Embed Size (px)
Citation preview

Il sistema nervoso
Il sistema nervoso è un network di cellule specializzate che recepiscono e comunicano informazioni riguardanti l’ambiente circostante, o parametri biochimici, per poter poi elaborarli e allestire una risposta, una reazione.Il sistema nervoso può essere diviso in
sistema nervoso centrale SNC sistema nervoso periferico SNP
Il sistema nervoso centrale è formato da encefalo (situato nel cranio) con le 12 paia di nervi cranici, e il midollo spinale (situato nel canale vertebrale) con i nervi spinali.L’encefalo da solo costituisce il 20% del consumo di O2, e consuma 150 gr di glucosio (700 Kcal) al giorno.Un altro tipo di classificazione suddivide il sistema nervoso in:
somatico afferenze somatiche e motilità volontaria autonomo afferenze viscerali, motilità viscerale e ghiandolare
il sistema autonomo è a sua volta suddiviso in simpatico e parasimpatico
Embriologia del sistema nervoso
La neurulazione è il processo per cui si sviluppa il sistema nervoso embrionale su stimolo differenziativo proveniente dalla notocorda, una struttura presente solo nei chordata.
Il SNC dei vertebrati si sviluppa da una striscia di neuroectoderma che appare sulla superfice dorsale dei vertebrati (placca neurale), al di sopra della notocorda.
Questa striscia si infossa verso l’interno della gastrula a formare una doccia circondata ai lati dalle creste neurali.
Le creste si fondono formando un tubo, il tubo neurale, nei giorni successivi la parte rostale (anteriore) del tubo neurale si allarga per formare tre vescicole: prosencefalo, mesencefalo e rombencefalo.

Prosencefalo, mesencefalo e rombencefalo sono deputati alla sviluppo di tutto l’encefalo, e riflettono la formazione di 3 sistemi sensori differenti.Il prosencefalo dà origine a chemorecettori, deputati al senso dell’olfatto, e a telencefalo e diencefalo.Il mesencefalo dà orgine a fotorecettori, per la vista, e al mesencefalo.Il rombencefalo dà origine a meccanocettori, per udito ed equilibrio, e a metencefalo (ponte e cervelletto) e mielencefalo (bulbo).
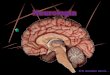
L’encefalo
L’encefalo è una struttura del sistema nervoso centrale completamente contenuta all’interno del cranio, si presenta come una massa gelatinosa, contiene come ordine di grandezza 100 miliardi di neuroni, con 10 mila connessioni sinaitiche per ognuno di essi.Le porzioni che costituiscono l’encefalo sono:
Cervello Tronco Cervelletto
Il cervello è costituito a sua volta dai 2 emisferi del telencefalo posti in connessione dalla falce (struttura fibrosa), più il diencefalo che comprende talamo, ipotalamo, ipofisi e nervi ottici.Il tronco invece comprende mesencefalo ponte e bulbo.Il cervelletto è quella porzione che alloggia nella fossa cranica posteriore.L’encefalo è avvolto dalle meningi che sono un sistema di 3 membrane che hanno funzione protettiva. Le meningi sono 3 e sono chiamate dura madre, aracnoide e pia madre (dall’esterno verso l’interno). Un’estensione della dura madre separa il cervelletto dai lobi occipitali del cervello, questa struttura è chiamata tentorio, e distingue le strutture dell’enfefalo in sopratentoriali (cervello) e sottotentoriali (tronco e cervelletto).
Sostanza bianca e sostanza grigia
Il SNC presenta un'organizzazione o citoarchitettonica caratterizzata dalla presenza di aree di sostanza grigia nelle quali sono prevalentemente collocati i corpi cellulari (o pirenofori o pericaria) dei neuroni, i dendriti, i tratti iniziali degli assoni (privi, questi ultimi di guaina mielinica) e le cellule gliali, e da aree di sostanza bianca, nelle quali sono prevalentemente localizzati gli assoni mielinizzati. Il suo colore bianco brillante è dovuto ai lipidi che formano la guaina mielinica. Nel midollo spinale la sostanza grigia occupa la porzione centrale, assumendo un caratteristico aspetto a farfalla, mentre la sostanza bianca occupa la porzione periferica. Nell'encefalo le aree di sostanza grigia sono organizzate in cortecce (corteccia cerebrale e cerebellare) sulla superficie esterna e in nuclei, con i neuroni organizzati in agglomerati, immersi in aree di sostanza bianca, in profondità (zona esterna grigia, zona media bianca, zona interna grigia).I nuclei della base ono formazioni grigie situate profondamente rispetto alla sostanza bianca telencefalica, in stretto rapporto con il talamo. Essi comprendono[10]:

il claustro l'amigdala il corpo striato, comprendente a sua volta:
il nucleo caudato il nucleo lenticolare, definizione che viene usata per indicare due nuclei strettamente
connessi, il pallido ed il putamen. Il corpo striato è un voluminoso insieme di nuclei che deve il nome alla sua particolare organizzazione strutturale: esso è infatti composto da un'alternanza di formazioni grigie intersecate da fasci di sostanza bianca, che conferiscono alla struttura quel particolare aspetto "striato". Il corpo striato viene anatomicamente assegnato al lobo frontale.Il corpo striato comprende nucleo caudato e nucleo lenticolare. Il nucleo caudato ha una forma di una virgola ed è suddiviso in testa, corpo e coda.Il nucleo lenticolare comprende altri 2 nuclei connessi tra loro: il pallido e il putamen.L'amigdala è una formazione grigia di forma globosa, situata in profondità rispetto al pallido, da cui è separata per l'interposizione delle fibre della capsula interna, e connessa con l'apice della coda del nucleo caudato; caudalmente, essa continua con l'uncus della circonvoluzione dell'ippocampo. Anatomicamente l'amigdala può essere assegnata al lobo temporale. Il claustro è una sottile lamina di sostanza grigia, in continuità anteriormente con l'amigdala ed addossata alla corteccia insulare, dalla quale è separata soltanto per l'interposizione della capsula estrema; medialmente, la capsula esterna lo separa invece dal putamen. Il suo significato funzionale non è del tutto noto. La figura sembra non rispettare la posizione di alcuni nuclei come l'amigdala, infatti un'unica sezione coronale è riduttiva perché in tre dimensioni la coda del nucleo caudato fa un giro fino a connettersi con l'amigdala.
LOBI CEREBRALI
La corteccia cerebrale per aumentare la superficie di connessione si ripiega formando circumvoluzioni e solchi. Alcuni di questi sono molto profondi e sono caratteristici della struttura della corteccia: la suddividono in lobi, che non sono una mera suddivisione anatomica, ma presentano una correlazione con le funzioni del cervello.

I solchi principali sono due: Solco centrale o scissura di Rolando Solco laterale o scissura di Silvio
La scissura di Rolando separa il lobo frontale dal lobo parietale. Il solco laterale, invece, delimita il lobo temporale. Nella parte posteriore è presente infine il lobo occipitale.Sono presenti altri 2 lobi, interni, nascosti al di sotto dei lobi già descritti, essi sono il lobo limbico, che si
estende solo sulla faccia mediale degli emisferi ed é una parte del cervello sviluppatasi dal vecchio cervello olfattivo, e l’insula di Reil, si trova in profondità in corrispondenza della scissura di Silvio.Il lobo frontale si occupa delle funzioni motorie, capacità di progettazione e funzioni raziocinanti.Il lobo parietale riceve le sensazioni somatiche.Il lobo occipitale si occupa della visione.Il lobo temporale si occupa di udito, apprendimento e memoria.
Il lobo limbico contiene amigdala e ippocampo e si occupa dei comportamenti emozionali.L’insula del Reil si occupa dell’empatia ed emozioni sociali.
LIQUOR E VENTRICOLI
Tutto il SNC è immerso in un liquido, chiamato liquor o liquido cefalorachidiano, ha funzione di protezione e di sostengo, perché il cervello letteralmente ci galleggia dentro riducendo il suo peso a soli 25 gr.Il liquor si trova nello spazio sub-aracnoideo, quindi tra aracnoide e pia madre, più nei ventricoli, che sono aree vuote all’interno dell’encefalo. Essi sono uno per emisfero (forami di Monro), che si congiungono in un terzo collegato poi ad un quarto tramite l’acquedotto di Silvio, da qui attraverso i forami di Lushka e

Magendie si riversa nella cisterna magna e poi continua nello spazio sub aracnoideo e nel canale del midollo.Il liquido cefalorachidiano circola con movimenti dinamici propri ma ritmati dall’attività cardiaca.All’interno dei ventricoli ci sono delle strutture deputate alla produzione di liquido cefalorachidiano, chiamati plessi corioidei. Questi producono giornaliermente 800 mL di liquor, che viene costantemente riassorbito nelle granulazioni di Pacchioni, il volume totale di liquido cefalorachidiano è 125 mL.E’ possibile prelevare una certa quantità di liquor per condurre analisi biochimiche, questa procedura chiamata rachicentesi viene effettuata per sicurezza tramite una puntura tra la 4° e la 5° vertebra lombare, in quanto il midollo si arresta alla 2° vertebra.
CERVELLETTO
Il cervelletto è una parte del sistema nervoso centrale; è posto in posizione dorsale rispetto al tronco encefalico, con il quale è collegato tramite 3 coppie di peduncoli:
peduncoli cerebellari superiori (che lo collegano con il mesencefalo) peduncolo cerebellari medi (i più voluminosi, che lo collegano con il ponte) peduncoli cerebellari inferiori (che lo collegano con il bulbo)
Viene quindi ad essere separato dal tronco dal quarto ventricolo cerebrale. Si trova collocato nella fossa endocranica posteriore. La parte superiore viene in rapporto con il tentorio del cervelletto, che lo separa dagli emisferi cerebrali, mentre la porzione inferiore con le fosse dell'osso occipitale. Il volume cerebellare costituisce il 10% del volume totale dell'encefalo ma contiene più della metà dei neuroni cerebrali. Macroscopicamente possiamo riconoscere una porzione centrale, il verme, e due emisferi cerebellari di destra e di sinistra.
Porzioni del cervelletto Emisferi e verme Mantello corticale (folia) Parte interna midollare Nuclei profondi (fastigio, globoso, emboliforme, dentato) Peduncolo cerebellare inferiore, medio e superiore
Funzioni del cervelletto Coordinazione muscolare Apprendimento degli schemi motori Mantenimento del tono, della postura e dell’equilibrio Funzioni cognitive e comportamentali? Linguaggio?
Organizzazione filogeneticaLobo flocculonodulare, archicerebellum
equilibrio, posizione del capo, movimenti oculariLobo anteriore, paleocerebellum
tono, posturaLobo posteriore, neocerebellum

coordinazione e pianificazione del movimento, apprendimento motorio
NERVI CRANICI
I nervi cranici, o nervi encefalici, sono un gruppo di nervi che invece di avere origine dal midollo spinale, partono direttamente dal tronco encefalico. Nell'anatomia umana ci sono dodici paia di nervi cranici pari (destri e sinistri), numerati dall'alto verso il basso con numeri romani:
1. nervo olfattivo (I) 2. nervo ottico (II) 3. nervo oculomotore (III) 4. nervo trocleare (IV) 5. nervo trigemino (V) 6. nervo abducente (VI) 7. nervo faciale (VII) 8. nervo vestibolococleare (VIII) 9. nervo glossofaringeo (IX) 10. nervo vago (X) 11. nervo accessorio (XI) 12. nervo ipoglosso (XII)
Hanno le stesse caratteristiche dei nervi spinali in quanto presentano fibre motrici e fibre sensitive, viscerali e somatiche. La differenza sta nel fatto che i nervi encefalici possono avere anche solo una di queste caratteristiche, mentre i nervi spinali sono nervi misti.Il nervo olfattivo (I) è di tipo esclusivamente sensitivo, la sua funzione è quella di recepire gli stimoli olfattivi che giungono nelle cavità nasali e trasportarli all'encefalo sotto forma di impulsi elettrici.Il nervo ottico (II) è il nervo che trasmette le informazioni visive dalla retina al cervello. E’ un nervo cranico infatti le fibre sono ricoperte dalla mielina prodotta dagli oligodendrociti, e il nervo ottico è avvolto nelle
meningi (dura madre, aracnoide, pia madre) e poco dopo essere entrato nella cavità cranica, attraverso il
foro ottico, le fibre si incrociano parzialmente nel chiasma ottico. Tecnicamente, assieme al nervo olfattivo, non sono nervi ma una continuazione del sistema nervoso centrale.I nervi oculomotore, trocleare e abducente (III, IV, VI) si occupano del movimento degl i occhi.Il nervo trigemino (V) ha 2 componenti: una sensitiva e una motrice. La componente sensitiva raccoglie stimoli riguardanti la sensibilità esterocettiva e propriocettiva di testa, faccia e meningi. La componente motrice innerva ad esempio muscoli masticatori e il muscolo tensore del timpano. Si divide in 3 branche: orbitale, massetere e mandibolare.Il nervo faciale (VII) comprende due distinti nervi: il nervo faciale propriamente detto ed il nervo intermedio (del Wrisberg). Il primo contiene fibre motrici somatiche per i muscoli mimici ed altri derivati del secondo arco branchiale, il secondo invece comprende fibre sensitive somatiche e viscerali.Il nervo vestibolo-cocleare (VIII) è nervo sensitivo, trasmette le informazioni codificate a livello dell'orecchio: ha un ramo cocleare che porta le sensazioni uditive, e un ramo vestibolare che porta sensazioni relative all’equilibrio.Il nervo glossofaringeo (IX) ha una componente parasimpatica con cui controlla le ghiandole salivari non controllate dal settimo nervo cranico (nervo facciale). Innerva il terzo arco branchiale e i suoi derivati.

Trasmettono le sensazioni del gusto tramite la loro componente sensitiva e con la componente motoria innervano la lingua e la faringe.Il nervo vago (X) parte dal midollo allungato e si porta verso il basso nel torace e nell'addome. I 2 nervi vaghi destro e sinistro sono tra i più importanti del corpo nonché il più lunghi ed i più ramificati tra i nervi cranici. Esso controlla tutta la muscolatura liscia non controllata dai nervi oculomotore, facciale e glossofaringeo e dai nervi spinali. In particolare innerva cuore, polmoni, intestino, stomaco.Il nervo accessorio (XI) innerva 2 muscoli deputati al movimento di collo e spalla: lo sternocleidomastoideo e il trapezio.Il nervo ipoglosso (XII) è esclusivamente motore e innerva muscoli del collo e la lingua.
MIDOLLO
Il midollo spinale è una porzione del sistema nervoso centrale; ha forma pressoché cilindrica, leggermente schiacciata in senso antero-posteriore, con 2 rigonfiamenti a livello cervicale e lombare, in quanto passano più nervi per connettere gli arti superiori ed inferiori. Il midollo spinale è posto nel canale vertebrale.Da ogni lato del midollo spinale emergono 31 paia di nervi spinali composti da 31 radici anteriori e 31 radici posteriori.Nel midollo spinale vi è la materia grigia (a forma di “h” o farfalla) circondata dalla materia bianca. Dall'esterno della materia grigia emergono due radici (o corna) dorsali (una dal lato sinistro ed una dal lato destro) e due radici ventrali. Le radici dorsali contengono gli assoni sensoriali e quelle ventrali contengono gli assoni motori.Le radici dorsali di ogni lato continuano verso l'esterno formando lungo il percorso il ganglio dorsale (detto anche ganglio spinale), qui alloggia il corpo cellulare dei neuroni “a T” (cioè con un solo prolungamento che poi si biforca, formando una T, e un ramo costituisce il dendrite e un ramo l’assone), che collegano l’assone sensitivo al neurone afferente sensitivo nel midollo. Nella radice ventrale tale ganglio non c’è, e il corpo del motoneurone alloggia direttamente nella materia grigia del midollo. Quindi le radici anteriori sono costituite essenzialmente da dagli assoni dei motoneuroni α e γ che si trovano nella testa del corno anteriore del midollo spinale. Le radici posteriori sono formate dalle fibre centrali dei neuroni a T, collocati nel ganglio spinale, che trasportano impulsi sensitivi, viscerali e somatici provenienti dalla periferia. Le radici ventrali e dorsali dopo essere uscite dalla vertebra si fondono in un punto successivo al ganglio. A questo punto prendono il nome di nervi spinali misti.I rami anteriori dei nervi spinali si anastomizzano tra di loro a formare sei plessi nervosi:

plesso cervicale (I-IV C) plesso brachiale (V-VII C, I T) plesso lombare (I-IV L) plesso sacrale (IV-VL, I-III S) plesso pudendo (III-IV S) plesso coccigeo (IV-V S, I Co)
A ciascuna radice corrisponde un territorio di innervazione o metamero. Per ogni metamero esiste un rispettivo cutaneo detto dermatomero. Per quanto riguarda i visceri la suddivisione metamerica è meno definita; tuttavia sono identificabili anche qui aree di corrispondenza tra afferenze splancniche e metameri che vengono chiamate aree di Head.In altre parole, una malattia che colpisce un organo interno può avere conseguenze sulla zona riflessa che ad esso corrisponde. Un esempio abbastanza noto è quello dell'infarto, che causa dolori nella parte interna del braccio sinistro.
NEURONE
Il neurone è una cellula specializzata nella conduzione di impulsi elettrici, possiamo (sempre relativamente) affermare, che è caratterizzata da una morfologia che riflette bene la sua funzione: possiede un corpo cellulare o soma, da cui originano numerosi prolungamenti citoplasmatici di forma ramificata, i dendriti e un unico grosso filamento molto più esteso degli altri, l'assone.Questa asimmetria cellulare, riflette la sua polarizzazione e la direzione della conduzione degli impulsi, che originano nei dendriti e vengono trasmessi fino alla terminazione assonale.

L'albero dendritico ha appunto il compito di contattare più neuroni possibili in maniera da ricevere il numero maggiore di stimoli, per questo motivo, ogni dendrite possiede delle spine dendritiche, e cioè ulteriori prolungamenti in maniera da aumentare la superficie di ricezione.A causa della sua peculiare funzione, il corpo cellulare del neurone possiede un imponente apparato di Golgi (visibile mediante impregnazione argentica), che si estende tutto intorno al nucleo, per la produzione di vescicole che immagazzinano i neurotrasmettitori, numerosi mitocondri per fornire energia alle pompe ioniche per la conduzione dell'impulso, e un citoscheletro caratteristico per il mantenimento della morfologia della cellula. I neuroni abbondano anche in lisosomi, perossisomi, per l'eliminazione di proteine mal ripiegate, e reticolo endoplasmico liscio e ruvido (evidenziabile tramite colorazione tigroide di Nissl).L'assone origina dal corpo nel monticello assonale, e al termine della sua estensione si sfrangia nelle terminazioni assoniche che vanno a contattare altri neuroni o organi.
FISIOLOGIA NEURONALE
Probabilmente a causa dell'elevata specializzazione e della morfologia complessa del neurone, questa cellula è incapace di dividersi, ad eccezione dei neuroni olfattivi che invece si dividono.Quindi, poiché tale cellula non può essere rimpiazzata, deve essere capace di durare per tutta la vita. Per avere una così lunga durata, il neurone deve essere capace di degradare in maniera molto efficiente sia le proteine extracellulari, che quelle intracellulari mal ripiegate.Le proteine extra-cellulari vengono internalizzate in vescicole endocitotiche, che poi si fondono con un lisosoma primario che contiene enzimi litici. Tale vescicola matura in un fagosoma o lisosoma secondario. I residui degradati vengono riciclati dalla cellula, tutti i materiali non degradabili vengono invece immagazzinati in lisosomi terziari (visibili come granuli di lipofucsina).Le proteine intra-cellulari mal ripiegate, o destinate alla degradazione vengono marcate con una coda di poli-ubiquitina. Il complesso che lega l'ubiquitina è composto da 3 enzimi: E1 attiva l'ubiquitina, E2 la coniuga ed E3 la lega. La proteina marcata viene così riconosciuta dal proteasoma, che è un complesso enzimatico di numerosi enzimi litici, capace di degradare la proteina. Talvolta alcune proteine assumono una conformazione tale da essere riconosciute dal proteasoma, ma non venire degradate, pertanto restano sottoforma di aggregati proteici.Quindi entrambi i sistemi non sono efficaci al 100% e l'accumulo di sostanze non degradate nel neurone, a lungo andare, ne causa la morte. Alterazioni in questo meccanismo sono spesso alla base delle malattie neurodegenerative.
Il citoscheletro è particolarmente sviluppato nei neuroni e le proteine del citoscheletro costituiscono un terzo del totale. Esso è importante per il mantenimento della forma, per la compartimentazione intracellulare e per il trasporto assonale. Il citoscheletro submembranoso è costituito essenzialmente da fodrina che funziona come proteina d’ancoraggio per i canali ionici ed è uno dei principali costituenti delle densità subsinaptiche. E’ anche concentrata nelle terminazioni sinaptiche ed in corrispondenza dei nodi di Ranvier. Nei neuroni il citoscheletro si estende anche nella profondità del corpo cellulare ed è costituito da tre tipi di neurofibrille: microtubuli (o neurotubuli), neurofilamenti (o filamenti intermedi) e microfilamenti.
I microtubuli sono composti da 13 filamenti di subunità alfa e beta, più altre proteine come MAP e tau, disposte a formare un cilindro cavo. Sono organizzate longitudinalmente negli assoni ma disordinatamente

nel pericarion, servono al movimento delle vescicole all'interno del neurone. I neurofilamenti sono costituiti da subunità a bastoncello, non si trovano nei dendriti ma sono abbondanti nei grossi assoni. I microfilamenti sono nient'altro che i filamenti di actina, sono piuttosto dispersi, ma aiutano a mantenere l'organizzazione immediatamente sottostante la membrana plasmatica, e la morfologia “fine” della membrana dei terminali presinaptici e delle spine dendritiche. L'actina è essenziale nei di coni di accrescimento cioè nella formazione di nuovi prolungamenti citoplasmatici (sprouting).La proteina Tau favorisce la polimerizzazione dei microtubuli, è abbondante nell'assone ma assente nei dendriti. Mutazioni in questa proteina causano demenza fronto-temporale. Si ipotizza un suo coinvolgimento anche nella malattia di Alzheimer, in quanto i paired helical filaments, che costituiscono i neurofibrillary tangles tipici della malattia, sono formati da proteina Tau.
TRASPORTO ASSONALE
Il monticello assonale contiene abbondante sostanza di Nissl e quindi ribosomi, mitocondri, neurofilamenti, è la sede di orgine del potenziale d'azione, perché è la zona in cui si sommano tutti gli impulsi provenienti dal neurone e infatti quelli innescati in zone spazialmente più vicine al monticello assonale hanno più peso nella generazione del potenziale d'azione.L'assone vero e proprio contiene mitocondri, RE liscio, e soprattutto microtubuli (talora cross-linked) e neurofilamenti per il trasporto di vescicole.La terminazione assonale contiene le vescicole presinaptiche.Il trasporto assonale può procedere in senso anterogrado o retrogrado.Il trasporto anterogrado va dal soma verso la terminazione assonale, e può essere rapido (400mm/die) per il trasporto di microorganelli con proteine, vescicole e mitocondri, o lento (1-4mm/die) per il trasporto di proteine enzimatiche e strutturali del citoscheletro nel caso venga reciso e coincide infatti col tempo richiesto alla ricostituzione dell'assone.Il trasporto retrogrado (200-300mm/die) porta sotanze di riciclo e fattori di crescita verso il corpo cellulare dalla sinapsi ed è anche sfruttato da virus, e proteine prioniche.Le chinesine sono alla base di quello anterogrado, mentre le dineine di quello retrogrado. Le proteine responsabili del trasporto anterogrado lento non sono conosciute, ma si ipotizza siano dineine, tubulina, clatrina, calmodulina.Si pensa che alterazioni del trasporto assonale siano causa di neuropatie dying-back in cui il neurone degenera a partire dalla periferia dell'assone, o sia coinvolta nella paresi spastica in quanto i neuroni colpiti sono quelli con assone più lungo.
VESCICOLE SINAPTICHE
La trasmissione dell'impulso nervoso da un neurone all'altro avviene tramite la sinapsi, che costituisce una interazione specializzata tra 2 cellule. Questa può essere elettrica o chimica.Nella sinapsi elettrica gli ioni passano direttamente da un neurone all'altro tramite gap junctions tra le 2 cellule, è molto veloce, ma non permette una modulazione/regolazione del passaggio.Nella sinapsi chimica, il segnale passa da un neurone all'altro sottoforma di intermedio chimico, rappresentato da piccole molecole o peptidi.

Questi vengono immagazzinati in vescicole, che sono tanto più grandi quanto maggiore è il loro contenuto in neurotrasmettitore.La fusione delle vescicole con la membrana e il conseguente rilascio di neurotrasmettitore nella sinapsi è intimamente connesso allo spostamento ionico della depolarizzazione. Infatti la fusione è innescata dalla presenza di Ca, che entra con la depolarizzazione. Lo ione calcio attiva le PKA e la CAM chinasi 2, la sinapsina 1 viene fosforilata, questa riduce la sua affinità per l'actina, e le vescicole così migrano verso la membrana, e viene innescata l'esocitosi.Dopo il rilascio del NT nella sinapsi questo o viene degradato o ricaptato.Lo stimolo del neurotrasmettitore può essere eccitatorio o inibitorio.Se eccitatorio il legame del NT col recettore causa un aumento di permeabilità al sodio e potassio causando depolarizzazione, mentre se inibitorio si ha un aumento della permeabilità al cloro causando iperpolarizzazione.
RECETTORI
1° gruppoRecettori composti da subunità multiple che costituiscono canali ionici i quali per azione del neurotrasmettitore vanno incontro a modifiche conformazionali che ne facilitano l’apertura. Azione molto rapida2° gruppoRecettori che hanno un dominio con attività di guanil-ciclasi e che generano cGMP quando attivati dal neurotrasmettitore .3° gruppoRecettori che hanno intrinseca attività chinasica e che si autofosforilano quando attivati dal neurotrasmettitore dando luogo successivamente ad una catena di reazioni metaboliche. 4° gruppoRecettori la cui azione è mediata da una G protein.
GLIA
La glia è costituita da tutte le cellule non neuronali del sistema nervoso.Gli astrociti sono le principali cellule della glia, fungono da sostegno, hanno un ruolo trofico e metabolico, inducono e mantengono le tight junctions delle cellule endoteliati a formare la barriera emato-encefalica, proliferano in seguito a lesioni.Gli oligodendrociti sono piccole cellule con modesti prolungamenti che svolgono la funzione di rivestimento isolante degli assoni dell'SNC, come le cellule di Schwann nel sistema nervoso periferico.La microglia, infine, consiste nel sistema di sorveglianza immune del SNC, infatti sono cellule a funzione fagocitaria e originano dal mesoderma e non dall'ectoderma.
POTENZIALE D'AZIONE

Esiste un particolare equilibrio di concentrazioni ioniche tra l'interno e l'esterno della cellula. Questo causa una differenza di potenziale, o potenziale di riposo, che solitamente è di 50-70 mV, questa è dovuta alla presenza di grossi anioni organici (proteine e amminoacidi), che non sono capaci di attraversare la membrana, buona permeabilità a K e Cl e non al Na, pompa Na/K, che trasporta Na all'esterno e K all'interno.Quindi alla fine il citosol risulta ricco di proteine (negative) e potassio, mentre al di fuori c'è molto Na e un lieve eccesso di Cl.Rispetto al potenziale di riposo, possiamo calcolare il potenziale di equilibrio di ogni ione che è +50 per il Na, -70 per Cl e -90 per K, quindi il sodio è molto spinto a spostarsi attraverso la membrana a differenza del cloro, che è praticamente immobile.Il potenziale d'azione consiste in una repentina inversione del potenziale di membrana, a causa dell'apertura di canali ionici che lasciano passare liberamente gli ioni positivi dall'esterno verso l'interno della membrana.Tali canali possono essere aperti da stimoli chimico/metabolici o anche stimoli elettrici, per cui un'apertura localizzata dei canali, comporta un'inversione del potenziale altrettanto localizzata, ma la diffusione degli ioni immediatamente sotto la membrana, causa l'apertura di altri canali nei dintorni, e un'ulteriore entrata di ioni, a catena. In questo modo la depolarizzazione della membrana cammina lungo la membrana, in una ondata che una volta innescata, non può più fermarsi. Poiché poi la parte di membrana appena depolarizzata non può farlo nuovamente, e siccome l'impulso inizia sempre nei dendriti, anche se il flusso di corrente tenta di diffondersi in tutte le direzioni, l'impulso può viaggiare solo verso la parte di membrana ancora non-depolarizzata e quindi sempre dai dendriti verso l'assone.A causa del meccanismo intrinseco che sfrutta, il potenziale d'azione infatti costituisce un fenomeno del tipo tutto-o-nulla, quindi esiste una soglia di potenziale, oltre la quale la depolarizzazione locale è capace di causare un'entrata di ioni tale da poter innescare l'ondata.Quando gli assoni sono mielinizzati la corrente non può propagarsi dove c’è il rivestimento di mielina e salta al successivo nodo di Ranvier (conduzione saltatoria) che non è rivestito, facendo progredire l'impulso con una velocità molto maggiore.
Dal punto di vista dei movimenti ionici quando l’impulso raggiunge un certo valore soglia (depolarizzazione di 20-30mV) i canali del Na+ si aprono per un periodo di circa 0.7 ms e poi si chiudono (influsso di Na+)I canali per il K+ di tipo ritardato si aprono più lentamente e rimangono aperti per tutto il tempo in cui la cellula è depolarizzata (efflusso di K+)Altri canali per il K+ di tipo rapido entrano in funzione per piccole depolarizzazioni della membrana.Nel periodo refrattario si ha attivazione dei canali
del K+ ed inattivazione dei canali del Na+, ciò causa iperpolarizzazione ed ineccitabilità del neurone. La pompa Na/K ATPasi ripristina il potenziale di riposo agendo in senso contrario a come gli ioni Na e K si sono spostati.

DEMENZA
Compromissione acquisita e persistente di multiple funzioni cognitive, di entità tale da interferire con le attività della vita quotidiana, in assenza di disturbi della coscienza. Le sindromi demenziali sono frequenti e per lo più croniche, irreversibili e progressive.Acquisita → perdita di funzionePersistente → duratura nel tempo a differenza di amnesia globale transitoriaMultiple funzioni cognitive → la tendenza a scordare le cose con la vecchiaia non è demenza (benign forgetfulness)Frequente → in vita adulta e in anziani, soprattutto in occidenteNeurodegenerativa → progressiva sofferenza del neurone fino a morte dello stesso
Le sindromi demenziali più diffuse sono: Malattia di Alzheimer (lobo temporale = memoria, più del 50% dei casi) D. fronto temporale (funzioni cognitive, capacità di eseguire, progettare = fronto; memoria =
temporale) D. con corpi di Lewy (tipici dell'Alzheimer ma non sono Alzheimer) D. vascolare (causata da arteriosclerosi)
MALATTIA DI ALZHEIMER
Malattia progressiva, a decorso fatale, caratterizzata da deficit della memoria recente, del linguaggio, delle funzioni visuospaziali, disturbi psichiatrici e comportamentali. Nelle fasi iniziali l’esame neurologico è negativo.
Fatale → l'allettamento in sé porta alla morte in ~10 anniMemoria recente → si intende ultime 24h, ultimi gg, il passato lo ricorderà benissimoLinguaggio → anomia, sfuggono le parole, uso di poche parole, di linguaggio poveroVisuospaziali → difficoltà ad orientarsi, fino a non riconoscere più le stanze di casaComportamento → inversione ritmo sonno/veglia, allucinazioni, delirio
E' una malattia progressiva divisa in 3 fasi: la fase iniziale dura 2-4 anni e mostra solo sintomi lievi riguardanti la memoria, segue poi una fase intermedia di altri 2-10 anni in cui i sintomi aumentano e peggiorano fino ad arrivare a una fase avanzata di 1-3 anni in cui il paziente è allettato, alimentato artificialmente, ha un pannolone, è completamente dipendente.
Prevalenza 4% dopo i 60 anni; 30-40% dopo gli 85 anni; >85 raddoppia ogni 5 anni.Incidenza (n° di nuovi casi / anno) // Prevalenza (casi totali / popolazione)L'Alzheimer è la forma di demenza più comune, la malattia neurodegenerativa più frequente.Le donne sono più colpite, il loro rischio aumenta esponenzialmente con l'età.Tra i fattori di rischio individuati ci sono genotipo e4 dell'APO-E (per la forma ad esordio tardivo), bassa scolarità, storia familiare, malattia coronarica, traumi cranici.

Esistono fattori genetici accertati per alcune forme.La forma ad esordio precoce (<65 anni) ha trasmissione autosomica dominante e costituisce il 5-10% dei casi di Alzheimer. Tra i geni probabilmente responsabili ci sono la presenilina 1 e 2 (coinvolti nel catabolismo dell'amiloide) e APP (precursore dell'amiloide che si accumula nei pazienti). Per quella ad esordio tardivo c'è una certa clusterizzazione familiare ma nessuna chiara ereditarietà.Interessante notare come le persone affette da sindrome di Down, dopo i 30 anni sviluppano Alzheimer nel 90% dei casi, e il fatto che il gene APP si trova sul cromosoma 21.
La diagnosi si fa tramite RMI e PET, la risonanza mostra atrofia corticale e sotto-corticale (non sempre segno di alzheimer) e atrofia dell'ippocampo, la PET invece ipometabolismo dei lobi parietali.
MALATTIA DI ALZHEIMER - PATOGENESI
La malattia è causata da atrofia cerebellare e perdita neuronale. Si riscontrano placche neuritiche costituite da accumuli di beta-amiloide nel neuropilo; alterazioni neurofibrillari a causa di accumulazione intracitoplasmatica di proteine Tau abnormemente fosforilate. L'accumulo di amiloide è il meccanismo più indagato e approfondito e sembra essere causato dal core di beta-sheet che innesca l'aggregazione e in seguito la precipitazione degli aggregati insolubili.
Mutazioni del precursore dell’amiloide causano AD ad esordio precoce e tutte le mutazioni di APP associate ad AD causano aumentata produzione di amiloide (nella S. di Down c'è triplicazione del gene).La beta amiloide è tossica in vitro causando morte cellulare e topi transgenici con aumentata espressione di beta amiloide presentano accumuli, placche neuritiche e deficit di memoria ed apprendimentoIl genotipo ε4 del gene ApoE è fattore di rischio per AD e provoca aumentata deposizione di amiloide in quanto sembra agire come uno chaperone del frammento alfabeta (vedi in seguito).La produzione di anticorpi anti amiloide migliora topi transgenici e pazienti con AD.L’accumulo di beta amiloide nelle placche neuritiche è quindi l’evento primario: la formazione di grovigli neurofibrillari, ossidazione, eccitotossicità glutammatergica, infiammazione, e attivazione della apoptosi sono probabilmente eventi secondari dell'accumulo.
APP è la proteina precursore dell'amiloide, è di 700 Aa e ha 2 domini dentro e fuori il Golgi.Questa proteina può essere clivata da 3 enzimi: alfa, beta e gamma secretasi. L'alfa secretasi è l'unica a fare un clivaggio normale, che non porta problemi. Beta e gamma secretasi creano nella maggior parte dei casi un frammento di 40 Aa che aggregano lentamente, e con una certa frequenza creano frammenti di 42 Aa che aggregano più velocemente. In condizioni fisiologiche i frammenti non aggregano e restano solubili, ma una risposta infiammatoria o altri fattori che alterno omeostasi ionica e stress ossidativo possono favorirne la deposizione. Per una normale solubilità dei frammenti deve esserci un rapporto alterato di alfa-beta42/alfa-beta40.Mutazioni del gene APP coprono il 10-15% dei casi di AD ad esordio precoce, mentre la presenilina 1 il 20-70% dei casi. La presenilina 1 è la componente essenziale per la catalisi della gamma-secretasi. Agisce probabilmente con effetto dominante negativo. Sono state ritrovate sia mutazioni loss of function con abbassamento dell'attività della gamma-secretasi, sia gain of function con aumento del rapporto alfa-beta42/alfa-beta40.

I grovigli neurofibrillari (neurofibrillary tangles) sono un altro segno distintivo della malattia, abbiamo detto che sono formati da aggregati di proteina tau abnormemente fosforilati. Tale anomalia nella fosforilazione causa il distacco di Tau dai microtubuli e la sua aggregazione in filamente elicoidali appaiati (paired helical filaments) e precipitazione.La proteina Tau è probabilmente coinvolta anche nella demenza frontotemporale, che somiglia all'AD per certi versi ma esordisce con anomalie del comportamento e non della memoria.
VIA PIRAMIDALE
Il sistema piramidale (o via piramidale) è un sistema di vie nervose che provvedono ai movimenti volontari dei muscoli, permettendo la pianificazione del movimento attraverso un circuito neuronale costituito da due neuroni. Il sistema è costituito da fibre motrici che originano dalla parte posteriore dei lobi frontali della corteccia cerebrale e terminano nel midollo allungato, nelle corna anteriori del midollo spinale; tali fibre a livello del bulbo si incrociano parzialmente. E' una via monosinaptica in quanto la cellula piramidale della corteccia contatta direttamente il neurone nel midollo che stimola il muscolo e questo garantisce velocità nell'esecuzione dei comandi, esistono poi altre vie polisinaptiche che sono deputate al controllo del movimento.L'area del cervello deputata al movimento volontario è la circonvoluzione frontale ascendente o Area 4, organizzata somatotopicamente (ovvero partendo dall'apice della circonvoluzione e procedendo verso il basso troviamo i neuroni deputati al movimento dei muscoli del piede, arto inferiore, tronco arto superiore, mano, collo e testa).

PARALISI CENTRALE
La paralisi centrale è causata dall'interruzione della via piramidale. Se viene interrotta a livello del primo neurone di moto (cellula piramidale e suo assone) la paralisi è di tipo spastico (i muscoli sono tonici), e può essere tetraplegica o paraplegica a seconda di dove l'interruzione avviene (a monte o a valle delle vertebre da cui partono i nervi che controllano gli arti); se l'interruzione è nell'area della corteccia A4, la paralisi è flaccida.Tetraplegia: paralisi dei quattro artiEmiplegia: paralisi di una metà del corpoDiplegia: paralisi degli arti superioriParaplegia: paralisi degli arti inferioriMonoplegia: paralisi di un solo arto
SISTEMA EXTRAPIRAMIDALE
l sistema extrapiramidale è un insieme di vie e di centri nervosi che agiscono direttamente o indirettamente sulla corretta azione motoria, controllando le reazioni istintive orientate e adattandole al movimento volontario, coordinato dal sistema piramidale.Le connessioni del sistema extrapiramidale sono molto complesse: gli impulsi periferici, giunti al talamo, sono smistati al corpo striato, vero complesso organizzatore degli impulsi, trasmessi poi alle formazioni extrapiramidali sottostanti. Grazie ai rapporti con la corteccia, queste strutture possono regolare la motilità piramidale, di origine corticale. Il sistema extrapiramidale è quindi un sistema a più sinapsi, che influenza in ultima istanza i motoneuroni spinali, regolando il tono muscolare e la motilità. In sostanza la corteccia con la via piramidale indica i movimenti, questo crea sensazioni relative al movimento che vengono inviate al talamo, il talamo trasferisce questi impulsi ai nuclei della base che fanno tutte le correzioni necessarie per controllare al meglio il movimento, e inibiscono la corteccia fino ad ottenere il movimento corretto. Queste vie utilizzano tutte connessioni glutammatergiche ad eccezione di una GABAergica.La substantia nigra (che è una sotto-struttura del talamo) inibisce a sua volta i nuclei della base per evitare una azione inibitoria sulla corteccia troppo spinta e facilitare il movimento. Questa via nigrostriatale utilizza come neurotrasmettitore principale la dopamina.
Le sindromi che colpiscono il sistema extrapiramidale possono essere:ipercinetiche distonia, corea, ballismo, ticipocinetiche parkinsonismo
COREA DI HUNTINGTON
Il morbo di Huntington è una malattia degenerativa del sistema extrapiramidale che rientra nel capitolo delle sindromi ipercinetiche. Tale patologia si presenta la corea come sua caratteristica principale e cioè una serie di movimenti bruschi ed involontari, migranti, afinalistici ed aritmici. E' causata dal danno a neuroni striatali, soprattutto del caudato, ma successivamente anche di quelli della corteccia, infatti porta

anche demenza (di tipo sottocorticale). Altri disturbi psichici sono depressione, disturbi del comportamento, allucinazioni e paranoia. Negli stadi avanzati il paziente diventa bradicinetico, rigido e distonico. Il paziente muore 15-20 anni dopo l’insorgenza dei primi sintomi. L’età d’esordio si colloca attorno ai 40-50 anni.
PATOGENESI
È una malattia rara, con prevalenza di circa 3 - 7 casi per 100.000 abitanti con discendenza europea occidentale e 1 per 1.000.000 con discendenza asiatica, trasmessa con modalità autosomica dominante a penetranza completa. La malattia è causata dalla degenerazione dei neuroni striatali GABAergici, con morte prima dei neuroni D2 che proiettano al globo pallido esterno (via indiretta), e poi del neuroni D1 che proiettano a globo pallido interno / substantia nigra (via diretta).Il ruolo di questi neuroni GABAergici era di inibire gli stimoli al movimento provenienti dalla corteccia, per cui la loro assenza causa una sindrome ipercinetica.
Tutti i pazienti affetti dal morbo di Huntington presentano una mutazione del gene per l'Htt, situato come detto sul braccio corto del cromosoma 4. Il gene normale presenta una sequenza trinucleotidica ripetitiva CAG ripetuta da 11 a oltre 34 volte.
Il numero delle ripetizioni negli affetti è aumentato e la malattia è tanto più precoce quanto maggiore è il numero delle ripetizioni. Esiste una soglia più o meno precisa nel numero di triplette oltre la quale gli individui manifestano la malattia. 10-26 ripetizioni sono considerate wt.27-35 intermedi pre-mutati36-41 penetranza ridotta>42 penetranza completa
La sequenza ripetuta è instabile, per cui durante la replicazione del DNA può amplificarsi ulteriormente o contrarsi.Se questa l'amplificazione avviene durante la spermatogenesi il nascituro con l'allele mutato svilupperà prima la malattia. Quindi l'instabilità germinale è all'origine del fenomeno dell'anticipazione della malattia, che si verifica se trasmessa da parte paterna.
L'huntingtina è una proteina di 3140 Aa e 67 esoni. E' espressa in tessuti sia neurali che non neurali, ma le espansioni di triplette causano sofferenza soprattutto nei neuroni.E' una proteina citoplasmatica e non ha omologia con proteine precedentemente caratterizzate. L'unico indizio è la presenza di un dominio HEAT per l'interazione proteina-proteina a valle delle ripetizioni.Infatti l'Htt sembra interagire con proteine coinvolte in endocitosi mediata da clatrina, apoptosi, trasporto vescicolare, signaling, morfogenesi.La Htt wild-type è anche anti-apoptotica e favorisce la produzione del BDNF.
In un lavoro del 2004 pubblicato su Cell, gli studi condotti hanno mostrato un importante coinvolgimento della Htt nel meccanismo di trasporto vesciloare assonico. Essa fungerebbe da acceleratore del complesso della Dineina, e la sua

mutazione va a limitare se non annullare questo effetto propulsivo, sebbene non sia stato possibile comprendere a fondo come l'elevato numero di Glutammine incida su questa deficienza. Ciò che invece sembra certo è il composto proteico maggiormente incisivo sulla neurodegenrazione, ossia il BDNF (Brain Derived Neuronic Factor). Questo, prodotto dalla corteccia cerebrale, è un composto che mantiene in vita i neuroni evitandone l'apoptosi. Il suo trasferimento dalla corteccia alla zona dello striatum per esempio, non può che avvenire tramite il trasporto assonico, perciò se intercorre una mutazione dell'Htt, tale fattore non arriva a destinazione e causa in breve tempo sia accumulo di materiale proteico che conseguente morte cellulare.
Le ripetizioni CAG sono in frame e corrispondono a ripetizioni di poliglutammina, comunque non alterano i processi di trascrizione o traduzione.Il KO del gene IT 15 del topo omologo dell'Htt non causa malattia, mentre il doppio KO non sopravvive durante lo sviluppo. L'espressione di forme mutate di IT 15 causano degenerazione neuronale.Le ripetizioni di poliglutammina sembrano essere tossice di per sé, in quanto anche se portate da un'altra proteina causano sofferenza neuronale.Le ripetizioni di poliglutammina possono essere substrato delle transglutaminasi che potrebbero formare residui non degradabili nei proteasomi o aggregati proteici. Inoltre i tratti di PoliG si comportano come un dominio zipper, aggregandosi tra loro.Tutto ciò è infatti confermato dalla presenza di aggregati proteici nel citoplasma, evidenziabili con anticorpi anti ubiquitina e anti poliQ, anche se non c'è un grande overlap tra neuroni con inclusioni e neuroni degeneranti.I tratti di poliG (a differenza delle ripetizioni nucleotidiche) possono interferire nella trascrizione e in altri processi cellulari in quanto possono sequestrare proteine e fattori di trascrizione, possono interferire col turn-over cellulare, e possono ostacolare il trafficking assonale. Inoltre l'huntingtina mutata lega quella wild-type probabilmente ostacolandone la funzione tramite un effetto dominante negativo.
MALATTIA DI PARKINSON
Il morbo di Parkinson è una malattia degenerativa del sistema extrapiramidale che colpisce i neuroni della substantia nigra, causando una sindrome ipocinetica. L'eziologia è sconosciuta: si crede a una collaborazione tra fattori ambientali e genetici.Incidenza 10-20/100,000prevalenza 100-150/100,000 dopo i 50 1/100Età di esordio 60-65 anniNon ci sono sessi, etnie, luoghi o classi sociali più colpite, anche se i dati sono controversi; esiste una certa incidenza familiare.
La substantia nigra è composta da soli 400 mila neuroni. Normalmente si perde l'1% di questi all'anno, mentre chi è affetto da Parkinson ne perde il 6-7%. La sintomatologia si sviluppa solo quando si sono persi il 60% dei neuroni.Quindi il principale fattore di rischio è l'invecchiamento, o l'allungamento della vita media.La deplezione è selettiva per i neuroni dopaminergici, e si è visto che anche se la substantia nigra è la zona del cervello che è evidentemente più colpita, c'è perdita neuronale anche nei nuclei caudato, putamen e pallido.La diagnosi può essere certa solo per via anatomopatologica, in cui si riscontra questa perdita neuronale con annessa depigmentazione della substantia nigra e la presenza di corpi di lewy.

Il corpi di lewy sono il gold standard per la diagnosi di parkinson anche se esiste un tipo di parkinson genetico che non li presenta, e un tipo di demenza diverso dal parkinson che li ha. I corpi di lewy sono inclusioni citoplasmatiche con centro eosinofilo ed alone più chiaro.La tomoscintigrafia con farmaci DAT permette di evidenziare la perdita selettiva di neuroni dopaminergici nel cervello, per cui ci aiuta nella diagnosi di parkinsonismo (e non della malattia di parkinson).
Nei gangli della base lo striato rappresenta la stazione di ingresso del segnale (dalla corteccia motoria), GP interno e SN pars reticularis rappresentano le stazioni di uscita del segnale (a talamo e corteccia motoria)La funzione dei gangli della base è di controllo sull’attività di talamo e corteccia motoriaLa degenerazione della SN pars compacta provoca aumentato output al talamo dei nuclei della base e conseguente aumento dell’inibizione talamo-corticaleIl tremore è dovuto ad un’attività ritmica di cellule talamo-corticali.In sintesi la substantia nigra esercita un ruolo inibitorio sui gangli della base, per cui la degenerazione di questa parte comporta un aumento dell'attività dei nuclei e poiché l'attività di questi è ritmica, si nota il tremore ritmico.
La triade dei sintomi cardine della malattia è costituita da: tremore, rigidità ed acinesia, con variabile gravità. L'acinesia (o ipocinesia) è la complessiva riduzione della motilità volontaria ed involontaria, e di regola si associa a lentezza dei movimenti (bradicinesia). Scompaiono i movimenti automatici come l'ammiccamento, la deglutizione, gesticolazione, mimica facciale.Il tremore è tipicamente "a riposo" scompare durante i movimenti volontari, è assente durante il sonno. Nelle fasi iniziali è localizzato soprattutto alle mani e alle dita.La rigidità è un segno caratteristico e costante e a volte costituisce per lungo tempo il solo segno di malattia. Si apprezza aumentata resistenza al movimento passivo.Per eseguire movimenti il paziente necessita di molta concentrazione e tipicamente la gestualità e la mimica sono molto scarse. La mimica facciale è scarsa, l'espressione impassibile. La deambulazione è tipicamente a piccoli passi, strisciati.Nelle fasi ultime della malattia ci sono anche disturbi psichici e declino cognitivo.
TERAPIA
La terapia consiste nel somminsitrare levo-dopa di cui il paziente è ovviamente deficitario, tuttavia poiché la terapia abbia effetto è necessaria comunque la presenza di neuroni dopaminergici nella substantia nigra. Poiché la dopammina non attraversa la BEE si somministra levo-dopa, ma siccome questa può essere metabolizzata anche a livello periferico, per evitare che questo avvenga si somministrano anche inibitori delle decarbossilasi (che non passano la BEE e quindi agiscono solo a livello periferico). Altri tipi di terapie:Somministrazione di agonisti della dopammina, che non hanno bisogno della presenza di neuroni dopaminergici per funzionare e ritardano la sindrome da assunzione cronica, ma hanno maggiori effetti collaterali.Anticoligergici o amantidina: riducono l'iper-stimolazione da parte dei nuclei e aiutano nei sintomi psichiatrici.Inibitori COMT: prolungano l'azione della levo-dopa

Nessuna terapia è definitiva in quanto nessuna di queste ferma la perdita neuronale. Questo significa che col tempo l'efficacia farmacologica diminuisce sempre più e aumentano gli effetti collaterali.Esistono anche terapia di tipo chirurgico in quanto si è visto che lesionando i nuclei della base migliorava la sintomatologia perché appunto viene interrotto il circuito iperstimolante.Autotrapianto surrenalico: elevata mortalità, scarsa efficaciaTrapianto di mesencefalo fetale: efficace, rare le complicanze gravi, problemi eticiStimolazione dei nuclei profondi (VIM, Gpi, NST) (deep brain stimulation): efficace, reversibile, si tratta di stimolazioni con elettrodi.
FATTORI DI RISCHIO GENETICI ED AMBIENTALI
FATTORI AMBIENTALIEsistono altre forme di Parkinsonismo che non sono la malattia di Parkinson la mimano e possono aiutarci a comprendere il meccanismo della malattia.Il parkinsonismo da manganese è uno di questi e si manifesta in seguito a intossicazione da manganese, ad esempio in lavoratori esposti in miniere, saldatori, batterie, insetticidi. Alla RMI si riscontra una iperintensità dei nuclei pallidi.Il meccanismo patogenetico ipotizzato è quello di una incrementata auto-ossidazione della levo-dopa.
Durante l'epidemia spagnola ci fu un ulteriore complicazione dovuta probabilmente ad un ulteriore virus che causava encefalite letargica (o sindrome di Von Economo), come esplose con la spagnola così sparì con essa. Era letale, ma chi sopravvisse ad essa rimaneva in stato letargico (dormiente) per tutta la vita. Accanto a questa letargia c'erano altri sintomi di tipo parkinson, e questa similitudine spinse un medico a trattare questi pazienti con levo-dopa. Essi si risvegliarono a distanza di 30 anni, tuttavia a causa del danno permanente (ai centri del sonno) dopo un certo tempo ritornavano letargici , e la levo-dopa dopo un po' causava sindrome da somministrazione cronica.
Un tipo di parkinsonismo molto interessante è quello causato da MPTP.L'MPTP è una sostanza chimica con cui si può venire realmente a contatto in quanto è un contaminante dell'eroina sintetizzata in laboratori clandestini, ed esistono inoltre molecole MPTP-simili nell'ambiente. Questa sostanza causa una sindrome parkinsoniana, con assenza di Lewy bodies.L'MPTP causa morte selettiva dei neuroni dopaminergici, e la somministrazione di questa sotanza in animali crea modelli di parkinsonismo abbastanza fedeli.L'MPTP non è tossico di per sé, ma è capace di attraversare la BEE, ed entrare nelle cellule g liali. Qui diviene substrato dell'enzima MAO B e trasformato in MPP+. L'MPP+ viene riconosciuto dal DAT e trasportato selettivamente all'interno del neurone dopaminergico (in quanto è l'unico a possedere il DAT). Qui inibisce il complesso I della fosforilazione ossidativa, incrementando la produzione di radicali liberi, che porta a morte il neurone.
I fattori di rischio per il parkinson sono tali e tanti che è difficilissimo orientarsi fra essi e capire in che modo contribuiscano: agenti infettivi, trauma cranico, erbicidi, pesticidi, metalli, solventi, campi magnetici, alcune professioni... Il fumo sembra invece avere un effetto protettivo. L’invecchiamento è il principale fattore di rischio Agenti virali potrebbero essere causa di parkinsonismo

Il manganese può causare parkinsonismo atipico L’ambiente rurale potrebbe essere un fattore di rischio Gli idrocarburi potrebbero essere un fattore di rischio Il fumo potrebbe avere un effetto protettivo Tossine mitocondriali possono indurre parkinsonismo
FATTORI GENETICILo studio di fattori genetici ha permesso di individuare 13 loci PARK su 8 geni in totale.Però il parkinson è ritenuto a causa monogenica solo nel 5% dei casi, c'è clustering familiare nel 15%, mentre è considerato sporadico nel restante 80% dei casi.Comunque, alcuni di questi geni causano un parkinson tipico, altri un parkinson aggressivo, altri uno giovanile (povero Marty McFly).I geni PARK 3,5,8,13 causano un parkinson tipico a trasmissione autosomica dominante, ma penetranza incompleta. L'esordio è intorno ai 60, sono presenti corpi di Lewy e i pazienti hanno buona risposta alla levo-dopa.I geni PARK 1,4 causano un parkinson aggressivo, trasmissione autosomica dominante e penetranza completa. Esordio verso i 50, anche qui ci sono corpi di Lewy e risposta a levo-dopa, ma il decorso è più rapido e porta anche demenza.I geni PARK 2,6,7 causano un parkinson giovanile, la trasmissione è autosomica recessiva, esordio prima dei 50, progressione lenta, buona risposta a levo-dopa, ma presenta numerosi altri sintomi psichiatrici aggiuntivi.
Siccome questi geni hanno anche una penetranza molto alta, significa che il meccanismo di morte neuronale deve essere legato intrinsecamente al difetto genetico.I meccanismi principali sono disfunzione del sistema ubiquitina-proteasoma, e disfunzioni mitocondriali con stress ossidativo e apoptosi.Questi dati sembrano indicare che lo stress da radicali liberi e da accumulo di proteine che tendono all’aggregazione potrebbe spiegare la morte di neuroni di per sé vulnerabili a questi fattori anche nell’anziano normale.
SISTEMA UBIQUITINA-PROTEASOMARiguardo le disfunzioni del sistema ubiquitina-proteasoma, la proteina alfa-sinucleina è di fondamentale importanza. Infatti i Lewy-bodies e le inclusioni citoplasmatiche gliali sono costituite da alfa-sinucleina. Il locus PARK-1 comprende il gene codificante per l'alfa-sinucleina, mentre PARK-4 è correlato alla sua iper-produzione. L'alfa-sinucleina ha una struttura prevalentemente di alfa-eliche, ma fattori ambientali come stress ossidativo, interazioni con altre proteine possono farla cambiare in struttura a beta-sheet, causandone l'aggregazione nelle inclusioni. Le mutazioni nel gene PARK-1 (alfa-sinucleina) causano:
aumentata tendenza all’aggregazione e alla formazione di fibrille favoriscono la depolarizzazione mitocondriale favoriscono l’attivazione dell’apoptosi inibiscono l’attività del proteasoma e i meccanismi di autofagia
PARK 2 invece è una E3-ligasi.
STRESS OSSIDATIVO E APOPTOSI

PARK6 codifica per la proteina PINK-1, che è una chinasi mitocondriale della famiglia della calmodulina, protegge i neuroni dallo stress indotto dalla disfunzione mitocondriale e dalla apoptosi.PARK7 codifica per DJ-1 coinvolta in risposta a stress ossidativo e protezione stress cellulare, forse ha un ruolo nella trasmissione dopaminergica.Le mutazioni in questi 2 geni causano parkinson giovanile e sono di tipo loss-of-function e sono disponibili test genetici per le famiglie colpite.PARK 8 è una chinasi, mutazioni in questo gene ne aumentano l'attività. La frequenza di queste è elevata in alcune popolazioni e causa parkinson con penetranza del 30%.
SINDROMI CEREBELLARI
La funzione principale del cervelletto è quella di coordinare le uscite motorie, in base a sensazioni propriocettive, vista e cocleo-vestibolari (udito ed equilibrio): infatti, le lesioni cerebellari compromettono la coordinazione dei movimenti degli arti e degli occhi ma anche l'equilibrio. Mantiene tono, postura ed equilibrio.Apprende gli schemi motori: si ritiene che il cervelletto riceva dalla corteccia cerebrale una "copia" del comando motorio che un soggetto intende volontariamente eseguire, e che riceva dagli arti informazioni relative all'effettivo svolgimento dello schema motorio impartito dalla corteccia cerebrale. Qualora sussistano delle differenze tra il movimento programmato e quello effettivamente realizzato, il cervelletto è in grado di correggere, con un meccanismo di feedback negativo, il movimento durante il suo realizzarsi. Per tale motivo si dice che il cervelletto è un "comparatore". Ricerche recenti hanno ipotizzato che tale compito di coordinazione del cervelletto sia generalizzabile anche alle funzioni cognitive. Un danno cerebellare quindi non impedisce l'utilizzo di una determinata funzione ma ne riduce l'efficienza.
Le sindromi cerebellari possono comprendere:Atassia della marcia e della stazione erettaDisartriaIpotoniaAsinergia e scomposizione del movimento (a scatti)Dismetria (mancanza di precisione nel movimento)Adiadococinesia (incapacità di effettuare con un ritmo rapido dei movimenti in direzioni opposte)Tremore intenzionaleFrenage (fenomeno del rimbalzo)Nistagmo (movimento oscillatorio occhi)Alterazioni movimenti oculariDifficoltà del linguaggio (in quanto necessita molta coordinazione)Tutti questi sintomi sono facilmente riconducibili ad alterazioni nel funzionamento del cervelletto. Le malattie cerebellari principali sono:Sclerosi multipla (o a placche)Degenerazione spino-cerebellareTumori fossa cranica posterioreemato/infartoAtassia di FriedreichSCA (spino cerebellar ataxia)

Esistono numerosi tipi di atassie genetiche. Alcune hanno causa metabolica, altre sono associate a difetti nel riparo del DNA, altre ancora sono degenerative collegate a folding e degradazione delle proteine, altre sono canalopatie.Le atassie a trasmissione autosomica recessiva hanno insorgenza prima dei 20 anni, ma non è una regola assoluta. E comunque anche le atassie a trasmissione autosomica dominante, X-linked e matrilineare hanno esordio prima dei 20 anni.
ATASSIA DI FRIEDREICH (friidraichh)L'atassia di Friedreich FRDA è una atassia genetica ad insorgenza infanto-giovanile, solitamente pubertà, è la più frequente tra quelle genetiche, prevalenza 1:50000 portatori 1:100. Colpisce anche il sistema piramidale e il secondo motoneurone. Nella sua patogenesi sembra implicata la deficienza di una proteina, la fratassina, che avrebbe il compito di smaltire i metaboliti di rifiuto dei processi energetici all'interno del mitocondrio: triplette GAA nel primo introne impediscono l'adeguato "srotolamento" del DNA che favorisce la sintesi della fratassina e che infatti è deficitaria. Maggiore è il numero di queste minore è l'esordio, l'espansione della tripletta è più frequente nella mdare. Anche mutazioni non-senso e missenso sono state descritte.Siccome il problema è all'interno del mitocondrio, può essere classificata come atassia mitocondriale.I neuroni del cervelletto sono soprattutto neuroni di Purkinje. L'etanolo è selettivamente tossico per questi neuroni, infatti quando ci si ubriaca si va incontro ad una atassia acuta: l'ubriaco ha difficoltà di equilibrio, parlato e coordinazione dei movimenti.
ATASSIA-TELEANGIECTASIAE' una atassia causata da mutazioni nel gene ATM (ataxia-teleangiectasia mutated) che è coinvolto nella riparazione del DNA.La perdita di neuroni di Purkinje nel cervelletto e in altri distretti neuronali è molto precoce, tant'è che l'atassia si instaura già entro i 3 anni. Quelli neuronali sono solo una parte dei sintomi perché la perdita di ATM comporta gravi squilibri nel controllo del ciclo cellulare e nella stabilità genomica.
ATAXIA OCULOMOTOR APRAXIAE' un'altra atassia collegata alla riparazione del DNA, stavolta a singolo filamento. Il sintomo principale è l'atassia oculo-motrice per cui il movimento dell'occhio non segue quello della testa.
ARSACSAutosomal recessive spastic ataxia charlevoix-saguenayIl gene SACS è quello responsabile, codifica per la sacsina, che è una chaperone.
SCA – SPINO CEREBELLAR ATAXIALa SCA non è una malattia, piuttosoto un gruppo di atassie tutte a trasmissione autosomica dominante caratterizzate da una patogenesi comune: la degenerazione selettiva del cervelletto e delle sue connessioni afferenti ed efferenti.Si manifestano in età adulta e sono tutte causate da tratti di poli-glutammina, ne sono state descritte 40 forme:La SCA-2 è la più frequente in Italia (50%)La SCA-3 assente in Italia è la più frequente in assoluto.

La SCA-6 agisce tramite effetto dominante-negativo, il gene codifica per un canale del Ca specifico delle cellule del Purkinje. Esistono modelli di SCA-1.Le triplette sono CAG sono tossiche di per sé come nella corea di Huntington, anche qui c'è instabilità mitotica e meiotica con anticipazione più probabile se a trasmettere l'allele è il padre. La grossa variabilità dell'espansione sembra poter spiegare la variabilità intra e interfamiliare.E' disponibile l'analisi genetica e prenatale . Siccome non c'è terapia il test predittivo si può effettuare solo con supporto psicologico se post-natale, e solo in caso i genitori vogliano abortire se pre-natale. In qualunque caso non si fornisce previsione sull'età di esordio.
ICTUS
L'ictus, o apoplessia, o stroke è l'improvvisa comparsa di segni e/o sintomi riferibili a deficit globale (coma) o focale delle funzioni cerebrali di durata >24 ore o ad esito infausto (altrimenti è un ictus transitorio) non attribuibile ad altra causa apparente se non a vasculopatia cerebrale.E' la terza causa di morte al mondo dopo cardiopatia ischemica e tumori. Nel 75% dei casi si verifica in persone >65 anni.Il recupero attualmente può essere discreto o buono, oppure può portare a grave disabilità (causa più comune di disabilità in adulti), oppure può essere letale.Esistono vari tipi di ictus:
Ictus ischemico 80% (infarto cerebrale) consiste nell'occlusione di unvaso per trombosi o embolia (fibrillazione atriale) o meno frequentemente da improvvisa e grave riduzione della pressione di perfusione del circolo ematico (per esempio in seguito a un infarto del miocardio, anche se il cervello è capace di regolare bene ipo e iperperfusione).
Ictus emorragico 15-20% dovuto a una emorragia intracerebrale non traumatica come nel caso di un aneurisma o angiopatia amiloidea.
Attacco ischemico transitorio (TIA) che dure <24h solitamente pochi minuti, probabilmente dovuto a un trombo che poi viene degradato mediante fibrinolisi.
Emorragia subaracnoidea 3%.
CIRCOLAZIONE CEREBRALE
Il cervello consuma il 28% del glucosio e il 20% dell'ossigeno giornaliero. Viene perfuso da 850 mL di sangue al minuto, cioè il 15% del totale di cui 700 dal circolo carotideo e 150 dal circolo vertebro-basilare.La circolazione cerebrale è divisa in un circolo anteriore e uno posteriore.Il circolo anteriore parte dall'arteria carotide comune (sono 2 destra e sinistra) che origina dall'arco aortico a sx, e dal tronco brachiocefalico a dx.L'arteria carotide comune si divide in arteria carotide interna ed esterna.L'arteria carotidea interna si ramifica nell'arteria cerebrale media e arterie cerebrali anteriori, quella esterna si occupa di altri organi.Dalle arterie cerebrali anteriori originano le comunicanti anteriori che collegano i circoli destro e sinistro.Il circolo posteriore parte dalle arterie vertebrali o succlavie.

Le succlavie si riuniscono nell'arteria tronco basilare.L'arteria tronco basilare si ramifica in arteria cerebrale posteriore, cerebellare antro-posteriore e superiore che costituiscono il circolo interno: irrorano le strutture del ponte e cervelletto.L'arteria cerebrale posteriore ha circoli anastomotici le comunicanti posteriori che la mettono in comunicazione con la carotide interna, che costituiscono il circolo esterno: irrorano il cervello.Il complesso di questi 7 vasi anastomotici che mettono in comunicazione circoli anteriore e posteriore, e sinistro e destro si chiama eptagono di willis.
Le cause di ischemia possono essere occlusioni di questi grossi vasi a causa di arteriosclerosi o emolie.Malattie dei piccoli vasi possono causare ictus lacunari.Altre cause sono le vasculiti e una riduzione sistemica del flusso.Gli effetti dell'ictus sono:
alterazione metabolica (30’’) disfunzione neuronale (1’) infarto irreversibile (5’) necrosi e rammolimento (giorni/settimane) gliosi (settimane/mesi)
La zona più colpita è chiamata nucleo ischemico e per quella non c'è nulla da fare, il danno è irreversibile. Esiste poi tutta la zona circostante, chiamata di penombra ischemica,
I fattori di rischio sono gli stessi dell'infarto coronarico:Ipertensione (danneggia parete arteriosa)
Fumo Diabete Obesità Ipercolesterolemia (x10) Cardiopatie (infarto recente) Familiarità
Fattori preventivi sono invece l'attività fisica e l'alcool
Una TAC al cranio basta per fare diagnosi di ictus. Si nota una zona densa bianca che rappresenta un versamento di globuli rossi, oppure una zona ipodensa, che rappresenta necrosi o edema. Talvolta si nota una zona ipodensa con al centro una densa che rappresenta la necrosi da infarto con sanguinamento.
Il trattamento è volto a salvare la zona di penombra ischemica, ma per farlo deve essere possibile intervenire entro 3 ore.Durante la fase acuta dell'infarto si mira a correggere i parametri ematici alterati (glucosio ed elettroliti), l'ipoperfusione e la concentrazione di ossigeno.Si prevengono ulteriori complicanze spingendo il paziente a una mobilizzazione precoce e al recupero delle sue attività.La riabilitazione passa per fisio e logoterapia.Per eliminare la causa dell'infarto si somministra aspirina e attivatore tissutale del plasminogeno per eliminare il trombo che occlude il vaso, nel caso di ictus ischemico.Si procede all'evacuazione chirurgica in caso di ictus emorragico, o somministrazione di fattore VIIa ricombinante.

In caso di TIA raramente si interviene chirugicamente in quanto si crea poi stenosi in >70% dei casi. Si preferisce somministrare aggreganti come aspirina, ticlopidina, clopidogel oppure anticoagulanti per i casi più gravi, in più si controllano fattori di rischio.
L'ischemia consiste essenzialmente in una ridotta perfusione cerebrale, che vuol dire meno glucosio e O2 a disposizione per il cervello.L'ischemia può essere focale nel caso di trombi, traumi ed emorragie, o globale in caso di infarto o ipotensione.Il core ischemico rappresenta la zona di massimo danno, priva di circolazione collaterale e perciò totalmente senza nutrimento. Qui il danno è marcato e le cellule muoiono per necrosi in seguito all'incapacità di mantenere l'omeostasi osmotica.La zona di penombra ischemica, invece, possiede una irrorazione residua, ha un danno limitato ed è possibile migliorarne il recupero con terapia neuroprotettiva. Qui i neuroni più sofferenti a causa di ROS, citochine infiammatorie, danno genotossico innescano il processo di apoptosi, che sappiamo è possibile bloccare se trattato in tempo.Esistono anche fenomeni a metà strada tra necrosi ed apoptosi indicati col nome di “necroptosi”.
Altre cause di suscettibilità possono essere particolari caratteristiche della circolazione cerebrale, elevata attività metabolica, limitate riserve energetiche, dipendenza dal metabolismo aerobico del glucosio.La corteccia, il cervelletto e quindi le cellule neuronali piramidali corticali e neuroni di purkinje sono quelli più suscettibili.Svariati meccanismi di danno cellulare (squilibri ionici, neurotossicità glutammatergica, stress ossidativo, attivazione di signalling intracellulare, rottura della BEE, fenomeni infiammatori), ma anche meccanismi di neuroprotezione si connettono e si influenzano reciprocamente durante l'ischemia. L'esito può essere la lisi cellulare aspecifica, la morte programmata o la soppravvivenza del neurone. La conoscenza parziale di questi eventi molecolari ha permesso di sviluppare molteplici strategie di intervento terapeutico neuroprotettivo in modelli animali, ma non è stato possibile trasferirle sull'uomo.Il deficit energetico causa un improvviso calo di ATP nella cellula. Siccome il 50% dell'energia del neurone è impiegata per la Na/K ATPasi, questa pompa è la prima a soffrirne. Il suo mancato funzionamento porta alla depolarizzazione della membrana, e apertura dei canali ionici specialmente del potassio.La bassa pressione di O2 causa lo shift verso metabolismo anaerobico con caduta del pH.Questi ultimi 2 eventi causano l'entrata di Ca nella cellula e efflusso di glutammato. Il Ca entra a causa della depolarizzazione e dell'attivazione del recettore NMDA del glutammato, esso attiva proteasi (calpaina danneggia citoscheletro), lipasi (PL-A2 → acido arachidonico), chinasi ca-calmodulina dipendenti, endonucleasi, produzione di ROS, liberazione di NO, fino alla morte cellulare.Il glutammato viene rilasciato in risposta all'influsso di calcio, a causa dell'alterata omeostasi non viene più recuperato né dalle cellule neuronali né da quelle gliali. Questo neurotrasmettitore libero ovviamente causa depolarizzazione dei neuroni circostanti. (spreading depression). L'iperattivazione del recettore NMDA causa un ulteriore aumento del Ca intracellulare.Se da un lato Ca e Glutammato causano numerosi danni, dall'altro attivano anche la cascata delle MAPK, che è di cruciale importanza per favorire la sopravvivenza neuronale: ERK infatti stimola neuroprotezione tramile CREB e BDNF, mentre JNK e p38 inibiscono l'apoptosi.
RADICALI LIBERIOltretutto quando il tessuto colpito viene riperfuso si crea ulteriore danno per produzione di radicali liberi

La ripresa del metabolismo mitocondriale, l'attività della NOS, della PL-A2, e l'ossidazione della xantina in ipoxantina (xantinossidasi), sono tutti sorgenti di ROS, che causano perossidazione lipica, ossidazione di proteine e DNA, attivazione MAPK attivazione metalloproteinasi di matrice.I radicali più importanti:Reactive Oxygen Species (ROS)
anione superossido (O2¯· ) radicale idrossilico (·OH) perossido di idrogeno (H2O2) perossidi lipidici
Reactive Nitrogen Species (RNS) ossido nitrico (NO·) perossinitrito (ONOO-)
NO· reagisce con O2¯· e forma ONOO- che provoca perossidazione dei lipidi e nitrosilazione delle proteine
Reazioni che incrementano la produzioen di radicali liberi:
Molti enzimi possono produrre ROS. L’anione superossido (O2¯· ) si converte in H2O2, che è più stabile, spontaneamente o tramite la superossido dismutasi (SOD). H2O2 è decomposta in H2O ed O2 dalla catalasi o dalla perossidasi. ·OH è generato tramite la reazione di Fenton
I neuroni sono più suscettibili ai ROS per una serie di motivi: elevato contenuto in lipidi, substrato per la perossidazione, elevato contenuto in Ferro, soprattutto in substantia nigra, che favorisce la reazione di Fenton, elevata attività metabolica mitocondriale, metabolismo della levo-dopa e bassi libelli di antiossidanti.Le difese dai ROS sono ovviamente SOD e catalasi che da un lato producono ROS, ma dall'altro, in ultima analisi, favoriscono la trasformazione di questi in H2O e O2. Glutatione e glutatione perossidasi, il cui ruolo è appunto quello di contrastare l'effetto dei ROS, e infine antiossidanti come Acido ascorbico, vit E e flavonoidi.
In caso di ischemia causata da danno microvascolare, sorge un ulteriore problema e cioè quello dell'infiammazione. Infatti un vaso danneggiato richiama sul posto piastrine e leucociti, stimola la produzione di molecole di adesione, citochine infiammatorie e l'attivazione di metalloproteinasi. Anche gli astrociti sono spinti a produrre citochine, ma al tempo stesso secernono fattori neuroprotettivi.Questo però finisce per danneggiare la BEE e permettere l'entrata di leucociti e cellule normalmente tenute fuori, e di acqua causando edema.

Inoltre le citochine IL-1,6, TNFa, TGFb, stimolano il rilascio di Acido arachidonico, NOS e aumentano l'eccitotossicità da glutammato e la produzione di ROS.Viene stimolata la proliferazione delle cellule della microglia che mostrano anche una spiccata attività fagocitotica, è interessante notare come alcuni monociti provenienti dal sangue poi differenzino a loro volta in cellule della microglia.
STRATEGIE NEUROPROTETTIVEServono ad impedire la morte apoptotica dei neuroni nella zona di penombra ischemica e limitare il danno al minimo.Si può prima di tutto somministrare trombolitici però entro 3 ore in quanto solo entro questo tempo si evitano i danni da riperfusione che abbiamo descritto.Altre strategie applicate solo in modelli animali sono:Poi si possono utilizzare anti-ossidanti per limitare i danni dei ROS.Si possono somministrare antagonisti dell'NMDA ed AMPA/KA in modo da contrastare l'eccitotossicità da glutammato.Somministrazione di anti-apoptotici.
MIASTENIA GRAVIS
E' una malattia autoimmune caratterizzata da progressiva debolezza muscolare e affaticamento dei muscoli scheletrici causata da trasmissione alterata al livello della giunzione neuromuscolare.Esiste anche una forma neonatale transitoria dovuta alla passaggio degli Ab dalla madre miastenica al feto attraverso la placenta. Ovviamente dopo la nascita scompare per degradazione degli Ab a conferma che si tratta di una malattia autoimmune.Colpisce le donne in 2-3 decade, mentre gli uomini in 6-7.Esiste anche una forma congenita causata da mutazioni dominanti e recessive di proteine della placca neuromuscolare.La patogenesi è dovuta all'alterazione della giunzione neuromuscolare, tuttavia non ti tratta di una patologia del neurone, infatti l'alterazione è post-sinaptica e consiste in un ridotto numero di recettori acetilcolinergici alla giunzione, inoltre è visibile un cambiamento morfologico: mentre la giunzione wt possiede numerose invaginazioni della membrana per aumentare la superficie di membrana e quindi il numero di recettori disponibili, nella giunzione mutata questi ripiegamenti sono assenti.Esiste una correlazione tra riduzione del numero dei recettori e gravità.Il recettore per l'Ach, ricordiamo, è formato da 2 subunità alfa, 1 subunità beta più una subunità gamma o eta. Il canale si apre quando entrambe le subunità alfa legano l'Ach.Gli Ab dosabili presenti nell'80-90% dei casi si legano ai recettori Ach e se trasferiti in modelli animali causano miastenia in questi, al tempo stesso indurre immunosoppressione o plasmaferesi migliora la malattia.Gli Ab causano un danno perché inducono il legame del complemento, che si lega covalentemente ad essi, ne aumenta l'endocitosi e la degradazione infine possono anche causare un blocco funzionale.I pazienti sieronegativi, che cioè non presentano Ab anti recettore Ach, nel 40% dei casi presentano anticorpi anti MuSk, che è una chinasi muscolo specifica.Il 75% dei pazienti miastenici ha alterazioni, anomalie al timo, la timectomia si dimostra utile in terapia. Un ipotesi patogenetica prevede che il timo possa contenere cellule mioidi che portano il recettore Ach sulla

membrana e indurre la risposta autoimmune. Spesso questi pazienti sono affetti anche da altre patologie autoimmuni.
La diagnosi è possibile effettuarla tramite:Test dell'aceticolinesterasi con cloruro di eprofonio: il cloruro di epofronio è un inibitore delle AchE per cui aumenta la [Ach] in circolo e si ha un miglioramento improvviso dei sintomi anche se molto breve, in quanto è un farmaco instabile.Anti-Ach-rec Ab assay: per ovvi motivi.Stimolazione ripetitiva nervi: a ogni impulso segue un potenziale muscolare senza variazioni, mentre in miastenici si riduce l'ampiezza del potenziale ad ogni stimolazione.Single-fiber EMG: boh.
Il trattamento si effettua tramite:Agenti anti-AchE (piridostigmina): diminuiscono il catabolismo dell'AchImmunosoppressione (ciclosporina, azatioprina,e soprattutto corticosteroidi): impedisce la risposta T autoimmunePlasmaferesi (in casi di miastenia acuta o grave)Infusione intravenosa di Ig: provoca riduzione della produzione di Ab endogeni(l'effetto di questi ultimi 2 dura solo 2-3 mesi)Timectomia
SCLEROSI MULTIPLA
La sclerosi multipla o a placche è una malattia demielinizzante infiammatoria multifocale del sistema nervoso centrale a patogenesi disimmune ed a decorso progressivo.Demielinizzante: perdita del rivestimento isolante di mielina degli assoni, che nell'SNC è prodotto dagli oligodendrociti e non dalle cellule di schwann.Infiammatoria: C'è presenza di reazione del sistema immunitarioMultifocale: presenza lesioni multiple come dimostrato dalla RMIDisimmune: la causa non è un alterazione del sistema immunitario, sebbene sia coinvolto.Progressiva: con il tempo peggiora, anche se la velocità di progressione è molto variabile
E' la più frequente delle malattie demielinizzantifreq 2-4/100000 prevalenza 50/100000 picco incidenza 30 anni (15-45) F/M = 2/1La triade si sintomi principali è: nistagmo, tremore intenzionale, parola scandita.
REPERTI DIAGNOSTICIA livello macroscopico:La patologia mostra rottura della BEE, infiammazione multifocale, e placche, cioè aree di demielinizzazione, che si presentano come aree di sostanza bianca di colore diverso in corpo calloso, cervelletto e midollo spinale, spesso in vicinanza di una piccola vena.A livello microscopico:L'infiammazione è causata da presenza di linfociti T e B e macrofagi oltre la BEE.

La demielinizzazione è causata sia dalla distruzione attiva delle guaine mieliniche (da parte dei macrofagi) e deficit di oligodendrociti, per cui è assente la produzione di mielina de novo.I disturbi neuronali sono causati dalla perdita assonale probabilmente dovuta all'infiammazione e perdita del rivestimento di mielina.La sclerosi ossia le cicatrici sono causate da proliferazione astrocitaria e produzione di fibre gliali per rimpiazzare le zone di necrosi.
DECORSO CLINICOIl decorso clinico è caratterizzato da una progressiva disfunzione neurologica progressiva.
85% forma recidivante-remittente (relapsing-remitting): è caratterizzata da episodi di distrubi neuronali isolati e diversi che si risolvono spontaneamente, i sintomi sono dovuti alla formazione di placche che poi scompaiono quando la placca si risolve. Le ricadute ci sono in media ogni 2 anni, i primi episodi non causano deficit permanente, ma il soggetto guarisce via via meno bene ad ogni recidiva fino al recupero incompleto.
Dopo circa 25 anni di malattia il 90% sviluppa la forma secondariamente progressiva in cui non c'è più remissione ma solo un peggioramento progressivo.
La forma primariamente progressiva è particolare in quanto inizia direttamente senza remissioni.Molte placche possono crearsi anche in zone silenti per cui non ci sono segni clinici e il paziente non se ne accorge.I sintomi clinici possono riguardareCervello disturbi motori, cognitivi, sensitivi, affettiviN. ottico neuritecervelletto atassiaMidollodisturbi motori, sensitivi, sfinterici, genitaliTronco disfagia, disartria
DIAGNOSILa RMI permette di fare diagnosi: le placche appaiono come aree di elevato segnale. Ci sono 2 criteri principali spaziale e temporale: spaziale perché la malattia deve presentare multifocalità e quindi si devono vedere più aree colpite, temporalità perché queste lesioni guariscono e quindi si notano in più episodi diversi.L'esame del liquido cefalo-rachidiano (normalmente trasparente, color ghiaccio-sciolto) mostra infiammazione e presenza di Ig oligoclonali.Indagini neurofisiologiche come la misura dei potenziali neuronali visivi, ottici, ecc, mostra alterazioni nella conduzione centrale.
TERAPIALa terapia è in parte sintomatica, per esempio si somministra baclofen in casi di spasticità, e in parte patogenetica, in cui si mira a sopprimere l'infiammazione.Nalizumab: è un anticorpo anti-molecole di adesione che impedisce ai leucociti di attraversare la BEE. Non è più usato perché causava encefalite virale.Bolo steroideo: cortisone ad altissima quantità per indurre immunosoppressione (così è più tollerato)IFNbeta: riduce il numero di ricadute però non c'è un gran miglioramento
EZIOLOGIA

Esiste concordanza nel 31% dei gemelli monozigoti e 5% nei gemelli dizigoti, inoltre i pazienti di 1° grado hanno un rischio 20-40 volte maggiore: questo fa pensare a un substrato genetico, probabilmente legato all'allele HLA-DR2.Esistono aree di prevalenza in emisfero nord e Australia, sono state registrate anche epidemie di SM, come sulle isole Faroe in seguito all'arrivo degli inglesi nella WWII: questo fa pensare alla probabile presenza di un virus che facilita lo sviluppo della SM.Il rischio di un'area è acquisito in caso di migrazione prima della pubertà (esposizione a virsu in periodo pre-puberale?).Esistono alcune somiglianze con l'encefalite allergica sperimentale causata dall'inziezione in modelli animali di tessuto cerebrale con adiuvante di freund, che causa una risposta immune contro la proteina basica della mielina.
PATOGENESIDanno mielinico e assonale:Il danno mielinico è caratteristico della malattia, mentre quello assonale è la causa dei disturbi e correla con la gravità. Il danno assonale è un fenomeno precoce nella malattia e la densità degli assoni è ridotta del 50% nelle aree di sostanza bianca apparentemente normale. La perdita assonale potrebbe essere correlata all'atrofia cerebrale osservata alla RMI.LinfocitiSi pensava che i linfociti coinvolti fossero i linfociti T, ma si è notata poi la produzione di Ig oligoclonali all'interno della teca cranica, ad esempio la neurite ottica è causata da Ab anti aquaporina. I linf T CD4+ mantengono comunque un ruolo come nella encefalite allergica sperimentale.Tappe del processo patogenetico:
attivazione dei linfociti T in periferia legame alla BEE passaggio della BEE attacco immunitario regolazione della risposta immunitaria rigenerazione e rimielinizzazione riorganizzazione funzionale danno assonale
La risposta dei linfociti T probabilmente è attivata da: proteina basica della mielina, proteina proteolipidica, glicoproteina mielinica oligodendrocitica. In effetti gli Ab per queste proteine presentano una certa cross reaction con antigeni di vari virus: HSV, HPV, EBV, Ad12 (molecular mimicry).Tappe dell'attacco immunitario:
le cellule della microglia (presentazione dell’antigene; attivazione di cellule che secernono citochine proinfiammatorie; rilascio di composti neurotossici; eliminazione delle cellule danneggiate)
le cellule T-helper CD4+ (rilascio di prodotti proinfiammatori, si trovano soprattutto negli infiltrati perivascolari)
i linfociti citotossici CD8+ (lisi cellulare, invadono il parenchima cerebrale) i macrofagi (fagocitosi e presentazione antigene) i mastociti (rilascio istamina ed altre sostanze)
LEGAME ALLA BEE

Ovviamente per l'attraversamento della BEE sono necessarie molecole di adesione. L'IFNbeta è capace di migliorare la prognosi dei pazienti in quanto inibisce l'adesione dei linfociti e l'attraversamento della barriera, allo stesso modo il Natalizumab (anti VLA4) spiazza l'interazione linfocita-cellula endoteliale.Per il passaggio della BEE è necessaria anche l'attivazione delle metalloproteinasi di matrice per la degradazione della membrana basale sottostante l'epitelio. Possono essere prodotte sia da cell immuni che gliali e nei pazienti SM la loro attività è aumentata.Cortisone e IFNbeta inibiscono la loro attivazione.
RIGENERAZIONE E DANNO ASSONALEIl glatiramer acetato (GA) stimola l’azione di cellule Th2 e Th3 che riducono l’infiammazione e stimolano rigenerazione e riparazione. In particolare le cellule Th2 rilasciano molecole regolatorie e producono BDNF che induce la crescita assonale, la mielinizzazione, la rigenerazione.
La capacità rigenerativa dei neuroni è poco nota, comunque non significativa, quindi è opportuno stimolare la sopravvivenza e il riparo di quelli restanti. Il neurotrophin brain-derived neurotrophic factor (BDNF) può prevenire il danno assonale e neuronale e stimolare la crescita assonale.
La rimielinizzazione può ristabilire la conduzione nervosa ed avere un effetto protettivo sull’assone, cellule progenitrici degli oligodendrociti rimpiazzano quelli perduti. Il processo è più intenso nelle lesioni iniziali e decade nelle fasi avanzate.
La plasticità corticale potrebbe avere un ruolo utile nella riduzione della disabilità. Studi di RM funzionale hanno dimostrato fenomeni di riorganizzazione funzionale nella SM
Il danno assonale è stato dimostrato non solo nella placca ma anche nel resto del cervello apparentemente normale. C'è infiltrazione di macrofagi e accumulo di proteine nell'assone che recede degradandosi fino a lasciare l'involucro vuoto degli oligodendrociti. Nei neuroni demielinizzati invece l'assenza dei nodi di ranvier causa una ridistribuzione dei canali del sodio lungo tutto l'assone con alterata omeostasi Na/Ca, entrata di Ca e danno neuronale.
EPILESSIA
L'epilessia è la predisposizione ad avere crisi epilettiche.La crisi epilettica è un disturbo transitorio della funzione cerebrale dovuto ad un'abnorme scarica ipersincrona dell'attività elettrica di una popolazione di neuroni (hughlings jackson); ne possono derivare effetti motori, sensoriali, mentali, che dipendono dal focolaio da cui orgina il firing.
Il focolaio epilettogenico è un insieme di neuroni, solitamente della corteccia, che effettua uno scoppio di attività elettrica, chiamato firing. Sono deafferentati (i dendriti non sono connessi a nessun assone), in stato di depolarizzazione parziale, hanno aumentata permeabilità agli ioni, sono cirdondati da neuroni GABAergici inibitori.

All'EEG si notano le scariche periodiche come punte che aumentano progressivamente ampiezza e frequenza. Quando la scarica coinvolge i neuroni della corteccia e i nuclei sottocorticali si ha una manifestazione clinica in relazione alle aree coinvolte.
La convulsione è un attacco parossistico di contrazioni muscolari involontarie, che rappresenta solo una del range di manifestazioni epilettiche. Può essere sintomatica cioè dovuta a febbre, ipertermia, ipoglicemia, ecc.
La sindrome epilettica consiste in un insieme di caratteri clinici che tendono a ricorrere insieme come il tipo di crisi, l'età di esordio, alterazione neurofisiologiche e elettroencefalografiche tipiche, eziologia.
La prevalenza è dell'1%, ne sono affetti 40-50 individui su 100000, esordisce già dai 2/3 anni in infanzia o dopo i 60 anni.
Tipi di epilessia: idiopatica (primaria) sintomatica (secondaria)
Oppure, altra classificazione: parziale esordio in area focale primariamente generalizzata esordio contemporaneo nei 2 emisferi secondariamente generalizzata esordio focale poi si estende a corteccia
CRISI EPILETTICA PRIMARIAMENTE GENERALIZZATASono varie:
Grande Male caratterizzato da crisi tonico e/o cloniche: contrazioni prolungate e ritmiche. Solitamente c'è una fase tonica di pochi secondi, seguita da una clonica di circa 1 minuto.
Piccolo Male semplice e complesso (assenze) Sindrome Lennox-Gastaut Epilessia mioclonica giovanile Spasmi infantili (tic di salam) Crisi atoniche
CRISI EPILETTICHE FOCALI (PARZIALI) Semplici - il paziente resta cosciente, assiste alla crisi, sa defnirla, manifestazioni
somatomotorie o somatosensitive e sensoriali (sensazioni di sogno, particolari sapori) Complesse - alterazioni non gravi della coscienza temporali (+ freq) o frontali Crisi tonico-cloniche secondariamente generalizzate
CRISI EPILETTICHE SECONDARIEPossono avere molteplici cause: tumori, malformazioni, traumi cranici, sostanze tossiche, farmaci, ictus, malattie degenerative, sospensione acuta alcool.
DIAGNOSIC'è bisogno di fare diagnosi differenziale da altre patologie come lipotimia (sincope), drop attack, cataplessia, stupor, coma, crisi psicogena.Bisogna inolte descrivere bene il tipo di crisi e verificare le alterazioni all'EEG, RM, TC, esame del liquor.

TERAPIALo scopo della terapia è quella di rendere il paziente libero dalle crisi senza effetti collaterali. Questo obiettivo è raggiunto nel 60% dei casi, nel restante 40% si parla di epilessie refrattarie alla terapia. Solitamente 1 solo farmaco basta nel fermare le crisi, raramente si usano combinazioni di essi.Prima si utilizzavano barbiturici, fenitoina o carbamazepina, oggi sono disponibili altre strategie:
Prolungamento del tempo di recupero da inattivazione dei canali Na+ voltaggio-dipendenti (carbamazepina, fenitoina, valproato)
Aumento dell'inibizione da parte del GABA (benzodiazepine, barbiturici) Inbizione del flusso di Ca nei canali T (etosuccimide, valproato)
Il trattamento farmacologico è consigliato se il paziente ha avuto più di una crisi, inoltre esistono 3 fattori di rischio principali per la ricorrenza delle crisi: alterazioni alla RMI, anomalie EEG dopo veglia protratta, crisi focale.Senza nessuno di questi fattori il rischio di ricorrenza è 15%, con tutti 80%.
PATOGENESILa caratteristica principale delle crisi epilettiche è la massiccia depolarizzazione e firing ad alta frequenza di un gruppo di neuroni.In modelli animali si è notata riduzione dell'inibizione gabaergica, e di fatti antagonisti gabaergici causano convulsioni, mentre le benzodiazepine che hanno effetto opposto sono anti-convulsivanti.Lo stesso vale per agonisti del glutammato che causano crisi, e viceversa antagonisti del glutammato.Esistono modelli animali di epilessia sia spontanei, sia knock-in e knock-out, sia un modello particolare chiamato Kindling, sia stati epilettici indotti chimicamente o elettricamente.Nell'uomo esistono predisposizioni genetiche all'epilessia, in particolare >50% delle mutazioni correlate causano canalopatie, colpiscono le subunità recettoriali alfa e beta della nicotina, canali del sodio, del calcio, recettore del gaba. <5% hanno trasmissione mendeliana.
CONVULSIONI NEONATALI FAMILIARI BENIGNESi tratta di una patologia a trasmissione autosomica dominante, che porta convulsioni nei primi giorni di vita che poi recedono spontaneamente: si pensa siano coinvolti geni per i canali del K+, con diminuito flusso di K+ e allungata depolarizzazione.
PICCOLO MALECi sono evidenze che suggeriscono la collaborazione tra fattori ambientali e 1 o 2 geni di suscettibilità. La causa è nel talamo o nella corteccia cerebrale, perché si tratta di alterazioni nel circuiti talamo-corticale, che regolano lo stato di coscienza/sonno.Ci sono 3 popolazioni neuronali coinvolte in questi circuiti:
neuroni talamici di relay neuroni reticolari talamici neuroni corticali piramidali
I neuroni talamici di relay possiedono canali del Ca di tipo T, che sono a bassa intensità di corrente e transienti, ma possono attivarsi in seguito a minime variazioni di voltaggio (favorendo lo spike), consentendo la modalità burst.Questi possono attivare i neuroni piramidali in 2 modalità: tonica (veglia e sonno REM) o burst (sonno non REM).

I neuroni reticolari sono capaci di iperpolarizzare i neuroni di relay mediante recettori GABA B, e di inibire sé stessi mediante recettori GABA A.Molti dei farmaci anti-epilettici agiscono sui recettori del GABA: etosuccimide, utilizzata soprattutto nelle assenze, e acido valproico causano il blocco dei canali T, inibendo il firing dei neuroni di relay; mentre le benzodiazepine inibiscono l'attivazione del GABA-A, lasciando i neuroni reticolari liberi di iperpolarizzare quelli di relay.Poiché durante il sonno si ha un'attivazione corticale ritmica così come nelle assenze, si pensa che siano proprio alterazioni questo circuito ad essere responsabili del piccolo male, a causa di anomalie nei canali T o nei recettori del GABA.
KINDLINGIl Kindling è un modello sperimentale di epilessia del lobo temporale indotta da somministrazione periodica di brevi stimoli elettrici all'amigdala o altre strutture del lobo limbico. Con le scariche ripetute man mano si hanno manifestazioni motorie fino a una crisi tonico-clonica. Una volta instaurata la sensibilità allo stimolo, questo permane nel tempo e basta una piccola scarica per provocare la crisi.Alla base di questo fenomeno c'è l'instaurazione di nuove connessioni neuronali.
STATO EPILETTICO INDOTTODifferisce dalle condizioni “normali” in quando non ci sono intervalli tra le crisi. Si ottiene mediante somministrazione di kainato o pilocarpina, o mediante stimolazione elettrica continua.
EPILESSIA DEL LOBO TEMPORALELe crisi parziali complesse a partenza dal lobo temporale sono la forma di epilessia più frequente. La crisi ha origine nella zona mesiale del lobo temporale, in cui si nota perdita neuronale e gliosi. La resezione di quest'area può controllare la crisi.I neuroni ippocampali piramidali CA3 sono particolarmente suscettibili a un firing di tipo burst e sono inibite dalle cellule dei granuli.Effettivamente si è notato sprouting di fibre nelle cellule dei granuli e formazione di nuove cellule dei granuli. Le fibre nate in eccesso vanno a connettere altre cellule dei granuli, creando un circuito che si auto-alimenta.