Embed Size (px)
Citation preview

Monitorizacion deconcentraciones plasmaticas (MCP)y farmacogenética del tratamientoantirretroviral
Dr. Daniel González de Requena(Laboratorio de Farmacocinética del Hospital Carlos III. Madrid)
libro_jornadas_VIH_2002 29/6/05 15:14 Página 73

[INTRODUCCIÓN]
Desde la introducción de la terapia antirretroviral de altaeficacia (HAART) se ha producido una importante reducciónen la morbilidad y mortalidad asociada a la infección por VIH.Sin embargo, el fracaso virológico se mantiene en unos rangosdel 20-67% en pacientes que inician por primera vez estetratamiento antirretroviral. Esta variabilidad en la respuestatradicionalmente ha sido atribuida a factores virológicos einmunológicos. Sin embargo, en los últimos años ha surgidoun gran interés entorno a los factores farmacológicos quepueden afectar la acción de los fármacos antirretrovirales.Entre estos, se diferencian dos campos. La farmacodinamia, o“qué hace el fármaco al cuerpo”, que engloba tanto la medidade la eficacia del fármaco (disminución de la carga viral,tiempo en alcanzar carga viral indetectable, incremento enCD4+) como la toxicidad del mismo (cambios en parámetrosde laboratorio, efectos adversos no cuantificables enlaboratorio).
Por otro lado, la farmacocinética refleja de manera cuantitativa“qué hace el cuerpo al fármaco” después de su administraciónen términos de exposición al fármaco. Uno de los principiosfundamentales de la farmacología es que existe una relacióncuantificable entre las concentraciones de fármaco(farmacocinética) y la eficacia y/o toxicidad(farmacodinamia). Así, la base de la monitorización deconcentraciones plasmáticas de los fármacos reside en elintento de conocer cuáles de los parámetros que describen lafarmacocinética de un fármaco pueden explicar suspropiedades farmacodinámicas, pudiendo ajustar las dosisadministradas para obtener máxima eficacia con el mínimo de
toxicidad.
[JUSTIFICACIÓN PARA LA MCP DEL TRATAMIENTO ARV]
Aproximadamente el 35% de los pacientes pueden presentarniveles subóptimos de inhibidores de la proteasa (IP) y, entreestos, el 50% podría experimentar fracaso virológico1. Sobre labase de que con las dosis estándar puede no alcanzarse las
74
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 74

75
concentraciones deseables en todos los pacientes, el papel demedir la exposición de cada fármaco para asegurar unasupresión de la carga viral duradera y prevenir el desarrollode cepas resistentes, así como prevenir la toxicidad asociadaa los fármacos, se ha convertido en un campo de gran interés.Sin embargo, no todos los fármacos son susceptibles de sermonitorizados. Han de cumplir una serie de requisitos:
• Debe existir una clara relación entre los parámetrosfarmacocinéticos y los farmacodinámicos;
• Se observa una gran variabilidad interindividual en laexposición al fármaco,
• Estudios clínicos han documentado el rango terapéutico delos fármacos (rango terapéutico estrecho).
• Las concentraciones plasmáticas reflejan la concentraciónde fármaco en el sitio de acción
• La técnica usada para la determinación de lasconcentraciones debe ser precisa, exacta y específica,requerir un volumen pequeño de muestra, obtenerresultados rápidamente y ser relativamente barata.
Basándose en estos criterios vamos a ver cómo, aunque se hadescrito una relación concentración de fármaco-respuestapara la mayoría de los fármacos usados en la actualidad, notodas las familias de antirretrovirales disponibles tienen lasmismas posibilidades de ser monitorizados2. Sus diferentescaracterísticas farmacocinéticas y farmacodinámicas3 así comola distinta disponibilidad de técnicas adecuadas para sudeterminación, nos llevan a considerarlas de distinta manera.En la tabla 1 se muestran los principales parámetrosfarmacocinéticos que han demostrado reflejar cambios enparámetros farmacodinámicos para los distintosantirretrovirales.
Inhibidores de la transcriptasa inversaanálogos de nucleósido (ITIAN)
Para poder ejercer su acción, los inhibidores de latranscriptasa inversa análogos de nucleósido (ITIAN) han de
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 75

incorporarse a la cadena de ADN en forma de nucleótidotrifosfato. Para esto, la molécula de fármaco ha de sufrir unpaso de activación intracelular consistente en una triplefosforilación llevada a cabo por las quinasas celulares delpaciente. Varios estudios han demostrado la importancia defactores celulares del huésped como causa del fracasovirológico. En concreto, la actividad de la timidin kinasa seha asociado con una disminución en la susceptibilidad aITIAN en pacientes expuestos a estos fármacos. Así,mientras que la medida de las concentraciones plasmáticasde ITIAN no reflejaría la actividad antiviral del fármacoactivado, la determinación de las concentracionesintracelulares de las formas trifosfato si ha demostradotener una buena correlación con la respuesta al
76
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
Tabla 1.Relación ente parámetros farmacocinéticos yfarmacodinámicos para los distintos antirretrovirales.
FÁRMACO PARÁMETRO PARÁMETRO REFERENCIASFARMACOCINÉTICO FARMACODINÁMICO
ZDV
3TC
SQV
RTV
IDV
NFV
APV
LPV
EFV
NVP
CZVD-TP
C3TC-TP
AUC
Cmin
CminCmin, AUC
Cmax, Cmin
Cmax, Cmin, AUC
CminCmin
Cmin, AUC, Cmax
CminAUC
Cmin
IQ Cmin
C12h
C12h
CminC12h
HIV-RNA
HIV-RNA
HIV-RNA
HIV-RNA y grado demutaciones
HIV-RNAGrado de mutacion
Toxicidad gastrointestinaly neurológicaCambios en triglicéridos
HIV.RNAHIV-RNA
Nefrolitiasis
HIV-RNAHIV-RNA
HIV-RNA
HIV-RNA
HIV-RNAToxicidad SNCToxicidad SNC
HIV-RNAToxicidad hepática
Fletcher, AIDS 2000
Fletcher, AIDS 2000
Gieschke, ClinPharmacokinet 1999Shapiro, Ann Intern Med 1996
Dumon, Ther Drug Monit 2000Molla, Nat Med 1996
Gatti, AIDS 1999
Hsu, Antimicrob AgentsChemoter 1997
Acosta, Pharmacotherapy 1999Murphy, J Infect Dis 1999.
Burger, 8th CROI 2001.
Seminari, JAIDS 1999.Fletcher, 8th CROI 2001
Hernando, 2nd Workshop onClinical Pharmacology 2001.
Kempf. Antiviral Therapy 2000Boffito, AIDS 2002
Marzolini, AIDS 2001
Nuñez, JAIDS 2001.
Veldkamp, AIDS 2001González de Requena, AIDS2002.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 76

77
tratamiento.5 Sin embargo, algunos estudios handemostrado la utilidad de mantener unas concentracionesplasmáticas controladas de ZDV en términos de mejorrespuesta virológica y mayores concentracionesintracelulares de las formas trifosfato.6
Por otro lado, las técnicas usadas para la determinación de lasconcentraciones intracelulares de las formas trifosfato sontécnicas muy laboriosas que precisan de un equipamientoque no está al alcance de todos los laboratorios defarmacocinética clínica.
Inhibidores de la proteasa
Los inhibidores de la proteasa (IP) son la familia deantirretrovirales para la que mayor justificación parece tener eluso de la MCP. Se trata de un grupo de fármacos con vidasmedias relativamente cortas, no precisa pasos de activaciónintracelular y se dispone de técnicas analíticas precisas, fiablesy baratas para su cuantificación. Numerosos estudios hasta lafecha han demostrado que existe una clara relación entre lasconcentraciones plasmáticas de IP y la eficacia y toxicidad delos mismos (tabla 1). A su vez, varios estudios clínicos handemostrado el beneficio de mantener unas concentracionesplasmáticas dentro del margen terapéutico, expresado comomenor tasa de fracaso virológico y menor número deinterrupciones del tratamiento por toxicidad asociada alfármaco monitorizado 42, 43.
Sin embargo, como revisaremos más adelante, diversosfactores pueden comprometer la penetración de estosfármacos a los compartimentos donde tienen que actuar, demanera que las concentraciones plasmáticas pueden, enciertas situaciones, no reflejar la concentración de fármacoen el lugar de acción. Además, estos fármacos sonampliamente metabolizados por el citocromo P450,comportándose en distinto grado como inhibidores delmismo y produciendo complicadas interacciones convariaciones, a veces impredecibles, en sus concentracionesplasmáticas.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 77

Inhibidores de la Transcriptasa InversaNo Análogos de Nucleósido (ITINAN)
Aunque inicialmente se pensó que esta familia de fármacos noera una clara candidata para la monitorización de susconcentraciones plasmáticas debido a su larga vida media y aque alcanzan concentraciones plasmáticas muy superiores a laIC95 del VIH para estos fármacos, recientes estudios handemostrado que existe una correlación entre los nivelesplasmáticos de estos fármacos y su eficacia y toxicidad. Encontra de lo que se pensaba inicialmente, las concentracioneseficaces in vivo no son tan inferiores a la Cmin alcanzada porestos fármacos. Por otro lado, gracias a su vida media larga ya la poca variabilidad intraindividual que presentan en susconcentraciones plasmáticas, el conocer las concentracionesplasmáticas que alcanza en cada paciente nos podría permitirajustar la dosis y evitar la rápida aparición de mutaciones queconfieren alto grado de resistencia para esta familia defármacos asegurando una supresión viral duradera. Al igualque los IP, estos fármacos son ampliamente metabolizadospor el sistema enzimático del citocromo P450, siendo tambiénsusceptibles de interacciones metabólicas que hagan variarsus concentraciones plasmáticas al coadministrar otrosfármacos metabolizados por esta vía.
[FACTORES QUE INFLUYEN LA ABSORCIÓN,ELIMINACIÓN Y DISTRIBUCIÓN DE LOSFÁRMACOS. FUENTES DE VARIABILIDADINTERINDIVIDUAL Y FARMACOGENÉTICA]
Uno de los factores más importantes que contribuyen a labiodisponibilidad de los fármacos es el efecto de primer paso.Este se produce tanto a nivel intestinal como hepático. Porun lado, a nivel intestinal se expresa la bomba detransmemembrana conocida como glicoproteína P (gp-P).Este transportador evita que se absorba el fármacotransportándolo desde el interior de los enterocitos a la luzintestinal. Por otro lado, la expresión de enzimasmetabolizadoras del citocromo 450 a nivel intestinal hace queel fármaco se metabolice antes de absorberse. Una vezabsorbido el fármaco es transportado por la vena porta hasta
78
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 78

79
el hígado, donde parte de la dosis que se ha podido absorbersufre un segundo proceso metabólico antes de incorporarse ala circulación sistémica.
Parte del fármaco es también excretado por bilis. Una vez queel fármaco ha alcanzado la circulación sistémica, ha de poderdistribuirse al resto de compartimentos corporales. Pero sólo lafracción libre (no unida a proteínas plasmáticas) es capaz deatravesar las membranas biológicas y distribuirse a loscompartimentos no sanguíneos. En este sentido la expresiónde la gp-P juega un papel esencial. Este transportador, ademásde a nivel intestinal se expresa en la barrera hematoencefálica,en testículos, en linfocitos y macrófagos, riñones y conductosbiliares. Así, aún alcanzando unas concentraciones plasmáticasóptimas, una sobreexpresión de esta molécula impediría ladistribución del fármaco a los distintos compartimentos.
Simultáneamente con estos procesos de distribución a losdistintos compartimentos, parte de la fracción no unida aproteínas plasmáticas sufre un 2º proceso de metabolismohepático. A este nivel, la variabilidad en la expresión delCYP450 puede provocar variaciones en las concentracionesplasmáticas entre los pacientes.
Unión a proteínas plasmáticas
Los IP (excepto IDV) y EFZ son fármacos que presentan un altogrado de unión a proteínas plasmáticas4. Las principalesproteínas plasmáticas responsables de esta unión son laalbúmina y la alfa-1-glicoproteína ácida. Esta última fracción esuna proteína inducible de fase aguda cuya concentraciónplasmática puede aumentar en más de 5 veces en infeccionesagudas y crónicas, así como en procesos inflamatorios ocancerígenos. Durante la infección por VIH, la concentración deAlfa-1-Flucoproteina ácida (AGP) puede verse aumentada conreplicación viral incontrolada o bajo infecciones oportunistas. Laalbúmina también puede sufrir variaciones en su concentraciónsérica en cuadros de insuficiencia hepática o síndromesnefróticos, desnutrición, enteropatías intestinales, hemorragias oquemaduras (hipoalbuminemias) así como en cuadros dedeshidratación (hiperalbuminemias).
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 79

Al determinar las concentraciones plasmáticas de losfármacos, lo que estamos determinando es la concentraciónplasmática total (fracción libre + fracción unida a proteínasplasmáticas). Únicamente la fracción libre o no unida aproteínas plasmáticas es capaz de penetrar en las células ydistribuirse desde el plasma a los tejidos y, por lo tanto,ejercer su acción.
Glicoproteína P.
La gp-P funciona como un mecanismo de transportelocalizado en la membrana que tiene la capacidad debombear activamente todos los IP desde el citoplasma celularal exterior. El efecto de esto es una disminución en labiodisponibilidad oral así como una disminución en lacapacidad de estos fármacos para atravesar la barrerahematoencefálica, de penetrar a testículos y de pasar al feto.La actividad de la gp-P depende del nivel de expresión delgen MDR1 así como de la funcionalidad de la proteínacodificada. La amplificación de genes, la inducción porfármacos como RTV y EFZ7, y probablemente otros factoresprovocan sobreexpresión de MDR1.
El polimorfismo en el exón 26 (C3435T) de MDR1 estásignificativamente correlacionado con los niveles de expresióny la función del MDR1. Individuos homocigotos para estepolimorfismo (alelo T/T) mostraban menor expresión delMDR1 a nivel intestinal.8 Todos los IP usados actualmente(IDV, SQV, RTV, LPV, APV, NFV) son transportados por la gp-P, sacando estos fármacos de la célula e impidiendo sudistribución.9 A parte de substratos, los IP han mostrado ser, endistinto grado, inhibidores de la gp-P. Según esta distintaafinidad y el distinto efecto de la unión a proteínas plasmáticasse ha descrito distinto grado de acumulación intracelular paracada uno de los IP10, así como diferencias en el paso acompartimentos no sanguíneos11 (tabla 2).
Un aumento en la expresión de gp-P podría limitar también lapenetración de los fármacos a distintos compartimentoscorporales12 creando “santuarios” para la replicación del VIH.Estos aspectos tienen implicaciones obvias para la efectividad
80
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 80

81
de los tratamientos con IP. La expresión de la gp-P tambiénpuede afectar la infectividad del VIH.
La sobreexpresión de la gp-P produjo una reducción de lainfectividad tanto a nivel de la fusión de las membranas viraly plasmática como en otros pasos del ciclo del VIH13. Asímismo se ha observado un incremento en la expresión de Pgpen CD4+ infectados por el VIH en relación con los controlesVIH-14. También se ha descrito que los IP tienen afinidad porotros transportadores como MRP-1, MRP-2, 15,10 o MRP-4 para elnucleósido AZT.16
Un estudio reciente da evidencias de que la variabilidadgenética en proteínas de transporte de fármacos y en enzimasmetabolizadoras pueden producir diferencias en lasconcentraciones plasmáticas entre individuos. En pacientesrecibiendo tratamiento con IP o con EFV se vio que aquellospacientes con concentraciones plasmáticas más altas teníanmás probabilidad de presentar el alelo CC para gp-P (wt) y/ogenotipo de pobre metabolizador para la isoenzima CYP2D6.Pacientes con las concentraciones de fármaco más bajastenían más probabilidad de presentar el alelo TT para la gp-Py/o un genotipo de metabolizador ultrarápido para laCYP2D617. Sin embargo, la introducción de la farmacogenéticaen el manejo rutinario del tratamiento antirretroviral ha de sercuidadosamente estudiado ya que niveles plasmáticos altos obajos se pueden identificar con métodos más sencillos.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
Tabla 2.Penetración de IP y ITINAN a distintos compartimentos nosanguíneos*.
RATIO CSF RATIO SEMEN NÓDULOS RATIO FLUIDOLINFÁTICOS CERVICOVAGINAL
IDV
NFV
LPV
RTV
APV
NVP
EFV
0,14
0
0
0
0,1
0,45
0,01
1,9
0,07
0,07
0,21
buena
0,6
0,01
2,07
0,58
0,21
0,64
Nd
Nd
Nd
0,7-1,45
ND
0,08-0,12
Nd
0,52-0,58
0,8-1,3
Nd
*: Lajevillade A. HIV Clinical Trials 2002; 3:27-35
libro_jornadas_VIH_2002 29/6/05 15:14 Página 81

Citocromo P 450
Tanto los IP como los ITINAN son extensamentemetabolizados por este sistema enzimático, en concreto por lafamilia del CYP3A. En la tabla 3 se muestran las principalesvías de metabolización para cada uno de los IP y ITINAN y elefecto que sobre cada una ejercen estos fármacos. Para todaslas vías usadas por estos fármacos se han descritopolimorfismos genéticos18.
Los polimorfismos en el metabolismo de fármacos dividen ala población en al menos dos fenotipos: metabolizadoresrápidos (EM) y metabolizadores lentos (PM). A nivel hepáticola más importante es la isoenzima CYP3A4. Supone el 20-29%del contenido de CYP450 del hígado. Otras isoenzimasimportantes y expresadas polimórficamente son la CYP1A2,2C9 (3-5% de caucasianos y 15-20% asiaticos PM), 2C19 (1-3%caucasianos PM)y 2D6 (5-10% caucasianos PM). Otra isoformaimportante es la CYP3A5. Se expresa polimórficamente en el25% de los adultos y representa el 15-32% del CYP3Ahepático. Para esta isoforma se han observado diferencias ensu Vmax de 2-21 veces y en la Km de 1-9 veces. A nivelintestinal también se expresa CYP450.
La familia mayoritaria es la del CYP3A, que supone el 70% delcontenido total de CYP450 de la mucosa intestinal. La
82
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
Tabla 3.Metabolismo de IP e ITINAN por las distintas isoenzimas delCYP450.
1A2 2B6 2C9 2C19 2D6 3A4 3A5 2E1 REF.
SQV
RTV
IDV
NFV
APV
LPV
NVP
EFV
2 1
2
X2
X2
X1
X
X
X1
X
X2
X
1
X
1
X1
X2
X1
X
X
X1
X
X1
X1
X1
X1
X1
X1
X2
X3
X1
X1
X
X
[19]
[19]
[19]
[19]
[19]
[20]
[21]
[21]
1: Efecto inhibidor. 2:Efecto inductor. 3:Efecto inductor/inhibidor mixto.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 82

83
isoforma CYP3A4 presenta una variación entre individuos dehasta 10 veces. De igual manera, el CYP3A5 se expresapolimórficamente en el 70% de la población, en contraste conel 25% a nivel hepático. También se ha detectado la presenciaa nivel intestinal de las familias CYP1A y CYP 2C.
En la mayoría de los casos se ha encontrado una basegenética que explique esta diferencia en la actividadenzimática de estas isoformas. Por otro lado, la expresión dedistintas isoformas puede verse inducida por otros fármacos.
En este aspecto se ha visto que diversos receptores nuclearescomo el receptor X de pregnano (PXR)22 o el receptor CAR,23
regulan la expresión de diversas isoformas tales comoCYP3A, 2B y 2C. Para estos receptores también se handescrito polimorfismos genéticos que pueden afectar suexpresión y su actividad. Por otro lado no hay que olvidarque los enzimas de fase II también están involucrados en elmetabolismo de los antirretrovirales. Así, los polimorfismosen los genes que codifican para estos enzimas de fase IIcomo la glucuronil transferasa, también pueden serimportantes.22
[MONITORIZACIÓN DE CONCENTRACIONESPLASMÁTICAS DE ANTIRRETROVIRALES ENLA PRÁCTICA CLÍNICA]
¿Qué parámetro farmacocinético es el másadecuado para la MCP?
A la hora de seleccionar qué parámetro farmacocinéticovamos a elegir como predictor de eficacia y/o toxicidad,hemos de tener en cuenta una serie de factores como son conqué parámetro farmacodinámico presenta mejor correlación y,si su cálculo u obtención es complejo. Entre los parámetrosfarmacocinéticos disponibles hay tres que han demostrado serindicadores de eficacia y/o toxicidad.
El Área Bajo la Curva (AUC) representa la exposición alfármaco a lo largo del intervalo de dosificación, siendorepresentativo de la cantidad de fármaco que se ha absorbido.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 83

Diversos estudios clínicos han demostrado que, sin duda, esel parámetro que mejor refleja la eficacia y la toxicidad altratamiento con IP y ITINAN. Además, la muestra de plasmapuede extraerse en cualquier punto del intervalo dedosificación. Sin embargo se trata de un parámetro difícil decalcular. Para su cálculo es necesaria la caracterizacióncompleta de la curva concentraciones plasmáticas-tiempo alo largo del intervalo de dosificación, o bien lacaracterización de la farmacocinética poblacional para esefármaco y la posterior aplicación de programas informáticospara su cálculo.
La Concentración máxima (Cmax) representa la máximaconcentración plasmática obtenida a lo largo del intervalo dedosificación. Este parámetro ha demostrado ser buen indicadorde la toxicidad, pero sin embargo ofrece poca o ningunainformación sobre eficacia. Además, para la obtención de lamuestra de plasma se precisa esperar un tiempo variabledespués de la toma del fármaco por el paciente hasta quealcance su concentración máxima (Tmax). Este hecho complicamucho su utilización en análisis de rutina.
La concentración mínima (Cmin) representa la concentraciónplasmática mínima alcanzada a lo largo del intervalo dedosificación. La Cmin ha demostrado ser un predictor fiablede la eficacia y de la adherencia al tratamiento. Diversosestudios han demostrado también su valor como predictor dela toxicidad para ciertos fármacos. Sin duda es el parámetromás fácil de obtener, al tratarse de la muestra de plasma antesde la toma de la siguiente dosis. Sin embargo, hay fármacosque, debido a su larga vida media, se administran enregímenes de una vez al día y la toma de la dosis se realizavarias horas antes de la extracción de la muestra. Este es elcaso de EFV, para el que la concentración plasmática a las 12horas también resulta ser buen predictor de eficacia y detoxicidad. Así, en la aplicación rutinaria de la MCP,elegiremos la Cmin como parámetro para caracterizar laefectividad y, según el fármaco que se monitorice, latoxicidad al tratamiento. No hay que olvidar que una únicadeterminación en un día aislado no siempre es indicativa deque el paciente esté tomando la medicación, pero enpacientes que se sabe que son buenos cumplidores si quetiene fiabilidad una única determinación.
84
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 84

85
¿Cuáles son las concentraciones mínimaseficaces (CME) para los IP y losITINAN?
Métodos de calculo.Diversos métodos se han usado para determinar lasconcentraciones plasmáticas eficaces (CME) para cada uno delos fármacos.
Estudios fenotípicos in vitro. A la hora de determinar lasconcentraciones necesarias para inhibir la replicación delvirus (IC50 o IC95) tradicionalmente se ha recurrido a ensayosfenotípicos in vitro en medios de cultivo que conteníanúnicamente un 5-10% de suero fetal bovino. Sin embargo, losIP (excepto IDV) y EFV son fármacos que presentan un altogrado de unión a proteínas plasmáticas.
Como ya vimos anteriormente, al determinar las concentracionesplasmáticas de los fármacos, lo que estamos determinando es laconcentración plasmática total (fracción libre + fracción unida aproteínas plasmáticas). Pero únicamente la fracción libre o nounida a proteínas plasmáticas es capaz de penetrar en las célulasy distribuirse desde el plasma a los tejidos y, por lo tanto, ejercersu acción. De esta forma, los ensayos in vitro que no incluyenproteínas séricas humanas podrían sobrestimar la actividad invivo de estos fármacos puesto que no tienen en consideración elalto grado de unión a proteínas plasmáticas. Varios estudios handemostrado que añadiendo proteínas plasmáticas a los mediosde cultivo, los valores de IC50 se veían incrementados variasveces. En la tabla 4 se muestran los incrementos en la IC50 en losIP y EFZ al añadir proteínas plasmáticas al medio de cultivo. Porotro lado, la estimación in vitro de la IC95 tampoco tiene encuenta los procesos de distribución del fármaco a loscompartimentos corporales y la influencia que sobre estos tienenlos distintos mecanismos de transporte del fármaco a través delas barreras fisiológicas (membrana celular, barrerahematoencefálica, placenta…), así como los equilibrios dedistribución existentes en la unión proteína-fármaco tanto a nivelplasmático como tisular.
Estudios clínicos. El mejor método para determinar la CMEconsiste en la determinación de los valores eficaces y tóxicosmediante estudios clínicos en pacientes recibiendo terapia
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 85

combinada. Este método tiene en consideración todas losfactores fisiológicos que pueden afectar la distribución de losfármacos por el cuerpo, dando un valor mucho más real quecualquiera de los métodos anteriores. Sin embargo, habría queconsiderar la distinta susceptibilidad de las cepas virales entrelos pacientes, así como la preexistencia y el desarrollo decepas con mutaciones de resistencia.
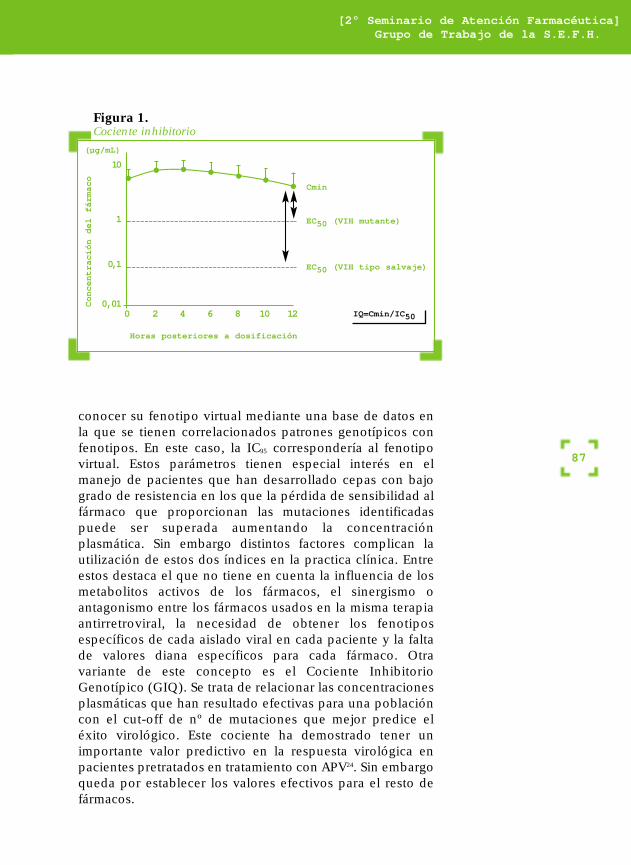
Cociente inhibitorio (IQ). El conocimiento de los parámetrosfarmacocinéticos (Cmin, Cmax) de un fármaco administradoa unas dosis determinadas nos permitiría conocer lapotencia relativa de esa dosificación mediante el cálculo delcociente inhibitorio (IQ). El IQ expresa la relación existenteentre la concentración mínima alcanzada por el fármacodurante el intervalo de dosificación y la IC95 de ese fármacopara una cepa viral determinada y ajustada según el gradode unión a proteínas plasmáticas mediante ensayosfenotípicos (IQ=Cmin/IC95). Nos indica cuantas veces laCmin está por encima de la IC95 para esa cepa (figura 1).Este IQ debe ser siempre mayor que 1, aunque aún no estáestipulado qué valor debe tener este IQ para cada uno delos fármacos. Diversos estudios han confirmado la utilidaddel IQ como predictor de una mejor respuesta virológica.Una variante de este parámetro es lo que se llama elcociente inhibitorio virtual o VIQ. En esta variante del IQ,tras conocer el genotipo de la cepa mayoritaria, podemos
86
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
Tabla 4.Aumento en la IC50 al añadir proteínas plasmáticas al mediode cultivo en ensayos fenotípicos in vitro
FARMACO ADICION SUERO HUMANO AAG 45 mg/ml50% HSO 100% HSb (1-2 mg/ml)C ALBÚMINA
0,5 mg/ml AAGd
RITONAVIR
SAQUINAVIR
NELFINAVIR
AMPRENAVIR
INDINAVIR
LOPINAVIR
EFAVIRENZ
20
25
35
6
2
5
-
25
28
90,6
16.5
3.5
14.2
-
37
30
37
42
2
-
-
-
-
-
-
-
-
17
a: Molla A.Virology 1998; 250: 255-62. b: Limoli K. 9th CROI. Abstract 583-W. c: Zhang X. J Infect Dis 1999; 180:1833. d: Becheler L. 37th ICAAC.Toronto 1997. Abstract I-115.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 86
87
conocer su fenotipo virtual mediante una base de datos enla que se tienen correlacionados patrones genotípicos confenotipos. En este caso, la IC95 correspondería al fenotipovirtual. Estos parámetros tienen especial interés en elmanejo de pacientes que han desarrollado cepas con bajogrado de resistencia en los que la pérdida de sensibilidad alfármaco que proporcionan las mutaciones identificadaspuede ser superada aumentando la concentraciónplasmática. Sin embargo distintos factores complican lautilización de estos dos índices en la practica clínica. Entreestos destaca el que no tiene en cuenta la influencia de losmetabolitos activos de los fármacos, el sinergismo oantagonismo entre los fármacos usados en la misma terapiaantirretroviral, la necesidad de obtener los fenotiposespecíficos de cada aislado viral en cada paciente y la faltade valores diana específicos para cada fármaco. Otravariante de este concepto es el Cociente InhibitorioGenotípico (GIQ). Se trata de relacionar las concentracionesplasmáticas que han resultado efectivas para una poblacióncon el cut-off de nº de mutaciones que mejor predice eléxito virológico. Este cociente ha demostrado tener unimportante valor predictivo en la respuesta virológica enpacientes pretratados en tratamiento con APV24. Sin embargoqueda por establecer los valores efectivos para el resto defármacos.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
Figura 1.Cociente inhibitorio
0,01
0,1
1
10
0 2 4
Horas posteriores a dosificación
Cmin
EC50 (VIH mutante)
EC50 (VIH tipo salvaje)
IQ=Cmin/IC50
Concentración del fármaco
6 8 10 12
(µg/mL)
libro_jornadas_VIH_2002 29/6/05 15:14 Página 87

Importancia de mantener un IQ elevado. Diversos factoreshacen aconsejable el mantener un IQ elevado. Por un lado, enun mismo individuo se pueden producir variaciones en lasconcentraciones plasmáticas (variabilidad intraindividual)según la dieta, la hora de administración del fármaco, ciclomenstrual o influencia de los ritmos circadianos. Manteniendoun IQ elevado, estas variaciones no llegarían a situar lasconcentraciones plasmáticas en niveles subterapeúticos,permitiendo el desarrollo de cepas resistentes. Por otra parte,la adherencia al tratamiento antirretroviral ha sido identificadacomo uno de los responsables del fracaso al tratamiento.Manteniendo un IQ elevado, un retraso en la toma de lamedicación no supondría situarse en niveles subterapeúticosen aquellos pacientes con peores perfiles de absorción y/oeliminación del fármaco, haciendo algo menos estricto elhorario de dosificación. En último lugar, hay una granheterogeneidad en la población viral en un mismo paciente,observándose variabilidad en la susceptibilidad a losfármacos entre las distintas cuasiespecies que pueden infectara un mismo paciente. También hay que tener en cuenta lapreexistencia de cepas resistentes en pacientes no expuestospreviamente a un fármaco. Con un IQ alto aseguramosalcanzar niveles de fármaco eficaces para todas las cepas.
Para conseguir aumentar las concentraciones plasmáticas defármaco se puede recurrir a aumentar la dosis de fármaco, conel consiguiente riesgo en aumentar la toxicidad, o también sepuede inhibir selectivamente su metabolismo intestinal yhepático, así como bloquear los sistemas de transporte demembrana, como la gp-P. En este aspecto la adición depequeñas dosis de RTV al régimen con otro IP ha demostradodar unos resultados excelentes. RTV es un potente inhibidorde distintas isoenzimas del citocromo P 450, así como de laglicoproteína P y otros transportadores de membrana. Así, aparte de aumentar la absorción de fármaco y disminuir sumetabolismo, se consigue una mayor distribución a tejidospermitiendo que el fármaco llegue a su lugar de acción. En elcaso de coadministración con SQV, el efecto consiste en unadisminución del efecto de primer paso intestinal y hepáticoaumentando la absorción (aumento en Cmax), pero tambiénse produce una disminución en la eliminación hepáticaproduciendo un aumento en la Cmin. En el caso de IDV seproduce una disminución en la eliminación sin apenas efecto
88
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 88

89
sobre la absorción intestinal, con el consiguiente aumento enla Cmin. El caso más significativo es la formulación delopinavir con ritonavir, que permite alcanzar unos nivelesplasmáticos de LPV de hasta 70 veces la CME.
Valores diana en la practica clínica.
Como hemos visto al analizar los métodos para calcular la CME,los ensayos clínicos ofrecen la información más fiable para fijarlos valores diana a alcanzar en nuestros pacientes. Sin embargohemos de tener en cuenta la presencia y el número demutaciones de resistencia para que estos valores sean aplicables.En la tabla 5 se recogen los valores eficaces y tóxicosencontrados en distintos estudios clínicas para IP y ITINAN.
Perspectivas de la MCP
Diversos estudios hasta el momento han demostrado elbeneficio de la MCP en el manejo de la terapiaantirretroviral.40-42 Su uso rutinario podría mejorar la respuestaal tratamiento antirretroviral tanto en pacientes naive comopretratados, y disminuir la incidencia de efectos adversos. Elinterés creciente por este campo ha llevado a publicar variasrevisiones en las que se trata cómo y cuándo se podríarecurrir a esta herramienta para lograr un manejo de la terapiaantirretroviral.44,45 Aunque aún no se dispone deconcentraciones diana para todos los antirretrovirales, la
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
Tabla 5.Concentración mínima efectiva y tóxicas para IP y ITINAN.
IP CME NAIVE CME PRETRATADOS CONC.TOXICA REF.(mg/ml) (Nº MUTACIONES (mg/ml)
RESPONDEDORES) (EFECTO)
Ritonavir
Saquinavir
Indinavir
Nelfinavir
Lopinavir
Amprenavir
ITINAN
Nevirapina
Efavirenz
2,1
0,.05
0,15
0,8
0,07a
0,28
3,4
1,1
ND
0,1
ND
1.25
3.55.7
1.25
ND
2.2
(3)
(2)
(2)(4.6)
(2)
¿?
ND
ND
Cmin>0,7(nefrotoxicidad)
ND
ND
ND
C12h>6(hepatotoxicidad)
4 (SNC)
[43]
[35, 36]
[32, 33]
[30, 31]
[25-27]
[32, 24]
[38, 39]
[34-37]
*: Ajuste por adición de 50% de suero humano.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 89

introducción de la MCP en la rutina clínica puede ofrecergrandes beneficios en las siguientes situaciones:
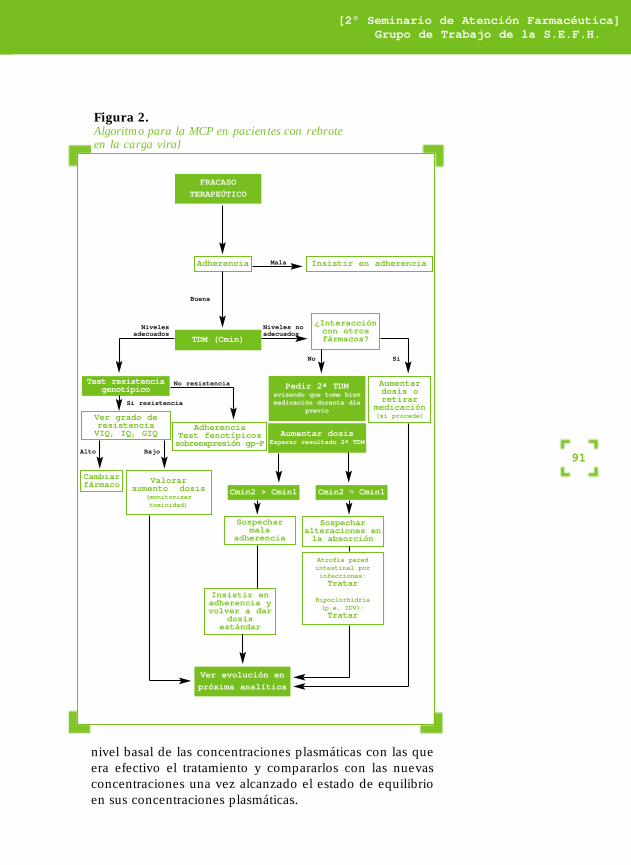
• Pacientes con buena adherencia al tratamientoantirretroviral con una respuesta virológica inicial pobre oque aunque sea adecuada inicialmente, es transitoria. Enesta situación, los valores de Cmin podrían ayudar paradeterminar qué pacientes tienen un alto o bajo gradometabólico determinado genéticamente, bajo grado deabsorción o presentan interacciones medicamentosas,pudiendo situar las concentraciones plasmáticas en nivelesóptimos y evitar el desarrollo de resistencias. En la figura 2se propone un algoritmo para el manejo de este grupo depacientes con rebrote en la carga viral.
• Pacientes que inician tratamiento con ITINAN. En estegrupo de fármacos que presentan una vida mediaplasmática larga y una variabilidad intraindividual pequeña,permitiría identificar a aquellos pacientes conconcentraciones subóptimas y situarlas en un rango óptimopara así evitar el desarrollo de mutaciones que confierenalta resistencia y así asegurar una respuesta virológicaduradera.
• Pacientes con signos clínicos de toxicidad en el caso defármacos con toxicidad concentración plasmática-dependiente. Este es el caso de la nefrolitiasis por IDV, latoxicidad en SNC para EFV, hepatotoxicidad por NVP enpacientes VHC+, o aumento en lípidos séricos por LPV y RTV.
• En terapias de rescate, junto con el genotipo, nos permitiríaoptimizar las dosis de fármaco de acuerdo al perfilgenotípico de la cepa infectante. En este sentido senecesitan más estudios que permitan determinar el valor dela Cmin eficaz según el patrón genotípico.
• Subgrupo de pacientes como embarazadas, niños, ancianosy pacientes con insuficiencia renal o hepática, que puedentener unos parámetros farmacocinéticos alterados yaltamente variables, y pueden tener un alto riesgo de tenerconcentraciones plasmáticas subóptimas.
• Pacientes que responden bien al tratamiento antirretroviraly van a iniciar un nuevo tratamiento con otro fármaco conel que potencialmente presenta interaccionesmedicamentosas. En este caso, habría que establecer el
90
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 90
91
nivel basal de las concentraciones plasmáticas con las queera efectivo el tratamiento y compararlos con las nuevasconcentraciones una vez alcanzado el estado de equilibrioen sus concentraciones plasmáticas.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
Figura 2.Algoritmo para la MCP en pacientes con rebrote en la carga viral
Adherencia Mala
Buena
Alto Bajo
Nivelesadecuados
No resistencia
Si resistencia
Niveles noadecuados
No Si
Insistir en adherencia
¿Interacción con otros fármacos?
Aumentardosis oretirar
medicación(si procede)Ver grado de
resistencia VIQ, IQ, GIQ
Cambiar fármaco
Sospecharmala
adherencia
Insistir enadherencia yvolver a dar
dosisestándar
Sospecharalteraciones enla absorción
Atrofia paredintestinal porinfecciones:Tratar
Hipoclorhidria(p.e. IDV):Tratar
Valorar aumento dosis
(monitorizartoxicidad)
Adherencia Test fenotípicossobreexpresión gp-P
TDM (Cmin)
FRACASO
TERAPEÚTICO
Ver evolución en
próxima analítica
Cmin2 ≈ Cmin1
Aumentar dosisEsperar resultado 2ª TDM
Pedir 2ª TDMavisando que tome bienmedicación durante día
previo
Cmin2 > Cmin1
Test resistenciagenotípico
libro_jornadas_VIH_2002 29/6/05 15:14 Página 91

• Identificar a aquellos pacientes que por su perfilfarmacocinético presentan unas concentraciones excesivascon los actuales regímenes de dosificación y podríanbeneficiarse de una simplificación a una pauta de una vezal día.
Limitaciones para la implantación de la MCP. Aunque todaslas técnicas disponibles para la determinación de lasconcentraciones plasmáticas son precisas, exactas, rápidas yasequibles para todos los laboratorios de farmacocinética, seimpone la necesidad de estandarizar estas técnicas parareducir a variabilidad existente entre los distintos laboratorios,así como un consenso internacional sobre quéconcentraciones plasmáticas son las que se han de alcanzar encada caso. Otros factores que pueden hacer variar losresultados obtenidos en cada laboratorio son la degradaciónde las muestras congeladas a lo largo del tiempo, el envío dela muestra de un lugar a otro y las condiciones del mismo ylos fármacos usados como estándar en el ensayo.
Sin duda alguna, la MCP va a tener gran aplicación y va a serde gran utilidad en el manejo del tratamiento antirretroviral,pero aún quedan algunos puntos por perfilar y es en esa líneasobre la que hay que trabajar en futuros ensayos clínicos.
[BIBLIOGRAFÍA]
1. Hill A, Stirnadel HA. Are clinical trials of TDM underpowered to detecteffects on HIV RNA? Three stage modeling of samples sizes. Program andabstracts of the 2nd International Workshop on Clinical Pharmacology ofHIV Therapy; April 2-4,2001; Noordwijk, The Netherands. Abstract 6.9.
2. González de Requena D, Jiménez-Nácher, Soriano V. Therapeutic DrugMonitoring for Antiuretroviralherapy: Usefulness and limitations. AIDS Rev2000; 2: 67-75.
3. Hoetelmans R. Clinical Pharmacokinetics of antirretroviral Drugs. AIDSRev 1999; 1: 156-66.
4. Turriziani O, Antonelli G, Verri A, et al. Alteration of thymidin kinaseactivity in cells treated with an antiviral agent. J Biol Regul HomeostAgents 1995; 9:47-51.
5. Fletcher C, Kawle S, Kakuda T, et al. Zidovudine triphosphate andlamivudine triphosphate concentration response relationship in HIV-infected persons. AIDS 2000;14:2137-44.
92
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 92

93
6. Fletcher C, Acosta E, Henry K, et al. Concentration-controlled zidovudinetherapy. Clin Pharmacol Ther 1998; 64:331-8.
7. Almond L, Chandler B, Hoggard P et al, EFV, but mot NVP, boosted P-gpexpression on blood cells. XIV International AIDS Conferencs, Barcelona2002. Abstract TuPeB4569.
8. Hoffmeyer O, Burk O, et al. Functional polymorphism of the humanmultidrug-resistance gene: Multiple sequence variations and correlation ofone allele with P-glycoprotein expression and activity in vivo. Proc NatlAcad Sci USA 2000;97(7):3473-78.
9. Lee C, Gottesman M, Cardarelli C et al. HIV.1 protease inhibitors are substratesfor the MDR1 multidrug transporter. Biochemistry 1998; 37:11 3594-601.
10. Jones K, Hoggard P, Sales S. et al. Differences in the intracellularaccumulation of HIV protease inhibitors in vitro and the effect of activetransport. AIDS 2001; 15:675-81
11. Jones K, Bray P, Khoo S, et al. P-Glycoprotein and transporter MRP1reduce protease inhibitor uptake in CD4 cells: potential for acceleratedviral drug resistance?AIDS 2001; 15:1353-8
12. Chaillou S, Durant J, Garraffo R, et al. Intracellular penetration of proteaseinhibitors in HIV infected patients: correlation with MDR-1 expressiongene, HIV-RNA and low dose of ritonavir. 2nd Workshop on ClinicalPharmacology of HIV Therapy, Noordwijk 2001. Abstract 2.1.
13. Flexner C, Speck RR. Role of multidrug transporters in HIV pathogenesis.8th CROI, Chicago 2001. Abstract S4.
14. Andreana A, Aggarwal S, et al. Abnormal expression of a 170 kD P-glycoprotein encoded by MDR1 gene, a metabolically active efflux pumpin CD4+ and CD8+ T cells from patients with human immunodeficiencyvirus type 1 infection. AIDS Res Hum Retrovirusas 1996;12(15):1457-62.
15. Huisman M, Smit J, Crommentuyn K, et al. Multidrug resistance protein 2(MRP2) transports HIV protease inhibitors, and transport can be enhancedby other drugs. AIDS 2002; 16:2295-301.
16. Schuetz J, Connelly M, Sun D, et al. MRP4: A previously unidentified factorin resistance to nucleoside-based antiviral drugs. Nat Med 1999; 5:1048-51.
17. Fellay J, Marzolini C, Meaden E, et al. Response to antirretroviral treatmentin HIV-1-infected patients with allelic variants of the multidrug resistancetransporter 1: a pharmacogenetics study. Lancet 2002; 359: 30-6.
18. Hasler J, Estarbrook R, Murray M, et al. Human cytochrome P450.Molecular Aspects of Medicine 1999;20:1-137.
19. Williams GC, Sinko P. Oral absorption of the HIV protease inhbitors: acurrent update. Adv Drug Deliv Rev 1999;39:211-38
20. Kumar GN, Jayanti V, Lee RD, et al. In vitro metabolism of the HIV-1protease inhibitor ABT-378: species comparison and metaboliteidentification. Drug Metab Dispos 1999;27(1): 86-91.
21. Smith PF, DiCenzo R, Morse GD. Clinical pharmacokinetiks of non-nucleoside reverse transcriptase inhibitors. Clin Pharmacokinet 2001:40(12): 893-905.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 93

22. Pirmohamed M, Back D. The pharmacogenomics of HIV therapy. ThePharmacogenomics Journal 2001; 1: 243-53.
23. Honkakoski P, Zelko I, Sueyoshi T, et al. The nuclear orphan receptor CAR-retinoir X receptor heterodimer activates the phenobarbital-responsiveenhancer module of the CYPB gene. Mol Cell Biol 1998; 18:5652-8.
24. Peytavin G, Lamotte C, Marcelin A et al. Predictivity of Amprenavir (APV)plasma concentrations on virological response in HIV-infected patientstreated with APV/RTV containing regimen: a Genophar substudy. 3rdInternational Workshop on Clinical Pharmacology of HIV Therapy,Washington DC, 2002. Abstract 7.7
25. Breilh D, Pellegrin I, Munck M et al. Virological response to lopinavir-based regimens: plasma and intracellular lopinavir concentrations anddrug resistance mutations (Kalephar study). Antiviral Therapy 2002; 7:S123
26. Boffito M, Arnaudo I, Raiteri R et al. Clinical use of lopinavir/ritonavir ina salvage therapy setting: pharmacokinetics and pharmacodinamics. AIDS2002; 16: 2081-3
27. Padilla S, Navarro A, Gallego J et al. The relationship between lipidelevations and lopinavir concentrations in HIV-infected patients, 42ndICAAC, San Diego 2002. Abstract H-1915.
28. Pellegrin I, Breilh D, Montestruc F et al. Virologic response to nelfinavir-based regimens: pharmacokinetic and drug resistance mutations(VIRAPHAR study). AIDS 2002; 16:1331-40.
29. Reijers M, Weigel H, Hart A et al. Toxicity and drug exposure in aquedruple drug regimen in HIV-1 infected patients participating in theADAM study. AIDS 2000; 14:59-68
30. Acosta E, Baken L, Page L, et al. Indinavir concentrations and antiviraleffect. Pharmacotherapy 1999, 19: 708-712.
31. Lamotte C, Peytavin G, Perre P, et al. Increasing adverse events withindinavir dosages and plasma concentrations in four different ritonavir-indinavir containing regimens in HIV-infected patients. 2nd Workshop onClinical Pharmacology of HIV Therapy, Noordwijk 2001. Abstract 4.1.
32. Sadler B, Gillotin C, Lou Y, et al. Pharmacokinetic and pharmacodinamicstudy of the human immunodeficiency virus protease inhibitor amprenavirafter multiple oral dosing. Antimicrob Agents Chemoter 2001;45:30-7.
33. Hoetelmans R, Van Heeswijk R, Meenhorst P. Plasma concentrations ofsaquinavir determine HIV-RNA response over a 48-week period. Int ConfAIDS 1998; 12: 33 [Abstract 42261].
34. Valer L, de Mendoza C, Gonzalez de Requena et al. Impact of HIVgenotyping and drug levels on the response to salvage therapy withsaquinavir/ritonavir. AIDS 2002; 16: 1964-7
35. Marzolini C, Talenti A, Decosterd L, et al. Efavirenz plasma levels canpredictc treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 2001;15:71-5.
36. Nuñez M, González de Requena D, Jimenez-Nácher I, et al. Higherefavirenz plasma levels correlate with development of insomnia. JAIDS2001;28:399.
94
Monitorizacion de concentraciones plasmaticas (mcp) y farmacogenética del tratamiento antirretroviral
libro_jornadas_VIH_2002 29/6/05 15:14 Página 94

95
37. González de Requena D, Gallego O, Briones C et al. Attainment of higherEfavirenz plasma levels allow to regain complete virus supression inpatients carrying NNRTI resistance. 9th Conference on Retroviruses andOpportunistic Infections, Seattle 2001. Abstract 454-W.
38. Veldkamp A, Gerrit J, Lange J, et al. High exposure to nevirapine inplasma is associated with an improved virological response in HIV-1infected individuals. AIDS 2001;15:1089-95.
39. González de Requena D, Nuñez M, Jiménez-Nácher I, et al. Liver toxicitycaused by nevirapine. AIDS 2002; 16:290-1
40. Durant J, Clevenbergh P, Garraffo R, et al. Importance of proteaseinhibitors plasma levels in HIV-infected patients trated with genotypic-guided therapy: pharmacological data from the Viradapt Study. AIDS2000;14:1333-39.
41. Burger D, Hugen P, Droste J, et al. Therapeutic drug monitoring (TDM) ofNelfinavir (NFV) and Indinavir (IDV)in treatment-naive patients improvestherapeutic outcomes after 1 year: results from ATHENA. 1st IASConference, Buenos Aires, 2001. Abstract 30.
42. Lamotte C, Descamps D, Joly V, et al. Validation of an algorithm forindinavir (IDV) dosage adjustment in patients with high IDV troughplasma concentrations (Cmin) to prevent adverse effects in a easy andpotent IDV/RTV containing regimen. 3rd International Workshop onClinical Pharmacology of HIV Therapy, Washington DC, 2002. Abstract 4.3
43. Dumon C, Solas C, Thuret I, ey al. Relationship between efficacy,tolerance and plasma drug concentration of ritonavir in children withadvanced HIV infection. Ther Drug Monit 2000;22: 402-8.
44. Flexner C, Piscitelli s. Therapeutic drug monitoring in HIV infection:current status and future directions. AIDS 2002; 16 Supplement 1.
45. Acosta E, Gerber J. Position paper on therapeutic drug monitorin of HIVinfection. AIDS Res Hum Retrov 2002; 18(12):825-34.
[2º Seminario de Atención Farmacéutica]Grupo de Trabajo de la S.E.F.H.
libro_jornadas_VIH_2002 29/6/05 15:14 Página 95

libro_jornadas_VIH_2002 29/6/05 15:14 Página 96





























