Embed Size (px)
Citation preview

___________________________________________________________________________DEA Environnement 2001 - 1 -
Phylogénie
Une première lecture de livres ou publications sur la phylogénie laisse une impression de
discipline confuse. Une relecture accentue cette impression et l'on peut dire sans se tromper
qu'ils existent au moins autant de méthodes d'approches que de phylogénéticiens. On peut
quand-même tenter une classification en mettent en exergue leurs ressemblances et leurs
différences.
La phylogénie peut se définir comme "le cours historique de la descendance des êtres
organisés" s'appuyant sur le concept de base de la descendance avec modification.
Le support formel de la représentation, admise par tous, est l'arbre phylogénétique.
- Les feuilles y représentent les "espèces" étudiées ou les taxons
- les noeuds internes, les ancêtres virtuels ayant divergé
- les arêtes, les liens de filiation. Les arêtes sont valuées.
En général, les arbres sont ternaires (noeud interne a trois arêtes) et peuvent être enracinés;
dans ce cas l'arbre est "orienté", il possède une origine qui est un noeud unaire appelé racine
ou ancêtre.
Un dendrogramme est un arbre additif enraciné, dont tous les chemins de la racine à chacune
des feuilles ont la même longueur.
DendrogrammeA B C
A
C
D
B
E
Arbre sans racine
noeud
arête
feuille arbre : graphe connexe sans cycle
X-arbre : on ajoute aux sommets réels (lestaxons) des sommets latents qui sont desancêtres virtuels (intermédiare virtuel)
arbre additif : arbre phylogénétique dont lesarêtes sont valuées
Cette représentation est le support formel de la "ressemblance" ou de la différence entre les
objets d'étude (espèces, organismes) de la phylogénie. Là est le problème crucial et là se
situent les différences entre phylogénéticiens. De l'anatomie comparée à l'étude des caractères
biologiques moléculaires, en passant par l'ontogénie et la paléontologie, le concept de
ressemblance a été approché de manières différentes selon l'école de systématique.
Depuis les œuvres des grands évolutionnistes du XIXe siècle, Lamarck, Darwin, Haeckel
jusqu'à la définition de la méthode cladistique par l'entomologiste Willi Hennig (1950), la
construction phylogénétique obéissait au principe du "triple parallélisme" :

___________________________________________________________________________DEA Environnement 2001 - 2 -
- Le triple parallélisme
- l'anatomie comparée : l'homologie repose essentiellement sur la morphologie, les
connexions de l'organe avec d'autres organes
- l'ontogénie : embryologie comparée en particulier sur les stades les plus précoces du
développement.
- la paléontologie : informations directement issues du temps géologique qui permet
d'établir les relations chronologiques.
- pour les cladistes, il convient d'identifier les états primitif (plésiomorphe) et dérivé
(apomorphe) des caractères homologues
- pour les systématiciens phénéticiens, adepte de la taxinomie numérique, la combinaison de
taxons ne peut être fondée que sur la base de similitude globale exprimée par un indice de
similitude.
- pour les sytématiciens évolutionnistes, la similitude globale seule ne peut fournir la base de
la reconstruction phylogénétique en raison de "fausses homologies" ou homoplasies
(convergence : apparition indépendante de caractères proches et réversion : apparition d'un
caractère ayant l'apparence de la morphologie ancestrale) et l'on doit prendre en compte
uniquement la similitude de traits homologues.
Le cladisme
La sytématique phylogénétique cherche à établir des groupes monophylétiques ; c'est à dire
des groupes dont le plus récent ancêtre commun à tous les éléments ne soit l'ancêtre d'aucun
élément d'un autre groupe. Cela apparaitra comme un sous-arbre dans l'arbre phylogénétique.
En ce sens, on cherche à rejeter les groupes paraphylétiques, dont le plus récent ancêtre
commun aux éléments d'un groupe est aussi un ancêtre plus ancien d'autres groupes, et les
groupes polyphylétiques, dont le plus récent ancêtre commun aux éléments du groupe est
aussi le plus récent ancêtre commun d'éléments d'un autre groupe.
A : les deux groupes sont monophylétiquesB : le groupe en vert est paraphylétiqueC : les deux groupes sont polyphylétiques
A B C

___________________________________________________________________________DEA Environnement 2001 - 3 -
Les définitions et les concepts importants sont :
Etat des caractères
De manière générale un caractère sera significatif s'il possède au moins deux états possibles :
- état plésiomorphe (état ancestral)
- état apomorphe (état dérivé de l'état plésiomorphe par spéciation)
Notion d'homologie et d'homoplasie
Afin de structurer les classifications que sur la base de monophylies, il faut pouvoir distinguer
les similitudes héritées d'un ancêtre commun des similitudes qui ne le sont pas.
Les premières sont appelées homologies et témoignent d'une réalité évolutive commune.
Les secondes sont appelées homoplasies et témoignent d'apparitions indépendantes de
caractères similaires chez des groupes n'étant pas nécessairement apparentés. Les
homoplasies sont aujourd'hui divisées en deux types : les convergences et les réversions. Une
convergence correspond à l’apparition d'un caractère indépendamment chez deux taxons (ou
davantage). Une réversion représente une réapparition chez un taxon d'un caractère identique
à celui de la forme ancestrale.
x (état plésiomorphe) -> x' (état apomorphe)
Parcimonie
Lors de la construction des arbres, seuls seront retenus les arbres dont le nombre total de
changement d'états seront minimaux. Cela amène les remarques suivantes :
1- Dans la "théorie de l'évolution" la nature est fort économe!
A B C
x
x'
x x'x'
Homologie
A B C
x
x
x' x'x
Convergence
A B C
x
x'
x xx'
Réversion
Homoplasie

___________________________________________________________________________DEA Environnement 2001 - 4 -
2- Cela permet de trouver des méthodes de construction des arbres.
3- Cette méthode est idéale pour le cas où les homoplasies n'existent pas
Les biologistes disposent de plusieurs algorithmes ou heuristiques pour soumettre la
construction des arbres à des ordinateurs. Avant de passer à la construction phylogénétique
moléculaire, il faut disposer d'un alignement des séquences puisque chaque position sera un
caractère putatif.
Pour une séquence protéique ou d'ADN chaque site "informatif" de la séquence est considéré
comme un caractère. Un site informatif est une poisition pour laquelle on trouve au moins
deux états et que l'on retrouve chacun dans au moins 2 séquences.
La complexité algorithmique du problème (NP-complet) limite l'évaluation de toutes les
combinaisons possibles (un site informatif pour 10 séquences représentent l'évaluation de 34
459 425 arbres).
L'algorithme le plus connu et le plus utilisé pour les séquences d'ADN (nombre de séquences
inférieur à 30) est celui désigné sous le nom de "Branch and bound". Celui-ci trouve une
solution minimale sans explorer l'ensemble de toutes les combinaisons.
Les méthodes probabilistes
Elles sont appliquées à un ensemble de caractères pour chacun desquels, une probabilité de
transition entre les divers états est définie.
A partir de là, la phylogénie est représenté par l'arbre dont la vraisemblance calculée à partir
des probabilités est maximale. La difficulté réside dans l'évaluation des probabilités de
chaque événement.
Par exemple la méthode du maximum de vraissemblance analyse l'ordre des branchements et
la longueur des branches en terme de probabilité

___________________________________________________________________________DEA Environnement 2001 - 5 -
Phénétique moléculaire
Nous allons nous intéresser plus particulièrement à deux méthodes phénétiques que vous
trouvez dans le programme "clustalv", le plus utilisé par les biologistes moléculaires, et qui
permettent d'exprimer des relations de proximité à partir d'un indice de similarité basé sur un
seul caractère : la comparaison des séquences primaires pour une protéine appartenant à la
même famille (par exemple la lipase pancréatique). La définition de la ressemblance de
séquences et les problèmes posés seront décrits plus loin.
Nous supposons que nous pouvons définir et calculer une distance entre deux séquences
protéiques (distance d'édition, etc.), et donc par exemple construire le tableau suivant pour 5
séquences:
1 2 3 4 5
2 d12 0
3 d13 d23 0
4 d14 d24 d34 0
5 d15 d25 d35 d45 0
6 d16 d26 d36 d46 d56
indice d'écart : d(i,j) = d(j,i)d(i,j) ≥ 0 et d(i,i)=0
distance : d(i,j) = 0 ⇒ i = j
distance métrique : d(i,j) ≤ d(i,k) + d(k,j)
distance ultramétrique :d(i,j) ≤ max(d(i,k), d(j,k))
Nous voulons passer d'une représentation d'un graphe complet (tableau double entrée) à la
représentation par un arbre phylogénétique.
Intuitivement nous allons passer d'un espace à (n-1) dimensions (ici 4) à un espace de
dimension 2 : cela ne peut se faire que par une approximation.
Plusieurs méthodes peuvent être appliquées : probabiliste, analyse en composante principale,
classifications hiérarchiques. Les plus employées sont les dernières, basées soit sur la
meilleure approximation tableau <-> arbre, ou encore sur l'application d'une récurrence :
Meilleure approximation
- par exemple approximation des moindres carrés entre le tableau et l'arbre additif à
construire
Récurrence
- la classification hiérachique descendante, basée sur la dichotomie : l'ensemble de
séquences est partagé en deux sous-ensembles, chacun regroupant les séquences les plus
proches entre elles. A chacun des deux sous-ensembles, on applique la même règle
précédente et cela jusqu'à épuisement.
- la classification hiérachique ascendante : une paire de séquences (ou OTU) sont regroupées
suivant un critère, ce regroupement définit un nouvel objet séquence (OTU) et l'on

___________________________________________________________________________DEA Environnement 2001 - 6 -
recommence la même opération avec les (n-1) objets restants après avoir recalculé un
nouveau tableau, et ainsi de suite jusqu'à épuisement. Les méthodes vont se différencier par le
critère de regroupement et par la méthode de calcul pour passer d'un tableau de n objets à un
tableau de (n-1) objets.
Dans le programme "clustalv", vous avez deux constructions par classification hiérarchique
ascendante :
- UPGMA : à partir du tableau, on définit une ultramétrique (lien moyen : l'arbre construit
sera un dendrogramme) et la règle de regroupement concerne les deux OTU les plus proches
(valeur minimale dans le tableau). L'ultramétrique sous entend la notion d'horloge
moléculaire.
- NJ : la règle de regroupement concerne les deux OTU qui produise l'arbre minimal
Lien moyen (UPGMA)
Cette méthode rentre dans le cadre des distances ultra-métriques. Pour passer d'un tableau
de n objets, à un tableau de (n-1) objets où on a regroupé les objets 1 et 2 par exemple, un
élément du nouveau tableau est calculé ainsi :
d(i,(12)) = (n1*d(i,1) + n2*d(i,2))
n1 + n2 où nk est le nombre d'éléments dans le sous-arbre (ou
OTU) k
on peut aussi définir :
- lien simple d(i,(12)) = min [ d(i,1) , d(i,2) ]
- lien complet d(i,(12)) = max [ d(i,1) , d(i,2) ]
Cette méthode qui pose que les séquences à regrouper sont les plus proches (valeur minimale
dans le tableau) sous-entend l'hypothèse de "l'horloge moléculaire" : taux de mutation
proportionnelle au temps évolutif et identique sur toutes les branches de l'arbre
Exemple :
a b c d ea 0b 1 0 on regroupe a et b en Ac 2,1 1,2 0d 3,3 2,3 1,2 0e 2,8 3 2,5 3,4 0-------------------------------------------------- A c d eA 0c 1,65 on regroupe c et d en Bd 2,8 1,2e 2,75 2,5 3,4-------------------------------------------------- A B eAB 2,225 on regroupe A et B en Ce 2,75 2,95-------------------------------------------------- C eCe 2,85

___________________________________________________________________________DEA Environnement 2001 - 7 -
d'où le dendrogramme ou encore
a b c d e
A BC
0,5 0,6
1,421,11
a b c d e
Méthode NJ (Neighbor-joining : Saitou N. and Nei M. 1987)
C'est une méthode de construction d'un arbre phylogénétique sans racine à partir d'un indice
d'écart (par exemple distance ou dissimilarité entre séquences). Elle est basée sur la recherche
d'une paire d'OTU (operational taxonomic units) qui minimisent la longueur totale des
branches de l'arbre et ceci à chaque étape de regroupement (parcimonie).
Nous avons un indice de proximité D sur un ensemble X de n séquences, représenté par un
tableau à deux entrées. Nous voulons passer à une représentation par un X-arbre additif libre
telle que la valuation sur l'arbre L (longeur des chemins entre deux feuilles) soit une "bonne
approximation" de D. (Cela est possible sans problème si l'indice est une distance métrique)
Indice de proximité (D) Arbre phylogénétique additif (L)
1 2 3 4 5
2 d12
3 d13 d23
4 d14 d24 d34
5 d15 d25 d35 d45
6 d16 d26 d36 d46 d56
→→→→1
5
42
3
6
a b c d
Si D distance métrique : OK
Figure 1
L15 = L1a + L5a etc .. et Lij pour i, j ∈{1..5} soit une bonne approximation deDij
Pour faire ceci, nous allons procéder en (n-2) cycles en répétant le processus suivant :
- regrouper les deux OTU les plus proches en créant une arête interne entre cette paire et le
reste des OTU suivant le critère de minimisation de la longueur de l'arbre obtenu

___________________________________________________________________________DEA Environnement 2001 - 8 -
- calculer la valuation intermédiaire
- regrouper la paire dans le tableau de dissimilarité et recalculer celui-ci par le lien moyen
Critère de sélection d'une paire d'OTU les plus proches
Pour tous les couples d'OTU, la valeur de Sij, longueur de l'arbre pour une topologie où i et j
sont regroupés en une paire, est calculée à partir des valeurs de dissimilarité Dkl.
Toutes ces longueurs d'arbre sont calculées en adaptant la formule d'un arbre en étoile à un
arbre avec une arête interne.
Pour un arbre en étoile : S0 = ∑i=1
N
LiX = 1
N-1 ∑i<j
Dij d'où pour un arbre avec une arête
interne X-Y et un regroupement en une paire de i et j, nous obtenons :
Sij = LXY + (LiX + LjX ) + ∑k=1, k≠i, k≠j
N
LkY
La paire choisie est celle qui minimise la valeur de Sij .
La valeur de Sij peut être calculée à partir des Dkl :
Sij = 1
2(N-2) ∑k=1, k≠i, k≠j
N
(Dik + Djk ) + 12 Dij +
1N-2 ∑
k<l, k,l ≠i, k,l≠j
N
Dkl
A partir du tableau d'indice de dissimilarité des OTU, le tableau de tous les Sij est calculé :
Indice de dissimilarité Valeur de Sij (longueur de l'arbre pourune toplogie où i et j sont regroupés
1 2 3 4 5
2 d12
3 d13 d23
4 d14 d24 d34
5 d15 d25 d35 d45
6 d16 d26 d36 d46 d56
→→→→
1 2 3 4 5
2 S12
3 S13 S23
4 S14 S24 S34
5 S15 S25 S35 S45
6 S16 S26 S36 S46 S56
et la paire sélectionnée pour le regoupement est celle qui minimise la valeur de Sij.

___________________________________________________________________________DEA Environnement 2001 - 9 -
Regroupement d'une paire d'OTU et création d'une arête interne
Schématiquement nous passons de la topologie en étoile à une topologie où une arête interne
est créée (ici c'est S15 qui est supposé minimum).
Etoile Création d'une arête interne
X
1
2
34
5
6
→→→→X Y
1
5
6
4
32
a
Figure 2Valuation de L1a et L5a
Estimation de la longueur des arêtes LiX , LjX , LXY
LXY = 1
2(N-2) [ ∑k=1, k≠i, k≠j
N
(Dik + Djk ) - (N-2) D12 - 2
N-3 ∑k<l, k,l ≠i, k,l≠j
N
Dkl ]
LiX et LjX sont calculés à partir de la méthode de Fitch et Margoliash :
LiX = (Dij + DiZ - DjZ ) / 2
LjX = (Dij + DjZ - DiZ ) / 2
où DiZ = 1
N-2 ∑k=1, k≠i, k≠j
N
Dik et DjZ = 1
N-2 ∑k=1, k≠i, k≠j
N
Djk
Remarque : seuls LiX et LjX sont calculés pour chaque cycle, sauf au dernier pour lesquel
on a une trichotomie.
Préparation du tableau de dissimilarité pour le cycle suivant
Nous passons d'un tableau avec 6 OTU à un tableau à 5 OTU où la paire i, j est regroupée en
un seul OTU (dans le cas de la figure 1 c'est 1 et 5 qui sont regroupés). Les nouveaux indices
de similarité sont recalculés à l'aide de la distance de Fitch et Margoliash :
___________________________________________________________________________DEA Environnement 2001 - 10 -
D(i-j)k = ( Dik + Djk - Dij) / 2
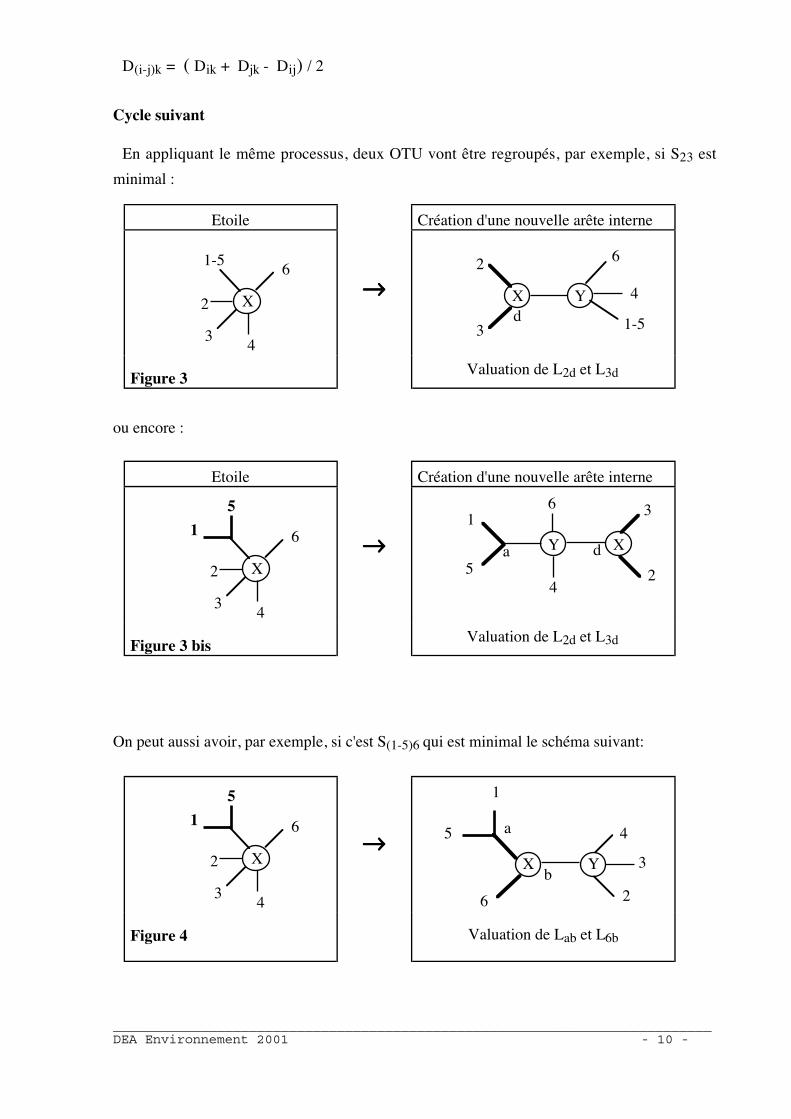
Cycle suivant
En appliquant le même processus, deux OTU vont être regroupés, par exemple, si S23 est
minimal :
Etoile Création d'une nouvelle arête interne
X
1-5
2
34
6→→→→ X Y
2
3
6
4
1-5d
Figure 3Valuation de L2d et L3d
ou encore :
Etoile Création d'une nouvelle arête interne
X2
34
61
5
→→→→ XY
1
5
6
4
a
3
2
d
Figure 3 bisValuation de L2d et L3d
On peut aussi avoir, par exemple, si c'est S(1-5)6 qui est minimal le schéma suivant:
X2
34
61
5
→→→→X Y
1
5 a
3
2b
6
4
Figure 4 Valuation de Lab et L6b

___________________________________________________________________________DEA Environnement 2001 - 11 -
Deux propriétés importantes doivent être soulignées en rapport avec la minimalité de Sij :
Critère d'un arbre d'évolution minimum
Pour un arbre additif, le critère d'une paire d'éléments les plus voisins est :
Les éléments 1 et 2 sont les plus proches si les relations d'inégalité suivantes sur l'indice
d'écart sont satisfaites :
∀ i, j (i≠ 1, 2 et j≠ 1,2) : D12 + Dij < D1i + D2j et D12 + Dij < D1j + D2i
Remarquons que ce crirère n'implique pas que D12 soit l'élèment minimal des Dij .
Soit S12 la longueur de l'arbre où la paire (1-2) est regroupée par ajout d'une arête interne
(voir pages précédentes)
Théorême : La condition de minimalité de S12 est équivalente à la condition des plus proches
voisins sur l'arbre additif.
Estimation des longueurs de branches par les moindres carrés
Nous avons vu que nous voulons passer d'une représentation par un indice de proximité D
sur un ensemble X de n séquences, représenté par un tableau à deux entrées à une
représentation par un X-arbre additif libre telle que la valuation sur l'arbre L (longeur des
chemins entre deux feuilles) soit une bonne approximation de D. Le problème est d'obtenir
une matrice A "la meilleur possible" telle que AL = D. La solution de cette équation par les
moindres carrés est de minimiser l'expression :
1
2(N-2) ∑k=1, k≠i, k≠j
N
(Dik + Djk ) + 12 Dij +
1N-2 ∑
k<l, k,l ≠i, k,l≠j
N
Dkl
qui est exactement égale à Sij . Or dans le choix de la création d'une arête interne, c'est le
critère de minimisation qui a été choisi. De plus la valuation intermédiaire des LiX , calculée
par la méthode de Fitch et Margoliash correspond à cette approximation.

___________________________________________________________________________DEA Environnement 2001 - 12 -
Les problèmes posées par ces méthodes
Les problèmes de construction d'arbres.
L'un des problèmes les plus récurrents est la "stabilité" d'un arbre phylogénétique. Par
exemple l'introduction d'une nouvelle séquence ou l'enlèvement d'une séquence donneront-
elles un arbre similaire, c'est à dire dont la topologie sera identique.
Pour éprouver la robustesse d'une solution, on applique la technique de re-échantillonage :
- "bootstrap"
On suppose que les caractères évoluent de manière indépendante : on crée un nouveau jeux de
données en changeant pour chaque caractère (site ou position pour une séquence d'ADN) de
manière aléatoire l'état (base pour une séquence d'ADN) et on compare l'arbre obtenu avec
l'original. On répète au moins 100 fois l'opération et on cacule pour chaque nœud le
pourcentage où il est trouvé dans la même topologie que dans l'arbre initial.
- "jackknife"
La méthode est similaire au bootstrap, avec toutefois une élimination de 20 % des caractères
(site ou position pour une séquence d'ADN)
Ces deux méthodes sont utilisées aussi bien dans le cas de méthodes cladistiques que
phénétiques ou autres.
Les problèmes de saturation
Pour un site dans une séquence, après plusieurs mutations successives on peut retrouver un
état ancestral (homoplasie), ce qui masquera le "message phylogénétique".
Ce phénomène interviendra d'autant plus souvent que le nombre d'états possibles sera faible
et que les séquences appartiendront à des taxons très éloignés.
Les problèmes d'orthologie-paralogie
Deux gènes sont orthologues s'ils ont acquis leur autonomie évolutive après un événement de
spéciation (ils appartiennent obligatoirement à 2 organismes différents). Ils ont un ancêtre
commun.
Deux gènes sont paralogues s'ils ont acquis leur autonomie évolutive après une duplication
génique et non un événement de spéciation.
Pour une reconstruction phylogénique, il faut disposer des gènes ou des produits de gènes
orthologues.
L'évolution ne se fait pas au hasard

___________________________________________________________________________DEA Environnement 2001 - 13 -
La notion de hasard sous-tend celle d'équiprobabilité indépendante des évènements survenant
lors de la spéciation.
Ceci signifie que dans toutes les méthodes utilisées, on devra s'attacher à se poser les
questions suivantes pour les séquences moléculaires:
- influence de la position dans une séquence
- pour les séquences nucléiques de géne codant des protéines : mutation sur la troisième
base du codon
- pour des séquences nucléiques : problème de transition (purine : A<->G ou pyrimidine
C<->T) et de transversion (purine<->pyrimidine)
- mutation des amino-acides : construction de matrice de substitution
Les problèmes spécifiques aux méthodes utilisées
Méthode cladistique
Il faut disposer d'un alignement de séquences. Chaque position de l'alignement sera un
caractère putatif. Pour les séquences de même longueur, cela ne pose aucun problème. Pour
les séquences ayant subi des insertions ou délétions lors de l'évolution, cela posera un
problème. Nous ne disposons d'aucune méthode pour construire le "bon" alignement.
Plusieurs méthodes donnant des résultats différents sont utilisées.
Méthode phénétique
Mesurer la ressemblance de séquences moléculaires ne conduit pas à un seul modèle et donc à
une seule méthode : voir le paragraphe suivant

___________________________________________________________________________DEA Environnement 2001 - 14 -
Ressemblance de séquences
La caricature du biologiste moléculaire, la plus actuelle, montrerait un biologiste ayant
"péché" une séquence et s'exclamerait à quoi tu ressembles ou en quoi diifères-tu!
Nous ne poserons pas la question de la pertinence de tout cartographier et de tout séquencer,
sainte quête, en vue d'obtenir le secret de la vie, mais simplement nous allons feuilleter
quelques pages du bréviaire.
Deux points de vue pour répondre à la question de la similarité entre deux séquences :
1 - l'analyse du mathématicien qui considère une séquence comme un mot construit à partir
d'un alphabet et qui a des méthodes opératoires pour établir des fontions de mesure
2 - l'analyse ou aussi l'expertise du biologiste qui se référera, au delà des réponses
précédentes, à d'autres connaissances que la séquence primaire : toutes les propriétés
biologiques
Voici quelques classiques de la biologie moléculaire : aucune démonstration ne sera donnée,
vous les trouverez dans les articles originaux. Dans cette catégorie de programmes, nous
trouvons : alignement de 2 séquences, de plusieurs séquences, alignement d'une séquence
contre une banque, recherche de consensus
La première image
Une représentation matricielle de points (dot-matrix), construite ainsi :
- une croix si xi=yj (où xj est un élément de la première séquence et yj un élément de la
deuxième séquence, sinon rien. La vision d'une suite croix consécutives dans une diagonale
souligne des identités entre des parties des deux séquences
M Q N W E T T A T T N Y E Q H N A W Y N N x x x x W x x E x x T x x x x V ? T x x x x T x x N x x Y x x D ? Q x x H x
L'expert biologiste peut indiquer qu'une substitution de valine par alanine ne change pas les
propriétés biologiques (de même E par D).

___________________________________________________________________________DEA Environnement 2001 - 15 -
Méthodes globales
La première analyse mathématique
Levenshtein introduit en 1965 (Levenshtein 1965) deux concepts très proches de distance
entre deux séquences A et B; l'un définit la distance comme le nombre minimum de
substitutions, de délétions ou insertions requises pour transformer A en B, l'autre comme le
nombre minimum de délétions ou insertions. Une distance définie ainsi a les propriétés d'une
distance métrique, à savoir:
° d(A,B) ≥ 0
° d(A,B)=0 si A=B
° d(A,B)= d(B,A)
° d(A,B) + d(B,C) ≥ d(A,C)
Cette distance se calcule de manière récursive (on parle de programmation dynamique) :Soient les séquences A de n lettres et B de m lettres; d(A,B)=d(an,bm) avec
d(ao,bo)=0, si i<0 ou j<0 alors d(ai,bj) est infini: et d(ai,bj) défini
récursivement ainsi
d(ai-1, bj) + w(ai, -) délétion de aid(ai,bj)= minimum { d(ai-1, bj-1) + w(ai,bj) substitution ai/bj d(ai, bj-1) + w(-, bj) insertion de bj
où w est une fonction poids: cette fonction a les valeurs suivantes: w(ai,-)=1,
w(ai,bj)=0 si ai=bj ou w(ai,bj)=1 si ai≠bj, w(-, bj)=1.
On définit simultanément un pointeur qui indique la position de la valeur minimum
précédente de cette manière:
(i-1, j) ou
p(i,j) = minimum { (i-1, j-1) ou
(i, j-1)
On peut facilement écrire un programme qui réalise cet algorithme et calcule une distance
entre deux séquences. Un alignement métrique entre les deux séquences s'obtient en partant
du pointeur p(n,m) et en remontant en arrière (backtrack) jusqu'à la position précédant
p(o,o).

___________________________________________________________________________DEA Environnement 2001 - 16 -
Exemple pédagogique:
A C G T G C G C p(8,6)= (7,6) p(7,6)= (6,5) 0 1 2 3 4 5 6 7 8 p(6,5)= (5,4) p(5,4)= (4,3) C 1 1 1 2 3 4 4 5 5 p(4,3)= (3,2) p(3,2)= (2,1) G 2 2 2 1 2 2 3 3 4 p(2,1)= (1,0) p(1,0)= (0,0) A 3 2 3 2 2 3 3 4 5 G 4 3 3 2 3 2 3 3 4 La distance est de 4: on peut obtenir B à
C 5 4 3 3 3 3 2 3 3 partir de A par 2 délétions et
T 6 5 4 4 3 4 3 3 4 2 substitutions.
L'alignement métrique proposé est:
A C G T G C G C LCS = CGGC (longest common subsequence)
| | ^ | | ^
- C G A G C T -
Le premier algorithme pour biologiste (Needleman S. and Wunsch C. 1970)
Basé aussi sur la programmation dynamique, il ne cacule pas la différence entre deux
séquences mais la similarité. Considérons deux séquences A(1,n) B(1,m)
A B C N J R O C L C R P M A J Le jeu est de finir de remplir ce tableau C J N R C 3 3 4 3 3 3 3 4 3 3 1 0 0 K 3 3 3 3 3 3 3 3 3 2 1 0 0 C 2 2 3 2 2 2 2 3 2 3 1 0 0 R 2 1 1 1 1 1 2 1 1 1 2 0 0 B 1 2 1 1 1 1 1 1 1 1 1 0 0 P 0 0 0 0 0 0 0 0 0 0 0 1 0
Le tableau est rempli ligne après ligne (en partant de la dernière) et pour chaque ligne colonne
après colonne (en partant de la dernière) en obéissant à la règle suivante :
- Le score S(i,j) est le nombre maximun de correspondance entre les deux parties de
séquences A(i,n) et B(j,m) (en prenant tous les chemins possibles à partir de (i,j)) et en
appliquant la valuation suivante :
- score pour une identité 1- score pour une substitution, une insertion ou délétion 0
La formule de récurrence est :

___________________________________________________________________________DEA Environnement 2001 - 17 -
si ai= bj+1 S(i,j+1) -1 + s(ai,bj), si non S(i,j+1) + s(ai,bj)S(i,j) = max si ai+1= bj+1 S(i+1,j+1) -1 + s(ai,bj), si non S(i+1,j+1) + s(ai,bj)
si ai+1= bj S(i+1,j) -1 + s(ai,bj), si non S(i+1,j) + s(ai,bj)avec évidemment S(n+1, j) = S(i, m+1) = 0
La similarité entre les deux séquences est égale à la valeur de S(1,1) et l'alignement est un
graphe qui a pour origine S(1,1) et parcourt la matrice pour des i et j croissant en recherchant
l'élément maximal voisin.
C'est une heuristique en ce sens qu'aucune démonstration n'indique qu'il calcule le maximum
de similarité. De plus, le nombre de séquences primaires augmentant, deux problèmes se sont
rapidement posés :
- la connaissance de séquences de famille de protéines montre qu'un score de substitution
identique pour toutes les paires d'amino acides n'est pas réaliste (par exemple substituer un
acide aspartique par un acide glutamique n'entraîne pas les mêmes conséquences biologiques
que substituer un acide aspartique par une lysine, ...) : voir plus loin le paragraphe sur les
matrices de substitution.
- le score à attribuer à une série d'insertion ou de délétion est-il la somme simple des scores
de chacun des phénomèmes.
Ceci définit des formules de score s(ai,bj) pour chaque paire d'amino acides plus
sophistiquées et l'introduction de score de délétion ou insertion faisant intervenir la longueur
de segment délété ou inséré par une formulaire linéaire (a +b(longueur)).
A partir de là, l'algorithme précédent a été modifié et d'autres ont apparu (en particulier, celui
de Sellers (1974) qui est une modification de celui de Levenstein). Smith F. et Waterman
(1991) ont comparé ces différents algorithmes et indiqué l'équivalence de certains (en
particulier celui de Sellers et celui de Needleman S. et Wunsch C) et ils ont défini les
conditions que doivent respecter les scores pour que la mesure (différence ou similarité) soit
une métrique (inégalité triangulaire respectée). Ils ont montré que si les scores utilisés pour
chaque paire d'amino acide est une métrique, alors la similarité ou la différence entre deux
séquences, calculées suivant les formules de récurrence précédentes est une métrique. A
moins de modifications importantes, cet algorithme n'est pas utlisé pour l'alignement sur une
banque.
Méthodes locales
L'algorithme le plus utilisé est celui de Smith et Waterman (1981) qui identifie les sous-
séquences maximales de deux séquences par programmation dynamique. Un matrice de score
est construite à l'aide d'une formule de récurrence (en reprenant les mêmes notations):
S(i-1, j-1) + s(ai, bj)S(i,j) = max S(i-k, j) - Wk
S(i, j-k) - Wk0avec évidemment S(i, 0)= S(0, j) = 0

___________________________________________________________________________DEA Environnement 2001 - 18 -
S(i,j) est le maximum de similarité entre deux segments se terminant en Ai et Bj. Une
séquence maximale est identifiée en trouvant l'élément maximal du tableau et en le
parcourant pour des indices décroissants jusqu'à la valeur nulle et en recherchant l'élément
voisin maximal. Dans l'article original, le score d'une identité est nul, celui d'un mismatch -
1/3 et Wk est égal à 1+ (1/3)k
Méthodes approchées
Lorsque l'alignement d'une séquence est réalisé contre une banque, le problème du temps de
calcul devient prédominant. Les algorithmes précédents sont trop gourmands en ressource. La
façon de voir la ressemblance a été posée d'une manière différente pour contourner cet
obstacle et des heuristiques ont été proposées.
FASTA (Lipman D. and Pearson W - Wilbur W.)
Ces deux auteurs ont fait les remarques suivantes :
- les ressemblances recherchées au "niveau biologique" concernent des fragments de
séquences
- de plus, dans ces fragments, la fréquence de substitution est beaucoup plus grande
que celle d'insertion ou délétion
Leur programme est basé sur la méthode de la diagonale, que l'on peut approcher
intuitivement par la représentation "dot-matrix". La ressemblance se définit par comparaison
de paire de fragments de chacune des séquences (fragment : partie de même longueur de
chacune des deux séquences, en dot-matrix c'est un morceau d'une diagonale). Ces deux
parties contiennent des mots communs séparés par des zones de substitution. Un fonction
score est attribué pour un fragment et la ressemblance est mesurée par le fragment de score
maximum.
L'algorithme se divise en 4 étapes :
- précodage des séquences en k-uple : mots de longeur k (4 acides nucléiques, 2 pour les
protéines). Ceci permet une efficacité beaucoup plus grande pour la deuxième étape
- recherche du fragment de plus haut score pour chaque diagonale qui est le score de la
diagonale (fragment = suite de mots séparés par de régions de substitution dont la longueur
maximale est prédéfinie (paramètre M))
- les scores des dix meilleures diagonales vont être recalculés en utilisant une matrice de
subtitution (PAM 250). C'est ce score qui est listé dans les résultats sous l'appellation
init1dans les programmes antécédents à FASTA (FASTP ..). Pour FASTA, ce score (initn) est
recalculé en essayant d'enchaîner à partir de la meilleure diagonale les fragments restants des
9 autres diagonales en tenant compte des insertions ou délétions dues au changement de
diagonale.

___________________________________________________________________________DEA Environnement 2001 - 19 -
- Les résulats sont classés par rapport au score précédent, et pour les meilleurs, un
alignement et un nouveau score (Opt) entre la séquence requête et la séquence de la banque,
sont calculés à partir de l'algorithme de Needleman et Wunsch légèrement modifié.
BLAST (Altschul S. et al. - Karlin S. and Altschul S.)
L'idée originale est de combiner à l'algorithme de recherche de paires de segments
homologues entre deux séquences une statistique qui permet de les classer.
La recherche se fait en plusieurs étapes :
- recherche de mots communs (w-mer) entre la séquence requête et les séquences de la
banque (celle-ci peut se faire à l'aide de matrice de substitution). Par défaut c'est Blosum62
qui est utilisée
- pour les mots "communs" supérieurs à un certain score (Expected), le mot est étendu si
possible à droite et à gauche et si le nouveau score obtenu est supérieur au "cut-off" S, le
segment est alors retenu (mis dans la liste des HSP : high scoring pair)
- pour une séquence de la banque pour laquelle au moins un segment satisfait à la condition
précédente, une valeur ("p-value"), basée sur une statistique de Poisson, qui évalue la
probablité que deux séquences "au hasard" aient en commun ces HSP, est calculée et les
résultats sont triés par valeur croissante.
Il est à remarquer que les valeurs de E et S sont en relation. La valeur par défaut de S est
calculée à partir de la valeur de E. Quant à la valeur par défaut de E, elle est le nombre de fois
qu'un tel segment est attendu "au hasard" dans la banque (10 pour la valeur par défaut). Il est
évidemment dépendant de la taille de la banque, de la fréquence des lettres (acides
nucléiques, ammino acides)
Le fait d'utiliser des matrices de substitution dans la première étape, pose un problème pour
des segments déclarés ressemblant entre deux séquences alors qu'au niveau biologique cela
n'a peut être pas de sens. Deux filtres sont prédéfinis
- région de faible complexité (SEG)
- région de répétitions internes (XNU)
Ces filtres vous permettent par exemple d'éliminer des ressemblances dues uniquement à des
régions acides ou basiques, etc.
Matrice de substitution
La recherche de ressemblances entre séquences, leur alignement mettent en jeu des relations
entre entre les constituants élémentaires des séquences, les nucléotides et les amino acides.
Les matrices de substitution sont écrites pour exprimer de telles relations. Quels sont les
critères les plus utilisés :
- identité : matrice unitaire
- code génétique

___________________________________________________________________________DEA Environnement 2001 - 20 -
- propriétés chimiques des amino acides
- substitutions observées dans des structures 3D supperposables (matrice de Risler)
- substitutions observées dans des alignements de protéines (matrice PAM, Blosum)
Le dernier critère, qui est le plus utilisé, a un défaut originel qui vient du fait que pour
fabriquer des alignements, on utilise très souvent des programmes qui font intervenir une
matrice de substitution. Les deux catégories de matrices les plus utilisées sont PAM et
Blosum.
PAM (Dayhoff M. et al.)
Ces matrices sont construites par étude de segments proches (moins de 15 % de différences)
de séquences de protéines homologues. Les fréquences observées de substitutions (ou
probabilité conditionelle : appelées "odd") sont transformées en logarithme de probabilité,
normalisé en unité d'évolution. Le logarithme est utilisé pour que dans les programmes de
recherche de ressemblance, la somme de ces éléments donne le logarithme de la probabilité
pour la séquence entière (le modèle étant Markovien : indépendance de fréquences de
substitution).
Les éléments diagonaux de la matrice indique une évolution sans substitution.
Pour PAM1, leur somme est telle qu'elle correspond à une probabilité de 99/100 (1 mutation
pour 100 résidus : d'où le nom PAM acepted point mutation)
L'indépendance des fréquences et les éléments de la matrice étant des logarithmes de
fréquences, nous pouvons calculer PAM(N) en élevant PAM1 à la puissance N, par exemple
pour PAM120, il faut multiplier PAM1 par elle-même 120 fois.
BLOSUM (Henikoff S. and Henikoff J.)
Ces matrices sont construites par étude de séquences de protéines et les séquences sont
découpées en blocs (2000 au total) par rapport au pourcentage d'amino acides inchangés.
Une matrice "d'odds" est calculée à partir des blocs d'alignement pour chaque valeur de
similitude, et ensuite chaque élément est transformé en unité d'information en prenant le
logarithme du rapport de la valeur observée à la valeur qu'on obtiendrait au hasard. Cette
matrice est ensuite normalisée. Ces matrices sont désignées par blosum(N) (blosum pour
blocks substitution matrix) où N représente le pourcentage de similitude. Les
correspondances entre blosum et PAM, basées sur la théorie de l'information sont :
- PAM250 -> blosum45 PAM160 -> blosum62 PAM120 -> blosum80

___________________________________________________________________________DEA Environnement 2001 - 21 -
BibliographieRessemblance- Altschul, Stephen F., Warren Gish, Webb Miller, Eugene W., Myers, and David J.Lipman (1990). Basic local alignment search tool. J. Mol. Biol. 215:403-10.
- Karlin, Samuel and Stephen F. Altschul (1990). Methods for assessing the statisticalsignificance of molecular sequence features by using general scoring schemes. Proc. Natl.Acad. Sci. USA 87:2264-68.
- Karlin, Samuel and Stephen F. Altschul (1993). Applications and statistics for multiplehigh-scoring segments in molecular sequences. Proc. Natl. Acad. Sci. USA 90:5873-7.
- Levenshtein V. (1965) Binary codes capable of correcting deletions, insertions, reversals.Cybernetics and Control Theory 10 (8): 707-710
- Needleman S. And Wunsch C. (1970) A general method applicable to the search forsimilarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443-453
- Pearson W and Lipman D. (1988) Improved tools for biological sequence comparison.Proc. Natl. Acad. Sci. USA 85, 2444-2448
- Sellers P. (1974) On the theory and computation of evolutionary distances. SIAM J. Appl.Math. 26, 787-793
- Smith T. and Waterman M. (1981) Identification of common molecular subsequence. J.Mol. Biol . 147, 195-197
- Smith T. Waterman M. and W. Fitch (1981) Comparative biosequence metrics. J. Mol. Evol.18, 38-46
- Wilbur W. and Lipman D. (1983) Rapid similarity searches of nucleic acid and protein databanks. Proc. Natl. Acad. Sci. USA 80, 726-730
- Wilbur W. and Lipman D. (1984) The context dependant comparison of biologicalsequences. SIAM J. APPL. MATH. 44, 557-567
Matrices de substitution- Dayhoff M., Barker W. and Hunt L. (1983) Establishing homologies in protein sequences.Methods in Enzymol. 91, 524-545
- Henikoff S. and Henikoff J. (1992) Amino acid substitution matrices from protein blocks.Proc. Nat. Acad. Sci. USA 89, 10915-10919
- Risler J., Delorme M., Delacroix H and Henaut A. (1988) Amino acid substitutions instructurally related proteins - A pattern approach : determination of a new and efficientscoring matrix. J. Mol. Biol. 204, 1019-1029
Phylogénie- Darlu P. et Tassy P. (1993) Reconstruction phylogénétique. Collection Biologie Théorique
: Ed Masson
- Fitch W. and Margoliash E (1967) Construction of phylogenetic trees. Science 155, 279-284
- Saitou N. and Nei M. (1987) The Neighbor-joining method : a new method forreconstructing phylogenetic trees. Mol. Biol. Evol. 4 , 406-425
![phylo 10May11 [互換モード]isw3.naist.jp/IS/Kawabata-lab/LECDOC_KINDAI/2010/phylo...系統樹(二分岐樹)のデータ構造 イイストースト ノード(node)と枝 (branch)からなるグラフ](https://img.pdfslide.tips/doc/110x75/5f940acb4fa4266f3b342b78/phylo-10may11-fffisw3naistjpiskawabata-lablecdockindai2010phylo.jpg)













![phylo 09May12 print [互換モード] - isw3.naist.jpisw3.naist.jp/IS/Kawabata-lab/LECDOC_KINDAI/2009/phylo_09May12.pdf · 3 系統樹(二分岐樹)のデータ構造 イースト](https://img.pdfslide.tips/doc/110x75/5e06643697ee6a778e0bdddd/phylo-09may12-print-fff-isw3naist-3-ccoeffe.jpg)














