Embed Size (px)
Citation preview

NORMATIVIDAD DE
MEDICAMENTOS DE ORIGEN
BIOLÓGICO EN COLOMBIA
DIRECCIÓN DE MEDICAMENTOS Y
PRODUCTOS BIOLÓGICOS
2016
ASS-ESA-DI129 V0 04/08/2016
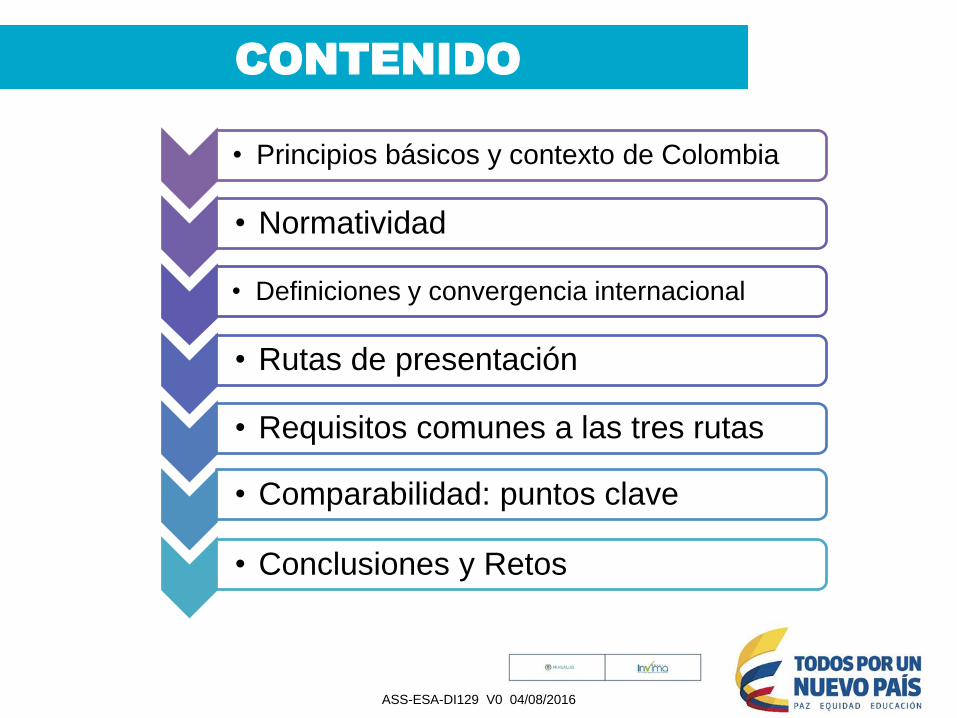
CONTENIDO
• Principios básicos y contexto de Colombia
• Normatividad
• Definiciones y convergencia internacional
• Rutas de presentación
• Requisitos comunes a las tres rutas
• Comparabilidad: puntos clave
• Conclusiones y Retos
ASS-ESA-DI129 V0 04/08/2016

PRINCIPIOS BÁSICOS
¿Qué son los medicamentos de origen biológico?
• Se obtienen a partir de organismos vivos o de sus tejidos.
• Las fuentes y métodos de producción incluyen cultivos de células o de
microorganismos o la extracción a partir de tejidos o de fluidos biológicos como
la sangre.
¿Qué diferencia hay entre los medicamentos de origen químico y
de origen biológico?
• Las características de calidad, seguridad y eficacia de los medicamentos de
origen biológico dependen del material biológico de origen, la complejidad
de su estructura y los procesos tecnológicos de su obtención.
Fuente: Min salud- ABC de los Biológicos y Biotecnológicos 2014
ASS-ESA-DI129 V0 04/08/2016

PRINCIPIOS BÁSICOS
¿Qué son lo medicamentos biotecnológicos?
Son un subconjunto especial de los medicamentos de origen biológico.
Comprenden una amplia variedad de medicamentos.
Los medicamentos de origen biológico se producen mediante procesos más
sencillos sin uso de información genética. Los biotecnológicos usan información
genética y tecnologías especiales para que las células actúen como fábrica de
sustancias para luego convertirlas en medicamentos.
¿Por qué una regulación especial para los medicamentos
biotecnológicos?
• Los desarrollos científicos y tecnológicos para la producción y las técnicas
analíticas de caracterización, avanzan aceleradamente y deben ser incorporadas
a la normatividad sobre registro sanitario y vigilancia.
Fuente: Min salud- ABC de los Biológicos y Biotecnológicos 2014
ASS-ESA-DI129 V0 04/08/2016

PRINCIPIOS BÁSICOS
¿Qué diferencia hay entre un medicamento biotecnológico y un biocomparable?
• Los biocomparables son también medicamentos biotecnológicos.
• Los medicamentos biotecnológicos nuevos, innovadores, pioneros o de marca
son aquellos a los cuales se les han hecho todas las etapas de investigación y
desarrollo y ensayos clínicos completos.
• Los biotecnológicos competidores, de entrada subsiguiente, biosimilares,
biocomparables.
• La diferencia entre los primeros y los segundos es que los nuevos por lo general
entran primero al mercado y están patentados porque representan innovaciones
tecnológicas.
• Los biotecnológicos competidores solo pueden entrar al mercado una vez las
patentes de lo biotecnológicos nuevos se han vencido porque son copias de los
mismos.
Fuente: Min salud- ABC de los Biológicos y Biotecnológicos 2014
ASS-ESA-DI129 V0 04/08/2016

CONTEXTO DE COLOMBIA
¿Cuáles son las razones de Colombia para expedir la nueva
legislación?
El Decreto se expide por razones legales, técnicas y de funcionamiento del
sistema de salud.
• Legal: Reformas en el sistema de Salud y Políticas Publicas Farmacéuticas y
desarrollo de la biotecnología
• Técnica: Actualización y ajuste regulación registro de medicamentos con
enfoque en biológicos, aumento solicitudes de ingreso de medicamentos
nuevos o biológicos genéricos ( vencimiento de patentes)
• Funcionamiento: sostenibilidad del sistema – productos biotecnológicos
segmento de mercado farmacéutico que más crece en Colombia ( 30% del
total). Promoción de la competencia y reducción de los precios.
Fuente: Min salud- ABC de los Biológicos y Biotecnológicos 2014
ASS-ESA-DI129 V0 04/08/2016
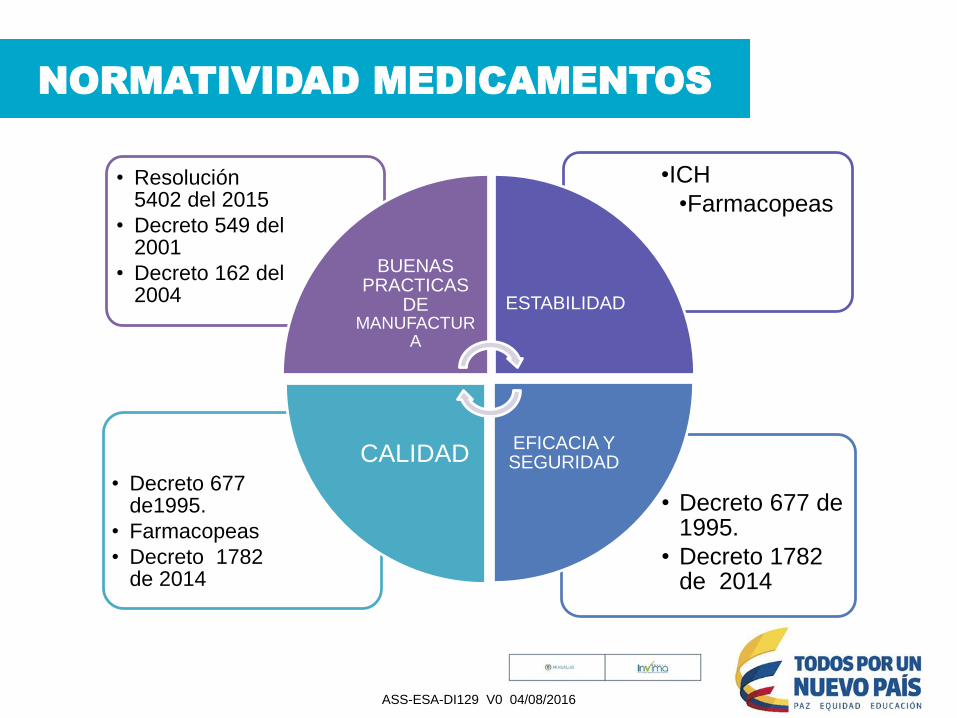
NORMATIVIDAD MEDICAMENTOS
• Decreto 677 de 1995.
• Decreto 1782 de 2014
• Decreto 677 de1995.
• Farmacopeas
• Decreto 1782 de 2014
•ICH
•Farmacopeas• Resolución
5402 del 2015
• Decreto 549 del 2001
• Decreto 162 del 2004
BUENAS PRACTICAS
DE MANUFACTUR
A
ESTABILIDAD
EFICACIA Y SEGURIDADCALIDAD
ASS-ESA-DI129 V0 04/08/2016
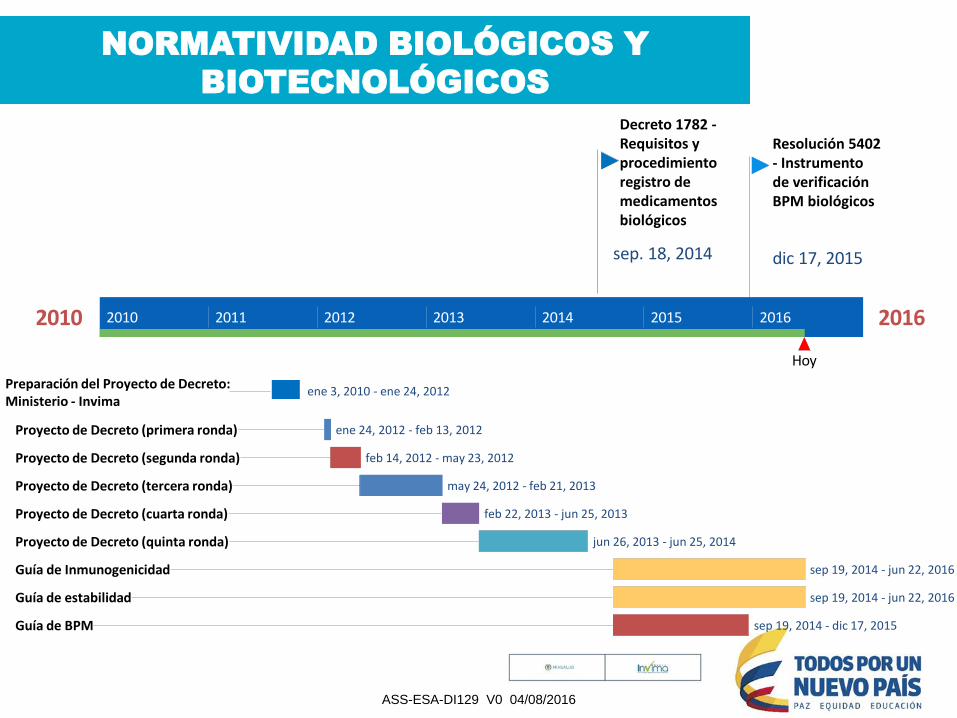
NORMATIVIDAD BIOLÓGICOS Y
BIOTECNOLÓGICOS
2010 2016
Hoy
2010 2011 2012 2013 2014 2015 2016
Decreto 1782 -Requisitos y procedimiento registro de medicamentos biológicos
sep. 18, 2014
Resolución 5402 - Instrumento de verificación BPM biológicos
dic 17, 2015
ene 3, 2010 - ene 24, 2012Preparación del Proyecto de Decreto: Ministerio - Invima
ene 24, 2012 - feb 13, 2012Proyecto de Decreto (primera ronda)
feb 14, 2012 - may 23, 2012Proyecto de Decreto (segunda ronda)
may 24, 2012 - feb 21, 2013Proyecto de Decreto (tercera ronda)
feb 22, 2013 - jun 25, 2013Proyecto de Decreto (cuarta ronda)
jun 26, 2013 - jun 25, 2014Proyecto de Decreto (quinta ronda)
sep 19, 2014 - jun 22, 2016Guía de Inmunogenicidad
sep 19, 2014 - jun 22, 2016Guía de estabilidad
sep 19, 2014 - dic 17, 2015Guía de BPM
ASS-ESA-DI129 V0 04/08/2016

DEFINICIONES COLOMBIA
Medicamentos biológicos
• Medicamentos derivados de organismos o células vivas o sus partes.
• Se pueden obtener de fuentes tales como tejidos o células, componentes de la sangre
humana o animal (como antitoxinas y otro tipo de anticuerpos, citoquinas, factores de
crecimiento, hormonas y factores decoagulación), virus, microorganismos y productos
derivados de ellos como las toxinas.
• Estos productos son obtenidos con métodos que comprenden, pero no se limitan a
cultivo de células de origen humano o animal, cultivo y propagación de
microorganismos y virus, procesamiento a partir de tejidos o fluidos biológicos humanos
o animales, transgénesis, técnicas de Ácido Desoxirribonucleico (ADN) recombinante, y
técnicas de hibridoma.
• Los medicamentos que resultan de estos tres últimos métodos se denominan
biotecnológicos.
[Decreto 1782 de 2014 ]
ASS-ESA-DI129 V0 04/08/2016

DEFINICIONES COLOMBIA
Medicamento de referencia
• Medicamento biológico cuyo registro sanitario ha sido autorizado por el INVIMA u
otra agencia sanitaria de referencia, mediante un expediente completo y que se
utiliza como comparador.
[Decreto 1782 de 2014]
Producto Bioterapéutico Comparable (PBC)
• Producto bioterapéutico comparable en términos de calidad, seguridad y eficacia a
un producto bioterapéutico de referencia (PBR) autorizado previamente.
[Invima]
ASS-ESA-DI129 V0 04/08/2016

CONVERGENCIA INTERNACIONAL
Prospectiva tecnológica
Algunos de los países han considerado establecer rutas de aprobación alternativas
a la de la comparabilidad.
Las condiciones para el uso de estas rutas:
• Se haya demostrado un alto grado de similaridad entre el medicamento
competidor y el estándar de referencia en términos de las especificaciones de
calidad
• Disponibilidad de suficiente información clínica en el dominio público.
Asamblea Mundial de Salud 2014 – Resolución WHA 67.21
• Presentada por Colombia, Argentina y otros países de UNASUR
• Incluye lenguaje a la regulación colombiana
• Insta a la Directora para convocar al Comité de Estarización de Biológicos – actualizar
Guías 2009 considerando necesidades y capacidades nacionales
Vaca, Claudia Et al. 2016. El debate de la regulación de medicamentos biotecnológicos:
Colombia en el contexto internacional.
ASS-ESA-DI129 V0 04/08/2016

CONVERGENCIA INTERNACIONAL
Regulaciones expedidas en Latinoamérica y Europa
Brasil, Chile, Colombia, Ecuador y Uruguay
• Expidieron regulaciones que permiten reducir la extensión y complejidad de pruebas
clínicas comparativas con diferentes restricciones y condiciones.
• Mecanismos de registros con tipo y extensión (o reducción) de las pruebas clínicas de los
competidores.
Argentina y México: Normativas especializadas
Regulaciones europeas
• 2013: EMA CHMP/437/04 Rev. 1, modifica la guía de registro de biosimilares 2006. Entró en
vigencia en abril de 2015.
• Permite la reducción de ensayos clínicos -aproximación simplificada. Establece las
condiciones necesarias sobre la eficacia y seguridad del biosimilar para la excepción a los
estudios clínicos confirmatorios.
Vaca, Claudia Et al. 2016. El debate de la regulación de medicamentos biotecnológicos:
Colombia en el contexto internacional.
ASS-ESA-DI129 V0 04/08/2016

CONVERGENCIA INTERNACIONAL
Regulaciones expedidas en Latinoamérica y Europa
Regulaciones europeas
• En circunstancias específicas, un estudio clínico confirmatorio puede no ser necesario. Esto
requiere que la similaridad de la eficacia y la seguridad puedan ser claramente deducidas
de la similaridad de las características físico-químicas, actividad biológica/potencia y los
perfiles PK y/o PD del biosimilar y el producto de referencia. Adicionalmente se requiere que
el perfil de impurezas y la naturaleza de los excipientes del biosimilar no den lugar a
preocupaciones. [EMA CHMP/437/04 Rev. 1]
• La EMA enuncia se exigirían, como mínimo, estudios farmacocinéticos y farmacodinámicos
comparativos.
• Contenidos de esta guía -elementos concordantes con la regulación colombiana:
• permiten la reducción de los ensayos clínicos en función de la demostración de
similaridad de las características fisicoquímicas, la actividad biológica y la potencia.
Vaca, Claudia Et al. 2016. El debate de la regulación de medicamentos biotecnológicos:
Colombia en el contexto internacional.
ASS-ESA-DI129 V0 04/08/2016

CONVERGENCIA INTERNACIONAL
Estados Unidos - Norma General FDA
ObamaCare: Mandato de diseñar una ruta abreviada (abbreviated pathway) para el
registro de biotecnológicos competidores.
- Incorporada en la “Public Health Service Act” (ley PHS) en la sección 351 (K) (30).
- Adopta el estándar de la comparabilidad: una ruta abreviada comparativa que otorga a la
FDA la discrecionalidad de reducir o eximir cualquier requisito (351(k)(2)(A)(ii)):
‘‘(ii) DETERMINATION BY SECRETARY.—The Secretary may determine, in the
Secretary’s discretion, that an element described in clause (i)(I) is unnecessary in an
application submitted under this subsection".
ADDITIONAL INFORMATION.—An application submitted under this subsection—
‘‘(I) shall include publicly-available information regarding the Secretary’s previous
determination that the reference product is safe, pure, and potent; and
‘‘(II) may include any additional information in support of the application, including publicly-
available information with respect to the reference product or another biological product.
ASS-ESA-DI129 V0 04/08/2016

Guías de
la FDA
Fuente: Vaca, Claudia: pautas para la implementación de la regulación: colombia en el contexto internacional. PPT.
Seminario internacional de productos biotecnológicos de referencia y biosimilares agosto 10-11 de 2015
ASS-ESA-DI129 V0 04/08/2016
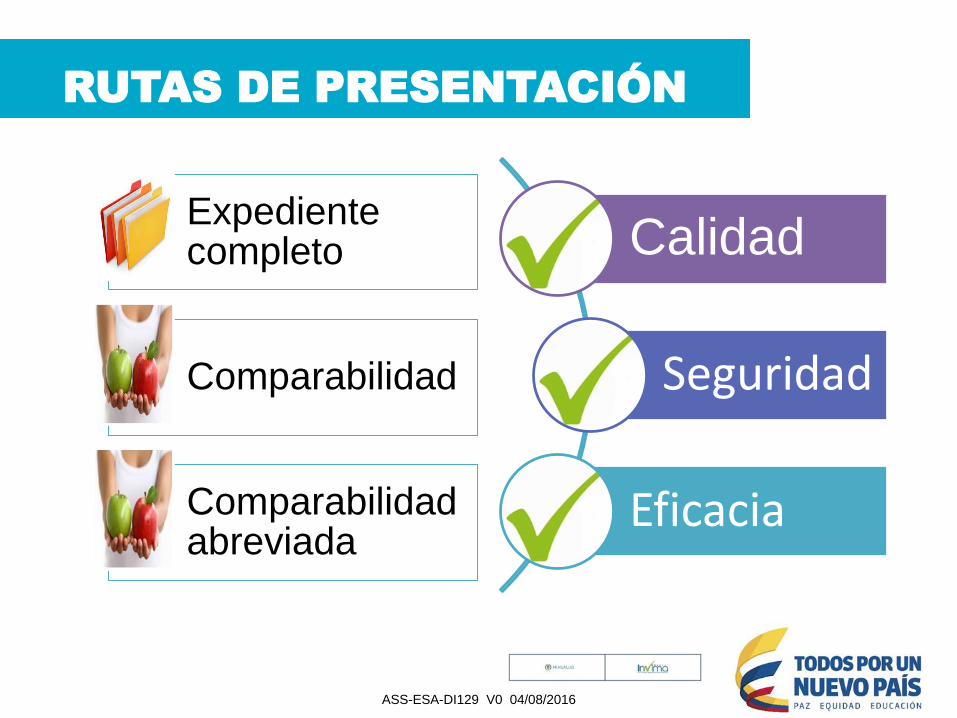
RUTAS DE PRESENTACIÓN
Expediente completo
Comparabilidad
Comparabilidad abreviada
Calidad
Seguridad
Eficacia
ASS-ESA-DI129 V0 04/08/2016
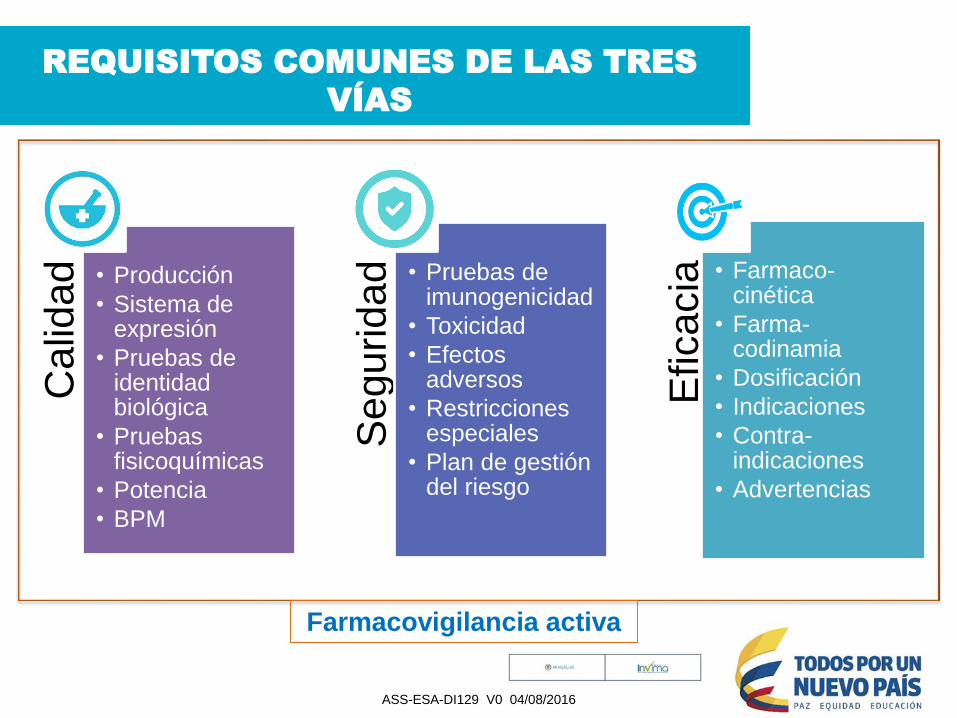
REQUISITOS COMUNES DE LAS TRES
VÍAS
Calid
ad • Producción
• Sistema de expresión
• Pruebas de identidad biológica
• Pruebas fisicoquímicas
• Potencia
• BPM
Seguridad • Pruebas de
imunogenicidad
• Toxicidad
• Efectos adversos
• Restricciones especiales
• Plan de gestión del riesgo
Eficacia • Farmaco-
cinética
• Farma-codinamia
• Dosificación
• Indicaciones
• Contra-indicaciones
• Advertencias
Farmacovigilancia activa
ASS-ESA-DI129 V0 04/08/2016

COMPARABILIDAD: PUNTOS CLAVE
• Concentración, vía de administración y dosis.
• Mismo mecanismo de acción.
• Evaluación completa (Colombia o Países de referencia)
Definición de medicamento de referencia
• Caracterización (fisicoquímica y biológica) del ingrediente farmacéutico activo.
• Perfil farmacocinética y farmacodinamia.
• Evaluación pre-clínica y clínica.
Ejercicio de comparabilidad
ASS-ESA-DI129 V0 04/08/2016

La denominación no se asocia con disminución de la rigurosidad en la evaluación
• Denominación común en la jerga farmacéutica que permiteabreviar experimentos con humanos y usar información pública sobreseguridad y eficacia de un medicamento no patentado (libre deprotección) para no exponer innecesariamente sujetos humanos aexperimentos que respondan preguntas de eficacia y seguridad queya han sido respondidas.
• No se trata de una ruta abreviada clásica para la aprobación demedicamentos genéricos pues se debe aportar información propiaresultante de pruebas imprescindibles y que son mucho más ampliasy específicas que las que se pedirían a un genérico de síntesisquímica.
COMPARABILIDAD: PUNTOS CLAVE
Fuente: Vaca, Claudia: pautas para la implementación de la regulación: colombia en el
contexto internacional. PPT. Seminario internacional de productos biotecnológicos de
referencia y biosimilares agosto 10-11 de 2015
ASS-ESA-DI129 V0 04/08/2016

• Colombia reconoce que tiene una postura de avanzada por su decisión de
hacer explícita y visible la ruta abreviada de comparabilidad.
• La normatividad, las guías y procedimientos que aplicará el Invima establece
los criterios bajo los cuales se podrían exceptuar o reducir ensayos clínicos
propios:
- moléculas ampliamente conocidas,
- suficientemente caracterizadas y
- altamente similares con el medicamento o estándar de referencia.
• La regulación de Colombia sigue una tendencia global
• Las regulaciones internacionales se acercan a reconocer un mecanismo
abreviado o simplificado (Europa, USA, Brasil, México, Ecuador) para
moléculas menos complejas y mejor caracterizadas (altamente similares).
CONCLUSIONES
ASS-ESA-DI129 V0 04/08/2016
Nuevo sistema para revisión integrando
diferentes grupos y disciplinas.
Nuevas guías
Inmunogenicidad
Plan de gestión de riesgo.
Fortalecer el sistema de farmacovigilancia
Fortalecer la comunicación con las
diferentes agencias.
RETOS
ASS-ESA-DI129 V0 04/08/2016

www.invima.gov.coGrupo de Registros Sanitarios de
Medicamentos y Productos BiológicosCarrera 10 No 64-28 - Bogotá D.C., Colombia.
Teléfono: (1) 2948700 Ext. 3919
GRACIAS
GRACIAS
ASS-ESA-DI129 V0 04/08/2016





























