Embed Size (px)
Citation preview

SECCIÓN 1 - COMO PLANEAR UN PROGRAMA DE SONDAJE
CONTENIDO
1 COMO PLANEAR UN PROGRAMA DE SONDAJE 1-1 1.1 General 1-1 1.2 Equipos para Sondaje 1-1 1.3 Sistemas Convencionales de Sondaje 1-2 1.4 Sistemas Especiales de Sondaje 1-3 1.5 Sondaje de Paredes Laterales Wireline 1-4 1.6 Sondaje Orientado 1-5 1.7 Brocas para Sondaje 1-5 1.8 Descarga de Fluido Característica de las Brocas para Sondaje 1-6 1.9 Colectores de Muestras 1-6 Tablas 1-1 Sistemas Convencionales de Sondaje 1-2 1-2 Sistemas Especiales de Sondaje 1-3 1-3 Métodos de Orientación de Sondaje 1-5 1-4 Guía General de Brocas para Sondaje 1-6 1-5 Colectores de Muestras 1-6

Prácticas Recomendadas para el Análisis de Núcleos
1 Como Planear un Programa de Sondaje 1.1 GENERAL 1.1.1 Alcance Esta sección trata de las complejidades de la planeación de un programa de sondaje, las decisiones a tomar, y los factores que influyen en las elecciones. 1.1.2 Principio Un programa de sondaje es similar a muchos proyectos de ingeniería. Se empieza con la premisa que una inversión cosechará una recompensa. Se avanza por una fase de exploración de fuentes alternas de información - pruebas de pozos, registros de sucesos, núcleos anteriores, y muestras o núcleos de paredes laterales. La planeación empieza con un listado de los objetivos del programa de sondaje. El mejor equipo para hacer esto es aquel que tenga personal de petrofísica, yacimientos, geología, perforación y producción. Cuando se discuten los objetivos, cada gasto debe resultar finalmente en la producción de más crudo o gas a menor costo unitario. Se indicarán las restricciones en presupuesto, lugar, y tiempo en el programa. El tamaño de la perforación, el ángulo de perforación, temperatura, presión, y tipo de roca influirán en la selección de las herramientas de sondaje. La planeación se vuelve un proceso interactivo donde se construye un consenso y se formula un programa detallado. La planeación y la comunicación son las claves para una operación exitosa de sondaje. 1.1.3 Objetivo El objetivo de cada operación de sondaje es recolectar información que resulte en una producción mas eficiente de crudo o gas. Algunas tareas específicas pueden incluir los: a. Objetivos geológicos:
1. información litológica: (a) Tipo de roca (b) Ambiente deposicional (c) Tipo de poros (d) Mineralogía/geoquímica 1. Mapas geológicas 2. Orientación de fracturas
a. Ingeniería petrofísica y de yacimientos: 1. Información de permeabilidad: (a) Correlación de permeabilidad/porosidad (b) Permeabilidad relativa 1. Datos de presión capilar 2. Datos para refinar los cálculos en los registros
de sucesos (a) Propiedades eléctricas (b) Densidad de granos (c) Registro de gamma de núcleos (d) Mineralogía y la capacidad de intercambio de
catión
1. Estudios del recobro mejorado de crudos 2. Estimación de reservas:
(a) Porosidad (b) Saturación de fluidos
a. Perforación y terminación: 1. Estudios de la compatibilidad de
fluido/formación 2. Datos del tamaño de grano para el diseño de
relleno de grava 3. Datos de la mecánica de la roca
1.1.4 Fluidos para el Sondaje 1.1.4.1 La selección de un fluido para sondaje debe
basarse en cuatro puntos: a. Seguridad. b. El objetivo principal del programa de sondaje. c. Intereses ambientales. d. Costo. 1.1.4.2 La seguridad tiene prioridad sobre todos los demás factores. El fluido de perforación debe diseñarse para soportar las presiones esperadas de la formación como también limpiar, lubricar, y estabilizar la perforación. Los objetivos del programa de sondaje deben influir en la selección del fluido de sondaje/perforación. Todos los fluidos para sondaje deben ser diseñados para tener una pérdida de filtro API de baja estática y muy baja pérdida de arranque dinámico para minimizar la purga de núcleos. 1.1.4.3 Los intereses ambientales también deben considerarse e incluirse en el presupuesto. Esto puede significar el uso de un sistema más costoso de fluido para perforación para cumplir con los objetivos ambientales, o proporcionar equipos adicionales para el manejo de fluido para perforación para asegurar su contención. 1.1.4.4 El costo es importante - aún así, es una buena práctica revisar el costo de todo el programa de análisis de núcleos y los beneficios esperados mientras se cotizan los sistemas de fluidos para perforación. Los ahorros en fluidos para perforación pueden incrementar el costo de los análisis de núcleos, y pueden poner la precisión de los estudios de núcleos en riesgo. 1.1.4.5 La pregunta acerca de cuál fluido de perforación sea mejor para el sondaje no puede contestarse directamente. Se han utilizado fluidos de perforación con base en agua, en aceite, espuma, y aire/vapor para cortar núcleos exitosamente. La mejor recomendación es seguir los criterios indicados arriba. Una evaluación de las necesidades del programa de perforación y análisis de núcleos resultará en una selección apropiada. 1.2 EQUIPOS PARA SONDAJE 1.2.1 Alcance

Esta sección presenta un resumen de herramientas para sondaje, incluyendo las pautas para seleccionar herramientas de sondaje para aplicaciones específicas. Se debe obtener los detalles de sistemas particulares de sondaje, y recomendaciones de sondaje para propósitos específicos de las empresas de servicio apropiadas. 1.2.2 Principio Los equipos de sondaje están diseñados para recuperar muestras de roca desde la profundidad de la tierra para estudios geológicos y de ingeniería. Las herramientas hacen un excelente trabajo de recuperar material para núcleos, y se han desarrollado equipos especializados para colectar fluidos en yacimientos y hasta encerrar la presión de fondo. 1.2.3 Aparato Con varias excepciones notables, los sistemas de sondaje consisten de un sacanúcleo interior suspendido de un montaje giratorio dentro de un sacanúcleo exterior conectado a la cadena del taladro. Se conecta una barrena cortanúcleos al fondo del cilindro exterior y se adapta un colector de muestras en el fondo del cilindro interior. Se bombea el fluido para perforación por la cadena del taladro, a través del montaje giratorio, por la corona circular entre los cilindro interior y exterior, y sale por la broca del taladro. 1.3 SISTEMAS CONVENCIONALES DE
SONDAJE 1.3.1 Sacanúcleos Convencional Existen herramientas convencionales de sondaje para cortar núcleos con diámetros exteriores de 1.75 a 5.25 pulgadas (44.5 a 133.4 milímetros). La longitud del núcleo puede variar de 1.5 pies (.46 metros) para aplicaciones de pozos horizontales de radio corto hasta mas de 400 pies (121.9 metros) para formaciones consolidadas gruesas y uniformes. El tamaño de la perforación, el ángulo de perforación, fuerza de la roca, y litología controlarán el diámetro y la longitud del núcleo que puede ser cortado en un solo recorrido. La selección final de un sistema particular dependerá de la formación, ubicación, y los objetivos del programa de sondaje. La Tabla 1-1 resume las opciones convencionales de sondaje disponibles. 1.3.2 Sacanúcleos Convencionales Reforzados Se han desarrollado herramientas de sondaje especiales para trabajo pesado para trabajar en
formaciones más duras que las normales, y cortar núcleos de longitud extendido. Los hilos reforzados permiten que se aplique mas par de torsión en la broca, y mejora el margen de seguridad contra fallas en las herramientas. Diseñados para cortar núcleos hasta 5.25 pulgadas (133.4 milímetros) en diámetro, estas herramientas son especialmente atractivas en situaciones donde el tiempo de montaje es el gasto más grande de sondaje. Se utilizan los sistemas de sondaje reforzados para mejor ventaja cuando se sondean longitudes mas largas de formaciones homogéneas o cuando se anticipan cargas de par de torsión más altas que las normales. El sacanúcleos marino fue el precursor a los sacanúcleos de trabajo pesado de la generación de hoy en día. Desarrollado para ser mas fuerte que los sistemas de sondaje existentes, la herramienta fue desarrollada para uso en aplicaciones mar adentro. El sacanúcleos marino incrementa el margen de seguridad contra fallas en las herramientas, pero está limitado a cortar núcleos de 3 pulgadas (76.2 milímetros) de diámetro. 1.3.3 Forros de los Sacanúcleos El uso de un forro en un cilindro interior de acero tiene dos funciones principales: mejorar la calidad del núcleo soportando el material de núcleo físicamente durante su manejo y servir como un sistema de preservación de núcleos. Se han usado plásticos PVC y ABS, fibra de vidrio, y aluminio como forros de cilindro interiores. Los forros se deslizan en un cilindro interior convencional y son agarrados por el montaje del colector de muestras y fricción. Los forros típicamente son de 30 pies (9.14 metros). Se pueden cortar para aplicaciones especiales, pero su longitud máxima es rara vez mas de 30 pies (9.14 metros) debido a las limitaciones de fabricación y el manejo de materiales. Los forros son indicados a menudo cuando se hace sondaje en formaciones no consolidadas o fracturadas. También son apropiados cuando se corta roca dura en lugares remotos y mar adentro cuando se requiere una preservación de núcleos inmediata. Los forros plásticos son adecuados para temperaturas hasta 180ºF (82.2ºC). Los forros de fibra de vidrio pueden utilizarse hasta 250ºF (121ºC), 350ºF (176.7ºC) si se utiliza una resina especial para temperaturas altas. Se recomienda el aluminio por lo general cuando se esperan temperaturas mayores a 250ºF (121ºC). La desventaja de los forros de sacanúcleos es que ellos reducen el diámetro efectivo del cilindro interior por aproximadamente 0.5 pulgadas (12.7 milímetros).
Tabla 1-1 - Sistemas Convencionales de Sondaje
Cilindro Interior Longitud del Núcleo Características Especiales Acero dulce 30 a 120 pies (9.14 a 36.58 m) Sistema preparado para la preservación de núcleos.
Aplicaciones de temperaturas altas Acero dulce 1.5 pies (.46 m) Diseñado para el sondaje de radio corto.

Acero forjable 120 a >400 pies (36.38 a >121.9 m)
Sacanúcleos mas fuerte, incluye una estabilización adicional de cilindro interior y exterior.
Fibra de vidrio 30 a 90 pies (9.14 a 27.43 m) Sistema preparado para la preservación de núcleos. Utilizado para formaciones consolidadas y no consolidadas. Temperaturas máximas de operación: resina normal 250ºF (121ºC), resina para altas temperaturas 350ºF (176.7ºC).
Aluminio 30 a 90 pies (9.14 a 27.43 m) Sistema preparado para la preservación de núcleos. Aplicaciones de altas temperaturas, máximo 350ºF (176.7ºC)
Acero con forro plástico
30 pies (9.14 m) Sistema preparado para la preservación de núcleos. Temperatura máxima de 180ºF (82.2ºC). Reduce el diámetro de núcleo por 1/2 pulgada (12.7 mm).
Acero con forro de fibra de vidrio
30 pies (9.14 m) Sistema preparado para la preservación de núcleos. Temperatura máxima de 250ºF (121ºC). Reduce el diámetro de núcleo por 1/2 pulgada (12.7 mm).
Acero con forro de aluminio
30 pies (9.14 m) Sistema preparado para la preservación de núcleos. Temperatura máxima de 350ºF (176.7ºC). Reduce el diámetro de núcleo por 1/2 pulgada (12.7 mm).
1.3.4 Cilindros Interiores Desechables Los cilindros interiores desechables sirven para los mismos propósitos generales que los forros. Estos mejoran la calidad del núcleo soportando el material de núcleo físicamente durante el manejo y sirven como sistema de preservación de núcleos. Además, el diámetro exterior del núcleo no es reducido, como sería el caso con un forro de cilindro interior. Existen cilindros interiores desechables de aluminio, fibra de vidrio, y acero dulce, y son fabricados de varios tamaños para adaptarse a la mayoría de los sistemas convencionales de sondaje. Además, el cilindro interior de fibra de vidrio tiene un bajo coeficiente de fricción que permite que el núcleo se deslice mas fácilmente en el sacanúcleos, así reduciendo el riesgo de atascamiento. 1.3.5 Sondaje de Pozos Horizontales o de
Ángulo Elevado Los pozos de radio medio [radios de 290 a 700 pies (88.4 a 213.4 metros)] y aquellos con longitud extendida pueden sondearse con sacanúcleos convencionales alimentados desde el tablero giratorio o por un motor dentro del pozo. La mayoría de los núcleos serán cortados sin el uso de un motor dentro del pozo, pero habrá casos donde se justifique el uso de un motor de lodo. El uso de un motor dentro del pozo permitirá que se realice el sondaje sin girar la cadena del taladro. Se colocaría típicamente un sacanúcleos convencional largo de 30 pies (9.14 metros) adelante del motor de lodo dentro del pozo. Los motores de lodo producen un alto par de torsión a una baja velocidad giratoria para una fuerza óptima de sondaje. La longitud del sacanúcleos y el diámetro del núcleo pueden variarse para acomodar las restricciones de perforación. El cilindro interior es estabilizado adaptándolo con un cojinete de rodillos especial o casquillos para centralizar el cilindro interior. Se puede colocar un "drop ball sub" especial entre el motor y el sacanúcleos para permitir que el fluido de perforación fluya a través del cilindro interior, limpiando los escombros antes del sondaje. La activación del sub desvía el flujo de fluido de
perforación entre el cilindro interior y el exterior para el sondaje. En algunas instancias durante el sondaje, puede ser necesario mantener un control estricto sobre el ángulo del pozo. Un sondaje sin el motor dentro del pozo puede mejorar el control del ángulo del pozo. 1.4 SISTEMAS ESPECIALES DE SONDAJE 1.4.1 General Han evolucionado sistemas especiales de sondaje para llenar las necesidades especificas de sondaje. Los sacanúcleos de presión retenida y de esponja surgieron de una necesidad para mejores datos de saturación de crudo. Los sistemas de sondaje de manga de caucho y de cierre completo fueron desarrollados específicamente para mejorar la calidad de los núcleos cortados de formaciones no consolidadas. Otros sistemas especiales de sondaje tienen capacidades únicas, las cuales los hacen útiles para los ingenieros y los geólogos que los emplean. La Tabla 1-2 resume algunas de las opciones especiales de sondaje disponibles. 1.4.2 Sondaje de Presión Retenida Los sacanúcleos de presión retenida son diseñados para recoger núcleos mantenidos en condiciones de presión de yacimiento. Aceptado como el mejor método para obtener datos de saturación de crudo basados en los núcleos, los núcleos de presión retenida también capturan gases de yacimientos. La herramienta es especialmente útil para estudiar la viabilidad de proyectos de recobro mejorado y para calcular el contenido de metano en carbón. Existen sacanúcleos de presión retenida en dos tamaños: 6 pulgadas (152.4 milímetros) y 8 pulgadas (203.2 milímetros) de diámetro exterior que cortan núcleos de 2.50 y 3.75 pulgadas (63.5 y 95.3 milímetros) de diámetro exterior, respectivamente. El cilindro de diámetro exterior corta hasta 20 pies (6.1 metros) de núcleo de 2.5 pulgadas (63.5 milímetros) de diámetro mientras mantiene una presión máxima de 10.000 psi (69 Mpa). El cilindro de diámetro exterior de 8 pulgadas (203.2 milímetros) corta 10 pies

(3.05 metros) de núcleo de 3.75 pulgadas (95.3 milímetros) de diámetro mientras retiene un máximo de 5.000 psi (34.5 Mpa) de presión interna. La máxima temperatura de operación recomendada es de 180ºF (82ºC).
Los sacanúcleos de presión son herramientas sofisticadas que requieren unas instalaciones en el sitio para hacer mantenimiento al cilindro y manejar los núcleos presurizados. Los procedimientos para el manejo de núcleos se encuentran en 2.2.5.
Tabla 1-2 Sistemas Especiales de Sondaje
Sistema de Sondaje Dimensiones Máximas del Núcleo Aplicaciones Especiales Presión retenida 3.75 pulg. X 10 pies (5000 psi) [95.3 mm
x 3.05 m (34.5 Mpa)] 3.76 2.5 pulg. X 20 pies (10000 psi)
[63.5 mm x 6.1 m (69 Mpa)]
Análisis de presión retenida, saturaciones de fluido, volumen y composición de gases.
Forrado con esponja 3.5 pulg. X 30 pies (88.9 mm x 9.1 m) Saturaciones de fluido De cierre completo 4.0 pulg. X 60 pies (101.6 mm x 18.3 m) Recobro de formaciones no
consolidadas Manga de caucho 3.0 pulg x 20 pies (76.2 mm x 6.1 m) Recobro de formaciones no
consolidadas, fracturadas o conglomereradas
Wireline recobrable 2.75 pulg. X 30 pies (69.9 mm x 9.1 m) El sondaje es posible sin un tubo disparador
Wireline pared lateral de percusión 1 pulg x 1.75 pulg (25.4 mm x 44.5 mm) Muestras obtenidas después de perforación y registro
Wireline pared lateral perforada .94 pulg x 1.75 pulg (23.9 mm x 44.5 mm) Muestras obtenidas después de perforación y registro
Sacanúcleos de pared lateral 2.5 pulg x 10 pies (63.5 mm x 3.05 m) Núcleo obtenido después de perforación y registro
1.4.3 Sistema de Sondaje Forrado con Esponja El sistema de sondaje forrado con esponja fue desarrollado para mejorar la precisión de los datos de saturación de crudo basados en núcleos. Un sistema de sondaje de esponja no atrapa los gases de un yacimiento. En lugar de esto, el sistema atrapa el crudo expulsado cuando se saca el núcleo a la superficie. La información de saturación es muy útil cuando se evalúan los proyectos mejorados de recobro de crudo. Un sistema de sondaje de esponja tiene la ventaja de ser menos costoso para operar que un sistema de sondaje de presión retenida, mientras ofrece una oportunidad para mejorar la precisión de los datos de saturación de crudo basados en los núcleos. La esponja es estable a una temperatura de 350ºF (176.7ºC). El sistema de sondaje de esponja es limitado a cortar un máximo de 30 pies (9.14 metros) de núcleo de 3.5 pulgadas (88.9 milímetros) de diámetro por recorrido. 1.4.4 Sistemas de Sondaje de Cierre Completo Los sistemas de sondaje de cierre completo fueron desarrollados para mejorar el recobro de formaciones no consolidadas. Estos sistemas utilizan forros para sacanúcleos o cilindros interiores desechables, y un sistema especial de colección de muestras para recobrar las rocas dificultosas. La tecnología de sondaje de cierre completo permite al cilindro interior deslizarse suavemente por encima del núcleo blando con un mínimo de perturbación, y luego sellar el núcleo dentro del sacanúcleos. Esto se hace utilizando un montaje de colección de muestras de cierre completo que permite una entrada del núcleo al cilindro interior sin obstrucciones, y luego después
del sondaje sella el fondo del cilindro interior. Los sistemas de sondaje de cierre completo están limitados actualmente a cortar núcleos de 3.5 pulgadas (88.9 milímetros) o 4 pulgadas (101.6 milímetros de diámetro. La longitud recomendada de núcleos es de 30 pies (9.14 metros). El diámetro interior liso y la ausencia de un colector de muestras expuesto pueden resultar en núcleos perdidos si se levanta la herramienta del fondo antes de activar al colector de muestras de cierre completo. 1.4.5 Sacanúcleos de Manga de Caucho El sistema de sondaje de manga de caucho fue el primer sistema desarrollado para mejorar las posibilidades de recobrar arenas no consolidadas, conglomerados, y formaciones duras fracturadas. El sacanúcleos de manga de caucho es único porque la parte superior del cilindro interior no se mueve con respecto al núcleo durante el sondaje. El cilindro exterior es perforado alrededor de una columna de roca que es encerrada progresivamente en una manga de caucho. La manga de caucho es más pequeña que el diámetro del núcleo. Esta se estira ajustadamente alrededor del núcleo, envolviéndolo firmemente y protegiendo de la fricción del fluido para perforación. El núcleo es soportado por la manga de caucho, así ayudando en el recobro de las formaciones blandas que no soportarían su propio peso. Solo existe un tamaño de sacanúcleos de manga de caucho, que corta 20 pies (6.1 metros) de núcleo de 3 pulgadas (76.2 milímetros) de diámetro por recorrido. La manga de caucho está limitada a temperaturas de no más de 200ºF (93ºC). No se recomienda la herramienta para uso en perforaciones de mas de 45 grados de inclinación. Además, el sondaje debe

pararse aproximadamente cada dos pies para permitir el reinicio de la herramienta. Esto puede resultar en el atascamiento del núcleo en formaciones fracturadas. El sistema funciona mejor en estructuras fijas de perforación, pero puede operarse en equipos flotantes si el movimiento de los equipos es mínimo. 1.4.6 Sacanúcleos Recobrable Wireline Las herramientas de sondaje recobrable son operacionalmente similares a los sistemas convencionales de sondaje excepto que están diseñados para sacar el cilindro interior a la superficie por wireline. Esto acelera la operación de sondaje eliminando la necesidad de interrumpir toda la cadena del taladro para cada núcleo. Se bombea una nueva sección de cilindro interior por la cadena del taladro y esta es asegurada en su lugar para el sondaje adicional, o un tapón de taladro es bombeado para facilitar la perforación mas adelante. Las herramientas de sondaje recobrable son por lo general mas pequeñas y mas livianas que los sistemas convencionales de sondaje. Esto es una ventaja cuando tienen que ser transportadas a lugares remotos o por helicóptero. Desafortunadamente, los diámetros de núcleos son limitados porque todo el montaje del cilindro interior debe pasar por la cadena del taladro. También, se debe tener cuidado para evitar "fregar" crudo o gas en el pozo mientras se recobra el cilindro interior. 1.5 SONDAJE DE PAREDES LATERALES
WIRELINE 1.5.1 General Se desarrollaron los sistemas de sondaje de paredes laterales wireline para obtener muestras de núcleos de un pozo después de que este haya sido perforado y registrado, y antes de pasar el entubado. Estas herramientas pueden ubicarse en zonas de interés utilizando datos de los registros gamma o de potencial espontáneo como guías. Las muestras ofrecen pequeñas partes de material de formaciones, adecuados para estudios geológicos y de ingeniería. 1.5.2 Sondaje de Percusión de Paredes
Laterales La mayoría de los núcleos de paredes laterales wireline se obtienen con sistemas de sondaje de percusión de paredes laterales. Estas herramientas disparan balas cilíndricas huecas y recobrables en la pared de una perforación sin entubado. La herramienta (pistola) es bajada a la profundidad deseada en un wireline, y luego es disparada por impulsos eléctricos controlados desde la superficie. Las balas permanecen conectadas a la pistola por medio de alambres, y el movimiento de la pistola saca las balas, que contienen las muestras, de la pared de la perforación. Hasta 66 muestras de 1 pulgada (25.4 milímetros) en diámetro por 1 3/4 pulgadas 44.5 milímetros) de longitud, pueden tomarse durante un recorrido en el pozo. Hay diferentes diseños de "sacanúcleos" de balas para formaciones blandas, y medianas a duras. Es una buena idea tener mas de
un tipo de sacanúcleos en el sitio hasta que se pueda mostrar un recobro de núcleos aceptable. Las ventajas del sondaje de percusión de paredes laterales son velocidad, bajo costo, y la capacidad de sacar muestras en zonas de interés después de correr registros en perforaciones abiertas. La desventaja es que la bala usualmente altera la formación, fracturando la roca mas dura o comprimiendo los sedimentos mas blandos. Esto reduce el valor cuantitativo de los datos de análisis de los núcleos de paredes laterales. El recobro por percusión de núcleos de paredes laterales tiende a ser bajo en roca muy dura o fracturada, y en arenas muy permeables sin consolidar. 1.5.3 Sondaje de Paredes Laterales por
Perforación La herramienta giratoria o de perforado para paredes laterales fue diseñada para recobrar muestras de núcleos en paredes laterales wireline sin el impacto destructivo del sistema de percusión. Apropiada para roca dura-a-friable, la herramienta giratoria para el sondaje de paredes laterales utiliza un taladro con punta de diamante para cortar muestras individuales. El efecto de palanca aplicado en el taladro saca la muestra de la pared lateral. El taladro y la muestra son retraídos al cuerpo de la herramienta donde se deposita la muestra. La herramienta es trasladada a un nuevo lugar después de depositar cada muestra. Se puede tomar un máximo de 30 muestras, 15/16 pulgadas (23.9 milímetros) de diámetro por 1 3/4 pulgadas (44.5 milímetros) de longitud en cada recorrido. Una ventaja del sistema giratorio de sondaje de paredes laterales es que este produce muestras de roca dura adecuadas para el análisis cuantitativo de núcleos. Una de las desventajas es que es mas costoso que el sondaje de percusión en paredes laterales en cuanto a costos por el tiempo de instalación, y el recobro de muestras tiende a ser bajo en formaciones no consolidadas. 1.5.4 Sistemas de Sondaje de Paredes
Laterales Algunos nuevos sistemas de sondaje de paredes laterales están entrando en el mercado, y merecen discusión por dos razones. Primero, están diseñados para adquirir una muestra de núcleos mas grande y mas continua de un pozo perforado y registrado que lo posible con las herramientas existentes para el sondaje de paredes laterales. Segundo, la aparición de nuevas herramientas confirma que aun queda lugar para mejoras en el área de adquisición de muestras de núcleos de alta calidad y bajo costo. El primer sistema es similar al sacanúcleos convencional. El sistema de sondaje de paredes laterales está diseñado para cortar hasta 10 pies (3.05 metros) de núcleos de 2 1/2 pulgadas (63.5 milímetros) de diámetro. La herramienta se conecta a una cadena de taladro convencional y se baja a la zona de interés. Allí, un brazo integral empuja el sacanúcleos contra un costado del pozo. De ahí en adelante, la herramienta funciona como un sacanúcleos convencional. El segundo sistema utiliza

un mango de látigo removible para guiar un sacanúcleos convencional en la formación. Ambos sistemas llenan la necesidad de adquirir muestras de núcleos de calidad después del registro de pozos. 1.6 SONDAJE ORIENTADO 1.6.1 Generalidades Los núcleos orientados son utilizados para orientar fracturas, campos de esfuerzo, y tendencias de permeabilidad. Las operaciones de exploración, producción, y perforación utilizan la información para la búsqueda de yacimientos fracturados, el diseño de inundaciones de agua, y la planeación de pozos horizontales. Los núcleos orientados se cortan típicamente utilizando un sacanúcleos convencional adaptado con un anillo especial de trazado, y un aparato para registrar la orientación de la cuchilla de trazado principal en relación con el norte magnético. Los métodos de laboratorio utilizados para orientar núcleos son correlación del núcleo con los registros de imagen de pozos y el método paleomagnético. La Tabla 1-3 indica los métodos usualmente utilizados para orientar núcleos.
Tabla 1-3 - Métodos de Orientación de Núcleos Método Lugar Comentarios Levantamiento por disparos múltiples
Pozo Debe parar la perforación para tomar la lectura
Levantamiento electrónico
Pozo Registra orientación vs. tiempo
Método paleomagnético
Laboratorio Orienta un intervalo continuo
Correlaciones de registro
Laboratorio Requiere capacidad recíproca en el núcleo y el pozo.
1.7 BROCAS PARA SONDAJE 1.7.1 Generalidades Las brocas para sondaje son una parte básica del sistema de sondaje. Desafortunadamente para los expertos y los principiantes igual, las brocas de sondaje vienen en una confusa variedad de estilos. Afortunadamente, existen pautas generales de brocas/formaciones de los fabricantes para ayudar en la selección de la broca apropiada. Con un poco de información básica, es posible tomar decisiones informadas sobre los tipos de cortadores, perfiles de brocas, y consideraciones hidráulicas para el margen de condiciones de sondaje anticipadas. La selección final de brocas debe ser guiada por los objetivos del programa de sondaje, junto con una confirmación que la broca ha sido aprobada en el campo para aplicaciones similares. La dureza (fuerza compresiva), abrasividad, y variabilidad de las rocas a sondear tendrá la influencia mas grande sobre la selección de cortadores. Las
pautas generales sugieren el uso de cortadores mas pequeños, mas resistentes a impactos entre mas duras sean las formaciones. Las brocas de taladro de descarga frontal con baja invasión diseñadas para formaciones de resistencia no consolidada-a-mediana pueden ser utilizadas en rocas mas duras o mas abrasivas, pero la vida útil de la broca puede ser reducida drásticamente. La información presentada en la Tabla 1-4 ofrece un resumen de los tipos de brocas para sondaje disponibles. Se debe obtener los detalles específicos sobre las brocas de sondaje y las recomendaciones para aplicaciones particulares de las empresas de servicios. 1.7.2 Brocas de Diamantes Naturales Se utilizan brocas de taladro de diamante natural cuando la formación es demasiado dura (alta resistencia compresiva) y/o abrasiva para otro tipo de elementos cortadores. Se pueden montar diamantes naturales grandes en una matriz de carburo de tungsteno, o se pueden dispersar recortes finos de diamantes en una matriz para formar lo que se llama una broca impregnada de diamantes. Las brocas impregnadas de diamantes naturales son para aplicaciones en formaciones ultra-duras. 1.7.3 Cortadores Compactos de Diamantes
Policristalinos (CDP) Los cortadores (CDP) compactos de diamantes policristalinos son materiales de diamantes artificiales que consisten de una capa de arenilla de diamantes del tamaño de un micrón sinterizada y adherida a espigas de carburo de tungsteno. El grosor de la capa de diamantes policristalinos es solo de 0.020 a 0.060 pulgadas (.51 a 1.52 milímetros). Las brocas CDP se utililzan para sondear formaciones que varían de muy blandas a medio duras. Las brocas son diseñadas para cortar por cizallamiento resultando en una alta velocidad de penetración. Debido a la geometría del cortador CDP, estos son susceptibles a daños por impacto, y por lo tanto no son recomendados para formaciones muy duras, altamente fracturadas, o de cuarzos. 1.7.4. Diamantes Térmicamente Estables (PTE) El producto (de diamantes) térmicamente estables, PTE, es similar a los CDP en que también es un material de diamantes artificiales. La diferencia principal en el material PTE es que tiene un margen mas alto de estabilidad térmica debido al filtrado del catalizador metálico utilizado en el proceso de sinterización de fabricación. Estos cortadores son apropiados para formaciones considerados por lo general demasiado duras y/o abrasivas para los cortadores CDP. Estos no son recomendados para formaciones blandas. 1.7.5 Brocas de Conos Giratorios La broca de taladro de conos giratorios utiliza cuatro conos giratorios montados con piezas insertadas de carburo de tungsteno o cortadores de diente triangular

para propósitos de sondaje. Los cortadores en los conos giran y se incrustan en el fondo del pozo y rompen la formación en compresión con una acción cinceladora. Debido a la lenta acción cortadora (rompimiento compresivo cincelador) y la cantidad de partes móviles, el uso de las brocas de taladro de conos giratorios no es común. 1.8 CARACTERÍSTICA DE DESCARGA DE
FLUIDOS DE CORTANÚCLEOS 1.8.1 Descarga por la Entrada Los cortanúcleos de descarga por la entrada están diseñados para tener el 100 por ciento del fluido pasar entre el anillo cortante y el diámetro interior del cortanúcleos (la "entrada"). Las brocas de descarga por la entrada están diseñadas para limpiar el diámetro interior del cortanúcleos, removiendo los recortes de esta área para asegurar una entrada muy uniforme del núcleo al sacanúcleos. La acción limpiadora reduce la tendencia a atascarse de las formaciones duras y/o quebradizas. 1.8.2 Descarga Frontal Los cortanúcleos de descarga frontal están diseñados para desviar algún fluido que normalmente pasaría a través de la entrada de la broca al frente de la broca. Esto limpia la superficie de la broca y reduce la cantidad de fluido que puede friccionar el núcleo mientras entra en el sacanúcleos. Se recomiendan las brocas de descarga frontal para uso en formaciones blandas y friables. 1.8.3 Perfil de Baja Invasión
Los cortanúcleos con perfil de baja invasión están diseñados para maximizar la velocidad de penetración, y minimizar la invasión de filtrado de fluido de perforación en el núcleo. El diseño incorpora aberturas de descarga frontal, una reducida cantidad de cortadores, y un espacio libre disminuido entre el cilindro interior y la superficie de la broca. Se recomienda el uso de cortanúcleos con perfil de baja invasión para formaciones de resistencia blanda a mediana. Las formaciones mas duras disminuirían la velocidad de penetración y posiblemente dañaría los cortadores. 1.9 COLECTORES DE MUESTRAS 1.9.1 Generalidades La parte mas crítica de cada sistema de sondaje es el colector de muestras que mantiene el núcleo en el cilindro mientras es llevada a la superficie. La Tabla 1-5 indica los colectores de muestras disponibles y sugieren aquellos mas apropiados para tipos de roca específicos. Muchas situaciones requieren una combinación de dos o mas colectores para asegurar el éxito. Las secuencias de arena friable intercalada con esquisto pueden requerir colectores tanto de tipo deslizante como de tipo plegadizo. Los colectores de cierre completo funcionan principalmente para asegurar buenos resultados cuando se hace sondaje en arenas no consolidadas, también incorporan colectores de anillo partido o de tipo deslizante para mejorar el recobro de núcleos en extremos de roca dura.
Tabla 1-4 - Guía General de Cortanúcleos
Propiedades de la Roca Tipo de Roca Cortanúcleos Roca abrasiva ultra-dura Rocas ígneas, cuarcita Impregnado de Diamante Natural
Roca abrasiva dura Arenisca, Esquisto, Aluvión Diamantes naturales montados en la superficie o cortadores PTE
Roca dura no abrasiva Caliza, Dolomita, Anhidrita Cortadores PTE Roca mediana a dura con capas
abrasivas Arenisca, Caliza, Esquisto PTE o diamantes naturales montados
en la superficie Roca de resistencia blanda a mediana Arenisca, Yeso, Esquisto Cortadores CDP, diseño de baja
invasión de fluidos Rocas blandas, sin capas pegajosas Sal, Anhidrita, Esquisto CDP o cortadores de conos giratorios
Roca blanda pegajosa Suelo Arcilloso Cortadores CDP, descarga frontal
Tabla 1-5 - Colectores de Muestras
Tipo Uso Recomendado Anillo partido, resorte Formaciones consolidadas
Collar Donde las características de la formación son desconocidas Deslizante Formaciones consolidadas, normalmente funciona con colector plegadizo o
con cuchillas orientadoras Dobladizo o plegadizo Formaciones consolidadas, fracturadas y no consolidadas donde la geología
es desconocida Canasta Formaciones no consolidadas, normalmente funciona con otro tipo de
colector de muestras Cierre completo Formaciones friables o no consolidadas para proporcionar un cierre

completo positivo

CONTENIDO
2 PROCEDIMIENTOS DE MANEJO Y PRESERVACIÓN DE NÚCLEOS EN LAS INSTALACIONES DE POZOS 2-1
2.1 Generalidades 2-1 2.2 Procedimientos de Manejo de Núcleos 2-2 2.3 Muestreo y Análisis en el Campo 2-6 2.4 Tipos de Rocas y Consideración Especial en su Manejo 2-7 2.5 Preservación de Núcleos para Análisis 2-12
2.6 Recomendaciones para el Manejo de Núcleos para Preservar su humectabilidad 2-15 2.7 Precauciones 2-15 2.8 Bibliografía 2-16
Figuras 2-1 Marcado de Núcleos 2-3 2-2 Datos de las Instalaciones del Pozo en el Análisis de Núcleos 2-8 2-3 Datos Básicos de Laboratorio en el Análisis de Núcleos 2-9

Prácticas Recomendadas para el Análisis de Núcleos
2 Procedimientos de Manejo y Preservación de Núcleos en las Instalaciones de Pozos
2.1 GENERALIDADES 2.1.1 Las recomendaciones incluidas en este documento pueden implicar el uso de materiales, operaciones y equipos peligrosos. Este documento no trata de todos los problemas de seguridad asociados con su uso. Es la responsabilidad del usuario de establecer las prácticas apropiadas de seguridad y salud y determinar la aplicabilidad de las limitaciones reglamentarias antes de usarlos. 2.1.2 Los procedimientos de manejo y preservación de núcleos en las instalaciones de pozos deben seguir las mejores prácticas posibles porque el valor de todo análisis de núcleos está limitado por esta operación inicial. Los objetivos de un programa de manejo de núcleos son los siguientes: a. Obtener material de roca representativa de la
formación. b. Minimizar la alteración física del material de roca
durante el manejo y el almacenamiento del núcleo.
Los problemas más grandes enfrentados por aquellos que manejan y preservan rocas de yacimientos para el análisis de núcleos son los siguientes: a. Selección de un material no reactiva de
preservación y un método para prevenir la pérdida de fluido o la adsorción de contaminantes.
b. Aplicación de métodos apropiados de manejo y preservación de núcleos basados en el tipo de roca, grado de consolidación, y tipo de fluido.
Los diferentes tipos de roca pueden requerir precauciones adicionales para obtener datos representativos de los núcleos (ver 2.4). Todo el material de núcleos debe ser preservado en las instalaciones del pozo tan pronto sea posible después de recobro para minimizar su exposición a las condiciones atmosféricas. 2.1.3 La terminología que se ha desarrollado para describir el estado de preservación de núcleos es históricamente importante, pero puede ser confuso porque a menudo no es usada consistentemente. Por ejemplo, el término "estado nativo" se ha utilizado a menudo para referirse a un núcleo perforado con lodo en base de aceite o crudo "lease" para tomar mediciones exactas de saturación de agua. De manera similar, "estado fresco" se ha utilizado a menudo para indicar que el núcleo fue perforado con fluido de perforación blando con base en agua y preservado en las instalaciones del pozo para limitar las pérdidas por evaporación. Este término también ha sido utilizado para incluir los núcleos cortados con
lodo en base de aceite. Para propósitos de consistencia, se recomienda la siguiente terminología: 2.1.3.1 núcleo fresco: Cualquier material de núcleo recobrado preservado tan pronto como sea posible en las instalaciones del pozo para prevenir pérdidas por evaporación y exposición al oxígeno. El tipo de fluido utilizado para sondaje debe registrarse, e.g., estado fresco (fluido de perforación en base de aceite), estado fresco (fluido de perforación en base de agua). 2.1.3.2 núcleo preservado: Similar al núcleo fresco, pero este implica algún periodo de almacenamiento. El núcleo preservado está protegido de alteraciones por cualquiera de una variedad de técnicas (ver 2.5). 2.1.3.3 núcleo limpio: Núcleo del cual los fluidos han sido removidos por solventes. El proceso de limpieza (secuencia de solventes, temperatura, etc.) debe especificarse. 2.1.3.4 núcleo de estado restaurado: Núcleo que ha sido limpiado, luego expuesto nuevamente a fluidos del yacimiento con la intención de restablecer la condición de humectabilidad del yacimiento. Este es a menudo la única alternativa disponible, pero no hay garantía que se restaure la humectabilidad del yacimiento. Las condiciones de exposición al crudo, especialmente en saturación inicial de agua, temperatura y tiempo, pueden afectar la humectabilidad final. 2.1.3.5 núcleo con presión retenida: Material que ha sido mantenido, hasta donde sea posible, en la presión del yacimiento con el fin de evitar cambios en las saturaciones de fluido durante el proceso de recobro. Ninguno de estos términos individualmente pueden describir adecuadamente el estado del núcleo. Se requiere una descripción completa de lodo de perforación, manejo, preservación y tratamiento subsecuente. 2.1.4 Para pruebas, se debe tomar una muestra del núcleo. Con el fin de obtener un análisis representativo de los núcleos de una formación de interés, se recomienda tomar muestras de todo el núcleo. Se debe retener toda la sección del núcleo. La toma de muestras de núcleos en las instalaciones de pozos puede ser importante por una variedad de razones (ver 2.3.1). Si se requiere una toma de muestras del núcleo, esta debe realizarse con el conocimiento que el procedimiento puede tener efectos sobre los esfuerzos y resultados de futuros análisis de núcleos. La toma de muestras en las instalaciones de pozos debe ser mínima para mantener la integridad del núcleo. Las muestras para la descripción de litología, por ejemplo, pueden tomarse de algunos pedazos quebrados del núcleo sin dañar ninguna parte de la roca intacta. Si está intacta, se remueven longitudes mensurables del núcleo, se debe dejar una nota o registro en su lugar

describiendo la longitud de la muestra, litología, la razón por la cual fue removida, y cualquier otra información pertinente. Si se requieren muestras de adentro de un segmento de núcleo intacto, se debe emplear un método de muestreo no percusivo. El objetivo de un procedimiento estándar de muestreo de núcleos es obtener muestras bajo un procedimiento uniforme para que los resultados sean independientes de sesgo humano. La selección de muestras es bastante sencilla para formaciones uniformes. Sin embargo, si una formación contiene una litología muy variada y tipos de porosidad heterogénea (tal como conglomerados, variedades de cuarzos, yacimientos fracturados, y esquistos y arenas intercalados), la selección apropiada de muestras representativas requiere mayor cuidado. Una persona calificada (ingeniero, geólogo, etc.) debe seguir un procedimiento de muestreo establecido para minimizar el sesgo estadístico. 2.1.5 Los procedimientos para el manejo y la preservación de núcleos prescritos son aplicables para todo material de roca convencionalmente sondeado. Muchas de las mismas prácticas aplican para núcleos de paredes laterales y recortes de perforado. Estos procedimientos recomendados han sido seleccionados como aquellos que producirán materiales de núcleos para el análisis de núcleos más confiable y representativo. El éxito de cualquier técnica dada es directamente relacionada con las propiedades de la roca del núcleo. Los procedimientos de manejo también deben basarse en la tecnología utilizada para recobrar el material de roca los objetivos del programa de sondaje. También se presenta una revisión de los materiales de preservación de núcleos. Cada trabajo de sondaje y yacimiento debe ser examinado cuidadosamente antes de diseñar un programa de manejo y preservación de las instalaciones de pozos. 2.2 PROCEDIMIENTOS DE MANEJO DE
NÚCLEOS 2.2.1 Generalidades Existen varios métodos para la adquisición de núcleos. Se pueden dividir las técnicas de sondaje continuas convencionales de diámetro completo en dos grupos: aquellas que emplean un cilindro interior estándar de acero para uso repetido, y aquellas que utilizan cilindros interiores desechables o forros. Otros métodos de sondaje tales como los aparatos de paredes laterales y los aparatos de sondaje recobrados por wireline obtienen material de roca utilizando equipos especiales. Los procesos especiales de sondaje, incluyendo los métodos de presión retenida y de esponja, están disponibles para obtener resultados de análisis de núcleos y fluidos más representativos de las condiciones in situ. El material de núcleos consolidado obtenido con un cilindro interior estándar de uso repetido debe sacarse del cilindro tan pronto como sea posible después de llegar a la superficie para minimizar la imbibición de fluido de perforación. Entre los posibles efectos
indeseables de la imbibición de fluidos se encuentran los siguientes: a. Cambios en las saturaciones de fluido, equilibrio
geoquímico y de soluciones de gas. b. Cambios en humectabilidad. c. Movilización de arcillas intersticial y minerales de
grano fino. d. Dilatación de arcilla y la degradación asociada de
propiedades mecánicas. Se debe reportar cualquier demora en la remoción del núcleo del cilindro. Varios tipos de roca y métodos de sondaje requieren niveles variables de atención y pueden dividirse en dos categorías principales: a. Manejo básico - Esta categoría requiere una
capacitación y/o experiencia mínima e incluye: 1. Un cilindro interior estándar de acero para uso
repetido utilizado para obtener núcleos en rocas consolidadas moderadamente homogéneas.
2. La adquisición de núcleos de paredes laterales por wireline con sondaje de percusión o giratorio.
b. Manejo especial - Esta categoría requiere una
capacitación extensiva y/o equipos especiales e incluye:
1. Cilindros interiores desechables y sacanúcleos orientados utilizados para obtener núcleos de rocas fracturadas o no consolidadas que pueden requerir una estabilización mecánica (Skopec, et al., 1992).
2. Sacanúcleos retenida a presión para mantener el núcleo en la presión del yacimiento para minimizar la expansión de fluido de la reducción en presión y la expulsión de fluido mientras se lleva el núcleo a la superficie (Sattler, et al., 1988)
3. Sacanúcleos de aluminio con un forro de esponja dentro de un cilindro interior de acero para atrapar los fluidos durante la expansión por la reducción en presión mientras se lleva el núcleo a la superficie (Park, 1983).
El uso de cualquier forro de cilindro interior para sondaje reduce el diámetro del núcleo resultante. 2.2.2 Remoción del Núcleo de un Cilindro
Interior Estándar de Acero para Uso Repetido
El núcleo debe removerse del cilindro interior en una posición horizontal cuando sea posible. Se debe tener cuidado para minimizar la sacudida mecánica durante la extracción. Se debe permitir que el núcleo se deslice del sacanúcleos ligeramente elevando el extremo superior del sacanúcleos. Si el núcleo no se desliza, se puede utilizar una vara para empujar el núcleo del cilindro. Puede ser necesario golpear el sacanúcleos suavemente con un martillo para iniciar el movimiento del núcleo. Sin embargo, no se debe martillar el sacanúcleos de una manera que cause una sacudida mecánica en el núcleo. En toda manipulación física se debe intentar exponer el núcleo al mínimo esfuerzo mecánico posible. Si no se puede

remover el núcleo con el método anterior, este debe sacar por bombeo con fluido. Si esto es necesario, se debe utilizar un arreglo adecuado de pistones que prevendrá que los fluidos tengan contacto directo y contaminen el núcleo. Se debe utilizar el fluido de sondaje si es necesario bombear directamente con fluidos. Se debe evitar el uso de agua fresca u otros fluidos extraños para el núcleo. Si se presiona agua por el pistón y este entra en contacto con el núcleo, se pueden obtener valores erróneamente altas de saturación de agua en un análisis subsecuente de núcleos porque cualquier presión excesiva en el cilindro puede hacer que el fluido penetre el núcleo. Se debe anotar cualquier dificultad o irregularidad encontrada cuando se remueve el núcleo del cilindro, e.g., la presión utilizada si fue sacado con fluido, pérdida de material del núcleo, etc. 2.2.3 Clasificación y Registro de Núcleos Se debe extender y empacar el núcleo en el piso de las instalaciones si hay espacio disponible. Alternativamente, el soporte de tubería puede utilizarse para este propósito. La clasificación y registro del núcleo no debe interferir con la operación de perforación y/o sondaje. Si se toma la decisión de manejar el núcleo en el piso de las instalaciones, hay que colocar bandejas, cajas, o piletas apropiadamente marcadas cerca del sacanúcleos. Si se va a extender el núcleo en el pasadizo, se prepara un área despejada y se coloca el núcleo entre dos trozos de tubería de perforación. Se debe tener cuidado para mantener la orientación, y preservar la secuencia correcta de los pedazos de núcleo. El punto clave es que el núcleo debe clasificarse y marcarse de tal manera que todo el intervalo de núcleo pueda volverse a ensamblar en el futuro. Se debe proteger al núcleo de temperaturas extremas, humedad, y deshidratación, i.e., sol directo, motores calientes, lluvia, vientos fuertes, y baja humedad relativa. Los materiales y equipos de preservación de núcleos deben estar cerca del área de manejo de núcleos para facilitar una operación rápida. Mediciones precisas de recobro deben ser tomadas y registradas. Se debe reportar cualquier recobro además del recorte del núcleo, así como el no recobro. Hay que escribir no recobro o recobro adicional en la parte de debajo de cada núcleo, a menos que alguna observación especial indique que se debe hacer una excepción. Todas las excepciones deben anotarse. Los siguientes datos y observaciones pueden ser útiles para determinar el origen de recobro adicional y la falta de recobro: a. Parámetros de perforación - tiempo de
perforación, velocidad de penetración, presión de bombeo, etc.
b. Condiciones generales del núcleo - continuidad, secciones quebradas, fracturas inducidas, etc.
c. Condición del equipo de sondaje de perforaciones de fondo.
Marque las profundidades de los núcleos de arriba abajo e indique en la parte de abajo si son de recobro adicional o falta de recobro. La parte de arriba del siguiente núcleo debe tener la profundidad perforada.
Esto quiere decir que en el caso de recobro adicional, habrá la misma profundidad en dos núcleos. Sin embargo, estos núcleos serán distinguibles uno del otro por sus números. Las profundidades de los núcleos deben ajustarse a las profundidades registradas antes de que se puedan hacer correlaciones entre las propiedades registradas y las propiedades del núcleo y entre los pozos sondeados y sin sondear. El ajuste núcleo-a-registro puede hacerse utilizando descripciones detalladas de núcleos o barridos de núcleos. Se recomienda que todos los sacanúcleos se extiendan en el pasadizo o el piso de las instalaciones antes de remover el núcleo. Las siguientes son las pautas apropiadas para extender y marcar el núcleo: a. La parte de abajo del núcleo sale del cilindro
primero y la primera pieza del núcleo debe colocarse en el fondo de una bandeja, caja, o pileta, y cada pieza siguiente se coloca mas cerca de la parte de arriba.
b. Se debe tener mucho cuidado para mantener la secuencia apropiada y la orientación del núcleo para asegurar que los segmentos individuales del núcleo no estén fuera de lugar o al revés. Cualquier porción del núcleo que esté muy partida debe meterse en bolsas de plástico y colocarse en su posición apropiada.
c. Arme el núcleo para que los extremos irregulares casen, luego mida la totalidad del recobro.
d. No lave el núcleo (ver 2.4, 3.5 y 3.6). Si hay demasiado fluido de perforación en la superficie del núcleo, se puede limpiar con un trapo limpio saturado en fluido de perforación, y este se puede exprimir tan a menudo como sea necesario.
e. Con marcadores indelebles rojos y negros, pegados con cinta, marque el núcleo de arriba abajo con líneas paralelas (ver Figura 2-1). La línea roja debe estar en el lado derecho si el individuo que marca se encuentra mirando de la parte de abajo del núcleo hacia arriba. Se deben utilizar flechas apuntando hacia arriba para evitar confusiones.
f. Con un marcador indeleble o pintura, empezando de arriba, dibuje una línea a través del núcleo a cada pie de distancia, y marque cada línea con la profundidad apropiada.
g. Para obtener un análisis confiable del núcleo, la velocidad es esencial en remover, extender, marcar y preservar el núcleo para minimizar las alteraciones debidas a exposición (ver 2.5).
h. El núcleo debe preservarse (ver 2.5) y colocarse en recipientes numerados para ser transportado al laboratorio. Se recomienda que todo el intervalo del núcleo sea preservado en las instalaciones del pozo, reservando el muestreo para las condiciones controladas en el laboratorio.
Basta unos pocos minutos de exposición, dependiendo de las condiciones atmosféricas, para causar una pérdida significativa de agua fracciones de hidrocarburo liviano en los núcleos. Si el núcleo es lavado con agua accidentalmente, dejado en el

sacanúcleos, o reposado antes de la preservación, entonces esta información debe ser anotada. 2.2.4 Manejo de Forros y Cilindros Interiores
Desechables El uso de forros interiores de sacanúcleos y cilindros interiores desechables mejoran el recobro de las formaciones fracturadas o de consolidación deficiente. Estos son hechos de plástico, fibra de vidrio, o aluminio y están clasificados para varias temperaturas. Cuando se hace sondeo en formaciones no consolidadas o de consolidación deficiente, elija el forro o el cilindro desechable para soportar la temperatura circulante. Los estratos duros tal como el esquisto son sondeados mejor utilizando fibra de vidrio o aluminio para prevenir el atascamiento y consecuentemente un recobro de núcleos deficiente. Ciertos aditivos de fluido para sondaje tal como el cáustico reaccionan con cilindros de aluminio causando la descarga de iones de aluminio, que pueden reaccionar con el núcleo para alterar las propiedades de su superficie. Cuando se sondea una formación de consolidación deficiente, para evitar la solidificación de la roca es aconsejable cortar longitudes cortas, 30 pies o menos dependiendo de la resistencia de la roca. En longitudes largas, la sección inferior del núcleo puede ser consolidada en exceso y dañado por el peso del material de encima. Un núcleo dañado es de uso limitado para el análisis de núcleos. Cuando se sondean formaciones fracturadas, las longitudes cortas de núcleos también pueden ser útiles para disminuir el riesgo de atascamiento. El sacanúcleos debe ser llevado a la superficie suavemente. Durante los últimos quinientos pies, el núcleo debe llevarse a la superficie lentamente para minimizar la expansión de gas que puede dañar el núcleo no consolidado severamente si la presión es reducida muy rápidamente. Cuando se esperan daños por expansión de gas, se puede utilizar un forro perforado o un cilindro interior desechable perforado para proporcionar un medio de escape para el gas. Todas las perforaciones deben ser selladas si el forro o el cilindro interior desechable es utilizado como recipiente de preservación de núcleos. Alternativamente, toda la sección perforada puede ser colocada en bolsas de plástico para prevenir la pérdida de fluidos. Un forro con el núcleo adentro puede bajarse en el pasadizo, dentro del cilindro interior metálico utilizando un sistema de poleas sujetado al extremo del pasadizo. El cilindro no debe golpear el equipo y debe bajarse suavemente al pasadizo. Los cilindros interiores con núcleos adentro se encorvan, especialmente aquellos hechos de fibra de vidrio o plástico, y deben ser soportados por una varilla. La varilla debe ser pegada al cilindro interior mientras cuelga del castillete. a. Acuñe el sacanúcleos para prevenir rotación y
remueva el colector de muestras. Transfiera el material del colector de muestras a un forro o un cilindro interior desechable de la longitud adecuada. El material del colector de muestras
es por lo general demasiado agitado para ser utilizado en el análisis cuantitativo de núcleos.
b. Se puede utilizar una extensión del pasadizo para remover todo el forro con el núcleo del cilindro interior sin encorvarse.
1. Si se remueve todo el forro o si se está manejando un cilindro interior desechable, busque adentro y ubique la parte superior del núcleo. Corte el forro en este punto. Marque el núcleo con líneas de orientación (rojo a la derecha y negro a la izquierda), profundidades, y otra identificación (ver 2.2.3). Marque las profundidades a cada pie empezando de arriba.
2. (Método Preferido) Si no hay espacio para remover todo el forro, saque de a de 3 pies (0.91 metros) utilizando el soporte adecuado para prevenir encorvamiento. Marque cada longitud de tres pies (0.91 metros) con líneas de orientación y con un número para representar su posición en la secuencia de longitudes cortadas. Marque las profundidades en las longitudes de tres pies (0.91 metros) una vez se procese todo el intervalo recobrado y se ubique la parte de arriba del núcleo.
c. Corte los forros y el núcleo en longitudes de 3
pies (0.91 metros) utilizando una sierra circular de aire o eléctrica. Los forros de fibra de vidrio y aluminio deben ser cortados utilizando una sierra circular fija. Tenga cuidado de evitar vibración y rotación del núcleo. Se deben utilizar abrazaderas en el sacanúcleos con mucho cuidado para evitar daños en la roca. Alternativamente, toda la longitud de 30 pies (9.1 metros) del núcleo puede taparse y transportarse con una varilla pegada al forro o el cilindro interior para evitar encorvamiento. Luego el núcleo puede ser cortado en el laboratorio en cualquier longitud especificada. Esto minimiza el manejo del núcleo en el pozo. Sin embargo, esto dificulta la preservación, muestreo, y los procedimientos de transporte.
d. Estabilice las longitudes de 3 pies (0.91 metros) físicamente utilizando un material de fundición no reactivo (e.g., epoxi) para llenar el espacio anular entre el núcleo y el forro. Alternativamente, la corona circular puede llenarse con fluido no reactivo con el fin de prevenir evaporación. Como precaución mínima, las longitudes de 3 pies (0.91 metros) deben sellarse con capacetes estándar.
e. Pase las longitudes a cajas marcadas y colóquelos sobre cojines para su transporte al laboratorio. Utilice tornillos para pegar las tapas de madera a las cajas de núcleos para evitar daños en los núcleos causados por el martilleo.
2.2.5 Núcleo retenida a presión Los sacanúcleos retenida a presión son diseñados para obtener la mejor saturación de fluido posible in situ. Este método de sondaje ofrece una alternativa al sacanúcleos convencional que pierde presión a su recobro a la superficie. Para permitir la medición de saturaciones de fluido en el laboratorio, el núcleo debe pasar por un manejo extensivo. El equipo del

sacanúcleos se coloca en una unidad especial de mantenimiento de núcleos y el fluido de perforación es descargada de la corona circular entre el cilindro interior y exterior utilizando un fluido no reactivo mientras se mantiene la contrapresión apropiada en todo el sistema. Luego se coloca todo el equipo del sacanúcleos en una caja-congeladora llena de hielo seco (2.5.2.2). Hasta este punto en el manejo de un núcleo retenida a presión, el trabajo debe ser realizado por personal de la empresa de servicios capacitado a sus especificaciones. Los siguientes procedimientos realizados sobre núcleos congelados deben ser supervisados por la empresa operadora: a. Remover el cilindro interior retenida a presión del
hielo, colocar en una cubierta de seguridad, y cortar en los largos deseados.
b. Colocar cada sección nuevamente en el hielo, mientras se va cortando. Tener cuidado de asegurar que las secciones son extendidas de tal manera que la parte superior y la parte inferior y la posición en la sección del núcleo se pueda identificar claramente (ver 2.2.3).
c. Levantar un extremo de la sección del núcleo a la vez y colocar la tapa del núcleo con etiqueta a cada extremo, asegurándolo con una abrazadera de manguera (ver 2.2.4).
d. Las etiquetas deben ser marcados con el nombre de la empresa, presión recuperada, ubicación legal, margen de profundidad del núcleo, y la profundidad del núcleo procesada.
e. Colocar las secciones del núcleo procesadas en cajas de embarque aisladas y empacar con hielo seco. Marcar la caja con los números y las profundidades del núcleo junto con la información sobre la empresa, ubicación, y embarque. Si las cajas de embarque aisladas están en tránsito por mas de 24 horas, puede ser necesario empacar hielo seco adicional.
2.2.6 Núcleo Esponjoso El equipo de sondaje esponjoso está diseñado para mejorar la medición de las saturaciones de fluido en yacimientos. Cuando el núcleo es llevado a la superficie, el fluido que se perdería de lo contrario por expulsión debido a la reducción en presión es atrapado por una esponja de poliuretano absorbente que rodea el núcleo. El equipo de sondaje consiste de 6 largos pre-cortados de 5 pies (1.52 metros) de forro de aluminio dentro de un cilindro interior estándar de acero. El núcleo esponjoso es, por lo general manejado de acuerdo con los procedimientos establecidos en 2.2.4. En la mayoría de los casos, el forro debe ser sacado del equipo de sondaje por bombeo. El forro pre-cortado es almacenado y preservado en tubos de embarque de PVC llenos de un fluido no reactivo adecuado. El tubo de embarque de PVC es sellado con una tapa rígida y una tapa de caucho de expansión de gas. Para propósitos de orientación, cada sección del forro es biselado por un extremo. Una vez el núcleo esponjoso llegue al laboratorio, este es abierto con fresa y tanto el núcleo y la esponja son
extraídos de todos los fluidos del yacimiento (ver 4.3.4). 2.2.7 Sondaje de Paredes Laterales Wireline Los núcleos de paredes laterales wireline son extraídos de la formación por varios medios. El sondaje de paredes laterales por percusión implica el uso de una carga explosiva que impulsa un proyectil hueco en la formación. Debido a las fuerzas producidas por la entrada de la bala en la formación, ocurre la compactación, fractura, y desorden de los granos de la roca. Se debe tener mucho cuidado cuando se maneja este material de núcleos. De manera alternativa, los núcleos de paredes laterales wireline pueden perforarse mecánicamente de la formación con una broca giratoria. Se minimizan los daños con esta técnica. Sin embargo, este método no es factible en todos los tipos de roca. Si la muestra se parte durante la remoción de la herramienta de sondaje, esta debe armarse por partes y se debe anotar cualquier daño. También existen otros aparatos de muestreo de paredes laterales que implican el uso de sacamuestras activados a presión. Cuando se utilizan técnicas de sondaje de paredes laterales, las muestras frágiles deben colocarse en tarros de plástico o de vidrio con tapas metálicas. No se debe colocar papel u otros materiales capaces de absorber líquidos dentro de los tarros para actuar como material de amortiguación para las muestras. Los laminados plásticos sellados a calor son una técnica aceptable de preservación para las muestras de paredes laterales perforadas. Todas las muestras deben estabilizarse y amortiguarse durante su transporte al laboratorio (ver 2.5.2.1) y deben ser marcadas con precisión (ver 2.2.3). 2.2.8 Sondaje Continuo Recobrado de Wireline En las operaciones de sondaje continuas recobradas de wireline (WRC), el sacanúcleos es recobrado mientras la cadena del taladro permanece dentro de la perforación. El tiempo de viaje es reducido y consecuentemente, el método puede ser menos costoso que el sondaje convencional. Típicamente, se sondean largos intervalos verticales continuamente, y ciertos pozos pueden ser sondeados desde la superficie hasta su profundidad completa. 2.2.8.1 Marcado de Profundidad en Núcleos WRC Una buena comunicación entre el personal de recobro de núcleos y el perforador en el campo es necesaria para el marcado exacto de profundidades en núcleos WRC. En el sondaje convencional, el perforador suministra la profundidad superior e inferior del intervalo sondeado. Esto puede causar alguna confusión en la asignación de las profundidades del núcleo, porque los núcleos convencionales son relacionados desde la parte superior del intervalo sondeado. Como el sondaje WRC es continuo, es mejor utilizar la sección inferior de la parte sondeada previa como la sección superior de la parte subsecuente. Se sugiere una supervisión para verificar un posible relleno entre secciones para minimizar errores causados por esto. El mantener

procedimientos exactos de contabilidad de núcleos en la forma de una hoja electrónica en tiempo real debe minimizar el potencial de errores en la determinación de la parte superior del siguiente intervalo. La información en la hoja electrónica debe incluir el número de sección del núcleo, la parte superior del intervalo sondeado, la parte inferior del intervalo sondeado, el porcentaje de recobro, y una columna que indique el punto desde el cual se empezó a marcar el núcleo. Todo el núcleo WRC debe ser marcado y etiquetado de acuerdo con 2.2.3, con la excepción que la asignación de profundidad debe modificarse de acuerdo con los siguientes procedimientos. Los procedimientos convencionales para asignar las profundidades del núcleo no siempre son apropiados para el núcleo WRC. En el sondaje WRC, el núcleo metido en una sección puede sacarse en la siguiente sección, así que la verdadera profundidad del núcleo actual puede estar en el intervalo previamente sondeada. El recobro de "núcleo metido" también significa que el volumen del núcleo sobre-recobrado es más grande que el comúnmente encontrado en el sondaje convencional. Dados los volúmenes más grandes de núcleo, es más importante utilizar un procedimiento de marcado de núcleos que evite asignar la misma profundidad a mas de una parte del núcleo. Si ocurre un sobre-recobrado, el marcado de la profundidad del núcleo puede comenzar en la parte inferior del núcleo, y la profundidad de esa parte del núcleo asignado como el fondo se reporta por el perforador. El marcado de núcleos luego debe moverse de abajo hacia arriba. Si se obtiene un recobro del 100 por ciento o más en la sección previa, las asignaciones de profundidad deben seguir los procedimientos convencionales, con el marcado de profundidad empezando en la parte superior. Para un recobro deficiente, se debe utilizar la siguiente ecuación para contar el núcleo faltante: Núcleo faltante = Profundidad Perforada - Longitud del Núcleo = [CDD - PDD] - [CBL + PC - VOID] (1) Donde: MC = longitud del núcleo faltante, pies (metros) CDD= profundidad del perforador actual, pies (metros) PDD=profundidad del perforador anterior, pies (metros) CBL = longitud del sacanúcleos, pies (metros) PC = núcleo prominente VOID = vacío en el sacanúcleos, pies (metros) En un recobro deficiente, las profundidades del núcleo deben marcarse de arriba hacia abajo, asignando recobro deficiente a la sección inferior del intervalo sondeado. 2.2.8.2 Método Alternativo para el Marcado de Profundidad en Núcleos WRC Una alternativa al método mencionado en 2.2.8.1 es asignar profundidades de la misma manera que los núcleos convencionales (ver 2.2.3). En cuanto a los núcleos convencionales, puede haber un recobro deficiente o un sobre-recobro. Empezando desde
arriba, marque el núcleo con marcas de profundidad cada pie hasta el final del núcleo. No se debe hacer ningún intento para resolver los intervalos de recobro deficiente o sobre-recobro antes del ajuste núcleo-a-registro. Si las profundidades están consistentemente marcadas desde la parte superior de cada sección y cada sección tiene un número único, puede haber la misma "profundidad" en dos núcleos consecutivos, pero serán distinguidos por su número de sección. Si se intenta ajustar un sobre-recobro en el campo, esto puede resultar en una tremenda confusión con los mismos núcleos etiquetados muchas veces. 2.2.8.3 Lavado de Núcleos WRC En cuanto al sondaje convencional, se debe tener cuidado para evitar daños en la roca; e.g., lavándola con fluidos no apropiados. Si existe alguna ambigüedad acerca de los efectos dañinos, elija la precaución, por ejemplo, evitando el lavado del núcleo. 2.2.8.4 Análisis de los Núcleos WRC El punto hasta el cual se analiza el núcleo WRC varia en cada operador. Los procedimientos de análisis de núcleos WRC difieren significativamente de aquellos utilizados para núcleos convencionales. En las operaciones de sondaje convencional, el núcleo es devuelto a un laboratorio para su análisis y, en algún momento futuro, se ajustan los resultados del análisis del núcleo para las profundidades perforadas. Con el núcleo WRC, mucho del análisis es realizado en el campo, y en algunos casos, el núcleo no puede archivarse nunca. Típicamente, un geólogo de campo debe describir la sección con los detalles litológicos suficientes para permitir el ajuste núcleo-a-registro y su correlación. El geólogo debe anotar las muestras de hidrocarburo, intervalos porosos, y los cambios en facies. Algunos operadores utilizan laboratorios móviles a través de los cuales realizan análisis bastante sofisticados del núcleo WRC, incluyendo la exploración con gamma, fotografías ultravioletas, cromatografía de gas, y mediciones de porosidad, densidad, susceptibilidad magnética, mineralogía y propiedades acústicas. 2.2.9 Sacanúcleos Orientado La orientación del sacanúcleos se logra utilizando instrumentos electrónicos de disparos múltiples y equipos especializados de trazado de núcleos. Alternativamente, la signatura de roca paleomagnética puede utilizarse para propósitos de orientación de núcleos. Se deben seguir procedimientos estrictos de manejo para asegurar que los datos de orientación son correlacionadas positivamente con la profundidad y apareados con la sección apropiada de material de núcleo. Esto es particularmente crítico en unidades de roca fracturadas donde comúnmente se utilizan cilindros interiores y forros desechables. 2.3 MUESTREO Y ANÁLISIS EN CAMPO 2.3.1 Generalidades

En general, no se recomienda el muestreo del material de núcleos recobrado en el pozo. Si es necesario tomar muestras inmediatamente, se deben tomar precauciones para minimizar el tiempo de exposición del núcleo. El muestreo debe ser rápido, eficiente, y realizado de acuerdo con las prácticas correctas de seguridad. Siempre obtenga muestras utilizando el método menos dañino o menos contaminante disponible. Todo el núcleo debe ser guardado en todos casos. Algunas razones específicas para el muestreo en el campo incluyen, pero no están limitadas a a) muestreo de recortes para la descripción litológica y/o determinación mineralógica, b) medición de las propiedades básicas de la roca, c) pruebas de compatibilidad-terminación de fluidos, d) estudios de humectabilidad, e) observación de fluorescencia/corte del crudo, f) mediciones de recobro de tensión inelástica, y g) estudios de desorción de metano para el análisis de carbón. Todas las secciones removidas del intervalo sondeado deben ser registradas en la hoja de datos de campo (ver 2.3.3) y representadas físicamente en las secciones de núcleo continuas utilizando espaciadores rígidos. La muestra removida debe ser preservada, marcada, y empacada de una manera consistente con la prueba deseada. Los datos adicionales pertinentes deben acompañar las muestras al laboratorio o deben estar disponibles para análisis en el pozo. Otras técnicas especiales de análisis de núcleos en el pozo son posibles si existen las instalaciones móviles adecuadas para realizar las pruebas bajo condiciones controladas. El martilleo puede dañar el núcleo y puede imposibilitar el análisis de núcleos. Si es posible, las muestras de recortes deben tomarse desde las roturas que ocurren naturalmente en el núcleo o con una sierra de guarnición de precisión. El tamaño de la muestra debe mantenerse en el mínimo necesario para realizar el análisis deseado. Coloque las muestras en bolsas y preserve la saturación de fluido con una técnica de preservación apropiada (ver 2.5). Si se va a realizar una revisión detallada de núcleos en el pozo, el muestreo y el manejo del núcleo deben hacerse rápidamente, y solo si la revisión es crítica para el éxito del objetivo del pozo. 2.3.2 Transporte y Logística El método de transporte debe ser práctico y debe ofrecer protección contra daños por cambios ambientales, vibraciones mecánicas, y el maltrato. Otros factores importantes a considerar cuando se elige el modo de transporte incluyen a) distancia del pozo al laboratorio, b) condiciones y terreno en tierra y mar adentro, c) competencia del material del núcleo, d) condiciones del clima, e) tipo de preservación o empaque, y f) costo. En todos los casos, se deben tomar precauciones para estabilizar el material del núcleo sin riesgos. En el transporte aéreo, puede ser que el depósito de almacenamiento no esté presurizado, y este puede ser un factor en la preservación del núcleo. No apile los núcleos de tal manera que se dañe el material del núcleo. Se deben tener cuidado con las empresas transportadoras comerciales que no están acostumbradas a transportar materiales frágiles. Por
razones de seguridad, puede ser necesario tratar a los núcleos empacados en hielo seco como "productos químicos" para propósitos de transporte. En todos los métodos de transporte, una carta de remisión o un formulario de documentación de contenidos con la información de embarque pertinente debe acompañar el envío. Se debe enviar una copia separada de esta carta al recipiente vía correo o fax. Se deben seguir todos los reglamentos aplicables del Departamento de Transportes de EE.UU. en el envío de materiales de núcleos. Cuando se utilizan cajas estándares para núcleos, estas pueden ser paletizadas, atadas y enviadas así mismo. Los núcleos, especialmente aquellos de materiales no consolidados, pueden ser congelados o refrigerados en el campo para preservación y estabilización durante su transporte y almacenamiento. Si se congelan, los núcleos deben ser completamente congelados antes del envío para evitar daños mecánicos. El núcleo congelado por lo general es empacado en recipientes aislados y empacados con hielo seco. Los núcleos refrigerados usualmente son enviados en unidades refrigeradoras independientes. Un aparato para supervisar y registrar la temperatura debe acompañar el núcleo para asegurar que se mantengan las condiciones deseadas durante su transporte. 2.3.3 Hoja de Datos Se debe conseguir una hoja de datos adecuada y esta debe ser diligenciada por el ingeniero o el geólogo del pozo, para proporcionar un registro mas completo de las condiciones del sondaje. Esta información será valiosa para la cualificación de la interpretación de los datos de análisis del núcleo. Además, este registro puede implicar que se tenga que realizar ciertas pruebas adicionales para complementar las pruebas básicas, o que otras pruebas no producirían datos significativos. Esto resultará en el análisis más útil en el menor tiempo con menor costo. Las Figuras 2-2 y 2-3 son formularios ejemplos, y se recomienda el uso de estos u otros similares. Es importante tener tantos datos pertinentes sean posibles para acompañar el material de núcleos. La siguiente es una lista de información deseable: a. Identificación del pozo, número API del pozo,
elevación, números y contactos del vendedor, como también sus teléfonos, números de fax y direcciones.
b. Tipo de fluido de perforación, contenidos, y datos medidos.
c. Tipo de núcleo y equipos utilizados. d. La(s) formacion(es) sondeadas, con la
profundidad del perforador superior e inferior. e. Indicación de la información crítica de sondaje y
cualquier nota pertinente, i.e., tiempo total de sondaje/viaje, dificultades, y recobro.
f. Salinidad de la formación de agua y los datos del fluido de producción.
g. Pautas de preservación. Tiempo de exposición. h. Análisis solicitado. i. Registro de sondaje y registros de perforación. j. Una descripción del núcleo. k. Registros de pozo y registros del lodo.

2.4 TIPOS DE ROCA Y CONDICIONES ESPECIALES DE MANEJO
2.4.1 Generalidades El término "tipo de roca" se utiliza para describir las características que distinguen el material del núcleo. Este puede referirse al grado de consolidación, la presencia de fracturas o vugs, composición (esquisto), o propiedades físicas (e.g., baja permeabilidad) de la roca. Las descripciones geológicas de la roca son más complejas y se han trazado esquemas de clasificación para categorizar tipos de roca específicos con respecto a textura, tipo de cementación, tamaño de grano, etc. Se deben tener en cuenta muchas consideraciones especiales cuando se diseña un programa para el manejo de núcleos en el campo. Los párrafos 2.4.2 a 2.4.13 incluyen las pautas generales para varios tipos de roca. 2.4.2 Roca Consolidada Las rocas consolidadas son duras como resultado de cimentación. No necesitan tratamiento especial en el pozo. La cimentación en rocas es definida como el proceso de precipitación de materiales que consolidan alrededor de las superficies del grano sólido. Las rocas pueden describirse como consolidadas, mal consolidadas (friable), o no consolidada, dependiendo del grado de compactación y cimentación. Las rocas consolidadas comunes incluyen caliza, dolomita, arenisca y una variedad de cuarzo. 2.4.3 Roca No Consolidada Las rocas no consolidadas tienen poco cemento, o no lo tienen y son esencialmente sedimentos compactados. Las rocas mal consolidadas tienen menos cemento pero no suficiente para endurecerlas. Es menor sondear estas rocas utilizando un forro en el cilindro interior o un cilindro interior desechable (ver 2.2.4). Se debe tener cuidado para prevenir el desmoronamiento del núcleo. Esto incluye el asegurar que el núcleo es llevado a la superficie, extendido suavemente, y preservado de tal manera que sobrevivirá el transporte (ver 2.5.2). 2.4.4 Roca No Consolidada - Aceite Liviano y Gas Es crítico preservar núcleos no consolidados que contienen aceite liviano de una manera eficiente y apropiada. Se deben evitar los movimientos innecesarios del núcleo. Los dos métodos comúnmente utilizados para preservar este tipo de roca comprenden métodos ambientales, tales como la congelación o refrigeración y estabilización mecánica con epoxi, resina de espuma, etc. El núcleo no consolidado que contiene aceite liviano es susceptible a pérdidas significativas de fluido durante el manejo en superficie. Como es el caso con la mayoría de los tipos de roca, cuando el núcleo ha llegado a la superficie, ya ha pasado por un desahogo de esfuerzo mecánico debido a la remoción de la sobrecarga de presión y ha tenido diferentes cantidades de expansión de gas cuando se pierde la presión de
poros interna. El grado a la cual estos efectos alterarán al núcleo depende de la profundidad, la presión del yacimiento, gravedad del aceite, propiedades del fluido, tipo de sedimento, y procedimientos de sondaje. Se debe tener cuidado para asegurar que no se acumule presión dentro del sacanúcleos durante el manejo. El forro del sacanúcleos o el cilindro interior desechable pueden perforarse previamente con orificios [1/8 pulgada (3.18 milímetros) de diámetro] para evitar la acumulación de presión. Los intervalos sondeados deben ser limitados en longitud para prevenir posibles daños en la roca bajo su propio peso. Mientras se sube el núcleo a través de los 500 pies superiores (152 metros) del pozo, el sacanúcleos debe sacarse lentamente para minimizar la probabilidad de desbaratar la roca y causar daños en el núcleo. Si se utiliza la congelación para estabilizar materiales no consolidados, el núcleo no debe ser transportado hasta que no esté completamente congelado, porque la congelación parcial puede causar daños estructurales en el núcleo (ver 2.5.2.2). Se recomienda llenar la corona circular entre el cilindro interior desechable o el forro y el núcleo con fluido para sondaje para estabilizar cuando no se utiliza epoxi o un material de fijación permanente. Sin embargo, este procedimiento puede alterar la saturación de fluido de la roca y sus características de humectabilidad. Cuando se utiliza epoxi, resina, o la inyección de espuma, el fluido de perforación debe ser completamente desalojado o drenado de la corona circular. El material de moldeo debe ajustarse y encajarse a la superficie del núcleo. 2.4.5 Roca No Consolidada - Aceite Viscoso La dificultad más grande en el manejo de rocas no consolidadas que contienen aceite viscoso es la prevención o la minimización de la expansión de núcleo retardada. La expansión es el resultado de la lenta emisión de gas desde el aceite viscoso, sin posibilidad de un drenaje de corto plazo debido a la baja movilidad del gas. La dilatación de la roca puede continuar fácilmente hasta que la fase de gas se vuelva continua, y esto puede requerir la expansión volumétrica de mas de 6 al 8 por ciento. En los forros, las areniscas de aceite viscoso no consolidadas se expandirán radialmente para llenar el espacio anular desocupado. Una vez el núcleo dilatado esté ajustado en el forro, la emisión adicional de gas puede causar una acción del pistón, causando que el núcleo se salga de los extremos de las secciones del forro resultando en sobre-recobros hasta del 5 por ciento. Es tentador sencillamente recortar el núcleo que sobresale y descartarlo, pero esto debe evitarse. El material que sobresale se mantiene en el extremo del forro del cual salió, y las tapas plásticas pueden ayudar para mantenerlo retenido. El mejoramiento de la calidad del núcleo en areniscas de aceite viscoso no consolidadas requiere las siguientes consideraciones: a. Proporcionar restricción mecánica a la
expansión. b. Proporcionar un medio para permitir el drenaje
del gas.

c. Proporcionar resistencia mecánica al núcleo. El ítem a comprende el uso de forros que tienen un diámetro interno ligeramente más grande [1/8 pulgada (3.18 milímetros)] que la broca del núcleo, para reducir la expansión radial. Durante el manejo y la preservación, se deben evitar el doblado del forro, calentamiento del núcleo, y exposición prolongada de los extremos del forro. La restricción axil ayudará a reducir la tendencia a salirse del núcleo. Esta restricción puede proporcionarse de diferentes maneras, incluyendo: a. Tapas de forro rígidas en lugar de tapas de
caucho, con las tapas engrapadas al forro en diferentes puntos, y aseguradas con abrazaderas roscadas .
b. Cortar segmentos de forro en largos exactos y colocarlos en una caja para núcleos fuerte para que los extremos de la caja proporcionen una restricción axil. Alternativamente, los núcleos pueden ser acuñados en las cajas con tablas de madera cortadas.
c. También se pueden utilizar técnicas especiales de manejo trazados por diferentes operadores, incluyendo cilindros de almacenamiento especiales o métodos de retención axil.
El gas es emitido del aceite viscoso lentamente, y continuará durante meses. Se recomienda el uso de forros pre-perforados en todas las áreas para reducir el recorrido del flujo de gas y eliminar los efectos de pistón. Los orificios pre-perforados [1/8 pulgada (3.18 milímetros) en diámetro] deben espaciarse a menos del radio del forro. No se recomienda el perforado posterior de los forros como alternativa aceptable porque esto prolonga el manejo del núcleo y puede resultar en daños. Si es necesario, el forro y los segmentos del núcleo pueden colocarse en cilindros y pueden volverse a presurizar con un gas inerte (N2) para demorar o parar la emisión de gas, y para evitar oxidación. La congelación de núcleos de aceite viscoso no consolidados puede ser necesaria, aunque en general, la congelación no se entiende muy bien. La congelación tiene los siguientes efectos: a) reducción de la velocidad de emisión y el volumen de gas, b) aumento de viscosidad del aceite lo cual restringe la expansión, y c) congelación del agua intersticial lo cual da al núcleo algo de esfuerzo mecánico para restringir la expansión y las fracturas. Como el agua de poros es por lo general salina, se tendrá que reducir la temperatura a menos de -40ºF (-40ºC) para asegurar todos los beneficios mecánicos (ver 2.5.2.2). El manejo de núcleos durante transporte y almacenamiento para materiales no consolidados que contienen aceite viscoso debe mantener la restricción mecánica y baja temperatura. Cuando se preparan secciones de núcleo para análisis, se debe permitir que la temperatura se aumente lentamente, para que el gas emitido se pueda disipar. Se debe mantener la restricción mecánica totalmente hasta que se equilibre el núcleo, un proceso que puede demorarse semanas por la alta viscosidad del aceite y la baja permeabilidad relativa.
2.4.6 Carbonatos "Vuggy" Los vugs grandes pueden debilitar el material del núcleo y causar dificultades en el recobro. En muchos casos, el recobro del núcleo es reducido en intervalos "vuggy" friables. Se pueden utilizar métodos estándares para la preservación de núcleos consolidados en este tipo de roca (ver 2.5). 2.4.7 Evaporados Las rocas salinas son por lo general bastante competentes y, excepto por su solubilidad, pueden considerarse rocas consolidadas. El núcleo que contenga sales en secuencias continuas o como rellenos de vugs y fracturas no debe lavarse con agua dulce bajo ninguna circunstancia. Como las propiedades físicas de las rocas salinas pueden ser alteradas por pequeños cambios en el contenido de humedad, los núcleos que contienen sales deben limpiarse inmediatamente hasta lograr un estado seco y preservarse. El transporte y el almacenamiento de núcleos que contienen sales siempre debe realizarse manteniendo la naturaleza soluble del material en cuestión. Los núcleos de evaporados, anhídridos, yeso y calcita no presentan problemas especiales de manejo. 2.4.8 Roca Fracturada Muchas rocas de yacimientos son naturalmente fracturadas. Se recomienda el uso de cilindros interiores desechables o forros de aluminio o fibra de vidrio para el sondaje de roca fracturada (ver 1.6 y 2.2.4). Un núcleo orientado puede ser útil para determinar la dirección de fractura y la dirección de esfuerzo in situ (ver 1.6 y 2.2.4). 2.4.9 Rocas Ricas en Minerales de Arcilla Puede haber minerales de arcilla en pequeñas cantidades en las rocas, y a pesar de esto tener un impacto profundo sobre las propiedades de las rocas. Algunas de las principales preocupaciones en las rocas que contienen minerales de arcilla incluyen: a. La presencia de esmectita (un mineral de arcilla
que se dilata), aunque en muy pequeñas cantidades (1 por ciento), tiene importancia en el manejo de núcleos por el potencial de dilatación, la alta capacidad de intercambio de cationes, y el potencial de succión osmótica.
b. Los minerales de arcilla intersticiales pueden ser movilizados físicamente por cambios en contenido de fluido, química, o alteración mecánica, resultando en el bloqueo de secciones de paso o cambios en las características de humectabilidad de la superficie de los poros u otros cambios físicos.
c. Los minerales de arcilla en contacto con sus fluidos naturales están el equilibrio termodinámico, y la exposición a otros fluidos alterará esto resultando en cambios en la actividad de minerales de arcilla, cationes intercambiables, y cambios consecuentes en comportamiento mecánico y de flujo.

d. Los esquistos y areniscas esmécticos pueden dilatarse cuando se remueve el esfuerzo restrictivo si hay agua libre, aun si el agua libre tiene propiedades idénticas a los fluidos intersticiales.
Los fluidos sobrantes o el lodo debe limpiarse inmediatamente de los núcleos de materiales esmécticos y los materiales ricos en minerales de arcilla, seguido por una preservación inmediata (ver 2.5.2). 2.4.10 Esquisto Además de la recomendación para las rocas que contienen minerales de arcilla (ver 2.4.9), existen asuntos especiales relacionados al manejo de esquistos altamente rajadizos. Estos materiales tienen planos de rajadura de baja resistencia que pueden partirse espontáneamente, aun si se maneja el núcleo con mucho cuidado. Una vez un núcleo de esquisto rajadizo se haya partido, puede ser imposible obtener muestras lo suficientemente grandes para un análisis de núcleo. Se recomienda que los núcleos de esquisto rajadizo se manejen de la siguiente manera: a. Evite el manejo o el movimiento excesivo del
núcleo. b. Remueva el agua sobrante. c. Preserve inmediatamente para parar la
desecación. d. Se pueden envolver los segmentos del núcleo
perpendiculares a los planos rajadizos con cinta de enmascarar o de fibra de vidrio para reducir la partición adicional. Alternativamente, se pueden utilizar plásticos que se encogen con el calor.
Los esquistos tienen baja permeabilidad y la lenta transferencia interna de humedad ocurrirá inevitablemente entre los estratos de diferente mineralogía y textura durante el almacenamiento de largo plazo. Si el núcleo está totalmente sin restringir, esto puede resultar en una partición demorada aun sin desecación o manejo. Los esquistos rajadizos son excepcionalmente sensibles a los cambios en temperatura, y deben mantenerse en una temperatura constante durante el transporte y almacenamiento. No se debe permitir la congelación de esquistos porque esto resulta en microfisuras masivas y un movimiento de la humedad interna. Los esquistos de aceite con volúmenes químicos orgánicos de mas del 20 por ciento son sensibles a la temperatura y la oxidación y deben ser preservados con particular velocidad si se requiere un análisis detallado. Los esquistos de aceite más fuertes soportados por matrices son típicamente más cuarzosos y con volúmenes químicos orgánicos de menos del 20 por ciento y en general no requieren un manejo especial. 2.4.11 Roca de Baja Permeabilidad La evaporación de fluidos, un problema en todos los materiales de núcleos, es una dificultad especial en los núcleos de baja permeabilidad y de baja porosidad
donde el cambio porcentual en saturación puede ser mucho más grande para el mismo volumen de fluido evaporado. El periodo de tiempo antes de proteger el núcleo de evaporación es crítico para estas muestras. La presencia de minerales de arcilla puede causar daños por evaporación irreversible en algunas muestras (ver 2.4.9). 2.4.12 Carbón El contenido de gas, el comportamiento de sorción del gas, la permeabilidad, permeabilidad relativa, análisis de clivajes y fracturas, composición del núcleo, y comportamiento mecánico in situ son los principales intereses en el análisis de carbón para la producción de metano en estratos de carbón. Se pueden realizar estudios de desorción de gas en el pozo con cajas de desorción. Los procedimientos para el manejo de núcleos de carbón deben incluir instrucciones para estos estudios especiales. Se han utilizado sacanúcleos recobrados por wireline, sacanúcleos con cilindros interiores desechables o forros, y los sacanúcleos retenidos a presión para cortar núcleos de carbón. El contenido de gas y la velocidad de desorción de gas son medidos comúnmente con métodos de desorción utilizando núcleos convencionales, núcleos de paredes laterales mecánicamente perforados, núcleos continuos recobrados por wireline, o recortes de perforación. Los segmentos de carbón son sellados en una caja y conservados isotérmicamente mientras se mide el volumen de gas emitido de la muestra. La medición del contenido de gas por desorción de caja requiere un estimado del volumen de gas perdido mientras se lleva el núcleo a la superficie y antes de sellar las muestras en la caja. Se debe normalizar el contenido de gas a una base material libre de minerales . Como el gas emitido no va a ser 100 por ciento metano, se debe analizar la composición del gas. La tecnología de núcleos retenidos a presión, la cual no requiere un estimado de gas perdido, ofrece un medio más exacto para determinar el contenido total de gas in situ de un estrato de carbón. El volumen de gas emitido del núcleo de carbón en el cilindro de presión se mide como función de tiempo, temperatura, y presión. Si se utilizan métodos de retención a presión para el sondaje de carbón, se incorpora un detector especial de temperatura interna, y no se lleva a cabo la purga de fluidos (ver 2.2.5). En un procedimiento, se lleva todo el cilindro retenido a presión nuevamente a la temperatura del fondo de la perforación y se permite que este se equilibre durante varios días antes de permitir que la presión se disipe y se desarme el aparato. Durante la fase de disipación de temperatura y presión, los cambios deben ser graduales para minimizar tanto la magnitud de la gradiente de presión como la gradiente de temperatura. La primera puede causar que el carbón se fracture internamente, mientras un enfriamiento rápido puede causar la abertura de grietas tractivas en la parte exterior del núcleo de carbón. Estas pueden afectar tanto la permeabilidad como el comportamiento mecánico. Alternativamente, el núcleo de carbón retenido a presión puede ser cortado en secciones de un pie de largo y colocado en cajas

de desorción para la medición del contenido de gas y la velocidad de desorción de gas. Se debe manejar el núcleo de carbón con cuidado en el pozo por su naturaleza heterogénea, y porque el metano encontrado en los pequeños poros dentro del núcleo está bajo presión. Un tratamiento negligente, el doblado del forro, o un golpe fuerte en el sacanúcleos puede causar la fragmentación del núcleo, convirtiendo el núcleo en algo inútil para el análisis. La presión de gas interna puede aumentar esta tendencia de deteriorarse, y se requiere tiempo para permitir la disipación de gas. Los fluidos de sondaje que están en contacto con el carbón para pruebas mecánicas o del flujo de fluido no deben contener materiales capaces de alterar la estructura del carbón. El carbón fresco se oxida cuando es expuesto al aire, potencialmente cambiando sus propiedades de superficie y sus características de sorción. Se debe minimizar el tiempo de exposición al aire, y el manejo especial puede incluir cajas para recibir segmentos de núcleo de carbón, seguido por la purga de los cilindros con un gas inerte o metano. Se deben utilizar lapiceros amarillos o pintura para marcar la superficie de las longitudes del núcleo. Como es el caso con cualquier otro núcleo, se aconseja la preservación de humedad y la minimización de tiempo de exposición. No se recomienda la congelación del núcleo de carbón, ni se considera necesaria. 2.4.13 Diatomita Las diatomitas son por lo general rocas de alta porosidad y baja permeabilidad compuestas de fases de cuarzo opalino con cantidades variables de material detrítico. Las diatomitas son sondeadas con cilindros interiores desechables o forros (ver 2.2.4). La diatomita puede ser preservado por medios ambientales, envoltura, etc. No se recomienda la congelación de diatomita. Se debe controlar la temperatura para mantener una temperatura constante de 35 a 40ºF (1.67 a 4.44ºC) durante las operaciones en el pozo y de transporte. 2.5 PRESERVACIÓN DE NÚCLEOS PARA
ANÁLISIS 2.5.1 Generalidades La preservación de un núcleo es un intento para mantenerlo, antes de su análisis, en la misma condición que existió en el momento de su remoción del sacanúcleos. En el proceso de cortar un núcleo, recobrarlo, y llevarlo a la superficie, el contenido de fluido de la roca es alterada por los cambios inevitables en presión, temperatura, etc. los métodos de núcleos retenidos a presión intentan minimizar estos efectos (ver 2.2.5). Las prácticas negligentes o incorrectas en el manejo y la preservación causan alteraciones adicionales en el núcleo y sus fluidos, así produciendo un núcleo aun menos representativo de la formación. La preservación y el empaque de núcleos pueden variar dependiendo de las pruebas requeridas, la cantidad de tiempo antes de pruebas, y la posibilidad
de realizar pruebas en el pozo. Si las muestras del núcleo han de utilizarse para determinar la saturación de fluidos o para un análisis de núcleos especial, es necesario preservarlas para su transporte al laboratorio. Se debe evitar la evaporación y la migración de fluidos como también la oxidación dentro de la muestra para obtener un análisis de núcleos confiable. Un objetivo adicional del programa de preservación es el de prevenir la rotura de los núcleos durante su envío y almacenamiento. El núcleo consolidado puede ser lo suficientemente durable para no requerir procedimientos especiales de manejo. Sin embargo, se debe tener cuidado especial con rocas no consolidadas fracturadas, etc. (ver 2.4). No se recomienda el uso de frascos de vidrio desprotegidos, plásticos fácilmente deformables (si no son estabilizados apropiadamente), cartones, recipientes no rígidos, y latas herméticas para propósitos de preservación de núcleos. 2.5.2 Métodos de Preservación de Núcleos No existe un método de preservación mejor que otro. La experiencia puede ayudar a determinar el método mas satisfactorio para el tipo de roca en cuestión. La elección del método dependerá de la composición, grado de consolidación, y las características distintivas de la roca. Por lo tanto, el uso general de un método específico de preservación no aplicará para todos los tipos de roca. Las técnicas requeridas para preservar núcleos para pruebas pueden depender de la cantidad de tiempo de transporte, almacenamiento, y la naturaleza de la prueba a realizar. Alguna variación en el método de preservación puede depender del análisis local de los núcleos o si deben ser preparados para envíos de larga distancia. Los métodos preferidos para preservar núcleos para análisis de laboratorio incluyen uno o mas de los siguientes: a. Estabilización mecánica. b. Preservación ambientalmente controlada
utilizando refrigeración, humedad regulada, o congelación, si es necesario (ver 2.5.2.2).
c. Laminados de plásticos sellados a calor. d. Bolsas plásticas. e. Baños y revestimientos. f. Sellado en cilindros interiores desechables,
forros, y tubos. g. Frascos anaeróbicos. 2.5.2.1 Estabilización Mecánica Todos los tipos de roca deben ser estabilizados mecánicamente antes de enviarlos al laboratorio. Esto es particularmente cierto para rocas no consolidadas (ver 2.4.4 y 2.4.5). El núcleo que ha sido cortado utilizando cilindros interiores con forros plásticos, de fibra de vidrio, o de aluminio o desechables puede ser encapsulado utilizando resina, cera, o espuma para llenar el espacio anular entre el núcleo y la manga. La resina tiene una viscosidad baja y llenará las fracturas finas. Sin embargo, esta es vaciada en la corona circular y no está bajo la presión suficiente para desalojar los fluidos de poros en la roca y por lo tanto no se impregna en el núcleo.

La estabilización mecánica para núcleos bien consolidados también puede ser tan sencillo como es envolver el núcleo en una envoltura de burbujas u otros materiales acolchados adecuados. Todo el material del núcleo debe considerarse frágil y debe ser manejado con cuidado (ver 2.3.2). Se debe tener cuidado para evitar la alteración de núcleos mal consolidados o fracturados antes de la estabilización mecánica. 2.5.2.2 Preservación Ambiental El control de las condiciones ambientales a las cuales el núcleo es sometido por refrigeración o manteniendo un ambiente húmedo pueden ayudar a preservar el núcleo (consultar la información sobre la preparación de núcleos en la Sección 3). La refrigeración de núcleos se usa principalmente para minimizar la evaporación de fluido y para proporcionar una estabilización mecánica. Esta técnica es útil para evitar que el núcleo se seque. Sin embargo, su eficacia está sujeta al tipo de fluido de sondaje y las propiedades de la roca y el fluido del yacimiento. Cuando se refrigera el núcleo, sigue siendo necesario estabilizar la roca mecánicamente para transportarlo al laboratorio (ver 2.5.2.1). Los núcleos preservados por congelación deben ser congelados aplicando hielo seco, nitrógeno líquido, o colocándolos dentro de una unidad congeladora eléctrica. La congelación puede resultar en la migración o la difusión de fluidos dentro de la estructura del núcleo o la rotura del núcleo. La congelación puede causar pérdidas significativas por evaporación a través de la sublimación. Los núcleos no consolidados que han sido congelados pueden ser empacados con una salmuera congelada de 1/4 pulgada (6.35 mm) de grosor (capa de hielo superficial) para reducir el proceso de sublimación. Esta medida es crítica si se utiliza la congelación para almacenamiento de largo plazo. Pueden ocurrir daños estructurales en el núcleo si este se deshidrata mientras está congelado. La práctica de congelar el núcleo es la mas común para rocas no consolidadas (ver 2.4.4 y 2.4.5). Se desconoce el efecto completo de congelación sobre las propiedades petrofísicas del núcleo. La congelación de un núcleo consolidado con agua intersticial no se entiende muy bien. La expansión de los cristales de hielo puede causar daños estructurales irreversibles al núcleo. La congelación puede afectar las propiedades de la roca purgada con agua dulce mas que aquellas purgadas con el líquido salino filtrado de perforación. Estos efectos se disminuirán con la reducción de la saturación de agua. Si es necesario permitir que el núcleo se caliente a la temperatura ambiente antes de pruebas, se debe prevenir la condensación de la humedad de la atmósfera en la superficie del núcleo. El deshielo del núcleo puede causar alguna redistribución de los fluidos dentro de la matriz del núcleo. La saturación de fluido y las propiedades (minerales) del yacimiento también pueden ser preservadas controlando la humedad relativa del ambiente del núcleo con hornos especialmente diseñados. Esta técnica tiene una aplicabilidad muy amplia y es mas efectiva con rocas que contienen minerales de arcilla
sensibles a la humedad y/o agua químicamente confinado en los minerales (ver 2.4.9). 2.5.2.3 Laminados Plásticos Sellados a Calor Existen diferentes laminados plásticos que se pueden sellar a calor. Se puede utilizar papel aluminio o Mylar para agregar rigidez al laminado. El empaque laminado de preservación de núcleos debe actuar como una barrera impenetrable al vapor de agua y gases, y debe ser resistente a la alteración química y degradación por fluidos. Los laminados son fáciles de usar y el proceso de preservación puede ser realizado rápidamente. Se debe tener cuidado para evitar desgarres o perforaciones en el laminado. Se requiere una superficie limpia y plana para alisar el laminado antes de sellar. Todo el núcleo debe ser envuelto previamente y pegado con un plástico durable u otro material para tapar los extremos del núcleo y los bordes afilados. Se debe marcar el segmento empacado con la información del pozo y la profundidad. El proceso de sellado a calor es crítico para el éxito de este método de preservación. El sellador a calor debe fijarse en la temperatura apropiada de acuerdo con las especificaciones del fabricante para obtener un sellado efectivo. Cualquier discontinuidad en el sellado anulará las propiedades barreras del material. Existen algunos laminados en forma tubular que requieren el sellado de dos extremos en lugar de cuatro. El espacio de la parte superior en el paquete de preservación debe ser minimizado. Sin embargo, se debe utilizar suficiente material para prevenir el debilitamiento si el paquete va a ser abierto y sellado nuevamente. En algunos casos, puede ser aconsejable evacuar el espacio de gas donde la pérdida de hidrocarburos livianos no es problema. Un gas inerte tal como el nitrógeno puede cobijar la roca para minimizar la oxidación. Cuando el material del núcleo se desgasifica, el paquete del laminado se inflará. Esto no presentará ningún problema si el paquete está sellado apropiadamente. Si es necesario, se puede tomar muestras del gas emitido directamente con una jeringa de gas estándar y luego se vuelve a sellar el paquete. El paquete de preservación debe ser marcado y mecánicamente estabilizado para su envío (ver 2.5.2.1). No se debe someter el paquete de preservación a temperaturas extremas en ningún momento. 2.5.2.4 Bolsas Plásticas Se recomiendan bolsas plásticas únicamente para la preservación de corto plazo. Las muestras de núcleos deben tener un espacio mínimo de aire entre el núcleo y las paredes de la bolsa. La parte sobrante de la bolsa puede ser doblada contra la pared del núcleo y pegada con cinta para asegurar un encaje ajustado. Como siempre, se deben seguir los procedimientos de marcado claro y estabilización apropiada. 2.5.2.5 Baños y Revestimientos Se utilizan baños y revestimientos cuando los núcleos van a ser probados después de pocas horas o días y cuando el material va a ser transportada por largas

distancias. También pueden utilizarse con laminados plásticos para agregar integridad mecánica. CUIDADO: Los núcleos nunca deben ser bañados directamente con cera derretida o material plástico derretido. Todo el núcleo debe ser previamente envuelto en un laminado o una película plástica sellado a calor y papel aluminio antes de bañarse. Todos los segmentos del núcleo deben ser marcados con la información del pozo y la profundidad. El propósito de la envoltura de la película plástica es de prevenir el contacto del núcleo y los fluidos de poros con la envoltura exterior de papel aluminio. Tal contacto puede causar oxidación del papel aluminio y la pérdida de sus propiedades de barrera de humedad y oxígeno. Se deben seguir los siguientes procedimientos con el método de envoltura y baño: a. Prepare una tina de calentamiento para el baño-
revestimiento varias horas antes de preservar el núcleo. Observe todas las precauciones de seguridad. Siga las recomendaciones de manejo del fabricante del baño. El recalentamiento del baño puede dañar la eficacia del revestimiento.
b. Envuelva el núcleo ajustadamente en una película plástica que se amoldará a la superficie de la roca, arrugando los extremos libres. Se aconsejan varias capas de película plástica de alta calidad para prevenir perforaciones.
c. Envuelva el núcleo con varias capas de papel aluminio, arrugando los extremos libres. Evite la perforación de la envoltura de aluminio.
d. Amarre un alambre alrededor del núcleo para formar una manija.
e. Sumerja la muestra del núcleo envuelto en papel aluminio en un material de revestimiento derretido. Una cantidad abundante de revestimiento debe encerrar el núcleo. Se recomienda un revestimiento de 1/8 a 1/4 pulgada (3.18 a 6.35 mm) de grosor. Esto se logra por medio del uso de múltiples sumersiones, permitiendo que cada revestimiento se endurezca antes de la aplicación de material adicional. Se recomienda que el revestimiento se deje endurecer suspendiendo el núcleo en el aire por medio de la manija de alambre.
f. La manija de alambre debe cortarse a ras con el revestimiento. El baño adicional debe aplicarse al extremo del alambre para eliminar un camino para oxidación o evaporación.
El material de revestimiento debe tener las siguientes propiedades: a. Debe ser dimensionalmente estable durante
largos periodos de tiempo. b. No debe reaccionar con aceite o agua y no debe
contener ácidos, aceites, solventes u otros líquidos que puedan exudar cuando se asiente.
c. La permeabilidad a gases, aceites y agua debe ser baja cuando se asiente.
d. Debe tener un bajo punto de fusión, preferiblemente debajo de 200ºF (93.3ºC) máximo y debe tener una viscosidad bastante
baja cuando se derrita. Los puntos de fusión mas altos son aceptables si se minimiza el tiempo de exposición al baño.
e. Cuando se remueva del calor y se exponga a condiciones ambiente, este debe estar seco y se debe acomodar "tack-free" dentro 5 a 15 segundos.
f. Cuando se asiente, este debe ser duro pero manejable, ligeramente elástico pero con buen resistencia a la tracción, y no debe derretirse a temperaturas por debajo de 180ºF (82.2ºC).
Como con todos los métodos de preservación de núcleos utilizados actualmente, la eficacia de largo plazo de baños y revestimientos sigue siendo incierta. 2.5.2.6 Cilindros Interiores Desechables, Forros y Tubos Rígidos Un medio conveniente de preservación de núcleos es posible cuando se utilizan cilindros interiores desechables o forros hechos de plástico, aluminio o fibra de vidrio (ver 2.2.4). El núcleo puede preservarse tal como está sellando los extremos del cilindro interior o el forro cortado. Esto no se recomienda como método de preservación de largo plazo, pero esto permitirá que el núcleo se procese rápidamente y sin equipos especiales. Los orificios en el cilindro interior deben sellarse antes de envío. El núcleo puede transportarse rápidamente al laboratorio para muestreo y pruebas, y si es necesario, es posible la preservación adicional de los recipientes sellados (ver 2.5.2.2). Cuando se utiliza un sacanúcleos convencional, se pueden utilizar tubos de acero, aluminio o plástico con acopladores, tapas o sellos de anillo adecuados para preservar el núcleo. Para utilizarlos eficazmente, el tubo y las piezas de los extremos deben ser no reactivos con el núcleo y sus fluidos. Los segmentos pueden estabilizarse de acuerdo con las pautas establecidas en 2.5.2.1 para minimizar los daños en el envío. 2.5.2.7 Frasco Anaeróbico La inmersión del núcleo en líquido dentro de un frasco anaeróbico puede utilizarse para prevenir la oxidación, evaporación secado durante el manejo del núcleo. El recipiente anaeróbico es un frasco alargado con una tapa sellable, en el cual se puede introducir un líquido y se puede remover el oxígeno libre. El líquido de inmersión debe ser compatible con los fluidos del núcleo y de poros, y debe ser capaz de mantener la humectabilidad actual de la muestra (ver 2.6). Típicamente, los siguientes fluidos son utilizados para inmersión: a. Salmuera de formación desoxigenada o salmuera
de formación sintética con insecticida. b. Crudo. c. Aceite mineral refinado despolarizado. Como siempre, siga todas las precauciones de seguridad cuando se utilicen frascos anaeróbicos para la preservación de muestras de un yacimiento.

2.6 RECOMENDACIONES PARA EL MANEJO DE NÚCLEOS PARA PRESERVAR LA HUMECTABILIDAD Una alteración de humectabilidad puede ocurrir durante el sondaje, el tratamiento de núcleos en el pozo, o durante el periodo de almacenamiento antes de realizar las mediciones en el laboratorio. Aquí solo hablamos de tratamiento en el pozo, aunque las medidas recomendadas únicamente pueden ser exitosas para preservar la humectabilidad si se toman las precauciones adecuadas en cada paso del proceso de recobro y análisis de núcleos. La validez de muchas pruebas de laboratorio depende del mantenimiento o el restablecimiento de condiciones de humectación del yacimiento. Sin embargo, muy rara vez hay un punto de referencia inicial para definir la humectabilidad in situ. Las prácticas recomendadas aquí pretenden proporcionar medidas de humectabilidad tan pronto sea posible después de la remoción del núcleo de la formación para que se pueda establecer un punto de referencia para la humectabilidad. Los núcleos no deben ser expuestos al aire por mas tiempo del necesario. Las pruebas de humectabilidad deben realizarse en el campo. Una simple observación de la imbibición de gotas de agua y aceite colocadas sobre la superficie del núcleo debe registrarse rutinariamente. En muchos casos, este será el alcance de las pruebas de humectabilidad en el campo, pero se pueden iniciar pruebas mucho mas completas en el campo también. Se requerirán algunas instalaciones especiales, incluyendo: a. La capacidad de cortar tapones de núcleos de los
núcleos enteros recién recobrados. Cualquier demora para cortar los tapones de núcleo permite que la capilaridad y la difusión distribuyan componentes activas de fluido de perforación a las porciones del núcleo que no han sido invadidas. Se debe utilizar una salmuera sintética o un aceite de laboratorio refinado como fluido reductor
b. Se necesitan portanúcleos y equipos de purga de núcleos para permitir la purga de tapones. Puede ser posible evitar daños causados por una fase de aceite inestable (en donde se depositan asfaltenos o parafinas debido a cambios en presión y temperatura) si la fase de aceite es rápidamente reemplazada por un aceite estable, no dañino.
c. También se necesitan instalaciones para pruebas de imbibición porque las pruebas de velocidad y alcance de imbibición son las mejores indicadores de humectabilidad en medios porosos.
Los requerimientos para mantener la humectabilidad variarán de un yacimiento a otro y tendrán que ser determinados experimentalmente hasta cierto grado. Se indican los métodos adecuados de preservación de núcleos para controlar la pérdida de fluidos en 2.5.2. 2.7 PRECAUCIONES 2.7.1 Generalidades
El objetivo fundamental de análisis de núcleos es el de obtener datos representativos de las propiedades de rocas en yacimientos in situ. El sondaje, el manejo y la preservación deben realizarse de tal manera para prevenir tanto la pérdida de fluidos intersticiales como también la contaminación con fluidos extraños. No se debe lavar el núcleo con agua o aceites antes de la preservación. Se deben adoptar procedimientos apropiados de sondaje y manejo para obtener datos de laboratorio significativos. Una alteración de la roca puede ocurrir durante el sondaje, manejo, preservación, muestreo, y preparación antes o durante el análisis. Para una determinación confiable del contenido del fluido de los núcleos, se debe diseñar un procedimiento uniforme para el manejo y la preservación. Se debe resaltar que un programa apropiadamente diseñado será para el beneficio no solo del usuario actual, sino también los usuarios futuros del material del núcleo. Las siguientes son algunas precauciones para el manejo y el empaque de muestras: a. Todos los núcleos deben ser preservados tan
pronto sea posible después de su remoción del sacanúcleos. Aún después de la preservación, las muestras no deben ser expuestas a condiciones extremas. Se desconoce la eficacia a largo plazo de los materiales de preservación de núcleos que se utilizan actualmente. Se debe utilizar un material probado y confiable si el núcleo va a ser almacenado durante largos periodos.
b. Siempre hay que minimizar el espacio sobrante de gas en los recipientes de preservación de núcleos para prevenir pérdidas por evaporación durante el almacenamiento del núcleo. Este procedimiento también minimizará las pérdidas por condensación en la superficie interior del recipiente y ayudará a prevenir la rotura de muestras mas sueltamente consolidadas durante el envío.
c. Para minimizar la pérdida de fluidos, el núcleo no debe entrar en contacto con telas, papel, ni otros materiales secos con vasos capilares finos.
d. No bañe ni cubra el núcleo directamente con ningún fluido.
e. Siga instrucciones estrictas de manejo para el procesamiento y la preservación de núcleos no consolidados (ver 2.2.4 y 2.5).
f. No preserve una roca no consolidada u otro tipo de roca en el mismo recipiente que una roca de litología muy diferente. Esto minimizará el potencial de daños mecánicos en muestras débiles.
g. En el caso que algún núcleo sea expuesto a condiciones severas de manejo o al lavado, esto debe quedar registrado. Todos los datos pertinentes deben acompañar al núcleo (ver Figuras 2-2 y 2-3).
h. Marque cada recipiente de preservación de manera apropiada. En los casos donde confidencialidad sea un problema, estos datos deben ser codificados con números e indicados en una lista maestra.
i. Se recomienda tener presente a un representante de la empresa que tenga

conocimiento del manejo y la preservación durante la operación de sondaje. Si esto no es posible, se deben dar instrucciones escritas explícitas al representante de la empresa de servicios responsable de esta actividad. En el momento de llegar al laboratorio, los núcleos deben permanecer en estado de preservación hasta que estén listos para análisis.
j. SE DEBEN SEGUIR TODOS LOS REGLAMENTOS DE SEGURIDAD APLICABLES EN EL MANEJO DE EQUIPOS DE SONDAJE Y MATERIALES DE SONDAJE. El personal del pozo debe tener la protección necesaria contra la exposición a materiales peligrosos utilizando ropa de trabajo, guantes, lentes protectoras, etc. También se recomiendan cascos, zapatos de punta de acero, y protección para los oídos. Cuando hay gases tóxicos presentes tal como el sulfuro de hidrógeno, debe haber aparatos personales de respiración. Se requiere un entrenamiento de seguridad para todos los trabajadores del campo antes de manejar equipos de sondaje, equipos de procesamiento, maquinaria, y material de núcleos.

CONTENIDO
1 SELECCIÓN DE NÚCLEOS Y PREPARACIÓN DE NÚCLEOS 3-1 3.1 Generalidades 3-1 3.2 Descripción de Núcleos 3-1
3.3 Registros Gamma de Núcleos y Registros Gamma Espectral de Núcleos 3-2
3.4 Formación de Imágenes de Núcleos 3-2 3.5 Muestreo de Núcleos y Preparación de Núcleos (Análisis
Básico de Núcleos) 3-4 3.6 Limpieza de Núcleos 3-5 3.7 Secado 3-7 3.8 Preservación de Muestras 3-7 Tablas 3-1 Solventes Seleccionados y Su Uso 3-6 3-2 Métodos de Secado de Muestras de Núcleos 3-7

Prácticas Recomendadas para el Análisis de Núcleos 3 Selección de Núcleos y Preparación de
Núcleos
3.1 GENERALIDADES Las recomendaciones incluidas en este documento pueden comprender el uso de materiales, operaciones, y equipos peligrosos. Este documento no trata todos los problemas de seguridad asociados con su uso. Es la responsabilidad del usuario el establecer prácticas apropiadas de seguridad y salud antes de usarlos y el cumplir con todos los requerimientos reglamentarios aplicables con respecto al uso y la eliminación de los materiales y equipos. 3.2 DESCRIPCIÓN DE NÚCLEOS 3.2.1 Fundamento El propósito de la inspección y la descripción de núcleos es el reconocimiento de características litológicas, deposicionales, estructurales, y diagenéticas de núcleos enteros o tajados. Las descripciones cualitativas y cuantitativas de núcleos proporcionan la base para el muestreo regular del análisis de núcleos, análisis de facies, y otros estudios de yacimientos tales como la calidad del yacimiento y análisis de núcleos suplementarios. Una descripción proporciona un registro accesible del núcleo. 3.2.2 Aparatos y Suministros Se recomiendan los siguientes equipos para uso en las descripciones estándar del núcleo: a. Formulario de registro para la recolección
sistemática de datos. b. Microscopio o lupa de mano. c. Regla para la medición de longitud. d. Regla para el tamaño de grano. e. Productos químicos apropiados tales como:
1. Agua o salmuera para mejorar la visibilidad de estructuras geológicas.
2. Ácido HCl diluido para identificar minerales carbónicos.
3. Alizarina roja para diferenciar calcita y dolomita.
4. Solventes de hidrocarburos para facilitar la detección de la fluorescencia de aceite bajo luces ultravioletas.
f. Registro de sondaje, informe de perforación, registros de lodo, información del pozo sobre núcleos perdidos.
g. Registro gamma de núcleos. h. Luz ultravioleta. 3.2.3 Precauciones Se deben observar las siguientes precauciones: a. Si las muestras que van a ser descritas han sido
preservadas para pruebas especiales, se debe evitar la exposición de las muestras al aire y a
productos químicos hasta que se hayan terminado las pruebas sobre el núcleo preservado.
b. Elija un formato apropiado para el registro. El objetivo de un formato para registro debe ser el de representar el núcleo con precisión. Cuando se describe el núcleo, es importante recolectar y registrar los datos de manera sistemática.
3.2.4 Procedimientos Se deben utilizar los siguientes procedimientos: a. Extienda el núcleo a analizar sobre una mesa de
observación. b. Revise la cantidad de núcleo con el informe de
sondaje y asegúrese que no se ha perdido material del núcleo durante el transporte. Anote cualquier daño o alternación al núcleo durante el manejo y transporte en el campo.
c. Revise la numeración y el orden de las cajas o recipientes contra la profundidad acumulativa.
d. Revise la continuidad y la orientación del núcleo con respecto a la parte superior del núcleo. Si el núcleo ha sido marcado de acuerdo con 2.2.3, la parte superior del núcleo está apuntando hacia arriba cuando la marca roja está al lado derecho.
e. Revise el orden de los segmentos del núcleo en las cajas. Busque "roturas" que hacen juego o las marcas de una pieza a la siguiente y de una caja a la siguiente.
f. Mida y marque la longitud en pies en cada caja. Marque el núcleo hasta la última 1/2 pulgada (1 centímetro).
g. Si hay registros de rayos gamma del pozo disponibles, se debe hacer una comparación con los registros de los rayos gama del núcleo (ver 3.3) para verificar que las profundidades de intervalos sondeados sean consistentes con las profundidades del registro de pozo.
h. Evalúe toda la secuencia sondeada antes de comenzar. Busque características distintivas tales como unidades, contactos, marcadores únicos (e.g., bentonita, carbon).
i. Registre las características principales tales como los siguientes utilizando nomenclatura y abreviaturas estandarizadas (ver Sección 8): 1. Litología del núcleo (esquisto, arenisca,
caliza, etc.) 2. Color. 3. Estratificación (grosor, contactos de los
estratos, marcadores de erosión). 4. Estructuras sedimentarias obvias. 5. Textura (tamaño de grano,
angulosidad/redondez y distribución). 6. Composición (granos, cemento, fósiles). 7. Tipos de porosidad. 8. Características diagenéticas y tectónicas. 9. Anote la cualquier mancha de aceite y
fluorescencia relacionada. j. Minimice las descripciones centímetro-por-
centímetro. Algunas características de escala fina pueden ser importantes y deben registrarse.

k. Registre las rocas que no sean del yacimiento, e.g., rocas de lodo, además de las posibles secuencias provechosas. Los cambios sutiles en litología pueden asistir en la correlación entre los registros de núcleo y pozo y la definición de zonificación pertinente.
l. Registre la información de fractura tal como ancho, extensión, densidad, orientación de fractura si el núcleo está orientado, y la presencia de cimentación o lodo. Donde sea posible, diferencie entre las fracturas naturales y aquellas inducidas por el sondaje.
3.3 REGISTROS DE RAYOS GAMMA DE
NÚCLEOS Y REGISTROS DE RAYOS GAMMA ESPECTRALES DE NÚCLEOS
3.3.1 Fundamento Los emisores de rayos gamma que ocurren naturalmente (hijas radiogénicas de uranio y torio junto con potasio-40) dan una respuesta de rayos gamma medible que puede registrarse con profundidad. Si este registro medido en la superficie se compara con las lecturas de rayos gamma tomadas de un registro de rayos gama en el pozo, los resultados pueden ser utilizados frecuentemente para ajustar la profundidad del núcleo para coincidir con las profundidades de perfil del pozo-abierto y para identificar las zonas donde se han perdido partes del núcleo. 3.3.2 Aparatos El aparato recomendado consiste de un transportador para un núcleo en movimiento, blindaje de plomo para reducir la radiación gamma del medio ambiente, y los detectores de rayos gama adecuados. Un sistema detector típico consiste de un cristal de escintilación blindado junto con un fotomultiplicador. El cristal de escintilación normalmente es yoduro de sodio revestido con talio [NaI(Tl)]. Otros cristales de escintilación incluyen yoduro de cesio (CsI) y germanio de bismuto (BiGeO). Se procesan las señales de los detectores y los eventos de rayos gamma son clasificados por energía y contados. Las unidades estándar de rayos gamma deben proporcionar cuentas totales de rayos gamma en unidades APIU. Con las unidades de rayos gamma espectrales, estas cuentas son convertidas en concentraciones de potasio, uranio y torio, y unidades API estándar de registros por rayos gamma. Se muestran las señales en una pantalla de computador o son ploteadas en formatos de registro y escala de pozo para una comparación directa con los registros de pozo. 3.3.3 Procedimientos Se deben utilizar los siguientes procedimientos: a. Ajuste la lectura del aparato registrador para que
corresponda con la escala utilizada con el registro por rayos gamma del pozo.
b. Registre la respuesta de rayos gamma del núcleo de abajo hacia arriba. Esto cumple con la secuencia utilizada en los registros de pozo.
c. El perfil de rayos gamma de los núcleos debe realizarse uniformemente, sin interrupción, en el orden correcto y con espacios entre las roturas de núcleo armadas.
d. Registre una sección repetida cerca de la parte inferior del núcleo para establecer la reproducibilidad de la medida.
3.3.4 Ventajas El perfil de rayos gamma de núcleos es ampliamente disponible y es utilizado en la práctica general para correlacionar la profundidad del núcleo con la profundidad del registro. El aparato de rayos gamma espectrales diferenciará las concentraciones de uranio, torio y potasio y puede utilizarse para identificar y diferenciar el esquisto, particularmente en los núcleos de arenisca con grandes cantidades de feldespato y mica de potasio. 3.3.5 Limitaciones Esta técnica no es capaz de detectar la baja actividad de rayos gamma y puede sufrir de interferencias significativas de fondo. El análisis de muestras para la respuesta de rayos gamma espectrales requiere un registro mas lento que aquel utilizado para determinar la respuesta total de rayos gamma. 3.3.6 Calibración El aparato debe ser calibrado antes de evaluar el material de núcleos. Las calibraciones son sensibles al tamaño del núcleo y el alcance de energía de rayos gamma. La calibración debe ser realizada midiendo la respuesta de rayos gamma a los tubos de calibración que contienen muestras puras de los elementos de interés. El aparato de rayos gamma total requiere un tubo de calibración que contiene potasio (K-40), uranio (U-238), y torio (T-232) de actividades conocidas. El procedimiento de calibración para la unidad de rayos gamma espectrales requiere los mismos elementos pero en tubos individuales. Un tubo de calibración en blanco debe ser medido para asegurar que una cantidad mínima de rayos gamma de fondo están interfiriendo con el aparato. La velocidad del transportador debe ser lo suficientemente lenta para proporcionar una relación aceptable de señal al ruido de fondo. Una relación de tres veces la raíz cuadrada de la velocidad de conteo mayor que uno es aceptable. 3.3.7 Precisión La precisión de la medición varia con la raíz cuadrada de la velocidad de conteo. 3.4 FORMACIÓN DE IMÁGENES DE NÚCLEOS Una imagen registrada del núcleo es esencial. Este registro proporcionará información que puede utilizarse si la observación del núcleo no es posible.

El registro puede incluir imágenes visuales de las características de la superficie el núcleo utilizando técnicas fotográficas, representaciones visuales de las estructuras internas del núcleo tales como radiografías, tomografías computarizadas de rayos x, imágenes de resonancia magnética, o imágenes acústicas. Todo registro debe incluir información sobre la profundidad del núcleo junto con una escala o explicación indicando el margen de intensidad de la imagen registrada. Además de las representaciones fotográficas, las técnicas modernas de formación de imágenes proporcionan información cuantificable para el núcleo que puede utilizarse como la base para el muestreo de rutina de análisis de núcleos, análisis de facies, estudios de la calidad de yacimientos, análisis suplementarios de núcleos, e interpretaciones de registros de pozos. 3.4.1 Fotografía 3.4.1.1 Fundamento El núcleo normalmente es fotografiado bajo la luz natural (5.500 K) o la luz ultravioleta (254-365 nm) junto con una escala de colores estándar. Las fotografías de luz natural muestran la litología y las estructuras sedimentarias y permiten la inspección de características específicas registradas en la descripción de núcleos. Las fotografías de luz ultravioleta pueden resaltar las zonas que contienen hidrocarburos causando la fluorescencia de la mayoría de aceites en sombras que varían de un marrón anaranjado para aceites viscosos a un amarillo brillante para aceites de alta gravedad. Los condensados pueden ser entre blanco muy claro y blanco azulado. Las zonas que no contienen hidrocarburos serán las regiones violetas, aunque algunos minerales como las calizas cretáceas también son violetas. 3.4.1.2 Ventajas La fotografía de núcleos proporciona un registro visual del núcleo que puede utilizarse para reconstruir partes del núcleo dañado, minimizar el manejo del núcleo, e identificar la ubicación de muestras si son fotografiadas después de la toma de muestras. 3.4.1.3 Limitaciones Los colores fotográficos pueden ser diferentes a los verdaderos colores del núcleo. El relieve de características puede requerir la humectación de la superficie del núcleo para la fotografía. Se requiere una escala de colores y una barra cromática si se necesitan ajustes. 3.4.2 Técnicas de Rayos X 3.4.2.1 Fundamento Las técnicas de rayos x puede utilizarse de manera no invasiva para examinar la naturaleza interna de un núcleo. Un haz de rayos x es dirigido hacia el núcleo y se miden las variaciones en la atenuación de
incidencia. En orden de definición creciente, estas técnicas son: fluoroscopio, radiografía x, y tomografía computarizada (exploración CT). La utilidad de estos métodos depende de su sensibilidad a los contrastes de densidad dentro del núcleo. Consecuentemente, las áreas densas, no porosas, serán contrastadas contra las áreas porosas de baja densidad. La alta definición de las variaciones de densidad dentro del núcleo requiere mediciones multi-direccionales o la separación espacial de las áreas contrastadas. 3.4.2.2 Fluoroscopio En esta técnica, se mueve el núcleo a través de una fuente de rayos x. El haz, atenuado por el núcleo, se choca contra una pantalla fluorescente, se intensifica, y es registrado por una cámara de vídeo. La imagen capturada puede observarse en un monitor, registrarse en un cassette de vídeo, y convertirse a un formato digital para un procesamiento y observación posterior. El producto de esta técnica es una imagen continua por todo el largo del núcleo. Como el núcleo está en movimiento, la resolución de la imagen que resulta puede ser disminuida. Sin embargo, se pueden identificar las variaciones de densidad, algunas estructuras geológicas, y áreas de núcleo perdido dentro de mangas plásticas o de fibra de vidrio. También es útil en la identificación de zonas fracturas o muy quebradas. 3.4.2.3 Radiografías X Se coloca una muestra de núcleo entre una fuente de rayos x y una película sensible a rayos x. Se captura la atenuación del haz de rayos x por el núcleo en la película. La película revelada también puede observarse en una mesa de luz, o utilizada para hacer una impresión en blanco y negro. Es posible obtener múltiples imágenes de la misma parte del núcleo y exponerlas lado-a-lado. Primero se debe tomar la radiografía x para obtener una imagen inicial y luego se rota 90 grados con el fin de obtener una segunda imagen. Esto es útil para orientar el núcleo dentro de la manga antes de tajarlo porque así se establece la dirección de estratificación. Durante una radiografía x, la muestra de núcleo es inmóvil y proporciona una imagen de más alta resolución que el método de fluoroscopio. La radiografía x permite la detección y evaluación de estructuras geológicas internas tales como planos de estratificación, fracturas y nódulos, cambios litológicos, y densidad de masa. 3.4.2.4 Tomografía Computarizada (CT) En CT, el núcleo es explorado por un haz de rayos x altamente alineado. Los detectores en los extremos opuestos del núcleo miden la intensidad del haz transmitido. La fuente de rayos x y/o los detectores giran o se trasladan alrededor de la muestra. Se toma una serie de medidas de atenuación de rayos x y estas son reconstruidas numéricamente para dar la distribución espacial de coeficientes de atenuación de rayos x dentro de la muestra.

La resolución de la imagen depende del grosor del haz de rayos x, la cantidad de detectores en el explorador, y el tamaño del arreglo de pixeles utilizado para reconstruir la imagen. El grosor del haz de rayos x varia de 2 milímetros a 10 milímetros. La CT permite la detección y la evaluación de estructuras geológicas internas tales como planos de estratificación, fracturas y nódulos, cambios litológicos, y densidad de masa. La combinación de exploraciones x-y secuenciales por el eje del núcleo puede proporcionar imágenes por tajadas o imágenes tridimensionales del núcleo. 3.4.2.5 Ventajas Las técnicas de rayos x proporcionan representaciones cuantificadas y objetivas del núcleo. Estas representaciones pueden proporcionar ventajas similares a los indicados para fotografías de núcleos en 3.4.1.2 sin la necesidad de exponer una superficie del núcleo. 3.4.2.6 Limitaciones La resolución de las imágenes es menor que aquella proporcionada por fotografías. La atenuación de rayos x puede variar con la mineralogía, dependiendo de la energía del haz de rayos x. Algunas aplicaciones aun están en la etapa de desarrollo. El desarrollo futuro de las relaciones entre las rocas y la atenuación de rayos x puede extender estos métodos a otras áreas de análisis de núcleos. 3.4.3 Resonancia Magnética Nuclear (RMN) 3.4.3.1 Fundamento La formación de imágenes RMN se utiliza para proporcionar una imagen reconstruida de fluidos dentro de una muestra de núcleo. Las mediciones RMN se basan en la observación que cuando se aplica la energía de excitación de la frecuencia de radio apropiada en un conjunto de núcleos, una porción de núcleos en un nivel de energía más bajo puede ser promocionado a un nivel de energía mas alto. Cuando se remueve la excitación, la velocidad a la cual los núcleos excitados vuelven al nivel inferior puede ser detectado y medido. La energía de excitación es suministrada por un campo magnético oscilante en resonancia con los núcleos. La respuesta de los núcleos se mide a través de una bobina receptora sintonizada. Se puede determinar la ubicación de los núcleos excitados con la aplicación de gradientes del campo magnético durante los procesos de excitación y detección. El núcleo principal utilizado en las aplicaciones de análisis de núcleos es 1H. Otros núcleos que pueden formar imágenes incluyen, pero no están limitados a, 2H, 31P, 23Na, 13C. 3.4.3.2 Ventajas Las imágenes de resonancia magnética no son invasoras y proporcionan una imagen que muestra las ubicaciones de fluido dentro de una muestra. Las ventajas indicadas en 3.4.2.5 aplican siempre que la
muestra sea contenida dentro de un soporte adecuado no metálico. 3.4.3.3 Limitaciones Esta técnica no es descriptiva del núcleo sino de los líquidos dentro del núcleo. La técnica requiere una alta densidad de núcleos resonantes para una señal adecuada. Por lo tanto, los núcleos de baja porosidad darán señales débiles. Los minerales pamagnéticos o ferromagnéticos pueden inhibir o degradar la señal medida y pueden distorsionar la imagen. 3.5 MUESTREO DE NÚCLEOS Y
PREPARACIÓN DE NÚCLEOS (ANÁLISIS BÁSICO DE NÚCLEOS)
3.5.1 Fundamento El procedimiento de muestreo para el análisis básico de núcleos es determinado por el tipo de información requerida. El muestreo por lo general tendrá en cuenta uno o más de los siguientes: a. Distribución litológica. b. Variaciones de porosidad y permeabilidad dentro
de las unidades litológicas. c. Distribución de hidrocarburos. 3.5.1.1 Muestras de Tapones También se refiere a ellos como tapones. Estos deben ser removidos de secciones de núcleos enteros que están orientados vertical u horizontalmente con respecto al eje completo del núcleo o con respecto a la normal de los planos de estratificación. Estos tapones proporcionarán datos sobre las propiedades de la matriz. Ver 3.5.2.4 para los procedimientos para la remoción de tapones de núcleo. 3.5.1.2 Muestras de Diámetro Completo Las muestras de diámetro completo (secciones de núcleos enteros), además de muestras de tapones, deben ser tomadas de los siguientes tipos de zonas o donde existen heterogenidades significativas de grande escala que son diferentes a las propiedades de la matriz. Estos pueden ocurrir en los siguientes: a. Carbonatos "vuggy" - Los vugs pueden sesgar la
medición de porosidad dentro de una muestra. Se debe tomar muestras para obtener una representación de la proporción relativa de vugs-a-roca en el yacimiento.
b. Yacimientos fracturados - Puede ser difícil tomar muestras en las zonas fracturadas debido a la naturaleza frágil de la roca. Sin embargo, las fracturas naturales pueden tener un impacto positivo significativo en la permeabilidad del yacimiento y se debe tomar muestras de donde sea posible o apropiado.
c. Conglomerados - Dentro de un conglomerado, si el tamaño del guijarro es grande con respecto al tamaño de la muestra, el tamaño de la muestra puede sesgar las mediciones de las propiedades

de la roca en gran cantidad. Para obtener una muestra representativa de una unidad de conglomerado, es necesario que el tamaño de la muestra sea suficiente para incluir todos los tamaños de guijarros.
3.5.2 Corte, Arreglo, y Montura de Muestras 3.5.2.1 Fundamento El núcleo debe ser cortado y arreglado para proporcionar muestras de formas regulares, mas comúnmente cilindros rectos. Puede ser necesario montar las muestras no consolidadas, que se están desintegrando, y muy friables antes de las pruebas. A pesar del grado de litificación, se debe mantener el manejo de las muestras en un mínimo. 3.5.2.2 Aparatos y Suministros Los siguientes equipos y suministros son comúnmente utilizados en las operaciones de corte y arreglo de muestras de núcleos: a. Sierra de tajada grande con hoja de diamante. b. Sierra de guarnición con hoja de diamante. c. Prensa taladradora con brocas de núcleos de
diamante, capaces de perforar muestras cilíndricas.
d. Afilador para cuadrar los extremos de los tapones.
e. Bombas de fluido para llevar diferentes refrigerantes (salmuera, aceite, aire, agua, N2 líquido) a las superficies de cortado.
f. Medios de marcado indeleble tal como tinta India. g. Mangas de plomo, aluminio, o plástico que
encoge con el calor para montar muestras blandas, no consolidadas, o muy frágiles.
La siguiente información debe estar disponible antes de las operaciones de corte, arreglo, o montura: a. La cantidad total de muestras requeridas. b. Tamaño y orientación necesaria. c. Ubicaciones de la profundidad exacta para las
muestras y la manera que deben ser marcadas. d. Fluidos a utilizar para cortar las muestras
(núcleos que contienen arcillas o laminaciones de esquistos sensibles a agua dulce se deteriorarán a menos que se utilicen los fluidos de perforación apropiados.
e. Imágenes de núcleos disponibles. f. Si es necesario, los materiales para el método
preferido de preservación tanto para los tapones como para el núcleo del cual se cortaron los tapones.
3.5.2.2 Precauciones Se deben tener las siguientes precauciones durante las operaciones de corte, arreglo y montura de muestras: a. Los operadores de sierras y prensas taladradoras
deben utilizar protectores de oídos, gafas
protectoras, guantes y otros implementos de seguridad necesarios.
b. El flujo de lubricante/refrigerante debe ser suficiente para enfriar la broca de taladro o la hoja de la sierra y remover los recortes sin desgastar la muestra.
3.5.2.4 Procedimientos Se deben seguir los procedimientos indicados a continuación para preparar diferentes tipos de muestras de núcleos: a. Muestras de tapones:
1. Perfore los tapones en puntos especificados utilizando la broca del tamaño apropiado. Se debe tener cuidado para perforar tapones rectos. Si se aplica demasiada presión durante la operación de perforación, la broca se doblará causando una deformación del tapón.
2. Recorte los tapones hasta la longitud requerida asegurando que los extremos quedan paralelos. Guarde y marque los extremos recortados.
3. Marque, preserve, y/o almacene las muestras, según la necesidad.
b. Muestras de diámetro completo: 1. corte las secciones del núcleo seleccionado
para análisis ligeramente mas largo que lo necesario para permitir las operaciones finales de molido.
2. Remueva las púas y alise los bordes astillados tratando los extremos. Los extremos del cilindro deben ser lo mas paralelos posible.
3. Marque las muestras claramente y presérvelas o almacénelas según la necesidad.
c. Muestras no consolidadas - Existen dos métodos distintos para preparar núcleos no consolidados. Los procedimientos dependen de la estabilización del núcleo por congelación, impregnación con epoxi, o ambos, o si no está estabilizado. Los núcleos estabilizados por lo general son perforados mientras los núcleos no estabilizados son cortados a ras. 1. Núcleos estabilizados - Los núcleos
congelados son obtenidos utilizando las prácticas indicadas para las muestras de tapones, con las modificaciones apropiadas para evitar la descongelación del núcleo. a. Enfríe el núcleo previamente con hielo
seco y perfore con refrigerante líquido N2.
b. Perfore los tapones en los puntos especificados utilizando la broca del tamaño apropiado. Se debe tener cuidado para perforar tapones rectos. Si se aplica demasiada presión durante la operación de perforación, la broca se doblará causando una deformación del tapón. Asegúrese que hay un flujo adecuado de nitrógeno líquido para que la muestra se mantenga congelada y para que se remuevan los recortes.

c. Recorte los tapones a la longitud requerida utilizando una hoja de sierra enfriada con N2 líquido, asegurando que los extremos están paralelos. Guarde y marque los extremos arreglados.
d. Monte los tapones en una manga previamente cargada con filtros previamente cargadas y de tamaño apropiado.
e. Marque, preserve, y/o almacene las muestras según la necesidad.
Se puede congelar y perforar los núcleos estabilizados con epoxi utilizando los procedimientos indicados en a-e arriba o por los procedimientos indicados para muestras de tapones (ver 3.5.2.4.a).
2. Núcleos no estabilizados - los núcleos no congelados pueden ser cortados utilizando una técnica de corte a ras. La cortadora debe ser introducida lentamente en el núcleo y removido. El tapón cortado debe ser removido suavemente de la cortadora e insertada directamente en una manga cargada previamente. Luego se recortan los extremos del tapón.
3.6 LIMPIEZA DE NÚCLEOS 3.6.1 Introducción Antes de la mayoría de las mediciones de porosidad y permeabilidad de laboratorio, los fluidos originales
deben ser completamente removidos de la muestra del núcleo. Esto por lo general se logra por medio de la purga, el desagüe, o el contacto con diferentes solventes para extraer hidrocarburos, agua y salmuera. 3.6.2 Aparatos y Suministros Se describen varias técnicas y aparatos para limpieza en los siguientes procedimientos y en la Sección 4. Se indican algunos solventes utilizados para propósitos de extracción de hidrocarburos en la Tabla 3-1. Los solventes mencionados son aquellos utilizados mas frecuentemente para extraer muestras para análisis de rutina. Algunos son preferidos para aplicaciones específicas: e.g., se ha encontrado que el cloroformo es excelente para muchos crudos Norte Americanos y que tolueno útil para crudos asfálticos. Antes de limpiar muestras con propiedades desconocidas, se debe probar una sub-muestra con diferentes solventes para verificar su eficacia para limpiar. Como los cristales residuales de sal afectan la porosidad y la permeabilidad medidas, las muestras de núcleos que contienen un agua de formación con alta salinidad pueden necesitar una extracción adicional para remover la sal. La sal puede ser removida con alcohol metílico u otros solventes en los cuales la sal es soluble. Los diferentes solventes utilizados para extraer muestras de núcleos pueden ser obtenidos por métodos físicos y químicos muy conocidos. Tales técnicas de recobro pueden hacer práctico el uso de un solvente costoso.
Tabla 3-1 - Solventes Seleccionados y Su Uso
Solvente Punto de Ebullición, ºC Solubilidad Acetona
Cloroformo/metanol azeotrope Ciclohexano
Cloruro de etilo Hexano Metanol
Cloruro de Metileno Nafta
Tetracloroetileno Tetrahidrofurano
Tolueno Tricloroetileno
Xileno
56.5 53.5 81.4 83.5
49.7-68.7 64.7 40.1
160.0 121.0 65.0
110.6 87.0
138.0-144.4
Aceite, agua, sal Aceite, agua, sal
Aceite Aceite, agua limitada
Aceite Agua, sal
Aceite, agua limitada Aceite Aceite
Aceite, agua, sal Aceite,
Aceite, agua limitada Aceite
3.6.3 Precauciones Se deben observar las siguientes precauciones durante las operaciones de limpieza de muestras de núcleos. a. Cuando se utilizan solventes, es la
responsabilidad del usuario establecer las prácticas apropiadas de seguridad y salud antes de su uso y de cumplir con todos los requerimientos reglamentarios con relación al uso y la eliminación de los materiales.
b. El solvente seleccionado no debe atacar, alterar o destruir la estructura de la muestra.
c. El cloroformo puede hidrolizar durante la extracción, formando ácido hidroclórico como resultado.
d. No todos los solventes son compatibles con todas las configuraciones de equipos. Se deben considerar las posibles reacciones solvente/equipos en la selección de equipos y solvente para uso en las operaciones de limpieza de núcleos.
e. Se deben utilizar calentadores eléctricos de tipo cerrado cuando se usan solventes inflamables.

Se deben observar las precauciones de seguridad tal como la ventilación adecuada del laboratorio y la accesibilidad fácil de extinguidores de incendios y duchas de seguridad.
f. Se debe realizar la extracción debajo de capuchas de ventilación dotadas con ventilación de aspiración forzada.
g. Se debe considerar los efectos de la temperatura sobre las muestras. Ver 3.7 para recomendaciones con relación al secado de las muestras de núcleos.
3.6.4 Procedimientos 3.6.4.1 Purga de Solvente por Presión Directa La extracción de hidrocarburos y sal de las rocas de yacimientos puede lograrse inyectando uno o más solventes en la muestra de núcleo bajo presión y a temperatura ambiente. La presión utilizada debe ser dependiente de la permeabilidad de la muestra y puede variar de 10 a 1.000 psi. Las muestras de núcleos pueden colocarse en una manga de caucho bajo una presión de sobrecarga o en un aparato portanúcleos adecuado que permitirá el flujo de solvente a través de la matriz de la muestra. El volumen del solvente requerido para remover los hidrocarburos completamente en la muestra del núcleo es dependiente de los hidrocarburos presentes en la muestra y el solvente utilizado. El núcleo se considera limpio cuando la corriente saliente está limpia. En algunos casos, se puede necesitar mas de un solvente para remover crudos pesados de tipo asfáltico. 3.6.4.2 Purga por Centrífuga Una centrífuga con una cabeza especialmente diseñada es utilizada para rociar solvente limpio y tibio (de un destilador) contra las muestras de núcleos. La fuerza centrífuga hace que el solvente fluya a través de las muestras desplazando y extrayendo el aceite (y el agua). Se debe variar la velocidad de rotación de unos cientos a unos miles de revoluciones por minuto (rpm), dependiendo de la permeabilidad y el grado de consolidación del núcleo. Se pueden utilizar la mayoría de los solventes comunes. Conley y Burrows describen el aparato y el procedimiento.1 3.6.4.3 Extracción por Solvente a Gas (U.S. Patente 2,617,719) En este procedimiento, un núcleo es sujeto a ciclos repetidos de impulso interno disuelto o solución-gas hasta limpiar el núcleo de hidrocarburos. Se remueven el solvente restante y el agua por medio de un horno de secado. Cuando se lleva un núcleo de una formación petrolífera a la superficie y este se despresuriza, el gas disuelto en el aceite sale de la solución y desaloja alguna cantidad de aceite y agua del núcleo. Esto resulta en algunos espacios de poros llenos de gas a presión atmosférica. Los espacios llenos de gas en el núcleo pueden llenarse casi completamente con
solvente rodeando el núcleo con un solvente adecuado que contenga un gas disuelto y aplicando la presión hidráulica suficiente. Bajo esta condición, el solvente se mezcla con el aceite en el núcleo y la subsecuente despresurización a la presión atmosférica remueve alguna cantidad del aceite residual. El gas de bióxido de carbono es excelente para este propósito por el poco peligro de incendio o explosión y la alta solubilidad en la mayoría de los solventes. Algunos solventes que se pueden utilizar son nafta, tolueno, o mezclas de solventes. Con ciertos tipos de crudo, se puede reducir el tiempo de limpieza si la cámara del núcleo se calienta con un baño de agua, baño de vapor, o con calentadores eléctricos. Una aplicación exitosa de este método para la limpieza de rutina de núcleos utiliza bióxido de carbono y tolueno a 200 psig, con una presión hidráulica de 1.000 psig. Se utilizan ciclos de aproximadamente 30 minutos. Stewart2 describe el aparato y el procedimiento. 3.6.4.4 Método de Extracción por Destilación Se pueden utilizar un extractor Soxhlet y un solvente o solventes adecuado(s) para disolver y extraer aceite y salmuera. La extracción puede arreglarse en un colector para que el solvente cargado de agua y aceite se pase a través de un sifón de cada extractor hacia un destilador común desde el cual se destila, condensa y se distribuye de nuevo un solvente fresco a todos los extractores. La limpieza de la muestra se determina del color del solvente que pasa a través de los sifones periódicamente desde el extractor. La extracción debe seguir hasta que el extracto salga transparente. La no-luminiscencia del extracto bajo la luz fluorescente es un buen criterio para determinar la extracción completa del aceite para un solvente dado. Se debe observar que la extracción completa de ciertos aceites de las muestras de núcleos pueden requerir mas de un solvente, y el hecho que un solvente esté transparente después de contacto con la muestra no necesariamente significa que el aceite haya sido removido completamente de la muestra. Se pueden encontrar detalles de un aparato y procedimiento en 4.3. 3.6.4.5 Extracción por Gas Licuado La extracción por gas licuado utiliza un extractor Soxhlet presurizado y un solvente polar condensado de un punto de ebullición bajo. El proceso es un procedimiento de extracción por destilación que utiliza solvente presurizado para limpiar el núcleo. El solvente se vuelve a generar a través de la destilación a baja temperatura. Como la extracción es realizada a una temperatura ambiente o menor, este puede practicarse sobre núcleos sensibles a calor tales como aquellos que contienen yeso. 3.6.5 Ventajas La limpieza de un núcleo remueve los fluidos originales, preparándolo para pruebas adicionales que no requieren esos fluidos.

3.6.6 Limitaciones Las condiciones de pruebas individuales pueden requerir una técnica particular para resultados óptimos. Algunas técnicas pueden ser más aplicables a tipos de roca específicos o algunos requerimientos de limpieza. 3.7 SECADO Las muestras de núcleos convencionales pueden ser secados por los métodos indicados en la Tabla 3-2:
Tabla 3-2 - Métodos para el Secado de Muestras de Núcleos
Tipo de Roca Método Temperatura, ºC
Arenisca (bajo contenido de arcilla) Arenisca (alto contenido de arcilla) Carbonato Con Yeso Esquisto u otra roca con alto contenido de arcilla
Horno convencional u horno al vacío Horno de humedad, humedad relativa de 40% Horno convencional u horno al vacío Horno de humedad, humedad relativa de 40% Horno de humedad, humedad relativa de 40% Horno convencional
116 90
63
116 90
60
60
Se debe secar cada muestra de núcleo hasta que el peso se vuelva constante. Los tiempos de secado pueden variar sustancialmente, pero por lo general son de mas de cuatro horas. 3.7.1 Precauciones Algunas precauciones que se deben observar en el secado de muestras para mediciones de núcleos de rutina son: a. Las muestras que contienen arcillas no deben
deshidratarse durante la preparación. Se debe tener cuidad en el secado de estas muestras. En algunos casos, se deben utilizar temperaturas menores que aquellas indicadas en la Tabla 3-2 para prevenir la deshidratación de arcillas.
b. Las muestras que contienen yeso requerirán procedimientos específicos durante la preparación. Se debe tener mucho cuidado para evitar la pérdida de agua y el cambio en la estructura cristalina.
c. Las muestras deben ser protegidas de la erosión por el goteo de solvente limpio cuando se utiliza la técnica de extracción por destilación.
d. La técnica de extracción no debe causar daños físicos en el núcleo.
e. El criterio usual para la limpieza de muestras es un extracto limpio. Sin embargo, muchos solventes no son solventes completos para algunos tipos de aceites y un extracto limpio puede reflejar la solubilidad del aceite y no la extracción completa.
f. Las muestras que contienen aceites asfálticos viscosos pueden requerir ciclos de mas de un solvente.
g. A menudo se puede lograr una limpieza de núcleos más eficaz con la combinación de solventes.
h. Permita que las muestras cargadas de solvente se ventilen en una capucha de vapores antes de colocarlas en un horno de secado cerrado.
3.8 PRESERVACIÓN DE MUESTRAS La preservación de muestras en el laboratorio dependerá de la cantidad de tiempo entre pruebas y el tipo de pruebas a realizarse. Cualquier técnica de almacenamiento o preservación debe asegurar la integridad estructural y evitar el secado, evaporación y oxidación no deseados.

CONTENIDO
4 MÉTODOS DE SATURACIÓN DE FLUIDOS 4-1 4.1 Introducción 4-1 4.2 Método Retorta a Presión Atmosférica 4-1 4.3 Método de Extracción por Destilación 4-7 4.4 Lavado de Solvente 4-23 4.5 Métodos de Barrido 4-25 4.6 Análisis de Carbón 4-26 4.7 Oil Shales 4-27 4.8 Núcleos que Contienen Yeso 4-28 4.9 Aspectos Históricos 4-31 4.10 Referencias 4-31 4.11 Bibliografía 4-31 Figuras
4-1 Retorta de Horno y Baño de María de Acero Inoxidable 4-3 4-2 Curva de Corrección de aceite de Retorta 4-4 4-3 Vaso de Retorta - Muestra de Pared 4-6 4-4 Aparato Dean-Stark para la Determinación Volumétrica de Agua 4-8
Aparato de Extracción Dean Stark para la Determinación Gravimétrica De Agua 4-8
4-6 Sistema de Recolección de Gas/Reducción de Presión 4-14 4-7 Muestra de Tapón y Rosca para Estudio de Invasión de Filtrado 4-21 Tablas
4-1 Tipo de Roca y Métodos Recomendados para la Prueba de Saturación de Fluidos 4-1
4-2 Capacidades de Medición de las Técnicas de Saturación de Barrido 4-26

4 Métodos de Saturación de Fluidos 4.1 INTRODUCCIÓN Esta sección documenta las técnicas especializadas desarrolladas para el análisis de muestras de núcleos con diferentes características físicas y tamaños, obtenidos con diferentes métodos de corazonamiento. La Tabla 4-1 indica las técnicas principales para determinar las saturaciones de fluidos de núcleos. Los párrafos 4.2 a 4.5 describen los siete métodos principales de análisis de saturación indicados en la Tabla 4-1 para núcleos de diámetro completo y tapones/núcleos de pared. Los tapones son definidos como muestras perforadas o formadas de núcleos recobrados con cualquiera de los varios aparatos de corazonamiento de diámetro completo. Los núcleos de pared se definen como núcleos obtenidos después de que el pozo haya sido creado por la broca de taladro o algún tipo de aparato de corazonamiento de orificio completo. Los párrafos 4.6 a 4.8 hablan de los métodos de análisis de saturación de fluidos para carbón, oil shale, y muestras que contienen yeso. Las muestras de oil shale y de carbón pueden obtenerse con aparatos de corazonamiento de diámetro completo. Sin embargo, el oil shale y el carbón también pueden ser extraídos de formaciones superficiales o poco profundos, y requieren procedimientos de muestreo no rutinarias diferentes a aquellos típicamente utilizados para núcleos de yacimientos. El formato de informes de saturaciones de fluidos para estos materiales difiere de las rocas típicas de yacimientos en que las saturaciones pueden ser expresadas como porcentaje de peso o galones por tonelada en lugar de porcentaje de volumen poroso. Se incluyen métodos de exploración que son capaces de medir saturaciones de fluidos con técnicas no destructivas en 4.5. Además, se incluye una sección histórica (ver 4.9) en la cual se describen, pero no son recomendados, algunos métodos previamente aceptables para la determinación de saturación de fluidos. Una tarea de evaluación de la formación difícil, si no imposible, es el recobrar un núcleo que tenga la misma saturación y distribución de fluidos como las de la formación antes del corazonamiento. Frecuentemente ocurren cambios en el contenido y distribución de fluidos durante las fases de corazonamiento, recobro, preservación y transporte. Los procesos de laboratorio como son el manejo, muestreo y prueba pueden causar alteraciones adicionales. Estos problemas no son tratados en detalle en esta sección, pero el lector deben estar atento de la cantidad de complicaciones que influyen en los valores finales de saturación de fluidos suministrados en un informe de laboratorio de análisis de núcleos. El análisis de saturación de fluidos puede alterar la humectabilidad del núcleo y así afectar los análisis adicionales sobre el material del núcleo.
4.2 MÉTODO RETORTA A PRESIÓN ATMOSFÉRICA
4.2.1 Procedimiento Básico 4.2.1.1 Fundamentos de Análisis Se obtienen las saturaciones de fluidos de agua y aceite con un proceso de retorta en el cual el aceite y el agua contenidos en una muestra fresca de material de núcleo triturado son vaporizados, condensados y recolectados en probeta calibrada. La saturación de gas se determina en una muestra adyacente litológicamente similar colocándola en una bomba de mercurio y midiendo la cantidad de mercurio inyectado con el agua y/o aceite en sus lugares. Tabla 4.1 - Tipo de Roca y Método Recomendado de
Saturación de Fluidos
Tipo de Roca Métodos de Prueba Recomendados
Carbonatos, clásticos consolidados No consolidadas (aceite liviano) No consolidadas (aceite pesado) Carbonatos con vugs Fracturadas De Arcilla Evaporados Baja permeabilidad Carbón Shales Oil shale Biatomita
a,b,c,d,e,f, c,d,e c,c(*),e b,d,e,f a,b,d a,c(*),e g,e a,b,c,d,e,f, h a,b,c a(*) c,e,
Clave: a=retorta a presión atmosférica b=extracción por destilación (diámetro completo) c=extracción por destilación (tapón) d=método de núcleo con presión retenida e=lavado con solvente/Karl Fischer f=método de núcleo de esponja g=método de contenido de yeso h=método de carbón (*)=procedimiento modificado
4.2.1.2 Aparato Los siguientes ítems describen los aparatos adecuados utilizados en el método básico de retorta: a. Retortas de acero inoxidable - Ver Figura 4-1.
Estas retortas están diseñadas para contener de 100 a 175 gramos de material de núcleo triturado. Cada retorta tiene una tapa roscada, que a su vez tiene un empaque para prevenir el escape de gases condensables. Se conecta un tubo de condensación largo de acero inoxidable al lado opuesto de la retorta. Se coloca una malla gruesa en el fondo de la retorta para evitar que el material de núcleo triturado entre en el tubo de condensación.

b. Horno - Ver Figura 4-1. Estos hornos son capaces de contener múltiples retortas. Se prefieren los elementos eléctricos de calentamiento tipo cinta. También se requieren un regulador de temperatura, un termopar, y un aparato de lectura de temperatura.
c. Baño de María - Ver Figura 4-1. Un baño de María debe ubicarse para que los tubos de condensación pasen por él, así intensificando la condensación de los gases emitidos.
d. Tubo de recepción de vidrio calibrado - Por lo general es aceptable un tubo de centrífuga común de 15 ml, aunque se puede requerir un tubo de mayor capacidad. El tamaño requerido depende de la cantidad de material de retorta y el total de fluidos contenido dentro de ese material. El tubo de recepción se conecta al extremo del tubo de condensación con un tapón de caucho.
e. Martillo y triturador de roca - Se puede utilizar una pica de geólogo o un martillo de mampostería. Los trituradores típicos son de la variedad "ardilla".
f. Sierra de diamante - Se puede utilizar una variedad de sierras comerciales con hojas de diamante.
g. Bomba de mercurio - Las típicas bombas de mercurio vienen con un arreglo de celda, tapa y válvula. Las celdas típicas pueden acomodar una muestra con un volumen de 10 a 15 cm3. La bomba de mercurio está dotada con una reglilla calibrada tipo nunio graduado en incrementos de 0.1 cm3. Se conecta un manómetro capaz de indicar presiones hasta de 1.000 psi (6.895 Mpa) a este sistema.
h. Centrífuga - Una centrífuga manual capaz de contener cuatro tubos de centrífuga de 15 ml es suficiente. Se pueden utilizar otras centrífugas de medidas iguales al tubo y fuerzas centrífugas necesarias.
4.2.1.3 Procedimientos/Precauciones El procedimiento básico normalmente es realizado en muestras tomadas de núcleos de diámetro completo. Se realizan análisis típicos pie por pie, excepto que algunas litologías como son el shale, anhidrita, yeso y arcillas pesadas se describen y no se tienen en cuenta para este tipo de análisis. Una muestra representativa de aproximadamente 2 pulgadas de longitud se parte
de cada pie utilizando un martillo de geólogo u otro similar. Esta muestra se parte en mitades a lo largo. Una mitad se utiliza para la muestra de saturación de fluidos y la otra mitad se utiliza para obtener un tapón para determinar la permeabilidad y/o la porosidad de la ley de Boyle (ver 5.3.2.1.1 y 5.3.2.2.1). Alternativamente, un fragmento de 1 a 2 pulgadas (25.4 a 50.8 milímetros) se puede partir adyacente al fragmento seleccionado para pruebas de saturación de fluidos y el tapón perforado. La ventaja de este último método de muestreo es que se puede sacar un tapón más grande más largo, así reduciendo el potencial de contaminación de fluidos de perforación. La muestra (10 a 15 gramos) para la medición de espacio de gas debe recortarse o moldearse a un tamaño conveniente, i.e., dimensiones que permitan colocarlo en la celda de la bomba de mercurio. Además, la muestra debe ser bien redondeada para permitir la adaptación con el mercurio. El peso de la muestra es registrado y se determina el volumen de esta muestra por el desplazamiento del mercurio para uso posterior en el cálculo por sumatoria de fluidos (ver 5.3.2.2.2 y 4.2.1.4). Se cierra la válvula de la celda y se incrementa la presión de 750 a 1.000 psi (5.171 a 6.895 Mpa) dependiendo de la permeabilidad y/o porosidad de la muestra. Se registra la cantidad de mercurio inyectado (corregido apropiadamente con los procedimientos de calibración de la bomba, ver 4.2.1.8). La muestra para la medición de agua y aceite debe ser triturado en fragmentos de 1/4 pulgada (6.4 centímetros). El material de núcleo triturado es seleccionado utilizando una malla Tyler 3 o 4 [aproximadamente 0.25 pulgadas (6.4 milímetros)] para remover los finos creados en el proceso de trituración. Una cantidad de este material (usualmente con precisión de 100 a 175 gramos) se pesa 0.01 gramos y se vierte en la retorta. Se aprieta la tapa de la retorta y ésta, junto con otras retortas preparadas de manera similar, se coloca dentro del horno. La temperatura del horno se mantiene inicialmente en 350ºF (177ºC) (Hensel 19821) hasta que las muestras dejen de producir agua. Los volúmenes de agua son registradas y luego la temperatura del horno se aumenta a una temperatura entre 1.000 y 1.200ºF (538 y 649ºC). Se supervisan los tubos de recolección de fluidos y cuando todas las muestras dejan de despedir fluidos, el proceso es considerado completo. El tiempo de retorta por lo general varia de 20 a 45 minutos. Se registran los volúmenes totales de agua y aceite. El ajuste inicial de 350ºF (177ºC) para la temperatura inicial del horno es seleccionado para remover el agua de poros, el agua absorbida, agua de arcilla entre capas (e.g., esmectita), y las aguas de hidratación (e.g., CaSO4), pero no agua de arcilla hidroxilo. Sin embargo, este método no es apropiado para muestras que contienen yeso o grandes cantidades de montmorrillonita. Los procedimientos de análisis de núcleos que contienen yeso se describen en 4.8 como un procedimiento para el análisis de núcleos especiales. Aunque las muestras que contienen montmorillonita pueden causar problemas en la determinación de saturación exacta de fluido, el calentamiento a 350ºF (177ºC) (Brown, 19612; Mackenzie, 19703) es documentado como la menor

temperatura en la cual se desorben aguas de hidróxilo. Aunque la pérdida de peso de agua de hidróxilo en arcilla en esta temperatura puede ser tan baja como el 5 por ciento, este puede tener impacto sobre los valores finales de saturación de agua y aceite. Este método es superior a las otras técnicas de retorta, e.g., el método meseta (Hensel, 19821). El método meseta sugiere que la representación gráfica de los datos del agua recolectada versus el tiempo, mientras se aplica calor continuamente, producirá mesetas donde se puede determinar un tiempo apropiado para la lectura de únicamente el agua de poros para cada formación probada. La temperatura final de retorta de 1.000 a 1.200ºF (538 a 649ºC) puede producir volúmenes de aceite incorrectos. Algunos materiales de núcleos pueden contener hidrocarburos sólidos, e.g., kerógeno, asfalto de Utah, etc. que se descomponen a estas temperaturas. El analista debe tener en cuenta la composición mineral y puede elegir hacer correcciones arbitrarias y sensatas para el aceite recolectado. En casos extremos, el procedimiento de retorta no debe utilizarse y se deben emplear otros métodos, e.g., procedimientos para la extracción por destilación (Dean-Stark) (ver 4.3). Otras fuentes de error en la medición de volúmenes de aceite surgen de: (a) restricción mecánica en los tubos de condensación, (b) sellos de retorta con escapes, y (c) coqueo y craqueo de ciertos aceites. Se deben emplear habitualmente las curvas de corrección de volúmenes de aceite (ver Figura 4-2).
Figura 4-1 Retorta de Horno y Baño de María de Acero Inoxidable
La cantidad "total" de agua recolectada después de todo el proceso puede ser valiosa para determinar si el agua de "poros" puede haber sido retenido mecánicamente en el tubo de condensación. El analista puede observar una densidad de grano calculada para una muestra que es irregular para la tendencia general. Una comparación entre el volumen inicial de agua (de poro) y el volumen final (total) de agua indica una diferencia mas grande que lo esperado basado en las diferencias de volúmenes de agua similares de muestras adyacentes. Luego el personal de laboratorio puede elegir hacer correcciones sensatas en los volúmenes de agua iniciales para que se puedan obtener datos razonables. El reporte de datos obviamente irregulares no le conviene al usuario final. Estos datos corregidos deben marcarse y se debe insertar una nota de pie de página. El material de núcleo triturado es susceptible a la pérdida de fluidos debido a la evaporación, especialmente de agua, debido a la alta área superficial del núcleo triturado. Se debe minimizar el tiempo de exposición para el material de núcleo triturado. Nota: Como se requiere el manejo de mercurio para este método de saturación de fluido, todas las normas existentes de seguridad y salud deben seguirse. Consulte los reglamentos ambientales y las leyes locales pertinentes. 4.2.1.4 Cálculos
El aceite corregido (ver Figura 4-2) y el agua de poro recolectado para cada muestra junto con el volumen de gas puede expresarse como un porcentaje de volumen total de la siguiente manera: Figura 4-2 - Curva de Corrección de aceite de Retorta
NDHg = WtHg/BvHg BvRet = WtRet/NDHg GB = (HgInj x 100)/BvHg OB = (OilVol x 100/BvRet WB = WatVol x 100)/BvRet GS = (GB x 100)/(GB + OB + WB) OS = (OB x 100)/(GB + OB + WB) WS = (WB x 100)/(GB + OB + WB) Donde: NDHg = densidad natural de una muestra para bomba de mercurio, g/cm3. WtHg = peso inicial de una muestra para bomba de mercurio, g. BvHg = volumen total de la muestra de la bomba de mercurio, cm3. WtRet = peso de la muestra de retorta, g. HgInj = volumen de mercurio inyectado corregido por factores de calibración de la bomba, cm3. OilVol = volumen del aceite recobrado corregido con curvas de calibración de aceite, cm3. WatVol = volumen de agua inicial recobrada, cm3. BvRet = volumen total de la muestra de retorta triturada, cm3. GB = saturación de gas como porcentaje del volumen OB = saturación de aceite como porcentaje del volumen WB = saturación de aceite como porcentaje del volumen GS = saturación de gas como porcentaje del volumen del volumen poroso OS = saturación de aceite como porcentaje del volumen del volumen poroso WS = saturación de agua como porcentaje del volumen del volumen poroso

La densidad de granos puede calcularse con los datos obtenidos durante el procedimiento de prueba (Hensel, 19821). Estos datos de densidad de granos son de valor primario como medio de control de calidad para asegurar que no se hayan tomado lecturas erróneas, que todos los fluidos se hayan determinado apropiadamente, y que no se han cometido errores en los cálculos. Esto no es el medio mas exacto para determinar la verdadera densidad de granos del material de roca. Sin embargo, el valor obtenido debe ser razonablemente representativo de la litología en prueba. Por lo tanto, el cálculo de la densidad de granos puede ser considerado una herramienta cualitativa para evaluación de datos. 4.2.1.5 Ventajas Las ventajas del método básico de retorta incluyen: a. Los fluidos son recolectados de muestras
relativamente grandes asegurando la mejor representación de la litología e incrementando la potencial precisión de todas las mediciones.
b. El proceso analítico es rápido y proporciona los datos de saturación requeridos dentro de pocas horas. Muchas muestras pueden ser evaluadas en corto tiempo y de manera económica, si hay suficientes retortas y hornos disponibles.
c. Los volúmenes de fluidos son medidos directamente a diferencia de otras técnicas que dependen de las combinaciones de volúmenes de fluidos y la pérdida de peso total.
d. La pérdida de granos, comúnmente asociado con areniscas friables y algunos carbonatos, no afecta los datos de saturación de fluidos.
4.2.1.6 Limitaciones Las limitaciones del método básico de retorta incluyen: a. La saturación de agua (y porosidad) puede ser
demasiado alto si las muestras contienen grandes cantidades de montmorillonita o yeso que se descomponen en altas temperaturas. Esto a su vez resultará en saturaciones de aceite demasiado bajas porque el volumen de aceite se expresaría como un porcentaje de un volumen de volumen poroso demasiado alto.
b. La saturación de aceite (y porosidad) puede ser demasiado alto si la muestra (e.g., algunos shales) contiene hidrocarburos sólidos que se descomponen en altas temperaturas. Esto a su vez resultará en saturaciones de agua demasiado bajas porque el agua de poro se expresa como porcentaje de un volumen de volumen poroso demasiado alto.
c. Se requieren curvas de corrección del volumen de aceite y puede que no sea posible obtener las correcciones para el aceite en cuestión. Puede ser necesario utilizar curvas generalizadas de corrección, basadas en gravedades API reportadas o supuestas.
d. Se requiere un segundo fragmento del material de núcleo para determinar el volumen total, densidad natural y el volumen poroso lleno de
gas. Esta muestra debe ser litológicamente similar a la muestra triturada para los datos de aceite y agua.
e. Los líquidos destilados pueden formar emulsiones.
4.2.1.7 Exactitud/Precisión La exactitud/precisión del método básico de retorta es: a. Utilizando una curva de corrección de volumen
de aceite, la exactitud del valor de aceite obtenido está dentro de +5% y la reproducibilidad está dentro de +2.5% de los volúmenes medidos.
b. La exactitud de los volúmenes de agua obtenidos con este método es del +2.5% de los volúmenes medidos.
4.2.1.8 Calibración Los puntos de calibración para el método básico de retorta incluyen: a. La probeta graduada debe ser revisado para
verificar su precisión utilizando una microbureta con agua. La limpieza de la probeta puede tener impacto sobre las lecturas debido a una falta de humectación completa y meniscos irregulares.
b. La bomba de mercurio utilizada para obtener volúmenes totales debe ser calibrada con patrones de acero de volúmenes conocidos. Además, se debe determinar un factor de bomba/sistema en la presión utilizada para determinar el volumen poroso lleno de gas. Se deben utilizar curvas y/o ecuaciones de corrección en ambos casos.
c. Todas las balanzas deben pasar por mantenimiento de rutina.
d. Las retortas deben ser sujetas a pruebas al vacío periódicamente para asegurar que los sellos están funcionando apropiadamente.
4.2.2 Núcleos de Pared por Percusión 4.2.2.1 Fundamentos de Análisis Este método es una modificación del procedimiento básico de retorta (ver 4.2.1). La diferencia principal es que las mediciones de los volúmenes de gas, aceite y agua son realizadas sobre la misma muestra a diferencia del procedimiento básico en donde se requieren fragmentos de núcleos adyacentes. En el procedimiento para muestras de pared las muestras son más pequeñas y por lo tanto los equipos son reducidos a escala para proporcionar una precisión comparable. Se puede aplicar este método en los núcleos de pared por rotación, pero esto daña las muestras para futuras pruebas. 4.2.2.2 Aparatos Los aparatos adecuados para la retorta de núcleos de pared incluyen: a. Retortas de acero inoxidable - Ver figura 4-3. El
diseño general es similar a las retortas utilizadas en el procedimiento básico, i.e., la retorta está

compuesta de una capa larga que contiene la muestra, un arreglo de empaque/tapa, y un tubo largo de condensación. La capa es de tamaño muy reducido. Se puede utilizar una tapa de botella en lugar de una tapa roscada, colocada con un aparato de tapado estándar. El tubo de condensación es mas corto y sus diámetros interior y exterior menores que los tubos utilizados para retortas mas grandes.
b. Horno - El horno para muestras de pared está compuesto de los mismos componentes que el horno utilizado en el procedimiento básico (ver 4.2.1.2, b). Se adapta el horno para núcleos de pared para acomodar múltiples retortas de pared.
c. Tubos receptores de vidrio calibrado - No se recomienda el tubo de centrífuga de 15-ml, mas bien el tubo receptor debe ser diseñado para acomodar muy pequeños volúmenes de fluidos(e.g., un máximo de 5 ml). Las graduaciones deben ser tales que sea posible obtener lecturas/interpolaciones dentro de los 0.01 ml mas cercanos.
d. Bomba de mercurio - Se puede utilizar la misma bomba de mercurio usado en el Procedimiento Básico (ver 4.2.1.2, g). Sin embargo, una cámara de muestras pequeña mejorará las mediciones de volumen total y espacio de gas.
e. El aparato para perforar la tapa del frasco es opcional (ver 4.2.2.3).
f. Un detector de gas de alambre caliente capaz de la medición cualitativa de gases combustibles es opcional (ver 4.2.2.3).
g. Un cromatógrafo de gas es opcional (ver 4.2.2.3). 4.2.2.3 Procedimientos/Precauciones 4.2.2.3.1 Procedimientos Las muestras deben recibirse en el laboratorio sellados en frascos de vidrio con tapas roscadas. Las muestras deben ser organizadas en orden de profundidad descendiente. Antes de abrir el frasco, muchos laboratorios emplean un procedimiento en donde se perfora la tapa del frasco y se realiza una medición del contenido de gases. No se hablará de este procedimiento en detalle en esta sección. Sin embargo, este procedimiento opcional mide los gases combustibles con un aparato de detección de gas de alambre caliente y algunos laboratorios determinan la composición de gas del espacio superior por cromatografía. Se indica el recobro de muestras (longitud de muestra), mientras sigue dentro del frasco, por las marcas de calibración en el costado del frasco. Luego se saca cada muestra del frasco y es limpiada de los sólidos de fluidos de perforación utilizando una cuchilla sencilla o una navaja. Se registra una breve descripción de la muestra incluyendo el tipo de roca, tamaño de grano o cristalino, si es lodoso, esquistoso, fósiles, color, intensidad y distribución de fluorescencia, y otras características litológicas megascópicas. La muestra es pesada con una precisión de 0.01 gramo. Algunos laboratorios pueden dividir la muestra preparada tal que suministre una porción para una medición de permeabilidad y otras medidas y/u otras,
distribución de tamaño de partículas, difracción de rayos x, etc. Esto obviamente está limitado a las muestras iniciales de gran tamaño, e.g., 2 pulgadas (50.8 milímetros) de longitud. Aun así los datos de saturación de fluidos son comprometidos por la prueba en una muestra mas pequeña. La mayoría de los laboratorios emplean un enfoque empírico en los datos de permeabilidad de pared. Ninguna de estas prácticas será discutida en esta sección.
Figura 4-3 - Cubeta de Retorta - Muestra de Pared Se coloca la muestra en la celda de una bomba de mercurio precalibrada, y se determina el volumen total por desplazamiento de mercurio (ver 5.2.1). Con la muestra sumergida en mercurio, se cierra una válvula y se aumenta la presión a 750 psi (5.171 Mpa). En muestras compactas (baja permeabilidad), se aumenta la presión a 1.000 psi (6.895 Mpa). Este procedimiento mide el volumen poroso llenos de gas suponiendo que la compresibilidad del agua y/o aceite presentes en los otros espacios de poros es mínima. La muestra es removida de la celda y dividida en fragmentos mas pequeños para observar el patrón de penetración de mercurio. El patrón de penetración es útil para pronosticar la producción probable. Se sigue el procedimiento de retorta descrita en 4.2.1.3, incluyendo el registro de agua inicial (de poros) y total recolectada y el uso de curvas de corrección de volumen de aceite (ver Figura 4-2). 4.2.2.3.2 Precauciones El material de núcleos de pared está sujeto a la pérdida de fluidos debido a evaporación así que se debe minimizar el tiempo de exposición a condiciones ambiente. Nota: Como se requiere el manejo de mercurio en este método de saturación de fluido, se deben seguir todas las normas de seguridad y salud existentes. Consulte los reglamentos ambientales pertinentes y las leyes locales.

4.2.2.4 Cálculos El volumen poroso llenos de aceite, agua de poros, y gas puede ser expresado como porcentaje de volumen total de la siguiente manera: GB = (HgInj x 100)/BV OB = (OilVol x 100)/BV WB= (WatVol x 100)/BV GS = (GB x 100)/(GB + OB + WB) OS = (OB x 100)/(GB + OB + WB) WS = (WB X 100)/(GB + OB + WB) Donde BV = volumen total de la muestra, cm3 HgInj = volumen de mercurio inyectado corregido por factores de calibración de la bomba, cm3 OilVol = volumen de aceite recobrado corregido con curvas de calibración de aceite, cm3 WatVol = volumen del agua recobrada inicial, cm3 GB = saturación de gas como porcentaje del volumen total OB = saturación de aceite como porcentaje del volumen total WB = saturación de agua como porcentaje del volumen total GS = saturación de gas como porcentaje del volumen poroso OS = saturación de aceite como porcentaje del volumen poroso WS = saturación de agua como porcentaje del volumen poroso Se puede calcular la densidad de grano con los datos obtenidos durante la prueba haciendo ligeras modificaciones en el método propuesto por Hensel, 1982.1 La densidad volumétrica, a menudo utilizado en varios laboratorios, puede ser calculado dividiendo el peso inicial de la muestra (entera) por el volumen total. 4.2.2.5 Ventajas Las ventajas del procedimiento de retorta para núcleos de pared incluyen: a. Se realizan las mediciones de gas, aceite y agua
utilizando una muestra. b. El método es relativamente rápido. c. Este es un método "directo" de medición donde
las mediciones de gas, aceite y agua son realizadas independientemente.
4.2.2.6 Limitaciones Las limitaciones del procedimiento de retorta para núcleos de pared incluyen: a. Los líquidos destilados pueden formar
emulsiones. b. Se requiere una calibración de aceite y agua c. Los volúmenes de aceite <0.1 ml son difíciles de
determinar con exactitud.
d. Para arenas muy friables o no consolidadas el volumen poroso llenos de gas puede ser alto debido a la expansión de la muestra cuando se libera la presión de sobrecarga.
e. Las muestras deben ser manejadas con cuidado para minimizar la pérdida de líquidos, especialmente agua. En una atmósfera de baja humedad, la pérdida de agua puede ser sustancial.
f. Los valores de porosidad y saturación de fluidos pueden ser erróneos si hay yeso o arcillas hidratables (e.g., montmorillonita) presentes y no se toman las precauciones para registrar únicamente el agua "de poros".
g. La calibración de todos los equipos es crítico porque las muestras son típicamente pequeñas y pueden contener igualmente pequeñas cantidades de aceite y agua. Mínimas diferencias en los volúmenes de fluidos registrados pueden resultar en rangos significativos de valores de saturación y porosidad.
4.2.2.7 Exactitud/Precisión La exactitud/precisión para el procedimiento de retorta para núcleos de pared es: a. La exactitud del método es fuertemente
dependiente del tamaño de la muestra, especialmente cuando se determinan los volúmenes de aceite.
b. Se debe utilizar discreción analítica en las pruebas de muestras con un peso inicial menor que 3 gramos.
c. Con la utilización de curvas de corrección de volumen de aceite, la precisión del valor de aceite obtenida está dentro del +5 por ciento de los volúmenes medidos.
d. La precisión de los volúmenes de agua está dentro del +3 por ciento de los volúmenes medidos.
4.2.2.8 Calibración Consulte la sección 4.2.1.8 del procedimiento básico de retorta. 4.3 METODO DE EXTRACCIÓN
POR DESTILACIÓN (DEAN STARK)
4.3.1 Muestras de Tapones 4.3.1.1 Fundamentos de Análisis Este procedimiento es apropiado para muestras de tapones y para núcleos de pared por rotación. El método de extracción por destilación (Dean Stark) para determinar la saturación de fluidos depende de la destilación de la fracción de agua, y la extracción de solvente de la fracción de aceite de la muestra. Se pesa la muestra y la fracción de agua es vaporizada por un solvente en ebullición. Se condensa el agua y

se recolecta en un recipiente calibrado. El solvente vaporizado también se condensa, remoja la muestra y extrae el aceite. La muestra se seca al horno y se pesa. El contenido de aceite es determinado por diferencia gravimétrica.
4.3.1.2 Aparatos y Reactivos Los siguientes aparatos son adecuados para este método y son ilustrados en la Figura 4-4. El aparato debe ser instalado en una campana de humos o un cuarto adecuado para evacuar los vapores del solvente. a. General - El aparato consiste de un manto
eléctrico o un aparato calentador con controles termostáticos. La unidad de destilación/extracción para una muestra consiste de un frasco de ebullición, un cartucho, sifón o trampa calibrada, y un condensador.
b. Frasco - El frasco es de boca ancha, cuello largo, y puede contener hendiduras en la base del cuello para soportar el cartucho de extracción.
c. Sifón - El sifón o la trampa tiene una sección graduada marcada en divisiones de 0.1 ml. El sifón graduado puede ser mas grande o mas pequeño para acomodar las muestras con diferentes volúmenes de agua. Junto a la sección graduada está un tubo o la trampa de vidrio doblado en ángulo recto con una articulación de vidrio en el extremo. La articulación de vidrio tiene una punta de gotera y la abertura en la punta debe ser diseñada para que el solvente que gotea se dirija al centro del frasco para asegurar el remojo del tapón de núcleo que se encuentra debajo. Una modificación en el sifón (Figura 4-5) permite determinar el agua gravimétricamente.
d. Condensador - El condensador es refrigerado por agua, de reflujo, de tubo de vidrio, con una envoltura condensadora de aproximadamente 11.8 pulgadas (300 milímetros) de largo y una cámara de aire (ver Figura 4-4). El fondo tiene una punta de gotera y el tubo interior debe ser vertical para reducir la dificultad para remover el agua de las superficies del condensador y la trampa.
e. Soporte desecante - Se conecta un soporte desecante de tubo de vidrio en la parte superior del condensador cuando se están extrayendo muestras y se utiliza un tapón de caucho cuando el aparato está inactivo.
f. Cartuchos de extracción - Se recomienda un cartucho de vidrio con fondo de vidrio poroso para sostener la muestra de tapón. Alternativamente, se puede utilizar un cartucho de celulosa. Sin embargo, los tipos de materiales de cartuchos diferentes a vidrio pueden causar errores en peso por la absorción de agua atmosférica. Además, los cartuchos de vidrio permiten ver el tapón para asegurar que el solvente está goteando y remojando la muestra para una extracción completa. Se puede utilizar un pequeño fragmento de lana de vidrio para cubrir el tapón de núcleo para prevenir erosión por el reflujo de solvente del condensador.
Figura 4-4 - Aparato Dean Stark para la Determinación
Volumétrica de Agua
Figura 4-5 - Aparato de Extracción Dean Stark para la Determinación Gravimétrica de Agua
g. Vaso de extracción - Se pueden utilizar vasos
adaptadas con sifones para sostener los cartuchos y permitir ciclos alternantes de inmersión/drenaje. Esto puede incrementar la eficiencia del proceso de extracción.
h. Horno de secado - Se puede utilizar un horno de convección o de vació que tenga un control de temperatura de +2ºC. Se prefiere un modelo a prueba de explosión, pero se pueden utilizar modelos que no son a prueba de explosión. Para muestras que contienen grandes cantidades de montmorillonita y otras arcillas, se puede utilizar un horno de humedad para ayudar a preservar una capa de agua sencilla o doble para aproximarse mejor al estado de hidratación esperado de las arcillas en el yacimiento. (Los niveles de humedad durante los procedimientos de secado son importantes para mantener las aguas de hidratación apropiadas.)
i. Perlas de ebullición - Se pueden colocar glóbulos de vidrio o pequeños recortes de material de alundum en el fondo del frasco de ebullición. Estos ayudan a reducir la tendencia del solvente a "golpearse" cuando se vuelve mas saturado de aceite. Si ocurre un golpe o un calentamiento excesivo, se crea un proceso de ebullición indeseable, contaminando la muestra y extendiendo el tiempo requerido para la fase inicial del proceso de extracción por destilación.
j. Solvente - Tolueno (calidad reactivo) u otro solvente adecuado.

4.3.1.3 Procedimientos/Precauciones 4.3.1.3.1 Procedimientos Se preparan las muestras utilizando un fluido compatible con el fluido utilizado para cortar el núcleo entero o los núcleos de pared perforados en el pozo. Esto asegura la preservación de aceite residual o la saturación de agua dependiendo del uso del fluido base aceite o agua. En el caso de las muestras congeladas, se debe utilizar nitrógeno líquido. El fluido restante que permanece en la muestra después del recorte de los extremos debe limpiarse utilizando un material que removerá el fluido superficial pero no extraerá el fluido del interior como una esponja. Este proceso se asemeja a la acción de una escobilla de goma. La superficie de la muestra debe tener una apariencia seca (con un brillo opaco) y no brillante con un exceso de fluido. La muestra de tapón debe pesarse con una exactitud de un miligramo (0.001 gramos) con una balanza analítica. El proceso debe realizarse rápidamente para minimizar la evaporación de los fluidos en la muestra. Después del peso final, las muestras y/o cartuchos deben colocarse dentro del aparato inmediatamente, o deben almacenarse en un recipiente para prevenir la evaporación hasta que se coloquen dentro del aparato. Este periodo de almacenamiento debe mantenerse al mínimo. La necesidad de pesar cada componente es para asegurar la explicación para la pérdida de peso. Cuando se utilizan cartuchos de extracción, estos se deben encontrar a temperatura ambiente y deben estar completamente secos. Las muestras son extraídas con un solvente que no aporta ni absorbe el agua recolectada. Se puede acondicionar el solvente agregando al menos 1 por ciento de agua por volumen al solvente e hirviendo el solvente previamente hasta que se estabilice la concentración de agua. Algunos laboratorios agregan el 15 por ciento de agua por volumen al solvente. El agua debe estar en exceso de la cantidad requerida para llevar a los solventes comunes al equilibrio cuando se evapora el agua por ebullición. El soporte del desecante asegura que la humedad atmosférica no afecta el agua recolectada. El agua puede adherirse al condensador y al costado de la trampa. Este fluido puede causar un error apreciable, especialmente para una muestra de tapón de baja porosidad. El agua adherida se puede desalojar con un chorro rígido de solvente de una botella de presión o un alambre de calibre pequeño con un anillo muy pequeño al final. Se utiliza el alambre para desalojar físicamente las partículas de agua que no se pueden remover con el chorro de solvente. A veces se utiliza detergente para aplanar la zona interfacial solvente/agua para una precisión mejorada. Pero por lo general no se recomienda el uso de detergente porque los efectos de una interacción de fluidos trifásica son impredecibles y se alterará la humectabilidad de la muestra. El proceso de destilación/extracción continua durante un mínimo de 48 horas. Se deben supervisar los niveles de agua a diario y se debe parar el proceso únicamente cuando no hay cambio en el volumen de agua recobrada en 24 horas. Se pueden requerir
periodos mas largos dependiendo del tamaño de la muestra de tapón y su permeabilidad. Esto es para asegurar que el solvente utilizado ha extraído todo el aceite posible del material de tapón. Cuando se trata de aceite viscoso (baja gravedad, alto asfalteno), se requiere otro tipo de solvente (ver 3.1) para limpiar el tapón completamente. Se debe guardar un registro completo de los volúmenes de agua recolectados. Se puede incrementar la velocidad de extracción y la eficiencia utilizando vasos de extracción dotados de sifones. Los métodos alternativos que aceleran la extracción implican el uso del aparato descrito hasta que la cantidad de agua recolectada se encuentra en un punto final estable, y luego se realiza uno de los siguientes procedimientos con las muestras y los cartuchos: a. Coloque las muestras en una unidad de fases de
vapor para terminar la extracción de aceite. b. Coloque las muestras en un limpiador de núcleos
a presión con CO2-tolueno para terminar la extracción de aceite. Esto debe limitarse a la muestras competentes de baja permeabilidad que no serán alteradas físicamente por este proceso.
c. Coloque la muestra en un extractor Soxhlet para alternar la extracción de aceite de tipos inmersión y drenaje.
d. Coloque la muestra en un aparato de extracción de aceite de flujo continuo.
e. Alterne entre tipos de solvente (e.g., tolueno y metanol).
La eficiencia de extracción se evalúa tratando la muestra con cloroteno bajo una fuente de luz ultravioleta para determinar si aún existe aceite, que despide rayos de luz fluorescente, o midiendo la densidad del grano de la muestra. Si la densidad de grano es menor que la anticipada para el tipo de roca, la muestra puede necesitar una extracción adicional. Luego se debe secar la muestra/el cartucho hasta lograr un peso estable. Las muestras con altas saturaciones de solventes inflamables deben ser secados en un horno de convección o de vacío a prueba de explosión, hasta que el solvente adicional se evapore antes de meterse al horno. Esto evita una posible explosión o incendio. Cuando se seca la muestra/el cartucho, se debe permitir que se enfríe a temperatura ambiente en un recipiente sellado tal como un desecador y luego se pesa. No se debe utilizar un horno que agregue humedad a la muestra para este paso. Sin embargo, cuando se van a medir porosidad y permeabilidad, las muestras que contienen grandes cantidades de montmorillonita y otras arcillas pueden requerir el secado en un horno de humedad para preservar el estado hidratado encontrado en el yacimiento. (El nivel de humedad en el horno es importante para mantener las aguas de hidratación apropiadas para medir permeabilidad y porosidad). Puede haber errores si no se tiene en cuenta un cartucho perforado, la pérdida de pequeñas partículas, y/o la perdida en la superficie de la muestra por la sal precipitada de salmueras concentradas. En el caso que ocurra una precipitación de sal pesada, se remueve la sal de la muestra por extracción con

metanol u otro solvente similar. Luego se seca y se pesa la muestra. El peso del agua recolectada en la trampa se resta de la pérdida total de peso líquido para determinar el peso del aceite extraído de la muestra de tapón. Un flujo de agua apropiado en el condensador mantendrá la temperatura lo suficientemente fresca para que los vapores se condensen en la tercera parte del fondo de la columna del condensador. 4.3.1.3.2 Precauciones Se deben seguir los reglamentos locales de seguridad con respecto al uso de reactivos. En general, sin embargo, se deben tener en cuenta las siguientes consideraciones de seguridad. a. El aceite puede contener compuestos que
exhiben propiedades cancerígenos y este puede ser inflamable.
b. El tolueno es moderadamente tóxico por absorción por la piel e inhalación. Este posee propiedades irritantes y anestésicos y es altamente inflamable.
c. Muchos otros solventes, mientras son eficaces para el proceso de remoción de aceite, pueden ser peligrosos y tóxicos. Consulte los reglamentos ambientales pertinentes y las leyes locales que controlan el uso de este solvente.
d. Los vapores del solvente deben condensarse en la tercera parte inferior del intercambiador de calor refrigerado por encima de la trampa de agua.
e. El analista debe estar atento al cambio en el punto de ebullición del solvente con altura o el cambio en el punto de ebullición del agua debido a sal en la solución. El punto de ebullición del solvente debe revisarse para asegurar una temperatura adecuada para la destilación de agua. Cuando se utilizan fluidos de perforación base KCl, el agua filtrado tendrá una concentración de sal del orden de 300.000 ppm, y por consiguiente, hervirá a una temperatura mucho mas alta que agua mas dulce. Se sugiere el uso de ortoxileno como reemplazo de tolueno en estos casos.
4.3.1.4 Cálculos Los siguientes cálculos son apropiados para este método: Porcentaje en peso de Agua (Gravimétrico) = (Peso del Agua) x 100
Peso inicial de la Muestra
ó Porcentaje en peso de Agua
(Volumétrico) = (Volumen del Agua) x (Densidad del Agua) x 100 / Peso inicial de la Muestra
Porcentaje en peso de Sólidos
(Gravimétrico) = Peso de la muestra seca x 100 / Peso inicial de la Muestra
Porcentaje en peso de Aceite
(Gravimétrico) = (Peso Inicial - Peso Seco - Peso del Agua) x 100 / Peso Inicial de la Muestra
Las saturaciones normalmente son expresadas como porcentajes de espacio poroso de la muestra. Por lo tanto, se requieren la porosidad de la muestra, la densidad del agua, y la densidad del aceite. Si el agua connata es una solución salina altamente concentrada, la densidad del agua debe corregirse para la sal en la solución. Se aplican los siguientes cálculos: % Agua = Volumen del volumen poroso
Volumen de Agua x 100
% Aceite=( Volumen de Volumen poroso
Peso Aceite)/(Densidad Aceite) x 100
El contenido líquido de la muestra es reportado con una exactitud del 0.1 por ciento de espacio poroso, e.g., 22.1 por ciento de aceite y 43.7 por ciento de agua. Conociendo la salinidad y la densidad de la salmuera, uno puede calcular el volumen de salmuera que se encontraba en el núcleo del volumen de agua destilada recobrada de la siguiente manera: Vbr = [(Vw)(ρw)/ρb] [1.000.000/(1.000.000 - Cs)] Donde: Vbr = volumen de salmuera correspondiente al volumen de agua destilada recobrada del tapón, cm3. Vw = volumen de agua destilada recobrada del tapón (e.g. Dean-Stark), cm3. ρw = densidad del agua destilada, g/cm3. ρb = densidad de la salmuera que tiene una concentración de Cs de sal, g/cm3. Cs = la concentración de sales disueltos en la salmuera =
Peso de la Salmuera ppm (1.000.000)(Peso de la Sal)
4.3.1.5 Ventajas Las ventajas de este método incluyen: a. Las determinaciones del volumen de agua
por lo general son bastante exactas. b. Generalmente, la muestra no se daña y
puede utilizarse para pruebas adicionales. Sin embargo, su humectabilidad puede ser alterada y ciertas arcillas (ejemplo, montmorillonita) o yeso también pueden ser sujetos a cambios.
c. Se utilizan temperaturas relativamente bajas [212ºF (100ºC)]; por lo tanto, se remueve poco o nada de las aguas de hidróxilo en la arcilla.
d. El procedimiento es sencillo y requiere poca atención durante la destilación.
4.3.1.6 Limitaciones Las limitaciones de este método incluyen:

a. Pueden aparecer errores en la
determinación de agua debido a los siguientes:
1. El agua atmosférica se condensa en el condensador cuando la humedad atmosférica es alta. Se pueden utilizar tubos disecantes para evitar este problema.
2. El agua se evapora de la muestra a temperatura ambiente cuando esta no se instala inmediatamente en el extractor con la circulación de agua de condensador.
3. Las perlas de agua se adhieren al vidrio sucio en el brazo lateral o el condensador.
4. Se puede precipitar la sal dentro de la muestra por salmueras connatas (agua intersticial salina). Esto puede resultar en cambios significativos en la porosidad y/o permeabilidad. Se puede remover la sal de la muestra con una extracción por metanol.
5. Se requiere una corrección por la mayor densidad del agua salada cuando la concentración total de sólidos se pasa de 20.000 ppm (ver 4.3.1.4, Ecuación 5).
6. El secado incompleto de solventes. 7. La pérdida de agua debido a que las
uniones en el frasco de extracción no son herméticas, o que las temperaturas de extracción sean demasiado altas, o de un flujo de agua insuficiente en el condensador.
8. Se debe considerar la flotabilidad de la densidad del aire solo cuando la muestra se pesa con una precisión de 0.1 mg.
9. El tiempo de extracción puede ser insuficiente.
10. La saturación de agua puede ser demasiado alta si las muestras contienen grandes cantidades de yeso (ver 4.8) o arcillas de montmorillonita (agua de hidratación). Los valores de permeabilidad y porosidad también pueden alterarse si las aguas de hidratación presentes en el yacimiento son removidos durante los procedimientos de extracción y secado (se prefiere el secado en un horno de humedad).
11. Si no se conoce la verdadera densidad del aceite, se introduce un error en el cálculo de saturación de aceite porque se debe suponer un valor para la densidad del aceite.
b. Los volúmenes de aceite no se encuentran directamente y pueden ser erróneos debido a los siguientes:
1. Agua adicional recolectado o perdido de la muestra mencionada arriba.
2. Pérdida de sólidos. 3. Limpieza incompleta del aceite. 4. Secado a una temperatura más alta que la
temperatura de extracción puede remover agua de hidratación adicional y dar un valor exagerado para el volumen de aceite.
c. La humectabilidad de la roca puede alterarse.
d. El material de la arcilla puede alterarse, y esto puede resultar en unas mediciones de permeabilidad erróneas.
4.3.1.7. Exactitud/Precisión No existen normas documentadas y la exactitud de los métodos no puede evaluarse. Sin embargo, con los procedimientos de calibración mencionadas en 4.3.1.8, la reproducibilidad del volumen de agua si se puede evaluar. Unos procedimientos similares evalúan la exactitud del volumen de aceite. En muestras relativamente pequeñas o muestras que contienen una alta saturación de gas con volúmenes residuales de aceite y agua, el error porcentual para las saturaciones de líquidos puede ser +50 por ciento de los valores medidos. El error porcentual será significativamente menor cuando se incrementan los volúmenes de líquidos. 4.3.1.8 Calibración Se debe revisar la exactitud de la medición de agua regularmente para asegurar que no haya ningún error sistemático que afecte los resultados. a. Para determinación gravimétrica - Los pesos
conocidos de agua agregado a los extractores son comparados con los pesos del agua recobrada por extracción bajo condiciones idénticas a aquellas utilizadas en la extracción de una muestra de arena de aceite. Los factores de corrección de agua pueden cambiar debido a la eficiencia de los condensadores utilizados en el equipo. Abajo se enumeran los valores típicos:
Peso Corregido Agua = (Peso Agua x a) + b
Donde: a y b, respectivamente, son la pendiente y la intersección de la ecuación de calibración, es decir, a = 1.003, +0.001; b = 0.090, +0.009. b. Para determinación volumétrica - Se utiliza una
bureta calibrada para dar volúmenes de agua conocidos en la trampa de agua. Las trampas deben tener un error de escala máximo de 0.002 ml. Se pueden calcular y aplicar los factores de corrección de volumen si es necesario.
4.3.2 Núcleos de Diámetro Completo 4.3.2.1 Principios de Análisis El método de extracción por destilación para determinar las saturaciones de fluidos en muestras de diámetro completo es gobernado por los mismos principios y procedimientos que aquellos utilizados en las muestras de tapón (ver 4.3.1.1). A continuación se tratan las diferencias en el procedimiento y equipos: 4.3.2.2 Equipos A continuación se relaciona el equipo necesario para realizar este método:
a. Recipiente de vidrio – El recipiente utilizado para acomodar la muestra con su diámetro total debe ser más grande que el recipiente de análisis de

tapones con un volumen de solvente más grande para la extracción de petróleo.
b. Trampa – La trampa o brazo tiene que ser lo suficientemente grande como para acomodar el volumen de agua presente en muestras más grandes. La sección graduada de este instrumento debe tener divisiones de 0.1 ml.
c. Protector de Extracción de la Muestra – En términos generales no se utilizan cartuchos para trabajar con muestras de diámetro completo. En su lugar se puede utilizar un material envolvente hecho de algodón no blanqueado para prevenir la pérdida de finos del núcleo de diámetro completo. Los cartuchos normalmente utilizados en el análisis de tapones no son fabricados en tamaños que se ajusten a los núcleos de diámetro completo.
4.3.2.3 Procedimientos Los procedimientos son iguales a aquellos para las muestras de tapón con la excepción de que los pesos de las muestras deben aproximarse a la décima de gramo (0.1). También, el tiempo de limpieza de 48 horas se puede extender con el fin de limpiar totalmente las muestras de volumen mayor. Frecuentemente se requiere mas tiempo y una limpieza adicional.
4.3.2.4 Cálculos Ver ecuaciones en 4.3.1.4.
4.3.2.5 Ventajas Consulte 4.3.1.5. Existen las siguientes ventajas adicionales cuando se realiza esta prueba en núcleos de diámetro total:
a. No se requiere una balanza analítica de alta precisión debido a que los pesos se calculan únicamente hasta la décima de gramo (0.1).
b. La pérdida de granos durante los procedimientos de manejo y prueba no es tan crítica como lo es con tapones, pero aun así debería minimizarse.
c. Los volúmenes de agua recolectada son bastante grandes en comparación con aquellos asociados con los análisis de tapones. Por lo tanto, las lecturas de agua necesitan registrarse aproximados al 0.1 ml.
d. Puesto que los volúmenes de petróleo son determinados por diferencia gravimétrica, los valores finales no se afectan en la misma extensión por pequeños errores en medición como aquellos determinados en tapones, excepto con muy bajas saturaciones de petróleo.
4.3.2.6 Limitaciones Existen las siguientes limitaciones cuando se analizan núcleos de diámetro completo:
a. Se requieren equipos más costosos como por ejemplo material de vidrio, hornos, sierras.
b. Se necesita un mayor espacio en el laboratorio. c. Se utilizan volúmenes más grandes de solventes
y por lo tanto la inversión es mayor. Las
logísticas de almacenamiento y/o disposición son más complejas.
d. Se necesitan períodos más largos para el proceso de destilación y limpieza de las muestras y por lo tanto se disminuye la puntualidad en el reporte de datos.
e. Se deben supervisar y observar muy cuidadosamente las normas de salud y seguridad debido a los grandes volúmenes de vapores y solventes comprendidos.
4.3.2.7 Exactitud / Precisión Consultar 4.3.1.7. 4.3.2.8 Calibración Consultar 4.3.1.8. 4.3.3 Análisis de Núcleos con Presión
Retenida 4.3.3.1 Introducción El objetivo del análisis de núcleos con presión retenida es proporcionar datos de saturación de fluidos sobre núcleos para los cuales se ha minimizado la expulsión de fluidos durante la recuperación de barril evitando la disminución de presión del fondo del hueco a condiciones de superficie. Una válvula de bola sella el barril y evita la caída de presión natural mientras se lleva el barril a la superficie. Además, el barril está diseñado para mantener la presión predeterminada durante la recuperación con una provisión de presión que compensa la pérdida de presión debido a una temperatura menor en la superficie.
Se minimiza la purga del núcleo con fluido filtrado de perforación antes de que el núcleo entre en el barril con un fluido de perforación especial que disminuye la pérdida de agua, tiene una alta tasa de penetración de corazonamiento, un bajo balance excesivo de fluido de perforación, y un diseño especial de broca. Además, antes de corazonar, el cilindro interior es por lo general llenado con un gel de baja invasión. Este material minimiza la invasión adicional de filtrado de fluido de perforación por medio de imbibición desplazando el fluido de perforación de la superficie del núcleo mientras el núcleo entra en el barril.
La saturación de fluidos es la cantidad de aceite y agua presente en el núcleo en condiciones de laboratorio. La alteración de las saturaciones de fluido originales por medio de purgas solo se pueden definir cualitativamente. Cualquier invasión de filtrado de fluido de perforación altera las saturaciones de los valores in situ a menos que las saturaciones in situ estén a condiciones residuales de invasión de agua. En yacimientos de baja presión, la invasión de filtrado se minimiza utilizando espuma como fluido de perforación. Con las precauciones anteriores, las saturaciones bajo estas condiciones pueden reflejar saturaciones cercanas a los valores in situ.
Se indican los procedimientos para el manejo en el sitio de perforación.

4.3.3.2 Principios del Análisis
Los núcleos se mantienen congelados con hielo seco hasta que se inicia el análisis de laboratorio. Esto reduce la presión de poros, congela el agua, inmoviliza el petróleo y atrapa el gas que no se licúa ni se congela a la temperatura del hielo seco.
Mientras se permite que el núcleo se descongele en las celdas de recolección de gas, el gas sale de la solución y expulsa tanto aceite como agua que son atrapados en un tubo receptor en el fondo de la celda. El gas es recolectado en el espacio vacío dentro de la celda. Este volumen de fluido no necesariamente refleja la cantidad de fluido producido por la disminución de presión de un núcleo convencional porque ambas condiciones de presión y temperatura diferentes existen en el yacimiento.
La pérdida de granos debe mantenerse a un mínimo para una mayor exactitud dado que los volúmenes de petróleo son determinados por los pasos del análisis de extracción por destilación y la extracción de solventes empujados por gas.
Si no hay gas libre presente en la zona corazonada y el núcleo se captura por encima del punto de burbuja del petróleo, la saturación total de líquido debe ser igual al espacio poroso después de aplicar un factor de formación volumétrico en el petróleo. Las saturaciones no deben variar mas del +5 por ciento.
El análisis de la invasión del filtrado nos permite evaluar la alteración de la saturación de fluidos de sus valores in situ. Las curvas de flujo fraccional de permeabilidad relativa ayudan mucho en este proceso de evaluación, si están disponibles. En la sección 4.3.7 se incluye un análisis más detallado respecto a estos principios de invasión.
4.3.3.3 Equipos Los siguientes equipos son necesarios para el análisis del núcleo de presión retenida. Estos ítems son considerados exclusivos para el análisis de núcleos de presión retenida. No se incluyen dispositivos utilizados para el análisis estándar de núcleos.
a. Cajas para el almacenamiento de núcleos - Cajas utilizadas para el almacenamiento del núcleo congelado hasta iniciar el análisis. Las cajas deben tener el tamaño suficiente para contener secciones de núcleo de 5 pies (1.52 metros) en su cilindro interior y suficiente hielo seco para mantener el núcleo en estado congelado. Estas cajas deben ser aisladas para minimizar la sublimación del hielo seco.
b. Hielo seco – Se utiliza para mantener la muestra o núcleo congelada hasta que se inicie el análisis.
c. Nitrógeno Líquido – Se utiliza para cortar los barriles que contienen el corazón y también durante su preparación, en los procesos de limpieza, perforación de tapones y revestimiento de muestras de diámetro completo.
d. Máquina de Corte – Utilizada para cortar ranuras diametralmente opuestas por todo el largo del
cilindro interior para facilitar la remoción de la muestra congelada.
e. Frasco dewar de Dos Litros de Capacidad – Utilizado para sumergir la muestra de manera periódica dentro del nitrógeno líquido para ayudar a la limpieza y para mantener el corazón congelado.
f. Herramientas para la remoción del Fluido de perforación o del Gel de baja invasión – Se incluyen varias herramientas como cuchillos, guantes aislantes, un martillo pequeño etc.
g. Celdas para la Recolección del Gas – Son celdas equipadas con un medidor de presión para determinar los volúmenes de gas que se han escapado del barril y de la muestra misma y que permiten análisis cromatográficos de gas.
h. Cilindros para la Recolección de Gas (250 centímetros cúbicos) – Se utilizan para la toma de muestras del gas presente en la celda con destino al análisis cromatográfico.
i. Centrífuga – Se utiliza para centrifugar los tubos de recepción presentes en la celda de recolección de gas para de esta manera obtener una buena separación de las fases petróleo/agua/sólidos.
j. Lámpara de Calentamiento – Se utiliza para mezclar el gas dentro de la celda de recolección antes de tomar la muestra.
k. Bomba de vacío portátil – se utiliza para hacer vacío en la celda de recolección de gas y en los cilindros de 250 centímetros cúbicos antes de la recolección del gas.
l. Tubos de Acero Inoxidable – [de diámetro de 3 pulgadas (76.2 milímetros) con fondos de malla] – las muestras de núcleo se colocan en estos tubos y de esta manera se evita que haya pérdidas de granos durante los procesos de recolección de gas y pasos de extracción por destilación del análisis.
m. Material Quirúrgico Envolvente – Utilizado para encapsular las muestras de núcleo durante el proceso de extracción con CO2 para de esta manera minimizar la pérdida de granos.
4.3.3.4 Procedimientos 4.3.3.4.1 Procedimientos para la Preparación
del Núcleo Los procedimientos para la preparación de la muestra incluyen los siguientes:
a. Las muestras de corazón, encapsuladas en tubos de acero y congelados en recipientes de hielo seco, se llevan del pozo al laboratorio.
b. Cada núcleo congelado y encapsulado en tubos se coloca en un recipiente lleno de hielo seco unido a una máquina de corte. Se cortan dos ranuras diametralmente opuestas a lo largo del tubo de acero a una profundidad ligeramente menor que el grosor de la pared del tubo. El nitrógeno líquido es dirigido hacia el punto de corte para asegurar una temperatura de tubo y corazón igual o menor que la temperatura del hielo seco.

c. Se separa el tubo en dos mitades y se retira la muestra congelada.
d. Se retira también el gel de baja invasión que se encuentra congelado con procedimientos de raspado y cepillado. Las secciones de muestra se sumergen en nitrógeno líquido periódicamente para de esta manera garantizar que permanece congelado.
e. Las muestras se examinan visualmente para determinar características litológicas y se seleccionan las muestras para el análisis.
f. Los extremos de cada segmento de corazón seleccionado son cortados con una sierra diamantada utilizando nitrógeno líquido como refrigerante. Los segmentos cortados de núcleo congelado se envuelven en plástico y papel aluminio y se almacenan bajo hielo seco hasta que se inicien las pruebas de laboratorio. La envoltura plástica y el papel aluminio protegen al núcleo de daños por sublimación
g. Un cuidado especial mientras el cortador abre el barril asegura un adecuado manejo del corazón. El uso de una malla pesada sobre el barril durante el proceso de corte previene heridas al analista si el barril se llegara a desprender debido a la excesiva presión interna causada por la expansión del fluido de perforación durante la congelación. Los bolsillos de gas natural donde el corazón se perdió dentro del barril pueden encenderse en el aire. El ambiente de dióxido de carbono y nitrógeno que rodean el barril durante el corte deben evitar que esto suceda.
4.3.3.4.2 Procedimientos para la Recolección
del Gas
Los procedimientos para recolección del gas incluyen los siguientes:
a. La muestra de núcleo cortada y congelada se coloca en un cilindro metálico de paredes delgadas con una malla fina en el fondo, se pesa rápidamente y se coloca en el sistema de recolección de gas (ver figura 4-6)
b. Se ensambla inmediatamente el sistema y se evacua durante 45 segundos para retirar el aire sin sacar el gas de la muestra. Se permite la descongelación de la muestra a temperatura ambiente.
c. El agua y el petróleo que salen por causa del gas en movimiento se recogen en un tubo graduado que está unido a la celda de recolección de gas.
d. El gas que sale se recoge en la celda de recolección respectiva. El sistema viene equipado con un medidor de presión que permite el monitoreo de la presión dentro de la celda. Si la presión en la celda excede los 0 psig, se abre una celda secundaria la cual ha sido evacuada con anterioridad para de esta manera recoger cualquier cantidad de gas adicional.
e. Se registran periódicamente los valores de presión barométrica, temperatura ambiente, presión del sistema y volúmenes de líquido producido. Se considera que la muestra se descongeló completamente cuando las lecturas
consecutivas nos indiquen equilibrio en el sistema.
f. Las muestras de gas emitido se pueden recoger en más de una celda. La muestra de gas que se recoja en una celda distinta a la primaria se analiza para determinar la gravedad y el porcentaje molar del gas. Se miden también los volúmenes de petróleo y agua que se recojan. Se determina el contenido de cloruro y de bromuro en el agua y se halla la gravedad del petróleo.
g. Se retiran de la celda la muestra y el cilindro correspondiente, se pesan y se colocan en un aparato de destilación que se ha preparado con anterioridad.
4.3.3.4.3 Procedimientos de Extracción
Destilación (Dean – Stark).
Los procedimientos de Extracción Destilación incluyen los siguientes pasos:
a. Se coloca la muestra en un aparato de Extracción Destilación (ver sección 4.3.1.2), se ensambla el sistema y se aplica calor para destilar el agua restante que permanece en la muestra y para extraer el petróleo que queda.
b. Cuando se completa el proceso de destilación, se registra el volumen de agua, el cual se determina por lecturas consecutivas. Se retiran la muestra y el dedal y se colocan en un horno de vacío a 240 oF (116 oC) para de esta manera retirar el solvente de extracción. Cuando se complete el proceso de secado, se sacan del horno la muestra y el dedal, se dejan enfriar en presencia de un desecante y luego se vuelven a pesar.
c. Se determina el volumen de petróleo adicional extraído de manera gravimétrica, utilizando la densidad del petróleo la cual ha sido determinada a partir del petróleo producido en el paso de recolección de gas. Se corrige el volumen del agua destilada para de esta manera obtener el volumen de equivalente de agua que tiene la misma salinidad que la del agua presente recogida durante la fase de recolección de gas. Si el agua expulsada durante la recolección del gas se contamina con filtrado de fluido de perforación, el agua recuperada durante la fase de recolección de gas puede que no tenga la misma salinidad si se le compara con agua de formación menos contaminada localizada en el centro de la muestra.
d. La muestra junto con todos los granos perdidos se envuelve con material quirúrgico envolvente para minimizar la pérdida de granos antes de hacer procedimientos adicionales de extracción.
4.3.3.4.4 Saturación y Extracción de Solvente
Empujado por Gas Los procedimientos de saturación y extracción de solvente empujado por gas incluyen:
a. Se pesan las muestras en el material envolvente **y se colocan en el extractor de solventes empujado por gas donde son sometidas a una limpieza posterior utilizando tolueno cargado con

dióxido de carbono y calentado a 180 oF (82 oC). Esta fase tiene como objetivo retirar cualquier aceite remanente. La pérdida de peso resultante de este proceso de extracción representa el peso del petróleo adicional que ha sido retirado y que se convierte en volumen de petróleo utilizando la densidad calculada previamente. En términos prácticos, para determinar el volumen de petróleo se utiliza la perdida de peso total que se obtiene a partir de los procesos de destilación extracción y extracción de solvente al cual se le resta el peso del agua recuperada.
b. Las muestras se colocan en un horno de convección (también denominado horno de vacío) y se someten a un secado a 240 oF (116
oC) hasta que el peso se estabilice. Para aquellas muestras que tienen grandes cantidades de montmorillomita y otras arcillas, es posible utilizar un horno de humedad para de esta manera mantener la capa de agua sencilla o doble y de esta manera aproximarse a las interacciones arcilla / agua que se espera encontrar en el yacimiento (es importante mantener los niveles de humedad durante el secado para mantener las aguas propias de hidratación).
c. Se miden las porosidades de diámetro completo junto con las permeabilidades al aire horizontales en cada segmento de la muestra. En los casos donde se combinen más de una muestra en un pie de análisis, estos valores se pesan en porciones representativas para un pie y se promedian para llegar a un valor promedio.
d. Las saturaciones de líquido en las condiciones del tanque se calculan utilizando el volumen poroso total de todos los segmentos que componen la muestra y el total de petróleo y de agua que se recuperaron de estos segmentos. El volumen de gas que se recogió de las muestras se determina a partir del volumen conocido del sistema de recolección de gas, el cual se corrige por los volúmenes de grano y la tara y a partir del contenido total de líquido y sal. Este volumen de gas se corrige posteriormente a condiciones estándar.
4.3.3.5 Cálculos Se utilizan los siguientes cálculos en este caso: a. El volumen de gas a temperatura y presión
estándar y el peso del gas se calculan así: Volumen de Gas =
14.7 x (460 + temperatura)
Volumen celda de recolección de gas x presión de la celda x 520 x (1- fracción aire)
Peso del Gas = Volumen del gas x 0.0012232 x
Gravedad del Gas Donde: El volumen de la celda se expresa en ml La presión de la celda se expresa en psia La temperatura se expresa en grados F El peso de gas se expresa en g.
El volumen del gas se expresa en ml a condiciones estándar
La gravedad del gas = (densidad del gas, g/ml) / (densidad del aire, g/ml)
Densidad del Aire a condiciones estándar = 0.0012232 g/ml
Las condiciones estándar – STP – son la temperatura a 0 grados centígrados y la presión a una atmósfera. El término (1-fracción de aire) es un factor de corrección. La evacuación de la celda de recolección retira todo el contenido de aire del sistema excepto un 5% y se pueden formar fugas pequeñas en el sello de la tapa que hace que entre aire al sistema. La cantidad de aire presente se determina haciendo un análisis de una muestra de gas obtenida a partir de la celda de recolección. El peso del gas se utiliza para revisar el balance material de la pérdida de peso total que resulta al extraer el gas, el petróleo y el agua de la muestra teniendo en cuenta la pérdida de peso que ya se ha calculado. b. Saturaciones: Saturación del Petróleo =
Volumen Poroso
(gcc Volumen Petroleo+vol Petróleo dest. Extr + Vol CO2 Extrac. Petról) x 100
Saturación de Agua =
Volumen Poroso
(gcc Volumen de agua + Volumen de agua por Dest Extrac) x 100
Donde: gcc Volumen de agua = volumen de agua por recolección de gas, en cm3 gcc Volumen de petróleo = volumen de petróleo por recolección de gas, en cm3
La saturación del petróleo se expresa como porcentaje de volumen poroso La saturación de agua se expresa como porcentaje de volumen poroso Todos los volúmenes de petróleo y agua son en ml. c. Volumen de Agua a partir de la destilación
extracción – ver la sección 4.3.2 d. Volumen poroso y Corrección de la densidad de
grano – cuando no hay remoción de sales de las muestras, las correcciones de volumen poroso y de densidad del grano se efectúan con base en la salinidad del agua, la cual se determina a partir del agua producida durante la colección de gas y la depleción de presión. También se puede utilizar metanol para remover la sal de la muestra.
Volumen poroso (corregido) = Volumen poroso + C
Volumen de agua x salinidad NaCl

Donde: El volumen del poro se expresa en ml. El volumen del agua a partir del proceso de destilación extracción se expresa en ml. La salinidad del NaCl se expresa en g / ml de agua destilada C = 2.165 g/ml (densidad de NaCl). Densidad de Grano (corregido) =
Volumen Total - Volumen Poroso (corregido)
Peso de la muestra - (Volumen de Agua x Salinidad NaCl)
Donde: La densidad de grano está en g/ml El volumen de agua por destilación-extracción está en ml. La salinidad de NaCl está en g/ml Volumen total está en ml El volumen poroso está en ml. 4.3.3.6 Ventajas y Limitaciones: Las ventajas / limitaciones de este método son: a. El corazonamiento con presión retenida es una
operación costosa, meticulosa y toma su tiempo realizarlo. Se requiere un hueco recto y limpio que contenga una espuma o fluido de perforación especialmente formulado.
b. Se debe ubicar un laboratorio que incluya los equipos especiales necesarios y supervisores expertos tan cerca al pozo como sea posible dado que puede ser difícil transportar con seguridad un núcleo congelado por grandes distancias.
c. El análisis suministra saturaciones de fluido que son más indicativas de los valores de saturación in situ si se le compara con otros análisis convencionales. La capacidad para evitar una expulsión de fluido durante el retiro de la muestra (caída de la presión) impide que haya alteraciones en la saturación durante el retiro de la muestra. Consulte las ventajas y limitaciones que tiene el proceso de limpieza en la destilación extracción (secciones 4.3.1.5 y 4.3.1.6) para de esta manera entender el impacto sobre la determinación de la saturación.
d. Minimizar el lavado del espacio poroso durante el corazonamiento es otro factor que debe considerarse para obtener valores cercanos de los valores de saturación in situ. Los procedimientos de corazonamiento con presión retenida están diseñados para evitar alteraciones por la caída de presión, pero no por el lavado. Como resultado, las saturaciones de los núcleos recuperados normalmente en fluidos de perforación base agua nos indican valores de saturación de aceite in situ solo si el lavado del filtrado es mínimo (ver 4.3.7). Si se presenta una gran cantidad de lavado, las saturaciones medidas indican mas bien los valores residuales. Esto es importante para la evaluación de proyectos de recobro terciario.
e. El lavado se minimiza cuando se disminuye el sobre balance de fluidos de perforación durante el corazonamiento. Esto se consigue utilizando fluidos de perforación de pérdida baja y penetrando la formación tan pronto como sea posible manteniendo al mismo tiempo un buen nivel de recobro. En ciertas situaciones, como por ejemplo presión depletado en el fondo del pozo, el utilizar una espuma estabilizada como fluido de perforación nos minimiza el lavado. No obstante, si la espuma se rompe durante el corazonamiento, el corazón está sujeto a altas pérdidas de fluido surfactante y ocurre un apreciable lavado.
f. La capacidad que se tenga para capturar agua y petróleo en la muestra proporciona una ventaja muy clara con respecto a los análisis convencionales y de esponja. Si se captura toda el agua que existía en la muestra al completar el corazonamiento, es posible determinar el volumen filtrado del gas, petróleo o agua que se desplazó durante las operaciones de muestreo. Si se conocen las saturaciones medidas y la cantidad de filtrado, la alteración de la saturación durante el corazonamiento se puede evaluar de manera cualitativa utilizando las características de flujo fraccional de permeabilidad relativa de la formación.
Por su parte, las muestras convencionales y de esponja permiten únicamente la determinación de filtrado residual una vez se ha completado el corazonamiento debido a que los fluidos se expulsan durante el retiro de la muestra. Puesto que la mayoría del filtrado se localiza en la periferia de la muestra, la medición del filtrado es erróneamente baja. En una muestra convencional, la saturación del petróleo también se reduce durante el proceso de depleción de la presión, aunque este petróleo puede quedarse en la esponja y luego analizarse. Si las saturaciones del petróleo están cercanas a las residuales, el valor de Kg / Ko es tal que usualmente muy poco petróleo es expulsado por empuje de gas. g. Puesto que el gas se retiene dentro de esta
muestra, es posible medir el volumen del gas así como su composición. Estos valores son importantes cuando se evalúen las eficiencias de barrido a partir de la inyección del gas. Las saturaciones residuales en zonas con un barrido alto permiten la evaluación de varias propiedades petrofísicas de la roca a saturación residual una vez se ha inyectado el gas.
h. Las relaciones de gas – petróleo, cuando se promedian en un intervalo, han concordado favorablemente con las mediciones de producción.
i. La gravedad del petróleo puede determinarse por cada pie en todo el intervalo de la muestra para así determinar si esta gravedad cambia de acuerdo con la profundidad. Los cambios de gravedad deben ser apreciables para ser observados por este método.
j. El diámetro de los núcleos de presión es en la actualidad 2.5 a 3.75 pulgadas (64.5 – 95.3 milímetros). El diámetro más pequeño aumenta

la proporción que hay entre el área de superficie y el volumen. El efecto de la invasión del filtrado del fluido de perforación y la alteración de la saturación es mayor en este caso que en un corazón con diámetro más grande. Al igual que en cualquier medición o análisis, el grado de exactitud disminuye a medida que se disminuye el tamaño de la muestra. Por lo tanto, a medida que se aumenta el diámetro del corazón pequeños errores de medida tendrán menos impacto sobre los análisis en corazones de presión retenida.
4.3.3.7 Exactitud / Precisión La exactitud / Precisión de este método es: a. Saturaciones de Fluido – Los límites de exactitud
para la determinación de fluidos para cada fase del análisis se indican a continuación:
1. Recolección de gas: Los volúmenes de fluido son ±0.5 ml. Los volúmenes de agua (destilación extracción) son ±0.5 ml.
2. Extracción con Solvente empujado por gas: Volumen del petróleo es ±0.1 g o ±0.1 ml.
b. Volumen del gas – El volumen del gas se
determina utilizando la Ley de Boyle. El volumen de gas resultante es ±2% del volumen real. Las variables que se utilizan en el cálculo del volumen de gas tienen una exactitud que se describe así:
Volumen de la Celda = ±5 ml Temperatura de la Celda = ±0.5 Fahrenheit Presión de la Celda = ±0.2 psia Fracción de aire = ±0.5 molar % 4.3.3.8 Calibración Los aspectos relacionados con la calibración incluyen
los siguientes: a. Los tubos de recepción ubicados en las celdas
de recolección de gas y los aparatos de destilación extracción deben calibrarse con agua desionizada en una balanza que puede medir centésimas de gramo (0.01 g). Los pesos hallados en cada intervalo se convierten a volúmenes utilizando la densidad del agua a la temperatura adecuada. Se deben determinar volúmenes en los tubos de recepción para cada lectura de 10 ml. Se interpola para determinar los volúmenes reales entre los volúmenes calibrados.
b. Los volúmenes en las celdas de recolección de gas se deben calibrar utilizando un gasómetro o un medidor exacto. Cada celda se evacua y se determina el volumen. El grado de repetibilidad
de los volúmenes ya medidos debe estar en un valor de 5 ml.
c. Las balanzas se deben revisar a intervalos regulares de tiempo para garantizar la exactitud en las mediciones.
4.3.4 Análisis de Corazones en Esponja 4.3.4.1 Principios de Análisis: En esta técnica la muestra entra en una manga hecha
con esponja de Poliuretano o de celulosa / poliuretano de media pulgada de espesor la cual se coloca dentro de un tubo de aluminio. A medida que la muestra se acerca a la superficie, los gases en expansión desplazan el crudo que se captura por medio de una manga hecha de esponja (el agua es capturado por una manga hecha de esponja mojado por agua). La manga hecha de esponja y humedecida con petróleo está hecha de poliuretano y tiene una porosidad de aproximadamente el 70%. Tiene la capacidad de comprimirse hasta llegar a niveles de porosidad casi nula. La esponja se satura con salmuera antes de introducirla en el pozo. Si se conocen las propiedades de la salmuera de la formación, esta solución utilizada para saturar la esponja debe corresponder con la salinidad y densidad del agua en la formación a ser corazonada. Cualquier cantidad de petróleo que salga de la muestra va a desplazar el agua presente en la esponja y se distribuirá en una capa delgada sobre las paredes de los poros de la esponja. La esponja mojado por el agua está hecha de una mezcla de fibra de celulosa con poliuretano. La esponja se satura con aceite mineral seco antes de introducirse en el pozo a corazonar, con un fluido de perforación base aceite. El objetivo es capturar cualquier cantidad de agua que escape de la muestra en el momento en que sale a la superficie. 4.3.4.2 Sistemas 4.3.4.2.1 Equipo Una sierra de mesa de alta velocidad y al menos una hoja de por lo menos 10 pulgadas (25.4 centímetros) de diámetro hecha de carburo de diamante y un motor de 1.5 caballos de potencia para abrir el barril de aluminio. 4.3.4.2.1.1. Equipo para el Análisis de la Esponja impregnada de Petróleo Se requiere el siguiente equipo adicional para el análisis de esponja mojada por petróleo: a. Recipiente para sostener la esponja (una lata
metálica, un Soxhlet grande etc.) b. Un espectrómetro para determinar la cantidad de
crudo en solución, es decir: 1. Un espectrómetro visible (detecta la
intensidad del color del crudo) 2. Un espectrómetro de Fluorescencia
Ultravioleta (detecta aromáticos)

3. Un espectrómetro infrarrojo próximo (detecta los enlaces C – H y los asfaltenos).
4. Espectrómetro de resonancia Magnética Nuclear (detecta el cambio en C-H)
5. Balanza analítica para preparar los estándares de extractos de crudos que se utilizan en la calibración de la respuesta espectroscópica.
6. Cromatógrafo de gas (opcional). 4.3.4.2.1.2 Equipo para el Análisis de la
Esponja Mojada por Agua Además de 4.3.4.2.1, el análisis de esponja mojada por agua requiere un aparato de extracción por destilación estándar (por ejemplo, Dean-Stark) para determinar el volumen de agua recobrado (ver 4.3.2). 4.3.4.2.2 Reactivos 4.3.4.2.2.1 Análisis de Esponja Mojada por Aceite Se utiliza cualquier buen solvente de crudo que: a. No tenga color visible, si se utiliza el
espetrómetro visible. b. No tenga aromáticos, si se utiliza el
espectrómetro de fluorescencia ultravioleta. c. Preferiblemente que no tenga enlaces C-H, si se
utiliza el espectrómetro de infrarrojo próximo o el espectrómetro de resonancia magnética (por ejemplo, clorocarbonos o clorofluorcarbonos libres de hidrógeno).
4.3.4.2.2.2 Análisis de la Esponja Mojada por Agua Cualquier solvente estándar para destilación extracción, por ejemplo el tolueno. 4.3.4.3 Procedimiento/Precauciones 4.3.4.3.1 Manejo en el Sitio de Perforación En el sitio de perforación, se debe tener cuidado para no sacudir los extremos del forro de la esponja, porque esto puede causar cambios de posición del núcleo de roca en el barril resultando en una falla en el apareamiento entre la esponja y la roca. Aunque el núcleo de esponja debe mantenerse fresco, congelarlo con la roca aun adentro puede resultar en el riesgo de inducir fracturas en el núcleo de roca. Para ciertas formaciones, como por ejemplo los carbonatos que tienen fisuras o que están fracturados, el núcleo puede romperse y acuñarse en la esponja causando amontonamiento. El corazón de esponja de diámetro mas grande es menos susceptible a este problema. Inmediatamente después del recobro, los corazones pueden cortarse en secciones (típicamente alrededor de 5 pies) y almacenarse en recipientes para transporte con el mismo fluido utilizado para saturar la esponja. 4.3.4.3.2 Análisis de Esponja Mojada por Aceite
Debido a que la esponja se satura previamente con agua y es altamente compresible, cualquier análisis de la cantidad de aceite en la esponja debe medir el aceite directamente, y no debe inferir el volumen de aceite de una medición del agua en la esponja y la porosidad (como se hace en el método de extracción por destilación para medir las saturaciones de aceite en rocas). Debido a que el petróleo en la esponja se ha distribuido uniformemente en una capa delgada sobre la esponja mojada por aceite, la compresión mecánica de la esponja no constituye un buen método para tratar de recobrar el aceite que ha sido capturado allí. Se puede utilizar un instrumento de Resonancia Magnética Nuclear de diámetro interior grande, como por ejemplo un detector de imagen NMR, directamente para determinar la cantidad de aceite en la esponja, siempre y cuando la manga de esponja se retire del forro de aluminio conductor de electricidad. Este método directo de NMR se volverá cada vez más atractivo a medida que el costo de medición disminuya y la disponibilidad de NMR aumente. Desde 1993, el método menos costoso y mas ampliamente aplicado, y el que se recomienda aquí, es la disolución del solvente del crudo capturado seguida de una determinación espectroscópica de la cantidad de crudo en solución. Como la esponja puede estar sujeta al ataque de solventes durante el paso de disolución, el se deben seleccionar solventes que sean suaves con la esponja o asegurarse que los análisis posteriores no se afecten por los componentes de la esponja disueltos al mezclarse con el crudo disuelto. Para la determinación espectroscópica, se debe elaborar una curva de calibración inyectando volúmenes o pesos conocidos de crudo en partes de la esponja y luego extraer la esponja con el solvente para determinar la respuesta espectroscópica de los extractos. Las muestras de esponja deben tomarse del mismo lote de esponja que utilizado en el corazonamiento. Si se utilizan lotes diferentes de esponjas durante el corazonamiento, se recomienda elaborar una nueva curva de calibración. 4.3.4.3.3 Análisis de Esponja Mojada por Agua La esponja mojada por agua es higroscópica, así que se debe tener especial cuidado para evitar que la esponja absorba humedad del ambiente porque esto podría tener un impacto en el análisis final para la saturación de agua. Se pueden realizar análisis estándar de destilación por extracción en la esponja mojada por agua para determinar el contenido de agua. 4.3.4.3.4 Separación de la Manga de Esponja y el Forro de Aluminio del Corazón de la Roca Con una sierra de mesa se hacen dos cortes separadas 180 grados a lo largo del forro de aluminio (eje longitudinal) que envuelve la manga de la esponja. Fije la profundidad del corte en 0.25 pulgadas (6.35 mm) para que la cuchilla de la sierra

corte únicamente el recipiente de aluminio y no la manga de esponja. También haga el corte entre un par de aletas centralizadoras (estas son las aletas que mantienen la muestra perfectamente centrada dentro del forro de aluminio), en lugar de hacerlo en la parte superior de la aleta. Luego, corte a través de la esponja utilizando un cuchillo y retire cada sección de la muestra del forro de aluminio para desbaste. Observe cualquier evidencia que indique que la muestra a cambiado de posición con respecto al recipiente. Marque el recipiente para que pueda cortarse en la misma longitud de la sección correspondiente de corazón desbastado y marque las piezas de núcleo completo y el forro. Complete el proceso de corte y sellado de la roca y las muestras de esponja en 30 minutos para así minimizar su exposición al medio ambiente. Esto minimiza la evaporación de los fluidos del núcleo, la oxidación del crudo líquido y los errores que pueden resultar en la cuantificación de la cantidad de crudo. Si la esponja no se analiza de inmediato, presérvelo cuidadosamente hasta que se pueda analizar (ver 2.2.6 – métodos de preservación). La saturación total corregida de aceite (o de agua) se obtiene sumando el volumen de aceite (o agua) en cada pieza de corazón desbastado con el volumen de aceite (o agua) en la manga de esponja correspondiente y dividiendo este resultado por el volumen poroso total. 4.3.4.3.5 Extracción por Solvente y Análisis Espectroscópico (esponja mojada por aceite) El siguiente es el análisis de esponja mojada por aceite: a. Prepare las muestras de esponja que tengan
cantidades conocidas de crudo y extraiga la esponja con el solvente elegido. Calibre la respuesta espectroscópica utilizando estos extractos.
b. Coloque el crudo del corazón de la esponja en la solución de cualquiera de las siguientes maneras: 1. Extracción con Soxhlet 2. Saturación por sumersión y agitación fuerte 3. Saturación por sumersión y desagregado de
la esponja en una licuadora. Para opciones 2 y 3, será necesario separar la manga de esponja del forro de aluminio. 4.3.4.4 Cálculos 4.3.4.4.1 Análisis de Esponja Mojada por
Aceite Los cálculos para el análisis de la esponja mojada por aceite son los siguientes: a. Determine la respuesta espectroscópica de una
alícuota del extracto de solvente.
b. Conociendo la cantidad total de extracto de solvente y haciendo una calibración con estándares preparados, determine la cantidad total de crudo que fue absorbida originalmente por la esponja.
Vtc = Wtc / ρc = (Ra / Rpwf) (Wts / ρc) Donde: Rpwf = respuesta del espectrómetro por fracción de peso del crudo absorbido por la esponja en el extracto de solvente final. Esta calibración de respuesta se realiza utilizando los estándares preparados que consisten de extractos de solventes de muestras de esponjas (del lote actual) que han sido inyectadas con cantidades conocidas de crudo. (Por ejemplo, la absorbencia o la fluorescencia del solvente por fracción de peso del crudo absorbido por la esponja cuando se utiliza una celda dada a una longitud de onda de luz dada). Las unidades de Rpwf deben ser iguales a las unidades de Ra Ra = Respuesta del espectrómetro para una alícuota del extracto solvente recobrado de un pedazo de esponja. La respuesta se obtiene bajo las mismas condiciones (es decir, utilizando la misma celda de la muestra, longitud de onda, etc.) que se utilizaron para calibrar la respuesta del espectrómetro con estándares preparados. Las unidades de Ra deben ser iguales a las unidades de Rpwf Wts = Peso de la solución total de crudo recobrado de un pedazo de esponja. El peso se mide antes de retirar la pequeña alícuota, g. Wtc = Peso total del crudo que estaba en un pedazo de corazón de esponja, g. Vtc = Volumen total del crudo que estaba en un pedazo de corazón de esponja, cm3. ρc = Densidad del crudo, g/ cm3. 4.3.4.4.2 Análisis de Esponja Mojada por
Agua Si se conoce la salinidad de la salmuera y la densidad, el volumen de la salmuera presente en la esponja puede calcularse a partir del volumen de agua destilada recobrada de la esponja, así: Vbr = [(Vw) (ρw) / ρb] [ 1.000.000 / (1.000.000 – Cs) Donde: Vbr = Volumen de la solución salina que corresponde al volumen de agua destilada recolectada de una parte de la esponja, cm3. Vw = Volumen de agua destilada recolectada de un pedazo de esponja (por ejemplo, Dean Stark), cm3. ρw = densidad de agua destilada, g/cm3

ρb = densidad de solución salina que tiene una concentración, Cs, de sal, g/ cm3. Cs = Concentración de sales disueltas en la solución salina = (1.000.000) (peso de sal) / (peso de solución salina, ppm. 4.3.4.5 Ventajas/Limitaciones El corazón de esponja es una alternativa menos costosa al método de presión retenida (descrita en la sección 4.3.3) y es más sencillo desde el punto de vista operacional. La cantidad de aceite (o de agua) capturada en la esponja se agrega a la cantidad de aceite (o agua) que permanece en el corazón para obtener valores de saturación de aceite (o agua) más exactos. A diferencia de los corazones de presión, los gases de hidrocarburos que se liberan no son retenidos, y por lo tanto no se encuentran disponibles para el análisis. De igual manera, con el corazón de esponja, se debe elegir entre la determinación de valores corregidos de la saturación de aceite del corazón o la determinación de los valores corregidos de la saturación de agua. Ambas saturaciones corregidas no pueden ser determinadas con el mismo corazón esponjoso. 4.3.4.6 Exactitud / Precisión Para una combinación de análisis espectroscópicos y de extracción, el volumen de aceite tomado del análisis de la esponja mojado por aceite está dentro de un 5% de exactitud del volumen real de aceite o 0.2 ml de aceite, la cantidad que sea mayor. El volumen de agua del análisis de corazón de esponja mojado por agua se encuentra dentro del 0.1 ml con respecto al volumen real, a partir del cual se puede calcular el volumen correspondiente de solución salina, si se conoce la salinidad y la densidad. El volumen de agua se determina con mayor exactitud que el volumen de aceite porque el agua es determinado por una medición directa del volumen. Por el contrario la muestra de aceite se encuentra altamente diluida en solvente y solo una alícuota de la solución diluida es analizada espectroscópicamente. 4.3.4.7 Calibración Consultar 4.3.1.8. Se deben seguir los procedimientos de calibración estándar para los equipos comerciales. 4.3.5 Shale Productiva 4.3.5.1 Principios de Análisis Este párrafo cubre las consideraciones especiales para medir saturaciones de fluido en espacios porosos en "shale productora de petróleo” - rocas compuestas de partículas de tamaño que varía desde la arcilla hasta el limo y son potencialmente productoras de petróleo. El análisis de muestras tomadas de este tipo de formación pueden requerir un manejo especial debido a su baja permeabilidad, una posible presencia de agua densa y presencia potencial de orgánicos
sólidos. No se recomienda un proceso de retorta para muestras que contienen orgánicos sólidos. Los orgánicos sólidos en temperaturas utilizadas en este proceso generarán petróleo produciendo altos volúmenes. Se debe utilizar un proceso de destilación para determinar los datos de saturación de fluido. El principio de la técnica es igual que la sección 4.3. Se debe utilizar este método si los orgánicos sólidos presentes no van a incluirse como volumen de petróleo. Si se requieren tiempos de análisis más cortos, la remoción de hidrocarburos puede agilizarse aplastando la muestra. La muestra aplastada también agilizará el tiempo de equilibrio para las mediciones de volumen de grano. 4.3.5.2 Equipo El equipo para extracción por destilación es igual que en 4.3.1.2. Para el aplastamiento de muestras se necesita un almirez con su mano. 4.3.5.3 Procedimientos/Precauciones Los procedimientos son iguales a los de 4.3.1.3. Si las muestras van a ser aplastadas para agilizar la remoción de hidrocarburos o la determinación de volúmenes de grano, se deben entonces medir los volúmenes totales y las muestras deben pesarse antes de someterlas a este proceso de aplastamiento. Se aplasta la muestra con el mortero y el mano y luego se pesa nuevamente. Si existe una perdida de peso durante el aplastamiento, se debe efectuar una corrección de volumen de roca o de fluido. Si la pérdida de peso es alta, se debe descartar la muestra y se debe probar otra. Un control cuidadoso de la pérdida de muestras durante todo el procedimiento es crítico para lograr mediciones de volumen muy exactas. 4.3.5.4 Cálculos Los cálculos deben realizarse de la manera estipulada en 4.3.1.4. 4.3.5.5 Ventajas Las ventajas de este método incluyen: a. Todas las mediciones pueden realizarse sobre la
misma muestra. b. Los orgánicos sólidos no son representados en
volúmenes de petróleo. c. Se pueden medir densidades de grano exactas. 4.3.5.6 Limitaciones Las limitaciones de este método incluyen: a. Los volúmenes de petróleo son sensibles a las
técnicas de secado. b. Se requieren largos tiempos de extracción debido
a la baja permeabilidad de este tipo de material rocoso.
c. La pérdida de la muestra es crítica para la determinación del contenido de petróleo.

4.3.5.7 Exactitud/Precisión La aplicación de las técnicas apropiadas de destilación y de secado deben permitir una determinación del contenido de agua y petróleo con una precisión de 2% de los valores medidos. Se debe tener un cuidado especial en el secado de las muestras por la capacidad de rehidratación de las rocas, lo cual ocasionará la medición de bajos volúmenes de petróleo que no corresponden a la realidad. 4.3.5.8 Calibración Las técnicas de calibración deben realizarse de la manera descrita en 4.3.1.8. 4.3.6 Análisis de Arena de Brea (Petróleo) 4.3.6.1 Principios de Análisis El método de extracción por destilación puede utilizarse para determinar las saturaciones de fluido en arenas de brea (petróleo) no consolidadas. Sin embargo se requieren ciertas modificaciones en el procedimiento y la presentación de datos. Los cambios necesarios se describen en los siguientes párrafos. 4.3.6.2 Equipo Se pueden utilizar los mismos equipos básicos descritos en 4.3.1.2. 4.3.6.3 Procedimiento/Precauciones El corazón no consolidado puede ser congelado antes de obtener la muestra. El tapón debe ser perforado utilizando nitrógeno líquido como lubricante. Este tapón congelado se coloca dentro de una manga, por lo general en politetrafluoroetileno (PTFE), que se encoge con el calor, con el fin de mantener el tapón intacto cuando se remueve el alquitrán. Luego se coloca la muestra en el equipo de extracción por destilación (ver Figuras 4-4 y 4-5) utilizando tolueno como solvente. En algunos casos, el corazón no puede congelarse, o si la muestra se obtiene durante el proceso de extracción de un depósito de arena de brea, la muestra de prueba puede analizarse basado en volumen/peso. En estos casos, la muestra es transferida a un cartucho de extracción apropiado como los que se mencionaron en 4.3.1.2 (f). Hay que evitar una ebullición violenta suministrando un calor mínimo para hervir el solvente. Se cambia el tolueno, cuando sea necesario, para evitar una acumulación de la brea y una subsecuente "ebullición intermitente" o sobrecalentamiento. Esta última condición, en el caso que se presente, puede frenar el proceso de destilación/extracción o posiblemente anular toda la prueba. Si el alquitrán tiene un punto de ebullición inicial mayor que 392oF (200 oC) el contenido de aceite puede determinarse directamente. Para una
determinación directa, los fondos de tolueno-alquitran son transferidos a un frasco volumétrico y se agrega tolueno hasta que el volumen total sea igual al volumen del frasco volumétrico. Se toma una alícuota del frasco y se coloca en un papel de filtro de fibra de vidrio. El tolueno se evapora y se pesa el alquitrán. Luego se calcula el peso total del alquitrán. Este método es conocido como el Método Modificado de Dean – Stark (destilación por extracción) o AOSI –35734. 4.3.6.4 Cálculos Los contenidos líquidos de una muestra por lo general son reportados como porcentaje de peso y no como porcentaje de volumen poroso. Contenido de Agua = [(Vw x ρw ) / peso de la muestra] x 100 Contenido de Aceite =
Peso de la Muestra
[(Peso muestra) – (Vw x ρw )- Peso seco] x 100
Donde: Contenido de Agua = Porcentaje de peso del peso de la muestra Contenido de Aceite = Porcentaje de peso del peso de la muestra Vw = Volumen de agua recobrado, ml. ρw = Densidad del Agua, g/cc Peso de la muestra = Peso original (neto) de la muestra, gm; (es decir, se excluye el peso de la manga) Peso seco de la muestra seca y limpia, g. Nota: Para propósitos prácticos, se puede suponer que la densidad del agua es de 1.00 g/cc. Para los cálculos del método ACOSA, ver 4.10, Referencia 4). 4.3.6.5 Ventajas/Limitaciones Las ventajas/limitaciones de este método incluyen: a. Los valores de contenido líquido son
determinados en una muestra b. El método es bastante rápido. c. Los cálculos no son complejos. d. Si la muestra tiene un alto contenido de brea
(petróleo), puede ser necesario parar el proceso de destilación para que el tolueno sucio en el frasco de ebullición pueda reemplazarse con tolueno limpio.
4.3.6.6 Exactitud/Precisión Los valores porcentuales de peso deben reportarse con una precisión del 0.1 por ciento. Aunque no existen estándares industriales, los valores porcentuales de peso deben caer dentro del margen de ±0.5% del valor calculado. Este nivel de exactitud debe ser aceptable en la mayoría de las aplicaciones.

4.3.6.7 Calibración Consulte la sección 4.3.1.8. 4.3.7 Análisis de Invasión de Filtrado (Corazones
de Presión) 4.3.7.1 Principios de Análisis El objetivo del análisis de invasión de filtrado es el de cuantificar la cantidad de filtrado de fluido de perforación que invade el corazón durante las operaciones de corazonamiento. Esto se logra agregando una sustancia rastreadora en cantidades conocidas al sistema de fluidos de perforación y midiendo el contenido de esta sustancia en el corazón. El grado de concentración/ actividad/dilución de este rastreador permite determinar la cantidad de filtrado de fluido de perforación en el corazón. El conocimiento de la cantidad de filtrado de fluido de perforación en el corazón permite una evaluación cualitativa de la alteración en la saturación que ocurre cuando se hace lavado durante el corazonamiento y el cálculo la salinidad del agua de formación (ver 7.7). Se pueden planear los estudios de invasión de filtrado para tipos de corazones distintos a los de presión utilizando una gran variedad de fluidos de corazonamiento base agua y base aceite. Los procedimientos para estos estudios no serán tratados en esta publicación. La amplia gama de tipos de corazones junto con la gran variedad de fluidos de corazonamiento y materiales potenciales de rastreo impide la recomendación de procedimientos estándares para estas técnicas. La utilización de características de permeabilidad relativa de flujo fraccional en la formación mejora la evaluación de alteración en la saturación de fluidos. El uso de brocas de corazonamiento con un perfil de baja invasión minimiza la invasión de filtrado de fluido para perforación y los cambios de saturación debidos al lavado. El sistema de fluidos de perforación se marca con un material rastreador. Para los fluidos de perforación base agua, se recomienda el uso de agua tritiada y esta es la base de las siguientes descripciones de procedimientos. Para el fluido de perforación base aceite, se ha utilizado hexadecano tritiado. El sistema de fluido de perforación es de manera suficiente para permitir un mezclado uniforme con el rastreador. Una “regla de oro” es la de hacer circular el fluido de perforación "volteándolo" tres veces. Durante el corazonamiento, se toma una muestra del fluido de perforación periódicamente para uso como referencia de base para la actividad/concentración de rastreador en el fluido de perforación para calcular la concentración de filtrado en el corazón. Se perfora un tapón vertical a ciertos intervalos resultando en muestras “tapón” y muestras “rosquilla” (ver Figura 4.7). Se extrae el agua de estas muestras y se determina la actividad del tritio con análisis de centelleo líquido. La disminución en la concentración/ actividad del rastreador comparado con el filtrado de
fluido de perforación es igual a la dilución del filtrado de fluido de perforación ocasionada por el agua de formación. La cantidad de invasión de filtrado de fluido de perforación puede determinarse para el exterior y el interior del corazón como también para la invasión total de filtrado de fluido de perforación. Para minimizar el grado al cual el filtrado de fluido de perforación se pasa al interior del corazón por difusión o imbibición, las muestras deben ser cortados en el pozo tan pronto como sea posible después del recobro del corazón. Si esto no es posible, el cilindro interior debe ser congelado y preservado rápidamente. Gidman y Conner en 19925 han reportado que el congelamiento de corazones causa una migración de fluidos que puede resultar en una evaluación incorrecta de la cantidad de invasión de filtrado de fluido de perforación. 4.3.7.2 Equipo Se utiliza el siguiente equipo: a. Prensa taladradora, capaz de cortar muestras
utilizando nitrógeno líquido como lubricante de la broca.
b. Equipo de destilación por extracción de 4.3.1.2 para el análisis de tapones.
c. Prensa de filtro del fluido de perforación para la extracción del filtrado de fluido de corazonamiento.
4.3.7.3 Procedimientos/Precauciones Los procedimientos y precauciones incluyen: Uso de los procedimientos de seguridad establecidos. Determinación de los intervalos para el estudio de
invasión de filtrado. Por lo general se toma una muestra aproximadamente cada 4 pies (1.22 metros) a lo largo de toda la sección corazonada.
Se debe tener especial cuidado para evitar la evaporación de agua durante la manipulación del corazón, los tapones y las rosquillas.
Los intervalos de corazón de diámetro deben aplanarse en cilindros rectos de aproximadamente 2-3 pulgadas (50.8 – 76.2 mm) de largo. Se perfora un tapón vertical del centro de cada muestra para proporcionar porciones de tapón y de rosquilla (ver Figura 4-7). Existen otros métodos para determinar la extensión espacial de la invasión de filtrado de fluido de perforación, como por ejemplo, cortar un tapón perpendicular al eje del corazón entero. Luego se rebana este tapón en muchos segmentos y se analiza cada segmento para detectar la presencia del rastreador.
Se pesan los tapones y las rosquillas y se colocan en equipos separados de extracción por destilación de Dean Stark para destilar el agua del corazón y extraer el petróleo. Se deben utilizar cartuchos para minimizar la perdida de granos. Se mide el agua removida de cada muestra, se embotella y se marca. Debido a que el agua tritiada tiene casi las mismas propiedades físicas y químicas

del agua común y corriente, esta no es separada durante el proceso de destilación.
Las muestras son secadas en un horno de convección a 240ºF (117ºC) hasta que los pesos se estabilicen. Se registra el peso seco de cada muestra.
Figura 4-7 - Muestra de Tapón y Rosquilla para el
Estudio de Invasión de Filtrado Se determina la porosidad con las mediciones
realizadas a las muestras. Se asume que la rosquilla tiene la misma porosidad que el tapón correspondiente. El volumen poroso de la rosquilla complemento se determina con la relación del peso del tapón al peso de la rosquilla.
Se calcula la saturación de agua para las muestras de tapones y rosquillas.
Se determina la cantidad de filtrado de fluido de perforación en el agua que extraído de cada par de tapones y rosquillas. Se calcula la proporción de filtrado de fluido de perforación con respecto a la cantidad de agua total en cada muestra y esta se expresa como porcentaje del volumen poroso de cada muestra.
El analista debe observar las medidas de seguridad apropiadas cuando utilice agua tritiada.
4.3.7.4 Cálculos Los siguientes cálculos son apropiados para este método. a. Saturación de agua en el tapón y la rosquilla. Sw (del tapón) = [Swt (del tapón) / Pv (del tapón)] x 100 Sw (de la rosquilla)=[Swt(rosquilla) / Pv (tapón) x (Peso
rosquilla/ Peso Tapón] x 100 Donde: Sw (del tapón) = Saturación de agua del tapón
expresada como porcentaje del volumen poroso Sw (de la rosquilla) = Saturación de agua de la
rosquilla expresada como porcentaje del volumen poroso
Swt (del tapón) = Volumen total de agua en el tapón, ml
Swt (de la rosquilla) = Volumen total de agua en la rosquilla, ml.
Pv (del tapón) = Volumen poroso del tapón, ml Los pesos de tapones y rosquillas se expresan en g. b. Invasión de filtrado en el tapón y en la rosquilla: Filtrado (Fluido de perforación) =
Actividad de Tritio (en el fluido de perforación)
Actividad de Tritio (en la muestra) x 100
Filtrado (PV) = Filtrado (fluido de perforación) x Sw /
100 Donde:
Filtrado (fluido de perforación) = Filtrado de fluido de
perforación como porcentaje de agua total. Filtrado (PV) = Volumen poroso, porcentaje. Sw = Saturación de agua, porcentaje del volumen
poroso. La actividad del tritio está en pCi / ml. 4.3.7.5 Ventajas/Limitaciones Las ventajas y limitaciones de este método incluyen: a. La ventaja de determinar la invasión de filtrado
de fluido de corazonamiento en corazones con presión retenida, a diferencia de corazones convencionales o de esponja, es que se retiene toda la fase de agua dentro del corazón durante el recobro porque la presión del barril se mantiene a la presión original de corazonamiento. Por lo tanto, se puede determinar cuantitativamente la cantidad de filtrado de fluido de perforación contenida en los segmentos del corazón al final de las operaciones de corazonamiento antes de volver a la superficie. Los fluidos son expulsados de los corazones convencionales o de esponjas a medida que el barril es recuperado del pozo a la superficie. Puesto que los corazones convencionales contienen un porcentaje mayor de filtrado de fluido de perforación en el periferia del corazón, este filtrado es el primer fluido acuoso que se expulsa cuando cae la presión durante el retiro del barril. El volumen de filtrado de fluido de perforación se reduce con esta expulsión de fluido, y la cantidad de filtrado de fluido de perforación medido en este tipo de corazones será únicamente el valor residual. Se puede elegir un forro para la esponja que retenga agua o aceite. El agua expulsado por la evolución de gas es recuperada con una esponja mojada por agua y puede ser analizado para verificar la presencia del rastreador de filtrado de fluido de perforación. Los fluidos de corazón también pueden ser analizados para detectar rastreadores utilizando la extracción de Dean – Stark y el análisis de las aguas recuperadas.
b. Es posible que la saturación in situ no pueda determinarse de manera cualitativa aunque se conozca la cantidad de filtrado de fluido de perforación contenido en los segmentos de corazón. El filtrado de fluido de perforación desplaza tanto el aceite como el agua. La cantidad de cada fase desplazada depende de la saturación in situ, las propiedades del fluido y de las propiedades de la formación. Si se encuentra in situ, la formación está en un nivel irreducible de saturación de agua. Por lo tanto el filtrado va a desplazar principalmente petróleo o gas. De manera inversa, si está in situ la formación se encuentra cerca de la saturación residual del aceite, y por lo tanto, el desplazamiento que se hace será principalmente de agua o de gas. Entre estos dos extremos, el desplazamiento de agua o de aceite depende de las permeabilidades relativas respectivas. Se deben

utilizar curvas fraccionales de flujo de permeabilidad relativa para evaluar la las alteraciones de saturación de fluido durante las operaciones de corazonamiento.
c. Se recomienda la utilización de agua tritiada como el rastreador de fluido de perforación porque ésta puede ser recuperada por el método de extracción de agua de Dean – Stark. Si en lugar de agua tritiada se van a utilizar rastreadores iónicos o éstos se utilizan junto con el agua tritiada, el criterio que debe usarse para la selección del rastreador es que no sean química ni biológicamente alterados por la roca del yacimiento. Los rastreadores aniónicos que se han utilizado con diferentes grados de éxito en fluidos de perforación base agua incluyen el bromuro, yoduro y sales de nitrato. Muchas aguas de yacimientos contienen bromuro y el yoduro en cantidades significativas, así que un análisis exacto de la composición del agua en el yacimiento es importante para seleccionar el rastreador apropiado. El rastreador de ion nitrato requiere el uso de bactericidas en el fluido de perforación para prevenir la reducción microbiológica de nitratos a nitritos. Los rastreadores iónicos deben ser recuperados de los corazones con un método de extracción de agua, lo cual requiere más tiempo y esfuerzo que la extracción de agua tritiada. Se indican los procedimientos en 7.7. Los rastreadores iónicos nos dan resultados de invasión de agua semi-cuantitativos.
d. Como los tapones y rosquillas se colocan congelados en el equipo de destilación por extracción de Dean Stark, se evita que el gas escape de las muestras. Las pérdidas de peso ocasionadas por la liberación de gas durante la extracción se incluyen en las pérdidas de peso atribuidos a la extracción de aceite. El petróleo se determina de manera gravimétrica. Si hay suficiente gas presente, puede haber un error significativo en la determinación de la saturación de aceite. Por lo tanto, solo se reporta la saturación de agua en los estudios de invasión de filtrado.
4.3.7.6 Exactitud/Precisión La precisión de las mediciones de la cantidad de invasión de filtrado de fluido de perforación en el corazón es de 5-20%. La utilización simultánea de más de un rastreador de fluido de perforación aumenta el nivel de confianza en el reporte de invasión de filtrado. No existe información sobre la exactitud de este método porque no hay un valor de referencia aceptado para la invasión de filtrado de fluido de perforación en el pozo. 4.3.7.7 Calibración La calibración del equipo de centelleo líquido, del equipo de detección del rastreador aniónico, y del cromatógrafo iónico es esencial para obtener resultados consistentes. 4.4 LAVADO CON SOLVENTE
4.4.1 Titulación de Karl Fischer 4.4.1.1 Principios de Análisis Se limpian las muestras de tapón de núcleo por medio de un desplazamiento miscible dinámico con una secuencia apropiada de solventes y el contenido de agua de los efluentes producidos analizados por la titulación de Karl Fischer (consultar ASTM D1364-906 y ASTM D4377-887). 4.4.1.2 Equipos Los equipos apropiados para este método incluye: a. General – El equipo incluye un sistema de flujo
para las inyecciones de solvente y un aparato de titulación de Karl Fischer para el análisis del efluente. El sistema de flujo de corazón consta de un soporte de núcleos, un dispositivo para la distribución del solvente y el hardware respectivo. El análisis de Karl Fischer requiere un aparato de titulación, los reactivos Karl Fischer apropiados, una balanza analítica y jeringas.
b. Portanúcleos – Un portanúcleos tipo Hassler o hidrostático, mangas y extremos y una fuente de presión apropiada para soportar las presiones de flujo y de confinamiento esperadas.
c. Dispositivo para la distribución del solvente – Se puede utilizar una bomba cromatográfica para líquidos de alta presión o un sistema de transferencia impulsado por gas. La bomba puede ser una jeringa o un sistema de flujo continuo. El método de presión constante requiere una botella de gas de alta presión, un regulador y un recipiente tal como un acumulador de pistón flotante.
d. Hardware afín – Consiste de tuberías resistentes a la corrosión, válvulas y accesorios para conectar el portanúcleos con el sistema de distribución del solvente.
e. Aparato de Titulación de Karl Fischer – Aparato volumétrico o Coulomático.
f. Balanzas Analíticas – que puedan medir de 0.1 mg a 1000 g
g. Jeringas –0.1 a 10 ml de capacidad, de plástico o de vidrio.
h. Materiales y Reactivos: Reactivos Karl Fischer: Utilice los reactivos
apropiados de acuerdo con el aparato de titulación escogido.
Tolueno, grado reactivo Metanol, grado Karl Fischer Frascos de vidrio, de 25 ml de capacidad
con tapas forradas con politetrafluoroetileno (PTFE).
Cristalería, recipientes con tapón de 1000 ml de capacidad y tubos receptores de 10 ml.
Nitrato de plata, 0.1 Normal. Solución de control, 0.1% de agua en
metanol estándar. 4.4.1.3 Procedimientos/Precauciones 4.4.1.3.1 Procedimientos

Este método es adecuado para la determinación de saturación de agua en núcleos enteros y muestras de tamaño de tapón y puede ser utilizado como alternativa al método de extracción por destilación (Dean Stark). a. Se pesa la muestra con una precisión de 0.1 g
(suponiendo el uso de una muestra de tamaño de tapón), y esta es cargada al portanúcleos. Se aplica el esfuerzo restrictivo deseado y se recogen los fluidos producidos en un tubo receptor para registrar estos volúmenes.
b. Se realiza una secuencia de inyección que cambia de metanol a tolueno, empezando con el solvente que sea miscible con la fase móvil, es decir, metanol si la solución salina es móvil o tolueno si el aceite es móvil.
c. Todos los frascos de almacenamiento, jeringas o los recipientes de recolección deben ser pesados o tarados antes de utilizarlos.
d. Se llena un frasco de vidrio con el reactivo inicial a inyectar y se sella. Luego se inyecta el solvente en la muestra de corazón en proporciones o presiones adecuadas para el material del corazón y el efluente es recolectado en un frasco con tapón. Se debe tener cuidado especial para minimizar la exposición del solvente a la humedad ambiental.
1. Para una inyección de metanol, se
recoge el efluente periódicamente y se le realizan pruebas con nitrato de plata para detectar la presencia de sales. La inyección se termina cuando una gota de nitrato de plata deja de formar precipitados en la alícuota de efluente.
2. Para el tolueno, la inyección se continua hasta que (a) el efluente aparezca claro o (b) el índice de refracción del efluente sea igual al de la solución inyectada.
3. La titulación de Karl Fischer se realiza lo más pronto posible para de esta manera minimizar los cambios en el contenido de agua debido a la absorción de humedad (consulte los pasos g-i).
e. Se repite el paso d utilizando el solvente alterno. f. Se repite el paso d utilizando el solvente inicial. g. Se vuelven a pesar los frascos de
almacenamiento y los recipientes con el efluente y se calculan los pesos del solvente.
h. Utilizando una jeringa, una porción de la solución que se está inyectando es analizada para detectar el contenido de agua con el método de titulación de Karl Fischer de acuerdo con los procedimientos recomendados por el fabricante. Se vuelve a pesar la jeringa para averiguar el peso exacto del solvente analizado. Una vez se está seguro de que la muestra del efluente es uniforme, se determina el contenido de agua de la manera descrita anteriormente. Los resultados son reportados en porcentaje de peso o equivalente de ppm.
i. Se descarga la muestra, secada utilizando las técnicas apropiadas, se pesa, y se determina el volumen poroso por el método de inyección de helio.
4.4.1.3.2 Precauciones Las precauciones para la titulación de Karl Fischer incluyen: a. Se deben consultar las normas de seguridad de
materiales para los procedimientos de manejo apropiados para todos los productos químicos requeridos, incluyendo los reactivos de Karl Fischer.
b. El metanol es altamente susceptible a la absorción de humedad atmosférica como también de las probetas no tratadas. Como es sumamente difícil cuantificar estas variables, se debe minimizar la exposición al aire, y las titulaciones deben realizarse lo más pronto posible.
c. La titulación de Karl Fischer requiere solo una pequeña cantidad de solvente. Por lo tanto, la muestra analizada debe ser representativa de toda la solución para producir resultados exactos.
4.4.1.4 Cálculos Se deben hacer los siguientes cálculos: a. Saturación de Agua : Sws = Wes x (Swc - Swi) Sws = Sumatoria de Swes Swbl = Sws / [(1-A) x ρb] Swb2 = (Swbl x 100) / PV Donde: ρb = densidad de la solución salina, g/ml. Swes = Contenido de agua del solvente efluente, g. Wes = Peso del solvente efluente, g. Swc = Contenido de agua en el efluente, g/g. Swi = Contenido de agua en el inyectante, g.g. Sws = Contenido de agua en la muestra, g. Sumatoria de Swes = Sumatoria de los contenidos de
agua en los solventes efluentes, g. Swbl = Contenido de solución salina en la muestra, ml. Swb2 = Saturación de solución salina en la muestra,
porcentaje de volumen poroso. A = Contenido de sal en la solución salina, gramos de
sal por gramos de solución salina. PV = Volumen poroso de la muestra, ml. b. Saturación de Aceite: Si la muestra fue saturada completamente, la
saturación de aceite puede determinarse por diferencia volumétrica:
So = 100 - Swb2 Si la muestra contenía una saturación inicial de gas, la saturación de aceite puede determinarse por diferencia gravimétrica: Wo = Wi – Wd - [Sws / (1 – A)]

Vo = Wo / ρo So = PV
Vo x 100
Donde: So = Saturación de aceite como porcentaje del
volumen poroso (PV). Swb2 = Saturación de la solución salina en la muestra,
porcentaje del volumen poroso. Wo = Peso del aceite, g. Wi = Peso inicial de la muestra, g. Wd = Peso de la muestra seca, g. Sws = Contenido de agua en la muestra, g. Vo = Volumen del aceite, ml. ρo = Densidad del aceite, g/ml. A = Contenido de sal en la solución salina, gramos de sal por gramo de solución salina. PV = Volumen poroso de la muestra, ml. c. Reporte de datos – las saturaciones de son
reportadas con una precisión de porcentaje 0.1 del espacio de poro.
4.4.1.5 Ventajas Las ventajas de este método incluyen: a. Todos los niveles de saturación pueden ser
determinados. b. La titulación de Karl Fischer es muy exacta. c. Se minimiza el daño a los minerales sensibles. d. El procedimiento remueve las sales de la
muestra. e. Las saturaciones pueden ser determinadas en
pruebas especiales de corazones, en las cuales los efectos del histéresis de estrés prohiben la descarga de la muestra.
4.4.1.6 Limitaciones Las limitaciones de este método incluyen: a. El metanol absorbe fácilmente la humedad del
medio ambiente. b. La exactitud del método depende del manejo de
solventes y las técnicas de almacenamiento. c. La muestra analizada debe ser representativa del
efluente total. d. La técnica es más compleja y más costosa que el
método de destilación por extracción. e. Las saturaciones de petróleo son determinadas
indirectamente. La saturación de aceite por diferencia gravimétrica asume que no hay pérdida de granos en la muestra.
f. Esta técnica no es apropiada para las muestras que contienen halita, azufre u otros minerales que son solubles en metanol.
4.4.1.7 Exactitud/Precisión Las unidades de titulación de Karl Fischer tienen una precisión de ± 0.5 % del valor medido. Sin embargo, la exactitud de la técnica se limita a la capacidad de manejo y almacenamiento de los solventes sin alterar su contenido de agua.
4.4.1.8 Calibración La operación de instrumentos es revisada con el análisis de soluciones estándar con un contenido de agua conocido. Las muestras contaminadas deben producir porcentajes de recuperación entre 90-110%. Se deben realizar análisis en duplicado sobre el 10% del total de las muestras analizadas y deben tener un porcentaje de diferencia relativa menor del 2%. 4.5 MÉTODOS DE EXPLORACIÓN 4.5.1 Introducción Se han reportado varias técnicas de laboratorio para el barrido de mediciones de saturaciones de agua, aceite y gas en corazones. Estas técnicas incluyen: (a) absorción lineal de rayos X; (b) absorción de microondas; (c) tomografía asistida por computador (axial) CT; (d) absorción lineal de rayos gamma y (e) resonancia magnética nuclear (NMR). Los métodos (a)-(d) pueden considerarse tecnologías emergentes para la medición de saturación. Otras técnicas de exploración que se han utilizado para la determinación de saturación son la resonancia de ondas de radio y la radiografía de atenuación neutrónica. La radiografía de neutrones tiene un buen potencial para determinar la saturación de fluidos porque los neutrones son atenuados mucho mas por fluidos (protones) que por rocas. Por lo tanto, es posible que la tomografía de neutrones no necesite la adición de agentes marcadores en los fluidos residentes para la determinación de saturación. Estas técnicas no son utilizados ampliamente en el análisis de corazones. 4.5.2 Principios Las técnicas de rayos X, CT y rayos gamma miden la absorción de una radiación electromagnética de alta energía por los fluidos marcados con agentes de alta absorción (por lo general elementos de alto peso atómico) para determinar la saturación del fluido. La técnica de absorción de microondas se basa en la absorción de energía de microondas por las moléculas de agua. La técnica de NMR se basa en la detección de fluidos que contengan protones (H1), carbono (C13), Sodio (Na23), fósforo (P31) y Flúor (F19) por campos magnéticos que se alternan a determinadas frecuencias de radio, mientras se ubica la muestra en un campo magnético grande y constante. 4.5.3 Equipos Los equipos que se utilizan para estas técnicas son generalmente sofisticados y costosos. Los equipos de rayos X y CT fabricados para uso médico son a menudo adecuados para estas mediciones de saturación. Los equipos médicos de NMR rara vez son adecuados para las mediciones de saturación. Los equipos de absorción de rayos gama y microondas usualmente han sido construidos especialmente para estos análisis.

4.5.4 Procedimientos/Precauciones Se deben considerar varios parámetros para una determinación exacta de la saturación cuando se utilizan métodos de exploración, tales como el ajuste del nivel de energía, el material de marcado y su concentración, y la interacción fluido - roca y fluido – fluido (consultar la bibliografía para conocer los procedimientos y precauciones para las técnicas individuales). 4.5.5 Cálculos Como todas las técnicas (excepto la NMR) generan señales tanto de la matriz sólida como de los fluidos, se requieren mediciones al mínimo de dos saturaciones conocidas (normalmente 0 y 100 por ciento) para establecer una línea de calibración. Las saturaciones desconocidas del sistema de aceite – solución salina son calculadas con base en la siguiente ecuación: Sw = (Usat - Wo ) / (Uw – Uo) Donde: Sw = Saturación desconocida de solución salina, como fracción. Usat = Atenuación del corazón en la saturación desconocida (Sw). Uw = Atenuación del corazón en una saturación del 100% de solución salina. Uo = Atenuación del corazón en una saturación del 100% de aceite. (Las unidades de Usat, Uw y Uo deben ser todas iguales). 4.5.6 Ventajas Una ventaja importante de estos métodos es su habilidad para suministrar información sobre la distribución espacial de la saturación de fluidos. Otra ventaja de estas técnicas es que las mediciones se hacen de manera no invasiva y no destructiva. No se requiere la extracción de fluidos antes de realizar las pruebas de flujo en corazones preservados (esto puede ser difícil en corazones de tamaño completo o ajustados). Las saturaciones pueden supervisarse sin terminar el procedimiento de laboratorio. Una ventaja que tiene las técnicas de imagen CT y NMR es que proporcionan la distribución de saturación de fluidos en tres dimensiones. Una ventaja de las técnicas de microondas y NMR es su capacidad de determinar la saturación de agua en el corazón sin la adición de agentes marcadores. Se pueden utilizar las técnicas de microondas y de NMR para determinar saturaciones en el análisis estándar de corazones porque no requieren ningún agente marcador y las curvas de calibración son establecidas después de los procesos de limpieza y resaturación. 4.5.7 Limitaciones
Una limitación de la técnicas de rayos X, CT y rayos gamma es que pueden determinar saturaciones en corazones siempre y cuando estén saturados con fluidos marcados. Por lo tanto, es posible que no sean adecuadas para los análisis básicos de corazones. Una limitación de la técnica NMR es la incapacidad de manejar corazones que contengan cantidades significativas de materiales ferromagnéticos, arcillas o gas. 4.5.8 Exactitud/Precisión La exactitud y capacidad de algunas de las técnicas de exploración para la medición de saturación se relacionan en la Tabla 4-2. La exactitud indicada en esta tabla representa valores que se logran únicamente en condiciones óptimas de calibración y medición. Tabla 4- 2 Capacidades de Medición de la Técnicas de Barrido Técnica Dimensiones Necesidad de
Marcador Exactitud, unidades de saturación
Resolución espacial, cm
Tamaño máximo de la muestra, pulg.
Fluidos medidos
Rayos x 2 Si 1 1.0 12 Petróleo Agua Gas
Tomografía 3 Si 1 0.2 12 Petróleo Agua Gas
Rayos Gamma 2 Si 3 0.1 12 Petróleo Agua Gas
Microondas 2 No 1 2.0 2 Agua Resonancia 3 No 1 0.1 4 Petróleo
Agua 4.5.9 Calibración Las técnicas de absorción requieren mediciones de intensidad de señales al mínimo de dos niveles de saturación para establecer una curva de calibración para relacionar la respuesta del instrumento a las saturaciones. La calibración depende del instrumento. 4.6 ANÁLISIS DE CARBON 4.6.1 Principios de Análisis Las técnicas básicas de análisis de corazones, como el análisis de retorta y de extracción de solvente por destilación no son adecuados para determinar saturaciones de fluidos en muestras de carbón. Se han desarrollado métodos alternativos para medir la saturación de fluido del carbón. En el caso del carbón el fluido de principal importancia es el agua. La saturación de agua, o más bien, el contenido de humedad es una propiedad fundamental del carbón que debe determinarse con exactitud para evaluar un estrato de carbón apropiadamente. El método descrito aquí para determinar el contenido de humedad en el carbón incluye el secado del agua en una muestra

aplastada en un horno de convección y tomar las mediciones sucesivas de peso hasta que se logre el equilibrio. La pérdida de peso es equivalente a la cantidad de agua sacada, y el contenido de humedad es reportado como porcentaje de peso de humedad con respecto al peso de la muestra de carbón mojado. 4.6.2 Equipos Los siguientes equipos son apropiados para este método: a. Trituradora – se utiliza una trituradora de tipo
mandíbula o giratoria para reducir el carbón al tamaño retenido en un tamiz No. 8 (2.38 mm).
b. Recipientes de secado – Los recipientes deben ser de un tamaño suficiente para acomodar las muestras trituradas extendidas en un grosor menor de 2.5 cm. Los lados de los recipientes no deben tener una altura mayor de 3.8 cm. Los recipientes deben ser hechos de un material estable que no se corroerá en la temperatura utilizada.
c. Horno para el secado – Se debe utilizar un horno de aspiración forzada capaz de mantener una temperatura constante de 225ºF, ± 5ºF (107ºC, ± 3ºC). La velocidad de la ventilación forzada en el horno debe ser lo suficientemente baja para no alterar la muestra. Se prefiere utilizar un horno que tenga un puerto para gas capaz de acomodar una manguera de un tanque de nitrógeno, porque la circulación de nitrógeno, en lugar de aire, en el horno puede ayudar a minimizar la oxidación de la muestra de carbón.
d. Balanza – La balanza utilizada para pesar las muestras debe tener una precisión de 0.1 gramo y tener una capacidad lo suficientemente grande para acomodar tanto la muestra como el recipiente.
e. Un tamiz No.8 (2.33 mm) – el tamiz debe ser lo suficientemente grande como para contener toda la muestra preparada mientras separa las partículas grandes.
f. Desecador de vidrio – el desecador debe ser lo suficientemente grande para acomodar la muestra preparada y el recipiente. Se recomienda el uso de un desecante fresco con código de color porque algunos desecantes pueden agregar humedad a la muestra si no se encuentran frescos.
4.6.3 Procedimiento/Precauciones 4.6.3.1 Muestreo 4.6.3.1.1 Trituración Se utiliza una trituradora de tipo mandíbula u otro adecuado o algún instrumento para reducir al tamaño retenido por un tamiz No. 8 (2.38 mm). Al menos un 95% de la muestra triturada debe pasar a través de un tamiz No. 8. 4.6.3.1.2 Tamaño de la Muestra
La muestra utilizada para la determinación de contenido de humedad debe tener un peso mínimo de 500 gramos. 4.6.3.2 Pesaje 4.6.3.2.1 Recipiente de Secado Inmediatamente después de completar la preparación de la muestra, mida y registre el peso del recipiente de secado limpio y seco. Vierta el carbón triturado y cernido en el recipiente de secado y extienda a un grosor no mayor de 2.5 cm. 4.6.3.2.2 Peso Inicial de la Muestra Mida y registre el peso de la muestra de carbón húmedo y el recipiente de secado. Coloque el recipiente dentro del horno a una temperatura de 225ºF, ± 5ºF (107ºC, ± 3ºC). 4.6.3.2.3 Mediciones del Peso Seco Después de que la muestra se ha secado durante un periodo de 1 hora, sáquela del horno y colóquela en un desecador de vidrio para que se enfríe. No se recomienda el uso un secante. Pese la muestra inmediatamente después de enfriarse y registre el peso. Vuelva a colocar la muestra en el horno. Repita este proceso de pesaje en intervalos de media hora hasta que la pérdida de peso entre mediciones sucesivas sea menor del 0.05 % del peso de la muestra. La ultima medición tomada es considerada el peso seco de la muestra más el peso del recipiente. 4.6.4 Precaución No seque en exceso la muestra de carbón. Ocurre una oxidación del carbón si este se seca demasiado tiempo en el aire. 4.6.5 Cálculos Calcule el contenido de humedad de la muestra
utilizando la siguiente ecuación: M = [{W2 – W3) / (W2 – W1)] x 100 Donde: M = Contenido de humedad en la muestra de carbón,
%. W1 = Peso del recipiente vacío, g. W2 = Peso del carbón húmedo más el recipiente, g. W3 = Peso del carbón seco más el recipiente, g. 4.6.6 Ventajas La ventaja principal de este método es su simplicidad. El procedimiento no requiere una inversión muy grande de capital en equipos especializados. Tampoco se necesita un entrenamiento especial para realizar las mediciones. Existen técnicas más sofisticadas para determinar el contenido de humedad en el carbón (consultar 4.10, Referencias 8, 9 y 10). Sin embargo, en el contexto de los análisis básicos de

corazones, la técnica descrita aquí se considera adecuada. 4.6.7 Limitaciones Algunas clases de carbón tienen la tendencia a oxidarse cuando se utiliza la técnica descrita. Los resultados del contenido de humedad serán demasiado bajos si ocurre una oxidación; la oxidación añade peso y causará un peso seco muy alto. Se recomienda mantener los tiempos de secado dentro de los lineamientos indicados para minimizar la posibilidad de oxidación. La circulación de nitrógeno, en lugar de aire, en el horno también podrá reducir el potencial de oxidación 4.6.8 Exactitud La exactitud de esta técnica es muy buena porque depende únicamente de la exactitud de la balanza. Por lo tanto, esta técnica tiene una precisión de ±0.05 %. 4.6.9 Precisión Los resultados de las determinaciones de contenido de humedad realizados en duplicado sobre la misma muestra por el mismo técnico utilizando el mismo equipo no deben variar mas de +0.3%. Los resultados de las mediciones del contenido de humedad realizados por diferentes laboratorios en muestras adyacentes no deben variar más de ±0.5 %. 4.6.10 Calibración Se recomienda el uso de un instrumento de calibración de peso para asegurar que la balanza funciona de manera apropiada. También un técnico calificado debe realizar una calibración periódica en la balanza en intervalos regulares de acuerdo con las especificaciones de fábrica para asegurar la exactitud de las mediciones. 4.7 OIL SHALES 4.7.1 Principios de Análisis Se define el oil shale como una roca compuesta de partículas que varían en tamaño entre arcilla y cieno que contienen cantidades varias de material orgánico sólido (ejemplo: kerógeno). Los orgánicos sólidos presentes en estas muestras por lo general son licuados para generar petróleo. El análisis de este tipo de formación, con baja permeabilidad y potencial para orgánicos sólidos requiere de procedimientos especiales. Se obtienen las saturaciones de petróleo y agua utilizando un proceso de retorta de alta temperatura. No se hace ningún intento para medir el volumen poroso lleno de gas. Las saturaciones de fluido son reportadas en galones por tonelada. 4.7.2 Equipos
Los equipos son iguales a aquellos utilizados para el Método Retorta a Presión Atmosférica (ver 4.2.1.2). Los fluidos son recolectados en tubos de centrífuga de 15 cm3. Por lo general, se emite el gas durante el proceso de retorta. Si se llega a requerir la recolección/medición de estos gases, se pueden utilizar probetas modificadas que consisten de una sección calibrada receptora de líquido con la porción superior de la probeta adecuada con un brazo lateral para permitir que los gases producidos sean dirigidos a las celdas de recolección o al equipo de medición. 4.7.3 Procedimientos/Precauciones El procedimiento es similar a los procedimientos descritos en 4.2.1.3, con las siguientes excepciones: a. La muestra representativa de aproximadamente
dos pulgadas de longitud no es dividida en dos. No se considera la permeabilidad en el análisis de oil shale.
b. No se hace ningún intento para tamizar la muestra triturada para excluir los finos creados durante el proceso de trituración.
c. No se prepara una muestra adyacente para las pruebas con bomba de mercurio descritas en 4.2.1.3, porque no se requiere el volumen poroso lleno de gas ni la porosidad.
d. Se ignoran las pérdidas de petróleo por coqueo y craqueo. Si estos valores son significativos, se debe efectuar una corrección para la retención mecánica.
e. Se permite que el horno se caliente a un ritmo consistente con los elementos de calentamiento y el número de retortas en operación. Es decir, no se hace ningún intento para medir el agua del poro en la temperatura inicial del horno. La temperatura del horno se fija inicialmente en 1000oF (538 oC). Cuando el horno se ha estabilizado en esta temperatura y todos los volúmenes en los tubos de recolección permanecen constantes, se considera terminado el proceso de retorta.
f. Bajo ciertas condiciones (ejemplo: las muestras que contienen diferentes variedades de kerógeno) el volumen del petróleo recuperado puede variar con la tasa de calentamiento.
g. Se utiliza únicamente el agua total recolectada en el cálculo final del contenido de agua.
h. Las operaciones de retorta deben llevarse a cabo con la ventilación adecuada (bajo campanas de humos), porque los aceites típicos recuperados de los kerógenos tienen un alto contenido de azufre y emiten vapores nocivos.
4.7.4 Cálculos Los contenidos de agua y aceite deben calcularse de la siguiente manera: Contenido de Aceite =
Peso de la muestra
Volumen del Aceite Recuperado x 239.7
Contenido de Agua =
Peso de la Muestra
Volumen del Agua Recuperada x 239.7

Donde: El volumen del petróleo está en mililitros. El volumen del agua está en mililitros. El peso de la muestra está en gramos. El contenido de aceite está en galones/toneladas. El contenido del agua está en galones/toneladas. 4.7.5 Ventajas Las ventajas de este método incluyen: a. Se pueden utilizar grandes volúmenes de
muestra. b. El método es bastante rápido. c. Todas las mediciones se hacen de directamente
e independiente de las otras. d. Se pueden utilizar procedimientos y equipos de
retorta estándares. e. No se necesita tener precauciones para
diferenciar el agua de poro del agua total. f. No se requieren curvas para la corrección del
volumen de aceite. g. Se pueden obtener con mayor facilidad las
muestras duplicadas debido a que se puede preparar un gran volumen de material triturado, y las muestras se pueden obtener por medio de la técnica de “cono y cuarto”.
4.7.6 Limitaciones Las limitaciones de este método incluyen: a. La cantidad de aceite producido puede depender
de la tasa de calentamiento. b. Debido a la presencia de emulsiones, puede
haber dificultad en la lectura del menisco en las probetas de recolección.
4.7.7 Exactitud/Precisión La exactitud/precisión de este método es: a. La exactitud de los volúmenes de agua y aceite
es de ± 2.5% de los volúmenes medidos. b. La reproducibilidad de la prueba debe caer
dentro de un rango de ± 0.5 gal/ton para muestras de bajo rendimiento (entre 3-10 galones/tonelada) y ± 1.0 gal/ton para muestras de alto rendimiento (20 – 40 galones/ tonelada).
4.7.8 Calibración Para los procedimientos de calibración de probetas, consultar 4.2.1.8 4.8 CORAZONES QUE CONTIENEN YESO 4.8.1 Principios de Análisis Los corazones que contienen yeso (CaSO4.2H2O) a menudo provienen de formaciones de carbonato. Tales formaciones son frecuentemente heterogéneas con cavidades/fracturas así que se prefieren los procedimientos de análisis para corazones de
diámetro completo. Sin embargo, no se recomienda la determinación de saturación de fluidos en corazones que contienen yeso con el método de extracción por destilación (Dean Stark) para los corazones de diámetro completo (ver 4.3.2) utilizando el tolueno como solvente. El yeso se deshidrata en el punto de ebullición del tolueno. Si el corazón contiene una cantidad significativa de yeso, se van a obtener volúmenes erróneos de agua y valores incorrectos de volumen poroso. El volumen poroso utilizado para determinar la saturación también estará errado por el incremento en el volumen poroso debido a la deshidratación del yeso. El volumen de aceite extraído de la muestra no se ve afectado por la presencia de yeso. Existen tecnologías emergentes, como por ejemplo la NMR, que representan buenas posibilidades para la cuantificación de yeso, y en el futuro estos servicios se podrán ofrecer dentro del paquete de servicios básicos de laboratorios comerciales. Se puede utilizar el método de titulación de Karl Fischer (ver 4.4.1) para analizar los corazones que contienen yeso determinando el contenido de agua en los solventes lavados durante la extracción de solventes fríos. Sin embargo, este método por lo general no está disponible en empresas de servicios para el análisis de una gran cantidad de muestras de corazones. El método cualitativo descrito en el siguiente párrafo puede ser realizado por laboratorios comerciales. El método cualitativo presentado aquí está basado en el ajuste de saturaciones de retorta para la deshidratación del yeso comparando las porosidades y los pesos de la retorta con las de baja temperatura sobre muestras adyacentes. Se limpia una muestra de tapón con solventes cíclicos de presión a bajas temperaturas para extraer el aceite y el agua de la muestra sin remover el agua que se encuentra químicamente retenido dentro del yeso. Luego se mide la porosidad de la ley de Boyle en la muestra limpia. Después de determinar la porosidad de la ley de Boyle, la muestra de tapón es replicada para determinar la cantidad de agua retenida en la muestra. Se utiliza la diferencia en porosidad entre las extracciones de baja y alta temperatura para calcular el volumen total del yeso contenido en la muestra de corazón. El volumen de agua emitido en la deshidratación de yeso es aproximadamente 1.27 veces el incremento en el volumen poroso que resulta del encogimiento del yeso (densidad del grano = 2.32 g/c3) a medida que se convierte en anhídrido (2.89 – 2.98 g/c3). Las mediciones de saturación de retorta se obtienen en los extremos recortados de la muestra de tapón o en el material adyacente de donde se taladró el tapón. El método de retorta a presión atmosférica (ver 4.2) remueve los fluidos en la muestra, y el agua de cristalización en el yeso. La porosidad de la sumatoria de fluidos se determina con los datos de la retorta. Luego se ajustan las saturaciones de agua determinadas por el método de retorta. El ajuste resultará en una disminución en la saturación de agua. Este ajuste es cualitativo porque los carbonatos son conocidos por ser heterogéneos; así que es posible que las muestras complementarias no contengan la misma cantidad de yeso.

4.8.2 Equipos Los equipos apropiados para este método incluyen: a. Equipos de retorta para temperaturas altas (ver
4.2.1.2) b. Aparato de limpieza a baja temperatura, es decir,
un saturador a presión lo suficientemente grande para hacer que el solvente circule en ciclos al mismo tiempo por varias muestras.
c. Porosímetro basado en la Ley de Boyle. 4.8.3 Procedimientos El corazón que será analizado se debe extender y encajar en una mesa como en la práctica estándar (ver 3.2.4). De cada pie de corazón a analizar, perfore una muestra de tapón. Si se utilizan tapones de diámetro de 1 pulgada, las muestras deben tener una longitud de por lo menos una pulgada. Después de recortar los tapones a la longitud deseada, estos deben pesarse. Revise la fluorescencia de la muestra y regístrela. El cálculo visual inicial de saturación de hidrocarburos le permitirá al analista evaluar mejor la muestra durante la fase de limpieza a presión. El tapón debe limpiarse con un método de bajas temperaturas, lo cual minimiza la alteración del yeso. Se determina entonces la saturación de fluido de los extremos y/o del material adyacente que permaneció en la muestra principal a partir de la cual se obtuvo este tapón por medio del método de retorta a presión atmosférica (ver sección 4.2.1.3). Utilice la máxima cantidad de material posible para de esta manera lograr una mayor precisión. Este proceso nos dará como resultado datos de saturación de fluido a altas temperaturas y se puede calcular la Sumatoria de la porosidad de fluidos (ver secciones 5.3.2.2.2 y 4.2.1). Las muestras perforadas deben limpiarse en una vasija de presión utilizando una mezcla azeotrópica de 1/3 de acetona, 1/3 de ciclohexano y 1/3 de metanol. Se colocan las muestras en la vasija de presión y se sumergen completamente en esta mezcla. Se aplica presión de aire o nitrógeno en la parte superior de la mezcla impulsándola en los poros de las muestras de tapón. Se hace circular la presión para impulsar la mezcla adentro y afuera de los tapones. Después de aproximadamente 24 horas, se debe revisar la fluorescencia de la muestra para verificar que la extracción se ha completado. Los tapones que tengan fluorescencia de aceite deben devolverse al recipiente de presión para otro ciclo de limpieza. Se considera que las muestras están limpias si no se detecta ninguna fluorescencia de aceite. Se colocan las muestras en un horno al vacío a 120ºF (49 ºC) hasta que se estabilice el peso. Las muestras deben permanecer en el horno por un período mínimo de 24 horas para remover la mezcla azeotrópica de las muestras. Se utilizan temperaturas bajas durante las fases de limpieza y secado para que este proceso minimice las alteraciones del yeso en las muestras de tapón. Después de limpiar y secar las muestras, se determina la porosidad de la ley de Boyle. Después de medir la porosidad de la ley de Boyle en la muestra de tapón, se retira este para remover el
agua retenida en el yeso. Después del proceso de retorta, se vuelve a pesar el tapón y se vuelve a medir la porosidad de la muestra de tapón con la ley de Boyle. Una comparación de la porosidad de retorta con la porosidad de baja temperatura identifica las zonas que contienen yeso. Cuando la porosidad de alta temperatura es mucho mas alta que la porosidad de baja temperatura, quiere decir que el contenido de yeso es muy alto. Cuando las dos porosidades concuerdan, es porque el contenido de yeso es bajo. El volumen de petróleo extraído por el método de retorta, no se ve afectado por la presencia de yeso. Sin embargo, el volumen poroso utilizado para calcular la saturación de aceite debe ser corregido por la presencia de yeso. La saturación de aceite puede ser calculada con el volumen de aceite y el volumen poroso corregido. 4.8.4 Precauciones Se debe tener cuidado en el proceso de secado de las muestras tomadas del saturador a presión para evitar la deshidratación del yeso presente. El monitor del horno de vació debe tener una sonda que mida la temperatura. 4.8.5 Cálculos Consulte el Método de Retorta a Presión Atmosférica (ver 4.2) y los Cálculos para la Porosidad por Sumatoria de Fluidos (ver 5.3.2.2.2) Consulte la sección 5.3.2.1.1 “Cálculos de porosidad utilizando la Ley de Boyle”. S
oc= V o / (Vbr x φ
lt) Donde: V
o = volumen de aceite obtenido de la retorta de los extremos recortados o el material adyacente a la muestra de tapón, cm3. Vbr = Volumen total del material de retorta, cm3. φ
lt = Porosidad de tapón después de extracción a baja temperatura, fracción. S
oc= Saturación ajustada de aceite, fracción. Si se asume que el valor de saturación del gas es cero, la saturación del agua S
wc es: S
wc = 1 - S oc
Si el valor de saturación del gas es un valor diferente a cero, la saturación de agua puede ser calculado asumiendo una densidad de grano para el anhídrido: ∆V w = (ρa / ρ g) x (Vbr x ∆φ) ∆Vcorr = (Vw - ∆V w) S wc = Vwcorr / (Vbr x φ lt) Donde: S wc = Saturación de agua ajustada para la deshidratación del yeso, fracción.

∆V w = Volumen de agua tomado de la deshidratación del yeso, cm3. ρa = Densidad de grano del anhídrido , en g/ cm3. ρ g = Densidad del grano del yeso, g/cm3. ∆φ = Diferencia en la porosidad entre las extracciones realizadas a alta y baja temperatura, fracción. ∆Vcorr = Volumen de agua ajustado por la deshidratación del yeso, en cc. (Vw = Volumen de agua obtenido de la retorta de los extremos recortados o el material adyacente a la muestra de tapón, cm3. De manera alternativa, las saturaciones se pueden expresar en términos de peso de las muestras: ƒwat = Wwat / (Wwat + Woil) Wewat = (Wpo – Wpr) x ƒwat Weoil = (Wpo - Wpr) x (1-ƒwat) Wgyp = Wpt – Wpr Vwcorr = (Wewat / ρbr) – (Wgyp / ρgw) Soc = (Weoil / ρoil) Vplt Swc = Vwcorr / Vplt Donde: Wpo = Peso del tapón después de perforación, g. Wpt = Peso del tapón después de la extracción a baja temperatura, g. Wpr = Peso del tapón después de retorta,g. Wwat = Peso del agua determinado por la extracción de retorta de los extremos de la muestra o el material adyacente, g. Woil = Peso del aceite determinado por la extracción de retorta de los extremos o del material adyacente, g. ƒwat = Fracción de peso del agua extraído por retorta, fracción. Wewat = Contenido estimado del agua en la muestra de tapón, incluyendo el agua retenido en el yeso, g. Weoil = Contenido estimado de aceite de la muestra de tapón, g. Wgyp = Agua de hidratación en la muestra de tapón, g. ρbr = Densidad de la solución salina de la formación, g/ cm3. ρgw = densidad del agua retenida en el yeso, en g/ cm3. Vwcorr = Volumen de agua ajustado por la deshidratación del yeso, cm3. ρoil = Densidad del aceite de la formación, g/ cm3. Vplt = Volumen poroso de la muestra de tapón después de la limpieza a temperatura baja, cm3. Soc = Saturación de aceite calculado para la muestra de tapón contabilizando el cambio experimentado en volumen poroso debido a la deshidratación del yeso, fracción. Swc = Saturación del agua calculado para el tapón contabilizando el agua retenido en el yeso y el cambio en volumen poroso debido a la deshidratación del yeso, fracción.
4.8.6 Ventajas Las ventajas de este método incluyen: a. Se minimizan los daños al yeso cuando se limpia
la muestra con la circulación de presión. Las mediciones de volumen poroso utilizadas para calcular las saturaciones son más representativas si el yeso no presenta daños.
b. Se puede calcular el contenido de yeso estableciendo comparando las porosidades.
4.8.7 Limitaciones Las limitaciones de este método incluyen: a. El proceso de limpieza a baja temperatura puede
ser demorado dependiendo de la gravedad del aceite y de la permeabilidad del material del corazón.
b. Los valores de saturación de aceite y agua de retorta no se determinan de la misma manera que las muestras que no contienen yeso.
4.8.8 Exactitud/Precisión La exactitud /precisión de este método es: a. Si se preserva el agua de cristalización durante la
circulación de presión, la determinación del volumen poroso tendrá una exactitud estándar.
b. La precisión de los cálculos de saturación de agua y de aceite por el método de retorta será disminuida debido al estimado del contenido de yeso por comparaciones de porosidades.
c. Las porosidades obtenidas utilizando el método del helio de la Ley de Boyle son más confiables que las porosidades obtenidas por el método de sumatoria de fluidos. Cuando los valores de porosidad hallados por la ley de Boyle y la sumatoria de fluidos concuerdan, se asume que el resultado de saturación es más confiable que en aquellos casos donde se presentan grandes diferencias entre las dos porosidades.
]4.8.9 Calibración El equipo utilizado para medir la porosidad por ambos métodos debe estar de acuerdo con los estándares para el método de porosidad apropiado. 4.9 HISTORIA 4.9.1 Método de Retorta al Vacío El método de retorta al vacío para obtener las saturaciones de fluido en corazones es una técnica de diámetro completo . Se utiliza este método de manera muy extensiva en las regiones de “rocas duras”, como es el caso de la Región Occidental de Texas, donde la producción proviene principalmente de formaciones carbonatadas. Aunque varias empresas que analizan corazones aún mantienen equipos para realizar este procedimiento, no es un procedimiento recomendable, habiendo sido reemplazado por el método de destilación por extracción (ver 4.3).

El procedimiento consiste en destilar los fluidos en el poro a una temperatura máxima de 450ºF (232ºC), manteniendo un vacío parcial en el sistema. Las probetas para la recolección de fluidos deben sumergirse en una baño de alcohol/hielo seco a una temperatura aproximada de -75ºF ( -59 ºC) para evitar la pérdida de vapores a través de la bomba al vacío. El procedimiento se presta para la determinación del volumen poroso por medio del método de sumatoria de fluidos. Esto es posible porque el primer paso consiste en saturar con agua dulce los espacios porosos que no están ocupados o que están llenos de gas. Se determina el volumen poroso por diferencia gravimétrica. Se emplea un factor de corrección para el volumen de aceite y el volumen del agua en el poro se halla por diferencia. Los tres valores comprenden el volumen poroso y los fluidos recolectados pueden expresarse de acuerdo a esto. Se puede calcular una porosidad utilizando un volumen total determinado en la misma muestra utilizada para el proceso de destilación. Una de las principales desventajas de este método es la condición en que quedan las muestras después del proceso de destilación. No es raro que las muestras queden negras debido al coqueo del aceite y ningún procedimiento de limpieza conocido puede restaurar las muestras de tal manera que puedan volver a utilizarse en otros procedimientos, como por ejemplo la porosidad de la ley de Boyle, o para pruebas de presión capilar o permeabilidad relativa. Por esta y otras razones es que el Subcomité de autores de este documento decidió no incluir esta técnica como un procedimiento recomendable.

CONTENIDO 5. DETERMINACION DE POROSIDAD
5.1 Información General 5.2 Medición del Volumen Total 5.3 Mediciones del Volumen Poroso 5.4 Procedimientos Históricos 5.5 Rocas Ricas en Sustancia Orgánica 5.6 Referencias
Figuras: 5-1 Volúmenes Porosos Totales y Efectivos conforme a las Definiciones de los Analistas de
Núcleos y de Registros 5-2 Aparato para Inmersión en Mercurio de Arquímedes 5-3 Bomba Volumétrica de Desplazamiento de Mercurio 5-4 (Flotabilidad) de Arquímedes con Fluidos Diferentes al Mercurio - Aparato 5-5 Porosímetro de Doble Celda - Ley de Boyle 5-6 Gráfico de una Celda de Carga Isostática para la Determinación Directa del Volumen
Poroso 5-7 Gráfico de Carga en el Laboratorio 5-8 Gráfico de Cargar en Laboratorio y Yacimiento 5-9 Método de Medición por Adsorción Volumétrica Tablas: 5-1 Guía de Volumen Total, Volumen de Grano y Volumen Poroso 5-2 Materiales de Envoltura para Muestras de Núcleos de Consolidación Deficiente/Sin
Consolidar 5-3 Densidad del Mercurio vs. Temperatura.

PRACTICAS RECOMENDADAS PARA EL ANÁLISIS DE NÚCLEOS 5. Determinación de Porosidad 5.1 INFORMACIÓN GENERAL 5.1.1 Comentarios Introductorios/Ecuaciones 5.1.1.1 La porosidad, definida como la relación del volumen poroso respecto al volumen total del material, es una propiedad intrínseca de todas las rocas de yacimientos. Se debe conocer la cantidad de espacio vacío que puede ser ocupado por hidrocarburos o agua en un yacimiento para obtener un cálculo inteligente de la cantidad inicial de petróleo/gas en el sitio. El nivel de precisión con el cual se puede determinar la porosidad es en general una función de los métodos utilizados en estas mediciones. Varias herramientas de registro utilizadas en los métodos eléctricos, nucleares, sónicos o de densidad son utilizadas para la determinación continua de porosidad en el pozo. Las mediciones obtenidas con estas herramientas de registro deben ser calibradas con las porosidades del núcleo determinadas bajo condiciones subterráneas simuladas, teniendo en cuenta el volumen de roca investigado por las herramientas de registro. Se deben considerar también los efectos de las desviaciones en las muestras de núcleo, la frecuencia de muestra y el volumen de muestra. Con una sola excepción, siempre se refiere a la determinación de porosidad que va de cero (0) a <400 psi (2760 kPa) de esfuerzo restrictivo. Se explica el método para determinar la porosidad a presiones excesivas simuladas en 5.3.2.2.1.2 5.1.1.2 A menos que se especifique lo contrario, los métodos de análisis descritos son aplicables a tapones de núcleo cilíndricos perforados de rocas consolidadas y relativamente homogéneas. La porosidad obtenida con el análisis de núcleos puede ser averiguada por varios métodos diferentes (ver Tabla 5-1). La medición del volumen total (BV) y del volumen del Grano (GV) nos da el volumen poroso (PV) por diferencia, y porosidad (∅) así: PV = (BV - GV) (1) ∅ = (BV - GV)/BV La medición directa del volumen poroso (PV) y del volumen del Grano (GV) da como resultado: ∅ = PV/(PV+GV) (2) La medición directa del volumen del poro (PV) y del volumen total (BV) da como resultado: ∅ = PV/BV (3) 5.1.1.4 En el laboratorio, por lo general se mide uno
de dos tipos de porosidad: estos son la porosidad efectiva o la porosidad total. Existe
una diferencia entre la definicion actual utilizada por los analistas de registros. Esto se complica aún más con las técnicas de análisis de núcleo diseñadas para dejar varios estratos moleculares de agua con arcilla o minerales dentro del espacio poroso (ver Figura 5-1).
5.1.1.4 Históricamente, la porosidad efectiva ha sido definida como una medida de los vacíos conectados. Se deriva a partir de la diferencia entre las determinaciones del volumen total y el volumen aparente del grano, o con una medición directa del espacio vacío conectado. El volumen medido del espacio vacío conectado puede variar con la preparación de la muestra y el método analítico utilizado. La porosidad total es una medida del espacio vacío total, tanto conectado como aislado, en la muestra de roca. Este puede ser determinado con la medición del volumen total, el peso seco y el volumen del grano en una muestra desagregada. 5.1.1.5 En el campo de la interpretación de registros, la porosidad efectiva se ha definido como el volumen poroso interconectado ocupado por los fluidos libres. En esta definición, la porosidad efectiva no incluye el volumen del agua ligada a arcilla o a minerales y en algunos enfoques analíticos, no incluye tampoco el agua adicional contenido dentro del shale por fuerzas capilares. Además, en el campo de la interpretación de registros, la porosidad total es ese volumen que ocupan todos los fluidos (espacio poroso conectado y aislado) en la roca, incluyendo el volumen ocupado por el agua unida a la arcilla (ver Figura 5-1). 5.1.1.6 La experiencia en el análisis de núcleos indica que para la mayoría de las rocas de yacimiento existen pocos poros aislados, y por lo tanto existe muy poco (o no existe) diferencia medible en la porosidad total y efectiva históricamente definida (Figura 5-1). Por lo tanto, con estas definiciones, la porosidad efectiva determinada con los análisis de núcleo en una muestra totalmente seca entre 210o y 240oF (99o a 116oC) corresponde más aproximadamente a la porosidad total definida por los analistas. En ciertas circunstancias, tal como el de las rocas volcánicas, puede haber una diferencia medible entre la porosidad efectiva (conectada) y la total (conectada y aislada) a partir del análisis del núcleo. Además, las técnicas de secado del núcleo pueden diseñarse de tal manera que se deja cierta cantidad de agua ligada en superficies minerales. 5.1.1.7 No existe un acuerdo universal entre los analistas de registros sobre la definición de porosidad efectiva (ver Figura 5-1). El menor valor para este parámetro se obtiene si todo el agua asociado con los shales (agua libre de aniones/adsorbida más el agua contenida en los capilares dentro del shale) se excluye como espacio poroso no efectivo. Se obtiene un valor mas alto si se excluye únicamente el agua libre de

aniones/adsorbida (definida como una función de la salinidad y también la capacidad de intercambio catiónico o la concentración de counteriones en la arcilla por unidad de volumen poroso, meq/cm3). Las porosidades de núcleos basados en muestras secas con humedad controlada por lo general se encuentran entre este valor mas alto y el valor de la porosidad total. En este momento es difícil seleccionar las condiciones de laboratorio que produzcan una porosidad efectiva en las muestras de núcleo de conformidad con las definiciones de los analistas. 5.1.1.8 Otro aspecto de la medición de porosidad, que en realidad se aplica a todo el campo del análisis de núcleos, es la selección entre un análisis convencional (muestra pequeña) o un análisis de diámetro completo. La decisión sobre el tipo que se debe utilizar preferiblemente se basaría únicamente en la homogeneidad de la formación que se está analizando. Muchas piedras areniscas son lo suficientemente homogéneas para que una muestra pequeña pueda considerarse representativa del incremento del análisis. Por otro lado, cuando la formación es heterogénea en cuanto a la estructura de los poros o la litología, como es el caso de los carbonatos vugulares o fracturados, las técnicas de núcleo análisis de núcleo de diámetro total. Por lo tanto, el tamaño de la muestra requerido para representar la estructura de poros y la litología del material del núcleo adecuadamente, como tambien la estrategia de muestreo, deben controlar el tipo de análisis utilizado.
5.1.2 Muestras de Núcleo con Consolidación
Deficiente o Sin Consolidar 5.1.2.1 Las muestras con consolidación deficiente o no consolidadas presentan requisitos únicos. El término “deficientemente consolidado” incluye una amplia gama de materiales de núcleos, desde muestras friables hasta muestras que no tienen ninguna consolidación, sin una cementación aparente entre los granos. Para efectos de esta discusión, el término “deficientemente consolidado” se refiere a cualquier muestra de tapón que requiere un montaje en un material envolvente apropiado para soportar el proceso de limpieza, preparación y medición. 5.1.2.2 Las muestras de tapón son usualmente envueltos utilizando un cilindro de metal o de polímero alrededor de la circunferencia de la muestra. La Tabla 5-2 relaciona algunos de los materiales envolventes comunes junto con sus ventajas y desventajas. 5.1.2.3 Para permitir el flujo, se cubren las caras del tapón con tamices u otros materiales porosos. El tamaño de la malla debe ser lo suficientemente pequeño para evitar la pérdida de granos, pero lo suficientemente grande para evitar taponamientos por finos movedizos.
Tabla 5-1: Indice de Volúmenes: total, de Grano y Poroso Número del Método
Titulo Propósito
5.2.1 5.2.2 5.2.3 5.2.4 5.2.5 5.3.1 5.3.2.1.1 5.3.2.2.1 5.3.2.2.2 5.3.2.2.3 5.4 5.5
Inmersión en Mercurio (Flotabilidad)de Arquímedes Desplazamiento de Mercurio Medidor (Flotabilidad de) Arquímedes con fluidos diferentes al mercurio Volumen total sumando volumen poroso y de grano Volumen poroso total de la densidad del grano de la muestra desagregada Método de Doble Celda de la Ley de Boyle Método de Celda Sencilla de la Ley de Boyle Sumatoria de Porosidad de Fluidos Método de Saturación de Líquidos Procedimientos Históricos Rocas ricas en materia orgánica
Medición del Volumen total Medición del Volumen total Medición del Volumen total Medición del Volumen total Determinación del Volumen total Medición del Volumen del Grano Medición del Volumen del Grano Medición Directa del Volumen del Poro Resumen de las Mediciones del volumen total y de Poro Medición directa del volumen de poro Medición del volumen total, de grano y de poro Volumen de poro y gas adsorbido
Típicamente se utilizan las mallas de tamaño 200 o 120. Las muestras algo consolidadas requieren un solo tamiz. Las muestras menos consolidadas pueden requerir tratamientos finales más sofisticados. Por ejemplo, se pueden utilizar dos tamices. Un tamiz es de malla más fina para evitar la pérdida de granos y se utiliza un segundo tamiz menos flexible y más gruesa para darle fuerza mecánica. El material escogido para los tamices debe ser inerte a los solventes y las salmueras utilizadas
para limpieza y pruebas. Se recomienda un acero inoxidable de alta calidad como el 316 o Monel. 5.1.2.4 El volumen poroso y de grano aparente del material envolvente y de los tamices en los extremos debe determinarse y contabilizarse como parte de la medición de porosidad. El volumen del material envolvente en una muestra individual puede determinarse aplicando el Método de Doble Celda de la

Ley de Boyle para el volumen de grano (ver 5.3.2.1.1) o el método de Arquímedes para el volumen total, utilizando ya sea mercurio (donde sea apropiado, ver 5.2.1) u otros solventes (ver 5.2.4). En la práctica, es mas fácil determinar la densidad del material envolvente
y luego determinar el peso de este material aplicado a cada muestra. Con estos datos se puede hallar el volumen del material. Este método asume que la densidad del material es constante de pieza en pieza.
Tabla 5-2 Material Envolvente para Muestras de Núcleo Poco Consolidadas/No consolidadas Material Envolvente Ventajas Desventajas Mangas de Plomo Cinta de Teflón Tubo de Teflón Encogimiento Térmico Manga de Aluminio Pintura Epóxica u Otra Cobertura Estaño
Maleable, se adhiere bien a la muestra Inerte Inerte, fácil del Aplicar Maleable, se adhiere razonablemente bien a la muestra Económica, fácil de Aplicar Maleable, se adhiere bien a la muestra
Interactúa con el mercurio y algunas salmueras. Difícil de aplicar, porosa y permeable Puede que no se adhiera bien a la muestra en bajos niveles de esfuerzo restrictivo, posible alteración de la muestra por el calor y por el estrés radial aplicado mientras se encoge el tubo Interactua con algunas salmueras y puede que no se adhiera bien a bajos niveles de esfuerzo restrictivo Interactua con solventes, fuerza mecánica deficiente, puede ser absorbido en la muestra Interactua con el mercurio y algunas salmueras
5.1.2.5 También se debe determinar el volumen de “grano” y el "poroso” de los tamices. El volumen de grano puede determinarse tomando una cantidad de tamices idénticos y midiendo su volumen sólido en un aparato de doble celda aplicando la Ley de Boyle. El valor determinado se divide por el número de tamices probados y este valor promedio se aplica a cada muestra. El volumen poroso es más sensible al método de prueba aplicado. Para el método de la Ley de Boyle, el volumen poroso o vacío en los tamices puede determinarse montando un tapón sólido, no poroso de la misma manera en que se preparan las muestras reales. El método de Celda-Sencilla de la Ley de Boyle da volúmenes vacío de los tamices. 5.1.2.6 Una vez se empaca la muestra con este material envolvente y tamices, se pueden realizar pruebas utilizando muchas de las mismas técnicas aplicadas en las muestras consolidadas. En las siguientes secciones se explica la aplicabilidad de cada método de medición para las muestras envueltas de poca consolidación. 5.1.2.7 Como las propiedades físicas de las muestras con poca consolidación son más sensibles al esfuerzo restrictivo que las muestras consolidadas, las mediciones realizadas a niveles de esfuerzo por debajo del esfuerzo efectivo del yacimiento in situ, no serán tan representativas de la roca del yacimiento. Por lo tanto, el método recomendado para determinar la porosidad de
muestras de consolidación deficiente es el método de Celda-Sencilla de la Ley de Boyle para la medición de volumen poroso en esfuerzo restrictivo elevado (ver 5.3.2.2.1.2) y el método de Doble-Celda de la Ley de Boyle para el volumen de grano. 5.2 MEDICION DEL VOLUMEN TOTAL
(TAMAÑO DE TAPON) Se requiere el volumen total de una muestra de roca para determinar la porosidad de esa muestra. El volumen total de un tapón de núcleo puede ser determinado por varios métodos. Las técnicas de medición incluyen la inmersión de Arquímedes, el desplazamiento de mercurio y por medidor. Los volúmenes totales también pueden determinarse sumando las mediciones directas del volumen de grano y el volumen poroso. El volumen total de una muestra seleccionada para la medición de porosidad debe medir preferiblemente por lo menos 10 cm3. Normalmente, las muestras son cilindros rectos con diámetros desde los 2.54 cm hasta 3.81 cm y longitudes de por lo menos 2.54 cm y 3.81 cm respectivamente. Se pueden utilizar formas irregulares tomando las precauciones adecuadas si no se pueden obtener muestras de dimensiones regulares. 5.2.1 Inmersión en Mercurio (Flotabilidad) de
Arquímedes

5.2.1.1 Principio Se sumerge un tapón de núcleo en mercurio y el volumen del mercurio desplazado por la muestra se determina gravimétricamente (Principio de Arquímedes). 5.2.1.2 Ventajas Las ventajas de este método incluyen: a. Las muestras pueden utilizarse para tests posteriores siempre que no ocurra una penetración del mercurio. b. El método es exacto si se utiliza una técnica cuidadosa y se hacen mediciones precisas. 5.2.1.3 Limitaciones Las limitaciones de este método incluyen: a. La retención de aire alrededor de las muestras creará errores y producirá volúmenes totales demasiado altos. b. Las muestras con cavidades o que tengan una permeabilidad extremadamente alta serán penetradas por el mercurio, resultando en valores de volumen total bajos y dejando las muestras inservibles para pruebas adicionales. Las muestras que tienen una superficie con cavidades o que contengan fracturas abiertas no son recomendadas para el análisis de volumen total por inmersión de mercurio. Sin embargo, si es necesario analizar este tipo de muestras utilizando esta técnica, la superficie de la muestra debe sellarse o se deben llenar las cavidades para evitar la penetración del mercurio. Es posible que este no sea un tratamiento adecuado. 1. Se pueden utilizar materiales tales como tubería de Teflón de termoencogible para recubrir las superficies cilíndricas de una muestra para tapar las fisuras. El volumen de cualquier cobertura debe restarse del volumen total medido, y no deben existir espacios de aire entre la tubería y la roca en superficies que no tengan fisuras. 2. Se pueden llenar las fisuras en la superficie (con arcilla, parafina o un epoxi), lo cual permitirá una medición directa del volumen total cuando se sumerja la muestra en el mercurio. Por lo general, el llenado de las fisuras es una prueba destructiva y debe ser el último paso en el análisis. 3. El calibrado de la longitud y el diámetro de la muestra, y el cálculo del volumen total (ver 5.2.3) es un método utilizado para superficies que tienen fisuras. La experiencia ha demostrado que los volúmenes totales calculados a menudo son demasiado altos. Las técnicas de calibración presentadas en 5.2.3.6 mejoran la exactitud de los datos. c. El método es inadecuado para las muestras con
consolidación deficiente o sin consolidación que están montadas en manguitos de plomo por la amalgamación del plomo y el mercurio. Además el
mercurio puede retenerse entre los materiales envolventes y la muestra. Como consecuencia, este no es un procedimiento recomendado para las muestras forradas
5.2.1.4 Exactitud La medicion del peso puede repetirse con una precisión de ± 0.015 gramos si no hay aire retenido cuando se sumerja la muestra y la temperatura permanezca constante. Las mediciones de volumen total pueden repetirse con una precisión de ± 0.01 cm3. 5.2.1.5 Equipos Los siguientes equipos son apropiados para este método: a. Balanza analítica electrónica de una sola bandeja, con una precisión de ± 0.01 g. b. Recipiente de mercurio lo suficientemente grande para sumergir completamente el tapón en posición horizontal sin tocar los lados del recipiente. d. Mercurio para llenar el recipiente para sumergir los
tapones completamente. e. Tenedor ajustable con marca de referencia. f. Termómetro. 5.2.1.6 Procedimiento El volumen total (BV) se mide por el método de Inmersión en Mercurio de Arquímedes con el aparato que se muestra en la Figura 5-2. Se coloca un vaso de mercurio sobre la balanza electrónica de bandeja sencilla, y se sumerge el tenedor hasta la marca de referencia. La marca se ubica de tal manera que la parte superior del tapón de núcleo quede sumergido de 3 a 7 milímetros dentro del mercurio. Luego se tara la balanza. Se retira el tenedor del mercurio, y el tapón que se va a medir flota a lo largo en el mercurio con el tenedor en la misma marca de referencia. La muestra no debe tocar los lados del recipiente de mercurio. El peso resultante representa la masa del mercurio desplazado. El alambre de acero inoxidable de calibre 18 (1.0 milímetro de diámetro) es un material adecuado para el tenedor. Se ha observado que el uso del alambre de mayor calibre resulta en mediciones de peso inestables (hay cambios de peso con el paso del tiempo) y tiempos prolongados de equilibrio. 5.2.1.7 Cálculos El volumen total se calcula utilizando la siguiente ecuación: BV =
Densidad del Mercurio a la temperatura de medición
Masa de Mercurio desplazada
5.2.1.8 Precauciones:

Una base pesada para sostener el ensamblaje del tenedor es esencial para mantener la muestra en su lugar para asegurar la medición precisa de pesos. Es importante utilizar la densidad correcta de mercurio para su temperatura. Una variación de 5ºC provocará un error sistemático de 0.02% en el volumen total (ver Tabla 5-3) Tabla 5-3: Densidad del Mercurio Vs. Temperatura oC Densidad del Mercurio g/cm3 oC Densidad del Mercurio g/cm3 18.0 19.0 20.0 21.0 22.0 23.0 24.0 25.0 26.0
13.5512 13.5487 13.5462 13.5438 13.5413 13.5389 13.5364 13.5340 13.5315
27.0 28.0 29.0 30.0 31.0 32.0 33.0 34.0
13.5291 13.5266 13.5242 13.5217 13.5193 13.5168 13.5144 13.5119
Con el fin de lograr pesos estables, el tenedor debe tener un diseño tal que permita un punto de contacto sobre la muestra. Si el volumen del tenedor es demasiado grande en relación con el volumen del tapón, se obtendrán pesos erróneos. Los pesos inestables pueden indicar que el tenedor no es el adecuado o que hubo invasión de mercurio. La contaminación de mercurio puede confirmarse comparando los pesos de las muestras antes y despues de la inmersión en mercurio. 5.2.2 Desplazamiento de Mercurio (Bomba de
Desplazamiento Volumétrico) 5.2.2.1 Principio El volumen total de la muestra se mide por desplazamiento de mercurio utilizando una bomba de desplazamiento volumétrico a la cual se conecta una cámara de acero inoxidable. 5.2.2.2 Ventajas Las ventajas de este método incluyen: a. Este procedimiento permite ejecutar mediciones rápidas. b. La técnica se utiliza como parte de la medición de porosidad por Sumatoria de Fluidos. c. Las muestras pueden utilizarse en tests posteriores si no ocurre una penetración o adsorción de mercurio. 5.2.2.3 Limitaciones Las limitaciones de este método incluyen:
a. El aire atrapado alrededor de la muestra producirá volúmenes totales demasiado altas. b. Las muestras con fisuras o con permeabilidades extremadamente altas pueden ser penetrados por el mercurio resultando en bajos valores de volumen total y dejando las muestras inservibles para pruebas adicionales (ver 5.2.3). c. En la mayoría de las bombas de desplazamiento de mercurio, la muestra se sumerge aproximadamente 50 milímetros en el mercurio. La altura del mercurio provoca una presión de cerca de 1 psi (6.9 kPa) en la parte superior de la muestra. Esto puede causar que los volúmenes totales sean sistemáticamente bajas debido a 1) conformidad con la aspereza de la superficie microscópica o 2) penetración en los poros grandes. La profundidad de la inmersión se minimiza (3-7 milímetros) en el método de Inmersión en Mercurio de Arquímedes (ver 5.2.1). d. Este método no es el adecuado para muestras no consolidadas montadas en manguitos de plomo debido a la amalgamación de plomo-mercurio. El mercurio también puede quedar atrapado entre los materiales envolventes y la muestra. Por lo tanto, este método no es recomendable para muestras forradas. 5.2.2.4 Exactitud La medición puede reproducirse con una precisión de 0.01 cm3 si la bomba se ha calibrado y se ha ajustado en cero para cada muestra. 5.2.2.5 Equipos La figura 5-3 muestra una bomba volumétrica de desplazamiento de mercurio de alta presión. La cámara para muestras podrá contener tapones hasta aproximadamente 25cm3 de volumen. 5.2.2.6 Procedimiento/Cálculos El volumen total de una muestra de núcleo de forma regular o irregular se obtiene por medio de desplazamiento de mercurio. La cámara que no contenga ninguna muestra, se llena con mercurio hasta el nivel de referencia. El aparato de lectura del instrumento se coloca en cero. Luego se baja el nivel de mercurio, se introduce la muestra y se llena la cámara nuevamente hasta el nivel de referencia. Se obtiene la lectura del volumen del instrumento. La diferencia entre esta lectura y el cero del instrumento es el volumen total de la muestra. 5.2.2.7 Precauciones El mercurio y la cámara deben ser limpios y libres de películas superficiales, sólidos y aceites. 5.2.2.8 Calibración

La bomba de mercurio se calibra con lingotes de acero de volumen conocido. 5.2.3 Medidor 5.2.3.1 Principio Las muestras que son cilindros rectos u otras formas regulares pueden ser calibrados para obtener el volumen total. Se puede utilizar un micrómetro o un calibre vernier (de nonio), el cual pueda leerse con una precisión de 0.002 cm, para medir longitud y diámetro. Se recomienda un mínimo de cinco mediciones. Nota : Las muestra con fisuras superficiales y/o fracturas abiertas pueden ser analizados utilizando este método, si se calibran los datos de volumen total en las muestras utilizando la técnica indicada en 5.2.3.6. 5.2.3.2 Ventajas Las ventajas de este método incluyen: a. La muestra puede utilizarse en pruebas posteriores. c. Es un procedimiento rápido. 5.2.3.3 Limitaciones Las limitaciones de este método incluyen: a. Las muestras de formas irregulares no pueden medirse por este método. b. Las irregularidades en las superficies de las muestras pueden dar volúmenes totales demasiado altos. c. Los volúmenes totales calibrados por lo general no son recomendados para métodos de porosidad donde PV = (BV-GV). La exactitud del volumen total no es tan crítica donde ƒ = PV/BV y el volumen poroso es determinado por medición directa (ver 5.3.2.2). En los casos donde el tipo de roca (con fisuras o fracturada) no requiera el uso de la técnica del medidor, los datos deben calibrarse de la manera indicada en 5.2.3.6. d. No se recomienda este método para muestras que necesiten forro, muestras de consolidación deficiente o sin consolidación alguna. Si se utiliza de todas maneras, se deben aplicar correcciones para el grosor del tamiz y el forro en los valores de longitud y diámetro. 5.2.3.4 Exactitud Se ha mostrado que las mediciones de longitud y diámetro en muestras reales se pueden reproducir con un nivel de confiabilidad del 99% (dentro de 3 desviaciones estándar) con una precisión de 0.15 mm para longitud y 0.04 mm para diámetro. Los volúmenes totales pueden repetirse con una precisión de 0.15 cm3. Ver 5.2.3.6 para mejorar este nivel de precisión. 5.2.3.5 Equipos
Se pueden utilizar los medidores digitales o de vernier. 5.2.3.6 Procedimiento La longitud y el diámetro de un cilindro o las dimensiones de una muestra de forma regular se miden en por lo menos cinco posiciones diferentes para definir cualquier irregularidad en la forma. Las pequeñas desviaciones en la forma se pueden promediar. Varias muestras sin fisuras y/o fracturas abiertas, cuyas dimensiones brutas cubran el alcance de muestras a analizar, deben tener su volumen total determinado utilizando una técnica de inmersión adecuada. Luego se debe determinar su volumen total utilizando la técnica exacta de medidor que se va a utilizar para la medición de muestras subsecuentes. Los factores de corrección pueden entonces determinarse para así calibrar los volúmenes totales con respecto a aquellos medidos por inmersión. Estos factores se aplican entonces a las mediciones subsecuentes de volumen total calibrado para corregirlas hasta llegar a un volumen de inmersión equivalente. Si se utilizan medidores digitales, se requiere menos tiempo y se podrán tomar más lecturas. Comparados con los medidores de vernier, las mediciones no son tan susceptibles al error humano porque las lecturas se muestran en forma digital. Los medidores digitales se pueden conectar con un computador para registrar las lecturas automáticamente. Se pueden tomar 10 lecturas de longitud o en menos de 60 segundos. 5.2.3.7 Cálculos El área transversal de la muestra cilíndrica se calcula a partir del diámetro promedio y se multiplica por la longitud promedio para obtener el volumen total. Se puede utilizar una fórmula apropiada para muestras no cilíndricas. 5.2.3.8 Precauciones Las precauciones de este método incluyen: a. Las mediciones de diámetro deben tomarse en por lo menos cinco posiciones de igual distancia a lo largo de la muestra. b. Las mediciones de longitud también deben obtenerse de por lo menos cinco posiciones tomadas alrededor de la periferia de la muestra. c. Asegúrese de que los medidores están en cero entre mediciones sucesivas. d. Cuando se midan diámetros o longitudes de
muestras con irregularidades en la superficie, asegúrese de que los medidores no penetren las fracturas o fisuras resultando en mediciones menores que las dimensiones reales.
5.2.4 Flotabilidad (Arquímedes) Con Fluidos Diferentes al Mercurio (ejemplo, Salmuera, Aceite Refinado o Tolueno)

5.2.4.1 Principio Un cuerpo colocado en un líquido flota por una fuerza igual al peso del líquido desplazado. 5.2.4.2 Ventajas Las ventajas de este método incluyen: a. Se pueden lograr valores exactos si se utiliza la técnica adecuada. b. La muestra puede ser totalmente saturado con líquido para otras pruebas que así lo requieran e. Si la muestra está 100% saturada de un solo fluido
antes de medir el volumen total, se pueden calcular el volumen poroso, el volumen del grano y la densidad del grano con los pesos registrados.
5.2.4.3 Limitaciones Las limitaciones de este método incluyen: a. El líquido puede ser inadecuado para pruebas posteriores y tendrá que removerse. b. Los núcleos que tengan fisuras no deben medirse por este método. c. No deben utilizarse líquidos que lixivien la muestra o que causen hinchazón de la matriz. d. Las fisuras o poros grandes en muestras con permeabilidad extremadamente alta se llenarán mientras la muestra se encuentra sumergida en el líquido resultando en bajos valores de volumen total (ver 5.2.2.3.b). Llenar estas fisuras o poros grandes antes de la medición permitirá una medición directa del volumen total cuando se tome el peso sumergido. f. Esta no es una técnica recomendable para
muestras envueltas con consolidación deficiente o no consolidadas debido a la posibilidad de atrapar un volumen de fluidos extraños entre la superficie del tapón y el forro.
5.2.4.4 Exactitud La medición del peso puede repetirse con una precisión de ±0.015 gramos si no haya aire atrapado cuando se sumerja la muestra y si la temperatura permanece constante. La exactitud de las mediciones de volumen total variará dependiendo de la densidad y la volatilidad del líquido utilizado en las mediciones. Una buena técnica debe dar un volumen total repetible con una precisión de 0.01 cm3. 5.2.4.5 Equipos Se requiere una balanza analítica con una precisión de un miligramo, un soporte de alambre fino, un recipiente para líquido y un termómetro. La Figura 5-4 muestra dos posibles configuraciones experimentales. 5.2.4.6 Procedimiento
5.2.4.6.1 Se satura una muestra con un líquido de densidad conocida, tal como una no dañina, un aceite refinado ligero o un solvente de un alto punto de ebullición. La muestra puede ser esencialmente 100% saturada con líquido evacuando el espacio poroso, introduciendo el saturante, y siguiendo con una saturación de presión (ver 5.3.2.2.3.6). 5.2.4.6.2 Se remueve el exceso de líquido cuidadosamente (evitando la pérdida de grano) de la muestra y luego la muestra saturada se pesa en el aire. Cuando se remueve el exceso de líquido de la superficie de la muestra, se deben tomar precauciones para asegurar que no se remueven los fluidos de los poros expuestos en la superficie. Se debe evitar el uso de materiales (como toallas secas) que remueven el líquido de los poros de la superficie por acción capilar, como también por método mecánico tal como la agitación violenta. Los métodos aceptables para remover el exceso de líquido son el de rodar la muestra sobre un trapo húmedo libre de pelusa o sobre una toalla de papel mojados con el líquido saturante, o de limpiar la muestra cuidadosamente con un trapo húmedo o con los dedos. 5.2.4.6.3 Se llena un cubilete con líquido saturante. Un soporte de alambre fino (de diámetro máximo de 1.0 milímetro) conectado al estribo de una balanza se sumerge en el líquido hasta una marca de referencia, y la balanza se coloca en cero. Luego, se coloca la muestra en el soporte, se sumerge hasta la marca de referencia y se obtiene el peso sumergido de la muestra. (ver Figura 5-4A). 5.2.4.6.4 La figura 5-4 B muestra un procedimiento alterno. Se llena un cubilete con el líquido saturante y se coloca en una balanza. Luego se baja el soporte de alambre fino en el líquido hasta la marca de referencia y se ajusta la balanza en cero. La muestra saturada se coloca luego en el soporte, se sumerge hasta la marca de referencia y se obtiene el peso inmerso de la muestra. 5.2.4.6.5 Se puede hacer una revisión al volumen poroso tomando (a) la diferencia en peso de la muestra 100% saturada en el aire y el peso seco y b) dividiendo por la densidad del líquido saturante. Nota: No es indispensable que la esté 100% saturada con el líquido para poder determinar el volumen total. La técnica da un volumen total exacto siempre y cuando la muestra no esté absorbiendo líquido cuando se toma el peso mientras está suspendido bajo el líquido. Sin embargo, si la muestra no está 100% saturada, los resultados de mediciones como el volumen de poro, el volumen poroso, el volumen de grano, y la densidad de grano calculados con los pesos serán incorrectos. 5.2.4.7 Cálculos

El volumen total calculado con el aparato mostrado en la Figura 5-4 A es igual al peso inicial de la muestra saturada (o parcialmente saturada con peso estabilizado) en el aire menos el peso cuando está sumergida, dividido por la densidad del líquido de inmersión. El volumen total calculado con el aparato mostrado en la figura 5-4B es igual al peso inmerso de la muestra saturada dividido por la densidad del fluido de inmersión.
A B
BV = Peso saturado - Peso inmerso; BV= Densidad del Fluido de Inmersión Densidad del Fluido de Inmersión
Peso Inmerso
5.2.4.8 Precauciones Las precauciones para este método incluyen: a. Se debe tener cuidado en el diseño del aparato. El arreglo debe ser tal que se sumerjan totalmente ambas muestras y el soporte de alambre se sumerjan dentro del líquido durante la medición, que no se encuentren apoyadas por el recipiente de inmersión y que sólo cierta longitud de alambre recto (diámetro máximo de 1.0 milímetro) penetre la superficie del líquido. El alambre y el fluido deben estar limpios. b. Es esencial utilizar la densidad correcta de fluido para la temperatura del líquido en el momento de la medición del peso sumergido. La densidad del fluido debe conocerse con una precisión de 0.005 g/cm3. Un error de 0.005 g/cm3 resultaría en una desviación de porosidad de 0.5 puntos (porosidad expresada como porcentaje) y una desviación en la densidad de grano de 0.01 g/cm3. La densidad del fluido puede medirse con un picnómetro, un medidor digital electrónico de densidad, varias balanzas de gravedad específica o puede también calcularse utilizando un estándar de silicio disponible del Instituto Nacional de Estándares y Tecnología. Vss = Wss /ρss (7) Donde : Vss = volumen estándar de silicio. Wss = peso al aire del estándar del silicio. ρss = Densidad estándar del silicio del Instituto Nacional de Estándares y Tecnología. ρF = Wss - Wiss /ρss Donde: ρF = densidad del fluido en el cual se sumerge el silicio. Wss = Peso inmerso del estándar de silicio.
c. Es probable que unos pesos inestables decrecientes indiquen que el fluido se está absorbiendo en la muestra, lo cual resultará en un volumen total demasiado bajo. d. Las fluctuaciones en temperatura deben minimizarse porque la densidad del líquido de inmersión podría variar. e. Se debe tener cuidado cuando se limpian las
superficies de la muestra saturada para evitar la pérdida de grano o una remoción excesiva de saturante. Ver 5.2.4.6.2 para las técnicas adecuadas.
5.2.5 Volumen Total Calculado Por Sumatoria de
Mediciones Directas de Volumen de Grano (5.3.2.1.1) y Volumen Poroso (5.3.2.2.1)
5.2.5.1 Cálculos El cálculo básico es: BV = GV + PV (9) 5.3 MEDICIÓN DE VOLUMEN POROSO El volumen poroso total es el espacio vacío total, tanto conectado como aislado, en una muestra de roca. 5.3.1 Volumen Poroso Total Calculado de la Densidad de Grano en Muestras Desagregadas El volumen poroso total es igual a la diferencia entre el volumen total de la muestra y el volumen de grano desagregado. La separación se realiza con el fin de exponer cualquier volumen poroso aislado. 5.3.1.1 Método Seco para Determinar la Densidad
de Grano/Volumen de Grano. 5.3.1.1.1 Principio Primero se determina el volumen total de una muestra limpia (ver 5.2). Luego, la muestra se seca, se pesa y se desagrega. Despues de la separación, se coloca una porción debidamente pesada dentro de un porosímetro de la Ley de Boyle para determinar el volumen de grano (ver 5.3.2.1.1). El volumen de grano en la muestra total se calcula utilizando la proporción del peso seco de la muestra consolidada con respecto al peso seco de la muestra desagregada que se coloca en el porosímetro. 5.3.1.1.2 Ventajas Las ventajas de este método son: a. Es rápido. b. La muestra se puede utilizar para mediciones complementarias que requieren una muestra desagregada. Ver Sección 7, Pruebas Complementarias. 5.3.1.1.3 Limitaciones

Las limitaciones de este método son: a. Se necesita realizar un proceso de secado. b. No es viable para las rocas que contienen yeso. c. No es viable para las rocas que contienen minerales tales como la halita que son solubles en el fluido limpiador. d. La técnica supone que cualquier volumen poroso aislado permanece aislado durante el tiempo en que se seca la muestra. e. Si el agua llena el espacio poroso y no se remueve mientras se seca la muestra, el volumen de grano calculado (GV) será erróneamente alto. 5.3.1.1.4 Exactitud La técnica es capaz de calcular la densidad de grano con una precisión de ± 0.01 g/cm3
. No hay datos de porosidad comparativos disponibles, pero se espera que la técnica calcula porosidades con una precisión de ± 0.4 unidades de porosidad, o aún mejores. 5.3.1.1.5 Equipos Los siguientes equipos son apropiados para este método: a. Pulverizador con tolerancia ajustable entre los platos moledores. b. Tamiz de malla 60 en conformidad con la Oficina de Estándares de los Estados Unidos. c. Balanza analítica, con una precisión de 0.1 miligramos a. Porosímetro de la Ley de Boyle modificado para el
manejo de muestras en polvo. (1) tapa para el vaso (para mantener la muestra en polvo dentro del vaso).
5.3.1.1.6 Procedimiento El procedimiento para este método es el siguiente: a. Preparación de la muestra. 1. Limpiar. 2. Secar a 225oF (107oC). 3. Enfriar en el Disecador. 4. Pesar la muestra y registrar el peso (W). 5. Repetir los pasos anteriores (del 2 al 4) hasta que se estabilice peso (W). 6. Determinar el Volumen Total (BV) (ver 5.2). b. Triture la muestra y pásela por un tamiz de calibre 60. c. Seque una porción de la muestra para medir el Volumen de Grano entre 210º y 240ºF (99ºC a 116ºC) hasta llegar a un peso constante (Wd). d. Determine el Volumen de Grano (GVd ) de la porción desagregada de la muestra por el método de Doble Celda de la Ley de Boyle (ver 5.3.2.1.1).
1. Calibre el porosímetro con la tapa del vaso de la muestra en su puesto. 2. Coloque un peso conocido (Wd) de las muestras trituradas y secas dentro de la cubeta de la muestra. 3. Mida el Volumen de Grano de la muestra
desagregada y seca con la tapa en su lugar. 5.3.1.1.7 Cálculos Los cálculos para este método son los siguientes:
PV = BV - GV (10) Donde: PV = Volumen poroso total. BV = Volumen total de la muestra original consolidada. GV = Volumen de grano de la muestra calculada por la ecuación b.
GV = GVd x W/Wd Donde: GVd = Volumen de grano medido en la porción de la muestra desagregada colocada en el porosímetro. W = Peso original de la muestra Wd = Peso de la porción de la muestra desagregada y seca colocada en el vaso del porosímetro.
GD = Wd/GVd (12) Donde: GD = Densidad de grano de la porción de la muestra. 5.3.1.1.8 Precauciones Las precauciones para este método incluyen: a. La porción de roca seleccionada para la medición de densidad de grano debe ser representativa de la muestra total. b. Ver 5.3.2.1.1.8, Precauciones para el método de
Doble Celda de la Ley de Boyle (cubeta matriz) para el volumen de grano, items a) al f).
5.3.1.1.9 Calibración Ver 5.3.2.1.1.9, Calibración para el método de Doble Celda de la Ley de Boyle (cubeta matriz), para el volumen de grano. 5.3.2 Volumen Poroso Efectivo de Muestras Agregadas El volumen poroso efectivo puede calcularse restando el volumen de grano del volumen total de la muestra, o por medición directa del volumen vacío de la muestra. 5.3.2.1 Medición del Volumen de Grano 5.3.2.1.1 Método de Doble Celda de la Ley de Boyle (Cubeta Matriz) para el Volumen de Grano

5.3.2.1.1.1 Principio Ley de Boyle: Cuando la temperatura permanece constante, el volumen de una masa dada de gas ideal varia inversamente con su presión absoluta. V1/V2 = P2/P1 o P1V1 = P2V2 (13) Se requiere una extensión de la ecuación para contabilizar la variación de temperatura y el comportamiento que tiene un gas no ideal para lograr una determinación exacta del volumen de gas. P1V1 / z1T1 = P1V1 / z2 T2 (14) Esta última ecuación debe utilizarse con un dispositivo de doble celda cuando se determina el volumen de grano de una muestra (GV). El gas entra en una celda de referencia que tiene un volumen conocido (V) a una presión de referencia predeterminada (100 o 200 psig). Luego se pasa el gas de la celda de referencia a una cámara conectada de volumen conocido que contiene una muestra del núcleo. Esto resulta en una menor presión de equilibrio, de la cual se calcula GV. Se resta posteriormente el GV del volumen total para determinar el volumen poroso y de ahí la porosidad. 5.3.2.1.1.2 Ventajas Las ventajas de este método incluyen: a. No se daña la muestra de ninguna manera y por lo tanto puede utilizarse para otras mediciones. b. La operación es rápida, sencilla, y tiene un excelente nivel de repetibilidad. c. Se pueden probar muestras que tengan formas irregulares o fisuras. 5.3.2.1.1.3 Limitaciones Las limitaciones de este método incluyen: a. Se requiere una calibracion extremadamente cuidadosa y frecuente para poder obtener un buen nivel de exactitud. b. Los cambios en temperatura o presión barométrica deben ser contabilizados en los cálculos. c. El valor de porosidad resultante será mas alto que el valor de porosidad verdadero si se adsorbe el gas en las superficies de la muestra. El uso de helio minimiza esta posibilidad. d. Este método es adecuado para muestras forradas de consolidación deficiente o no consolidadas si se utilizan técnicas adecuadas. El volumen del material envolvente y los tamices en los extremos debe determinarse con precisión y debe restarse del volumen de grano aparente medido. El material envolvente y el volumen sólido del tamiz se pueden determinar 1) por medición directa en el vaso matriz de la Doble Celda de la Ley de Boyle antes de utilizarla, o 2) el volumen del material envolvente se
puede calcular del peso medido, y de la densidad del material envolvente previamente determinada. 5.3.2.1.1.4 Exactitud Un sistema bien calibrado determinará el volumen de grano con una precisión de ± 0.2 % del valor real. Esto corresponde a aproximadamente ±0.03 cm3 en muestras de una pulgada de diámetro y aproximadamente 1 pulgada de largo y ±0.1 cm3 en muestras de 1 ½ pulgadas de diámetro y 2 pulgadas de largo. 5.3.2.1.1.5 Equipos El volumen de grano se mide en un aparato que consiste de dos cámaras conectadas de volúmenes conocidos. La figura 5-5 muestra un ejemplo de tal aparato. Nota : Para obtener mediciones precisas es importante a) incorporar una válvula que tenga un volumen cero de desplazamiento (por ejemplo, una válvula de bola) entre el volumen de referencia y la cámara de muestras, o b) se debe conocer el volumen de desplazamiento de la válvula. La válvula de bola debe ventilarse siempre a presión atmosférica antes de colocarla en su posición cerrada. Si se utiliza de esta manera, el volumen de la válvula se incorpora dentro del volumen de la cámara calibrada y las mediciones serán precisas. De lo contrario, el volumen de desplazamiento de la válvula debe ser medido y contabilizado en los cálculos para obtener una medición exacta del volumen del grano. 5.3.2.1.1.6 Procedimiento Primero, se calibra primero el porosímetro, obteniendo el volumen de referencia de la cámara (Vr) y el volumen de la cámara de muestras (Vc). Luego, se coloca la muestra en la cámara respectiva. Se deja entrar el helio en la cámara de referencia a una presión predeterminada, típicamente 100-200 psig (690-1380 kPa). Se deben permitir alrededor de 30 segundos para que haya equilibrio en la presión, y luego se registra p1 (la presión indicada por el transductor digital). Se permite que el gas se expanda dentro de la cámara de muestras. Se mide la presión disminuida resultante (p2) una vez que el sistema haya llegado a su equilibrio (ver 5.3.2.1.1.8 e). Se calcula el volumen de grano de la muestra utilizando las ecuaciones de la ley de los gases indicadas en 5.3.2.1.1.7. 5.3.2.1.1.7 Cálculos El volumen del grano de la muestra se calcula con la presión inicial de referencia de la cámara y la presión final del sistema por medio de la ecuación de la ley de los gases. El volumen poroso es la diferencia entre el volumen total y el volumen de grano.

La siguiente ecuación para volumen de grano resulta del balance masivo de gas dentro de las cámaras de referencia y de muestras. P1Vr + Pa(Vc - Vg) = P2Vr + P2(Vc - Vg + Vv) (z1T1r zaT1c z2T2r z2T2c
15)
Donde : P1 = presión absoluta del volumen de referencia inicial P2 = presión absoluta expandida Pa = presión atmosférica absoluta inicialmente en la cámara de muestras z1 = factor z del gas a P1 y T1 z2 = factor z del gas a P2 y T2 za = factor z del gas a T1 y a presión atmosférica. T1r = temperatura absoluta del volumen de referencia a P1. T1c = temperatura absoluta de la cámara de muestras a P1. T2r = temperatura absoluta del volumen de referencia despues de que P2 se haya estabilizado. T2c = temperatura absoluta de la cámara de muestras despues de que P2 se haya estabilizado. Vg = volumen de grano. Vc = volumen de la cámara de muestras. Vr = volumen de la cámara de referencia. Vv = volumen de desplazamiento de la válvula (de la posición cerrada a la abierta). p1 = presión manométrica del volumen de referencia inicial. p2 = presión manométrica final del sistema. Si existen condiciones isotérmicas (T1 = T2) y si los valores de z se asumen iguales a 1.0, la ecuación anterior se reduce a: Vg = Vc - Vr ( P1 - P2)/(( P2 - Pa) + Vv (P2)/( P2 - Pa)(16) Si las presiones absolutas P1 y P2 se expresan como equivalentes a las presiones manométricas (es decir, P1 = (p1 + Pa) y se sustituyen en la ecuación anterior, resulta lo siguiente: Vg = Vc - [Vr ( P1/P2) - 1] + [Vv (1 + Pa/P2 (17) Si se utiliza un válvula de bola con volumen de desplazamiento cero, y si la válvula siempre se ventila a la atmósfera antes de cerrarla, Vv se incluye en el volumen de la cámara de muestras, Vv = 0.0 y la ecuación se simplifica aún más, así : Vg = Vc - [Vr ( P1/P2) - 1 (18) Nota : Los cálculos que ignoran los factores z o el volumen de la válvula en cantidades tan pequeñas como 0.1 cm3, pueden resultar en errores de porosidad de aproximadamente 0.5 puntos de porosidad. 5.3.2.1.1.8 Precauciones
Las precauciones para este método incluyen: a. El vaso matriz debe diseñarse para garantizar que tendrá el mismo volumen interno cada vez que se ensamble. b. El sistema debe recalibrarse si hay cambios en la temperatura o la presión barométrica. c. La tapón de núcleo debe secarse completa y apropiadamente. Algunas muestras requieren de técnicas especiales de secado. d. Cuando la salinidad del agua de poro (del agua de la formación o de filtrado de lodos) es superior a 100.000 mg/l, las muestras deben lixiviarse con metanol para remover las sales. Si se ignora el paso de cuando la salinidad del agua de poro se excede de 100.000 mg/l, se pueden desviar las porosidades por debajo en 0.4 puntos de porosidad hacia el nivel inferior (típicamente 20% de porosidad y una saturación de agua del 50% en el espacio poroso). e. La presión de equilibrio es esencial para lograr un volumen de grano exacto. El equilibrio por lo general se logra en 1-2 minutos, aunque los núcleos con baja permeabilidad y porosidad requieren mayor tiempo de estabilización (desde 30 minutos hasta varias horas). Un tiempo prolongado permite mayor probabilidad de cambios en la temperatura y en la presión barométrica. f. Los volúmenes muertos o extraños (el volumen no ocupado por la muestra en la cámara de muestras) debe mantenerse en el mínimo, o de lo contrario se determinarán volúmenes de grano erróneos. Se pueden agregar cilindros sólidos de acero inoxidable de volumen conocido para llenar el volumen vacío presente en la cámara de muestras que dejan las muestras mas cortas. Cuando se coloquen estos cilindros, se deben restar del volumen de grano calculado para obtener el volumen de grano de la muestra. Nota : Si hay cilindros de acero en el vaso matriz durante la calibración para obtener Vc y Vp, sus volúmenes no deben restarse. f. El volumen poroso de la muestra a medir debe
aproximarse al volumen de la celda de referencia para obtener mejores resultados analíticos. Se puede revisar la exactitud del método en pequeñas muestras irregulares determinando la densidad de grano en muestras, más grandes, más pequeñas y luego de tamaño similar de una densidad de grano conocida (como la Arena de Ottawa).
5.3.2.1.1.9 Calibración La calibración del porosímetro varía con el instrumento. En principio, se hacen dos o más mediciones; una con la cámara de muestras llena de cilindros sólidos de acero inoxidable de volumen conocido. Luego se hacen mediciones posteriores despues de remover uno o más cilindros que representan el 80, 60, 40, 20% de la cámara de volumen de muestras. Para mayor precisión, se deben retirar los cilindros suficientes para reducir la p1 a la mitad.

Se permite entrar el helio en la cámara de referencia a un presión predeterminada, típicamente 100 - 200 psig (690-1380 kPa). Se debe permitir unos 30 segundos para el equilibrio de la presión, y luego se registra el valor de p1 (presión de la celda de referencia indicada en la lectura del transductor digital). El gas se extiende luego a la cámara de muestras. Se mide la presión más baja que resulta (p2) despues de que el sistema haya alcanzado el equilibrio (aproximadamente 30 segundos). Con un volumen de la válvula Vv en cero, una temperatura constante, y el Vg conocido por el volumen del cilindro de acero en la cámara, Vr y Vc pueden calcularse por la solución simultánea de la ecuación apropiada presentada en 5.3.2.1.1.7. Cuando Vv no es igual a cero, o cuando la temperatura varía, la solución se vuelve más compleja, pero el principio permanece igual. Nota 1: En algunos sistemas se vuelven a ordenar las ecuaciones, y los datos son adecuados por regresión lineal por el margen de volúmenes de cilindros para obtener Vc y Vr. Nota 2: Los transductores de presión deben calibrarse a intervalos de 5 psi con un medidor de peso muerto o un transductor de presión estandar secundario 0.02% o de escala completa. La capacidad del transductor puede ajustarse a “presión verdadera” con un polinomio de cuarto grado utilizando técnicas de regresión. La revisión de la calibración puede realizarse midiendo el volumen de cada cilindro de acero y comparándolo con el volumen conocido. El resultado debe ser igual o menor de 0.03 cm3 para asegurar que la porosidad tenga una precisión de 0.5 puntos de porosidad en los volúmenes de grano medidos. 5.3.2 Medición del Volumen Vacío 5.3.2.2.1 Método de Celda Sencilla de la Ley de Boyle para la Medición Directa de Volumen Poroso. 5.3.2.2.1.1 Bajo Esfuerzo Restrictivo 5.3.2.2.1.1.1 Principio El volumen poroso se determina en un aparato que consiste de una celda de referencia de presión inicial volumen conocido, que luego se ventila en el volumen poroso de una muestra. La muestra se mantiene en un portanúcleos que utiliza una manga elastomérica y tapones en los extremos. Estos se ajustan a la muestra cuando se ejerce un esfuerzo restrictivo en sus superficies externas. La manga y los tapones de los extremos a su vez ejercen un esfuerzo compresivo sobre la muestra de núcleo. El volumen poroso por lo tanto se determina directamente utilizando la Ley de Boyle. Este en contraste con el método de doble celda donde se
determina el volumen de grano y se calcula el volumen poroso restando el volumen de grano del volumen total. El portanúcleos debe ser una celda de carga Hassier, isostática, biaxial o triaxial. Se utilizan bajos esfuerzos restrictivos, que son generalmente 400 psig o menos. En las rocas duras y consolidadas, el tipo de portanúcleos es de poca importancia debido a que ocurre una reducción mínima de espacio poroso en bajos esfuerzos. Aunque típicamente se les denomina “mediciones ambiente,” es importante anotar que algún nivel finito de esfuerzo restrictivo es necesario, y que el esfuerzo puede tener efectos muy significativos sobre las rocas friables o no consolidadas. Se permite al helio entrar en la celda de referencia de volumen conocido (Vr) a una presión de referencia predeterminada (100-200 psi). Luego se ventila el gas de la celda de referencia en el volumen poroso de la muestra. Esto resulta en una presión de equilibrio más baja, a partir de la cual se calcula el volumen poroso. 5.3.2.2.1.1.2 Ventajas Las ventajas de este método incluyen: a. La muestra, si está limpia y seca al inicio de la prueba, estará limpia al final de la prueba y lista para mediciones posteriores. b. La porosidad y la permeabilidad pueden determinarse de manera secuencial cargando la muestra solamente una vez. c. La operación es rápida y sencilla. d. Se eliminan las reacciones dañinas entre la roca y el fluido saturante utilizando un gas no reactivo. e. La medición directa del volumen poroso elimina la sensibilidad del volumen poroso a errores en la medición de BV o GV, donde BV y los GV son números grandes con respecto al volumen poroso, y PV = BV - GV. 5.3.2.2.1.1.3 Limitaciones Las limitaciones de este método incluyen: a. El sistema debe calibrarse cuidadosamente para el volumen muerto. b. Los cambios en temperatura y presión barométrica deben contabilizarse en los cálculos c. La muestra debe ser un cilindro recto de buena calidad sin fisuras superficiales ni esquinas desportilladas (las fisuras darán como resultado un volumen poroso demasiado bajo y los extremos no paralelos darán volúmenes porosos demasiado altos). Un error en la porosidad que se exceda de 1.5 unidades de porosidad puede resultar de una sola cara no paralela, lo cual resulta en una reducción en la longitud de la muestra de 1.0 mm en un lado de un cilindro de una pulgada de diámetro por un tapón recto cilíndrico de una pulgada de longitud.

d. La muestra debe estar libre de hidrocarburos y seca, o de lo contrario se determinarán volúmenes porosos demasiado bajos. e. El gas utilizado no debe adsorberse en los sitios de minerales activos tales como arcillas o materiales carbonosos. La adsorción resulta en volúmenes porosos demasiado altos, aunque puede minimizarse con la utilización de helio. f. Algunas rocas que tienen baja permeabilidad (< 0.01 md) pueden requerir un largo período (de treinta minutos hasta varias horas) para alcanzar la presión de equilibrio. Estos largos períodos permiten mayor oportunidad para cambios en temperatura y presión barométrica. g. Cuando la salinidad del agua del poro (del agua de la formación o por filtrado de lodo) son mayores de 100.000 mg/l, las muestras deben lixiviarse con metanol para remover las sales. Si se ignora este paso de lixiviación cuando la salinidad del agua del poro excede los 100.000 mg/l puede producir desviaciones en la porosidad hasta 0.4 puntos por debajo (típicamente 20% de porosidad y saturación de agua de 50% del espacio poroso). h. Este método es adecuado para muestras forradas de consolidación deficiente o no consolidadas si se observan las precauciones apropiadas. Se debe determinar el volumen vacío de los tamices o las placas porosas adheridas a las caras de la muestra. Este volumen debe restarse del volumen vacío medido en la muestra forrada para obtener el volumen poroso de la muestra. Se puede preparar y probar como muestra un tapón forrado sólido, no poroso igual al diámetro de la muestra. Esto dará el volumen del tamiz o el volumen vacío de la placa porosa, así como cualquier volumen que resulte al traslaparse el material envolvente en los extremos de la muestra. i. La porosidad se determina en un esfuerzo
restrictivo bajo, lo cual resulta en una porosidad mas alta que la presente en el yacimiento.
5.3.2.2.1.1.4 Equipos El aparato básico mostrado en la figura 5-6 es igual al ilustrado para el porosímetro de doble celda de la Ley de Boyle en la Figura 5-5 (ver 5.3.2.1.1.5). La diferencia principal es el diseño de la cámara de muestras, la cual elimina el volumen alrededor de la periferia de la muestra. 5.3.2.2.1.1.5 Procedimientos El porosímetro se calibra primero, dando el volumen de la cámara de referencia (Vr) y el volumen muerto del sistema (Vd). Luego se inserta un tapón de núcleo seco en una manga elastomérica. Una espiga con diámetro igual al del tapón se pone en contacto con cada extremo de la muestra. Un esfuerzo restrictivo de 400 psi o menos se aplica en la superficie externa del elastómero. Si la muestra se confina en un contenedor isostático, un
esfuerzo restrictivo igual se aplica en la superficie externa de las espigas (ver Figura 5-6). Se deja que entre gas helio a la cámara de referencia del porosímetro (Vr) a una presión predeterminada (p1), la cual generalmente es 100-200 psig (690 a 1380 kPa). Se registra la presión y la camara de presión se evacúa en el volumen del vacío de la muestra. Se registra entonces la presión más baja de equilibrio que resulta. Se calcula el volumen del poro de la muestra se calcula utilizando las ecuaciones de la sección 5.3.2.2.1.1.6. 5.3.2.2.1.1.6 Cálculos Se determina el volumen de la muestra expandiendo el helio desde una celda de referencia a una presión inicial conocida directamente en la roca porosa. Se deriva la siguiente ecuación de volumen poroso por el equilibrio de la masa de gas dentro de la celda de referencia, el volumen muerto del sistema, el volumen de la válvula y el volumen poroso de la muestra. P1Vr + Pa(Vp + Vd) = P2(Vr + Vp + Vd + Vv)Z1T1r ZaT1 Z2T2
(19)
Donde : P1 = presión absoluta del volumen inicial de referencia P2 = presión absoluta expandida Pa = presión absoluta atmosférica inicialmente en la muestra Z1 = factor de desviación de gas a P1 y T1 Z2 = factor de desviación de gas a P2 y T2 Za = factor de desviación de gas a Pa y T1 T1r = temperatura absoluta del volumen de referencia a P1 T1 = Temperatura absoluta del volumen poroso de la muestra a Pa T2 = temperatura absoluta del volumen de referencia y de la muestra despues de que se haya estabilizado P2 Vr = volumen de la cámara de referencia Vp = volumen poroso de la muestra Vv = volumen de desplazamiento de la válvula (desde la posición cerrada a la posición abierta). Vd = volumen muerto del sistema Si existen condiciones isotérmicas, (T1 = T2 = T1r = Tl) y: P1Vr + Pa(Vp + Vd) = P2(Vr + Vp + Vd + Vv) Z1 Za Z2
(20)
al agrupar los términos, tenemos: Vp = Vr ([ (P1Z2)/(P2Z1)] -1) - vv
1 - (PaZ2)/P2Za
- Vd (21)
Nota : Esta ecuación es válida si no ocurre ningún cambio en el volumen poroso mientras que la presión porosa de la muestra se incrementa de Pa a P2, es decir, a) cuando el esfuerzo restrictivo efectivo neto

permanece constante, o b) cuando no es constante pero el cambio de volumen poroso y el cambio de esfuerzo restrictivo son despreciables. 5.3.2.2.1.1.7 Precauciones Las precauciones para este método incluyen: a. La presión restrictiva en la superficie externa de la manga elastomérica debe aplicarse utilizando líquido y no gas. El gas puede difundirse dentro de la manga alargando el tiempo de duración de la prueba. b. El durómetro de la manga elastomérica debe estar lo suficientemente bajo (se sugiere un máximo de 50) para que la manga selle bien sobre los extremos metálicos de las espigas y se adhiera apropiadamente a la superficie de la muestra al bajo esfuerzo restrictivo utilizado. c. Las muestras con extremos que se desvíen ligeramente de los lados paralelos pueden probarse con éxito utilizando dos discos de goma deformables. Cada disco debe ser blando, con un diámetro igual al del extremo y debe tener un orificio perforado en el centro. Se debe insertar un disco entre cada espiga metálica ubicada en el extremo y la cara de la muestra. El disco se comprimirá y se adherirá para llenar el espacio vacío entre la muestra y la espiga extrema. Nota : Se recomienda este procedimiento únicamente cuando los extremos de la muestra no pueden volverse cuadrados. Este procedimiento no reemplaza las muestras de buena calidad. 5.3.2.2.1.1.8 Calibración La calibración de equipos utiliza (a) lingotes de volumen conocido que son agregados o retirados de una cámara especial que se introduce únicamente para la calibración, o b) una serie de tapones de acero perforados con precisión para cubrir la gama de volúmenes porosos a medir. Se carga el volumen de referencia en la presión inicial y se envía a cámara especial o al volumen vacío conocido que tiene el tapón de calibración confinado. Se repite la medición utilizando múltiples lingotes o tapones de calibración. Las ecuaciones del volumen poroso se pueden resolver simultáneamente para obtener el volumen de la celda de referencia Vr y el volumen muerto. Estos valores luego se convierten en una constante para cálculos subsecuentes de volumen poroso. El volumen muerto (Vd) del sistema también se puede determinar utilizando un lingote de acero sólido como muestra dentro del portanúcleos o colocando las espigas extremas adyacentes en la manga elastomérica y aplicando el mismo esfuerzo restrictivo bajo que será utilizado para la medición de muestras. Se desahoga la celda de referencia cargada (Vr) en el lingote de acero o a las espigas extremas adyacentes, despues de lo cual se utiliza la ecuación de volumen poroso para calcular Vd. Este valor tambien permanece constante mientras no ocurran cambios en el sistema. Se pueden medir los
tapones de acero como revisión final de la operación del sistema y de los valores Vr y de Vd apropiados. Estos tapones de revisión también deben utilizarse como confirmación diaria del desempeño apropiado de los equipos. 5.3.2.2.1.1.9 Exactitud/Precisión Un sistema apropiadamente calibrado da volúmenes porosos con una precisión de ±0.03 cm3 en cilindros perfectos. Las mediciones en las muestras de núcleos reales indican una dispersión de aproximadamente 0.1 cm3 en muestras que tienen 50 cm3 de volumen total, lo cual da una desviación de porosidad de ±0.2 unidades de diferencia del valor real. 5.3.2.2.1.2 Esfuerzo Restrictivo Elevado 5.3.2.2.1.2.1 Principio El principio es similar al indicado para bajo esfuerzo restrictivo; sin embargo, en esta medición, se determina la porosidad a un nivel de estrés elevado. Los datos de laboratorio a su vez son utilizados para calcular la porosidad del yacimiento en condiciones de estrés in situ. A diferencia de las mediciones de bajo esfuerzo restrictivo, el conocimiento de (a) la magnitud elevada, y b) el método de aplicación (esfuerzo isostático, esfuerzo triaxial, esfuerzo biaxial, o esfuerzo uniaxial) debe conocerse para medir los resultados en relación con las condiciones del yacimiento (ver figuras 5-7 y 5-8). Históricamente, la mayoría de las mediciones rutinarias de volumen poroso en esfuerzos elevados se han realizado con un esfuerzo restrictivo isostático, porque este es un método fácilmente aplicado y relativamente más económico. Los datos son presentados subsecuentemente como porosidad vs. esfuerzo isostático efectivo (presión restrictiva menos presión de poro). En algunos yacimientos, se debe medir una compresibilidad de volumen poroso más compleja y más demorada para determinar con mayor precisión la porosidad bajo las condiciones del yacimiento. Estas mediciones de compresibilidad requieren típicamente que haya líquido presente en el espacio poroso para simular los efectos de interacción entre el líquido y la roca, si existe,1 y que están fuera del alcance de este documento (ver referencias en 5.6). 5.3.2.2.1.2.2 Ventajas Las ventajas de este método incluyen: a. Las ventajas son similares a aquellas para el bajo esfuerzo restrictivo (ver 5.3.2.2.1.1.2). b. Las mediciones realizadas a un elevado esfuerzo restrictivo representan más exactamente la porosidad original del yacimiento que las mediciones realizadas a un nivel bajo o a un valor cero. 5.3.2.2.1.2.3 Limitaciones

Las limitaciones de este método incluyen: a. Las limitaciones son similares a los ítems a - h para bajo esfuerzo restrictivo (ver 5.3.2.2.1.1.3). b. La penetración de la manga elastomérica en las depresiones entre los granos de arena o las fisuras superficiales de la muestra a medida que aumenta el esfuerzo restrictivo puede indicar una reducción en el volumen poroso, aunque no ocurra ninguna compresión de la muestra. Esta penetración rara vez supera los 0.2 unidades de porosidad para la arenisca sin fisuras (por ejemplo, de 10% a 9.8%), y aunque esté presente, normalmente no tiene un impacto significativo en los valores de porosidad. Para las muestras con fisuras, este error si puede ser significativo. Para reducirlo, se puede utilizar una manga interna de mylar o de metal. Los efectos en la superficie pueden minimizarse incrementando el radio de la muestra (r) para que la relación del área de superficie en relación con el volumen total sea igual a 2/r y disminuya a medida que el radio de la muestra aumenta. c. El esfuerzo restrictivo inicial debe ser lo suficientemente alto para sellar la manga elastomérica a la muestra y a los extremos del portamuestras, y para presionar los extremos del portamuestras contra la cara de la muestra. El sellado del elastómero a la superficie de la muestra depende de la dureza del elastómero y de la aspereza de la muestra. La fuerza de sellado también debe ser lo suficientemente alta para evitar el paso de gas si se va a medir permeabilidad en conjunto con la porosidad. Consecuentemente, pueden ocurrir dos efectos opuestos sobre la porosidad. Una adhesión deficiente de los extremos del portamuestras a la muestra, lo cual da valores de porosidad demasiado altos, puede ser compensada por la compresión del volumen poroso de la muestra. El uso de muestras cilíndricas rectas de buena calidad con extremos paralelos minimiza las dificultades de ajuste. d. La porosidad resultante a cualquier esfuerzo elevado puede depender de la tasa de aplicación del esfuerzo, del tiempo de estabilización y de los antecedentes del esfuerzo.2 e. Se debe conocer o se debe calcular el esfuerzo efectivo del yacimiento para utilizar los datos de porosidad de esfuerzo elevado determinados en el laboratorio. Los esfuerzos reales del yacimiento que causan reducciones en el volumen poroso por lo general no son isostáticos. El esfuerzo principal máximo en la mayoría de los yacimientos es vertical. Este esfuerzo efectivo vertical es igual al peso por unidad de área de roca de recubrimiento menos la presión del yacimiento. El esfuerzo de recubrimiento se puede calcular de la profundidad y un registro de densidad de pozo, o suponiendo que la densidad de la roca es igual a 1.0 psi/pies3. Se puede medir la presión del yacimiento, o en áreas de presión normal, se supone que es la profundidad multiplicada por el gradiente de agua, lo cual es aproximadamente 0.45 - 0.5 psi/pie.
Nota : Los yacimientos submarinos tienen un componente de esfuerzo de recubrimiento que resulta de la profundidad del agua y la densidad, y también de la roca de recubrimiento. Típicamente, los esfuerzos horizontales constituyen alguna fracción del esfuerzo vertical, y pueden calcularse de los tratamientos hidráulicos de fracturas o pruebas de integridad de la formación (filtración). El esfuerzo efectivo medio del yacimiento es un promedio de los esfuerzos horizontales y verticales. Consecuentemente, el esfuerzo efectivo medio del yacimiento es típicamente menor que un esfuerzo isostático igual al esfuerzo efectivo vertical. f. Se ha demostrado que la presencia de agua dentro del espacio poroso debilita algunas rocas, así incrementando la reducción de volumen poroso cuando se aplica el esfuerzo restrictivo. Como las mediciones rutinarias, indicadas aquí, se hacen sobre muestras de roca limpias y secas, si el yacimiento contiene agua intersticial, se introduce cierto nivel de incertidumbre. Mientras que este efecto es mínimo en las rocas duras y bien consolidadas, la importancia del efecto puede cuantificarse realizando pruebas especializadas en las muestras que contienen agua. g. Los núcleos friables o poco consolidados pueden
mostrar una compresión significativa en esfuerzos restrictivos bajos.
5.3.2.2.1.2.4 Equipos El equipo es similar al indicado para la técnica de bajo esfuerzo restrictivo. Sin embargo, las mediciones de alto esfuerzo restrictivo requieren un material para la manga hecho de un elastómero fuerte que pueda soportar las fuerzas impuestas durante la aplicación de esfuerzo restrictivo, y que aún se adhiera a la muestra en un esfuerzo bajo. (Texto de las gráficas de la página 5-16) Esfuerzo isostático: Bajo la carga de esfuerzo isostático, se aplica en la muestra un esfuerzo igual en todas las direcciones, y puede ocurrir tensión en todos los ejes de la muestra. Una reducción excesiva de porosidad ocurre cuando el esfuerzo isostático impuesto es igual al esfuerzo vertical del yacimiento (es decir, el esfuerzo de recubrimiento) Esfuerzo triaxial: Bajo condiciones de verdadero esfuerzo triaxial, se aplica un esfuerzo desigual a los tres ejes principales de la muestra. Por lo general, las tensiones serán diferentes en cada eje. Típicamente, se utilizará una muestra en forma de cubo o prisma rectangular. Esfuerzo biaxial: Las condiciones de esfuerzo biaxial constituyen un caso especial de carga de esfuerzo triaxial. Cuando se carga un cilindro con esfuerzo

biaxial, el esfuerzo paralelo al eje del cilindro es diferente al esfuerzo aplicado alrededor de la circunferencia de la muestra. Las tensiones pueden ocurrir paralelo al eje y al diámetro del cilindro. Tensión Uniaxial: La compresión de tensión uniaxial es un caso especial de carga de esfuerzo biaxial. El esfuerzo aplicado a la circunferencia es suficiente para mantener el diámetro constante mientras se incrementa el esfuerzo paralelo al eje del cilindro. La tensión ocurre únicamente paralelo al eje del cilindro. 5.3.2.2.1.2.5 Procedimientos Los procedimientos para las mediciones de esfuerzo restrictivo elevado son similares a los indicados para bajo esfuerzo restrictivo en 5.3.2.2.1.1.5. Típicamente, el volumen poroso de una muestra se determina a niveles seleccionados esfuerzo restrictivo creciente. La presión de equilibrio dentro del espacio poroso de la muestra a cada esfuerzo restrictivo normalmente es desahogado antes de la medición subsecuente de volumen poroso. Se utilizan las ecuaciones mostradas en 5.3.2.2.1.1.6 para calcular los volúmenes porosos. 5.3.2.2.1.2.6 Cálculos Las ecuaciones básicas para volumen poroso (PV) son las mismas que las indicadas para mediciones de bajo esfuerzo restrictivo, pero el cálculo de la porosidad también debe contabilizar la reducción del volumen total (BV) de la muestra mientras se incrementa el esfuerzo restrictivo. Los enfoques que se pueden utilizar incluyen los siguientes: Porosidad = PV/(BV - ∆PV sin esfuerzo) (22) Donde ∆PV = reducción en el volumen poroso entre la PV inicial y la PV en cualquier nivel de esfuerzo. Esto supone que la reducción en el volumen total en cualquier nivel de esfuerzo es igual a la reducción en volumen poroso en el mismo nivel de esfuerzo. Porosidad = PV (GV + PV) (23) Donde PV = volumen poroso en cualquier nivel de esfuerzo. Esto asume el volumen de grano (GV) determinado a un nivel de cero esfuerzo es constante en todos los niveles de esfuerzo restrictivo aplicados. Porosidad = PV/BV calculado (24) Mida el volumen poroso (PV), el diámetro de la muestra, y su longitud en cada nivel de esfuerzo impuesto y calcule el volumen total. Estos procedimientos no se hacen de manera rutinaria. 5.3.2.2.1.2.7 Calibración
La calibración del equipo utiliza los mismos principios indicados para bajos esfuerzos restrictivos (ver 5.3.2.2.1.1.8). La validez de la operación del sistema en un esfuerzo restrictivo elevado se puede confirmar midiendo los volúmenes porosos del tapón de control en los esfuerzos restrictivos seleccionados para la determinación de volumen poroso de la muestra. 5.3.2.2.1.2.8 Exactitud/Precisión Un sistema calibrada apropiadamente tiene una precisión de volumen poroso de la misma magnitud observada en bajos esfuerzos restrictivos (ver 5.3.2.2.1.1.9), dando una dispersión de porosidad de ±0.2 o 0.3 unidades de porosidad. 5.3.2.2.2 Resumen de la “Sumatoria de Porosidad de Fluidos” La técnica de “Sumatoria de Fluidos“ para la determinación de porosidad se discute con mayor detalle en Métodos de Saturación de Fluidos, bajo “Método de Retorta a Presión Atmosférica” (ver 4.2). Esto proporciona datos de porosidad rápidamente puesto que las muestras no requieren limpieza ni secado. El volumen poroso es calculado midiendo y sumando los volúmenes de aceite, gas y agua presentes en una muestra de núcleo recientemente recobrada. La porosidad se determina dividiendo el volumen poroso por el volumen total de la muestra de roca. Cuando se siguen los procedimientos apropiados, y si las rocas son razonablemente uniformes y no contienen minerales hidratables que complican la determinación de los volúmenes de agua, las porosidades de la ley de Boyle en un núcleo adyacente por lo general estarán de acuerdo con una precisión de ± 0.5 unidades de porosidad.6 Sin las precauciones y técnicas especiales, la sumatoria de la porosidad de fluidos puede resultar demasiado alta debido al exceso de recobro de agua de los minerales hidratables. Esta técnica no es válida en núcleos meteorizados de los cuales los líquidos se han evaporado. Las mediciones en rocas meteorizadas dan un volumen de gas demasiado bajo, y en consecuencia una porosidad también baja, porque el mercurio inyectado a la presión utilizada no llena los pequeños poros de los cuales se ha evaporado el agua. Se puede medir la densidad de grano con los volúmenes medidos de rocas y fluidos. Esta puede compararse con una densidad de grano estimada basada en la litología. La magnitud de la diferencia entre los valores calculados y los estimados es la base del control de calidad que mostrará las mediciones erróneas o los errores en el cálculo de datos. Esto no es válido cuando hay minerales extraños (pesados o livianos) presentes en la roca. Estas condiciones hacen que la densidad del grano sea variable y difícil de medir con exactitud. La retorta que se realice en la roca destruye la muestra, así que se debe utilizar el núcleo adyacente para las mediciones de permeabilidad. Estas muestras de

permeabilidad se pueden someter a pruebas para determinar la porosidad utilizando el procedimiento de la Ley de Boyle, y comparándolo con la sumatoria de valores de fluidos como prueba de control de calidad. No se espera que haya una concordancia perfecta entre las muestras, pero los datos no deben mostrar ninguna desviación. 5.3.2.2.3 Método de Saturación de Líquidos 5.3.2.2.3.1 Principio La medición de la porosidad (espacio poroso conectado) por el método de saturación de líquidos implica una determinación gravimétrica del volumen poroso obteniendo: a) el peso de muestra de núcleo seca y limpia, b) el peso de la muestra saturada con un líquido de densidad conocida, y c) el peso de la muestra saturada sumergida en el mismo líquido. 5.3.2.2.3.2 Ventajas Las ventajas de este método incluyen: a. Se pueden manipular muchas muestras a la vez. b. Nos da una medición directa del volumen poroso. c. Consulte 5.2.4.2, a y b. 5.3.2.2.3.3 Limitaciones Las limitaciones de este método incluyen: a. El procedimiento es lento con relación al tiempo total transcurrido de medición. b. El procedimiento es básicamente preciso, pero limitado a las muestras que pueden ser 100% saturadas y determinarse con éxito el peso saturado. c. No se recomiendan las muestras que contienen fisuras superficiales para la determinación de porosidad por este método debido a la pérdida potencial de líquido de las fisuras durante el proceso de pesaje. Si se pierde líquido, este volumen no sería incluido en el volumen poroso, y se calculará una porosidad demasiado baja. Sin embargo, el volumen de grano de núcleos con fisuras puede determinarse con precisión con este método. d. Consulte 5.2.4.3, a y c. 5.3.2.2.3.4 Exactitud El método debe determinar la porosidad (espacio poroso conectado) con una precisión de 0.5 unidades de porosidad y un volumen de grano de ± 0.2 % del valor real si se utiliza la técnica apropiada. 5.3.2.2.3.5 Equipo Los equipos apropiados para este método incluyen: a. Balanza analítica con una precisión de 1 miligramo.
b. Recipientes adecuados que se pueden utilizar para contener líquido sin airear al vacío. c. Desecador al vacío y saturador de presión. d. Una fuente adecuada de vacío capaz de mantener menos de 0.1 mm de presión de mercurio. e. Un líquido filtrado de baja viscosidad y baja presión de vapor de una densidad conocida con el fin de saturar las muestras de núcleos. Algunos líquidos que se han utilizado son: 1) salmueras 2) aceites refinados de laboratorio 3) decano y 4) tolueno. f. Recipientes adecuados que puedan utilizarse para contener muestras saturadas bajo líquido sin airear. 5.3.2.2.3.6 Procedimiento Obtenga el peso de la muestra limpia y seca. La muestra debe desecarse sobe un material deshidratante adecuado, tal como CaCl2 o gel de sílice, antes de determinar el peso seco. Coloque la muestra seca ya pesada en una cámara (desecador al vacío y saturador de presión) y aplique alto vacío durante aproximadamente 8 horas. El período de evacuación puede ser seguido de una inyección de CO2 para retirar el aire absorbido en la roca. Esto reemplaza los gases adsorbidos como O2 o N2 con CO2, por lo general un gas más soluble en los líquidos típicamente usados. Se pueden requerir varios ciclos de evacuación y de inyección de CO2 para muestras de baja permeabilidad. Se requiere un mayor tiempo de contacto con CO2 y ciclos de vacío mas largos para desplazar el N2 en la roca más apretada. Para muestras de muy baja permeabilidad, el período de evacuación debe ser hasta 12 a 18 horas (de la noche a la mañana). El líquido utilizado para saturar la muestra limpia debe estar libre de aire disuelto. Por lo tanto, el líquido se debe desaerear el líquido antes de introducirlo en la muestra. Se debe tener mucho cuidado para evitar que el líquido entre en contacto con el aire despues de la desaereación. Los hidrocarburos son fluidos más fluidos mas humectantes que las salmueras y se prefieren sólo si se desea obtener mediciones de porosidad y las muestras saturadas con salmuera no se requieren para pruebas adicionales. El líquido desaereado se vacía en el recipiente que contiene la muestra de núcleo. Se permite que el núcleo se sature. Despues de sumergir la muestra completamente en el líquido, se continua el vacío durante 30 minutos a 1 hora más. El método preferido es someter el líquido que rodea la muestra a una presión de 2000 - 3000 psi (13800 - 20700 kPa) durante 4 horas por lo menos para asegurar una saturación completa. La saturación de presión debe resultar en el ingreso del líquido en los vacíos adicionales que no se encuentran bien conectados o donde no ha ocurrido una imbibición espontánea con el líquido. Luego se remueve la muestra del recipiente saturador y se pesa sumergida en el líquido saturante. El exceso de líquido se remueve de la muestra cuidadosamente (evitando la pérdida de grano) y se pesa la muestra saturada en el aire. Cuando se

remueve el exceso de líquido de la superficie de la muestra, se deben tomar todas las precauciones para asegurar que no se remueven los líquidos de los poros expuestos en la superficie. Se debe evitar el uso de materiales (tales como las toallas secas) que remueven líquido de los poros superficiales por acción capilar, así como cualquier método mecánico como el sacudido violento. Rodar la muestra sobre un trapo libre de pelusa o sobre una toalla de papel mojada con el líquido saturante, o limpiarla cuidadosamente con el trapo humedecido o los dedos son técnicas adecuadas para remover el exceso de líquido. Despues de pesar cada lote de muestras, se determina la densidad del líquido saturante desaereado utilizado en la cámara de saturación. Esto se debe hacer con exactitud, puesto que la densidad del saturante es probablemente la mayor fuente de errores a menos que se utilice la Ecuación 5.3.2.2.3.8e. Consulte los procedimientos indicados en la 5.2.4.6 para la determinación del volumen total. Cuando se une al peso seco original y la densidad del fluido, el volumen de grano puede calcularse directamente (ver Ecuación 5.3.2.2.3.8c). Para el almacenamiento de muestras antes de usarlas, los tapones de núcleo por lo general se pasan a frascos de tapa rosca u otros recipientes adecuados que se llenan de líquido desaereado con el fin de minimizar la presencia de aire en el recipiente. 5.3.2.2.3.7 Precauciones Las precauciones para este método incluyen: a. Algunas precauciones especiales son necesarias para asegurar una saturación completa. Los núcleos de baja permeabilidad pueden quedar no completamente saturados si se utiliza este método. c. Para lograr una exactitud aceptable, es
indispensable utilizar la densidad correcta del fluido en el momento de determinar el peso saturado de la muestra.
c. Pueden resultar errores en el volumen poroso por una limpieza inapropiada de la muestra antes de determinar el peso saturado. 5.3.2.2.3.8 Cálculos Los cálculos para este método son los siguientes: a. PV = Peso saturado - Peso seco
Densidad del Saturante
b. BV = Peso saturado - Peso inmerso
Densidad del Líquido de Inmersión (ver 5.2.4)
c. GV =
Densidad del Líquido de Inmersión Peso seco - Peso inmerso
d. Revisión del Volumen Poroso PV = BV - GV (PV por diferencia debe concordar con el PV calculado en a.)
Porosidad = PV/BV =
Peso saturado-Peso sumergido
Peso saturado-Peso seco/Densidad del Saturante
Densidad del líquido de inmersión Si la temperatura es constante y la densidad del saturante y el líquido de inmersión son iguales, la densidad del fluido se cancela y no se necesita en esta ecuación para calcular la porosidad. Porosidad =
Peso saturado - Peso inmerso Peso saturado - Peso seco
f. Revisión de porosidad: Porosidad =
BV BV - GV
(La porosidad debe concordar con la calculada en e.) g. Densidad del grano = Peso seco/GV Donde : PV = volumen poroso GV = volumen de grano BV = volumen total 5.4 Procedimientos Históricos Esta sección relaciona y resume las técnicas que se han utilizado siempre para medir la porosidad, el volumen total, el volumen de grano, y la densidad de grano. Estas técnicas por lo general han sido reemplazadas por otras técnicas más nuevas. Mayor información acerca de estos procedimientos se puede encontrar en la Primera Edición de API RP 40, API Prácticas Recomendadas para el Procedimiento de Análisis de Núcleos, Agosto de 1960. 5.4.1 Desplazamiento de Mercurio Esta técnica consiste en sumergir una muestra de núcleo seca en mercurio dentro de un picnómetro calibrado. Se pesa el volumen del mercurio desplazado por la muestra para determinar el volumen total de la muestra (API RP 40, 1960, Sección 3.311, p. 15; Sección 3.56, p. 26.) 5.4.2 Medidor del Volumen Total Este procedimiento también implica sumergir una muestra en mercurio. El volumen del mercurio desplazado por la muestra a su vez desplaza un segundo líquido en un tubo de vidrio inclinado y graduado. Se lee el volumen total de la muestra directamente del tubo de vidrio graduado (API RP 40, 1960, Sec 3.312, p. 15; Sección 3.57, p. 26.) 5.4.3 Método de Celda Sencilla de la Ley de
Boyle Utilizando el Porosímetro de Kobe

El Porosímetro de Kobe consiste de una bomba de mercurio, un medidor de presión, válvulas de entrada y salida de gas, y una cámara para muestras. La determinación del volumen total de la muestra requiere llenar la cámara de muestras que contiene la muestra con mercurio. Esto deja la muestra sumergida en mercurio. El volumen total se determina restando el volumen del mercurio necesario para llenar la cámara vacía con el volumen de mercurio necesario para llenar la cámara que contiene la muestra. El volumen de grano se calcula colocando la bomba de mercurio en un volumen “predeterminado”. Durante esta parte de la prueba, no se sumerge la muestra en mercurio. Se desahoga la presión en la celda a presión atmosférica (P1). Se bombea el mercurio en la celda de la muestra (con la válvula de salida de gas cerrada) y se registra esta nueva presión (P2). Conociendo los volúmenes de mercurio en la celda, el volumen vacío predeterminado, y las presiones, se puede utilizar la Ley de Boyle para calcular el volumen de grano (P1 x V1 = P2 x V2). La porosidad se calcula a partir del volumen total y el volumen del grano. La densidad de grano se calcula con el peso de la muestra y del volumen de grano (API RP 40, 1960, Sección 3.32211, p. 17, Sección 3.5.10, p. 28.) 5.4.4 Método de Washburn - Bunting El porosímetro de Washburn - Bunting mide el volumen poroso de la muestra. El aparato consiste de una cámara para muestras con un tubo capilar graduado en la parte superior y una llave de paso para abrir y cerrar el sistema hacia la atmósfera. Se conecta un tubo a la parte inferior de la cámara de muestras, el cual se conecta a una ampolla de vidrio llena de mercurio. Se coloca una muestra en la cámara respectiva. El procedimiento se inicia subiendo la ampolla de mercurio para mandar el mercurio a la cámara de muestras inundando la muestra y subiendo al capilar calibrado. Cuando el mercurio se encuentre por encima de la llave de paso, esta se cierra. Se baja entonces la ampolla de mercurio hasta que la muestra quede flotando sobre el mercurio dentro de la cámara de muestras. La muestra se encuentra ahora al vacío, y el aire de los poros de la muestra llena la cámara de muestras y el capilar calibrado. Despues de unos minutos, se restituye la presión atmosférica al aire que ha escapado subiendo la ampolla de mercurio hasta que su nivel sea igual al que se encuentra en el capilar. El volumen del aire en el capilar calibrado es igual al volumen poroso de la muestra de roca. Se puede mejorar la precisión restando el volumen de aire medido utilizando un lingote sólido en lugar de la muestra para contabilizar el aire adsorbido en la superficie de la muestra (API RP 40, 1960, Sección 3.3221, p. 17, Sección 3.5.12, p. 30.) 5.4.5 Método Seco para la Densidad de Grano y
el Volumen Poroso
En este procedimiento, se extrae la roca, se seca, se tritura y se cuela. La porción de la roca triturada que pasa a traves de un tamiz de calibre 60 y queda atrapada en un tamiz de malla de calibre 100, se pesa y se coloca en la taza para muestras del aparato. Luego se utiliza una bomba de mercurio para bombear el mercurio a la taza para muestras. Cuando la presión del sistema alcance el valor de calibración preestablecida, se registra el volumen del mercurio inyectado. Con el cálculo del volumen de la taza vacía, se puede determinar el volumen total de la muestra en la taza. Utilizando el peso de la muestra, se puede calcular la densidad de grano. Utilizando una muestra de roca complementario, se calcula el volumen de grano dividiendo el peso seco de la muestra complementaria por la densidad de grano calculada de la muestra molida. El volumen poroso puede determinarse restando el volumen de gran del volumen total medido de la muestra. (API RP 40 1960, Sección 3.3211, p. 16, Sección 3.59, p. 26.) 5.4.6 Método Húmedo para la Densidad de Grano
y el Volumen Poroso Se mide el volumen total y se pesa y se tritura la muestra seca. La muestra triturada se coloca en un frasco volumétrico calibrado que contiene un volumen conocido de un fluido humectante adecuado (como tolueno, agua, etc.). El aumento del volumen es igual al volumen de grano de la muestra triturada. La densidad de grano se puede calcular dividiendo el peso de la muestra triturada por su volumen medido. El volumen poroso puede calcularse restando el volumen de grano del volumen total. (API RP 40, 1960, Sección 3.3212, p. 16; Sección 3.59, p. 27.) 5.4.7 Sumatoria de Diámetro Total de los Fluidos
Utilizando el Método Retorta-Vacío Este método fue utilizado extensamente en las regiones de “rocas duras” (Texas Occidental) donde la producción proviene predominantemente de las formaciones de carbonatos. Cada muestra de diámetro completo se pesaba y luego el espacio poroso lleno de gas se saturaba con agua a presión. La muestra se pesaba nuevamente, y el incremento del peso era igual al volumen de gas. Los fluidos dentro del espacio poroso se destilaban del núcleo sometido a un vacío parcial, a una temperatura máxima de 450oF (232 oC). Se colectaban los fluidos en una probeta sumergida en un baño de alcohol/hielo seco a -75oF (-59oC). Esto se hacía para condensar vapores y evitar pérdidas a lo largo del sistema de vacío. Se leían los volúmenes condensados y se empleaba un factor de corrección del aceite. El agua de los poros de la muestra se calculaba restando el volumen del gas del total de agua recolectada en el tubo de condensación. El volumen poroso se calculaba sumando los volúmenes del gas, del aceite corregido y del agua de los poros. Un volumen total determinado de la muestra se utilizaba para el

cálculo de porosidad. (API RP 40, 1960, Sección 4.21, p. 39; y 4.52, p. 42). Una de las principales desventajas de este método era la condición en que quedaban los núcleos después del proceso de destilación. Casi siempre las muestras se volvían negras del aceite coqueado y ningún proceso de limpieza podía restaurar las muestras a una condición donde se consideraran adecuados para usarse nuevamente en pruebas adicionales (por ejemplo, Porosidad con la Ley de Boyle, Pruebas de Presión Capilar, Permeabilidad Relativa). Por esta y otras razones, el subcomité encargado de editar esta Práctica Recomendada #40 decidió no registrarlo como un procedimiento recomendable. 5.5 Rocas Ricas en Materia Orgánica En los últimos años, ha habido un interés considerable en el desarrollo de yacimientos de shale de gas y metano carbonado. Los shales de gas y el carbón son típicamente formaciones fracturadas altamente orgánicas, en las cuales mucho del gas contenido es adsorbido en el material orgánico. Debido a que el gas adsorbido comprende una porción significativa del gas contenido en estos yacimientos, es necesario determinar el contenido del gas adsorbido y la manera que se libera el gas (isotermas de adsorción/desorción) para hacer cálculos de las reservas y hacer predicciones respecto a la producción. Los shales de aceite contienen material orgánico sólido, típicamente licuada por la aplicación de calor para generar aceite. Se explican las técnicas para evaluar estos tipos de depósitos en 4.7. La porosidad, indicada en esta sección, normalmente no se requiere. 5.5.1 Carbón No existen métodos generalmente aceptados en la industria para mediciones de porosidad, permeabilidad o permeabilidad de muestras de carbón en el laboratorio. Tampoco existen mediciones hechas en laboratorios publicadas que sean aceptadas como estándares. Las técnicas de laboratorio estándares para la medición de porosidad en rocas de yacimientos que contienen aceite o gas deben, por lo tanto, modificarse cuando se aplican al carbón utilizado en los estudios de metano en eras de carbón. A continuación se encuentran comentarios y referencias y comentarios que reflejan las prácticas actuales. En la producción de metano a partir de vetas de carbón, la “porosidad” puede definirse en términos de tres parámetros precisos. Para el calculo del gas metánico contenido en eras de carbón, son los siguientes: a) “el contenido del gas in situ” del carbón, el cual es equivalente al volumen de gas contenido en la porosidad llena de gas en un yacimiento convencional de gas. Además, el carbón es un sistema de porosidad dual que consta de b) una porosidad matricial (microporo) en donde el metano es adsorbido en la superficie del carbón, y c) porosidad del listón (porosidad de la
microfractura que ocurre naturalmente). La red del listón proporciona el camino y la permeabilidad para el flujo de los fluidos. 5.5.1.1 Contenido de Gas In Situ El único método por el cual se puede medir directamente el contenido total de gas in situ de las eras de carbón es el uso de la tecnología de núcleo a presión.7 El volumen de gas emitido de un núcleo de carbón en un sacanúcleos a presión se mide como función del tiempo, la temperatura y la presión. A menudo se determina el contenido de gas y el porcentaje de desorción por métodos de desorción en caja.8 Las muestras de carbón utilizadas incluyen las los núcleos obtenidos utilizando el corazonamiento convencional, corazonamiento de pared, recobro con alambre, y recortes de carbón. Los núcleos de carbón (o recortes) son sellados en la caja y se mide el volumen de gas emitido de la muestra de la manera indicada.7 La determinación del contenido de gas por desorción en caja requiere un cálculo del volumen de gas perdido mientras se llevan las muestras a la superficie y antes de sellar las muestras en la caja. El contenido de gas obtenido de muestras convencionales y de pared por el método de desorción en caja es mucho menos costoso que el corazonamiento a presión, y es la técnica que más se utiliza. Además, los núcleos de presión son colocadas a menudo en cajas de desorción para completar el proceso de desorción. El contenido de gas obtenido de diferentes métodos o muestras puede compararse despues de normalizar el carbón a una base libre de cenizas. Como el gas emitido no siempre es 100% metano,11 la composición del gas debe analizarse para determinar el contenido de metano. 5.5.1.2 Porosidad Matricial La porosidad matricial del carbón es por lo general más grande que la porosidad del listón. Sin embargo, como el metano es adsorbido en la matriz en lugar de existir como un gas libre, rara vez se utilizan las mediciones de porosidad matricial convencional de aceite o gas para aplicaciones de metano de eras de carbón. Si se requiere el volumen poroso convencional, se pueden utilizar las técnicas de equilibrio de humedad12 y la porosidad con helio de la Ley de Boyle13. Nota : Cualquier gas que se adsorba producirá valores de porosidad matricial erróneos con la técnica de la Ley de Boyle. A menudo se utiliza el helio, el cual no se adsorbe de manera apreciable. 5.5.1.3 Porosidad del Listón Aunque el total de la porosidad del carbón es la porosidad matricial, la red de listones proporciona el camino y la permeabilidad para el flujo de los fluidos. La medición de la porosidad del listón en el carbón es, por

lo tanto, importante para la determinación de la compresibilidad del volumen poroso, como también para las saturaciones para hallar la permeabilidad relativa de gas - agua o las mediciones de presión capilar. La porosidad del listón es función de la presión restrictiva neta. La porosidad del listón se debe medir con el núcleo de carbón sometida a una presión restrictiva uniforme. Los materiales envolventes comúnmente utilizados para las muestras no consolidadas16 son los que se utilizan en los núcleos de carbón. Las mediciones de la porosidad del carbón se han realiazado en muestras de carbón secados en hornos al vacío.14 Los estudios realizados sobre el desgaste del carbón indican que este secado altera la estructura del carbón,15 dando resultados erróneos. Un procedimiento adecuado para medir la porosidad del listón en núcleos que no han sido sometidos al proceso de secado es la técnica de impulso mezclable. En este procedimiento, el fluido que satura el núcleo es desplazado por un segundo fluido con propiedades físicas diferentes, pero que es mezclable con el fluido saturante.16 La técnica de impulso mezclable puede utilizar concentración de un indicador en el agua. Un indicador que es adsorbido producirá resultados erróneos. Dentro de los márgenes del error experimental, el desplazamiento no mezclable con helio saturado con vapor de agua de un núcleo de carbón saturado con agua, produce los mismos resultados que la técnica del indicador en el impulso mezclable. Nota : Los núcleos de carbón a menudo tienen metano residual en la matriz, lo cual puede afectar las mediciones de porosidad. Se ha utilizado la saturación de núcleos de carbón con helio (saturado con vapor de agua), seguido por una evacuación, para remover el metano residual. 5.5.2 Shale Productor de Gas En los gas shales, como es el caso del Devonian, el gas puede almacenarse como gas libre en la matriz o en las fracturas, y tambien puede existir como gas adsorbido en las superficies orgánicas y de arcilla. El gas adsorbido puede representar hasta el 80% del gas que contenido, enfatizando la necesidad de isotermas de gas adsorbido. Los volúmenes de gas adsorbidos en el shale van de 1/5 a 1/50 de lo que corresponde al carbón, y los equipos y los procedimientos de medición deben diseñarse para obtener datos exactos. Como se dijo en el caso del carbón, no existen procedimientos estándar en la industria que posibiliten efectuar las mediciones en este caso. Los resultados reportados sobre mediciones de isotermas de gas adsorbido en muestras de shale indican que actualmente existe una gran disparidad en las condiciones entre los laboratorios que realizan pruebas.18 Estas disparidades resultan en resultados de adsorción variables, y enfatizan la necesidad de las
personas que utilizan los datos de entender claramente lo que se está midiendo y lo que se está reportando. Los simuladores utilizados en el modelado de yacimientos, a menudo utilizan como entrada el contenido de metano adsorbido comparado con los datos de presión. Los datos se reportan generalmente en unidades de scf/ton. Por consecuencia, es importante saber si la isoterma medida y reportada se refiere únicamente al gas adsorbido, o si representa una isoterma de sorción total, la cual incluye el gas adsorbido como también al gas almacenado como gas libre en la matriz y/o en las fracturas. La presencia de agua reduce los volúmenes de gas adsorbidos. Por lo tanto, las pruebas de adsorción en roca seca van a sobrestimar las reservas de gas, y por esto se prefiere hacer las pruebas en muestras de roca que tengan un equilibrio en la humedad. Nota : Aún no se han definido las técnicas para establecer condiciones apropiadas de equilibrio en la humedad. Por lo tanto, muchas de las isotermas de adsorción medidas han sido sobre shale "tal cual" o en roca seca. Aunque la tecnología de medición aún se está evolucionando, ciertos elementos importantes que afectan los resultados de medición y datos reportados han sido identificados.18 Los ingenieros que utilizan estos datos deben decidir con el laboratorio de medición los siguientes elementos claves. Se deben establecer procedimientos que tengan más probabilidades de producir datos representativos del yacimiento y que sean requeridos en el simulador matemático que se va a utilizar. 5.5.2.1 Elementos Claves que Afectan las
Mediciones Los factores que afectan las mediciones incluyen: a. Técnicas para el recobro de núcleos (incluyendo el fluido de corazonamiento) b. Técnica para la preservación de la muestra. c. Tamaño y forma de la muestra (triturada? colada hasta que tamaño?) d. Condiciones de secado de la muestra:
1. “Tal como se recibe” 2. Seca (a qué temperatura y por cuanto tiempo?) 3. En condiciones de equilibrio (a qué temperatura y por cuanto tiempo?)
e. Temperatura y presión estándar utilizadas para reportar los volúmenes de gas medidos en scf/ton. f. Toneladas métricas o toneladas americanas de la muestra “tal como se recibió”, seca o roca de peso equilibrado utilizada como base para scf/ton. g. Gas total absorbido (incluyendo la porosidad del gas libre) o el gas adsorbido únicamente isotermo.

h. Técnica de calibración de la celda. i. Medición de temperatura y control de la celda. j. Densidad reportada de la muestra (total o de grano; seca o húmeda). k. Se alcanzó el equilibrio en cada nivel de adsorción, o se terminó este paso de la prueba en el tiempo de adsorción seleccionado? l. Volumen de la muestra probada. m. Muestra evacuada antes de la prueba. n. El gas utilizado en la medición de la adsorción (usualmente metano). o. Gas utilizado en la medición de la porosidad del “volumen de gas libre” (usualmente helio). 5.5.2.2 Equipo Se muestra un aparato diseñado para determinar la adsorción volumétrica en la Figura 5-9.19 Se parece a un porosímetro de doble celda de uso común. 5.5.2.2.1 Porosidad Libre (de Fractura y Matricial) El volumen vació matricial y de fractura se puede determinar utilizando el porosímetro de doble celda de la Ley de Boyle (ver figura 5-5) con el helio como fase de gas. 5.5.2.2.2 Isotermas de Adsorción Primero se completa una medición de isoterma con helio. Esto produce un espacio de gas libre con una adsorción mínima. Luego se carga el metano en la celda de referencia y se expande hacia la cámara de muestras. La cámara de referencia se aísla nuevamente de la celda de muestras y se carga con gas a una presión más alta. Se repite el procedimiento, y se calcula el gas adsorbido a la presión más alta. Este proceso se repite hasta alcanzar los niveles de presión deseados. La diferencia entre las isotermas de helio y metano representa el gas adsorbido. Ya se presentaron las ecuaciones el cálculo de isotermas, como tambien los factores que identifican el análisis de error que afectan la exactitud de las mediciones.19 5.5.3 Oil Shale La evaluación de los oil shales típicamente no requieren mediciones de la porosidad de la roca, porque el aceite recobrado es generado de materiales orgánicos sólidos sujetos al calor. Ver 4.7 para la determinación del contenido de aceite.

Volumen de Grano Total Volumen de Arcilla Seca Volumen de Grano No Arcilloso Volumen de Agua Contenido en el Núcleo (Típicamente mayor que el volumen en núcleos secos por humedad) Volumen de Arcilla Seca Agua Contenida Volumen Poroso Aislado Total φ Efectivo φ
Volumen Poroso Conectado Núcleo Efectivo φ Total φ Núcleo totalmente seco (definición histórica) Núcleo seco por humedad para dejar el agua contenido (Ver texto - el agua contenido es típicamente menor que el valor derivado del núcleo) Volumen poroso aislado
Figura 5-1 - Volumenes Porosos Totales y Efectivos Definidos por Analistas de Núcleos y Registros

SECCIÓN 6 - DETERMINACIÓN DE PERMEABILIDAD
CONTENIDO
6 DETERMINACIÓN DE PERMEABILIDAD 6.1 Introducción 6.2 Teoría 6.3 Aplicaciones Prácticas para Mediciones de Permeabilidad en Régimen Permanente 6.4 Teoría y Aplicación de Determinaciones de Permeabilidad de Régimen Variable 6.5 Exactitud y Precisión 6.6 Calibración de Instrumentos 6.7 Referencias 6.8 Apéndices Figuras 6-1 Ilustración del Potencial de Flujo vs. Diferencias en Presión 6-2 Radio de Deslizamiento de Gas Roca-Dependiente rb, como Función de Permeabilidad
y Porosidad 6-3 Resistividad Inercial, ß, como Función de Permeabilidad 6-4 Máximos Gradientes de Presión Generales Permitidos para Uso de la Ecuación Darcy
con Deslizamiento Corregido 6-5 Tipo de Gráfica Utilizada para Obtener el Factor Klinkenberg de Deslizamiento de Gas,
b 6-6 Flujo Descendente de Liquido con Cabeza Impulsora, h 6-7 Flujo Ascendente de Líquido con Cabeza Impulsora, h 6-8 Flujo Ascendente de Líquido con una Diferencia de Presión Impuesta, ∆p 6-9 Reducción de Permeabilidad con Esfuerzo Hidrostático Neto Creciente 6-10 Esquema del Aparato de Permeabilidad para el Flujo Axil de Gas 6-11 Diagrama de Flujo Simplificado para Baja Presión, Mediciones de Permeabilidad del
Flujo Axil de Gas 6-12 Soporte de Núcleos de Baja Presión Tipo Hassler 6-13 Soporte de Núcleos de Alta Presión para Esfuerzos Hidrostáticos 6-14 Soporte de Núcleos de Alta Presión para Carga Hidrostática o Biaxil 6-15 Esquema de Permeámetro Explorador de Régimen Permanente 6-16 Factor Geométrico Sin Dimensiones para Mediciones con el Permeámetro Explorador 6-17 Configuración de Flujo Transversal para Gases o Líquidos 6-18 Factor Geométrico Sin Dimensiones para el Flujo Transversal 6-19 Soporte de Núcleos Tipo Hassler para Medidas de Permeabilidad Transversal 6-20 Esquema para el Flujo Radial de Entrada 6-21 Permeámetro de Flujo Radial de Diámetro Completo 6-22 Esquema de Permeámetro de Gas de Caída de Presión 6-23 Esquema de Permeámetro Explorador de Caída de Presión 6-24 Aparato de Caída del Impulso para el Flujo Axil de Gases 6-25 Esquema de Permeámetro de Caída de Impulso de Líquidos 6-26 Esquema para la Calibración de Volúmenes en Permeámetros de Régimen Variable A.6-1 Diagrama para Determinar el Máximo ∆p Permitido en Medidas de Permeabilidad de
Líquidos de Flujo Axil B.6-1 Construcción de Variables de Flujo yn y pgn B.6-2 Factores de Corrección Sin Dimensiones para el Flujo de Masa No Constante en
Mediciones de Permeabilidad de Caída de Presión B.6-3 Variación de K∞, β y Error Típico con b como Dato de Caída de Presión

D.6-1 Relación de Primera Raíz para Ecuación de Caída del Impulso D.6-2 Diagrama de Caída del Impulso para Encontrar ƒ1 de ƒ0 con Relaciones Conocidas de V2/V1 (para Gases) o S2/S1 (para Líquidos) Tablas 6-1 Constantes en la Ecuación Forcheimer o Darcy con Varios Conjuntos de Unidades 6-2 Selección Rápida y Guía de Referencia para Medidas de Permeabilidad Utilizando
Gases 6-3 Selección Rápida y Guía de Referencia para Medidas de Permeabilidad Utilizando
Líquidos 6-4 Viscosidad de Aire (para Grados F y C) en Una Atmósfera 6-5 Viscosidad de Nitrógeno (para Grados F y C) en Una Atmósfera 6-6 Viscosidad del Helio (para Grados F y C) en Una Atmósfera 6-7 Factores de Desviación de las Leyes del Gas para el Aire, Calculados con la Ecuación
de Estado Beattie-Bridgemann 6-8 Factores de Desviación de las Leyes del Gas para el Nitrógeno, Calculados con la
Ecuación de Estado Beattie-Bridgemann 6-9 Factores de Desviación de las Leyes del Gas para el Helio, Calculados con la Ecuación
de Estado Beattie-Bridgemann D.6.1 Constantes de Caída de Impulso cuando a=b (V1=V2 o S1=S2)

6 Determinación de Permeabilidad 6.1 INTRODUCCIÓN La permeabilidad es una propiedad de un medio poroso y es una medida de su capacidad de transmitir fluidos. La medición de permeabilidad de una roca porosa, o estrato, es una medición de la conductividad fluida del material específico. La permeabilidad es el equivalente en flujo de fluidos de la conductividad eléctrica o térmica. El recíproco de permeabilidad representa la resistividad viscosa que el medio poroso ofrece al flujo de fluidos cuando prevalecen velocidades de circulación bajas. Esta condición de flujo es comúnmente llamado "flujo viscoso", o formalmente "flujo de Stokes". La medición del flujo de fluido a través de una muestra en alguna dirección específica produce la permeabilidad de la muestra en esa dirección. La permeabilidad de un medio homogéneo isótropo es igual en todas partes, y en todas las direcciones. Sin embargo, las rocas reales no son ni perfectamente homogéneas ni isótropas. 6.1.1 Definiciones 6.1.1.1 Flujo de Darcy Los experimentos y estudios de Darcy a mediados del siglo diecinueve resultaron en una expresión empírica de las relaciones entre las variables comprendidas en el flujo de fluidos a través de medios porosos, conocido ahora como la Ley de Darcy. En resumen, la Ley de Darcy dice que la velocidad volumétrica de flujo por unidad de área de la sección transversal de un medio poroso (el flujo volumétrico) es directamente proporcional al gradiente de potencial, e inversamente proporcional a la viscosidad del fluido. El coeficiente de proporcionalidad es la permeabilidad. Por lo tanto, el gradiente de potencial requerido para el Flujo de Stokes es igual al producto de la viscosidad del fluido y su flujo volumétrico, dividido por la permeabilidad de la roca. El gradiente de potencial para el flujo horizontal de líquidos, o en cualquier dirección para medidas prácticas con gas de baja densidad es igual al gradiente de presión. 6.1.1.2 Resistencia Inercial de Forchheimer El trabajo de Forchheimer al principio del siglo veinte mostró que la Ley de Darcy es un caso limitante, restringido a bajos flujos volumétricos. En flujos más altos, Forchheimer observó que el gradiente de potencial requerido para un flujo volumétrico dado es mayor que aquel pronosticado por la Ley de Darcy por una cantidad proporcional al producto de la densidad del fluido y el cuadrado de su flujo volumétrico. La disipación de energía inercial es debida a innumerables aceleraciones (i.e., rapidez de cambio en la dirección y magnitud de velocidad) que experimenta un fluido mientras pasa por caminos tortuosos en un medio poroso. Estas aceleraciones causan patrones de flujo secundarios, en los cuales parte del flujo de energía se convierte en calor por esfuerzo viscoso.
En un yacimiento de petróleo, especialmente lejos de un pozo productor o de inyección, los flujos volumétricos son por lo general tan bajos que la Ley de Darcy se puede aplicar. Sin embargo, los altos flujos cerca de los pozos asociados con los altos gradientes de presión pueden causar efectos inerciales significativos conocidos como "superficies sensibles a la velocidad". Estos son particularmente frecuentes cerca de los pozos productores de gas, y en perforaciones donde la baja viscosidad del gas permite flujos muy altos. En mediciones de laboratorio de muestras de alta permeabilidad (donde los efectos inerciales se observan con la mayor frecuencia), los bajos flujos volumétricos requieren bajas gradientes de presión, las cuales pueden ser difíciles de medir con precisión. Flujos mas altos causarán un bajo cálculo de permeabilidad si se emplea la Ley de Darcy. 6.2.1.3 presenta un procedimiento para estimar la máxima gradiente de presión permitida como función de permeabilidad, para evitar errores significativos de permeabilidad debidos a la resistencia inercial. Una alternativa es tomar múltiples medidas de velocidad de flujo y emplear la ecuación de Forchheimer para obtener tanto la permeabilidad como la resistividad inercial de cada muestra. Como asunto práctico, esto se logra mas rápidamente con la técnica de variación de presiones presentada en 6.4.1.1. 6.1.1.3 Deslizamiento de Gas de Klinkenberg Aun cuando se toman en cuenta los efectos inerciales de manera apropiada, la permeabilidad de un medio poroso a gas depende del recorrido libre medio del gas que fluye, y por lo tanto entre otras cosas, de su presión absoluta. Esto es debido a un fenómeno conocido como el deslizamiento, un hecho indicado primero a la industria petrolera por Klinkenberg. El deslizamiento de gas ha sido pasado por alto o ignorado por los investigadores de permeabilidad anteriores a pesar de la teoría y los datos experimentales de deslizamiento en el flujo de gases a través de pequeños tubos capilares presentados en literatura científica tan temprano como 1875. Cuando se ignora el deslizamiento de gas, la permeabilidad calculada con la ecuación Forchheimer, o de la Ley de Darcy (siempre que la resistencia inercial sea insignificante), es mas alta que aquella obtenida utilizando un líquido no-reactivo. Cuando se expresa como porcentaje, esta diferencia es pequeña para las muestras de alta permeabilidad, pero se vuelve progresivamente más grande con la permeabilidad descendente. Esta se minimiza utilizando altas presiones de poros medias en las mediciones de permeabilidad de gas. Para evitar el problema de obtener permeabilidades de gas dependientes de la presión de poros, Klinkenberg presentó un método en el cual las medidas de permeabilidad de gas tomadas en diferentes presiones de poros medias pueden extrapolarse hasta una presión de poros infinita. Él demostró que esta permeabilidad de gas extrapolada (ahora llamada la "permeabilidad Klinkenberg", k∞) es igual a la permeabilidad obtenida utilizando un líquido

no reactivo, tal como un hidrocarburo limpio y refinado. La permeabilidad de las muestras de rocas, especialmente aquellas que contienen ciertas arcillas, pueden alterarse cuando son limpiadas y secadas en preparación para mediciones de permeabilidad de gas. A menudo, k ∞ es mas alto en estas muestras que la permeabilidad medida utilizando soluciones acuosas. Pueden requerirse técnicas especiales de limpieza y/o secado. 6.1.1.4 Líquidos El uso de líquidos para mediciones de permeabilidad elimina el problema del deslizamiento de gas, y a velocidades de flujo razonables y usuales, la resistencia inercial por lo general es insignificante. Por lo tanto, la Ley de Darcy puede utilizarse directamente para calcular la permeabilidad desde una sola medida de velocidad de flujo. Sin embargo, la alteración potencial de permeabilidad de la interacción de los componentes de roca y líquidos (especialmente soluciones acuosas), refina el movimiento, y el taponamiento micróbico requiere atención especial. Además, puede ser necesario remover el líquido restante en una muestra antes de que se puedan realizar otras mediciones. Debido a estos problemas, la mayoría de las mediciones rutinarias de permeabilidad se han hecho utilizando gases. Sin embargo, para algunas muestras, tales como aquellas sensibles a las técnicas de secado, las mediciones de permeabilidad líquidas son consideradas la única alternativa aceptable. 6.1.1.5 Dependencia de Esfuerzo Restrictivo La permeabilidad de un medio poroso es sensible a la magnitud de esfuerzos compresivos netos a los cuales es sujeto el medio, y a su historia de esfuerzo. Mientras se incrementan los esfuerzos restrictivos en un portamuestras, la permeabilidad de la roca se disminuye. La disminución observada, cuando se incrementan los esfuerzos restrictivos de 200 o 300 psi a varios miles psi, varia considerablemente - desde solo un pequeño porcentaje para muestras razonablemente permeables, bien consolidadas, hasta una magnitud o más para muestras de baja permeabilidad que contienen micro-fracturas. En general, es aconsejable aproximar los esfuerzos de yacimientos en las mediciones de permeabilidad en laboratorio para obtener valores más cercanos a los esperados en las permeabilidades de yacimientos en su lugar de origen. También pueden observarse disminuciones de permeabilidad adicionales más pequeñas como función de tiempo después de elevarse los esfuerzos restrictivos debido a escurrimiento. El escurrimiento es común en esfuerzos compresivos justo mas bajos que aquellos que causan fallas en las muestras. 6.1.2 Unidad de Permeabilidad 6.1.2.1 Unidad Tradicional Con el fin de hacer uso práctico del concepto que los medios porosos poseen la propiedad medible de la
permeabilidad, se ha definido una unidad estándar arbitraria, el "darcy". Un medio poroso tiene una permeabilidad de un darcy cuando un fluido monofásico de una viscosidad de un centipoise que llena completamente los vacíos del medio pase a través de él bajo "condiciones del flujo de Stokes" a una velocidad de 1 cm3/s por centímetro cuadrado de área de la sección transversal bajo una presión o una gradiente hidráulica equivalente de 1 atm/cm. "Condición del flujo de Stokes" significa simplemente que "la velocidad de flujo será lo suficientemente baja para ser directamente proporcional a la presión o la gradiente hidráulica". La unidad de permeabilidad (el darcy), como coeficiente de proporcionalidad entre cantidades físicas, posee las dimensiones de longitud cuadradas. La permeabilidad representa una propiedad del medio y es independiente del fluido, excepto como se califica de aquí en adelante. Por conveniencia la sub-unidad milidarcy (igual a 0.001 darcy) puede utilizarse. La ortografía de las formas plurales de la unidad ha sido estandarizada y establecida en la literatura como "darcys" y "milidarcys". Aunque no se recomiendan otras sub-unidades, el microdarcy (igual a 0.001 milidarcy o 10-6 darcy) se utiliza a menudo en conjunto con arenas de gas de baja permeabilidad. Además, se refiere a menudo al nanodarcy (igual a 0.001 microdarcy o 10-9 darcy) para rocas muy apretadas, tales como el granito micro-fracturado. 6.1.2.2 Unidad SI La unidad fundamental SI de permeabilidad, m2, se define de la siguiente manera: una permeabilidad de un metro cuadrado permitirá un flujo de 1 m3/s de fluido de una viscosidad de 1 Pa·s a través de un área de 1 m2 bajo una gradiente de presión de 1 Pa/m. Un darcy es igual a 0.986923 x 10-12m2. Debido al exponente tan pequeño, esta es una unidad difícil para uso común. La unidad de permeabilidad preferida por la Sociedad de Ingenieros de Petróleos de AIME es el micrómetro cuadrado (µm2). Un darcy (la unidad tradicional es igual a 0.986923 µm2. 6.1.2.3. Unidades Típicas Utilizadas en Hidrología La mayoría de los ingenieros ambientales tienen conocimientos en hidrología, en la cual se utilizan los términos permeabilidad intrínseca, conductividad hidráulica, y transmisividad. La física del flujo de fluidos que manejan los ingenieros ambientales, los analistas de núcleos, o los ingenieros de petróleos es básicamente igual, pero el lenguaje utilizado por un grupo es a menudo extraño para otro grupo. Esto surge de las diferentes aplicaciones. Los hidrólogos muy a menudo tratan con el flujo de aguas subterráneas en acuíferos, en los cuales la viscosidad, densidad y compresibilidad del agua demuestran variaciones bastante pequeñas. En estos acuíferos, a menudo es conveniente determinar las diferencias en el potencial de flujo midiendo las diferencias en las presiones líquidas - las elevaciones a las cuales sube el agua en pequeños pozos de prueba perforados en el acuífero. En el laboratorio, una técnica de medición análoga sería anexar una serie de manómetros de agua a una muestra de núcleos a través de la cual

fluye agua. La altura del agua en el manómetro mas cerca al extremo corriente arriba del núcleo sería la mayor, y las elevaciones en el nivel de agua (desde una referencia horizontal) sería progresivamente menor corriente abajo. En los yacimientos de petróleo, las propiedades de fluidos varían mucho, usualmente mas que una fase de fluido está presente, y las presiones a menudo son demasiado grandes para tomar medidas de presión manométricas. Las presiones son medidas directamente con manómetros o transductores. Por lo tanto, las ecuaciones para estas aplicaciones tratan con presiones, no carga. Los potenciales de flujo deben calcularse desde presiones, velocidades de flujo, y las propiedades de fluidos y rocas a través de ecuaciones diferenciales parciales apropiadas. La solución a menudo requiere una aproximación numérica de diferencias finitas de estas ecuaciones. 6.1.2.3.1 Diferencias en el Potencial de Flujo vs.
Diferencias de Presión Es importante comprender que la diferencia en el potencial de flujo, no la diferencia de presión, es la fuerza impulsora para el flujo de fluidos, saber qué es el flujo potencial, y averiguar cuando las diferencias de presión se pueden utilizar legítimamente en las ecuaciones de flujo. Las relaciones entre potencial de flujo y presión se muestran en la figura 6-1. Dos tanques, cada uno abierto a la presión atmosférica en la parte superior y equipado con tubos manométricos en varias profundidades, están conectados por un pequeño tubo. Cada uno está parcialmente lleno de agua hasta la misma profundidad. La presión de cada tanque en la zona interfacial aire-agua es atmosférica, y aumenta con la profundidad. Observamos que el nivel de agua en cada tubo manométrico es igual, y excepto por un aumento capilar muy pequeño en cada tubo, es igual al nivel de agua dentro de los tanques. Este nivel de agua en los tubos manométricos es una medida del potencial de flujo dentro de los tanques, que es igual en cada profundidad, aunque la presión aumenta con la profundidad. Si la presión, por sí sola, fuera la fuerza impulsora para el flujo, el agua de alta presión del fondo del tanque fluiría hacia arriba, hacia la presión menor. Este no es el caso. Si suponemos que la densidad del agua en todas partes del tanque es constante (una aproximación muy cercana), entonces el potencial de flujo, ϕ, en todas partes es:
ϕ = p - ρgz/C4 (1) Donde:
p = presión a la profundidad z (debajo de la
superficie del agua, z aumenta hacia abajo) ρ = densidad del agua g = aceleración local de gravedad C4 = factor de conversión (encontrada en la Tabla
6-1) para que las unidades sean consistentes.
Sin embargo, la presión en cualquier profundidad del tanque es: p = p1 + ρgz/C4 (2) Donde p1 es la presión en la zona interfacial aire-agua, la presión atmosférica en este caso. Por lo tanto, si combinamos las Ecuaciones 1 y 2, vemos que ϕ = p1, y el potencial de flujo es igual en cualquier profundidad. También es igual en ambos tanques, así que no ocurre ningún flujo dentro de ni entre los tanques. Condición 1: a. Se llenan ambos tanques con agua hasta la
misma profundidad, y se abren a la presión atmosférica.
b. La presión del agua aumenta en ambos tanques con la profundidad.
c. Sin embargo, el potencial de flujo (indicado por los niveles de agua en los tubos manométricos) es igual para todas las profundidades, y es igual en ambos tanques.
d. Por lo tanto, no ocurre ningún flujo dentro de ni entre los tanques.
Condición 2: a. Se aumenta la presión de aire por encima
del agua en el tanque izquierdo. Luego se tapa el respiradero.
b. El potencial de flujo en el tanque izquierdo se aumenta de la manera indicada con niveles de agua en los tubos manométricos allí.
c. El agua fluye del tanque izquierdo al tanque derecho hasta que los niveles de agua en los tubos manométricos están iguales para ambos tanques.
Figura 6-1 - Ilustración del Potencial de Flujo vs. Diferencias de Presión

Tabla 6-1 - Constantes en la Ecuación Forchheimer o Darcy con Varios Conjuntos de Unidades
Variable o Constante Unidades o Valores de las Constantes
SI SPE Preferido Tradicional Uso Común A, área perpendicular al flujo m2 m2 cm2 cm2 b, Factor de deslizamiento Klinkenberg Pa Pa atm psi D, diámetro de la muestra M M cm cm g, aceleración gravitacional m/s2 m/s2 cm/s2 cm/s2 k, permeabilidad m2 m2 darcy milidarcy L, longitud de la muestra M m cm cm M, peso molecular del gas kg/kg-mole kg/kg-mole g/g-mole g/g-mole P, presión absoluta Pa Pa atm psia p, presión manométrica Pa Pa atm psig ∆p, diferencia de presión Pa Pa atm psi q, velocidad volumétrica de flujo r, radio de la muestra o el sello M m cm cm S, almacenamiento compresivo s, distancia de la dirección del flujo M m cm cm T, temperatura absoluta K K K K V?, volumen del poro V?, volumen total del yacimiento v?, flujo volumétrico m/s m/s cm/s cm/s β, resistividad inercial m-1 m-1 cm-1 pies-1 ρ, densidad del gas o el líquido kg/m3 kg/m3 g/cm3 g/cm3 µ, viscosidad del gas o el líquido Pa·s Pa·s cp cp R, constante de la ley del gas 8314 8314 82.05 1205.8 C1, en la ecuación Darcy o Forch. 1.0 1.0 1.0 6.8046E-2 C2, en la ecuación Darcy o Forch. 1.0 1.0E+12 1.0 1000. C3, constante en la ecuación Forch. 1.0 1.0 9.8692E-7 3.2379E-8 C4, constante en ecuaciones con g 1.0 1.0 1.0133E+6 68950. Ahora, suponga que se bombea aire al tanque izquierdo a través de su respiradero para que la presión de aire por encima del agua es mayor que la presión atmosférica. Luego se tapa el respiradero. Observaríamos que los niveles del agua en todos los manómetros conectados al tanque izquierdo han subido por igual. Excepto por la cantidad de agua requerida para aumentar los niveles en los tubos, el nivel del agua dentro del tanque no ha cambiado. Inicialmente, las condiciones en el tanque derecho no cambian. De los niveles manométricos superiores en el tanque izquierdo (indicados por las líneas punteadas en la Figura 6-1), relativos a aquellos en el tanque derecho, concluimos que el potencial de flujo es superior en el primero, aunque los niveles del agua dentro de los dos tanques son iguales. Efectivamente, el agua fluye del tanque izquierdo al derecho hasta que los niveles manométricos son iguales para ambos tanques. Cuando termina el flujo, el nivel del agua dentro del tanque izquierdo será menor que en el tanque derecho. En las mediciones monofásicas de permeabilidad de gas, las diferencias en la "presión de gas" son por lo general mas pequeñas que la precisión de las mediciones de presión, y se pueden ignorar. Por lo tanto las diferencias en presión o las gradientes de presión son aproximaciones muy cercanas de las diferencias o gradientes en el potencial de flujo en cualquier orientación de flujo (horizontal o vertical). Se explican en detalle las condiciones para las mediciones monofásicas de líquidos en 6.2.2. Las diferencias o gradientes de presión pueden sustituirse
con las diferencias o gradientes de potencial de flujo únicamente en situaciones especiales y restringidas. 6.1.2.3.2 Permeabilidad Intrínseca La permeabilidad intrínseca, k, utilizada por los hidrólogos es idéntica a la permeabilidad definida en 6.1.2.2, aunque se define en términos de cargas y viscosidad cinemática, v, en lugar de la viscosidad dinámica, µ, utilizada en la ecuación Darcy definida en 6.1.2.1. La viscosidad cinemática es igual a la viscosidad dinámica dividido por la densidad del fluido. Un medio poroso tiene una permeabilidad intrínseca de una unidad de longitud cuadrada si puede transmitir en tiempo de unidad un volumen de una unidad de fluido de viscosidad cinemática a través de una área unidad de sección transversal medida en ángulos rectos a la dirección de flujo bajo una gradiente de potencial de unidad. Si se utilizan unidades SI, la permeabilidad intrínseca tiene la unidad de m2. 6.1.2.3.3 Conductividad Hidráulica La conductividad hidráulica, K, no es solo una propiedad de un medio poroso. Esta depende también de la viscosidad cinemática del fluido corriente, y es apropiado únicamente para acuíferos de aguas subterráneas. Sus unidades son aquellas de velocidad, por lo general pies por día o metros por día. Un medio tiene conductividad hidráulica de longitud de unidad por tiempo de unidad si puede

transmitir en tiempo de unidad un volumen de unidad de agua subterránea a la viscosidad predominante a través de una área unidad de sección transversal medida en ángulos rectos a la dirección de flujo bajo una gradiente hidráulica de cambio de carga a través de una longitud de flujo. La conductividad hidráulica está relacionada con la permeabilidad (o la permeabilidad intrínseca) por: K =
µ (3) kρg
Donde k es la permeabilidad, y los otros símbolos están definidos arriba. Se puede utilizar cualquier conjunto consistente de unidades en la Ecuación 3. Con unidades SI, K tendrá la unidad de metros por segundo. 6.1.2.3.4 Transmisividad La transmisividad, T, de un acuífero de aguas subterráneas incluye el grosor del acuífero, b, y la viscosidad cinemática del agua confinada. La transmisividad es la velocidad a la cual el agua de la viscosidad cinemática predominante se transmite a través de un ancho de una unidad del acuífero bajo una gradiente hidráulica de una unidad. Esta tiene una unidad de longitud cuadrada por unidad de tiempo, a menudo pies2/día, o m2/día. La transmisividad es igual a Kb. Esta se puede calcular desde la permeabilidad así: T =
µ (4) kρgb
Las definiciones para permeabilidad intrínseca, conductividad hidráulica, y transmisividad son tomadas de Lohman. 42 Se pueden encontrar los factores de conversión para varios conjuntos de unidades en Ground Water (1993). 43 6.1.3 Terminología 6.1.3.1 Permeabilidad Específica La definición de la unidad estándar de permeabilidad requiere que el medio poroso contenga solo un liquido homogéneo monofásico. La propiedad del medio determinado así es llamada apropiadamente la permeabilidad específica, y es señalada por su símbolo k. Con esta definición, el término permeabilidad se convierte en una propiedad únicamente del medio poroso, y el valor numérico de la permeabilidad es constante e independiente del fluido utilizado en la medición. Cuando ciertos minerales y fluidos entran en contacto y resulta una interacción, o se deposita algún material del fluido, este fenómeno, efectivamente, produce un nuevo medio. La permeabilidad del nuevo medio puede ser diferente a la del medio inalterado. Cuando esto ocurre, se debe indicar claramente con referencias apropiadas para el medio y el fluido, e.g., "la permeabilidad de arena Woodbine al agua salada (agua dulce, crudo, heptano, etc., según el caso)."
Las permeabilidades de gas no corregidas son excluidas de la definición anterior debido a los efectos del deslizamiento de gas. Las permeabilidades de gas, que serán llamadas kg (o kaire, kN2, kHe, etc.) dependen del gas específico utilizado en la determinación de permeabilidad, y de su presión de poros media. Ambos deben especificarse para definir la permeabilidad del medio. Solo la permeabilidad Klinkenberg, con deslizamiento corregido, cuando no es afectado por efectos de alta velocidad (Forchheimer) es constante e independiente del fluido utilizado en la medición. Como la condición de esfuerzo bajo la cual se toma la medida puede tener un efecto significativo sobre la permeabilidad, esta también debe especificarse. Algunos ejemplos son kaire con una presión de poros media de 32.3 psia y un esfuerzo restrictivo radial de 400 psig: o k∞ a un esfuerzo hidrostático neto de 5.000 psi. La primera especificación indica que una permeabilidad de gas fue obtenida utilizando aire a una presión de poro absoluta promedio de 32.3 psia, y que se aplicó una presión de 400 psig a la bota de caucho del portamuestras. Por lo tanto el esfuerzo radial neto es 400 + 14.7 - 32.3, o alrededor de 382 psi. El esfuerzo axil neto es desconocido. En el segundo ejemplo, una permeabilidad Klinkenberg (con deslizamiento corregido) fue obtenido que es independiente del gas utilizado o su presión de poros media (excepto que la presión de poros afecta el esfuerzo neto). Se aplicaron esfuerzos netos radiales y axiles ("hidrostáticos") de magnitudes iguales de 5.000 psi durante la medición. 6.1.3.2 Permeabilidad Efectiva y Relativa Este manual de prácticas rutinarias recomendadas solo trata con las mediciones monofásicas de permeabilidad. Por lo tanto, solo se definirán la permeabilidad efectiva y la permeabilidad relativa. La permeabilidad efectiva de un medio poroso es una medida de su conductividad fluida a una fase específica de un sistema polifásico de fluidos que reside dentro del medio, donde se especifica la saturación de cada fase (la fracción de su volumen de poros efectivo total llenado por cada fase). La permeabilidad relativa es la relación de la permeabilidad efectiva de una fase específica del fluido, a saturaciones definidas de todas las fases, a alguna permeabilidad arbitraria de referencia. La permeabilidad de referencia puede ser la permeabilidad específica de la muestra, su permeabilidad Klinkenberg, la permeabilidad efectiva de una de las fases del fluido en una condición de saturación específica, etc. La permeabilidad de referencia utilizada tiene que especificarse. El libro tres de History of Petroleum Engineering (API, 1961) 39 de cinco volúmenes, es una rica fuente de referencia sobre los primeros trabajos en mediciones de permeabilidad en laboratorios, incluyendo los esfuerzos experimentales de Fancher, Lewis, y Barnes, 40 y de Wyckoff, Botset, Muskat, y Reed. 41 6.2 TEORÍA Las mediciones de permeabilidad monofásicas pueden separarse en cuatro categorías principales:
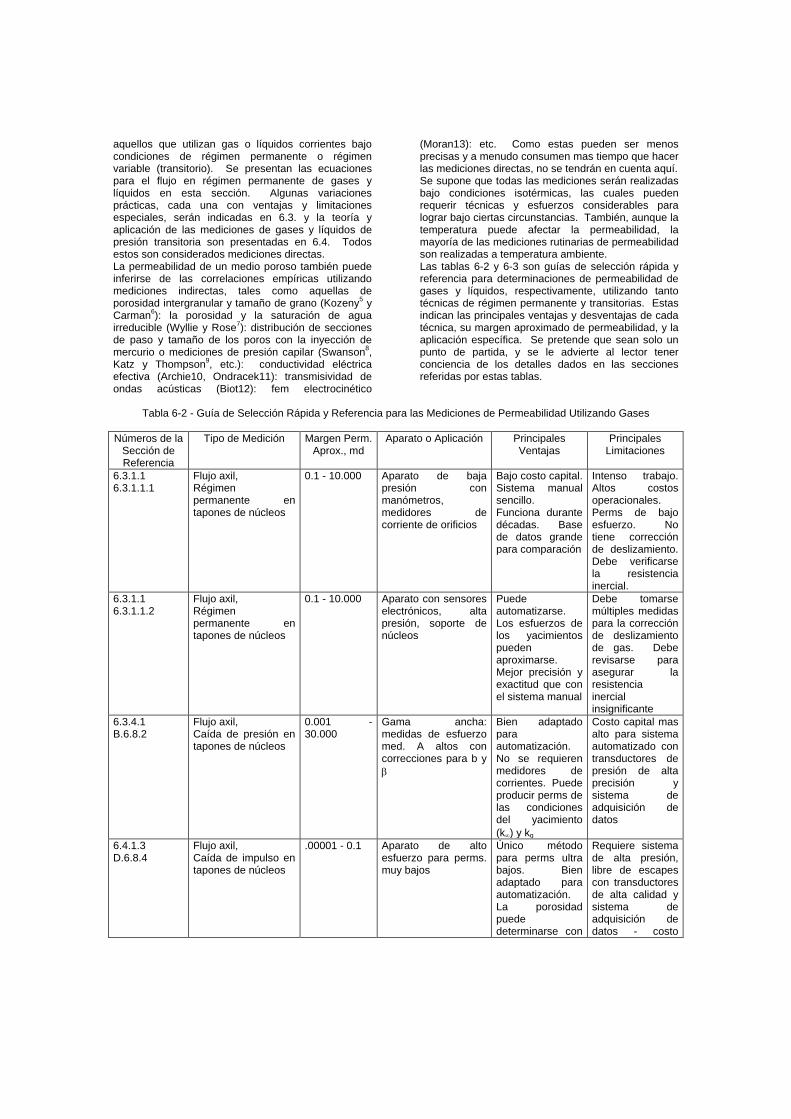
aquellos que utilizan gas o líquidos corrientes bajo condiciones de régimen permanente o régimen variable (transitorio). Se presentan las ecuaciones para el flujo en régimen permanente de gases y líquidos en esta sección. Algunas variaciones prácticas, cada una con ventajas y limitaciones especiales, serán indicadas en 6.3. y la teoría y aplicación de las mediciones de gases y líquidos de presión transitoria son presentadas en 6.4. Todos estos son considerados mediciones directas. La permeabilidad de un medio poroso también puede inferirse de las correlaciones empíricas utilizando mediciones indirectas, tales como aquellas de porosidad intergranular y tamaño de grano (Kozeny5 y Carman6): la porosidad y la saturación de agua irreducible (Wyllie y Rose7): distribución de secciones de paso y tamaño de los poros con la inyección de mercurio o mediciones de presión capilar (Swanson8, Katz y Thompson9, etc.): conductividad eléctrica efectiva (Archie10, Ondracek11): transmisividad de ondas acústicas (Biot12): fem electrocinético
(Moran13): etc. Como estas pueden ser menos precisas y a menudo consumen mas tiempo que hacer las mediciones directas, no se tendrán en cuenta aquí. Se supone que todas las mediciones serán realizadas bajo condiciones isotérmicas, las cuales pueden requerir técnicas y esfuerzos considerables para lograr bajo ciertas circunstancias. También, aunque la temperatura puede afectar la permeabilidad, la mayoría de las mediciones rutinarias de permeabilidad son realizadas a temperatura ambiente. Las tablas 6-2 y 6-3 son guías de selección rápida y referencia para determinaciones de permeabilidad de gases y líquidos, respectivamente, utilizando tanto técnicas de régimen permanente y transitorias. Estas indican las principales ventajas y desventajas de cada técnica, su margen aproximado de permeabilidad, y la aplicación específica. Se pretende que sean solo un punto de partida, y se le advierte al lector tener conciencia de los detalles dados en las secciones referidas por estas tablas.
Tabla 6-2 - Guía de Selección Rápida y Referencia para las Mediciones de Permeabilidad Utilizando Gases
Números de la
Sección de Referencia
Tipo de Medición Margen Perm. Aprox., md
Aparato o Aplicación Principales Ventajas
Principales Limitaciones
6.3.1.1 6.3.1.1.1
Flujo axil, Régimen permanente en tapones de núcleos
0.1 - 10.000 Aparato de baja presión con manómetros, medidores de corriente de orificios
Bajo costo capital. Sistema manual sencillo. Funciona durante décadas. Base de datos grande para comparación
Intenso trabajo. Altos costos operacionales. Perms de bajo esfuerzo. No tiene corrección de deslizamiento. Debe verificarse la resistencia inercial.
6.3.1.1 6.3.1.1.2
Flujo axil, Régimen permanente en tapones de núcleos
0.1 - 10.000 Aparato con sensores electrónicos, alta presión, soporte de núcleos
Puede automatizarse. Los esfuerzos de los yacimientos pueden aproximarse. Mejor precisión y exactitud que con el sistema manual
Debe tomarse múltiples medidas para la corrección de deslizamiento de gas. Debe revisarse para asegurar la resistencia inercial insignificante
6.3.4.1 B.6.8.2
Flujo axil, Caída de presión en tapones de núcleos
0.001 - 30.000
Gama ancha: medidas de esfuerzo med. A altos con correcciones para b y β
Bien adaptado para automatización. No se requieren medidores de corrientes. Puede producir perms de las condiciones del yacimiento (k∞) y kg
Costo capital mas alto para sistema automatizado con transductores de presión de alta precisión y sistema de adquisición de datos
6.4.1.3 D.6.8.4
Flujo axil, Caída de impulso en tapones de núcleos
.00001 - 0.1 Aparato de alto esfuerzo para perms. muy bajos
Único método para perms ultra bajos. Bien adaptado para automatización. La porosidad puede determinarse con
Requiere sistema de alta presión, libre de escapes con transductores de alta calidad y sistema de adquisición de datos - costo
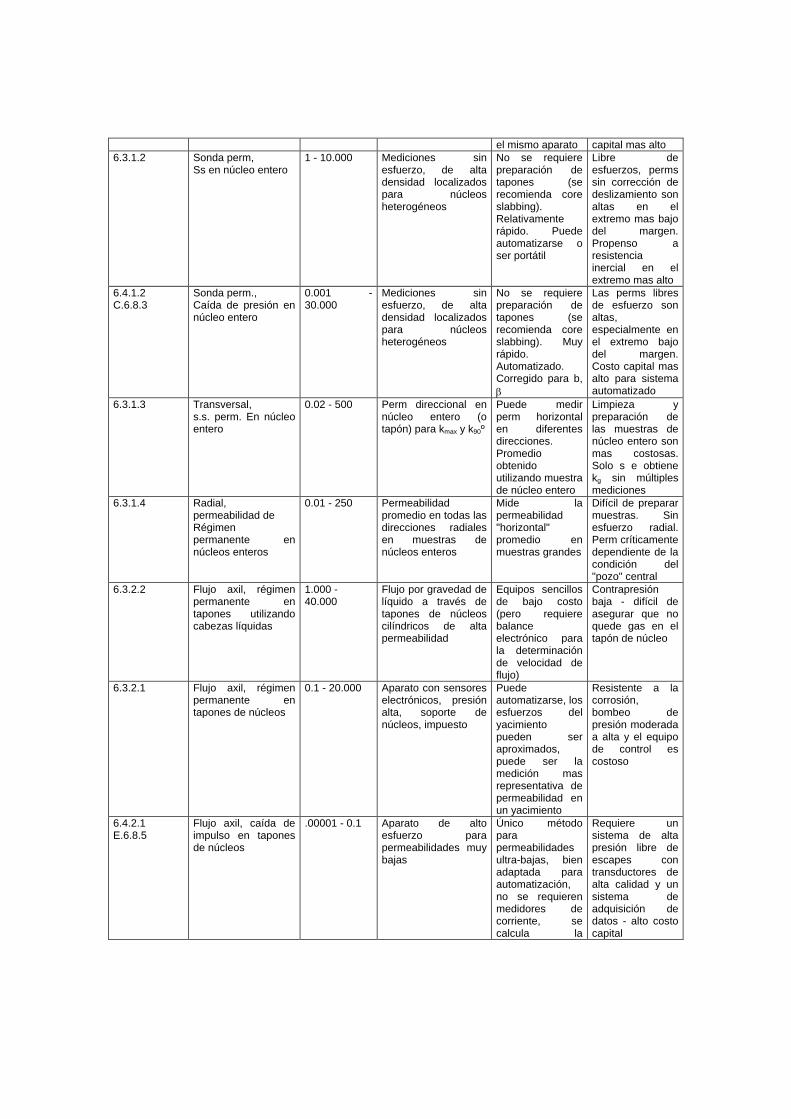
el mismo aparato capital mas alto 6.3.1.2 Sonda perm,
Ss en núcleo entero 1 - 10.000 Mediciones sin
esfuerzo, de alta densidad localizados para núcleos heterogéneos
No se requiere preparación de tapones (se recomienda core slabbing). Relativamente rápido. Puede automatizarse o ser portátil
Libre de esfuerzos, perms sin corrección de deslizamiento son altas en el extremo mas bajo del margen. Propenso a resistencia inercial en el extremo mas alto
6.4.1.2 C.6.8.3
Sonda perm., Caída de presión en núcleo entero
0.001 - 30.000
Mediciones sin esfuerzo, de alta densidad localizados para núcleos heterogéneos
No se requiere preparación de tapones (se recomienda core slabbing). Muy rápido. Automatizado. Corregido para b, β
Las perms libres de esfuerzo son altas, especialmente en el extremo bajo del margen. Costo capital mas alto para sistema automatizado
6.3.1.3 Transversal, s.s. perm. En núcleo entero
0.02 - 500 Perm direccional en núcleo entero (o tapón) para kmax y k90º
Puede medir perm horizontal en diferentes direcciones. Promedio obtenido utilizando muestra de núcleo entero
Limpieza y preparación de las muestras de núcleo entero son mas costosas. Solo s e obtiene kg sin múltiples mediciones
6.3.1.4 Radial, permeabilidad de Régimen permanente en núcleos enteros
0.01 - 250 Permeabilidad promedio en todas las direcciones radiales en muestras de núcleos enteros
Mide la permeabilidad "horizontal" promedio en muestras grandes
Difícil de preparar muestras. Sin esfuerzo radial. Perm críticamente dependiente de la condición del "pozo" central
6.3.2.2 Flujo axil, régimen permanente en tapones utilizando cabezas líquidas
1.000 - 40.000
Flujo por gravedad de líquido a través de tapones de núcleos cilíndricos de alta permeabilidad
Equipos sencillos de bajo costo (pero requiere balance electrónico para la determinación de velocidad de flujo)
Contrapresión baja - difícil de asegurar que no quede gas en el tapón de núcleo
6.3.2.1 Flujo axil, régimen permanente en tapones de núcleos
0.1 - 20.000 Aparato con sensores electrónicos, presión alta, soporte de núcleos, impuesto
Puede automatizarse, los esfuerzos del yacimiento pueden ser aproximados, puede ser la medición mas representativa de permeabilidad en un yacimiento
Resistente a la corrosión, bombeo de presión moderada a alta y el equipo de control es costoso
6.4.2.1 E.6.8.5
Flujo axil, caída de impulso en tapones de núcleos
.00001 - 0.1 Aparato de alto esfuerzo para permeabilidades muy bajas
Único método para permeabilidades ultra-bajas, bien adaptada para automatización, no se requieren medidores de corriente, se calcula la
Requiere un sistema de alta presión libre de escapes con transductores de alta calidad y un sistema de adquisición de datos - alto costo capital

velocidad con ∆b & t
6.3.2.1 6.3.1.3.1
Flujo transversal, régimen permanente en núcleos enteros
0.005 - 500 Permeabilidad direccional en núcleo entero (o tapón) para kmax y k90º
Puede medir permeabilidad "horizontal" en diferentes direcciones, promedio obtenido utilizando muestra de núcleo entero
Igual que para el flujo axil de régimen permanente (arriba), limpieza, preparación y manejo de muestras de núcleo entero mas costoso
6.3.1.4 6.3.1.4.2
Flujo radial de régimen permanente en núcleos enteros
0.002 - 250 Permeabilidad promedio en todas las direcciones radiales en muestras de núcleo entero
Mide permeabilidad "horizontal" promedio en muestras grandes
Igual que arriba, difícil de preparar las muestras, sin esfuerzo radial, permeabilidad críticamente dependiente de la condición del "pozo" central
*Principales ventajas de utilizar gas en lugar de líquido a. Fácil de usar - no requiere técnicas especiales de saturación b. No-reactivo con roca - no corrosivo para los equipos c. No se requiere limpieza después de mediciones d. Menos propenso que el líquido a movilizar finos en muestra de rocas e. No soporta el crecimiento microbiano, ni requiere filtración especial Principales desventajas: a. Requiere corrección de deslizamiento de gas - especialmente con permeabilidades mas bajas b. Propenso a resistencia inercial de alta velocidad significativo en rocas de alta permeabilidad c. Es mas difícil de lograr hermeticidad necesaria que con líquidos d. En algunos casos, puede ser menos representativo a la permeabilidad en el yacimiento. 6.2.1 Ecuaciones de Permeabilidad de Régimen
Permanente para Gases La ecuación general para mediciones de permeabilidad de régimen permanente para gases será presentada primero. Esta incluye provisión para efectos de deslizamiento de gas y resistencia inercial, que se vuelven algo complicados y requieren mas mediciones que lo generalmente posible utilizando mediciones de régimen permanente. Sin embargo, sirve para unificar la teoría de mediciones de gas y ofrece principios y pautas para el uso de la ecuación Darcy, la cual es la base para los casos prácticos de régimen permanente que siguen. A diferencia de los líquidos, los gases son altamente compresibles. Además, bajo condiciones normales de laboratorio, la densidad de gas es lo suficientemente bajo para que los efectos de la gravedad se puedan ignorar en la toma de medidas de permeabilidad. La viscosidad de aire, nitrógeno, o helio (ver Tablas 6-4, 6-5 y 6-6 respectivamente) es menor que 1/50 que la de agua a temperatura ambiente. Por lo tanto, los flujos volumétricos de gases son correspondientemente mas altas para una gradiente de presión dada, y pueden desviar de las condiciones del flujo de Stokes, requerida por la Ley de Darcy.
Finalmente, el fenómeno del deslizamiento de gas, o el efecto Klinkenberg, requiere un tratamiento especial. Debido a estas diferencias, el punto de partida para desarrollar ecuaciones que pertenecen a la medición de permeabilidad utilizando gases es la forma diferencial de la ecuación de Forchheimer, en la cual la relación Klinkenberg será insertada: -dP = C2µqs + ds C1Akg C1A2
C3βρ2
Donde: S = distancia en la dirección de flujo qs = velocidad volumétrica del flujo de gas a
través del área A por unidad de tiempo A = área perpendicular al flujo de gas dP/ds = gradiente de presión en s a la cual se refiere
qs / A µ = viscosidad dinámica de gas kg = permeabilidad aparente del medio a un gas
específico β = coeficiente de resistividad inercial ρ = densidad del gas Las constantes C1, C2 y C3 permiten el uso de varios conjuntos de dimensiones en las ecuaciones de flujo

de gas que siguen. La tabla 6-1 muestra los valores de estas constantes para SI, SPE preferidas, tradicionales, o dimensiones de uso común. Observe que aparece una P mayúscula en la ecuación arriba. Este símbolo no-estándar indica que la presión es absoluta. Una p minúscula se usa para indicar una presión manométrica o una diferencia de presión en cualquiera de las siguientes ecuaciones. Estas convenciones han sido adoptadas en este documento por la confusión entre las dos presiones, y el error frecuente cometido cuando una presión manométrica medida no es convertida a una presión absoluta cuando es necesario. Por lo tanto, para seguir con esta convención:
P = p + Pa (6) Donde Pa es la verdadera presión barométrica ambiente, convertida en las unidades apropiadas (no la presión barométrica del aeropuerto local, que ha sido convertida en la presión a nivel del mar). La densidad de un gas real es: ρ = zRT (7)
MP
Tabla 6-4 - Viscosidad de Aire (para Grados F y C) a Una Atmósfera* * Calculada con la ecuación de Sutherland: µaire = T + 120
14.969T1.5
Donde: T es temperatura absoluta, grados Kelvin (grados Celsius + 273), y µaire es la viscosidad de aire a la presión de una atmósfera, en micropoises. Multiplicar por 1.0 E-04 para convertir a centipoises, o por 1.0 E-07 para convertir a Pa·s. Las constantes están basadas en el trabajo de Montgomery35 y Birge36.
Tabla 6-4 - Viscosidad de Nitrógeno (para Grados F y C) a Una Atmósfera*
* Calculada con la ecuación de Sutherland: µN2 = T + 102
13.85T1.5
Donde: T es temperatura absoluta, grados Kelvin (grados Celsius + 273), y µN es la viscosidad de nitrógeno a la presión de una atmósfera, en micropoises. Multiplicar por 1.0 E-04 para convertir a centipoises, o por 1.0 E-07 para convertir a Pa·s. Las constantes están basadas en el trabajo de Licht y Stechert37. La viscosidad del nitrógeno a temperaturas razonablemente cercanas a 25ºC y presiones de 253 atmósferas pueden caclularse con: µN2 [T,P] = µN2 [T,1] - 0.12474 + 0.123688P + 1.0542E-03P2 - 1.5052E-06P3 Donde µN2 [T,1] es la viscosidad de nitrógeno a la presión de una atmósfera (de las tablas arriba), micropoises, y P es presión, atmósferas. La dependencia de presión es adaptada a los datos de Gracki, et al34 dentro de una desviación máxima de 0.1%. La dependencia de presión a 0ºC y 50ºC es casi igual a 25ºC.
Tabla 6-4 - Viscosidad de Helio (para Grados F y C) a Una Atmósfera* *Calculada con: µHe = 187.0 (T/273.1)0.685 Donde: T es temperatura absoluta, grados Kelvin (grados Celsius + 273), y µHe es la viscosidad de helio a la presión de una atmósfera, en micropoises. Multiplicar por 1.0 E-04 para convertir a centipoises, o por 1.0 E-07 para convertir a Pa·s. Las son de Chapman y Cowling38. De acuerdo con los datos de Gracki, et al 34, la viscosidad del helio a 25ºC pasa por un mínimo muy poco profundo con presión. A 37 atm es de 0.43 por ciento menos que a 1 atm y a 158 atm su viscosidad es 0.17 por ciento mas alto que a 1 atm. Donde z es el factor de desviación de gas, el cual es igual a 1.0 para un gas ideal. Los valores de z como funciones de temperatura y presión están dadas en las Tablas 6-7, 6-8 y 6-9 para aire, nitrógeno, y helio, respectivamente. Son calculados con la ecuación de estado Beattie-Bridgeman14. Ver Tabla 6-1 para valores de la constante universal de los gases, R, para los diferentes conjuntos de unidades. M es el peso molecular del gas. La temperatura en la Ecuación 7 es absoluta, en grados Kelvin.
T = ºC + 273.15 = (ºF - 32)/1.8 + 273.15 (8) 6.2.1.1 Tratamiento del Factor de Deslizamiento de
Gas La relación Klinkenberg, escrita como una función puntual (i.e., relacionado con un punto específico en una muestra, no un valor promedio) es:

kg = k∞ (1 + b/P) (9) Observe que kg depende de la presión. Por lo tanto, no puede tratarse como una constante cuando se integra la ecuación Forchheimer (o Darcy). En la Ecuación 9, b tiene que tener las mismas unidades que aquellas de la presión usadas. Desafortunadamente, el factor de deslizamiento de gas, b, es en parte una propiedad de roca y en parte una propiedad de gas, lo cual ha causado confusión y algunos problemas. Por ejemplo, un b medido utilizando aire, pero no indicado como tal, puede ser mal empleado en los cálculos con un gas diferente con propiedades significativamente diferentes. Es un procedimiento bastante sencillo para separar las propiedades del gas del factor de deslizamiento, para que solo quede la propiedad dependiente de la roca. De acuerdo con Klinkenberg3, b está relacionado con el recorrido libre medio de moléculas de gas, λ, con la relación: λ =
4cP (10)
br
Pero, desde la teoría de gas cinético15, el recorrido libre medio es dada por:
λ = 1.881µ P M (11)
RT
En las ecuaciones 10 y 11, se puede utilizar cualquier unidad consistente. De estas dos ecuaciones, y ajustando para las unidades dadas a continuación, obtenemos: b = [99.5µ T/M](c/r) (12) Donde: P = presión absoluta de gas R = constante universal de los gases B = factor de deslizamiento de gas de
Klinkenberg, psi. (La constante 99.5 se convierte a 6.77 si b tiene la unidad de atm).
µ = viscosidad de gas, cp.
Tabla 6-7 - Factores de Desviación de la Ley de Gas para Aire, Calculados de la Ecuación de Estado Beattie-Bridgemann14
Tabla 6-8 - Factores de Desviación de la Ley de Gas para Nitrógeno, Calculados de la Ecuación de Estado Beattie-Bridgemann14
Tabla 6-7 - Factores de Desviación de la Ley de Gas para Helio, Calculados de la Ecuación de Estado Beattie-
Bridgemann14
T = temperatura absoluta del gas durante la medición de b, en grados K
M = peso molecular del gas c = "coeficiente de aceptación" sin dimensiones
que es ligeramente menos que 1.0 de acuerdo con Klinkenberg.3
r = radio de tubos capilares distribuidos al azar (espacialmente) que se aproximan a un medio poroso en el modelo de Klinkenberg, µm.
En la ecuación 12, el factor dentro de corchetes depende únicamente de las propiedades del gas, y el factor entre paréntesis es dependiente de la roca. Si definimos el factor roca-dependiente, r/c, como el "radio efectivo de deslizamiento de gas", rb, y lo resolvemos cambiando la ecuación 12: Rb = 99.5µ b M (13)
T
Entonces este es el factor recomendado de deslizamiento de gas a reportar, el cual es independiente del gas utilizado y la temperatura. Este se debe reportar además de b, o en lugar de b. La analogía de tubos capilares distribuidos al azar de Klinkenberg relaciona rb a permeabilidad y porosidad con: rb = Cb φ
k∞
donde k∞ es la permeabilidad Klinkenberg, (µm2), y φ es la porosidad (fracción). En el modelo de Klinkenmberg, Cb es igual a √24/c, o aproximadamente 5. Si relajamos este modelo "demasiado sencillo", se puede encontrar Cb empíricamente. En la figura 6-2, rb es representado vs. √ k∞ /φ. La pendiente de la línea trazada en este gráfico doblemente logarítmico es unidad. Desde su posición, se calcula que Cb es aproximadamente 11. Los datos, obtenidos utilizando helio en varios tapones de núcleos, muestran una dispersión considerable. Excepto por el valor de la constante, estos apoyan el modelo de Klinkenberg cualitativamente. Se anticipa que los parámetros de deslizamiento de gas, b o rb, son funciones de la estructura de poros, lo cual puede explicar la dispersión. 6.2.1.2 Flujo de Gas de Régimen Permanente - La
Ecuación Forchheimer El régimen permanente se logra cuando las presiones corriente arriba y corriente abajo y la velocidad de flujo todos se vuelven invariables con el tiempo. En un régimen permanente, la velocidad de flujo de masa es constante por toda la muestra y no cambia con el tiempo. Por lo tanto:
ρq3 = ρrqr (15)
Figura 6-2 - Radio de Deslizamiento de Gas Roca-Dependiente, rb, como Función de Permeabilidad y Porosidad

El subíndice r se refiere a las condiciones de referencia de temperatura y presión bajo las cuales se mide la velocidad de flujo de masa. Por lo tanto, en condiciones isotérmicas de régimen permanente, donde la temperatura de referencia es igual a la siguiente temperatura de gas corriente: Pq3 = z zr (16)
Prqr
Sustituir las ecuaciones 7, 9 y 16 en la ecuación 5 produce la forma diferencial de la ecuación Forchheimer para el flujo de gases isotérmico de régimen permanente, con deslizamiento de gas corregido: C1(-dP) = C2µPrqrz + ds Ak∞z1(P+b) A2RTzr
2P (17) C3βMPr
2qr2z
La forma integrada de esta ecuación puede utilizarse para obtener k∞, b y β. Esto requiere un mínimo de tres, y preferiblemente por lo menos seis mediciones en las cuales la presión corriente arriba (y/o opcionalmente la presión corriente abajo) es muy variada para causar variaciones sustanciales en la presión de poros media y la velocidad de flujo. Para mediciones rutinarias, esto no es viable (por costos) bajo condiciones de régimen permanente. Sin embargo, para la mayoría de los casos, una información adecuada para el cálculo de estos parámetros puede obtenerse de un solo ciclo de caída de presión (ver 6.4.1.1 y 6.4.1.2). 6.2.1.3 La Ecuación Darcy con Corrección de
Deslizamiento Cuando su último término se vuelve insignificantemente pequeño, la ecuación 17 se reduce a la forma diferencial de la Ley de Darcy, con deslizamiento corregido, y es la base para la mayoría de las mediciones prácticas rutinarias. Pequeñas gradientes de presión resultan en bajas velocidades de flujo, que son cuadradas en el último término, y por lo tanto son pequeñas. La magnitud del término también depende de los valores de β y la densidad del gas. Densidad es una función de temperatura, presión y peso molecular del gas utilizado. La figura 6-3 muestra los valores típicos de β como función de permeabilidad para arenisca (círculos abiertos) y rocas de carbonato (cuadrados) (datos de Jones16), y para arenas homogéneas sin consolidar (círculos sólidos, datos de Geertsma17). Se cree que la considerable dispersión está relacionada con la heterogeneidad en la roca.16 Las muestras con fracturas o finas capas de material de permeabilidad superior paralelas a la dirección de flujo tienen βs mas altas que la roca homogénea, debido a las velocidades localmente altas en estas "bandas" de alta permeabilidad. La figura 6-3 contiene dos curvas punteadas que rodean la mayoría de los puntos, y una curva central que "mejor se adapta". La Figura 6-4, la cual es basada en los datos de la figura 6-3, presenta máximas gradientes de presión general permitidas, ∆p/L, que son lo suficientemente pequeños para permitir el uso de la ecuación Darcy
con deslizamiento corregido para el cálculo de permeabilidad sin incurrir en errores significantes debido a resistencia inercial. Las curvas indicadas son para aire a 72ºF, y para presiones corriente abajo de 14.7, 50, 100 y 200 psia. Debido a la dispersión en β, estas curvas son aproximadas, y deben interpretarse de la siguiente manera - Si el β real para una muestra específica por casualidad se encuentra en la línea punteada superior en la Figura 6-3, y el máximo ∆p/L indicado en la Figura 6-4, para las condiciones utilizadas, fueron aplicados para la medición de permeabilidad, entonces la permeabilidad calculada sería bajo por un 5 por ciento si el término final en la ecuación 17 fuera ignorado. Si el β real fuera dos veces el valor indicado en la línea punteada superior de la figura 6-3, y se aplicara el mismo ∆p/L, entonces la permeabilidad calculada sería demasiado bajo por 10 por ciento. Por otro lado, si el β real fuera un factor de 10 menor que el indicado por la línea punteada superior (por lo tanto, mas cercano a la alta densidad de puntos), el error de permeabilidad sería únicamente 0.5 por ciento, etc. El uso de los valores máximos para ∆p/L indicados en la Figura 6-4 resultará en errores (debido a la resistencia inercial) de menos del 2 por ciento para la mayoría de las muestras cuando se utiliza la ecuación Darcy con deslizamiento corregido para el cálculo de permeabilidad. Empezando con las permeabilidades mas altas para la presión corriente debajo de 14.7 psia en la Figura 6-4, la máxima caída de presión permitida aumenta con una permeabilidad decreciente, de la manera esperada. Sin embargo, para permeabilidades por debajo de 0.3 milidarcys, ∆pmax/L disminuye con una permeabilidad decreciente. Este resultado inesperado es una consecuencia del deslizamiento de gas. El deslizamiento es mayor en las permeabilidades mas bajas, causando velocidades de flujo de gas mas altas que las que ocurriría sin deslizamiento. Excepto por las permeabilidades muy bajas, la máxima gradiente general permitida se reduce cuando se aumenta la presión corriente abajo. La contrapresión aumentada incrementa la densidad del gas, así aumentando la significación del término final en la ecuación 17. Este resultado debe reconocerse cuando se hacen mediciones múltiples con presiones de poros cada vez mas altas para la corrección de deslizamiento de gas de Klinkenberg. Las permeabilidades aparentes de gas medidas en altas presiones de poros pueden ser demasiado bajas debido a la inadvertida introducción de resistencia inercial que no se puede despreciar. Los valores máximos de ∆p/L son específicos para gas. Los valores para nitrógeno y aire son similares, pero aquellos para helio son mas altos. La viscosidad del helio es mas alto, y su peso molecular es menor que el del aire. Las ecuaciones para calcular ∆p/L para otros gases y líquidos son desarrollados en A.6.8.1. La condición del flujo de Stokes se cumple cuando el último término de la ecuación 17 es inapreciable. La ecuación Darcy restante con deslizamiento corregido puede integrarse y utilizarse para calcular permeabilidades Klinkenberg para varias configuraciones de flujo frecuentemente usados:

k∞ =
Donde C1 y C2 son constantes de conversión de la Tabla 6-1 para permitir diferentes conjuntos de
unidades para las variables. P1 y P2 son las presiones absolutas de inyección y de descarga de gas, respectivamente.
2C2µPrqrzm C1zrGf(P1-P2)(P1+P2+2b) (18)
Nota: Datos de Jones 16 y Geertsma17.
Figura 6-3 Resistividad Inercial, β, como Función de Permeabilidad
Para realizar esta integración, se supuso que z en la ecuación 17 era reemplazable por su valor medio, zm. Los factores de desviación de gas zm y zr se calculan en la presión media de poros, Pm (ver ecuación 20 mas adelante) y la presión absoluta de referencia, P1, en la cual se mide qr, respectivamente, y en la temperatura del gas, Gf es un factor geométrico que tiene la dimensión de longitud. Se indica el factor específico para cada una de las diferentes configuraciones de flujo en las secciones siguientes a la 6.3.1. El factor de deslizamiento de gas de Klinkenberg, b, debe determinarse con múltiples mediciones, o con una correlación. Si es obtenido con una correlación, esto debe indicarse claramente. Para obtener b, del cual se calcula rb utilizando la ecuación 13, la permeabilidad aparente del gas, kg, se mide un mínimo de tres (y preferiblemente por lo menos cuatro) diferentes presiones medias de poros. Cada permeabilidad se calcula con: kg =
Y es trazada contra el recíproco de la presión media de poros ilustrado en la Figura 6-5. La presión media de poros es definida como:
Pm = 1/2 (P1+P2) (20) La intersección de este trazado gráfico es k∞, y su pendiente es igual a bk∞, de acuerdo con la relación Klinkenberg: kg = k∞(1 + b/Pm) (21) La pendiente y la intersección pueden encontrarse utilizando la regresión lineal. Es esencial asegurar que la resistencia inercial es insignificante para cada medición. De otra manera, el kg será bajo, especialmente con valores Pm mas altos. La partida de linealidad ofrece una autoverificación de consistencia.
2C2µPrqrzm C1zrGf(P1-P2)(P1+P2) (19)
A menudo, para mediciones rutinarias de permeabilidad, se reportan únicamente las permeabilidades de gas (sin corrección de deslizamiento de gas), de la manera calculada en la ecuación 19. Para estas mediciones, es esencial reportar el gas utilizado y su presión media de poros. De lo contrario, las permeabilidades reportadas no están completamente especificadas.
Nota: Para aire a 72ºF.
Figura 6-4 - Máximas Gradientes de Presión General para Uso de la Ecuación Darcy
Figura 6-5 - Tipo de Gráfica Utilizado para Obtener el Factor Klinkenberg de Deslizamiento de gas, b
6.2.2 Ecuaciones de Permeabilidad de Régimen Permanente para Líquidos
Los líquidos tienen compresibilidades mas bajas, densidades mas altas, y viscosidades mas altas que las de gas. Para mediciones de régimen permanente que impliquen pequeñas diferencias en presión, los líquidos pueden ser tratados como fluidos incompresibles. Debido a las densidades mas altas de líquidos, los efectos de la gravedad normalmente no pueden ignorarse en las mediciones de permeabilidad, excepto por el flujo horizontal. La partida de la Ley de Darcy rara vez es un problema con los líquidos. Los viscosidades mas altas resultan en flujos volumétricos significantemente mas bajos que aquellas observadas para gases en las mismas gradientes de presión. Pero sus densidades mas altas compensan en cierto modo el efecto de viscosidad en relación con la resistencia inercial. Ver
A.6.8.1 para el cálculo de los valores ∆b/L máximos permitidos a usar para mediciones de permeabilidad de líquidos cuando se utiliza la Ley de Darcy. El recorrido libre medio entre las moléculas de líquidos corrientes es tan pequeño que no se observa deslizamiento. Sin embargo, los líquidos - especialmente las soluciones acuosas - pueden reaccionar con componentes de roca porosa, alterando su permeabilidad. También, se debe tener cuidado para evitar movilizar finos en una roca con altos flujos de líquido. La Ley de Darcy, incluyendo los efectos de gravedad, para el flujo Stokes de líquidos a través de medios permeables puede expresarse en términos generales como: vs = q = -C1k (dp - pg dz A C2µ (ds C4 ds) (22)
)

Donde: s = distancia en la dirección de flujo vs = flujo volumétrico (volumen de flujo a través
de una unidad de área del medio poroso por unidad de tiempo)
z = coordenada vertical (aumentando hacia abajo)
ρ = densidad del fluido dp/ds= gradiente de presión en s en el punto
referido µ = viscosidad del líquido k = permeabilidad del medio q = régimen de volumen de flujo A = área de la sección transversal perpendicular
a las líneas de flujo C1, C2 y C4 son constantes, encontradas en la Tabla 6-
1, que hacen que las unidades sean consistentes.
6.2.2.1 Casos Especiales para el Flujo de Régimen
Permanente de Líquidos Incompresibles La ecuación 22 puede reducirse a formas mas sencillas bajo condiciones especiales de flujo. Algunas de las mas importantes son las siguientes. 6.2.2.1.1 Flujo Horizontal de Líquidos Debido a que no hay componente vertical de flujo, dz/ds = 0, y la integración de la ecuación 22 resulta en: k = C1Gf(p1-p2) (23)
C2qµ
Donde Gf es un factor geométrico que tiene la dimensión de longitud. Los factores para flujo axil, transversal, o radial se dan en las ecuaciones 27, 29, o 30, respectivamente, después de 6.3.1. 6.2.2.1.2 Flujo Vertical de Líquidos Surgen dos sub-casos, dependiendo si el flujo es descendente o ascendente, y dz/ds es igual a +1 o -1, respectivamente: a. Flujo descendente con una cabeza impulsora h
(definida en la Figura 6-6):
k = C1Aρg(h+L) (24)
C2 C4qµL
Donde L es la longitud de la muestra y h es la altura de la cabeza impulsora. Si la cabeza impulsora, h, es cero, tenemos un flujo descendente libre, en el cual la velocidad de flujo es independiente de la longitud de la muestra. b. Flujo ascendente con cabeza impulsora, h,
(definida en la Figura 6-7):
k = C1Aρgh (25)
C2 C4qµL
Si se logra un flujo ascendente de régimen permanente con una diferencia de presión impuesta, ∆p, la cual se mide con un transductor de diferencia de presión con ambos puertos en la misma elevación (de la manera indicada en 6-8) y ambas líneas
transductivas completamente llenas con el mismo líquido, entonces:
k = C1A∆p (26)
C2 qµL
Después de lograr el régimen permanente (i.e., la ∆p y la velocidad de flujo son invariables con el tiempo), las válvulas de entrada y salida deben cerrarse simultáneamente, y se debe permitir que el transductor de diferencia de presión alcance un equilibrio. La lectura de equilibrio debe ser cero. Si no es cero, esta lectura debe restarse de la lectura de régimen permanente. 6.2.3 Disminución de Permeabilidad en Esfuerzos
Netos Elevados La permeabilidad de la roca de yacimientos disminuye con esfuerzos netos crecientes. El esfuerzo neto para permeabilidad usualmente es definido como la diferencia entre esfuerzo restrictivo y presión media de poros. La Figura 6-9 ilustra la reducción típica de permeabilidad con esfuerzos hidrostáticos netos crecientes. Las reducciones en porcentaje son por lo general mayores con tapones de permeabilidades mas bajas. Por ejemplo, las permeabilidades sin esfuerzo de tapones 6-2, 8-2 y 11-2 en la Figura 6-9 son alrededor de 700, 40, y 10 milidarcys, respectivamente. La permeabilidad de una arena de gas compacta (elg., 0.01 milidarcy o menos a cero esfuerzo) puede reducirse al 10 por ciento o menos de su permeabilidad sin esfuerzo con altos esfuerzos compresivos netos. 6.3 APLICACIONES PRÁCTICAS PARA
MEDICIONES DE PERMEABILIDAD DE RÉGIMEN PERMANENTE
Se describen varias configuraciones prácticas para la medición rutinaria en régimen permanente, primero para gases, luego para líquidos. Cada uno puede tener ventajas y limitaciones específicas (e.g., velocidad, costo, precisión, margen de permeabilidad que puede acomodarse, niveles de esfuerzo alcanzables, mediciones direccionales, susceptibilidad a error, etc.) que son explicadas en detalle. Se presentan ciertas precauciones para evitar peligros graves. Aunque calculamos la permeabilidad de una muestra por último, la cual es una medida de su conductividad fluida, es la conductancia de la muestra que impone su comportamiento de flujo fluido. La conductancia depende de la longitud del recorrido y el área disponible para flujo, como también la permeabilidad de la muestra. Por lo tanto, el margen de permeabilidad mensurable puede extenderse en algunas configuraciones de flujo con la elección sensata de dimensiones de muestra. Por ejemplo, para mantener velocidades razonables de flujo con

flujo axil cuando las permeabilidades son muy altas, se pueden preparar muestras largas de pequeño diámetro. A la inversa, para muestras de baja permeabilidad, los tapones cortos de diámetro mas grande son convenientes. Otras condiciones siendo iguales, la permeabilidad máxima que se puede medir con precisión con un tapón de tres pulgadas de longitud es tres veces mas grande que la que se puede medir en otro tapón idéntico de una pulgada. Las muestras presentadas para medición a menudo varían en permeabilidad desde menos de 0.001 a mas
de 30.000 milidarcys - una relaciona de mas de 3x107. Para acomodar esta variación, aparatos de medición y/o control de múltiples márgenes de presión y velocidad de flujo deben ser adecuados para ser insertados en un permeámetro, o para elegir entre múltiples aparatos incorporados. Para simplificar, se ilustra únicamente un conjunto de aparatos para cada configuración a continuación.
Figura 6-6 - Flujo Descendente de Líquido con Cabeza Impulsora, h
Figura 6-7 - Flujo Ascendente de Líquido con Cabeza Impulsora, h
Figura 6-8 - Flujo Ascendente de Líquido con Diferencia de Presión Impuesta, ∆p
Figura 6-9 - Reducción de Permeabilidad con Esfuerzo Hidrostático Neto Creciente
6.3.1 Gases 6.3.1.1 Flujo Axil de Gases de Régimen
Permanente Se muestra un arreglo gráfico para el flujo axil de gases en la Figura 6-10. Se monta un tapón de núcleo cilíndrico limpio y seco o una muestra de núcleo entero de longitud L y diámetro D en un portamuestras. El portamuestras contiene una manga flexible con el fin de hacer una junta hermética al gas en las paredes cilíndricas de la muestra, y para aplicar esfuerzos restrictivos radiales. El esfuerzo axil puede transmitirse a la muestra aplicando fuerza a uno o ambos tapones extremos, por medios mecánicos, neumáticos, o hidráulicos. Si las magnitudes del esfuerzo radial y el esfuerzo axil son iguales, se dice que la muestra es isostáticamente o hidrostáticamente esforzada. Si las magnitudes son desiguales, la muestra es biaxilmente esforzada. Cuando se utilizan gases a presiones relativamente bajas (hasta unos cientos psig), los efectos de la gravedad son insignificantes, y el portamuestras puede orientarse horizontal o verticalmente. Los dos tapones extremos tienen un puerto axil para transportar gas hacia adentro y hacia fuera de la muestra. Cada uno debe tener ranuras radiales y circulares u otros medios para distribuir gas a toda su superficie de inyección, y para recolectar gas de todas partes de su superficie de salida. Cada tapón extremo, preferiblemente, también contiene un segundo puerto para medir las presiones corriente arriba y corriente abajo, P1 y P2, respectivamente. La presión corriente arriba y la diferencia de presión, ∆p, ilustrada en la Figura 6-10, o la presión corriente abajo y la diferencia de presión. Sin embargo, las líneas para conectar los aparatos que miden presión pueden ser derivadas de las líneas de flujo cerca de los puertos axiles, siempre que sean lo suficientemente grandes para no causar caídas de presión significativas entre cada conexión y la superficie correspondiente de la muestra. Es esencial que la ramificación de cada conexión (no el recorrido) sea conectada al transductor de presión. De lo contrario, los efectos de presión dinámica pueden influir sobre las medidas de presión.
Observe que ∆p, utilizado en todo este documento siempre es un número positivo. Por lo tanto, es igual a P1-P2 o a p1-p2. De la manera indicada anteriormente, una P mayúscula indica una presión absoluta, y una p minúscula es una diferencia de presión o una presión manométrica. La línea de salida puede ventilarse directamente a la presión atmosférica (cuando el medidor de corriente queda corriente arriba de la muestra), conectada a un aparato medidor de la velocidad de flujo, o a un regulador de contrapresión para propósitos de crear presiones medias de poros elevadas. En el segundo caso, el medidor de corriente puede estar corriente arriba de la muestra o corriente abajo del regulador de contrapresión. La velocidad de flujo volumétrico, qr, puede medirse en la presión corriente arriba o corriente abajo, o en alguna otra presión, que en todos los casos es simbolizado como Pr, una presión absoluta. Se supone que la temperatura en la cual se mide la velocidad de flujo es igual a la temperatura de gas corriente. Alternativamente, el grupo (qrPr)/(zrTr), el cual es proporcional a la velocidad de flujo de masa, puede determinarse con un medidor de corriente de masas. Ver 6.6.2 para la calibración de medidores de flujo de masa y la aplicación de resultados en el cálculo de permeabilidad. 6.3.1.1.1 Aparatos 6.3.1.1.1.1 Bajo Esfuerzo Restrictivo La Figura 6-11 muestra un diagrama de flujo simplificado para las mediciones de baja presión. La Figura 6-12 muestra un soporte de núcleos tipo Hassler de baja presión, en el cual se aplica típicamente un esfuerzo restrictivo radial de 400 psi y un esfuerzo axil de magnitud desconocida en un tapones de núcleos de 3 pulgadas (19- a 76- milímetros) de longitud con un diámetro de 1.0- o 1.5- pulgadas (25- o 38-milímetros). Estas dimensiones y esfuerzos se han vuelto virtualmente estándar en el análisis convencional de núcleos de bajo esfuerzo. Se transmite un esfuerzo radial al tapón cuando se aplica una presión neumática o hidráulica al espacio anular entre la manga de caucho y el soporte de núcleos. El

esfuerzo axil es típicamente transmitido desde un tornillo que restringe el tapón del extremo mas bajo. Normalmente, este esfuerzo no se mide, y es algo dependiente del operador. Los soportes de núcleos tipo Fancher (ver página 35 de API RP 40, 1960) no son recomendados debido al sellado dependiente del operador y no reproducible y los esfuerzos restrictivos producidos. 6.3.1.1.1.2 Esfuerzo Restrictivo Elevado La Figura 6-13 ilustra un soporte de núcleos para aplicar esfuerzos hidrostáticos, típicamente hasta un máximo de 10.000 psi. Por seguridad se recomienda una minimización de difusión de gas a través de la manga, y para prevenir corrosión, se recomienda aceite hidráulico o un aceite mineral pesado para generación de esfuerzo. El área de la sección transversal del tubo que conecta el tapón extremo interno con el recipiente a presión externo reduce el esfuerzo axil en este soporte de núcleos. Para tubería O.D. de 1/8 pulgada, la reducción no es grande, pero con diámetros mas grandes, el esfuerzo axil puede ser significativamente menor que el esfuerzo radial generado. Para operación a temperatura ambiente, se recomienda un durómetro 40 a 70 (dureza Shore A), nitrilo (Buna-N) caucho en grosores de manga de 0.125 a 0.25 pulgadas. Un menor durómetro y grosor pueden utilizarse para presiones menores. Ocurre un flujo frío con mayor esfuerzo y temperatura. En esfuerzos por encima de 6.500 psi, los durómetros superiores a 70 son deseables para extender la vida de la manga. La Figura 6-14 ilustra un soporte de núcleos de alta presión que puede utilizarse para aplicar cargas hidrostáticas o biaxiles a los tapones de núcleo. El esfuerzo axil es generado por un intensificador de esfuerzo. Una presión neumática o hidráulica empuja sobre un pistón de diámetro mas grande que el del tapón extremo. La fuerza resultante es transmitida por el tapón extremo inferior a la muestra. La presión requerida es inversamente proporcional a la relación de áreas. Por ejemplo, si el pistón mas grande tiene un área 10 veces la del tapón extremo, una presión de 600 psig aplicada al pistón grande transmitiría un esfuerzo de 6.000 psi al tapón de núcleo. Si el
esfuerzo axil es igual a la presión hidráulica aplicada a la manga de caucho, entonces el tapón de núcleo es hidrostáticamente esforzado. De lo contrario, es biaxilmente esforzado. Además de su capacidad de proporcionar esfuerzos desiguales, la mayor ventaja de este tipo de soporte de núcleos es que no se requiere un desensamble para cambiar de muestras. La presión restrictiva es descargada (y a menudo se aplica un aspirador para agrandar la manga), el tapón extremo inferior es retirada, se remueve la muestra anterior, y se inserta la muestra nueva. Se deben observar las estrictas precauciones de seguridad tanto en el diseño como en la operación de los equipos de alta presión. Ninguna de las partes que contienen altas presiones pueden sufrir deformación permanente a presiones al menos iguales a 1.5 veces la presión máxima de operación, o de lo contrario, deben cumplir con los códigos locales, nacionales, o internacionales que aplican para recipientes de alta presión. 6.3.1.1.2 Cálculos Se calcula la permeabilidad de gas, kg, con la ecuación 19 (ver 6.2.1.3). Si se hacen múltiples mediciones a diferentes presiones medias de poros, se obtienen k∞ y b de un diagrama Klinkenberg (ver Figura 6-5), o una regresión lineal descrita arriba. Si se encuentra b con una correlación, entonces k∞ se calcula con la ecuación 18. Estas afirmaciones también aplican para todas las demás configuraciones de flujo de gas indicadas mas adelante. El factor geométrico para el flujo axil en las ecuaciones 18 y 19 es: Gf =
Tradicionalmente, cuando un tapón de núcleo o un núcleo entero es cortado para que su eje quede paralelo a las superficies de asiento o la dirección de permeabilidad, la permeabilidad medida es llamada permeabilidad "horizontal", kH. De igual manera, una permeabilidad "vertical", kv, es obtenida cuando el eje de la muestra es perpendicular a las superficies de asiento.
πD2 4L (27)
Figura 6-10 - Gráfico del Aparato de Permeabilidad para el Flujo Axil de Gas
Figura 6-11 - Diagrama de Flujo Simplificado para Baja Presión, Mediciones de Permeabilidad del Flujo Axil de Gas
Figura 6-12 - Soporte de Núcleos de Baja Presión Tipo Hassler
Figura 6-13 - Soporte de Núcleos de Alta Presión para Tensión Hidrostática
Figura 6-14 - Soporte de Núcleos de Alta Presión para Carga Hidrostática o Biaxil
6.3.1.1.3 Ventajas Las ventajas de este método incluyen: a. El método experimental es sencillo y ha sido un
modelo de la industria durante muchos años. Por
consiguiente, existe una gran base de datos históricos para comparación directa.
b. El gas no reacciona con roca, y las muestras son limpias al final de las mediciones.
c. La técnica se presta para una amplia variedad de equipos. Con un soporte de núcleos de baja presión tipo Hassler, manómetros, aparatos de flujo volumétrico de tipo orificio, y la adquisición

manual de datos, los costos capitales son bajos. Sin embargo, se recomienda un aparato mejorado que incluye un soporte de núcleos hidrostático o biaxil de alta presión y sistemas automatizadas de control y de adquisición de datos.
d. Los medidores de corriente y los transductores de presión pueden intercambiarse fácilmente para extender el margen de permeabilidad.
6.3.1.1.4 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. El limite práctico inferior de permeabilidad con
esta técnica es alrededor de 0.1 milidarcy. En rocas de baja permeabilidad el tiempo requerido para lograr un régimen permanente se vuelve largo, y las mediciones de flujo se pueden volver algo inexactos.
b. La desventaja principal de mediciones de bajo esfuerzo sin corrección de deslizamiento (típicamente, esfuerzo restrictivo radial de 400 psi, y esfuerzo axil desconocido) es que sobrestiman la permeabilidad de yacimientos en el sitio, especialmente en muestras compactas. La corrección de deslizamiento Klinkenberg puede hacerse con mediciones en diferentes presiones medias de poros, pero las mediciones rutinarias normalmente se hacen en una sola presión, y esta presión rara vez es reportada. Si no se reporta esta presión, es difícil normalizar la permeabilidad, y no está especificada completamente. Se debe reportar el gas utilizado y la presión media de poros. Si se hace una corrección de deslizamiento de gas con una correlación, se debe reportar este hecho.
c. La consecución del flujo Stokes, especialmente con muestras de alta permeabilidad, pueden ser difíciles de averiguar con mediciones de una sola punta. Las mediciones deben revisarse según la sección 6.2.1.3 para asegurar que no se ha pasado de la máxima ∆p/L permitida.
d. Es crítico cortar extremos perfectamente cuadrados en los tapones de núcleos, especialmente con altos esfuerzos restrictivos. Un punto alto recibirá un esfuerzo desproporcionadamente alto, lo cual podría aplastar esta porción del tapón. Esto, por supuesto, aplica para todas las técnicas que impliquen tapones de núcleo cilíndricos.
6.3.1.2 Mediciones de Permeabilidad de Sondas
de Régimen Permanente con Gases Cuando el gas fluye del extremo de un tubo de diámetro pequeño (o "sonda") sellado contra las superficies de una muestra de núcleo entero tajada o sin tajar, o un afloramiento de roca permeable, el patrón de flujo es algo similar al del flujo hemisférico (ver Figura 6-15). Esta configuración es utilizada mucho para hacer mediciones de permeabilidad de gas poco costosas, no destructivas sin esfuerzo dentro de reducidos radios de influencia. Si los núcleos no están limpios y secos, una medición
produce una permeabilidad efectiva en alguna saturación de aceite y agua desconocida. Estas mediciones pueden hacerse a espacios cercanos para determinar la variación de permeabilidad en una formación heterogénea. Los instrumentos para tomar estas medidas por lo general son llamados "permeámetros exploradores", o "mini-permeámetros". Una verdadera geometría de flujo hemisférico requeriría (a) una muestra isotrópica grande y homogénea con una superficie superior plana, (b) una cavidad hemisférica debajo de esta superficie, inmediatamente debajo de la sonda, con un radio, ri, igual al radio interior del ocluidor de la sonda, (c) una extensión muy grande (infinita) de la muestra en todas las direcciones debajo de la superficie de contacto, y (d) un ocluidor en todas partes de esta superficie excepto por encima de la cavidad hemisférica. Como estas condiciones no son nada prácticas, Goggin, et al.19 realizó numerosos cálculos para producir factores de flujo geométrico sin dimensiones que compensa la falta de una cavidad hemisférica en la superficie de una pequeña muestra finita, y un ocluidor de sonde de un pequeño radio finito interior y exterior. Estos factores sin dimensiones son ilustrados en la Figura 6-16 como función de la relación del radio exterior, ro, del ocluidor de la sonda con su radio interior, ri. Para el verdadero flujo hemisférico semi-infinito, G0 sería igual a 2π. La relación mostrada en la Figura 6-16 es para muestras con una superficie superior plana y la extensión y profundidad lateral suficiente para que los límites exteriores no tengan influencia sobre las mediciones de permeabilidad. Para propósitos prácticos, los factores mostrados pueden utilizarse con poco error sobre muestras que tienen una profundidad al menos igual a cuatro veces el radio interior del ocluidor de la sonda, y donde el límite lateral mas cercano es al menos 4ri del eje de la sonda. Además, se presenta poco error si se coloca la sonda sobre una superficie cilíndrica de la muestra, siempre que se haga una junta hermética al gas positiva, el radio de la muestra es de al menos 12ri, y se observen las restricciones de límites indicadas para la muestra. Ver Goggin, et al.19 para los factores geométricos sin dimensiones relacionados con los límites mas cercanos. 6.3.1.2.1 Aparatos Para mediciones de la permeabilidad de gas (ver Figura 6-15), la sonda debe tener un ocluidor de caucho blando que tenga ri y ro dimensionalmente estables (o reproducibles) cuando se aplique presión sobre la muestra, y medios para producir una fuerza de sellado constante y reproducible. Esta fuerza de sellado debe ser adecuada para prevenir escapes de gas en alguna parte de la superficie de contacto entre el ocluidor y la roca. El radio interior del ocluidor es típicamente de 2 a 3 milímetros, y la relación de su radio interior al exterior es típicamente de 1.5 a 2.5. Los radios y las relaciones mas pequeñas producen mas resolución con relación a la heterogeneidad a pequeña escala, y se utilizan los valores mas grandes para obtener mas promediación de permeabilidad. Estas dimensiones también deben tener una escala común con la aspereza de la textura de la roca.

Se posiciona la sonda arriba de un punto deseado en una muestra, el cual debe ser relacionado con la profundidad y posición relativa al eje del núcleo o algún otro punto de referencia. Se baja la sonda a la superficie de la roca, y se hace oclusión contra ella. Se envía gas a la presión P1 de la sonda a la muestra. Este sale de la muestra a la presión atmosférica. Se debe tomar medidas para regular la presión dentro de la muestra, y para medir la presión y la velocidad volumétrica del flujo de gas a esta presión. Se selecciona el margen de los medidores de corriente basado en la permeabilidad investigada. Se registra la presión y la velocidad de flujo cuando ambos sean constantes. 6.3.1.2.2 Cálculos De estas mediciones, mas las dimensiones de los ocluidores, la presión atmosférica, y la viscosidad de gas, kg puede calcularse de la Ecuación 19, donde, si p1 es la presión manométrica corriente arriba: P1 = p1 + Pa, P2 = Pa, Pr = P1, Y Gf = Gori (28) La permeabilidad obtenida es cargada mas fuertemente (en muestras no-homogéneas) dentro de un radio igual al radio del ocluidor interior, y progresivamente menos con radios crecientes. Debido al pequeño volumen de roca investigada, la permeabilidad a escala macro debe obtenerse con las técnicas apropiadas de promediación. 6.3.1.2.3 Ventajas Las ventajas de este método incluyen: a. El método no es destructivo en el sentido
que no hay necesidad de cortar los tapones de núcleo. Es rápido y tiene bajos costos, así permitiendo el muestreo de alta densidad.
b. La sonda investiga solo un pequeño volumen de roca. La medición es apropiada para la investigación de la variación de permeabilidad espacial en núcleos que contienen laminaciones delgadas y heterogeneidad a pequeña escala. Se puede medir la variación de permeabilidad direccional alrededor de la circunferencia de un núcleo entero.
c. El aparato puede diseñarse para el servicio portátil de pozos, y para uso sobre afloramientos, siempre que se pueda lograr una oclusión adecuada.
d. Con múltiples medidores de corriente, se puede medir un margen de permeabilidad de 1 a 10.000 milidarcys.
6.3.1.2.4 Precauciones y Limitaciones
Las precauciones y limitaciones de este método incluyen: a. Longitudes mínimas de recorrido de flujo en
una muestra (de ri a ro) varían de 0.2 a 0.4 cm. para ocluidores típicos de puntas de sonda. La mayoría de la caída total de presión ocurre dentro de esta distancia, la cual es un orden de magnitud mas corto que la longitud del recorrido para el flujo axil a través de la mayoría de los tapones de núcleo. La presión de inyección, p1, debe ser reducido correspondientemente para evitar una resistencia inercial de flujo significativo. Aunque Goggin, et al.19 propuso una "corrección de flujo de alta velocidad" utilizando una correlación para obtener β, esta corrección solo puede aproximarse con mediciones de una sola velocidad, porque β varía por lo menos + un orden de magnitud en una permeabilidad dada para la mayoría de los márgenes de permeabilidad. Sin embargo, esta corrección es mas pequeña entre mas baja sea la presión de inyección.
b. Se aplica casi cero esfuerzo a la roca durante una medición. Por lo tanto, las permeabilidades tienden a ser muy optimistas, especialmente en muestras compactas. Si no se hacen las correcciones Klinkenberg (con una correlación), la permeabilidad se sobrestima aún mas, especialmente en roca de baja permeabilidad.
c. A menos que la punta de sonda sea restringida lateralmente, y sea esencialmente plana en el fondo (en la condición sin esfuerzo) su factor geométrico puede ser muy sensible al esfuerzo de sellado aplicado. Este esfuerzo, por lo tanto, debe ser altamente reproducible.
d. Aunque la técnica puede utilizarse sobre segmentos de núcleo entero, se recomienda el tajado para remover la invasión de sólidos de barro y otra contaminación de superficie, y para tener una superficie plana y lisa para el sellado de punta de sonda, lo cual es difícil de garantizar de lo contrario.
e. Si la superficie del núcleo no está seco, la permeabilidad medida puede ser erróneamente baja debido a los efectos severos de permeabilidad relativa. La permeabilidad puede aumentarse con el tiempo, y luego estabilizarse cuando los líquidos móviles se evaporan o se alejan de la punta de la sonda.
f. Los ocluidores de punta de sonda deben revisarse con regularidad para verificar que no haya desgaste, deterioro, y granos de arena incrustados. Además, deben cambiarse cuando sea necesario. Es útil realizar pruebas de escapes periódicas sobre una superficie impermeable lisa y plana para detectar defectos o inclusiones.

g. Debido al pequeño volumen de roca investigada por esta técnica, las permeabilidades en escala macro deben obtenerse utilizando las técnicas apropiadas de promediación.
6.3.1.3 Flujo Transversal de Gases en Régimen
Permanente Esta técnica se utiliza principalmente para medir la permeabilidad direccional "horizontal" en (pero no limitado a) las muestras de núcleo entero. El gas fluye desde una malla de entrada que cubre un ángulo subtendido θ y toda la longitud L de una muestra de núcleo entero (o tapón de núcleo), a través de la muestra, y hasta una malla de salida similar colocada diametralmente a través de la malla de entrada (ver Figura 6-17). El patrón de flujo es complejo, y el área normal en las líneas de corriente para el flujo de gas es variable por todo el recorrido de flujo. Collins20, utilizando una transformación de representación conforme, calculó un factor geométrico sin dimensiones, Gθ. Este se muestra en la Figura 6-18, y es función del ángulo subtendido por las mallas. La permeabilidad se mide normalmente en dos direcciones - una dando el valor máximo (usualmente en la dirección de ruptura principal o la dirección de permeabilidad), y la otra a 90 grados al máximo. 6.3.1.3.1 Aparatos y Cálculos El sistema y las mediciones de flujo requeridos para el cálculo de las permeabilidades de flujo transversal son similares a aquellos para el flujo axil en régimen permanente. La Figura 6-19 muestra un soporte de núcleos tipo Hassler para las mediciones de permeabilidad transversal. Se permite al gas entrar y salir de la muestra a través de aberturas diametralmente opuestas a través de la manga. Estas se conectan a dos mallas tejidas que cubren toda la longitud de la muestra y un ángulo subtendido conocido sobre su circunferencia. Si el ángulo subtendido de cada malla es de 90 grados, entonces el factor geométrico sin dimensiones, Gθ, es igual a 1.0. Los materiales de sellado no deben penetrar en el tejido ni restringir el flujo. Ambos extremos de la muestra son sellados con discos de caucho blando. Figura 6-17 - Configuración de Flujo Transversal para
Gases o Líquidos
Figura 6-18 - Factor Geométrico Sin Dimensiones para
El Flujo Transversal
Figura 6-19 - Soporte de Núcleos Tipo Hassler para Mediciones de Permeabilidad Transversal
El factor geométrico dimensional a usar en las Ecuaciones 18 o 19 para flujo transversal es: Gf = Gθ (29)
L
Una muestra de núcleo entero puede colocarse en un soporte de núcleos grande para mediciones de permeabilidad "vertical" longitudinal, antes o después de las mediciones transversales. Los cálculos son aquellos para flujo axil (ver 6.3.1.1.2). 6.3.1.3.2 Ventajas Las ventajas de este método incluyen: a. Las mediciones sobre muestras de núcleo
entero implican grandes volúmenes de roca, así permitiendo una considerable promediación de permeabilidad. Esto minimiza el efecto de heterogeneidad a pequeña escala.
b. En muestras orientadas de núcleo entero, se pueden hacer mediciones ortogonales para determinar la permeabilidad direccional.
6.3.1.3.3 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. Una muestra de núcleo entero muy rara vez es
cortada precisamente normal o paralela a las superficies de la roca. Por lo tanto, las permeabildades "verticales" y "horizontales" definidas de manera convencional no podrán obtenerse exactamente con mediciones longitudinales y transversales, en estas muestras.
b. Las muestras mas grandes se demoran mucho mas para limpiar, secar y llegar a régimen permanente. Por lo tanto, los experimentos son mas demorados y mas costosos que aquellos sobre tapones de núcleos.
c. Se deben observar las mismas precauciones relacionadas con el deslizamiento de gas y la resistencia inercial, indicadas en 6.3.1.1.4 para el flujo axil.
6.3.1.4 Flujo de Gases Radial en Régimen
Permanente La Figura 6-20 muestra la configuración de flujo para el flujo radial en régimen permanente. La superficie superior y la inferior de una muestra cilíndrica limpia y seca de longitud L y radio exterior re en la cual se corta un diámetro interior axil del radio rw, son sellados en todas partes excepto por encima del diámetro interior central. El gas a la presión P1 fluye desde la superficie cilíndrica sin sellar radialmente hasta el diámetro interior central, y sale a una presión P2. De lo contrario, el sistema y las mediciones de flujo son iguales que los del flujo axil. En principio, se puede invertir la dirección de flujo para que el gas sea inyectado en el diámetro interior central a la presión P, y salga a través de la superficie cilíndrica exterior a la presión P. Sin embargo, por lo general no se recomienda una inyección al pozo porque su área de flujo mas reducido es mas propenso a daños por rastros de partículas en los fluidos inyectados. 6.3.1.4.1 Aparatos y Procedimiento

El permeámetro radial de diámetro completo, mostrado en la Figura 21, consiste de tres partes; la célula, que es lo suficientemente grande para mantener una presión de entrada uniforme; un pistón para aplicar la fuerza de sellado; y el equipo de placa flotante que consiste de una placa fija inferior, un balín de pivote, tres resortes a 120 grados cada uno, y la placa flotante superior. Se coloca el núcleo sobre un empaque de caucho sólido de 1 pulgada de grosor pegado a la placa flotante inferior. Luego se levanta el núcleo contra la tapa cerrada, el orificio central del núcleo alineado con el del empaque superior. Mientras se incrementa la presión del pistón, la placa flotante inferior se ajusta automáticamente si los extremos no son paralelos. Para verificar si existe un escape de aire entre los extremos del núcleo y los empaques de caucho, se incrementa la presión del pistón. Una disminución en la velocidad de flujo indica que ha existido un escape. Este prueba se repite hasta que no se note ningún cambio en la velocidad de flujo. 6.3.1.4.2 Cálculos Las velocidades de flujo y las caídas de presión son medidas y utilizadas de manera igual a las mediciones de flujo axil. Se logra el régimen permanente cuando ambos se vuelven invariables con el tiempo. La permeabilidad, kg, se calcula con la Ecuación 19, en donde el factor geométrico para flujo radial es: Gf = 2πL ln[re/rw] (30)
La permeabilidad promedio de las muestras no homogéneas es cargada mas fuertemente por la permeabilidad en el radio interior del pozo, rw (donde la gradiente de presión es mayor), y progresivamente menos en radios mas grandes. 6.3.1.4.3 Ventajas La permeabilidad medida se promedio por toda la longitud de la muestra y en todas las direcciones radiales. 6.3.1.4.4 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. La permeabilidad es críticamente dependiente
del radio interno del diámetro interior del pozo. Pequeños errores en su valor, especialmente con orificios de diámetros pequeños, causarán grandes errores de permeabilidad. De manera similar, las rupturas inducidas o los daños de superficie cera de este orificio afectarán la permeabilidad aparente enormemente.
b. La preparación de muestras para esta medición no es fácil. Por lo tanto, la prueba tiende a ser algo mas costoso que las configuraciones alternativas.
c. Los esfuerzos axiles no son balanceados por los esfuerzos radiales. El método no se adapta
fácilmente a la aplicación de esfuerzos elevados para la medición de una permeabilidad realista de yacimientos in situ.
d. Una gran parte del material de núcleo es alterada por el orificio central requerido para esta técnica, y la muestra usualmente no puede utilizarse para otras pruebas.
Figura 6-20 - Esquema para el Flujo Radial de Entrada
6.3.2 Líquidos Los aparatos de permeabilidad para líquidos en régimen permanente tienen muchas características en común con aquellos para gas. Las diferencias entre líquidos y gases que pueden requerir modificaciones de equipos son: la viscosidad y densidad de líquidos son considerablemente mayores y la compresibilidad es mucho menor que las propiedades correspondientes para gas. Además, los líquidos (especialmente soluciones acuosas que contienen sales) pueden ser corrosivos, apoyar la actividad microbiana, y reaccionar con los componentes de arcilla y minerales en la roca. Se observa a menudo que la permeabilidad a los líquidos es menor que la permeabilidad de gas con corrección de deslizamiento. Esto puede ser debido en parte a la interacción líquido-arcilla (especialmente con soluciones acuosas), migración de finos con altas velocidades de flujo líquido, o una saturación líquida incompleta. 6.3.2.1 Mediciones de Permeabilidad de
Líquidos en Régimen Permanente con un ∆p Impuesto
6.3.2.1.1 Aparatos Este tipo de aparato de permeabilidad no depende de las cabezas de líquido para la entrega de fluidos. Es compuesto de tres unidades separadas: un portamuestras, un sistema de medición de presión, y un sistema de distribución de fluidos. Excepto por la resistencia a corrosión y la compatibilidad de la manga de caucho con un líquido específico, el portamuestras, las líneas de distribución, y los equipos de medición de presión para las mediciones de permeabilidad de líquidos no son diferentes a los sistemas para gases. La mayor densidad de los líquidos requiere mayor atención a las cabezas de líquido. Estos normalmente se cancelan con el flujo horizontal, siempre que ambas aberturas del transductor de diferencia de presión utilizado estén a la misma elevación, y que todas las líneas de medición de presión estén completamente llenas con el mismo fluido que el fluido corriente, y que estén a la misma temperatura. En este caso, cuando termina el flujo a través de la muestra, la lectura ∆p debería llegar a cero. Bajo estas circunstancias, la Ecuación 23 (ver 6.2.2.1.1), la cual aplica para flujo horizontal, también es válido para flujo vertical ascendente y descendente (ver comentarios después de la Ecuación 26 en 6.2.2.1.2). Debido a la viscosidad multiplicada por 50 (o mas) de la mayoría de los líquidos en comparación a la de gases, las velocidades de flujo de líquidos son correspondientemente menores en la misma gradiente

de presión. El sistema de distribución de fluidos es normalmente una fuente primaria velocidad de flujo constante o una fuente de presión contante que puede regularse para resultar en una velocidad de flujo constante. Las bombas volumétricas de desplazamiento positivo de un tiempo ofrecen el medio mas preciso para distribuir un líquido a una velocidad constante controlada. El uso de estas bombas excluyen la necesidad de recolectar los líquidos producidos de un núcleo para determinar la velocidad de flujo. Sin embargo, el volumen contenido en una bomba de un tiempo puede ser insuficiente para lograr el régimen permanente. Para eliminar los trastornos de presión y las discontinuidades de velocidad de flujo completamente, se recomienda dos bombas y un sistema de control sofisticado para un sistema de flujo de "paso único", y tres son convenientes para un sistema de flujo recirculatorio. Un logro lento de régimen permanente, siempre que no cambie la permeabilidad de la muestra debido a un taponamiento progresivo, interacción de arcilla o minerales, etc., se debe al alto almacenamiento compresivo en el sistema. El almacenamiento compresivo es el producto del volumen un la compresibilidad efectiva de cada componente en el sistema. La compresibilidad efectiva es la suma de las compresibilidades del fluido y el recipiente. La compresibilidad fluida puede ser reducido notablemente (si hay gas presente) aplicando una alta contrapresión en el sistema, así disolviendo las burbujas de gas. Se debe tener cuidado para remover el gas atrapado en el sistema, y para degasificar los líquidos aplicando un aspirador. La compresibilidad del recipiente se reduce haciendo los recipientes mas rígidos. Los volúmenes del sistema deben minimizarse donde sea posible. El uso de recipientes de transferencia (donde, por ejemplo, el aceite de una bomba desplaza una solución salina inyectada a un tapón de núcleo) al menos duplica el volumen, en comparación con el desplazamiento directo de solución salina de la bomba. Sin embargo, las consideraciones de corrosión pueden evitar el bombeo directo. Cuando el almacenamiento compresivo es casi cero y la permeabilidad permanece constante, el logro de un régimen permanente es virtualmente instantáneo. Cuando se utiliza una bomba de tipo cromatografía, el operador debe asegurar que su distribución sea probada en todo el alcance a emplear. Para calcular la permeabilidad, las velocidades de flujo deben medirse independientemente. Esto puede lograrse recolectando y pesando el líquido producido en un recipiente pesador sobre una balanza electrónica a intervalos frecuentes temporizados. Se calcula la velocidad de flujo volumétrico dividiendo la velocidad de acumulación de masa por la densidad del líquido. Los sistemas de presión constante utilizan una fuente de gas o líquido para desplazar el líquido degasificado de un recipiente y a través de la muestra. El líquido a desplazar es contenido en un acumulador de pistón o una bolsa plegable de caucho. El fluido que desplaza pasa a través de un regulador de presión. En el caso de las bombas de tipo cromatografía, la velocidad de flujo debe determinarse independientemente. 6.3.2.1.2 Procedimientos y Cálculos
El líquido a utilizar debe filtrarse bien a través de un filtro fino poco antes de usarse. Un filtro de 0.2 micrones removerá bacterias como también partículas sólidas. Una muestra de prueba completamente saturada debe cargarse cuidadosamente en un portamuestras apropiado, asegurando que no queda aire atrapado durante el proceso. Luego se aplican esfuerzos restrictivos. En la ausencia de otros datos de permeabilidad, se debe aplicar un ∆p bajo o una baja velocidad de flujo inicialmente para permitir que el operador haga una aproximación inicial de permeabilidad, y así fijar las condiciones de velocidad de flujo y caída de presión, y elegir los equipos apropiados para estas mediciones. Ver A.6.8.1 para determinar la máxima ∆p/L para evitar una resistencia inercial significativa. Sin embargo, esto por lo general no es un problema grave con líquidos. Se debe tener cuidado para evitar la movilización de finos de roca. Se debe recolectar y observar la corriente que sale para verificar si hay contaminación. Los esfuerzos restrictivos deben ajustarse para obtener el esfuerzo neto deseado. Se debe mantener un control estricto sobre la temperatura para asegurar una viscosidad constante, y para evitar cambios en la velocidad de flujo volumétrico con la expansión o contracción de líquido. Esto es especialmente crítico cuando se trata de recipientes grandes, acompañados por bajas velocidades de desplazamiento de una bomba volumétrica de desplazamiento positivo, con tapones de baja permeabilidad. Grandes variaciones en la velocidad de flujo pueden ocurrir en esta situación con pequeños cambios en temperatura sobre cortos intervalos de tiempo. El factor crítico es la velocidad de expansión volumétrica térmicamente inducida, ∆q:
∆q = βTV(dT/dt)
relativa a la velocidad de desplazamiento de la bomba. Donde: βT = coeficiente de expansión térmica volumétrica de líquidos, ºC-1 o ºF-1. V = volumen de líquido que se está desplazando. dT/dt = velocidad del cambio de temperatura Para ilustrar, suponga que una bomba volumétrica de desplazamiento positivo contiene 1.000 c3 de decano que se está inyectando a un tapón de baja permeabilidad a una velocidad de desplazamiento nominal de 1.000 cm3/hr, y que la temperatura del decano se incrementa a 0.002ºF/minuto, debido a cambios en la temperatura ambiente. El coeficiente de expansión térmica del decano es alrededor de 5.5 x 10-4 volúmenes por volumen por grado F. Por lo tanto, la expansión térmica de decano es de 5.5 x 10-4 x 1000 , 0.002 = 0.011 cm3/min. Con la velocidad de desplazamiento de la bomba de 0.0166 cm3/min., esto causa un error en la velocidad de flujo del 66 por ciento de la velocidad nominal! Si se incrementara la velocidad de desplazamiento por un factor de 10, el error se disminuiría al 6.6 por ciento. Además, si se redujera el volumen de la bomba a 10 cm3, el error

sería solo 0.066 por ciento. Otra manera de reducir la expansión volumétrica es incrementar la masa térmica de la bomba, y mejorar su aislamiento. Con un sistema de distribución con presión constante (en el cual la velocidad de flujo debe determinarse independientemente), la expansión térmica de líquidos en su recipiente de desplazamiento no es un problema, siempre que el regulador de presión corriente arriba sea de descarga automática. En este caso la presión permanecerá constante a pesar de la expansión térmica. Después de que la velocidad de flujo y la diferencia de presión se hayan estabilizado, estas deben ser leídas y registradas, y luego se debe terminar el flujo. Las líneas transductoras deben llenarse completamente con el líquido de prueba y ambas aberturas deben estar a la misma elevación. Después de que la diferencia de presión se haya estabilizado con el flujo en cero, esta se lee. Si no es cero, la lectura debe restarse de la ∆p corriente estabilizada. La permeabilidad de líquidos se calcula con la Ecuación 23, utilizando el factor geométrico dimensional apropiado, Gf. Estos factores para flujo axil, flujo transversal, y flujo radial son dados en las Ecuaciones 27, 29 y 30, respectivamente. Con las anteriores restricciones para líneas transductoras, la Ecuación 23 aplica para el flujo axil vertical ascendente o descendente como también al flujo horizontal para todas las tres configuraciones geométricas. Después de que se haya completada la medición y se vaya a remover la muestra para análisis adicional, se debe tener cuidado para asegurar que, como la muestra se dilata mientras se disminuye el esfuerzo restrictivo, se sature de líquido y se evite el ingreso de aire. 6.3.2.1.3 Ventajas Las ventajas de este método incluyen: a. Diferencias de presión para el flujo de Stokes con
líquidos son mayores y mas fáciles de medir que aquellas con gases, especialmente para muestras de alta permeabilidad.
b. No se requiere la corrección de deslizamiento de gas.
c. Las mediciones de permeabilidad de líquidos tomadas cuidadosamente pueden ser mas representativas de las permeabilidades del yacimiento.
d. No se requiere el secado de muestras, una operación potencialmente dañoso.
e. Las mediciones de permeabilidad de líquidos pueden ser beneficiosos si los análisis adicionales requieren que el tapón se sature con el mismo líquido.
6.3.2.1.4 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. La saturación y preparación de la muestra de
roca, y la preparación y manejo de líquidos es por lo general mas difícil y demorado que las preparaciones para mediciones de gas.
b. Los equipos resistentes a la corrosión, de bombeo de alta presión y los de control son costosos.
c. Se debe tener cuidado para evitar fluidos que afectan a los componentes de la roca.
d. Las mediciones de líquidos en régimen permanente con tapones de baja permeabilidad pueden ser difíciles de realizar con precisión, y requieren largos periodos para lograr el régimen permanente. Se recomiendan pequeños volúmenes, cortas longitudes de tapones, almacenamientos compresivos bajos, y un control de temperatura muy estricto.
6.3.2.2 Mediciones de Permeabilidad de
Líquidos en Régimen Permanente Utilizando Cabezas de Líquido
Las Figuras 6-6 y 6-7 (Sección 6.2.2.1.2) muestran dos configuraciones para mediciones de flujo axil en las cuales el líquido es suministrado a una velocidad y presión constante por medio de cabezas de líquido constantes. La configuración de la Figura 6-7 es especialmente útil para mediciones precisas de bajo consto con tapones de núcleo de permeabilidad moderada a alta. No se requiere ninguna bomba o transductor de presión. El depósito de entrada con altura ajustable y el depósito fijo de salida ambos tienen desagües de rebose. Se permite que el líquido entre al depósito de entrada desde otro recipiente de suministro a una elevación mas alta a través de una válvula de medición a una velocidad ligeramente mayor a la que acepta la muestra de roca. El exceso de líquido fluye por el desagüe y es recolectado para reciclado futuro en el recipiente de suministro. El líquido que rebosa del depósito de salida cae en un recipiente pesador sobre una balanza electrónica. La velocidad de flujo volumétrico, q, se calcula dividiendo la velocidad de acumulación de masa (de mediciones de peso e intervalos) por la densidad del líquido, la cual debe determinarse. La permeabilidad se calcula con la Ecuación 25, donde ρ es la densidad del líquido en la temperatura de medición, g es el valor local de aceleración gravitacional, y h es la diferencia en elevación entre las superficies libres de las dos cabezas de líquido. Como se emplea muy poca contrapresión, se debe tener mucho cuidado para excluir aire de todas las porciones del sistema, incluyendo el tapón de núcleo. Para muestras de muy alta permeabilidad (10 a 40 darcys), la configuración de la Figura 6-7 es útil porque el desagüe de rebose en el recipiente corriente abajo minimiza la variación en la cabeza de líquido debido a fuerzas retentivas capilares. Estas fuerzas pueden ser significativas en la punta de boquillas o aberturas de salida de pequeño diámetro, creando una gran variación relativa en la ∆p. 6.4 TEORÍA Y APLICACIÓN DE
DETERMINACIONES DE PERMEABILIDAD EN RÉGIMEN VARIABLE
La llegada de sistemas de adquisición de datos de alta velocidad, transductores de presión precisos, y computadores digitales han hecho que sea no solo

factible, sino conveniente medir permeabilidades bajo condiciones de flujo transitorias o de régimen variable. Las mediciones transitorias emplean depósitos de volumen fijo para gas o líquido. Estos pueden ubicarse corriente arriba de la muestra - de los cuales el gas o el líquido fluye a la muestra a medir, o corriente abajo - a los cuales fluye desde la muestra, o en ambas partes. Cuando el fluido fluye desde un depósito corriente arriba, su presión en ese depósito disminuye con el tiempo. De manera similar, cuando el fluido fluye a un depósito corriente abajo, la presión allí aumenta con el tiempo. Velocidades de flujo instantáneas pueden calcularse del volumen del depósito y la velocidad instantánea de cambio de presión, obviando la necesidad de un aparato de medición de velocidad de flujo. Cuando el fluido se expande, este realiza trabajo de flujo a costa de la pérdida de energía interna. Esto se observa como una disminución en su temperatura. De manera similar, cuando se comprime el fluido, se realiza trabajo sobre él, y su temperatura se incrementa. Como las velocidades de flujo instantáneas son calculadas de las velocidades de cambios de presión, es obligatorio mantener condiciones isotérmicas, o medir temperaturas instantáneas y utilizar las formulaciones matemáticas apropiadas. Las ecuaciones presentadas en este documento asumen condiciones isotérmicas. 6.4.1 Técnicas de Presión Transitoria para
Gases Afortunadamente, el gas tiene una baja capacidad térmica, y los cambios de temperatura relacionadas con el trabajo pueden ser virtualmente eliminados fabricando depósitos de material de alta conductividad térmica y empacándolos con tubos de cobre paralelos al eje de flujo. También, donde se anticipan altas velocidades de flujo (con muestras de alta permeabilidad), el helio es la mejor elección. Su coeficiente de difusión es mucho mas alto que el de aire o nitrógeno. Otros problemas térmicos potenciales pueden surgir de los cambios en la temperatura ambiente en mediciones de larga duración de muestras de baja permeabilidad, y de la expansión Joule-Thomson mientras el gas fluye a través de una muestra. El primero se minimiza con excelente aislamiento térmico, un control perfeccionado de la temperatura ambiente, y reduciendo el tiempo de medición. La expansión Joule-Thomson se maneja idealmente utilizando helio, el cual aumenta en temperatura (a condiciones ambiente) al expandirse, a diferencia de todos los demás gases excepto el hidrógeno. Este calentamiento tiende a neutralizar el enfriamiento relacionado con el trabajo. Han surgido dos categorías principales de técnicas de presión transitoria para medir permeabilidad en el laboratorio. Una se llama el método de "caída de impulso". Este se caracteriza por el uso de depósitos tanto corriente arriba como corriente abajo, los cuales son relativamente pequeños en volumen. Estos y la muestra se llenan con gas a una presión bastante alta, 1.000 a 2.000 psig, la cual reduce el deslizamiento y la compresibilidad del gas. Después de lograr un equilibrio de presión en todo el sistema, se incrementa
la presión en el depósito corriente arriba, típicamente por 2 a 3 por ciento de la presión inicial, causando una impulsión de presión a fluir a través de la muestra. Esta técnica es apropiada para muestras de baja permeabilidad, 0.1 milidarcy a 0.01 microdarcy. Pequeñas diferencias de presión y bajas permeabilidades virtualmente eliminan la resistencia inercial de flujo. Solo se hablará de técnicas "de retardo" en este documento. Estas producen permeabilidades generales que son comparables con los valores de régimen permanente. Los variaciones transitorios "tempranos" proporcionan información relacionada con heterogeneidad en las muestras, la cual está mas allá del alcance propuesto para este documento. La otra técnica se llama el método de "caída de presión". Este se caracteriza por el uso de solo depósitos corriente arriba. El extremo corriente debajo de la muestra se abre a la presión atmosférica. La máxima presión corriente arriba utilizada es bastante baja, 10 a 250 psig (variando inversamente con la permeabilidad a medir). Una sola caída de presión transitoria produce datos para 6 a 30 cálculos separados de permeabilidad, cada uno en una velocidad de flujo y una presión media de poros diferentes. Una variación adecuada de las condiciones de flujo durante una prueba transitoria hace posible el cálculo de la permeabilidad con corrección de deslizamiento (Klinkenberg) (k∞), el factor de deslizamiento Klinkenberg (b), y la resistividad inercial (β) del medio poroso. Esta técnica, la cual tiene un alcance útil de permeabilidad de 0.001 a 30.000 milidarcys (por medio del uso de múltiples depósitos de gas corriente arriba y transductores de presión), complementa el método de caída de impulso. Para muestras de alta permeabilidad (>1.000 md), donde b es pequeño, es difícil determinar b con precisión cuando una muestra es abierta a la presión atmosférica. Este es mas confiablemente aproximada con una correlación. Además de las diferencias físicas entre los sistemas de flujo, los enfoques para la derivación de ecuaciones de flujo son bastante diferentes para las técnicas de caída de impulso y de caída de presión. En la primera, la ecuación de Darcy y la ecuación de continuidad (la cual es una manifestación de la conservación de la masa) se resuelven simultáneamente. En el método de caída de presión, la solución en régimen permanente de la ecuación Forchheimer con corrección de deslizamiento se utiliza como punto de partida. Se deriva esta solución, la cual no da cuenta del aumento en el flujo de masa con distancia en la longitud de una muestra en algún instante específico (con flujo de régimen variable). Luego esta es insertada en la ecuación de continuidad, la cual es integrada para proporcionar una corrección (y perfeccionamiento) en la ecuación final de flujo. Este proceso se repite iterativamente hasta que se puedan satisfacer tanto la ecuación de Forchheimer como la de continuidad. La desviación entre las soluciones de régimen permanente y de caída de presión obtenida así depende de la relación del volumen de poros de la muestra con el volumen del depósito de gas corriente arriba. Cuando esta relación es pequeña, la solución de régimen permanente es casi exacta. Mientras se

aumenta la relación (PV mas grande o volumen de depósito mas pequeño), la corrección que debe aplicarse a la solución de régimen permanente se aumenta progresivamente. 6.4.1.1 Caída de Presión, Flujo Axil de Gas 6.4.1.1.1 Aparatos y Procedimiento El aparato de caída de presión (ver Figura 6-22) emplea un distribuidor de gas corriente arriba que es conectado a un portamuestras capaz de aplicar esfuerzos hidrostáticos a una muestra cilíndrica de diámetro D y longitud L. Un depósito de gas corriente arriba de volumen calibrado puede conectarse, o aislarse de, el volumen calibrado del distribuidor por medio de una válvula. (Se utilizan múltiples volúmenes en el depósito para acomodar una amplia gama de permeabilidades). La abertura de salida del portamuestras se abre a la atmósfera. Esta abertura tiene una válvula que se puede cerrar para propósitos de arranque. Se conecta un transductor exacto de presión que mide la presión manométrica al distribuidor inmediatamente corriente arriba del portamuestras. Se llena el depósito, el distribuidor, y la muestra - en la válvula de salida - con gas. Después de unos pocos segundos para equilibrio térmico, se abre la válvula de salida para iniciar la variación transitoria de presión. Cuando la presión corriente arriba ha caído a alrededor del 85 por ciento de la presión de llenado, tiempo durante el cual se establece un perfil plano de presión por toda la longitud de la muestra, se empieza la recolección de datos. Se leen y registran las presiones en intervalos seleccionados y los tiempos transcurridos correspondientes. 6.4.1.1.2 Cálculos Se indican los procedimientos para calcular k∞, b y β de los datos de caída de presión axil en B.6.8.2. 6.4.1.1.3 Ventajas Las ventajas de este método incluyen: a. Se puede determinar la permeabilidad con
deslizamiento corregido, k∞, el factor de deslizamiento de gas de Klinkenberg, b (hasta 1.000 md), y el coeficiente Forchheimer de resistividad inercial, β, con una sola prueba de caída de presión.
b. La permeabilidad sin corrección de deslizamiento, kg, puede calcularse (ver 6.4.1.1.5) con estos resultados para cualquier gas deseado en cualquier presión media de poros para propósitos de comparación con mediciones convencionales.
c. No se requieren medidores de corriente para el método. Se calculan las velocidades de flujo con las mediciones de presión-tiempo.
d. Para un tapón de núcleo de una permeabilidad dada, el tiempo requerido para que la presión caiga de una presión a otra es directamente proporcional al volumen del depósito de gas corriente arriba. Por lo tanto, los tiempos de
medición pueden controlarse para una amplia gama de permeabilidades proporcionando varios volúmenes de depósitos - pequeños para bajas permeabilidades y mas grandes para permeabilidades mas altas.
e. El margen práctico de permeabilidad es de 0.001 a 30.000 milidarcys si se utilizan múltiples volúmenes de depósito y márgenes transductores.
f. Una desviación del flujo de Stokes no tiene importancia. Esta es compensada en el esquema de análisis de datos.
g. Las mediciones de permeabilidad con corrección de deslizamiento en altos esfuerzos restrictivos reflejan las condiciones de depósitos con mas precisión, especialmente para muestras de baja permeabilidad.
Figura 6-22 - Esquema para Permeámetro de Gas de
Caída de Presión
6.4.1.1.4 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. Para minimizar los efectos térmicos con los
tapones de alta permeabilidad, se deben utilizar bajas presiones de llenado (<25 psig), diámetros pequeños (1 pulgada), y longitudes largas (>2 pulgadas).
b. Para minimizar el tiempo de medición con muestras de baja permeabilidad, el diámetro del tapón debe ser grande (1.5 pulgadas) y su longitud corta (> 1.5 pulgadas).
c. La técnica requiere transductores de presión de alta calidad, equipos rápidos de adquisición de datos, y una alta demanda de computo. Por lo tanto, como asunto práctico, se requiere un computador digital.
d. Los depósitos de gas deben tener una masa térmica considerable y una alta capacidad de transferencia de calor para evitar cambios de temperatura relacionados con el trabajo durante la expansión de gas, especialmente con las muestras de alta permeabilidad. Los cambios de temperatura se minimizan aún mas utilizando helio, y reduciendo la presión de arranque para estas pruebas de caída. Las muestras de alta permeabilidad también demandan aberturas de diámetro grande en el sistema de distribución de gas para minimizar las diferencias de presión internas.
e. Las muestras de baja permeabilidad requieren un sistema libre de escapes (para esta o cualquier otra técnica), y no puede haber desvíos en el portamuestras (después de la manga de caucho). Se debe disponer de un aislamiento térmico para minimizar los cambios de temperatura inducidos por los cambios en el ambiente.
f. Las muestras de baja permeabilidad a menudo muestran una sensibilidad extrema al esfuerzo. Cuando la presión media de poros se disminuye, el esfuerzo neto se incrementa (con un esfuerzo restrictivo constante), y la permeabilidad disminuye. En los cálculos, se supone que la

permeabilidad permanece constante durante toda la caída de presión. Esta suposición puede resultar en el cálculo de un β bajo o negativo en muestras muy sensibles al esfuerzo. Como los cambios mas grandes en permeabilidad ocurren en esfuerzos netos bajos, la sensibilidad al esfuerzo se minimiza realizando las mediciones a esfuerzos netos mas altos (>2000 psi), y disminuyendo la variación en la presión de poros durante una prueba.
6.4.1.1.5 Cálculo de la Permeabilidad de Gas con
la Permeabilidad de Klinkenberg Debido a la frecuencia histórica de las mediciones de permeabilidad axil de gas en régimen permanente realizadas sin la corrección Klinkenberg, grandes bases de datos se han desarrollado con estas mediciones. A menudo se desea comparar nuevos datos con mediciones anteriores en el mismo depósito. Para hacer esto, k∞ debe estar "sin corrección" para el deslizamiento de gas. La permeabilidad sin corrección de deslizamiento, kg, para cualquier gas en cualquier temperatura y presión media de poros puede calcularse con k∞ y b medidos de la siguiente manera: a. Calcular el "radio de deslizamiento de gas" de la
muestra, rb, (con la Ecuación 13 en 6.2.1.1) utilizando los valores apropiados para peso molecular, viscosidad, y el b obtenido para el gas utilizado en la medición en la temperatura absoluta de la medición.
b. Con este rb, encontrar b de la Ecuación 12 para el nuevo gas, utilizando su peso molecular y la viscosidad en la temperatura deseada.
c. Con el nuevo b, el k∞ medido, y la presión media de poros deseada, calcular kg para el nuevo gas con la Ecuación 21.
Estos pasos pueden combinarse en una sola ecuación:
31
Donde el subíndice m se refiere a la medición de gas o condición, y c se refiere al gas o condición deseado para el cual kg será calculado. Para ilustrar el uso de la Ecuación 31, suponga que para una roca específica, el k∞ y b medidos son 10.62 milidarcys y 6.57 psi, respectivamente, utilizando helio a 72ºF. Para propósitos de comparar esta medición con los datos antiguos tomados sobre la misma formación, se desea calcular kaire a una presión media de poros de 18.0 psia y una temperatura de 75ºF. El peso molecular del helio es 4.0026, y su viscosidad a 72ºF es 0.01967 cp. El peso molecular promedio del aire es 29.0, y su viscosidad a 75ºF es 0.01837 cp. Por lo tanto, de la Ecuación 31, kaire a 75ºF y 18.0 psia es:
(ecuación)
Una nota de advertencia: muchas de las mediciones anteriores fueron tomadas en bajos niveles de esfuerzo, típicamente a 250 o 400 psi. Aún con la
corrección indicada, los valores kaire antiguos sobre una roca comparable pueden ser mas altos si las nuevas mediciones fueran tomadas en esfuerzos significativamente mas altos. Además, es posible que las bases de datos antiguas no incluyan la presión media y la temperatura requerida para realizar estos cálculos. 6.4.1.2 Caída de Presión, Mediciones de
Permeabilidad de Gases con Sonda La técnica de caída de presión se adapta fácilmente para mediciones con permeámetro explorador para proporcionar permeabilidades de gas rápidas y no destructivas corregidas tanto para el deslizamiento de gas como para la resistencia inercial. Como en el caso de aparatos de régimen permanente, las mediciones son tomadas típicamente en altas densidades de muestreo y casi cero esfuerzo sobre muestras de núcleo entero tajadas o sin tajar. 6.4.1.2.1 Aparato y Procedimiento La Figura 6-23 muestra esquemáticamente el instrumento descrito por Jones22. Para operarlo, los volúmenes corriente arriba se llenan con gas (usualmente el nitrógeno seco) hasta una presión alrededor de 10 psig. Se coloca la sonda encima de una muestra, y luego se baja y se sella contra su superficie con una presión neumática fija en el pistón operador de la sonda. Se inicia la caída de presión abriendo la válvula interna mas cercana a la sonda. Se selecciona el volumen apropiado del depósito de gas para la medición determinada de permeabilidad por medio de las válvulas internas en el distribuidor. Esta selección se hace con base en la velocidad de caída de presión cuando todos los volúmenes internos estén conectados a la sonda. Si la velocidad es menor que un valor predeterminado, se cierran las válvulas apropiadas para los tanques mas grandes. Se lee y se registran las presiones a intervalos preseleccionados y los tiempos correspondientes para el cálculo de k∞. 6.4.1.2.2 Cálculos Se habla de los procedimientos de cálculo en C.6.8.3. 6.4.1.2.3 Ventajas Las ventajas de este método incluyen: a. El margen práctico de permeabilidad de esta
técnica es de 0.001 a 30.000 milidarcys. b. Los tiempos de medición en este margen varían
de 35 a 2 segundos. c. No se requieren medidores de corriente. Solo se
hacen mediciones de tiempo-presión. d. Se obtienen permeabilidades con deslizamiento
corregido (k∞) y sin corrección de deslizamiento (kg) con una sola prueba de caída de presión.
e. Los efectos de resistencia inercial de flujo, los cuales pueden ser severos aún con pequeñas diferencias de presión y muestras de baja permeabilidad, son eliminados del cálculo de permeabilidad con esta técnica.

f. El método no es destructivo en el sentido que no hay necesidad de cortar los tapones de núcleo. Esto permite un muestreo de alta densidad en secciones que exhiben una variación de permeabilidad frecuente y severa.
g. El volumen de la muestra de roca investigada por una sola prueba puede variarse un poco variando el tamaño de las dimensiones de sellado de la punta de sonda. Sin embargo, en general este volumen es bastante pequeño (la permeabilidad medida es localizada). Esta afirmación también aplica para las mediciones en régimen permanente (ver 6.3.1.2.3).
6.4.1.2.4 Precauciones y Limitaciones Excepto por los problemas de resistencia inercial y deslizamiento de gas que son manejados por los procedimientos de cálculo, las precauciones y limitaciones que aplican para las mediciones de permeabilidad con sonda en régimen permanente (ver 6.3.1.2.4) también aplican para las mediciones de caída de presión. 6.4.1.3 Caída de Impulso, Flujo Axil de Gas 6.4.1.3.1 Aparato El aparato de caída de impulso, mostrado en la Figura 6-24, consiste de un depósito de gas corriente arriba de volumen V1, un portamuestras capaz de aplicar altos esfuerzos restrictivos (usualmente isostáticos), el cual contiene la muestra del volumen nominal de poros Vp, y un depósito corriente debajo de volumen V2. Un transductor de diferencia de presión mide la diferencia de presión entre los depósitos, y un segundo transductor mide la presión absoluta en el depósito corriente abajo. 6.4.1.3.2 Procedimiento Con las válvulas 1 y 2 abiertas, se llenan ambos depósitos y la muestra con gas (usualmente nitrógeno seco) hasta una presión típicamente entre 1.000 y 2.000 psig. El periodo de llenado debe permitir el tiempo adecuado para que el gas se difunda en la muestra (típicamente) de baja permeabilidad. Después del periodo de llenado, se cierra la válvula y se supervisa la presión hasta que no se observe ningún cambio adicional, indicando un equilibrio térmico y de presión. Esta presión estabilizada es P2[O]. Todas las válvulas deben ser del tipo que no tiene cambios en volumen interno cuando se abren o se cierran. Después del equilibrio, se cierran las válvulas 1 y 2, y se incrementa la presión en el depósito corriente arriba por ∆p1, la cual es el 2 o 3 por ciento de P2[O]. Después de estabilizarse la presión en V1, i.e., cuando ∆p1 se vuelve constante, se abre la válvula 1, iniciando la porción de presión transitoria de la medición. 6.4.1.3.3 Cálculos Ver D.6.8.4. para los detalles.
Figura 6-24 - Aparato de Caída de Impulso para el Flujo Axil de Gases
6.4.1.3.4 Ventajas Las ventajas de este método incluyen: a. La técnica de caída de impulso es aplicable a la
medición de permeabilidades muy bajas, 0.1 milidarcy a cerca de 0.01 microdarcy, y posiblemente se pueda extender en cualquier dirección con la selección cuidadosa de volúmenes del depósito de gas y los márgenes del transductor de presión.
b. No se requieren medidores de corriente. Solo se hacen mediciones de tiempo-presión.
c. Estas mediciones se adaptan bien para la aplicación de esfuerzos de depósito-condición para proporcionar permeabilidades representativas.
d. Se puede medir la porosidad simultáneamente o por separado en el mismo aparato.
6.4.1.3.5 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. Debido a las permeabilidades tan bajas que
pueden ser medidas con esta técnica, es de suma importancia que el aparato sea libre de escapes. El control de variaciones en la temperatura ambiente también es crítico.
b. Aunque se emplean altas contrapresiones, las permeabilidades obtenidas no tienen corrección de deslizamiento de gas y pueden ser algo altas. Por ejemplo si el b para el nitrógeno fuera 100 psi para un tapón determinado, y la presión media de poros fuera 1.000 psia, la permeabilidad de gas obtenida sería 10 por ciento mas alto que k∞.
6.4.2 Técnicas de Presión Transitoria para
Líquidos 6.4.2.1 Caída de Impulso, Flujo Axil de Líquidos A menos que se impongan grandes gradientes de presión (100 a 1.000 psi/cm o mas), las permeabilidades líquidas de rocas compactas (k<0.1 milidarcy) son difíciles de medir o al menos demorados con métodos de régimen permanente. Sin embargo, las permeabilidades hasta 0.1 microdarcy pueden medirse utilizando técnicas de caída de presión. Se calculan velocidades de flujo muy bajas con las velocidades de cambio de presión en los depósitos de líquido de almacenamiento compresivo conocido - mientras el líquido se expande de un depósito corriente arriba o es comprimido en uno corriente debajo de la muestra. Por lo tanto, los parámetros importantes en estas mediciones son almacenamientos compresivos efectivos de los depósitos corriente arriba y corriente abajo y de la muestra de roca. El almacenamiento compresivo efectivo de cada depósito es su volumen interno multiplicado por la suma de la compresibilidad de

líquido y la compresibilidad del recipiente. El almacenamiento compresivo de la muestra es el producto de su volumen de poros y la suma de las compresibilidades del líquido y el PV. El aparato de caída de impulso de líquido debe tener la posibilidad de medir almacenamiento compresivo de los volúmenes corriente arriba y corriente abajo (ver E.6.8.5.1). 6.4.2.1.1 Procedimiento Para una medición de permeabilidad de líquidos de caída de presión (ver Figura 6-25), se carga una muestra de roca completamente saturada de líquido en el portamuestras, el cual se presuriza, preferiblemente a un esfuerzo restrictivo bastante alta (usualmente hidrostático). El alto esfuerzo minimiza la variación de permeabilidad por los cambios en esfuerzo neto debidos a la presión de poros variante. Se llena el sistema de líquido, de la manera indicada en E.6.8.5., y se presuriza hasta la presión de poros inicial. Se deja la válvula de llenado abierta el tiempo suficiente para permitir que el líquido a alta presión se difunda en la muestra. Con las válvulas 1 y 2 aun abiertas, se cierra la válvula de llenado, y se supervisa el transductor corriente abajo hasta que no se observe ningún cambio adicional en la presión. Luego se cierran las válvulas 1 y 2 y la impulsión de presión, ∆p1, es generada apretando la válvula de aguja. Todas las válvulas, excepto por la válvula de aguja, deben ser del tipo que no tiene cambios en volumen interno cuando se abren o se cierran. La caída de impulso se inicia abriendo la válvula 1. La diferencia de presión inicial en toda la muestra, ∆p[O], será ligeramente menor que ∆p1, la cual fue generada por la válvula de aguja con la válvula 1 cerrada, así excluyendo el volumen muerto corriente arriba, Vd, del volumen total corriente arriba, V1. Por lo tanto, ∆p[O] debe ser calculada de la manera indicada en E.6.8.5. 6.4.2.1.2 Indicios de una Muestra Bastante
Homogénea La presión corriente arriba se disminuye mientras la impulsión de presión recorre la longitud de la muestra, pero la presión corriente abajo permanece constante hasta que el impulso "surge". De ahí en adelante, la presión corriente arriba sigue disminuyendo, y la presión corriente abajo se eleva. Si la muestra es razonablemente homogénea y los almacenamientos compresivos corriente arriba y corriente abajo son iguales, entonces poco después del periodo de propagación de impulso la disminución en la presión corriente arriba es compensada por un aumento igual corriente abajo, y la presión media de poros permanecerá constante mientras la ∆p[t] sigue disminuyendo linealmente - en una caída semi- logarítmica. 6.4.2.1.3 Indicios de una Muestra Heterogénea Sin embargo, si la muestra no es homogénea, pero se caracteriza por un sistema de fractura de relativamente alta conductividad fluida y una porosidad matriz de baja conductividad, y si el
almacenamiento compresivo de la muestra es casi tan grande, o mas grande que aquellos en los depósitos corriente arriba y corriente abajo (depósitos pequeños), la presión media de poros continuará disminuyendo aún después de que ∆p[t] haya alcanzado cero, y la linealidad de la caída de presión semi-logarítmica puede haberse distorsionado. En este ∆p[t] caso es rápidamente dispersado por el sistema de fractura, pero el líquido sigue difundiéndose lentamente en la matriz compacta. Si los almacenamientos del depósito son grandes en comparación con los de la matriz, la disminución en la presión media de poros, debido al movimiento de líquido a la matriz, es apenas perceptible. Para una discusión mas completa de la medición de heterogeneidad en muestras de núcleos, ver Kamath, et al.29 6.4.2.1.4 Cálculos Ver E.6.8.5.3 para los detalles de los cálculos para mediciones de caída de presión axil de líquidos. 6.4.2.1.5 Ventajas Las ventajas de este método incluyen: a. No se requieren correcciones de deslizamiento
para las mediciones de líquidos. b. Las mediciones de permeabilidad de líquidos de
alto esfuerzo pueden ser mas representativas de las condiciones de depósitos que las mediciones de gas.
c. Esta técnica es útil para la medición de rocas de baja permeabilidad (alrededor de 0.01 microdarcy a 0.1 milidarcy).
d. El trabajo de expansión o compresión en sistemas líquidos es pequeño. Por lo tanto los cambios en temperatura relacionados con el trabajo son correspondientemente pequeños.
6.4.2.1.6 Precauciones y Limitaciones Las precauciones y limitaciones de este método incluyen: a. Mantenimiento de condiciones isotérmicas es de
suma importancia. Pequeños cambios en la temperatura ambiente pueden causar grandes cambios de presión en sistemas líquidos de alta presión.
b. Se requieren transductores de presión y sistemas de adquisición de datos de alta calidad.
c. Un sistema hermético es esencial para las mediciones de alta presión de tapones de baja permeabilidad.
d. Se deben observar las precauciones usuales de resistencia de corrosión, compatibilidad fluida, una filtración cuidadosa de líquidos, y la exclusión de gas del sistema.

6.5 EXACTITUD Y PRECISIÓN 6.5.1 Primera Parte 6.5.1.1 Introducción Los datos de permeabilidad representan mediciones cuantitativas, y es importante tener valores correctos. Todos los métodos analíticos son sujetos a una dispersión aleatoria y a errores del operador, los equipos y aquellos relacionados con la técnica, los cuales, tomados en conjunto, comprenden una incertidumbre experimental. Para distinguir entre una tendencia real en un conjunto de datos y una variación debido a la incertidumbre experimental, se recomienda que los datos de análisis de núcleos sean reportados junto con un informe de la incertidumbre con la cual se registraron estos datos. 6.5.1.2 Definiciones 6.5.1.2.1 precisión (o reproducibilidad) de una medición: Una expresión de los límites dentro de los cuales la medición, con una probabilidad indicada, puede ser reproducida siempre que la medición sea sujeta únicamente a una variación fortuita. Se determina la precisión con mediciones repetidas sobre una sola muestra, y normalmente es dada como una desviación estándar o una desviación estándar relativa, también conocida como el coeficiente de desviación. 6.5.1.2.2 exactitud de una medición: Una expresión de la fidelidad entre el valor obtenido experimentalmente y el valor verdadero. La diferencia entre el valor medido y el valor verdadero se llama el error. Los resultados que caigan fuera del máximo error permitido o la tolerancia pueden ser causados por errores operativos o errores sistemáticos. 6.5.1.2.3 error asimétrico (o sistemático): Una desviación que persiste durante un ciclo de medición y afecta todas las mediciones de la misma manera. El error sistemático se debe normalmente a la desviación instrumental, o una calibración defectuosa, o procedimientos operativos. 6.5.1.2.4 aplicabilidad: El concepto de aplicabilidad, además de precisión y exactitud, es apropiado en este caso. Las mediciones de permeabilidad han sido realizadas con alta precisión y exactitud, produciendo resultados cercanos a los valores verdaderos de permeabilidad para las condiciones reportadas bajo las cuales fueron medidas. Pero, estos valores precisos y exactos pueden ser muy diferentes a aquellos que se hubieran obtenido para condiciones que existen en la formación de la cual se tomaron las muestras. Las razones para las diferencias incluyen diferentes condiciones de esfuerzo, interacción roca-fluido, diferencias en la presión de poros, o la composición del gas si se ignora el deslizamiento de gas, etc. 6.5.1.3 Afirmaciones de Error en las Mediciones
de Permeabilidad
El concepto de exactitud requiere que el valor verdadero de una muestra sea conocido. Esto por lo general no es el caso. En su lugar, se obtienen materiales certificados de referencia estándar para uso científico o industrial de instituciones estándar reconocidos. Estos materiales son analizados en intervalos regulares junto con muestras rutinarias. Luego se obtiene la exactitud comparando el valor medio, calculado con análisis repetidos sobre el material estándar, con el valor certificado para ese prototipo. En la medición de permeabilidad, algunos materiales estándar apropiados pueden ser rocas naturales o materiales sintéticos permeables. Sin embargo, las muestras sintéticas aun no pueden ser producidos de tal manera que permita una pre-determinación exacta de permeabilidad. Por lo tanto, cada prototipo debe pasar un programa extensivo de pruebas antes de ser certificado, y después existe el riesgo que el prototipo cambie durante el manejo regular en el laboratorio. Hasta ahora no se han certificado prototipos de permeabilidad en instituciones estándar reconocidos. Por lo tanto, se recomienda un enfoque diferente para determinar la exactitud de mediciones de permeabilidad. 6.5.1.4 Estándares Internos Se presenta el concepto de estándares internos de permeabilidad para determinar exactitud, y como una facilidad en los equipos de pruebas para errores asimétricos o sistemáticos. Los estándares internos pueden ser muestras sintéticas y/o naturales que cubren un amplio margen de valores de permeabilidad, que han sido analizadas repetidamente por diferentes operadores en diferentes aparatos (cuidadosamente calibrados) utilizando el fluido requerido. Cuando se han recolectado muchas medidas, se calcula un valor medio y una desviación estándar para la muestra. Si la variación acerca del medio es pequeña, i.e., la desviación estándar es baja, se acepta la muestra como un estándar interno, y este puede utilizarse rutinariamente para verificar la exactitud de los equipos y corregir el error sistemático en los resultados. Existen medios porosos y permeables sintéticos en diferentes materiales, e.g., tal como cerámicas y vidrios porosos, y bolitas sinterizadas de plástico, vidrio o metal. Estos materiales a menudo son suministrados por el fabricante con un valor especificado de permeabilidad, el cual, sin embargo, debe considerarse aproximado. 6.5.1.5 Errores Metodológicos Todas las ecuaciones para calcular permeabilidad presentadas en este documento asumen un flujo fluido a través de muestras homogéneas e isotrópicas bajo condiciones isotérmicas. Por lo tanto, estas definen una permeabilidad promedio o efectivo en la dirección general de flujo dictado por el experimento (i.e., axil, radial, transversal, etc.). A menos que se determine la estructura interna macroscópica de cada muestra por medios independientes, es virtualmente imposible modelar el flujo de otra manera. Como las rocas reales casi nunca son isotrópicas u homogéneas, la

suposición que sí lo son, distorsionará invariablemente los patrones reales de flujo, y en consecuencia, la permeabilidad calculada, y también el factor de resistividad inercial y el de deslizamiento de gas, cuando estos aplican. Las ecuaciones para análisis de régimen variable requieren corrección para el flujo de masa no constante (con distancia desde el punto de inyección) en cualquier instante durante la variación de presión. Por lo tanto, se debe determinar el volumen de poros de una muestra, independientemente o en conjunto con la misma medición de permeabilidad. Los errores en la estimación de volumen de poros resultará en errores de permeabilidad, especialmente cuando la PV es casi tan grande, o mas grande que el volumen del recipiente de gas. Los errores pequeños en PV son inconsecuentes cuando los volúmenes de depósitos son relativamente grandes. Además, las técnicas transitorias asumen una permeabilidad constante por toda la duración de una prueba. Los cambios en la presión de poros cambian el esfuerzo neto a la cual se sujeta la muestra, así cambiando ligeramente su permeabilidad durante una prueba. En una medición de caída de presión, esto puede distorsionar el cálculo de resistividad inercial (β). Estos errores son minimizados limitando la magnitud de la caída de presión, e incrementando el esfuerzo restrictivo (donde los cambios en esfuerzo neto tienen menos efecto sobre la permeabilidad - según la Figura 6-9). Pueden resultar errores graves cuando las condiciones de operación son tales que la ley de Darcy no aplica debido a una resistencia inercial significativa. Estos son frecuentes en mediciones de gas para flujo axil en régimen permanente sobre tapones de alta permeabilidad (varios darcys), o mediciones de permeabilidad con sonda en régimen permanente sobre muestras de 10 milidarcys o mas. Se minimiza estos errores utilizando valores muy pequeños. Se pueden presentar otros errores utilizando técnicas de preparación de muestras inapropiadas, tal como una limpieza adecuada, o si no se remueven los cristales de sal en las muestras extraídas que anteriormente contenían solución salina casi saturada. El secado inapropiado de muestras con alto contenido de arcilla puede aumentar o reducir su permeabilidad. Los finos incrustados por cortar o moler los extremos de las muestras pueden reducir la permeabilidad. Los picos o los extremos no cuadrados de un tapón de núcleo pueden ser aplastados bajo altos esfuerzos axiles, alterando la permeabilidad. Los líquidos incompatibles o sucios causan taponamiento. Los fluidos de alta velocidad (especialmente líquidos) pueden causar la migración de finos y taponamiento. Las permeabilidades de gas (sin corrección de deslizamiento de gas), medidas en bajo esfuerzo restrictivo, si son reportadas apropiadamente (incluyendo el gas utilizado, presión media de poros, y esfuerzo neto), son especificadas apropiadamente y no constituyen un error per se. Sin embargo, si se espera que estas pronostiquen el rendimiento de un depósito con precisión sin corrección, puede que estas no sean aplicables, especialmente en muestras de mas baja permeabilidad, donde las correcciones son substanciales.
6.5.1.6 Errores de los Componentes de Equipos Las fuentes principales de error asociados con los equipos de prueba son escapes, desviación de muestras, error de transductores de presión, error de los medidores de corriente, y la variación en la temperatura ambiente. Además, la expansión de fluido no isotérmico, la calibración incorrecta de volúmenes de depósitos, y resolución y velocidad inadecuadas de adquisición de datos pueden causar errores en mediciones de régimen variable. Con permeámetros exploradores, la precisión de la posición de la sonda y la repetibilidad de la fuerza de sellado también son importantes. 6.5.1.6.1 Escapes y Desvío de la Muestra Todos los tipos de permeámetros son sujetos a escapes en la tubería, accesorios, válvulas, transductores, medidores de corriente, y soportes de núcleos. El desvío de fluido entre un tapón de núcleo y la manga de caucho del portamuestras, o entre una muestra y el ocluidor de la sonda de un permeámetro explorador, también constituye un escape. La magnitud de error causado por un escape por lo general se incrementa mientras la permeabilidad se disminuye. Por ejemplo, un pequeño escape en la medición de un tapón de 3 darcys apenas afectará su permeabilidad calculada, mientras el mismo escape con un tapón de 0.001 milidarcys sería desastroso. Se pueden localizar los escapes grandes en permeámetros de gas con la ayuda de una solución de jabón que forma burbujas cuando es aplicada en un escape. Los escapes pequeños se encuentra muy fácilmente por medio de mediciones de presión. Varias porciones del sistema de flujo son presurizados con gas, luego supervisados por el transductor de presión del sistema. El escape es localizado a través del proceso de eliminación. Puede ser necesario cargar un tapón impermeable de acero en el portamuestras para aislar las porciones corriente arriba y corriente abajo del aparato. Algunos sistemas incorporan rutinas automatizadas de detección de escapes. El desvío de muestras usualmente puede ser eliminado incrementando el esfuerzo restrictivo y/o disminuyendo la dureza (durómetro) del caucho. 6.5.1.6.2 Error del Transductor de Presión La precisión de un transductor de presión es especificado usualmente como la suma raíz de cuadrados (SRC) de las desviaciones de su producto de las presiones reales por todo su margen como un porcentaje de su valor máximo de escala (VME). Por lo general existen precisiones SRC de 0.5 VME o mejor. La mayoría de los transductores tienen al menos dos ajustes - cero y sensibilidad. La graduación cero es un equivalente para permitir un rendimiento eléctrico nulo a cero presión aplicada. El ajuste de sensibilidad cambia el aumento o el multiplicador del transductor que convierte su rendimiento eléctrico en una presión. Algunos transductores también tienen uno o mas ajustes

internos de linealidad. Los transductores de presión también son afectados por los cambios en temperatura, y tienen compensación de temperatura. A menudo, el precio es casi proporcional a la calidad de la compensación de temperatura. El usuario debe tener en cuenta las consecuencias de la especificación de "exactitud a plena escala". Por ejemplo, un transductor de 0.5 por ciento, utilizado al 10 por ciento de plena escala, puede tener un error del 5 por ciento en el valor de su lectura de presión, y estar dentro de la especificación. En vista de la definición de SRC, el error particular en un punto dado puede ser mayor que el porcentaje SRC dado del valor máximo de la escala. Con las técnicas apropiadas de calibración que permite una capacidad matemática de poner nuevamente en ceros y un adaptador de curvas alineales, indicado en 6.61, la repetibilidad de un transductor está entre sus características mas importantes. Existen transductores de alta calidad con 0.01 o 0.02 por ciento de repetibilidad VME. Para minimizar los efectos de histérisis, un transductor siempre debe estar calibrado en la dirección de su uso anticipado. Por ejemplo, para mediciones de caída de presión, un transductor debe estar calibrado hasta su presión máxima de escala primero, seguido por una secuencia de presiones descendentes. Las técnicas de régimen variable tienen demandas adicionales para las mediciones de presión. Se calculan las derivadas presión-tiempo. Por lo tanto, se debe minimizar el ruido, o pequeñas fluctuaciones en voltaje y la resolución de la lectura no debe ser truncada por un convertidor de tensión análogo-a-digital (ADC) con muy pocos bits. El ruido se minimiza empezando con un transductor de buena calidad con poco ruido de salida y cables blindados. Se puede utilizar un filtro análogo pasabajos en conjunto con un filtrado digital. Con un ADC razonablemente rápido (1.000 conversiones por segundo) se pueden tomar múltiples lecturas, y luego se promedian. Un ADC integrador lento (60 conversiones por segundo) saca el promedio automáticamente. Se recomienda un ADC de 15 bits (0.0031 por ciento de la resolución máxima de escala) como mínimo para mediciones de régimen variable para minimizar el error por truncamiento. Existen tarjetas temporizadoras de alta calidad con exactitudes con alcances de partes por millón. Es imperativo consultar la tarjeta temporizadora inmediatamente antes o después de leer el ADC, sin comandos que se interpongan en el programa operativo, para que los intervalos de presión correspondan exactamente a los intervalos de tiempo para el cálculo de derivadas presión-tiempo. Además, los intervalos de presión deben ser lo suficientemente grandes para que el ruido en las mediciones no destruyan la precisión del cálculo de la derivada. Es posible que se tenga que determinar los intervalos óptimos por ensayo y error para algún sistema particular. Una gráfica desigual de derivadas presión-tiempo vs. presiones medias geométricas de los datos de caída de presión indica un exceso de ruido o un truncamiento en las lecturas de tiempo o presión. El problema usualmente se elimina incrementando los intervalos de presión y/o tiempo, incrementando la
resolución (número de bits) del ADC, o mejorando la calidad del transductor. La rutina de muestreo debe escribirse en lenguaje de montaje para velocidad máxima. 6.5.1.6.3 Error del Medidor de Corriente Las técnicas de régimen permanente dependen de una determinación exacta de la velocidad de flujo. Los aparatos volumétricos de lectura directa en conjunto con lecturas de tiempo pueden producir exactitudes mejores que el 1 por ciento. Los aparatos indirectos (orificios calibrados, medidores y controladores de corriente) normalmente se pueden ajustar para rendir una precisión mejor que el 1 por ciento, pero la exactitud es totalmente dependiente de la calibración. Se requieren varios medidores de corriente para cubrir todo el alcance de permeabilidad. Cada uno debe utilizarse únicamente dentro de su alcance lineal de trabajo. Ver 6.6 para la calibración de medidores de corriente masivo. 6.5.1.6.4 Variación en la Temperatura Ambiente Los cambios en la temperatura afectan tanto la volumétrica como la viscosidad del fluido. La temperatura debe mantenerse constante durante una medición de permeabilidad. Nunca coloque el aparato cerca de aberturas de calentamiento o enfriamiento o cerca de una ventana donde la radiación del sol pueda causar grandes cambios en temperatura. En los equipos de régimen variable, la velocidad del cambio de temperatura dentro de los depósitos de fluido debe ser tal que los cambios en presión inducidos por temperatura sean pequeños con relación a las disminuciones de presión causadas por el flujo de fluido de un depósito. En las aplicaciones de campo de mediciones con permeámetro explorador, los cambios en temperatura afectan la viscosidad del fluido, la velocidad de flujo y las mediciones de presión. 6.5.2 Segunda Parte 6.5.2.1 Afirmaciones de Error en los Informes La presentación de datos medidos con los límites de error asociados en un informe de análisis de núcleos puede basarse en dos enfoques diferentes: (a) se utilizan los valores medidos para calcular y reportar un valor medio con los límites de error asociados de la manera indicada a continuación, y (b) los valores medidos se dan junto con una afirmación de exactitud, la cual el laboratorio puede basar en mediciones repetidas de estándares internos. 6.5.2.1.1 Límites de Error Calculados Suponga que una gran cantidad de mediciones repetidas (normalmente mas de 100) son realizadas sobre una sola muestra. Si las mediciones están sujetas únicamente a una variación fortuita, se cree generalmente que los datos tienen una distribución normal o gausiano con un valor medio X:
Ecuación

Y una desviación estándar, s:
Ecuación
Donde (X1-X) es la desviación de una sola medición de la media y n es el número total de mediciones. El resultado se reporta como X + s. La desviación estándar es una medida de la dispersión o la variación de los datos con respecto a la media. Una distribución normal tiene una probabilidad de que el 68.3 por ciento de las observaciones caerán dentro de los límites de mas o menos una desviación estándar de la media. El coeficiente de variación, CV, es la desviación estándar relativa expresada como un porcentaje:
CV = 100 (s/X)
A menudo, solo un número limitado de mediciones son registradas. Esto quiere decir que estamos tratando con estadísticas de muestras pequeñas y se debe aplicar la distribución-t. A menudo es mas pertinente definir los intervalos de certeza de la media en lugar de indicar la desviación. El intervalo de certeza es una estimación de los límites dentro de los cualesmuy probablemente se encuentra el valor. Cuando se reportan los resultados del análisis del núcleo se recomienda expresar los resultados de la siguiente manera: Ecuación
que representa el intervalo de certeza. Los valores de t se encuentran consignados en la mayoría de los libros de estadística o en tablas como es el caso del texto de Fisher y Yates. Estos valores inclusive se dan para “varios grados de libertad”, para n-1 y para valores diferentes de “niveles de confiabilidad” o probabilidad. Se utiliza comúnmente un nivel de confiabilidad del 95 % (el cual corresponde a las desviaciones estándar, ajustadas de acuerdo al número pequeño de observaciones). A continuación se da un ejemplo que ilustra estos conceptos. Se realizaron tres mediciones sobre la misma muestra. La permeabilidad se calculó en cada caso en los valores 3.35, 3.18 y 3.42 milidarcys, a partir de los cuales se calculó el promedio y la desviación estándar: X = 3.32 md s = 0.12 md El valor de t con dos grados de libertad (= n-1, donde n = tres mediciones) y un nivel de confiabilidad de 95 % es de 4.303. Por lo tanto, el intervalo de confiabilidad es: ± 4.303 (0.12 / 3-1) = 3.32 ± 0.37 md Dicho de otro modo, estos cálculos implican que considerando una probabilidad del 95%, la permeabilidad real se ubica dentro de un intervalo que va desde 2.95 hasta 3.69 millidarcys, siempre y cuando no haya desviaciones en las mediciones.
La permeabilidad de Klinkenberg se calcula a partir de las mediciones en la permeabilidad del gas, realizando un análisis de regresión lineal donde se compara la permeabilidad con el promedio recíproco de los valores de presión de poro. El resultado que se observa en la línea de regresión se puede evaluar analizando el coeficiente de correlación r, el cual se especifica con el valor de la permeabilidad. Si deseamos un nivel de confiabilidad del 95%, el valor de r debe ser mayor o igual a 0.95 en el caso de una línea de regresión con cuatro (4) puntos y por lo menos debe tener un valor de 0.997 en una línea de regresión de tres (3) puntos. Las muestras que arrojen valores de r más bajos deben revisarse para detectar fracturas u otras irregularidades. En caso de encontrarse irregularidades, éstas deben reportarse en el listado de datos. De igual manera, las mediciones deben revisarse para detectar la ocurrencia de resistencia inercial no despreciable cuando las presiones promedio del poro sean altas. 6.5.2.1.2 Límites de Error Reportados por el Laboratorio En un análisis rutinario del núcleo, se puede acumular un gran número de determinaciones realizadas a los tapones estándar en un período de tiempo dado. Es perfectamente válido que el laboratorio prepare una tabla que ilustre la exactitud y precisión para los diferentes rangos de permeabilidad, los cuales se basan en mediciones estándar. Thomas Pugh ha estudiado la exactitud que puede obtenerse en las mediciones de permeabilidad. Utilizando mediciones convencionales realizadas a conjuntos de tapones estándar a ritmo constante, efectuadas por varios laboratorios ubicados por todo el mundo, los autores en mención establecieron intervalos de confiabilidad derivados de manera estadística con una probabilidad del 99%, a partir del cual se calcularon los otros intervalos de confianza que se relacionan en la siguiente tabla: Intervalo de Confiabilidad, %, con unan probabilidad de: Rango kx, Milidarcys 68.3%
(= promedio CV) 95% 99%
0.01-0.1 0.1-1.0 1-50 50-1000
±8% 8 5 3
±16% 16 10 6
±21% 21 13 8
Las cifras que se muestran en esta tabla se pueden interpretar de la siguiente manera. Supongamos que cierto tapón de núcleo tiene una permeabilidad de 10.0 milidarcys, el cual se ubica en el rango de 1-50 de la tabla. Si los laboratorios ubicados por todo el mundo realizaran un total de 1/1000 mediciones, se esperaría que un número aproximado de 683 de las permeabilidades que se reporten (esto es, 68.3% del total) estará en el rango que entre 9.5 y 10.5 millidarcys (±5%) del valor real). De igual manera, 950 de estas mediciones estarán entre 9.0-11.0 millidarcys y 990 en el rango que va de 8.7 a 11.3 millidarcys (con 10 mediciones ubicadas por fuera de este rango).

6.5.2.1.3 Número de Cifras Significativas El número de cifras que se deben escribir en un reporte debe respetar el error asociado con el valor de medición. Por ejemplo, 4.2357, ± 0.0327 md debe reportarse en la siguiente forma: 4.24 ± 0.03 md. Es decir, se espera que si hay error, éste se presente en el último decimal de la cifra que se ha redondeado. Considerando la tabla ya mencionada, se justifican únicamente dos cifras significativas (es decir, 4.2 md), a menos que un laboratorio en particular obtenga mejores resultados de manera significativa que los de la tabla. 6.6 INSTRUMENTOS DE CALIBRACION 6.6.1 Calibración utilizando un Transductor de Presión Esta sección se relaciona con la utilización de un calibrador que actúa como Transductor de presión, el cual debe mostrar una lectura de cero cuando entra en contacto con la presión atmosférica del ambiente. También se relaciona con un Transductor absoluto, siempre y cuando se aplique un alto grado de vacío para el valor de lectura a “presión cero (absoluto)”. Sea cual fuere el Transductor de presión, éste debe calibrarse de manera periódica. Aunque en términos generales se sabe que la calibración de un Transductor se realiza en la fábrica utilizando tolerancias ya establecidas, debe recalibrarse la configuración del sistema de flujo utilizando un convertidor análogo / digital (ADC). Esto se logra con un estándar primario de alta calidad, como es el caso de un tester de peso muerto o un manómetro de precisión y un catetómetro o con un Transductor secundario de presión estándar (preferiblemente con exactitud del 0.01 al 0.02 a escala total), el cual pueda llevarse a estándar primario. Se aplica una presión estándar y se lee el voltaje resultante o salida ADC. Este procedimiento se repite varias veces, en intervalos que tengan aproximadamente el mismo espacio en todo el rango que vaya a calibrarse, incluyendo la presión cero. El valor deseable es una calibración de 21 puntos (a intervalos del 5% de la escala total). 6.6.1.1 Cálculos Estos datos (si son razonablemente lineales - hasta un 0.1 - 0.5 % de la escala total) se ajustan en forma de polinomio, usando la técnica de los mínimos cuadrados. Una regla de oro para escoger el orden del polinomio es la siguiente: el número de puntos de datos que se toma en la calibración debe ser por lo menos el doble del número de coeficientes calculados a partir del ajuste polinomial. Para un valor de 21 puntos o más, una buena alternativa sería un polinomial de cuarto orden. En términos generales, es casi imposible ajustar el cero en el potenciómetro del Transductor de tal manera que los resultados del Transductor sean exactamente de voltaje cero a una presión de calibración de cero. Además, algunos ADC leen únicamente voltajes positivos (los voltajes negativos salen como cero). En este caso, el ajuste cero del
Transductor debe ser los suficientemente positivo como para que un arrastre del cero no ocasione nunca un voltaje negativo de la presión cero. Un polinomio de cuarto orden con el cual se ajustan datos de calibración tiene la siguiente forma: p = a0 + a1v + a2v2 + a3v3 + a4v4 (37) donde p es la presión actual (determinada a partir del estándar) y v es el voltaje que se ha tomado del Transductor para cada punto de datos. Si el voltaje de presión cero es positivo, entonces a0 con la regla de mínimos cuadrados va a ser negativo. Es de suma importancia tener calibraciones que pasen por cero a una presión cero. Para ilustrar este caso, supongamos que se ha presentado un cambio de voltaje a cero presión de un 5% de la escala total. - con los cambios de temperatura y tiempo - a partir del voltaje de presión cero en el momento de calibración. Este cambio en la magnitud de cero no es raro y no parece representar un peligro. No obstante, aunque tengamos un ajuste de campo perfecto, esto nos representa un 5% de error en las lecturas de presión a un 10% de la escala total o un 25% de error en una lectura de presión realizada a un 2% de la escala total. Para evitar este problema, el software operativo debe tener un comando que ponga en “cero matemático” todos los transductores. A menos que el valor de a0, tomado a partir de la calibración, tenga un valor idéntico a cero en la ecuación 37, el arrastre del cero no puede corregirse simplemente restando el voltaje presente a presión cero de todos los voltajes subsecuentes y luego utilizar estas diferencias en la ecuación 37. En vez de hacer esto, la ecuación 37 debe transformarse matemáticamente en: p = A0 + A1(v-v0) + A2(v-v0)2 + A3(v-v0)3 + A4(v-v0)4 (38) Para lograr esta transformación, se debe encontrar el “mejor ajuste” a voltaje de presión cero (v0) en el momento de efectuar la calibración. Este voltaje se calcula a partir de todos los puntos en la calibración, no solamente a partir del valor leído cuando se aplica una presión cero. Un primer estimativo se calcula a partir de la siguiente ecuación: v0 = -a0 / a1 (39) Este valor de v se inserta en la ecuación 37 para de esta manera calcular cual es el “mejor ajuste” de presión (p0) que corresponde a este voltaje. Si el valor de v0 fuese el correcto, entonces el valor de (p0) sería exactamente igual a cero. Este estimativo se perfecciona utilizando la siguiente ecuación: v0 [nuevo] = v0[viejo] - p0 / a1 (40) Este nuevo valor de voltaje a presión cero se inserta en la ecuación 37 para calcular un valor perfeccionado de p0. Este valor de v0 y de p0 se utilizan en la ecuación 40 para perfeccionar aún más v0. Este

proceso se continua desarrollando hasta que p0 ≤ 1 x 10-10 psig. A continuación, los coeficientes de A0....A4 se determinan por el método de los mínimos cuadrados de los datos de calibración originales utilizando la ecuación 38, determinando el valor final de v0 tal como se ha descrito. Las ecuaciones 37 y 38 nos producen exactamente la misma calibración, excepto que la Ecuación 38 (después de determinar sus coeficientes) nos permite la eliminación del arrastre cero insertando el valor actual de v0. Los coeficientes A0....A4 para un Transductor en particular deben colocarse en un archivo adecuado en ese mismo programa operativo del Transductor. El valor actual de v0 debe determinarse periódicamente utilizando el programa operativo de un instrumento en particular. Se logra ejerciendo una presurización a la máxima presión con respecto a la que normalmente se utiliza en el instrumento. Se ventila luego a presión atmosférica y se deja estabilizar (generalmente por 30 segundos). Una vez transcurrido este tiempo, se lee v0 y se almacena el valor para subsecuentes cálculos utilizando la ecuación 38. El ajuste realizado con un polinomial de cuarto orden reduce el promedio de desviación absoluta en un factor de 4 a 20, comparado con el método lineal de los mínimos cuadrados. La ecuación 38 también nos elimina el arrastre de cero. La mejora total (especialmente a presiones bajas) es significativo si se le compara con una calibración que no haya sido corregida para el arrastre de cero. El coeficiente A0 en la ecuación 38 en teoría debe ser igual a cero. Generalmente es menor que 1 x 10-8 y se calcula y reporta con el único fin de garantizar la transformación de la ecuación 37 en ecuación 38 se hizo bien. Si el objetivo es calcular la presión de salida de un ADC, se debe borrar el término A0 de la ecuación 38. 6.6.2 Calibración de las Medidores de Flujo de Masas Los medidores de flujo de masa responden a la tasa de flujo de masa y no a la tasa de flujo volumétrico. La tasa de flujo volumétrico en condiciones dadas de temperatura y presión (generalmente, la temperatura ambiente del momento y la presión atmosférica) de un gas que fluye a través de un medidor de flujo de masa se mide con un dispositivo independiente (como medidores de burbujas en el caso de tasas de flujo bajas, los cuales están conectados en serie con el medidor de flujo de masa). De manera alterna, se puede utilizar un pistón flotante con sellos de mercurio dentro de un dispositivo de desplazamiento volumétrico de precisión. El voltaje (o la lectura a escala que sea) tomada del medidor de flujo de masa se calibra con frecuencia contra esta tasa de flujo volumétrica, lo cual suministra una ecuación a partir de la tasa de flujo volumétrico que puede calcularse a partir del voltaje adquirido. Este procedimiento no es el correcto y limita de manera innecesaria la calibración !. Esta tasa de flujo volumétrico es valida únicamente si el medidor de flujo de masa se utiliza a exactamente la temperatura de calibración y presión. Los cambios ya sea en presión o
temperatura ocasionan errores en la calibración, los cuales pueden evitarse efectuando cambios simples de procedimiento. Debido a que la salida del medidor de flujo de masa es proporcional a la tasa de flujo de masa de un determinado gas, las tasas de flujo volumétrico que pasan a traves del medidor de burbujas durante un proceso de calibración debe multiplicarse por la presión absoluta en el medidor estándar (y no en el medidor de flujo de masa). Esto representa en términos generales la presión atmosférica actual que puede leerse utilizando un barómetro de mercurio de buena calidad que tenga las correcciones adecuadas. Esta presión debe convertirse a las mismas unidades que las utilizadas en el calculo de la permeabilidad (ejemplo: psia, Pa o atm). Además, las tasas de flujo volumétrico deben dividirse por la temperatura absoluta, (459.67 + oF) o (273.15 + oC) en el medidor estándar y por z , que es el factor de desviación del gas, calculado a la temperatura y presión que tiene el medidor estándar. El resultado de esta manipulación se define como “factor de flujo de masa” ymf. Ymf = qPcall
zcalTcal
(41)
Los subíndices cal se refieren a la presión y temperaturas absolutas que corresponden a la tasas de flujo volumétrico medidas durante la calibración. La ventaja de este procedimiento es que la calibración no se limita o restringe a ser exactamente la misma presión y temperatura medidas durante el tiempo de operación. El medidor de flujo de masa puede operarse a cualquier presión y temperatura que esté dentro de las especificaciones, excepto si ocurriesen cambios pequeños en el instrumento. Esto nos permite, por ejemplo, que el medidor de flujo se pueda utilizar en condiciones de contra corriente. La operación contra corriente tiene sus ventajas: (a) no va a entrar al medidor ni arena ni piedrecillas provenientes del núcleo lo cual hace que su exactitud disminuya y (b) virtualmente no se observa resistencia de flujo entre el medidor de masa y el tapón del núcleo, lo cual permite que se logre de manera más rápida las condiciones estables. Debido a que hay cambios en el instrumento, el medidor de flujo de masa debe calibrarse a diferentes presiones y temperaturas para de esta manera lograr resultados más precisos. Recuerde que aunque se reporten calibraciones para las condiciones en el medidor de flujo de masa, la presión y la temperatura en el medidor de burbujas (u otro dispositivo volumétrico) se utiliza para calcular los factores de flujo de masa. En condiciones normales, los efectos de los cambios de temperatura y presión son pequeños y pueden interpolarse fácilmente. 6.6.2.1 Cálculos En caso de que sea necesario ajustar los datos de calibración, se debe utilizar el mismo procedimiento descrito parra los transductores de presión para

producir la ecuación de calibración que pueda corregirse a un arrastre de cero. En las ecuaciones 37, 38 y 39, los valores de p y p0 se reemplazan por los valores ymf - ymf0, respectivamente. Si se calibran únicamente 6-8 tasas de flujo a cada presión, a continuación se debe reemplazar el polinomio de cuarto orden por un polinomio de segundo orden. Se debe obtener un mínimo de 11 puntos equidistantes para el polinomio de cuarto orden. Sea cual fuere el polinomio que se utilice, se debe probar con detenimiento calculando entre 50-100 puntos a lo largo del rango total para así garantizar que la ecuación final no nos va a producir resultados inesperados entre los puntos de calibración. El voltaje (o la lectura del instrumento) a una tasa cero de flujo de masa se determina en el tiempo de funcionamiento cerrando las válvulas en los extremos ascendente y descendente del medidor de flujo (en su presión actual) y asegurándose de que no hay flujo pasando. Una vez transcurridos 30 segundos necesarios para la estabilización, se debe registrar el dato a la salida del ADC, vo, y este valor debe restarse del voltaje flujo de salida, en el equivalente de flujo de la ecuación 38. Los siguientes son los factores de flujo de masa que se utilizan en el cálculo de la permeabilidad del gas: kg = Gf(P1-P2) (P1+P2) (42)
29392 µzmymfT
Donde: Kg = Permeabilidad de gas no corregida en millidarcys. = Viscosidad del gas en cp zm= Factor de desviación de la ley de los gases, calculado en la presión promedio de presión en el poro a la temperatura actual ymf = Factor de flujo de masa en (cm3/s) (psia/oR) o en (cm3/s) (psia/oK) T = Temperatura absoluta en el tiempo de corrida. Se utilizan las mismas unidades que en Tcal, oR o oK Gf = Factor geométrico dimensional para una configuración de flujo dada. En cm P1 = Presión ascendente absoluta en psia P2 = Presión descendente absoluta en psia. Dicho de otro modo, ymfT se utiliza en lugar de la expresión qrPr/zr en el cálculo de la permeabilidad. Si se utilizan unidades diferentes a las anteriores en la ecuación 42, la cifra 29392 se reemplaza por 2C2/C1 (ver tabla 6-1). 6.6.3 Calibración del Volumen de Referencia Los medidores de permeabilidad de gases que se encuentran en estado inestable contienen volúmenes de reserva y volúmenes muertos que deben calibrarse de la manera más precisa. Un método muy conveniente consiste en usar la expansión de gas junto con la Ley de Boyle, utilizando siete u ocho tapones de acero cilíndricos, cada uno de los cuales contiene un hueco axial cuidadosamente medido que se extiende a todo lo largo. La longitud y diámetro de cada hueco debe medirse con respecto a la pulgada
0.0001 más cercana. Estos tapones deben ajustar dentro del soporte de muestra y deben estar sellados perfectamente con una manga de goma que tiene el soporte. Los huecos pueden variar en volumen desde un valor cercano a cero hasta el volumen del depósito o reservorio que se va a calibrar. Los cambios de volumen del hueco de un tapón al siguiente deben ser aproximadamente iguales. El Transductor de presión debe calibrarse con mucho cuidado antes de efectuar la calibración volumétrica y debe leerse cero a un valor de cero psig. Los cambios de temperatura ambientales deben minimizarse durante la calibración volumétrica. 6.6.3.1 Cálculos Debido a que la calibración de cada sistema puede variar ligeramente, a continuación se ilustran únicamente los principios de calibración de acuerdo con la Ley de Boyle. La Ley de Boyle especifica que el total de las masas de gas en cada parte del sistema antes de una expansión es igual a la masa total después de la expansión, es decir, que no entra ni salle gas del sistema. Bajo condiciones isotérmicas, el grupo VP/z es proporcional a la masa de gas en el volumen V, donde P representa la presión absolutas y z el factor de desviación del gas a la presión y temperatura del gas contenido en el volumen V. Con respecto a la figura 6-26, supongamos que se ha cargado uno de los varios tapones de acero dentro del receptáculo y se ha sellado con una manga de goma. El depósito o reservorio de volumen Vo, que incluye el agujero de aislamiento de la válvula en forma de bola, el volumen muerto superior V1 que incluye el volumen dentro del Transductor y el tapón del extremo superior, el volumen conocido del hueco en el tapón de acero VH, y el volumen muerto inferior V2 están llenos de helio o de nitrógeno a una presión p1 (presión del calibrador). Para lograr un trabajo más preciso, este gas debe descargarse del sistema para eliminar el aire. El ciclo de descargue debe repetirse y el sistema se debe llenar por tercera vez. Luego se debe cerrar la válvula de aislamiento y el Vo se elimina a presión atmosférica. Una vez completado este procedimiento se cierra la válvula de eliminación. Una vez haya equilibrio en la presión, se lee uy se registra la presión final p1. Se procede a abrir entonces la válvula de aislamiento, expandiéndose el gas desde las porciones inferiores del sistema hacia Vo. Una vez se alcance el equilibrio, se lee y se registra la presión final p2 en todas las partes del sistema. Se carga cada uno de los tapones de acero restantes dentro del sostenedor y se repite el procedimiento. La presión atmosférica, pa debe leerse con un barómetro de mercurio que de buenos niveles de precisión y correcciones adecuadas. Se ingresa ahora en el lado izquierdo de la ecuación siguiente la masa de gas en cada parte del sistema antes de la expansión y las masas después de la expansión se escriben en el lado derecho, así:

(V1+V2+VHi) (P1+Pa) + (V0)(Pa/za) = (v1+v2+VH1+V0) ( z1 z2
P2+Pa)
Una ecuación similar se obtiene para cada uno de los tapones de acero. Estas ecuaciones se reorganizan para tener: (44) Los volúmenes V0 y - (V1 + V2) se consideran ahora la pendiente y el intercepto respectivamente a partir de la regresión lineal, donde VH es la variable y la cantidad dentro del paréntesis es la variable x para cada tapón. El volumen muerto superior V1 se halla insertando un tapón de acero sólido dentro del soporte del núcleo. El gas se expande a partir de este volumen hasta llegar el volumen que ahora es conocido para nosotros, Vo. Una ecuación similar a la ecuación 43 se escribe para la nueva configuración y se soluciona para el término V1. El volumen del depósito o reservorio Vr para una medición pasajera de la permeabilidad es igual a V1 cuando la válvula de aislamiento se encuentre cerrada o a (vo + V1) cuando se encuentre abierta. 6.8 Apéndices A.6.8.1 Derivada de Ecuaciones para calcular el ∧P y así aplicar la Ley de Darcy La “condición de flujo Stokes” que es un requisito para la utilización legítima de la ecuación de Darcy, se satisface cuando la resistencia al flujo de inercia es despreciable con relación a la resistencia de flujo viscosa. El propósito de este apéndice es presentar las ecuaciones para estimar el ∧P máximo permisible que se va a utilizar en las mediciones de permeabilidad de flujo axial. Si no se excede el máximo, la ecuación de Darcy puede utilizarse de una forma segura en la mayoría de los casos. Las ecuaciones se van a derivar para los gases y a continuación se escribe una ecuación para el flujo de líquidos. A.6.8.1.1 Gases Cuando la ecuación 17 (ver 6.2.1.2), - ecuación diferencial de Forchheimer para el flujo estable de gases - se corrige para pérdida de gas y se integra con respecto a la longitud, se convierte en: (A-1) Cuando el segundo término del lado derecho es pequeño en relación con el primer término, fFo es virtualmente igual a 1.0. Si asumimos este valor, si ajustamos todos los factores de desviación de gas a 1.0 (que una suposición válida para las presiones bajas) y si escogemos la presión media aritmética Pm para que sea la presión referencia, Pr, para la cual se calcula el valor q, la ecuación A-1 se simplifica así:
(A-2) donde vm, la velocidad Darcy, es igual a qm / A. Esto se puede escribir de otra manera de la siguiente forma: (A-3) En el numerador del segundo término dentro del paréntesis, Kg reemplaza el grupo de términos Km(1+b/Pm) para la ecuación 21. Este segundo término por completo no tiene dimensiones y puede considerarse como el “numero Reynolds para flujo a través de un medio poroso”. Ruth y Ma sugieren que este número debe llamarse el “numero de Forchheimer”. Los factores a la derecha de la ecuación A-4 se agrupan en varios componentes del número de Forchheimer, el cual es la proporción de inercia para la resistencia viscosa. El primer factor contiene las constantes (ver tabla 6-1) que hacen las dimensiones consistentes. El siguiente factor es la longitud característica, el cual es análogo al diámetro del tubo en el número de Reynolds para un fluido que pasa por tubos. El siguiente factor puede reconocerse como la densidad del gas a la presión del poro promedio. El factor final contiene la velocidad del fluido dividido por su viscosidad. La velocidad se obtiene dividiendo la velocidad Darcy por la porosidad de la roca. De esta manera, se basa entonces en el promedio actual de área disponible para flujo y no en el área superficial. El producto de esta velocidad y densidad de gas es el flujo de masa de gas a través del tapón. La porosidad incluida en la longitud característica cancela la porosidad en el factor final. A diferencia del flujo por un tubo, cuando haya una transición repentina de flujo laminar a turbulento en un rango estrecho en el número Reynolds, la resistencia de inercia asociada con el flujo a través del medio poroso se aumenta gradualmente y suavemente a media que el número de Forchheimer aumenta. Esta diferencia puede explicarse de una manera muy sencilla. En el flujo laminar dentro de un tubo, los fluidos se encuentran en posición paralela, unidireccional sin prácticamente ninguna aceleración. De repente, a cierta velocidad crítica, el flujo se vuelve inestable y sufre varios cambios de dirección, lo cual conlleva a la presentación de aceleraciones, las cuales aumentan en magnitud a medida y de manera suave a medida que el flujo de masa aumenta. De igual manera, hay todo un espectro de tamaños de tubos equivalentes en un medio poroso. Por lo tanto, no se observan transiciones repentinas de flujo laminar a turbulento, con un aumento considerable en el gradiente de presión. Cuando el número de Forchheimer es cero, toda la resistencia de flujo se debe a la viscosidad y la ley de Darcy se aplica completamente. Cuando el número es igual a uno, la mitad de la resistencia total del flujo se debe a la viscosidad. Si el número de Forchheimer es igual a nueve, únicamente el diez por ciento de la resistencia total del flujo equivale a la viscosidad y el otro noventa por ciento es resistencia por inercia.

El asterisco que aparece en la ecuación A-4 se utiliza para diferenciar el número de Forchheimer del número definido en la ecuación B-5 que contiene km en lugar de kg, Es correcto como se utiliza en las ecuaciones B-6 y B-7, aunque se necesita el término N*
fo en los siguientes cálculos. Si se pudiera encontrar un valor ∧p para un tapón de núcleo de tal manera que N*
fo fuese igual a 0.005 (es decir, el valor dentro del paréntesis en la ecuación A-3 es igual a 1.005), entonces la ecuación de Darcy (o lo que es equivalente a la ecuación A-3 con un valor de 1.000 dentro de los paréntesis) sub estimaría el término km en solamente un 0.5 %. Hallamos el valor de ∧p fijando primero el valor de N*
fo en la ecuación A-3 en 0.005 y luego resolviendo vm. (A-5) Este y el valor escogido para N*
fo se reemplazan en la ecuación A-4, la cual se resuelve para ∧p, teniendo en cuenta que Pm = p2 + ½ ∧p.: (A-6) La tarea ahora es encontrar los valores apropiados para β (en vista de la gran cantidad de dispersión que se observa cuando se plotea β vs k∞). Se tomó un enfoque un poco más conservador: los valores de beta que estaban en la curva superior punteada de la figura 6-3 se dividieron por 10. Esto corresponde a una curva que se ubica entre la curva sólida central por un factor de aproximadamente 3 para los valores bajos de km y gradualmente cae por debajo de la curva central a las permeabilidades más altas. Las betas en este caso se esperaría que fuesen de algún modo menores que los valores escogidos y el error en que se incurre de calcular la permeabilidad utilizando la ecuación de Darcy (siempre y cuando ∧p max calculada de la misma manera a como se hizo en la ecuación A-6 no se supera durante la medición de la permeabilidad) sería menor de 0.5 %. De otro lado, si el valor beta del tapón se ubica en la línea punteada superior, la utilización del mismo ∧p max resultaría en una permeabilidad calculada que es baja en un 5%. Si el valor beta del tapón fuese el doble el valor mostrado en la parte superior de la curva punteada, el error sería del 10 % y a sí sucesivamente. El cálculo de ∧p max no se obtiene de manera directa. La ecuación A-6 contiene los valores b, kg y k∞. Con el fin de hacer la figura 6-4, b se halló a partir de una correlación propuesta por Jones para los datos ubicados en una gran cantidad de tapones utilizando helio. bHt = 16.4 k∞
-.0382 (A-7) donde el término bHt representa el valor Klinkenberg para pérdida de gas para el helio en psi y k∞ es la permeabilidad corregida de escape en millidarcys. Para calcular bair la constante 16.4 en la ecuación A-7 se convierte en 5.71 utilizando la técnica que se
muestra en la ecuación 31. Los valores para C1, C2, C3 y R para las unidades deseadas se encuentran en la tabla 6-1. El kair que se necesita se calcula a partir de la siguiente ecuación: (A-8) Debido a que se desconoce le valor del ∧p max, utilice el k∞ como la primera opción para saber kg en la ecuación A-6, y solucione el valor de ∧p max. Inserte este valor en la ecuación A-6 para el término kg y continúe de esta manera para definir los estimativos hasta que no haya más cambios en el valor de ∧p max. La convergencia se logra generalmente en 4 -10 cálculos. Los resultados de estos cálculos se muestran en la figura 6-4 para las presiones descendentes de 14.7, 50, 100 y 2000 psia para el aire a temperatura ambiente. Los valores de ∧p / L se reportan en forma de “psi por pulgada de núcleo”. Aunque estas proporciones dependen ligeramente de la longitud y se calcularon para tapones que tenían 2 pulgadas de largo, los resultados son muy aproximados para nuestro propósito en tapones que medían entre 1-3 pulgadas. Si empezamos con las permeabilidades más altas para una presión de 14.7 psia de presión descendente, la máxima caída de presión permitida aumenta a medida que la permeabilidad disminuye, tal como se espera. Sin embargo, para permeabilidades que se ubican por debajo de 0.3 millidarcys, ∧p max /L disminuye al disminuir la permeabilidad. Este resultado tan inesperado se debe a pérdidas de gas. Las pérdidas de gas por escapes son mayores cuando las permeabilidades son más bajas, ocasionando tasas más altas de flujo de gas que las que se encuentran sin escapes. Se disminuye cuando el promedio de presión de poro se aumenta a presiones superiores. Cuando hay permeabilidades más altas, los valores de ∧p max / L se reducen al aumentar la presión posterior. Esto se debe principalmente a un incremento en la densidad del gas, lo cual aumenta el flujo de masa y por lo tanto, resistencia de la inercia para una velocidad dada. A.6.8.1.2 Líquidos Los líquidos se consideran casi incompresibles y no están sujetos a escapes. Por lo tanto la derivada para la máxima caída de presión para mantener las condiciones del flujo de Stokes es más simple que lo que se ha descrito para los gases. Escojamos de nuevo un valor de 0.005 para el número de Forchheimer y la misma relación de beta vs permeabilidad que se utilizó en los gases. La ecuación para los líquidos sería:

(A-9) donde: ∧p max = Máxima caída de presión para el flujo de Stokes, en psi. L: Longitud del tapón del núcleo, en pulgadas µL = Viscosidad del líquido, en cp ρL = Densidad del líquido en g/ centímetros cúbicos β = Coeficiente de resistividad inercial, en pies a la menos uno kL = Permeabilidad del tapón al líquido, en millidarcys. Dados beta y kL, el lado izquierdo de la ecuación A-9 se aplica para un núcleo de cualquier longitud. Los resultados se grafican en la figura A 6-1. La utilización de esta figura se ilustra de la siguiente manera: supongamos que se ha determinado la permeabilidad de un tapón de núcleo de longitud 2.50 pulgadas con una caída de presión de 35.8 psi y un líquido con viscosidad de 1.53 Contrato de Compra y densidad 0.816 g / cm3. Se ha calculado una permeabilidad de 99.8 millidarcys utilizando la ley de Darcy. Se considera válida la utilización de la ley de Darcy ? Si tomamos la figura A6-1, una permeabilidad de 99.8 nos da un valor de aproximadamente 6.36, valor que es igual al lado izquierdo de la ecuación A-9. ∧pmaxρl / Lµ2 = 6.36 Por lo tanto, ∧pmax = 6.36 x 2.50 x 1.532 / 0.816 = 45.6 psi debido a que se utiliza ∧p para la medición, y que 35.8 fue menor que el máximo valor permitido, concluimos que la ecuación de Darcy nos dio resultados válidos. B6.8.2 Calculo de la Permeabilidad a partir de mediciones de Flujo de Gas Axial y de Caída de Presión La ecuación para flujo axial utilizada junto con el método de caída de presión propuesto por Jones, comienza con la ecuación es estado estable de Forchheimer para el flujo isotérmico axial, corregida para el escape de gas. Se obtiene integrando la ecuación 17 (ver 6.2.1.2) lo cual da como resultado: (B-1) donde: (B-2) (B-3) (B-4) (B-5) (B-6) y (B-7) El número de Forchheimer, NF0 (ecuación B-5) que es la proporción sin dimensiones de la inercia sobre
resistividad viscosa, constituye un “número Reynolds para el flujo a través de un medio”. Esto se discute con mayor detalle en el apéndice A. El “Factor de interacción de Forchheimer” ƒF0 (ecuación B-7) es un número sin dimensiones que tiene su máximo valor de 1.0 (siempre que b o NF0 tienda a cero). Su valor mínimo para cualquier caso práctico es 0.95 La tasa de flujo instantáneo que ingresa a la muestra se determina a partir del depósito o reservorio total conectado y el volumen múltiple ascendente en la abertura del núcleo Vr y la derivada presión - tiempo: (B-8) donde: (B-9) representa una corrección para los gases no ideales. El subíndice 1 en esta ecuación se refiere a las condiciones ascendentes de la abertura de la muestra. Debido a que todas las tasas de flujo, las presiones (excepto la presión atmosférica) y factores de desviación de gas se refieren a esta localización, este subíndice se disminuye en las ecuaciones consignadas más adelante. El subíndice n se refiere a tiempo, no a posición. En términos generales es más útil medir la presión del calibrador que la presión absoluta que se requiere en la ecuación B-8. Debido a que la muestra se desocupa directamente en la presión atmosférica, la presión del calibrador es igual a ∧p a lo largo de una muestra en cualquier instante. Si multiplicamos y dividimos la ecuación B-8 por p y consideramos dp = dP, obtenemos lo siguiente: (B-10) Podemos definir una función de tasa de flujo instantánea y así: (B-11) La derivada del a ecuación B-11 se aproxima con precisión a partir de cualquier par de puntos de presión - tiempo adyacentes: (B-12) Esta aproximación equivale a la pendiente verdadera (derivada) del ln [p] vs la curva de tiempo (ver figura B 6-1) en el punto medio de presión del intervalo, que corresponde a la media geométrica de las dos presiones: (B-13) Por lo tanto, ya se calcula para el instante cuando la presión ascendente es pxn. La presión media del poro en la muestra es: (B-14)

Ahora, excepto por una sola complicación, cualquiera de los varios conjuntos de valores para yn, pxn, pmn y ƒF0 obtenidos a partir de caídas de presión se pueden utilizar para calcular las variables de flujo en la ecuación. Con un flujo estable, donde la presión en cualquier punto de la muestra no varía con el tiempo, la tasa de flujo de masa es constante a lo largo de la muestra. La complicación es que ese concepto no es cierto para flujos inestables. Cada vez que la presión en cualquier punto de la muestra disminuye con el tiempo, la densidad del gas en ese punto disminuye proporcionalmente lo cual da como resultado la depleción de masa de gas. Debido a que la presión ascendente disminuye continuamente durante la caída de la presión, la presión en toda la longitud de la muestra también disminuye, excepto en el extremo donde se ubica la salida, donde siempre se registra la presión atmosférica. La tasa de depleción de masa del gas residente en la muestra está acompañada por un incremento igual en la proporción de masa del gas que fluye. Por lo tanto, en un momento dado mientras ocurre un descenso de presión transitorio, la tasa de flujo de masa que sale de la muestra es mayor que la proporción de masa que está ingresando a la muestra. Si el volumen del gas del depósito o reservorio , Vr, es grande comparado con el volumen del poro de la muestra, Vp, el incremento relativo es pequeño. Si estos volúmenes son comparables, el incremente es bastante significativo. En el límite, un volumen infinito en el depósito o reservorio nos produce un flujo estable - sin incremento. En el extremo opuesto, cuando el volumen del depósito o reservorio es cero, todo el gas que sale de la muestra se origina a partir de la misma muestra. Debido a que el flujo de masa no es constante a lo largo de la muestra en cualquier momento durante la presión transitoria, no podemos utilizar la solución estable directamente. La solución correcta puede lograrse utilizando una técnica repetida en la que la ecuación de continuidad se separa de la ecuación de flujo aunque al final ambas ecuaciones se satisfacen dentro de una tolerancia aceptable: la derivada parcial de la densidad del gas respecto al tiempo en cualquier longitud tomada a partir de la solución estable de la ecuación de flujo, se inserta en la ecuación de continuidad a partir de la cual se deriva la variación aproximada del flujo de masa con la longitud. Este proceso de derivación se hace para varias condiciones de presión, magnitud del factor de escape y proporciones de volumen de poro con respecto a volumen del depósito o reservorio. El flujo de masa corregido y no constante se inserta en la forma diferencial de la ecuación de flujo, a la cual se la hace una re - integración con respecto a la longitud. El proceso se repite hasta que los cambios se consideren despreciables. Se consideraron cuatro repeticiones como el número adecuado en todos los casos prácticos. La tasa de flujo corregida que puede insertarse entro de la ecuación de flujo integrada para permitir el incremento del promedio de peso en el flujo de masa con una longitud en cualquier momento es: (B-15)
donde: (B-16) y: (B-17) Los factores de corrección para un flujo de masa de gas que nos s constante Gm[cn,ϒ] se muestra como función de c para los diferentes valores de ϒ en la figura B6-2. Podemos ahora definir una “función de tasa de flujo instantánea” ϒcn así: (B-18) donde el término ƒzn (ecuación B-9) se evalúa a una presión Pn, donde Pn = pgn + Pa. Esta función puede insertarse en la ecuación B-1 para producir la forma integrada de la ecuación de Forchheimer para flujo temporal: (B-19) El numero de Forchheimer (Ecuación B-5) se debe calcular para cada ƒFon de la ecuación ya descrita. En términos de la función de flujo corregido, esto equivale a: (B-20) En (ecuación B-6) debe calcularse para cada punto de datos que se obtiene para de esta manera calcular ƒFon (ecuación B-7). Los coeficientes A1 y A2 de la ecuación B-19, los cuales nos dan km y beta respectivamente, se determinan a partir del conjunto de ecuaciones correspondiente a cada punto de datos, n, utilizando la regresión lineal de la siguiente manera: 1. Calcule un valor inicial para el factor de escape, b. Para el helio esta dado por la siguiente ecuación: (B-21) donde ycm representa la función de flujo que corresponde al valor más bajo de pg que se ha obtenido. 2. Fijar ƒFo para todos los n. 3. Calcular cn (ecuación B-17) y ycn (ecuaciones B-12
y B-18) para cada n. 4. Calcular los términos de la mano izquierda y
derecha en la ecuación B-19 para cada n 5. Hacer una regresión lineal para obtener A1 y A2. 6. A partir de estos valores, calcular Nfo (ecuación B-
20), E (ecuación B-6) y ƒFo (de la ecuación B-7) para cada n.
7. Repetir los pasos 4 al 6 hasta que el cambio de cualquiera de los ƒF0 no exceda el valor

0.001tomando el valor de la repetición pasada. Se recomienda hacer tres repeticiones.
8. Calcular un error estándar a partir de la última regresión lineal.
9. Escoger un nuevo b que es 10% mayor que el primer b que se calculó.
10. Repetir los pasos 3-8 utilizando el conjunto de valores que se ha calculado para ƒF0 y el nuevo valor de b.
11. Si el nuevo error estándar es menor que el anterior, aumente b en un 10%. Repita el paso 10.
12. Continúe aumentando o disminuyendo el valor de b, repitiendo el paso 10 hasta que encuentre el error estándar mínimo. Este corresponde a los mejores valores para b, A1 y A2, considerando los mínimos cuadrados. Calcule km y beta a partir de A1 y A2 (ecuaciones B-2 y B-3 respectivamente).
En la figura B 6-3 se muestran los cambios típicos que se presentan en k∞, β y el error estándar SE con respecto a B (la escala para el SE se omite para mayor claridad). A medida que se incrementa el valor de b disminuyen los valores calculados para k∞, β. El valor correcto en este ejemplo para b, el cual se observa en el error estándar mínimo es 60.6 psi. El valor calculado de β se convierte en cero a un factor de escape de 61.5 psi y es negativo para valores de bs mayores. Si el calculo da como resultado un valor negativo para β, esto significa que el valor de b es demasiado alto. Sin embargo, valores negativos de β también se pueden obtener por sensibilidad extrema de la permeabilidad ante cambios en el estrés neto. Los cambios en km, β con respecto a b que se muestran en la figura B 6-3 se consideran generales, en el sentido de que siempre disminuyen al aumentar b. Con respecto a los cálculos por computador, podemos afirmar que hay mayor trabajo en la técnica de caída de presión comparado con los cálculos que se hacen para condiciones estables. Sin embargo, el procedimiento completo que hemos descrito se realiza casi que de manera instantánea en un computador pequeño. Las rocas de baja permeabilidad a diferenciales pequeños con frecuencia muestran poca resistencia inercial. En estos casos, el término en la ecuación B-19 que se multiplica por A2 es pequeño, ƒF0 es casi igual a 1 y la ecuación B-19 se puede aproximar de la siguiente manera: (B-22) Para que esta aproximación sea válida, la gráfica ploteada de yznzmn vs Pmn debe ser lineal. La ecuación B-22 es la ecuación de Darcy, corregida para escapes de gas para las mediciones de caída de presión. Su pendiente e intercepto son igual a 1 / A1 y b / A1 respectivamente. Por lo tanto, los valores de km y b se calculan a partir de: (B-23)
y b = (Intercepto) / (Pendiente) (B-24) La desviación de la propiedad lineal, especialmente cuando el valor de Pmn aumenta, indica que la resistencia inercial no es despreciable en estas condiciones. C.6.8.3 Cálculo de la Permeabilidad a partir de Mediciones de Caída de Presión y de Flujo de Gas en la sonda Las funciones de tasa de flujo instantáneo yn y la media geométrica de las presiones pg se calculan a partir de la ecuación B-12 y B-13, respectivamente. Estos valores se insertan en: (C-1) donde: (C-2) y: (C-3) El factor dimensional geométrico para el término de Forchheimer GƒF0 se debe determinar por modelamiento numérico. Si se puede determinar el factor geométrico, el término β puede calcularse a partir de la ecuación C-3. Utilice la ecuación C-1 para corregir la resistencia inercial, sin interesar si se puede calcular β. Nótese que a excepción de los diversos factores geométricos que tienen los término A3 y A4, la ecuación C-1 es virtualmente idéntica al ecuación B-19 (para el flujo axial) excepto por la realización de ciertas simplificaciones en las ecuaciones del permeametro - sonda. Estas simplificaciones son: (a) se omiten factores como la desviación de gas z y la interacción de Forchheimer ƒF0. (b) Los valores de yn no están corregidos para el flujo de masa no constante (espacial). Con la utilización de las dimensiones sonda - sello, el volumen del poro que influye la medición de la permeabilidad es tan pequeño, con relación al volumen aún del más pequeño depósito o reservorio de gas, que la suposición de un estado estable de flujo de masa constante a través de una superficie isopotencial no presenta ningún error apreciable en el cálculo de la permeabilidad. Por ejemplo, el error con un radio de sonda de 3.8 mm y un volumen de depósito o reservorio de 5.2 cm3 es menor del 0.1 %. El término b* que aparece en la ecuación C-1 es un factor de escape de gas que se toma a partir de una correlación. Aunque en teoría b no puede determinarse directamente a partir de los datos de caída de presión, el rango de las presiones de poro promedio medidas (con una presión de llenado de 10 psig o menos) es demasiado pequeño para determinar el valor de `b de manera confiable. La correlación de

b* se obtiene utilizando la técnica iterativa o de repeticiones. La primera aproximación (para nitrógeno en psi) se calcula utilizando: (C-4) donde ym corresponde al valor más bajo de pg obtenido a partir los datos de caída de presión. Este primer valor hallado se utiliza en la ecuación C-1 para calcular los términos a la izquierda y a la derecha para cada valor yn y pgn que se obtienen. A continuación, se hallan los coeficientes A3 y A4 a partir de la ecuación C-1 utilizando la regresión lineal. El primer estimativo de la permeabilidad de Klinkenberg se obtiene reacomodando la ecuación C-2 así: (C-5) Utilizando este k∞, se puede hallar un mejor estimativo de b*N2 teniendo en cuenta la correlación: b*N2 = 6.9 k∞ -0832. (C-6) Este nuevo valor de b* se vuelve a insertar en la ecuación C-1 y el proceso se repite hasta que el cambio en b* de una repetición a otra sea menor de 0.1 psi. La convergencia se obtiene al cabo de 2-4 repeticiones. La resistencia inercial virtualmente desaparece para las muestras que tienen permeabilidades menores a 0.1 millidarcys para 0presiones iniciales de menos de 10 psig. Para estas muestras, se puede ignorar el término A4 en la ecuación C-1 y la ecuación Forchheimer reduce la ecuación de escape corregido de Darcy: (C-7) Esta ecuación puede utilizarse cuando se obtienen únicamente tres puntos y - pg para muestras de baja permeabilidad . El término y* y pg* se refieren a cualquiera de estos puntos. El calculo se hace de manera iterativa nuevamente. El estimativo inicial de b* se calcula a partir de la ecuación C-4. El estimativo se perfecciona calculando K∞ (ecuación C-7) el cual se utilizará luego en la ecuación C-6. El proceso se continua hasta que el cambio de b* de una operación a la otra sea menor de 0.1 psi. La convergencia se obtiene generalmente al cabo de 2-4 iteraciones. La permeabilidad del gas kg para un gas dado a cualquier presión de poro promedio puede calcularse a partir de k∞ y b* como se ha descrito para 6.4.1.1.5 en el caso del flujo axial. D.6.8.4 Cálculo de Permeabilidad a partir de Mediciones de Disminución del Pulso y Flujo de Gas Axial La diferencia presión inicial a lo largo de la muestra ∧p [0] que es ligeramente menor que el ∧p 1, (ver 6.4.1.3.2) debe calcularse a partir de un balance de masa de gas: (D-1)
V1, el volumen ascendente total, incluye: los volúmenes internos del depósito o reservorio ascendente, la cámara ascendente del Transductor de presión diferencial, las líneas conectoras al Transductor en la válvulas 1, 2 y la válvula de llenado, el volumen muerto ascendente Vd (volumen dentro de la válvula 1), el tapón del extremo ascendente y la línea que los conecta. El volumen total descendente, V2 incluye los volúmenes del depósito o reservorio descendente, el transductor y el tapón del extremo, la cámara descendente del Transductor diferencial y las líneas que lo conectan incluyendo la línea a la válvula 2. Los volúmenes V1, V2 y Vd deben hallarse con una calibración cuidadosa utilizando las técnicas de Boyle (ver sección 6.6.3.1). La presión diferencial y la presión descendente se monitorean como funciones del tiempo. Los datos se analizan a partir de la solución de la ecuación de difusividad, que se deriva combinando la forma diferencial de la Ley de Darcy con la ecuación de continuidad. La muestra se supone que inicialmente está a una presión uniforme del poro. Luego en el tiempo t = 0, un pulso de presión ligeramente mayor (∧p[0] se aplica a su extremo ascendente a partir del depósito o reservorio ascendente. A medida que el gas fluye de V1 a la muestra, la presión V1 disminuye. La presión en V2 permanece constante por un período corto de tiempo hasta que el pulso de presión ha atravesado la longitud de la muestra. Luego aumenta la presión en V2. Debido a la disminución de P1[t] y al aumente de P2[t], el ∧p[t] continua disminuyendo y gradualmente se aproxima a cero a medida que las presiones ascendentes y descendentes se igualan. La tasa de disminución de la presión depende de la permeabilidad: entre más baja sea la permeabilidad, más baja será la disminución. La solución general para la diferencia de presión como función del tiempo la presenta Dicker y Smits, los cuales se basaron en un trabajo original hecho por Brace et al., la solución de error-función propuesta por Bourbie y Walls, la solución analítica general de Hsieh et al y en los trabajos de otros autores como Chen y Stagg, Haskett et al y otros. El resultado es el siguiente: (D-2) donde θm son las raíces de la ecuación: (D-3) a es la proporción del almacenamiento compresivo (el producto del volumen por la compresibilidad) del volumen del poro de la muestra de tal manera que la del depósito o reservorio es: (D-4) b es la proporción de almacenamiento compresivo del volumen del poro de la muestra de tal manera que la del depósito o reservorio descendente es: (D-5)

y tD es el tiempo sin dimensión representado por: (D-6) donde C1 y C2 son constantes de conversión dadas en la tabla 6-1 que se necesitan para hacer Td sin dimensión. Los términos cg, cpv, cv y cv2 son las compresibilidades del gas, el volumen del poro de la muestra y los depósito o reservorios ascendente y descendente respectivamente. La compresibilidad del gas es: (D-7) en la ecuación D-7, hemos identificado ƒz como el valor del grupo dentro de los paréntesis. Que es el responsable de la desviación del comportamiento de gas ideal. A ciertas presiones, esta desviación no es despreciable. Jones suministra los valores de ƒz para el nitrógeno a 72oF. Si el sistema se construye de manera rígida, es decir, con vasos de paredes gruesas y si los volúmenes de desplazamiento de los transductores son pequeños en relación con V1, y V2 entonces cv1 y cv2 son pequeños comparados con cg y pueden ignorarse. La compresibilidad del volumen de poro varía considerablemente entre muestras de diferentes litologías. Sin embargo, se minimiza utilizando niveles de estrés alto en confinamiento. Generalmente es menor de 2 x 10-5 psi-1 para niveles de estrés de 5000 psi o más. Un cpv de 1 x 10-5 psi-1 es igual a 1% de la compresibilidad de gas a una presión de 1000 psia o 2% a 2000 psia. Las diferencias de presión al cuadrado (que se aplica únicamente a los gases) que aparecen a mano izquierda de la ecuación D2 son: (D-8) La ecuación D-2 se aplica a todos los valores de a, b y tp siempre y cuando se utilicen todas las raíces de la ecuación D-3 y sean constantes todas las compresibilidades. Haskett y colaboradores minimizan el efecto de la restricción incorporando la compresibilidad que varía con el tiempo dentro del “pseudo tiempo ajustado”. Sin embargo, con las condiciones experimentales apropiadas, ninguna de las restricciones representa un problema serio, como se verá más adelante. La utilización de diferencias de presión al cuadrado en la ecuación D-2 es casi equivalente a la utilización de pseudo-presiones como lo sugirió Haskett y colaboradores. Debido a que el cambio en la presión media del poro es tan pequeño en la técnica ya descrita, los cambios son pequeños en el factor z y en la viscosidad del gas. Las proporciones de estos factores entre el numerador y el denominador de la ecuación D-9 son casi iguales a la unidad. Para las condiciones experimentales diferentes a las descritas más adelante, donde las variaciones en el promedio de presión de poro son mayores, se recomienda la utilización de pseudo - presiones. La escogencia de tamaños de depósito o reservorio no se encuentra limitada directamente por la ecuación D-2. Algunos investigadores han utilizado vasos
ascendentes grandes (a≡0) o vasos descendentes grandes (b≡0). Otros están a favor de la utilización de depósito o reservorios que son similares en magnitud al volumen del poro de la muestra. (a o b ≡0.2 a 5). En términos generales, para mantener los tiempos de medición razonablemente cortos con muestras de baja permeabilidad, por lo menos uno de los depósito o reservorios debe ser pequeño. Dicker y Smits nos presentan evidencias contundentes que hace que los volúmenes ascendentes y descendentes sean idénticos y relativamente pequeños. Los volúmenes iguales crean una situación deseable de simetría. A continuación del período en donde el pulso de presión atraviesa la longitud de la muestra, la disminución de presión en el vaso ascendente trae como consecuencia un incremento de presión descendente de igual magnitud, manteniéndose de esta manera constante el promedio de presión de poro y reduciéndose los cambios compresionales en el volumen total de poro hasta llegar casi a cero. Debido a que la presión promedio en la mitad ascendente (de una muestra homogénea) disminuye con el tiempo, mientras que la presión promedio en la mitad descendente se incrementa en la misma cantidad, la tasa de flujo que ingresa a la muestra es igual a aquella que sale de la muestra en ese mismo instante. La tasa máxima en cualquier momento ocurre en la mitad del trayecto de una muestra razonablemente homogénea. Otra consecuencia de la simetría (donde a = b) es que todas las raíces pares de la ecuación D-3 se cancelan en la ecuación D-2. La primera raíz de la ecuación D-3, θ1 es ligeramente menor que la raíz cuadrada de (a + b) aún para los valores pequeños de a y de b (en grandes depósito o reservorios). Si definimos ƒ1 como: (D-9) entonces, ƒ1 = 1000 cuando a + b = 0 y gradualmente disminuye a medida que a + b se vuelve más grande, tal como se muestra en la figura D. 6-1. En esta figura, Vs es el valor más pequeño entre V1 y V2 y VL es el más grande de los dos volúmenes de depósito o reservorios. Se halla el cuadrado de la primera raíz de la ecuación D-3, así: θ1
2 = ƒ1 (a + b) (D-10) Las raíces más altas de la ecuación D-3 son iguales a: θm = (m -1) ∏, para (a+b) = 0 (D-11) y se incrementa gradualmente a medida que (a+b) aumenta. Exceptuando los primeros momentos cuando tD es pequeña, únicamente el primer término de la suma(ecuación D-2) es significativo. El segundo término es cero cuando a es igual a b. Con un valor de tD de 0.1, todos los términos superiores aumentan únicamente 0.16% a la contribución del primer término

cuando a= b =1 y cantidades menores para valores más pequeños de a y de b. Por lo tanto, si no se utilizan tiempos iniciales, la ecuación D-2 se reduce a un exponencial simple, el cual se puede escribir de la siguiente manera: (D-12) donde: (D-13) De esta manera, si se plotean los valores experimentales en el ln[∧p[t])2 /∧p[0])2 ] vs tiempo, nos daría como resultado una línea recta (excepto en los tiempos iniciales), la cual se intercepta en ln [ƒ0] y una pendiente (slope) negativa de valor absoluto: (D-14) donde los términos C1 y C2 son constantes tomadas de la tabla 6-1 que se utilizan para hacer consistentes las unidades. Si cv1 y cv2 son despreciables comparados con cg, entonces: (D-15) La presión promedia de poro en la ecuación D-15, Pm, es la presión absoluta. Cuando a = b y la muestra de roca es homogénea ( o si a y b son pequeños, es decir, menores de 0.3), Pm no debe cambiar durante la porción lineal de la caída semi-logarítmica. Se calcula a partir de: Pm [t] = P2[t] + ½ ∧p [t] (D-16) El cálculo de la pendiente puede ejecutarse durante la medición utilizando la más reciente de las dos lecturas de tiempo - presión. (D-17) El valor absoluto de esta pendiente será inicialmente alto (si a ≡ b) y descenderá de manera relativamente rápida, volviéndose constante a medida que el tiempo aumenta. Una vez que la pendiente se ha vuelto constante para varias lecturas, se puede obtener el intercepto y la pendiente de la porción lineal del descenso con la regresión lineal de: (D-18) donde Ao es el intercepto y A1 es la pendiente. Unicamente se van a utilizar en la regresión aquellos puntos de datos que nos den una pendiente constante (a partir de la ecuación D-17). De la ecuación D-12 vemos que ƒo se puede obtener a partir del intercepto: ƒo = exp [Ao]. (D-19) La ecuación D-13 nos muestra que ƒo está en función de a y de b, de igual manera que θ1. Cuando a = b, ƒ1 es ligeramente mayor que ƒo: 0.0055% para a = 0.1 ; 0.12% para a = 0.5 y o.44% para a = 1.0. Los valores
de θ12, ƒ1 y ƒo se relacionan en la tabla D.6-1 donde se
consideran varios valores de a, cuando a = b. Estos valores pueden calcularse a partir de las ecuaciones D-3, D-10 y D-13 cuando los depósito o reservorios no son de igual volumen (o para ser más exactos, de igual almacenamiento compresivo). La figura D.6-2 nos muestra un ƒ1 como función de ƒo para diferentes proporciones V2 / V1. Debido a que V1 y V2 se determinan por procesos de calibración, ƒ1 puede determinarse únicamente a partir de ƒo y la permeabilidad se calcula a partir de: (D-20) Utilizando el valor de ƒo obtenido, el volumen de poro de la muestra puede estimarse a partir de: (D-21) donde el valor de a se halla a partir de la tabla 6-10, o de la figura D.6-1. Si se estima el cpv como igual a 8x10-6psi-1, lo cual puede ser demasiado alto o demasiado bajo por un factor de 1 - 4 comparado con el valor real a un estrés efectivo neto alto (4000-5000 psi)el error máximo de la su[posición es aproximadamente 1.6% para Pm = 1000 psia o 3.2% para Pm = 2000 psia. A valores netos de estrés más bajos, se esperaría que cpv fuese más alto y variable. El transductor de presión diferencial debe ajustarse a cero por medios matemáticos o eléctricos antes de cerrar la válvula No. 2 (en el momento en que la verdadera presión diferencial es cero en P2[0]). Si el Transductor mantiene su linearidad, aunque su ganancia o sensibilidad pueda cambiar ligeramente desplazándose de una presión de referencia baja a una alta, (es decir, cuando P2 se sube de la presión atmosférica a 1000-2000 psia) no se incurre en ningún error (excepto en el cálculo de Pm) debido a que todos los valores de ∧p son relativos, se presentan como proporciones. Tabla D.6-1 Constantes de Descenso de Pulsos cuando a=b (V1=V2, o S1=S2) Página 6-57 El rango práctico de permeabilidad para la medición del descenso del pulso es aproximadamente entre 0.1 millidarcy a 0.01 microdarcy (1 E-4 a 1 E-8 µm2). El tiempo de descenso del pulso es casi proporcional a la longitud de la muestra e inversamente proporcional a la permeabilidad y a la presión del poro promedio. El tiempo también depende de cuanto se ha medido del descenso del pulso. Como guía general, los volúmenes de los depósito o reservorios V1 y V2 debe ser por lo menos tan grandes como el volumen más grande del poro y por lo tanto 2-10 veces el volumen del poro de la mayoría de las muestras que se van a examinar. El ascenso de la porción semilogarítmica lineal del pulso ocurre casi al mismo tiempo transcurrido, sin interesar la proporción PV - volumen del depósito o reservorio. El trabajo

realizado por Kamath y colaboradores nos muestra que la sensibilidad a la heterogeneidad en la permeabilidad se reduce a medida que a y b se vuelven más pequeñas. (depósito o reservorios más grandes). El límite de permeabilidad superior se puede aumentar incrementando el volumen de los depósito o reservorios. Sin embargo, la sensibilidad (y por lo tanto la precisión) de la determinación del volumen del poro (Ecuación D-21) se reduce con volúmenes más grandes. Quizá la parte que más tiempo conlleva realizar en esta técnica es la de permitir que el sistema alcance la verdadera presión de equilibrio antes de la generación del pulso de presión. A medida que la presión se hace uniforme en todas partes del sistema, las fuerzas se aproximan a cero. Si el volumen del poro de una muestra se determina de manera independiente, el paso para equilibrar la presión se puede eliminar, tal como lo sugiere Jones. E.6.8.5 Cálculo de Permeabilidad a partir del descenso de Pulso y Mediciones de Flujo de Líquido Axial E.6.8.5.1 Aparato y Medición de Almacenamiento Compresivo. Un método conveniente para medir el almacenamiento compresivo en el aparato para las mediciones de descenso en el pulso es incluir una válvula de aguja con tornillo micrométrico (como se ve en la figura 6-25), el cual cuenta con un volumen de desplazamiento calibrado total (área corte transversal del émbolo en la parte alargada del sello multiplicada por la longitud de su golpe variable) de casi 0.02 a 0.05 cm3. Las válvulas 1 y 2 deben de tal configuración que no tengan cambios internos de volumen cuando se abran o se cierren. Para medir los almacenamientos, cargue un cilindro de acero sólido dentro del sostenedor de la muestra y aplique un estrés de confinamiento. Con las válvulas 1 y 2 abiertas, aplique un vacío a la válvula marcada fill / vacuum (llenado / vacío) para retirar todo el aire del aparato. Luego llene el sistema por completo con líquido libre de gas hasta el 80% aproximadamente de la escala total de presión en el transductor de presión descendente. Una vez que se haya estabilizado la presión, cierre las válvulas 1 y 2. La válvula de aguja, válvula 3, debe estar casi abierta. El Transductor de presión diferencial debe leer cero y la lectura p2 no debe cambiar cuando las válvulas 1 y 2 están cerradas. Registre la lectura del micrómetro. Luego atornille el micrómetro, disminuyendo el volumen del sistema ascendente hasta que el Transductor de presión diferencial marque la escala total, o sea 100 psi. Este aumento de presión desde una lectura cero inicial se denomina ∧p1, y el descenso de volumen, calculado a partir de la lectura del micrómetro es ∧V1. Debido a que se excluyó el volumen entre la válvula 1 y el extremo ascendente del tapón de acero en el soporte de la muestra (“volumen muerto ascendente”) de la medición de compresión, el “almacenamiento
compresivo ascendente” S1 menos el “almacenamiento compresivo del volumen muerto” Sd se calcula así: (E-1) Para determinar Sd, abra la válvula 1. Una vez que se estabilice la lectura de la presión diferencial en ∧p2, calcule el almacenamiento del volumen muerto ascendente a partir de: (E-2) y el almacenamiento compresivo ascendente a partir de: (E-3) Para determinar el almacenamiento descendente, primero que todo desatornille la válvula de aguja hasta pasar - y luego regresar - a la lectura inicial. Revise que el Transductor inicial debe leer cero nuevamente y que el Transductor descendente debe mostrar su lectura inicial. Abra la válvula No. 2. Lea la presión del Transductor descendente p2[1]. Atornille la válvula de aguja hasta que la presión descendente estabilizada p2[2] sea casi igual a la lectura a escala total de este Transductor. Calcule la disminución de volumen ∧V2 a partir de las lecturas micrométricas y el almacenamiento compresivo descendente a partir de: Estas calibraciones de almacenamiento compresivo deben realizarse para cada líquido diferente que se utilice. E.6.8.5.2 Procedimiento para la Medición de la Permeabilidad. Para efectuar la medición del descenso de permeabilidad del líquido, se carga una muestra de roca saturada completamente de líquido dentro del soporte de la muestra, el cual es entonces presurizado , preferiblemente a un estrés de confinamiento alto (usualmente hidrostático). El sistema se llena con líquido, como se ha descrito, y se presuriza a la presión de poro inicial. La válvula de llenado se deja abierta por el tiempo necesario para permitir que el líquido a alta presión se difunda en la muestra. Se cierran entonces las válvulas 1 y 2 y el pulso de presión y el pulso de presión se genera atornillando la válvula aguja. Una vez que se han estabilizado todas las presiones, se calcula S1 - Sd a partir de la ecuación E-1. Este resultado debe tener el mismo valor que el obtenido de la calibración. El descenso del pulso se inicia abriendo la válvula 1. La diferencia de presión a través de la muestra ∧p [0] será ligeramente menor que ∧p [1], la cual se generó con la válvula aguja con la válvula 1 cerrada, por lo tanto excluyendo el volumen muerto ascendente del volumen ascendente. Por lo tanto ∧p [0] debe calcularse utilizando ∧p [1] y ∧V [1] que corresponde al generación en el tiempo de corrido de pulso de

presión y el Sd que se determinado a partir de la calibración. (E-5) E. 6.8.5.3 Cálculo de Descenso de Pulso Las ecuaciones D-2 y D-3 se aplica a las mediciones de descenso de pulso en líquido y en gases. Sin embargo, las diferencias de presión en el lado izquierdo de la ecuación D-2 deben reemplazarse por las diferencias de presión de primera potencia para líquidos: (E-6) Los términos a y b se refieren a las proporciones de almacenamiento compresivo de líquidos. (E-7) y (E-8) y kg y cg en la ecuación D-6 se reemplazan por kL y cL, respectivamente. Una vez finalizada la porción temprana de descenso en la presión, resultará una línea recta si graficamos el logaritmo natural de ∧p [t] ∧p [0] contra el tiempo. (E-9) o de manera equivalente, (E-10) Ao y A1 que representan el intercepto y la pendiente de la línea recta, se hallan a partir de la ecuación E-10 utilizando la regresión lineal en la porción lineal únicamente en los datos de descenso de presión. El valor de ƒo definido por la ecuación D - 14 se calcula a partir del intercepto: (E-11) El parámetro ƒ1 que es necesario para los cálculos de permeabilidad, se halla a partir de ƒo y la proporción de almacenamientos ascendentes y descendentes, S1 y S2 utilizando la figura 6.D-2. La permeabilidad se calcula con la siguiente ecuación: (E-12) Amaefule y colaboradores utilizaron presiones de poro que iban desde 500 a 4500 psi. Kamath y colaboradores recomiendan presiones de poro de aproximadamente 1000 psi y tamaños de pulso de casi el 20% de la presión del poro, o mejor, 10% del estrés neto de confinamiento. Los volúmenes de los depósito o reservorios ascendentes y descendentes deben ser iguales si es posible, por las razones enunciadas en la sección de descenso de pulso de
gas. Deben ser ligeramente menores o del mismo volumen que el volumen de poro de la roca que se va a medir si se va a investigar la heterogeneidad o 5-10 veces más grande o más si se buscan permeabilidades promedio comparables con los valores estables. Consulte las publicaciones de Kamath y colaboradores con respecto a “soluciones de tiempos iniciales” si desea investigar la heterogeneidad, la cual no hace parte del tema de este documento.

CAPITULO 7 PRUEBAS COMPLEMENTARIAS
INDICE 7. PRUEBAS COMPLEMENTARIAS 7.1 Conceptos Generales 7.2 Caracterización Petrográfica 7.3 Distribución del Tamaño del Grano 7.4 Gravedad del Petróleo 7.5 Caracterización del Petróleo 7.6 Solubilidad Acida 7.7 Determinación de la Salinidad del Agua en la Muestra 7.8 Referencias Bibliográficas 7.9 Bibliografía Figuras 7-1 Ejemplos de formas gráficas para el Reporte de los Datos del Análisis del
Tamiz 7-2 Diferencia del Indice de Refracción (RI) vs Salinidad (RI de la solución – RI del
Agua Destilada). 7-3 Tabla de conversión de Resistividad Salina vs Temperatura y Salinidad Tablas 7-1 Tamaño de tamices disponibles 7-2 Ejemplo de Datos obtenidos del Análisis del Tamiz

PRACTICAS QUE SE RECOMIEDAN PARA EL ANALISIS DE LA MUESTRA 7. PRUEBAS COMPLEMENTARIAS
7.1 ASPECTOS GENERALES
Junto con los análisis de la muestra es posible encontrar datos complementarios como la solubilidad ácida o calcimetría, la gravedad del petróleo, la salinidad del agua en la muestra, la distribución del tamaño del grano y la caracterización básica del petróleo. Por lo tanto, se han incluido estos tests debido a que los datos así obtenidos son de gran utilidad en la comprensión e interpretación de los resultados del análisis practicado a la muestra. 7.2 CARACTERIZACION PETROGRAFICA La caracterización Petrográfica constituye una parte importante del análisis de la muestra. Los resultados conjuntos de la información Petrográfica y los análisis básicos de la muestra nos suministran un marco de referencia integrado para la evaluación del depósito o reservorio, incluyendo la interpretación de los registros. Utilizando datos geológicos es posible ampliar los resultados de los análisis de la muestra hacia partes del reservorio o depósito donde no se han realizado mediciones. El análisis petrográfico también es la base para la interpretación de los análisis de la muestra, especialmente de aquellos que tuvieron un comportamiento inesperado o anómalo. Este tipo de resultados pueden tener validez y su origen puede radicar en las variaciones que tiene la textura de la roca y/o en su mineralogía. 7.2.1 La caracterización Petrográfica se ejecuta con mayor precisión utilizando el
mismo tapón de muestra que se usó en los análisis de rutina, de tal manera que los datos obtenidos se pueden aplicar de manera directa. En aquellos casos en los que no pueda utilizarse la mima muestra, se puede tomar las secciones de los extremos que se sacaron al preparar las muestras o se puede obtener material del mismo lecho rocoso de donde salieron las muestras. Se recomienda utilizar mínimo una (1) pulgada cúbica [40 gramos aproximadamente), los cuales deben guardarse desde el principio para ser destinados a esta caracterización Petrográfica.
7.2.2 La descripción de las muestras y su análisis nos suministran una información valiosa respecto a la calidad y carácter de la muestra. Las descripciones deben hacerse tomando las muestras extraídas con la ayuda de un buen microscopio binocular. Se deben registrar los datos referentes a la porosidad visual y el tipo de poro (intergranular, fisura, móldico), aspectos inducidos por el proceso de muestreo [perforación] como por ejemplo, fracturas inducidas, invasión de fluido durante la perforación y aspectos relacionados con la heterogeneidad natural (láminas de arcilla, variación litológica normal en el lecho, fracturas mineralizadas etc.). La caracterización Petrográfica debe incluir información de la textura y de los principales observaciones a nivel mineralógico. Los datos de la textura incluyen la información respecto al tamaño

del grano, el ordenamiento, redondeo y forma de este grano. Las observaciones mineralógicas incluyen el tipo de grano y su abundancia. Las cantidades aproximadas de los componentes de cemento y matriz pueden calcularse en la mayoría de los casos utilizando un microscopio binocular.
7.2.3 Puede obtenerse información Petrográfica cuantitativa y semicuantitativa a
partir de cortes delgados, difracción con rayos X (XRD), análisis químico de la roca (generalmente utilizando la fluorescencia con rayos X – XRF) y la espectroscopía de Transformación infrarroja de Fourier (FTIR). La microscopía electrónica de barrido (SEM) es de gran utilidad en la determinación de la distribución espacial de los minerales arcillosos dentro de la muestra.
7.3 DISTRIBUCION DEL TAMAÑO DEL GRANO
Las aplicaciones de los datos respecto a la distribución del tamaño del grano son de gran utilidad en las siguientes áreas: (a) a nivel de ingeniería, en programas de completamiento de pozos en sedimentos friables y sin consolidación. (b) a nivel geológico, para la evaluación de la heterogeneidad de la arena e interpretación del ambiente deposicional en sedimentos clásticos consolidados y no consolidados. (c) a nivel petrofísico, en las fases de evaluación de las formaciones para poder entender las repuestas de los registros. La distribución del tamaño del grano también se utiliza generalmente para los análisis de las paredes de la muestra y así derivar la permeabilidad. Los métodos que se utilizan generalmente incluyen el tamizado en húmedo y en seco, análisis del tamaño de las partículas que se quedaron en el tubo, análisis del tamaño del grano en secciones delgadas y el análisis del tamaño de las partículas por difracción láser . 7.3.1 Análisis con Tamiz (Agitador Mecánico)
7.3.1.1 Principio Se somete a vibración mecánica una masa de peso conocido, desagregada, haciéndola pasar por pantallas organizadas en hileras donde las aperturas se hacen cada vez más pequeñas. Los pesos parciales de la muestra que se quedan en las pantallas individuales se utilizan para el análisis del tamiz tabulando tamaño de la apertura vs porcentaje de muestra retenida.
7.3.1.2 Equipo Se recomienda la utilización del siguiente equipo: a. Dispositivo para tamizar de manera mecánica b. Tamices de estándares US o Tyler (los tamices deben estar diseñados de
acuerdo con la norma E-11 de la ASTM, Especificaciones de los Tamices de Alambre destinados a ser Utilizados en Pruebas.
c. Balanza que mida 0.01 gramos d. Mortero con un mango de punta suave

e. Cepillo de cerdas suaves f. Horno para secar. 7.3.1.3 Procedimiento Los tamices deben estar limpios, secos, su peso debe ser conocido y deben estar ordenados de acuerdo a su tamaño, desde el más grande al más pequeño. Se debe colocar una bandeja en la parte inferior del ensamblaje. La tabla 7-1 muestra los tamices disponibles y su equivalente en tamaño. Tabla 7-1 Tamaños de Tamices Disponibles Tamiz – Estándar US Tamaño en milímetros Gravilla Gravilla Gravilla Gravilla Gravilla Arena muy Gruesa Arena muy Gruesa Arena muy Gruesa Arena muy Gruesa Arena Gruesa Arena Gruesa Arena Gruesa Arena Gruesa Arena Media Arena Media Arena Media Arena Media Arena Fina Arena Fina Arena Fina Arena Fina Arena muy Fina Arena muy Fina Arena muy Fina Arena muy Fina Arenisca Arenisca Arenisca
5 6 7 8 10 12 14 16 18 20 25 30 35 40 45 50 60 70 80 100 120 140 170 200 230 270 325 430
4.00 3.36 2.83 2.38 2.00 1.68 1.41 1.19 1.00 0.84 0.71 0.59 0.50 0.42 0.35 0.30 0.25 0.21 0.18 0.15 0.13 0.11 0.09 0.07 0.06 0.05 0.04 0.03
Se extrae una muestra representativa y se le retiran los hidrocarburos y la sal. Se seca hasta lograr un equilibrio en su peso retirando el solvente y el agua. La muestra se desagrega suavemente, se pesa hasta el 0.01 gramo más cercano (una muestra en términos generales debe tener 100 gramos si se van a utilizar
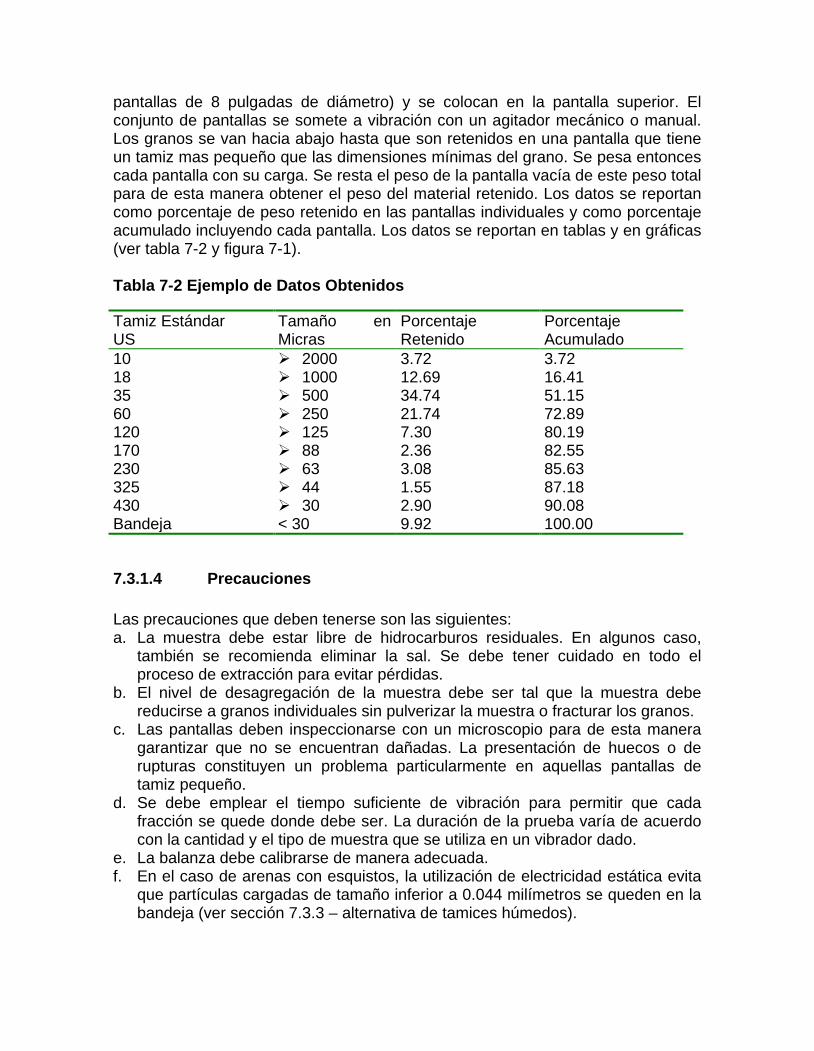
pantallas de 8 pulgadas de diámetro) y se colocan en la pantalla superior. El conjunto de pantallas se somete a vibración con un agitador mecánico o manual. Los granos se van hacia abajo hasta que son retenidos en una pantalla que tiene un tamiz mas pequeño que las dimensiones mínimas del grano. Se pesa entonces cada pantalla con su carga. Se resta el peso de la pantalla vacía de este peso total para de esta manera obtener el peso del material retenido. Los datos se reportan como porcentaje de peso retenido en las pantallas individuales y como porcentaje acumulado incluyendo cada pantalla. Los datos se reportan en tablas y en gráficas (ver tabla 7-2 y figura 7-1). Tabla 7-2 Ejemplo de Datos Obtenidos Tamiz Estándar US
Tamaño en Micras
Porcentaje Retenido
Porcentaje Acumulado
10 18 35 60 120 170 230 325 430 Bandeja
2000 1000 500 250 125 88 63 44 30 < 30
3.72 12.69 34.74 21.74 7.30 2.36 3.08 1.55 2.90 9.92
3.72 16.41 51.15 72.89 80.19 82.55 85.63 87.18 90.08 100.00
7.3.1.4 Precauciones Las precauciones que deben tenerse son las siguientes: a. La muestra debe estar libre de hidrocarburos residuales. En algunos caso,
también se recomienda eliminar la sal. Se debe tener cuidado en todo el proceso de extracción para evitar pérdidas.
b. El nivel de desagregación de la muestra debe ser tal que la muestra debe reducirse a granos individuales sin pulverizar la muestra o fracturar los granos.
c. Las pantallas deben inspeccionarse con un microscopio para de esta manera garantizar que no se encuentran dañadas. La presentación de huecos o de rupturas constituyen un problema particularmente en aquellas pantallas de tamiz pequeño.
d. Se debe emplear el tiempo suficiente de vibración para permitir que cada fracción se quede donde debe ser. La duración de la prueba varía de acuerdo con la cantidad y el tipo de muestra que se utiliza en un vibrador dado.
e. La balanza debe calibrarse de manera adecuada. f. En el caso de arenas con esquistos, la utilización de electricidad estática evita
que partículas cargadas de tamaño inferior a 0.044 milímetros se queden en la bandeja (ver sección 7.3.3 – alternativa de tamices húmedos).

7.3.1.5 Cálculos Los cálculos de este método son: % Retenido en cada pantalla = [(Peso pantalla cargada – peso pantalla vacía) / Peso total muestra] x 100 (1) % Retenido Acumulado = suma del porcentaje retenido en cada pantalla (hasta la pantalla deseada, incluyendo todas las pantallas que se encuentren por encima de la misma). 7.3.1.6 Ventajas Las ventajas de este método son: a. Se considera un método aceptable para clasificar los tamaños del grano que
tengan más de 0.044 milímetros, en arenas con cementación mínima. Este método es estándar y preciso.
b. La muestra puede volverse a examinar, De esta manera se garantiza la repetibilidad.
c. La muestra puede mezclarse nuevamente o mantenerse en fracciones separadas para después realizar otras pruebas.
d. Es una prueba sencilla que se puede realizar con un equipo mínimo.
7.3.1.7 Limitaciones Es un método que requiere de cierto tiempo para realizarlo (se recomienda una hora por muestra, en pantallas que tengan un diámetro de 8 pulgadas), se necesita una cantidad de muestra relativamente alta y nos puede dar resultados inexactos en arenas con esquistos y en arenas consolidadas.
7.3.1.8 Exactitud y Precisión Si el test se ejecuta de manera adecuada y se toman todas las precauciones enunciadas, se espera que la desviación sea del 3% en la muestra total y de 0.5% en cada pantalla.
7.3.1.9 Calibración Los documentos utilizados en este caos son el ASTM C-136 Método Estándar para el Análisis de Tamices y Agregados Gruesos y el ASTM C-702 Práctica para la reducción de la Muestra agregada hasta llegar al nivel utilizado en la muestra. 7.3.2 Análisis del Tamiz (Agitador Sónico)
7.3.2.1 Principio

Consulte la sección, 6.2.1, con la salvedad de que las muestras preparadas se someten a vibración utilizando energía acústica.
7.3.2.2. Equipo Se recomienda la utilización del siguiente equipo: a. Aparato para tamizado sónico b. Tamices estándar US o Tyler (consulte la sección 7.3.1.2.b.) c. Balanza que mida 0.001 gramos d. Mortero y mango con punta suave e. Cepillo de cerdas suaves f. Diseminador de muestra g. Horno para secar
7.3.2.3 Procedimiento La preparación de la muestra y los procedimientos de material son similares a los descritos en la sección 7.3.1. S e colocan entre cinco y quince gramos de muestra en la pantalla superior. Se ajustan las amplitudes para el proceso de tamizado y el pulso para que se agiten los granos. El tiempo del procedimiento varía entre 10-20 minutos y depende del tamaño de la muestra y del grado de finura.
7.3.2.4 Precauciones Las precauciones que deben tenerse en este método son: a. Consulte la sección 7.3.1.4, puntos a al f. b. Se debe tener en cuenta que las determinaciones de peso deben ser más
exactas en este método puesto que las muestras son más pequeñas.
7.3.2.5 Cálculos Consulte la sección 7.3.1.5
7.3.2.6 Ventajas Las ventajas que se tienen con este método son las siguientes: a. Se considera un método aceptable para clasificar los tamaños del grano que
tengan más de 0.044 milímetros, en arenas con cementación mínima b. Es una prueba sencilla que se puede realizar con un equipo mínimo. c. Este test es más rápido de hacer que el método donde se utiliza el agitador
mecánico (se demora aproximadamente 15 minutos en cada muestra). d. Se necesita menos muestra (5-15 gramos)

e. La muestra puede mezclarse nuevamente para efectuar el procedimiento otra vez, guardarse en fracciones separadas o se puede utilizar en tests adicionales.
7.3.2.7 Limitaciones Las limitaciones que se observan en este método son: a. Se debe tener precaución para obtener una muestra representativa debido a
que su tamaño es muy pequeño. b. Debido a que los marcos de las pantallas son de plástico, es posible que se
observe retención de partículas cargadas lo cual representa un inconveniente. Esto da como resultado que se deba aumentar el tiempo de tamizado en arenas finas. En gran parte de los caso, este aumento significativo de tiempo no se considera apropiado para el desplazamiento de partículas < 0.044 milímetros a la bandeja.
7.3.2.8 Precisión Consulte la sección 7.3.1.8
7.3.2.9 Calibración Consulte la sección 7.3.1.9 7.3.3 Análisis con Tamiz (Combinación Húmeda y Húmeda/Seca) 7.3.3.1 Contrastando con la técnica en seco en la cual la muestra se agita
utilizando vibradores mecánicos o sónicos, el uso de un tamiz húmedo se refiere a la técnica de lavar una muestra representativa a través de tamices individuales. Esta técnica requiere de más tiempo para su ejecución pero se prefiere cuando se va a trabajar con arenas finas.
7.3.3.2 La combinación de una análisis en húmedo / seco, como su nombre lo dice, es una combinación de técnicas húmedas y secas. Es una buena alternativa cuando se va a trabajar con arenas finas. Se pesa la muestra y luego se hace pasar de forma húmeda por un tamiz 325 y de esta manera se retira la fracción < 0.044 mm. El material que ha quedado retenido en el tamiz 325 se somete a un proceso de secado, se pesa y se tamiza de la manera descrita en las secciones 7.3.1 o 7.3.2 (con un agitador mecánico o sónico).
7.3.4 Análisis del Tamaño de las Partículas (Difracción Láser)
7.3.4.1 Principio

Se coloca una muestra extraída y desagregada en un fluido apropiado (que puede ser oleoso o acuoso, depende del criterio del test). Este líquido se mezcla, se revuelve o se somete a recirculación para de esta manera garantizar la dispersión de las partículas. Se proyecta un rayo láser a través de la muestra dispersa y por medio de un computador se analizan los patrones de difracción. El principio utilizado para calcular la distribución del tamaño del grano es la teoría de difracción de Fraunhofer. Los ángulos de la luz incidente son inversamente proporcionales al tamaño de las partículas y la intensidad de la luz distribuida por las partículas es directamente proporcional a la cantidad de partículas.
7.3.4.2 Equipo e Reactivos Se recomienda el siguiente equipo para la ejecución de este método: a. Dispositivo láser / computador adecuado. b. Diseminador de la muestra c. Balanza que mida 0.0001 gramos. d. Mortero y mango de punta suave e. Equipo para sonificación f. Solución desfloculante g. Horno para efectuar el secado.
7.3.4.3 Procedimiento Se extrae una muestra representativa y se le retiran los hidrocarburos y se somete a un proceso de secado para retirarle el solvente. La dispersión de partículas se hace antes de la medición con la ayuda de un equipo de sonificación. Se carga el depósito del fluido con el fluido respectivo y se hace una medición de referencia. Se introduce la muestra sobre la cual se agrega una solución surfactante (que con frecuencia es el hexametafosfato de sodio) para de esta manera reducir la tensión de la superficie y estimular la desfloculación de las partículas. Esta mezcla de fluido con la muestra se somete a un proceso de recirculación o se revuelve para garantizar que la dispersión de las partículas sea uniforme. Durante la fase de medición, se proyecta un haz de rayo láser de longitud de onda conocida de tal manera que atraviese la mezcla fluido / muestra. El rayo de luz difractado hace impacto en un fotodetector y posteriormente se utiliza la teoría de difracción de Fraunhofer para calcular la distribución del tamaño de las partículas a partir de la luz difractada. Los datos obtenidos se reportan en forma de tabla o gráfica hasta el rango de submicras (ver tabla 7-2 y figura 7-1).
7.3.4.4 Precauciones Las precauciones que deben tenerse en este caso son: a. Consulte la sección 7.3.1.4, puntos a y b. b. Los Equipos deben estar calibrados de manera apropiada (incluyendo la
alineación del láser, la calibración del fotodetector etc.).

c. El fluido que sirve de transportador no debe tener burbujas ni partículas extrañas en todo el período de medición.
d. La muestra debe encontrarse dispersa de manera uniforme en el fluido de transporte y debe desflocularse durante la fase de medición.
e. Se debe tener especial cuidado en la obtención de la muestra, la cual debe ser representativa.
7.3.4.5 Cálculos Los algoritmos que se utilizan para calcular la distribución del tamaño del grano a partir de patrones de difraccion son de naturaleza compleja y dependen del fabricante. De todas maneras se pueden utilizar las siguientes ecuaciones para ilustrar las relaciones existentes entre el ángulo del patrón de difracción, el tamaño de las partículas, intensidad de la luz y cantidad de las partículas. El ángulo del patrón de difracción está dado por la siguiente ecuación: Seno de θ = 122 λ / d (3) Donde: θ = Medio ángulo medido hasta el primer mínimo del patrón, en grados. λ = Longitud de onda de la luz incidente, en micras d = diámetro de las partículas, en micras. La intensidad total de la luz distribuida está dada por la ecuación:
I = Knd2 I = Luz difractada total, en watt K = Constante de calibración del Equipo, en watt / micras2 n = número de partículas d = diámetro de las partículas, en micras.
7.3.4.6 Ventajas Las ventajas de este método son: a. El rango de medición del tamaño del grano nos da una clasificación de las
partículas hasta el nivel de micras incluido (es decir, areniscas muy finas y partículas arcillosas).
b. Los datos se pueden repetir, es decir, la reproducibilidad de la prueba es alta. c. La técnica requiere cantidades mínimas de muestra (< 1.0 gramos) d. La medición es un procedimiento rápido (va desde 30 segundos hasta varios
minutos).

7.3.4.7 Limitaciones Las limitaciones de este método son: a. Existe un límite en cuanto al tamaño por encima del cual no se pueden realizar
mediciones. Este límite está en la fracción de la arena gruesa. Si el tamaño de las partículas supera este límite, se necesita realizar un pre – tamizado y se deben combinar los conjuntos de datos.
b. El equipo y el hardware acompañante es costoso.
7.3.4.8 Calibración Los requisitos de calibración dependen del fabricante del Equipo y del Equipo en si. Generalmente incluye una alineación correcta del láser, una calibración del fotodetector y una calibración del patrón de difracción utilizando un estándar de precisión.
7.3.4.9 Precisión del Método No se dispone de información suficiente en este momento para determinar la exactitud del método. 7.3.5 Medición del Tamaño del Grano en Secciones Delgadas (Técnica Visual)
7.3.5.1 Principio Esta medición del tamaño del grano utilizando secciones delgadas se puede efectuar en muestras que van desde la arena sin consolidación hasta la roca que está bien consolidada. Es el método de elección en el caso de las rocas consolidadas debido a que estas muestras no pueden someterse a buenos procesos de desagregación sin que ocurra rompimiento de los granos o fallas en la separación de granos adyacentes bien cementados. La técnica se basa en la preparación de una sección delgada y en la medición del tamaño de los granos individuales ya sea de manera visual o con la ayuda de un sistema de análisis de imagen, utilizando un microscopio petrográfico.
7.3.5.2 Equipos e Reactivos Se recomienda el siguiente equipo para la realización de este método: a. Microscopio petrográfico equipado con un micrómetro ocular. b. Equipo para la preparación de la sección delgada. c. Laminas de vidrio d. Resina epóxica coloreada.

7.3.5.3 Procedimiento Las muestras se impregnan con resina epóxica coloreada y se montan sobre una lámina de vidrio para proceder al corte de las láminas, las cuales deben tener 30 micras de grosor. Este grosor permite que la luz se transmita a través de la mayoría de los granos de arena. El tamaño del grano se mide utilizando el eje más largo de las secciones transversales de los granos individuales en una sección bidimensional. En gran parte de los casos, las muestras que no tienen consolidación alguna necesitan dispersión en aceite, el cual se coloca sobre la lámina de vidrio. Generalmente se restringe la medición del tamaño a una especie mineral específica (casi siempre cuarzo en el caso de la arena), aunque se pueden incluir otras especies (como el feldespato). En términos generales se miden entre 200 – 300 granos. El número de granos en realidad depende del ordenamiento. Las arenas que tengan un ordenamiento pobre necesitan más mediciones que las arenas cuyo ordenamiento sea bueno. Los granos se seleccionan al azar utilizando la técnica de conteo de punto descrita por Chayes (1956). Las mediciones se expresan en milímetros y posteriormente convertidas a las unidades de preferencia (ya sea pulgadas, unidades pi, etc.).
7.3.5.4 Precauciones Las precauciones que deben tenerse con este método son: a. Las secciones delgadas permiten que la medición del tamaño del grano se
efectúe en dos dimensiones únicamente. Teniendo en cuenta que los granos tienen formas irregulares, no tienen un tamaño uniforme y que casi todos los sedimentos que están compuestos de partículas no – esféricas muestran anisotropía dimensional (es decir, hay una orientación definida del grano), es necesario hacer suposiciones que simplifiquen el proceso puesto que de lo contrario no existiría una solución única para determinar la distribución del tamaño del grano a partir de secciones delgadas (Blatt et al, 1980).
b. Los aparatos deben calibrarse c. Las secciones delgadas deben ser representativas del intervalo del depósito. d. Se debe tener especial cuidado en la obtención de la muestra representativa
debido a que se van a medir relativamente pocos granos. e. Deben existir suficientes granos disponibles para la medición en la sección
delgada para de esta manera garantizar que puede haber confiabilidad estadística.
f. Para las arenas laminadas que tienen un tamaño de grano que contrasta entre las láminas, se considera que es difícil la selección de un tamaño de grano sin que se presente ninguna desviación.
7.3.5.5. Cálculos El tamaño del grano se mide utilizando unidades no dimensionales que son visibles a través del ocular del microscopio. Estas unidades se llevan a milímetros

utilizando un factor de conversión que depende del grado de magnificación que se utilice. La distribución del tamaño se calcula con base en la frecuencia del volumen.
7.3.5.6 Ventajas Las ventajas que tiene este método son: a. El rango de medición generalmente se toma a partir de un tamiz tipo US entre
10-400 b. La medición de la distribución del tamaño del grano en la sección delgada se
puede aplicar perfectamente a la mayoría de las rocas que se encuentran en los depósitos o reservorios (areniscas y rocas más gruesas).
c. La técnica permite una medición estandarizada del tamaño del grano en los reservorios caracterizados por una consolidación rocosa.
7.3.5.7 Limitaciones Las limitaciones de este método son las siguientes: a. La técnica es dispendiosa, toma mucho tiempo realizarla y está limitada a una
visión bidimensional. b. El equipo y los Equipos que se utilizan son costosos. c. La obtención del promedio del tamaño del grano y de la distribución del tamaño
a partir de la sección delgada no es un procedimiento directo y puede estar sometido a variaciones. Los datos se consideran relativamente apropiados (Kellerhals et al).
d. La resolución de la imagen (que depende de la magnificación y del grosor de la sección) limita la medición de partículas muy pequeñas (areniscas y fracciones arcillosas).
7.3.5.8 Exactitud y Precisión Los datos tienen una ligera desviación hacia las partículas pequeñas. La precisión está en función del número de granos medidos.
7.3.5.9 Calibración Es posible efectuar una calibración del tamaño medido en la sección delgada utilizando el tamizado y otras técnicas (Rosenfeld et al, 1953). 7.3.6 Medición del Tamaño del Grano en Secciones Delgadas – Análisis de la Imagen
7.3.6.1 Principio

La medición del tamaño del grano a partir de arenas no consolidadas o consolidadas se puede efectuar utilizando procedimientos automatizados o semi-automatizados de análisis de imagen. La información que se consigna en este caso se refiere al cálculo del tamaño del grano en arenas no consolidadas.
7.3.6.2 Equipo e Reactivos Se recomienda el siguiente equipo: a. Analizador de imágenes b. Microscopio petrográfico con fuente de luz c. Equipo para la preparación de secciones delgadas d. Láminas de vidrio e. Resina epóxica coloreada. 7.3.6.3 Procedimiento (Arenas No Consolidadas) Se dispersan los granos de arena sobre la lámina de vidrio de tal manera que pocos se toquen entre sí. El área de proyección o diámetro de cada grano se puede hallar utilizando un aparato de análisis de imagen que consta de una cámara de vídeo adherida a un microscopio (Mazullo y Kennedy, 1985). Los granos se separan del fondo utilizando una diferencia de intensidad para el fondo (más brillante) y para los granos (más oscuro) como criterio para establecer el umbral. mas oscuro) como criterio para establecer el umbral. En términos generales se necesitan 200-500 granos que a su vez requieren de 5-15 minutos para su procesamiento. 7.3.6.4 Procedimiento (Arenas Consolidadas) La determinación de la distribución del tamaño del grano utilizando técnicas de análisis de imagen en arenas consolidadas es un procedimiento complicado por la característica misma de las muestra: hay gran contacto entre los grano. Sin embargo, existen técnicas que utilizan las distancia entre los centros de los poros intergranulares para calcular el tamaño del grano.
7.3.6.5 Precauciones Las precauciones que deben tenerse en este caso son: a. Consulte la sección 7.3.5.4 b. Se pueden presentar mediciones inexactas si los granos que se van a medir
están en contacto unos con otros. Esto da como resultado la obtención de diámetros mayores que los reales.

7.3.6.6 Ventajas Las ventajas que se tienen con este método son: a. La obtención de los primeros datos se hace de una manera relativamente
rápida y los datos se almacenan en formato digital. b. Se pueden analizar muestras muy pequeñas
7.3.6.7 Limitaciones Las limitaciones que tiene este método son: a. Se debe tener cuidado en que los granos sean representativos. Es por lo tanto
necesario que la separación de la muestra y la selección de los granos se realice de manera muy cuidadosa.
b. La adquisición del sistema de análisis de imagen junto con el software respectivo y la cámara de vídeo puede ser más costoso que la técnica visual.
7.3.6.8 Cálculos Los datos deben convertirse a frecuencia de volumen. Esto se efectúa de la siguiente manera: (a) definiendo el número de intervalos de clase basado en el diámetro del grano. (b) elevando al cubo el radio de todos los granos en cada intervalo y agregando los volúmenes dentro de cada intervalo. (c) sumando todos los volúmenes de los intervalos para de esta manera obtener el volumen total. La fracción de volumen en cada intervalo se calcula dividiendo por este total.
7.3.6.9 Exactitud y Precisión Se ha demostrado que los datos son precisos y que se desvían ligeramente de los datos del tamiz (la diferencia se debe al hecho de que los tamices son selectivos tanto para forma como para tamaño).
7.3.6.10 Calibración Debe definirse el tamaño del pixel o punto para una magnificación dada al utilizar este sistema. 7.3.7 Análisis del Tamaño de las Partículas – Ley de Stokes
7.3.7.1 Principio Se introduce una muestra representativa dentro de una columna líquida y se determinan las velocidades de sedimentación de la partículas monitoreando el incremento de peso que sufre la bandeja que se encuentra suspendida en la columna. Se calcula el diámetro de la sedimentación de las partículas (el diámetro

de una esfera de igual densidad que tiene una velocidad igual de sedimentación) utilizando ya sea la Ley de Stoker, una forma modificada de la misma o la Fórmula de Gibbs (Gibbs et al 1971). Los datos se reportan de la misma manera que los datos de un tamiz estándar.
7.3.7.2 Equipo El equipo que se recomienda en este caso es el siguiente: a. Mortero y mango de punta suave b. Cepillo de cerdas suaves c. Balanza de cargue inferior (de 0.001 gramos) o transductor de resistencia d. Columna de decantación y hardware relacionado. e. Horno para secar
7.3.7.3 Procedimiento La preparación de la muestra debe seguir el protocolo que se ha resumido en la sección 7.3.1. Se pesan la muestra de manera muy precisa (hasta 0.001 gramos) y se introduce en la columna de decantación. Las partículas empiezan a caer a través del líquido en proporción a su tamaño, forma y densidad. Las partículas se acumulan en la bandeja que se encuentra suspendida en una balanza de precisión o un transductor de resistencia, la cual se ubica a una distancia conocida tomando como punto de referencia la parte superior de la columna. Se toman los datos de peso y de tiempo y se registran en un computador y se procede a calcular las velocidades de sedimentación. La duración del test varía desde varios minutos hasta 20 minutos y está relacionado con el tipo de equipo / Equipo y con el tamaño de las partículas. Se utiliza una forma modificada de la Ley de Stoke o la Fórmula de Gibbs para calcular los diámetros de sedimentación. Si no se recupera toda la muestra al final del test, la diferencia se clasifica como más fina que el diámetro equivalente de sedimentación en la última medición.
7.3.7.4 Precauciones Las precauciones que deben tenerse en este método son: a. Consulte la sección 7.3.1.4, puntos a, b y c. b. La columna de sedimentación debe estar limpia (la densidad del fluido debe
ser constante y la medición de las partículas suspendidas al efectuar otra medición debe darnos una cantidad despreciable).
c. Las partículas deben estar uniformemente dispersas apenas se introduzcan en la columna y deben estar desfloculadas.
d. Puesto que la cantidad de muestra es muy pequeña, se debe tener especial cuidado en el pesaje de la misma.
e. Se deben eliminar las corrientes de convección.

7.3.7.5 Cálculos Se utiliza la Ley de Stoke, una forma modificada de la misma o la Fórmula de Gibbs para calcular el radio de los granos a partir de los datos de velocidad de sedimentación. La Ley de Stoke: V = 2gr2 (ρg - ρƒ ) / 9µ (5) La Fórmula de Gibbs: V = - 3µ + √ 9µ2+ gr2 ρƒ (ρg - ρƒ ) (0.015476+ 0.19841r) / ρƒ ( 0.011607+014881r) Donde: V = Velocidad de sedimentación, en cm / seg. g =aceleración de la gravedad, en cm / seg2 r = Radio del grano, en cm ρg = Densidad del grano, en g / cc ρƒ = Densidad del Fluido, en g /cc µ = Viscosidad del fluido, en poise.
7.3.7.6 Ventajas Las ventajas de este método son las siguientes: a. Es un test rápido (hasta 20 minutos) b. El equipo no es costoso c. El rango de medición es relativamente amplio (a través de un tamiz 500,
estándar US).
7.3.7.7 Exactitud y Precisión No hay información disponible al respecto
7.3.7.8 Calibración El procedimiento de calibración para este caso se describe en la publicación de Gibbs et al 1971. 7.4 GRAVEDAD DEL PETROLEO
Es necesario conocer la gravedad específica del petróleo para poder convertir el peso del petróleo en volumen dentro del método de destilación – extracción que determina la saturación de fluidos (ver la sección 4.3). La gravedad del petróleo puede determinarse por medio del método de la gota de petróleo, por

el método del índice refractivo, utilizando un método gravimétrico, el densitómetro digital o un método de resonancia magnética nuclear.
7.4.1 Método de la Gota de Petróleo
7.4.1.1 Principio Este método consiste en suspender una gota de petróleo en un medio líquido cuya gravedad se puede medir con un hidrómetro o con una balanza para gravedad específica.
7.4.1.2 Equipo El Equipo que se utiliza en este método es el siguiente: a. Cilindro de vidrio o de plástico transparente b. Hidrómetro API o balanza para gravedad específica
7.4.1.2 Procedimiento Se coloca una gota de petróleo que ha sido recuperada de la muestra durante la prueba de saturación de fluido en un cilindro de vidrio o plástico que contiene una solución de alcohol metílico y agua. Posteriormente se ajusta la gravedad de esta solución añadiendo alcohol o agua hasta que la gota de aceita permanezca suspendida, es decir, que cuando después de agitar la solución, la gota no se va al fondo ni sube. La gravedad de la solución se mide entonces con la ayuda de un hidrómetro API o una balanza para gravedad específica. El procedimiento correcto para medir la gravedad utilizando un hidrómetro se encuentra en la norma ASTM D 287-55 Método para hallar la Gravedad API del Petróleo. La gravedad que se observe a temperatura ambiente se corrige a temperatura 60 grados Fahrenheit y se convierte en gravedad específica utilizando las tablas 5 y 3 respectivamente que se encuentran en la publicación ASTM- IP Tablas de medición de petróleo.(Edición Norteamericana). La balanza para medir gravedad específica permite realizar una lectura directa de la gravedad. Una tabla simple que ayude en la conversión de unidades de gravedad en líquidos y densidad puede ser de gran utilidad en este caso.
7.4.1.4 Precauciones Las precauciones que deben tenerse con este método son: a. Se deben retirar todas las burbujas que se encuentren adheridas a la superficie
de la gota antes de ajustar la gravedad en la solución de alcohol y agua.

b. La solución debe agitarse después de agregar alcohol o agua para de esta manera garantizar que la solución es uniforme y una detección efectiva de la gravedad.
7.4.1.5 Ventajas No se necesita un equipo demasiado elaborado o complejo.
7.4.1.6 Limitaciones No se dispone en el momento de información suficiente al respecto.
7.4.1.7 Exactitud y Precisión No se dispone en el momento de información suficiente al respecto. 7.4.2 Método del Indice de Refracción
7.4.2.1 Principio El método del índice de refracción para determinar la gravedad API se utiliza generalmente para determinar la gravedad del petróleo. Este petróleo se recupera de las muestras mientras se efectúa la sumatoria de la prueba de fluidos de saturación. Esta técnica también se utiliza para determinar la gravedad API de un tanque de petróleo, prueba de formación, prueba de perforación etc. Y en los casos en que se disponga de muy poca cantidad de petróleo. Se necesitan unas muestras de petróleo de gravedad API conocidas pues es la base para el rango donde se van a ubicar el petróleo sometido al test. Se miden los índices de refracción del petróleo antes y después del procedimiento de sumatoria de fluidos. Se plotea el índice de refracción versus la gravedad dl petróleo. La gravedad del petróleo se determina o calcula utilizando una regresión lineal simple.
7.4.2.2 Equipo El equipo que se necesita en este caso es: a. Hidrómetros API b. Material de vidrio variado c. Pipeta de vidrio d. Refractometro (rango del índice de refracción 1.30-1.90) 7.4.2.3 Procedimiento

La gravedad API de una muestra de petróleo se determina con el hidrómetro API utilizando la norma ASTM D287-55: Método para hallar la gravedad API del Petróleo o utilizando una balanza para gravedad específica. Con la ayuda de una pipeta de vidrio se toma una muestra de petróleo del tanque y se coloca en el Refractómetro. Se determina el índice de refracción de la muestra. La muestra se somete entonces a una fase de análisis de la sumatoria de fluidos (retorta) (ver sección 4.2 Método de retorta para Determinación de Saturación con Fluidos, análisis de muestras convencionales y laterales). Se determina el índice de refracción del petróleo recuperado. Se plotea el índice de refracción antes y después de la retorta versus la gravedad API. Se determinan las gravedades API de manera gráfica o se calculan matemáticamente. 7.4.2.4 Precauciones Las precauciones en este método son las siguientes: a. La operación del Refractómetro debe verificarse midiendo el índice de
refracción de un líquido conocido. b. El prisma del Refractómetro y la placa de vidrio del ocular deben estar
completamente limpias. c. El prisma del Refractómetro debe colocarse y asegurarse contra el ocular
cuando se efectúen las mediciones. d. Antes de efectuar las mediciones, se debe retirar todo el material contaminante
acuosos y sólidos. e. El petróleo crudo debe tener un color claro y se debe utilizar un Refractómetro
de película delgada. 7.4.2.5 Cálculos Se puede hacer una regresión lineal simple de las gravedades y de los índices de refracción. Para calcular la gravedad API se utilizan la pendiente, el índice de refracción y el intercepto. 7.4.2.6 Ventajas Las ventajas que tiene este método son: a. La técnica es sencilla y rápida y los datos obtenidos tienen buena precisión . b. No se necesita de un equipo complejo c. La prueba requiere de muy poca cantidad de muestra (0.03 cc). 7.4.2.7 Exactitud y Precisión La gravedad que se determine con este método debe estar ± 1 grado API con respecto a la gravedad determinada con el ASTM D287-55, si se efectúan las calibraciones adecuadas y se toman las medidas de precaución necesarias. 7.4.3 Método Gravimétrico (Método del Micro – Picnómetro)

7.3.4.1 Principio La densidad del petróleo se determina utilizando un picnómetro pequeño y se calcula entonces la gravedad API. 7.4.3.2 Equipo El equipo que se necesita en este método es: a. Balanza (que mida 0.001 gramos) b. Picnómetros calibrados – los picnómetros pueden estar hechos de vidrio
delgado en forma de U. Se pueden preparar varios tamaños de muestra cambiando la longitud o el diámetro interno del tubo. Los volúmenes deben calibrarse con un líquido de densidad conocida, con el picnómetro completamente llenos o llenos hasta la línea de referencia (se puede utilizar agua destilada libre de gas).
7.4.3.3 Procedimiento Se pesa un picnómetro seco y limpio. Una porción del petróleo que se va a analizar se coloca en el picnómetro. Es preferible utilizar la cantidad más grande de la muestra para la determinación de la gravedad. Se pesa nuevamente el picnómetro y se calcula el peso del petróleo y se divide por el volumen capilar para determinar la densidad. La gravedad específica se calcula dividiendo la densidad del petróleo por la densidad del agua a temperatura ambiente. Se calcula la gravedad API. 7.4.3.4 Precauciones El peso del picnómetro debe minimizarse con respecto al peso del petróleo. De esta manera se reduce el error a medida que la diferencia de peso se hace más precisa. 7.4.3.5 Cálculos En este método se realizan los siguientes cálculos: Densidad del Petróleo = Peso Picnómetro cargado (gr.) – Peso inicial Picnómetro / Volumen Picnómetro
Gravedad específica = Densidad del petróleo (g / cc) / Densidad del Agua (g/cc)
Gravedad API (deg) = (141.5 / Gravedad específica) – 131.5 7.4.3.6 Ventajas Las ventajas que tiene este método son: a. Se pueden obtener datos precisos si el picnómetro se calibra de manera
apropiada.

b. La determinación es rápida. 7.4.4. Método del Densitómetro Digital 7.4.4.1 Principio El principio para la medición utilizando el densitómetro digital se basa en el cambio de la frecuencia natural de un oscilador hueco cuando se encuentra lleno de un fluido. El cambio en la frecuencia del oscilador está relacionado con el cambio de masa del oscilador que ocurre después de colocar la muestra y, por lo tanto, está también relacionada con la densidad de la muestra. 7.4.4.2 Equipo Se necesita un densitómetro digital. 7.4.4.3 Procedimiento Se determinan las constantes de calibración del Equipo midiendo las frecuencias del oscilador cuando tiene aire y cuando tiene agua destilada. Se inyecta la muestra en el oscilador y se determina la frecuencia. La gravedad API se calcula de acuerdo a lo indicado en la sección 7.4.3.5. 7.4.4.4 Ventajas La técnica es rápida y precisa. 7.4.4.5 Limitaciones Se necesitan cantidades relativamente altas de petróleo (1-2 cc). 7.4.4 Método de la Resonancia Magnética Nuclear (NMR) 7.4.5.1 Principio El método de la NMR para determinar la viscosidad del petróleo crudo se basa en la relación de Stoke que existe entre la viscosidad y la difusión molecular rotacional y translacional que ocasiona la relajación del giro NMR. Se puede utilizar ya sea 1H o 13C NMR. La principal ventaja de este método es que puede aplicarse a una gran cantidad de petróleo crudo y a emulsiones de petróleo crudo y agua, al petróleo que esta dentro de los tapones de muestra o en la muestra total o al petróleo que se encuentra disuelto en solventes. 7.4.5.2 Equipo

El equipo es un espectrómetro NMR de transformación de Fourier con capacidad para cambios químicos 1H o 13C. Para las mediciones de la difusión, se necesita una sonda NMR de gradiente de pulso, teniendo en cuenta que los gradientes deben estar por encima de los 100 gauss / cm. 7.4.5.3 Procedimiento El tiempo de relajación de la rejilla 1H o 13C (T1) se mide con una secuencia de recuperación inversa o de saturación. La viscosidad y la gravedad API se miden a partir de correlaciones que ya están publicadas en reportes. Se prefiere utilizar el 13C NMR para el análisis del petróleo dentro de las muestras puesto que no se necesita la separación por cambio químico entre las fases del petróleo y el agua. La aplicación de un campo magnético amplio y de pulso constante hace posible que se hagan mediciones directas de difusión translacional de 1H NMR en líquidos. La secuencia de pulso es generalmente un eco que produce una vuelta de 90o – T – 180o, aplicándose un gradiente de pulso en el campo magnético después de los pulsos RF de 90o y de 180o. La difusión en el gradiente de campo aplicado ocasiona una pérdida irreversible en la amplitud del eco debido a que únicamente aquellos núcleos que no se difunden a una región donde hay un campo magnético diferente son los que se van a enfocar nuevamente. El tiempo entre pulsos (T) se mantiene muy corto (aproximadamente un milisegundo) para evitar que se presente una difusión restringida en las paredes del poro. Las señales de agua y petróleo se separan ya sea por medio de espectroscopía para cambios químicos o agregando Mn ADTA al agua para eliminar las señales respectivas. El coeficiente de difusión del petróleo se relacionan con la viscosidad por medio de la Ley de Stoke y la gravedad API se calcula a partir de reportes que ya están publicados entre la viscosidad y la gravedad API. 7.4.5.4 Ventajas Las ventajas que tiene este método son: a. La técnica es rápida, no invasiva y puede automatizarse b. La técnica se aplica al petróleo ya sea que se encuentre en grandes
cantidades, en muestras o en extractos líquidos. c. Se requiere una preparación mínima de la muestra d. Las mediciones se pueden hacer en emulsiones de petróleo / agua e. Se necesita muy poca cantidad de petróleo para la NMR 1H (< 0.1 cc). 7.4.5.5 Limitaciones Las limitaciones que tiene este método son: a. El espectrómetro NMR es costoso. b. Se puede aplicar el método T1 para las muestras de petróleo que tengan hasta
1000 cp (aproximadamente 15 grados de API) debido al aplanamiento del T1

versus la correlación de la viscosidad. Esta limitación no se aplica a las mediciones de difusión con NMR, las cuales se limitan únicamente al nivel de fuerza del gradiente de campo aplicado.
7.5 CARACTERIZACION DEL PETROLEO 7.5.1 Caracterización del Petróleo (Método Cromatográfico / Vaporización de
Hidrocarburos) Los hidrocarburos residuales que se quedan en los cortes del taladro y en las muestras se pueden analizar para cuantificar el volumen de hidrocarburo presente y las características de composición del petróleo. Estos datos se pueden interpretar para de esta manera obtener información que es útil en las siguientes áreas: a. Procesos de Alteración – La calidad del crudo está determinada por uno o más
procesos de alteración que afectan el petróleo durante el su acumulación en el reservorio. Los cambios resultantes de los procesos de alteración se reflejan en la composición del hidrocarburo residual que se encuentra en los espacios o poros de la roca.
b. Tipo de Producción de Hidrocarburos – La composición del hidrocarburo residual en la matriz de la roca ubicada en la superficie refleja la composición original del reservorio menos los componentes que se han escapado durante el proceso de depleción ocasionada por la presión. La distribución de los componentes intermedios y más pesados se puede utilizar para predecir el tipo de producción de hidrocarburos (no productiva, gas, condensado o petróleo).
c. Cambios en las características de la roca (Permeabilidad) - El escape de los componentes durante la depleción causada por la presión se encuentra limitado por el tamaño molecular y por la volatilidad del hidrocarburo, aunque no se descarta que está influida en gran medida por la permeabilidad. El contraste composicional puede relacionarse con la permeabilidad y la variación de la misma.
d. Contaminación (Fluidos de perforación y/o aditivos) – Los fluidos de perforación y/o los aditivos se pueden analizar utilizando el mismo procedimiento que el utilizado en las muestras de roca. Se puede utilizar el contraste composicional y las técnicas relacionadas con la proporción de hidrocarburos para distinguir entre hidrocarburos nativos y contaminantes que entran en la perforación.
e. En la correlación de petróleo producido con respecto a los cortes o muestras. 7.5.1.1 Principio Este método involucra la vaporización de los hidrocarburos residuales en los cortes de perforación o en las muestras directamente en una columna de cromatografía. Los componentes separados por la columna se detectan utilizando un detector de ionización de llama (FID). Los resultados nos permitirán encontrar los picos y las áreas.

7.5.1.2 Equipo El equipo recomendado en este caso es el siguiente: a. Cromatógrafo – el cromatógrafo debe contener una columna capilar grande (ID
0.5 milímetros) que tiene una fase estacionaria que es adecuada para la separación y un detector de ionización de llama.
b. Celda de vaporización (nivel bajo de volumen muerto, control exacto de la temperatura y distribución uniforme de temperatura).
c. Bote para la muestra (tubo de cuarzo) d. Integrador e. Balanza (0.001 gramos). 7.5.1.3 Procedimiento Se selecciona una muestra representativa (0.01 a 1 gramo) y se coloca en un bote especial para la muestra que el cual se ha tarado (los cortes deben lavarse con agua para retirar el fluido producto de la perforación y se seca al aire entre dos y cuatro horas antes de colocarse en el bote). La cantidad de la muestra utilizada depende de la saturación del petróleo, la capacidad del detector y las limitaciones que tenga la columna. Se pesa el bote con la muestra y se coloca en la celda de vaporización. Se considera que la grasa o aceita que sale de la piel humana es un contaminante y por lo tanto se debe tener cuidado y evitar en lo posible manipular la muestra directamente. Se vaporizan los hidrocarburos a una temperatura tal que no se induzca el cracking, teniendo en cuenta que temperaturas superiores a 320 grados centígrados van a ocasionar el cracking de moléculas grandes. El gas resultante se pasa directamente a la columna sin separarlo. Se puede aumentar el nivel de resolución incrementando la temperatura de la columna en una proporción programada. El producto del detector de ionización de llama se puede convertir en milivoltios (altura) y en milivoltios x segundos (área). Se identifican los picos específicos de acuerdo al tiempo de retención. 7.5.1.4 Precauciones Las precauciones que se deben tener en este método son: a. La muestra debe ser representativa b. Las muestras más grandes pueden aumentar el nivel de repetibilidad pero al
mismo tiempo pueden restringir la resolución del pico. c. La alta saturación de agua ligada puede ocasionar una supresión de la
respuesta del detector, particularmente en los componentes más livianos. d. Se necesita exactitud en los pesos (± 0.001 gramos)

e. Se debe tener cuidado para evitar el calentamiento durante el proceso de secado de la muestra y de esta manera evitar pérdidas de los componentes más livianos.
7.5.1.5 Cálculos 7.5.1.5.1 Cuantificación del volumen de Hidrocarburo residual:
Area total normalizada (Ant) = Peso de la muestra (gramos) Area total (At)
7.5.1.5.2 Procesos de Alteración – Los índices pueden tabularse para
diferenciar cambios en la composición. A continuación se relacionan varios índices que se utilizan generalmente para determinar el tipo, el origen y los procesos de alteración: a. n – C15 + (%) = > area n – C15
At
x 10
b. n – alcano (%) = n – c área componentesAt
x 100
c. Proporción n-C17 + n-C18 con respecto al pristano + fitano = (Altura n-C17 + altura n-C18) / (Altura del pristano + altura del fitano)
d. Proporción pristano / fitano = Altura del pristano
Altura del fitano
e. Indice de Preferencia del Carbono = [Σalturas impares n –C17-C31] / [Σalturas pares n –C16-C30] + [Σalturas impares n –C17-C31] / [Σalturas pares n –C18-C32]
7.5.1.5.3 Tipo de Producción de Hidrocarburo El porcentaje de area dentro de cada grupo (liviano, intermedio y pesado) de carbono se calcula de la siguiente manera: a. % de los componentes livianos = < Area C8
At x 100
b. % de componentes intermedios = < Area C13At
x 100
c. % de componentes pesados = < Area C8At
x 100
La predicción de la producción de hidrocarburos se puede basar en las siguientes normas:

a. No – hidrocarburo Productivo – porcentaje relativamente alto de componentes livianos, area total relativamente baja.
b. Productivo de Gas – Porcentaje relativamente alto de componentes más livianos con porcentajes bajos de componentes intermedios. Area total relativamente alta.
c. Productivo de Petróleo – Porcentaje relativamente alto de componentes intermedios y pesados. Area total grande.
7.5.1.6 Ventajas Las ventajas que tiene este método son: a. Se pueden utilizar muestras extremadamente pequeñas (0.01 – 1.0 gramos) b. No se necesita extracción del solvente y partición de la muestra c. Se puede monitorear de manera simultánea la composición del petróleo y el
contenido de hidrocarburos residuales. 7.5.1.7 Limitaciones El agua ligada puede suprimir la respuesta del detector, particularmente de los componentes más livianos. 7.5.1.8 Precisión Si se hacen pesos exactos de la muestras y se lleva un control cuidadoso de las condiciones de operación, se producirán volúmenes reproducibles en un 3%. La reproducibilidad de la distribución de los componentes depende del mantenimiento de las condiciones operativas y de la condición de la columna. Esto puede verificarse corriendo nuevamente la prueba. 7.5.1.9 Calibración La calibración del equipo es muy importante para obtener resultados consistentes. Las siguientes áreas se consideran críticas: a. Identificación de los componentes – La identificación de los picos individuales
se basa en parte en el tiempo de retención. El tiempo de retención depende de la tasa de flujo del gas transportador, de la temperatura de la columna y de las condiciones de la fase estacionaria de la columna. Se debe calibrar el flujo del gas transportador y el sistema en general debe revisarse de manera rutinaria para evitar las goteras. Se pueden medir los tiempos de retención para los picos específicos. La muestra se puede inyectar con componentes específicos que ayudan en la calibración como es el caso de alcanos normales.
b. Calibración de la Respuesta del detector – Se debe utilizar un estándar para revisar la resolución del pico y el ruido del sistema. Las áreas relativas deben ser consistentes. Es típico encontrar una desviación estándar del 2-3%.
c. Reproducibilidad – La reproducibilidad puede evaluarse haciendo pruebas repetidas sobre la misma muestra. Los índices específicos que se utilizan deben revisarse junto con el área total normalizada por el peso.

7.6 SOLUBILIDAD ACIDA 7.6.1 Principio Las pruebas de solubilidad ácida efectuadas en cortes o muestras se utilizan para evaluar el HCl y/o la reactividad del lodo ácido con la roca del reservorio (HCl / HF). Estos datos se utilizan para evaluar el éxito potencial de la estimulación ácida, retiro del daño cerca al pozo, para evaluar la sensibilidad del daño de la formación, para determinar la proporción de calcita / dolomita en los carbonatos y para determinar el contenido de cuarzo en los carbonatos. Los procedimientos de la prueba se pueden adaptar para determinar los volúmenes ácidos que se necesitan para el tratamiento a nivel de campo y para determinar los coeficientes de respuesta al ácido. El test de solubilidad ácida es una prueba gravimétrica que se ejecuta para determinar la sensibilidad que tiene la formación al ácido. Se coloca una masa conocida de muestra seca en exceso de ácido durante un período de tiempo específico. Posteriormente la muestra se seca y se vuelve a pesar. La diferencia en peso se utiliza para determinar el porcentaje de material soluble en ácido. 7.6.2 Equipo y Materiales
El equipo que se utiliza para la determinación de la solubilidad ácida incluye: a. Balanza (0.001 gr.). b. Material de vidrio variado para solubilidad de HCl (vasos de precipitado,
embudos, cilindros graduados etc.). c. Vasos de precipitado de polietileno o polipropileno, embudos, cilindros
graduados etc. del mismo material para determinar la solubilidad del lodo ácido.
d. Mortero y mango e. Tamiz US de pantalla 80 f. Horno para secar g. Papel de filtro resistente al ácido h. HCl, 15% en peso. i. Lodo ácido (HCl 12% en peso – HF 3% en peso) j. Equipo de seguridad industrial apropiado (máscara, delantal etc. ). 7.6.3 Procedimiento (Solubilidad en HCl ; contenido de calcita) Se saca una muestra representativa y se le retiran los hidrocarburos. Se procede a secar la muestra hasta que se alcance un peso contante garantizándose de esta manera la remoción de solvente y de agua. La muestra se desagrega de manera suficiente para producir varios gramos de material más fino que el tamiz 80. Se pesa aproximadamente un (1) gramo de muestra y se coloca en 150 cc de HCl 15% por un tiempo mínimo de 60 minutos y un máximo de 65 minutos sin revolverla. Se prepara el equipo de filtración y se pesa el papel de filtro. La

muestra ácido / muestra se filtra y se enjuaga con agua desionizada. La muestra restante se seca y se pesa. La diferencia en peso se utiliza para calcular el porcentaje de material soluble en ácido.
7.6.4. Procedimiento (Solubilidad en HCl ; Contenido de Calcita + Dolomita).
La dolomita es soluble en HCl a 80 grados centígrados. Aplicando el procedimiento descrito en la sección 7.6.3 y a esta temperatura se obtiene el valor considerado como el valor total de carbonato (calcita + dolomita). 7.6.5 Procedimiento (Solubilidad en Lodo Acido – Contenido Total de
Material Soluble en Acido) Los carbonatos y el material silíceo como por ejemplo los feldespatos y las arcillas se disuelven en una mezcla de HCl y HF. Esta mezcla se denomina lodo ácido. Aunque se utilizan varias concentraciones, se recomienda la mezcla de 12% en peso de HCl y 3% en peso de HF. Se aplica el procedimiento ya descrito utilizando un Equipo que tolere el HF y el resultado será la solubilidad ácida total (es decir, carbonatos, feldespatos, óxidos de hierro, arcillas etc. ). 7.6.6 Precauciones Es un procedimiento gravimétrico y por lo tanto requiere que los procedimientos se sigan de manera estricta y con una buena técnica de laboratorio para de esta manera garantizar la reproducibilidad. PRECAUCION. Se deben emplear todas las precauciones necesarias en el manejo de ácidos en los procedimientos contenidos en las secciones 7.6.3, 7.6.4 y 7.6.5. 7.6.7 Cálculos A continuación se detallan los cálculos utilizados en este método: Contenido de Calcita (HCl, Temperatura ambiente) % Soluble en Acido =
Peso Inicial de la muestra Peso Inicial muestra – Peso final de muestra x 100
Contenido Total de Carbonato (HCl, 80 grados centígrados) % Soluble en Acido =
Peso Inicial de la muestra Peso Inicial de muestra – Peso final de la muestra x 100

% de Dolomita = % Contenido Total de Carbonato - % contenido de calcita Contenido Total Soluble en Acido (Lodo Acido): % Soluble en Acido =
Peso Inicial de la muestra Peso Inicial de muestra – Peso final de la muestra x 100
7.6.8 Ventajas La técnica no tiene complicación alguna y el equipo para realizarla es simple. 7.6.9 Limitaciones Las limitaciones que tiene este método son: a. El procedimiento es indicativo de la reactividad máxima de la roca puesto que
la muestra se encuentra desagregada y el test se realiza en exceso de ácido. b. La siderita es soluble en HCl a temperatura ambiente y por lo tanto puede
ocasionar distorsiones en el cálculo del contenido de calcita. 7.6.10 Exactitud y Precisión No se cuenta con información al respecto. 7.6.11 Calibración La balanza debe estar calibrada. 7.7 DETERMINACION DE LA SALINIDAD DEL AGUA PRESENTE EN LA
MUESTRA Es aconsejable realizar una determinación del agua presente en la muestra debido a que esto podría ser de gran ayuda en la interpretación de los datos que resulten del análisis practicado a la muestra y en la evaluación de los registros eléctricos. La salinidad, también denominada sólidos disueltos totales (TDS) se define como la suma de todos los materiales disueltos en términos de miligramos de sal por kilogramo de solución (mg/kg.), lo cual es equivalente a parte por millón (ppm). La salinidad se calcula a partir de la cantidad de ion cloro presente en el agua de la muestra y se expresa como la cantidad equivalente de cloruro de sodio. No obstante, si se calcula la salinidad a partir de mediciones de resistividad, ésta se representa como el equivalente de NaCl en todos los iones solubles (electrolitos). Las mediciones que se hagan de la salinidad del agua se basan en la suposición de que todos los iones químicos y los gases presentes en la muestra a nivel de reservorio son los mismos presentes en la muestra a condiciones de medio ambiente. Es posible que esto no pueda aplicarse a la salmuera con

concentraciones iónicas cercanas a los límites de la solubilidad, es decir, soluble a temperatura y presión del reservorio pero insoluble a condiciones ambientales. Durante la operación de muestreo, el filtrado del fluido (lodo) puede invadir la muestra o núcleo y diluir la formación de formación de agua. Por lo tanto se deben tener en cuanta las siguientes precauciones: el material de la muestra debe obtenerse utilizando una técnica poco invasiva, el fluido del a muestra debe tener bajas pérdidas de fluido API, las muestras del núcleo destinadas al análisis deben obtenerse en el sitio del pozo, justo después de las superficies del núcleo. Los tapones que se destinen a análisis de la salinidad del agua deben ser los ubicados en el centro de la muestra, lo más lejos posible del perímetro donde se presenta la más alta filtración de fluido. Con el fin de evitar la evaporación de agua a partir del material, el tapón o la muestra deben sellarse justo después de haber sido retiradas (ver sección 2.5). No se recomienda como técnica alterna a la recolección de la muestra en el sitio del pozo, el congelar toda la muestra una vez se ha retirado del barril para evitar migraciones (trazas) y saturación contra corriente. El frente de salinidad se desarrolla a medida que la muestra se congela, concentrándose la sal por delante de la línea de congelamiento. Si se conoce la salinidad del agua de formación y del filtrado, el grado de lavado que experimenta el fluido de la muestra se puede calcular por medio de la salinidad del agua presente en la muestra (aunque puede afectarse por distribuciones no uniformes). Aun si se desconoce la salinidad del agua de formación, el grado de lavado se puede calcular si el fluido de la muestra contiene un químico que pueda ser rastreado como por ejemplo el agua tritiada, bromuro o yoduro que se asumen bajos en el agua de formación. Si el fluido de la muestra contiene altas concentraciones de potasio, debido a que el cloruro de potasio es la fuente principal de la salinidad, se puede rastrear el ion potasio. En el agua de formación, la proporción de sodio : potasio es mayor que uno (1). Un análisis catiónico (de sodio y de potasio) del agua de la muestra nos proporciona un método para poder determinar se la muestra se encuentra relativamente libre de fluido. Si se rastrea el potasio se podrá únicamente obtener un indicativo cualitativo del grado de invasión de fluido en la muestra. Es más cuantitativo rastrear químicos como el agua tritiada, el bromuro o el yoduro. Estos aniones no se atenúan químicamente por efectos de la matriz de la roca como si puede sucederle a los aniones como el potasio.
7.7.1 Recuperación del Agua de la Muestra Para medir la salinidad del agua presente en la muestra se debe primero separar esta agua de la muestra. Esto puede lograrse utilizando medios mecánicos, sacando el agua con lavados de fluidos no miscibles o con centrifugación. Los medios mecánicos no reportan buenos resultados cuando el agua de la muestra se encuentra cerca de un nivel de saturación en el cual no puede reducirse. Cuando no pueda sacarse salmuera utilizando medios mecánicos, las sales pueden sacarse con agua a partir de una muestra seca. La salinidad se calcula a partir de la cantidad de sal extraída y de la cantidad de agua que estaba inicialmente presente en la muestra.

7.7.1.1 Centrifugación
7.7.1.1.1 Principio Los fluidos presentes en la muestra, incluyendo el agua de formación, pueden extraerse al centrifugar la muestra.
7.7.1.1.2 Equipos El equipo que se recomienda en este caso es: a. Centrífuga y contenedores para la muestra b. Material de vidrio variado.
7.7.1.1.3 Procedimiento La centrífuga debe ser de tal tamaño y manejarse de tal manera que se pueda generar una fuerza de por lo menos 1000 veces la fuerza de la gravedad en el centro de la muestra. El recipiente donde se va a colocar la muestra debe ser una unidad con poco aire, perforada en la parte inferior donde se ubica la cámara de recepción. Las muestras se colocan de maneras simétrica, de tal manera que correspondan al tamaño de los recipientes. Se someten luego a un proceso mínimo de centrifugación de una (1) hora o se deja toda la noche si la permeabilidad de la muestra es menos que 100 md. Se recoge la salmuera con la ayuda de un embudo separador o utilizando una pipeta. Se debe guardar esta muestra para la determinación de la salinidad.
7.7.1.1.4 Precauciones Las precauciones que deben tenerse con este método son: a. La temperatura de la centrifuga debe controlarse para de esta manera reducir
la pérdida potencial de agua b. Se debe tener cuidado en evitar la evaporación de agua en cualquiera de los
pasos descritos que comienzan con la manipulación de la muestra hasta el traslado de la muestra de salmuera al contenedor y la determinación de su salinidad.
7.7.1.1.5 Ventajas Las ventajas que se observan con este método son: a. No es necesario que la muestra tenga forma cilíndrica o que esté consolidada b. La presencia de petróleo crudo en la muestra no interfiere con este
procedimiento ni con la validez de los resultados.

7.7.1.1.6 Limitaciones Las limitaciones que tiene este método son: a. El tiempo necesario para la centrifugación puede prolongarse en las muestras
con baja permeabilidad b. El procedimiento funciona únicamente su la muestra contiene un nivel de
saturación de sal lo suficientemente alto que permita la remoción de una porción a la velocidad más alta posible.
c. La fuerza centrífuga sobre la muestra puede alterar de manera irreversible la matriz de la roca, cambiando la porosidad y la permeabilidad de tal manera que la muestra no puede utilizarse en otros análisis.
d. La salinidad del agua de la muestra que ha sido extraída puede no ser representativa de la salinidad del agua en las condiciones de presión y temperatura que se observan en el ámbito de reservorio
7.7.1.1.7 Exactitud / Precisión La precisión de la técnica de separación se encuentra limitada por la capacidad que se tenga de preservar la muestra desde el sitio del pozo hasta el laboratorio y el posterior manejo que se le haga durante los procesos de extracción para no afectar la salinidad del agua. La evaporación afecta las muestras pequeñas o las muestras que tengan saturaciones bajas de sal, Sw = < 5% aumentando su salinidad. El porcentaje de error se reduce significativamente a medida que la cantidad de volumen de agua aumenta. Si se tienen todos los cuidados necesarios, el error en la detección de salinidad será menor del 2% total.
7.7.1.2 Lavado de Fluido No Miscible 7.7.1.2.2 Principio Se puede expulsar el agua de formación utilizando un lavado de desplazamiento con material no miscible.
7.7.1.2.2 Equipo El equipo que se utiliza en este caso es: a. Recipiente para la muestra tipo Hassler o triaxial b. Bomba solvente c. Material de vidrio variado
7.7.1.2.3 Procedimiento Se monta la muestra en una manga de goma tipo Hassler o Triaxial que permite que el fluido salga a través de la matriz de la muestra. Los detalles de este procedimiento se dan en la sección 3.6.4.1. Como fluido no miscible en agua se

puede utilizar el aceite mineral o un solvente orgánico como el tolueno. Entre más alta sea el contraste de la viscosidad entre el fluido insoluble en agua y el agua, más probabilidades habrá de que se produzca agua de formación durante el lavado. Se separa una muestra de la solución salina producida utilizando un embudo de separación o utilizando una pipeta y tomando la muestra de la capa de solución con sal. Esta muestra debe guardarse para efectuar con ella la determinación de la salinidad.
7.7.1.2.4 Precauciones Las precauciones que deben tenerse en este método son las siguientes: a. El fluido utilizado para expulsar el agua de formación debe tener una baja
solubilidad en agua para que no se cambie la salinidad del agua extraída. b. Se debe tener un cuidad especial para evitar la evaporación del agua durante
la manipulación de la muestra, el lavado y la transferencia de la solución al recipiente donde se analizará la salinidad.
7.7.1.2.5 Ventajas La presencia de petróleo crudo del reservorio en la muestra no interfiere con este procedimiento ni afecta la validez de los resultados.
7.7.1.2.6 Limitaciones Las limitaciones de este método incluyen las siguientes: a. Para las muestras que tengan baja permeabilidad es posible que se prolongue
el tiempo de lavado. b. Este procedimiento arroja buenos resultados siempre y cuando la muestra
contenga una saturación salina que se reduce por la caída de presión aplicada, es decir, la muestra proviene de zonas de transición de agua o de agua – petróleo.
c. Si la presión hidrostática neta utilizada para confinar la muestra durante el proceso de lavado supera la presión neta del reservorio, la porosidad de la muestra puede sufrir compactamientos irreversibles y por lo tanto esta muestra queda inservible para ser utilizada en otros análisis
d. La muestra debe tener forma cilíndrica e. Es posible que la salinidad del agua extraída no sea representativa de la
salinidad presente en el reservorio donde hay otras condiciones de presión y de temperatura.
7.7.1.2.7 Exactitud – Precisión
Consulte la sección 7.7.1.1.7

7.7.2 Extracción Acuosa de la Sal presente en la Muestra Se extraen las sales del agua presente en la muestra seca.
7.7.2.1 Equipo El equipo a utilizar en este caso es: a. Mortero con mango b. Balanza c. Equipo de Desecación d. Material de vidrio variado.
7.7.2.2 Procedimiento Se selecciona una muestra de aproximadamente 50 gramos. Se debe conocer la saturación de agua. El método de elección es utilizar una porción de la muestra que ya ha sido utilizada en un test para determinar la saturación. No es tan recomendable seleccionar la muestra del material situado en un punto que se encuentra lo más cercano posible a la muestra utilizada en el test de medición de saturación. La muestra debe estar libre de hidrocarburos. Se considera adecuado utilizar muestras que han pasado por un proceso de extracción con Tolueno Dean - Stark. Se maceran las muestras en un mortero hasta lograr un tamaño de tamiz 16 y se seca en el horno hasta lograr un peso constante. Una vez se enfría en el equipo de desecación, se pesa y se transfiere a un recipiente de vidrio. Se agregan 100 ml de agua destilada y se mezcla vigorosamente durante varios minutos. La agitación se continúa de manera intermitente hasta completar un mínimo de 1 hora. La solución salina resultante se filtra o se decanta y se guarda para la determinación de salinidad.
7.7.2.3 Precauciones Las precauciones que deben tenerse con este método son: a. Si se sospecha que la muestra contiene material arcilloso, durante la etapa de
secado se debe utilizar un horno para humedad constante a una temperatura de 140 grados Fahrenheit (60 grados centígrados) y 45% de humedad para determinar el contenido de agua en la muestra. Esto se efectúa para evitar la remoción de agua ligada ya sea química o físicamente (agua que no contribuye a la salinidad). De esta manera se evita hacer sobre estimaciones de la cantidad de agua presente en la muestra, lo cual da como resultado una sub estimación de la salinidad. Puesto que el proceso de secado a humedad constante como el que se realiza en estas condiciones no deshidrata las arcillas, no se utilizará esa fracción de 100 ml de agua que se recomendaba para rehidratarlas.

b. Se debe tomar medidas para evitar la evaporación del agua en todos los pasos, comenzando con el proceso de guardar la muestra y luego con su transferencia al contenedor donde se va a determinar la salinidad.
7.7.2.4 Ventajas Se trata de un procedimiento simple que puede realizarse con ayuda de pocos Equipos en un período corto de tiempo.
7.7.2.5 Limitaciones Las limitaciones que se detectan en este método son las siguientes: a. La presencia de hidrocarburos en la muestra interfiere con la determinación del
contenido de agua en la misma. Durante el proceso de secado, se pierden los componentes más volátiles de los hidrocarburos y por lo tanto se puede sobre estimar la cantidad de agua presente.
b. Si la muestra contiene minerales en forma de sulfuro, como la pirita que ha sufrido oxidación durante la manipulación, almacenamiento o extracción, se puede correr el riesgo de incurrir en una sobre estimación de las sales extraídas. La oxidación de la pirita produce sales de sulfato de hierro que son solubles en agua.
c. Si la matriz de roca in situ incluye sales solubles en su fase sólida (que están en un equilibrio de solubilidad con respecto a la formación de la solución salina), como por ejemplo el caso de la Anhidrita o del cloruro de sodio, durante la fase de secado y extracción estas sales pueden también extraerse debido a que existe una proporción alta agua : roca, lo cual nos puede producir una exagerada salinidad del agua en la muestra.
d. La muestra se destruye. e. Es posible que la salinidad calculada del agua presente en la muestra no sea
representativa con respecto a la presión del reservorio y a las condiciones de temperatura existentes allí.
7.7.3 Determinación de la Salinidad del Agua presente en la Muestra La salinidad del agua se determina utilizando (a) determinación química del ión cloruro; (b) el índice de refracción o (c) medición de la resistividad. La salinidad se expresa como parte de cloruro de sodio por millón de agua (aunque otras sales estén presentes), lo cual equivale a miligramos de cloruro de sodio por kilogramo de solución (cloruro de sodio y agua). Existen otras técnicas analíticas para medir el contenido de sal del agua. Estas técnicas incluyen la cromatografía de iones, la absorción atómica y la espectroscopía de plasma apareado. La combinación de estos métodos puede determinar de manera precisa la composición aniónica y catiónica del agua presente en la muestra aunque se debe considerar que el equipo que se utiliza es

mucho más costoso que el que se describe para los procedimientos en 7.7.3.1, 7.7.3.2 y 7.7.3.3.
7.7.3.1 Determinación Química del Ion Cloruro La concentración de cloruro se mide en la solución salina no diluida que sale de la muestra o a partir el filtrado que se obtiene por la separación de la muestra fraccionada y el aguas utilizada en el proceso de lavado. La salinidad del agua se expresa en forma de partes por millón de cloruro de sodio en el agua de la muestra (mg/kg.).
7.7.3.1.1 Equipos y Procedimiento Los aparatos a utilizar, los reactivos y el procedimiento se describen en detalle en la forma API RP 45 Prácticas Recomendadas para el Análisis de Aguas en los Pozos de Petróleo y también en la publicación ASTM D512-89 Métodos Estándar para Determinar el Ion Cloruro en Agua. De los cuatro métodos ASTM que se relacionan, el método A (Titriado Mercurimetrico), el Método B (Titriado con Nitrato de Plata), el Método C (Colorimétrico) y el Método D (Electrodo selectivo a ciertos iones), se recomienda utilizar el Método B. Los cálculos que se describen en esta publicación dará la salinidad en términos de iones cloruro en lugar de cloruro de sodio. Para calcular el cloruro de sodio, consulte la sección 7.7.3.1.3.
7.7.3.1.2 Precauciones Las precauciones que deben tenerse son las siguientes: a. La solución salina debe diluirse para ubicarla dentro del rango óptimo que
necesitan los procesos de tritiado y los equipos. b. Realice determinaciones de cloruro en blancos para corregir cualquier cloruro
que se encuentre en el material de vidrio y en agua que se utilicen durante la prueba.
7.7.3.1.3 Cálculos Se utilizan los siguientes cálculos: a. La salinidad del agua presente en la muestra una vez extraída puede
calcularse de manera directa. La normalidad de la solución salina que se mide con el tritiado con cloro puede convertirse a un valor de concentración de cloruro de sodio en mg/kg. de la siguiente manera:
Conc. De NaCl (mg/kg.) = 1000 x N1 x MWNaCl / Dw Donde :
N1 = V2 x N2 / V1 N1 = Normalidad de la solución salina tritiada, en meq/ml V1 = Volumen de la solución salina tritiada, en ml

N2 = Normalidad del reactivo cloruro (por ejemplo Nitrato de plata), en meq/ml V2 = Volumen del reactivo cloruro al final del proceso de tritiado, en ml Dw = Densidad del agua en la muestra, en g/ml MWNaCl = Peso equivalente del NaCl = 58.5 g/equ b. Cuando no pueda sacarse el agua de la muestra y las sales se extraen con
agua, la salinidad del agua presente originalmente en la muestra se puede calcular yendo hacia atrás en el proceso. La salinidad determinada por el método químico se convierte en miligramos totales de cloruro de sodio que sale de la muestra. Este número representa la sal total en el agua presente en la muestra. Los datos referentes a la sal total en miligramos y al peso del volumen del agua de la muestra que se obtiene con las pruebas de saturación se utilizan para calcular la concentración de sal en el agua de la muestra. Este valor se expresa en partes por millón de cloruro de sodio.
Los miligramos de cloruro de sodio que se lixiviaron de la muestra se calculan de la siguiente manera (se parte del modo del método de Titriado con cloruro).
Peso del NaCl (mg) = N1 x 58.5 x Vw Donde: N1 = V2 x N2 / V1 N1 = Normalidad de la solución salina tritiada. V1 = Volumen de la solución salina tritiada.
N2 = Normalidad del reactivo cloruro (por ejemplo Nitrato de plata). V2 = Volumen del reactivo cloruro al final del proceso de tritiado, en ml Vw = Volumen del agua utilizada para lixiviar o extraer la muestra, en ml. El peso del agua presente en la muestra se puede obtener directamente por diferencia, restando el peso de la muestra antes y después del secado, si no hay petróleo presente. El proceso de secado debe seguir las instrucciones dadas en la sección 5.3.2.2.3.6 La salinidad (mg/kg.) del agua presente se obtiene aplicando: Mg NaCl/kg. agua en muestra = mg NaCl g de agua en la muestra + g de NaCl
x 1000.0 (27)
Si la muestra contiene petróleo, se utiliza la siguiente fórmula que nos dan una aproximación al valor de la salinidad. La saturación del fluido y la porosidad deben determinarse utilizando ya sea esta misma muestra o una tomada de un núcleo adyacente. La cantidad de agua de la muestra se calcula de la siguiente manera: Peso del agua en la muestra, g. = Vb x θ x Sw x Dw (28) Donde : Vb = [(W/Dg / 1 - θ)] (29) W = Peso seco de la muestra, en g Dg = Densidad del grano, en g/cc θ = Porosidad como fracción Vb = Volumen total de la muestra, cc Sw =Saturación del agua, como fracción del volumen del poro Dw = Densidad del agua en la muestra, en g/ml

Si no se ha determinado la densidad del grano, se utiliza un valor representativo para la formación que sea pertinente. La densidad del agua en la muestra se asume que es inicialmente 1.00. Una vez que se ha calculado la salinidad con la ecuación 27, el peso del agua en la muestra obtenido con la ecuación 28 se vuelve a calcular utilizando una nueva densidad que se basa en la salinidad ya calculada. Se calcula entonces una nueva salinidad utilizando la ecuación 27.
7.7.3.1.4 Ventajas Las ventajas que tiene este método son: a. Es rápido y conveniente b. La determinación de cloruro es precisa.
7.7.3.1.5 Limitaciones Las limitaciones que tiene este método son: a. El resultado se expresa en forma de cloruro de sodio. Los aniones diferentes al
cloruro, como por ejemplo el sulfato y el bicarbonato no se detectan aunque estén presentes en la solución.
b. Otros iones haluros como es el caso del bromuro y el yoduro se determinan como cloruros.
7.7.3.1.6 Exactitud / Precisión
Al recuperar el agua de la muestra utilizando procesos como la centrifugación o lavado con materiales no miscibles, la exactitud en la determinación de la salinidad (en mg/kg.) está controlada por la técnica que se utilice para la determinación de la salinidad, tritiado con cloro, en la muestra que tiene aproximadamente ±1-2% de la salinidad total. La practica API 45 que se recomienda reporta que la precisión es de aproximadamente un 1% y la exactitud es de casi 2% de la cantidad presente. Cuando las sales del agua presente en la muestra se tienen que extraer de un a muestra seca, la exactitud en la determinación de la salinidad está limitada por la determinación del contenido de agua. McCoy et al publicaron reportes donde las determinaciones de la salinidad (cloruro) en lodos con base de petróleo (OBM) en contacto con la fase petróleo – agua concordaban en un 1% en promedio con la salinidad del agua producida. Las mediciones de concentración de cloruro se obtuvieron de extractos de agua en muestras de aproximadamente 2000 OBM (utilizando procedimientos similares a los descritos en las secciones 7.7.2 y 7.7.3).
7.7.3.2. Medición del Indice de Refracción Los constituyentes iónicos totales del agua se pueden calcular midiendo el índice de refracción. Este índice de refracción varía de forma directa con la concentración iónica de cloruro de sodio y otras sales. A partir de los reportes que se encuentran en la literatura, se pueden preparar gráficas estándar que muestran el índice de

refracción de las soluciones de cloruro de sodio a varias salinidades y temperaturas (ver figura 7-2). Si se conoce el índice de refracción, se puede determinar la salinidad del agua presente en la muestra que se expresa en forma de partes por millón de cloruro de sodio (mg/kg.) con esta gráfica y utilizando el índice de refracción conocido.
7.7.3.2.1 Equipo. El equipo que se utiliza en este método es el siguiente: a. Refractómetro b. Material de vidrio diverso
7.7.3.2.2 Procedimiento Se debe calibrar el Equipo con concentraciones conocida de soluciones de cloruro de sodio, lo cual da como resultado una gráfica que es similar a la que se muestra en la figura 7-2. Los detalles del Equipo y de los procedimientos se encuentran consignados en la publicación ASTM D542-85, Método Estándar para la Extracción del Agua presente en el Poro y Determinación del Contenido Soluble de Sal de los Suelos Utilizando un Refractómetro. El índice de refracción se mide un una muestra de solución salina. Si se utiliza la gráfica estándar (ver figura 7-2) y los cálculos apropiados, el índice de refracción se puede convertir en un valor de salinidad.
7.7.3.2.3 Precauciones Se debe corregir el valor del índice de refracción a una temperatura estándar.
7.7.3.2.4 Ventajas Las ventajas que tiene este método son las siguientes: a. La determinación es rápida b. Se determina la concentración equivalente total de cloruro de sodio.
7.7.3.2.5 Limitaciones Todos los iones que se encuentren en el agua se calculan como cloruro de sodio.
7.7.3.2.6 Exactitud / Precisión Consulte la sección 7.7.3.1.6 donde se consignan unos comentarios generales al respecto. De conformidad con la norma ASTM D4542, la determinación de la salinidad utilizando el Refractómetro tiene una exactitud de ± 300 mg/kg. de salinidad.

7.7.3.3 Medición de la Resistividad Se pueden calcular los constituyentes iónicos totales del agua por medio de la medición de resistividad. La resistividad varía de forma inversa con la concentración iónica de cloruro de sodio y otras sales en una gama amplia de concentraciones. Las gráficas estándar como la que se muestra en la figura 7-3, muestra el valor de resistividad para varios niveles de salinidad y temperatura de soluciones de cloruro de sodio. Si se conoce la resistividad, se puede determinar la salinidad del agua presente en la muestra, la cual se expresa en forma de partes por millón de cloruro de sodio (mg/kg.). Esto se realiza con la ayuda de la gráfica y utilizando un valor de resistividad ya calculado.
7.7.3.3.1 Equipo El equipo que se utiliza en este caso es: a. Celda de resistividad b. Medidor de resistividad c. Material de vidrio variado.
7.7.3.3.2 Procedimiento Se debe calibrar la celda de resistividad con concentraciones conocidas de cloruro de sodio, lo cual nos dará una gráfica que es similar a la que se muestra en la figura 7-3. Se toma una porción de la solución obtenida por lixiviación de la muestra y se coloca en la celda. La resistividad se mide con un medidor apropiado y se calcula en Ohmetros. Utilizando la gráfica estándar ya descrita (figura 7-3) y los cálculos pertinentes, el valor de resistividad se convierte en valor de salinidad. Para mayor información respecto al aparato y al procedimiento, consulte la publicación ASTM D1125-82, Método para Probar la Conductividad Eléctrica del Agua.
7.7.3.3.3 Precauciones Las precauciones que deben tenerse con este método son las siguientes: a. Se debe calibrar el medidor de resistividad y la celda b. El valor de resistividad debe corregirse a temperatura estándar c. Asegúrese de que los electrodos de la celda de resistividad estén limpios antes
de tomar las mediciones. d. Calibre el electrodo con una solución salina estándar durante la utilización de la
técnica para determinar / revisar la constante de la celda, tal como lo describe Worthington et al.
e. El sulfuro de hidrógeno puede ocasionar cambios en la calibración de ciertos electrodos.

7.7.3.3.4 Cálculos Los cálculos que se utilizan en este método son los siguientes: a. Se puede calcular directamente la salinidad del agua que sale de la muestra.
La resistividad de la solución salina se puede convertir directamente a concentración de miligramos de sal por kilogramo de solución, utilizando una tabla de conversión del tipo que se ilustra en la figura 7-3.
b. Cuando no se pueda extraer el agua de la muestra y la sales se saquen con agua, se debe calcular la salinidad del agua que estaba presente originalmente en la muestra. La salinidad obtenida a partir de la medición de resistividad se convierte en miligramos totales de cloruro de sodio lixiviado de la muestra. Este número representa el total de sal presente en el agua de la muestra. La sal total en miligramos y el peso del volumen de agua de la muestra que se obtiene con la prueba de saturación se utiliza para calcular la concentración de sal en el agua de la muestra y este valor se expresa en partes por millón de cloruro de sodio. La salinidad (mg/kg.) del agua se obtiene con los cálculos de la sección 7.7.3.1.3 b.
7.7.3.3.5 Ventajas Las ventajas que tiene este método son: a. Rápida determinación b. Los datos de resistividad se relacionan directamente con los registros eléctricos c. Se determina la concentración total de cloruro de sodio equivalente.
7.7.3.3.6 Limitaciones Todos los iones que se encuentren presentes en el agua se calculan en forma de cloruro de sodio.
7.7.3.3.7 Exactitud / Precisión Consulte los comentarios generales que se encuentran en la sección 7.7.3.1.6 y la publicación ASTM D1125, si desea información específica respecto a la resistividad. 7.7.3.4 Determinación de Cationes El análisis directo de sodio, potasio, calcio y magnesio permite la determinación de la salinidad del agua y puede representar un método para conocer la salinidad del agua y ver si está libre de fluido de la muestra. Otros cationes participan en la formación de aguas pero representan menos del 5% de la salinidad. La salinidad del agua presente en la muestra se puede calcular a partir de la concentración de estos cuatro iones.

La concentración de estos iones puede medirse rápidamente, de manera simultánea y muy precisa utilizando un espectrómetro de emisión atómica acoplando de manera inductiva (ICPES). Se pueden determinar también de manera individual utilizando un espectrómetro de absorción atómica (AAS). Algunos fluidos en la actualidad presentan altas concentraciones de potasio. En la mayoría de las aguas de formación, la proporción de sodio : potasio es mayor que uno. De esta manera se puede afirmar que para los fluidos que tengan como base el potasio, la invasión puede calcularse con base en la proporción de potasio : sodio en el agua de la muestra. 7.7.3.4.1 Equipo y Procedimiento El equipo y el procedimiento para la determinación de cationes utilizando AAS se encuentra detallada en la publicación ASTM D4191 en el caso del sodio, ASTM D4192, para el potasio, ASTM E508 para el calcio. El aparato y el procedimiento para la determinación de Cationes utilizando el ICPES se describe en la publicación ASTM D1976. 7.7.3.4.2 Precauciones Las precauciones que deben tenerse con este método son: a. Para el cálculo de la salinidad, se asume que el anion es cloruro aunque se
encuentran cantidades significativas de iones sulfato y carbonato y cantidades más pequeñas de otros iones.
b. El cálculo de la invasión de fluido utilizando la proporción Na/K requiere de un conocimiento respecto a la interacción entre el potasio y la arcilla sódica de bentonita en el fluido de la muestra y con otras arcillas a nivel de roca de formación.
c. Se necesita tener un conocimiento claro de las evaporitas locales y sus efectos en la preparación de la muestra.
d. Unicamente personal entrenado en estas técnicas puede realizar el análisis, el cual debe hacerse en un laboratorio que tenga el equipo instalado de manera permanente.
7.7.3.4.3 Cálculos Se pueden utilizar los métodos descritos en la sección 7.7.3.1.3 para calcular la concentración de cloruro de sodio en la solución salina no diluida y el lixiviado se puede utilizar para calcular la concentraciones de Cationes reemplazando el pesos molecular del cloruro de sodio por el peso atómico del cation. 7.7.3.4.4. Ventajas Las ventajas que tiene este método incluyen las siguientes:

a. Tanto el ICPES como el AAS son métodos analíticos muy precisos. b. Con estos datos es posible determinar la invasión del fluido de la muestra. c. El análisis de los cationes puede suministrar una imagen más completa de la
salinidad en lugar de asumir que el sodio es el único cation presente, que es lo que se hace cuando la determinación de la salinidad se basa únicamente en el análisis del cloruro.
7.7.3.4.5 Limitaciones Las limitaciones son la falta de disponibilidad que tiene la técnica y el alto costo que tiene el ICPES y el AAS. 7.7.3.4.6 Exactitud / Precisión Consulte la sección 7.7.3.1 si desea unos comentarios generales y la publicación ASTM D4191, D4192, E508 y D1976 si desea más detalles. En términos generales, el AAS y el ICPES deben determinar la concentración de estos Cationes con una exactitud y precisión del 1% – 2%





























