Embed Size (px)
DESCRIPTION
ppok patogenesis
Citation preview

SARI PUSTAKA
PATOGENESIS DAN PATOFISIOLOGI
PENYAKIT PARU OBSTRUKTIF KRONIK (PPOK)
Pembimbing
dr. Khalif Munir
disusun oleh
Ardytia Lesmana 080100049
William Wiryawan 080100059
Muliadi Limanjaya 080100083
T. Amira Raihan Nst 080100127
Sofie Zalitha Hsb 080100376
Citra Aryanti 080100050
Marianto 080100112
Gembira Ira Hutahaean 080100163
Yunita Manurung 080100255
Novita Y Pangaribuan 080100371
DEPARTEMEN PULMONOLOGI & ILMU KEDOKTERAN RESPIRASI
FAKULTAS KEDOKTERAN
UNIVERSITAS SUMATERA UTARA
RUMAH SAKIT UMUM PUSAT HAJI ADAM MALIK
2012

ii
KATA PENGANTAR
Puji dan syukur kami panjatkan ke hadirat Tuhan yang Maha Esa atas
berkat dan hidayah-Nya sehingga sari pustaka ini dapat kami selesaikan tepat
pada waktunya.
Pada sari pustaka ini kami menyajikan judul mengenai patogenesis dan
patofisiologi PPOK. Adapun tujuan penulisan sari pustaka ini adalah untuk
memenuhi tugas kepaniteraan klinik Departemen Pulmonologi dan Ilmu
Kedokteran Respirasi, Rumah Sakit Umum Haji Adam Malik Medan.
Pada kesempatan ini, kami ingin menyampaikan pula terima kasih yang
sebesar-besarnya kepada dr. Khalif Munir atas kesediaan beliau sebagai
pembimbing kami dalam penulisan sari pustaka ini. Besar harapan kami, melalui
tulisan ini, pengetahuan dan pemahaman kita mengenai penyakit paru obstruktif
kronik semakin bertambah.
Penulis menyadari bahwa penulisan sari pustaka ini masih belum
sempurna, baik dari segi materi maupun tata cara penulisannya. Oleh karena itu,
dengan segala kerendahan hati, penulis mengharapkan kritik dan saran yang
membangun demi perbaikan tulisan ini. Atas bantuan dan segala dukungan dari
berbagai pihak baik secara moral maupun spiritual, penulis ucapkan terima kasih.
Semoga sari pustaka ini dapat memberikan sumbangan bagi perkembangan ilmu
pengetahuan khususnya kesehatan.
Medan, 6 Desember 2012
Penulis

iii
DAFTAR ISI
HALAMAN JUDUL..................................................................................... iKATA PENGANTAR.................................................................................. iiDAFTAR ISI................................................................................................. iii
BAB I PENDAHULUAN........................................................................ 11.1. Latar Belakang................................................................... 11.2. Tujuan................................................................................ 21.3. Manfaat.............................................................................. 2
BAB II TINJAUAN PUSTAKA............................................................... 32.1 Definisi dan Patogenesis PPOK......................................... 32.1.1. Faktor Genetik dalam Patogenesis PPOK.......................... 32.1.2. Reaksi Inflamasi dalam Patogenesis PPOK....................... 42.1.3. Stres Oksidatif pada PPOK................................................ 82.2. Patofisiologi PPOK............................................................ 9
BAB III KESIMPULAN............................................................................ 13
DAFTAR PUSTAKA

1
BAB I
PENDAHULUAN
1.1. Latar Belakang
Penyakit Paru Obstruktif Kronik (PPOK) adalah penyakit paru kronik
yang ditandai oleh hambatan aliran udara di saluran napas yang bersifat progresif
non-reversibel atau reversibel parsial. PPOK merupakan salah satu penyumbang
kesakitan dan kematian di dunia yang cukup tinggi. Penyakit ini berhubungan
dengan respons inflamasi paru abnormal dan progresif terhadap gas atau partikel
yang berbahaya.1,2 Secara epidemiologi, PPOK merupakan penyebab kematian
keempat tertinggi di dunia dan diperkirakan akan menjadi penyebab kematian ke-
3 di dunia pada tahun 2020 dan juga sebagai peringkat empat penyakit penting
yang menimbulkan kecacatan.3,4 Pada tahun 2004 diestimasi terdapat 64 juta
penderita PPOK di seluruh dunia, dan lebih dari 3 jutanya meninggal pada tahun
2005, setara dengan 5% dari total kematian global di tahun tersebut. Hampir 90%
dari seluruh kematian karena PPOK terjadi di negara miskin dan berkembang.5
Di Indonesia sendiri, tidak ada data yang akurat tentang kekerapan PPOK.
Hasil survei penyakit tidak menular oleh Dirjen PPM & PL di 5 rumah sakit
propinsi di Indonesia (Jawa Barat, Jawa Tengah, Jawa Timur, Lampung, dan
Sumatera Selatan) pada tahun 2004, menunjukkan PPOK menempati urutan
pertama penyumbang angka kesakitan (35%), diikuti asma bronkial (33%), kanker
paru (30%) dan lainnya (2%).6
Berbeda dengan asma, penyakit PPOK menyebabkan obstruksi saluran
pernapasan yang non-reversibel. Pada penderita PPOK terdapat gangguan
mekanis dan pertukaran gas di sistem pernapasan dan mengakibatkan menurunnya
aktivitas fisik pada kehidupan sehari-hari. Obstruksi saluran napas yang kronis
mengakibatkan volume udara keluar dan masuk tidak seimbang sehingga terjadi
air trapping. Kondisi obstruksi saluran pernapasan yang terus menerus ini akan
menyebabkan diafragma mendatar, gangguan kontraksi saluran pernapasan
sehingga fungsinya sebagai otot utama pernapasan berkurang. Sebagai

2
kompensasinya, terjadi pemakaian terus menerus otot-otot interkostal dan otot
inspirasi tambahan sehingga menimbulkan gejala sesak napas pada pasien PPOK.7
Penyakit paru obstruktif kronik merupakan penyakit sistemik yang
mempunyai hubungan antara keterlibatan metabolik, otot rangka dan genetika
molekuler. Keterbatasan aktivitas merupakan keluhan utama penderita PPOK
yang sangat mempengaruhi kualitas hidup. Inflamasi sistemik, penurunan berat
badan, peningkatan risiko penyakit kardiovaskular, osteoporosis, dan depresi
merupakan manifestasi sistemik PPOK yang sering dijumpai.8 Oleh karena itu,
perlu perhatian khusus dalam dasar perjalanan penyakit PPOK sehingga dapat
disusun strategi baru yang baik dan komprehensif serta dapat memberikan kondisi
dan prognosis yang lebih baik.
1.2. Tujuan Penulisan
Adapun tujuan penulisan sari pustaka ini adalah memahami teori
mengenai patogenesis dan patofisiologi PPOK dan untuk memenuhi persyaratan
dalam mengikuti kegiatan Kepaniteraan Klinik Senior (KKS) Rumah Sakit Umum
Pusat Haji Adam Malik, Departemen Pulmonologi dan Kedokteran Respirasi,
Fakultas Kedokteran Universitas Sumatera Utara.
1.3. Manfaat Penulisan
a. Memperkukuh landasan teori ilmu kedokteran di bidang pulmonologi,
khususnya penyakit paru obstruktif kronik.
b. Sebagai bahan informasi bagi pembaca yang ingin mendalami topik-topik
lebih lanjut yang berkaitan dengan penyakit paru obstruktif kronik.

3
BAB II
TINJAUAN PUSTAKA
2.1. Definisi dan Patogenesis PPOK
Penyakit paru obstruktif kronik (PPOK) menurut Global Initiative for
Chronic Obstructive Lung Disease (GOLD) adalah penyakit kronik yang ditandai
oleh hambatan aliran udara yang tidak sepenuhnya reversibel. Keterbatasan aliran
udara ini berhubungan dengan respons inflamasi paru abnormal dan progresif
terhadap gas atau partikel yang berbahaya.1,2 Hambatan aliran udara ini akibat
respons inflamasi abnormal terhadap partikel gas yang berbahaya. Faktor risiko
PPOK yaitu kebiasaan merokok, polusi udara, hipereaktivitas bronkus, riwayat
infeksi saluran napas bawah berulang, defisiensi alfa-1 antitripsin, dan nutrisi
yang buruk. Merokok merupakan penyebab PPOK terbanyak (95% kasus) di
negara berkembang.1
2.1.1. Faktor Genetik dalam Patogenesis PPOK
Penyakit Paru Obstruktif Kronis (PPOK) dapat disebabkan oleh asap
rokok maupun gas noksius lainnya ditambah dengan adanya faktor risiko genetik.
Selain dugaan hubungan genetik pada gen alfa antitripsin, pada penelitian
diidentifikasi adanya beberapa lokus kromosom yang spesifik dalam timbulnya
PPOK, diantaranya: kromosom 12p: MGST1, MGP, kromosom 19p: LTB4
omega hidrolase (CYP4F2), tropelastin, kromosom 22q: heme oksigenase 1,
TIMP3, kromosom 2q: IL-8α.9 Faktor-faktor pemicu ini akan merangsang
patogenesis PPOK melalui rentetan proses inflamasi yang saling tumpang tindih
dan membentuk lingkaran setan. Pada berbagai penelitian ditemukan bahwa
pelepasan sitokin pasien PPOK lebih tinggi dibanding perokok asimptomatik.
Sitokin sputum pasien dengan eksaserbasi lebih dari 3 kali dibanding yang
memiliki eksaserbasi lebih dari 2 kali10 karena pada PPOK eksaserbasi, toksin
bakteri memperburuk reaksi inflamasi yang ada dan bila disebabkan oleh virus,
virus langsung mendestruksi sel epitel saluran nafas.11

4
2.1.2. Reaksi Inflamasi pada PPOK
Pada awal PPOK, dihipotesiskan bahwa asap rokok menyebabkan stres
oksidatif berulang yang akan merusak DNA sel epitel saluran nafas sehingga
terjadi mutasi somatik,12-14 dan mengganggu kemampuan gen perbaikan DNA.12,13
Sel epitel yang telah bermutasi tersebut dikenali sebagai antigen asing sehingga
terjadi rentetan reaksi inflamasi dengan tujuan menghancurkan antigen asing
tersebut. Fragmen-fragmen dari sisa reaksi inflamasi nantinya dapat memicu
reaksi inflamasi sehingga terjadi seperti suatu lingkaran setan. Tahap pertama
yang terjadi adalah rekruitmen sel dendritik ke sel epitel tersebut yang dimediasi
kemokin 7.15 Sel dendritik menjadi inisiator reaksi inflamasi selanjutnya melalui
kemotaksis sel NK, makrofag, neutrofil, dan limfosit. Hubungan ini bersifat
timbal balik di mana para leukosit dan sel inflamasi saling memicu rekruitmen
satu sama lainnya. Asap rokok sendiri juga dapat menjadi sebab primer
kemotaksis sel-sel inflamasi walaupun dihipotesiskan bahwa sel dendritik menjadi
inisiator dari proses inflamasi yang terjadi. Sel inflamasi kedua yang berperan
awal dalam proses inflamasi PPOK adalah makrofag terutama makrofag
subepitelial CD68+. Sel makrofag juga akan melakukan fagosit pada bahan yang
terinhalasi, apabila memungkinkan, akan makrofag akan mendestruksi bahan ini.16
Pada penelitian Safwat et al., ditemukan bahwa konsentrasi makrofag meningkat
5-10 kali pada bilasan bronkus dan sputum pasien PPOK.17-19 Makrofag yang
ditemukan pada bilasan paru juga tampaknya memiliki ukuran yang lebih kecil
dan bersifat imatur dari makrofag paru normal. Ini disebabkan karena pelepasan
prekursor monosit yang prematur. Makrofag mempunyai jangka hidup yang lebih
lama pada perokok, bahkan bisa bertahan sampai lebih dari 2 tahun. Hal ini
kemungkinan disebabkan klirens mukosiler yang kurang efektif pada perokok.
Klirens limfatik juga menurun disebabkan karena kerusakan jaringan dan/atau
kehilangan struktur limfatik paru, atau ketidakmampuan sistem limfatik paru yang
untuk mengimbangi influks pulmonal.16 Makrofag akan melepaskan berbagai
mediator-mediator inflamasi yang akan merangsang kemotaksis sel-sel inflamasi
lainnya. Leukotrien B4, IL-1, IL-8, dan GRO-α dapat menduduki reseptor
CXCR1 dan CXCR2 untuk rekrutimen neutrofil. MCP-1, GRO-α, ENA-78

5
merangsang monosit melalui reseptor CXCR2. IP-10, Mig, I-TAC memicu sel
CD8+ melalui reseptor CXCR3.20 Makrofag juga berperan dalam proses
elastolisis melalui sekresi MMP-1, MMP-9, MMP-12, dan katepsin K, L, S
dengan target destruksi elastin pada dinding alveolus sehingga rekoil dari alveolus
akan berkurang, alveolus kolaps, udara saat ekpirasi terperangkap, dan dinding
dada dapat menjadi hiperinflasi.21-23 Dalam penelitian, ditemukan juga penekanan
TIMP yang mendukung proses elastolisis yang terjadi. Makrofag ditemukan juga
dapat mesekresikan TGF-α dan TGF-β yang melalui EGFR, CTGF akan memicu
Smad3 dalam ekspresi fibroblas, prokolagen, dan antiproteinase yang akan
berperan dalam proses fibrosis dalam remodelling saluran nafas. EGFR juga dapat
merangsang MAP kinase sehingga terjadi peningkatan MUC5AC, MUCB,
MUC5B yang akan memicu hiperplasia sel goblet dan kelenjar mukus.24,25
Neutrofil dan limfosit CD8+ menjadi suatu sel inflamasi yang berperan penting
dalam patogenesis PPOK. Neutrofil melepaskan kemoatraktan seperti TNF-α, IL-
1, IL-2, IL-3, IL-8, LTB4 untuk perbanyak rekruitmen neutrofil itu sendiri, dan
sel-sel inflamasi lainnya. Proses inflamasi paling utama yang ditimbulkan
neutrofil dalam hubungannya dengan patogenesis PPOK yaitu sekresi mediator
proteolisis seperti MMP-1 (proteinase), MMP-3 (proteinase), MMP-8
(kolagenase), MMP-9 (elastase), MMP-12 (elastase), neutrofil elastase, katepsin
G, serin protease, dan sistein proteinase.9 Struktur protein dan matriks
ekstraselular penyusun saluran nafas dan alveolus akan terdestruksi lebih cepat
dalam hitungan jam-hari dibanding proses pembentukannya dalam hitungan hari-
bulan.26,27 Hipersekresi mukus oleh sel goblet dan kelenjar mukus akan terangsang
secara berlebihan sebagai suatu proses kompensasi yang bahkan akan
memperburuk patogenesis penyakit. Selain itu, neutrofil ditemukan meningkatkan
molekul adhesi seperti ICAM-1 dan E-selektin sehingga meningkatkan perlekatan
sel inflamasi pada epitel saluran nafas. Terlebih lagi, proses remodeling berupa
fibrosis di mana saluran nafas kehilangan fungsi asli dan elastisitasnya,
patogenesis PPOK akan terus berlanjut dan menjadi semakin buruk. Pada
penelitian, telah banyak dibuktikan penemuan limfosit CD8+ di dinding dan otot
polos saluran nafas.28 Maturasi sel dendritik yang terganggu juga akan

6
menyebabkan sel T regulatori yang mengatur rasio sel T CD4+/CD8+ terganggu.
Akibatnya, CD8+ akan lebih banyak dibanding CD4+ karena waktu yang
dibutuhkan sel dendritik untuk memicu proliferasi CD8+ lebih cepat.29 Sel
limfosit CD8+ berperan penting dalam mekanisme sekresi perforin dan granzim B
yang mengaktifkan jalur apoptosis ligan Fas-Fas30 sehingga akan terjadi apoptosis
sel epitel saluran nafas dan destruksi alveolus. Chrysofakis, et al. menunjukkan
bahwa sel T CD8+ pada pasien PPOK lebih sitotoksik dan kandungan enzim litik
lebih banyak dibanding orang normal. Limfosit CD4+ kadarnya tidak terlalu
bermakna PPOK, di mana peningkatan limfosit CD4+ berhubungan dengan asma,
dapat memicu reaksi autoimun minimal pada sel epitel saluran nafas melalui
carbonyl modified antibody.27,31,32
Gambar 1. Inflamasi pada PPOK31
Sel epitel saluran napas penting sebagai pertahanan dan produksi mukus
oleh sel goblet akan melindungi saluran napas terhadap bakteri dan partikel
terinhalasi.33 Sel epitel mengeluarkan defensins dan peptida kation dengan efek
anti bakteri yang merupakan sistem pertahanan alami yang berperan dalam proses
perbaikan jaringan.34 Sel epitel saluran nafas sendiri juga dapat memicu
rekruitmen mediator-mediator inflamasi sebagai suatu mekanisme pertahanan

7
yang salah. Mediator yang dapat dirangsang adalah TNF-α, TGF-β, IL-1, IL6, IL-
8, IL-9, IL-11, IL-13, MCP-1, VEGFR, FGF-1, FGF2 terutama di epitel bronkial,
bronkiolar, dan alveolar dan makrofag bronkiolar, otot polos saluran nafas, dan
otot polos vaskular sebagai respon pertahanan terhadap kerusakan epitel dan
endotel.11,35 TNF-α akan mengaktivasi NF-KB yang menjadi dalang dari
rekruitmen, transkripsi dan aktivasi hampir seluruh sitokin dan kemokin. TNF-α
juga akan meningkatkan ICAM-1, aktivasi makrofag untuk memproduksi MMP,
aktivasi sel epitel bronkus untuk memproduksi tenaskin. IL-1 merangsang
proliferasi fibroblas, meningkatkan sekresi prostaglandin dan kolagenase,
meningkatkan sekresi fibronektin dan kolagen, ICAM-1, dan IL tipe lainnya. IL-6,
IL-8, dan IL-9 menyebabkan metaplasia mukus dan fibrosis subepitelial. IL-11
akan memicu proliferasi miofibroblas menyebabkan hiperplasia otot polos saluran
nafas, memicu sekresi PDGF, dan TGF-β. IL-13 menyebabkan inflamasi dan
hiperplasia sel goblet. VEGFR memicu angiogenesis dan mitogenesis sel endotel
vaskular.28 GM-CSF juga akan disekresikan oleh sel epitel untuk merekrut sel
inflamasi lainnya.32
Gambar 2. Remodeling Saluran Napas Akibat Rokok/Polutan32
2.1.3. Stres Oksidatif pada PPOK

8
Pada keadaan normal terdapat keseimbangan antara oksidan dan
antioksidan. Asap rokok yang mengandung berbagai radikal bebas seperti O2-,
H2O2, OH-, ONOO-. Pada pasien PPOK dengan risiko faktor genetik, ditemukan
adanya ketidakseimbangan antara oksidan dan antioksidan sehingga radikal bebas
pada asap rokok tidak dapat ternetralisir. Enzim NADPH yang ada dipermukaan
makrofag dan neutrofil akan mentransfer satu elektron ke molekul oksigen
menjadi anion superoksida dengan bantuan enzim superoksid dismutase. Zat
hidrogen peroksida (H2O2) yang toksik akan diubah menjadi OH dengan
menerima elektron dari ion feri menjadi ion fero, ion fero dengan halida akan
diubah menjadi anion hipohalida (HOCl). Hal ini akan menyebabkan stres
oksidatif sehingga terjadi aktivasi p38MAP kinase yang memicu NF-KB,
meningkatkan rekruitmen neutrofil karena perangsangan GM-CSF di sum-sum
tulang, merangsang sekresi mukus, memicu proteolisis melalui supresi anti
protease, meningkatkan molekul adhesi, meningkatkan proses remodeling fibrosis
melalui TGF-β, inaktivasi fungsi α1-antritripsin, dan menyebabkan
bronkokonstriksi serta kebocoran plasma melalui mediator isoprostan.11,25
Penelitian Barrerio et al. menunjukkan bahwa adanya mekanisme peroksidasi
lipid, oksidasi protein dan tiol, serta oksidasi DNA pada pasien PPOK.36
Makrofag menghasilkan ROS dan nitric oxide (NO) membentuk peroxynitrite
mengakibatkan resistensi steroid. Protein inflamasi pada PPOK diatur oleh
transcription factor nuclear factor-kB (NFkB) terutama saat eksaserbasi.32

9
Gambar 3. Efek Rokok pada PPOK32
Faktor genetik juga disinyalir menyebabkan produksi terganggunya fungsi
antiprotease sehingga tidak cukup untuk menetralisir efek berbagai protease.
Baru-baru ini, terdapat hipotesis baru mengenai keterlibatan adenovirus,
ketidakseimbangan asetilasi histon dan deasetilasi histon yang menyebabkan
remodelling kromatin sehingga memicu ekspresi gen-gen inflamasi.11 Walaupun
begitu, tetaplah proses inflamasi yang menjadi dasar dari penyakit PPOK
sehingga harus ditatalaksana dengan baik dan komprehensif.
2.2. Patofisiologi PPOK
. Emfisema, penyebab morbiditas dan mortalitas utama penderita PPOK,
ditandai dengan hilangnya struktur alveoli karena inflamasi berlebihan dan
destruksi jaringan sehingga mengakibatkan pembesaran parenkim paru yang
terlihat dari berkurangnya daya elastisitas paru.37-39 Keterbatasan arus ekspirasi
yang merupakan tanda khas dari PPOK dan terjadi karena emfisema dan disfungsi
saluran pernapasan (fibrosis, edema mukosa, dan penyumbatan mukus).
Emfisema dapat menyebabkan menurunnya tekanan elastis paru sehingga akan

10
mengurangi arus tekanan ekspirasi udara yang melalui saluran pernapasan yang
sempit dimana terjadi peningkatan resistensi.40
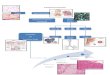
Gambar 3. Proses patologi pada PPOK dan perubahan yang terjadi41
Banyak definisi PPOK menekankan pada emfisema dan bronkitis kronik.
Hal ini tidak berlaku lagi. Emfisema, atau destruksi permukaan tempat
penggantian gas pada paru (alveoli), merupakan istilah patologis yang sering
(tetapi kurang tepat) digunakan secara klinis dan menggambarkan hanya salah
satu dari beberapa kelainan struktural yang terjadi pada PPOK.1
Pada individu normal, volume relaksasi sistem respirasi merupakan
keseimbangan antara daya antara tekanan elastisitas paru dari dalam dan tekanan
keluar dari dinding dada. Pada PPOK, terjadi peningkatan komplians paru yang
disebabkan emfisema sehingga terjadi peningkatan volume relaksasi daripada
individu normal.40 Selain itu, terjadi overekspansi dari dinding toraks yang
memendeknya otot inspirasi sehingga terjaid penurunan kapasitas tekanan otot-
otot inspirasi.42
Sesak karena PPOK dapat disebabkan juga karena menyempitnya dan
meningkatnya resistensi saluran pernapasan. Hal ini akan menyebabkan impedansi
dari ventilasi yang meningkat yang akan diimbangi peningkatan usaha bernapas
dengan peningkatan output motorik pada pusat pernapasan.42
Bronkitis kronik, yang ditandai dengan batuk dan produksi sputum selama
3 bulan atau lebih, terjadi pada 50% perokok.43 Bronkitis kronik terjadi karena

11
hipersekresi mukus (seperti yang dijelaskan sebelumnya) yang disebabkan
hipertrofi/hiperplasia kelenjar mukosa bronkus dan hiperplasia/metaplasia sel
goblet, rusaknya epitel dan metaplasia mukosa, remodeling mikrovaskular
bronkus, hipertrofi/hiperplasia otot polos dan fibrosis ECM.44,45,46 Sekresi mukus
merupakan faktor risiko potensial menyebabkan turunnya fungsi paru yang
terlihat dengan penurunan volume ekspirasi paksa detik pertama (VEP1). Batuk
pada PPOK sendiri terjadi karena adanya agen-agen tusif seperti prostaglandin43
atau substansi lain yang secara tidak langsung mengaktivasi refleks batuk dan
sekresi mukus seperti takikinin.47 Selain itu, pengeluaran sitokin-sitokin akan
memicu inflamasi saluran napas, hipersekresi mukus, dan disfungsi silia yang
akan merangsang refleks batuk. Hal ini didukung dari penelitian Smith et al
(2006) yang menyatakan bahwa >80% penderita PPOK akan mengalami
penurunan prevalensi batuk setelah 5 tahun berhenti merokok.43
Fibrosis pada bronkus penderita PPOK diawali dengan perubahan
fibroblas. Fibroblas merupakan tipe sel yang sebagian besar mengatur produksi
ECM dan pengaturan turnover dari ECM dengan menstimulasi pemecahan
protein.49 Fibroblas dipengaruhi oleh molekul-molekul yang berbeda seperti
sitokin dan faktor pertumbuhan. TGF-β merupakan sitokin utama yang
menstimulasi fibroblas untuk memproduksi ECM dengan menggunakan jalur
Smad, sedangkan TNF-α dan IFN-γ merupakan inhibitor fibroblas. Pada keadaan
patologis seperti PPOK, terjadi perubahan turnover ECM. Terjadi proses
patologis berupa proteolisis parenkim paru, disertai dengan deposisi ECM
berlebihan (fibrosis) pada bronkus dan bronkiolus. Fibroblas diketahui
mempunyai berbagai fenotip seperti fibroblas non kontraktil dan miofibroblas
kontraktil (fibroblas aktif). Miofibroblas merupakan sel yang berperan dalam
remodeling saluran napas karena kemampuannya untuk menghasilkan dan
menimbun komponen ECM baru (terutama kolagen), dan mengatur turnover
ECM dengan mensekresikan matriks metalloproteinase (MMP).50 Miofibroblas
diperkirakan merupakan perubahan dari fibroblas dan sel ASM pada keadaan
profibrosis.51 Hipotesa lain mengatakan bahwa miofibroblas berasal dari epitel
yang mengalami transdiferensiasi menjadi fibroblas yang selanjutnya menjadi

12
miofibroblas. Hal ini terjadi apabila sel epitel terpapar dengan TGF-β1 dengan
upregulasi TNF-α.52
Hipersekresi mukus kronik pada PPOK berat berhubungan dengan
mortalitas dan menggambarkan peningkatan risiko infeksi lanjut. Penelitian
histopatologis PPOK menunjukkan keterlibatan saluran napas perifer (bronkiolus)
dan parenkim paru yaitu obstruksi dan fibrosis pada bronkiolus. Penyempitan
saluran napas kecil yang ireversibel, emfisema dan obstruksi lumen dengan
sekresi mukus dapat menyebabkan hambatan aliran udara pada PPOK. Penebalan
saluran napas kecil dengan peningkatan pembentukan folikel limfoid dan
penimbunan kolagen di bagian luar saluran napas menghambat pembukaan
saluran napas. Lumen saluran napas kecil berkurang karena penebalan mukosa
berisi eksudat sel radang yang meningkat sejalan dengan beratnya penyakit.
Hambatan aliran udara pada PPOK disebabkan oleh beberapa derajat penebalan
dan hipertrofi otot polos pada bronkiolus respiratorius.53

13
BAB III
KESIMPULAN
Penyakit Paru Obstruktif Kronik (PPOK) adalah penyakit paru kronik
yang ditandai oleh hambatan aliran udara di saluran napas yang bersifat progresif
non-reversibel atau reversibel parsial. Perjalanan penyakit PPOK disebabkan oleh
genetik dan asap rokok atau polutan yang menyebabkan perubahan epitel dan
aktifnya sel dendritik yang ikuti dengan adanya reaksi amplifikasi inflamasi oleh
neutrofil, makrofag, dan epitel saluran napas. Reaksi inflamasi yang terjadi
melibatkan berbagai jenis sitokin dan proteinase. Selain reaksi inflamasi yang
terjadi, terdapat peran stres oksidatif dari zat yang terinhalasi dan rokok yang
menyebabkan apoptosis dan inflamasi yang lebih lanjut. Keseluruhan hal ini akan
menyebabkan remodeling saluran napas dan hipersekresi mukus akibat
hipertrofi/hiperplasia kelenjar mukosa bronkus, dan berkurangnya elastisitas
alveoli secara permanen dengan gejala yang tampak pada penderita PPOK berupa
sesak napas yang persisten dan batuk (non) produktif kronik.

14
DAFTAR PUSTAKA
1. Global Initiative for Chronic Obstructive Lung Disease. Pathogenesis,
pathology and pathophysiology. In: Global strategy for diagnosis,
management and prevention of chronic obstructive lung disease. NHLBI
Publication; Updated 2011.p.27-39.
2. Tim Pokja PPOK. Definisi. Dalam: PPOK pedoman praktis diagnosis dan
penatalaksanaan di Indonesia. Jakarta: PDPI; 2004.p.1-2.
3. American Thoracic Society dan European Respiratory Society, 2004.
Standards for the Diagnosis and Management of Patients with COPD.
Available from http://www.copd-ats-ers.org/copddoc.pdf [Accessed 3
December 2012]
4. Lopez AD, Murray CC. The global burden of disease 1990-2020. Nat Med
1998; 4: 1241-3.
5. World Heart Organization, 2011. Chronic obstructive pulmonary disease
(COPD): Fact sheet. Available from
http://www.who.int/mediacentre/factsheets/fs315/en/ [Accessed 3
December 2012].
6. Depkes RI, 2008. Keputusan Menteri Kesehatan, Pedoman Pengendalian
Penyakit Paru Obstruktif Kronik. Available from
http://www.depkes.go.id/downloads/Kepmenkes/pengendalian_ppok.pdf
[Accessed 2 December 2012].
7. Agustin, H., dan Yunus, F., 2008. Proses Metabolisme Pada Penyakit Paru
Obstruktif Kronik (PPOK). Dalam: Jurnal Respirologi Indonesia. 2008.
Jakarta: Perhimpunan Dokter Paru Indonesia, 155-161.
8. Agusti AGN, Noguera A, Sauleda J, Sala E, Pons J, Busquets X. Systemic
effect of chronic obstructive pulmonary disease. Eur Respir J 2003;
21:347-60.
9. Shapiro S. 2003. The pathophysiology of copd: what goes wrong and why.
Adv Stud Med 3(2B):S91-S98.

15
10. Wedzicha JA, Seemungal TA, MacCallum PK, et al. Acute exacerbations
of chronic obstructive pulmonary disease are accompanied by elevations
of plasma fibrinogen and serum IL-6 levels [In Process Citation]. Thromb
Haemost 2000; 84: 210–215)
11. MacNee W. Pathogenesis of Chronic Obstructive Pulmonary Disease.
Proc Am Thorac Soc 2005; 2: 258-266
12. Anderson GP, Bozinovski S. Acquired somatic mutations in the molecular
pathogenesis of COPD. Trends Pharmacol Sci 2003; 24: 71–76
13. Chang CL, Marra G, Chauhan DP, et al. Oxidative stress inactivates the
human DNA mismatch repair system. Am J Physiol Cell Physiol 2002;
283: C148–C154
14. Wistuba II, Lam S, Behrens C, et al. Molecular damage in the bronchial
epithelium of current and former smokers. J Natl Cancer Inst 1997; 89:
1366–1373.
15. Vermaelen K, Pauwels R. Pulmonary dendritic cells. Am J Respir Crit
Care Med 2005; 172: 530–551.
16. Tetley TD. 2002. Macrophages and the Pathogenesis of COPD. Chest
121:5
17. Safwat T, Saeed A, Dina AF,2 Weaam AM1. The Phagocytic Activity of
Peripheral Blood Macrophages in COPD Patients. Egypt J Bronchol 2008;
2(2): 243-252
18. Vestbo J, Lange P. GOLD provide information of prognostic value in
chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;
166:329-32
19. Pesci A, Balbi B, Majori M, et al. Inflammatory cells and mediators in
bronchial lavage of patients with chronic obstructive pulmonary disease.
Eur Respir J 1998; 12:380-6
20. Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, Chung KF. Balance of
matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from
alveolar macrophages in cigarette smokers: regulation by interleukin-10.
Am J Respir Crit Care Med 2000; 162: 1355–1360.

16
21. Viegi G, Pistelli F, Sherrill DL et al Definition, epidemiology and natural
history of COPD. Series ‘‘Comprehensive Management of End-Stage
COPD’’ Edited by N. Ambrosino and R. Goldstein Number 1 in this
Series. Eur Respir J 2007;30:993- 1013.
22. Wan ES, Silverman EK. Genetics of COPD and Emphysema. Chest
2009;136:859-866.
23. MacNee. Oxidants/Antioxidants and COPD. Chest 2000;117:303S–317S.
24. Bonniaud, P., Kolb, M., Galt, T., et al. Smad3 null mice develop airspace
enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J.
Immunol. 2004; 173: 2099–2108.
25. Barnes PJ. Mediators of Chronic Obstructive Pulmonary Disease,
Pharmacol Rev 2004;56: 515-548.
26. Jeffery PK. Structural and inflammatory changes in COPD: a comparison
with asthma. Thorax 1998;53:129–136.
27. Brashier BB, Kodgule R. Risk Factors and Pathophysiology of Chronic
Obstructive Pulmonary Disease (COPD). JAPI 2012; 60: 17-21.
28. Boer WI, Alaagappan VKT, Sharma HS. Molecular Mechanisms in
Chronic Obstructive Pulmonary Disease Potential Targets for Therapy.
Cell Biochemistry and Biophysics 2007; 47:131-148
29. van Stipdonk MJ, Hardenberg G, Bijker MS, et al. Dynamic programming
of CD8+ T lymphocyte responses. Nat Immunol 2003; 4: 361–365.
30. Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in
the lungs of smokers and their relation to emphysema. Eur Respir J
2001;17:946–953.
31. Stebbins KJ, Evans JF and Lorrain DS. DP2 Receptor Antagonists: Novel
Therapeutic Target for COPD. Mol Cell Pharmacol 2010;2(3):89-96.
32. Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, Fan CK.Balance of
matrix metalloprotease-9 and tissue inhibitor ofmetalloprotease-1 from
alveolar macrophages in cigarette smokers. regulation by interleukin-10.
Am J Respir Crit CareMed 2000; 162: 1355-60.

17
33. Vermeeren MA, Creutzberg EC, Schols AM, Postma DS, Pieters WR,
Roldaan AC, Wouters EF. Prevalence of nutritional depletion in a large
out-patient population of patients with COPD. RespirMed 2006; 10:23-5.
34. Roisin R, MacNee W. Pathophysiology of chronic obstructive pulmonary
disease. Eur Respir Mono 1998; 3: 107-26.
35. Fooladi AAI, Yazdani S, Nourani MR. Lung and Systemic Inflammation
in COPD. Intl Journal COPD 2007:2(4); 452-462
36. Barreiro E, Peinado VI, Galdiz JB, Ferrer E, Marin-Corral J, Sa´nchez F,
et al. Cigarette Smoke–induced Oxidative Stress A Role in Chronic
Obstructive Pulmonary Disease Skeletal Muscle Dysfunction. Am J Respir
Crit Care Med 2010; 182(4): 477-488
37. Falk JA, Martin UJ, Scharf S, Criner GJ. Lung elastic recoil does not
correlate with pulmonary hemodynamics in severe emphysema. Chest
2007; 132:1476–1484
38. Murarescu ED, Eloae-Zugun F, Mihailovici MS. Experimental COPD
induced by solid combustible burn smoke in rats: a study of the
emphysematous changes of the pulmonary parenchyma. Rom J Morphol
Embryol 2008; 49:495–505
39. Inoue K, Koike E, Yanagisawa R, Takano H. Extensive analysis of
elastase-induced pulmonary emphysema in rats: ALP in the lung, a new
biomarker for for disease progression? J Clin Biochem Nutr 2010; 46:168–
176
40. O’Donnel DE, Banzett RB, Carrieri-Kohlman V, Casaburi R, Davenport
PW, Gandevia S, et al. Pathophysiology of Dsypnea in Chronic
Obstructive Pulmonary Disease. Proc Am Thorac Soc 2007; 4(2):145-
168.)
41. O’Donnel DE, Banzett RB, Carrieri-Kohlman V, Casaburi R, Davenport
PW, Gandevia S, et al. Pathophysiology of Dsypnea in Chronic
Obstructive Pulmonary Disease. Proc Am Thorac Soc 2007; 4(2):145-168.

18
42. American Thoracic Society. Dyspnea: Mechanisms, Assessment, and
Management: A Consensus Statement. Am J Respir Crit Care Med 1999;
159: 321-340.
43. Smith J, Woodcock A. Cough and Its Importance in COPD. Int J Chron
Obstruct Pulmon Dis 2006; 1(3): 305-314.
44. Saetta M, Turato G, Baraldo S, Zanin A, Braccioni F, Mapp CE, et al.
Goblet cell hyperplasia and epithelial inflammation in peripheral airways
of smokers with both symptoms of chronic bronchitis and chronic airflow
limitation. Am J Respir Crit Care Med 2000; 161: 1016–1021.
45. Atzori L, Lucattelli M, Scotton CJ, Laurent GJ, Bartalesi B, De Cunto G,
et al. Absence of proteinase-activated receptor-1 signaling in mice confers
protection from fMLP-induced goblet cell metaplasia. Am J Respir Cell
Mol Biol 2009; 41:680–687
46. Zanini A, Chetta A, Saetta M, Baraldo S, Castagnetti C, Nicolini G, et al.
Bronchial vascular remodelling in patients with COPD and its relationship
with inhaled steroid treatment. Thorax 2009; 64:1019–1024.
47. Joos GF, De Swert KO, Schelfhout V, Pauwels RA. The Role of Neural
Inflammation in Asthma and Chronic Pulmonary Disease. Ann N Y Acad
Sci 2003; 992: 218-230.
48. Dekkers BG, Maarsingh H, Meurs H, Gosens R. Airwaystructural
components drive airway smooth muscle remodeling in asthma. Proc Am
Thorac Soc 2009; 6:683–692.
49. Parameswaran K, Willems-Widyastuti A, Alagappan VK, Radford K,
Kranenburg AR, Sharma HS. Role of extracellular matrix and its
regulators in human airway smooth muscle biology. Cell Biochem
Biophys 2009; 44:139–146
50. Westergren-Thorsson G, Larsen K, Nihlberg K, Andersson- Sjoland A,
Hallgren O, Marko-Varga G, et al. Pathological airway remodelling in
inflammation. Clin Respir J 2010; 4(Suppl 1):1–8.
51. Salasar LM, Herrera AM. 2011. Fibrotic Response of Tissue Remodelling
in COPD. Lung 189: 101-109.

19
52. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence
that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest
2002; 110:341–350.
53. Lamb D, McLean A, Gillooly M, Warren PM, Gould GA, MacNee W.
Relation between distal airspace size, bronchiolar attachments, and lung
function. Thorax 1993; 48:1012-7.