Embed Size (px)
Citation preview

FARMACEUTSKA INSPEKCIJSKA KONVENCIJA
KOOPERACIJSKA SHEMA FARMACEUTSKE INSPEKCIJE
PE 010-4
1. ožujka 2014.
PIC/S NAČELA I SMJERNICE DOBRE
PRAKSE ZA PRIPREMU LIJEKOVA U
USTANOVAMA ZDRAVSTVENE
SKRBI
© PIC/S ožujak 2014.
Zabranjeno reproduciranje u komercijalne svrhe.
Reproduciranje za internu uporabu je dozvoljeno ako
je priznat izvor.
Urednik: PIC/S tajništvo
e-mail: [email protected]
web stranica: http://www.picscheme.org

Sadržaj
Sadržaj: Broj stranice:
SADRŽAJ ...................................................................................................................................... 2
POVIJEST DOKUMENTA ........................................................................................................... 5
A. UVOD ............................................................................................................................... 5
A.1 NAMJENA ........................................................................................................................ 5
A.2 PODRUČJE PRIMJENE ................................................................................................... 5
A.3 POVEZNICA NA NAČELA I SMJERNICE I NAČELA DOBRE PROIZVOĐAČKE PRAKSE
(DPP) U INDUSTRIJI .................................................................................................................... 5
B. POJMOVNIK .................................................................................................................... 6
1. SUSTAV OSIGURANJA KAKVOĆE .......................................................................... 10
1.1 NAČELA .........................................................................................................................10
1.2 OSIGURANJE KAKVOĆE ............................................................................................10
1.3 DOBRA PRAKSA PRIPREME LIJEKOVA ................................................................ 10
1.4 PROVJERA KAKVOĆE ................................................................................................11
2. OSOBLJE ........................................................................................................................11
2.1 NAČELA .........................................................................................................................11
2.2 OPĆI ZAHTJEVI ............................................................................................................11
2.3 OSPOSOBLJAVANJE I TRAJNA IZOBRAZBA ........................................................ 12
2.4 HIGIJENA .......................................................................................................................12
3. PROSTORI I OPREMA ..................................................................................................13
3.1 NAČELA .........................................................................................................................13
3.2 OPĆI ZAHTJEVI ............................................................................................................13
3.3 PROIZVODNI PROSTORI ............................................................................................13
3.4 SKLADIŠNI PROSTORI ................................................................................................14
3.5 PROSTORI ZA PROVJERU KAKVOĆE ......................................................................14
3.6 POMOĆNI PROSTORI ..................................................................................................14
3.7 OPREMA ........................................................................................................................15
4. DOKUMENTACIJA ....................................................................................................... 15
4.1 NAČELA .........................................................................................................................15
4.2 OPĆI ZAHTJEVI ............................................................................................................15
4.3 DOKUMENTACIJA ZA MAGISTRALNE PRIPRAVKE ........................................... 16
4.4 DOKUMENTACIJA ZA GALENSKE PRIPRAVKE
4.4.1 Specifikacije.....................................................................................................................17
4.4.2 Upute ...............................................................................................................................17
4.4.3 Zapisi ...............................................................................................................................18
4.5 OPĆI POSTUPCI I DODATNA DOKUMENTACIJA ................................................. 19
5. PROIZVODNJA .............................................................................................................19
5.1 NAČELA .........................................................................................................................19
5.2 OPĆI ZAHTJEVI ............................................................................................................20
5.3 SPRJEČAVANJE UNAKRSNE KONTAMINACIJE .................................................. 20
5.4 PROCJENA RIZIKA ZA PROIZVOD I DOKAZIVANJE PRIKLADNOSTI............. 20
5.5 POLAZNI MATERIJALI ...............................................................................................21
5.6 POSTUPCI IZRADE ......................................................................................................22
5.7 PAKOVNI MATERIJAL ................................................................................................22
5.8 PAKIRANJE ...................................................................................................................22
5.9 ODBIJENI, PRERAĐENI I VRAĆENI MATERIJALI I PROIZVODI ....................... 23

6. PROVJERA KAKVOĆE ................................................................................................23
6.1 NAČELA .........................................................................................................................23
6.2 OPĆI ZAHTJEVI ...........................................................................................................23
6.3 UZORKOVANJE ............................................................................................................24
6.4 ISPITIVANJE .................................................................................................................24
6.5 PUŠTANJE U PROMET ................................................................................................25
7. UGOVARANJE POSLOVA ...........................................................................................25
7.1 NAČELA .........................................................................................................................25
7.2 OPĆI ZAHTJEVI ............................................................................................................26
7.3 DAVATELJ UGOVORA ................................................................................................26
7.4 PRIMATELJ UGOVORA ..............................................................................................26
8. REKLAMACIJE I POVLAČENJE PROIZVODA ........................................................ 26
8.1 NAČELA .........................................................................................................................26
8.2 PROBLEMI S KAKVOĆOM .........................................................................................27
8.3 POVLAČENJE PROIZVODA ........................................................................................27
9. SAMOINSPEKCIJE ....................................................................................................... 27
9.1 NAČELA .........................................................................................................................27
DODATAK 1: SMJERNICE O NORMAMA POTREBNIM ZA PRIPREMU STERILNIH
LIJEKOVA
UVOD .......................................................................................................................................... 28
OSOBLJE ..................................................................................................................................... 29
PROSTORI I OPREMA ...............................................................................................................30
ODJEĆA ....................................................................................................................................... 33
ČIŠĆENJE .................................................................................................................................... 33
DOKUMENTACIJA ....................................................................................................................34
STERILNA PRIPREMA ..............................................................................................................34
PRIPREMA PROIZVODA KOD KOJIH SE STERILIZACIJA PROVODI NA KRAJU
PROIZVODNJE .......................................................................................................................... 35
STERILIZACIJA VLAŽNOM TOPLINOM ..............................................................................35
ASEPTIČKA PRIPREMA ...........................................................................................................36
PROVJERA KAKVOĆE .............................................................................................................37
PRAĆENJE .................................................................................................................................. 38
KLASIFIKACIJA “U MIROVANJU” .........................................................................................39
PRAĆENJE OKOLIŠA “TIJEKOM RADA” ............................................................................ 39
GRANICE ISPITIVANJA ZA PRAĆENJE OKOLIŠA ..............................................................41
DODATAK 2: SMJERNICE O NORMAMA POTREBNIM ZA PRIPREMU NESTERILNIH
TEKUĆINA, KREMA I MASTI 44
UVOD .......................................................................................................................................... 44
NAČELO ...................................................................................................................................... 44
PROSTORI I OPREMA ...............................................................................................................44
PROIZVODNJA .......................................................................................................................... 45
DODATAK 3: DOBRE PRAKSE ZA PRIPREMU RADIOFARMACEUTIKA U
USTANOVAMA ZDRAVSTVENE SKRBI 47
UVOD .......................................................................................................................................... 47
NAMJENA ................................................................................................................................... 48
PODRUČJE PRIMJENE .............................................................................................................. 48
OSOBLJE ..................................................................................................................................... 48
PROSTORI I OPREMA ...............................................................................................................49
DOKUMENTACIJA ....................................................................................................................51
PROIZVODNJA .......................................................................................................................... 51
PROVJERA KAKVOĆE .............................................................................................................53

NADZOR ..................................................................................................................................... 54
LITERATURA .............................................................................................................................56
POVIJEST REVIZIJE .................................................................................................................. 56

Povijest dokumenta
Usvojilo povjerenstvo PICS-a 19. studenoga 2007.
Stupanje na snagu verzije PE 010-1 1. travnja 2008.
Stupanje na snagu verzije PE 010-4 1. ožujka 2014.
A. Uvod A.1 Namjena
Ova načela i smjernice namijenjene su davanju uputa o dobrim praksama za pripremu lijekova za primjenu kod ljudi.
A.2 Područje primjene
Dok se PIC/S-ove smjernice PE 009 odnose na industrijsku proizvodnju lijekova za distribuciju, osnovni zahtjevi prikazani u ovim načelima i smjernicama odnose se na pripremu lijekova koja se obično obavlja u ustanovama zdravstvene skrbi za izravno izdavanje pacijentima.
U vrijeme izdavanja ovaj je dokument odražavao trenutačnu najvišu razinu ove pripreme. Osim ovdje navedenih načela postoje i druge prihvatljive metode, kojima se mogu ostvariti ova načela i smjernice. Ova načela i smjernice nisu namijenjene ograničavanju razvoja alternativnih sustava, novih zamisli ili novih tehnologija koje osiguravaju razinu osiguranja kakvoće najmanje jednakovrijednu onoj postavljenoj ovim načelima i smjernicama.
Uvijek kad se određuje stupanj do kojeg su odredbe utvrđene ovim dokumentom obvezujuće potrebno je pozvati se na nacionalno zakonodavstvo i regulatorne politike koje utvrđuje odgovarajuće nadležno tijelo. Ovaj je dokument samostalan dokument i treba ga koristiti za nadzor vezan za PIC/S.
A.3 Poveznica na načela i smjernice dobre proizvođačke prakse
(DPP) u industriji
Ova načela i smjernice podijeljene su na 9 glavnih poglavlja, čime slijede strukturu Načela i Smjernica dobre proizvođačke prakse u industriji . Glavni tekst upotpunjen je dodacima koji dopunjuju glavni dio ovih načela i smjernica, u kojima se navode općenita pravila za pripremu posebnih grupa lijekova, kao što su sterilni proizvodi (Dodatak 1) i nesterilne tekućine, kreme i masti (Dodatak 2). Navođenje općih pravila može se sastojati od isticanje važnih točaka glavnog dijela, kao i upotpunjavanja preciznijim smjernicama za posebne prilike koje taj dodatak pokriva.

B. Pojmovnik
Mnoge od definicija u ovom pojmovniku identične su definicijama iz Načela i smjernica dobre proizvođačke prakse. Ovdje su uvrštene radi lakšeg razumijevanja teksta.
1. Djelatni farmaceutski sastojak Bilo koja tvar ili kombinacija tvari kojoj se pripisuje učinak gotovog lijeka ili koja djeluje kao takva.
2. Serija Određena količina polaznih materijala, pakovnih materijala ili proizvoda obrađenih u jednom ili nizu postupaka tako da se može očekivati da će biti homogena.
3. Serijski/kontrolni broj Karakteristična kombinacija brojeva, simbola i/ili slova kojom se nedvosmisleno identificira neka serija.
4. Proizvod na veliko Bilo koji proizvod koji je prošao sve faze izrade, ali bez završnog pakiranja.
5. Umjeravanje Skup radnji kojima se pod određenim uvjetima uspostavlja odnos između vrijednosti koje pokazuje mjerni instrument ili mjerni sustav, odnosno vrijednosti koju predstavlja materijalna mjera i odgovarajućih poznatih vrijednosti poredbenog standarda.
6. Čisti prostori Prostor s definiranom provjerom čistoće od onečišćenja česticama i mikroorganizmima tako osmišljeno na način a se smanji unos stvaranje i zadržavanje onečišćenja unutar prostora.
7. Zatvoreni postupak Postupak kojim se sterilni farmaceutski proizvod priprema prijenosom sterilnih sastojaka ili otopina u prethodno sterilizirani zataljeni spremnik, izravno ili pomoću uređaja za sterilni prijenos, pri čemu se otopina ne izlaže utjecaju vanjskog okoliša.
8. Kontrolirani radni prostor Zatvoren radni prostor,opremljen prikladnim sustavima za dobavu i filtraciju zraka, konstruiran i korišten na takav način da bi se unos, stvaranje i zadržavanje onečišćenja smanjilo na unaprijed definiranu razinu. Kontrolirani radni prostor također se može koristiti za zaštitu vanjskog okoliša od materijala kojima se u njemu rukuje, npr. cjepiva ili citotoksične tvari.
9. Kritično područje Onaj dio kontroliranog radnog prostora u kojem se otvaraju spremnici i proizvod se izlaže utjecaju okoliša. Onečišćenje česticama i mikroorganizmima treba smanjiti na razine u skladu s namjenom.
10. Unakrsna kontaminacija Onečišćenje materijala ili proizvoda nekim drugim materijalom ili proizvodom.

11. Izvještaj o odstupanju Izvještaj o odstupanju je izvještaj o bilo kakvom odstupanju od uobičajenih postupaka i dokumentacije koje se pojavi tijekom postupka pripreme kao i izvještaj o posljedičnoj popravnoj radnji.
12. Magistralni pripravak Proizvod koji se izdaje odmah nakon pripreme i ne čuva se na zalihi.
13. Rok valjanosti Završetak razdoblja roka valjanosti, u nekodiranom obliku, nakon kojega se lijek više ne bi trebao koristiti. Izražava se također kao: najbolje upotrijebiti do..
14. Gotov proizvod Lijek koji je prošao sve faze proizvodnje, uključujući pakiranje u svoj konačni spremnik.
15. Ustanove zdravstvene skrbi Ustanove koje opskrbljuju svoje pacijente lijekovima u skladu s nacionalnim zakonima.
16. Međuproizvod Djelomično obrađen materijal koji bi trebao proći daljnje korake pripreme.
17. Rok valjanosti nakon otvaranja Završetak razdoblja primjene u kojem se lijek može uzeti ili primijeniti nakon što je pakiranje otvoreno, odnosno nakon što je prva doza lijeka uzeta iz pakiranja.
18. Pakiranje Sve radnje, uključujući punjenje i označivanje, kojima proizvod na veliko treba podvrgnuti kako bi postao gotov proizvod. Napomena: sterilno doziranje obično se ne smatra dijelom pakiranja, jer su sterilni proizvodi na veliko napunjeni u primarne spremnike, ali još nisu konačno upakirani.
19. Pakovni materijal Svaki materijal uključen u pakiranje polaznog materijala, međuproizvoda ili gotovog proizvoda, pri čemu je isključeno svako vanjsko pakiranje koje se koristi za transport ili pošiljke. Pakovni materijali nazivaju se primarnima ili sekundarnima, ovisno o tome jesu li ili nisu namijenjeni za izravni kontakt s proizvodom.
20. Postupak pripreme Sve radnje nabave materijala i proizvoda, proizvodnje, provjere kakvoće, puštanja u promet, skladištenja, dostave lijekova i odnosne provjere. Napomena: Jednostavno izdavanje lijekova prema odobrenim uputama te bez potrebe farmaceutskog tehničkog znanja, pri čemu su lijekovi spremni za neposrednu primjenu (npr. otapanje praška za neposrednu primjenu prema uputama za pacijenta koje su priložene uz odobreni proizvod), obično se ne smatra pripremom.
21. Izrada Onaj dio pripreme lijeka koji uključuje izradu farmaceutskog oblika lijeka.

22. Proizvodnja Obuhvaća sve postupke i radnje u pripremi lijeka, od zaprimanja materijala, preko izrade i pakiranja, do njegova dovršetka kao gotovog proizvoda.
23. Rukovoditelj proizvodnje Osoba odgovorna za nadgledanje proizvodnje koja treba biti u odjelu u kojem se odvija proizvodnja. Ta osoba treba biti upućena u sve što se događa i treba moći osigurati da se postupak odvija na propisani način.
24. Proizvodi za neposrednu uporabu Proizvodi koji će se primijeniti neposredno nakon pripreme i koji ne prolaze čuvanje ili skladištenje.
25. Farmaceutski izolator Zaštitni uređaj koji koristi tehnologiju barijere kako bi osigurao zatvoreni, kontrolirani radni prostor.
26. Kvalifikacija Sustavni i dokumentirani postupak temeljen na procjeni rizika kojim se dokazuje da postrojenja, prostorije ili oprema rade ispravno, da su prikladni za svoju namjenu i u stvarnosti daju očekivane rezultate.
27. Karantena Status polaznih ili pakovnih materijala, materijala i tvari, međuproizvoda, proizvoda na veliko ili gotovih proizvoda izoliranih fizički ili na drugi učinkovit način dok čekaju odluku o njihovu puštanju u promet ili odbijanju.
28. Osoba zadužena za puštanje serije lijeka u promet Osoba koja pušta u promet pripremljene lijekove. Ova osoba može biti i „odgovorna osoba“.
29. Odgovorna osoba (za puštanje serije lijeka u promet) Osoba koja je u konačnici odgovorna za sve korake u pripremi lijekova, uključujući puštanje istih u promet. Ova osoba mora imati dovoljno znanstvenog i tehničkog obrazovanja i iskustva za obavljanje opisane dužnosti.
30. Procjena rizika Sastoji se od identifikacije opasnosti, te analize i ocjene rizika povezanih s izlaganjem tim opasnostima. Procjene rizika za kakvoću počinju dobro definiranim opisom problema ili pitanjem o riziku., Koji je odgovarajući alat za upravljanje rizikom i vrste informacija potrebne za postavljanje pitanja i rješavanje rizika bit će lakše prepoznati kada je dotični rizik dobro definiran. Kao pomoć u jasnom definiranju rizika (ili više njih) u svrhu procjene rizika, često pomažu tri temeljna pitanja:
1. Što bi moglo poći po krivu? 2. Kolika je vjerojatnost da će poći po krivu? 3. Koje su posljedice (ozbiljnost)?
31. Samoinspekcija Procjena napravljena pod odgovornošću iste organizacije kako bi pratila valjanost sustava osiguranja kakvoće i usklađenost s ovim načelima i smjernicama. Može ju voditi imenovana nadležna osoba (ili više njih) iz organizacije ili uz pomoć vanjskih stručnjaka.

32. Specifikacije Pogledati poglavlje 4.
33. Polazni materijal Tvar koja se koristi za pripremu lijeka; pakovni materijal ne ubraja se u polazne materijale.
34. Galenski pripravak Proizvod koji se priprema za zalihu i raspoloživ je za izdavanje.
35. Uređaj za prijenos Fiksni ili pokretni uređaj koji omogućuje da se materijal prenosi u spremnik i iz spremnika ili farmaceutskog izolatora bez izlaganja istoga utjecaju vanjskog okoliša.
36. Validacija Sustavni, dokumentirani postupak temeljen na procjeni rizika, kojim se dokazuje da je određeni postupak usklađen s dobrom proizvođačkom praksom (DPP) i u stvarnosti reproducibilno dovodi do zahtijevanih rezultata.
37. Radni ciklus Definirano razdoblje za koje raspoloživi dokazi pokazuju održavanje prikladnih radnih uvjeta.

1. Sustav osiguranja kakvoće 1.1 Načela
Radi zaštite javnog zdravlja, lijekovi trebaju biti visoke kakvoće, neškodljivi djelotvorni. Treba ih pripremiti na takav način da budu u skladu sa svojom namjenom i da njihova kakvoća stalno udovoljava postavljenim zahtjevima. Za pouzdano ostvarenje ovog cilja potrebno je osmisliti i pravilno primijeniti opsežan sustav osiguranja kakvoće, koji u sebi uključuje načela i smjernice Dobre prakse pripreme kako su opisana u ovom dokumentu. Sustav osiguranja kakvoće treba dokumentirati i njegova se djelotvornost treba pratiti.
1.2 Osiguranje kakvoće 1. Osiguranje kakvoće predstavlja ukupni zbroj organiziranih radnji provedenih s ciljem da osiguraju kakvoću lijeka prema traženoj namjeni Njegovu djelotvornost i prikladnost treba redovito procjenjivati.
2. Osiguranje kakvoće osigurava da se:
a. Lijekovi razvijaju i pripremaju u skladu s najnovijim saznanjima.
b. Proizvodni i kontrolni postupci jasno navode i primjenjuju u skladu s načelima i smjernicama Dobre prakse pripreme.
c. Lijekovi isporučuju samo ako su ispravno proizvedeni, provjereni i skladišteni u skladu s definiranim postupcima i ako ih je u promet pustila odgovarajuća nadležna osoba (odnosno, odgovorna osoba ili osoba zadužena za puštanje serije lijeka u promet).
d. Primjenjuju odgovarajuće mjere, čime se osigurava da se lijekovi puštaju u promet, skladište i da se njima rukuje na takav način koji osigurava potrebnu kakvoću tijekom cijelog roka valjanosti i do isteka roka valjanosti nakon otvaranja.
e. Upotrebljavaju i održavaju sustavi dokumentacije.
1.3 Dobra praksa pripreme lijekova 1. Dobra praksa pripreme onaj je dio sustava osiguranja kakvoće koji osigurava da se proizvodi stalno pripremaju prema odgovarajućim normama kakvoće. 2. Da bi se pripremili lijekovi stalne kakvoće, potrebno je zadovoljiti sljedeće osnovne zahtjeve:
a. Osoblje treba biti obrazovano i osposobljeno u skladu sa svojom funkcijom. Odgovornosti i sposobnosti trebaju biti jasno definirane.
b. Prostori i oprema trebaju biti u skladu sa svojom namjenom.
c. Svim postupcima osiguranja kakvoće treba procijeniti prikladnost i opisati ih u odgovarajućim uputama i postupcima.

d. Postupke vezane uz pripremu lijekova treba provoditi u skladu s načelima Dobre prakse pripreme kako je opisano u ovim smjernicama. Zapisi trebaju dokazivati da su svi potrebni koraci završeni. Dokumentacija treba pokazivati kompletnu povijest lijeka.
e. Potrebno je procijeniti kakvoću pripremljenih proizvoda. Procjenu je potrebno dokumentirati. Dokumentacija obično obuhvaća:
● Pregled dokumentacije o pripremi
● Usporedbu rezultata ispitivanja, rezultata praćenja okoliša i specifikacija, kada je prikladno
● Procjenu svih odstupanja
f. Lijekovi se ne puštaju u promet prije nego što odgovarajuća nadležna osoba (odnosno odgovorna osoba ili djelatnik odgovoran za puštanje proizvoda u promet) potvrdi da udovoljavaju svim navedenim zahtjevima.
g. Lijekovima, polaznim i pakovnim materijalima potrebno je rukovati i skladištiti ih na način koji osigurava očuvanje kakvoće tijekom cijelog roka valjanosti. Reklamacije na proizvode potrebno je procijeniti, a uzroke neispravnosti u kakvoći potrebno je istražiti. Potrebno je poduzeti odgovarajuće mjere u pogledu pogrešne pripreme te primijeniti mjere opreza koje će spriječiti ponavljanje manjkavosti.
1.4 Provjera kakvoće
Provjera kakvoće je onaj dio Dobre prakse pripreme koji se bavi uzorkovanjem, specifikacijama i ispitivanjem te organiziranjem, dokumentiranjem i odobravanjem postupaka, koji osiguravaju da se provode sva potrebna i odgovarajuća ispitivanja te da polazni i pakovni materijali, kao i međuproizvodi nisu odobreni za uporabu, a gotovi proizvodi pušteni u promet sve dok njihova kakvoća nije ocijenjena zadovoljavajućom.
2. Osoblje 2.1 Načela
Uspostava i održavanje sustava osiguranja kakvoće i ispravna priprema lijekova ovise o osoblju. Zato je potreban dovoljan broj zaposlenika odgovarajuće osposobljenih za izvršavanje svih zadataka. Pojedinačne odgovornosti potrebno je dokumentirati, a pojedinci na koje se odnose trebaju ih jasno razumjeti. Cjelokupno osoblje treba biti upoznato s načelima i smjernicama Dobre prakse pripreme i sustavom osiguranja kakvoće. Osoblju je potrebno osigurati početno i trajno osposobljavanje, koje također treba obuhvaćati neophodne upute o higijeni.
2.2 Opći zahtjevi 1. Odgovorna osoba odgovorna je za kakvoću pripremljenih lijekova i za usklađenost s ovim Načelima i smjernicama. Određene dužnosti mogu se delegirati odgovarajuće osposobljenim osobama (npr. djelatniku za puštanje serije lijeka u promet,

rukovoditelju proizvodnje). U odsutnosti Odgovorne osobe, potrebno je imenovati zamjenika.
2. Ustanova koja priprema proizvod treba imati dovoljan broj odgovarajuće osposobljenog osoblja, tako da se nabava, skladištenje, proizvodnja, provjera i puštanje u promet farmaceutskih proizvoda odgovarajuće i u potpunosti provjeravaju.
3. Razina kompetencija osoblja ovisi o dužnostima i zahtjevima djelatnosti koju organizacija obavlja. 4. Organizacija koja priprema proizvod treba imati organizacijsku shemu koja jasno prikazuje nivoe odgovornosti.
5. Dužnosti i odgovornosti cijelog osoblja, uključujući sve zamjenike, trebaju biti definirane opisom poslova.
2.3 Osposobljavanje i trajna izobrazba 1. Novo osoblje treba nakon zapošljavanja i dalje trajno osposobljavati na svim područjima koja su potrebna za ispunjavanje njihovih dužnosti. 2. Osoblju treba omogućiti trajnu, dokumentiranu izobrazbu, a može se odvijati unutar ili izvan organizacije.
2.4 Higijena 1. Potrebno je osigurati upute za higijensko ponašanje te za prikladno odijevanje osoblja. Potrebno je provesti izobrazbu osoblja u skladu s time. Odjeća treba odgovarati radnjama koje se obavljaju. 2. Rizik od onečišćenja proizvoda od strane osoblja potrebno je odgovarajućim metodama smanjiti na minimum. Osoblje treba obavijestiti rukovoditelja proizvodnje o zaraznim bolestima i otvorenim ranama na izloženoj površini tijela. Rukovoditelj proizvodnje odlučuje o prikladnosti dotične osobe za obavljanje radnji u proizvodnim prostorima ili o posebnim zaštitnim mjerama koje je potrebno poduzeti da bi se izbjeglo onečišćenje proizvoda. Ako odgovarajuća zaštita nije moguća, osobi ne treba dozvoliti sudjelovanje u radnjama pripreme lijeka.
3. Potrebno je osigurati da nema rizika od onečišćenja ni za osoblje ni za proizvode. U proizvodnim prostorima potrebno je zabraniti uzimanje hrane, pića ili pušenje. 4. Potrebno je poduzeti odgovarajuće mjere opreza kako bi se spriječilo onečišćenje proizvoda putem kontakta s radnikom. Dodatne zaštitne mjere (npr. sanitacija ruku, nošenje rukavica) potrebno je poduzeti za lijekove kod kojih je povećani rizik od mikrobiološkog onečišćenja.

3. Prostori i oprema 3.1 Načela
Prostori i oprema trebaju biti prikladni za planirane radnje i ne smiju predstavljati nikakvu opasnost za kakvoću proizvoda.
3.2 Opći zahtjevi 1. Prostori i oprema trebaju biti odgovarajuće dizajnirani, izgrađeni, korišteni, održavani i obnavljani, kako bi se osigurala njihova prikladnost za planirane radnje i rizik od pogrešaka smanjio na minimum. Kapacitet treba biti dovoljan da omogući logičan slijed postupaka i prikladno razdvajanje radnji. 2. Potrebno je koristiti prikladno dizajnirane prostore i opremu, kao i odgovarajuće tehnike rada da bi se smanjio rizik od onečišćenje, primjerice unakrsnom kontaminacijom ili nakupljanjem prašine ili nečistoća. Dizajn treba biti takav da omogućuje temeljito čišćenje. Posebna pažnja potrebna je kod uzimanja uzoraka ili čišćenja opreme , a po potrebi kod dezinfekcije nakon popravaka ili održavanja. 3. Potrebno je poduzeti odgovarajuće mjere za zaštitu od ulaza insekata i drugih životinja (kontrola štetočina).
4. Radnje pranja i čišćenja same po sebi ne bi trebale biti izvor onečišćenja.
5. Pristup proizvodnim, skladišnim i prostorima za provjeru kakvoće potrebno je omogućiti samo ovlaštenom osoblju.
6. Potrebno je definirati i pratiti okolišne uvjete (temperatura, vlaga, svjetlo) tijekom proizvodnje, provjere kakvoće i skladištenja (uključujući hladno skladištenje), i, po potrebi, provjeravati. Rezultate praćenja potrebno je dokumentirati, procjenjivati i čuvati. Kad uvjeti izađu izvan određenih granica, potrebno je poduzeti odgovarajuće popravne radnje. 7. Svi prostori trebaju biti čisti, uredni i dobro osvijetljeni.
3.3 Proizvodni prostori 1. Proizvodni prostori trebaju omogućavati odgovarajuće odvajanje od drugih radnji.
2. Potrebno je razmotriti odvajanje prostora za posebne farmaceutske oblike (npr. postrojenja za suhu i vlažnu proizvodnju). Ako odvajanje prostora za posebne farmaceutske oblike nije moguće, potrebno je provesti dokumentiranu procjenu rizika i poduzeti odgovarajuće mjere prije istovremenog rukovanja različitim farmaceutskim oblicima.
3. Potrebno je osigurati posebne prostorije za opasne proizvode, npr. citostatike, peniciline, proizvode biološkog podrijetla radiofarmaceutike, pripravke iz krvi. U

iznimnim slučajevima može se prihvatiti kampanjski način rada, uz uvjet da su poduzete posebne mjere opreza i provedene sve potrebne procjene rizika. 4. Materijale i proizvode potrebno je skladištiti i njima rukovati na takav način da je rizik od miješanja različitih proizvoda ili njihovih sastojaka minimalan, da se izbjegne unakrsna kontaminacija i da se smanji rizik od propuštanja ili neispravnog obavljanja nekog koraka postupka.
5. Prostori za vaganje i uzorkovanje proizvoda trebaju biti dovoljno odijeljeni od proizvodnih prostora kako bi se izbjegla unakrsna kontaminacija.
3.4 Skladišni prostori 1. Skladišni prostori trebaju biti dostatni za uredno skladištenje raznih vrsta materijala i proizvoda. Primjeri su: polazni i pakovni materijali, poluproizvodi, međuproizvodi i gotovi proizvodi, proizvodi u karanteni, proizvodi pušteni u promet, odbijeni, vraćeni ili proizvodi povučeni iz prometa.
2. Polazne i pakovne materijale obično je potrebno čuvati izvan proizvodnog prostora, osim u slučaju kad su odgovarajuće odvojeni.
3. Odbijene, vraćene i povučene, te materijale i proizvode u karanteni potrebno je čuvati u odvojenim prostorima i jasno označiti njihov status kao takav.
4. Potrebno je navesti i pratiti uvjete skladištenja (npr. temperatura, relativna vlaga) neophodne za sprječavanje štetnog djelovanja na kakvoću materijala ili proizvoda. Prikladna provjera treba omogućiti da se svi dijelovi odgovarajućeg skladišnog prostora održavaju unutar navedenih uvjeta. Skladišne prostore potrebno je opremiti uređajima za bilježenje ili drugim uređajima za praćenje koji će pokazati kad uvjeti izađu izvan navedenih granica, tako da se događaji izvan specifikacija mogu procijeniti i poduzeti odgovarajuće mjere.
3.5 Prostori za provjeru kakvoće
Radnje provjere kakvoće obično se provode u namjenskim prostorima. Ako se to ne može postići, potrebno je poduzeti korake kojima bi se izbjegle greške i onečišćenja.
3.6 Pomoćni prostori 1. Prostorije za odmor i okrjepu potrebno je odvojiti od drugih prostora.
2. Sanitarni čvorovi i prostorije za presvlačenje i pranje trebaju biti lako dostupne i primjerene broju korisnika. Sanitarni čvorovi ne smiju imati izravan pristup iz proizvodnih ili skladišnih prostora.

3.7 Oprema 1. Opremu za pripremu potrebno je dizajnirati, smjestiti i održavati na način koji odgovara njezinoj namjeni. 2. Oprema treba biti izvedena na način koji omogućava lako i temeljito čišćenje. Opremu je potrebno čuvati u čistim i suhim uvjetima 3. Oprema za mjerenje, vaganje i kontrolu treba imati potrebnu preciznost: potrebno ju je umjeravati kao i provjeravati ispravnost funkcioniranja te umjeravanje ponavljati u odgovarajućim intervalima.
4. Neispravnu opremu potrebno je ukloniti iz proizvodnih prostora i prostora za provjeru kakvoće ili je barem jasno označiti kao neispravnu.
4. Dokumentacija 4.1 Načela
Dobra dokumentacija u papirnatom ili elektroničkom obliku ključan je dio sustava osiguranja kakvoće. Lako čitljiva i jasno napisana dokumentacija sprječava pogreške u usmenoj komunikaciji i omogućuje sljedivost pripremljenog lijeka.
4.2 Opći zahtjevi 1. Podatke relevantne za kakvoću, uključujući procjene rizika, potrebno je dokumentirati. 2. Izraz dokumentacija sažima poimenično:
a. Specifikacije Trebaju postojati prikladno odobrene i datirane specifikacije za polazne materijale, pakovne materijale i gotove proizvode; kad je primjereno, one također trebaju biti raspoložive i za poluproizvode, međuproizvode ili proizvode na veliko.
b. Upute za određeni proizvod Trebaju biti raspoložive upute za izradu, pakiranje, provjeru kakvoće i puštanje u promet koje opisuju sastav, navodeći sve korištene polazne i druge materijale te navode sve radnje izrade i pakiranja, kao i ispitivanja u svrhu provjere kakvoće te radnje puštanja u promet.
c. Zapise Dokumenti o izradi, pakiranju i provjeri kakvoće koji bilježe činjenice
povijesti lijeka tijekom pripreme koje su povezane s kakvoćom.

d. Opću dokumentaciju o postupcima i dodatnu dokumentaciju Upute za provođenje normiranih radnji i drugi dokazi koji dokumentiraju povijest i kakvoću lijeka. Primjeri su: opis primitka robe, uzorkovanja, poredbenih uzoraka pripremljenih proizvoda, ispitivanja, puštanja, odbijanja, umjeravanja, čišćenja, dezinficiranja, provođenja higijenskih radnji, osposobljavanja osoblja i rukovanja opremom.
3. Sve specifikacije, upute i postupci trebaju biti odobreni, potpisani i datirane od strane odgovorne osobe ili osobe koju imenuje odgovorna osoba.
4. Svi pisani dokumenti trebaju biti čitljivi, jasni, jednoznačni i ažurirani. Elektroničke zapise potrebno je odgovarajuće zaštititi od neovlaštenih promjena i gubitka podataka. Čitljivost elektronički čuvanih podataka potrebno je osigurati tijekom cijelog razdoblja njihova čuvanja. 5. Ukupnost ovih dokumenata treba omogućiti potpunu sljedivost postupka pripreme lijeka.
6. Svaku izmjenu dokumenta potrebno je potpisati i datirati. Izmjenu je potrebno provesti na takav način da omogući čitanje prvotne informacije. Razlog izmjene treba biti očigledan. Jednakovrijedne mjere potrebno je poduzeti i za elektroničke zapise.
7. Zapise je potrebno čuvati dovoljno dugo da to bude u skladu sa zahtjevima nacionalnog zakonodavstva. U svakom slučaju, zapise treba čuvati najmanje jednu godinu nakon isteka roka valjanosti odgovarajućeg gotovog proizvoda. Postupke i upute za pripremu (uključujući recepture) potrebno je čuvati najmanje pet godina nakon prestanka njihove upotrebe.
4.3 Dokumentacija za magistralne pripravke 1. Za magistralne pripravke, potrebno je navesti najmanje naziv i jačinu pripravka te rok valjanosti. Potrebno je koristiti odobrene polazne materijale za koje treba biti dostupna odgovarajuća dokumentacija.
Recept može sadržavati upute za izradu i pakiranje. Ako nema posebnih uputa, potrebno je koristiti opće upute za izradu svake vrste pripravka, npr. priprema kapsula, masti.
3. Potrebno je sačuvati zapis koji prikazuje glavne postupke izrade i pakiranja, uključujući ime osobe odgovorne za svaki postupak. Ovaj zapis treba biti usklađen s poglavljem 4.4.3, kad je primjenjivo.
4.4 Dokumentacija za galenske pripravke 1. Za lijekove koji su obuhvaćeni područjem primjene ovih smjernica, obično ne postoji registracijski dosje koji su odobrila regulatorna tijela. Stoga je potrebno sačuvati dokumentaciju specifičnu za pripravak (dosje pripravka). To se odnosi na specifikacije, upute i zapise.

2. Da bi se uspostavila specifikacija novog pripravka, uputa i postupaka, potrebno je prije početka izrade pripravka provesti farmaceutsku procjenu opravdanosti terapijske primjene, navesti podatke o neškodljivosti, toksičnosti, biofarmaceutskim svojstvima, stabilnosti i podatke o razvoju pripravka.
3. Dosje pripravka, ako se isti koristi opetovano ili tijekom duljih vremenskih razdoblja također treba sadržavati podatke o ispitivanju kakvoće, podatke o stabilnosti, podatke o validaciji i sl.
4.4.1 Specifikacije
1. Trebaju postojati odobrene specifikacije (primjerice, poveznice na farmakopeju) za polazni i pakovni materijal, kao i za poluproizvode, međuproizvode i gotove pripravke. 2. Specifikacije polaznih materijala i, kad je primjenjivo, pakovnih materijala, trebaju sadržavati:
a. Naziv (uključujući poveznicu na farmakopeju, kad je primjenjivo)
b. Opis
c. Postupke za uzorkovanje i ispitivanje s poveznicama
d. Kvalitativne i kvantitativne zahtjeve s granicama prihvatljivosti
e. Kad je primjenjivo, uvjete skladištenja i mjera opreza
f. Rok valjanosti
3. Specifikacije za poluproizvode, međuproizvode ili gotove pripravke trebaju sadržavati:
a. Naziv
b. Opis farmaceutskog oblika i jačinu
c. Sastav
d. Opis pakiranja
e. Upute za uzorkovanje i ispitivanje ili poveznicu na postupke
f. Kvalitativne i kvantitativne zahtjeve s granicama prihvatljivosti
g. Uvjete skladištenja, mikrobiološke zahtjeve i bilo kakve posebne mjere opreza tijekom rukovanja, kad je primjenjivo
h. Rok valjanosti
4.4.2 Upute Upute za izradu
1. Upute za izradu trebaju sadržavati:
a. Naziv proizvoda
b. Opis farmaceutskog oblika i jačinu

c. Veličinu serije
d. Vrstu i količinu svih korištenih polaznih materijala e. Očekivano iskorištenje poluproizvoda, međuproizvoda ili gotovog pripravka
f. Detaljne opise svih koraka izrade
g. Upute za provjere tijekom izrade s granicama prihvatljivosti
h. Uvjete skladištenja (također za poluproizvode i međuproizvode) i mjere opreza, kad je primjenjivo.
Upute za pakiranje
2. Upute za pakiranje trebaju sadržavati:
a. Naziv proizvoda
b. Farmaceutski oblik i jačinu
c. Veličinu pakiranja
d. Tekst za označavanje ili osnovnu naljepnicu
e. Popis svih potrebnih pakovnih materijala, s podacima o vrsti, specifikaciji, veličini i količini
f. Detaljne opise svih koraka u pakiranju
g. Upute za provjere tijekom pakiranja s granicama prihvatljivosti
h. Uvjete skladištenja (također za poluproizvode i međuproizvode) i mjere opreza, kad je primjenjivo
4.4.3 Zapisi Zapisi o izradi i pakiranju
1. Zapisi o izradi i pakiranju trebaju sadržavati:
a. Kvalitativne i kvantitativne informacije o svim korištenim materijalima kao što su serijski broj korištenog materijala ili druge poveznice koje omogućuju sljedivost do daljnjih dokumenata koji se odnose na kakvoću (npr. pripravak, broj analize, broj certifikata)
b. Identitet pripravka (uključujući serijski broj i sastav) i datum pripreme
c. Informacije o svim radnjama i zapažanjima, kao što je dokumentacija o čišćenju, raščišćavanju linije, vaganju, iskorištenjima u međukoracima, očitanjima i izračunima, kao i o uzorkovanju
d. Zapise procesnih provjera određenih serija i dobivene rezultate
e. Inicijale ili potpise odgovornih izvršitelja značajnih postupaka izrade i provjere
f. Sva odstupanja od odobrenih uputa za izradu
g. Iskorištenje gotovog pripravaka
h. Primjerak korištene naljepnice
i. Usuglašavanje konačnog broja naljepnica
j. Kad je primjenjivo, ime pacijenta ili identitet
2. Zapise o izradi na kraju treba ocijeniti i odobriti Odgovorna osoba ili djelatnik odgovoran za puštanje serije lijeka u promet, potpisom uz naznaku datuma. Zapisi provjere kakvoće

3. Zapisi provjere kakvoće trebaju sadržavati:
a. Naziv pripravka
b. Farmaceutski oblik i jačinu
c. Kontrolni /serijski broj
d. Ime osobe odgovorne za pripremu lijeka ili naziv dobavljača
e. Metodu ispitivanja; sva odstupanja od metode potrebno je opravdati
f. Rezultate ispitivanja; certifikat analize od osobe odgovorne za pripremu lijeka ili dobavljača koja uključuje datum ispitivanja, kad je primjenjivo
g. Rok valjanosti polaznog materijala
h. Datum ispitivanja
i. Inicijale osobe koja provodi ispitivanje
j. Odluku o puštanju u promet ili odbijanju s inicijalima Odgovorne osobe ili djelatnika odgovornog za puštanje serije lijeka u promet
4.5 Opći postupci i dodatna dokumentacija 1. Posebni pisani postupci trebaju biti dostupni za:
a. Zaprimanje, uzorkovanje i puštanje u proizvodnju polaznih i pakovnih
materijala
b. puštanje u daljnju fazu izrade ili u promet i odbijanje
poluproizvoda/međuproizvoda i gotovih pripravaka, uključujući hitno puštanje u
promet
c. Povlačenje gotovih pripravaka iz prometa
d. Umjeravanje i kvalifikaciju opreme (npr. autoklava, sterilizatora suhom
toplinom, termometara, vaga, opreme za određivanje tališta)
e. Validaciju procesa
f. Čišćenje, dezinfekciju i održavanje opreme (npr. opreme za
demineralizaciju vode, opreme za destilaciju, hladnjaka) i prostora
g. Osposobljavanje osoblja (npr. u odnosu na ostvarivanje higijenskih mjera)
h. Rukovanje opremom, kad je primjenjivo
i. Postupke za nadziranje, uključujući praćenje trendova
j. Postupak o radnjama koje je potrebno poduzeti u slučaju odstupanja i
reklamacija
k. Samoinspekcije
2. Provođenje ovih radnji treba zabilježiti primjerice u dokumentaciju o seriji, na
posebnom formularu ili u laboratorijskom dnevniku.
5. Proizvodnja 5.1 Načela

Proizvodnim radnjama potrebno je osigurati zahtijevanu kakvoću pripravaka.
Proizvodnju trebaju obavljati i nadzirati odgovarajuće osposobljene osobe.
5.2 Opći zahtjevi 1. Proizvodnju treba obavljati osposobljeno osoblje. 2. Polazni materijali trebaju biti odobreni prije upotrebe. Identitet, težinu i volumen svih polaznih materijala treba nezavisno provjeriti druga osoba ili validirani kompjutorizirani sustav (npr. provjera bar koda). 3. Osim magistralnih pripravaka, proizvodnja se treba odvijati na temelju pisanih uputa u kojima su detaljno izloženi svi odgovarajući postupci. 4. Potrebno je poduzeti sve potrebne tehničke i organizacijske mjere da bi se izbjegle zamjene. 5. Potrebno je zabilježiti sve provedene postupke.
6. Oprema i materijal koji se koriste u svim postupcima radnjama trebaju biti prikladni za svoju namjenu.
7. Sve pripravke i materijale u svim postupcima pripreme treba zaštititi od mikrobiološkog i drugih onečišćenja.
8. U svako doba tijekom izrade svi pripravci trebaju biti identificirani. Naljepnice ili oznake na spremnicima i opremi trebaju biti jasne i nedvosmislene. 9. U svako doba tijekom izrade operativni status prostora i opreme (npr. očišćen, u upotrebi), prostorija i opreme treba biti jasan.
5.3 Sprječavanje unakrsne kontaminacije
Potrebno je poduzeti neophodne tehničke i organizacijske mjere da bi se izbjegla unakrsna kontaminacija.
5.4 Procjena rizika pripravka i dokazivanje prikladnosti 1. Mogući rizik od štete po zdravlje u slučaju grešaka (npr. manjkavosti u kakvoći) različit je kod različitih grupa pripravaka, stoga je potrebno da ga odgovarajuće osposobljena osoba procijeni i dokumentira. Na mogući rizik uglavnom utječe:
a) Vjerojatnost pojavljivanja greške.
Primjeri povezanih faktora rizika su:
● Niska koncentracija neotopljenog djelatnog sastojka (rizik od nepravilnog doziranja zbog nehomogenosti)
● Velika podložnost rastu mikroorganizama (rizik od mikrobiološkog rasta)

● Dulja razdoblja skladištenja ili upotrebe (rizik od kemijske razgradnje ili mikrobiološkog rasta) ● Vrsta objekta u kojoj se pripravak izrađuje (rizik od onečišćenja u slučaju nekontroliranog radnog okoliša) ● Loša tehnika rada (rizik od zamjena ili onečišćenja)
b) Vjerojatnost otkrivanja moguće pogreške.
Primjeri povezanih faktora rizika su:
● Nedostatak kontrolnih mehanizama, npr. praćenja, procesnih i završnih provjera (rizik od neuočavanja pogrešaka ili manjkavosti)
c) Posljedice moguće greške (rizik po zdravlje). Primjeri povezanih faktora rizika su:
● Opseg radnji (rizik od zahvaćanja većeg broja pacijenata zbog široke primjene) ● Grupe izrađenih pripravaka i put primjene, npr. sterilni pripravci za primjenu u venu (rizik zbog sustavnih posljedica mikrobiološkog onečišćenja)
Više informacija o provođenju procjene rizika moguće je naći u ICH-ovim smjernicama Q9 (Upravljanje rizicima u kakvoći).
2. Potrebno je poduzeti mjere neophodne za odgovarajuće rješavanje prepoznatog mogućeg rizika i za osiguranje tražene kakvoće.
3. Potreba za dokazivanje prikladnosti poduzetih mjera ovisi o prepoznatom mogućem i riziku i potrebno ga je procijeniti. 4. Kad se dokazivanje prikladnosti pokaže nužnim, potrebno je provesti odgovarajuće kvalifikacije i validacije. Načela kvalifikacije i validacije opisana su u Dodatku 15 PIC/S dokumenta PE 009. Ako se isti postupak primjenjuje na niz pripravaka (npr. aseptičko doziranje sličnih pojedinačnih pripravaka), validaciji se može pristupiti kroz provođenje jednog ispitivanja za najgori mogući slučaj, uvažavajući odgovarajuće kriterije za sve srodne pripravke. Takav pristup u literaturi naziva se “bracketing”. 5. Utjecaj promjena na kakvoću pripravka kod već kvalificiranih objekata prostorija i opreme, utjecaj izmjena u sastavu ili u kakvoći polaznih materijala i utjecaj promjena kod već validiranih procesa treba procijeniti odgovarajuće osposobljena osoba i to u odnosu na potrebu i opseg ponovne kvalifikacije ili ponovne validacije i to prije nego se izmjena provede. 6. Prikladnost postojećih validacija potrebno je provjeravati u odgovarajućim razdobljima, u skladu s unaprijed utvrđenim postupkom. Proces je potrebno ponovno validirati ako validacija više nije prihvatljiva - primjerice zbog niza malih izmjena koje se odvojeno ne bi smatrale relevantnima, ali kombinirane dobivaju na značaju.
5.5 Polazni materijali

1. Polazni materijali koji se koriste za izradu pripravaka trebaju odgovarati zahtjevima odgovarajućih specifikacija. 2. Polazne materijale treba skladištiti u originalnim spremnicima. Ako se prenose u druge spremnike, isti trebaju biti čisti i označeni svim informacijama specifičnima za tu seriju. U tom pogledu, kakvoća treba biti zajamčena tijekom cijelog razdoblja uporabe. Zabranjeno je miješanje različitih serija. 3. Za polazne materijale s kratkim rokom valjanosti potrebno je naznačiti datum prvog otvaranja.
4. Polazni materijal kojem je istekao rok valjanosti ili je zastario treba uništiti, a njegovo zbrinjavanje zabilježiti.
5.6 Postupci izrade 1. Prije početka bilo kakvog postupka izrade, važno je osigurati (i dokumentirati) da su radni prostori i oprema čisti i da ne sadrže nikakve polazne materijale i proizvode koji nisu potrebni za trenutni postupak te da sva oprema funkcionira na zadovoljavajući način. Moguće poteškoće potrebno je prijaviti ključnom osoblju. 2. Poluproizvode i međuproizvode je potrebno skladištiti u prikladnim uvjetima i nedvosmisleno ih označiti.
3. Suvišan materijal preostao nakon proizvodnje obično treba uništiti. Na zalihu ga treba
vraćati samo nakon pažljive provjere. O tome je potrebno voditi bilješke.
5.7 Pakovni materijal 1. Pakovni materijal smije se koristiti samo ako je prikladan za dotičnu namjenu. To znači da spremnik ili sustav za zatvaranje ne smiju svojim negativnim utjecajem prouzročiti rizik za kakvoću lijekova. Kad je primjenjivo, pakovni materijal koji se koristi treba omogućavati antimikrobno djelovanje i dostatnu zaštitu od vanjskih utjecaja i mogućeg onečišćenja. 2 Naljepnice/signature trebaju udovoljavati zahtjevima nacionalnih propisa i obično uključuju sljedeće informacije:
a. Naziv pripravka
b. Farmaceutski oblik
c. Djelatni farmaceutski sastojak/sastojke i količinu/količine
d. Sadržaj (količina, npr. grami, broj tableta)
e. Serijski/kontrolni broj
f. Rok valjanosti i, po potrebi, datum primjene
g. Priprematelj
3. Pakovni materijal kojem je istekao rok valjanosti ili je zastario treba uništiti, a njegovo zbrinjavanje zabilježiti.
5.8 Pakiranje

1. Spremnici trebaju biti čisti prije uporabe. 2. Da bi se isključile zamjene ili pogrešno označivanje, označivanje treba uslijediti odmah nakon punjenja i zatvaranja. U suprotnom, potrebno je provesti odgovarajuće sigurnosne mjere
5.9 Odbijeni, prerađeni i vraćeni materijali i pripravci 1. Odbijene materijale i pripravke potrebno je kao takve označiti i skladištiti u odvojenim prostorima.
2. Prerada i dorada nesukladnih pripravaka mogu se provoditi samo iznimno kad ih odobri Odgovorna osoba. Potrebno ih je provesti u skladu s pisanim uputama i o tome voditi zapise. Potrebno je provesti procjenu rizika kojom će se procijeniti moguće posljedice na kakvoću i rok valjanosti pripravka, kao i potrebe za dodatnim ispitivanjima. 3. Odgovorna osoba ili djelatnik odgovoran za puštanje serije lijeka u promet treba odlučiti hoće li se pustiti u promet prerađen ili dorađen pripravak, nakon što procijeni svu odgovarajuću dokumentaciju (i rezultate dodatnih ispitivanja).
4. Izdane pripravke koji su vraćeni i koji su bili izvan kontrole organizacije koja ih je izradila potrebno je uništiti, osim ako ne postoji nikakva sumnja u njihovu kakvoću. U iznimnim slučajevima može se razmotriti njihova prerada ili dorada nakon što ih je, u skladu s pisanim postupkom, kritički procijenila Odgovorna osoba ili djelatnik odgovoran za puštanje serije lijeka u promet. Ako se pojavi bilo kakva sumnja u kakvoću pripravka, isti se ne smije smatrati prikladnim za ponovno izdavanje ili ponovnu uporabu. Svaku poduzetu radnju potrebno je odgovarajuće zabilježiti.
6. Provjera kakvoće 6.1 Načela 1. Provjera kakvoće osigurava da su zadovoljeni svi zahtjevi kakvoće.
2. Pobliže rečeno, ona osigurava provođenje potrebnih ispitivanja te puštanje proizvoda u promet samo ako su u skladu sa zahtjevima kakvoće.
3. Obujam ispitivanja kakvoće potrebno je definirati na temelju procjene rizika (usporediti s poglavljem 5.4). i uvažavanja informacija o stabilnosti i fizičkim svojstvima proizvoda. 4. Provjera kakvoće i radnje puštanja u promet trebaju biti neovisne o radnjama pripreme.
6.2 Opći zahtjevi

1. Oprema za ispitivanje treba biti u skladu s namjenom. 2. Sve radnje potrebno je obavljati u skladu s definiranim postupcima i zabilježiti ih.
3. Bilješke o ispitivanjima potrebno je čuvati najmanje jednu godinu nakon isteka roka valjanosti polaznih materijala ili gotovog proizvoda, ovisno o tome koji rok je dulji.
6.3 Uzorkovanje 1. Uzorci uzeti za analizu trebaju predstavljati reprezentativni dio materijala koji se ispituje. 2. Kad se gotovi proizvodi podvrgnu analitičkim provjerama, potrebno je zadržati prikladan broj analitičkih poredbenih uzoraka tijekom odgovarajućeg vremenskog razdoblja nakon isteka roka valjanosti.
6.4 Ispitivanje Ispitivanje sirovina
1. Zahtjevi kakvoće i ispitivanja trebaju biti u skladu s važećom farmakopejom. Ako farmakopeja ne sadržava odgovarajuću monografiju, mogu se primijeniti druge farmakopeje. U suprotnom je potrebno koristiti formularije ili profesionalne norme koje bi mogla priznati nadležna tijela. Ako ne postoje službeno priznate norme, normu je potrebno postaviti na temelju lokalnih istraživanja ili na temelju specijalizirane literature, u kojem je slučaju metodu potrebno validirati.
2. Procjenom rizika u svrhu definiranja ispitivanja sirovina potrebno je uzeti u obzir da je potvrda identiteta sadržaja svakog spremnika od posebne važnosti. U svakom slučaju potrebno je provjeriti naljepnicu i nepovredivost otvaranja svakog spremnika. Na certifikate analiza serija potrebno je pozvati se samo u slučajevima kad je pouzdanost proizvođača ili dobavljača koji izdaju certifikat provjerena. 3. Gotovi proizvodi koji su pušteni u promet, a koji se koriste kao polazni materijali obično se ne ispituju.
Ispitivanje gotovih proizvoda
4. Procjenom rizika u svrhu definiranja ispitivanja gotovih proizvoda posebno je potrebno uvažavati svojstva proizvoda, upotrebu proizvoda, kao i rizike koji se povezuju s njegovom pripremom. 5. Obično se ne provodi ispitivanje kakvoće magistralno pripremljenih pripravaka. Laboratorijski reagensi koji se koriste za ispitivanje
6. Laboratorijske reagense koji se pripremaju za zalihu potrebno je označiti datumom pripreme i rokom valjanosti.

6.5 Puštanje u promet 1. Odgovorna osoba u konačnici je odgovorna za kakvoću pripremljenih i u promet puštenih pripravaka. Stvarno puštanje u promet može se delegirati drugoj odgovarajuće osposobljenoj osobi (odnosno djelatniku odgovornom za puštanje serije u promet).
2. Postupak puštanja serije u promet treba obuhvaćati provjeru je li pripravak pripremljen u skladu s važećim postupcima i načelima Dobre prakse za pripremu, opisanim u ovim smjernicama, te udovoljava li zahtjevima važećih specifikacija.
7. Ugovaranje poslova 7.1 Načela 1. Ovisno o lokalnim okolnostima i nacionalnom zakonodavstvu, posao koji organizacija za pružanje zdravstvene skrbi ugovara s vanjskim ugovarateljem može obuhvaćati radnje koje se izravno tiču pripreme, kao što su izrada, pakiranje ili provjera kakvoće, ali i usluge koje nisu izravno povezane s pripremanjem, ali koje ipak imaju značajan učinak na kakvoću izrađenih pripravaka ili na bilo kakve rezultate dobivene provjerom kakvoće. Takve usluge koje se često ugovaraju s vanjskim ugovarateljem s nekim drugim odjelom ili organizacijom, mogu obuhvaćati:
a. održavanje sustava dobave zraka, sustava za vodu ili drugih pomoćnih sustava
b. održavanje ključne opreme kao što su izolatori, kabineti s laminarnim strujanjem zraka, sterilizatori, vage
c. sterilizaciju sastavnica i potrošne robe kao što su perači poda, odjeća, posuđe
d. usluge nadzora okoliša
e. dobavljanje mikrobiološke potrošne robe (npr. taložne pločice)
f. rukovanje otpadom
g. kontrolu štetočina
2. Svaki posao koji može utjecati na kakvoću izrađenih pripravaka , a koji se ugovara s vanjskim ugovarateljem treba biti uređen pisanim tehničkim ugovorom.
3. U hitnom slučaju, pojedini magistralno izrađeni pripravak može se pribaviti bez pisanog ugovora. Ovo treba biti samo iznimna pojava.

7.2 Opći zahtjevi 1. Ugovorom (sporazumom) o razini tehničke usluge potrebno je precizirati detalje posla koji će se obaviti, specifikaciju koju je potrebno zadovoljiti i odgovornosti svake strane.
2. Ugovor trebaju odobriti i potpisati primatelj ugovora (odnosno ugovaratelj treće strane) i odgovorna osoba davatelja ugovora.
7.3 Davatelj ugovora 1. Davatelj ugovora treba u ugovoru točno specificirati koju razinu usluge traži i prema kojoj specifikaciji.
2. Davatelj ugovora treba se uvjeriti da je primatelj ugovora odgovarajuće osposobljen i – po potrebi – ovlašten za uspješno obavljanje usluge. Stupanj do kojeg se primatelji ugovora provjeravaju potrebno je definirati na temelju procjene rizika. Procjena rizika treba obuhvaćati postojeće dokaze da primatelj ugovora udovoljava zahtjevima ugovora i zakonskim zahtjevima (npr. Dobre prakse pripreme). Provjere primatelja ugovora treba provesti Odgovorna osoba ili netko koga imenuje Odgovorna osoba.
3. Sva izvješća koja primatelj ugovora dostavi, u kojima se sažimaju rezultati ili obavljeni posao, davatelj ugovora treba formalno pregledati i prihvatiti kao usklađene s potrebnim specifikacijama. Ovaj pregled i formalno prihvaćanje trebaju biti detaljno opisani u postupcima sustava kakvoće, a postupcima je potrebno propisati tko je ovlašten za pregled i prihvaćanje ovih izvješća.
7.4 Primatelj ugovora 1. Svaki je posao potrebno obaviti u skladu s ugovorom.
2. O svakoj usluzi ili rezultatima koji nisu u skladu s potrebnom specifikacijom potrebno je obavijestiti Odgovornu osobu davatelja ugovora. 3. Primatelj ugovora ne smije proslijediti trećoj strani niti jedan dio posla koji mu je prema ugovoru povjeren bez prethodne ocjene i odobrenja takvih dogovora od strane davatelja ugovora. Dogovori napravljeni između primatelja ugovora i neke treće strane trebaju osigurati da su priprema i ključne informacije raspoloživi na isti način kao i između originalnog davatelja ugovora i primatelja ugovora.
8. Reklamacije i povlačenje pripravka 8.1 Načela
Sve greške, manjkavosti, reklamacije i drugi znakovi problema s kakvoćom potrebno je pažljivo pregledati u skladu s pisanim postupkom. Da bi bilo moguće hitno i učinkovito povlačenje izrađenih pripravaka koji imaju ozbiljne nedostatke, potrebno je razviti odgovarajući postupak.

8.2 Problemi s kakvoćom 1. Greške, manjkavosti, reklamacije i druge znakove koji ukazuju na probleme s kakvoćom potrebno je istražiti. Potrebno je uspostaviti odgovarajuće mjere koje osiguravaju poduzimanje učinkovite popravne radnje. Uzrok i sadržaj nedostataka, poduzete popravne mjere i obavljena ispitivanja treba dokumentirati pisanim putem i dodati u zapise o izradi pripravka. 2. Kod prijave manjkavosti pripravka, potrebno je provjeriti pojavnost istog problema i kod ostalih pripravaka i obustaviti isporuku dok se problem u potpunosti ne istraži.
8.3 Povlačenje pripravka 1. Kada su nedostaci potencijalno štetni za zdravlje, potrebno je odmah započeti povlačenje pripravka i smjesta o tome obavijestiti nadležno tijelo. 2. Treba postojati pisani postupak za povlačenje.
3. Povučene pripravke potrebno je kao takve označiti i skladištiti u odvojenim prostorima. Potrebno je osigurati da se isti ne mogu greškom isporučiti.
4. Tijek i napredovanje povlačenja potrebno je bilježiti. Potrebno je izdati konačni izvještaj u kojem se daje obračun isporučenih i povučenih količina pripravka. Izvještaj čuvati pet godina, osim ako nacionalni propisi ne zahtijevaju drugačije vrijeme čuvanja.
9. Samoinspekcije 9.1 Načela 1. Sustav osiguranja kakvoće, uključujući pitanja osoblja, prostora, opreme, dokumentacije, izrade, provjere kakvoće, distribucije pripravaka, dogovora za rješavanje reklamacija i poslova ugovorenih s vanjskim ugovarateljem, potrebno je pregledavati u redovnim intervalima kako bi se provjerila njihova usklađenost s načelima Dobre prakse pripreme opisane u ovim smjernicama.
2. Potrebno je utvrditi program samoinspekcija koji uvažava vrstu i složenost obavljanih radnji. Program obuhvaća i godišnji plan samoinspekcija s bilješkama i dokazima da se poduzimaju odgovarajuće popravne radnje.
3. Samoinspekcije trebaju na neovisan i detaljan način provoditi određene odgovarajuće osposobljene osobe.
__________________

DODATAK 1
SMJERNICE ZA KORIŠTENJE NORMI POTREBNIH ZA PRIPREMU
STERILNIH PRIPRAVAKA
Uvod
1. Sterilna izrada pripravaka obuhvaća:
● izrada pripravaka kod kojih se sterilizacija provodi na kraju izrade
● Aseptička izrada pripravaka
2. Ovaj Dodatak dodaje se glavnom dijelu ovih smjernica. U njemu se navode dodatna pravila za izradu sterilnih pripravaka. Poglavlja ovog Dodatka u početku navode pravila važeća za sve vrste gore navedenih sterilnih pripravaka, nakon čega, po potrebi, slijede poglavlja koja sadrže posebne smjernice primjenjive na samo jednu grupu pripravaka.
3. Sterilni pripravci smatraju se grupom proizvoda visokog rizika, primjerice zbog:
● Povećane mogućnosti mikrobiološkog onečišćenja pripravaka izrađenih u nekontroliranim uvjetima;
● Veće količine mikrobioloških onečišćenja u nekontroliranim uvjetima;
● Povećanog rizika od sustavne infekcije povezane s pripravcimaizrađenim u nekontroliranim uvjetima; ● Povećanog rizika od medikacijskih grešaka kad se injekcije pripremaju bez stručnog ljekarničkog nadzora.
Izrada se treba odvijati u dobro kontroliranim uvjetima koristeći dobro utvrđene postupke u sustavu osiguranja kakvoće. Tako se znatno smanjuje rizik povezan s ovim pripravcima.
4. Kod pojedinačnih vrsta pripravaka primjeri za njihove specifične faktore rizika su:
Citotoksici i radiofarmaci: Visoka razina opasnosti za rukovatelja koji izrađuje pripravak te visoki rizik od grešaka u izradi.
Otopine za potpunu parenteralnu prehranu: Mogu biti jako složene, ovisno o recepturi i broju dodavanja; također postoji visoki rizik od mikrobiološkog onečišćenja i visok rizik od greške u izradi.
Epiduralne i kardioplegijske otopine: Visoki rizik povezan s mikrobiološkim onečišćenjem. Uređaji za infuziju i ambulantni uređaji (npr. analgezija pod kontrolom pacijenta): Rizik od mikrobiološkog rasta neki pripravci mogu se davati tijekom duljeg vremenskog razdoblja na temperaturama tijekom primjene jednakim ili bliskim tjelesnoj temperaturi; tehnička složenost također je rizik.

Infuzije, štrcaljke i infuzijske vrećice: Rizik od grešaka u izradi i mikrobiološkog onečišćenja. Neke otopine mogu pospješiti rast bakterija i/ili gljivica. Neke otopine mogu se primjenjivati tijekom duljih vremenskih razdoblja.
Irigati(isključujući oftalmološke: Trajanje primjene.
Pripravci za oko – sa ili bez konzervansa: Rizik od mikrobiološkog rasta; složenost; rizik od greške u izradi
Ostalo (npr. biološki pripravci, faktor VIII): Potrebno je procijeniti na temelju pojedinačnih pripravaka
DIO 1
Osoblje
5. Odgovorna osoba treba imati odgovarajuće znanje te važeće praktično i teorijsko iskustvo u izradi sterilnih pripravaka te odgovarajuće osposobljavanje iz mikrobiologije.
6. Svu sterilnu izradu treba provoditi odgovarajuće osposobljeno osoblje. Osoblje koje nadzire radnje sterilne izrade treba imati odgovarajuće kompetencije i pisana ovlaštenja od strane Odgovorne osobe.
7. Cjelokupno osoblje koje radi u sterilnim postupcima treba u potpunosti biti svjesno mogućih posljedica svakog odstupanja od validiranih postupaka, kako za integritet pripravka tako i za integritet pacijenta. Potrebno je redovito podsjećati na rizičnu prirodu ovog postupka.
8. Prije početka sterilnog rada, cjelokupno osoblje treba proći odgovarajuću izobrazbu,
nakon čega je potrebno procijeniti njihovu kompetenciju. Osoblje koje radi s radiofarmaceuticima treba biti prikladno osposobljeno u skladu s nacionalnim zakonima glede propisa o ionizirajućem zračenju.
9. Osoblje treba proći osposobljavanje koje će im pružiti:
a) odgovarajuće znanje o Dobroj proizvođačkoj praksi ili Dobroj praksi pripreme
b) znanje o lokalnim praksama, uključujući o zdravlju i sigurnosti c) kompetenciju za potrebne sterilne vještine d) znanje o farmaceutskoj mikrobiologiji e) praktično znanje o odjelu, pripravcima i uslugama koje pružaju.
10. Potrebno je redovito provoditi procjenu kompetencije svakog člana osoblja u vezi
obavljanja radnji na sterilan način te, po potrebi, omogućiti reviziju ili ponovno osposobljavanje.
Posebni zahtjevi za aktivnosti aseptičke izrade
11. Nadzorno osoblje unutar odjela za aseptičku izradu treba dobro poznavati čiste prostore i tehnologiju uređaja za pročišćavanje zraka, te također temeljito poznavati sve pojedine značajke dizajna u njihovu odjelu, npr. sustave za ventilaciju, položaj i klase HEPA filtara, vrste radnih jedinica, dizajn izolatora.

12. Osoblje uključeno u aseptičke postupke treba imati specifičnu kompetenciju i vještine u aseptičkoj tehnici. Njihovu aseptičku tehniku potrebno je periodički procjenjivati provođenjem simulacija aseptičkog doziranja korištenjem hranjive podloge (usporediti s dijelom 4). Opravdanost i učestalost ovih periodičkih provjera trebaju biti dokumentirane Navedeno je potrebno dopuniti redovitim praćenjem aseptičke tehnike kako bi se osiguralo da rukovatelj može pripremiti pojedinačne doze precizno i sigurno.
DIO 2
Prostori i oprema
13. Prostori se trebaju nalaziti u okolišu koji, zajedno s mjerama za zaštitu pripravaka, predstavlja minimalan rizik za onečišćenja materijala ili pripravaka. U slučaju izrade citostatika i radiofarmaceutika, potrebno je također poduzeti mjere za zaštitu osoblja od materijala kojima rukuje.
Prema potrebnim značajkama okoliša (usporediti s dijelom 6) čisti prostori za izradu sterilnih pripravaka svrstavaju se u 4 klase (A, B, C i D). Razinu klase prostora potrebno je specificirati prema radnjama koje se u njoj obavljaju i pripravcima koji se izrađuju.
U skladu s time, za svaki čisti prostor ili blok čistih prostora trebaju biti specificirani uvjeti “u radu” (instalacija funkcionira na određeni način rada s navedenim brojem osoblja u radu) i uvjeti „u mirovanju“ (kompletna instalacija s opremom za izradu, ali bez osoblja, odnosno, bez ljudi). Kako bi se postigli specificirani uvjeti potrebno je definirati odgovarajuću filtraciju zraka (završni HEPA filtri za klase A, B i C) te dovoljan broj izmjena zraka (usporediti s dijelom 6). Da bi se postigli uvjeti “u radu”, ovi prostori trebaju biti izvedeni na način da uvjete „u mirovanju“ postižu nakon razdoblja „čišćenja“ od 15-20 minuta (okvirna vrijednost) po završetku rada
14. Sterilnu izradu potrebno je provoditi u namjenskim čistim prostorima koji imaju zračno zaustavne ('airlock') propusnike koji omogućavaju ulazak osoblja, materijala i opreme. Prostorije za presvlačenje trebaju biti izvedene kao zračno zaustavni ('airlock') propusnici. 15. Položaji korištenje slivnika potrebno je pažljivo osmisliti zbog mogućnosti mikrobiološkog onečišćenja. Slivnici ili objekti za pranje ruku zabranjeni su unutar prostora za izradu i dijelu prostora za završno presvlačenje. Ako se nalaze u susjednim prostorima, potrebno ih je redovito dezinficirati i nadzirati uvjete u njima. 16. Za svu opremu koja se koristi za izradu trebaju postojati i primjenjivati se pisani standardni operativni postupci.
17. Kad je primjenjivo, opremu je potrebno redovito umjeravati i provjeravati točnost uređaja za mjerenje volumena.
Posebni zahtjevi za izradu pripravaka koji se završno steriliziraju: 18. Kako bi se rizik od onečišćenja mikroorganizmima i česticama smanjio,
priprema sastavnica i većine pripravka treba se odvijati u okolišu najmanje klase D. Ako postoji neuobičajen mikrobiološki rizik za pripravak, primjerice zato što isti aktivno podržava rast mikroorganizama ili se čuva dulje vrijeme prije sterilizacije ili se ne izrađuje u zatvorenim posudama, izradu je potrebno provoditi u okolišu klase C.

Punjenje pripravka za završnu sterilizaciju potrebno je raditi u okolišu najmanje klase C.
Kad je pripravak pod neuobičajenim rizikom od onečišćenja iz okoliša, primjerice zato što je postupak punjenja spor ili su spremnici širokoga grla ili su nužno izloženi više od nekoliko sekundi prije brtvljenja, punjenje je potrebno obavljati u prostoru klase A s pozadinom najmanje klase C. Izradu i punjenje masti, krema, suspenzija i emulzija prije završne sterilizacije općenito je potrebno provoditi u okolišu klase C.
Tablica 2.1 daje primjere radnji za pripravke koji se završno steriliziraju, a trebaju se obaviti u različitim klasama prostora.
Tablica 2.1
Klasa Primjeri radnji za pripravke koji se završno steriliziraju
A Punjenje pripravaka, kada su pod neuobičajenim rizikom
C Izrada otopina, kada su pod neuobičajenim rizikom. Punjenje pripravka
D Izrada otopina i sastavnica za naknadno punjenje
Posebni zahtjevi za radnje aseptičke izrade: 19. Nakon pranja, sastavnicama je potrebno rukovati u okolišu najmanje klase D. Rukovanje sterilnim polaznim materijalima i sastavnicama, osim ako se kasnije u postupku podvrgavaju sterilizaciji ili filtraciji kroz filtar koji zadržava mikroorganizme, treba se provoditi u okolišu klase A.
Izradu otopina koje će se sterilno filtrirati tijekom postupka potrebno je provoditi u okolišu klase C; ako se otopine neće filtrirati, pripremu materijala i pripravka potrebno je provoditi u okolišu klase A. Rukovanje i punjenje aseptički izrađenih pripravaka (otvoreni i zatvoreni postupci) potrebno je provoditi u okolišu klase A u kabinetu s laminarnim strujanjem (LFC) ili izolatoru s pozitivnim tlakom. Prostorija treba imati pozitivan tlak (idealno 10 - 15 paskala) i protok zraka u odnosu na okolne prostore niže klase kako bi se pripravak zaštitio od onečišćenja.
Tablica 2.2 daje primjere radnji za aseptičku izradu koje će se provoditi u različitim klasama.

Tablica 2.2
Klasa Primjeri radnji za aseptičke pripravke
A Aseptička izrada i punjenje
C Izrada otopina za filtraciju
D Rukovanje sastavnicama nakon pranja
20. Izradu pod negativnim tlakom, čime se od onečišćenja štite rukovatelj i okoliš, potrebno je koristiti samo za pripremu opasnih farmaceutskih pripravaka (npr. citotoksičnih lijekova, radiofarmaceutika i radioobilježenih pripravaka iz krvi), zajedno s odgovarajućim mjerama opreza protiv onečišćenja pripravka(npr. prikladna kakvoća zraka u pozadini, propusnici s pozitivnim tlakom).
Kabineti s horizontalnim laminarnim strujanjem (LFC) nisu prikladni za pripremu opasnih lijekova. Umjesto toga trebaju se koristiti biozaštitni kabineti (eng. biohazard safety cabinets, BSC), s okomitim, prema dolje usmjerenim strujanjem zraka koji se okomito usisavaiz kabineta, a ne prema rukovatelju.
21. Budući da se aseptički pripravci ne steriliziraju završno, mikrobiološka kakvoća okoliša u kojoj se izrađuju iznimno je važna. Zato okoliš treba kontrolirati, a pristup treba dozvoliti samo ovlaštenim osobama. Za kabinete s laminarnim strujanjem (LFC) i biozaštitne kabinete (BSC), pozadina treba udovoljavati zahtjevima klase B, a klasa D za izolatore, osim ako nije drugačije opravdano
Bilo kakvo opravdanje za odabir pozadine niže klase treba se temeljiti na dokumentiranoj procjeni rizika koju je potrebno obaviti vrlo pažljivo. Mogući faktori koji se mogu uzeti u obzir u takvoj procjeni rizika obuhvaćaju:
● Vrijeme između izrade i upotrebe
● Korištenje zatvorenog sustava (pogledati pojmovnik)
● Prirodu i sastav pripravka
Tablica 2.3 daje pregled preporučenih minimalnih klasa prostora.
Tablica 2.3
Radni okoliš Okoliš u pozadini
LFC/BSC Klasa A Klasa B
Izolatori Klasa A Klasa D
22. Da bi se rizik od unakrsne kontaminacije smanjio na minimum, prostori trebaju biti namjenski. Potrebno je osigurati prostore za opasne proizvode, npr. citostatike, peniciline, biološke proizvode, radiofarmake i pripravke iz krvi. U iznimnim slučajevima može se prihvatiti kampanjski način rada, uz uvjet da se poduzmu posebne mjere opreza i da su obavljene potrebne procjene rizika.

Odjeća
23. Odjeća i njezina kakvoća trebaju odgovarati postupku i klasi radnog prostora. Odjeću je potrebno nositi na takav način da zaštiti pripravak od onečišćenja.
U prostorima koji se koriste za izradu pripravaka iz krvi, radiofarmaka i živih virusa potrebno je nositi namjensku odjeću. Opis odjeće potrebne za svaku klasu prostora naveden je ispod:
- Klasa D: Kosu, ruke i, kad je primjenjivo, bradu i brkove treba pokriti. Potrebno je nositi opće zaštitno odijelo i prikladnu obuću ili navlake za obuću. Potrebno je poduzeti odgovarajuće mjere da bi se izbjeglo bilo kakvo onečišćenje koja dolazi izvan čistih prostora.
- Klasa C: Kosu, ruke i, kad je primjenjivo, bradu i brkove treba pokriti. Potrebno je nositi jednodijelno ili dvodijelno odijelo s hlačama, skupljeno na zglobovima i s visokim ovratnikom, te odgovarajuću obuću ili navlake za obuću. Oni ne smiju otpuštati doslovno nimalo vlakana ili čestica.
- Klasa A/B: Pokrivalo za glavu treba potpuno pokrivati kosu i, kad je primjenjivo, bradu i brkove; treba biti utaknuto u ovratnik odijela; potrebno je nositi masku za lice koja sprječava širenje kapljica. Potrebno je nositi odgovarajuće sterilne, gumene ili plastične rukavice bez talka i steriliziranu ili dezinficiranu obuću. Nogavice hlača potrebno je utaknuti unutar obuće, a rukave odijela u rukavice. Zaštitna odjeća ne smije otpuštati doslovno nimalo vlakana ili čestica, niti zadržavati čestice koje padaju s tijela.
24. Odjeća za boravak izvan čistih prostora ne smije se unositi u prostorije za presvlačenje koje vode u prostore klase B i C. Za svakog djelatnika u klasi A/B, potrebno je osigurati čisto sterilno (sterilizirano ili odgovarajuće sanitirano) zaštitno odijelo za svaki radni ciklus. Rukavice je potrebno tijekom postupaka redovito dezinficirati. Maske i rukavice potrebno je mijenjati najmanje prilikom svakog radnog ciklusa.
Posebni zahtjevi za aseptički rad
25. Važno je vizualno provjeriti da je odijelo u dobru stanju i da su šavovi zatvoreni. Potrebno je razmotriti periodičku provjeru broja čestica i bioopterećenja (kontaktne ploče) (usporediti s dijelom 6). Obrazloženje za učestalost ovakvih periodičkih ispitivanja treba biti dokumentirano. Učestalost pranja odjeće potrebno je uskladiti s aktivnostima koje se obavljaju, a biocidna sredstva za pranje ili gama-zračenja treba koristiti za područja klase C, odnosno klase B.
Čišćenje
26. Čiste prostore potrebno je redovito čistiti u skladu s dokumentiranim i odobrenim postupkom. Svo osoblje koje obavlja poslove čišćenja mora se prethodno dokumentirano osposobiti, uključujući i o odgovarajućim područjima Dobre proizvođačke prakse. Dozvola za samostalan rad osobama se ne može izdati prije nego su prethodno ocijenjeni kao odgovarajuće osposobljeni.

27. Potrebno je koristiti namjensku opremu i čuvati je na način da se mikrobiološko onečišćenje smanji na minimum. Glave perača poda potrebno je baciti ili ponovno sterilizirati nakon svakog ciklusa čišćenja.
28. Sredstva za čišćenje i dezinfekciju ne smiju sadržavati žive mikroorganizme, a ona koja se koriste u prostorima klase A i B trebaju biti sterilna i bez spora.
29. Učinkovitost čišćenja potrebno je rutinski dokazivati mikrobiološkim uzorkovanjem s površina, npr. kontaktnim pločama ili brisevima.
30. Potrebno je razmotriti periodičku primjenu sporocidnih sredstava za čišćenje da bi se smanjila onečišćenja mikroorganizmima koji stvaraju spore.
31. Potrebno je koristiti virucidna sredstva za dekontaminaciju prostora u kojima se rukuje pripravcima iz krvi ili virusima.
32. Za sterilne alkoholne sprejeve i druge materijale koji se unose u čiste prostore potrebno je definirati rok valjanosti nakon otvaranja.
DIO 3
Dokumentacija
Općenita pitanja
33. Opće smjernice DPP-a o dokumentaciji potrebno je primijeniti na sve sustave kakvoće povezane sa sterilnim postupcima.
Upute za rad i zapisi o radu
34. Upute za izradu i zapisi uzeti iz 'glavnog obrasca' moraju biti odobreni prije korištenja. Iste trebaju biti dovoljno detaljne da omoguće nadzor, sljedivost polaznih materijala i sastavnica pripravka.
35. Kompletirane zapise o radu potrebno je čuvati tijekom dovoljno dugog razdoblja koje udovoljava zakonskim zahtjevima. U svakom slučaju, zapise je potrebno čuvati najmanje jednu godinu nakon isteka roka valjanosti odgovarajućeg gotovog pripravka. Postupke i upute za pripremu (uključujući recepte) potrebno je čuvati najmanje pet godina nakon prestanka njihove uporabe.
36. Upute za rad i zapisi bit će osmišljeni za svaku radnu jedinicu i trebaju biti osmišljene na način koji mogućnost grešaka kod prepisivanja smanjuje na minimum. Upute za rad i zapisi mogu se kombinirati u jedan dokument („radni listovi“). Dokumentaciju o radu potrebno je uskladiti sa zahtjevima danim u poglavlju 4.4 glavnoga dijela ovih smjernica.
DIO 4
Sterilna priprema 37. Sve korake rukovanja u sterilnom postupku potrebno je kontrolirati opsežnim standardnim operativnim postupcima koji osiguravaju da je rezultat procesa sterilan pripravak potrebne kakvoće.

38. Sve sterilizacijske postupke potrebno je validirati. Djelotvornost svakog novog postupka potrebno je validirati, a validaciju potvrditi u planiranim intervalima na temelju povijesti učinkovitosti ili kada se provede značajna izmjena u postupku ili opremi.
39. Posebna pažnja potrebna je kad usvojena sterilizacijska metoda nije opisana u važećem izdanju farmakopeje ili kad se koristi za pripravak koji nije jednostavna vodena ili uljna otopina. 40. Potrebno je izbjegavati istovremenu izradu različitih pripravaka, različitog sastava u istoj radnoj jedinici. Prije započinjanja sljedeće radnje, potrebno je provesti raščišćavanje linije, odnosno sav materijal mora se ukloniti iz prostora da bi se spriječila unakrsna kontaminacija i zamjene. Kada se tijekom istog radnog ciklusa priprema slijed sličnih pripravaka za niz pacijenata (npr. različite koncentracije citotoksičnog pripravka), potreban je osobit oprez da bi se izbjegle greške. 41. Kad u prostoriji postoji više od jedne radne jedinice, potrebno je provesti dokumentiranu procjenu rizika i poduzeti odgovarajuće mjere prije početka istovremenog rukovanja različitim pripravcima.
Izrada pripravaka koji se završno steriliziraju
42. Potrebno je poduzeti mjere opreza da bi se onečišćenja tijekom svih faza izrade smanjila na minimum.
43. Mikrobiološko onečišćenje polaznih materijala treba biti minimalno.
44. Materijale podložne otpuštanju vlakana potrebno je u minimalnoj količini čuvati u čistim prostorima
45. Kada je prikladno, potrebno je poduzeti potrebne mjere da se oneščićenje završnog pripravka česticama smanji na minimum.
46. Nakon završnog procesa čišćenja sastavnicama, spremnicima i opremom potrebno je rukovati na takav način da se spriječi ponovna kontaminacija.
Sterilizacija vlažnom toplinom
47. Zapisi o sterilizaciji trebaju biti dostupni za svaki ciklus sterilizacije. Oni se odobravaju kao dio postupka puštanja u promet.
48. Za učinkovitu sterilizaciju, cjelokupan materijal potrebno podvrgnuti potrebnoj obradi, a postupak treba provesti na način koji osigurava postizanje iste. Validaciju postupka potrebno je provesti na početku, kao i redovito naknadno, u skladu s rizikom, te nakon provođenja značajnijih izmjena opreme ili procesa.
49. Potrebno je uspostaviti validirane obrasce punjenja. Preporučuje se u postupcima koristiti fotografije ili detaljne crteže kako bi se osigurao konzistentan način punjenja.
50. Temperaturu i tlak treba bilježiti tijekom svakog ciklusa sterilizacije i periodički provjeravati pomoću tablica pare. Nezavisne mjerače temperature i tlaka na autoklavu potrebno je pratiti i bilježiti tijekom srednjeg dijela ciklusa te radi sličnosti usporediti s očitanjima u grafu.

51. Ispitivanja uklanjanja zraka i testove curenja na komori potrebno je također učestalo provoditi za cikluse s poroznim materijalom.
52. Kad se očekuje kontakt s kritičnim površinama potrebno je koristiti čistu paru. Periodički je potrebno provjeravati kakvoću pare, uključujući pregrijanu paru, suhoću i ispitivanja na nekondenzirajuće plinove.
53. Potrebno je koristiti indikatore topline koji će pokazati je li autoklavirani materijal steriliziran (kako bi se izbjegla zamjena s nesterilnim pripravkom).
Aseptička izrada
54. Ključni elementi aseptičkog postupka obuhvaćaju:
a) Održavanje cjelovitosti aseptičkog područja rada i brigu za radnu jedinicu i njezin okoliš.
b) Rukovanje i priprema polaznih materijala, posebice sve postupke dezinfekcije.
c) Ulazak materijala u prostore izrade
d) Standardne tehnike aseptičkog rada, uključujući rad bez dodirivanja kritičnih površina, ispravno smještanje materijala unutar laminarne struje zraka i upotrebu posebnih dijelova opreme te redovitu sanitaciju rukavica.
e) Odvajanje i kretanje materijala kojim se osigurava da ne dođe do slučajne unakrsne kontaminacije ili zamjene recepata ili pripravaka. f) Uklanjanje pripravaka i otpadnih materijala iz prostora izrade.
g) Cijeli postupak aseptičke izrade treba provoditi odgovarajuće osposobljeno osoblje, ovlašteno za obavljanje svog rada od strane Odgovorne osobe.
h) Broj ljudi prisutnih u prostoriji treba biti minimalan (međutim, tijekom simulacije aseptičkog doziranja treba biti prisutan najveći dozvoljeni broj osoba, kako bi se ispitalo najveće opterećenje).
i) Samo sterilni materijali smiju se unositi u prostore klase A ili B, npr. taložne ploče, brisevi i materijali za čišćenje. Prije unošenja u prostore klase A ili B, otopine pripravaka koje nisu sterilne treba filtrirati kroz sterilni filtar nazivne veličine pora od 0,22 mikrona (ili manje) . Kada to nije moguće, potrebno je provesti odgovarajuće postupke dekontaminacije.
55. Validaciju aseptičkog postupka izrade potrebno je provesti pomoću hranjive podloge kojom se simulira aseptički postupak (simulacija aseptičkog doziranja korištenjem hranjive podloge 'media fills'). Potrebno ju je provesti na početku, kao i redovito naknadno, u skladu s rizikom i kad god se provedu značajne izmjene opreme ili postupka. Simulacijom postupka potrebno je što vjernije oponašati rutinske aseptičke postupke (odnosno rukovanje koje se obično izvodi) i obuhvatiti sve ključne korake izrade. Odabir hranjive podloge treba se temeljiti na farmaceutskom obliku lijeka te selektivnosti, bistrini, koncentraciji i prikladnosti hranjive podloge za sterilizaciju.

56. Bočice za simulaciju aseptičkog doziranja korištenjem hranjive podloge potrebno je inkubirati na odgovarajućoj temperaturi, vodeći brigu da se spremnici povremeno preokrenu kako bi podloga došla u dodir sa svim površinama. Daljnje smjernice dane su u PIC/S-ovom dokumentu PI 007. Svako onečišćenje potrebno je istražiti u potpunosti, čak i ako postoji sumnja u cjelovitost spremnika.
57. Sve intervencije koje se pojave tijekom postupka izrade potrebno je zabilježiti u dokumentaciju o seriji lijeka. Potrebno je odrediti intervencijska pravila s odobrenim intervencijama koje simuliraju aseptičko doziranje.
58. Rok valjanosti nakon otvaranja svake otopine na veliko koja se koristi kao sastojak (npr. vrećica parenteralne infuzije ili bočica citotoksičnog sredstva) potrebno je opravdati. Bilo kakvi spremnici nekonzerviranih pripravaka koji se koriste kao polazni materijali ne smiju se koristiti nakon 24 sata od njihova prvog otvaranja. Svo vrijeme potrebno ih je zaštititi od onečišćenja ili kvarenja.
59. Sterilne jednokratne komponente kao što su filtri, igle, cijevi ne smiju se upotrebljavati u više od jednog radnog ciklusa i trebaju se ukloniti na kraju svakoga dana ili ciklusa.
60. Kod punjenja spremnika za višekratnu uporabu, potrebno je ispitati cjelovitost filtara na svakoj seriji i voditi brigu da kapacitet filtra kod pripravka s visokim biološkim opterećenjem ili zbog filtracije prekomjernih volumena nije premašen. Filtar treba biti kompatibilan s pripravkom.
61. Prijenos materijala u radnu jedinicu klase A obično se provodi dezinfekcijom ili sanitacijom radije nego sterilizacijom, stoga je važno za ovaj postupak imati pisani, validirani standardni operativni postupak. Ovu metodu neophodno je validirati praktičnim ispitivanjima koja pokazuju učinkovito uklanjanje vijabilnih organizama sa svih površina. Raspršivanje i brisanje smatraju se djelotvornijima za sanitaciju površina nego samo raspršivanje. 62. Preporučuje se nabava sastavnica steriliziranih gama-zračenjem ili sterilnih sastavnica na veliko u dvostruko/trostruko omotanom pakiranju umjesto raspršivanja po velikom broju pojedinačnih komponenti u klasi A (npr. pakiranja štrcaljki). 63. Postupkom čišćenja također treba učinkovito ukloniti ostatke pripravka s površina radne jedinice.
DIO 5
Provjera kakvoće
64. Sve sastavnice, polazne i pakovne materijale potrebno je vizualno provjeriti prije upotrebe kako bi se osiguralo da udovoljavaju zahtjevima specifikacije
65. Ako su sami polazni materijali odobreni lijekovi, tad ih obično nije potrebno ispitivati prije upotrebe, međutim, za neke materijale kao što su radiofarmaceutici neka ispitivanja mogu biti potrebna
66. Ako se pripravak izrađuje za pojedinačnog pacijenta, pretpostavlja se da neće biti potrebno završno ispitivanje pripravka, osim za radiofarmaceutike, kod kojih se mjeri aktivnost svake doze.

67. U kojem obujmu se provode fizička, kemijska i mikrobiološka ispitivanja kakvoće potrebno je definirati na temelju procjene rizika (usporediti s poglavljem 5.4 glavnog dijela ovih smjernica) i treba biti u skladu sa zahtjevima navedenim u poglavlju 6 glavnog dijela ovih smjernica.
68. Uzorci za fizičku, kemijsku i mikrobiološku analizu mogu se dobiti od:
a) Neupotrijebljenih pripravaka b) Dodatnih uzoraka koji se posebno izrađuju c) Uzoraka uzetih na kraju postupka miješanja pripravka, a
prije konačnog zatvaranja i prije iznošenja iz kritičnog područja. 69. Mikrobiološka analiza nije potrebna za svaku seriju. Kao druga mogućnost
prihvatljiv je program redovite mikrobiološke analize jedinica proizvedenih tijekom određenog vremenskog razdoblja ili redoviti program simulacije aseptičkog doziranja (odnosno, validacija postupka pomoću hranjive podloge).
70. Svaki porast potrebno je istražiti i dokumentirati u izvještaju o odstupanjima.
71. Uzorkovanje zadnjeg izrađenog spremnika,, a prije izdavanja, može ugroziti cjelovitost i stoga se ne preporučuje. Međutim, spremnike zatvorene taljenjem, npr. staklene ili plastične ampule, potrebno je sto posto ispitati na cjelovitost zatvaranja.
72. Laboratorij koji provodi ispitivanje treba biti u potpunosti upoznat s tehničkom pozadinom i zahtjevima sterilne izrade te imati validirane metode analize pripravaka i uzoraka. Odgovorna osoba treba osigurati da ispitni laboratorij ima opsežno znanje o mikrobiologiji i da se sustavi osiguranja kakvoće redovno provjeravaju. Kontrolne laboratorije na drugim lokacijama potrebno je redovito nadzirati.
73. Metode analize trebaju ukazivati na stabilnost pripravka. Potrebno ih je validirati na odgovarajući način.
DIO 6
Praćenje
74. Pored simulacija aseptičkog doziranja korištenjem hranjive podloge (usporediti s poglavljem 4), provodi se praćenje uvjeta da bi se dobili dokazi da su postupak, rukovatelji i organizacija pod kontrolom. Praćenje se sastoji od kvalifikacijskih radnji (klasifikacija „u mirovanju“) i praćenja okoliša u korištenim jedinicama (praćenje uvjeta okoliša „u radu“). Za farmaceutske svrhe, glavni kriterij prema kojem se procjenjuju sterilni prostori treba biti rizik od mikrobiološkog onečišćenja pripravka. Međutim, zbog nepreciznosti i varijabilnosti mikrobioloških metoda ispitivanja, preporučuje se dopuniti mikrobiološku provjeru okoliša praktičnijim praćenjem fizičkih parametara.
75. U kojem obujmu se provodi praćenje potrebno je definirati i temeljiti na procjeni rizika (usporediti s poglavljem 5.4 glavnog dijela ovih smjernica). Ovaj dio obuhvaća preporuke o učestalosti praćenja. Lokalni postupci mogu odstupati od ovih preporuka i uvijek ih je potrebno opravdati.

Osim faktora rizika navedenih u poglavlju 5.4 glavnog dijela ovih smjernica, sljedeće okolnosti mogu dovesti do povećane učestalosti praćenja (odnosno, češće nego ovaj dio preporučuje):
● Uočena odstupanja (npr. rezultati praćenja izvan specifikacije) ● Izmjene
● Intervencije u okolišu (npr. građevinski radovi) ● Povećani obujam poslova (više radnji koje je potrebno pratiti)
Moguće okolnosti koje mogu opravdati smanjenu učestalost praćenja (npr. rjeđe nego što ovaj dio preporučuje) uključuju:
● Upotrebu zatvorenih sustava tijekom izrade
● Upotrebu izrađenih pripravaka neposredno nakon izrade
● Završnu sterilizaciju pripravka ● Smanjenje obujma rada (manje radnji koje potrebno je pratiti)
76. Pisani izvještaj o rezultatima ispitivanja koji ukazuje značenje rezultata i preporučenu radnju potrebno je dati na znanje cjelokupnom osoblju na koje se odnosi te ukupne zapise čuvati u arhivi za buduće poveznice.
Klasifikacija “u mirovanju”
77. Odgovorna osoba treba procijeniti sva područja povezana s postupkom sterilne izrade radi provjere usklađenosti s odgovarajućom klasom čistih prostora u stanju bez prisutnosti ljudi:
a) Prilikom puštanja u rad b) Nakon izmjena ili postupaka održavanja, kako je primjereno c) Rutinski, u skladu s dogovorenom učestalosti
78. Klasifikacijska ispitivanja Preporučena učestalost za klasifikacijska ispitivanja (Tablica 6.1)
Kabineti s laminarnim strujanjem (LFC) / Biozaštitni kabineti (BSC):
Brojanje čestica 1 godišnje
Izmjena zraka u prostoriji po satu 1 godišnje
Brzine strujanja zraka u radnim jedinicama 1 godišnje
Provjere cjelovitosti HEPA filtara 1 godišnje
Izolatori:
Ispitivanje funkcionalnosti alarma izolatora 1 godišnje
Test curenja izolatora 1 godišnje
Provjere cjelovitosti HEPA filtara 1 godišnje
Praćenje okoliša “tijekom rada”
79. Redovito praćenje okoliša, postupka i gotovog pripravka ključan je dio osiguranja kakvoće svih sterilno izrađenih pripravaka

Dostupne su Norme i smjernice za mnoge od fizičkih i mikrobioloških aspekata (usporediti PIC/S i EU vodič za dobru proizvođačku praksu za industrijsku proizvodnju). Odgovorna osoba i ključno osoblje trebaju se pozivati na ove dokumente te iste razumjeti, s posebnim naglaskom na dijelove koji se odnose na sterilne postupke.
80. Osobitu važnost potrebno je posvetiti dobivanju smislenih rezultata praćenju trendova, te postavljanju kućnih (internih) standarda i granicama poduzimanja mjera Informacije je potrebno aktivno i umješno procijeniti, a ne samo popunjavati u svrhu vođenja zapisa.
81. Svaka jedinica treba imati program periodičkog ispitivanja (npr. prema dnevnim ciklusima, tjedno, mjesečno, kvartalno i godišnje), pri čemu se svi rezultati dokumentiraju i čuvaju za inspekciju. Preporučene učestalosti fizičkog i mikrobiološkog praćenja prikazane su kao smjernice u tablicama 6.2 i 6.3. Optimalna učestalost ispitivanja ovisit će o pojedinoj jedinici i radnjama koje se izvode. Programom praćenja potrebno je potvrditi da okoliš udovoljava odgovarajućoj normi. On nije zamjena za stalni oprez rukovatelja u osiguravanju ispravnog funkcioniranja cjelokupne opreme.
82. Praćenje fizičkih parametara
Preporučena učestalost praćenja fizičkih parametara (tablica 6.2)
Kabineti s laminarnim strujanjem (LFC) / Biosigurnosni kabineti BSC):
Razlike tlakova između prostorija Prije početka rada, obično svakodnevno daily
Razlike tlakova svih HEPA filtara (radna stanica)
Prije početka rada, obično svakodnevno
Brojanje čestica Kvartalno u stanju rada
Izolatori:
Razlike tlakova kroz svih HEPA filtre
Prije početka rada, obično svakodnevno
Cjelovitost rukavica izolatora Vizualne provjere kod svakog ciklusa
Ispitivanje održavanja tlaka u izolatoru(s pričvršćenim rukavicama)
Tjedno
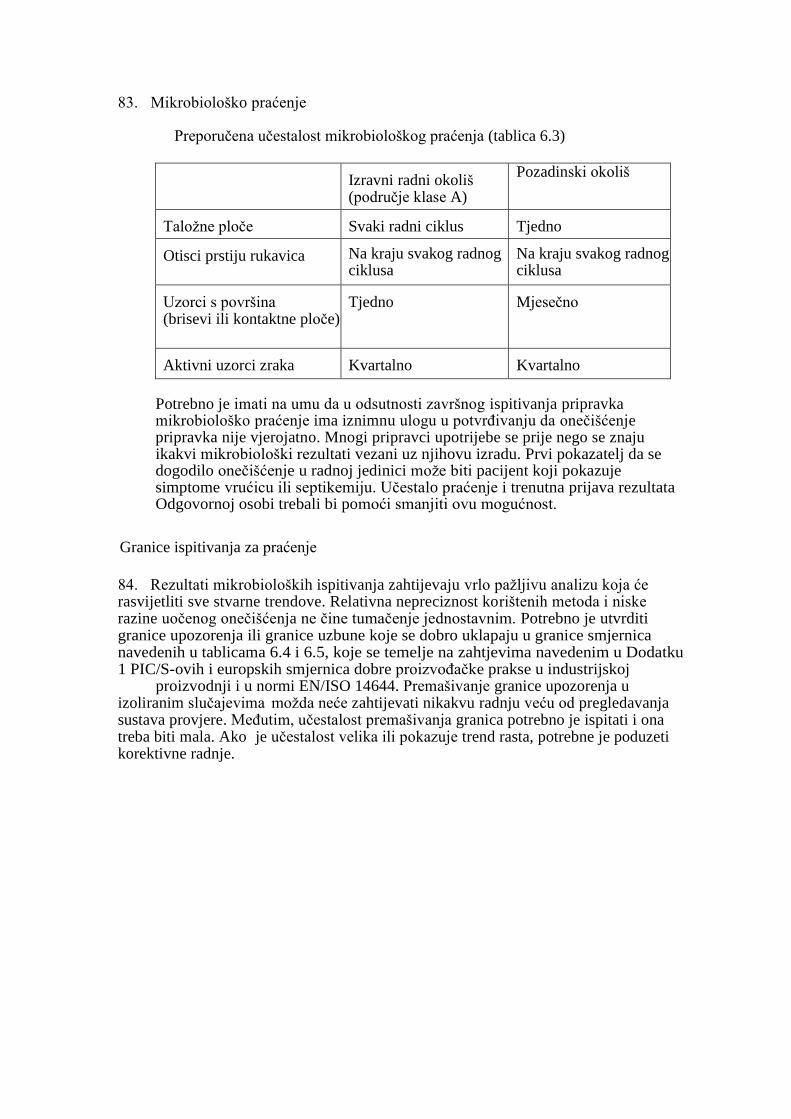
83. Mikrobiološko praćenje
Preporučena učestalost mikrobiološkog praćenja (tablica 6.3)
Izravni radni okoliš (područje klase A)
Pozadinski okoliš
Taložne ploče Svaki radni ciklus Tjedno
Otisci prstiju rukavica Na kraju svakog radnog ciklusa
Na kraju svakog radnog ciklusa
Uzorci s površina (brisevi ili kontaktne ploče)
Tjedno Mjesečno
Aktivni uzorci zraka Kvartalno Kvartalno
Potrebno je imati na umu da u odsutnosti završnog ispitivanja pripravka mikrobiološko praćenje ima iznimnu ulogu u potvrđivanju da onečišćenje pripravka nije vjerojatno. Mnogi pripravci upotrijebe se prije nego se znaju ikakvi mikrobiološki rezultati vezani uz njihovu izradu. Prvi pokazatelj da se dogodilo onečišćenje u radnoj jedinici može biti pacijent koji pokazuje simptome vrućicu ili septikemiju. Učestalo praćenje i trenutna prijava rezultata Odgovornoj osobi trebali bi pomoći smanjiti ovu mogućnost.
Granice ispitivanja za praćenje
84. Rezultati mikrobioloških ispitivanja zahtijevaju vrlo pažljivu analizu koja će rasvijetliti sve stvarne trendove. Relativna nepreciznost korištenih metoda i niske razine uočenog onečišćenja ne čine tumačenje jednostavnim. Potrebno je utvrditi granice upozorenja ili granice uzbune koje se dobro uklapaju u granice smjernica navedenih u tablicama 6.4 i 6.5, koje se temelje na zahtjevima navedenim u Dodatku 1 PIC/S-ovih i europskih smjernica dobre proizvođačke prakse u industrijskoj proizvodnji i u normi EN/ISO 14644. Premašivanje granice upozorenja u izoliranim slučajevima možda neće zahtijevati nikakvu radnju veću od pregledavanja sustava provjere. Međutim, učestalost premašivanja granica potrebno je ispitati i ona treba biti mala. Ako je učestalost velika ili pokazuje trend rasta, potrebne je poduzeti korektivne radnje.

85. Praćenje fizičkih parametara
Granice za praćenje fizičkih parametara kontroliranih područja i uređaja (tablica 6.4)
Klasa Maksimalni dozvoljeni broj čestica u zraku/m3 jednak
ili veći od
Izmjene
zraka (broj na
sat)
Brzina
strujanja zraka
(m/s
+- 20%)
Razlika tlaka u
odnosu na susjednu prostoriju niže klase
(Pa) u mirovanju tijekom rada
0.5 μm 5.0 μm 0.5 μm 5.0 μm
A 3 520 20 3 520 20 N/A 0.45 HLF
0.30 VLF
N/A LFC
>15 izolator
B 3 520 29 352 000 2 900 >20 N/A >10
C 352 000 2 900 3 520 000 29 000 >20 N/A >10
D 3 520 000 29 000 Nije
definirano
Nije
definirano
>10 N/A >10
Napomene:
N/A = nije primjenjivo
LFC = kabinet s laminarnim strujanjem HLF = vodoravno laminarno strujanje; VLF = okomito laminarno strujanje
U svrhu klasifikacije u područjima klase A, potrebno je uzeti uzorak od najmanje 1 m3 po mjestu
uzorkovanja. Tako će se osigurati da na proces klasifikacije ne utječu negativno lažna brojanja koja
potječu od elektroničkih šumova, zalutale svjetlosti, itd. Za klasu A klasifikacija čestica iz zraka je
ISO 4.8 određena granicom za čestice od ≥ 5.0 µm. Za klasu B, klasifikacija čestica iz zraka je ISO
5 za obje veličine čestica na koje se odnosi. Za razred C, klasifikacija čestica iz zraka je ISO 7
odnosno ISO 8. Za klasu D, klasifikacija čestica iz zraka je ISO 8. U svrhu klasifikacije,
metodologija norme EN/ISO 14644-1 definira i minimalni broj mjesta uzorkovanja i veličinu uzorka
na temelju granice klase za najveću veličinu čestice u klasi i metode vrednovanja prikupljenih
podataka.
U svrhu klasifikacije potrebno je koristiti prenosive brojače čestica s kratkom dužinom cijevi za
uzorkovanje zbog relativno više stope gubitka čestica od ≥ 5.0µm u sustavima udaljenog
uzorkovanja s duljim cijevima. U sustavima s jednosmjernim strujanjem zraka potrebno je
koristiti izokinetičke glave za uzorke.
Praćenje “u radu” može se provoditi tijekom uobičajenih radnji, simuliranih radnji ili simulacije
aseptičkog doziranja, budući da je pri tome potrebna simulacija najgoreg slučaja. Norma EN ISO
14644-2 daje informacije o ispitivanju u svrhu dokazivanja stalnog udovoljavanja zahtjevima
dodijeljenih klasa čistoće.

86. Mikrobiološko praćenje
Preporučene granice za mikrobiološko praćenje čistih prostora u radu
(Tablica 6.5)
Preporučene granice za mikrobiološka onečišćenja (a)
Klasa Uzorak zraka
(cfu/m3)
Taložne ploče,
promjer 90mm
(cfu/4 h) b)
Kontaktne ploče,
promjer 55mm
(cfu/pločica)
Otisak rukavice,
5 prstiju
(cfu/rukavica)
A <1 <1 <1 <1
B 10 5 5 5
C 100 50 25 -
D 200 100 50 -
Napomene:
(a) Ovo su prosječne vrijednosti
(b) Pojedinačne taložne ploče mogu se izlagati kraće od 4 sata, a u tom slučaju granice je potrebno proporcionalno smanjiti.
_____________

DODATAK 2
SMJERNICE O NORMAMA POTREBNIMA ZA PRIPREMU
NESTERILNIH TEKUĆINA, KREMA I MASTI
Uvod
1. Ovaj je Dodatak dopuna glavnom dijelu ovih smjernica i navodi općenita pravila opisana u istome za pripremu nesterilnih tekućina, krema i masti. Kad se za neposrednu primjenu izrađuju samo pojedinačni spremnici, moguće je smanjiti neke od donjih zahtjeva.
Načelo
2. Tekućine, kreme i masti tijekom pripreme mogu naročito biti podložni mikrobiološkimi drugim onečišćenjima. Stoga je potrebno poduzeti posebne mjere za sprječavanje svakog onečišćenja.
Prostori i oprema
3. Da bi se pripravak zaštitio od onečišćenja preporučuje se upotreba zatvorenih sustava za izradu i prijenos. Prostore u kojima se izrađuju pripravci ili otvoreni spremnici izlažu utjecaju okoliša obično je potrebno učinkovito prozračivati filtriranim zrakom.
4. Prostori za izradu ne smiju se koristiti za druge radnje. 5. Potrebno je poduzeti mjere za smanjenje rizika od onečišćenja, a one mogu obuhvaćati:
a) Upotreba namjenske odjeće i pokrivala za kosu.
b) Kod otvorenih postupaka preporučuje se lokalna filtracija zraka i nošenje rukavica.
c) Čišćenje opreme neposredno nakon korištenja
d) Ispiranje opreme koja dolazi u dodir s pripravkom vodom prikladnog stupnja kakvoće (pročišćena voda ili voda za injekcije u boci može biti prikladna ako se koristi u roku od 24 h nakon otvaranja).
e) Osiguranje uklanjanja ostataka sredstava za čišćenje i sanitaciju (npr. hipoklorita).
f) Provjera je li oprema čista i suha prije skladištenja.
g) Pažljivo skladištenje očišćene opreme.
h) Svi materijali uneseni u prostore za izradu trebaju biti čisti.
i) Sanitacija kritičnih površina alkoholom prije upotrebe.
j) Provjera spremnika i poklopaca u svrhu provjere jesu li čisti i suhi prije upotrebe.
k) Spremnici gotovog pripravka ne smiju se višekratno koristiti.
l) Perači poda i krpe ne smiju otpuštati vlakna, potrebno ih je sanitirati svaki dan ako se koriste višekratno i ne smiju se koristiti za čišćenje drugih prostora.
m) Ako se u prostorima za izradu istovremeno obavlja više od jedne radnje, potrebno je osigurati odgovarajuće odvajanje da bi se spriječila unakrsna kontaminacija i zamjene. Potrebno je provesti procjenu rizika.

n) Preporučuje se korištenje namjenske opreme za visoko djelatne tvari, peniciline, cefalosporine, nadražujuća sredstva, citotoksične tvari, ektoparaziticide i druge tvari koje su jako opasne ili teško se čiste. Ove materijale potrebno je identificirati i provesti procjenu rizika.
6. Sve vrste spremnika, cjevovode i crpke potrebno je dizajnirati i instalirati tako da se mogu lako čistiti i, po potrebi, sanitirati. Izuzetno je važno da dizajn opreme ima minimalan broj „mrtvih kutova“ ili mjesta u kojima se mogu nakupljati ostaci i tako poticati rast mikroorganizama.
7. Potrebno je izbjegavati upotrebu staklene opreme kad god je to moguće. Nehrđajući čelik visoke kakvoće često je prvi izbor materijala za dijelove koji dolaze u dodir s pripravkom. Kad se koristi staklena oprema, prije i nakon upotrebe potrebno je provjeriti ima li oštećenja.
Izrada
8. Kemijska i mikrobiološka kakvoća vode koja se koristi u izradi treba biti specificirana i pod nadzorom.
Kakvoća treba biti specificirana prema “Napomeni za smjernice o kakvoći vode za farmaceutsku uporabu” koju je izdala Europska agencija za lijekove (EMA) i treba udovoljavati zahtjevima farmakopeje.
Kad se koristi odgovarajuće certificirana sterilna voda za injekcije ili ispiranje u boci, nije potrebno provoditi mikrobiološka ili kemijska ispitivanja. Za rutinsko praćenje sustava za vodu, potrebno je periodički (obično tjedno) bilježiti opća ispitivanja, kao što su biološko opterećenje (ukupni broj živih aeroba), vodljivost, ukupni organski ugljik (TOC) ili slična očitanja. Povremeno (obično tromjesečno) potrebno je provesti posebnu kemijsku analizu. Potrebno je brinuti o održavanju sustava za vodu da bi se izbjegao rizik od rasta mikroorganizama. Nakon svake kemijske sanitacije sustava za vodu, potrebno je provesti validirani postupak ispiranja kako bi se osiguralo da je sredstvo za sanitaciju učinkovito uklonjeno.
9. Materijali koji će vjerojatno otpuštati vlakna ili druga onečišćenja, poput kartona ili izloženog neobrađenog drva, ne smiju biti prisutni u prostorima u kojima se izrađuju pripravci ili se čisti spremnici izlažu utjecaju okoliša.
10. Potrebno je voditi brigu o održavanju homogenosti smjesa, suspenzija itd. tijekom punjenja. Postupke miješanja i punjenja mogu se validirati, a vrijeme i brzine miješanja potrebno je zabilježiti. Posebno pažljiv treba biti na početku postupka punjenja, nakon zastoja i na kraju postupka kako bi se održala homogenost mješavina. 11. Kad se gotovi pripravak ne pakira odmah, maksimalno razdoblje skladištenja i uvjeti skladištenja moraju biti navedeni i potrebno je pridržavati ih se. Preporučuje se da se pripravak pakira što je prije moguće (isti dan).
12. U kojem obujmu se provode fizička, kemijska i mikrobiološka ispitivanja kakvoće potrebno je definirati na temelju procjene rizika (usporedi poglavlje 5.4 glavnog dijela ovih smjernica). Kad je to moguće, uzorke gotovog pripravka potrebno je vizualno pregledati prije puštanja u promet.

13. Potrebno je odrediti i opravdati rok valjanosti (krajnji datum uporabe) za neotvoreni pripravak. Preporučuje se odrediti rok valjanosti nakon otvaranja.
_________________

DODATAK 3
DOBRE PRAKSE ZA PRIPREMU RADIOFARMACEUTIKA U
ORGANIZACIJAMA ZDRAVSTVENE SKRBI
Uvod
1. Postoje razne vrste radiofarmaceutika koji se izrađuju u organizacijama zdravstvene skrbi, uključujući:
● Sterilne pripravke s odobrenjem za stavljanje u promet aseptički pripremljene u organizaciji zdravstvene skrbi. Oni se u pravilu rutinski koriste u dijagnostičke svrhe u nuklearnoj medicini i obuhvaćaju: tehnecij-99m radiooznačene ligande dobivene kombinacijom komponenti iz kompleta s [
99mTc]pertehnetatom iz
radionuklidnog generatora. ● Radionuklidne prekursore s odobrenjem za stavljanje u promet, primjerice itrij-90 koji se koristi kao polazni materijal za sintezu. ● Sterilne pripravke bez odobrenja za stavljanje u promet koji se sintetiziraju,
radiooznačavaju, pročišćavaju i formuliraju za dijagnostičku ili terapijsku primjenu. Radionuklidi, osobito oni kratkoživući, mogu se izrađivati u organizaciji s ciklotronom, primjerice: fluorin-18, kisik-15 ili ugljik-11. Druga je mogućnost da se isporuče kao radioaktivne kemikalije, primjerice jod-123, jod-124, lutecij-177.
Generatorskim sustavom radionuklida germanij-68 / galij-68 također se može u organizaciji proizvesti galij-68 kao PET radionuklid.
Potrebno je obratiti pozornost na neradioaktivni hladni kemijski prekursor koji se u većini slučajeva dobiva od vanjskih dobavljača kao kemijska tvar. Kako ovaj kemijski prekursor u prvom redu određuje farmakokinetičko ponašanje radiooznačene djelatne tvari, posebnu pažnju potrebno je posvetiti identitetu i čistoći. Nečistoće bi također mogle maskirati radionuklide umjesto kemijskih prekursora.
● Oralni pripravci sa ili bez odobrenja za stavljanje u promet koji se izrađuju kao kapsule ili otopine koje pacijent uzima u svrhu dijagnoze ili terapije (npr. jod-131 za liječenje štitnjače). ● Inhalacijski pripravci sa ili bez odobrenja za stavljanje u promet, poput plinova, aerosolova ili čestica (primjerice [
81m]kripton, [
99m[Tc]-DTPA, [
99m[Tc]-grafit).
2. Radiofarmaceutici su drugačiji od ostalih farmaceutskih pripravaka zbog sigurnosnih pitanja svojstvenih ovim pripravcima te trebaju udovoljavati svim nacionalnim propisima za zaštitu od zračenja i dobroj proizvođačkoj praksi (DPP). Štoviše, dijagnostičke radiofarmaceutike potrebno je pripremiti, provjeriti i uporabiti unutar kratkog razdoblja od nekoliko sati ili čak minuta zbog fizičkog poluvremena života radionuklida, što predstavlja pravi izazov zbog svoje tehničke zahtjevnosti.

3. Neke zemlje možda su već utvrdile okvir za izdavanje dozvole za pripremu radiofarmaceutika u organizacijama zdravstvene skrbi. Često se na temelju nacionalnih propisa u redovitim intervalima provode rutinski nadzori.
Namjena
4. Namjena ovog Dodatka jest:
● Dopuniti i poboljšati važeće smjernice za pripremu radiofarmaceutika. Ovaj Dodatak ne ponavlja nijednu smjernicu za izradu sterilnih pripravaka koja je već sadržana u Dodatku 1 ovih smjernica
● Dopuniti bilo koju smjernicu koju su već razvila nacionalna nadležna tijela.
Namjena nije:
● Dati detaljne tehničke informacije o radiofarmaceuticima koji su dostupni u drugim literaturnim izvorima.
● Zamijeniti bilo koju smjernicu koju su već razvila nacionalna nadležna tijela.
● Mijenjati bilo koje specifikacije i postupke provjere kakvoće za radiofarmaceutike koji su već navedeni u nacionalnoj farmakopeji.
● Dati detaljne upute o sigurnosti i zaštiti od zračenja.
Područje primjene
5. Ovaj Dodatak obuhvaća isključivo izradu radiofarmaceutika u organizacijama zdravstvene skrbi.
On ne obuhvaća:
● Proizvodnju radiofarmaceutika s odobrenjem za stavljanje u promet.
● Proizvodnju radiofarmaceutika za istraživanja ili klinička ispitivanja jer neke organizacije zdravstvene skrbi i sveučilišta mogu dijeliti zajedničke prostore ili imati posebna nacionalna ovlaštenja glede pripreme ovih proizvoda.
DIO 1
Osoblje
6. Osoblje, uključujući ugovorno osoblje, treba imati podrobno znanje o rizicima rada s ionizirajućim zračenjem te imati znanje o zaštiti od zračenja. Osoblje je potrebno redovito kontrolirati te njihovo izlaganje ionizirajućem zračenju smanjiti na minimum.

7. Izradu i puštanje pripravka u promet treba provoditi odgovarajuće kvalificirano i osposobljeno osoblje s podrobnim poznavanjem Dobre prakse izrade, aseptičkog rada i radiokemije.
8. Zbog malog broja djelatnika koji rade u organizacijama zdravstvene skrbi, mora postojati jasna podjela odgovornosti za provjeru kakvoće (QC) i postupaka izrade. Neprihvatljivo je da jedna osoba obavlja oba zadatka za isti pripravak. Također trebaju postojati posebni postupci za puštanje lijeka u promet izvan redovitog radnog vremena. Napomena: Za pripravke koji se izrađuju iz odobrenih kompleta i generatora u skladu s proizvođačevim uputama, kad provjera kakvoće nije obavezna, jedna bi osoba mogla izraditi pripravak, ali puštanje u svrhu izdavanja pacijentu treba provesti nezavisna osoba.
9. Potreban je dovoljan broj radnika kako bi izrada mogla teći bez ugrožavanja kakvoće pripravaka. Čimbenici, kao što su broj raspoloživih radnika i vrijeme raspoloživo za izradu, trebaju se koristiti kod planiranja kapaciteta kojim se osigurava da sustav kakvoće ne bude preopterećen.
10. Neke jedinice u sklopu sveučilišta ili velikih kliničkih centara mogu pripremati doze za davanje pacijentima kao i za istraživanja. Važno je da osoblje koje provodi istraživanja ne priprema rutinske doze za pacijente osim ako nije izričito osposobljeno prema Pravilima dobre proizvođačke prakse za što postoje pisani zapisi.
DIO 2
Prostori i oprema
11. Radiofarmaceutike je potrebno izrađivati u posebnim prostorijama.
12. Radiofarmaceutski pripravci za injekcije trebaju se izrađivati u okolišu koji pripravak štiti od mikrobiološkog onečišćenja. Istovremeno, okoliš također treba zaštititi od ionizirajućeg zračenja. 13. Kada se na istoj lokaciji provode i istraživačke radnje i izrada, te radnje potrebno je fizički i organizacijski razdvojiti jednu od druge, kako istraživačko osoblje ne bi utjecalo na zadatke koji se provode prema pravilima dobre proizvođačke prakse (DPP).
14. Potrebna je odgovarajuća oprema za otkrivanje i praćenje ozračenosti osoblja.
15. Zbog zdravstvenih i sigurnosnih potreba, da bi se osiguralo brzo uklanjanje onečišćenja s osoblja, odgovarajuće metode kao što su slivnici i/ili tuševi možda će se morati nalaziti bliže čistim prostorima nego u slučaju uobičajenog aseptičkog rada, ovisno o nacionalnim zahtjevima za sigurnost. Potrebno je razmotriti odgovarajuću provjeru izvedbe i održavanje slivnika i/ili tuševa. U skladu s lokalnim zahtjevima mogu se također primjenjivati i druge metode.
16. Označivanje krvnih stanica potrebno je obavljati u prostoriji u kojoj se ne provode radnje izrade pripravaka. Potrebno je procijeniti i dokumentirati rizik od korištenja zajedničkih prostorija za presvlačenje za dvije radnje.

17. Kad god je to moguće, izradu pripravaka potrebno je provoditi zatvorenom metodom, kao što je prijenos iz jednog zabrtvljenog sterilnog spremnika u drugi. Aseptički probod čepa bočice potrebno je obavljati što rjeđe.
18. Radnje izrade potrebno je obavljati ili u radnim jedinicama s laminarnim strujanjem, vrućim komorama ili u zatvorenim radnim jedinicama sa zaštitom od zračenja. Ako se koriste otvorene radne jedinice, primjerice zaštitni kabineti, zrak u okolišu treba biti više klase, ovisno o tome izrađuje li se pripravak aseptički ili se završno sterilizira.
19. Prije upotrebe potrebno je vizualno provjeriti rukavice za izolatore i narukavlje za rukovanje vrućim komorama. Ako je moguće, test curenja na vrućim komorama te izolatorima potrebno je obavljati u razdobljima definiranim u Dodatku 1.
20. Očekuje se da se svaka otvorena metoda izrade i rukovanja aseptičnim pripravcima obavlja u okolišu klase A. U slučaju punjenja gotovog pripravka u pojedinačni spremnik za neposrednu primjenu zatvorenom metodom pripreme, za radnje punjenja odgovarajući može biti prostor klase C, temeljeno na procjeni rizika.
21. Pripravke koji se mogu završno sterilizirati potrebno je puniti u okolišu klase C. Za korake prije punjenja, npr. radiokemijsku sintezu, potrebno je koristiti okoliš klase C ili je u protivnom drugu klasu potrebno opravdati procjenom rizika.
22. Kad je primjenjivo, materijale je potrebno prenijeti u radnu jedinicu pomoću međusobno povezanih otvora za prijenos.
23. Generatore je potrebno smjestiti u odgovarajući prostor ili radnu jedinicu pored radne jedinice za izradu. Tražena klasa okoliša treba se temeljiti na procjeni rizika kako bi se moguće onečišćenje tijekom eluiranja smanjilo na minimum.
24. Kad se za izradu radiofarmaceutika koristi izolator, općenito nije dozvoljena recirkulacija zraka
25. Klasa okolnog prostora ovisit će o raznim čimbenicima koje treba ocijeniti u dokumentiranoj procjeni rizika, a obuhvaća vrstu sustava zaštite radne jedinice te metodu izrade pripravka. U donjoj tablici dani su minimalni očekivani standardi za cijeli raspon aktivnosti.
Metoda pripreme Otvorena radna jedinica
Zatvorena radna jedinica
Otvorena aseptička B *Napomena D Zatvorena aseptička C D Otvorena sa završnom sterilizacijom D D
Zatvorena sa završnom sterilizacijom
D D
Zatvorene radne jedinice definiraju se kao izolatori i vruće komore koje se mogu ispitati na curenje. Otvorene radne stanice definiraju se kao kabineti s laminarnim strujanjem zraka koji su djelomično otvoreni sprijeda.

Ako se aseptičko izrađenim radioaktivnim lijekom u otvorenoj bočici, u bilo kojem trenutku tijekom izrade, rukuje izvan radne jedinice, okolni prostor treba udovoljavati zahtjevima klase B.
* Napomena: Kad se lijek primjenjuje neposredno nakon izrade, ovisno o procjeni rizika, može se dozvoliti korištenje okoliša klase C.
26. Radioaktivne materijale potrebno je čuvati u zaštitnom vanjskom pakiranju kao što su posude od olova/volframa ili jednakovrijedni spremnici za zaštitu od zračenja.
27. Ovisno o mikrobiološkom riziku za pripravak, izradu kapsula i drugih nesterilnih pripravaka potrebno je obavljati u kontroliranom okolišu s odgovarajućom zaštitom od zračenja.
DIO 3
Dokumentacija
28. Važeći su opći zahtjevi za dokumentaciju iz ovih smjernica.
29. Kada se koristi proizvod s odobrenjem za stavljanje u promet, npr. bočice u koje se dodaje tehnecij, potrebno je slijediti zahtjeve iz upute o lijeku.
30. Kad se koriste elektronički dokumenti, potrebno je čuvati integritet podataka. Mora postojati dovoljna kontrola pristupa dokumentaciji, a dokumenti moraju biti dostupni u razumnom vremenu. Isti opći zahtjevi primjenjuju se također na dokumente u papirnatom obliku.
DIO 4
Proizvodnja
31. Iako u Dodatku 1 ovih smjernica postoji dio o polaznim materijalima, uvrštena su neka daljnja pojašnjenja za radiofarmaceutike. Polazni materijali uključuju generatore, kemikalije za sintezu poput manoze, acetonitrila, smole za ionsku izmjenu i sl., sastavnica za pakiranje i potrošnu robu poput štrcaljki i igala. Sve materijale potrebno je zaprimiti u skladu s nacionalnim zahtjevima i svaki formalni postupak zaprimanja treba obuhvaćati provjeru dokumentacije, potvrdu da su materijali dobiveni od odobrenog dobavljača i provjeru kakvoće, kad je prikladno.
Sljedeće dodatne smjernice daju se za različite vrste sastavnica:
● Neka potrošna roba u nekim zemljama može biti medicinski proizvod s odobrenjem, što je potrebno potvrditi prilikom zaprimanja. ● Očekuje se, iako su neki generatori proizvodi s odobrenjem, da će se neka ispitivanja obaviti prilikom zaprimanja da bi se potvrdila sukladnost sa specifikacijom, npr. izbijanje molibdena i aluminija, ako to već nije naznačeno u uputi o lijeku.
● Kemikalije poput manoze za sintezu/proizvodnju radiofarmaceutika mogu biti već unaprijed pakirane u kasete ili isporučene na veliko, te je potrebno obaviti

kemijsku provjeru identiteta ispitivanja čistoće osim, ako na drugi način nije opravdano. Kemikalije u unaprijed pakiranim kasetama možda neće trebati ispitivati ako u organizaciji zdravstvene skrbi postoji siguran postupak odobravanja dobavljača, pri čemu je proveden nadzor kod dobavljača.
● Budući da će neradioaktivni kemijski prekursor odrediti farmakokinetičko ponašanje gotovog proizvoda, potrebno je na odgovarajući način obaviti provjeru identiteta i ispitivanje čistoće kemijskog prekursora kao što je kvalifikacija dobavljača, ispitivanje prilikom zaprimanja i sve dokumentirati. ● Uputa o lijeku sadrži važne informacije o pripremi proizvoda. Stoga je vrlo važno uspostaviti postupak prilikom zaprimanja kojim se potvrđuje da u uputama za pripremu nisu provedene izmjene.
● Kad je potrebno razrjeđivanje, koriste se i već odobrena sredstva za razrjeđivanje. 32. Postupak sinteze za neke se radiofarmaceutike smatra prvim dijelom pripreme. Budući da se jedinice sinteze mogu koristiti za različite proizvode, potrebno je razmotriti rizik od unakrsnog onečišćenja i poduzeti odgovarajuće mjere za sprječavanje istoga uporabom namjenskih sastavnica i opreme ili procjenom učinkovitosti čišćenja, kad je to prikladno. 33. Postupke sinteze najbolje je kompjutorski kontrolirati, a kompjutorizirani sustav
potrebno je validirati. Ako se postupci sinteze kontroliraju ručno, potrebno je izraditi detaljne pisane upute za svaki korak postupka.
34. Kad god je moguće, potrebno je koristiti generator ili proizvod s odobrenjem, međutim, zbog kliničke potrebe pacijenta i raspoloživosti zaliha, to možda neće biti moguće. U takvim okolnostima, potrebno je provesti procjenu rizika da bi se odredilo mogu li se koristiti proizvodi iz drugih zemalja, uz uvažavanje svih nacionalnih regulatornih zahtjeva.
35. Da bi se smanjio rizik od onečišćenja treba postojati postupak za prijenos i sanitaciju generatora i bilo kojih drugih materijala u prostor za pripremu. 36. Da bi se održala sterilnost generatore je potrebno koristiti u skladu s uputama proizvođača ili, ako se koristi generator bez odobrenja, na temelju odgovarajućih praksi.
37. Broj sastavnica ili količinu potrošne robe koji se čuvaju u radnoj jedinici potrebno je održavati minimalnima da ne bi ometali strujanja zraka.
38. Sve sastavnice koje se koriste za aseptički rad, poput cijevi, filtara, moraju prije uporabe biti sterilne i koristiti se jednokratno. Sastavnice je, po mogućnosti, potrebno sastaviti i sterilizirati prije upotrebe da bi se broj aseptičnih spojeva smanjio na minimum.
39. Da bi se spriječila međusobna zamjena proizvoda, različiti proizvodi ne smiju se pripremati istovremeno u istoj radnoj jedinici. Kad se pripremaju prašci ili plinovi u prostoriji sa više radnih jedinica potrebno je razmotriti mogućnost unakrsne kontaminacije.

40. Proizvod se obično eluira ili puni u sterilne bočice koje isporučuje proizvođač ili se lokalno nabavljaju. Sterilnost tih bočica potrebno je potvrditi kroz dokumentaciju o zaprimanju.
41. Prilikom označivanja radiofarmaceutika moraju se uzeti u obzir specifični zahtjevi unutar svih nacionalnih regulatornih ili farmakopejskih zahtjeva.
42. Kad je to potrebno, bočice koje se koriste u pripremi potrebno je označiti. Posude od olova/volframa ili jednakovrijedne spremnike potrebno je nakon pripreme označiti odgovarajućim informacijama. 43. Spremnike koji se koriste za zaštitu bočice ili štrcaljke od zračenja potrebno je provjeriti kako bi se osiguralo da su sve prethodne naljepnice uklonjene prije uporabe. 44. Spremnike koji se koriste za zaštitu bočice ili štrcaljke od zračenja treba očistiti i sanitirati prije ponovne uporabe u radnoj jedinici. Volfram ili slični materijali imaju prednost pred olovom koje otpušta čestice.
45. Za proizvode koji se aseptički pune mora se obaviti ispitivanje cjelovitosti filtara. Ispitivanje cjelovitosti prije filtracije možda neće biti moguće za radiofarmaceutike, osobito ako se koristi filtar s ventilom, ali ispitivanje cjelovitosti nakon filtriranja potrebno je obaviti za sve proizvode. 46. Kad se nakon pripreme koriste sustavi automatske isporuke, primjerice za PET proizvode, mora se osigurati da se bočica ne ošteti ili ne pukne tijekom prijenosa u odredišni spremnik. Sve brtve na dnu spremnika koje štite bočicu od oštećenja ili reflektori trebaju biti prisutne.
47. Čišćenje kritičnih područja kao što su radne jedinice treba obavljati osoblje zaduženo za proizvodnju. Prostorije mogu čistiti ugovorni radnici koji su prikladno osposobljeni za dobru proizvođačku praksu (DPP). Za dodatne informacije o učestalosti čišćenja pogledati Dodatak 1.
48. Postojanost sredstava za umjeravanje doza potrebno je provjeravati svakodnevno ili prije uporabe. Također ih je potrebno provjeriti prema nacionalnim normama i u redovitim intervalima potvrđivati linearnost.
DIO 5
Provjera kakvoće 49. Potrebno je provesti odgovarajuća ispitivanja generatora bez odobrenja, kao što je izbijanje molibdena i aluminija. Ispitivanje sterilnosti može se provesti tijekom životnog vijeka generatora, kad je primjereno, ovisno o uporabi, o tome je li generator s odobrenjem ili bez odobrenja i svim drugim relevantnim čimbenicima. Proizvodi pripremljeni iz generatora i kompleta s odobrenjem obično ne zahtijevaju provjeru kakvoće (QC), osim ako je tako navedeno u proizvođačevim uputama.
50. Kada je dostupna farmakopejska monografija, provjera kakvoće (QC) radiofarmaceutika bez odobrenja treba se temeljiti na istoj ili je provjeru kakvoće potrebno opravdati na drugi način na temelju vjerojatnih nečistoća, ostatnih otapala iz procesa proizvodnje i rizika za pacijenta. Također potrebno je razmotriti metodu

proizvodnje dotičnog generatora jer ona može biti drugačija od one koja se koristi za pripremu materijala koji se upotrijebio tijekom razvoja monografije.
51. Poluvrijeme života proizvoda zahtijeva da se provjera kakvoće mora završiti unutar kratkog vremenskog razdoblja, tako da se neka dugotrajnija ispitivanja, kao što je ispitivanje sterilnosti, moraju dovršiti retrospektivno. Rezultati praćenja ciklusa (pogledati dio 6) također možda nisu raspoloživi u vrijeme puštanja serije u promet. Važno je, stoga, sakupljati povijesne podatke o prihvatljivoj učinkovitosti čime će se osigurati pouzdanost pri puštanju proizvoda u promet za sigurnu primjenu u bolesnika.
52. Potrebno je prepoznati da za neke radiofarmaceutike može postojati više koraka puštanja u promet; puštanje za isporuku do mjesta primjene, puštanje u promet za primjenu na pacijentima i konačno puštanje u promet kada su dostupni svi rezultati ispitivanja, uključujući ispitivanje sterilnosti.
53. Na mjestu pripreme proizvoda te u odredišnoj organizaciji zdravstvene skrbi, ako je udaljena od mjesta pripreme, treba uspostaviti sustav kojim se osigurava da se radiofarmaceutici ne primjenjuju prije nego je obavljeno puštanje lijeka za primjenu. Proizvode je potrebno transportirati u skladu s lokalnim zakonima o zračenju, a odgovornosti se trebaju definirati tehničkim ugovorom s transportnom tvrtkom, ako se takva koristi.
54. Početno detaljno istraživanje rezultata izvan specifikacije (eng. Out of Specification, OOS) možda neće biti moguće unutar kratkog vremenskog roka do puštanja u promet, pa najprikladnija odluka u tom slučaju može biti odbijanje dotične serije i ponovna priprema. 55. Potreban je postupak kojim se opisuju mjere koje Odgovorna osoba treba poduzeti ako se rezultati izvan specifikacije (OOS) dobiju nakon puštanja lijeka za primjenu. Takve događaje treba istražiti da bi se odredile popravne i zaštitne radnje kako se isto ne bi ponovilo u budućnosti. Ovaj postupak mora se dokumentirati, a odgovornog liječnika obavijestiti što je prije moguće, da bi se odredio utjecaj na pacijenta.
56. Za proizvode koji se primjenjuju prije konačnog puštanja u promet, npr. izrada PET-trendova proizvodnih parametara i rezultati provjere kakvoće (QC) važan su dio puštanja serije u promet kojima se potvrđuje da je serija unutar prihvatljivih granica i da se može pustiti u promet za primjenu na pacijentima. Rezultate izvan trendova potrebno je istražiti te opravdati svako puštanje proizvoda u promet nakon toga.
57. Dovoljan broj zadržanih uzoraka potrebno je pohraniti kroz vremensko razdoblje koje je znanstveno opravdano kao korisno. Obično nisu potrebni uzorci za proizvode pripremljene iz kompleta i generatora s odobrenjem.

DIO 6
Praćenje
58. Za aseptične proizvode za injiciranje, simulacija aseptičkog doziranja mora točno simulirati metodu pripreme i uglavnom obuhvaća prijenos proizvoda štrcaljkom iz jednog spremnika u drugi te razrjeđivanje ili uklanjanje uzoraka iz bočice s proizvodom. Obično nije potrebno provesti simulaciju aseptičkog doziranja ni za koji korak sinteze. Ako postoje neke razlike u načinima na koje se rukuje kompletima s odobrenjem, npr. postoji dodatni korak razrjeđivanja, to je potrebno uzeti u obzir prilikom izrade programa simulacije aseptičkog doziranja.
59. Potrebno je provoditi praćenje okoliša tijekom pripreme nesterilnih proizvoda, ako postoje bilo kakvi mogući rizici za pacijente.
60. Očekuje se da se praćenje okoliša provodi prema učestalosti definiranoj u Dodatku 1 ovih smjernica.
61. Cilj treba biti provođenje brojanja čestica tijekom punjenja.
POJMOVNIK (Uključuje samo terminologiju koja nije već spomenuta u ovim smjernicama.) Radna jedinica - Namjensko mjesto na kojem se odvija rad u zaštićenom okolišu. To može biti izolator ili vruća komora koja je potpuno izolirana od okolnog prostora ili zaštitni kabinet koji je otvoren prema okolnom prostoru, budući da prednji dio možda ne mora biti potpuno zatvoren. Otvorena metoda pripreme – priprema u kojoj se materijalom može rukovati ili ga se može prenositi pomoću otvorenih bočica, pri čemu se proizvod izlaže utjecaju okoliša. Zatvorena metoda pripreme – pogledati glavni dokument no, radi pojašnjenja, elucija iz generatora u zabrtvljenu bočicu, a zatim izvlačenje sadržaja u štrcaljku kroz septum, svrstavala bi se u zatvorenu metodu pripreme.

Literatura:
(1) PIC/S Guide PE 009: Guide to Good Manufacturing Practice for Medicinal
http://www.picscheme.org
(2) Eudralex Volume 4 - Medicinal Products for Human and Veterinary Use:
EU Guidelines to Good Manufacturing Practice http://ec.europa.eu/enterprise/pharmaceuticals/eudralex
(3) ICH Guideline Q9: Quality Risk Management, International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for http://www.ich.org
(4) EN/ISO 14644: Clean rooms and associated controlled environments, European
http://www.cen.eu
(5) Alison M. Beaney (Editor): Quality Assurance of Aseptic Preparation Services,
http://www.pharmpress.com
(6) Note for guidance on quality of water for pharmaceutical use, European
http://www.ema.europa.eu
Povijest revizije
Datum Broj verzije Razlozi revizije
1. travnja 2008. PE 010-2 Ispravak tiskarske greške u točci 54
Dodatka 1
1. listopada 2008. PE 010-3 Ispravak tiskarske greške u točci 85
Dodatka 1
1. ožujka 2014. PE 010-4 Dopuna Dodatka 3





























