Embed Size (px)
Citation preview

Präparation und Kristallisation von
ribosomalen Proteinen mit dem
Kernimportfaktor Importin 7
Diplomarbeit
vorgelegt von
Christian Schiller
aus
Kassel
angefertigt
im Institut für Mikrobiologie und Genetik an der Biologischen Fakultät
der Georg-August-Universität zu Göttingen
2006

Referent: Prof. Dr. Ralf Ficner
Korreferent: Prof. Dr. Oliver Einsle
Tag der Abgabe der Diplomarbeit: 29. Juni 2006
Letzter Tag der mündlichen Diplomprüfung: 6. Juli 2005
Hiermit erkläre ich, dass die vorliegende Diplomarbeit „Präparation und Kristallisation
von ribosomalen Proteinen mit dem Kernimportfaktor Importin 7“ selbstständig verfasst
und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt wurden.
Göttingen, den 29. Juni 2006

Inhaltsverzeichnis
III
INHALTSVERZEICHNIS
1. EINLEITUNG ....................................................................................................................................1
1.1 DER ZELLKERN..........................................................................................................................1
1.2 GRUNDLAGEN DES KERNTRANSPORTS......................................................................................1
1.2.1 DER KERNPORENKOMPLEX.....................................................................................................1
1.2.2 REZEPTOREN DER IMPORTIN ß/ß-KARYOPHERIN-SUPERFAMILIE ...........................................3
1.2.3 KARYOPHERIN-VERMITTELTER KERNIMPORT........................................................................5
1.2.4 DAS RANGTP-SYSTEM...........................................................................................................6
1.3 KERNIMPORT DER RIBOSOMALEN PROTEINE L5 UND L23A.....................................................7
1.3.1 BIOCHEMISCHE DATEN ZU RPL5 ............................................................................................7
1.3.2 BIOCHEMISCHE DATEN ZU RPL23A AUS HOMO SAPIENS........................................................9
1.3.3 KARYOPHERIN-VERMITTELTER KERNIMPORT VON RPL5 UND RPL23A .................................9
1.3.4 RIBOSOMALE BIOGENESE.....................................................................................................11
1.3.5 FUNKTIONELLE ROLLE VON RPL5 BEI TRANSPORTPROZESSEN DER 5S RNA......................12
1.4 AUFGABENSTELLUNG UND ZIELSETZUNG ..............................................................................14
2 MATERIAL UND METHODEN............................................................................................15
2.1 MATERIAL................................................................................................................................15
2.1.1 FEINCHEMIKALIEN ................................................................................................................15
2.1.2 GERÄTE.................................................................................................................................15
2.1.3 KIT-SYSTEME........................................................................................................................17
2.1.4 ORGANISMEN........................................................................................................................17
2.1.5 PLASMIDE..............................................................................................................................17
2.1.6 ENZYME UND INHIBITOREN ..................................................................................................18
2.1.7 GRÖßENSTANDARDS.............................................................................................................18
2.1.8 DNA-OLIGONUKLEOTIDE.....................................................................................................19
2.1.9 KRISTALLISATIONSSCREENS.................................................................................................20
2.1.10 COMPUTERPROGRAMME.....................................................................................................20
2.2 METHODEN..............................................................................................................................21
2.2.1 MOLEKULARBIOLOGISCHE METHODEN................................................................................21
2.2.1.1 Nukleinsäurebiochemische Methoden...............................................................................21
2.2.1.1.1 Polymerase-Kettenreaktion.............................................................................................21
2.2.1.1.2 Spaltung von DNA durch Restriktionsendonukleasen ...................................................23
2.2.1.1.3 DNA-Dephosphorylierung am 5´-Ende..........................................................................23

Inhaltsverzeichnis
IV
2.2.1.1.4 DNA-Ligation.................................................................................................................24
2.2.1.1.5 DNA-Sequenzierung.......................................................................................................24
2.2.1.1.6 Agarosegelelektrophorese...............................................................................................26
2.2.1.1.7 Visualisierung von Nukleinsäuren mit Ethidiumbromid ................................................26
2.2.1.1.8 DNA-Isolierung aus Agarosegelen.................................................................................27
2.2.2 ZELLBIOLOGISCHE METHODEN............................................................................................27
2.2.2.1 Transformation chemisch kompetenter Zellen ..................................................................27
2.2.2.2 Präparation von Plasmid-DNA..........................................................................................28
2.2.2.3 Expression rekombinanter Proteine...................................................................................28
2.2.2.3.1 Expression rekombinanter Proteine in Escherichia coli im kleinen Maßstab ................28
2.2.2.3.2 Überexpression von rekombinanten Proteinen in Escherichia coli................................29
2.2.2.3.3 Ernte und Aufschluss einer Expressionskultur ...............................................................29
2.2.3 PROTEINBIOCHEMISCHE METHODEN....................................................................................31
2.2.3.1 Einengen von Proteinlösungen durch Zentrifugation ........................................................31
2.2.3.2 Aufbereitung von Zellproben für eine SDS-PAGE ...........................................................31
2.2.3.3 Diskontinuierliche Polyacrylamidgelelektrophorese von Proteinen..................................31
2.2.3.4 Visualisierung von Proteinen durch Coomassie Brilliant Blue.........................................33
2.2.3.5 Proteolytische Spaltung von GST-Fusionsproteinen.........................................................33
2.2.4 CHROMATOGRAPHISCHE METHODEN...................................................................................34
2.2.4.1 Affinitätschromatographische Reinigung von GST-Fusionsproteinen..............................34
2.2.4.2 Affinitätschromatographische Reinigung über Ni+-NTA-Sepharose ................................35
2.2.4.3 Kationenaustauschchromatographie über SP-Sepharose...................................................36
2.2.4.4 Ausschlusschromatograpisches Umäquilibrieren von Proteinen.......................................37
2.2.4.5 Präparative Ausschlusschromatographie von Proteinen....................................................38
2.2.4.6 Analytische Ausschlusschromatographie von Proteinen...................................................39
2.2.4.7 Kalibrierung einer Auschlusschromatographiesäule .........................................................40
2.2.5 ISOTHERME TITRATIONSKALORIMETRIE ..............................................................................40
2.2.6 SPEKTROSKOPISCHE METHODEN..........................................................................................42
2.2.6.1 Absorption .........................................................................................................................42
2.2.6.1.1 Konzentrationsbestimmung von Nukleinsäure-Lösungen..............................................43
2.2.6.1.2 Konzentrationsbestimmung von Protein-Lösungen........................................................43
2.2.7 KRISTALLOGRAPHISCHE METHODEN...................................................................................45
2.2.7.1 Kristallisation von Proteinen .............................................................................................45
2.2.7.2 Micro-Seeding zur Verbesserung der Kristallordnung ......................................................47

Inhaltsverzeichnis
V
3 ERGEBNISSE...........................................................................................................................48
3.1 BIOCHEMISCHE ARBEITEN MIT DEM PROTEIN RPL5 AUS XENOPUS LAEVIS .........................49
3.1.1 KLONIERUNG UND EXPRESSION VON RPL5 ..........................................................................49
3.1.1.1 Subklonierung von rpL5 aus Xenopus laevis in den Vektor pGEX-6P-1..........................49
3.1.1.2 Expression von rpL5 mit GST-Affinitätssequenz..............................................................49
3.1.1.3 Zellernte und Aufschluss von GST-rpL5...........................................................................50
3.1.2 REINIGUNG VON RPL5 AUS XENOPUS LAEVIS........................................................................51
3.1.2.1 Affinitätschromatographische Reinigung über Glutathion-Sepharose ..............................51
3.1.2.2 Spaltung des Fusionsproteins GST-rpL5 durch PreScission Protease...............................52
3.1.2.3 Kationenaustauschchromatographische Reinigung von rpL5 ...........................................53
3.1.2.4 Finales Umpuffern von rpL5 .............................................................................................54
3.1.2.5 Ausschlusschromatographische Analyse des gereinigten rpL5.........................................56
3.1.3 KRISTALLISATIONSVERSUCHE MIT DEM KOMPLEX IMP7/RPL5............................................58
3.1.3.1 Präparation eines Komplexes aus rpL5 und Importin 7.....................................................58
3.1.3.2 Versuch einer Kristallisation des Imp7/rpL5-Komplexes .................................................59
3.1.4 KLONIERUNG VERKÜRZTER FRAGMENTE VON RPL5............................................................60
3.2 BIOCHEMISCHE ARBEITEN MIT DEM PROTEIN RPL23A AUS HOMO SAPIENS ........................62
3.2.1 EXPRESSION, ZELLERNTE UND AUFSCHLUSS VON RPL23A ..................................................62
3.2.1.1 Subklonierungen von rpL23a in die Vektoren pGEX-6P-1 und pQE80 ...........................62
3.2.1.2 Expression von rpL23a......................................................................................................63
3.2.1.3 Zellernte und Aufschluss der Expressionskulturen............................................................64
3.2.2 REINIGUNG VON RPL23A AUS HOMO SAPIENS......................................................................64
3.2.2.1 Versuch einer Reinigung von rpL23a mit Deca-Histidin-Sequenz ...................................64
3.2.2.2 Affinitätschromatographische Reinigung über Glutathion-Sepharose ..............................65
3.2.2.3 Kationenaustauschchromatographische Reinigung von rpL23a........................................67
3.2.2.4 Finales Umpuffern von rpL23a..........................................................................................69
3.2.2.5 Ausschlusschromatographische Analyse des gereinigten rpL23a .....................................70
3.2.3 KRISTALLISATIONSVERSUCHE MIT DEM KOMPLEX IMP7/RPL23A .......................................71
3.2.3.1 Präparation eines Komplexes aus rpL23a und Importin 7.................................................71
3.2.3.2 Kristallisationsversuche mit Imp7/rpL23a.........................................................................73
3.2.4 FUNKTIONELLE UNTERSUCHUNGEN DER BINDUNG VON RPL23A DURCH
KERNIMPORTREZEPTOREN................................................................................................................74
3.2.4.1 Kartierung der Importin ß-Bindedomäne von rpL23a.......................................................74
3.2.4.1.1 Kartierung durch analytische Ausschlusschromatographie ............................................74
3.2.4.1.2 Kartierung durch isotherme Titrationskalorimetrie ........................................................76

Inhaltsverzeichnis
VI
3.2.4.2 Funktionelle Untersuchung der Bindung von rpL23a und Importin 7 ..............................79
3.2.5 PRÄPARATION DER BIB-DOMÄNE VON RPL23A...................................................................80
3.2.5.1 Klonierung der BIB-Domäne in den Vektor pGEX-6P-1..................................................81
3.2.5.2 Expression, Zellernte und Aufschluss ...............................................................................81
3.2.5.3 Affinitätschromatographische Reinigung über Glutathion-Sepharose ..............................81
3.2.5.4 Ausschlusschromatographische Reinigung der BIB-Domäne...........................................82
3.2.5.5 Komplexpräparationen von rpL23aBIB und den Kernimportrezeptoren Imp7 und Impß 83
3.2.5.6 Versuch einer Ko-Kristallisation der BIB-Domäne mit Imp7 und Impß...........................85
4 DISKUSSION............................................................................................................................87
4.1 BIOCHEMISCHE ARBEITEN MIT RPL23A.................................................................................88
4.1.1 PRÄPARATION VON RPL23A UND RPL23ABIB .....................................................................88
4.1.2 KRISTALLISATIONSVERSUCHE FÜR KOMPLEXE MIT RPL23A UND RPL23ABIB ...................89
4.1.3 CHARAKTERISIERUNG DER BINDUNG VON IMPß AN RPL23A ...............................................91
4.1.4 FUNKTIONELLE ARBEITEN MIT IMP7 UND RPL23A...............................................................97
4.2 BIOCHEMISCHE ARBEITEN MIT RPL5.....................................................................................99
4.2.1 PRÄPARATION VON RPL5......................................................................................................99
4.2.2 KRISTALLISATIONSVERSUCHE MIT IMP7/RPL5 UND RPL5..................................................100
4.2.3 DIE ROLLE VON IMPORTREZEPTOREN BEI IMPORTPROZESSEN MIT RPL5...........................101
5 ZUSAMMENFASSUNG........................................................................................................105
6 SUMMARY ..............................................................................................................................106
7 LITERATURVERZEICHNIS...............................................................................................107
8 ABKÜRZUNGSVERZEICHNIS ..........................................................................................116
9 DANKSAGUNG .....................................................................................................................119

Einleitung
1
1. Einleitung
1.1 Der Zellkern
Der Zellkern enthält fast die gesamte genetische Information einer eukaroytischen Zelle
und ist der Ort ihrer Replikation und Transkription. Gegen das Cytoplasma ist der Zellkern
durch eine zweischichtige Kernmembran abgegrenzt. Diese Kompartimentierung
ermöglicht eukaroytischen Zellen differenziertere Stoffwechselvorgänge, als dies in
prokaryotischen Zellen möglich wäre, erfordert aber auch die Existenz komplexer
Transportmechanismen für den geregelten Stoffaustausch zwischen den Kompartimenten.
So sind in eukaryotischen Zellen Transkription und Translation räumlich und zeitlich
voneinander getrennt. Die mRNA muss aus dem Zellkern in das Cytoplasma zum Ort der
Proteinbiosynthese, den Ribosomen, exportiert werden. Diese bestehen aus verschiedenen
rRNAs und Proteinen. Die Biogenese der ribosomalen Untereinheiten findet zu großen
Teilen in den Nukleoli des Zellkerns statt und erfordert daher wiederum einen spezifischen
Import der im Cytoplasma synthetisierten ribosomalen Proteine. Ebenso ist für die
ribosomale Biogenese eine genaue Modulation der Transkription nötig, was den Import
von Transkriptionsfaktoren beinhaltet.
Der Zellkern benötigt darüber hinaus eine Vielzahl weiterer Moleküle wie z.B. die
Proteine des Replikations- und Transkriptionsapparates oder die verschiedenen
Untereinheiten der Nukleosome. Diese und weitere Transportprozesse werden in
eukaryotischen Zellen durch eine äußerst komplexe, spezifische und hoch regulierte
Transportmaschinerie vermittelt.
1.2 Grundlagen des Kerntransports
1.2.1 Der Kernporenkomplex
Der Stoffaustausch zwischen Zellkern und Cytoplasma wird durch einen Porenkomplex
(nuclear pore complex – NPC) vermittelt, der in die Doppelmembran des Zellkerns
eingebettet ist. Eine proliferierende somatische Zelle besitzt etwa 3000 bis 4000
Einleitung
2
Kernporen (Görlich und Kutay, 1999) und an jeder Kernpore können Hunderte von
Transportprozessen pro Minute stattfinden (Görlich und Mattaj, 1996). Der NPC von
Vertebraten hat eine ungefähre molekulare Masse von 125 MDa (Reichelt et al., 1990) und
besteht aus cirka 30 verschiedenen Proteinen, die Nukleoporine (Nups) genannt werden.
Einige dieser Nukleoporine liegen als Kopien in einer Anzahl vor, die dem Vielfachen der
Zahl 8 entspricht (Cronshaw et al., 2002).
Die Struktur des NPC ist zwischen Vertebraten und Hefen konserviert und weist eine
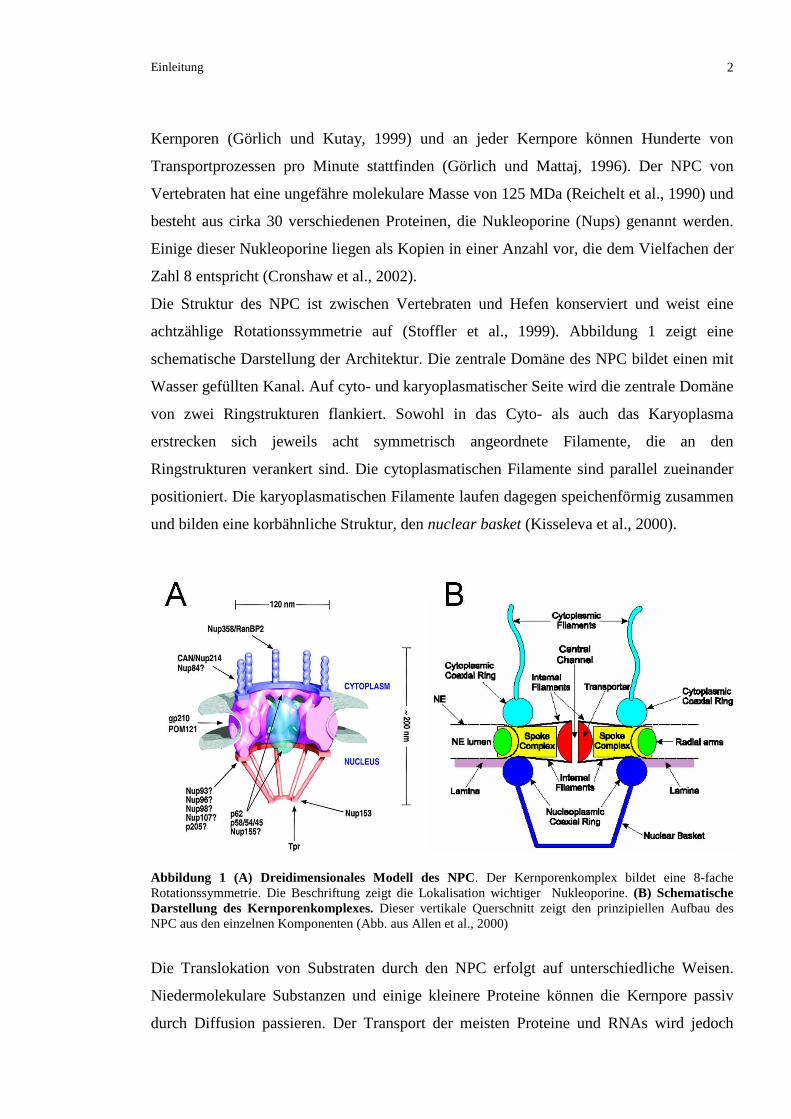
achtzählige Rotationssymmetrie auf (Stoffler et al., 1999). Abbildung 1 zeigt eine
schematische Darstellung der Architektur. Die zentrale Domäne des NPC bildet einen mit
Wasser gefüllten Kanal. Auf cyto- und karyoplasmatischer Seite wird die zentrale Domäne
von zwei Ringstrukturen flankiert. Sowohl in das Cyto- als auch das Karyoplasma
erstrecken sich jeweils acht symmetrisch angeordnete Filamente, die an den
Ringstrukturen verankert sind. Die cytoplasmatischen Filamente sind parallel zueinander
positioniert. Die karyoplasmatischen Filamente laufen dagegen speichenförmig zusammen
und bilden eine korbähnliche Struktur, den nuclear basket (Kisseleva et al., 2000).
Abbildung 1 (A) Dreidimensionales Modell des NPC. Der Kernporenkomplex bildet eine 8-fache Rotationssymmetrie. Die Beschriftung zeigt die Lokalisation wichtiger Nukleoporine. (B) Schematische Darstellung des Kernporenkomplexes. Dieser vertikale Querschnitt zeigt den prinzipiellen Aufbau des NPC aus den einzelnen Komponenten (Abb. aus Allen et al., 2000)
Die Translokation von Substraten durch den NPC erfolgt auf unterschiedliche Weisen.
Niedermolekulare Substanzen und einige kleinere Proteine können die Kernpore passiv
durch Diffusion passieren. Der Transport der meisten Proteine und RNAs wird jedoch

Einleitung
3
signalabhängig über spezifische Transportrezeptoren vermittelt. Mittlerweile sind
allerdings auch Transportsubstrate wie der Transkriptionsfaktor PU.1 bekannt, die durch
direkte Interaktion mit dem NPC transloziert werden (Zhong et al., 2005).
Ein Drittel der identifizierten Nukleoporine bei Vertebraten und Hefen besitzt eine Reihe
seriell angeordneter Domänen mit den Aminosäuremotiven FXFG oder GLFG (engl. FG-
repeats) (Doye und Hurt, 1997; Chook und Blobel, 2001). Diese FG-repeats-tragenden
Nukleoporine interagieren im Verlaufe der Translokation spezifisch mit Transport-
rezeptoren (Bednenko et al., 2003). Es existieren verschiedene Modelle, die den
rezeptorvermittelten Kerntransport durch den NPC beschreiben. Ein Modell geht z.B. von
einer Translokation durch nacheinander folgende Bindungen des Rezeptors an FG-repeats
entlang eines ansteigenden Affinitätsgradienten aus (Ben-Efraim und Gerace, 2001).
1.2.2 Rezeptoren der Importin ß/ß-Karyopherin-Superfamilie
Die in dieser Diplomarbeit untersuchten Importrezeptoren Importin 7 (Imp7) und
Importin ß (Impß) gehören zur Importin-ß/ß-Karyopherin-Superfamilie (kurz
Karyopherine). Der Kerntransport der meisten bekannten Transportsubstrate wird durch
Mitglieder dieser Rezeptor-Superfamilie vermittelt (Weis, 2003).
Bis heute wurden 20 verschiedene Karyopherine von Homo sapiens und 14 von
Saccharomyces cerevisae identifiziert. Die Zahl der Import-spezifischen Rezeptoren
überwiegt dabei. So sind bei S. cerevisae 10 Karyopherine auf Import und drei auf
Exportprozesse beschränkt. Nur von dem Rezeptor Msn5 sind sowohl Import- als auch
Exportfunktionen bekannt. (Mosammaparast et al., 2004).
Karyopherine sind relativ große Proteine (95-145 kDa) mit einem isoelektrischen Punkt im
sauren Bereich und ähnlicher Domänenstruktur (Mosammaparast et al., 2004; Fried und
Kutay, 2003; Weis, 2003) Sie besitzen eine N-terminale Ran-Bindestelle, eine oder
mehrere Nukleoporin-Bindestellen sowie eine oder mehrere Substratbindestellen
(Cingolani et al., 2002; Conti, 2002; Chook und Blobel, 2001; Görlich und Kutay, 1999).
Ihre prinzipielle Architektur besteht aus einer Reihe von tandemartig angeordneten
Sekundärstrukturmotiven, den so genannten HEAT-repeats (von „Huntingtin-elongation-
A-subunit-TOR“). Jeder HEAT-repeat ist aus zwei, durch eine Schleife miteinander

Einleitung
4
verbundene, antiparallele α-Helices (A und B) aufgebaut. Benachbarte HEAT-repeats sind
gegeneinander relativ beweglich, und so angeordnet, dass Importin-ß-Superfamilien-
proteine eine flexible superhelicale Tertiärstruktur besitzen (Cingolani et al., 1999;
Fukuhara et al., 2004).
Verschiedene Komplexstrukturen von Impß zeigen, dass die B-Helices hier mehr auf der
konkaven, inneren Seite des Rezeptors liegen und in die Bindung verschiedener Substrate
wie z.B. PTHrP (parathyroid hormone-related protein) und RanGTP involviert sind. Die
A-Helices sind mehrheitlich auf der konvexen äußeren Seite lokalisiert und interagieren
mit den Nukleoporinen des NPC (siehe Abbildung 2) (Cingolani et al., 2002; Conti et al.,
2006).
Abbildung 2: Schematische Darstellung der Domänenorganisation von Impß. Jeder HEAT-repeat wird in Form von zwei Kreisen symbolisiert, die jeweils für A- und B-Helix stehen. Eingezeichnet sind die an den A-Helices lokalisierten Bindeseiten von RanGTP, PTHrP und der IBB-Domäne von Imp� sowie die auf der Seite der B-Helices gelegene Nukleoporin-Bindeseite (Abb. aus Cingolani et al., 2002).
Ihre Flexibilität verleiht den Karyopherinen die Fähigkeit zu großen Änderungen der
Konformation und ermöglicht ihnen damit, eine Vielzahl unterschiedlicher Importsubstrate
jeweils spezifisch über einen induced-fit-Mechanismus zu binden (Conti et al., 2006; Lee
et al., 2005; Matsuura und Stewart, 2004; Fukuhara et al., 2004; Cingolani et al., 1999). In
Abbildung 3 sind verschiedene Strukturen von Impß allein bzw. im Komplex mit
Bindungspartnern dargestellt. Während ungebundenes Impß in einer relativ gestreckten
Form mit niedriger superhelicaler Windungszahl vorliegt, nimmt es je nach Bindungs-
partner verschiedene kompaktere Konformationen an.
.

Einleitung
5
Abbildung 3: Strukturelle Flexibilität von Karyopherinen am Beispiel von Impß. v.l.n.r.: Struktur von freiem Impß auf Basis von small-angle X-ray scattering (SAXS) (Fukuhara et al., 2004). Desweiteren sind die Kristallstrukturen der Komplexe Impß-Ran (Lee et al. 2005), Impß-SREBP-2 (Lee et al., 2003), Impß-Nup1p (Liu und Stewart, 2005) und Impß-IBB� (Cingolani et al., 1999) abgebildet (Abbildung aus Conti et al., 2006).
1.2.3 Karyopherin-vermittelter Kernimport
Die Fähigkeit der Kerntransportmaschinerie, eine Vielzahl von chemisch unterschiedlichen
Substraten spezifisch zu translozieren, wurde durch die Entwicklung verschiedener
Mechanismen erreicht. Zum einen sind Transportrezeptoren aufgrund ihrer strukturellen
Flexibilität in der Lage, eine Vielzahl verschiedener Substrate zu binden. Zum anderen
wurden im Laufe der Evolution unterschiedliche Mechanismen des Kerntransports
entwickelt. Bis zum heutigen Zeitpunkt sind drei verschiedene prinzipielle Import-
mechanismen von Karyopherinen bekannt:
1. Der Ein-Rezeptor-Weg: Hierbei bindet ein einzelner Import- oder Exportrezeptor das
Substrat und vermittelt die Translokation durch den NPC. Ein Beispiel hierfür ist der
Import der ribosomalen Proteine L5 und L23a (Rout et al., 1997; Jäkel und Görlich, 1998).
2. Der Ein-Rezeptor-Ein-Adapter-Weg: Ein Transportrezeptor bindet das Substrat und
fungiert anschließend als Adapter für einen zweiten Transportrezeptor, der den
eigentlichen Transport durch den NPC vermittelt. Ein bekanntes Beispiel ist der Import
von Substraten mit klassischer Kernlokalisationssequenz (engl. nuclear localisation signal
– kurz NLS) durch das Impα/Impß-Heterodimer (Adam et al., 1994; Moroianu et al., 1995)
3. Der Zwei-Rezeptoren-Ein-Substrat-Weg: Der Transport des Substrates wird durch ein
zuvor gebildetes Heterodimer aus zwei verschiedenen Rezeptoren vermittelt. Ein Beispiel
ist der Import des Linker-Histons H1 durch das Impα/Imp7-Heterodimer (Jäkel et al.,
1999; Bäuerle et al., 2002).

Einleitung
6
Bei allen beschriebenen Importmechanismen erfolgt die Bindung des Substrats durch den
oder die Rezeptoren im Cytoplasma, gefolgt von einer rezeptorvermittelten Translokation
durch den Zentralkanal des NPC. Die Freilassung des Substrats am nuclear basket des
NPC oder im Karyoplasma beinhaltet die Bindung von RanGTP und / oder die Beteiligung
weiterer karyoplasmatischer Faktoren.
1.2.4 Das RanGTP-System
Eine Schlüsselrolle bei Karyopherin-vermittelten Import- oder Exportprozessen spielt der
Zyklus der kleinen ras-verwandten GTPase Ran. Dieses System bestimmt die
Direktionalität von Transportprozessen und stellt durch Hydrolyse von GTP die benötigte
Energie zur Verfügung. (Nigg et al., 1999; Melchior et al., 1993; Moore und Blobel, 1993;
Drivas et al., 1990).
Ran kann durch seine GTPase-Domäne gebundenes GTP zu GDP hydrolysieren. Die
Verteilung von GDP- bzw. GTP-gebundenem Ran innerhalb der verschiedenen
Kompartimente von eukaryotischen Zellen ist nicht homogen. Vielmehr besteht ein
RanGTP/GDP-Gradient zwischen Zellkern und Cytoplasma. So ist die Ran-GTP-
Konzentration im Kern hoch und im Cytoplasma niedrig. Genau entgegengesetzt hierzu
verhält sich der Gradient der RanGDP-Konzentration zwischen beiden Kompartimenten.
(Melchior et al., 1993; Moore und Blobel, 1993; Görlich et al., 1996 b). Erzeugt wird
dieser Gradient durch eine Reihe von regulatorischen Proteinen mit ebenfalls
kompartimentierter Verteilung. RanGTP wir durch verschiedene Karyopherine aus dem
Kern exportiert. Im Cytoplasma erfolgt die Hydrolyse von RanGTP zu RanGDP. Dabei
wird die intrinsische GTPase-Aktivität von Ran durch ein Zusammenspiel der Proteine
RanGAP1 (Ran-GTPase-activating-protein) sowie RanBP1 (Ran-binding-protein) und
RanBP2 erheblich gesteigert (Bischoff et al., 1994, 1995; Yokoyama et al., 1995; Seewald
et al., 2002). Der Transport von freiem RanGTP in den Kern wird durch den Rezeptor
NTF2 vermittelt (Moore und Blobel, 1994; Paschal und Gerace, 1995; Smith et al., 1998;
Ribbeck et al., 1998). Im Kern erfolgt dann der Nukleotidaustausch von Ran-gebundenem
GDP gegen GTP durch den Austauschfaktor RanGEF (früherRCC1) (Bischoff und
Postingl, 1991, 1995).

Einleitung
7
Abbildung 4: Schematische Darstellung des Ran-Zyklus. Ran wechselt zwischen einer GDP- und einer GTP-gebundenen Form und zirkuliert zwischen dem Nukleus und dem Cytoplasma. RCC1 katalysiert den Nukleotidaustausch im Karyoplasma (aus Görlich et al., 2003). Der Export erfolgt nach der Bindung an Importrezeptoren oder Exportkomplexe. Die Hydrolyse von RanGTP zu RanGDP erfolgt entweder durch direkte Interaktion von RanGAP oder durch vorheriges Binden von RanBP1. Der Zyklus wird durch den Import von RanGDP durch NTF2 vollendet (Abb. aus Görlich et al., 2003).
1.3 Kernimport der ribosomalen Proteine L5 und L23a
Die beiden ribosomalen Proteine rpL5 und rpL23a sind Transortsubstrate der Import-
rezeptoren Imp7 und Impß. Beide ribosomalen Proteine besitzen gemeinsame
Eigenschaften, wie eine z.B. eine hohe Basizität und die Fähigkeit RNA zu komplexieren.
Sie unterscheiden sich jedoch in strukturellen und funktionellen Eigenschaften.
1.3.1 Biochemische Daten zu rpL5
Das ribosomale Protein rpL5 aus X. laevis besitzt drei verschiedene, räumlich
unterschiedliche und unabhängig voneinander funktionelle Kernlokalisationssequenzen
(NLS) (Claußen et al., 1999; Rudt und Pieler, 2002). In Abbildung 5 ist eine Kartierung
der bekannten Transportsignale von rpL5 dargestellt:

Einleitung
8
NLS-1 (AS 1-25): Diese N-terminale NLS umfasst die ersten 25 Aminosäuren von L5.
Die Sequenz ähnelt dem klassischen, von Nucleoplasmin bekannten bpNLS (bipartite
NLS) (Robbins et al., 1991). Hierbei sind zwei kurze basische Sequenzen durch eine
sogenannte „Spacer-Region“ getrennt. Innerhalb desselben Sequenzabschnitts liegt auch
Kernexportsignal (NES), das eine physiologische Rolle beim Export von rpL5 im
Komplex mit 5S rRNA als 5S Ribonucleoprotein (RNP) spielt (Murdoch et al., 2002).
NLS-2 (AS 32-253): Dieses Signal umfasst Sequenzelemente, die über die Aminosäure-
sequenz verstreut sind. Es gehört daher zu keiner bekannten Klasse von NLS-Konsensus-
sequenzen.
NLS-3 (AS 261-285): Die C-terminale NLS ähnelt ebenfalls dem von Nucleoplasmin
bekannten bpNLS.
Abbildung 5: Kartierung der verschiedenen Import und Exportsignale von L5: NLS-1 /NES (AS 1-25, blau), NLS-2 (AS 32-253, gelb), NLS-3(AS 261-285, rot) (Kartierung nach Claußen et al., 1999; Rout und Pieler, 2001; Murdoch et al., 2002).
Nur NLS-1 und NLS-3 sind in der Lage den Import eines 5S RNPs zu vermitteln. Die
Bindung von 5S rRNA erfordert sowohl N- als auch C-terminale Reste (AS 17-296).
Möglicherweise wird NLS-2 durch die gebundene RNA maskiert. NLS-1 ist in vitro in der
Lage, die Karyopherine Importin α, Importin ß, Importin 7 oder Transportin zu binden.
Die funktionellen Importrezeptoren von NLS-2 und NLS-3 sind unbekannt (Claußen et al.,
1999). Der Import von rpL5 über NLS-1 und NLS-2 ist Ran-abhängig (Rudt und Pieler,
2001). Daher ist eine Beteiligung von Karyopherinen an NLS-2 involvierenden
Importprozessen wahrscheinlich. Der Import von rpL5 über NLS-3 ist unabhängig von
RanGTP und bekannten Importrezeptoren, kann jedoch bei in vitro Import-Assays durch
Energiedepletion inhibiert werden (Rudt und Pieler, 2001).

Einleitung
9
1.3.2 Biochemische Daten zu rpL23a aus Homo sapiens
rpL23a gehört zur Gruppe I der ribosomalen Proteine (Wool et al., 1995). Es hat eine
Länge von 156 Aminosäuren und ein Molekulargewicht von 17,7 kDa. Das Protein ist
äußerst basisch und enthält 30 Lysin- und 11 Arginin-Reste. Der mit dem Programm
ProtParam berechnete theoretische isoelektrische Punkt liegt bei pH 10,44.
Das humane L23a ist ein Ortholog der ribosomalen Proteine L25 aus S. cerevisae, L23 bei
dem Archaeon Methanococcus vannielii und L23 bei dem Eubakterium E. coli. (Suzuki
und Wool, 1993; McIntosh und Bonham-Smith, 2001) In der Kristallstruktur der großen
ribosomalen Untereinheit von Haloarcula marismortui (Auflösungsgrenze 2.4 Å)
interagiert L23 mit den Domänen I und III der 23S rRNA (Ban et al., 2002).
rpL23a besitzt eine Kernlokalisationssequenz, welche die Aminosäurereste 32-74 umfasst
und BIB-Domäne (für beta-like import receptor binding domain) genannt wird. Alle bisher
als funktionell charakterisierten Importrezeptoren binden diese NLS (Jäkel und Görlich,
1998). Die BIB-Domäne ist äußerst basisch mit einem berechneten pI von 12,2 und einer
Nettoladung von +18. Im Vergleich zu den klassischen SV40-Typ-NLS (Kalderon et al.,
1984) oder bpNLS (Robbins et al., 1991) zeigt die BIB-Domäne einen komplexeren
Aufbau.
1.3.3 Karyopherin-vermittelter Kernimport von rpL5 und rp L23a
Sowohl rpL23a als auch rpL5 können durch verschiedene Mitglieder der Importin ß-
Superfamilie in den importiert werden. So konnte durch in vitro Import-Assays der Import
beider Proteine durch die Transportrezeptoren Impß, Imp7, Imp5 und Transportin
nachgewiesen werden (Jäkel und Görlich, 1998). Der Mechanismus entspricht dem Ein-
Rezeptor-Weg (vgl. 1.2.3). Abbildung 6 zeigt eine schematische Darstellung eines solchen
Importweges für den Transportrezeptor Imp7:

Einleitung
10
Abbildung 6: Modell eines Ein-Rezeptor-Imports mit Imp7. rpL5 oder rpL23a (hier cargo genannt) werden im Cytoplasma durch Imp7 gebunden. Nach der Translokation durch den NPC erfolgt die Dissoziation des Importkomplexes durch Bindung von RanGTP an Imp7 (Abbildung mit freundlicher Genehmigung entliehen von D. Wohlwend, Model nach Jäkel und Görlich, 1998).
Neben ihrer Importfunktion erfüllen die Importrezeptoren von ribosomalen Proteinen auch
noch eine chaperonartige Schutzfunktion. Viele ribosomale Proteine besitzen große
basische Oberflächen, die z.B. eine funktionelle Rolle bei der Bindung von rRNA oder
Transportrezeptoren spielen. Sie sind daher aber auch in der Lage, unspezifische
Komplexe mit Nukleinsäuren zu bilden (siehe hierzu Abbildung 7) (Jäkel und Görlich,
2002).
Abbildung 7: Schematische Illustration des basischen Chaperonproblems. Basische ribosomale Proteine (rot dargestellt) können durch multivalente ionische Interaktionen große Aggregate mit Polyanionen wie z.B. Nukleinsäuren formen (aus Jäkel und Görlich, 2002).

Einleitung
11
Importrezeptoren können durch spezifische Bindung große Teile dieser basischen
Oberflächen von der Umgebung abschirmen. Diese chaperonartige Schutzfunktion
verhindert die unspezifische Aggregation von ribosomalen Proteinen mit Polyanionen wie
z.B. Nukleinsäuren (Jäkel und Görlich, 2002).
1.3.4 Ribosomale Biogenese
Der Import von ribosomalen Proteinen wie L5 oder L23a ist ein Bestandteil der Biogenese
von Ribosomen. Dieser Prozess findet bei Eukaryoten zu einem großen Teil in einer
definierten Substruktur des Zellkerns, dem Nukleolus, statt.
Der Nukleolus ist aus verschiedenen Substrukturen aufgebaut: der dichten fibrillären
Komponente (DFC), der granulären Komponente (GC) und dem fibrillären Zentrum (FC).
Frühe Prozessierungsschritte der ribosomalen Biogenese finden im DFC statt (Milkereit et
al., 2001), bevor die prä-ribosomalen Partikel zunächst zur granulären Komponente und
schließlich ins Karyoplasma transportiert werden. Von hier werden die ribsomalen
Untereinheiten schließlich durch die Kernpore ins Cytoplasma exportiert (Lei und Silver,
2002).
Die ribosomalen Untereinheiten setzen sich sowohl aus rRNA als auch einer Vielzahl von
ribosomalen Proteinen zusammen: In Eukaryoten besteht die kleine 40S-Untereinheit des
Ribosoms aus einer einzelnen 18S rRNA und je nach Organismus aus 30 bis 50
verschiedenen ribosomalen Proteinen (RPs). Die große 60S-Untereinheit des eukaryo-
tischen Ribosoms ist aus drei verschiedenen rRNA-Komponenten (5S, 5,8S und 23S
rRNAs) und 40 bis 50 verschiedenen ribosomalen Proteinen aufgebaut (Woolford und
Warner, 1991; Planter und Mager, 1998). Alle rRNAs bis auf die 5S rRNA werden
zunächst in Form einer einzelnenen Vorläufer- RNA durch die RNA-Polymerase I
transkribiert. Die hierfür codierende rDNA ist in Form von tandemartigen
Transkriptionseinheiten aufgebaut (Cheutin et al., 2002). Bei allen Eukaryoten wird die 5S
rRNA unabhängig durch die RNA-Polymerase III im Nukleus transkribiert (Lee und
Nazar, 2003).
In allen eukaryotischen prä-RNAs sind interne (ITS1, ITS2) und externe (5`ETS und
3`ETS) nichtcodierende „Spacer“-Sequenzen inseriert, die codierende Sequenzabschnitte

Einleitung
12
voneinander trennen (Nazar, 2004). Nach dem Ende bzw. in der späten Phase der
Transkription beginnt die Prozessierung des rRNA-Primärtranskripts durch eine geordnete
Reihe von endo- und exonucleolytischen Spaltungen, wobei diese „Spacer“-Regionen
entfernt werden. In Abbildung 8 sind die rRNA-Primärtranskripte, sowie die zugehörigen
Nuklease-Schnittstellen dargestellt.
Abbildung 8: Schematische Darstellung der bei den meisten Eukaryoten vorkommenden rRNA-Primärtranskripte. Kleine Pfeile markieren Schnittstellen von bekannten, prozessierenden Nukleasen, große Pfeile markieren Schnittstellen von bisher noch nicht identifizierten Nukleasen (Abbildung aus Nazar 2004).
Im Laufe des Reifeprozesses wird die prä-rRNA auch an zahlreichen Stellen kovalent
modifiziert, z.B. durch Methylierung von 2’-OH-Gruppen. Ein Großteil dieser
Modifikationen findet schon während der Transkription der Vorläufer rRNA statt (Maden,
1990). An der Biogenese der ribosomalen Untereinheiten sind neben Nukleasen,
Methyltransferasen und anderen chemisch modifizierenden Proteinen des weiteren auch
eine Vielzahl von Helikasen, RNA-Chaperonen und snoRNAs (engl. small nucleolar
RNAs) beteiligt. Nach der Wanderung in das Karyoplasma erfolgt der rezeptorvermittelte
Export der maturierten Untereinheiten. Die 60S Untereinheiten werden unter Beteiligung
des Karyopherins CRM1 und des Adaptermoleküls NMD3 exportiert. NMD3 bindet dabei
an leucinreiche NES in ribosomalen Proteinen (Johnson et al., 2002).
1.3.5 Funktionelle Rolle von rpL5 bei Transportprozessen der 5S RNA
Das ribosomale Protein L5 ist ein wichtiger Bindungspartner der 5S rRNA innerhalb der
60S Untereinheit des Ribosoms. Es spielt darüber hinaus eine bedeutende Rolle bei
zyklischen Transportprozessen der 5S rRNA im Laufe der Zellentwicklung.
Studien mit Oocyten von Xenopus laevis haben gezeigt, dass die 5S rRNA zeitlich vor
anderen ribosomalen Komponenten transkribiert wird (Murdoch et al., 2002). Nach der

Einleitung
13
Transkription kann die 5S rRNA z.B. durch den Transkriptionsfaktor TFIIIA gebunden
werden, wobei ein 7S RNP-Komplex ensteht (Allison et al., 1991; Guddat et al., 1990;
Wischnewski et al., 2004). Dieser
7S RNP-Komplexe werden zu einem beträchtlichen Teil in das Cytoplasma exportiert
(Guddat et al., 1990). Ein Reimport der 7S RNPs ist nicht möglich, wahrscheinlich, weil
das Importsignal von TFIIIA durch die 5S rRNA maskiert wird (Rudt und Pieler, 1996).
Die cytoplasmatischen 7S RNPs können als eine Speicherform der 5S rRNA angesehen
werden (Pieler und Rudt, 1997). Während der späteren Entwicklungsphasen von Oocyten
aus Xenopus laevis steigt der Bedarf an Ribosomen auf ein maximales Maß, und die 5S
rRNA wird für die ribosomale Biogenese im Nukleolus benötigt. In der Folge kommt es zu
einer Ersetzung von TFIIIA durch rpL5, wobei 5S RNPs entstehen (Allison, et al., 1995).
Diese RNPs werden durch Interaktion von rpL5 mit Komponenten der Kernimport-
maschinerie in den Zellkern importiert (Claußen et al., 1999; Murdoch und Allison, 1996).
Hiefür kommen auch die in die Importrezeptoren Imp7 und Impß in Betracht (Jäkel und
Görlich, 1998; Rudt und Pieler, 2001). Ein wiederholter Export von 5S RNPs unter
Beteiligung des Adaptermoleküls Nukleophosmin und CRM1-RanGTP ist ebenfalls
möglich (Yu et al., 2006). Der letztgenannte Transportweg könnte auch eine Rolle beim
Export von ribosomalen 60S Untereinheiten spielen, was bisher jedoch noch nicht
bewiesen werden konnte.

Einleitung
14
1.4 Aufgabenstellung und Zielsetzung
Die Aufgabenstellung dieser Diplomarbeit bestand zum einen in der Etablierung der
Expression und Präparation der ribosomalen Proteine L5 aus Xenopus laevis und L23a aus
Homo sapiens. Neben den Volllängeproteinen sollten auch funktionelle Fragmente
kloniert, exprimiert und präpariert werden.
Desweiteren sollten die gereinigten ribosomalen Proteine im Anschluss als Komplexe mit
dem Kernimportrezeptor Importin 7 aus Xenopus laevis präpariert werden. Ein Hauptziel
war es, diese Kernimportkomplexe zu kristallisieren, um mittels Röntgenbeugungs-
experimenten ihre Struktur aufklären zu können.
Zusätzlich sollte in funktionellen Untersuchungen die Bindung von rpL23a durch die
Kernimportrezeptoren Importin ß und Importin 7 charakterisiert werden. Im Vordergrund
standen hierbei die räumliche Lokalisierung der Bindestellen der Rezeptoren für rpL23a,
sowie eine Untersuchung von Bindungsenergien und Bindungsaffinitäten.

Material und Methoden
15
2 Material und Methoden
2.1 Material
2.1.1 Feinchemikalien
Alle Feinchemikalien und organischen Substanzen werden von den Firmen AppliChem
(Darmstadt), BioRad (München), Fluka (Buchs), Merck (Darmstadt), Mettler-Toledo
(Steinbach), MWG Biotech (München), Oxoid (Basingstoke, GB), Roth (Karlsruhe) oder
Sigma-Aldrich (Steinheim) bezogen und besitzen den Reinheitsgrad pro analysis. Dabei
wird in der Regel der günstigste Anbieter gewählt.
2.1.2 Geräte
AbiPrism 3100 DNA Sequencer Applied Biosystems, Darmstadt
Agarose-Gelelektrophoresekammer BioRad, München
Äkta Prime General Electrics Health Care, München
Äkta Purifier General Electrics Health Care, München
Binokulare Carl Zeiss, Jena
Inkubator Mytron Schütt, Göttingen
Fraktionssammler Frac900 General Electrics Health Care, München
GelDoc Geldokumentationsgerät BioRad, München
Gelschüttler Promax 1020 Heidolph, Schwabach
Heizbad IKA, Staufen
Heizblock Dri-Block CB-2A Techne, Minneapolis, USA
Innova 4230 Schüttelinkubator New Brunswick Scientific, Nürtingen
Magnetrührer IKAMAG REO IKA, Staufen
Microfluidizer 110 S Microfluidics, USA
Microtip 102 C Branson, USA
PCR-Thermocycler Biometra, Göttingen
pH-Meter Beckman Coulter, Krefeld

Material und Methoden
16
Photometer Biometra, Göttingen
Pipettierhilfe Accu-Jet Brand, Wertheim
Röntgendiffraktometer RU-H3R Rigaku, Japan
Rotationsschüttler Karl Hecht, Staufen
Rotor JA-20 / JA-30.50 Ti Beckman Coulter, Krefeld
Rotor JLA 8.1000 Beckman Coulter, Krefeld
Rotor S4180 Beckman Coulter, Krefeld
SDS-Gelelektrophoresekammer Biometra, Göttingen
Vakuumkonzentrator 5301 Eppendorf, Hamburg
Spektralphotometer General Electrics Health Care, München
Auftragsschleifen (10 ml, 50 ml, 150 ml) General Electrics Health Care, München
Taumelrollenmischer RM5 Schütt, Göttingen
Thermomixer comfort Eppendorf, Hamburg
Tischzentrifuge 5417 R Eppendorf, Hamburg
Tischzentrifuge Micro centrifuge II Sylvania, Ohio, USA
Ultraschallbad Sonorex Super RK 510 Bandelin, Berlin
Unitron Schüttelinkubatoren Infors, Einsbach
Vortexer Schütt, Göttingen
VP-ITC Mikrokalorimetersystem Microcal Inc., Northhampton, USA
Zentrifuge Allegra 21R Beckman Coulter, Krefeld
Zentrifuge Avanti J-20 XPIJA-20 Beckman Coulter, Krefeld
Zentrifuge Avanti J-30 I Beckman Coulter, Krefeld
Zentrifuge Avanti JA-20 Beckman Coulter, Krefeld
Chromatographiesäulen
Einweg-Leersäulen BioRad, München
GSH-Sepharose Säule (30ml) General Electrics Health Care, München
HisTrap Chelating Ni-NTA-Sepharose (5ml) General Electrics Health Care, München
HiPrep 26/60 Desalting (53ml) General Electrics Health Care, München
SP-Sepharose FF (24ml) General Electrics Health Care, München
Superdex 75 (26/60, 10/300) General Electrics Health Care, München
Superdex 200 (26/60, 10/300) General Electrics Health Care, München

Material und Methoden
17
2.1.3 Kit-Systeme
Big Dye Terminator v1.1 Mix Applied Biosystems, Darmstadt
Finnzymes Phusion High-Fidelity PCR Kit Finnzymes, Finnland
NucleoSpin Plasmid Macherey-Nagel, Düren
QIAquick Gelextraktionskit QIAGEN, Hilden
QIAquick PCR Reinigungskit QIAGEN, Hilden
QIAGEN Plasmid Mini Kit QIAGEN, Hilden
2.1.4 Organismen
E. coli BL21(DE3)
E. coli BL21(DE3) RP
E. coli Rosetta 2 (DE3)
E. coli BL21(DE3) STAR Stammsammlung der AG Ficner
E. coli HMS174 (DE3)
E. coli Origami
E. coli SG13009 (pREP4)
E. coli M15
E. coli XL1-Blue
2.1.5 Plasmide
pGEX-5X-1-rpL5 T. Pieler, Universität Göttingen
pGEX-6P-1-rpL5
pGEX-6P-1-rpL5/nostop* (AS 1-48)
pGEX-6P-1-rpL5/nostop* (AS 1-189)
pGEX-6P-1-rpL5/nostop* (AS 1-234)
pGEX-6P-1-rpL5 (AS 48-296)
pGEX-6P-1-rpL5 (AS 189-296)
pGEX-6P-1-rpL5 (AS 234-296)

Material und Methoden
18
pQE70-rpL23a D. Doenecke, Universität Göttingen
pQE80-rpL23a
pGEX-6P-1-rpL23a
pGEX-6P-1-rpL23a /nostop* (AS 32-74)
* Translationsstop am Stopcodon der Multiple Cloning Site (= +7 Aminosäuren)
2.1.6 Enzyme und Inhibitoren
Calf Intestinal Alkaline Phosphatase (CIP) New England Biolabs, Frankfurt
Phusion-DNA-Polymerase Finnzymes, Finnland
Pfu-DNA-Polymerase New England Biolabs, Frankfurt
PreScission-Protease Universität Göttingen
Protease Inhibitor Cocktail Tabletten
„EDTA-free“
Roche, Mannheim
Restriktionsenzyme
(BamHI, XhoI, HindIII)
Fermentas, St. Leon-Rot
T4-DNA-Ligase Fermentas, St. Leon-Rot
Taq-DNA-Polymerase Universität Göttingen
2.1.7 Größenstandards
BR(broad range)-Protein-Standard New England Biolabs, Frankfurt
DNA-Standard „1kb-ladder” New England Biolabs, Frankfurt
Gel Filtration Standard Bio-Rad Laboratories, München

Material und Methoden
19
2.1.8 DNA-Oligonukleotide
Die Berechnung der Schmelztemperaturen Tm für die verwendeten DNA-Oligonukleotide
erfolgte nach den unten angegebenen Formeln. Die DNA-Oligonukleotide werden von 5´
nach 3´ dargestellt.
Tm = 81,5 + 0,41 [%GC] – (675/N)
N = Anzahl der Nukleotide
Alle Oligonukleotide stammen von der Firma MWG Biotech, München:
pGEX_f GCT GGC AAG CCA CGT TTG GT
pGEX_r CGT CTC CGG GAG CTG CAT GT
PQE80_for GTG AGC GGA TAA CAA TTT CAC ACA G
pQE80_rev CGC CCG GCG GCA ACC GAG CGT TCT
rpL23a_BamHIfor CGG GAT CCA TGG CGC CGA AAG CGA AG
rpL23a_XhoIrev CCG CTC GAG TTA GAT GAT CCC AAT TTT GTT G
rpL23a_HindIIIrev CCC AAG CTT TTA GAT GAT CCC AAT TTT GTT G
BIB_BamHI_for CGG GAT CCG TCC ACA GCC ACA AAA AG
BIB_XhoI_rev CCG CTC GAG ATA GTG GTC AAG CTT GTT TC
rpL5_BamHIfor CGG GAT CCA TGG GGT TCG TAA AGG
rpL5_XhoIrev CCG CTC GAG TTA GCT GTC TGC CTT CTG
rpL5_N48rev CCG CTC GAG CTT GGG AGT ATT GTA CTT G
rpL5_N189rev CCG CTC GAG TTC TTT GCT TTC AGA GTC ATA G
rpL5_N234rev CCG CTC GAG ATC TGC TGC GAC ACC ATT C
rpL5_C48for CGG GAT CCA AGT ACA GGA TGA TTG TAC G
rpL5_C189for CGG GAT CCG AAT TCA ATG CTG AGG TC
rpL5_C234for CGG GAT CCG ATC AGT TGG AAG ACA TAT AC

Material und Methoden
20
2.1.9 Kristallisationsscreens
Die nachfolgend aufgelisteten Screens werden von Mitarbeitern der Abteilung für
Molekulare Strukturbiologie an der Universität Göttingen angesetzt.
Crystal Screen 1 Hampton Research, USA
Crystal Screen 2 Hampton Research, USA
Crystal Screen Lite Hampton Research, USA
Crystal Screen Cryo Hampton Research, USA
Crystal Screen PEG/Ion Hampton Research, USA
JB Screens 1-10 Jena Bioscience, Jena
Magic Screens 1-4 Biogenova, Kanada
Footprint Screens 1-3 Stura et al., 1999
Structure Screens 1-3 Molecular Dimensions, England
Folgende käuflich erwerbbare, bereits fertig angesetzte Screens wurden verwendet:
JCSG+ Nextal Biotechnologies, Kanada
Opti Salts Nextal Biotechnologies, Kanada
PACT Nextal Biotechnologies, Kanada
pHClear Nextal Biotechnologies, Kanada
The MPDs Nextal Biotechnologies, Kanada
2.1.10 Computerprogramme
DeepView 3.7 http://www.expasy.org/spdbv/
Lasergene (Protean, Seqman, Megalign) DNAStar, Madison, US
ProtScale http://www.expasy.ch/tools/protscale.html
PSIPredict http://bioinf.cs.ucl.ac.uk/psipred/
SWIFT II Biochrom Ltd., England
Swiss Model http://swissmodel.expasy.org/

Material und Methoden
21
2.2 Methoden
2.2.1 Molekularbiologische Methoden
2.2.1.1 Nukleinsäurebiochemische Methoden
2.2.1.1.1 Polymerase-Kettenreaktion
Der Prozess der Polymerase-Kettenreaktion (engl. polymerase chain reaction, PCR) wurde
1983 von Kary Mullis entwickelt. Mit Hilfe dieser Technik ist es möglich, ein spezifisches
DNA-Fragment zu amplifizieren, das zwischen zwei Regionen bekannter Nucleotid-
sequenzen innerhalb einer längeren DNA-Matrize liegt.
Für die Amplifikation der DNA wird eine hitzestabile DNA-Polymerase verwendet. DNA-
Polymerasen sind nicht in der Lage, Nukleinsäureketten de novo zu synthetisieren. Daher
benötigt man für eine PCR kurze DNA-Oligonukleotide (20-30 Basen), die zu den
bekannten, das 3`- und das 5`-Ende der Ziel-DNA flankierenden Sequenzen,
komplementär sind. In einem PCR-Ansatz muss außerdem eine ausreichende Menge aller
Desoxyribonukleosidtriphosphate (dNTPs) vorhanden sein.
Ein PCR-Prozess besteht aus einer Anzahl von 25 bis 40 Zyklen, die in einem
Thermocycler durchgeführt werden. Jeder Zyklus besteht aus drei Schritten:
1. Denaturierung: Die doppelsträngige DNA wird auf 95°C erhitzt, um die beiden
komplementären Stränge zu trennen. Dabei werden die Wasserstoffbrücken-
bindungen, die die beiden DNA-Stränge zusammenhalten, aufgebrochen.
2. Primerhybridisierung: Durch die Senkung der Temperatur auf 4 °C unterhalb des
Schmelzpunktes der DNA-Oligonukleotide können sich diese hochspezifisch an die
zu amplifizierende DNA-Sequenz anlagern (Hybridisierung).
3. Elongation: Die DNA-Synthese erfolgt durch Änderung der Temperatur auf das
Optimum der jeweilig verwendeten DNA-Polymerase (Elongation). Die Zeit, die
dieser Schritt benötigt, ist abhängig von der verwendeten DNA-Polymerase und der
Länge des DNA-Fragments, das vervielfältigt werden soll.

Material und Methoden
22
Durch Wiederholung der Zyklen wird eine nahezu exponentielle Vervielfältigung der Ziel-
DNA erreicht, weil jeder Zyklus theoretisch die Matrizenanzahl verdoppelt, die dann für
den folgenden Zyklus zur Verfügung steht.
Für die Klonierung in Vektoren enthalten die Primer normalerweise bereits die Sequenzen
für die 5’- und die 3’-Restriktionsschnittstellen, mit deren Hilfe eine Insertion in den
Zielvektor gelingt.
Im Folgenden wird ein typischer Ansatz für eine Polymerase-Kettenreaktion dargestellt:
1-150 ng DNA (Matrize oder Zielsequenz)
1x PCR-Puffer
10 mM je dATP, dGTP, dCTP, dTTP
0-10 % (v/v) DMSO
10 pmol DNA-Oligonukleotide (Primer)
1-2 U Phusion-Polymerase / Pfu-Polymerase / Taq-Polymerase
ad 50 µl H2O
Die Amplifikations-PCR von rpL5 und rpL23a mit der Phusion-DNA-Polymerase wurde
nach folgendem Schema durchgeführt:
1. Denaturierung 98 °C 2 Minuten
2. Denaturierung 95 °C 30 Sekunden
3. Primer-Hybridisierung X °C 30 Sekunden
4. Elongation 72 °C 1-3 Minuten
40 Zyklen
5. Abschlußelongation 72 °C 10 Minuten
Die Temperatur „x“, bei der die Hybridisierung der DNA-Oligonukleotide stattfindet,
hängt von der Schmelztemperatur derselben ab. Für die Hybridisierungstemperatur wird
für gewöhnlich ein Wert 4° C unterhalb des Schmelzpunkts der Oligonukleotide gewählt.
Die optimale Elongationstemperatur variiert zwischen den Polymerasen. Für die ebenfalls
verwendete Pfu-Polymerase beträgt sie 68°C. Abweichungen von diesem Programm und
die Spezifizierung der Hybridisierungstemperatur werden im Ergebnisteil angegeben.

Material und Methoden
23
2.2.1.1.2 Spaltung von DNA durch Restriktionsendonukleasen
Restriktionsendonukleasen erkennen spezifische Basensequenzen in DNA-Doppelhelices
und hydrolysieren die Phosphodiesterbindungen zwischen zwei Nukleotiden. Man findet
diese Enzyme in Prokaryoten, wo sie dem Abbau von Fremd-DNA dienen. Dabei bleibt
die eigene DNA aufgrund von Methylierungen an spezifischen Stellen ungeschnitten.
Die Nukleasen erkennen dabei Sequenzen mit zweifacher Rotationssymmetrie
(Palindrome) und schneiden die Doppelhelix meist so, dass überhängende Enden (sticky
ends) entstehen. Diese überhängenden DNA-Enden hybridisieren leicht mit
komplementären Sequenzen. So wird das gerichtete Einsetzen der mit bestimmten
Restriktionsenzymen geschnittenen Zielsequenz in einen mit denselben Enzymen
geschnittenen Vektor ermöglicht.
Ein typischer Ansatz von 20 µl enthält:
1 µg DNA
1x Restriktionsenzympuffer
1 U Restriktionsendonuklease für 3’ Schnittstelle
1 U Restriktionsendonuklease für 5’ Schnittstelle
ad 20 µl H2O
Der Ansatz wird für eine Stunde bei 37 C inkubiert und zur anschließenden Inaktivierung
der Restriktionsenzyme für 5 Minuten bei 95 C belassen. Danach werden die
geschnittenen Fragmente durch Agarosegelelektrophorese (2.2.1.1.6) analysiert und
gegebenenfalls für folgende Schritte aus dem Gel isoliert (2.2.1.1.8).
2.2.1.1.3 DNA-Dephosphorylierung am 5´-Ende
Um Rezirkularisierungen von bereits geschnittenem Vektor zu vermeiden, wird jeweils das
5´-Phosphat der überhängenden Enden entfernt. Die Religation kann ausgeschlossen
werden, da die T4-DNA-Ligase nur 5´-Phosphate mit 3´-Phosphaten verknüpfen kann. So
wird die Ligationseffizienz des gewünschten Fragmentes erheblich gesteigert.

Material und Methoden
24
Nach der Spaltung des Vektors mit den entsprechenden Restriktionsendonukleasen werden
zu dem hitzeinaktivierten Ansatz 2 U CIP (calf intestinal alkaline phosphatase) pro µg
Vektor-DNA hinzugegeben und für 30 Minuten bei 37 °C inkubiert. Danach erfolgt die
Isolation des geschnittenen und dephosphorylierten Vektors durch Agarosegelelektro-
phorese (2.2.1.1.6) und anschließende Elution der DNA aus dem Agarosegel (2.2.1.1.8).
2.2.1.1.4 DNA-Ligation
DNA-Ligasen katalysieren die Bildung einer Phosphodiesterbindung benachbarter
Nukleotide. Dabei werden zwei DNA-Einzelstränge zu einem einzigen verknüpft. Bei
Klonierungen benutzt man diese Enzyme zur Verknüpfung von Fragment und Vektor nach
der Hybridisierung der überliegenden Enden. Meistens wird hierfür die Ligase des
Bakteriophagen T4 verwendet. Die von der Ligase katalysierte Kondensationsreaktion ist
energetisch an die Hydrolyse von ATP zu AMP und Pyrophosphat gekoppelt, weshalb im
Ligationsansatz auch immer ATP enthalten sein muss.
Ein typischer Ligationsansatz setzt sich wie folgt zusammen:
2 U T4-DNA-Ligase
x M Plasmid
y M Zielsequenz
1x T4-DNA-Ligase-Puffer
ad 20 µl H2O
Dabei wird das Zielgen in 4-fach molarem Überschuss in Relation zum Plasmid eingesetzt.
2.2.1.1.5 DNA-Sequenzierung
Das Ketten-Abbruch-Verfahren nach Sanger et al., (1977) beruht auf der in vitro-Synthese
eines DNA-Strangs mit Hilfe einer DNA-Polymerase. Die Synthese wird nur an der Stelle
initiiert, wo ein genau definiertes Oligonukleotid („Primer“) an das Template bindet. Die
Synthese-Reaktion wird durch den Einbau von Nukleotid-Analoga, Fluoreszenz-markierte
2`,3`-Didesoxynukleosid-5`-triphosphate (ddNTPs), abgebrochen. Diesen Analoga fehlen

Material und Methoden
25
die 3`-OH Gruppen, die für die Elongation der DNA-Ketten notwendig sind. Wird eine
geeignete Mischung aus dNTPs und einem der vier ddNTPs verwendet, dann bricht die
enzymkatalysierte Reaktion statistisch an allen Stellen ab, wo das entsprechende ddNTP
eingebaut werden kann. Der Polymerase, die für die Sequenzierung eingesetzt wird, fehlt
die 5’,3’-Exonuclease-Aktivität, daher bricht die Synthese ab, und es entstehen Fragmente
definierter Länge, die elektrophoretisch aufgetrennt und detektiert werden können.
Mit dem Seq-Mix BigDye Terminator v1.1 von Applied Biosystems kann die komplette
Reaktion in einem einzigen Ansatz durchgeführt werden.
Für einen typischen Sequenzierungsansatz werden pipettiert:
200 ng zu sequenzierendes Fragment (Template)
8 pmol DNA-Oligonukleotid (Primer)
1 µl Seq-Mix
1 µl Seq-Puffer
ad 10 µl H2O
Das Programm für Sequenzierungsreaktionen ist dem unter 2.2.1.1.1 genannten Programm
analog, wobei die optimale Temperatur für die Polymerisation bei 60 °C liegt. Die
Hybridisierungstemperatur ist abhängig von den verwendeten DNA-Oligonukleotiden und
die Elongationszeit von der Länge des zu sequenzierenden DNA-Fragmentes.
Nach der Reaktion werden die Produkte für den Sequenzierungsautomaten gereinigt, um
störende Faktoren wie DNA-Oligonukleotide, Polymerase und restliche ddNTPs zu
entfernen.
Hierzu wird dem Ansatz hinzupipettiert:
1 µl 0,125 M EDTA
1 µl 3 M Natriumacetat
50 µl Ethanol (96 %)
Der Ansatz wird vorsichtig durchmischt, für 5 Minuten inkubiert und anschließend bei
16100 xg für 15 Minuten und bei 4 °C zentrifugiert. Der Überstand wird abgenommen, das

Material und Methoden
26
Pellet in 70 µl Ethanol (70 %) gewaschen und erneut für 5 Minuten zentrifugiert. Das
Pellet wird 2 Minuten an der Luft getrocknet und schließlich in 30 µl Wasser
aufgenommen. Die gereinigten DNA-Fragmente werden in einem Kapillarsequenzierer
analysiert.
2.2.1.1.6 Agarosegelelektrophorese
Die Agarose-Gelelektrophorese wird zur Reinigung, Trennung und Identifizierung von
zirkulärer Plasmid-DNA und DNA-Fragmenten benutzt. Aufgrund der negativen Ladung
ihrer Phosphatgruppen wandern die DNA-Moleküle im elektrischen Feld. Die
Auftrennung erfolgt nach der Größe aufgrund der unterschiedlichen Mobilität
verschiedener DNA-Fragmente innerhalb der gleichmäßig vernetzten Agarose. Es wurden
im Zuge dieser Arbeit ausschließlich 1 %ige Agarosegele benutzt. Die Agarose wird dazu
mit TBE-Puffer versetzt und in der Mikrowelle zum Kochen gebracht. Für das Gel wird
eine Flachbettkammer abgedichtet, die Agaroselösung hineingegossen und ein
Probenkamm in das Gel gesteckt. Nach etwa einer halben Stunde ist das Gel erstarrt und
wird in eine mit 1x TBE gefüllte Elektrophoresekammer gelegt. Zum Laden der DNA-
Proben werden diese mit Probenpuffer versetzt und in die Geltaschen pipettiert. Die
elektrophoretische Trennung erfolgt bei 12 mA/cm2 Gelfläche.
Laufpuffer (TBE) DNA-Probenpuffer (6x)
0,09 M TrisBase 0,5 % (w/v) Bromphenolblau
0,09 M Borsäure 0,5 % (w/v) Xylencyanol FF
0,01 M EDTA pH 8,0 60 % (v/v) Glycerin
2.2.1.1.7 Visualisierung von Nukleinsäuren mit Ethidiumbromid
Elektrophoretisch getrennte Nukleinsäuren können mit Ethidiumbromid im UV-Licht
sichtbar gemacht werden. Hierzu wird das Gel ca. 30 Minuten in einem
Ethidiumbromidbad (1 µg/ml Ethidiumbromid) inkubiert. Ethidiumbromid interkaliert
dabei zwischen den innerhalb der DNA-Helix gestapelten Basen und fluoresziert intensiv

Material und Methoden
27
orange. Die DNA wird durch monochromes Licht mit 365 nm Wellenlänge DNA
detektiert.
2.2.1.1.8 DNA-Isolierung aus Agarosegelen
Um die DNA von Enzymen und anderen Verunreinigungen, die im weiteren Verlauf der
experimentellen Prozedur stören könnten, zu befreien, wird sie nach der
elektrophoretischen Trennung gereinigt.
Hierfür wird ein QIAquick Gelextraktionskit der Firma Qiagen verwendet. Die durch
Ethidiumbromid angefärbte DNA (2.2.1.1.7) wird zunächst möglichst knapp aus dem
Agarosegel ausgeschnitten. Dann wird die DNA aus der Agarose herausgelöst und
anschließend an eine Einweg-Säule gebunden. Bei den hier verwendeten Säulen handelt es
sich um schwache Anionenaustauscher.
Nach einem Waschschritt wird die DNA von der Säule eluiert. Alle Arbeitsschritte werden
nach Angaben des Herstellers durchgeführt.
2.2.2 Zellbiologische Methoden
2.2.2.1 Transformation chemisch kompetenter Zellen
Zum Einbringen von modifizierten Vektoren in Bakterien-Zellen werden
Transformationen durchgeführt. Da jedoch E. coli nicht, wie zum Beispiel Bacillus
subtilis, eine natürliche Kompetenz aufweist, müssen die Zellen mit bestimmten Puffern
chemisch kompetent gemacht werden.
Zu 50 µl kompetenter Zellen werden 20 µl Ligationsansatz oder 1 µg Plasmid-DNA
pipettiert und gemischt. Die Zellen werden dann 20 Minuten auf Eis inkubiert. Für die
DNA-Aufnahme werden die Zellen bei 42°C für 45-60 Sekunden inkubiert und
anschließend 2 Minuten auf Eis belassen. Nach Zugabe von 450 µl antibiotikafreiem 2-
YT-Medium werden die Zellen 60 Minuten bei 37°C unter Schütteln inkubiert. 100 µl des
Transformationsansatzes werden je auf einer Selektionsplatte ausgestrichen, an der Luft
getrocknet und die Platten umgedreht über Nacht bei 37°C inkubiert.

Material und Methoden
28
2.2.2.2 Präparation von Plasmid-DNA
Zur Amplifikation ganzer Plasmide werden während dieser Arbeit ausschließlich
Plasmidpräparationen im kleinen Maßstab (Mini-Präparation) durchgeführt. Dabei werden
die Vektoren in E. coli XL1-Blue-Zellen transformiert (2.2.2.1) und diese wachsen
gelassen. Die Zellen werden nach den Angaben des Herstellers für Plasmid-Isolations-Kits
geerntet und aufgeschlossen. Die Plasmid-DNA wird von der genomischen DNA und
anderen Zelltrümmern sowie Proteinen über Einweg-Säulen befreit.
Für die Präparation im kleinen Maßstab werden 5 ml Übernacht-Kultur aufgeschlossen
und die DNA nach Herstellerangaben präpariert.
2.2.2.3 Expression rekombinanter Proteine
2.2.2.3.1 Expression rekombinanter Proteine in Escherichia coli im kleinen Maßstab
Für die Etablierung der Expression eines Proteins in Escherichia coli ist es sinnvoll, die
Expressionsrate des Zielproteins in verschiedenen Expressionsstämmen zu vergleichen.
Das rekombinante Plasmid wir hierfür in die zu testenden Zellen transformiert (2.2.2.1).
20ml 2YT-Medium werden mit den entsprechenden Antibiotika in einem sterilen
Glasgefäß vorgelegt und von einer angewachsenen Transformationsplatte angeimpft. Die
Kultur wird bei 37°C auf einem Schüttler inkubiert und die Expression bei einer optischen
Dichte von OD600 = 0,8 mit IPTG gestartet. Bei den verwendeten pGEX-6P-1-Konstrukten
erfolgt die Induktion durch 500 µM IPTG. Bei Konstrukten mit dem Plasmid pQE80
beträgt die Konzentration c(IPTG)= 100 µM.
Für den Vergleich der Expressionsrate mittels SDS-PAGE (2.2.3.3) wird jeweils eine
Zellprobe zum Zeitpunkt der Induktion sowie drei Stunden nach Induktion genommen.

Material und Methoden
29
2.2.2.3.2 Überexpression von rekombinanten Proteinen in Escherichia coli
Für die Expression von rekombinanten Proteinen in E.coli wird zunächst eine Vorkultur
mit kleinem Volumen angezogen, die später zur Inokulierung der Expressionskultur dient.
Hierzu werden 20ml 2YT-Medium mit den entsprechenden Antibiotika in einem sterilen
Glasgefäß vorgelegt und von einer angewachsenen Transformationsplatte angeimpft. Die
Vorkultur wird bei 30°C über Nacht anwachsen gelassen. Am nächsten Morgen werden
jeweils 500 ml 2YT-Medium mit den entsprechenden Antibiotika versetzt und mit der
Vorkultur im Verhältnis 1:200 angeimpft. Diese Kultur wird bei 37°C im
Inkubationsschüttler bis zu einer OD600 = 1,6 anwachsen gelassen und dann mit 500 ml
4 °C kaltem 2YT-Medium verdünnt. Dieses frische Medium enthält Di-
Kaliumhydrogenphosphat (K2HPO4) mit einer Endkonzentration von 30mM. Durch die
Pufferwirkung von K2HPO4 und die Anhebung des pH-Werts auf pH 8 sollen die
Expressionsbedingungen verbessert werden.
Nach dem Verdünnen wird die Expressionskultur bei OD600 = 0,8 mit IPTG induziert. Bei
Expressionen von in pGEX-6P-1 einklonierten Genen beträgt die benötigte IPTG-
Konzentration 500 µM. Expressionskulturen mit dem Vektor pQE80 werden mit 100 µM
IPTG induziert. Die Expression findet bei 16°C für 16-18 Stunden im Inkubationsschüttler
statt.
Die Ernte der Zellen erfolgt durch Zentrifugation bei 5.000 x g für 20 Minuten bei 4 °C.
2 YT-Medium
1,6 % (w/v) Trypton
1 % (w/v) Hefeextrakt
0,5 % (w/v) NaCl
Sterilisation durch Autoklavieren
2.2.2.3.3 Ernte und Aufschluss einer Expressionskultur
Um rekombinante Proteine aus Bakterienzellen zu isolieren, müssen die Zellen zunächst
geerntet und aufgebrochen werden. Die Ernte erfolgt durch Zentrifugation bei 5.000 xg für
20 Minuten und bei 4 °C. Das Bakterien-Pellet wird einmalig in kaltem PBS (phosphate

Material und Methoden
30
buffered saline) resuspendiert und erneut bei 4000 xg für 25 Minuten bei 4 °C
zentrifugiert. Nach dem Dekantieren des Überstandes werden die Zellen entweder sofort
aufgeschlossen oder in flüssigem Stickstoff gefroren und gelagert.
Vor dem Aufschluss der Bakterien-Pellets werden diese in 4° C kaltem Lysispuffer
resuspendiert. Dabei werden für ein Pellet von einem Liter Schüttelkultur (2.2.2.3.2) 20ml
Lysispuffer eingesetzt. Um proteolytische Enzyme zu inaktivieren, wird dem Ansatz eine
halbe Tablette Protease-Inhibitor (protease inhibitor cocktail complete EDTA-free)
zugegeben und diese gelöst. Bei frühen Reinigungsversuchen wurden dem Lysispuffer
3U/ml Benzonase beigefügt, um RNA abzubauen. Hierdurch sollte an Proteine gebundene
RNA entfernt werden. Wegen einer zu niedrigen Effizienz dieser Methode wurde bei
späteren Reinigungen auf eine Zugabe von Benzonase verzichtet. Der Zellaufschluss im
pneumatischen Zelldesintegrator (Fluidizer) wird bei 80psi in 7 Zyklen durchgeführt.
Nach dem Aufbruch der Zellen wird die Suspension in JA30-Zentrifugenröhrchen
überführt und bei 30000 xg für 40 Minuten bei 4 °C zentrifugiert. Nach der Trennung des
Lysates in Zelltrümmer und Überstand werden die Proben über SDS-
Polyacrylamidgelelektrophorese analysiert.
Der Überstand wird je nach Art des exprimierten Proteins weiterverarbeitet.
PBS Lysispuffer
0,14 M NaCl 2,2 M LiCl
0,01 M NaH2PO4 / Na2HPO4 pH 7,4 50mM Tris-HCl pH 7,5
5 mM MgCl2
2 mM ß-Mercaptoethanol
Die weitere Verarbeitung und Trennung des Überstandes erfolgt durch die in 2.2.4
beschriebenen Methoden.

Material und Methoden
31
2.2.3 Proteinbiochemische Methoden
2.2.3.1 Einengen von Proteinlösungen durch Zentrifugation
Proteinlösungen werden durch Zentrifugation mit Ultrafiltrationssäulen konzentriert. Diese
Säulen enthalten Filter, welche Substanzen, die kleiner als die Porengröße der Filter sind,
durchlassen und größere Moleküle, in diesem Fall das gewünschte Protein, zurückhalten.
Die Ultrafiltrationssäulen wurden vor Gebrauch mit H20 bei der vorgeschriebenen g-Zahl
gespült. Die Zentrifugationen erfolgen bei 4000 xg bei 4° C. Nach der Zentrifugation wird
jeweils die Konzentration der Proteinlösung bestimmt (2.2.6.1.2).
2.2.3.2 Aufbereitung von Zellproben für eine SDS-PAGE
Die Analyse der Expression eines Zielproteins in einer E.coli-Kultur geschieht mit Hilfe
einer SDS-PAGE (2.2.3.3). Hierzu wird zum gewünschten Zeitpunkt eine Probe mit einem
Volumen von V= 0,1 bis 0,5 ml aus der Kultur entnommen.
Die in der Probe enthaltenen Bakterien werden durch Zentrifugation in einer Eppendorf-
Mikrozentrifuge für 5 min bei 16100 xg abzentrifugiert und der Überstand verworfen.
Anschließend wird das Pellet mit SDS-Probenpuffer (2.2.3.3) resuspendiert. Dabei wird
nach folgender Formel vorgegangen:
0,2 * oD = Menge an SDS-Probenpuffer [ml] (bei 1ml Probe)
Vor dem Laden des SDS-Gels wird die Probe für 5 min bei 95°C auf einem Heizblock
inkubiert.
2.2.3.3 Diskontinuierliche Polyacrylamidgelelektrophorese von Proteinen
Die SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE - nach Laemmli, 1970) ist eine
elektrophoretische Methode zur Auftrennung von Proteingemischen nach ihrer Größe.
Die Proteinprobe wird hierfür zunächst mit Natriumdodecylsulfat (SDS) und ß-Mercapto-
ethanol versetzt und hitzedenaturiert. ß-Mercaptoethanol reduziert Disulfid-brücken
zwischen Cystein-Seitenketten. Das Detergens SDS denaturiert Proteine, wobei es die

Material und Methoden
32
ursprüngliche Ladung durch die negative Ladung seiner Sulfatgruppe weitgehend
überdeckt, so dass alle Proteine ein nahezu gleiches Verhältnis von Ladung zu Größe
besitzen. Die Trennung der Proteine im elektrischen Feld erfolgt hauptsächlich nach dem
Molekulargewicht durch den Molekularsiebeffekt des Polyacrylamidgels.
Bei der diskontinuierlichen SDS-PAGE nach Laemmli werden die Proteine außerdem
innerhalb des Sammelgels fokussiert. Das Prinzip dieser Fokussierung beruht auf der
Entstehung eines Feldstärkegradienten in Laufrichtung der Probe. Das untere Trenngel
enthält einen Puffer mit einem pH-Wert von 8,8. Der pH-Wert des darüber geschichteten
Sammelgels ist mit pH 6.8 deutlich niedriger. Beim Anlegen eines Stromflusses bewegen
sich die im Sammelgel vorhandenen Chloridionen mit der höchsten Geschwindigkeit
Richtung Anode. Hinter den Chloridionen bildet sich eine Zone geringerer Ionendichte
und erhöhter Feldstärke, was zur Beschleunigung von Proteinen und Folgeionen führt. Das
aus dem Laufpuffer nachfolgende Glycin liegt bei pH 6.8 als Zwitterion mit niedriger
Mobilität vor, so dass sich ein Feldstärkegradient innerhalb des Sammelgels bildet. Die
Geschwindigkeit der Proteine ist größer als die des Folgeions Glycin, aber langsamer als
die der Leitionen. Es kommt zu einer Fokussierung der Proteine als scharfe Front
innerhalb des Feldstärkegradienten. Beim Auftreffen auf das Trenngel ändern die
Folgeionen aufgrund des erhöhten pH-Wertes ihre Ladung und damit ihre
Ionenbeweglichkeit. Sie überholen die Proteine, die sich dann wieder in einem Gebiet
konstanter Feldstärke bewegen. Die Folge ist eine Auftrennung der am Trenngel
komprimierten Proteinfront. Das Trenngel wirkt dabei als Molekularsieb, da die Proteine
durch die weit engeren Poren des Trenngels in Abhängigkeit ihrer Masse verlangsamt und
so ihrer Größe nach getrennt werden.
Für die Vorbereitung einer SDS-PAGE werden vier Gele in einer Mehrfachgießkammer
gegossen. Hierbei wird nach der Anleitung des Herstellers vorgegangen.
Die Proteinproben werden 1:1 (v/v) mit SDS-Probenpuffer versetzt und in die Taschen des
Gels geladen. Zellsuspensionsproben werden wie unter 2.2.3.2 beschrieben für die SDS-
PAGE aufbereitet. Nach dem Auftragen der Proben erfolgt die elektrophoretische
Auftrennung bei einer angelegten Stromstärke von 30mA.

Material und Methoden
33
Trenngel (15 %) Sammelgel (5 %)
15 % Acrylamid, 0,4 % Bisacrylamid 5% Acrylamid, 0,13 % Bisacrylamid
0,375 M Tris/HCl pH 8,8 0,125 M Tris/HCl pH 6,8
0,1 % (w/v) SDS 0,1 % (w/v) SDS
0,1 % (v/v) TEMED 0,1 % (v/v) TEMED
0,05 % (w/v) Ammoniumpersulfat 0,05 % (w/v) Ammoniumpersulfat
Protein-Laufpuffer SDS-Probenpuffer (2x)
0,025 M Tris-HCl 0,0625 M Tris-HCl pH 6,8
0,192 M Glycin 0,07 M SDS
0,1 % (w/v) SDS 50 % (v/v) Glyzerin
0,1 % (w/v) Bromphenolblau
5 % (v/v) 2-Mercaptoethanol
2.2.3.4 Visualisierung von Proteinen durch Coomassie Brilliant Blue
Elektrophoretisch getrennte Proteine können mittels einer Coomassie Brilliant Blue G
250/R250-Lösung sichtbar gemacht werden, wobei die Nachweisgrenze je nach Protein bei
cirka 200-400 ng/0,5cm Bande liegt. Dabei wird das Gel nach der Elektrophorese für
mindestens 30min in einem Färbebad geschwenkt. Das Entfärben erfolgt danach für
mehrere Stunden in Wasser. Durch wiederholtes Färben und Entfärben kann eine
kontrastreichere Bandenfärbung erzielt werden.
Färbelösung
10 % (v/v) Ethanol (96 %)
5 % (v/v) Essigsäure
0,002 % (w/v) Coomassie G/R250
2.2.3.5 Proteolytische Spaltung von GST-Fusionsproteinen
Die ausgehend von dem Vektor pGEX-6P-1 exprimierten Proteine besitzen eine N-
terminale Glutathion-S-Transferase-Affinitätssequenz (GST-Sequenz). Nach der

Material und Methoden
34
Reinigung über eine GSH-Sepharose wird die GST-Sequenz proteolytisch vom Zielprotein
abgespalten. Zwischen dem Zielprotein und der Glutathion-S-Transferase-Affinitäts-
sequenz befindet sich eine Erkennungssequenz für die PreScission-Protease. Diese besteht
aus den sieben Aminosäuren Leu-Glu-Val-Leu-Phe-Gln-Gly-Pro. Die PreScission-
Protease schneidet hochspezifisch zwischen den Aminosäuren Gln und Gly.
Für einen beliebigen proteolytischen Ansatz wird pro mg GST-Fusionsprotein etwa 50 µg
PreScission-Protease eingesetzt und die Lösung für mindestens 10 Stunden bei 4 °C auf
dem Taumelrollenmischer inkubiert.
Nach der Behandlung mit der Protease wird die Vollständigkeit der proteolytischen
Spaltung durch SDS-Polyacrylamidgelelektrophorese (2.2.3.3) analysiert.
2.2.4 Chromatographische Methoden
2.2.4.1 Affinitätschromatographische Reinigung von GST-Fusionsproteinen
GST-Fusionsproteine besitzen eine Glutathion-S-Transferase-Sequenz, mit deren Hilfe es
möglich ist, das Fusionsprotein affinitätschromatografisch über Glutathion-Sepharose 4B
zu reinigen. Das Enzym Glutathion-S-Transferase bindet spezifisch an das Substrat
Glutathion (γ-Glutamylcysteinylglycin).
GSH-Sepharose-Säulen besitzen kovalent an die Säulenmatrix gekoppelte Glutathion-
gruppen, an die das gewünschte Fusionsprotein über seine GST-Sequenz bindet. Nach
einem Waschschritt wird das immobilisierte Zielprotein durch Zugabe von reduziertem
Glutathion von der Säule eluiert.
Die in dieser Arbeit verwendete 30 ml GSH-Sepharose (General Electrics Health Care)
muß zunächst mit Puffer A equilibriert werden. Anschließend wird der Überstand des
zentrifugierten Aufschlusses (30000 x g, 4 °C, 40 Minuten) auf die Säule geladen. Es folgt
ein Waschschritt mit dem für den Aufschluss verwendeten Lyispuffer (2.2.2.3.3), um
gebundene RNA zu entfernen. Die kompetitive Verdrängung, und damit die Elution,
erfolgt durch 20 mM Glutathion in Puffer C. Das Eluat wird fraktioniert. Alle
Affinitätschromatographien von GST-Fusionsproteinen mit GSH-Sepharose 4B konnten
auch bei 20°C durchgeführt werden.

Material und Methoden
35
Laufpuffer A Waschpuffer B ( = Lysispuffer vgl. 2.2.2.3.3)
50 mM Tris-HCl pH 7,5 50 mM Tris-HCl pH 7,5
500 mM NaCl 2,2 M LiCl
2 mM ß-Mercaptoethanol 5mM MgCl2
2mM ß-Mercaptoethanol
Elutionspuffer C
50 mM Tris-HCl pH 7,5 (pH 8,0 für GST-rpL23a)
500 mM NaCl
20 mM Reduzierte Glutathion
2 mM ß-Mercaptoethanol
2.2.4.2 Affinitätschromatographische Reinigung über Ni+-NTA-Sepharose
Bei dieser affinitätschromatographischen Methode nutzt man die Eigenschaft von
Histidin-Seitenketten, stabile Komplexbindungen mit zweiwertigen Nickel-Kationen
auszubilden. Proteine, die mit N- oder C-terminaler His-Sequenz exprimiert werden, lassen
sich so selektiv reinigen. Die Nickel-Ionen sind meist an Nitrilotriessigsäure-Sepharose
(NTA) immobilisiert. Die beiden verbleibenden freien Koordinationsstellen stehen für die
Bindung von mobilen Liganden zur Verfügung.
Das in dieser Arbeit verwendete Protein rpL23a, exprimiert aus dem Vektor pQE80,
besitzt eine N-terminale Sequenz aus 10 Histidinen. Die Imidazol-Gruppen dieser
Histidin-Seitenketten binden hochaffin an die Nickel2+-Ionen.Die Elution des Proteins
erfolgt durch einen Imidazol-Gradienten. Imidazol verdrängt die Histidin-Seitenketten mit
steigender Konzentration kompetitiv von den Nickel-Ionen und sorgt so für die Elution des
Proteins von der Säule.
In dieser Arbeit wurden HisTrapChelating Ni-NTA-Sepharose-Säulen (5 ml, General
Electrics Health Care) benutzt. Alle Chromatographien dieser Art wurden bei 4 °C
durchgeführt. Die Säule wird zunächst mit Puffer A equliibriert. Anschließend wird der
Überstand des zentrifugierten Aufschlusses (30000 x g, 4 °C, 40 Minuten) auf die Säule
geladen. Es folgt ein Waschschritt mit Puffer B, um unspezifische gebundene Proteine zu
entfernen (2.2.2.3.3). Im Anschluss wird die Säule in Puffer C umequilibriert. Die

Material und Methoden
36
kompetitive Verdrängung, und damit die Elution, erfolgt durch einen Imidazolgradienten
(Puffer C – Puffer D). Das Eluat wird fraktioniert.
Puffer A Puffer C
50 mM HEPES pH 7,0 50 mM Tris pH 7,5
1M LiCl 300 mM NaCl
5 mM MgCl2 2 mM 2-Mercaptoethanol
2 mM 2-Mercaptoethanol
Puffer B wie Puffer A
+ 20 mM Imidazol
Puffer D wie Puffer C
+ 800 mM Imidazol
2.2.4.3 Kationenaustauschchromatographie über SP-Sepharose
Bei der Ionenaustauschchromatographie werden geladene Moleküle nach ihrer Ionenstärke
bei einem bestimmten pH-Wert aufgetrennt. Die Trägermatrix eines Ionentauschers besitzt
kovalent gebundene positiv geladene (Anionenaustauscher) oder negativ geladene
Gruppen (Kationenaustauscher). Entgegengesetzt geladene Moleküle, die in einer
flüssigen Phase gelöst sind (in diesem Fall Proteine), können über elektrostatische
Wechselwirkungen an die funktionellen Gruppen der Säulenmatrix binden. Ausmaß und
Stärke der Bindung an den Ionentauscher hängen von pH und Ionenstärke des Puffers, dem
isoelektrischen Punkt des Proteins und der Dichte der Ladungen auf der Matrix ab. Die
Auftrennung von unterschiedlichen, an die Gegenionen der Matrix gebundenen Molekülen
erfolgt über kompetitive Verdrängung durch stärkere Ionen im Elutionspuffer. Schwach
geladene Moleküle eluieren früher als stark geladene. Auf diese Weise können z. B.
Proteine nativ nach ihrem isoelektrischen Punkt pI getrennt werden.
Der Substituent der SP-Sepharose, eine Sulphopropylgruppe, besitzt relativ starke
anionische Eigenschaften, da die negative Ladung der funktionellen Sulphonat-Gruppe
mesomeriestabilisiert ist. Daher werden Kationen mit hoher Affinität an die Säulenmatrix
gebunden. Der pH-Wert des Puffers wird so eingestellt, dass er mindestens 2 Einheiten
unterhalb des isoelektrischen Punkts des Zielproteins liegt, um sicherzustellen, dass das

Material und Methoden
37
Zielprotein als Kation mit hoher Nettoladung vorliegt und über basische
Aminosäureseitenketten an die Sulphonat-Gruppen binden kann. Für Arbeiten während
dieser Diplomarbeit wurde eine 24ml SP-Sepharose FF-Säule von GE Healthcare
verwendet.
Vor der Benutzung wird mit 5 Säulenvolumina Puffer A äquilibriert. Die Elution erfolgt
über einen ansteigenden NaCl-Gradienten (Puffer A – Puffer B). Hierbei kommt es zu
einer kompetitiven Verdrängung der an die Sulphonatgruppen gebundenen basischen
Aminosäureseitenketten durch Cl--Ionen und somit zu einer zeitversetzten Elution der
verschiedenen gebundenen Proteine.
Es werden 5 ml-Fraktionen gesammelt, die später via SDS-PAGE analysiert werden. Die
Fraktionen mit dem gesuchten Protein werden vereinigt und für weitere Reinigungsschritte
bei 4° C aufbewahrt.
Puffer A Puffer B
50 mM Tris-HCl pH 7,7 für rpL5
pH 8,4 für rpL23a
50 mM Tris-HCl pH 7,7 für rpL5
pH 8,4 für rpL23a
300 mM NaCl 1 M NaCl
2 mM ß-Mercaptoethanol (nur für rpL5) 2mM ß-Mercaptoethanol (nur für rpL5)
2.2.4.4 Ausschlusschromatograpisches Umäquilibrieren von Proteinen
Proteine, bei denen eine Affinitätschromatographie über einen Ionentauscher den finalen
Reinigungsschritt darstellt, müssen vor dem letzten Schritt der Präparation in den finalen
Puffer überführt werden. Hierfür wird eine HiPrep 26/60 Desalting-Entsalzungssäule
verwendet. Hierbei handelt es sich im Prinzip um eine ausschlusschromatographische
Säule. Auf Grund der grobporigen Beschaffenheit des Säulenmaterials (Sephadex™ G-25
Fine) werden größere Moleküle, wie Proteine, jedoch kaum noch nach ihrer Masse
getrennt.
Zunächst wird die Säule mit dem dreifachen Säulenvolumen an Puffer äquilibriert. Bis zu
10 ml Probe können pro Säulenlauf über eine Auftragungsschleife geladen werden. Die
Proben des Absorptionsmaximums werden nach dem Lauf vereinigt, konzentriert (2.2.3.1)
und die Proteinkonzentration bestimmt (2.2.6.1.2). Eine anschließende Qualitätskontrolle

Material und Methoden
38
des präparierten Proteins, z.B. zum Nachweis von Polymeren, mittels einer analytischen
Ausschlusschromatographie (2.2.4.6), ist notwendig. Für längere Lagerung wird das
Protein in Aliquots von 100 µl in flüssigem Stickstoff weggefroren und bei -80°C gelagert.
Finaler Puffer für rpL5 und rpL23a Finaler Puffer für rpL23aBIB
20 mM Tris-HCl pH 7,5 20 mM Tris-HCl pH 7,5
300 mM NaCl 100 mM NaCl
2 mM ß-Mercaptoethanol 2 mM ß-Mercaptoethanol
2.2.4.5 Präparative Ausschlusschromatographie von Proteinen
Die HiPrep 26/60 Superdex 200 Säule (General Electrics Health Care) eignet sich zur
Trennung von Proteinen mit einer Molekülmasse von 10 bis 600 kDa. Ihre Gelmatrix
besteht aus Allyldextran, das kovalent zu N,N’-Methylenbisacrylamid verknüpft ist.
Für die Präparation von Proteinkomplexen werden die jeweiligen Proteine vor Beginn des
Säulenlaufs in einem definierten Verhältnis (siehe Ergebnisse) und einem Gesamtvolumen
von 5 ml für eine Stunde bei 4° C inkubiert. Im Anschluss wird eventuell vorhandenes
Präzipitat durch Zentrifugation bei 13000 xg für 10 min und bei 4° C in einer Eppendorf-
Tischzentrifuge entfernt.
Nach Äquilibrierung der Säule mit 1,5-fachem Säulenvolumen des entsprechenden Puffers
wird die Probe über eine 5 ml-Auftragsschleife injiziert. Die Proteinprobe wird von der
Säule mit Puffer eluiert und in 3 bis 5ml großen Fraktionen gesammelt. Die
Ausschlusschromatographie erfolgt für alle Proteine bei 4 °C und einer Fließgeschwin-
digkeit von 1,5 ml/Minute. Die Analyse der Fraktionen erfolgt anschließend durch SDS-
Polyacrylamidgelelektrophorese (2.2.3.3).

Material und Methoden
39
Puffer für den Komplex
Imp7/rpL23a
Puffer für den Komplex
Imp7/rpL5
20 mM Tris-HCl pH 7,5 20 mM Tris-HCl pH 7,5
200 mM NaCl 300 mM NaCl
2 mM ß-Mercaptoethanol 2 mM ß-Mercaptoethanol
Puffer für die Präparation von
rpL23a-BIB
Puffer die Komplexe
Imp7/rpL23aBIB
Imp7C598/rpL23aBIB
Impß 127-641/rpL23aBIB
20 mM Tris-HCl pH 7,6 20 mM Tris-HCl pH 7,6
100 mM NaCl 100 mM NaCl
1mM MgCl2 2 mM ß-Mercaptoethanol
2 mM ß-Mercaptoethanol
2.2.4.6 Analytische Ausschlusschromatographie von Proteinen
Analytische Ausschlusschromatographiesäulen sind für die Präparation von Proteinen im
großen Maßstab aufgrund der geringen auftragbaren Probevolumina (> 500µl) eher
ungeeignet. Sie eignen sich jedoch gut für die Analyse von Proteinkomplexen. Da stabile
Komplexe während der Elution in der Regel intakt bleiben, lassen sich aus ihrem
Elutionsvolumen Rückschlüsse auf Eigenschaften wie z.B. das Molekulargewicht ableiten.
Für die analytische Gelfiltration wird eine Superdex S200 10/300 GL Säule von General
Electrics Health Care verwendet.
Für die Analyse von Komplexen der ribosomalen Proteine L5 bzw. L23a mit
Importrezeptoren werden die jeweiligen Proteine vor Beginn des Säulenlaufs in einem
Verhältnis von 2:1 (Rezeptor : ribosomales Protein) und einem Gesamtvolumen von
250 µl für eine halbe Stunde bei 4° C inkubiert. Im Anschluss wird eventuell vorhandenes

Material und Methoden
40
Präzipitat durch Zentrifugation bei 16100 xg für 10min und bei 4° C in einer Eppendorf-
Tischzentrifuge entfernt. Danach wird die Probe auf die zuvor mit Puffer gewaschene
Säule aufgetragen und eluiert. 0,5 ml große Fraktionen werden gesammelt und im SDS
Polyacrylamidgel analysiert.
Elutionspuffer
50 mM Tris/HCl pH 7.5
500 mM NaCl
2.2.4.7 Kalibrierung einer Auschlusschromatographiesäule
Das Retentionsvolumen eines Proteins korreliert bei einer ausschlusschromatographischen
Säule innerhalb eines bestimmten Trennbereichs direkt mit dem Molekulargewicht. So ist
es möglich, aus den Retentionsvolumina einer Reihe von globulären Proteinen mit
bekanntem Molekulargewicht eine Kalibrierungsgerade für eine Ausschlusschromato-
graphiesäule zu erstellen. Die Retentionsvolunina der Proteine sind hierbei proportional
zum Logarithmus des Molekulargewichts. Aus der Auftragung des Logarithmus der
Molekulargewichte der Proteine gegen die Retentionsvolumina lässt sich durch lineare
Regression die Funktion einer Kalibrierungsgerade errechnen. Hierfür wurden 25 µl Gel
Filtration Standard der Firma Bio-Rad (2.1.7) auf eine Superdex S200 10/300 GL-Säule
geladen und mit einer Flussgeschwindigkeit von 0,5 ml/min in dem unter 2.2.4.6
angegebenen Puffer eluiert. Die Kalibrierungsfunktion der Supderdex S200 10/300 GL-
Säule ist unter 3.1.2.5 angegeben.
2.2.5 Isotherme Titrationskalorimetrie
Die Methode der isothermischen Titrationskaloriemetrie (ITC) ermöglicht die
thermodynamische Charakterisierung von Protein-Protein-Interaktionen. Fast jede
chemische Reaktion ist mit einer Freisetzung oder Absorption von Wärme verbunden. Mit
Hilfe der ITC ist es möglich, diese Wärme hochsensitiv über einen Leistungskompen-
sationsmechanismus im µcal·s-1-Bereich zu messen. Dabei wird die Wärmeenthalpie

Material und Methoden
41
bestimmt, die bei der Assoziation eines Liganden mit seinem Bindepartner umgesetzt
wird. Die Titrationsexperimente wurden in einem VP-ITC Mikrokalorimetersystem
(Microcal Inc., MA) durchgeführt.
Das ITC besteht aus einer Referenzzelle (RZ) und einer Messzelle (MZ), die jeweils ein
Volumen von ca.1.4 ml fassen. Sie sind von einem adiabatischen Mantel umgeben, der den
Temperaturausgleich mit ihrer Umgebung verhindert. Während die RZ den Puffer der
Probe enthält, wird die MZ mit der Lösung des ersten Reaktionspartners befüllt. In die
MZ wird eine Spritze eingeführt, welche die Lösung des zweiten Reaktanden enthält.
Diese Spritze fasst ein Volumen bis zu 285 µl. Das Ende der Spritzennadel dient als
Rührpaddel und ermöglicht eine Durchmischung des Reaktionsvolumens. In festgelegten
Zeitintervallen wird so ein definiertes Volumen des zweiten Reaktanden computergesteuert
in die MZ injiziert.
Die RZ wird mit einer geringen Leistung im µW-Bereich beheizt. Dieses Heizsystem wird
als „Referenz-Offset“ bezeichnet. Sowohl zwischen der RZ und MZ als auch zwischen den
Zellen und dem Mantel des Gerätes werden ständig die Temperaturdifferenzen ∆T1 und
∆T2 ermittelt. Die Temperatur der Zellen und des Mantels werden jeweils auf die der
Referenzzelle durch die Aktivierung eines weiteren Heizsystems, dem „Jacket Feedback“,
abgestimmt. Kommt es aufgrund einer Reaktion innerhalb der MZ zu einer Absorption
oder Freisetzung von Energie in Form von Wärme, ist eine Temperaturdifferenz ∆T1
detektierbar. Dies führt zur Erzeugung eines dem ∆T1-Wert proportionalen elektrischen
Signals, welches über ein Rückkopplungsglied das sogenannte „Cell Feedback“ aktiviert.
Dies ist das dritte Heizsystem, welches durch eine Reduktion (exotherme Reaktion in der
MZ) oder Verstärkung (endotherme Reaktion in der MZ) der Heizleistung zu einem
Temperaturausgleich zwischen RZ und MZ (∆T1=0) führt. Dieses elektrische
Kompensationssignal wird als Funktion der Zeit aufgezeichnet.
In der vorliegenden Arbeit wurde die ITC dazu genutzt, die bei der Bildung von
Komplexen aus Kernimportrezeptoren (Impß und Imp7) und ribosomalen Proteinen
umgesetzten Bindungsenthalpien, -entropien und -affinitäten zu detektieren sowie die
auftretenden Stöchiometrien zu ermitteln. Dabei wurden die Importrezeptoren in eine
Messzelle vorgelegt und das jeweilige ribosomale Protein hinzutitriert. Die verwendeten
Proteinkonzentrationen sind in den jeweiligen Ergebnisteilen angegeben. Die
Proteinkonzentrationen der Importrezeptoren wurden nach Bearden und die der
ribosomalen Proteine über die Absorption bei 280 nm bestimmt (2.2.6.1.2).

Material und Methoden
42
Alle Lösungen wurden vor Gebrauch bei 8 °C entgast (ThermoVac, Microcal) und unter
Vermeidung von Luftblasen in die mehrmals mit Puffer gespülte Messzelle des
Titrationskalorimeters (VP-ITC, MicroCal) eingebracht. Die Puffer- und Konzentrations-
bedingungen der einzelnen Experimente sind im jeweiligen Ergebnisteil aufgeführt. Die
Referenzzelle enthielt stets den gleichen Puffer wie die Messzelle. Beim Befüllen der
computergesteuerten Injektionseinheit wurde darauf geachtet, dass keine Luftblasen in das
System gelangten und der als Rührer dienende abgeflachte Spritzenkopf gut gereinigt
wurde. Alle ergebnisrelevanten Messungen wurden bei 10 °C durchgeführt, da einige der
entstehenden Proteinkomplexe zum Präzipitieren neigten und dadurch die Messsignale
verfälschten. Zu Beginn jedes ITC-Experiments wurden zunächst 4 µl injiziert. Die
darauffolgenden Injektionsvolumina waren jeweils 10 µl groß. Die Auswertung der
Rohdaten erfolgte über MicroCal Origin Version 7.0.
Folgende Puffersysteme wurden verwendet:
ITC-Puffer 1 (NaCl/Tris Puffer) ITC-Puffer 2 (Kaliumacetat-Puffer)
20 mM Tris / HCl pH 7,5 20 mM Kaliumphosphat pH 7,5
300 mM NaCl 20 mM Tris / HCl pH 7,5
1mM MgCl2 300 mM Kaliumacetat
1mM MgCl2
2.2.6 Spektroskopische Methoden
2.2.6.1 Absorption
Alle Absorptionsmessungen werden an einem Ultraspec2100pro Spektrophotometer in
100µl Küvetten durchgeführt.

Material und Methoden
43
2.2.6.1.1 Konzentrationsbestimmung von Nukleinsäure-Lösungen
Die Konzentration wässriger Nukleinsäurelösungen wird quantitativ im Photometer bei
260 nm gegen einen Leerwert bestimmt. Folgende Umrechnungswerte werden hierbei
herangezogen:
1 A 260nm = 50 µg/ml doppelsträngige DNA
1 A 260nm = 40 µg/ml einzelsträngige DNA
1 A 260nm = 33 µg/ml Oligonukleotid
Die Reinheit der Nukleinsäurelösung wird über den Quotient der Extinktionen bei 260 nm
und 280 nm ermittelt.
Während die aromatischen Basen der Nukleinsäuren ein Absorptionsmaximum bei 260 nm
besitzen, absorbieren die aromatischen Aminosäuren der Proteine vor allem im Bereich
von 280 nm. Reine DNA liegt bei einem Quotienten von 1,8 bis 2,0 vor. Mit Proteinen
verunreinigte DNA-Lösungen besitzen einen kleineren Quotienten der Absorptionen bei
260 nm bzw. 280 nm.
2.2.6.1.2 Konzentrationsbestimmung von Protein-Lösungen
Es wurden drei verschiedene Methoden zur quantitativen Bestimmung von
Proteinlösungen verwendet:
Methode nach Bearden (1978) („Bradford-Assay“):
Proteine lassen sich in phosphorsaurer Lösung mit dem Farbstoff Coomassie Brilliant Blue
anfärben. Die Proteinkonzentration kann mittels Absorption bei 595 nm photometrisch
bestimmt werden, da sich das Absorptionsspektrum des Farbstoffes in einem Bereich von
OD595 0,1 – 0,9 proportional zur Proteinkonzentration ändert.
20 µl Proteinprobe werden mit 1 ml 1:5 in Wasser verdünnter Bradfordreagens (1 mg/ml
Coomassie Brilliant Blue G 250 in 85%iger H3PO4) versetzt und nach 10 Minuten die
Absorption bei 595 nm gegen einen Leerwert gemessen. Aus dem Vergleich zu einer
durch verschiedene Konzentrationen an BSA erstellten Eichkurve lässt sich die
Proteinmenge der unbekannten Probe ermitteln.

Material und Methoden
44
Proteinbestimmung über die Absorption bei 280 nm:
Bei dieser Methode wird die Proteinkonzentration durch Messung der Absorption bei einer
Wellenlänge von 280 nm ermittelt. Hierfür wird der native Extinktionskoeffizient (ε280)
benötigt. Extinktionskoeffizienten nativer Proteine bei 280 nm (ε280) werden mit einem
modifiziertem Protokoll nach Gill und von Hippel, ausgehend vom Lambert-Beerschen
Gesetz errechnet:
dcA ××= ε
Dabei entspricht ε dem molaren Extinktionskoeffizienten [M-1cm-1], c der Konzentration
[M] und d der Schichtdicke der Küvette [cm]. Der Extinktionskoeffizient eines
denaturierten Proteins (ε280denat) lässt sich von der Aminosäuresequenz ableiten
(Protparam, Expasy), während die Absorption bei 280 nm (A280) einer nativen und einer
denaturierten Proteinprobe gleicher Konzentration (c) in einer Küvette gleicher
Schichtdicke (d) messbar sind. Dabei gilt:
denat
natdenatnat A
A×=
εε
Die Konzentrationen der Proteinlösungen müssen dabei so eingestellt werden, dass die
Absorption A280 zwischen 0,2 und 0,8 beträgt und damit im linearen Bereich liegt. Für die
Messung wird bei allen Proteinen jeweils der finale Präparationspuffer (vgl. 2.2.4.4,
2.2.4.5) verwendet. Die zu denaturierten Proben werden zusätzlich mit 6 M
Guanidiniumhydrochlorid versetzt und für zwei Stunden bei 37 °C inkubiert. Die
Messungen werden jeweils drei Mal und bei 10 °C durchgeführt. Ist der native
Extinktionskoeffizient eines Proteins bei 280 nm (ε280) bekannt, so kann die
Proteinkonzetration über Messung der Absorption bei 280 nm errechnet werden. Nach
dem Lambert-Beerschen Gesetz gilt:
d
Ac
×=
ε
Dabei entspricht ε dem molaren Extinktionskoeffizienten [M-1cm-1], c der Konzentration
[M] und d der Schichtdicke der Küvette [cm].

Material und Methoden
45
Methode nach Ehresmann et al. (1973):
Diese Methode ist nützlich für die Konzentrationsmessung von Proteinen, die mit RNA
verunreinigt sind. Da RNA bei Wellenlängen von 228,5 und 234,5 nm eine identische
Absorption zeigt, ist es möglich, ihre Interferenz durch einen Korrekturfaktor zu
eliminieren. Dadurch fließt nur noch die Absorption des Polypeptidrückrades der Proteine
in die Berechnung mit ein. Es gilt:
14,35,2345,228 AA
c−
=
Dabei entspricht A228,5 der Absorption bei einer Wellenlänge von 228,5 nm, A234,5 der
Absorption bei einer Wellenlänge von 234,5 nm und c der Proteinkonzentration [mg/ml].
2.2.7 Kristallographische Methoden
2.2.7.1 Kristallisation von Proteinen
Bei der Kristallisation eines Proteins wird mit der Hilfe von Salzen, Puffern und
Präzipitantien eine Proteinlösung in den übersättigten Zustand überführt. In diesem
metastabilen Zustand gibt es zwei Möglichkeiten, wie sich ein Protein verhalten kann:
1. Es fällt aus, die Konzentration an gelöstem Protein sinkt dadurch drastisch ab.
Dieser Vorgang ist stark exergonisch, da so der höchste Grad an Entropie erreicht
werden kann.
2. Das Protein bildet Kristallisationskeime aus, aus denen Proteinkristalle wachsen
können. Dieser Prozeß geschieht in der Regel deutlich langsamer als die
Präzipitation und ist energetisch auch nicht so günstig, da nicht die maximale
Entropie erreicht wird.
In der Kristallographie werden Bedingungen gesucht, die den zweiten Fall ermöglichen.
Dazu werden zu Beginn initiale Kristallisationstests durchgeführt, um erste
Kristallisationsbedingungen für ein Protein zu finden. Hierbei handelt es sich um eine
Sammlung von empirisch ermittelten Eingangsbedingungen, die relativ häufig zur
Kristallisation verschiedener Proteine geführt haben. Das gereinigte Protein wird dabei zu
Anfang meist in einer Konzentration von mehr als 5mg/ml eingesetzt.

Material und Methoden
46
Sobald Bedingungen gefunden sind, die für eine Kristallisation günstig sind, wird in einem
Raster um die Bedingungen herum optimiert. Dabei können die Konzentrationen der
enthaltenen Salze, Puffer und Präzipitantien variiert werden, es können aber auch
Substitutionen getestet werden.
Für die Kristallisation im so genannten sitzenden Tropfen werden Kristallisationsplatten
mit 24 Ansätzen verwendet, deren einzelne Kammern eine zentrale Säule mit darin
liegender Vertiefung, dem so genannten well, und ein darunter liegendes Reservoir
besitzen. In das Reservoir wird eine Bedingung vorgelegt, die dann in der zentralen
Vertiefung mit dem Protein zu einem Tropfen vermischt wird. Es besteht also ein
Konzentrationsgradient an Salzen und Präzipitantien zwischen Tropfen und Reservoir, da
der Tropfen neben dem Protein nur 50 % der Salz- und Präzipitans-Konzentration des
Reservoirs enthält. Durch die Dampfdiffusion des Wassers aus dem Tropfen in das
Reservoir wird das Volumen des Tropfens in der zentralen Vertiefung nun langsam
verringert und das enthaltene Protein konzentriert. Dabei wird der Punkt der Übersättigung
erreicht und es kann als Ausweg aus diesem metastabilen Zustand eine
Proteinkristallisationskeimung stattfinden.
Zunächst werden 500 µl der zu testenden Kristallisationsbedingung in das Reservoir
pipettiert. Aus diesem wird nun 1 µl entnommen und in die Vertiefung überführt.
Anschließend wird 1 µl der Proteinlösung hinzupipettiert und die gesamte
Kristallisationsplatte mit Klarsichtklebeband verschlossen, um einem Austrocknen
vorzubeugen. Der Tropfen wird in regelmäßigen Abständen auf beginnende Kristallisation
kontrolliert. Bilder der verschiedenen Tropfen werden mit einer Digitalkamera, die auf ein
Binokular aufgesetzt wird, aufgenommen und am Computer bearbeitet.
Ein Teil der in dieser Diplomarbeit durchgeführten Kristallisationsansätze wurde mit
einem Kristallisationsroboter der EMBL-Outstation in Hamburg pipettiert (EMBL
Hamburg c/o DESY, Notkestraße 85, 22603 Hamburg, Germany). Dabei handelte es sich
ebenfalls um eine „Kristallisation im sitzenden Tropfen“. Allerdings wurden bei dieser
automatisierten Technik 200 nl Reservoir mit 200 nl Protein gemischt. Die Auswertung
der pipettierten Screens erfolgte über das Internet. In regelmäßigen Abständen wurden von
einem vollautomatischen Lagerungs- und Überwachungssystem digitale Fotos aller
Bedingungen aufgenommen und auf einen per Passwort verfügbaren Internet-Server
geladen.

Material und Methoden
47
2.2.7.2 Micro-Seeding zur Verbesserung der Kristallordnung
Wenn sich Kristalle formiert haben, die eine ungünstige innere Ordnung besitzen z. B. von
unregelmäßiger Gestalt und geringer Auflösung im Röntgendiffraktometer sind, so können
Bruchstücke dieser Kristalle in verdünnten Lösungen (z. B. 1:200 in einer frischen
Proteinlösung) Kristallisationskeime für Kristalle höherer Ordnung sein.
Hierzu werden Kristalle aus der Originalbedingung in ein Mikrozentrifugenröhrchen mit
einer Lösung der Originalbedingung überführt und mechanisch durch einen Vortexer in
Kristallisationskeime zerlegt.
Diese Kristallisationskeime werden mithilfe eines Katzenschnurrhaares in neue Tropfen
überführt, „gesät“. Deren Inhalte sind analog zum ersten, mit einer Ausnahme: Die
Präzipitans- oder die Proteinkonzentration in diesen Tropfen müssen gegenüber dem ersten
verringert sein, da es sonst zu einer sofortigen Präzipitation des Proteins in den neuen
Tropfen kommen kann.

Ergebnisse
48
3 Ergebnisse
Der Ergebnisteil ist in zwei Abschnitte gegliedert.
Im ersten Abschnitt werden zunächst die Ergebnisse der Expression und Reinigung des
ribosomalen Proteins rpL5 aus Xenopus laevis dargestellt. Des Weiteren wird die
Präparation eines Komplexes von rpL5 mit dem Kernimportrezeptor Importin 7 (ebenfalls
X. laevis) sowie der Versuch einer Kristallisation dieses Komplexes beschrieben.
Der zweite Abschnitt behandelt zunächst die Ergebnisse der Expression und Reinigung des
ribosomalen Proteins rpL23a aus Homo sapiens. Auch dieses Protein wurde als Komplex
mit Importin 7 präpariert und für Kristallisationsversuche eingesetzt.
Ferner werden die Ergebnisse einer funktionellen Untersuchung der Bindung zwischen
rpL23a und den Importrezeptoren Impß und Imp7 dargestellt. Hierfür wurden die
Methoden der analytischen Ausschlusschromatographie und der isothermen
Titrationskalorimetrie eingesetzt.
Der letzte Teil des zweiten Abschnitts beschreibt zunächst die Klonierung und Präparation
der Bindedomäne von rpL23a (AS 32 - 74) für die Importrezeptoren Imp7 und Impß. Im
Anschluss wird die Präparation von Komplexen der Bindedomäne mit den
Importrezeptoren Imp7 (Volllänge), Imp7C598 (AS 598 - 1038) und Impß127-641 (AS
127-641) erläutert. Auch diese Komplexe wurden in Kristallisationsexperimenten
eingesetzt.

Ergebnisse
49
3.1 Biochemische Arbeiten mit dem Protein rpL5 aus Xenopus laevis
3.1.1 Klonierung und Expression von rpL5
3.1.1.1 Subklonierung von rpL5 aus Xenopus laevis in den Vektor pGEX-6P-1
Zu Beginn der Diplomarbeit wurde das codierende Gen für rpL5 aus Xenopus laevis
(accession number BC042258; NCBI), kloniert in den Expressionsvektor pGEX-5X-1,
von T. Pieler (Universität Göttingen) zur Verfügung gestellt. Dieses Konstrukt ermöglicht
die Expression von rpL5 als Fusionsprotein mit einer N-terminalen Gluathion-S-
Transferase (GST)-Affinitätssequenz. Die ausgehend von pGEX-5X-1 exprimierten
Fusionsproteine besitzten zur Entfernung der GST-Sequenz jedoch nur eine Schnittstelle
für die Protease Faktor Xa. Diese Protease war nicht verfügbar und im Labor nicht
praxiserprobt. Daher wurde eine Subklonierung in den Vektor pGEX-6P-1 durchgeführt.
Die Expressionsprodukte dieses Vektors sind ebenfalls Fusionsproteine mit einer N-
terminalen GST-Sequenz. Sie besitzen jedoch zwischen Affinitätssequenz und Zielprotein
eine Schnittstelle für die Protease PreScission.
Für die Subklonierungs-PCR (2.2.1.1.1) wurden die DNA-Oligonukleotide
rpL5_BamHIfor und rpL5_XhoIrev (2.1.8) verwendet und das amplifizierte Gen über
BamHI- und XhoI-Schnittstellen in pGEX-6P-1 inseriert. Das Vektorkonstrukt pGEX-6P-
1-rpL5 wurde in E. coli XL1-Blue transformiert (2.2.2.1) und in einer anschließenden
Mini-Präparation (2.2.2.2) amplifiziert.
Zur Kontrolle der Subklonierung wurde eine Sequenzanalyse (2.2.1.1.5) unter
Verwendung der DNA-Oligonukleotide pGEX_f und pGEX_r (2.1.8) durchgeführt.
3.1.1.2 Expression von rpL5 mit GST-Affinitätssequenz
Ein erstes Ziel war es zunächst, einen geeigneten Expressionsstamm sowie geeignete
Wachstumsbedingungen für eine maximale Expressionsausbeute von rpL5 zu finden. Da

Ergebnisse
50
das Protein für Kristallisationsansätze verwendet werden sollte, wurde eine Ausbeute von
mehreren mg/L Expressionskultur angestrebt.
Mehrere E.coli-Stämme (BL21(DE3), BL21(DE3) RP, Rosetta 2 (DE3), BL21(DE3)
STAR) wurden mit dem zuvor amplifizierten Plasmid pGEX-6P-1-rpL5 transformiert
(2.2.2.1). Die Expression erfolgte nach dem unter 2.2.2.3.1 beschriebenen Protokoll. Die
stärkste Expression des Zielproteins zeigte sich in den Stämmen BL21(DE3) RP und
BL21(DE3) Rosetta 2. Beide Stämme wurden für die Überexpression eingesetzt und
lieferten etwa gleiche Mengen des Fusionsproteins GST-rpL5. Abbildung 9 zeigt eine
SDS-PAGE mit Proben der Expression von GST-rpL5 in Rosetta 2 (DE3).
Die Überexpression von GST-rpL5 im präparativen Maßstab erfolgte wie unter 2.2.2.3.2
beschrieben.
Abbildung 9: SDS-PAGE der Expression von GST-rpL5 in pGEX-6P-1 in E.coli Rosetta 2 (DE3). (vI = vor Induktion, nI = nach Induktion, M=Größenstandard BR, 15%iges SDS-Gel)
3.1.1.3 Zellernte und Aufschluss von GST-rpL5
Die Expressionskulturen wurden durch Zentrifugation bei 5000 xg für 20 Minuten und bei
4 °C geerntet und wie unter 2.2.2.3.3 beschrieben aufgeschlossen. Bei den ersten
Versuchen einer Affinitätsreinigung mit einer GSH-Sepharose-Säule waren größere
Mengen RNA aus E. coli unspezifisch an rpL5 gebunden (Daten nicht gezeigt). Mit Hilfe
einer hohen Konzentration von LiCl war es jedoch möglich, die gebundene RNA von rpL5
zu lösen. Daher wurde ein Lysispuffer mit 2,2 M LiCl für den Aufschluss der
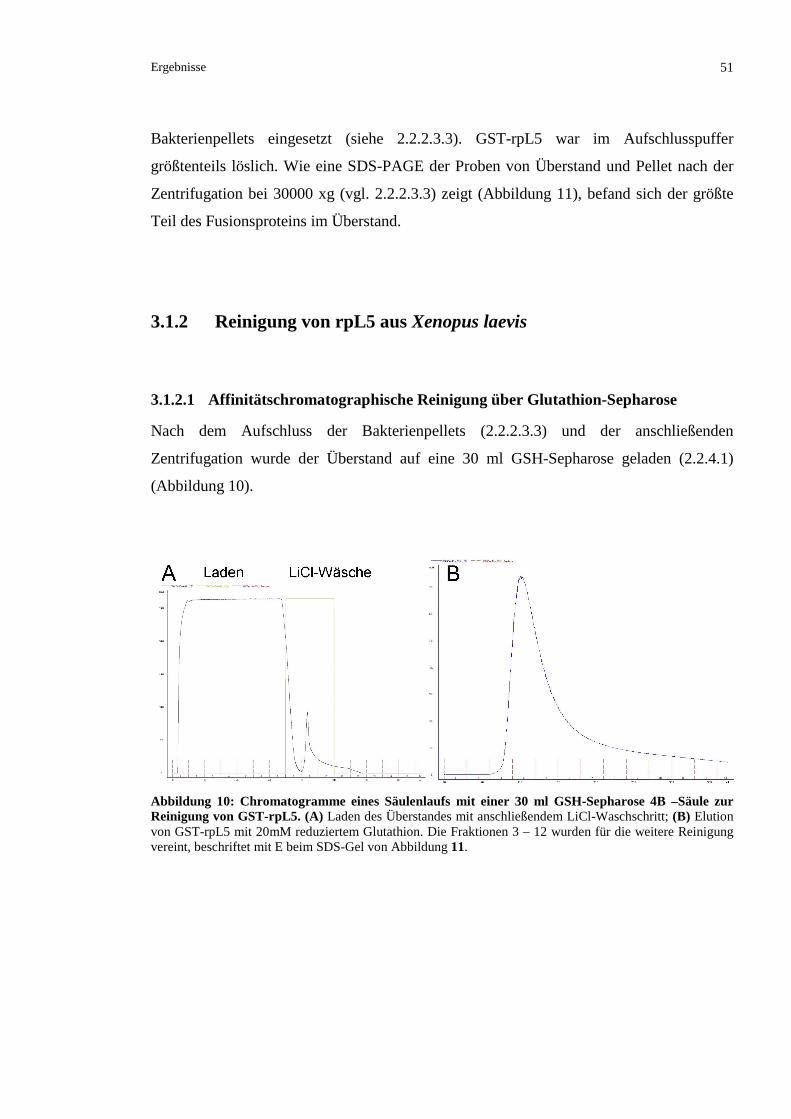
Ergebnisse
51
Bakterienpellets eingesetzt (siehe 2.2.2.3.3). GST-rpL5 war im Aufschlusspuffer
größtenteils löslich. Wie eine SDS-PAGE der Proben von Überstand und Pellet nach der
Zentrifugation bei 30000 xg (vgl. 2.2.2.3.3) zeigt (Abbildung 11), befand sich der größte
Teil des Fusionsproteins im Überstand.
3.1.2 Reinigung von rpL5 aus Xenopus laevis
3.1.2.1 Affinitätschromatographische Reinigung über Glutathion-Sepharose
Nach dem Aufschluss der Bakterienpellets (2.2.2.3.3) und der anschließenden
Zentrifugation wurde der Überstand auf eine 30 ml GSH-Sepharose geladen (2.2.4.1)
(Abbildung 10).
Abbildung 10: Chromatogramme eines Säulenlaufs mit einer 30 ml GSH-Sepharose 4B –Säule zur Reinigung von GST-rpL5. (A) Laden des Überstandes mit anschließendem LiCl-Waschschritt; (B) Elution von GST-rpL5 mit 20mM reduziertem Glutathion. Die Fraktionen 3 – 12 wurden für die weitere Reinigung vereint, beschriftet mit E beim SDS-Gel von Abbildung 11.
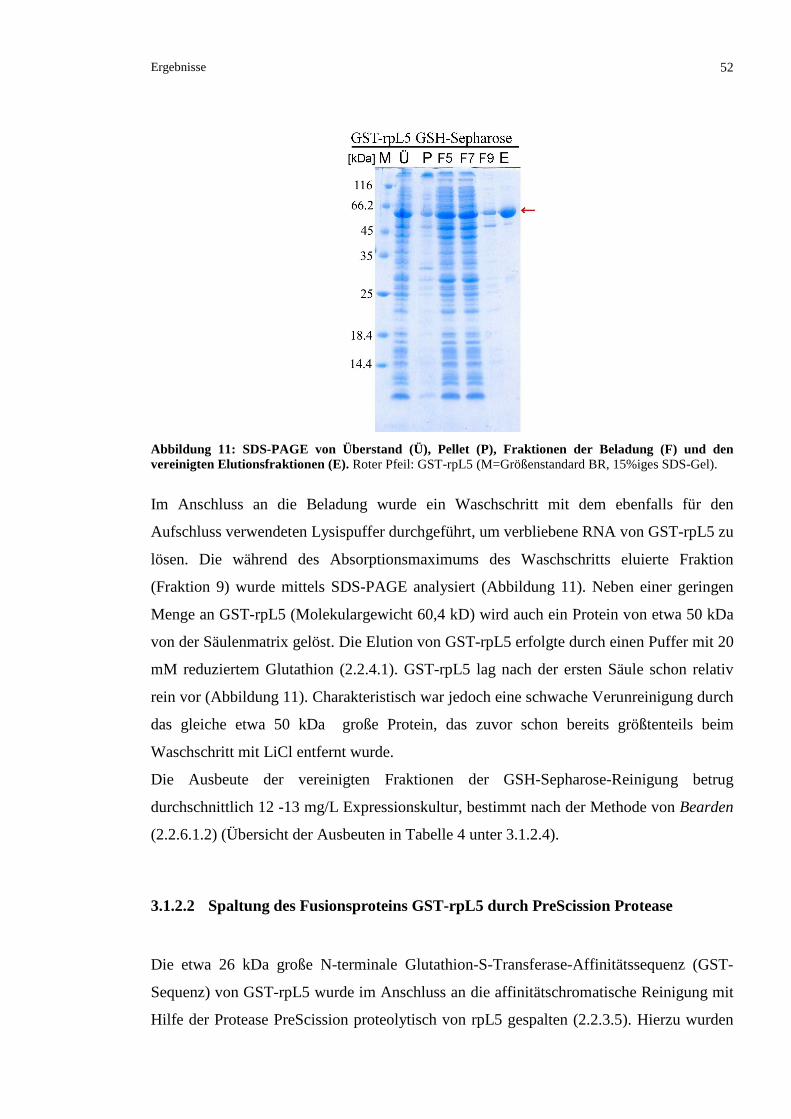
Ergebnisse
52
Abbildung 11: SDS-PAGE von Überstand (Ü), Pellet (P), Fraktionen der Beladung (F) und den vereinigten Elutionsfraktionen (E). Roter Pfeil: GST-rpL5 (M=Größenstandard BR, 15%iges SDS-Gel).
Im Anschluss an die Beladung wurde ein Waschschritt mit dem ebenfalls für den
Aufschluss verwendeten Lysispuffer durchgeführt, um verbliebene RNA von GST-rpL5 zu
lösen. Die während des Absorptionsmaximums des Waschschritts eluierte Fraktion
(Fraktion 9) wurde mittels SDS-PAGE analysiert (Abbildung 11). Neben einer geringen
Menge an GST-rpL5 (Molekulargewicht 60,4 kD) wird auch ein Protein von etwa 50 kDa
von der Säulenmatrix gelöst. Die Elution von GST-rpL5 erfolgte durch einen Puffer mit 20
mM reduziertem Glutathion (2.2.4.1). GST-rpL5 lag nach der ersten Säule schon relativ
rein vor (Abbildung 11). Charakteristisch war jedoch eine schwache Verunreinigung durch
das gleiche etwa 50 kDa große Protein, das zuvor schon bereits größtenteils beim
Waschschritt mit LiCl entfernt wurde.
Die Ausbeute der vereinigten Fraktionen der GSH-Sepharose-Reinigung betrug
durchschnittlich 12 -13 mg/L Expressionskultur, bestimmt nach der Methode von Bearden
(2.2.6.1.2) (Übersicht der Ausbeuten in Tabelle 4 unter 3.1.2.4).
3.1.2.2 Spaltung des Fusionsproteins GST-rpL5 durch PreScission Protease
Die etwa 26 kDa große N-terminale Glutathion-S-Transferase-Affinitätssequenz (GST-
Sequenz) von GST-rpL5 wurde im Anschluss an die affinitätschromatische Reinigung mit
Hilfe der Protease PreScission proteolytisch von rpL5 gespalten (2.2.3.5). Hierzu wurden

Ergebnisse
53
die nach der GSH-Sepharose vereinigten Fraktionen für mindestens 12 Stunden bei 4° C
mit der Protease PreScission im Massenverhältnis 1:200 inkubiert. Die GST-Sequenz
wurde fast vollständig abgespalten, wie anhand einer mittels SDS-PAGE (Abbildung 12)
analysierten Probe ersichtlich ist.
Bei ersten Reinigungsversuchen kam es im Verlaufe des Schneideprozesses zu starker
Präzipitatbildung in Folge einer zu niedrigen NaCl Konzentration (300mM NaCl). Dieser
Effekt konnte jedoch durch Verwendung eines Puffers mit 500mM NaCl weitestgehend
eliminiert werden (2.2.4.1).
Als wichtig erwies sich, dass alle auf die proteolytische Abspaltung der GST-Sequenz
folgenden Reinigungsschritte bei möglichst niedrigen Temperaturen durchgeführt werden
(4°C), da rpL5 bei Raumtemperatur zur Präzipitation neigt. Wichtig war auch die Zugabe
von Protease-Inhibitoren vor dem Laden der Probe auf die nachfolgende SP-Sepharose-
Säule (2.2.4.3), da rpL5 sensitiv für einen Abbau durch Proteasen ist (Daten nicht gezeigt).
3.1.2.3 Kationenaustauschchromatographische Reinigung von rpL5
Nach der proteolytischen Abspaltung der N-terminalen GST-Sequenz von rpL5 wurde als
nächstes eine chromatographische Reinigung mit Hilfe eines Sulphopropyl-Sepharose-
Kationentauschers (SP-Sepharose) durchgeführt (2.2.4.3). Hauptziel dieses Reinigungs-
schrittes war die Trennung der abgespaltenen GST-Affinitätssequenz von rpL5. GST
bildet unter den gegebenen Pufferbedingungen (2.2.4.1) Dimere mit einem
Molekulargewicht von ca. 52 kD und kann daher durch Gelfiltrationssäulen nur schwierig
von rpL5 (34,4 kDa) getrennt werden. Der pI (isoelektrischer Punkt) von GST liegt bei pH
6,8, weshalb das Protein bei pH 7,5 in Puffer A (vgl.2.2.4.3) eine negative Nettoladung
besitzt und nach dem Beladen der Säule im Durchfluss eluiert wird.
Die Auftrennung der gebundenen Proteine erfolgte über einen Salzgradienten von 0,3 bis 1
M NaCl (Abbildung 12). Bei einer Konzentration von etwa 750-800 mM NaCl wurde rpL5
von der Säulenmatrix verdrängt. Eine Analyse von Proben des Absorptionsmaximums
mittels SDS-PAGE zeigte, dass rpL5 in hochreiner Form vorlag. Die Fraktionen 28 bis 34
wurden vereinigt und für die weitere Verarbeitung bei 4° C gelagert.
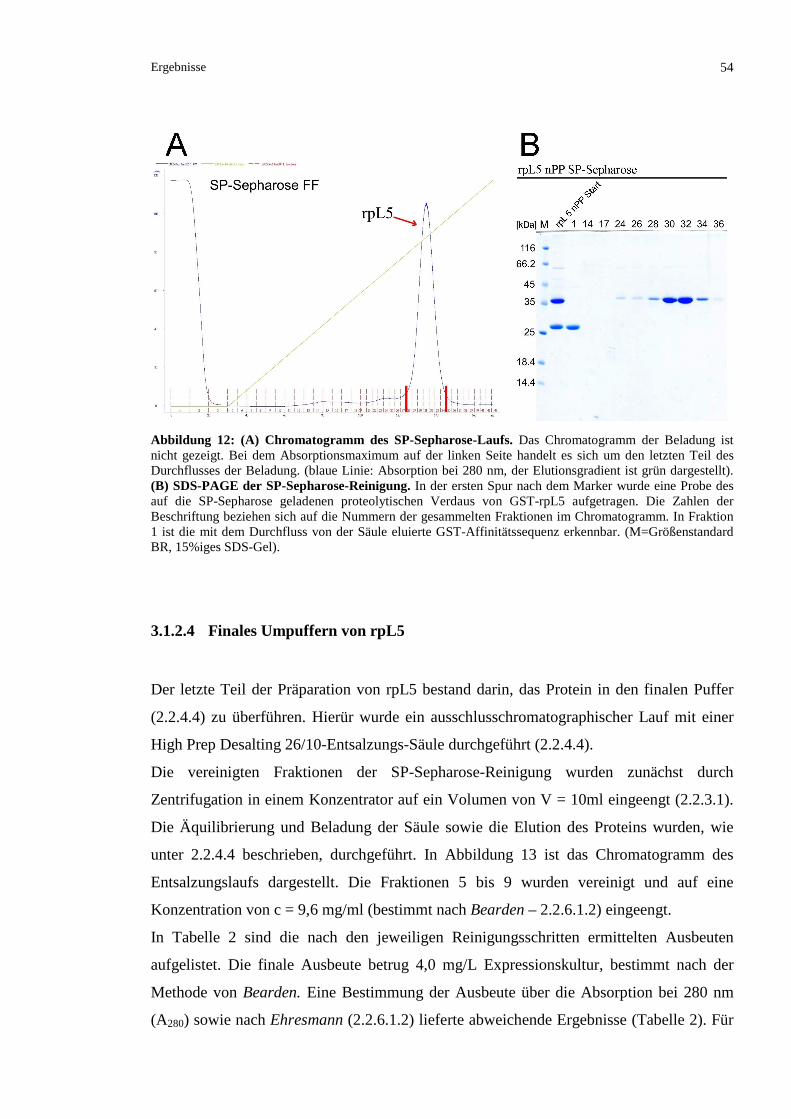
Ergebnisse
54
Abbildung 12: (A) Chromatogramm des SP-Sepharose-Laufs. Das Chromatogramm der Beladung ist nicht gezeigt. Bei dem Absorptionsmaximum auf der linken Seite handelt es sich um den letzten Teil des Durchflusses der Beladung. (blaue Linie: Absorption bei 280 nm, der Elutionsgradient ist grün dargestellt). (B) SDS-PAGE der SP-Sepharose-Reinigung. In der ersten Spur nach dem Marker wurde eine Probe des auf die SP-Sepharose geladenen proteolytischen Verdaus von GST-rpL5 aufgetragen. Die Zahlen der Beschriftung beziehen sich auf die Nummern der gesammelten Fraktionen im Chromatogramm. In Fraktion 1 ist die mit dem Durchfluss von der Säule eluierte GST-Affinitätssequenz erkennbar. (M=Größenstandard BR, 15%iges SDS-Gel).
3.1.2.4 Finales Umpuffern von rpL5
Der letzte Teil der Präparation von rpL5 bestand darin, das Protein in den finalen Puffer
(2.2.4.4) zu überführen. Hierür wurde ein ausschlusschromatographischer Lauf mit einer
High Prep Desalting 26/10-Entsalzungs-Säule durchgeführt (2.2.4.4).
Die vereinigten Fraktionen der SP-Sepharose-Reinigung wurden zunächst durch
Zentrifugation in einem Konzentrator auf ein Volumen von V = 10ml eingeengt (2.2.3.1).
Die Äquilibrierung und Beladung der Säule sowie die Elution des Proteins wurden, wie
unter 2.2.4.4 beschrieben, durchgeführt. In Abbildung 13 ist das Chromatogramm des
Entsalzungslaufs dargestellt. Die Fraktionen 5 bis 9 wurden vereinigt und auf eine
Konzentration von c = 9,6 mg/ml (bestimmt nach Bearden – 2.2.6.1.2) eingeengt.
In Tabelle 2 sind die nach den jeweiligen Reinigungsschritten ermittelten Ausbeuten
aufgelistet. Die finale Ausbeute betrug 4,0 mg/L Expressionskultur, bestimmt nach der
Methode von Bearden. Eine Bestimmung der Ausbeute über die Absorption bei 280 nm
(A280) sowie nach Ehresmann (2.2.6.1.2) lieferte abweichende Ergebnisse (Tabelle 2). Für
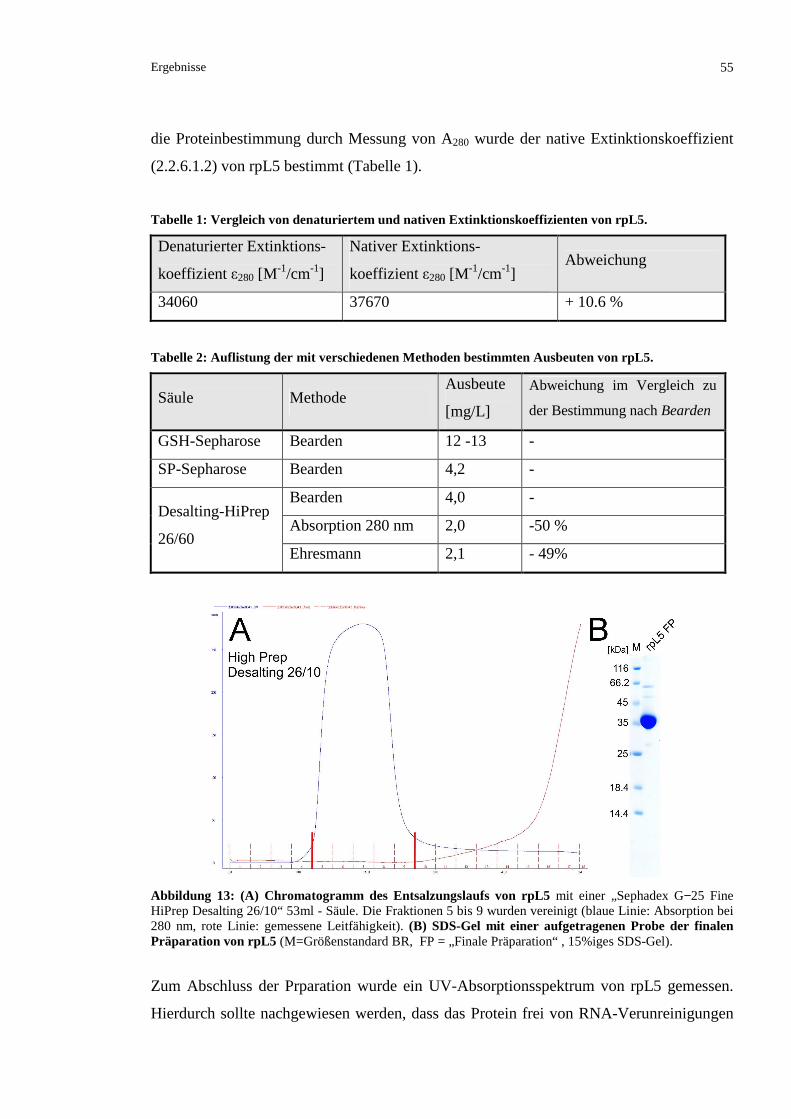
Ergebnisse
55
die Proteinbestimmung durch Messung von A280 wurde der native Extinktionskoeffizient
(2.2.6.1.2) von rpL5 bestimmt (Tabelle 1).
Tabelle 1: Vergleich von denaturiertem und nativen Extinktionskoeffizienten von rpL5.
Denaturierter Extinktions-
koeffizient ε280 [M-1/cm-1]
Nativer Extinktions-
koeffizient ε280 [M-1/cm-1]
Abweichung
34060 37670 + 10.6 %
Tabelle 2: Auflistung der mit verschiedenen Methoden bestimmten Ausbeuten von rpL5.
Säule Methode Ausbeute
[mg/L]
Abweichung im Vergleich zu
der Bestimmung nach Bearden
GSH-Sepharose Bearden 12 -13 -
SP-Sepharose Bearden 4,2 -
Bearden 4,0 -
Absorption 280 nm 2,0 -50 % Desalting-HiPrep
26/60 Ehresmann 2,1 - 49%
Abbildung 13: (A) Chromatogramm des Entsalzungslaufs von rpL5 mit einer „Sephadex G−25 Fine HiPrep Desalting 26/10“ 53ml - Säule. Die Fraktionen 5 bis 9 wurden vereinigt (blaue Linie: Absorption bei 280 nm, rote Linie: gemessene Leitfähigkeit). (B) SDS-Gel mit einer aufgetragenen Probe der finalen Präparation von rpL5 (M=Größenstandard BR, FP = „Finale Präparation“ , 15%iges SDS-Gel).
Zum Abschluss der Prparation wurde ein UV-Absorptionsspektrum von rpL5 gemessen.
Hierdurch sollte nachgewiesen werden, dass das Protein frei von RNA-Verunreinigungen
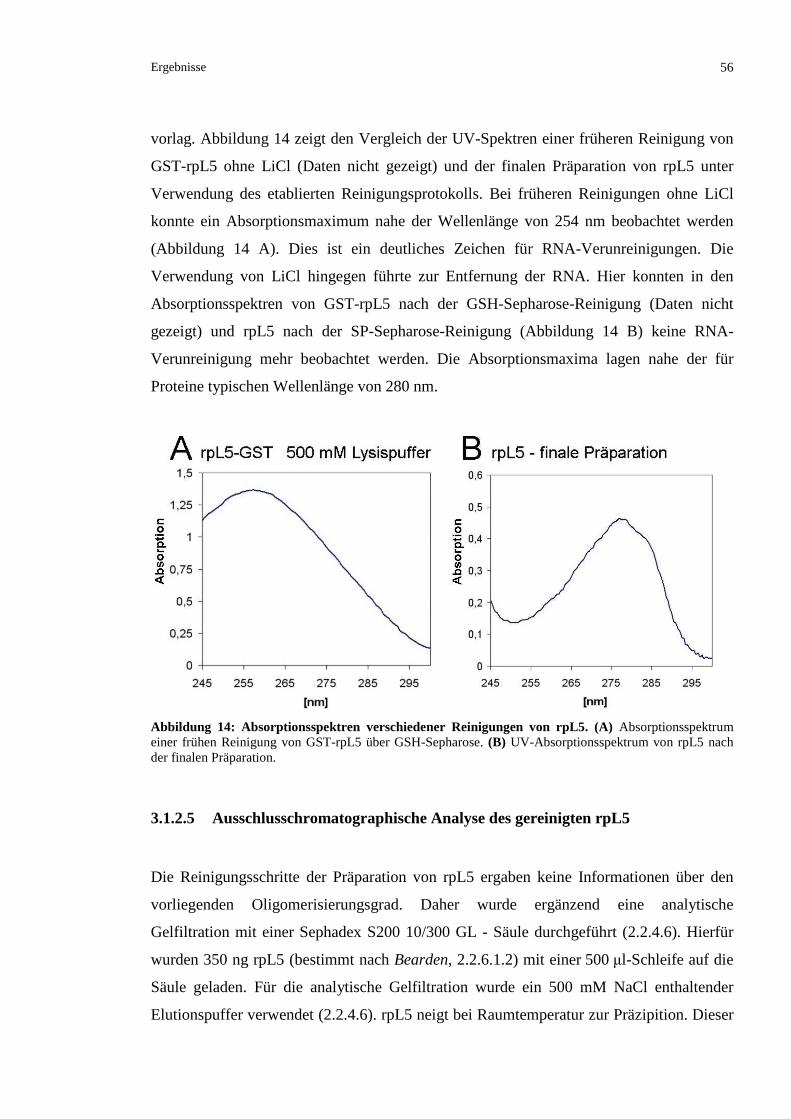
Ergebnisse
56
vorlag. Abbildung 14 zeigt den Vergleich der UV-Spektren einer früheren Reinigung von
GST-rpL5 ohne LiCl (Daten nicht gezeigt) und der finalen Präparation von rpL5 unter
Verwendung des etablierten Reinigungsprotokolls. Bei früheren Reinigungen ohne LiCl
konnte ein Absorptionsmaximum nahe der Wellenlänge von 254 nm beobachtet werden
(Abbildung 14 A). Dies ist ein deutliches Zeichen für RNA-Verunreinigungen. Die
Verwendung von LiCl hingegen führte zur Entfernung der RNA. Hier konnten in den
Absorptionsspektren von GST-rpL5 nach der GSH-Sepharose-Reinigung (Daten nicht
gezeigt) und rpL5 nach der SP-Sepharose-Reinigung (Abbildung 14 B) keine RNA-
Verunreinigung mehr beobachtet werden. Die Absorptionsmaxima lagen nahe der für
Proteine typischen Wellenlänge von 280 nm.
Abbildung 14: Absorptionsspektren verschiedener Reinigungen von rpL5. (A) Absorptionsspektrum einer frühen Reinigung von GST-rpL5 über GSH-Sepharose. (B) UV-Absorptionsspektrum von rpL5 nach der finalen Präparation.
3.1.2.5 Ausschlusschromatographische Analyse des gereinigten rpL5
Die Reinigungsschritte der Präparation von rpL5 ergaben keine Informationen über den
vorliegenden Oligomerisierungsgrad. Daher wurde ergänzend eine analytische
Gelfiltration mit einer Sephadex S200 10/300 GL - Säule durchgeführt (2.2.4.6). Hierfür
wurden 350 ng rpL5 (bestimmt nach Bearden, 2.2.6.1.2) mit einer 500 µl-Schleife auf die
Säule geladen. Für die analytische Gelfiltration wurde ein 500 mM NaCl enthaltender
Elutionspuffer verwendet (2.2.4.6). rpL5 neigt bei Raumtemperatur zur Präzipition. Dieser
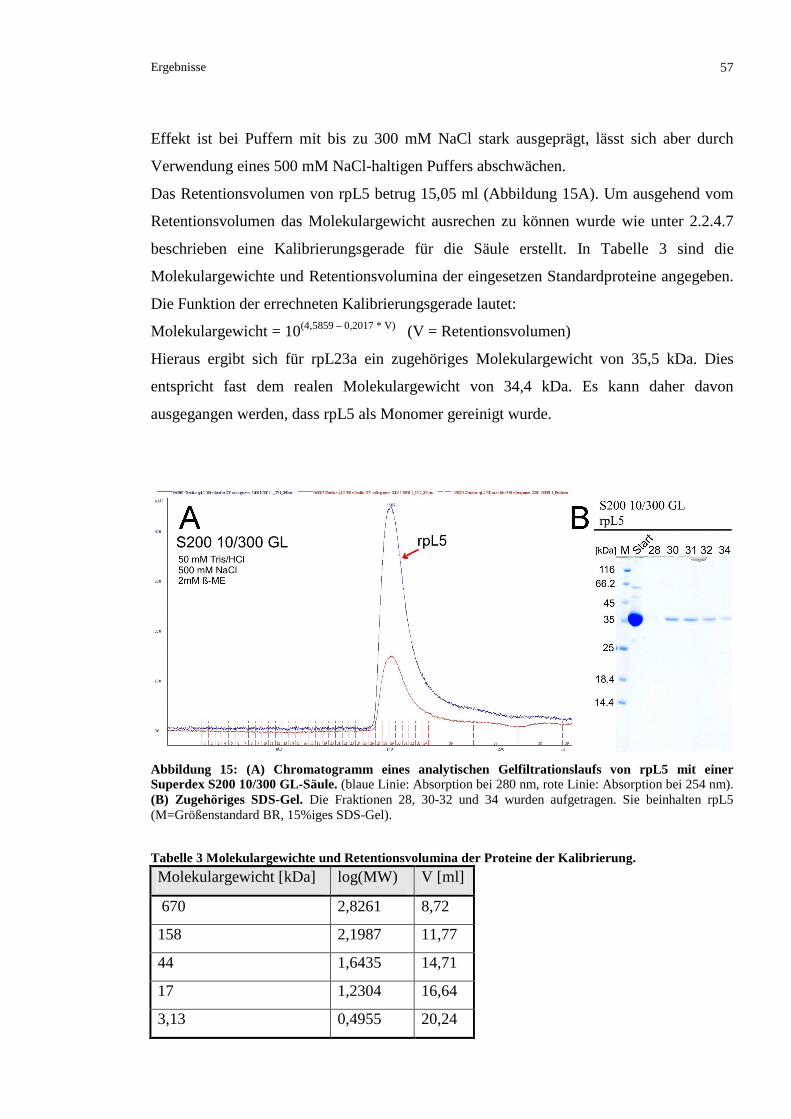
Ergebnisse
57
Effekt ist bei Puffern mit bis zu 300 mM NaCl stark ausgeprägt, lässt sich aber durch
Verwendung eines 500 mM NaCl-haltigen Puffers abschwächen.
Das Retentionsvolumen von rpL5 betrug 15,05 ml (Abbildung 15A). Um ausgehend vom
Retentionsvolumen das Molekulargewicht ausrechen zu können wurde wie unter 2.2.4.7
beschrieben eine Kalibrierungsgerade für die Säule erstellt. In Tabelle 3 sind die
Molekulargewichte und Retentionsvolumina der eingesetzen Standardproteine angegeben.
Die Funktion der errechneten Kalibrierungsgerade lautet:
Molekulargewicht = 10(4,5859 – 0,2017 * V) (V = Retentionsvolumen)
Hieraus ergibt sich für rpL23a ein zugehöriges Molekulargewicht von 35,5 kDa. Dies
entspricht fast dem realen Molekulargewicht von 34,4 kDa. Es kann daher davon
ausgegangen werden, dass rpL5 als Monomer gereinigt wurde.
Abbildung 15: (A) Chromatogramm eines analytischen Gelfiltrationslaufs von rpL5 mit einer Superdex S200 10/300 GL-Säule. (blaue Linie: Absorption bei 280 nm, rote Linie: Absorption bei 254 nm). (B) Zugehöriges SDS-Gel. Die Fraktionen 28, 30-32 und 34 wurden aufgetragen. Sie beinhalten rpL5 (M=Größenstandard BR, 15%iges SDS-Gel).
Tabelle 3 Molekulargewichte und Retentionsvolumina der Proteine der Kalibrierung.
Molekulargewicht [kDa] log(MW) V [ml]
670 2,8261 8,72
158 2,1987 11,77
44 1,6435 14,71
17 1,2304 16,64
3,13 0,4955 20,24

Ergebnisse
58
3.1.3 Kristallisationsversuche mit dem Komplex Imp7/rpL5
Ein Hauptziel dieser Diplomarbeit war die Untersuchung der Interaktionen der
ribosomalen Proteine rpL5 und rpL23a mit dem Kernimportrezeptor Importin 7 aus
Xenopus laevis. Zu diesem Zweck wurde ein Komplex der rekombinant gewonnenen
Proteine rpL5 und Importin 7 präpariert. Der gereinigte Imp7/rpL5-Komplex sollte zur
Kristallisation gebracht werden, um eine Aufklärung der dreidimensionalen Struktur durch
Röntgenbeugungsexperimente zu ermöglichen.
3.1.3.1 Präparation eines Komplexes aus rpL5 und Importin 7
Der Kernimportfaktor Imp7 aus Xenopus laevis wurde nach einem Protokoll von Daniel
Wohlwend (Universität Göttingen) gereinigt. Für die Präparation des Imp7/rpL5-
Komplexes wurden 57.6 nmol rpL5 (2,0 mg - bestimmt nach Bearden, 2.2.6.1.2) und
40 nmol Imp7 (4,8 mg - bestimmt nach Bearden, 2.2.6.1.2) für 60 Minuten auf Eis
inkubiert. Der gebildete Komplex wurde mit Hilfe einer präparativen Gelfiltration mittels
einer Superdex S200 XK 26/60-Säule bei 4° C isoliert (2.2.4.5). Hierfür wurde die Säule
zunächst mit einem Säulenvolumen „Komplexpuffer Imp7/rpL5“ (2.2.4.5) equilibriert.
Anschließend wurde der Ansatz über eine 5 ml Auftragungsschleife auf die Säule geladen
und der Säulenlauf mit „Komplexpuffer Imp7/rpL5“ durchgeführt. Ein Absorptions-
maximum nach 164 ml Elutionsvolumen enthielt den isoliert vorliegenden Imp7/rpL5-
Komplex (Abbildung 16). Die Fraktionen 14 bis 20 wurden vereinigt und auf 9,4 mg/ml,
bestimmt nach Bearden (2.2.6.1.2), konzentriert (2.2.3.1). Aus der SDS-PAGE einer Probe
der Präparation ging hervor, dass Imp7 einen stabilen Komplex mit rpL5 gebildet hatte
(Abbildung 16). Der präparierte Komplex wurde bei 4° C gelagert und für
Kristallisationsexperimente verwendet (3.1.3.2).
Ergebnisse
59
Abbildung 16: (A) Chromatogramm der Komplexpräparation von Imp7/rpL5 mit einer Superdex S200 XK 26/60 - Säule. (B) Zugehöriges SDS-Gel. Es wurden 5 �l (links) und 10 �l (rechts) Imp7/rpl5 in 1:1 SDS-Probenpuffer (2.2.3.3) aufgetragen (M=Größenstandard BR, 15%iges SDS-Gel).
3.1.3.2 Versuch einer Kristallisation des Imp7/rpL5-Komplexes
Ein Ziel der vorliegenden Diplomarbeit sollte die Kristallisation des Komplexes aus
Importin 7 und rpL5 sein. Alle hierfür mit dem Komplex pipettierten
Kristallisationsansätze sind in Tabelle 4 aufgeführt. Zu Beginn wurden die
Kristallisationsscreens Footprint I und II (2.1.9) bei 20°C mit einer Konzentration von 9,4
mg/ml pipettiert, um das Präzipitationsverhalten zu analysieren (2.2.7.1). Nachdem der
Imp7/rpL5-Komplex in etwa 90% aller Bedingungen von Footprint I und II präzipitierte,
wurde die Proteinkonzentration auf 4,8 mg/ml verdünnt. Ein Großteil der folgenden
Kristallisationsscreens wurde mit einem Kristallisationsroboter der EMBL-Outstation in
Hamburg pipettiert. Die Auswertung dieser Kristallisationsansätze erfolgte über das
Internet (2.2.7.1)
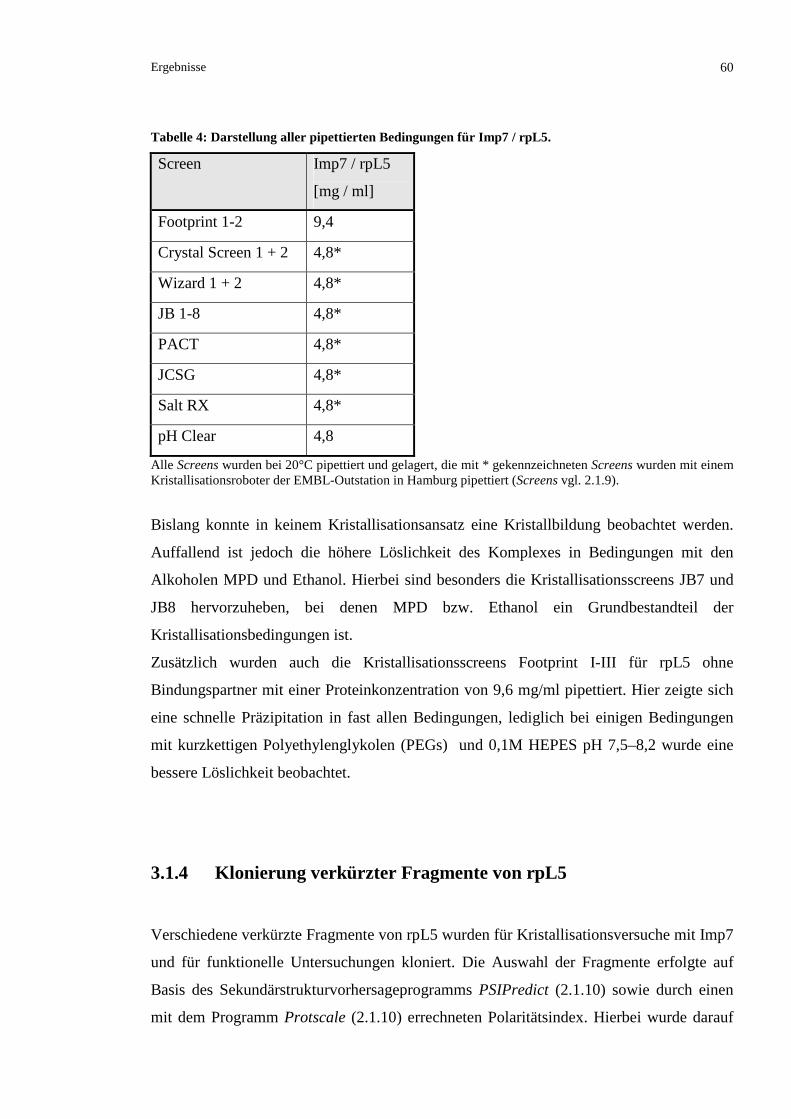
Ergebnisse
60
Tabelle 4: Darstellung aller pipettierten Bedingungen für Imp7 / rpL5.
Screen Imp7 / rpL5
[mg / ml]
Footprint 1-2 9,4
Crystal Screen 1 + 2 4,8*
Wizard 1 + 2 4,8*
JB 1-8 4,8*
PACT 4,8*
JCSG 4,8*
Salt RX 4,8*
pH Clear 4,8
Alle Screens wurden bei 20°C pipettiert und gelagert, die mit * gekennzeichneten Screens wurden mit einem Kristallisationsroboter der EMBL-Outstation in Hamburg pipettiert (Screens vgl. 2.1.9).
Bislang konnte in keinem Kristallisationsansatz eine Kristallbildung beobachtet werden.
Auffallend ist jedoch die höhere Löslichkeit des Komplexes in Bedingungen mit den
Alkoholen MPD und Ethanol. Hierbei sind besonders die Kristallisationsscreens JB7 und
JB8 hervorzuheben, bei denen MPD bzw. Ethanol ein Grundbestandteil der
Kristallisationsbedingungen ist.
Zusätzlich wurden auch die Kristallisationsscreens Footprint I-III für rpL5 ohne
Bindungspartner mit einer Proteinkonzentration von 9,6 mg/ml pipettiert. Hier zeigte sich
eine schnelle Präzipitation in fast allen Bedingungen, lediglich bei einigen Bedingungen
mit kurzkettigen Polyethylenglykolen (PEGs) und 0,1M HEPES pH 7,5–8,2 wurde eine
bessere Löslichkeit beobachtet.
3.1.4 Klonierung verkürzter Fragmente von rpL5
Verschiedene verkürzte Fragmente von rpL5 wurden für Kristallisationsversuche mit Imp7
und für funktionelle Untersuchungen kloniert. Die Auswahl der Fragmente erfolgte auf
Basis des Sekundärstrukturvorhersageprogramms PSIPredict (2.1.10) sowie durch einen
mit dem Programm Protscale (2.1.10) errechneten Polaritätsindex. Hierbei wurde darauf
Ergebnisse
61
geachtet, dass die Enden der Fragmente nicht innerhalb von sekundärstrukturtragenden
Sequenzabschnitten lokalisiert sind. Dadurch sollte eine Strukturstabilisierung der
Fragmente erreicht werden. Des Weiteren wurde darauf geachtet, dass die neue terminale
Aminosäure des Fragments eine ionische Seitenkette besitzt. Dies sollte die Löslichkeit der
Fragmente verbessern.
Für die Klonierung der rpL5-Fragmente wurde das in den Vektor pGEX-6P-1 klonierte
Volllänge-Gen als Matrize verwendet (3.2.1.1). Die Amplifikations-PCR (2.2.1.1.1)
erfolgte unter Verwendung der in Tabelle 5 angegebenen Oligonukleotide (2.1.8). Die
amplifizierten Genfragmente wurden über die Endonukleasen BamHI und XhoI in pGEX-
6P-1 inseriert (2.2.1.1.2) und ligiert (2.2.1.1.4). Zur Kontrolle der Klonierung wurde eine
Sequenzanalyse (2.2.1.1.5) unter Verwendung der DNA-Oligonukleotide pGEX_f und
pGEX_r (2.1.8) durchgeführt. Die Konstrukte pGEX-6P-1-rpL5/nostop* (AS 1-48),
pGEX-6P-1-rpL5/nostop* (AS 1-189) und pGEX-6P-1-rpL5/nostop* (AS 1-234) werden
bis zum Stopcodon der multiple cloning side translatiert, so dass nach den rpL5-
Fragmenten die Aminosäuresequenz LERPHRN folgt.
Tabelle 5 Übersicht über die Vektorkonstrukte der verkürzten Fragmente von rpL5.
Vektor-Konstrukte (2.1.5) Verwendete Oligonukleotide (2.1.8)
pGEX-6P-1-rpL5/nostop* (AS 1-48) rpL5_BamHIfor, rpL5_N48rev
pGEX-6P-1-rpL5/nostop* (AS 1-189) rpL5_BamHIfor, rpL5_N189rev
pGEX-6P-1-rpL5/nostop* (AS 1-234) rpL5_BamHIfor, rpL5_N234rev
pGEX-6P-1-rpL5 (AS 48-296) rpL5_C48for, rpL5_XhoIrev
pGEX-6P-1-rpL5 (AS 189-296) rpL5_C189for, rpL5_XhoIrev
pGEX-6P-1-rpL5 (AS 234-296) rpL5_C234for, rpL5_XhoIrev

Ergebnisse
62
3.2 Biochemische Arbeiten mit dem Protein rpL23a aus Homo sapiens
3.2.1 Expression, Zellernte und Aufschluss von rpL23a
3.2.1.1 Subklonierungen von rpL23a in die Vektoren pGEX-6P-1 und pQE80
Zu Beginn der Diplomarbeit wurde das Gen von rpL23a aus Homo sapiens (accession
number BC014459; NCBI), in dem Expressionsvektor pQE70, von D. Doenecke
(Universität Göttingen) zur Verfügung gestellt (2.1.5). rpL23a wird ausgehend von pQE70
als Fusionsprotein mit einer N-terminalen 4z-Affinitätssequenz exprimiert. Mit „z“ wird
hierbei die IgG-Bindedomäne des Proteins A von Staphylococcus aureus bezeichnet, mit
deren Hilfe eine affinitätschromatografische Reinigung des Fusionsproteins über eine IgG-
Sepharose-4B-Säule möglich ist (Jäkel und Görlich, 1998). Da eine solche Affinitätssäule
nicht verfügbar war, musste eine Subklonierung, des für rpL23a codierenden Gens, in
geeignete Expressionsvektoren durchgeführt werden.
Es wurden zwei verschiedene Strategien zur Reinigung von rpL23a geplant:
Zum einen sollte das Protein mit einer N-terminalen Deca-Histidin-Affinitätssequenz,
ausgehend von dem Vektor pQE80 (2.1.5) exprimiert werden, um mittels einer Ni+-NTA-
Sepharose-Säule gereinigt werden zu können. Des Weiteren sollte, wie für rpL5 auch, eine
Affinitätsreinigung mittels GSH-Sepharose etabliert werden. Hierfür wurde eine
Subklonierung in den Vektor pGEX-6P-1 (2.1.5) durchgeführt.
Für die Subklonierung von rpL23a aus pQE070 in den Vektor pQE80 (2.1.5) wurde eine
Subklonierungs-PCR (2.2.1.1.1) mit den DNA-Oligonukleotiden rpL23a_BamHIfor und
rpL23a_HindIIIrev (2.1.8) durchgeführt. Das amplifizierte Gen wurde über BamHI- und
HindIII-Schnittstellen in pQE80 inseriert. Das Vektorkonstrukt pQE80-rpL23a wurde in
E. coli XL1-Blue transformiert (2.2.2.1) und in einer anschließenden Mini-Präparation
(2.2.2.2) amplifiziert. Zur Kontrolle der Subklonierung wurde eine Sequenzanalyse
(2.2.1.1.5) unter Verwendung der DNA-Oligonukleotide pQE80_for und pQE80_rev
(2.1.8) durchgeführt. Analog erfolgte die Subklonierung von rpL23a in den Vektor pGEX-
6P-1 (2.1.5). Hierfür wurden die Oligonukleotide rpL5_BamHIfor und rpL5_BamXhoIrev
eingesetzt ((2.1.8) und das amplifizierte Gen über die BamHI- und XHhoI-Schnittstellen in

Ergebnisse
63
pGEX-6P-1 inseriert. Die Sequenzanalyse (2.2.1.1.5) erfolgte unter Verwendung der
DNA-Oligonukleotide pGEX_f und pGEX_r (2.1.8).
3.2.1.2 Expression von rpL23a
Nach der Klonierung wurden zunächst Testexpressionen verschiedener E. coli-Stämme für
die beiden verwendeten Vektorkonstrukte durchgeführt. Mehrere E. coli-Stämme wurden
mit den Plasmiden pQE80-rpL23a bzw. pGEX-6P-1-rpL23a transformiert (2.2.2.1). Die
Expression erfolgte nach dem unter 2.2.2.3.1 beschriebenen Protokoll.
Für die Expression von N(His)10-rpL23a wurden die E.coli-Stämme M15 und SG3009
(pREP4) getestet. Das Expressionssignal war in beiden Stämmen etwa gleich stark (Daten
nicht gezeigt). M15 wurde für die Überexpression von N(His)10-rpL23a weiterverwendet.
Für die Expression von GST-rpL23a wurden die E.Coli-Stämme BL21 (DE3), BL21
(DE3) Star, BL21 (DE3) RP, BL21 (DE3) Rosetta 2 und Origami getestet (Daten nicht
gezeigt). Die Expression war in BL21 (DE3) RP und BL21 (DE3) Rosetta 2 am stärksten,
weshalb diese Stämme für die Überexpression von GST-rpL23a ausgewählt wurden.
Die Überexpression von N(His)10-rpL23a und GST-rpL23a im präparativen Maßstab
erfolgte wie unter 2.2.2.3.2 beschrieben. In Abbildung 17 sind die SDS-Gele für die
Proben der Expression von N(His)10-rpL23a und GST-rpL23a abgebildet. Es wurde
jeweils eine Probe zu Expressionsbeginn sowie zum Zeitpunkt der Zellernte aufgetragen.
Abbildung 17: (A) SDS-PAGE der Expression von N(His)10-rpL23a in pQE80 in E. coli M15. (B) SDS-PAGE der Expression von GST-rpL23a in pGEX-6P-1 in E.coli Rosetta 2 (DE3). (roter Pfeil = GST-rpL23a bzw. N(His)10 N(His)10-rpL23a, vI = vor Induktion, nI = nach Induktion, M = Größenstandard BR, 15%iges SDS-Gel)

Ergebnisse
64
3.2.1.3 Zellernte und Aufschluss der Expressionskulturen
Die Zellernte der Expressionskulturen für N(His)10-rpL23a und GST-rpL23a erfolgte
analog zur Zellernte der GST-rpL5-Expressionskulturen (vgl. 3.1.1.3) nach dem unter
2.2.2.3.3 beschriebenen Protokoll. Die Analyse des Aufschlusses von N(His)10-rpL23a
durch SDS-PAGE ist in Abbildung 18 (3.2.2.1) abgebildet. N(His)10-rpL23a befindet sich
nach der Zentrifugation bei 30000 xg für 40 min bei 4° C fast vollständig im Überstand.
Das SDS-Gel des Aufschlusses von GST-rpL23a ist in Abbildung 20 (3.2.2.2) dargestellt.
Auch GST-rpL23a befindet sich nach der Zentrifugation fast vollständig im Überstand.
3.2.2 Reinigung von rpL23a aus Homo sapiens
3.2.2.1 Versuch einer Reinigung von rpL23a mit Deca-Histidin-Sequenz
Zu Beginn der Diplomarbeit wurde versucht, N(His)10-rpL23a durch Affinitätschromato-
graphie mit einer HisTrapChelating Ni-NTA-Sepharose-Säule nach dem unter 2.2.4.2.
beschriebenen Protokoll zu reinigen. Das Protein konnte mit 450 – 600 mM Imidazol von
der Säule eluiert werden. Problematisch war jedoch eine starke Verunreinigung mit RNA,
die dazu führte, dass N(His)10-rpL23a im Laufe von 24 Stunden teilweise präzipitierte und
bei nachfolgenden Gelfiltrationen (Daten nicht gezeigt) ausschließlich im Ausschluss-
volumen eluiert wurde. Abbildung 19 zeigt ein UV-Spektrum der vereinigten Fraktionen
von N(His)10-rpL23a nach der Nickel-Säule. Auffällig ist die hohe Absorption bei 254 nm,
die charakteristisch für RNA ist.

Ergebnisse
65
Abbildung 18: (A) Chromatogramm der Elution von rpL 23a(His)10 von einer HisTrapChelating Ni-NTA-Sepharose-Säule. Die roten Balken markieren die vereinigten Fraktionen 15-25 (blau: UV-Absorption 280 nm, rot: Leitfähigkeit, grün: Anteil Elutionspuffer). (B) Zugehöriges SDS-Gel. (F = Probe nach Aufschluss, Pellet, Ü = Überstand, D = Durchfluss, W = Waschen mit Lysispuffer, 1-40 = Elutions-fraktionen, Gua = Abschließende Reinigung der Säule mit 6M Guanidiniumhydrochlorid)
(His)10-rpL23a nach HisTrap-Säule
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1,8
245 250 255 260 265 270 275 280 285 290 295 300
[nm]
Abs
orpt
ion
Abbildung 19: UV-Spektrum von (His)10-rpL23a nach der Reinigung mit einer HisTrapChelating Ni-NTA-Sepharose-Säule
3.2.2.2 Affinitätschromatographische Reinigung über Glutathion-Sepharose
Die Reinigung des Fusionsproteins GST-rpL23a exprimiert aus dem Vektor pGEX-6P-1
erfolgte weitgehend analog zur Reinigung von GST-rpL5 (3.1.2).

Ergebnisse
66
In Abbildung 20 ist neben den Chromatogrammen auch ein SDS-Gel mit Proben von
Aufschluss, Beladung, Durchfluss, Waschschritt und Elution abgebildet. Bei dem nach der
Beladung durchgeführten Waschschritt mit Lysispuffer wurde eine kleine Menge des
gebundenen GST-rpL23a von der Säule gelöst. Die Elution von GST-rpL23a erfolgte
durch einen Puffer mit 20 mM reduziertem Glutathion (vgl. 2.2.4.1). GST-rpL23a lag in
den vereinigten Elutionsfraktionen noch schwach verunreinigt vor (mehrere Banden
unterhalb von 40 kDa). Die Ausbeute der vereinigten Fraktionen der GSH-Sepharose-
Reinigung betrug durchschnittlich 12 mg/L, bestimmt nach der Methode von Bearden
(2.2.6.1.2) (Übersicht der Ausbeuten in Tabelle 7 unter 3.2.2.4).
Die Verwendung von LiCl in Lysis- und Waschpuffer (2.2.2.3.3 und 2.2.4.1) führte zu
einer Verbesserung der Löslichkeit im Vergleich zu frühen Reinigungsversuchen ohne
LiCl (Daten nicht gezeigt). Eine Messung eines Absorptionsspektrums für GST-rpL23a
nach der GSH-Sepharose-Reinigung deutete jedoch darauf hin, dass immer noch RNA-
Verunreinigungen vorhanden waren (siehe Abbildung 23 unter 3.2.2.3).
Zur Entfernung der GST-Affinitätssequenz wurde GST-rpL23a, analog zu GST-rpL5,
(vgl.2.2.3.5 und 3.1.2.2) mit der Protease PreScission in einem Masseverhältnis von 1:200
für mindestens 12 Stunden über Nacht bei 4° C inkubiert. Eine im Anschluss durch SDS-
PAGE analysierte Probe zeigte die vollständige Spaltung des Fusionsproteins (siehe
Abbildung 22 unter 3.2.2.3).
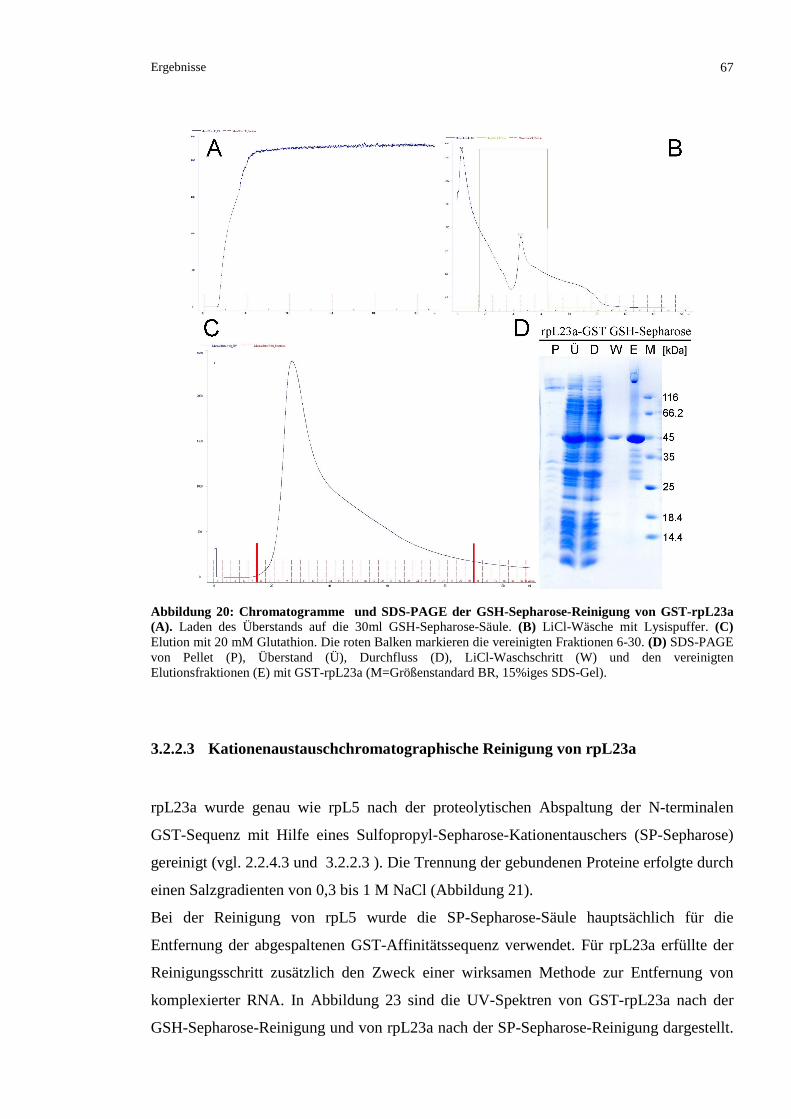
Ergebnisse
67
Abbildung 20: Chromatogramme und SDS-PAGE der GSH-Sepharose-Reinigung von GST-rpL23a (A). Laden des Überstands auf die 30ml GSH-Sepharose-Säule. (B) LiCl-Wäsche mit Lysispuffer. (C) Elution mit 20 mM Glutathion. Die roten Balken markieren die vereinigten Fraktionen 6-30. (D) SDS-PAGE von Pellet (P), Überstand (Ü), Durchfluss (D), LiCl-Waschschritt (W) und den vereinigten Elutionsfraktionen (E) mit GST-rpL23a (M=Größenstandard BR, 15%iges SDS-Gel).
3.2.2.3 Kationenaustauschchromatographische Reinigung von rpL23a
rpL23a wurde genau wie rpL5 nach der proteolytischen Abspaltung der N-terminalen
GST-Sequenz mit Hilfe eines Sulfopropyl-Sepharose-Kationentauschers (SP-Sepharose)
gereinigt (vgl. 2.2.4.3 und 3.2.2.3 ). Die Trennung der gebundenen Proteine erfolgte durch
einen Salzgradienten von 0,3 bis 1 M NaCl (Abbildung 21).
Bei der Reinigung von rpL5 wurde die SP-Sepharose-Säule hauptsächlich für die
Entfernung der abgespaltenen GST-Affinitätssequenz verwendet. Für rpL23a erfüllte der
Reinigungsschritt zusätzlich den Zweck einer wirksamen Methode zur Entfernung von
komplexierter RNA. In Abbildung 23 sind die UV-Spektren von GST-rpL23a nach der
GSH-Sepharose-Reinigung und von rpL23a nach der SP-Sepharose-Reinigung dargestellt.
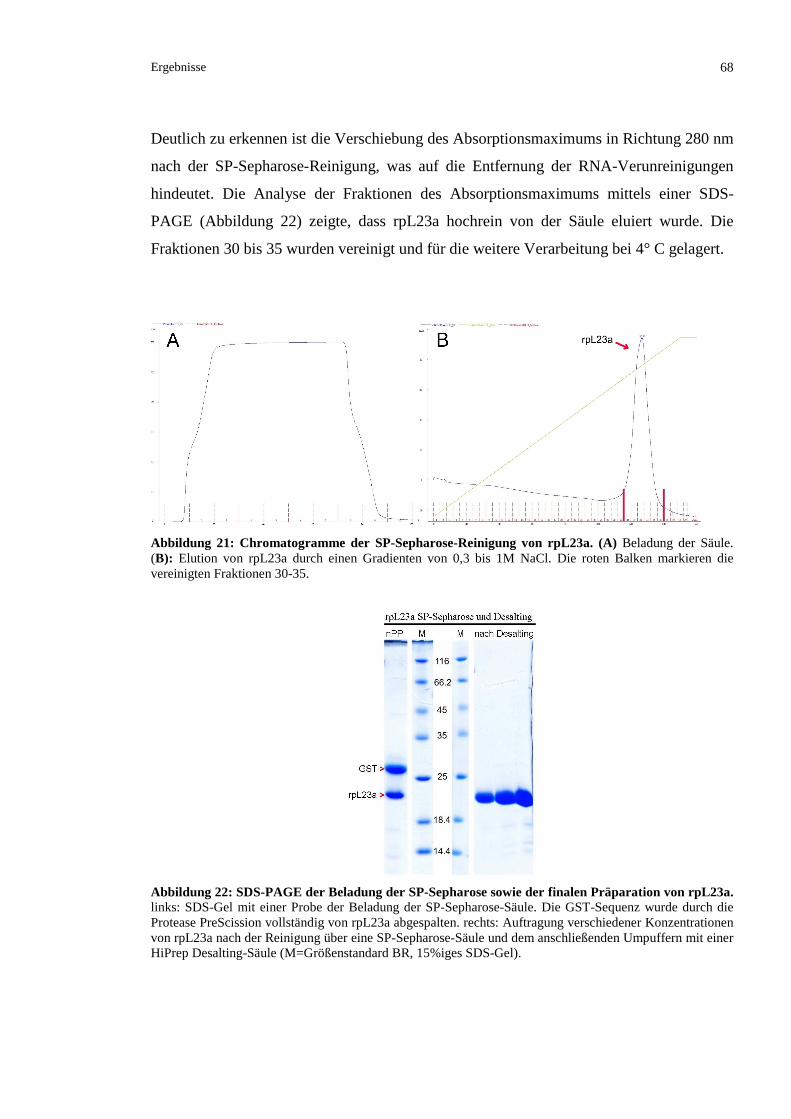
Ergebnisse
68
Deutlich zu erkennen ist die Verschiebung des Absorptionsmaximums in Richtung 280 nm
nach der SP-Sepharose-Reinigung, was auf die Entfernung der RNA-Verunreinigungen
hindeutet. Die Analyse der Fraktionen des Absorptionsmaximums mittels einer SDS-
PAGE (Abbildung 22) zeigte, dass rpL23a hochrein von der Säule eluiert wurde. Die
Fraktionen 30 bis 35 wurden vereinigt und für die weitere Verarbeitung bei 4° C gelagert.
Abbildung 21: Chromatogramme der SP-Sepharose-Reinigung von rpL23a. (A) Beladung der Säule. (B): Elution von rpL23a durch einen Gradienten von 0,3 bis 1M NaCl. Die roten Balken markieren die vereinigten Fraktionen 30-35.
Abbildung 22: SDS-PAGE der Beladung der SP-Sepharose sowie der finalen Präparation von rpL23a. links: SDS-Gel mit einer Probe der Beladung der SP-Sepharose-Säule. Die GST-Sequenz wurde durch die Protease PreScission vollständig von rpL23a abgespalten. rechts: Auftragung verschiedener Konzentrationen von rpL23a nach der Reinigung über eine SP-Sepharose-Säule und dem anschließenden Umpuffern mit einer HiPrep Desalting-Säule (M=Größenstandard BR, 15%iges SDS-Gel).
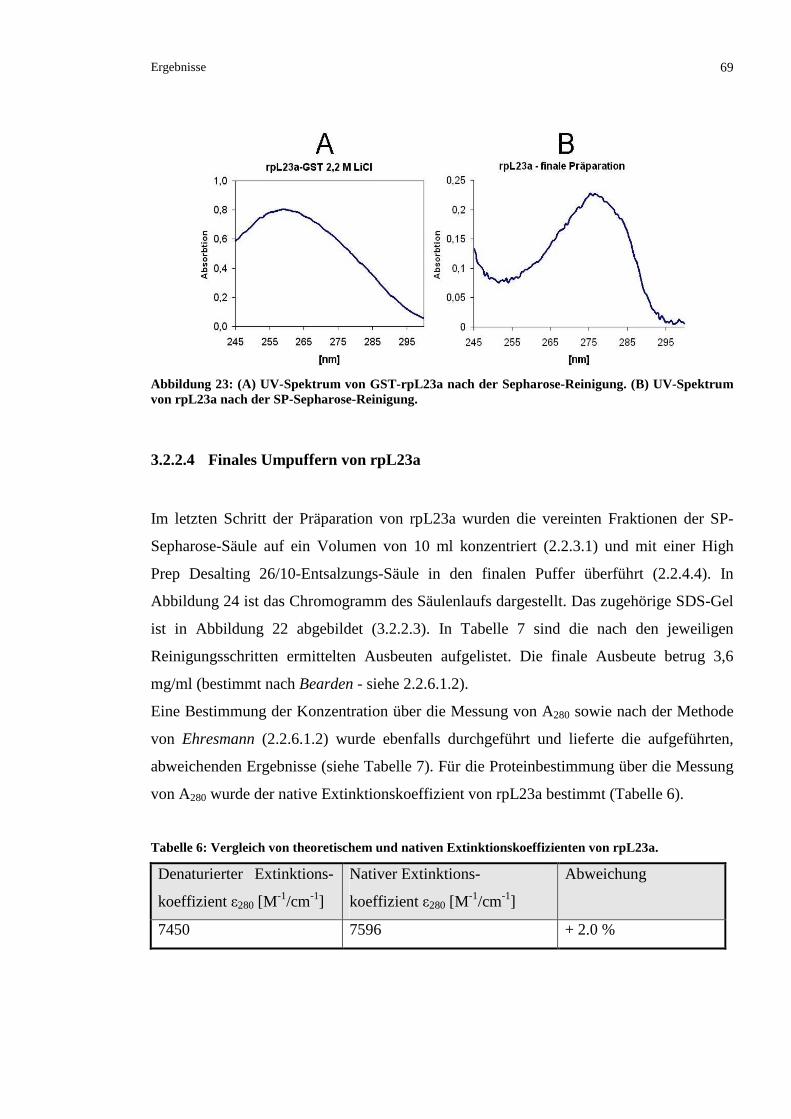
Ergebnisse
69
Abbildung 23: (A) UV-Spektrum von GST-rpL23a nach der Sepharose-Reinigung. (B) UV-Spektrum von rpL23a nach der SP-Sepharose-Reinigung.
3.2.2.4 Finales Umpuffern von rpL23a
Im letzten Schritt der Präparation von rpL23a wurden die vereinten Fraktionen der SP-
Sepharose-Säule auf ein Volumen von 10 ml konzentriert (2.2.3.1) und mit einer High
Prep Desalting 26/10-Entsalzungs-Säule in den finalen Puffer überführt (2.2.4.4). In
Abbildung 24 ist das Chromogramm des Säulenlaufs dargestellt. Das zugehörige SDS-Gel
ist in Abbildung 22 abgebildet (3.2.2.3). In Tabelle 7 sind die nach den jeweiligen
Reinigungsschritten ermittelten Ausbeuten aufgelistet. Die finale Ausbeute betrug 3,6
mg/ml (bestimmt nach Bearden - siehe 2.2.6.1.2).
Eine Bestimmung der Konzentration über die Messung von A280 sowie nach der Methode
von Ehresmann (2.2.6.1.2) wurde ebenfalls durchgeführt und lieferte die aufgeführten,
abweichenden Ergebnisse (siehe Tabelle 7). Für die Proteinbestimmung über die Messung
von A280 wurde der native Extinktionskoeffizient von rpL23a bestimmt (Tabelle 6).
Tabelle 6: Vergleich von theoretischem und nativen Extinktionskoeffizienten von rpL23a.
Denaturierter Extinktions-
koeffizient ε280 [M-1/cm-1]
Nativer Extinktions-
koeffizient ε280 [M-1/cm-1]
Abweichung
7450 7596 + 2.0 %
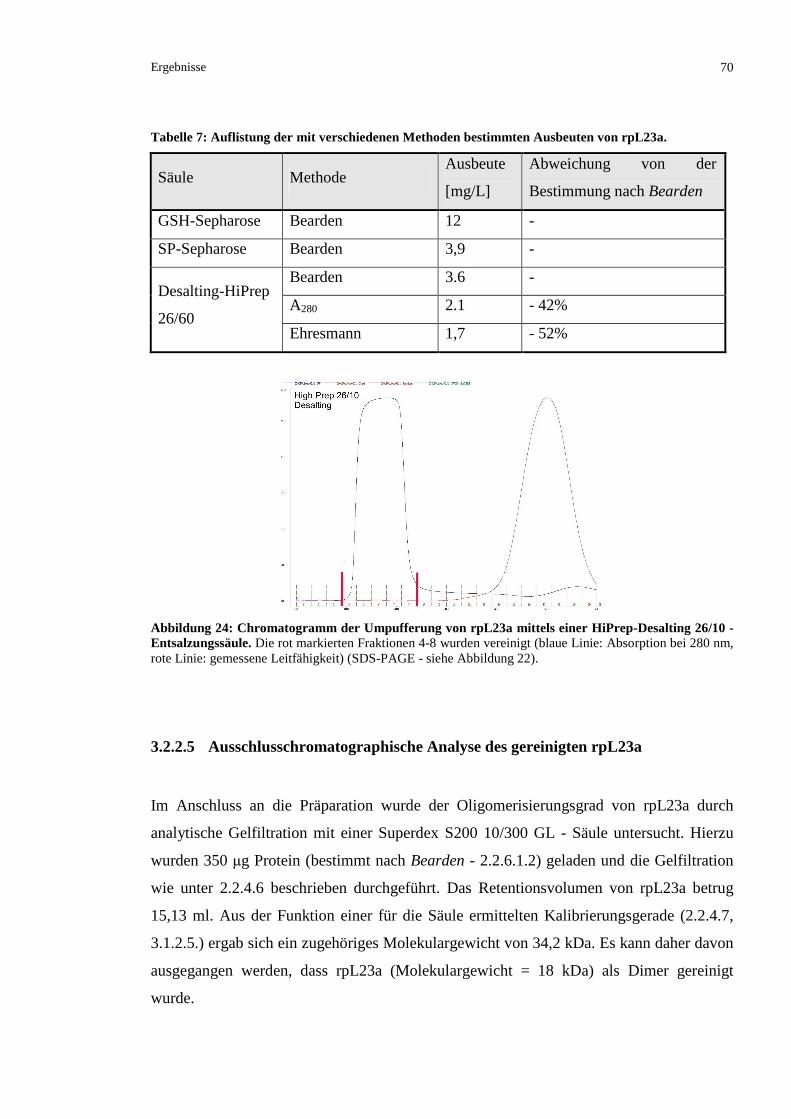
Ergebnisse
70
Tabelle 7: Auflistung der mit verschiedenen Methoden bestimmten Ausbeuten von rpL23a.
Säule Methode Ausbeute
[mg/L]
Abweichung von der
Bestimmung nach Bearden
GSH-Sepharose Bearden 12 -
SP-Sepharose Bearden 3,9 -
Bearden 3.6 -
A280 2.1 - 42% Desalting-HiPrep
26/60 Ehresmann 1,7 - 52%
Abbildung 24: Chromatogramm der Umpufferung von rpL23a mittels einer HiPrep-Desalting 26/10 - Entsalzungssäule. Die rot markierten Fraktionen 4-8 wurden vereinigt (blaue Linie: Absorption bei 280 nm, rote Linie: gemessene Leitfähigkeit) (SDS-PAGE - siehe Abbildung 22).
3.2.2.5 Ausschlusschromatographische Analyse des gereinigten rpL23a
Im Anschluss an die Präparation wurde der Oligomerisierungsgrad von rpL23a durch
analytische Gelfiltration mit einer Superdex S200 10/300 GL - Säule untersucht. Hierzu
wurden 350 µg Protein (bestimmt nach Bearden - 2.2.6.1.2) geladen und die Gelfiltration
wie unter 2.2.4.6 beschrieben durchgeführt. Das Retentionsvolumen von rpL23a betrug
15,13 ml. Aus der Funktion einer für die Säule ermittelten Kalibrierungsgerade (2.2.4.7,
3.1.2.5.) ergab sich ein zugehöriges Molekulargewicht von 34,2 kDa. Es kann daher davon
ausgegangen werden, dass rpL23a (Molekulargewicht = 18 kDa) als Dimer gereinigt
wurde.
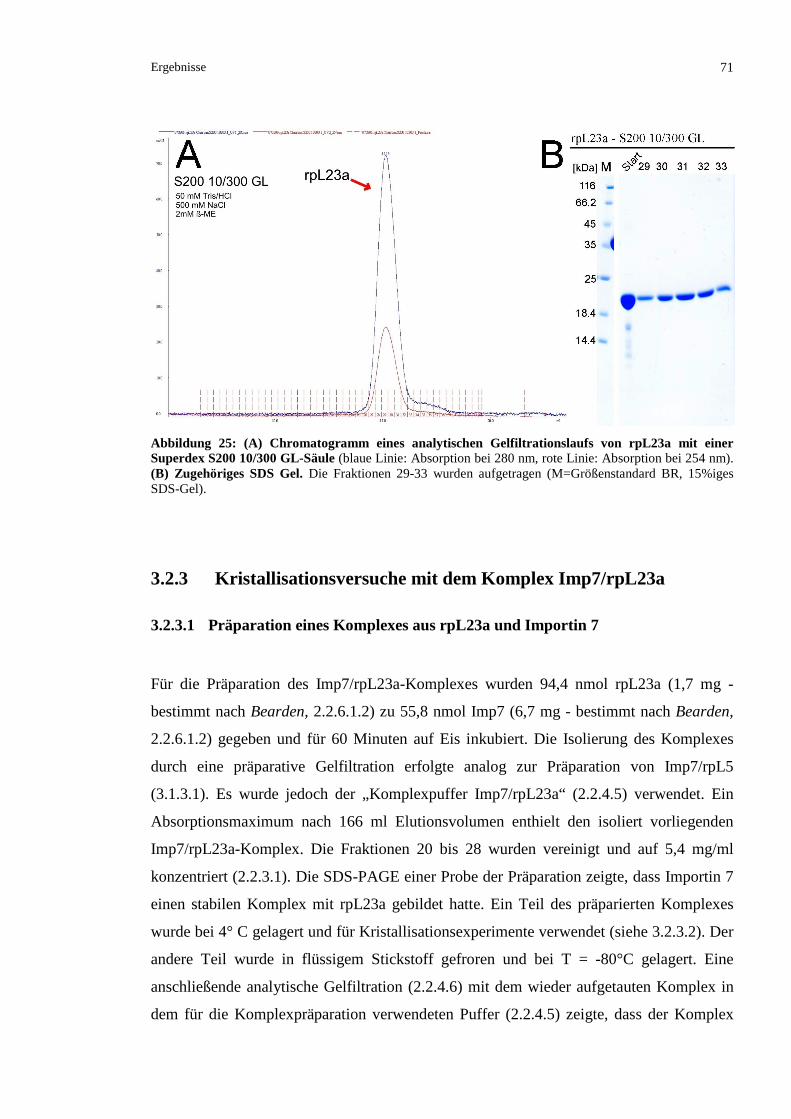
Ergebnisse
71
Abbildung 25: (A) Chromatogramm eines analytischen Gelfiltrationslaufs von rpL23a mit einer Superdex S200 10/300 GL-Säule (blaue Linie: Absorption bei 280 nm, rote Linie: Absorption bei 254 nm). (B) Zugehöriges SDS Gel. Die Fraktionen 29-33 wurden aufgetragen (M=Größenstandard BR, 15%iges SDS-Gel).
3.2.3 Kristallisationsversuche mit dem Komplex Imp7/rpL23a
3.2.3.1 Präparation eines Komplexes aus rpL23a und Importin 7
Für die Präparation des Imp7/rpL23a-Komplexes wurden 94,4 nmol rpL23a (1,7 mg -
bestimmt nach Bearden, 2.2.6.1.2) zu 55,8 nmol Imp7 (6,7 mg - bestimmt nach Bearden,
2.2.6.1.2) gegeben und für 60 Minuten auf Eis inkubiert. Die Isolierung des Komplexes
durch eine präparative Gelfiltration erfolgte analog zur Präparation von Imp7/rpL5
(3.1.3.1). Es wurde jedoch der „Komplexpuffer Imp7/rpL23a“ (2.2.4.5) verwendet. Ein
Absorptionsmaximum nach 166 ml Elutionsvolumen enthielt den isoliert vorliegenden
Imp7/rpL23a-Komplex. Die Fraktionen 20 bis 28 wurden vereinigt und auf 5,4 mg/ml
konzentriert (2.2.3.1). Die SDS-PAGE einer Probe der Präparation zeigte, dass Importin 7
einen stabilen Komplex mit rpL23a gebildet hatte. Ein Teil des präparierten Komplexes
wurde bei 4° C gelagert und für Kristallisationsexperimente verwendet (siehe 3.2.3.2). Der
andere Teil wurde in flüssigem Stickstoff gefroren und bei T = -80°C gelagert. Eine
anschließende analytische Gelfiltration (2.2.4.6) mit dem wieder aufgetauten Komplex in
dem für die Komplexpräparation verwendeten Puffer (2.2.4.5) zeigte, dass der Komplex
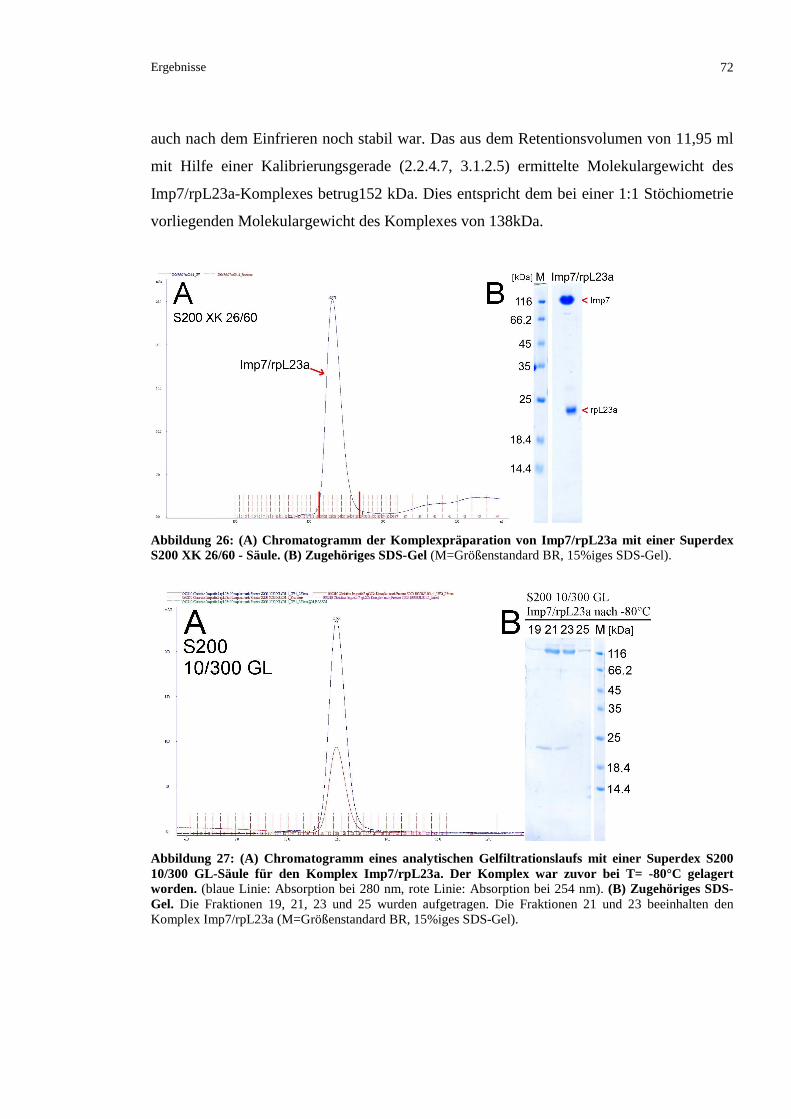
Ergebnisse
72
auch nach dem Einfrieren noch stabil war. Das aus dem Retentionsvolumen von 11,95 ml
mit Hilfe einer Kalibrierungsgerade (2.2.4.7, 3.1.2.5) ermittelte Molekulargewicht des
Imp7/rpL23a-Komplexes betrug152 kDa. Dies entspricht dem bei einer 1:1 Stöchiometrie
vorliegenden Molekulargewicht des Komplexes von 138kDa.
Abbildung 26: (A) Chromatogramm der Komplexpräparation von Imp7/rpL23a mit einer Superdex S200 XK 26/60 - Säule. (B) Zugehöriges SDS-Gel (M=Größenstandard BR, 15%iges SDS-Gel).
Abbildung 27: (A) Chromatogramm eines analytischen Gelfiltrationslaufs mit einer Superdex S200 10/300 GL-Säule für den Komplex Imp7/rpL23a. Der Komplex war zuvor bei T= -80°C gelagert worden. (blaue Linie: Absorption bei 280 nm, rote Linie: Absorption bei 254 nm). (B) Zugehöriges SDS-Gel. Die Fraktionen 19, 21, 23 und 25 wurden aufgetragen. Die Fraktionen 21 und 23 beeinhalten den Komplex Imp7/rpL23a (M=Größenstandard BR, 15%iges SDS-Gel).

Ergebnisse
73
3.2.3.2 Kristallisationsversuche mit Imp7/rpL23a
Das Präzipitationsverhalten des Imp7/rpL23a-Komplexes wurde zunächst durch Ansetzen
der Footprint-Screens mit 5,4 mg/ml (bestimmt nach Bearden, 2.2.6.1.2) überprüft.
Nachdem sich noch am selben Tag in der großen Mehrheit der Ansätze (> 90 %) Präzipitat
gebildet hatte, wurde die Proteinkonzentration für weitere Kirstallisationsansätze auf 2,5
mg/ml verdünnt.
Tabelle 8: Darstellung aller pipettierter Bedingungen
Kristallisationsscreen Proteinkonzentration
[mg/ml]
Footprint 1-3 5,4 / 3,8
Crystal Screen 1-2 3,0 / 2,5
Crystal Screen Lite 2,5
JB Screen 1-7, 9-10 2,5
Crystal Screen Cryo 2,5
Structure Screen 2,5
Magic Screen 1-4 2,5
PEG-Ion Screen 2,5
Nextal: The MPDs 2,5
Alle Ansätze wurden bei 20°C und mit
einem 1:1 Verhältnis von Präzipitans und
Protein pipettiert.
In Bedingung B1 des Kristallisationssreens JB 9 waren nach einem Tag Protein-
Sphärolithe gewachsen (Abbildung 28).
Abbildung 28: Protein-Sphärolithe in Bedingung B1 Screens JB 9. Zusammensetzung: 0,1 M Hepes, 0,2 M Tri-NaCitrat, 15% (w/v) Isopropanol, Kristallisationversuch bei 20°C im „sitzenden Tropfen“, Konzentration Imp7/rpL23a: c = 2,5 mg/ml, 1:1 Verhältnis von Präzipitans und Protein.

Ergebnisse
74
Eine Reproduktion dieser Sphärolithe konnte jedoch bisher nicht erreicht werden:
Folgende Parameter der Originalbedingung wurden hierfür variiert: pH-Wert,
Proteinkonzentration, Tropfengröße, Konzentration von Tri-NaCitrat und Isopropanol.
Ebenfalls durchgeführt wurde ein micro-seeding (2.2.7.2) mit Sphärolithen aus der
Originalbedingung sowie eine Präparation der Komplexe ohne Gelfiltration. Bei letzteren
wurden sowohl die molaren Verhältnisse der Komplexpartner als auch die
Proteinkonzentrationen variiert.
3.2.4 Funktionelle Untersuchungen der Bindung von rpL23a durch
Kernimportrezeptoren
3.2.4.1 Kartierung der Importin ß-Bindedomäne von rpL23a
Für die funktionelle Charakterisierung des Komplexes wurden zwei verschiedene
Methoden verwendet. Durch analytische Gelfiltrationsläufe (2.2.4.6) mit verkürzten
Fragmenten von Impß sollte die Bindungsstelle für rpL23a in Impß lokalisiert werden.
Ergänzend sollten die Bindungseigenschaften ausgewählter Komplexe durch isotherme
Titrationskalorimetrie (2.2.5) bestimmt werden.
3.2.4.1.1 Kartierung durch analytische Ausschlusschromatographie
Für die Kartierung der rpL23a-Bindungsstelle in Importin ß wurden verkürzte Fragmente
von Importin ß auf ihre Fähigkeit hin untersucht, stabile Komplexe mit rpL23a zu bilden.
Hierfür wurden die verschiedenen Impß-Fragmente jeweils mit rpL23a inkubiert und die
Entstehung von Komplexen durch analytische Gelfiltrationsexperimente (2.2.4.6) mit einer
Superdex S200 10/300 GL-Säule überprüft. In
Tabelle 9 ist eine Übersicht der untersuchten Impß-Fragmente aufgeführt. Die
Beschriftung bezieht sich dabei auf die nicht verkürzte Seite der Fragmente (N = Amino-
Terminus, C = Carboxy-Terminus). Beispielsweise umfasst das Fragment ImpßN641 die
Aminosäuren 1-641 des Volllängenproteins, vom N-Terminus aus betrachtet. Das
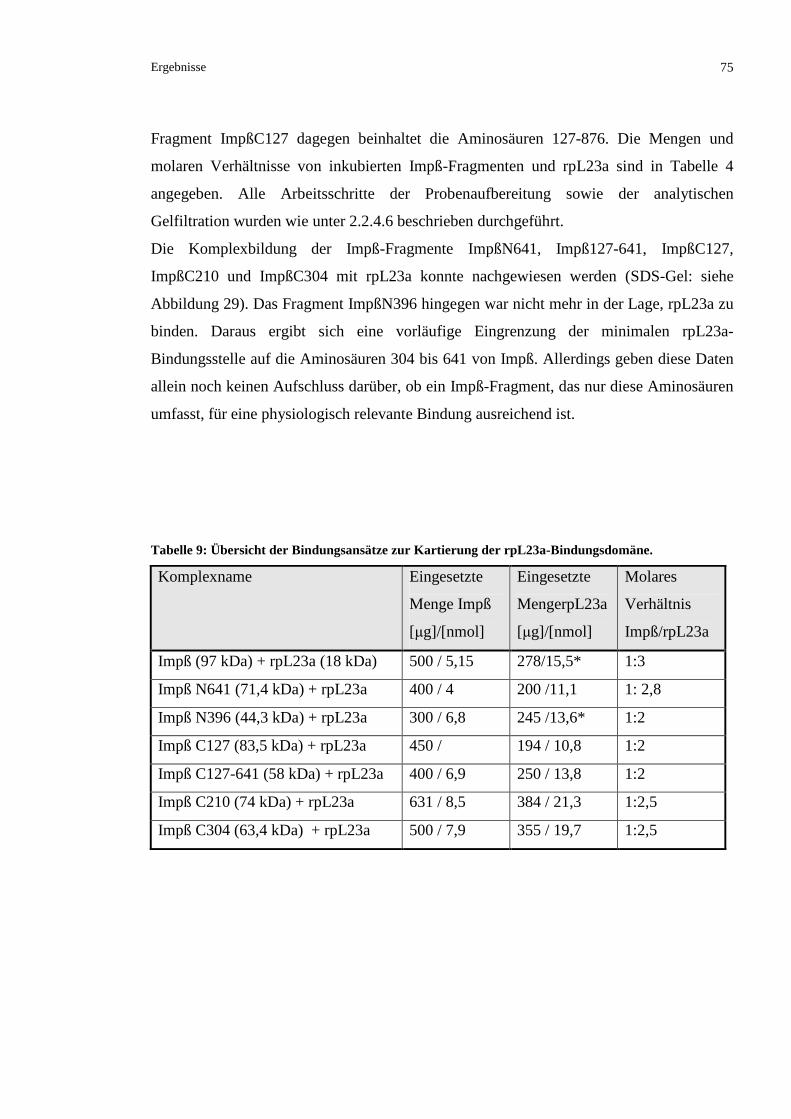
Ergebnisse
75
Fragment ImpßC127 dagegen beinhaltet die Aminosäuren 127-876. Die Mengen und
molaren Verhältnisse von inkubierten Impß-Fragmenten und rpL23a sind in Tabelle 4
angegeben. Alle Arbeitsschritte der Probenaufbereitung sowie der analytischen
Gelfiltration wurden wie unter 2.2.4.6 beschrieben durchgeführt.
Die Komplexbildung der Impß-Fragmente ImpßN641, Impß127-641, ImpßC127,
ImpßC210 und ImpßC304 mit rpL23a konnte nachgewiesen werden (SDS-Gel: siehe
Abbildung 29). Das Fragment ImpßN396 hingegen war nicht mehr in der Lage, rpL23a zu
binden. Daraus ergibt sich eine vorläufige Eingrenzung der minimalen rpL23a-
Bindungsstelle auf die Aminosäuren 304 bis 641 von Impß. Allerdings geben diese Daten
allein noch keinen Aufschluss darüber, ob ein Impß-Fragment, das nur diese Aminosäuren
umfasst, für eine physiologisch relevante Bindung ausreichend ist.
Tabelle 9: Übersicht der Bindungsansätze zur Kartierung der rpL23a-Bindungsdomäne.
Komplexname Eingesetzte
Menge Impß
[µg]/[nmol]
Eingesetzte
MengerpL23a
[µg]/[nmol]
Molares
Verhältnis
Impß/rpL23a
Impß (97 kDa) + rpL23a (18 kDa) 500 / 5,15 278/15,5* 1:3
Impß N641 (71,4 kDa) + rpL23a 400 / 4 200 /11,1 1: 2,8
Impß N396 (44,3 kDa) + rpL23a 300 / 6,8 245 /13,6* 1:2
Impß C127 (83,5 kDa) + rpL23a 450 / 194 / 10,8 1:2
Impß C127-641 (58 kDa) + rpL23a 400 / 6,9 250 / 13,8 1:2
Impß C210 (74 kDa) + rpL23a 631 / 8,5 384 / 21,3 1:2,5
Impß C304 (63,4 kDa) + rpL23a 500 / 7,9 355 / 19,7 1:2,5

Ergebnisse
76
Abbildung 29: SDS-PAGE der über eine Superdex S200 10/300 GL - Säule nachgewiesenen Komplexe von Impß-Fragmenten mit rpL23a. v.l.n.r.: Impß Volllänge/rpL23a, ImpßN641/rpL23a, ImpßN396/rpL23a, ImpßC127/rpL23a, Impß127-641/rpL23a, ImpßC210/rpL23a, ImpßC304/rpL23a (M=Größenstandard BR, 15%iges SDS-Gel).
3.2.4.1.2 Kartierung durch isotherme Titrationskalorimetrie
Ziel dieser Experimente war eine Aufklärung der Energetik und Affinität der Bindung von
Impß und rpL23a mit Hilfe von isothermer Titrationskalorimetrie (ITC) (2.2.5). Mit dieser
Methode ist es es möglich, die Bindungsenthalpien und Bindungskonstanten (Kd-Werte)
von geeigneten Bindungspartnern zu ermitteln.
Erste ITC-Experimente für die Messung der Bindungsenthalpie von Impß und rpL23a
wurden in verschiedenen Puffern durchgeführt, die 100-500mM NaCl und 20mM Tris
pH 7,5 enthielten, wie z.B. „ITC-Puffer 1“ (siehe 2.2.5). In diesen Puffern kam es jedoch
im Verlaufe der ITC-Messungen zu starker Präzipitatbildung. Eine Grundvorraussetzung
für die Vergleichbarkeit der ITC-Daten von verschiedenen Proteinkomplexen ist aber, dass
alle Bindungspartner in genau demselben Puffersystem vorliegen. Bindungsenthalpien, die
in verschiedenen Puffersystemen gemessenen wurden, können je nach pH-Wert und
Ionenzusammensetzung der verwendeten Puffer stark voneinander abweichen. Es war
daher notwendig ein System zu finden, in dem alle zu vergleichenden Proteinkomplexe
löslich waren. Durch die Einführung eines Puffersystems mit 300 mM Kaliumacetat und

Ergebnisse
77
20 mM Kaliumphosphat pH 7,5, (siehe „ITC-Puffer 2“ 2.2.5), konnten schließlich ITC-
Messungen ohne merkliche Präzipitation durchgeführt werden (2.2.5.).
Zunächst wurde die Bindung von Impß an rpL23a mittels ITC untersucht. Für eine
Eingrenzung der vollständigen Bindungsstelle in Impß wurde zusätzlich die Bindung des
Fragments Impß127-641 analysiert. Für diese ITC-Experimente wurde die
Proteinkonzentration von rpL23a durch Messung von A280 bestimmt (2.2.6.1.2). Die
Konzentrationsbestimmung von Impß und Impß127-641 erfolgte nach Bearden (2.2.6.1.2).
In Tabelle 10 sind die eingesetzten Proteinkonzentrationen, die ermittelten Stöchiometrien
(N), die Bindungsenthalpien (∆H), Dissoziationskonstanten (Kd) und Entropien (∆S) für
die Komplexe Impß/rpL23a und Impß127-641/rpL23a angegeben. Dabei wurden für
Impß/rpL23a (N ≈ 0,88) und Impß127-641 (N ≈ 0,36) unterschiedliche Stöchiometrien
ermittelt. Die real vorliegenden Stöchiometrien waren nicht eindeutig bestimmbar, da je
nach angewendeter Proteinbestimmungsmethode unterschiedliche Proteinkonzentrationen
für rpL23a gemessen wurden (3.2.2.4). Daher besitzen die vorliegenden Ergebnisse der
ITC-Messung nur eine eingeschränkte Aussagekraft (4.1.3).
Deutlich erkennbar ist jedoch die Tendenz einer niedrigeren Affinität von Impß (AS 127-
641) für rpL23a im Vergleich zu Impß in voller Länge (siehe Kd-Werte in Tabelle 10). Die
gemessenen Bindungsenthalpien ∆H sind positiv, was bedeutet, dass es sich bei der
Bindung von Impß an rpL23a um einen endothermen Prozess handelt. Für beide
Komplexe konnte zudem eine Zunahme der Entropie ∆S beobachtet werden.
Ausgehend von einem Modell der Bindung von Impß an rpL23a mit einer 1:1
Stöchiometrie sowie von Impß127-641 mit einer 1:2 Stöchiometrie wurde eine
willkürliche Anpassung der rpL23a-Konzentrationen durchgeführt. Die sich hieraus
ergebenen ITC-Daten sind in Tabelle 10 in Klammern aufgeführt.
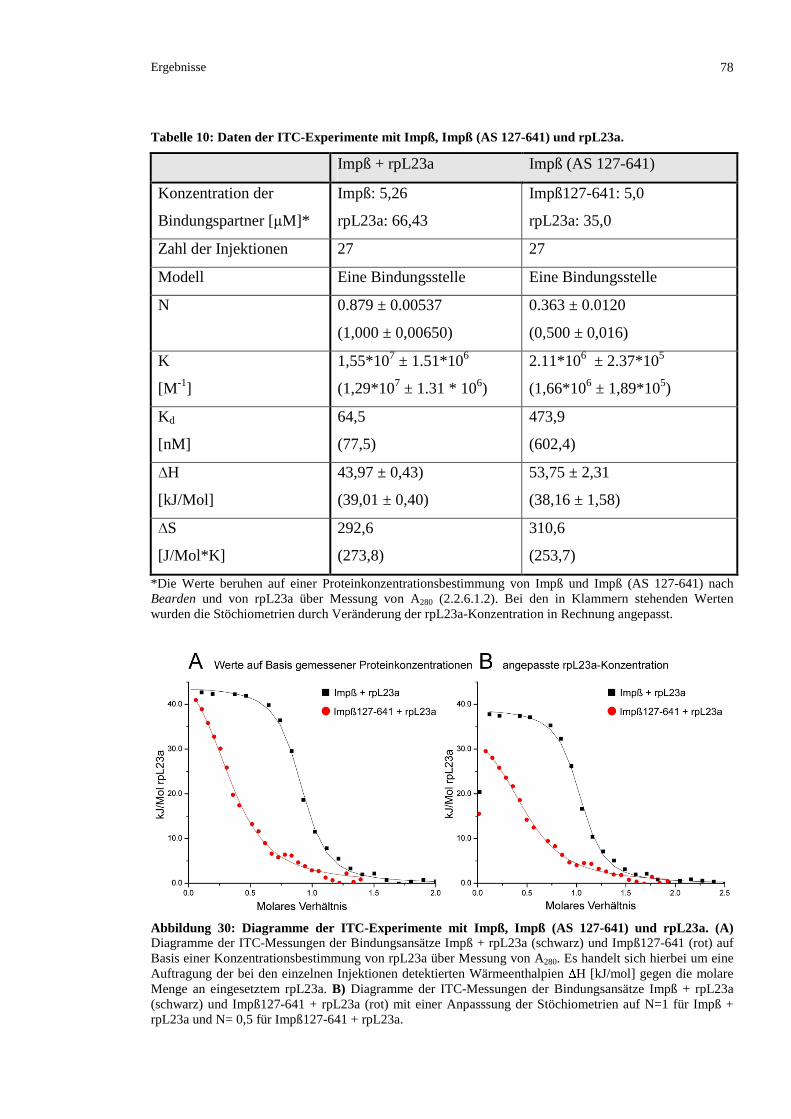
Ergebnisse
78
Tabelle 10: Daten der ITC-Experimente mit Impß, Impß (AS 127-641) und rpL23a.
Impß + rpL23a Impß (AS 127-641)
Konzentration der
Bindungspartner [µM]*
Impß: 5,26
rpL23a: 66,43
Impß127-641: 5,0
rpL23a: 35,0
Zahl der Injektionen 27 27
Modell Eine Bindungsstelle Eine Bindungsstelle
N 0.879 ± 0.00537
(1,000 ± 0,00650)
0.363 ± 0.0120
(0,500 ± 0,016)
K
[M -1]
1,55*107 ± 1.51*106
(1,29*107 ± 1.31 * 106)
2.11*106 ± 2.37*105
(1,66*106 ± 1,89*105)
Kd
[nM]
64,5
(77,5)
473,9
(602,4)
∆H
[kJ/Mol]
43,97 ± 0,43)
(39,01 ± 0,40)
53,75 ± 2,31
(38,16 ± 1,58)
∆S
[J/Mol*K]
292,6
(273,8)
310,6
(253,7)
*Die Werte beruhen auf einer Proteinkonzentrationsbestimmung von Impß und Impß (AS 127-641) nach Bearden und von rpL23a über Messung von A280 (2.2.6.1.2). Bei den in Klammern stehenden Werten wurden die Stöchiometrien durch Veränderung der rpL23a-Konzentration in Rechnung angepasst.
Abbildung 30: Diagramme der ITC-Experimente mit Impß, Impß (AS 127-641) und rpL23a. (A) Diagramme der ITC-Messungen der Bindungsansätze Impß + rpL23a (schwarz) und Impß127-641 (rot) auf Basis einer Konzentrationsbestimmung von rpL23a über Messung von A280. Es handelt sich hierbei um eine Auftragung der bei den einzelnen Injektionen detektierten Wärmeenthalpien �H [kJ/mol] gegen die molare Menge an eingesetztem rpL23a. B) Diagramme der ITC-Messungen der Bindungsansätze Impß + rpL23a (schwarz) und Impß127-641 + rpL23a (rot) mit einer Anpasssung der Stöchiometrien auf N=1 für Impß + rpL23a und N= 0,5 für Impß127-641 + rpL23a.
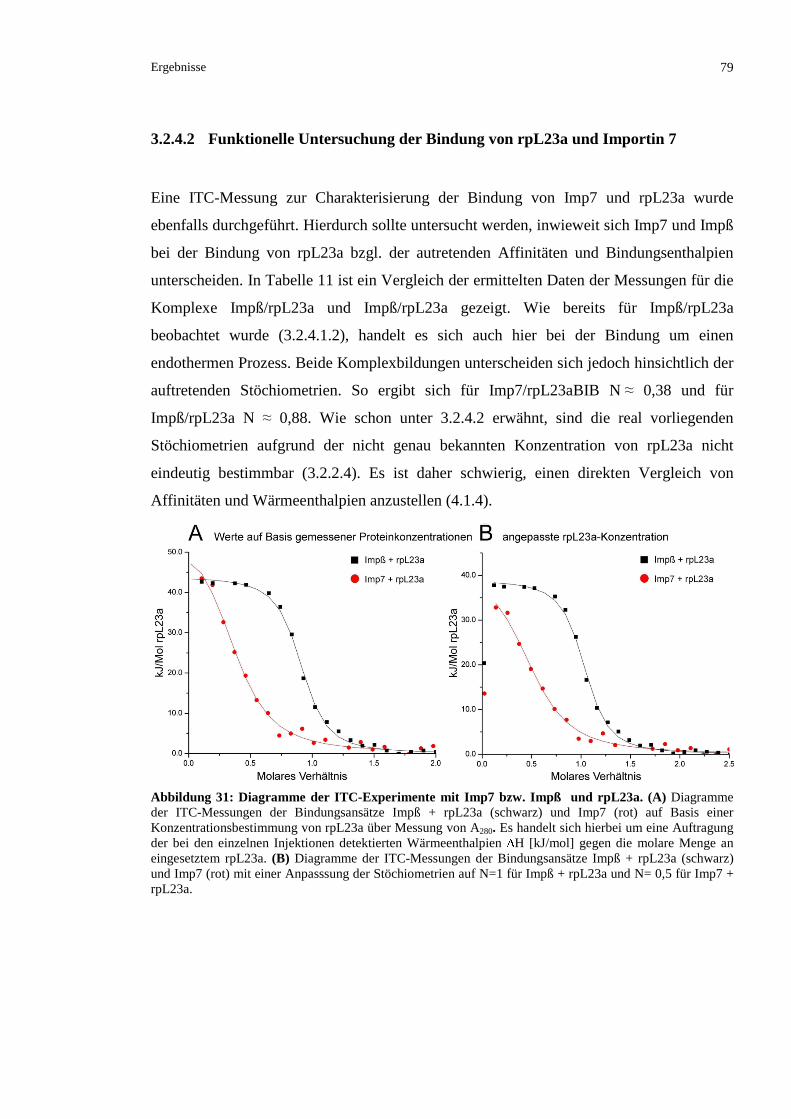
Ergebnisse
79
3.2.4.2 Funktionelle Untersuchung der Bindung von rpL23a und Importin 7
Eine ITC-Messung zur Charakterisierung der Bindung von Imp7 und rpL23a wurde
ebenfalls durchgeführt. Hierdurch sollte untersucht werden, inwieweit sich Imp7 und Impß
bei der Bindung von rpL23a bzgl. der autretenden Affinitäten und Bindungsenthalpien
unterscheiden. In Tabelle 11 ist ein Vergleich der ermittelten Daten der Messungen für die
Komplexe Impß/rpL23a und Impß/rpL23a gezeigt. Wie bereits für Impß/rpL23a
beobachtet wurde (3.2.4.1.2), handelt es sich auch hier bei der Bindung um einen
endothermen Prozess. Beide Komplexbildungen unterscheiden sich jedoch hinsichtlich der
auftretenden Stöchiometrien. So ergibt sich für Imp7/rpL23aBIB N ≈ 0,38 und für
Impß/rpL23a N ≈ 0,88. Wie schon unter 3.2.4.2 erwähnt, sind die real vorliegenden
Stöchiometrien aufgrund der nicht genau bekannten Konzentration von rpL23a nicht
eindeutig bestimmbar (3.2.2.4). Es ist daher schwierig, einen direkten Vergleich von
Affinitäten und Wärmeenthalpien anzustellen (4.1.4).
Abbildung 31: Diagramme der ITC-Experimente mit Imp7 bzw. Impß und rpL23a. (A) Diagramme der ITC-Messungen der Bindungsansätze Impß + rpL23a (schwarz) und Imp7 (rot) auf Basis einer Konzentrationsbestimmung von rpL23a über Messung von A280. Es handelt sich hierbei um eine Auftragung der bei den einzelnen Injektionen detektierten Wärmeenthalpien �H [kJ/mol] gegen die molare Menge an eingesetztem rpL23a. (B) Diagramme der ITC-Messungen der Bindungsansätze Impß + rpL23a (schwarz) und Imp7 (rot) mit einer Anpasssung der Stöchiometrien auf N=1 für Impß + rpL23a und N= 0,5 für Imp7 + rpL23a.

Ergebnisse
80
Tabelle 11: Daten der ITC-Experimente von Imp7 mit rpL23a und Impß mit rpL23a im Vergleich .
Imp7+ rpL23a Impß + rpL23a
Konzentration der
Bindungspartner [µM]*
Imp7: 4,0
rpL23a: 50,5
Impß: 5,26
rpL23a: 66,43
Zahl der Injektionen 27 27
Modell Eine Bindungsstelle Eine Bindungsstelle
N 0,376 ± 0,0141
(0,500 ± 0,00201)
0.879 ± 0.00537
(1,000 ± 0,00650)
K
[M -1]
3,65*106 ± 5,87*105
(2,56*106 ± 4,21*105)
1,55*107 ± 1.51*106
(1,29*107 ± 1.31*106)
Kd
[nM]
274,0
(390,1)
64,5
(77,5)
∆H
[kJ/Mol]
55,47 ± 2,79
(42,64 ± 2,30)
43,97 ± 0,43)
(39,01 ± 0,40)
∆S [J/Mol*K] 321,4
(273,0)
292,6
(273,8)
*Die Werte beruhen auf einer Proteinkonzentrationsbestimmung von Imp7 und Impß nach Bearden und von rpL23a über Messung von A280 (2.2.6.1.2). Bei den in Klammern stehenden Werten wurden die Stöchiometrien durch Veränderung der rpL23a-Konzentration in der Rechnung angepasst.
3.2.5 Präparation der BIB-Domäne von rpL23a
Die vollständige Kernlokalisationssequenz von rpL23a für den Kernimport durch Imp7
oder Impß umfasst die Aminosäuren 32-74 und wird BIB-Domäne genannt (Jäkel und
Görlich, 1998). Im folgenden Abschnitt wird die Klonierung und Präparation der BIB-
Domäne beschrieben. Das Hauptziel dieser Reinigung war die Präparation von Komplexen
der BIB-Domäne mit Importin 7, Imp7C598 und Impß127-641 für
Kristallisationsversuche. Die BIB-Domäne stellt hierbei einen im Komplex möglichst
unflexiblen Bindungspartner dar. Dies sollte zu einer Erhöhung der Stabilität der
Komplexe in einem möglichen Kristallgitter führen und damit die Wahrscheinlichkeit
einer Kristallisation erhöhen.

Ergebnisse
81
3.2.5.1 Klonierung der BIB-Domäne in den Vektor pGEX-6P-1
Für die Klonierung der BIB-Domäne wurde das in den Vektor pGEX-6P-1 klonierte
Volllänge-Gen als Matrize verwendet (3.2.1.1). Die Amplifikations-PCR (2.2.1.1.1)
erfolgte unter Verwendung der Primer BIB_BamHI_for und BIB_XhoI_rev (2.1.8). Das
amplifizierte Genfragment wurde anschließend über die Endonukleasen BamHI und XhoI
in pGEX-6P-1 inseriert (2.2.1.1.2) und ligiert (2.2.1.1.4). Zur Kontrolle der Klonierung
wurde eine Sequenzanalyse (2.2.1.1.5) unter Verwendung der DNA-Oligonukleotide
pGEX_f und pGEX_r (2.1.8) durchgeführt. Das Konstrukt pGEX-6P-1-rpL23a-
BIB/nostop wird bis zum Stopcodon der multiple cloning side translatiert, so dass nach der
BIB-Domäne die Aminosäuresequenz LERPHRN folgt.
3.2.5.2 Expression, Zellernte und Aufschluss
Die Expression der BIB-Domäne als GST-Fusionsprotein erfolgte in dem E. coli-Stamm
BL21(DE3) RP analog zu Volllänge-GST-rpL23a (3.2.1.2). In Abbildung 32 (siehe
3.2.5.3) ist eine SDS-PAGE der Vor- und Nachinduktionsproben abgebildet. Das 32 kDa
große Fusionsprotein wurde in einem deutlichen Maß exprimiert.
Die Zellernte und der Aufschluss der Zellen erfolgte ebenfalls analog zum
Volllängeprotein (3.2.1.3). Das Fusionsprotein GST-rpL23aBIB befindet sich nach dem
Aufschluss fast vollständig im Überstand (SDS-Gel: siehe 3.2.5.3 Abbildung 32).
3.2.5.3 Affinitätschromatographische Reinigung über Glutathion-Sepharose
Der erste Reinigungssschritt der BIB-Domäne bestand in einer Affinitätsreinigung über
eine GSH-Sepharose-Säule. Hierbei wurde analog zur Reinigung von rpL23a unter
Verwendung der gleichen Puffer vorgegangen (vgl.3.2.2.2). Zur Entfernung der GST-
Affinitätssequenz wurde rpL23aBIB mit der Protease PreScission im Massenverhältnis

Ergebnisse
82
1:200 für mindestens zwölf Stunden bei 4° C inkubiert (vgl.2.2.3.5.). Dabei blieb fast die
gesamte Proteinmenge löslich.
Abbildung 32: (A) SDS-PAGE der Expression von rpL23aBIB in pGEX-6P-1 in E. coli BL21 (DE3) RP (vI = vor Induktion, nI = nach Induktion, M=Größenstandard). (B) SDS-Gel der Reinigung von GST-rpL23aBIB über GSH-Sepharose. V.l.n.r.: Pellet (P), Überstand (Ü), Durchfluss (DF) und vereinigte Elutionsfraktionen (E) (M=Größenstandard BR, 15%iges SDS-Gel).
3.2.5.4 Ausschlusschromatographische Reinigung der BIB-Domäne
Der zweite und letzte Schritt der Präparation der BIB-Domäne bestand in einer
ausschlusschromatographischen Reinigung mittels einer Superdex S75 XK 26/60
Gelfiltrationssäule. Hierfür wurde die Probe, wie unter 2.2.4.5 beschrieben, vorbereitet, auf
die Säule geladen und eluiert. Abbildung 33 zeigt das SDS-Gel der analysierten Proben der
Elution. Die BIB-Domäne lag nach der Präparation hochrein vor.
Nach der Elution wurden die rpL23aBIB enthaltenden Fraktionen vereinigt und die
Proteinkonzentration mit den drei unter 2.2.6.1.2 beschriebenen Methoden vermessen.
Dabei konnten große Abweichungen der ermittelten Konzentration je nach angewandter
Methode beobachtet werden. Für die Proteinbestimmung über die Messung von A280
wurde der native Extinktionskoeffizient (2.2.6.1.2) von rpL23aBIB bestimmt (siehe
Tabelle 12).
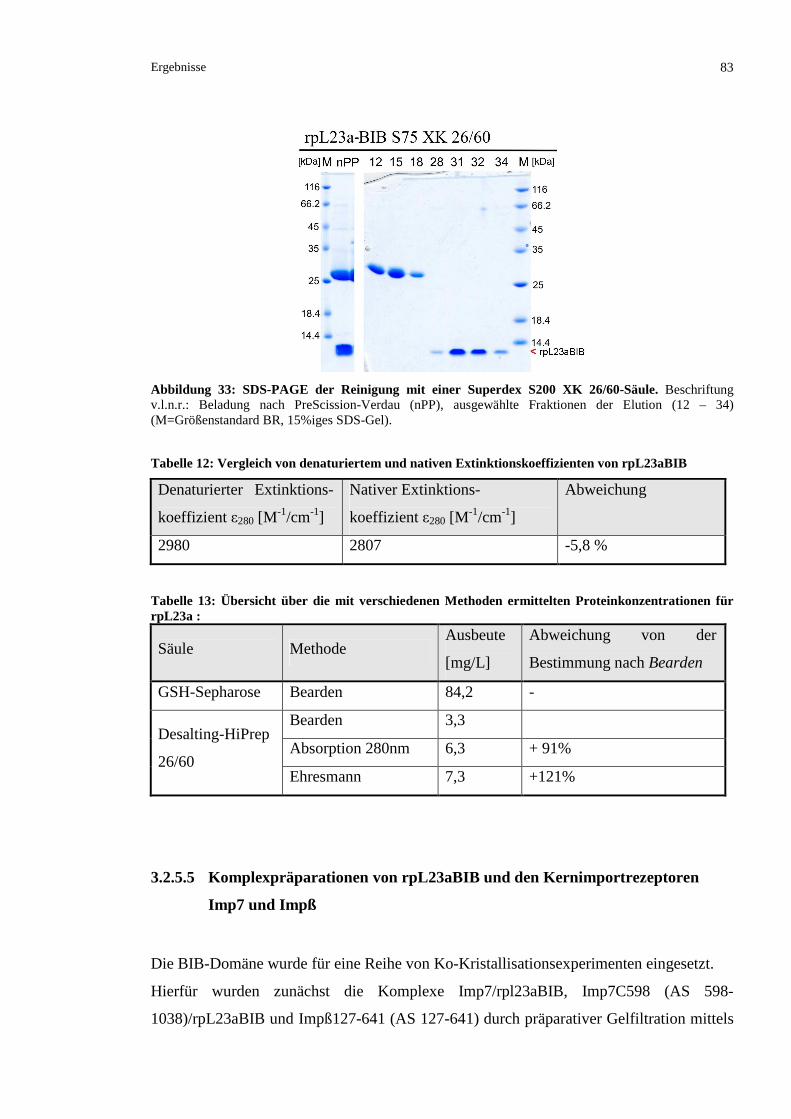
Ergebnisse
83
Abbildung 33: SDS-PAGE der Reinigung mit einer Superdex S200 XK 26/60-Säule. Beschriftung v.l.n.r.: Beladung nach PreScission-Verdau (nPP), ausgewählte Fraktionen der Elution (12 – 34) (M=Größenstandard BR, 15%iges SDS-Gel).
Tabelle 12: Vergleich von denaturiertem und nativen Extinktionskoeffizienten von rpL23aBIB
Denaturierter Extinktions-
koeffizient ε280 [M-1/cm-1]
Nativer Extinktions-
koeffizient ε280 [M-1/cm-1]
Abweichung
2980 2807 -5,8 %
Tabelle 13: Übersicht über die mit verschiedenen Methoden ermittelten Proteinkonzentrationen für rpL23a :
Säule Methode Ausbeute
[mg/L]
Abweichung von der
Bestimmung nach Bearden
GSH-Sepharose Bearden 84,2 -
Bearden 3,3
Absorption 280nm 6,3 + 91% Desalting-HiPrep
26/60 Ehresmann 7,3 +121%
3.2.5.5 Komplexpräparationen von rpL23aBIB und den Kernimportrezeptoren
Imp7 und Impß
Die BIB-Domäne wurde für eine Reihe von Ko-Kristallisationsexperimenten eingesetzt.
Hierfür wurden zunächst die Komplexe Imp7/rpl23aBIB, Imp7C598 (AS 598-
1038)/rpL23aBIB und Impß127-641 (AS 127-641) durch präparativer Gelfiltration mittels
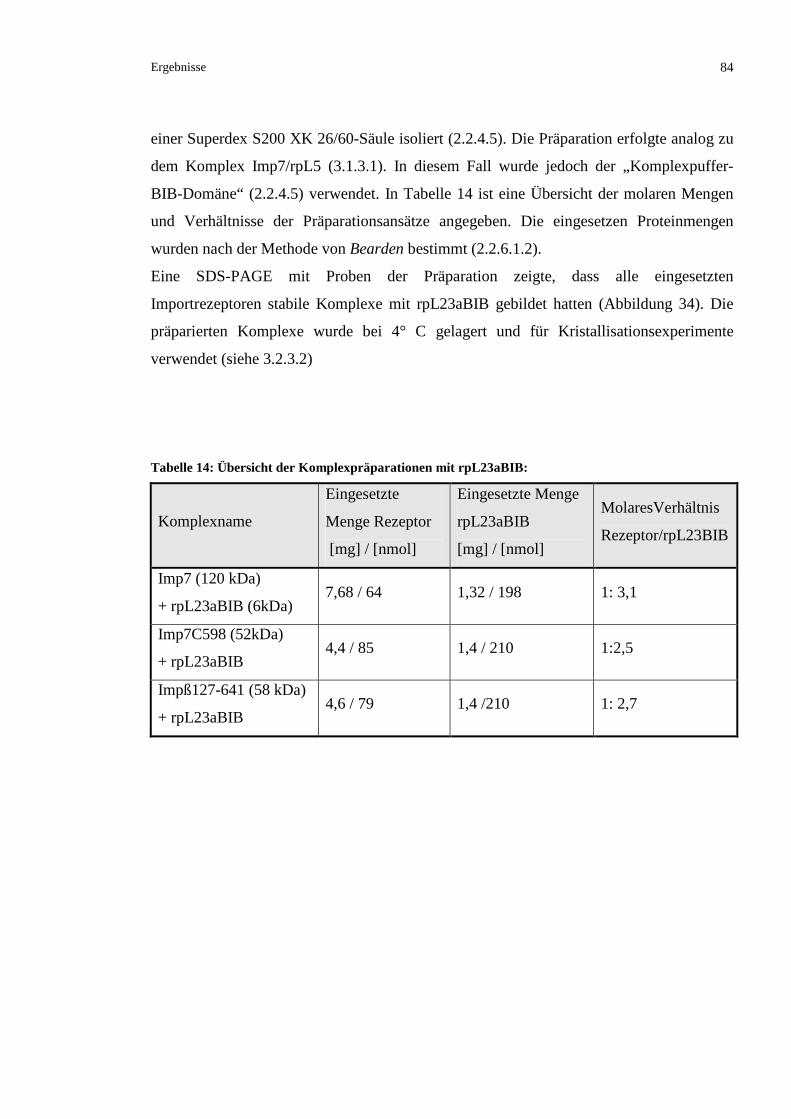
Ergebnisse
84
einer Superdex S200 XK 26/60-Säule isoliert (2.2.4.5). Die Präparation erfolgte analog zu
dem Komplex Imp7/rpL5 (3.1.3.1). In diesem Fall wurde jedoch der „Komplexpuffer-
BIB-Domäne“ (2.2.4.5) verwendet. In Tabelle 14 ist eine Übersicht der molaren Mengen
und Verhältnisse der Präparationsansätze angegeben. Die eingesetzen Proteinmengen
wurden nach der Methode von Bearden bestimmt (2.2.6.1.2).
Eine SDS-PAGE mit Proben der Präparation zeigte, dass alle eingesetzten
Importrezeptoren stabile Komplexe mit rpL23aBIB gebildet hatten (Abbildung 34). Die
präparierten Komplexe wurde bei 4° C gelagert und für Kristallisationsexperimente
verwendet (siehe 3.2.3.2)
Tabelle 14: Übersicht der Komplexpräparationen mit rpL23aBIB:
Komplexname
Eingesetzte
Menge Rezeptor
[mg] / [nmol]
Eingesetzte Menge
rpL23aBIB
[mg] / [nmol]
MolaresVerhältnis
Rezeptor/rpL23BIB
Imp7 (120 kDa)
+ rpL23aBIB (6kDa) 7,68 / 64 1,32 / 198 1: 3,1
Imp7C598 (52kDa)
+ rpL23aBIB 4,4 / 85 1,4 / 210 1:2,5
Impß127-641 (58 kDa)
+ rpL23aBIB 4,6 / 79 1,4 /210 1: 2,7
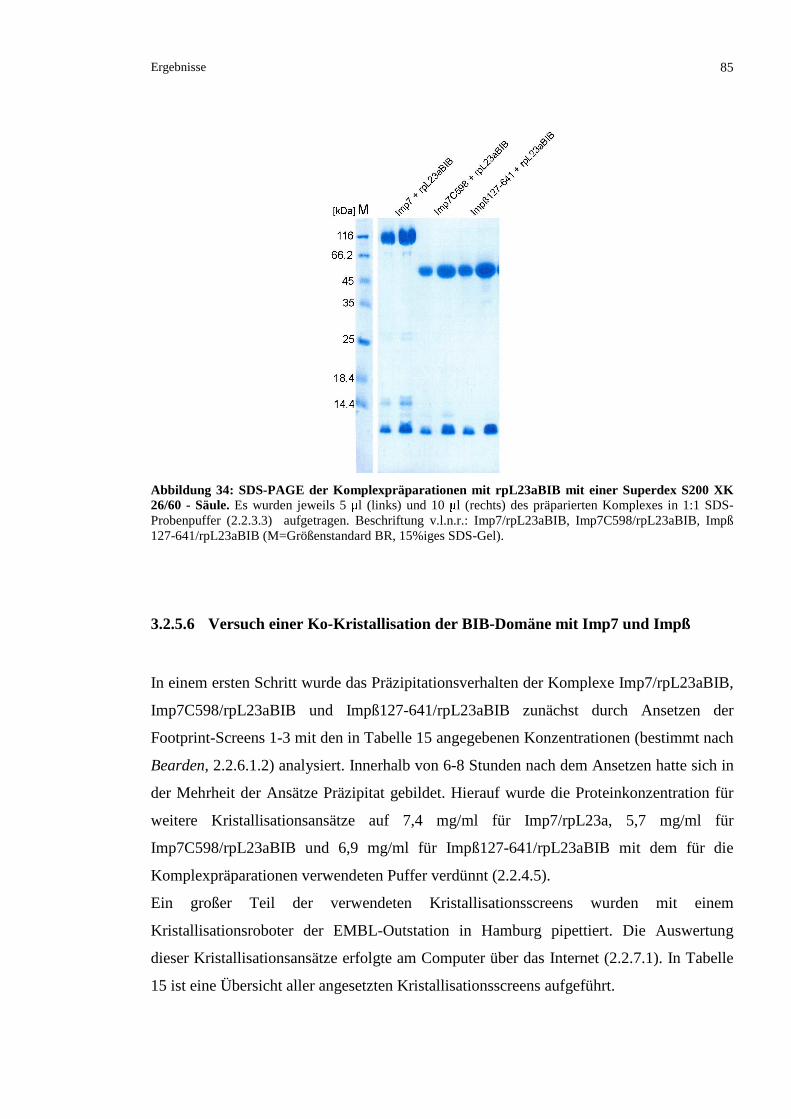
Ergebnisse
85
Abbildung 34: SDS-PAGE der Komplexpräparationen mit rpL23aBIB mit einer Superdex S200 XK 26/60 - Säule. Es wurden jeweils 5 �l (links) und 10 �l (rechts) des präparierten Komplexes in 1:1 SDS-Probenpuffer (2.2.3.3) aufgetragen. Beschriftung v.l.n.r.: Imp7/rpL23aBIB, Imp7C598/rpL23aBIB, Impß 127-641/rpL23aBIB (M=Größenstandard BR, 15%iges SDS-Gel).
3.2.5.6 Versuch einer Ko-Kristallisation der BIB-Domäne mit Imp7 und Impß
In einem ersten Schritt wurde das Präzipitationsverhalten der Komplexe Imp7/rpL23aBIB,
Imp7C598/rpL23aBIB und Impß127-641/rpL23aBIB zunächst durch Ansetzen der
Footprint-Screens 1-3 mit den in Tabelle 15 angegebenen Konzentrationen (bestimmt nach
Bearden, 2.2.6.1.2) analysiert. Innerhalb von 6-8 Stunden nach dem Ansetzen hatte sich in
der Mehrheit der Ansätze Präzipitat gebildet. Hierauf wurde die Proteinkonzentration für
weitere Kristallisationsansätze auf 7,4 mg/ml für Imp7/rpL23a, 5,7 mg/ml für
Imp7C598/rpL23aBIB und 6,9 mg/ml für Impß127-641/rpL23aBIB mit dem für die
Komplexpräparationen verwendeten Puffer verdünnt (2.2.4.5).
Ein großer Teil der verwendeten Kristallisationsscreens wurden mit einem
Kristallisationsroboter der EMBL-Outstation in Hamburg pipettiert. Die Auswertung
dieser Kristallisationsansätze erfolgte am Computer über das Internet (2.2.7.1). In Tabelle
15 ist eine Übersicht aller angesetzten Kristallisationsscreens aufgeführt.
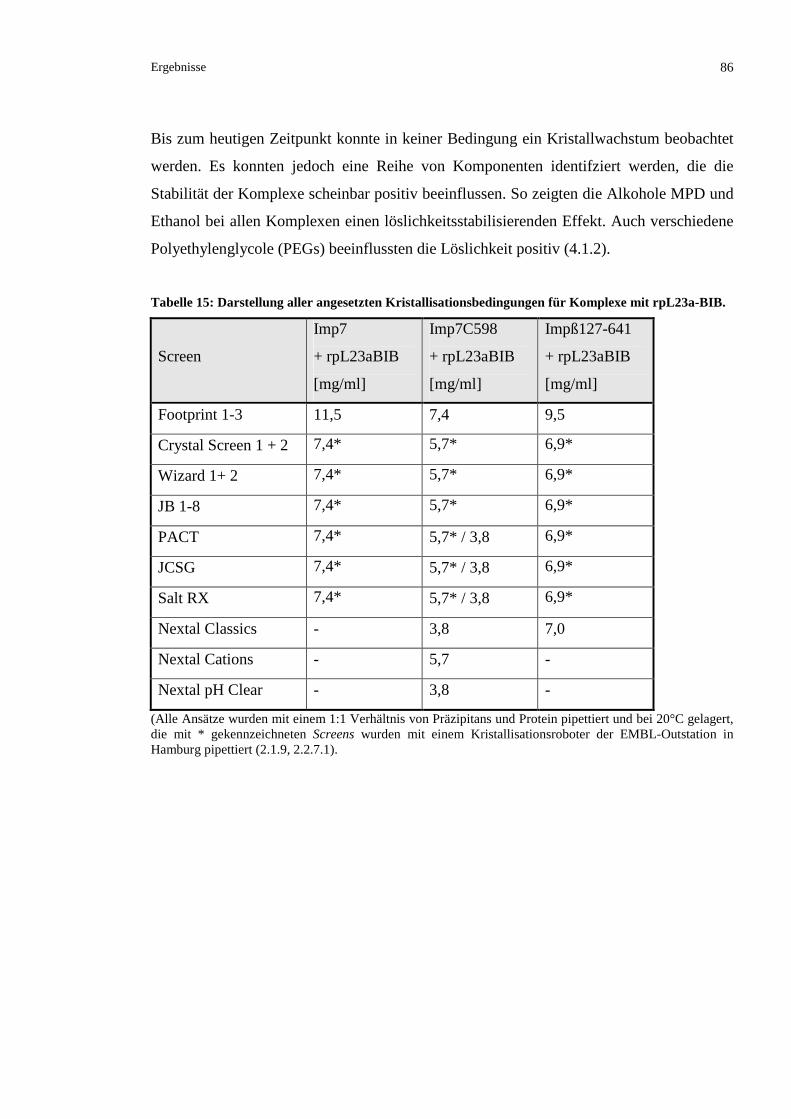
Ergebnisse
86
Bis zum heutigen Zeitpunkt konnte in keiner Bedingung ein Kristallwachstum beobachtet
werden. Es konnten jedoch eine Reihe von Komponenten identifziert werden, die die
Stabilität der Komplexe scheinbar positiv beeinflussen. So zeigten die Alkohole MPD und
Ethanol bei allen Komplexen einen löslichkeitsstabilisierenden Effekt. Auch verschiedene
Polyethylenglycole (PEGs) beeinflussten die Löslichkeit positiv (4.1.2).
Tabelle 15: Darstellung aller angesetzten Kristallisationsbedingungen für Komplexe mit rpL23a-BIB.
Screen
Imp7
+ rpL23aBIB
[mg/ml]
Imp7C598
+ rpL23aBIB
[mg/ml]
Impß127-641
+ rpL23aBIB
[mg/ml]
Footprint 1-3 11,5 7,4 9,5
Crystal Screen 1 + 2 7,4* 5,7* 6,9*
Wizard 1+ 2 7,4* 5,7* 6,9*
JB 1-8 7,4* 5,7* 6,9*
PACT 7,4* 5,7* / 3,8 6,9*
JCSG 7,4* 5,7* / 3,8 6,9*
Salt RX 7,4* 5,7* / 3,8 6,9*
Nextal Classics - 3,8 7,0
Nextal Cations - 5,7 -
Nextal pH Clear - 3,8 -
(Alle Ansätze wurden mit einem 1:1 Verhältnis von Präzipitans und Protein pipettiert und bei 20°C gelagert, die mit * gekennzeichneten Screens wurden mit einem Kristallisationsroboter der EMBL-Outstation in Hamburg pipettiert (2.1.9, 2.2.7.1).

Diskussion
87
4 Diskussion
Im Vordergrund dieser Arbeit stand die strukturelle und funktionelle Untersuchung der
Bindung von Imp7 und Impß an die ribosomalen Proteine rpL23a und rpL5.
Hiefür wurde zunächst eine Expressions- und Reinigungsstrategie für rpL5 und rpL23a
entwickelt. Beide Proteine konnten präpariert werden. Es wurden ausreichende Ausbeuten
sowohl für Kristallisationsversuche als auch für funktionelle Untersuchungen erzielt. Des
Weiteren wurde die Importrezeptorbindedomäne von rpL23a (BIB-Domäne oder
rpL23aBIB) kloniert, exprimiert und gereinigt.
Ein Ziel der Arbeit war die strukturelle Aufklärung von Komplexen aus ribosomalen
Proteinen mit dem Kernimportrezeptor Imp7. Hierfür wurden Komplexe von rpL5, rpL23a
und rpL23aBIB mit Imp7 präpariert und für Kristallisationsversuche eingesetzt. Die
Kristallisation stellt die Vorraussetzung für röntgenkristallographische Strukturanalysen
der Komplexe dar. Die ribosomalen Proteine sollten einen stabilisierenden Einfluss auf die
Konformation von Imp7 im Komplex ausüben und somit die Wahrscheinlichkeit einer
Kristallisation erhöhen. Die Kristallisationsansätze der präparierten Komplexe führten
jedoch bisher nicht zur Bildung von Proteinkristallen.
Ein weiterer Schwerpunkt lag in funktionellen Untersuchungen der Interaktion von
Kernimportrezeptoren mit ribosomalen Proteinen. Hierfür wurde hauptsächlich die
Bindung von Impß und rpL23a untersucht. Dabei wurde die Bindungsstelle für rpL23a in
Impß grob lokalisiert. Ein weiteres Ziel war die Aufklärung der Energetik und Affinität
dieser Bindung mit Hilfe von isothermer Titrationskalorimetrie (ITC). Es konnte gezeigt
werden, dass die Bindung von Impß und Imp7 an rpL23a einen endothermen Charakter
besitzt. Für eine genaue Lokalisierung der minimalen Bindungsstelle für rpL23a in Impß
müssten jedoch noch weitere ITC-Messungen durchgeführt werden.

Diskussion
88
4.1 Biochemische Arbeiten mit rpL23a
4.1.1 Präparation von rpL23a und rpL23aBIB
Die Präparation von rpL23a als Fusionsprotein von GST konnte erfolgreich etabliert
werden. Hierfür wurde rpL23a zunächst affinitätschromatographisch über GSH-Sepharose
gereinigt (3.2.2.2) und im Anschluss über Kationenaustauschchromatographie hochrein
isoliert (3.2.2.3).
Zu Beginn der Diplomarbeit bereitete die Präzipitation von rpL23a durch RNA große
Probleme. Die RNA führte durch Bindung an das basische rpL23a zur Bildung von großen
Aggregaten. Diese Eigenschaft, auch unspezifische Komplexe mit RNA bilden zu können,
ist auch von anderen basischen ribosomalen Proteinen bekannt (Jäkel und Görlich; 2002).
Die Entfernung der RNA von rpL23a konnte durch den Einsatz von LiCl (3.2.1.3, 3.2.2.3)
und durch Kationenaustauschchromatographie (3.2.2.3) erreicht werden. Ein Teil der
gebundenen RNA wurde durch die Verwendung eines Lysis- und Waschpuffers, der LiCl
enthielt, entfernt. Lithiumionen bilden scheinbar auf Grund ihrer geringen Größe und
niedrigen Polarisierbarkeit stabilere Komplexe mit den Phosphatgruppen der RNA als
beispielsweise Natriumionen. Die Lithiumionen sind somit in der Lage, ionische
Wechselwirkungen zwischen den Phosphatgruppen der RNA und basischen Seitenketten
von rpL23a aufzulösen, was zur Dissoziation der RNA führt. Die Verwendung von LiCl
führte zu einer Verbesserung der Löslichkeit von rpL23a nach der GSH-
Sepharosereinigung (3.2.2.2). Es zeigte sich aber, dass rpL23a trotzdem noch durch
gebundene RNA verunreinigt war (3.2.2.3). Durch Kationenaustauschchromatographie mit
einer SP-Sepharose-Säule wurde auch diese restliche RNA schließlich entfernt (3.2.2.3).
Hierbei scheinen die negativ geladenen Sulfopropylgruppen der SP-Sepharose die RNA
kompetitiv von rpL23a zu verdrängen. Verschiedene andere Reinigungsstrategien zur
Entfernung der RNA scheiterten, wie z.B. die Spaltung der RNA durch das Enzym
Benzonase (2.2.2.3.3), die Dissoziation durch hohe NaCl-Konzentrationen (Daten nicht
gezeigt) oder die Fällung durch Streptomycinsulfat (Oxenburgh und Snoswell, 1965).
Eine analytische Gelfiltration ergab, dass rpl23a wahrscheinlich in Form eines Dimers
präpariert worden war (3.2.2.5). Als problematisch erwies sich die Bestimmung der

Diskussion
89
Proteinkozentration, da hier je nach verwendeter Methode große Unterschiede festgestellt
werden konnten (3.2.2.4).
Ein Versuch, rpL23a mit einer Deca-Histidin-Sequenz zu reinigen, wurde zu einem frühen
Zeitpunkt der Diplomarbeit aufgegeben (3.2.2.1). Das Protein konnte zwar mit einer
Nickel-NTA-Affinitätssäule gereinigt werden, neigte jedoch aufgrund von RNA-
Verunreinigungen zur Präzipitation. Infolge desssen wurde es bei Gelfiltrationen mit dem
Ausschlussvolumen eluiert (Daten nicht gezeigt). Eine Entfernung der RNA mit einem
ähnlichen Protokoll wie für GST-rpL23a (3.2.1.3, 3.2.2) ist aber auch bei diesem
Konstrukt möglich.
Eine frühere Untersuchung von rpL23a (Jäkel und Görlich, 1998) hatte gezeigt, dass die
Kernlokalisationssequenz für die Importrezeptoren Imp7 und Impß in einem
Sequenzabschnitt lokalisiert ist, der die Aminosäuren 32 bis 74 beinhaltet. Diese, auch als
BIB-Domäne (für beta-like import receptor binding domain) bezeichnete
Aminosäuresequenz, wurde ebenfalls für Kristallisationszwecke im Rahmen der
Diplomarbeit präpariert (3.2.5).
4.1.2 Kristallisationsversuche für Komplexe mit rpL23a und
rpL23aBIB
Die Strukturaufklärung von Komplexen aus ribosomalen Proteinen und Imp7 stand im
Vordergrund der Diplomarbeit. Zu diesem Zweck wurden auch Komplexe von rpL23a und
rpL23aBIB mit Imp7 präpariert (3.1.3.1) und für Kristallisationsversuche eingesetzt
(3.1.3.2). Hierbei sollten die ribosomalen Proteine eine Stabilisierung der Konformation
von Imp7 im Komplex bewirken. Eine Studie hatte gezeigt, dass ungebundene
Karyopherine eine hohe strukturelle Flexibilität besitzen (Fukuhara et al., 2004). Eine
Bindung von Substraten führte bei den beobachteten Fällen zu einer Stabilisierung der
Konformation. Zutreffenderweise handelt es sich bei den meisten bisher veröffentlichten
Kristallstrukturen von Karyopherinen um Komplexe mit anderen Bindungspartnern
(Madrid und Weis 2005, Conti et al. 2006).
Für erste Kristallisationsversuche wurde die gereinigte BIB-Domäne von rpL23a (3.2.5.4)
verwendet. Hierfür wurden Komplexe der BIB-Domäne mit Imp7, dem C-terminalen

Diskussion
90
Imp7-Fragment Imp7C598 und dem Importin-ß-Fragment Impß127-641 präpariert
(3.2.5.5) und für Kristallisationsversuche eingesetzt (3.2.5.6). Die BIB-Domäne stellt
hierbei einen im Komplex möglichst unflexiblen Bindungspartner dar. Dies sollte zu einer
Erhöhung der Stabilität der Komplexe in einem möglichen Kristallgitter führen und damit
die Wahrscheinlichkeit einer Kristallisation erhöhen.
Ein Großteil der Kristallisationsscreens für die Komplexe Imp7/rpL23aBIB.
Imp7C598/rpL23aBIB und Impß127-641/rpL23aBIB wurde mit einem
Kristallisationsroboter der EMBL-Outstation in Hamburg angesetzt (3.2.5.6). Bisher
konnte in keiner der pipettierten Bedingungen eine Bildung von Proteinkristallen
beobachtet werden.
Ausgehend von den pipettierten Kristallisationansätzen lassen sich einige Aussagen über
den Einfluss bestimmter Komponenten auf das Präzipitationsverhalten der verschiedenen
Komplexe machen. Eine Eigenschaft, die für alle Komplexe beobachtet werden konnte,
war der positive Einfluss von Alkoholen wie MPD und Ethanol auf die Löslichkeit. So
zeigte der Komplex Imp7/rpL23aBIB eine erhöhte Löslichkeit in 15-50% MPD. Auch
scheinen pH-Werte im Bereich von pH 7,5-8,5 eine stabilisierende Wirkung auf den
Komplex auszuüben. Auch verschiedene Polyethylenglycole (PEGs) besitzen einen
positiven Einfluss auf die Löslichkeit. So konnte ein löslichkeitsstabilisierender Effekt für
PEG 400 und PEG 4000 in Kombination mit einem basischen pH-Wert beobachtet
werden. Der Komplex Imp7C598/rpL23aBIB zeigte im Bezug auf die eben genannten
Komponenten ein ähnliches Löslichkeitsverhalten. Auffällig war zusätzlich der positive
Effekt von Glycerin für die Löslichkeit. Der Komplex Impß127-641/BIB scheint durch 10-
60% MPD oder auch Ethanol, sowie in verschiedenen PEGs, wie z.B. PEG 400 und
PEG10000, stabilisiert zu werden.
Ein Komplex von rpL23a in voller Länge mit Imp7 wurde ebenfalls in einer Vielzahl von
Kristallisationsbedingungen eingesetzt (3.2.3.2). Auffällig war hier zunächst eine deutliche
Reduzierung der Löslichkeit im Vergleich zu den Komplexen Imp7/rpL23aBIB und
Imp7C598/rpL23aBIB. Möglicherweise führen die zusätzlichen Aminosäuren von rpL23a
in voller Länge zu einer niedrigeren Stabilität des Komplexes. Es konnte keine
Kristallbildung beobachtet werden. Lediglich in der Bedingung JB9-B1 kam es zur
Entstehung von Protein-Sphärolithen (Abbildung 28 unter 3.2.3.2). Diese waren relativ
rund und zeigten nur leichte Ansätze einer Kantenbildung. Eine Beobachtung von
Salzkristallen kann nahezu ausgeschlossen werden. Die Sphärolite zerfielen bei leichter

Diskussion
91
Berührung sofort. Aus den entstandenen Bruchstücken wuchsen innerhalb weniger
Stunden eine Vielzahl neuer Sphärolithe. Bis zum heutigen Zeitpunkt war es jedoch trotz
einer umfangreichen Variation einzelner Komponenten der Bedingung sowie eines micro-
seeding nicht möglich, diese Sphärolithe zu reproduzieren. Auch für den Imp7/rpL23a-
Komplex können einige Aussagen über den Einfluss einiger einzelner Komponenten im
Bezug auf die Löslichkeit gemacht werden. So scheinen hier bestimmte Alkohole wie z.B.
Isopropanol die Löslichkeit ebenso positiv zu beeinflussen wie leicht basische pH-Werte
im Bereich von pH 7,0 – 8,5. Stabilisierend ist auch der Einfluss von Carbonsäure-Salzen
wie NaFormiat, NaAcetat und Tri-NaCitrat.
Für weitere Kristallisationsversuche mit Imp7 erscheint es sinnvoll, im besonderen Maße
verkürzte Fragmente von rpL23a wie z.B. die BIB-Domäne einzusetzen. Diese Komplexe
zeigten eine wesentlich höhere Löslichkeit als der Komplex mit rpL23a in voller Länge.
Eine weitere Möglichkeit bestände darin, die Komplexe bei anderen Temperaturen wie
z.B. 4°C oder 30°C zu pipettieren.
4.1.3 Charakterisierung der Bindung von Impß an rpL23a
Parallel zu den Kristallisationsversuchen mit Komplexen aus Imp7 und den ribosomalen
Proteinen L5 und L23a wurde der Versuch unternommen, durch funktionelle
Untersuchungen mittels analytischer Gelfiltrationsläufe und isothermer
Titrationskalorimetrie mehr über die Eigenschaften der Bindung von Importrezeptoren mit
rpL23a herauszufinden. Hierfür wurde der Importrezeptor Impß verwendet. Bei Impß
handelt es sich um einen äußerst universellen Rezeptor, der eine bedeutende Rolle bei
einer Vielzahl von zellulären Transport- und Entwicklingsprozessen spielt (Harel und
Forbes, 2004).
Ein bereits veröffentlichtes Pull-down-Experiment mit verkürzten Fragmenten von Impß
und der BIB-Domäne von rpL23a grenzte die minimale Bindungsstelle von rpL23aBIB
auf die Aminosäuren 286-462 in Impß ein (Jäkel und Görlich, 1998). Von bereits
kristallisierten Komplexen von Impß mit verschiedenen Bindungspartnern ist jedoch
bekannt, dass sich die Bindungsstellen in Impß oft aus mehreren Komponenten
zusammensetzen. Diese müssen in der Primärsequenz nicht zwangsläufig benachbart sein

Diskussion
92
(Lee et al., 2005, 2003, Liu und Stewart, 2005; Cingolani et al., 2002, 1999). Die
Ergebnisse der Studie lieferten keine Hinweise darauf, inwieweit es sich bei den
Aminosäuren 286-462 um die vollständige, physiologisch relevante Bindungsstelle von
Impß für rpL23a in voller Länge handelt.
Ziel der praktischen Arbeiten mit Impß und rpL23a im Rahmen dieser Diplomarbeit sollte
es daher sein, durch Kombination von analytischen Gelfiltrationsläufen und isothermer
Titrationskalorimetrie sowohl die Lokalisation der vollständigen Bindungsstelle zu
bestimmen, als auch die Energetik der Bindung aufzuklären.
Für die Kartierung der rpL23a-Bindungsstelle in Importin ß wurden zunächst verkürzte
Fragmente von Importin ß auf ihre Fähigkeit hin untersucht, stabile Komplexe mit rpL23a
zu bilden (Abbildung 35). Auf diese Weise wurde die rpL23a-Bindungsstelle zunächst auf
die Aminsoäuren 304 bis 641 von Impß eingegrenzt. Die Ergnisse der analytischen
Gelfiltrationen wurden als Basis für weiterführende ITC-Experimente verwendet.
Abbildung 35: Schematische Darstellung der Kartierung der rpL23a-Bindungsstelle in Impß. (Grafik
modifiziert nach Wohlwend et al., 2006 in Vorbereitung)
Die isotherme Titrationskalorimetrie eignet sich in Kombination mit analytischen
Gelfiltrationsexperimenten als Methode für eine genaue Bestimmung von Bindungsstellen
in Proteinen. Mit dieser Methode ist es möglich, die bei der Bindung zwischen Proteinen
umgesetzte Wärmenthalpie ∆H, die Stöchiometrie N, die Dissoziationskonstante (Kd)
sowie die Gesamtentropie (∆S) zu bestimmen.

Diskussion
93
Bei den ersten ITC-Experimenten mit rpL23a und Impß wurden NaCl-haltige Puffer
eingesetzt, die jedoch eine teilweise Präzipitation der entstandenen Proteinkomplexe im
Verlauf der Titration verursachten (3.2.4.1.2). Schon eine geringe Präzipitatbildung führt
jedoch zu Schwankungen der Basislinie des Wärmedetektors und somit zu einer
Verfälschung der Messwerte. Erst durch die Verwendung eines Kaliumacetat und
Kaliumphosphat enthaltenden Puffers (2.2.5) konnten schließlich ITC-Experimente ohne
störende Präzipitation durchgeführt werden.
Ein Problem bei der Interpretation der gesammelten Daten stellen die Werte der
errechneten Stöchiometrien dar. Wie unter 3.2.2.4 beschrieben wurde, ergeben sich je nach
verwendeter Proteinbestimmungsmethode große Abweichungen für die ermittelte
Konzentration von rpL23a. Für die ITC-Experimente wurde die Konzentration von rpL23a
über die Absorption bei 280 nm bestimmt (2.2.6.1.2). Die Bestimmung der
Konzentrationen der Importrezeptoren erfolgte nach der Methode von Bearden (2.2.6.1.2).
Unter Einsetzung dieser Konzentrationen ergab sich für die Bindung von Impß an rpL23a
eine Stöchiometrie von N ≈ 0,88 (Tabelle 10 unter 3.2.4.1.2). Am nächsten käme diesem
Wert eine 1:1- Stöchiometrie von Impß und rpL23a. Für die Bindung von Impß (AS 127-
641) mit rpL23a ergab sich dagegen eine Stöchiometrie von N ≈ 0,36 (Tabelle 10 unter
3.2.4.1.1). Hierbei könnte es sich sowohl um eine 1:2- als auch um eine 1:3-Stöchiometrie
handeln. Im Falle eines 1:2- Verhältnisses würde dies bedeuten, dass zwei Moleküle
Impß127-641 ein Molekül rpL23a binden würden. Die gleiche Stöchiometrie ergibt sich
auch für die Bindung von Imp7 an rpL23a mit N ≈ 0,38 (Tabelle 11 unter 3.2.4.2).
Auffällig ist auch die Tendenz einer deutlich verringerten Affinität der Bindung von
Impß127-641 an rpL23a im Vergleich zu Impß. Möglicherweise ist Impß127-641 nicht
mehr in der Lage, die Kernlokalisationssequenz (NLS) von rpL23a vollständig zu binden,
so dass stattdessen zwei Moleküle Impß127-641 gemeinsam in unspezifischer Weise an
die NLS binden. Genauere Aussagen auf Basis der ermittelten Kd-Werte sind nicht
sinnvoll, solange kein Nachweis über die vorliegenden Stöchiometrien erbracht ist. Für
eine Klärung der vorliegenden Stöchiometrien wäre es sinnvoll, analytische
Gelfiltrationsläufe mit dem für ITC-Messungen verwendeten Puffer durchzuführen.
Hierfür könnten auch die bei ITC-Experimenten entstehenden Komplexe direkt durch
Gelfiltrationen analysiert werden.

Diskussion
94
Eine vollständige Kartierung der rpl23a-Bindungsstelle in Impß konnte in Rahmen der
Diplomarbeit nicht erreicht werden. Die ITC-Experimente mit dem Impß-Fragment
Impß127-641 haben gezeigt, dass die Aminosäuren 127-641 von Impß wahrscheinlich
nicht die vollständige Bindungsstelle beinhalten. Eindeutig beobachtbar war die Tatsache,
dass es sich bei der Bindung von Impß oder Imp7 an rpL23a unter den gegebenen
Bedingungen um eine endotherme Reaktion mit positiven ∆H handelt. Zudem konnte eine
Zunahme der Entropie während der Bildung der untersuchten Komplexe beobachtet
werden. Diese Entropiezunahme könnte eine Erklärung dafür sein, warum die Bindung der
Importrezeptoren an rpL23a trotz ihrer endothermen Eigenschaften spontan abläuft. Die
Kompensation der umgesetzten Wärmeenthalpie ∆H durch die Entropie ∆S ist eine
allgemeine Eigenschaft von Protein-Protein-Interaktionen. Damit eine Reaktion spontan
ablaufen kann, muss ihre freie Energie ∆G negativ sein. Die Definition von ∆G lautet:
∆G = ∆H – T∆S (∆G = Änderung der freien Energie, ∆H = Änderung der
Wärmeenthalphie, T = Temperatur, ∆S = Änderung der Entropie ).
Daraus folgt, dass Reaktionen, bei denen die umgesetzte Wärmeenthalpie ∆H ein positives
Vorzeichen besitzt, nur bei einer entsprechenden, gleichzeitigen Zunahme der Entropie
stattfinden können. Die beobachtete Entropiezunahme ∆S setzt sich aus mehreren
Teilbeträgen zusammen, deren genaues Verhältnis unbekannt ist. Eine Möglichkeit für die
beobachtete Entropiezunahme wäre das Freiwerden von proteingebundenen Salzionen im
Verlaufe dieser Bindung. So binden die Importrezeptoren Imp7 und Impß an die basische
BIB-Domäne von rpL23a (AS 32-74) (Jäkel und Görlich 1998). Durch einen
Verdrängungsprozess infolge neu entstehender Salzbrücken zwischen geladenen
Seitenketten der beiden Proteine würden die zuvor gebundenen Salzionen in Lösung gehen
und somit eine Zunahme der Entropie verursachen.
Ein weiterer Teilbetrag der Entropieänderung kann in einer Änderung der Freiheitsgrade
der Atome innerhalb der Proteine während der Bindung bestehen. Durch small-angle X-
ray scattering (SAXS)-Studien mit Impß und dem Importrezeptor Transportin (Fukuhara
et al., 2004) und durch Kristallstrukturen von Impß mit verschiedenen Bindungspartnern
(Lee et al., 2005, 2003, Liu und Stewart, 2005; Cingolani et al., 2002, 1999) ist bekannt,
dass Karyopherine große konformelle Änderungen während der Bindung von Substraten
oder RanGTP eingehen können. Eine aktuelle Studie stellt ein Modell auf, wonach

Diskussion
95
Karyopherine bei der Bindung von Substraten Energie speichern können (Conti et al.,
2006). Vereinfacht kann man sich die Karyopherine hierbei als Moleküle mit einer
federartigen Architektur vorstellen (siehe Abbildung 36).
Abbildung 36: Schematische Illustration des federartigen Aufbaus von ß-Karyopherinen. Jede Windung der Feder besteht aus einem HEAT-repeat, der wiederum aus zwei �-Helices aufgebaut ist (A und B) (Abbildung nach Stewart, 2003).
Eine Veränderung der relaxierten Struktur durch Änderung der Konformation bei der
Bindung von Substraten oder RanGTP führt zu einer Erhöhung der inneren Energie der
Karyopherine in der Art einer Federspannung. Die verhältnismäßigen Anteile von
enthalpischen und entropischen Effekten bei einer solchen Änderung der inneren Energie
sind unbekannt. Es könnte jedoch sein, dass die Bindung von Impß an rpL23a durch eine
Konformationsstabilisierung zu einer Verringerung der Freiheitsgrade der Proteinatome
führt. Dadurch würde dieser Prozess einen negativen Teilbeitrag zur Gesamtentropie
leisten, die insgesamt jedoch trotzdem zunimmt.
Auf der Grundlage von Kartierungsexperimenten (Jäkel und Görlich, 1998; sowie 3.2.4.2),
der postulierten Chaperonfunktion von Importrezeptoren (Jäkel und Görlich 2002) sowie
den bereits bekannten Komplexen von Impß mit anderen Bindungspartnern (Lee et al.,
2005, 2003, Liu und Stewart, 2005; Cingolani et al., 2002, 1999) können einige
Vermutungen über die Art der chemischen Wechselwirkungen des Impß/rpL23a-
Komplexes angestellt werden. Die BIB-Domäne von rpL23a besitzt einen äußerst
basischen Charakter. Es ist daher anzunehmen, dass Impß und auch andere funktionelle
Kernimportrezeptoren wie z.B. Imp7 über eine Reihe von sauren Aminosäuren an diese
basischen Reste binden. Impß besitzt eine ungeordnete Schleife von sauren Aminosäuren
(AS 331-341, engl. acidic loop), die zwischen der A- und der B-Helix vom HEAT-repeat 8

Diskussion
96
inseriert ist (Cingolani et al., 1999). Sie liegt damit innerhalb der Sequenz der minimalen
rpL23a-Bindungsstelle, die durch analytische Gelfiltrationen (3.2.4.2) und Pull-down-
Experimente (Jäkel und Görlich, 1998) bekannt ist. Eine Beteiligung dieser sauren
Aminosäuren an der Bindung der Kernlokalisationssequenz ist daher wahrscheinlich. Aus
den Strukturen von Impß im Komplex mit der IBB-Domäne von Impα (Cingolani et al.,
1999) und PTHrP (Cingolani et al., 2002) ist bekannt, dass die Bindung von Impß zur
Ausbildung von Sekundärstrukturmotiven innerhalb der Bindungsstellen der
Interaktionspartner führt. Eine Sekundärstrukturvorhersage von rpL23a mit dem
Programm PSI-Predict (Abbildung 37) zeigte, dass die BIB-Domäne (AS-32-74) eine
relativ ungeordnete Struktur mit wenigen kurzen ß-Strängen besitzt. Eine Entstehung von
Sekundärstrukturmotiven innerhalb der BIB-Domäne infolge einer Bindung von
Importrezeptoren durch einen induced-fit-Mechanismus ist daher vorstellbar.
Abbildung 37: Sekundärstrukturvorhersage für rpL23a durch PSIpredict. Grüne Zylinder, H: Helices, gelbe Pfeile, E: Faltblätter, C: Schleife, Höhe der blauen Balken: Konfidenz der Vorhersage, die Aminosäurepositionen sind unterhalb der Sekundärstruktur nummeriert, rote Pfeile: Anfang und Ende der BIB-Domäne (AS 32-74).

Diskussion
97
Die hier dargestellten Kartierungsexperimente mit Imp7 konnten teilweise zur Aufklärung
der Lokalisation der Bindungsstellen, sowie der Energetik und Kinetik der Bindung
genutzt werden. Für eine genaue Aufklärung der chemischen Wechselwirkungen der
beiden Bindungspartner ist jedoch eine strukturelle Aufklärung des Impß/rpL23a-
Komplexes nötig. Die Identifizierung der vollständigen Bindungsstellen von Impß und
Imp7 für rpL23a ist jedoch ein nützliches Hilfsmittel für eine mögliche spätere
Strukturaufklärung. Kristallisationsversuche mit solchen Rezeptor-Fragment/rpL23a-
Komplexen könnten die Kristallisation postiv beeinflussen, da diese Komplexe
wahrscheinlich eine stabilere Konformation besitzen.
4.1.4 Funktionelle Arbeiten mit Imp7 und rpL23a
Angesichts der Tatsache, dass mehrere Rezeptoren den Kernimport von rpL23a vermitteln
können stellt sich die Frage, inwieweit zwischen den verschiedenen Rezeptoren
Unterschiede bzgl. der Affinität und Bindungsenthalpie bestehen. Für diesen Zweck wurde
auch die Bindung von Imp7 an rpL23a durch eine ITC-Messung untersucht (3.2.4.2). Wie
bereits für Impß beobachtet (3.2.4.1.2) handelt es sich auch bei dieser Bindung um einen
endothermen Prozess. Die ermittelte Stöchiometrie zeigte mit N ≈ 0,38 eine große
Abweichung zur Bindung von Impß an rpL23a (N ≈ 0,88). Der Wert ähnelt eher der
beobachteten Stöchiometrie für das Impß-Fragment Impß127-641 (N ≈ 0,36). Es könnte
sich also auch hier sowohl um eine 1:2- als auch eine 1:3-Stöchiometrie handeln. Dies
würde bedeuten, dass entweder zwei oder sogar drei Moleküle Imp7 ein Molekül rpL23a
binden. Dieser Beobachtung wiederspricht jedoch einer analytischen Gelfiltration des
Komplexes Imp7/rpL23a (3.2.3.1). Das hierbei aus dem Retentionsvolumen ermittelte
Molekulargewicht des Imp7/rpL23a-Komplexes beträgt 152 kDa. Dies entspricht dem bei
einer 1:1 Stöchiometrie vorliegenden Gewichts des Komplexes von 138kDa. Es muss
jedoch gesagt werden, dass die Gelfiltration mit einem anderen Puffer durchgeführt wurde
(2.2.4.6). Es ist nicht auszuschließen, dass sich mit dem für ITC-Messungen verwendeten
Puffer (2.2.5) eine andere Stöchiometrie ergibt. Für die Klärung dieses Problems wäre es,
wie bereits vorgeschlagen (4.1.3), sinnvoll, analytische Gelfiltrationsläufe mit dem für
ITC-Messungen verwendeten Puffer durchzuführen.

Diskussion
98
Auf die Frage nach der Lokalisation der Bindungsstelle für rpL23a in Imp7 gibt es erste
Hinweise. Die erfolgreiche Präparation des Komplexes Imp7C598/rpL23aBIB (3.2.5.5)
grenzt den Bereich einer minimalen Bindungsstelle für rpL23a in Imp7C598 auf den C-
terminalen, die Aminosäuren 598 bis 1038 umfassenden Bereich, von Imp7 ein
(Abbildung 38).
Abbildung 38: (A) Schematische Darstellung von Imp7 mit der Kartierung der bekannten Bindeseiten. (B) Schematische Darstellung des Imp7-Fragments Imp7C598. (Beschriftung: saure Schleife 1 = rot, saure Schleife 2 = orange, Impß-Bindedomäne = gelb) (Grafik modifiziert nach Wohlwend et al., 2006 in Vorbereitung) Innerhalb dieses Sequenzbereichs sind zwei ungeordnete Schleifenstrukturen lokalisiert,
die ein hohe Dichte von sauren Aminosäuren aufweisen (Schleife 1: AS 882-912 und
Schleife 2: AS 927-957). Wie bereits für den Komplex Impß/rpL23a besprochen wurde,
(4.1.3) könnten diese sauren Aminosäuren auch bei der Bindung von Imp7 an rpL23a eine
wichtige Rolle spielen. Hierbei wird eine Bindung der sauren Seitenketten von Imp7 an
basische Bereiche der Kernlokalisationssequenz angenommen.
Eine zusätzliche Bindung von basischen und auch anderen Resten außerhalb der NLS von
rpL23a wäre ebenfalls möglich. Dies könnte in verstärktem Maße der Fall sein, wenn man
davon ausgeht, dass Imp7 eine chaperonartige Funktion für das Importsubstrat rpL23a
besitzt, wie von Jäkel und Görlich postuliert wurde (1.3.3) (Jäkel und Görlich, 2002). Die
Autoren stellten die Theorie auf, dass Imp7 eine effizientere Chaperonfunktion besitzt als
Impß. Basis dieser Vermutung war ein Experiment, bei dem die Komplexe Impß/rpL23a,
Transportin/rpL23a, Imp5/rpL23a und Imp7/rpL23a auf einen CM-Sepharose-
Kationentauscher geladen wurden. Während Impß/rpL23a und Transportin/rpL23a
teilweise an den Kationentauscher gebunden wurden, zeigten die Komplexe Imp5/rpL23a
und Imp7/rpL23a keine Bindung. Daraus schlossen die Autoren, dass Imp7 und Imp5 im

Diskussion
99
Gegensatz zu Impß und Transportin zusätzliche Bereiche außerhalb der BIB-Domäne
gebunden hatten und somit ionische Wechselwirkungen von rpL23a mit dem
Kationentauscher verhinderten. Die Autoren postulierten, dass aus einer Reihe von
funktionellen Importrezeptoren nur bestimmte Rezeptoren eine effiziente
Chaperonfunktion besitzen. Allerdings ist dieses Experiment noch kein direkter Nachweis
für eine Chaperonfunktion von Importrezeptoren. Ein vorstellbares Experiment für einen
Vergleich der Chaperonfunktionen von Impß und Imp7 könnte in einem Nachweis der
RNA-Bindefähigkeit der Komplexe bestehen. Beispielweise wäre es möglich, die
Komplexe Imp7/rpL23a und Impß/rpL23a zunächst an einer Affinitätssäule zu
immobilisieren und im Anschluss mit einer Zelllysatprobe zu inkubieren. Nach einem
Waschschritt und der anschließenden Elution könnte eine spektrometrische Untersuchung
der Komplexe durchgeführt werden. Das Auftreten von gebundener RNA wäre ein
Hinweis auf eine unzureichende Chaperonfunktion des Importrezeptors.
4.2 Biochemische Arbeiten mit rpL5
4.2.1 Präparation von rpL5
Die Präparation von rpL5 konnte erfolgreich etabliert werden (3.1.2). Hierfür wurde rpL5
genau wie rpL23a durch Affinitätschromatographie über GSH-Sepharose und
anschließend über Kationenaustauschchromatographie gereinigt. Auch bei der Reinigung
von rpL5 trat zunächst das Problem auf, dass das Protein infolge von gebundener RNA
präzipitierte (3.1.1.3 und 3.1.2.1). Durch die Verwendung eines Lysis- und Waschpuffers,
der LiCl enthielt, gelang es jedoch schließlich, die gebundene RNA zu entfernen (3.1.2.4).
Auffällig war auch die Anfälligkeit von rpL5 für Proteasen (3.1.2.2). rpL5 weist nach der
Entfernung der GST-Affinitätssequenz eine Temperaturinstabilität auf, so dass es bei 25°C
zur Präzipitation neigt (3.1.2.2). Die Verwendung eines Puffers, der 500 mM NaCl
enthielt, verringerte diesen Effekt, konnte ihn jedoch nicht vollständig eliminieren
(3.1.2.5). Diese Temperaturempfindlichkeit ist auch von früheren Untersuchungen mit

Diskussion
100
rpL5 bekannt (Scripture und Huber, 1995, DiNitto und Huber, 2003). Daher ist es, wie
beobachtet, kaum möglich, analytische Gelfiltrationen bei Raumtemperatur durchzuführen.
4.2.2 Kristallisationsversuche mit Imp7/rpL5 und rpL5
Ein Großteil der Kristallisationsscreens für den Komplex Imp7/rpL5 wurde mit einem
Kristallisationsroboter der EMBL-Outstation in Hamburg pipettiert (3.1.3.2). Bis zum
heutigen Zeitpunkt konnte in keiner der angesetzten Bedingungen eine Kristallbildung
beobachtet werden. Die Alkohole MPD (2-Methyl-2,4-pentandiol) oder Ethanol zeigten
jedoch einen positiven Einfluss auf die Löslichkeit des Komplexes. So konnte in einem
Teil der Bedingungen mit MPD oder Ethanol über einen Zeitraum von fast 4 Monaten
keine Präzipitation von Protein festgestellt werden. Für weitere Kristallisationsversuche
erscheint es daher sinnvoll, die MPD- und Ethanol-haltigen Kristallisationsscreens JB7
und JB8 mit höheren Proteinkonzentrationen als 4,8 mg/ml zu pipettieren. Eine weitere
Möglichkeit bestünde darin, den Komplex bei anderen Temperaturen, wie z.B. 4°C oder
30°C, zu pipettieren. Des Weiteren erscheint die Verwendung von verkürzten N-
terminalen Fragmenten für die Kristallisation sinnvoll, da die von Imp7 erkannte
Kernlokalisationssequenz nur die Aminosäuren 1-25 von rpL5 umfasst (Claußen et. al.,
1999; Rudt und Pieler, 2001).
In einem ergänzenden Versuch wurde rpL5 ohne Bindungspartner in
Kristallisationsansätzen bei 20°C eingesetzt. Hierbei konnte in den meisten Bedingungen
eine rasche Präzipitation beobachtet werden. Ein möglicher Grund hierfür ist eine zu hoch
eingesetzte Proteinkonzentration. Ein weiterer Grund könnte auch die beobachtete
Temperaturinstabilität und niedrige Strukturierung des Proteins sein (4.2.3). Sekundär- und
Tertiärstrukturvorhersagen zeigten, dass rpL5 weite N- und C-terminale Sequenzabschnitte
mit einer geringen Strukturierung besitzt, die eine Kristallisation des Proteins ohne
Bindungspartner behindern könnten (4.2.3).

Diskussion
101
4.2.3 Die Rolle von Importrezeptoren bei Importprozessen mit rpL5
Wie unter 1.3.5 beschrieben wurde, ist der Reimport von im Cytoplasma gespeicherter 5S
rRNA an rpL5 gekoppelt. Angesichts der Tatsache, dass die Biogenese der ribosomalen
Untereinheiten im Nukleolus und im Nukleus stattfindet, stellt sich die Frage, warum im
Verlaufe der Evolution überhaupt ein solcher Transportzyklus für die 5S RNA entwickelt
wurde. Es gibt zum jetzigen Zeitpunkt einige Hinweise aber keine wirklichen Antworten
auf diese Frage. In einer Studie wurde postuliert, dass die Bindung an rpL5 eine
Vorbedingung für den Einbau der 5S rRNA in die große ribosomale Untereinheit darstellt
(Steitz et al., 1988). Eine andere Untersuchung beschreibt ein Modell, indem die Bindung
der 5S rRNA an rpL5 in Form einer Feedback-Regulierung die eigene Transkriptionsrate
reguliert (Pittman et al., 1999). Eine Studie von Lin et al. schließlich stellte die Theorie
auf, dass die 5S rRNA eine chaperonartige Rolle als Faltungshelfer für rpL5 während der
Translation spielt. Die Autoren vermuten weiter, dass es sich hierbei um einen
entwicklungsgeschichtlich sehr alten Mechanismus handeln könnte. Dieser könnte aus
einer Zeit stammen, in der RNA-Moleküle eine Vielzahl von Funktionen besaßen, die
heute von Proteinen ausgeübt werden (Lin et al., 2001).
rpL5 ist ein temperatursensibles Protein. DiNitto und Huber zeigten mit Hilfe von
zirkulärer Dichroismus-Spektroskopie, dass rpL5 aus Xenopus laevis ohne Bindungs-
partner in vitro einen Denaturierungspunkt von Tm= 27°C besitzt. Ein Komplex aus 5S
rRNA und rpL5 besitzt hingegen einen Denaturierungspunkt von Tm = 44°C. Die Autoren
identifizierten größere Sequenzabschnitte in rpL5, vor allem an den N- und C-terminalen
Enden, die eine niedrige Strukturierung aufweisen. Der niedrige Ordnungsgrad scheint für
die Temperaturinstabilität des Proteins verantwortlich zu sein (DiNitto und Huber, 2003).
Ein mit dem Programm Swiss-Model (2.1.10) errechnetes Modell der Aminosäuren 10-232
ist in Abbildung 39 dargestellt. Deutlich erkennbar ist hier eine geringe Strukturierung der
N-terminalen Region sowie einiger zentraler Sequenzabschnitte. Der C-terminale Teil (AS
233-296) fehlt in diesem Modell. Eine Untersuchung der Aminosäuresequenz durch
DiNitto und Huber mit dem Programm PONDR sagt auch für den hier nicht dargestellte C-
terminalen Teil einen niedrigen Strukturierungsgrad vorher (DiNitto und Huber, 2003).

Diskussion
102
Abbildung 39 Schematisches Modell der Struktur von rpL5 (AS 10 – 232) aus Xenopus laevis errechnet mit dem Programm Swiss-Model (2.1.10). (A) Schematisches Modell von rpL5 (AS-10 – 232). Die Sekundärstrukturen sind in einem Farbspektrum von blau (N-Terminus) nach rot (C-Terminus) gefärbt. Die rosa gefärbten Aminosäuren 48, 189 und 234 wurden als Grenzen für die Klonierung von rpL5-Fragmenten ausgewählt (AS 234 nicht dargestellt – stattdessen ist die letzte dargestellte Aminosäure AS232 angefärbt) (3.1.4). (B) Schematisches Model von rpL5 (AS 10-232) mit einer farbigen Illustration der chemischen Eigenschaften der Seitenketten. Beschriftung: basische Reste (blau), saure Reste (rot), polare Reste (gelb), unpolare Reste (grau).
Die Autoren postulierten, dass die Bindung der 5S-RNA in Form eines gegenseitigen
induced-fit-Mechanismus zu einer Stabilisierung von zentralen und C-terminalen
Sequenzabschnitten in rpL5 führt. Besonders die Wechselwirkungen zwischen
aromatischen, im zentralen Bereich der Primärsequenz lokalisierten Aminosäuren mit
verschiedenen Basen der 5S rRNA scheint für die Stabilisierung von rpL5 eine wichtige
Rolle zu spielen (DiNitto und Huber, 2003, 2001). Die N-terminalen Aminosäuren spielten
hingegen keine nachweisbare Rolle bei der Bindung der RNA. Diese Region ist
wahrscheinlich innerhalb des 5S RNPs relativ exponiert und vermittelt verschiedene
Transportprozesse (DiNitto und Huber, 2003). Die ersten 25 N-terminalen Aminosäuren
von rpL5 beinhalten sowohl eine Kernlokalisationssequenz für verschiedene
Importrezeptoren wie Imp7 und Impß als auch eine Kernexportsequenz für einen CRM1
vermittelten Export (1.3.1) (Murdoch et al., 2002; Rudt und Pieler, 2001; Claußen et al.,
1999). Des Weiteren wird vermutet, dass die N-terminale Region von rpL5 eine wichtige
Rolle beim Einbau von 5S RNPs in die große ribosomale Untereinheit spielt (Lin et al.,
1999).
Auch andere Interaktionspartner von rpL5 wie z.B. das MDM-2 Onkoprotein (Marechal et
al., 1994) oder der eukaryotische Initiationsfaktor 5A (Schatz et al., 1998) könnten einen
strukturstabilisierenden Einfluss auf rpL5 haben. Die Tatsache, dass rpL5 in vivo meist in
Form von 5S RNPs oder in Komplexen mit anderen Bindungspartnern vorkommt, spielt
auch für die Kristallisation von rpL5 mit Importrezeptoren eine wichtige Rolle. Es stellt

Diskussion
103
sich die Frage, welche physiologische Rolle ein Imp7/rpL5- oder Impß/rpL5-Komplex
ohne gebundene 5S rRNA besitzt. Um eine Stabilisierung der Struktur von rpL5 zu
ermöglichen, müssten die Importrezeptoren wahrscheinlich mehr als nur die ersten 25
Aminosäuren binden. Impß und das Impß-Fragment ImpßC127 (AS 127-876) konnten
rpL5 bei analytischen Gelfiltrationen binden, zeigten jedoch eine deutliche
Temperaturinstabilität, so dass ein Teil des Komplexes auf der Säule präzipitierte (Daten
nicht gezeigt). Der Komplex Impß/rpL5 neigte bei einem ITC-Experiment zu starker
Präzipitation (Daten nicht gezeigt). Es erscheint daher möglich, dass Impß in vitro keinen
strukturstabilisierenden Einfluss auf rpL5 ausübt. Dies könnte bedeuten, dass Impß
lediglich mit der N-terminalen NLS-tragenden Region von rpL5 interagiert. Für den
Komplex Imp7/rpL5 fehlen Beobachtungen über eine eventuelle Temperaturinstabilität, da
dieser Komplex ausschließlich bei 4°C präpariert wurde (3.1.3.1).
Wie Untersuchungen des Kernimports von rpL5 in der Vergangenheit gezeigt hatten,
besitzt rpL5 neben einer N-terminalen NLS (AS 1-25) auch eine C-terminale NLS (AS
261-285) sowie eine zentrale, weite Sequenzabschnitte umfassende NLS (AS 31-253). Die
beiden letztgenannten sind nicht in der Lage, den Kernimport eines 5S RNPs zu vermitteln
(Claußen et al., 1999). Während die C-terminale NLS in einem unbekannten Ran-
unabhängigen Mechanismus den Import mediiert, ist der Kernimport über die zentrale
NLS Ran-abhängig (Rudt und Pieler, 2001). Diese Ran-Abhängigkeit ist ein Indiz für den
Kerntransport über ein bisher nicht identifiziertes Karyopherin. Die Tatsache, dass die
zentrale NLS keinen 5S RNP Import vermitteln kann, deutet darauf hin, dass sich die
Bindung des unbekannten Rezeptors und der 5S rRNA gegenseitig ausschließen. Aufgrund
der Größe der NLS (AS 31-253) ist es vorstellbar, dass der hierfür spezifische, unbekannte
Rezeptor eine stabilisierende Rolle, ähnlich der der 5S rRNA, für rpL5 spielt und den
Kernimport von rpL5 ohne 5S rRNA vermittelt. Der Nachweis eines
strukturstabilisierenden, chaperonartigen Einflusses von Importrezeptoren auf rpL5 könnte
ein Ziel weiterer Untersuchungen sein.
Für weitere Arbeiten mit Imp7 und Impß, insbesondere die Kristallisation betreffend, wäre
es wichtig, einen Nachweis für die Strukturstabilisierung von rpL5 zu erbringen. Wie auch
schon für rpL23a vorgeschlagen wurde, könnte der Nachweis der RNA-Bindefähigkeit von
Importrezeptor/rpL5-Komplexen hierfür Hinweise liefern. Auch die Überprüfung der
Temperaturinstabilität von Imp7/rpL5 wäre wichtig. Für weitere Kristallisationsversuche
mit Komplexen aus Importrezeptoren und rpL5 erscheint es in jedem Fall sinnvoll, auch

Diskussion
104
verkürzte N-terminale Fragmente von rpL5 zu verwenden. Dies träfe in besonderem Maße
zu, wenn sich zeigen würde, dass Imp7 keinen strukturstabilisierenden Einfluss auf rpL5
ausübt. Anhand einer Sekundärstrukturvorhersage mit dem Programm PSIPredict (2.1.10)
wurden bereits drei Expressionskonstrukte für die rpL5 Fragmente rpL5 (AS 1-48), rpL5
(AS 1-189) und rpL5 (AS 1-234) kloniert (Abbildung 39, 3.1.4). Das Fragment rpL5 (AS
1-48) konnte bereits gereinigt werden, eine Nachweis der Bindung durch Imp7 steht
jedoch noch aus (Daten nicht gezeigt).
Die Kristallisation von Komplexen aus Imp7 und ribosomalen Proteinen ist von hohem
Interesse, da bis zum heutigen Zeitpunkt keine Struktur von Imp7 aufgeklärt werden
konnte. Darüber hinaus gibt es keine veröffentlichte Struktur eines Kernimportrezeptors im
Komplex mit einem ribosomalen Protein. So könnten durch die Strukturaufklärung solcher
Komplexe auch Antworten auf die Frage nach der Rolle von Importrezeptoren als
Chaperone von ribosomalen Proteinen gefunden werden.

Zusammenfassung
105
5 Zusammenfassung
Im Verlaufe der Biogenese von Ribosomen wird eine Vielzahl von ribosomalen Proteinen
in den Zellkern importiert und nachfolgend zusammen mit verschiedenen rRNAs im
Nukleolus in neu entstehende ribosomale Untereinheiten integriert. Der Kernimport der
ribosomalen Proteine wird dabei durch Importrezeptoren der Importin-ß-Superfamilie
vermittelt. Die Rezeptoren binden die ribosomalen Proteine im Cytoplasma und mediieren
die Kernporenpassage des entstandenen Komplexes. Bekannte Mitglieder dieser Protein-
Superfamilie sind Impß und Imp7. Beide Rezeptoren sind in der Lage, den Import der
ribosomalen Proteine rpL5 und rpL23a zu vermitteln.
In dieser Arbeit wurde die Bindung der ribosomalen Proteine rpL5 und rpL23a an die
funktionellen Importrezeptoren Imp7 und Impß untersucht. Für diesen Zweck wurden
rpL5, rpL23a und die als BIB-Domäne bezeichnete Importrezeptorbindedomäne von
rpL23a kloniert, exprimiert und in hochreiner Form isoliert.
Die Untersuchung von Komplexen aus Imp7, Impß und den zuvor gereinigten ribosomalen
Proteinen, wurde über verschiedene Herangehensweisen angegangen. So wurde versucht
verschiedene Komplexe, bestehend aus Imp7 und den genannten ribosomalen Proteien, zu
kristallisieren, um eine Strukturaufklärung zu ermöglichen. Bisher konnte jedoch keine
Kristallbildung beobachtet werden.
Ferner konnten durch funktionelle Untersuchungen Erkenntnisse über die Interaktion von
Kernimportrezeptoren mit ribosomalen Proteinen gewonnen werden. So wurde durch
Kartierungsexperimente die Bindungsstelle für rpL23a in Impß eingegrenzt. Mit Hilfe von
isothermer Titrationskalorimetrie konnte gezeigt werden, dass die Bindung von Impß und
Imp7 an rpL23a in vitro einen endothermen Charakter besitzt. Des Weiteren wurden
Hinweise darauf gefunden, dass bestimmte Ansammlungen von sauren Aminosäuren in
niedrig strukturierten Sequenzabschnitten von Impß und Imp7 eine wichtige Funktion bei
der Bindung des ribosomalen Proteins rpL23a einnehmen. Diese sauren Aminosäuren
könnten auch eine Rolle bei der postulierten Chaperonfunktion von Importrezeptoren für
basische ribosomale Proteinen spielen.

Zusammenfassung
106
6 Summary
The process of ribosome biogenesis includes the import of ribosomal proteins into the
nucleus. The subsequent integration of both ribosomal proteins and different rRNAs into
the nascent ribosomal subunits takes place in the nucleolus. The nuclear import of
ribosomal proteins is mediated by import receptors of the importin-ß-superfamily. These
receptors bind to ribosomal proteins in the cytoplasm and mediate the passage of the
formed complexes through the nuclear pore complex. Prominent members of this protein-
superfamily are Imp7 and Impß. Both receptors are capable of importing the ribosomal
proteins rpL5 and rpL23a.
In this diploma thesis, the binding of the ribosomal proteins rpL5 and rpL23a to their
functional import receptors Imp7 and Impß was the focus of investigations. For this
purpose, the two ribosomal proteins and the import receptor binding domain (BIB-domain)
of rpL23a were cloned, expressed and isolated ultrapure. The investigation of complexes
consisting of Imp7, Impß and the previously purified ribosomal proteins was tackled by
different approaches. Crystallisation trials were carried out with complexes of Imp7 and
either rpL5, rpL23a or the BIB-domain in order to obtain crystals for structure
determination. However, a formation of crystals has not been observed so far.
Furthermore, functional investigations enlightened some properties of interactions between
importreceptors and ribosomal proteins. The binding site for rpL23a within Impß could be
mapped roughly. Using isothermal titration calorimetry, it could be shown that binding of
Impß and Imp7 to rpL23a is an endothermic process in vitro. Additionally, the interaction
studies with truncated fragments of Impß and Imp7 indicate the involvement of acidic
loops of Imp7 and Impß in the binding of the ribosomal protein rpL23a: These loops might
also be important for the postulated function of import receptors as chaperones for exposed
basic domains of ribosomal proteins.

Literaturverzeichnis
107
7 Literaturverzeichnis
Adam, E.J., Adam, S.A., 1994: Identification of cytosolic factors required for nuclear
location sequence-mediated binding to the nuclear envelope. J Cell Biol 125, 547-555.
Allison, L.A., North, M.T., Neville, L.A., 1995: Differential binding of oocyte-type and
somatic-type 5S rRNA to TFIIIA and ribosomal protein L5 in Xenopus oocytes:
specialization for storage versus mobilization. Dev Biol 168, 284-295.
Allison, L.A., Romaniuk, P.J., Bakken, A.H., 1991: RNA-protein interactions of stored 5S
RNA with TFIIIA and ribosomal protein L5 during Xenopus oogenesis. Dev Biol 144,
129-144.
Ban, N., Nissen, P., Hansen, J., Moore, P.B., Steitz, T.A., 2000: The complete atomic
structure of the large ribosomal subunit at 2.4 A resolution. Science 289, 905-920.
Bäuerle, M., Doenecke, D., Albig, W., 2002: The requirement of H1 histones for a
heterodimeric nuclear import receptor. J Biol Chem 277, 32480-32489.
Bearden, J.C., Jr., 1978: Quantitation of submicrogram quantities of protein by an
improved protein-dye binding assay. Biochim Biophys Acta 533, 525-529.
Ben-Efraim, I., Gerace, L., 2001: Gradient of increasing affinity of importin beta for
nucleoporins along the pathway of nuclear import. J Cell Biol 152, 411-417.
Bischoff, F.R., Klebe, C., Kretschmer, J., Wittinghofer, A., Ponstingl, H., 1994: RanGAP1
induces GTPase activity of nuclear Ras-related Ran. Proc Natl Acad Sci U S A 91, 2587-
2591.
Bischoff, F.R., Krebber, H., Smirnova, E., Dong, W., Ponstingl, H., 1995: Co-activation of
RanGTPase and inhibition of GTP dissociation by Ran-GTP binding protein RanBP1.
Embo J 14, 705-715.

Literaturverzeichnis
108
Bischoff, F.R., Ponstingl, H., 1991: Catalysis of guanine nucleotide exchange on Ran by
the mitotic regulator RCC1. Nature 354, 80-82.
Bischoff, F.R., Ponstingl, H., 1995: Catalysis of guanine nucleotide exchange of Ran by
RCC1 and stimulation of hydrolysis of Ran-bound GTP by Ran-GAP1. Methods Enzymol
257, 135-144.
Cheutin, T., O'Donohue, M.F., Beorchia, A., Vandelaer, M., Kaplan, H., Defever, B.,
Ploton, D., Thiry, M., 2002: Three-dimensional organization of active rRNA genes within
the nucleolus. J Cell Sci 115, 3297-3307.
Chook, Y.M., Blobel, G., 2001: Karyopherins and nuclear import. Curr Opin Struct Biol
11, 703-715.
Cingolani, G., Bednenko, J., Gillespie, M.T., Gerace, L., 2002: Molecular basis for the
recognition of a nonclassical nuclear localization signal by importin beta. Mol Cell 10,
1345-1353.
Cingolani, G., Petosa, C., Weis, K., Müller, C.W., 1999: Structure of importin-beta bound
to the IBB domain of importin-alpha. Nature 399, 221-229.
Claußen, M., Rudt, F., Pieler, T. 1999: Functional modules in ribosomal protein L5 for
ribonucleoprotein complex formation and nucleocytoplasmic transport. J Biol Chem 274,
33951-33958.
Conti, E., 2002: Structures of importins. Results Probl Cell Differ 35, 93-113.
Conti, E., Müller, C.W., Stewart, M. 2006: Karyopherin flexibility in nucleocytoplasmic
transport. Curr Opin Struct Biol 16, 237-244.
Cronshaw, J.M., Krutchinsky, A.N., Zhang, W., Chait, B.T., Matunis, M.J., 2002:
Proteomic analysis of the mammalian nuclear pore complex. J Cell Biol 158, 915-927.

Literaturverzeichnis
109
DiNitto, J.P., Huber, P.W. 2001: A role for aromatic amino acids in the binding of
Xenopus ribosomal protein L5 to 5S rRNA. Biochemistry 40, 12645-12653.
DiNitto, J.P., Huber, P.W., 2003: Mutual induced fit binding of Xenopus ribosomal protein
L5 to 5S rRNA. J Mol Biol 330, 979-992.
Doye, V., Hurt, E., 1997: From nucleoporins to nuclear pore complexes. Curr Opin Cell
Biol 9, 401-411.
Drivas, G.T., Shih, A., Coutavas, E., Rush, M.G., D'Eustachio, P. 1990: Characterization
of four novel ras-like genes expressed in a human teratocarcinoma cell line. Mol Cell Biol
10, 1793-1798.
Ehresmann, B., Imbault, P., Weil, J.H., 1973: Spectrophotometric determination of protein
concentration in cell extracts containing tRNA's and rRNA's. Anal Biochem 54, 454-463.
Fried, H., Kutay, U., 2003: Nucleocytoplasmic transport: taking an inventory. Cell Mol
Life Sci 60, 1659-1688.
Fukuhara, N., Fernandez, E., Ebert, J., Conti, E., Svergun, D., 2004: Conformational
variability of nucleo-cytoplasmic transport factors. J Biol Chem 279, 2176-2181.
Gill, S.C., von Hippel, P.H., 1989: Calculation of protein extinction coefficients from
amino acid sequence data. Anal Biochem 182, 319-326.
Görlich, D., Kutay, U., 1999: Transport between the cell nucleus and the cytoplasm. Annu
Rev Cell Dev Biol 15, 607-660.
Görlich, D., Mattaj, I.W., 1996: Nucleocytoplasmic transport. Science 271, 1513-1518.
Görlich, D., Pante, N., Kutay, U., Aebi, U., Bischoff, F.R., 1996: Identification of different
roles for RanGDP and RanGTP in nuclear protein import. Embo J 15, 5584-5594.

Literaturverzeichnis
110
Görlich, D., Seewald, M.J., Ribbeck, K., 2003: Characterization of Ran-driven cargo
transport and the RanGTPase system by kinetic measurements and computer simulation.
Embo J 22, 1088-1100.
Guddat, U., Bakken, A.H., Pieler, T., 1990: Protein-mediated nuclear export of RNA: 5S
rRNA containing small RNPs in xenopus oocytes. Cell 60, 619-628.
Harel, A., Forbes, D.J., 2004: Importin beta: conducting a much larger cellular symphony.
Mol Cell 16, 319-330.
Jäkel, S., Albig, W., Kutay, U., Bischoff, F.R., Schwamborn, K., Doenecke, D., Görlich,
D., 1999: The importin beta/importin 7 heterodimer is a functional nuclear import receptor
for histone H1. Embo J 18, 2411-2423.
Jäkel, S., Görlich, D., 1998: Importin beta, transportin, RanBP5 and RanBP7 mediate
nuclear import of ribosomal proteins in mammalian cells. Embo J 17, 4491-4502.
Jäkel, S., Mingot, J.M., Schwarzmaier, P., Hartmann, E., Görlich, D., 2002: Importins
fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed
basic domains. Embo J 21, 377-386.
Johnson, A.W., Lund, E., Dahlberg, J., 2002: Nuclear export of ribosomal subunits. Trends
Biochem Sci 27, 580-585.
Kalderon, D., Richardson, W.D., Markham, A.F., Smith, A.E., 1984: Sequence
requirements for nuclear location of simian virus 40 large-T antigen. Nature 311, 33-38.
Kiseleva, E., Goldberg, M.W., Cronshaw, J., Allen, T.D., 2000: The nuclear pore complex:
structure, function, and dynamics. Crit Rev Eukaryot Gene Expr 10, 101-112.
Lee, S.J., Sekimoto, T., Yamashita, E., Nagoshi, E., Nakagawa, A., Imamoto, N.,
Yoshimura, M., Sakai, H., Chong, K.T., Tsukihara, T., Yoneda, Y., 2003: The structure of

Literaturverzeichnis
111
importin-beta bound to SREBP-2: nuclear import of a transcription factor. Science 302,
1571-1575.
Lee, Y., Nazar, R.N., 2003: Terminal structure mediates 5 S rRNA stability and integration
during ribosome biogenesis. J Biol Chem 278, 6635-6641.
Lei, E.P., Silver, P.A., 2002: Protein and RNA export from the nucleus. Dev Cell 2, 261-
272.
Lin, E., Lin, S.W., Lin, A., 2001: The participation of 5S rRNA in the co-translational
formation of a eukaryotic 5S ribonucleoprotein complex. Nucleic Acids Res 29, 2510-
2516.
Lin, E., Liu, S.R., Lin, A., 1999: Biochemical analysis of a 5S rRNA-associated sub-
particle from trypsinized eukaryotic ribosomes. J Biochem (Tokyo) 125, 1029-1033.
Liu, S.M., Stewart, M., 2005: Structural basis for the high-affinity binding of nucleoporin
Nup1p to the Saccharomyces cerevisiae importin-beta homologue, Kap95p. J Mol Biol
349, 515-525.
Maden, B.E., 1990: The numerous modified nucleotides in eukaryotic ribosomal RNA.
Prog Nucleic Acid Res Mol Biol 39, 241-303.
Marechal, V., Elenbaas, B., Piette, J., Nicolas, J.C., Levine, A.J., 1994: The ribosomal L5
protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol 14, 7414-
7420.
Matsuura, Y., Stewart, M., 2004: Structural basis for the assembly of a nuclear export
complex. Nature 432, 872-877.
McIntosh, K.B., Bonham-Smith, P.C., 2001: Establishment of Arabidopsis thaliana
ribosomal protein RPL23A-1 as a functional homologue of Saccharomyces cerevisiae
ribosomal protein L25. Plant Mol Biol 46, 673-682.

Literaturverzeichnis
112
Melchior, F., Paschal, B., Evans, J., Gerace, L., 1993: Inhibition of nuclear protein import
by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as
an essential transport factor. J Cell Biol 123, 1649-1659.
Milkereit, P., Gadal, O., Podtelejnikov, A., Trumtel, S., Gas, N., Petfalski, E., Tollervey,
D., Mann, M., Hurt, E., and Tschochner, H., 2001: Maturation and intranuclear transport
of pre-ribosomes requires Noc proteins. Cell 105, 499-509.
Moore, M.S., Blobel, G., 1993: The GTP-binding protein Ran/TC4 is required for protein
import into the nucleus. Nature 365, 661-663.
Moore, M.S., Blobel, G., 1994: Purification of a Ran-interacting protein that is required for
protein import into the nucleus. Proc Natl Acad Sci U S A 91, 10212-10216.
Moroianu, J., Blobel, G., Radu, A., 1995: Previously identified protein of uncertain
function is karyopherin alpha and together with karyopherin beta docks import substrate at
nuclear pore complexes. Proc Natl Acad Sci U S A 92, 2008-2011.
Mosammaparast, N., Pemberton, L.F., 2004: Karyopherins: from nuclear-transport
mediators to nuclear-function regulators. Trends Cell Biol 14, 547-556.
Murdoch, K., Loop, S., Rudt, F., Pieler, T., 2002: Nuclear export of 5S rRNA-containing
ribonucleoprotein complexes requires CRM1 and the RanGTPase cycle. Eur J Cell Biol
81, 549-556.
Murdoch, K.J., Allison, L.A., 1996: A role for ribosomal protein L5 in the nuclear import
of 5S rRNA in Xenopus oocytes. Exp Cell Res 227, 332-343.
Nazar, R.N., 2004: Ribosomal RNA processing and ribosome biogenesis in eukaryotes.
IUBMB Life 56, 457-465.
Nigg, E.A., Baeuerle, P.A., Lührmann, R., 1991: Nuclear import-export: in search of
signals and mechanisms. Cell 66, 15-22.

Literaturverzeichnis
113
Oxenburgh, M.S., Snoswell, A.M., 1965: Use of streptomycin in the separation of nucleic
acids from protein in a bacterial extract. Nature 207, 1416-1417.
Pante, N., 2004: Nuclear pore complex structure: unplugged and dynamic pores. Dev Cell
7, 780-781.
Paschal, B.M., Gerace, L., 1995: Identification of NTF2, a cytosolic factor for nuclear
import that interacts with nuclear pore complex protein p62. J Cell Biol 129, 925-937.
Pemberton, L.F., Rosenblum, J.S., Blobel, G., 1999: Nuclear import of the TATA-binding
protein: mediation by the karyopherin Kap114p and a possible mechanism for intranuclear
targeting. J Cell Biol 145, 1407-1417.
Pittman, R.H., Andrews, M.T., Setzer, D.R., 1999: A feedback loop coupling 5 S rRNA
synthesis to accumulation of a ribosomal protein. J Biol Chem 274, 33198-33201.
Planta, R.J., Mager, W.H., 1998: The list of cytoplasmic ribosomal proteins of
Saccharomyces cerevisiae. Yeast 14, 471-477.
Reichelt, R., Holzenburg, A., Buhle, E.L., Jr., Jarnik, M., Engel, A., Aebi, U., 1990:
Correlation between structure and mass distribution of the nuclear pore complex and of
distinct pore complex components. J Cell Biol 110, 883-894.
Ribbeck, K., Lipowsky, G., Kent, H.M., Stewart, M., and Görlich, D., 1998: NTF2
mediates nuclear import of Ran. Embo J 17, 6587-6598.
Robbins, J., Dilworth, S.M., Laskey, R.A., Dingwall, C., 1991: Two interdependent basic
domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite
nuclear targeting sequence. Cell 64, 615-623.
Rosorius, O., Fries, B., Stauber, R.H., Hirschmann, N., Bevec, D., Hauber, J., 2000:
Human ribosomal protein L5 contains defined nuclear localization and export signals. J
Biol Chem 275, 12061-12068.

Literaturverzeichnis
114
Rout, M.P., Blobel, G., Aitchison, J.D., 1997: A distinct nuclear import pathway used by
ribosomal proteins. Cell 89, 715-725.
Rudt, F., Pieler, T., 1996: Cytoplasmic retention and nuclear import of 5S ribosomal RNA
containing RNPs. Embo J 15, 1383-1391.
Rudt, F., Pieler, T., 2001: Cytosolic import factor- and Ran-independent nuclear transport
of ribosomal protein L5. Eur J Cell Biol 80, 661-668.
Schatz, O., Oft, M., Dascher, C., Schebesta, M., Rosorius, O., Jaksche, H., Dobrovnik, M.,
Bevec, D., Hauber, J., 1998: Interaction of the HIV-1 rev cofactor eukaryotic initiation
factor 5A with ribosomal protein L5. Proc Natl Acad Sci U S A 95, 1607-1612.
Scripture, J.B., Huber, P.W., 1995: Analysis of the binding of Xenopus ribosomal protein
L5 to oocyte 5 S rRNA. The major determinants of recognition are located in helix III-loop
C. J Biol Chem 270, 27358-27365.
Seewald, M.J., Korner, C., Wittinghofer, A., Vetter, I.R., 2002: RanGAP mediates GTP
hydrolysis without an arginine finger. Nature 415, 662-666.
Smith, A., Brownawell, A., Macara, I.G., 1998: Nuclear import of Ran is mediated by the
transport factor NTF2. Curr Biol 8, 1403-1406.
Steitz, J.A., Berg, C., Hendrick, J.P., La Branche-Chabot, H., Metspalu, A., Rinke, J.,
Yario, T., 1988: A 5S rRNA/L5 complex is a precursor to ribosome assembly in
mammalian cells. J Cell Biol 106, 545-556.
Stewart, M., 2003: Structural biology. Nuclear trafficking. Science 302, 1513-1514.
Stoffler, D., Fahrenkrog, B., Aebi, U., 1999: The nuclear pore complex: from molecular
architecture to functional dynamics. Curr Opin Cell Biol 11, 391-401.

Literaturverzeichnis
115
Suzuki, K., Wool, I.G., 1993: The primary structure of rat ribosomal protein L23a. The
application of homology search to the identification of genes for mammalian and yeast
ribosomal proteins and a correlation of rat and yeast ribosomal proteins. J Biol Chem 268,
2755-2761.
Weis, K., 2003: Regulating access to the genome: nucleocytoplasmic transport throughout
the cell cycle. Cell 112, 441-451.
Wilson, D.N., Nierhaus, K.H., 2005: Ribosomal proteins in the spotlight. Crit Rev
Biochem Mol Biol 40, 243-267.
Wischnewski, J., Rudt, F., Pieler, T., 2004: Signals and receptors for the nuclear transport
of TFIIIA in Xenopus oocytes. Eur J Cell Biol 83, 55-66.
Wool, I.G., Chan, Y.L., Gluck, A., 1995: Structure and evolution of mammalian ribosomal
proteins. Biochem Cell Biol 73, 933-947.
Yokoyama, N., Hayashi, N., Seki, T., Pante, N., Ohba, T., Nishii, K., Kuma, K.,
Hayashida, T., Miyata, T., Aebi, U., et al., 1995: A giant nucleopore protein that binds
Ran/TC4. Nature 376, 184-188.
Yu, Y., Maggi, L.B., Jr., Brady, S.N., Apicelli, A.J., Dai, M.S., Lu, H., Weber, J.D., 2006:
Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol 26,
3798-3809.
Zhong, H., Takeda, A., Nazari, R., Shio, H., Blobel, G., Yaseen, N.R., 2005: Carrier-
independent nuclear import of the transcription factor PU.1 via RanGTP-stimulated
binding to Nup153. J Biol Chem 280, 10675-10682.

Abkürzungsverzeichnis
116
8 Abkürzungsverzeichnis
α alpha
A Absorption
A280nm Absorption bei 280 nm
Å Angström [1 Å = 0.1 nm]
AS Aminosäuren
ATP Adenosintriphosphat
Β beta
bp Basenpaare
bzgl. bezüglich
c Konzentration [M]
C Cytosin
° C Grad Celsius
cal Kalorie
CIP calf intestine phosphatase
cm Zentimeter
d Tage
Da Dalton [g/mol]
ddH2O bidestilliertes Wasser
ddNTP Didesoxynukleotidtriphosphat
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
dsDNA doppelsträngige DNA
E. coli Escherichia coli
EDTA N, N, N´, N´ Ethylendiamintetraactetat
et al. et altera
EtOH Ethanol
γ gamma
G Guanin
∆G Änderung der freien Energie [J/mol]
GSH reduziertes Glutathion

Abkürzungsverzeichnis
117
GST Glutathion-S-Transferase
GTP Guanosintriphosphat
h Stunden
∆H Änderung der Wärmeenthalpie [J/mol]
HEPES 4-(2-Hydroxyethyl)-piperazin-1-ethansulfonsäure
H. sapiens Homo sapiens
Imp Importin
IPTG Isopropyl-β-D-isothiogalactosid
ITC Isotherme Titrationskalorimetrie
k kilo (als präfix)
kb Kilobasen
Kd Dissoziationskonstante [M-1]
J Joule
λ Wellenlänge [nm]
µ mikro (als Präfix)
m milli (als präfix)
M Molarität
mAU Milliabsorptionseinheiten
mRNA Messenger RNA
MTase Methyltransferase
MW Molekulargewicht [g/mol]
n nano (als Präfix)
NPC nuclear pore complex, Kernporenkomplex
nPP nach PreScission-Protease(-Verdau)
OD Optische Dichte
PBS Phosphat-gepufferte Salzlösung
PCR Polymerasekettenreaktion
prä-rRNA rRNA-Vorläufer
Ran Ras-related nuclear antigen
RanBP Ran binding protein
RanGAP Ran GTPase activating protein
RanGEF Ran guanine nucleotide exchange factor
RNA Ribonukleinsäure

Abkürzungsverzeichnis
118
rpL ribosomal protein of the large subunit
rRNA ribosomal RNA
s Sekunde
∆S Änderung der Entropie [J/mol * K]
S. cerevisiae Saccharomyces cerevisiae
SDS Natriumdodecylsulfat
SDS-PAGE SDS-Polyacrylamidgelelektrophorese
snoRNA small nucleolar RNA
RNP Ribonucleoprotein
Taq Thermus aquaticus
TBE Tris-Borsäure-EDTA-Puffer
TEMED N, N, N´, N´ Tetramethylethylendiamin
Tm Schmelztemperatur
Tris Tris(hydroxymethyl)-aminomethan
tRNA transfer RNA
U Unit (Einheit der Enzymaktivität)
UV Ultraviolett
V Volumen
vgl. vergleiche
v/v Volumenprozent (volume per volume)
w/v Gewichtsprozent (weight per volume)
X. laevis Xenopus laevis
xg Vielfaches der Erdbeschleunigung (g = 9,81 m/s2)
z.B. zum Beispiel
ε molarer Extinktionskoeffizient [M-1 cm-1]

Danksagung
119
9 Danksagung
Herrn Prof. Dr. Ralf Ficner danke ich für die Überlassung dieses interessanten Themas, die
hervorragenden Arbeitsbedingungen und die Förderung der Arbeit durch wertvolle
Diskussionen und fachliche Anregungen.
Herrn Prof. Dr. Oliver Einsle danke ich sehr herzlich für die Übernahme des Korreferates.
Dr. Achim Dickmanns bin ich für die stets motivierende Begleitung und fachliche
Unterstützung dieser Arbeit dankbar.
Mein herzlicher Dank gilt Daniel Wohlwend für die hervorragende Betreuung dieser
Arbeit, die vielen fachlichen Diskussionen, Ratschläge, Ideen und aufmunternden Worte.
Frau Dr. Anja Strasser und Thomas Monecke danke ich für die Durchsicht des
Manuskriptes sowie für die vielen großen und kleinen Hilfen und die nette Atmosphäre im
Laboralltag.
Winfried Lehndeckel danke ich für die Ratschläge bei vielen technischen Fragen.
Mein ganz persönlicher Dank gilt besonders Britta.

Danksagung
120





























