Embed Size (px)
Citation preview

RECRISTALIZARECRISTALIZAÇÇÃO ÃO e e
PURIFICAPURIFICAÇÇÃO ÃO da da
ACETANILIDAACETANILIDA

INTRODUÇÃO
A RECRISTALIZAÇÃO consiste na dissolução de uma substância sólida num solvente, a quente, e depois, por resfriamento, obtém-se novamente o estado cristalino mais puro. O tamanho desses cristais varia de acordo com a velocidade de resfriamento, isto é, quanto mais lenta, mais eficiente será a recristalização.

RECRISTALIZAÇÃO EPURIFICAÇÃO DA
ACETANILIDA
Os testes de solubilidade consistirão em misturar, em tubo de ensaio, uma mesma quantidade de acetanilida (0,1 g) com 3 mL de: água, etanol, éter etílico, clorofórmio, acetona e benzeno, agitando vigorosamente.
Após teste a frio deve-se fazer o mesmo aquecendo o sistema em banho-maria (se o solvente apresentar alguma periculosidade, efetuar aquecimento em capela).
Escolher o melhor solvente
Fluxograma:RECRISTALIZAÇÃO E
PURIFICAÇÃO DaAcetanilida
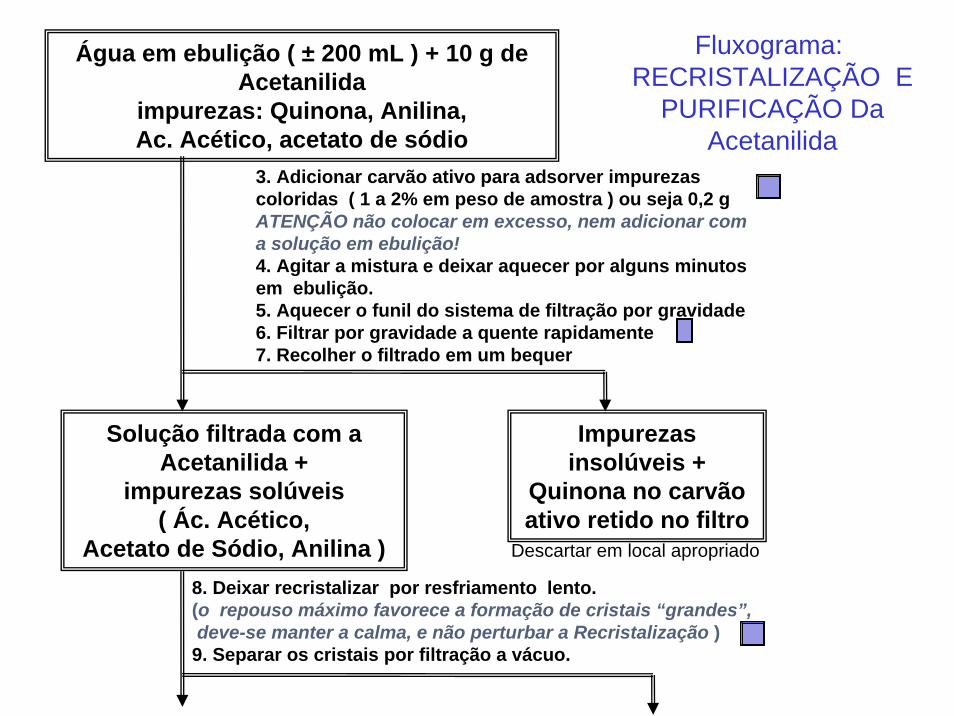
Água em ebulição ( ± 200 mL ) + 10 g de Acetanilida
impurezas: Quinona, Anilina, Ac. Acético, acetato de sódio
3. Adicionar carvão ativo para adsorver impurezas coloridas ( 1 a 2% em peso de amostra ) ou seja 0,2 gATENÇÃO não colocar em excesso, nem adicionar com a solução em ebulição!4. Agitar a mistura e deixar aquecer por alguns minutos em ebulição.5. Aquecer o funil do sistema de filtração por gravidade6. Filtrar por gravidade a quente rapidamente7. Recolher o filtrado em um bequer
Solução filtrada com a Acetanilida +
impurezas solúveis ( Ác. Acético,
Acetato de Sódio, Anilina )
Impurezas insolúveis +
Quinona no carvão ativo retido no filtro
8. Deixar recristalizar por resfriamento lento.(o repouso máximo favorece a formação de cristais “grandes”,deve-se manter a calma, e não perturbar a Recristalização )
9. Separar os cristais por filtração a vácuo.
Descartar em local apropriado
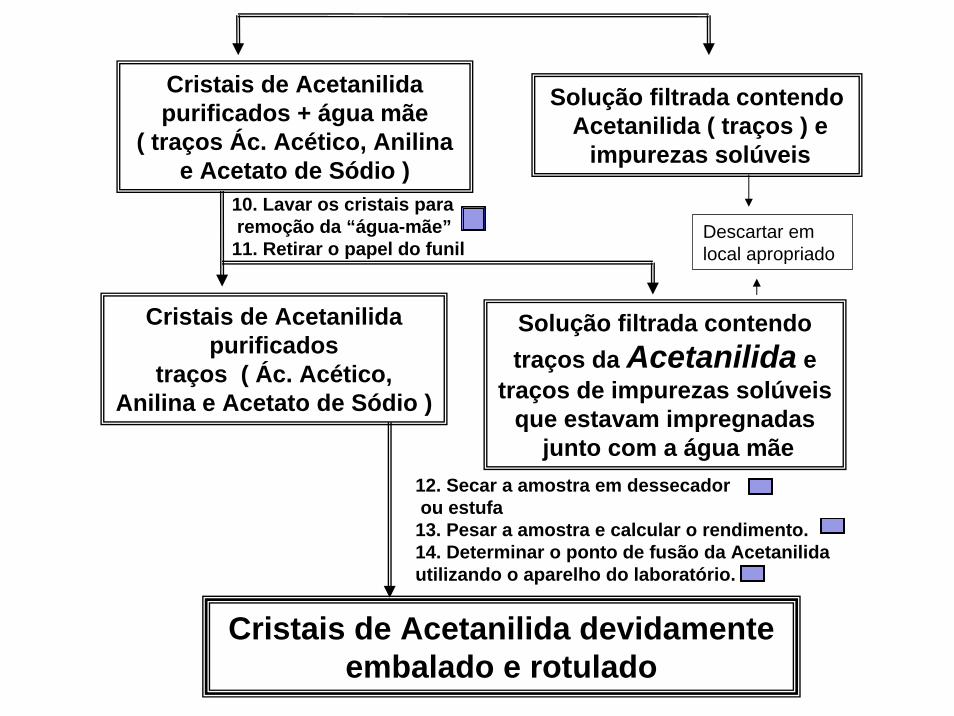
Cristais de Acetanilida purificados + água mãe
( traços Ác. Acético, Anilina e Acetato de Sódio )
Solução filtrada contendoAcetanilida ( traços ) e
impurezas solúveis
Cristais de Acetanilida purificados
traços ( Ác. Acético, Anilina e Acetato de Sódio )
Cristais de Acetanilida devidamenteembalado e rotulado
Solução filtrada contendo traços da Acetanilida e
traços de impurezas solúveis que estavam impregnadas
junto com a água mãe12. Secar a amostra em dessecadorou estufa
13. Pesar a amostra e calcular o rendimento.14. Determinar o ponto de fusão da Acetanilida utilizando o aparelho do laboratório.
10. Lavar os cristais pararemoção da “água-mãe”
11. Retirar o papel do funilDescartar em local apropriado

BibliografiaALLINGER, N. L., CAVA, M. P., DEJONGH, D. C., JOHNSON, C. R.,
LEBEL, N. A.,STEVENS, C. L., Química Orgânica, 2º ed., Guanabara Dias, RJ, 1985
FESSENDEN, R. J., FESSENDEN, J. S., FEIST. P., Organic Laboratory Techniques, 3º Ed., Brooks Cole, Canada, 2000
CAMPOS. M.M., Química Orgânica, USP, SP, 1976VOGEL, A. I., Química Orgânica, 3a ed., USP, RJ, 1981Budavari, S., et all, The Merck Index, 12th , USA, 1996.PAVIA, D. L., LAMPMAN, G. M., KRIZ, G. S., Introduction to
LaboratoryTechniques : a microscale approach, 2nd ed., Philadelphia: Saunders College, 1995.

Escolha do SolventeO sucesso da recristalização depende da escolha correta do
solvente, onde devemos considerar os seguintes critérios:
• Verificação da polaridade.
• O solvente deve dissolver grande quantidade da substância em temperatura elevada e pequena quantidade em temperaturas baixas.
• Ao ser esfriado, o solvente deve produzir cristais bem formados do sólido purificado, e deve ser facilmente removível.
• O solvente não deve reagir com o sólido.
• Outros fatores como a facilidade de manipulação, a volatilidade, ainflamabilidade, o caráter tóxico e o custo também deve ser levado em conta.
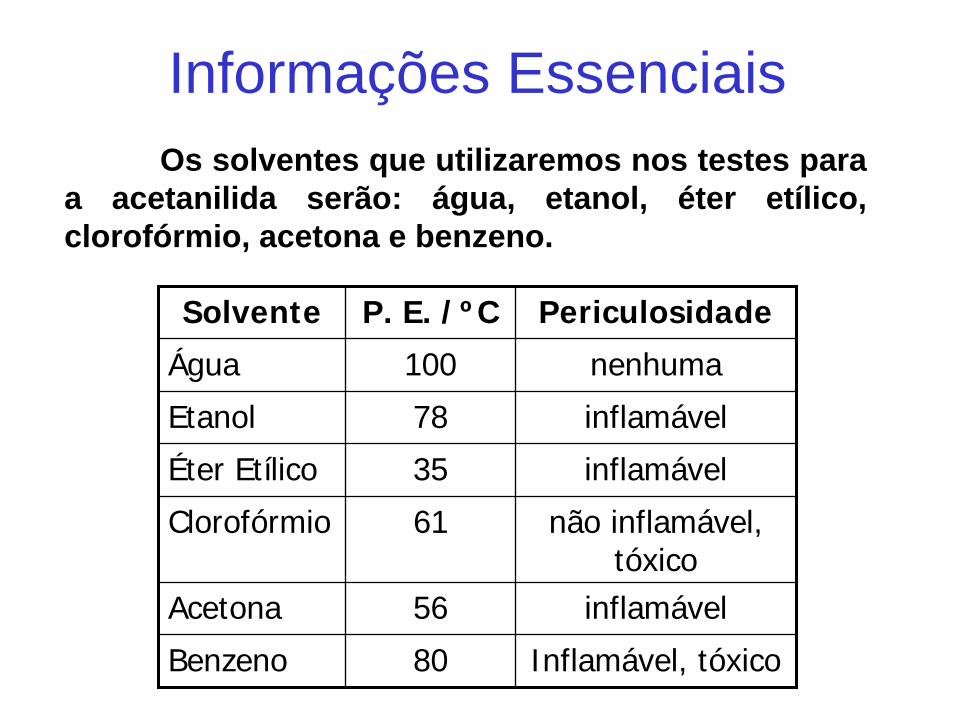
Os solventes que utilizaremos nos testes para a acetanilida serão: água, etanol, éter etílico, clorofórmio, acetona e benzeno.
Informações Essenciais
Solvente P. E. /ºC Periculosidade
Água 100 nenhuma
Etanol 78 inflamável
Éter Etílico 35 inflamável
Clorofórmio 61 não inflamável, tóxico
Acetona 56 inflamável
Benzeno 80 Inflamável, tóxico

Tabela de Teste de SolubilidadeSolvente Solúvel a
frio?Solúvel a quente ?
Temperatura máxima permitida
Água ~100ºC
Etanol ~78ºC
Éter etílico ~35ºC
Clorofórmio ~61ºC
Acetona ~56ºC
Benzeno ~80ºC
Se o solvente dissolver todo o material a frio, não servirá.Se o solvente não dissolver o material mesmo a quente, não servirá.Se o material dissolveu-se pouco a frio e completamente a quente, este pode ser um provável solvente a ser utilizado para recristalização.
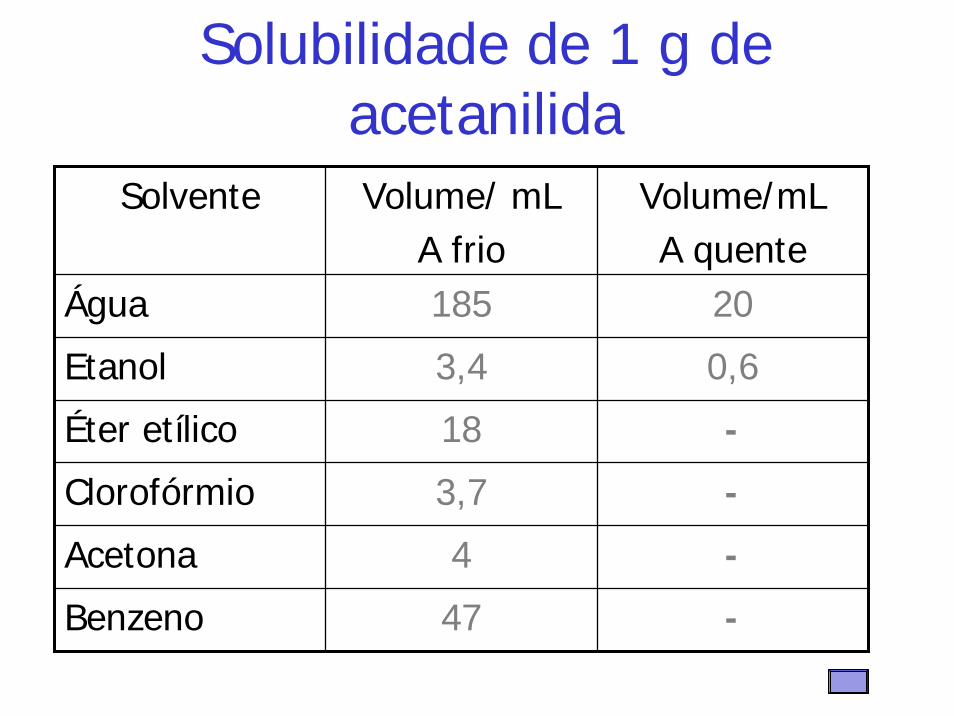
Solubilidade de 1 g de acetanilida
Solvente Volume/ mL A frio
Volume/mLA quente
Água 185 20
Etanol 3,4 0,6
Éter etílico 18 -
Clorofórmio 3,7 -
Acetona 4 -
Benzeno 47 -

REMOÇÃO DE IMPUREZAS
Impurezas coloridas ou resinosas podem ser removidas pela adição de pequenas quantidades de carvão ativo à solução a ser aquecida. As impurezas adsorvidas na superfície das partículas de carvão são removidas durante a filtração. O uso de excesso de carvão ativo leva a perdas do material a ser purificado.

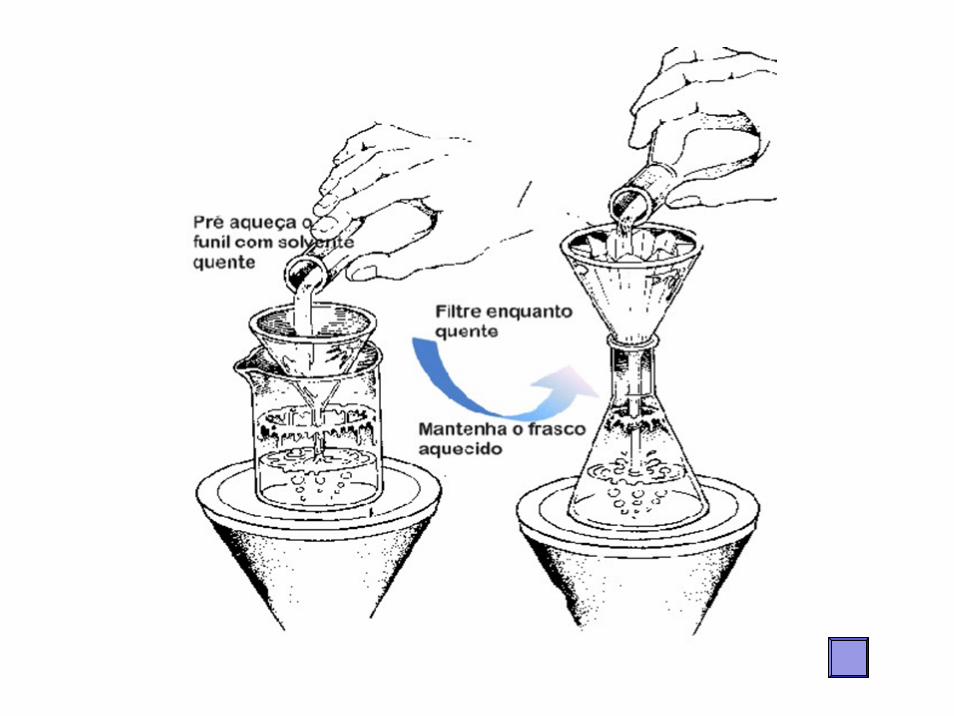
Filtração a QuenteFiltração à quente com papel de filtro
pregueado. A solução a ser filtrada é levada àebulição e transferida para o sistema filtranteem pequenas proporções
A operação deve ser feita rapidamente, evitando a cristalização da acetanilida no filtro ou no funil.(durante o processo o funil pode esfriar).
Tomar bastante cuidado durante a realização!!

Recristalização• Deve ser feita em um erlenmeyer para evitar a contaminação por poeira, ou em um béquer tampado ;
• O resfriamento deve ser lento (a temperatura ambiente) para quese formem cristais puros e perfeitos, pois se ocorrer o contrário acontecerá a precipitação e nesta a rede cristalina é formada tão rapidamente que as impurezas são presas dentro da rede. Por essemotivo não é utilizado o banho de gelo.
• Caso não ocorra a cristalização espontânea deve-se arranhar o interior do recipiente com um a bastão de vidro ou adicionar alguns cristais do produto.

Lavagem dos CristaisDepois que o principal filtrado tiver sido
removido, deve-se lavar os cristais a fim de remover a “água-mãe” que, na secagem, contaminaria os mesmos.
O líquido de lavagem será a água (fria) e deve ser usada na menor quantidade possível.
Aplica-se novamente a sucção e os cristais são pressionados com um bastão de vidro com borracha. A lavagem é repetida, caso seja necessário.

SECAGEM DOS CRISTAISA secagem pode ser feita em uma estufa
(verificar antes o ponto de fusão do material) ou simplesmente por exposição ao ar livre sobre um vidro de relógio, o qual deve estar coberto com um papel de filtro (Tem a vantagem de não decompor o produto, mas é demorado e pode gerar contaminações.
Pode-se, também, utilizar um dessecador a vácuo na presença de um agente secante (sílica gel) ou ainda, um dessecador sem vácuo.

PONTO DE FUSÃOO ponto de fusão reflete o grau de pureza
de uma substância e é a temperatura na qual o sólido cristalino se tornou completamente líquido.
Correção do ponto de fusão
ΔT =0,00012 (760 - p)(t +273 )
t760 = ΔT + t

Correção do ponto de fusãoObservações
O ponto de fusão teórico da acetanilida é em torno de 114 °C, o valor encontrado pode ser menor que o valor da literatura.
Tem-se observado valores em torno de 84 °C, devido a formação de um sistema binário entre água e acetanilida.
Isso significa uma provável presença de impurezas, pois a mistura de dois sólidos que formam ponto eutético sempre teráponto de fusão menor que uma espécie pura.
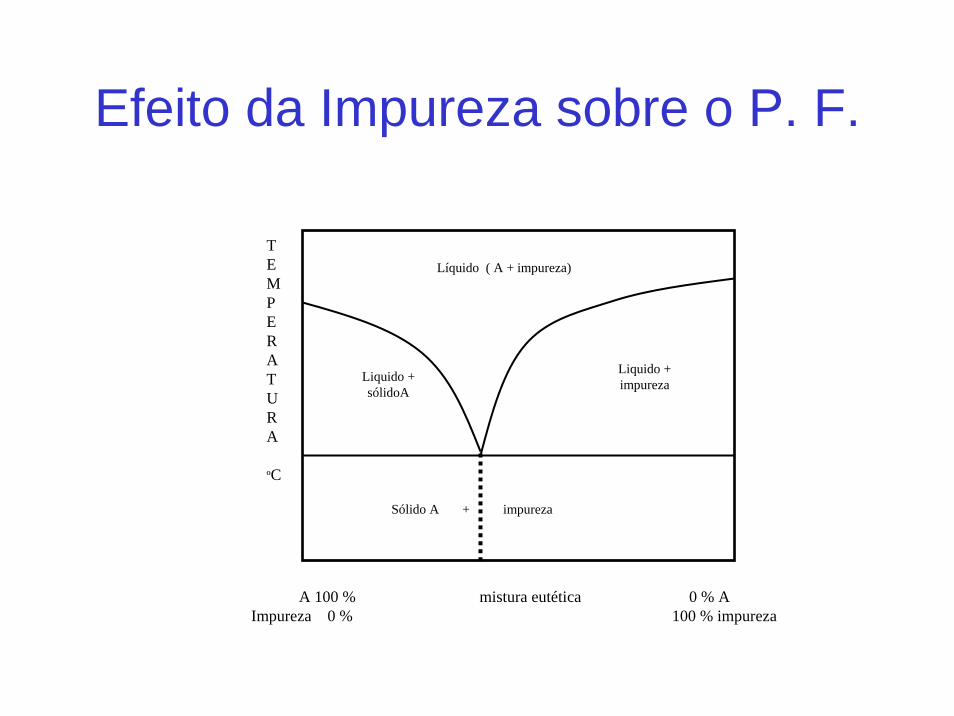
Efeito da Impureza sobre o P. F.
TEMPERATURA
ºC
Liquido + sólidoA
Líquido ( A + impureza)
Liquido + impureza
Sólido A + impureza
A 100 % mistura eutética 0 % AImpureza 0 % 100 % impureza

VERIFICAÇÃO DO RENDIMENTO DA ACETANILIDA OBTIDA
massa da acetanilida final x 100
massa da acetanilida inicialRendimento =
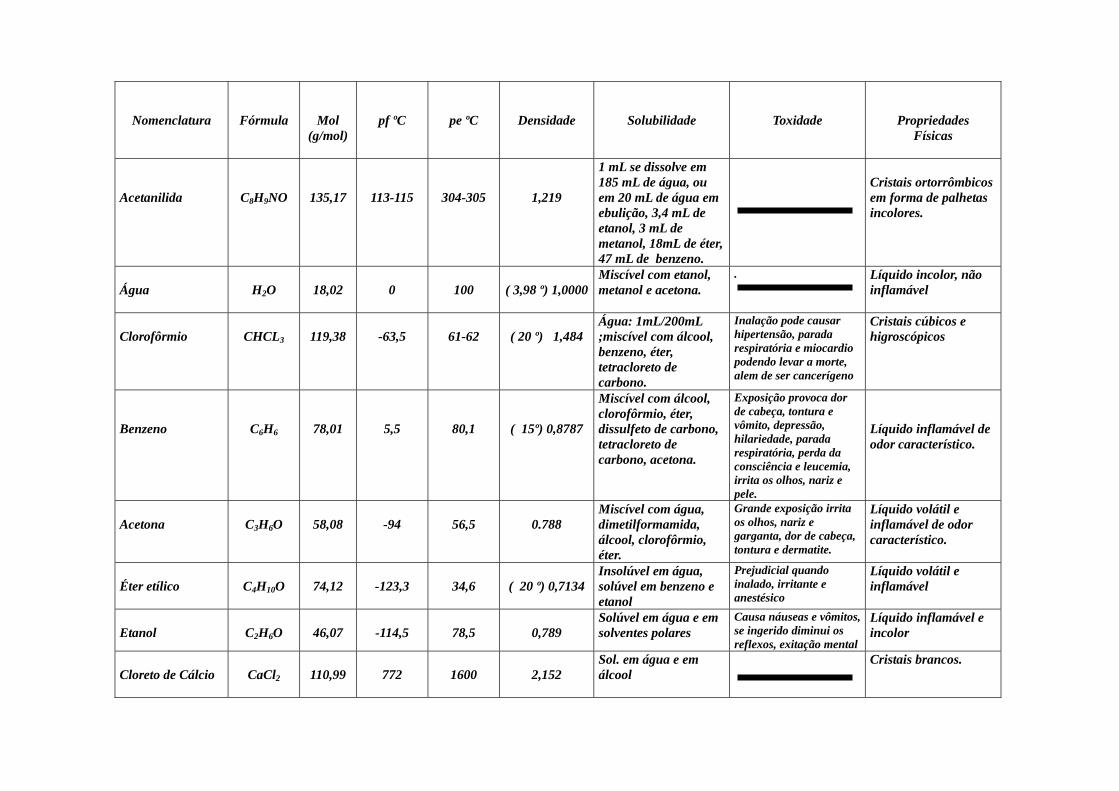
Nomenclatura
Fórmula
Mol (g/mol)
pf ºC
pe ºC
Densidade
Solubilidade
Toxidade
Propriedades Físicas
Acetanilida
C8H9NO
135,17
113-115
304-305
1,219
1 mL se dissolve em 185 mL de água, ou em 20 mL de água em ebulição, 3,4 mL de etanol, 3 mL de metanol, 18mL de éter, 47 mL de benzeno.
Cristais ortorrômbicos em forma de palhetas incolores.
Água
H2O
18,02
0
100
( 3,98 º) 1,0000
Miscível com etanol, metanol e acetona.
.
Líquido incolor, não inflamável
Clorofôrmio
CHCL3
119,38
-63,5
61-62
( 20 º) 1,484
Água: 1mL/200mL ;miscível com álcool, benzeno, éter, tetracloreto de carbono.
Inalação pode causar hipertensão, parada respiratória e miocardio podendo levar a morte, alem de ser cancerígeno
Cristais cúbicos e higroscópicos
Benzeno
C6H6
78,01
5,5
80,1
( 15º) 0,8787
Miscível com álcool, clorofôrmio, éter, dissulfeto de carbono, tetracloreto de carbono, acetona.
Exposição provoca dor de cabeça, tontura e vômito, depressão, hilariedade, parada respiratória, perda da consciência e leucemia, irrita os olhos, nariz e pele.
Líquido inflamável de odor característico.
Acetona
C3H6O
58,08
-94
56,5
0.788
Miscível com água, dimetilformamida, álcool, clorofôrmio, éter.
Grande exposição irrita os olhos, nariz e garganta, dor de cabeça, tontura e dermatite.
Líquido volátil e inflamável de odor característico.
Éter etílico
C4H10O
74,12
-123,3
34,6
( 20 º) 0,7134
Insolúvel em água, solúvel em benzeno e etanol
Prejudicial quando inalado, irritante e anestésico
Líquido volátil e inflamável
Etanol
C2H6O
46,07
-114,5
78,5
0,789
Solúvel em água e em solventes polares
Causa náuseas e vômitos, se ingerido diminui os reflexos, exitação mental
Líquido inflamável e incolor
Cloreto de Cálcio
CaCl2
110,99
772
1600
2,152
Sol. em água e em álcool
Cristais brancos.
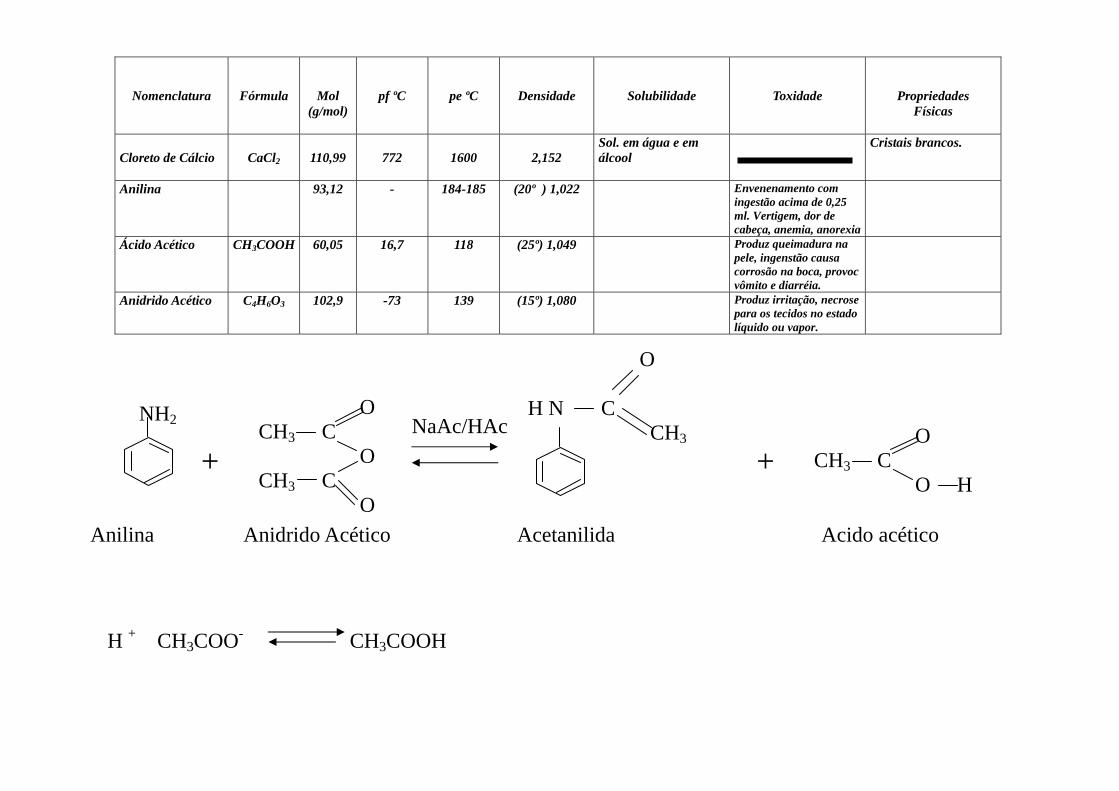
Nomenclatura
Fórmula
Mol (g/mol)
pf ºC
pe ºC
Densidade
Solubilidade
Toxidade
Propriedades Físicas
Cloreto de Cálcio
CaCl2
110,99
772
1600
2,152
Sol. em água e em álcool
Cristais brancos.
Anilina 93,12 - 184-185 (20º ) 1,022 Envenenamento com ingestão acima de 0,25 ml. Vertigem, dor de cabeça, anemia, anorexia
Ácido Acético CH3COOH 60,05 16,7 118 (25º) 1,049 Produz queimadura na pele, ingenstão causa corrosão na boca, provoc vômito e diarréia.
Anidrido Acético C4H6O3 102,9 -73 139 (15º) 1,080 Produz irritação, necrose para os tecidos no estado líquido ou vapor.
NH2
+
O CH3 C O CH3 C O
O H N C CH3
+ O CH3 C O H
NaAc/HAc
Anilina Anidrido Acético Acetanilida Acido acético
H + CH3COO- CH3COOH
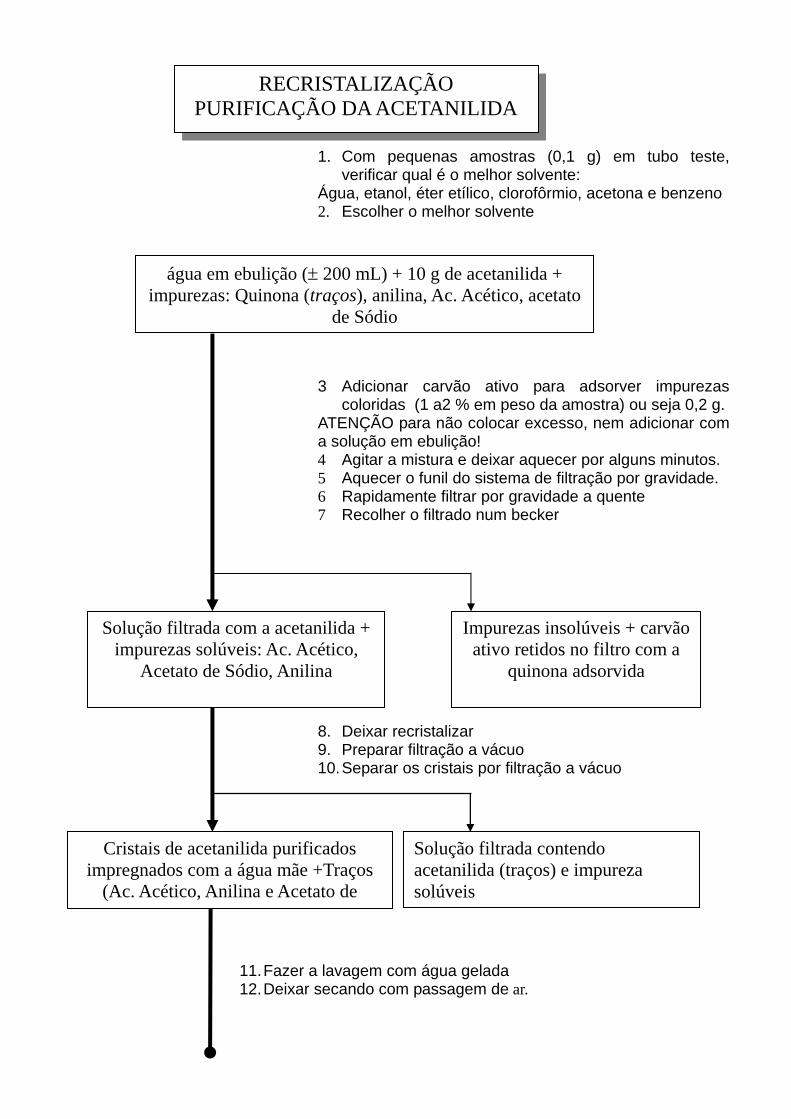
RECRISTALIZAÇÃO PURIFICAÇÃO DA ACETANILIDA
1. Com pequenas amostras (0,1 g) em tubo teste, verificar qual é o melhor solvente:
Água, etanol, éter etílico, clorofôrmio, acetona e benzeno 2. Escolher o melhor solvente
água em ebulição (± 200 mL) + 10 g de acetanilida + impurezas: Quinona (traços), anilina, Ac. Acético, acetato
de Sódio
3 Adicionar carvão ativo para adsorver impurezas coloridas (1 a2 % em peso da amostra) ou seja 0,2 g.
ATENÇÃO para não colocar excesso, nem adicionar com a solução em ebulição! 4 Agitar a mistura e deixar aquecer por alguns minutos. 5 Aquecer o funil do sistema de filtração por gravidade. 6 Rapidamente filtrar por gravidade a quente 7 Recolher o filtrado num becker
Solução filtrada com a acetanilida + impurezas solúveis: Ac. Acético,
Acetato de Sódio, Anilina
Impurezas insolúveis + carvão ativo retidos no filtro com a
quinona adsorvida
8. Deixar recristalizar 9. Preparar filtração a vácuo 10. Separar os cristais por filtração a vácuo
Cristais de acetanilida purificados impregnados com a água mãe +Traços
(Ac. Acético, Anilina e Acetato de
Solução filtrada contendo acetanilida (traços) e impureza solúveis
11. Fazer a lavagem com água gelada
12. Deixar secando com passagem de ar.
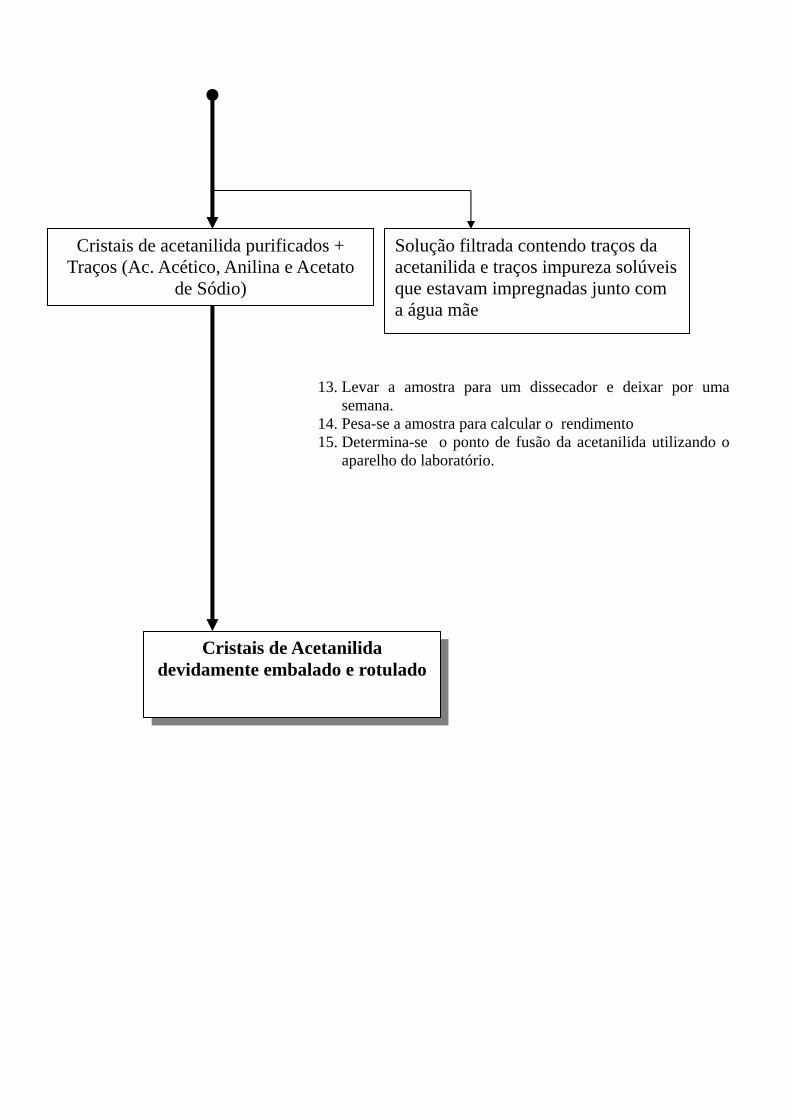
Cristais de acetanilida purificados + Traços (Ac. Acético, Anilina e Acetato
de Sódio)
Solução filtrada contendo traços da acetanilida e traços impureza solúveis que estavam impregnadas junto com a água mãe
13. Levar a amostra para um dissecador e deixar por uma semana.
14. Pesa-se a amostra para calcular o rendimento 15. Determina-se o ponto de fusão da acetanilida utilizando o
aparelho do laboratório.
Cristais de Acetanilida devidamente embalado e rotulado

Introdução a Recristalização Compostos sólidos a temperatura ambiente são geralmente
purificados por recristalização. A técnica geral envolve dissolução do material a ser purificado a
quente e resfriamento lento da solução, que possibilitará a formação e precipitação de cristais.
Se fizer um processo lento, ocorre uma cristalização seletiva, se for feito de maneira rápida, o processo é não seletivo.
A cristalização é um processo de equilíbrio no qual produz compostos muito puros.
Um pequeno núcleo é formado e depois segue depositando camada por camada onde o cristal praticamente seleciona as moléculas que formarão o retículo cristalino.
Por isso que não pode ser feito de maneira rápida, pois pode arrastar impurezas por coprecipitação:
• • •
oclusão – mecânica , isomórfica e não isomórfica inclusão adsorsão
Em contra partida um processo muito lento também se torna inviável, um tempo ideal é entre 10 mim a horas, ou de um dia para o outro, e não segundos ou dias.
Devemos nos atentar então para não cometer dois erros principais: 1. Resfriamento muito rápido da solução 2. Escolhe um solvente inadequado
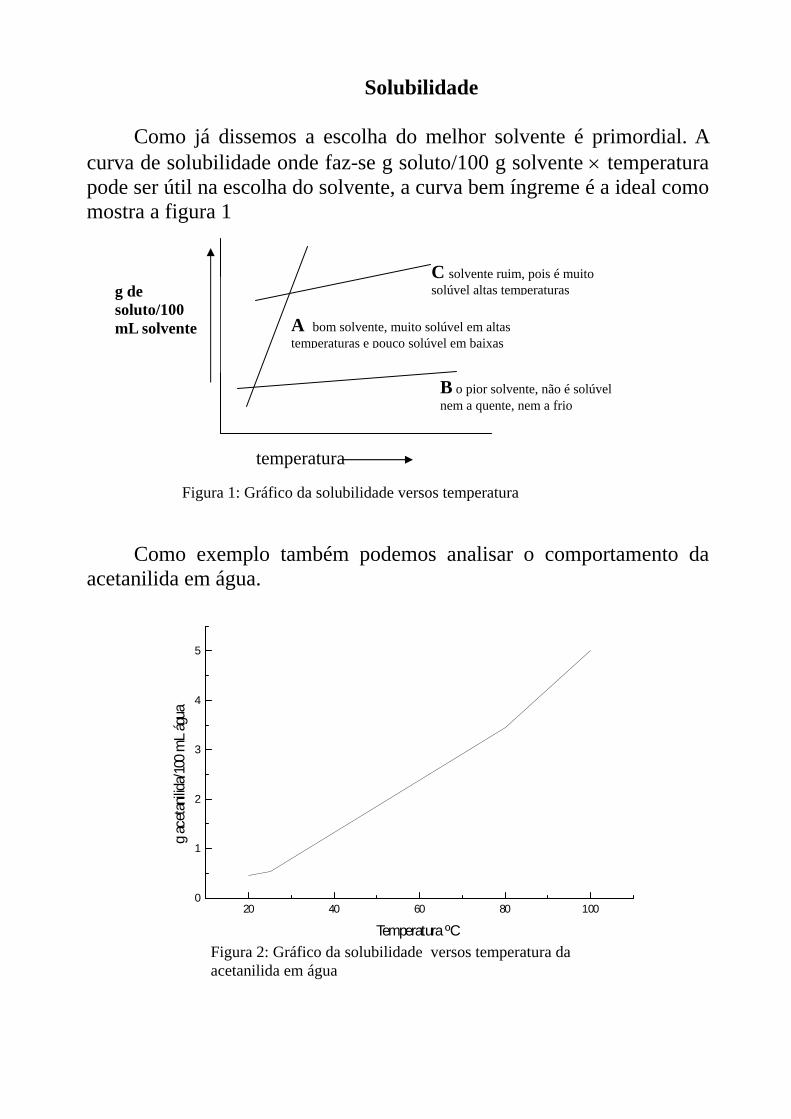
Solubilidade
Como já dissemos a escolha do melhor solvente é primordial. A curva de solubilidade onde faz-se g soluto/100 g solvente × temperatura pode ser útil na escolha do solvente, a curva bem íngreme é a ideal como mostra a figura 1
B o pior solvente, não é solúvel nem a quente, nem a frio
A bom solvente, muito solúvel em altas temperaturas e pouco solúvel em baixas
C solvente ruim, pois é muito solúvel altas temperaturas
temperatura
Figura 1: Gráfico da solubilidade versos temperatura
g de soluto/100 mL solvente
Como exemplo também podemos analisar o comportamento da
acetanilida em água.
Figura 2: Gráfico da solubilidade versos temperatura da acetanilida em água
20 40 60 80 1000
1
2
3
4
5
g ac
etan
ilida/
100
mL
água
Temperatura ºC

A solubilidade de um solvente é função também da polaridade, semelhante dissolve semelhante, (polar – polar; apolar – apolar; as funções orgânicas como --OH, --NH, --CONH, --COOH, são mais solúveis em solventes hidroxilados polares como água, metanol e etanol, do que em hidrocarbonetos como benzeno e hexano; mas se a função não é a parte principal a situação se inverte, pois numa molécula como CH3(CH2)11OH por exemplo é insolúvel em água pois devido ao seu tamanho tem características de molécula apolar, e pode se dissolver em hidrocarbonetos.
Tabela 1: Ordem decrescente de polaridade do Solvente
H20 Água
RCOOH Acidos organicos (ácido acético)
RCONH2 Amidas (N,N-dimetilformamida)
ROH Alcool ( metanol, etanol)
RNH2 Aminas ( trietilamina, piridina)
RCOR Aldeidos, cetonas (acetona)
ROCOOR Esteres (Acetatode etila)
RX Haletos organicos (CH2Cl2>CHCl3>CCl4)
ROR Eteres ( eter dietilico)
ArH Aromáticos (benzeno, tolueno)
RH Alcanos (hexano, éter petróleo)
Como Ocorre a Cristalização
Compostos orgânicos isolados, raramente são puros, pois são contaminados por impurezas. O sucesso de uma recristalização depende da grande diferença de solubilidade entre o soluto dissolvido no solvente a quente e a frio.
Mecanismo de precipitação: As condições de precipitação é que vão influenciar as
características físicas do solvente entre elas a temperatura, a concentração a solubilidade do precipitado entre outras.
Estes fatores são melhores compreendidos quando a solução esta supersaturada, ou seja que é um estado instável onde a partir desse ponto começa a precipitar
i.exe
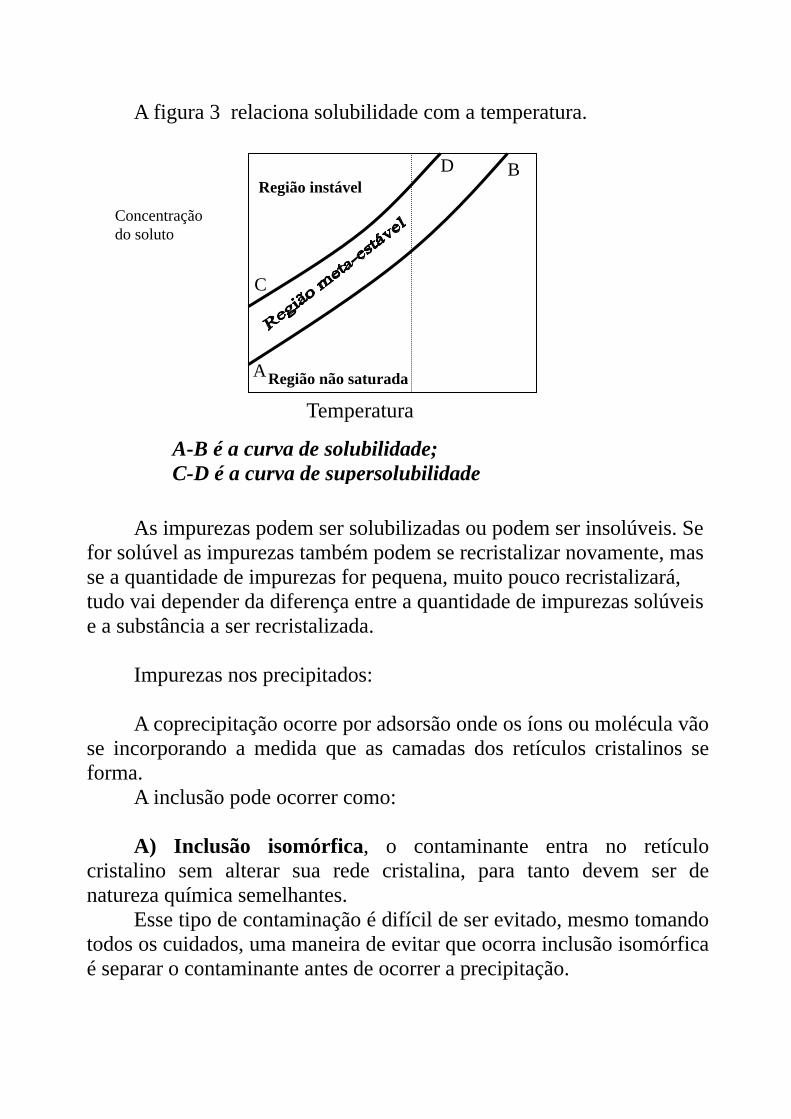
A figura 3 relaciona solubilidade com a temperatura.
A-B é a curva de solubilidade; C-D é a curva de supersolubilidade
Temperatura
Região instável
Região não saturada
Concentração do soluto
A
D
C
B
As impurezas podem ser solubilizadas ou podem ser insolúveis. Se
for solúvel as impurezas também podem se recristalizar novamente, mas se a quantidade de impurezas for pequena, muito pouco recristalizará, tudo vai depender da diferença entre a quantidade de impurezas solúveis e a substância a ser recristalizada.
Impurezas nos precipitados: A coprecipitação ocorre por adsorsão onde os íons ou molécula vão
se incorporando a medida que as camadas dos retículos cristalinos se forma.
A inclusão pode ocorrer como: A) Inclusão isomórfica, o contaminante entra no retículo
cristalino sem alterar sua rede cristalina, para tanto devem ser de natureza química semelhantes.
Esse tipo de contaminação é difícil de ser evitado, mesmo tomando todos os cuidados, uma maneira de evitar que ocorra inclusão isomórfica é separar o contaminante antes de ocorrer a precipitação.

B) Inclusão não isomórfica, o contaminante entra no retículo cristalino sem alterar a sua rede cristalina até certo limite, isso ocorre por que os dois compostos apresentam a mesma forma cristalina, mas com espaços diferentes dentro da rede cristalina. Portanto se estiver em pequena quantidade, o comportamento é semelhante a inclusão isomórfica, em grande quantidade provoca grandes deformações nos cristais que estão sendo formados.
C) Oclusão, o contaminante é incorporado de forma a formar
grandes alterações na rede cristalina. Um contaminante isomórfico nunca se acomoda tão bem na rede como o composto que está sendo precipitado, por isso quando adsorvido temporariamente pelo cristal pode ser expulso pelo processo de crescimento dos cristais por dessorção. Mas se a precipitação ocorrer rapidamente e não se der o tempo necessário para o crescimento dos cristais o contaminante é adsorvido pelo cristal e forma-se então imperfeições na rede.
A oclusão pode ser diminuída sensivelmente pelo processo de crescimento e digestão dos cristais lentamente em contado coma a água mãe.
Pode também ocorrer oclusão de água no processo de crescimento dos cristais, se o solvente utilizado for a água. Não pode ser removida por simples aquecimento, mas se o ponto de fusão do cristal for alto, pode-se aquecer a temperaturas elevadas e evaporar a água se a pressão interna for grande o bastante para romper a estrutura do cristal.
Rendimento da técnica: O processo de recristalização também ocasiona perda de material,
por isso se for repetido, mais puro se tornará o composto, mas o resultado e uma quantia menor do que a amostra inicial.
Ex: Impureza B 1 g, composto A 10 g, dissolvido em 100 mL solvente onde a solubilidade de ambos é 1 g/100 mL a 20 ºC e 10 g/100 mL a 100 ºC, quando a solução for resfriada a 20 ºC, o soluto a precipitará 9 g e a impureza permanecerá na solução, 1 g de soluto também ficará na solução.

Segunda cristalização
Primeira cristalização
Impuro (9g A+2g B)
Puro (8g A +1g B)
Bem puro (7 g A) Solução (1 g A+1 g B)
Solução (1 g A+1 g B)
Figura 4: purificação por recristalização.
Técnica de Recristalização Um bom solvente deve dissolver uma grande quantidade de soluto em temperatura elevada ou mesmo no ponto de ebulição, e dissolver pouco do mesmo soluto quando estiver a temperatura ambiente ou temperatura mais baixas, (quanto mais insolúvel a temperatura ambiente melhor). Características do solvente: 1. Ser facilmente removido quando o composto purificado estiver seco. 2. Permitir uma boa formação dos cristais (recristalizaçao) 3. Não reagir quimicamente com a substância a ser purificada
A escolha do melhor solvente não é um procedimento fácil de realizar. Para recristalização de compostos conhecidos a literatura pode fornecer alguns dados, mas nem sempre essas informações podem ser úteis se impurezas diferentes estão envolvidas no processo de contaminação.
A melhor maneira de se escolher o solvente adequado é realizar testes preliminares em tubos de ensaios com 0,1 g de material a ser recristalizado e dissolve-lo no solvente que está sendo, repetindo este procedimento para vários tipos de solventes.
i.exe

Tabela 3: Solubilidade da Acetanilida
solvente Temperatura ºC
g soluto/100 mL solução
Água 100 5 80 3,45 50 0,84 25 0,54 20 0,46 Etanol 20 29 78 167 Metanol 20 33 clorofôrmio 20 27 Acetona 20 25 Glicerol 20 20 Dioxano 20 12,5 Éter etílico 20 5,6
Um solvente pouco solúvel deve ser descartado porque por
exemplo se for gasto 0.80 g/ 100 mL para dissolve-lo, amostras maiores precisaria de uma quantidade enorme de solvente, o que tornaria inviável à prática.
O solvente então que diluir o soluto no ponto de ebulição mesmo que seja necessário sua adição, e permitir uma boa recristalização poderá ser escolhido para efetuar a recristalização.
Deve-se tomar cuidado para não escolher um solvente que tenha ponto de ebulição mais alto que o composto a ser cristalizado, pois o composto pode se fundir formando uma espécie de óleo que não se recristaliza quando o solvente esfriar, formando um líquido super resfriado, se isso ocorrer, deve-se então tentar dissolver novamente o óleo e deixar resfriar novamente para tentar a recristalização. Soluções supersaturadas ou certos tipos de impurezas também provocam a formação desse óleo, por exemplo a acetanilida em água numa concentração maior que 5,5 % pode formar o óleo.
Um solvente com baixo ponto de ebulição permite ser removido com mais facilidade do soluto purificado depois de filtrado e lavado. Se for muito volátil deve se tomar os devidos cuidados.
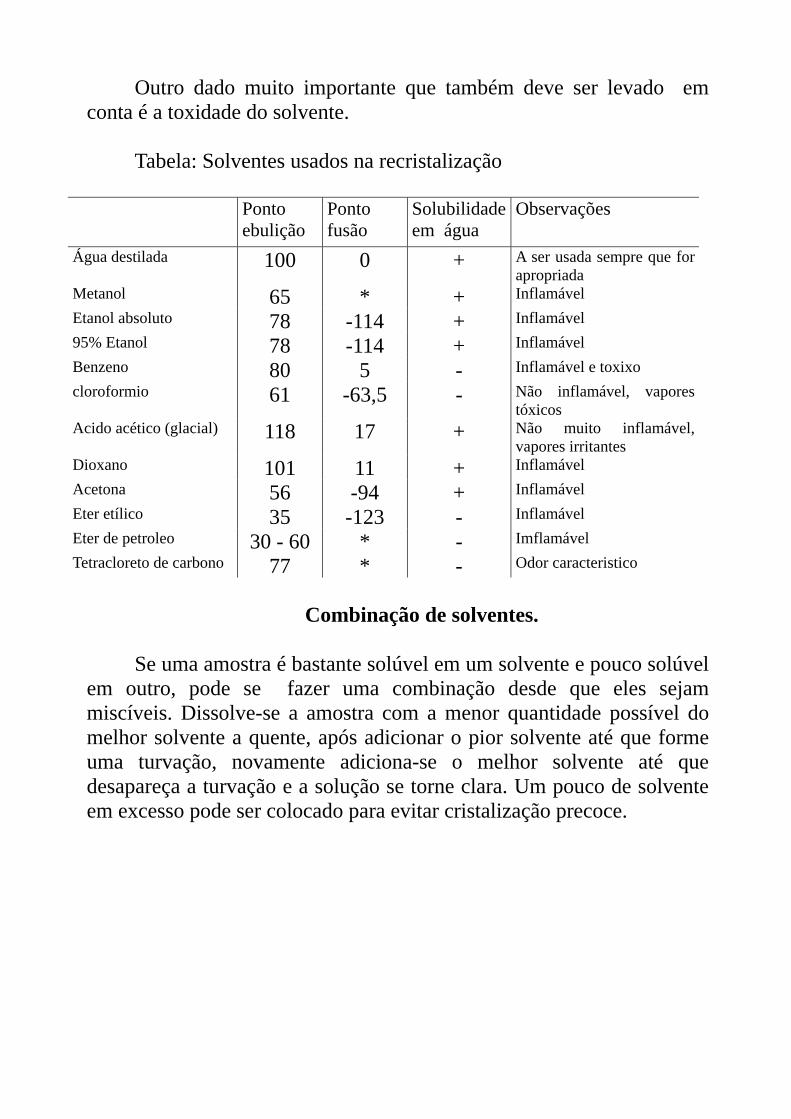
Outro dado muito importante que também deve ser levado em conta é a toxidade do solvente.
Tabela: Solventes usados na recristalização
Ponto ebulição
Ponto fusão
Solubilidade em água
Observações
Água destilada 100 0 + A ser usada sempre que for apropriada
Metanol 65 * + Inflamável Etanol absoluto 78 -114 + Inflamável 95% Etanol 78 -114 + Inflamável Benzeno 80 5 - Inflamável e toxixo cloroformio 61 -63,5 - Não inflamável, vapores
tóxicos Acido acético (glacial) 118 17 + Não muito inflamável,
vapores irritantes Dioxano 101 11 + Inflamável Acetona 56 -94 + Inflamável Eter etílico 35 -123 - Inflamável Eter de petroleo 30 - 60 * - Imflamável Tetracloreto de carbono 77 * - Odor caracteristico
Combinação de solventes.
Se uma amostra é bastante solúvel em um solvente e pouco solúvel
em outro, pode se fazer uma combinação desde que eles sejam miscíveis. Dissolve-se a amostra com a menor quantidade possível do melhor solvente a quente, após adicionar o pior solvente até que forme uma turvação, novamente adiciona-se o melhor solvente até que desapareça a turvação e a solução se torne clara. Um pouco de solvente em excesso pode ser colocado para evitar cristalização precoce.

Tabela 3 Combinações de solventes miscíveis usados na recristalização
Metanol-Água Eter-acetona Etanol-Água Eter-Eter de petróleo Ácido Acético- Água Benzeno-ligroína Acetona-Água Cloreto de metila-Metanol Éter-Metanol Dioxano-Água
Procedimento experimental 1. Testar os solventes: Água, etanol, éter, cloroformio, acetona e
benzeno. 2. Pode ser feito o teste em grupos ou individual. 3. Separar 0,1 g de amostra por tubo para realizar o teste. 4. Adicionar o solvente gota a gota e verificar sua solubilidade a frio,
adicionar até aproximadamente 1 mL. 5. Se não diluir, aquecer até a ebulição, (tomar cuidado com os solventes
voláteis, pois estes não podem ser trabalhados diretamente com o bico de gás, e sim usar uma manta aquecedora ou banho maria).
6. Se com pequeno aquecimento já se dissolver, pode-se descartar o solvente, mas se não dissolver, quando chegar no ponto de ebulição adicionar solvente até a dissolução, se completar aproximadamente 3 mL, caso ainda não dilua o composto, também descarta-se o solvente pois é pouco solúvel naquele solvente.
7. Se dissolver deixar esfriar para verificar se ocorre recristalização, pode-se atritar com um bastão de vidro não polido para auxiliar a recristalização.
Dissolução da amostra
Escolhido o melhor solvente, deve-se então pesar uma porção da amostra e dissolver no solvente a quente. Deve-se minimizar as perdas de soluto saturando ao máximo a solução, mas tomando cuidado para não formar o óleo.
Coloca-se o soluto num erlermayer com uma quantidade de soluto ligeiramente menor do que a quantidade necessária para dissolução.

Quando estiver em ebulição, adiciona-se pequenas quantidades de solvente até a dissolução se complete.
Deve-se porem tomar cuidado para não confundir impurezas sólidas insolúveis e fibra de papel com a substancia, pois isso levaria a adição de mais solvente e diminuindo o rendimento da recristalização, o teste preliminar de escolha do solvente ajudará a reconhecer como são as impureza insolúveis.
Quando o solvente for muito volátil, inflamável, toxico ou caro aconselha se usar um condensador de refluxo.
Usar também pedras de porcelana para evitar que o frasco trinque ou quebre, e também tornar uniforme o aquecimento da solução.
Carvão ativo (descoramento)
Quando a substância se dissolveu por completo, para eliminar
impurezas coloridas, materiais resinosos, usa se o carvão ativo para adsorver as impurezas.
Usa se uma pequena quantidade de carvão ativo, pois o excesso pode levar o carvão a adsorver também a substancia a ser cristalizada.
A quantidade ideal é em torno de 1 a 2 % (p/p) do total pesado da amostra, se não for suficiente vai se adicionando mais 0,5 % até que limpe todas as impurezas.
Deve-se te o CUIDADO para não colocar carvão ativo sobe a solução em ebulição, pois provocaria uma grande agitação da solução levando ao derramamento e perda de material.
Tomar cuidado também com o pó muito fino do carvão ativo pois faz mal aos pulmões quando inalado.
Tipos de carvão ativo: Animal: mais baratos, porém não tão eficaz. Carvão obtido da madeira vendidos com as marcas, Norit* (obtido
de madeira de vidoeiro), Darco*, Nuchar*.
Procedimento experimental 1. Pesar 10 g de acetanilida impura 2. Dissolver em becker de 500 Ml, com aproximadamente 190 a
200 mL

3. Adicionar 0,2 g aproximadamente de carvão ativo, não esquecendo de antes esperar a solução esfriar pelo menos um minuto.
4. Agitar a solução com o carvão ativo 5. Preparar o equipamento de filtração por gravidade
(a) (b) (c) (d)
Filtração por gravidade Filtração efetuada para separar a solução com amostras dissolvidas
das impurezas insolúveis de do carvão ativo. Deve ser feita de maneira rápida e eficiente para não ocorrer cristalização no papel de filtro que além de perder material pode obstruir o funil.
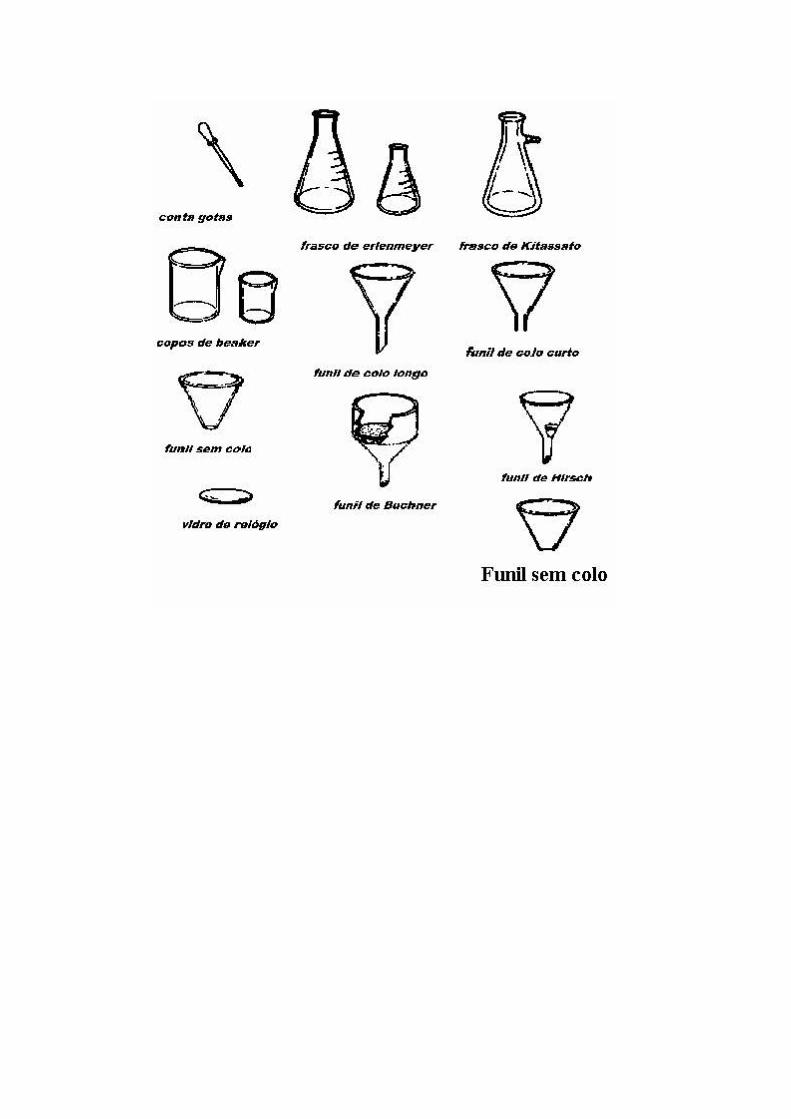
Para a filtração por gravidade pecisa se de um funil sem colo, papel de filtro pregueado e o recipiente para receber o filtrado.
A solução pode esfriar enquanto está sendo filtrada, para evitar uma cristalização precoce pode se tomar algumas precauções:
Colocar uma quantidade extra de solvente, lembrando que depois da filtração o excesso de solvente deve ser removido por evaporação
•
•
•
Aquece-se o funil antes de realizar a filtração com solvente quente (que será descartado logo após a operação), e depois filtrar rapidamente mantendo o funil aquecido com uma manta como mostra a figura Usa se um funil sem colo, e quando aquece-lo (vide fig.), ao invés de recolher o filtrado no erlenmeyer, faz-se num becker como mostra a figura 6.
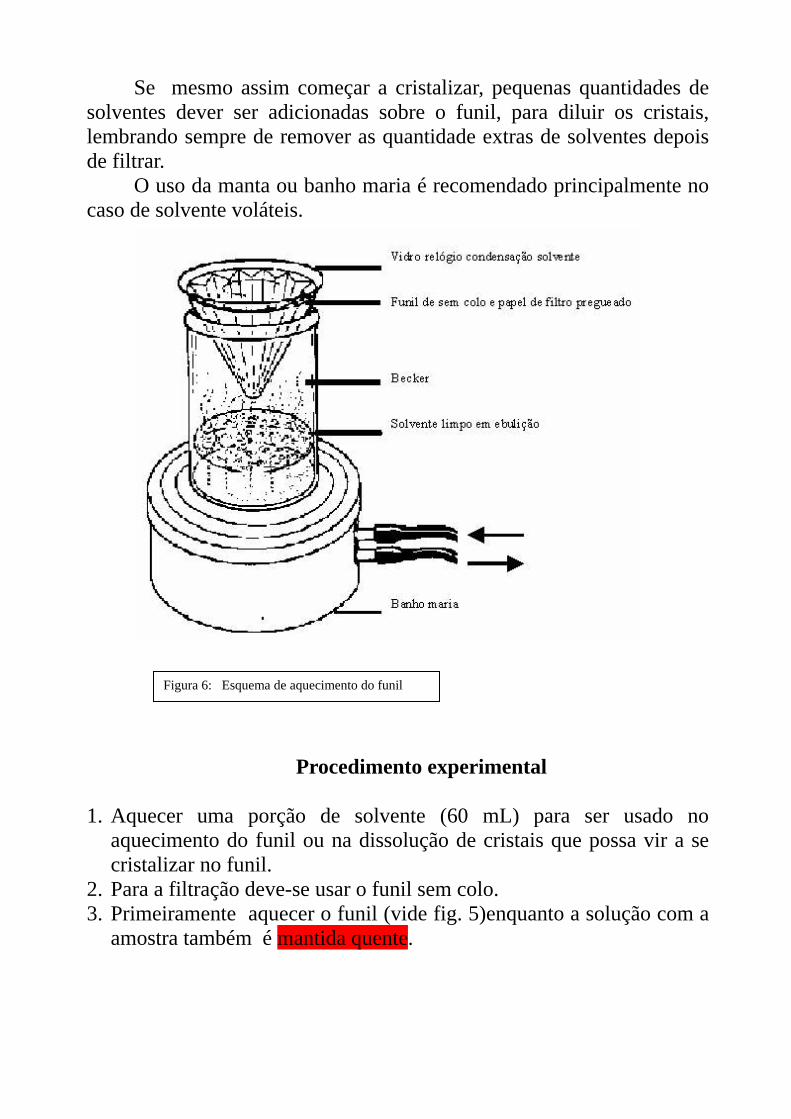
Se mesmo assim começar a cristalizar, pequenas quantidades de solventes dever ser adicionadas sobre o funil, para diluir os cristais, lembrando sempre de remover as quantidade extras de solventes depois de filtrar.
O uso da manta ou banho maria é recomendado principalmente no caso de solvente voláteis.
Figura 6: Esquema de aquecimento do funil
Procedimento experimental
1. Aquecer uma porção de solvente (60 mL) para ser usado no aquecimento do funil ou na dissolução de cristais que possa vir a se cristalizar no funil.
2. Para a filtração deve-se usar o funil sem colo. 3. Primeiramente aquecer o funil (vide fig. 5)enquanto a solução com a
amostra também é mantida quente.

4. Com auxilio de um bastão de vidro, filtra-se a solução rapidamente a quente (fig. 7).
5. restante sólido dentro do becker pode ser removido com um bastão de vidro e fazendo lavagem com pequenas quantidade da água mãe.
6. Colocar um pedaço de arame entre o funil e o becker para evitar pressão.
Recristalização
A recristalização é um processo lento e delicado. Depois de filtrar,
ajusta-se a quantidade de solvente e deixa-se esfriar para recristalizar. Melhor se for feito em um erlermeyer para evitar contaminação por pó, se fizer em um becker, melhor deixar tampado por um vidro relógio enquanto ocorre a recristalização.
A taxa de resfriamento é que termina o tamanho dos cristais, se quiser obter cristais grandes, deixa-se resfriar lentamente, não esquecendo que cristais muito grandes facilitam a oclusão e se quiser obter cristais pequenos, acelera-se o resfriamento lembrando também do risco de obstruir o funil e dificuldade de lavagem além de que um resfriamento rápido provoca o arraste de impurezas junto com a precipitação dos cristais.
O ideal seria obter cristais entre 2 a10 mm

Eventuais problemas com a recristalização
o novam
r novamente lentamente sob agitação até formar os prime
, usa se no
alcatrão ou outra substância viscosa que age como um coloi
1.
parede do frasco e servirão de semente para iniciar a
2.
ços da amostra original ou de
3. Relátil, usa se o condensador de
refluxação pode variar de 10 min a horas
ou de um dia para
Substancia com baixo ponto de fusão, se forem recristalizados
rapidamente podem também formar o líquido oleoso. Se isso acontecer dissolva novamente adicionando um pouco de solvente e aquecend
ente a solução agitando bastante até a dissolução da substância. Deixar resfriairos cristais. Se formar o óleo devido a impurezas e a recristalização falharvamente carvão ativo e tenta-se novamente a recristalização. Também pode acontecer dos cristais resistirem a cristalização, isso
ocorre quando a solução está muito saturada ou está sendo impedida por impurezas como
de protetor. Deve-se então induzir a cristalização
Arranha-se a parede do frasco com um bastão de vidro não polido. Esse procedimento provoca a formação dos cristais pela frequência que emite ou por arrastar para dentro da solução pequenos cristais que estavam nanucleação. Coloca-se na solução pequenos cristais da amostra para servir de semente. Para obter os cristais retira-se algumas gotas da solução coloca-se numa lâmina e força-se a cristalização por resfriamento rápido, ou utiliza-se de pequenos pedacompostos já purificados e guardados.
sfria-se a solução em banho de gelo Lembrando que se o solvente for voo para evitar evaporação (Figura 8). O tempo ideal para recristaliz
o outro.
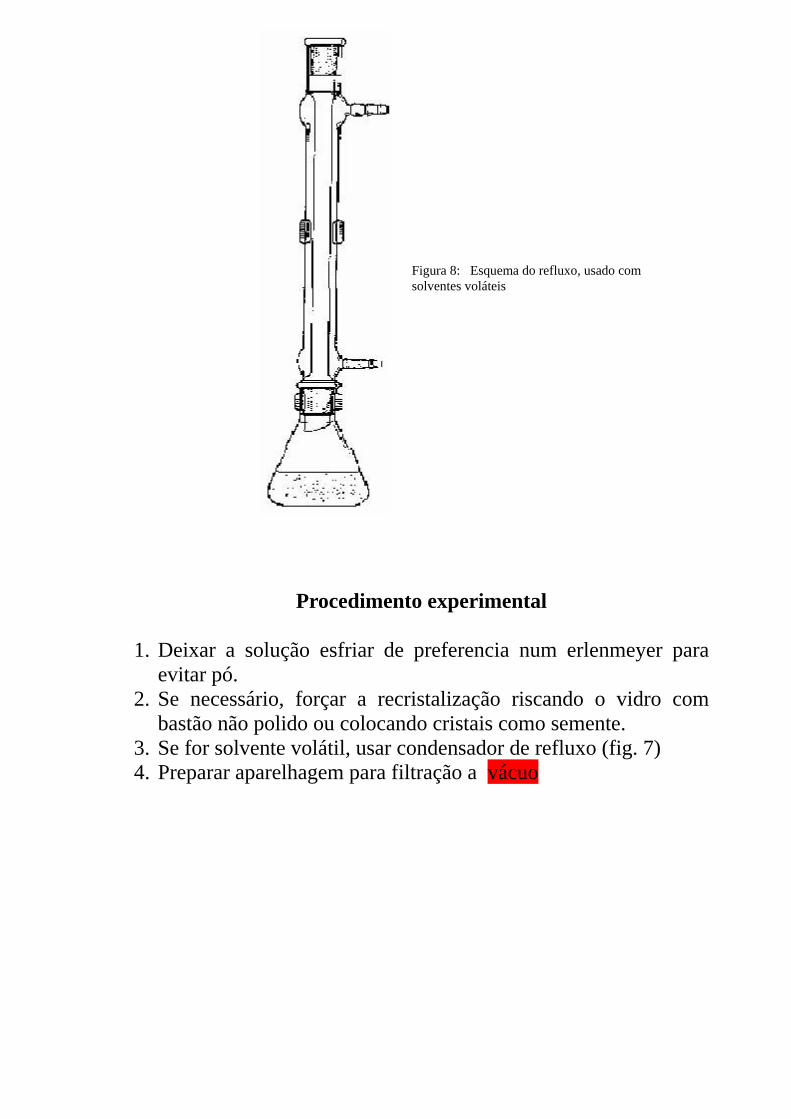
Procedimento experimental 1. Deixar a solução esfriar de preferencia num erlenmeyer para
evitar pó. 2. Se necessário, forçar a recristalização riscando o vidro com
bastão não polido ou colocando cristais como semente. 3. Se for solvente volátil, usar condensador de refluxo (fig. 7)
Figura 8: Esquema do refluxo, usado com solventes voláteis
4. Preparar aparelhagem para filtração a vácuo
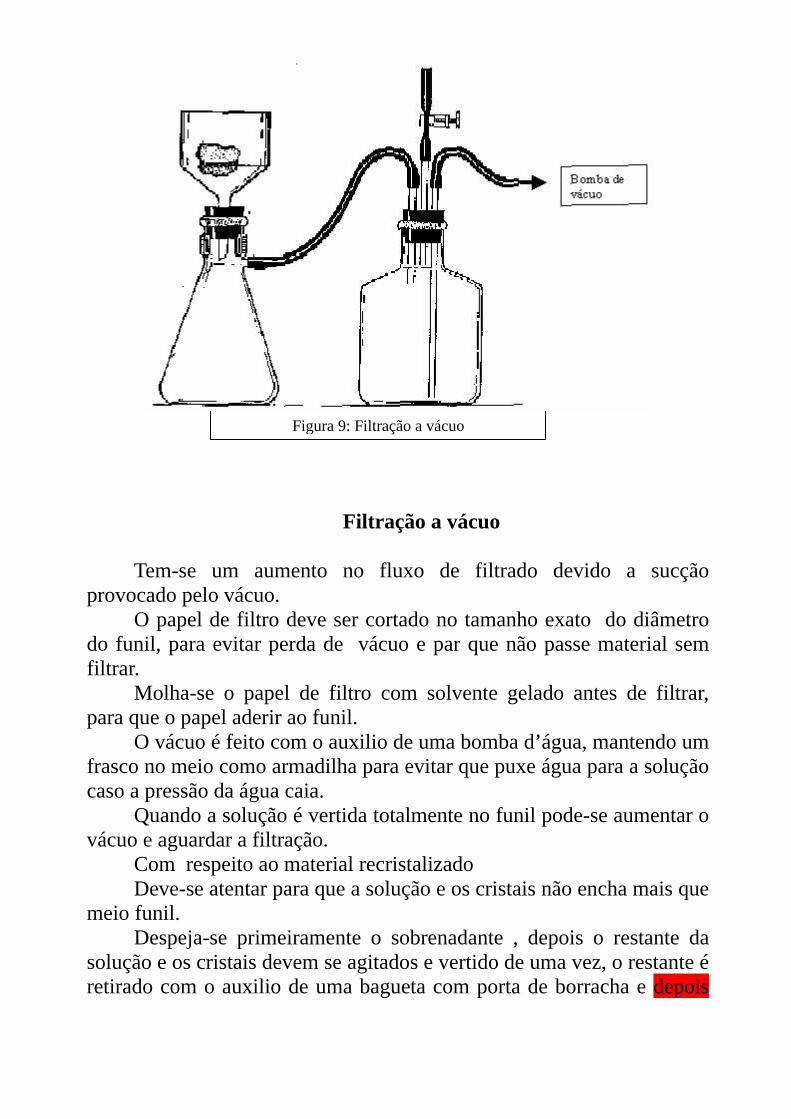
Figura 9: Filtração a vácuo
Filtração a vácuo
Tem-se um aumento no fluxo de filtrado devido a sucção provocado pelo vácuo.
O papel de filtro deve ser cortado no tamanho exato do diâmetro do funil, para evitar perda de vácuo e par que não passe material sem filtrar.
Molha-se o papel de filtro com solvente gelado antes de filtrar, para que o papel aderir ao funil.
O vácuo é feito com o auxilio de uma bomba d’água, mantendo um frasco no meio como armadilha para evitar que puxe água para a solução caso a pressão da água caia.
Quando a solução é vertida totalmente no funil pode-se aumentar o vácuo e aguardar a filtração.
Com respeito ao material recristalizado Deve-se atentar para que a solução e os cristais não encha mais que
meio funil. Despeja-se primeiramente o sobrenadante , depois o restante da
solução e os cristais devem se agitados e vertido de uma vez, o restante é retirado com o auxilio de uma bagueta com porta de borracha e depois

lavado com um pouco da água mãe filtrada ou solvente gelado para retirar todos os cristais do frasco.
Procedimento experimental
1. Recorte o papel de filtro exatamente do tamanho da boca do
funil de buchner 2. Acione a bomba de vácuo 3. Importante colocar entre o kitassato e a bomba de vácuo um
frasco como armadilha, para evitar que seja aspirado água caso caia a pressão de água.
4. Recolher o restante dos cristais com uma bagueta de ponta de borracha
5. Lave o restante com um pouco de água mãe ou solvente gelado
Lavagem dos Cristais
Depois de filtrado a substância, antes da secagem é preciso lavar a substância para evitar que impurezas dissolvidas na solução que encharca os cristais fiquem aderidas nos cristais quando o solvente evaporar.
Faz-se a lavagem com um solvente limpo e gelado, podendo ser lavado por 2 vezes se a substância não for muito solúvel no solvente gelado. Se os cristais não estiverem amontoados num bolo sólido, pode-se fazer vácuo normalmente para auxiliar a lavagem, caso contrário faz-se vácuo normalmente para auxiliar o processo de lavagem.
Uma boa alternativa para uma segunda lavagem é usar um solvente mais volátil e miscível com o primeiro e que não dissolva os cristais a frio, pois sua melhor evaporação facilitará a secagem dos cristais.
Deixa-se então os cristais secando com passagem de ar, é aconselhável cobrir o funil com um papel de filtro para evitar contaminação com pó.
Procedimento Experimental
1. Fazer a lavagem com solvente gelado para eliminar impurezas no solvente.
2. Lavar 2 vezes, verificar condições dos cristais para decidir a intensidade de vácuo a ser usado.

3. Deixa-se ficar passando ar para secagem do solvente 4. Prepara-se para a secagem
Secagem.
Um método comum é colocar em um vidro relógio e secar ao ar livre. Este método tem a vantagem de não se correr o risco de decompor o produto, mas é demorado alem de substância higroscópicas certamente se hidrolizarem no contato com o ar atmosférico.
Também pode-se secar os cristais usando papel absorvente, mas
corre-se o risco de contaminar os cristais com fibras do papel. Outra maneira é colocando num vidro relógio e colocando em uma
estufa para secar, mas deve-se tomar muito cuidado para não exceder o ponto de fusão do cristal, nem colocar cristais que se decompõe com o calor ou sublimam facilmente.
Outra maneira, talvez a melhor é utilizar um dissecador a vácuo na presença de um agente secante, ou se preferir deixar dentro do dissecador sem fazer vácuo, por um longo período como por exemplo uma ou duas semanas.
No caso da nossa experiência vamos deixar dentro do dissecador por uma semana usando como secante o cloreto de cálcio anidro.
Precauções no uso do dissecador se for usado vácuo
• •
•
•
Cuidado com amostras que sublimam facilmente Cuidado para não implodir o dissecador, então por medida de segurança deve-se usar uma gaiola protetora Para tornar o dissecador livre de ar, untar a superfície esmerilhada da tampa e do corpo com vaselina. Usar um kitassato de segurança entre o dissecador e a bomba de vácuo.

Maneira universal de encher um dissecador. Acido Sulfúrico concentrado na metade inferior, hidróxido de
sódio nas saliências do dissecador, cobrindo-se parte inferior com uma tela de zinco ou um chapa porcelana perfurada. Éter, cloroformio, tetracloreto de carbono, benzeno, tolueno e vapores similares, deve-se colocar aparas de parafinas recentemente cortadas sobre o hidróxido de sódio, ou cloreto de cálcio ou sílica gel podem ser colocados alternativamente.
Ao usar o dissecador sob pressão, coloca-se a substância com um vidro relógio invertido para impedir que cristais pequenos sejam varridos do dissecador.
Procedimento experimental
1. Depois que a substância já secou bem no funil de Buchner, onde se deixou passando ar por algum tempo, retira-se os cristais e leva-se a um dissecador, onde deixará a acetanilida secando por uma semana.
2. Deixar os cristais dentro de um cadinho, ou um vidro relógio, pois não tendo vácuo, não há perigo de pequenos cristais serem sugados e perdidos.
3. Para finalizar pesar a amostra e determinar o ponto de fusão
Teoria do ponto de fusão.
O ponto de fusão de um sólido cristalino é a temperatura na qual o sólido começa a se tornar um líquido sob a pressão de uma atmosfera.
Um composto orgânico cristalino puro tem um ponto de fusão bem definido com um intervalo menor de 0,5 ºC, presença de impurezas miscíveis ou parcialmente miscíveis faz com que essa diferença entre a primeira formação do líquido e a fusão total aumenta bastante, também causa o início do ponto de fusão a uma temperatura mais baixa.
L M L’ Ponto de fusão
Pressão de vapor
Temperatura ºCFigura 8: razão da fusão do sólido cristalino ser constante
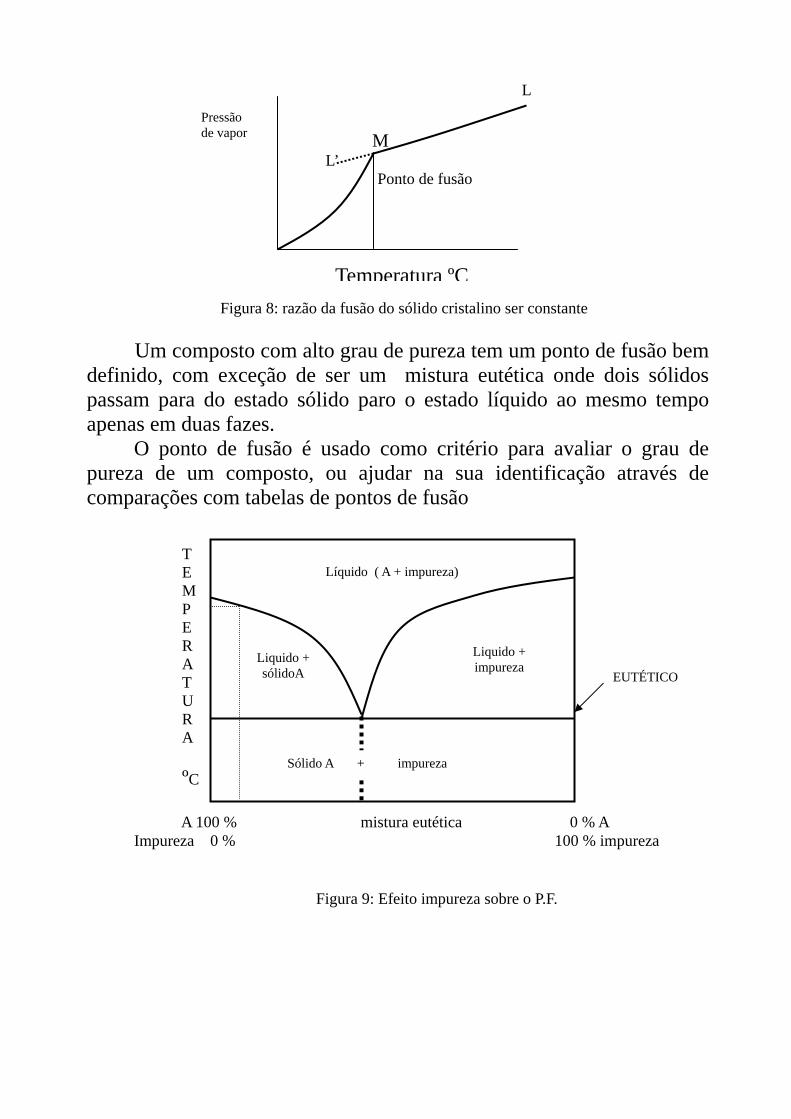
Um composto com alto grau de pureza tem um ponto de fusão bem
definido, com exceção de ser um mistura eutética onde dois sólidos passam para do estado sólido paro o estado líquido ao mesmo tempo apenas em duas fazes.
O ponto de fusão é usado como critério para avaliar o grau de pureza de um composto, ou ajudar na sua identificação através de comparações com tabelas de pontos de fusão
A 100 % mistura eutética 0 % A Impureza 0 % 100 % impureza
EUTÉTICO
TEMP ERATURA ºC
Liquido + sólidoA
Líquido ( A + impureza)
Liquido + impureza
Sólido A + impureza
Figura 9: Efeito impureza sobre o P.F.

Métodos de determinar o ponto de Fusão
Há vários métodos de determinar o ponto de fusão, enter eles uns são via experimentos de aquecimentos da solução em banho com substância com ponto de ebulição maior, ou aparelhos de resistência elétrica equipados com termômetros.
Um método comum é o de aquecer uma pequena quantidade de
substância que está num tubo capilar, próximo a um termômetro dentro de um banho num líquido que tenha um ponto de ebulição maior que o ponto de fusão da substância.

Outro método é utilizando um aparelho aquecido eletricamente
como o da figura abaixo:
etro enrolado por um fio de resist
rre o ponto de fu
capilares para determinar o ponto de fusão, e sim lamínulas de vid
anual que está
Determina até 2 capilares simultaneamente, um bloco de cobre com buracos para capilares e o termôm
ência elétrica coberto por um vidro. Pode-se manter vários velocidade de aquecimentos. Há um aparato para se observar o momento em que ocosão, onde os capilares são iluminados por um luz de 6V. No caso da nossa experiência utilizaremos um outro aparelho que
não utilizaro.
Para utiliza-lo corretamente deve se utilizar o mjunto com o aparelho tal. ou no procedimento experimen

Procedimento experimental
endimento
3 aparelho através da Chave (liga/Desliga), na parte de trás do
4 2 minutos para estabilização dos circuitos eletrônicos do
5 ma lamínula sobre a superfície do bloco de alumínio do
6 ma pequena amostra do produto a ser analisado sobre a
9
tec
. Se este valor é ultrapassado, o valor inicia novamente em 1
. Se este valor é ultrapassado, o valor inicia novamente em 30 ºC
a], permanecendo piscando por mais alguns segundos o valor ajusta
óximo ao ponto de fusão do produto de ensaio (quando
namicamente durante o ensaio permitindo maior velocidade no mesmo;
1. Pesa-se a amostra e calcula-se o r2 Determinação do ponto de fusão
Ligar o mesmo Aguardar aparelho. Colocar uaparelho Colocar ulamínula.
7 Colocar outra lamínula sobre a amostra. 8 Ajustar o foco da lente para uma boa observação.
Selecionar a taxa de aquecimento (em graus centígrados/min) desejada para o ensaio, na tecla [Taxa] aquecimento. Pressionando-se esta tecla rapidamente para o lado esquerdo ou direito, o valor da taxa de aquecimento é mostrado piscando no display. Soltando-se a
la por alguns segundos, o valor da temperatura retorna ao display. Para aumentar ou diminuir o valor da taxa de aquecimento, deve-
se manter pressionada esta tecla para o lado direito ou esquerdo, respectivamente. Se a tecla é pressionada para o lado direito, o valor da taxa de aquecimento permanece piscando e aumentando de 0,5 ºC/min até 30 ºC/min
ºC/min. Se a tecla é pressionada para o lado esquerdo, o valor da taxa de
aquecimento permanece piscando e diminuindo de 0,5 em 0,5 ºC/min até 1 ºC/min
/min. Selecionado o valor da taxa de aquecimento desejada, solta-se a
tecla [Taxdo. Obs.: a)Para maior precisão na determinação do ponto de fusão é
recomendado a utilização de uma taxa de aquecimento baixa (1 a 3 ºC/min), praquecido); b)A taxa de aquecimento pode ser alterada di

c)Deve ser considerada uma pequena inércia térmica, quando da mudança da taxa de aquecimento durante o ensaio. Pressionar a tecla (liga/desliga) aquecimento para iniciar o aquecimento na taxa selecionada no item anterior. Observar a amostra até que a mesma inicie a fusão. Neste instante, pressionar a tecla (fixa) para armazenar o ponto de início da fusão, quando então o valor no display pisca rapidamente. Prosseguir observando a amostra ate que a mesma esteja totalmente fundida. Neste instante, pressionar a tecla (fixa ) novamente para armazenar o ponto de Fim da fusão. Obs: Quando a tecla (fixa) é pressionada para armazenar o ponto de Fim da fusão, o processo de aquecimento é desligado automaticamente. Pressionando-se a tecla (fixa) podem sem observados os valores de início e fim da fusão, armazenados na memória do aparelho. Obs: Os valores armazenados permanecem piscando, enquanto estão sendo mostrados no display. Pressionar a tecla (liga/desliga) resfriamento, para resfriar o bloco de alumínio do aparelho até a temperatura ambiente. Obs: Durante o resfriamento pode ser observado o ponto de cristalização da amostra. Pressionar a tecla (liga/Desliga) aquecimento, para limpar os valores armazenados na memória e retornar a temperatura ao display. Retirar a amostra e reiniciar o procedimento, se desejado. Em ambientes com forte circulação de ar, ensaios de ponto de fusão em temperaturas altas, de uma mesma amostra, podem ter variação de alguns graus (± 1 ºC). Neste caso, utilizar a tampa de alumínio sobre as lamínulas com as amostras evitando-se que a temperatura da mesma não sofra influência do ambiente, obtendo se com isto maior homogeneidade nos ensaios. Esta tampa deve ser colocada de forma que o “rasgo” da mesma fique alinhado com o sistema de iluminação. A tampa deve ser retirada antes do resfriamento pois esta permanece aquecida por algum tempo, caso isto precise ser feito, deve ser manipulado com muito cuidado.

Segunda Recristalização
Pode-se concentrar o filtrado através de evaporação, e forçar uma nova recristalização, mas sempre levando em conta que o produto obtido não é menos puro que o da primeira recristalização.

APARELHAGEM UTILIZADA NA TÉCNICA