Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Ueber die Konfiguration der Kohlenstoffatome 23 und 24 bei denTriterpenen der β-Amyrin-Oleanolsäure-Gruppe
Author(s): Vogel, Arnold
Publication Date: 1952
Permanent Link: https://doi.org/10.3929/ethz-a-000090059
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2158
I. lieber die Konfiguration der Kohlenstoffatonie
23 und 24 bei den Triterpenender /S-Amyrin-Oieanolsäure-Gruppe
II. lieber Umsetzungen im Ringe C des oc-Amyrins
Von der
Eidgenössischen Technischen Hochschule in Zürich
zur Erlangung
der Würde eines Doktors der Technischen Wissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
ARNOLD VOGEL
dipl. Ingenieur-Chemiker
von Dachsen (Kt. Zürich)
Referent: Herr Prof. Dr. L. Ruzicka
Korreferent: Herr Priv.-Doz. Dr. 0. Jeger
1952
Juris-Verlag, Zürich

MEINEN LIEBEN ELTERN
IN DANKBARKEIT
GEWIDMET

Meinem sehr verehrten Lehrer,
Herrn Prof. Dr. L. Ruzicka,
unter dessen Leitung die vorliegende Arbeit ausgeführt wurde,
möchte ich für seine Unterstützung und sein Interesse herz¬
lich danken.
Herrn Priv.-Doz. Dr. 0. Jeger bin ich für die vielen
Ratschläge und Anregungen und das mir stets entgegengebrach¬
te WohlwqAlen zu besonderem Dank verpflichtet.
Der Rockefeiler Foundation in New York und der Georg-
Lunge-Stiftung danke ich für die finanzielle Unterstützung
dieser Arbeit.

INHALTSVERZEICHNIS
Einleitung 5
I. Teil. Beitrag zur Stereochemie der pentacyclischen
Triterpene der /3-Amyrin-Oleanolsäure-Gruppe .... 9
ï^S2£§ïi§2^êE_ÏSU 9
A. Bisherige Kenntnisse über die Stereochemie der
pentacyclischen Triterpene 9
B. Zusammenhänge im Bau der Ringe A und B bei der
Oleanolsäure und den tricyclischen Diterpensäuren .20
C. Die Konstitution und Konfiguration der Ringe A und
B einiger Diterpensäuren 25
D. Eigene Arbeiten 30
1. Allgemeines zur Bestimmung der Konfiguration der
Kohlenstoffatome 23 und 24 bei den Triterpenender ^-Amyrin-Oleanolsäure-Gruppe 30
2. Versuche in der Gypsogenin-Reihe 32
3. Umsetzungen in der «-Boswellinsäure-Reihe • • -34
4. Diskussion der Versuchsergebnisse 38
£S2eEiSeB£§li§£_T§il 39
Zusammenfassung 56
II. Teil. Zur Konstitution des Ringes C des a-Amyrins -57
ï^S2£SÎiS£^êî!_ï2ii 57
A. Einleitung 57
B. Die Konstitution des Ringes C 60
C. Eigene Arbeiten 64
§ïE2riS§S£§ii£I_ïêii 70
Zusammenfassung 79

EINLEITUNG
Die Triterpene sind eine in der Natur weitverbreite¬
te Gruppe von stickstoffreien Verbindungen, deren Koh-
lenstoffgertist 30 Kohlenstoffatome enthält und aus 6 Iso-
pentanresten aufgebaut ist. Die Triterpene kommen vor
allem in Pflanzen, vereinzelt auch in Tieren vor, und
zwar in freier Form, oder als Ester bzw. als Glykoside.
Die Konstitution der meisten Vertreter dieser Klasse von
Naturverbindungen ist heute bekannt.
Bei der Konstitutionsaufklärung der Triterpene ka¬
men verschiedene Arbeitsmethoden zur Anwendung, von de¬
nen die wichtigsten genannt seien: die Dehydrierung mit
Selen oder Palladium, der systematische oxydative Abbau,
die thermische Spaltung geeigneter Abbauprodukte und die
ümwandlungsreaktionen. Ausserdem spielte die konsequente
Anwendung der Isoprenregel eine wichtige Rolle. Mit Hilfe
der Umwandlungsreaktionen gelang es, verschiedene Tri-
terpenverbindungen ineinander überzuführen und dadurch
die zahlreichen Vertreter dieser Klasse von Naturstof¬
fen in wenige Gruppen einzuordnen. Die Verbindungen ei¬
ner Gruppe besitzen das gleiche Kohlenstoffgerüst und
unterscheiden sich nur in der Art, Zahl oder Stellung
ihrer Punktionen.
Einteilung der Triterpene
A) Aliphatische Triterpene
Squalen'
1) Karrer und Helfenstein, Helv. 14, 78 (1931)

- 6 -
B) Tricyclische Triterpene1)
Ambrein '
C) Tetracyclische Triterpene2) 3)
Die Lanosteringruppe ', die Elemisäuren ', die Euphor-
biumalkohole ', Onocerin ', die Polyporensäuren ',
Basseol, Butyrospermol , Sojasapogenol D
D) Pentacyclische Triterpene9)
a. Die y3_Amyrin-01eanolsäure-Gruppe :
/8-Amyrin, Oleanolsäure, Hederagenin, Gypsogenin, Ery-
throdiol, Glycyrrhetinsäure, a-Boswellinsäure, Siare-
sinolsäure, Sumaresinolsäure, Echinocystsäure, Genin A,
Maniladiol, Quillajasäure ', Sojasapogenole A und C
Sojasapogenol B ', Germanicol,Morolsäure
b. Die a-Amyrin-Ursolsäure-Gruppe:
o-Amyrin ,Ursolsäure ', ^-Boswellinsäure '',
Brein l8), Uvaol19^
20) 21) 22)c. Die Lupeol-Heterobetulin-Gruppe :
lupeol, Betulin, Betulinsäure, Heterobetulin, Tara-
xasterol, Arnidiol, Faradiol
1) Jeger, Durst & Ruzicka, Helv. _3£, 1859 (1947)2) Voser, Mijovic, Jeger & Ruzicka, Helv. 31, 1585 (1951)3) Arnold, Koller & Jeger, Helv. 34, 555 (1951)4) Christen, Jeger & Ruzicka, Helv. _34, 1675 (1951)5) Zimmermann, Helv. 21, 853 (1938)6) Cross, Eliot, Heilbron & Jones, Soc. 1940, 632
7) Seitz & Jeger, Helv. _3_2, 1626 (1949)8) Meyer, Jeger~& Ruzicka, Helv. 33, 1835 (1950)9) Bischof, Jeger & Ruzicka, Helv. 3_2, 1911 (1949)10) Ruzicka, Bischof, Taylor, Meyer & Jeger, Coli.trav.Tch.
15_, 893 (1950)11) Meyer, Jeger & Ruzicka, Helv. 33, 672 (1950)12) Meyer. Jeger & Ruzicka, Helv. 33, 687 (1950)13) David, Bull.Socchim. 43, 155 TÎ949)14) Barton & Brooks, Soc. 1951, 257
15) Meiseis, Jeger & Ruzicka, Helv. 33_, 700 (1950)16) Dreiding, Jeger & Ruzicka, Helv. 33, 1325 (1950)17) Ruzicka, Jeger & Ingold, Helv. 27, 1859 (1944)18) Bttchi, Jeger & Ruzicka, Helv. 29, 442 (1946)19) Orr, Parks, Dunker & Uhl, J.Am.Pharm.Assoc. 34,, 39 (1945)20} Lardelli, Diss. ETH, Zürich 194921) Koller, Hiestand, Dietrich & Jeger, Helv. 33, 1050 (1950)22) Barton & Holness, Soc. 1952, 78
- 7 -
d. Keiner Gruppe zugeordnet:
Friedelin,Cerin ', Chinovasäure i', Aescigenin
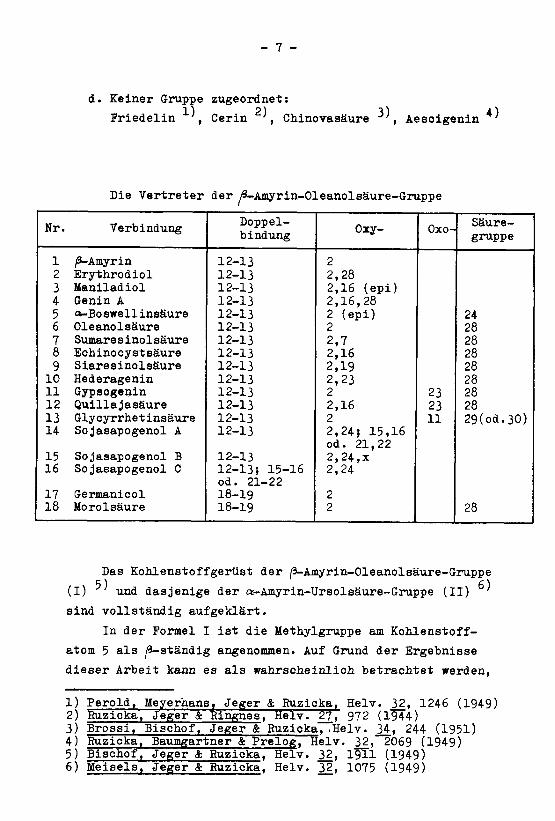
Die Vertreter der /9-Amyrin-Oleanolsäure-Gruppe
Nr. VerbindungDoppel¬bindung
Oxy- Oxo-Säure¬
gruppe
1 /9-Amyrin 12-13 2
2 Erythrodiol 12-13 2,283 Maniladiol 12-13 2,16 (epi)4 Genin A 12-13 2,16,285 a-Boswellinsäure 12-13 2 (epi) 246 Oleanolsäure 12-13 2 28
7 Sumaresinolsäure 12-13 2,7 28
8 Echinocystsäure 12-13 2,16 28
9 Siaresinolsäure 12-13 2,19 28
10 Hederagenin 12-13 2,23 28
11 Gypsogenin 12-13 2 23 28
12 Quillajasäure 12-13 2,16 23 28
13 Glycyrrhetinsäure 12-13 2 11 29(od.30)14 Sojasapogenol A 12-13 2,24? 15,16
od. 21,2215 Sojasapogenol B 12-13 2,24,x16 Sojasapogenol C 12-13; 15-16
od. 21-222,24
17 Germanicol 18-19 2
18 Morolsäure 18-19 2 28
Das Kohlenstoffgerüst der /a-Amyrin-Oleanolsäure-Gruppe
(I) 'und dasjenige der «-Amyrin-Ursolsäure-Gruppe (II)
sind vollständig aufgeklärt.
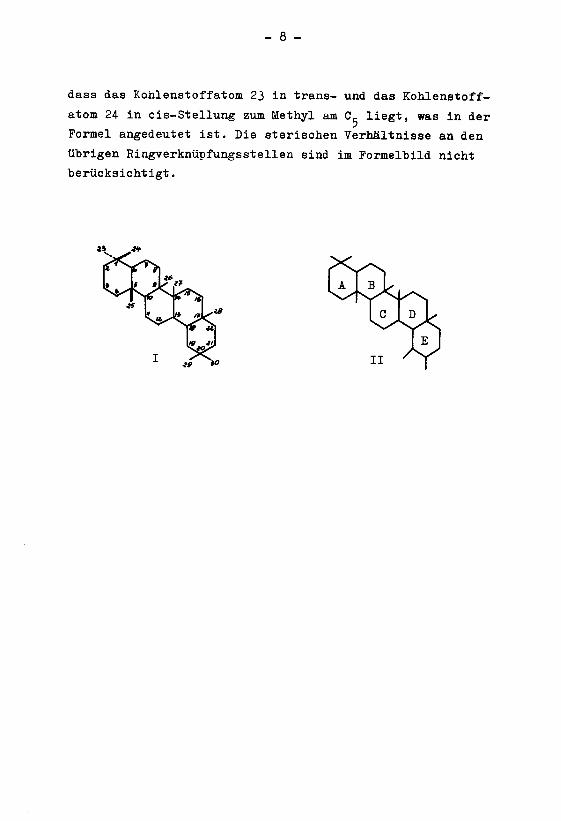
In der Formel I ist die Methylgruppe am Kohlenstoff¬
atom 5 als /3-ständig angenommen. Auf Grund der Ergebnisse
dieser Arbeit kann es als wahrscheinlich betrachtet werden,
1) Perold, Meyerhans, Jeger & Ruzicka, Helv. 3_2, 1246 (1949)2) Ruzicka, Jeger & Ringnes, Helv. 27, 972 (1944)3) Brossi, Bischof, Jeger & Ruzicka,.Helv. _34, 244 (1951)4) Ruzicka, Baumgartner & Prelog, Helv. 12_, 2069 (1949)5) Bischof, Jeger & Ruzioka, Helv. _3_2, 1911 (1949)6) Meiseis, Jeger & Ruzicka, Helv. .32, 1075 (1949)
- 8 -
dass das Kohlenstoffatom 23 in trans- und das Kohlenstoff¬
atom 24 in cis-Stellung zum Methyl am C- liegt, was in der
Formel angedeutet ist. Die sterischen Verhältnisse an den
übrigen Ringverknüpfungsstellen sind im Formelbild nicht
berücksichtigt.

- 9 -
I. I E I I. BEITRAG ZUR STEREOCHEMIE DER PENTACYCLISCHEN
TRITERPENE DER /3-AMYRIN-OLEANOLSAEURE-GRUPPE
THEORETISCHER TEIL
A. BISHERIGE KENNTNISSE ÜEBER DIE STEREOCHEMIE DER
PENTACYCLISCHEN TRITERPENE1^
Aus seinen röntgenographischen Messungen der Molekül¬
dimensionen der Oleanolsäure und des Gypsogenins schloss
2)Giacomello
,dass das Kohlenstoffgerüst dieser Verbindun¬
gen vollkommen flach sein müsse, woraus sich eine trans-
anti-trans-Verknüpfung aller 5 Ringe ergibt. Die Hydroxyl¬
gruppe am Cp wurde von Giacomello als senkrecht zur Ebene
des flachen Ringsystems stehend angenommen, und diese Kon¬
figuration wurde von Ruzicka und Gubser,durch Messung
der Verseifungsgeschwindigkeiten der Acetate des /3-Amyrins
und des epi-/S-Amyrins und durch ihre Interpretation der Ka¬
lottenmodelle dieser beiden Verbindungen, bestätigt.
Aus den Eigenschaften der Tricarbonsäure C,,H,gOg, die
bei der energischen Oxydation der Abietinsäure erhalten wur¬
de (siehe Seite 25), kann geschlossen werden, dass die Rin¬
ge A und B der Abietinsäure und damit auch die gleichen Rin¬
ge der Oleanolsäure ( die experimentelle Verknüpfung der
Oleanolsäure mit der Abietinsäure folgt im nächsten Kapitel,
Seite 20) trans-verknüpft sind, vorausgesetzt dass bei der
Bildung der C,,-Tricarbonsäure keine Epimerisierung stattge-
1) Zusammenfassende Abhandlung: Barton & Holness, Soc. 1952, 782) Giacomello, G. 68, 363 (1938)3) Ruzicka & Gubser, Helv. 28, 1054 (1945)

- 10 -
funden hat . Was die Hydroxylgruppe am C„ betrifft, so er¬
gab ein erneuter Vergleich des 0-Amyrins mit dem epi-^-Amyrin
in Bezug auf die relativen Stabilitäten, den relativen Hin¬
derungsgrad und die Abspaltungsreaktionen, im Sinne der mo-
2)dernen Auffassung der polaren und äquatorialen Bindungen ,
dass diese Hydroxylgruppe im /3-Amyrin eine äquatoriale Lage
einnehmen muss, dass sie also die entgegengesetzte Konfigu¬
ration zu derjenigen besitzt, die ihr ursprünglich zugeschrie¬
ben wurde '. Für den Ring A des /3-Amyrins ergibt sich somit
die Teilformel III, die die sterischen Verhältnisse relativ
zu einer willkürlich gewählten Konfiguration am C angibt.
Als nächstes soll die Stereochemie der Ringe D und E
der Oleanolsäure besprochen werden, die sich hauptsächlich
auf die Ergebnisse der Untersuchungen von Barton stützt. Um
die lactonisierung der Oleanolsäure zu erklären, muss ange¬
nommen werden, dass die Carboxylgruppe am C-7 im stereoche¬
mischen Sinne polar ist. Diese Annahme ist aber sowohl mit
einer eis- als auch mit einer trans-Verknüpfung der Ringe
D/E vereinbar. Dass diese Ringe cis-verknüpft sind, wurde
durch die folgenden Reaktionen wahrscheinlich gemacht.'
Der Acetyl-11-keto-oleanolsäure-methylester (IV; R=Ac) lässt
sich sowohl durch Behandlung mit Bromwasserstoff in Eisessig
als auch mit Alkali isomerisieren. Durch Enolisierung der
a-,/3-ungesättigten Ketogruppe kann das Asymmetriezentrum am
C.» oder dasjenige am C,g epimerisiert werden. Die Ketogrup¬
pe der erhaltenen Verbindung (V; R=Ac) konnte nun, analog der
katalytischen Hydrierung des Acetyl-11-keto-oleanolsäure-
methylesters (IV;,R=Ac) zum Acetyl-oleanolsäure-methylester
(VI; R=Ac, R'=Me), wegreduziert werden. Das Reduktionspro¬
dukt (VII; R=Ac, R'=Me) erwies sich als Acetyl-18-iso-olea-
1) Barton & Schmeidler, Soc. 1948, 1197? 1949, 232
2) Barton & Rosenfelder, Soc. 1951, 1048
3) Barton, Exper. 6, 316 (1950"T^4) Barton & Holness, Soc. 1952, 785) Kitasat0, Acta phytochimica j8, 1 (1934)

- 11 -
nolsäure-methylester, denn bei der Oxydation mit Selendioxyd
lieferte es den bekannten Acetyl-dehydro-oleanolsäure-methyl-
ester (VIII; R=Ac, R'=Me) ', der auch bei der Oxydation des
Acetyl-oleanolsäure-methylesters (VI; R=Ao, R'=Me) mit Selen-
2)
dioxyd entsteht '. Diese Reaktionen zeigen, dass in der Olea-
nolsäure die Konfiguration am C,g unbeständig ist. Daher müs¬
sen die Ringe D/E cis-verknüpft sein. Die Bildung von VIII
(R=Ac, R'=Me) bei der Reduktion von IV (R=Ac)^'
und von V
(R=Ac) mit Natrium und Alkohol und nachfolgender Acetylie-
rung ist ein weiterer Beweis dafür, dass die Inversion am
C,g stattfindet und nicht am C,Q. Ausserdem lässt sich das
Dienon IX (R=Ac), in dem das Asymmetriezentrum am C,g aufge¬
hoben ist, mit Alkali nicht isomerisieren. - Diese Versuche
zeigen weiterhin, dass die Ringe B und C stabil mit einander
verknüpft sind.
Ein weiterer Hinweis auf die cis-Verknüpfung der Ringe
D/E der Oleanolsäure ist folgender. Wird die Oleanolsäure
(VI; R=R'=H) in Chloroformlösung mit Chlorwasserstoff behan¬
delt, so stellt sich fast augenblicklich ein Gleichgewicht
der freien Säure (76 #) und ihres lactons (X; R=H) (24 j6)
ein. Ein Gemisch der gleichen Zusammensetzung erhält man bei
gleicher Behandlung des Lactons. Im Gegensatz dazu geht die
18-iso-Oleanolsäure (VII; R=R'=H) unter den gleichen Bedin¬
gungen irreversibel in das entsprechende Lacton (XI; R=H)
über, das daher thermodynamisch viel stabiler sein muss. Wird
nun die Oleanolsäure oder ihr echtes lacton mit Bromwasser¬
stoff in Eisessig behandelt (diese Mischung ist ein viel
stärkerer Protonen-Donator als Chlorwasserstoff in Chloro¬
form) ,so bildet sich das 18-iso-Lacton.
1) Barton & Brooks, Soc. 1951, 257
2) Ruzicka, Grob & van der Sluys-Veer, Helv. £2, 788 (1939)3) Bilham, Kon & Ross, Soc. 1942, 540
H
1
1Pp.
et
7p
BCDB-O
CO
CD
1et
et
te
1piP
NP.
H-
H-
PB
BP
uç?
ro
p.
et
CD
Oet
CD
CD
et
wCO
HB
H-
Hu
CD
et
0>)
CD
0t)
M
oPi
mH
0«J
•O
BH
oa
>*•
o
1a>
OP
Bo
P0
«=:
BO
a1
H4
P-
OH)
1p.
MH-
4H
CD
P.
WB"
•^^
0M
to
P.
Bg
P.
CD
|O
rv>
P-
.—*
(0
«<j
PO
•d
Mp.
P^-^
tf
4B"
et
H-
BH>
«O
<<!
B1
4(B
P
Pp
et
OPi
P-
MH
<D
4P-
ÖB
PP-
P2
P.
OP
O<
piW
O4
et
cm
P1
B1
p:
PH-
P?
?P4
B"O
H4o
4CD
CBCD
PB-
CO
M(D
P.
Sp.
P-
XS
et
P.
H-
OO
W
PHj
ÏCD
oCO
P-
P.
P-
B(D
BP.
BH-
a1
CO
4P
(D
M-
<p-
4o
H,
p.
4B
MP.
CD
HCD
pW
Ba
PCD
<<!
«<!
HO«
)P
Bp
3O
PiO
0>1
B"
p.
O4
fl>
p-
pW
Bet
p1
PP
P.
pet
B"
et
CD
CD
c!4
P-
M-
B4
OCD
CO
H-
'SB
M-
OÖ
M(D
^1
••
B"
M-
H-
H-
PCD
1B
BOU
\j\
Pi:
Pp.
MCD
H-
14
Mro
aHj
H4
CD
44
gi-D
BP
O
IIP
(X)
CO
PP-
CD
ëci
H(D
P.
Hj
-
wCK
)P
H3
Cp:
H~
H4
CD
PiO
CD
CO
OOP
aW
i-b
vn
>••
pH-
g«
et
OH
BH
CD
Pi£
UD
l-H
o"
p.
CD
O<••
4P
PH
MFü
•B
P.
B4
P-
-J
>4
4CD
OB
CD
OB"
Het
S3"
4•
V.
'—
a4
et
p-
p.
Bet
V4
Op
o
wP!
>H)
B1
H-
Ol
pi3
o
IIt
P:
1CD
CD
HtH
Mg
pN
en
p.
es
«a1
0«5
<<!
fcs.
Ml
wH
Het
et
(D
(D
P.
«•
PiIl
CD
oCO
CD
oCD
P-
B0>)
et
Bto
p:
7?
•d
Bp
H)
O0
P-
4B
F!K
ta
VP
p.
P>
a>
Bo
>-*
.«
Pt*
en
Bcm
o1
oB
oo
M3
HCO
CD
0P
OB
&h-
n>
44
Mff
tO
CO
HP
OP-
p.
bd
l-H
MP
o0
et
2—'
—4
o
<^
4P.
SB1
StV
CD
On
P»>
^^
MCD
O0
H-
>tt>
34
oP.
CD
H-
4P
MII
Mp
PP
a1
pCD
CO
CD
CD
<<!
wtd
BO
p.
44
CD
H-
P.
p.
Wet
Pet
piP
P-
CD
et
OCD
<••
<»
et
4B
p:
OP.
œCD
a1
B-
o1-1
MH
PM
aet
H-
Po
>M
MP
td
HB
CD
et
P-
w<<
44
MM
B*
Op
H-
4CD
4M
nCO
MX
4^—'
B4
4P
MCD
OP.
p.
^^
.—,
(D
H
- 13 -
re wurde in die A ' -2-Aeetoxy-19-oxo-oleanen-28-säure
übergeführt, die beim Erhitzen leicht decarboxylierte, wobei
die Doppelbindung in 17,18-Stellung wanderte. Das so gewonne¬
ne £r ' -2-Acetoxy-19-oxo-nor-oleanen lieferte bei der Be¬
handlung mit Lithiumaluminiumhydrid und nachfolgender Ace-
tylierung die Verbindung XIII (R=Ac). XIII (R=Ac) wurde also
auf 2 Wegen erhalten. Derjenige aus der Morolsäure berührt
das Asymmetriezentrum am C...nicht. Der zweite aus der Sia-
resinolsäure enthält eine Decarboxylierung. Die Siareslnol-
säure hat die Carboxylgruppe in der gleichen räumlichen La¬
ge wie die Oleanolsäure, da sie auch in die Dehydro-oleanol-
säure (VIII; R=R'=H) übergeführt werden konnte1' 2'. Bei
der erwähnten Decarboxylierung muss nun die C-H-Bindung, die
am CL, gebildet wird, auf der gleichen Seite des Moleküls
liegen wie die Carboxylgruppe, und dies trifft auch für die
unberührte Konfiguration am C,, in der Morolsäure zu. Die
Partialsynthese der Morolsäure ausgehend von der 12,13-Di-
hydro-12-keto-siaresinolsäure (XV; R=H) *'durch Reduktion
nach Tolff-Kishner und Wasserabspaltung zeigt, dass die Konfi¬
guration am C.., in der Morolsäure thermodynamisch stabil ist.
Das gleiche muss bei der 12,13-Dlhydro-12-keto-oleanolsäu-
re (XVI; R=R'=H) der Fall sein, denn sie lässt sich mit Al¬
kali nicht isomerisieren.
X̂II XIII
OOH
1) Bilham, Kon & Ross, Soc. 1942, 540
2) Ruzicka, Grob, Egli & Jeger, Helv. 2£, 1218 (1943)
3) Barton, Brooks & Holness, Soc. 1951. 278
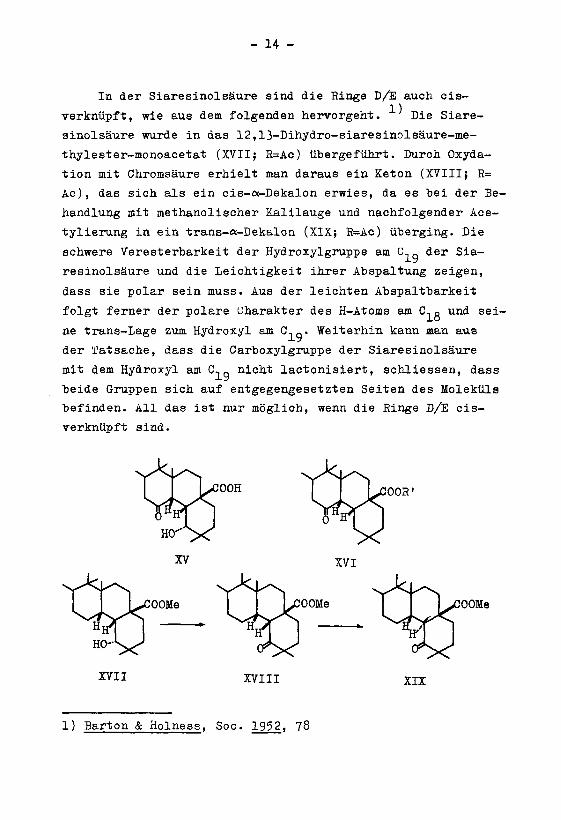
- 14 -
In der Siaresinolsäure sind die Ringe D/E auch cis-
verknüpft, wie aus dem folgenden hervorgeht. Die Siare¬
sinolsäure wurde in das 12,13-Dihydro-siaresinolsäure-me-
thylester-monoacetat (XVII; R=Ac) übergeführt. Durch Oxyda¬
tion mit Chromsäure erhielt man daraus ein Keton (XVIII; R=
Ac), das sich als ein cis-oc-Dekalon erwies, da es bei der Be¬
handlung mit methanolischer Kalilauge und nachfolgender Ace-
tylierung in ein trans-«-Dekalon (XIX; R=Ac) überging. Die
schwere Veresterbarkeit der Hydroxylgruppe am C,0der Sia-
resinolsäure und die Leichtigkeit ihrer Abspaltung zeigen,
dass sie polar sein muss. Aus der leichten Abspaltbarkeit
folgt ferner der polare Charakter des H-Atoms am C.q und sei¬
ne trans-Lage zum Hydroxyl am C,q. Weiterhin kann man aus
der Tatsache, dass die Carboxylgruppe der Siaresinolsäure
mit dem Hydroxyl am C,q nicht lactonisiert, schliessen, dass
beide Gruppen sich auf entgegengesetzten Seiten des Moleküls
befinden. All das ist nur möglich, wenn die Ringe D/E cis-
verknüpft sind.
00H 00R'
XV XVI
I JL^OOMe T LcOOMe T j^COOMe
"TOXVII XVIII XIX
1) Barton & Holness, Soc. 1952, 78

C...amHydroxylzumtrans-LagediesichergibtC,-amAtom
H-polaredasFürC,7-Carboxylgruppe.diewieliegenküls
Mole¬desSeitegleichenderaufC,,amHydroxylgruppedie
mussDaherKonfiguration.entgegengesetztediestituenten
Sub-polarebenachbartezweinunhabenCyclohexansdesform
Sessel¬derInbildet.SesselformeineCRingderdassden,
wer¬gedeutetsomussDiesstabil.12,13-Dihydro-oleanolsäure
derinC/DundB/CRingederVerknüpfungendiesindwähnt,
er¬schonWieWasserabspaltung.(trans-)derLeichtigkeit
derundAcetylierbarkeitschwerenihrerwegensein,lar
po¬mussXXIIIVerbindungderin-HydroxylgruppeC-...Die
werden.nen
gewon¬AcetylierungnachfolgendeundLithiumaluminiumhydrid
mitReduktiondurch(R=Ac)XXIausauchkonnteR=Ac)(XXIV;
-2,28-Diacetoxy-oleanen'AerhalteneDasliess.spalten
ab¬Wasserleichtsichdemaus(R=Ac),XXIIITriol-diacetat
dasergabAcetylierung,einervongefolgtminiumhydrid,
Lithiumalu¬mit(R=Ac)XXIIvonReduktionDie(R=Ac).XXII
KetondasWasserstoffperoxydmitOxydationdurchletzteren
demausundR=Ac)(XXI;Acetoxy-oleanen-28-säuremethylester
-2-'Aden(R=Ac)XXausmanerhieltAcetylierungund
MethylierunganschliessendeundWolff-Kishnernachduktion
Re¬durchoderHydrierungkatalytischeDurchübergeführt.
(R=Ac)XXKeton«^-ungesättigtedasinBrommithydrierung
De¬durchwurdeR'=Me)R=Ac,(XVI;nan-28-säuremethylester
2-Acetoxy-12-keto-olea-DerReaktionen.folgendenaussich
ergebenuC^undC.0amVerhältnissesterischenDie
ist.dargestelltXX
Formelderindaswieliegen,C/DRingederEbenederSeiten
entgegengesetztenauf.
C,amMethyldasundC.amboxylgruppe
Car-diedasssich,ergibtHieraussind.trans-verknüpftC/D
Ringediedasswerden,geschlossenkannXVIundXVbindungen
Ver¬denin,
CamKonfigurationderStabilitätderAus
-15-
en
H
MMM
11H
Bp
1P-
41P
71
p
B*O
P-
B«<
P-
pP
Pp
1P-
Pp
p-
PB
Oet
P.
H4
p.
P4
HP
O4
P.
P-
PP-
ro
B0<
)1
piH
p<<
B"
4
C-J.
et
OB
00
Op:
p-
PB
B"
H-
BO
P4
pt»
PB
a>
et
pp
p-
et
pp
PB-
et
Pp.
WP
p-
P4
C0>
)p
pC
4.
p
P.
<<;
Pp.
Ö(V
PB
Bp:
HP.
XP
H1
et
O)
OB"
P.
Pp
*—-
et
OP.
(gp-
•K>
pg
2B
Bo
BH-
»B
Hp.
Pbd
>*_y
H•d
OP
p:o
P-
<p
p-
\J\
B4
et
Pw
1H
<P-
y>
BP
$B-
P<o
PB-
B4
ifH
PO
Mp-
1p
4P-
HO
p.
Pp-
P<<
lM
P*<
pP
Bp
B*
PB
^^.
Het
a-
et
wet
et
p4
BCD
Wp-
op-
P4
BII
ro
4et
p4
PP
4m
-f*
pet
ro
piW
oP
B"
Op
mW
P3
<to
VJl
Bbd
B<t
t>
P.
pP>
«3
mP.
PP
BH
Pro
•••
B<x
>P-
mB
HH
pB
Mra
p4
4p.
W0<
5p-
P2
BH
CD
xa
p<q
PB"
—'
PP
HB
mB
p
O4
ro
p.
rS
PO
ro
P.
et
Ow
p-
4^
P-
•P
p-
4W
P!g
BB
S4
2p
om
(^
et
pp:
öCD
^^
BP
IIP
piP
pp
B-
BP-
Pp
p<
O4
td
CK)
Po1
p-
p-
4•
4p.
B•
Pp-
^^
HH
PS
mp
4p
<ip
Oet
Bet
et
Op
OP-
ta
PB
HÏ
sa
4S
4O
Pp-
<q
H)
P1
^-^
41
p.
Op
P-
B-
IIp
et
P.
BM
B"
4H
pp
OP
wH>
PO
Çd
p-
O«<
1M
opi
^S
BP
BP4
Bp.
a>
PM
pp:
et
u"p-
P-
P
oB
et
>••
H-
HO
B4
gM
CD
•d
Otj
4f
Wp
p-
<q
41
OP
ow
MB
4p-
Pw
CK)
BH
CD
ro
B-
et
et
P.
et
!»
HCD
BP.
O
4H
h>d
B4
OO
CO
Bp-
1P
Pp.
PH-
PO
ro
*—\
opi
4P
PPi
Bo
tu
P-
P.
C-J.
H-
N
p.
H)
4m
P:
piH
Hm
IIP.
B4
PP
PB
p-
mp.
H«<
)P-
00
P-
WP
P-
O*
ffCK
)H)
p:
OP-
OB1
HB
3pr
et
Pi
B0
pet
pP
P.
Bet
PB
co
••a
pm
et
ft>
Pe
HO
pH
4P
p1*
HP
Mo
PP
B-
H)
oP
p-
oP-
PB
MP.
P-
*4
4H
MH
P4
•d
Oq
BH
PB
*H
p-
!»
Bp«
p:o
«<!
P0«
)P
PS
BP
1p
Po
OP
BM
BB1
3>1
BP
ro
pS
CD
C*>
1B
B"
p-
*->v
et
4p
PX
HH
4H
Bo
4o
pB
gp
P«
<^
4CD
oP
Bo
Pi4
P«1
bd
»B
PB
•d
PCK
)1
p:
CD
1P.
p.
Öp
OC*
lp
p-
3*
<H
p4
HB
<<!
C«)
B"
•d
et
B*d
UP-
2B
iji
«<
p-
p«<
H-
B1
BO
•d
OP:
>d
PCK)
BP
oet
p.
et
o1
P-
B4
PCK
)P
PH
H-
ro
I-1
pp
4P»
P
g"pi
2H
42
P-
4<<
}g
Öo
oo
p1
et
4p.
CK)
o<f
cÖ
PH
et
iS»
Bw
!»
<U>
Mp-
44
<i
M<t
Opi
P4
1H
P<
B1
PP
<<i
o"
5?
B4
Pp
op
*B"
BO
et
et
BM
44
ro
ro
Het
mB*
ro
4H-
PiW
pp
OP
P*—.
0«)
p.
Om
P-
oH
PW
PM
pm
o1
4O
H
- 17 -
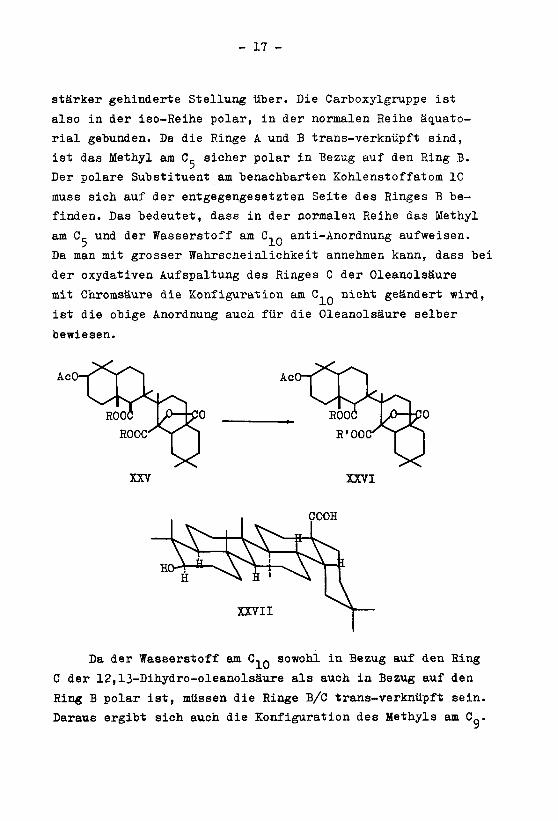
stärker gehinderte Stellung über. Die Carboxylgruppe ist
also in der iso-Reihe polar, in der normalen Reihe äquato¬
rial gebunden. Da die Ringe A und B trans-verknüpft sind,
ist das Methyl am C, sicher polar in Bezug auf den Ring B.
Der polare Substituent am benachbarten Kohlenstoffatom 10
muss sich auf der entgegengesetzten Seite des Ringes B be¬
finden. Das bedeutet, dass in der normalen Reihe das Methyl
am Cj- und der Wasserstoff ai C.. anti-Anordnung aufweisen.
Da man mit grosser Wahrscheinlichkeit annehmen kann, dass bei
der oxydativen Aufspaltung des Ringes C der Oleanolsäure
mit Chromsäure die Konfiguration am C._ nicht geändert wird,
ist die obige Anordnung auch für die Oleanolsäure selber
bewiesen.
XXV XXVI
COOH
XXVII
Da der Wasserstoff am C,Q sowohl in Bezug auf den Hing
C der 12,13-Dihydro-oleanolsäure als auch in Bezug auf den
Ring B polar ist, müssen die Ringe B/C trans-verknüpft sein.
Daraus ergibt sich auch die Konfiguration des Methyls am Cq.

- 18 -
Somit lassen sich die stereochemischen Verhältnisse der
12,13-Dihydro-oleanolsäure wie in der Formel XXVII darstel¬
len, sofern der Ring C, wie von Barton angenommen wurde, ei¬
ne Sesselform bildet.
Die Ueberführung des Lupeols in <P-Amyren (XXVIII)
und die Ueherführung der Betulinsäure (XXIX; R=H) in Mora-
21diol (XXXII; R=R'=H) zeigen, dass die Konfigurationen an
den C-Atomen 2, 5, 6, 9» 10, 14 und 17 im Lupeol und im/3-
Amyrin identisch sind, und dass die Konfiguration am C, -. im
Lupeol die gleiche sein muss wie in der Morolsäure und in
der 12,13-Dihydro-oleanolsäure. Bei der Behandlung der Betu¬
linsäure (XXIX) mit Bromwasserstoff in Eisessig geht sie in
das Lacton XXX über. Durch Spaltung des letzteren mit Li¬
thiumaluminiumhydrid und nachfolgende Acetylierung wurde
das Triol-diacetat XXXI (R=R'=Ac) hergestellt, aus dem sich
leicht Wasser abspalten liess, wobei Jioradiol-diacetat
(XXXII; R=R'=Ac) gebildet wurde. Die Leichtigkeit der Was¬
serabspaltung aus der Verbindung XXXI (R=R'=Ao) und die
schwere Acetylierbarkeit des Hydroxyls am Cln zeigen, dass
sowohl der Wasserstoff am C,g
als auch das Hydroxyl am C,q
polar sind und sich in trans-Stellung befinden. Die Bildung
von XXXI (R=R'=H) aus dem Lacton XXX führt zum Schluss, dass
die Oxymethylgruppe am C,„ zum polaren Hydroxyl am G,- cis-
ständig ist. Daraus folgt, dass die Oxymethylgruppe am C,„
und der Wasserstoff am C,g anti-Anordnung aufweisen, d.h.
dass die Ringe D/E trans-verknüpft sind '. Dies konnte
durch die Partialsynthese von XXXI (R=R'=Ac) bestätigt wer¬
den, die durch Reduktion der aus der Siaresinolsäure gewon¬
nenen Verbindung XIX (R=Ac) mit Lithiumaluminiumhydrid und
1) Arnes, Halsall & Jones, Soc. 1951, 450
2) Davy, Halsall & Jones, Soc. 1951, 2696
3) Davy, Halsall, Jones & Meakins, Soc. 1951, 2702
- 19 -
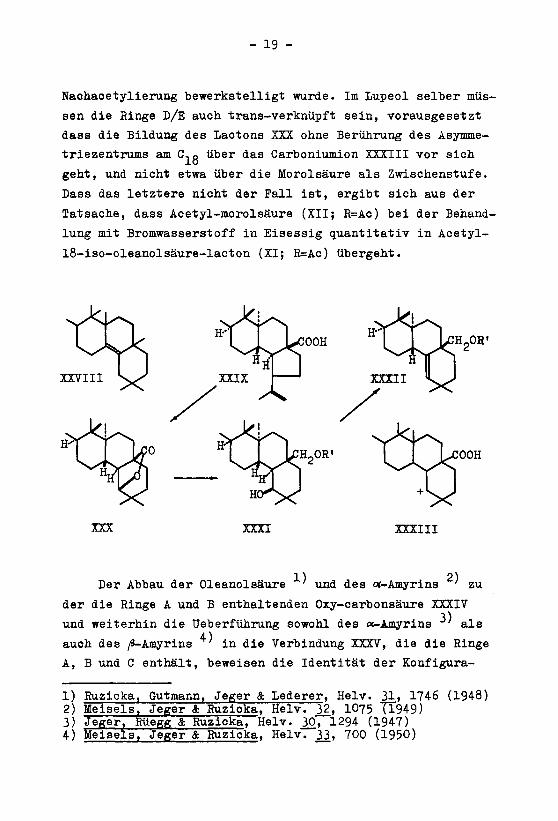
Nachacetylieruug bewerkstelligt wurde. Im Lupeol selber müs¬
sen die Hinge D/E auch trans-verknüpft sein, vorausgesetzt
dass die Bildung des Lactons XXX ohne Berührung des Asymme¬
triezentrums am C,q über das Carboniumion XXXIII vor sich
geht, und nicht etwa über die Morolsäure als Zwischenstufe.
Dass das letztere nicht der Fall ist, ergibt sich aus der
Tatsache, dass Acetyl-morolsäure (XII; R=Ac) bei der Behand¬
lung mit Bromwasserstoff in Eisessig quantitativ in Acetyl-
18-iso-oleanolsäure-lacton (XI; R=Ac) übergeht.
OOH
H2OR'
XXX XXXI XXXIII
1) 2)
Der Abbau der Oleanolsäure ' und des a-Amyrins'zu
der die Ringe A und B enthaltenden Oxy-carbonsäure XXXIV
und weiterhin die üeberführung sowohl des «-Amyrins' als
4)
auch des /3-Amyrins' in die Verbindung XXXV, die die Ringe
A, B und C enthält, beweisen die Identität der Konfigura-
1) Ruzicka, Gutmann, Jeger & Lederer, Helv. 31, 1746 (1948)2) Meiseis, Jeger & Ruzicka, Helv. _3_2, 1075 TÏ949)3) Jeger, Rüegg & Ruzicka, Helv. 30, 1294 (1947)4) Meiseis, Jeger & Ruzicka, Helv. 13, 700 (1950)

- 20 -
tionen der Kohlenstoffatome 2, 5» 6 und 9 im «- und /S-Amy-
rin und machen eine Uebereinstimmung am Asymmetriezentrum
10 wahrscheinlich. Weitere Kenntnisse über die Stereochemie
des «-Amyrins besitzt man vorläufig nicht.
C00H'
Il C
XXXIV
XXXV
B. ZUSAMMENHAENGE IM BAU DES RINGE A UND B BEI DEH OLEA-
NOLSAEURE UND DEN TRICYCLISCHEN DITERPENSAEUREN
Durch experimentelle Verknüpfung der Oleanolsäure mit
dem tricyclischen Triterpen Ambrein ', des Ambreins mit dem
2)
bicyclischen Diterpen Manool ' und des letzteren mit der
tricyclischen Abietinsäure'konnte die Uebereinstimmung
der Konstitution und Konfiguration der Ringe A und B bei den
Triterpenen der ^-Amyrin-Oleanolsäure-Gruppe, den bicycli¬
schen Diterpenen der Manool-Sclareol-Gruppe und den tricyc¬
lischen Diterpenen der Abietinsäure-Gruppe bewiesen werden.
Die Uebereinstimmung im Bau der Ringe ü'B der Oleanol¬
säure mit dem bicyclischen Teil des Ambreins ^XLI) ergab
sich durch die Herstellung identischer Abbauprodukte dieser
beiden Verbindungen, die die Ringe A/B enthielten K Bei
1) Jeger, Durst à Ruzicka, Helv. .30, 1859 (1947)2) Jeger, Durst & Büchi, Helv. 30, 1853 (194-7)3) Ruzicka & Sternbach, Helv. _2J5, 1036 (1942)4) Ruzicka, Gutmann, Jeger & lederer, Helv. j&, 1746 (1948)

- 21 -
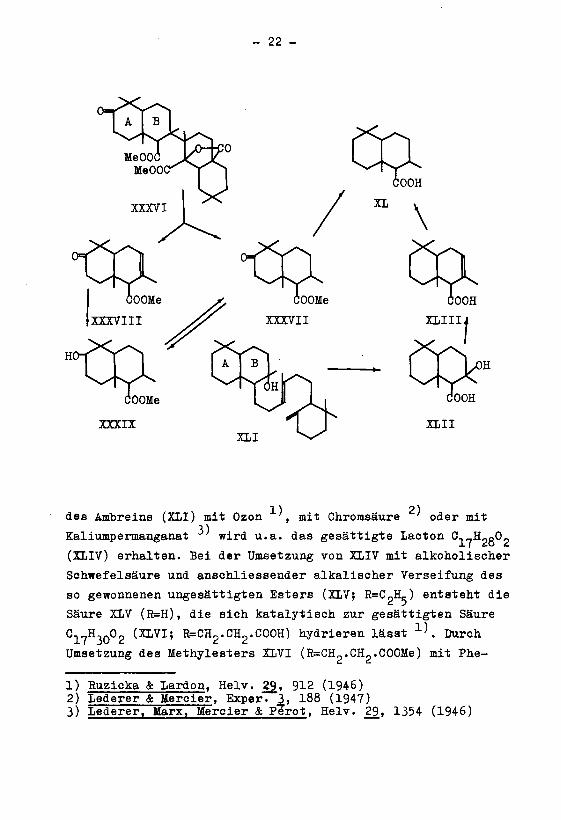
der thermischen Spaltung des iso-Oleanon-disäure-dimethyl-
ester-lactons (XXXVI), eines bekannten Abbauproduktes der
Oleanolsäure ', zerfällt das Molekül in einen ketonischen,
die Ringe A und B und einen nichtketonischen, die Ringe D
und E enthaltenden Anteil. Der ketonische Anteil, der durch
Behandlung des Pyrolyseproduktes mit Girard-Reagens T abge¬
trennt werden kann, besteht aus einem nicht trennbaren Ge¬
misch des gesättigten (XXXVII) und des ungesättigten Keto-
esters (XXXVIII) (oder Isomere), welches bei der katalyti¬
schen Hydrierung den gesättigten Oxy-ester XXXIX liefert.
Diese Verbindung wird durch vorsichtige Einwirkung von Chrom¬
säure zum Keto-ester XXXVII zurückoxydiert. Die anschlies¬
sende Reduktion nach Wolff-Kishner führt zur gesättigten
Säure C, ^„,0,, (XL), welche die Ringe a/B der Oleanolsäure
enthält.
Die Säure XL liess sich auch aus dem Ambrein gewinnen.
Bei der Oxydation des Ambreins (XLI) mit Kaliumpermanganat
entsteht in geringer Menge die gesättigte Oxy-säure C-j-H-gO,(XLII), aus der beim Kochen mit methanolischer Schwefelsäure
Wasser abgespalten wird, wobei sich die ungesättigte Säure
C^j-Hp.O« (XLIII) bildet. Diese liefert bei der katalytischen
Hydrierung die gesättigte Säure Clt-H?fi0p (XL), welche die
Ringe A/B des Ambreins enthält und mit dem Abbauprodukt der
Oleanolsäure identisch ist. Die Identität konnte durch den
Vergleich der Methylester und Anilide weiter gestützt werden.
Der übereinstimmende Bau des bicyclischen Teils des Am¬
breins mit den beiden Ringen des Diterpens Manool (XLVIII)
wurde durch Ueberführung dieser beiden Verbindungen in iden-
2)tische Abbauprodukte bewiesen '. Bei dem oxydativen Abbau
1) Ruzicka & Hofmann, Helv. 19, 114 (1936); Gutmann, Jeger& Ruzicka, Helv. 34, 1154"Tl951)
2) Ruzicka, Durst & Jeger, Helv. 30, 353 (1947)
- 22 -
XII
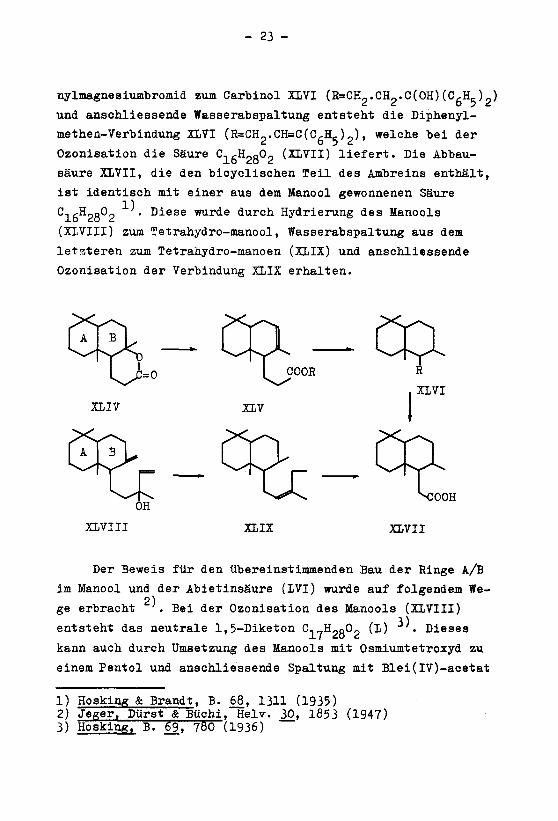
des Ambreins (XII) mit Ozon1)
mit Chromsäure2)
oder mit
3)wird u.a. das gesättigte lacton C. H?qO„Kaiiumpermanganat
(XIIV) erhalten. Bei der Umsetzung von XIIV mit alkoholischer
Schwefelsäure und anschliessender alkalischer Verseifung des
so gewonnenen ungesättigten Esters (XIV; R=C„H,-) entsteht die
Säure XIV (R=H), die sich katalytisch zur gesättigten Säure
C17H3002 (ELVI; R=CH2.CH2.C00H) hydrieren lässt 1K Durch
Umsetzung des Methylesters XIVI (R=CHp.CH2.C00Me) mit Phe-
1) Ruzicka & lardon, Helv. £9, 912 (1946)2) lederer & Mercier, Exper. 3, 188 (1947)3) lederer, Marx, Mercier & Plrot, Helv. ^9, 1354 (1946)
- 23 -
nylmagnesiumbromid zum Carbinol XLVI (R=CH2.CHp.C(OH)(C,H5)_)und anschliessende Wasserabspaltung entsteht die Diphenyl-
methen-Verbindung XLVI (R=CH2.CH=C(C6H,-)2), welche bei der
Ozonisation die Säure C-igHgoOg (XLVII) liefert. Die Abbau¬
säure XLVII, die den bicyclischen Teil des Ambreins enthält,
ist identisch mit einer aus dem Manool gewonnenen Säure
C, ,H„o02 • Diese wurde durch Hydrierung des Manools
(XLVIII) zum Tetrahydro-manool, Wasserabspaltung aus dem
letzteren zum Tetrahydro-manoen (XLIX) und anschliessende
Ozonisation der Verbindung XLIX erhalten.
XLIV
COOH
XLV
XLVI
Q^r _ Q>r _ Ca
XLVIII XLIX XLVII
Der Beweis für den übereinstimmenden Bau der Ringe A/B
im Manool und der Abietinsäure (LVI) wurde auf folgendem We-
2)ge erbracht . Bei der Ozonisation des Manools (XLVIII)
entsteht das neutrale 1,5-Diketon C17H2g02 (L) . Dieses
kann auch durch Umsetzung des Manools mit Osmiumtetroxyd zu
einem Pentol und anschliessende Spaltung mit Blei(IV)-acetat
1) Hosking & Brandt, B. 68, 1311 (1935)2) Jeger, Durst & Büchi, Helv. i^t 1853 (1947)3) Hosking, B. 69, 780 (1936)
- 24 -
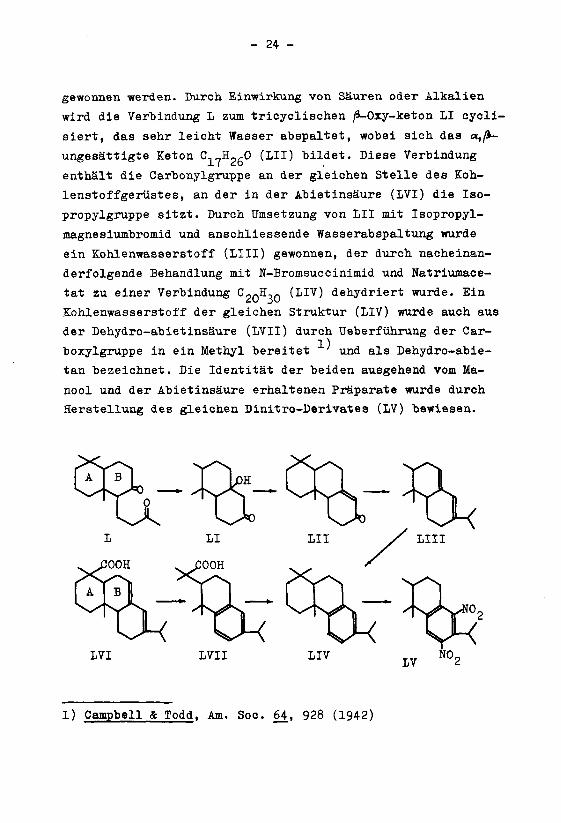
gewonnen werden. Durch Einwirkung von Säuren oder Alkalien
wird die Verbindung L zum tricyclischen /3-0xy-keton LI cycli-
siert, das sehr leicht Wasser abspaltet, wobei sich das a,^-
ungesättigte Keton C,7H2,0 (III) bildet. Diese Verbindung
enthält die Carbonylgruppe an der gleichen Stelle des Koh-
lenstoffgerüstes, an der in der Abietinsäure (IVI) die Iso-
propylgruppe sitzt. Durch Umsetzung von LH mit Isopropyl-
magnesiumbromid und anschliessende Wasserabspaltung wurde
ein Kohlenwasserstoff (LUI) gewonnen, der durch nacheinan-
derfolgende Behandlung mit N-Bromsuccinimid und Natriumace-
tat zu einer Verbindung C20H,0 (LIV) dehydriert wurde. Ein
Kohlenwasserstoff der gleichen Struktur (LIV) wurde auch aus
der Dehydro-abietinsäure (LVII) durch Ueberführung der Car-
boxylgruppe in ein Methyl bereitet' und als Dehydro-abie-
tan bezeichnet. Die Identität der beiden ausgehend vom Ma-
nool und der Abietinsäure erhaltenen Präparate wurde durch
Herstellung des gleichen Dinitro-Derivates (LV) bewiesen.
%
LVII
1) Campbell & Todd, Am. Soc. 64, 928 (1942)

- 25 -
C. DIE KONSTITUTION UND KONFIGURATION DER RINSE A
UND B EINIGER DITEHPENSAEUREN
Die trans-Verkntipfung der Ringe A/B der Ahietinsäure
(1VI) wurde auf folgendem Wege abgeleitet. Bei der Oxy¬
dation der Ahietinsäure mit Kaliumpermanganat oder Salpe-2 ) ^1
tersäure entsteht eine Tricarbonsäure O-j-H-gOg (LVIII) '
J/,
die nur aus dem Hing A hervorgehen kann, da sie die Methyl¬
gruppe, die bei der uehydrierung der Ahietinsäure zum fieten
4 )
Ü18H18 ^^) erhalten bleibt, enthält, zwei der Uarboxyl-
gruppen müssen Reste des Ringes B darstellen, während die
dritte der ursprünglichen Carboxylgruppe der Ahietinsäure
entspricht. Von den beiden Methylgruppen entspricht eine dem
Methyl am C, der Ahietinsäure und die zweite, auch tertiär
gebundene, die bei der Dehydrierung der Ahietinsäure abge¬
spalten wird, muss einer Methylgruppe am C-Atom 12 entspre¬
chen, da bei der Dehydrierung der Tricarbonsäure LVIII mit21
Selen m-Xylol entsteht '. Die läge der Carboxylgruppe am
C-Atom 1 in der Ahietinsäure wurde durch 2 Versuchsreihen
bewiesen. Abietinsäure-äthylester wurde nach Bouveault-
Blanc zum Abietinol (LX) reduziert, und dieses einerseits
durch Wasserabspaltung mit Phosphorpentachlorid unter Re-
tropinacolinumlagerung zu LXI und anschliessende Dehydrie¬
rung mit Schwefel in l-Aethyl-7-isopropyl-phenanthren C. 0H0_
(LXII) übergeführtJ
,andererseits zum Abietinal (IXIII)
oxydiert, das bei der Reduktion nach Wolff-Kishner den Koh¬
lenwasserstoff LUI lieferte, der durch Dehydrierung in
Reten (LIX) verwandelt wurde
Die C11-Tricarbonsäure LVIII ist optisch inaktiv. Es
muss in ihr also eine Mesoform (Symmetrieebene) vorliegen.
Für eine solche gibt es nur 2 Möglichkeiten (Formeln a und b),
1) Ruzicka & Sternbaoh, Helv. ,25, 1036 (1942)2) Ruzicka, Goldberg, Huyser & Seidel, Helv. 14., 545 (1931)3) Ruzicka & Sternbach, Helv. 21, 565 (1938)4) Vesterberg, B. 36"T~4200 (1903); Ruzicka & Waldmann,
Helv. 16, 842 (1933)5) Ruzicka, de Graaff & Müller, Helv. 15, 1300 (1932);
Haworth, Soc. 1932, 2717
6) Ruzicka, Waldmann, Meier & Hösli, Helv. 16_, 169 (1933)
- 26 -
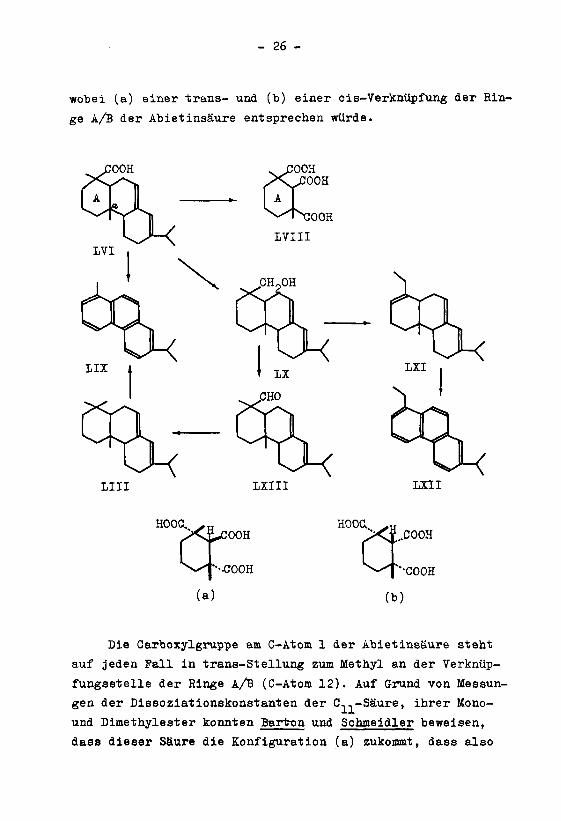
wobei (a) einer trans- und (b) einer cis-Verknüpfung der Rin¬
ge A/B der Abietinsäure entsprechen würde.
00H
00H
LVIII
IUI IXIII LXII
HOOG. Hooa.
C;[...COOH
'"•COOH
(b)
Die Carboxylgruppe am C-Atom 1 der Abietinsäure steht
auf jeden Fall in trans-Stellung zum Methyl an der Verknüp¬
fungsstelle der Ringe A/B (C-Atom 12). Auf Grund von Messun¬
gen der Dissoziationskonstanten der C^-Säure, ihrer Mono-
und Dimethylester konnten Bartron und Schmeidler beweisen,
dass dieser Säure die Konfiguration (a) zukommt, dass also

- 27 -
die Ringe A/B der Abietinsäure trans-verknüpft sind, voraus¬
gesetzt dass bei der Bildung der C,.,-Säure keine Epimerisie-1)
rung stattgefunden hat
Die Agathendisäure (IXIV) besitzt den gleichen Bau der
Ringe A/B wie die Abietinsäure (trans-Verknüpfung), nur befin¬
det sich die Carboxylgruppe am C, in cis-Stellung zum Methyl
an der Ringverknüpfungsstelle. Diese Tatsachen konnten wie
folgt abgeleitet werden. Bei der Dehydrierung liefert die
Agathendisäure Agathalin (1,5,6-Trimethyl-naphthalin) und
2)Pimanthren (1,7-Dimethyl-phenanthren) . Um die Lage der
schwer verseifbaren Carboxylgruppe festzulegen, wurde der
tricyclische iso-nor-Agathensäure-methylester (LXV) nach Bou-
veault-Blanc reduziert, aus dem entstandenen Alkohol Wasser
abgespalten und der erhaltene Kohlenwasserstoff (LXVI) de¬
hydriert. Man erhielt Homo-pimanthren (l-Aethyl-7-methyl-
phenanthren) (LXVII), wodurch die läge dieser Carboxylgrup¬
pe am C-Atom 1 bewiesen war '. Die trans-Verknüpfung der
Ringe A/B wurde durch üeberführung der Agathendisäure in
1,1,7,12-Tetramethyl-octahydro-phenanthren (LXVIII) bewie¬
sen, da diese Verbindung auch aus dem Manool hergestellt wer¬
den konnte,welches den gleichen Bau der Ringe A/B auf¬
weist wie die Abietinsäure. Die cis-Stellung der Carboxyl¬
gruppe am C-Atom 1 der Agathendisäure zum Methyl an der Ring¬
verknüpfungsstelle ergibt sich schon aus der, verglichen mit
der Abietinsäure, viel schwereren Verseifbarkeit ihrer Ester,
was auf eine sterische Hinderung hinweist. Bewiesen wurde die¬
se Konfiguration durch oxydativen Abbau der Agathendisäure
mit Kaliumpermanganat, der eine optisch aktive Tricarbon-
säure c-itH.,,-C> lieferte, die in Form ihres optisch aktiven
1) Barton & Schmeidler, Soc. 1948, 1197; 1949, 232
2) Ruzicka, de Graaff & Hosking, Helv. 14, 233 (1931)3) Ruzicka & Jacobs, B. 57, 509 (1938)4) Ruzicka, Zwicky & Jeger, Helv. 31, 2143 (1948)
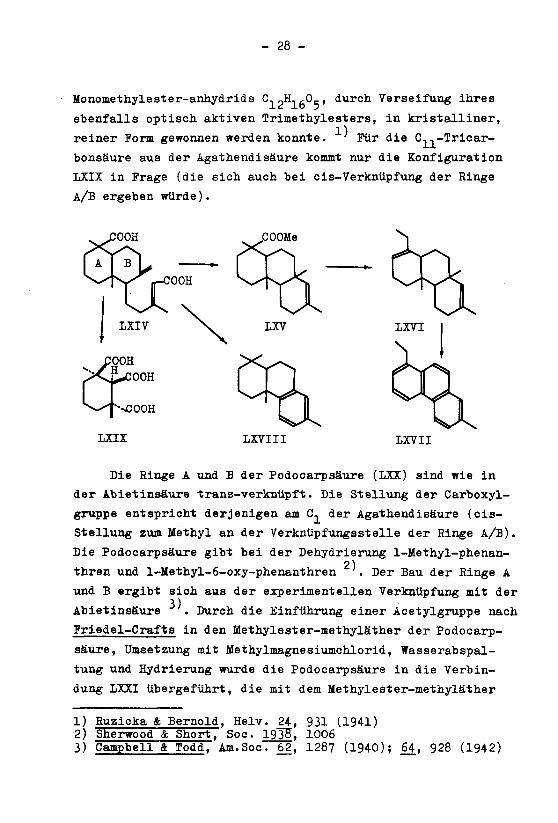
- 28 -
Monomethylester-anhydrids C.?H, ,-0,-, durch Verseifung ihres
ebenfalls optisch aktiven Trimethylesters, in kristalliner,
reiner Form gewonnen werden konnte. ' Für die C,,-Tricar-
bonsäure aus der Agathendisäure kommt nur die Konfiguration
LXIX in Frage (die sich auch bei eis-Verknüpfung der Ringe
A/B ergeben würde).
f A bL -
COOMe
LXIV LXV
LXVIII LXVII
Die Ringe A und B der Podooarpsäure (LXX) sind wie in
der Abietinsäure trans-verknüpft. Die Stellung der Carboxyl-
gruppe entspricht derjenigen am C, der Agathendisäure (cis-
Stellung zum Methyl an der Verknüpfungsstelle der Ringe A/B).
Die Podooarpsäure gibt bei der Dehydrierung 1-Methyl-phenan-
thren und l-Methyl-6-oxy-phenanthren2)
Der Bau der Ringe A
und B ergibt sich aus der experimentellen Verknüpfung mit der
Abietinsäure . Durch die Einführung einer Acetylgruppe nach
Friedel-Crafts in den Methylester-methyläther der Podooarp¬
säure, Umsetzung mit Methylmagnesiumchlorid, Wasserabspal¬
tung und Hydrierung wurde die Podooarpsäure in die Verbin¬
dung LXXI übergeführt, die mit dem Methylester-methyläther
1) Ruzicka & Bernold, Helv. 24, 931 (1941)2) Sherwood & Short, Soc. 193%, 1006
3) Campbell & Todd, Am.Soc. £2, 1287 (1940); 64_, 928 (1942)
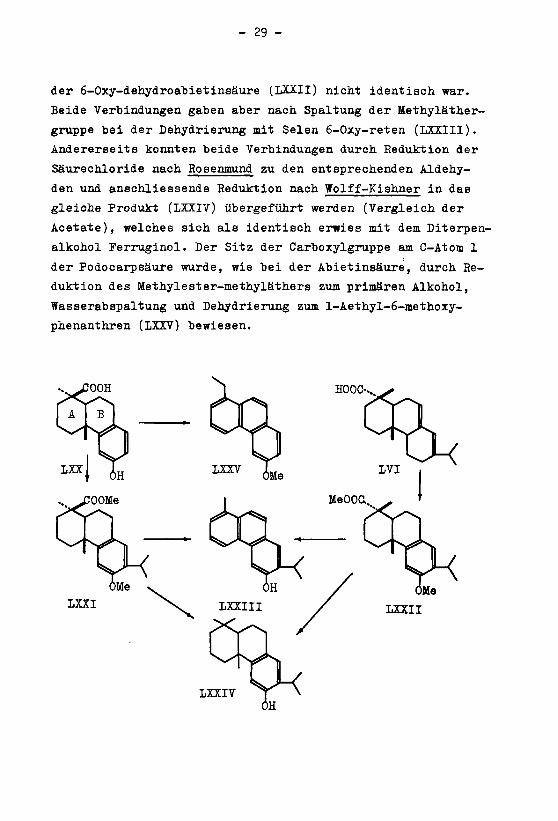
- 29 -
der 6-Oxy-dehydroabietinsäure (LXXII) nicht identisch war.
Beide Verbindungen gaben aber nach Spaltung der Methyläther¬
gruppe bei der Dehydrierung mit Selen 6-Oxy-reten (LXXII1).
Andererseits konnten beide Verbindungen durch Reduktion der
Säurechloride nach Rosenmund zu den entsprechenden Aldehy¬
den und anschliessende Reduktion nach Wolff-Kishner in das
gleiche Produkt (LXXIV) übergeführt werden (Vergleich der
Acetate), welches sich als identisch erwies mit dem Diterpen-
alkohol Perruginol. Der Sitz der Carboxylgruppe am C-Atom 1
der Podocarpsäure wurde, wie bei der Abietinsäure, durch Re¬
duktion des Methylester-methyläthers zum primären Alkohol,
Wasserabspaltung und Dehydrierung zum l-Aethyl-6-methoxy-
phenanthren (LXXV) bewiesen.

- 30 -
Die Verbindungen LXXI und LXXII unterscheiden sich also
nur durch die Konfiguration der Substituenten am C-Atom 1,
wobei die Carboxylgruppe der Verbindung IXXI (aus Podocarp-
säure) sich wie bei der Agathendisäure in cis-Stellung zum
Methyl an der Verknüpfungsstelle der Ringe A/B befindet und
stark gehindert ist. Die Ester der Podocarpsäure sind äus¬
serst schwer verseifbar.
D. EIGENE ABSEITEN
1. Allgemeines zur Bestimmung der Konfiguration der
Kohlenatoffatome 23 und 24 bei den Triterpenen der
ft-Amyrin-Oleanolsäure-Gruppe
Während bei den Diterpenverbindungen die sterischen Ver¬
hältnisse der beiden am C-Atom 1 im Ringe A gebundenen Sei¬
tenketten untersucht worden sind, war man bei den Triterpe¬
nen über die Konfiguration der entsprechenden, als C?, und
C„. bezeichneten Atome noch nicht orientiert. Eine Bestimmung
der räum!jenen Lage dieser Atome ist mit allem Vorbehalt auf
Grund folgender Ueberlegungen möglich. Die beiden epimeren
Hethoxy-carbonsäure-methylester LXXI und LXXII, die sich le¬
diglich durch eine Isomerie am C-Atom 1 unterscheiden, wei¬
sen ein stark verschiedenes optisches Drehungsvermögen auf.
Der Ester LXXII (aus Abietinsäure) ist um ca. 40° schwächer
rechtsdrehend als das Epimere LXXI (aus Podocarpsäure).
L.F. Fieser und M. Fieser'haben diesen Unterschied im
optischen Drehungsvermögen von LXXI und LXXII besonders her¬
vorgehoben und die Regel aufgestellt, dass das schwächer
1) Natural Products Related to Phenanthrene, 3rd Edition,S. 74, 1949

- 31 -
rechtsdrehende Isomere eines Epimerenpaares vom Typus LXXI
und LXXII am C-Atom 1 die gleiche Konfiguration aufweisen
soll wie die Abietinsäure. Eine solche Schlussfolgerung
kann aber nicht endgültig sein, da auch vom C-Atom 1 ent¬
fernte Asymmetriezentren seinen Drehungsbeitrag beeinflus¬
sen können. Daher kann der Vergleich des optischen Drehungs¬
vermögens nur Anhaltspunkte zur vorläufigen Konfigurations¬
zuteilung geben, die erst auf ihre Zuverlässigkeit durch
eindeutige Methoden geprüft werden müssen.
Ein Epimerenpaar vom geforderten Typus ist in der ß-
Amyrin-Oleanolsäure-Gruppe bekannt. Es wird gebildet von
den beiden Isomeren 4 ' -2,23-Dioxy-oleanen und A12'1-*-
2,24-Dioxy-oleanen (IXXVI und LXXVII). In der Literatur sind
die beiden Verbindungen als Hederadiol und epi-a-Boswellen-
diol bezeichnet. Die Oxygruppe am C-Atom 2 befindet sich in
beiden in der gleichen räumlichen Lage.
Hederadiol wurde aus Hederagenin (LXXVIII) durch Re¬
duktion des Säurechlorids nach Rosenmund und anschliessende
Reduktion des erhaltenen Aldehyds nach Wolff-Kishner gewon¬
nen . Es besitzt eine spez. Drehung (<*)> = +87 .
Epi-«-Boswellendiol wurde aus a-Boswellinsäure (LXXIX)
durch Oxydation des Hydroxyls zur Ketogruppe, Reduktion des
»-Bowellenonsäure-methylesters mit Platin und Wasserstoff zu
epi-cr-Boswellinsäure-methylester und anschliessende Reduktion2)
nach Bouveault-Blanc gewonnen '. Für diese Verbindung wurde
eine spez. Drehung (°0D = +87 angegeben. Das mit dem epi-«-
Boswellendiol identische Dihydro-sojasapogenol C soll eine
spez. Drehung («)_ = +95° aufweisen '.
Auf jeden Fall sind die Unterschiede im optischen Dre¬
hungsvermögen der beiden Isomeren LXXVI und LXXVII, wahr¬
scheinlich wegen der gegenseitigen Beeinflussung der beiden
1) Ruzicka & Marxer, Helv. _23, 144 (1940)2) Wirz, Diss. ETH, Zürich 1942
3) Meyer, Jeger & Ruzicka, Helv. 13, 672 (1950)
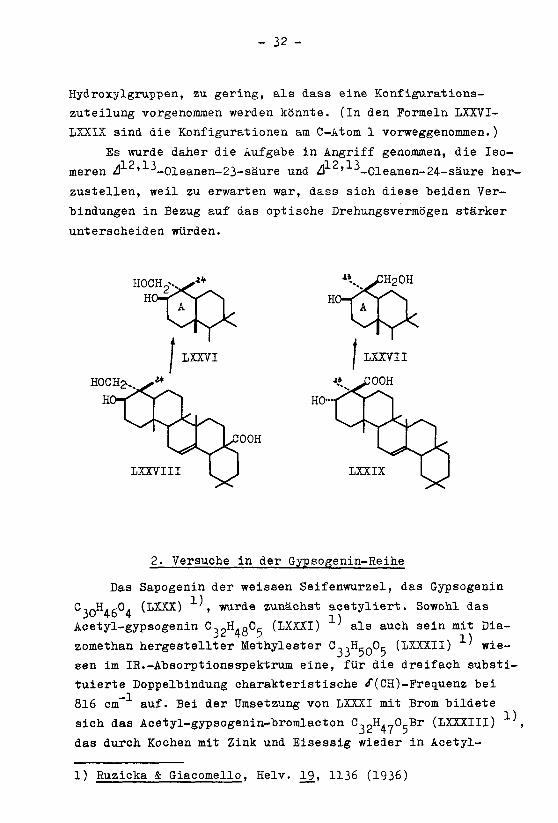
- 32 -
Hydroxylgruppen, zu gering, als dass eine Konfigurations-
zuteilung vorgenommen werden könnte. (In den Formeln IXXVI-
LXXIX sind die Konfigurationen am C-Atom 1 vorweggenommen.)
Es wurde daher die Aufgabe in Angriff genommen, die Iso--IQ-IT -] Q "I "3
meren à ' -Oleanen-23-säure und Ù ' -Oleanen-24-säure her¬
zustellen, weil zu erwarten war, dass sich diese beiden Ver¬
bindungen in Bezug auf das optische Drehungsvermögen stärker
unterscheiden würden.
OOH
2. Versuche in der Gypsogenin-Reihe
Das Sapogenin der weissen Seifenwurzel, das Gypsogenin
C^H^O, (LXXX) ,wurde zunächst acetyliert. Sowohl das
30 46 4 -,\
Acetyl-gypsogenin C-.2H.gOj- (LXXXI) ;als auch sein mit Dia-
zomethan hergestellter fflethylester C^H^Ot, (LXXXII) ' wie¬
sen im IR.-Absorptionsspektrum eine, für die dreifach substi¬
tuierte Doppelbindung charakteristische <f(CH)-Frequenz bei
816 cm auf. Bei der Umsetzung von LXXXI mit Brom bildete
sich das Acetyl-gypsogenin-bromlacton C^-H.-Oj-Br (LXXXIII) ,
das durch Kochen mit Zink und Eisessig wieder in Acetyl-
1) Ruzicka & Giacomello, Helv. 19, 1136 (1936)

- 33 -
gypsogenin (LXXXI) zurückgeführt werden konnte. Das durch
reduktive Oeffnung des Bromlactons gewonnene Präparat war
mit dem Acetylierungsprodukt des Gypsogenins nach Schmelz¬
punkt, Mischprobe und dem IR.-Spektrum identisch. Hierauf
wurde die Verbindung LXXXIII mit Chromtrioxyd in Eisessig
zum Acetyl-gypsogeninsäure-bromlacton C.pH.-OgBr (LXXXIV)
oxydiert, und der mit Diazomethan hergestellte Methylester
C,,H.o0^Br (LXXXV) ' des letzteren durch Reduktion mit55 49 o
Zink und Eisessig in den Acetyl-gypsogeninsäure-monomethyl-
ester C-.-,Hc»0^ (LXXXVI) übergeführt. Der, wiederum durch Um-33 ?<-> b °
setzung mit Diazomethan bereitete Acetyl-gypsogeninsäure-
dimethylester C^H^Og (IXXXVII) ;zeigte im IR.-Spektrum
die für die dreifach substituierte Doppelbindung charakte¬
ristische f(CH)-Frequenz bei 817 cm" . Der Disäure-mono-
methylester LXXXVI lieferte bei der Einwirkung von Thionyl-
chlorid das Säurechlorid C-.,H.o0cCl (LXXXVIII), das nach55 4y P
Rosenmund zum Aldehyd C.,Hc-.Oc (LXXXIX) reduziert wurde. Die55 ?Q P
Reduktion des Aldehyds LXXXIX nach Wolff-Kishner mit nach¬
folgender Methylierung mit Diazomethan ergab, allerdings in
kleiner Ausbeute, den 4 '-2-Oxy-oleanen-23-säuremethyl-
ester C^r&zcP-, (XCII). Andererseits wurde der Aldehyd LXXXIX
durch Umsetzung mit Aethylendithioglykol in das Aethylendi-
thioketal XC übergeführt, das bei der reduktiven Entschwe¬
felung mit Raney-Nickel in nahezu quantitativer Ausbeute den
412»1^_2-Acetoxy-oleanen-23-säuremethylester C,,H5204 (XCI)
lieferte. Das IR.-Spektrum dieser Verbindung weist wiederum
die für eine dreifach substituierte Doppelbindung typische
f(CH)-Frequenz bei 817 cm" auf, woraus die unveränderte Lage
der Doppelbindung hervorgeht. Dass die Doppelbindung bei der
oben geschilderten Ueberführung einer Carboxylgruppe in ein
1) Ruzioka & Giacomello, Helv. 20, 299 (1937)2) Ruzicka, Giacomello & Grob, Helv. 21, 83 (1938)

- 34 -
Methyl nicht wandert, ergibt sich auch aus Analogie zum fol¬
genden Modellversuch. Acetyl-oleanolsäure wurde in ihr Säure¬
chlorid übergeführt und dieses nach Rosenmund zum Acetyl-
oleanol-aldehyd reduziert. Letzterer ging bei der Umsetzung
mit Aethylendithioglykol und nachfolgender Entschwefelung
mit Raney-Nickel in /3-Amyrin-acetat über, das nach Schmelz¬
punkt, Mischprobe und spezifischer Drehung mit einem authen¬
tischen Präparat identisch war.
Der durch alkalische Verseifung des Acetats XCI gewon-
nene ^ '-2-Oxy-oleanen-23-säuremethylester (XCII) war mit
dem durch Reduktion des Aldehyds LXXXIX nach Wolff-Kishner
hergestellten Präparat (siehe oben) identisch. Der Oxy-ester
XCII wurde nun zum à ' -2-0xo-oleanen-23-säuremethylester
C,,H.qO, (XCIII) oxydiert, dieser in das AethylendithioketalaL2 1}
XCIV übergeführt und letzteres mit Raney-Nickel zum tr~ '-
Oleanen-23-säuremethylester C,-.H,-0Op (XCV) reduziert. Durch
Verseifung des Esters XCV im Einschlussrohr bei 200° wurde
das gesuchte Endprodukt dieser Reaktionsfolge, die a' ->~
Oleanen-23-säure C-,QH.g02 (XCVI) gewonnen, die noch durch
Umsetzung ihres Säurechlorids mit Ammoniak in das Säureamid
C3()H.g0N (XCVII) übergeführt wurde.
3» Umsetzungen in der a-Boswellinsäure-Reihe
Die o-Boswellinsäure (XCVIII) 'wurde in ihren Methyl¬
ester (XCIX) übergeführt und dieser zum ^12,1^-2-0xo-oleanen-24-säuremethylester ^jH.gO, («-Boswellenonsäure-methylester,
1) Ruzicka & Wirz, Helv. 23, 132 (1940)
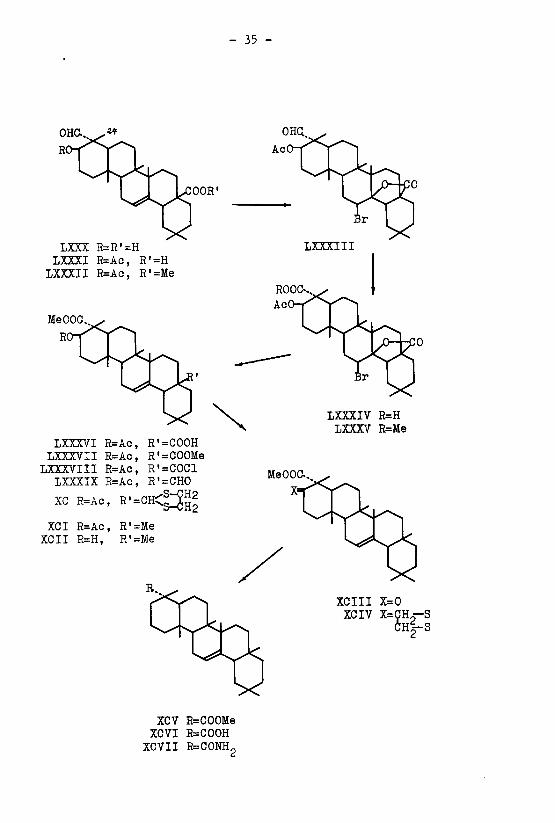
- 35 -
OHQ
OOR'
LXXX R=R'=H
LXXXI R=Ac, R'=H
LXXXII R=Ac, R'=Me
LXXXIII
LXXXIV R=H
LXXXV R=Me
LXXXVI R=Ac, R'=COOH
LXXXVII R=Ac, R'=COOMe
LXXXVIII R=Ac, R'=COCl
LXXXIX R=Ac, R'=CHO
XC R=Ac, R'=CH<m|2XCI R=Ac, R'=Me
XCII R=H, R'=Me
XCIII X=0
XCIV X=ÇH_—SCH%-S
XCV R=COOMe
XCVI R=COOH
XCVII R=CONH„

- 36 -
1) 2)
CI) oxydiert. Das UV.-Spektrum' dieses Ketons weist ein
Maximum bei 290 nyi, loge = 1,46, auf . Die Umsetzung von
CI mit Aethylendithioglykol lieferte das Aethylendithioketal
CII, aus dem durch reduktive Entschwefelung mit Raney-Nickel
der ^r ' -01eanen-24-säuremethylester C^1H5002 (CI11) gew011-
nen wurde. Das IB.-Spektrum dieses Esters weist ein stark
ausgeprägtes Absorptionsmaximum bei 813 cm" auf, das der
,f(CH)-Frequenz einer dreifach substituierten Doppelbindung
entspricht. CHI konnte auch auf einem zweiten Wege aus der
»-Boswellinsäure hergestellt werden. Beim Erhitzen des Ben-
zoyl-«-boswellinsäure-methylesters (C) auf 330-340 wurde
Benzoesäure abgespalten, unter Bildung des Û »J' ' -Olea-
dien-24-säuremethylesters O^H.gOp (CVI), der bei der Hydrie¬
rung mit Platin und Wasserstoff in die Verbindung CHI über¬
ging. Die beiden Präparate von CHI waren nach Schmelzpunkt,
Mischprobe und IR.-Spektrum identisch. Durch Verseifung von
CHI mit 15-proz. methanolischer Kalilauge bei 180-190 wur¬
de die Ar ' i-01eanen-24-säure C,0H.gO„ (CIV) gewonnen, und
diese durch Einwirkung von Ammoniak auf ihr Säurechlorid in
das 412,13-01eanen-24-säureamid C,0H.g0N (CV) übergeführt.
Um ganz sicher zu sein, dass in der, auf oben beschrie¬
benem Wege hergestellten /r ' -Oleanen-24-säure (CIV) der
Ring A noch als 6-Ring vorliegt, wurde diese Verbindung in
das bekannte a' -Oleanen (/9-Amyren) übergeführt. Zu die¬
sem Zwecke wurde die Säure CIV mit Thionylchlorid zum Ar *-
Oleanen-24-säurechlorid umgesetzt und dieses nach Rosenmund
zum Ar ' -24-0xo-oleanen reduziert. Durch Umsetzung des
1) Wirz, Diss. ETH, Zürich 1942; vgl. auch Helv. .33, 686(1950)2) Die UV.-Absorptionsspektren wurden, wenn nichts anderes
vermerkt, in alkoholischer Lösung aufgenommen.3) Dieses Spektrum wurde in Chloroform-Lösung aufgenommen.
- 37 -
letzteren mit Aethylendithioglykol und nachfolgendes Verko¬
chen mit Raney-Nickel wurde das #2,13-01eanen C3QH Q(CVII)1)
erhalten, das nach Schmelzpunkt, Mischprobe, spez. Drehung
und IR.-Spektrum mit einem aus /r ' -2-Oxo-oleanen (/9-Amy-2)
ron) 'durch Reduktion nach Wolff-Kishner gewonnenen Präpa¬
rat identisch war. Das IR.-Spektrum von CVII zeigte wiederum
das Absorptionsmaximum der dreifach substituierten Doppel¬
bindung bei 814 cm~ .
R'O-
300Me
XCVTII R=R'=H
XCIX R=Me, R'=H
C R=Me, R'=CcHcCO' 6 5
OOMe
CI X=0
CII X=ÇHx-SCHS-S
CVI
CHI R=COOMe
CIV R=COOH
CV R=CONH2CVII R=Me
1) Mnterstein & Stein, A. 502, 223 (1934); Ruzicka, Schel¬
lenberg & aoldberg, Helv. 20, 791 (1937)2) Vesterberg, B. 24, 3836 (lB91); Ruzicka & Wirz, Helv. 24,
24Ö (1941)

- 38 -
4* Diakussion der Versuchsergebnisse
In der vorliegenden Arbeit sind zwei Vertreter der ß-Amyrin-Oleanolsäure-Gruppe, das Gypsogenin (LXXX) und die
o-Boswellinsäure (XCVIII), welche an den C-Atomen 23 oder 24
eine Aldehyd- bzw. eine Säure-Gruppe enthalten, in das Epi-
merenpaar 412,13-01eanen-23-säure (XCVI) und à1 ' ^-Oleanen-
24-säure (CIV) übergeführt worden. Wie aus der Tabelle 1 her-
vorgeht, besitzen die aus dem Gypsogenin bereitete a~ '
Oleanen-23-säure (XCVI), ihr Methylester XCV und Amid XCVII,
eine um ca. 40° geringere positive Drehung als die entspre¬
chenden, aus der «-Boswellinsäure hergestellten epimeren Ver¬
bindungen CIV, CHI und CV mit einer funktionellen Gruppe am
C0,. Anhand dieser Beobachtungen kann man, bei Anwendung der
Regel von L.F. Fieser und M. Fieser (siehe Seite 30), mit Vor-
behalt annehmen, dass die (Jarboxylgruppe der à ' -Oleanen-
23-säure die gleiche sterische Lage einnimmt wie die Carboxyl-
gruppe der Abietinsäure, also in trans-Stellung zum Methyl am
Cr liegt. Eine endgültige Konfigurationszuteilung wird jedoch
erst auf Grund der Bestimmung der Verseifungsgeschwindigkei-
ten der Ester XCV und CHI möglich sein, da sich diese bei
der Verseifung analog verhalten müssen wie das Verbindungs¬
paar Abietinsäure-methylester - Podocarpsäure-methylester, in
dem die räumliche Lage der Carboxylgruppen bekannt ist.
Tabelle 1
Verbindung <«>D «D
ü-2'13_01eanen-23-säuremethylester(XCV)uL2'1^_01eänen-24-säuremethylester(CIII)
+ 97°;+ 96°
+138°;+135°+441°}+437°
+627°;+613°
d-2,13-01eanen-23-säure (XCVI)
<^2'13-01eanen-24-säure (CIV)
+108°
+144°
+475°
+634°
6L2»13_01eanen-23-säureamid (XCVII)
sL2'13_01eanen-24-säureamid (CV)
+ 91°
+140°
+400°
+615°

- 39 -
EXPERIMENTELLER TEIL1
A. Versuche in der Gypsogenin-Reihe
Reinigung des Sapogeningemisches der weissen Seifenwurzel.
Das rohe Gypsogenin wurde zunächst über das Kaliumsalz gerei¬
nigt. Zu diesem Zwecke wurde das Sapogeningemisch in sieden¬
dem Alkohol gelöst und mit einer alkoholischen Lösung der be¬
rechneten Menge Kalilauge versetzt. Der durch Eindampfen der
Lösung erhaltene Brei wurde im Dampftrockenschrank getrocknet,
und das nunmehr als harte Masse vorliegende Kaliumsalz gepul¬
vert und im Soxleth mit Aether extrahiert. Der ungelöste Rück¬
stand wurde mit verd. Salzsäure in die freie Säure übergeführt
und diese in Essigester aufgenommen. Die Essigesterlösung
wurde zur Kristallisation eingeengt und die auf diese Weise
gewonnene Substanz noch aus Alkohol umkristallisiert.- Das
nach dieser Methode gereinigte Gypsogenin wies Schmelzpunkte
auf, die zwischen 273° und 288° lagen (bei leichter Zerset¬
zung), und stellte noch keine einheitliche Verbindung dar.
Jiine bessere Reinigung war jedoch für die Weiterverarbei¬
tung unnötig.- in der Jî'olge wurde das rohe Sapogeningemisch
nur durch Umkristallisation aus Methanol gereinigt und dann
weiterverarbeitet.
Acetyl-gypsogenin (LXXXI) . 5,7 g Gypsogenin vom
Schmelzpunkt 278-280° wurden in 60 cur Pyridin und 60 cnr
1) Die Schmelzpunkte sind korrigiert und wurden in einer im
Hochvakuum evakuierten Kapillare bestimmt. Die optischenDrehungen wurden in Chloroform in einem Ronr von 1 dm Län¬
ge gemessen.
2) Vgl. Ruzicka & Giacomello, Helv. 19, 1136 (1936)

- 40 -
Acetanhydrid gelöst und über Nacht stehen gelassen. Das nach
der üblichen Aufarbeitung erhaltene Rohprodukt wurde aus Me¬
thanol kristallisiert. Man erhielt so 3 g, nadeiförmige Kri¬
stalle bildende Substanz, die bei ca. 160 unscharf schmolz,
bei höherer Temperatur wieder erstarrte, um dann bei 241-242
erneut zu schmelzen. Zur Analyse gelangte ein 4mal aus Metha¬
nol umkristallisiertes und 5 Tage im Hochvakuum bei 105 ge¬
trocknetes Präparat vom Schmelzpunkt 241-242°.
4,151 mg Subst. gaben 11,378 mg C02 und 3,517 mg H20
C32H4g05 Ber. C 74,96 H 9,44 i-
Cef. C 74,80 H 9,48 f.
Acetyl-gypsogenin-methylester (LXXXII) '. 220 mg Ace-
tyl-gypsogenin wurden in Methanol gelöst und mit ätherischer
Diazomethanlösung umgesetzt. Das nach üblicher Aufarbeitung
erhaltene Rohprodukt kristallisierte aus Methanol in Nadeln,
die scharf bei 202 schmolzen. Zur Analyse gelangte ein 4mal
umkristallisiertes und 44 Tage bei 105 im Hochvakuum getrock¬
netes Präparat vom Schmelzpunkt 203 •
3,666 mg Subst. gaben 10,082 mg C0„ und 3,130 mg H„0
C33H5005 Ber. C 75,24 H 9,57 %
Gef. C 75,09 H 9,65 #
(«)D = +82° (c = 0,84)
Aoetyl-gypsogenin-bromlacton (LXXXIII) '. 500 mg Ace-
tyl-gypsogenin vom Schmelzpunkt 241-242° wurden in 8 cm
heissem Methanol gelöst. Nach kurzem Abkühlen tropfte man
eine Lösung von 160 mg Brom (genau 1 Mol) in 5 cur Tetra¬
chlorkohlenstoff langsam zu, verdünnte hierauf mit Aether
1) Vgl. Ruzicka & Giacomello, Helv. 19, 1136 (1936)

- 41 -
auf ca. 100 cm und wusch die Lösung ein paarmal mit Wasser.
Das schaumige Rohprodukt kristallisierte heim Bespritzen mit
Methanol. Die Kristalle schmolzen hei 229-230 und gaben mit
Tetranitromethan keine Farbreaktion. Das rohe Bromlacton
wurde durch eine Säule aus 18 g Aluminiumoxyd (Akt.II) chro-
matographiert.
Frakt. Lösungsmittel Menge eluierter Substanz
1-5
6-8
9-10
11-12
13-16
500 cm3 Päth.-Bzol(4:l)
300 cm3 Päth.-Bzol(2:1)
200 cm3 Päth.-Bzol(2:l)
200 cm3 Päth.-Bzol(l:l)
500 cm3 Päth.-Bzol(l:l)
50 mg Krist. Smp.239-242°
140 mg Krist. Smp.236-248°60 mg Krist. Smp.248-260°
80 mg Krist. Smp.256-258°
50 mg Krist. Smp.250-260°
Die Fraktionen 7-16 wurden vereinigt und aus Methylen¬
chlorid-Methanol kristallisiert. Das Acetyl-gypsogenin-brom-
lacton bildete entweder dünne Nadeln oder Blättchen und
schmolz unter Zersetzung, was die grosse Empfindlichkeit des
Schmelzpunktes erklärt. Zur Analyse gelangte ein 2mal umkri¬
stallisiertes und 48 Stunden im Hochvakuum bei 80-90 ge¬
trocknetes Präparat vom Schmelzpunkt 253° und ein zweites,
durch 5maliges Umlösen gereinigtes und anschliessend im Hoch¬
vakuum getrocknetes Präparat vom Schmelzpunkt 250-251 ••
3,658 mg Subst. gaben 8,683 mg C02 und 2,600 mg H„0
3,795 mg Subst. gaben 9,051 mg COp und 2,668 mg H-0
3,816 mg Subst. gaben 1,226 mg AgBr
2,852 mg Subst. gaben 0,911 mg AgBr
C,2H 05Br Ber. C 64,96 H 8,01 Br 13,51 1'
Gef. C 64,78 H 7,95 Br 13,67 1>
Gef. C 65,09 H 7,87 Br 13,59 1»
(oc)D = +74°; +77° (c = 0,88? 0,85)

- 42 -
Reduktion des Acetyl-gypsogenin-bromlactons (LXXXIII)
mit Zink und Eisessig '. 300 mg Substanz vom Schmelzpunkt
246-247° (Zers.) wurden in 10 cm Eisessig gelöst und mit
2 g Zinkstaub 6i Stunden unter Rückfluss erhitzt. Es wurde
sodann mit Aether verdünnt, vom Zink abfiltriert und die Lö¬
sung mit Wasser gewaschen. Man erhielt 290 mg Rohprodukt.
Nach 2maligem Umkristallisieren aus Methanol schmolzen die
nadeiförmigen Kristalle bei 240-242° (nach unscharfem Schmel¬
zen bei 160-170° und Wiedererstarren) und gaben mit dem Ace-
tylierungsprodukt des Gypsogenins (LXXXI) gemischt keine De¬
pression. Zur Analyse wurde noch 2mal aus Methanol umkristal¬
lisiert und 2i Tage bei 90° im Hochvakuum getrocknet; Smp.241 •
3,897 mg Subst. gaben 10,660 mg C02 und 3,268 mg H„0
C32H48°5 Ber' C 74'96 H 9'U *
Gef. C 74,65 H 9,38 f>
Das IR.-Absorptionsspektrum ist mit demjenigen des Ace-
tyl-gypsogenins (LXXXI) identisch.
13,28-Lacton der 2-Acetoxy-13-oxy-12-brom-oleanan-23,28-———
?~1
disäure (Acetyl-gypsogeninsäure-bromlacton, LXXXIV) '.
300 mg Acetyl-gypsogenin-bromlacton vom Schmelzpunkt 246-247°
(Zers.) wurden in 8 cm Eisessig gelöst und bei Zimmertempe¬
ratur eine Lösung von 210 mg Chromtrioxyd in 7 cur Eisessig
und 0,16 cm konz. Schwefelsäure zugefügt. Nach 2i-tägigem
Stehen wurde in üblicher Weise aufgearbeitet. Nach 5maligem
Umkristallisieren aus Methanol schmolz die Säure, die in Bü¬
scheln vereinigte Prismen bildete, unscharf bei 315-320°.
Das Analysenpräparat wurde 48 Stunden im Hochvakuum bei 100°
getrocknet.
1) Vgl. Ruzioka, Giacomello & Grob, Helv. 21, 83 (1938)2) Vgl. Ruzicka & Giacomello, Helv. 20, 299 (1937)

- 43 -
3,726 mg Subst. gaben 8,598 mg C02 und 2,592 mg H203,725 mg Subst. gaben 8,620 mg C02 und 2,640 mg HpO
C H.„06Br Ber. C 63,25 H 7,80 #
Gef. C 62,97 H 7,78 Jt
Gef. C 63,15 H 7,93 *
13,28-Lacton des 2-Acetoxy-13-oxy-12-brom-oleanan-23,28-
disäure-monomethylesters (Acetyl-gypsogeninsäure-methylester-
bromlacton, LXXXV) '. 1,12 g der Säure LXXXIV wurden in Me¬
thanol unter Zusatz von wenig Methylenchlorid gelöst und mit
ätherischer Diazomethanlösung umgesetzt. Der Ester kristalli¬
sierte aus Methylenchlorid-Methanol in rechteckigen Nadelbü¬
scheln, die bei 254-256 (unter Zersetzung) schmolzen. Zur
Analyse gelangte ein 7mal umkristallisiertes und 84 Stunden
im Hochvakuum bei 100 getrocknetes Präparat.
3,665 mg Subst. gaben 8,552 mg CO« und 2,623 mg HpO4,946 mg Subst. gaben 1,688 mg AgBr
4,422 mg Subst. verbrauchten bei der Methoxylbestimmung nach
Vieböck und Brecher 2,129 cur 0,02-n. Na„S20,C H OgBr Ber. C 63,76 H 7,95 Br 12,86 1 OCH, 4,99 £
Gef. C 63,68 H 8,01 Br 12,80 1 OCH, 4,98 #
(«)D = +80° (c = 1,16)
A ' -2-Acetoxy-oleanen-23-3äuremethylester-28-säure
(Acetyl-gypsogeninsäure-monomethylester, LXXXVI). 1,46 g
13,28-Lacton des 2-Acetoxy-13-oxy-12-brom-oleanan-23,28-di-
säure-monomethylesters (LXXXV) löste man in 35 cm Eisessig
und erhitzte die Lösung nach Zugabe von 6 g Zinkstaub 5è
Stunden am Rückfluss. Darauf wurde vom unverbrauchten Zink
abfiltriert, die Lösung im Vakuum auf ein kleines Volumen
1) Vgl. Huzicka & Giacomello, Helv. 20, 299 (1937)

- 44 -
eingeengt und in üblicher Weise aufgearbeitet. Aus Methylen¬
chlorid-Methanol erhielt man Nadeln, die scharf bei 258-259°
schmolzen und mit Tetranitromethan eine gelbe Farbreaktion
gaben. Zur Analyse wurde die Substanz 4mal umkristallisiert
und 52 Stunden im Hochvakuum bei 90 getrocknet; Smp. 259 •
3,670 mg Subst. gaben 9,806 mg C02 und 2,996 mg H^O
5,065 mg Subst. verbrauchten bei der Methoxylbestimmung nach
Vieböck und Brecher 2,837 cnr 0,02-n. Na^SpO,
C33H5006 Ber. C 73,03 H 9,29 1 0CH3 5,72 £
Gef. C 72,92 H 9,13 1 OCH, 5,79 ft
(«)D = +72°; +74° (c = 0,89; 0,73)
Acetyl-gypsogeninsäure-dimethylester (LXXXVII) '.
500 mg des Mono-esters LXXXVI wurden in wenig Methylenchlo¬
rid gelöst und mit ätherischer Diazomethanlösung versetzt.
Das Rohprodukt wurde durch eine Säule von 15 g Aluminiumoxyd
(Akt.II) chromatographiert. Mit Petroläther-Benzol-Gemischen
und Benzol Hessen sich insgesamt 410 mg Substanz eluieren,
die aus Methylenchlorid-Methanol in Nadeln kristallisierte.
Der scharf bei 175° schmelzende Diester wurde zur Analyse
4mal umkristallisiert und 3^ Tage bei 60-80 im Hochvakuum
getrocknet.
3,984 mg Subst. gaben 10,717 mg C02 und 3,295 mg H20
C34H52°6 Ber' G 73'34 H 9'41 *
Gef. C 73,41 H 9,25 1>
TO T "}
& ' -2-Acetoxy-oleanen-23-3äuremethylester-28-säure-
chlorid (LüJÜLVIII). 410 mg des Disäure-monomethylesters
LXXXVI vom Schmelzpunkt 259° wurden in wenig absolutem Ben¬
zol gelöst, die Lösung mit 5 Tropfen Pyridin und 2,5 g Thio-
1) Vgl. Ruzicka, Giacomello & Grob, Helv. 21, 83 (1938)

- 45 -
nylchlorid versetzt und bei Zimmertemperatur 24 Stunden ste¬
hen gelassen. Darauf dampfte man die Lösung im Vakuum zur
Trockene ein und kristallisierte den Rückstand 4mal aus ab¬
solutem Aceton. Die erhaltenen Nadeln, die bei 237-239° (un¬
ter Zersetzung) schmolzen, wurden zur Analyse im Hochvakuum
48 Stunden bei 80-90° getrocknet.
3,755 mg Subst. gaben 9,684 mg CO« und 2,998 mg H„0
033H49°5C1 Ber' ° 70'62 H 8'80 *
Gef. C 70,38 H 8,90 $>
a~ ' -2-Acetoxy-28-oxo-oleanen-23-säuremethylester
(LXXXIX). 410 mg des Säurechlorids LXXXVIII wurden in 20 cm
absolutem Xylol gelöst und nach Hosenmund in Gegenwart von
200 mg eines 5-proz. Palladium-Bariumsulfat-Katalysators bei
140° reduziert. Nach 4 Stunden waren 70 # der berechneten
Menge Chlorwasserstoff abgespalten, worauf die Reduktion un¬
terbrochen wurde. Das nach üblicher Aufarbeitung gewonnene
Rohprodukt wurde durch eine Säule aus 12 g Aluminiumoxyd
(Akt.II) chromatographiert. Mit Petroläther-Benzol (9:1)-,
(4:1)- und (2:1)-Gemischen liessen sich insgesamt 210 mg
Substanz eluieren, die aus Methylenchlorid-Methanol in glän¬
zenden Blättchen vom Schmelzpunkt 261-262° kristallisierte.
Zur Analyse gelangte ein 3mal umkristallisiertes und im Hoch¬
vakuum 15 Stunden bei 150° getrocknetes Präparat vom
Schmelzpunkt 263-264°.
3,610 mg Subst. gaben 9,929 mg COp und 3,061 mg H„0
C33H50°5 Ber* C 75'24 H 9'57 *
Gef. C 75,06 H 9,49 %
(«) = +68° (c = 0,89)
3

- 46 -
Reduktion des Aldehyds LXXXIX nach Wolff-Kishner.
200 mg Substanz wurden mit 5 g Hydrazin und 5 cm absolutem
Alkohol unter Zusatz von wenig absolutem Toluol 45 Minuten
am Rückfluss gekocht. Hierauf wurde das Gemisch mit einer Lö¬
sung von 1 g Natrium in 10 cm absolutem Alkohol vereinigt
und im Einschlussrohr 12 Stunden auf 210° erhitzt. Das nach
der üblichen Aufarbeitung gewonnene Rohprodukt (180 mg) wur¬
de mit ätherischer Diazomethanlösung umgesetzt und dann
durch eine Säule aus 6 g Aluminiumoxyd (Akt.II) chromatogra-
phiert. Mit 300 cm Petroläther-Benzol (9sl) liessen sich
130 mg einer öligen Substanz eluieren, die mit Tetranitro-
methan eine stark gelbe Färbung gab, jedoch nicht zur Kri¬
stallisation gebracht werden konnte. Das Aether-Eluat (20 mg)
wurde 2mal aus Methanol umkristallisiert und die nadeiförmi¬
gen Kristalle zur Analyse im Hochvakuum bei 180° sublimiert.
Das Präparat schmolz bei 205-206°.
2,610 mg Subst. gaben 7,547 mg C02 und 2,596 mg H„0
C31H50°3 Ber* C 79'09 H 10'71 *
Gef. C 78,91 H 11,13 $>
12 1 ^Es liegt der A '
-2-Oxy-oleanen-23-säuremethylester
(XCII) vor.
a' -2-Acetoxy-oleanen-23-säuremethylester (XCI).
12 13Durch eine Lösung von 150 mg des â ' -2-Acetoxy-28-oxo-
oleanen-23-säuremethylesters (LXXXIX) in 2 cm absolutem
Methylenchlorid und 0,3 cm Aethylendithioglykol wurde unter
Eiskühlung während 75 Minuten ein trockener Chlorwasserstoff¬
strom geleitet. Die Lösung wurde dann in der Kälte im Vakuum
eingedampft, das so bereitete amorphe Aethylendithioketal
XC in 25 cm absolutem Dioxan gelöst, die Lösung mit Raney-
Nickel (hergestellt aus 3 g Legierung) versetzt und 6 Stun¬
den am Rückfluss erhitzt. Anschliessend wurde vom Raney-

OxydationsproduktdaswurdeAufarbeitungüblichenderNach
geschüttelt.gutOxydationsmischungbereitetenWasserom270
undSchwefelsäurekonz.g80Natriumdichromat,g60auseiner
cmJ2mitMinuten90ZimmertemperaturbeiundgelöstEisessig
cm2
undBenzolcm10inwardenXCIIOxy-estersdesmg1303
cm321
(XCIII),J12'13-2-Oxo-oleanen-23-säuremethylester
0,77)=(c+97°=(«)
1>10,57H78,68CGef.
*10'71H79'09CBer'C31H50°3
H^Omg3,326undC0„mg10,154gabenSubst.mg3,522
Schmelzpunktsdepression.keine
Oxy-methylestergewonnenenWolff-KishnernachLXXXIXAldehyds
desReduktiondurchdemmitgabund206-207°beischmolzEs
sublimiert.180beiHochvakuumimundumkristallisiertmisch
Ge¬gleichemaus4malwurdeAnalysenpräparatDasschmolzen.
203-204°beidieNadeln,feineMethylenchlorid-Methanolaus
manerhieltAufarbeitungüblichenderNachverseift.hitze
Siede¬beiStunden3Kalilaugemethanolischer5-proz.cm50
mitundgelöstBenzolweniginwurdenXCIAcetyl-methylester
mg320(XCII).J12>13-2-0xy-oleanen-23-säuremethylester
1,12)=(c+84°=(«)D
<jL10,52H77,03CGef.
*10'22H77'29CBer*C33H52°4
HpOmg3,354undC0„mg10,060gabenSubst.mg3,565
.246Smp.sublimiert;190°beikuum
Hochva¬imundumkristallisiert3malwurdeAnalysenpräparat
Das246°.SchmelzpunktkonstantenvomBlättchenglänzendenol
Methylenchlorid-Metha¬ausbildeteRohproduktkristallineDas
eingedampft.VakuumimFiltratdasundabfiltriertNickel
-47-

- 48 -
aus Methylenchlorid-Methanol umkristallisiert ; schöne Nadeln
vom Schmelzpunkt 183-183,5 • Zur Analyse gelangte ein 4mal
umkristallisiertes und im Hochvakuum bei 165-170° sublimier-
tes Präparat vom Schmelzpunkt 183,5-184 •
3,530 mg Subst. gaben 10,250 mg COp und 3,304 mg H-0
C31H4g03 Ber. C 79,4-3 H 10,30 +•
Gef. C 79,24 H 10,47 i>
(«)D = +98° (c = 1,11)
J12'13-01eanen-23-säuremethylester (XCV). 160 mg412»13-
2-Oio-oleanen-23-säuremethylester (XCIII) wurden in 5 cm abso¬
lutem Methylenchlorid und 0,5 cm Aethylendithioglykol gelöst
und durch die Lösung unter Eiskühlung während 1 Stunde ein
trockener Chlorwasserstoffström geleitet. Das Reaktionsge¬
misch wurde dann im Vakuum bei Zimmertemperatur eingedampft,
das kristalline, in reiner Form nicht isolierte Aethylendi-
thioketal XCIV in 45 cm absolutem Dioxan gelöst und die Lö¬
sung nach Zugabe von Raney-Nickel (hergestellt aus 15 g Le¬
gierung) 15 Stunden am Rückfluss gekocht. Nach der Aufarbei¬
tung löste man das Reduktionsprodukt in Petroläther und chro-
matographierte über eine Säule aus 5 g Aluminiumoxyd (Akt.I).
Mit 1,2 1 Petroläther und 600 cur Petroläther-Benzol (9:1)-
Gemisch liessen sich insgesamt 140 mg Substanz eluieren. Zur
Analyse wurden die bei 156-157 schmelzenden Petroläther-
Praktionen 4mal aus Methylenchlorid-Methanol umkristallisiert
und die erhaltenen feinen Nadeln im Hochvakuum bei 150° subli-
miert; Smp. 157-157,5°.
3,528 mg Subst. gaben 10,573 mg C02 und 3,523 mg HgO
C31H50°2 Ber" c 8l»88 H 11,08 #
Gef. C 81,78 H 11,18 #
(«)D = +96°; +97° (c = 1,109; 1,064)

- 49 -
J12»13-01eanen-23-säure (XCYI). 290 mg des J12'13-
Oleanen-23-säuremethylesters (XCV) wurden durch 16i-stündi-
ges Erhitzen mit 10 cm 15-proz. methanolischer Kalilauge
im Einschlussrohr auf 190-200° verseift. Das kristalline
Rohprodukt bildete aus Methylenchlorid-Methanol umgelöst
lange Blättchen vom Schmelzpunkt 282-285°• Zur Analyse ge¬
langte ein 5mal umkristallisiertes und im Hochvakuum 68 Stun¬
den bei 100-105° getrocknetes Präparat vom Schmelzpunkt 285-
288° (nach schwachem Sintern).
3,677 mg Subst. gaben 11,014 mg C02 und 3,597 mg HgO0 0H4802 Ber. C 81,76 H 10,98 <f>
Gef. C 81,77 H 10,95 1>
(«)D = +108° (c = 1,20)
J12»13-01eanen-23-säureamid (XCYII). Eine Lösung von
170 mg J1 ' 3-01eanen-23-saure (XCVT) in ca. 10 cm3 absolu¬
tem Benzol wurde mit einigen Tropfen Pyridin und 5 g Thio-
nylchlorid versetzt und 15 Stunden bei 20° stehen gelassen.
Hierauf wurde im Vakuum bei Zimmertemperatur zur Trockene
eingedampft, das kristalline, in reiner Form nicht isolierte,
£r ' -Oleanen-23-säurechlorid in einem Gemisch gleicher Vo-
lumenteile Benzol und Methylenchlorid gelöst und durch die
Lösung bei 20° ein trockener Ammoniakstrom geleitet. Das
Reaktionsgemisch wurde dann mit Aether verdünnt und die äthe¬
rische Lösung mit Wasser gewaschen. Aus Methylenchlorid-
Petroläther kristallisierten dünne, verfilzte Nadeln, die für
die Analyse 4mal umkristallisiert und anschliessend im Hoch¬
vakuum bei 200° sublimiert wurden. Das Präparat schmolz bei
269-270°.

- 50 -
3,510 mg Subst. gaben 10,498 mg CO« und 3,481 mg HpO
3,253 mg Subst. gaben 0,096 cm3 N2 (23°/723 mm)
G30H49ON Ber% G 81»94 H i:l»23 » 3,19 °h
Gef. C 81,62 H 11,10 N 3,24 <f>
>D(«)_ = +91° (c = 1,40)
B. Ueberführung der Acetyl-oleanolsäure in /ä-Amyrin-acetat
Acetyl-oleanolsäure-chlorid. 2 g Acetyl-oleanolsäure
vom Schmelzpunkt 265-266 wurden in absolutem Benzol gelöst,
mit 6 Tropfen Pyridin und 5 g Thionylchlorid versetzt und die
Lösung 21 Stunden bei 20 stehen gelassen. Das Reaktionsge¬
misch wurde im Vakuum eingedampft und der kristalline Rück¬
stand 2mal aus absolutem Aceton umkristallisiert. Die erhal¬
tenen Nadeln schmolzen bei 231-232° (unter Zersetzung).
Acetyl-oleanol-aldehyd. 300 mg Acetyl-oleanolsäure-
chlorid wurden in 20 cm absolutem Xylol gelöst und nach
Rosenmund in Gegenwart von 300 mg eines 5-proz. Palladium-
Bariumsulfat-Katalysators und 5 mg Chinolin-Schwefel bei
140 hydriert. Nach 4 Stunden waren 56 f> des berechneten
Chlorwasserstoffes abgespalten, üs wurde vom Katalysator
abfiltriert und die Lösung eingedampft. Das kristalline Roh¬
produkt wurde durch eine Säule von 10 g Aluminiumoxyd (Akt.II)
chromatographiert. Mit Petroläther-Benzol-Gemischen wurden
insgesamt 110 mg Substanz eluiert, die nach einmaligem Um¬
kristallisieren aus Methylenchlorid-Methanol bei 251°
schmolz. Der Aldehyd bildete glänzende Blättchen.
|ft-Amyrin-acetat. 100 mg Acetyl-oleanol-aldehyd wurden
in 1 cm absolutem Methylenchlorid gelöst, mit 0,2 cm Aethy-

- 51 -
lendithibl versetzt und durch die Lösung 1 Stunde unter Eis¬
kühlung trockenes Chlorwasserstoffgas geleitet. Hierauf wur¬
de im Vakuum zur Trockene eingedampft, das ölige Produkt in
20 cm absolutem Dioxan gelöst und die Lösung mit Raney-Nickel
(hergestellt aus 3 g Legierung) 6 Stunden am Rückfluss ge¬
kocht. Das nach üblicher Aufarbeitung erhaltene Rohprodukt
war kristallin und bildete aus Methylenchlorid-Methanol schö¬
ne Nadeln vom Schmelzpunkt 241-242°, die, mit /3-Amyrin-acetat
vom gleichen Schmelzpunkt gemischt, keine Depression gaben.
Zur Analyse wurde die Substanz 2mal umkristallisiert und an¬
schliessend im Hochvakuum bei 210 sublimiert. Smp. 242 .
3,500 mg Subst. gaben 10,507 mg C02 und 3,495 mg H20
C32H52°2 Ber# C 81'99 H 11,18 #
Gef. C 81,92 H 11,16 f>
(«)D = +86° (c = 0,68)
C. Umsetzungen in der «-Boswellinsäure-Reihe
u~ ' -2-0xo-oleanen-24-säuremethylester («-Boswellen-
onsäure-methylester, CI) '. 500 mg «-Boswellinsäure-methyl-
ester (XCIX) vom Schmelzpunkt 215-216° wurden in 15 cm Ben¬
zol und 2 cm Eisessig gelöst und mit 5 om einer aus 60 g
Natriumdichromat, 80 g konz. Schwefelsäure und 270 cm Wasser
hergestellten Oxydationsmischung 1^ Stunden bei Zimmertempe¬
ratur geschüttelt. Das nach der üblichen Aufarbeitung erhal¬
tene kristalline Produkt schmolz nach einmaligem Umlösen aus
Methylenchloria-Methanol bei 208-209°. Das Analysenpräparat
wurde noch 2mal umkristallisiert und im Hochvakuum bei 180
sublimiert; Smp. 210-211°.
1) Wirz, Diss. ETH, Zürich 1942; vgl. auch Helv. 33, 686 (1950)

- 52 -
3,564 mg Subst. gaben 10,351 mg COp und 3,267 mg HpO
C31H48°3 Ber* C 79'43 H 10'30 f°
Gef. C 79,25 H 10,26 f>
(«)D = +115° (c = 0,73)
#2'13-01eanen-24-säuremethylester (CHI). 400 mg
jl2,13_2_0xo_oleanen_24_Säuremethylester (CI) vom Schmelz¬
punkt 209-210° wurden in 3 cm absolutem Methylenchlorid
und 0,4 cm Aethylendithioglykol gelöst und durch die mit
Eis gekühlte Lösung 75 Minuten ein trockener Chlorwasser¬
stoffström geleitet. Nach der üblichen Aufarbeitung erhielt
man in quantitativer Ausbeute das Aethylendithioketal CII,
welches bei 232-233° schmolz. Dieses wurde in 20 cm abso¬
lutem Dioxan gelöst und mit Raney-Nickel, hergestellt aus
7 g Legierung, 5i Stunden zum Sieden erhitzt. Nach Filtra¬
tion und Eindampfen der Lösung wurde das kristalline Reduk¬
tionsprodukt (330 mg) durch eine Säule von 12 g Aluminium¬
oxyd (Akt.II) chromatographies. Mit 1,2 1 Petroläther lies-
sen sich 300 mg Substanz eluieren, die aus Methylenchlorid-
Methanol umkristallisiert glänzende Blättchen vom Schmelz¬
punkt 178-179 bildete. Das Analysenpräparat wurde 4mal um¬
kristallisiert und anschliessend im Hochvakuum bei 150°
sublimiert. Smp. 179-180°.
3,658 mg Subst. gaben 10,957 mg CO- und 3,604 mg HpO
C31H50°2 äer' C 8l'88 H 11,08 #
Gef. C 81,75 H 11,02 £
(«)D = +138°; +135° (c = 0,854; 0,919)
^2,3;12,13-01eadien-24-säuremethylester (CVI). 540 mg
Benzoyl-a-boswellinsäure-methylester'(C) vom Schmelzpunkt
140 wurden in 3 Ansätzen im offenen Glasrohr 1 Stunde auf

- 53 -
330-340 erhitzt, wobei sich im oberen Teil des Rohres Ben¬
zoesäure in langen Nadeln absetzte. Nach dem Erkalten wurde
das braun gefärbte Produkt 2 Stunden mit 5-proz. methanoli¬
scher Kalilauge gekocht und das Reaktionsgemisch dann wie
gewohnt aufgearbeitet. Man erhielt 320 mg stark gefärbte
Substanz, die durch eine Säule aus 10 g Aluminiumoxyd (Akt.I-
II) chromatographiert wurde. Mit Petroläther Hessen sich
170 mg einer kristallinen Verbindung eluieren, die nach ein¬
maligem Umkristallisieren aus Chloroform-Methanol bei 202
schmolz. Das Analysenpräparat wurde 2mal umkristallisiert
,und im Hochvakuum bei 180 sublimiert. Smp. 204 •
3,571 mg Subst. gaben 10,771 mg COp und 3,432 mg ÏÏL0
C31H48°2 Ber* C 82'24 H 10»69 #
Gef. C 82,31 H 10,75 1>
Hydrierung des d2'-^12'1 -01eadien-24-säuremethyl-
esters (CVI). 60 mg Substanz vom Schmelzpunkt 204 wurden
in 25 oi Eisessig gelöst und mit 100 mg vorhydriertem Pla¬
tinoxyd-Katalysator 15 Stunden in einer Wasserstoffatmo¬
sphäre geschüttelt. Das nach der üblichen Aufarbeitung er¬
haltene kristalline Reduktionsprodukt schmolz nach einmali¬
gem Umlösen aus Chloroform-Methanol bei 177-178 . Zur Ana¬
lyse gelangte ein 2mal umkristallisiertes und im Hochvakuum
bei 160 sublimiertes Präparat vom Schmelzpunkt 180 . Es gab
mit der, durch reduktive Entschwefelung des Aethylendithio-
ketals CII gewonnenen gleich schmelzenden Verbindung keine
Erniedrigung des Schmelzpunktes. Die IR.-Spektren der beiden
Präparate des Û ' -01eanen-24-säuremethylesters waren iden¬
tisch.
3,588 mg Subst. gaben 10,750 mg C02 und 3,573 mg H20
C31H50°2 Ber* C 8l»88 H l:L.08 #
Gef. C 81,76 H 11,14 #

- 54 -
Es liegt der ZT" ' -Oleanen-24-säuremethylester (CHI)
vor.
AL2,13-Qleanen-24-säure (civ). 500 mg des J12'13-01ea-
nen-24-säuremethylesters (CHI) wurden durch 21-stündiges
Erhitzen mit 20 cm 15-proz. methanolischer Kalilauge im
Einschlussrohr auf 180-190° verseift. Die Säure kristalli¬
sierte aus Methylenchlorid-Methanol in flachen Prismen, die
scharf bei 296-297 schmolzen. Zur Analyse gelangte ein 5mal
umkristallisiertes Präparat, welches 3 Tage im Hochvakuum bei
90-100° getrocknet wurde. Smp. 298-299°.
3,660 mg Subst. gaben 10,956 mg COp und 3,573 mg H_0
C30H48°2 Ber' C 8l>76 H 10»98 f°
Gef. C 81,70 H 10,92 #
(«)D = +144° (c = 1,08)
d> ' -01eanen-24-säureamid (CV). Das Säureamid CV wur¬
de in gleicher Weise bereitet wie das isomere /r ' -Oleanen-
23-säureamid (XCVII). Es kristallisierte aus Methylenchlorid-
Petroläther in dünnen, verfilzten Nadeln. Zur Analyse gelang¬
te ein 3mal umkristallisiertes und im Hochvakuum bei 200°
sublimiertes Präparat vom Schmelzpunkt 264-265°.
3,648 mg Subst. gaben 10,952 mg C02 und 3,660 mg HpO3,768 mg Subst. gaben 0,108 cm3 N2 (20°/727 mm)
C30H ON Ber. C 81,94 H 11,23 N 3,19 #
Gef. C 81,93 H 11,22 N 3,20 <$>
(«) = +140° (c = 1,23)

- 55 -
D. Ueberführung der a'
-Oleanen-24-säure (CIV) in
J12,13-01eanen (CVII)
610 mg der oben 'beschriebenen u' -Oleanen-24-säure
wurden in absolutem Benzol gelöst, die Lösung mit 5 Tropfen
Pyridin und 5 g Thionylchlorid versetzt und 15 Stunden bei
Zimmertemperatur stehen gelassen. Hierauf wurde das Reak¬
tionsgemisch im Vakuum zur Trockene eingedampft und das
/f ' -01eanen-24-säurechlorid aus Aceton umkristallisiert.
Man erhielt 400 mg Substanz vom Schmelzpunkt 170-173°•
Diese wurden in absolutem Xylol gelöst und in bekannter
Weise in Anwesenheit eines Palladium-Bariumsulfat-Katalysa¬
tors nach Rosenmund bei 140 reduziert. Das Reduktionspro¬
dukt wurde durch eine Säule aus Aluminiumoxyd (Akt.I) sorg¬
fältig chromâtographiert. Die dabei gewonnenen Petroläther-
Benzol- und Benzol-Eluate (insgesamt 130 mg) wurden in 3 cm
absolutem Methylenchlorid gelöst und durch die Lösung, nach
Zugabe von 0,3 cm Aethylendithioglykol, 50 Minuten unter
Eiskühlung trockenes Chlorwasserstoffgas geleitet. Das Reak¬
tionsgemisch wurde im Vakuum in der Kälte vom Lösungsmittel
befreit, in Methanol aufgenommen und zur Trockene einge¬
dampft. Das teilweise kristalline Produkt wurde in 25 cm
absolutem Dioxan gelöst und mit Raney-Nickel (hergestellt
aus 4 g Legierung) Ai Stunden am Rückfluss gekocht. Nach der
Aufarbeitung wurde das Reduktionsprodukt (110 mg) durch eine
Säule von 6 g Aluminiumoxyd (Akt.I) chromâtographiert. Mit
50 cm Pentan liessen sich 70 mg kristalline Substanz vom
Schmelzpunkt 149-151° eluieren. Diese wurde ein zweites Mal
an 6 g Aluminiumoxyd (Akt.I) chromatographiert. Schon mit
35 cm Pentan kam die gesamte Menge des Kohlenwasserstoffes
(70 mg), der jetzt bei 150-153° schmolz. Dieser kristalli¬
sierte aus Methylenchlorid-Methanol in gut ausgebildeten
Nadeln. Zur Analyse wurde 5mal umkristallisiert und an-

- 56 -
schliessend im Hochvakuum bei 130° sublimiert. Das Präparat
schmolz bei 159° und gab mit dem u~ ' -Oleanen (CVII) ,
das durch Reduktion des â1 ' -2-Oxo-oleanens nach ffolff-
Kishner erhalten worden war, keine Erniedrigung des Schmelz¬
punktes.
3,310 mg Subst. gaben 10,652 mg C02 und 3,637 mg H.,0
C30H50 Ber. C 87,73 H 12,27 $
Gef. C 87,82 H 12,30 <f>
(«)D = +99° (c = 0,75)
ZUSAMMENFASSUNG
Die pentacyclischen Triterpene Gypsogenin (LXXX) und
oc-Boswellinsäure (XCVIII) wurden in das Epimerenpaar à -
Oleanen-23-säure (XCVI) und 2l12,13-01eanen-24-säure (CIV)
übergeführt. Die aus Gypsogenin bereitete Verbindung XCVI so¬
wie ihre Derivate XCV und XCVII weisen eine um ca. 40° gerin¬
gere positive Drehung auf als die entsprechenden, aus oc-Bos¬
wellinsäure gewonnenen Isomeren CIV, CHI und CV.
Anhand dieser optischen Drehungen lässt sich provisorisch
folgern, dass die Aldehydgruppe des Gypsogenins (C-0 in trans-
und die Carboxylgruppe der «-Boswellinsäure (02A) in cis-
Stellung zum Methyl am C- liegt.
1) Winterstein & Stein, A. 502, 223 (1934); Ruzicka, Schellen¬
berg & Goldberg, Helv. 20, 791 (1937)2) Vesterberg, B. 2±, 3836~Tl891); Ruzicka & Wirz, Helv. 24,
24Ö (1941)

- 57 -
II. T E I L. ZUR KONSTITUTION DES RINGES C DES «-AMYRINS
THEORETISCHER TEII****************************»****#*
A. EINLEITUNG
Bevor auf die Konstitution des Ringes C des ce-Amyrins
eingegangen wird, sollen die charakteristischen Reaktionen,
die an der Doppelbindung durchgeführt worden sind, bespro¬
chen werden.
Die Doppelbindung des «-Amyrins ist ausserordentlich
reaktionsträge. Sie reagiert nicht mit Persäuren,lässt
sich nicht katalytisch hydrieren und lagert auch kein Brom
2)an . Bei der Oxydation des a-Amyrin-benzoats (CVIIIc) mit
Wasserstoffperoxyd oder Ozon entsteht das «-Amyrin-benzoat-
oxyd C,„H,-.0, (CIXc) ', welches sich leicht mit Salzsäu¬
re zum oc-Amyranonol-benzoat (CXc) isomerisieren lässt . Die
Oxydo-gruppe der Verbindung CIXc ist auch gegen Alkalien
nicht sehr stabil, doch gelingt es, durch milde alkalische
Verseifung und nachfolgende Acetylierung at-Amyrin-acetat-
oxyd C-^Hj-pCU (ClXb) zu gewinnen ', welches auch durch Oxy¬
dation des cv-Amyrin-acetats (CVIIIb) mit Ozon oder Wasser-
stoffperoxyd gebildet wird'
. Das Acetat ClXb lässt sich
analog dem Benzoat CIXc mit Salzsäure leicht zum «-Amyranon-
ol-acetat (CXb) umlagern'
,dessen UV.-Spektrum ein Maxi¬
mum bei 282 m/t, log 6 = 2,35, aufweist. Die Ketogruppe in den
1) Ruzicka, Silbermann & Furter, Helv. 15, 482 (1932)2) Vesterberg, B. 23, 3189 (1890)3) Seymour, Sharpies & Spring, Soc. 1939, 1075
4) Mclean, Silverstone & Spring, Soc. 1951, 935
5) Ruzicka, Jeger, Redel & Volli, HelvTjËE, 199 (1945)

- 58 -
Verbindungen CXb und CXc ist sehr reaktionsträge; sie lässt
sich weder mit Ketonreagenzien umsetzen, noch nach Wolff-
Kishner oder Clemmensen reduzieren.
Sowohl oc-Amyranonol-benzoat (CXc) als auch «-Amyrin-ben-
zoat-oxyd (CIXc) geben bei der Reduktion mit Natrium und Amyl¬
alkohol unter Verseifung der Benzoylestergruppe «-Amyrandiol1) 2)
C^0H52°2 (CXIa) und bei der Umsetzung mit Brom in Eis¬
essig das Bromid CXIIc,, das durch Bromwasserstoffabspaltung
in das iso-a-Amyrenonol-benzoat C,„HcoO, (CXIIIc) übergeht1) 2) ~i) il 5 i
' ' '. Die «,a-ungesättigte Ketogruppe der Verbindungen
CXIIIb und CXIIIc lässt sich mit Platin und Wasserstoff nicht
hydrieren. Mit Natrium und Amylalkohol wird dagegen die Car-
bonylgruppe zur sekundären Hydroxylgruppe reduziert; die ent¬
stehende Verbindung geht aber sofort unter Wasserabspaltung
in das «-Amyradienol C,0H.gO (CXIVa) über,das 2 konju¬
gierte Doppelbindungen in einem Hing enthält (Maximum im UV.-
Spektrum bei 280 m.u, log e = 4,05). Das Acetat des a-Amyra-
dienols C,2H^o02 (CXIVb) entsteht bei der partiellen Dehydrie¬
rung des «-Amyrin-acetats (CVIIIb) mit Schwefel oder N-Brom-5)
succinimid '.
Durch Oxydation des «-Amyrin-acetats mit Chromsäure wird
in «-Stellung zur Doppelbindung eine Ketogruppe eingeführt ';
es entsteht das «-Amyrenonol-acetat C-j2H-00, (CXVb), dessen
UV.-Spektrum ein Maximum bei 250 ny/, log e = 4,1, besitzt.
Das Carbonyl dieser Verbindung lässt sich mit Platin und Was-
7)serstoff wieder wegreduzieren . Mit Natrium und Amylalkohol
bildet sich wiederum «-Amyradienol (CXIVa) '.
1) Seymour, Sharpies & Spring, Soc. 1939, 1075
2) McLean, Silverstone & Spring, Soc. 1951, 935
3) Seymour & Spring, Soc. 1941, 319
4) Ewen & Spring, Soc. 1940, 1196
5) Jacobs & Fleck, J.Biol.Chem. 88, 137 (1930); Ruzicka,Jeger & Redel, Helv. 26, 1235~Tl943)
6) Vesterberg, B. 24, 3836" (1891); Spring & Vickerstaff,Soc. 1937, 249
7) Ruzicka, Leuenberger & Schellenberg, Helv. 20, 1271 (1937)8) Ruzicka, Müller & Schellenberg, Helv. £2, 7ÏÏÏÏ (1939); Ewen,
Spring & Vickerstaff, Soc. 1939, 1303? Ruzicka, Volli &
JegerT Helv. 28, 1628 (1945T^

- 59 -
Die Oxydation des «-Amyradienol-acetats (CXIVb) mit Per-
benzoesäure ergibt ein Gemisch, aus dem sich die beiden Iso¬
meren or-Amyrenonol-acetat (CXVb) und iso-a-Amyrenonol-acetat
(CXIIIb) isolieren lassen . Wird hingegen das Acetat GXIVb
mit Wasserstoffperoxyd oder auch mit Ozon bei 0 oxydiert, so
erhält man ein Gemisch, aus dem sich ot-Amyrenonol-acetat (CXVb)
und ein isomeres <y,/3-ungesättigtes Keton, das iso-a-Amyrenonol-1)2)
II-acetat (CXVIb) abtrennen lassen '. Die Oxydation des
Benzoats CXIVc mit Wasserstoffperoxyd ergibt ein analoges Ge¬
misch. Das iso-of-Amyrenonol-II-acetat (CXVIb) zeigt im UV.-
Spektrum ein Maximum bei 250 m/t, log 6 = 4,04. Es lässt sich
durch längere Behandlung mit starkem Alkali quantitativ in
iso-a-Amyrenonol (CXIIIa) überführen ' und unterscheidet
sich von diesem wahrscheinlich durch die räumliche Stellung
des Wasserstoffatoms am C,,.
Mit Natrium und Amylalkohol geht1) 2)
es wie dieses in «-Amyradienol (CXIVa) über'
'. Im Gegen¬
satz zur Verbindung CXIIIb lässt sich das iso-c*-Amyrenonol-II-
acetat (CXVIb) mit Platin und Wasserstoff zum gesättigten Ke¬
ton w-Amyranonol-II-acetat C,2HS20, (CXVIIb) hydrieren .
Bei der Oxydation des «-Amyranonol-acetats (CXb) mit
Chromsäure in siedendem Eisessig wird ein «-Diketon C,oH,-_0.
,.
B 32 50 4
(CXVIIIb) erhalten,das sich auch bei der Ozonisation des
«-Amyradienol-acetats (CXIVb) bei 22° bildet '.
1) McLean, Ruff & Spring, Soc. 1951, 1093
2) Ewen & Spring, Soc. 1940, 1196
3) Ruzicka, Jeger, Redel & Volli, Helv. _28, 199 (1945)
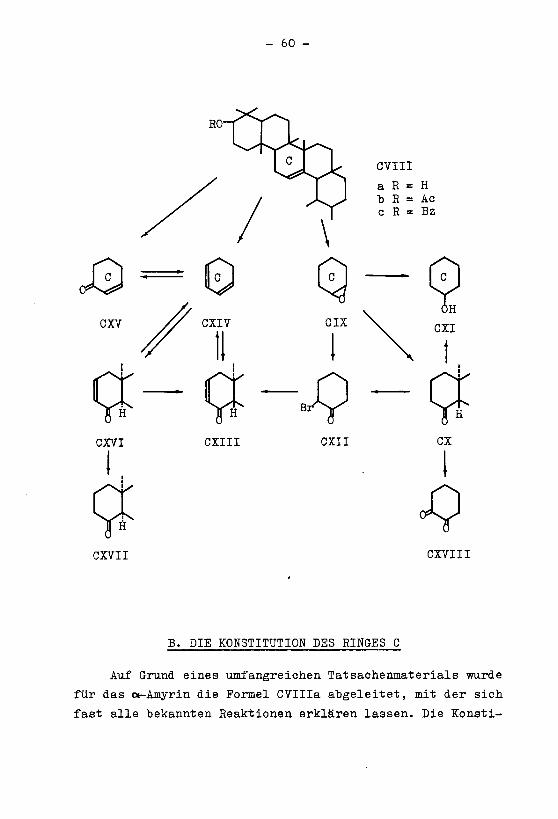
- 60 -
CVIII
a R = H
b R = Ac
c R = Bz
CXVIII
B. DIE KONSTITUTION DES RINGES C
Auf Grund eines umfangreichen Tatsachenmaterials wurde
für das cx-Amyrin die Formel CVIIIa abgeleitet, mit der sich
fast alle bekannten Reaktionen erklären lassen. Die Konsti-

- 61 -
tution einer Naturverbindung kann aber erst dann als voll¬
ständig aufgeklärt betrachtet werden, wenn die läge jedes
einzelnen Atoms bewiesen ist. Hierbei kommt der Synthese der
Verbindung oder von Spaltstücken der Verbindung eine ausschlag¬
gebende Bedeutung zu. Hauptsächlich zwei Arbeitsmethoden, die
Dehydrierung und die thermische Spaltung geeigneter Abbau¬
produkte, wurden angewendet, um aus den Triterpenen leich¬
ter synthetisierbare Verbindungen herzustellen. Beide Metho¬
den erfordern hohe Temperaturen (ca. 300-350 ), bei denen Um-
lagerungen des Kohlenstoffgerüstes nicht ausgeschlossen sind.
Was nun den Ring C des a-Amyrins betrifft, so wurde aus
den Ergebnissen der Dehydrierung und anderen bekannten Reak¬
tionen geschlossen, dass er ögliedrig ist und an den C-Ato-
men 9 und 14 je eine Methylgruppe trägt. Da die Dehydrierun¬
gen aber bei Temperaturen von ca. 350 durchgeführt werden,
darf die Möglichkeit nicht ausser Betracht gelassen werden,
dass dabei eine Ringverengerung stattfinden könnte, wobei
sich gleichzeitig eine oder zwei Methylgruppen bilden wür¬
den.
Pur die Konstitutionsaufklärung des ot-Amyrins besonders
wichtig war die pyrolytische Spaltung des aus dem ot-Amyrin
zugänglichen Keto-dimethylesters CXIX, die zu 2 Paaren von
bicyclischen Spaltprodukten (XXXVII, XXXVIII, CXX und CXXI)
führte, deren Konstitution eindeutig bestimmt werden konnte .
Es wäre auch in diesem Falle denkbar, dass sich von den beiden
in der Verbindung CXIX an den C-Atomen 9 und 14 angenommenen
Methylen eines oder beide erst bei der Pyrolyse aus einer,
die beiden bicyclischen Molekelhälften verbindenden Kohlen¬
stoffkette bilden würden.
Es soll nun eine Reaktionsfolge besprochen werden, die
sich nur mit dem Vorhandensein eines Methyls am C-Atom 14
des Triterpengerüstes erklären lässt. Bei der Oxydation des
1) Meiseis, Jeger & Ruzicka, Helv. 32, 1075 (1949)
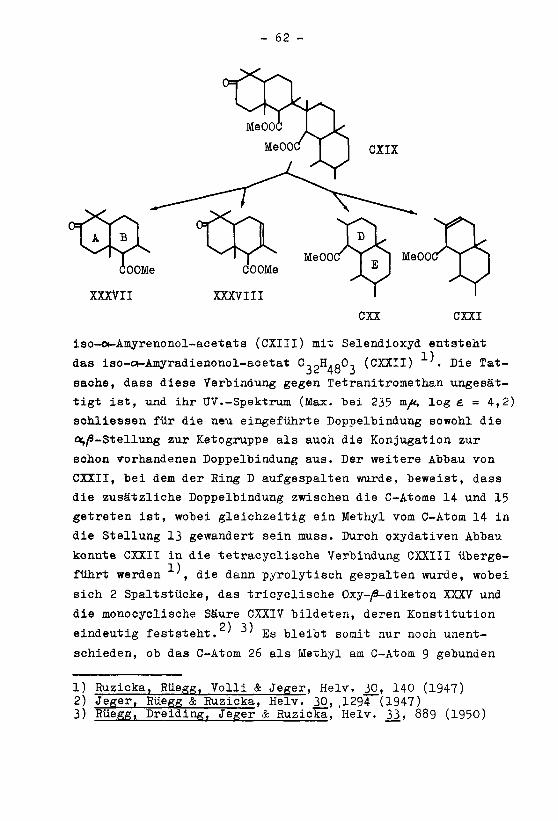
- 62 -
XXXVII
CXX CXXI
iso-cx-Amyrenonol-acetats (CXIII) mit Selendioxyd entsteht
das iso-a-Amyradienonol-acetat c:j2H4.803 (CXXI1) • Die Tat¬
sache, dass diese Verbindung gegen Tetranitromethan ungesät¬
tigt ist, und ihr ÜV.-Spektrum (Max. bei 235 ny<, log £ = 4,2)
schliessen für die neu eingeführte Doppelbindung sowohl die
Q^/9-Stellung zur Ketogruppe als auch die Konjugation zur
schon vorhandenen Doppelbindung aus. Der weitere Abbau von
CXXII, bei dem der Ring D aufgespalten wurde, beweist, dass
die zusätzliche Doppelbindung zwischen die C-Atome 14 und 15
getreten ist, wobei gleichzeitig ein Methyl vom C-Atom 14 in
die Stellung 13 gewandert sein muss. Durch oxydativen Abbau
konnte CXXII in die tetracyclische Verbindung CXXIII überge¬
führt werden,die dann pyrolytisch gespalten wurde, wobei
sich 2 Spaltstücke, das tricyclische 0xy-/3-diketon XXXV und
die monocyclische Säure CXXIV bildeten, deren Konstitution
2) 3)
eindeutig feststeht. ' ' Es bleibt somit nur noch unent¬
schieden, ob das C-Atom 26 als Methyl am C-Atom 9 gebunden
1) Ruzicka, Rüegg, Volli & Jeger, Helv. 3£, 140 (1947)2) Jeger, Rüegg & Ruzicka, Helv. ^30, ,1294 (1947)3) Rüegg, Dreiding, Jeger & Ruzicka, Helv. j^3, 889 (1950)

- 63 -
ist oder aber als Glied des Ringes C vorliegt (im letzteren
Falle wäre der Ring C 7gliedrig). Es ist nämlich nicht voll¬
kommen unmöglich, dass bei der pyrolytischen Herstellung der
Verbindung XXXV bei 290° eine Ringverengerung stattfindet
unter Bildung eines Methyls an der Ringverknüpfungsstelle der
Ringe 3/C.
Das Ergebnis einer neueren, physikalischen Untersuchung
des Problems spricht für eine 6-Gliedrigkeit des Ringes C '.
Es konnte nämlich gezeigt werden, dass die Lage der Carbo-
nylbande im IR.-Spektrum des oi-Amyranonol-acetats (CXb) und
des aus diesem hergestellten C-Nor-ketons einem 6-Ring- bzw.
einem 5-Ring-Keton entspricht.
Das iso-o-Amyradienonol-acetat (CXXII) wird bei der Be¬
handlung mit Chlorwasserstoff in Eisessig sehr leicht isome-
risiert, wobei eine Verbindung C,pH.gO, erhalten wird, die
mit Tetranitromethan keine Färbung mehr gibt und deren UV.-
2}Spektrum gegenüber CXXII unverändert ist '. Es kann dieser
Verbindung die Formel CXXV zukommen. Bei ihrer Bildung müss-
te die Methylgruppe, die sich in CXXII in Stellung 13 befin¬
det, an das C-Atom 14 zurückwandern.
Bei der katalytischen Hydrierung des iso-ce-Amyradienon-
ol-acetats (CXXII) wurde u.a. auch das iso-«-Amyrenonol-ace-
tat (CXIII) und das c^Amyradienol-acetat (CXIVb) erhalten '
. Diese Tatsache wurde zunächst als Beweis betrachtet, dass
CXXII noch das unveränderte Kohlenstoffgerüst des «-Amyrins
besitzt. Dies steht aber im Widerspruch zum oben geschilder¬
ten Abbau von CXXII, bei dem der Ring D geöffnet wurde.
Es war daher wünschenswert, das iso-ot-Amyradienonol-
acetat nochmals näher zu untersuchen und insbesondere die
Hydrierung dieser Verbindung mit vollkommen reinem Material
1) Meyer, Jeger, Prelog & Ruzicka, Helv. ^34, 74-7 (1951)2) Ruzicka, Rüegg, Volli & Jeger, Helv. .30, 140 (1947)3) Volli, Diss. ETH, Zürich I948
- 64 -
zu wiederholen, weil es nicht unmöglich schien, dass das
früher beobachtete Auftreten von CXIII und CXIV als Hydrie¬
rungsprodukte von CXXII auf Verunreinigungen der zur Hydrie¬
rung gelangenden Substanz zurückzuführen ist.
OOH
CXXV
XXXVCXXIV
C. EIGENE ARBEITEN
Da sich aus dem Ergebnis dieser Arbeit noch keine ein¬
deutigen Schlüsse in Bezug auf die Konstitution der herge¬
stellten Verbindungen ziehen lassen, sollen die UV.- und IR.-
1) 2}
Spektren' ' dieser Verbindungen eingehend besprochen wer¬
den.
1) Siehe Pussnote 2) Seite 36.
2) Die Aufnahme und Diskussion der IR.-Absorptionsspektrenverdanke ich Hrn. Dr. Hs.H. Günthard. Die Spektren wur¬
den, wenn nichts anderes angegeben, in Nujol-Paste auf¬
genommen.

- 65 -
Ausgehend vom «-Amyrin-benzoat (CVIIIc) wurde zunächst
über die Zwischenstufen des oKAmyrin-benzoat-oxyds (CIXc)
und des iso-oeAmyrenonol-benzoats (CXIIIc) das iso-ot-Amyren-
onol-acetat C,2H500, (CXIIIb)1^ '
hergestellt. Das UV.-
Spektrum dieser Verbindung (Max. bei 248 m/<, log e = 4,02)
beweist, dass ein «.^-ungesättigtes Keton vorliegt. Daher
muss für die 3 Banden, die in der 1600 cm -Region des IR.-
Spektrums auftreten, folgende Zuordnung als sehr wahrschein¬
lich gelten: 1733 cm" tf(CO) der Acetoxygruppe; 1655 cm-1
^)(C0) und 1605 cm-1 v(CC) der Gruppierung >C=CH-C=0 .
Die v(C0)- und v(CC)-Frequenzen der er,/9-ungesättigten
Ketogruppe sind abnormal tief (normal wären 1680 bzw. 1620
cm ).
Die Oxydation von CXIIIb mit Selendioxyd in siedendem
Eisessig'lieferte 2 Produkte: (a) das iso-a-Amyradienon-
ol-acetat C32H48°3 (CXXI:0> das mit Tetranitromethan eine
gelbe Farbreaktion gab, und (b) als Nebenprodukt eine iso¬
mere Verbindung, die nach Schmelzpunkt, Mischprobe und IR.-
Spektrum mit dem durch Isomerisierung von CXXII erhaltenen
Dienon CXXV identisch war.
Mit Hilfe des UV.-Spektrums des iso-o-Amyradienonol-
acetats (CXXII), nach dem ein o,/9-ungesättigtes Keton vor¬
liegt (Max. bei 236 m^<, log e = 4,00), lässt sich auch das
IR.-Spektrum einigermassen sicher deuten. Den 4 Banden der
Doppelbindungsregion kann man die folgende, mit der ange¬
nommenen Strukturformel in Uebereinstimmung stehende Deu¬
tung geben: 1733 cnT1 (1248 cm-1) y(C0) Acetoxybande; 1675
cm" v(C0) und 1618 cm~ v(CC) der Gruppierung >C=CH-C=0 ;
1645 cm- v(CC) der 3fach substituierten Doppelbindung im
Ringe D.
1) Seymour, Sharpies & Spring, Soc. 1939, 1075
2) Mclean, Silverstone & Spring, Soc. 1951, 935
3) Ruzicka, Rüegg, Volli & Jeger, Helv. 30, 140 (1947);
vgl. auch Rüegg, Diss. ETH, Zürich 1948"

- 66 -
Die durch Umlagerung des iso-a-Àmyradienonol-acetats
mit Chlorwasserstoff in Eisessig'
gewonnene Verbindung
C,?H.qO-. (CXXV) war gegen Tetranitromethan gesättigt. Dem
UV.-Spektrum nach (Max. bei 236 m/t, log e = 4,03) steht das
Vorhandensein einer a,ß-ungesättigten Ketogruppe fest. Im
IR.-Spektrum treten in der 1600-1700 cm -Region 3 Banden
bei 1730 bzw. 1664 und.1615 cm- auf, die wahrscheinlich
der Acetoxygruppe bzw. der Gruppierung ^C=CH-C0-C=CC^ zukom¬
men.
Die Oxydation von GXXII mit Selendioxyd in Dioxan bei
210° lieferte, analog der unter den gleichen Bedingungen
durchgeführten Oxydation des entsprechenden /*-Amyrin-Deri-
vates,ein Dien-dion 0\?H.fi0. (CXXVI), das mit Tetrani¬
tromethan keine Färbung mehr gab. Das IR.-Spektrum dieser
Verbindung lässt sich nur unter gewissen Voraussetzungen mit
der angenommenen Formel vereinbaren. Es weist in der 1600
cm- -Region (im Spektrum der zu 5 # in Schwefelkohlenstoff
gelösten Substanz) 4 Banden auf bei 1733, 1686,1658 und 1613
cm- sowie eine sehr schwache Schulter bei 1623 om" . Von
diesen dürfte die 1733 cm" -Bande der Acetoxygruppe zuzuord¬
nen sein (siehe auch die charakteristische Absorption bei
1250 cm- ); bei den beiden intensiven Banden bei 1686 und
1658 cm- dürfte es sich um die v(C0)-Frequenzen der beiden
o^-ungesättigten Ketogruppen handeln. Die letztere der bei¬
den liegt ungewöhnlich tief. Die Schulter bei 1623 cm- und
die 1613 cm" -Bande dürften die ->;(CC)-Frequenzen der beiden
ay3-ungesättigten Ketogruppen darstellen.
Das IR.-Spektrum der festen Substanz zeigt teilweise
etwas andere Frequenzen für die Absorptionsbanden in der
1600-1700 cm-1-Region: i?(C0) Acetoxygruppe 1733 cm-1; v(C0)
bzw. v(CC) der ov9-unges. Ketogruppe im Ring C 1642 bzw. 1610
1) Ruzicka, Rüegg, Volli & Jeger, Helv. 30, 140 (1947)2) Jeger & Ruzicka, Helv. 28, 209 (1945)

- 67 -
cm (sehr schwache Schulter); v(CO) bzw. v(CC) der a,/Z-un-
ges. Ketogruppe im Ring D 1698 bzw. 1621 cm .
Mit diesen Annahmen steht das UV.-Spektrum in Einklang,
soweit es die Existenz der «,/3-ungesättigten Ketogruppen be¬
trifft (breites Maximum bei 240 mu, log £ = 4,16). Zudem aber
weist das UV.-Spektrum noch eine breite Absorptionsbande bei
332 m/u (log £ = 2,53) auf, für die eine Deutung fehlt.
Bei der Hydrierung des iso-a-Amyradienonol-acetats(CXXII)
mit Platin und Wasserstoff in Eisessig entstand als Haupt¬
produkt ein gesättigtes Keton C,2H520, (CXXVII), dessen UV.-
und IR.-Spektrum mit der angenommenen Struktur in Einklang
stehen. Das UV.-Spektrum besitzt ein Maximum bei 290 mu,
log e = 1,82. Im IR.-Spektrum treten bei 1718 und 1693 cm
Absorptionsbanden auf, die der Acetoxy- und der Ketogruppe
zuzuordnen sind. Die Frequenz der letzteren liegt ca. 10
cm- tiefer, als normalerweise für ein gesättigtes 6-Ring-
Keton beobachtet wird.
In kleiner Menge wurde bei der Hydrierung von CXXII
auch das iso-oc-Amyrenonol-acetat (CXIII) erhalten. Es ist
unwahrscheinlich, dass dieses schon in der zur Hydrierung
gelangenden Substanz enthalten war, da diese sorgfältig
chromatographisch gereinigt war. Ein Modellversuch, in dem
ein Gemisch des ungesättigten Ketons CXIII und des Dienons
CXXII chromatographiert wurde, zeigte, dass eine saubere
Trennung der beiden Verbindungen auf diesem Wege möglich
ist; dabei wurde unerwarteterweise zuerst das iso-os-Amyra-
dienonol-acetat (CXXII) eluiert.
Bei der katalytischen Hydrierung des Isomeren CXXV wur¬
de die Ketogruppe wegreduziert unter Bildung eines Diens
C12H50°2 (CXXVIII)> das mi't Tetranitromethan eine hellgelbe
Färbung gab. Daneben konnte nur noch unverändertes Ausgangs-
material aber kein iso-Qi-Amyrenonol-acetat (CXIII) festge¬
stellt werden. Es scheint daher ausgeschlossen, dass die

- 68 -
Bildung von CXIII bei der Hydrierung von CXXII über die Ver¬
bindung CXXV als Zwischenstufe verlaufen könnte. Das IR.-
Spektrum des Diens CXXVIII zeigt ausser der Acetoxybande
(1727 om" ) in der Doppelbindungsregion nur sehr schwache
Banden (bei 1695 ? und 1669 ? cm" ). Die Annahme einer 3fach
substituierten Doppelbindung steht mit dem Auftreten von Ab¬
sorptionsbanden zwischen 810 und 840 cm~ in Uebereinstim-
mung . Eine Ketonbande kann nicht beobachtet werden.
Die Isomerisierung von CXXVIII mit Chlorwasserstoff in
Eisessig-Chloroform ergab ein Produkt, das mit Tetranitrome-
than eine dunkelbraune Färbung gab, jedoch infolge Mangels
an Substanz nicht mehr gereinigt werden konnte.
um zu prüfen, ob das bei der Hydrierung von CXXII er¬
haltene gesättigte Keton CXXVII nicht mit dem «-Amyranonol-2)
II-acetat (CXVII) ' identisch sei, wurde diese Verbindung
hergestellt. Dazu wurde «-Amyrin-benzoat mit N-Bromsuccin-
imid zum a-Amyradienol-benzoat (CXIVc) dehydriert, und die¬
ses mit Wasserstoffperoxyd oxydiert. Das durch fraktionier¬
te Kristallisation aus dem Oxydationsgemisch gewonnene iso-
ot-Amyrenonol-II-benzoat (CXVIc) wurde durch Verseifung und
Acetylierung in das iso-ot-Amyrenonol-II-acetat C-.pH,-n0,(CXVIb) übergeführt. IR.- und UV.-Spektrum dieser Verbindung
zeigen übereinstimmend das Vorhandensein einer or,y9-unges.
Ketogruppe an (Max. im UV. bei 252 m.u, log e = 4,06). Die
i>(CO)-Prequenz liegt ausserordentlich tief (1647 cm ), da¬
gegen weicht die zugehörige v(CC)-Frequenz (1615 cm- ) nur
wenig vom normalen Wert ab. Die Acetatbande (1731 cm ) be¬
sitzt eine für feste Verbindungen dieser Reihe normale Fre-
1) Im allgemeinen gibt eine 4fach substituierte Aethylen-bindung mit unter sich ähnlichen Substituenten zu keiner
wahrnehmbaren Absorption im 1600 cm~l-Gebiet Anlass.
2) McLean, Ruff & Spring, Soc. 1951, 1093
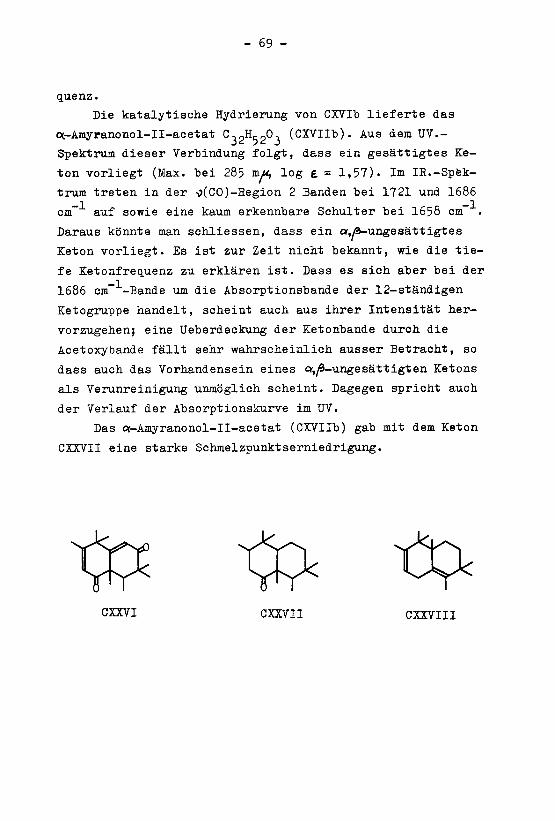
- 69 -
quenz.
Die katalytische Hydrierung von CXVIb lieferte das
oc-Amyranonol-II-acetat G32H520:} (CXVIIt)* Aus dem UV.-
Spektrum dieser Verbindung folgt, dass ein gesättigtes Ke¬
ton vorliegt (Max. bei 285 mu, log e = 1.57). Im IR.-Spek¬
trum treten in der -j(CO)-Region 2 Banden bei 1721 und 1686
cm" auf sowie eine kaum erkennbare Schulter bei 1658 cm~ .
Daraus könnte man schliessen, dass ein a,/3-ungesättigtes
Keton vorliegt. Es ist zur Zeit nicht bekannt, wie die tie¬
fe Ketonfrequenz zu erklären ist. Dass es sich aber bei der
1686 cm" -Bande um die Absorptionsbande der 12-ständigen
Ketogruppe handelt, scheint auch aus ihrer Intensität her¬
vorzugehen; eine Ueberdeckung der Ketonbande durch die
Acetoxybande fällt sehr wahrscheinlich ausser Betracht, so
dass auch das Vorhandensein eines ^^-ungesättigten Ketons
als Verunreinigung unmöglich scheint. Dagegen spricht auch
der Verlauf der Absorptionskurve im UV.
Das or-Amyranonol-II-acetat (CXVIIb) gab mit dem Keton
CXXVII eine starke Schmelzpunktserniedrigung.
*fr ^ tfçxCXXVI CXXVII CXXVIII

- 70 -
EXPERIMENTELLER TEIL1)
ct-Amyrin-benzoat-oxyd (CIXc) ' ;. 30 g «-Amyrin-
benzoat vom Schmelzpunkt 190 wurden in 1 Liter nichtsta-
bilisiertem Eisessig gelöst und zur siedenden Lösung im
Verlaufe einer Stunde eine Mischung von 180 cm 30-proz.
Wasserstoffperoxyd und 180 cm Eisessig zugetropft, wobei
kein Niederschlag ausfiel. Nach weiterem 2-stündigem Ko¬
chen wurde die heisse Lösung mit Wasser versetzt, bis sie
trübe wurde und dann zur Kristallisation 3 Stunden bei 0°
stehen gelassen. Das abgenutschte und gut abgesogene Roh¬
produkt wurde einmal aus Methylenchlorid-Methanol umkri¬
stallisiert. Das Oxyd (10 g) bildete lange Blättchen, die
bei 216-217 schmolzen und mit Tetranitromethan keine Farb¬
reaktion gaben.
2) 3)
Iso-oc-Amyrenonol-benzoat (CXIIIc) ' '. 18 g «-Amy-
rin-benzoat-oxyd wurden in 900 cm Eisessig gelöst und zur
Lösung, nach Zugabe von 5 Tropfen einer gesättigten Brom¬
wasserstofflösung in Eisessig, bei 85-90 während 15 Minu¬
ten unter heftigem Rühren 57,5 cm einer Lösung von Brom
in Eisessig (101 mg/cm ; entsprechend 1,1 Mol) zugetropft.
Es fiel kein Niederschlag aus. Darauf wurde noch 3è Stun¬
den unter Rühren auf 95-100 erhitzt.- Die dunkelbraune
Reaktionslösung wurde in 3 Liter Wasser gegossen und aus-
geäthert. Das Rohprodukt war grösstenteils kristallin.
38 g auf diese Weise in 2 Ansätzen erhaltene Substanz
wurde durch eine Säule von 500 g Aluminiumoxyd (Akt.III)
chromatographiert. Mit Petroläther-Benzol-Gemischen und Ben-
1) Siehe Pussnote 1) Seite 39.
2) Seymour, Sharpies & Spring, Soc. 1939, 1075
3) McLean, Silverstone & Spring, Soc. 1951, 935

- 71 -
zol liessen sich 38 g Substanz eluieren. Das iso-c*-Amyren-
onol-benzoat kristallisierte aus Methylenchlorid-Methanol
in dichten, oft federartig verwachsenen Kristallen vom
Schmelzpunkt 212°. Ausbeute 28 g.
Iso-o-Amyrenonol-acetat (CXIIIb). 48O mg des Benzoats
CXIIIc wurden durch Kochen mit 10-proz. methanolischer Kali¬
lauge (unter Zusatz von etwas Benzol) verseift. Das nach üb¬
licher Aufarbeitung erhaltene Rohprodukt (420 mg) wurde mit
Pyridin-Acetanhydrid bei Zimmertemperatur acetyliert. Das
Acetat bildete aus Methylenchlorid-Methanol glänzende Blätt¬
chen vom Schmelzpunkt 294-295 • Zur Analyse gelangte ein 4mal
umkristallisiertes und anschliessend im Hochvakuum bei 250°
sublimiertes Präparat.
3,567 mg Subst. gaben 10,395 mg C02 und 3,314 mg H20
C32H5003 Ber. C 79,61 H 10,44 $
Gef. C 79,53 H 10,40 #
Oxydation von iso-Qfr-Amyrenonol-acetat (CXIIIb) mit Se¬
lendioxyd . 2g iso-a-Amyrenonol-acetat wurden in 100 cm
stabilisiertem Eisessig gelöst und mit 6 g Selendioxyd 23
Stunden am Rückfluss gekocht. Die rot gefärbte Lösung wurde
durch Piltration vom ausgeschiedenen Selen befreit, mit Was¬
ser verdünnt und ausgeäthert. Die ätherische Lösung wurde
4mal mit verd. Natronlauge, einmal mit verd. Schwefelsäure
und dann mit Wasser gewaschen. Das amorphe, braun gefärbte
Rohprodukt (1,61 g) kristallisierte beim Bespritzen mit Me¬
thanol. Es wurde durch eine Säule aus 50 g Aluminiumoxyd
(Akt.II) chromatographiert.
1) Ruzicka, Rüegg, Volli & Jeger, Helv. 30, 140 (1947);
vgl. auch Rüegg, Diss. ETH, Zürich 194ÏÏ

- 72 -
Frakt Lösungsmittel Menge eluierter Substanz
1
2-4
5-6
7-9
10
11
12
13
300 cm3 Päth.-Bzol(9:1)
900 cm3 Päth.-Bzol(4:l)
600 cm3 Päth.-BzoK2:1)
900 cm3 Päth.-Bzol(l:l)
300 cm3 Päth.-Bzol(l:l)
300 cm3 Benzol
300 cm Benzol
300 cm3 Bzol-Aeth.(9:l)
10 mg Krist.
80 mg Krist. Smp.219°
160 mg Krist. Smp.219-220°
20 mg Krist. Smp.220-221°
70 mg Krist. Smp.226-227°
50 mg Krist. Smp.234-236°
250 mg Krist. Smp.256-257°
Die vereinigten Fraktionen 3-10 kristallisierten aus
Methylenchlorid-Methanol in Nadeln, die mit Tetranitromethan
eine gelbe Färbung gaben. Das Analysenpräparat wurde 4mal
umkristallisiert und im Hochvakuum bei 180-190 sublimiert;
Smp. 220°.
3,688 mg Subst. gaben 10,805 mg COp und 3,318 mg HpO
C32H4803 Ber. C 79,95 H 10,07 #
Gef. C 79,95 H 10,08 f°
(«)D = +10° (c = 1,79)
Es liegt iso-a-Amyradienonol-acetat (CXXII) vor.
Die Fraktion 13 schmolz nach 6maligem Umkristallisie¬
ren aus Methylenchlorid-Methanol bei 257-259°• Die erhalte¬
nen nadelförmigen Kristalle vrarden zur Analyse im Hochvakuum
bei 210° sublimiertj das Präparat schmolz bei 261-262° und
gab mit Tetranitromethan eine ganz schwache Gelbfärbung.
3,782 mg Subst. gaben 11,061 mg C02 und 3,430 mg HgO
C32H48°3 Ber' C 79'95 H 10'07 ^
Gef. C 79,80 H 10,14 f>
(«)D = +122° (c = 1,29)
Es lag die Verbindung CXXV vor, die jedoch, nach der
Farbreaktion mit Tetranitromethan und der optischen Drehung

- 73 -
zu schliessen, noch verunreinigt war. Jedenfalls gab das
Präparat mit der durch Isomerisierung des iso-o-Amyradien-
onol-acetats (CXXII) erhaltenen Verbindung CXXV keine Sehmelz-
punktsdepression und die IR.-Spektren waren praktisch iden¬
tisch.
Isomerisierung von iso-Qf-Amyradienonol-acetat (CXXII). '
100 mg iso-Of-Amyradienonol-acetat vom Schmelzpunkt 219-220°
wurden in 25 cm Eisessig gelöst und in die Lösung trockener
Chlorwasserstoff bis zur Sättigung eingeleitet. Nach 3-tägigem
Stehen bei Zimmertemperatur wurde die Lösung im Vakuum ein¬
gedampft. Das kristalline Produkt bildete aus Methylenchlo¬
rid-Methanol Nadeln, die bei 263 schmolzen und gegen Tetra-
nitromethan gesättigt waren. Zur Analyse wurde 2mal umkri¬
stallisiert und anschliessend im Hochvakuum bei 210° subli-
miert. Das Präparat schmolz bei 264 •
3,598 mg Subst. gaben 10,535 mg CO« und 3,234 mg HJD
C32H48°3 Ber- C 79'95 H 10»07 *
Gef. C 79,89 H 10,06 <f>
(«)D = +167° (c = 1,08)
Es liegt die Verbindung CXXV vor.
Oxydation von iso-ot-Amyradienonol-acetat (CXXII) mit
Selendioxyd bei 210 '. 250 mg Substanz vom Schmelzpunkt
219-220 wurden in 17 cm Dioxan suspendiert und mit 750 mg
Selendioxyd im Einschlussrohr 18 Stunden auf 210° erhitzt.
Nach dem Erkalten wurde vom ausgeschiedenen Selen abfiltriert,
eingedampft, in Aether aufgenommen und die ätherische Lö¬
sung mit verd. Natronlauge und Wasser gewaschen. Das Oxyda-
1) Ruzicka, Rttegg, Volli & Jeger, Helv. 30, 140 (1947)2) Vgl. Jeger & Ruzicka, Helv. 28, 209 (1945)

- 74 -
tionsprodukt kristallisierte aus Methylenchlorid-Methanol
in glänzenden Blättchen, die bei 237-238° schmolzen und mit
Tetranitromethan keine Farbreaktion gaben. Es wurde durch
eine Säule aus 8 g Aluminiumoxyd (Akt.II) chromatographiert.
Mit 650 cm Petroläther-Benzol (9:1) Hessen sich 130 mg
Substanz eluieren, die zur Analyse 2mal umkristallisiert
und im Hochvakuum bei 200 sublimiert wurde; das Präparat
schmolz bei 240-241°.
3,687 mg Subst. gaben 10,494 mg C02 und 3,070 mg H20
C32H46°4 Ber* C 77'69 H 9'37 *
Gef. C 77,68 H 9,32 Jl
(«)D = +14° (c = 1,39)
Es liegt die Verbindung CXXVI vor.
1) 2)Hydrierung von iso-Qfr-Amyradienonol-acetat (CXXII)
450 mg Substanz vom Schmelzpunkt 219-220° wurden in 50 cm
Eisessig gelöst und in Gegenwart von Platin, hergestellt
durch Vorhydrierung von 150 mg Platindioxyd, in einer Was¬
serstoffatmosphäre geschüttelt. Nach 20 Stunden waren genau
2 Mol Wasserstoff aufgenommen. Die durch Filtration vom Pla¬
tin befreite Lösung wurde im Vakuum eingedampft. Das kri¬
stalline Rohprodukt schmolz unscharf bei 199-212 . Es wurde
durch eine Säule von 20 g Aluminiumoxyd (Akt.I) chromato¬
graphiert .
Frakt Lösungsmittel Menge eluierter Substanz
1-2
3-4
5-6
7
8
9-10
600 cm3 Päth.-Bzol(2:l)
600 cm3 Päth.-Bzol(l:l)
600 cm Benzol
300 cm3 Bzol-Aeth.(9:l)
300 cm3 Bzol-Aeth.(9:1)
600 cm3 Aether
10 mg Krist. Smp.175-177°
20 mg Krist. Smp.204-214°
50 mg Krist. Smp.219°160 mg Krist. Smp.211-212°
10 mg Krist. Smp.295°70 mg Oel
1) Ruzicka, Rüegg, Volli & Jeger, Helv. 30, 140 (1947)2) Rüegg, Diss. ETH, Zürich 1948; Volli, Diss. ETH, Zur.1948

- 75 -
Die Fraktion 8 kristallisierte aus Methylenchlorid-Me¬
thanol in glänzenden Blättchen, die bei 295° schmolzen und
gegen Tetranitromethan gesättigt waren. Ein Mischschmelz-
punkt mit iso-Of-Amyrenonol-acetat (CXIIIb) ergab keine De¬
pression.
Die Fraktionen 5-7 wurden vereinigt und durch eine Säu¬
le von 6 g Aluminiumoxyd (Akt.II) chromatographiert.
Frakt. Lösungsmittel Menge eluierter Substanz
1-5
6
7-8
9
550 cm3 Petroläther
150 cm3 Päth.-Bzol(9:l)
400 cm3 Päth.-Bzol(9:1)
200 cm3 Päth.-Bzol(l:l)
90 mg Krist. Smp.219-222°
40 mg Krist. Smp.219-220°
40 mg Krist. Smp.209-212°
40 mg Krist. Smp.208°
Die Fraktionen 1-6 wurden vereinigt und 4mal aus Methy¬
lenchlorid-Methanol umkristallisiert. Das Hydrierungsprodukt
bildete lockere glänzende Blättchen und gab mit Tetranitro¬
methan keine Färbung. Das Analysenpräparat wurde im Hoch¬
vakuum bei 180-190° sublimiert; es schmolz bei 222-223°.
3,732 mg Subst. gaben 10,852 mg C02 und 3,681 mg HgO
C32H52°3 Ber* C 79'28 H 10>81 1>
Gef. C 79,35 H 11,04 t
(«)D = +96°; +99° (c = 1,04; 1,13)
Es liegt die Verbindung CXXVII vor.
Hydrierung der Verbindung CXXV. 380 mg Substanz vom
Schmelzpunkt 261-263° wurden in 50 'cm Eisessig mit Platin,
hergestellt durch Vorhydrierung von 160 mg Platindioxyd, in
einer WasserstoffatmoSphäre geschüttelt. Nach 16 Stunden be¬
trug die gemessene Wasserstoffaufnähme 1 Mol; die Lösung war
durch ausgeschiedene Substanz getrübt. Nach der üblichen Auf¬
arbeitung wurde das kristalline Rohprodukt durch eine Säule

- 76 -
von 12 g Àluminiumoxyd (Akt.II) chromatographiert.
Prakt Lösungsmittel Menge eluierter Substanz
1-5
6
7
8-9
10
1300 cm Petroläther
300 cm3 Päth.-Bzol(9:l)
300 cm3 Päth.-Bzol(9:l)
600 cm3 Path.-Bzol(1:1)
300 cm Benzol'
170 mg Krist. Smp.252-254°
30 mg Krist.
30 mg Krist. Smp.261-262°80 mg Krist. Smp.261-263°
4-0 mg Krist.
Die Fraktionen 1-5 gaben mit Tetranitromethan eine hell¬
gelbe Färbung. Sie wurden vereinigt und 3mal aus Methylen¬
chlorid-Methanol umkristallisiert; glänzende Blättchen. Zur
Analyse gelangte ein im Hochvakuum bei 200° sublimiertes
Präparat vom Schmelzpunkt 254° •
3,600 mg Subst. gaben 10,846 mg C02 und 3,489 mg HgO
C32H5002 'Ber. C 82,34 H 10,80 jt
Gef. C 82,22 H 10,85 $
(«)D = +90° (c = 1,60)
Es liegt die Verbindung CXXVIII vor.
Die Fraktionen 7-9 kristallisierten aus Methylenchlo¬
rid-Methanol in Nadeln, die gegen Tetranitromethan gesättigt
waren und mit dem Ausgangsmaterial (Verbindung CXXV) keine
Erniedrigung des Schmelzpunktes gaben.
oc-Amyradienol-benzoat (CXIVc) '. 10 g ex-Amyrin-ben-
zoat wurden mit 8 g 93-proz. N-Bromsuccinimid in 300 cm ab¬
solutem Tetrachlorkohlenstoff li Stunden am Rückfluss ge¬
kocht. Darauf wurde die rote Lösung vom Succinimid abfil¬
triert, eingedampft, der Rückstand in Aether aufgenommen
und die ätherische Lösung mit verd. Katronlauge und verd.
Salzsäure gewaschen. Das stark gefärbte, schaumige Rohpro-
1) Vgl. Ruzicka, Jeger & Redel, Helv. 26_, 1235 (1943)

- 77 -
dukt (10 g) wurde durch eine Säule aus 200 g Aluminiumoxyd
(Akt.II) chromatographiert. Mit 1,8 Liter Petroläther und
600 cm Petroläther-Benzol (9:1) liessen sich 7,3 g farblo¬
ser Substanz eluieren, die aus Methylenchlorid-Methanol in
glänzenden Blättchen kristallisierte. Die Verbindung schmolz
bei 175-176 und gab mit Tetranitromethan eine dunkelbraune
Färbung.
Iso-ct-Amyrenonol-II-benzoat (CXVIc) . 5 » 9 g «-Amy-
radienol-benzoat (CXIVc) wurden in 270 cm nichtstabilisier-
tem Eisessig gelöst. Hierauf wurde unter heftigem Rühren und
Erwärmen auf dem Wasserbade während 20 Minuten eine Mischung
von 25 cm 30-proz. Wasserstoffperoxyd und 25 cm Eisessig
zugetropft. Es fiel ein kristalliner Niederschlag aus, der
sich nach 2-stündigem weiterem Rühren und Erwärmen wieder
löste. Nach erneutem Zutropfen einer Mischung von 25 cm 30-
proz. Wasserstoffperoxyd und 25 cm Eisessig im Verlaufe von
10 Minuten wurde noch 1 Stunde gerührt und erhitzt, dann die
Lösung mit heissem Wasser verdünnt, bis sie trüb blieb und
über Nacht abkühlen gelassen. Der feinkristalline Nieder¬
schlag wurde abgenutscht, gut abgesogen und dann einer frak¬
tionierten Kristallisation aus Methylenchlorid-Methanol un¬
terworfen. Aas der heissen Lösung schied sich zuerst das
iso-a-Amyrenonol-II-benzoat in dicken Nadeln aus. Nach 4ma-
ligem Umkristallisieren wurden 870 mg dieser Verbindung er¬
halten, die bei 241° schmolz und gegen Tetranitromethan ge¬
sättigt war.
Iso-ot-Amyrenonol-II-acetat (CXVIb). 870 mg des Benzo-
ates CXVIc wurden in wenig Benzol gelöst und mit 100 cm
5-proz. methanolischer Kalilauge 2 Stunden am Rückfluss ge-
1) Vgl. McLean, Ruff & Spring, Soc. 1951, 1093

- 78 -
kocht. Das nach der üblichen Aufarbeitung erhaltene Versei-
fungsprodukt kristallisierte aus Methylenchlorid-Methanol
in dünnen, verfilzten Nadeln vom Schmelzpunkt 211 . Das rohe
iso-oKÄmyrenonol-II (760 mg) wurde mit Pyridin-Acetanhydrid
bei Zimmertemperatur acetyliert. Das Acetat bildete aus Me¬
thylenchlorid-Methanol ungelöst Blättchen, die konstant bei
200-201° schmolzen. Zur Analyse gelangte ein 3mal umkristal¬
lisiertes und im Hochvakuum bei 180° sublimiertes Präparat.
3,74-2 mg Subst. gaben 10,932 mg COp und 3,4-32 mg H„0
C32H50°3 Ber* C 79'51 H 10'44 *
Gef. C 79,73 H 10,26 #
(«)D = +18° (c = 1,15)
ot-Amyranonol-II-acetat (CXVIIb) . 350 mg iso-a-Amy-
renonol-II-acetat (CXVIb) vom Schmelzpunkt 200-201° wurden
in 55 cm Eisessig mit Platin, hergestellt aus 180 mg Pla¬
tindioxyd, in einer Wasserstoffatmosphare geschüttelt. Nach
21 Stunden entsprach die gemessene Wasserstoffaufnahme 1,2
Mol. Das nach Abfiltrieren des Platins und Eindampfen der
Lösung im Vakuum erhaltene kristalline Produkt wurde 4mal
aus Methylenchlorid-Methanol umgelöst. Es bildete flache
Nadeln, die konstant 232-233 schmolzen und mit Tetranitro-
methan keine Färbung gaben. Der Mischschmelzpunkt mit dem,
durch Hydrierung des iso-cx-Amyradienonol-acetats (CXXII) ge¬
wonnenen, gesättigten Keton (CXXVII) ergab eine starke De¬
pression. Das Analysenpräparat wurde im Hochvakuum bei 200
sublimiert.
3,777 mg Subst. gaben 10,986 mg C02 und 3,636 mg HgO
C32H52°3 Ber' C 79,28 H 10,81 <f>
Gef. C 79,38 H 10,77 <f>
(a)D = +64° (c = 1,40)
1) Vgl. McLean, Ruff & Spring, Soc. 1951, 1093

- 79 -
Das auf oben beschriebenem Wege erhaltene a-Amyranonol-
II-acetat war mit einem, von Prof. Dr. F. S. Spring in freund¬
licher Weise aus Glasgow gesandten Präparat dieser Verbin¬
dung nach Schmelzpunkt, Mischprobe und spezifischer Drehung
identisch.
Herrn W. Manser, unter dessen Leitung die Mikroanaly¬
sen ausgeführt wurden, danke ich für seine wertvolle Mit¬
arbeit.
ZUSAMMENFASSUNG-
Im Verlaufe von Untersuchungen, die für den strengen
Beweis der Konstitution des Ringes C des ot-Amyrins, bzw. für
die Lokalisierung der C-Atome 26 und 27 von Bedeutung sind,
wurde das bekannte iso-ct-Amyradienonol-acetat (CXXII) berei¬
tet und dieses einerseits zu einem gesättigten Keton (CXXVII)
reduziert, das mit keinem bekannten Keton der ot-Amyrin-Reihe
identisch ist, andererseits zu einem Dien-dion (CXXVI) oxy¬
diert. Die UV.- und IR.-Spektren dieser 3 Verbindungen ste¬
hen mit der angenommenen Struktur (C-Atom 27 als Methyl am
C, ,) nicht im Widerspruch.
Das iso-«-Amyradienonol-acetat (CXXII) wurde zu einer
schon bekannten Verbindung (CXXV) isomerisiert und diese zu
einem Dien (CXXVIII) reduziert. Die Spektren und sonstigen
Eigenschaften der Verbindungen CXXV und CXXVIII lassen sich
mit der angenommenen Strukturformel (C-Atom 27 als Methyl am
C,.) vereinbaren.

LEBENSLAUF
Ich wurde am 9. August 1924 in Bukarest (Rumänien) ge¬
boren. Nach Absolvierung der Primarschule und 6-£ Jahren
Mittelschule in Bukarest trat ich im Frühjahr 1942 in. die
Kantonale Oberrealschule Zürich ein und verliess diese im
Jahre 1944 mit dem Maturitätszeugnis Typus C. Im Herbst des
gleichen Jahres begann ich mein Chemie-Studium an der Eid¬
genössischen Technischen Hochschule und erwarb im Herbst
1948 das Diplom als Ingenieur-Chemiker. Seit anfangs 1949
arbeitete ich im organisch-chemischen Laboratorium der ETH
unter der Leitung von Herrn Prof. Dr. L. Ruzicka an der
vorliegenden Arbeit.
Zürich, Mai 1952.




























![[p] Une nouvelle de David Ruzicka](https://img.pdfslide.tips/doc/110x75/62170012c4f78d50fe52adb9/p-une-nouvelle-de-david-ruzicka.jpg)
