Embed Size (px)
Citation preview

Yosuke TanakaNobutaka Hirokawa*Dept of Cell Biology and
Anatomy, Graduate School
of Medicine, University of
Tokyo, Hongo, Tokyo
113-0033 Japan.
*e-mail: hirokawa@
m.u-tokyo.ac.jp
http://www.trends.com S39
Trends in Genetics Mouse Models of Human Diseases | Review
0167-7799/02/$ – see front matter ©2002 Elsevier Science Ltd. All rights reserved. PII: S0168-9525(02)02839-1
IntroductionCharcot-Marie-Tooth disease (CMT) is the most common
of hereditary peripheral neuropathies with a prevalence
of 1:2500.The disease is inherited in an autosomal domi-
nant, recessive, or X-linked manner, and is included in a
disease category of hereditary motor-sensory neuropathy
(HMSN). Due to an impairment of peripheral neurons,
affected people progressively develop a weakness of pe-
ripheral muscles and become unable to walk. Muscular at-
rophy of distal muscles of the legs gives a characteristic
feature to the ankles called ‘inverted Champagne bottle
sign’. Weakness of peroneal muscle also makes the foot
arch higher, and induces a skeletal anomaly called ‘pes
cavus’ (or claw foot). Since its first description by three
great neurologists, Jean Martin Charcot (1825–1893),
Pierre Marie (1853–1940) and Howard Henry Tooth
(1926–1956), the primary causes of this disease have
been obscure; that is, until the past decade, when posi-
tional cloning of the responsible genes was achieved.
Orthopedic surgery has been almost the only effective
therapeutic approach for the symptoms of this disease.
The peripheral nervous system (PNS) mainly consists of
neurons and Schwann cells. A Schwann cell is a peripheral
form of glia, which produces a long protrusion called
myelin sheath that enrolls the neuronal axons. Schwann
cells also provide neurons with nutrition and neuro-
trophins, which are essential for neuronal survival and
function.Thus, neurons themselves, as well as the support-
ing Schwann cells, can be the primary site of peripheral
neuropathy.Although each of these could finally cause neu-
ronal degeneration and muscular atrophy, these different
pathologies can be distinguished by measuring the motor
nerve conduction velocity (MNCV), because a demyelina-
tion causes the loss of the electrical insulation provided by
the myelin sheath, impairing the saltatory conduction of
the axons. Using this criterion, CMT has been classified
into two major categories: patients with myelin impair-
ment (myelinopathy) have a low MNCV; whereas those
with neuronal impairment (axonopathy) maintain normal
MNCV. The former is mainly classified to CMT type I
(CMT1; HMSNI), and the latter is classified to CMT type II
(CMT2; HMSNII). Some other subtypes of CMT develop
hearing loss or vocal cord paralysis as well as characteristic
symptoms (see review by Benstead and Grant1).
Using linkage mapping for patient pedigrees, the re-
sponsible gene loci have been mapped on human chro-
mosomes, based on which, the subtypes of the disease
have been further classified. In the past five years, postge-
nomic approaches to the study of the human genome
have successfully determined the candidate genes for the
major subtypes, as summarized in Table 1. In this review,
we focus on the latest findings obtained from studies
using mouse models of CMT, including our recent discov-
ery of the gene responsible for CMT2A, and the involve-
ment of axonal transport in the pathogenesis of CMT2.
CMT1 mouse models with dysfunction ofSchwann cellsCMT1A models: PMP22 spontaneous mutantand transgenic linesCMT1A is one of the most extensively studied CMTs, with
the onset of the clinical symptoms occurring at approxi-
mately 12 years of age, and caused by overexpression or
Mouse models of Charcot-Marie-Tooth diseaseYosuke Tanaka and Nobutaka HirokawaA common peripheral neuropathy, Charcot-Marie-Tooth disease, progressively develops with distal muscle atrophy. Severalgenes expressed in Schwann cells and neurons have been identified to be responsible for this hereditary disease, and usedin generating transgenic and knockout mice. Such mice are good disease models for cell biological and therapeutic studies.
ALS: amyotrophic lateral sclerosisCH: congenital hypomyelination CMT: Charcot-Marie-Tooth diseaseCNS: central nervous systemDSS: Dejurine-Sottas syndromeGjb1: gap junction membrane channel protein beta 1HMSN: hereditary motor-sensory neuropathyKIFs: kinesin superfamily proteinsMNCV: motor nerve conduction velocityMPZ: myelin protein zeroPMP22: peripheral myelin protein 22PNPP: peripheral nerve pressure palsyPNS: peripheral nervous systemPRX: periaxinNFH: neurofilament, heavy polypeptideNFL: neurofilament, light polypeptideNFM: neurofilament, medium polypeptide
Abbreviations
In association with MKMD
http://research.bmn.com/mkmd
http://www.trends.comS40
Trends in GeneticsReview | Mouse Models of Human Diseases
point mutation of a myelin integral membrane protein,
peripheral myelin protein 22 (PMP22).
CMT1A patients with duplication of genomic loci
encoding PMP22 develop peripheral neuropathy with a de-
creased MNCV2. Their peripheral nerves show a character-
istic onion-bulb appearance (Fig. 1), that is, rings of myelin
sheath around the neuronal axons, representing repeated
cycles of demyelination and remyelination. Homozygous
duplication and some point mutations of PMP22 also cause
a more severe form, Dejurine-Sottas syndrome (DSS or
HMSN III), which is defined by early onset, low MNCV and
severe disability. Conversely, those with a haploinsufficiency
of PMP22 develop a distinct entity of neuropathy, peripheral
nerve pressure palsy (PNPP)3, which develops after pro-
longed kneeling, and has a characteristic tomacula or a
sausage-like degeneration of peripheral myelin sheath.
PMP22 mouse models have been successfully gener-
ated using both forward and reverse genetic approaches
as described below.
Using classical forward genetics, Trembler (tre) and
Trembler-Jackson (tre-J) spontaneous mutant mice have
long been treated as a good peripheral neuropathy
model with abnormal gait. They were found to carry
mutations of G150D and L16P, respectively, in the
PMP22 protein4. Vallat et al.5 have analyzed the levels of
various myelin proteins in tre/+ mice, and found that
the PMP22 level is slightly lower than the normal level,
so that this point mutation would result in the gain-of-
function of PMP22. Using another forward genetics ap-
proach, Isaacs et al.6 performed a large-scale mouse ENU
mutagenesis screening, and found that two strains Tr-
m1H and Tr-m2H suffering from resting tremor carry
mutations in the PMP22 gene. Interestingly, the Tr-m1H
strain had a mutation of the same amino acid found in a
family with DSS (H12R).
Using reverse genetics, several lines of transgenic
mouse and rat models overexpressing PMP22 have been
established7–9, all of which develop an abnormal gait with
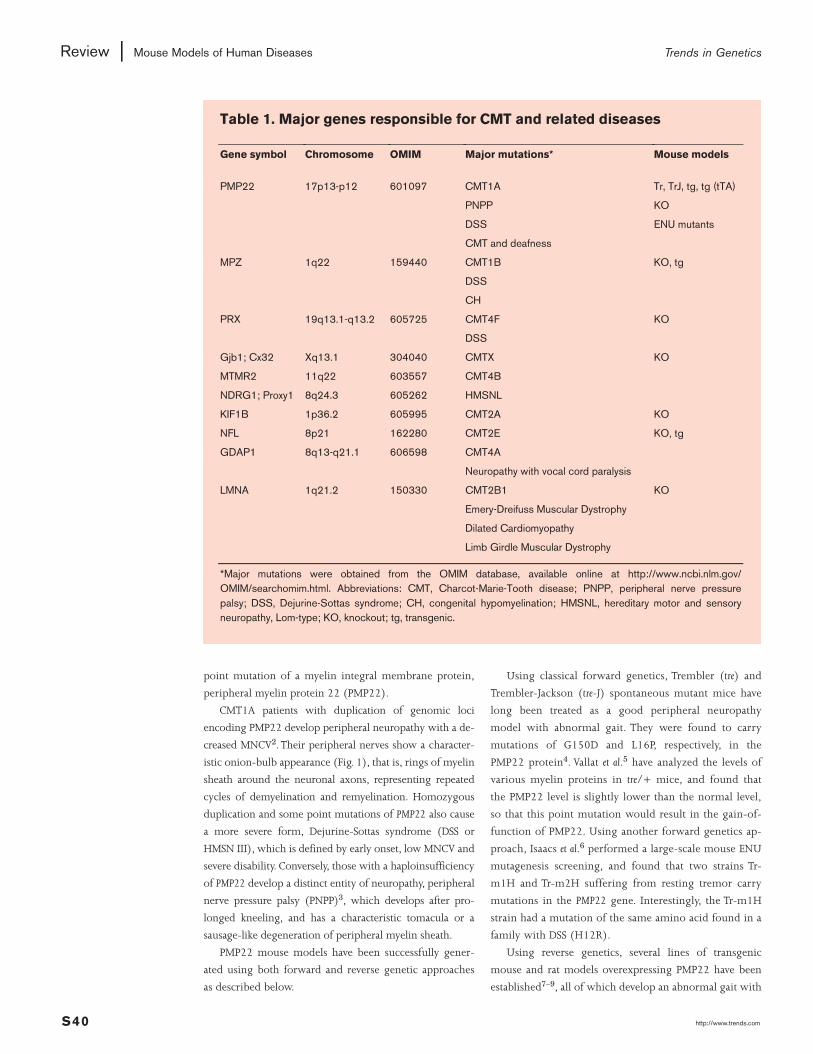
Table 1. Major genes responsible for CMT and related diseases
Gene symbol Chromosome OMIM Major mutations* Mouse models
PMP22 17p13-p12 601097 CMT1A Tr, TrJ, tg, tg (tTA)
PNPP KO
DSS ENU mutants
CMT and deafness
MPZ 1q22 159440 CMT1B KO, tg
DSS
CH
PRX 19q13.1-q13.2 605725 CMT4F KO
DSS
Gjb1; Cx32 Xq13.1 304040 CMTX KO
MTMR2 11q22 603557 CMT4B
NDRG1; Proxy1 8q24.3 605262 HMSNL
KIF1B 1p36.2 605995 CMT2A KO
NFL 8p21 162280 CMT2E KO, tg
GDAP1 8q13-q21.1 606598 CMT4A
Neuropathy with vocal cord paralysis
LMNA 1q21.2 150330 CMT2B1 KO
Emery-Dreifuss Muscular Dystrophy
Dilated Cardiomyopathy
Limb Girdle Muscular Dystrophy
*Major mutations were obtained from the OMIM database, available online at http://www.ncbi.nlm.gov/OMIM/searchomim.html. Abbreviations: CMT, Charcot-Marie-Tooth disease; PNPP, peripheral nerve pressurepalsy; DSS, Dejurine-Sottas syndrome; CH, congenital hypomyelination; HMSNL, hereditary motor and sensoryneuropathy, Lom-type; KO, knockout; tg, transgenic.

Figure 1. Onion-bulbappearance ofperipheral nerve inhypertrophicneuropathy
A cross section of peripheralnerve of a 10-year-old girlpresumably suffering fromCMT1A. Upon repeatedregeneration process of theaffected myelin sheath, three tofive layers of Schwann cellprocesses (asterisks) werefound to be circumferentiallyarranged around a small axon(Ax) with a myelin sheath (M).The processes are markedly flatand electron dense, and areseparated by connective tissue.Reproduced, with permission,from Schröder, J. M. (2001)Pathology of Peripheral Nerves,Springer-Verlag.
http://www.trends.com S41
Trends in Genetics Mouse Models of Human Diseases | Review
demyelinating progressive peripheral neuropathy, reveal-
ing the toxicity of excess PMP22 protein in Schwann
cells. The PMP22 knockout mouse shows irregular, volun-
tary stretching of the hindlimbs and trembling, which
may mimic PNPP.
Recently, Perea et al.10 generated transgenic mice that
conditionally overexpress the PMP22 protein using the
tet-off system. They generated two transgenic lines with:
(1) a Schwann-cell-specific expression of tTA, which is
composed of tetracycline repressor (TetR) and the VP16
activation domain; and (2) a PMP22 expression regulated
by the tetracycline operator (PhCMV*-1-PMP22); and
then crossed the two lines together. The resulting double
transgenic mice overexpress PMP22 under normal condi-
tions and develop neuropathy, but upon administration of
tetracycline-containing foods, the transgene can be
switched off. Using this system, they showed that inactiva-
tion of the transgene induces remyelination of peripheral
nerves of mice that had suffered from neuropathy.
Other models of myelin-based CMTsCMT1-related myelinopathic peripheral neuropathy is
also caused by mutations of several structural proteins in
Schwann cells (Fig. 2). Myelin protein zero (MPZ; P0),
periaxin (PRX) and gap junction membrane channel
protein beta 1 (Gjb1/connexin 32) are responsible for
CMT1B, CMT4F and CMTX, respectively, and mouse
models have been generated as reported in the following
text. MPZ is an immunoglobulin-related 28-kD integral
membrane glycoprotein of Schwann cells. It is a major
protein of the PNS, but is not expressed in the central ner-
vous system (CNS). K96E and D90E point mutations were
identified in pedigrees of autosomal dominant CMT1B
patients11, and the Mpz knockout mouse has served as a
model of CMT1B12 — both in homozygous and het-
erozygous states. Mpz(−/−) mice are deficient in normal
motor coordination and exhibit tremors and occasional
convulsions, and have severely impaired myelin formation
in peripheral nerve axons. In the case of Mpz(+/−) mice,
the myelin sheath seems normally formed initially but it
gradually thins after four weeks of age13.
PRX is a constituent of the dystroglycan-dystrophin-re-
lated protein-2 complex, which links the Schwann cell
cytoskeleton to the extracellular matrix14, and its muta-
tions cause a recessive demyelinating neuropathy, CMT4F.
Similar to the Mpz(+/−) mice, Prx(−/−) mice initially
myelinate normally but later develop a demyelinating peri-
pheral neuropathy15. Prx(−/−) mice show a pronounced
unsteadiness in the gait by six to nine months of age, los-
ing the body weight and suffering from neuropathic pain.
Gjb1 is a component of the gap junction, a major
intercellular channel for transmitting small molecules or
ions. In Schwann cells, Gjb1 is localized near the nodes of
Ranvier and in Schmitt-Lanterman incisures, which may
connect adjacent layers of the myelin sheath. Bergoffen et
al.16 described that a point mutation of Gjb1 is responsi-
ble for an X-linked dominant demyelinating neuropathy,
CMTX. A knockout mouse model for CMTX has been
established by Nelles et al.17. Gjb1(−/−) mice develop sig-
nificant reduction in body and brain weight, enhanced
intrinsic excitability of neurons, and demyelinating pe-
ripheral neuropathy after four to six months of age,
showing an onion-bulb appearance of the myelin sheath,
thinned myelin layers, and abnormally thick periaxonal
collars18. The sensory impairment is more moderate than
that in Mpz(+/−) mice18. Some other responsible genes
are myotubularin-related protein-2 (MTMR2) for CMT4B
or N-myc downstream-regulated gene 1 (NDRG1) for
HMSN-Lom (see Table 1), for which mouse models have
not yet been generated.
A transcription factor with three tandem zinc finger
motifs was found to be involved in CMT-like demyelinat-
ing peripheral neuropathy: mutations of early growth re-
sponse 2 (EGR2) have been detected in patients of CMT1,
DSS, and congenital hypomyelination (CH). Its mouse
homolog Krox20 was found to function in hindbrain
development, as seen through marked expression in
rhombomeres 3 and 5, and to also function in PNS
myelinogenesis. Krox20(−/−) mice show various pheno-
types in hindbrain segmentation, bone formation, growth
retardation, and neurology, with retarded performance
and tremors. The differentiation of peripheral Schwann
cells is affected and shows an amyelination19,20, as
expected from the localization of Krox20.
Finally, a xenograft experiment has been performed by
Sahenk and Chen21. They grafted CMTX patients’ sural
nerve segments into cut ends of the sciatic nerves of a nude

http://www.trends.comS42
Trends in GeneticsReview | Mouse Models of Human Diseases
mouse. Since the nerve segments contained the nuclei of
Schwann cells, but not those of neurons, the regeneration
of murine sciatic nerve would mainly reflect on the activ-
ity of patients’ Schwann cells. As the nude mouse cannot
generate mature T lymphocytes due to a lack of the thy-
mus, it was able to serve as a recipient of the xenograft.
Results showed that the myelin sheath from axonopathic
CMT2 patients as well as from normal controls could suc-
cessfully induce the innervation of murine nerves, while
myelin sheath from myelinopathic CMTX patients could
not, suggesting that the myelin sheath from CMTX pa-
tients is indeed essential for developing neuropathy. This
could also be considered as an interesting experimental
model of demyelinating neuropathy.
CMT2 mouse models with dysfunction of neuronsA molecular motor for axonal transport —KIF1Bββ — is responsible for CMT2AIn contrast to the CMT1 diseases arising from Schwann
cell dysfunction, CMT2 occurs due to the dysfunction of
neurons themselves. The cytoskeleton is a filamentous
protein network that is essential for cellular morphogene-
sis and intracellular transport22.
Recently, two proteins associated with axonal cyto-
skeleton have been identified as being involved in the
pathogenesis of CMT2 (Fig. 3).
We have recently found that axonal transport is in-
volved in CMT2 pathogenesis through our research of
molecular motors. Neurons transport essential organelles
and protein complexes to their axons, using microtubule
motors that include approximately 50 kinds of kinesin
superfamily proteins (KIFs)23,24. A recently identified KIF
isoform, KIF1Bβ, is the motor for synaptic vesicle precur-
sors and responsible for CMT2A pathogenesis25. kif1B(−/−)
mice suffer from severe neuronal degeneration in the CNS,
which causes fatal newborn apnea. Of the two splicing
isoforms of the kif1B gene, only the KIF1Bβ isoform can
rescue neuronal death in primary culture.
kif1B(+/−) mice develop progressive muscle weakness
with a MNCV within the normal range, resembling the
symptoms of CMT2. Through a cell biological approach,
we found that KIF1Bβ binds to membrane organelles
containing synaptic vesicle proteins. We then compared
the level of synaptic vesicle proteins that are transported
to nerve endings by immunoblotting sciatic nerve lysate,
and found that the levels of both the cargo synaptic vesi-
cle proteins and the KIF1Bβ motor indeed decreases in
the peripheral axons of kif1B(+/−) mice. Since the human
KIF1B gene was mapped within the interval in which
CMT2A had been mapped, we collaborated with clinical
departments to analyze the genome of patient pedigrees,
and found a loss-of-function mutation of the motor
domain (Fig. 4). Since this dysfunctional motor did not
tightly bind to the track microtubules, loss-of-function as
opposed to a dominant negative mechanism explains the
development of CMT2A in this patient.
Accordingly, in CMT2A patients and kif1B(+/−) mice,
haploinsufficiency of the KIF1Bβ motor results in a defi-
ciency of the cargo proteins being transported by this
motor, including synaptic vesicle proteins in nerve axons
and endings, bringing about progressive dysfunction of
peripheral neurons.
Neurofilament L protein is involved in CMT2ERecently, point mutations of the neurofilament L (NFL)
protein have been found to be responsible for a domi-
nantly inherited CMT2E neuropathy26. Neurofilament
triplets (NFH, NFM, NFL) copolymerize to generate bun-
dles of intermediate filaments that are important compo-
nents of the neuronal cytoskeleton; thus, it is reasonable
to assume that mutations of NFL can cause a peripheral
neuropathy. Knockout mice for NFH and NFM have already
been established by several groups, although their pheno-
types are slightly different from each other, possibly
reflecting the difference in the gene targeting strategy
used (see review by Hirokawa and Takeda27). Moreover,
the mouse overexpressing the human NFH gene was es-
tablished by Julien and colleagues28 and has long been
treated as a classical mouse model for amyotrophic lateral
sclerosis (ALS), which causes neuronal swelling.
Julien and colleagues29 further established an NFL
knockout mouse but its phenotype was not so severe as
expected from human patients. When they axotomized
peripheral nerves, regeneration of myelinated axons was
Figure 2. Schematicrepresentation of across section of amyelinated axon
A process from a Schwann cellenrolls neuronal axons, formingthe myelin sheath. Mutations instructural proteins of the myelinsheath and a transcription factorEGR2 are responsible for themyelinopathic CMT1.

Figure 3. Axonalcytoskeleton isessential for CMT2pathogenesis
Two major components of theaxonal cytoskeleton, microtubule(MT) and neurofilament (NF) aresupporting unique protrudingmorphology of an axon. They arecrosslinked together like aladder with microtubule-associated proteins andsubunits of neurofilaments.Synaptic vesicle proteins aretransported via the MT trackswith a membrane organellecalled synaptic vesicleprecursor (SVP) by a molecularmotor KIF1Bβ. Mutations ofNFL and KIF1Bβ areresponsible for CMT2E andCMT2A, respectively. Adapted,with permission, from Wiley(Ref. 22).
http://www.trends.com S43
Trends in Genetics Mouse Models of Human Diseases | Review
found to be delayed. Thus, this simple knockout mouse
does not completely mimic the CMT patients’ pheno-
type. Future genetic and biochemical approaches, such
as introducing subtle mutations that have been charac-
terized from patients, would greatly facilitate the exami-
nation of how those mutations can alter the kinetics of
neurofilament assembly.
Future perspectivesHere, we have briefly reviewed the recent progress made
in positional cloning of candidate genes responsible for
CMT along with generation of their mouse models. Most
of the genes are found to encode structural and functional
proteins in Schwann cells and neurons. PMP22, MPZ,
GJB1 and PRX are the proteins localized in the myelin
sheath, and EGR2 may regulate the expression of essential
proteins in Schwann cells. KIF1Bβ and NFL are, respec-
tively, the microtubule motor and neuronal cytoskeletal
component that are highly expressed in neurons.
On the other hand, conventional criteria for the differ-
ential diagnosis between HMSN I, II, and others, are es-
sentially based on the symptoms of patients. For example,
DSS or HMSN III is an early onset and more severe form
of CMT. However, recent molecular approaches have iden-
tified that the same gene, for example, PMP22, with differ-
ent point mutations, is responsible for multiple diagnoses
of HMSN as summarized in Table 1. Moreover, the distinc-
tion between myelinopathy and axonopathy is sometimes
obscure, because some proteins are essential for both
neurons and Schwann cells30. Conversely, patients with
the same HMSN diagnosis could carry different mutations
in different genes. These multiple relationships between
differential diagnoses and candidate genes should be pre-
cisely redefined through accumulation of point mutation
data obtained by high-throughput SNP genotyping of
CMT patients.
Most of the current mouse models have been gener-
ated using transgenic techniques and homologous recom-
bination. Because, in many cases, the gene dosage affects
the severity of the disease, as in the case of PMP22, the
homologous recombination technique is more favorable
for a stable gene expression level, although this takes more
effort and time than simple transgenics. However, as in the
case of NFL, simple knockouts cannot always mimic the
disease phenotype because of the existence of a dominant
negative effect. Thus, generation of a series of knock-in
mice with a subtle mutation or SNP that has been charac-
terized from patient pedigrees will be the next step. For
introducing such subtle mutations, the cre-mutated-loxP
technology will enable us to easily introduce different
expression units into a specific site that is primarily
defined by a single homologous recombination31.
Two strains of different mouse models can be crossed
together. Suter and colleagues generated Mpz(+/−);
Gjb1(−/−) and Mpz(−/−);Gjb1(−/−) mice to demonstrate
that the demyelination process is accelerated in these dou-
ble knockout mice32. Although a single mutation would
be sufficient for the occurrence of human CMT, a double
mutation study in the mouse will still be fruitful to reveal
functional relationship between the two genes.
How will the mouse models be used in future studies?
They can be used for both basic and application studies.
For basic research, mutations could be a key to analyzing
the protein function. As was applied to the KIF1B study25,
biochemical and cell biological approaches will help in
determining how certain residues function, such as the
catalytic core, protein–protein interaction site or modifi-
cation site. Cell lines or primary cultured cells from the
mouse models will be a powerful tool for analyses that
will further clarify molecular machines that power the
functions of neurons and Schwann cells.
Regarding the clinical application of mouse models,
information on the mutated residues will be applied to
gene diagnosis and therapeutic trials. The study of a con-
Figure 4. Dysfunctional motor proteins with a CMT2A mutation
Immunofluorescence of Vero cells transfected with either wild-type or Q98L mutant KIF1Bβ constructs.Red and green signals indicate KIF1Bβ and tubulin, respectively. Healthy motors drive to and accumulateat the cell periphery where the microtubule tracks end, while the Q98L mutant motor remains at the cellcenter (arrows). Thus, the KIF1Bβ-Q98L mutant motor cannot transport synaptic vesicle precursors tothe axons. Bar: 15 µm. Reproduced, with permission, from Cell (Ref. 25).

http://www.trends.comS44
Trends in GeneticsReview | Mouse Models of Human Diseases
ditional transgenic mouse for PMP22 suggests that some
forms of CMT1A can be reversed10, that is, silencing of
PMP22 overexpression can induce remyelination of pe-
ripheral nerves. This interesting finding should stimulate
the application of the tetracycline system on other types
of CMT mouse models, to test whether the pathological
defects are irreversible or not. These mice and their pri-
mary cultured cells will be useful for developing new
drugs for peripheral neuropathy. Antisense drugs will also
be potentially effective, but sufficient care should be taken
because an excessive reduction of the protein expression
level by antisense oligos could cause a PNPP neuropathy
as an adverse effect.
CMT has long been considered an incurable disease of
unknown cause. In this postgenomic era, great successes in
positional cloning studies have finally revealed the genes
responsible for this disease. The series of mouse models
generated to date are opening the door to its gene therapy,
as well as providing further knowledge concerning the
molecular and cell biological mechanism of how Schwann
cells and neurons work together to maintain the PNS.
AcknowledgementsWe thank Dr. Michael Schroeder (University Clinic
RWTH, Aachen, Germany) for permitting us to reproduce
an eletronmicrograph from his atlas.The study on CMT2A
was supported by a COE grant-in-aid from the Ministry
of Education, Culture, Sports, Science and Technology of
Japan to N.H.
References1 Benstead,T.J. and Grant, I.A. (2001) Progress in clinical
neurosciences: Charcot-Marie-Tooth disease and related inheritedperipheral neuropathies. Can. J. Neurol. Sci. 28, 199–214
2 Patel, P.I. et al. (1992) The gene for the peripheral myelin proteinPMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat.Genet. 1, 159–165
3 Silander, K. et al. (1996) A de novo duplication in 17p11.2 and a novelmutation in the Po gene in two Dejerine-Sottas syndrome patients.Hum. Mutat. 8, 304–310
4 Suter, U. et al. (1992) Trembler mouse carries a point mutation in amyelin gene. Nature 356, 241–244
5 Vallat, J.M. et al. (1999) Expression of myelin proteins in the adultheterozygous Trembler mouse. Acta Neuropathol. (Berl.) 98, 281–287
6 Isaacs, A.M. et al. (2000) Identification of two new Pmp22 mousemutants using large-scale mutagenesis and a novel rapid mappingstrategy. Hum. Mol. Genet. 9, 1865–1871
7 Huxley, C. et al. (1996) Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YACDNA. Hum. Mol. Genet. 5, 563–569
8 Magyar, J.P. et al. (1996) Impaired differentiation of Schwann cells intransgenic mice with increased PMP22 gene dosage. J. Neurosci. 16,5351–5360
9 Sereda, M. et al. (1996) A transgenic rat model of Charcot-Marie-Tooth disease. Neuron 16, 1049–1060
10 Perea, J. et al. (2001) Induced myelination and demyelination in aconditional mouse model of Charcot-Marie-Tooth disease type 1A.Hum. Mol. Genet. 10, 1007–1018
11 Hayasaka, K. et al. (1993) Charcot-Marie-Tooth neuropathy type 1B isassociated with mutations of the myelin P(0) gene. Nat. Genet. 5,31–34
12 Giese, K.P. et al. (1992) Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, anddegeneration of myelin and axons. Cell 71, 565–576
13 Martini, R. et al. (1995) Protein zero (P0)-deficient mice show myelindegeneration in peripheral nerves characteristic of inherited humanneuropathies. Nat. Genet. 11, 281–286
14 Guilbot, A. et al. (2001) A mutation in periaxin is responsible forCMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease.Hum. Mol. Genet. 10, 415–421
15 Gillespie, C.S. et al. (2000) Peripheral demyelination and neuropathicpain behavior in periaxin-deficient mice. Neuron 26, 523–531
16 Bergoffen, J. et al. (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042
17 Nelles, E. et al. (1996) Defective propagation of signals generated bysympathetic nerve stimulation in the liver of connexin32-deficientmice. Proc. Natl.Acad. Sci. U. S.A. 93, 9565–9570
18 Anzini, P. et al. (1997) Structural abnormalities and deficientmaintenance of peripheral nerve myelin in mice lacking the gapjunction protein connexin 32. J. Neurosci. 17, 4545–4551
19 Schneider-Maunoury, S. et al. (1993) Disruption of Krox-20 results inalteration of rhombomeres 3 and 5 in the developing hindbrain. Cell75, 1199–1214
20 Topilko, P. et al. (1994) Krox-20 controls myelination in theperipheral nervous system. Nature 371, 796–799
21 Sahenk, Z. and Chen, L. (1998) Abnormalities in the axonalcytoskeleton induced by a connexin32 mutation in nerve xenografts.J. Neurosci. Res. 51, 174–184
22 Hirokawa, N. (1994) The neuronal cytoskeleton: roles in neuronalmorphogenesis and organelle transport. In Molecular Neurobiology:Mechanisms Common to Brain, Skin, and Immune System. pp. 117–143.Wiley-Liss, Inc.
23 Hirokawa, N. (1998) Kinesin and dynein superfamily proteins andthe mechanism of organelle transport. Science 279, 519–526
24 Miki, H. et al. (2001) All kinesin superfamily protein, KIF, genes inmouse and human. Proc. Natl.Acad. Sci. U. S.A. 98, 7004–7011
25 Zhao, C. et al. (2001) Charcot-Marie-Tooth disease type 2A caused bymutation in a microtubule motor KIF1Bβ. Cell 105, 587–597
26 Mersiyanova, I.V. et al. (2000) A new variant of Charcot-Marie-Toothdisease type 2 is probably the result of a mutation in theneurofilament-light gene. Am. J. Hum. Genet. 67, 37–46
27 Hirokawa, N. and Takeda, S. (1998) Gene targeting studies begin toreveal the function of neurofilament proteins. J. Cell Biol. 143, 1–4
28 Cote, F. et al. (1993) Progressive neuropathy in transgenic miceexpressing the human neurofilament heavy gene: a mouse model ofamyotrophic lateral sclerosis. Cell 73, 35–46
29 Zhu, Q. et al. (1997) Delayed maturation of regenerating myelinatedaxons in mice lacking neurofilaments. Exp. Neurol. 148, 299–316
30 Vance, J.M. (2000) The many faces of Charcot-Marie-Tooth disease.Arch. Neurol. 57, 638–640
31 Araki, K. et al. (1997) Targeted integration of DNA using mutant loxsites in embryonic stem cells. Nucleic Acids Res. 25, 868–872
32 Neuberg, D.H. (1998) Accelerated demyelination of peripheral nervesin mice deficient in connexin 32 and protein zero. J. Neurosci. Res. 53,542–550





























