Embed Size (px)
Citation preview

Métabolisme normal et pathologiquedes acides aminés
L2 – UE8
Pr H Puy

Maladies héréditairesdu métabolisme des acides aminés
= aminoacidopathies
I. Introduction
♦ 2 catégories :
- anomalies du transport membranaire
- enzymopathies touchant le métabolisme des AA
♦ 50aine aminoacidopathies♦ maladies métaboliques
(mutation sur le gène codant pour l’enzyme).
Conséquences :1/ diminution des produits en aval (déficit fonctionnel )et/ou2/ augmentation des produits en amont (toxicité )

Molécules organiques associant une fonction acide et une fonction amine
H
R
Cα COO-H3N
+
OrigineENDOGENE
synthèse de novo
renouvellement des protéines
EXOGENE : alimentation
1/ Structure des acides aminés

Rôle biosynthétique Précurseurs : - des protéines- d’hormones- de neurotransmetteurs- de nucléotides- de l’hème
Rôle énergétique
substrats énergétiques (comme le glucose ou les acides gras) :
1/ leur catabolisme alimente le cycle de Krebs
2/ certains sont des substrats de la néoglucogenèse
(AA glucoformateurs : Ala , Gly, Ser, Cys…)
CH2 CH2 COO-H3N
+CH2
certains, à l’état libre, ont une activité biologique propre
ex : acide γ-aminobutyrique G.A.B.A
Rôle fonctionnel
2/ Importance des acides aminés

Aptitude à synthétiser les AA n’est pas identique chez tous les organismes :
- la plupart des bactéries et des plantes peuvent synthétiser les 20 AA,
- mammifères : environ la moitié des AA sont synthétisés
AA indispensables : synthèse impossible, source dans alimentation
AA non indispensables : synthèse endogène
Acide aspartiqueAsparagine
Acide glutamiqueGlutamine
AlanineCystéineTyrosineGlycineProlineSérine
Arginine*Histidine* Leucine
Thréonine Lysine
TryptophanePhénylalanine
ValineMéthionineIsoleucine
Non indispensables dans l’alimentation
Indispensables dans l’alimentation
* AA «semi-indispensables chez l’enfant» : peuvent être synthétisés chez l’adulte mais sont synthétisés à un taux insuffisant chez l’enfant (en particulier pour sa croissance)
3/ Besoins en acides aminés chez l’homme
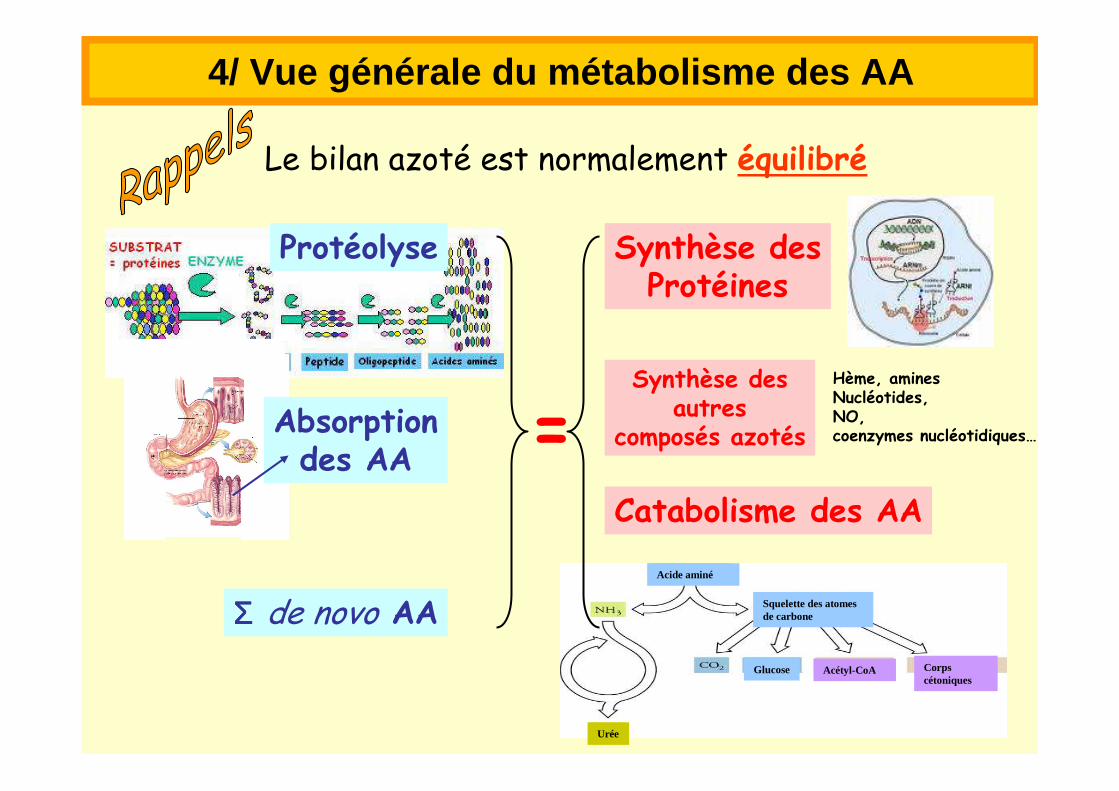
Le bilan azoté est normalement équilibré
Protéolyse Synthèse desProtéines
Acide aminé
Squelette des atomes de carbone
Glucose Acétyl-CoA Corps cétoniques
Urée
Catabolisme des AA
Synthèse desautres
composés azotés
Hème, aminesNucléotides, NO,coenzymes nucléotidiques…
Absorptiondes AA
ΣΣΣΣ de novo AA
=
4/ Vue générale du métabolisme des AA

PHE����
TYR
Pyruvate����
ALA
ALATOxaloacétate
����
ASP����
ASN
ASAT
aCG����
GLU
GLN PRO ARG
3 PhosphoGlycerate����
SER
CYS GLY
Biosynthèse des acides aminés NON essentiels
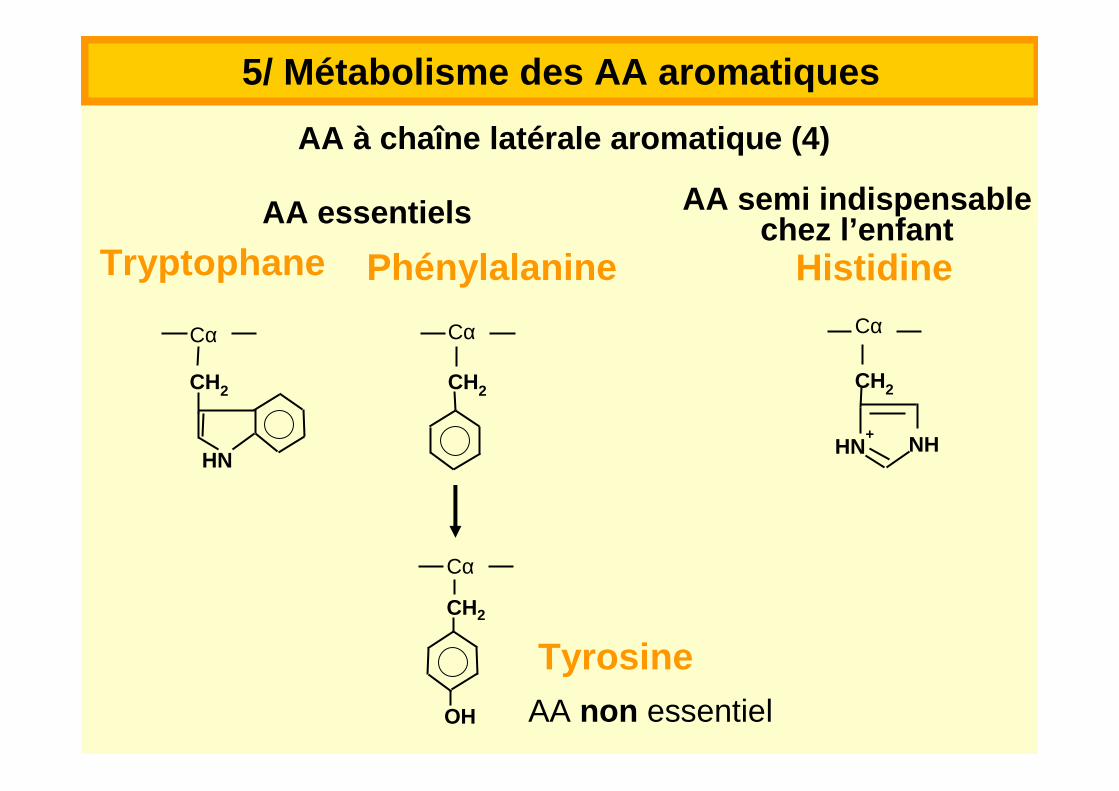
TryptophaneAA essentiels
5/ Métabolisme des AA aromatiques
HistidinePhénylalanine
TyrosineAA non essentiel
AA semi indispensablechez l’enfant
AA à chaîne latérale aromatique (4)
HN
Cα
CH2
Cα
CH2
OH
Cα
CH2
NH
CH2
Cα
HN+

II. Métabolisme du tryptophane
NH
CHCH2
NH2
COOH
Tryptophane
Sérotonine
NAD+
AcétoacétylCoA
AA essentiel- Alimentation- Catabolisme prot. endogènes NADP+
Mélatonine
bactériesintestinales
IndolScatol
Catabolisme à noyau ouvert
Catabolisme à noyau fermé

NH
CHCH2
NH2
COOH
Tryptophane
Cynurénineformylase
COOH
N
3 OH anthraniliqueoxydase
acide nicotinique
CONH2
N
nicotinamide
NAD+
+ PRPP+ ATP
NH2
CH2CO
NH2
CH COOH
cynurénine- CHO
NH2
CH2CO
NH2
CH COOH
OH
Cynuréninehydroxylase
3 hydroxycynurénine+ O2
NH2
COOH
OH
Cynuréninase
acide 3 hydroxy-anthranilique
- alanine
1/ Biosynthèse du NAD +II. Trp
NH
CH2CO
NH2
COOHCHO
CHTrp
pyrrolaseformyl cynurénine+ O2
Foie

TRPFOIE
Catabolisme à noyau ouvert
NAD+ NADP+
TRP pyrrolase
Insuline – NADPH,H+
TRP, Cortisol, Glucagon
Alimentation +++Acide nicotinique
Vit PP
acétoacétylCoAALA
B6

NH
CHCH2
NH2
COOH
NH
CH2CO
NH2
COOHCHO
CH
NH2
CH2CO
NH2
CH COOH
NH2
CH2CO
NH2
CH COOH
OHNH2
COOH
OH
COOH
N
CONH2
N
Tryptophane
Trppyrrolase
formyl cynurénine
Cynurénineformylase
cynurénine
Cynuréninehydroxylase
3 hydroxycynurénine
Cynuréninase 3OH anthraniliqueoxydase
acide 3 hydroxy-anthranilique
acide nicotinique nicotinamide
NAD+
+ PRPP+ ATP
+ O2 - CHO
+ O2
- alanine
AcétoacétylCoA
Catabolisme à noyau ouvert

NH
CHCH2
NH2
COOH
NH
CHCH2
NH2
COOHHO
NH
CH2CH2
NH2
HO
Trp hydroxylase
5 hydroxyTrp décarboxylaseCO2
O2
Tryptophane
5 hydroxytryptophane
5 hydroxytryptamine = Sérotonine
2/ Biosynthèse de la sérotonineII. Trp
IntestinSNCPlaquettes

Catabolisme à noyau fermé
TRP SerotonineIntestin, cerveau, plaquettes
1% Vasoconstricteur+++
NeuromodulateurSNC
5HIAA urines
Ac. 5 HydroxyIndolAcétique
Monoamine Oxydase(MAO) Pathologie : ��� 5HIAA
Tumeurs carcinoïdes du grêle
Pharmacologie :IMAO (antidépresseur)
Hydroxylation+ décarboxylation

Catabolisme à noyau fermé
TRP Serotonine1%
Hormone centrale de régulation des rythmes chronobiolo giques
Sécrétée dans la glande pinéale, variations nycthémérales
Jour << Nuit : pics (1-3h +++)
Régulée +++
Sécrétion inhibée par la lumière
MELATONINE
N acétylation + méthylation
NH
CH2CH2
NH2
HO
NH
CH2CH2
NH
H3CO
CO CH3
3/ Biosynthèse de la mélatonineII. Trp
Cerveau

Voie ���� ���� en pathologie
Acide Indoleacétique
Dérivés indoliques
TRP
Bactéries intestinales
NH
CH3
NH
Indol Scatol
(selles)
4/ Catabolisme intestinalII. Trp
Catabolisme à noyau fermé
NH
CHCH2
NH2
COOH

Carence en vit PP ( = ac. nicotinique = vit. B3 = ni acine)⇒⇒⇒⇒ Pellagre et érythème pellagroïde
L’Homme peut synthétiser la quantité requise de vit PP lorsque
l’apport alimentaire de TRP est suffisant
Sources TRPViandes/Œufs
Lait
Noix de coco
Sources vit PPViande/Poisson/Œuf
Légumes secs
Noix, amandes
⇒⇒⇒⇒ Pays pauvres +++ (maïs, riz)
4/ PathologieII. Trp

Pellagre
Physiopathologie:- vit. PP � NAD, NADP indispensables dans les réactionsd’oxydo-réduction cellulaires, la chaîne respiratoir e et la ΣΣΣΣ des AG
- les signes cliniques sont les conséquences des bas niveauxde NAD-NADP
Zones photoexposées
Les besoins εεεε sont + importants pour réparer les altérations produites par l’exposition solaire
Tissu cérébral où les dépenses εεεε sont importantes
Tissus à renouvellement rapide : Tube Digestif et PEAU

Pellagre
Signes cliniques : Règle des « 3D » Dermatose (zones exposées)Diarrhée Chronique
Démence
Diagnostic biologique :Dosage des métabolites urinaires de la niacine (seul indicateur)
� Méthylnicotinamide URINAIRE
Traitement : vit PP + polyvitamines
DesGroseilliers JP & Shiffman NJ., 1976
Pellagre

60% du TRP absorbé est détourné vers la ΣΣΣΣ de la sérotonine (en situation normale : 1% du TRP est utilisépour synthèse de la sérotonine)
Pellagre et érythème pellagroïde
Etiologies : Causes primairesCarence nutritionnelleMaladie d’Hartnup
Causes secondaires :Alcoolisme chroniqueSyndrome de malabsorption (Crohn, Maladie coeliaque ...)
Médicaments : Isoniazide, 5FU, phénobarbital …(inhibition métabolisme de la vit PP)
Tumeurs carcinoïdes

Maladie de HARTNUP
Traitement : administration de VIT PP
Diagnostic : détection d’Amino Acidurie spécifique avec recherche des
dérivés du catabolisme du TRP par la flore intestin ale
Signes cliniques : début vers 3 – 7 ans
rashs cutanés pellagroïdes sur les parties découvertes
épisodes neurologiques (ataxie)
�aminoacidurie spécifique
�rétention d’AA , en particulier TRP �, dans les intestins
Autosomique récessive : mutation du gène SLC6A19
Due à un trouble de la réabsorption tubulaire rénale et du transport intestinal des AA
monoamino-monocarboxyliques donnant lieu à

II. Métabolisme du tryptophane
NH
CHCH2
NH2
COOH
Tryptophane
Sérotonine
NAD+
AcétoacétylCoA
AA essentiel- Alimentation- Catabolisme prot. endogènes NADP+
Mélatonine
bactériesintestinales
IndolScatol
Catabolisme à noyau ouvert
Catabolisme à noyau fermé
5/ ConclusionII. Trp
IntestinSNCPlaquettes
SNC
Foie
Selles
PellagreMaladie de Hartnup
95%
1%

PHE: AA essentiel
III. Métabolisme de la phénylalanine
Synthèse hépatique +++
PHEhydroxylase
Dihydropteridinereductase
CofacteurH4-bioptérine
O2
irréversible
NADP NADPH,H+

PHE: AA essentiel
1/ Phénylcétonurie classique
PHEhydroxylase
Dihydropteridinereductase
CofacteurH4-bioptérine
O2
irréversible
����Phénylcétonurie
NADP NADPH,H+
III. Phe
Synthèse hépatique +++

Synthèse hépatique +++
Voie alternative du catabolisme de PHEau cours de la phénylcétonurie
PHE
Phenylacetate Phenyllactate
Phenylpyruvate
transaminase
PYR
ALA
Directement toxique pour les neurones +++

CH
CH2
Phénylalanine
NH3+
COO-
CO
CH2
COO-
CHOH
CH2
COO-
CO2
CH2
COO-
Ala
CH
CH2
Tyrosine
COOH
OH
PHEhydroxylase
phénylpyruvate
NH3+
phényllactate
phénylacétate
PyruvateTransaminase
Catabolisme alternatifpathologique
de Phe

La phénylcétonurie classique= de type I
1/ 25 000Autosomique récessive (mutation gène codant pour la PHE hydroxylase
Signes cliniques : à la fin de la 1ère année arriération mentale +++ irréversibleretard psychomoteur, de croissance, microcéphalietroubles de la pigmentation (peau et cheveux clairs)⇒⇒⇒⇒ état grabataire
Signes biologiques1- PHE ������������ dans le sang
métabolisme dévié vers les voies secondairestransamination ⇒ phénylpyruvate , phényllactate � ⇒⇒⇒⇒ URINES
2- Anomalies de TYR� Catécholamines, � Mélanine, altération des H. thyroïdiennes
3- Anomalies de TRP (anomalie associée par compétition )� Sérotonine

La phénylcétonurie classique
Test de dépistage dans les 1ers jours de vie: TEST DE GUTHRIEObligatoire en France
Ensemencement de Bactéries (ne peuvent pousser sans PHE) sur un milieu
contenant un analogue de PHE qui arrête toute croissance.
On ajoute une goutte de sang du Nné:
si [PHE] est ��� ⇒ croissance proportionnelle des bactéries
Confirmation : dosage de PHE
Pronostic : très favorable si le Nné est traité dès la naissanc e :
Régime strict pauvre en PHE, supplémenté en TYR, jus qu’à 6 ans
d’où intérêt

N. B :
La jeune femme phénylcétonurique souhaitant avoir un
enfant doit auparavant se replacer sous un régime très strict
pour éviter l’embryofœtopahie (malformations osseuses,
cardiaques et occulaires, microcéphalie et arriération
mentale).

PHE: AA essentiel
Synthèse hépatique +++
PHEhydroxylase
Dihydropteridinereductase
CofacteurH4-bioptérine
O2
irréversible
���� PCU maligneNADP NADPH,H+
2/ Phénylcétonurie maligneIII. Phe

Déficit de la dihydroptéridine réductase
déficit en tétrahydrobioptérine
Maladie autosomique récessive, rare
Signes cliniques de la phénylcétonurie classique
Traitement : tétrahydrobioptérine
La phénylcétonurie maligne= de type II
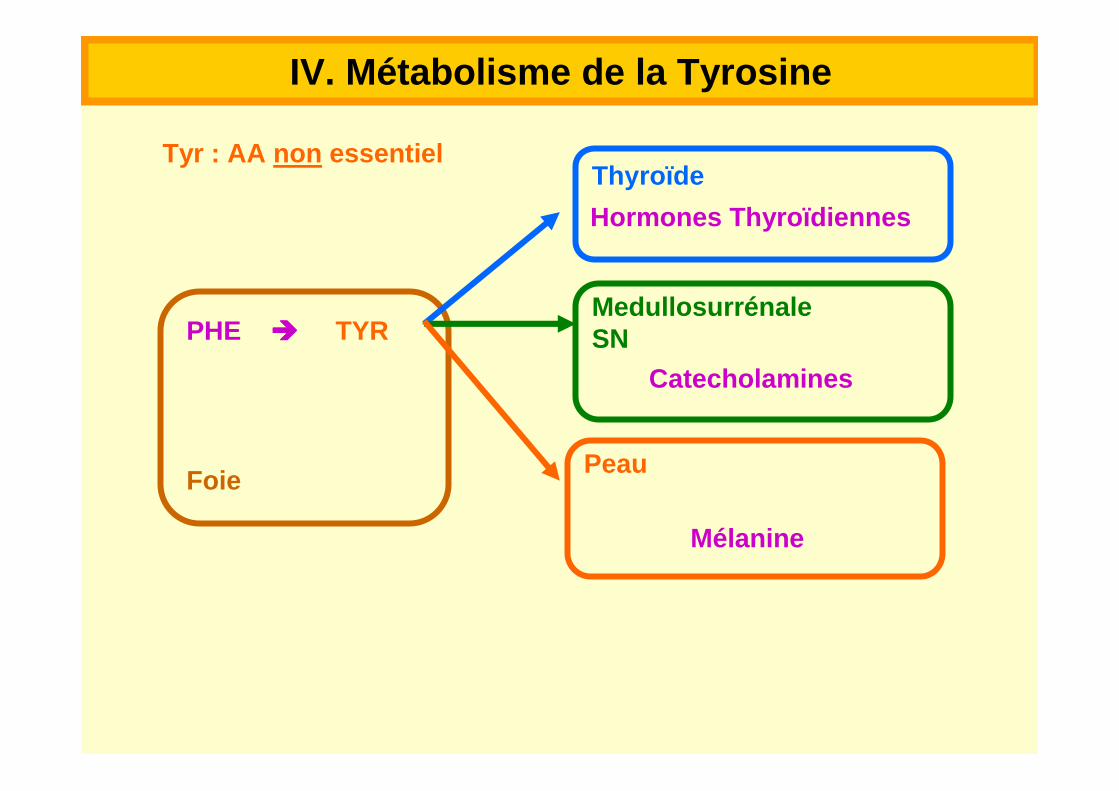
Tyr : AA non essentiel
PHE ���� TYR
Foie
Thyroïde
Hormones Thyroïdiennes
MedullosurrénaleSN
Catecholamines
Peau
Mélanine
IV. Métabolisme de la Tyrosine

Synthèse à partir de PHE
SurrénalesSN
TYR
DOPATYR hydroxylase (H4-bioptérine)
DopamineDOPA décarboxylase
Neurotransmetteur SNC
NoradrénalineDopamine hydroxylase
NeurotransmetteurSN Sympathique Périphérique
VMAAc vanillylmandelique
AdrénalineN méthyltransférase
HormoneNeurotransmetteur (SNC)
medullosurrénale
1/ Synthèse des catécholaminesIV. Tyr

TYR
DOPATyrosinase
Mélanines
Mélanocytes (peau)
� Albinisme
2/ Synthèse de la mélanineIV. Tyr
Polymérisation

TYR
3/ Synthèse des hormones thyroïdiennesIV. Tyr
Biochimie métabolique,Borg & Reeber
Triiodothyronine (T3)Thyroxine T4

Bergeron et al, 2003
Tyr transaminase
p OH phénylpyruvate oxydase
Homogentisate oxydase
MAA isomérase
Fumaryl acétoacétase
ique = alcaptone
cycle de Krebsnéoglucogenèse corps cétonique
4/ Catabolisme hépatiqueIV. Tyr

TYROSINE
P-Hydroxyphenylpyruvate
Tyrosine transaminase
Acide homogentisique (alcaptone)
p OH phenylpyruvate oxydase
Acide maleylacetoacetique
Homogentisate oxydase
Acide fumarylacetoacétique
Fumarate Acétoacétate
Fumaryl acetoacetase
TYROSINOSES
Succinylactone (inhibiteur ALAD )
� II Ulcères cornéensHyperkératose
� III Retard mental
� Alcaptonurie Arthrites
� I Anomalies hépatiques& rénales
MAA isomérase
5/ Pathologies : vue d’ensembleIV. Tyr

Bergeron et al, 2003
Tyr transaminase
p OH phénylpyruvate oxydase
Homogentisate oxydase
MAA isomérase
Fumaryl acétoacétase
ique = alcaptone
cycle de Krebsnéoglucogenèse corps cétonique
Alcaptonurie
6/ AlcaptonurieIV. Tyr

Alcaptonurie
- déficit en homogentisate oxydase = homogentisate 1,2-dioxygénase
- maladie héréditaire autosomique récessive
- accumulation d’acide homogentisique (alcaptone)
- éliminé dans les urines
- coloration brune des urines (formation de pigment
par oxydation de l’ac. homogentisique)
- dépôt de pigment (ocre / bleu) dans tissu conjonctif : ochronose
apparition vers 20 – 30 ans
- atteintes articulaires vers 40 – 60 ans

Ladjouze-Rezig, 2006
Alcaptonurie

Tyrosinose hépato rénale (I) déficit en Fumaryl acétoacétase
Forme Aiguë vomissements, diarrhées ++
mort en 6 mois - tableau de cirrhose
Forme Chronique Insuffisance Hépatique progressive , rachitisme
Crises de porphyrie : douleurs abdominales, signes neurologiques
Accumulation de Succinylactone (inhibiteur ALAD ) � ALA+++
Mort < 10 ans
Pronostic: fatal sans traitement
Age du diag. est important : avant l’âge de 2 mois: 75 à 96 % de survie
après l’âge de 2 mois : 29% survie
7/ Tyrosinose de type IIV. Tyr

Bergeron et al, 2003
Tyr transaminase
p OH phénylpyruvate oxydase
Homogentisate oxydase
MAA isomérase
ique
Fumaryl acétoacétase
Tyrosinose de type I
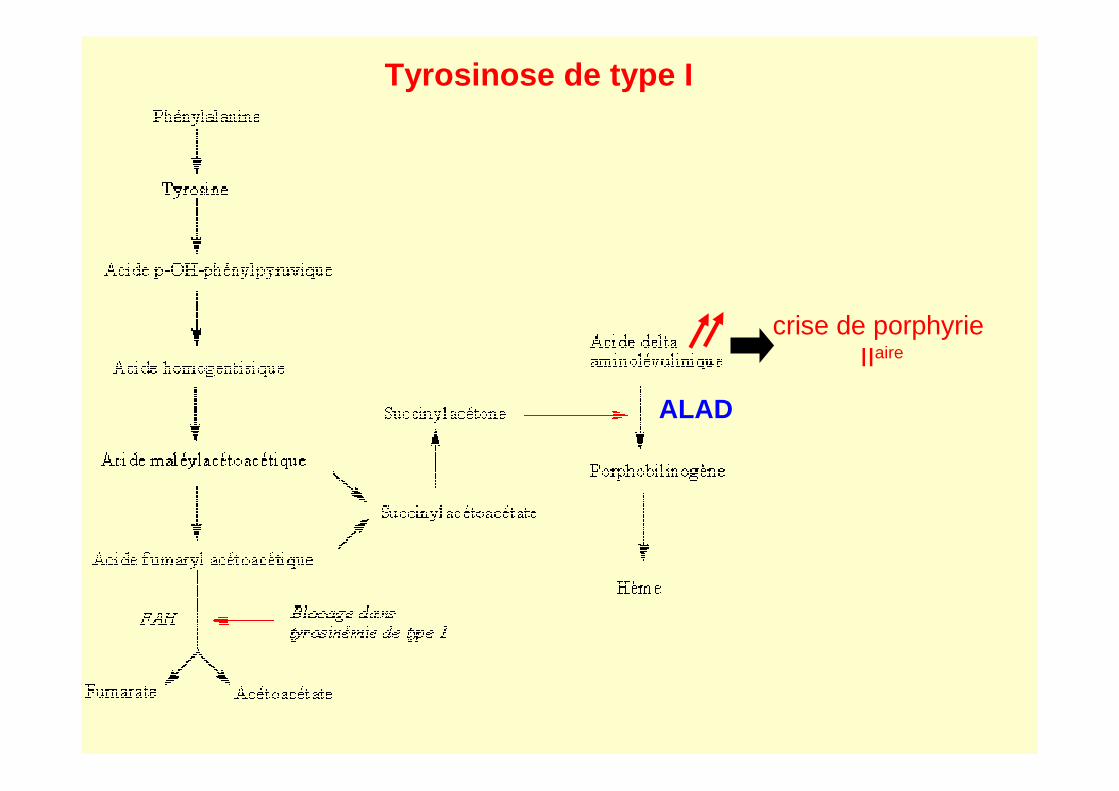
Tyrosinose de type I
ALAD
crise de porphyrieIIaire

Succinyacétone Acide δ amino-lévulinique

Diagnostic biologique :
� Chromatographie des AA plasmatiques et urinaires
� Dosage de la succinylacétone et de l’acide δ-aminolévulinique dans les urines
La présence dans le plasma et surtout dans les urines de succinylacétone est le signe pathognomonique de la tyrosinémie de type I
Tyrosinose de type I
Incidence: 1/ 120 000 naissances dans le monde
(1/ 17 000 au Québec ⇒⇒⇒⇒ dépistage)

Traitement :
But du traitement de la tyrosinémie de type I = réduction, voire l'abolition de la production de la succinylacétone.
� Le régime restreint en phénylalanine et tyrosine représente depuis longtemps la base du traitement de la maladie.
� Traitement fondé sur l'inhibition de la 2l'inhibition de la 2 èèmemeenzyme duenzyme ducatabolisme de la tyrosine,catabolisme de la tyrosine, p OH phénylpyruvate oxydase
par la NITISINONE = NTBC (herbicide)

Bergeron et al, 2003
Tyr transaminase
p OH phénylpyruvate oxydase
Homogentisate oxydase
MAA isomérase
ique
Fumaryl acétoacétase
Tyrosinose de type I
NITISINONE

Tyrosinose occulo-cutanée (II) (Sd de RICHNER-HANHART)
Déficit en Tyrosine transaminase
Maladie autosomique récessive
Accumulation de TYR ds le sang et les tissus
ulcérations oculaires
bulles, érosions cutanées
retard mental
Tyrosinémie ��������
Traitement: régime pauvre en PHE et TYR +++
8/ Tyrosinose de type IIIV. Tyr

Tyr transaminase
p OH phénylpyruvate oxydase
Homogentisate oxydase
MAA isomérase
Fumaryl acétoacétase
ique = alcaptone
cycle de Krebsnéoglucogenèse corps cétonique
Tyrosinosede type II

CHROMATOGRAMME PLASMATIQUE

Phénylalanine Tyrosine
Phénylacetate Phényllactate
Phénylpyruvate
Hormones thyroïdiennes
Dopa
Dopamine
Noradrénaline
Adrénaline
Mélanines
AlbinismePhénylcétonurie
PHEhydroxylase
P-Hydroxyphenylpyruvate
Acidehomogentisique
Acide maleylacetoacetique
Acide fumarylacétoacétique
Fumarate Acetoacétate
Alcaptonurie
Tyrosinose II
Tyrosinose I
Tyrosinose III

Leucinose= maladie des urines à odeur de sirop d’érable
due au déficit en de la déshydrogénase des acides α-cétoramifiés produitspar la transamination des acides aminés ramifiés.
Augmentation +++ des 3 AA ramifiés (Val Ile et Leu)et par l’apparition d’un 4ème : alloisoleucine





























