Embed Size (px)
Citation preview

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
1
Relatório de Resultados e Desdobramentos do
9º Encontro Nacional de Inovação em Fármacos e Medicamentos
9º ENIFarMed
Coordenação geral, compilação, e revisão ortográfica: Mariana Sandroni Coordenação de programa: Carlos Tagliati
Em setembro de 2015
Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
2
SUMÁRIO
Item N. Pág.
Agradecimentos 3
1) Resumo 4
2) Introdução 5
3) Resultados 6
3.1) 1ª Plenária “Políticas de fomento, uso do poder de compras, e encomendas: perspectivas diante do cenário atual”
6
3.2) 2a Plenária “Internacionalização dos laboratórios farmacêuticos nacionais: acesso a novos medicamentos”
8
3.3) 3ª Plenária Internacional “Cenário Global da inovação farmacêutica: enfoque no Brasil”
10
3.4) 4ª Plenária “Regulação do registro de inovações e de preços” 12
3.5) Sessão Temática 1 “Ações para a produção de insumos da biodiversidade brasileira”
14
3.6) Sessão Temática 2 “Modelos de gestão para acelerar a inovação e a integração de políticas públicas para doenças que afetam populações negligenciadas”
16
3.7) Sessão Temática 3 “Biotecnologia e a intercambialidade para biossimilares” 19
3.8) Sessão Temática 4 “Pré-clínicos: custo ou investimento? Quais as melhores práticas e alternativas/”
21
3.9) Sessão Temática 5 “Criando os elos fundamentais da cadeia de fitoterápicos: extratos e marcadores”
23
3.10) Sessão Temática 6 “Premiação Reconhecimento Técnico” 25
3.11) Sessão Temática 7 “Pesquisa Clínmica: como alavancar esta importante fase do desenvolvimento de medicamentos?”
26
3.12) Sessão Temática 8 “Eficácia e segurança de produtos contendo nanotecnologia no setor cosmético e no farmacêutico”
29
3.13) Sessão Temática 9 “Prospecção em patentes: cenário e perspectivas em câncer”
33
4) Conclusões 36
5) Desdobramentos 39
Programa realizado do 9º ENIFarMed 41

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
3
AGRADECIMENTOS:
Agradeço a todos os participantes do 9º ENIFarMed pelo amplo debate sobre temas de suma importância para a inovação no setor saúde e para a competitividade do Brasil e pela validação de propostas e sugestões debatidas durante o Encontro, destacadas nos itens Conclusões e Desdobramentos deste relatório. Também agradeço à equipe da Sociedade Brasileira Pró-Inovação Tecnológica (Protec) e do Instituto de Pesquisa e Desenvolvimento em Fármacos e Produtos Farmacêuticos (IPD-Farma) pelo apoio na divulgação e realização do evento. Aos parceiros, agradeço e peço manutenção do relacionamento institucional. Cabe um agradecimento especial aos patrocinadores e apoiadores do 9º ENIFarMed, além dos associados do IPD-Farma, pois sem eles nada seria possível. Para finalizar, tenho a honra de destacar e agradecer profundamente a excelente qualidade das relatorias técnicas entregues pelos profissionais listados abaixo, por ordem alfabética, material esse que deu base e viabilizou a elaboração do presente relatório.
Ana Claudia Dias, Abifina
Caio Victor França, Biozeus
Carlos Martins, Interface CTI
Carlos Vitor, Aché
Carolina Reis, CellSeq
Dyeison Antonow, FM/PUCRS
Eliziane Patricio, UNIPLAC
Fernando Marcussi, Alanac
Heloisio Rodrigues, PHASE Pharma
Henrique Menezes, UFPB
João Carlos Gabaglia, Agência inovação UFRJ
Luiz Villarinho, ENSP/Fiocruz
Marco Torronteguy, MMK Advogados
Patrícia Teixeira, IE/UFRJ
Pedro Ohara, Avanti
Sylvia Loloma, ThomsonReuters
Vera Lucia Luiza, ENSP/Fiocruz
Yuri Tukoff, IPT-SP

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
4
1) Resumo O acúmulo de competências é o principal resultado de uma política industrial de sucesso, que
resulta de um conjunto de ações coordenadas entre entes públicos e privados com o objetivo de aumentar competitividade. Ainda verificam-se gargalos antigos, como a necessidade de fomentar a capacitação em áreas de interesse como fitoterápicos, biossimilares, testes clínicos e pré-clínicos, e a integração da academia com a indústria para gerar produtos concretamente. Embora as perspectivas no cenário atual sejam de contingenciamento orçamentário e de restrição do espaço fiscal no curto prazo, as tendências da política industrial devem priorizar o efeito sistêmico, a inovação da produção e a competitividade.
A inovação farmacêutica no contexto internacional está fortemente orientada para o desenvolvimento de novos medicamentos, desde suas primeiras etapas, enquanto que para uma grande parcela dos pesquisadores brasileiros a inovação se caracteriza na realização de ensaios clínicos de produtos que já estão em estágio avançado de desenvolvimento em outros países. Por outro lado, o Brasil apresenta atividades de inovação e um processo inicial de internacionalização pela indústria farmacêutica brasileira, que ainda carece de foco, disciplina e ritmo. Tomando-se em consideração que a inovação se dá a nível local, políticas de Estado são fundamentais para promover a criação de clusters de inovação em algumas regiões brasileiras.
O monitoramento do mercado é uma alternativa à regulação e ao tabelamento de preços de medicamentos, mas não há previsão quanto às conclusões desse debate no âmbito do governo.
Os maiores gargalos para a fitoterapia têm três principais frentes: (1) Farmacotécnico, (2) Governamental, e (3) Clínico. Pelo lado técnico, é necessário trabalhar a padronização dos medicamentos fitoterápicos pois a identificação química já está bem estabelecida mas a linha clínica ainda é limitada e o médico ainda não acredita na fitoterapia. Outro ponto importante é definir a diferença entre extratos padronizados e extratos quantificados. No que tange o acesso ao patrimônio genético e à repartição de benefícios no uso da biodiversidade brasileira, o grande desafio que estamos vivenciando atualmente é a construção de uma agenda para definição dos termos do Decreto Presidencial que regulamentará a Lei.
Os estudos de biossimilares são complexos, longos e de elevado custo. Portanto, o processo de adoção de novos métodos dependerá da harmonização de práticas entre diferentes entidades internacionais. O mesmo acontece para os ensaios pré-clínicos, pois há falta de certificação das instituições nacionais, o que leva as empresas nacionais a repetir ensaios em ambientes certificados, fora do Brasil. Há falta de: formação de desenvolvedores de novas pesquisas pré-clínicas, incentivo, padronização da execução dos projetos pré-clínicos, além de uma forte alavancagem dos projetos de pesquisa clínica e pré-clínica de desenvolvimento nacional.
Com relação à pesquisa clínica, foi apontada a necessidade de um desenvolvimento e padronização das análises feitas pela ANVISA, redução progressiva dos tempos de análise dos estudos, verificação e acompanhamento do cumprimento dos prazos estipulados pelas RDCs # 9 e 10 e, acima de tudo, a continuidade do processo de formação de técnicos capacitados para analise das submissões recebidas.
Os setores com maior potencial em nanotecnologia para o Brasil são os de produtos farmacêuticos, químicos e cosméticos, além do setor de energia. O foco dos pesquisadores e da indústria deve estar sempre voltado em como a dimensão da partícula pode influenciar a segurança e a eficácia de fármacos, cosméticos e demais produtos fabricados em tais dimensões pois quanto menor a partícula, melhor é a sua absorção pelo organismo. Assim, as vantagens de se usar nanotecnologia na área farmacêutica são: levar a substância ativa ao local de ação; diminuir a competição com tecidos não específicos; aumentar a

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
5
especificidade de ação; aumentar a penetração celular do fármaco; reduzir efeitos adversos; aumentar a estabilidade química da substância ativa. Por outro lado, os principais problemas dos fármacos atuais consistem em sua fraca solubilidade, na remoção do ativo do corpo antes de sua efetiva ação no tratamento de moléstias, baixa efetividade e crescente toxicidade, ação em locais do corpo que não precisam de tratamento, o alvo de tratamento é muitas vezes bloqueado por barreiras biológicas do organismo e resistência do corpo contra a ação do ativo.
O grande problema de um país burocrático ocorre quando a atividade meio se torna mais importante do que a atividade fim. Portanto, nos falta um ponto focal que orquestre a questão sanitária e a questão do Instituto Nacional da Propriedade Industrial (INPI), com sua falta de estrutura e seu enorme backlog, pois a demora no exame dos pedidos gera uma insegurança jurídica muito grande. É preciso atentar para o fato de que a empresa processa o examinador, e não processa o INPI, o que gera uma enorme pressão nos examinadores.
O presente relatório traz nos itens Conclusões e Desdobramentos as principais propostas relativas aos temas abordados, e seus respectivos interlocutores.
2) Introdução A cada edição, o ENIFarMed traz propostas novas, as quais são destacadas nos Relatórios de Resultados
e Desdobramentos emitidos desde a quarta edição, em 2010. O 8º ENIFarMed mostrou que a trajetória da inovação no Brasil é recente e tem na saúde um grande potencial de desenvolvimento. Assim, a saúde deve ser tratada como um sistema produtivo e, neste ponto, é preciso pensar em reconstruí-lo trazendo a indústria, serviços e produtos para debater de forma conjunta como podemos capitalizar a cadeia de valor e amenizar gargalos como (1) Regulação; (2) Preço; (3) Pesquisa clínica; e (4) Carga tributária. Foi demonstrado que as empresas nacionais precisam utilizar estratégias de diferenciação sustentáveis, catch-
up em biotecnologia bem sucedida, e inserções internacionais (que será tema da 2ª Plenária de hoje), junto a uma consolidação de estruturas de governança e a uma articulação virtuosa entre a necessidade de saúde e a política industrial (tema da 1ª Plenária de hoje). O debate e as propostas sobre estes e demais temas hiper relevantes, junto à integração e à sinergia promovidas pelo Encontro, são a base do sucesso do ENIFarMed, Encontro que aproxima anualmente o governo com a indústria e a academia em torno de questões vitais para o Complexo Industrial da Saúde. Nesta nona edição, foram mantidas a ExpoFarMed e a Sessão de Pôsteres, que evidencia os trabalhos mais inovadores, com foco no mercado e relevância social através da Premiação Reconhecimento Técnico e reúne projetos que, a cada edição, apresentam mais qualidade e maturidade. Em 2014, o ENIFarMed recebeu um representante da Câmara de Deputados para discutir questões relativas ao uso da biodiversidade brasileira. Foi um passo muito importante. Em 2015, na Sessão Temática sobre Biodiversidade, o ENIFarMed recebeu o Deputado Alceu Moreira para dar continuidade ao debate sobre o tema.
O 9º ENIFarMed é uma realização do Instituto de Pesquisa e Desenvolvimento em Fármacos e Produtos Farmacêuticos - IPD-Farma e da Sociedade Brasileira Pró-Inovação Tecnológica – Protec. O Encontro foi viabilizado por instituições e empresas parceiras há bastante tempo. Nesta nona edição, o ENIFarMed recebeu o oferecimento por parte do BNDES, e foi patrocinado pelas empresas associadas ao IPD-Farma Biolab e Cristália, além do GrupoFarmaBrasil e do Sebrae. Como apoiadores, o 9º ENIFarMed contou com as associadas Blanver e Nortec Química, além da Capes, da Interfarma, e da Universidade Federal de Minas Gerais (UFMG). O evento também recebeu o apoio institucional da Abafarma, Abifina, Abifisa, Aintec, Abrifar, Axonal, Biominas, Biotec AHG, Cienp, CMQV, CRF-SC, Febrafar, Grupo Midia, IATS, INCT-

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
6
Inofar, IFRJ, INT, IVB, NUPEEC, Nanobusiness, PróGenéricos, RETS, SBMF, SBBiotec, Sindifar, Sinfacope, Tecnoparq, UNESP, UEZO, e UNICAMP. O evento reuniu 225 pessoas e teve a Sessão de Abertura presidida por Dante Alario Junior, presidente do Conselho Deliberativo do IPD-Farma, CEO da Biolab e um dos nossos grandes parceiros na luta pela promoção da cultura da inovação. Em seguida, o diretor-presidente da Protec e do IPD-Farma, Roberto Nicolsky, proferiu a Palestra de Abertura que evidenciou os pontos vitais da “Agenda de crescimento para o Brasil”.
3) RESULTADOS 3.1) 1ª Plenária - Políticas de fomento, uso do poder de compras, e encomendas: perspectivas diante do cenário econômico atual COMPOSIÇÃO DA MESA - Moderador: Lelio Maçaira, CEO da Laborvida; Palestra Magna, Reginaldo Arcuri, presidente do Grupo FarmaBrasil. Debatedores: Pedro Palmeira, chefe do Departamento Defarma do BNDES; Marcus Simões o Coordenador-Geral das Indústrias Químicas e de Transformados Plásticos da Secretaria de Desenvolvimento da Produção (SDP/MDIC); Sergio Frangioni, Diretor-presidente da Blanver. Plateia convidada: Jamaira Giora, consultora. Relatores: Heloisio Rodrigues, CEO da PHASE Pharma e Patrícia Teixeira, do Instituto de Economia da UFRJ.
Apesar de haver resultados ruins na indústria de transformação como um todo, os indicadores da
indústria farmacêutica são positivos. Enquanto o PIB, o emprego e a massa salarial vem caindo de um mês para outro, os indicadores da indústria farmacêutica vem subindo, tanto em volume quanto em valor faturado. Na variação mensal junho/maio do índice de produção por setor industrial enquanto a indústria de transformação apresenta 0.1%, praticamente estagnado, as farmoquímicas e farmacêuticas cresceram 2.4%, o que evidencia o fato de que o setor produtivo farmacêutico é muito importante para o equilíbrio do Brasil, e gera benefícios como a geração de empregos qualificados.
A criação da política das PDPs – Parceria do Desenvolvimento Produtivo – tem como um de seus objetivos racionalizar o poder de compra do Estado, mediante a centralização seletiva dos gastos na área da saúde para reduzir custos de aquisição do SUS e para fomentar a produção no país e promover o desenvolvimento tecnológico. Estima-se que as PDPs geraram uma economia de R$ 2,5 bilhões nos últimos 5 anos. O sucesso das PDPs está pautado em três elementos: racionalização, parceiros adequados e escolhas corretas. Das 98 PDPs assinadas, apenas 27 foram efetivadas, o que merece reflexão e, eventualmente, ações corretivas. E, para o monitoramento contínuo que assegure a viabilidade e perenidade dessa política, sugere-se a criação de um Grupo de Trabalho (GT) com especialistas para realizar essas avaliações. O uso do poder de compras pelo governo como fator de fomento do complexo industrial da saúde apresenta perspectivas positivas e otimistas que passam por escolhas corretas, implementação de novo marco regulatório, instituição da defesa oral para aprovação dos projetos e criação de comitês técnicos interministeriais. Essas novas disposições devem gerar um novo modelo para as novas PDPs, especialmente para produtos biológicos, permitindo, por exemplo, que empresas menores com tecnologias mais avançadas possam participar. Cabe lembrar o setor de equipamentos médicos hospitalares, onde é preciso investimentos. Foi sugerida a encomenda programada para as PDPs, com planejamento do orçamento e ampliação do acesso aos medicamentos. Temos como exemplo o programa Farmácia Popular, que trabalha com a compra centralizada, fortalecendo o poder de compra governamental.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
7
Foi destacado que o Brasil, quando “quis”, tornou-se competitivo no agronegócio e na aviação civil pois existiram condições necessárias para o sucesso de políticas públicas de qualidade e de longa duração, articulação entre produção de conhecimento científico e produção industrial e a existência de empresariado nacional inovador nos segmentos citados. No caso do agronegócio, houve crédito abundante, política de preços, seguro, políticas específicas para produtos e insumos e, na área da articulação do conhecimento, a criação da Embrapa foi fundamental. Na aviação, os pontos principais foram a decisão do Estado de criar uma indústria aeronáutica de nível mundial, política de compras e decisão de privatização, programa de apoio do BNDES para consolidação da empresa e financiamento das vendas e o projeto AMX de transição para tecnologia de jato. A articulação do conhecimento científico foi marcado pela criação e capacitação do Instituto Tecnológico de Aeronáutica (ITA), incentivo a pesquisa e formação de recursos humanos de excelência. Hoje a Embraer é a 3a maior empresa aeronáutica civil do mundo. E é por isso que verifica-se uma excelente janela de oportunidade para a indústria farmacêutica, que já apresenta aumento físico de produção da ordem de 7.18% e crescimento médio faturamento de cerca de 14% ao ano. Os farmoquímicos não comprometem tanto a balança comercial mas os medicamentos sim, sendo que só os biológicos representam 35.4% do total importado. Outro fator importante são os investimentos em empresas de biotecnologia como Bionovis, Orygen, Cristália, Libbs, e outras, que já totalizam R$1.4 bilhão.
A articulação intersetorial está sendo feita em poder de compra, acesso a medicamentos de alto custo, produção, regulação e inovação. Só o avanço no financiamento da assistência farmacêutica cresceu 70% no período 2011-2015, de R$8.4 bilhões para R$14 bilhões. Do mesmo modo, as compras centralizadas já geraram economia de R$1.3 bilhões no período 2010-2014. Tudo isso pode ser representado pela assinatura de 98 acordos de PDPs entre 19 laboratórios públicos e 55 laboratórios privados abrangendo 91 produtos que representam 12% dos medicamentos comprados pelo Ministério da Saúde (MS) e representam 61% dos gastos. Outro ponto a ser destacado são as cooperações interministeriais como o Inova Saúde, entre MS, BNDES, FINEP, representando investimento total aprovado de R$7 bilhões com contrapartida de R$2 bilhões.
Se, por um lado, o apoio da Anvisa é importante como fator de desenvolvimento da indústria farmacêutica tanto em vigilância sanitária quanto no desenvolvimento da indústria farmacêutica nacional, pois a experiência da Anvisa tem impacto direto nos negócios da empresa e na ampliação do acesso da população a medicamentos mais baratos, seguros e eficazes, por outro lado a política nacional é um meio para chegar ao desenvolvimento em longo prazo. Sociedade e governo devem se conscientizar sobre o que é ser competitivo, ter preços para negociação, como lidar com o mercado mundial e como preparar-se para a nova onda da internacionalização. Neste sentido, a política nacional deverá ser alicerçada em três pontos: Fomento, Poder de compra e Inovação. A política de fomento pode ser representada pela nova política industrial de promover adensamento da cadeia das indústrias farmoquímica e farmacêutica para minimizar as perdas para importações, incrementar a participação das indústrias farmoquímica e farmacêutica nas cadeias de produção de tecnologia, incrementar a internacionalização, incentivar a inovação incremental e radical por via do ajuste de preços pela CMED, consolidar as PDPs alinhadas ao novo marco regulatório, e a integração da ANVISA com o INPI como entidades indutoras de inovação.
Ainda verificam-se gargalos antigos, como a necessidade de fomentar a capacitação em áreas de interesse como fitoterápicos, biosimilares, testes clínicos e pré-clínicos, e a integração da academia com a indústria para gerar produtos concretamente. O poder de compra deve ser indutor da inovação incremental que venha ao encontro do interesse do Estado, com benefícios para a população e para amenizar o perfil epidemiológico do país e as doenças negligenciadas. Embora as perspectivas no cenário

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
8
atual sejam de contingenciamento orçamentário e de restrição do espaço fiscal no curto prazo, sem possibilidade de desoneração, as tendências da política industrial devem priorizar o efeito sistêmico, a inovação da produção e a competitividade. E aí cabe modernizar o marco legal, disponibilizar recursos, centralizar compras governamentais, encorajar a internacionalização, fortalecer centros de P&D e criar instrumentos para superar dificuldades e barreiras. Ressalta-se que o acúmulo de competências é o principal resultado da política industrial, como conjunto de ações coordenadas entre entes públicos e privados com o objetivo de aumentar competitividade. Essa política passa pela relação de confiança e respeito a contratos, e deve assegurar que o Estado não seja capturado em função de objetivos de curto prazo. Por isso, o BNDES atua há 13 anos e sempre com estratégias discutidas e acertadas com a indústria farmacêutica, de que maior força não é o financiamento em si e sim a indução de uma visão do setor com uma trajetória evolutiva que começou desde o início do programa com competências em bioequivalência, passando por competências para inovação incremental e radical, até chegar em competências para protocolos pré-clínicos e clínicos.
Ainda percebe-se a necessidade de se unificar conceitos de inovação incremental, o que já está sendo discutido pela FINEP, BNDES e ANVISA, conjuntamente. Outro grupo de trabalho está discutindo as PDPs de desenvolvimento e encomendas tecnológicas, que não podem ser vulgarizadas e, portanto, devem receber maior atenção para projetos de maior importância e maior risco. As PDPs de biotecnologia representam um segmento nascente e já contam com financiamentos aprovados de R$1.5 bilhões pelo BNDES, além de outros R$1.5 bilhões da FINEP, para empresas como Orygen, Bionovis, Cristália, Libbs, Axisbiotec, além de laboratórios oficiais. Porém, esse modelo também deve levar em conta o principal gargalo, que é o esgotamento da capacidade dos laboratórios oficiais para absorver toda a tecnologia que está sendo ofertada. Então, resta definir qual a solução para que os laboratórios públicos e o MS consigam resolver esse impasse. O desafio para os laboratórios públicos é hercúleo. Para melhorar a atual situação, foi sugerido que uma parte da receita dos laboratórios oficiais seja investida em capacitação de recursos humanos. Há quem acredite na necessidade de se trazer cientistas do exterior para capacitar nossos jovens profissionais. Também foi apontada a oportunidade representada pelo mercado de fitoterápicos, com nova regulamentação que deve ser aprovada em 90 dias.
3.2) 2ª Plenária - Internacionalização dos laboratórios farmacêuticos nacionais: acesso a novos
mercados COMPOSIÇÃO DA MESA - Moderador: Marcio Falci, Assessor para Presidência Científica da Biolab. Palestrantes: Igor Bueno, Superintendente da Finep em São Paulo, José Correia, presidente da Abiquifi e Eduardo Cruz, CEO da Axisbiotec. Debatedores: João Transmontano, presidente da Biolotus Biotech e Maria Luisa Campos Leal, Diretora de Desenvolvimento Tecnológico e Inovação da ABDI. Plateia convidada: Claudio Maurício de Souza, Diretor Científico do IVB. Relatores: Henrique Menezes, Professor do Departamento de Relações Internacionais da Universidade Federal da Paraíba (UFPB) e Fernando Marcussi, Coordenador Técnico Regulatório da Alanac.
Foi apresentado um panorama das iniciativas governamentais para estimular a inovação e a internacionalização dos laboratórios brasileiros, assim como os programas específicos das agências presentes à mesa. Em termos gerais, o processo de construção, desconstrução e tentativa de retomada da indústria farmacêutica brasileira vem desde o fim da Segunda Guerra Mundial. A política de abertura para as grandes multinacionais estrangeiras, a partir dos anos 1950, e a desastrosa abertura comercial dos anos

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
9
1990 impuseram uma severa crise à industria farmacêutica genuinamente brasileira. Hoje, com um conjunto de políticas públicas bem organizadas e efetivas, está havendo uma importante transformação: a transição das atividades baseadas na cópia para atividades de inovação, assim como as empresas brasileiras estão iniciando um processo de internacionalização - não apenas buscando acesso a novas tecnologias produzidas internacionalmente, mas também entrar em mercados estrangeiros. Isso estaria vinculado, de um lado, ao estímulo governamental, mas também à melhoria na gestão de recursos humanos e ao aumento da competitividade das empresas.
A dinâmica dos mercados globais de medicamentos tem um elemento chave: uma eventual crise e reorganização mundial da Pesquisa, Desenvolvimento e Inovação (P&D&I) na indústria farmacêutica. Essa crise está relacionada à incompatibilidade entre o aumento do custo e do investimento em P&D e a apresentação e aprovação pelas autoridades sanitárias de novas moléculas. O vertiginoso aumento da P&D está, na realidade, atrelado a uma diminuição no número de novas moléculas. Ainda sobre o cenário do mercado global, duas outras questões mereceram destaque. Uma tendência no aumento da comercialização de medicamentos não apenas nas economias centrais, mas especialmente nos mercados emergentes, o que abre ótimas oportunidades para a indústria brasileira. Ainda, o mercado de genéricos, apesar de significativo, tende a ser menos relevante e dinâmico diante das inovações radicais e de fármacos de especialidades clínicas. Diante desse cenário, há necessidade de se buscar novos arranjos para o modelo centralizado de inovação no setor farmacêutico – parcerias, criação de fundos específicos de capital de risco, etc.
O desempenho recente da indústria farmacêutica nacional apresenta um dado importante: o mercado farmacêutico brasileiro cresce mais do que a economia como um todo, assim como tem aumentado a participação das empresas de capital nacional nesse mercado. O elemento importante é o crescente esforço de internacionalização das empresas brasileiras. Além da busca por mercados estrangeiros, a internacionalização da P&D passa a ser vista como uma saída e um estímulo à competitividade no setor. Entretanto, mesmo com todo esse forço, se analisarmos comparativamente, a indústria brasileira ainda está muito aquém das economias avançadas e mesmo de países emergentes como China e Índia. Aumentar a competitividade da indústria farmacêutica brasileira demanda ainda um maior “salto para fora” e as agências de financiamento, como é o caso da FINEP, atualmente, têm mecanismos importantes para contribuir com isso. A internacionalização, nesse sentido, seria importante, dadas as características do mercado global: uma tendência global de aumento do gasto per capita com medicamentos, tendo em vista a tendência de crescimento desse mercado, de expansão das importações de medicamentos por países desenvolvidos e emergentes e a necessidade de aumentar a competitividade e atingir o mercado de fármacos inovadores, apesar da importância econômica do mercado de genérico.
Destacam-se as oportunidades em oncologia que deverá se tornar, em 2020, a principal área terapêutica para a indústria farmacêutica, e que cresce acima de 11% ao ano; além dos genéricos: em mercados menos desenvolvidos, já representam mais da metade do mercado farmacêutico, e ainda há muito espaço para crescimento, em especial nos países mais desenvolvidos.
O projeto da Abiqufi com a APEX, o Brazilian Pharma Solutions: the effective choice, engloba outras associações, empresas e organizações do setor farmacêutico e tem 5 eixos:
Projeto imagem sanitária: visa garantir que o Brasil continue sendo visto como um país “regulatoriamente sério” – tendo grande destaque a ANVISA;
Projeto de Competitividade Internacional: esse projeto é feito em parceria com o Sindusfarma, com a consultoria do Instituto Farma de Governança Operacional – IFGO e com apoio institucional do BNDES, FINEP, Apex-Brasil e ABDI;

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
10
Ações de Promoção Comercial: exposição das empresas brasileiras as grandes convenções internacionais do setor farmacêutico;
Ações de inteligência comercial;
Projeto Comprador: para trazer clientes para negociar diretamente com as empresas brasileiras. Quanto às potencialidades da internacionalização, temos dois eixos. De um lado, as questões internas
das empresas, ou seja, que produtos têm maior potencial para internacionalização; que estrutura corporativa permite sustentar o projeto de internacionalização; a existência de uma cultura externa e inovadora; o elevado conhecimento do sistema regulatório local. De outro lado, questões externas: o conhecimento do ambiente internacional; a identificação de sinergias com potenciais parceiros; a certificação BPF de acordo com os critérios internacionais; o alto conhecimento do sistema de patentes; e o elevado conhecimento do sistema regulatório internacional. Fármacos tradicionais são importantes para a balança comercial e para tratamentos “normais” mas não se aplicam para um processo efetivo de internacionalização de empresas em um ambiente competitivo. O diagnóstico final é que o mercado internacional está aberto para internacionalização pois “aceita” e “reconhece” as empresas brasileiras.
A Agência Brasileira de Desenvolvimento Industrial (ABDI) apresentou dois projetos da agência para fomentar a internacionalização no setor. O primeiro, “Agendas Tecnológicas Setoriais”, no qual identifica-se setores e tecnologias potenciais e relevantes para nossa competitividade e que o país tem competência para efetivamente concretizar. O segundo projeto é o “Projeto Diáspora Brasileira”, em que são identificados brasileiros em posição de destaque no exterior, com o propósito de estimular contato e cooperação com grupos no país.
Uma questão importante apontada se refere ao marco regulatório nacional, e envolve, por exemplo, o risco de se tratar questões e tecnologias relativas à medicina regenerativa como inconstitucional, tendo em vista a utilização de material humano na pesquisa e desenvolvimento. Com a incerteza, há uma diminuição na propensão a investir nesse setor. Com isso, há a necessidade de clarear essa questão no Supremo Tribunal Federal e, eventualmente, no Congresso.
Para finalizar, alguns pontos cruciais foram levantados: a seleção de parceiros na internacionalização é uma etapa crítica, pois os projetos são de longo prazo e os impactos de uma seleção incorreta só serão detectados tardiamente, atrasando enormemente o projeto. A precificação da inovação no Brasil coloca-se como um entrave. Os investimentos em inovação são desconsiderados no momento da concessão do preço pela CMED. Parcerias com universidades são extremamente burocráticas e, por vezes, a assinatura de um contrato demora mais de 1 ano. A FINEP tem linhas de financiamento específicas para projetos de parcerias entre universidades e empresas. Entretanto, os recursos para investimentos em novos projetos têm sido contingenciados ano a ano. 3.3) 3ª Plenária - Cenário global da inovação farmacêutica: enfoque no Brasil COMPOSIÇÃO DA MESA - Moderador: Lauro Moretto, presidente da Academia Nacional de Farmácia. Palestrante: João Sanches, CEO da NVS Holding, Senior Advisor da ASG no Brasil, diretor senior da MSD. Debatedor: Henry Suzuki, Diretor da Axonal. Plateia convidada: Marcus Soalheiro, Diretor da Nortec Química. Relatora: Carolina Reis, Diretora de Novos Projetos da CellSeq.
Embora o Brasil tenha tido avanços no cenário local de inovação farmacêutica, tais avanços têm acontecido de forma mais limitada, mais lenta e menos efetiva do que em grande parte do mundo. Assim,

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
11
o que se observa é que, apesar do país estar indo para frente, está ficando cada vez mais pra trás, fato que precisa ser revertido. A Plenária distinguiu três pontos principais da situação atual do Brasil no que se refere à inovação pela indústria farmacêutica: a falta de foco, disciplina e ritmo. Os setores envolvidos no ecossistema farmacêutico no Brasil não atuam em rede e por sua vez não estão inseridos no cenário global. Falta uma interação entre academia, indústria, agências financiadoras e regulatórias, e ações isoladas refletem a falta de foco e estratégia para se atingir objetivos concretos. A inovação se dá a nível local, por isso políticas de Estado são fundamentais para promover a criação de clusters de inovação em algumas regiões brasileiras. Embora o Brasil tenha evoluído na tentativa de inovar nos últimos anos, o ritmo que isso ocorre ainda é muito lento comparando-se ao resto do mundo, de modo que o Brasil tem se distanciado dos demais países. É preciso estimular a mentalidade empreendedora, apostar no jovem, em pequenas empresas e startups, que são fonte de boas ideias, assumem riscos e consequentemente trazem a inovação em seus pilares. Deve-se criar a cultura da inovação nas empresas existentes e nas que serão abertas, através da solidificação dos pilares da cultura de inovação: os recursos humanos, processos, valores, comportamentos, ambiente e sucesso, aos quais estão inerentes o propósito, talento, criatividade, trabalho em equipe, recompensa, entre outros fatores que precisam ser cultivados para o fortalecimento dessa cultura de inovação.
O Marco Regulatório, a Lei do Bem, a Lei de Inovação formam um ambiente em franca evolução no Brasil na busca pela inovação, mas ainda verifica-se uma fraca inserção do país no cenário global associado à falta de um ecossistema farmacêutico que atue em rede. Falta uma interação entre academia, indústria, agências financiadoras e regulatórias, e ações isoladas refletem a falta de foco e estratégia para se atingir objetivos concretos. Embora o Brasil tenha evoluído na tentativa de inovar nos últimos anos, o ritmo que isso ocorre ainda é muito lento comparando-se ao resto do mundo. São necessárias ações mais focadas a fim de promover de fato a inovação. O setor público investe o suficiente, mas a interação ente universidades e empresas ainda é muito baixa, e ambos apresentam-se deficitários. O ambiente de proteção da propriedade intelectual é fraco, a burocracia e a morosidade são praticamente impeditivos de promover avanços nesse setor. Além disso, métricas baseadas apenas em número de patentes não representam uma realidade de inovação. É essencial que o setor privado busque ações junto à Fapesp, por exemplo, que possui fundos para dar suporte à boas ideias, assim como outras iniciativas dentro da Universidade, a fim de estreitar esse tipo de relação. Outro ponto é que a inovação se dá a nível local, por isso políticas de Estado são fundamentais para promover a criação de clusters de inovação em algumas regiões brasileiras. Além disso, políticas de atração de grandes empresas para o Brasil são necessárias para se desenvolver o setor de P&D no cenário nacional, atrair recursos humanos, conhecimento e não só a tecnologia já desenvolvida.
Como resposta a este quadro, sugere-se o estabelecimento de um plano estratégico de inovação para o país que inclua a priorização e racionalização de investimentos, a desoneração de atividades de PD&I, e a melhoria no ensino em todos os níveis. O Plano deve ser mantido independentemente do grupo que esteja no poder, ou seja, um Projeto de Estado. E o setor de saúde pode alicerçar este processo mas precisa urgente de reformas nos sistemas nacionais de saúde, mudanças em agências regulatórias, privilegiando o desenvolvimento de produtos para necessidades ainda não supridas (“unmet medical
needs”), em detrimento ao registro produtos sem custo-benefício comprovado, remuneração da cadeia de valor, com base nos resultados (“outcomes”). Diante deste quadro de inovação farmacêutica, talvez seja melhor falar sobre formas de atuação para empresas e profissionais, onde verifica-se um estágio de “êxodo”, já que um grande número de empresas “nacionais” passaram a realizar atividades de pesquisa e desenvolvimento no exterior. A questão do “êxodo” também parece ter sido opção natural para

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
12
pesquisadores e profissionais brasileiros que hoje atuam no exterior. Em época de “vacas gordas” em solo brasileiro, muitos para cá voltaram. Mas será que continuarão voltando ou que ficarão por aqui, com os atuais cortes de investimentos e de atrasos de pagamentos? “Migrar” para ecossistemas mais favoráveis pode resolver questões individuais, mas não são alternativa para todos. Então, o que podemos dizer em relação a empresas, ICTs e profissionais que optaram por continuar por aqui (ou que não tiveram opção)?
Fazer mais do mesmo, não parece ser opção razoável. Trabalhar nas “soluções” apontadas neste relatório é importante, mas levará muito tempo. Terá vantagem quem aproveitar “oportunidades globais”, através de uma mudança de postura e também de muita capacitação, pois é preciso conhecer temas como propriedade intelectual, aspectos regulatórios internacionais, e mercados estrangeiros. Importar profissionais e programas de capacitação nessas áreas pode ser um bom começo, considerando que o planejamento estratégico e as futuras ações para o país e para o setor dependem de tais competências. O fechamento de torneiras de onde vinham jorrando recursos é um convite, se não um ultimatum, para uma mudança.
Assim, pensar na internacionalização é inverter a lógica de apenas substituir importação e assumir um papel relevante no setor de exportação de tecnologia e inserção no mercado global. A inovação gera liberdade econômica para o país que por sua vez avança e gera impacto real no ecossistema como um todo. A inovação farmacêutica no contexto internacional está fortemente orientada para o desenvolvimento de novos medicamentos, desde suas primeiras etapas, enquanto que para uma grande parcela dos pesquisadores brasileiros a inovação se caracteriza na realização de ensaios clínicos de produtos que já estão em estágio avançado de desenvolvimento em outros países. É muito útil desenvolver alguns outros eixos temáticos para a inovação no Brasil, com foco em medicamentos. Dentre esses novos eixos temáticos pode-se destacar as pesquisas orientadas para dar continuidade a estudos preliminares contido em teses e trabalhos de prospecção já praticamente abandonados. Também considera-se muito relevante desenvolver pesquisas para aperfeiçoar medicamentos tradicionais através de novas tecnologias, que permitam obter produtos com maior desempenho terapêutico e com redução de custos promovidos por tecnologias diferenciadas, além da dedicação ao desenvolvimento de recursos humanos para gerenciamento de pesquisadores, com vistas a torná-los mais eficazes em suas atividades. 3.4) 4ª Plenária - Regulação do registro de inovações e preços COMPOSIÇÃO DA MESA - Moderador: Marcelo Quintão, economista da área técnica do GrupoFarmaBrasil. Palestrantes: Leandro Safatle, Secretário Executivo da CMED e Bruno Abreu, Gerente de Regulação de Mercados do Sindusfarma. Debatedores: Ogari Pacheco, Diretor Presidente do Cristália e Pedro Bernardo, Diretor de Acesso ao Mercado da Interfarma. Plateia convidada: Fernando Marcussi, representando a Alanac. Relator: Marco Torronteguy, advogado da Mattos Muriel Kestener ou MMK Advogados.
A Plenária ocorrida em 2012 sobre Registro e Preços para produtos inovados e Compras Públicas evidenciou que a carga tributária tem grande parcela de culpa na dificuldade de inovação da indústria nacional e que a solução perpassa por definir três condições: o que é inovação de fato, que inovações incrementais valem ganhos de preço, e o que é inovação no sentido econômico. Com base na definição desses conceitos, será possível definir critérios para valoração de tecnologias. O relatório do 6º ENIFarMed menciona que, mesmo precisando de ajustes, a legislação precisa ser seguida pela sociedade, assim como

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
13
pelos servidores da Anvisa, que analisam por ano três mil apresentações de medicamentos novos, similares e genéricos. Para a Anvisa, os preços dos medicamentos no Brasil devem continuar sendo regulados, pois o balizamento de preços protege, de certo modo, a indústria nacional, fazendo com que essas não sejam atropeladas pelas empresas multinacionais. Por outro lado, o setor industrial percebe a autorização de preço como uma etapa que reduz sua competitividade e traz insegurança para a produção de medicamentos inovados, em função do risco e do valor do investimento da inovação não ser considerado no preço final aprovado.
A Câmara de Regulação do Mercado de Medicamentos, ou CMED, é o órgão regulador de preços no
Brasil que busca contribuir para o aperfeiçoamento da regulação de preços no país. Os objetivos institucionais da CMED e sua composição pluriministerial (envolvendo os Ministérios da Saúde, Justiça, Fazenda, Casa Civil e Desenvolvimento, Indústria e Comércio Exterior) permitem a análise a partir de diferentes enfoques. O Conselho de Ministros, o Comitê Técnico Executivo e a Secretaria Executiva (Anvisa) têm como objeto de atuação o nosso mercado, que apresenta assimetria de informação e baixa elasticidade no preço/demanda em razão da essencialidade dos produtos regulados (medicamentos são bens de consumo inelástico). As características do mercado de medicamentos justificam a regulação de preços desses produtos. Entre os benefícios da regulação pela CMED destacam-se os seguintes: ela tem evitado que os preços de medicamentos subam acima da inflação e tem garantido a redução nos preços de entrada no mercado brasileiro. A regulação de preços promove convergência entre os preços nacionais e internacionais. A CMED está estudando e comparando a regulação de preços de medicamentos no Brasil e em outros países, atenta às recentes alterações da regulação de preços em diversos países. O objetivo será a revisão da Resolução CMED nº 2/2004. O caso brasileiro tem peculiaridades: a concentração de mercado é uma importante característica do mercado brasileiro de medicamentos. O mercado brasileiro, com a pretensão de atender ao sistema público que acolhe mais de 100 milhões de pessoas, tende a crescer nos próximos anos, também em razão da recente ascensão da classe média, pressionando a demanda por produtos nacionais e importados. Considerando esse cenário, a CMED está atenta ao tema da inovação. O Brasil tem como referência o preço externo, o que dificulta a captação inicial de informações para definição de preço de inovações nacionais. A CMED está avaliando o melhor modo de precificar a inovação realizada no mercado interno brasileiro, atenta à política de incentivar inovações incrementais que tragam benefícios ao paciente e ao sistema de ciência e tecnologia no país.
É crescente o número de tecnologias disponíveis no mercado, com rápida velocidade de difusão, alto custo e demanda por incorporação ao SUS, que é uma demanda gerada pela oferta. Verifica-se um aumento dos “casos omissos”, que não se encaixam em nenhuma das seis categorias para regulação de preços definidas pela Resolução CMED nº 2/2004. Vários produtos ficam à margem das categorias definidas pela regulação. O aumento de “casos omissos” mostra a necessidade de se aperfeiçoar o sistema de precificação no país. É necessário atualizar a Resolução CMED nº 2/2004, para atualizar as seis categorias de preços. Além disso, falta disciplinar, p.ex., a precificação de biossimilares, da nanotecnologia, de medicamentos específicos, de medicamentos para doenças raras, de dispositivos médicos intrínsecos aos medicamentos. É importante refletir que o aumento de “casos omissos” implica o aumento da discricionariedade da CMED.
À luz desse cenário, foram apresentados diagnósticos e sugestões para o aperfeiçoamento da regulação de preços no Brasil. Destacam-se as sugestões para revisão da Resolução CMED nº 2/2004; aumento da equipe técnica da CMED; maior detalhamento das rotinas de análise; assessoramento do Comitê Técnico por um comitê ad hoc, com maior expertise técnica, para elevar o debate técnico nos

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
14
recursos administrativos; revisão do critério atual de “cesta de países”, para que sejam utilizados como comparadores países com realidades mais próximas ao Brasil, talvez avaliando a possibilidade de se retornar ao sistema de média de 3 países.
Os debates ponderaram a importância da inovação garantir ganho terapêutico ou real benefício ao paciente. Houve concordância sobre a necessidade de revisão da sistemática de precificação adotada pelos regulamentos da CMED. Foi sugerida a possibilidade de que as empresas discutam preço com o órgão regulador antes de investir na inovação, ao invés de fazê-lo depois de anos de investimento. Essa mudança poderia incentivar a inovação no país. Foram debatidos os critérios de análise pela CMED. Antigamente o país controlava o preço pela oferta (controle do custo da indústria farmacêutica), passando a controlar o preço pelo lado da demanda (controle do custo para o paciente, a partir de uma escala de comparações de preços de medicamentos em diferentes países). Desde 2003, o país se esforça para fundir a política de preços com a análise de custo-benefício. Nesse modelo, a ausência de comparação para diversos medicamentos é um problema para a precificação. A regulação brasileira não deveria olhar apenas para o “custo-minimização” (comparação de preços de um medicamento contra outro medicamento), mas deveria considerar de modo mais amplo o “custo-benefício” (avaliação sistêmica da economia e dos custos indiretos).
A inovação envolve riscos econômicos e a precificação estimula (ou não) a assunção de riscos pela indústria nacional. A discussão sobre a revisão da Resolução CMED nº 2/2004 evidenciou a importância de se estabelecer regras claras para que se tenha transparência e segurança para investimentos no país. A CMED está consolidando sua pesquisa sobre comparação de preços em diferentes países e em breve o setor regulado será ouvido com relação à proposta de aperfeiçoamento da Resolução CMED nº 2/2004. Também foi questionada a possibilidade de liberação de preços de medicamentos isentos de prescrição (MIPs). Já se sabe que a estrutura de mercado dos MIPs é mais concorrencial e menos concentrada do que outros setores do mercado farmacêutico. A variável “preço” é mais relevante e o mercado de MIPs é mais elástico que o mercado de medicamentos em geral, razão pela qual se estuda a possibilidade de uma regulação diferenciada. O monitoramento do mercado pode ser uma alternativa à regulação e ao tabelamento de preços. A CMED está estudando o tema internamente, mas não há previsão quanto às conclusões desse debate. Também se questionou os desafios da precificação de medicamentos vis-à-vis as diferenças regionais do custo de distribuição de medicamentos no país, notadamente em razão da imposição do preço CIF para os detentores de registro sanitário. A distribuição é um desafio para regulação de preços, que poderá rever a margem linear de distribuição atualmente aplicada. A CMED debate o tema, considerado nas discussões relativas à atualização da Resolução CMED nº 2/2004. 3.5) Sessão Temática 1 - Ações para a produção de insumos da
biodiversidade brasileira COMPOSIÇÃO DA MESA - Moderadora: Maria Behrens, Chefe do Departamento de Produtos Naturais de Farmanguinhos/FIOCRUZ. Palestrantes: Alceu Moreira, Deputado Federal do PMDB/RS; Henry Novion, Departamento de Patrimônio Genético do Ministério do Meio Ambiente; Benilson Barreto, do Departamento de Assistência Farmacêutica (DAF) do Ministério da Saúde. Debatedores: Ricardo Dias, Diretor Técnico Científico do Grupo Centroflora; e João Carlos Basílio, presidente da Abihpec. Relatores: Carlos Vitor, Coordenador de Inovação Radical do Aché e Ana Claudia Dias, técnica da Abifina.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
15
Cabe destacar que o tema vem sendo discutido desde a 5ª edição do ENIFarMed, em 2011, quando recebemos o então representante do MMA, Braulio Dias. No ano seguinte, além do debate em uma sessão do ENIFarMed, foi realizado um Workshop sobre Biodiversidade através uma parceria com o INPI e do apoio da Biolab. No ano passado, o 8º ENIFarMed recebeu pela primeira vez um representante do Poder Legislativo, o Deputado Alexandre Roso, do PSB-RS, para debater o questão da biodiversidade, o que foi um grande passo. Cabe lembrar que o Brasil esteve sob a Medida Provisória 2.186/2001 por 14 anos, e que entraves desta magnitude não podem perdurar por tanto tempo. Depois de 6 tentativas de melhoria no arcabouço legal por diversos autores, incluindo a ex-ministra Marina Silva, o Projeto de Lei 7735 foi encaminhado ao Congresso Nacional em junho de 2014, aprovado na Câmara naquele ano, e aprovado no Senado e pela presidente Dilma em 2015. Espera-se para novembro deste ano o início da regulamentação da lei, sendo este o ponto chave do debate do 9º ENIFarMed.
A sessão temática sobre biodiversidade do 9º ENIFarMed abordou os avanços obtidos com a recente aprovação da Lei 13.123/2015 que estabeleceu um novo marco legal de acesso à biodiversidade brasileira. Desde junho de 2000 o acesso à biodiversidade nacional era regulado pela MP 2.186-16, medida esta que trouxe grande insegurança jurídica tanto para o setor industrial quanto para os pesquisadores que conduziam linhas de pesquisa na área de produtos naturais. O objetivo da sessão foi apresentar os avanços obtidos e debater as ações a serem tomadas com a aprovação da Lei e sua entrada em vigor em novembro de 2015. Os quatorze anos de experiência sobre o que efetivamente não funcionou durante a vigência da MP 2186-16 baseou a definição, mais precisa possível, dos conceitos da Lei 13.123/2015. A MP acima referenciada não conseguiu cumprir seus objetivos, o que pode ser evidenciado pelo pequeno número de contratos de repartição de benefícios firmados. A medida, que visava proteger a biodiversidade contra a biopirataria acabou protegendo-a inclusive dos próprios brasileiros com um grau de exigência que, na prática, não fazia sentido. Embora houvesse um grande esforço do Conselho de Gestão do Patrimônio Genético (CGEN) e do próprio Ministério de Meio Ambiente (MMA) em destravar e desburocratizar o processo de acesso à biodiversidade através de mecanismos infralegais, a própria MP em si era um grande limitante. O novo modelo de gestão do acesso à biodiversidade migra para um processo mais ágil, fácil e integrado. Além de definições muito bem construídas que facilitam a interpretação e aplicação da Lei, o foco está nos resultados e não no processo. Em outras palavras, o objetivo passa a ser o desenvolvimento de produtos e sua posterior comercialização e repartição de benefícios justa e equitativa, em prol da conservação da biodiversidade. O CGEN passará a ser mais democrático, com a participação da sociedade civil e setores empresariais para auxiliar e trazer transparência às tomadas de decisão.
O Brasil detém a maior biodiversidade do planeta com cerca de 50 mil espécies superiores identificadas. Esta riqueza biológica, no entanto, não foi ainda explorada em sua totalidade. Estima-se que apenas 1% deste total tenha sido química e farmacologicamente estudada. Se considerarmos apenas a floresta amazônica brasileira, cerca de 30 mil espécies foram catalogadas e apenas 2% delas foram submetidas a algum tipo de investigação científica. O mercado farmacêutico mundial fatura ao ano em torno de USD 1 trilhão, sendo que apenas 7% deste faturamento é resultante de produtos advindos de medicamentos fitoterápicos e plantas medicinais. O cenário no Brasil é ainda pior. Dos R$ 60 bilhões faturados pela indústria farmacêutica nacional menos de 2% refere-se a produtos fitoterápicos. A falta de investimentos e, consequentemente, de inovação é apontada como grande responsável por esta pequena participação e pela falta de crescimento do segmento frente ao mercado farmacêutico total (MFT). De acordo com um estudo inédito realizado pelo Grupo Farmabrasil, o segmento de medicamentos fitoterápicos representava em 2013 apenas 1,18% do MFT.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
16
Em relação às políticas públicas, como a Rename e a Renisus, a PNPMF e as PDPs, cabe destacar que estas ainda não incluem fitoterápicos, ou seja, outro ponto que merece ser discutido é a ausência de fitofármacos e medicamentos fitoterápicos na Política de Desenvolvimento Produtivo (PDP), o que acaba dificultando a inserção de produtos no Sistema Único de Saúde (SUS). A indústria de cosméticos e produtos de higiene pessoal também sofreu as consequências do atual marco legal da biodiversidade. Embora o potencial do setor seja de grande relevância, com faturamento que já ultrapassa os US$ 40 bilhões, somente no Brasil representando cerca de 10% do mercado mundial, ele não explora convenientemente as oportunidades da biodiversidade brasileira devido aos gargalos trazidos pela Medida Provisória 2.186-16. Segundo a Ministra do MMA, Isabella Teixeira, será reduzida a burocracia para o desenvolvimento de novos produtos e a biodiversidade será um ativo. Porém, as oportunidades de melhorias não se limitam à desburocratização dos procedimentos de acesso à biodiversidade. Cerca de 80% dos insumos farmacêuticos ativos utilizados pela indústria brasileira são importados e suscetíveis a intensas variações cambiais. Como gargalo temos a dependência de importação de insumos farmacêuticos ativos. As oportunidades estão no fortalecimento do mercado e na independência de importação de matéria-prima. O mercado de fitoterápicos está caindo (1,18%), mas o novo marco deverá trazer mais investimentos. Para tal, foi sugerido repensar o poder de compra governamental, pois a compra descentralizada não vai fazer aumentar a fitoterapia no SUS.
A cadeia produtiva dos fitoterápicos, que envolve muitas áreas além do financiamento, vem sendo incentivada através da inclusão no SUS de fitoterápicos: em 2007 dois fitoterápicos, em 2009 seis medicamentos, em 2012 mais dois e em 2015, doze fitoterápicos. Já está sendo elaborada a Rename de 2016. Para que qualquer medicamento esteja na Rename é necessário que o medicamento tenha registro. Assim, é fundamental que a indústria produza mais medicamentos. Desde 2012, o MS tem criado editais para auxiliar no fortalecimento dos fitoterápicos. Um deles foi a criação dos Arranjos Produtivos Locais (APLs), dentro de um conceito ampliado de saúde. Além dos APLs, o MS apoia 36 projetos de Assistência Farmacêutica em Plantas Medicinais e Fitoterápicos. No ano passado, o MS começou a apoiar os laboratórios públicos em Alagoas e em Minas Gerais. Outra questão que tem gerado polêmica é o financiamento para elaboração de monografias. Os principais desafios são: a gestão compartilhada, Brasil com situação de saúde bem heterogênea, necessidade de articulação com área produtiva.
A construção do texto do novo marco de acesso à biodiversidade pode ser considerada uma das mais ricas experiências políticas vivenciadas por todos os atores envolvidos no processo, através da
mediação. A participação de todas as partes interessadas, desde representantes das comunidades tradicionais, passando por diversos segmentos da indústria e pastas governamentais foram fundamentais para os consensos alcançados. A repercussão do novo marco legal de acesso à biodiversidade foi tamanha que três dias após sua publicação no Diário Oficial da União (DOU) trinta e sete países solicitaram acesso ao texto. A aprovação da Lei 13.123/2015, novo marco legal de acesso à biodiversidade, promete reduzir a burocracia para empresas e pesquisadores que pretendem fazer uso desta rica fonte de novos produtos, seja retomando projetos que foram interrompidos devido à falta de segurança jurídica ou realizando novos acessos. Os mecanismos claros de repartição de benefícios garantem que a pesquisa e o desenvolvimento de produtos sejam liberados e a definição de conceitos importantes como patrimônio genético, conhecimento tradicional associado, repartição de benefícios, consentimento prévio e acesso, garantem a inclusão da pesquisa e do desenvolvimento tecnológico em novos produtos. A nova legislação legaliza e incentiva as pesquisas, estimula a inovação e o lançamento de novos produtos, além de garantir uma repartição de benefícios mais justa, para conservação da biodiversidade, e com descontos para repartição não monetária. Além da atualização de conceitos, houve foco nos resultados e não no processo, integração

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
17
de bancos de dados públicos - simplicidade e rastreabilidade, isenções para micro e pequenas empresas e microempreendedor individual, dispensa de consentimento prévio para o acesso ao patrimônio genético e licenciamento de direitos de propriedade intelectual e produtos intermediários não repartem benefícios. Cada setor específico é tratado de forma específica pela lei. Empoderem povos e comunidades tradicionais: o consentimento informado é prévio, obrigatório e formalizado segundo os usos, costumes e tradições. Protocolos comunitários são formalmente reconhecidos como instrumentos de manifestação de vontade, valorizados o seu uso e difusão. Adicionalmente, a lei valoriza as empresas que a respeitam, reduz a burocracia e o tempo para desenvolvimento de novos produtos, fato gerador de repartição de benefícios (RB), com o objetivo de só ocorrer se houver exploração econômica, ou seja, RB sem efeito cascata. A nova legislação reconhece o setor agrícola e mantém a RB apenas no material reprodutivo, destravando a pesquisa e a inovação, pois valoriza pesquisadores, inovadores e instituições de pesquisa e desenvolvimento. O desafio é regulamentar sem burocratizar, manter o sistema ágil e integrado, garantir o estímulo, fazer do CGEN um novo conselho e garantir a repartição de benefícios. A Consulta Pública da Regulamentação de Lei de Biodiversidade foi publicada, ficará aberta até o dia 16 de outubro de 2015 e, até novembro, teremos um cenário regulamentado. Portanto, o grande desafio que estamos vivenciando atualmente é a construção de uma agenda para definição dos termos do Decreto Presidencial que regulamentará a Lei. Precisamos regulamentar sem, no entanto, burocratizar o processo. 3.6) Sessão Temática 2 - Modelos de gestão para acelerar a inovação e a integração de políticas
públicas para doenças que afetam populações negligenciadas COMPOSIÇÃO DA MESA - Moderador: Eric Stobbaerts, Diretor da DNDi. Palestrantes: Ana Rabello, Coordenadora do Programa colaborativo de P&D de novas alternativas terapêuticas e de diagnóstico para doenças negligenciadas SCTIE-MS/Fiocruz/DNDi; Luiz Carlos Dias, Professor Titular do Instituto de Química, Universidade de Campinas (Unicamp); e Walter Britto, da Universidades Aliadas por Medicamentos Essenciais (UAEM). Relatores: Vera Lucia Luiza, Professora da ENSP e Henrique Menezes, Professor adjunto do Departamento de Relações Internacionais da UFPB.
A segunda sessão temática do 9º ENIFarMed teve como objetivo geral debater modelos de gestão e integração de políticas públicas para fomentar a produção e acesso a novos medicamentos e tratamentos voltados à populações e doenças negligenciadas (DN), cuja definição perpassa a noção de doenças incidentes em populações negligencias (doenças das pobrezas; doenças em que há inovação, mas não há acesso). Além disso, é importante mostrar que existem modelos de inovação criativas e tentativas de sucesso de parcerias público-privadas para acelerar a inovação no âmbito das DN. Dessa perspectiva que se depreende a necessidade de se falar em ‘inovação para o acesso’ e não apenas o desenvolvimento de tecnologias longe do acesso das pessoas.
A experiência da Fiocruz no desenvolvimento de novas tecnologias para o tratamento de DN pode ser dividido em três eixos: 1) Projeto de desenvolvimento da combinação Artesunato + Mefloquina (ASMQ), que tem demonstrado grande sucesso para o tratamento da Malária; 2) Projeto com a FUNTEC para desenvolvimento de medicamentos e diagnostico em DN; e 3) Estudos em parceria FIOCRUZ - DNDI para prospecção de medicamentos para 14 DN. O primeiro projeto começou em 2002 por iniciativa da OMS pela necessidade de ampliar as opções disponíveis de tratamentos alternativos para a Malária que

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
18
melhorassem os tratamentos existentes, especialmente com a ampliação de opções existentes, melhoria na administração e redução de custos. Parte fundamental da inovação nesse projeto era justamente a junção de dois medicamentos já existentes para a formulação de um novo que possa ser ministrado em poucas doses e poucos dias. O movimento coordenado pelo TDR, OMS e DNDI tinha a finalidade de desenvolver medicamentos mais simples de administrar e baratos. Várias instituições nacionais assim como vários países participaram deste esforço em suas diferentes etapas. Obteve-se o registro em 2008 e, neste ano, está sendo concluída a pré-qualificação. Num momento subseqüente houve o processo de registro do novo medicamento e a transferência da tecnologia para a empresa CIPLA. Os estudos clínicos mostraram impacto positivo na eficácia no tratamento da doença, posologia cômoda, melhora da adesão, melhora terapêutica, e redução da transmissão. A aproximação da DNDi com a Fiocruz sensibilizam os pesquisadores para a importância de produtos (outptus) para além da publicação de artigos científicos ou de patentes. Mas a barreira para fazer a distribuição via OMS/OPAS está agora sendo vencida com a pré-qualificação. No Brasil, após o evento na Fiocruz, frente à informação de que a decisão da Câmara Técnica de restringir a indicação do ASMQ para áreas não amazônicas com bases na resistência da mefloquina isolada, se decidiu fazer a busca e, se necessário, conduzir um teste de Resistência ao Choque Térmico (RCT) para avaliar a resistência ao ASMQ.
O segundo eixo trata da necessidade de aproximação e cooperação entre diversos atores nacionais para o transbordamento da produção científica para a produção de produtos úteis. O projeto FUNTEC para o desenvolvimento de medicamentos e diagnóstico em DN tem esse propósito, de articulação de órgãos públicos, Ministérios e financiamento do BNDES. Prevê a necessidade de trabalhar a partir de demandas sociais específicas para a produção de inovações e tecnologias específicas e a produção de Efavirenz pediátrico em Farmanguinhos; além de uma formulação oral de anfotericina B, em parceria com a UFMG; e outros estudos para Doença de Chagas e Leishmaniose.
O terceiro eixo trata de um acordo entre a Secretaria de Ciência e Tecnologia para Insumos Estratégicos (SCTIE) do Ministério da Saúde, a Fiocruz e a DNDi para a prospecção de medicamentos para quatorze DN e para o estudo focado na magnitude da doença, produtos disponíveis e sua efetividade. Um exemplo dado foi o tratamento da leishimaniose viceral, mas ainda é importante fortalecer políticas que busquem a autonomia nacional e a adequação da pesquisa para o desenvolvimento de novos tratamentos à situação concreta do país e dos nossos pacientes. Estão estudando a leishmaniose visceral onde há basicamente apenas 2 produtos disponíveis: o antimoniato e a anfotericina. O tratamento com anfotericina lipossomal custa R$33.000 cada. Com a negociação de preços, para uso em leishmaniose visceral, o tratamento sai por cerca de R$1.000, contra cerca de R$300 com o antimoniato. O MS optou por ampliar as situações de uso da anfotericina lipossomal resultando na falta do produto. Como alternativa para lidar com a situação, uma nota técnica recente recomenda o uso de outras formas de anfotericina, sem estudo clínico suficiente que suporte a decisão. Verifica-se, aqui, que a falta de estudos clínicos está impactando as políticas públicas. As iniciativas mencionadas buscam superar o desafio de busca de mecanismos de financiamento assim como buscam modelos de gestão inovadores.
A proposta da DNDi é desenvolver parcerias com países endêmicos, caso do Brasil para várias das doenças alvo da iniciativa, de maneira a apoiar o desenvolvimento de capacidades locais. A DNDi tem acumulado várias experiências exitosas para malárias, doença do sono, além do tratamento pediátrico para chagas. Aspectos importantes a se considerar nos produtos são a distribuição em todas as áreas, com comodidade posológica, baixo preço e garantia de oferta que fuja do monopólio. O maio desafio atual é aumentar a solubilidade dos produtos para chagas sem perder potência. Destaca-se o exemplo do modelo de gestão na parte da descoberta, com grande apoio da química no Brasil. Tem havido internalização de

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
19
competências. Houve uma grande negociação para que se pudesse fazer ciência num modelo mais aberto, que não se limitasse à patenteabilização, dada à urgência de fazer o produto chegar ao mercado.
As atividades de pesquisa e desenvolvimento para chegar com um novo medicamento são muito longas, podendo ultrapassar os 15 anos. Os casos LOLA (DNDi) e a Brazil Heterocycles (MMV) mostram que o objetivo dessas organizações é acelerar o processo, através da cooperação entre múltiplos atores envolvidos no processo da produção até a comercialização do medicamento. Assim, o objetivo dos projetos e das pesquisas é desenvolver, em até 5 anos, candidatos clínicos para Chagas e Malária. Apesar da parceria com importantes universidades, não se trata de um projeto acadêmico com a finalidade de produção de artigos. Foca-se em inovações úteis à populações vulneráveis, através do desenvolvimento de novas moléculas para substituir tratamentos antigos, com menos efeitos colaterais, e adaptados às regiões. O LOLA é um consórcio da DNDi para desenvolvimento de novos fármacos. Prepara-se as moléculas no laboratório da Unicamp; depois vão para outros laboratórios para realização de vários estudos e testes em vários laboratórios no mundo.
A malária ainda causa alta mortalidade na África, sobretudo em crianças. O projeto de Malária MMV surgiu da necessidade de combinações para tratar a malária devido ao aumento da resistência ao componente isolado e por isso não se trata mais malária com apenas um fármaco: é necessário ao menos dois, inclusive para evitar transmissão, assim como é preciso o desenvolvimento de várias formulações diferentes para a mesma doença, para evitar a resistência e controlar a transmissão. Há um grande conjunto de parceiros que recebem os produtos desenvolvidos no laboratório da Unicamp (academia e empresas). As big pharmas que participam não injetam recursos, mas emprestam expertise e laboratórios. A despeito de vários produtos com indicação para malária, o artesunato é um ótimo candidato, mas não se pode correr o risco de perder a artemisinina.
A Universities Allied for Essential Medicines (UAEM) é uma organização de base estudantil iniciada em 2001 e que trabalha no sentido de promover acesso a medicamentos por meio de ações e práticas de patenteamento. No âmbito da Organização Mundial de Saúde (OMS) há uma ampla discussão sobre os dilemas entre inovação para doenças negligenciadas e propriedade intelectual. Analisando-se a cronologia das discussões e processos políticos acerca da inovação tecnológica, propriedade intelectual e acesso a conhecimento, destaca-se o documento Public Health innovation and IPRs (WHA 59.24) da Assembleia Geral da OMS, que tinha como temática justamente as doenças de tipo II e III (classificação apresentada no documento do MSF “Fatal Imbalance”, que são as que afetam principalmente os países mais pobres) e, como implementação, um modelo de gestão e financiamento que drible a falha do mercado farmacêutico. Existem dois importantes projetos caminhando atualmente: 1) Demonstration projects (projetos que visam o desenvolvimento de tecnologias de saúde como medicamentos, diagnósticos, dispositivos médicos, vacinas, etc.); 2) Global health observatory. Verifica-se ainda a necessidade de continuar a discussão sobre um Tratado Global para P&D em DN. Esse tratado asseguraria contribuição obrigatória de 0,01% do PIB dos países membros em um pool gerenciado pela OMS; e incorporaria às discussões já correntes sobre a possibilidade de financiar a inovação na origem e não a posteriori com o aumento dos preços (delinkage – conceito proposto pelo CWG, que desvincula o financiamento do desenvolvimento do preço de venda). Este princípio está contido em duas resoluções, uma relacionada à vacinação, de importância na preservação da saúde, e também no tema de resistência microbiana, que teve grande atenção na OMS deste ano. A discussão é que, se nada for feito, haverá um retrocesso de 70 anos no enfrentamento das doenças infecciosas.
Alunos da UEAM, UFMG, PUC de Minas, UFRJ, Fundação Getúlio Vargas e UNICAMP atualmente estudam as práticas de licenciamento das universidades brasileiras e como podem contribuir para o

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
20
acesso. Sobre a iniciativa o University Report Card mostrou que é feito, por preenchimento voluntário de várias universidades no mundo, uma classificação da capacidade de inovação em direção ao acesso. A base dessa mobilização está em uma ação do UEAM quanto à estavudina. O produto foi desenvolvido por 2 pesquisadores de Yale em 2001, sendo licenciado para uma grande empresa farmacêutica. A empresa o ofertava na África subsaariana pelo mesmo preço que nos Estados Unidos. Os estudantes de direito de Yale, ao saberem, procuraram a direção da universidade, que disse que não podia fazer nada por razões contratuais com a empresa produtora. Depois de negociações propiciadas pela mobilização dos estudantes, se conseguiu a redução dos preços praticados na África. A importância do lucro deve ser reconhecida, mas os modelos de gestão devem garantir o lucro justo e não desmesurado.
O desafio é pensar o que se pode fazer, sobretudo para as doenças do tipo I e II, para mudar o cenário tradicional e pensar outras coisas. Assim, a mesa apresentou modelos de gestão no desenvolvimento de produtos novos, em fase de bancada, de produtos na fase de produção industrial, o que envolve os desafios de registro e colocado no produto no mercado até sua chegada ao usuário final e a iniciativa do UEAM, voltado à discussão da propriedade intelectual (patentes) e como um modelo inovador, iniciado por um grupo da sociedade civil. Cada uma dessas situações foi exemplificada com casos concretos, permitindo reflexões quanto ao potencial de cada iniciativa assim como dos desafios envolvidos.
3.7) Sessão Temática 3 - Biotecnologia e a intercambialidade para biossimilares COMPOSIÇÃO DA MESA - Moderador: Thiago Mares Guia, Gerente Médico e Científico da Bionovis. Palestrantes: André Abrahão, Diretor Médico da Merck Serono; Priscila Scheinberg, Gerente de Assuntos Regulatórios da Orygen; Daniela Marreco Cerqueira, Gerente de Produtos Biológicos da Anvisa,; Dirceu Barbano, Consultor e ex-presidente da Anvisa. Relatores: Carlos Martins, da Interface CTI e Eliziane Patricio, da UNIPLAC.
Assim como já ocorreu em outros países, o Brasil empenha-se hoje em adotar uma regulamentação que defina o que são medicamentos biossimilares. Um dos pontos relevantes é saber quando o medicamento pode ser considerado intercambiável com o produto que lhe serviu de referência. A sessão temática “Biotecnologia e Intercambialidade para biossimilares” trouxe uma contribuição importante ao reunir representantes da indústria e agência reguladora. O desenvolvimento e a produção industrial de medicamentos biológicos demandam um grau de domínio tecnológico muito maior do que os produtos originados de fármacos sintéticos.
Quanto à intercambialidade de biossimilares, o tema ainda precisa ser amplamente discutido pela classe médica, governo e sociedade. As aplicações dos medicamentos biológicos são amplas, mas principalmente voltadas para o tratamento de doenças crônicas. Devido à complexidade dos medicamentos biológicos, o custo com desenvolvimento torna-se elevado, e somam-se a esse fator barreiras mercadólogicas. Desse modo, a escolha do tratamento com medicamentos biológicos acaba sendo restringida pela capacidade financeira do paciente quando o governo não se responsabiliza pelos gastos. Na Noruega, foi desenvolvido um estudo clínico para intercambialidade para várias indicações. Cabe saber se o modelo de estudo apresentado seria aplicável em todos os casos de aprovação de biossimilares intercambiáveis devido ao elevado custo. Assim torna-se questão-chave para intercambialidade: considerando-se que os estudos são complexos, longos e de elevado custo, seriam

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
21
necessários estudos para todas as indicações? Ou seja, o tema de intercambialidade é de extrema importância e precisa de definições o mais breve possível, visto é de interesse da sociedade para que os pacientes possam ter acesso ao tratamento.
Quanto à extrapolação de indicações, os biossimilares não são cópias idênticas, mas precisam apresentar comparabilidade de qualidade, segurança, e eficácia clínica com os produtos de referência. Contudo, até mesmo os produtos biológicos inovadores já passaram por vários processos de mudanças. Dessa forma, os produtos disponíveis no mercado já não são idênticos aos da época do registro, mas precisam ter a mesma eficácia e segurança em todas as indicações terapêuticas que o produto registrado. Qual é o racional científico para que possa ser realizada extrapolação de indicação? A totalidade de evidências de comparação não-clínica e clínica e com base na literatura disponível. Contudo, a decisão se a extrapolação de múltiplas indicações é aceitável (ou não) é determinada caso a caso pelas agências regulatórias. Todavia, ainda há muito receio da comunidade médica científica sobre a indicação de biossimilares, principalmente para casos em que não há estudos clínicos, devido ao desconhecimento do rigor utilizado para registrar um fármaco como biossimilar. A aprovação de indicações baseadas na extrapolação de dados não são bônus, nem conduzido por considerações econômicas para diminuir o custo de biossimilares, mas sim em embasamento científico. Extrapolação é uma consequência lógica do conceito biossimilar. Extrapolação vem sendo exercida por muitos anos com mudanças no processo de fabrico de produtos biológicos de origem, fato nem sempre conhecido pelos clínicos, raramente comunicado. A geração de dados redundantes ou simplesmente reconfortantes deveria ser evitada, uma vez que a extrapolação deve ser sempre baseada em critérios científicos sólidos e objetivos.
A Anvisa apresentou sua classificação do que pode ser registrado como produto biológico no Brasil, evidenciou as diferenças entre produtos inovadores e não-inovadores e exibiu a relação dos 15 principais medicamentos com maior volume de vendas no mercado mundial, dos quais 50% são medicamentos biológicos. A apresentação fez um balanço da legislação que regula o setor no país, a partir de 1976 e dos mecanismos para registro de medicamentos biológicos, adotados com base na Resolução de Diretoria Colegiada, RDC 55/2010. O país dispõe de uma base regulatória para a nomeclatura dos medicamentos biológicos em consonância com a legislação internacional, e deve ter regras claras para avaliar a intercambialidade dos fármacos. Esse trabalho tem como referência a legislação já existente em outros países, cenário ilustrado com os exemplos da União Europeia, dos Estados Unidos e Canadá. Na União Europeia, as decisões sobre intercambialidade entre biossimilares e os biológicos de referência não são do EMA (European Medicines Agency) mas de cada país. Nos Estados Unidos o FDA, Food and Drugs
Administration, pode designar um biossimilar como intercambiável com o produto de referência, mas os estados regulam as leis relacionadas à substituição entre drogas. O Health Canada, por sua vez, não declara a intercambialidade para genéricos nem para biossimilares. A intercambialidade permanece uma decisão clínica ou local.
Cabe destacar que o debate realizado hoje no Brasil é muito semelhante ao que vem ocorrendo no cenário internacional. Não há consenso global entre as agências, mesmo entre as que atuam mais conjuntamente como a da União Europeia, do Japão, Canadá ou Austrália. Essa falta de consenso existe mesmo em algumas questões que poderiam ser simples. Por exemplo: o que é biossimilar? Há pequenas, às vezes não tão pequenas, diferenças de conceito e isso acaba tendo decorrências importantes. Outra discussão em evidência na Europa é a necessidade de uma maior compreensão, mais coletiva e aprofundada, sobre o que significa o medicamento biológico. Quando alguém vai discutir com médicos e pacientes sobre a diferença entre um produto biológico e um sintético? E que os biossimilares não são idênticos mas também são efetivos? Sob este quadro, temos um tensionamento da indústria mundial,

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
22
entre quem produziu e desenvolveu e quem vai entrar no mercado fazendo cópia. As agências reguladoras colocam-se no meio desse conflito. Agrava-se o problema com o receio da classe médica ao uso dos biossimilares intercambiáveis, considerando que esse fato ainda ocorre para os genéricos, os quais já estão consolidados no mercado nacional há anos. A divulgação de informação sobre os biossimilares para a comunidade médica e associação de pacientes é de interesse das empresas de biotecnologia. As empresas podem assegurar a qualidade dos produtos que serão produzidos no Brasil, considerando a forma como a transferência da tecnologia de produção dos biossimilares das empresas parceiras está ocorrendo, ou seja, sob criterioso controle de qualidade. A promoção de discussões convidando a comunidade científica, pacientes e a agência reguladora é um caminho para estabelecer o acesso dos pacientes a esse tipo de tratamento de forma cientificamente embasada. Por outro lado, a promoção do uso de biossimilares não deve estar apenas a cargo das indústrias farmacêuticas. Há uma grande responsabilidade da agência regulatória, considerando o respeito que essa possui perante a sociedade. Sendo assim, caberia também à ANVISA promover o diálogo com a sociedade sobre o assunto. Se determinado biossimilar teve seu registro aprovado e passou pelo crivo técnico da agência regulatória, isso pode ser utilizado. Sendo assunto de interesse público, as sociedades médicas também devem possuir papel ativo, discutindo esse tema para que haja conhecimento da classe médica e consequentemente dos pacientes e da sociedade em geral.
3.8) Sessão Temática 4 - Pré-clínicos: custo ou investimento? Quais as melhores práticas e
alternativas? COMPOSIÇÃO DA MESA - Moderador: Carlos Tagliati, Professor da UFMG, apoiadora do evento e sócio fundador da empresa InVitroCells. Palestrantes: Norberto Rech, Consultor e ex-diretor da Anvisa; Simone Fanan, Diretora Executiva da Tridskin e da Socieda Brasileira de Métodos Alternativos (SBMAlt; Joel Majerowitz, que também está acompanhando a questão dos métodos alternativos na Anvisa. Debatedores: Silvia Berlanga, Professora da USP, e Octávio Pesgrave, do Dep. Farmacologia e Toxicologia do INCQS / Fiocruz. Relatores: Silvia Barreto Ortiz, Consultora e Caio Victor França, da Biozeus.
A discussão acerca da qualidade dos estudos pré-clínicos permeia o amplo debate sobre a queda da eficiência em P&D na indústria farmacêutica global e a escalada dos custos de desenvolvimento de novos medicamentos. A tendência que tem origem no final dos anos 1990 é alvo de ampla investigação que, até o momento, revela uma série de fatores, desde aspectos técnico-científicos até modelos de gestão em prática no setor. Nesse contexto, confiabilidade e reprodutibilidade dos resultados obtidos em ensaios pré-clínicos são, sem dúvida, um dos fatores associados à alta taxa de insucesso na busca por fármacos e medicamentos inovadores. Nesta nona edição, a abordagem da Sessão Temática sobre pré-clínicos analisou a importância de ensaios pré-clínicos para o processo de desenvolvimento de novos fármacos, de elaborar e implementar modelos de análise capazes de substituir o uso de animais e, também, de estabelecer uma rede brasileira de prestadores de serviço credenciados à realização de ensaios atendendo aos padrões de qualidade internacional. Estudos de toxicologia pré-clínica com novas entidades moleculares têm papel de destaque na redução do risco de reprovação em fases posteriores por questões de segurança. Com o crescimento da escala de estudos em curso em todo o setor farmacêutico, o uso racional de animais de experimentação tornou-se matéria de discussão em órgãos sanitários e entidades internacionais envolvidas na regulamentação do desenvolvimento de novos medicamentos. Autoridades internacionais cogitam a possibilidade de eliminar por completo estudos envolvendo animais. Diversas

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
23
técnicas têm sido desenvolvidas no sentido de aumentar o poder preditivo de sistemas in vitro no que diz respeito à segurança e eficácia de novos fármacos. O Conselho Nacional de Controle de Experimentação Animal (Concea) já reconhece 17 métodos alternativos envolvendo modelos baseados em células ou metodologias in vivo executadas com menor número de animais. Tal reconhecimento foi estendido pela Anvisa.
Apesar dos possíveis avanços no sentido de alcançar maior nível de participação de testes in vitro no programa de desenvolvimento pré-clínico de novos candidatos terapêuticos, há que se manter em mente que a substituição completa de modelos animais é um cenário distante. Parece haver claro consenso de que testes farmacológicos e toxicológicos in vivo continuarão sendo imprescindíveis ao cumprimento das diretrizes das agências regulatórias, ao menos nos próximos anos. As técnicas que permitirão a substituição do uso de animais ainda estão em desenvolvimento e há poucas metodologias reconhecidas no que diz respeito ao teste de medicamentos. O processo de adoção de novos métodos dependerá da harmonização de práticas entre diferentes entidades internacionais. A Organização para Cooperação e Desenvolvimento Econômico (OCDE) foi citada como um dos órgãos que atua de forma proativa no sentido de estabelecer protocolos padronizados que permitam a certificação. No cenário brasileiro, a demanda por animais de experimentação certificados é mal atendida, dificultando a ampla atuação de prestadores de serviço na área. Contudo, não há ainda evidências suficientes para total confiança na substituição de ensaios clássicos por métodos alternativos. Também há um trabalho conjunto de instituições brasileiras ligadas à elaboração, estabelecimento e acreditação de padrões técnicos, como o Instituto Nacional Controle Qualidade em Saúde (INCQS – Fiocruz), Laboratório Nacional de Biociências (LNBIO) e Instituto Nacional de Metrologia, Qualidade e Tecnologia (INMETRO), no sentido de desenvolver metodologias alternativas baseadas em sistemas in vitro.
O Brasil vem buscando, talvez de forma ainda tímida, organizar uma rede de serviços de P&D em fármacos, principalmente no que tange ensaios pré-clínicos. Sob este aspecto, o mapeamento dos centros já existentes no Brasil pode auxiliar no direcionamento dos investimentos voltados à estruturação deste tipo de serviço no país. Ao mesmo tempo, a capacitação de mão de obra adequada é imprescindível à prestação de serviços de qualidade. De certa forma, pode-se afirmar que a baixa inserção de grupos brasileiros em redes internacionais de ensaios pré-clínicos é devido à escassez de profissionais capacitados à disposição dos centros brasileiros.
A deficiência dos estudos realizados no Brasil dá-se por conta da falta de certificação das instituições nacionais. Percebe-se uma posição negativa das agências regulatórias de outros países em relação aos estudos realizados no Brasil. Como consequência, empresas que constroem seus dossiês com resultados gerados por prestadores de serviço brasileiros por vezes necessitam repetir ensaios em ambientes certificados, fora do Brasil. Verifica-se, ainda, o atraso das autoridades brasileiras em alcançar a capacidade de certificar prestadores de serviço nacionais, dificultando a entrada dos mesmos em redes internacionais de ensaios pré-clínicos. A participação da Anvisa (e do INMETRO) para acreditar os centros nacionais como operadores nas boas práticas laboratoriais será essencial ao reconhecimento da confiabilidade dos prestadores de serviço nacionais. Ademais, o posicionamento claro da autoridade sanitária brasileira reforçará o pleito do Brasil a participar nos comitês internacionais que norteiam as melhores práticas no setor farmacêutico.
A fim de participar das cadeias internacionais de desenvolvimento de novos fármacos, as empresas brasileiras terão de se adequar aos padrões internacionais, não apenas nos quesitos técnicos, mas também em termos de velocidade de execução dos serviços. O estabelecimento de grande parte das companhias nacionais em instituições acadêmicas torna a dinâmica dos serviços burocrática e lenta. Somando-se à

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
24
pequena escala do fluxo de trabalho, que encarece a atividade, e a desconfiança por parte de agências sanitárias de outros países, prestadores de serviço brasileiros têm grande dificuldade de enfrentar a concorrência global.
Em resumo, a questão pode ser encarada, ao menos, sob duas óticas distintas. Do ponto de vista das empresas farmacêuticas, que necessitam construir dossiês de qualidade para alcançar o registro de seus produtos, a realização de estudos dentro dos padrões internacionais representa um gasto inevitável que, por isso, deve ser executado da forma mais racional e direta possível. Por outro lado, sob a ótica da gestão pública, fomentar a capacitação e garantir meios para certificação das empresas nacionais corresponde a um importante investimento, que proverá suporte à estratégia de inserção do Brasil em redes globais de inovação em fármacos e medicamentos. A estruturação de uma cadeia brasileira de prestadores serviços pré-clínicos acreditados em nível internacional dará suporte à indústria nacional e à participação de instituições brasileiras em estudos pré-clínicos promovidos por multinacionais.
3.9) Sessão Temática 5 - Criando os elos fundamentais da cadeia de Fitoterápicos: extratos e
marcadores COMPOSIÇÃO DA MESA - Moderador: Luis Carlos Marques, Professor e orientador da Universidade Anhanguera de São Paulo – UNIAN. Palestrantes: Frederico Silva, da empresa LAS do Brasil; e Fábio Perazzo, Professor da UNIFESP. Debatedoras: Luzia Franco, Coordenadora de PD&I da Herbarium; e Vanderlan Bolzani, Diretora da Agência de Inovação da UNESP (AUIN), Coordenadora do Núcleo de Bioensaio, Biossíntese e Ecofisiologia de Produtos Naturais do IQ-Ar (NuBBE) e Cris Ropke, da Phytobios. Relatora: Sylvia Loloma, Gerente de desenvolvimento de negócios da ThomsonReuters.
Na área de fitoterápicos ainda há nichos a serem explorados pois, apesar dos profundos debates, a
Fitoterapia ainda necessita de articulação política. Desde 2009, os temas abordados no ENIFarMed foram diversos, desde doenças negligenciadas, fitoterapia no mundo, problemas de qualidade, inovação e regulamentação. Mas, de forma concreta, pouco resultado foi alcançado nestes fóruns de discussão em função da falta de articulação dos pontos técnicos discutidos e a sua capitalização em temas estratégicos. O mercado de fitoterapia brasileiro representa um mercado de US 1,2 milhões e vem, ao longo dos últimos anos, caindo, refletindo a falta de coordenação entre os anseios e ações concretas públicas.
A questão do risco e do investimento em inovação requer ousadia e, para se obter resultados frente à biodiversidade brasileira, é necessária uma ação coordenada entre todas as áreas. O investimento em extratos presentes no European Medicines Agency (EMA) pode não configurar interesse imediato devido ao custo e necessidade de investimento para o empresariado brasileiro, mas extratos da biodiversidade brasileira ainda apresentam oportunidades e um trabalho coordenado entre fabricante de extrato e fabricante de medicamento pode resultar em casos de sucesso.
Destacam-se alguns gargalos:
• Poucas empresas brasileiras com extratos padronizados. O mercado mundial impõe documentação robusta a preços altamente competitivos. Como o foco é a qualidade, as empresas devem se adequar a esta realidade;
• Faltam dados mercadológicos;
• Farmacopéia Brasileira não tem ainda fitoterapia inclusa, sendo necessário trabalhar a respeito;
• A mão-de-obra especializada não é um problema;

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
25
• A Renasus foi criada para o interesse nacional e não efetivou seu objetivo;
• Os APL´s do Ministério da Agricultura em APL´s não tem rastreabilidade. Este ponto precisa ser discutido para o produto ser mais efetivo.
Em suma, os maiores gargalos têm três principais frentes: (1) Farmacotécnico: como necessidade dos fabricantes nacionais elaborarem documentos robustos, extratos padronizados desde a cadeia do plantio, a preços competitivos equiparados com o mercado global e padronização dos medicamentos, (2) Governamental: como revisão das APL´s e conferir um ambiente regulatório previsível, e (3) Clínico: investimentos em estudos clínicos estruturados atendendo à demanda da classe médica e dos pacientes de terem segurança e eficácia. Uma das soluções propostas foi a de que o BNDES fomente o mercado com editais para extratos de plantas nativas com marcadores.
A questão crítica é trabalhar a padronização dos medicamentos fitoterápicos pois a identificação química já está bem estabelecida mas a linha clínica ainda é limitada e o médico ainda não acredita na fitoterapia. É necessário organizar resultados clínicos de forma estruturada voltada para o racional médico. Nos anos de 1985 a 1993, houve 7.405 casos de intoxicação e naquele momento foi necessário calcular o fator de risco x benefício na utilização das plantas medicinais. Os estudos em fitoterapia e a padronização dos extratos se intensificaram e, hoje, esta questão já é uma tendência do setor. Mas a indústria ainda tem dificuldade com o quesito estudo de segurança e eficácia, pois há o questionamento do “Porque investir, se o paciente não tem a percepção da diferença clínica entre o medicamento e o Fitoterápico?”, sendo que somente com o estudo clínico é possível comprovar os diferenciais dos mesmos e, às vezes, até mesmo mostrar menor evento adverso.
Outro ponto se relaciona à definição extrato: (1) Extratos padronizados (ajuste teor com adição de excipientes inertes ou mistura de lotes) e (2) Extratos quantificados (ajuste teor com adição mistura de lotes de extratos). Futuramente pode haver críticas até pertinentes ao Good Manufacture Practice (GMP) e à padronização de lote, já que os extratos padronizados seriam mais rastreáveis em termos de processo comparados aos extratos quantificáveis. Sugere-se fornecer apoio às startups e ter mais parcerias com as indústrias, fortalecendo as linhas de pesquisa.
Atualmente, 50% do consumo da empresa Herbarium vêm de extratos importados. A empresa possui procedimentos internos de rastreabilidade do extrato: do produtor ao produto acabado. Deve-se ter em mente que o processo de auditoria será facilitado com a elevação do número de indústrias nacionais e, ao estabelecer linhas de parcerias, é preciso focar em projetos que apresentem valor agregado.
Dentre as principais dificuldades, pode-se destacar: - Marcos regulatórios que impactam no desenvolvimento de novos produtos; - Dificuldades na padronização do lote a lote; - Semelhança dos extratos.
Alguns pontos críticos na escolha de um fornecedor, como a troca de um fornecedor ou a inserção de uma segunda fonte, somados a problemas como falta de especificação com marcadores, diferença nos limites de solventes e diferenças nos pesticidas aos aprovados, além dos custos, geram problemas farmacotécnicos e de suprimentos que, geralmente, são impossíveis de se inserir uma segunda fonte por falta de similaridade e competitividade, o que causa um questionamento se vale a pena mudar o fornecedor existente de extrato.
Em um pipeline de projetos de inovação, há 2 tipos de projetos: - Extratos Novos, listados no EMA: sendo estes mais fácil de comprar o extrato do exterior, pois certamente já estará padronizado e terá menor custo;

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
26
- Extratos Inovadores Brasileiros: neste caso, é mais fácil trabalhar com o fornecedor local, buscando padronizar o extrato e conduzindo o processo de acordo com a necessidade, realizando estudo clínico e adequações de dossiê de acordo com a legislação vigente.
O mercado de bioprodutos (substâncias derivadas da biodiversidade) é um setor que requer uma visão organizada e que oferece muitas oportunidades. A Lei do Bem auxilia a realização de inovações no Brasil e é um recurso que deve ser usado. Como exemplo, temos a região amazônica, que apresenta o maior índice de patentes em eletrônica e não na biodiversidade. Logo, um trabalho coordenado é essencial. Em termos científicos, já é possível fazer análise dos extratos de forma completa.
Assim, as barreiras que existem são: - Padronização dos plantios e das coletas; - A cadeia produtiva deve ser mais ousada; - As APL´s e sua aplicação devem ser revistas; - Para a indústria, a customização dos extratos gera custos muito altos e não possibilita escala industrial; - O governo poderia ajudar com demandas especificas; - Melhor discussão das novas RDC´s; - Falta de previsibilidade regulatória. 3.10) Sessão Temática 6 - Premiação Reconhecimento Técnico: integração entre pesquisadores e o
setor farma Apresentador: Jorge Luis Audy, representando a PUC-RS, a Anprotec, a Capes e o CNPq.
Membros do Comitê Científico-Tecnológico de Seleção para a Premiação Reconhecimento Técnico: Carlos Tagliati, Presidente do Comitê, professor na UFMG e sócio fundador da empresa InVitroCells João Carlos Gabaglia, Agência Inovação da UFRJ Julia Paranhos, UFRJ Luis Lopez Martinez, NAPesq - Núcleo de Apoio à Pesquisa Clínica /HCFMUSP Maria Behens, Farmanguinhos/Fiocruz Roberto Nicolsky, UEZO e Protec Rubens Alves Pereira, NUPEEC-UFPel Sergio Jose Mecena da Silva Filho, UFF/Escola de Engenharia Thais Helena Gasparoto, Faculdade Odontologia/USP William Waissmann, ENSP/Fiocruz
Premiados e os respectivos títulos dos trabalhos: 1º - Wanise Barroso, Fiocruz - Subsídio ao exame de pedidos de patente, uma solução para o acesso aos medicamentos de Hepatite C 2º - Marcelo Davanço, UNESP e UFPE - Estudo farmacocinético pré-clínico de novos comprimidos de benznidazol para o tratamento da doença de Chagas. 3º - Ricardo Dalla Costa, Thermo Fisher Scientific - Development and Validation of a Scalable Next-Generation Sequencing System for Assessing Relevant Somatic Variants in Solid Tumors 4º - Silvia Cuffini, UNIFESP - Correlation between crystalline microstructure and bioequivalence in Anti-HIV Drug Efavirenz

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
27
5º - Livia Deris Prado, Farmanguinhos -Desenvolvimento de formulação de efavirenz para o incremento da dissolução 6º - Petrônio Athayde-Filho, UFPB - Modulação da resistência a drogas por derivados do ácido selenoglicólico em staphylococcus aureus
3.11) Sessão Temática 7 - Pesquisa Clínica: como alavancar esta importante fase do desenvolvimento
de medicamentos? COMPOSIÇÃO DA MESA - Moderador: João Massud, Diretor Científico da SBMF e sócio da TrialsConsulting. Palestrantes: Ricardo Eccard, Especialista em regulação e vigilância sanitária da Anvisa; Jorge Venâncio, da Comissão Nacional de ética em Pesquisa (CONEP), Mirian Franco, Assessora Científica Comitê de Ética em Pesquisa-CEP da UNIFESP. Debatedores: João Batista Calixto, Professor Titular da UFSC, e Gilberto de Nucci, Professor Titular da Unicamp, Professor da USP, e consultor da Biolab. Relatores: Pedro Ohara, Gerente de Operações Clínicas da Avanti e Dyeison Antonow, Professor/Pesquisador do Instituto de Pesquisas Biomédicas da Faculdade de Medicina da PUCRS.
Cabe destacar que finalmente o fórum obteve êxito em conseguir uma excelente composição dos
palestrantes e debatedores, incluindo no debate a Agência Nacional de Vigilância Sanitária (ANVISA) e a Comissão Nacional de Ética em Pesquisa (CONEP). A importância da pesquisa clínica em diversos avanços na saúde se dá através do aumento da expectativa de vida, controle da AIDS, cura de alguns tipos de câncer, aumento da estimativa e qualidade de vida de outros além da diminuição/controle proporcional dos eventos cardiovasculares fatais. Apesar de ocupar a 9ª posição mundial em termos de economia e 6º lugar no mercado de medicamentos, o Brasil possuiu uma reduzida produção de pesquisa e desenvolvimento, que leva à diminuição da capacitação dos profissionais e da descoberta e do desenvolvimento de novas tecnologias.
A avaliação metodológica dos projetos de pesquisa clínica implementadas pela Resolução de Diretoria Colegiada da ANVISA - RDC #9/2015 busca: harmonizar os processos da ANVISA com os processos das agências internacionais (Food and Drug Administration - FDA e European Agency for the Evaluation
of Medicinal Products - EMEA, por exemplo); solidificar os procedimentos das Inspeções de Boas Práticas Clínicas realizadas pela ANVISA; reduzir os prazos para aprovação de estudos clínicos; evitar a avaliação múltipla de documentos iguais por diferentes relatores (por exemplo, brochuras do medicamento/produto em teste que, na legislação antiga, era analisada para cada um dos estudos clínicos, mesmo que esses fossem realizados com o mesmo medicamento/produto em teste) - o objetivo é ter um relator analisando todos os estudos clínicos de um mesmo medicamento/produto em teste; definir escopos de análise diferentes para os setores internos da ANVISA, separando os medicamentos (RDC #9/2015) dos produtos para saúde (RDC #10/2015); facilitar os processos de importação de produtos para estudos clínicos, isentando-os da necessidade de aprovação prévia ao embarque das licenças de importação pela sede da ANVISA (Brasília) - a análise e o deferimento serão realizados localmente pelo Posto Aeroportuário (PAP) do aeroporto de entrada; avaliação do plano de desenvolvimento do medicamento e/ou produto em estudo para verificação da forma global do desenvolvimento e da antecipação de possíveis riscos e fragilidades do fármaco ou medicamento em teste; implementação da verificação do controle de qualidade da produção dos produtos em teste; verificação dos estudos não clínicos quanto à sua execução de acordo com as Boas Práticas de Laboratório e validação dos resultados.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
28
Em julho de 2015 todas as análises iniciais dos estudos submetidos à ANVISA sob a RDC 39/2008 foram finalizadas. A partir de julho, todas as análises iniciais pela Coordenação de Pesquisa Clínica em Medicamentos e Produtos Biológicos (COPEC) são de protocolos submetidos sob a RDC #9/2015. Ficou bastante clara a necessidade de um desenvolvimento e padronização das análises feitas pela ANVISA, redução progressiva dos tempos de análise dos estudos, verificação e acompanhamento do cumprimento dos prazos estipulados pelas RDCs # 9 e 10 e, acima de tudo, a continuidade do processo de formação de técnicos capacitados para analise das submissões recebidas.
Em relação às atividades da CONEP, destacam-se: abertura do processo de diálogo com todos os centros de pesquisa; análises dos projetos visando a defesa ética dos participantes de pesquisa clínica porém sem impactar drasticamente a condição do projeto; redução significativa dos prazos de análise nos últimos 24 meses, reduzindo o número de estudos aguardando a primeira análise de 180 para 90 dias; implementação da acreditação de Comitês de Ética em Pesquisa (CEPs) para redução da dupla análise de estudos clínicos de risco leve a moderado ao participante de pesquisa. A CONEP torna-se, então, a última instância para avaliação de casos em que os CEPs acreditados julgam não serem capazes de analisar e de estudos de elevado risco ao paciente. A classificação de risco será automática e feita com base nas informações iniciais inseridas na Plataforma Brasil; distribuição de um manual aos CEPs cadastrados na Plataforma Brasil listando os erros mais comuns verificados pela CONEP e informando sua resolução; lançamento da Plataforma Brasil 3.0 e os planos de atualização – 3.1 (outubro de 2015) inserção da entrada de eventos adversos diretamente na Plataforma Brasil e 3.2 (dezembro de 2015) inserção da listagem de Biobancos e controle dos mesmos através da Plataforma Brasil. Como pontos de melhoria e desenvolvimento, foram listados a diminuição contínua do tempo de análise inicial dos projetos e a necessidade de padronização das análises entre os relatores da CONEP.
O fato da UNIFESP possuir um CEP único para analisar todos os projetos de todas as especialidades faz com que haja um volume muito grande de projetos avaliados mensalmente pelo CEP da UNIFESP. Está em estudo a criação de outros CEPs devido à expansão da UNIFESP para outro campus. Destacam-se: profissionalização dos CEPs e dos seus membros, visando uma melhor qualidade de análise dos estudos clínicos e principalmente agilidade da análise; dedicação exclusiva dos membros do CEP às atividades de análise dos estudos clínicos. Atualmente, os membros dos CEPs têm a atividade nos CEPs como uma atividade extra e não remunerada; treinamento dos membros do CEP de acordo com um plano de ensino estabelecido pela CONEP; acreditação dos CEPs pela CONEP só será possível após a implementação do plano de ensino e treinamento definido pela CONEP para harmonizar o conhecimento e procedimento de cada uma dos CEPs acreditados, evitando analises dispares; problemas da Plataforma Brasil.
Há necessidade de maior desenvolvimento da pesquisa pré-clínica no Brasil que, por sua vez, incentiva e embasa o desenvolvimento de projetos de pesquisa clínica nacionais. Um dos pontos falhos da pesquisa pré-clínica no Brasil é a falta de formação de desenvolvedores de novas pesquisas pré-clínicas. A nova RDC #9/2015 da ANVISA exige a padronização dos estudos pré-clínicos com as boas práticas de laboratório, evitando assim que estudos sem qualidade sejam utilizados como base de estudos clínicos desenvolvidos futuramente com os resultados obtidos. Além disso, destaca-se que a padronização e o maior controle da ANVISA nos testes pré-clínicos permitem que o foco seja direcionado cada vez mais para a qualidade de novos projetos analisados, ficando assim as questões de segurança pré-avaliadas pelas normas de Boas Práticas de Laboratório (BPL). Pontos de melhoria e desenvolvimento a serem resolvidos: maior incentivo e desenvolvimento de estudos pré-clínicos; padronização da execução dos projetos pré-clínicos; alavancagem dos projetos de pesquisa clínica e pré-clínica de desenvolvimento nacional.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
29
A melhor avaliação pela ANVISA de projetos propostos com medicamentos que já se encontra em comercialização (nova dose ou apresentação) foi exemplificada através de um projeto proposto com uma determinada molécula que já é comercializada na dose de 10 mg. Este mesmo medicamento, que já possui estudos pré-clínicos e estudos clínicos fases I, II III e IV desenvolvidos e com resultados publicados e registrados na ANVISA, ao ser proposto na dose de 5 mg em uma nova apresentação e com comprovada diminuição da biodisponibilidade do fármaco, recebeu a solicitação de um estudo fase I da ANVISA para avaliação de sua tolerabilidade. Tal solicitação não faz sentido, uma vez que a tolerabilidade já foi previamente analisada em dose superior e com maior biodisponibilidade no registro da dosagem registrada de 10 mg.
Pontos finais de discussão: a ANVISA possui capacidade plena de avaliar estudos de fase I, II, III ou IV, apesar do número reduzido dos dois primeiros que seguem as diretrizes internacionais de avaliação de estudos não-clínicos. A questão sobre a acreditação dos CEPs gerou a discussão sobre um possível conflito de interesses na avaliação de estudos entre instituições de grande porte. Foi citado o exemplo de que a acreditação pode gerar medidas diferentes de análises pelos CEPs credenciados quando estes forem selecionados para analisar os projetos da instituição que, historicamente, compete com a instituição proponente. Foi citado o exemplo da possibilidade do CEP da Faculdade de Medicina da Universidade de São Paulo (FMUSP), ao ser selecionado como CEP acreditado para a segunda verificação dos estudos da UNIFESP, utilizar pesos e medidas mais rigorosos e não aplicáveis na avaliação do projeto que usaria na análise de projetos internos e vice-versa. Como resultado desta discussão, ficou claro que o processo de acreditação precisará ser desenvolvido em diversos âmbitos, especialmente na qualificação e instrução dos relatores para padronização das análises realizadas. O ponto final de discussão apresentado foi sobre a necessidade da CONEP focar suas atenções em questões éticas de análise e não se ater a questões relacionadas à semântica das palavras e, acima de tudo, definição clara dos padrões a serem utilizados nas avaliações para que os estudos possam ser elaborados de forma correta, contribuindo ainda mais com a redução dos tempos de avaliação e aprovação. 3.12) Sessão Temática 8 - Eficácia e segurança de produtos contendo nanotecnologia no setor
cosmético e no farmacêutico COMPOSIÇÃO DA MESA - Moderadora: Natália Cerize, Pesquisadora do IPT-SP. Palestrantes: Adriano Marim de Oliveira, Pesquisador IPT-SP, Simone Sotto Mayor, Consultora técnica especializada da Mayor Consultoria, e Renata Raffin, Diretora de Inovação da Inventiva. Relator: Yuri Tukoff, da Coordenadoria de Planejamento e Negócios IPT-SP.
A nanotecnologia, apesar de ser uma área relativamente recente, é transversal, pode ser aplicada em vários campos do conhecimento como eletrônica, energia e saúde e possui relevante penetração de mercado em diversos segmentos, em produtos já incorporados no cotidiano, como lentes de óculos, protetores solares, embalagens de alimentos, roupas, revestimentos aplicados na construção civil, dentre outros. Além disso, é vista como oportunidade de desenvolvimento dos países. Em 2011, a Federação das Indústrias do Estado de São Paulo (FIESP) publicou um trabalho que apresentava dados da importância da área no Brasil: 1/3 dos cientistas, 50% da produção de nanotecnologia na América Latina, 1099 empresas no setor e potencial de mercado de US$ 33 bilhões em 2020. O mesmo estudo indicava que os setores com maior potencial em nanotecnologia para o Brasil eram os de produtos farmacêuticos, químicos e cosméticos, além do setor de energia. A combinação de recursos terapêuticos e de diagnóstico (Theranostics) é a atual fronteira do conhecimento em nanotecnologia, com pesquisas iniciadas, mas

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
30
mercado ainda incipiente. Outras áreas de interesse recente são: nanocarreadores empregando conjugações de polímeros, lipídeos, e micelas; diagnóstico e liberação direcionada; co-entrega de fármacos; conjugação de ligantes biológicos; liberação estímulo-responsiva e terapia de combinação sinérgica, dentre outros desenvolvimentos. Outro aspecto fundamental a ser levantado é o dimensional. O debate não deve ser centrado em explicar se nanotecnologia são partículas ou cápsulas abaixo de 100 nanômetros ou 1 micrômetro. O foco dos pesquisadores e da indústria deve estar sempre voltado em como a dimensão da partícula pode influenciar a segurança e a eficácia de fármacos, cosméticos e demais produtos fabricados em tais dimensões.
O debate perpassou a eficácia de produtos contendo nanotecnologia e trouxe uma perspectiva de “desenvolvedor” de processos/produtos empregando a tecnologia com uma visão de uma instituição de pesquisa, desenvolvimento e apoio à inovação das empresas que atende a indústria nacional e aos setores público e privado. Também evidenciou a diferenciação entre nanociência e nanotecnologia, além de mostrar conceitos relativos à nanotecnologia farmacêutica (que engloba nanomateriais e dispositivos para liberação controlada de ativos farmacêuticos, diagnósticos, imagens e biossensores) e à nanomedicina (que consiste no uso tratamento, diagnóstico, monitoramento e controle de sistemas biológicos por meio de módulos submicrométricos). Mas, “porque nano?”. Existe um certo modismo no uso do termo “nano”, e seus efeitos associados à diminuição do tamanho de partículas, cápsulas e estruturas permitem técnicas tais como: mudança de cor em função do tamanho da partícula (efeito quântico), aumento da área de reação da partícula e penetração de sistemas de liberação controlada (efeito de borda). Mudar a escala de tamanho de uma partícula modifica as propriedades de um material e amplia seu potencial de aplicação e, assim, verifica-se vantagens da nanotecnologia aplicada nas ciências da vida. A nanoescala aplicada em fármacos aumenta a biodisponibilidade oral e é mais aceita pelo paciente já que sua absorção pelo organismo aumenta em comparação com remédios administrados por via oral e produzidos em escala maior do que a nanométrica. Em relação ao seu uso em fármacos, outros aspectos positivos apresentados foram: aumento de área superficial, modificação de solubilidade, aumento de taxa de dissolução, doses menores, ação mais rápida e menor variabilidade entre pacientes. Quanto às formas de se fabricar nanoestruturas, foi explicado que a construção top-down funciona por subtração de um material bruto, esculpindo-o. De maneira antagônica, a construção bottom-up consiste na deposição camada a camada de material, até se formar a estrutura desejada, tal como é feito na manufatura aditiva, popularmente conhecida como “Impressão 3D”.
Foram apresentados seis processos de produção de produtos em escala nanométrica: Emulsificação e Difusão de Solvente, Emulsificação e Evaporação de Solvente, Polimerização Interfacial, Polimerização In-Situ, Secagem por Spray-Dryer e Homogeneização de Alta Pressão. O desenho do processo é a rotina do Instituto de Pesquisas Tecnológicas (IPT) e esses processos já eram feitos pela instituição sem que recebessem a denominação de processos nanotecnológicos. Como exemplos de projetos concluídos pelo IPT, foram citados o projeto de desenvolvimento de mecanismos microfluídicos para redução do padrão cristalino e aumento da taxa de dissolução de um ativo e o projeto de um protetor solar nanoestruturado com controle do filtro nas faixas de radiação solar UVA e UVB. E, para ilustrar a cadeia produtiva simplificada da nanotecnologia, foram apresentados os três estágios da nanotecnologia. O primeiro estágio é o de nanomateriais, que são estruturas em nanoescala não processadas. Como exemplos dessa etapa da cadeia produtiva podem ser citados nanopartículas, nanotubos, pontos quânticos, fulerenos, dendrímeros, materiais nanoporosos, dentre outros. Na segunda etapa da cadeia de nano estão os nanointermediários, que consistem em produtos intermediários com componentes em nanoescala. São exemplos desta etapa revestimentos, tecidos, chips de memória, componentes óticos, materiais biocombustíveis e fios

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
31
supercondutores. Por fim, foram mostrados os nanoprodutos, definidos como bens de consumo que incorporam nanotecnologia. Esses são os produtos de uso cotidiano como celulares, automóveis, roupas, cosméticos, dentre outros. Para a fabricação dos componentes de cada uma das três etapas supracitadas, utilizam-se nanoferramentas, ou seja, equipamentos e programas de computador que possuem as funções de visualizar, manipular e modelar matéria em nanoescala. Em suma, “quem faz nano, faz nanomateriais; quem vende nano, vende nanoproduto”. Destaque para a iniciativa do Governo Federal, por meio do Ministério da Ciência, Tecnologia e Inovação (MCTI), com a operação do Sistema de Laboratórios em Nanotecnologia (SisNano), do qual o IPT é associado. Outra iniciativa de destaque foi a instituição do Núcleo de Bionanomanufatura no IPT. O Núcleo congrega, em suas instalações, laboratórios nas áreas de Nanotecnologia, Biotecnologia, Microtecnologia e Metrologia dimensional de alta precisão. Em nanotecnologia, foi ressaltada a experiência do IPT com a resolução de problemas da indústria no que se refere à nanoencapsulação de ativos, síntese de nanofibras, síntese de nanocompósitos, síntese de nanopartículas cerâmicas, modificação de superfície de nanoestruturas, sistemas de liberação controlada e técnicas de caracterização avançadas.
Quanto às especificidades dos aspectos regulatórios relacionados à nanotecnologia em diferentes países e regiões do mundo, foi apresentada a caracterização das diferentes agências, institutos e órgãos reguladores nos Estados Unidos. Em relação à colaboração entre as 25 agências que coordenam ações voltadas para a nanotecnologia, foi apresentada a Nano.gov (National Nanotechnology Initiative); para financiamento de projetos que envolvem nanotecnologia em saúde, foi citada a NIH (National Institutes of
Health). Sobre o FDA (U.S. Food and Drug Administration), foi relevado seu papel na promoção da segurança de alimentos, biofármacos, dispositivos médicos e vacinas, embora a agência não adote definições regulatórias de “nanotecnologia”, “nanoescala” ou termos relacionados. O FDA tem um caráter muito mais voltado à colaboração do que à punição e sempre recomenda que se consulte a agência antes de se desenvolver produtos que envolvam nanotecnologia. Em junho de 2014, a agência lançou um guia para a segurança de produtos cosméticos envolvendo nanotecnologia ou utilizando nanomateriais, que preconiza que a segurança de biomateriais deve ser analisada sob a ótica da caracterização do nanomaterial e de sua toxicologia.
Como exemplo de outros países, foram citadas as políticas do órgão canadense Health Canada. Tomando-se como base o que é preconizado pela política canadense “Policy Statement on Health Canada's
Working Definition for Nanomaterial” de outubro de 2011, os nanomateriais são aqueles que apresentam efeitos técnicos diferenciados em função da escala da partícula. Quanto à segurança, o órgão canadense considera que os nanomateriais não são mais perigosos para a saúde humana ou para o meio ambiente do que outras substâncias químicas. Visão similar é defendida pelo REACH (Registration, Evaluation,
Authorisation and Restriction of Chemicals) órgão da região europeia que considera que substâncias químicas são tão seguras ou perigosas quanto a nanotecnologia. Outra iniciativa europeia para regulação, desta vez em nível internacional, é o NanoReg. O Brasil é membro, sendo um dos poucos países de fora da União Europeia a participar da iniciativa. A coordenação científica do NanoReg no Brasil é do Instituto Nacional de Metrologia, Qualidade e Tecnologia (INMETRO). Já a gestão fica a cargo do Ministério da Ciência, Tecnologia e Inovação (MCTI). O NanoReg tem importante papel na intermediação entre Instituições Científicas e Tecnológicas (ICTs) e agências reguladoras nacionais. Mais outra iniciativa europeia de regulação e parametrização da nanotecnologia apresentada foi a NanoForce. A recomendação geral da NanoForce é de que a tecnologia precisa de regulamentação baseada em nanociência para evitar o efeito negativo de restrições, por meio da desaceleração do desenvolvimento da nanotecnologia, considerando que ela já produz impacto sobre a economia de países como Alemanha e Coreia do Sul e

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
32
gera soluções tecnológicas importantes, dentre outros aspectos. Quatro grandes diretrizes foram apresentadas sobre essa iniciativa: (1) testes padronizados preexistentes para toxicidade de materiais não são adaptáveis para nanomateriais e, sendo assim, novos testes precisam ser desenvolvidos; (2) a nanociência ajudará no desenvolvimento de novos testes para nanoprodutos; (3) impactos no meio ambiente e na saúde causados por nanoprodutos devem ser avaliados caso a caso; (4) os nanomateriais, na maioria dos casos, vão se aglomerar ou se dissolver, não agindo sobre seres vivos como nanoobjetos.
O norteador do desenvolvimento da nanotecnologia é o princípio da precaução, apresentado originalmente na Rio-92. Por meio dessa diretriz, deve se desacelerar o desenvolvimento enquanto não sejam sanados potenciais efeitos colaterais indesejados. A conclusão é de que existem muitas dúvidas quanto à segurança e à toxicidade de nanoprodutos. Soluções podem estar na resolução de lacunas apontadas pela NanoForce no que se refere à definição uniforme, protocolos e parâmetros uniformizados entre os países no desenvolvimento da nanotecnologia. E o prazo para essas resoluções já foi dado: 2020. O panorama brasileiro atual é centrado em iniciativas como o NanoReg, o Fórum de Competitividade em Nanotecnologia, o Projeto de Lei referente à rotulagem de produtos que envolvem nanotecnologia (PL 5133/ 2013) e a Política Nacional de Nanotecnologia (PL 6741/ 2013). O projeto de rotulagem prevê a colocação no rótulo de frases como “Obtido por Processo Nanotecnológico” e “Contém Ingrediente Nanotecnológico”, além da inserção de símbolos de advertência. Já a Política Nacional de Tecnologia foi considerada como “burocrática”. Diante das políticas nacionais para regulação da nanotecnologia levanta-se a questão: “Nossas políticas serão preventivas ou restritivas?”.
É ampla a perspectiva das novas tecnologias no desenvolvimento de produtos como, por exemplo, a vetorização de ativos farmacêuticos para tratamento eficaz de moléstias e redução de efeitos colaterais em tratamentos de saúde. Vetorizar consiste em levar o ativo para o local exato a ser tratado. Sendo assim, foram apresentadas três estratégias para vetorizar um ativo: (1) pró-fármacos (planejamento e síntese); (2) reconhecimento molecular (biologia molecular); (3) métodos físicos (farmacotecnia). As vantagens de se usar nanotecnologia na área farmacêutica são: levar a substância ativa ao local de ação; diminuir a competição com tecidos não específicos; aumentar a especificidade de ação; aumentar a penetração celular do fármaco; reduzir efeitos adversos; aumentar a estabilidade química da substância ativa. Por outro lado, os principais problemas dos fármacos atuais consistem em sua fraca solubilidade, na remoção do ativo do corpo antes de sua efetiva ação no tratamento de moléstias, baixa efetividade e crescente toxicidade, ação em locais do corpo que não precisam de tratamento, o alvo de tratamento é muitas vezes bloqueado por barreiras biológicas do organismo e resistência do corpo contra a ação do ativo. Assim, na nanotecnologia o foco é o local do tratamento e será possível retomar estudos com ativos que foram abandonados por causar severos efeitos colaterais quando ministrados por via oral, por exemplo, e utilizá-los sob a ótica da vetorização. As dimensões e efeitos técnicos decorrentes da miniaturização de partículas também foram discutidos. Quanto menor a partícula, melhor é a sua absorção pelo organismo. Ademais, sob dimensões entre 100 e 200 nanômetros, ativos têm baixo potencial de permeação para outros órgãos. Outros aspectos positivos da nanotecnologia são a eficácia superior de fármacos, potencial maior de tempo de vida no mercado de produtos nanotecnológicos, a nanoencapsulação pode proporcionar novas patentes, dentre outras. Sob o aspecto de custos, processos nanotecnológicos são menos dispendiosos do que processos de síntese química, por exemplo. Adicionalmente, a nanopartícula de um fármaco puro tem um valor menor das partículas encontradas em medicamentos convencionais. E, para diferenciar entre nanomaterial e nanotecnologia, há que se entender os conceitos. Nanomaterial é qualquer material insolúvel ou biopersistente com, pelo menos, uma dimensão inferior a 100 nm. Nanotecnologia, por sua vez, são todas as tecnologias que se utilizam das propriedades físico-químicas e biológicas alteradas pela

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
33
escala nanométrica. Como exemplos de nanopartículas lábeis temos lipossomas, nanoesferas, nanocápsulas, nanopartículas lipídicas e nanoemulsões. Um comparativo entre fármacos convencionais e nanofármacos nas faixas tóxica, terapêutica e subterapêutica, mostra a estabilidade em dose única dos ativos farmacêuticos com nanotecnologia e a necessidade de várias doses com remédios tradicionais, que podem variar entre as faixas tóxicas e subterapêuticas de maneira cíclica por dose. A regulação de cosméticos na Europa define nanomateriais, lista as caracterizações necessárias para nanocosméticos, estabelece rotulagem específica e propõe análise de risco em cosméticos.
E, para a reflexão de todos, foram colocadas algumas perguntas: (1) os testes de segurança de medicamentos e cosméticos de hoje se aplicam para produtos em dimensões nanométricas? (2) os testes de eficácia de medicamentos e cosméticos de hoje se aplicam para produtos em dimensões manométricas? (3) resultados de tais testes são confiáveis no contexto da nanotecnologia? (4) É ou não factível a instituição de uma classificação de patentes específica para nanotecnologia? Outros questionamentos foram colocados e respondidos. A aprovação da doxorrubicina perpassou testes clínicos, e a farmacocinética ainda é feita em cães, mas a doxorrubicina não é produzida ainda no Brasil. Aliás, não há nenhum medicamento aprovado no Brasil utilizando-se de nanotecnologia. O fato dos desenvolvimentos serem em nanoescala dificulta um pouco a identificação dessas moléculas mas a nanotecnologia pode melhorar a estabilidade de fármacos. Então, como comprovar se os ativos farmacêuticos atingirão o local desejado do corpo para o tratamento de moléstias? Em animais comprova-se por meio dos seguintes processos: doseamento nos órgãos, microdiálise no tecido específico e posologia específica. A nanotecnologia pode trazer benefícios como a redução do efeito colateral dos fármacos. O uso de técnicas analíticas apropriadas permite identificar se o ativo atingido foi encapsulado mas o processo de quantificação compreende a etapa adicional de extração do ativo para a compreensão da quantidade de ativo encapsulado e possibilita a avaliação de estabilidade química relacionando fração encapsulada versus não encapsulada. E, quanto aos produtos que estão sendo analisados, a classe de medicamento que mais se avalia são os antitumorais. Esta é a classe terapêutica que lidera as pesquisas e os lançamentos de produtos em nível mundial, justamente pela vantagem da fenestração de tecido. Quando se administra um antitumoral, seja pela via parenteral ou por via intravenosa, ele tem um tempo de circulação muito maior e torna-se possível aumentar o intervalo entre as doses a serem administradas. Adicionalmente, ele se localiza preferencialmente no tumor, tendo sua distribuição seletiva por conta de seu tamanho. Hoje, é muito difícil dizer que uma nanopartícula não funcione para o tratamento do câncer. O debate é: qual é a melhor estratégia para melhorar eficácia e a degradabilidade dos materiais empregados nos antitumorais? A nanotecnologia foi “desenhada” para o câncer, pelas características de tamanho e organização do tecido tumoral. No caso do tratamento de cardiopatias, a nanotecnologia não é tão promissora, já que o espaçamento entre as células do coração é muito pequeno, ao contrário do que ocorre com os tumores. A ANVISA está registrando produtos nanotecnológicos e a eficácia de tais produtos é confirmada por meio de testes pré-clínicos e clínicos. O estágio que se encontra a nanotecnologia ainda apresenta muitos desafios de comparabilidade, eficácia e segurança, já que não é possível ter certeza se uma dose encapsulada possui relação com outro volume de dose, só que sem encapsulação. Além disso, pesquisadores e a indústria deverão estar atentos para novos efeitos colaterais decorrentes do uso de nanotecnologia em fármacos e cosméticos. 3.13) Sessão Temática 9 - Prospecção em patentes: cenário e perspectivas em câncer COMPOSIÇÃO DA MESA - Moderadora: Luciene Amaral, Especialista sênior da Academia de Propriedade Intelectual do INPI. Palestrantes: Carlos Gil Moreira, Coordenador da Rede Nacional de Pesquisa Clínica em

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
34
Câncer - RNPCC/SCTIE/DECIT/MS e diretor do INCA; Alexandre Lourenço, Chefe da Divisão de Observatório Tecnológico do INPI. Debatedores: Arystóbulo Freitas, responsável pela Área de Patentes da PróGenéricos; Letícia Covesi, da EMS. Relatores: João Carlos Gabaglia, Advogado Especialista em Propriedade Intelectual da Agência Inovação UFRJ e Luiz Villarinho, Pesquisador da ENSP/Fiocruz.
A sessão foi composta para apresentar o estudo realizado dentre os 4 tipos principais de câncer no
Brasil: mama, útero, pulmão e próstata. Quanto às patentes, o lado que todos conhecem é o lado do monopólio da patente no mercado, mas tem um lado da patente que não é muito conhecido que é o lado da informação técnica que as patentes proporcionam. E o estudo apresentado tem como objetivo a prospecção desse tipo de informação altamente valiosa. É possível prospectar as patentes estrangeiras e permitir que se atue nas lacunas tecnológicas encontradas, sendo este o papel do observatório. A patente permite localizar quem produziu qual tecnologia, de forma estratégica, além de verificar tendências tecnológicas, ver o que há de novo e o que já é consolidado. A patente é muito criticada como indicador de inovação pois está entre a pesquisa básica e a pesquisa clínica, mas seu papel de indicador não é absoluto: a cada 15 mil novas moléculas desenvolvidas, apenas uma fração chega ao mercado. Então, a pergunta a ser feita é: o que de fato é relevante? São várias as abordagens possíveis, como por exemplo, as tendências do mercado. A tecnologia relevante vai estar nas mãos dos principais depositantes, as grandes empresas, que fazem monitoramento das principais tecnologias em desenvolvimento. Uma patente farmacêutica tem um escopo difuso, como por ex., o diclofenato, que poderia ser usado como filtro solar.
O foco do observatório foram as patentes no tratamento do câncer. Os dados deveriam auxiliar a responder perguntas tais como: Quais são os principais mercados? A resposta vem rápido: os mercados americano, japonês e europeu são os principais mercados. Por outro lado, as doenças negligenciadas não são o foco das “big players” farmacêuticas.
São diversas as ferramentas para se atingir o nº de 84.351 documentos de patentes. O período analisado foi o de 2001 a 2011; a base de dados utilizada foi a do derwent innovations índex. Desejava-se saber qual é a principal tecnologia. O resultado indicou que trata-se de uma pergunta importante, mas que é necessário reduzir a amostragem de 3.000 documentos para um número mais reduzido. Muitos documentos têm interseção com vários tipos de câncer. Para o câncer de mama, foram encontrados 1.435 documentos. O critério utilizado foi a partir dos principais depositantes, além de outros critérios de busca. A patente farmacêutica tem valor enquanto está em sigilo, ou seja, a patente tem valor de mercado apenas no 1º ano de depósito. Portanto, a patente farmacêutica tem que ser negociada muito rápido. Para o câncer de pulmão, foram localizados 919 documentos e para cada tipo de mecanismo de ação e tecnologia, foi feito um corte específico. O antimonoclonal tem um alto valor agregado e quando se fala em diagnósticos, biomarcadores estão muito presentes, bem como a combinação de biomarcadores com outras técnicas diagnósticas.
Há hoje uma revolução tecnológica completa no tratamento do câncer. Não é mais necessário focar apenas no medicamento, pois já existem outras formas de tratamento. Foram duas revoluções, a dos antimonoclonais e a dos sintéticos. O número de alvos já foi identificado entre o ano 2000 e 2008. Para onde a oncologia aponta: para a imuno-oncologia e para a terapia personalizada, diagnóstico personalizado molecular de câncer. É um mercado estimado em 20 bilhões de dólares já em 2018, no mundo. O investimento de um fármaco genuinamente nacional é possível. Hoje são em torno de quase 1.000 novas moléculas anti-câncer, sendo que o diagnóstico também será revolucionado. A empresa Roche Diagnostica está muito bem colocada no mercado de oncologia, pois apostou no diagnóstico.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
35
O Instituto Nacional de Câncer (INCA) possui a rede nacional de pesquisa clínica em câncer. O câncer de pulmão é o maior mercado da indústria farmacêutica hoje, principalmente causada pelo tabagismo. Um dos grandes alvos é o câncer de colo uterino, bem como o câncer de próstata, com um desenvolvimento crônico de evolução na doença. O novo paradigma em oncologia é a imunoterapia, pois hoje a imunoterapia é muito mais ativa, com os imunoinibidores muito mais avançados. A imuno-oncologia, com modelos de parceria e cooperação, ainda é possível no Brasil, mas os pacientes tratados no SUS sofrem um gap tecnológico de 10 a 20 anos nos tratamentos disponíveis, dependendo do tipo de câncer. Existem estratégias terapêuticas que, no Brasil, podem fazer a diferença. Em terapia celular, o Brasil está muito bem posicionado, sendo uma área estratégica de investimento. O estudo foi muito importante para embasar a tomada de decisão, principalmente na parte de fomento e financiamento da indústria nacional.
Quando foi questionada a liberdade de operar e se, dentre os depositantes, havia algum nacional e se há algum caminho para a indústria nacional explorar, a resposta foi de que temos apenas 11 patentes em câncer de universidades nacionais. Ou seja, a indústria nacional não tem conseguido explorar o mercado de câncer. Porém, a indústria nacional deve investir no setor oncológico, sobretudo em Antibody Drug Conjugates (ADC) e nanotecnologia, ou seja, novos carreadores, e no diagnóstico molecular. O modelo coreano/chinês pode ser adotado como paradigma mas, para tanto, o modelo regulatório deve ser aprimorado, bem como deve-se desenhar uma nova forma de registro de fármaco, sendo que os asiáticos aprenderam a desenhar com os big players. No caso de terapia celular, foi defendido o co-desenvolvimento.
Os genéricos dependem de inovação, portanto os seus produtores estão muito preocupados com inovação, bem como a proteção legal do sistema patentário. Mas há sérios abusos no sistema patentário nacional, sendo que a inovação invariavelmente é estendida no prazo legal (art. 40 da LPI). Existem algumas teses aventureiras tais como a aplicação literal do acordo TRIPS no sistema legal nacional sem passar por um crivo legislativo nacional; o abuso do direito de petição; equívocos do sistema sanitário (particularmente no registro sanitário), etc. É inegável o impacto dos genéricos no acesso aos medicamentos oncológicos para a indústria nacional. E é uma preocupação da Anvisa o fato de eventualmente representar uma barreira para a oncologia nacional. O grande problema de um país burocrático ocorre quando a atividade meio se torna mais importante do que a atividade fim. Portanto, falta alguém ou alguma instituição que orquestre a questão sanitária e a questão do INPI, com sua falta de estrutura. O objetivo tecnológico brasileiro deve ser o de passar da mera tecnologia incremental para a disruptura, ou seja, para a inovação radical.
O backlog do INPI representa de 600 a 700 exames de patentes por cada examinador que a instituição dispõe. O depósito do pedido, ou melhor, a demora no exame dos pedidos, gera uma insegurança jurídica muito grande, sendo deficitária a estrutura do INPI por falta de pessoal inclusive. Desde 2010 foi feito um estudo que de 700 a 1.000 patentes são relevantes pois têm impacto direto no SUS. Mas qual é a importância de uma patente farmacêutica que não está relacionada com o mercado? A Portaria interna do INPI diz que o Ministério da Saúde tem prerrogativa para definir o que é prioritário de ser examinado: seria um fast track para patentes relevantes para o SUS e outros interesses públicos. A Colômbia não tem backlog: eles têm um quadro de pessoal proporcionalmente 4 vezes maior que o do Brasil. É preciso atentar para o fato de que a empresa processa o examinador, e não processa o INPI. Portanto, há uma enorme pressão nos examinadores. São necessários 3 anos para treinar um examinador de patentes. Já se passaram 8 oito anos para se formular uma diretriz de exame que ainda não foi feita. O princípio da interpretação evolutiva, no caso das patentes, significa que o examinador pode mudar de

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
36
opinião de acordo com os documentos apresentados, pois a própria ciência pode evoluir, mas tal direcionamento tem que ser definido por uma diretriz e não por uma lei. Neste sentido, o INPI precisa ser fortalecido, e novos examinadores são necessários. Destaca-se o fato de que o INPI é capaz de realizar o exame prévio, basta que haja uma diretriz definida para tanto.
4) CONCLUSÕES Há uma tendência no aumento da comercialização de medicamentos nos mercados emergentes, o
que abre ótimas oportunidades para a indústria brasileira. O mercado farmacêutico brasileiro, utilizando-se desta “janela”, tem crescido mais do que a economia como um todo, com nítido aumento da participação das empresas de capital nacional. A Política de Desenvolvimento Produtivo (PDP) do setor saúde gerou, nos últimos 5 anos, uma economia de R$ 2,5 bilhões. O sucesso está pautado na racionalização, parceiros adequados e escolhas corretas e alicerçado no Fomento, no Poder de compra e na Inovação. Para o monitoramento contínuo que assegure a viabilidade e perenidade das PDPs, sugere-se a criação de um Grupo de Trabalho (GT) com especialistas para realizar essas avaliações. Foi sugerida a encomenda programada para as PDPs, com planejamento do orçamento e ampliação do acesso aos medicamentos. Outros pontos relevantes perpassam modernizar o marco legal, disponibilizar recursos, centralizar compras governamentais, encorajar a internacionalização, e fortalecer centros de P&D. Para amenizar a atual situação dos laboratórios oficiais, foi sugerido que uma parte da receita seja investida em capacitação de recursos humanos, além de trazer cientistas do exterior para capacitar nossos jovens profissionais. Outra questão, sobretudo em função do mercado de genéricos, é a necessidade de se buscar novos arranjos para o modelo centralizado de inovação no setor farmacêutico – parcerias, e criação de fundos específicos de capital de risco. O Marco Regulatório, a Lei do Bem, e a Lei de Inovação formam um ambiente em franca evolução no Brasil na busca pela inovação, mas ainda verifica-se uma fraca inserção do país no cenário global associado à falta de um ecossistema farmacêutico que atue em rede. Falta uma interação entre academia, indústria, agências financiadoras e regulatórias. Ações isoladas refletem a falta de foco e estratégia para se atingir objetivos concretos e aqui cabe mencionar o papel do fórum ENIFarMed: integrar não apenas a indústria com a academia, mas também promover uma sinergia entre as ações do governo (leia-se Poderes Legislativo, Executivo e Judiciário) e o ambiente inovativo brasileiro.
Desde 2003, o país se esforça para fundir a política de preços com a análise de custo-benefício. Nesse modelo, a ausência de comparação para diversos medicamentos é um problema para a precificação. Sugere-se considerar de modo mais amplo o “custo-benefício” através da avaliação sistêmica da economia e dos custos indiretos. Também foi sugerida a possibilidade de que as empresas discutam preço com o órgão regulador no início do processo de inovação, ao invés de fazê-lo depois de anos de investimento, de forma a incentivar a inovação no país. Outras sugestões foram: revisão da Resolução CMED nº 2/2004; aumento da equipe técnica da CMED; maior detalhamento das rotinas de análise; assessoramento do Comitê Técnico por um comitê ad hoc, com maior expertise técnica, para elevar o debate técnico nos recursos administrativos; revisão do critério atual de “cesta de países”, para que sejam utilizados como comparadores países com realidades mais próximas ao Brasil, talvez avaliando a possibilidade de se retornar ao sistema de média de 3 países. Também foi questionada a possibilidade de liberação de preços de medicamentos isentos de prescrição (MIPs), já que a variável “preço” é mais relevante para os MIPs e o mercado de MIPs é mais elástico que o mercado de medicamentos em geral. Assim, o monitoramento do mercado deve ser uma alternativa à regulação e ao tabelamento de preços.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
37
Dos R$ 60 bilhões faturados pela indústria farmacêutica nacional, menos de 2% refere-se a produtos fitoterápicos. Em relação às políticas públicas, como a Rename e a Renisus, a PNPMF e as PDPs, estas ainda não incluem fitoterápicos. Já que as oportunidades estão no fortalecimento do mercado e na independência de importação de matéria-prima (leve-se em consideração que a maioria dos fitoterápicos comercializados no Brasil são plantas exógenas), foi sugerido repensar o poder de compra governamental, considerando-se que a compra descentralizada não vai elevar o uso da fitoterapia no SUS. Alguns pontos críticos na escolha de um fornecedor, como a troca de um fornecedor ou a inserção de uma segunda fonte, somados a problemas como falta de especificação com marcadores, diferença nos limites de solventes e diferenças nos pesticidas aos aprovados, além dos custos, geram problemas farmacotécnicos e de suprimentos que, geralmente, são impossíveis de se substituir, pois há dificuldade de se inserir uma segunda fonte por falta de similaridade e competitividade, o que causa dúvida quanto à troca do fornecedor existente de extrato. Uma das soluções propostas foi a de que o BNDES fomente o mercado com editais para extratos de plantas nativas com marcadores. Também verificou-se a necessidade de organizar resultados clínicos de forma estruturada voltada para o racional médico, além de definir os conceitos de “extrato”.
O uso dos fitoterápicos será fortalecido pelo marco legal da biodiversidade recém constituído. Cabe destacar que a repercussão do novo marco legal de acesso à biodiversidade foi tamanha que, três dias após sua publicação no Diário Oficial da União (DOU), trinta e sete países solicitaram acesso ao texto. A Consulta Pública da Regulamentação de Lei de Biodiversidade já foi publicada, ficará aberta até o dia 16 de outubro de 2015 e, até novembro, teremos um ambiente regulamentado. Portanto, o grande desafio que estamos vivenciando atualmente é a construção de uma agenda para definição dos termos do Decreto Presidencial que regulamentará a Lei. É preciso regulamentar sem, no entanto, burocratizar o processo.
Do ponto de vista das empresas farmacêuticas, que necessitam construir dossiês de qualidade para alcançar o registro de seus produtos, a realização de estudos pré-clínicos dentro dos padrões internacionais representa um gasto inevitável que, por isso, deve ser executado da forma mais racional e direta possível. Por outro lado, sob a ótica da gestão pública, fomentar a capacitação e garantir meios para certificação das empresas nacionais corresponde a um importante investimento, que proverá suporte à estratégia de inserção do Brasil em redes globais de inovação em fármacos e medicamentos. A estruturação de uma cadeia brasileira de prestadores serviços pré-clínicos acreditados em nível internacional dará suporte à indústria nacional e à participação de instituições brasileiras em estudos pré-clínicos promovidos por multinacionais. Deve-se ressaltar que a Índia é um exemplo de sucesso em testes pré-clínicos, promovido por políticas públicas simples e eficientes.
No que concerne a pesquisa clínica, ficou bastante clara a necessidade de um desenvolvimento e padronização das análises feitas pela ANVISA, redução progressiva dos tempos de análise dos estudos, verificação e acompanhamento do cumprimento dos prazos estipulados pelas RDCs # 9 e 10 e, acima de tudo, a continuidade do processo de formação de técnicos capacitados para análise das submissões recebidas. E, em relação às atividades da Comissão Nacional de Ética em Pesquisa (CONEP), destacam-se: abertura do processo de diálogo com todos os centros de pesquisa; análises dos projetos visando a defesa ética dos participantes de pesquisa clínica porém sem impactar drasticamente a condição do projeto; redução significativa dos prazos de análise nos últimos 24 meses, reduzindo o número de estudos aguardando a primeira análise de 180 para 90 dias; implementação da acreditação de Comitês de Ética em Pesquisa (CEPs) para redução da dupla análise de estudos clínicos de risco leve a moderado ao participante de pesquisa. A CONEP torna-se, então, a última instância para avaliação de casos em que os CEPs acreditados julgam não serem capazes de analisar e de estudos de elevado risco ao paciente. A

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
38
classificação de risco será automática e feita com base nas informações iniciais inseridas na Plataforma Brasil; distribuição de um manual aos CEPs cadastrados na Plataforma Brasil listando os erros mais comuns verificados pela CONEP e informando sua resolução; lançamento da Plataforma Brasil 3.0 e os planos de atualização – 3.1 (outubro de 2015) inserção da entrada de eventos adversos diretamente na Plataforma Brasil e 3.2 (dezembro de 2015) inserção da listagem de Biobancos e controle dos mesmos através da Plataforma Brasil. Como resultado desta discussão, ficou claro que o processo de acreditação precisará ser desenvolvido em diversos âmbitos, especialmente na qualificação e instrução dos relatores para padronização das análises realizadas. O ponto final de discussão apresentado foi sobre a necessidade da CONEP focar suas atenções em questões éticas de análise e não se ater a questões relacionadas à semântica das palavras e, acima de tudo, definição clara dos padrões a serem utilizados nas avaliações para que os estudos possam ser elaborados de forma correta, contribuindo ainda mais com a redução dos tempos de avaliação e aprovação.
Existe um certo modismo no uso do termo “nano”, e seus efeitos associados à diminuição do tamanho de partículas, cápsulas e estruturas. Em relação ao seu uso em fármacos, os aspectos positivos apresentados foram: aumento de área superficial, modificação de solubilidade, aumento de taxa de dissolução, doses menores, ação mais rápida e menor variabilidade entre pacientes. Soluções podem estar na resolução de lacunas apontadas pela NanoForce no que se refere à definição uniforme, protocolos e parâmetros uniformizados entre os países no desenvolvimento da nanotecnologia, cujo prazo é 2020. Sendo assim, foram apresentadas três estratégias para vetorizar um ativo: (1) pró-fármacos (planejamento e síntese); (2) reconhecimento molecular (biologia molecular); (3) métodos físicos (farmacotecnia). Outros aspectos positivos da nanotecnologia são a eficácia superior de fármacos, potencial maior de tempo de vida no mercado de produtos nanotecnológicos, a nanoencapsulação pode proporcionar novas patentes, dentre outras. Sob o aspecto de custos, processos nanotecnológicos são menos dispendiosos do que processos de síntese química. Porém, ainda não há nenhum medicamento aprovado no Brasil utilizando-se de nanotecnologia. O estágio que se encontra a nanotecnologia ainda apresenta muitos desafios de comparabilidade, eficácia e segurança, já que não é possível ter certeza se uma dose encapsulada possui relação com outro volume de dose, só que sem encapsulação. Além disso, pesquisadores e a indústria deverão estar atentos para novos efeitos colaterais decorrentes do uso de nanotecnologia em fármacos e cosméticos. O futuro está no setor oncológico, sobretudo os Antibody Drug Conjugates (ADC) e a nanotecnologia, ou seja, novos carreadores, além do diagnóstico molecular. As quatro grandes diretrizes são: (1) testes padronizados preexistentes para toxicidade de materiais não são adaptáveis para nanomateriais e, sendo assim, novos testes precisam ser desenvolvidos; (2) a nanociência ajudará no desenvolvimento de novos testes para nanoprodutos; (3) impactos no meio ambiente e na saúde causados por nanoprodutos devem ser avaliados caso a caso; (4) os nanomateriais, na maioria dos casos, vão se aglomerar ou se dissolver, não agindo sobre seres vivos como nanoobjetos.
O depósito do pedido de patente junto ao INPI, ou melhor, a demora no exame dos pedidos, gera uma insegurança jurídica muito grande, sendo deficitária a estrutura do INPI por falta de pessoal, inclusive. Em 2010 foi feito um estudo que revelou que de 700 a 1.000 patentes são relevantes pois têm impacto direto no SUS. Mas qual é a importância de uma patente farmacêutica que não está relacionada com o mercado? A Portaria interna do INPI diz que o Ministério da Saúde tem prerrogativa para definir o que é prioritário de ser examinado: seria um fast track para patentes relevantes para o SUS e outros interesses públicos. Assim, o INPI precisa ser fortalecido e novos examinadores são necessários. Destaca-se o fato de que o INPI é capaz de realizar o exame prévio, basta que haja uma diretriz definida para tanto. Uma questão importante apontada se refere ao marco regulatório nacional, e envolve, por exemplo, o risco de

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
39
se tratar questões e tecnologias relativas à medicina regenerativa como inconstitucional, tendo em vista a utilização de material humano na pesquisa e desenvolvimento. Com a incerteza, há uma diminuição na propensão a investir nesse setor. Com isso, há a necessidade de clarear essa questão no Supremo Tribunal Federal e, eventualmente, no Congresso.
5) DESDOBRAMENTOS De forma absolutamente sucinta, as principais sugestões sobre todos os temas debatidos
encontram-se abaixo enumeradas com os respectivos interlocutores destacados. Cabe ao Poder Executivo, em especial ao Ministério da Saúde, em consonância com demais Ministérios tangenciados e a Casa Civil, garantir a perenidade e o sucesso das PDPs através das seguintes sugestões:
1) Criação de um Grupo de Trabalho (GT) com especialistas para realizar as avaliações das parcerias e dos respectivos contratos das PDPs.
2) Realização de um planejamento do orçamento e ampliação do acesso aos medicamentos através da encomenda programada para as PDPs.
3) Fortalecimento dos laboratórios oficiais através da capacitação de recursos humanos, eventualmente com cientistas vindos do exterior.
E, para garantir um bom ambiente à inovação, o Poder Executivo, incluindo as agências reguladoras, de patentes, e órgãos de fomento, devem buscar meios para dar suporte às seguintes ideias:
4) Estabelecer um plano estratégico de inovação para o país que inclua a priorização e racionalização de investimentos, a desoneração de atividades de PD&I, e a melhoria no ensino em todos os níveis.
5) Disponibilizar recursos, centralizar as compras governamentais, e buscar novos arranjos para o modelo centralizado de inovação no setor farmacêutico – parcerias, e criação de fundos específicos de capital de risco.
6) Fortalecer centros de P&D&I de inovação em algumas regiões brasileiras em consonância com as aptidões regionais e com as demandas de mercado.
7) Fortalecer a integração da academia com a indústria para gerar produtos, processos e serviços inovados. Neste ponto, cabe observar que é preciso que a capacitação leve em conta temas como propriedade intelectual, aspectos regulatórios internacionais, e mercados estrangeiros.
8) Desenvolver eixos temáticos para a inovação no Brasil, com foco em medicamentos, como por exemplo as pesquisas orientadas para dar continuidade a estudos preliminares contido em teses e trabalhos de prospecção já praticamente abandonados, e desenvolver pesquisas para aperfeiçoar medicamentos tradicionais através de novas tecnologias, que permitam obter produtos com maior desempenho terapêutico e com redução de custos promovidos por tecnologias diferenciadas, além da capacitação e formação de recursos humanos para gerenciamento de pesquisadores, com vistas a torná-los mais eficazes em suas atividades.
9) Apoiar a internacionalização das empresas com capital nacional.
10) Atualizar o arcabouço regulatório, frente ao marco legal que dispomos, de forma a agilizar processos e, assim, promover um ambiente mais propício à inovação e à competitividade.
11) Modernizar as agências âncora da inovação, como o INPI, que precisa ser fortalecido e novos examinadores devem ser contratados para reduzir o backlog.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
40
12) Agilizar processos da Anvisa, sem prejuízo para a qualidade e a segurança dos serviços da agência, privilegiando o desenvolvimento de produtos para necessidades ainda não supridas (“unmet
medical needs”), em detrimento ao registro produtos sem custo-benefício comprovado, remuneração da cadeia de valor, com base nos resultados (“outcomes”).
13) Reformular o processo de precificação para que as empresas possam definir o preço máximo com o órgão regulador no início do processo de inovação. Considerar de modo mais amplo o “custo-benefício” através da avaliação sistêmica da economia e dos custos indiretos. Elevar o debate técnico nos recursos administrativos. Revisar o critério atual de “cesta de países”, para que sejam utilizados como comparadores países com realidades mais próximas ao Brasil. Liberar os preços de medicamentos isentos de prescrição (MIPs), utilizando-se de um monitoramento do mercado.
Cabe ao Poder Legislativo, com o apoio das agências reguladoras e da sociedade civil:
14) Construir uma agenda para definição dos termos do Decreto Presidencial que regulamentará a Lei 13.123/2015, sobre o uso da biodiversidade brasileira.
15) Clarear as questões relativas às tecnologias de medicina regenerativa, tendo em vista a utilização de material humano na pesquisa.
O debate técnico revelou que ainda verificam-se gargalos antigos, como a necessidade de fomentar a capacitação em áreas de interesse como fitoterápicos, biossimilares, testes clínicos e pré-clínicos. Para estas áreas, foram destacados os pontos mais urgentes e importantes.
16) Os maiores gargalos para a fitoterapia têm três principais frentes: (1) Farmacotécnico, com necessidade de definição do termo extrato, padronização de extratos e marcadores, e inclusão da fitoterapia na Farmacopéia Brasileira; (2) Governamental, pois as políticas públicas não incluem fitoterápicos; e (3) Clínico, pois há que se desenvolver resultados clínicos voltados para o racional médico.
17) Harmonização de práticas entre diferentes entidades internacionais com o marco regulatório brasileiro através da adoção de novos métodos de análise de biossimilares e ensaios pré-clínicos.
18) Estruturação de uma cadeia brasileira de prestadores serviços pré-clínicos acreditados em nível internacional para dar suporte à indústria nacional e para incluir as instituições brasileiras em estudos pré-clínicos promovidos por multinacionais.
19) Com relação à pesquisa clínica, verifica-se a necessidade de um desenvolvimento e padronização das análises feitas pela Anvisa, redução progressiva dos tempos de análise dos estudos, verificação e acompanhamento do cumprimento dos prazos estipulados pelas RDCs # 9 e 10 e, acima de tudo, a continuidade do processo de formação de técnicos capacitados para análise das submissões recebidas. O processo de acreditação precisará observar a qualificação e instrução dos relatores para padronização das análises realizadas. A CONEP deve focar suas atenções em questões éticas de análise de forma a contribuir com a redução dos tempos de avaliação e aprovação.
20) A nanotecnologia ainda apresenta muitos desafios de comparabilidade, eficácia e segurança no setor farmacêutico, os quais precisam de solução técnica.

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
41
PROGRAMA REALIZADO
Dia 18 de agosto de 2015 – 3ª feira - Centro de Convenções Rebouças
08h00 08h30
Credenciamento
08h30 10h30
Sessão de Abertura – Sala Turquesa 3 Presidente de mesa: Dante Alario Jr, presidente do Conselho Deliberativo do IPD-Farma e da Biolab
Palestra de Abertura: Roberto Nicolsky, Protec - “Agenda de crescimento para o Brasil”
10h30 11h00
CAFÉ na ExpoFarMed - Salão Lilás
Exposição do vídeo de DoHyun Cho, da Medical Strategy – EUA/Korea, que proferiu presencialmente a palestra internacional do 6º ENIFarMed, em 2012 – Sala Turquesa 3
11h00 13h00
1ª Plenária – Políticas de fomento, uso do poder de compras, e encomendas: perspectivas diante do cenário econômico atual – Sala Turquesa 3
Moderador: Lelio Maçaira, Laborvida
Palestra Magna: Reginaldo Arcuri, Grupo Farma Brasil Debatedores: Plateia Convidada: Pedro Palmeira, BNDES Sergio Frangioni, Blanver Jamaira Giora, Consultora Marcus Simões, MDIC
Relatores: Heloisio Rodrigues, PHASE Pharma e Patrícia Teixeira, IE/UFRJ
13h00 14h00
BRUNCH - Salão Lilás
14h00 15h00
Sessão de Pôsteres na ExpoFarMed - Salão Lilás
15h00 16h30
Sessão Temática 1 –Sala Turquesa 3 Ações para a produção de insumos
da biodiversidade brasileira Moderadora: Maria Behrens,
Farmanguinhos/Fiocruz Palestrantes: Alceu Moreira, Dep. Federal (PMDB/RS) Henry Novion, DPG/MMA Benilson Barreto, DAF/MS Debatedores: Ricardo Dias, Centroflora João Carlos Basílio, Abihpec Relatores: Carlos Vitor, Aché e Ana Claudia
Dias, Abifina
Sessão Temática 2 –Sala Turquesa 2 Modelos de gestão para acelerar a
inovação e a integração de políticas públicas para doenças que afetam
populações negligenciadas Moderador: Eric Stobbaerts, DNDi
Palestrantes: Ana Rabello, Fiocruz Luiz Carlos Dias, Unicamp Walter Britto, Universidades Aliadas por Medicamentos Essenciais (UAEM)
Relatores: Vera Lucia Luiza, ENSP e Henrique Menezes, UFPB
Sessão Temática 3 –Sala Turquesa 1 Biotecnologia e a intercambialidade
para biossimilares Moderador: Thiago Mares Guia, Bionovis
Palestrantes: André Abrahão, Merck Serono Priscila Scheinberg, Orygen Daniela Cerqueira, Anvisa Dirceu Barbano, Consultor
Relatores: Carlos Martins, Interface CTI e Eliziane Patricio, UNIPLAC
16h30 17h00
CAFÉ na ExpoFarMed - Salão Lilás
17h00 18h30
2ª Plenária – Internacionalização dos laboratórios farmacêuticos nacionais: acesso a novos mercados - Sala Turquesa 3
Moderador: Marcio Falci, Assessor para Presidência Científica da Biolab Palestrantes: Debatedores: Plateia Convidada: Igor Bueno, Finep João Transmontano, Biolotus Biotech Claudio Maurício de Souza, IVB José Correia, Abiquifi Maria Luisa Campos Leal, ABDI Eduardo Cruz, Axisbiotec Relatores: Henrique Menezes, UFPB e Fernando Marcussi, Alanac
18h30 19h30
ENCERRAMENTO com lançamento do livro “Do protecionismo ao desenvolvimentismo - A indústria farmoquímica brasileira”, por Thaís Soares, Luciene Amaral, Helvécio Rocha, e Alexandre Lourenço - Salão Lilás
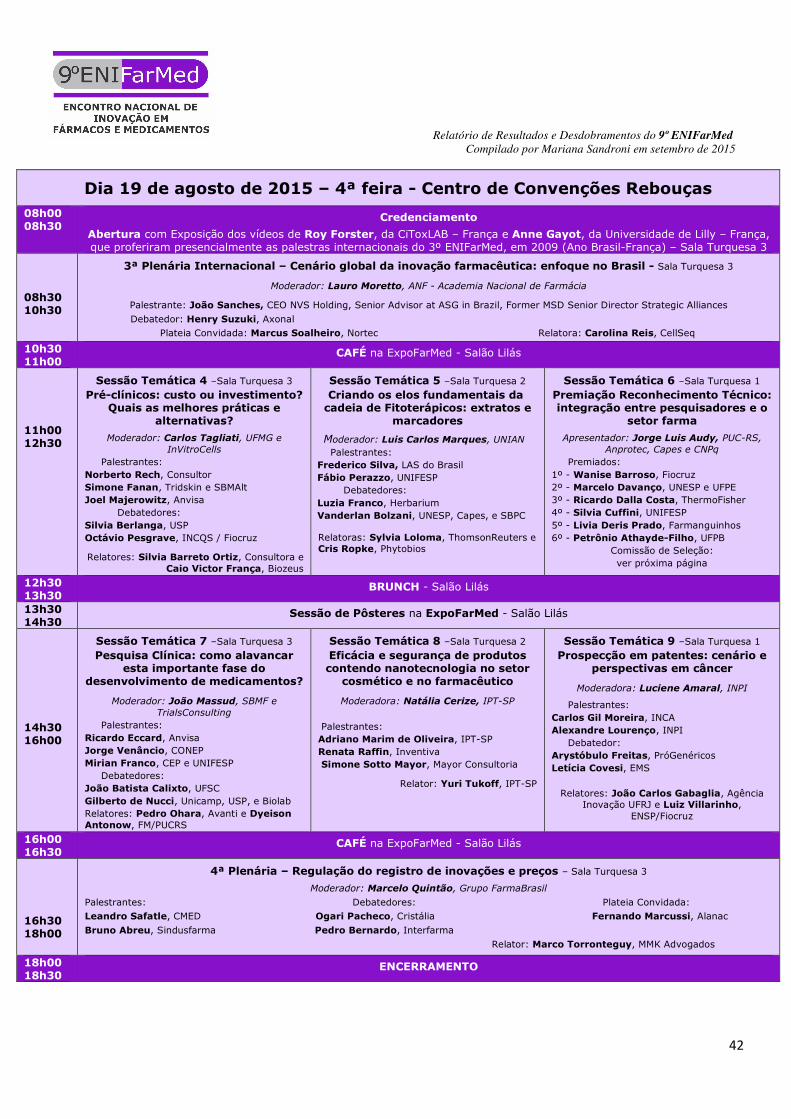
Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
42
Dia 19 de agosto de 2015 – 4ª feira - Centro de Convenções Rebouças
08h00 08h30
Credenciamento
Abertura com Exposição dos vídeos de Roy Forster, da CiToxLAB – França e Anne Gayot, da Universidade de Lilly – França, que proferiram presencialmente as palestras internacionais do 3º ENIFarMed, em 2009 (Ano Brasil-França) – Sala Turquesa 3
08h30 10h30
3ª Plenária Internacional – Cenário global da inovação farmacêutica: enfoque no Brasil - Sala Turquesa 3 Moderador: Lauro Moretto, ANF - Academia Nacional de Farmácia
Palestrante: João Sanches, CEO NVS Holding, Senior Advisor at ASG in Brazil, Former MSD Senior Director Strategic Alliances Debatedor: Henry Suzuki, Axonal Plateia Convidada: Marcus Soalheiro, Nortec Relatora: Carolina Reis, CellSeq
10h30 11h00
CAFÉ na ExpoFarMed - Salão Lilás
11h00 12h30
Sessão Temática 4 –Sala Turquesa 3 Pré-clínicos: custo ou investimento?
Quais as melhores práticas e alternativas?
Moderador: Carlos Tagliati, UFMG e InVitroCells
Palestrantes: Norberto Rech, Consultor Simone Fanan, Tridskin e SBMAlt Joel Majerowitz, Anvisa Debatedores: Silvia Berlanga, USP Octávio Pesgrave, INCQS / Fiocruz Relatores: Silvia Barreto Ortiz, Consultora e
Caio Victor França, Biozeus
Sessão Temática 5 –Sala Turquesa 2 Criando os elos fundamentais da cadeia de Fitoterápicos: extratos e
marcadores Moderador: Luis Carlos Marques, UNIAN
Palestrantes: Frederico Silva, LAS do Brasil Fábio Perazzo, UNIFESP Debatedores: Luzia Franco, Herbarium Vanderlan Bolzani, UNESP, Capes, e SBPC
Relatoras: Sylvia Loloma, ThomsonReuters e Cris Ropke, Phytobios
Sessão Temática 6 –Sala Turquesa 1 Premiação Reconhecimento Técnico: integração entre pesquisadores e o
setor farma Apresentador: Jorge Luis Audy, PUC-RS,
Anprotec, Capes e CNPq Premiados: 1º - Wanise Barroso, Fiocruz 2º - Marcelo Davanço, UNESP e UFPE 3º - Ricardo Dalla Costa, ThermoFisher 4º - Silvia Cuffini, UNIFESP 5º - Livia Deris Prado, Farmanguinhos 6º - Petrônio Athayde-Filho, UFPB
Comissão de Seleção: ver próxima página
12h30 13h30
BRUNCH - Salão Lilás
13h30 14h30
Sessão de Pôsteres na ExpoFarMed - Salão Lilás
14h30 16h00
Sessão Temática 7 –Sala Turquesa 3 Pesquisa Clínica: como alavancar
esta importante fase do desenvolvimento de medicamentos?
Moderador: João Massud, SBMF e TrialsConsulting
Palestrantes: Ricardo Eccard, Anvisa Jorge Venâncio, CONEP Mirian Franco, CEP e UNIFESP Debatedores: João Batista Calixto, UFSC Gilberto de Nucci, Unicamp, USP, e Biolab Relatores: Pedro Ohara, Avanti e Dyeison Antonow, FM/PUCRS
Sessão Temática 8 –Sala Turquesa 2 Eficácia e segurança de produtos contendo nanotecnologia no setor
cosmético e no farmacêutico Moderadora: Natália Cerize, IPT-SP
Palestrantes: Adriano Marim de Oliveira, IPT-SP
Renata Raffin, Inventiva Simone Sotto Mayor, Mayor Consultoria
Relator: Yuri Tukoff, IPT-SP
Sessão Temática 9 –Sala Turquesa 1 Prospecção em patentes: cenário e
perspectivas em câncer Moderadora: Luciene Amaral, INPI
Palestrantes: Carlos Gil Moreira, INCA Alexandre Lourenço, INPI Debatedor: Arystóbulo Freitas, PróGenéricos
Letícia Covesi, EMS
Relatores: João Carlos Gabaglia, Agência Inovação UFRJ e Luiz Villarinho,
ENSP/Fiocruz
16h00 16h30
CAFÉ na ExpoFarMed - Salão Lilás
16h30 18h00
4ª Plenária – Regulação do registro de inovações e preços – Sala Turquesa 3 Moderador: Marcelo Quintão, Grupo FarmaBrasil
Palestrantes: Debatedores: Plateia Convidada: Leandro Safatle, CMED Ogari Pacheco, Cristália Fernando Marcussi, Alanac Bruno Abreu, Sindusfarma Pedro Bernardo, Interfarma Relator: Marco Torronteguy, MMK Advogados
18h00 18h30
ENCERRAMENTO

Relatório de Resultados e Desdobramentos do 9º ENIFarMed
Compilado por Mariana Sandroni em setembro de 2015
43
Integrantes da Comissão de Programa e do Comitê de Seleção
Comissão Nacional de Programa:
Adriana Diaféria, Grupo FarmaBrasil Alaide de Sá Barreto, Centro Universitário Estadual da Zona Oeste – UEZO Carlos Tagliati, InVitro Cells Eric Stobbaerts, DNDi Henry Suzuki, Axonal Jackeline Barbosa, Herbarium Laboratório Botânico Ltda Jean Daniel Peter, Globe Química João Massud, Sociedade Brasileira de Medicina farmacêutica (SBMF) e TrialsConsulting José Gomes Temporão, ISAGS/UNASUR Luciene Amaral, INPI Luís Carlos Marques, UNIAN Maria Behens, Farmanguinhos/Fiocruz Mariana Sandroni, Marsan e Protec Natália Neto Pereira Cerize, IPT-SP Pedro Palmeira, BNDES Rubens Alves Pereira, NUPEEC-UFPel Sergio Jose Mecena da Silva Filho, UFF/Escola de Engenharia
Comitê Científico-Tecnológico de Seleção para a Premiação Reconhecimento Técnico:
Presidente do Comitê: Carlos Tagliati, UFMG / InVitroCells
João Carlos Gabaglia, Agência Inovação da UFRJ Julia Paranhos, UFRJ Luis Lopez Martinez, NAPesq - Núcleo de Apoio à Pesquisa Clínica /HCFMUSP Maria Behens, Farmanguinhos/Fiocruz Roberto Nicolsky, UEZO e Protec Rubens Alves Pereira, NUPEEC-UFPel Sergio Jose Mecena da Silva Filho, UFF/Escola de Engenharia Thais Helena Gasparoto, Faculdade Odontologia/USP William Waissmann, ENSP/Fiocruz





























