Embed Size (px)
Citation preview

UNIVERSIDADE DE ÉVORA
CENTRO DE QUÍMICA DE ÉVORA
BOLSA DE INVESTIGAÇÃO
Título do Projecto: CLAYCATS – Argilas do Porto Santo modificadas como catalisadores
de reacções orgânicas em fase líquida
Projecto nº: POCTI/CTM/47619/2002
Data de início da bolsa: 1 de Fevereiro de 2004 Duração: 12 meses
Bolseira: Patrícia Alexandra Ferreira Russo
Investigadora Responsável: Maria Manuela Lopes Ribeiro Carrott
Considerações gerais
O objectivo do trabalho desenvolvido durante a bolsa de investigação foi avaliar o
efeito da activação ácida de argilas do Porto Santo na estrutura e propriedades texturais
(porosidade e área superficial) dos materiais. O programa de trabalhos inicialmente
proposto desenvolver foi cumprido. As tarefas desenvolvidas foram as seguintes:
- pesquisa bibliográfica;
- estudo e optimização do método e das condições técnicas para obtenção de difractogramas
de raios X. Caracterização por DRX das amostras original e modificadas;
- determinação de isotérmicas de adsorção de azoto a 77 K das amostras original e
modificadas;
- caracterização de amostras seleccionadas por espectroscopia de IV;
- colaboração no desenvolvimento e calibração de uma instalação de vácuo gravimétrica
para estudos de adsorção em fase gasosa de compostos orgânicos;
- determinação de isotérmicas de adsorção de n-pentano a 298 K, por método gravimétrico,
em amostras seleccionadas;
1

- determinação de isotérmicas de adsorção de azoto a 77 K, antes e após pré-adsorção de n-
nonano e desgaseificação a várias temperaturas, na amostra original e em amostras
modificadas seleccionadas;
- análise das isotérmicas pelos métodos BET, s e DR;
- análise global dos resultados.
Parte dos resultados obtidos foram objecto de uma comunicação na XXIX Reunião
Ibérica de Adsorção, encontrando-se o resumo em anexo.
2

1. Introdução
Os minerais de argila são silicatos com estrutura lamelar. As lamelas são formadas
por folhetos constituídos por tetraedros de sílica ou octaedros de oxigénio e átomos
metálicos. Nas argilas 1:1 as lamelas são constituídas por um folheto octaédrico
directamente ligado a um tetraédrico enquanto que nas 2:1 um folheto octaédrico encontra-
se entre dois tetraédricos. Estes materiais são também classificados em dioctaédricos ou
trioctaédricos de acordo com a população dos folhetos octaédricos. Nos primeiros, 2/3 dos
octaedros estão ocupados essencialmente por catiões trivalentes, geralmente Al(III) ou
Fe(III), e nas argilas trioctaédricas a maior parte dos octaedros estão ocupados por catiões
divalentes, em geral, Mg(II) ou Fe(II).
A substituição isomórfica é comum nos minerais argilosos. Assim, nos tetraedros o
silício pode estar substituído por Al3+ ou Fe3+ e no folheto octaédrico os catiões podem ser
Al3+, Mg2+, Fe3+ ou Fe2+. Devido à substituição isomórfica pode surgir um défice de carga
positiva que é compensado pela presença de pequenos catiões de troca, situados entre as
lamelas.
Os minerais mais conhecidos do tipo 2:1 são os do grupo das esmectites. As
esmectites são muito utilizadas em vários ramos da indústria devido à sua elevada
capacidade de troca iónica, capacidade expansiva e elevada área superficial. Os sítios
octaédricos da montmorilonite, a esmectite dioctaédrica mais comum, estão ocupados
principalmente por Al(III) mas parcialmente substituído com Fe(III) e Mg(II).
Nos minerais argilosos naturais as partículas de argila encontram-se agregadas e
apresentam frequentemente outras impurezas minerais. Os tratamentos ácidos desagregam
as partículas de argila, eliminam impurezas, removem os catiões octaédricos e substituem
os catiões de compensação por protões. Desta forma, este tipo de tratamento pode
influenciar significativamente a actividade catalítica das argilas [1].
O objectivo do presente trabalho foi estudar as alterações estruturais e texturais
induzidas pelo tratamento com ácido clorídrico em uma esmectite da ilha do Porto Santo.
Foram analisados os efeitos da concentração do ácido, da temperatura e do tempo de
contacto da solução ácida com o sólido. Todas as amostras foram caracterizadas por
difracção de raios X e adsorção de azoto a 77 K. Amostras seleccionadas foram
caracterizadas por espectroscopia de infravermelho, adsorção de n-pentano a 298 K e pré-
adsorção de nonano seguida de adsorção de azoto. Para cada temperatura foram escolhidas
3

as amostras tratadas durante mais tempo com ácido de diferentes concentrações, ou seja, as
amostras que sofreram tratamento mais intenso, uma vez que o estudo destas amostras
permite também tirar algumas conclusões acerca da porosidade das restantes amostras.
4

2. Parte experimental
2.1. Preparação das amostras
A argila estudada é uma esmectite na forma sódica da Serra de Dentro da Ilha do
Porto Santo (Madeira) e amostras resultantes do tratamento desta com soluções de ácido
clorídrico de várias concentrações e a várias temperaturas. Todas as amostras foram
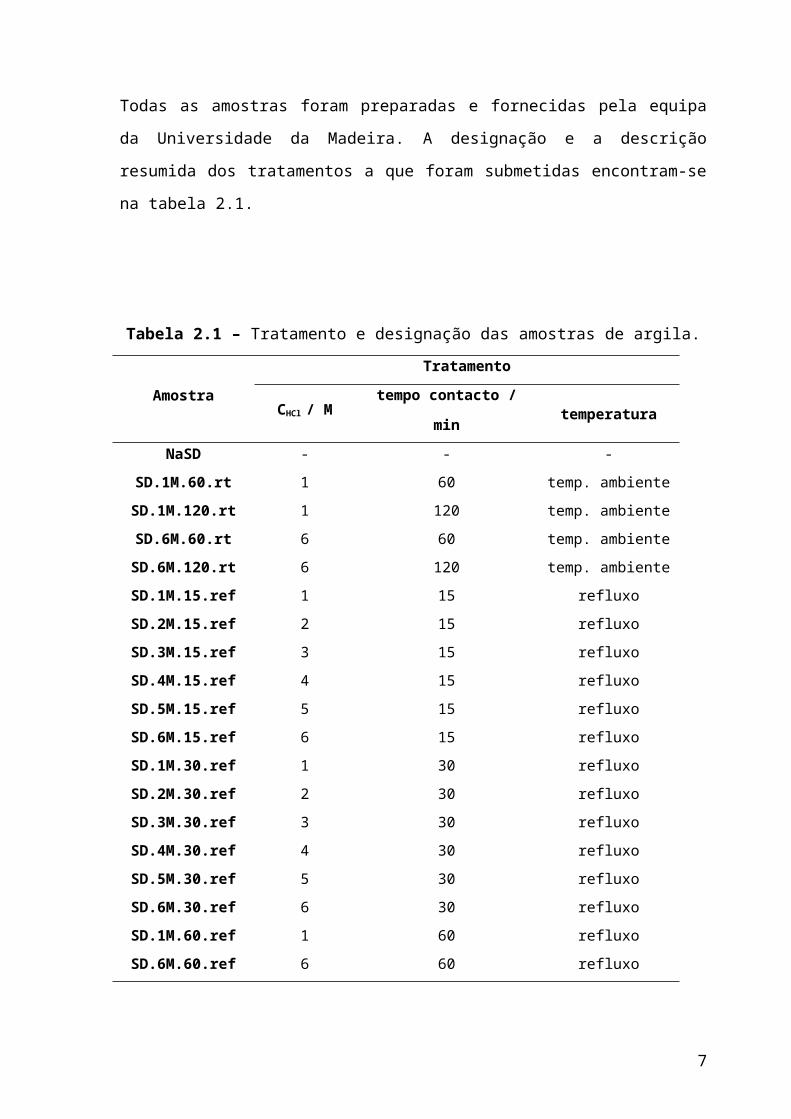
preparadas e fornecidas pela equipa da Universidade da Madeira. A designação e a
descrição resumida dos tratamentos a que foram submetidas encontram-se na tabela 2.1.
Tabela 2.1 – Tratamento e designação das amostras de argila.
AmostraTratamento
CHCl / M tempo contacto / min temperatura
NaSD - - -
SD.1M.60.rt 1 60 temp. ambiente
SD.1M.120.rt 1 120 temp. ambiente
SD.6M.60.rt 6 60 temp. ambiente
SD.6M.120.rt 6 120 temp. ambiente
SD.1M.15.ref 1 15 refluxo
SD.2M.15.ref 2 15 refluxo
SD.3M.15.ref 3 15 refluxo
SD.4M.15.ref 4 15 refluxo
SD.5M.15.ref 5 15 refluxo
SD.6M.15.ref 6 15 refluxo
SD.1M.30.ref 1 30 refluxo
SD.2M.30.ref 2 30 refluxo
SD.3M.30.ref 3 30 refluxo
SD.4M.30.ref 4 30 refluxo
SD.5M.30.ref 5 30 refluxo
SD.6M.30.ref 6 30 refluxo
SD.1M.60.ref 1 60 refluxo
SD.6M.60.ref 6 60 refluxo
5

2.2. Difracção de raios X
Os difractogramas de raios X foram obtidos num difractómetro Bruker AXS D8
Advance, usando radiação CuK1 ( = 0.1540598 nm). Todas as medições foram efectuadas
com 40 kV e 30 mA, tendo sido utilizadas fendas variáveis V20 (óptica secundária na fonte
e detector) e fendas fixas de 0.6 mm (detector). Para cada amostra foram traçados
difractogramas entre 2 e 80° (2θ), com incrementos de 0,02° e um intervalo de tempo de 2s
por incremento.
2.3. Espectroscopia de infravermelho
Os espectros de FTIR foram obtidos num espectrómetro Perkin-Elmer Paragon
1000 PC. Foram preparadas pastilhas de KBr com 0.8 mg de amostra e 200 mg de KBr que
foram colocadas a 150 ºC durante a noite, antes da realização dos espectros. Para cada
amostra foram realizados 128 varrimentos na região do espectro de 4000 a 400 cm -1, com
uma resolução de 4 cm-1.
2.4. Estudos de adsorção
2.4.1. Adsorção de azoto
As isotérmicas de adsorção de azoto a 77 K foram determinadas num aparelho
Sorptomatic 1990 da CE Instruments. Antes da determinação da isotérmica todas as
amostras foram desgaseificadas a 150 ºC durante 8 horas. As calibrações foram realizadas
com hélio, a 77 K. Foram utilizados azoto N55 (Air Liquide) e hélio (Linde) com pureza
99.999 %.
2.4.2. Adsorção de n-pentano
6
As isotérmicas de adsorção de n-pentano a 298 K foram obtidas por método
gravimétrico numa instalação de pyrex equipada com uma microbalança CI Electronics e
sensor de pressão Barocel 600AB. Antes da determinação da isotérmica as amostras foram
desgaseificadas a 200 ºC durante 5 horas. O n-pentano (99%, Riedel-de-Haen) foi
previamente purificado através de ciclos sucessivos de solidificação/fusão, em vácuo.
2.4.3. Pré-adsorção de n-nonano seguida de adsorção de azoto
As isotérmicas de adsorção de azoto a 77K foram determinadas numa instalação de
vácuo equipada com um manómetro Edwards Barocel 570 e unidade de controlo
Datametrics 1174. Os “volumes dos espaços mortos” foram calibrados com hélio.
Antes da adsorção de n-nonano as amostras foram desgaseificadas a 200 ºC durante
5 horas e determinada a isotérmica de adsorção de azoto a 77K. A pre-adsorção de n-
nonano foi realizada da seguinte forma: as amostras foram deixadas em contacto com o
vapor de n-nonano durante 1 hora à temperatura ambiente, após o que foram arrefecidas até
77K, temperatura a que permaneceram durante 30 minutos. Seguidamente foram
desgaseificadas a esta temperatura e depois 5 horas à temperatura ambiente. As amostras
foram depois desgaseificadas a 50 ºC e 75 ºC ( no caso de todo o n-nonano não ter sido
removido a 50 ºC). Após cada desgaseificação determinou-se a isotérmica de adsorção de
azoto a 77K.
Foram utilizados azoto C50 e hélio com pureza 99.999 %, ambos da Gasin, e n-
nonano (99 %, Sigma) previamente purificado por ciclos sucessivos de solidificação/fusão,
em vácuo.
3. Resultados e discussão
7

3.1. Caracterização por difracção de raios X
Na figura 3.1 é apresentado o difractograma de raios X da amostra de argila original
(NaSD). Nas figuras 3.2 a 3.4 são apresentados os difractogramas de raios X das amostras
que sofreram tratamento com ácido clorídrico.
Pode-se ver na figura 3.1 que a amostra original, em termos de minerais argilosos,
tem essencialmente esmectite mas também vestígios de outro mineral argiloso,
provavelmente caulinite. Além dos minerais argilosos, estão também presentes outros
minerais. O principal é o dióxido de titânio, na forma de anatase. Verifica-se que o sólido
possui óxido de ferro na forma de hematite. Aparece um pico de intensidade relativamente
fraca que indica a presença de uma pequena quantidade do mineral quartzo na amostra. A
amostra tem também vestígios de feldspatos do supergrupo da plagioclase, os quais podem
ser anortite ou albite. Como os valores de d destas espécies são muito próximos, e os
resultados da análise química mostram que a percentagem de sódio presente na amostra é
idêntica à de cálcio, não é possível distingui-los.
Fig. 3.1 – Difractograma de raios X da amostra original, NaSD (E - esmectite; C – caulinite; A-
anatase; Q – quartzo; F – feldspato; FA – fluorapatite; H – hematite).
8
20 40 60 802
Inte
ns
idad
e /
u.a
.
E
C
E
A
Q
F + E
FA
H
E
H
A
EA
H
E A
H
E
A AE
E

Fig. 3.2 – Difractogramas de raios X das
amostras SD.1M.120.rt e SD.6M.120.rt.
Fig. 3.3 – Difractogramas de raios X das
amostras SD.1M.30.ref, SD.2M.30.ref e
SD.3M.30.ref.
Na figura 3.2 são comparados os difractogramas de amostras tratadas com ácido de
diferente concentração, à temperatura ambiente, com o da amostra não tratada. Os
tratamentos à temperatura ambiente, mesmo com ácido de concentração mais elevada (6M),
não causam alterações estruturais significativas. Como a primeira fase do tratamento ácido
é a substituição dos catiões de compensação, neste caso sódio e cálcio, por protões, os picos
referentes aos feldspatos desaparecem mesmo para o tratamento mais fraco. Os tratamentos
deste tipo também eliminam grande parte da hematite pois os picos correspondentes a esta
espécie diminuem bastante de intensidade sendo a diminuição maior na amostra tratada
com ácido mais concentrado. Nestas amostras o pico de maior intensidade do difractograma
encontra-se ligeiramente deslocado (sendo o deslocamento idêntico em ambas as amostras)
para ângulos mais baixos em relação ao da amostra inicial, o que indica um espaçamento
interlamelar ligeiramente maior. Em ambas as amostras observa-se que o quartzo não é
totalmente removido.
O difractograma da amostra SD.1M.30.ref (figura 3.3) evidencia alguns danos
estruturais, mas pouco significativos, verificando-se que a estrutura da argila mantém-se e
que o pico de maior intensidade permanece aproximadamente no ângulo do das amostras
tratadas à temperatura ambiente. Os picos da hematite desaparecem completamente o que
9
20 40 60 802
Inte
ns
idad
e /
u.a
.
NaSD
SD.1M.120.rt
SD.6M.120.rt
0 20 40 60 802
Inte
ns
idad
e /
u.a
.
NaSD
SD.1M.30.ref
SD.2M.30.ref
SD.3M.30.ref

parece indicar que nesta amostra esta espécie foi completamente eliminada. Por outro lado,
o pico do quartzo desaparece quase por completo mas ainda existe uma quantidade
relativamente pequena deste mineral na amostra. Observa-se ainda que começa a surgir
uma banda larga na região de 2 ~23º que é característica da sílica amorfa e, portanto,
indica que, nestas condições, o tratamento produziu uma quantidade apreciável de sílica
amorfa.
Os tratamentos em refluxo com ácido de concentração superior a 1M têm efeitos
idênticos aos descritos anteriormente para a amostra SD.1M.30.ref, mas gradualmente mais
acentuados com o aumento da concentração do ácido. Assim, na amostra tratada com ácido
2M (figura 3.3) os vestígios de quartzo já desapareceram por completo e o pico mais
intenso do mineral de argila diminuiu bastante de intensidade. Nestas amostras (figuras 3.3
e 3.4) os picos da esmectite vão desaparecendo gradualmente enquanto que a banda
correspondente à sílica amorfa vai-se tornando cada vez mais evidente. Por outro lado, a
anatase é bastante resistente aos tratamentos ácidos continuando os correspondentes picos
bastante evidentes nestas amostras.
Fig. 3.4 – Difractogramas de raios X das amostras SD.4M.30.ref, SD.6M.30.ref e SD.6M.60.ref.
Nas amostras submetidas aos tratamentos mais intensos (figura 3.4), SD.6M.30.ref e
SD.6M.60.ref, basicamente deixam de existir os picos que identificam a esmectite e os
10
0 20 40 60 802
Inte
nsi
dad
e /
u.a
.
NaSD
SD.4M.30.ref
SD.6M.30.ref
SD.6M.60.ref

difractogramas são dominados pela banda da sílica amorfa e picos da anatase. Estes dois
tratamentos provocam a destruição completa do mineral inicial resultando numa mistura de
sílica amorfa e anatase.
3.2. Caracterização por espectroscopia de infravermelho
Na figura 3.5 são apresentados os espectros de FTIR da amostra de argila não
tratada e de amostras submetidas a tratamento ácido em diferentes condições. Os espectros
estão ordenados de baixo para cima por ordem de aumento de intensidade do tratamento, ou
seja, por aumento da temperatura e/ ou aumento da concentração de ácido. Apesar dos
espectros terem sido determinados entre 4000 e 400 cm-1, na figura 3.5 é apenas
apresentada a região do espectro com interesse para avaliar o efeito dos tratamentos na
estrutura.
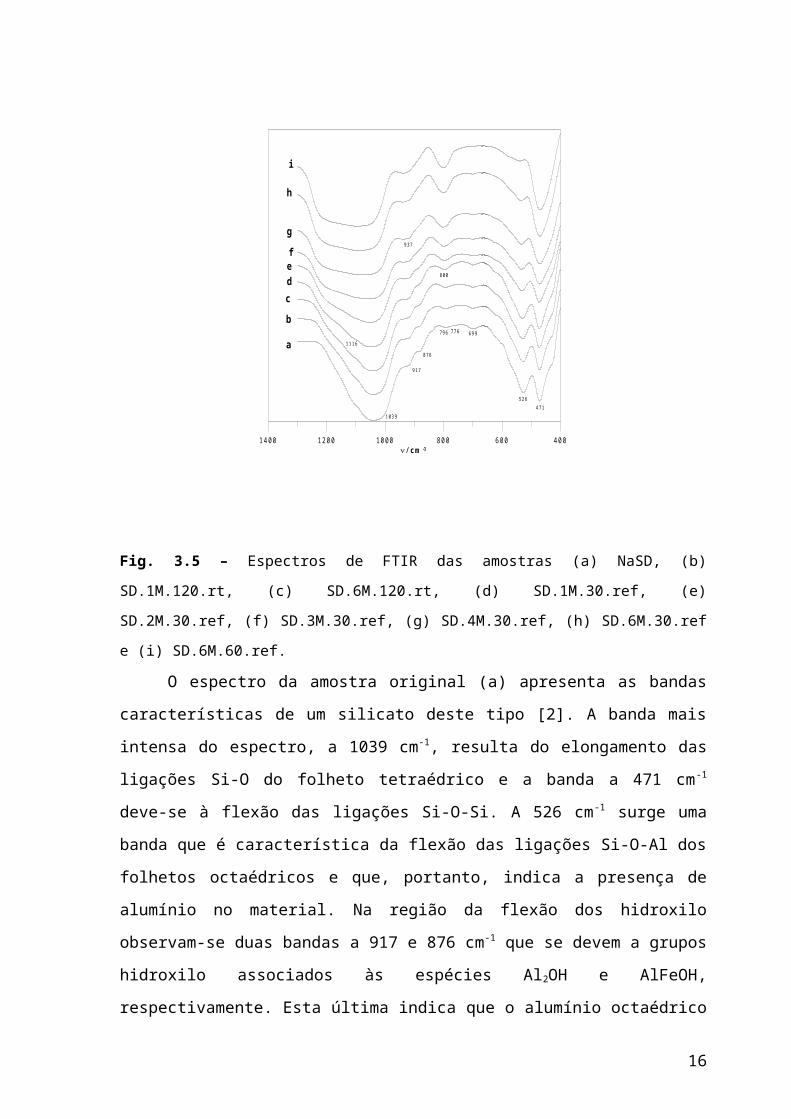
Fig. 3.5 – Espectros de FTIR das amostras (a) NaSD, (b) SD.1M.120.rt, (c) SD.6M.120.rt, (d)
SD.1M.30.ref, (e) SD.2M.30.ref, (f) SD.3M.30.ref, (g) SD.4M.30.ref, (h) SD.6M.30.ref e (i)
SD.6M.60.ref.
O espectro da amostra original (a) apresenta as bandas características de um silicato
deste tipo [2]. A banda mais intensa do espectro, a 1039 cm -1, resulta do elongamento das
11
1400 1200 1000 800 600 400 cm -1
a
b
c
d
ef
g
h
i
471
526
699796
876
917
1039
937
776
800
1116
ligações Si-O do folheto tetraédrico e a banda a 471 cm -1 deve-se à flexão das ligações Si-
O-Si. A 526 cm-1 surge uma banda que é característica da flexão das ligações Si-O-Al dos
folhetos octaédricos e que, portanto, indica a presença de alumínio no material. Na região
da flexão dos hidroxilo observam-se duas bandas a 917 e 876 cm-1 que se devem a grupos
hidroxilo associados às espécies Al2OH e AlFeOH, respectivamente. Esta última indica que
o alumínio octaédrico encontra-se parcialmente substituído por ferro. A 699 cm -1
observa-se uma banda de fraca intensidade que se deve à flexão, para fora do plano, das
ligações Si-O [1]. O dubleto de fraca intensidade a 776 e 796 cm-1 reflecte a presença de
quartzo na amostra, o que também foi detectado por DRX.
Uma análise global dos espectros da figura 3.5 permite dizer que o efeito mais
notório do tratamento ácido no espectro de IV é a alteração da posição e forma da banda
principal do espectro ( ~1039). Com o aumento da intensidade do tratamento ácido esta
banda desloca-se para maiores números de onda e surge uma banda acima de 1100 cm -1,
que se deve à vibração das ligações Si –O em sílica amorfa com estrutura tridimensional.
Outra banda característica da sílica amorfa, a 800 cm-1, surge no espectro da amostra
SD.1M.30.ref e aumenta progressivamente de intensidade. Por outro lado, as bandas
relacionadas com os catiões octaédricos diminuem gradualmente de intensidade, reflectindo
a eliminação destas espécies. Na região da flexão dos hidroxilo as bandas a 917 e 876 cm -1
vão desaparecendo e aparece uma banda a ~937 cm-1 que se deve à flexão de OH ligados a
sílica amorfa.
Verifica-se que os tratamentos mais suaves (à temperatura ambiente) não provocam
alterações significativas no espectro de IV do mineral argiloso, em relação ao da amostra
original. Nos espectros das amostras SD.1M.120.rt (b) e SD.6M.120.rt (c) ocorre apenas
um ligeiro deslocamento da banda de maior intensidade para números de onda maiores,
sendo o deslocamento maior no espectro da última. As bandas associadas aos catiões
octaédricos diminuem ligeiramente de intensidade, sendo mais notória a relacionada com o
ferro. Este resultado sugere que estes catiões foram removidos em pequena quantidade e
que o ferro é eliminado com mais facilidade.
Na amostra SD.1M.30.ref (d) os efeitos do tratamento ácido são mais evidentes, em
particular, há um maior deslocamento da banda principal do espectro para maiores
frequências e surge uma banda a 800 cm-1 que, como já se disse, deve-se a sílica amorfa. O
alargamento da banda de maior intensidade deve-se à sobreposição da banda acima dos
1100 cm-1 com a banda a ~1039 cm-1. Estes resultados reflectem a presença de uma
quantidade significativa de sílica amorfa na amostra. As bandas que identificam os catiões
12

octédricos sofrem uma diminuição mais acentuada em relação ao que foi observado para as
amostras activadas à temperatura ambiente, especialmente a associada ao ferro.
No espectro da amostra SD.2M.30.ref (e) a região da flexão dos hidroxilo é
dominada por uma banda a 937 cm-1, sendo pouco nítidas as bandas associadas ao ferro e
alumínio. Além disso, a banda a 526 cm-1, a mais sensível à presença de alumínio no
folheto octaédrico, encontra-se bastante diminuída. Nos espectros das amostras activadas
em refluxo com ácido de concentração superior as alterações em relação ao espectro da
amostra NaSD são idênticas às referidas para a SD.2M.30.ref, mas sucessivamente mais
pronunciadas. Na região do elongamento das ligações Si-O a banda referente à sílica
amorfa com estrutura tridimensional torna-se predominante e a banda a 800 cm-1 torna-se
cada vez mais intensa. Estes tratamentos provocam danos estruturais bastante acentuados.
Os espectros das duas amostras submetidas a tratamento mais forte, (h) e (i), são
bastante diferentes do da amostra original, evidenciando um elevado grau de alteração
estrutural. Estas amostras são basicamente estruturas porosas tridimensionais de sílica
amorfa, pois os seus espectros são dominados pelas bandas características da sílica e a
banda a 526 cm-1 tem uma intensidade muito baixa ou, no caso da amostra SD.6M.60.ref, é
praticamente inexistente.
3.3. Adsorção de n-pentano
Nas figuras 3.6 a 3.8 são apresentadas as isotérmicas de adsorção e de desadsorção
de n-pentano a 298 K na argila original e em amostras activadas a diferentes temperaturas e
com ácido de diferentes concentrações. Os valores de área superficial e C, obtidos por
aplicação do método BET às isotérmicas, estão registados na tabela 3.1. As áreas foram
calculadas admitindo que a área da molécula de n-pentano na superfície do sólido é 0.45
nm2 [3].
13

Fig. 3.6 – Isotérmicas de adsorção de n-
pentano a 25 ºC nas amostras NaSD,
SD.1M.120.rt e SD.6M.120.rt.
Fig. 3.7 – Isotérmicas de adsorção de n-
pentano a 25 ºC nas amostras
SD.1M.30.ref, SD.2M.30.ref e
SD.3M.30.ref.
Fig. 3.8 – Isotérmicas de adsorção de n-pentano a 25 ºC nas amostras SD.4M.30.ref, SD.5M.30.ref,
SD.6M.30.ref e SD.6M.60.ref.
Observando as figuras 3.6 a 3.8 verifica-se que a forma da isotérmica varia bastante
com a intensidade do tratamento, o que reflecte alterações gradualmente mais profundas na
textura da superfície dos materiais, ou seja, na área superficial e porosidade. Assim, as
14
0 0.2 0.4 0.6 0.8 1p/pº
0
0.4
0.8
1.2
1.6n
ad
s /
mm
ol g
-1
NaSDSD.1M.120.rtSD.6M.120.rt
0 0.2 0.4 0.6 0.8 1p/pº
0
0.5
1
1.5
2
2.5
3
nad
s / m
mo
l g-1
SD.1M.30.refSD.2M.30.refSD.3M.30.ref
0 0.2 0.4 0.6 0.8 1p/pº
0
1
2
3
4
5
nad
s / m
mo
l g-1
SD.4M.30.refSD.5M.30.refSD.6M.30.refSD.6M.60.ref

isotérmicas da figura 3.6 são próximas do tipo IIb da classificação da IUPAC[4], com
ciclos de histerese do tipo H4, os quais estão associados a poros estreitos em forma de
fenda, formados entre partículas lamelares. O aumento da intensidade do tratamento de
activação em relação aos que foram submetidas as amostras cujas isotérmicas se
apresentam na figura 3.6 origina gradualmente, por um lado, um aumento mais acentuado
da quantidade adsorvida na região da multicamada e, por outro lado, a alteração da
curvatura da isotérmica na região inicial de baixas pressões, de tal forma que as isotérmicas
das amostras submetidas a tratamento mais intenso (figura 3.8) estão mais próximas do tipo
IV. Também a forma do ciclo de histerese é alterada, passando a estar próximo do tipo H3.
Através da análise global das isotérmicas de adsorção de n-pentano conclui-se
imediatamente que as amostras de argila, mesmo as menos activadas, não possuem poros
que se comportem como microporos primários em relação às moléculas de hidrocarboneto.
As isotérmicas reflectem dois efeitos do tratamento ácido nas propriedades superficiais dos
sólidos, resultado da delapidação dos folhetos octaédricos. Por um lado, parece haver um
alargamento dos poros de todos os tamanhos e, consequentemente, um aumento da área
superficial. Por outro lado, a interacção entre as moléculas do adsorvato e a superfície do
adsorvente diminui de intensidade, de tal forma que as isotérmicas das amostras sujeitas a
tratamento mais intenso (figura 3.8) têm na parte inicial carácter de tipo III. A forma destas
isotérmicas indica que a interacção entre as moléculas de adsorvato e a superfície do
adsorvente é mais fraca dos que nas restantes amostras, o que está relacionado com a
alteração da composição química da superfície dos materiais. Ambos os efeitos
traduzem-se, quantitativamente, na diminuição, de uma forma geral, do valor de C (tabela
3.1) ao se passar da amostra original para a mais activada. A forma destas isotérmicas
indica que nas correspondentes amostras a interacção entre as moléculas de adsorvato e a
superfície do adsorvente é mais fraca dos que nas restantes amostras.
Como se vê na tabela 3.1, a área superficial, de uma forma geral, aumenta com o aumento
da intensidade do tratamento de activação. Tendo em conta que a interacção adsorvato-
adsorvente é alterada é natural que a área ocupada pela molécula de n-pentano na superfície
do adsorvente também varie, de forma que haverá um erro associado aos valores de ABET
provavelmente crescente com o aumento da intensidade do tratamento. Independentemente
deste facto, os valores evidenciam claramente uma tendência.
Os tratamentos à temperatura ambiente são os que menos alteram a textura
superficial das amostras. As isotérmicas das amostras SD.1M.120.rt e SD.6M.120.rt têm
forma idêntica à da amostra não tratada, mas a quantidade adsorvida é superior.
15
Inicialmente as isotérmicas são coincidentes mas a amostra SD.1M.120.rt adsorve maior
quantidade de pentano na região da multicamada, o que indica que esta amostra possui mais
área externa (que inclui área da superficie externa e paredes dos mesoporos). As áreas das
duas amostras activadas à temperatura ambiente são idênticas e cerca de 20 % superior à da
NaSD. Por outro lado, enquanto que o valor de C da SD.1M.120.rt é menor do que o da
argila inicial, o da SD.6M.120.rt é superior, sendo de facto de todas as amostras estudadas a
que apresenta C mais elevado. Estes resultados sugerem que o ácido mais concentrado
produz poros mais estreitos do que o de concentração 1M.
Tabela 3.1 – Resultados da aplicação do método BET às isotérmicas de adsorção de n-pentano a
298K nas amostras de argila original e tratadas com ácido clorídrico.
Amostra A / m2 g-1 C
NaSD 106 52
SD.1M.120.rt 129 45
SD.6M.120.rt 123 65
SD.1M.30.ref 223 30
SD.2M.30.ref 328 13
SD.3M.30.ref 399 9
SD.4M.30.ref 437 6
SD.5M.30.ref 425 6
SD.6M.30.ref 385 5
SD.6M.60.ref 344 4
Os tratamentos de activação em refluxo, mesmo o mais suave ( com ácido 1M),
alteram de forma significativa a área superficial das amostras. Através das figuras 3.7 e 3.8
verifica-se que nestas amostras o volume total de pentano adsorvido é bastante superior ao
da NaSD. Para concentrações de ácido relativamente baixas (1, 2 e 3 M) o aumento da
16

concentração de ácido conduz a grandes aumentos de área superficial. Ao se passar da
concentração de 3 M para 4 M a área superficial continua a aumentar mas não de forma tão
acentuada. As amostras SD.4M.30.ref e SD.5M.30.ref apresentam áreas idênticas, a desta
última ligeiramente inferior, ou seja, os dois tratamentos parecem ter efeitos semelhantes.
Nas duas amostras submetidas a tratamento mais forte (SD.6M.30.ref e SD.6M.60.ref)
ocorre um decréscimo da área superficial em relação ao valor máximo. Como se viu
anteriormente, estas amostras apresentam danos estruturais de tal forma acentuados que a
estrutura do mineral argiloso deixa de existir. Isto terá levado ao colapso de parte da
estrutura porosa e, consequentemente, à diminuição da área superficial.
3.4. Adsorção de azoto e pré-adsorção de n-nonano seguida de adsorção de
azoto
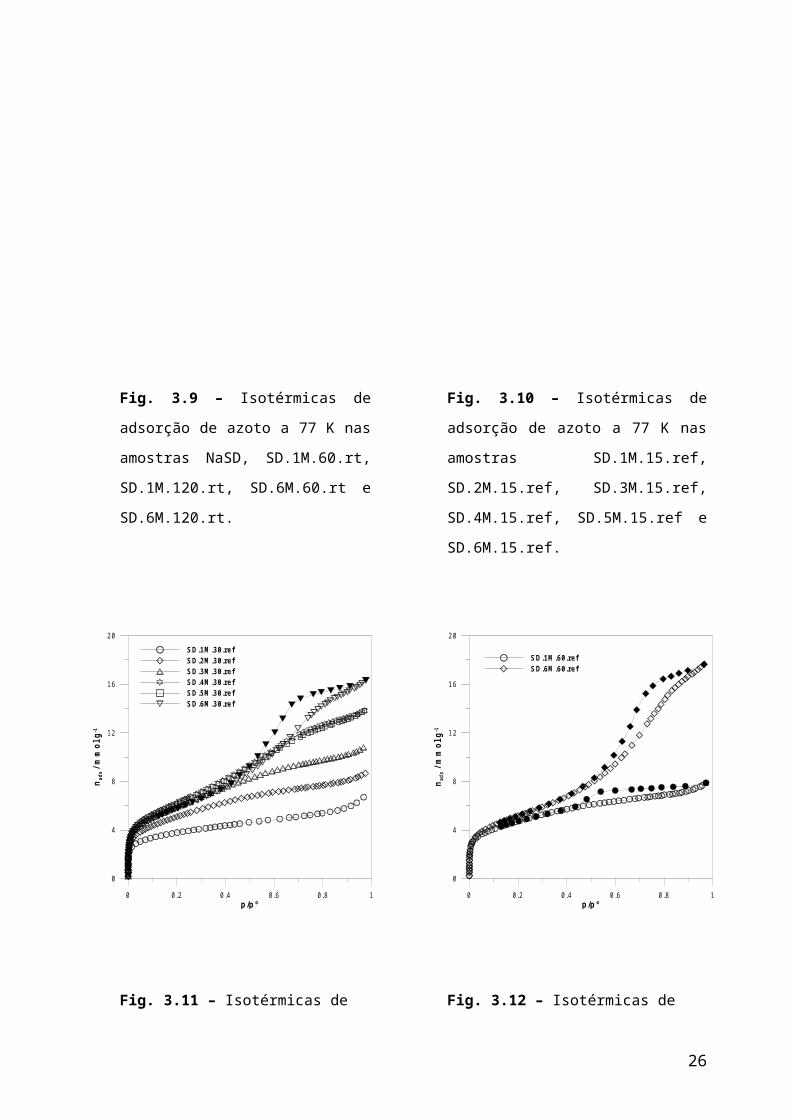
Nas figuras 3.9 a 3.12 são apresentadas as isotérmicas de adsorção de azoto a 77 K
na argila original e em amostras submetidas a diversos tratamentos com ácido clorídrico.
Nas figuras 3.15 a 3.23 estão representadas as isotérmicas de adsorção de azoto a 77 K em
amostras algumas das amostras, antes e após adsorção de n-nonano e desgaseificação a
várias temperaturas. Todas as isotérmicas possuem ciclo de histerese mas por questão de
simplicidade na apresentação dos resultados apenas para algumas amostras são
apresentadas as isotérmicas de desadsorção.
17
0 0.2 0.4 0.6 0.8 1p/pº
0
1
2
3
4
5
nad
s / m
mo
l g-1
NaSDSD.1M.60.rtSD.1M.120.rtSD.6M.60.rtSD.6M.120.rt
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
16
na
ds
/ m
mo
l g-1
SD.1M.15.refSD.2M.15.refSD.3M.15.refSD.4M.15.refSD.5M.15.refSD.6M.15.ref

Fig. 3.9 – Isotérmicas de adsorção de
azoto a 77 K nas amostras NaSD,
SD.1M.60.rt, SD.1M.120.rt, SD.6M.60.rt
e SD.6M.120.rt.
Fig. 3.10 – Isotérmicas de adsorção de
azoto a 77 K nas amostras SD.1M.15.ref,
SD.2M.15.ref, SD.3M.15.ref,
SD.4M.15.ref, SD.5M.15.ref e
SD.6M.15.ref.
Fig. 3.11 – Isotérmicas de adsorção de
azoto a 77 K nas amostras SD.1M.30.ref,
SD.2M.30.ref, SD.3M.30.ref,
SD.4M.30.ref, SD.5M.30.ref e
SD.6M.30.ref.
Fig. 3.12 – Isotérmicas de adsorção de
azoto a 77 K nas amostras SD.1M.60.ref e
SD.2M.60.ref.
A isotérmica de adsorção de azoto a 77 K na amostra original (NaSD) é mista entre
tipo I e tipo IIb da classificação da IUPAC [4]: a baixas p/pº a quantidade adsorvida é
elevada, ou seja, na região inicial a isotérmica tem carácter do tipo I, o que é indicativo de
microporosidade primária; após o preenchimento dos microporos não é atingido um
18
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
16
20
na
ds
/ m
mo
l g-1
SD.1M.30.refSD.2M.30.refSD.3M.30.refSD.4M.30.refSD.5M.30.refSD.6M.30.ref
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
16
20
na
ds /
mm
ol g
-1
SD.1M.60.refSD.6M.60.ref

patamar, mas a quantidade adsorvida aumenta gradualmente na região da multicamada, o
que significa que o sólido possui área externa associada à microporosidade; a isotérmica
apresenta ciclo de histerese do tipo H4 e, portanto, o aumento da quantidade adsorvida na
região da multicamada deve-se também a adsorção na superfície das paredes de mesoporos,
formados entre as partículas lamelares; o facto da isotérmica não ser completamente
vertical na região de baixas pressões mostra que a amostra possui microporosidade
secundária.
As isotérmicas das amostras activadas à temperatura ambiente e também as das
amostras tratadas em refluxo, para ambos os tempos de contacto, com ácido de
concentração relativamente baixa, têm forma semelhante à da NaSD.
De forma idêntica ao que se observou em relação à adsorção de n-pentano, também
a forma das isotérmicas de azoto variam de acordo com a intensidade do tratamento, de tal
forma que as isotérmicas das amostras sujeitas a tratamento ácido mais intenso já se
aproximam do tipo IV da classificação da IUPAC, correspondentes a materiais
mesoporosos.
Na tabela 3.2 estão registados os resultados da aplicação dos métodos BET, s e DR
a todas as isotérmicas de adsorção de azoto a 77 K determinadas. Observa-se que os valores
de VDR são bastante mais elevados do que os volumes de microporos obtidos pelo método
s e que variam de forma oposta, aumentando, de uma forma geral, com o aumento da
temperatura e/ou da concentração do ácido. Esta diferença deve-se ao facto dos volumes
obtidos por aplicação da equação DR contabilizarem a microporosidade mais estreita, a
microporosidade secundária e também mesoporosidade estreita, isto é, área externa,
enquanto que os Vmp obtidos através do método s correspondem apenas a
microporosidade. Desta forma, os valores de VDR não correspondem ao volume de
microporos da amostra e o facto de aumentarem com o aumento da intensidade do
tratamento deve-se à cada vez maior contribuição da área externa, que aumenta neste
sentido. Nas amostras submetidas a tratamento com ácido de concentração 6M a área
superficial diminui, o mesmo acontecendo com o valores de VDR.
As áreas obtidas pelo método s correspondem, no caso das amostras que
apresentam volume de microporos (Vmp) nulo, à área superficial total do sólido, enquanto
que para as restantes amostras corresponderão essencialmente à área da superfície externa e
das paredes dos mesoporos, podendo também incluir a área da superfície de alguma
microporosidade secundária. Nestas últimas amostras, as áreas BET são superiores às
obtidas pelo método s, devido à contribuição da microporosidade.
19

Os valores de C e Eo apresentam uma tendência decrescente com o aumento da
intensidade do tratamento ácido, talvez mais notória nos valores de C, que pode ser
resultado da modificação da composição química da superfície ou de um alargamento
progressivo dos poros ou ainda da conjunção de ambos os efeitos.
A análise global das isotérmicas apresentadas nas figuras 3.9 a 3.12 e dos valores
registados na tabela 3.2 permite afirmar que, como havia sido constatado através dos
estudos de adsorção de n-pentano, a activação à temperatura ambiente não provoca
alterações muito drásticas nas propriedades de superfície da argila, conduzindo a amostras
com volume de microporos superior ao da NaSD e áreas externas ligeiramente superiores.
Por outro lado, os tratamentos em refluxo conduzem a alterações mais significativas na
superfície do sólido. De uma forma geral, ocorre nestas amostras a diminuição do volume
de microporos havendo, por outro lado, um aumento significativo da área superficial. Os
referidos efeitos são, de uma forma geral, tanto maiores quanto maior é a intensidade do
tratamento.
Os resultados indicam que a temperatura é o factor que mais influência tem na
alteração da porosidade e da área superficial da argila, uma vez que mesmo a amostra
tratada em refluxo com o ácido de menor concentração tem uma capacidade de adsorção
bastante maior do que qualquer uma das amostras activadas à temperatura ambiente,
algumas delas tratadas com ácido clorídrico de concentração superior e durante mais
tempo. A utilização de ácido com diferente concentração e de diferentes tempos de
contacto no tratamento à temperatura ambiente não leva a resultados muito distintos pois,
como se vê na figura 3.9, as isotérmicas deste conjunto de amostras estão relativamente
próximas umas das outras. As amostras tratadas com ácido 1M possuem Vmp e As
ligeiramente superiores aos da amostra original, sendo os da amostra que esteve mais
tempo em contacto com a solução ácida ligeiramente superiores. Nas duas amostras
tratadas com ácido mais concentrado, o aumento do Vmp é maior mas a As da amostra
SD.6M.60.rt é idêntica à da SD.1M.60.rt enquanto que a da SD.6M.120.rt é inferior à de
ambas as amostras tratadas com ácido 1M. Estes resultados aparentemente são
concordantes com os da adsorção de n-pentano e, à primeira vista, estes resultados parecem
indicar que não existe uma relação linear entre a concentração do ácido, tempo de contacto
e o Vmp e As das amostras. No entanto, admitindo que a microporosidade é acessível através
de poros mais largos (mesoporos) isto pode ser explicado da seguinte forma: a remoção dos
catiões octaédricos por parte do ácido cria lacunas nas paredes dos mesoporos, algumas das
quais terão profundidade suficiente para se comportarem como microporos em relação às
20

moléculas de azoto, enquanto que outras constituirão apenas rugosidade na superfície das
paredes dos mesoporos, contribuindo assim para o aumento da mesoporosidade (As); o
ácido mais concentrado consegue criar lacunas mais profundas e, portanto, mais
microporosidade do que área das paredes dos mesoporos. O aumento do tempo de contacto
tem apenas como consequência um ligeiro acentuar de cada um destes efeitos.
Enquanto que todas as amostras activadas à temperatura ambiente são mais
microporosas do que a NaSD, nas amostras tratadas em refluxo isto apenas se observa na
SD.1M.15.ref, SD.2M.15.ref e SD.1M.30.ref, não sendo no entanto o aumento do Vmp tão
acentuado. Nas restantes amostras o Vmp é menor do que o da amostra original e diminui
com o aumento da concentração do ácido e tempo de contacto. Nas amostras submetidas a
tratamento mais intenso esta microporosidade não existe. Pelo contrário, a área superficial
aumenta com o aumento da concentração de ácido e tempo de contacto, pelo menos até
determinadas condições. Nas amostras activadas a temperatura mais elevada (refluxo), a
concentração do ácido clorídrico parece ter maior influência na porosidade e área
superficial do que o tempo de contacto pois verifica-se que há uma diminuição do Vmp e
aumento da As significativos quando, para um determinado tempo de contacto, se aumenta
a concentração do ácido, enquanto que, para uma mesma concentração, o aumento do
tempo de contacto conduz a alterações mais suaves. Nas amostras submetidas a tratamento
mais drástico, nomeadamente a SD.6M.30.ref e a SD.6M.60.ref, a tendência de aumento da
área superficial com o aumento da concentração do ácido e/ou tempo de contacto é
alterada, isto é, estas amostras possuem área inferior à da amostra cujo valor é máximo
(SD.5M.30.ref). Esta tendência foi também observada nos estudos de adsorção de n-
pentano e resulta da elevada decomposição estrutural que, provavelmente, provoca o
colapso de parte da estrutura porosa.
Nas figuras 3.13 e 3.14 são apresentadas as isotérmicas de adsorção de azoto a 77 K
e de n-pentano a 298 K nas amostras NaSD e SD.6M.60.ref, respectivamente, representadas
em volume de líquido adsorvido, admitindo que a densidade do adsorvato no interior dos
poros é idêntica à densidade normal do líquido à temperatura a que decorre a adsorção.
Observa-se que as isotérmicas dos dois adsortivos, na mesma amostra, não coincidem e que
têm claramente forma distinta (para as restantes amostras os resultados são idênticos e, por
21

isso, apenas se apresentam, como exemplo, os destas duas amostras). Isto pode ser devido a
alterações da densidade do adsorvato no interior dos poros, estando a densidade do azoto
mais próxima da do líquido, devido a estas moléculas empacotarem de forma mais
eficiente. No entanto, a diferente forma das isotérmicas na região de baixas p/pº na amostra
NaSD sugere que é mais provável a diferença dever-se a um efeito de peneiração
molecular. As moléculas de azoto, de menor dimensão do que as do hidrocarboneto,
conseguem entrar em microporos estreitos, inacessiveis às de n-pentano.
Fig. 3.13 – Isotérmicas de
adsorção/desadsorção de azoto a 77 K e n-
pentano a 298 K, expressas em termos de
volume de líquido adsorvido, na amostra
NaSD.
Fig. 3.14 – Isotérmicas de
adsorção/desadsorção de azoto a 77 K e n-
pentano a 298 K, expressas em termos de
volume de líquido adsorvido, na amostra
SD.6M.60.ref.
No caso da amostra SD.6M.60.ref, a diferença entre a forma das isotérmicas na
região inicial deve-se à interacção entre as moléculas de hidrocarboneto e a superfície do
sólido serem mais fracas do que o que acontece com o azoto. No entanto, o patamar da
isotérmica de adsorção de azoto a p/pº próximas da saturação está ligeiramente acima do da
isotérmica de pentano. Como já se viu, esta amostra não possui microporosidade primária
e, portanto, a existência de um efeito de peneiro molecular parece ser pouco provável . Este
resultado pode ser devido a alterações da densidade do adsorvato no interior dos poros,
sendo a densidade do hidrocarboneto no interior dos poros inferior ao valor do líquido
22
0.0 0.2 0.4 0.6 0.8 1.0p/pº
0.00
0.04
0.08
0.12
0.16
Va
ds /
cm3(l
iq)
g-1
NaSDN 2
n-C 5
0.0 0.2 0.4 0.6 0.8 1.0p/pº
0.0
0.2
0.4
0.6
0.8
Va
ds /
cm3(l
iq)
g-1
SD.6M .60.refN 2
n-C 5

devido a um empacotamento pouco eficiente das moléculas, já tendo sido obtidos
resultados semelhantes em outros materiais [5-9]. Por outro lado, pode dever-se a uma
aumento da densidade do azoto em relação ao valor no líquido. Em estudos realizados em
materiais mesoporosos do tipo MCM-41 [10,11] foi sugerido que a obtenção de volumes
porosos com azoto superiores aos de outros adsortivos se podia dever à sua densidade nos
poros ser superior à do líquido. Assim, os resultados obtidos nesta amostra podem dever-se
a um destes efeitos ou aos efeitos combinados da diminuição da densidade dos
hidrocarbonetos e aumento da densidade do azoto, em relação ao líquido.
Fig. 3.15 – Isotérmicas de adsorção de
azoto a 77K na amostra NaSD, antes e
após adsorção de n-nonano e
desgaseificação a várias temperaturas.
Fig. 3.16 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.1M.120.rt,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
23
0 0.2 0.4 0.6 0.8 1p/pº
0
1
2
3
na
ds /
mm
ol g
-1
NaSD200ºC
Tamb
50 ºC
75 ºC
0 0.2 0.4 0.6 0.8 1p/pº
0
1
2
3
4
nad
s / m
mo
l g-1
SD.1M.120.rt200 ºC
Tamb
50 ºC
75 ºC
0 0.2 0.4 0.6 0.8 1p/pº
0
1
2
3
4
nad
s / m
mo
l g-1
SD.6M.120.rt200 ºC
Tamb
50 ºC
75 ºC
0 0.2 0.4 0.6 0.8 1p/pº
0
2
4
6
na
ds /
mm
ol g
-1
SD.1M.30.ref200 ºC
Tamb
50 ºC
75 ºC

Fig. 3.17 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.6M.120.rt,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
Fig. 3.18 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.1M.30.ref,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
Fig. 3.19 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.2M.30.ref,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
Fig. 3.20 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.3M.30.ref,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
24
0 0.2 0.4 0.6 0.8 1p/pº
0
2
4
6
8
10
na
ds /
mm
ol g
-1
SD.2M.30.ref200 ºC
Tamb
50 ºC
75 ºC
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
na
ds /
mm
ol g
-1
SD.3M.30.ref200 ºC
Tamb
50 ºC
75 ºC
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
16
na
ds /
mm
ol g
-1
SD.4M.30.ref200 ºC
Tamb
50 ºC
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
16
na
ds /
mm
ol g
-1
SD.6M.30.ref200 ºC
Tamb
50 ºC

Fig. 3.21 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.4M.30.ref,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
Fig. 3.22 – Isotérmicas de adsorção de
azoto a 77K na amostra SD.6M.30.ref,
antes e após adsorção de n-nonano e
desgaseificação a várias temperaturas.
Fig. 3.23 - Isotérmicas de adsorção de azoto a 77K na amostra SD.6M.60.ref, antes e após adsorção
de n-nonano e desgaseificação à temperatura ambiente.
Tabela 3.2 – Resultados da aplicação dos métodos s, BET e DR às isotérmicas de adsorção de
azoto a 77K, obtidas nas amostras desgaseificadas a 200 ºC e com n-nonano pré-adsorvido, seguido
de desgaseificação à temperatura ambiente, 50 e 75 ºC.
Amostra Temp
s BET DR
Vmp /
cm3 g-1
A /
m2 g-1
A /
m2 g-1C
VDR /
cm3 g-1
Eo /
kJ mol-1
NaSD 200 0.019 100 145 427 0.059 16.2
tamb 0.000 84 85 112 0.032 12.8
25
0 0.2 0.4 0.6 0.8 1p/pº
0
4
8
12
16
20
na
ds /
mm
ol g
-1
SD.6M.60.ref200 ºC
Tamb
50 0.009 97 117 334 0.048 14.5
75 0.016 97 134 413 0.054 15.8
SD.1M.60.rt 200 0.022 116 168 426 0.070 14.8
SD.6M.60.rt 200 0.033 115 194 485 0.081 15.1
SD.1M.120.rt
200 0.027 123 184 477 0.078 14.3
tamb 0.000 122 122 129 0.050 12.2
50 0.017 120 159 336 0.068 13.4
75 0.023 120 174 384 0.074 13.9
SD.6M.120.rt
200 0.032 103 180 464 0.075 14.9
tamb 0.005 98 111 171 0.046 12.1
50 0.023 102 156 360 0.066 13.9
75 0.030 101 173 403 0.073 14.4
SD.1M.15.ref 200 0.023 223 276 226 0.113 14.5
SD.2M.15.ref 200 0.022 285 336 191 0.133 14.6
SD.3M.15.ref 200 0.012 372 400 149 0.153 14.6
SD.4M.15.ref 200 0.006 430 444 132 0.168 14.4
SD.5M.15.ref 200 0.000 455 455 131 0.174 14.2
SD.6M.15.ref 200 0.000 471 473 124 0.179 14.1
SD.1M.30.ref
200 0.025 247 304 270 0.123 14.5
tamb 0.000 225 226 122 0.086 13.4
50 0.014 247 277 227 0.112 14.0
75 0.022 248 298 224 0.120 14.3
SD.2M.30.ref
200 0.004 390 405 119 0.147 15.8
tamb 0.000 310 312 81 0.117 12.9
50 0.000 379 380 114 0.145 14.1
Tabela 3.2 – Resultados da aplicação dos métodos s, BET e DR às isotérmicas de adsorção de
azoto a 77K, obtidas nas amostras desgaseificadas a 200 ºC e com n-nonano pré-adsorvido, seguido
de desgaseificação à temperatura ambiente, 50 e 75 ºC (cont.).
Amostra Temp
s BET DR
Vmp /
cm3 g-1
A /
m2 g-1
A /
m2 g-1C
VDR /
cm3 g-1
Eo /
kJ mol-1
SD.3M.30.ref 200 0.000 463 465 134 0.180 13.7
tamb 0.000 397 403 68 0.149 13.1
26

50 0.000 442 443 111 0.169 13.6
SD.4M.30.ref200 0.000 494 493 122 0.187 13.9
tamb 0.000 416 431 92 0.164 12.6
SD.5M.30.ref 200 0.000 493 498 113 0.189 14.0
SD.6M.30.ref200 0.000 450 451 121 0.166 14.1
tamb 0.000 411 422 100 0.151 13.7
SD.1M.60.ref 200 0.010 356 379 155 0.147 14.4
SD.6M.60.ref200 0.000 428 425 144 0.158 13.5
tamb 0.000 420 410 141 0.157 13.9
Os estudos de pré-adsorção de n-nonano seguida de adsorção de azoto permitem
obter uma imagem mais detalhada acerca da porosidade das amostras de argila e de como
esta é afectada pelos tratamentos de activação. O método baseia-se na ideia de que, após
contacto do n-nonano com o adsorvente e desgaseificação deste à temperatura ambiente, as
moléculas do hidrocarboneto ficam retidas em poros onde a interacção entre adsorvato e
adsorvente é suficientemente forte, em princípio, nos microporos ou, no máximo, em
mesoporos estreitos. Para remover as moléculas do interior destes poros é necessário
desgaseificar a temperaturas superiores à temperatura ambiente. À medida que a
temperatura de desgaseificação aumenta vão sendo removidas moléculas de hidrocarboneto
de poros cada vez mais estreitos e a uma determinada temperatura é eliminado n-nonano de
poros com uma determinada dimensão. Isto é comprovado pelo aumento do valor de C e Eo
à medida que a temperatura de desgaseificação aumenta, o que indica que porosidade mais
estreita vai-se tornando acessível ao azoto. Nas amostras estudadas verifica-se que as áreas
obtidas por aplicação do método s às isotérmicas de adsorção de azoto determinadas antes
e após adsorção de nonano, das amostras com microporosidade apreciável, são idênticas, o
que significa que as moléculas de hidrocarboneto são removidas da superfície externa e dos
mesoporos durante a desgaseificação à temperatura ambiente. Estudos idênticos em zeólitos
BEA e em sílica não porosa [12] indicaram que as moléculas de n-nonano são removidas da
superfície externa e dos mesoporos durante a desgaseificação à temperatura ambiente.
Além disso, as áreas obtidas por aplicação do método s às isotérmicas após
desgaseificação à temperatura ambiente são em todos os casos idênticas às obtidas pelo
método BET, excepto para a amostra SD.6M.120.rt, o que significa que toda a
27

microporosidade está ocupada ou bloqueada por n-nonano, permanecendo apenas a
superfície externa e a mesoporosidade disponível para adsorção de azoto.
Verifica-se que, para todas as amostras em que este estudo foi realizado, excepto
para a SD.6M.120.rt, o Vmp obtido após adsorção de n-nonano e desgaseificação à
temperatura ambiente é nulo. Como o diâmetro das moléculas de n-nonano é idêntico ao
das moléculas de pentano, e se concluiu anteriormente que estas últimas não conseguem
aceder à microporosidade mais estreita, é legítimo assumir que o mesmo acontece com as
moléculas de n-nonano. Desta forma, o facto dos microporos primários não estarem
disponíveis para adsorção de azoto após desgaseificação à temperatura ambiente sugere que
estes estão acessíveis através de poros mais largos onde as moléculas de n-nonano estão
adsorvidas, bloqueando a “passagem” às moléculas de azoto.
Tendo em conta o raciocínio anterior, o facto da amostra SD.6M.120.rt apresentar
alguma microporosidade estreita após desgaseificação à temperatura ambiente poderá
significar que foram criados microporos directamente abertos para a superfície externa ou
para a superfície de mesoporos largos e onde as moléculas de nonano não entram, o que
parece confirmar a hipótese colocada anteriormente para explicar as alterações da
porosidade provocadas pelo tratamento à temperatura ambiente.
De forma a facilitar a compreensão dos resultados do estudo de pré-adsorção de
nonano seguida de adsorção de azoto apresenta-se na figura 3.24 a percentagem do volume
total disponível para adsorção de azoto após desgaseificação às várias temperaturas, para
cada uma das amostras.
28

0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
NaSD
SD.1M
.120
.rt
SD.6M
.120
.rt
SD.1M
.30.
ref
SD.2M
.30.
ref
SD.3M
.30.
ref
SD.4M
.30.
ref
SD.6M
.30.
ref
SD.6M
.60.
ref
% V
tota
l ace
ssív
el a
o N
2
Fig. 3.24 – Percentagem de volume, das amostras estudadas por pré-adsorção de nonano, acessível
às moléculas de azoto após aquecimento a várias temperaturas (azul – após desgaseificação à
temperatura ambiente; rosa – após aquecimento a 50 ºC; verde – após aquecimento a 75 ºC;
cinzento – após aquecimento acima de 75 ºC).
Como se disse anteriormente, as moléculas de n-nonano são imediatamente
removidas da superfície externa e paredes dos mesoporos aquando da desgaseificação à
temperatura ambiente. Assim, a região azul do gráfico corresponderá à percentagem de
quantidade de azoto adsorvido na superfície externa e paredes dos mesoporos e, portanto,
directamente relacionada com a quantidade deste tipo de superfície na amostra. As regiões
rosa, verde e cinzenta corresponderão a microporosidade primária e secundária. Nas
amostras que possuem microporos primários é impossível distinguir os dois tipos de
microporosidade dentro de cada uma das regiões e, provavelmente, todas elas terão uma
determinada proporção dos dois tipos de microporos uma vez que, como se assumiu
anteriormente, a remoção de moléculas de n-nonano de poros mais largos desbloqueará um
determinado número de microporos primários e o Vmp deixa de ser nulo imediatamente
após aquecimento a 50 ºC. Estas regiões estarão associadas, no entanto, a microporos
secundários de diferente dimensão. Assim, a desgaseificação a 50 ºC remove as moléculas
29

de hidrocarboneto dos microporos secundários mais largos que, por sua vez, desbloqueiam
alguns microporos primários. Nas amostras que apresentam Vmp nulo as porções destas
cores corresponderão apenas a microporos secundários de diferentes dimensões.
Por observação da figura 3.24 é claramente evidente que, de uma forma geral, a
microporosidade vai desaparecendo gradualmente com o aumento da intensidade do
tratamento, no sentido do aumento da área superficial, o que, mais uma vez, sugere que os
poros vão sofrendo um alargamento progressivo.
De acordo com a figura 3.24, a amostra NaSD possui além dos microporos
primários, parece possuir três tamanhos de microporos secundários. A maior parte da
microporosidade bloqueada e ocupada pelas moléculas de n-nonano fica disponível após
desgaseificação da amostra a 50 ºC e, portanto, tratar-se-á dos supermicroporos mais largos
e alguma microporosidade primária que ficou desbloqueada. Alguns microporos
secundários de dimensão intermédia são desgaseificados a 75 ºC desbloqueando mais
alguma microporosidade primária. As moléculas de nonano que são removidas por
aquecimento acima dos 75 ºC encontrar-se-ão adsorvidas em microporos secundários
bastante estreitos com dimensão, provavelmente, já próxima da dos microporos primários.
De notar que para esta amostra o valor de As após desgaseificação à temperatura ambiente é
inferior aos restantes valores. Estes resultados sugerem que, após desgaseificação à
temperatura ambiente, o nonano fica também a bloquear alguma mesoporosidade estreita e
que esta fica livre para adsorção de azoto após aquecimento a 50 ºC. Desta forma, a parte
azul do gráfico correspondente a esta amostra deveria ser maior. Nas amostras que
apresentam Vmp nulo ou próximo de zero as áreas são distintas entre si pois estas áreas são
as áreas totais dos sólidos e a sua diminuição reflecte o bloqueamento de microporos
secundários ou também de alguma mesoporosidade estreita.
Como já foi dito, o tratamento com ácido cloridríco a temperatura baixa aumenta o
Vmp das amostras em relação ao da NaSD, sendo esse aumento maior na amostra tratada
com a solução de ácido mais concentrada. À primeira vista a figura 3.24 não parece indicar
isso, principalmente em relação à amostra SD.1M.120.rt, mas é necessário ter em conta que
na amostra NaSD alguma mesoporosidade estreita ficou também bloqueada pelas
moléculas de hidrocarboneto. De qualquer forma, é evidente que o tratamento a esta
temperatura mantém a porosidade das amostras semelhante à da amostra inicial, sendo
necessários pelo menos três aquecimentos para remover o nonano de todos os poros.
A microporosidade mais estreita (desgaseificada acima de 75 ºC) é quase totalmente
eliminada no tratamento em refluxo mais suave (1M) e nos tratamentos mais intensos não
30

existe. Esta amostra apresenta um Vmp ainda superior ao da NaSD, mas inferior aos das
SD.1M.120.rt e SD.6M.120.rt que, portanto, corresponderá a microporosidade
relativamente larga, uma vez que as regiões verde e rosa aparentemente não sofreram
alterações significativas.
A amostra SD.2M.30.ref possui um Vmp muito reduzido e através do gráfico
apresenta apenas dois tamanhos de microporos que serão essencialmente microporos
secundários. O mesmo se observa para a amostra SD.3M.30.ref que, por sua vez, apresenta
menor quantidade de microporos secundários.
Os resultados das amostras activadas em refluxo evidenciam claramente a tendência
de desaparecimento dos poros mais estreitos com o aumento da concentração do ácido que,
em conjunto, com a tendência do aumento de área superficial neste sentido, indica que o
desaparecimento dos poros mais estreitos se deve à sua transformação em poros cada vez
mais largos devido a alargamento. Assim, as amostras SD.4M.30.ref e SD.6M.30.ref são já
essencialmente mesoporosas contendo ainda uma pequena quantidade de microporos
secundários largos, enquanto que a amostra SD.6M.60.ref é uma sílica mesoporosa.
31

4. Referências
1. Breen, C., Zahoor F. D., Madejová, J., Komadel, P., J. Phys. Chem. B, 101(1997)5324.
2. Madejová, J., Bujdák, J., Janek, M., Komadel, P., Spectrochimica acta A, 54(1998)1397.
3. T. Paryjczak, Gas Chromatography in Adsorption and Catalysis, Ellis Horwood, 1986.
4. F. Rouquerol, J. Rouquerol, K. S. W. Sing, Adsorption by Powders & Porous Solids,
Academic Press, London, 1999.
5. M. M. L. Ribeiro Carrott, Evolução das propriedades de superfície e transformações
estruturais envolvidas na decomposição térmica do hidróxido de magnésio, Tese de
Doutoramento, Universidade de Lisboa, 1990.
6. M. M. L. Ribeiro Carrott, P. J. M. Carrott, M. Brotas de Carvalho, K. S. W. Sing, J.
Chem. Soc. Faraday Trans., 87 (1991) 185.
7. P. J. M. Carrott, J. J. Freeman, Carbon, 29 (1991) 499.
8. P. J. M. Carrott, M. M. L. Ribeiro Carrott, I. P. P. Cansado, Stud. Surf. Sci. Catalysis,
128 (2000) 323.
9. M. M. L. Ribeiro Carrott, P. A. Russo, C. Carvalhal, P. J. M. Carrott, J. P. Marques, J.
M. Lopes, I. Gener, M. Guisnet, F. Ramôa Ribeiro, Micropor. Mesopor. Mater., in press.
10. M. M. L. Ribeiro Carrott, A. J. Candeias, P. J. M. Carrott, P. C. Ravikovitch, A. V.
Neimark, A. D. Sequeira, Micropor. Mesopor. Mater., 47 (2001) 323.
11. M. M. L. Ribeiro Carrott, P. J. M. Carrott, A. J. Candeias, K. K. Unger, K. S. W. Sing,
em Fundamentals of Adsorption 6 (F. Meunier, Ed.), Elsevier, Amesterdam, 1998, p. 69.
12. P. A. Russo, Avaliação da porosidade de zeólitos BEA desaluminados, Relatório de
estágio, Universidade de Évora, 2003.
32





























