Embed Size (px)
Citation preview

MICROREVIEW
DOI: 10.1002/ejoc.201402024
N-Heterocyclic Carbene Catalysis as a Tool for Gaining Access to the3,4-Dihydropyran-2-one Skeleton
Domenico C. M. Albanese[a] and Nicoletta Gaggero*[b]
Keywords: 3,4-Dihydropyran-2-ones / Michael addition / N-Heterocyclic carbenes / α,β-Unsaturated acyl azolium ions /Azolium enols / Azolium enolates / Oxygen heterocycles
An overview of the progress made in the synthesis of 3,4-dihydropyran-2-ones since the development of NHC cataly-sis is reported. Two distinct classes of species – electrophilicα,β-unsaturated acylazolium and nucleophilic azolium enol/enolate intermediates – can easily be produced from different
1. IntroductionSubstituted enol δ-lactones, better known as 3,4-di-
hydropyran-2-ones, are important moieties found in naturalproducts, pharmaceuticals and synthetic intermediates(Scheme 1).
The 3,4-dihydropyran-2-one system constitutes the keyfunctionalized subunit of isocoumarins, a family of naturaloccurring compounds that show a broad spectrum of phar-macological activities such as antifungal, anti-inflamma-tory, antimicrobial, phytotoxic and cytotoxic, to mentiononly a few. For example, the isocoumarin NM-3,[1] an
[a] Dipartimento di Chimica, Università degli Studi di Milano,via Golgi 19, 20133 Milano, Italia
[b] Dipartimento di Scienze Farmaceutiche, sezione di ChimicaGenerale e Organica “A. Marchesini”, Università degli Studi diMilano,via Venezian 21, 20133 Milano, ItaliaE-mail: [email protected]://www.unimi.it/chiedove/ENG/schedaPersonaXML.jsp?matricola=14768
Domenico Albanese received his Ph.D. degree in 1993 with Prof. Dario Landini, working on phase-transfer catalysis.After short stays at the Imperial College London and the Technical University of Denmark he obtained a permanentposition at the Università degli Studi di Milano, where he was appointed associate professor in 2008. His research interestsinclude developments of phase-transfer catalysis, green chemistry and development of new environmentally friendly anti-fouling agents.
Nicoletta Gaggero received her Ph.D. degree in 1992 working on stereoselective reactions with natural proteins, enzymesand models of enzymes. After working at the Laboratoire de Chimie de Coordination du CNRS in Toulouse, she obtaineda permanent position at the Università degli Studi di Milano. Her research interests cover the field of biocatalysis andasymmetric synthesis.
Eur. J. Org. Chem. 2014, 5631–5640 © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 5631
substrates through the intermediacy of NHC catalysis. Theyenable the construction of the 3,4-dihydropyran-2-one skel-eton, the core of natural and synthetic products with signifi-cant biological activities, through Michael additions, cyclo-additions and rearrangements.
analogue of the natural product cytogenin, inhibits thegrowth of human hendotelial cells in culture and tumourangiogenesis in human tumour xenograft models.[2] The iso-coumarin fragment is also a component in rubromycins, aclass of antitumour antibiotics attracting intensive syntheticefforts.[3] Moreover 3,4-dihydropyran-2-ones are importantbuilding blocks for the synthesis of a number of naturalproducts displaying a wide range of potent biological activi-ties.
Nepetalactone is the starting material for the total syn-thesis of englerin A, a guaiane sesquiterpene that selectivelyinhibits the growth of renal cancer cell lines.[4] Methyl(4aR,7R,7aS)-7-methyl-1-oxo-1,4a,5,6,7,7a-hexahydro-cyclopenta[c]pyran-4-carboxylate (A) is the key intermedi-ate for the total synthesis of (–)-7-deoxyloganin, a memberof the family of iridoids known for their medicinal ac-tivity.[5] The synthesis of α-herbertenol, a fungicidal sesqui-terpene found in a wide variety of liverworts, involves 6-methyl-3,4-dihydropyran-2-one.[6] Finally, 3,4-dihydro-

D. C. M. Albanese, N. GaggeroMICROREVIEW
Scheme 1. Selected examples of the employment of 3,4-dihydro-pyran-2-ones in the synthesis of natural products.
pyran-2-ones are widely recognized as useful buildingblocks for the synthesis of pyran-2-ones,[7] γ-lactones,[8] cy-clic enamines,[9] substituted benzenoids[10] and function-alized indene- and dihydrobenzofurancarboxylates(Scheme 2).[11]
Scheme 2. Selected examples of the use of 3,4-dihydropyran-2-onesin organic synthesis.
Michael addition of enolates to α,β-unsaturated carbonylcompounds, followed by intramolecular cyclization, is oneof the most straightforward, efficient and simple methodsfor constructing the 3,4-dihydropyran-2-one skeleton andrepresents the main synthetic pathway.
In 1997, Kobayashi[12] reported the first example, fol-lowed by Katritzky.[13] Mukayama utilized chiral quater-nary ammonium phenoxides derived from chinchona alka-loids in a new organocatalysed Michael process under
www.eurjoc.org © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2014, 5631–56405632
phase-transfer conditions.[14] Kanemasa utilized chiralLewis acid and amine catalysts.[15] Ma developed a highlyenantioselective method in water catalysed by a diarylprol-inol ether in combination with acetic acid.[16] More recently,Smith employed chiral isothioureas as catalysts.[11]
In spite of the importance of this class of compounds, tothe best of our knowledge their syntheses have never beenreviewed.
In this Microreview we outline the most efficient and ge-neral approaches to the synthesis of 3,4-dihydropyran-2-ones – both in racemic and in nonracemic form – that takeadvantage of N-heterocyclic carbene (NHC) catalysis.[17]
Since the discovery of new synthetic routes based on NHCcatalysis, the number of reports dealing with the synthesisof 3,4-dihydropyran-2-ones have increased enormously.NHC-catalysed polarity reversal of the aldehyde carbonylgroup has been known for a long time and is central to anumber of organic transformations. However, in the acti-vation of simple aldehydes for new carbon–carbon bondformation, only the carbonyl carbon atom could be used asa reactive nucleophilic site under NHC-catalysis conditions.Among the most notable applications in this area are thebenzoin and Stetter reactions.[18] More recent studies havehighlighted the fact that simple manipulations on Breslowintermediates[19] generated from enals or aldehydes bearingreducible α-functional groups give a number of reactive spe-cies that offer a multitude of possibilities for designing newand unconventional reactions.[20]
In particular, the reactive species involved in the synthe-sis of 3,4-dihydropyran-2-ones are α,β-unsaturated acyl-azolium ions and azolium enols/enolates (Scheme 3).
Scheme 3. Reactive NHC intermediates involved in the synthesis of3,4-dihydropyran-2-ones.
Both of these structures are the key intermediates in dif-ferent methodologies, including conjugate 1,4-addition re-actions, [4+2] cycloaddition reactions and rearrangements.The precise mechanisms of these ring-forming reactionshave been found to depend strictly on the reagents and reac-tion conditions. In fact, some authors describe the pathwayinvolving azolium enols/enolates as [4+2] cycloaddition,whereas others suggest a Michael addition/cyclization path-way to explain the formation of pyran-2-ones (Scheme 4,path B). Therefore, in this Microreview we choose to reportthe exact definitions given by the authors.

3,4-Dihydropyran-2-ones by N-Heterocyclic Carbene Catalysis
Scheme 4. Generation of 3,4-dihydropyran-2-ones from enals with the aid of NHC catalysis.
2. Generation and Reactivity of Azolium Enol/Enolate and α,β-Unsaturated AcylazoliumIntermediates
In 2004, Bode and Glorius independently discovered thatenals could serve to generate the corresponding α,β-unsatu-rated Breslow intermediates in the presence of catalyticNHC precursors.[17c,17d] Known as homoenolates, they ex-hibit nucleophilic reactivity at the β-position, and theirchemistry is widely illustrated in the literature.[20a,20b] Fur-ther studies highlighted the fact that homoenolate oxidationcauses a second umpolung at the β-position, giving rise toα,β-unsaturated acylazolium intermediates. The cationic na-ture of the α,β-unsaturated acylazolium ions, in combina-tion with steric shielding that hampers the reactivity of thecarbonyl carbon, could directly generate a species ideallysuited to act as a reactive conjugate acceptor in Michaeladdition reactions.
In fact, in the presence of an ambident nucleophile, suchas an enolate, α,β-unsaturated acylazolium ions undergo1,4-addition by the nucleophilic carbon atom, followed byintramolecular acylation, to give 4,5,6-trisubstituted 3,4-di-hydropyran-2-ones, at the same time releasing the NHC cat-alyst (Scheme 4, path A).
Activation of enals through NHC catalysis could also beaccomplished via the corresponding azolium enol/enolates(Scheme 4, path B). Access to enolate reactivity occursthrough β-protonation of the homoenolate in competitionwith homoenolate chemistry. Tuning the selectivity to fav-our either pathway depends on the nature of the enal andof the electrophile, the strength of the base used to generatethe active NHC catalyst, the reaction solvent and finally thestructure of the catalyst. The more electron-rich imid-azolium-derived NHCs promote the homoenolate pathway,whereas triazolium-derived NHCs favour enolate forma-tion.
Nucleophilic azolium enols or enolates thus enabled ap-parent [4+2] cycloaddition pathways as one of the preferen-
Eur. J. Org. Chem. 2014, 5631–5640 © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5633
tial strategies to access the 3,4,6-trisubstituted 3,4-di-hydropyranone skeleton.
3. Azolium Enol/Enolate Intermediates in theSynthesis of 3,4-Dihydropyran-2-ones
The use of enals as substrates is advantageous, becausemany are commercially available, stable compounds, al-though they tend to dimerize through a variety of pathways.However, other starting materials, such as α-halo and α-aroyloxy aldehydes, ketenes and simple aldehydes are alsocapable of generating NHC enolates (Scheme 5).
Scheme 5. Substrates for the synthesis of 3,4-dihydropyran-2-onesvia enols/enolates.
By this approach, chiral dihydropyridinones have beenobtained in good yields and with excellent enantioselectivi-ties from α-chloro aldehydes and α,β-unsaturated sulfon-ylimines,[21] whereas chiral 3,4,6-trisubstituted dihydro-pyran-2-ones have been generated from the same chloroaldehydes and enones bearing electron-withdrawinggroups.[22] In both cases the chiral NHC employed was ofparamount importance (Scheme 6).

D. C. M. Albanese, N. GaggeroMICROREVIEW
Scheme 6. Synthesis of chiral 3,4-dihydropyran-2-ones from α-chloro aldehydes by Bode.
Racemic α-chloro aldehydes reacted under mild condi-tions either with enones bearing electron-withdrawinggroups or with β,γ-unsaturated α-oxo esters only in thepresence of 0.5–2 mol-% of chiral NHC 1a.[22] The observednotable cis diastereoselectivities have been ascribed to thestereoselective formation of (Z)-enolates in the redox reac-tions of enals in association with a high preference for endocycloaddition.
α-Aroyloxyaldehydes have also recently been used asreadily prepared, stable alternatives to α-halo aldehydes.[23]
α-Chloro aldehyde bisulfite adducts could also be used asα-chloro aldehyde surrogates, being bench-stable and easilyavailable.[24] The reaction has been carried out under aque-ous/organic biphasic conditions with α-chloro aldehydes be-ing generated in situ with 1 m K2CO3 (aqueous solution).[25]
This approach provides a practical protocol for NHC-cata-lysed Diels–Alder reactions.
When α,β-unsaturated aldehydes are used as starting ma-terials, a crucial factor for the enolate generation was foundto be the amine employed to deprotonate the starting azol-ium salt. In particular, weak amine bases such as DMAP(conjugated acid pKa = 9.2) and N-methylmorpholine(NMM, conjugated acid pKa = 7.4) gave dihydropyranones,whereas excess of moderate bases such as Et3N (conjugatedacid pKa = 10.8) generated lactones as main products.[26] Ifthe conjugate acid of the catalytic base is acidic enough toprotonate the Breslow intermediate, an enolate equivalentis produced, and Diels–Alder products can be formed(Scheme 7).
Scheme 7. Synthesis of chiral 3,4-dihydropyran-2-ones by Bode.
More recently, inorganic bases in excess in combinationwith catalytic amounts of acid have been used to promotethe enolate [4+2] cycloaddition pathway from α,β-unsatu-rated aldehydes (2 equiv.) by increasing the effective protonconcentration.[27] Aryl- and alkylenals have been made toreact with various chalcones in the presence of KOAc (ex-cess) and 25 mol-% of CH3COOH (Scheme 8).
www.eurjoc.org © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2014, 5631–56405634
Scheme 8. Synthesis of chiral 3,4-dihydropyran-2-ones by Chi andLi.
In 2007, Scheidt disclosed the optimal conditions thatfavoured enolate generation and the subsequent intramolec-ular Michael reaction of the enolate with a pendant α,β-unsaturated ketone. In the presence of chiral 1a, this was ahighly diastereo- and enantioselective process. The initiallyformed dihydropyranones were then converted into the cor-responding open-chain esters or amides by treatment withalcohols or amines (Scheme 9).[28]
Scheme 9. Synthesis of chiral 3,4-dihydropyran-2-ones by Scheidt.
The proposed pathway for this process involves the for-mation of Breslow intermediate I from the α,β-unsaturatedaldehyde and NHC. β-Protonation of I affords the enol nu-cleophile II, which generates the enol lactone by intramo-lecular Michael addition followed by acylation (Scheme 10).
Scheme 10. Catalytic cycle proposed by Scheidt.
The use of a solvent that is not a Lewis base, such asdichloromethane, and a weak base such as iPr2NEt are cru-cial for the key β-protonation.
Nair developed an enol-mediated Michael addition/cycli-zation sequence based on enals and methyl vinyl ketone,using the weak amine base DMAP for the deprotonationof the NHC precatalyst (Scheme 11).[29]

3,4-Dihydropyran-2-ones by N-Heterocyclic Carbene Catalysis
Scheme 11. Synthesis of 3,4-dihydropyran-2-ones from vinylketones by Nair.
Oxidative generation of ester enolate equivalents fromunfunctionalized aldehydes under chiral NHC catalysisconditions was reported in two independent and almost si-multaneous papers (Scheme 12).
Scheme 12. Synthesis of chiral 3,4-dihydropyran-2-ones from un-functionalized aliphatic aldehydes.
Rovis obtained cis-lactones with excellent enantio-selectivities and good diastereoisomeric ratios by treatingsimple aldehydes and chalcones in the presence of K2CO3
(0.5 equiv.), phenazine as oxidant (1 equiv.) and 1a (20 mol-%).[30] Under analogous reaction conditions trans-lactamswere obtained by using ketimines instead of chalcones.
Chi employed Cs2CO3 (20 mol-%), 3,3�,5,5�-tetra-tert-butyldiphenoquinone (2, 1 equiv.) as oxidant and 1a(10 mol-%) to prepare 3,4,6-trisubstituted cis-3,4-dihydro-pyran-2-ones with enantiomeric excesses of 99% and di-astereoisomeric ratios of 20:1 from aliphatic aldehydes andchalcones.[31] Scaling up of the process was possible throughthe use of catalytic amounts of expensive 2 in the presenceof MnO2 in excess. Both methods have obvious advantagesover known functionalized enolate precursors.
The dihydropyran-2-one skeleton bearing a quaternarystereocenter has been efficiently constructed through formal[4+2] cycloaddition between disubstituted ketenes and en-ones in the presence of l-pyroglutamic acid derived triazol-ium salt 1c (Scheme 13).[32]
A variety of ketenes have been allowed to react with en-ones bearing electron-withdrawing and electron-donatinggroups to generate the corresponding substituted dihydro-pyran-2-ones in good yields and with high enantio-selectivity.[33] The best results have been obtained by slowaddition of ketenes to the reaction mixtures. It is worth not-ing that similar results have been obtained when keteneshad been generated in situ from acyl chlorides in the pres-ence of excess Et3N. The major trans isomers could be con-
Eur. J. Org. Chem. 2014, 5631–5640 © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5635
Scheme 13. Synthesis of chiral 3,4-dihydropyran-2-ones from ket-enes by Ye.
verted into the cis isomers by in situ LDA deprotonationafter completion of the NHC-promoted cycloaddition, fol-lowed by kinetically controlled protonation at –78 °C.
A range of β,γ-unsaturated α-oxo carboxylic esters andamides were employable in such reactions. However, dif-ferent results with regard to stereoselectivity and sensitivityto isomerisation in the presence of excess base were ob-tained by Smith, depending upon the presence of a γ-arylor γ-alkyl substituent.[34]
Ketenes and 3-aroylcoumarins have been used to synthe-size the corresponding dihydrocoumarin-fused dihydro-pyranones. When the NHC generated from l-pyroglutamicacid derived triazolium salt bearing a free hydroxy groupwas used as the catalyst, high yields and good to high dia-stereo- and enantioselectivities were obtained.[35]
4. α,β-Unsaturated Acylazolium Intermediates inthe Synthesis of 3,4-Dihydropyran-2-ones
A diverse array of precursors, not only α,β-unsaturatedaldehydes, have been utilized for the efficient preparation ofcatalytic α,β-unsaturated acylazolium intermediates as ac-ceptors in the Michael addition/cyclization pathway(Scheme 14).
Scheme 14. Starting substrates for the preparation of 3,4-di-hydropyran-2-ones via α,β-unsaturated acylazolium intermediates.
With regard to this, Lupton first disclosed the use of α,β-unsaturated acylazolium ions as acceptors for the synthesisof 3,4-dihydropyran-2-ones starting from α,β-unsaturatedacyl fluorides (Scheme 15).[36]

D. C. M. Albanese, N. GaggeroMICROREVIEW
Scheme 15. Synthesis of 3,4-dihydropyran-2-ones from α,β-unsatu-rated acyl fluorides by Lupton.
A range of conjugate acceptors, included some featuringβ-disubstitution, reacted with enolates to give mainly bicy-clic pyran-2-ones in good yields.
The proposed pathway for the process involves the ad-dition of α,β-unsaturated enol ester I to the NHC to gener-ate acyl azolium intermediate II with elimination of enolate.Subsequent conjugate addition provides azolium enolateIII. Proton transfer and acylation give pyranone V and re-generate the catalyst (Scheme 16).
Scheme 16. Catalytic cycle proposed by Lupton.
Using this protocol, Lupton developed the rearrange-ment of cyclic α,β-unsaturated ester 3 to access bicyclic di-hydropyranone 4, the key intermediate for the total synthe-sis of 7-deoxyloganin (Scheme 17).[5]
Scheme 17. Key intermediate en route to deoxyloganin.
Straight after the publication of Lupton’s work, Bodetook advantage of catalytically generated chiral α,β-unsatu-rated azolium ions to synthesize 3,4-dihydropyran-2-ones
www.eurjoc.org © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2014, 5631–56405636
with ees of up to 99% (Scheme 18) with the aid of Rovis’striazolium salt 1a (see Section 3).[37]
Scheme 18. Synthesis of chiral 3,4-dihydropyran-2-ones by Bode.
Bode carried out the reactions in neutral media and uti-lized stable enolic nucleophiles such as naphthol, kojic acidand dicarbonyl compounds. Detailed kinetic investigationscarried out under these new conditions are consistent witha Coates–Claisen rearrangement of a hemiacetal intermedi-ate II, generated by 1,2-addition of the stable enol oxygenatom VII, to the acylazolium ion I. The mechanism is de-picted in Scheme 19. The possibility of starting from enalsand stoichiometric amounts of oxidant was also ex-plored.[37b]
Scheme 19. Catalytic cycle proposed by Bode.
Xiao used 2-alkenylazolium ions obtained from alkynylaldehydes and 1b as Michael acceptors.[38] Conjugate ad-dition/cyclization with β-oxo esters or 1,3-diketones in thepresence of potassium tert-butoxide provided function-alized 3,4-dihydropyranones with yields up to 75%(Scheme 20).
Scheme 20. Synthesis of 3,4-dihydropyran-2-ones from alkynylaldehydes by Xiao.

3,4-Dihydropyran-2-ones by N-Heterocyclic Carbene Catalysis
The catalytic cycle might proceed through the formationof Breslow intermediate I, which could undergo proton-ation by a more acidic (than an alcohol) H-donor – a β-oxo ester or 1,3-diketone – to give enolate anion III and theelectrophilic acylazolium species II. Subsequent Michaeladdition, H-migration and acylation would furnish the 3,4-dihydropyranone ring (Scheme 21).
Scheme 21. Catalytic cycle proposed by Xiao.
Using chiral NHC 1a, Xiao developed a direct and ef-ficient stereoselective method to prepare pyran-2-ones fromynals and 1,3-dicarbonyl compounds (Scheme 22).[39]
Scheme 22. Asymmetric synthesis of chiral 3,4-dihydropyran-2-ones by Xiao.
Moreover, by simple addition of 3,3�,5,5�-tetra-tert-butyldiphenoquinone (2), the investigation was extended toα,β-unsaturated enals.
Studer used oxidatively generated acylazolium ions forthe synthesis of 3,4-dihydropyran-2-ones, starting from cin-namaldehyde, acetylacetone, 1,4-dimethyl-1H-1,2,4-triazol-4-ium iodide (1e, 2 mol-%) as a cheap carbene precursorand diazabicyclo[5.4.0]undecene (DBU). As an organic oxi-dant, 2 was added (Scheme 23).
Scheme 23. Synthesis of 3,4-dihydropyran-2-ones by Studer.
Eur. J. Org. Chem. 2014, 5631–5640 © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5637
The reactions proceeded efficiently with various 1,3-di-carbonyl compounds and with substituted cinnamaldehydederivatives. 3-Methylbut-2-enal and acetylacetone providedthe corresponding dihydropyranone in 73% yield, showingthat this method can be used to build up quaternary carboncentres. The suggested catalytic cycle is depicted inScheme 24. The reaction between enal and carbene gener-ates the Breslow intermediate. Two-electron oxidation anddeprotonation provide the redox-activated Michael ac-ceptor, which undergoes conjugate addition by deproton-ated 1,3-dicarbonyl compound. Proton transfer and cycliza-tion lead to pyran-2-one and carbene regeneration.[40]
Scheme 24. Catalytic cycle proposed by Studer.
The same approach was followed by You in an asymmet-ric version of the reaction. The combination of camphor-derived triazolium salt 1f (10 mol-%) with a catalyticamount of NaBF4 was found to be highly efficient, provid-ing pyran-2-ones with enantiomeric excesses of up to 96%(Scheme 25).[41]
Scheme 25. Synthesis of chiral 3,4-dihydropyran-2-ones from α,β-unsaturated enals by You.
In 2011 Ye reported the generation of α,β-unsaturatedacylazolium ions from α-bromoenals and NHCs.[42] Subse-quent Michael addition with 1,3-dicarbonyl compounds inthe presence of a base provided the corresponding pyran-2-ones (Scheme 26).

D. C. M. Albanese, N. GaggeroMICROREVIEW
Scheme 26. Synthesis of chiral 3,4-dihydropyran-2-ones from α-bromoenals by Ye.
A series of chiral NHC precursors, easily prepared froml-pyroglutamic acid, were tested in order to optimizestereoselectivity. It is interesting to note that enantio-selectivity could be reversed depending on the structure ofthe catalyst and the nature of the base. Two possible transi-tion states (TSs) A and B were proposed in order to explainthe stereochemical outcome of the reaction. In TS A, theenolate attacks from the less hindered face of the acylazol-ium intermediate generated from 1g. In TS B, a hydrogenbond between 1h and the enolate reverses the stereochemi-cal selectivity. However, when strong bases are used, thehydroxy group on the catalyst is deprotonated, and thehydrogen bond is not formed, so this reversal of stereo-chemistry is not observed (Figure 1).
Figure 1. Transition states suggested by Ye.
The possible catalytic cycle is depicted in Scheme 27. Ad-dition of the NHC to bromo aldehyde gives Breslow inter-mediate I, which tautomerizes to bromo ketone II. Elimi-nation of bromide provides intermediate III. Michael ad-dition of the 1,3-dicarbonyl compound gives IV, which fur-nishes pyran-2-one V by ring closure and regenerates theNHC.
In 2012 Yao reported NHC-catalysed reactions of α-bromo aldehydes or α,β-dibromo aldehydes with 1,3-di-carbonyl compounds in the presence of 10 mol-% of 1b andCs2CO3 in excess to generate highly functionalized 3,4-di-hydropyran-2-ones with yields up to 94%.[43]
Also, Biju reported the enantioselective synthesis of 3,4-dihydropyran-2-ones through the reaction between 2-
www.eurjoc.org © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2014, 5631–56405638
Scheme 27. Catalytic cycle proposed by Ye.
bromoenals and 1,3-dicarbonyl compounds under mildconditions and with a relatively low catalyst loading of 1a(5 mol-%).[44] The reaction had broad substrate scope andfurnished enantiomeric excesses of up to 97% and yields ofup to 96% (Scheme 28). The authors provided an explana-tion of the mechanism and the enantioselectivity of the pro-cess based on DFT calculations. In particular, these studieshighlighted that the 1,4-addition step is controlled by ahydrogen bond between the enol and carbonyl moieties.Moreover, they found that in the presence of LiOAc·2H2Oan energetically more favourable proton-transfer step beforecyclization would be involved.
Scheme 28. Synthesis of chiral 3,4-dihydropyran-2-ones from 2-bromoenals by Biju.
More recently, Biju reported the synthesis of 4,5-disubsti-tuted 3,4-dihydropyran-2-ones with ee values of up to 99%via α,β-unsaturated acylazolium intermediates generatedfrom α-bromoenals and 1a (5 mol-%), together with enoliz-able aldehydes in the presence of Na2CO3.[45]
An approach to 3,4-dihydropyran-2-ones from β-bromo-enals was disclosed by Ma (Scheme 29).[46] This result sug-gests that a route to α,β-unsaturated acylazolium ions fromenals with a reducible group at the β-position is likely in-volved; this is complementary to routes from α-reducible
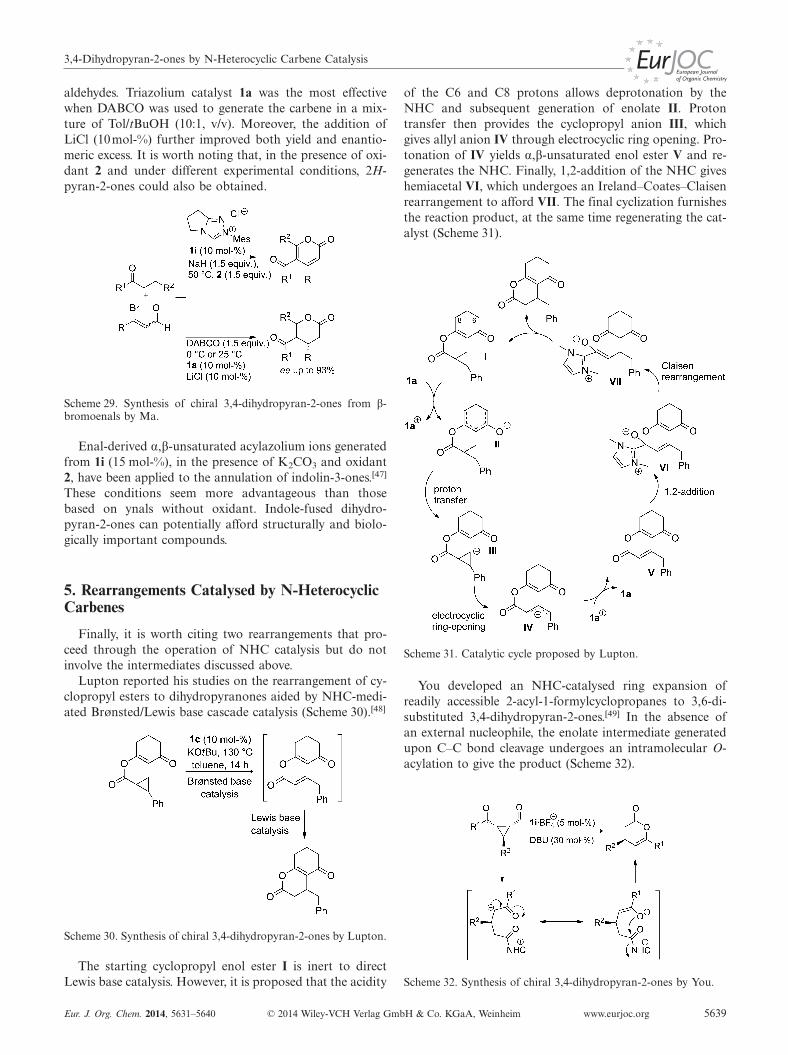
3,4-Dihydropyran-2-ones by N-Heterocyclic Carbene Catalysis
aldehydes. Triazolium catalyst 1a was the most effectivewhen DABCO was used to generate the carbene in a mix-ture of Tol/tBuOH (10:1, v/v). Moreover, the addition ofLiCl (10mol-%) further improved both yield and enantio-meric excess. It is worth noting that, in the presence of oxi-dant 2 and under different experimental conditions, 2H-pyran-2-ones could also be obtained.
Scheme 29. Synthesis of chiral 3,4-dihydropyran-2-ones from β-bromoenals by Ma.
Enal-derived α,β-unsaturated acylazolium ions generatedfrom 1i (15 mol-%), in the presence of K2CO3 and oxidant2, have been applied to the annulation of indolin-3-ones.[47]
These conditions seem more advantageous than thosebased on ynals without oxidant. Indole-fused dihydro-pyran-2-ones can potentially afford structurally and biolo-gically important compounds.
5. Rearrangements Catalysed by N-HeterocyclicCarbenes
Finally, it is worth citing two rearrangements that pro-ceed through the operation of NHC catalysis but do notinvolve the intermediates discussed above.
Lupton reported his studies on the rearrangement of cy-clopropyl esters to dihydropyranones aided by NHC-medi-ated Brønsted/Lewis base cascade catalysis (Scheme 30).[48]
Scheme 30. Synthesis of chiral 3,4-dihydropyran-2-ones by Lupton.
The starting cyclopropyl enol ester I is inert to directLewis base catalysis. However, it is proposed that the acidity
Eur. J. Org. Chem. 2014, 5631–5640 © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.eurjoc.org 5639
of the C6 and C8 protons allows deprotonation by theNHC and subsequent generation of enolate II. Protontransfer then provides the cyclopropyl anion III, whichgives allyl anion IV through electrocyclic ring opening. Pro-tonation of IV yields α,β-unsaturated enol ester V and re-generates the NHC. Finally, 1,2-addition of the NHC giveshemiacetal VI, which undergoes an Ireland–Coates–Claisenrearrangement to afford VII. The final cyclization furnishesthe reaction product, at the same time regenerating the cat-alyst (Scheme 31).
Scheme 31. Catalytic cycle proposed by Lupton.
You developed an NHC-catalysed ring expansion ofreadily accessible 2-acyl-1-formylcyclopropanes to 3,6-di-substituted 3,4-dihydropyran-2-ones.[49] In the absence ofan external nucleophile, the enolate intermediate generatedupon C–C bond cleavage undergoes an intramolecular O-acylation to give the product (Scheme 32).
Scheme 32. Synthesis of chiral 3,4-dihydropyran-2-ones by You.

D. C. M. Albanese, N. GaggeroMICROREVIEW
6. ConclusionsThe recent progress in N-heterocyclic carbene catalysis
has provided a powerful tool for the synthesis of 3,4-di-hydropyran-2-ones, which are known medicinal agents anduseful building blocks.
This Microreview outlines the flexibility of NHC chemis-try, which allows the generation of a number of reactiveintermediates. In particular, azolium enol/enolates and α,β-unsaturated acylazolium ions provide the title compoundsthrough different synthetic pathways. Either 4,5,6- or 3,4,6-trisubstituted pyran-2-ones can be obtained by startingfrom simple substrates under mild conditions and in astraightforward manner. Moreover, enantiopure α-pyr-anones could be obtained by using chiral NHC precursors.It is anticipated that the design of new NHCs, along withtheir versatility, will prompt numerous new organocatalyticapplications.
[1] This compound is in phase I clinical trials.[2] S. Pal, V. Chatare, M. Pal, Curr. Org. Chem. 2011, 15, 782–800.[3] M. Brasholz, S. Sörgel, C. Azap, H.-U. Reißig, Eur. J. Org.
Chem. 2007, 3801–3814.[4] M. Willot, L. Radtke, D. Konning, R. Forhlich, V. H. Gessner,
C. Strohmann, M. Christmann, Angew. Chem. Int. Ed. 2009,48, 9105–9108; Angew. Chem. 2009, 121, 9269.
[5] L. Candish, D. W. Lupton, Org. Lett. 2010, 12, 4836–4839.[6] D. C. Harrowven, J. C. Hannam, Tetrahedron 1999, 55, 9333–
9340.[7] a) T. Kume, H. Iwasaki, Y. Yamamoto, K. Akiba, Tetrahedron
Lett. 1988, 29, 3825–3828; b) D. Rosenthal, P. Grabowich, E. F.Sabo, J. Fried, J. Am. Chem. Soc. 1963, 85, 3971–3979.
[8] a) A. G. Schultz, L. Pettus, J. Org. Chem. 1997, 62, 6855–6861;b) A. K. Mandal, D. G. Jawalkar, J. Org. Chem. 1989, 54,2364–2369.
[9] S. H. Thang, D. J. Rigg, Synth. Commun. 1993, 23, 2355–2361.[10] J. A. Robl, Tetrahedron Lett. 1990, 31, 3421–3424.[11] D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin, A. D.
Smith, J. Am. Chem. Soc. 2011, 133, 2714–2720.[12] S. Kobayashi, M. Moriwaki, Synlett 1997, 5, 551–552.[13] A. R. Katritzky, O. V. Denisko, J. Org. Chem. 2002, 67, 3104–
3108.[14] T. Tozawa, H. Nagao, Y. Yamane, T. Mukaiyama, Chem. Asian
J. 2007, 2, 123–134 and references cited therein.[15] a) K. Itoh, S. Kanemasa, Tetrahedron Lett. 2003, 44, 1799–
1802; b) K. Itoh, M. Hasegawa, J. Tanaka, S. Kanemasa, Org.Lett. 2005, 7, 979–981; c) F. Ono, M. Hasegawa, S. Kanemasa,J. Tanaka, Tetrahedron Lett. 2008, 49, 5105–5107.
[16] J. Wang, F. Yu, X. Zhang, D. Ma, Org. Lett. 2008, 10, 2561–2564.
[17] For recent reviews on NHC catalysis see: a) D. Enders, O. Nie-meier, A. Henseler, Chem. Rev. 2007, 107, 5606–5655; b) N.Marion, S. Diez-Gonzalez, S. P. Nolan, Angew. Chem. Int. Ed.2007, 46, 2988–3000; Angew. Chem. 2007, 119, 3046; c) X. Bu-gau, F. Glorius, Chem. Soc. Rev. 2012, 41, 3511–3522; d) J.Mahatthananchai, J. W. Bode, Acc. Chem. Res. 2014, 47, 696–707.
[18] D. Enders, T. Balensiefer, Acc. Chem. Res. 2004, 37, 534–541.[19] See Scheme 4.[20] For recent reviews on NHC homoenolate chemistry see: a)
B. E. Maki, A. Chan, K. A. Scheidt, Synthesis 2008, 8, 1306–1315; b) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R.Paul, A. Jose, V. Sreekumar, Chem. Soc. Rev. 2011, 40, 5336–5346. For recent reviews on NHC enol/enolate chemistry see:
www.eurjoc.org © 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Eur. J. Org. Chem. 2014, 5631–56405640
c) J. Douglas, G. Churchill, A. D. Smith, Synthesis 2012, 44,2295–2309; d) H. U. Hora, P. Wheeler, T. Rovis, Adv. Synth.Catal. 2012, 354, 1617–1639. For a recent review on NHCacyl anion chemistry see ref.[18] For a recent review on NHCacylazolium chemistry see ref.[17d]
[21] M. He, J. R. Struble, J. W. Bode, J. Am. Chem. Soc. 2006, 128,8418–8420.
[22] M. He, G. J. Uc, J. W. Bode, J. Am. Chem. Soc. 2006, 128,15088–15089.
[23] K. B. Ling, A. D. Smith, Chem. Commun. 2011, 47, 373–375.[24] α-Chloro aldehydes are known to be sensitive to moisture and
oxygen; see: N. De Kimpe, R. Verhé in The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines, John Wileyand Sons, New York, 1988, chapter 3.
[25] M. He, B. J. Beahm, J. W. Bode, Org. Lett. 2008, 10, 3817–3820.[26] a) J. Kaeobamrung, M. Kozlowski, J. W. Bode, Proc. Natl.
Acad. Sci. USA 2010, 107, 20661–20665.[27] Z. Fu, H. Sun, S. Chen, B. Tiwari, G. Li, Y. R. Chi, Chem.
Commun. 2013, 49, 261–263.[28] E. M. Phillips, M. Wadamoto, A. Chan, K. A. Scheidt, Angew.
Chem. Int. Ed. 2007, 46, 3107–3110; Angew. Chem. 2007, 119,3167.
[29] V. Nair, R. R. Paul, K. C. S. Lakshmi, R. S. Menon, A. Jose,C. R. Sinu, Tetrahedron Lett. 2011, 52, 5992–5994.
[30] X. Zhao, K. E. Ruhl, T. Rovis, Angew. Chem. Int. Ed. 2012, 51,12330–12333; Angew. Chem. 2012, 124, 12496.
[31] J. M. Ruojie, Y. X. Chen, B. Tiwari, Y. R. Chi, Org. Lett. 2013,15, 50–53.
[32] Y.-R. Zhang, H. Lv, D. Zhou, S. Ye, Chem. Eur. J. 2008, 14,8473–8476.
[33] A single recrystallization from hexane/propan-2-ol (9:1) pro-vided an increase in diastereomeric ratios and enantiomeric ex-cess.
[34] S. M. Leckie, T. B. Brown, D. Pryde, T. Lebl, A. M. Z. Slawin,A. D. Smith, Org. Biomol. Chem. 2013, 11, 3230–3246.
[35] T.-Y. Jian, X.-Y. Chen, L.-H. Sun, S. Ye, Org. Biomol. Chem.2013, 11, 158–163.
[36] S. J. Ryan, L. Candish, D. W. Lupton, J. Am. Chem. Soc. 2009,131, 14176–14177.
[37] a) J. Kaeobamrung, J. Mahatthananchai, P. Zheng, J. W. Bode,J. Am. Chem. Soc. 2010, 132, 8810–8812; b) J. Mahatthanan-chai, J. Kaeobamrung, J. W. Bode, ACS Catal. 2012, 2, 494–503; c) J. Mahatthananchai, P. Zheng, J. W. Bode, Angew.Chem. Int. Ed. 2011, 50, 1673–1677; Angew. Chem. 2011, 123,1711; d) ref.[17d]
[38] Z.-Q. Zhu, J.-C. Xiao, Adv. Synth. Catal. 2010, 352, 2455–2458.[39] Z.-Q. Zhu, X.-L. Zheng, N.-F. Jiang, X. Wan, J.-C. Xiao,
Chem. Commun. 2011, 47, 8670–8672.[40] S. De Sarkar, A. Studer, Angew. Chem. Int. Ed. 2010, 49, 9266–
9269; Angew. Chem. 2010, 122, 9452.[41] Z.-Q. Rong, M.-Q. Jia, S.-L. You, Org. Lett. 2011, 13, 4080–
4083.[42] F.-G. Sun, L.-H. Sun, S. Ye, Adv. Synth. Catal. 2011, 353,
3134–3138.[43] C. Yao, D. Wang, J. Lu, T. Li, W. Jiao, C. Yu, Chem. Eur. J.
2012, 18, 1914–1917.[44] S. R. Yetra, A. Bhunia, A. Patra, M. V. Mane, K. Vanka, A. T.
Biju, Adv. Synth. Catal. 2013, 355, 1089–1097.[45] S. R. Yetra, T. Kaicharta, S. S. Kunte, R. G. Gonnade, A. T.
Biju, Org. Lett. 2013, 15, 5202–5205.[46] G. Wang, X. Chen, G. Miao, W. Yao, C. Ma, J. Org. Chem.
2013, 78, 6223–6232.[47] Y. Lu, W. Tang, Y. Zhang, D. Du, T. Lu, Adv. Synth. Catal.
2013, 355, 321–326.[48] L. Candish, D. W. Lupton, Chem. Sci. 2012, 3, 380–383.[49] G.-Q. Li, L.-X. Dai, S.-L. You, Org. Lett. 2009, 11, 1623–1625.
Received: February 6, 2014Published Online: July 4, 2014












![N-Heterocyclic Carbene Organocatalytic Transformations in ...which could lead to a highly enantioselective benzoin condensation in up to 83% yield and 95% ee.[27] In addition, Leeper](https://img.pdfslide.tips/doc/110x75/5fe758ff35fff30574689c6d/n-heterocyclic-carbene-organocatalytic-transformations-in-which-could-lead-to.jpg)
















