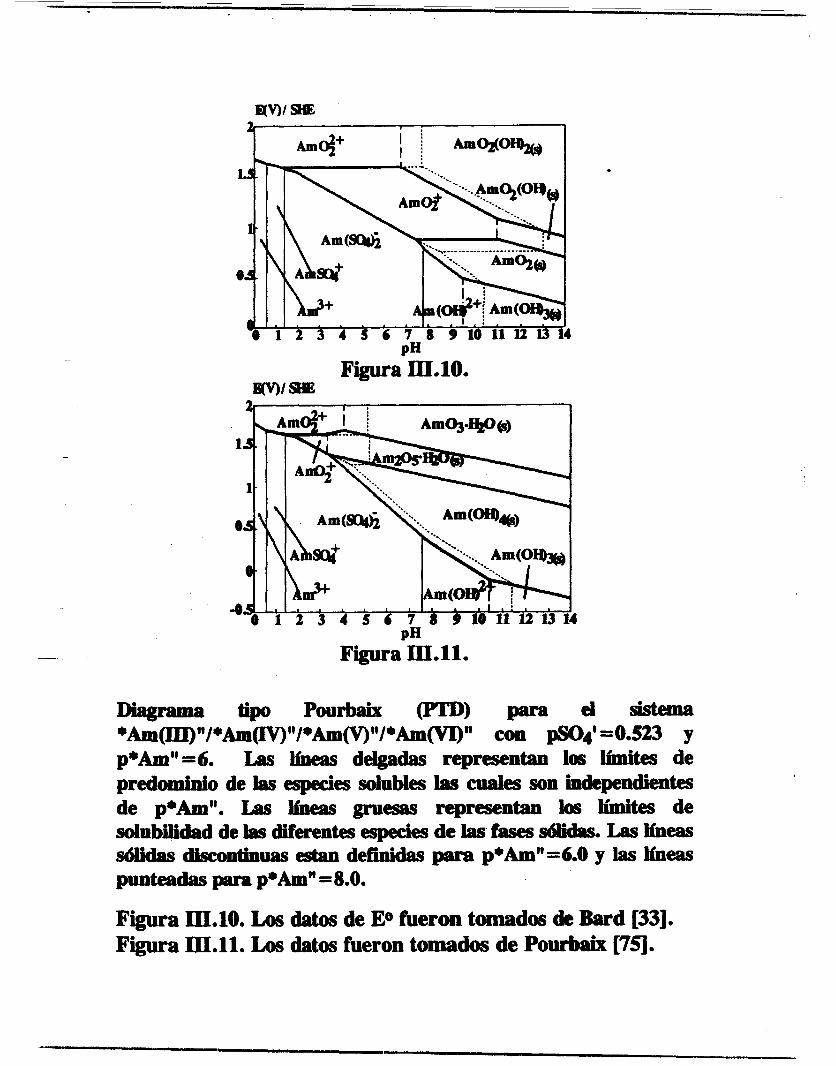
Figura III.9. Diagrama de existencia predominio (PED) para el sistema Am(VI)/H2SO4/H20 en el espacio p*Am"(VI)/pH para cualquier valor de pSO4' .

Am02(OH) ( s ) ' PH de la figura 111.8: 1 I
I I ' PH Anio2 (OH
2 (S 1 de la figura 111.9:
Los equilibrios generalizados utilizados para la construcción del PTD son:
E. G. b"(V1) + e - An"(V) - - * - - M:+ + e -mi
2H20 + Amo:' + e 4 Aaio2(OH) ( s )
H* + + e - Anro20H(s) + H20
-c-- -- - *
Am'") 4 Am(IV1 *
E. G.
E. G.
43

* * donde Am"(VI), An"(V1, An"(1V) y Am"(II1) representan todas
las especies formadas por hidrólisis y complejación con SO; y
H20. Por otra parte, el PTD se construye en función de E y pH,
utilizando los potenciales de la tabla 111.4.
Tabla 111.4. Potenciales estandar de reducción del americio [331
~
Eo, V
1.59
O. 82
2.62
-2.07
1.2
1.72
-0.9
o. 9
o. 7
o. 22
-2.53
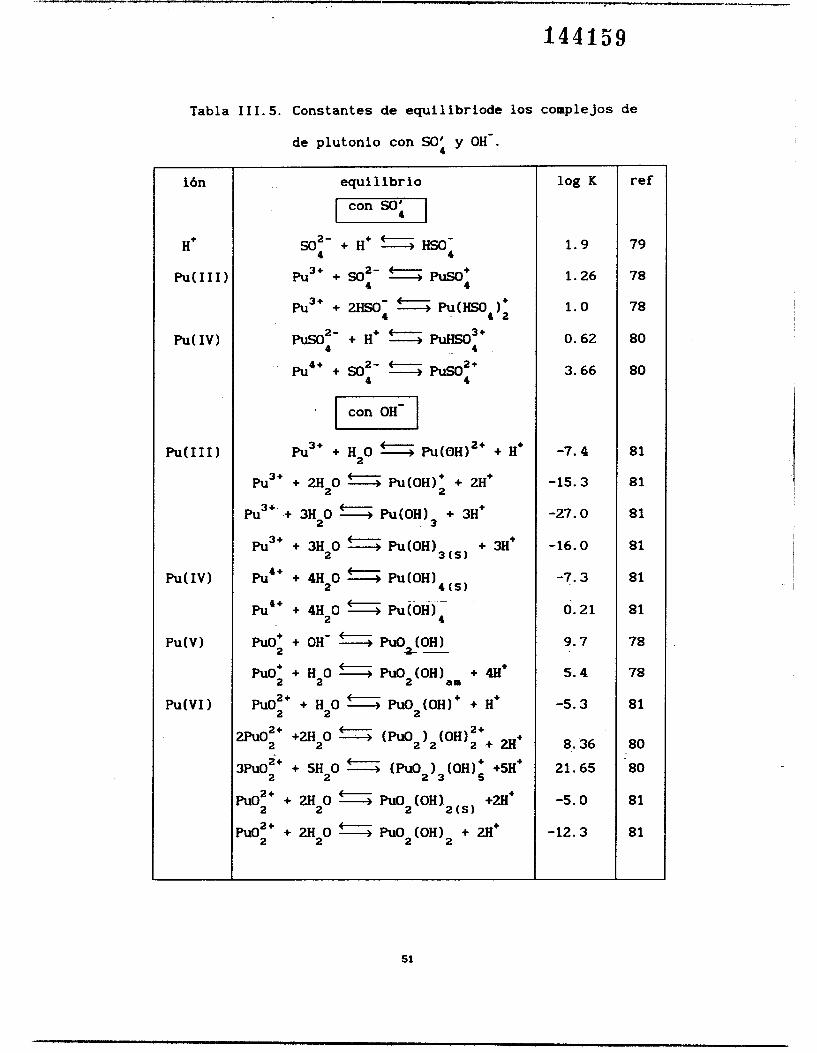
En l a figura 111.10 se muestran todas las especies solubles y las
fases sólidas para los diferentes estados de oxidacián del americio. Por ejemplo las líneas discontinuas representan el
límite inferior inicial (en la escala de pH) de la fase sólida,
cuando partiendo de una concentración de samario de M se
forma un depósito sólido y queda una solución con una
concentración final de lo-* M ( 1 íneas punteadas 1.
I
44

Si comparamos el PTD obtenido con los datos de Bard [331
publicados en 1982 (figura 111.10) con el PTD obtenido con los datos de Pourbaix I751 publicados en 1962 (figura III.11), se observan pocas diferencias. La diferencia más sobresaliente es la ausencia de la fase sólida AUI(OH)~(~, (datos de Pourbaix) en el diagrama hecho con los datos de Bard. Los datos de Bard de las especies solubles fueron obtenidos en medio ácido y los de las especies insolubles se obtuvieron en
medio básico; es decir que estos datos no son resultado de
extrapolaciones y por ello los valores tienen meJor precisión.
Este ejemplo hace énfasis en la importancia que tiene seleccionar
los datos adecuadamente.
Es importante señalar que en todos los diagramas donde aparece el estado de oxidación de h(1II) esta presente la especie aniónica de sulfato de americio, De manera análoga en los
diagramas de samario aparece la especie correspondiente -de
samario. También se observa que el límite inferior, en la escala
de pH, donde empieza a aparecer el hidróxido insoluble es 9.5 aproximadamente una unidad de pH arriba del correspondiente al samario.
Am(S04); .
111.2.3. Sis tm Pdácido
En el caso del plutonio definen como sigue:
suifúrico/agua/e-
las especies globales generalizadas se
* - Pu"(II1):
con respecto a condiciones de saturación:
45
PH Figura III.10.
m/=
Magrama tipo Pourbaix (PTD) para el sistema * A m ~ " / * A r n ( n ? " / S A m C V ) " / * A m ( V I ) " con pSO4' =OS23 y p*Am"=6. Lris beas deigadas representan los limites de predominio de las especies soiubles las cuaies son independientes de p*Am". Las líneas gruesas representan los limites de sdubilidad de las diferentes especies de las fases didas. Las líneas sdlldas discontinuas estan definidas para p8Am"=6.0 y las líneas punteadas para p*Am" = 8.0.
Figura III.10. Los datos de EO fueron tomados de Bard [33]. Figura III.11. Los datos fueron tomados de Pourbaix [75l.

con respecto a SO;:
Pu"(III)= Pu' (III)(s)
PU''(II1) ( c ) = Pu' (III)(,
con respecto a OH-:
Pu' (I I I )= Pu3++Pu(OHl2+
Puso;(III)= Puso;+ Ri(fw141;
Pu' (III)(s)= Pu(OH)31s)
* - Pu"(1V): i
con respecto a condiciones de saturación:
con respecto a SO::
Pu"(IV)= Pu' (IV)+ PSO;
* - Pu(V1:
con respecto a condiciones de saturación:

con respecto a SO':
Pu"(V)= hi' (VI 4
Pu"(V1 (s)= Pu' (VI (s)
con respecto a OH-:
(ai) Pu' (VI Pu02(OH)
* - PU"(V1):
con respecto a condiciones de saturación:
con respecto a SO;: -
Pu" (VI )= Pu' (VI I
PU"(VS(c?r Pu' (VI I (s)
con respecto a OH-:
Pu' (VI)= RJ):+ Pu02(OHI+
La relación entre las diferentes especies generalizadas se
expresa por medio de los siguientes equilibrios generalizados:
47

E. G.
E. G.
E. G.
48

E. G.
E. G.
E. G.
49

3PuO;'
3Pu02 (OH 1 +
Para la elaboración de los PZD y PED de los diferentes estados de oxidación del plutonio se emplearon los datos publicados por Baes [791, Sillén [801, Martell [781 y Vitorge 1811, los cuales se resumen en la tabla 111.5.
1 4 4 1 5 9
Tabla 111.5. Constantes de equilibriode los complejos de
de plutonio con SO; y OH-.
equilibrio
p5iiq-l
. I c on OH- I Pu3+ + H20 f-- - Pu(OH12* + H+
Pu3+ + 2H20 f-- Pu(OH)S + 2H*
Pu3+ + 3H20 t-- - Pu(OH)3(s, c-- Pu3+ + 3H20 j Pu(OHl3 + 3H+
+ 3H+
t Pul* + 4H O - PU(OHI~(~, Pul* + 4H20 - Pu(0HI4 PuOi + OH- - PuO (OH) PuOi + H20 - Pu02 (OH)
2 f-- ---
t-- a-
c-- + 4H+ a m
PuO:' + H20 f-- - PuOz(OH)+ + H+
?PuO:+ +2H20 t-- (Pu0212(0H):*+ a+
P u O p + 5H20 t-- (Pu0213(OHl~ +SH*
%O:' + 2H20 c-- - Pu02(OH)zcs, %O:' + 2H20 - t-- PuO2(0HI2 + 2H+
+a+
log K
1.9
1.26
1. o
O. 62
3.66
-7.4
-15.3
-27. O
-16. O
-7.3
o. 21 9.7
5.4
-5.3
8.36
21.65
-5. o -12.3
ref
79
78
78
80
80
81
81
81
81
81
81
78
78
81
80
80
81
81
51

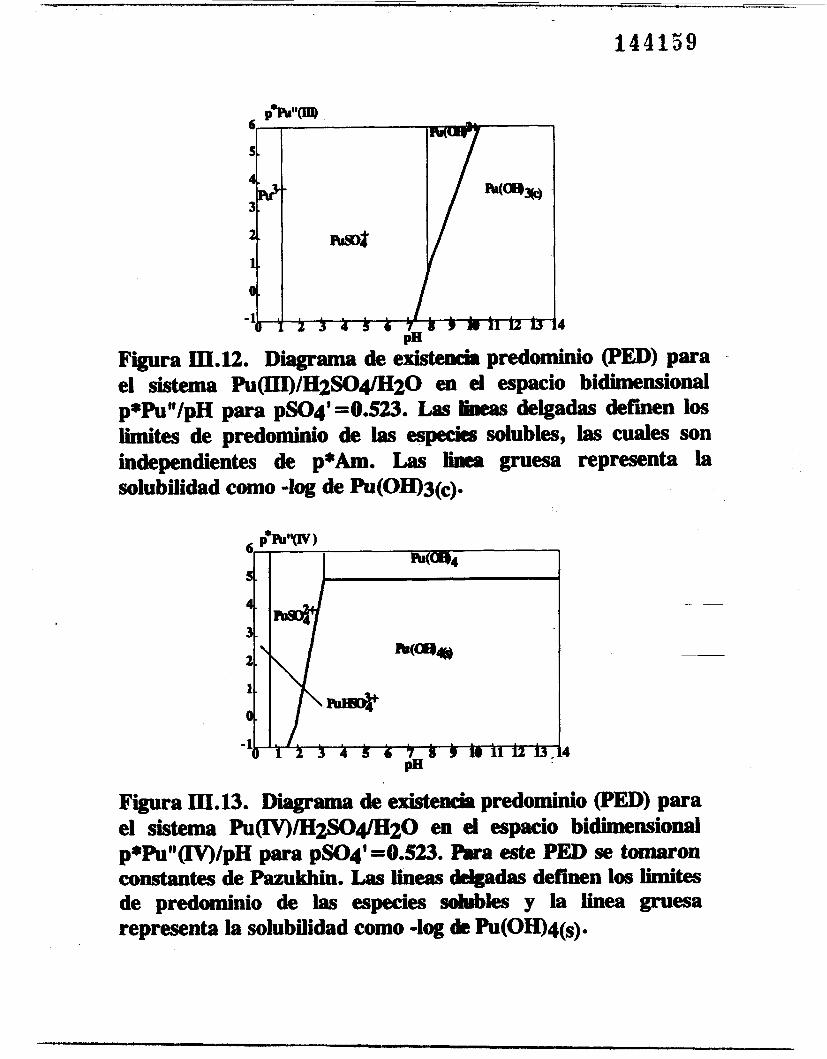
Se obtuvieron los PED en función de pPu" y pH. Las figuras
111.12111.13, 111.14 y 111.15 son los PED para los sistemas Pu(III)/CO~/H20, Pu(IV)/SO;/H20, Pu(V)/SO;/H20 y Pu(VI)/C0;/H20
respectivamente. En cada diagrama aparecen varias especies solubles y una sola fase sólida, es decir, PU(OH)~(~) para el
para el Pu(IV), Pu02(OH) para el Pu(1V) y
PUO~(OH)~(~, para el Pu(V1). Igualmente en un intervalo amplio de
pH (diferente en cada diagrama) la fase sólida esta presente.
Por otro lado para ilustrar las diferencias que hay en los datos de especies y constantes de equilibrio se obtuvieron dos PTD con los datos más recientes y más coherentes entre si. La figura
111.16 corresponde a datos de Baes 179, Sillén [801, krtell 1781
y Vitorge I811 y al figura 111.17 utiliza los mismos datos solo se sustituyen los que corresponde a Pu(IV) con datos de Pazukhin
[821 y los de Pu(V) con datos de Capdevila [831. Los equilibrios
generalizados y los diagramas PED unidimensionales generados con todos estos datos son muy numerosos, baste decir que siguiendo el proceso utilizado para los casos anteriores se obtienen los correspondientes diagramas PTD. Comparando ambos diagramas podemos darnos cuenta que las
diferencias son pequeñas en general tanto para las especies
solubles como para las fases sólidas. La- ifRica diferencia
importante radica en la especie Pu(OH),, pues mientras el primer
diagrama aparece como especie insoluble en 4L-segundo aparece
como especie soluble. Pazukhin asume que dicha especie es soluble hasta concentraciones de M como lo es el caso de los diagramas elaborados. También se observa que para la especie
trivalente no aparece ninguna especie aniónica como es el caso
Pu(III1, PuOztc1 am
-
del samario y del americio.
52
144159
6 If A
Figura III.112. Diagrama de existed predominio (PED) para el sistema Pu(III)/H2SO4/H20 en el espacio bidimensional p f h V p H para pSO4'=0.523. Las lineas delgadas defmen los limites de predominio de las especies solubles, las cuales son independientes de p+Am. Las iinea gruesa representa l a - solubilidad como -log de Pu(OH)3(c).
4
Figura III.13. Diagrama de existencia predominio (PED) para el sistema Pu~/"20 en el espacio bidimensional p*PU"(IV)/pH para pSO4'=0.523. h este PED se tomsron constantes de Pazukhin. Las iineas -das &€hen los limites de predominio de las especies sohrbles y la linea gruesa representa la solubilidad como -log dt Pu(OH)q(s).
-1 i 3-
2,
1,
O ,
1 2 3 4 5 6 'I 8 9 10111213 PH
1 2 3 4 5 6 'I 8 9 10111213 PH
.4
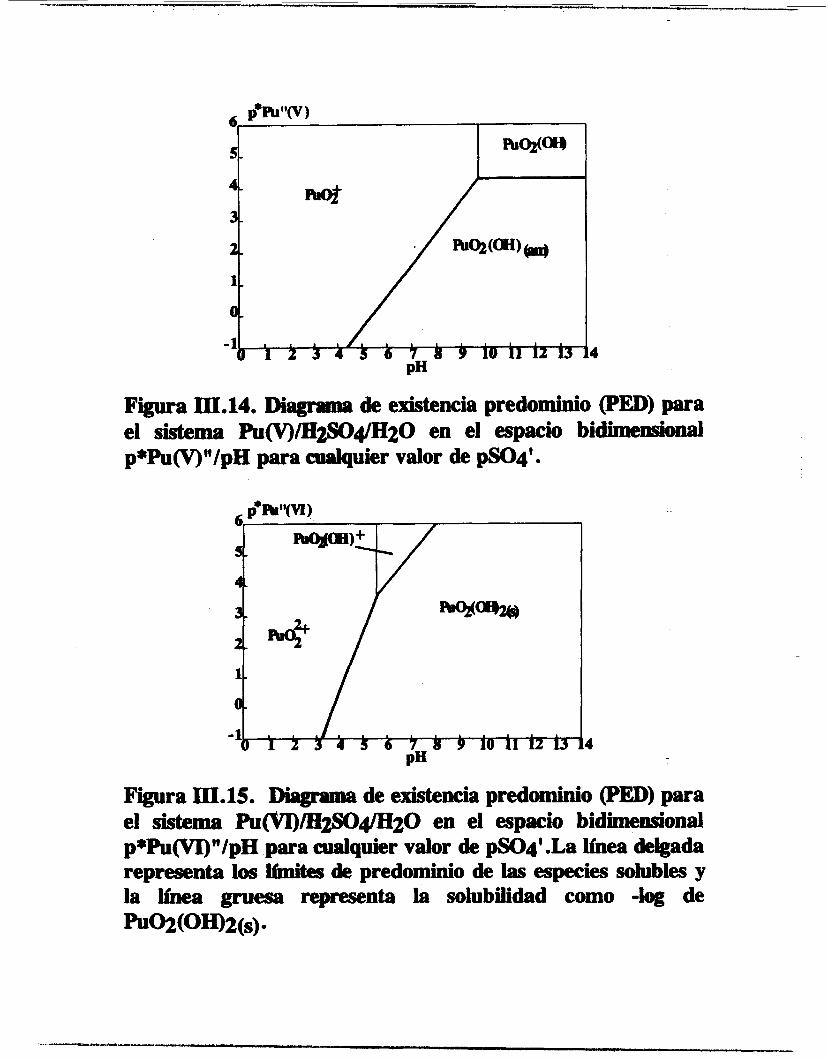
Figura III.14. Disgrrma de existencia predominio (PED) para el sistema Pu(V)/E~4/H20 en el espacio bidimeIisional p*Pu(V)"/pH para adquier valor de pS04'.
PIf .4
Figura III.15. Diagmma de existencia predominio (PED) para el sistema Pu($7I)/"20 en el espacio bidimensional p*Pu(VI)"/pH para cualquier valor de pSOq'.La h ea delgada representa 1- i€mites de predominio de las especies sohbles y la linea gruesa representa la solubilidad como -log de ~oZ(OH)2(s).
i
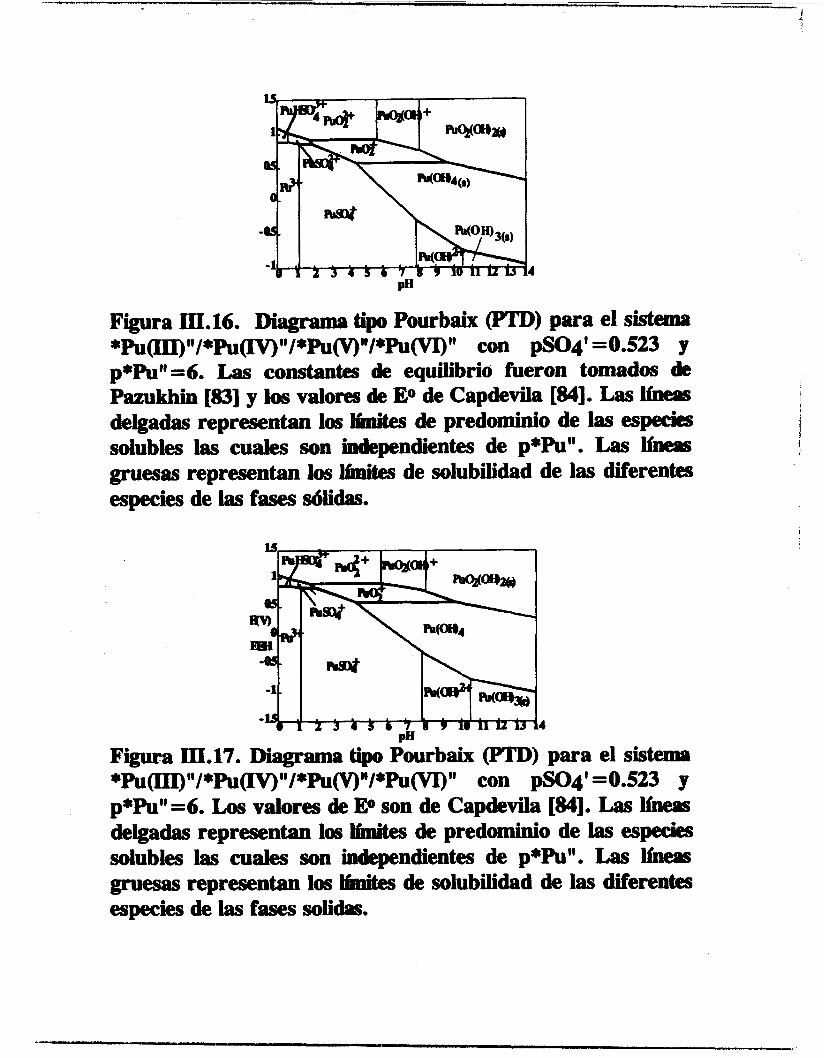
4
Figura III.16. Diagrama tipo Pourbaix 0) para el siStemri *Pu(III)"/*pU(n3"/*~~~/*~~" con pSO4' =OS23 y p+Pu"=6. Las constantes de equilibrio fueron tomados de Pazukhin [83] y los valores de EO de Capdevila [U]. Las lineis
solubles las cuales son independientes de p*Pu". Las liiieacs gruesas representan íos hites de solubilidad de las diferentes especies de las fases sólidas.
I
(
deigadas representan los bi tes de predominio de las especies 1 i
i
4
Figura III.17. Diagrama tipo Pourbaix (PTD) para el sistema *pU(III)"/*Pu(n3"/*Pu~"/*~~'' con pSO4' =OS23 y p*Pu"=6. Los valores de E@ son de Capdeda [U]. Las heas delgadas representan los h i tes de predominio de las especies solubles las cuales son independientes de p*Pu". Las Ifnezis gruesas representan los li'nnites de solubilidad de las diferentes especies de las fases solib.

IV. HODEL0 DE HANSEN
--
Hansen en 1959 [151 aborda desde un punto de vista teórico el depósito de lantánidos y de actínidos, como hidróxidos insolubles, sobre cátodos metálicos durante la reducción del agua en medio ácido. En 1960 [16] presenta resultados experimentales explicándolos con las predicciones teóricas de su modelo. Sin embargo, su desarrollo experimental presenta deficiencias que no permiten el uso estricto de su modelo, las deficiencias se presentan a nivel del tratamiento hidrodinámico para el diseño de la celda electroquímica y el experimento mismo. Hansen desarrolla su modelo partiendo de la presencia de la capa límite de difusión (presente en la interfase cátodo-solución) con un espesar constante (en el tiempo) y con el resto de la solución electrolítica bien agitada y de composición uniform?. Sin embargo, la celda diseñada por Hansen y su diseño experimental provocan la presencia de un vórtice, el cual a su vez induce el flujo turbulento de la solución. Además, la velocidad de rotación
del agitador (una barra magnética) no es constante, lo cual también contribuye a la inducción de flujo turbulento con una capa hidrodinámica de espesor no uniforme. Entonces, las condiciones hidrodinámicas no son las adecuadas para lograr un estado estacionario bien definido como se requiere para la aplicación del modelo de Hansen. Por otra parte, en la celda de Hansen las líneas de flujo del fluido de la solución hacia la superficie del cátodo no son perpendiculares con respecto a la superficie del electrodo. Esto se debe considerar en el desarrollo de un modelo puesto que se
presentan líneas de flujo en dos dimensiones . Esto ocurre cuando el cátodo sobre el que se deposita el material permanece inmóvil. De hecho la poca exactitud y las caídas en las curvas de
53

rendimiento de depósito reportadas por él mismo se explicarían en función de la presencia de un vórtice, de variaciones en la velocidad de agitación y la agitación adicional por la evolución
del hidrógeno gaseoso cuando al cátodo se le aplican grandes densidades de corriente (superiores a 25 mA. cm-2). El electrodo de disco rotatorio es una herramienta muy Útil para el estudio de problemas teóricos y experimentales en el caso de electrodos sólidos, debido a que los procesos se llevan a cabo en condiciones de estado estacionario. Esto es muy importante desde el punto de vista electroquímico porque en este caso el transporte de masa puede ser analizado matemáticamente (ah en el caso de reacciones donde intervenga un etapa química a d d s de la electroquímica). En el sistema diseñado el flujo de cualquier especie (electroactiva o no) de la solución hacia el disco, es independiente de la distancia con respecto al eje de rotación del electrodo. Por lo tanto las características electroquíiicas fundamentales como el espesor de la capa límite de difusión y la densidad de corriente se mantienen constantes sobre toda la superficie del disco. En el anexo 3 se resúmen las características hidrodinámicas de la celda a través de los parámetros como el espesor de la capa hidrodinámica límite, el espesw dela capa límite de difusión, el número de Reynolds y el número de Peclet. Además, se especifican los criterios numéricos que indican las Condiciones de estado estacionario con flujo laminar normal a la superficie del electrodo.
~
IV.l. Teoría
Una vez observadas las condiciones mencionadas arriba, es posible utilizar el modelo de Hansen el cual se describe a continuación en su aspectos más relevantes. El modelo se basa en la difusión de los iones hidroxilo e hidrógeno dentro de la capa límite de difusión en condiciones
54

estacionarias. Se considera el caso de un flujo unidimensional laminar normal a la superficie del cátodo. Se asegura que no hay transporte de materia electroactiva por migración, al adicionarse una sal que actua como electrolito soporte. Finalmente también se
asume que el transporte de materia en la solución es por convección forzada y en el interior de la capa límite de difusión por difusión pura. En el estado estacionario el transporte de los iones hidroxilo e hidrógeno obedecen a la siguientes ecuaciones:
(IV. 1 )
donde: ai y C ~ H son laszoncentraciones de los iones hidrógeno e hidroxilo dentro de la capa de difusión, DII y DOH son los coeficientes de difusión de los iones hidrógeno e hidroxilo, S(x) es el término "fuente" que describe la desaparición de uno de los iones por recombinación con el otro ión, x es la distancia entre un punto y la superficie del cá t odo .
Además: _- CHCOH = Kw (IV. 3)
donde 15i es la constante de autoionización del agua.
Considerando que la estequiometría de recombinación es de 1:1 ,
tenemos que:
55

Si tomamos COH= K ~ C H y sustituyendo en IV.3:
DH d2CH - dx2
d%w/CH dx2 ’
integrando IV. 5:
integrando IV. 6:
(IV. 5 )
expresando la ecuación IV.7 como un polinomio de segundo grado:
2 DOH CH - - KW - 2(ax+b)CH = O, DH (IV. 8)
de donde resolviendo para CH tomando la raíz positiva y
rear reglando :
De manera análoga podemos obtener la expresión para COH: -~
112 C ~ H = cx + d + [(cx + dI2 + DH Ku] ,
(IV. 9)
(IV. 101
donde a,b,c y d son constantes de integración.
Estableciendo las condiciones límite para x=O y x=i con i como el espesor de la capa límite de difusión en el anexo 3 1 4 1 ,
tenemos que en la interfase capa de difusiódsolución
electrolítica: t3=C para x=i. Con C como la concentración de
iones hidrógeno en la solución.
Por otro lado, puesto que la producción de iones hidroxilo por
56

reducción catódica del agua debe ser igual a la razón de remoción de los hidroxilos de la superficie del cátodo, tenemos que:
para x= O. (IV. 11)
Si la corriente, I, es transportada principalmente por los iones hidrógeno (en el límite entre la capa de difusión y la solución), la condición límite de la ecuación IV. 11 también puede ser expresada por analogía como:
para x= 1. dCH -1 = -m - d x '
(IV. 12)
También tomamos en cuenta que el proceso ocurre con una solución electrofitica ácida, entonces asumimos que: C2>> KH.
Sustituyendo IV.9 en IV. 11, derivando y tomando x= 1: -
(IV. 131 2 DOH DH = $[(ax+b) + [ (ax+b) + - DH Kw] ''1 x= 1'
integrando IV.13 y sustituyendo x por 1 tenemos:
I .[(. --
ai+b) + DOHKW/DH E = a + (IV. 14)
Por otro lado, sí reescribimos IV.9 sustituyendo CH por C y x por 1:
despe Jando:
D o i d ( W / D H al+b = 2 - 2c ,
(IV. 15)
(IV. 16)

sustituyendo IV. 16 en IV. 14:
despejando
T
"a" de IV.17:
(IV. 17)
y rearreglando
I DH
a = -
[ k2- DoHKw/DH] 2+
IV. 18:
. ( IV. 19) DOH KW
2 Si asumimos que C >> KW tenemos que IV.19 es:
I 2DH'
a = -
una solución aproximada de
~ ( IV.20)
Ahora bien si retomamos IV. 15, desarrollamos y despejamos a "b": -__
2 sustituyendo IV.20 en IV.21 y asumiendo que C >> Kw: :
b = - 1 (.-:I. 2
58
( IV. 22)

Con las ecuaciones de a, b y la ecuación 11.58 podemos calcular la distribución del pH en la capa de difusión, la función correspondiente de pH= f(x) presenta un punto de inflexión alrededor de pH=7. Este punto de inflexión estaría asociado al espesor de la capa de iones hidroxilo E OH, el cual puede ser calculado partiendo de la ecuación 11.12 integrando de lo^ a 1 y
de CH= C a CH= O:
CDH IOH = I - 7. (IV. 231
Cuando se incrementa l a concentración de iones hidrógeno, C, el punto de inflexión se desplaza hacia X= O cuando la densidad de corriente es constante. Es decir que con una densidad de corriente constante existe un valor de C= CMX donde 10~~0. De la ecuación IV.23 asumiendo que l o ~=O cuando C= ciiax:
I1 Gar= - DH' (IV. 241
Análogamente de la ecuación IV.24 podemos definir un valor de Iiín suficiente para tener IoH>O:
( IV. 25)
Por otro lado, también podemos calcular el valor de la concentración de iones hidrógeno para x=O expandiendo el término raíz cuadrada como una serie de Taylor de la ecuación IV.8 y
utilizando los dos primeros términos:
DOHKN I1 -CD" cH= - (IV. 26)
Las ecuaciones IV.23 a IV.26 tienen amplia utilidad práctica, sin embargo, se observan ciertas limitaciones en los casos de bajas densidades de corriente, las cuales se presentan como las óptimas
59

debido a que la producción de gas hidrógeno (elemento indeseable por participar en el desprendiriento del depósito arrastrado por
las burbujas) en el cátodo es proporcional a la densidad de corriente.
IV. 2. Resultados
Para ilustrar los resultados ~2 la aplicación CI los diagramas tipo PTD y del uso del cátodo rotatorio diseñado en este trabajo, en el modelo de Hansen, a continuación se describe un ejemplo con el Am.
Del diagrama PTD de la figura 111.10 (cap. 111) del Am se obtiene el valor de pH=9.5 a partir del cual aparece la fase sólida Am(OH)3(s). A valores de pH inferiores a 9.5 aparece una sola especie soluble predominante, Am(S04)~. La densidad de corriente mínima necesaria para que el depósito de americio se lleve a cabo, considerando que el pH en la superficie debe tener un valor de 9 . 5 , se calcula como sigue: Se parte de una solución de Am(II1) lo-%/ SO4 O. 3 M con un pH=5 ( datos experimentales de la optimización del método de depósito). Si calculamos I i i n (ecuación IV.25) a partir del modelo de Hansen obtenemos que Imin> 2.5 x mA (con pHsoiuciÓn=S, 1=2.82 x cm (ver anexo 3, con 1=0) y DF 7.3 x lo-’ cm2 s-l [SO1 y si la calculamos a partir de la ecuación de CH ( IV.26) despejando a I y haciéndola I i i n tomando el valor límite de CH como lo-* mol cme3 (pH=5)
241
241
DOH KW C DH rain= - + - m 1 1 ’
2 -1 tenemos que Imin= 7.9 x ioe2
[SO1 1. Experimentalmente se tiene que para las mismas condiciones con I= 7 x mA el rendimiento de depósito es del orden de 1.6%.
cm-2 (con DOH= 5 x 10:’ cm s
60

Por otro lado tomando la misma ecuación de Hansen para CH,
tomando una densidad de corriente de 0.8 mA cm-2 se obtiene un
valor calculado de pH para la superficie del cátodo de 10.6. Este valor de pH en el diagrama PTD del Am (figura 111.10) se encuentra dentro del intervalo donde predomina la especie insoluble AIR(OH)~(~, . Además este valor de pH correspondería a un rendimiento de depósito teórico de 99%, el cual es similar al valor del rendimiento de depósito experimental (97.5%). Para las mismas condiciones de depósito descritas arriba se pueden calcular adicionalmente 2 parámetros:
241
a. La máxima concentración de iones hidrógeno en la solución que permitirían obtener depósitos de americio aplicando un valor determinado de corriente. De la ecuación IV.24 con 1 4 . 8 mA se obtiene que caX= 3.18 x lo-' M, lo que equivale a un pH de solución de 3.5. En idénticas condiciones experimentales se ha observado que el depósito de americio inicia cuando la solución
tiene un pH aproximado de 3.
b. El espesor de la capa ]OH (en la superficie del &todo) como resultado del gradiente de pH que se genera cuando se aplica corriente al cátodo. Este espesor esta dado por la ecuación
-- IV.23. Con un valor de I= 1 mA se obtiene que Im= 2.73 x cm, dicho espesor corresponde al 97% del espesor total de la
-
-- capa límite de difusión calculada según el anexo 3.
Como se puede observar en este ejemplo las predicciones derivadas de los diagramas tipo PTD contribuyen significativamente a un uso mas preciso del modelo de Hansen para calcular condiciones óptimas de depósito de actínidos como el americio. También, se observa que el diseño del la celda y el cátodo rotatorio han permitido el uso mas formal de dicho modelo. Por otro lado, como se verá en el capítulo VII, existe correlación entre las predicciones del modelo de Hansen y los resultados experimentales (empleando la celda diseñada en este trabajo), torando en cuenta que el modelo de Hansen adquiere más
61

consistencia empleando en los cálculos los datos aportados por
los PTD’s.
62

v. TECNICAS EXPER1-m
V. 1. Equipos
V . l . 1 . Electrodos y Celdas Electroquíriw
Para el estudio electroquímico se utilizó una configuración de tres electrodos, un electrodo de referencia de mercurio sulfato de mercurio saturado (ECM, marca Tacussel), un contraelectrodo de carbón vitreo alojado en un compartimiento separado con una pared de vidrio poroso y un electrodo- de trabajo. Para la técnica de voltamperometría cíclica se empleó un electrodo de alambre de platino como electrodo de trabaJo y para las de voltamperometría en régimen estacionario y de impedancia electroquímica se utilizó un disco rotatorio de platino. El contraelectrodo fue hecho de la siguiente manera : una sección de barra de grafito de 0.3 _(2m. de diámetro y de 0.2 cm. de longitud fue incrustada en la cavidad de una pequeña barra de cobre. Se colocó un poco de -- estaño líquido en la cavidad de la barra de cobre y al introducir la barra de carbón vítreo en la cavidad, el estaño se solidificó dejando incrustada la barra de carbón. De esta manera, se evitó el uso del mercurio líquido, el cual presenta el riesgo de escapar y contaminar el electrolito soporte. En- el extremo opuesto de la barra de cobre fue conectado un alambre conductor cuyo extremo contrario, a su vez, tiene un conector tipo banana. El conjunto carbón vítreo-cobre-alambre conductor fueron introducidos en un tubo de vidrio, sellando con pasta de silicón ambos extremos del tubo, quedó expuesta una longitud de 1.5 cm. de la barra de carbón vítreo y el conector.
63

El electrodo de alambre de platino fue elaborado de la misma manera que el contraelectrodo con la diferencia de que el alambre de platino fue unido directamente al alambre conductor. La
longitud expuesta del alambre de platino es de 0.65 cm. con un diámetro de 0.04 cm. El electrodo de disco de platino utilizado es el ED1 de la marca
Tacussel, consiste en un cilindro de Teflón con una barra de platino empotrada concéntricamente, quedando expuesta una cara circular de platino de 0.2 cm. de diámetro. El electrodo de acero inoxidable fue elaborado con las mismas dimensiones que el de platino. Para el estudio electroquímico se utilizaron dos tipos de celda. Para la voltamperornetria cíclica se construyó una celda de vidrio de tipo semiesférico, tres bocas esmeriladas permiten introducir los electrodos, otra permite introducir el electrolito soporte previamente desoxigenado en un compartimiento anexo, otra permite el acceso del nitrógeno gaseoso y una auxiliar para adicionar
-
reactivos o retirar líquido. En la boca del contraelectrodo fue colocado un sello de agua para mantener una ligera sobrepresión cuando se trabaja en atmósfera inerte de nitrógeno. Para la voltamperornetria en régimen estacionario y para la impedancia electroquímica se construyó una celda de vidrirtipo cilíndrico con una tapa de polietileno de alta densidad, una cavidad en la tapa permitió inclinar el disco de platiw.-€Xras cavidades permitieron introducir los tres electrodos y el nitrógeno gaseoso. Para la obtención de los depósitos en el estudio de parámetros experimentales se diseñó una celda electroquímica y un cátodo rotatorio (figuras V. 1 y V . 2 1. La celda y el cátodo fueron diseñados utilizando el modelo hidrodinámico para el electrodo disco rotatorio (WE) como criterio de diseño. Memás se tomaron en cuenta aspectos como: distancia interelectrodos inferior a 1
cm. para evitar el uso de volúmenes de electrolito soporte superiores a los 15 ml. y para evitar una caída omhica
64
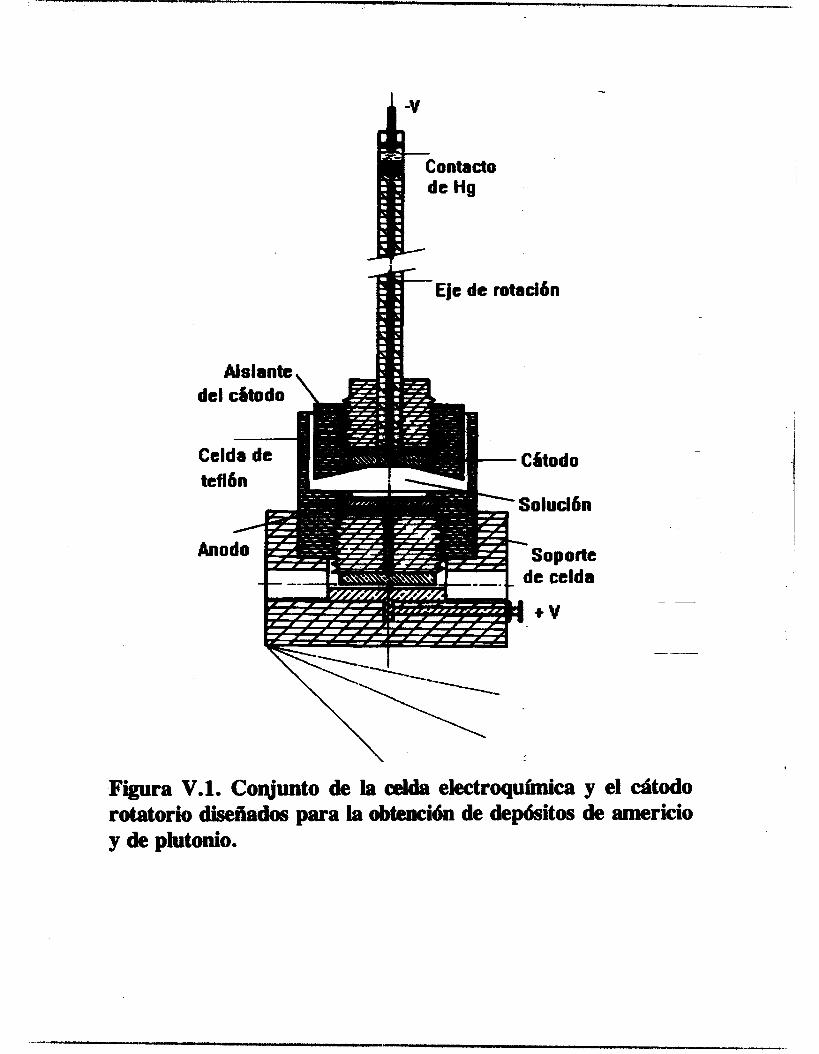
\
Figura V.1. Coqjunto de la alda electroquímica y el cátodo rotatorio diseñados para la obtención de depósitos de americio y de plutonio.
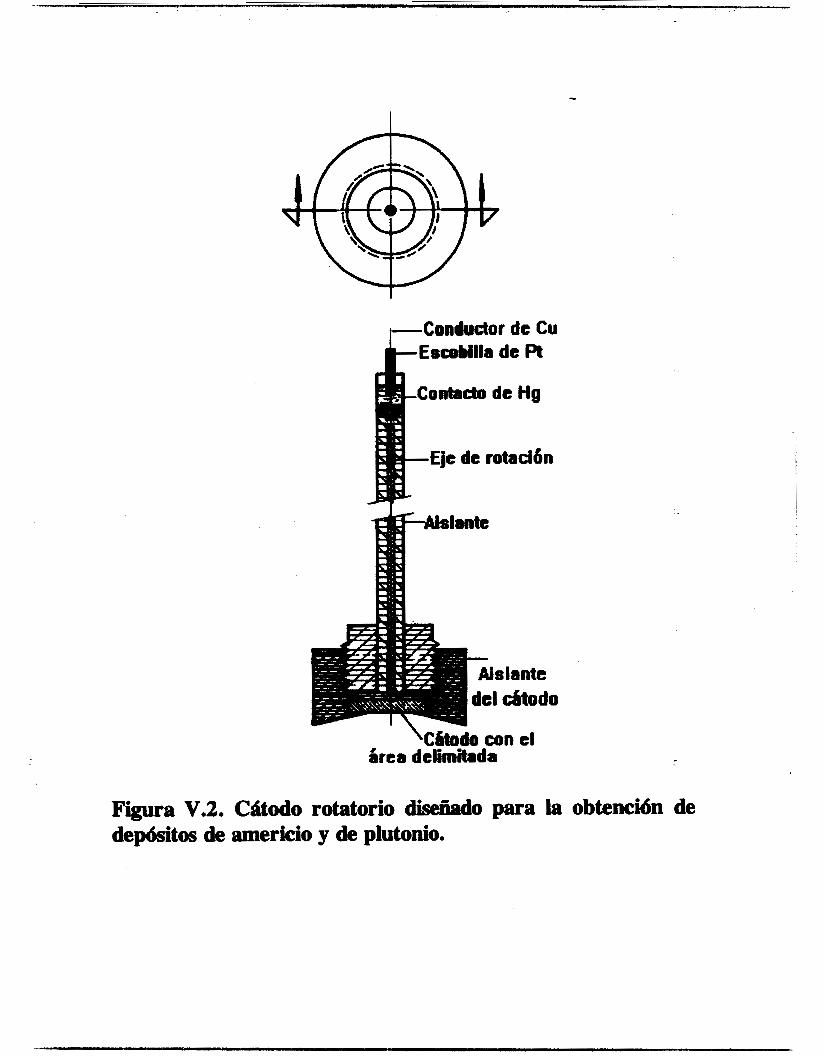
-Conductor de Cu EswQilla de pi
\Chdo can el área delimitada
Figura V.2. Cátodo rotatorio diseñado para la obtención de depósitos de americio y de plutonio.

importante, la relación de áreas del cátodo y del ánodo superior a 3 para obtener una distribución homogénea de la corriente, el material del ánodo electroquimicamente inerte para las condiciones de trabajo deseadas, la temperatura del sistema alrededor de 2OoC con variaciones inferiores al 10 %, material de
la celda y del aislante del cátodo inertes a los ácidos inorgánicos e hidrófobo. El material hidrófobo con la superficie pulida permite disminuir la fricción sobre las películas de
solución acuosa que se deslizan sobre su superficie contribuyendo así a mantener un flujo laminar en la periferia del disco rotatorio. La celda con forma cilíndrica fue elaborada en teflón. En el fondo se encuentra un disco de platino de forma circular inmovilizado entre la pared del fondo de la celda y un tapón roscado. El tapón roscado forma parte de la base de la celda sirviendo además como conductor, pues cuenta con un alma de latón recubierta de lucita. La celda descansa sobre un soporte de lucita en cuyo fondo se encuentra un disco de latón, el cual da continuidad como elemento conductor. Una barra del mismo material pone en contacto a un alambre conductor y al latón de la base del soporte de la celda. De esta manera se asegura la conducción de corriente desde el ánodo de platino hasta el alambre conductor de la fuente de corriente. La masa de bronce es 50 veces la masa del h o d 0 de platino, entonces si hubiera un incremento en la temperatura del platino ésta se distribuiría en todo el bronce, de tal manera que si la temperatura del disco se incrementara a 8OoC la temperatura, al equilibrio, del conjunto bronce-platino sería de 21.17’C. Además, en la base del soporte de la celda hay 6 canales que permitirían, por el intercambio de aire del exterior, enfriar la masa de bronce. El soporte de l a celda es orientable por lo que la celda puede ser incl inada. El cátodo rotatorio consiste en un aislante de teflón de forma
65

cilíndrica, en un disco metálico sobre el cual se obtienen los
depósitos y en una barra que, sujeta al rotor, permite girar el
cátodo. La cara del aislante, expuesta al ánodo, tiene una concavidad de tipo cilíndrico truncado, de tal manera que en el
fondo se define una horadación de forma circular concéntrica con
respecto al aislante. El disco metálico se encuentra sujeto entre el aislante de teflón y una tapa de lucita con un alma de latón,
de tal manera que la concavidad del aislante delimita la
-superficie circular sobre el disco. Una barra de lucita con un alma de latón solidaria de la tapa del aislante, funciona com
eje de rotación al ser ésta conectada al cabezal del motor. Eh el otro extremo de la barra hay un coigartiiiento con mercurio
metálico, en él se encuentra inmersa una barra de platino
conectada directamente al alma de latón del eje de rotación y ma
barra de platino conectada a un alambre conductor de la fuente de 1 I I
corriente. De esta manera se logra la conducción eléctrica desde i
el disco metálico hasta la fuente de corriente, sin utilizar
mercurio metálico en contacto directo con el cátodo. Por medio de
un multíiaetro fue medida l a resistencia (de la línea de
conducción de la corriente) entre los dos conductores poniendo en contacto directo el ánodo y el cátodo. El valor de la resistencia me de 5 n. Igualmente la masa de latón de la tapa del aislante y del alma
*eje de rotación permite disipar el calor generado en el disco
metálico, de manera semejante a l a del ánodo. Además, una serie
de perforaciones en todo lo largo de la lucita del eje de
rotación permite el intercambio de aire con el exterior enfriaido al latón.
El motor también es orientable como la celda, de tal manera que es posible obtener depósitos con la celda y el cátodo rotatorio
inclinados manteniendo las superficies del ánodo y del cátodo
paralelas equidistantes en cualquier punto de la superficie de
ambos.
Para impedir la vibración radial y axial del cátodo rotatorio el
66

eje de rotación tiene una alma de bronce de 0.4 cm. de diámetro.
Además, el eje está sujeto en 2 puntos fijos solidarios al motor,
un punto es el cabezal del motor (el cual se encuentra a 2 cm.
del aislante y del cátodo) y otro es el balero solidario al motor y que sujeta al eje en un punto a 20 cm. del aislante y del cátodo.
V. l .2 . Motor
La rotación del cátodo se logró por medio de un motor con control digital de velocidad (marca Jank & Kunkel 1KA-UERK modelo
RW-20Il2M) de 0-2000 RPM. Sobre la base del motor (figura V .3 1 se construyó un posicionador levadizo sobre el cual se encuentra la celda electroquímica. El posicionador permite colocar a la celda en la posición deseada incluida la inclinacibn con respecto al
plano posicionador.
La cabeza del motor gira sobre un eje solidario a la base del motor, se colocó en el eje un cuadrante, el cual permite inclinar el cátodo rotatorio en el ángulo deseado y con respecto a la
normal de la superficie del posicionador levadizo.
- __ V. 1.3. Caja de Faraday
Durante las mediciones de impedancia electroquímica, el
diFpTtivo celda electroquimica-electrodos fue protegido de
campos eléctricos externos generados por carga estática y de
campos magnéticos asociados a líneas de transmisión de corriente. La protección se logró manejando el dispositivo dentro de una
caja de Faraday elaborada en- hierro al carbón y recubierta con pintura de aluminio. Las señales provenientes de los electrodos pasan a través de cables conectados a conectores tipo BNC
(aislados de la caja de Faraday), y a su vez cables coaxiales
blindados tipo BNC recogen las señales y las conducen al
instrumento de medición. La caja de Faraday fue puesta a tierra a
67
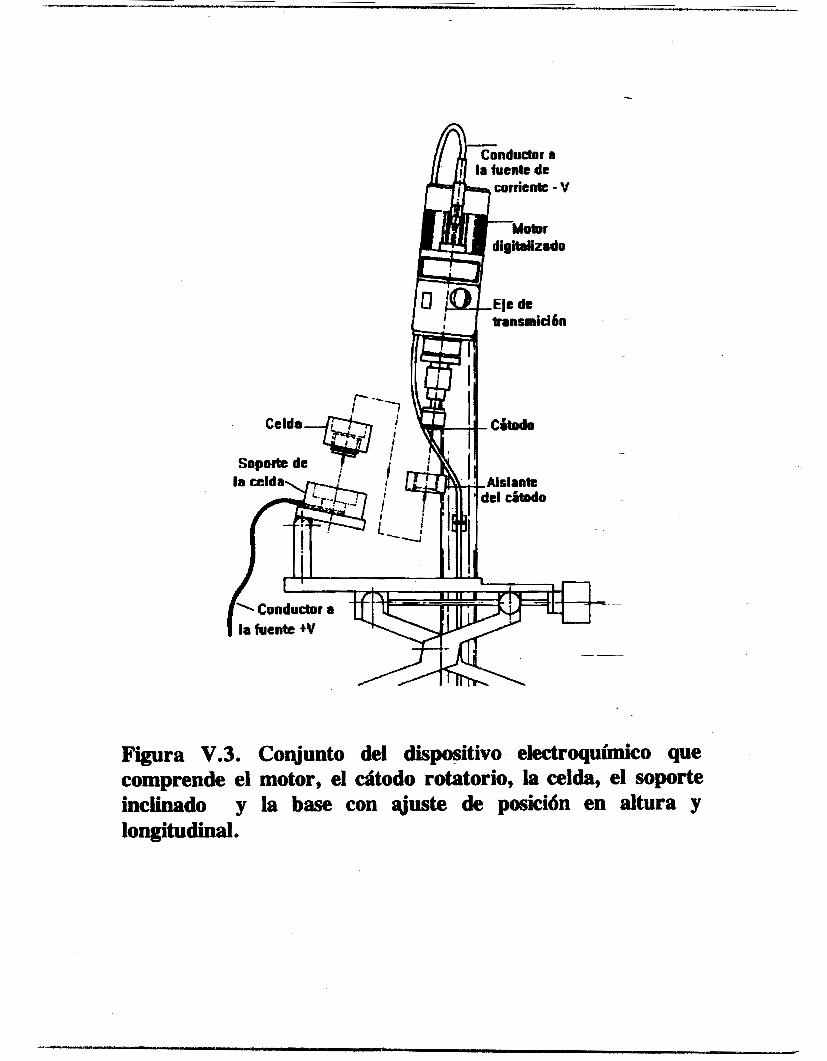
Conducbr a
corritntc
.EJe de
- V
,do
--
Figura V.3. Conjunto del dispositivo electroquúnico que comprende el motor, el Catodo rotatorio, la celda, el soporte inclinado y la base con ajuste de posición en altura y longitudinal.

través del blindaje del cable coaxial terminando en la tierra física del laboratorio.
V. 1.4. Potenciostato Para la voltamperometría cíclica se utilizó un potenciostato galvanostato modelo BIPAD marca Tacussel acoplado a un generador de señales modelo GSTP3 de la misma marca. Los voltamperogramas se obtuvieron en un graficador XY WX1100 marca Graphtec.
111.1.5. Sistema de medidas electroquímicas
Las mediciones de voltamperometria en régimen estacionario y de impedancia electroquímica se llevaron a cabo con el sistema computarizado 2-Computer marca Tacussel. Las mediciones se llevaron a cabo utilizando una ricrocomputadora 9826 de Hewlett Packard por medio de programas desarrollados por la compañia COLEA. El graficador 7470 A de Hewlett Packard acoplado a la microcomputadora generó los gráficos de voltamperometría y de impedancia.
-
V. 1.6. Disco rotatorio Para las técnicas de voltamperometria en régimen estacionario y
de impedancia se utilizó un disco rotatorio de 0.2 cm. de diámetro marca Tacussel, un motor compacto y ligero daba movimiento al disco, la velocidad de rotación es controlada por la unidad Controvit de Tacussel con intervalo de velocidades de 0-500 RPM.
____
V. 1.7. Medidores de pH El pH de las soluciones del electrolito soporte fueron realizadas con el medidor de pH modelo 240 de Corning, el de las soluciones radiactivas con un medidor modelo Metrohm 654 de Roucaire. Ambos medidores fueron calibrados con soluciones buffer marca Merck
68

cubriendo los intervalos de 3 a 7 y de 7 a 10.
V. l .8 . Balanzas
En la preparación del electrolito soporte de sulfato de sodio y de la solución de samario, las sales fueron pesadas con una
balanza modelo 52000 marca Bocch y las pesadas de las soluciones
radiactivas para preparar las diluciones de europio, americio y plutonio se llevaron a cabo con una balanza modelo AT200
electrónica marca Mettler. La balanza AT200 fue colocada dentro
de una caja de guantes con presión negativa.
V. 1.9. Cafa de guantes
La elaboración de las diluciones de material radiactivo se llevó
a cabo dentro de una caja de guantes con 2 compartimientos. Un compartimiento para la manipulación de la solución y otro para la
balanza AT200 para la pesada de las alícuotas de material radiac t ivo. Para la obtención de depósitos de europio, americio y plutonio,
el dispositivo electroquímico fue colocado dentro de otra caja de
guantes con presión negativa y sistema de extracción con filtros
absolutos.
V.l.10. Espectróretro de fluorescencia de rayos X
La determinación del samario depositado en cada muestra fue
realizada por medio de un espectrómetro de fluorescencia de rayos X. La excitación de la muestra se logró por medio de una fuente
de Am-241 (emisor a, X, y ) .
Se empleó un detector de estado sólido de germanio hiperpuro de 2 cm y 1 cm de diámetro. Las señales provenientes del detector fueron amplificadas por el módulo 514 de 1NEL.La digitalización de los pulsos se obtuvo con un convertidor analógico digital tipo Wilkinson de 100 t4hz de Nuclear Data. El análisis del espectro de
3
69

rayos X se realizó con una microcomputadora 286 GS Goupil de marca Bull. El software Inter PC instalado en la máquina permitió obtener los datos que se emplearon en el análisis cuantitativo.
V. 1.11. Espectrómetro gaiira
El europio 152, elemento radiactivo emisor S/r fue cuantificado por medio de un sistema de espectrometría gama. La radiación incidió sobre un detector de estado sólido de germanio dopado con litio de tipo coaxial de 3"x 3". El detector fue protegido de la
radiación cósmica y ambiental por medio de un blindaje de plomo de 5 cm de espesor. Los pulsos de voltaje generados pasaron a
través de un amplificador 7129 de Enertec y fueron digitalizados por el módulo Cosinus GS de Nuclear Data. El espectro de energías fue almacenado en una microcomputadora 386/20 e Marca Compaq y posteriormente analizado por el programa Inter PC.
V. 1.12. Espectrómetro alfa
Siendo emisores alfa el americio y el plutonio, fueron cuantificados por espectrometría alfa. El detector de estado sólido de silicio con unión implantada de iones se encontraba dentro de un compartimiento de vacío, el vacío se logró por medio de una bomba mecánica lográndose vacíos del orden de 10-2mBar. Para el análisis cuantitativo se empleó un detector de 450 mm y
para la caracterización de los espectros alfa se empleó un detector de 20 mm2 recibiendo un haz colimado de partículas alfa. Cada detector contó con un modelo que incluia una fuente de polarización, un amplificador y un convertidor analógico digital, los modelos son 7191 y 7185 de Enertec respectivamente. La adquisición de los espectros de energía se obtuvo con una microcomputadora 386/2Oe de Compaq y el programa Inter PC permitió obtener el análisis cuantitativo de los espectros.
2
70

V. 1.13. Analizador de imagenes
La distribución de los depósitos se obtuvo procesando un detector plástico de trazas nucleares, en un analizador de imagenes. Se utilizó un detector tipo CR39 de Kodak. El procesador incluye un digitalizador de imágenes y un programa de computadora del IMP
que permitió determinar la densidad de trazas nucleares por
unidad de área.
V. 2. Reactivos
V. 2.1. Electrolito soporte
El electrolito soporte fue preparado con sulfato de sodio anhidro grado análisis con un 99% de pureza marca Merck. Se empleó agua desionizada para la disolución, con una resistividad característica de 10l2 O. El ajuste del pH de las diferentes soluciones se logró agregando H2S0, al 96% grado analítico de Merck, o bien, agregando NaOH al 90% grado analítico de Merck. Las soluciones fueron almacenadas en frascos de pol ie t i leno. -
_- V.2.2. Solución de -io La solución de Samario fue preparada con sulfato de samario al 99.9% de pureza de Aldrich, utilizando agua desionizada.
V.2.3. Solución de europio
Se realizó la dilución de una solución madre de europio 152 radiactivo, tomando una alicuota y diluyéndola en una solución de sulfato de sodio. La solución madre de Eu-152 es producida por la compañia ORIS-INDUSTRIE de Francia.
71

V. 2.4. Solución de mricio A partir de una solución madre de Am-241 en ácido nítrico diluido, se obtuvo la solución por dilución de l a solución madre en una solución de sulfato de sodio. La solución madre de h-241
es producida por la compaAia ORIS INWSTRIE de Francia.
V. 2.5. Solución de plutonio Se elaboraron soluciones de plutonio 239íplutonio 240 y de plutonio 240 a partir de las soluciones madre correspondientes, comercializadas por ORIS-INMISTRIE, Francia. Alicuotas de cada solución fueron diluidas en soluciones de sulfato de sodio. Todas las soluciones empleadas fueron almacenadas en frascos de polietileno herméticamente cerradas para evitar la evaporación del agua y el aumento en la concentración del elemento de interés. Las soluciones de Am y de Pu tuvieron 2 semanas (máximo) de almacenamiento antes de ser utilizadas. Las soluciones fueron almacenadas a 18-Z1°C.
v. 3. Rocedimientos
V. 3.1. EitudToXectroquíiico
V. 3.1.1. Volt-tría cíclica a). La solución electrolítica (con diferente valor de pH y pC0;
para cada caso) fue desoxigenada por medio del burbujeo de gas nitrógeno durante 20 minutos. El gas nitrógeno antes de penetrar a la celda pasaba por una trampa de pirogalol y por una trampa de agua desionizada. Además, durante todos los experimentos se mantenía una atmósfera de nitrógeno dentro de la celda. Después se realizaba la electrólisis galvanostática (con diferentes
72

densidades de corriente y tiempos para cada caso) e
inmediatamente después se obtenían los voltamperogramas
(in-situ), dentro del intervalo de potenciales de
electroactividad. Posteriormente, la superficie del electrodo de
Pt era regenerada aplicando barridos rápidos (0.5 VIS) dentro del intervalo de potenciales de electroactividad durante 30 minutos y con burbujeo de Nitrógeno. Una vez regenerado el electrodo se agregaba la solución de Sm(II1) a la solución electrolítica y con burbujeo de gas
nitrógeno, se retiraban las trazas de oxígeno molecular disuelto.
La electrólisis con Sa(II1) era realizada en las mismas
condiciones de la solución electrolítica, al final se obtenía la serie de voltamperograms correspondientes (insitu con v=50
mV. s- l ) en igualdad de condiciones también.
b). Se empleó una solución desoxigenada (con burbujeo de gas nitrógeno) para caracterizar el electrodo de Pt dentro del
intervalo de potenciales de electroactividad. Después, el
-
electrodo de Pt permaneció dentro de un vaso de pprecipitados, inmerso en una solución electrolítica con Sm(II1) durante
períodos variados de tiempo (para cada caso).
Posteriormente el disco de Pt fue enjuagado con agua desionizada para retirar de la superficie-del-Pt las trazas de la solución de Sm(II1) e inmediatamente después fue colocado dentro de la celda con la solución previament+&xigenada y en atmósfera de
nitrógeno.
Finalmente se obtuvieron los voltamperogramas del electrodo de Pt dentro del intervalo de potenciales de electroactividad.
V. 3.1.2. Voltamperoiatría en régimen estacionario
a). Inicialmente la solución electrolítica fue desoxigenada por burbujeo de gas nitrógeno y manteniendo una atmósfera de nitrógeno en el interior de la celda se obtuvieron los voltamperogramas del electrodo de Pt, el cual rotaba a velocidad
73

144159
constante. La velocidad de barrido fue de 1 mV s e ' .
Después, programas de barrido de 0 . 5 V s-'
burbujeo de nitrógeno. Al agregar una alicuota de solución de Sm(II1). según el procedimiento descrito en el párrafo anterior. Finalmente a una velocidad de barrido de 1 mV s-l se obtenían los voltamperogramas con el electrodo rotando a una velocidad constante en una atmósfera inerte de nitrógeno gaseoso. Se obtuvieron voltamperogramas cubriendo un intervalo de valores de pH de 2 a 10.
b). El electrodo de Pt fue caracterizado por voltamperometria cíclica en la solución electrolitica desoxigenada y con atmósfera inerte de nitrógeno.
Después, el electrodo fue mantenido dentro de un recipiente cerrado, inmerso en una solución de electrolito soporte con Sm (111) con diferentes tiempos de inmersión y con diferentes
se regeneraba la superficie original del electrodo con durante 30 minutos y con
-
concentraciones de Srn (111). Al retirar el electrodo de Pt de la solución de Srn (III),éste era inmediatamente enjuagado e introducido en la celda de electrolito soporte desoxigenado y con atmósfera inerte de nitrógeno. Finalmente con el electrodo rotando se- obtenían los voltamperogramas del Pt.
V. 3.1.3. Irpedsncia electroquímica En la solución electrolítica desoxigenada y mantenida en atmósfera inerte de Nitrógeno se introdujo el electrodo de Pt (previamente caracterizado por voltamperornetria cíclica) y se obtuvo un primer diagrama de impedancia electroquímica aplicando un cierto sobrepotencial al electrodo de Pt. Después, se regeneraba la superficie original del Pt aplicando un programa de barridos cíclicos con una velocidad de 0.5 V/s durante 20 minutos y con burbujeo de nitrógeno.
74

Posteriormente a la solución electrolítica se agregaba Sm ( 1 1 1 ) y
con burbujeo de nitrógeno se eliminaban las trazas de oxígeno
disuelto en la solución. Finalmente, se aplicó una seflal sinusoidal sobrepuesta a un
sobrepotencial al electrodo de Pt y se obtuvo el correspondiente diagrama de impedancia electroquímica.
Durante las mediciones la celda fue mantenida dentro de la caja
de Faraday de hierro para evitar la influencia de cargas
estáticas y de campos electromagnéticos que pudieran generar
corrientes parásitas.
V.3.2. Optimización del método de depósito
El tratamiento de limpieza de los discos de acero inoxidable
(electropulido de fábrica) y de Platino utilizados consistió en
lo siguiente:
Los discos fueron sometidos a un baño ultrasónico inmersos dentro de una solución de extrán (detergente neutro, Merck) durante 4 horas. Cada disco fue inmovilizado en el fondo del recipiente para evitar que el contacto de uno con otro provocara rayaduras
-
en la superficie de éstos. /
Después, los discos fueron lavados enérgicamente con agua
desionizada para asegurar la ausencia de detergente en la
superficie. Entonces, en las condiciones descritas fueron
utilizados para efectuar los depósitos de los diferentes
elementos. Después de los depósitos el disco fue enjuagado con una solución de amoniaco al 1% para retirar las trazas de electrolito soporte
sin disolver el depósito. Posterior a un secado al aire libre el depósito fue calcinado a 4OO0C durante 20 minutos. La finalidad
del calcinado es obtener por deshidratación una película de óxido más adherente al sustrato.
Todos los depósitos fueron efectuados a temperatura (2OOC) y presión ( 1 atm. 1 ambientes.
75

Las condiciones iniciales de optimización del método son:
-Solución electrolítica de Na2C04 (empleando para el ajuste pH ácido sulfúrico e hidróxido de sodio) con un volumen de ml. -=todo de acero inoxidable y de platino con una área de 18 cm'.
del 12
-Anodo de Platino con una área de 5.3 cm'. -Distancia interelectrodos de 0.8 cm. -Inclinación del sistema cátodo rotatorio-celda de 15' con respecto a la normal de la plataforma donde se asienta la celda
y el soporte del motor del cátodo. -Concentración del elemento variable según cada caso oscilando entre y M.
La optimización del método de depósito se realizó estudiando el comportamiento del rendimiento de depósito en función del pH de la solución, de la concentración del electrolito soporte, de la densidad de corriente, de la velocidad de rotación del cátodo y
del tiempo de depósito. El estudio se realizó en el orden mencionado por lo que el rendimiento evolucionó con el número de parámetros. Debido al principio de depósito que es por pmpitación del hidróxido, se estudiaron en primer lugar los parámetros relacionados con la acidez de la solución y de fiterface metal disolucibn y después los parámetros clásicos en estudios cinéticos como son la velocidad de rotación y el tiempo de depósito. En lo que al rendimiento de depósito se refiere se reporta un promedio de 10 medidas con una desviación estándar en % relativa al rendimiento promedio.
V.3.2.1. pH y pSO; del electro l i to soporte
A partir de los resultados cualitativos obtenidos en el estudio
76

electroquímico y en el estudio fisicoquímico de las disoluciones, se definió el intervalo de pH a ser estudiado resultando ser de 2 a 7. Entonces se prepararon soluciones con valores de pH de 2, 3.5 , 4 . 5 , 6 y 7 , y con ellas se obtuvieron depósitos para el cálculo del rendimiento de depósito correspondiente a cada pH. Además, se estudió la influencia de la concentración del sulfato de sodio como electrolito soporte sobre el rendimiento de depósito. Se elaboraron soluciones con concentraciones de 1 , 0 .5 ,
0.31, 0 .2 y O. 1 M con los correspondientes valores de pH señalados arriba. De esta manera, se obtuvo el comportamiento del rendimiento de depósito en el plano pH de la solución-concentración del electrolito soporte.
111.3.2.2. Densidad de corriente Los valores de los parámetros tiempo de depósito y frecuencia de rotación del cátodo se mantuvieron como en el párrafo anterior y
los valores de pH de la solución y concentración del electrolito soporte se fijaron como resultado de la optimización tomando los valores de 5 y 0.3 M respectivamente. Se estudió el rendimiento de depósito dentro del intervalo de densidad de corriente de O a 25.4 El límite superior fue determinado en función de la eficiencia del sistema para evacuar el gas hidrógeno generado en la superficie del cátodo. Corrientes superiores a 25.5 mA producen un volumen de gas hidrógeno superior al que el sistema puede evacuar por lo que su acumulación en la superficie pasiva al catodo hasta aislarlo completamente de la solución electrolítica. Dentro del intervalo se obtuvieron depósitos aplicando densidades de corriente de: O , 0 .8 , 0.18, 0.34, 0.71, 1.41, 2.82, 5.65,
8.47, 16.95 y 25.4 mA cm-’.

V. 3.2.3. Velocidad de rotación del cátodo El parámetro tiempo de depósito se mantuvo fijo con el valor asignado en los párrafos anteriores. Los valores de los parámetros pH, concentración de electrolito soporte y densidad de corriente se fijaron como 5, 0.3 M y 0.5-1 mA (dependiendo del elemento) respectivamente. La convección forzada con flujo laminar a través de un cátodo rotatorio permite transportar masa, de la solución hacia la interfase cátodo-solución, en condiciones estacionarias. Experimentalmente el intervalo de estudio fue de 100 a 1200 rprn, donde el límite superior fue determinado al observar la aparición de espuma en la solución a velocidades superiores a 1200 rpm (siendo indeseable la presencia de espuma durante el depósito).Se obtuvieron depósitos para las velocidades de rotación de 100,
200, 400, 600, 800, 1000 y 1200 rprn.
V.3.2.4. Tiempo de depósito Siendo el Último parámetro sujeto de estudio, los restantes parámetros tomaron los valores correspondientes al término del estudio de cada uno. El pH de la solución electrolítica fue de 5,
la concentración del electrolito soporte fue de 0 . 3 M, la densidad de corriente de 0.5-1 nul cme2 (dependiendo del elemento) y la velocidad de rotación del cátodo de 1200 rpm. Uno de los elementos que condicionan el rendimiento de depósito es la cinética del proceso por lo que necesariamente habrá una dependencia con el tiempo. Se estudió el rendimiento de depósito obteniendo depósitos para los tiempos de 5, 15, 25, 35, 45, 65,
80 y 110 minutos.
78

V. 3.3. Espectrometría .
V. 3.3.1. Fluorescencia de rayos X
En la determinación cualitativa y cuantitativa del rendimiento de depósito del Samario se utilizó la técnica de fluorescencia de
rayos X debido a que el Samario no es radiactivo como el resto de
los elementos estudiados. La detección se llevó a cabo por
difracción por medio de una fuente exitadora de Am-241 de forma anular. El tipo de detector utilizado permite alcanzar mejor resolución del espectro hasta energías bajas puesto que el
detector no tiene ventana y el tratamiento del espectro se
realizó con un programa de computo para análisis de espectros de Rayos X (fabricado por Inter PC). Las muestras fueron medidas con
tiempos de detección del orden de 28 horas debido a las
cantidades de materia tan pequeñas (del orden de nanogramos).
Para el análisis de los depósitos de Samario se contó con la previa calibración del equipo utilizando fuentes disponibles comercialmente.
La identificación del Samario se basó en el espectro de energías
de emisión de rayos X publicados en la literatura y la
cuantificación _ _ del Sm se llevó a cabo por integración de áreas
bajo los picos característicos por comparación entre la masa
conocida de una muestra patrón (geometría idéntica a la de la
muestra) y la masa desconocida de cada muestra obtenida por vía elect roquírnica.
_-
V. 3.3.2. Espectrometría g a r a
Otro de los elementos estudiados fue el europio, utilizando el
isótopo radiactivo Eu-152 (emisor j3,r). Se utilizó un detector de buena resolución (1.0 Kev para el pico de 1332 Kev del Co-60) y un programa de computadora para el análisis del espectro gamma (fabricado por Inter PC), Las muestras fueron medidas con tiempos
79

de detección de 10 minutos. Con el equipo previamente calibrado en energía se procedió a la identificación de los picos con las energías características del Eu-152 y posteriormente con el
programa de análisis Inter PC se procedió al análisis cuantitativo por comparación de las áreas bajo los picos de una muestra patrón de Eu-152 con una cantidad conocida y de las muestras obtenidas electroquímicamente.
El conjunto de energías gamma empleadas en el análisis fue:
Er (Kev) Intensidad Relativa ( X I
1 1408. O 20.8
2 1112.1 13.5
3 967.5 14.6
4 778.9 12.9
5 121.8 28.4
Se seleccionaron aquefias energías que interfirieran lo menos posible con otras energías y que tuvieran la mayor intensidad relativa de emisión.
V. 3.3.3. Espectroretría alfa
El americio y el p1utoni-u fueron analizados y cuantificados por espectrometría alfa utilizando un detector de buena resolución (con una resolución intrínseca de 10.7 Kev empleando un colimador) y un detector de gran superficie para la cuantificación. Los espectros alfa fueron analizados con un equipo previamente calibrado en energías y eficiencia de detección, los radioisótopos fueron identificados en función de las energías características y aplicando el programa Inter PC se obtuvo el área bajo los picos para después calcular la actividad radiactiva de cada radioisótopo utilizando el factor de eficiencia de detección. Las muestras fueron medidas durante 60

mi nut os.
Las energías utilizadas en el análisis y cuantificación son:
Ey(Mev1 Intensidad relativa ( X I
Am-241 5.388 1.4
5.443 12.8
5.485 85.2
5.544 o. 3
Pu-239 5.105 11.7
5.143 15.1
5.156 73. o
El análisis de la estructura fina de emisión alfa del Am-241 y del Pu-239, para el cálculo de las probabilidades de emisión alfa se llevó a cabo utilizando espectros experimentales seleccionados. El trabajo de análisis espectral, convolución y
cálculo de probabilidades se efectuó en una microcomputadora.
- --
V.3.4. Caracterización Física de los depósitos
V. 3.4.1. Alfagrafía Para determinar la uniformidad con que se encuentra distribuido el depósito sobre la superficie del sustrato, se aprovechó la emisión de partículas alfa del AB-241 aplicando la técnica de trazas de partículas cargadas en materiales dieléctricos. En una cámara cerrada con un vacío de torr se irradió una película de plástico CR39 con una fuente de Ai-241 colocada a 0.2 cm de distancia durante 20 minutos. Después del grabado químico, la película fue analizada en un micróscopio óptico para determinar la densidad de
(Area de 9 cm2 y espesor de 300
81

las trazas por unidad de superficie.
V. 4.2. Perf ilometría Para la medida de la rugosidad de los cátodos empleados para los depósitos, se empleó un perfilómetro Sloan modelo Dektrak I1 A
con una punta de diamante de 25 de diámetro, la cual aplica un peso aproximado de 30 mg (0.25 niN) sobre la muestra. Se
realizaron barridos de 800 pm de longitud con una resolución vertical de 5 A. Las muestras se colocaron en la plataforma del instrumento y el resto de la operación se llevó a cabo automáticamente piloteada por la microcomputadora, la cual proporciona los datos de
rugosidad: La rugosidad es expresada en unidades de longitud y es el promedio de las diferencias que hay entre la altura media
(distancia entre el máximo y el mínimo desplazadentos verticales) y la distribución de los desplazamientos a lo largo de 1 barr ido.
82

VI. FSNDIO ELEcrriogvlnrco
Los lantánidos y los actínidos tienen potenciales electroquímicos de reducción mas negativos que el de descomposición del agua [331. A través de la generación de un gradiente de pH en la interfase del cátodo se logra superar el producto de solubilidad de los hidróxidos correspondientes E151 (del orden de El gradiente de pH en la superficie del cátodo se debe a la reducción de los iones hidrógeno de un medio acuoso ácido. Debido a esto el proceso de depósito de estos elementos en medio acuoso, involucra su precipitación como hidróxidos de manera selectiva sobre el cátodo. Otsuka 1661 reporta propiedades electrocatalíticas de algunos lantánidos en reacciones de síntesis orgánica. Recientemente Jacksik 1671 en am estudio teórico concluye en que los lantánidos poseen propiedades electrocatalíticas en la reacción de evolución de hidrógeno. De aquí que el estudio electroquímico del depósito de estos materiales, se basa en el efecto electrocatalítico sobre la reacción de evolución de hidrógeno durante la reducción catódica del ión hidrógeno en medio acuoso ácido. Por otro lado cabe recordar que debido a la similitud en el comportamiento electroquímico de los lantánidos y de los actínidos 1681, el presente estudio se realizó sustituyendo al americio y al plutonio por el samario. Mediante la técnica de voltamperometría en regimen de difusión estacionario sé estudió el depósito en función del efecto electrocatalítico sobre la reacción de evolución de hidrógeno. Posteriormente se aplicó la técnica de voltanperometría no estacionaria para observar el efecto sobre la adsorción y la desorción del ión hidrógeno sobre la superficie del electrodo y finalmente por análisis armónico de
83

la impedancia electroquímica se determinó la resistencia de transferencia de carga, la resistencia total y la capacidad de la doble capa.
V I . 1. Voltampemmetria en régimen de difusión estacionario
a. Reducción de H’. En un estudio teórico Jacksik [671 propone que las aleaciones de lantánidos con metales del grupo VIIIA,
como el platino, pueden presentar un efecto electrocatlitico en la reacción de evolución de hidrógeno. Con la finalidad de observar tal efecto se obtuvieron voltamperogramas de electrodos de platino en contacto con una solución electrolitica conteniendo samario loe3 M con una concentración total de sulfato de sodio de 0 .3 i4. La concentración de samario fue arbitraria mientras que la concentración de sulfato garantizó la ausencia del problema de la caída ohmica que pudiera perturbar las características de los voltamperogramas que sólo deben reflejar el efecto del proceso en estudio. Las curvas de respuesta corresponden a diferentes valores de pH de la solución. El límite inferior de pH fue seleccionado como el valor mínimo necesario para obtener depósito de lantánidos según Hansen [15,161, y el límite superior se obtuvo del diagrama tipo Pourbaix, el cual corresponde al limite inferior de la zona de predominio de la especie insoluble Sm(OH)3ts) (ver la sección de estudio fisicoquíiico). Las condiciones de medición fueron v= 1 mV s , E,=EI4,
Ef’Eevolución de Hz’ Las figuras VI.1, a VI.6 presentan la respuesta voltamperoiétrica para los valores de pH de 2 a 8 respectivamente. En todas las figuras la linea continua representa la respuesta sin saiario y
la línea discontinua la respuesta con samario. En el intervalo de pH de 2 a 5 la corriente límite de difusión es mayor en presencia de samario, a pH=5 se distinguen dos intervalos de potencial para la corriente límite . En el
-1
con el cátodo rotando a 200 rpm.
84
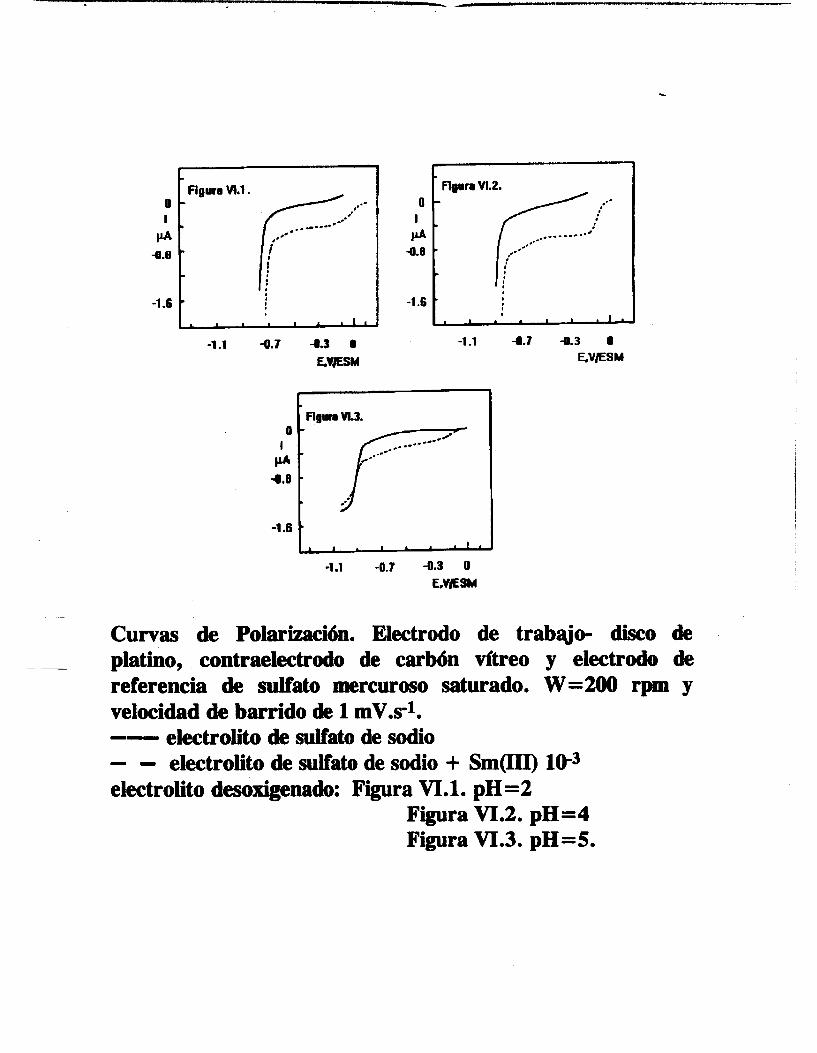
O I
PA -0.8
-1.6
+ ' " ' ' " ' -1 .l -0.7 -0.3 I
€W=M
I
-1.1
-1.1 4.7 -0.3 O EXIESM
4.7 4.3 o E.VíESM
-
Curvas de Polarización. Electrodo de trabajo- disco de -~ platino, contraektrodo de carbón vítreo y electrodo de
referencia de sulfato mercuroso saturado, W=200 rpm y velocidad de barrido de 1 mV.s-f. --- electrolito de sulfato de sodio - - electrolito de sulfato de sodio + S r n o 10-3 electrolito desoxigenado: Figura W.1. pH=2
Figura VI.2. pH=4 Figura VI.3. pH=5.
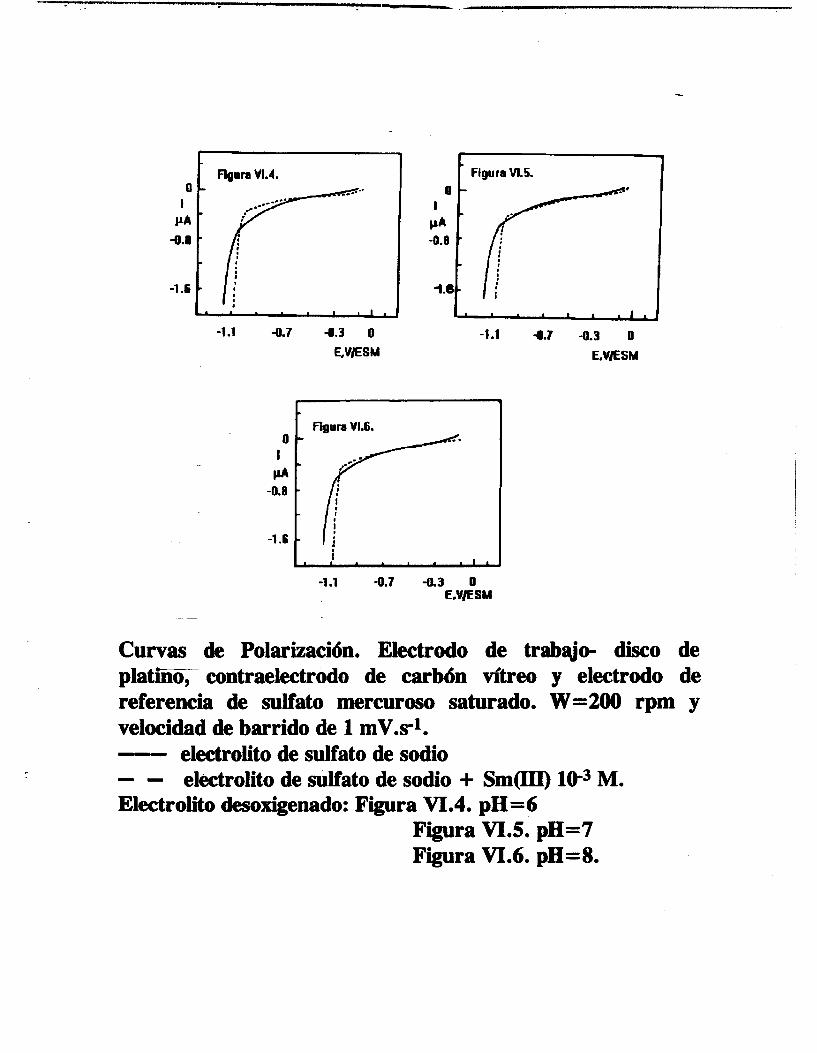
O I
PA 4.8
-1 .c
Fgura V1.6. -I/- I I
i I .
-1.1 6.7 4 . 3 o E,VIECM
O I PA
-0.8
-1 .c
1 FiguraVi.5.
c
-1.1 4.7 -0.3 O
E.ViESM
-1.1 -0.7 -0.3 O E.VESrn
_ _
Curvas de Polarización. Electrodo de trabajo- disco de plat- contraelectrodo de carbón vítreo y electrodo de referencia de sulfato mercuroso saturado. W=200 rpm y velocidad de barrido de 1 mV.s-1. --- electrolito de sulfato de sodio - - electrolito de sulfato de sodio + S m o 10-3 M. Electrolito desoxigenado: Figura VI.4. pH=6
Figura VI.5. pH=7 Figura VI.6. pH=8.

intervalo de 2 a 4 tenemos una corriente limite debida a la
adsorción de hidrógeno: H+ + e 4 H . A pH=5 observamos dos valores de corriente limite debidos a la adsorción de hidrógeno:
-f--
a c ad
-t-- - t - Had H + + e - H ~ H ~ O + e intervalo de 6 a 10 se presenta solo una de hidrógeno: H O + e - H + OH-.
Por otro lado el potencial de inicio de la es menos negativo cuando el samario potencial se desplaza hacia valores menos pH de la solución aumenta de 2 a 4.
a c ad
-f--
2 ad
+ OH- [69.701. En el corriente de adsorción
evolución de hidrógeno esta presente. Dicho
negativos conforme el
El estudio fisicoquímico (ver la sección correspondiente) sugiere la formación de una especie aniónica predominante, Sm(c04)~, tomando en cuenta las condiciones de trabaJo de pH y pS0;. Entonces con l a finalidad de observar la probable participación por adsorción de dicha especie (en ausencia del hidróxido de samario insoluble) sobre la reacción de adsorción y evolución de hidrógeno, se obtuvieron voltamperogramas de platino tratado
previamemente por inmersión en una solución ácida de samario. Los
voltamperogramas fueron obtenidos en una solución libre de samario. Las figuras VI.7 y V1.8 corresponden a los voltamperogramas de platino y de platino tratado por inmersión respectivamente. En Lsentido de los potenciales negativos se observa la corriente asociada a la carga de la doble capa y
después la correspondiente a la adsorción y a la del inicio de la evolución de hidrógeno. En el barrido de retorno se observa la corriente con una intensidad menor. En la figura VI.8 para el platino modificado con samario por inmersión durante 216 horas, se observa un desplazamiento del potencial de corriente nula en el-sentido de los positivos con respecto al valor del potencial del platino no modificado. Además, el valor de la corriente de adsorción y de evolución de hidrógeno se ve incrementada en el platino modificado con samario.
85
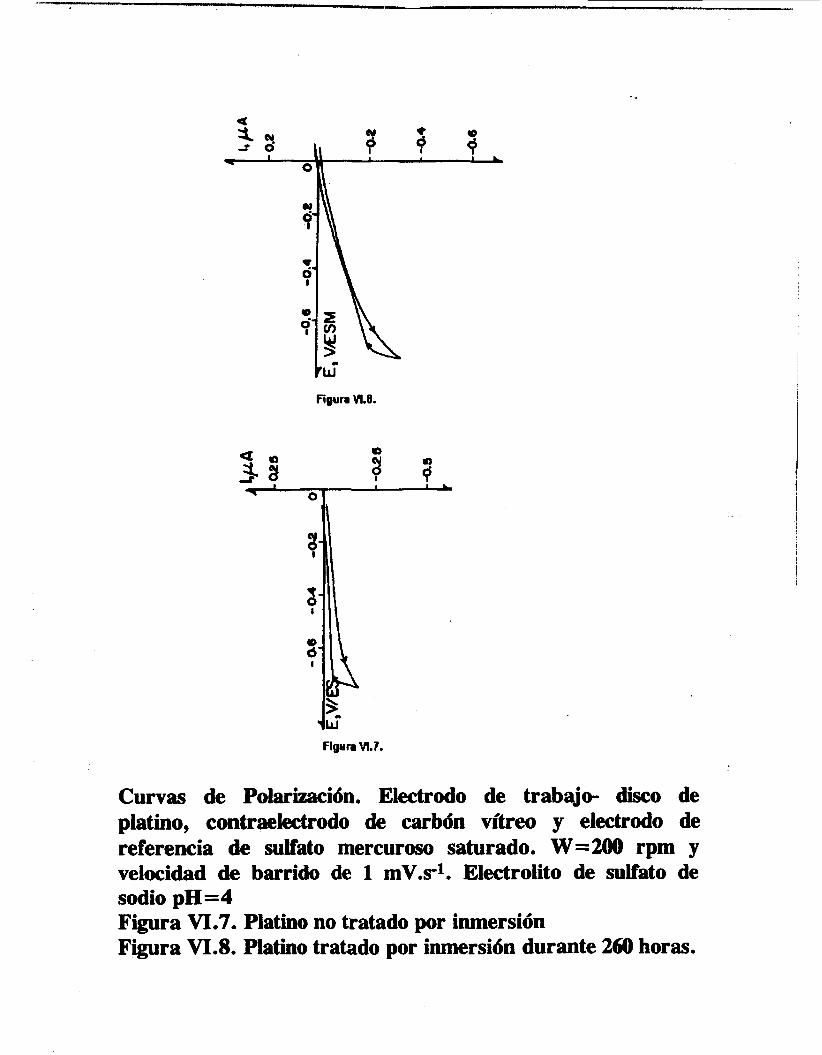
Fiwn W.8.
Figura Vi.?.
Curvas de Polarización. Electrodo de trabajo- disco de platino, contraelectrodo de carbón vítreo y electrodo de referencia de sulfato mercuroso saturado. W=200 rpm y velocidad de barrido de 1 rnV.s-1. Electrolito de sulfato de sodio pH=4 Figura VI.7. Platino no tratado por inmersión Figura VI.8. Platino tratado por inmersión durante 260 horas.

b. Reducción de 02. También se analizó la influencia del canario en la reacción de reducción de oxígeno molecular, disuelto en una
solución saturada. Las figuras VI. 9 a VI. 14 corresponden a voltamperogramas para valores de pH de solución de 2 a 10 respectivamente. La línea continua corresponde a la respuesta sin samario y la línea discontinua a la respuesta con samario. En todo el intervalo de pH estudiado la corriente de reducción de oxígeno es menor cuando el samario esta presente. En presencia de
samario la corriente de reducción de oxígeno es mayor en el intervalo de pH de 2 a 4 con respecto al intervalo de 6 a 8 y a pH=lO aumenta sensiblemente. A pH=4 se distinguen dos valores de
corriente de reducción de oxígeno y en el intervalo de pH de 6 a 10 se observa un solo valor de corriente de reducción de oxígeno. A pH=lO la corriente de reducción aumenta pues en estas condiciones el hidróxido de samario precipita masivamente en el seno de la solución impidiendo el depósito selectivo del hidróxido sobre la superficie del cátodo. Del análisis de los voltamperogramas se desprende que el desplazamiento en el sentido positivo del potencial de corriente nula y el del potencial de evolución de hidrógeno, y las diferencias en la intensidad de la corriente límite siendo mayor en presencia de samario (como hidróxido), ponen en evidencia el efecto electrocatalítico sobre la reacción de adsorción y de evolución de hidrógeno. Donde la maxima respuesta electrocatalítica se encuentra a pH=4 con la probable participación de una especie de samario adsorbida sobre el cátodo. En este sentido Jacksik [671 afirma en su estudio teórico que, por ejemplo, en una matriz de platino con samario presente en la superficie, la energía de activación del proceso mencionado disminuye teniendo consecuencias electrocatalíticas. En la reducción de oxígeno el samario podría pasivar la superficie del cátodo para la reacción de reducción de oxígeno,
por la probable formación de una película de hidróxido de samario en la superficie.
86
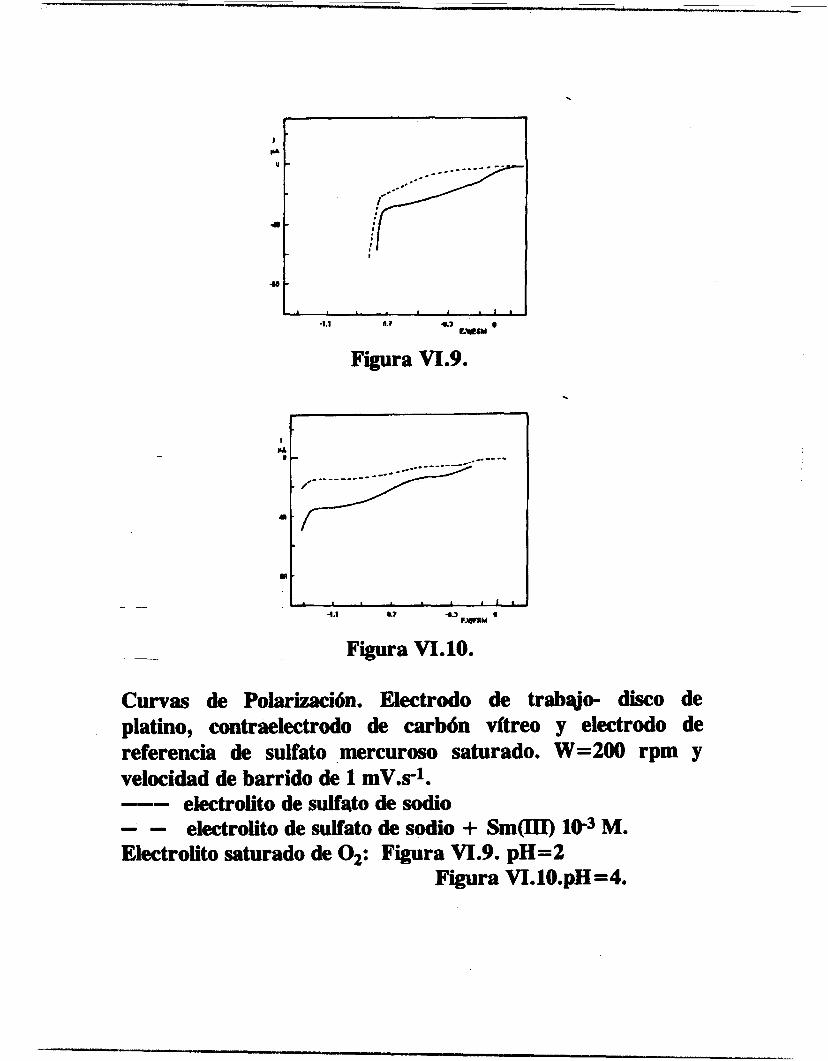
.
I
Figura VI.9.
-1.1 1.7 4.3 F . r p w
-- Figura VI.10.
.
Curvas de Polarización. Electrodo de trabav- dis o de platino, contraelectrodo de carbón vftreo y electrodo de referencia de sulfato mercuroso saturado, W=200 rpm y velocidad de barrido de 1 mV.s-1. --- eiectrolito de sulfato de sodio - - electrolito de sulfato de sodio + S m O 10.3 M. Electroiito saturado de 02: Figura VI.9. pH=2
Figura VI.lO.pH=4.
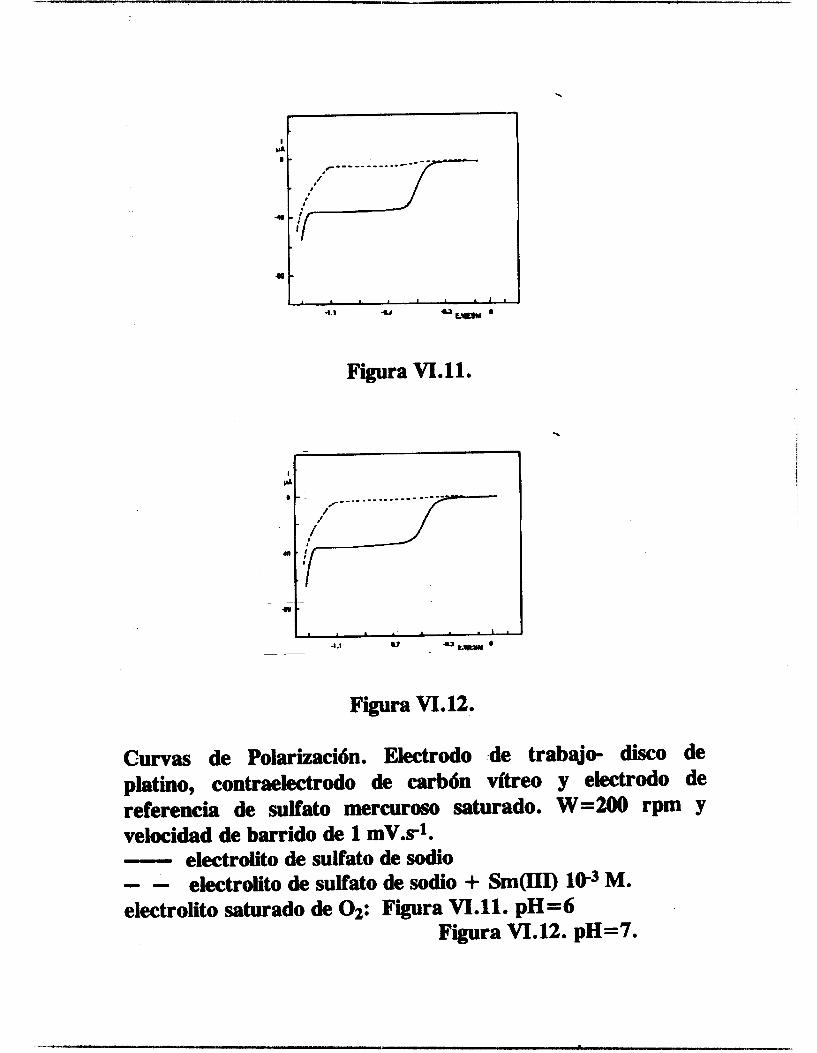
Figura VI.11.
Figura VI.12.
Curvas de Polarización. Electrodo .de trabajo- disco de platino, contraelectrodo de carbón vitreo y electrodo de referencia de sulfato mercurm saturado. W=200 rpm y velocidad de barrido de 1 mV.s-1. - electrolito de sulfato de sodio - - electrolito de sulfato de sodio + S r n o M. electrolito saturado de 02: Figura Ví.11. pH=6
Figura VI.12. pH=7.
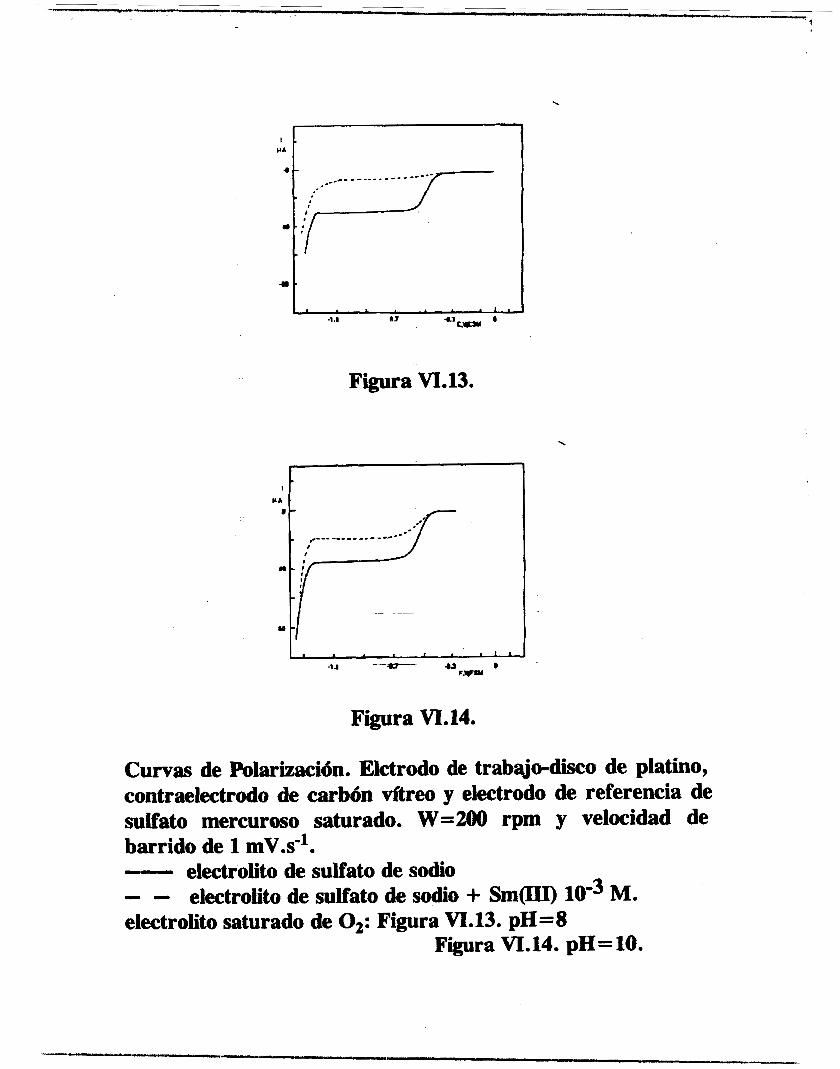
Figura W.13.
8
I . 1 . -1 .I -- u 8
F.*FIy
Figura VI.14.
Curvas de Polarización. Elctrodo de trabajoldisco de platino, contraelectrodo de carbón vítreo y electrodo de referencia de sulfato mercuroso saturado, W=200 rpm y velocidad de barrido de 1 mV.s". I-- electroiito de sulfato de sodio - - electrolito de sulfato de sodio + S m O loo3 M. electrolito saturado de 02: Figura VI.13. pH=8
Figura VI.14. pH= 10,

VI .2 . Voltampcrometría de barrido triangular Con la finalidad de observar detalladamente el efecto del samario sobre la adsorción de hidrógeno, se obtuvieron voltamperogramas de barrido triangular. En la figura VI.15 se muestra un voltamperograma de platino en ácido sulfúrico 0.5 M (como referencia), donde aparecen los picos característicos de adsorción y desorción de hidrógeno, en punteado se muestra el efecto del samario presente en solución sobre la forma del voltamperograma no observándose cambios significativos. La figura VI. 16 corresponde a una serie de voltamperogramas obtenidos en Na2C04 a pH=2.8 con un electrodo de platino, las curvas se obtienen insitu después de la electrólisis a potencial constante, en medio sulfato con samario en solución. La figura VI. 17 presenta un conjunto de voltamperogramas obtenidos en el mismo medio con un electrodo de platino con hidróxido de samario depositado en su superficie. Las curvas se obtuvieron insitu después de la electrólisis a E=constante en un medio de sulfato con samario en solución. La comparación de las figuras VI.16 y VI.17 permite observar un desplazamiento de los voltamperogramas hacia corrientes catódicas (con un valor casi constante), en el intervalo de -810 a 272 mV, la casi desaparición de los picos de desorción de hidrógeno y la aparición de un pico superpuesto en la región de formación de los Óxidos superficiales de platino, todo esto en el caso del platino con depósito de hidróxido de samario. El desplazamiento de la corriente en el sentido anódico de la figura VI.16 se interpreta como la respuesta la oxidación del hidrógeno molecular generado a -810 mV durante la electrólisis sin samario. Para observar la influencia de la especie aniónica de samario se obtuvieron voltamperogramas en ácido sulfUrico O. 5 M de platino tratado por inmersión en solución de samario/ácido sulfúrico 0.5
- _-
87
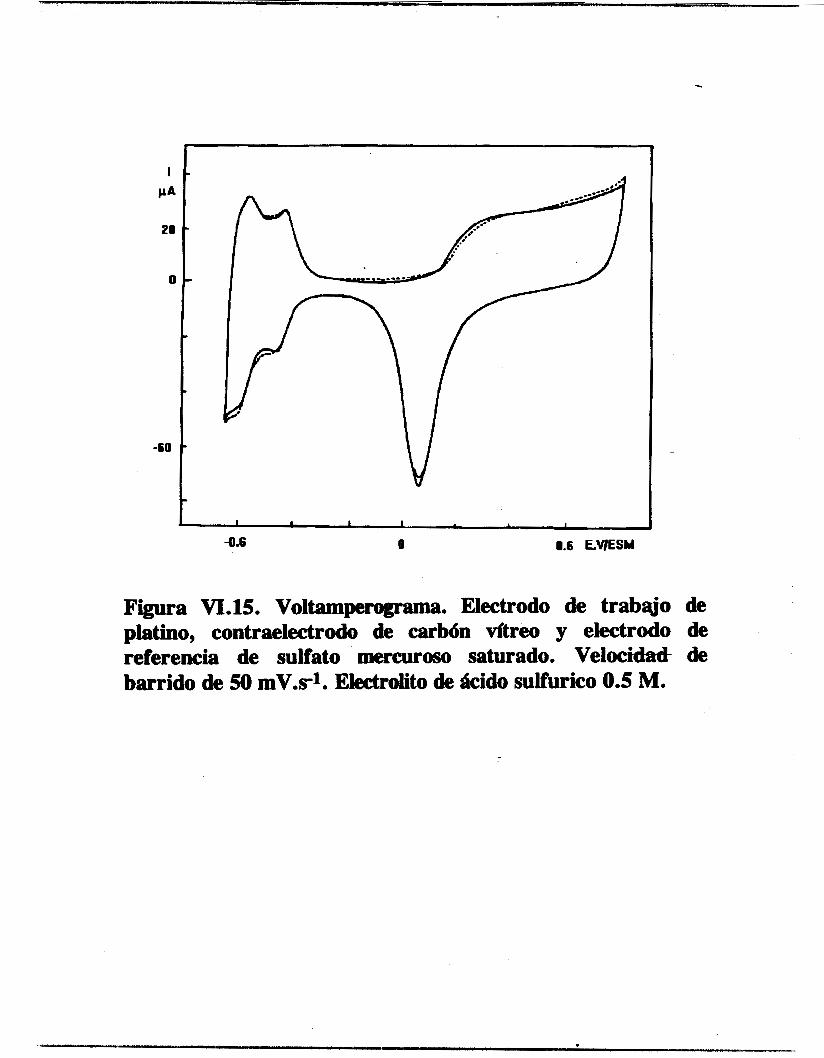
I . I I I
1.6 EWESM 9.6 O
Figura VI.15. Voltampeqrama. Electrodo de t r abw de platino, contraelectrodo de carbón vítreo y electrodo de referencia de sulfato mercuroso saturado, VelocWtf de barrido de 50 mV.s-1. Electrolito de ácido sulfuric0 0.5 M.
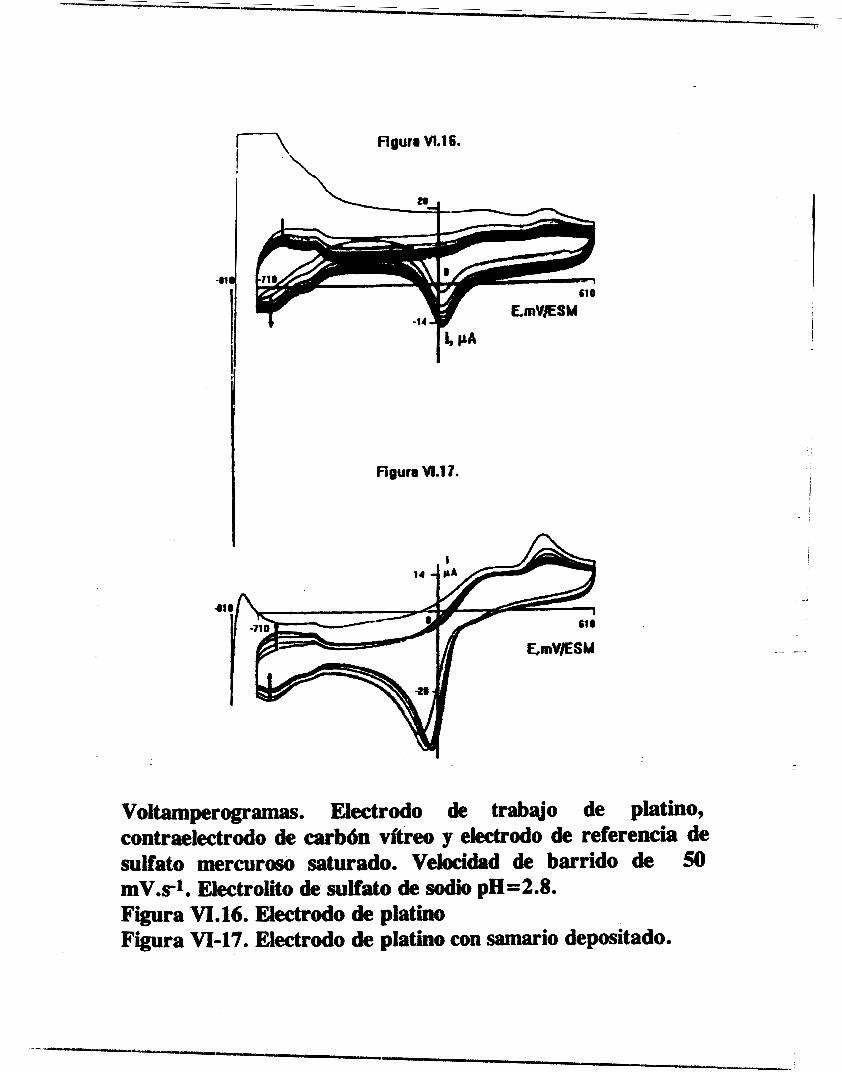
\ Figura VI. I o.
Figura Ul.17.
‘I _ -
Voltamprogramas. Electrodo de trabajo de platino, contrael&rodo de carbón vítreo y electrodo de referencia de sulfato mercurm saturado. Velocidad de barrido de 50 mV.s-f. Electrolito de sulfato de sodio pH=2.8. Figura VI.16. Electrodo de platino Figura VI-17. Electrodo de platino con samario depositado.

M. Este procedimiento evita la formación de hidróxido de samario (Sm(S04)a) en el catodo. En la figura VI. 18 aparecen una serie de voltamperogramas obtenidos después de haber tratado por inmersión (3400 horas) en solución de SMácido sulfúrico 0.5 M, un electrodo de platino. La evolución de los voltamperogramas (de "a" a "d") muestra la reactivación de la superficie de platino. Se observa que la adsorción de hidrógeno se presenta a potenciales mas positivos a partir del segundo barrido y que la corriente de evolución de hidrógeno (a -640 mV) es superior, todo esto con respecto al platino no tratado (figura VI.15).
También en el primer barrido de la figura VI. 18 se observa que los picos asociados a la formación de los óxidos superficiales de platino se encuentran inhibidos casi por completo. en los barridos subsecuentes dichos picos van reapareciendo junto con el de reducción de los Óxidos. En los - primeros barridos, en lugar de los picos de los óxidos superficiales aparece un solo pico muy intenso el cual va desapareciendo con el tiempo. En la figura VI. 19 aparece una serie de voltamperogramas función del tiempo de inmersión del platino en solución Cm(III)/H2S04 0.5 M. Se observa que la intensidad del pico oxidación presente en la región de los óxidos superficiales platino, depende del tiempo de inmersión. También la corriente
en de de de de
adsorción y desorción de hidrógeno disminuye con el aumento del tiempo de inmersión. En el Vomperograma de 3400 horas de inmersión desaparecen por completo los picos de formación de óxidos superificiales de platino y en todos los voltamperogramas no hay corriente de reducción de los Óxidos superficiales de platino. Tamblén se observa la inhibición de la reacción de oxidación del hidrógeno molecular. Los resultados descritos arriba indicarían que el hidróxido de samario depositado sobre el platino electrocataliza la adsorción irreversible de hidrógeno tomando en cuenta que en presencia del hidróxido la adsorción de hidrógeno se presenta a potenciales mas positivos y que la corriente de desorción es muy pequeña. Además,
88
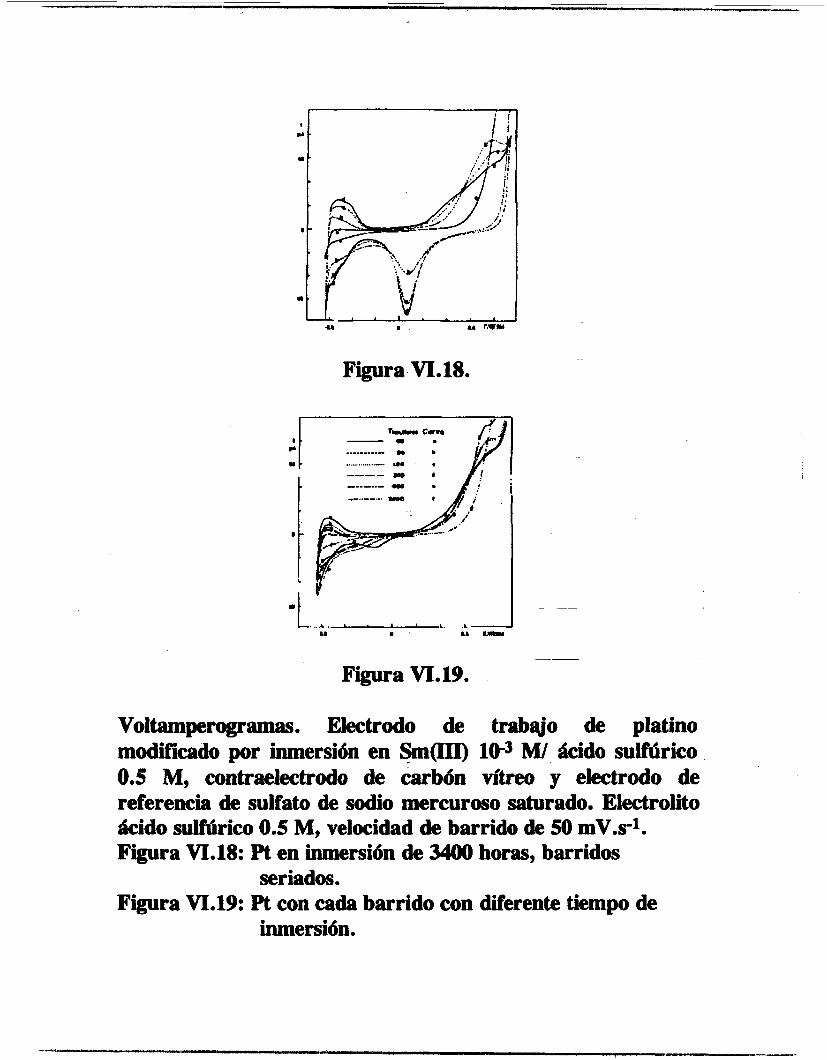
Figura VI.18.
"t I - - -
Figura VI.19.
Voltamperogramas. Electrodo de traba0 de platino modificado por inmersión en Sm(III) 10-3 M/ ácido sulfúrico 0.5 M, contraelectrodo de &bón vítreo y electrodo de referencia de suifato de sodio mercuroso saturado. Electrolito ácido sulfiirico 0.5 M, velocidad de barrido de 50 mV.s-1. Figura VI.18: Pt en inmersión de 3400 horas, barridos
Figura VI.19: Pt con cada barrido con diferente tiempo de seriados.
inmersión.

que el pico que aparece en la región de los Óxidos superficiales de platino estaría asociado a la oxidación de alguna especie
relacionada con la especie aniónica Sm(C04); presente de manera
predominante en las condiciones de pH y pC0; utilizadas. La especie de samario inicial en la solución podría inhibir la formación de los óxidos superficiales de platino ya que aparece
el pico de oxidación de la especie inicialmente soluble y al
mismo tiempo desaparecen los picos de oxidaciódreducción
reversible de platino.
VI.3. Análisis armónico de la impedancia electroquímica La medición de la impedancia electroquímica permite obtener de manera directa la caída ohmica, la resistencia de transferencia
de carga y la capacidad de la doble capa [72,73,741. Las figuras VI.20 y VI.21 son diagramas de impedancia tipo -
Nyquist del platino en medio sulfatos y de platino medio
sulfatos/samario respectivamente, polarizando a -700 mV/EcM con
un potencial alterno superpuesto de f10 mV aplicado en un intervalo de frecuencias de 10000 a 0.002 Hz. De ambos diagramas se obtuvo un valor de caída ohmica de 200 Q, una resistencia de transferencia de carga de mas de 40000 Q y una capacidad de la
doble capa del orden de 1.4 x F. Se observa que la adición -
de samario no modifica la caida ohmica ni la capacidad de la doble capa.La resistencia de transferencia de carga para la reacción de formación de hidrógeno es alta pues se encuentra a un valor de potencial donde no ocurre la reacción de formación de hidrógeno. Las figuras VI. 22 y VI. 23 corresponden a diagramas de impedancia en las mismas condiciones que la anteriores cambiando sólo el
valor del potencial de polarización el cual tiene un valor de
-1100 mV/ESM en estos dos casos. La caída ohmica en ambos casos es del orden de la centena de ohms y la capacidad de la doble capa es de 1.1 x F para el caso con samario y de O. 5 x lo-’
89
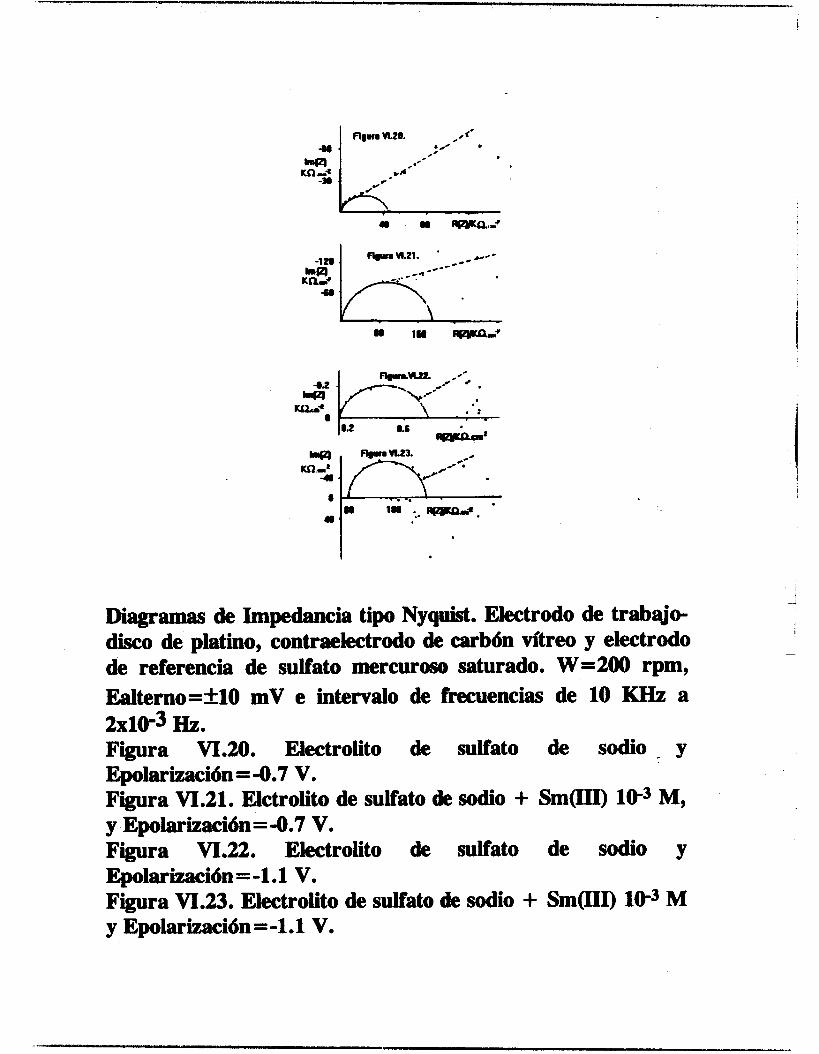
(r91 IcÍ1.d
-48
o
48 W 1" -.. Rclriuu.
i Diagrams de Impedancia tipo Nyquist. Electrodo de trabajo-
de referencia de sulfato mercurom saturado. W=200 rpm, Ealterno=+lO mV e intervalo de frecuencias de 10 KHz a
Figura VI.20. Electrolito de sulfato de sodio y Eplarización=-O.7 V. Figura VI.21. Elctrolito de sulfato de sodio + S m o 10-3 M, y Epolarización=-O.7 V. Figura VI.22. Electrolito de sulfato de sodio y Eplarización =-1 . 1 V. Figura VI.23. Electrolito de sulfato de sodio + Sm(III) 10-3 M y EpIarización=-l.l V.
I
i disco de platino, contraelectrodo de carbón vítreo y electrodo
2 x 1 ~ 3 HZ.
-

F para el caso sin samario. La resistencia de transferencia de carga para la reacción de formación de hidrógeno es de 550 R sin samario y de 150 R con samario. En este caso se observa que la resistencia de transferencia de carga para la reacción de formación de hidrógeno disminuyó en presencia de samario con un factor 3.6. Y la carga de la doble capa es del doble con samario. Los datos de resistencia de transferencia de carga indicarían que la presencia de samario cataliza la reacción de formación de hidrógeno disminuyendo la resistencia de transferencia de carga por disminución de la energía de activación del proceso. También la capacidad de la doble capa se ve modificada por la presencia de la especie de samario adsorbida específicamente. Directamente en el diagrama VI.23 se observa un efecto inductivo que podría ser imputable a fenómenos de adsorción específica. El conjunto de resultados del estudio electroquímico confirma el efecto electrocatalítico del samario en platino sobre la reacción de formación de hidrógeno, donde la etapa de adsorción se presenta de manera irreversible. En esta reacción parece intervenir una especie de samario adsorbida específicamente (Sn(S04)i), la cual reaccionaria, en la superficie del platino, con el hidrógeno adsorbido produciéndose la reducción del samario adsorbido. Una probable reacción del canario con el hidrógeno sería:
t Sm(0) + Pt - Sm-Pt 90

t-- 3 H* + 3 e- + Crn-Pt --+ 3/2 H + Sm-Pt
2
Las primeras dos reacciones muestran la irreversibilidad de la adsorción de hidrógeno y las reacciones dos a cuatro la formación
del enlace intermetálico Pt-Sm que electrocataliza la evolución de hidrógeno. Por otro lado el Sm-Pt se oxidaría en la región de potenciales donde ocurre la oxidación superficial reversible del platino, y
siendo un Óxido hidratado y/o un hidróxido impediría la formación de los Óxidos superficiales de platino. Además la voltamperoietria mostró ser una técnica muy sensible para la detección de hidróxido de samario depositado sobre los electrodos utilizados (platino). En efecto fue posible encontrar que con el aumento de la densidad de corriente se incrementa la cantidad de gas hidrógeno generado en el cátodo y que este gas
provoca la perdida parcial del depósito de hidróxido de samario por desprendimiento. Existe por lo tanto un compromiso entre la densidad de corriente y el pH de la solución para que se logre el depósito de samario minimizando la posibilidad de pérdida del mismo por desprendimiento. El pH de la solución debe ser lo suficientemente ácido como para mantener al samario en solución y para lograr el depósito aplicando una densidad de corriente lo más baja posible. El pH de la solución se situaría entonces alrededor de 5 y la densidad de corriente alrededor de 3 mA cm-2.
i
91

VII. ESlllDIO DE P- EXPERIMENTALES
En la optimización de un proceso donde se selecciona un parámetro respuesta para definirlo en función de otros parámetros experimentales, se requiere de un enfoque estadístico para que las conclusiones sean objetivas sobre la confiabilidad y la validez de los resultados. Debe tomarse en cuenta que los métodos estadísticos [84,851 sólo nos peniten asociar a un enunciado un cierto error y un nivel de confianza, donde las inferencias estadísticas deben ser interpretadas físicamente.
Diseño factorial de experimentos, definiciones La mayoría de los experimentos involucran el estudio de los efectos de uno o más parámetros. El diseño factorial se presenta como un método estadístico eficiente para experimentos multiparamétricos [84,851. Se entiende por diseño factorial el estudio de todas las combinaciones posibles de los niveles de los parámetros en cada prueba completa o en la replicación del experimento. Por ejemplo, si tenemos "a" niveles del parámetro A y "b" niveles del parámetro B, entonces cada replica contiene todas las combinaciones "ab". Cuando los parámetros estan dispuestos en diseño factorial se dice que estan cruzados. El efecto de un parámetro se define como el cambio en la respuesta producido por un cambio en el nivel del parámetro, a esto se le conoce como efecto principal porque se refiere a los parámetros primarios de interés en el experimento. En algunos experimentos podemos encontrar que la diferencia en la respuesta entre los niveles de un parámetro no es la misma en todos los niveles del otro parámetro. Cuando esto ocurre, se dice que hay interacción entre los parámetros. El diseño factorial tiene varias ventajas. Es más eficiente que los experimentos donde se maneja un parámetro a la vez. Además,
~-
-~
92

8
el diseño factorial es necesario cuando se presentan
interacciones entre los parámetros puesto que es posible estimar
los efectos de un parámetro a diferentes niveles de otro
parámetro, proporcionando conclusiones válidas sobre un cierto intervalo de los paránetros experimentales.
En el depósito de películas delgadas en medios acuosos por
métodos electroquímicos [86,87,88,891, la optimización consiste
en obtener un máximo en el parámetro respuesta R(rendimient0 de depósito), por ejemplo en función del pH de la solución, de la
concentración de electrolito soporte (pS04 en nuestro caso),
densidad de corriente (i), velocidad de rotación del cátodo (01 y
tiempo de depósito ( t ) . Los valores de rendimiento utilizados corresponden al proaedio de los obtenidos para Sm, Eu, Am y Pu. Como antecedente para el-desarrollo experimental se determinaron
los intervalos de estudio de cada parámetro como sigue:
9
1.- pH solución.- Mediante los estudios fisicoquímico y
electroquímico se determinó que en el intervalo de pH de la
solución de 2 a 7 se obtiene el depósito de hidróxido de canario. ~ __
2.-- pS0' solución.- De acuerdo con los criterios de distribución uniforme de la axriente (anexo 21 la concentración de
electrolito debe ser superior a 0.1 M para tener distribuida
uniformemente la corriente en el cátodo.
Por otro lado la celda-cátodo rotatorio diseñados en el presente
trabajo presentan aún limitaciones prácticas. Para velocidades de
rotación del orden de 1200 rpm con concentraciones de electrolito superiores a O. 5 M la vibración del cátodo (axial y radial) y el
aumento de la viscosidad del mismo electrolito es tal que por
cavitación se produce espuma.
4
93

3.- Densidad de corriente, i .- En el estudio electroquímico se mostró que densidades de corriente en el intervalo de 0 . 5 a 26
mA cm-’ se obtiene el depósito de hidróxido de samario sin pérdidas apreciables por arrastre de materia durante la evolución
de hidrógeno.
4.- Velocidad de rotación del cátodo, w .- Como resultado del estudio hidrodinámico realizado (anexo 31, se tiene que las variaciones en las vibraciones y en la velocidad de rotación del cátodo son admisibles dentro del intervalo de f 5%. El sistema diseñado cumple con dicha condición en el intervalo de velocidades de 100-1200 rpa.
5.- Tiempo de depósito, t .- Los tiempos de depósito en medio acuoso reportados en la literatura [l-321, tienen valores típicos
del orden de 60-120 minutos con una tendencia hacia los 100
minutos. El estudio de la relación R ( % I - t de depósito se
realizó en el intervalo de tiempo de 5 a 120 minutos.
En el diseño experimental para el estudio de la respuesta en función de los parámetros definidos anteriormente, se tomaron de manera “casi-formal“ criterios de disefío factorial (método estadístico para la minimización del mímero de experimentos en la optimización de un proceso). Los espacios geométricos de trabajo, definidos por los intervalos de los parámetros, son ligeramente asimétricos ( f SA), de ahí que la condición de simetría se cumple de manera casi-formal. En los análisis se palió dicha asimetría con la abundancia en el número de puntos experimentales. De acuerdo con Montgomery 1851 el diseño factorial es adecuado en estudios multiparamétricos porque éste contempla diferentes niveles de intensidad y las probables interacciones entre diferentes parámetros. Primero se observó la respuesta (R) en función de cada parámetro
94

por separado para determinar la dependencia de R con respecto a cada uno de ellos. Después para observar las interacciones entre los parámetros se hizo un diseño de 2 parámetros con 3 niveles de
intensidad probando todas las posibles combinaciones que en total
dan 9.
Con respecto a la confiabilidad de los resultados cabe aclarar
que las condiciones de replicación de cada punto experimental,
selección aleatoria de la secuencia de estudio de los parhetros
y la reproducibilidad en las condiciones de trabajo, fueron consideradas como requisito para un análisis estadistico de
resultados [851.
Para la obtención de la función analítica de cada caso se utilizó
el programa de computadora llamado SIGMAPLOT (Sandel Scientific,
versión 4.04) y para la obtención de las superficies de respuesta
el programa de computadora llamado EXCEL (Microsoft, v.4.0). El programa SIGMAPLOT [901 es vertical y adecuado para este tipo de casos puesto que la elaboración de la matríz necesaria para el ajuste de la función admite las asimetrías como las que se presentan en este caso al proporcionar a cambio más elementos
para la matriz. Además, el ajuste por mínimos cuadrados utiliza
el método iterativo 1 lamado "El compromiso de
Marquardt-Levenberg". Dicho método introduce una variante al
método de Gauss [851 , ya que desde la primera iteración se
restringen los valores de los coeficientes deg-plinomio
circunscribiéndolos a un elipsoide que tiene su centro geométrico en el punto inmediato anterior y el cual cambia de dimensiones en las subsecuentes iteraciones. De esta manera se evitan las
sobrestimaciones y, por lo mismo, se evita que se presente la
divergencia durante el ajuste.
También introduce criterios estadísticos para revisar la bondad
del ajuste y el error asociado a cada coeficiente del polinomio.
El programa inicia el ajuste de la función propuesta con los
criterios establecidos por el usuario:
95

a. Tolerancia.- Equivale al valor absoluto de la diferencia entre la norma (la norma es la raiz cuadrada de la suma de los cuadrados de los residuos (R experimental - R calculada)) de una iteración y la siguiente. Tolerancia especificada lom6.
b. Stepsize o "magnitud del paso".- Equivale al inverso del parametro de Marquardt. Dicho parámetro está relacionado con la minimización de la suma de los cuadrados y por lo mismo esta relacionado indirectamente con los coeficientes del polinomio. Magnitud de paso especificada
c. 1teraciones.- Se especifica el número límite de iteraciones permitidas por el usuario, sin embargo, el proceso puede detenerse en 2 casos: -cuando la norma alcanza el valor de la tolerancia, -cuando los parámetros ya no tienen cambios significativos,
-
siendo del orden de magnitud del "stepsize".
Cuando se alcanza alguno de estos valores se dice que hubo convergencia obteniéndose el valor de la norma, el valor de cada coeficiente del polinorio y la dependencia de la respuesta con cada uno de éstos. También se especifica el coeficiente de
variación o error relativo asociado a cada coeficiente. La dependencia se define como la relación entre la varianza del parámetro en estudio cuando éste varia con las demás constantes y
la varianza del mismo parámetro cuando éste se mantiene constante con los demás variando, queda definido como:
_ _
--
varianza P lcte. 'P2-PS variables Dep = 1 -
I
var ianza 'ívar i ab i e 1 P2-P5cte.
96

---
Entonces cuando Dep 4 1 la respuesta depende del parámetro en es t Ud i o. La norma expresa numéricamente la bondad del ajuste puesto que
cuando ésta tiende a cero es porque los residuos tienden a cero y los valores calculados son idénticos a los experimentales. Finalmente el coeficiente de variación o error relativo siendo la relación porcentual de la desviación estandar con el valor del
coeficiente, éste tenderá a un mínimo cuando el error sobre el coeficiente sea mínimo.
V I I. 1. R d tados
Las condiciones iniciales de los parámetros de depósito de
Sm,Eu,h y Pu son:
pH = 3.5
i = 5.6 mA/cm2
o = 41.9 s-l
= 900 s tdspó. i t o
El estudio se inició con la intensidad de la respuesta, en
función de la concentración de electrolito soporte.
-R = f ( [SO;] 1.- La concentración de electrolito soporte se expresa en unidades de Mol M-3 como obtenida es la siguiente:
-log[cO~l--psO~. La función
R ( X ) = -1.625 PSO; + 18.87
ERROR 60.8 12.5
DEPENDENCIA o. 8 o. 8
Los errores en el orden son 60.8% y 12.5% los cuales son
97

--
relativamente grandes y esto se debe a que la intensidad de la
respuesta, R, es del orden del 20% (115 de la intensidad máxima esperada). Sin embargo, la norma es de 0.72 lo que se traduce en un buen ajuste con los 4 puntos dentro del intervalo del 95% de confianza de la función obtenida. Estadísticamente las diferencias en la respuesta de cada punto no son significativas puesto que (XI = 22.72 2 0.63(1S) y todos los puntos están dentro de una desviación estandar. Sin embargo gráficamente (Figura VII.1) se observa que R disminuye ligeramente cuando la concentración de SO; disminuye. Esta tendencia poco pronunciada podría explicarse en términos de la formación de especies aniónicas las cuales se adsorben específicamente en el cátodo metálico. Dichas especies predominan
en función del aumento de la concentración de SO' desplazando la constante de equilibrio entre los aniones y las especies hidroxiladas hacia los valores de pH mayores. En este caso la formación de especies hidroxiladas (solubles e insolubles) tendrá lugar más cerca de la superficie del cátodo donde el pH es más elevado. La adsorción especifica y la formación de especies hidroxiladas cerca de la superficie del cátodo hace que las partículas del hidróxido insoluble formado se aglutinen en la superficie y en las cercanías del cátodo formando un gel con un pequeño espesor adherido a la superficie, proporcionándole estabilidad. Esta pequeña diferencia en rendimientos se podría explicar considerando que el proceso de adsorción específica es lento y sólo una pequeña cantidad de aniones se adsorben en los intervalos de tiempo estudiados. Matijevic 1911, señala que en la fase de coalescencia de las
4
partículas del sol para formar el gel la adición de sales inorgánicas aumenta la velocidad de dicho proceso. Los iones de la sal sirven como material "aglomerante" de las partículas del sol, debido a atracciones electrostáticas. Suponiendo que lo anteriormente dicho es cierto, la concentración Óptima de SO; es 0 . 5 M, sin embargo, como las diferencias son
98
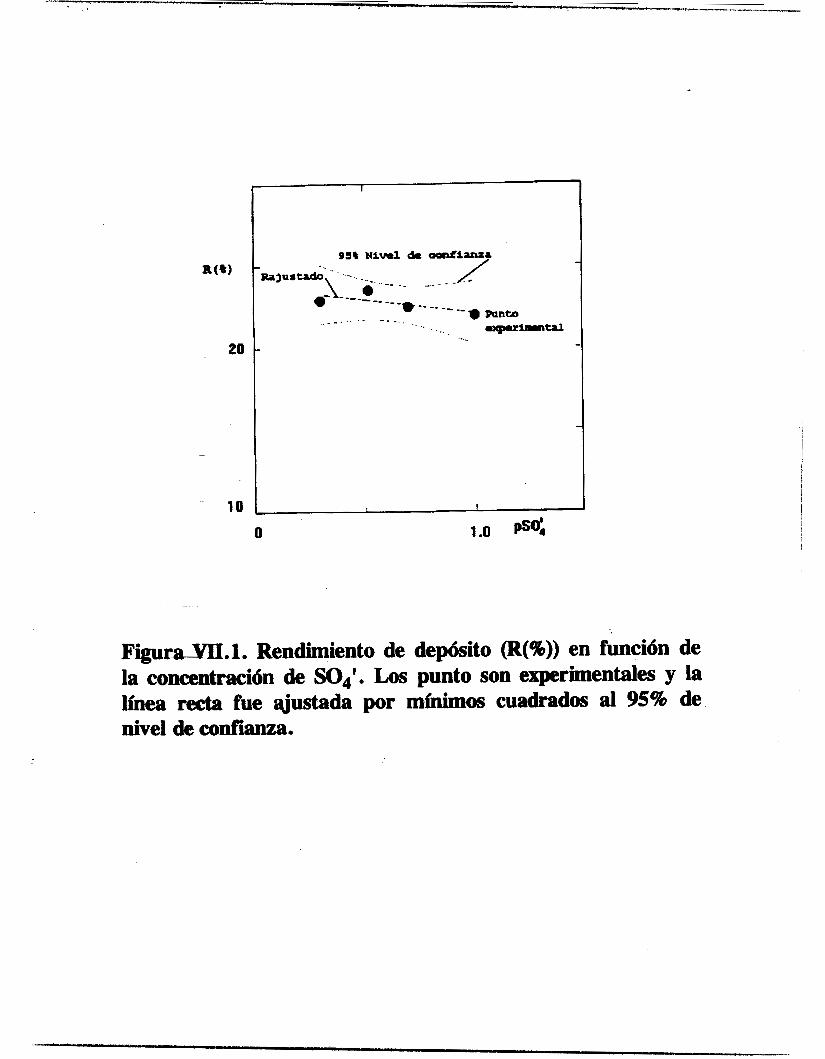
R (6)
20
-
10
I
-
O 1.0 psd,
FigwaYII.1. Rendimiento de depósito (R(%)) en función de la concentración de SO4'. Los punto son experimentales y la lhea recta fue ajustada por mínimos cuadrados al 95% de nivel de confianza.

pequeñas y como las concentraciones altas contribuyen a la formación de espuma, un valor confiable de la Concentración es 0.3 H.
-R = f ( [E'] 1.- La concentración de iones hidrógeno tiene unidades de bl/M y se expresa como -log [H'l = pH. La función
resultante se ajustó a los resultados experimentales con un valor de norma de 0.17, la función, las dependencias y los errores son:
3
R ( % I = - O. 2495 p$ + O. 3153 pH + 22.8
ERROR 26.4% 98% 1.3%
OEPENDENC I A O. 98 O. 98 O. 98
De acuerdo con los resultados el ajuste es bueno colo se observa gráficamente (figura VII .2 ) con los puntos experimentales distribuidos a lo largo de la curva dentro del intervalo de confianza del 95%. Una vez más la magnitud de los errores se debe a la intensidad de la respuesta R. En el intervalo analizado pH = 3.5 - 7 en el estudio la función es decreciente con el pH (disminución de la concentración de [H'l 1, con un máximo en 3.5 y un minirno en 7. El punto correspondiente a pH = 2 no fue incluido en el ajuste puesto que el rendimiento es-- ( c* 0.3) y los errores asociados a la función son RUY grandes resultando mal ajuste. Debe tomarse en cuenta que en el diseflo de experimentos la intensidad de la respuesta en todos los casos debe ser del mismo orden de magnitud. En la figura VI I .2 se ilustra el ajuste considerando al punto pH = 2, se observa que el ajuste no es adecuado y que los
intervalos de confianza son muy grandes.
Utilizando los valores iniciales de los parámetros de depósito se calculó la máxima concentración de iones hidrógeno en solución (Gáx o pE in imo ) a partir de la cual es posible tener un rendimiento con un valor diferente de cero. Con la ecuación de
-
99

GB~X de Hansen el valor obtenido es 2.63 x Mol, equivalente a pH = 2.58. Es decir que para las condiciones experimentales impuestas el depósito se lleva a cabo con valores de concentración de iones hidrógeno inferiores a 2.63 x M. La Figura V I I . 3 muestra el arreglo del conjunto de puntos experimentales y del punto calculado. Se observa que el cambio de
pendiente entre la región de rendimiento alrededor de O y la de máximo rendimiento alrededor de 22.5% es muy grande.
En el intervalo de 3.5C pH< 5 el espesor de la capa de iones hidroxilo generados en la interfase OH = 2.9 x - 3.299 x
cm) es inferior al espesor de l a capa límite de difusión, 6
(3.3 x cm). Los espesores de lo^ son calculados a partir de los desarrollos de Hansen [151 y el de la capa límite de difusión a partir de la teoría de electrodo-disco rotatorio [511. Para el intervalo de pH de 5.1 a 7 se tiene OH = a. Si la probabilidad de interacción entre las partículas del sol formado para coalescer y formar el gel aumenta con la disminución del volumen asociado al espesor OH y al diámetro del cátodo, entonces con la finalidad de aumentar dicha probabilidad se disminuye el espesor lon (con el diámetro del disco constante). Esto podría explicar que el máximo de rendimiento se tenga a pH =
3.5 puesto que el mínimo valor de fa está asociado a dicho pH. Con el aumento del pH y de lo^ disminuye el rendimiento en más del 10% con respecto al obtenidoaptL= 3.5.
Debido a que el rendimiento experimental a pH = 3.5 y 5 es similar, y a que la densidad de corriente necesaria para crear una capa de OH- es menor para el caso de un pH menos ácido, se tomó como valor del pH de la solución Óptima el valor de 5. Se busca que con el empleo de corrientes menores, disminuya la cantidad de hidrógeno generado durante el depósito que deba ser evacuado de la interfase.
100
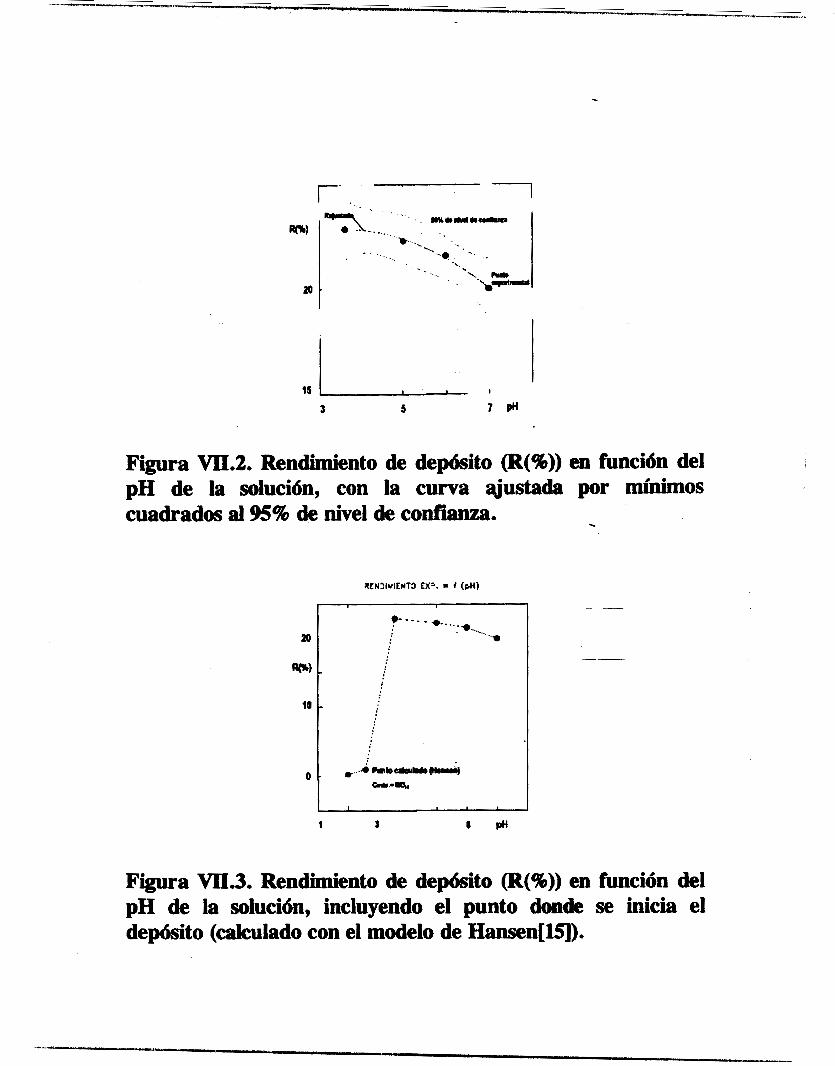
Figura VII.2. Rendimiento de depósito (R(%)) en función del pH de la solución, con la curva ajustada por mínimos cuadrados al %% de nivel de confianza.
5
Figura ViI.3. Rendimiento de depósito (R(%)) en función del pH de la solución, incluyendo el punto donde se inicia el depósito (cakulado con el modelo de Hansen[ls]).

-R = f(i) .- La función fue ajustada a la densidad de corriente en términos de un polinomio de segundo grado, se obtuvo un buen ajuste con un valor de norma de 0.92:
R ( X I = 2.822 x 10 i2 - 1.93 x i + 23.21
ERROR 89% 32% 1%
DEPENDENCIA 0.95 O. 96 O. 6
Todos los puntos se encuentran dentro del intervalo de confianza del 95%. Localmente en el intervalo de i de 1 . 8 a 14 mA.cm-2, se
encuentra un máximo ubicado en 3.4 mA.~ia-~, sin embargo no es claro puesto que no se diferencian al superponerse las barras de error asociadas a cada uno de estos puntos. En el intervalo restante, la función es decreciente con la densidad de corriente (Figura VII. 4) .
Al inicio de la curva se llega a una densidad de corriente tal que se alcanza un "máximo" de rendimiento cuando el pH en la interfase es superior al pH de precipitación de los hidróxidos y además se logra un cierto espesor de OH- superior al espesor del gel formado en la superficie del cátodo. Posteriormente el rendimiento disminuye probablemente debido a 2 efectos aditivos en cuanto a la baja del rendimiento, con el aumento de la --
densidad de corriente aumenta la cantidad de gas hidrógeno generado durante el depósito el cual al desprenderse como ___
burbujas podría arrastrar agregados del gel de hidróxido formado y, por otro lado, también aumenta el espesor de la capa lo^" disminuyendo la probabilidad de coalescencia de las partículas del sol formado. La densidad de corriente necesaria para crear las condiciones de depósito debe ser superior a un valor mínimo el cual puede ser calculado con el tratamiento de Hansen [ is] , que da un valor de Iiín > 0.2 W c m . Sin embargo, como se mostró en la sección del estudio fisicoquímico del proceso (diagramas tipo PE, PZ y PT)
-
2
101
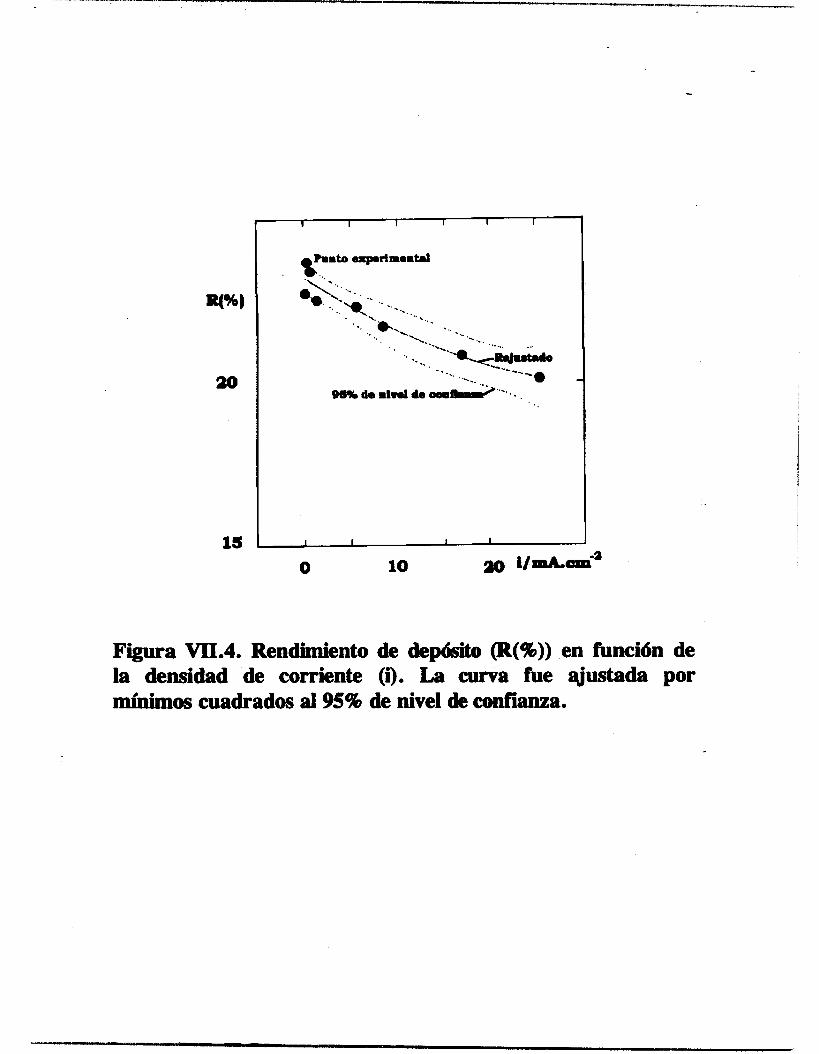
R(%)
20
15
h i t o oxporineitd . %..
O 10 #) i/mA.=-=
Figura VII.4. Rendimiento de dephito (R(%)) en función de la densidad de corriente (i). La curva fue ajustada por múiimoS cuadrados al 95% de nivel de d i a m a .

que utilizando el valor del límite inferior del pH de inicio de aparición de la fase sólida correspondiente al hidróxido, es posible calcular el valor de Imín correspondiente a cada caso concreto (tomando en cuenta el elemento y las especies químicas presentes en la solución). Para el presente caso, el valor de Imín calculado es de 0.68 mA cm-2, experimentalmente se observó que rendimientos significativos del orden del 1% o más se presentan a densidades de corriente del orden de 0 . 8 mA cm-2. Con este valor de corriente el pH calculado para la superficie del cátodo es de 11.9 y con un espesor correspondiente lo^ igual 3.29
x 10-3cm (99% de la capa límite de difusión). Si en un principio están dadas las condiciones para el depósito cuantitativo pero los rendimientos son del orden del 24% (máximo). Las pequeñas variaciones de rendimiento observadas en el intervalo de corriente estudiado no podrían explicarse por la variación del espesor lo^ como lo plantea Hansen [151, pues en todo el intervalo de corriente el espesor loti es idéntico al valor del espesor de la capa límite de difusión. Entonces podríamos pensar que el proceso de gelificación es lento y que tomando un valor de i optimizado de 1.0 mA cm-2 quedaría por observar parámetros de velocidad de rotación del cátodo y tiempo de depósito.
-R = f ( w 1 .- La función se presenta creciente con respecto a la velocidad de rotación del cátodo (01, sin presentarse un máximo en el intervalo de w estudiado (figura VII.5). En el límite superior de w encontramos la limitación del dispositivo diseñado en cuanto a que la producción de espuma por vibraciones axiales y
radiales del cátodo, inicia con valores superiores a 126 s-'. Un diseño mejorado permitiría, en principio, evitar las vibraciones y llegar a un máximo de R superior al que se podría obtener con el dispositivo utilizado.
102
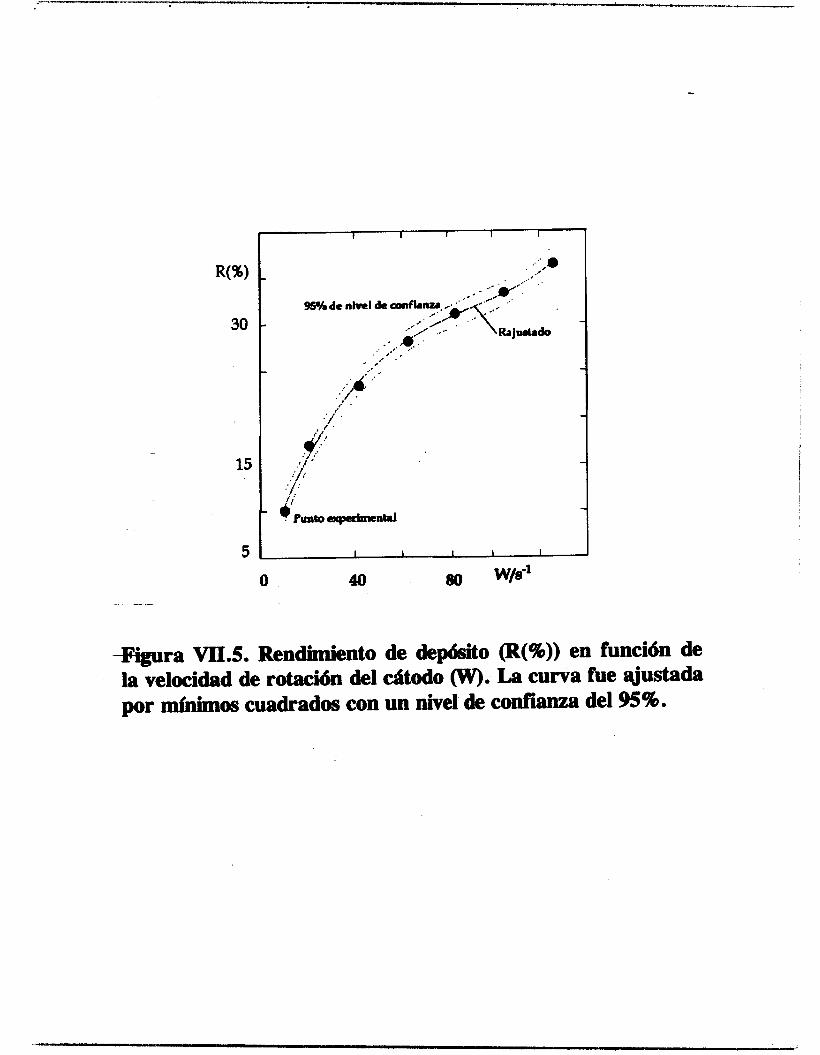
30 -
-
15
-
I I I I I
45gura VIIS. Rendimiento de depósito e(%)) en función de la velocidad de rotación del cátodo 0. La curva fue ajustada por minimoCr cuadrados con un nivel de confi8I1z8 del 95%.

.̂.-
La expresión de la función es (Figura V I I . 5 ) :
3 R ( % I = 2.699 x lo-' w - 7.17 x us + 0.74 w + 3.6
ERROR 27% 21% 12% 36x
DEPENDENCIA o. 99 o. 99 0.99 0.95
La bondad del ajuste se encuentra expresada en el valor de norma de 1.3 con la serie de puntos distribuidos aleatoriamente a lo largo de la curva y dentro del intervalo de confianza del 95%.
En la ecuación del espesor de la capa límite de difusión [151
encontramos que es inversamente proporcional a la raiz cuadrada de la velocidad de rotación del cátodo, por lo que en la Figura VII .6 se encuentran graficados el rendimiento y el espesor de la
capa de difusión (6) en función de la velocidad de rotación del cátodo. Se observa que el rendimiento corresponde a una función exponencial y el espesor al inverso de una exponencial de grado 1/2. Al espesor más pequefío corresponde el rendimiento más alto y
al espesor más grande el rendimiento más bajo con la relación de rendimientos Ri r ayo r /b o r de 3.65 y la relación de espesores Oi.ayor/Orenor de 3.46. Esto querria decir que la función de R con respecto a 6 es una inversa de segundo grado (Figura VII.7).
- --
ERROR 25% 10.6% 38%
DEPENDENCIA o. 99 o. 99 O. 98
La norma es de 1.25 con los puntos experimentales distribuidos a lo largo de la función dentro del intervalo de confianza del 95%.
El dispositivo experimental de Hansen [151 no permite emplear el modelo convectivo-difusiona1 para un estado estacionario como el que se tiene con un cátodo disco rotatorio, y por lo mismo, no le es posible estudiar con rigor el efecto de la velocidad de
103
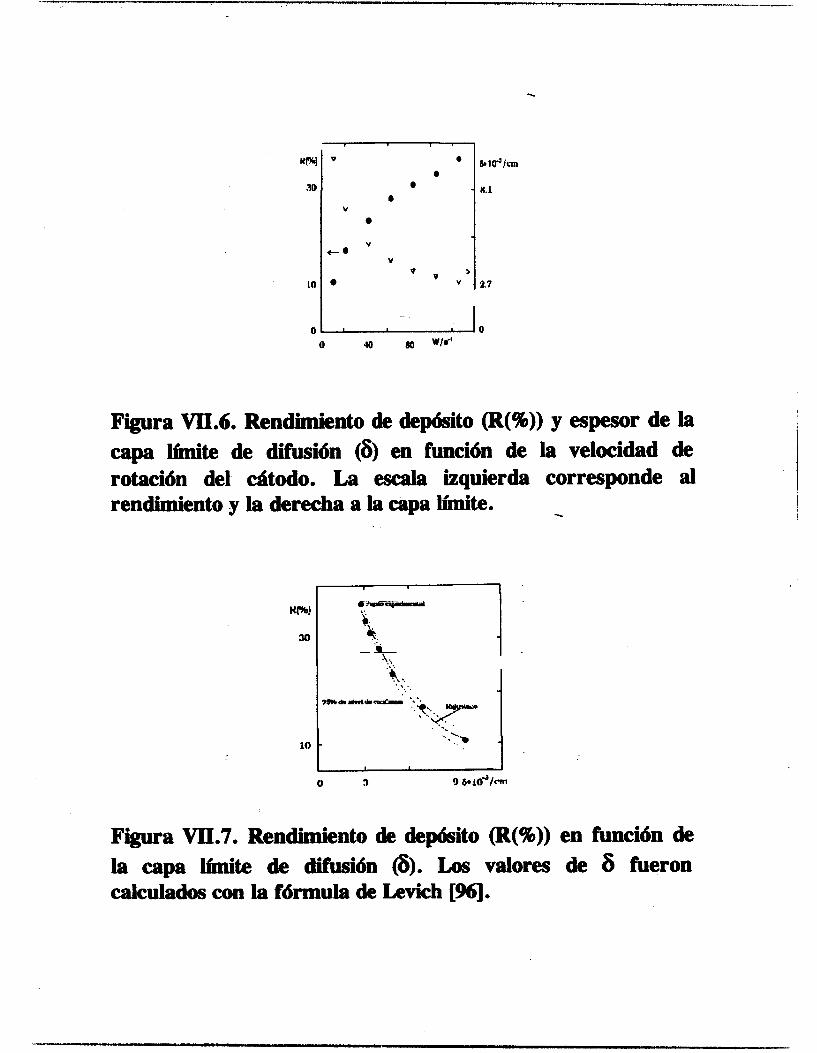
Y-J
30
IO
I .
0 e e
e
Y O
4-0 V
Y
e O V V
Figura VII.6. Rendimiento de &phito (R(%)) y espesor de la capa limite de difusión (6) en función de la velocidad de rotación del &todo. La escala izquierda corresponde al rendimiento y la derecha a la capa límite.
\
10
Figura VII.7. Rendimiento de depósito (R(%)) en función de la capa Wte de difiisión (6). Los valores de 6 fueron calculados con la fórmula de Levkh [%J.

rotación del catodo sobre el rendimiento. En este aspecto podríamos decir que la velocidad a la que la solución fluye hacia la superficie del disco, aumenta con la velocidad de rotación del cátodo y en el mismo sentido el transporte del lantinido o
actinido de la solución hacia la interfase. Por otro lado, tampoco toma en cuenta que al tener espesores similares 6 y lo^: evitamos que el transporte por difusión se realice entre el intervalo 6-loti y al mismo tiempo proaovemos el transporte por convección (más rápido que la difusión) hasta los limites de 1oH.
En el empleo del disco rotatorio para la obtención de depósitos de hidróxidos y/o óxidos el proceso está determinado por la transferencia de materia, por convección de la solución hacia la interfase y por difusión en la capa límite de difusión 1921 . El transporte por convección en la solución involucra velocidades del orden de 1 c d s y por difusión en la- capa de difusión velocidades del orden de 0.03 cds. Ambos parámetros, la convección forzada a velocidades de rotación mayores y la sirilitud de los espesores de OH y 6 podrian explicar el comportamiento del rendimiento con respecto a u donde se observa una fuerte dependencia. Para el estudio del tiempo de depósito se - L a 6 entonces como w
óptimo 126 s-' donde se observa una fuerte dependencia. -__
-R = f( t depósito).- La respuesta es una función creciente con el tiempo, con cambios de pendiente de mayor a menor hasta con un factor 10 aproximadamente. Se ajustó un polinomio de tercer grado, resultando un valor de norma de 2.7, con la siguiente forma (Figura VII .8) .
104
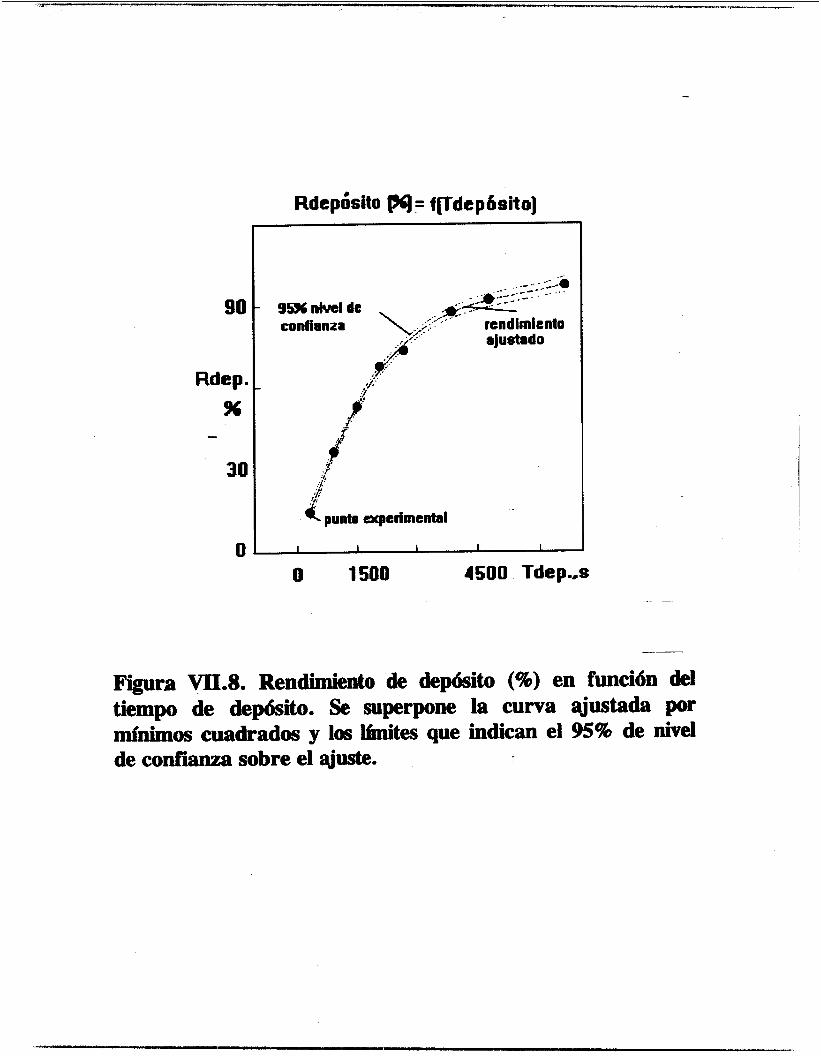
90
Rdep. 96
- 30
O
RdcpÓsRo In] = f ~ d c p b i t o )
, . _- -. -3.
rendimiento 8justado
.--- -.*--, _. .. . - :*:---. - . ._ *.*'. 5 -
.*.8 I
95K nlvei de ronfianza \?$;. .
,.,<- ,/a
f
O 1500 4500 Tdep.,s -
Figura VII.8. Rendimiento de depósito (96) en función del tiempo de depósito. Se superpone la curva ajustada por mínimos cuadrados y lw iímites que indican el 95% de nivel de confianza sobre el ajuste.

-2 R(%) = 4.5 x lo-'* td3 - 7.47 x td2 + 4.41 x 10 td + 2.2
ERROR 17% 10.6% 5.2% 7.8%
DEPENDENCIA o. 99 o. 99 o. 99 O. 92
Se observa que R depende de td y que el tiempo de depósito para
un rendimiento teórico del 100% tiene un valor de 7135 segundos (2 horas). La distribución de los puntos experimentales se
encuentra a lo largo de la curva teórica y dentro del intervalo correspondiente al 95% de nivel de confianza. Para un tiempo de 6600 segundos experimentalmente hay un rendimiento de 97.5 f2.5%
, para el mismo tiempo el R calculado es de 97.6%. Por otro lado, para un tiempo t=O segundos el rendimiento calculado es de 2.2% lo cual no tiene significancia estadística tonando en cuenta que el error experimental es del orden de 2.5%. Para todos los tiempos de depósito las condiciones experimentales son similares, por lo que los cambios en la velocidad de precipitación podrían deberse a una disminución en la
concentración del número de partículas coloidales formadas en la capa alcalina presente en la superficie del cátodo, dicha concentración es proporcional a la cantidad de materia que difunde de la solución hacia la capa alcalina. Entonces si disminuye la cantidad de materia y la cantidad de partículas coloidales, el tiempo de coalescencia para la formación del gel aumenta con la disminución de la probabilidad de interacción
interpartículas en el interior de la capa alcalina (de volumen constante).
-R = f(@, i1.- La interrelación entre la densidad de corriente aplicada (i) y el pH de la solución es puesta en evidencia por el modelo de Hansen [151 donde el pH en la interfase cátodo solución
y el espesor de iones hidróxido se definen matemáticamente en función de i y del pH de la solución. Sin embargo, la influencia
1 o5

de la interacción i-pH sobre el rendimiento de depósito no fue estudiada en los trabajos de Hansen. La interacción entre el pH de la solución fuente de H+ y la densidad de corriente aplicada fuente de OH- determina la concentración y espesor de OH- en la interface cátodo-solución necesarios para tener un depósito cuantitativo del hidróxido correspondiente. Dicha interacción fue estudiada en el intervalo de pH de 2 a 8.5 y de i de O a 25.4 (mA . cme2). A partir de dichos valores con los puntos experiaentales se obtuvo la superficie de respuesta en función de pH y de i
(Figura VII.9 1, y a partir de ésta se obtuvieron las curvas de isorespuesta (Figura VII. 10). En ambas figuras se observa que la
respuesta es función de la interacción pH - i y que la región de máxima respuesta se encuentra ubicada en los intervalos de pH de 3.5 a 6 y de i de O. 18 a 1.4 mA Los valores límite de la superficie de respuesta son 2C pH C8.5 y O. 18< i C25.42 .A
Se obtuvo la correspondiente función analítica con un valor de norma de 54.1 y es de la siguiente forma:
,
-2 2 €I(%)=-l. 385pH2+15. 29pH-3.35~10 i +O. 902 i +6.046x 10-3pHi- 24.8
ERROR(%) 14.3 13.9 48.8 52.8 829% 21.5
DEPENDEWCIAO. 99 O. 99 O. 94. O. 96 o. 91 o. 97
Como se puede observar los valores de dependencia de R son superiores a 0.9 marcando la influencia de la interacción sobre R. El valor de norma y el de error asociado al coeficiente del término pH-i son elevados mostrando que la función define dicha interacción aunque no completamente. El modelo coincide con el intervalo de máxima respuesta experimental en el caso del pH de 5
a 6 lo que no sucede en el caso del intervalo de i. La diferencia podría deberse a que a bajas densidades de corriente el cambio de intensidad de la respuesta es muy rápido en una fracción de intervalo muy pequeña. Las correspondientes superficies de respuesta y curvas de isorespuesta se muestran en las Figuras
106
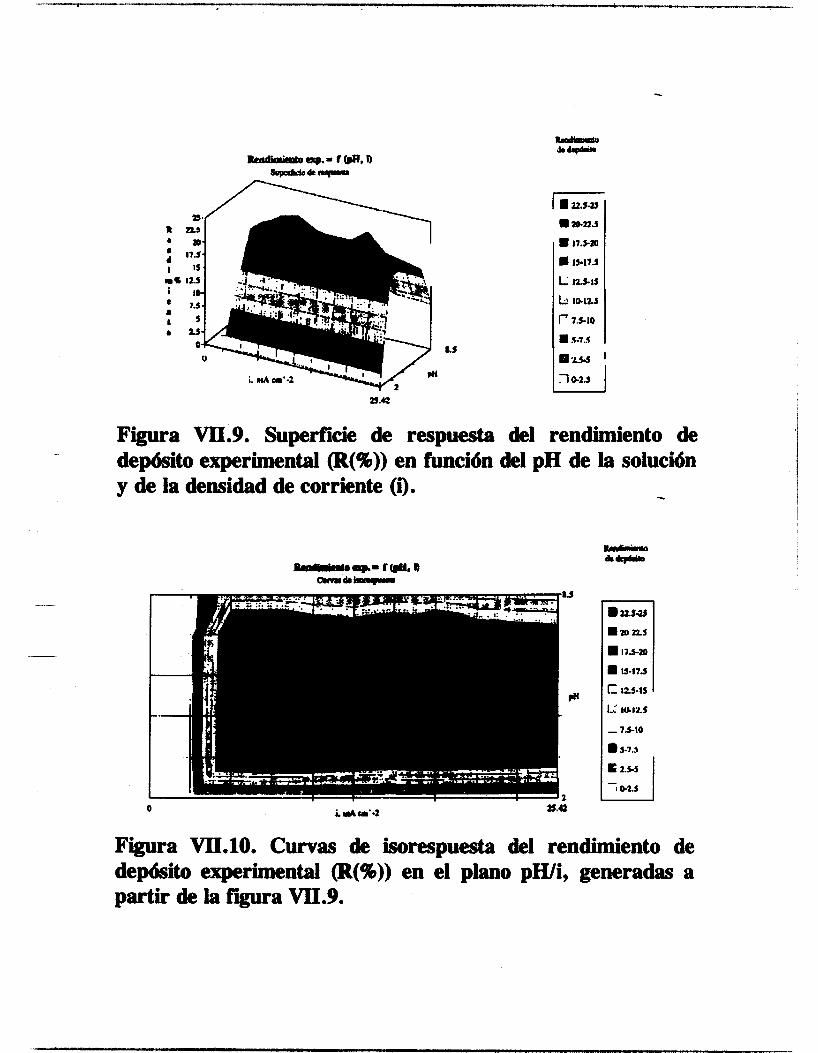
R.5
Figura VII.9. Superficie de respuesta del rendimiento de depósito experimental (R(%)) en función del pH de la solución y de la densidad de corriente (i).
%.O i rAer'.2 O
P.W
1 w n s 11.5-Ñ
fl U-17.5
125-15
I-: w11.5
- 7.CIO
3-1.5
E 2 5 5
i 02.5 -
Figura VII.10. Curvas de isorespuesta del rendimiento de depósito experimental (R(%)) en el plano pH/i, generadas a partir de la figura VII.9.

VII. 11 y VII. 12, donde se observa que el modelo reproduce las respuestas experimentales de manera aproximada con las diferencias mencionadas. En efecto, el modelo se ajusta mejor a
la respuesta experimental si los valores de i de O y 0.08 rnA
no son tomados en cuenta, las Figuras VII. 13 y VII. 14 son la superficie de respuesta y las curvas de isorespuesta respectivamente, las cuales no consideran dichos puntos. Se puede observar que ambas figuras semejan más a las experimentales V11.9 y VII. 10 tomando de las mismas el intervalo de i de O. 18 a 25.42 iA CEI-~. Las superficies de respuestas son similares y las curvas de isorespuesta definen una zona similar de máxima respuesta, la i va de 0.18 a 25.42 en ambos casos y el pH de 5
a 6 en el modelo y de 3.5 a 6 en los experimentos.
-R = f(pH, PSI;).- En la literatura ambos paribnetroc son estudiados por separado, considerando sólo su influencia sobre la conductividad del medio y sobre el rendimiento de depósito. Sin embargo, en el estudio fisicoquírnico de la solución electrolítica (Cap.111 1 se demuestra que es importante estudiar la interacción de ambos parámetros sobre el rendimiento ya que se trata de especies químicas en solución que como entorno quimico van a defiRir las características de los complejos formados y por consiguiente proporcionar información sobre el probable mecanismo de depósito de cada Lantánido o Actínido sobre todo si se presentan numerosos estados de oxidación como en el caso del pl ut oni o.
A partir de los resultados experimentales se ajustó un polinomio de segundo grado con interacción pH - pS0; , con un valor de norma de 16.8, el polinomio tiene la siguiente forra:
107
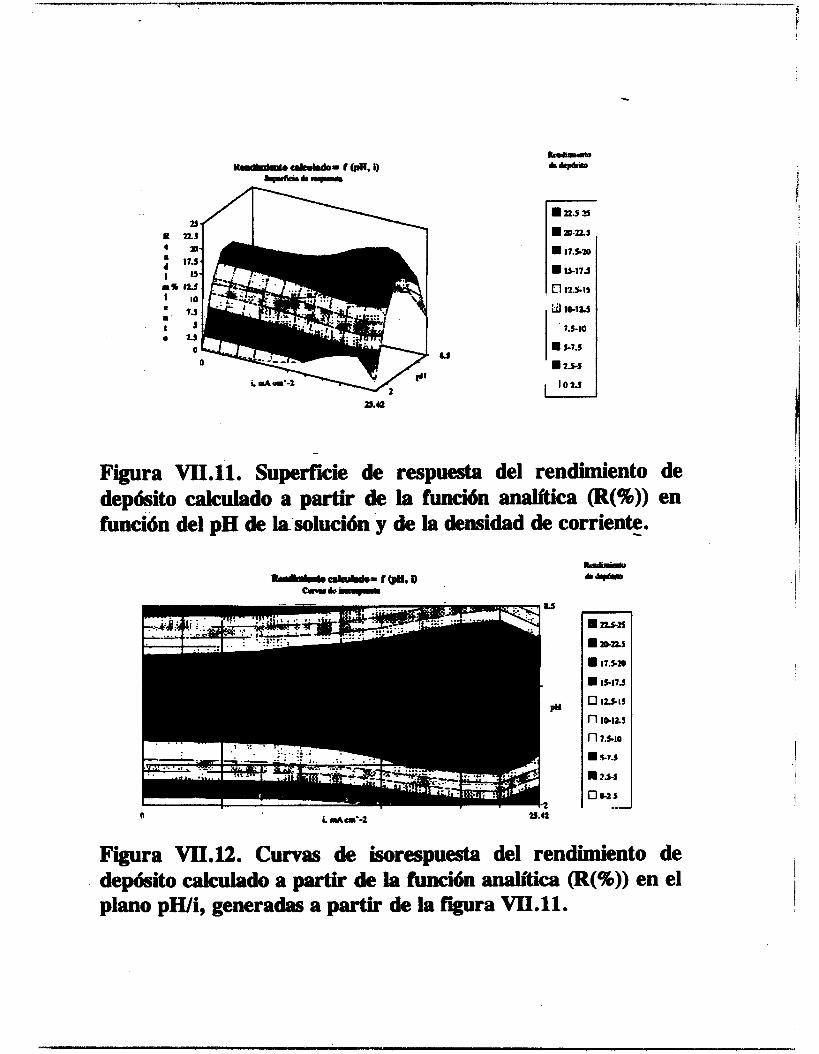
i
f1.a
u lD12.5
i s m 8 s-7.5
a 2~
I l 0 2 l J
-
Figura VII.11. Superficie de respuesta del rendimiento de depósito calculado a partir de la función d t i c a (I€(%)) en función del pH de la solución y de la densidad de corriente. z
'LI.42 i. rA ea'-2 n
Figura VII.12. Curvas de isorespuesta del rendimiento de depósito calculado a partir de la fuiición analítica (R(%)) en el plano pWi, generadas a partir de la figura VII.11.
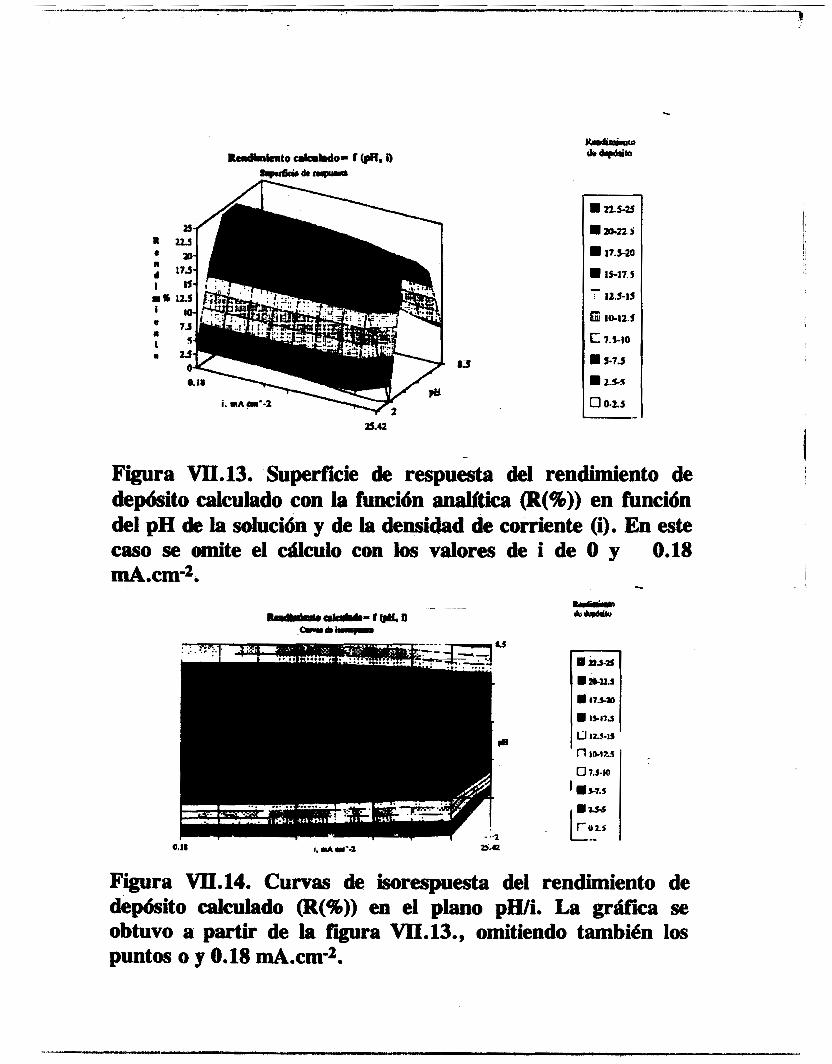
i Figura VII.13. Superficie de respuesta del rendimiento de depósito calculado con la función d t i c a (R(%)) en función del pH de la solución y de la densidad de corriente (i). En este caso se omite el cálculo con los valores de i de O y 0.18
I mA.cm-2. z 1
Figura VII.14. Curvas de isorespuesta del rendimiento de depósito calculado (R(%)) en el plano pWi. La grafica se obtuvo a partir de la f i i ra VII.13., omitiendo también los puntos o y 0.18 mA.cm-2.

R(%)=-l. 562pH *17.53pH-6.602pS0;+8.973pSO;-O. 3532pHpSOi-28. OS
ERROR ( X ) 10.5 11.3 113 145 350 26
DEPENDENCIA 0.99 0.99 0.96 o. 99 O. 97 o. 99
Como se puede observar los valores de dependencia son superiores a 0.95, indicando que el modelo con interacción propuesto
corresponde a la dependencia de R con respecto a dicha interacción. Las Figuras VII.15 y VII.16 muestran respectivamente la superficie de respuesta y las curvas de isorespuesta experimentales en función de pH y pS0; , se observa que ambos parametros definen una zona de máxima respuesta (con los intervalos de pH de 3.5 a 6.7 y de pS0; de 0.3 a 0.7) con una forma irregular. En la gráficamente se observa la influencia de la interacción sobre la respuesta con algunas irregularidades en
la zona de máxima respuesta. Por otro lado, la superficie de respuesta (Figura VII.17) y las curvas de isorespuesta (Figura VII. 18) obtenidas a partir del iodelo muestran un comportamiento similar aunque más regular con las mismas tendencias. La zona de maxima respuesta se encuentra ubicada en el intervalo de pH de 5 a 6 y de pC0; de 0.4 a 0.7 aproximadamente, la cual se encontraría dentro de la zona irregular experimental. De la misma
- _
manera se observa la influencia de la interacción mostrando además que la evolución de la respuesta en el eje de pS0; es monótona con pendiente negativa.
-Características físicas de l o s depósitos
La primera característica medida- fue la uniformidad del depósito. Como la distribución de la densidad de trazas alrededor de un promedio es función de la uniformidad del depósito, se calculó la densidad de trazas en una unidad de superficie (un cuadrado de 1.8 x cm 1 siguiendo un barrido lineal y radial con respecto al centro del círculo del depósito. El conjunto de valores
2
108

I M22.5 I 11.5-20
15-17.5
L 17"SlS
E IDI2.J
7.5-10
s7.5
I 2 5 5
L 023
8.5
Figura VII.15. Superficie de respuesta del rendimiento de depósito experimental (IR(%)) en función del pH y pS04' de la solución.
.
I 12.5-25
10-22.5
a 17.5-20
a Is-173
- 123-15 l E E - 7.s 1w2.1 10
B 5-73
2.S-5 J 3 0-2.5
Figura VII.16. Curvas de isorespuesta del rendimiento de depósito experimental (R(%)) en el plano pH/pS04', generadas a partir de la f w r a VIII.15.
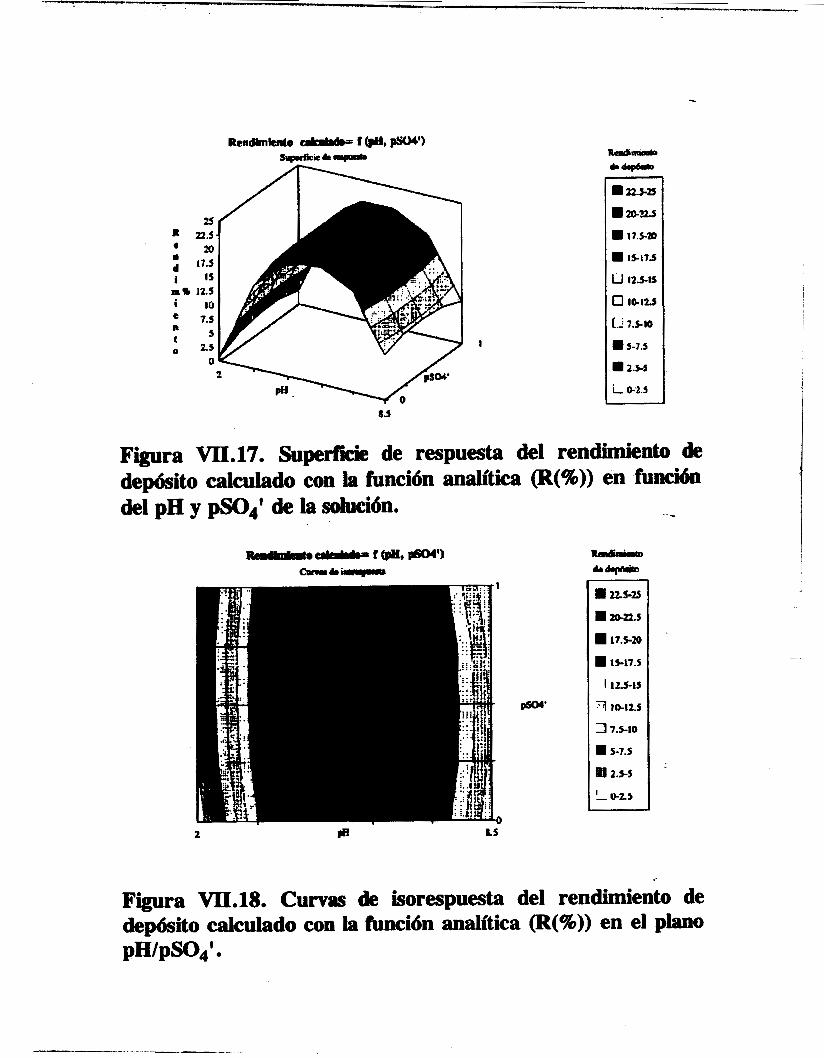
V 1
v
I
a T ~ S B
17.5-20
m 20-22.5
15-17.5
112.5-I5
Y 10-125
3 7.540
a 5-7.5
2.53
1- u-2.3
8.5
Figura VII.17. Supedkie de respuesta del rendimiento de depósito calculado con la función analítica (R(%)) en función de¡ pH y pS04' de la sokrción.
2
Fieura VII.18. Curvas de isorespuesta
- - -
del rendimiento de de-mito calculado con la función analítica (R(%)) en el piano PWPs04'

proporciona una densidad de trazas promedio y una desviación
estándar asociada, cabe señalar que se calculó el promedio de los
valores sin tomar en cuenta los valores de los extremos del
depósito (1, 2 12 y 13 de la Figura V I I . 19). De los valores obtenidos se constata que el depósito es uniforme con una
dispersión del material depositado de f 10% (desviación estándar relativa).
Por el método del análisis del espectro de un haz de protones monoenergéticos acelerados se buscó determinar el espesor del
depósito, sin embargo, la técnica no proporcionó resultados
consistentes. Por otro lado, si realizamos un cálculo teórico a partir de la actividad medida de una fuente de Am-241 y de la superficie de depósitos tenemos que con:
Actividad redida = 546 Bq p,,-241= 11.68 g cme3,
2 Area depóslto = 1.767 cm ,
el espesor teórico es de 2.38 x A lo cual es incoherente. Tal incoherencia podría ser explicada en función de una superficie recubierta por el depósito menor al área geométrica
del cátodo. Con la finalidad de explicar estas diferencias se
realizó un estudio de perfilometría para determinar la rugosidad del cátodo sin depósito. Los valores de rugosidad, Ra, son:
109
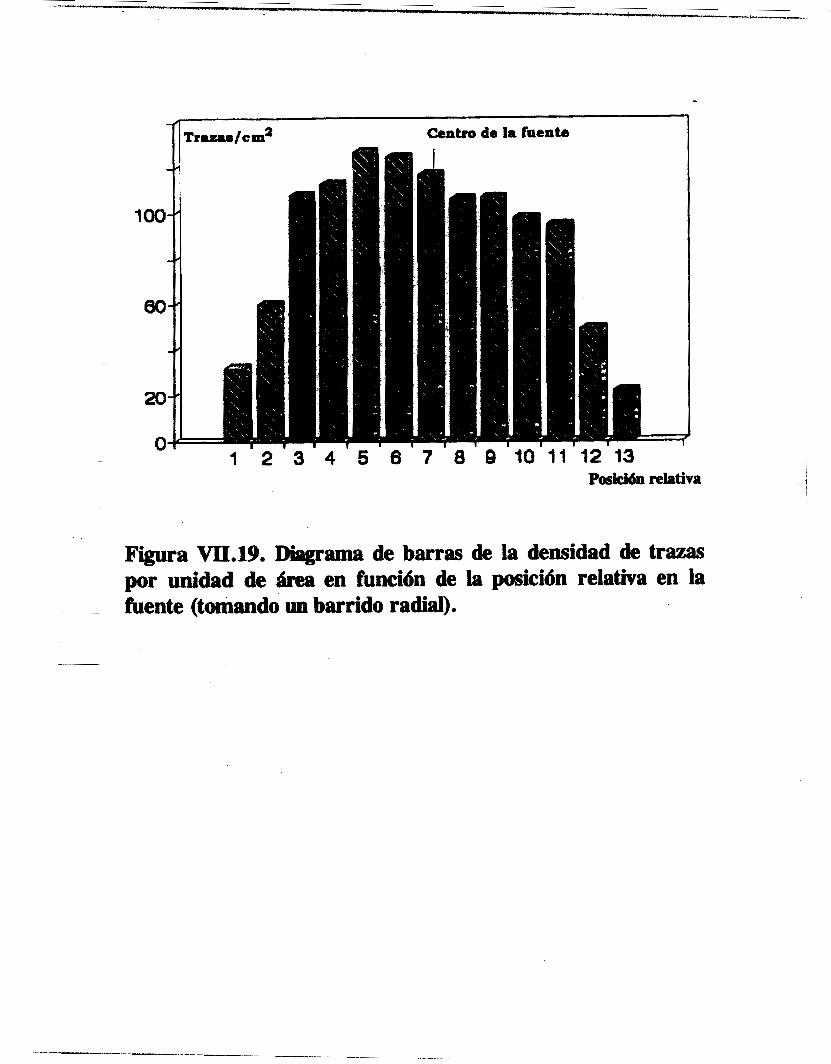
-nTruu,cml Centro de la fuente
1
13 Posición reiativa
Figura VII.19. Diagrama de barras de la densidad de trazas por unidad de giFea en función de la posición relativa en la fuente (tomando un barrido radial). ~
Cátodo
Pulido Electrolítico
Pulido Mecánico No. 400
Pulido Mecánico No. 800
Pulido Mecánico No. 1200
Ra (1)
287
Pulido Mecánico "brillante"
Rolado en frío 1 I I
919
1360
Estos valores de rugosidad son muy grandes si los comparamos con el espesor de depósitos C 50 reportados en la literatura 1931 . Es decir, - que las rugosidades medidas refuerzan la hipótesis de un área ocupada por el depósito inferior a la geometrica y mucho
menor a la real ya que el área efectiva para el depósito aumenta
en función de la rugosidad de una superficie. Hasta aquí entonces
el estado de la superficie, la uniformidad del depósito y las
cantidades depositadas permiten decir que el depósito consiste en
un conjunto de placas depositadas en toda la superficie del cátod0.-
Por otro lado, la máxima diferencia en rugosidad es de casi 600%
y síiienibargo, las diferencias en rendimiento de depósito son del
orden del 5% en el intervalo de rugosidad estudiado. Adeds,
tampoco existe diferencia en rendimiento entre los cátodos de
acero inoxidable y de platino.
Del estudio de la optirnización de parámetros de depósito se desprende que los resultados son reproducibles y consistentes con la teoría de disco rotatorio [50,511, con el modelo de Hansen de
depósito de hidróxidos [is1 y con la de formación de geles (911 . Para la realización del estudio se observaron las siguientes condiciones:
-Con el estudio fisicoquímico realizado se aseguró que el anión
110

(SO2-) del electrolito soporte no se descompone y, por lo mismo, no contamina la solución. Además, se predijo el comportamiento de la solución electrolítica. -El estudio hidrodinárnico para el diseño de la celda (ver anexo 3) permitió cumplir con las condiciones para el empleo del modelo de disco rotatorio. -La distribución de la corriente fue analizada (ver anexo 2) lo cual permitió asegurar condiciones de distribución uniforme de
4
la corriente, -La prueba de cumplimiento de la ley de Levich (ver anexo 1 )
permitió asegurar que el cátodo inclinado no modifica las condiciones de disco rotatorio expuestas en la teoría.
Una vez que se cumple con las condiciones enlistadas es posible utilizar la metodología expuesta en el presente trabajo para definir las condiciones - particulares para el depósito de los diferentes Lantánidos y Actínidos con comportamientos similares. Las condiciones a encontrar para cada caso son el pH de la solución y la densida¿ de corriente necesarios para Óptimos resultados en uniformidad y rendimiento de depósito. Los partímetros Óptimos para cada elemento son:
~ ~~
Para sarario europio y americio: pH=5 pso;..o. 5
i= 1 m~.cm-~ w= 126 s-l
t= 110 minutos
__-
Para el plutonio: pH=5 pco;=o. 5 i= 1.2 m ~ . w= 126 s-l
t= 110 minutos.
111

VI I I. ESPECTROIIETRfA
El análisis de espectros es uno de los problemas que se presentan en el cálculo de constantes nucleares como las probabilidades de emisión. Para medir con exactitud el area bajo la curva de cada linea espectral, es necesario ajustar los resultados experimentales a alguna forma de pico. Si varios picos se traslapan el ajuste es esencial para determinar la contribución de cada pico. Se han propuesto varias formas de curvas, de manera semiempírica, para el análisis de espectros donde cotnunmente se aplica un procedimiento de ajuste por mínimos cuadrados para encontrar los mejores parámetros que representen los espectros. Mas recientemente se han aplicado técnicas de minimización SIMPLEX [521 al ajuste de espectros [531. Esta aplicación depende del método de máxima semejanza, el cual maximiza la información cuando hay un número de eventos relativamente bajo 1541.
VIII.1. Métodos de ajuste
Se han propuesto diversas formas de curvas que toman en cuenta los diferentes factores que afectan la detección de partículas alfa. Utilizando un método de ajuste se calculan las áreas correspondientes a cada emisión de cada núclido en el espectro medido. Se hace notar que con este tipo de procedimientos se obtienen áreas correspondientes a cada emisión de uno o varios núclidos y por lo mismo se obtienen resultados de intensidades de emisión y de relaciones isotópicas para cada fuente radiactiva medida. El modelo mas simple que toma en cuenta los efectos de cola de las bajas energías de picos alfa fue propuesta por Acefía [551
y utilizada posteriormente por Williams 1561 para estudiar
!
11s

espectros de plutonio. La curva consiste en una gaussiana unida a una exponencial en la región de la cola de bajas energías. El siguiente paso en complejidad es el uso de una gaussiana unida a una exponencial de segundo grado en la parte de bajas energías del pico. Watzig y Westmeier 1571 analizaron espectros complejos de hasta 20 picos utilizando este tipo de ajuste de curva, además
desarrollaron una versión comercial llamada ALFüN. Baba [581
propuso una curva similar pero con una exponencial de tercer grado en al región de bajas energías, estudiando espectros de plutonio. Otro programa comercialmente disponible para analizar espectros alfa es "FIT, hecho por Welch [591, el cual utiliza en el análisis de espectros la forma de pico representada por una gaussiana a la cual se le puede agregar una función para el fondo, una exponencial para la cola de bajas energías o una función "step". -Garcia-Toraño [601 en su programa de ajuste NOLIN propone otro tipo de curva para ajustar picos alfa. La curva propuesta es una división de tres, dos gaussianas y una hipérbola en el intervalo de bajas energías. Otro &todo para encontar curvas adecuadas para ajustar picos alfa es aquel que utiliza la convolución de diferentes curvas para obtener el ajuste requerido. Este método
- --- fue aplicado por Bland [6llpara desarrollar el programa CALGARY de deconvolución de espectros. El método se basa en el algoritmo
---SIMPLEX [621. En la siguiente sección se describen los elementos importantes del método. En el caso mas general modelo se componen de las siguientes fases: a. Adquisición del espectro en forma discreta numérica:
los métodos de ajuste de un espectro a un
Ei, con 1'1,. . . ,n
siendo Ei el número de pulsos contados en el canal I, con n como el número de canales.
113

b. Construcción de una función de ajuste:
Fi(ai,. . ,a,) con i=l,. . ,n
son los siendo Fi el valor de la función en el canal I y al,. . , a, parámetros de ajuste. c. Selección del criterio que describa la calidad del ajuste de la función modelo al espectro experimental. Este criterio toma un valor extremo para el mejor ajuste. d. Búsqueda del conjunto de paráretros al,. , , a, que proporcionen el criterio extremo. e. Evaluación de la incertidumbre, sobre los parámetros de ajuste a los que concierne la adecuación del modelo, la calidad del ajuste y las fluctuaciones estadísticas del espectro medido.
VIII. 1.1. Mquisicibn de l a medida
Se adquiere el espectro experimental por medio de un analizador multicanal que genera un archivo ASCII cuyo número de orden de los valores corresponde al número de canal del espectro con un cierto número de pulsos asociados a cada canal.
VIII. 1.2. Rinción de ajuste
La función de ajuste se elige de tal nanera que se da cuenta de los fenómenos involucrados en la detección de las partículas alfa. La forma de un pico alfa esta influenciada por varios elementos dominantes: la trayectoria y la difusión de la partícula en la fuente, la colección de cargas, la influencia de la electrónica y la amplificación, y la dispersión de la energía en la ventana del detect or. La observación de un pico muestra la superposición de un pico casi gaussiano, debido a los defectos de colección y de la electrónica, con una cola a la izquierda del pico yendo hasta la energía cero, relacionada con los efectos de absorción y difusión
114

en la fuente y el soporte (figura VIII.l). La cola de bajas energías varía con el tipo de sistema de medición y con la calidad de la fuente radiactiva.
VIII. 1.2.1. Uodelo matemático
Los picos alfa se ven profundamente afectados por los efectos de cola, el tratamiento matemático torna en cuenta simultáneamente los efectos gaussianos y los efectos de alargamiento de las bajas energías; el modelo común utiliza la convolución de una
exponencial decreciente hacia las bajas energías y truncada en la energía del máximo del pico alfa con una gaussiana. Sin embargo, el programa CALGARY utiliza la suma de dos
exponenciales distintas cuando se convoluciona con la gaussiana; Este modelo mas versátil permite una mejor adaptación a los datos
experimentales [631. También -se agrega una constante con la finalidad de describir el efecto de los contenidos no nulos de los primeros canales.
La función analítica se escribe como un producto:
f(u)= G(u)*E(u)H(-u) (VIII. 1 )
_ _
donde u es la variable energía, G(u) la gaussiana normalizada, F(u)H(-u) es la exponencialdpa sección izquierda del pico, con H(-u) como la función “step”. Si consideramos f(E) como una función conjunta de probabilidad-densidad y dividimos u=x+y,
entonces
con c como la desviación estandar de la gaussiana, significa una función de probabilidad-densidad gaussiana simétrica (la cual toma en cuenta la propagación simétrica del pico en el espectro),
115
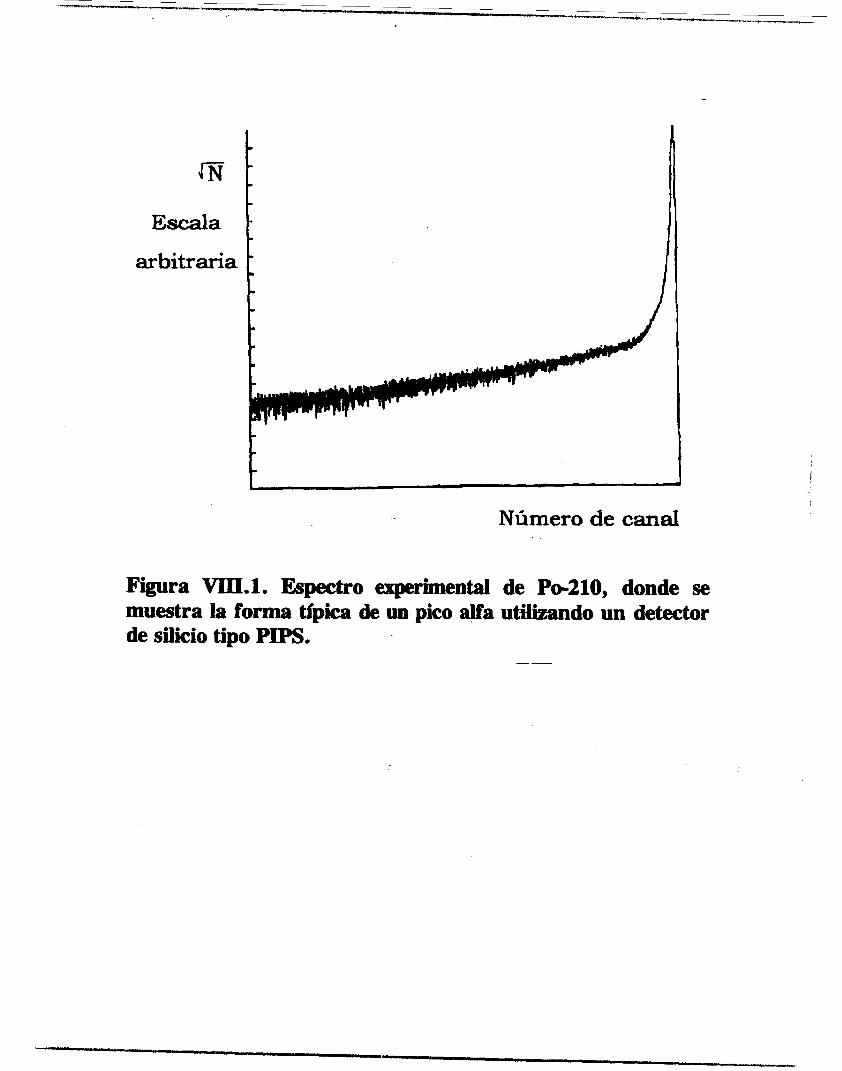
JÑ
Escala
arbitraria
Número de canal
Figura VIII.1. Espectrú experimental de Po-210, donde se muestra la forma t€ph de un pico alfa utiiiesuido un detector de silicio tipo PIPS.

Y
P,(y)= 1 exp(X/T] H(-y), con -a < y < a, (VIII.3)
con t como parámetro, es una función de probabilidad exponencial asimétrica (la región izquierda del pico), la cual toma en cuenta la propagación asimétrica del pico en las bajas energias del pico. La función conjunta de probabilidad-densidad es la convolución de las probabilidades precedentes [541:
sustituyendo e integrando se obtiene: -
-1/2 u 2
27
P (u)= 1 exp[ + \]erfc[z ( + )] (VIII.5) U
donde erfc(x) es la función complementaria de error: - -
(V111.6) _- erfc(x)=
En realidad los picos alfa tienen un area, A, diferente de la unidad y una posición media, p, diferente de cero, entonces:
112 u-p 2
f(u)= Ap (u]= A exp[r U-P + \]erfc[Z [o + :)l. (VII1.7) 2t U
116

Hasta este punto se ha descrito el método de L’Hoir j641. En lo
subsecuente se estudian las modificaciones introducidas por
Bortels [631 a este método.
Por convención tenemos:
sin la función step. Bortels divide la exponencial en 2 partes, una asociada a + 1’ la
cual toma en cuenta la asimetría en el pico (cerca de la moda) y
otra a #2, considerando la cola a energías más bajas. Entonces
de tal manera que:
donde O,[u) es la sura ponderada de 2 funciones (con el parhetro
t similar a Q en el orden de magnitud y t2 similar a 3u en orden de magnitud), con como factor de peso tenemos que: 1
y gi2 es una función constante con, en algunos casos un término extra para la función exponencial de largo alcance (con t3 mayor
que c en orden de magnitud), donde:
#,(u)= C + cE3(u). (VIII. 12)
Para un espectro real con m picos:
F(u)= fl(u)+ R(u), (VIII. 13) 1 =1
donde R(u) representa la distribución de los residuos del ajuste.
117

Entonces:
donde el segundo término de la primera parte es la función de la cola y el primer término de la segunda parte es la función de ajuste. Puesto que la función de la cola representa una corrección relativamente pequeña, se puede hacer la siguiente aproximación:
I
AIGl(u) F(u). I =1
(VIII. 15)
Además, en la sustracción de la cola sólo se considera el término constante de #2(u) poniendo c=O. Entonces:
FC(u)= F(u)-F(u)*C=Z AIG1(u)* (1-I))El(U)+VE2(u) + R(~).(VI11.16) I =1
El desarrollo de la función de ajuste lleva a
En el procedimiento de ajuste se utiliza el método de mínimos
cuadrados. El valor de X2 (chi cuadrada reducida) es utilizado para calificar el ajuste. Los parámetros de ajuste son cuatro (e, t
Cabe señalar que en la función de ajuste empleada por Martin-Sánchez [all, la función exponencial es en realidad una
A
t2,q) más rn áreas Al y rn medias p . 1' I
118

doble exponencial que sigue contemplando la asimetría cerca de la
moda del pico y la cola en las bajas energías a diferencia de
Bortels (631 que hace una simplificación. La exponencial da cuenta de la asimetría y considera una constante para las bajas
energías.
En el anexo 7 se expone la relación que existe entre los
parámetros de ajuste, la forma de la función analítica de
deconvolución de espectros alfa y el significado físico de las
interacciones que concurren durante la detección de las
partículas alfa emitidas por los núcleos atómicos.
VIII.1.3. Criterio de ajuste
El criterio de ajuste es una magnitud que da cuenta de la
similitud del modelo con el espectro experimental. Como los valores experimentales presentan fluctuaciones estadísticas, este
criterio también lo es y debe elegirse como un estimador del
máximo de similitud del ajuste. Como consecuencia de esto, la
selección de este criterio se apoya sobre hipótesis que tienen
que ver con la dispersión de los datos experimentales y la calidad del ajuste y al similitud del modelo solo deben ser
juzgados dentro del marco de estas hipótesis.
VIII.1.3.1. Criterio de minims cuadrados
Si se considera que el espectro experimental Ei es el resultado de fluctuaciones gaussianas, independientes, con una desviación
estandar,c, constante sobre todo el espectro del modelo Fi, se puede demostrar (anexo 4)[52] que el criterio de mínimos cuadrados:
2 n (Ei-FI)
M= 1 i=l 2r2 ’
(VIII. 18)
119

es un estimador del máximo de similitud del ajuste de F, a E,, con un valor mínimo del criterio asociado al mejor ajuste. Debe considerarse en todo momento que si la hipótesis citada arriba no se verifica (tendencias, valores aberrantes, parásitos, baja estadística de conteo) este criterio no sería un buen indicador del ajuste.
VIII.l.3.2. Criterio de x” Este criterio corresponde a una generalización de los mínimos cuadrados en el caso donde Q no puede ser considerado como una constante sobre todo el espectro. Se supone entonces una distribución fluctuaciones
aleatoria normal e independiente de las del número de pulsos por canal:
n 2 2 x”= (Ei-Fi 1 /ei,
i =I (VIII. 19)
La minimización de x“ conlleva, dentro del marco de las hipótesis enunciadas arriba, el mejor ajuste. Este valor de x” puede ser normalizado dividiendo por el número de grados de libertad del problema: _. ~-
A
x”= ?/f. (VIII. 20)
con f-(n-k-1) como el número de grados de libertad, siendo n el número de canales del espectro y k el número de paráaetros a Justables.
El ajuste es mejor cuando x” se acerca a 1 . Sin embargo, se puede demostrar que este valor depende de la estadística de conteo del
espectro experimental y que x” aumenta linealmente con el número de pulsos en el espectro (anexo 4) 1651.
A
A
120

Este hecho tiene dos implicaciones prácticas:
- las comparaciones entre los modelos deben ser hechas con espectros de la misma estadística,
A
- un valor de X2 cercano de 1 de un espectro de baja estadística tiene poco significado en cuanto a lo adecuado del modelo con respecto al espectro experimental.
VIII. 1.3.3. Criterio de Poisson La aplicación del criterio de x” presenta problemas con espectros que tienen canales de bajo contenido, pues la desviación estandar toma valores de cero y el cálculo conduce a divisiones por cero. En el caso donde se puede decir que la distribución de las fluctuaciones por canal responde a una estadística de Poisson, el criterio de Poisson definido por la ecuación (anexo 4):
n
C= C (F1-El*logFl), 1 =1
(VI I I . 21 1
indica el mejor ajuste cuando tiende a un mínimo. Se puede mostrar que para valores grandes de El, el criterio de Poisson converge hacia x” fanexo 5 ) .
VI I I. 1.4. Algorib- deajuste
Se vió anteriormente que la operación de ajuste de la función Fl(ai,..,a,) al espectro experimental Ei se podía reducir a la minimización de un criterio f (El, Fl (al, . . , ak) ) con respecto a los
Dicha minirnización se puede realizar por diferentes métodos, el más clásico consiste en buscar el valor de los parámetros anulando la derivada del criterio con respecto a los parámetros:
pwámetros a 1> * * , ak.
- - - (f(El,Fl(a l,..,ak)))= O. d a
1 (VIII. 22)
121

El sistema de las i ecuaciones se puede resolver por un método matricial. El inconveniente es que se requiere calcular las derivadas del criterio, debiendo modificar el algoritmo de cálculo de minimización. En este caso se debe modificar el algoritmo de cálculo para cada modificación de la función de ajuste F1: Sin embargo, este método presenta la ventaja de proporcionar la solución rapidamente. El método de resolución que se aplica en el programa utilizado, emplea un método diferente al matricial: El método SIMPLEX desarrollado por Nelder [62], empleado en aplicaciones agronómicas, consiste en construir, en un espacio de k
dimensiones, donde cada parámetro de ajuste esta asociado a cada eje, una figura con k+l puntos llamada SIMPLEX. Esta figura se deforma sucesivamente según tres procesos geométricos elementales: la reflexión, la contracción y la expansión. La función que se va a minimizar se calcula en cada punto y las deformaciones del SIMPLEX se suceden hasta que este converge hacia un punto del espacio donde la función es mínima. El método del SIMPLEX es un algoritmo robusto de minimización, sin embargo presenta el inconveniente de ser muy lento cuando el número de parámetros para ajustar es elevado. Dicho inconveniente es el precio que se paga para evitar el cálculo de las-derivadas
de la función a ser minimizada. El algoritmo SIMPLEX se describe en el anexo 5. --
VIII.1.5. Evaluación de l a incertididn de los parámetros de
ajuste Los parámetros de ajuste fluctúan álrededor de su valor promedio debido a las fluctuaciones del espectro experimental y de la eventual imprecisión del ajuste. Por esto es necesario determinar, además del valor promedio, su varianza asociada. Las matrices de varianzas-covarianzas asociadas a los valores más probables de los parámetros al,. . , ak son (L") con L definida
i J
122

según la siguiente ecuación (541:
aF aF E -F a ' ~ ( VI I I . 23 1 1 n n n n
J n E aai aa
La matriz de error que da cuenta de la calidad del ajuste puede ser evaluada en primera aproximación por medio de la siguiente ecuación:
(VIII. 24)
Si se considera una función g , utilizando los parámetros de ajuste (por ejemplo la superficie de un pico de acuerdo con el modelo de ajuste considerado), la varianza de esta función será del orden de:
(VI I I . 25 1
La determinación de las incertidumbres presupone entonces la inversión de una matriz de orden k2 cuyos componentes son derivadas parciales de la función de ajuste. Este método de evaluación conlleva cálculos pesados que hacen que se pierda el ~ --
interés esencial del método SIMPLEX que permite evitar calcular derivadas de la función de ajuste. --
El programa CALGARY emplea un método mas pragmático de evaluación de las incertidumbres. Se trata de un método de Monte Carlo consistente en generar varios espectros "experimentales" (los cuales son fluctuaciones poissonianas del espectro experimental original) y en observar la incidencia de estas -fluctuaciones sobre los parámetros de ajuste. Se hace notar que esta aprobciiación da cuenta de la varianza de los parámetros de ajuste relacionada con las fluctuaciones estadisticas del espectro experimental original y no considera la varianza relacionada con
la calidad del ajuste o del modelo. Entonces este método se debe
123

2 utilizar solo con espectros de buena estadistica cuyo valor de X
sea cercano de 1 .
VI1 I. Resu1 tados
El espectro experimental, obtenido por medio de un analizador multicanal, generalmente es el resultado del arreglo de amplitudes de pulsos eléctricos provenientes de un detector, a través de un sistema de tratamiento de señales. La forma espectral de los pulsos es el resultado, por una parte de fenómenos físicos intrínsecos al proceso observado (la duración de la vida de estados exitados, etc.) y por otra parte, de efectos debidos a la transferencia de la información ( 1a.ventana de entrada del detector, etc ... ) y del funcionamiento no ideal de cada componente del sistema instrumental de medida. El resultado de estos procesos aparece como una combinación lineal de los procesos elementales, convolucionados por el funcionamiento instrumental de cada elemento del sistema de medida.. Siendo los procesos elementales los de interés, la operación de "deconvolución de un espectro" se hace necesaria para extraer del espectro total las contribuciones debidas a los procesos elementales (picos, energías, continuo, etc. 1, con el fin de deducir la información que proviene de la fuente de radiación observada tal como la composición isotópica o los datos nucleares (probabilidades de emisión, etc ... I . En general, la operación de deconvolución del espectro no es un problema con una solución unívoca pues la deformación de la señal original debida al instrumento de medida conduce a una pérdida de información. La restauración de la señal se logra introduciendo hipótesis, restricciones o modelos sobre la forma del fenómeno observado lo que permite, hasta cierto límite, paliar la pérdida de información.
124

El método generalmente consiste en definir un modelo del fenómeno observado y aJustar los parámetros para que el modelo sea "semejante" lo más posible al espectro experimental, por lo que la operación es más un ajuste que una deconvolución del espectro. Entonces, el término deconvolución de espectros es impropio y la operación que se realiza sería más bien de sumación de espectros.
VIII. 2.1. Espectrometría alfa
En el análisis cualitativo y cuantitativo para el cálculo del rendimiento de depósito de los espectros se emplearon programas de computadora disponibles comercialmente, como es el caso del
~ programa Interalpha de Intertechnique. El análisis cuantitativo de precisión se llevó a cabo empleando varios programas no comerciales elaborados en el Departamento de Física de la Universidad de Calgary por el Dr. John Bland [611.
Dadas sus características definidas previamente, proporcionaron resultados muy precisos incluso tomando en cuenta la baja estadística de conteo en el caso del Am-241.
El programa FILECONV permitió convertir los archivos con extensión .CHN provenientes del programa comercial, a un formato predefinido con la extensión .DAT y el programa MONACO (subrutina
del principal SMENU) permitió seleccionar la región de interés del espectro correspondiente a la de los picos 01 sujetos de estudio. El programa MONACO genera 2 archivos diferentes: el primero es un archivo con extensión .Exp donde se encuentra grabada la zona de interés del espectro experimental. Considerando que los espectros responden a una estadística de tipo Poisson y que el espectro experimental es una variación de la función ajustada, se puede generar la función de ajuste del espectro, de esta manera se genera otra región de interés
- -
--
12s

producida por la función de ajuste y que se registra con la extensión .VAR. Ambos archivos .EXP y .VAR pueden ser analizados para calcular los errores asociados a los resultados de amplitudes y areas de picos alfa. El programa ALBION3 ajusta la función a los valores experimentales y a través del análisis de la función determina la posición, la amplitud de los picos identificados y las correspondientes areas, todo asociado a un valor de X reducida. Los datos de la o' de la gaussiana del modelo, las constantes de las exponencialec, la proporción de la primera exponencial y el ''fraction step", así COW las posiciones y amplitudes de los picos son utilizados por el programa R A Y W para obtener el gráfico del espectro experimental, de la función ajustada, de cada pico por separado y la historia de los residuos con la sura
total de los mismos residuos. Finalmente, el programa üLTRIX permite calcular las intensidades relativas de emisión partiendo de los parámetros de la función ajustada y las posiciones de los picos. Además calcula la varianza de las intensidades relativas. Ambas operaciones las puede realizar con el espectro obtenido como una variación de Poisson del espectro original.
2
-
VIII.2.1.1. Americio-241 El espectro experimental de la figura VIII.2 corresponde a una
fuente elaborada con el &todo desarrollado. En el espectro se
presentan cinco picos correspondientes a las energías de mayor intensidad de emisión, Tabla VIII.l.
126
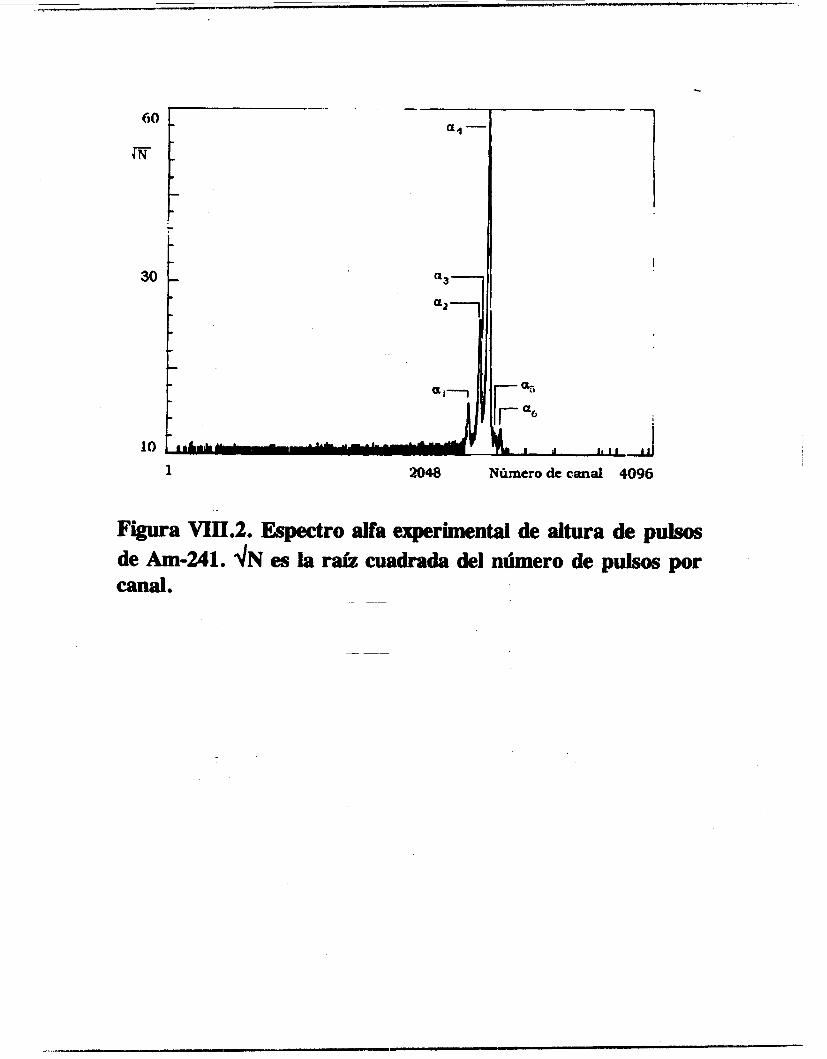
60
m
30 I
10 J 1 2048 Número de canal 4096
Figura VIII.2. Espectro alfa experimental de altura de pulsos . de Am-241. qN es la raíz cuadrada del número de pulsos por cand.
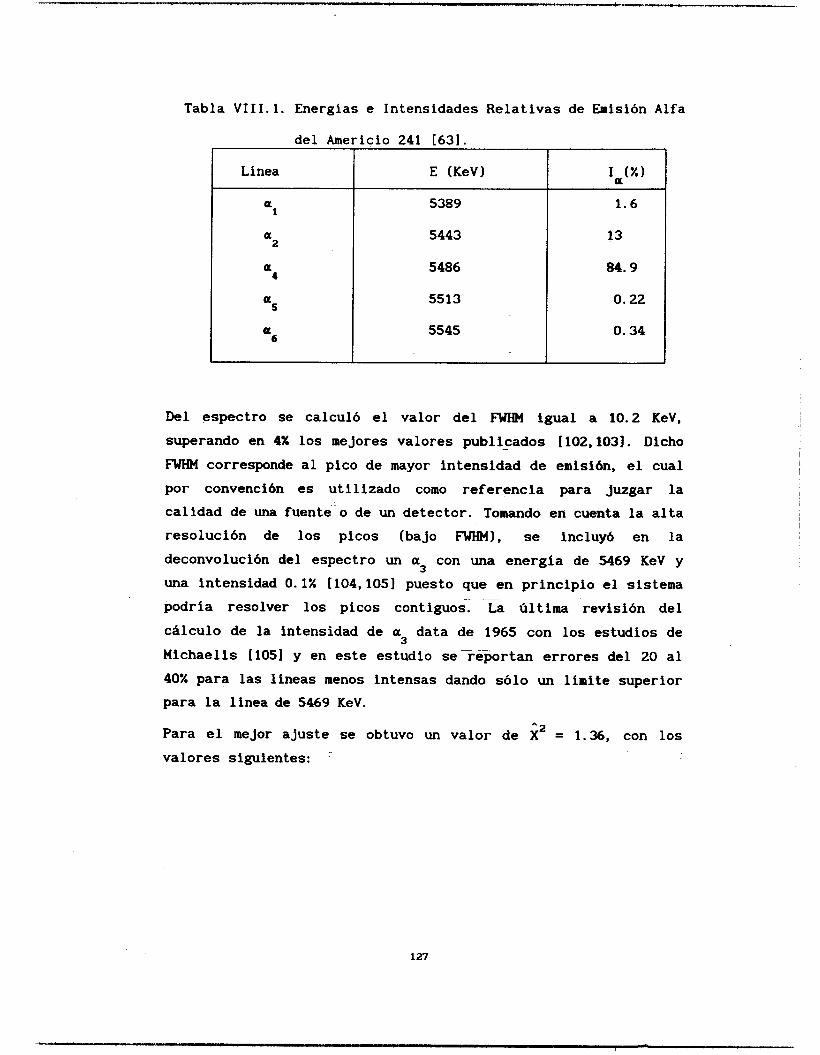
Tabla VIII.l. Energías e Intensidades Relativas de Emisión Alfa
5486
5513
5545
del Americio 241 [631.
84.9
o. 22
o. 34
Línea
a
a
a
a
a
1
2
4
s
6
E (KeV) I (XI a
5389 I 1.6
5443 13
Del espectro se calculó el valor del FWHM igual a 10.2 KeV, superando en 4% los mejores valores publicados - [102,1031. Dicho FWHM corresponde al pico de mayor intensidad de emisión, el cual por convención es utilizado como referencia para juzgar la calidad de una fuente-o de un detector. Tomando en cuenta la alta resolución de los picos (bajo FHM), se incluyó en la deconvolución del espectro un a con una energía de 5469 KeV y
una intensidad 0.1% [104,1051 puesto que en principio el sistema podría resolver los picos contiguos. La última revisión del cálculo de la intensidad de a3 data de 1965 con los estudios de Michaelis [lo51 y en este estudio sereportan errores del 20 al 40% para las lineas menos intensas dando sólo un límite superior para la linea de 5469 KeV.
Para el mejor ajuste se obtuvo un valor de #" = 1.36, con los valores siguientes:
3
__
127

Sigma de la gaussiana, = 3.57 Kev Constante de la primera exponencial, = 10.74 KeV-' Constante de la segunda exponencial, = 47.95 KeV-' Proporción de la primera exponencial, = 0.913
Fraction Step = 4.15 x lo-'
Grados de libertad, u = 334
En la figura VII I .3 aparecen los 6 picos del espectro original y
superpuesta la función ajustada según los parámetros. La función se ajusta bien a los 6 picos del Am-241, sin embargo el pico número 5 (en el espectro original) se encuentra menos definido que en el número 6 a pesar de que las intensidades son de magnitud similar. Como resultado de esto, la función no se ajusta
bien en esa región del espectro, lo cual puede ser aejor observado en la figura V I I I . 4 donde aparecen la función, los
picos convolucionados y en la parte inferior los residuos (diferencias entre Yexp-Ycal). En la región del espectro entre el pico 4 y el 5 aparecen valores de los residuos con una distribución no aleatoria, es decir que los residuos definen un pico. A pesar de lo expuesto se calcularon las probabilidades de emisión de las 6 líneas de Am-241 proporcionando valores que concuerdan con los publicados [63,106,1071 los cuales -recen en la tabla VIII .2 .
128
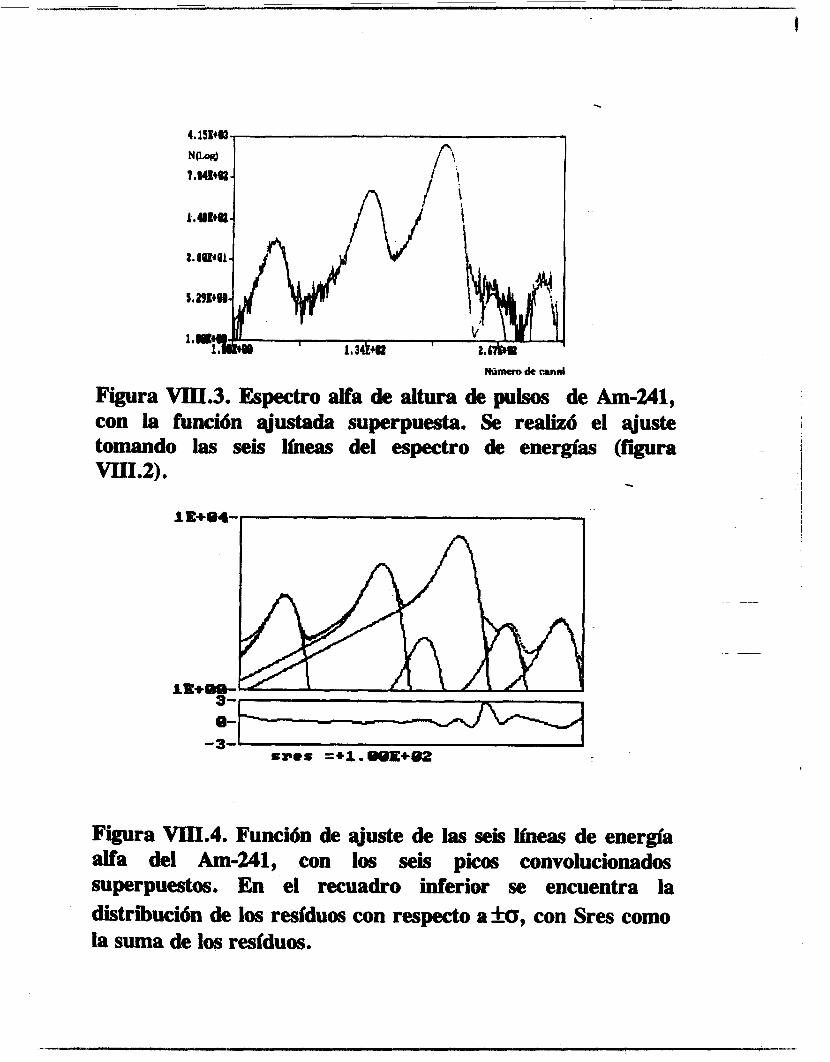
f
.
Numero ¿e n n d
Figura VIII.3. Espectro aifa Cae altura de pulsos de Am-241, con la función qjustada superpuesta. Se realizó el ajuste tomando las seis lineas del espectro de energí" (figura vIII.2) .
lEI@I-(
3-
e-- - -3-2
sllras =+l. oopi+@2
Figura VIII.4. Función de ajuste de las seis líneas de energía aifa del Am-241, con los seis picos convolucionados superpuestos. En el recuadro inferior se encuentra la distribución de los residuos con respecto a-, con Sres como la suma de los resíduos.
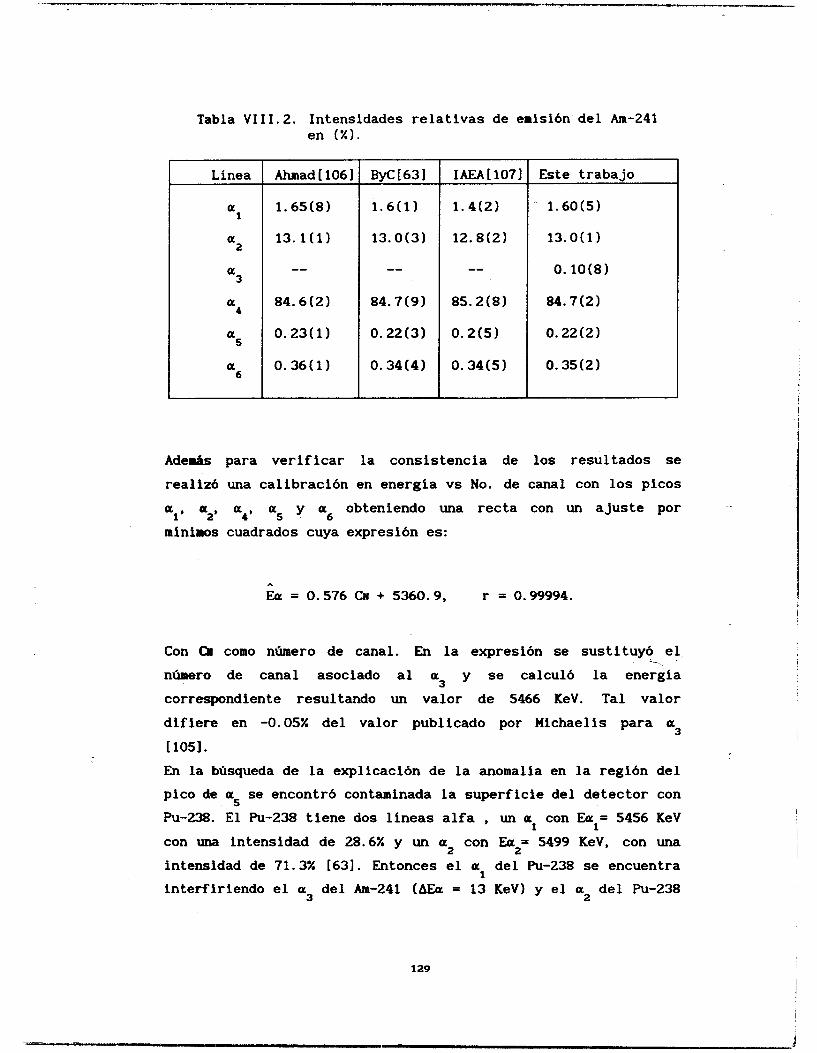
Tabla VIII.2. Intensidades relativas de emisión del Am-241 en ( X I .
Línea
a
a
a
a
a
a
1
2
3
4
5
6
Ahmad[1061
1.65(8)
13.1(1)
--
84.6(2)
0.23(1)
0.36(1)
ByC[631
1.6(1)
13.0(3)
-- 84.7(9)
O. 22(3)
O. 34(4)
IAF.A[ 1071 Este trabajo
1.60(5)
13.0(1)
O. lO(8
84.7(2)
0.22(2)
0.35(2)
Aden& para verificar la consistencia de los resultados se realizó una calibración en energía vs No. de canal con los picos a Q a a y a6 obteniendo una recta con un ajuste por minims cuadrados cuya expresión es:
1’ 2’ 4’ 5
L.
Ea = 0.576 CN + 5360.9. r = 0.99994.
Con Ur como número de canal. En la expresión se sustituyó el número de canal asociado al a y se calculó la energía correspondiente resultando un valor de 5466 KeV. Tal valor difiere en -0.05% del valor publicado por Michaelis para a3
[ 1051.
En la búsqueda de la explicación de la anomalia en la región del pico de a se encontró contaminada la superficie del detector con Pu-238. El Ri-238 tiene dos líneas alfa , un a con EOr = 5456 KeV con una intensidad de 28.6% y un a con Eh2= 5499 KeV, con una intensidad de 71.3% [631. Entonces el ai del Pu-238 se encuentra interfiriendo el a3 del Am-241 ( E a = 13 KeV) y el a2 del hi-238
-~
3
5
1 1
2
129
i

el a5 del Am-241 (AEa = 13 KeV). En el siguiente análisis espectral se consideró la presencia del a2 del Plutonio, con los siguientes parámetros se obtuvo un valor
de i2= 1.09:
Sigma de la gaussiana = 3.52 KeV
Constante de la primera exponencial = 10.70 KeV-'
Constante de la segunda exponencial = 43.84 KeV-'
Proporción de la primera exponencial = 0.903
Fraction Step = 4.15 x lo-' Grados de libertad, u = 334
La figura VIII.5 muestra el gráfico de la función resultante
superpuesto al espectro experimental. Se observa que en el flanco izquierdo del a5 del Am-241 la función se aproxima más a la forma
del espectro experimental. El flanco derecho del mismo as también
muestra alguna diferencia con respecto a la función. Esto podría
explicarse si se toma en cuenta que el Pu-238 emite electrones de
conversión en una banda de energias de 21 a 26 KeV [lo81 y que se
presentaría el efecto de sumación entre el a2 (Ea = 5499 KeV) del
Pu-238 y los electrones de conversión, lo que proporcionaría
eventos detectados en la región de 5520 a 5525 KeV localizada precisamente en el flanco derecho del a5 (Ea = 5513 KeV) del
Am-241 y antes del a6 (Ea = 5545 KeV) del Americio. Esta región
del espectro no podría ser convolucionada debido a que la energía
de los electrones no es discreta sino un continuo proporcionando
un pico suma con características diferentes a los valores de los
parámetros de los picos alfa.
La figura VIII.6 es un gráfico donde aparecen superpuestos, la
función ajustada, los picos convolucionados y el espectro
original. En la parte inferior se encuentra la distribución de
los residuos con la suma total Sres = 60. Se observa una vez más
que la función se aproxima de iejor manera al espectro, con los
130
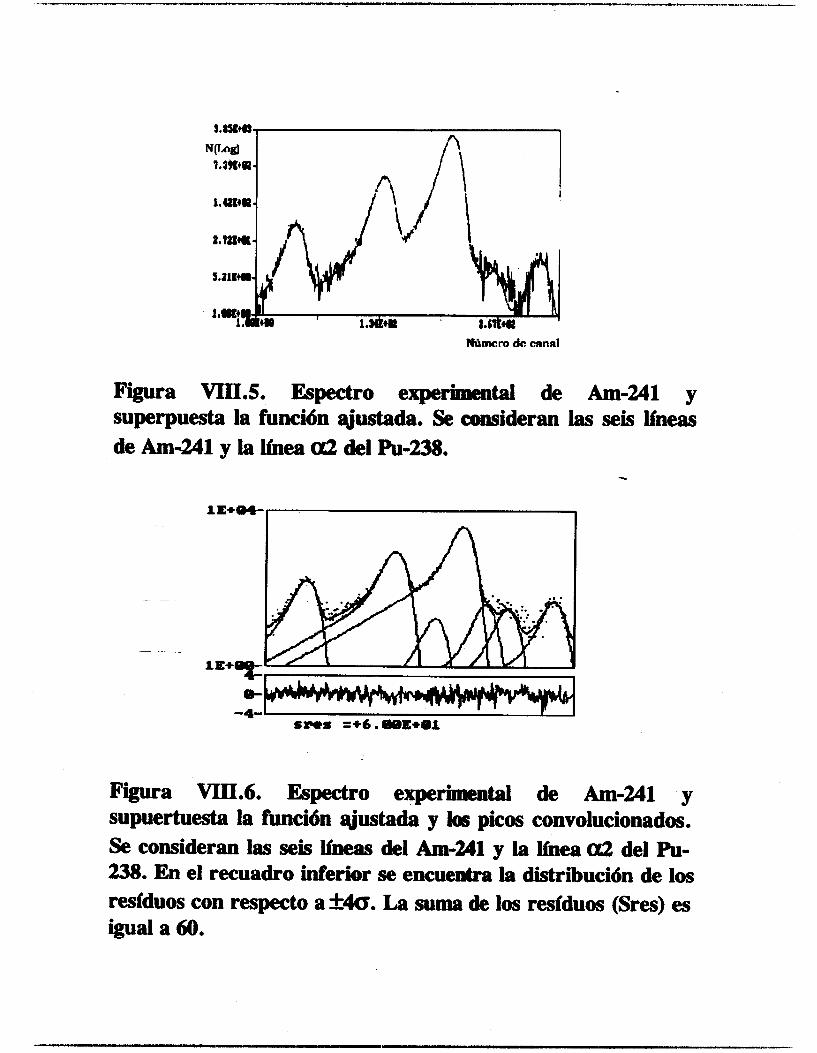
Figura VIII.5. Espectro experimental de Am441 y superpuesta la función ajustada. Se consideran las seis lineas de Am441 y la b e a a2 del Pu-238.
Figura VIII.6. Espectro experimental de Am-241 y supuertuesta la función ajustada y k~ picos convolucionados. Se consideran las seis limieas del Am441 y la heaa2 del Pu- 238. En el recuadro inferier se encuentra la distribución de los residuos con respecto a&&. La suma de los residuos (Sres) es igual a 60.

residuos por canal distribuidos de manera aleatoria alrededor del valor cero. Con los parámetros de ajuste obtenidos se calcularon las intensidades relativas de emisión y las desviaciones asociadas, las cuales se resumen en la Tabla VIII.3.
Tabla VIII.3. Intensidades relativas de emisión del Am-241
1 Línea a
a
a
a
a -
a
1
2
3
4
5
6
la ( X I
1.61 I 0.05 X
13.06 I 0.13 X
0.09 I 0.08 X
84.79 I 0.16 X
0.22 I 0.02 X
0.23 f 0.02 X
Los valores de intensidades de la Tabla VIII.3 son consistentes con los valores reportados [63,106,1071 excepto para la intensidad del a6 donde hay una diferencia del orden del 50%. Se revisaron los valores de intensidad calculándolos con el espectro de Am-241 generadocomo una variación de Poisson del espectro experimental y efectivamente vuelve a aparecer la misma diferencia para el a
En un tercer análisis de convolución de espectros se tomó en - cuenta la presencia de los picos a y a del Pu-238 y el pico
suma del a más electrones de conversión. Se convolucionaron 9 picos en total resultando un muy buen ajuste con un valor de x" =
1.045, los valores finales de los parámetros de ajuste se presentan en la Tabla VIII.4.
-- ~
6'
1 2
2
131

Tabla VIII.4. Valores de los parametros de ajuste para seis
líneas de Am-241 y tres de Pu-238
Sigma de la gaussiana = 3.55 KeV
Constante de la primera exponencial = 10.64 KeV-'
Constante de la segunda exponencíal = 43.57 KeV-'
Proporción de la primera exponencial = 0.902
Fraction Step = 4.179 x lo-'
Grados de libertad = 334
En la figura VIII.7 se encuentran superpuestos el espectro experimental y la función ajustada. Se puede observar que en este caso la función ajustada concuerda mejor con el espectro original incluyendo la región cercana al pico a del Am-241. Y en la figura VIII.8 aparecen el espectro, la función y los picos convolucionados, donde se observa la distribución aleatoria de los residuos en todo el espectro. Como un control adicional sobre el ajuste, con los picos a del Am-241 y los 3 del hi-238, se reand-una calibración en energía con los 5 picos más intensos del Am-241 y con los datos de calibración se calculó la energíatk4es picos señalados a partir de su posición en el espectro. Se obtuvo un coeficiente de correlación de la calibración de 0.9999 y los valores de energía de los picos asociados a las particulas alfa y a la suma alfa +
electrón de conversión presentan una desviación máxima de 0.04% con respecto a los reportados en la literatura [63,65,106,1071. Con los valores de los parámetros se calcularon los valores de intensidades de emisión relativas, considerando la presencia del hi-238 incluido el pico suma. En la Tabla VIII. 5 se enlistan los valores de intensidades medidas y para comparación se agregan los
5
3
132
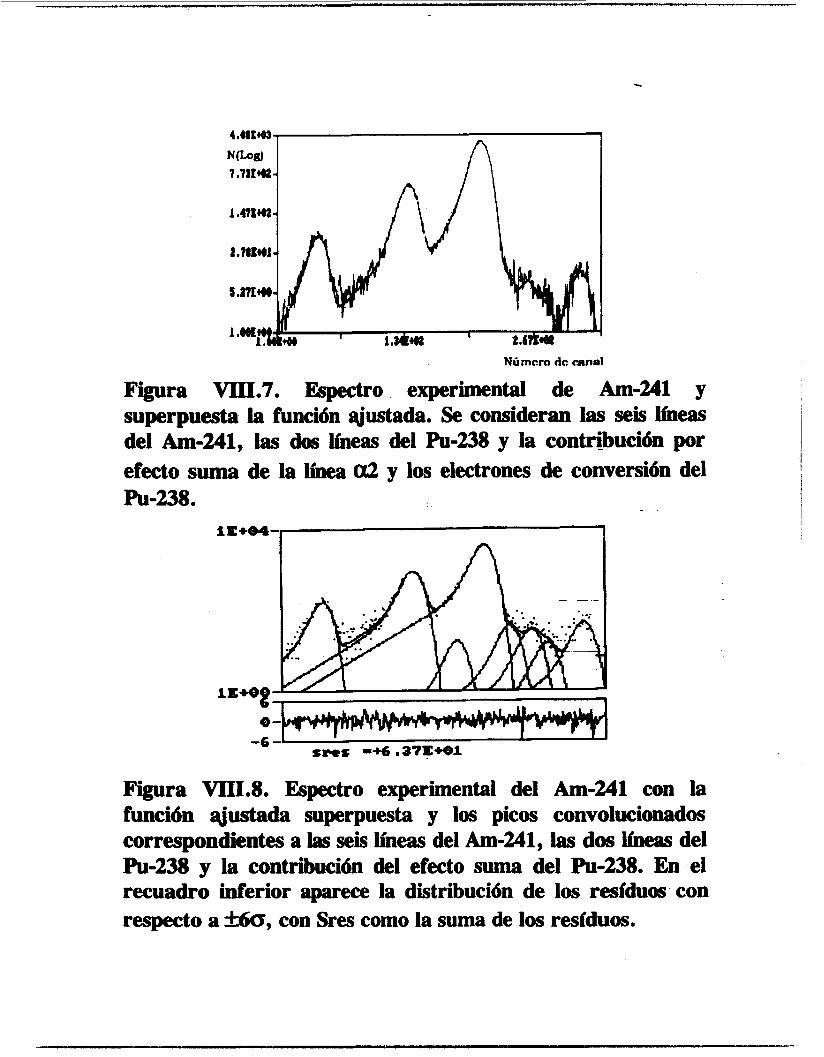
Nümcrn dc mnrrl
Figura VIII.7. Espectro experimental de Am-241 y superpuesta la función s@&ada. Se consideran las seis beas del Am-241, las dos beas del Pu-238 y la contribución por efecto suma de la lúiea a2 y los electrones de conversión del Pu-238.
a=+--
A
Figura VIII.8. Espectro experimental del Am-241 con la función ajustada superpuesta y los picos convolucionados correspondientes a las seis líneas del Am-241, las dos líneas del Pu-238 y la contribución del efecto suma del Pu-238. En el recuadro inferior aparece la distribución de los redduos con respecto a *, con Sres como la suma de los residuos.
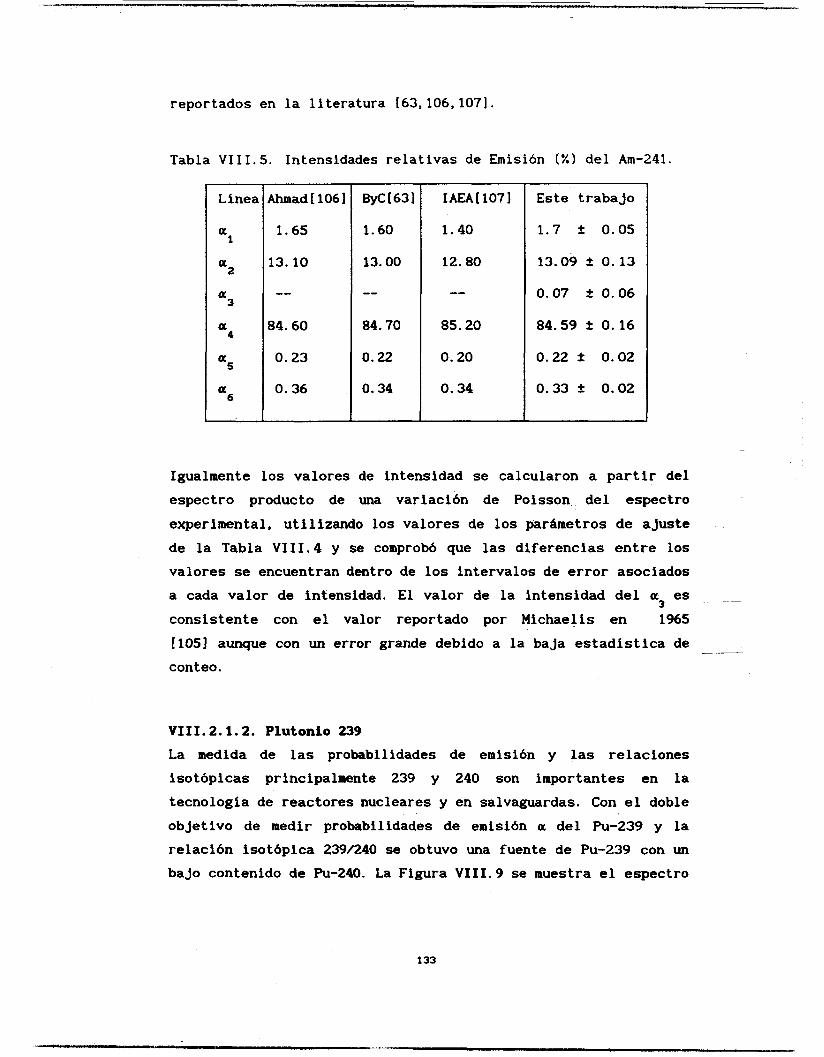
reportados en la literatura [63,106,1071.
Tabla VI I I .5 . Intensidades relativas de Emisión ( X I del Am-241.
Línea
a
a
a
a
a
a
1
2
3
4
5
6
Ahmad 1061
1.65
13.10
-- 84.60
O. 23
O. 36
ByCi631
1.60
13.00
-- 84.70
o. 22 O. 34
IAEA [ 107 1
1.40
12.80
-- as. 20 o. 20 o. 34
Este trabajo
1.7 f 0.05
13.09 I O. 13
0.07 f 0.06
84.59 t O. 16
0.22 2 0.02
0.33 f 0.02
Igualmente los valores de intensidad se calcularon a partir del
espectro producto de una variación de Poisson del espectro
experimental, utilizando los valores de los parámetros de ajuste
de la Tabla VIII.4 y se comprobó que las diferencias entre los
valores se encuentran dentro de los intervalos de error asociados
a cada valor de intensidad. El valor de la intensidad del a3 es
consistente con el valor reportado por Michaelis en 1965
[lo51 aunque con un error grande debido a la baja estadistica de conteo.
-~
_-
VIII. 2.1.2. Plutonio 239 La medida de las probabilidades de emisión y las relaciones
isotópicas principalmente 239 y 240 son importantes en la
tecnología de reactores nucleares y en salvaguardas. Con el doble
objetivo de medir probabilidades de emisión a del Pu-239 y la
relación isotópica 239/240 se obtuvo una fuente de Pu-239 con un bajo contenido de Pu-240. La Figura VIII.9 se muestra el espectro
133

experimental de una fuente de plutonio elaborada según el método anteriormente descrito. El espectro se obtuvo en condiciones e instalaciones idénticas a las del Am-241, con un ángulo sólido de 0.51% de 4ir se obtuvo un FWHM = 10.5 KeV y una calibración en energía de 0.58 KeV/canal. Dicho espectro fue utilizado para convolucionar los picos correspondientes al Pu-239 y al Pu-240, los parámetros de ajuste se enlistan en la Tabla VIII.6, los cuales proporcionaron un valor de 2 = 1.18.
Tabla VIII.6. Parámetros de ajuste del espectro alfa
del Pu-239/240.
Sigma de la gaussiana = 3.60 KeV
Constante de la primera exponencial = 3.61 KeV-'
Constante de la segunda exponencial = 13.80 KeV-'
Proporción de la primera exponencial = 0.88
Fraction Step = 0.0
Grados de libertad = 213
Con un procedimiento análogo al del Am-241 se determinaron las probabilidades de emisión, las cuales son comparadas con las citadas en la literatura 11071 en la Tabla VIII.7. El espectro experimental superpuesto y la función ajustada se muestran en la Figura VIII.10. Se observa que la función se ajusta al espectro en todo el intervalo de energías con una distribución aleatoria de los residuos alrededor del cero, sin mostrar tendencias anóma 1 as.
134
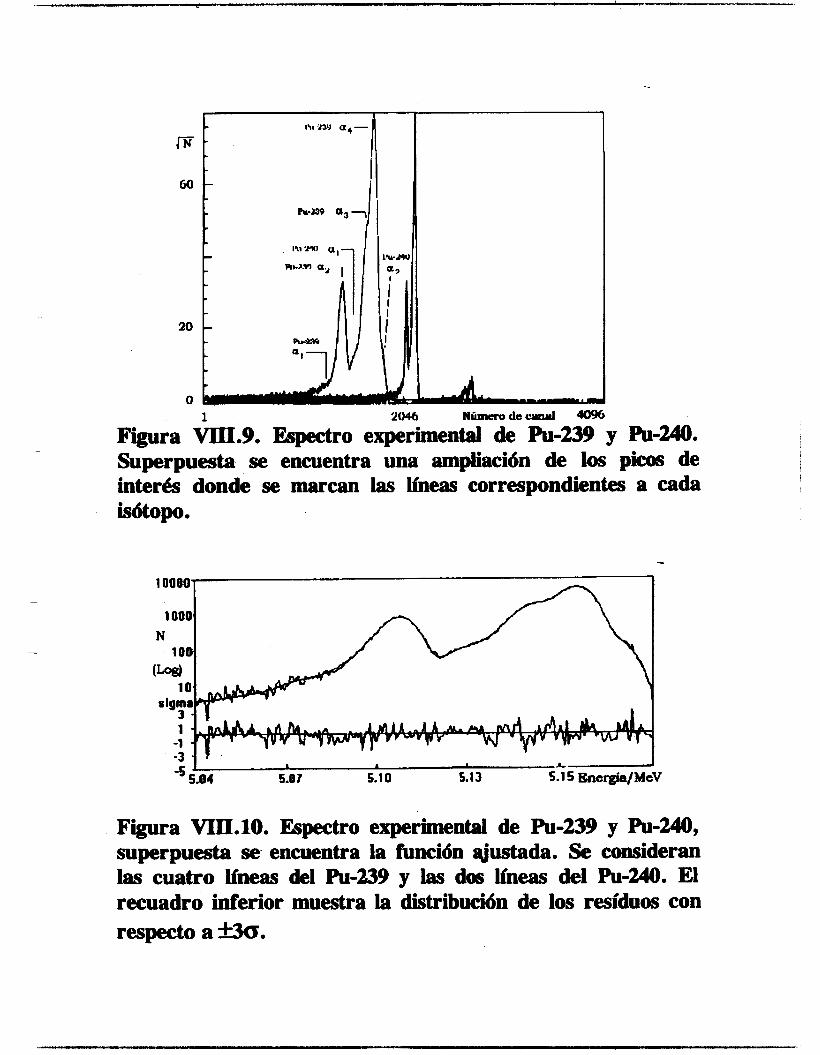
IN
Go
20
O 1 204a Núxnexv&cd 4096
Figura VIII.9, Espectro experimental de Pu-239 y h-244h Superpuesta se encuentra una ampliación de los picos de interés donde se marcan las líneas correspondientes a cada iS6tOpO.
'""""1 I
- 3 4 t - I a 1 .L I
-5 :.o4 5.0 7 5.10 5.13 5.1 5 Energia/MeV
Figura VIII.10. Espectro experimental de Pu-239 y Pu-240, superpuesta se- encuentra la función ajustada. Se consideran las cuatro heas del h-239 y las dos líneas del Pu-240. El recuadro inferior muestra la distribución de los resíduos con respecto a &3a.
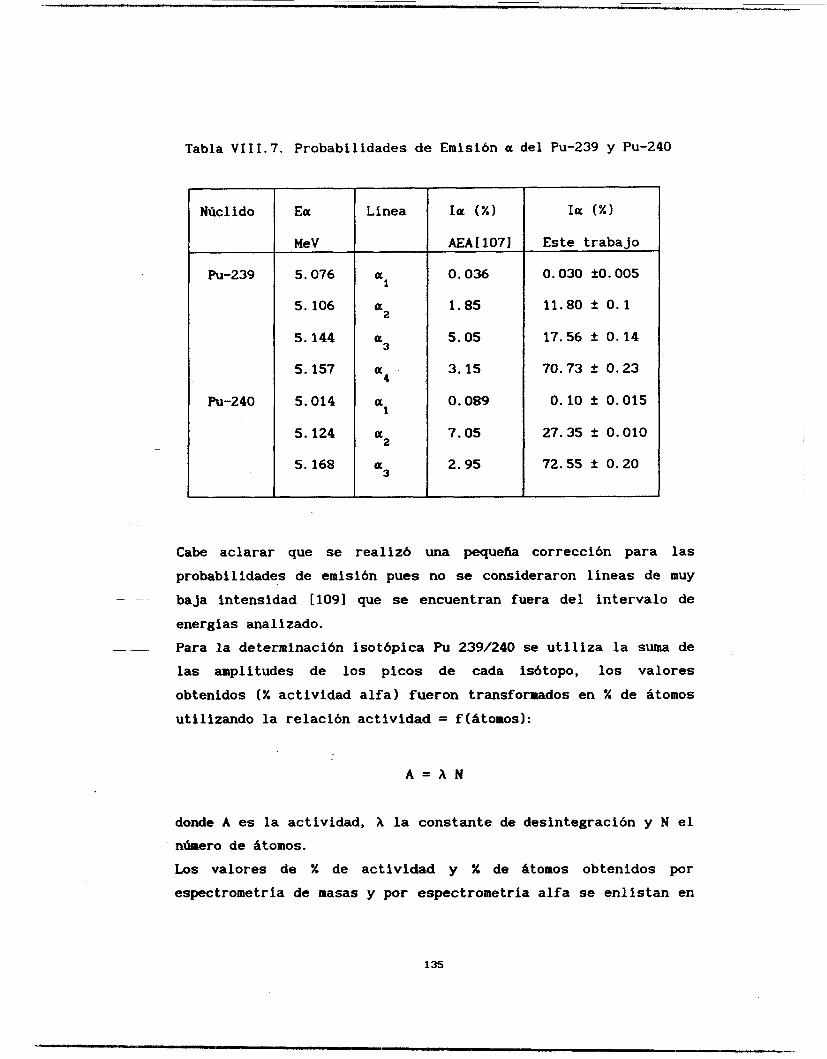
Tabla VIII.7. Probabilidades de Emisión a del Pu-239 y Pu-240
a 2
a 3
Núcl ido
1.85
5. o5
Pu-239
Pu-240
Ea
MeV
5.076
5.106
5.144
5.157
5.014
5.124
5.168
Línea Ia ( X I
O. 036
a 4
I a 1
I 7.05 a 2
I 2-95 a 3
Ia ( X I
Este trabajo
O. 030 fO.005
11.80 f 0.1
17.56 f O. 14
70.73 f 0.23
o. 10 f o. o15 27.35 f 0.010
72.55 f 0.20
Cabe aclarar que se realizó una pequeña corrección para las
probabilidades de emisión pues no se consideraron líneas de muy
~ _ _ baja intensidad [lo91 que se encuentran fuera del intervalo de
energías analizado.
-__ Para la determinación isotópica Pu 239/240 se utiliza la suma de las amplitudes de los picos de cada isótopo, los valores
obtenidos (X actividad alfa) fueron transformados en % de átomos
utilizando la relación actividad = f(átomos1:
A = A N
donde A es la actividad, A la constante de desintegración y N el número de átomos. Los valores de X de actividad y X de átomos obtenidos por
espectrometría de masas y por espectrometría alfa se enlistan en
135
la Tabla VIII. 8.
Tabla. VIII.8. Composición isotópica del espectro de Pu-2391240
Espectrometría de Masas Espectrometría alfa
(referencia)
Núclido X átomos Pu 239/240 X actividad X átomos Pu 2391240
Pu-239
Pu-240
Como se puede observar de la Tabla VIII . 8, la relación isotópica Pu 2391240 obtenida por Espectrometría alfa difiere en +0.22% del valor de referencia. Esta primera experiencia revela la
-
99.1 111.60
o. 888
posibilidad de emplear la espectrometría alfa (con los conceptos desarrollados en este trabajo) como método de control en la pureza isotópica del material de plutonio que se emplea en la elaboración de combustible nuclear mixto para los reactores de generación áe-eñergía eléctrica. El presente estudio de espectrometría pone en primer plano la calidad debeespectros obtenidos con resoluciones WWHM) que se encuentran limitadas por la calidad del sistema de detección y el "software" de análisis espectral empleado, el cual demostró ser el mejor actualmente desarrollado y citado en la literatura.
96.79
3.18
VIII.2.2. Espectrometría X y Gatma
Ambas espectrometrías permitieron determinar los rendimientos de depósito para el caso del samario y del Eu-152 respectivamente. El "software" convencional disponible comercialmente es adecuado para tal objetivo no requiriéndose de software especializado para
99. o9 111.84
O. 886
136

el análisis cuantitativo
IV. 2.2.1. Espectrometría X Uno de los métodos de análisis cualitativo y cuantitativo no
destructivo, para elementos no radiactivos, es la fluorescencia
de rayos X [1101. Los rayos X de fluorescencia del Samario se obtuvieron con una fuente excitadora de An-241. Se obtuvieron los
espectros de Samario con el analizador multicanal calibrado a
0.05 KeVIcanal. En la figura VIII.ll se muestra un espectro X del
Samar io. Las líneas de fluorescencia Koc y K del disco de platino que
contiene el depósito de samario no aparecen pues con energías B
promedio de 65.5
encuentran dentro contrario, los
excitadora) están
KeV y 76.9 KeV ill11 respectivamente, no se
del intervalo de energías de interés. Por el
picos correspondientes al Am-241 (fuente
distribuidos en el intervalo de interés, por -
ejemplo el pico de 43.4 KeV y los más intensos correspondientes a
picos de escape [1121. En el espectro aparecen los 2 picos más importantes (más intensos) del Samario; el de 39.58 KeV de energía promedio (conjunto de energías agrupadas en un solo pico)
con una intensidad relativa de 73.9% y el de 45.9 KeV con una
intensidad relativa de 17.3% [ill] . Ambos picos del Americio fueron utilizados para obtener los valores de rendimiento de
depósito integrando áreas-jo los picos empleando el software
convencional de la Intertechnique.
IV. 3.2.2. Espectrometría Gama
La medida del Eu-152 en el cálculo del rendimiento se realizó por
espectrometría G a m a pues se trata de un núclido emisor beta
gama. De la misma manera empleando el software convencional de
Intertechnique se calcularon las áreas de los fotopicos empleadas
en el cálculo de la actividad radiactiva. La Figura VIII. 12 muestra el espectro gamma del Eu-152 con todas sus líneas puesto
137
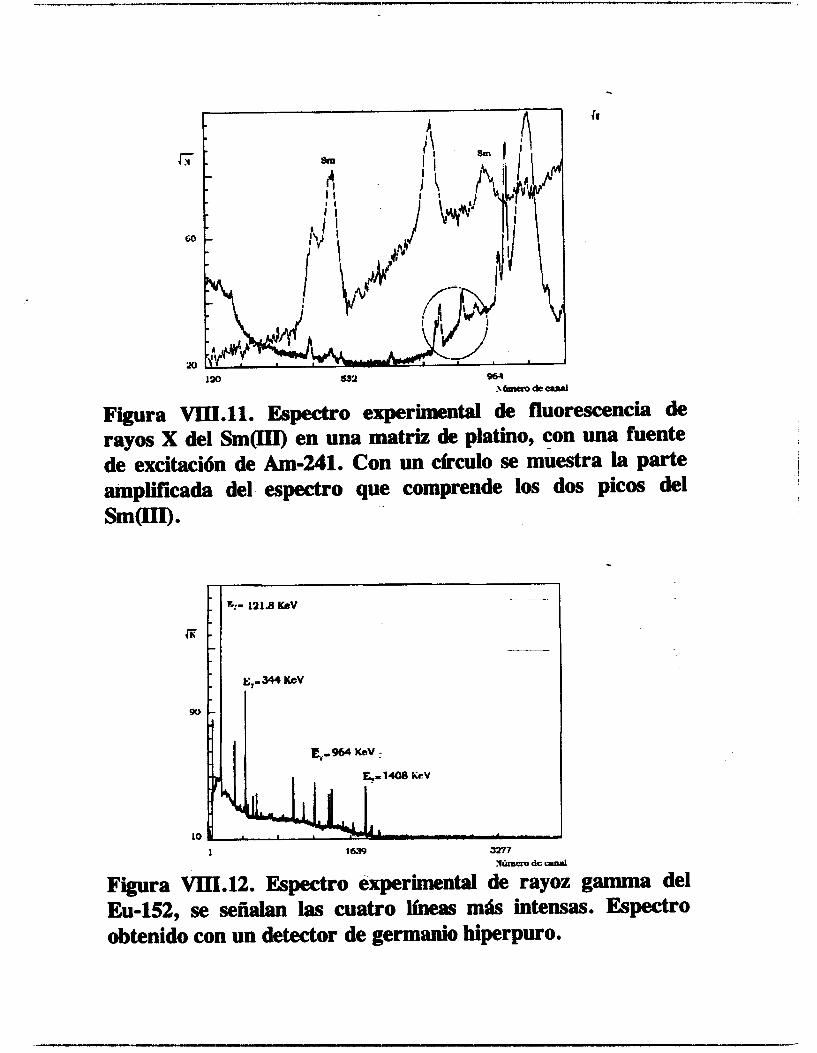
M
K -
120
~- ir.- l2lSKeV
- ___
E,- 344 Kev
JSZ
Figura VIII.11. Etspectro experimental de fluorescencia de rayos X del S r n O en una matriz de platino, con una fuente de excitación de Am-241. Con un circulo se mÜestra la parte amplificada del espectro que comprende los dos picos del Sm(III).

que es un emisor multigamma. En el espectro aparecen los fotopicos, el fondo natural, el fondo Compton, el pico de aniquilación de pares (electrón-positrón), picos de escape simple y doble. Se seleccionaron 5 fotopicos para el análisis cuantitativo, en función de su intensidad de emisión, de su distribución en el espectro y de la no interferencia de otros
picos. En la Tabla VIII.9 se enumeran las energías seleccionadas y sus respectivas intensidades relativas.
Tabla VIII. 9. Energías gamma del Eu-152 11081. !
E (Kev) r 121.8
778.9
%3.5
1112.1
1408. O
I ( X I 7
28.4
12.9
14.6
13.5
20.8
138

En este estudio se determinó que durante la inmersión del platino en la solución de Sm(III), se adsorbe la especie aniónica Sm(CO 1- en la superficie de este. En el depósito de samario en la forma Sm(OH)3(s, sobre el platino, la especie adsorbida Sm(S0 1- reacciona con el hidrógeno adsorbido formando un enlace intermetálico Pt-Sm 1671, el cual electrocataliza la reacción de evolución de hidrógeno con la etapa de adsorción de hidrógeno de
manera irreversible. El proceso electrocatalítico tiene cuatro etapas involucrando las especies H+, Hadsorbfdo, Ci(cO,);, Sm-Pt y H2. El recubrimiento parcial del platino por Ci-Pt electrocataliza la reacción de evolución de hidrógeno y el recubrimiento total pasiva la superficie inhibiendo la evolución de hidrógeno, la reducción de oxígeno y la misma formación de los óxidos superficiales de
4 2
4 2
p 1 at ino . La continuación de los estudios para verificar la formación de las especies mencionadas (no han sido citadas en la literatura) y la formación del enlace intermetálico por inmersión del electrodo en solución de Sm(III1 abriría un campo nuevo para la investigación de nuevos procesos y para la electrocatálisis en sintésis orgánica, por ejemplo, recientemente Quiroz et al [comunicación personal, 19941 han utilizado un electrodo de platino en presencia de Sm(II1) para electrocatalizar la reacción de hidrogenación de la molécula de benceno. También se predijo en el sistema del americio la especie anjónica Am(S04)i, en el del plutonio la especie aniónica correspondiente no esta presente. El plutonio presenta una fisicoquímica complicada por el número de especies y los desplazamientos de equilibrios por presencia de especies de plutonio suceptibles de reaccionar entre si (generalmente el plutonio existe en los
139

estados de oxidación IV,VI y 111). También es preciso considerar la dispersión de los datos de constantes de equilibrio y de solubilidad que se encuentran en la literatura. Por ejemplo los datos de solubilidad de la especie PU(OH),(~) no son
consistentes. Los datos de Pazukhin [821 son los que proporcionan diagramas con las predicciones de condiciones de depósito más congruentes con los resultados experimentales. Es conveniente continuar actualizando los datos de estas constantes con los publicados incluyendo los reportes de nuevas especies. Por otro lado, el comportamiento de samario, europio, americio y plutonio es similar en el proceso de depósito electroquimico por formación de los correspondientes hidróxidos insolubles. Los datos aportados por los diagramas (PZD,PED y PTD) de samrio, por el estudio electroquímico de y por el modelo de Hansen [151, han permitido optimizar de manera consistente el método de depósito uniforme (t 10x1 de americio, plutonio y europio con rendimientos de depósito de 97.5% 2 2.5%. Dichos depósitos han mostrado ser adherentes y estables sin haber pérdida de actividad notable. La aplicación directa del modelo de Hansen para predecir las condiciones de depósito de estos elementos (pH de l a solución y densidad de corriente) habría conducido a resultados inconsistentes como el mismo lo reporta 1161.
En este trabajo el modelo de Hansen fue empleado tomando las predicciones de los diagramas para las condiciones de aparición de la fase insoluble de hidróxido de los elementos correspondientes. Además, siendo los conceptos de capa límite de difusión, de régimen de difusión estacionario y de convección forzada elementos de base en su modelo, en este trabajo se empleó una celda electroquímica con un cátodo rotatorio diseñada en base a la teoría del electrodo disco rotatorio [SO], la cual contempla dichos conceptos. Las características de las películas depositadas han permitido obtener espectros de Am y de Pu/240Pu de alta resolución con un FHWM característico de 10.2 y 10.8 KeV
24 1 239
140
! -__I_- -

respectivamente. Estos valores de resolución indican que las características de las películas satisfacen los criterios de la función analítica empleada en la convolución de los picos alfa, es decir que las películas son los suficientemente delagadas como para asumir que la pérdida de energía de las partículas al atravesar la película es despreciable y lo suficientemente uniformes en espesor como para asegurar que la asimetría del pico por variaciones en el espesor de la película es también despreciable. Los factores que afectan la forma del pico son la interacción de las partículas alfa con el detector y la resolución de la electrónica, mismos que son considerados en la función analítica con la doble exponencial para la asimetría y la gaussiana para los fenómenos estadisticos. Con la función analítica se calcularon las intensidades de los picos convolucionados, mismas que son proporcionales a las intensidades relativas de emisión.
Am Los valores de probabilidades de emisión alfa del calculados con rayor precisión en este trabajo son similares a los publicados [63, 104,105,1071. También se calculó la probabilidad de emisión de la línea or3 del 24'Am cuyo último estudio data de 1965 [1051. A pesar de la interferencia del Pu el valor calculado de la línea a3 se encuentra dentro del intervalo reportado [1051, aunque con un error grande. Dados los alcances de la metodología descrita en este trabajo es recomendable continuar con el estudio del americio 241 para tener mayor exactitud y precisión en el cálculo, aumentando la estadística de conteo y utilizando un detector de alta resolución libre de cualquier contaminación. Con respecto al 239pu/240PuD las intensidades relativas de emisión calculadas con mayor precisión en este trabajo son
similares a las publicadas [1071, excepto los valores de las líneas a3 y a4 del Pu. La diferencia de los valores calculados en este trabajo con los publicados podría deberse al fenómeno de suma de electrones de conversión con partículas alfa de
241
238
239
141

determinada línea durante la medición de la fuente. Los autores
reportan haber utilizado grandes geometrías de medición por lo
que la probabilidad de detectar la suma de electrones de
conversión con partículas alfa no es despreciable puesto que el
U originado por la desintegración del Pu emite electrones
de conversión los cuales pueden coincidir con las partículas alfa
de mayor intensidad de emisión. Cabe señalar que en este trabajo
se utilizaron pequeñas geometrías de medición. Se sugiere revisar
los valores de probabilidades de emisión de las líneas a3 y a4
del 239Pu. La espectrometría alfa como método de control de calidad en la
235 239
elaboración de combustible nuclear (control de la pureza
isotópica) para reactores de potencia, encontraría en la
metodología expuesta en el presente trabajo una de las mejores
alternativas publicadas. El método de espectrometría alfa es
menos costoso y sofisticado y con la calidad de los resultados
para el control de pureza isotópica de combustible nuclear
comparable a la que se obtiene por espectrometría de masas.
142

ANEXO 1
Verificación de l a ley de Levich con un cátodo rotatorio
inclinado a 15'
Durante la obtención de los depósitos, el cátodo rotatorio y la celda electroquímica (incluido el &nodo) permanecen paralelos e inclinados 15 con respecto a la normal de la base del aparato. La inclinación de ambos podría causar perturbaciones en el tipo de flujo de la solución hacia el cátodo y en la estabilidad de la capa límite de difusión. Por otro lado, uno de los criterios utilizados para verificar la presencia de un flujo laminar de solución y la estabilidad de la capa límite de difusión es la verificación de la ley de Levich [96l,que establece la relación lineal entre la corriente límite de difusión (ii,c) y la raíz cuadrada de la frecuencia de rotación del cátodo (o . La verificación de esta relación se llevó a cabo utilizando la reacción de reducción deCu2+ a Cuo en medio sulfato de sodio a pH = 5 con una concentración de cobre 2.5 x
Se utilizó un dispositivo convencional de 3 electrodos, un contraelectrodo de carbón vítreo en compartimiento separado, un electrodo de referencia de sulfato de mercurio saturado en compartimiento separado y un electrodo de trabajo de cobre (disco rotatorio) con un predepósito de Cobre insitu para estabilizar la superficie con una granulometría uniforme. Primeramente se obtuvo la curva respuesta en corriente aplicando un programa de barrido lineal de -0.410 V/Esn hasta - 1 . 1 V/ESM con una velocidad de barrido de 12 mV.s-'. La respuesta es una curva que corresponde a la corriente límite de difusión. Se seleccionó un potencial de -0.59 V/ESM como potencial de depósito
O
1/2 ]
M.
143
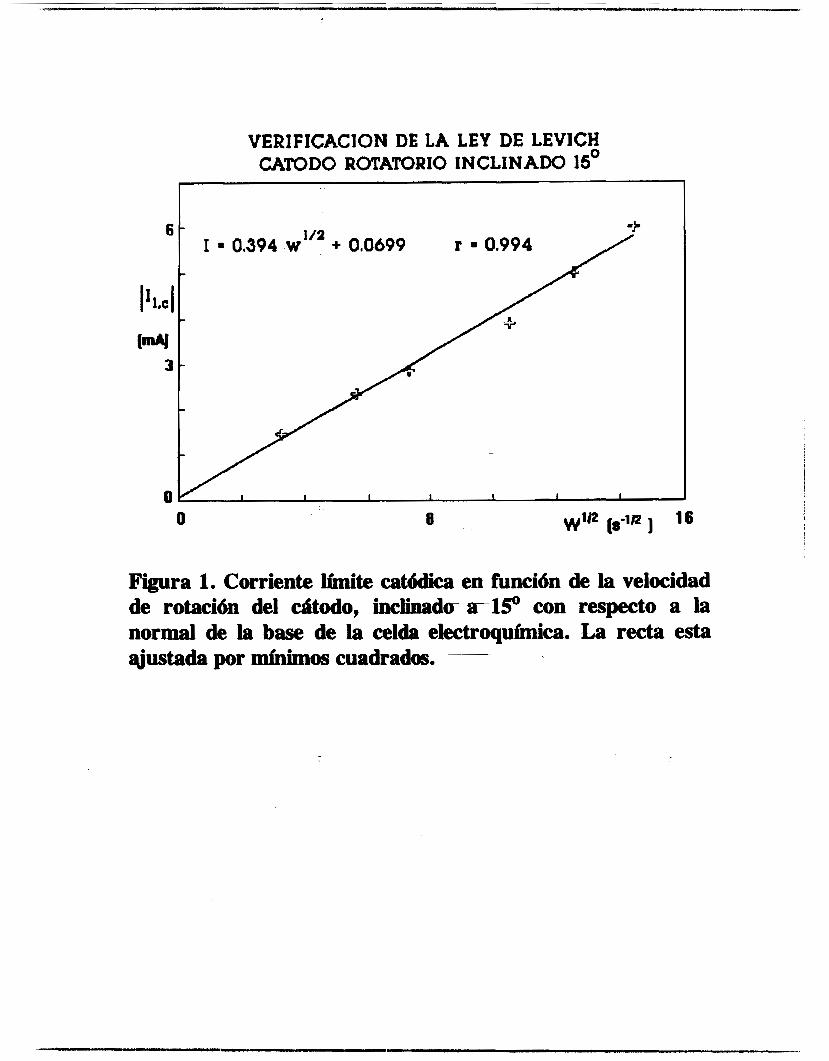
VERIFICACION DE LA LEY DE LEVICH CATODO ROTATORIO INCLINADO 15'
6
IW 3
o O 8 w'@ [s"" ] 16
Figura 1. Corriente límite catódica en función de la velocidad de rotación del cátodo, inclinadcr r15" con respecto a la normai de la base de la celda electroquíinica. La recta esta ajustada por mínimos cuadrah. -~

para obtener el valor de la corriente límite en función de la velocidad de barrido. Se obtuvieron valores de corriente limite para las velocidades de rotación de 100, 300, 500, 1000, 1500 y 2000 rpm. La velocidad de rotación fue convertida a la frecuencia de rotación w (o = 2nf) en s . Con estos datos se obtuvo el gráfico de I i , c = f(w 1 y por ajuste de mínimos cuadrados se obtuvo la pendiente (m), la ordenada al origen ( I i , ~ , o ) y el coeficiente de correlación r (figura 1 ) . El valor del coeficiente de correlación (0.994) muestra que ambas variables están correlacionadas de manera
-1
112
lineal como se deriva de la ecuación de Levich. Además, la ordenada al origen tiene un valor cercano a cero (diferente de cero probablemente debido a la contribución de la convección natural) correspondiendo una vez ds al tipo de ecuación de la ley de Levich. Estos resultados permitieron verificar que la inclinación de la celda y del cátodo a 15' no afecta el flujo
-
laminar de la solución hacia el cátodo manteniéndose estable el espesor de la capa límite de difusión.
144

ANEXO 2
Distribución de l a corriente.
Si se asume que la resistencia de una solución electrolítica es muy pequeña, entonces la densidad de corriente en la superficie del cátodo es uniforme, es decir, es independiente en el sentido radial del cátodo en forma de disco. Aunque en general es el caso
de los sistemas electroquímicos, la distribución real de la corriente depende de la resistencia de la solución, así como de la transferencia de carga y de masa. Newman [97,98,991 Albery y Hitchman [loo, 101,lOZl estudiando la distribución de la corriente la clasificaron de la siguiente manera : a) Distribución primaria de la corriente.- es aquélla que no toma en cuenta los componentes provenientes de la sobretensión de activación y de la sobretensión de concentración y esto a nivel de la superficie del cátodo. En este caso es posible admitir que en el cátodo tenemos una superficie equipotencial. En estas condiciones, para un disco de radio rl limitado en la periferia por un cuerpo aislante cuyo radio es grande con respecto a rl , la corriente eléctrica (líneas) se propagan en dirección normal a las superficies equipotenciales a condición de que el contraelectrodo se encuentre colocado en el infinito. La densidad de corriente no es uniforme en cada punto del disco, siendo mucho mayor en le periferia (r = r 1 con respecto al céntro del mismo (r = O ) .
Esto se explica por el hecho de que en la periferia del disco la
transferencia de masa se lleva a cabo por medio de un flujo normal a la superficie y también por un flujo lateral 1 oque no sucede en el centro del disco, colocándose en las condiciones de
-
1
- i I !
145

la ley de Ohm tenemos que: i = 4Krl(AE)
Donde: K es la conductividad específica de la solución A€ es la diferencia de potencial en el seno de la solución
entre el cátodo y el contraelectrodo, por lo que la resistencia total se expresa como:
1 1
4 q R R =
b) Distribución de corriente secundaria.- es aquélla que toma en cuenta las contribuciones por fenómenos cinéticos en el electrodo y por transferencia de masa. En este caso la distribución de la corriente es más uniforme que en el caso de la primaria. Albery y Hitchman demostraron que la distribución puede ser expresada en términos de un paránetro adimensional, p, definido como:
Donde: RE es la resistencia del electrodo proveniente de la transferencia de carga y de la polarización de concent ración.
Cuando p +an quiere decir que la resistencia del electrodo tiende a ser muy pequeña comparada con la resistencia total, lo que quiere decir que fenómenos de transferencia de carga y masa son muy pequeños, por lo que la distribución secundaria tiende a ser similar a la primaria. En el caso contrario, cuando p, O (la FUI
es pequefla y la RE grade) se obtiene una distribución casi uniforme. De acuerdo con Albery y Hytchman para evitar las distribuciones no uniformes, las condiciones experimentales deben ser tales que:
p < 0.1
146

Ahora bien, como R no es medible directamente es conveniente
expresar tal desigualdad en función de otros parámetros.
Si experimentalmente podemos medir la diferencia de potencial
entre el cátodo y el contraelectrodo y la corriente aplicada, ambos parámetros se encuentran relacionados de la siguiente manera :
E
di +RE “1 de 2 tenemos que
E - d i - “ I E - R n
sustituyendo 3 en 1 tenemos que:
RR < 0.1 =
de donde: “ 3 < - 1 11 RR E dE
lo cual en función de K y ri (parárnetros medibles) equivale a:
“1 < 0.36 Krl dE E
Para las condiciones experimentales de depósito tenemos que:
(2)
(3)
E = 3 V
i = i x A
r = 0.75 cm 1
1 47

y de la literatura [lo21 tenemos que para una solución acuosa de
sulfato de sodio 0.3 M la conductividad molar es:
2 cm
mo 1 A = 36.376 -
por lo que la conductividad específica es:
-1 -I K = A x [Na2S041 = 1.0913 x cm
Sustituyendo valores tenernos que:
di ] = 3.33 R-' < 2.946 s i 1 dE 3v
Dicho valor permite decir que en las condiciones experimentales
de depósito, la distribución de la corriente es uniforme.
148

ANMO 3
Estudio hidrodinhico de l a celda electroquímica
El electrodo disco rotatorio (RDE) es muy útil para estudiar problemas teóricos y experimentales relacionados con el uso de electrodos sólidos, debido a que los procesos en el RDE se llevan a cabo bajo condiciones estacionarias. Desde un punto de vista electroquímico es importante puesto que el transporte de masa puede ser analizado matemáticamente, aun si el proceso en el electrodo presenta - alguna reacción química homogenea o
heterogenea. En el sistema RDE el flujo de las especies (electroactivas o no) hacia cualquier punto del disco es independiente de la distancia entre tal punto y el eje de rotación del disco. Por lo tanto, características electroquímicas fundamentales como el espesor de la capa límite de difusión y la densidad de corriente se mantienen constantes sobre toda la superficie del disc;.
Balance de masa
La transferencia de masa hacia el electrodo involucra contribuciones por difusión, convección y migración. En un
- electrolito fluyendo a la velocidad V, la densidad de flujo de
masa Ji para cualquier especie iónica (con una concentración Ci, una difusividad Di y una mobilidad iónica Vi) puede ser expresada como :
i
+
149

+ + + + Ji = Ji dif + Ji,conv + Ji,migr.
= -Ai grad ( C i ) + CiV + ViCiE
i siendo E la intensidad del campo eléctrico.
i Para un fluido. incompresible (div V =O) con un flujo
estacionario, el balance diferencial de masa es:
i i i - - - AiACi - V grad (Ci) - Vi div (CiE) aci at (2)
Si despreciamos las variaciones de la difusividad iónica con la concentración, siendo ACi el laplaciano de las concentraciones.
Si además se asume que el transporte no depende del campo
eléctrico (es el caso de un exceso de electrolitol el balance de masa puede ser redefinido con la ecuación clásica de la
difusión-convectiva:
i + - = AiACi - V . grad ( C i ) at
la cual se reduce en condiciones estacionarias a: i i
AiACi = V . grad (Ci ) (4)
En la Última ecuación la sección izquierda de la igualdad
corresponde al transporte por difusión y la sección derecha al transporte por convección.
150

Características del flujo
El modelo del comportamiento hidrodinámíco del sistema RDE esta basado en las siguientes suposiciones, las cuales están presentes
usualmente en el desarrollo experimental. a) La velocidad angular, a, del electrodo es lo suficientemente baja como para evitar turbulencias. b) €1 disco de radio, Rd, es l o suficientemente largo como para despreciar efectos de borde. c ) El electrodo se encuentra inherso en un electrolito sin limites. En este caso deben considerarse 2 regiones en el elect rol i to:
c.1) La capa hidrodinámica lliite, la cual se encuentra en la vecindad del electrodo y donde la viscocidad actua de manera importante, lo que da origen a variaciones de la velocidad del fluido. Dichas variaciones se encuentran en el intervalo de la velocidad de rotación del electrodo y la velocidad del resto de la solución. c.2) El seno de la solución, en el cual la velocidad del electrolito es uniforme con variaciones menores a 1% . Von Karmán [941 y Cochran 1951 obtuvieron teóricamente la distribución de velocidades dentro de la capa hidrodinámica límite resolviendo simultáneamente las ecuaciones de Navier-Stockes y la de continuidad. Las velocidades axial, radial y angular, Vy, Vr, V#, pueden ser expresadas en función de una variable adimensional 2 la cual depende de la distancia, y, de la superficie del disco, quedando definida como:
__-
1/2 2 = [o /VI y (5)
siendo v la viscosidad cinemática del electrolito. Para valores de 2 mayores tie 3.6, se observó que todas las variaciones de las velocidades no exceden 1% (v SV, v =O,v = O ) .
Entonces el espesor, bH, de la capa hidrodinámica límite puede expresarse como:
Y r 9
151

112 = 3.6 [ v / u1 aH
Es importante seflalar que el espesor, 6” no depende de las coordenadas radiales por lo que se mantiene constante sobre toda
la superficie del electodo. A b más, se mostró que se pueden ignorar los efectos de borde cuando aH< O. 1 Rd y Rd >O. 5 m y que los efectos de las paredes de la celda sobre el flujo son despreciables cuando la relación 6WFId está próxima a cero. Generalmente el flujo se caracteriza por medio de un paráretro adimensional llamado número de Reynolds, Re, el cual está definido como la relación entre las fuerzas de inercia y de fricción. Para los sistemas RDE, este parámetro se define como:
Re = w Rd/v ( 7 )
Entre mayor sea el valor de Re, mayores son las fuerzas de inercia y las perturbaciones en el fluido. Entonces a grandes valores de Re corresponde un flujo turbulento y a bajos valores un flujo laminar. Esto se traduce en la existencia de un valor critico, Re,crit, de Re significando la transición entre estos dos tipos de flujo. Para el RDE, este valor crítico fue determinado por muchos autores, resultando un valor promedio de 2.4 x 10 2 19%. Sin embargo, algunas veces aparecen turbulencias con valores de Re inferiores a Re,crit debido a la presencia de vibraciones de tipo axial y/o radial (sólo si su amplitud excede de 0.Olmm) o a la rugosidad del electrodo (actuando la rugosidad como promotora de la turbulencia induciendo agitación extra). El uso de superficies electropulidas evita la presencia de turbulencia. Otro caso extremo corresponde para valores de Re donde se debe considerar la contribución de la convección natural a la agitación por convección forzada. Esto ocurre para valores de Re
__
5 _-
152

del orden de 10, donde el espesor de la capa hidrodinámica límite ya no es despreciable con respecto al radio del disco. Entonces
para limitar la convección natural y para evitar los efectos de las paredes de la celda y de cualquier objeto inmerso en la solución debe ser satisfecha la siguiente condición:
Características de la transferencia de masa.
Utilizando la distribución de las velocidades del fluido determinada por Von Karuh y Cochran, Levich 1961 encontró a partir de la ecuación y la expresión analítica de la densidad de fluJo de masa en la superficie del electrodo y obtuvo que el espesor, 6, de la capa límite de difusión está expresado por:
-
6 = 1.61 [ D ~ / W ] ” ~ [V/~I~/~ { 1 + O. O3539 [Di/ul
El espesor de la capa límite de.difusión también está relacionado con el coeficiente de transferencia de masa, K, de la siguiente manera:
~. -
y también puede ser expresado como una función de la difusividad y 2 parámetros adimensionales, los números de Reynolds y de Schmidt,Sc. El número Sc está definido a partir de las propiedades físicas del electrolito por:
Sc = u/Di . -’ (11)
Un tercer parámetro es el número de Peclet, Pe, el cual también puede ser utilizado para caracterizar la transferencia de masa en
153

la capa de difusión a traves de:
Pe = Re . Sc = wRd/Di (12)
Para los líquidos comunes (excepto los metales líquidos), la capa
límite de difusión es substancialmente más delgada que la capa hidrodinámica límite. Cuando Pe >1 el transporte de masa en la solución está gobernado sólo por agitación convectiva y los efectos por difusión son despreciables. Además, cuanto ids cerca se está del electrodo, más importante es la contribución de la difusión. Cuando las variaciones de la velocidad del disco son superiores al 5%, no es posible alcanzar el estado estacionario porque el espesor de la capa limite de difusión varía de acuerdo a la ecuación 9. Finalmente si 6 0.05 bH, la difusión convectiva está presente en el sistema electroquímico. El sistema celda-catodo disco rotatorio diseñado y construido para la realización de los experimentos esta caracterizado por los siguientes parhetros hidrodinámicos:
- Re= 6948 - Pe= 9.7 x 10
- 6- 3.24 x cm
S
- 6= 2.77 x cm
154

Anexo 4
CRITERIOS DE MAXIMA SiMILITUD
Consideremos un espectro experimental consti tuido de n valores, El, siendo i el número de canal. Sea F1(al,..,ak) la función para ajustarla al canal I , dependiente de los k parámetros al,..,ak. Un método intuitivo de apreciación de la similitud del espectro experimental El a los valores teóricos Fl, consiste en calcular la probabilidad de que el espectro El sea una fluctuación estadística de la ley Fi(ai,. . ,ak). La maximización de esta probabilidad, por ajuste de los parámetros al,. . ,aks nos lleva al ajuste óptimo. Calculemos la probabilidad de que El sea una fluctuación
estadística de Fl(al,. . ,ak). Este cálculo no es realizable en general aunque es necesario que se hagan hipótesis sobre la naturaleza de la distribución estadística de los valores de El con respecto al modelo Fl (al,. . ,ak). Consideremos una dsitribución normal con desviación estandar constante, una normal
con desviación estandar no constante y una poissoniana.
a. Estadística noriel con desviación estándar constante Si se supone que las fluctuaciones de cada canal son independientes, correspondientes a una distribución gaussiana con desviación estándar constante en todo el espectro, la probabilidad de ocurrencia de un estado Elkc a partir del modelo Fl(al,. . ,ak) esta dada por la ley normal:
El-Fl (al , . . , ak) P (El/Fl 1 = exp [-; [ u
siendo Q la desviación estandar de la solución y c el intervalo de tolerancia.
155

La probabilidad de ocurrencia del espectro El(l=l,.,n) es el producto de las probabilidades de cada canal:
n EI-FI (al,. . , akl P(E/F)espectro= fl exp - 1 2 (
I =1
experimental ocurrencia de
. & (2)
Maximizar el valor de Pespectro juzgando sobre los paráaetros
a . ,ak es equivalente a minimizar el logaritmo de -P (P es una probabilidad, entonces es una magnitud positiva inferior a 1 ) : 1'
2 [El-Fl (al, . . , ak) 1 - - n log E (31 log 'espectro 1 =l 2c2
Como por hipótesis n y c son constantes, minimizar -log Pespectro es equivalente a minimizar la expresión:
lo cual corresponde al criterio de mínimos cuadrados. Entonces el criterio de mínimos cuadrados corresponde de similitud del modelo si:
a un máximo ~-
- los errores de medida por canal son independientes, - los errores estan distribuidos se& una ley normal, ____
- la desviación estandar de las fluctuaciones es constante.
b. Estadística normal con desviación estándar no constante Si se consideran las hipótesis similares a las enunciadas en a. sin considerar la desviación estándar constante en todo el espectro, un razonamiento similar al precedente nos lleva a minimizar:
W 1 &
1 =l
156
(5)

la expresión 5 es.idéntica a la 4 solo que la desviación ectandar tiene subíndice 1, pues es relativa a cada canal. El criterio de
2 la expresión 5 corresponde al de X . Si consideramos que en general se puede considerar la varianza por canal igual al conteo entonces Q = E 2 dando:
1 I ’
Con la finalidad de limitar las grandes fluctuaciones relativas de los canales con bajos conteos es posible rempalzar et por q:
Por regla general el valor de x” es normalizado dividiendolo por el número de grados de libertad del problema:
A con: 9 como x“ reducida, n número de canales y k número de
parámetros ajustables del modelo.
c. Estadística Poissoniana Suponiendo que las fluctuaciones del número de cuentas por canal, Et, son independientes y responden a una estadística poissoniana, alrededor del valor promedio definido por la función de ajuste Fl, se puede calcular la probabilidad de que un espectro experimental sea una fluctuación poissoniana del espectro teórico. Aplicando el procedimiento anterior se obtiene:
157

Fll e ' . c (E/F )espectro (9)
Se supone que los errores por canal son independientes. El mejor
ajuste consiste en maimizar P (E/F)' entonces también -log P (E/F):
f (-EllogF1+ F1-logc + log(El! 1) (10) 1 =1
(EYF)= -log P
como los dos últimos términos de la ecuación 10 son constantes, el criterio a minimizar es:
n C= 1 (Fl- Eilog Fl)
1 =1 (11 1
este criterio de Poisson ya no tiene divisiones con valores probables nulos. Entonces este criterio se aplica bien en los casos donde hay bajos conteos. Nota: Si se supone que los valores de Ei son lo suficientemente importantes para que se pueda aplicar la fórmula de Stirling a E,!, la ecuación 10 se transforma en (despreciando el término
-log P (E/F)= f=l[-EllogFi + Fi- log& + EilogEi - (12)
de donde:
Fl n 1 (-EllogE + Fl- El- loge) i =1 1 (EYF)= -log P
por otro lado:
(13)
158

desarrollando 14 en serie:
(15)
Substituyendo 15 en 13
superior se obtiene:
Y despreciando los términos de orden
lo que equivale a l criterio de X2.
159

Anexo 5
A
Relación entre el criterio de 9 y la estadística de medida de los espectros
A El criterio de 3 esta definido por:
con: n como número de canales y k número de parámetros Aproximando ri a se obtiene: -
libres.
(2) 1 n-k-1
i =1
Poniendo para cada canal i , que p i es la esperanza matemática del
valor de conteo y definiendo ai como la diferencia, en el canal i, entre el valor medido y la esperanza matemática:
N = E(El) (3) i
sustituyendo 4 en 2 y separando- en 4 términos:
A A A A 3= x2 + g + x;
A (6)
160

con:
A 1 n a t XI= G=E=iJ c E i=l 1
( 7 )
( 8 )
( 9 )
A
x" describe la distribución según una ley de $ del valor
experimental alrededor de un promedio. Esta distribución converge hacia una ley gaussiana para grandes valores del número de grados
de libertad. Su valor promedio es 1, sin importar el número de grados de libertad:
A
-
A E(?)= 1 (10)
La desviación relativa, A, entre el espectro proredio y la
función de ajuste queda definida como:
_ _ (pi-Fi (al,. . , akl 1 A = (11)
*i i
entonces 3 queda expresada como: B
A
xB= & c 1=1 (.: ( ; )2Ei] por otro lado pi= El, de donde:
n 1 A
xB= o rxl d:
(12)
( 13)
n
I =1 con S=c E que representa el número total de pulsos contados en
1
161

el espectro. Si empleamos un término de espectro normalizado El/S podemos a su vez definir un término 7 que indique l a
desviación entre el espectro verdadero promedio y el modelo:
sustituyendo 14 en 12 tenemos que:
A
r s xB= 0 (15)
A A h
El término < es un cruzado de f y $. Se puede considerar que localmente, el término pi-Fl (al.. . ,ak) es de signo constante mientras que el término 6 cambia de signo de manera aleatoria. La
suma del producto de estos términos no tiene razón para crecer si
la función de ajuste no tiene discontinuidades muy pronunciadas.
Entonces el promedio de Xc es nulo: E(<)= O.
Entonces la esperanza matemática de $ es:
A h 2
A
es decir que en el límite:
A E(?) = 1 + ' 7 j n-k- 1 T S (17)
h
Si el ajuste es perfecti, t es nulo y el valor de 9 es igual a 1.
Si la estadística del espectro es baja (SbaSa) ? también es igual a 1, aún para valores de r no despreciables. Observando
esta relación se puede decir que 2 es más insensible al modelo
h
A
162

en la medida que el espectro tienda a una baja estadística.
Cabe señalar que, para un ajuste imperfecto pero correcto, el
valor de ? varía linealmente con el número de pulsos integrados del espectro.
A
163

Anexo 6
ALCORIRH) DEL SIMPLEX
Sea f(Ei,Fi(ai,.. ,ak)) la función para minimizar con los k
parámetros. Definamos un espacio de k dimensiones cuyos ejes tienen vectores de dirección de coordenadas (al, O, O, . . O ) ,
(O,az’. . ) (O, . . . O,ak). En este espacio, el SIMPLEX es una figura no degenerada de k+l puntos engendrando una hipersuperficie. Este SIMPLEX se construye a partir de un punto de inicio Po, teniendo por coordendas un juego de parhtros (al,. . ,ak) iniciales.cada uno de los k puntos restantes del SIMPLEX
construye utilizando una homotecia de la forma:
i Pi= PO + hi . a ( 1 )
donde hi es una constante y ai la coordenada unitaria por eje. En cada punto Pi, el valor de la función f (Ei,Fi (pi) ) puede ser
calculado y la selección de los valores de la función permite definir Firw y Pmin correspondientes a los puntos para los cuales la función es máxima. El baricentro del SIMPLEX, P igualmente es calculado. El principio de resolución consiste en deformar el SIMPLEX en el hiperespacio con la ayuda de tres operaciones geométricas elementales, la reflexión, la contracción y la expansión, con la finalidad de hacerlo converger hacia el punto para el cual la función es minima. Las diferentes fases del algoritmo son las siguientes: a. Reflexión. El punto PIIU~ se refleja sobre el eje Pmax-p según la ecuación:
164

* siendo P el punto reflejado y a la constante de reflexión (a>O),
P esta situado sobre la recta que une PMX a del lado de P, de tal manera que la distancia P*P sea igual a a veces la distancia
*
PmaxF. Si f(F) es inferior a f(PMX1 y superior a f(Pmlnl es posible hacer otra reflexión cambiando PMX.
b. Expansión. Si f(P 1 es inferior a f ( h i n ) la reflexión produjo un nuevo mínimo. En este caso P es expandido según:
*
** * P = TP + (1-r)P (3)
- a es el coeficiente de expansión (r>l), igual a la relación de distancia P**P sobre P*P. Si f(P 1 es inferior a f(Pmin), Pmax
es remplazado por P y el proceso continua. Si f(P es superior f(Pmin) la expansión no es satisfactoria y PMX es
** ** **
* reniplazado por P . c. Contracción. Si en el es superior a f(Pl) para se define un nuevo Pmax
-- -
-___ contracción según: **
* proceso de reflexión aparece que f (P 1 cualquier valor de i diferente de imx,
con P y se forma un punto P por * **
. . P = 6 Pliut + (1-#3)P (41
/3 es el coeficiente de contracción (O<fWl ) que representa la * *- ** relacion entre la distancia P P y la distancia Pp. El punto P ** remplaza a Pia~ y se prosigue el proceso hasta que f(P 1 sea superior a f ( b ) o a f(P ), es decir que en el caso donde el punto contraido corresponde a una fracción superior de Pi.ax o P . En este Útimo caso todos los puntos Pi son remplazados por los puntos respectivos -(P +PW] y se reinicializa el proceso.
*
1 2 i
165

7 ---
ANEXO 7
Procesos que determinan la respuesta de m detector PIPS
El espectro de partículas alfa monoenergéticas muestra una forma de pico asimétrica cuando es medido con un detector de silico como el PIPS. A continuación se describen los procesos que determinan la respuesta de un detector PIPS a las partículas alfa, particularmente la dependencia de la forma de la línea con la energía. Para transiciones alfa monoenergéticas la distribución de la altura de pulsos medida, muestra una forma asimétrica la cual ha sido reportada por mucho autores, por ejemplo L'Hoir 1641. Un modelo analítico, H, de la función de la forma asimétrica, ha sido expresado como la convolución de una función de densidad de probabilidad compuesta por una gaussiana y una o mas exponenciales I631 :
Aquí A y p corresponden al área y la posición del pico y Q es la desviación estándar de G(t1. vi es la intensidad relativa y (r es la constante característica del i-ésimo término exponencial. La distribución de altura de pulsos se calcula como la convolución de la distribución de energía, N, de las partículas emitidas por el detector y como la función respuesta R:
166

N(Es) se calcula para una fuente homogénea, la función R se obtiene a partir de un modelo de detector empleando métodos computacionales. Puesto que el espectro calculado puede ser aproximado por el modelo analítico (ecuación 1), los paráaietros de forma u, qi, T~
pueden ser interpretados sobre bases físicas y su dependencia con la energía puede ser deducida.
a. El espectro de l a fuente La distribución en energía de las partículas emitidas por fuentes delgadas (para espectrometría alfa) queda determinada por la pérdida de energía por interacción con los electrones del medio y
por la dispersión de -la misma pérdida o "straggling". La influencia de la pérdida de energía por interacción con núcleos es despreciada en el caso de partículas alfa de alta energía. La distribución de la energía de las partículas alfa que emergen de una fuente delgada se calcula asumiendo que:
- se considera el usrcie una fuente homogénea con un espesor uniforme, para l a cual la pérdida de energía es lo suficientemente -orno para permitir el uso de un poder de frenado constante !So= S(E0) donde Eo es la energía inicial de las partículas alfa, - se considera una distribución gaussíana del "straggling" con una varianza SI&,
la distribución de la energía de las partículas alfa emitidas perpendicularmente con respecto a la superficie de la fuente, esta dada por:
1 67

donde No es el número total de partículas detectadas, ds es el espesor de la fuente, x la profundidad a la que se emite la partícula alfa y E(x)=Eo-So.x es la ,media de la energía de las partículas emitidas a la profundidad x. En esta función (ecuación 3) la dispersión de la pérdida de energía no es exacta para pequeñas profundidades de emisión, esta
es la razón por la que algunas partículas aparentemente tienen una energía de emisión mayor que Eo (en el espectro, Eo se encuentra ligeramente a la izquierda del límite superior del pico de energía Eo). En situaciones reales el espectro de la fuente puede ser diferente de la aproximación de la ecuación 3 debido a diferentes efectos. El espesor no uniforme del depósito provoca variaciones en la longitud de la trayectoria de la partícula alfa dentro de la fuente. La composición elemental no homogénea de la fuente tiene como consecuencia variaciones en el cálculo del poder de
-
frenado. Finalmente, la forma del espectro puede ser influenciada por efectos geométricos, tales como la diferencia en la longitud de la trayectoria de las partículas alfa detectadas dentro de un amplio ángulo sólido. Estos efectos cambian la forma del espectro de emisión de manera impredecible incrementando la asimetría del espectro. ~ __
_- b. Función respuesta del detector La función respuesta del detector R(Es,h) se define como a probabilidad por intervalo de altura de pulso dh, de que una partícula que incide en el detctor con una energía cinética Es, de origen a un pulso con una altura entre h y dh+h. Por lo tanto. R(Es,h) es proporcional al espectro de altura de pulsos correspondiente a partículas monoenergéticas. Para los detectores tipo PIPS se asume que el detector puede ser modelado por una capa muerta delgada de espesor dd presente en el frente del detector y por un volumen sensible infinito del detector. Debido a la no homogeneidad del perfil de iones
168

implantados o bien durante el proceso de ataque químico y
recocido del silicio, dd puede variar en diferentes partes de la superficie del detector (con una desviación estándar 6 1.
Cuando partículas monoenergéticas de energía inicial Es penetran dd
en la capa muerta pierden una energía promedio Ed=S(Es)-dd, donde
S(Es)>O es el poder de frenado de los iones incidentes en el material del detector. Después de pasar la capa muerta, las partículas tienen una cierta energía con una desviación estándar aEd debida a cuatro contribuciones: "straggling" de la pérdida de energía por interacciones con los electrones del medio (6Ed,s), "straggling" de la pérdida de energía por interacción con los núcleos (am ), variaciones en al longitud de la trayectoria debida a múltiples dispersiones (am,.) y variaciones en el
espesor de la capa muerta (6 I . Para partículas alfa con Ed, t energías, Es, mayores que 100 KeV, las contribuciones de OD y
pueden ser despreciadas. Para partículas alfa de alta energía la pérdida de energía en una capa muerta con un espesor de 50 run, es lo suficientemente pequeña como para que la
pueda ser influencia de las variaciones de espesor, despreciada. Por lo tanto, la distribución de energía Dd,s de las partículas alfa que penetran en el volumen sensible del detector esta determinada por la distribución gaussiana del "straggling" de la pérdida de energía por interacción con los electrones. Sin embargo, es necesario mencionar que para las partícu-fa con energías cercanas al máximo de poder de frenado (alrededor de 400
KeV) la influencia de variaciones del espesor de la capa muerta pueden ser importantes. En la región sensible del detector las partículas transfieren su
,n
- ,n
%d,n
%,t'
energía promedio Es-Ed a los electrones por mecanismos de ionización y de excitación (frenado electrónico) y a los átomos del silico por transferencia de energía cinética (frenado nuclear). Sin embargo, parte de la energía transferida en las colisiones nucleares también contribuye a la ionización y
excitación electrónica debida a los átomos de silicio que tienen
169

energía de retroceso. La energía restante se invierte en daños al cristal y vibraciones de la red del mismo. cristal. Puesto que la energía Es-Ed esta estadísticamente dsitribuida entre ionización y excitación de los átomos de silicio (energia ES,e) y pérdida no electrónica (energía ES-Ed-Es,e) es de esperarse que halla fluctuaciones estadísticas de ES,U. Ce ha encontrado que la distribución &,e, no es gaussiana [641, esta es la razón por la cual se presenta la asimetría en un espectro obtenido con iones monoenergéticos de helio. La energia Es,e de los electrones se invierte en al producción de pares agujero-electrón y en vibraciones de la red. Se ha encontrado que, para Es.e=constante, la distribución estadística Dc,p de la energía Es,p
agujero-electrón creados, estándar
correspondiente al número de pares es' gaussiana con una desviación
aEC, p= 4 W ES, e* F (4)
donde W es la energía promedio necesaria para la producción de un par agujero-electrón y F es el factor Fano. La densidad de portadores de carga producida por partículas alfa es relativamente baja. Si el detctor es operado a saturación, es decir que la fuerza del campo eléctrico es lo suficientemente alta como para despreciar la atrapamiento y recombinación de electrones o agujeros en el volunen sensible del detector. En este caso la carga total producida por el proceso de detección es medida por un sistema amplificador sensible a la carga. La contribución del ruido del amplificador a la resolución en energía puede ser facilmente medida por medio de un generador de pulsos de precisión. La distribución en energía de este ruido es gaussiana con una desviación estándar 8
En la tabla 1 se presentan los procesos relevantes para la resolución en energía de un detector PIPS.
h'
170

Tabla 1 . Contribuciones a la resolución en energía y a la función respuesta de un detector PIPS.
Dd, t
Ds, e
Damp
Contribución -~ ~
"straggling" de la pérdida de en- ergía en la capa muerta variaciones del espesor de la capa muer ta ionización y excitación electróni- ca por partículas alfa
estadística del par agujero-electrón Resolución del sistema amplifi- cador
forma
gaussiana
gaussiana o asimétrica asimétrica
gaussiana
gaussiana
teoría
despre- ciada simulación en compu- tadora experimen-
experimen-
Si ahora consideramos todos los procesos estadísticamente independientes, la función respuesta, R, del detector se puede obtener por la convolución de las contribuciones individuales. Su desviación estándar esta
'Etot= ('Eh.s 2
La única contribución no
dada por:
detector es la distribución DC,e de la energía B , e , la cual es la energía transferida a los electrones en el detector de silicio (y por lo tanto disponible para la producción de pares agujero-electrón). La función respuesta, R, del detector se calcula como la convolución de Ds,e y distribuciones gaussianas. Por lo tanto, R, mismo puede ser aproximado a la convolución de distribuciones con una gaussiana y la suma de dos o mas exponenciales. El término gaussiano describe los procesos independientes como el "straggling" de la pérdida de energía electrónica en la capa muerta, al estadística del par agujero-electrón, ruido electrónico y el componente gaussiano de B , e . El término
. /
gaussiana a la función de respuesta del
(5)
171

acimétrico se debe al componente asidtrico de %,e . Por lo tanto
el modelo de ajuste de picos utilizado en este trabajo esta
justificado sobre bases de los procesos físicos, los cuales
determinan la función respuesta, R, del detector.
172

REFERENC I AS
1. J. P. Perrine. Reporte interno CEA-Saclay, Francia, 1976. 2. C. Bergey. Préparation des sources alpha. Session analyse par
spectrométrie alpha. INSTN, Francia, 1983. 3. B.F. Myasoedov, Y.M. Kulyako, I.S. Sklyarenko, J. Inor. and
Nucl. Chem., 38 (1976) 827.
4. Th. Handley, J.H. Cooper, Anal. Chem., 41 (1969) 381.
5. C. Bergey, J. Cesario, S. Deniaud, Analusis 8 (1980) 490. 6. S.M. Kim, R. Orins, 48 (1965). 7. N. Getoff, M. Bildstein, NIM, 36 (1965) 173. 8. D.C. Auaiann, G. Mullen, NIM, 126 (1968) 115. 9. A.E. Lally, K.M. Glover, NIM, 226 (1984) 223. 10. K. Kolodney, J. Electrochem. Soc., (Nov 1982) 2438. 11. H.W. Miller, R . J . Brouns, Anal. Chem., 24, No.3 (1952) 536. 12. F. L. More, G.W. Smith, Nucleonics, 13 No. 4, (1955) 66. 13. R. KO, Nucleonics, 14 No.5, (1956) 74 14. R.Ko, Nucleonics, 15 No. 1 (1957) 72. 15. P.G. Hansen, J. Inorg. Nucl. Chem., 12 (1959) 30
16. P.G. Hansen, J. INorg. Nucl. Chem., 17 (1960) 232.
-_ 17. A.C. Samartseva, Atomaya Energiya, 9 No.4 (1960) 324. 18. A.G. Deruyter, NIM, 15 (1962) 164.
-- 19. M. Y. Doman, E.K. Dukes, Anal. Chem., 36 No. 2 (1964) 393. 20. J . W . Morgan, Radiochim. Acta, 15 N0.4 (1971) 190. 21. N.A. Talvitie, Anal. Chem., 44 No. 2 (1972) 280. 22. K.W. Puphal, D.R. Olsen, Anal. Chem., 44 No. 2 (1972) 284 23. C.W. Sill, K.W. Puphal, Anal. Chem., 46 No. 12 (1974) 1275. 24. O. Frindik, Z. Anal. Chem., 276 (1975) 67.
25. I.K. Kressin, Anal. Chern., 49 No. 8 (1977) 642. 26. D. Roman, J. Radionanal. Chem., 60 (1980) 317. 27. B.G. Broda, H. Herz, U. Wenzel, Anal. Chimica Acta, 147
(1983) 105.
173

28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39. 40.
41.
42.
43.
44.
45.
46.
47.
E. Holm, Int. J. Appl. Radiat. Isot., 35 No. 4 (1984) 285. D, Roman, Int. J. Appl. Radiat. Isot., 36 No.8 (1985) 677. N. Trautmann, H. Folger, NIM, A282 (1989) 102. S.B. Jundhale, C.D. Lokhande, Bull. of Electrochem. , 7 No. 10
(1991) 452.
S.B. Junhale, C.D. Lokhande, Mat. Chem. and Phys., 27 (1991)
262.
A. Bard, R. Parsons, J. Jordan: Standard Potentials in Aqueous Solutions. IUPAC, Uarcel Decker, 1985. T. Moeller: The Chemistry of the Lanthanides. Reinhold Publ., New York, 1963. F. A. Cotton, G. Wilkinson: Advanced Inorganic Chemistry. John Wiley and Sons, New York, 1980.
D.E. Hobart, K. Samhouni, J.P. Young, V.E. Norvell, G. Mamantov, J.R.- Peterson, Inorg. Nucl. Chem. Lett., 16 (1980)
321.
G.R. Choppin, Radiochim. Acta, 32 (1983) 43.
C. Keller: The Chemistry of the Transuranium Elements. Verlag chemie, GMBH, Alemania, 1971. J.C. Sullivan et al, Inorg. Nucl. Chem. Lett. 12 (1976) 599.
B. F. Myasoedov: Actinides in Perspective. N. Edelstein ed. , Pergamon Press, Oxfortd, 1982. V.P. Shilov, Sov. Radiochei., 18 (1976) 567.
A.Rojas, f;(;onzález, Anal. aim. Acta, 187 (1986) 279.
- _ -
A. Rojas, M.T. Ramirez, J.G. Ibáñez, 1. González, J. Electrochem Soc., 138 (1991) 365.
A. Rojas, M.T. Ramírez, J.G. Ibáñez, I. González, Anal. Chim. Acta, 246 (1991) 435. A. Rojas, M.T. Ramírez, I . González, J.G. Ibáñez, Anal. Chim. Acta, 259 (1992) 95.
A. Rojas, M. T. Ramírez, 1. González, Anal. Chirn. Acta, 278
(1993) 321.
A. Rojas, M. T. Ramírez, I. González, Anal. Chim. Acta, 278
(1993) 335.
174

48. A. Ringboa: Complexation in Analytical Chemistry. Wiley Interscience, New York, 1963.
49. G. Charlot: Cows de Chimie Analytique Générale. Mascon, Paris, 1967.
50. Y. V. Pleckov, V. Y. Filinovskl: The Rotating Disc Electrode. Studies in soviet science, Plenum Press, USA, 1976.
51. F. OPekar, P. Betan, J. Electroanal. Chem., 69 (197611.
52. W.M. Press, B.P. Flannery, S.A. Teukolsky, U. Vetterling: Numerical Recipes, Cambridge UNiversity Press, New York, 1986.
53. C.J. Bland, A. Martin-Sánchez, NIM, A286 (19910) 375.
54. S. Brandt: Statistical and computational methods in data analysis. North Holland, Amsterdam, 1989.
55. M.L. Aceña, E. Garcia-Toraño, Determinación de isótopos de uranio por espectrometría alfa. reporte JEN 442, Madrid, 1979.
-
56.R.L. Williams, Int. J. Radiat. Appl. Isot., 35 (19841 271.
57. U. Watsig, U. Westmeier, NIM, 153 119781 517.
58. H. Baba, NIM, 148 (1978) 173.
59. R.B. Welch, F. Giger, D.T. Jost, H.R. von Gunten, U. Krahenbühl, NIM, A269 (19881 615.
60. E. Garcia-Toraño, M. L. -AceXa, NIM, A185 (1981 1 261.
61. A. Martin-Sánchez, F. Vera-Tomé, C.J. Bland: Data analysis of alpha-particle spectrafroni Low Level Sources, in: Low level measurements of man-made radionuclides in the environment. Garcia León M. & Madurga G., eds., World scientific Press, Singapore, 1991.
62. J.A. Nelder, R. Mead, Computer journal, 7 (19651 308.
63. G. Bortels, P. Collaers, Appl. Radiat. Isot. 38 No.10 (1987)
831.
64. A. L’Hoir, NIM, 223 (1984) 336.
65. Y. Isozumi, NIM, A235 (19851 164.
66. K. Otsuka, K. Jinno, Inorg. Chim. Acta, 121 (1986) 237.
67. M.H. Jacksik, High Temp. Science, 30 (1990) 19.
175

68. N.M. Greenwood, A. E. Earnshaw: Chemistry of the elements. Pergamon Press, Oxford, 1984.
69. A. J. Arvía, M. C. Gordano: Introducción a la electrocatalisis, monografía No. 27, OEA-PRDCT, Washington, 1984.
70. R. Woods: Electroanalytical chemistry: chemisorption at electrode. ed. A . J . Bard, 9, Marcel Decker, New York, 1976.
71. B.R. Hinton, D.R. Arnott, Microstructural science, 17 (1990) 311.
72. R. Durand, Tésis de doctorado, INP, Grenoble, Francia, 1978. 73. C. Gabrielli, reporte técico 4183, Solartron, Francia, 1984. 74 C. Cachet, E. Epelboin, M. Keddam, R. Wiart, J. Electroanal.
Chem., 100 (1979) 745.
75. M. Pourbaix: Atlas of electrochemical equilibria in aqueous solutions. CEBELCOR, Pergamon Press, Oxford, 1966.
76. E. Hogfeldt: Stability constants of metal-ion complexes, Part A: inorganic ligands, IUPAC chemical data series No. 21,
Pergamon Press, Pew York, 1979.
77. K. Kragten, L G . Decnop-Ueever, Talanta, 26 (1978) 1105. 78. R. Smith, A. Martell: Critical Stability Constants. Plenum
Press, 1975.
79. C.F. Baes, R.E. Mesmer: The Hydrolysis of Cations. Wiley, New ~ _ _
York, 1976.
80. L.G. Sillén: Stability Constants of Metal -ion Complexes. Special Publication No. 17, the chemicafsociety, London, 1971.
81. P. Vitorge, Radiochim. acta, (1993) en prensa. 82. E.M. Pazukhin, E.G. Kudryatsev, Radiokhimiya 32 No. 4 (1990)
318.
83. H. Capdevila, Tésis de doctorado, Universidad de Paris, Instituto de Fisica Nuclear, Francia, 1992.
84. G. P. Box, N. R. Draper: Empirical model-building and response surfaces- John Wiley & Sons, New York, 1987.
85. D.C. Montgomery: Design and analysis of experiments. John Wiley & sons, New York, 1984.
176

86. A. J. Bard, L.R. Faulkner: Experimental methods. John Wiley &
sons, New York, 1980. 87. U. Forker: Cinética electroquímica. EUDEBA, Buenos Aires,
1971.
88. S. Becson, J. Gitton: Manipulations de electrochimie. Mascon & Cie., Paris, 1972.
89. D. T. Sawyer, J. L. Roberts: Experimental electrochemistry for chemists. John Wiley & sons, New York, 1974.
90. JANDEL SCIENTIFIC, programa SIGMAPLOT, versión 4.04, manual del usuario, 1992.
91. E. Matijevic: Surface and Colloid science. Vo. 6, John Wiley and Sons, New York, 1973.
92. A.J. Bard, L.R. Faulkner: Electrochimie, principe, methodes et applications. Masson, Paris, 1983.
93. J. Truffy, Tésis de Ingeniero CNAM, Escuela de artes de Paris, 1990.
94. T. von Kármán, 2'. Angew. Math. Mec., 1 (1921) 233. 95. W.G. Cochran, Proc. Cambridge Phil. Soc., 30 (1934) 365.
96. V. G. Levich: Fizikokhiai tcheskaya Gidrodinamica, Moscu, 1959. 97. J. Newman, J. Electrochem. Soc., 113 (1966) 501. 98. J. Newman, J. Electrochem Soc., 113 No.12 (1966) 1235.
99. J. Newman, j. Phys. Chem., 70 (1966) 1327. 100. U. J. Albery, M. L. Hitchaan, Trans. Faraday Soc., 67 (1971)
2408.
101. W.J. Albery, M.L. Hitchman: Ring dis electrodes. Clarendon Press, Oxford, 1971.
102. R. C. Weast: Handbook of chemistry and Physics. 68th edition, CRC Press, 1987-1988.
102. J. Truffy, Reporte interno LPRi-DAMRI-CEA, FRancia, 1990. 103. U. Westeimer: Processing and computer evaluation of alpha
particle spectra from Si-detectors and ionisation chambers. Notas del curso interregional de entrenamiento, OIEA, Alemania, 1989

--
104. S. A. Baranov, V. M. KUlakov, V. N. Chatinsky, Nuclear Physics, 56 (1%4) 252.
105.W. Michaelis, 2. für Physik, 186 (1965) 42. 106. I. h a d , NIM, A244 (1984) 319. 107. IAEA: Decay data of tranactinium nuclides. Reporte técnico
No. 261, IAEA, Viena, 1986. 108. F. Lagoutine, N. Coursol, J. Legrand: Table of
radionuclides, LMRI-CEA, Francia, 1983. 109.
110.
111.
112.
LARA: Fichier de réferences LMRI pour la spectroietrie gamma et alpha, MI-CEA, Francia, 1987. M.C. Lépy, J. More1:deconvolution de spectres 7 et X, étude bibliographique, reporte interno M I 09/99, Francia, 1990.
E. Browne, R.B. Firestone: Table of radioactive isotopes, vol. 5, Shirley Wiley Interscience, 1986.
C.J. Bland, J. Morel, M. Etcheverry, H.C. Lépy: Determination of Pu-239 and Am-241 U-Ray intensities using a Simplex method for fitting, Appl. Radiat. Isot. (enviado 1.
178