Embed Size (px)
Citation preview

Katalyse mit NHCs –Metallkatalyse und
Organokatalyse
MC II Seminar Vortrag am 16.12.2010 von Roxana Lorenz, Tamara
Wittmann und Florian Koschitzki

Gliederung
1. Einführung
2. Kreuzkupplungsreaktionen
3. Goldkomplexe in der Katalyse
4. Metathese
5. Organokatalyse

Vergleich von NHCs mit tertiären Phosphanen als LigandenGemeinsamkeiten:
• Starker, neutrale σ-Elektronen Donoren
• Geringe π-Rückbindung von Metall zu Ligand -> hohe Elektonendichte
Unterschiede:
• P-C Bindung dissoziiert bei hohen Temperaturen – NHCs sind thermisch stabiler
• Phosphane sind oxidationsempfindlich
• Topologie: Phosphansubstituenten zeigen vom Metallzentrum weg („Kegel“) – NHC-Substituenten formen eine „Tasche“ um das Metallatom -> größerer Einfluss der Substituenten auf das Metallzentrum
• NHCs haben stärkere Donor-Eigenschaften
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813

Bindungsdissoziationsenergien
[3] N. M. Scott, S. P. Nolan Eur. J. Inorg. Chem. 2005, 1815-1828

Katalytischen Aktivität von NHC Liganden
• Sterische und elektronische Effekte können prinzipiell getrennt voneinander beeinflusst werden
• Sterisch anspruchsvolle Substituenten am Stickstoff - Elektronendichte des Carben-Kohlenstoff-Atoms wird nur gering beeinflusst
• Der heterozyklische Ring ist für die elektronischen Eigenschaften verantwortlich
• Die elektronischen Unterschiede zwischen verschiedenen NHCs sind sehr gering
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813

Gliederung
1. Einführung
2. Kreuzkupplungsreaktionen
3. Goldkomplexe in der Katalyse
4. Metathese
5. Organokatalyse

Katalysezyklus
1. Oxidative Addition– RX addiert sich an Pd
– Pd0 -> PdII
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
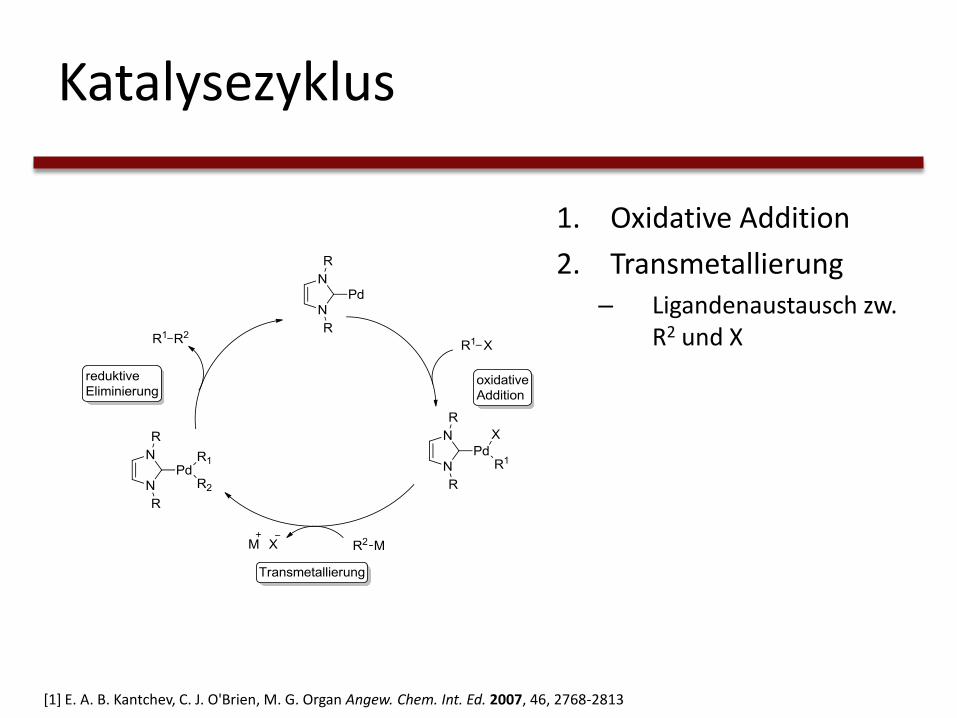
Katalysezyklus
1. Oxidative Addition
2. Transmetallierung– Ligandenaustausch zw.
R2 und X
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813

Katalysezyklus
1. Oxidative Addition
2. Transmetallierung
3. Reduktive Eliminierung– Bindungsbildung der
Liganden
– PdII -> Pd0
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813

Vorteile von NHC-Liganden
1. Starke s-Bindung der NHCs führt dazu, dass das Pd Zentrum zur oxidativen Addition fähig ist – auch mit Arylchloriden oder Alkylhaliden
2. Sterisch anspruchsvolle NHCs erleichtern die reduktive Eliminierung
3. Die starke Pd-NHC Bindung und die begrenzten möglichen Zerfallswege stellen sicher, dass das Metallatom in einem löslichen, katalytisch aktiven Zustand bleibt, auch wenn nur ein NHC-Ligand koordiniert ist
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
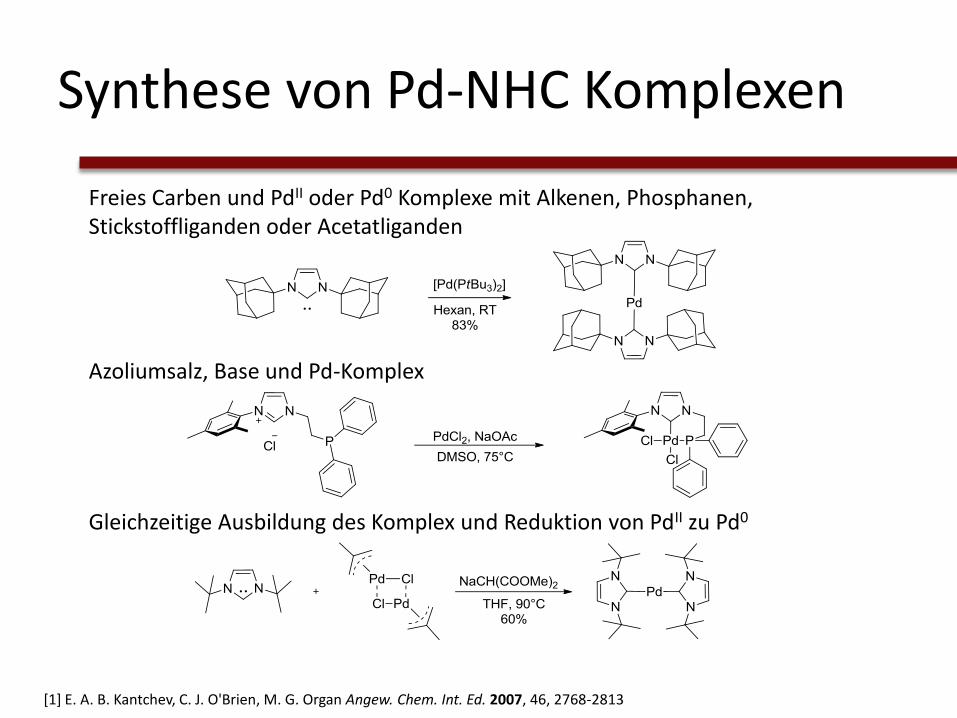
Synthese von Pd-NHC Komplexen
Freies Carben und PdII oder Pd0 Komplexe mit Alkenen, Phosphanen, Stickstoffliganden oder Acetatliganden
Azoliumsalz, Base und Pd-Komplex
Gleichzeitige Ausbildung des Komplex und Reduktion von PdII zu Pd0
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813

Suzuki-Miyaura Reaktion
•Große Anzahl stabiler Borverbindungen verfügbar•Ungiftige Nebenprodukte•Läuft in einer großen Auswahl von Lösungsmitteln ab•Aryliodide und –bromide sind reaktiv, Arylchloride eher unreaktiv
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
Bindung Mittlere Bindungsenergie
C-Cl 327 kJ/mol
C-Br 272 kJ/mol
C-I 214 kJ/mol
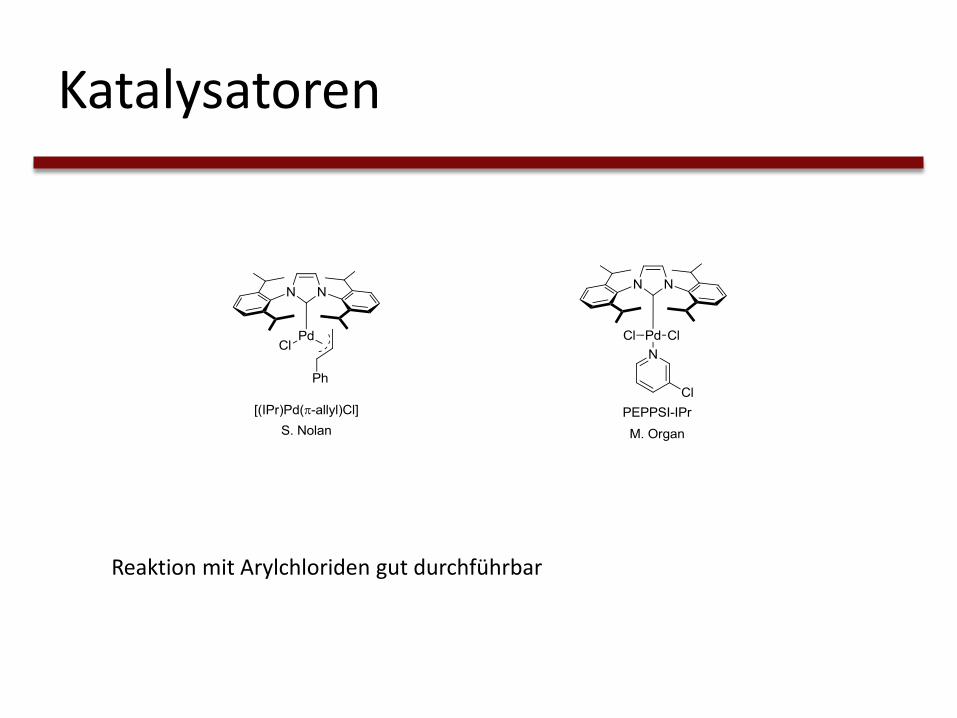
Katalysatoren
Reaktion mit Arylchloriden gut durchführbar
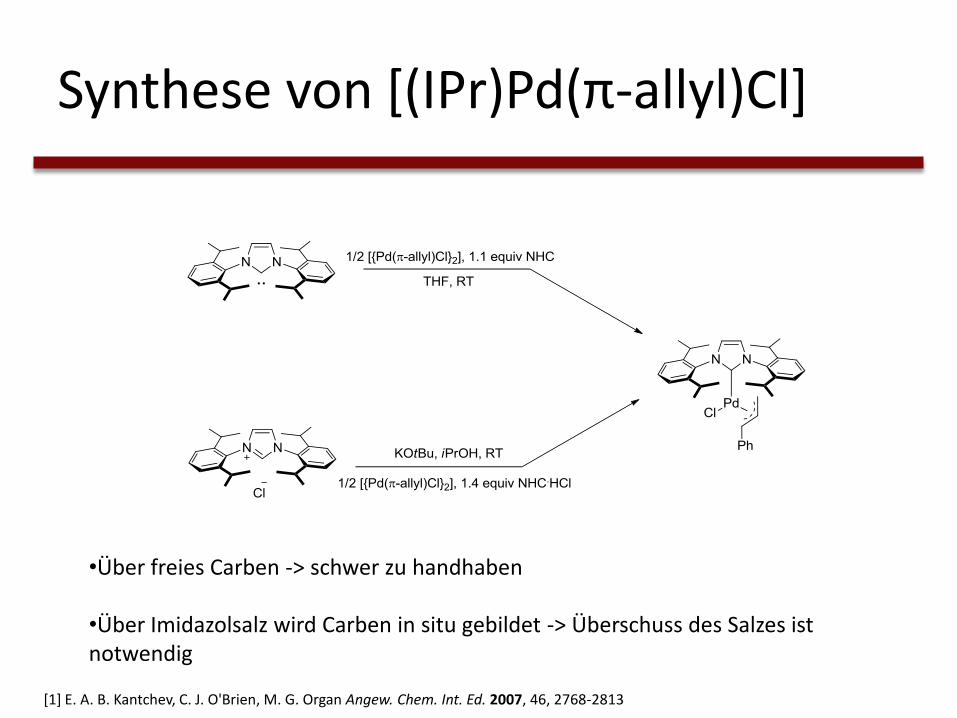
Synthese von [(IPr)Pd(π-allyl)Cl]
•Über freies Carben -> schwer zu handhaben
•Über Imidazolsalz wird Carben in situ gebildet -> Überschuss des Salzes ist notwendig
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
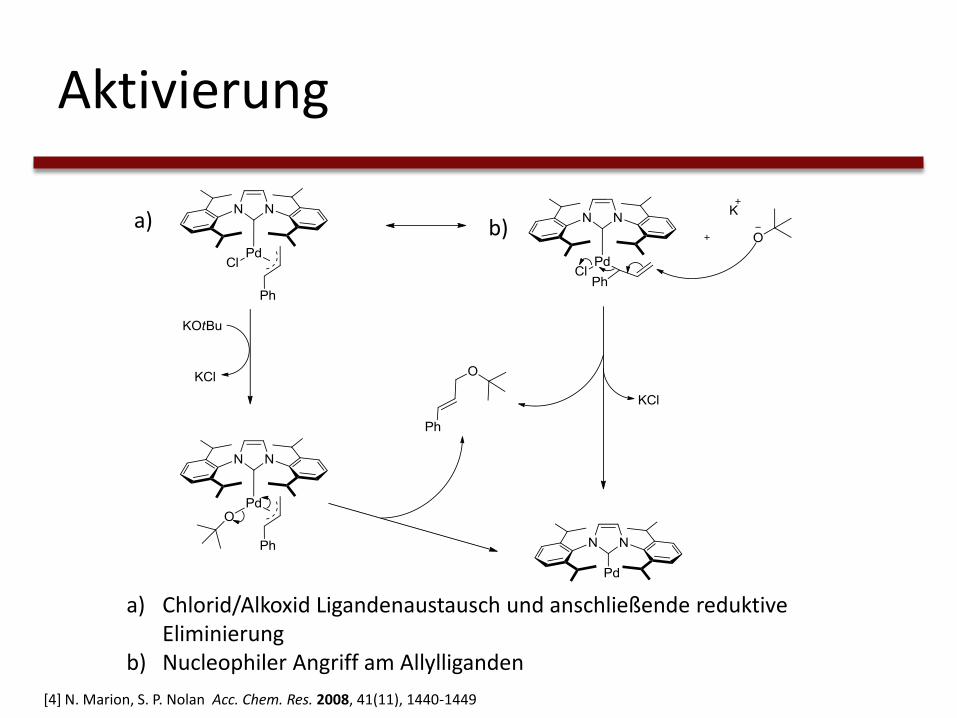
Aktivierung
a) Chlorid/Alkoxid Ligandenaustausch und anschließende reduktive Eliminierung
b) Nucleophiler Angriff am Allylliganden
a) b)
[4] N. Marion, S. P. Nolan Acc. Chem. Res. 2008, 41(11), 1440-1449
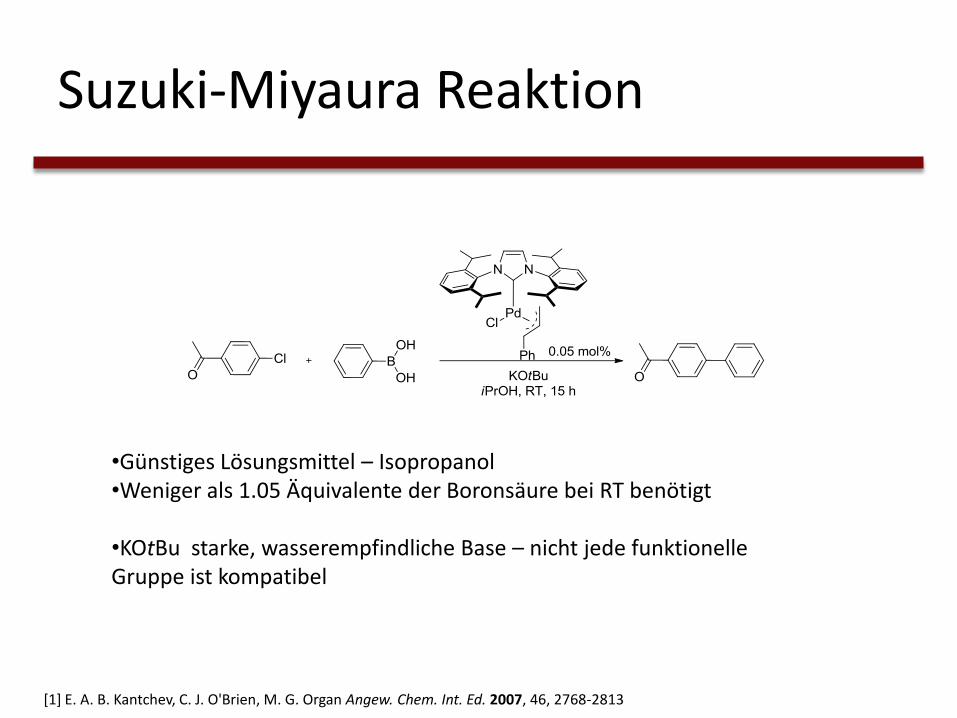
Suzuki-Miyaura Reaktion
•Günstiges Lösungsmittel – Isopropanol•Weniger als 1.05 Äquivalente der Boronsäure bei RT benötigt
•KOtBu starke, wasserempfindliche Base – nicht jede funktionelle Gruppe ist kompatibel
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
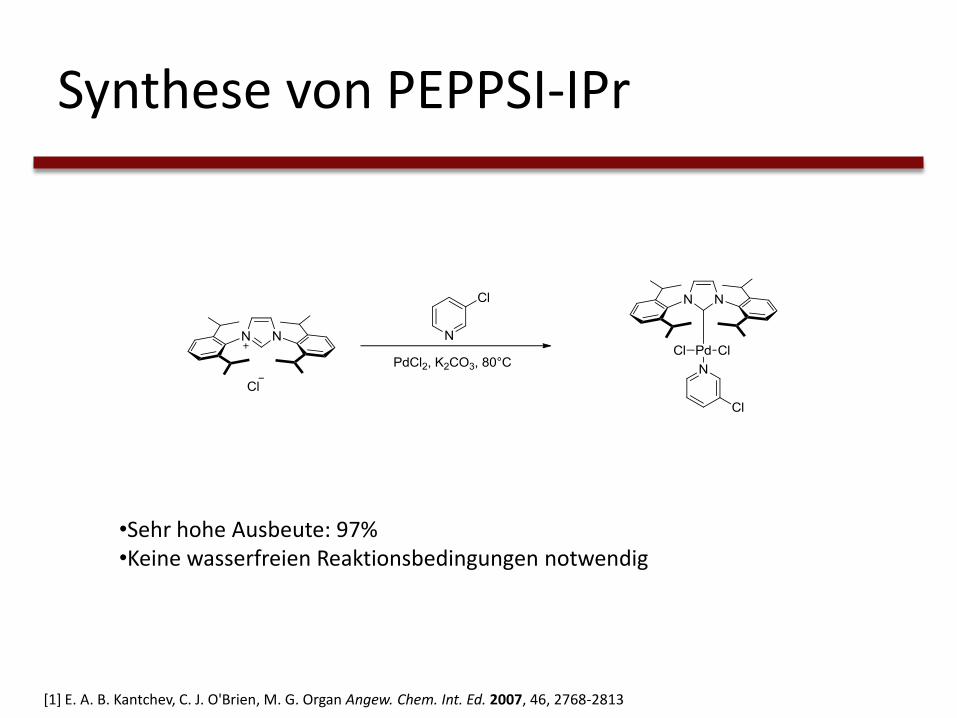
Synthese von PEPPSI-IPr
•Sehr hohe Ausbeute: 97%•Keine wasserfreien Reaktionsbedingungen notwendig
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
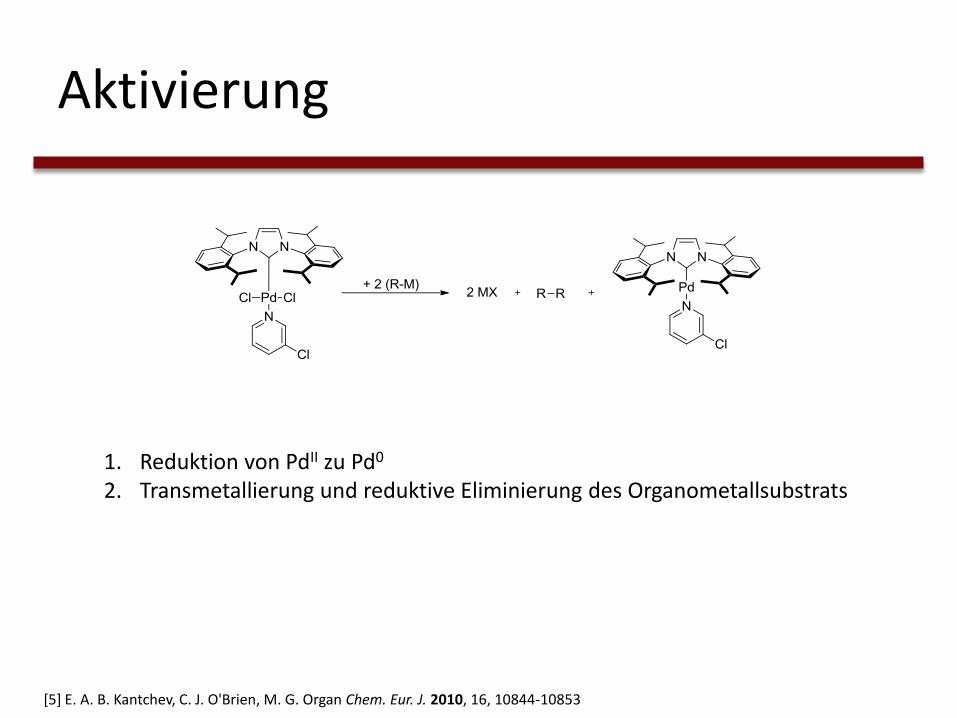
Aktivierung
1. Reduktion von PdII zu Pd0
2. Transmetallierung und reduktive Eliminierung des Organometallsubstrats
[5] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Chem. Eur. J. 2010, 16, 10844-10853
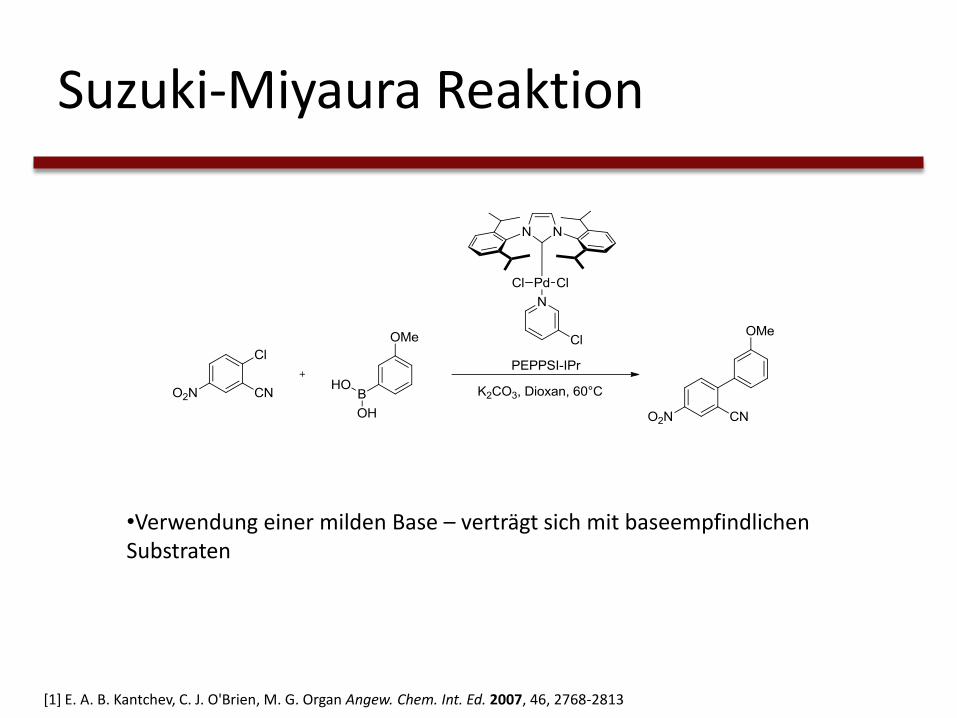
Suzuki-Miyaura Reaktion
•Verwendung einer milden Base – verträgt sich mit baseempfindlichen Substraten
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
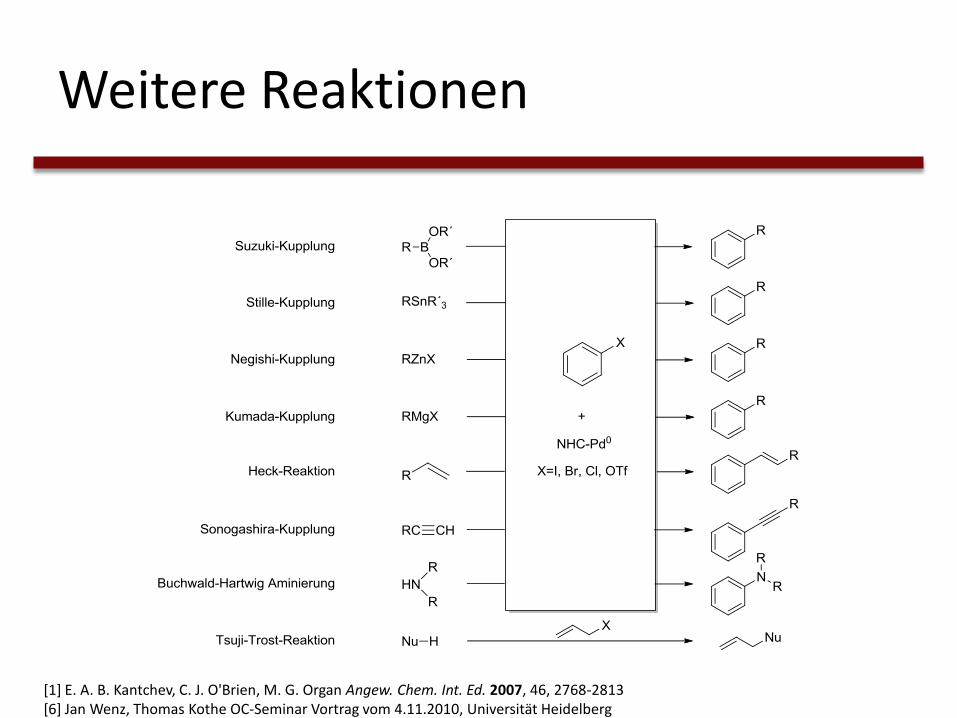
Weitere Reaktionen
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813[6] Jan Wenz, Thomas Kothe OC-Seminar Vortrag vom 4.11.2010, Universität Heidelberg

Gliederung
1. Einführung
2. Kreuzkupplungsreaktionen
3. Goldkomplexe in der Katalyse
4. Metathese
5. Organokatalyse
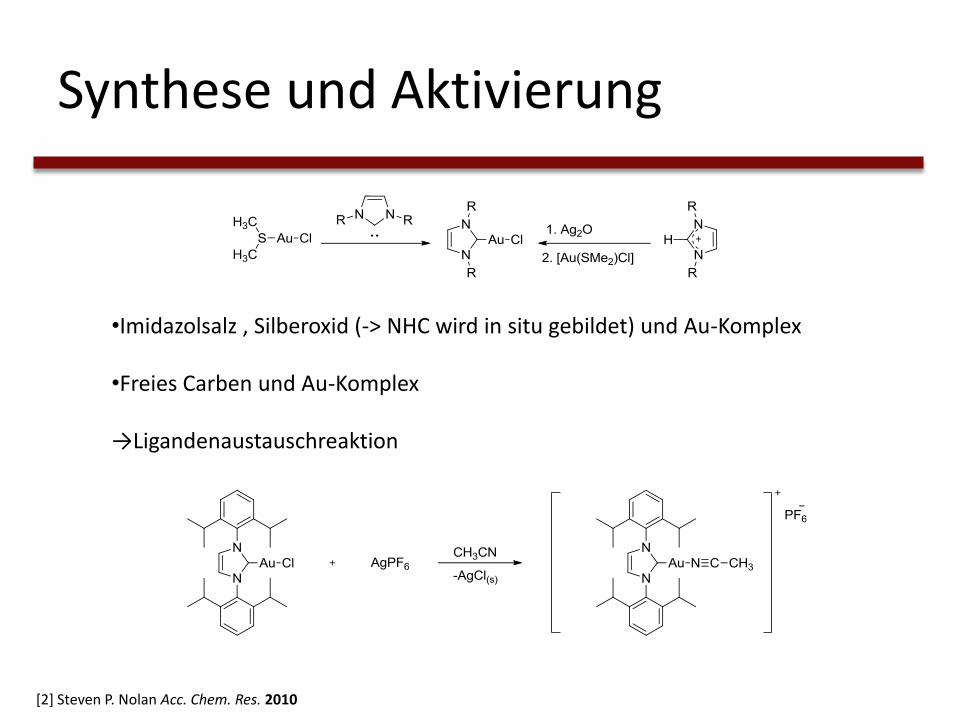
Synthese und Aktivierung
•Imidazolsalz , Silberoxid (-> NHC wird in situ gebildet) und Au-Komplex
•Freies Carben und Au-Komplex
→Ligandenaustauschreaktion
[2] Steven P. Nolan Acc. Chem. Res. 2010
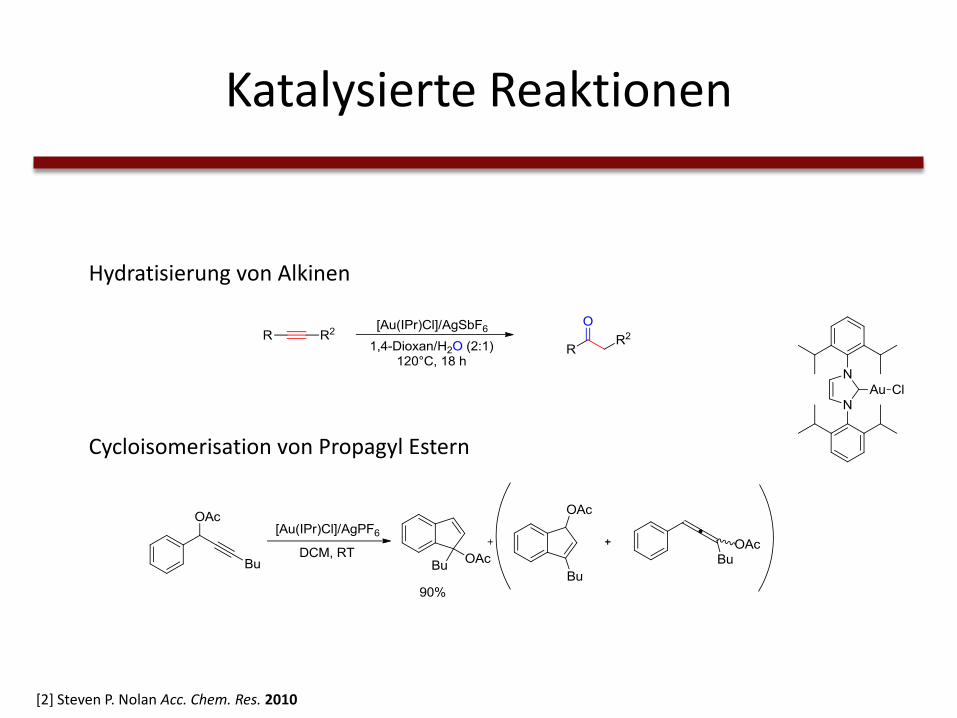
Katalysierte Reaktionen
Hydratisierung von Alkinen
[2] Steven P. Nolan Acc. Chem. Res. 2010
Cycloisomerisation von Propagyl Estern

Vorteile von NHC-Liganden in Au katalysierten Reaktionen
• Führen teilweise zu anderen Reaktivitäten/Selektivitäten als Phosphanliganden
• Komplexe sind thermisch stabiler -> extreme Reaktionsbedingungen sind unproblematisch
• Ganz neue Komplexe können synthetisiert werden, deren Phosphananaloga bis jetzt nicht synthetisiert werden konnten
[2] Steven P. Nolan Acc. Chem. Res. 2010

Gliederung
1. Einführung
2. Kreuzkupplungsreaktionen
3. Goldkomplexe in der Katalyse
4. Metathese
5. Organokatalyse

Metathese
• griechisch meta = Wechsel, thesis = Position
• Methode zur Neuknüpfung von C-C-Bindungen durch Austausch von Reste verschiedener Moleküle (Bsp. Alkenmetathese)
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787; [8] Advanced information on the Nobel Prize in Chemistry 2005, 2005, Kungl. Vetenskaakademien

Geschichte der Olefin-Metathese
• 1950: Ziegler => Polymerisation von Ethylen
Weitere Berichte folgten, aber Mechanismus immer unbekannt
• 1963: Banks und Bailey => „Olefindisproportionierung“
• 1967: N. Calderon : Polymerisation anderer Cycloolefine
• Calderon: Polymerisation von cyclischen Alkenen und die Disproportionierung acyclische Alkene gleicher Typ von Reaktion => „Olefinmetathese“
• 1970: Mechanismus von Chauvin und Herisson
• 2005: Nobelpreis Für Chauvin, Grubbs und Schrock für die Entwicklung neuer Katalysatoren
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787

• Austausch von Alkyl-Gruppen von Alkenen über eine [2+2]Cycloaddition/-reversion mit einem Metall-Carben-Komplex als Katalysator und einem Metallcyclobutan-Intermediat
• Un-/symmetrisch substituierte Alkene führen zu einem Produktgemisch ( E/Z-Isomere)
• 1. Schritt: Initiierung des aktiven Metall-Carben-Komplexes
1. Cycloaddition 2. Cycloreversion
[9] T. Laue, A. Plagens John Wiley, 2005, 0-470-01040-1
Mechanismus der Olefinmetathese
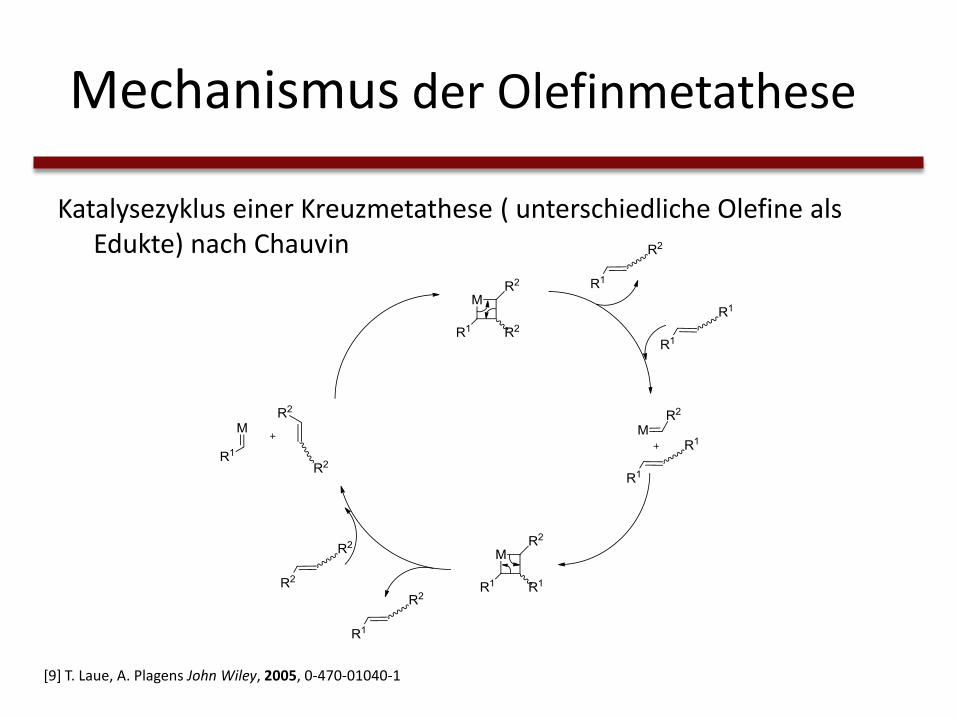
Katalysezyklus einer Kreuzmetathese ( unterschiedliche Olefine als Edukte) nach Chauvin
[9] T. Laue, A. Plagens John Wiley, 2005, 0-470-01040-1
Mechanismus der Olefinmetathese

• Katalysezyklus vereinfacht:
– die Ringöffnung in der Gegenrichtung wird nicht berücksichtigt
– Neben-/Produkte können auch weiter reagieren (E/Z-Isomere)
• Schritte im Zyklus sind reversibel -> Olefin-Produktgemisch; Ausbeute bestimmt durch thermodynamisches Gleichgewicht
• Ausbeute der gewünschten Produkte kann durch bestimmte Katalysatoren verbessert werden
• Entfernen von Reaktionsprodukten aus dem Reaktionsgemisch beeinträchtig Gleichgewicht (z.B. Entfernen von Ethylen -> Rückrk wird unterdrückt)
[9] T. Laue, A. Plagens John Wiley, 2005, 0-470-01040-1
Mechanismus der Olefinmetathese

• Früher: Multikomponenten Systeme
• Beanspruchten zu spezielle Reaktionsbedingungen und eine lange Aktivierungsdauer
• Moderne Katalysatoren Ruthenium- und Molybdänbasiert (stabile Metall-Carben-Komplexe & direkte WW mit der Doppelbindung des Substrates)
[10] C. Eischenbroich: Organonetallchemie Teubner, 1990, 3-519-23501-3
Katalysatoren

• Metall-Kohlenstoff-Doppelbindungen führen zu Metall-Carben-Komplexe
• Schrock-Carben: Triplett-Carben
σ-Hinbindung und π-Rückbindung jeweils durch ein Elektron des Metalls & Liganden
• Nucleophilie: starke M-π-C(Carben)-Rückbindung und Abwesenheit von –I-Substituenten am C-Atom
[10] C. Eischenbroich: Organonetallchemie Teubner, 1990, 3-519-23501-3
Schrock-Katalysatoren
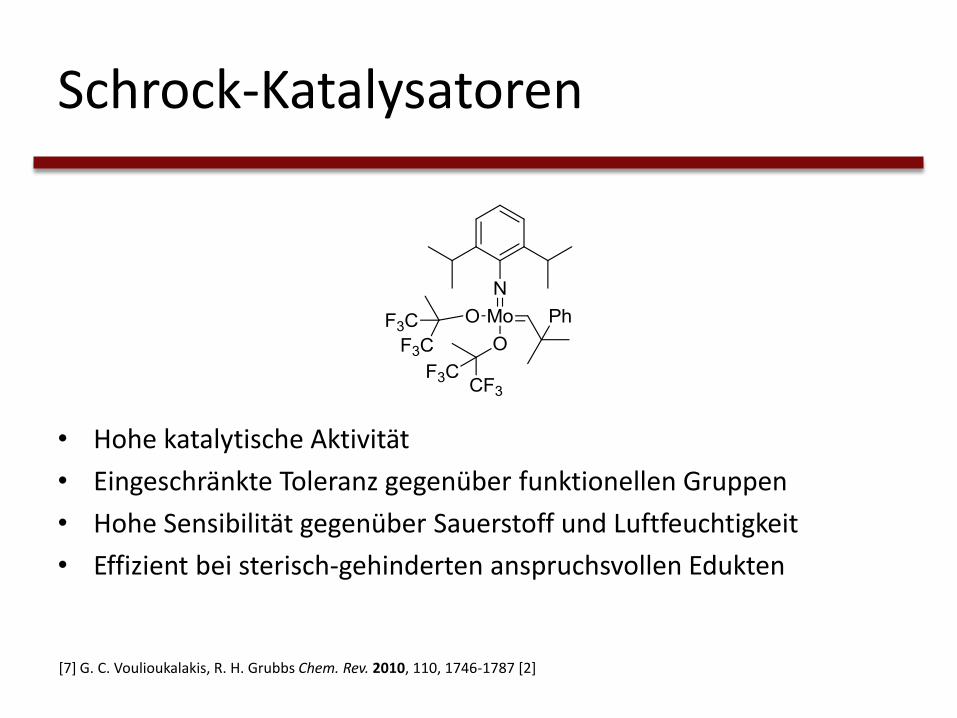
• Hohe katalytische Aktivität
• Eingeschränkte Toleranz gegenüber funktionellen Gruppen
• Hohe Sensibilität gegenüber Sauerstoff und Luftfeuchtigkeit
• Effizient bei sterisch-gehinderten anspruchsvollen Edukten
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787 [2]
Schrock-Katalysatoren

• Erste metatheseaktive Rutheniumalkylidkomplexe = Grubbs-Katalysatoren 1. Generation
• Geringere Aktivität
• Einfachere Herstellung und Handhabung
• Stabiler gegenüber Sauerstoff und Luftfeuchtigkeit
• Toleriert größere Anzahl an funktionellen Gruppen
[11] L.S. Hegedus: Transition metals in the synthesis of complex organic molecules-2nd ed., 1999, 1-891389-04-1
Grubbs-Katalysatoren
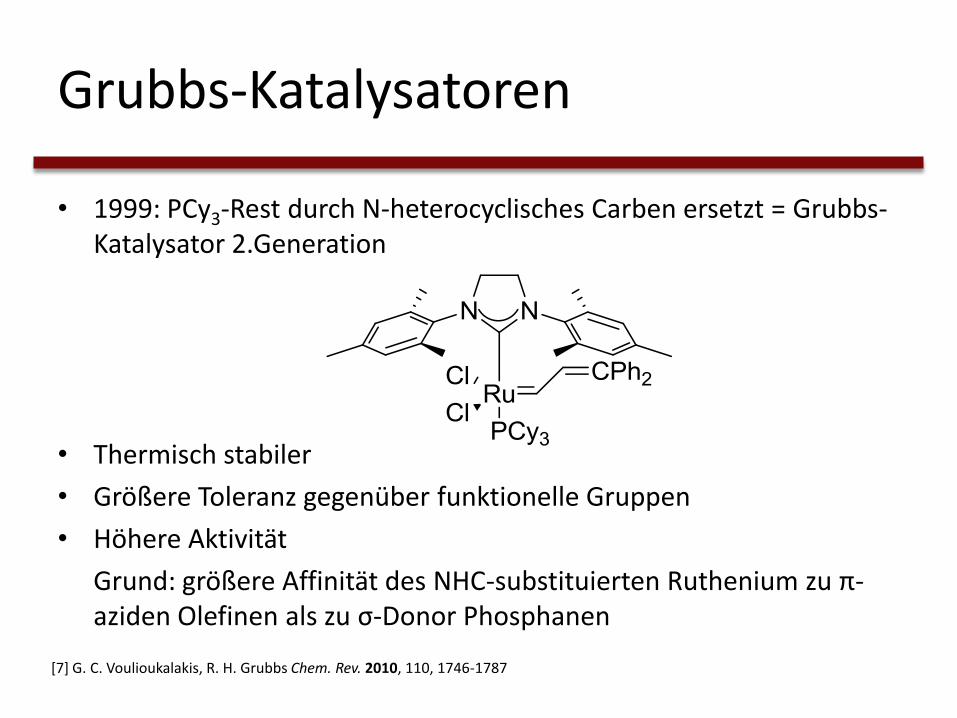
• 1999: PCy3-Rest durch N-heterocyclisches Carben ersetzt = Grubbs-Katalysator 2.Generation
• Thermisch stabiler
• Größere Toleranz gegenüber funktionelle Gruppen
• Höhere Aktivität
Grund: größere Affinität des NHC-substituierten Ruthenium zu π-aziden Olefinen als zu σ-Donor Phosphanen
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787
Grubbs-Katalysatoren
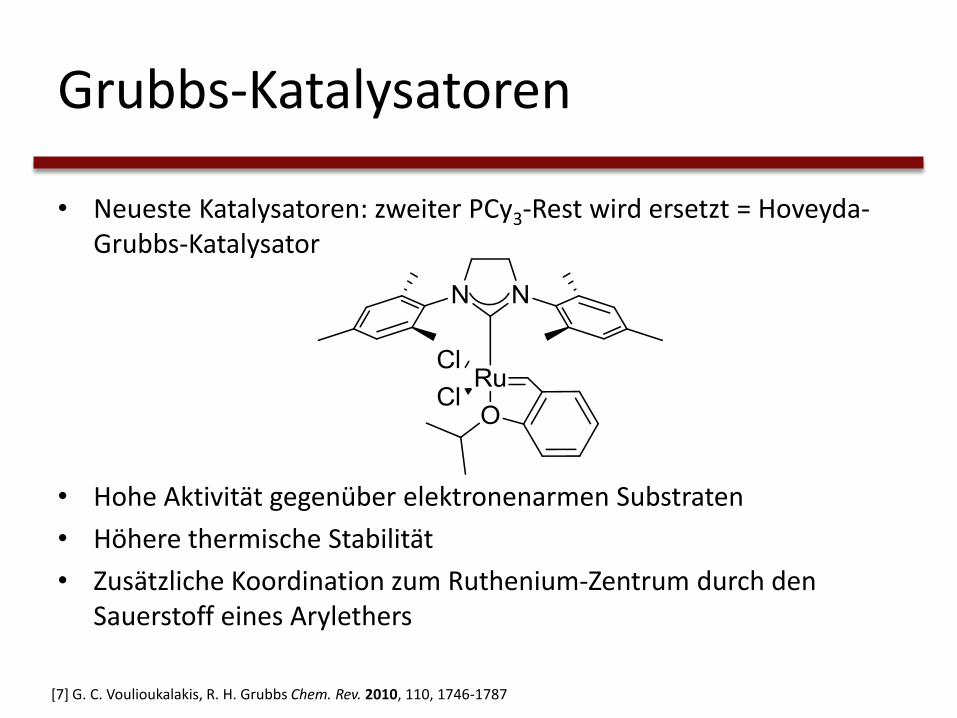
• Neueste Katalysatoren: zweiter PCy3-Rest wird ersetzt = Hoveyda-Grubbs-Katalysator
• Hohe Aktivität gegenüber elektronenarmen Substraten
• Höhere thermische Stabilität
• Zusätzliche Koordination zum Ruthenium-Zentrum durch den Sauerstoff eines Arylethers
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787
Grubbs-Katalysatoren
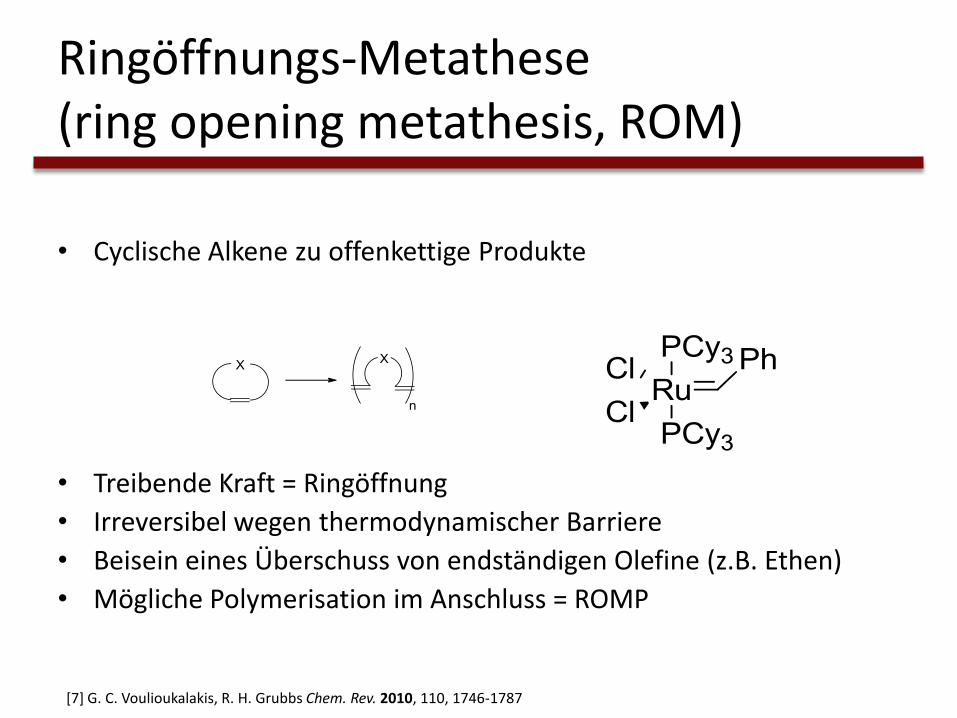
• Cyclische Alkene zu offenkettige Produkte
• Treibende Kraft = Ringöffnung
• Irreversibel wegen thermodynamischer Barriere
• Beisein eines Überschuss von endständigen Olefine (z.B. Ethen)
• Mögliche Polymerisation im Anschluss = ROMP
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787
Ringöffnungs-Metathese (ring opening metathesis, ROM)
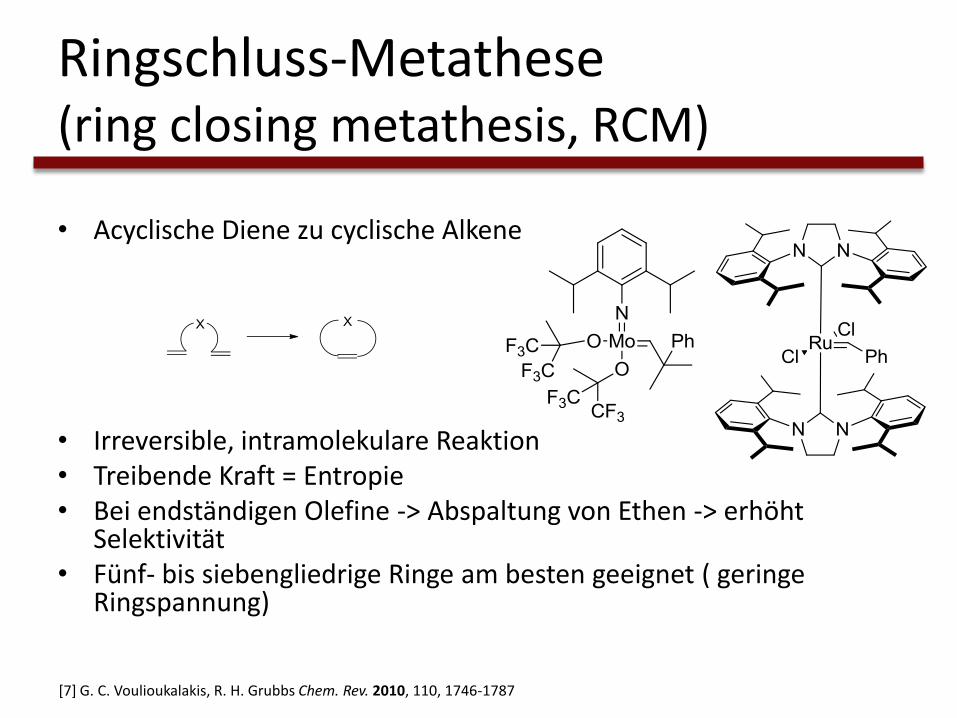
Ringschluss-Metathese(ring closing metathesis, RCM)
• Acyclische Diene zu cyclische Alkene
• Irreversible, intramolekulare Reaktion• Treibende Kraft = Entropie• Bei endständigen Olefine -> Abspaltung von Ethen -> erhöht
Selektivität• Fünf- bis siebengliedrige Ringe am besten geeignet ( geringe
Ringspannung)
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787
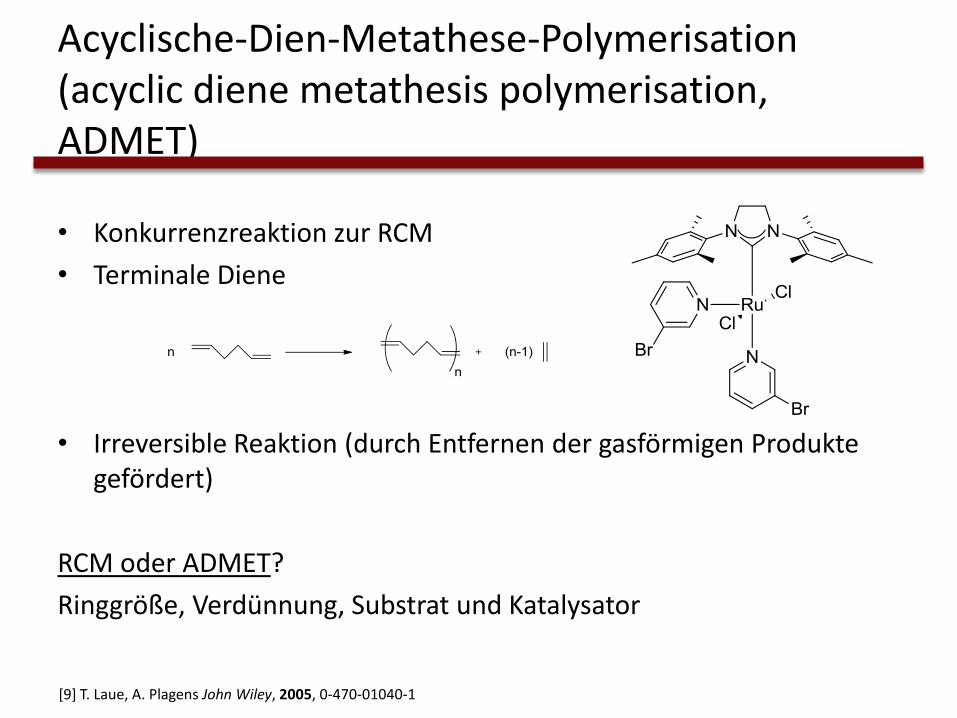
Acyclische-Dien-Metathese-Polymerisation (acyclic diene metathesis polymerisation, ADMET)
• Konkurrenzreaktion zur RCM
• Terminale Diene
• Irreversible Reaktion (durch Entfernen der gasförmigen Produkte gefördert)
RCM oder ADMET?
Ringgröße, Verdünnung, Substrat und Katalysator
[9] T. Laue, A. Plagens John Wiley, 2005, 0-470-01040-1
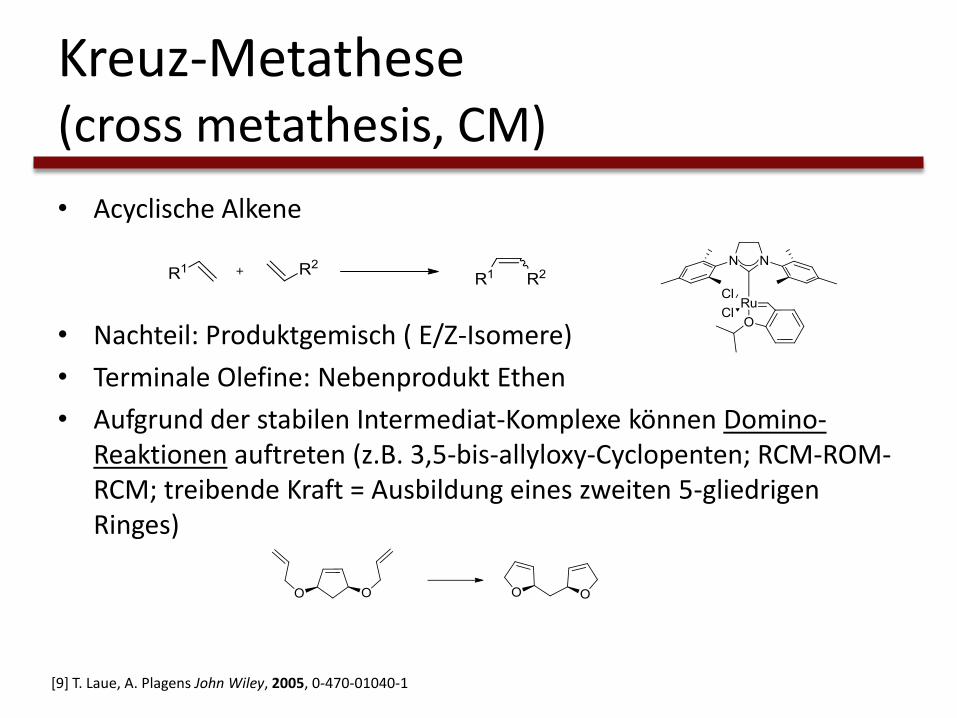
Kreuz-Metathese(cross metathesis, CM)
• Acyclische Alkene
• Nachteil: Produktgemisch ( E/Z-Isomere)
• Terminale Olefine: Nebenprodukt Ethen
• Aufgrund der stabilen Intermediat-Komplexe können Domino-Reaktionen auftreten (z.B. 3,5-bis-allyloxy-Cyclopenten; RCM-ROM-RCM; treibende Kraft = Ausbildung eines zweiten 5-gliedrigen Ringes)
[9] T. Laue, A. Plagens John Wiley, 2005, 0-470-01040-1

Anwendungen der Olefinmetathese
• Bildung von Ringsysteme
• Produktion von Schlüssel-Chemikalien für Polymer- und Petrochemie
• Zubereitung spezieller Polymere von Cycloalkanen durch ROMP
• Naturstoffchemie
[10] C. Eischenbroich: Organonetallchemie Teubner, 1990, 3-519-23501-3

N-heterocyclische Carbene in der Organokatalyse
Florian Koschitzki
16.12.2010

Inhaltsverzeichnis
• Allgemein
• Benzoin-Kondensation
• Stetter-Reaktion
• Morita-Baylis-Hillman Reaktion
• Fazit
• Quellen

Allgemein
Was ist Organokatalyse?

Allgemein
• Definition: Katalyse org. Reaktionen durch rein org. Moleküle
→ komplett metallfrei
• „Chemische Antwort auf das Vorbild der Natur“
• Bereits 1859 von Justus von Liebig entdeckt
• 1912 erste asym. org.kat. Reaktion von Bredig und Fiske
• Jedoch hervorragende Ergebnisse in der Metall-Katalyse
→ Organokatalyse vorerst in Schatten gestellt

Allgemein
Vorteile:
– Feuchtigkeits- und sauerstoffunempfindlich
– Bei Normalatmosphäre/ RT druchführbar
– Kostengünstig und leichterhältlich
– Geringere Toxizität, da metallfrei
→ relevant für Pharmaindustrie
Nachteil:
– Prozentual viel Kat. notwendig
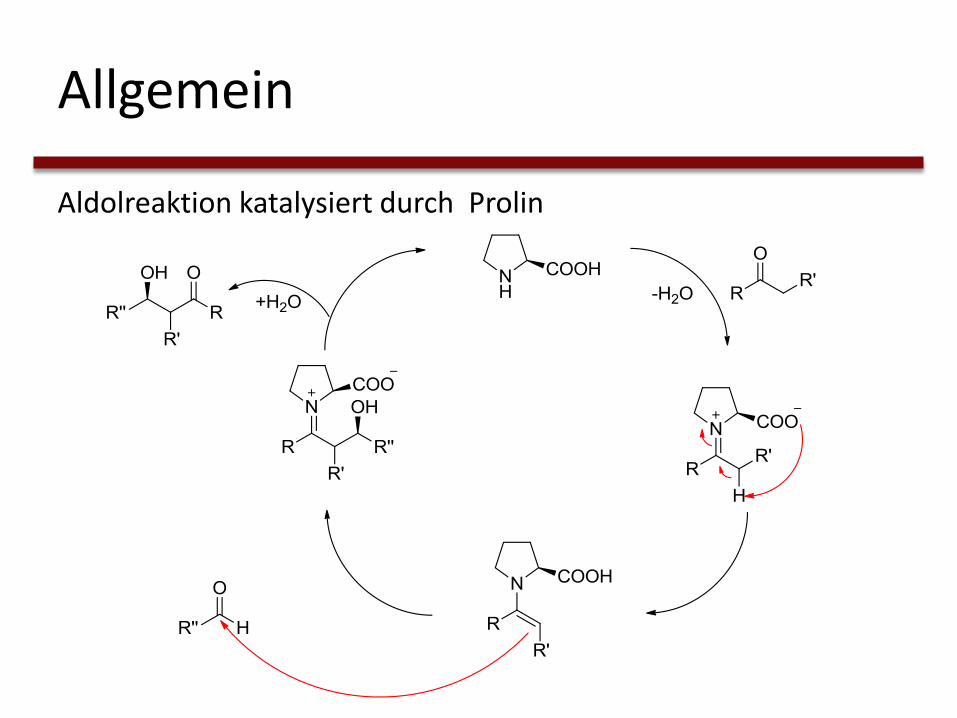
Allgemein
Aldolreaktion katalysiert durch Prolin
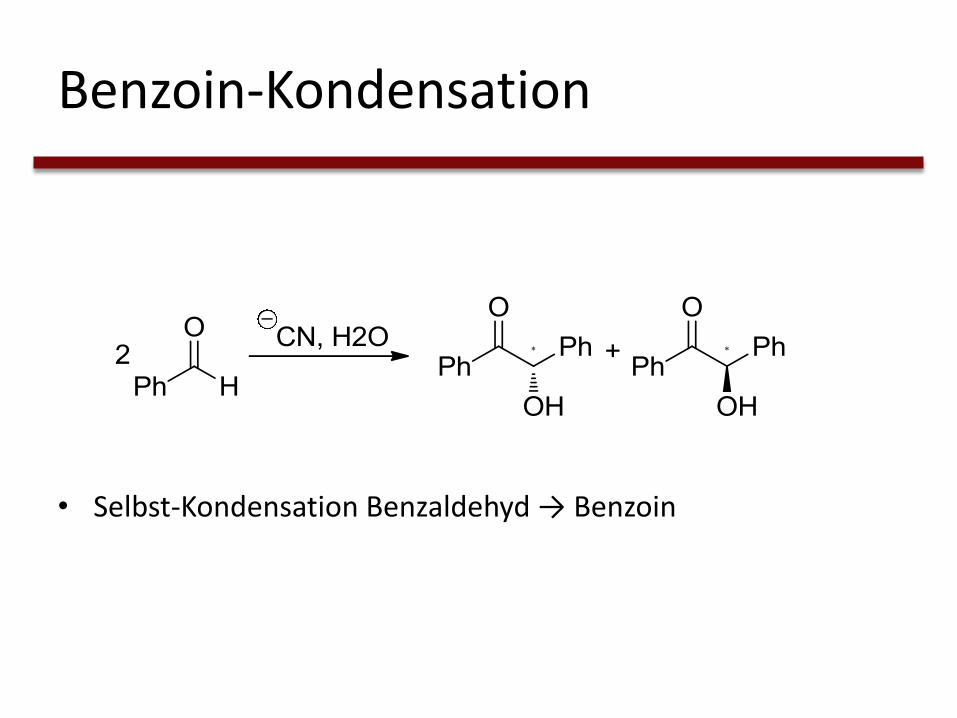
Benzoin-Kondensation
• Selbst-Kondensation Benzaldehyd → Benzoin
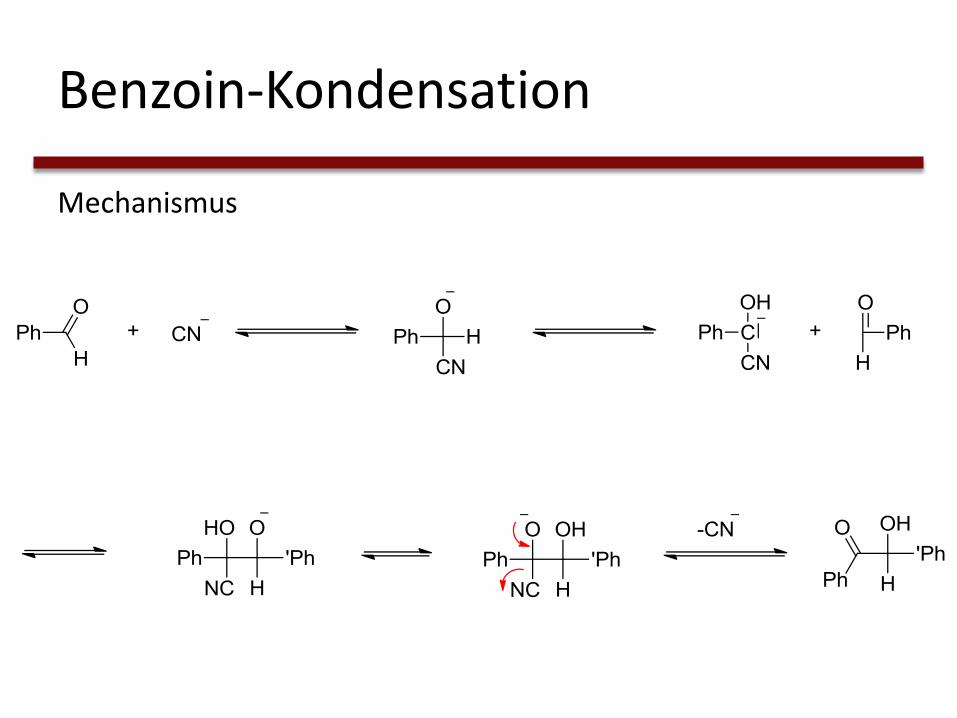
Benzoin-Kondensation
Mechanismus

Benzoin-Kondensation
• C-C knüpfende Reaktion → Katalyse von hohem Interesse
• Stereozentrum wird kreiert
Nachteile:
– Enatiomerengemisch
– Toxizität des Cyanids
– Nur mit arom. Aldehyden
→ Katalyse durch NHCs
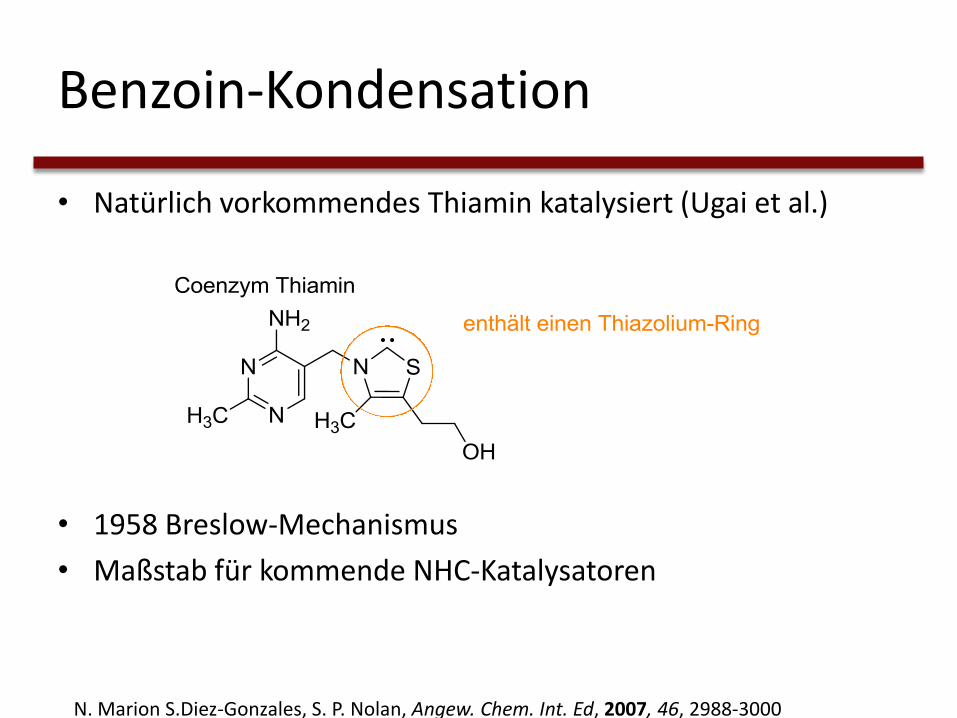
Benzoin-Kondensation
• Natürlich vorkommendes Thiamin katalysiert (Ugai et al.)
• 1958 Breslow-Mechanismus
• Maßstab für kommende NHC-Katalysatoren
N. Marion S.Diez-Gonzales, S. P. Nolan, Angew. Chem. Int. Ed, 2007, 46, 2988-3000
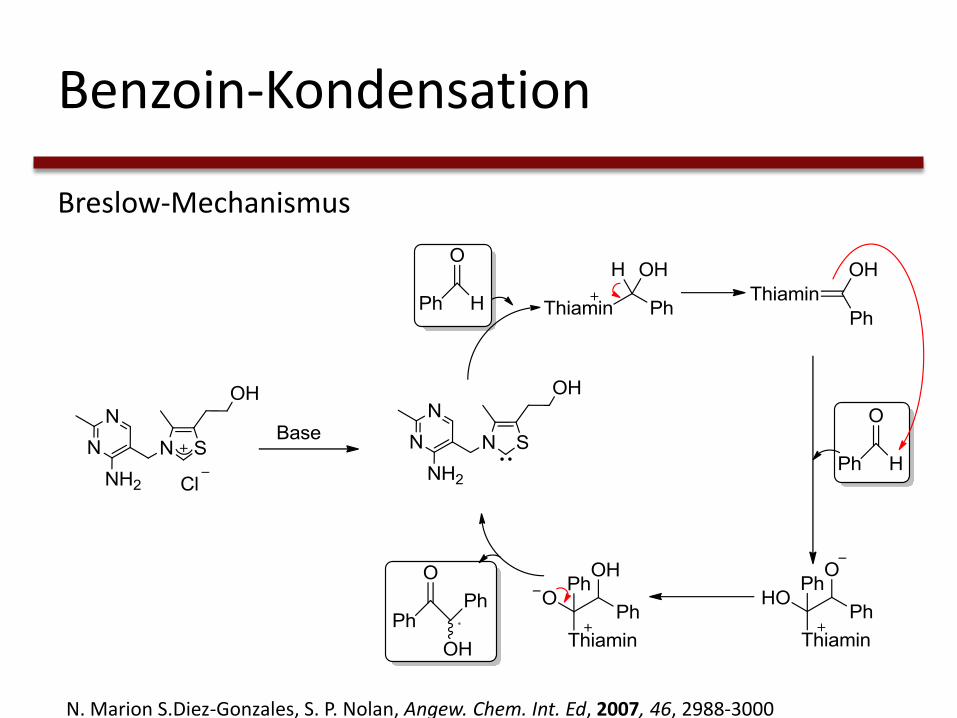
Benzoin-Kondensation
Breslow-Mechanismus
N. Marion S.Diez-Gonzales, S. P. Nolan, Angew. Chem. Int. Ed, 2007, 46, 2988-3000

Benzoin-Kondensation
→ Verbesserte Ausbeuten
→ Stereoselektivität durch asymmetrische NHCs
→ Auch mit aliphatischen Aldehyden
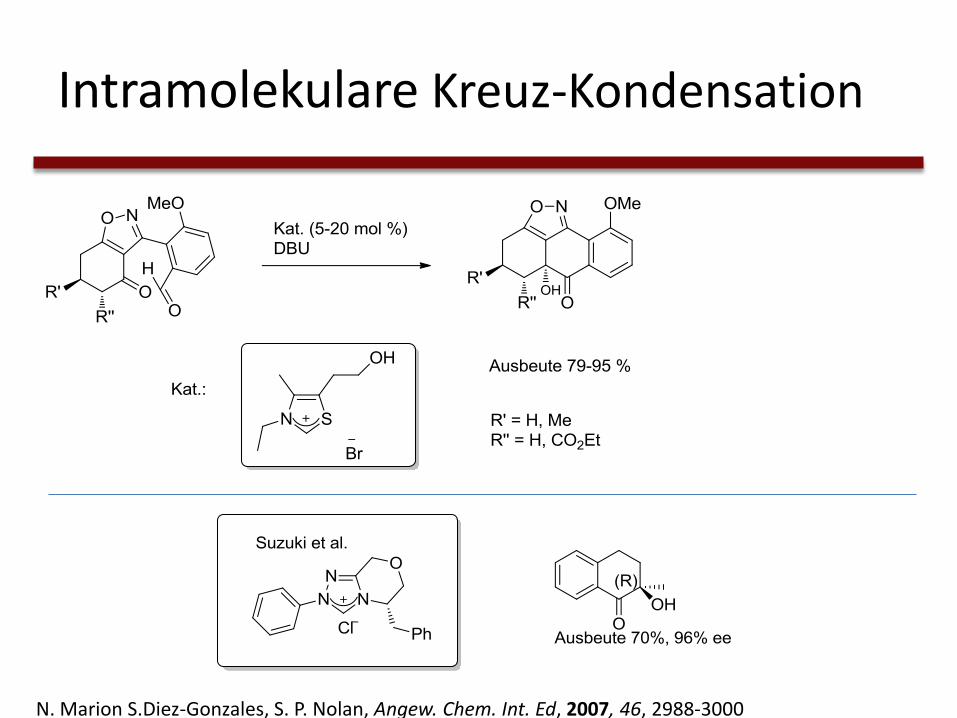
Intramolekulare Kreuz-Kondensation
N. Marion S.Diez-Gonzales, S. P. Nolan, Angew. Chem. Int. Ed, 2007, 46, 2988-3000
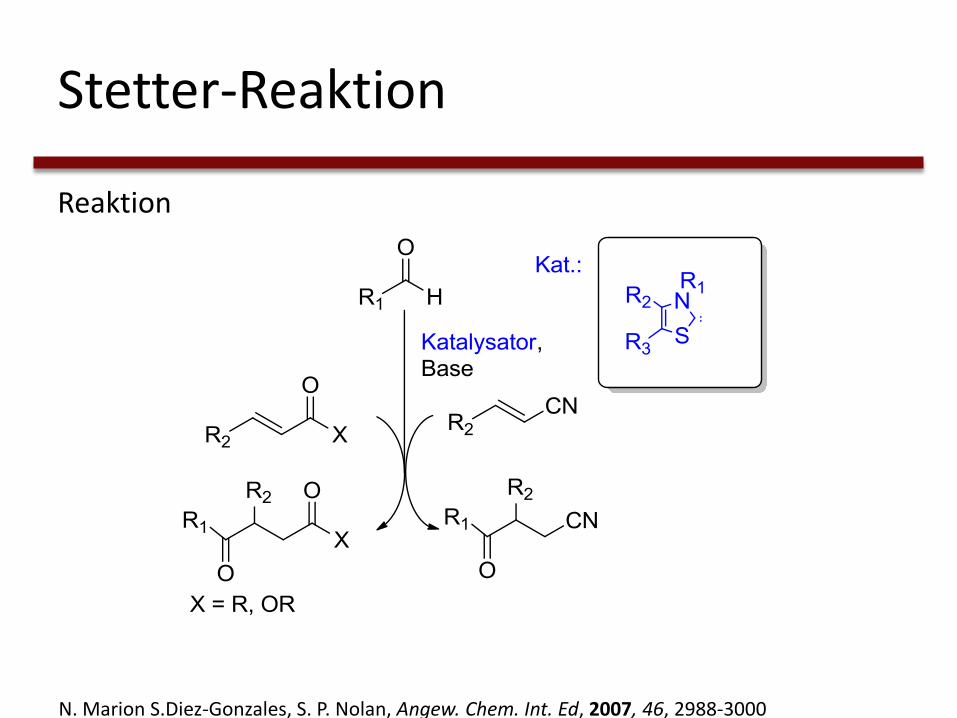
Stetter-Reaktion
Reaktion
N. Marion S.Diez-Gonzales, S. P. Nolan, Angew. Chem. Int. Ed, 2007, 46, 2988-3000
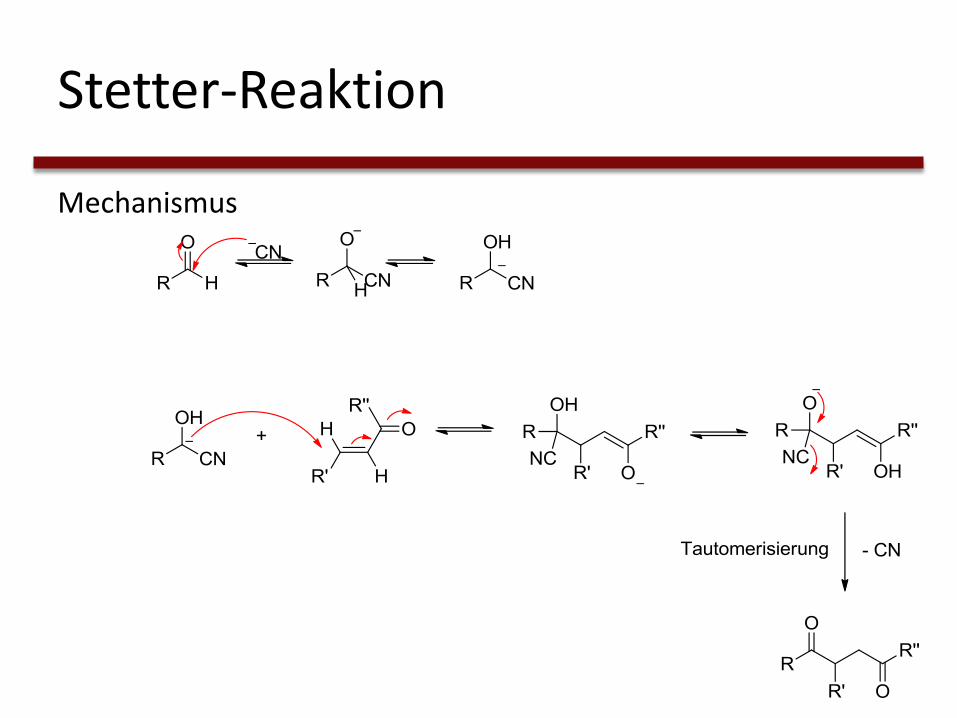
Stetter-Reaktion
Mechanismus

Stetter-Reaktion
• Präparative Darstellung von:
1,4-Diketonen, 4-Ketoestern und 4-Ketonitrilen
• Durch Cyanid katalysiert
• Nur aromatische Reste möglich
• Keine Stereoselektivität
→ NHCs
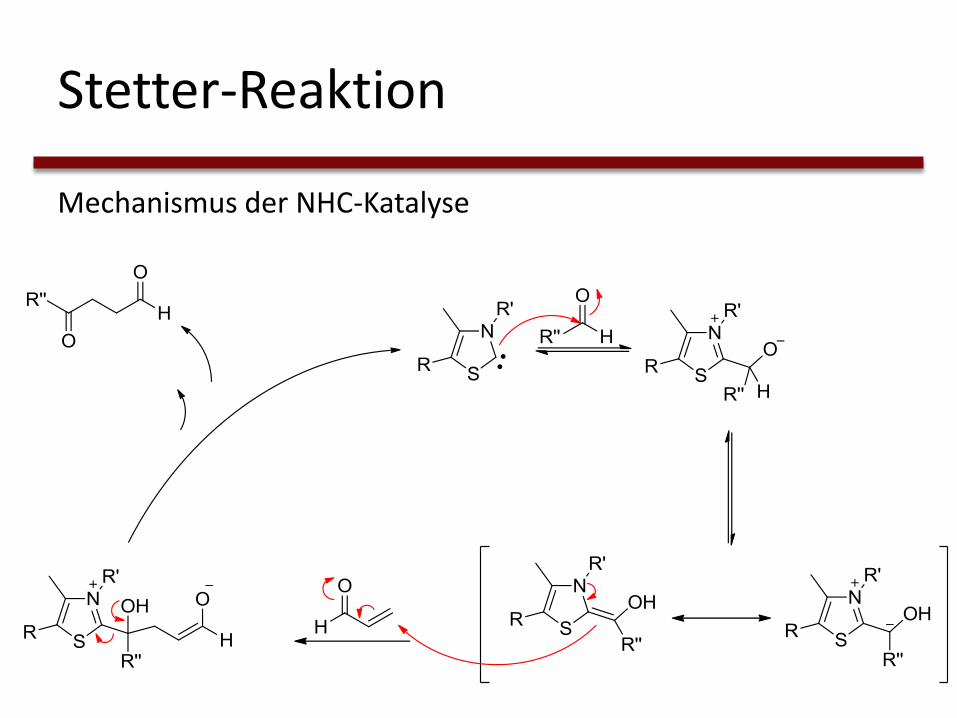
Stetter-Reaktion
Mechanismus der NHC-Katalyse
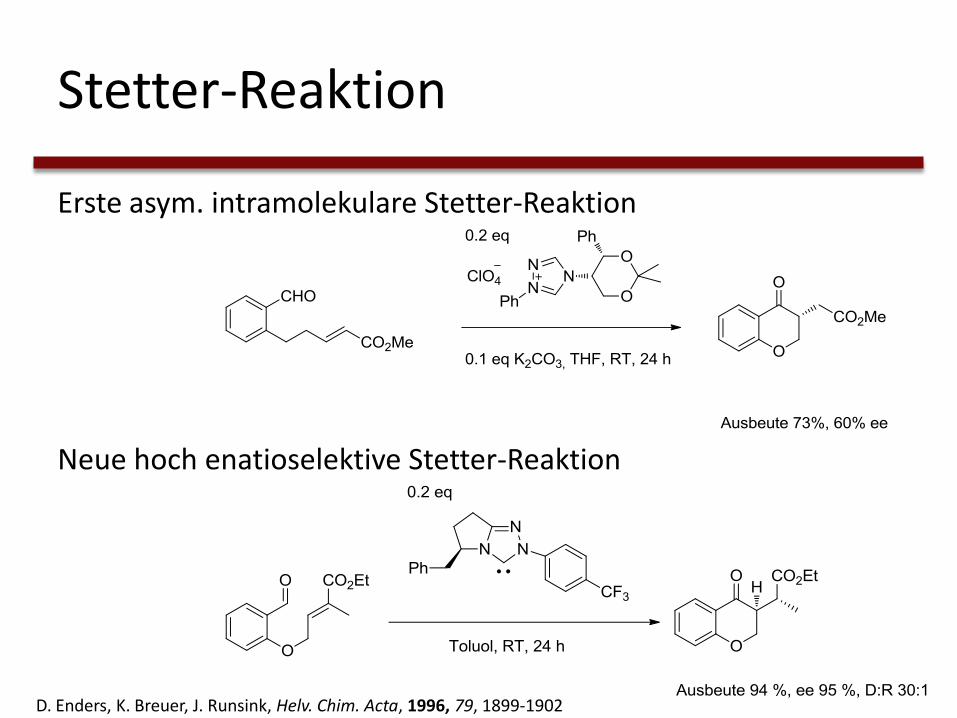
Stetter-Reaktion
Erste asym. intramolekulare Stetter-Reaktion
Neue hoch enatioselektive Stetter-Reaktion
D. Enders, K. Breuer, J. Runsink, Helv. Chim. Acta, 1996, 79, 1899-1902
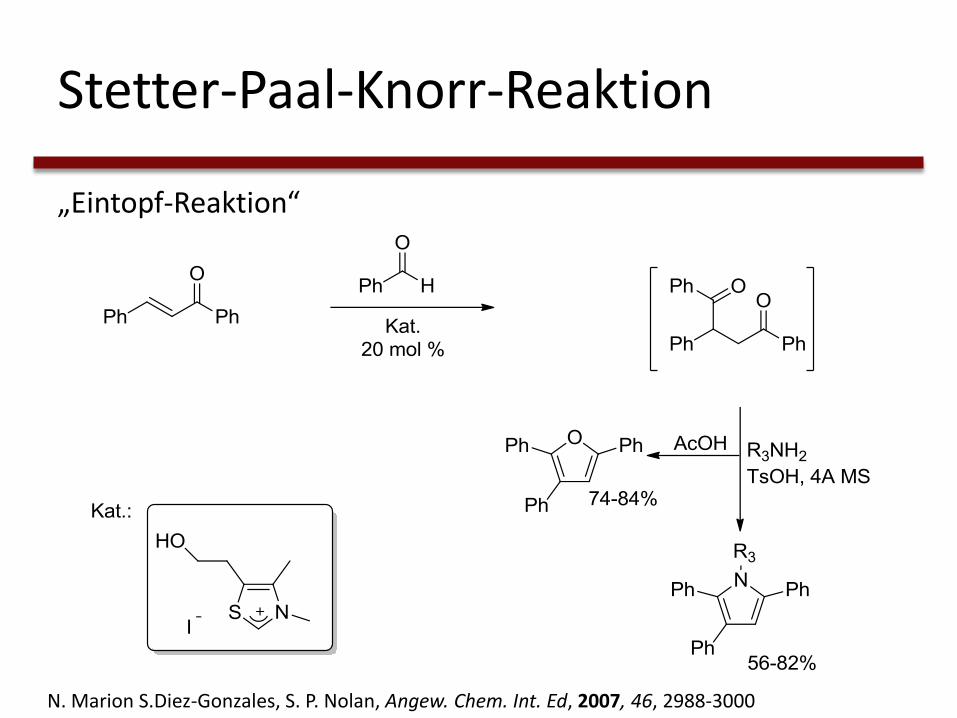
Stetter-Paal-Knorr-Reaktion
„Eintopf-Reaktion“
N. Marion S.Diez-Gonzales, S. P. Nolan, Angew. Chem. Int. Ed, 2007, 46, 2988-3000
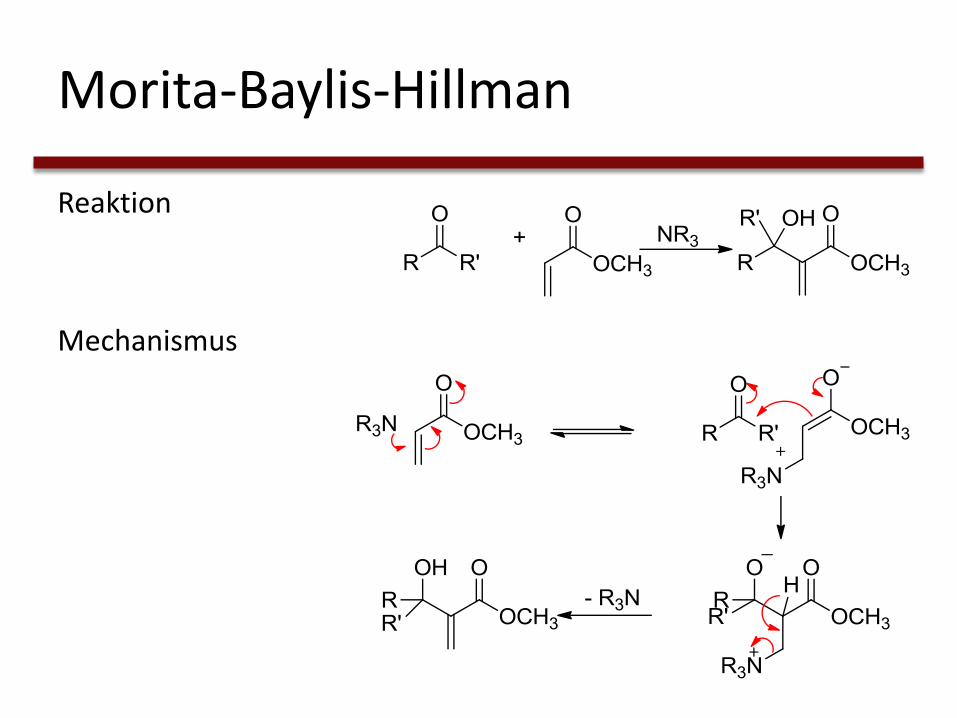
Morita-Baylis-Hillman
Reaktion
Mechanismus
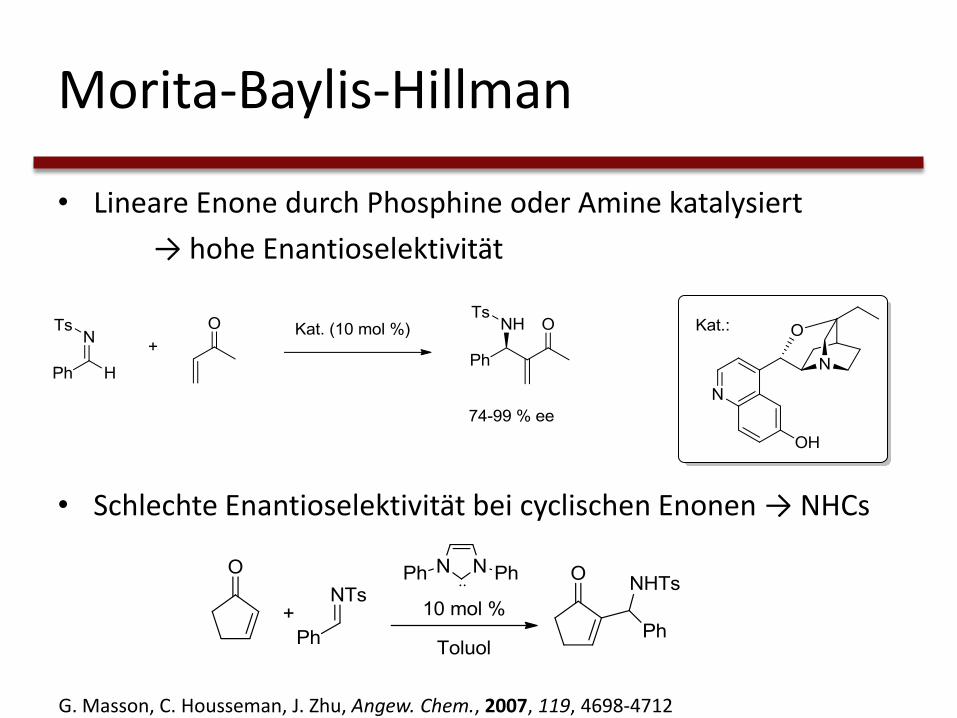
Morita-Baylis-Hillman
• Lineare Enone durch Phosphine oder Amine katalysiert
→ hohe Enantioselektivität
• Schlechte Enantioselektivität bei cyclischen Enonen → NHCs
G. Masson, C. Housseman, J. Zhu, Angew. Chem., 2007, 119, 4698-4712

Bandbreite
NHCs sind geeignet für:
• Kondensations-Reaktionen
• Generierung von Homoenolaten
• Ring öffnende Polymerisationen
• a3 zu d3 Umpolungen
• Transesterifizierung

Fazit
• Durch starke σ-Bindung elektronenreiches Metallatom
• Thermische Stabilität
• NHCs so variabel, dass es für die jeweilige Reaktion passt
-> Phosphankomplexe werden verdrängt
• Ökologisch und ökonomische Vorteile gegenüber Metall-Katalyse
• Rasante Entwicklung in den letzten 20 Jahren
• Große Vielseitigkeit an Reaktionen
• Verbesserung der Ausbeute und Stereoselektivität
• Durch Modifikation der NHCs gewünschte Produkte erhaltbar
• Vorher schwerzugängliche Produkte durch NHCs möglich

Quellen
[1] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Angew. Chem. Int. Ed. 2007, 46, 2768-2813
[2] Steven P. Nolan Acc. Chem. Res. 2010
[3] N. M. Scott, S. P. Nolan Eur. J. Inorg. Chem. 2005, 1815-1828
[4] N. Marion, S. P. Nolan Acc. Chem. Res. 2008, 41, 1440-1449
[5] E. A. B. Kantchev, C. J. O'Brien, M. G. Organ Chem. Eur. J. 2010, 16, 10844-10853
[6] Jan Wenz, Thomas Kothe OC-Seminar Vortrag vom 4.11.2010, Universität Heidelberg
[7] G. C. Voulioukalakis, R. H. Grubbs Chem. Rev. 2010, 110, 1746-1787
[8] Advanced information on the Nobel Prize in Chemistry 2005, 2005, Kungl. Vetenskaakademien
[9] T. Laue, A. Plagens John Wiley, 2005, 0-470-01040-1
[10]C. Eischenbroich: Organonetallchemie Teubner, 1990, 3-519-23501-3
[11]L.S. Hegedus: Transition metals in the synthesis of complex organic molecules-2nd ed., 1999, 1-891389-04-1

Quellen
• N. Marion S.Diez-Gonzales, S. P. Nolan, Angew. Chem. Int. Ed, 2007, 46, 2988-3000
• H. Takikawa, Y. Haschisu, J. W. Bode, K. Suzuki, Angew. Chem. Int. Ed, 2006, 45, 3492-3494
• J. Pesch, K. Harms, T. Bach, Eur. J. Org. Chem, 2004, 2025-2035
• D. Enders, O. Niemeier, T. Balensiefer, Angew. Chem., 2006, 1491-1495
• D. Enders, K. Breuer, J. Runsink, Helv. Chim. Acta, 1996, 79, 1899-1902
• J. Read de Alaniz, T. Rovis, J. Am. Chem. Soc., 2005, 127, 6284-6289
• G. Masson, C. Housseman, J. Zhu, Angew. Chem., 2007, 119, 4698-4712
• L. He, Y.-R. Zhang, X.-L. Huang, S. Ye, Synthesis, 2008, 17, 2825-2829
• L. He, T.-Y. Jian, S. Ye, J. Org. Chem., 2007, 72, 7466–7468





























