Embed Size (px)
Citation preview

UNIVERSITÀ DEGLI STUDI DI CATANIA
DOTTORATO DI RICERCA IN FISICA
ANTONINO MARIA DELFINO COSENTINO
SPETTROSCOPIA RAMAN PER I BENI CULTURALI. IL PROGETTO COLORAMAN
TESI PER IL CONSEGUIMENTO DEL TITOLO
XVII CICLO

UNIVERSITÀ DEGLI STUDI DI CATANIA
DOTTORATO DI RICERCA IN FISICA
ANTONINO MARIA DELFINO COSENTINO
SPETTROSCOPIA RAMAN PER I BENI CULTURALI. IL PROGETTO COLORAMAN
TESI PER IL CONSEGUIMENTO DEL TITOLO
Il Coordinatore (Prof. Antonio Insolia)
Il Tutor (Prof. Sebastiano Olindo Troja)
XVII CICLO

Sommario I
Sommario Introduzione............................................................................................................................... I Capitolo 1. La spettroscopia Raman per i Beni Culturali........................................................1
1.1 Le indagini fisiche per i Beni Culturali ............................................................................1 1.2 Approccio complementare................................................................................................1 1.3 I Database ........................................................................................................................2 1.4 Ambre e resine fossili .......................................................................................................4 1.5 Copali ...............................................................................................................................5 1.6 Resine ...............................................................................................................................5 1.7 Materiali organici .............................................................................................................6
Medicamenti .......................................................................................................................6 Protettivi lapidei .................................................................................................................6 Pergamene e vella...............................................................................................................7 Bio-deterioramento.............................................................................................................7 Mummificazione.................................................................................................................7
1.8 Prodotti della corrosione dei metalli.................................................................................7 1.9 Minerali e gemme.............................................................................................................8 1.10 Vetri e mosaici in pasta vitrea ........................................................................................9 1.11 Ossidiane ........................................................................................................................9 1.12 Ceramiche.......................................................................................................................9 1.13 Pigmenti........................................................................................................................11
Pigmenti puri ....................................................................................................................11 Pigmenti su pitture murali ...............................................................................................12 Pigmenti su vetro..............................................................................................................13 Pigmenti su manoscritti ....................................................................................................13 Pigmenti stesi ad olio........................................................................................................14 Pigmenti e inchiostri su carta ...........................................................................................14 Pigmenti su pietra .............................................................................................................15 Pigmenti su papiri.............................................................................................................15 Pigmenti dell’arte primitiva rupestre................................................................................15 Pigmenti stesi a tempera e ad acquarello..........................................................................16 Pigmenti su tessuti ............................................................................................................16 Pigmenti su elementi architettonici ..................................................................................16
1.14 Altre metodologie Raman.............................................................................................16 Capitolo 2. Introduzione all’effetto Raman............................................................................18
2.1 Introduzione....................................................................................................................18 2.2 L’energia molecolare......................................................................................................18 2.3 I gradi di libertà del moto molecolare ............................................................................18 2.4. Modi normali di vibrazione ...........................................................................................19 2.5 Modelli molecolari meccanici ........................................................................................19 2.6 Coordinate usate nella descrizione delle vibrazioni molecolari .....................................20 2.7 Formula classica per la frequenza vibrazionale di una molecola diatomica. .................21 2.8 Assorbimento infrarosso e variazione del momento di dipolo .......................................23 2.9 Anarmonicità e ipertoni ..................................................................................................24 2.10 La funzione potenziale vibrazionale.............................................................................25 2.11 Transizioni vibrazionali e assorbimento infrarosso......................................................25 2.12 La rotazione delle molecole lineari ..............................................................................26 2.13 Transizioni rotazionali e assorbimento infrarosso........................................................27 2.14 Intensità delle linee rotazionali.....................................................................................31

Sommario II
2.15 Spettro roto-vibrazionale. Spiegazione classica. ..........................................................32 2.16 Spettro Roto-vibrazionale: trattazione quanto-meccanica............................................33 2.17 Spettro Raman: generalità. ...........................................................................................36 2.18 Spettro Raman vibrazionale .........................................................................................37 2.19 Spettro Raman rotazionale............................................................................................39 2.20 L’origine vibrazionale delle frequenze di gruppo ........................................................43
Oscillatori diatomici .........................................................................................................43 Oscillatori accoppiati........................................................................................................43 Oscillatori accoppiati asimmetrici ....................................................................................46
2.21 La Fluorescenza............................................................................................................47 Capitolo 3. Strumentazione per Raman dispersivo ................................................................50
3.1 Introduzione....................................................................................................................50 3.2 Le sorgenti ......................................................................................................................50 3.3 Lo spettroscopio .............................................................................................................51 3.4 I sistemi di campionamento............................................................................................54 3.5 I rivelatori .......................................................................................................................57 3.6 I sistemi di gestione ed elaborazione dati.......................................................................58 3.7 Il problema della fluorescenza.......................................................................................58
Capitolo 4. Progetto ColoRaman: presentazione ...................................................................61 4.1 Introduzione....................................................................................................................61 4.2 Peaks finder ....................................................................................................................61 4.3 Il sito internet..................................................................................................................75 4.4 Apparato sperimentale....................................................................................................82
Capitolo 5. Progetto ColoRaman: risultati .............................................................................86 5.1 Introduzione....................................................................................................................86 5.2 Pigmenti e colori. Introduzione ......................................................................................86
5.2.1 Colore, pigmento e colorante...................................................................................86 5.2.2 Le lacche..................................................................................................................87
5.3 Il medium........................................................................................................................87 5.3.1 Leganti e film pittorico............................................................................................87 5.3.2 Vernici .....................................................................................................................89
5.4 Il database dei pigmenti pittorici stesi ..........................................................................89 5.4.1 Introduzione.............................................................................................................89 5.4.2. Preparazione dei campioni......................................................................................90 5.4.3. Risultati e discussione ............................................................................................93
5.5 Analisi P2S2 (Pigments’ Peaks’ Shifts Survey) ............................................................100 5.5.1 Introduzione...........................................................................................................100 5.5.2 Risultati e discussione ...........................................................................................103
5.6 La sezione on-line del progetto ColoRaman ................................................................107 Capitolo 6. Progetto ColoRaman: verifica............................................................................112
6.1 Introduzione..................................................................................................................112 6.2 Tempera ........................................................................................................................112
6.2.1 Chiesa di Sant’Antonio Abate in Avola ................................................................112 6.3 Affresco ........................................................................................................................118
6.3.1 Chiesa di S. Egidio ................................................................................................118 6.4 Olio ...............................................................................................................................119
6.4.1 San Gerolamo, olio su tela, museo di Militello in val di Catania..........................119 Conclusioni ............................................................................................................................121 Bibliografia ............................................................................................................................122

Introduzione I
Introduzione L’attività di ricerca svolta nel corso di questo dottorato ha avuto per obiettivo la definizione delle più opportune metodologie d’impiego della spettroscopia Raman per la conservazione, il restauro e lo studio dei Beni Culturali e lo sviluppo delle implementazioni volte a superarne gli attuali limiti di applicazione. La parte più importante di questo lavoro è stata denominata progetto ColoRaman. La spettroscopia Raman (RS) è utilizzata con successo per le indagini sui BB.CC. Perché essa, però, potesse affermarsi pienamente quale metodologia diagnostica, non sembrava più sufficiente la produzione di contributi scientifici riguardanti casi specifici, ma, piuttosto, era parso necessario fornire, ad esempio, dettagliate indicazioni sui materiali che potevano essere identificati, sul tipo di manufatti che era possibile analizzare e soprattutto, per ogni tipo di oggetto, sulla più opportuna implementazione dell’apparato strumentale. Lo sviluppo del progetto ColoRaman si proponeva di fornire queste informazioni relativamente ad una particolare classe di materiali d’interesse per lo studio dei Beni artistici, quella dei pigmenti pittorici. Una introduzione alla storia dell’applicazione della RS alle indagini sui BB.CC, è presentata nel cap. 1. Si ricorda come a partire dalla fine degli anni ’80, in seguito ai progressi tecnici, cominciarono ad apparire sulle riviste scientifiche review dei lavori realizzati su oggetti d’interesse storico-artistico. Gli strumenti diventarono, contemporaneamente, sempre più accessibili sia economicamente che come utenza e un numero sempre crescente di musei cominciò a dotarsi di almeno un sistema Raman oltre che di uno staff di tecnici specializzati. Il cap. 1 propone, inoltre, alcuni dei lavori più rappresentativi della ricerca compiuta in ambito accademico. Data la gran varietà d’archeomateriali e, in generale, di oggetti d’interesse storico artistico esaminati, si è preferito presentare questi lavori suddividendoli in base al materiale o al tipo di manufatto, così da estrapolare e offrire al lettore solo gli aspetti più interessanti. Il cap. 2 fornisce gli strumenti indispensabili per la comprensione del fenomeno fisico alla base di questa metodologia. Dopo aver affrontato la trattazione dell’interazione delle onde elettromagnetiche con la materia ed, in particolare, con le rotazioni e vibrazioni delle molecole, ci si sofferma sull’effetto Raman, fornendo qualche esempio dei metodi d’interpretazione vibrazionale degli spettri. Il cap. 3 descrive la strumentazione per spettroscopia Raman dispersiva e l’apparato sperimentale utilizzato per il progetto ColoRaman mentre il cap.4 introduce al lavoro complementare volto all’ottimizzazione e diffusione delle conoscenze acquisite tramite la realizzazione del sito internet e di utilities quali il software peaks finder. Il cap. 5 introduce il progetto ColoRaman il cui obiettivo è stato testare le effettive potenzialità di un sistema combinato per spettroscopia Raman e di fluorescenza, dotato di tre laser, per la caratterizzazione dei pigmenti pittorici secchi ma soprattutto dei relativi colori già stesi, intendendosi con colore la miscela di pigmento e legante. I pigmenti secchi, utilizzati come standard di riferimento, vengono caratterizzati nelle migliori condizioni ma non altrettanto è possibile quando li si vuole identificare già stesi su un manufatto come colore. Infatti, i leganti ed i supporti pittorici, quasi sempre di origine organica, sono responsabili di una fluorescenza in molti casi così intensa da coprire il debole segnale Raman originato dal pigmento, rendendo così impossibile l’identificazione di quest’ultimo. Si dimostra, tuttavia, che è spesso possibile stabilire per ogni pigmento e per ogni singola tecnica pittorica la lunghezza d’onda di eccitazione capace o di diminuire l’intensità della fluorescenza oppure di generare una intensa emissione Raman così da migliorare il rapporto segnale-rumore, essendo in questo caso il rumore costituito dalla emissione di fluorescenza.

Introduzione II
Nel capitolo vengono presentati i database di spettri Raman di pigmenti pittorici secchi e già stesi con le tecniche pittoriche dell’affresco, della tempera all’uovo, della tempera alla caseina e dell’olio. Oltre al database di spettri Raman è stato realizzato anche quello degli spettri di fluorescenza. Benché questi ultimi possano essere facilmente acquisiti, praticamente con lo stesso sistema adoperato per la spettroscopia Raman dispersiva, finora sono stati pochi i lavori che hanno indagato la caratterizzabilità dei pigmenti pittorici anche tramite i loro spettri di fluorescenza. Tra l’altro, questa metodologia può risultare particolarmente utile perché, diversamente da quanto avviene per il debole segnale Raman, per l’emissione di fluorescenza il rapporto segnale-rumore è, in genere, così alto da permettere l’identificazione del pigmento già steso. Il capitolo affronta anche il problema della compatibilità degli spettri dei pigmenti secchi con quelli dei pigmenti stesi. Infatti, era ragionevole aspettarsi che, a causa della sovrapposizione della fluorescenza del legante e del supporto, gli spettri di uno stesso pigmento secco e steso potessero presentare delle differenze in termini di posizione dei picchi. Inoltre, a causa della interazione pigmento-legante, era lecito aspettarsi, oltre agli spostamenti dei picchi, anche delle variazioni delle loro intensità relative. La corretta attribuzione degli spettri richiedeva di poter conoscere, nell’eventualità che fossero presenti, l’entità di queste differenze. Il passo successivo è stato il test dei risultati del progetto ColoRaman e delle sue implementazioni software su reali oggetti d’arte. Il cap. 6 propone degli esempi d’indagini, realizzate su manufatti decorati con alcune delle tecniche pittoriche esaminate, i cui risultati sono stati, di volta in volta, confrontati con quelli contenuti nei database.

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 1
Capitolo 1. La spettroscopia Raman per i Beni Culturali
1.1 Le indagini fisiche per i Beni Culturali Le indagini sui Beni Culturali (BB.CC.) si servono, in genere, di documenti e di fonti storiche. Da questi gli studiosi e in particolare gli storici dell’Arte riescono a trarre informazioni circa l’età e la provenienza di un manufatto nonché circa le tecnologie impiegate per crearlo. Nel caso che non ci siano, però, dati sufficienti o che questi siano contrastanti, le metodologie di indagine fisico-chimiche possono fornire informazioni scientificamente sicure. La spettroscopia Raman A partire dalla fine degli anni ’80, in seguito ai progressi tecnici, cominciarono ad apparire sulle riviste scientifiche review sulle possibili applicazioni della spettroscopia Raman (RS) [AA.VV 1989, Gerrard 1994, Huong 1996, Lawson et al. 1997, Lyin et al. 1998]. Una review specificamente dedicata all’indagine sui pigmenti pittorici comparve già nel 1992 [Best et al. 1992] e a questa ne seguirono molte altre [Clark 1999, Clark 2002, Vandenabeele 2004]. Gli strumenti per RS diventano sempre più accessibili sia economicamente che come utenza e un numero sempre crescente di musei possiede almeno un sistema Raman oltre che uno staff di tecnici specializzati. È ragionevole credere che nel prossimo decennio questo campo di ricerca continuerà a crescere grazie allo sviluppo, ad esempio, delle strumentazioni mobili a fibre ottiche ma anche dei software per l'elaborazione dei dati. In altre parole, mentre la RS fino a qualche tempo fa era l'appannaggio solo dei più sofisticati laboratori, oggi, le istituzioni accademiche lavorano sempre più a contatto con gli operatori culturali. Gli staff dei musei e gli storici dell'Arte sono interessati all’analisi di differenti categorie di oggetti e il numero di manufatti da analizzare è enorme. La cooperazione tra, da una parte, gli storici dell'Arte, gli operatori museali, gli scienziati addetti alla conservazione e gli archeologi e dall’altra, gli spettroscopisti, i chimici e i geologi sembra sempre più produttiva e indispensabile. Questo capitolo presenta alcuni dei lavori più rappresentativi dello sforzo compiuto in ambito accademico per comprendere le reali potenzialità della RS relativamente alle indagini sui BB.CC. Data la grande varietà di archeomateriali e, in generale, di oggetti d’interesse storico artistico esaminati, si è preferito presentare questi lavori suddividendoli in base al materiale o al tipo di manufatto, così da estrapolare e offrire al lettore solo gli aspetti più interessanti. Per ogni categoria, i lavori sono stati a loro volta divisi in due gruppi, relativamente al tipo di strumentazione adoperata, FT-Raman o Raman, intendendosi con questo ultimo termine il Raman dispersivo. È sembrato opportuno, infatti, permettere al lettore di riscontrare quale strumentazione è da considerarsi, al momento, più adatta per ciascun tipo di applicazione. Ovviamente, c’è da attendersi che con le nuove implementazioni hardware verranno a cadere molti dei limiti tecnici che attualmente costringono l’applicazione dell’FT-Raman o del Raman dispersivo solo ad alcuni campi. Ad esempio, sta emergendo da alcuni studi che l’FT-Raman, grazie allo sviluppo di teste di campionamento remote dotate di fibre ottiche, potrà essere applicato con successo anche in situ. Dell’applicazione delle altre metodologie Raman, che compaiono soltanto raramente e che attualmente non sono direttamente adatte alle indagini sui BB.CC, non verificando, in particolare, le condizioni di non-distruttività e/o di applicabilità in situ, si parlerà in un paragrafo a loro dedicato.
1.2 Approccio complementare È doveroso puntualizzare che, sebbene la spettroscopia Raman si presti ottimamente alle indagini per i BB.CC., la quantità e qualità di dati che se ne possono ottenere è certamente
Capitolo 1. La Spettroscopia Raman per i Beni Culturali 2
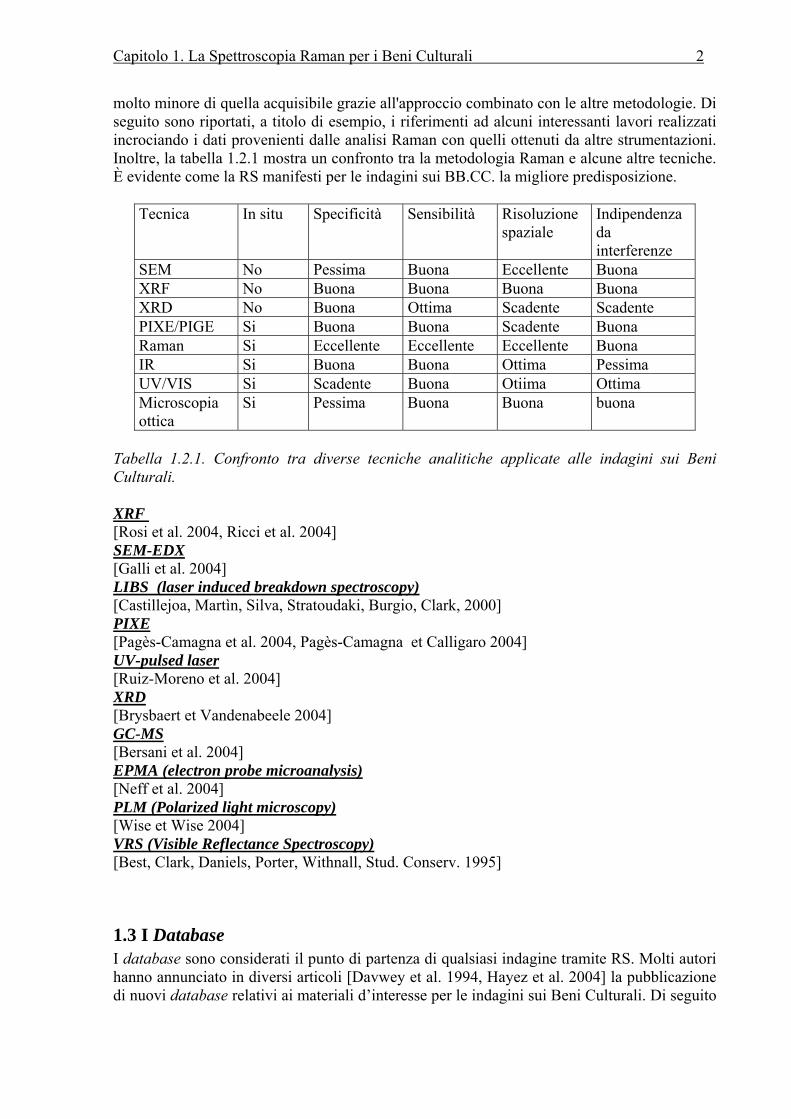
molto minore di quella acquisibile grazie all'approccio combinato con le altre metodologie. Di seguito sono riportati, a titolo di esempio, i riferimenti ad alcuni interessanti lavori realizzati incrociando i dati provenienti dalle analisi Raman con quelli ottenuti da altre strumentazioni. Inoltre, la tabella 1.2.1 mostra un confronto tra la metodologia Raman e alcune altre tecniche. È evidente come la RS manifesti per le indagini sui BB.CC. la migliore predisposizione.
Tecnica In situ Specificità Sensibilità Risoluzione spaziale
Indipendenza da interferenze
SEM No Pessima Buona Eccellente Buona XRF No Buona Buona Buona Buona XRD No Buona Ottima Scadente Scadente PIXE/PIGE Si Buona Buona Scadente Buona Raman Si Eccellente Eccellente Eccellente Buona IR Si Buona Buona Ottima Pessima UV/VIS Si Scadente Buona Otiima Ottima Microscopia ottica
Si Pessima Buona Buona buona
Tabella 1.2.1. Confronto tra diverse tecniche analitiche applicate alle indagini sui Beni Culturali. XRF [Rosi et al. 2004, Ricci et al. 2004] SEM-EDX [Galli et al. 2004] LIBS (laser induced breakdown spectroscopy) [Castillejoa, Martìn, Silva, Stratoudaki, Burgio, Clark, 2000] PIXE [Pagès-Camagna et al. 2004, Pagès-Camagna et Calligaro 2004] UV-pulsed laser [Ruiz-Moreno et al. 2004] XRD [Brysbaert et Vandenabeele 2004] GC-MS [Bersani et al. 2004] EPMA (electron probe microanalysis) [Neff et al. 2004] PLM (Polarized light microscopy) [Wise et Wise 2004] VRS (Visible Reflectance Spectroscopy) [Best, Clark, Daniels, Porter, Withnall, Stud. Conserv. 1995]
1.3 I Database I database sono considerati il punto di partenza di qualsiasi indagine tramite RS. Molti autori hanno annunciato in diversi articoli [Davwey et al. 1994, Hayez et al. 2004] la pubblicazione di nuovi database relativi ai materiali d’interesse per le indagini sui Beni Culturali. Di seguito

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 3
elenchiamo i database attualmente disponibili su rivista e/o su internet. Caratteristiche peculiari di questi database sono le lunghezze d’onda d’eccitazione utilizzate, la possibilità di scaricare gli spettri e i criteri per la loro ricerca nel caso dei database on-line. Database di spettri Raman di materiali d’interesse per i BB.CC. IRUG (http://www.irug.org/) Il portale dell’IRUG, Infrared and Raman users group permette di accedere al database di spettri Raman e infrarossi di vari materiali d’interesse per i BB.CC. (solo in formato PDF). Gli spettri sono messi a disposizione dai soci e quindi le lunghezze d’onda ed i sistemi sono diversi. Attualmente, però, gli spettri Raman sono molto pochi anche se il numero di spettri infrarossi è notevole. La ricerca avviene per nome. [Vandenabeele et al. 2000] È un database di spettri Raman ottenuti con eccitazione a 780 nm e raccoglie gli spettri di leganti e vernici utilizzati per la realizzazione di manufatti d’interesse storico-artistico. [Burgio et Clark 2001] Database di spettri FT-Raman (1064 nm) di minerali, pigmenti, leganti e vernici. [Bouchard et Smith 2003] Raccoglie gli spettri Raman ottenuti con eccitazione a 514.5 e 632.8 nm di 45 minerali d’interesse per la storia dell’Arte, l’Archeologia e, in particolare, lo studio della corrosione dei metalli e della realizzazione dei vetri colorati. [Edwards, Farwell, Daffner 2003] Spettri FT-Raman, ottenuti con eccitazione a 1064 nm, di cere e resine naturali. Database di minerali [Coleyshaw, Griffit, Bowell 1994] Spettri FT-Raman di minerali. Uno dei primi lavori realizzati sui minerali con questo sistema. CalTech, California Institute of Technology (http://minerals.gps.caltech.edu) Permette di scaricare gli spettri Raman, ottenuti con eccitazione a 514.5, 782, 785 nm di centinaia di minerali. Benché non concepito per le indagini sui BB.CC. costituisce un’ottima sorgente di informazioni. La ricerca avviene per nome. AIST, Advanced Industrial Science and Technology (http://www.aist.go.jp) Anche questo database non è stato concepito per le indagini sui Beni Culturali. Fornisce spettri Raman acquisiti solo a 514.5 nm ma il numero di minerali analizzati è molto vasto. Gli spettri sono scaricabili e la ricerca avviene per classe mineralogica. Dipartimento di Fisica, Università di Parma (http://www.fis.unipr.it ) Gli spettri, acquisiti a 632.8 nm, sono scaricabili e la ricerca avviene per classe mineralogica. Database di pigmenti pittorici Christopher Ingold Laboratories, Department of Chemestry, University College London (http://www.chem.ucl.ac.uk/resources/raman/speclib.html)

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 4
[Bell, Clark, Gibbs 1997] É il primo database di pigmenti pittorici ad essere stato pubblicato on-line ed è il punto di riferimento per questo tipo di indagini. Gli spettri sono ottenuti con eccitazione a 514.5 e 632.8 nm e sono scaricabili. La ricerca avviene per nome del pigmento LENS, European Laboratory for Non Linear Spectroscopy, Firenze (http://www.chim.unifi.it/raman) Gli spettri, ottenuti con eccitazione a 488, 514.5 e 647 nm, sono solo visualizzabili in PDF. La ricerca avviene per nome del pigmento. [Vandenabeele, Moens, Edwards, Dams 2000] Database di spettri Raman acquisiti con eccitazione a 780 nm di pigmenti moderni.
1.4 Ambre e resine fossili L’ambra è la resina fossilizzata del pinus succinifera e viene raccolta principalmente sulle spiagge del mar Baltico. Le resine non provenienti dal mar Baltico vengono dette genericamente resine fossili. L’ambra è la più dura resina conosciuta e può essere dispersa nell’olio per ottenerne una vernice. La sua formula viene spesso riportata come C10H16O [Mark et al. 1969] ma con il rapporto C:H:O variabile. L’esatta composizione chimica dell’ambra non è nota, ma si è certi della presenza dell’acido succinico nonché di zolfo fino all’1%. Molte tecniche analitiche sono state applicate allo studio delle ambre, in particolare, la spettroscopia infrarossa, la spettroscopia ultravioletta-visibile, la gas-cromatografia e la spettroscopia di massa [Beck et al. 1993, Galletti et al. 1993, Savkevich 1981, Beck 1968]. In particolare, la spettroscopia infrarossa è stata utilizzata con successo per l’identificazione e la caratterizzazione di ambre e resine fossili provenienti da diverse regioni, dal mar Baltico ai mari caraibici [Nissenbaum et al. 1993, Srebrodol’skii 1986]. Tutte queste tecniche richiedono però la manipolazione delle resine con solventi organici o la loro decomposizione tramite riscaldamento o acidi. La RS si propone, invece, come alternativa non distruttiva ed in situ in tutti quei casi che devono escludere la distruzione del campione. FT-Raman: caratterizzazione L’indagine tramite FT-Raman permette di ottenere dalle ambre spettri caratteristici [Edwards 1996, Moreno et al. 2000]. In particolare è possibile distinguere le ambre dalle altre resine fossili in quanto l’acido succinico ha delle bande osservabili ( 1420 ( δ (CH2) ) e 937 ( ν (CC) ) cm-1 ) così come anche lo zolfo ( 504 (ν (SS) ), 651 ( v ( CS ) ) cm-1 ). Le ambre e le resine fossili sono a loro volta distinguibili dalle resine contemporanee non maturate grazie alle seguenti caratteristiche:
1) Assenza della banda a 3020 cm-1 relativa alla presenza del gruppo –C=CH- 1) Minore intensità della banda a 2850 cm-1 dovuta alla diminuzione del contenuto in –
CH3 2) Assenza delle bande a 1612, 760, 740, 713 cm-1 3) Minore rapporto dell’intensità relativa del picco a 1646 cm-1 ( ν (C=C) ) rispetto a
quello a 1450 cm-1 ( δ (CH2) ) dovuto al progressivo deterioramento del legame ν (C=C) (il valore di questo rapporto è 1.7 per le resine di pino non maturate, 1.0 per le copali, 0.5-0.6 per le ambre).
È inoltre possibile distinguere, tramite l’analisi dei loro spettri, le ambre dalle loro imitazioni, in particolare, quelle realizzate a base di acrilati, come il pmma, o di uretani.

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 5
Oltre che delle resine fossili della I e II classe (terpenoidi) è documentata anche la caratterizzazione delle più rare resine della III classe, i polistireni naturali, quale la siegburgite [Winkler et al. 2003]. La determinazione dell’origine geologica (ambiente in cui è avvenuta la maturazione) e/o botanica delle resine è stata tentata, portando ad alcuni importanti risultati [Moreno 2001]. Tuttavia, alcuni autori hanno riscontrato che le caratteristiche degli spettri delle resine fossili devono essere messe in relazione, non solo alla loro origine botanica, ma contemporaneamente anche alle condizioni geologiche in cui è avvenuta la loro maturazione, cosicché, per tentare l’attribuzione geografica e botanica, sembra necessaria la realizzazione di un database [Jehlicka et al. 2004]. FT-Raman: datazione È stato osservato, innanzitutto, che è possibile riconoscere le ambre “giovani” grazie all’assenza delle bande a 3040 ( ν (CH) ) e 1000 ( ν (CC) ) cm-1 [Edwards 1996]. Molto più interessante è il metodo di datazione delle ambre e delle resine fossili, proposto a più riprese [Shen et al. 1997, Winkler et al. 1998, Winkler et al. 2001, Brody et al. 2001], che si basa sulla correlazione tra l’età della resina e l’intensità del rapporto ( 1640/1440 cm-1 ). La variazione dell’intensità di questo rapporto con l’età è stata accertata per resine fossili dal Cretaceo fino all’età contemporanea ma essa non può essere utilizzata come un metodo assoluto di datazione in quanto, come già detto, responsabili delle variazioni dell’intensità di questo rapporto possono essere anche altri fattori quali, ad esempio, le condizioni geologiche di maturazione della resina. Tuttavia, si può affermare, ancora una volta, che un database con spettri di riferimento potrebbe essere usato per una datazione relativa.
1.5 Copali Per copali s’intendono, in generale, le resine di origine vegetale. Talvolta però col termine copale vengono indicate anche le ambre non maturate. Raman Le indagini condotte con eccitazione a 785 nm permettono di ottenere spettri caratteristici, in particolare, di copali messicane [Vandenabeele et al. 2003].
1.6 Resine Col termine resine si intendono tutte quelle estratte da piante o da animali e che vengono usate come agenti decorativi, protettivi o coloranti. La conoscenza della natura della resina presente su di un manufatto è estremamente importante per l’identificazione dei processi chimici che possono aver luogo a seguito della loro degradazione dovuta, a sua volta, alle condizioni di interramento o di esposizione ambientale. L’identificazione delle resine può essere essenziale per la determinazione della provenienza e per la conoscenza delle vie commerciali, delle culture e delle tecnologie materiali. Lo studio delle resine antiche è inoltre utile ai tecnici del restauro che possono così adottare le più opportune metodologie per la loro rimozione o per il loro reintegro. Un caso a sé, particolarmente importante è quello dei leganti pittorici e delle vernici Molti problemi relativi alla conservazione dei dipinti nascono, infatti, dalla degradazione del legante pittorico e/o della vernice. In molti casi le analisi eseguite sui dipinti mirano solo all’identificazione dei pigmenti e per tale motivo esse spesso sono realizzate usando solo la XRF o metodologie correlate. Per quanto riguarda invece l’analisi dei leganti e delle vernici, i metodi a raggi X sono considerati

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 6
inadatti per questo tipo di indagini. Infatti, con queste tecniche gli elementi leggeri non possono essere rilevati e l’analisi elementale non è sufficiente a distinguere tra queste sostanze organiche. Tuttavia, è stato dimostrata la possibilità di caratterizzare diverse vernici tramite la concentrazione degli elementi più pesanti, ottenuta tramite la fluorescenza X a riflessione totale. Nonostante questo metodo vada bene per le vernici, cosiddette per violino, non è, però, adatto per le vernici dei dipinti dove gli elementi presenti nelle particelle dei pigmenti mascherano quelli delle vernici. Per quanto riguarda poi i metodi di separazione (come la gas-cromatografia, l’elettroforesi capillare e la cromatografia liquida) e la spettroscopia di massa, esse risultano molto efficienti per l’analisi dei leganti, ma l’ampio consumo di campione e la sua complessa preparazione ne costituiscono un grosso limite. In questo tipo di esami il campione viene distrutto, rendendo così impossibile ripetere e confermare le analisi. Concludendo, anche se i metodi spettroscopici vibrazionali (IR e Raman ) soffrono, rispetto a molte delle tecniche cromatografiche discusse, di limiti maggiori, essi risultano più adatti per le analisi non distruttive ed in-situ dei leganti. Raman Un database di resine ma anche di leganti e vernici pittoriche è stato realizzato con eccitazione laser a 780 nm. Sono state altresì forniti esempi di analisi su manufatti verniciati. [Vandenabeele et al. 2000]. FT-Raman Un database di molte tipologie di resine, in particolare di cere è stato presentato nel 1996 [Edwards, Farwell, Daffner 1996]. Studi specifici sono stati svolti sulle resine utilizzate dagli indiani d’America [Edwards 1997] e sulle resine archeologiche provenienti da giare funerarie vietnamite [Edwards, Sibley et al. 1997].
1.7 Materiali organici Esiste una ampia letteratura sull’analisi Raman e soprattutto FT-Raman di diversi archeomateriali organici.
Medicamenti FT-Raman Il contenuto di una cassa di medicamenti di una nave inglese del XVI secolo è stato studiato tramite FT-Raman permettendo l’individuazione di miscele di resine e di composti a base di solfati, carbonati e ossidi [Edwards et al. 2004].
Protettivi lapidei Il micro-consolidamento e la protezione totale delle superfici esterne sono i principali obiettivi di ogni intervento conservativo su superfici lapidee naturali o parte di oggetti d’arte. Alcune sostanze adesive sono usate per il micro-consolidamento con lo scopo di aumentare la coesione dei materiali e la loro resistenza meccanica al degrado. Molti sono i polimeri sintetici che sono stati utilizzati nel campo della conservazione tra i quali molti, purtroppo, senza alcuno specifico studio di qualità. Raman

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 7
È stato realizzato uno studio dettagliato sulla degradazione di alcuni polimeri acrilici usati come protettivi per i materiali porosi d’interesse artistico. In particolare sono stati indagati il grado di penetrazione di questi consolidanti, la loro stabilità fotochimica e la reversibilità del trattamento [Miliani et al. 2002].
Pergamene e vella FT-Raman Gli spettri Raman di antiche pergamene e vella (pelli più raffinate rispetto alle pergamene) sono stati acquisiti [Edwards et al. 2001, Edwards et Perez 2004] e costituiscono la base per futuri studi sulla loro interazione con i pigmenti e i leganti presenti nei manoscritti. In particolare, sono state studiate la presenza di tracce delle procedure utilizzate per la loro preparazione e in molti casi è stata rilevata la presenza di solfati e calce spenta. Inoltre, il loro grado di deterioramento può essere messo in relazione con l’osservazione delle variazioni della struttura della proteina –CONH- e della distruzione chimica del legame –CSSC-.
Bio-deterioramento FT-Raman Sono stati ottenuti gli spettri FT-Raman delle incrostazioni dovute al substrato di licheni su calcari [Edwards et al. 1995]. La presenza di ossalato di calcio diidrato è stata rilevata dall’osservazione delle bande del legame ν(CO) dello ione ossalato. Sono state eseguite anche misure quantitative per la misura dell’ammontare di cellulosa nei licheni, utilizzando la banda ν(CH), e per verificare la distribuzione, in profondità, dell’ossalato di calcio diidrato dalla superficie dell’incrostazione fino ai talli. Uno studio sulle colonie di licheni su rocce granitiche [Prieto et al. 1999] ha messo in evidenza la loro capacità di concorrere alla degradazione chimica dei graniti tramite la formazione di gesso.
Mummificazione FT-Raman Sono state investigate le modificazioni nella pelle mummificata di quattro mummie provenienti dalla Groelandia, XV secolo [Gniedecka et al. 1997]. Gli spettri della pelle delle mummie sono risultati molto simili, ma diversi da quelli ottenuti da campioni di pelle contemporanea congelata ed essiccata. In particolare gli spettri della pelle delle mummie indicavano variazioni nella struttura delle proteine. Simile caratteristica presentava la pelle di una mummia più antica, dimostrando così che queste modificazioni dovevano aver avuto luogo dopo la mummificazione, in un tempo relativamente breve.
1.8 Prodotti della corrosione dei metalli Raman Sono stati studiati i prodotti della corrosione, dovuta all’interramento, presente su manufatti in bronzo, specificatamente, monete bronzee cinesi [McCann et al. 1999]. Sono stati individuati i prodotti solitamente presenti nei manufatti antichi in bronzo, come la cuprite, la malachite e l’azurite. Inoltre, l’alta risoluzione spaziale del micro-Raman ha permesso di localizzare i prodotti di corrosione all’interno di strati di corrosione e anche all’interno di fasi metallografiche, permettendo così di indagare i meccanismi del deterioramento. Inoltre, è

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 8
stato possibile dedurre le condizioni ambientali in cui ha avuto luogo la corrosione, in quanto i diversi prodotti provenienti da ogni tipo di processo sono risultati distinguibili. La corrosione atmosferica dei manufatti in bronzo è stata invece investigata su gruppi statuari moderni [Hayez et al 2004]. In particolare, sono stati indagati i differenti tipi di solfato di rame, come l’antlerite, la brochantite, la posnjakite e la chalcanthite, prodotti dall’interazione dell’atmosfera con le diverse fasi del rame. Uno studio sui minerali atacamite e paratacamite, cloridi basici del rame, [Frost et al. 2002] ha dimostrato la facilità con cui gli idrossi cloridi del rame possono essere individuati e quindi ha confermato la possibilità di analizzare pigmenti degradati e manufatti in bronzo, rame e ottone corrosi. Infine, uno studio sulla corrosione degli archeomateriali in ferro propone un metodo di ricostruzione virtuale delle superfici corrose basato sullo studio dell’interazione del manufatto con l’ambiente in cui si è degradato [Neff et al. 2004].
1.9 Minerali e gemme Sono stati pubblicati molti lavori sui minerali di interesse per gli studi sui Beni Culturali, in particolare, quelli generati dalla corrosione dei metalli, quelli utilizzati per la realizzazione dei vetri colorati, quelli usati come pigmenti e quelli d’interesse perché parti integranti di archeomateriali o di manufatti come nel caso delle gemme preziose. Per quanto riguarda queste ultime, le indagini Raman non si limitano solo alla loro caratterizzazione ma soprattutto alla ricerca di metodi per distinguerle dalle loro imitazioni (le pietre preziose possono essere create, come il rubino sintetizzato già alla fine del XIX secolo, o assemblate, unendo lamine di differenti gemme, per creare un particolare effetto o gemme semplicemente di dimensione maggiore). È possibile, inoltre, rilevare i trattamenti subiti dalle pietre al fine di esaltarne le caratteristiche. I metodi più utilizzati sono il riscaldamento (l’ametista viene riscaldata per ottenere la citrina), la tintura (l’agata viene tinta per ottenerne di diverso colore), l’irraggiamento con raggi UV (i diamanti vengono irraggiati per variarne la trasparenza), la stabilizzazione (ottenuta facendo assorbire alla pietra oli naturali o sintetici, viene applicata agli smeraldi notoriamente soggetti a fratture), la diffusione (realizzata depositando un film sottile sulla superficie della pietra), e infine il laser drilling (utilizzato per rimuovere le impurità). Raman Sono stati pubblicati lavori dettagliati sul comportamento di singoli minerali, come ad esempio l’aragonite [Frech et Wang 1980], i polimorfismi del silicio [Kingma et Hemley 1994] e la goethite [Kustova et al. 1992], oppure, sono stati compilati dei cataloghi di spettri Raman di minerali d’importanza per le ricerche in storia dell’Arte e in Archeologia [Bouchard et Smith, 2003]. Per le gemme si ricordano, analogamente, lavori su singole pietre, come lo smeraldo [Moroz et al. 2000], tentando in questo caso anche una indagine sulla provenienza, o lavori comprendenti più categorie di gemme [Jenkins et Larsen, 2004]. Altrettanto numerosi sono infine gli studi su gemme e minerali presenti su archeomateriali, come ad esempio gli intaglios d’epoca romana [Smith et Robin 1997] o i reliquari medievali [Reiche 2004]. FT-Raman I minerali rispondono bene all’analisi Raman dispersivo e per questo motivo esiste una sola libreria di spettri FT-Raman di minerali [Coleyshaw et al. 1994].

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 9
1.10 Vetri e mosaici in pasta vitrea Tramite RS è possibile identificare gli agenti opacizzanti e coloranti e studiare il fenomeno del deterioramento. L’acqua condensa sulla superficie del vetro e reagisce con gli ioni dei metalli alcalini (sodio e potassio) che migrano sulla superficie del vetro. La soluzione alcalina risultante contribuisce alla dissoluzione della matrice silicica, formando uno strato di gel e così gli ioni dei metalli alcalini possono reagire con i gas inquinanti dell’ambiente circostante. Raman Sono stati indagati i fenomeni di degradazione che hanno colpito i vetri inglesi (XIX e XX secolo) del National Museum of Scotland. Su questi vetri, in particolare, è stata osservata la formazione di un deposito cristallino. Si è potuto concludere che questo deposito era dovuto agli alti valori di acido formico, e probabilmente di formaldeide, presente dentro le teche del museo e emesso dalle materie plastiche utilizzate per la loro realizzazione [Robinet et al. 2004]. Una classe particolare di vetri è quella costituita dalle paste vitree utilizzate per i mosaici. Grande attenzione è stata posta nello studio della tecnologia utilizzata per la loro creazione e, in particolare, sugli agenti coloranti e opacizzanti. È il caso di un lavoro sulle tesserae di mosaico provenienti da ville di epoca romana in Sicilia [Galli et al. 2004].
1.11 Ossidiane Le ossidiane, vetri naturali di origine vulcanica, sono state utilizzate fin dalla preistoria sia per la realizzazione di strumenti che per decorazione. La loro caratterizzazione, possibile con un gran numero di tecniche, permette la ricostruzione dei traffici commerciali preistorici. Queste metodologie sono, però, per la maggior parte, distruttive ed è per questo che la RS viene proposta come tecnica alternativa. Raman Anche se attualmente sono stati pubblicati pochi spettri Raman di ossidiane [ White et al. 1984], è già stata esaminata la possibilità di determinare l’origine geologica delle ossidiane tramite RS [Milleville et al. 2003]. Buoni risultati sono stati ottenuti per la caratterizzazione e la distinzione delle ossidiane provenienti da Lipari, Palmarola, Pantelleria e Monte Arci in Sardegna [Bellot-Gurlet et al. 2004].
1.12 Ceramiche L’esame dettagliato delle ceramiche può fornire informazioni utili alla loro caratterizzazione e identificazione. In scala micrometrica le ceramiche risultano spesso costituite da più fasi di cui la composizione, la distribuzione, la dimensione e l’orientamento dipendono dalle tecnologie utilizzate. L’analisi micro-Raman permette di identificare queste fasi e quindi di identificare i materiali ed i metodi utilizzati per la produzione delle ceramiche. Esistono diversi tipi di ceramiche. Le più diffuse sono le maioliche, le porcellane, il lustro e gli ingobbi. Si possono poi considerare anche le terrecotte decorate tramite semplice pigmentazione. La RS permette, non solo, di distinguere agevolmente tra i manufatti appartenenti a queste categorie ma anche di rilevare le differenze tra le diverse metodologie di produzione. Di seguito, sono presentati gli studi più importanti suddivisi per tipologie di manufatti.Tutti questi lavori sono stati realizzati con sistemi Raman dispersivi in quanto le ceramiche non presentano eccessiva fluorescenza e le fasi mineralogiche sono meglio osservabili con eccitazione laser ad alte frequenze.

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 10
Ceramiche ingobbiate Con questo termine s’intendono le ceramiche che presentano sulla superficie uno strato di composizione diversa rispetto a quella del corpo. Questo strato ha generalmente una struttura più raffinata che conferisce al manufatto un aspetto gradevole. Le ceramiche romane dette sigillata ne costituiscono un esempio. Ne è stata indagata la differenza in composizione tra i materiali utilizzati per il corpo e la superficie [Lofrumento et al. 2004]. Ceramiche pigmentate Sono stati individuati i pigmenti usati per la decorazione di figurine in ceramica delle tombe appartenenti alla dinastia Han (II secolo). In particolare, oltre all’ematite, al cinabro, alla goethite, alla calcite, all’azurite e al nero fumo è stato caratterizzato un nuovo pigmento viola (BaCuSi2O6) di cui non si conosceva l’uso [Zuo et al. 2003]. Si ricordano inoltre gli studi su varie figurine dipinte dal sito di Xishan [Zuo et al. 1999] Lustri La tecnica del lustro consiste nel depositare un film contenente argento e/o rame, nella forma metallica e ionica, appena sotto la superficie dello smalto. Fu prodotta a cominciare dal XI secolo nei paesi musulmani. È stato realizzato uno studio molto approfondito su lustri egiziani del XI-XII secolo [Colomban et Truong 2004]. Maioliche Le prime maioliche, dette proto-maioliche, originarie della Sicilia e dell’Italia del Sud, sono caratterizzate dall’uso di smalto stannifero. Sono state studiate le proto-maioliche della Puglia, permettendo l’identificazione del lapis lazuli [Clark, Curri, Laganara 1997, Clark, Curri, Henshaw 1997] e degli ossidi di ferro [Clark et Curri 1998], così come le maioliche di Andrea della Robbia (XV secolo) su cui è stato identificato il giallo di Napoli [Sakellariou et al. 2004]. Porcellane Gli studi condotti sulle porcellane mirano alla loro classificazione in base alla composizione dell’impasto, dello smalto e dei metodi di pigmentazione utilizzati. Impasti L’aggiunta di ossidi o di altri composti porta alla rottura dei legami Si-O, con conseguente variazione del grado di polimerizzazione e quindi anche dell’intensità dei modi di stretching e di bending del legame Si-O. È stato dimostrato che la costituzione di un database di spettri ottenuti da impasti prodotti con materiali e tecniche diverse permette di distinguerli più facilmente che con altre analisi chimiche [Colomban 2003]. Tra i lavori realizzati su specifici manufatti si ricorda lo studio della composizione dell’impasto delle proto-porcellane vietnamite del XIII-XIV secolo [Liem et al. 2000, Liem, et al. 2004]. L’impasto delle porcellane inglesi del XVIII secolo così come quello della manifattura francese di Sèvres può essere di due tipi, impasto fine e impasto grosso. Questi, dal contenuto mineralogico diverso, sono distinguibili tramite RS come dimostrato nei lavori condotti su porcellane inglesi di Rockingham, francesi di Sèvres e italiane del XVI secolo [Edwards, Colomban, Bowden 2004, Colomban et Treppoz 2001, Colomban, Milande, Lucas 2004]. Smalti Nello smalto si possono riconoscere due componenti fondamentali, la matrice vetrosa e il fondente. Se lo smalto non deve essere trasparente ma bianco si aggiunge un opacizzante. Se lo si vuole di una opportuna tonalità si aggiunge un colorante. È stata realizzata, una dettagliata indagine sui metodi per la colorazione degli smalti [Colomban et al. 2001] ma si trovano lavori anche su singoli agenti coloranti come la malayite [Faurel et al. 2003] e il blu di cobalto su porcellane cinesi del XVII secolo [De Waal 2004]. È stato dimostrato che la presenza di più strati di smalti colorati può essere facilmente individuata grazie all’indagine tramite micro-Raman [Colomban J. Raman Spectrosc. 2003].

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 11
Opacizzanti I primi opacizzanti utilizzati sono stati il piombo (PbO) (smalti piombiferi) e lo stagno (SnO2) (smalti stanniferi), nell’ultimo caso si parla di faience Tra i lavori realizzati su smalti stanniferi si ricordano quello sugli smalti Iznik (XV-XVII secolo) [Colomban et al. 2004] e quello su faience egiziani [Clark et Gibbs 1997].
1.13 Pigmenti I lavori mirati all’identificazione dei pigmenti sono al momento i più numerosi. Le analisi tramite RS hanno portato ad individuare pigmenti, leganti e preparazioni pittoriche di cui non si aveva notizia. Queste indagini hanno realmente permesso di migliorare la conoscenza sulla diffusione dei prodotti e delle tecniche antiche così come auspicato da Clark nei suoi lavori. Sul piano del restauro e della conservazione la RS ha apportato un notevole contribuito. Sono state documentate trasformazioni del colore dovute a miscele instabili, all'adozione di pigmenti sperimentali o alla reazione dei pigmenti con i materiali di supporto. Ad esempio, sono stati diffusamente verificati l’annerimento del carbonato basico di piombo attraverso il contatto con il bianco d'uovo usato nel medioevo come legante per i manoscritti e l’instabilità delle miscele di tedraossido di piombo e di cinabro esposte a forte illuminazione. La RS può anche essere adoperata per monitorare l’azione di interventi di restauro, potenzialmente distruttivi. Così, ad esempio, è stata esaminata l’azione della pulitura laser, in particolare, su elementi policromi del medioevo spagnolo. [Castillejo et al. 2003]. L’esperienza nel campo dei Beni Culturali, ormai più che ventennale, ha prodotto un patrimonio di informazioni che costituiscono, oramai, un punto di riferimento imprescindibile. Così, ad esempio, è stato dimostrato ampiamente che l’analisi dei pigmenti stesi deve essere eseguita evitando l’eventuale degradazione indotta dal laser. A dimostrazione di ciò citiamo un lavoro realizzato sul Trionfo d’amore di Botticelli. In un primo tempo era stata pubblicata la notizia dell’identificazione, tra i pigmenti, della plattnerite (PbO2) e della galena (PbS). Si è poi verificato, però, che questi composti provenivano dalla degradazione dovuta al laser dei pigmenti a base di piombo realmente utilizzati nel dipinto[Smith et al. 2001]. Di seguito vengono proposti alcuni esempi di indagini sui pigmenti. Sono presentati, per maggiore chiarezza, suddivisi in base al supporto pittorico, al legante o al particolare tipo di manufatto.
Pigmenti puri Forniamo di seguito una bibliografia di lavori pubblicati riguardanti indagini su pigmenti non ancora stesi, ovvero non miscelati con alcun legante. Diversamente dai database si tratta in questo caso di pubblicazioni mirate alla interpretazione degli spettri al fine di illustrarne particolari caratteristiche. Cinabro Raman [Peters et al. 1996, Taylor et al. 1970]. Blu ultramarino (PB 29) Raman [Clark et Franks 1975, Clark et Cobbold 1978, Clark et al. 1983]. Bianco di piombo (PW 1) Raman [Brooker et al. 1983, Durman et al. 1985] FT-Raman [Ciomartan et al. 1996]. Gesso Raman [Krishnamurthy et Soots 1971, Berenblut et al. 1973]. Indigo FT-Raman [Tatsch Schrader 1995]. Lazurite Raman [Guineau 1984]. Realgar e Pararealgar Raman [Trentelman et al. 1996]. Blu egiziano (PB 31) Raman [Pagès- Camagna et al. 1999, Baraldi et al. 2001]. Rosso di piombo Raman [Dalton 1977].

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 12
Magnetite Raman [Shebanova et Lazor 2003]. Malachite Raman [Guineau 1984, Schmidt et Lutz 1993]. Litargirio Raman [Dalton 1977]. Ocre Per ocre si intendono i pigmenti a base di terre, quali l’ocra rossa e gialla, la limonite, la goethite, la terra di Siena, la terra di Siena bruciata, il caput mortuum e l’ematite. È stato proposto un procedimento per distinguere le ocre tramite RS. Questo metodo si basa sulla osservazione dei rapporti tra i picchi più intensi, attribuibili alle maggiori componenti di solito presenti nelle ocre, e sul loro raffronto con delle tabelle contenenti i valori di questi rapporti misurati per le ocre di riferimento [Bikiaris et al. 1999]. Orpimento Raman [Razzetti et Lottici 1979]. Giallo hansa (PY 13) Raman [Trevor et H Onoh 2004]. Verde ossido di cromo (PG 17) Raman [Zuo et al. 1996]. Giallo oro (aurum musivum) FT-Raman [Edwards, Dixon 2003]. Giallo di piombo e zinco Raman [Clark 1995].
Pigmenti su pitture murali Per pitture murali si intendono quelle che per supporto pittorico utilizzano un intonaco. Si dividono in due categorie, l’affresco e la tempera. A loro volta all’interno di queste categorie esistono diversi tipi di tecniche che la spettroscopia Raman spesso può distinguere. Raman La RS viene spesso impiegata per risolvere quesiti emersi da precedenti indagini realizzate, per lo più, con spettroscopie elementali. Ad esempio, gli affreschi dipinti dal Perugino nel monastero di Santa Agnese a Perugia erano stati già oggetto di studio, ma le ulteriori analisi tramite RS [Ricci et al. 2004] hanno permesso di spiegare l’inattesa presenza di zinco nella terra marrone, utilizzata dal maestro nell’ultimo periodo della sua produzione. Essa è stata definitivamente interpretata come dovuta non ad un eventuale pigmento sconosciuto ma ad impurità di zinco presente nella terra. Analogamente, il manganese contenuto nelle aree rosse è stato attribuito alla presenza di una terra bruna mischiata all’ematite. Gli esempi di analisi tramite Raman sono abbondanti e spaziano su buona parte della produzione artistica anche extraeuropea su parete. Si và così da una tomba romana in Ucraina [Smith et Barbet 1999] ai dipinti murali di Pancelinos nella chiesa di Protaton sul monte Athos [Daniilia et al. 2000], dalle pitture del XV secolo nella cappella di S. Orso ad Aosta [Perardi et al. 2003] a quelle del Parmigianino (XVI secolo) nell’abbazia di S. Giovanni Evangelista e nella chiesa di S. Maria della Steccata a Parma [Bersani et al. 2003, Bersani et al. 2004], dalle pitture dalla tomba di Feng Hui nella provincia dello Shaanxi (X secolo) [Wang et al. 2004] alle pitture murali tibetane dal tempio di Thubchen Lakhang in Lo Manthang in Nepal [Mazzeo et al. 2004], dal buon fresco dei siti dell’età del bronzo della Grecia micenea [Brysbaert et Vandenabeele 2004] alle pitture di Napoleone Verga a Perugia [Rosi et al. 2004]. FT-Raman
Le analisi tramite FT-Raman danno anch’esse la possibilità di identificare i pigmenti consentendo la ricostruzione delle tecniche e dei materiali. Così, ad esempio, l’analisi degli affreschi medievali del convento della Peregrina in Spagna [Rull Perez et al. 1999] ha permesso di rilevare il rosso di piombo, la cui presenza è risultata di grande interesse in quanto le fonti storiche riconoscevano già allora l’instabilità delle miscele di questo pigmento. Allo stesso modo è possibile indagare sulle modalità di stesura dei pigmenti e scoprirne ad esempio l’uso gerarchico anche nell’affresco, così come è avvenuto, per la prima volta, in

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 13
occasione di una campagna di restauro e di analisi preventive su di un affresco a Basconcillos del Tozo (Spagna) [Edwards, Farwel, Rull Perez, 1999]. In alcuni casi è possibile ipotizzare la miniera da cui i pigmenti provengono se, come nel caso degli spettri ottenuti dai frammenti di affresco dal palazzo di Erode a Gerico [Edwards et al. 1999], questi mostrano, dall’assenza di una o più picchi, che il cinabro utilizzato non può provenire di certo dalle miniere di Almaden (Spagna) ma probabilmente dalla miniera di Tarna (Spagna). Non mancano, concludendo, esempi di lavori realizzati su manufatti di diverse epoche e tecniche, come le pitture murali inglesi del XII-XIII secolo dalla Sherborne Abbey [Edwards, Brooke, Tait 1997], quelle post-bizantine dalla chiesa di S. Charalambos in Atene [Chryssoulakis et Panayiotou 1997] e quelle di alcune ville romane in Inghilterra [Edwards, Middleton et al. 2003]. Nel campo dell’analisi delle pitture murali, L’FT-Raman viene, però, maggiormente apprezzato per le indagini sullo stato di degrado dovuto a biodeterioramento. Particolarmente esemplificativo è lo studio realizzato su frammenti di affresco rinascimentale del palazzo Farnese a Caprarola [Edwards et al. 1997]. Gli spettri FT-Raman ottenuti indicarono che le incrostazioni create dai licheni erano dei centri di attività chimica dove i prodotti della loro attività metabolica, principalmente acido ossalico, erano stati responsabili della trasformazione del substrato (carbonato di calcio) in ossalato idrato di calcio. In quella stessa occasione fu anche affrontato lo studio dei meccanismi adottati dai licheni per sopravvivere a contatto con i pigmenti utilizzati nell’affresco, molti dei quali a base di elementi tossici.
Pigmenti su vetro FT-Raman Sono stati analizzati vetri colorati di produzione inglese dal XIV al XIX secolo permettendo l’individuazione dei pigmenti ocra rossa e cinabro [Edwards et JKF Tait 1998].
Pigmenti su manoscritti La RS ha trovato nel campo delle indagini sui manoscritti il suo principale sviluppo. Il suo contributo è notevole per quanto riguarda la loro conservazione ed il loro restauro. Così, ad esempio, è stato indagato dettagliatamente il ruolo che l’inquinamento da H2S ha nell’annerimento dei pigmenti a base di rame, carbonato di piombo, ossidi, idrossidi e solfati, con particolare attenzione ai pigmenti utilizzati nei manoscritti oltre che negli acquarelli. [Clark et Gibbs 1998, Clark et Gibbs (Anal. Chem.) 1998, Burgau et al. 1999, Smith et Clark 2002]. Grazie a questi studi la presenza di biossido di piombo, ad esempio, su un manoscritto fiorentino del XV secolo può essere attribuita con certezza all’ossidazione del bianco di piombo trasformatosi in solfito di piombo e/o in biossido di piombo [Bicchieri et al. 2000]. La RS può contribuire inoltre al riconoscimento di precedenti interventi di restauro che, eseguiti con materiali e metodi non sufficientemente testati, possono compromettere l’integrità del manoscritto. Questo è quanto è stato fatto su di un manoscritto persiano del XIII secolo [Clark et Gibbs 1998]. Il manoscritto era in buone condizioni se non per il pigmento verde, identificato come verdigris e per quello blu, lazurite, che apparivano, invece, molto degradati. Il loro stato è stato attribuito principalmente al restauro che negli anni ’70 ha fatto uso di nylon solubile, una soluzione di N- metiossimetilnylon in metanolo. Questo intervento ha causato la dissoluzione del verdigris e l’avvio del processo di deterioramento della lazurite.

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 14
Raman Clark è uno degli autori che più hanno contribuito a diffondere con review su diverse riviste l’applicazione della RS sui manoscritti [Clark (J. Mol. Struct.) 1995]. I lavori realizzati sui manoscritti possono essere divisi in ulteriori tre categorie in dipendenza del tipo di supporto utilizzato. Infatti, supporti diversi implicano eventualmente diversi tipi di interazioni con i pigmenti e diversi leganti. Manoscritti su carta. Sono più recenti di quelli su pelli ma altrettanto preziosi. Sono stati già analizzati un manoscritto arabo del XIII secolo [Clark et Huxley 1996], tre manoscritti tedeschi del XV secolo [Burgio et al. (J. Mol. Struct.) 1997 ] e alcuni manoscritti persiani del XV secolo [Bruni et al. 2001]. Manoscritti su vellum. Ricordiamo un lavoro su alcuni manoscritti medievali latini del XV secolo [Burgio et al. 1997]. Manoscritti su pergamena. Fondamentali lo studio di una Bibbia del XIII secolo [Best et al. 1993], e quello di un codice delle leggi islandese del XIV secolo [Best et al. 1995]. Seguono poi documenti su pergamena del XV secolo [Bruni et al. 1998], manoscritti greci del XIII secolo [Magistro et al. 2001], manoscritti inglesi del VIII-IX secolo [Brown et Clark 2004, Brown et Clark (217-223) 2004, Brown et Clark (181-189) 2004, Clark et van der Weerd 2004], manoscritti turchi del XVI secolo [Jurado-Lopez et al. 2004] e manoscritti persiani del XVI secolo [Hayez et al. 2004]. FT-Raman Quando ancora l’FT-Raman era appena ai suoi esordi, fu pubblicata da un gruppo tedesco [Schrader et al 1993] una delle prime indagini su manoscritto. Seguì poi solo un lavoro su alcuni manoscritti su carta cinesi [Clark et al. 1997], infatti, la RS dispersiva, nel frattempo, aveva dato prova di essere più adatta per l’analisi di questo tipo di manufatti.
Pigmenti stesi ad olio Raman L’analisi non-distruttiva degli oli si scontra con molte difficoltà tecniche legate essenzialmente alla fluorescenza derivante dal legante e dalla vernice. I primi lavori realizzati su oli erano distruttivi in quanto si avvalevano di diclorometano per la rimozione della vernice e del legante [Davwey et al. 1994]. Per avere un esempio di analisi realmente non distruttiva si è dovuta aspettare la diffusione dei sistemi Raman dispersivi nel vicino infrarosso, con i quali è stato finalmente possibile studiare, ad esempio, vari dipinti ad olio italiani del XVII secolo [Ruiz-Moreno et al. 2003]. FT-Raman Un sistema FT-Raman dotato di fibre ottiche è stato testato con successo su alcuni oli di vari autori del novecento [Vandenabeele et al. 2001]. Questo sistema sembra attualmente il più promettente.
Pigmenti e inchiostri su carta Raman Fra i nuovi campi d’indagine quello degli inchiostri e dei pigmenti su carta sembra il più originale. In realtà il tipo di oggetti analizzabili sembra davvero infinito, si và, infatti, dalla

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 15
caratterizzazione degli inchiostri e delle matite utilizzate per le opere grafiche su carta da Gustave Moreau [Pages-Camagna et al. 2004], all’indagine sugli inchiostri utilizzati nelle opere pubblicitarie dei primi del novecento da artisti australiani [Wise et Wise 2004]. Seguono le analisi dei pigmenti utilizzati sui primi francobolli [Chaplin et al. 2004], sulla carta da parati del primo ottocento, ritrovata in un palazzo basco [Castro et al. 2004] e su una stampa cinese del IX secolo [Bell et al. 2000]. FT-Raman Per analisi su frammenti di carta da parati l’FT-Raman può essere usata con successo come nel caso dello studio sui pigmenti e sugli inchiostri utilizzati per la realizzazione delle carte da parati prodotte dalla tipografia Santa Isabel, nella regione basca (fine del XIX secolo) e ritrovate durante il restauro della Torre de los Varona [Castro et al. 2001].
Pigmenti su pietra I pigmenti possono essere stesi o direttamente sulla pietra o più comunemente sopra una preparazione. Raman Disponiamo di un solo esempio di questo tipo d’indagine, il lavoro realizzato su statuette egiziane in calcare pigmentate della 5°-6° dinastia [Ambers, 2004]. FT-Raman Anche con l’FT-Raman disponiamo di un solo esempio, la statua policroma in pietra del XII secolo in Santa Maria la Real, Sasamon, Spagna [Edwards, Farwell, Newton, Rull Perez, Jorge Villar 2000].
Pigmenti su papiri Raman I papiri egiziani originali possono essere distinti dalle loro contraffazioni tramite un’accurata analisi dei pigmenti utilizzati [Burgio et Clark 2001].
Pigmenti dell’arte primitiva rupestre L'effetto rovinoso dell’esposizione all'ambiente dei pigmenti è molto più evidente proprio nell'arte rupestre primitiva. Localizzate variazioni della temperatura, dell'umidità e dell'inquinamento dell'aria rovinano non solo i pigmenti ma anche il substrato. L'applicazione della RS all’analisi dell'arte primitiva rupestre ha fornito informazioni che altrimenti non sarebbero state accessibili. È stata rilevata la significativa eterogeneità fra campioni di pigmenti prelevati nello stesso sito, attribuibile non solo ai pigmenti stessi ma anche all’interazione con l’ambiente. La FT-Raman ha inoltre permesso di rilevare la presenza di materiali organici la cui presenza, oltre ad essere importante per la possibile datazione archeometrica, ha rilevanza per lo studio delle tecnologie primitive. Il numero limitato di pigmenti documentati nell’arte primitiva non deve portare a credere che non vi fossero altre fonti. Ad esempio, anche se il carbone costituiva la fonte di pigmento nero più facile da ottenere non và esclusa a priori la possibilità di riscontrare in studi futuri l’uso della magnetite o della pyrolusite, nonché l’uso del nero d’ossa.

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 16
FT-Raman Attualmente la FT-Raman è stata adoperata per l’identificazione dei pigmenti e delle loro miscele, la costituzione di database di spettri Raman di materiali utilizzati nell’arte primitiva e la caratterizzazione dei prodotti del biodeterioramento. Esempi di questi lavori sono quelli condotti sull’arte rupestre dei nativi americani del Seminole Canyon (3000-4200 AC) [Edwards, Drummopnd, Russ 1998], della regione del Big Bend in Texas [Edwards, Drummopnd, Russ 1999] e del Catamarca Cave in Argentina nonché sulle pitture paleolitiche delle grotte calcaree francesi [Smith et al. 1999].
Pigmenti stesi a tempera e ad acquarello Raman Una classe di manufatti decorati a tempera, particolarmente studiata, è quella delle icone. Icone post bizantine del XV-XVIII secolo [Burgio et al. 2003], del XVI secolo [Danilia et al. 2002], del XIX secolo [Burgio, Melessanaki et al. 2001]. Non mancano altri esempi, un busto di Nefertite [Wiedemann et al. 2002] e vari oggetti funerari egiziani [Edwards et al. 2004], manufatti policromi su legno del periodo rococò dalla chiesa di Escatròn, Zaragoza, Spagna [Castillejo et al. 2000, Castillejo, Matin, Silva, et al. 2000] e un crocifisso processionale del medioevo spagnolo [Centeno et al. 2004]. Tra gli acquarelli ricordiamo il Trionfo d’Amore di Botticelli [Andalò et al. 2001] e i ritratti in miniatura del tesoro di Elisabetta I ovvero l’Armada Jewel [Derbyshire et Withhall 1999].
Pigmenti su tessuti Raman È stata analizzata la pigmentazione su vari tipi di tessuti [Coupry et al 1997, Perardi et al. 2000].
Pigmenti su elementi architettonici Raman Per elementi architettonici si intendono sia elementi esterni che interni. Così, ad esempio, sono stati individuati i pigmenti e la preparazione di alcuni elementi architettonici in calcare di epoca medievale da Aquileia [Roascio et al. 2002] e dalla chiesa medievale di S. Clemente a Roma [Edwards 2003]. FT-Raman Sono state analizzate le pitture degli interni di una villa del XIX secolo [Kendix et al. 2004].
1.14 Altre metodologie Raman Le altre metodologie Raman non permettono attualmente misure non-distruttività e/o in situ e per tale motivo vengono utilizzate solo per analisi di sussidio a quelle realizzate direttamente sui manufatti con le metodologie Raman dispersivo e FT-Raman. Ad esempio la SERRS è

Capitolo 1. La Spettroscopia Raman per i Beni Culturali 17
stata utilizzata per l’analisi semi-quantitativa dell’indaco e dei pigmenti a base di alizarina e purpurina [Shadi et al. 2003, Shadi et al. 2004].

Capitolo 2. Introduzione all’effetto Raman 18
Capitolo 2. Introduzione all’effetto Raman
2.1 Introduzione Questo capitolo si propone di fornire gli strumenti indispensabili per capire i meccanismi dell’emissione Raman trattandone solo gli aspetti principali giacché una trattazione più dettagliata esulerebbe dallo scopo di questo capitolo. Solo dopo aver affrontato la trattazione dell’interazione delle onde elettromagnetiche con la materia ed, in particolare, con le rotazioni e vibrazioni delle molecole, ci si soffermerà sull’effetto Raman fornendo qualche esempio dei metodi d’interpretazione vibrazionale degli spettri.
2.2 L’energia molecolare Nell’energia totale di una molecola si distinguono quattro componenti: la traslazionale, la rotazionale, la vibrazionale e quella elettronica. In prima approssimazione i contributi di queste energie possono essere considerati separatamente. Le transizioni energetiche elettroniche danno luogo ad emissione o ad assorbimento nelle regioni dell’ultravioletto e del visibile dello spettro elettromagnetico. Le transizioni che coinvolgono solo stati rotazionali riguardano energie nella regione delle microonde oppure del lontano infrarosso, le transizioni vibrazionali invece sono responsabili dell’emissione o dell’assorbimento nella regione dell’infrarosso. La radiazione elettromagnetica è caratterizzata dalla sua lunghezza d’onda λ, la sua frequenza ν ed il suo numero d’onda ν . Il numero d’onda espresso in cm-1 rappresenta il numero d’onde in un treno d’onde lungo 1 cm, esso si può esprimere anche come:
ν =cν o
λν 1=
dove c è la velocità della luce. Gli stati vibrazionali e rotazionali molecolari possono essere studiati sia tramite la spettroscopia Raman che tramite quella infrarossa. Sebbene siano in relazione, le informazioni fornite dalle due spettroscopie non sono le stesse ma, permettendo ognuna di far luce su caratteristiche diverse delle molecole, risultano spesso complementari. In accordo alla teoria quantistica, l’energia dei fotoni Ep è data da: Ep = hν oppure Ep = hcν dove h è la costante di Plank. L’energia del fotone può essere assorbita o emessa da una molecola così da cambiare la sua energia rotazionale, vibrazionale o elettronica di una quantità ∆Em essendo ∆Em = Ep
Se la molecola acquista energia, ∆Em è positivo ed il fotone è assorbito. Se la molecola perde energia, ∆Em è negativo e viene emesso un fotone il cui numero d’onda è
hcEm∆
=ν .
2.3 I gradi di libertà del moto molecolare Lo studio delle vibrazioni molecolari può essere affrontato partendo dal modello classico che vede i nuclei come semplici punti matematici con assegnata massa. Le forze internucleari che tengono insieme la molecola, si assumono simili a quelle esercitate da una molla senza massa che tende a ristabilire le distanze e gli angoli della condizione d’equilibrio. Ogni massa richiede tre coordinate per definire la posizione.

Capitolo 2. Introduzione all’effetto Raman 19
Come conseguenza essa avrà tre indipendenti gradi di libertà, ad esempio x, y, z, in un sistema cartesiano, nelle corrispondenti direzioni del moto. Se la molecola è costituita da N nuclei atomici, ci saranno in totale 3N gradi di libertà. Il centro di gravità della molecola richiede tre coordinate per definire la sua posizione e quindi altrettanti gradi di libertà riguardanti il suo moto. Quando una molecola non lineare è nella sua configurazione d’equilibrio, essa richiede tre coordinate rotazionali per specificare l’orientamento della molecola attorno al centro di massa. Ad esempio, queste possono essere tre coordinate angolari che descrivono la rotazione attorno a tre assi mutuamente perpendicolari, ognuno passante per il centro di massa. Una molecola non lineare ha quindi tre indipendenti gradi di libertà rotazionali. Una molecola lineare ne ha solamente due attorno a due assi mutuamente perpendicolari, entrambi perpendicolari all’asse della molecola. Infatti, la rotazione di una molecola lineare attorno all’asse molecolare non viene considerata un grado di libertà in quanto non si verifica alcuno spostamento dei nuclei. Sottraendo i gradi di libertà rotazionali e traslazionali dal numero totale 3N, ci rimangono 3N-6 gradi di libertà interni per una molecola non lineare e 3N-5 per una lineare. La traslazione del centro di massa e le rotazioni della molecola attorno ad esso possono ovviamente aver luogo senza generare variazioni della configurazione della molecola stessa. I gradi di libertà interni cambiano, invece, la configurazione della molecola senza interessare il moto del centro di massa e la rotazione attorno ad esso.
2.4. Modi normali di vibrazione Può essere dimostrato che i 3N-6 gradi di libertà interni di una molecola non lineare corrispondono a 3N-6 modi normali di vibrazione. In ogni modo normale di vibrazione tutti gli atomi della molecola vibrano con la stessa frequenza e passano simultaneamente per le rispettive posizioni di equilibrio. Le relative vibrazioni dei singoli atomi possono differire in ampiezza e direzione ma il centro di gravità non si muove e la molecola non ruota. Se le forze che legano la molecola sono funzioni lineari dello spostamento dei nuclei dalla loro posizione d’equilibrio, allora le vibrazioni molecolari saranno armoniche. In questo caso ogni coordinata cartesiana di ogni atomo, potrà essere rappresentata in funzione del tempo come una funzione seno o coseno, vedi figura 3.1.
Figura 2.4.1. Modi normali di vibrazione di un modello palla-molla di una molecola diatomica come HCl. Il grafico dello spostamento di ogni massa rispetto al tempo è una funzione seno ed il centro di massa (linea tratteggiata) rimane immobile.
2.5 Modelli molecolari meccanici I modi normali vibrazionali possono essere rappresentati sperimentalmente usando modelli meccanici. Uno di questi sistemi è mostrato in figura 2.5.1.

Capitolo 2. Introduzione all’effetto Raman 20
Figura 2.5.1. Rappresentazione delle vibrazioni molecolari tramite il modello palla - molla di una molecola, in questo caso del tipo CO2. La sorgente dell’oscillazione è costituita da un motore la cui velocità di rotazione è regolabile. Un modello palla-molla come CO2 è sospeso da lunghi fili, uno per ciascuna palla. Ciò lascia il modello libero di muoversi nel piano orizzontale. Il motore è collegato opportunamente ad uno dei fili così da poter esercitare una debole forza periodica sul modello sia lungo che attraverso l'asse del modello. Variando la velocità del motore, varia la frequenza della forza di disturbo. Quando la frequenza della forza è uguale ad una delle frequenze naturali di vibrazione del modello, si manifesta la risonanza. Il modello reagisce vibrando in uno dei suoi modi normali. In tal caso, si osserva che tutte le masse si muovono di semplice moto armonico con la stessa frequenza e passano simultaneamente per le loro posizioni di equilibrio. In alcuni modi alcune masse possono vibrare con ampiezza zero, cioè non si muovono. Ad una diversa frequenza della forza di disturbo, può essere attivato un altro modo normale di vibrazione. Quando la frequenza della forza non coincide con quella di un modo normale di vibrazione, il modello rimane immobile. Se invece di essere eccitato con il motore, il modello viene semplicemente colpito con un martello, i moti risultanti non appaiono più armonici. In realtà, ad una più attenta analisi, si troverà che i moti consistono di combinazioni di tutti i modi normali di vibrazione eccitati, insieme a traslazioni e rotazioni. La molecola lineare di CO2 ha 3N-5 o 4 modi fondamentali di vibrazione. Il modello qui discusso ne ha solo tre essendo costretto a muoversi solo nel piano.
2.6 Coordinate usate nella descrizione delle vibrazioni molecolari Le vibrazioni di stretching del modello in figura. 2.5.1 sono illustrate in figura 2.6.1 insieme a diversi tipi di coordinate usate per la rappresentazione delle vibrazioni molecolari.

Capitolo 2. Introduzione all’effetto Raman 21
Figura 2.6.1. Vibrazioni di stretching di un modello triatomico lineare in diversi tipi di coordinate. Le coordinate cartesiane X tengono conto della dislocazione delle masse. Le coordinate interne S descrivono la variazione della lunghezza dei legami. Le coordinate cartesiane descrivono lo spostamento dalla posizione di equilibrio di ciascun nucleo. Ogni atomo ha il suo sistema di coordinate cartesiane con l’origine posta nella posizione di equilibrio del nucleo. Le coordinate interne indicano il cambiamento di forma della molecola rispetto alla sua configurazione d’equilibrio, senza quindi tenere in conto l’orientamento e la posizione della molecola nello spazio. I modi normali vibrazionali possono essere descritti usando uno o l’altro sistema di riferimento. Ricordiamo che in tutti i modi normali vibrazionali, tutte le coordinate variano periodicamente con la stessa frequenza e passano per l’equilibrio simultaneamente. La forma del modo normale è allora definita specificando le relative ampiezze (positive o negative) delle varie coordinate nel sistema di riferimento usato.
2.7 Formula classica per la frequenza vibrazionale di una molecola diatomica. Supponiamo che la molecola diatomica sia rappresentata da due masse m1 ed m2 connesse da una molla senza massa, figura 2.7.1.

Capitolo 2. Introduzione all’effetto Raman 22
Figura 2.7.1. Modello di molecola diatomica. Sulla destra sono riportate due soluzioni delle equazioni del moto riportando la posizione delle masse in funzione del tempo, per una traslazione e per una vibrazione. Per semplicità poniamo che le due masse possano muoversi solo lungo l’asse molecolare. Chiamiamo x1 ed x2 i rispettivi spostamenti dalla posizione di equilibrio. Ovviamente (x1-x2) sarà la variazione della lunghezza della molla rispetto all'equilibrio. Supponiamo che la forza rappresentata dalla molla verifichi la legge di Hooke, allora, possiamo esprimere la forza esercitata dalla molla su ciascun nucleo come:
21
2
112 )(dt
XdmXXF −=− e - 22
2
212 )(dt
XdmXXF =− (2.7.1)
Quando (X2-X1) è positivo (molla allungata) la forza su m1 è diretta nella direzione delle x positive mentre quella su m2 nella direzione opposta. Le soluzioni di queste equazioni del moto sono: X1= A1cos (2πνt + α) e X2= A2cos (2πνt + α) (2.7.2) Esse descrivono moti armonici semplici. Osserviamo che sebbene ogni massa oscilli con la propria ampiezza, la frequenza e la fase sono uguali per entrambe, ciò significa che le due masse passeranno contemporaneamente per l’equilibrio. Differenziando due volte rispetto al tempo otteniamo:
)2cos(4 2212
21 απννπ +−= tA
dtXd
(2.7.3)
)2cos(4 2222
22
απννπ +−= tAdt
Xd
Sostituendo X1 ed X2 (eq. 2.7.2) e le loro derivate seconde (eq. 2.7.3) nell’eq. 2.7.1 otteniamo, F(A2-A1)=-m1A1π2ν2 , F(A2-A1)=-m2A2π2ν2 (2.7.4) Queste equazioni possono dare il rapporto delle ampiezze A1/A2.
2212
1
4 νπmFF
AA
−= e
FmF
AA 22
2
2
1 4 νπ−= (2.7.5)
Eliminando A1/A2 e risolvendo rispetto a ν otteniamo 16π4m1m2ν4- 4π2F(m1+m2)ν2 + F2 = F2
così
)4
)((21
22124
mmmmF
πνν
+= (2.7.6)
In una soluzione otteniamo ν=0 che sostituita nell'equazione dell’ampiezza (eq. 2.7.5) dà A1=A2. Per ogni istante t si ha (eq. 2.7.2) X1= X2 , cioè una semplice traslazione della molecola lungo l’asse x.
L’altra soluzione è )11(21
21 mmF +=
πν (2.7.7)
Notiamo che la frequenza ν è indipendente dall’ampiezza. Sostituendo tale valore di ν in entrambe le equazioni (2.7.5) otteniamo
1
2
2
1
mm
AA −
= (2.7..8)
oppure per ogni istante t (dall’eq. 2.7.2) X1/X2 = -m2/m1. Ciò indica che il centro di gravità non si muove. Queste due soluzioni per ν corrispondono ai due gradi di libertà che le due masse hanno rispetto alla direzione individuata dall’asse della molla; (1) traslazione senza

Capitolo 2. Introduzione all’effetto Raman 23
vibrazione e (2) vibrazione senza traslazione (vedi figura 2.7.1). Spesso l’eq. 2.7.7 viene riscritta usando la massa ridotta u.
uF
πν
21
= (2.7.9)
dove 21
21
mmmmu+
=
Date la frequenza e la massa ridotta, la costante di forza può essere calcolata da F = 4π2ν2u
2.8 Assorbimento infrarosso e variazione del momento di dipolo Dal modello meccanico illustrato in figura 2.5.1 abbiamo visto che la molecola possiede certe frequenze vibrazionali naturali. Analogamente al motore, la radiazione infrarossa fornisce la forza oscillante che agendo sulla molecola la mette in vibrazione. Quando la frequenza della forza oscillante del motore si approssima a quella vibrazionale propria del modello, quest'ultimo assorbe energia dal motore e quindi accresce la propria energia vibrazionale aumentando l'ampiezza del moto. A frequenze diverse il modello non assorbe energia. Analogamente succede per la radiazione infrarossa: la molecola irradiata con un largo range di raggi infrarossi, assorbe solo quelli con frequenze uguali alle sue frequenze vibrazionali naturali (vedi figura 2.8.1).
Figura 2.8.1. Schema dell'assorbimento infrarosso. La molecola di HCl accresce la sua energia vibrazionale assorbendo energia da un fotone infrarosso di frequenza uguale a quella i vibrazione della molecola.
negativa si considera la carica totale degli elettroni concentrata nel loro centro di
del fotone in prossimità della molecola può essere considerato uniforme su tutta la molecola.
d Mentre la frequenza di assorbimento dipende dalle frequenze vibrazionali molecolari, l'intensità di assorbimento dipende dalla capacità effettiva della molecola di assorbire l'energia del fotone. Questa è legata alla variazione del momento di dipolo che si verifica in conseguenza della vibrazione. Il momento di dipolo è definito, nel caso di un dipolo semplice, come il prodotto della carica per la distanza tra le due cariche. In una molecola complessa questa definizione può essere conservata se la particella di carica positiva si assume uguale alla carica positiva totale dei protoni concentrata nel centro di gravità degli stessi e per la particellagravità. Dal momento che la lunghezza d'onda degli infrarossi è molto più grande della dimensione della maggior parte delle molecole, il campo elettrico

Capitolo 2. Introduzione all’effetto Raman 24
Tale campo esercita una forza attrattiva o repulsiva sulle cariche molecolari, cosicché il momento elettrico di dipolo della molecola oscilla con la stessa frequenza del fotone. A certe particolari frequenze il momento di dipolo ed i nuclei oscillano simultaneamente e la vibrazione molecolare può essere attivata. Più il momento di dipolo varia durante la vibrazione, più facilmente il campo elettrico del fotone può attivare la vibrazione. Se una vibrazione molecolare non causa variazione del momento di dipolo, allora l'oscillazione forzata del momento di dipolo non può attivare la vibrazione. Tutto ciò è sintetizzato nella seguente regola di selezione: Affinché possa assorbire la radiazione infrarossa, la vibrazione molecolare deve causare una variazione nel momento di dipolo della molecola. Può dimostrarsi che l'intensità di una banda di assorbimento infrarosso è proporzionale al quadrato della variazione del momento di dipolo causata dalla vibrazione molecolare che dà luogo alla banda di assorbimento. Nella molecola di HCl attorno all'atomo di cloro c'è un leggero eccesso di carica elettronegativa ed attorno all'atomo di H invece c'è un eccesso di carica positiva. Queste due regioni possono essere viste come costituenti un unico dipolo. Durante la vibrazione della molecola di HCl, le dimensioni del dipolo variano così come le distribuzioni della carica in eccesso, causando così la variazione del momento di dipolo. In queste condizioni, la vibrazione può assorbire infrarossi, infatti, il campo elettrico del fotone può interagire con il momento di dipolo variabile. Nella molecola di H2, invece è presente un centro di simmetria che implica un momento di dipolo nullo. Durante la vibrazione viene mantenuto il centro di simmetria cosicché il momento di dipolo rimane nullo. Questa vibrazione è insensibile agli infrarossi dal momento che non c'è una variazione del momento di dipolo con cui il campo elettrico possa interagire. Nella molecola di CO2 è presente un centro di simmetria che viene mantenuto simmetrico ed in fase, durante la vibrazione di stretching, cosicché la vibrazione è inattiva agli infrarossi. Se il centro della carica negativa si suppone posto a metà strada tra gli atomi di ossigeno e inoltre si suppone che il centro della carica positiva sia posto in corrispondenza dell'atomo di carbonio, allora può vedersi che la vibrazione antisimmetrica e fuori fase di stretching e la vibrazione di bending, causano una variazione del momento di dipolo e quindi sono sensibili agli infrarossi. Dovrebbe essere chiaro a questo punto che se una molecola nella sua configurazione di equilibrio ha un centro di simmetria, allora le vibrazioni che conservano inalterato tale centro saranno insensibili agli infrarossi.
2.9 Anarmonicità e ipertoni Fino ad ora abbiamo discusso solo le vibrazioni armoniche. Se le forze elastiche non sono linearmente proporzionali allo spostamento nucleare si verificano anarmonicità meccaniche. L'anarmonicità elettrica ha luogo se la variazione del momento di dipolo non è linearmente proporzionale allo spostamento nucleare. Se una vibrazione è armonica la funzione che rappresenta lo spostamento dei nuclei in funzione del tempo è un seno o un coseno. Se invece è anarmonica la funzione sarà sempre periodica ma non con un semplice andamento di tipo seno o coseno. Come risultato la frequenza vibrazionale non sarà più completamente indipendente dall'ampiezza come è invece nel caso armonico. Se si fa un grafico dell'andamento del momento di dipolo rispetto al tempo per una vibrazione classica, nel caso di anarmonicità meccaniche e/o elettriche ne risulterà un'onda periodica ma non sinusoidale. Tali funzioni periodiche possono però essere scomposte in termini di funzioni seno e coseno le cui frequenze sono multipli interi delle frequenze vibrazionali fondamentali (analisi di Fourier).

Capitolo 2. Introduzione all’effetto Raman 25
Ciò implica che se la vibrazione è anarmonica, il momento di dipolo oscillerà con la frequenza fondamentale e con i suoi multipli interi. Queste sono chiamate primo ipertono fondamentale, secondo ipertono... queste oscillazioni del momento di dipolo possono interagire con la radiazione elettromagnetica di frequenza uguale a quella fondamentale o ai suoi multipli interi. L'intensità dell'assorbimento di un ipertono dipende dalla quantità di anarmonicità nella vibrazione. Gli ipertoni possono essere rivelati nello spettro infrarosso ma generalmente sono molto deboli, ciò implica che sebbene le vibrazioni molecolari siano abbastanza anarmoniche, l'anarmonicità non è grande e può in prima approssimazione essere trascurata.
2.10 La funzione potenziale vibrazionale La funzione potenziale vibrazionale di una molecola diatomica rappresenta l'energia potenziale V rispetto alla distanza internucleare r. Le variazioni dell'energia potenziale internucleare sono causate da variazioni dell'energia repulsiva elettronica e nucleare, funzione a sua volta, della distanza internucleare. Dal momento che i nuclei che vibrano si muovono molto più lentamente degli elettroni, si assume che l'energia elettronica si riarrangi istantaneamente al variare della posizione dei nuclei (approssimazione di Born-Oppenheimer). A grandi distanze internucleari i due atomi si attraggono l'un l'altro, mentre a piccole distanze la repulsione internucleare predomina generando così una distanza di equilibrio re per i nuclei cui si raggiunge la minima energia potenziale. Generalmente non disponiamo di una espressione esatta per le forze che tengono i nuclei nelle loro posizioni di equilibrio ma possiamo usare la spettroscopia infrarossa e Raman proprio per determinare il campo delle forze molecolari.
2.11 Transizioni vibrazionali e assorbimento infrarosso L'unica variabile nell'equazione dell'energia vibrazionale è il numero quantico ν. Em=(ν + ½)hνm (2.11.1) Se esso varia di +1 allora la molecola assorbe l'energia ∆Em in modo tale che
mmm hhE νννν )21(]
21)1[( +−++=∆ (2.11.2)
mm hE ν=∆ (2.11.3) Il fotone che ha energia tale da aumentare il numero quantico vibrazionale di un’unità, ha la frequenza uguale a quella classica vibrazionale della molecola mp νν = . Se il numero quantico varia di -1 allora ∆Em=-hνm e la molecola perde energia emettendo un fotone della stessa frequenza. Negli oscillatori armonici quanto-meccanici il numero quantico vibrazionale può variare solo di ±1. Se la vibrazione è armonica tutte le altre transizioni sono proibite. Questa regola di selezione quanto-meccanica corrisponde al caso classico in cui l'energia
vibrazionale può variare solo quando il campo elettrico del fotone e il momento di dipolo molecolare vibrano alla stessa frequenza. Se il numero quantico varia di +2 allora
mmm hhE νννν )1(]1)2[( +−++=∆ (2.11.4) 22
Combinando l'eq. 2.11.4 e l'eq. 2.11.1 è chiaro che mp νν 2= (2.11.5)

Capitolo 2. Introduzione all’effetto Raman 26
Cosicché il fotone che ha energia opportuna per aumentare il numero quantico di 2 unità ha una frequenza doppia rispetto a quella della molecola. Se la vibrazione è armonica non c'è alcuna componente del momento del dipolo vibrante alla stessa frequenza e la transizione è proibita. Se la vibrazione è anarmonica, allora come abbiamo visto ci sarà una piccola componente del momento del dipolo vibrante a 2, 3, 4, ecc. volte la frequenza molecolare. Ciò corrisponde alla regola di selezione per cui in un oscillatore quanto-meccanico anarmonico il numero quantico può variare di ±1, ±2, ±3,… Poiché dalla funzione di distribuzione di Boltzmann la maggior parte delle molecole a temperatura ambiente si trova nel livello vibrazionale fondamentale ν=0, è predominante la transizione ν=1←ν=0 detta transizione fondamentale, responsabile della maggior parte dell'assorbimento infrarosso. Le altre transizioni permesse come ad esempio (ν=2←ν=1) daranno luogo ad assorbimento alla stessa frequenza della transizione fondamentale di un oscillatore armonico. Comunque le intensità relative saranno relativamente basse dal momento che i livelli ν=1 e ν=2 hanno basse popolazioni.
2.12 La rotazione delle molecole lineari La figura 2.12.1 mostra la molecola di HCl dove mH è la massa dell'atomo di idrogeno e mCl quella dell'atomo di cloro.
Figura 2.12.1 La molecola di HCl rappresentata come un rotore rigido. Sia G il centro di massa della molecola, d la distanza del cloro da G, e r0 la distanza di equilibrio internucleare. Sappiamo che
dmdrm ClH =− )( 0 (2.12.1)
ClH
H
mmrmd
+= 0
(2.12.2)
Se supponiamo che la molecola ruoti come un rotore rigido attorno all'asse b passante per il suo centro di massa e perpendicolare all'asse internucleare, allora il momento di inerzia Ib rispetto all'asse è dato da
220 )( dmdrmI ClHb +−= (2.12.3)
Sostituendo d dalla eq. 2.12.2

Capitolo 2. Introduzione all’effetto Raman 27
20r
mmmmI
ClH
ClHb += (2.12.4)
e richiamando la definizione di massa ridotta, u, otteniamo 2
0urI b = (2.12.5) dove
ClH
ClHHCl mm
mmu+
=
In altre parole la molecola ruotando attorno all'asse b ha lo stesso momento di inerzia di una particella di massa u che ruota con un raggio r0 L'energia classica rotazionale Erot di un corpo rotante attorno ad un asse è data da
2)2(21
rrot IE πν= (2.12.6)
Oppure
IpErot 2
2
=
Dove νr è la frequenza rotazionale e p è il momento angolare. L'energia rotazionale ed il momento angolare possono assumere, nel caso classico, qualsiasi valore. L'energia quanto-meccanica rotazionale è invece quantizzata. Per una molecola diatomica rigida i livelli energetici rotazionali Erot sono dati da:
brot I
JJhE 2
2
8)1(
π+
= (2.12.7)
dove J è il numero quantico rotazionale che può assumere solo valori interi 0,1,2,3,… Questa equazione può essere scritta esprimendo i livelli energetici in cm-1
)1( += JBJhcErot (2.12.8)
dove B è chiamata la costante rotazionale ed è definita da
cIhB
b28π
= (2.12.9)
2.13 Transizioni rotazionali e assorbimento infrarosso In figura 2.13.1 è illustrato il caso classico di un'onda elettromagnetica polarizzata in un piano il cui campo elettrico varia nelle direzioni delle x .

Capitolo 2. Introduzione all’effetto Raman 28
Figura 2.13.1. Interazione del campo elettrico di una radiazione elettromagnetica con il momento di dipolo ruotante di una molecola in rotazione. Supponiamo che l'onda incontri la molecola in rotazione con un momento di dipolo permanente µ. Per semplicità poniamo la direzione dell'asse x giacente nel piano individuato dal momento di dipolo rotante. Se l'angolo che il momento di dipolo intercetta con l'asse x è θ allora la componente del momento di dipolo nella direzione x è
θµµ cos=x (2.13.1) Per la molecola lineare θ varia periodicamente a causa della rotazione e può essere scritto come
trπνθ 2= (2.13.2) dove νr è la frequenza rotazionale. Combinando le eq. 2.13.1 e 2.13.2 otteniamo:
)2cos( trx πνµµ = (2.13.3) La componente del momento di dipolo µx nella direzione del campo elettrico della radiazione oscilla quindi con frequenza νr. Nel caso più generale la direzione x forma un angolo ϕ con il piano definito dal momento di dipolo rotante, in tal caso la massima ampiezza di µx, nell'eq. 2.13.3, non sarà µ ma bensì la costante µ cosϕ. Il campo elettrico della radiazione esercita delle forze sulle cariche molecolari nella direzione x e quindi fa sì che µx oscilli con la stessa frequenza del fotone νp. Quando νp e νr sono pressoché uguali, le forze esercitate dal campo elettrico della radiazione rimarranno sincronizzate con il momento di dipolo molecolare ruotante per un tempo sufficientemente lungo da causare un incremento dell’energia rotazionale a spese dell’energia della radiazione. Da quanto detto dovrebbe essere chiaro che, affinché possa assorbire radiazione infrarossa o microonde, la molecola in rotazione deve possedere un momento di dipolo permanente. Molecole diatomiche omonucleari come N2 ed O2 non hanno un puro spettro rotazionale infrarosso poiché il loro momento di dipolo permanente è zero. In accordo con la meccanica quantistica, un aumento dell’energia rotazionale molecolare ∆Erot deve essere uguale all’energia del fotone Ep il cui assorbimento ha causato l’aumento dell’energia rotazionale.
ppprot hchEE νν ===∆ (2.13.4)
Se il numero quantico rotazionale per una molecola lineare aumenta di uno la molecola acquista l’energia rotazionale ∆Erot data da

Capitolo 2. Introduzione all’effetto Raman 29
)1()]2)(1[( +−++=∆ JBhcJJJBhcErot (2.13.5) Questa eq. si ottiene sottraendo l’eq. 2.12.8 dalla stessa eq. scritta per (J+1). Da questa otteniamo
o l’eq.(2.13.4) con l’eq.(2.13.6) si ottiene ∆Erot=2Bhc(J+1) (2.13.6) Combinand
)1(2 += JBpν (2.13.7) Questa equazione dà la frequenza in cm-1 del fotone la cui energia è tale da
co rotazionale da J a J+1. Questa è l’unica transizione peaumentare il
numero quanti rmessa per
lecola diatomica.
a relazione tra le linee spettrali e i livelli energetici è evidenziata posizionando lo spettro in
a regola di selezione quanto-meccanica per l’assorbimento, ∆J=+1, corrisponde lassicamente all’osservazione che il campo elettrico della radiazione oscillante con frequenza
olecola il più a lungo possibile
dell’energia del fotone. Ciò è diverso dal
l’assorbimento. B può essere calcolata dai valori delle frequenze in cm-1. Ad esempio, tenendo presente l’eq. 2.13.7, la distanza tra due linee di assorbimento adiacenti è uguale a 2B. Da B possiamo calcolare il momento d’inerzia (eq. 2.12.9) e da questo la distanza internucleare (eq. 2.12.4). La figura 2.13.2 mostra i livelli energetici e le transizioni dello spettro di assorbimento rotazionale di una molecola diatomica.
Figura 2.13.2. Livelli energetici rotazionali e spettro rotazionale di una moLmodo che il punto corrispondente a lunghezza d’onda zero si trovi sul livello energetico da cui la transizione ha origine. Lcνp è capace di aumentare la frequenza rotazionale molecolare νr” fino ad una frequenza leggermente più alta νr’, quando νp è compreso tra νp” e νp’. In questo caso, il campo elettrico oscillante della radiazione, può rimanere sincronizzato nella propria fase con il momento di dipolo in rotazione della mmentre la frequenza rotazionale varia da νr” a νr’. Questo aumento della frequenza rotazionale è una naturale conseguenza dell’aumento dell’energia rotazionale causata dall’assorbimentocaso della variazione della energia vibrazionale dove la frequenza rimane inalterata.

Capitolo 2. Introduzione all’effetto Raman 30
La frequenza rotazionale classica νr” per una molecola lineare nel livello energetico J può
essere ottenuta dall’eq. 2.12.6, per l’energia rotazionale classica, e dall’eq. 2.12.7 per l’energia quanto-meccanica.
)1(8
)2(21
2
22" += JJ
IhI
brb π
πν (2.13.8)
)1(2)( " += JJBcJrν (2.13.9) La frequenza classica rotazionale νr” per il numero quantico J+1 si ottiene dall’eq. precedente sostituendo J+1 a J.
)2)(1(2)( 1' ++=+ JJBcJrν (2.13.10)
Dall’eq.(12.7) vediamo che la frequenza νp che il fotone deve possedere per causare la transizione da J a J+1 è:
)1(2 += JBcpν (2.13.11)
Dividendo l’eq. (12.9), (12.10), e (12.11) per 12 +JBc si può vedere che: 2:1:)(::)( 1
'" ++=+ JJJJrpJr ννν (2.13.12) Quando il numero quantico aumenta di uno, la frequenza del fotone assorbito deve essere compresa tra le frequenze rotazionali classiche iniziale e finale della molecola. Notiamo che il fotone che ha energia opportuna per causare l’incremento del numero quantico di due unità, ha una frequenza molto più alta di quelle classiche rotazionali coinvolte nella transizione. Esso, infatti, ha una frequenza leggermente più alta del doppio della frequenza rotazionale classica iniziale e sappiamo che non c’è alcuna componente del momento di dipolo oscillante a tale frequenza. Non c’è anarmonicità nella transizione quindi essa è proibita dal punto di vista quanto-meccanico. Il puro spettro rotazionale di HCl è stato misurato e i risultati sono mostrati in figura 2.13.3.
Figura 2.13.3. Bande d’assorbimento nell’infrarosso lontano per HCl e relative quantità calcolate. Le diminuzioni dei valori mostrati nell’ultima colonna sono causati dallo stretching centrifugo. Infatti, nei livelli rotazionali superiori la molecola ruota più velocemente allungando leggermente il legame. Ciò porta ad una diminuzione della distanza energetica tra

Capitolo 2. Introduzione all’effetto Raman 31
i livelli e ad un leggero incremento del momento d’inerzia. Se la piccola variazione causata dallo stretching centrifugo viene ignorata, allora 2B è uguale approssimativamente a 20.8 cm-1. Sostituendo nella eq. 2.12.9 otteniamo:
240102
27
2 1069.2109979.24.10)1416.3(8
106253.68
cmgrBc
hIb ⋅×=××××
×== −
−
π (2.13.13)
Dall’eq. (2.13.5) la distanza internucleare può essere calcolata come 216
24
402
0 10665.110616.11069.2 cmr −
−
−
×=××
= (2.13.14)
e quindi Acmr &29.11029.1 80 =×= −
2.14 Intensità delle linee rotazionali In una molecola diatomica non simmetrica, la transizione rotazionale (J+1←J) darà luogo ad una singola linea nel puro spettro rotazionale la cui intensità è proporzionale alla popolazione nJ del livello rotazionale J, così come descritto dalla funzione di distribuzione di Boltzmann.
KTEEJKTE
KTEJJ J
J
egg
egeg
nn /)(
0/
0
/
0
0
0
−−−
−
== (2.14.1)
Il fattore gJ rappresenta il numero di stati energetici con la stessa energia EJ. Nel caso rotazionale, il livello più basso (J=0) è uno solo, (g0=1), invece i livelli più alti hanno molteplicità (2J+1). Quindi per il livello J otteniamo, per nJ,
KTBhcJJJ eJnn /)1(
0
)12( +−+= (2.14.2)
nj è riportata in funzione della temperatura in figura 2.14.1.
Figura 2.14.1. Distribuzioni relative delle molecole di HCl nei differenti stati energetici rotazionali a due diverse temperature. Si può subito vedere che essa presenta un massimo per J uguale a
21
2max −=BhckTJ (2.14.3)
L’eq. si ottiene differenziando l’eq. 2.14.2), ponendo dnj/dJ = 0 e risolvendo rispetto a J.
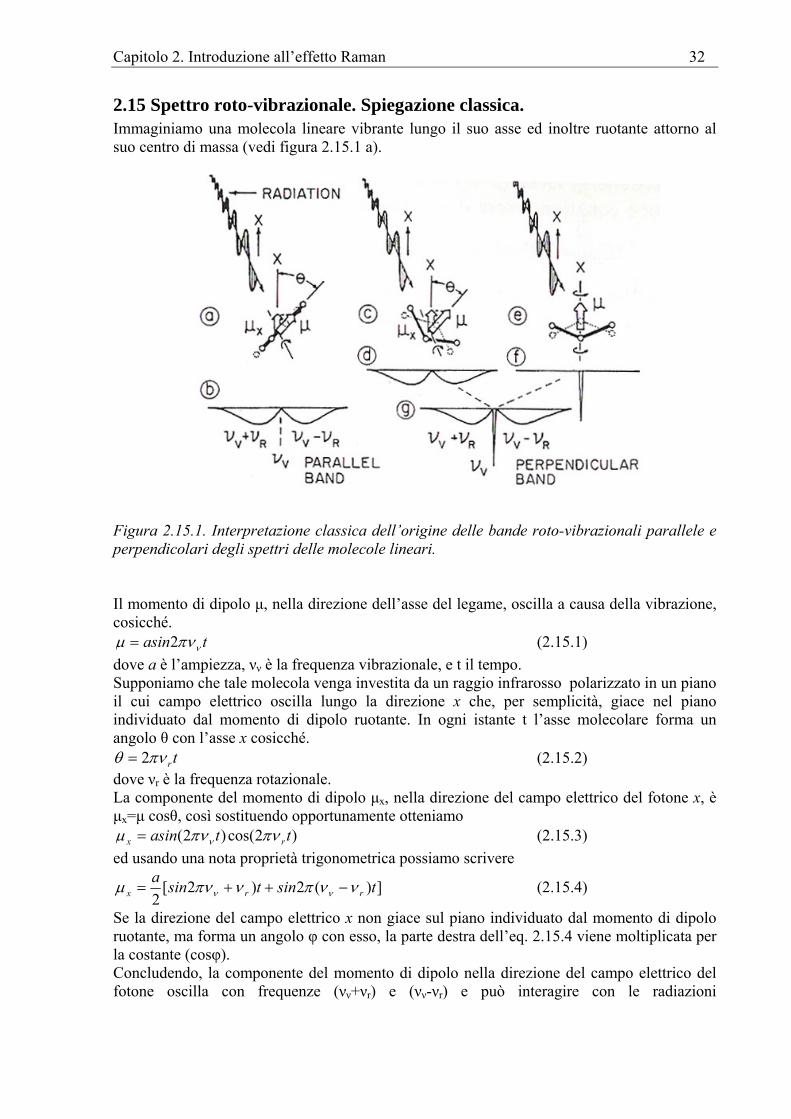
Capitolo 2. Introduzione all’effetto Raman 32
2.15 Spettro roto-vibrazionale. Spiegazione classica. Immaginiamo una molecola lineare vibrante lungo il suo asse ed inoltre ruotante attorno al suo centro di massa (vedi figura 2.15.1 a).
Figura 2.15.1. Interpretazione classica dell’origine delle bande roto-vibrazionali parallele e perpendicolari degli spettri delle molecole lineari. Il momento di dipolo µ, nella direzione dell’asse del legame, oscilla a causa della vibrazione, cosicché.
tasin νπνµ 2= (2.15.1) dove a è l’ampiezza, νν è la frequenza vibrazionale, e t il tempo. Supponiamo che tale molecola venga investita da un raggio infrarosso polarizzato in un piano il cui campo elettrico oscilla lungo la direzione x che, per semplicità, giace nel piano individuato dal momento di dipolo ruotante. In ogni istante t l’asse molecolare forma un angolo θ con l’asse x cosicché.
trπνθ 2= (2.15.2) dove νr è la frequenza rotazionale. La componente del momento di dipolo µx, nella direzione del campo elettrico del fotone x, è µx=µ cosθ, così sostituendo opportunamente otteniamo
)2cos()2( ttasin rx πνπνµ ν= (2.15.3) ed usando una nota proprietà trigonometrica possiamo scrivere
])(2)2[2
tsintsinarrx ννπνπνµ νν −++= (2.15.4)
Se la direzione del campo elettrico x non giace sul piano individuato dal momento di dipolo ruotante, ma forma un angolo φ con esso, la parte destra dell’eq. 2.15.4 viene moltiplicata per la costante (cosφ). Concludendo, la componente del momento di dipolo nella direzione del campo elettrico del fotone oscilla con frequenze (νν+νr) e (νν-νr) e può interagire con le radiazioni

Capitolo 2. Introduzione all’effetto Raman 33
elettromagnetiche che hanno le stesse frequenze. Se consideriamo un grande numero di molecole identiche di un gas, tutte vibreranno alla stessa frequenza νν , che dipende dalle masse e dalle costanti di forza, mentre la frequenza rotazionale νr è variabile e l’insieme delle molecole avrà frequenze rotazionali diverse. La distribuzione del numero di molecole riportata in funzione della frequenza rotazionale, avrà un massimo per una certa frequenza (vedi figura 2.14.1). Quando in queste funzioni di distribuzione sostituiamo a νr, le frequenze (νν+νr) e (νν-νr), ci si aspetta una banda di assorbimento tipo doppietto, (vedi figura 2.15.1 b). Questa previsione classica è pienamente verificata in uno spettro infrarosso a bassa risoluzione di un gas di una molecola diatomica non simmetrica dove la variazione del momento di dipolo avviene lungo l’asse molecolare. Una molecola lineare poliatomica come CO2 ha due tipi di vibrazioni attive agli infrarossi: - Quando il momento di dipolo varia lungo l’asse, come nelle vibrazioni di stretching antisimmetriche, alloro rifacendoci a quanto sopra ci si aspetta un largo doppietto. - Quando il momento di dipolo varia perpendicolarmente all’asse molecolare, come nelle vibrazioni di bending, si presenta una situazione diversa. Una molecola lineare può ruotare con uguale probabilità attorno a due assi mutuamente perpendicolari. Quando l’asse di rotazione è perpendicolare al piano nel quale la molecola è legata (vedi figura 2.15.1 c) allora la rotazione molecolare varia l’orientamento del momento di dipolo oscillante come visto prima, dando risultati spettrali simili (vedi figura 2.15.1 d). Quando la direzione dell’asse di rotazione è invece parallela alla direzione della variazione del momento di dipolo (vedi figura 2.15.1 e), la rotazione molecolare non influenza l’orientamento del momento di dipolo che invece oscilla semplicemente alla frequenza vibrazionale νν e dà luogo ad un solo picco (vedi figura 2.15.1 f). Dal momento che la molecola può ruotare con uguale probabilità attorno ad entrambi gli assi, la banda totale è una combinazione di un picco centrale e di due larghe bande, tutti con la stessa intensità integrale (vedi figura 2. 15.1 g). Nel caso di una vibrazione di stretching anti-simmetrica e fuori fase (vedi figura 2.15.1) non è possibile ruotare la molecola senza che vari l’orientamento del momento di dipolo oscillante, cosicché il picco centrale non si presenta. Queste previsioni classiche sono pienamente confermate dallo spettro della CO2 a bassa risoluzione, dove entrambe le due bande hanno una struttura a doppietto simile, ma solo una banda ha il picco centrale. Si vede che la forma della banda roto-vibrazionale può essere usata per verificare se la variazione del momento di dipolo avviene lungo o attraverso l’asse molecolare, ciò in questo caso è sufficiente ad assegnare le bande alla vibrazione di bending o di stretching, senza ambiguità. Se si aumenta la risoluzione dello spettro, le due bande si mostreranno costituite da linee discrete a causa della quantizzazione del momento angolare.
2.16 Spettro Roto-vibrazionale: trattazione quanto-meccanica Una banda roto-vibrazionale di uno spettro infrarosso di un gas ha una struttura complessa poiché una variazione dell’energia vibrazionale, Eν di una molecola, può essere accompagnata da una variazione dell’energia rotazionale, Er. Assumiamo inizialmente che tali energie siano additive. Combinando l’eq. (2.12.6) con l’espressione per l’energia dei livelli vibrazionali di un oscillatore armonico quanto-meccanico, Evib = (ν + ½)hν, per una molecola lineare l’energia totale è data da

Capitolo 2. Introduzione all’effetto Raman 34
)1()21( 0 +++=
+JBhcJhcE
rνν
ν (2.16.1)
Sia J” il numero quantico rotazionale nello stato vibrazionale fondamentale ν=0 e J’ sia il numero quantico rotazionale nel primo stato eccitato vibrazionale ν=1. Sottraendo l’eq. 2.16.1 dove ν=0 e J=J” dalla stessa eq. ma sostituendo ν=1 e J=J’ otteniamo
)]1()1([()( ""''0
1 +−++=−+ JJJJBcmhc
E r νν (2.16.2)
Una molecola lineare poliatomica può avere due tipi di bande roto-vibrazionali. Una banda parallela quando la variazione del momento di dipolo è parallela all’asse molecolare. Una banda perpendicolare quando la variazione del momento di dipolo è invece perpendicolare all’asse. Per la banda parallela la regola di selezione è ∆J = ±1. Per la banda perpendicolare è invece ∆J=0, ±1. Quando ∆J=0 e J’ =J”, l’eq. 2.16.2 diventa
01 )( νν == −+ cmQ
hcE r (2.16.3)
Questa eq., indipendente dal valore di J, dà la frequenza in cm-1 del cosiddetto ramo Q della banda perpendicolare. Quando ∆J=1 e J’ = J” +1, l’eq. (1.16.2) diventa
)1(2)( "0
1 ++==∆ −+ BJcmR
hcE r νν (2.16.4) ,....2,1,0" =J
Questa eq. fornisce le frequenze in cm-1 delle bande rotazionali che danno luogo al ramo R i cui valori dipendono da J” (il numero quantico rotazionale dello stato vibrazionale più basso). Quando ∆J = -1 e J’ = J”-1, l’eq. 2.16.4 diviene
)(2)( "0
1 JBcmPhcE r −==
Λ −+ νν (2.16.5) ,....3,2,1" =J
Questa è l’eq. per le frequenze delle bande rotazionali che danno luogo al ramo P i cui valori dipendono da J”. In questa eq. J” non può essere zero dal momento che J’ è di un intero più basso di J”(vedi figura 2.16.1). Le bande parallela e perpendicolare di una molecola lineare hanno identici rami P ed R con spaziatura di 2B tra le bande rotazionali, in accordo con queste eq. La banda perpendicolare ha un ramo Q centrale che lo distingue dalla banda parallela. A bassa risoluzione i rami P ed R della banda parallela appaiono come ampi doppietti la cui struttura fine non è risolta. La spaziatura dei doppietti, ∆ν , dei massimi di intensità dei rami P ed R sono dati da
IkT
cπν 1=Λ (2.16.6)
dove c è la velocità della luce, k è la costante di Boltzmann, T la temperatura assoluta ed I il momento d’inerzia. Questa eq. si ottiene sostituendo il valore di Jmax dato dall’eq. 2.14.3 nelle eq. (2.16.4 e 2.16.5 per i rami P ed R e quindi sottraendo l’eq. 2.16.5 dalla eq. 2.16.4. Nella banda roto-vibrazionale di HCl (figura 2.16.2) le singole bande rotazionali che insieme danno luogo alla banda più larga, non sono uniformemente spaziate, come dovrebbero essere secondo l’eq. presentata, ma diventano molto più vicine a più alte lunghezze d’onda.
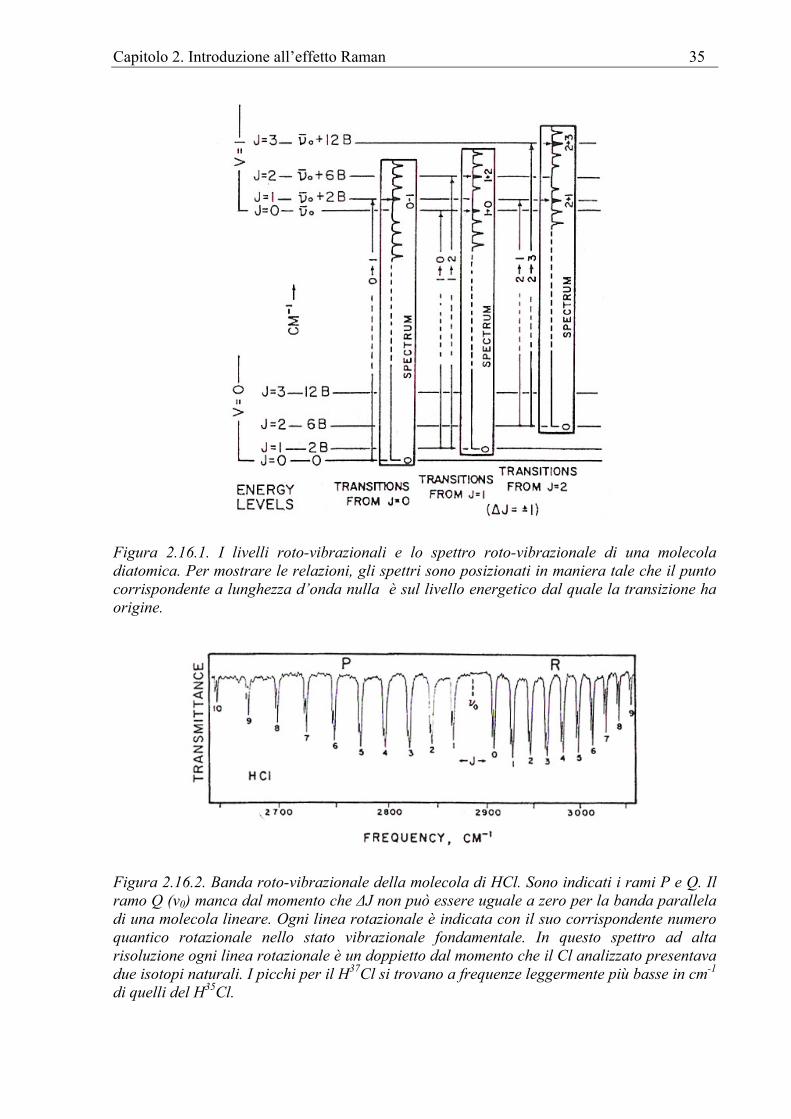
Capitolo 2. Introduzione all’effetto Raman 35
Figura 2.16.1. I livelli roto-vibrazionali e lo spettro roto-vibrazionale di una molecola diatomica. Per mostrare le relazioni, gli spettri sono posizionati in maniera tale che il punto corrispondente a lunghezza d’onda nulla è sul livello energetico dal quale la transizione ha origine.
Figura 2.16.2. Banda roto-vibrazionale della molecola di HCl. Sono indicati i rami P e Q. Il ramo Q (ν0) manca dal momento che ∆J non può essere uguale a zero per la banda parallela di una molecola lineare. Ogni linea rotazionale è indicata con il suo corrispondente numero quantico rotazionale nello stato vibrazionale fondamentale. In questo spettro ad alta risoluzione ogni linea rotazionale è un doppietto dal momento che il Cl analizzato presentava due isotopi naturali. I picchi per il H37Cl si trovano a frequenze leggermente più basse in cm-1 di quelli del H35Cl.

Capitolo 2. Introduzione all’effetto Raman 36
Nella vibrazione di stretching la distanza internucleare media e quindi il momento d’inerzia aumenta al crescere del numero quantico vibrazionale. Dal momento che la costante rotazionale è proporzionale al reciproco del momento d’inerzia, essa ha un valore più piccolo nello stato eccitato piuttosto che in quello fondamentale. La costante rotazionale B non è, quindi, costante. Sia B0 la costante rotazionale nello stato vibrazionale fondamentale e B1 nel primo stato eccitato. L’eq. 2.16.2 deve essere modificata per dare:
)]1([)]1([)]1([)( ""0
""0
''10
1 +−+−++=∆ −+ JJBJJBJJBcm
hcE r νν (2.16.7)
Per il ramo Q dove ∆J=0 e J’= J” otteniamo, ponendo J al posto di J” , Q(cm-1) = ν 0 +(B1 – B0)J2 + (B1 – B0)J J = 0, 1, 2, 3, … Per il ramo R dove ∆J = +1 e J’= J” + 1 otteniamo R(cm-1) = ν 0 + 2B1 + (3B1 – B0)J + (B1 – B0)J2 J = 0, 1, 2, 3, … Per il ramo P dove ∆J = -1 e J’ = J” - 1 otteniamo P (cm-1) = ν 0 – (B1 + B0)J + (B1 – B0)J2 J = 1, 2, 3, …. (2.16.8) In accordo con queste eq. non è necessario che i rami P ed R siano uniformemente spaziati. Il ramo Q (quando presente) non consiste più di una singola linea. Nella determinazione della costante rotazionale vengono selezionate un paio di linee rotazionali, la cui separazione dipende dalla costante rotazionale dello stato fondamentale (B0), e un altro paio di linee rotazionali la cui separazione dipende dalla costante rotazionale dello stato eccitato (B1). Due combinazioni particolarmente utili sono
)12(2 0)1()1( +=− +− JBPR JJ (2.16.9) )12(2 1 +=− JBPR JJ (2.16.10)
2.17 Spettro Raman: generalità. Dalla fisica classica sappiamo che la luce di qualsiasi lunghezza d’onda, può interagire elasticamente con una molecola, mantenendo cioè la sua frequenza inalterata (scattering di Rayleigh). Il fenomeno si spiega considerando che il vettore campo elettrico E
r dell’onda
luminosa incidente induce sulle shell elettroniche della molecola un momento elettrico di dipolo Ep
rr α= (α è la polarizzabilità della molecola) che oscilla con la stessa frequenza νp
della luce incidente, detta primaria (il pedice p sta per primaria). A sua volta il dipolo indotto oscillando emette radiazione sempre della stessa frequenza νp. Nel 1928 Raman osservò [Raman et Krishnan 1928] che oltre all’atteso scattering di Rayleigh, lo spettro presentava righe con frequenze maggiori e minori. Questo fenomeno, previsto teoricamente da Smekal nel 1923, è il cosiddetto effetto Raman. Uno spettro Raman è schematizzato in figura 2.17.1.

Capitolo 2. Introduzione all’effetto Raman 37
Figura 2.17.1. Schema di uno spettro Raman di una molecola diatomica. La linea di Rayleigh con numero d’onda pν della luce primaria è circondata dalle linee Raman rotazionali. Le linee Raman roto-vibrazionali dei rami Q, S, O, sono spaziate di una quantità corrispondente a quella dell’energia vibrazionale molecolare vibν . Si osservò poi che l’entità dello shift delle frequenze era legata alle frequenze vibrazionali e rotazionali della molecola scatteratrice ed era invece indipendente dalla frequenza della luce primaria. Affronteremo il problema dell’interpretazione dell’effetto Raman studiando separatamente il contributo dovuto al moto rotazionale e quello dovuto alle vibrazioni della molecola.
2.18 Spettro Raman vibrazionale
Nella teoria classica dello scattering elastico della luce si assume che la molecola non presenti né moto vibratorio né moto rotatorio. Quando la luce primaria di frequenza νp e quindi il corrispondente campo elettrico E=Eocos(2πνpt) colpiscono la molecola, un momento di dipolo p agisce sulle shell elettroniche che cominciano ad oscillare con la stessa frequenza νp di E, il campo elettrico. Avremo cioè: p(t)=αE0cos(2πνpt) (2.18.1) In realtà, la molecola sta vibrando con una delle sue frequenze caratteristiche. Poiché la polarizzabilità α della molecola varia in funzione della distanza internucleare R, avremo che le oscillazioni del momento indotto saranno a loro volta modulate sulla frequenza di vibrazione della molecola νvib. Possiamo esprimere α in serie di potenze di R centrata su R0 :
)()()( 00 RRdRdRR −+=ααα + Termini di ordine superiore (2.18.2)
A sua volta R, a causa della vibrazione molecolare, dipende dal tempo secondo l’espressione: R-Ro=qcos(2πνvibt) (2.18.3) Combinando questa ultima equazione con la precedente otteniamo :
)2cos()]2cos()([)( 00 tEtqdRdREtp pvib πνπνααα +== (2.18.4)

Capitolo 2. Introduzione all’effetto Raman 38
Usando poi note relazioni trigonometriche possiamo riscriverla come :
])(2cos[])(2cos[)2cos()()( 000 ttqEdRdtERtp vibpvibpp ννπννπαπνα −+++= (2.18.5)
Vediamo allora che il 2o membro contiene un termine dipendente da νp, uno da νp +νvib e l’altro da νp-νvib. Quindi il momento elettrico di dipolo vibra con tre diverse frequenze; cioè emette tre distinte linee spettrali. Abbiamo così spiegato l’origine del cosiddetto primo ordine vibrazionale dell’effetto Raman. Sperimentalmente si osservano, seppure con intensità decrescenti, anche linee con frequenze νp±2νvib, νp±3νvib, ….etc. Queste sono dovute ai termini di ordine superiore al primo dello sviluppo in serie di α(R) che avevamo precedentemente trascurato. Tali linee si definiscono effetto Raman del secondo, terzo,…ordine. Dall’eq. precedente vediamo subito che la condizione per il verificarsi dell’effetto Raman è
che 0≠dRdα cioè la polarizzabilità α della molecola deve variare con R. Questa condizione è
sempre verificata per molecole diatomiche, cosicché molecole omonucleari e quindi non polari (che non hanno un momento di dipolo permanente) come H2 e N2 presentano sempre effetto Raman. Il loro spettro rotazionale e vibrazionale, può essere osservato sfruttando l’effetto Raman, diversamente tali spettri non sarebbero accessibili utilizzando la spettroscopia infrarossa. Nelle molecole poliatomiche con centri di inversione, le due spettroscopie si completano a vicenda. In tali molecole i modi normali sensibili agli infrarossi non lo sono al Raman, e viceversa. Ciò può essere illustrato considerando la molecola di CO2: - La vibrazione di stretching simmetrica ν1 è insensibile agli infrarossi, dal momento che i centri della carica positiva e negativa, durante la vibrazione, si sovrappongono. Tuttavia la polarizzabilità varia a causa della vibrazione generando lo spettro Raman. - La vibrazione di stretching antisimmetrica ν2 al contrario, è sensibile agli infrarossi in quanto in questo caso è presente un momento di dipolo, ma non dà spettro Raman in quanto le variazioni della polarizzabilità dovute all’allungamento e all’accorciamento dei legami C-O si compensano a vicenda. La teoria classica fin qui descritta spiega molte delle osservazioni sperimentali, ma fallisce nel tentativo di interpretare il comportamento delle intensità delle singole linee. Classicamente infatti ci aspetteremmo che le linee distanziate della stessa quantità in più o in meno rispetto alla linea di Rayleigh presentino le stesse intensità. In realtà le linee di Stokes, cioè quelle con
pνν < sono molto più intense delle corrispondenti linee anti-Stokes, cioè quelle con pνν > . La spiegazione si trova in una interpretazione quanto-meccanica dell’effetto Raman visto come uno scattering anelastico del fotone. Supponiamo che inizialmente la molecola abbia numero quantico vibrazionale v = 0. Lo scattering anelastico fa sì che la molecola acquisti dal fotone uno o più quanti vibrazionali di energia, cosicché sarà v′ = 1, 2,… Il numero d’onda del fotone scatterato diverrà allora:
1,2,3...n =−= vibp nννν Queste non sono altro che le linee Stokes. Al contrario per le linee anti-Stokes si suppone che la molecola abbia un numero quantico vibrazionale v > 0 e nel processo di scattering questa volta sia il fotone ad acquistare dalla molecola uno o più quanti vibrazionali d’energia. Il suo numero d’onda sarà allora:
1,2,3...n =+= vibp nννν

Capitolo 2. Introduzione all’effetto Raman 39
Detto ciò risulta evidente che il rapporto tra le intensità delle linee Stokes e anti-Stokes, per esempio del primo ordine, dipenderà dal rapporto tra il numero di molecole nello stato vibrazionale fondamentale v = 0 e nel primo stato eccitato v = 1. Quindi il rapporto tra le intensità sarà dato da:
)0v()1v(
==
=−
nn
II
Stokes
Stokesanti
Questo rapporto può essere calcolato agevolmente con la distribuzione di Boltzmann per un sistema in equilibrio termico osservando che la differenza energetica tra i due stati è ∆E = hνvib :
kTh
Stokes
Stokesanti vibeI
I /ν−− = (2.18.6)
Notiamo che l’intensità delle linee anti-Stokes, al crescere della temperatura, tende ad essere uguale all’intensità delle linee Stokes, mentre con l’abbassarsi della temperatura le linee Stokes predominano nettamente.
2.19 Spettro Raman rotazionale Sperimentalmente attorno alla riga corrispondente allo scattering di Rayleigh si osservano due serie di righe tutte spaziate della stessa distanza. Come vedremo la spaziatura di queste ultime è legata alla frequenza rotazionale della molecola. Vedi figure 2.19.1 e 2.19.2.
Figura 2.19.1. Il diagramma mostra uno spettro Raman rotazionale. Alla sinistra e alla destra della riga di Rayleigh a pν , si hanno le linee Stokes ed anti-Stokes. Anche in questo caso si osservano delle serie di righe spettrali su entrambi i lati della riga di Rayleigh, ma ora con spaziatura corrispondente ai quanti rotazionali. Possiamo ancora spiegare molti aspetti riferendoci alla meccanica classica. La polarizzabilità di una molecola non sferica è anisotropa e deve essere trattata come un tensore con le polarizzabilità principali
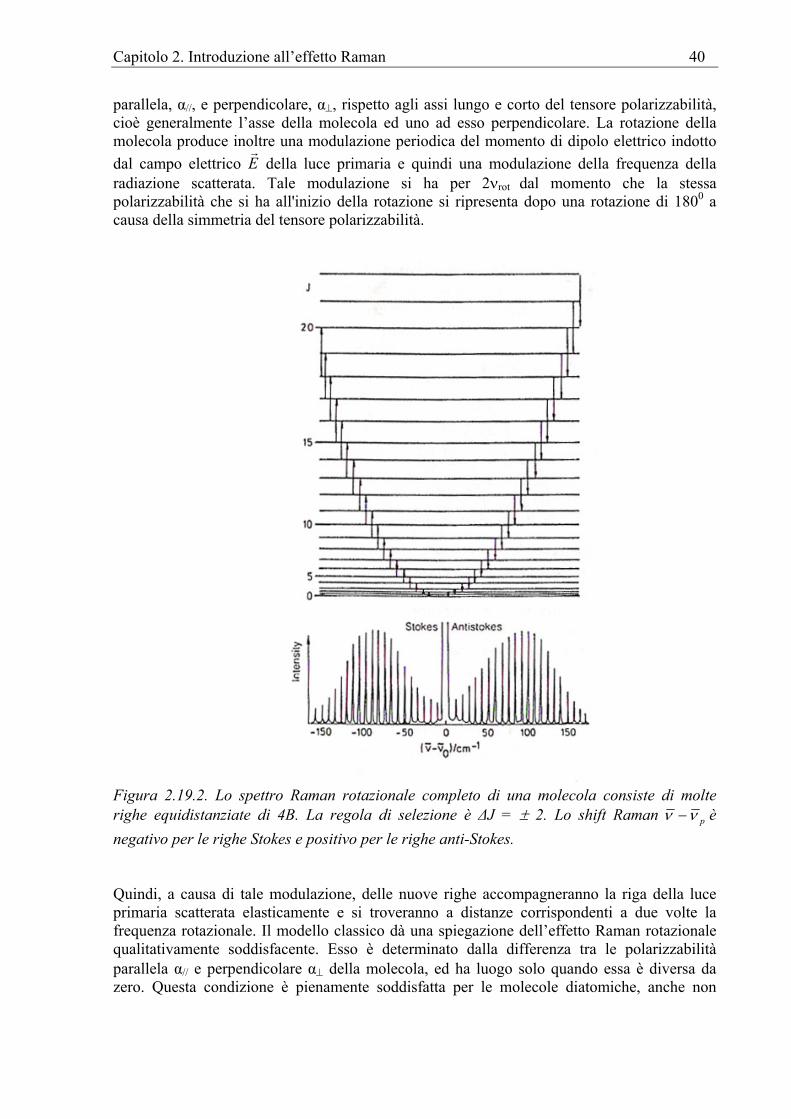
Capitolo 2. Introduzione all’effetto Raman 40
parallela, α//, e perpendicolare, α⊥, rispetto agli assi lungo e corto del tensore polarizzabilità, cioè generalmente l’asse della molecola ed uno ad esso perpendicolare. La rotazione della molecola produce inoltre una modulazione periodica del momento di dipolo elettrico indotto dal campo elettrico E
r della luce primaria e quindi una modulazione della frequenza della
radiazione scatterata. Tale modulazione si ha per 2νrot dal momento che la stessa polarizzabilità che si ha all'inizio della rotazione si ripresenta dopo una rotazione di 1800 a causa della simmetria del tensore polarizzabilità.
Figura 2.19.2. Lo spettro Raman rotazionale completo di una molecola consiste di molte righe equidistanziate di 4B. La regola di selezione è ∆J = ± 2. Lo shift Raman pνν − è negativo per le righe Stokes e positivo per le righe anti-Stokes. Quindi, a causa di tale modulazione, delle nuove righe accompagneranno la riga della luce primaria scatterata elasticamente e si troveranno a distanze corrispondenti a due volte la frequenza rotazionale. Il modello classico dà una spiegazione dell’effetto Raman rotazionale qualitativamente soddisfacente. Esso è determinato dalla differenza tra le polarizzabilità parallela α// e perpendicolare α⊥ della molecola, ed ha luogo solo quando essa è diversa da zero. Questa condizione è pienamente soddisfatta per le molecole diatomiche, anche non

Capitolo 2. Introduzione all’effetto Raman 41
polari come H2, N2 e CO2. Per molecole simmetricamente tetraedriche come CH4 e CCl4, al contrario, si ha che α⊥=α//, cosicché non c’è effetto Raman rotazionale. Quanto-meccanicamente il processo viene visto in termini di uno scattering anelastico del fotone che acquista o cede alla molecola due quanti rotazionali secondo la regola di selezione ∆J = 0, ±2 (dove J è il numero quantico rotazionale). In particolare nel caso più semplice di un rotore rigido i livelli di energia rotazionale sono dati da EJ = BJ(J+1). Si ottiene così che lo shift del numero d'onda delle linee Raman rotazionali è:
( )( ) ( )[ ] [ ]64132 +±=+−++±= JBJJJJBrotν (2.19.1) Quindi il numero d'onda di una linea Raman rotazionale è rotp ννν ±= . In una transizione con ∆J = +2, la molecola raggiunge uno stato rotazionale più alto poiché il fotone gli ha ceduto due quanti rotazionali di energia. Analogamente, per ∆J = -2 è vero il contrario. Risulta ora chiara la struttura dello spettro rotazionale Raman mostrato in figura 2.19.1. La prima riga rotazionale Raman con J = 0 è posta ad una distanza 6B da pν , la riga primaria, dopodiché le altre linee seguono con spaziatura costante di 4B. Per quanto riguarda le intensità delle righe rotazionali esse, analogamente a quanto detto per le linee vibrazionali, dipendono dalla popolazione termodinamica degli stati rotazionali. La figura 2.19.2 mostra che, a causa della piccolezza dei quanti rotazionali, il numero di linee rotazionali in uno spettro può essere molto grande in quanto grande è il numero dei diversi stati rotazionali eccitati occupabili. Inoltre, poiché la differenza tra le popolazioni termiche di stati rotazionali vicini è molto piccola in confronto a kT, risulta che le intensità delle linee Raman rotazionali Stokes e anti-Stokes sono pressoché uguali. Osserviamo che, nel caso di molecole più complesse di un semplice rotore lineare, la regola di selezione su ∆J cambia. Per esempio per CH3Cl abbiamo ∆J = 0, ±1, ±,2. Il suo spettro Raman rotazionale avrà molte più righe di quello di un rotore lineare. Si è ora in grado di comprendere uno spettro Raman rotazionale. Ogni riga vibrazionale è accompagnata da due serie di righe rotazionali che possono interpretarsi allo stesso modo che per la struttura rotazionale in uno spettro roto-vibrazionale infrarosso, ma tenendo conto delle diverse regole di selezione per le transizioni Raman. La figura 2.19.3 mostra, come esempio, una sezione dello spettro di 16O2 ovvero la riga vibrazionale a =eν 1556 cm-1, insieme alle sue righe rotazionali. La struttura dello spettro Raman corrisponde a quello di uno spettro vibrazionale infrarosso, ma la differenza sta nelle regole di selezione che per il Raman, sono ∆v = ±1, (±2,…) e ∆J = 0, ±2,. La figura 2.19.4 illustra la differenza tra spettri Raman e spettri roto-vibrazionali infrarossi. Confrontiamo ora lo spettro roto-vibrazionale infrarosso con il corrispondente spettro Raman, rifacendoci alla figura 2.19.4. In uno spettro roto-vibrazionale di una molecola diatomica, per ∆v = 1 abbiamo : Ramo R rotvib νν + , ∆J = 1, )1(2 += JBrotν Ramo P rotvib νν − , ∆J = -1, BJrot 2=ν e eventualmente Ramo Q vibν , ∆J = 0 Diversamente nello spettro Raman, per esempio per le linee Stokes con ∆v = +1 Ramo S ),( rotvibp ννν +− ∆J = 2 Ramo Q vibp νν − ∆J = 0 Ramo O ),( rotvibp ννν −− ∆J = -2
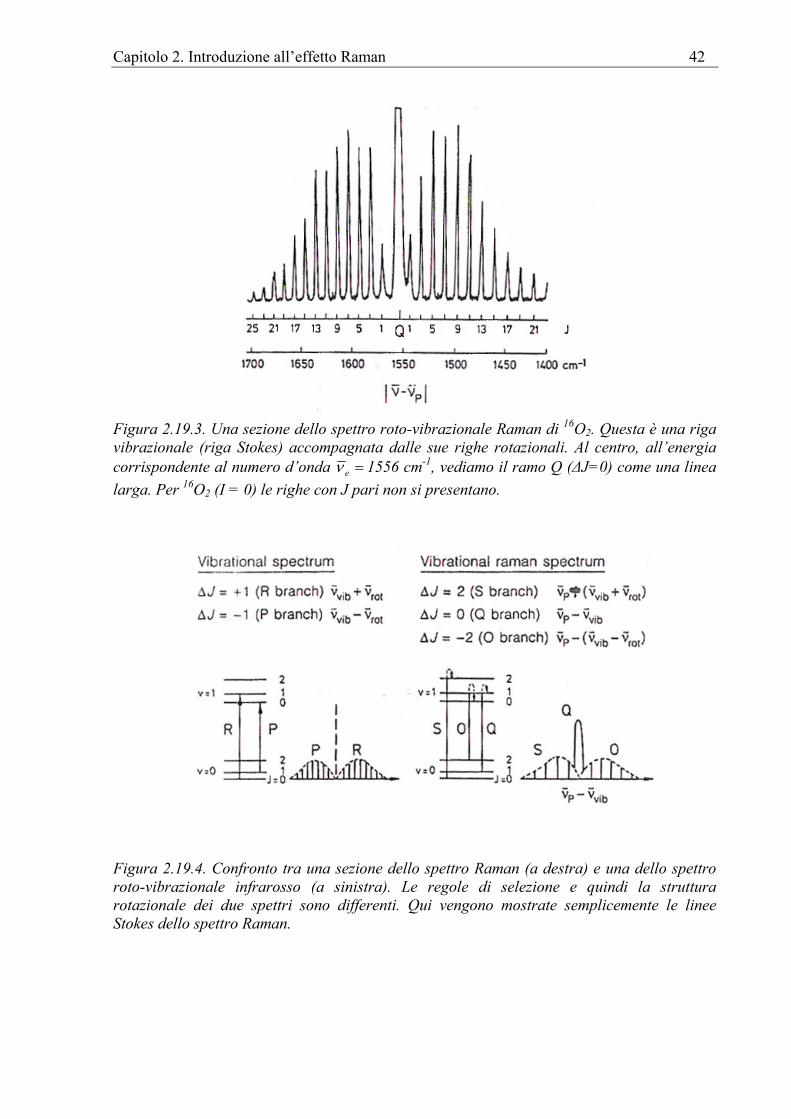
Capitolo 2. Introduzione all’effetto Raman 42
Figura 2.19.3. Una sezione dello spettro roto-vibrazionale Raman di 16O2. Questa è una riga vibrazionale (riga Stokes) accompagnata dalle sue righe rotazionali. Al centro, all’energia corrispondente al numero d’onda =eν 1556 cm-1, vediamo il ramo Q (∆J=0) come una linea larga. Per 16O2 (I = 0) le righe con J pari non si presentano.
Figura 2.19.4. Confronto tra una sezione dello spettro Raman (a destra) e una dello spettro roto-vibrazionale infrarosso (a sinistra). Le regole di selezione e quindi la struttura rotazionale dei due spettri sono differenti. Qui vengono mostrate semplicemente le linee Stokes dello spettro Raman.

Capitolo 2. Introduzione all’effetto Raman 43
2.20 L’origine vibrazionale delle frequenze di gruppo Date le masse, la geometria molecolare e le costanti di forza, esistono dei metodi matematici per il calcolo del tipo e della frequenza di tutti i modi normali di vibrazione limitatamente al caso di molecole relativamente semplici. Infatti, appena le molecole diventano più complesse, la difficoltà della trattazione matematica cresce enormemente, e al suo posto si preferiscono metodi empirici. Si è riscontrato sperimentalmente che certi gruppi di atomi submolecolari producono bande sempre in una caratteristica regione dello spettro vibrazionale. Queste bande sono dette frequenze di gruppo. Tutti gli alcani, ad esempio, avranno in comune alcune bande di frequenza, ma, tuttavia, ognuno di essi avrà un certo numero di bande caratteristiche della particolare specie molecolare. Gli effetti dell’interazione meccanica che controllano la forma della vibrazione sono relativamente uguali da molecola a molecola, rendendo così la frequenza facilmente prevedibile. Gli effetti dell’interazione meccanica possono aver luogo solo tra vibrazioni appartenenti alla stesso tipo di simmetria. Ad esempio, in una molecola con un piano di simmetria, le vibrazioni nel piano non potranno interagire con le vibrazioni fuori del piano. In questo paragrafo tratteremo come esempio, soltanto alcuni casi più semplici, ovvero gli oscillatori accoppiati simmetrici ed asimmetrici.
Oscillatori diatomici Si consideri un modello molecolare diatomico. La frequenza vibrazionale è data da
)11(21
21 MMF +=
πν (2.20.1)
dove M1 e M2 sono le masse in grammi e F è la costante di forza in dyne/cm. Se le masse vengono espresse in unita di massa atomiche (u) e la costante di forza è espressa in millidyne/angstrom, allora la frequenza in cm-1 è data da:
)11(130321 MM
F +=ν (2.20.2)
dove 1303 è uguale a (NA x 105)1/2/(2πc)e NA è il numero di Avogadro 6.02252 x 1023 mole-1.
Oscillatori accoppiati Una molecola complessa consiste di un certo numero di atomi legati. Usiamo il termine oscillatore per ogni legame tra due atomi che possono oscillare attorno ad una posizione di equilibrio sia in termini di lunghezza che di angoli di vibrazione. Due di questi oscillatori si dicono accoppiati se quando oscillano interagiscono in modo tale da esercitare reciprocamente forze l’uno sull’altro. Consideriamo il caso più semplice, il modello lineare M-M-M costituito da due oscillatori diatomici M-M accoppiati. L’accoppiamento ha luogo poiché il movimento della massa comune M a seguito della vibrazione di uno degli oscillatori M-M, inevitabilmente influenza anche l’altro oscillatore. La frequenza dell’oscillatore M-M, in cm-1, è data da:
21
21
)2()/(1303 MF=ν Un altro modello diatomico M-(M/2) dove la massa M è legata ad una massa uguale a (M/2) ha una frequenza, in cm-1, che è data da:
21
21
)3()/(1303 MF=ν

Capitolo 2. Introduzione all’effetto Raman 44
In figura 2.20.1 sono rappresentati due di questi modelli M-(M/2) vibranti con sfasamento di 180° uno accanto all’altro con uguali ampiezze. I due oscillatori separati vibrano con centri di gravità stazionari e le due masse (M/2) entrambe vibrano sinusoidalmente e rimangono alla stessa distanza durante la vibrazione. Dal momento che la distanza è costante, essa può essere posta a zero come in figura 2.20.1(b), dove è rappresentato un modello lineare M-M-M durante un modo di vibrazione normale di stretching fuori fase. Si osserva infatti che il centro di gravità è stazionario e tutti gli atomi si muovono sinusoidalmente con la stessa frequenza e passano attraverso il punto di equilibrio simultaneamente. La frequenza e le ampiezze sono uguali a quelle delle componenti diatomiche in figura 2.20.1(a).
Figura 2.20.1. Scomposizione delle vibrazioni di stretching del modello lineare triatomico M-M-M nelle componenti diatomiche M-(M/2) e M-(∞). Consideriamo ora un modello diatomico M-(∞), dove la massa M è legata ad una massa infinita. La frequenza di vibrazione, in cm-1, è:
21
21
)1()/(1303 MF=ν In figura 2.20.1(c) sono rappresentati due di tali oscillatori vibranti in fase con uguali ampiezze. Essendo le masse nel mezzo infinite, esse non si muovono e la loro distanza rimane costante durante la vibrazione. Questa distanza può essere posta a zero combinando praticamente queste due masse infinite in un’unica massa anch’essa infinita. Quando si guarda alla risultante delle due forze esercitate dalle due molle sulla massa centrale, si vede che questa forza è nulla in ogni istante. Questo significa che la massa centrale può avere qualsiasi valore da infinito a zero senza mai muoversi. In figura 2.20.1(d) la massa centrale è stata posta uguale a M ed è stato rappresentato un modello M-M-M durante un modo normale di vibrazione di stretching in fase la cui frequenza e le ampiezze sono le stesse delle componenti diatomiche in figura 2.20.1(c). Questi risultati possono essere così riassunti.
M-M 21
21
)2()/(1303 MF=ν

Capitolo 2. Introduzione all’effetto Raman 45
In fase M-M-M 21
21
)1()/(1303 MF=ν
Fuori fase M-M-M 21
21
)3()/(1303 MF=ν Se ignoriamo gli atomi di idrogeno, possiamo utilizzare questi risultati per confrontare lo stretch C=C dell’etilene H2C=CH2 a 1623 cm-1 con lo stretch in fase e fuori fase C=C=C dell’allene H2C=C=CH2 rispettivamente a 1071 e 1980 cm-1, dove i rapporti delle frequenze non sono molto diversi dai rapporti 21/2:11/2:31/2 previsti grazie a questo semplice modello. Da quanto detto possiamo concludere che i due legami C=C nell’allene non vibrano separatamente. Poiché uno dei legami C=C è esattamente uguale all’altro, entrambi sono eccitati vibrazionalmente allo stesso istante. Se si parte dall’equilibrio ed un legame comincia ad allungarsi, l’altro legame deve modificarsi in lunghezza in uno dei due possibili modi. Può allungarsi in fase con l’altro legame oppure contrarsi fuori fase rispetto all’altro legame. Durante lo stretch fuori fase rappresentato nella figura 2.20.1(b) le forze (rappresentate da frecce) esercitate dalle due molle sono dirette nella stessa direzione sulla massa centrale cosicché qui la risultante è nulla. Si può dire che le due forze cooperano nel ristabilire le lunghezze dei legami. La condizione di “cooperazione” tende ad innalzare la frequenza di combinazione dell’oscillatore rispetto alla frequenza di un singolo oscillatore M-M. Durante lo stretch in fase in figura 2.20.1(d) le forze delle due molle operano in opposizione sulla massa centrale cercando di ristabilire la lunghezza di equilibrio dei legami. La condizione di “opposizione” tende ad abbassare la frequenza di combinazione rispetto alle componenti di un singolo oscillatore M-M. Queste conclusioni ritornano utili quando si vuole analizzare il tipo di vibrazioni nelle frequenze di gruppo. Dal momento che è ora stato compreso che due oscillatori accoppiati ed uguali vibreranno insieme, in fase o fuori fase, si può tornare ad osservare le componenti di un oscillatore diatomico. In figura 2.20.1(b) e (d) ogni massa subisce una forza da ogni molla. Queste forze, secondo la legge di Hook, sono date da FS dove F è la costante di forza e S è l’allungamento della molla. Le figure 2.20.1(e) e (g) mostrano due singoli oscillatori estrapolati dalle figura 2.20.1(b) e (d), rispettivamente. Entrambi differiscono da un oscillatore diatomico convenzionale in quanto le forze sulle due masse sono diverse. Queste masse vibreranno con la stessa frequenza e stesse ampiezze delle masse degli oscillatori diatomici convenzionali rappresentati nelle figure 2.20.1(f) e (h) dove forze uguali (FS) agiscono su ogni massa, ma le masse sono state sostituite cosicché il rapporto forza/massa è lo stesso degli oscillatori rappresentati nelle figure sopra. Il moto vibrazionale di una massa è definito dalla sua accelerazione (forza/massa). Se questo rapporto non cambia il moto vibrazionale si mantiene uguale. Con quanto detto abbiamo scomposto le vibrazioni in fase e fuori fase del modello lineare M-M-M in componenti diatomiche le cui frequenze in cm-1 possono essere ottenute dalla eq. 2.20.2. Usando le stesse argomentazioni applicate per il modello M-M-M, le frequenze di stretching, in cm-1, per il modello simmetrico lineare M1-M2
-M1 risultano:
in fase M1-M2-M1 )1(13031M
F=ν (4.6)
fuori fase M1-M2-M1 )21(130321 MM
F +=ν (4.7)

Capitolo 2. Introduzione all’effetto Raman 46
Oscillatori accoppiati asimmetrici Le vibrazioni in fase e fuori fase di stretching del modello M-M-M sono rappresentate nuovamente nella figura 2.20.2(a) e (d). Il problema è capire come queste vibrazioni vengono modificate se i due oscillatori diventano diversi. In figura 2.20.2(b) e (c) è stata diminuita la frequenza dell’oscillatore a destra rispetto a quello a sinistra. Questo è stato ottenuto in figura 2.20.2(b) rendendo la costante di forza a destra nulla (nessuna molla) e in figura 2.20.2(c) rendendo la massa a destra infinita. Per questi due casi estremi si può immediatamente illustrare la forma della vibrazione fuori-fase dove si nota che mentre entrambe le lunghezze dei legami variano, la massa a destra dell’oscillatore a bassa frequenza è ferma (∆X=0). In figura 2.20.2(b) questo avviene perché le forze su questa massa sono nulle. In figura 2.20.2(e) e (f) si è innalzata la frequenza dell’oscillatore destro. Questo è stato ottenuto in figura 2.20.2(e) rendendo la costante di forza della forza a destra infinita (legame rigido) e in figura 2.20.2(f) rendendo la massa a destra nulla. Per questi due casi estremi si può immediatamente illustrare la forma della vibrazione a bassa frequenza dove si nota che la lunghezza del legame dell’oscillatore a destra ad alta frequenza non varia (∆S=0). In figura 2.20.2(f) la molla destra non può distorcersi (tranne che ad una infinitamente alta frequenza) a causa della massa nulla alla sua estremità. Naturalmente, se i cambiamenti operati su uno dei due oscillatori non sono così estremi come quelli discussi, gli effetti sulla forma della vibrazione saranno intermedi tra questi e quelli degli oscillatori simmetrici accoppiati in figura 2.20.2(a) e (d).
Figura 2.20.2. Le vibrazioni di stretching di modelli triatomici asimmetrici (le due righe in basso) confrontate con quelle di un modello simmetrico (riga superiore). Gli spostamenti degli atomi sono indicati da frecce e le variazioni dei legami sono indicate da S per gli allungamenti e da C per le contrazioni. Da quanto detto possiamo enunciare due importanti approssimazioni, che sono molto utili per l’analisi della forma delle vibrazioni delle frequenze di gruppo.

Capitolo 2. Introduzione all’effetto Raman 47
1. Nella vibrazione ad alta frequenza di un oscillatore asimmetrico, i soli atomi che si muovono sono quelli della componente dell’oscillatore a più alta frequenza.
2. Nella vibrazione a bassa frequenza di un oscillatore asimmetrico, l’unico legame (angolo) che viene deformato è quello dell’oscillatore a più bassa frequenza
Queste approssimazioni migliorano quando le frequenze delle componenti degli oscillatori sono molto diverse e peggiorano invece quando le frequenze si approssimano. La prima approssimazione indica che le vibrazione di stretching dell’idrogeno (X-H), le vibrazioni di stretching a triplo legame (C≡C, C≡N), e le vibrazioni di stretching a doppio legame (C=C, C=N, C=O, P=O, S=O) dovrebbero tutte avere distinte frequenze di gruppo quando questi gruppi sono uniti al resto della molecola tramite legami X-Y a bassa frequenza e l’atomo intermedio è carbonio o un atomo più pesante. Dal momento che l’atomo di giunzione è praticamente immobile, la vibrazione è meccanicamente insensibile alla natura del gruppo attaccato. Un gruppo come C=S, avendo una frequenza prossima a quelle dei legami C-N, C-O, oppure C-C che lo attaccano al resto della molecola, dovrebbe avere una frequenza di gruppo debole. La ragione è che può potrebbe esserci una forte interazione C=S con gli oscillatori a singolo legame attaccati. Questi possono a loro volta interagire con altri oscillatori \a legame singolo attaccati ad essi. Ciò rende la vibrazione C=S meccanicamente sensibile alla natura dei gruppi attaccati.
2.21 La Fluorescenza Lo studio degli spettri Raman è spesso reso difficile dalla presenza di un fondo dovuto all’emissione di fluorescenza. L'emissione della luce dai materiali ha origine da due meccanismi: l'emissione termica e la luminescenza. Mentre nel primo processo tutti gli atomi del solido partecipano all'emissione di luce, nella luminescenza solo un numero limitato di atomi (impurità o difetti del cristallo) vengono eccitati e prendono parte all'emissione di luce. Le impurità e/o i difetti insieme agli atomi circostanti formano un centro luminescente o centro ottico. L'assorbimento e l'emissione di luce da un atomo possono essere descritti, in uno schema molto semplificato, da un oscillatore armonico, come mostrato in figura (2.21.1), composto da una carica positiva (+e) fissata a z=0 e da un elettrone legato e oscillante attorno ad essa lungo l'asse z.

Capitolo 2. Introduzione all’effetto Raman 48
Figura 2.21.1. La radiazione elettromagnetica proveniente da un dipolo elettrico oscillante. La lunghezza della freccia è proporzionale all’intensità della radiazione in ciascuna direzione. Il momento elettrico di dipolo dell'oscillatore con frequenza angolare ω0, è dato da
)exp( 00 tiMezM ω== (2.21.1) e la sua energia, somma dell'energia cinetica e potenziale, è dove m2
022
0 )2/( Memeω e è la massa dell'elettrone. Tale dipolo elettrico vibrante trasferisce energia alla radiazione elettromagnetica con un rate medio di per secondo e quindi ha un rate di decadimento energetico totale dato da
20
30
40 )12/( Mcπεω
30
20
2
0 6 cme
Aeπε
ω= (2.21.2)
Quando la variazione di energia dell'oscillatore è espressa come una funzione esponenziale , la sua costante di tempo τ0/τte−
o è uguale Ao-1, che altro non è che la vita media
dell'oscillatore, cioè il tempo richiesto dall'oscillatore affinché la sua energia diminuisca fino a e-1 dell'energia iniziale. Si può dimostrare che la vita media di un oscillatore radiante con lunghezza d'onda di 600 nm è τo = 10-8sec. L’interpretazione quantistica del fenomeno è rappresentata nella figura 2.21.2 che riassume i principali processi di interazione tra la radiazione luminosa e le molecole.

Capitolo 2. Introduzione all’effetto Raman 49
Figura 2.21.2. (a) Diffusione Rayleigh; (b) Raman non risonante, Stokes ed anti-Stokes; (c) Effetto Raman pre-risonante; (d) Effetto Raman di risonanza; (e) Fluorescenza. Il caso e in figura 2.21.2 mostra, in particolare, la successione degli eventi che si concludono con la fluorescenza. L’assorbimento iniziale della radiazione incidente porta la molecola in uno stato elettronico eccitato. A questo punto, essa discende la scala dei livelli vibrazionali, cedendo energia alle molecole circostanti tramite urti, (decadimento non radiativo), fino a raggiungere il livello vibrazionale più basso dello stato elettronico eccitato. Si verifica adesso la transizione radiativa a partire dallo stato vibrazionale fondamentale dello stato elettronico superiore verso lo stato vibrazionale fondamentale dello stato elettronico inferiore. Poiché la transizione emissiva ha luogo dopo che si è scaricata nell’ambiente una certa quantità di energia vibrazionali, l’emissione di fluorescenza si verifica ad una frequenza più bassa rispetto a quella della radiazione eccitante. Il tempo per cui la molecola rimane nello stato eccitato si chiama tempo di vita e si indica con τ. Gli spettri di fluorescenza possono essere utilizzati, al pari di quelli Raman, per la caratterizzazione dei materiali.

Capitolo 3. Strumentazione per Raman dispersivo 50
Capitolo 3. Strumentazione per Raman dispersivo
3.1 Introduzione La figura 3.1.1 mostra la prima strumentazione che permise nel 1928 di scoprire l’effetto Raman all’omonimo fisico indiano.
Figura 3.1.1. La foto ritrae Raman nel 1928 insieme al primo apparato per la spettroscopia Raman Essenzialmente un sistema dispersivo per l’acquisizione di spettri Raman si serve di 5 componenti, 1) Sorgente monocromatica 2) Spettroscopio 3) Sistema di campionamento 4) Rivelatore 5) Sistema di gestione ed elaborazione Nei paragrafi successivi illustriamo brevemente le singole componenti.
3.2 Le sorgenti La possibilità di distinguere le righe Raman implica necessariamente che la sorgente eccitatrice sia la più monocromatica possibile, al fine di avere una sola e stretta riga di Rayleigh, da cui le righe Raman possano quindi ben distinguersi. I laser più comunemente usati in spettroscopia Raman vanno dal violetto (488nm) agli infrarossi (1060nm) nel qual caso si parla di FT-Raman. Benché lo shift Raman sia indipendente dalla lunghezza d’onda di eccitazione, λ, non è così per l’intensità delle bande. È nota la dipendenza dell’intensità delle righe Raman, IR, da νP (frequenza della luce primaria), IR = f(νP-∆ν)4, dove ∆ν è lo shift Raman. Si verifica che, sebbene IR cresca al diminuire della νp, la fluorescenza aumenta anch’essa, fino a poter coprire completamente il debole segnale Raman. Bisogna quindi, di volta in volta, scegliere opportunamente il laser più adatto, per ogni tipologia di campioni. Per un campione molto fluorescente converrà usare laser negli infrarossi (1060 nm) che stimolano pochissima

Capitolo 3. Strumentazione per Raman dispersivo 51
fluorescenza, in tal modo le righe Raman sebbene deboli riescono ad essere distinte. Segue una breve descrizione dei principi di funzionamento dei laser: I Laser I primi due diagrammi di figura 3.2.1 mostrano rispettivamente il fenomeno dell'emissione e dell'assorbimento spontaneo. Sia A* uno stato eccitato di A, i processi possono essere espressi come: A*→A+γ ed γ+A→A* dove γ rappresenta un fotone Il terzo diagramma invece introduce il fenomeno dell'emissione stimolata: γ + A* → A + γ + γ In questo fenomeno un fotone incidente di energia hν induce la diseccitazione dell'atomo con la contemporanea emissione di due fotoni entrambi con energia hν, costituendo così una sorgente di onde elettromagnetiche monocromatica e coerente, essendo i due fotoni in fase. Questi ultimi fotoni possono a loro volta interagire con altri atomi analogamente eccitati, causando così un processo a valanga che aumenta l'intensità della radiazione. La parola laser è infatti l'acronimo di Light Amplification by Stimulated Emission of Radiation.
Figura 3.2.1. Schema del fenomeno dell’emissione e dell’assorbimento spontaneo.
3.3 Lo spettroscopio Esistono diverse tipologie di spettroscopi. Oltre che disperdere lo spettro è necessario separare l’intenso scattering di Rayleigh da quello Raman. I primi spettroscopi Raman erano molto complessi e di difficile uso (spettroscopi a triplo stadio) ma oramai questi sono stati sostituiti da modelli molto più semplici che utilizzano i filtri notch per separare le due componenti scatterate, Raman e Rayleigh. Di seguito diamo qualche accenno sui filtri notch e sugli spettroscopi dispersivi (che usano cioè un reticolo di diffrazione). Il filtro notch Il filtro notch ha il compito di separare l’emissione Rayleigh da quella diffusa per effetto Raman perché altrimenti la diffusione di Rayleigh, da 106 a 10 12 volte più intensa di quella

Capitolo 3. Strumentazione per Raman dispersivo 52
Raman, coprirebbe totalmente quest’ultima. Il filtro blocca la luce con la stessa frequenza del laser utilizzato e fa passare le altre frequenze. Osserviamo però che la luce con shift del numero d’onda minore e maggiore di circa 100 cm-1 rispetto a quello del laser, viene ugualmente riflessa non permettendo quindi l’analisi della luce Raman di tali frequenze. La figura 3.3.1 mostra la curva di trasmissione per un filtro notch, un Holographic Super-Notch-Plus™ Filter prodotto dalla Kaiser Optical System Inc. e centrato a 532 nm.
Figura 3.3.1 Curva di trasmissione per un notch centrato a 532 nm Come può notarsi dalla figura il notch possiede effettivamente una stretta banda di filtraggio che impedisce la trasmissione della luce con frequenza vicina a 532 nm e garantisce invece un’ottima trasmissione (~80%) delle altre frequenze. I limiti imposti da tale notch non sono tuttavia così restrittivi. La maggior parte delle applicazioni della spettroscopia Raman richiede di lavorare in una regione tra 100 cm-1 e 4000 cm-1 pienamente coperta dal nostro filtro notch. Lo spettroscopio Diamo innanzitutto qualche accenno sul reticolo di diffrazione che è la componente fondamentale di ogni spettroscopio Esso è oggi prodotto fondamentalmente in due modelli ognuno con i propri vantaggi e svantaggi, il reticolo classico e quello olografico. Vediamo brevemente i principi su cui si basa il reticolo di tipo classico. Esso consiste di un supporto con un gran numero di fenditure parallele equi distanziate. La figura 3.3.2 mostra uno schema della sezione di un reticolo di diffrazione piano.

Capitolo 3. Strumentazione per Raman dispersivo 53
Figura 3.3.2 Sezione reticolo di diffrazione piano Un’onda piana incide da sinistra perpendicolarmente al piano del reticolo. Si usa una lente convergente per convergere i raggi nello stesso punto P. L’intensità della figura che si osserva nello schermo è il risultato degli effetti combinati dell’interferenza e della diffrazione. Ogni fenditura produce diffrazione e a loro volta i fasci diffratti interferiscono tra loro producendo la figura finale. Osserviamo che le onde che partono dalle fenditure sono tutte in fase, tuttavia, per una direzione arbitraria θ rispetto al piano del reticolo, le onde devono percorrere cammini di diversa lunghezza prima di arrivare su di un particolare punto P dello schermo. Dalla figura 3.3.2 si nota che la differenza di cammino tra onde provenienti da fenditure adiacenti è pari a dsenθ. Se questa differenza eguaglia una lunghezza d’onda o un suo multiplo intero le onde provenienti da tutte le fenditure saranno in fase in P e si osserverà una linea luminosa. Allora la condizione per avere i massimi nella figura d’interferenza all’angolo θ è: dsenθ = mλ (m=0,1,2,3,…) (3.3.1) m si definisce numero d’ordine del massimo mentre d è la spaziatura del reticolo detta passo reticolare. Se ora la radiazione incidente contiene diverse lunghezze d’onda, per ognuna di esse il massimo di ordine m si troverà ad un angolo specifico, mentre a θ = 0 si vedranno tutte le lunghezze d’onda in corrispondenza a m=0 (massimo d’ordine zero). La distribuzione d’intensità per il reticolo di diffrazione nel caso di una sorgente monocromatica è mostrata schematicamente in figura 3.3.3.
Figura 3.3.3. Distribuzione d’intensità per un reticolo di diffrazione Se la sorgente contiene diverse lunghezze d’onda si osserveranno altrettanti massimi per ogni ordine. A differenza delle larghe frange chiare prodotte da due fenditure, le frange di un

Capitolo 3. Strumentazione per Raman dispersivo 54
reticolo sono molto più strette e gli intervalli di zone scure molto più larghi. Possiamo mostrare un semplicissimo schema di uno spettroscopio a reticolo di diffrazione in figura 3.3.4.
Figura 3.3.4. Schema spettroscopio La luce da analizzare viene fatta passare attraverso una fenditura quindi, grazie ad un collimatore o ad uno specchio collimatore, si ottiene un fascio di luce parallelo che incide perpendicolarmente sul reticolo. La luce diffratta emerge da quest’ultimo ad angoli che soddisfano l’eq. 3.3.1. .L’osservazione di questi angoli permette di risalire alla lunghezza d’onda. Altra grandezza caratterizzante un reticolo è il suo potere risolutivo R R = λ / λ2-λ1 = λ / ∆λ Dove λ1 e λ2 sono due lunghezze d’onda tanto vicine da essere appena risolvibili secondo il criterio di Rayleigh col reticolo in oggetto e ∆λ = λ2-λ1, λ ≅ λ1,λ2 Quindi un reticolo con alto potere risolutivo può distinguere piccole differenze di lunghezza d’onda. Esso dipende dall’ordine con cui si lavora e dal numero N di fenditure illuminate. Si può infatti dimostrare che il potere risolutivo dell’m-esimo ordine è R = Nm
3.4 I sistemi di campionamento
Essenzialmente ci sono tre tipi di campionamento, macro, micro e remoto. Il macro permette di analizzare campioni macroscopici, il campionamento micro che prevede l’uso delle ottiche di un microscopio, campioni microscopici, mentre il sistema remoto usa una testa di campionamento per eseguire misure micro in remoto, sfruttando delle fibre ottiche. Campionamento macro I liquidi e i solidi campionati in macro, vengono irraggiati col laser e lo scattering Raman può essere osservato o a 90°, oppure in back scattering, come illustrato in figura 3.4.1
Figura 3.4.1. Sistemi di campionamento Macro.

Capitolo 3. Strumentazione per Raman dispersivo 55
campionamento micro Hirscfeld suggerì di applicare la spettroscopia Raman anche a
ebole, in presenza di un
stato allora ottimizzato con i seguenti accorgimenti: utilizzando collettori con
campionamento remoto o remoto basato sull'uso delle fibre ottiche costituisce un mezzo
el Raman
nel Micro-Raman
IlFu nel 1969 che Tomas campioni di dimensioni dell’ordine del micron con l’ausilio di un microscopio. L’idea era quella di focalizzare, eccitare il campione microscopico e quindi raccogliere in back scattering la radiazione Raman, il tutto attraverso le ottiche di un microscopio. Molti furono i dubbi sollevati circa l’effettiva realizzabilità di un tale sistema, in particolar modo preoccupava il rapporto segnale-rumore che se ne sarebbe ricavato. Ci si trovava nella necessità di rivelare un segnale Raman molto dnotevole scattering elastico e anelastico rispettivamente 1012 e 104 ordini di grandezza maggiori. Il sistema ègrande apertura numerica e minimizzando il contributo proveniente dallo scattering elastico e anelastico di tutte le possibili sorgenti diverse dal campione. Queste due condizioni si traducono nell’uso di obiettivi con la maggiore apertura numerica disponibile e di ottiche del microscopio e del supporto del campione che se eccitati emettano il minore segnale Raman possibile. IlIl sistema di campionamentversatile per l'esecuzione delle misure in svariati ambienti e per i più diversi scopi. I vantaggi di un sistema remoto sono importanti, in particolare, per l’applicabilità dai beni culturali. Infatti, non è sempre possibile e/o permesso estrarre campioni dai manufatti, o semplicemente prelevarli dal loro ambiente di conservazione. L’uso di un sistema remoto permette di superare tali limiti. Un caso particolare è quello dell’analisi di oggetti immersi dentro vari tipi di liquidi [Smith 2003]. Basi fisiche: fibre ottiche nel Raman e
he. Una volta che la luce penetra Diamo qualche accenno sul funzionamento delle fibre otticnella fibra entro un cono di semi angolo α, esso è totalmente trasmesso all'interfaccia tra i due mezzi di indici di rifrazione n1 ed n2, e α è dato da (vedi figura 3.4.2). senα = (n1
2 - n22 )1/2 /n0
igura 3.4.2. Schema di una fibra ottica
sserviamo che: ggio di luce emergente è anch'esso conico, con lo stesso semi angolo se
F OA) All'uscita il ral'indice di rifrazione del mezzo d'entrata e d'uscita sono uguali.

Capitolo 3. Strumentazione per Raman dispersivo 56
B) In base alla natura del core e del rivestimento interno della fibra le fibre ottiche in commercio presentano coni di accettazione con semiangolo compreso tra 120 e 340. Per quanto riguarda l’accoppiamento della fibra ottica con il mezzo, quando la fibra ottica è immersa in un mezzo, il raggio laser con potenza P produce all'uscita un locale irraggiamento I che varia rapidamente nello spazio. Per un campione sottile di superficie S direttamente a contatto con la fibra, l'irraggiamento I è massimo, I = I max =P/S (Watt/m2). Se ora invece consideriamo un volumetto dV posto a distanza z >> D (diametro fibra), lungo l'asse ottico, l'irraggiamento decresce rapidamente come 1/z2 , vedi figura 3.4.3.
Figura 3.4.3. La fibra ottica di eccitazione del campione. Più dettagliatamente se assimiliamo il raggio di luce ad un cono di radiazione con semiangolo α tangente al bordo della fibra, prodotto da una sorgente puntiforme localizzata dentro la fibra stessa, otteniamo: I(z) = I(0) z0
2 / ( z+ z02)
Considerazioni analoghe possono essere avanzate per la valutazione dell'efficienza di raccolta. Si può infatti dimostrare che il cono di luce con angolo solido Ω raccolto all'entrata della fibra, è massimo per un campione a diretto contatto con il bordo della fibra e anche l'efficienza di raccolta decresce rapidamente come 1/z2. Sono possibili diverse configurazioni tra le fibre per la raccolta e per l'eccitazione. Diversi sono i modi di utilizzare le fibre per la realizzazione di un sistema remoto: 1) Fibra singola: In un'unica fibra passano sia il fascio laser che le emissioni Raman. 2) Due fibre parallele: Una fibra guida l’emissione di eccitazione laser e una raccoglie il segnale Raman. Esse vengono montate parallelamente sulla testa remota e puntano due punti distinti del campione. La radiazione Raman raccolta è quella proveniente dalla zona d’intersezione tra la superficie irradiata dal laser e quella coperta dalla fibra di raccolta. 3)Due fibre convergenti: Analogo al caso sopra ma con le fibre che mirano ad un unico punto sul campione. Sistema della Dilor: E' il sistema, utilizzato nel nostro lavoro, brevettato dalla DILOR che sostanzialmente utilizza un opportuno filtro Notch, vedi figura 3.4.
Figura 3.4.4. Schema della testa di campionamento remoto prodotta dalla Dilor.

Capitolo 3. Strumentazione per Raman dispersivo 57
Utilizzando un sistema Raman remoto si presenta un problema legato agli spettri Raman delle fibre ottiche. Le numerose riflessioni della radiazione all'interno della fibra ottica generano infatti un ' intenso scattering Raman da parte del materiale costituente la fibra. Tale spettro Raman influisce sulle capacità analitiche del sistema, dal momento che il suo range è abbastanza ampio. La condizione peggiore si ha nel caso del sistema con una sola fibra, tuttavia anche nel caso in cui si usino fibre separate, per l'eccitazione e per la raccolta, lo spettro delle fibre viene acquisito insieme a quello del campione. Il sistema della DILOR costituisce una soluzione ottimizzata a tale problema: la radiazione laser trasportata dalla fibra ottica prima di raggiungere il campione viene filtrata da un filtro interferenziale cosicché il campione viene eccitato da radiazione alla sola frequenza del laser e non anche dallo scattering Raman della fibra. Il segnale Raman raccolto e separato dallo scattering di Rayleigh grazie al filtro notch, è, infine, generalmente tanto debole che possiamo trascurarne il contributo nella generazione di scattering Raman della fibra che attraversa per giungere fino allo spettroscopio. Un punto cruciale per la realizzazione delle misure riguarda l’adattamento dei vari obiettivi ai diversi campioni. La risoluzione spaziale e la profondità di osservazione sono strettamente legate alla lunghezza focale e alla apertura numerica dell'obiettivo, insieme al diametro delle fibre di entrata e di uscita che giocano il ruolo di diaframmi confocali.
3.5 I rivelatori Rivelatori monocanale Sono i fotomoltiplicatori otticamente connessi alla fenditura d’uscita dello spettrometro che muovendo il reticolo presenta alla fenditura una lunghezza d’onda alla volta. Tali rivelatori sono caratterizzati da elevata sensibilità ed ottima linearità di risposta, bassa velocità d’acquisizione, massima risoluzione. Rivelatori multicanale Sono le CCD. Esse sono costituite da una schiera (array) di fotodiodi al silicio e da un sistema di raffreddamento. Vengono accoppiate alla porta spettroscopica, cioè al dispositivo ottico d’interfaccia tra la fenditura d’uscita dello spettroscopio e la CCD. Quando l’otturatore viene aperto, ogni pixel che viene colpito da un fotone produce una carica. Le cariche di tutti i pixel vengono raccolte separatamente grazie ad una griglia di elettrodi. Quando dopo un tempo T di acquisizione, detto tempo di integrazione, l’otturatore viene chiuso, l’unità di controllo della CCD gestisce la tensione di ogni elettrodo così da convogliare la carica raccolta da ogni singolo pixel in un registro di lettura, Il segnale proveniente dalla CCD viene quindi elaborato, amplificato e convertito in digitale dall’elettronica dell’unità di controllo della CCD e da quest’ultima arriva alla memoria del PC. Si noti che, a causa della dispersione operata dal reticolo, i pixel orizzontalmente adiacenti ricevono fotoni di λ diverse. Ciò permette di acquisire con la CCD un range spettrale abbastanza ampio pur mantenendo fermo il reticolo e quindi con un’unica misura, che richieda ad esempio un tempo ∆t. Diversamente i fotomoltiplicatori, se n è il numero di intervalli in cui viene diviso lo stesso range spettrale, richiederebbero un tempo n∆t (supponendo uguale il tempo di acquisizione necessario per una misura con la CCD e con il fotomoltiplicatore). Risulta allora evidente che l’uso della CCD rende più spedita la misura. Ricordiamo la possibilità di utilizzo bidimensionale della CCD che permette usando due o più fibre ottiche e un opportuno adattatore ottico tra queste e la fenditura d’entrata dello spettrometro di eseguire misure simultanee di campioni diversi.
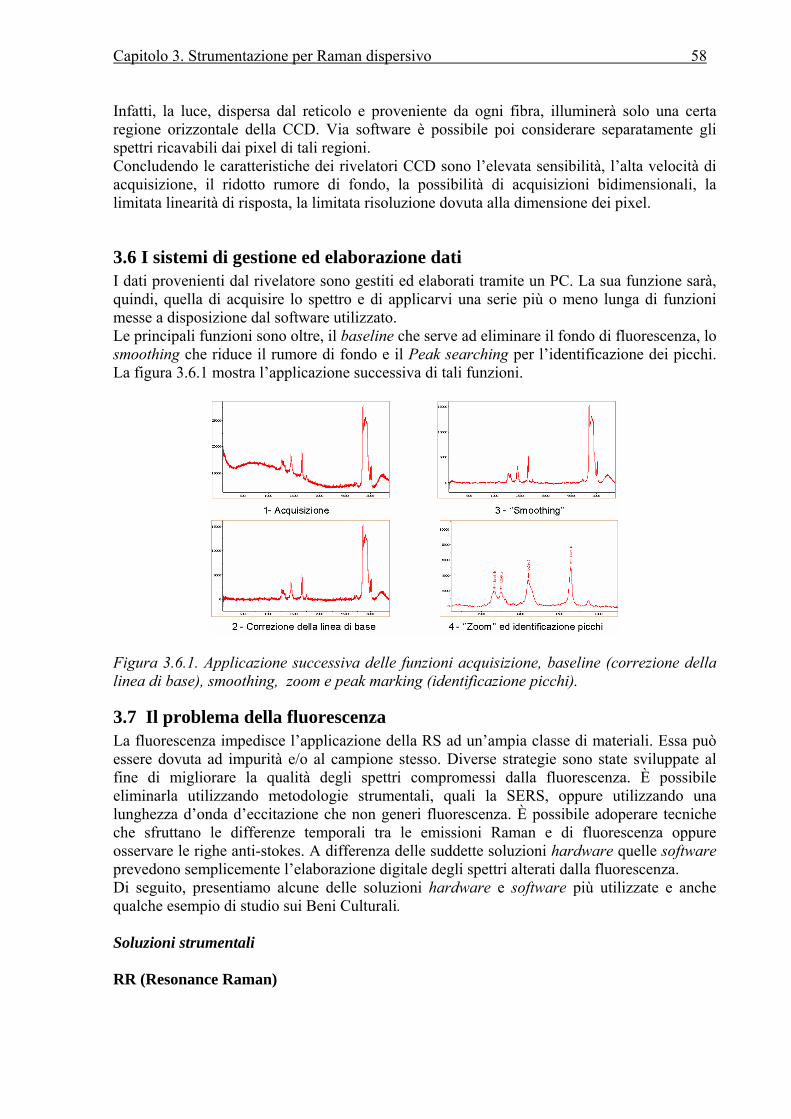
Capitolo 3. Strumentazione per Raman dispersivo 58
Infatti, la luce, dispersa dal reticolo e proveniente da ogni fibra, illuminerà solo una certa regione orizzontale della CCD. Via software è possibile poi considerare separatamente gli spettri ricavabili dai pixel di tali regioni. Concludendo le caratteristiche dei rivelatori CCD sono l’elevata sensibilità, l’alta velocità di acquisizione, il ridotto rumore di fondo, la possibilità di acquisizioni bidimensionali, la limitata linearità di risposta, la limitata risoluzione dovuta alla dimensione dei pixel.
3.6 I sistemi di gestione ed elaborazione dati I dati provenienti dal rivelatore sono gestiti ed elaborati tramite un PC. La sua funzione sarà, quindi, quella di acquisire lo spettro e di applicarvi una serie più o meno lunga di funzioni messe a disposizione dal software utilizzato. Le principali funzioni sono oltre, il baseline che serve ad eliminare il fondo di fluorescenza, lo smoothing che riduce il rumore di fondo e il Peak searching per l’identificazione dei picchi. La figura 3.6.1 mostra l’applicazione successiva di tali funzioni.
Figura 3.6.1. Applicazione successiva delle funzioni acquisizione, baseline (correzione della linea di base), smoothing, zoom e peak marking (identificazione picchi).
3.7 Il problema della fluorescenza La fluorescenza impedisce l’applicazione della RS ad un’ampia classe di materiali. Essa può essere dovuta ad impurità e/o al campione stesso. Diverse strategie sono state sviluppate al fine di migliorare la qualità degli spettri compromessi dalla fluorescenza. È possibile eliminarla utilizzando metodologie strumentali, quali la SERS, oppure utilizzando una lunghezza d’onda d’eccitazione che non generi fluorescenza. È possibile adoperare tecniche che sfruttano le differenze temporali tra le emissioni Raman e di fluorescenza oppure osservare le righe anti-stokes. A differenza delle suddette soluzioni hardware quelle software prevedono semplicemente l’elaborazione digitale degli spettri alterati dalla fluorescenza. Di seguito, presentiamo alcune delle soluzioni hardware e software più utilizzate e anche qualche esempio di studio sui Beni Culturali. Soluzioni strumentali RR (Resonance Raman)

Capitolo 3. Strumentazione per Raman dispersivo 59
Non permette la riduzione della fluorescenza, ma permette di migliorare il rapporto segnale rumore. Come esempio, uno studio su due pigmenti azotati [Dines et Onoh 2004] e sul blu ultramarino [Clark, Franks 1975]. Accoppiamento con un laser UV-pulsato In questo caso, in realtà, si tratta di una soluzione distruttiva, sebbene dell’ordine del micron. Un laser UV-pulsato permette la rimozione, grazie all’effetto di foto-ablazione non termica, degli strati di vernice che impediscono, per la loro fluorescenza, l’identificazione dei pigmenti [Ruiz-Moreno et al. 2004]. UVRS (Ultra-Violet Raman Spectroscopy) L’eccitazione nell’ultravioletto può ridurre notevolmente la fluorescenza ma sorgono problemi dovuti alla fotochimica. FT-Raman Come già illustrato nel capitolo 1, è oggi la tecnica più promettente. Il suo utilizzo per l’analisi dei campioni molto fluorescenti risale al 1986 [Chase 1986]. Raman dispersivi con eccitazione NIR e IR Sono oramai diffusi i sistemi per Raman dispersivo che utilizzano eccitazione laser nel vicino infrarosso (780-785 nm). A questi va aggiunta anche l’eccitazione a 1064 nm che in realtà tentata nel 1988, come alternativa al FT-Raman è stata in seguito abbandonata [Porterfield et Campion 1988]. SERS e SERRS (Surface Enhanced (Resonance) Raman Spectroscopy) Collocando la molecola sulla superficie di un metallo conduttore si inibisce la fluorescenza, ma la tecnica richiede una ampia preparazione del campione. Esempi sono la caratterizzazione dell’indigo, dell’alizarina e della purpurina [Shadi et al. 2003, Shadi et al. 2004]. Tecniche di modulazione della lunghezza d’onda Modulando la lunghezza d’onda d’eccitazione, le corrispondenti lunghezze d’onda Raman scatterate ( ma non il fondo di fluorescenza) sono anch’esse modulate. Cosicché, si osserva una modulazione dell’intensità dello scattering Raman che è proporzionale alla derivata della linea spettrale rispetto alla lunghezza d’onda. È così possibile ottenere uno spettro della derivata prima dello spettro Raman che è, essenzialmente, priva di fluorescenza. WSK (Wavelength Shift-Keyed) Uno spettrometro WSK per spettroscopia Raman permette di ottenere un risultato analogo a quello realizzato con la tecnica della modulazione della lunghezza d’onda ma servendosi soltanto di due lunghezze d’onda discrete. Si presenta quindi più semplice da gestire ed implementare [Stellman et Bucholtz, 1998]. Soluzioni software SSRS La metodologia SSRS (Subtracted Shifted Raman Spectroscopy) considera che nelle CCD ogni elemento del rivelatore ha una differente risposta di circa 1%, cosicché una CCD illuminata uniformemente dà uno spettro soggetto ad un rumore di fondo apparentemente casuale. L’acquisizione e la successiva sottrazione di uno spettro acquisito variando di pochissimo la posizione del reticolo di diffrazione, permette di ottenere spettri privi del rumore della CCD e quindi di distinguere i deboli picchi Raman sovrapposti alla fluorescenza [Bell et al. 1998, Bell et al. 2000]. Fuzzy Logic È stato creato un filtro basato su logica fuzzy per migliorare il rapporto segnale-rumore di uno spettro Raman disturbato da raggi cosmici e rumore elettronico. Il filtro sopprime il rumore e simultaneamente conserva le informazioni senza conoscere a priori la forma delle bande

Capitolo 3. Strumentazione per Raman dispersivo 60
Raman e le caratteristiche del rumore [Soneira et al. 2002]. Con logica fuzzy è stato compilato anche un sistema per la rilevazione dei picchi di uno spettro Raman, utilizzando come criteri quelli stessi che userebbe un osservatore che cercasse di esaminare visivamente uno spettro. Questo metodo si propone quindi di eliminare gli errori soggettivi dell’esame visivo [Perez-Pueyo et al. 2004]. PLS È stato dimostrato che il preprocessamento in derivata seconda tramite regressione multivariata (PLS) può diminuire l’interferenza dovuta alla fluorescenza. La normalizzazione degli spettri ottenuti in derivata seconda permette la loro rapida ed automatica identificazione. Questo metodo è stato testato su pigmenti sia puri che diluiti con un legante [Weis et al. 2004].

Capitolo 4. Progetto ColoRaman: presentazione 61
Capitolo 4. Progetto ColoRaman: presentazione
4.1 Introduzione L’ obiettivo di questo lavoro di ricerca è stato la realizzazione del progetto ColoRaman, ampiamente discusso nel capitolo seguente. In questo capitolo si vuole invece presentare il lavoro complementare volto all’ottimizzazione e diffusione delle conoscenze acquisite con lo sviluppo del progetto ColoRaman, l’ultimo paragrafo è dedicato invece alla presentazione dell’apparato sperimentale.
4.2 Peaks finder Esistono molti software che confrontano uno spettro con quelli contenuti in un database di spettri, trovando quello più simile. Questi database di spettri sono costituiti dagli spettri di ciascun composto, esaminato separatamente. Nelle applicazioni ai BB.CC., in molti casi, l’analisi deve essere condotta su oggetti costituiti da più composti e lo spettro che ne viene fuori è la somma dei loro contributi sia Raman che di fluorescenza. In questi casi, la ricerca all’interno di un database di spettri potrebbe essere inutile. Sono stati sviluppati, appositamente per le analisi sui BB.CC., dei software per la deconvoluzione degli spettri ottenuti da miscele di pigmenti [Coma et al. 2000, Breitman et al. 2003], tuttavia, a detta deglii stessi autori, questi metodi computazionali risentono del gran numero di variabili a cui è soggetta una misura su di un oggetto non preparato in laboratorio. Sembra, allora, che potrebbe essere molto più conveniente eseguire la ricerca in un database di picchi. Questo programma svolge proprio questa funzione. Si tratta di un software realizzato in C++ che permette di attribuire rapidamente i picchi di uno spettro servendosi di un database di picchi Raman. Esso compie la ricerca dentro un database i cui record, corrispondenti ad un composto, possono contenere fino a 20 picchi. Di seguito è presentato il listato del programma. Peak’s finder //---DIRETTIVE DEL PRECOMPILATORE----------------------------------------------- #include <iostream.h> #include <fstream.h> #include <cstdlib> const int PIGMENTI=100; //(Numero di pigmenti del database+1) //////////////////////////////////////////////////////////////////////////////// //----Definizione della classe "pigmento"---------------------------------------- class pig public: //---Costruttore----------------------------------------------------- pig () for (int i=0; i<20; i++) picchi532 [1][i]=0; picchi532 [2][i]=0; picchi633 [1][i]=0; picchi633 [2][i]=0; picchi780 [1][i]=0; picchi780 [2][i]=0; //////////////////////////////////////////////////////////////////// char intensity;

Capitolo 4. Progetto ColoRaman: presentazione 62
int id; int num_picchi; int picchi532 [2][20]; int picchi633 [2][20]; int picchi780 [2][20]; //---Member Function prendi_p532---------------------------------------- void prendi_p532() cin.clear(); cin.delbuf(); cout.clear(); num_picchi=0; do cout<<" How many peaks? (max 20): "; cin>>num_picchi; while(num_picchi>20); if (num_picchi<2) cout<<" Input peak's Raman shift (cm^-1):\n"; else cout<<" Input Raman shift of the "<< num_picchi<<" peaks (cm^-1):\n"; for (int j=0; j<num_picchi; j++) cout<<" Peak "<<j+1<<"="; cin >> picchi532 [1][j]; intensity532: cout<<" Input relative Intensity (w=Weak,m=Medium, s=Strong, b=Broad): "; cin>>intensity; switch (intensity) case 'w': picchi532 [2][j]=1;break; case 'm': picchi532 [2][j]=2;break; case 's': picchi532 [2][j]=3;break; case 'b': picchi532 [2][j]=4;break; default: cout<<" Please, choose w, m, s, or b !"; goto intensity532; break; return; //---Member Function prendi_p633---------------------------------------- void prendi_p633() num_picchi=0; do cout<<" How many peaks? (max 20): "; cin>>num_picchi; while(num_picchi>20); if (num_picchi<2) cout<<" Input peak's Raman shift (cm^-1):\n"; else cout<<" Input Raman shift of the "<< num_picchi<<" peaks (cm^-1):\n"; for (int j=0; j<num_picchi; j++) cout<<" Peak "<<j+1<<"="; cin >> picchi633 [1][j]; intensity633: cout<<" Input relative Intensity (w=Weak, m=Medium, s=Strong, b=Broad) ";

Capitolo 4. Progetto ColoRaman: presentazione 63
cin>>intensity; switch (intensity) case 'w': picchi633 [2][j]=1;break; case 'm': picchi633 [2][j]=2;break; case 's': picchi633 [2][j]=3;break; case 'b': picchi532 [2][j]=4;break; default: cout<<" Please, choose w, m, s, or b !"; goto intensity633; break; //---Member Function prendi_p780---------------------------------------- void prendi_p780() num_picchi=0; do cout<<" How many peaks? (max 20): "; cin>>num_picchi; while(num_picchi>20); if (num_picchi<2) cout<<" Input peak's Raman shift (cm^-1):\n"; else cout<<" Input Raman shift of the "<< num_picchi<<" peaks (cm^-1):\n"; for (int j=0; j<num_picchi; j++) cout<<" Peak "<<j+1<<"="; cin >> picchi780 [1][j]; intensity780: cout<<" Relative Intensity (w=Weak, m=Medium, s=Strong, b=Broad): "; cin>>intensity; switch (intensity) case 'w': picchi780 [2][j]=1;break; case 'm': picchi780 [2][j]=2;break; case 's': picchi780 [2][j]=3;break; case 'b': picchi532 [2][j]=4;break; default: cout<<" Please, choose w, m, s, or b !"; goto intensity780; break; //---Member Function dainullo532--------------------------------------- //Serve ad azzerare il buffer void dainullo532() int j=0; for ( j=0; j<20; j++) if(picchi532[1][j]!=0) cout<<picchi532[1][j]; switch (picchi532[2][j]) case 1: cout<<"w"<<endl;break; case 2: cout<<"m"<<endl;break; case 3: cout<<"s"<<endl;break; case 4: cout<<"b"<<endl;break;

Capitolo 4. Progetto ColoRaman: presentazione 64
else continue; //---Member Function dai_p532--------------------------------------- void dai_p532() cout<<" Record "<<id<<endl; cout<<" Peaks' Raman shift (cm^-1):\n"; int j=0; for ( j=0; j<20; j++) if(picchi532[1][j]!=0) cout<<picchi532[1][j]; switch (picchi532[2][j]) case 1: cout<<"w"<<endl;break; case 2: cout<<"m"<<endl;break; case 3: cout<<"s"<<endl;break; case 4: cout<<"b"<<endl;break; else continue; //---Member Function dai_p633--------------------------------------- void dai_p633() cout<<" Pigment "<<id<<endl; cout<<" Peaks' Raman shift (cm^-1):\n"; int j=0; for ( j=0; j<20; j++) if(picchi633[1][j]!=0) cout<<picchi633[1][j]; switch (picchi633[2][j]) case 1: cout<<"w"<<endl;break; case 2: cout<<"m"<<endl;break; case 3: cout<<"s"<<endl;break; case 4: cout<<"b"<<endl;break; else continue; //---Member Function dai_p780--------------------------------------- void dai_p780() cout<<" Pigment "<<id<<endl; cout<<" Peaks' Raman shift(cm^-1):\n"; int j=0; for ( j=0; j<20; j++) if(picchi780[1][j]!=0)

Capitolo 4. Progetto ColoRaman: presentazione 65
cout<<picchi780[1][j]; switch (picchi780[2][j]) case 1: cout<<"w"<<endl;break; case 2: cout<<"m"<<endl;break; case 3: cout<<"s"<<endl;break; case 4: cout<<"b"<<endl;break; else continue; /////////////////////////////////////////////////////////////// ; ///////////////////////////////////////////////////////////////////////////////// //-----------Definizione struttura pigRAM---------------------------------------- struct pigRAM int id_RAM, ricorrenza; ; ///////////////////////////////////////////////////////////////////////////////////////////////////////////// //---------------------MAIN----------------------------------------------------------------------------------- void main() /////////////////////////////////////////////////////////////////////////// //---Dichiarazioni locali MAIN--------------------------------------------- char risp='s'; int id_mod,i_ins; int num_pigmenti=PIGMENTI; int frequenza,frequenza_mostra, frequenza_ricerca; pig inserimento; pig blankpig; ///////////////////////////////////////////////////////////////////////// //---Dicharazione ed inizializzazione vettore di pigRAM------------------------------------------------ pigRAM pigmenti[PIGMENTI]; for(int n=0; n<PIGMENTI; n++) pigmenti[n].id_RAM=0; pigmenti[n].ricorrenza=0; ////////////////////////////////////////////////////////////////////////////////////////// //---GESTIONE FILE----------------------------------------------------------------------- fstream dati,datimod, datimostra; ///////////////////////////////////////////////////////////////////////////////////////// //---INTESTAZIONE GRAFICA----------------------------------------------------------------

Capitolo 4. Progetto ColoRaman: presentazione 66
cout<<"\n\n"; cout<<" *************************************************************************\n" <<" *"<<"\t\t\t\t\t\t\t\t\t *\n" <<" *"<<"\tLDL & BB.CC. *\n" <<" *"<<"\t\t\t\t\t\t\t\t\t *\n" <<" *"<<" \tLaboratory of Luminescence Dating and of *\n" <<" *"<<" \tPhysical Methodologies for Cultural Heritage *\n" <<" *"<<"\t\t\t\t\t\t\t\t\t *\n" <<" *"<<"\t\t\t\t\t\t\t\t\t *\n" <<" *"<<" \tPeaks Finder v.1/2004 *\n" <<" *"<<"\t\t\t\t\t\t Antonino Cosentino *\n" <<" *************************************************************************\n\n\n\n"; ///////////////////////////////////////////////////////////////////////////////////// //---SwitchBoard------------------------------------------------------------- switchboard: cout<<"\n"<<" ***************************************\n" <<" * SWITCHBOARD *\n" <<" ***************************************\n" <<" * *\n" <<" * 1\tSeek peaks *\n" <<" * 2\tInitialize peaks data file *\n" <<" * 3\tEdit peaks data *\n" <<" * 4\tShow peaks data *\n" <<" * 5\tExit *\n" <<" * *\n" <<" ***************************************\n"<<endl; cout<<" Choose an option (1,2,3,4,5): "; int scelta; cin>>scelta; cout<<"\n"; switch (scelta) case 1: cout<<"__________________________________________________________________________\n"; cout<<" Seeking peaks\n"; cout<<"--------------------------------------------------------------------------\n"; goto ricerca; break; case 2: cout<<"__________________________________________________________________________\n"; cout<<" Peaks data initialized\n"; cout<<"--------------------------------------------------------------------------\n"; goto inizializzazione; break; case 3: cout<<"__________________________________________________________________________\n"; cout<<" Editing peaks data\n"; cout<<"--------------------------------------------------------------------------\n"; goto modifica; break; case 4: cout<<"__________________________________________________________________________\n"; cout<<" Showing peaks data\n"; cout<<"--------------------------------------------------------------------------\n"; goto mostra; break; case 5: exit (1); goto switchboard; break; default:cout<<" Please. input 1,2,3,4,or 5. \n";

Capitolo 4. Progetto ColoRaman: presentazione 67
goto switchboard; /////////////////////////////////////////////////////////////////////////////// //---Modifica Dati Pigmento ---------------------------------------------- modifica: datimod.open ("peaks.dat",ios::out|ios::in| ios::binary); if (!datimod) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); do cout<<" Input record ID: "; cin>>id_mod; while(id_mod>PIGMENTI); /*do cout<<" Input laser excitation: [5 (531.5), 6 (632.8), 7 (780)] "; cin>>frequenza; while(frequenza!=5 && frequenza!=6 && frequenza !=7); */ switch (frequenza=5) //Ho modificato aggiungendo "=5" cosicchè viene usata sempre la case 5 case 5: datimod.seekp((id_mod-1)*sizeof(inserimento)); inserimento.prendi_p532(); inserimento.id=id_mod; datimod.write ((char*)(&inserimento), sizeof(inserimento)); datimod.close(); //----Codice per l'azzeramento del Buffer--- datimostra.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimostra) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); int index; index=0; do cout<<"__________________________________________________________________________\n"; datimostra.seekg((PIGMENTI-1)*sizeof(inserimento)); datimostra.read((char*)&inserimento, sizeof(inserimento)); inserimento.dainullo532(); cout<<"__________________________________________________________________________\n"; while (index--); datimostra.close(); break; case 6: datimod.seekp((id_mod-1)*sizeof(inserimento)); inserimento.prendi_p633(); inserimento.id=id_mod; datimod.write ((char*)(&inserimento), sizeof(inserimento)); datimod.close(); //----Codice per l'azzeramento del Buffer--- datimostra.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimostra)

Capitolo 4. Progetto ColoRaman: presentazione 68
cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); int index633; index633=0; do cout<<"__________________________________________________________________________\n"; datimostra.seekg((PIGMENTI-1)*sizeof(inserimento)); datimostra.read((char*)&inserimento, sizeof(inserimento)); inserimento.dainullo532(); cout<<"__________________________________________________________________________\n"; while (index633--); datimostra.close(); break; case 7: datimod.seekp((id_mod-1)*sizeof(inserimento)); inserimento.prendi_p780(); inserimento.id=id_mod; datimod.write ((char*)(&inserimento), sizeof(inserimento)); datimod.close(); //----Codice per l'azzeramento del Buffer--- datimostra.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimostra) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); int index780; index780=0; do cout<<"__________________________________________________________________________\n"; datimostra.seekg((PIGMENTI-1)*sizeof(inserimento)); datimostra.read((char*)&inserimento, sizeof(inserimento)); inserimento.dainullo532(); cout<<"__________________________________________________________________________\n"; while (index780--); datimostra.close(); break; default: cout<<" Please, input 5, 6, or 7 \n"; goto switchboard; ///////////////////////////////////////////////////////////////////////// //---Mostra Database----------------------------------------------------- mostra: datimostra.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimostra) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); do cout<<" Input record ID: "; cin>>id_mod; while(id_mod>PIGMENTI); /*do cout<<" Input laser excitation: [5 (531.5), 6 (632.8), 7 (780)] "; cin>>frequenza_mostra;

Capitolo 4. Progetto ColoRaman: presentazione 69
while(frequenza_mostra!=5 && frequenza_mostra!=6 && frequenza_mostra !=7); */ int indice; switch (frequenza_mostra=5) case 5: indice=0; do cout<<"__________________________________________________________________________\n"; datimostra.seekg((id_mod-1)*sizeof(inserimento)); datimostra.read((char*)&inserimento, sizeof(inserimento)); inserimento.dai_p532(); cout<<"__________________________________________________________________________\n"; datimostra.clear(); while (indice--); //----Codice per l'azzeramento del Buffer--- datimod.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimod) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); int index_mos532; index_mos532=0; do cout<<"__________________________________________________________________________\n"; datimod.seekg((PIGMENTI-1)*sizeof(inserimento)); datimod.read((char*)&inserimento, sizeof(inserimento)); inserimento.dainullo532(); cout<<"__________________________________________________________________________\n"; while (index_mos532--); datimod.close(); break; case 6: indice=0; do cout<<"__________________________________________________________________________\n"; datimostra.seekp((id_mod-1)*sizeof(inserimento)); datimostra.read((char*)&inserimento, sizeof(inserimento)); inserimento.dai_p633(); cout<<"__________________________________________________________________________\n"; datimostra.clear(); while (indice--); //----Codice per l'azzeramento del Buffer--- datimod.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimod) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); int index_mos633; index_mos633=0; do cout<<"__________________________________________________________________________\n"; datimod.seekg((PIGMENTI-1)*sizeof(inserimento)); datimod.read((char*)&inserimento, sizeof(inserimento)); inserimento.dainullo532(); cout<<"__________________________________________________________________________\n";

Capitolo 4. Progetto ColoRaman: presentazione 70
while (index_mos633--); datimod.close(); break; case 7: indice=0; do cout<<"__________________________________________________________________________\n"; datimostra.seekp((id_mod-1)*sizeof(inserimento)); datimostra.read((char*)&inserimento, sizeof(inserimento)); inserimento.dai_p780(); cout<<"__________________________________________________________________________\n"; datimostra.clear(); while (indice--); //----Codice per l'azzeramento del Buffer--- datimod.open ("peaks.dat", ios::out|ios::in| ios::binary); if (!datimod) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); int index_mos780; index_mos780=0; do cout<<"__________________________________________________________________________\n"; datimod.seekg((PIGMENTI-1)*sizeof(inserimento)); datimod.read((char*)&inserimento, sizeof(inserimento)); inserimento.dainullo532(); cout<<"__________________________________________________________________________\n"; while (index_mos780--); datimod.close(); break; default: cout<<" Please, input 5, 6, or 7 \n"; datimostra.close(); goto switchboard; ////////////////////////////////////////////////////////////////////////////// //---Inizializzazione Database------------------------------------------------- inizializzazione: dati.open ("peaks.dat", ios::out|ios::in| ios::binary); for(i_ins=0; i_ins<PIGMENTI; i_ins++) inserimento.id=i_ins+1; dati.seekp((i_ins)*sizeof(inserimento)); dati.write(reinterpret_cast<const char*>(&inserimento),sizeof(pig)); dati.close(); goto switchboard; ////////////////////////////////////////////////////////////////////// //---Acquisizione valori dei picchi dello spettro in esame ---------------------------- ricerca: int nump=0; do cout<<" Input how many peaks you want seek (max 20): "; cin>>nump;

Capitolo 4. Progetto ColoRaman: presentazione 71
while(nump>20); float p[20]=0; cout<<" Input peaks (cm^-1): \n"; for (int k=0; k<nump; k++) cout<<" Peak "<<k+1<<" = "; cin>>p[k]; /*do cout<<" Input laser excitation: [5 (531.5), 6 (632.8), 7 (780)] "; cin>>frequenza_ricerca; while(frequenza_ricerca!=5 && frequenza_ricerca!=6 && frequenza_ricerca !=7); */ ///////////////////////////////////////////////////////////////////////////////////////////////////////////// //-------Inserimento tolleranza,------------------------------------------------------------------------------ int tolleranza; cout<<"\n Input accuracy (cm^-1): "; cin>>tolleranza; cout<<"__________________________________________________________________________\n"; ///////////////////////////////////////////////////////////////////////////////////////////////////////////// //---Algoritmo per l'individuazione dei pigmenti------------------------------------------------ dati.open ("peaks.dat", ios::out|ios::in| ios::binary); dati.clear(); if (!dati) cerr<<"ERROR: It was not possible open the file\n\n"<<endl; exit(1); switch (frequenza_ricerca=5) //Ho aggiunto "=5" case 5: for(k=0;k<nump;k++) //parte il ciclo per ogni k-esimo picco indagato dati.clear(); dati.seekg(0); while(!dati.eof()) dati.read((char*)&inserimento, sizeof(inserimento)); for(int j=0;j<20;j++) //parte il ciclo per ogni j-esimo picco del pigmento if (p[k]<=inserimento.picchi532[1][j]+tolleranza && p[k] >=inserimento.picchi532[1][j]-tolleranza && inserimento.picchi532[1][j]!=0) cout<<"\n\n Peak at: "<<p[k]<<" (cm^-1)"<<endl; cout<<" Record "<<inserimento.id<<" has a peak at " << inserimento.picchi532[1][j]; switch (inserimento.picchi532[2][j]) case 1: cout<<"w";break; case 2: cout<<"m";break;

Capitolo 4. Progetto ColoRaman: presentazione 72
case 3: cout<<"s";break; case 4: cout<<"b";break; cout<<" / Seeked Peak-Record "<<inserimento.id<<" peak = "<<(p[k]-inserimento.picchi532[1][j])<<" (cm^-1)"<<endl; pigmenti[inserimento.id].ricorrenza++; pigmenti[inserimento.id].id_RAM=inserimento.id; break; case 6: for(k=0;k<nump;k++) //parte il ciclo per ogni k-esimo picco indagato dati.clear(); dati.seekg(0); while(!dati.eof()) dati.read((char*)&inserimento, sizeof(inserimento)); for(int j=0;j<20;j++) //parte il ciclo per ogni j-esimo picco del pigmento if (p[k]<=inserimento.picchi633[1][j]+tolleranza && p[k] >=inserimento.picchi633[1][j]-tolleranza && inserimento.picchi633[1][j]!=0) cout<<"\n\n Peak at:"<<p[k]<<" (cm^-1)"<<endl; cout<<" Record "<<inserimento.id<<" has a peak at " << inserimento.picchi633[1][j]; switch (inserimento.picchi633[2][j]) case 1: cout<<"w"<<endl;break; case 2: cout<<"m"<<endl;break; case 3: cout<<"s"<<endl;break; case 4: cout<<"b"<<endl;break; cout<<" / Seeked Peak-Record "<<inserimento.id <<" peak = "<<(p[k]-inserimento.picchi633[1][j])<<" (cm^-1)"<<endl; pigmenti[inserimento.id].ricorrenza++; pigmenti[inserimento.id].id_RAM=inserimento.id; break; case 7: for(k=0;k<nump;k++) //parte il ciclo per ogni k-esimo picco indagato dati.clear(); dati.seekg(0); while(!dati.eof()) dati.read((char*)&inserimento, sizeof(inserimento)); for(int j=0;j<20;j++) //parte il ciclo per ogni j-esimo picco del pigmento if (p[k]<=inserimento.picchi780[1][j]+tolleranza && p[k] >=inserimento.picchi780[1][j]-tolleranza && inserimento.picchi780[1][j]!=0) cout<<"\n\n Peak at: "<<p[k]<<endl; cout<<" Record "<<inserimento.id<<" has a peak at " << inserimento.picchi780[1][j]; switch (inserimento.picchi780[2][j])

Capitolo 4. Progetto ColoRaman: presentazione 73
case 1: cout<<"w"<<endl;break; case 2: cout<<"m"<<endl;break; case 3: cout<<"s"<<endl;break; case 4: cout<<"b"<<endl;break; cout<<" / Seeked Peak-Pigment's "<< inserimento.id<<" peak = "<<(p[k]-inserimento.picchi780[1][j])<<" (cm^-1)"<<endl; pigmenti[inserimento.id].ricorrenza++; pigmenti[inserimento.id].id_RAM=inserimento.id; break; default:cout<<" Please, input 5, 6, or 7 \n"; dati.close(); /////////////////////////////////////////////////////////////////////////////////////////////// //---Risultati Ricorrenza-------------------------------------------------------------<------ cout<<endl<<"--------------------------------------------------------------------------\n"; for(int i=0; i<PIGMENTI;i++) if(pigmenti[i].ricorrenza!=0) cout<<" ID:"<<pigmenti[i].id_RAM; cout<<" recourse:"<<pigmenti[i].ricorrenza<<endl; cout<<"__________________________________________________________________________\n"; //---bubble Sorting-------------------------------------------------------------------- /*Pigmenti diversi possono avere picchi alle stesse frequenze e quindi un solo picco può non essere sufficiente a definire con certezza la presenza di uno o l'altro pigmento. Ecco allora che il pigmento con il maggior numero di picchi sovrapponibili a quelli in esame è il più probabile */ pigRAM temp; for (i=1; i<PIGMENTI; i++) for (int j=0;j<PIGMENTI-i;j++) if (pigmenti[j].ricorrenza>pigmenti[j+1].ricorrenza) temp.id_RAM = pigmenti[j+1].id_RAM; temp.ricorrenza = pigmenti[j+1].ricorrenza; pigmenti[j+1].id_RAM=pigmenti[j].id_RAM; pigmenti[j+1].ricorrenza=pigmenti[j].ricorrenza; pigmenti[j].id_RAM=temp.id_RAM; pigmenti[j].ricorrenza=temp.ricorrenza; cout<<"\n\n PEAKS SEEKING RESULTS:"<<endl; for(int z=0; z<PIGMENTI; z++) if( pigmenti[z].ricorrenza !=0) cout<<"\n Record "<<pigmenti[z].id_RAM<<" shows "<<pigmenti[z].ricorrenza<<" peak(s) matching"<< endl; cout<<endl; for(i=0; i<PIGMENTI;i++) //ri-inizializzazione pigmenti[i].ricorrenza=0; pigmenti[i].id_RAM=0;
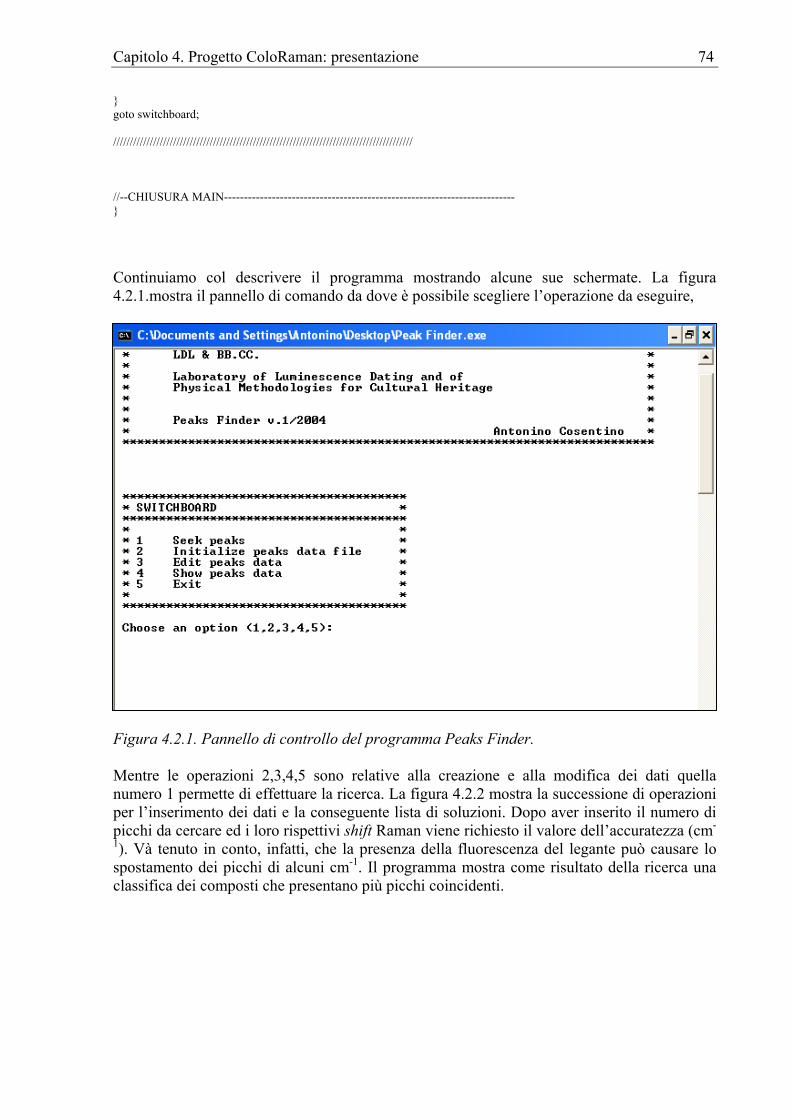
Capitolo 4. Progetto ColoRaman: presentazione 74
goto switchboard; ////////////////////////////////////////////////////////////////////////////////////////// //--CHIUSURA MAIN------------------------------------------------------------------------- Continuiamo col descrivere il programma mostrando alcune sue schermate. La figura 4.2.1.mostra il pannello di comando da dove è possibile scegliere l’operazione da eseguire,
Figura 4.2.1. Pannello di controllo del programma Peaks Finder. Mentre le operazioni 2,3,4,5 sono relative alla creazione e alla modifica dei dati quella numero 1 permette di effettuare la ricerca. La figura 4.2.2 mostra la successione di operazioni per l’inserimento dei dati e la conseguente lista di soluzioni. Dopo aver inserito il numero di picchi da cercare ed i loro rispettivi shift Raman viene richiesto il valore dell’accuratezza (cm-
1). Và tenuto in conto, infatti, che la presenza della fluorescenza del legante può causare lo spostamento dei picchi di alcuni cm-1. Il programma mostra come risultato della ricerca una classifica dei composti che presentano più picchi coincidenti.

Capitolo 4. Progetto ColoRaman: presentazione 75
Figura 4.2.2. Operazioni per la ricerca dei picchi all’interno del database.
4.3 Il sito internet La diffusione aggiornata dei dati è stata possibile grazie alla loro pubblicazione sul sito del gruppo, di ricerca, figura 4.3.1. La parte relativa alla spettroscopia Raman si divide a sua volta in otto sezioni, figura 4.3.2. Della sezione ColoRaman parleremo diffusamente nel capitolo 5. Di seguito, invece, presentiamo le altre sezioni. Oil gallery Questa sezione permette di osservare la stesura ad olio dei pigmenti del database così da verificarne le tonalità e la resa del colore, figura 4.3.3. Equipment Viene descritta sinteticamente la strumentazione per spettroscopia Raman a disposizione del laboratorio, figura 4.3.4. Painting La descrizione dei materiali e delle tecniche utilizzate per la stesura dei pigmenti viene documentata in questa sezione, figura 4.3.5.

Capitolo 4. Progetto ColoRaman: presentazione 76
Utilities Questa sezione contiene i link per accedere alle utilities di volta in volta sviluppate. Ovvero, al programma Peaks Finder e ai database di picchi Raman, ad una presentazione ragionata e referenziata (con riferimenti ai rispettivi articoli) e ad una bibliografia di 242 lavori attinenti l’applicazione della spettroscopia Raman per l’Arte e l’Archeologia, figura 4.3.6. Links Viene qui proposta una selezione di siti che forniscono risorse on-line gratuite riguardanti l’applicazione della spettroscopia Raman all’Arte e all’Archeologia. Sono quindi presenti i link ai database di spettri Raman di materiali d’interesse per i BB.CC., di minerali e di pigmenti pittorici. Ci sono anche i collegamenti aggiornati alle conferenze, alle riviste on-line ed ai siti dei fornitori di pigmenti, figura 4.3.7. Publications Sono qui raccolti i riferimenti bibliografici ai lavori del gruppo già pubblicati e riguardanti le applicazioni della spettroscopia Raman ai BB.CC. Ove possibile il materiale è anche consultabile, figura 4.3.8. La frequentazione del sito è visionabile tramite appositi servizi. In figura 4.3.9.è riportato l‘andamento delle visite del sito dal giugno 2003 al maggio 2004. Il sito veniva già visitato perché disponeva di un precedente database di spettri Raman e di fluorescenza di pigmenti pittorici secchi. La pubblicazione nel marzo 2004 del nuovo progetto ColoRaman così come del programma Peaks Finder nell’aprile 2004 hanno fatto registrare un incremento delle visite di più del 300%. Particolarmente importante è la frequentazione del sito dal Belgio, dall’Inghilterra e dalla Francia, paesi dove attualmente è più vivace la ricerca in questo campo, figura 4.3.10.
Capitolo 4. Progetto ColoRaman: presentazione 77
Figura 4.3.1.Home page del sito del gruppo di ricerca LDL & BB.CC.
Figura 4.3.2. Sezione del sito dedicata alla spettroscopia Raman ed in particolare al progetto coloRaman
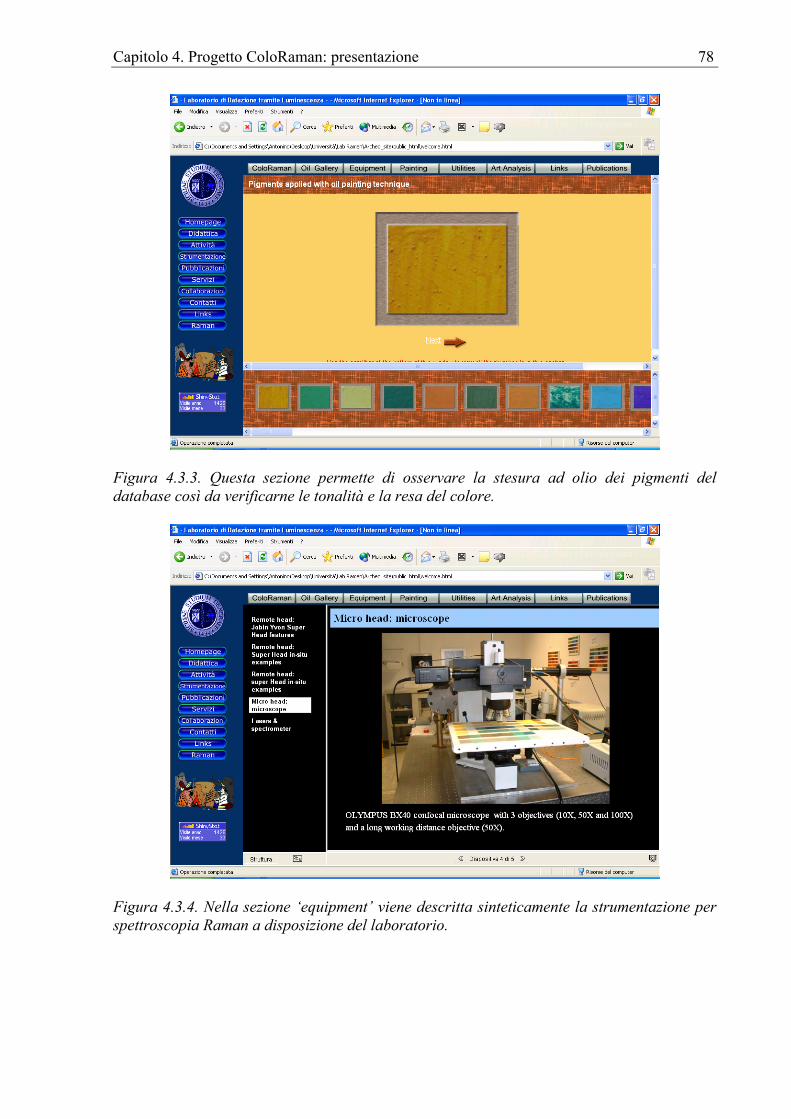
Capitolo 4. Progetto ColoRaman: presentazione 78
Figura 4.3.3. Questa sezione permette di osservare la stesura ad olio dei pigmenti del database così da verificarne le tonalità e la resa del colore.
Figura 4.3.4. Nella sezione ‘equipment’ viene descritta sinteticamente la strumentazione per spettroscopia Raman a disposizione del laboratorio.
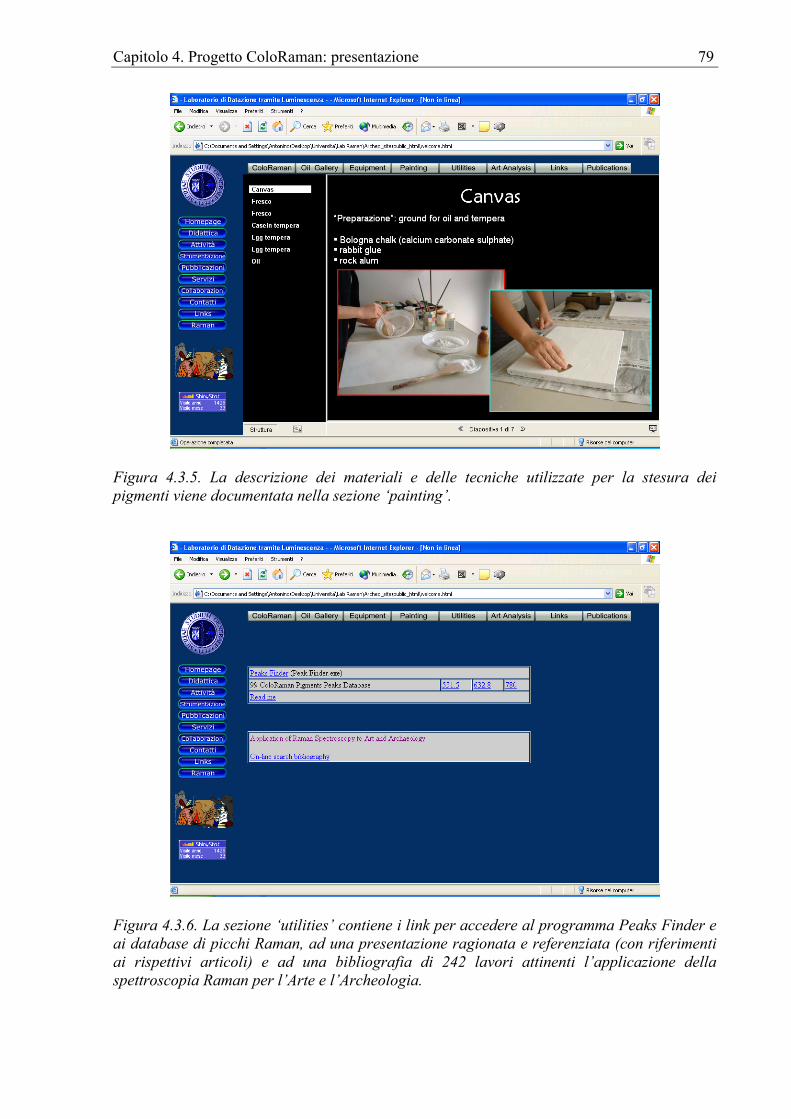
Capitolo 4. Progetto ColoRaman: presentazione 79
Figura 4.3.5. La descrizione dei materiali e delle tecniche utilizzate per la stesura dei pigmenti viene documentata nella sezione ‘painting’.
Figura 4.3.6. La sezione ‘utilities’ contiene i link per accedere al programma Peaks Finder e ai database di picchi Raman, ad una presentazione ragionata e referenziata (con riferimenti ai rispettivi articoli) e ad una bibliografia di 242 lavori attinenti l’applicazione della spettroscopia Raman per l’Arte e l’Archeologia.

Capitolo 4. Progetto ColoRaman: presentazione 80
Figura 4.3.7. Viene qui proposta una selezione di siti che forniscono risorse on-line gratuite riguardanti l’applicazione della spettroscopia Raman all’Arte e all’Archeologia. Sono quindi presenti, in particolare, i link ai database di spettri Raman di materiali d’interesse per i BB.CC.
Figura 4.3.8. Sono qui raccolti i riferimenti bibliografici ai lavori del gruppo già pubblicati e riguardanti le applicazioni della spettroscopia Raman ai BB.CC. Ove possibile il materiale è ancheconsultabile.

Capitolo 4. Progetto ColoRaman: presentazione 81
igura 4.3.9. Il sito veniva già visitato perché disponeva di un precedente database di spettri
igura 4.3.10. La frequentazione del sito in percentuale, per ogni Paese.
FRaman e di fluorescenza di pigmenti pittorici secch. La pubblicazione nel marzo 2004 del nuovo progetto ColoRaman così come del programma Peaks Finder nell’aprile 2004 hanno fatto registrare un incremento delle visite di più del 300%.
F

Capitolo 4. Progetto ColoRaman: presentazione 82
4.4 Apparato sperimentale
o lo schema e una foto della strumentazione usata per an. Di seguito illustriamo le caratteristiche delle sue
Figura 4.4.1. Schema strumentazione per spettroscopia Raman.
isponiamo di tre laser: un laser Nd YAG a 531.5 nm e potenza di 40 mW, un He-Ne a 632.8 a 780 nm e potenza di 19 mW.
lla radiazione aman
tto dell’accoppiamento di un microscopio allo spettrometro. Nel nostro caso
ione del campione necessaria alla sua centratura e focalizzazione è fornita da una minosa bianca.
Le figure 4.4.1 e 4.4.2 mostranl’acquisizione degli spettri Ramcomponenti:
Sorgente di eccitazione
I
Ottica di centratura
II
Campione III
Ottica di
raccolta
Spettroscopio V
Rivelatore VI
Sistema di gestione
VII
Figura 3.8.2. Foto dell’apparato sperimentale Ottica per l’eccitazione del campione: il laser Dnm con una potenza di 30mW e un laser diodo Ottica per la centratura e focalizzazione del campione e per la raccolta deRAbbiamo già detto che l’esecuzione di misure di microscopia Raman si avvalgono innanzitudisponiamo di un microscopio Olympus BX40 corredato di 4 obiettivi: 10x /0.25 (ingrandimento) / Apertura numerica 50x /0.75 100x /0.90 50x /0.50 L’illuminazsorgente lu

Capitolo 4. Progetto ColoRaman: presentazione 83
remoto er il nostro lavoro abbiamo utilizzato la testa remota prodotta dalla Dilor con un obiettivo da
di NA 0.50). La figura 3.7.3 è una foto della testa remota
Figura 3.8.3. Foto della testa di cam to remoto montata al museo Bellomo (SR)
Figura 3.8.4. La testa remota montata sul suo supporto è pronta per eseguire misure in situ.
Sistema di campionamentoPmicroscopio (ingrandimenti da 50X configurata per misure su superfici verticali, mentre la figura 3.8.4 la mostra montata per misure su superfici orizzontali.
pionamen
Lo spettrometro

Capitolo 4. Progetto ColoRaman: presentazione 84
tilizziamo uno spettrometro TRIAX 190 prodotto dalla ISA. Esso contiene 3 reticoli di ng) intercambiabili ognuno dei quali può ruotare attorno ad un asse passante
ret), l’otturatore (shutter) e lo epper-motor, che muove il reticolo, sono controllati via PC. La densità del grating cambia,
a CCD impiegata è prodotta dalla ISA. Viene raffreddata per effetto Peltier e quindi alla classe delle TE CCD (raffreddate Termo Elettricamente) che possono lavorare
CD 3000 Detector Controller )
Udiffrazione (gratiper il piano del reticolo stesso e ad esso parallelo. La luce proveniente dalla fibra ottica inserita nell’apposita fessura d’entrata (entrance slit) viene innanzitutto raccolta da uno specchio collimatore che la riflette sul reticolo. Qui viene scomposta nelle sue varie lunghezze d’onda e diretta verso uno specchio focalizzatore che la riflette verso la fessura d’uscita (exit slit) e quindi all’adattatore multicanale cui è interfacciata la CCD per la d’uscita (exit slit) e quindi all’adattatore multicanale cui è interfacciata la CCD per la rivelazione, figura 3.8.5.
Figura 3.8.5. Veduta dall’alto dello spettrometro TRIAX 190. Le slit, cioè le fessure, la torretta con i reticoli (grating's turstsia la risoluzione che la larghezza dello spettro. Il grating va quindi scelto in modo da avere la migliore risoluzione e la maggiore sensibilità possibile nel range delle lunghezze d’onda d’interesse. La torretta alloca tre grating uno da 1800g/mm, uno da 1200g/mm e l’altro da 150 g/mm. Per ottimizzare il rapporto segnale/rumore è importante che lo specchio collimatore non sia investito eccessivamente dalla luce infatti ciò aumenterebbe la possibilità di stray light sul rivelatore. Così è bene limitare la larghezza della slit d’entrata. La CCD. Lappartienesenza interruzioni alla temperatura di lavoro fissata. Il rivelatore a CCD (CCD Detector Head) è montato sull’apposita porta spettrografica d’uscita dello spettrometro. Alcuni parametri importanti forniti dalla casa costruttrice : Dark current <250 elettroni /pixel/ora Temperatura di lavoro -550 C Unità di controllo della CCD (C

Capitolo 4. Progetto ColoRaman: presentazione 85
are sul PC cui è collegata. Essa controlla la CCD secondo i comandi impostati via softwL’unità di controllo della CCD, SPECTRUM ONE, fornisce l’alimentazione per il sistema di raffreddamento e per la polarizzazione dei sensori della CCD ed inoltre ha il compito di amplificare e digitalizzare il segnale. Esso poi trasferisce, tramite la porta IEEE-488, i segnali digitalizzati al personal computer che grazie al software SPECTRAMAX visualizza ed elabora gli spettri acquisiti.

Capitolo 5. Progetto ColoRaman: risultati 86
Capitolo 5. Progetto ColoRaman: risultati
5.1 Introduzione L'analisi dei pigmenti di un manufatto può permettere di: Caratterizzazione. Determinare la località di estrazione dei pigmenti inorganici di origine mineraria. Storia dell’Arte Determinare la tavolozza tipica di ogni artista. Indagini micrometriche Discernere gli elementi costitutivi di una miscela di pigmenti e di identificare gli strati di pittura differenti per composizione o per dimensione delle particelle. Restauro Individuare i pigmenti originali al fine di riparare aree danneggiate e riscontrare eventuali cambiamenti del colore verificatisi nel tempo. Conservazione. Individuare gli agenti responsabili del degrado e quindi di suggerire lo sviluppo delle più opportune tecniche per preservare i manufatti. Autenticazione e Datazione Avanzare una datazione indiretta dei manufatti grazie alle informazioni storiche sull’uso dei pigmenti.
5.2 Pigmenti e colori. Introduzione Presentiamo di seguito una raccolta di informazioni di base necessarie alla comprensione della terminologia in uso per l’analisi dei pigmenti.
5.2.1 Colore, pigmento e colorante Ogni tecnica pittorica si avvale di sostanze coloranti dette pigmenti che possono essere di natura vegetale, minerale o animale; un legante detto anche medium che costituisce il tessuto connettivo dei pigmenti, un veicolo, caratterizzato da compatibilità chimica con il medium e che viene utilizzato allo scopo di far rinvenire il colore sulla tavolozza e di rendere agevole l'operazione di trasferimento del colore dalla tavolozza alla superficie da colorare. Già in epoche remote, nella terminologia relativa ai colori si distingueva tra color e pigmentum. Con il primo termine si intendeva la tinta in generale, e con il secondo la sostanza da cui si otteneva il colore. Plinio distingueva i pigmenti in due grandi categorie: colori floridi e colori austeri. La caratteristica principale dei colori floridi è la trasparenza, fra essi Plinio cita il minio, il cinabro, l'indaco. I colori austeri sono invece colori coprenti fra essi le ocre, le terre, la cerussa, la sandracca. La stessa distinzione, secondo criteri chimici e fisici obiettivi, è stata elaborata in epoca contemporanea, la distinzione sostanziale è quella fra pigmenti e coloranti. I pigmenti, costituiti da polveri colorate, più o meno fini, hanno colore e consistenza, i coloranti, invece, non hanno consistenza ma solo colore. Colour index name Il C.I name consiste di una lettera per la definizione della categoria (N = natural, P = pigment, D = dye) di una o due lettere per la definizione approssimativa del colore (B = blue, Bk = black, Br = brown, G = green, O = orange, R = red, V = violet, W = white, Y = yellow) ed infine di un numero seriale di due cifre assegnato in relazione alla composizione chimica.

Capitolo 5. Progetto ColoRaman: risultati 87
Questo sistema di classificazione dei pigmenti e delle tinture, elaborato e gestito dalla Society of Dyers and Colorists ed internazionalmente riconosciuto, permette di distinguere i pigmenti e le tinture non in base al loro nome, che come è noto non è univoco ed è anzi causa di equivoci, ma in base alla loro composizione chimica. E’ stato adottato in questo lavoro in quanto permette di rendere più agevole e preciso la consultazione del lavoro stesso.
5.2.2 Le lacche. L'impiego più conosciuto dei coloranti in pittura è nella composizione delle Lacche. Le lacche sono pigmenti, ottenuti da coloranti organici di origine animale o vegetale, fissati su sostanze inorganiche quali il carbonato di calcio o l'idrossido di alluminio, per mezzo di procedimenti chimici. In tal modo le lacche prendono consistenza assumendo il corpo della sostanza inorganica. Le lacche così ottenute possono essere utilizzate come un normale pigmento. Esse mantengono per loro natura, la caratteristica della semitrasparenza ragione per la quale vengono utilizzate soprattutto nella pittura ad olio sotto forma di velature sottili.
5.3 Il medium. I pigmenti allo stato naturale sono polveri colorate macinate. Il loro impiego in pittura prevede che le stesse vengano impastate con un medium o legante. Il medium caratterizza la tecnica pittorica, nel senso che ogni tecnica implica l'uso di un proprio medium, che costituisce il tessuto connettivo dei microgranuli contenuti nel pigmento. Il pigmento mescolato con il medium forma la pellicola pittorica che viene comunemente applicata sulla preparazione cioè il supporto. Nello strato pittorico così creato, è il pigmento che conferisce il colore, mentre il legante (olio, acqua, colla, caseina, etc.) costituisce una sostanza trasparente che assume il colore del primo.
5.3.1 Leganti e film pittorico Si è già visto che la pellicola pittorica è composta dalla mescolanza del pigmento con il medium e che, di tale combinazione, il pigmento costituisce la materia colorante. Il medium, che prende il nome di legante pittorico, per la funzione aggregante che ha sui microgranuli dei colore, è una sostanza trasparente caratterizzata da proprietà fisiche, chimiche, ottiche e meccaniche specifiche per ciascuna classe di legante. La più importante funzione di un legante consiste nella formazione di una sostanza filmogena e cioè di uno strato sottilissimo e solido. Essa viene sollecitata da fenomeni fisici e chimici quali l'evaporazione, la solidificazione, l'ossidazione, e la polimerizzazione e determina in ciascun legante la proprietà coesiva e adesiva. La coesione consiste nella capacità di aggregazione dei microgranuli del pigmento, il quale si presenta come polvere incoerente. L'adesione invece, è la capacità che ha l'impasto, sostenuto dal legante, di restare saldato al supporto. La formazione del film pittorico coincide con l'essiccamento del legante. Leganti organico-proteici I leganti proteici sono di origine animale, quelli maggiormente utilizzati sono le colle animali, l'uovo, la caseina ed il latte. Essi sono solubili in acqua allo stato fluido. In seguito alle modificazioni che subiscono nel corso dell'essiccamento per fenomeni chimici (formazione di legami peptidici), diventano più resistenti. Possono essere aggrediti per via enzimatica e attaccati da muffe e batteri in presenza di umidità. Per tale motivo e ai fini di una buona conservazione i leganti proteici vanno protetti

Capitolo 5. Progetto ColoRaman: risultati 88
con sostanze antifermentative contro l'attacco dei microrganismi. Il normale invecchiamento delle proteine può essere alterato da fattori chimici e fisici che danno luogo al fenomeno della denaturalizzazione, consistente in una modificazione che non comporta necessariamente il loro degrado. D’altronde, tale fenomeno determina una diminuzione della solubilità in acqua, che nel caso di proteine utilizzate come legante, finisce addirittura per giovare allo strato pittorico. Le colle animali si ottengono dalle pelli, dalle cartilagini e/o dalle ossa di alcuni animali che vengono messe a bollire, purificate ed essiccate. La sostanza proteica delle colle animali, che assicura loro buone proprietà adesive e coesive, è il collagene. Le colle animali essiccando formano una pellicola con buone proprietà statiche. Le colle animali sono state impiegate nella tecnica a tempera come medium e nelle preparazioni dei supporti alla pittura (imprimiture e mestiche) come legante, misto a gesso. L'uovo ed in particolare il tuorlo, viene impiegato nella tempera a uovo come medium. Nella miniatura invece viene impiegato il l’albume. Questo ultimo talvolta viene impiegato anche come fissativo per la pittura su tavola. Sia l’albume che il tuorlo sono sostanze che si diluiscono in acqua. Essiccando il tuorlo presenta oltre che sostanze proteiche, anche molte sostanze lipidiche che lo rendono simile ad un grasso e quindi insolubile. L’albume invece nell'asciugare forma un film abbastanza fragile e sensibile all'acqua. I grassi contenuti nel rosso d'uovo non sono di natura siccativa, perciò, se da un lato presentano lo svantaggio di tardare ad asciugare, d'altro lato hanno il pregio di non avere la tendenza a polimerizzare e dunque a subire modificazioni. Il tuorlo, infatti, grazie alla presenza di tali grassi forma pellicole con buone capacità di coesione, adesione, flessibilità e stabilità alla luce caratteristica quest'ultima che fa sì che le pellicole non ingialliscano. La caseina è una proteina contenuta nel latte che si ottiene mediante un complesso procedimento chimico. Essa viene impiegata nella tempera su tavola ma soprattutto nella tempera su parete, oltre che per la preparazione di emulsioni con olio. La caseina non ha buone proprietà meccaniche: essa non è molto flessibile perché asciugando diventa molto fragile e tende a sgretolarsi. Il latte, invece, è più stabile e resistente della caseina perché ha una composizione più grassa. Leganti organico-vegetali I leganti organici vegetali provengono da essudazioni di piante e/o di miscele vegetali ottenute dal succo dei frutti e dei semi. Posiamo distinguere gli oli siccativi e le gomme vegetali. Oli siccativi. Per oli siccativi si intendono solamente quelli che nell'essiccare riescono, in un periodo di tempo limitato, a formare una sostanza filmogena. Gli oli che rispondono a tale requisito sono pochi; e fra essi il più impiegato in pittura è l'olio di lino. Esso, al tatto, asciuga da solo in due giorni; mentre l'essiccamento totale, fino agli strati più profondi, dura decine d'anni, nel corso delle quali hanno luogo fenomeni di ossidazione e di polimerizzazione. Il totale essiccamento dell'olio di lino porta alla formazione di una nuova sostanza: la linossina. Per accelerare tale processo vengono utilizzati dei catalizzanti. Nel passato l'olio di lino veniva mescolato, per favorirne l'essiccamento, con biacca e/o litargirio (pigmenti a base di piombo noti per le loro proprietà siccative). L'invecchiamento dell'olio di lino e la sua trasformazione in linossina, conferiscono alla struttura pigmento-legante maggiore stabilità e solidità. Il difetto maggiore dell'olio di lino è che invecchiando tende a ingiallire. Tale fenomeno, che è irreversibile, può imprimere a tutto il dipinto, nelle sue conseguenze più estreme, un appiattimento cromatico. Gomme vegetali. Le gomme vegetali sono sostanze naturali vegetali prodotte dall'essudazione di alcune piante. Sono sostanze idrofile essendo solubili e gonfiabili in acqua. Formano una pellicola

Capitolo 5. Progetto ColoRaman: risultati 89
abbastanza fragile che viene resa più elastica aggiungendo della glicerina o del miele. Come leganti sono state utilizzate nell'acquerello e nella miniatura al posto dell'uovo La più comune fra le gomme è la gomma arabica che si scioglie in acqua facilmente. Fra le altre gomme ricordiamo la gomma adragante e la gomma-di ciliegio, di susino e di pesco.
5.3.2 Vernici In campo pittorico e in particolare nel restauro, con il termine vernice si intende uno strato sottile, trasparente e il più delle volte incolore, applicato sulla superficie pittorica alla quale aderisce. La funzione principale delle vernici consiste nella protezione della pellicola pittorica dall'aggressione degli agenti esterni. La loro prerogativa è quella di formare pellicole sottili (film) con alta capacità adesive. Per la composizione di vernici vengono impiegate, sin dall'antichità, varie sostanze scelte fra le resine, gli oli e le cere. Le vernici hanno anche una funzione di carattere estetico in quanto, rispondendo a particolari requisiti ottici, riescono a influire positivamente sulla resa ottica della superficie pittorica. Le vernici costituiscono, infatti, uno strato omogeneo e otticamente uniforme che riduce di molto la diffusione della luce della superficie pittorica non verniciata, che si configura invece come una superficie disomogenea in ragione degli ispessimenti delle pennellate e del colore. Per tali caratteristiche la vernice rinforza le cromie dei colori aumentandone il contrasto.
5.4 Il database dei pigmenti pittorici stesi
5.4.1 Introduzione Il problema dell’identificazione dei pigmenti già stesi. Come ampiamente dimostrato nel capitolo 1, la RS viene utilizzata con successo per le indagini sui BB.CC. Tuttavia, perché la RS possa affermarsi pienamente quale metodologia diagnostica, non è ora più sufficiente la produzione di contributi scientifici riguardanti casi specifici, ma è necessario fornire, ad esempio, dettagliate indicazioni sui pigmenti che possono essere identificati, sul tipo di manufatti che è possibile analizzare e soprattutto, per ogni tipo di oggetto, sulla più opportuna implementazione dell’apparato strumentale. Il progetto ColoRaman si è proposto quindi di testare le effettive potenzialità di un sistema combinato per spettroscopia Raman, dotato di tre laser come sorgenti di eccitazione (531.5 nm, 632.8 nm, 780 nm), per la caratterizzazione dei pigmenti pittorici secchi ma soprattutto dei relativi colori già stesi, intendendosi con colore la miscela di pigmento e legante. I pigmenti secchi, utilizzati come standard di riferimento, vengono caratterizzati nelle migliori condizioni ma non altrettanto è possibile quando li si vuole identificare già stesi su un manufatto come colore. Infatti, i leganti ed i supporti pittorici, quasi sempre di origine organica, sono responsabili di una fluorescenza in molti casi così intensa da coprire il debole segnale Raman originato dal pigmento e rendere così impossibile l’identificazione di quest’ultimo. E’ possibile, tuttavia, stabilire per ogni pigmento e per ogni singola tecnica pittorica la lunghezza d’onda di eccitazione capace o di diminuire l’intensità della fluorescenza oppure di generare una intensa emissione Raman così da migliorare il rapporto segnale-rumore, essendo in questo caso il rumore costituito dalla emissione di fluorescenza. Poiché i diversi materiali impiegati per la realizzazione dei leganti e dei supporti possono contribuire in maniera diversa a mascherare il segnale Raman é necessario condurre questa indagine distintamente per ogni tecnica pittorica.

Capitolo 5. Progetto ColoRaman: risultati 90
Questo lavoro presenta i database di spettri Raman di pigmenti pittorici secchi già stesi con le tecniche pittoriche dell’affresco, della tempera all’uovo, della tempera alla caseina e dell’olio. Tutte le informazioni relative ai 99 pigmenti oggetto dell’indagine vengono fornite con le modalità discusse sopra. L’obiettivo è quello di rendere, così facendo, questi dati agevolmente confrontabili con gli eventuali lavori che saranno realizzati con gli stessi criteri. Oltre al database di spettri Raman viene pubblicato anche quello degli spettri di fluorescenza. Benché questi ultimi possano essere facilmente acquisiti, praticamente con lo stesso sistema adoperato per la spettroscopia Raman dispersiva, finora sono stati pochi i lavori che hanno indagato la caratterizzabilità dei pigmenti pittorici anche tramite i loro spettri di fluorescenza [Wallert 1986, Guineau 1989, ]. Tra l’altro, questa metodologia può risultare particolarmente utile perché, diversamente da quanto avviene per il debole segnale Raman, per l’emissione di fluorescenza il rapporto segnale-rumore è, in genere, così alto da permettere l’identificazione del pigmento già steso. Inoltre, anche se i pigmenti caratterizzabili tramite la loro fluorescenza sono solo una piccola parte, nei casi in cui il segnale Raman non sia misurabile, l’identificazione dei pigmenti potrebbe essere comunque possibile tramite la loro fluorescenza. Si fa notare, infine, che il database raccoglie solo gli spettri di fluorescenza i cui picchi siano stati ritenuti sufficientemente caratteristici. Standardizzazione dei database Numerosi sono i database di spettri Raman di pigmenti pittorici pubblicati o disponibili on-line. Poche informazioni vengono però, in genere, fornite circa la provenienza del pigmento analizzato. Questo dato, tuttavia, risulta essere di fondamentale importanza in quanto è stato osservato che pigmenti prodotti da fornitori diversi e commercializzati con lo stesso nome, possono presentare spettri Raman differenti se non addirittura incompatibili [Peréz et al. 2002]. Sarebbe allora auspicabile la standardizzazione dei database dei pigmenti, fornendo informazioni dettagliate sui prodotti analizzati. Per i pigmenti commercializzati: tutte le caratteristiche del pigmento analizzato sufficienti a contraddistinguere inequivocabilmente ogni prodotto (ditta fornitrice, nome e codice del prodotto). Per i pigmenti prodotti dai laboratori di ricerca: la formula chimica, le sostanze utilizzate e la procedura seguita per la preparazione dei pigmenti. Si potranno così confrontare tra loro i database di spettri Raman, permettendo così, ad esempio, di confrontare i dati ottenuti con sistemi Raman diversi e di riscontrare le eventuali differenze tra i prodotti commercializzati con lo stesso nome ma prodotti da ditte diverse. Inoltre, se realizzati con gli accorgimenti suggeriti, con altrettanta immediatezza si potranno confrontare anche i database di dati raccolti con altre tecniche analitiche. In questo modo, per ogni prodotto commerciale, si potrebbe avere a disposizione una quantità di informazioni decisamente più ampia.
5.4.2. Preparazione dei campioni I database di spettri Raman e di fluorescenza sono stati compilati raccogliendo gli spettri di 99 diversi pigmenti pittorici acquistati dalla ditta Kremer (Germania) e dalla Zecchi di Firenze (Italia). Per l’elaborazione dei database di spettri Raman e di fluorescenza dei pigmenti già stesi si è proceduto, in laboratorio, alla stesura dei pigmenti, secondo le ricette antiche [Thompson 1936, Doerner 1949, Wehlte 1975, Seymour 1995]. E’ stato così possibile eseguire le misure su riproduzioni realizzate utilizzando materiali e tecniche accuratamente selezionate. Si sono volute ricreare, cioè, le stesse condizioni di una reale misura in situ su

Capitolo 5. Progetto ColoRaman: risultati 91
manufatti originali. Le tele sono state prodotte con dimensioni tali da poter essere facilmente esaminate direttamente con il microscopio, figura 5.4.2.12. Ricette e prodotti per la realizzazione delle quattro tecniche pittoriche sono descritte di seguito: Preparazione: supporto pittorico per l’olio e le tempere, è stata realizzata miscelando gesso di Bologna, colla di coniglio e allume di Rocca. Nelle figure 5.4.2.1 e 5.4.2.2 alcuni momenti della preparazione delle tele.
Figura 5.4.2.1, 5.4.2.2. Preparazione dell’impasto di gesso di Bologna, colla di coniglio e allume di rocca. Legante per Tempera all’uovo: un cucchiaino di aceto, un cucchiaino di miele, un cucchiaino di allume di Rocca e un albume (per i colori freddi) o un tuorlo (per i colori caldi).Figure 5.4.2.3, 5.4.2.4, 5.4.2.5.
Figura 5.4.2.3, 5.4.2.4. Mescolatura del pigmento secco e del legante.

Capitolo 5. Progetto ColoRaman: risultati 92
Figura 5.4.2.5. Stesura del colore sulla preparazione Legante per tempera alla caseina: il legante caseina è prodotto dalla Zecchi (Firenze) art. 2050. Figure 5.4.2.6 e 5.4.2.7.
Figure 5.4.2.6, 5.4.2.7 Miscelatura del pigmento con la tempera alla caseina e stesura del colore. Affresco: calce (grassello), sabbia calcarea.
Figura 5.4.2.8, 5.4.2.9, 5.4.2.10 Preparazione dell’affresco e stesura dei pigmenti. Legante per l’olio: olio di lino chiarificato della Lefranc & Bourgeois (Francia). Figura 5.4.2.11

Capitolo 5. Progetto ColoRaman: risultati 93
Figura 5.4.2.11. Stesura del pigmento miscelato con olio di lino.
Figura 5.4.2.12. Le tele possono essere esaminate direttamente con il microscopio.
5.4.3. Risultati e discussione Di seguito, in tabella 5.4.3.1, vengono riportati, per ognuno dei 99 pigmenti analizzati, il nome dell’articolo, la ditta produttrice (K = Kremer, Z = Zecchi) ed il relativo codice del prodotto. Per snellire il testo ad ogni pigmento è stato associato un numero identificativo, ID (prima colonna) che sarà utilizzato, d’ora in poi, al posto del nome del pigmento. Le lettere indicano se è stato possibile ottenere il corrispondente spettro Raman o di fluorescenza. Per ognuna delle tre lunghezze d’onda di eccitazione, le lettere indicano, inoltre, in quali tecniche pittoriche è stato possibile identificare il pigmento ed è stata adottata la seguente convenzione: P = Pigmento, F = Affresco, C = Caseina, E = Uovo, O = Olio. Le lettere maiuscole si riferiscono agli spettri Raman, mentre le corrispondenti lettere minuscole fanno riferimento agli spettri di fluorescenza. La tabella è anche pubblicata on line, con tutti gli spettri disponibili, al sito: http://www.ct.infn.it/~archeo/
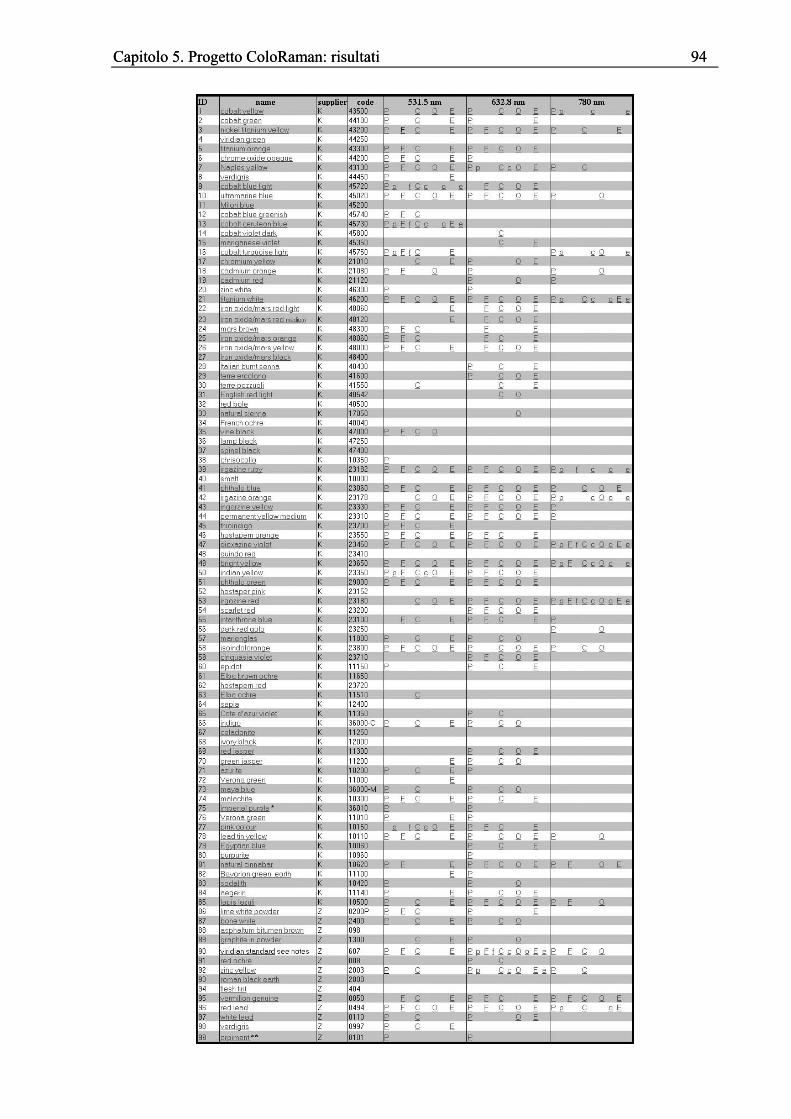
Capitolo 5. Progetto ColoRaman: risultati 94
Capitolo 5. Progetto ColoRaman: risultati 94

Capitolo 5. Progetto ColoRaman: risultati 95
Tabella 5.4.3.1. Risultati dl progetto ColoRaman. La tabella riporta, per ogni pigmento, il numero d’identificazione (ID), il nome inglese del prodotto, il fornitore (K = Kremer, Z = Zecchi) e il codice prodotto. La presenza di una lettera indica se è stato possibile ottenere uno spettro Raman o di fluorescenza caratterizzante. Le lettere maiuscole si riferiscono agli spettri Raman, le lettere minuscole agli spettri di fluorescenza. Per ognuna delle tre lunghezze d’onda di eccitazione, le lettere indicano in quale tecnica pittorica è stato possibile identificare il pigmento. È stata adottata la seguente convenzione P = pigmento secco, F = Affresco, C = Tempera alla caseina, E = Tempera all’uovo, O = olio di lino. * La porpora non è stata stesa perché non va usata con nessuna delle quattro tecniche pittoriche studiate. ** L’orpimento non è stato steso con nessuna delle quattro tecniche perché non attualmente disponibile in quantità sufficiente. Pigmenti caratterizzabili solo con una lunghezza d’onda di eccitazione. Alcuni pigmenti sono caratterizzabili tramite spettroscopia Raman solo con una delle tre lunghezza d’onda testate. Utilizzando le altre radiazioni laser, l’emissione Raman è troppo debole oppure la fluorescenza dello stesso pigmento è troppo intensa. Di seguito, vengono riportati i pigmenti che è stato possibile caratterizzare solo con una delle tre lunghezze d’onda di eccitazione disponibili: Eccitazione a 531.5 nm 8,9,12,13,35,38,45,98 Eccitazione a 632.8 nm 17, 28, 29, 54, 59, 65, 69, 70, 77, 79, 80, 82, 89, 91, 94. Eccitazione a 780 nm 56 Si comprende subito che una strumentazione operante con queste tre lunghezze d’onda permette di caratterizzare più pigmenti rispetto ad un sistema operante con una o due sole lunghezze d’onda di eccitazione. E cosa ancor più importante, come sarà discusso più avanti, una strumentazione così equipaggiata consente, spesso, di identificare i pigmenti già stesi sui manufatti. Risonanza, fluorescenza e tecniche pittoriche: selezione della radiazione laser di eccitazione. Come mostrato nel paragrafo precedente, alcuni pigmenti possono essere caratterizzati solo utilizzando una delle tre lunghezze d’onda disponibili. Questo dato, da solo, è sufficiente a incentivare l’uso di un apparato Raman equipaggiato con almeno tre laser. Questo sistema risulta, però, indispensabile per l’identificazione dei pigmenti sui manufatti. Si può osservare dalla tabella 5.4.3.1, che alcuni pigmenti, caratterizzabili utilizzando tutte e tre le lunghezze d’onda, una volta stesi, possono essere identificati utilizzando solo una delle tre radiazione laser. Frequenze di eccitazione alte, come quella a 531.5 nm, generano segnali Raman intensi, ma possono indurre, contemporaneamente, una emissione di fluorescenza del legante così forte da coprire l’emissione Raman del pigmento. Analogamente, una frequenza bassa, come quella a 780 nm, riduce l’emissione di fluorescenza del legante ma contemporaneamente, anche quella Raman. Va tenuto in conto, infine, che l’eventuale emissione di risonanza Raman può essere così intensa da essere rilevabile a discapito della pur forte fluorescenza. Di seguito, vengono riportati alcuni spettri per illustrare quanto detto. Tranne che per l’esempio relativo all’affresco, vengono riportati sovrapposti solamente gli spettri che hanno elementi utili all’identificazione del pigmento. Si sottintende quindi che se lo spettro ottenuto con una certa lunghezza d’onda non è riportato esso non è risultato utile all’identificazione del pigmento. Affresco
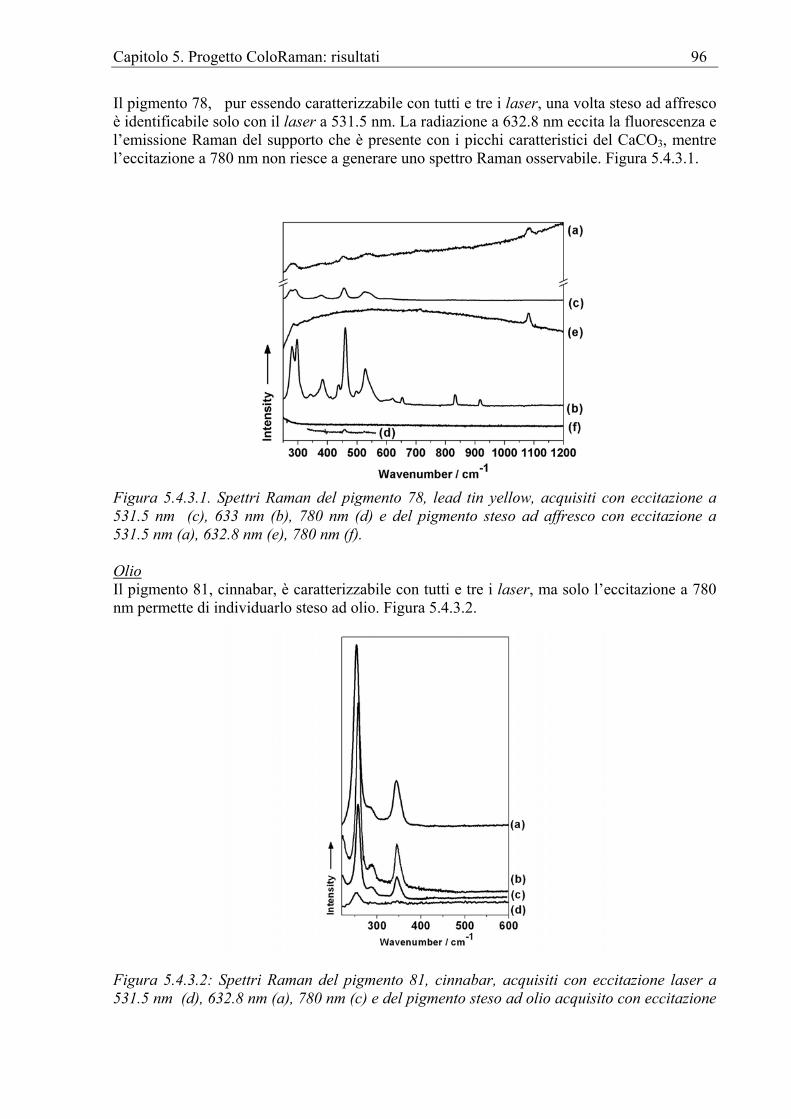
Capitolo 5. Progetto ColoRaman: risultati 96
Il pigmento 78, pur essendo caratterizzabile con tutti e tre i laser, una volta steso ad affresco è identificabile solo con il laser a 531.5 nm. La radiazione a 632.8 nm eccita la fluorescenza e l’emissione Raman del supporto che è presente con i picchi caratteristici del CaCO3, mentre l’eccitazione a 780 nm non riesce a generare uno spettro Raman osservabile. Figura 5.4.3.1.
Figura 5.4.3.1. Spettri Raman del pigmento 78, lead tin yellow, acquisiti con eccitazione a 531.5 nm (c), 633 nm (b), 780 nm (d) e del pigmento steso ad affresco con eccitazione a 531.5 nm (a), 632.8 nm (e), 780 nm (f). Olio Il pigmento 81, cinnabar, è caratterizzabile con tutti e tre i laser, ma solo l’eccitazione a 780 nm permette di individuarlo steso ad olio. Figura 5.4.3.2.
Figura 5.4.3.2: Spettri Raman del pigmento 81, cinnabar, acquisiti con eccitazione laser a
531.5 nm (d), 632.8 nm (a), 780 nm (c) e del pigmento steso ad olio acquisito con eccitazione
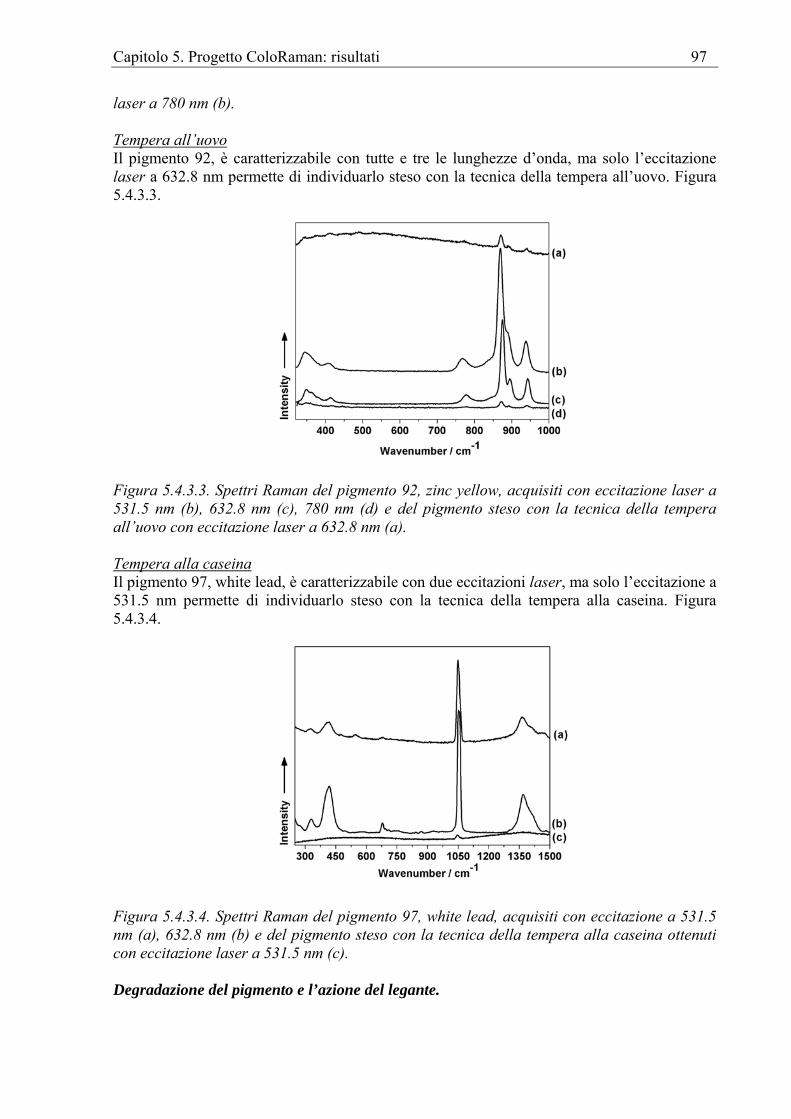
Capitolo 5. Progetto ColoRaman: risultati 97
empera all’uovo
laser a 780 nm (b). T
caratterizzabile con tutte e tre le lunghezze d’onda, ma solo l’eccitazione
igura 5.4.3.3. Spettri Raman del pigmento 92, zinc yellow, acquisiti con eccitazione laser a
empera alla caseina
Il pigmento 92, è laser a 632.8 nm permette di individuarlo steso con la tecnica della tempera all’uovo. Figura 5.4.3.3.
F531.5 nm (b), 632.8 nm (c), 780 nm (d) e del pigmento steso con la tecnica della tempera all’uovo con eccitazione laser a 632.8 nm (a). T
ead, è caratterizzabile con due eccitazioni laser, ma solo l’eccitazione a
igura 5.4.3.4. Spettri Raman del pigmento 97, white lead, acquisiti con eccitazione a 531.5
egradazione del pigmento e l’azione del legante.
Il pigmento 97, white l531.5 nm permette di individuarlo steso con la tecnica della tempera alla caseina. Figura 5.4.3.4.
Fnm (a), 632.8 nm (b) e del pigmento steso con la tecnica della tempera alla caseina ottenuti con eccitazione laser a 531.5 nm (c). D
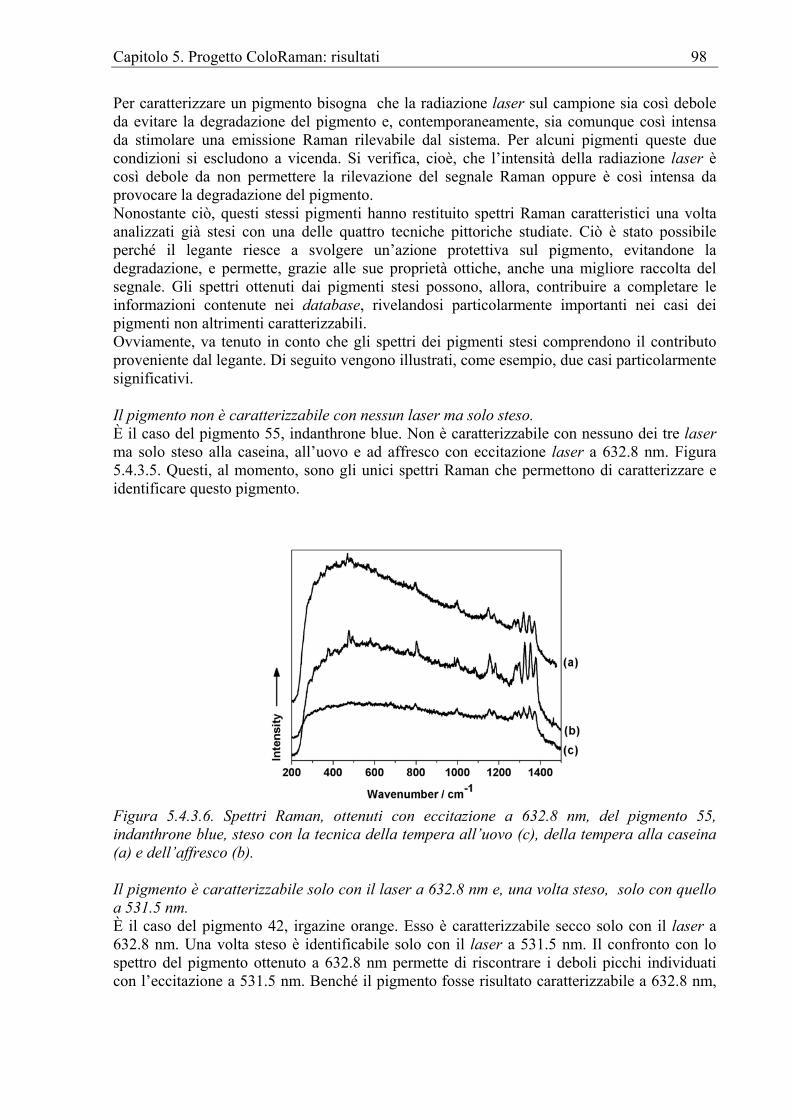
Capitolo 5. Progetto ColoRaman: risultati 98
azione laser sul campione sia così debole
nno restituito spettri Raman caratteristici una volta
spettri dei pigmenti stesi comprendono il contributo
pigmento non è caratterizzabile con nessun laser ma solo steso. e con nessuno dei tre laser
,
pigmento è caratterizzabile solo con il laser a 632.8 nm e, una volta steso, solo con quello
pigmento 42, irgazine orange. Esso è caratterizzabile secco solo con il laser a
Per caratterizzare un pigmento bisogna che la radida evitare la degradazione del pigmento e, contemporaneamente, sia comunque così intensa da stimolare una emissione Raman rilevabile dal sistema. Per alcuni pigmenti queste due condizioni si escludono a vicenda. Si verifica, cioè, che l’intensità della radiazione laser è così debole da non permettere la rilevazione del segnale Raman oppure è così intensa da provocare la degradazione del pigmento. Nonostante ciò, questi stessi pigmenti haanalizzati già stesi con una delle quattro tecniche pittoriche studiate. Ciò è stato possibile perché il legante riesce a svolgere un’azione protettiva sul pigmento, evitandone la degradazione, e permette, grazie alle sue proprietà ottiche, anche una migliore raccolta del segnale. Gli spettri ottenuti dai pigmenti stesi possono, allora, contribuire a completare le informazioni contenute nei database, rivelandosi particolarmente importanti nei casi dei pigmenti non altrimenti caratterizzabili. Ovviamente, va tenuto in conto che gli proveniente dal legante. Di seguito vengono illustrati, come esempio, due casi particolarmente significativi. IlÈ il caso del pigmento 55, indanthrone blue. Non è caratterizzabilma solo steso alla caseina, all’uovo e ad affresco con eccitazione laser a 632.8 nm. Figura 5.4.3.5. Questi, al momento, sono gli unici spettri Raman che permettono di caratterizzare e identificare questo pigmento.
Figura 5.4.3.6. Spettri Raman, ottenuti con eccitazione a 632.8 nm, del pigmento 55indanthrone blue, steso con la tecnica della tempera all’uovo (c), della tempera alla caseina (a) e dell’affresco (b). Ila 531.5 nm. È il caso del632.8 nm. Una volta steso è identificabile solo con il laser a 531.5 nm. Il confronto con lo spettro del pigmento ottenuto a 632.8 nm permette di riscontrare i deboli picchi individuati con l’eccitazione a 531.5 nm. Benché il pigmento fosse risultato caratterizzabile a 632.8 nm,
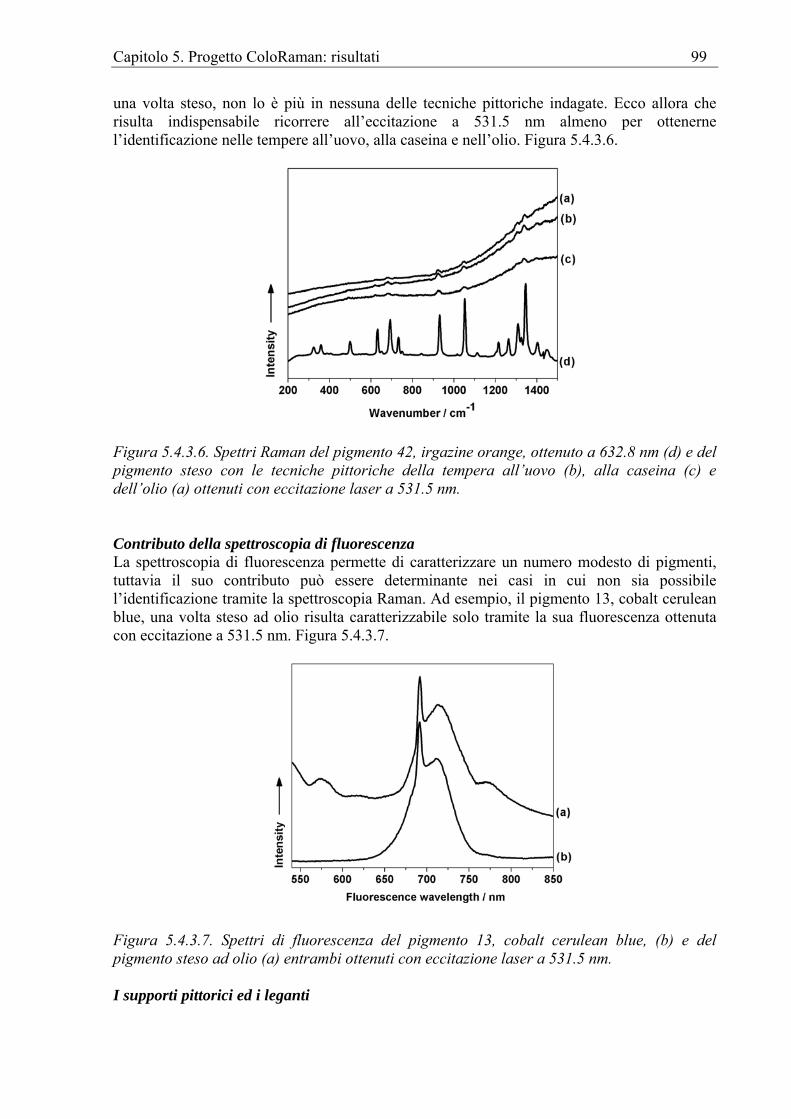
Capitolo 5. Progetto ColoRaman: risultati 99
una volta steso, non lo è più in nessuna delle tecniche pittoriche indagate. Ecco allora che risulta indispensabile ricorrere all’eccitazione a 531.5 nm almeno per ottenerne l’identificazione nelle tempere all’uovo, alla caseina e nell’olio. Figura 5.4.3.6.
igura 5.4.3.6. Spettri Raman del pigmento 42, irgazine orange, ottenuto a 632.8 nm (d) e del
ontributo della spettroscopia di fluorescenza atterizzare un numero modesto di pigmenti,
igura 5.4.3.7. Spettri di fluorescenza del pigmento 13, cobalt cerulean blue, (b) e del
I supporti pittorici ed i leganti
Fpigmento steso con le tecniche pittoriche della tempera all’uovo (b), alla caseina (c) e dell’olio (a) ottenuti con eccitazione laser a 531.5 nm. CLa spettroscopia di fluorescenza permette di cartuttavia il suo contributo può essere determinante nei casi in cui non sia possibile l’identificazione tramite la spettroscopia Raman. Ad esempio, il pigmento 13, cobalt cerulean blue, una volta steso ad olio risulta caratterizzabile solo tramite la sua fluorescenza ottenuta con eccitazione a 531.5 nm. Figura 5.4.3.7.
Fpigmento steso ad olio (a) entrambi ottenuti con eccitazione laser a 531.5 nm.
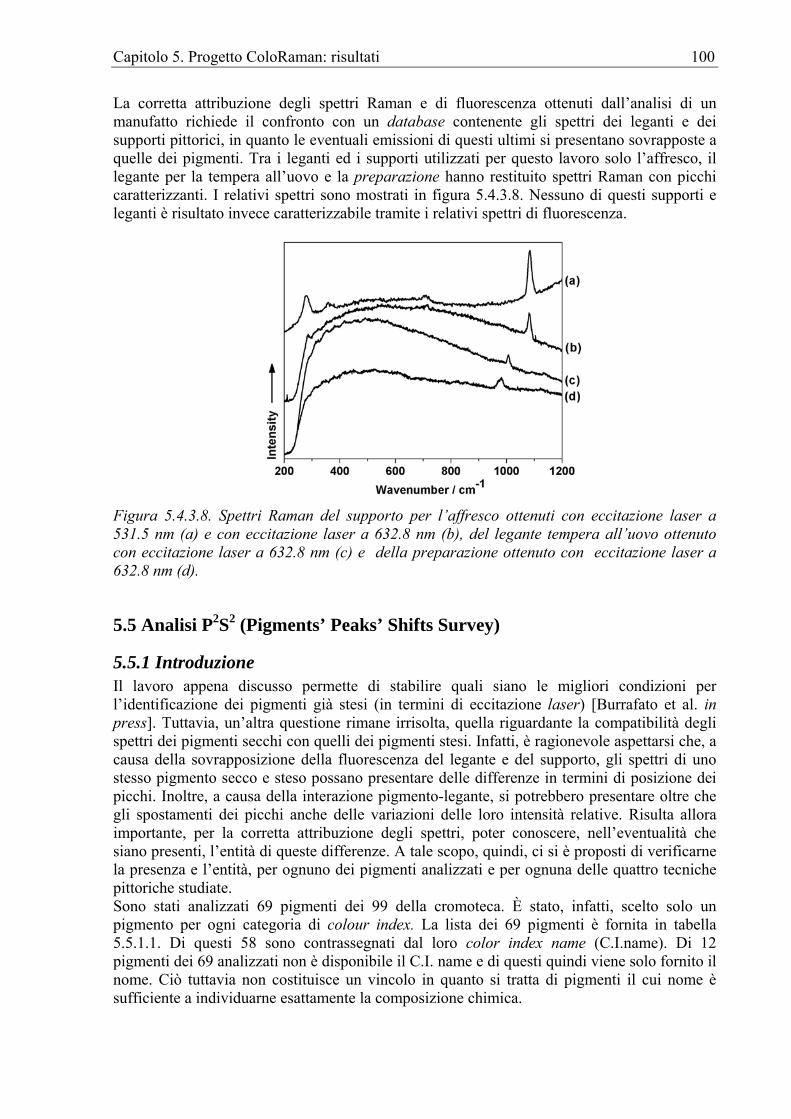
Capitolo 5. Progetto ColoRaman: risultati 100
a corretta attribuzione degli spettri Raman e di fluorescenza ottenuti dall’analisi di un o con un database contenente gli spettri dei leganti e dei
ottenuto
.5 Analisi P2S2 (Pigments’ Peaks’ Shifts Survey)
ette di stabilire quali siano le migliori condizioni per gmenti già stesi (in termini di eccitazione laser) [Burrafato et al. in
categoria di colour index. La lista dei 69 pigmenti è fornita in tabella
sufficiente a individuarne esattamente la composizione chimica.
Lmanufatto richiede il confrontsupporti pittorici, in quanto le eventuali emissioni di questi ultimi si presentano sovrapposte a quelle dei pigmenti. Tra i leganti ed i supporti utilizzati per questo lavoro solo l’affresco, il legante per la tempera all’uovo e la preparazione hanno restituito spettri Raman con picchi caratterizzanti. I relativi spettri sono mostrati in figura 5.4.3.8. Nessuno di questi supporti e leganti è risultato invece caratterizzabile tramite i relativi spettri di fluorescenza.
Figura 5.4.3.8. Spettri Raman del supporto per l’affresco ottenuti con eccitazione laser a 531.5 nm (a) e con eccitazione laser a 632.8 nm (b), del legante tempera all’uovocon eccitazione laser a 632.8 nm (c) e della preparazione ottenuto con eccitazione laser a 632.8 nm (d).
5
5.5.1 Introduzione Il lavoro appena discusso perml’identificazione dei pipress]. Tuttavia, un’altra questione rimane irrisolta, quella riguardante la compatibilità degli spettri dei pigmenti secchi con quelli dei pigmenti stesi. Infatti, è ragionevole aspettarsi che, a causa della sovrapposizione della fluorescenza del legante e del supporto, gli spettri di uno stesso pigmento secco e steso possano presentare delle differenze in termini di posizione dei picchi. Inoltre, a causa della interazione pigmento-legante, si potrebbero presentare oltre che gli spostamenti dei picchi anche delle variazioni delle loro intensità relative. Risulta allora importante, per la corretta attribuzione degli spettri, poter conoscere, nell’eventualità che siano presenti, l’entità di queste differenze. A tale scopo, quindi, ci si è proposti di verificarne la presenza e l’entità, per ognuno dei pigmenti analizzati e per ognuna delle quattro tecniche pittoriche studiate. Sono stati analizzati 69 pigmenti dei 99 della cromoteca. È stato, infatti, scelto solo un pigmento per ogni 5.5.1.1. Di questi 58 sono contrassegnati dal loro color index name (C.I.name). Di 12 pigmenti dei 69 analizzati non è disponibile il C.I. name e di questi quindi viene solo fornito il nome. Ciò tuttavia non costituisce un vincolo in quanto si tratta di pigmenti il cui nome è

Capitolo 5. Progetto ColoRaman: risultati 101
i dei pigmenti dry e dei
resente, sono stati segnati i suoi picchi. Altrimenti,
rto segnale-rumore.
rrispondente al legante utilizzato per la loro
Note suLe tabeshifts d t’ultimo non è presente gli shifts sono
che si presenta con maggiore intensità. Questo ultimo viene
il fornitore ((K = Kremer, Z = Zecchi) e il odice prodotto. La presenza di una lettera indica se è stato possibile ottenere uno spettro
La tabella 5.5.1.1 riporta inoltre per ciascuno dei 69 pigmenti analizzati il nome inglese di ciascun prodotto, il fornitore (K = Kremer, Z = Zecchi) ed il codice del prodotto. La tabella è on-line e permette di consultare sia gli spettri sovrappostcorrispondenti pigmenti già stesi sia le corrispondenti tabelle dei picchi. La presenza di shift maggiori di 4cm-1 è indicata da lettere in grassetto. Note sulla rappresentazione degli Spettri Gli spettri sono rappresentati con linee guida corrispondenti alla posizione dei picchi. Si è adottata la seguente prassi.
1) Se lo spettro del pigmento secco è psono state indicate le posizioni dei picchi di quello spettro, ottenuto dai pigmenti stesi, con il migliore rappo
2) Se lo spettro del pigmento secco è presente e negli spettri dei pigmenti stesi sono presenti picchi non presenti in quello secco, quest’ultimi sono stati segnalati aggiungendo alla loro etichetta la sigla costesura, secondo la notazione descritta sopra. lle tabelle dei picchi lle dei picchi riportano le posizioni dei picchi dei pigmenti secchi e stesi e i relativi ei picchi rispetto al pigmento dry. Se ques
calcolati rispetto allo spettrosegnalato da un asterisco. Gli spettri acquisiti con eccitazione a 531.5 o 632.8 nm sono indicati aggiungendo rispettivamente ‘5’ o ‘6’. Tabella 5.5.1.1. Lista dei 69 pigmenti analizzati. La tabella riporta per ogni pigmento il colour index name (CI name), il nome inglese,cRaman. Per ognuna delle due lunghezze d’onda di eccitazione, le lettere indicano in quale tecnica pittorica è stato possibile identificare i pigmenti. È stata adottata la seguente convenzione. P = Pigmento secco, F = Affresco, C = Tempera alla caseina, E = Tempera all’uovo, O = olio di lino. La presenza negli spettri di shift maggiori di 4 cm-1 è indicata dall’uso di lettere in grassetto.
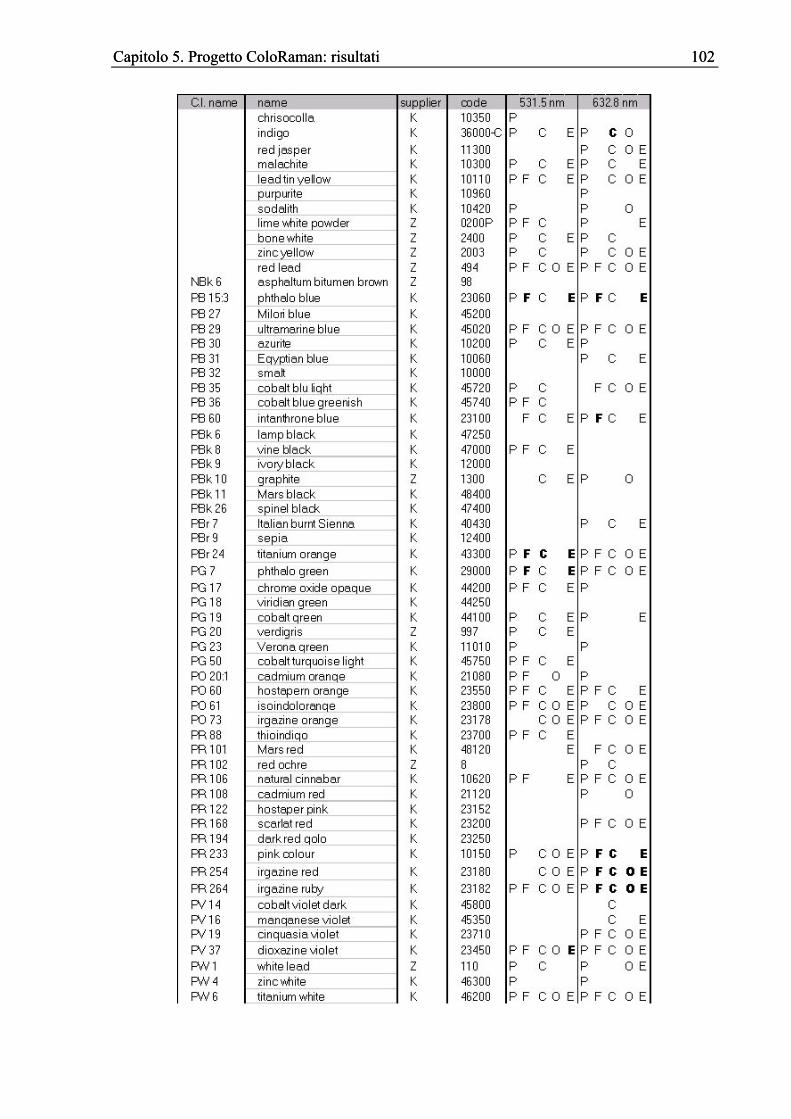
Capitolo 5. Progetto ColoRaman: risultati 102
Capitolo 5. Progetto ColoRaman: risultati 102
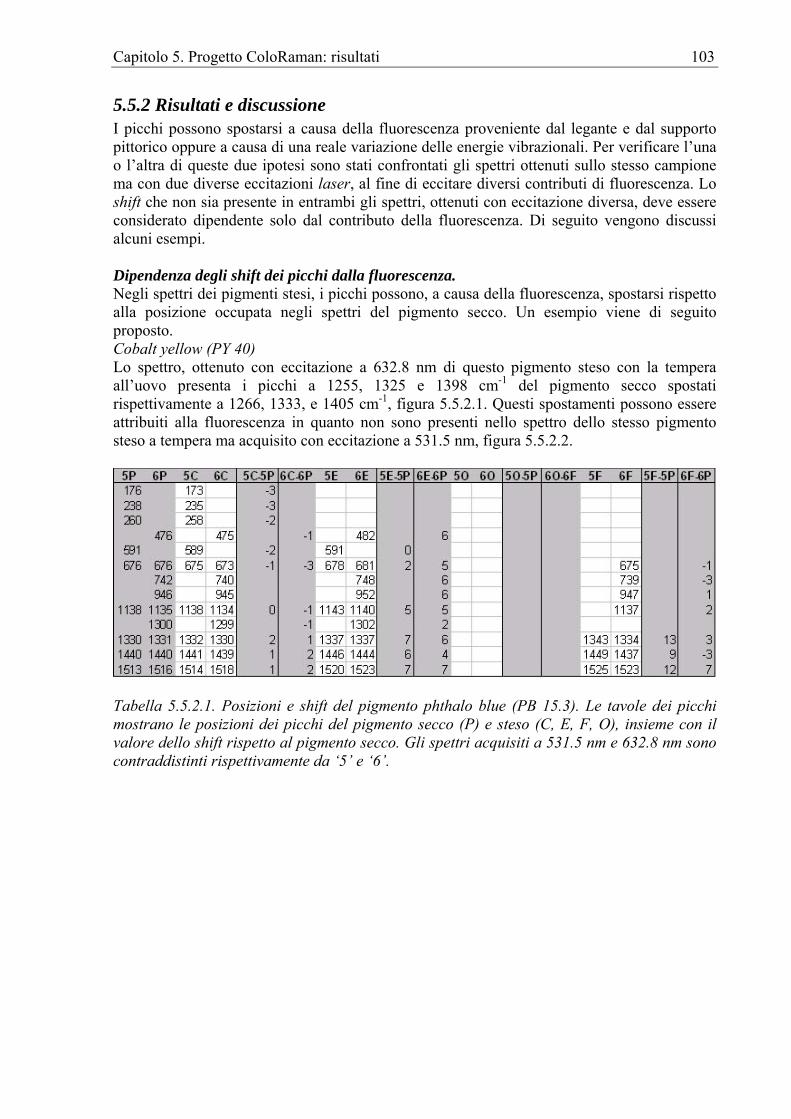
Capitolo 5. Progetto ColoRaman: risultati 103
5.5.2 Risultati e discussione I picchi possono spostarsi a causa della fluorescenza proveniente dal legante e dal supporto pittorico oppure a causa di una reale variazione delle energie vibrazionali. Per verificare l’una o l’altra di queste due ipotesi sono stati confrontati gli spettri ottenuti sullo stesso campione ma con due diverse eccitazioni laser, al fine di eccitare diversi contributi di fluorescenza. Lo shift che non sia presente in entrambi gli spettri, ottenuti con eccitazione diversa, deve essere considerato dipendente solo dal contributo della fluorescenza. Di seguito vengono discussi alcuni esempi. Dipendenza degli shift dei picchi dalla fluorescenza. Negli spettri dei pigmenti stesi, i picchi possono, a causa della fluorescenza, spostarsi rispetto alla posizione occupata negli spettri del pigmento secco. Un esempio viene di seguito proposto. Cobalt yellow (PY 40) Lo spettro, ottenuto con eccitazione a 632.8 nm di questo pigmento steso con la tempera all’uovo presenta i picchi a 1255, 1325 e 1398 cm-1 del pigmento secco spostati rispettivamente a 1266, 1333, e 1405 cm-1, figura 5.5.2.1. Questi spostamenti possono essere attribuiti alla fluorescenza in quanto non sono presenti nello spettro dello stesso pigmento steso a tempera ma acquisito con eccitazione a 531.5 nm, figura 5.5.2.2.
Tabella 5.5.2.1. Posizioni e shift del pigmento phthalo blue (PB 15.3). Le tavole dei picchi mostrano le posizioni dei picchi del pigmento secco (P) e steso (C, E, F, O), insieme con il valore dello shift rispetto al pigmento secco. Gli spettri acquisiti a 531.5 nm e 632.8 nm sono contraddistinti rispettivamente da ‘5’ e ‘6’.
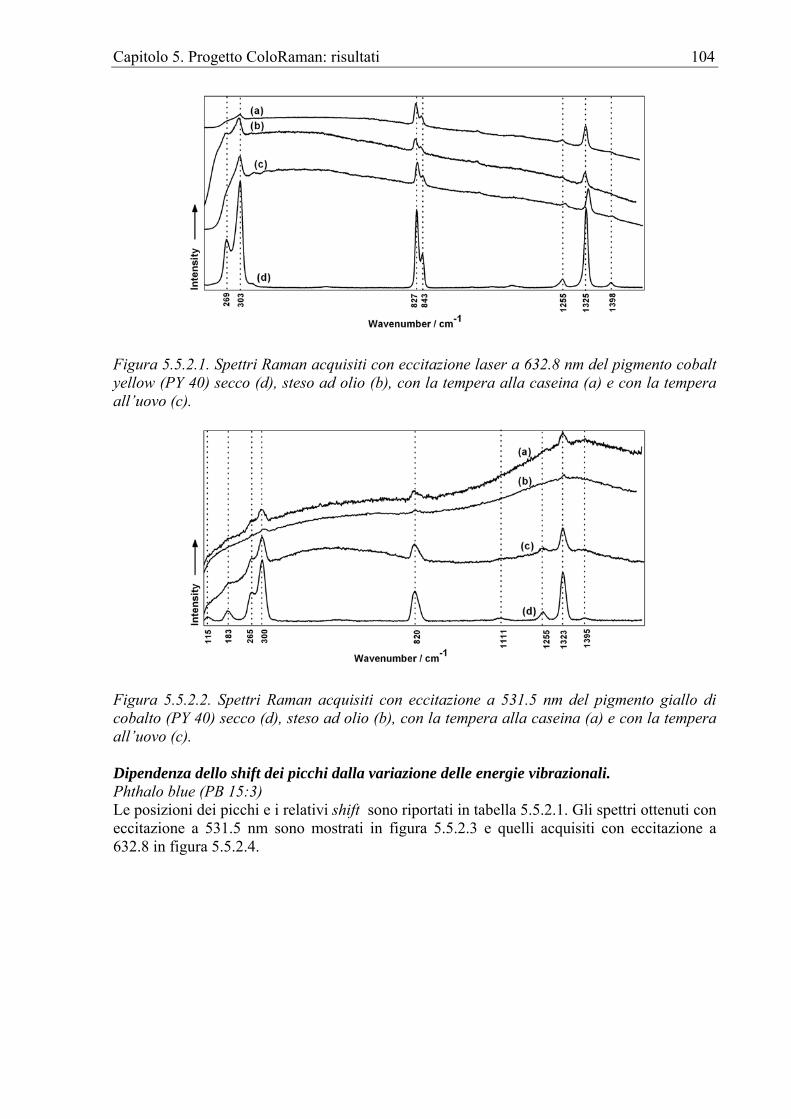
Capitolo 5. Progetto ColoRaman: risultati 104
Figura 5.5.2.1. Spettri Raman acquisiti con eccitazione laser a 632.8 nm del pigmento cobalt yellow (PY 40) secco (d), steso ad olio (b), con la tempera alla caseina (a) e con la tempera all’uovo (c).
Figura 5.5.2.2. Spettri Raman acquisiti con eccitazione a 531.5 nm del pigmento giallo di cobalto (PY 40) secco (d), steso ad olio (b), con la tempera alla caseina (a) e con la tempera all’uovo (c). Dipendenza dello shift dei picchi dalla variazione delle energie vibrazionali. Phthalo blue (PB 15:3) Le posizioni dei picchi e i relativi shift sono riportati in tabella 5.5.2.1. Gli spettri ottenuti con eccitazione a 531.5 nm sono mostrati in figura 5.5.2.3 e quelli acquisiti con eccitazione a 632.8 in figura 5.5.2.4.
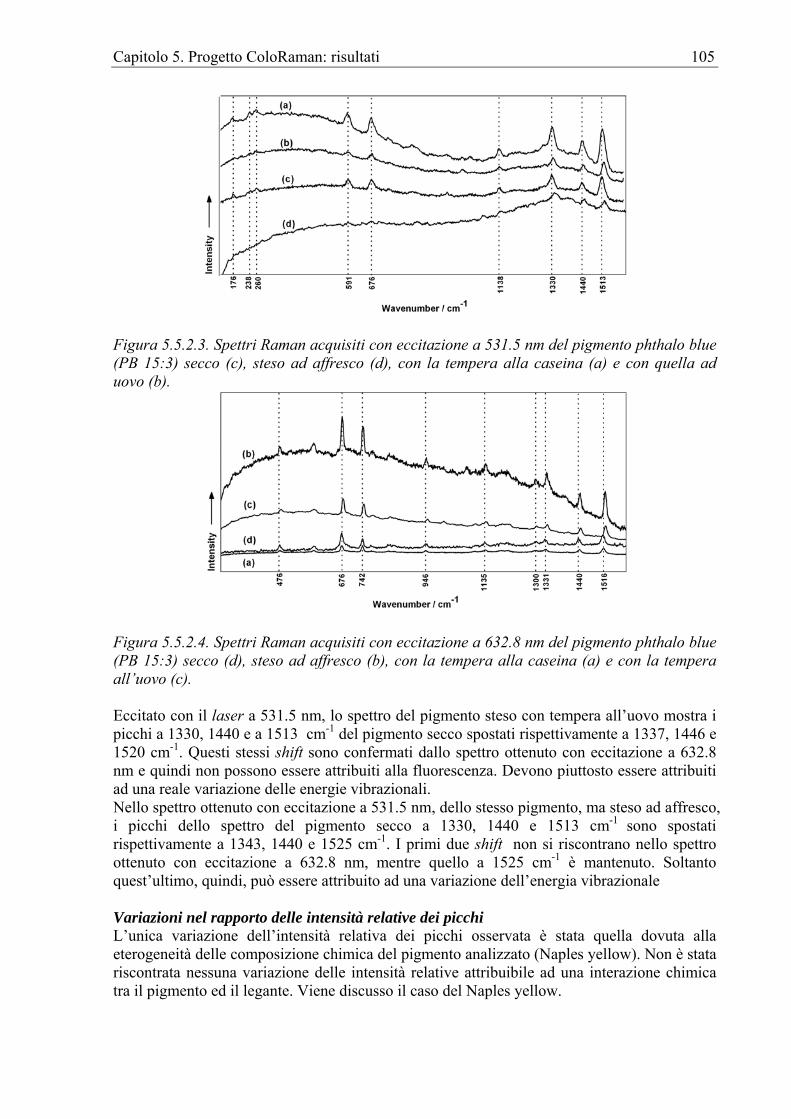
Capitolo 5. Progetto ColoRaman: risultati 105
Figura 5.5.2.3. Spettri Raman acquisiti con eccitazione a 531.5 nm del pigmento phthalo blue (PB 15:3) secco (c), steso ad affresco (d), con la tempera alla caseina (a) e con quella ad uovo (b).
Figura 5.5.2.4. Spettri Raman acquisiti con eccitazione a 632.8 nm del pigmento phthalo blue (PB 15:3) secco (d), steso ad affresco (b), con la tempera alla caseina (a) e con la tempera all’uovo (c). Eccitato con il laser a 531.5 nm, lo spettro del pigmento steso con tempera all’uovo mostra i picchi a 1330, 1440 e a 1513 cm-1 del pigmento secco spostati rispettivamente a 1337, 1446 e 1520 cm-1. Questi stessi shift sono confermati dallo spettro ottenuto con eccitazione a 632.8 nm e quindi non possono essere attribuiti alla fluorescenza. Devono piuttosto essere attribuiti ad una reale variazione delle energie vibrazionali. Nello spettro ottenuto con eccitazione a 531.5 nm, dello stesso pigmento, ma steso ad affresco, i picchi dello spettro del pigmento secco a 1330, 1440 e 1513 cm-1 sono spostati rispettivamente a 1343, 1440 e 1525 cm-1. I primi due shift non si riscontrano nello spettro ottenuto con eccitazione a 632.8 nm, mentre quello a 1525 cm-1 è mantenuto. Soltanto quest’ultimo, quindi, può essere attribuito ad una variazione dell’energia vibrazionale Variazioni nel rapporto delle intensità relative dei picchi L’unica variazione dell’intensità relativa dei picchi osservata è stata quella dovuta alla eterogeneità delle composizione chimica del pigmento analizzato (Naples yellow). Non è stata riscontrata nessuna variazione delle intensità relative attribuibile ad una interazione chimica tra il pigmento ed il legante. Viene discusso il caso del Naples yellow.
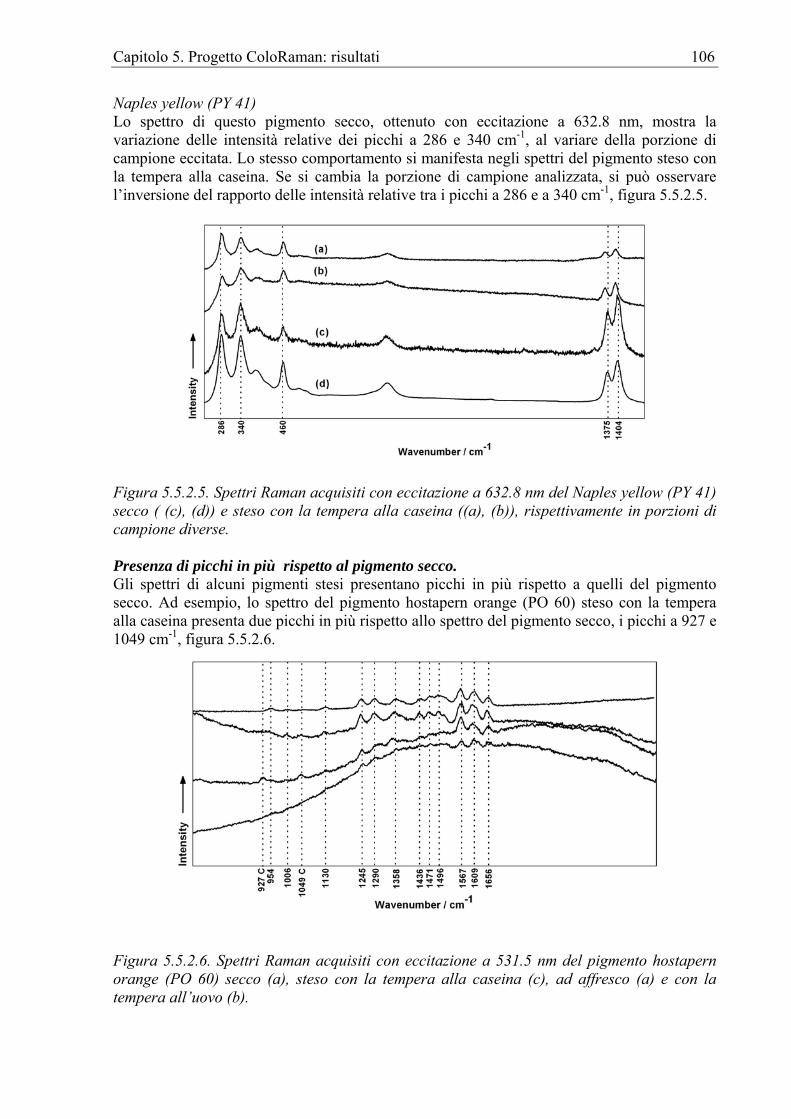
Capitolo 5. Progetto ColoRaman: risultati 106
Naples yellow (PY 41) Lo spettro di questo pigmento secco, ottenuto con eccitazione a 632.8 nm, mostra la variazione delle intensità relative dei picchi a 286 e 340 cm-1, al variare della porzione di campione eccitata. Lo stesso comportamento si manifesta negli spettri del pigmento steso con la tempera alla caseina. Se si cambia la porzione di campione analizzata, si può osservare l’inversione del rapporto delle intensità relative tra i picchi a 286 e a 340 cm-1, figura 5.5.2.5.
Figura 5.5.2.5. Spettri Raman acquisiti con eccitazione a 632.8 nm del Naples yellow (PY 41) secco ( (c), (d)) e steso con la tempera alla caseina ((a), (b)), rispettivamente in porzioni di campione diverse. Presenza di picchi in più rispetto al pigmento secco. Gli spettri di alcuni pigmenti stesi presentano picchi in più rispetto a quelli del pigmento secco. Ad esempio, lo spettro del pigmento hostapern orange (PO 60) steso con la tempera alla caseina presenta due picchi in più rispetto allo spettro del pigmento secco, i picchi a 927 e 1049 cm-1, figura 5.5.2.6.
Figura 5.5.2.6. Spettri Raman acquisiti con eccitazione a 531.5 nm del pigmento hostapern orange (PO 60) secco (a), steso con la tempera alla caseina (c), ad affresco (a) e con la tempera all’uovo (b).
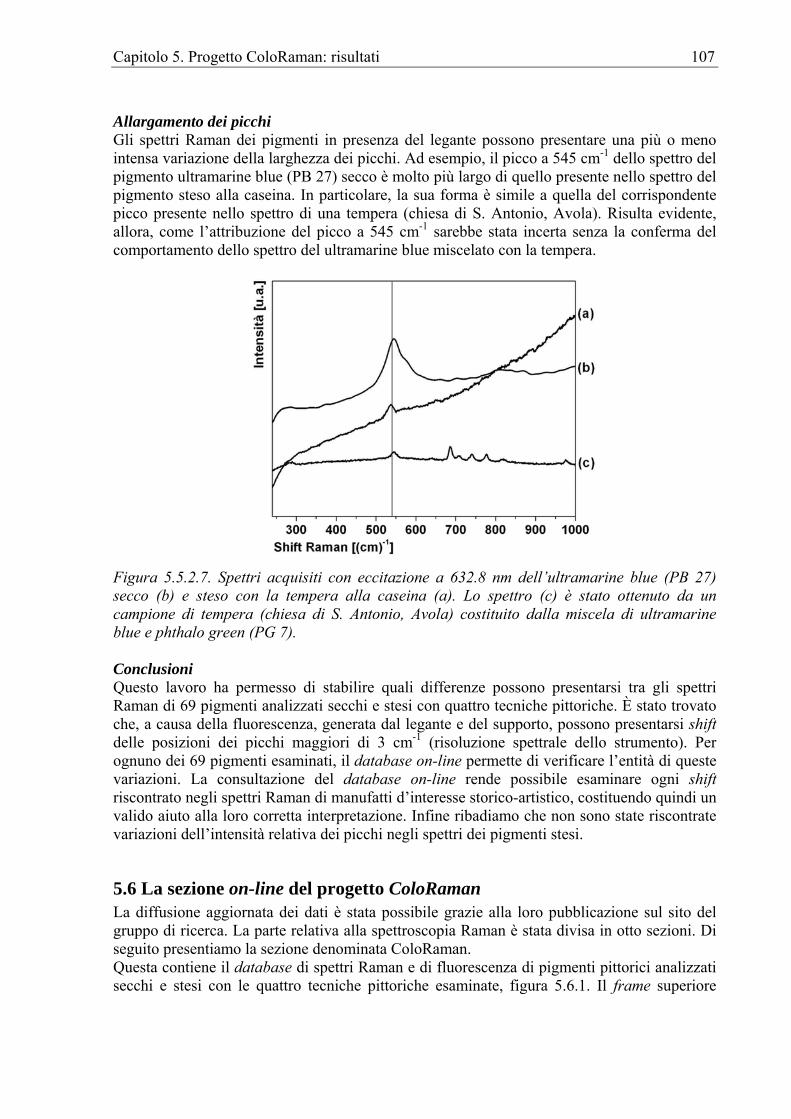
Capitolo 5. Progetto ColoRaman: risultati 107
pigmenti in presenza del legante possono presentare una più o meno tensa variazione della larghezza dei picchi. Ad esempio, il picco a 545 cm-1 dello spettro del
(PB 27) secco è molto più largo di quello presente nello spettro del
) to da un
campione di tem ultramarine
di stabilire quali differenze possono presentarsi tra gli spettri aman di 69 pigmenti analizzati secchi e stesi con quattro tecniche pittoriche. È stato trovato
ella fluorescenza, generata dal legante e del supporto, possono presentarsi shift
a diffusione aggiornata dei dati è stata possibile grazie alla loro pubblicazione sul sito del gruppo di ricerca. La parte relativa alla spettroscopia Raman è stata divisa in otto sezioni. Di seguito presentiamo la sezione denominata ColoRaman. Questa contiene il database di spettri Raman e di fluorescenza di pigmenti pittorici analizzati secchi e stesi con le quattro tecniche pittoriche esaminate, figura 5.6.1. Il frame superiore
Allargamento dei picchi Gli spettri Raman dei inpigmento ultramarine bluepigmento steso alla caseina. In particolare, la sua forma è simile a quella del corrispondente picco presente nello spettro di una tempera (chiesa di S. Antonio, Avola). Risulta evidente, allora, come l’attribuzione del picco a 545 cm-1 sarebbe stata incerta senza la conferma del comportamento dello spettro del ultramarine blue miscelato con la tempera.
Figura 5.5.2.7. Spettri acquisiti con eccitazione a 632.8 nm dell’ultramarine blue (PB 27secco (b) e steso con la tempera alla caseina (a). Lo spettro (c) è stato ottenu
pera (chiesa di S. Antonio, Avola) costituito dalla miscela diblue e phthalo green (PG 7). Conclusioni Questo lavoro ha permesso Rche, a causa ddelle posizioni dei picchi maggiori di 3 cm-1 (risoluzione spettrale dello strumento). Per ognuno dei 69 pigmenti esaminati, il database on-line permette di verificare l’entità di queste variazioni. La consultazione del database on-line rende possibile esaminare ogni shift riscontrato negli spettri Raman di manufatti d’interesse storico-artistico, costituendo quindi un valido aiuto alla loro corretta interpretazione. Infine ribadiamo che non sono state riscontrate variazioni dell’intensità relativa dei picchi negli spettri dei pigmenti stesi.
5.6 La sezione on-line del progetto ColoRaman L

Capitolo 5. Progetto ColoRaman: risultati 108
di interesse tramite il suo colour
ittoriche
permette di accedere agli spettri selezionando il pigmentoindex oppure il suo nome. Cliccando sul colour index desiderato si ottiene, nel frame in alto, la lista dei corrispondenti pigmenti attualmente esaminati, figura 5.6.2. È possibile da questo frame accedere direttamente agli spettri, ottenuti con le tre diverse eccitazioni laser, dei pigmenti stesi nelle quattro tecniche pittoriche, cliccando negli appositi campi. Cliccando invece sul nome del pigmento ne vengono visualizzate, nel frame in basso, le informazioni chimico-commerciali procurate dal fornitore. Gli spettri sono visualizzabili oltre che singolarmente anche sovrapposti. In particolare, è possibile osservare gli spettri, ottenuti con diverse eccitazioni laser, del pigmento secco o steso con una certa tecnica pittorica. I campi contrassegnati dalle sigle CC, CE, CF, CO, CP permettono di visualizzare queste immagini, rispettivamente, per la tempera alla caseina, la tempera all’uovo, l’affresco, l’olio di lino, ed i pigmenti secchi, figura 5.6.3. Diversamente, i campi 5F, 6F e 7F permettono di visualizzare sovrapposti gli spettri di fluorescenza, ottenuti con un solo laser, del pigmento secco e steso con tutte le tecniche pittoriche, figura 5.6.4. I campi contrassegnati dalle sigle A5, A6, A7 consentono invece di osservare sovrapposti gli spettri Raman, ottenuti con una sola lunghezza d’onda d’eccitazione, del pigmento secco e steso con tutte le tecniche pittoriche. In questo caso è stato adottato il formato PDF che consente di ingrandire agevolmente le immagini, figura 5.6.5. Cliccando su T5 e T6 si ottengono le tabelle dei picchi. È così possibile osservare contemporaneamente gli spettri e le posizioni dei relativi picchi, figura 5.6.6.
Figura 5.6.1. Sezione ColoRaman. Questa contiene il database di spettri Raman e di fluorescenza di pigmenti pittorici analizzati secchi e stesi con le quattro tecniche pesaminate. Il frame superiore permette di accedere agli spettri selezionando il pigmento di interesse tramite il suo colour index oppure tramite il suo nome.
Capitolo 5. Progetto ColoRaman: risultati 109
so colour index.
Il fram
Figura 5.6.3. Cliccando ad esempio, su CC è possibile osservare gli spettri sovrapposti e ottenuti con diverse lunghezze d’onda d’eccitazione di un pigmento steso alla caseina.
Figura 5.6.2. Il frame in alto mostra la lista dei pigmenti esaminati con lo stese in basso raccoglie le informazioni chimiche e commerciali procurate dal fornitore.
Capitolo 5. Progetto ColoRaman: risultati 110
igura 5.6.4. I campi 5F, 6F e 7F permettono di visualizzare sovrapposti gli spettri di
igura 5.6.5. I campi A5, A6, A7 permettono di visualizzare sovrapposti gli spettri, ottenuti
Ffluorescenza, ottenuti con un solo laser, del pigmento secco e steso con tutte le tecniche pittoriche.
Fcon una sola lunghezza d’onda d’eccitazione, del pigmento secco e steso con tutte le tecniche
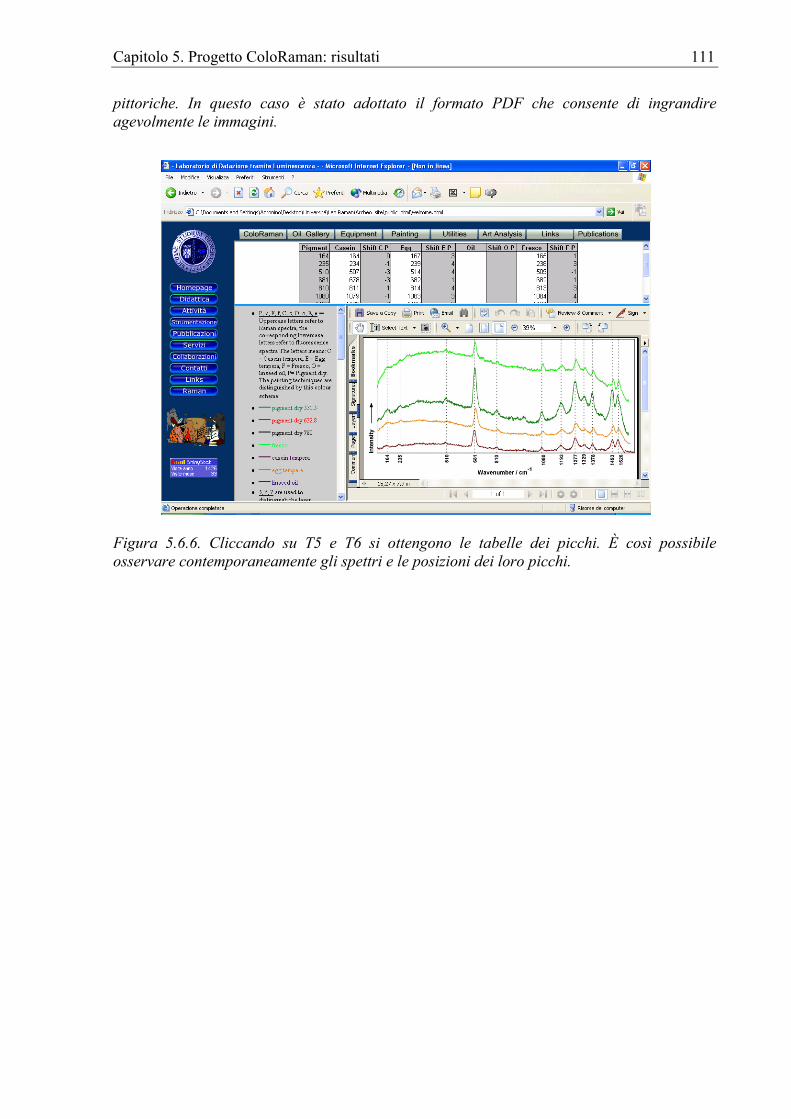
Capitolo 5. Progetto ColoRaman: risultati 111
pittoriche. In questo caso è stato adottato il formato PDF che consente di ingrandire agevolmente le immagini.
Figura 5.6.6. Cliccando su T5 e T6 si ottengono le tabelle dei picchi. È così possibile osservare contemporaneamente gli spettri e le posizioni dei loro picchi.

Capitolo 6. Progetto ColoRaman: verifica 112
Capitolo 6. Progetto ColoRaman: verifica
6.1 Introduzione Il progetto ColoRaman si proponeva di conoscere e ottimizzare le prestazioni di un sistema Raman equipaggiato con tre laser e predisposto per le indagini sui Beni Culturali. Questo lavoro è stato realizzato utilizzando dei campioni preparati in laboratorio ed, il più possibile, conformi, in quanto a materiali e tecniche, ai manufatti originali. Il passo successivo è stato il test su reali oggetti d’arte del sistema ColoRaman e delle sue implementazioni software. Di seguito, saranno proposti degli esempi d’indagini, realizzate su manufatti decorati con alcune delle tecniche pittoriche esaminate, i cui risultati verranno, di volta in volta, confrontati con quelli contenuti nel progetto ColoRaman ed, inoltre, sarà illustrato come utilizzare il software on-line e off-line.
6.2 Tempera
6.2.1 Chiesa di Sant’Antonio Abate in Avola Era stato richiesto un intervento per la caratterizzazione dei pigmenti utilizzati nelle tempere presenti sulle pareti laterali dell’abside della chiesa di S. Antonio ad Avola (SR). La scelta dell’immagine da esaminare è caduta su quella rappresentante S. Marco, posta sulla parete sinistra. Nel tentativo di identificare i pigmenti presenti nella tavolozza dell’autore e di accertare eventuali interventi di restauro, cinque campioni sono stati prelevati dalla parte destra del dipinto e uno dalla parte sinistra. La figura 6.2.1.1 riporta l’immagine della tempera e la localizzazione dei prelievi effettuati. In tabella 6.2.1.2 sono riportate le posizioni e i colori dei sei campioni esaminati. I campioni sono stati analizzati con tutte e tre le lunghezze d’onda. Nessuno spettro caratteristico è stato ottenuto per i campioni 1_1, 1_2, 1_3 e 1_5. L’intensità della radiazione di fluorescenza emessa dal legante, dal supporto pittorico e dal pigmento utilizzato è stata tale da impedire l’acquisizione del segnale Raman. Non è stato quindi possibile acquisire gli spettri dei pigmenti bruni e rossi. Diversamente, il campione verde, 1_4, e quello verde-blu, 1_6, hanno restituito spettri Raman caratteristici, di seguito, discussi in dettaglio. Campione 1_4 Solo con l’eccitazione a 632.8 nm è stato possibile ottenere uno spettro dal campione 1_4. Questo spettro è mostrato in figura 6.2.1.3 insieme all’immagine del campione al microscopio (10X). I picchi più intensi sono quelli a 679, 1204 e 1527 cm-1. Utilizziamo il programma Peaks Finder per attribuirli ad un pigmento. In figura 6.2.1.4 è mostrata la schermata del programma relativa all’operazione d’inserimento dei valori dei picchi e alla ricerca all’interno del database on-line di spettri. Si osserva che il pigmento più probabile è il numero 51.Questo a differenza degli altri pigmenti proposti presenta ben due picchi coincidenti. Utilizzando il database on-line è possibile verificare che il pigmento 51 è il verde phthalo (PG 7). Bisogna ora decidere quale degli spettri del pigmento steso va utilizzato per il confronto. Va ricordato, infatti, (vedi capitolo 5) che è opportuno confrontare lo spettro di un campione non con quelli dei pigmenti secchi ma con quelli dei pigmenti stesi con la stessa tecnica pittorica. Ovviamente, delle quattro tecniche pittoriche esaminate vanno immediatamente escluse l’olio e l’affresco giacché sappiamo che il dipinto in esame è una tempera. Non abbiamo notizia, però, del tipo di legante utilizzato. Possiamo affermare con certezza che non si tratta di

Capitolo 6. Progetto ColoRaman: verifica 113
tempera all’uovo, poiché non si è osservato il suo caratteristico picco a 982 cm-1. Non rimane che confrontare lo spettro del campione 1_4 con quello del verde phthalo (PG 7) steso con la tempera alla caseina. È, infatti, molto probabile che si sia usata la caseina, anche se non è possibile affermarlo con certezza poiché non è stato ancora possibile caratterizzare questo legante. La figura 6.2.1.5 riporta sovrapposti gli spettri del verde phthalo (PG 7) steso alla caseina cm-1 e del campione 1_4. Tutti i picchi sono sovrapponibili. È allora possibile confermare con sicurezza l’identificazione del verde phthalo (PG 7). Campione 1_6 La figura 6.2.1.6 mostra, a sinistra, gli spettri del campione 1_6, ottenuti con eccitazione a 632.8 nm e 780 nm. Lo spettro ottenuto a 632.8 nm è molto più intenso di quello a 780 nm. Ciò coincide con quanto ricavabile dalla consultazione del database on-line ColoRaman, figura 6.2.1.6 a destra, riguardo all’analisi del pigmento verde phthalo (PG 7) steso a tempera (CC). La precedente analisi del campione 1_4 ci suggerisce di attribuire anche questi spettri al verde phthalo (PG 7). La figura 6.2.1.7 mostra a sinistra l’ingrandimento (10X) del campione 1_6 e a destra il suo spettro sovrapposto a quello del verde phthalo (PG 7) steso alla caseina, a quello del blu oltremare ( PG 27) e a quello della calcite. È evidente che lo spettro del campione 1_6 è costituito dalla loro sovrapposizione. I picchi del carbonato di calcio provengono dal supporto pittorico. Per l’attribuzione del picco a 542 cm-1 ci si è avvalsi del programma Peaks Finder. La figura 6.2.1.8 mostra il risultato della ricerca. Dei cinque pigmenti candidati solo il pigmento 85, blu ultramarino (PB 27), e il 96, rosso di piombo (red lead) presentano un picco intenso (s) intorno a 542 cm-1. Il rosso di piombo (red lead) può essere escluso perché l’immagine al microscopio (10X) del campione 1_6 (figura 6.2.1.7) presenta evidenti grani di pigmento blu immersi nel fondo verde. L’analisi con il laser a 780 nm restituisce anch’essa dei picchi Raman, anche se molto deboli, figura 6.2.1.9, e lo spettro ottenuto corrisponde a quello del verde phthalo (PG 7). Sono così confermati i risultati ottenuti con l’eccitazione laser a 632.8 nm. Conclusioni Sono stati identificati due pigmenti, il blu ultramarino (PB 27) e il verde phthalo (PG 7). L’identificazione di questo ultimo assume rilevanza notevole nella ricostruzione delle vicende vissute dal dipinto. A differenza del blu oltremare, sintetizzato già nel medioevo, il verde phthalo fu, infatti, brevettato solo nel 1927 (Wehlte 1975), circa 150 anni dopo la realizzazione della tempera che risulta, quindi, interessata da procedure di restauro relativamente recenti.
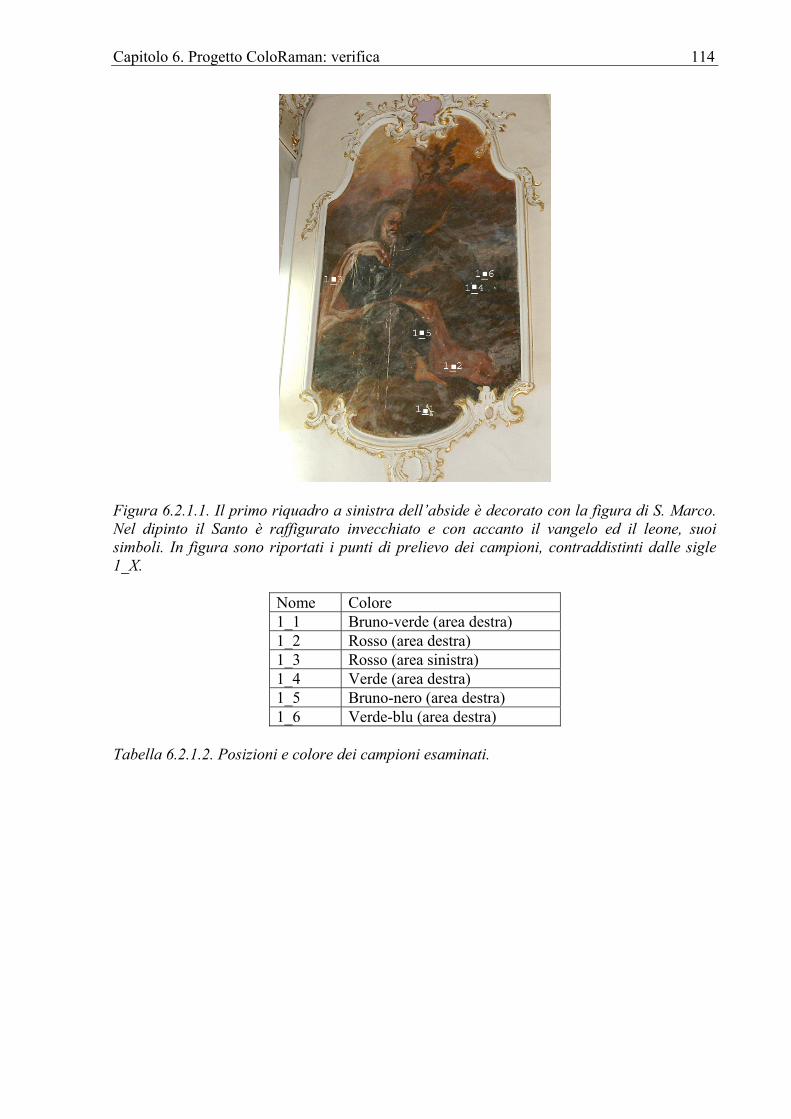
Capitolo 6. Progetto ColoRaman: verifica 114
Figura 6.2.1.1. Il primo riquadro a sinistra dell’abside è decorato con la figura di S. Marco. Nel dipinto il Santo è raffigurato invecchiato e con accanto il vangelo ed il leone, suoi simboli. In figura sono riportati i punti di prelievo dei campioni, contraddistinti dalle sigle 1_X.
Nome Colore 1_1 Bruno-verde (area destra) 1_2 Rosso (area destra) 1_3 Rosso (area sinistra) 1_4 Verde (area destra) 1_5 Bruno-nero (area destra) 1_6 Verde-blu (area destra)
Tabella 6.2.1.2. Posizioni e colore dei campioni esaminati.
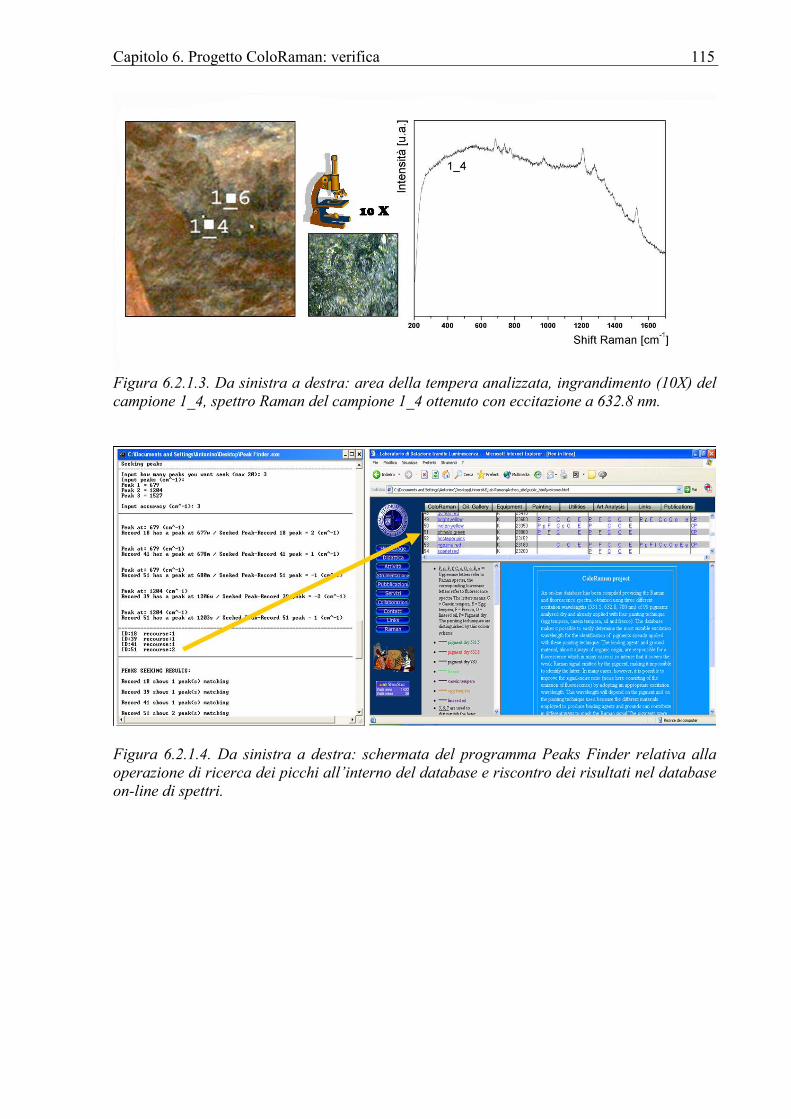
Capitolo 6. Progetto ColoRaman: verifica 115
Figura 6.2.1.3. Da sinistra a destra: area della tempera analizzata, ingrandimento (10X) del campione 1_4, spettro Raman del campione 1_4 ottenuto con eccitazione a 632.8 nm.
Figura 6.2.1.4. Da sinistra a destra: schermata del programma Peaks Finder relativa alla operazione di ricerca dei picchi all’interno del database e riscontro dei risultati nel database on-line di spettri.
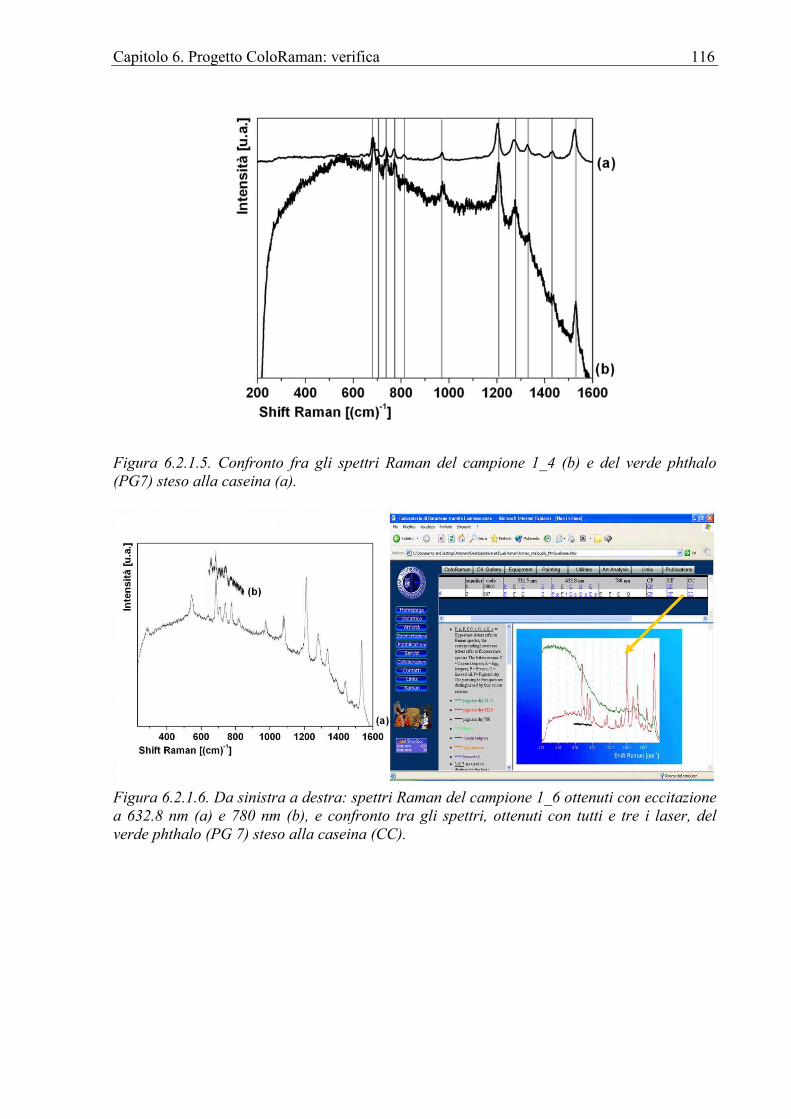
Capitolo 6. Progetto ColoRaman: verifica 116
Figura 6.2.1.5. Confronto fra gli spettri Raman del campione 1_4 (b) e del verde phthalo (PG7) steso alla caseina (a).
Figura 6.2.1.6. Da sinistra a destra: spettri Raman del campione 1_6 ottenuti con eccitazione a 632.8 nm (a) e 780 nm (b), e confronto tra gli spettri, ottenuti con tutti e tre i laser, del verde phthalo (PG 7) steso alla caseina (CC).
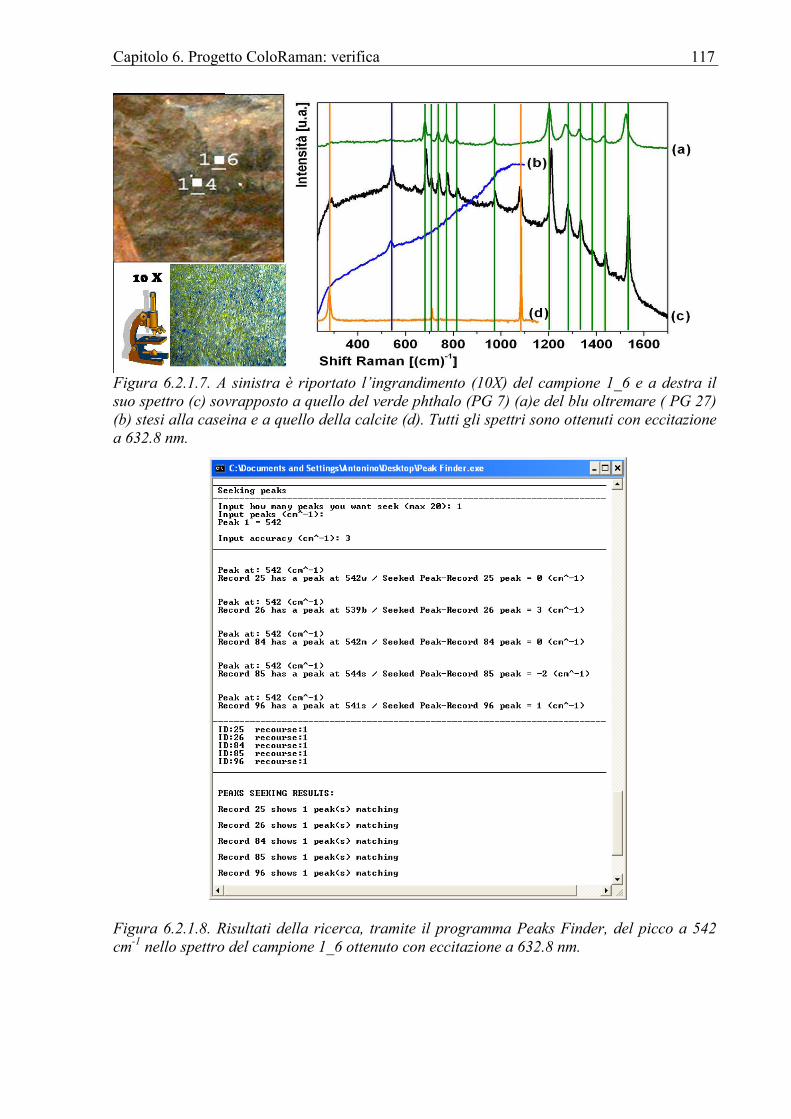
Capitolo 6. Progetto ColoRaman: verifica 117
Figura 6.2.1.7. A sinistra è riportato l’ingrandimento (10X) del campione 1_6 e a destra il suo spettro (c) sovrapposto a quello del verde phthalo (PG 7) (a)e del blu oltremare ( PG 27) (b) stesi alla caseina e a quello della calcite (d). Tutti gli spettri sono ottenuti con eccitazione a 632.8 nm.
Figura 6.2.1.8. Risultati della ricerca, tramite il programma Peaks Finder, del picco a 542 cm-1 nello spettro del campione 1_6 ottenuto con eccitazione a 632.8 nm.
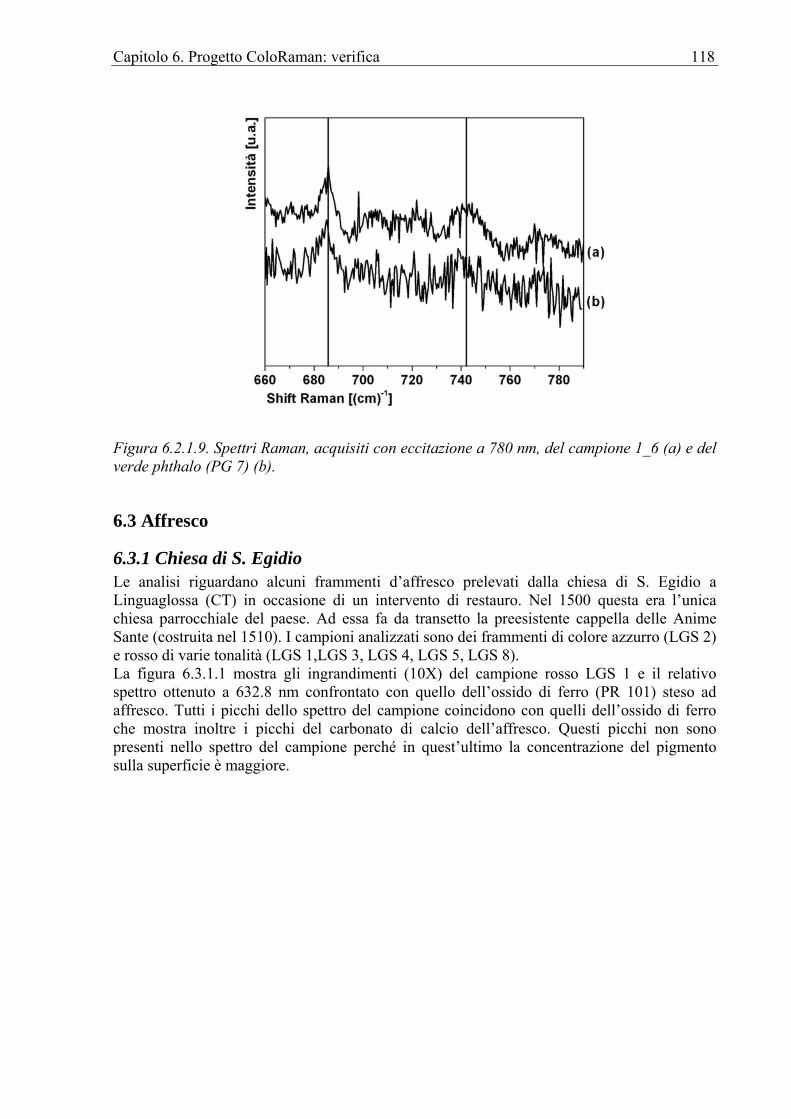
Capitolo 6. Progetto ColoRaman: verifica 118
Figura 6.2.1.9. Spettri Raman, acquisiti con eccitazione a 780 nm, del campione 1_6 (a) e del verde phthalo (PG 7) (b).
6.3 Affresco
6.3.1 Chiesa di S. Egidio Le analisi riguardano alcuni frammenti d’affresco prelevati dalla chiesa di S. Egidio a Linguaglossa (CT) in occasione di un intervento di restauro. Nel 1500 questa era l’unica chiesa parrocchiale del paese. Ad essa fa da transetto la preesistente cappella delle Anime Sante (costruita nel 1510). I campioni analizzati sono dei frammenti di colore azzurro (LGS 2) e rosso di varie tonalità (LGS 1,LGS 3, LGS 4, LGS 5, LGS 8). La figura 6.3.1.1 mostra gli ingrandimenti (10X) del campione rosso LGS 1 e il relativo spettro ottenuto a 632.8 nm confrontato con quello dell’ossido di ferro (PR 101) steso ad affresco. Tutti i picchi dello spettro del campione coincidono con quelli dell’ossido di ferro che mostra inoltre i picchi del carbonato di calcio dell’affresco. Questi picchi non sono presenti nello spettro del campione perché in quest’ultimo la concentrazione del pigmento sulla superficie è maggiore.
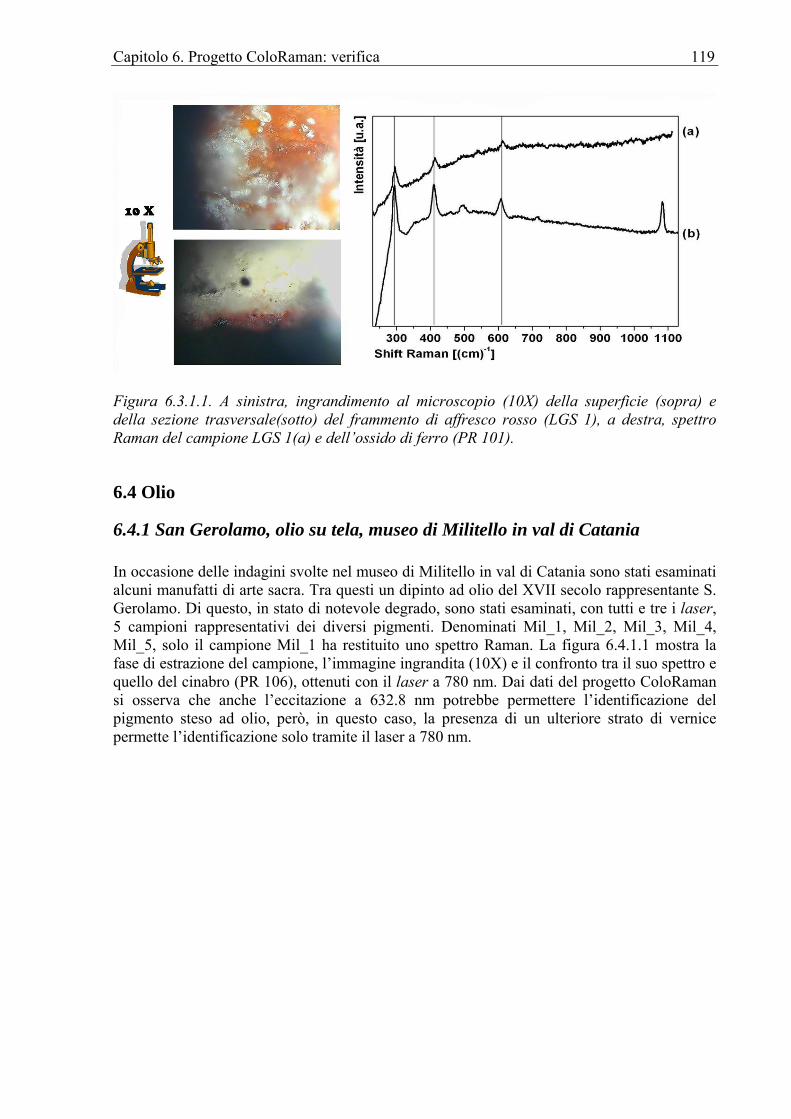
Capitolo 6. Progetto ColoRaman: verifica 119
Figura 6.3.1.1. A sinistra, ingrandimento al microscopio (10X) della superficie (sopra) e della sezione trasversale(sotto) del frammento di affresco rosso (LGS 1), a destra, spettro Raman del campione LGS 1(a) e dell’ossido di ferro (PR 101).
6.4 Olio
6.4.1 San Gerolamo, olio su tela, museo di Militello in val di Catania In occasione delle indagini svolte nel museo di Militello in val di Catania sono stati esaminati alcuni manufatti di arte sacra. Tra questi un dipinto ad olio del XVII secolo rappresentante S. Gerolamo. Di questo, in stato di notevole degrado, sono stati esaminati, con tutti e tre i laser, 5 campioni rappresentativi dei diversi pigmenti. Denominati Mil_1, Mil_2, Mil_3, Mil_4, Mil_5, solo il campione Mil_1 ha restituito uno spettro Raman. La figura 6.4.1.1 mostra la fase di estrazione del campione, l’immagine ingrandita (10X) e il confronto tra il suo spettro e quello del cinabro (PR 106), ottenuti con il laser a 780 nm. Dai dati del progetto ColoRaman si osserva che anche l’eccitazione a 632.8 nm potrebbe permettere l’identificazione del pigmento steso ad olio, però, in questo caso, la presenza di un ulteriore strato di vernice permette l’identificazione solo tramite il laser a 780 nm.
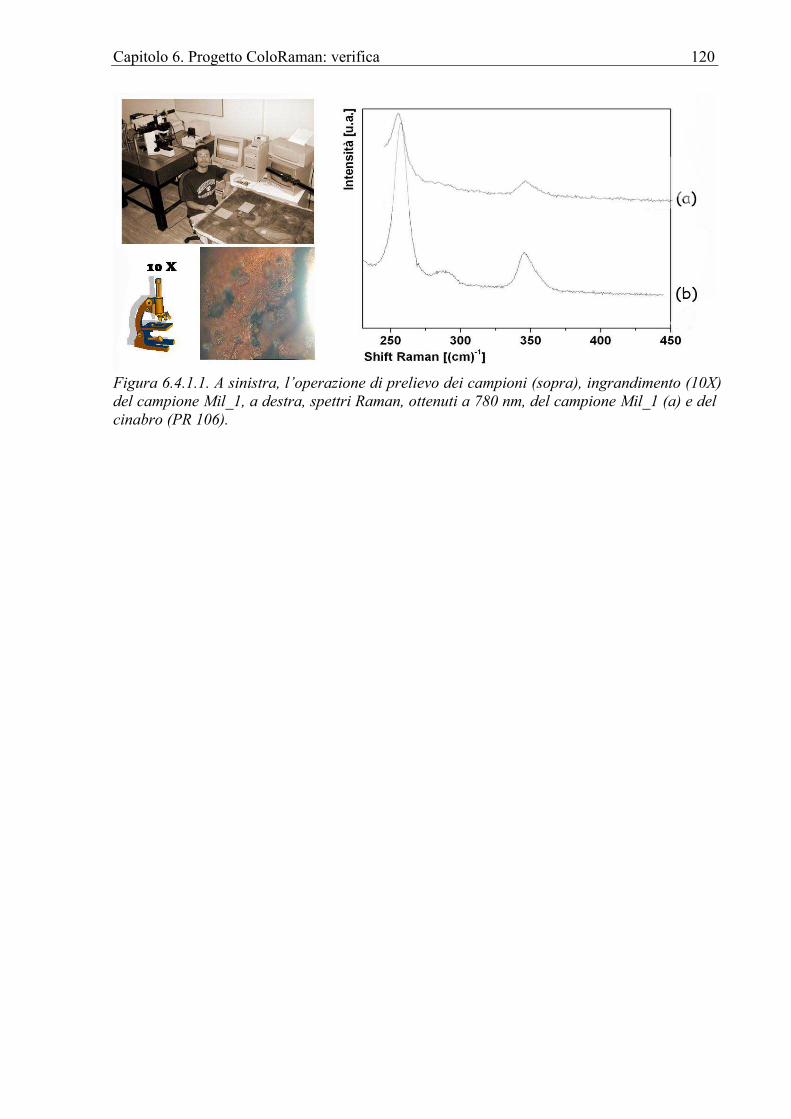
Capitolo 6. Progetto ColoRaman: verifica 120
Figura 6.4.1.1. A sinistra, l’operazione di prelievo dei campioni (sopra), ingrandimento (10X) del campione Mil_1, a destra, spettri Raman, ottenuti a 780 nm, del campione Mil_1 (a) e del cinabro (PR 106).

Conclusioni 121
Conclusioni L’attività di ricerca svolta nel corso di questo dottorato ha permesso di sviluppare delle implementazioni volte a superare alcuni dei limiti che ostacolavano l’applicabilità della spettroscopia Raman alle indagini sui Beni artistici e storici. Il progetto ColoRaman, grazie al lavoro d’ottimizzazione e diffusione delle conoscenze acquisite tramite la realizzazione del sito internet e di utilities quali il software peaks finder, potrà contribuire alla diffusione e alla piena affermazione della spettroscopia raman quale metodologia diagnostica particolarmente adatta alle indagini sui Beni artistici e storici. Sono state testate le effettive potenzialità di un sistema combinato per spettroscopia Raman, dotato di tre laser come sorgenti d’eccitazione, per la caratterizzazione dei pigmenti pittorici secchi ma soprattutto dei relativi colori già stesi, Si è potuto dimostrare che è spesso possibile stabilire per ogni pigmento e per ogni singola tecnica pittorica la lunghezza d’onda di eccitazione capace o di diminuire l’intensità della fluorescenza oppure di generare una intensa emissione Raman così da migliorare il rapporto segnale-rumore. Sono stati compilati i database di spettri Raman di pigmenti pittorici secchi e già stesi con le tecniche pittoriche dell’affresco, della tempera all’uovo, della tempera alla caseina e dell’olio. Oltre al database di spettri Raman è stato realizzato anche quello degli spettri di fluorescenza ed è stato dimostrato che questa metodologia può risultare particolarmente utile perché, diversamente da quanto avviene per il debole segnale Raman, per l’emissione di fluorescenza il rapporto segnale-rumore è, in genere, così alto da permettere l’identificazione del pigmento già steso. È stato affrontato, inoltre, il problema della compatibilità degli spettri dei pigmenti secchi con quelli dei pigmenti stesi, verificandone caso per caso il comportamento. Infine, l’affidabilità dei risultati del progetto ColoRaman e delle sue implementazioni software è stata testata con successo su reali oggetti d’arte.

Bibliografia 122
Bibliografia AA.VV. Br.25 (1989) 589-621 C Andalò, M Bicchieri, P Bocchini, G Casu, GC Galletti, PA Mandò, M Nardone, A Sodo, M. Plossi Zappalà, Anal. Chim. Acta 429 (2001) 279-286 J. Ambers, J. Raman Spectrosc. 35 (2004) 768-773 P Baraldi, F Bondioli, C Fagnano, AM Ferrari, A Tinti, M Vinella, Annali di Chimica 91 (2001) 679-692 CW Beck, Quad. Merceol., 7 (1968) 1 CW Beck, EC Stout, B Kosmowska-Ceranowicz, Geol.Foeren, Stockholm Foerh., 115 (1993) 145 SEJ Bell, ESO Bourguignon, A Dennis, Analyst 123 (1998) 1729-1734 SEJ Bell, ESO Bourguignon, AC Dennis, JA Fields, JJ McGarvey, KR Seddon, Anal. Chem. 72 (2000) 234-239 IM Bell, RJH Clark, PJ Gibbs, Spectrochim. Acta, Part A 53 (1997) 2159-2179 L Bellot-Gurlet, FX Le Bourdonnec, G Poupeau and S Dubernet, J. Raman Spectrosc. 35 (2004) 671-677 BJ Berenblut, P Dawson, GR Wilkinson, Spectrochim. Acta, Part A 20A (1973) 29-36 D Bersani, G Antonioli, PP Lottici, A Casoli, Spectrochim. Acta, Part A 59 (2003) 2409-2417 D Bersani, PP Lottici, G Antonioli, E Campani, A Casoli, C Violante, J. Raman Spectrosc. 35 (2004) 694-703 S Best, R Clark, M Daniels, RT Withnall, Chem. Br. 29 (1993) 118-122 SP Best, RJH Clark, MAM. Daniels, CA Porter, R Withnall, Stud. Conserv. 40 (1995) 31-40 SP Best, RJH Clark, PJ Gibbs, R Withnall, Endeavour 16 (1992) 66-73 M Bicchieri, M Nardone, A Sodo, J. Cult. Heritage 1 (2000) 277-279 D Bikiaris, S Daniilia, S Sotiropoulou, O Katsimbiri, E Pavlidou, AP Moutsatsou, Y Chryssoulakis, Spectrochim. Acta, Part A 56 (1999) 3-18 M Bouchard , DC Smith, Spectrochim. Acta, Part A 59 (2003) 2247-2266 M Breitman, S Ruiz-Moreno , R Pérez-Pueyo, J. Cult. Heritage 4 (2003) 314s-316s

Bibliografia 123
A Brysbaert, P Vandenabeele, J. Raman Spectrosc. 35 (2004) 686-693 RH Brody, HGM. Edwards, AM Pollard, Spectrochim. Acta, Part A 57 (2001) 1325 MH Brooker, S Sunder, P Taylor, VJ Lopata, Can. J. Chem. 61 (1983) 494-502 KL Brown, RJH Clark, J. Raman Spectrosc. 35 (2004) 4-12 KL Brown, RJH Clark, J. Raman Spectrosc. 35 (2004) 181-189 KL Brown, RJH Clark, J. Raman Spectrosc. 35 (2004) 217-223 S Bruni, F Cariati, F Casadio, V Guglielmi, J. Cult. Heritage 4 (2001) 291-296 S Bruni, F Cariati, F Casadio, L Toniolo, Spectrochim. Acta, Part A 55 (1999) 1371-1377 L Burgio, DA Ciomartan, RJH. Clark, J. Raman Spectrosc. 28 (1997) 79-83 L Burgio, DA Ciomartan, RJH Clark, J. Mol. Struct. 405 (1997) 1-11 L Burgio, RJH Clark, Spectrochim. Acta, Part A 57 (2001) 1491-1521 L Burgio, RJH Clark, J. Raman Spectrosc. 31 (2000) 395-401 L Burgio, RJH Clark, PJ Gibbs, J. Raman Spectrosc. 30 (1999) 181-184 L Burgio, RJH Clark, K Theodoraki, Spectrochim. Acta, Part A 59 (2003) 2371-2389 L Burgio, K Melessanaki, M Doulgeridis, RJH Clark, D Anglos, Spectrochim. Acta, Part B 56 (2001) 905-913 Burrafato G., Calabrese M., Cosentino A, Gueli A.M., Troja S.O., ZuccarelloA, Journal of Raman Spectroscopy in press. M Castillejo, M Martín , M Oujja , E Rebollar ,C Domingo , JV García-Ramos, S Sánchez-Cortés, J. Cult. Heritage 4 (2003) 243-249 M Castillejo, M Martìn, D Silva, T Stratoudaki, D Anglos, L Burgio, RJH Clark, J. Mol. Struct. 550-551 (2000) 191-198 M Castillejo, M Matin, D Silva, T Stratoudaki, D Anglos, RJH. Clark, L Curri, J. Cult. Heritage 1 (2000) 297-302 K Castro, MD Rodriguez-Laso, LA Fenandez, JM Madariaga, J. Raman Spectrosc. 33 (2001) 17-25 SA Centeno, D Mahon, MT Wypyski, J. Raman Spectrosc. 35 (2004) 774-780

Bibliografia 124
TD Chaplin, A Jurado-Lopez, RJH Clark, DR Beech, J. Raman Spectrosc. 35 (2004) 600-604 DB Chase, J. Am. Chem. Soc. 108 (1986) 7485-7488 Y Chryssoulakis, C Panayiotou, Revue d'Archéometrie 21 (1997) 97-101 DA Ciomartan, RJH Clark, LJ McDonald, M Odlyha, J. Chem. Soc., Dalton Trans. (1996) 3639-3645 RJH Clark, Chem. Soc. Rev. (1995) 1187-196 RJH Clark, C. R. Chimie 5 (2002) 7-20 RJH Clark, J. Mol. Struct. 347 (1995) 417-428 RJH Clark, J. Mol. Struct. 480-481 (1999) 15-20 RJH Clark, DG Cobbold, Inorg. Chem. 17 (1978) 3169-3174 RJH Clark, L Criland, BM Kariuki, KDM Harris, R Withhnall, J. Chem. Soc., Dalton Trans. (1995) 2577-2582 RJH Clark, L Curri, J. Mol. Struct. 440 (1998) 105-111 RJH Clark, ML Curri, GS Henshaw,C Laganara, J. Raman Spectrosc. 28 (1997) 105-109 RJH Clark, ML Curri, C Laganara, Spectrochim. Acta, Part A 53 (1997) 597-603 RJH Clark, TJ Dines, M Kurmoo, Inorg. Chem. 22 (1983) 2766-2772 RJH Clark, ML Franks, Chem. Phys. Lett. 34 (1975) 69-72 RJH Clark, PJ Gibbs, J. Raman Spectrosc. 28 (1997) 99-103 RJH Clark, PJ Gibbs, J. Archaeological Science 25 (1998) 621-629 RJH Clark, PJ Gibbs, Anal. Chem. 70 (1998) 99A-103A RJH Clark, PJ Gibbs, Chem. Commun. (1997) 1003-1004 RJH Clark, PJ Gibbs, KS Seddon, NM Brovenko, YA Petrosyan, J. Raman Spectrosc. 28 (1997) 91-94 RJH Clark, K Huxley, Science and technology for cultural heritage 5(2) (1996) 95-101 RJH Clark et J van der Weerd, J. Raman Spectrosc. 35 (2004) 279-283 P Colomban, J. Non- Cristalline solids, 323 (2003) 180-187

Bibliografia 125
P Colomban, J. Raman Spectrosc. 34 (2003) 420-423 P Colomban, V Milande, L Le Bihan, J. Raman Spectrosc. 35 (2004) 527-535 P Colomban, V Milande, H Lucas, J. Raman Spectrosc. 35 (2004) 68-72 P Colomban, G Sagon, X Faurel, J. Raman Spectrosc. 32 (2001) 351-360 P Colomban, C Truong, J. Raman Spectrosc. 35 (2004) 195-207 P Colomban, F Treppoz, J. Raman Spectrosc. 32 (2001) 93-102 EE Coleyshaw, WP Griffit, R Bowell, Spectrochim. Acta, Part A 50A (1994) 1909-1918 L Coma, M Breitman, S Ruiz-Moreno, J. Cult. Heritage 1 (2000) S273-S276 A Cosentino, AM Gueli, SO Troja,A Zuccarello, Art2000 proceedings, (2002). C Coupry, G Sagon, P Gorguet-Ballesteros, J. Raman Spectrosc. 28 (1997) 85-89 JCS Dalton, J. Chem. Soc., Dalton Trans.(1977) 1096-1103 S Daniilia, D Bikiaris, L Burgio, P Gavala, RJH Clark, Y Chryssoulakis, J. Raman Spectrosc. 33 (2002) 807-814 S Daniilia, S Sotiropoulou, D Bikiaris, C Salpistis, G Karagiannis, Y Chryssoulakis, BA Price, JH Carlson, J. Cult. Heritage, 1 (2000) 91-110 R Davwey, DJ GarDiner, BW Singer, M Spokes, J. Raman Spectrosc. 25 (1994) 53-57 A Derbyshire, R Withnall, J. Raman Spectrosc. 30 (1999) 185-188 TJ Dines, H Onoh, J. Raman Spectrosc. 35 (2004) 284-298 M Doerner, The materials of the artist and their use in painting, Harcourt Brace & Company, San Diego, 435, (1949). R Durman, UA Jayasooriya, SFA Kettle, J. Chem. Commun. (1985) 916-917 HGM Edwards, J. Mol. Struct. 661-662 (2003) 271-277 HGM Edwards, CJ Brooke, JKF Tait, J. Raman Spectrosc. 28 (1997) 95-98 HGM Edwards, P Colomban, B Bowden, J. Raman Spectrosc. 35 (2004) 656-661 HGM Edwards, EL Dixon, IJ Scowen, F Rull Perez, Spectrochim. Acta, Part A 59 (2003) 2291-2299 HGM Edwards, L Drummopnd, J Russ, Spectrochim. Acta, Part A 54 (1998) 1849-1856

Bibliografia 126
HGM Edwards, L Drummopnd, J Russ, J. Raman Spectrosc. 30 (1999) 421-428 HGM Edwards and MJ Falk, J. Raman Spectrosc. 28 (1997) 211-218 HGM Edwards, DW Farwell, Spectrochim. Acta, Part A 52 (1996) 1119-1125 HGM. Edwards, DW Farwell, L Daffner, Spectrochim. Acta, Part A 52 (1996) 1639-1648 HGM Edwards, DW Farwell, EM Newton, F Rull Perez, S Jorge Villar, J. Raman Spectrosc. 31 (2000) 407-413 HGM Edwards, DW Farwell , EM Newton , FR Perez , SJ Villar, Spectrochim. Acta, Part A 57 (2001) 1223-1234 HGM Edwards, DW Farwell, S Rozenberg, J. Raman Spectrosc. 30 (1999) 361-366 HGM Edwards, ER Gwyer, JKF Tait, J. Raman Spectrosc. 28 (1997) 677-684 HGM Edwards, DW Farwel, F Rull Perez, SJ Villar, J. Raman Spectrosc. 30 (1999) 307-311 HGM Edwards, PS Middleton, SEJ Villar, DLA. de Faria, Anal. Chim. Acta 484 (2003) 211-221 HGM Edwards, EM Newton, J Russ, J. Mol. Struct. 550 (2000) 245-256 HGM Edwards, FR Perez, J. Raman Spectrosc. 35 (2004) 754-760 HGM Edwards, NC Russel, MDR Seaward, D Slarke, Spectrochim. Acta, Part A 51 (1995) 2091-2100 HGM Edwards, MG Sibley, C Heron, Spectrochim. Acta, Part A 53 (1997) 2373-2382 HGM Edwards, MG Sibley, B Derham and CP Heron, J. Raman Spectrosc. 35 (2004) 746-753 HGM Edwards, JKF Tait, Appl. Spectrosc. 52 (1998) 679-683 HGM Edwards, SEJ Villar, AR David, DLA de Faria, Anal. Chim. Acta 503 (2004) 223-233 X Faurel, A Vanderperre, P Colomban, J. Raman Spectrosc. 34 (2003) 290-294 R Frech, EC Wang, Spectrochim. Acta 36A (1980) 915-919 RL Frost, W Martens, JT Kloprogge and PA Williams, J. Raman Spectrosc. 33 (2002) 801-806 GC Galletti e R Mazzeo, Rapid Commun. Mass Spectrom. 7 (1993) 646.

Bibliografia 127
S Galli, M Mastelloni, R Ponterio, G Sabatino, M Triscari, J. Raman Spectrosc. 35 (2004) J. Raman Spectrosc. 35 (2004) DL Gerrard, Anal. Chem. 66 (1994) 547R-557R M Gniedecka, HC Wulf, O Faurskof nielsen, DH Chistensen, J. Raman Spectrosc. 28 (1997) 179-184 B Guineau, Stud. Conserv. 29 (1984) 35-41 B Guineau, Stud Conserv 34 (1989) 38-44. V Hayez, S Denoel, Z Genadry, B Gilbert, J. Raman Spectrosc. 35 (2004) 781-785 V Hayez, J Guillaume, A Hubin and H Terryn, J. Raman Spectrosc. 35 (2004) 732-738 PV Huong, Vibrational Spectroscopy 11 (1996) 17-28 J. Jehlicka, SE Jorge Villar and HGM.Edwards, J. Raman Spectrosc. 35 (2004) 761-767 A Jenkins, RA Larsen, Spectroscopy 19(4) (2004) 20-25 A Jurado-Lopez, O Demko, RJH Clark, D Jacobs, J. Raman Spectrosc. 35 (2004) 119-124 E Kendix, OF Nielsen and MC Christensen, J. Raman Spectrosc. 35 (2004) 796-799 KJ Kingma, RJ Hemley, Am. Mineral. 79 (1994) 269-273 N Krishnamurthy, V Soots, Can. J. Phys. 49 (1971) 885-896 GN Kustova, EB Burgina, VA Sadykov, SG Poryvaev, Phys. Chem. Miner. 18 (1992) 379-382 EE Lawson, BW Barry, AC Williams, J. Raman Spectrosc. 28 (1997) 111-117 NQ Liem, G Sagon, VX Quang, HV Tan, P Colomban, J. Raman Spectrosc.31 (2000) 933-942 NQ Liem, NT Thanh, P Colomban, J. Raman Spectrosc.33 (2002) 287-294 LA Lyin, CK Keating, AP Fox, BE Baker, L HE, SR Nicewarner, SP Mulvaney, MJ Natan, Anal. Chem. 70 (1998) 341R-361R C Lofrumento, A Zoppi, EM Castellucci, J. Raman Spectrosc. 35 (2004) 650-655 F Magistro, D Majolino, P Migliardo, R Ponterio, J. Cult. Heritage 2 (2001) 191-198 HF Mark, JJ McKelta and DF Othmer (Eds.), Kirk-Othmer’s Encyclopedia of Chemical Technology, 2nd edn., Vol 17, Natural resins, Wiley-Interscience, New York, 1969, pp. 397

Bibliografia 128
R Mazzeo, P Baraldi, R Lujan, C Fagnano, J. Raman Spectrosc. 35 (2004) 678-685 LI McCann, K Trentelman, T Possley, B Golding, J. Raman Spectrosc. 30 (1999) 121-132 C Miliani, M Ombelli, A Morresi, A Romani, Surface & Coatings Technology 151-152 (2002) 276-280 A. Milleville, L Bellot-Gurlet, B Champignon, Santallie Revue d’Archéometrie 27 (2003) 123 YM Moreno, DH Christensen, OF Nielsen, Asian Chem. Lett. 5(3) (2001) 117-123 I Moroz, M Roth, M Boudeulle, G Panczer, J. Raman Spectrosc. 31 (2000) 485-490 D Neff, S Reguer, L Bellot-Gurlet, P Dillmann, R Bertholon, J. Raman Spectrosc. 35 (2004) 739-745 A Nissenbaum, Hakiyama’iy beYisrael, 12 (1993) 32 S Pagès- Camagna, S Colinart, C Coupry, J. Raman Spectrosc. 30 (1999) 313-317 S Pages-Camagna, A Duval, H Guicharnaud, J. Raman Spectrosc. 35 (2004) 628-632 S Pages-Camagna, T Calligaro, J. Raman Spectrosc. 35 (2004) 633-639 A Perardi, L Appolonia, P Mirti, Anal. Chim. Acta 480 (2003) 317-325 A Perardi, A Zoppi, E Castellucci, J. Cult. Heritage 1 (2000) 269-272 R Perez-Pueyo, MJ Soneira, S Ruiz-Moreno, J. Raman Spectrosc. 35 (2004) 808-812 M Pérez., K Castro, MD Rodrìguez, MA Olazabal, JM Madariaga, Art2002 proceedings, 2002. MJ Peters, LE McNeil, K Dy, Solid State Commun. 97 (1996) 1095-1099 DR Porterfield, A Campion, J. Am. Chem. Soc. 110 (1988) 408-410 B Prieto, MDR Seaward, HGM. Edwards, T Rivas, B Silva, Spectrochim. Acta, Part A 55 (1999) 211-217 CV Raman, KS Krishnan, Nature, 121 (1928) 501 C Razzetti, PP Lottici, Solid State Commun. 29 (1979) 361-364 I Reiche, S Pages-Camagna and L Lambacher, J. Raman Spectrosc. 35 (2004) 719-725 C Ricci, I Borgia, BG Brunetti, C Miliani, A Sgamellotti, C Seccaroni, P Passalacqua, J. Raman Spectrosc. 35 (2004) 616-621

Bibliografia 129
S Roascio , A Zucchiatti , P Prati , A Cagnana, J. Cult. Heritage 3 (2002) 289-297 L Robinet, K Eremin, B Cobo del Arco, LT Gibson, J. Raman Spectrosc. 35 (2004) 662-670 F Rosi, C Miliani, I Borgia, B Brunetti, A. Sgamellotti, J. Raman Spectrosc. 35 (2004) 610-615 S Ruiz-Moreno, A Lopez-Gil, A Gabaldon, C Sandalinas, J. Raman Spectrosc. 35 (2004) 640-645 S Ruiz-Moreno, R Perz-Pueyo, A Gabaldon, M Soneira, C Sandalinas, J. Cult. Heritage 4 (2003) 309s-313s F Rull Perez, HGM Edwards, A Rivas, L Drummond, J. Raman Spectrosc. 30 (1999) 301-305 K Sakellariou, C Miliani, A Morresi, M Ombelli, J. Raman Spectrosc. 35 (2004) 61-67 SS Savkevich, Phys. Chem. Mineral, 7 (1981) 1. M Schmidt, HD Lutz, Phys. Chem. Miner. 20 (1993) 27-32 B Schrader, G Baranovic, A Epding, GG Hoffmann, PJM Van Kan, S Keller, P Hildebrandt, C Lehner, J Sawatzki, Appl. Spectrosc. 47 (numero 9) (1993) 1452-1456 P Seymour, A short book about oil painting, Lee Press,Yorkshire, 87, (1995). IT Shadi, BZ Chowdhry, M.J Snowden, R Withnall, Spectrochim. Acta, Part A 59 (2003) 2213-2220 IT Shadi, BZ Chowdhry, MJ Snowden, R Withnall, J. Raman Spectrosc. 35 (2004) 800-807 ON Shebanova and P Lazor, J. Raman Spectrosc. 34 (2003) 845-852 ZX Shen, SL Yee, TS Tay, L Quin, SH Tang, Asian J. Spectrosc. 1 (1997) 127 DC Smith, Spectrochim. Acta Part A 59 (2003) 2353-2369 DC Smith, A Barbet, J. Raman Spectrosc. 30 (1999) 319-324 DC Smith, M Bouchard, M Lorblanchet, J. Raman Spectrosc. 30 (1999) 347-354 GD Smith, RJH Clark, J. Cult. Heritage 3 (2002) 101-105 GD Smith, L Burgio, S Firth, RJH. Clark, Anal. Chim. Acta 440 (2001) 185-188 DC Smith, S Robin, J. Raman Spectrosc. 28 (1997) 189-193 MJ Soneira, R Perz-Pueyo, S Ruiz-Moreno, J. Raman Spectrosc. 33 (2002) 599-603 BI Srebrodol’skii, Doklady Akud, Nauk. SSSR, 291 (1986) 1223.

Bibliografia 130
CM Stellman, F Bucholtz, Spectrochim. Acta, Part A 54 (1998) 41-47 E. Tatsch, B. Schrader, J. Raman Spectrosc. 26 (1995) 467-473 W Taylor, A Pinczuk, E Burstein, Phys. Rev. B: Condens. Matter 1 (1970) 4058-4070 DV Thompson, The practice of tempera painting, Dover Pubblications, inc., New York, 141, (1936) K Trentelman, L Stodulski, M Pavlosky, Anal. Chem. 8 (1996) 1755-1761 JD Trevor, H Onoh, J. Raman Spectrosc. 35 (2004) 284-298 P. Vandenabeele, J. Raman Spectrosc. 35 (2004) 607-609 P Vandenabeele, DM Grimaldi, HGM Edwards, L Moens, Spectrochim. Acta, Part A 59 (2003) 2221-2229 P Vandenabeele, L Moens, HGM Edwards, R Dams, J. Raman Spectrosc. 31 (2000) 509-517 P Vandenabeele, F Verpoort, L Moens, J. Raman Spectrosc. 32 (2001) 263-269 P Vandenabeele, B Wehling, L Moens, HGM Edwards, M De Reu, G Van Hooydonk, Anal. Chim. Acta 407 (2000) 261-274 D de Waal, J. Raman Spectrosc. 35 (2004) 646-649 A Wallert, Stud Conserv 31 (1986) 145-155 X Wang, C Wang, J Yang, L Chen, J Feng, M Shi, J. Raman Spectrosc. 35 (2004) 274-278 K Wehlte, The materials and techniques of painting, Kremer, New York, 672, (1975). TL Weis, Y Jiang, ER Grant, J. Raman Spectrosc. 35 (2004) 813-818 WB White, DG Minser, J. Non-Cryst. Solids 67 (1984) 45 HG Wiedemann, E Arpagaus, D Muller, C Marcolli, S Weigel, A Reller, Thermochimica Acta 382 (2002) 239-247 W Winkler, ECh Kirchner, A Asenbaum and M Musso, J. Raman Spectrosc. 32 (2001) 59-63 W Winkler, ECh Kirchner, M Musso, JA Asenbaum, Mitt. Osterr. Miner. Ges. 143 (1998) 398 W Winkler, M Musso, ECh Kirchner, J. Raman Spectrosc. 34 (2003) 157-162 D Wise, A Wise, J. Raman Spectrosc. 35 (2004) 710-718

Bibliografia 131
A Zoppi, C Lofrumento, EM Castellucci, MG Migliorini, Spectrosc. Europe 14/5 (2002) 16.-20 J Zuo, C Xu, B Hou, C Wang, Y Xie, Y Qian, J. Raman Spectrosc. 27 (1996) 921-923 J Zuo, C Xu, C Wang, Z Yushi, J. Raman Spectrosc. 30 (1999) 1053-1055 J Zuo, X Zhao, R Wu, G Du, C Xu, C Wang, J. Raman Spectrosc. 34 (2003) 121-125





























