Embed Size (px)
Citation preview
1
BIOCHIMIE DU SANG1. Métabolisme de l’erythrocyte2. Production et élimination d’éléments
cellulaires: synthèse de hème: pathologies3. Metabolisme et Transport du Fe4. Composants du Plasma (structure &
fonction)
PorphyriesDéficience aquise ou hereditaire de la synthèse du heme - accumulation et excretion augmentée des précurseursmetaboliques (each unique) - toutes les porphyries sont autosomales dominantes, saufles porphyries congenitales erythropoietiques (recessives)
origines: hepatique (=aïgue ou chronique) ou erythropoietique
Certains intermédiaires tetrapyrrole sont photosensibles - formation de radicaux superoxide - croutes sur la peau, prurites - peau peut s’assombrir, pilosité - hypertrichosis
-
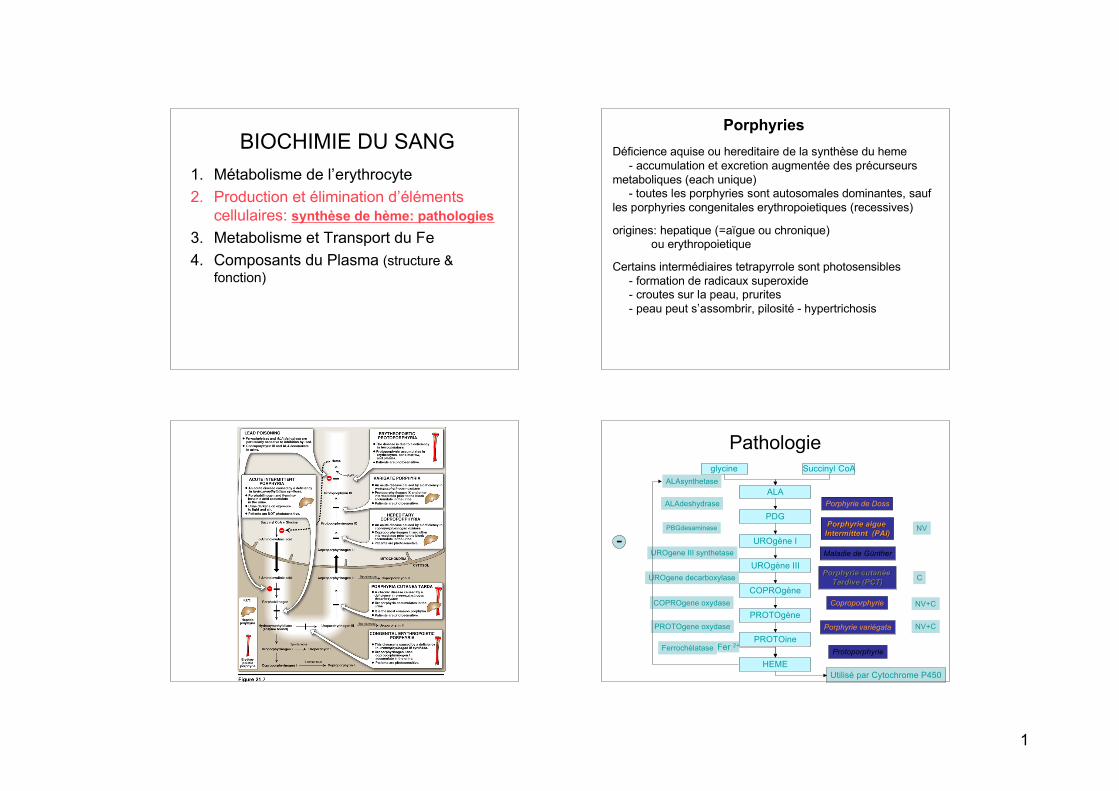
PathologieSuccinyl CoAglycine
PROTOine
PROTOgène
COPROgène
UROgène III
UROgène I
PDG
ALA
HEME
Fer 2+
ALAsynthetase
ALAdeshydrase
PBGdesaminase
UROgene III synthetase
UROgene decarboxylase
COPROgene oxydase
PROTOgene oxydase
Ferrochélatase
Porphyrie de Doss
Porphyrie aigue Porphyrie aigue Intermittent (PAI)Intermittent (PAI)
Maladie de Günther
Porphyrie cutanée Porphyrie cutanée Tardive (PCT)Tardive (PCT)
Coproporphyrie
Porphyrie variégata
Protoporphyrie
Utilisé par Cytochrome P450
NV
C
NV+C
NV+C
2
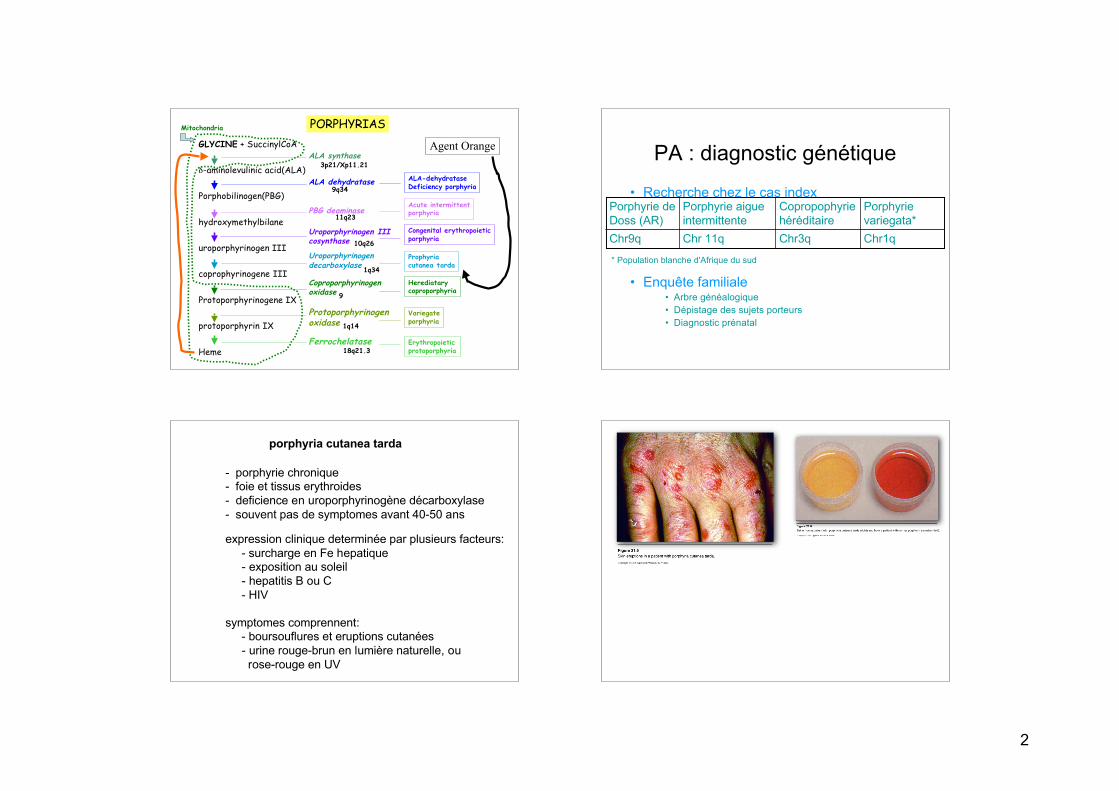
PORPHYRIASGLYCINE + SuccinylCoA
δ-aminolevulinic acid(ALA)
Porphobilinogen(PBG)
hydroxymethylbilane
uroporphyrinogen III
coprophyrinogene III
Protoporphyrinogene IX
protoporphyrin IX
Heme
ALA synthase
ALA dehydratase
PBG deaminase
Uroporphyrinogen IIIcosynthase
Uroporphyrinogendecarboxylase
Coproporphyrinogenoxidase
Protoporphyrinogenoxidase
Ferrochelatase
ALA-dehydrataseDeficiency porphyria
Acute intermittentporphyria
Congenital erythropoieticporphyria
Prophyria cutanea tarda
Herediatarycoproporphyria
Variegateporphyria
Erythropoieticprotoporphyria
Mitochondria
9q34
11q23
10q26
1q34
9
1q14
18q21.3
3p21/Xp11.21
Agent Orange PA : diagnostic génétique
• Recherche chez le cas index
• Enquête familiale• Arbre généalogique• Dépistage des sujets porteurs• Diagnostic prénatal
Chr1qChr3qChr 11qChr9q
Porphyrievariegata*
Copropophyriehéréditaire
Porphyrie aigueintermittente
Porphyrie deDoss (AR)
* Population blanche d'Afrique du sud
porphyria cutanea tarda
- porphyrie chronique- foie et tissus erythroides - deficience en uroporphyrinogène décarboxylase- souvent pas de symptomes avant 40-50 ans
expression clinique determinée par plusieurs facteurs: - surcharge en Fe hepatique - exposition au soleil - hepatitis B ou C - HIV
symptomes comprennent: - boursouflures et eruptions cutanées - urine rouge-brun en lumière naturelle, ou rose-rouge en UV

3
Acute hepatic porphyrias
- porphyrie aigue intermittente (hydroxymethylbilane synthase)- coproporphyrie héréditaire (coprophyrinogen oxidase)
- variagate porphyrie (protoporphyrinogen oxidase)
-symptomes similires - attaques aïgues et douleureusesde nature gastrointestinales,neurologiques/psychologiques, cardiovasculaires
-souvent augmentée- par médicaments, barbituriques, infections, jeûnes, alcool
- activateurs du système p450 - utilise le heme, - augmente ALA synthase, augmente les métabolitespathologiques
erythropoietic porphyrias- porphyrie congenitale erythropoietique (uroporphyrinogen III synthase)
- protoporphyrie erythropoietique (ferrochelatase)
symptomes : - boursouflures et eruptions cutanées dans l’enfance - cirrhose hépatique cholestatique et déficit hépatiqueprogressif
Traitement
-soutien medical contre la douleur et vomissements- hemine: diminue synthèse de ALA synthase- éviter la lumière du soleil et certains médicaments,drogues, etc.,
Roi George III - “Mad King George”
Crises aigues de douleurs abdominales et confusionmentale - sans doute porphyrie - avec complications en raison de tous les médicamentsque les médecins lui administraient
vampires et loups-garous? - on pense que ce sont des porphyries mal interprétéesau moyen-age - photosensibilité, sang rouge (aussi dents) hypertrichosis
The Madness of Inbreeding
George III : fortes douleurs abdominales, confusion mentale, urinesombre.
4
Porphyries acquises
- hexochlorobenzene comme fongicide en Turquie en 1950s - des milliers d’enfants ont mangé du pain de blé traité
- ont développé une porphyria cutanea tarda due à l’inhibition de la uroporphyrinogène décarboxylase
-due à une hypertrichosis- “monkey children”
Porphyries acquises
Empoisonnement au plomb
- inhibition de ferrochelatase, ALA dehydratase - désplace le Zn+2 sur le site actif de l’enzyme
enfants - troubles du development - baisse de IQ - hyperactivité - insomnie - nombreux autres problèmes de santé
adultes - fortes douleurs abdominales - confusion mentale - nombreux autres symptomes
Heme stimulates hemoglobin synthesis in reticulocytes
HCI = heme controlledinhibitor
Reduced initiation of translation

5
***
Dans Erythrocyte, la synthèse de heme est regulatée au niveau desenzymes ferrochelatase* and porphobilinogen deaminase** TOXICITE au Plomb
Symptomes• Irritibilité • faible appetit• Lethargie • douleurs abdominales• insommnie (+ vomissements)• maux de tête • Constipation
Pathophysoiologie• le Pb se lie sur de nombreux groupes sulfhydryl• Inhibe de multiples reactions enzymatiques, en particulier
celles de la biosynthesis du heme (PBG synthase & ferrochelatase)
• principal symptome d’intoxication au Pb: élévation de 5-ALA sans augmentation concomitante de PBG
BIOCHIMIE DU SANG1. Métabolisme de l’erythrocyte2. Production et élimination d’éléments
cellulaires3. Metabolisme et Transport du Fe4. Composants du Plasma (structure &
fonction): bilirubine - dégradation du hème
6
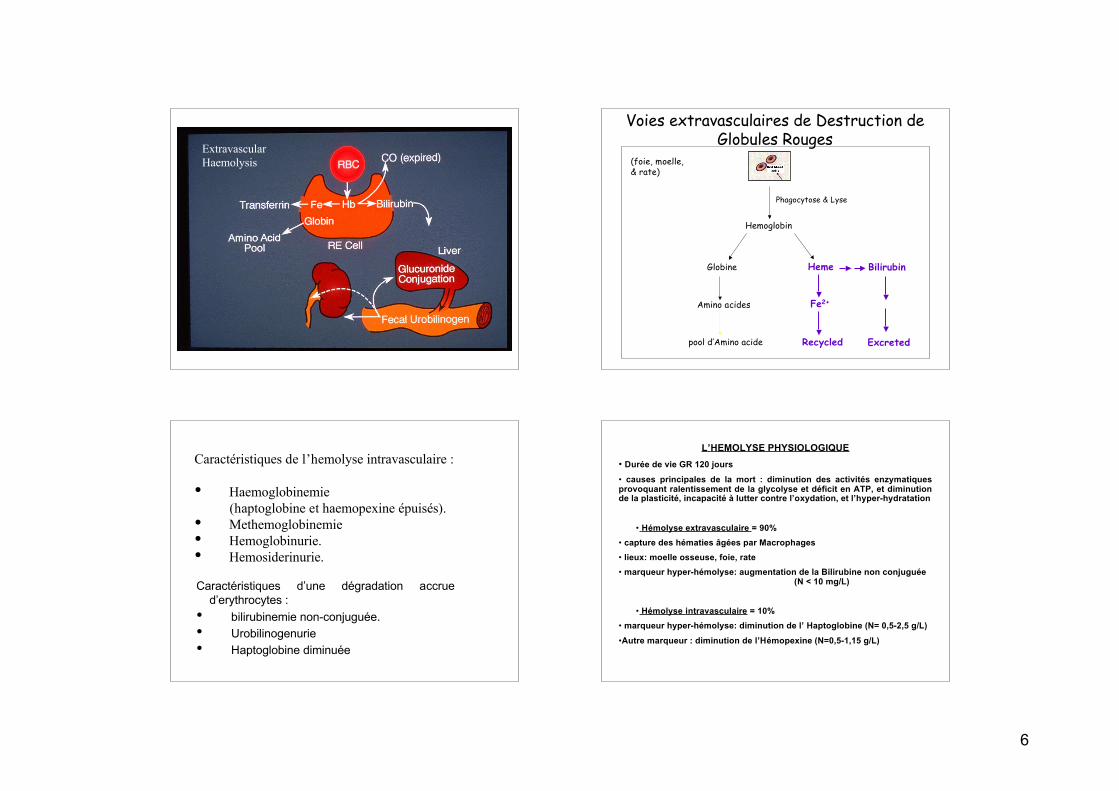
ExtravascularHaemolysis
Voies extravasculaires de Destruction deGlobules Rouges
(foie, moelle, & rate)
Hemoglobin
Globine
Amino acides
pool d’Amino acide
Heme Bilirubin
Fe2+
Excreted
Phagocytose & Lyse
Recycled
Caractéristiques de l’hemolyse intravasculaire :
• Haemoglobinemie(haptoglobine et haemopexine épuisés).
• Methemoglobinemie• Hemoglobinurie.• Hemosiderinurie.
Caractéristiques d’une dégradation accrued’erythrocytes :
• bilirubinemie non-conjuguée.• Urobilinogenurie• Haptoglobine diminuée
L’HEMOLYSE PHYSIOLOGIQUE• Durée de vie GR 120 jours
• causes principales de la mort : diminution des activités enzymatiquesprovoquant ralentissement de la glycolyse et déficit en ATP, et diminutionde la plasticité, incapacité à lutter contre l’oxydation, et l’hyper-hydratation
• Hémolyse extravasculaire = 90%• capture des hématies âgées par Macrophages• lieux: moelle osseuse, foie, rate• marqueur hyper-hémolyse: augmentation de la Bilirubine non conjuguée
(N < 10 mg/L)
• Hémolyse intravasculaire = 10%• marqueur hyper-hémolyse: diminution de l’ Haptoglobine (N= 0,5-2,5 g/L)
•Autre marqueur : diminution de l’Hémopexine (N=0,5-1,15 g/L)
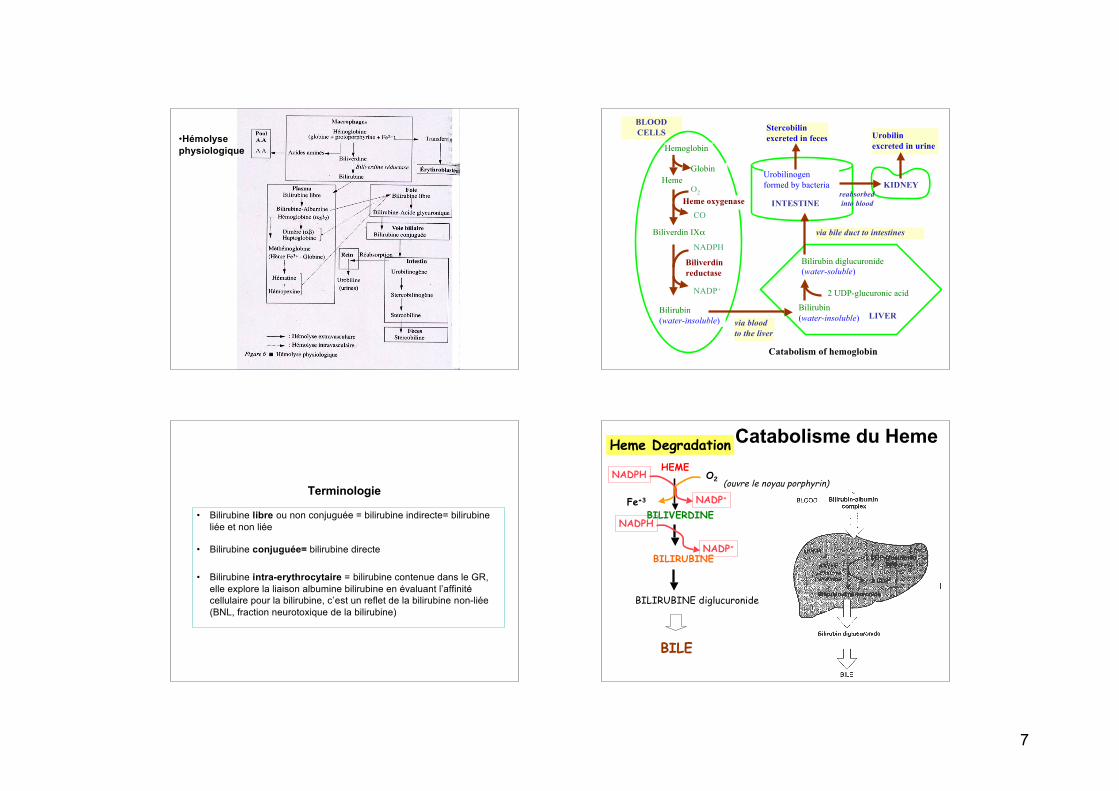
7
•Hémolysephysiologique
BLOODCELLS
LIVER
Bilirubin diglucuronide(water-soluble)
2 UDP-glucuronic acid
via bile duct to intestines
Stercobilinexcreted in feces
Urobilinogenformed by bacteria KIDNEY
Urobilinexcreted in urine
CO
Biliverdin IXα
Heme oxygenaseO2
Bilirubin(water-insoluble)
NADP+
NADPH
Biliverdinreductase
HemeGlobin
Hemoglobin
reabsorbed into blood
Bilirubin(water-insoluble)via blood
to the liver
INTESTINE
Catabolism of hemoglobin
Terminologie
• Bilirubine libre ou non conjuguée = bilirubine indirecte= bilirubineliée et non liée
• Bilirubine conjuguée= bilirubine directe
• Bilirubine intra-erythrocytaire = bilirubine contenue dans le GR,elle explore la liaison albumine bilirubine en évaluant l’affinitécellulaire pour la bilirubine, c’est un reflet de la bilirubine non-liée(BNL, fraction neurotoxique de la bilirubine)
Catabolisme du HemeHeme DegradationHEME
BILIVERDINE
O2
Fe+3
NADPH
NADP+
(ouvre le noyau porphyrin)
BILIRUBINE
NADPH
NADP+
BILIRUBINE diglucuronide
BILE

8
Traitement de Hemoglobin libre (Intravasculaire)
Buts: 1. Scavenge iron2. Empêcher des pertes majeures de Fe3. Complexer le heme libre (très toxique)
• Haptoglobine : le complex hemoglobin-haptoglobin est rapidementmétabolisé par le foie et la rate, formant un complexe Fe-globiune etbilirubine. Empêche une perte de Fe dans l’urine.
• Hemopexine: lie heme libre. Le complexe heme-hemopexin est capté parle foie et le Fe est stoqué par la ferritine
• Methemalbumine: complexe le heme oxidé et albumin.
Dégradation du hème
Dégradation du Hème
L’essentiel du hème est d’origine erythrocytaire (85%) - le rest provient de turnover de cytochromes,
p450s, erythrocytes immature
GR dure 120 jours, degradé par système reticuloendothelial (RE)
Formation de BilirubineRE : heme oxygenase microsomale - requiert NAPDH, O2 - hydroxyle le pont methenyl - Fe+2 oxidé en Fe+3 - 2ème reaction clive le noyau - CO libéré avec Fe+3
produit biliverdin = pigment vertréduit en bilirubine (rouge-orange) = pigments de bile
Dégradation du Heme
Capture de bilirubine par le foiehydrophobe - transporté par l’albumine - des médicaments anioniquespeuvent displacer la bilirubin, dommages du SNC chez le nourisson (salicylates)lie des protéines intracellulaires dans le foie - ligandin
formation de bilirubine diglucuronideconjugation de 2 molecules de glucuronate - augmente solubilité - bilirubin glucuronyltransferase - l’acide UDP-glucuronique est le doneur - la bilirubine conjuguée lie aussi l’albumine,mais très faiblement

9
Degradation du Heme
excretion de bilirubine dans la biletransport actif dans la bile
-requiert de l’energie - susceptible de troubles hépatiques - bilirubine non-conjuguée n’est pas excretée
formation d’urobilinedans l’intestin
- bilrubine diglucuronide est hydrolysée et reduite par bacteries en urobilinogène
- urobilinogene oxidée par bacteries en stercobiline - couleur brune desmatières fecales
- urobilinogène est en partie reabsorbée dans le sang
- cycle enterohépatique del’urobilinogène (capté par le foie et re-excreté dans la bile)
- urobilnogène: dans le rein - convertie en urobiline (jaune) -ceci donne à l’urine sa couleur caractéristique
Terminologie• Bilirubine libre ou non conjuguée = bilirubine indirecte= bilirubine liée et non liée• Bilirubine conjuguée= bilirubine directe• Bilirubine intra-erythrocytaire = bilirubine contenue dans le GR , elle explore la
liaison albumine bilirubine en évaluant l’affinité cellulaire pour la bilirubine, c’estun reflet de la BNL ( fraction neurotoxique de la bilirubine)

10
Jaunissee (icterus)
hyperbilirubinemie - provoque coloration jaune de la peau, ongles et sclerose
- pas une maladie, mais un symptome de troubles sous-jacents
HYPER-BILIRUBINEMIES•concentrations plasmatiques augmentées de bilirubine (> 3 mg/dL)lorsqu’il y a déséqulilbre entre production et excretion•Cliniquement reconnu comme jaunisse
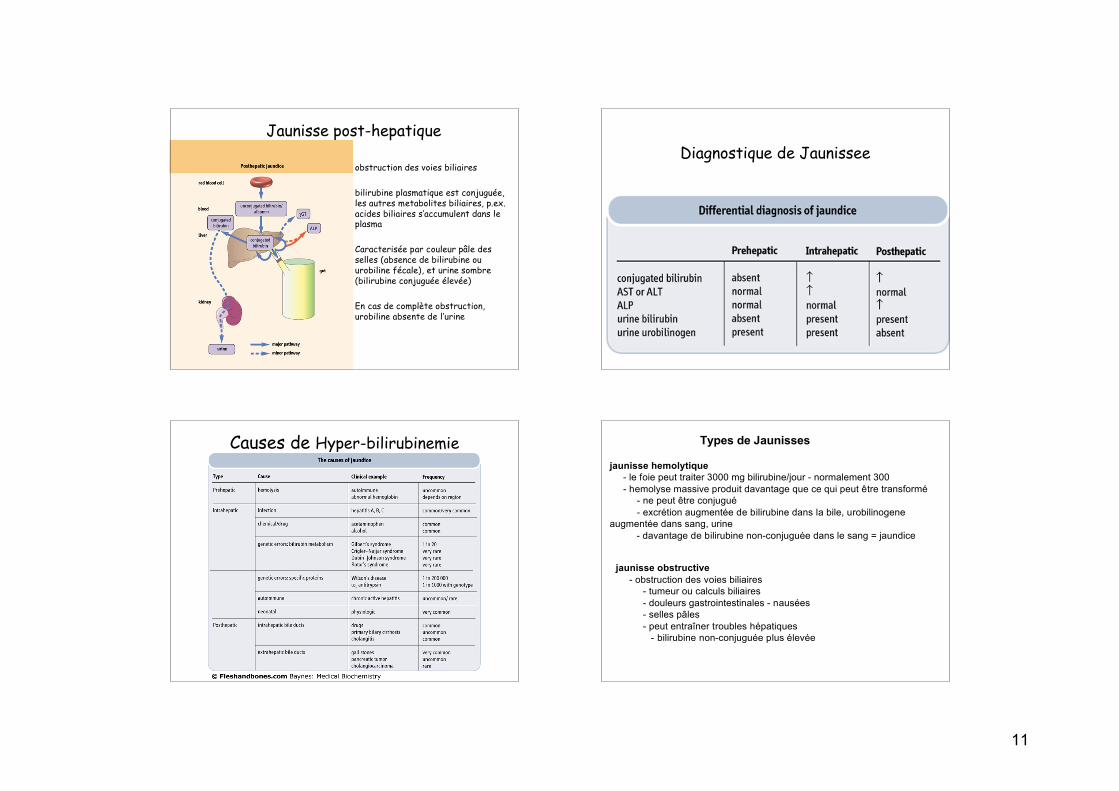
Jaunisse Pré-hépatique (hemolytique)
• production excessive de bilirubine(supérieure aux capacités deconjuguaison du foie) suite àhémolyse
• Lyse excessive d’érythrocytessouvent dûe à une maladieautoimmune; maladie hemolytique dunouveua-né (incompatibilité Rh- ouABO-); érythrocytesstructurallement anormaaux (Sicklecell disease); ou dégradationextravasculaire
• concentrations plasmatiques élevéesde bilirubins non-conjuguée(concentration normale ~0.5 mg/dL)
Jaunisse intra-hépatique
• Trouble de capture,conjugation, ou secretion oubilirubine
• Reflète une dysfonctiongeneralisée du foie(hepatocyte)
• Dans ce cas,hyperbilirubinemie estnormallement accompagnéepar d’autres troubles desmarqueurs biochemiques dela fonction hépatique
11
Jaunisse post-hepatique
• obstruction des voies biliaires
• bilirubine plasmatique est conjuguée,les autres metabolites biliaires, p.ex.acides biliaires s’accumulent dans leplasma
• Caracterisée par couleur pâle desselles (absence de bilirubine ouurobiline fécale), et urine sombre(bilirubine conjuguée élevée)
• En cas de complète obstruction,urobiline absente de l’urine
Diagnostique de Jaunissee
Causes de Hyper-bilirubinemie Types de Jaunisses
jaunisse hemolytique - le foie peut traiter 3000 mg bilirubine/jour - normalement 300 - hemolyse massive produit davantage que ce qui peut être transformé - ne peut être conjugué - excrétion augmentée de bilirubine dans la bile, urobilinogeneaugmentée dans sang, urine - davantage de bilirubine non-conjuguée dans le sang = jaundice
jaunisse obstructive - obstruction des voies biliaires - tumeur ou calculs biliaires - douleurs gastrointestinales - nausées - selles pâles - peut entraîner troubles hépatiques
- bilirubine non-conjuguée plus élevée

12
Jaunisse hepatocellulaire - endommagement du foie (cirrhose ou hepatite)provoque une élévation de bilirubine dans le sang, suite àmanque de conjuguaison - bilirubine conjuguée n’est pas exportée dans la bile donc diffuse dans le sang - urobilinogene augmentée dans circulation entero-hépatique - => urine sombre, selles claires - ASAT & ALAT elevatées - nausées et anorexie
Types de Jaunisses Jaunisse du nouveau-né
Les prematurés accumulent souvent de la bilirubineCause: expression tardive de bilirubine glucuronyl-transferase - expression maximum (niveau adulte) à ~ 4 semaines - bilirubine en exces peut provoquer encephalopathietoxique (kernicterus) - traitement. lumière fluorescente bleue - convertit la bilirubine en composé plus polaire - sera excreté dans la bile sans conjugation - syndrome de Crigler-Najjar: deficience de bilirubine glucuronyltransferase
Anémie hémolytique
excesshemolysis
bilirubine non conjuguée (dans le sang) bilirubine conjuguée
(libérée dans lescanalicules biliaires)
Hepatite
bilirubine nonconjuguée (dans le sang) bilirubin conjuguée (dans le sang)
Calcul biliaire
bilirubine non conjuguée (dans le sang) bilirubin conjuguée (dans le sang)
Exemples d’hyperbilirubinemies
Troubles génétiques du Metabolisme de Bilirubine
Jaunisse modéréebilirubineconjuguée ⇑⇑
transport anormal debilirubine conjuguée dans lesysteme biliaire
syndromeDubin-Johnson
Très faible jaunisedurant maladie
Bilirubine non-conjuguée ⇑
activité réduite deUDP-glucuronyltransferase
syndromeGilberts
Profonde jaunisseBilirubine non-conjuguée ⇑⇑⇑
Forte déficience enUDP-glucuronyltransferase
syndromeCrigler-Najjar
Tableau cliniqueBilirubineDéficienceCondition

13
Cycle de la bilirubine
Hb hème
Globineet fer
Biliverdine + CO
bilirubine
bilirubine liée
Bilirubine conjuguée
Stercobilinogène et urobilinogène
MyoglobineCytochromecatalase
Hème oxygénase
réductase
albumineHEPATOCYTES
Ligandines Y & Z
Ac glucuronique
Glucuronyl Tase
bactéries
β-glucuronidase
Bilirubine non liée
Terminologie• Bilirubine libre ou non conjuguée = bilirubine
indirecte= bilirubine liée et non liée
• Bilirubine conjuguée= bilirubine directe
• Bilirubine intra-erythrocytaire = bilirubinecontenue dans le GR , elle explore la liaisonalbumine bilirubine en évaluant l’affinitécellulaire pour la bilirubine, c’est un reflet dela BNL ( fraction neurotoxique de la bilirubine)
Les ictères à bilirubine libre
ICTÈRE DU NOUVEAU NE
BILIRUBINE BILIRUBINELIBRE CONJUGUÉE
Hémolytique Non hémolytique : ictère simple : ictère au lait de mère
: maladie de Gilbert /Crigler-Najjar: Hypothyroïdie
Corpusculaire Extra corpusculaire1°) anomalie de la membrane: allo-immunisation : ABO et Rh spherocytose
infectieux2°) anomalies enzymatiques:
deficit en G6PD et en PK
NB la thalassémie et la drépanocytose n’ont pas de révélation néonatale du fait de l’importancedu contingent d’ hémoglobine F
ICTERES NONHEMOLYTIQUES

14
Ictère simple• 1- hémolyse physiologique plus importante chez le NN, (masse
globulaire plus élevée et durée de vie des GR plus courte.
• 2- augmentation du taux d’ hème oxygénase, ( endotoxinesbactériennes, stress post hypoxique.
• 3- immaturité de la glucuronyl Transférase
• 4- flore bactérienne se fait progressivement après la naissance.
• 5- protéine Y & Z synthétisée en post natal.
• 6- medicaments qui se lient à l’albumines: caféine, diazépam,furosemide, digoxine, aminosides.
ictère au lait de mère• Début vers J5• Secondaire à l’existence dans le lait d’une lipoprotéine
lipase, inhibant la glycuroconjugaison par libérationexcessive d’acide gras inhibiteurs compétitifs de labilirubine
• Disparition de l’ictère à J3 de sevrage, ou aprèschauffage du lait à 56° C
• En pratique « on s’en fout… » enfin on explique à lamaman qui est inquiète
Maladie de Gilbert
• Déficit partiel en glucuronyl transférase• Maladie autosomique dominante• ictère modéré en période néonatale
•Trouble hépatique bénin
• transmissible dans 50% des individus concernés
•Faible augmentation de bilirubine non-conjuguée• dûe à une baisse de la capacité du foie de conjuguer labilirubine•souvent en corrélation avec une maladie ou un jeûne
•plus fréquent chez Hommes que chez Femmes•début des symptomes dans l’adolescence, et entre 20-30 ans
•traité par faibles doses de phenobarbital pour stimulerl’activité UDP glucuronyl transferase
Syndrome de Gilbert

15
• Trouble de secretion biliaire de bilirubineconjuguée
• Accompagné d’hyperbilirubinemie faible
Syndromes de Dubin-Johnson et RotorMaladie de Crigler-Najjar
• Type 1– Déficit complet et létal de la glucuronyl transférase– Autosomique récessive– Notion de consanguinité
• Type 2– Déficit partiel en glucuronyl transférase– Autosomique dominant à pénétrance variable
• Autosomal recessive
• très rare < 200 cases dans le monde – frequence genique < 1:1000
• complete absence de forte reduction de conjuguaison de bilirubine
• accompagné de severe hyperbilirubinemie non-conjuguée,normalement présente dès la naissance
• individus présentent un grand risque kernicterus
• Condition fatale si l’enzyme est completement absent
• Treatement par phototherapie (10-12 hrs/day) et transplantationhépatique vers 5 ans
Syndrome Crigler-Najjar
Ictères hémolytiques

16
Ictère hémolytique d’origine corpusculaire
• Anomalie constitutionnelle de lamembrane des GR• Anomalie enzymatique de la membrane
des GR
Maladie de Minkowski-Chauffardou sphérocytose héréditaire
• Transmission autosomique dominante degravité variable (pas de prédisposition depopulation)
• Grande cause d’anémie constitutionnellefréquente dans la population française
Maladie de Minkowski Chauffardou sphérocytose héréditaire
• Physiopathologie– Anomalie protéique membranaire au niveau de la spectrine– Perturbation de la fonction ATP ase, avec flux exagéré de
Na ds la cellule entraînant une hyper H2Oballonnement de la Cellule sphérocyte (pertede phosphoPL membranaire perte de surface
microsphérocyte irréversible dense et rigide se déformantmal pour traverser les petits capillaires de la rate (cordonsde Billroth) HEMOLYSE ICTERE
Autres anomalies constitutionnellesde la membrane des GR
• Elliptocytose familiale• Stomatocytose héréditaire et anomalies
d’échanges des cations• Acanthocytose constitutionnelle

17
Déficit en G6PD• 1OO millions de porteurs (pourtour méditerranéens, Afrique, Asie)• Physiopathologie
– >130 mutations (g situé sur le Chr X déficit surtout chez les hommes– Fonction anormales:instabilité de la molécule qui se dégrade prématurément dans le GR– Enzyme déficitaire: modification du site enzymatique - l’affinité pour le substrat est
réduite– Accumulation de peroxyde toxique que la glutathion peroxydase ne pourra éliminer
Hémolyse• Diagnostic: dosage enzymatique• Evolution parfois mortelle• Traitement: transfusion et interdire les médicaments oxydants qui augmentent les
peroxydes ( APS, sulfamides, chloramphénicol, vit K)
Déficit en pyruvate kinase• Maladie autosomique récessive• Physiopathologie
– Défaut de régénération de l’ATP déficit de la pompe Na et anomales des échangeslipidiques hémolyse chronique avec poussées
• Diagnostic– dosage enzymatique
• Traitement– Transfusion– Splénectomie parfois si destruction splénique
Incompatibilité ABO• Mère O, enfant A ou B• Passage d’agglutinines irrégulières anti A ou B d’origine maternelle
dans la circulation fœtale, acquis lors d’une transfusion ou d’uncontact antérieur avec l’antigène
• COMMBS direct souvent (-), présence d’agglutinines anti A ou B
Incompatibilité Rhésus• Mère Rh (-), enfant Rh (+)• Physiopathologie
– Passage trans-placentaire d’hématies fœtales porteuse de l’Ag D,chez une mère Rh (-) au cours
• D’une grossesse antérieure• secondaire à
– Décollement trophoblastique– HRP, FCS, IVG, GEU– Prélèvements ovulaires (biopsie de trophoblaste, amniocentèse, PSF)– Chirurgie abdo-pelvienne– Traumatisme abdominal
Ictère hémolytique infectieux• Agression directe de l’hématie par l’agent
infectieux
Ictère à bilirubine conjuguée• Physiopathologie
– Cholestase= déficit de sécrétion hépatique des acidesbiliaires
• Mécanisme– Obstacle intra ou extra-hépatique
• Clinique– Selles décolorées, urines foncées, +/- HM
Cholestase extra hépatique

18
Atrésie des voies biliaires• 1/ 10 000 enfants• Précocité du diagnostic (échographie) et chirurgie < 6 semaines ou
risque d’évolution vers la cirrhose (transplantation)
Cholestase extra hépatique
• Syndrome d’Alagille ou paucité ductulaire syndromatique– Cholestase et syndrome polymalformatif– Autosomique dominant (Chr 20)– Raréfaction des voies biliaires intra hépatique– 10 % évolution vers la cirrhose
• Infection (E.Coli) foetopathie (rubéole, CMV), postnatal ( EBV, EchoV,Coxsakiie, adenoV)
• Métabolique tyrosinémie, galactosémie, intolérance au fructose• Endocrine déficit en cortisol, Hypoplasie congénitale des surrénales)
• Génétiques: syndrome de Zellweger, mucoviscidose, déficit en α1−anti-trypsine
• Autres: cholangite sclérosante, hypoxie hépatique
Cholestase intra hépatique





























