Embed Size (px)
Citation preview

Ce
ntr
o d
e E
stu
dio
s d
e P
ostg
rado
Más
ter
Uni
vers
itario
en
Oliv
ar y
ace
ite d
e ol
iva
Universidad de Jaén
Centro de Estudios de Postgrado
Trabajo Fin de Máster
Trabajo Fin de Máster
DISEÑO E INSTALACIÓN
DE UN LABORATORIO DE
ANÁLISIS DE ACEITUNA, ACEITES Y DERIVADOS
Alumno/a: Caracuel Pareja, Elizabeth
Tutor/a: Prof. D. Rafael Pacheco Reyes
Profa. Dña. M. Dolores La Rubia García
Dpto: Ingeniería Química, Ambiental y de los
Materiales

DISEÑO DE UN LABORATORIO DE ANÁLISIS DE ACEITUNA, ACEITES Y DERIVADOS
Esta memoria constituye el Trabajo Fin de Máster y se
presenta a la Comisión Evaluadora en Jaén a 14 de
diciembre del año 2017.
Fdo. Elizabeth Caracuel Pareja
RAFAEL PACHECO REYES, PROFESOR TITULAR DEL
DEPARTAMENTO DE INGENIERÍA QUÍMICA,
AMBIENTAL Y DE LOS MATERIALES DE LA
UNIVERSIDAD DE JAÉN.
Como TUTOR de Dña. Elizabeth Caracuel Pareja, en el
Máster Universitario en Olivar y Aceite de Oliva, durante el
curso 2016-2017
INFORMA: Que el presente trabajo fin de máster, Diseño
de un laboratorio de análisis de aceituna, aceites y
derivados, ha sido realizado en el Departamento de
Ingeniería Química, Ambiental y de los Materiales, por la
Licenciada Dña. Elizabeth Caracuel Pareja, para la
obtención del Título de Máster Universitario en Olivar y
Aceite de Oliva por la Universidad de Jaén, bajo la dirección
de los Dres. D. Rafael Pacheco Reyes, y Dña. Mª Dolores
La Rubia García.
Jaén, diciembre de 2017
Fdo.: Rafael Pacheco Fdo. Mª Dolores La Rubia

AGRADECIMIENTOS
A Rafael Pacheco, por su dedicación, disponibilidad y por su gran ayuda en todo
momento. Con personas como él es un auténtico placer aprender.
A Paco, mi compañero de ilusiones y mi marido, por el apoyo incondicional y por
su esfuerzo para que persiga todos mis sueños sin importar el alcance de ellos.
A mi futuro pequeñín, por permitirme cerrar una etapa antes de empezar otra.
A mis padres, por facilitarme el camino para conseguir todos mis propósitos.
A todos mis compañeros del Máster por acompañarme en esta experiencia y en
especial a Marilú, gran persona y mejor amiga.

ÍNDICE

ELIZABETH CARACUEL PAREJA
II
1 RESÚMENES ............................................................................................. 1
1.1 RESUMEN ............................................................................................ 2
1.2 SUMMARY ............................................................................................ 3
2 ESTUDIO DE VIABILIDAD .......................................................................... 4
2.1 JUSTIFICACIÓN DEL PROYECTO ...................................................... 5
2.1.1 Antecedentes. ................................................................................. 5
2.1.1.1 Superficie nacional de olivar .................................................... 5
2.1.1.2 Superficie andaluza de olivar ................................................... 6
2.1.2 Localización .................................................................................... 7
2.1.2.1 Clima ........................................................................................ 8
2.1.2.2 Entorno..................................................................................... 8
2.2 ESTUDIO DE MERCADO ..................................................................... 9
2.2.1 Almazaras y cooperativas ............................................................... 9
2.2.2 Competencia directa ..................................................................... 10
3 MEMORIA ................................................................................................. 11
3.1 INTRODUCCIÓN ................................................................................ 12
3.1.1 Marco contextual .......................................................................... 12
3.1.2 El olivo y su fruto .......................................................................... 19
3.1.2.1 Características principales ..................................................... 19
3.1.2.2 Condiciones de cultivo ........................................................... 20
3.1.3 El aceite de oliva ........................................................................... 21
3.1.3.1 Situación actual ...................................................................... 21
3.2 PROCESO DE ELABORACIÓN DEL ACEITE DE OLIVA .................. 22
A) En la explotación agrícola ................................................................ 22
3.2.1 Recolección de aceituna ............................................................... 22
3.2.1.1 Ordeño ................................................................................... 22
3.2.1.2 Vareo ..................................................................................... 23

ELIZABETH CARACUEL PAREJA
III
3.2.1.3 Vibración ................................................................................ 24
3.2.1.4 Recogida del Suelo ................................................................ 24
B) En la almazara ................................................................................. 25
3.2.2 Recepción de la aceituna ............................................................. 25
3.2.3 Molienda ....................................................................................... 26
3.2.4 Batido ........................................................................................... 27
3.2.5 Extracción ..................................................................................... 29
3.2.5.1 Extracción por presión ........................................................... 30
3.2.5.2 Extracción continua por centrifugación .................................. 30
3.2.5.3 Extracción de aceite de orujo ................................................. 32
3.2.6 Proceso de refinado ...................................................................... 32
3.2.7 Clasificación y almacenamiento .................................................... 33
3.2.8 Envasado del aceite de oliva ........................................................ 34
3.3 CLASIFICACIÓN DE LOS ACEITES DE OLIVA ................................. 35
3.3.1 Aceites de oliva vírgenes .............................................................. 35
3.3.2 Aceite de oliva no vírgenes ........................................................... 36
3.4 NORMATIVA VIGENTE PARA ANÁLISIS DE ACEITE....................... 38
4 DISEÑO DEL LABORATORIO .................................................................. 98
4.1 PLANO GENERAL .............................................................................. 99
4.2 SALA DE RECEPCIÓN ..................................................................... 100
4.3 DESPACHOS .................................................................................... 100
4.4 SALA DE JUNTAS ............................................................................ 100
4.5 PATIO DE RECEPCIÓN Y ALMACENAMIENTO DE MATERIA PRIMA
………………………………………………………………………………101
4.6 SALA DE MOLTURACIÓN ................................................................ 101
4.7 SALA DE ANÁLISIS .......................................................................... 103
4.8 ALMACÉN DE MUESTRAS DE ACEITE .......................................... 106

ELIZABETH CARACUEL PAREJA
IV
5 ÁREA COMERCIAL ................................................................................ 107
5.1 SERVICIOS OFERTADOS ............................................................... 108
5.1.1 Análisis de aceituna .................................................................... 108
5.1.1.1 Índice de madurez ................................................................ 108
5.1.1.2 Peso medio .......................................................................... 109
5.1.1.3 Relación pulpa-hueso .......................................................... 110
5.1.1.4 Humedad.............................................................................. 110
5.1.1.5 Contenido graso total ........................................................... 110
5.1.2 Análisis de aceite ........................................................................ 113
5.1.2.1 Toma de muestra ................................................................. 113
5.1.2.2 Grado e índice de acidez ..................................................... 114
5.1.2.3 Índice de peróxidos .............................................................. 116
5.1.2.4 Medida espectrofotométrica de la absorción en la región
ultravioleta de un aceite ....................................................................... 119
5.1.2.5 Medida de la composición de ácidos grasos ........................ 122
5.1.2.6 Medida del contenido en ceras mediante cromatografía de
gases con columna capilar .................................................................. 125
5.1.2.7 Medida de la concentración y composición de esteroles ..... 128
5.1.2.8 Medida de la concentración en eritrodiol y uvaol ................. 134
5.1.2.9 Medida del contenido en estigmastadienos en aceites vegetales
…………………………………………………………………….139
5.1.2.10 Humedad y volátiles ............................................................ 143
5.1.2.11 Impurezas solubles en éter de petróleo ............................... 145
5.1.2.12 Índice de refracción ............................................................. 146
5.1.2.13 Índice de yodo. Método de Wijs .......................................... 147
5.1.2.14 Índice de saponificación ...................................................... 150
5.1.3 Análisis de subproductos ............................................................ 152
5.1.3.1 Humedad en orujos (UNE 55031) ........................................ 152

ELIZABETH CARACUEL PAREJA
V
5.1.3.2 Materia grasa en orujos (UNE 55032) .................................. 152
5.1.3.3 Materia grasa en alpechines ................................................ 153
5.1.4 Análisis foliar............................................................................... 154
5.1.4.1 Toma de muestra ................................................................. 154
5.1.4.2 Época de muestreo .............................................................. 156
5.1.4.3 Preparación de la muestra ................................................... 156
5.1.4.4 Mineralización de la muestra ................................................ 157
5.1.4.5 Determinación de nitrógeno ................................................. 158
5.1.4.6 Determinación de fósforo ..................................................... 160
5.1.4.7 Determinación de sodio ....................................................... 162
5.1.4.8 Determinación de potasio..................................................... 163
5.1.4.9 Síntomas visuales de deficiencias nutritivas en el olivo ....... 165
5.2 PLAN DE VENTAS............................................................................ 167
6 SEGURIDAD Y SALUD ........................................................................... 168
6.1 PREVENCIÓN DE RIESGOS LABORALES EN LABORATORIOS .. 169
6.1.1 Legislación .................................................................................. 169
6.1.2 Normas generales de seguridad ................................................. 169
6.2 PLAN DE EMERGENCIA .................................................................. 171
6.2.1 Actuación en caso de emergencia .............................................. 171
6.2.2 Almacenamiento de reactivos ..................................................... 176
6.3 GESTIÓN DE RESIDUOS ................................................................ 179
6.3.1 Pretratamiento de residuos ......................................................... 181
6.3.2 Normativa aplicable .................................................................... 182
6.3.2.1 Sustancias peligrosas .......................................................... 182
6.3.2.2 Preparados peligrosos ......................................................... 182
7 ESTUDIO ECONÓMICO FINANCIERO .................................................. 183
7.1 INVENTARIO .................................................................................... 184

ELIZABETH CARACUEL PAREJA
VI
7.2 ESTUDIO ECONÓMICO ................................................................... 187
8 CONCLUSIONES ................................................................................... 191
9 BIBLIOGRAFÍA ....................................................................................... 193


1 RESÚMENES

ELIZABETH CARACUEL PAREJA
2
1.1 RESUMEN
A partir de los conocimientos adquiridos durante el Máster Universitario en Olivar
y Aceite de oliva, se ha diseñado este proyecto que aborda toda la información
necesaria para el diseño e instalación de un laboratorio dentro del sector oleícola.
Con este proyecto se pretende tener un manual a seguir para todo el personal
que trabaje en el sector, así como una herramienta informativa de los distintos
métodos de análisis de aceituna, del aceite de oliva y de sus subproductos.
En el mismo se abordan las características principales del cultivo del olivar, el
proc eso de elaboración de aceite de oliva, el diseño efectivo del laboratorio
con todas sus zonas de trabajo, los servicios ofertados, así como los planes de
seguridad, emergencias y gestión de residuos de dicha empresa, para finalizar
con un estudio económico-financiero.
A su vez, dicho trabajo sirve como orientación sobre las instalaciones,
instrumental, material y protocolos necesarios para poder desempeñar un puesto
de trabajo en una empresa como tal.
Por otra parte, destacar que este manual está apoyado principalmente en la
normativa vigente relativa al análisis de aceite.
Del análisis económico realizado, se deduce que el proyecto que se presenta es
viable desde el punto de vista económico y social.

ELIZABETH CARACUEL PAREJA
3
1.2 SUMMARY
Based on the knowledge acquired during the Master's Degree in Olive Grove and
Olive Oil, this project has been designed to address all the necessary information
for the design and installation of a laboratory within the olive sector.
This project aims to have a manual to follow for all the personnel working in the
sector, as well as an informative tool of the different methods of analysis of olive,
olive oil and its by-products.
This proyect includes the main characteristics of olive growing, the process of
preparing olive oil, the effective design of the laboratory with all work zones, the
services offered, as well as the company's safety, emergency and waste
management plans, to finalize with an economic-financial study.
In turn, this work serves as guidance on facilities, instruments, materials and
protocols necessary to be able to perform a job in a laboratory.
On the other hand, it should be noted that this manual is mainly supported by the
current regulations regarding oil analysis.
From the economic analysis carried out, it can be deduced that the project
presented is viable from the economic and social point of view.

2 ESTUDIO DE VIABILIDAD
ELIZABETH CARACUEL PAREJA
5
2.1 JUSTIFICACIÓN DEL PROYECTO
Hacer un estudio de la viabilidad del proyecto de implantación de un laboratorio
de análisis es fundamental para su futura puesta en marcha. Toda inversión
requiere de una garantía para poder llevarse a cabo. A través de este proyecto
se pretende justificar la necesidad de implantación de un nuevo laboratorio en el
territorio seleccionado. Además, se abordarán todos los recursos necesarios, los
medios disponibles, los servicios comerciales ofertados y la clientela a la que irá
dirigida la nueva empresa.
2.1.1 Antecedentes.
2.1.1.1 Superficie nacional de olivar
Aunque es en Andalucía, y más concretamente en Jaén y la cuenca alta del rio
Guadalquivir, donde se concentra la mayor parte del cultivo del olivo y de la
producción de aceites de oliva, existen otras áreas geográficas en España en las
que se pueden encontrar explotaciones olivareras.
El olivar español está presente en 34 provincias de 13 Comunidades Autónomas.
Ocupa una superficie de 2.494.328 has, que incluyen las de doble uso (almazara
y mesa), de las que el 96% corresponden a variedades de aceituna para
almazara (2.394.554 has) y el 4% restante a variedades para mesa (99.774 has)
(Fig.2.1).
Tabla 2.1. Superficie Nacional de Olivar. Elaboración propia
COMUNIDAD AUTÓNOMA SUPERFICIE, Has %
Andalucía 1.534.127 61,37
Extremadura 262.700 10,53
Castilla-La Mancha 358.706 14,38
Cataluña 122.889 4,92
Aragón 47.462 1,90
Resto 173.521 6,95
TOTAL 2.494.328 100

ELIZABETH CARACUEL PAREJA
6
Figura 2.1. Distribución geográfica nacional de las variedades de olivo más
representativas.
2.1.1.2 Superficie andaluza de olivar
El cultivo del olivo ocupa más del 60% de la superficie agraria de Andalucía,
adquiriendo una importancia relevante en la provincia de Jaén, el sur de
Córdoba, el noroeste de Granada, el norte de Málaga y el sudeste de
Sevilla(Fig.2.1).
Tabla 2.2. Distribución Provincial de la superficie de olivar andaluz (2016).
Fuente: SIGPAC. Consejería de Agricultura, Pesca y Desarrollo Rural.
Elaboración propia
PROVINCIA HECTÁREAS %
JAÉN 586.173 38,20
CÓRDOBA 351.735 22,92
SEVILLA 206.932 13,48
GRANADA 184.073 11,99
MÁLAGA 127.490 8,31
HUELVA 36.512 2,37
CÁDIZ 23.133 1,50
ALMERÍA 18.079 1,17
TOTAL ANDALUCÍA 1.534.127 100

ELIZABETH CARACUEL PAREJA
7
Jaén y Córdoba son las principales provincias olivareras, concentrando ambas
en torno al 60% de la superficie de olivar de Andalucía. Les siguen en
importancia Sevilla, Granada y Málaga, mientras que el conjunto de Cádiz,
Huelva y Almería contabiliza apenas el 5,1% del olivar andaluz.
2.1.2 Localización
Teniendo en cuenta la valoración de la superficie de olivar que hay en España,
el laboratorio será implantado en Priego de Córdoba, ciudad ubicada en el
sureste de la provincia de Córdoba, a unos 100 kilómetros de la capital, con
buenos accesos y una población de 23.000 habitantes (Fig.2.2).
Priego de Córdoba se encuentra localizada en pleno corazón de las Sierras
Subbéticas, limitando con dos provincias, al este con Jaén y al sureste con
Granada, y tres partidos judiciales, al noroeste con Baena, al suroeste con Rute
y al oeste con Cabra (Fig.2.3).
Figura 2.2. Situación geográfica de Priego de Córdoba. Fuente: D.O.
Figura 2.3. Olivar de Priego de Córdoba.

ELIZABETH CARACUEL PAREJA
8
2.1.2.1 Clima
Priego de Córdoba posee un clima mediterráneo continentalizado de influencia
atlántica, propiciado por la lejanía del litoral y favorecido por la escasa influencia
marítima, en el que las precipitaciones que antaño fueron abundantes, en la
actualidad se manifiestan de forma irregular en invierno y muy escasas en
verano.
La temperatura media anual suele ser elevada, en torno a los 19 °C, con
inviernos suaves en los que se presentan bajadas de temperaturas de forma
brusca, llegando a alcanzar grados bajo cero, con probabilidad de nieve, y
veranos muy calurosos y secos, con temperaturas de hasta 44 °C.
2.1.2.2 Entorno
El término municipal de Priego de Córdoba está dedicado casi en su totalidad a
la agricultura del olivar para la elaboración del aceite, del que ha obtenido la
denominación de origen, en parte debido a la climatología y geología que la
rodea. Con un relieve muy accidentado, y un clima que han propiciado su
desarrollo, aunque también dedicado al cultivo del cereal, leguminosas y otros
cultivos (Fig.2.4).
Posee una orografía montañosa entre los olivares milenarios y centenarios que
conforman el jardín que rodea a esta Comarca, un olivar peculiar, de sierra
resistente a las adversidades climatológicas del tiempo.
Figura 2.4. Olivar en Priego de Córdoba

ELIZABETH CARACUEL PAREJA
9
2.2 ESTUDIO DE MERCADO
Con el objetivo de encontrar un hueco en el mercado, hay que evaluar las
diferentes empresas de la zona que trabajan en el sector del olivar, ya que son
las que mejor podrían abastecer al laboratorio para poder desarrollar sus
funciones. También hay que tener en cuenta la competencia directa que tendría
la implantación del laboratorio en Priego de Córdoba, es decir, hay que evaluar
la presencia de otros laboratorios que puedan estar trabajando para las mismas
empresas que podrían ser potenciales clientes de la nueva empresa.
2.2.1 Almazaras y cooperativas
Priego de Córdoba
- Aceites Barranco La Palma, S.L.
- Almazara de Muela, S.L.
- S.C.A. Ntra. Sra. De La Cabeza
- S.C.A. Olivarera La Purísima
- Almazara Gomeoliva
- S.C.A. Olivarera Ntra. Sra. del Carmen (Zamoranos)
- Sucesores de Morales Morales, S.L. (Zamoranos)
Almedinilla
- Aceites Fuente Grande, S.A.
- Manuel Molina Muñoz e Hijos, S.L.
- S.C.A. Ntra. Sra. del Carmen (Brácana)
- S.L. Aceites el Vizconde
Carcabuey
- Aceite Aroden Hispania, S.L.
- Marín Serrano El Lagar, S.L.
- S.C.A. Almazaras de la Subbética

ELIZABETH CARACUEL PAREJA
10
2.2.2 Competencia directa
Laboratorios Oleosur
Laboratorios Oleosur S.L., están especializados en análisis de calidad y pureza
del aceite de oliva y en rendimiento industrial de la aceituna cuyo objetivo es
ofrecer un servicio innovador y de calidad al sector aceitero.
Optimum Quality
Optimum Quality es una Empresa de Base Tecnológica cuya principal actividad
se desarrolla en el área de la Seguridad y Calidad Alimentaria. Están
especializados en la realización de análisis de calidad y pureza para aceituna y
aceites, además de en sistemas de Gestión y Seguridad Alimentaria.

3 MEMORIA

ELIZABETH CARACUEL PAREJA
12
3.1 INTRODUCCIÓN
En este apartado, se realizará una revisión de las características principales del
cultivo, las etapas del proceso de elaboración de aceite de oliva, clasificación y
la normativa aplicable.
3.1.1 Marco contextual
Ley 5/2011, de 6 de octubre, del olivar de Andalucía. BOJA núm.205, de 19
de octubre de 2011.
Extracto de la mencionada ley:
Exposición de motivos:
I
El olivar es el agrosistema más representativo y simbólico de Andalucía, y desde
los poderes públicos y el conjunto de la sociedad andaluza es considerado como
estratégico. Enraizado en el territorio de nuestra comunidad desde su prehistoria,
el olivo silvestre se doméstica en época fenicia. A partir de entonces el paisaje
de olivar ha dado forma tanto a las campiñas como a las sierras andaluzas. La
importancia del cultivo del olivar ha estado impulsada por la intervención de los
hombres, que han encontrado en su conformación como bosque ordenado y en
su excelente adaptación a las muy diversas y pobladas comarcas andaluzas
motivos más que suficientes para una expansión discontinua, pero prolongada e
inacabada, de este cultivo, a lo que habría que añadir la importancia social y
económica de la transformación y distribución de sus producciones y el
aprovechamiento de sus subproductos. Por todo ello, puede decirse que, si hay
un cultivo arraigado en la cultura milenaria de Andalucía, ese es sin duda el
olivar, que ha sido fuente de inspiración literaria, musical y pictórica de nuestros
artistas, además de seña de identidad de muchos de los grandes movimientos
sociales que se han desarrollado en nuestra región.
En la actualidad, Andalucía mantiene, desde un punto de vista económico, un
claro liderazgo mundial en el olivar, resultando ser un elemento imprescindible
de cohesión social y territorial de sus comarcas que posee, además, un alto valor
medioambiental. Así, representa la tercera parte del olivar europeo; produce el
40% del aceite y el 20% de la aceituna de mesa en el mundo; es lugar de asiento

ELIZABETH CARACUEL PAREJA
13
de más de ochocientas almazaras, más de doscientas entamadoras, unas treinta
y cinco extractoras de orujo y casi seiscientas envasadoras de aceite; constituye
la principal actividad de más de trescientos pueblos andaluces en los que viven
más de doscientas cincuenta mil familias de olivareros, y proporciona más de
veintidós millones de jornales al año.
La superficie de olivar en Andalucía es aproximadamente de un millón quinientas
mil hectáreas, el 60% de la superficie oleícola española y el 30% de la superficie
europea, que se distribuye por todas las provincias andaluzas, pero que adquiere
una importancia relevante en la provincia de Jaén, el sur de la de Córdoba, el
noroeste de la de Granada, el norte de la de Málaga y el sudeste de la de Sevilla,
que conforman el denominado «Eje del olivar de Andalucía». La producción de
aceite de oliva en la Comunidad Autónoma fluctúa en torno al millón de
toneladas, y la de aceituna de mesa alrededor de las cuatrocientas mil toneladas.
El valor de la producción de aceite de oliva y de la aceituna de mesa percibido
por los olivicultores supone aproximadamente el 24% de la producción en valor
de la rama agraria andaluza.
II
Las políticas sectoriales han impulsado, y deben continuar promoviendo, un
olivar rentable, eficiente, competitivo y sostenible.
La modernización del sistema industrial a partir de la adhesión de España a las
Comunidades Europeas ha sido de una importancia manifiesta. El apoyo público
al sector mediante las líneas de ayuda a la industrialización, transformación y
comercialización modernizó el sistema industrial y, junto al plan de mejora de la
calidad del aceite de oliva y al esfuerzo del sector, dio lugar a una elaboración
adecuada para la obtención de aceites de magnífica calidad. La mejora en la
recepción de la aceituna, la disminución de los tiempos de atrojamiento, el lavado
del producto, el sistema continuo y el almacenamiento en acero inoxidable han
supuesto la consecución de una mayor parte de los aceites de las categorías
virgen y virgen extra, en contraste con tiempos pretéritos, en los que la mayoría
del aceite era lampante, había que refinarlo y el mercado estaba ocupado por el
denominado entonces «aceite puro de oliva», mezcla de aceite refinado
encabezado con aceite virgen. En la mejora de la calidad de los aceites hay que

ELIZABETH CARACUEL PAREJA
14
considerar también como un elemento fundamental la actividad de las
denominaciones de origen que se han constituido en Andalucía en los últimos
años.
Por otro lado, las nuevas plantaciones orientadas a la búsqueda de la
productividad espacial y temporal y a la mecanización de la recolección, junto
con el riego de olivares tradicionales, son la base del espectacular aumento de
la producción oleícola. Estos nuevos olivares, junto a la gran expansión de
almazaras y entamadoras plenamente integradas en la vía de la modernización,
representan hoy una sólida plataforma tecnológica de futuro que debe
impulsarse de un modo firme y decidido. Para ello, son precisas acciones
políticas que garanticen el desarrollo de la investigación, la innovación y la
formación, la promoción de la calidad tanto para la salud como para el consumo,
la vertebración del sector en asociaciones interprofesionales eficientes, la
promoción de estructuras de comercialización bien integradas y adecuadamente
dimensionadas; en suma, de instrumentos para una modernización permanente
del sector.
Por otra parte, el riesgo de abandono de los olivares menos productivos pone de
manifiesto la relevancia de las funciones no comerciales de este sector, tales
como la provisión de bienes públicos y de productos saludables y de calidad y el
mantenimiento de la población y de los sistemas locales de producción, así como
la vigilancia de los territorios, a lo que habría que añadir la contribución de este
cultivo a la lucha contra la erosión, a la prevención y reducción de la incidencia
de incendios forestales, a la fijación de notables cantidades de dióxido de
carbono (CO2) que ayuden a mitigar el cambio climático, a la preservación de
paisajes agrarios tradicionales y al mantenimiento de la diversidad biológica.
III
Existe una demanda social, contrastada en numerosos estudios y en las últimas
reformas de la política agrícola común (PAC), para que la agricultura en general
y el olivar en particular generen bienes y servicios públicos, de utilidad no solo
para los agricultores, sino también para el conjunto de la sociedad rural y para
los habitantes del medio urbano.

ELIZABETH CARACUEL PAREJA
15
Las administraciones públicas, los titulares de las explotaciones olivareras y
todos los agentes vinculados al sector del olivar, en un marco de colaboración y
conforme a lo establecido en el Plan Director del Olivar, deben promover
actuaciones públicas y privadas que garanticen el derecho de la sociedad sobre
estos bienes públicos, evitando actuaciones que los mermen, y promoviendo
acciones que los provean en mayor medida.
Con ese objetivo, los poderes públicos deben emprender acciones para
garantizar el desarrollo sostenible de los territorios de olivar, teniendo en cuenta
su carácter multifuncional y poniendo en valor los diversos productos y servicios
que el olivar ofrece. Se requiere, por tanto, una acción positiva, integral,
multidisciplinar y coordinada por parte de los agentes públicos y privados
afectados, con objeto de promover la competitividad y sostenibilidad de los
territorios olivareros y del sector oleícola en su conjunto, considerando los
aspectos económicos, ambientales, sociales y culturales.
La actual política agrícola de la Unión Europea contiene varios instrumentos,
especialmente dentro del Reglamento (CE) núm. 1698/2005 del Consejo, de 20
de septiembre de 2005, relativo a la ayuda al desarrollo rural a través del Fondo
Europeo Agrícola de Desarrollo Rural (FEADER), que pueden contribuir a
avanzar hacia una correcta provisión de bienes públicos por parte del olivar y
que están contenidos en el Programa de Desarrollo Rural Sostenible de
Andalucía para el periodo 2010-2014, siguiendo las líneas de actuación de la Ley
45/2007, de 13 de diciembre, para el desarrollo sostenible del medio rural, cuyo
objetivo fundamental es alcanzar, en los ámbitos rurales de España, un
desarrollo económico y social, de manera que aumente la calidad de vida de la
población, se logre un mayor progreso social asociado a la mejora de la renta
agraria y no abandone el territorio y a su vez se realice de forma sostenible,
objetivo que coincide con el olivar y su cultivo en Andalucía al ocupar aquel gran
parte de su medio rural.
En este sentido, el Estatuto de Autonomía para Andalucía determina, en su
artículo 185, que corresponde a la Comunidad Autónoma la gestión, planificación
y ejecución de los fondos europeos y, en general, de los que se canalicen a
través de los programas europeos asignados a la Comunidad, en especial los
aprobados en aplicación de los criterios de convergencia o derivados de la

ELIZABETH CARACUEL PAREJA
16
situación específica de Andalucía, posibilitando que aquellos puedan ser
modulados con criterios sociales y territoriales, dentro del respeto a las normas
europeas aplicables.
IV
El Estatuto de Autonomía para Andalucía, en su artículo 48.3.a), establece que
corresponde a la Comunidad Autónoma de Andalucía la competencia exclusiva,
de acuerdo con las bases y la ordenación de la actuación económica general, y
en los términos de lo dispuesto en los artículos 38, 131 y 149.1.11.ª, 13.ª, 16.ª,
20.ª y 23.ª de la Constitución, en materia de ordenación, planificación, reforma y
desarrollo de los sectores agrario, ganadero y agroalimentario, y, de forma
especial, la mejora y ordenación de las explotaciones agrícolas, ganaderas y
agroforestales, así como el desarrollo rural integral.
Esta ley debe suponer el paso definitivo para garantizar el mantenimiento y
mejora del olivar tradicional en un contexto de provisión múltiple de servicios
económicos, sociales, ambientales y culturales. Debe asegurar, promover y
valorizar un patrimonio acumulado por Andalucía durante centenares de años,
que es seña de identidad, de pertenencia y de liderazgo.
La Ley se plantea los siguientes objetivos: a) avanzar en la eficiencia de nuestros
territorios y del sector del olivar de forma equitativa y sostenible, b) ser un
instrumento esencial para el asentamiento de las personas, la generación de
empleo, un mayor progreso del medio rural y de sus habitantes y una superior
calidad de vida, y la cohesión social y territorial, c) orientar nuestros productos
hacia el mercado y propiciar estabilidad al sector, d) impulsar la mejora del
modelo productivo, en base a la industria agroalimentaria y la transparencia en
la cadena de valor, e) aumentar nuestra capacidad de respuesta ante los
cambios de los mercados y los cambios tecnológicos, y ante las amenazas
climáticas, y f) contribuir al mantenimiento de la singularidad de los paisajes y de
los efectos ambientales positivos asociados al olivar.
La presente ley tiene en consideración la evolución de la política agrícola y de
desarrollo rural de la Unión Europea, y en especial de las políticas de apoyo a
las rentas agrarias, de regulación de mercados, de calidad y seguridad
alimentaria y de medio ambiente. Asimismo, en la elaboración de esta ley se ha

ELIZABETH CARACUEL PAREJA
17
tenido en consideración lo dispuesto en la Ley 12/2007, de 26 de noviembre,
para la promoción de la igualdad de género en Andalucía, respecto al principio
de igualdad de oportunidades entre mujeres y hombres.
La ley se articula en un título preliminar y cinco títulos ordinarios.
El Título Preliminar determina el objeto de la ley, el espacio geográfico de su
aplicabilidad, que el texto legislativo denomina como «territorio de olivar», el
ámbito de actuación desde la perspectiva sectorial que emana del cultivo y sus
productos, los fines que propugna para conseguir el objeto de la ley y los
principios inspiradores en que se basa el texto normativo.
El Título I determina los instrumentos de gestión sostenible del olivar, siendo el
Plan Director del Olivar el principal de ellos. Este se configura como el principal
instrumento de coordinación e integración de las acciones a desarrollar por el
conjunto de actores implicados en los territorios y, en particular, de los que
participan activamente en la cadena de valor del olivar y sus productos.
Contiene también este título dos tipos contractuales para una mejor gestión de
los territorios del olivar. El primero de ellos es el Contrato Territorial de Zona
Rural, contemplado en la Ley de desarrollo rural sostenible, por el cual un grupo
de explotaciones olivareras de una zona determinada, de forma voluntaria,
suscribe contratos con la Administración para unos determinados fines,
principalmente de carácter productivo y ambiental. El segundo tipo es el Contrato
Territorial de Explotación, por el cual la persona titular de una explotación
olivarera se obliga a unos compromisos respecto a su actividad, y la
Administración a otros, especialmente en cuanto a la concesión de ayudas,
compensaciones y servicios.
Se configura además un sistema propio para el sector olivarero de información y
apoyo a la toma de decisiones, de manera que haya la máxima transparencia en
el sector y la accesibilidad de las personas interesadas al conocimiento de la
situación de los subsectores del olivar y del mercado.
Por último, este título instituye el Consejo Andaluz del Olivar como órgano asesor
de la Administración.

ELIZABETH CARACUEL PAREJA
18
El Título II, de medidas para el fomento del olivar, contiene el conjunto de
actuaciones a realizar por el sector productor con el apoyo de los poderes
públicos para mantener y mejorar la renta de los olivicultores. Acciones de
reestructuración del olivar existente, mejora de los regadíos y nuevas puestas en
riego, siempre con sistemas ahorradores de agua, aprovechamiento energético
de los residuos del olivar, apoyo al olivar de producción integrada y ecológica,
fomento de la multifuncionalidad de las explotaciones, investigación y
capacitación son, entre otras, medidas de apoyo al olivar que tradicionalmente
se cultiva en Andalucía.
En todo el conjunto de medidas propuestas como en las correspondientes al
título siguiente, la ley propone la cooperación público-privada para conseguir
sinergias de desarrollo.
El Título III hace referencia a la transformación, promoción y comercialización de
los productos del olivar. Por el conjunto de estudios científicos realizados en
varias universidades y centros de investigación nutricional de varios países son
conocidas las condiciones del aceite de oliva como alimento saludable y también
las condiciones de excelencia gastronómica del aceite de oliva y de la aceituna
de mesa de calidad, ambos, productos constituyentes de la dieta mediterránea,
que tiene una alta consideración, entre otras, nutricional. Por ello, en el título se
articulan medidas para seguir mejorando la estructura productiva de las
almazaras y del sector entamador, aun reconociendo el importante esfuerzo
realizado por el sector y la Administración a partir de la adhesión de España a
las Comunidades Europeas en 1986, que deriva en que actualmente las
condiciones de elaboración del aceite de oliva y de la aceituna de mesa son
buenas y se logran, por ejemplo, unos aceites de magnífica calidad y unas
aceitunas de mesa reconocidas internacionalmente.
Se fomenta la calidad de los productos derivados del olivar de manera que se
obtenga, además de aceite y aceituna de mesa de calidad contrastada,
productos de máxima excelencia gastronómica.
De otra parte, en el título se articulan medidas de promoción y comercialización
del aceite de oliva y de la aceituna de mesa, en aras de incrementar su consumo
tanto en el ámbito nacional como internacional.

ELIZABETH CARACUEL PAREJA
19
El Título IV articula medidas para la coordinación y la vertebración del sector,
considerando el Plan Director del Olivar como el instrumento básico para dicho
fin. Tanto en el sector productor como en el transformador y el comercializador
son necesarias actuaciones de integración para eliminar y reducir costes de
producción, concentrar la oferta de productos ante la situación de preeminencia
de los operadores de compra en el mercado y articular actuaciones
promocionales para una mejor comercialización del aceite de oliva y de la
aceituna de mesa. A estos fines se articula fundamentalmente el título, al
considerar la vertebración del sector y la acción colectiva, en los que el
cooperativismo agroalimentario y otras formas de asociacionismo tienen una
función primordial, como un elemento esencial para el buen fin del cultivo del
olivar en Andalucía.
El Título V, por último, se refiere a la tutela del patrimonio natural olivarero y a la
cultura del aceite de oliva, y hace referencia a su importancia histórica en
Andalucía y a la necesidad que existe de darle, por los valores que comporta, un
tratamiento específico, sin perjuicio de la cobertura general que la Ley de
Patrimonio Histórico de Andalucía da a este tipo de bienes.
3.1.2 El olivo y su fruto
El olivo (olea europaea) es un árbol que pertenece a la familia botánica
Oleaceae, y dentro de esa familia es la única especie con fruto comestible.
Procede del acebuche (Olea europaea var. Sylvestris), que es la misma planta
en estado silvestre. El olivo se encuentra expandido por toda la cuenca
mediterránea, siendo Andalucía la zona olivarera por excelencia a nivel mundial.
3.1.2.1 Características principales
Sus hojas son verde oscuro por el haz, con un característico brillo debido a la
existencia de una gruesa cutícula y blanquecinas por el envés, simples, de forma
lanceolada y bordes enteros. Es un árbol perenne y las hojas suelen vivir dos o
tres años.
Las flores son blancas y pequeñas dispuestas en racimos que florecen hacia el
mes de mayo.
El tronco es grueso y su corteza grisácea.

ELIZABETH CARACUEL PAREJA
20
El fruto es la aceituna, una pequeña drupa ovoide de sabor muy amargo, color
verde amarillento, pulpa oleosa una vez que ha llegado a la madurez y con un
hueso que encierra la semilla.
El olivo, la aceituna, el aceite y sus derivados, han sido muy importantes para
muchas civilizaciones a lo largo de la historia de la humanidad, pues han
ayudado a su supervivencia, principalmente como fuente de alimentación y
salud.
El olivo se ramifica a escasa altura y sus ramas tienden a dispersarse. Requiere
mucho sol y rehuye la humedad. El suelo debe ser profundo pero seco.
Es un árbol centenario. La producción se inicia al octavo o noveno año y va
incrementándose al aumentar la edad hasta llegar a los 35 o 40 años. Su
productividad se estabiliza entre los 65 y 80 años y a partir de esa edad los
rendimientos decrecen.
Son árboles de crecimiento lento, pero tienen una gran cualidad: cavando
cuidadosamente y empaquetando sin dañar sus raíces, se puede trasladar sea
cual sea su edad; de hecho, hay empresas dedicadas a la venta de olivos adultos
para la decoración.
Alcanza una altura de 20 a 25 metros en edad adulta y con condiciones
favorables, y un diámetro de 8 a 10 metros. No obstante, lo habitual es que se
practique una poda cada dos o tres años, manteniéndolo entre los 4 y los 8
metros de altura.
La madera del olivo es muy dura pero fácil de pulir e ideal para tallar utensilios.
Es en definitiva un árbol que aúna la elegancia con la robustez.
En cuanto al fruto, cabe citar que las aceitunas destinadas para la obtención de
aceite se recolectan maduras (Normalmente desde finales de diciembre hasta
mediados de febrero, dependiendo de la zona), y las destinadas al consumo a
medio madurar (Finales de noviembre y principios de diciembre).
3.1.2.2 Condiciones de cultivo
El cultivo del olivo se inició hace más de 6000 años. Sus orígenes se encuentran
en el Oriente Medio, en una zona que comprendería Creta, Egipto, Palestina,

ELIZABETH CARACUEL PAREJA
21
Siria, Irak e Irán. Conforme se fueron implantando los olivos, el aceite fue
sustituyendo a las grasas animales que se utilizaban en la alimentación y
también para otros usos variados.
En España se cultiva especialmente en la cuenca mediterránea y en las regiones
de clima suave. Actualmente, el 95% del terreno mundial cultivado se encuentra
en el área mediterránea.
El olivo es sensible al frío y al calor extremo, el clima más adecuado es el
templado y generalmente se cultiva en zonas donde la temperatura media del
mes más cálido está comprendida entre 22ºC y 33ºC y la del mes más frío es
superior a 4 ºC.
En España las zonas olivareras reciben una media de precipitaciones que oscila
entre los 500 y los 650 litros/m2. Se adapta bien a los suelos pobres, pero prefiere
los suelos calcáreos y permeables, así como las zonas de poca pendiente y
agradece que la separación entre unas plantas y otras sea alrededor de los 10
metros. La poda ha de favorecer la recolección y para ello se debe rebajar la
altura de las primeras ramas, reducir el diámetro de la copa y airear las ramas
para facilitar la floración. El abonado y los diferentes cuidados del árbol también
influyen y son los que propician su mayor o menor producción.
3.1.3 El aceite de oliva
3.1.3.1 Situación actual
Como se ha comentado anteriormente, el país que más olivos posee es España
(más de 300 millones de olivos), seguido a gran distancia por Grecia e Italia y un
poco más atrás se encuentran situadas Túnez, Turquía, y Siria. Es el primer país
en el ranking de producción mundial de aceite de oliva, con una producción
media anual de 700.000-800.000 toneladas, llegando a alcanzar 1.000.000 en
recientes campañas, y superando ampliamente esta cantidad como es el caso
de la campaña 2001-2002 con una producción de 1.300.000 toneladas.
También es el primer país exportador mundial.
A nivel nacional, el mayor volumen de producción de aceite de oliva se encuentra
en la región de Andalucía (aprox. un 80%), seguido de Castilla la Mancha (6-

ELIZABETH CARACUEL PAREJA
22
7%), Extremadura (5%) y Cataluña (4%), estando el resto (4%) integrado
principalmente por las comunidades Valenciana y Aragonesa.
En España, la propiedad de los olivares está bastante dispersa, hay gran
cantidad de personas con explotaciones de tamaño pequeño o mediano, a
menudo situadas en zonas de baja productividad, de tal forma que en la mayoría
de los casos, los rendimientos obtenidos con la venta de la aceituna no son más
que una pequeña ayuda para los propietarios, que buscan mantener las
explotaciones a menudo heredada de antepasados.
3.2 PROCESO DE ELABORACIÓN DEL ACEITE DE OLIVA
A) En la explotación agrícola
3.2.1 Recolección de aceituna
La recolecta de la aceituna es la labor agrícola que culmina el proceso anual de
cultivo y cuidados del olivo. El objetivo del cultivo del olivo es obtener el mayor
número posible de aceitunas en las mejores condiciones y calidad posibles para
maximizar así el beneficio del producto final, sea aceite de oliva o aceitunas de
mesa. La técnica de recogida, el tipo de producto y calidad a que se destina el
fruto, las herramientas, la cantidad y experiencia de la mano de obra, el estado
de madurez del fruto, los cuidados en el manipulado y transporte son aspectos
esenciales para la labor de recogida de aceituna.
De la elección del sistema de recolección de aceituna depende el daño que
pueda sufrir el fruto, ya que las roturas ocasionadas serán el lugar de penetración
de hongos que deterioran el aceite y la puerta de salida de grasa en el proceso
de lavado. También hay que valorar el daño que puede causar el sistema de
recolección al olivo, ya que es la fuente de producción de los siguientes años.
3.2.1.1 Ordeño
Se trata de recoger la aceituna del árbol de forma manual, desprendiéndola de
las ramas con las manos sin que entre en contacto con el suelo. Esta técnica es
la más utilizada cuando el fruto se destina a aceituna de mesa. El fruto no debe
estar muy maduro, en otro caso el fruto habrá caído al suelo y el ordeño ya no
tiene sentido. Requiere mucho esfuerzo en mano de obra y una mínima o nula

ELIZABETH CARACUEL PAREJA
23
mecanización. Con esta técnica se preservan las aceitunas de cualquier tipo de
agresión por manipulación mecánica y el olivo no sufre daño alguno. Para la
elaboración de ciertos aceites de oliva vírgenes extra de máxima calidad también
se usa esta técnica ya que preserva el fruto intacto de cara a la extracción de su
aceite en la almazara, sin embargo, los costes en mano de obra son muy altos,
pues es una técnica muy laboriosa (Fig. 3.1).
Figura 3.1. Ordeño de Aceituna
3.2.1.2 Vareo
En esta técnica, el olivicultor se sirve de una vara de longitud variable (hasta 4
metros) para derribar la aceituna cimbreando y golpeando las ramas del olivo.
La aceituna se derriba sobre lienzos o bien cae al suelo. En esta técnica el
olivo sufre bastante, pues pierde muchas de sus ramas y hojas en el golpeo y
generalmente se considera que acentúa la vecería (alternancia de la
producción, un año mucho, otro poco). Sin embargo, es un método muy
extendido por su alta productividad (Fig.3.2).
Figura 3.2. Vareo de Aceituna

ELIZABETH CARACUEL PAREJA
24
3.2.1.3 Vibración
En la actualidad se realiza de forma mecánica con vibradores autopropulsados,
pinzas vibradoras acopladas al tractor o con vibradoras motorizadas de mano
que actúan sobre el tronco o rama del olivo haciendo caer las aceitunas. Es una
evolución del “sacudido” manual en el que se agitaba manualmente las ramas
para desprender la aceituna. La maquinaria vibradora más evolucionada aúna
un elemento vibrador autopropulsado y un paraguas recolector para recoger la
aceituna conforme se desprende. También se está investigando para adaptar los
peines vibradores de cítricos para el derribo de la aceituna. La vibración
multidireccional es la más eficaz (Fig.3.3).
Este método es el más utilizado en la actualidad, pues disminuye los costos de
recolección y reduce los daños que se ocasionan al olivo con el sistema de vareo.
Va siempre acompañado del vareo, debido a la cantidad considerable de
aceitunas que no caen al suelo simplemente usando la vibración.
Figura 3.3. Vibración con paraguas del olivo.
3.2.1.4 Recogida del Suelo
Sea como fuere el derribo de la aceituna (natural por maduración, vareo o
vibración), si no hay un mecanismo para recoger la aceituna como mantas o
lienzos, o que el propio mecanismo recolector tenga un paraguas para capturar
la aceituna, la aceituna caerá al suelo de donde hay que recogerla para su
transporte a la almazara. Si se va a realizar la recogida del suelo, es de vital

ELIZABETH CARACUEL PAREJA
25
importancia aplanar y cercar el suelo bajo y alrededor del olivo para facilitar la
labor posterior en la recogida, esta preparación previa se llama “hacer suelos”.
El método de recogida de suelo más rudimentario es la recogida manual una por
una de las aceitunas, muy tediosa. También se puede barrer, aspirar o soplar la
aceituna para amontonarla y luego cargarla en los cestos a mano. En cualquier
caso, al caer al suelo la aceituna se mezcla con polvo, arena, piedras y otras
impurezas que dañan el fruto iniciando su degradación, además la aceituna de
suelo requiere en la almazara un proceso de criba y lavado para eliminar la
suciedad y las piedras, además de las ramas y hojas, por lo que la aceituna de
suelo es de inicio una aceituna que generará un aceite de oliva de menor calidad.
B) En la almazara
3.2.2 Recepción de la aceituna
Las aceitunas recolectadas se transportan a la almazara para su molienda. Los
métodos de transporte deben mantener intacta la integridad del fruto, por lo que
se debe evitar transportar aceituna ensacada.
El transporte de las aceitunas del suelo y del árbol debe hacerse por separado,
en medios de transporte limpios; y recepcionarse en la almazara diferenciadas.
Para obtener un aceite de calidad, la aceituna debe procesarse cuanto antes,
como máximo en las 24h. siguientes a su recogida.
Primero se clasifica el fruto recepcionado. Esta ordenación se suele hacer en
función de la variedad, del grado de maduración, del estado sanitario. Después
se procede a realizar el proceso de limpieza se eliminan mediante una criba los
residuos que traen las aceitunas, como son las hojas, tallos, tierra o pequeñas
piedras. Posteriormente las aceitunas se lavan con agua fría para eliminar otras
impurezas como el polvo, barro o restos de herbicidas (Fig. 3.4).
Una vez limpia se transporta la aceituna a la báscula para pesarla, tomar
muestras que faciliten la trazabilidad y análisis. El peso de la aceituna en la
báscula una vez limpia permite valorar el importe que se le pagará al agricultor
por la aceituna vertida en la tolva de recepción. De la báscula la aceituna pasa a
la tolva de almacenamiento donde esperará su turno para iniciar el proceso de
molturación. El almacenamiento en esta tolva ha de ser el mínimo posible y

ELIZABETH CARACUEL PAREJA
26
siempre menor de 24 horas para evitar la fermentación de la aceituna y por lo
tanto la disminución de la calidad del aceite generado.
Figura 3.4 Recepción y lavado de aceituna en una almazara.
3.2.3 Molienda
Con esta operación se inicia el proceso de separación del aceite de los tejidos
del fruto; el objetivo es romper las células de la pulpa para que el aceite salga
libremente de las vacuolas, permitiendo la formación de gotas mayores que
puedan ser separadas de las otras fases. Por lo tanto, durante la molienda se
produce la rotura de la aceituna mediante unos martillos de acero inoxidable u
otro metal inerte hasta que sean inferiores al diámetro de la criba. El diámetro de
paso de la criba influye directamente en la liberación del aceite y por lo tanto en
el rendimiento en aceite de la aceituna molturada, también influye en el tiempo
de molturado.
La molturación por martillos rotatorios sustituye a los molinos de piedra de los
sistemas tradicionales. Los molinos de piedra (Fig.3.5), además de la rotura de
la aceituna propiciaban la agregación de las gotitas de aceite liberado por la
fricción y el lento arrastre de la pasta oleosa por lo que en el sistema tradicional
no era necesario el batido de la masa antes de la extracción. En cambio, la
molturación a alta velocidad dispersa las gotitas de aceite en la pasta oleosa y
las deja en un tamaño poco apropiado para su extracción directa y se hace
necesaria una fase de batido para aglutinar las gotas de aceite en otras más
grandes y facilitar la extracción.
Normalmente, los molinos de martillo suelen ser de eje horizontal. En ellos se va
introduciendo la aceituna de forma automatizada y recibe el impacto de los

ELIZABETH CARACUEL PAREJA
27
martillos metálicos que giran a gran velocidad. El molino cuenta con una criba de
un diámetro determinado, cuando las partículas lo alcanzan, pasan, si no, siguen
dentro del molino hasta que lo hagan (Fig.3.6).
Figura 3.5. Molino de piedra
Figura 3.6. Molino de martillos industrial
3.2.4 Batido
La operación de batido tiene como objetivo reunir las gotas dispersas de aceite
agrupándolas formando unas de mayor tamaño para facilitar la separación
sólido-líquido. Esta fase se lleva a cabo mediante termobatidoras, máquinas

ELIZABETH CARACUEL PAREJA
28
dotadas de unas palas que mueven, de manera lenta pero continua, la pasta en
unos recipientes semicilíndricos.
Las batidoras son depósitos horizontales o verticales cuya misión es calentar y
voltear la masa para propiciar la agregación de las gotas de aceite por fricción y
facilitar de esa manera la extracción del aceite (Fig.3.7). En el batido, es
necesario calentar la masa para disminuir la viscosidad del aceite y así, facilitar
la formación de la fase oleosa y su extracción. Para calentar la masa batida, las
termobatidoras poseen una camisa por la que se hace circular agua caliente que
a su vez facilita la extracción del aceite. Según la variedad de la aceituna, el
índice de madurez con el que se haya recogido, y el diámetro de la criba de
molturación, pueden aparecer las llamadas “pastas difíciles”, en estos casos está
autorizado el uso de microtalco natural y otras sustancias que mejoran las
características de las pastas y que se adicionan al principio del batido.
Para obtener aceites de calidad, la temperatura de la masa de batido no debe
superar los 27ºC - 29ºC, y el tiempo de batido no superar los 90 minutos. Lo
recomendable sería trabajar a 25ºC durante 60 minutos, ya que esta temperatura
es suficiente para facilitar la extracción del aceite, disminuir su viscosidad y
favorecer la formación de la fase oleosa. A temperaturas y tiempos inferiores se
producen fenómenos de intercambio entre los constituyentes de la masa molida
que tienen escasa repercusión en los caracteres organolépticos. A temperaturas
y tiempos superiores aumentan las velocidades de reacción de todos los
procesos de intercambio y degradación de los componentes lábiles,
responsables de los aromas frutados frescos y armónicos, apareciendo flavores
a calentado, cocido o quemado, y que en función del tiempo de preparación
puede también detectarse como fondo aromático y regusto, la sensación a
madera, orujo o alpechín, a causa de los intercambios con los otros
constituyentes de la masa.
En los sistemas continuos de dos fases, al no incorporar agua en el batido, se
aumenta el tiempo empleado para conseguir la homogeneidad deseada. Si se
aumenta en exceso el tiempo de batido se provoca una disminución de
polifenoles, k225, y de la estabilidad, aumentando la intensidad del color, e incluso
aparecen olores anómalos por excesivo tiempo de contacto del aceite con el
agua de vegetación.

ELIZABETH CARACUEL PAREJA
29
Figura 3.7. Termobatidora de eje vertical
3.2.5 Extracción
La extracción es la fase en la que se separa el aceite, contenido en la masa que
sale de la batidora, del resto de componentes de la aceituna; agua, hueso, piel,
etc (Fig.3.8).
Figura 3.8. Elaboración de aceite de oliva.

ELIZABETH CARACUEL PAREJA
30
3.2.5.1 Extracción por presión
Es el sistema tradicional. La pasta molida se coloca entre capachos de esparto
y se somete a presión, para expulsar el mosto oleoso (mezcla de aceite y agua),
que se decanta para luego obtener por flotación, por diferencia de densidad, el
aceite flotante. Actualmente, este sistema está en deshuso.
3.2.5.2 Extracción continua por centrifugación
Es el más utilizado a nivel industrial por resultar más eficiente y económico. En
él, la pasta, una vez batida, se centrifuga, siempre sin añadir productos químicos
ni calor. Gracias a la distinta densidad de los líquidos, los productos extraídos se
separan en niveles, quedando en la parte más exterior de la centrifugadora los
más pesados (agua y orujo) y, más hacia el centro, los menos pesados (aceite).
Se denominan sistemas continuos porque al contrario que el tradicional en el que
la prensa tiene que parar para descargarse y cargarse de nuevo, en estos la
obtención es continua, la centrífuga no para de ser alimentada por un extremo y
por el contrario sale el aceite y los subproductos.
Hay diferentes procesos de centrifugado:
- Centrifugación de 3 fases
La extracción de aceite de oliva se efectúa mediante una operación que se
realiza en plantes modulares que trabajan en continuo.
En ella, las fases líquidas se separan de la fase sólida por medio de la aplicación
de fuerzas centrífugas que aumentan las diferencias entre las densidades
especiales del aceite, agua de vegetación y la materia sólida, realizándose en
una centrifugadora horizontal, que consiste en un rotor cilíndrico-cónico y un
rascador helicoidal del eje hueco, que gira coaxialmente en el interior del mismo.
La diferencia de velocidad entre uno y otro hace que los sólidos se adosen en la
parte interior del rotor y sean arrastrados hacía un extremo por el tornillo sinfín.
Los líquidos (el aceite y la fase acuosa) forman anillos concéntricos más
interiores según su densidad y salen al exterior por diferentes conducciones.
Para una mejor separación de los componentes es necesario agregar agua a la
pasta que viene de la batidora. La cantidad de agua que se adiciona influye en

ELIZABETH CARACUEL PAREJA
31
el rendimiento de la extracción. Del decantador centrífugo salen dos tipos de
líquidos, uno de color verde formado por el aceite y algo de fase acuosa y el otro
de color marrón constituido por la fase acuosa y algo de aceite. Dependiendo del
tamaño las máquinas en el mercado pueden procesar entre 0.5 y 4.0 toneladas
de aceitunas/hora.
El mayor problema del proceso a tres fases es el elevado volumen de agua
residual que se genera (alpechín) (1,2-1,3 litros/Kg. de aceituna procesada),
debido a la adición de agua que se realiza a la pasta de aceitunas antes de entrar
en el decantador horizontal.
Estos líquidos tienen una alta contaminación y una elevada concentración en
polifenoles que impiden su depuración por medios convencionales, con lo cual
se deben de depositar en balsas para su evaporación con el aumento en costos,
problemas de salud e impacto visual.
- Centrifugación de 2 fases:
Debido al elevado volumen de agua residual generada por el proceso a tres
fases, surgieron los nuevos modelos de decantadores centrífugos horizontales
que eran capaces de separar la fase aceitosa de la pasta de aceituna sin requerir
la adición de agua caliente. Esto implicaba que no se produjeran vertidos líquidos
teniendo el residuo sólido un mayor contenido en humedad.
En la extracción del aceite de oliva por el sistema de centrifugación de dos fases
se obtienen separadamente, por una parte, el aceite y, por otra, una pasta fluida
(alperujo) que contiene el agua de vegetación y la pulpa.
Las dos primeras fases se realizan de forma similar a las del sistema de tres
fases. La fase aceitosa se separa de la aceituna (pulpa + agua de vegetación)
por efecto de la fuerza centrífuga que aumenta la diferencia entre las densidades
específicas del aceite y el alperujo (materia sólida más el agua de vegetación).
Esta operación se realiza en un decantador centrífugo horizontal similar al
empleado en el sistema de tres fases.
En este caso no es necesario agregar agua para conseguir una mejor separación
del aceite. Como la fase oleosa que sale del decantador puede llevar partículas

ELIZABETH CARACUEL PAREJA
32
sólidas, es conveniente disponer a la salida del aceite un tamiz vibratorio para
separar los pequeños trozos de pulpa o hueso.
La fase oleosa se somete a una centrifugación en una centrifuga de platos, en
esta operación se agrega cierta cantidad de agua al aceite con objeto de lavarlo
y poder retirarle parte de la humedad que traía del decantador, esta agua es el
único vertido que se genera en este proceso de extracción del aceite.
El centrifugado a dos fases tiene un rendimiento superior que el de tres fases
debido al hecho de que en el nuevo sistema no se añade agua a la pasta y se
evita la formación de emulsiones aceite/agua. No produce apenas vertidos, sólo
se origina un pequeño volumen del agua añadida en el lavado del aceite en la
centrifugación, evitándose los problemas de medioambientales que se originan
por las grandes dimensiones que tienen que tener las balsas de evaporación. La
calidad del aceite producido es superior a la obtenida en el proceso de tres fases,
debido a la gran cantidad de poli fenoles y/o di fenoles. Ello implica que dicho
aceite es más estable durante el almacenamiento como lo indican los elevados
valores del tiempo de estabilidad oxidativa que se tienen en los aceites obtenidos
por este sistema (RuedaOliva,2005).
3.2.5.3 Extracción de aceite de orujo
La obtención del aceite que aún permanece en el producto secundario sólido
(orujo) que se obtiene después de la molturación y prensado de la aceituna se
realiza normalmente, al igual que en todos los aceites de semillas, con el uso de
disolventes, normalmente hexano. Este proceso no se realiza en las almazaras
sino en las así llamadas orujeras a donde se lleva este producto.
3.2.6 Proceso de refinado
Es el proceso químico y físico al que se someten los aceites de oliva vírgenes
que por sus características organolépticas y de acidez no son aptos para el
consumo, y los aceites de orujo de aceituna. La refinación puede llevarse a cabo
por dos vías: física o química; esta última se recomienda para aceites de no muy
elevada acidez para evitar las pérdidas por neutralización.
Durante el refinado se realizan las siguientes operaciones:

ELIZABETH CARACUEL PAREJA
33
Depuración o desgomado. Con la adición de agua y ácido fosfórico se eliminan
algunos compuestos como los fosfolípidos y las gomas que pueden ser
causantes de la formación de mucílagos y cuerpos gomosos en los envases, de
apariencia poco atractiva para el consumidor. En el proceso se eliminan
inevitablemente otros compuestos deseables como algunas proteínas.
Neutralización. Es el proceso para la eliminación de la acidez. Con sosa se
separan los ácidos grasos libres del aceite, dejando la acidez prácticamente a
cero. Este proceso implica la eliminación de una gran parte de los carotenos,
precursores de la Vitamina A, presente en los aceites vírgenes.
Decoloración. Consiste en la eliminación de las sustancias responsables del
color. Estas son principalmente los carotenos y la clorofila. El aceite se trata con
arcilla activada a 100 ºC.
Desodorización. Se eliminan mediante calor todos los compuestos
responsables de los olores y sabores desagradables. Como la mayoría de estos
compuestos son volátiles, se eliminan haciendo pasar el aceite por una corriente
de aire a 200-250 ºC. De forma secundaria también se eliminan los ácidos grasos
libres que pudieran quedar. En este proceso se eliminan también la mayoría de
tocoferoles, esteroles y polifenoles, y sus importantes propiedades antioxidantes.
Winterización. Este proceso tiene como objeto eliminar los triglicéridos de punto
de fusión más elevado, con lo que se consigue que el aceite se mantenga líquido
a temperaturas más bajas. Simplemente se enfría el aceite con agua, los
triglicéridos a eliminar se solidifican y después se separan. Su función, al igual
que la Depuración, es mejorar la apariencia del producto.
Este proceso se realiza en las refinerías, no en las almazaras.
3.2.7 Clasificación y almacenamiento
El aceite es almacenado en la almazara antes de que sea envasado y llegue al
mercado en perfectas condiciones de consumo. En esta fase experimentará
cambios favorables ya que pierde parte de los aromas amargos mientras gana
en matices y sensaciones dulces y agradables.
Los aceites deben clasificarse y almacenarse de manera diferenciada en función
de su calidad y procedencia (suelo o árbol).

ELIZABETH CARACUEL PAREJA
34
El aceite obtenido se almacena en trujales o depósitos en las bodegas de las
almazaras (Fig.3.9). El material de éstos debe ser inerte y la temperatura idónea
debe estar entre los 18ºC - 20ºC, para permitir una maduración de los aceites sin
favorecer la oxidación y es necesario realizar una escrupulosa limpieza de los
depósitos antes de su llenado.
Las impurezas acumuladas en los fondos deben ser eliminadas, pues junto a la
tierra y polvo, se encuentran sustancias proteicas y azucaradas que fermentan
fácilmente, comunicando al aceite olores y sabores desagradables a moho-
humedad, avinagrado, sucio, borras, tierra, etc.; y también elevan la acidez. Por
ello, los depósitos deben purgarse periódicamente, incluso el trasvasado a otro
depósito limpio.
Figura 3.9. Almacenamiento del aceite en tanques de acero inoxidable.
3.2.8 Envasado del aceite de oliva
Antes del envasado de los aceites de oliva vírgenes (virgen extra ó virgen), el
aceite es filtrado para eliminar restos de impurezas y humedad que pueda llevar
(también pueden envasarse sin previo filtrado, llamado “aceite en rama” o "aceite
sin filtrar", lo que provocará que con el tiempo se formen pequeños posos en el
fondo del envase).
En las plantas envasadoras, se llenan y etiquetan los envases de aceite de oliva
virgen extra, o virgen, para su comercialización.

ELIZABETH CARACUEL PAREJA
35
Para que quede listo para su consumo no sirve cualquier material para envasar
aceite. Los más utilizados son PET (plástico), vidrio, lata y cartón revestido
(Fig.3.10). Es recomendable la utilización de envases opacos que no dejen pasar
la luz para no alterar las excelencias que esconde el aceite en su interior.
Figura 3.10. Envasado de aceite en PET
3.3 CLASIFICACIÓN DE LOS ACEITES DE OLIVA
La legislación de la Unión Europea ha establecido en el Convenio Internacional
del Aceite de Oliva del año 1986 la denominación de "Aceite de oliva" únicamente
al aceite procedente del fruto del olivo, con exclusión de aquellos obtenidos por
disolventes o mediante mezcla con aceites de otra naturaleza distinta. Esta
denominación tampoco es aplicable al aceite obtenido del orujo de la aceituna.
La legislación distingue los siguientes tipos de aceite de oliva para su
comercialización, que se consideran a continuación y cuyo esquema general se
recoge en la figura 3.11.
3.3.1 Aceites de oliva vírgenes
Es aquel aceite obtenido exclusivamente por procedimientos mecánicos o por
otros medios físicos en condiciones, especialmente térmicas, que no produzcan
la alteración del aceite, que no haya tenido más tratamiento que el lavado, la
decantación, la centrifugación y el filtrado. Es un producto natural que conserva
el sabor, los aromas y las vitaminas de la fruta.
Tiene la personalidad de la zona de donde procede. A su vez se clasifican en:
Aceite de Oliva Virgen Extra: sinónimo de máxima calidad, es aquel que
conserva intactas todas sus características sensoriales y propiedades para la
salud. Se puede considerar zumo de aceitunas sin aditivos ni conservantes, ha

ELIZABETH CARACUEL PAREJA
36
de tener una acidez menor de 0,8% y presentar unas características sensoriales
agradables e identificables. La puntuación organoléptica, dada por un panel de
cata cualificado, debe ser igual o superior a 6,5 puntos.
Aceite de Oliva Virgen: sin la palabra “Extra” sigue siendo zumo de aceituna sin
aditivos ni conservantes, pero presenta algún defecto sensorial por mínimo que
sea. Su acidez ha de ser menor del 2% y la puntuación obtenida por un panel de
cata cualificado debe ser igual o superior a 5,5 puntos. En otras palabras, los
defectos deben ser prácticamente imperceptibles para el consumidor.
Aceite de Oliva Lampante: Denominado así porque en la antigüedad era el que
se empleaba para las lámparas de aceite que iluminaban los hogares. Es un
aceite de oliva virgen con una acidez libre, expresada en ácido oleico, superior
a 3,3º y con un gusto defectuoso
3.3.2 Aceite de oliva no vírgenes
Dichos aceites se clasifican:
Aceite de Oliva Refinado: Es el obtenido por refinación de aceites de oliva
vírgenes y con acidez no superior a 0,3º, mediante técnicas de refinado que no
producen alteración en la estructura glicerídica inicial. Es bastante habitual, que
proceda de aceite de oliva lampante que no puede envasarse para consumo
humano por su elevada acidez, pero que tras pasar por este proceso de
refinación química se logra disminuir la acidez por debajo de 0,3º y puede
envasarse y distribuirse sin ningún problema.
Aceite de Oliva: Aceite de oliva constituido por una mezcla de aceite de oliva
refinado y de aceites de oliva vírgenes distintos del aceite lampante, cuya acidez
libre no podrá ser superior a 1º y cuyas otras características son conformes a las
establecidas para esta categoría. Este es el aceite de oliva que se puede
comprar a un precio menor (aunque en los últimos tiempos, la diferencia de
precio con el aceite de oliva virgen es menor) y es el que más se vende en
España.
Aceite de Orujo de Oliva Crudo: Aceite obtenido mediante a partir del orujo de
oliva mediante tratamiento con disolvente o por medios físicos, o que

ELIZABETH CARACUEL PAREJA
37
corresponda, con excepción de algunas características determinadas, a un
aceite de oliva lampante.
Aceite de orujo de oliva refinado: Aceite obtenido mediante refino de aceite de
orujo de oliva crudo, cuya acidez libre no podrá ser superior a 0,3º y cuyas otras
características son conformes a esta categoría.
Aceite de orujo de oliva: Aceite constituido por una mezcla de aceite de orujo
de oliva refinado y de aceites de oliva vírgenes distintos del lampante, cuya
acidez libre no podrá ser superior a 1º.
Figura 3.11. Clasificación de los aceites de oliva. Fuente: Oleo Estepa

ELIZABETH CARACUEL PAREJA
38
3.4 NORMATIVA VIGENTE PARA ANÁLISIS DE ACEITE
Nº Diario Oficial: L_ _ Página_ Fecha_ _ _
►M1 Reglamento (CEE) no 3682/91 de la Comisión de 17 de diciembre de 1991
L 349 36 18.12.1991
►M2 Reglamento (CEE) no 1429/92 de la Comisión de 26 de mayo de 1992 L
150 17 2.6.1992
►M3 Reglamento (CEE) no 1683/92 de la Comisión de 29 de junio de 1992 L 176
27 30.6.1992
►M4 Reglamento (CEE) no 1996/92 de la Comisión de 15 de julio de 1992 L 199
18 18.7.1992
►M5 Reglamento (CEE) no 3288/92 de la Comisión de 12 de noviembre de 1992
L 327 28 13.11.1992
►M6 Reglamento (CEE) no 183/93 de la Comisión de 29 de enero de 1993 L 22
58 30.1.1993
►M7 modificado por el Reglamento (CEE) no 826/93 de la Comisión de 6 de abril
de 1993 L 87 6 7.4.1993
►M8 Reglamento (CEE) no 620/93 de la Comisión de 17 de marzo de 1993 L 66
29 18.3.1993
►M9 Reglamento (CE) no 177/94 de la Comisión de 28 de enero de 1994 L 24
33 29.1.1994
►M10 Reglamento (CE) no 2632/94 de la Comisión de 28 de octubre de 1994 L
280 43 29.10.1994
►M11 Reglamento (CE) no 656/95 de la Comisión de 28 de marzo de 1995 L 69
1 29.3.1995
►M12 Reglamento (CE) no 2527/95 de la Comisión de 27 de octubre de 1995 L
258 49 28.10.1995
►M13 Reglamento (CE) no 2472/97 de la Comisión de 11 de diciembre de 1997
L 341 25 12.12.1997

ELIZABETH CARACUEL PAREJA
39
►M14 Reglamento (CE) no 282/98 de la Comisión de 3 de febrero de 1998 L 28
5 4.2.1998
►M15 Reglamento (CE) no 2248/98 de la Comisión de 19 de octubre de 1998 L
282 55 20.10.1998
►M16 Reglamento (CE) no 379/1999 de la Comisión de 19 de febrero de 1999 L
46 15 20.2.1999
►M17 Reglamento (CE) no 455/2001 de la Comisión de 6 de marzo de 2001 L
65 9 7.3.2001
►M18 Reglamento (CE) no 2042/2001 de la Comisión de 18 de octubre de 2001
L 276 8 19.10.2001
►M19 Reglamento (CE) no 796/2002 de la Comisión de 6 de mayo de 2002 L
128 8 15.5.2002
►M20 Reglamento (CE) no 1989/2003 de la Comisión de 6 de noviembre de 2003
L 295 57 13.11.2003
►M21 Reglamento (CE) no 702/2007 de la Comisión de 21 de junio de 2007 L
161 11 22.6.2007
Últimas modificaciones
►M22 Reglamento (CE) 640/2008, de 4 de julio de 2008, L 178, de 05.07.2008
►M23 Reglamento (UE) 61/2011, de 24 de enero de 2011, L 023, de 27.01.2011
►M24 Reglamento de Ejecución (UE) 299/2013, de 26 de marzo de 2013, L 090,
de 28.03.2013
►M25 Reglamento (UE) nº 1310/2013 del Parlamento Europeo y del Consejo de
17 de diciembre de 2013 L 347 865 20.12.2013
►M26 Reglamento Delegado (UE) 2015/1830, de 8 de julio de 2015, L 266, de
13.10.2015
►M27 Reglamento de Ejecución (UE) 2015/1833, de 12 de octubre de 2015 L
266, de 13.10.2015

ELIZABETH CARACUEL PAREJA
40
►M28 Reglamento (UE) 2016/791 Parlamento Europeo y del Consejo de 11 de
mayo de 2016 L 135 1 24.5.2016
►M29 Reglamento Delegado (UE) 2016/1166 de la Comisión de 17 de mayo de
2016 L 193 17 19.7.2016
►M30 Reglamento Delegado (UE) 2016/1226 de la Comisión de 4 de mayo de
2016 L 202 5 28.7.2016
Rectificado por:
►C1 Rectificación, DO L 347 de 28.11.1992, p. 69 (91/2568)
►C2 Rectificación, DO L 176 de 20.7.1993, p. 30 (93/183)
►C3 Rectificación, DO L 63 de 4.3.1998, p. 34 (97/2472)
►C4 Rectificación, DO L 189 de 27.6.2014, p. 261 (1308/2013)
►C5 Rectificación, DO L 130 de 19.5.2016, p. 45 (1308/2013)
►C6 Rectificación, DO L 222 de 17.8.2016, p. 114 (2016/791)
A continuación, se recoge un extracto del mencionado Reglamento.
Artículo 1
1. Se considerarán aceites de oliva vírgenes, con arreglo a las letras a) y b) del
punto 1 del anexo del Reglamento no 136/66/CEE, los aceites cuyas
características se ajusten a las indicadas, respectivamente, en los puntos 1 y 2
del anexo I del presente Reglamento.
2. Se considerará aceite de oliva lampante, con arreglo a la letra c) del punto 1
del anexo del Reglamento no 136/66/CEE, el aceite cuyas características se
ajusten a las indicadas en el punto 3 del anexo I del presente Reglamento.
3. Se considerará aceite de oliva refinado, con arreglo al punto 2 del anexo del
Reglamento no 136/66/CEE, el aceite cuyas características se ajusten a las
indicadas en el punto 4 del anexo I del presente Reglamento.
4. Se considerará aceite de oliva compuesto exclusivamente por aceites de oliva
refinados y aceites de oliva vírgenes, con arreglo al punto 3 del anexo del

ELIZABETH CARACUEL PAREJA
41
Reglamento no 136/66/CEE, el aceite cuyas características se ajusten a las
indicadas en el punto 5 del anexo I del presente Reglamento.
5. Se considerará aceite de orujo de oliva crudo, con arreglo al punto 4 del anexo
del Reglamento no 136/66/CEE, el aceite cuyas características se ajusten a las
indicadas en el punto 6 del anexo I del presente Reglamento.
6. Se considerará aceite de orujo de oliva refinado, con arreglo al punto 5 del
anexo del Reglamento no 136/66/CEE, el aceite cuyas características se ajusten
a las indicadas en el punto 7 del anexo I del presente Reglamento.
7. Se considerará aceite de orujo de oliva, con arreglo al punto 6 del anexo del
Reglamento no 136/66/CEE, el aceite cuyas características se ajusten a las
indicadas en el punto 8 del anexo I del presente Reglamento.
Artículo 2
1. Las características de los aceites, previstas en el Anexo I, se determinarán
empleando los métodos de análisis siguientes:
— para determinar los ácidos grasos libres, expresados en porcentaje de ácido
oleico, el método recogido en el Anexo II,
— para determinar el índice de peróxidos, el método recogido en el Anexo III,
— para determinar el contenido de ceras, el método recogido en el anexo IV,
— para determinar el contenido de esteroles, el método recogido en el Anexo V,
— para determinar el eritrodiol + uvaol, el método recogido en el Anexo VI,
— para determinar el porcentaje de monopalmitato de 2-glicerilo, el método
recogido en el anexo VII,
— para el análisis espectrofotométrico, el método recogido en el Anexo IX,
— para determinar la composición de ácidos grasos, el método recogido en los
Anexos X «A» y X «B»,

ELIZABETH CARACUEL PAREJA
42
— para determinar los solventes halogenados volátiles, el método recogido en el
Anexo XI,
— para la valoración de las características organolépticas de los aceites de oliva
vírgenes, el método recogido en el Anexo XII, que se aplicará de conformidad
con el apartado 2,
— para determinar los estigmastadienos, el método recogido en el Anexo XVII,
— para determinar la composición de los triglicéridos de ECN42, el método que
figura en el Anexo XVIII,
— para determinar el contenido de alcoholes alifáticos, el método recogido en el
anexo XIX.
2. La comprobación de las características organolépticas de los aceites de oliva
vírgenes por parte de las autoridades nacionales o sus representantes correrá a
cargo de paneles de catadores autorizados por los Estados miembros.
Las características organolépticas del aceite de oliva mencionado en el apartado
1 se considerarán conformes a la categoría declarada si un panel de catadores
autorizado por el Estado miembro interesado confirma dicha clasificación.
En caso de que el panel autorizado no confirme tal declaración en lo que
respecta a las características organolépticas de la categoría de aceite de oliva
declarada, las autoridades nacionales o sus representantes ordenarán que se
proceda, a petición del interesado, a dos análisis contradictorios por otros
paneles autorizados; al menos uno de ellos deberá ser realizado por un panel de
cata autorizado por el Estado miembro productor.
Las características en cuestión se considerarán conformes a las declaradas si
ambos análisis contradictorios confirman la clasificación declarada. En caso
contrario, los gastos generados por los análisis contradictorios, sin perjuicio de
otras posibles sanciones, correrán por cuenta del interesado.

ELIZABETH CARACUEL PAREJA
43
ANEXOS
Índice
Anexo I: Características de los aceites de oliva
Anexo I ter: Esquema de decisiones
Anexo II: Determinación de los ácidos grasos libres, método en frío
Anexo III: Determinación del índice de peróxidos
Anexo IV: Determinación del contenido de ceras mediante cromatografía de
gases con columna capilar
Anexo V: Determinación de la composición y del contenido de esteroles mediante
cromatografía de gases con columna capilar
Anexo VI: Determinación del eritrodiol y uvaol
Anexo IX: Prueba espectrofométrica en el ultravioleta
Anexo XV: Contenido de aceite de los orujos de aceituna
Anexo XVI: Determinación del índice de yodo
Anexo XVII: Método para la determinación de estigmastadienos en los aceites
vegetales
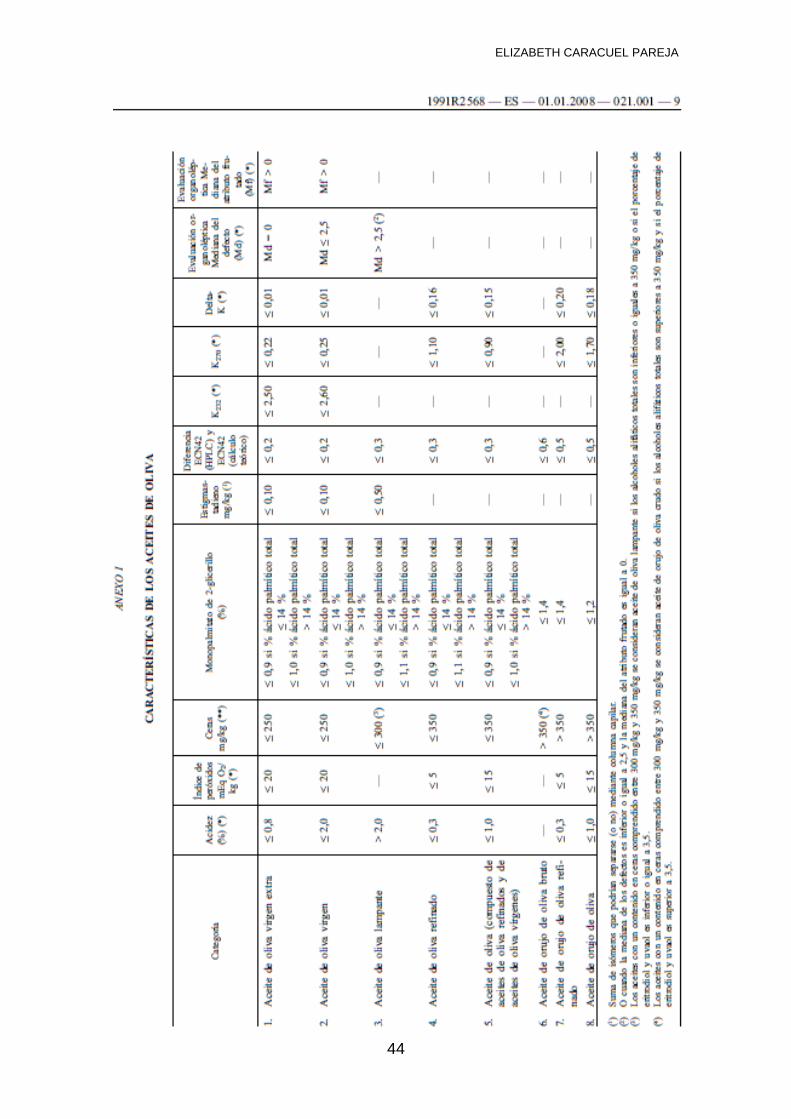
ELIZABETH CARACUEL PAREJA
44

ELIZABETH CARACUEL PAREJA
45

ELIZABETH CARACUEL PAREJA
46
ANEXO I ter
ESQUEMA DE DECISIONES PARA LA COMPROBACIÓN DE LA
CONFORMIDAD DE UNA MUESTRA DE ACEITE DE OLIVA CON LA
CATEGORÍA DECLARADA
El análisis de la conformidad de un aceite de oliva o de orujo de oliva con la
categoría declarada podrá efectuarse:
a) mediante la realización, en cualquier orden, de los análisis previstos para
comprobar que se reúnen las características mencionadas en el anexo I, o bien
b) mediante la realización, según el orden indicado en el esquema de decisiones,
de los análisis previstos en el mismo, hasta la adopción de una de las decisiones
mencionadas en dicho esquema.
Los análisis relativos a los contaminantes, necesarios para comprobar la
conformidad con las normas de la Comunidad Europea, se realizarán por
separado.
El esquema de decisiones se aplicará a todas las categorías de aceite de oliva y
de orujo de oliva. Se compone de cuadros numerados del 1 al 11, que deberán
seguirse en el orden indicado en el cuadro general en función de la categoría de
aceite declarada.
Para la lectura de los cuadros general a 11:
— la línea doble (=) indica el camino que debe seguirse en caso de conformidad
(respuesta positiva) con las condiciones previstas en la casilla anterior; la línea
de puntos (…) indica, por el contrario, el camino que debe seguirse en caso de
no conformidad,
— los títulos de las casillas que figuran en los cuadros 1 a 11 se refieren a los
análisis previstos en el presente Reglamento, según las correspondencias
mencionadas en el apéndice 1 del presente anexo,
— las letras que figuran en los círculos de decisión negativa de los cuadros 1 a
11 remiten a datos indicativos mencionados en el apéndice 2 del presente anexo
y no implican por sí mismas ninguna obligación de continuar los análisis o la
certeza de la mencionada presunción.

ELIZABETH CARACUEL PAREJA
47
Cuadro 1
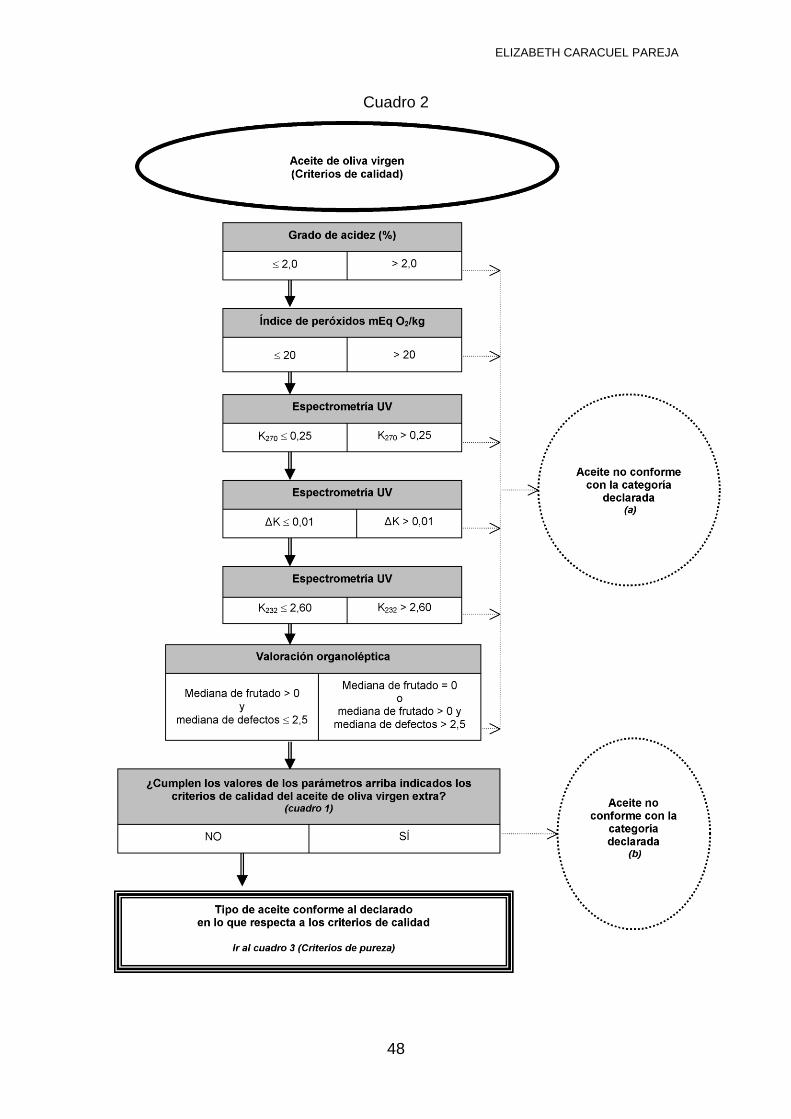
ELIZABETH CARACUEL PAREJA
48
Cuadro 2

ELIZABETH CARACUEL PAREJA
49
Cuadro 3
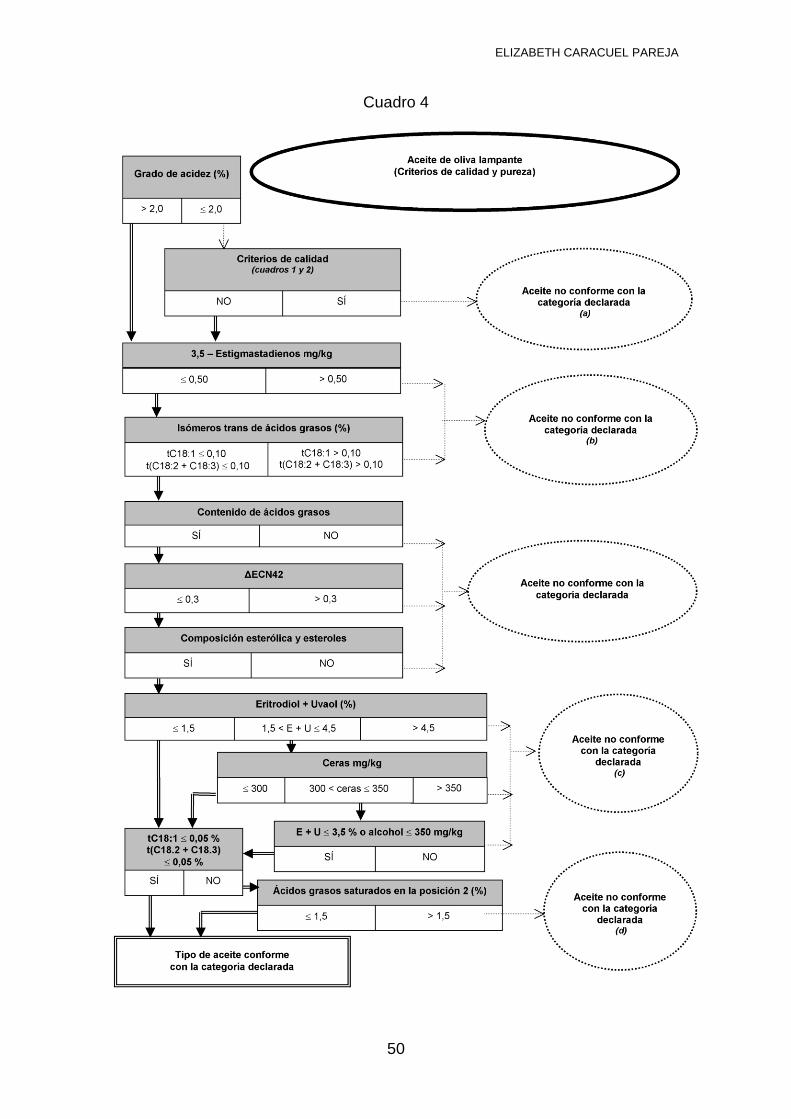
ELIZABETH CARACUEL PAREJA
50
Cuadro 4
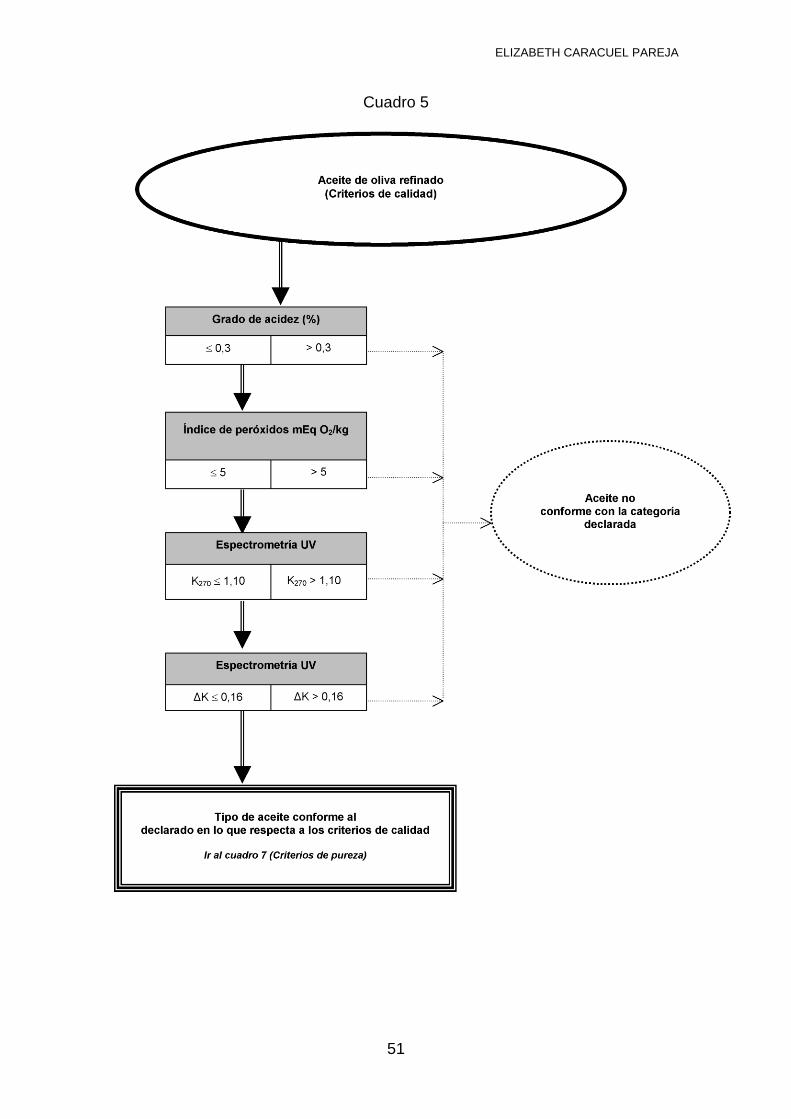
ELIZABETH CARACUEL PAREJA
51
Cuadro 5

ELIZABETH CARACUEL PAREJA
52
Cuadro 6
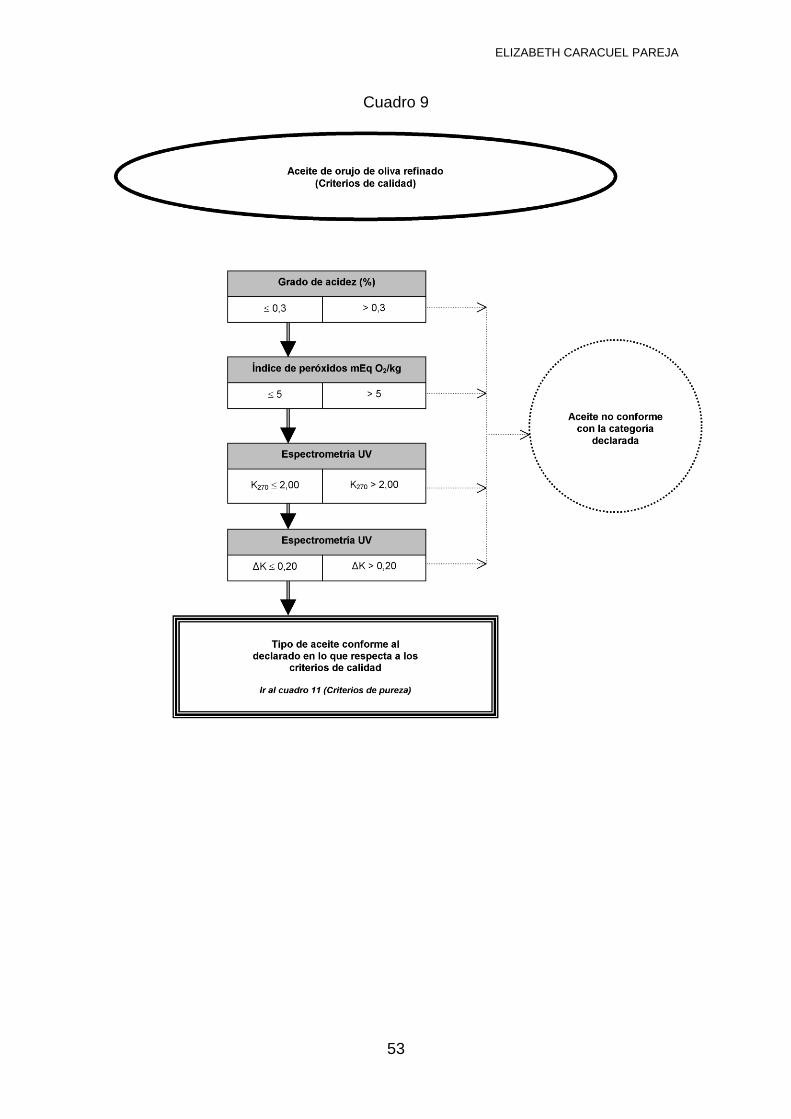
ELIZABETH CARACUEL PAREJA
53
Cuadro 9

ELIZABETH CARACUEL PAREJA
54
ANEXO II
DETERMINACIÓN DE LOS ÁCIDOS GRASOS LIBRES, MÉTODO EN FRÍO
1. OBJETO
Determinar los ácidos libres en los aceites de oliva. El contenido en ácidos
grasos libres se expresa mediante la acidez calculada según el método
convencional.
1.1. Principio
Disolución de la muestra en una mezcla de disolventes y valoración de los ácidos
grasos libres mediante una solución etanólica de hidróxido potásico.
1.2. Reactivos
Todos los reactivos deben ser de calidad analítica reconocida y el agua utilizada
debe ser agua destilada o de una pureza equivalente.
1.2.1. Mezcla de éter dietílico y etanol de 95 % (V/V), en proporción de
volumen 1:1.
Nota: El éter dietílico es muy inflamable y puede formar peróxidos explosivos.
Debe utilizarse tomando especiales precauciones. Debe neutralizarse
exactamente en el momento de su utilización con la solución de hidróxido
potásico (1.2.2) en presencia de 0,3 ml de la solución de fenolftaleína (1.2.3) por
cada 100 ml de mezcla.
Nota: Si no es posible utilizar éter dietílico, puede sustituirse por una mezcla de
disolventes formada por etanol y tolueno. Si fuera necesario, el etanol podría
sustituirse, a su vez, por 2-propanol.
1.2.2. Solución etanólica valorada de hidróxido potásico, = 0,1 M o, en caso
necesario, = 0,5 M. Nota 1.
Debe conocerse, y comprobarse inmediatamente antes de su utilización, la
concentración exacta de la solución etanólica de hidróxido potásico. Debe
utilizarse una solución que haya sido preparada por lo menos cinco días antes y
decantada en un frasco de vidrio marrón cerrado con tapón de goma. La solución
debe ser incolora o de color amarillo paja.

ELIZABETH CARACUEL PAREJA
55
Nota: Se puede preparar una solución incolora y estable de hidróxido potásico
de la manera siguiente: Llevar a ebullición, y mantener ésta a reflujo durante una
hora, 1 000 ml de etanol con 8 g de hidróxido potásico y 0,5 g de virutas de
aluminio. Destilar inmediatamente.
Disolver en el destilado la cantidad requerida de hidróxido potásico. Dejar
reposar durante varios días y decantar el líquido claro sobrenadante,
separándolo del precipitado de carbonato potásico.
La solución también puede prepararse de la manera siguiente sin efectuar la
destilación: añadir 4 ml de butilato de aluminio a 1 000 ml de etanol y dejar
reposar la mezcla durante algunos días. Decantar el líquido sobrenadante y
disolver en él la cantidad necesaria de hidróxido potásico. La solución está lista
para ser utilizada.
1.2.3. Solución de 10 g/l de fenolftaleína en etanol de 95-96 % (V/V) o
solución de 20 g/l de azul alcalino (en caso de aceites de oliva muy coloreados)
en etanol de 95-96 % (V/V).
1.3. Material
Material habitual de laboratorio, y en particular:
1.3.1. Balanza analítica
1.3.2. Matraz erlenmeyer de 250 ml de capacidad
1.3.3. Bureta de 10 ml de capacidad, con graduación de 0,05 ml
1.4. Procedimento
1.4.1. Preparación de la muestra para la prueba
La determinación se efectuará en una muestra filtrada. Si el contenido global de
humedad e impurezas es inferior al 1 %, se utilizará la muestra tal cual.
1.4.2. Muestra para la prueba
Tomar la muestra, según el grado de acidez previsto, de acuerdo con el cuadro
siguiente:

ELIZABETH CARACUEL PAREJA
56
Grado de acidez
previsto
Peso de la muestra,gr Precisión de la pesada
de la muestra, gr
<1 20 0,05
1 a 4 10 0,02
4 a 15 2,5 0,01
15 a 75 0,5 0,001
> 75 0,1 0,0002
Pesar la muestra en el matraz erlenmeyer (1.3.2)
1.4.3 Determinación
Disolver la muestra (1.4.2) en 50 a 150 ml de la mezcla de éter dietílico y etanol
(1.2.1), previamente neutralizada.
Valorar, agitando, con la solución de hidróxido potásico de 0,1 M (1.2.2) (véase
nota 2) hasta el viraje del indicador (la coloración rosa de la fenolftaleína debe
permanecer al menos durante 10 segundos).
Nota 1: La solución etanólica valorada de hidróxido potásico (1.2.2) puede
sustituirse por una solución acuosa de hidróxido potásico o sódico siempre que
el volumen de agua añadido no provoque una separación de las fases.
Nota 2: Si la cantidad necesaria de la solución de hidróxido potásico de 0,1 M
supera los 10 ml, debe utilizarse una solución de 0,5 M.
Nota 3: Si la solución se enturbia durante la valoración, añadir una cantidad
suficiente de la mezcla de disolventes (1.2.1) para que la solución se aclare.
1.5 Expresión de la acidez en porcentaje de ácido oleico
La acidez, expresada en porcentaje de ácido oleico es igual a:
siendo:
V: volumen en ml de la solución valorada de hidróxido potásico utilizada.

ELIZABETH CARACUEL PAREJA
57
c: concentración exacta, en moles por litro, de la solución de hidróxido potásico
utilizada.
M: peso molecular del ácido en que se expresa el resultado (ácido oleico= 282).
P: peso en gramos de la muestra utilizada.
Se tomará como resultado la media aritmética de dos determinaciones

ELIZABETH CARACUEL PAREJA
58
ANEXO III
DETERMINACIÓN DEL ÍNDICE DE PERÓXIDOS
1. OBJETO
La presente norma describe un método para la determinación del índice de
peróxidos de los aceites y grasas.
2. ÁMBITO DE APLICACIÓN
La presente norma es aplicable a los aceites y grasas animales y vegetales.
3. DEFINICIÓN
El índice de peróxidos es la cantidad (expresada en miliequivalentes de oxígeno
activo por kg de grasa) de peróxidos en la muestra que ocasionan la oxidación
del yoduro potásico en las condiciones de trabajo descritas.
4. PRINCIPIO
La muestra problema, disuelta en ácido acético y cloroformo, se trata con
solución de yoduro potásico. El yodo liberado se valora con solución valorada de
tiosulfato sódico.
5. APARATOS
Todo el material utilizado estará exento de sustancias reductoras u oxidantes.
Nota: No engrasar las superficies esmeriladas.
5.1. Navecilla de vidrio de 3 ml
5.2. Matraces con cuello y tapón esmerilados, de 250 ml de capacidad
aproximadamente, previamente secados y llenos de gas inerte puro y seco
(nitrógeno o, preferiblemente, dióxido de carbono).
5.3. Bureta de 25 o 50 ml, graduada en 0,1 ml.
6. REACTIVOS
6.1. Cloroformo para análisis, exento de oxígeno por borboteo de una corriente
de gas inerte puro y seco.
ELIZABETH CARACUEL PAREJA
59
6.2. Ácido acético glacial para análisis, exento de oxígeno por borboteo de una
corriente de gas inerte puro y seco.
6.3. Solución acuosa saturada de yoduro potásico, recién preparada, exenta
de yodo y yodatos.
6.4. Solución acuosa de tiosulfato sódico 0,01 N o 0,002 N valorada
exactamente; la valoración se efectuará inmediatamente antes del uso.
6.5. Solución de almidón, en solución acuosa de 10 g/l, recién preparada con
almidón soluble.
7. MUESTRA
La muestra se tomará y almacenará al abrigo de la luz, y se mantendrá
refrigerada dentro de envases de vidrio totalmente llenos y herméticamente
cerrados con tapones de vidrio esmerilado o de corcho.
8. PROCEDIMIENTO
El ensayo se realizará con luz natural difusa o con luz artificial. Pesar con
precisión de 0,001 g en una navecilla de vidrio (5.1) o, en su defecto, en un
matraz (5.2) una cantidad de muestra en función del índice de peróxidos que se
presuponga, con arreglo al cuadro siguiente:
Índice de Peróxidos que se supone
(meq de O2/kg)
Peso de la muestra problema
(en gr)
0 a 12 5 a 2
12 a 20 2 a 1,2
20 a 30 1,2 a 0,8
30 a 50 0,8 a 0,5
50 a 90 0,5 a 0,3
Abrir un matraz (5.2) e introducir la navecilla de vidrio que contenga la muestra
problema. Añadir 10 ml de cloroformo (6.1). Disolver rápidamente la muestra
problema mediante agitación. Añadir 15 ml de ácido acético (6.2) y, a
continuación, 1 ml de solución de yoduro potásico (6.3). Cerrar rápidamente el

ELIZABETH CARACUEL PAREJA
60
matraz, agitar durante 1 minuto y mantenerlo en la oscuridad durante 5 minutos
exactamente, a una temperatura comprendida entre 15 y 25 °C.
Añadir 75 ml aproximadamente de agua destilada. Valorar (agitando al mismo
tiempo vigorosamente) el yodo liberado con la solución de tiosulfato sódico (6.4)
(solución 0,002 N si se presuponen valores inferiores a 12 y solución 0,01 N si
se presuponen valores superiores a 12), utilizando la solución de almidón (6.5)
como indicador.
Efectuar dos determinaciones por muestra.
Realizar simultáneamente un ensayo en blanco. Si el resultado del ensayo en
blanco sobrepasa 0,05 ml de la solución de tiosulfato sódico 0,01 N (6.4), sustituir
los reactivos.
9. EXPRESIÓN DE LOS RESULTADOS
El índice de peróxidos (I.P.), expresado en miliequivalentes de oxígeno activo
por kg de grasa se calcula mediante la fórmula siguiente:
siendo:
V: ml de solución valorada de tiosulfato sódico (6.4) empleados en el ensayo,
convenientemente corregidos para tener en cuenta el ensayo en blanco.
N: normalidad exacta de la solución de tiosulfato sódico (6.4) empleada.
P: peso, en gramos de la muestra problema.
El resultado será la media artimética de las dos determinaciones efectuadas.

ELIZABETH CARACUEL PAREJA
61
ANEXO IV
DETERMINACIÓN DEL CONTENIDO DE CERAS MEDIANTE
CROMATOGRAFÍA DE GASES CON COLUMNA CAPILAR
1. OBJETO
El presente método describe un procedimiento para la determinación del
contenido de ceras en los aceites de oliva. Las ceras se separan en función del
número de átomos de carbono. El método puede utilizarse, en particular, para
distinguir el aceite de oliva obtenido por presión del obtenido mediante extracción
(aceite de orujo de oliva).
2. PRINCIPIO
La materia grasa, a la que se habrá añadido un patrón interno adecuado, se
fracciona mediante cromatografía en columna de gel de sílice hidratado; se
recupera la fracción eluida en primer lugar en las condiciones de ensayo (cuya
polaridad es inferior a la de los triglicéridos) y, a continuación, se analiza
directamente mediante cromatografía de gases en columna capilar.
3. MATERIAL
3.1. Matraz Erlenmeyer de 25 ml.
3.2. Columna de vidrio para cromatografía de gases, de 15,0 mm de diámetro
interior, entre 30 y 40 cm de longitud, y provista de una llave.
3.3. Cromatógrafo de gases adecuado para utilizarse con columna capilar,
dotado de un sistema de introducción directa en la columna, que incluya los
siguientes elementos:
3.3.1. Recinto termostático para las columnas, provisto de un programador
de temperatura.
3.3.2. Inyector en frío para introducción directa en la columna.
3.3.3. Detector de ionización de llama y convertidor-amplificador.
3.3.4. Registrador-integrador que pueda funcionar con el convertidor
amplificador (3.3.3), con un tiempo de respuesta inferior o igual a un segundo y
velocidad variable del papel. (También se pueden utilizar sistemas

ELIZABETH CARACUEL PAREJA
62
informatizados que contemplen la obtención de los datos de cromatografía de
gases mediante un ordenador.).
3.3.5. Columna capilar de vidrio o sílice fundida, de 8 a 12 m de longitud y de
0,25 a 0,32 mm de diámetro interior, recubierta interiormente de fase
estacionaria, con un espesor uniforme que oscile entre 0,10 y 0,30 μm. (Se
encuentran en el comercio fases estacionarias adecuadas listas para su empleo,
del tipo de la SE-52 o SE-54.).
3.4. Microjeringa para introducción directa en columna, de 10 μl de capacidad,
provista de una aguja cementada.
3.5. Vibrador eléctrico.
3.6. Rotavapor.
3.7. Horno de mufla.
3.8. Balanza analítica con una precisión de ± 0,1 mg.
3.9. Material de vidrio normal de laboratorio.
4. REACTIVOS
4.1. Gel de sílice de una granulometría comprendida entre 60 y 200 μm.
El gel de sílice debe mantenerse durante un mínimo de 4 horas en el horno a
500 °C. Tras su enfriamiento, se añade un 2 % de agua respecto a la cantidad
de gel de sílice tomada. Se agita bien para homogeneizar la masa. Se mantiene
al abrigo de la luz durante al menos 12 horas antes de su uso.
4.2. n-hexano, de calidad para cromatografía.
4.3. Éter etílico, de calidad para cromatografía.
4.4. n-heptano, de calidad para cromatografía.
4.5. Solución patrón de araquidato de laurilo al 0,1 % (m/v) en hexano (patrón
interno). (También puede utilizarse palmitato de palmitilo o estearato de
miristilo.).
4.5.1. Sudán 1 (1-fenil-azo-2-naftol).

ELIZABETH CARACUEL PAREJA
63
4.6. Gas portador: hidrógeno o helio puro, de calidad para cromatografía de
gases.
4.7. Gases auxiliares:
— hidrógeno puro, de calidad para cromatografía de gases,
— aire puro, de calidad para cromatografía de gases.
5. PROCEDIMIENTO
5.1. Preparación de la columna cromatográfica
Suspender 15 g de gel de sílice (4.1) en n-hexano (4.2) e introducirlo en la
columna (3.2). Completar la sedimentación espontánea con ayuda de un
vibrador eléctrico (3.5), a fin de obtener una capa cromatográfica más
homogénea. Hacer pasar 30 ml de n-hexano para eliminar las posibles
impurezas. Pesar exactamente con ayuda de la balanza (3.8) 500 mg de la
muestra en el matraz Erlenmeyer de 25 ml (3.1) y añadir la cantidad adecuada
del patrón interno (4.5), en función del contenido previsto de ceras; por ejemplo,
añadir 0,1 mg de araquidato de laurilo si se trata de aceite de oliva y de 0,25 a
0,50 mg si se trata de aceite de orujo.
Transferir la muestra así preparada a la columna cromatográfica, con ayuda de
dos porciones, cada una de 2 ml, de n-hexano (4.2).
Dejar fluir el disolvente hasta que se sitúe a 1 mm por encima del nivel superior
del absorbente, y a continuación hacer pasar 70 ml más de nhexano para
eliminar los n-alcanos presentes de forma natural. Comenzar entonces la elución
cromatográfica recogiendo 180 ml de la mezcla de nhexano/éter etílico en la
proporción 99:1, manteniendo un flujo aproximado de 15 gotas cada 10
segundos. La elución de la muestra debe efectuarse a una temperatura ambiente
de 22 °C ± 4 °C.
Notas:
— La mezcla n-hexano/éter etílico (99:1) debe prepararse cada día.
— Para controlar visualmente la elución correcta de las ceras, es posible añadir
a la muestra en solución 100 μl de Sudán al 1 % en la mezcla de elución. Como

ELIZABETH CARACUEL PAREJA
64
el colorante tiene una retención intermedia entre las ceras y los triglicéridos,
cuando la coloración llega al fondo de la columna cromatográfica hay que
suspender la elución por haberse eluido ya todas las ceras.
Secar la fracción resultante en el rotavapor (3.6) hasta que se haya eliminado
prácticamente todo el disolvente. Eliminar los dos últimos mililitros del disolvente
con ayuda de una débil corriente de nitrógeno; añadir a continuación de 2 a 4 ml
de n-heptano.
5.2. Cromatografía de gases
5.2.1. Operaciones preliminares
Instalar la columna en el cromatógrafo de gases (3.3), conectando el terminal de
entrada al sistema en columna y el terminal de salida al detector. Efectuar los
controles generales del cromatógrafo de gases (funcionamiento de los circuitos
de gas, eficacia del detector y del registrador, etc.).
Si la columna se utiliza por vez primera, es conveniente acondicionarla.
Hacer pasar a través de la columna una corriente ligera de gas y, a continuación,
encender el cromatógrafo de gases. Calentar gradualmente hasta alcanzar, al
cabo de unas 4 horas, la temperatura de 350 °C. Mantener esta temperatura
durante al menos 2 horas y, a continuación, ajustar el aparato a las condiciones
de trabajo [regulación del flujo del gas, encendido de la llama, conexión con el
registrador electrónico (3.3.4), regulación de la temperatura del recinto para la
columna, regulación del detector, etc.] y registrar la señal con una sensibilidad al
menos dos veces superior a la prevista para efectuar el análisis. La línea de base
deberá ser lineal, sin picos de ningún tipo ni deriva.
Una deriva rectilínea negativa indica que las conexiones de la columna no son
correctas; una deriva positiva indica que la columna no ha sido acondicionada
adecuadamente.
5.2.2. Elección de las condiciones de trabajo
En general, las condiciones de trabajo deben ser las siguientes:
— temperatura de la columna:

ELIZABETH CARACUEL PAREJA
65
20ºC/min 5ºC/min 20ºC/min
al inicio
80ºC(1’)
240ºC 325ºC
(6’)
340ºC
(10’)
— temperatura del detector: 350 °C;
— cantidad inyectada: 1 μl de la solución (2-4 ml) de n-heptano,
— gas portador: helio o hidrógeno a la velocidad lineal óptima para el gas
seleccionado (véase el apéndice),
— sensibilidad del instrumento: capaz de responder a las condiciones siguientes:
Estas condiciones pueden modificarse en función de las características de la
columna y del cromatógrafo de gases, de modo que se obtenga la separación de
todas las ceras, una resolución satisfactoria de los picos y un tiempo de retención
del patrón interno C32 de 18 ± 3 minutos. El pico más representativo de las ceras
debe haber medido al menos el 60 % del fondo de la escala.
Determinar los parámetros de integración de los picos de manera que se obtenga
una evaluación correcta de las áreas de los picos considerados.
Nota: Ante lo elevado de la temperatura final, se admite una deriva positiva que
no debe ser superior al 10 % del fondo de la escala.
5.3. Realización del análisis
Tomar 1 μl de la solución con la microjeringa de 10 μl; sacar el émbolo de la
jeringa hasta que la aguja esté vacía. Introducir la aguja en el sistema de
inyección e inyectar rápidamente después de 1 o 2 segundos; dejar pasar unos
5 segundos y extraer lentamente la aguja.
Llevar a cabo el registro hasta que las ceras se hayan eluido completamente. La
línea de base debe satisfacer siempre las condiciones exigidas.
5.4. Identificación de los picos
Identificar los distintos picos sobre la base de los tiempos de retención,
comparándolos con mezclas de ceras analizadas en las mismas condiciones y
cuyos tiempos de retención sean conocidos.

ELIZABETH CARACUEL PAREJA
66
La figura presenta un cromatograma de las ceras de un aceite de oliva virgen.
5.5. Determinación cuantitativa
Calcular con ayuda del integrador las áreas de los picos correspondientes al
patrón interno y a los ésteres alifáticos de C40 a C46.
Determinar el contenido en ceras de cada uno de los ésteres, en mg/kg de
materia grasa, aplicando la siguiente fórmula:
siendo:
Ax = el área del pico de cada éster, en milímetros cuadrados
As = el área del pico del patrón interno, en milímetros cuadrados
ms = la masa del patrón interno que se ha añadido, en miligramos
m = la masa de la muestra tomada para la determinación, en gramos.
6. EXPRESIÓN DE LOS RESULTADOS
Indicar la suma de los contenidos de las distintas ceras de C40 a C46, en mg/kg
de materia grasa (ppm).
Nota: Los componentes que se deben cuantificar hacen referencia a los picos
con un número par de carbonos, comprendidos entre los ésteres C40 y C46,
según el ejemplo del cromatograma de las ceras el aceite de oliva indicado en la
figura siguiente. Si el éster C46 aparece dos veces, para identificarlo conviene
analizar la fracción de las ceras de un aceite de orujo de oliva en que el pico C46
sea fácil de identificar por ser claramente mayoritario.
Los resultados deben expresarse con una cifra decimal.

ELIZABETH CARACUEL PAREJA
67
ANEXO V
DETERMINACIÓN DE LA COMPOSICIÓN Y DEL CONTENIDO DE
ESTEROLES MEDIANTE CROMATOGRAFÍA DE GASES CON COLUMNA
CAPILAR
1. OBJETO
El presente método describe un procedimiento para la determinación del
contenido de esteroles en las materias grasas, expresado como contenido de
cada uno de los esteroles analizados y como contenido total de esteroles.
2. PRINCIPIO
Saponificación de la materia grasa, a la que se habrá añadido α-colestanol como
patrón interno, con una solución etanólica de hidróxido potásico; a continuación,
extracción del insaponificable con éter etílico.
Separación de la fracción de esteroles del insaponificable extraído mediante
cromatografía en placa de gel de sílice básica; los esteroles recuperados del gel
de sílice se transforman en trimetilsililéteres y se analizan mediante
cromatografía de gases con columna capilar.
3. MATERIAL
3.1. Matraz de 250 ml, provisto de refrigerante de reflujo con juntas
esmeriladas.
3.2. Embudos de separación de 500 ml.
3.3. Matraces de 250 ml.
3.4. Equipo completo de cromatografía en capa fina, utilizando placas de vidrio
de 20 × 20 cm.
3.5. Lámpara ultravioleta de una longitud de onda de 366 o 254 nm.
3.6. Microjeringa de 100 μl y 500 μl.
3.7. Embudo cilíndrico filtrante con filtro poroso G3 (porosidad 15-40 μm), de 2
cm de diámetro y 5 cm de altura, aproximadamente, con un dispositivo adecuado
para la filtración en vacío y una junta esmerilada macho 12/21.

ELIZABETH CARACUEL PAREJA
68
3.8. Matraz cónico para vacío de 50 ml, con junta esmerilada hembra 12/21
acoplable al embudo filtrante (3.7).
3.9. Probeta de 10 ml de fondo cónico con tapón hermético.
3.10. Equipo de cromatografía de gases que pueda funcionar con columna
capilar, provisto de un sistema de división de flujo formado por:
3.10.1. Horno para la columna, que pueda mantener la temperatura deseada
con precisión de ± 1 °C.
3.10.2. Inyector con elemento vaporizador de vidrio tratado con persilano.
3.10.3. Detector de ionización de llama y convertidor-amplificador.
3.10.4. Registrador-integrador que pueda funcionar con el convertidor
amplificador, con un tiempo de respuesta no superior a 1 segundo y con
velocidad de papel variable.
3.11. Columna capilar de vidrio o sílice fundida, de 20 a 30 m de longitud y de
0,25 a 0,32 mm de diámetro interno, recubierta interiormente de líquido SE-52,
SE-54 o equivalente, con un espesor uniforme que oscile entre 0,10 y 0,30 μm.
3.12. Microjeringa de 10 μl para cromatografía de gases, con aguja endurecida.
4. REACTIVOS
4.1. Hidróxido potásico, solución etanólica 2N aproximadamente: disolver,
enfriando al mismo tiempo, 130 g de hidróxido potásico (valoración mínima del
85 %) en 200 ml de agua destilada y completar hasta un litro con etanol.
Conservar la solución en botellas de vidrio oscuro bien cerradas.
4.2. Éter etílico de calidad para análisis.
4.3. Sulfato sódico anhidro de calidad para análisis.
4.4. Placas de vidrio recubiertas con gel de sílice, sin indicador de
fluorescencia, de 0,25 mm de espesor (disponibles en el comercio ya preparadas
para el uso).

ELIZABETH CARACUEL PAREJA
69
4.5. Hidróxido potásico, solución etanólica 0,2N: disolver 13 g de hidróxido
potásico en 20 ml de agua destilada y completar hasta un litro con etanol.
4.6. Benceno para cromatografía.
4.7. Acetona para cromatografía. (Véase 5.2.2).
4.8. Hexano para cromatografía. (Véase 5.2.2).
4.9. Éter etílico para cromatografía. (Véase 5.2.2).
4.10. Cloroformo de calidad para análisis.
4.11. Solución patrón para cromatografía en capa fina: colesterol o fitosteroles,
solución al 2 % en cloroformo.
4.12. Solución de 2,7-diclorofluoresceína al 0,2 % en etanol. Para hacerla
ligeramente básica se añaden algunas gotas de solución alcohólica 2N de
hidróxido potásico.
4.13. Piridina anhidra para cromatografía.
4.14. Hexametildisilazano.
4.15. Trimetilclorosilano.
4.16. Solución problema de trimetilsililéteres de los esteroles: preparar en el
momento del uso a partir de esteroles puros o de mezclas de esteroles obtenidos
de aceites que los contengan.
4.17. α-colestanol, disolución al 0,2 % (m/V) en cloroformo (patrón interno).
4.18. Gas portador: hidrógeno o helio de calidad para cromatografía de gases.
4.19. Gases auxiliares:
— hidrógeno de calidad para cromatografía de gases,
— aire de calidad para cromatografía de gases.
5. PROCEDIMIENTO
5.1. Preparación del insaponificable.

ELIZABETH CARACUEL PAREJA
70
5.1.1. Con la microjeringa de 500 μl introducir en el matraz de 250 ml un
volumen de disolución de α-colestanol al 0,2 % en cloroformo (4.17) que
contenga una cantidad de α-colestanol correspondiente al 10 %
aproximadamente del contenido de esteroles en la alícuota de la muestra para
la determinación. Por ejemplo, para 5 g de muestra añadir 500 μl de la solución
de α-colestanol al 0,2 %, si se trata de aceite de oliva, y 1 500 μl, si se trata de
aceite de orujo de oliva.
Evaporar en corriente de nitrógeno hasta sequedad y, a continuación, pesar con
precisión, en el mismo matraz, 5 g de muestra seca y filtrada.
En el caso de aceites con un alto contenido de colesterol puede producirse un
pico cuyo tiempo de retención sea idéntico al del colestanol. En tal caso el
análisis de la fracción de esteroles debe realizarse dos veces: con patrón interno
y sin él o hay que utilizar betulinol en lugar de colestanol.
5.1.2. Añadir 50 ml de solución etanólica de hidróxido potásico 2N, adaptar
el refrigerante de reflujo y calentar en baño María con ligera ebullición, agitando
enérgica e ininterrumpidamente hasta que se produzca la saponificación (la
solución se vuelve límpida). Calentar durante 20 minutos más y, a continuación,
añadir 50 ml de agua destilada por la parte superior del refrigerante; separar el
refrigerante y enfriar el matraz a 30 °C aproximadamente.
5.1.3. Transvasar cuantitativamente el contenido del matraz a un embudo de
separación de 500 ml, mediante varios lavados con un total aproximado de 50
ml de agua destilada. Agregar 80 ml aproximadamente de éter etílico, agitar
enérgicamente durante unos 30 segundos y dejar reposar hasta la separación
de las fases (nota 1).
Separar la fase acuosa inferior pasándola a un segundo embudo de separación.
Efectuar otras dos extracciones de la fase acuosa por el mismo procedimiento,
utilizando cada vez de 60 a 70 ml de éter etílico.
Nota 1: Las posibles emulsiones podrán eliminarse añadiendo pequeñas
cantidades de alcohol etílico o metílico con un pulverizador.

ELIZABETH CARACUEL PAREJA
71
5.1.4. Reunir las fracciones etéreas en un mismo embudo de separación y
lavarlas con agua destilada (50 ml cada vez) hasta que el agua de lavado
presente reacción neutra.
Una vez eliminada el agua de lavado, secar con sulfato sódico anhidro y filtrar
sobre sulfato sódico anhidro a un matraz de 250 ml previamente pesado, lavando
el embudo y el filtro con pequeñas cantidades de éter etílico.
5.1.5. Destilar el éter hasta que queden unos pocos ml; a continuación, secar
con un vacío ligero o en una corriente de nitrógeno, completando el secado en
una estufa a 100 °C durante 15 minutos aproximadamente; dejar enfriar en un
desecador y pesar.
5.2. Separación de la fracción de esteroles.
5.2.1. Preparación de las placas básicas: sumergir completamente las placas
con gel de sílice (4.4) en la solución etanólica 0,2N de hidróxido potásico (4.5)
durante 10 segundos; dejar secar las placas en campana durante dos horas y,
por último, mantenerlas en una estufa regulada a 100 °C durante una hora.
Sacarlas de la estufa y conservarlas en un desecador de cloruro de calcio hasta
el momento del uso (las placas sometidas a este tratamiento deberán utilizarse
en un plazo de quince días como máximo).
Nota 2: Si se utilizan placas básicas de gel de sílice para la separación de la
fracción de esteroles, ya no es necesario tratar el insaponificable con alúmina.
De este modo, todos los compuestos de naturaleza ácida (ácidos grasos y otros)
quedan retenidos en la línea de aplicación, y la banda de los esteroles aparece
perfectamente diferenciada de la banda de los alcoholes alifáticos y triterpénicos.
5.2.2. Introducir en la cubeta de desarrollo de las placas una mezcla de
benceno-acetona 95:5 (v/v) hasta una altura de 1 cm aproximadamente. Puede
utilizarse como alternativa una mezcla de hexano y éter etílico 65:35 (v/v). Cerrar
la cubeta con su correspondiente tapa y dejar transcurrir media hora como
mínimo, de forma que se alcance el equilibrio líquido-vapor. En las caras
interiores de la cubeta pueden colocarse tiras de papel de filtro que se sumerjan
en el eluyente: de esta manera el tiempo de desarrollo se reduce casi un tercio
y se obtiene una elución más uniforme y regular de los componentes.

ELIZABETH CARACUEL PAREJA
72
Nota 3: Para que las condiciones de elución sean perfectamente reproducibles,
la mezcla de desarrollo deberá cambiarse en cada prueba.
5.2.3. Preparar una solución de insaponificable (5.1.5) en cloroformo al 5 %
aproximadamente y, con la microjeringa de 100 μl, depositar 0,3 ml de dicha
solución en una placa cromatográfica (5.2.1) a unos 2 cm de uno de los bordes,
formando una línea lo más fina y uniforme posible. A la altura de la línea de
aplicación se depositan, en un extremo de la placa, de 2 a 3 μl de la solución de
referencia de esteroles (4.11) para poder identificar la banda de esteroles una
vez efectuado el desarrollo.
5.2.4. Introducir la placa en la cubeta de desarrollo, preparada como se indica
en el punto 5.2.2. Deberá mantenerse una temperatura ambiente entre 15 y 20
°C. Tapar inmediatamente la cubeta y dejar que se produzca la elución hasta
que el frente del disolvente se sitúe a 1 cm aproximadamente del borde superior
de la placa. Sacar la placa de la cubeta y evaporar el disolvente en una corriente
de aire caliente o dejando la placa bajo una campana unos minutos.
5.2.5. Pulverizar la placa ligera y uniformemente con la solución de 2,7-
diclorofluoresceína. Al examinar la placa a la luz ultravioleta puede identificarse
la banda de los esteroles mediante comparación con la mancha obtenida a partir
de la solución de referencia; marcar con lápiz negro los límites de la banda a lo
largo de los márgenes de fluorescencia.
5.2.6. Rascar con una espátula metálica el gel de sílice contenido en el área
delimitada. Introducir el material obtenido, finamente triturado, en el embudo
filtrante (3.7); añadir 10 ml de cloroformo caliente, mezclar cuidadosamente con
la espátula metálica y filtrar en vacío, recogiendo el filtrado en el matraz cónico
(3.8) acoplado al embudo filtrante. Lavar el residuo en el embudo tres veces con
éter etílico (empleando cada vez unos 10 ml), recogiendo cada vez el filtrado en
el mismo matraz cónico acoplado al embudo. Evaporar el filtrado hasta obtener
un volumen de 4 a 5 ml, transvasar la solución residual al tubo de ensayo de 10
ml (3.9) previamente pesado, evaporar hasta sequedad mediante calentamiento
suave en corriente ligera de nitrógeno, recoger con algunas gotas de acetona,
evaporar de nuevo hasta sequedad, introducir en una estufa a 105 °C durante
unos 10 minutos, dejar enfriar en el desecador y pesar.

ELIZABETH CARACUEL PAREJA
73
El residuo que queda en el tubo de ensayo está formado por la fracción de
esteroles.
5.3. Preparación de los trimetilsililéteres.
5.3.1. Agregar al tubo que contiene la fracción de esteroles el reactivo de
silanización formado por una mezcla de piridina-hexametildisilazano
trimetilclorosilano 9:3:1 (v/v/v) (nota 4), a razón de 50 μl por miligramo de
esteroles, evitando toda absorción de humedad (nota 5).
Nota 4: Existen soluciónes comerciales listas para el uso. Además, también
existen otros reactivos de silanización, como el bis-trimetilsililacetamida + 1 %
de trimetilclorosilano, que se diluye en el mismo volumen de piridina anhidra.
5.3.2. Tapar el tubo y agitar cuidadosamente (sin invertir) hasta la completa
disolución de los esteroles. Dejar reposar un cuarto de hora, como mínimo, a
temperatura ambiente y centrifugar durante algunos minutos; la solución límpida
queda lista para el análisis mediante cromatografía de gases.
Nota 5: La formación de una ligera opalescencia es normal y no ocasiona
ninguna interferencia. La formación de una floculación blanca o la aparición de
una coloración rosa son indicios de presencia de humedad o de deterioro del
reactivo. En este caso deberá repetirse la prueba.
5.4. Cromatografía de gases.
5.4.1. Operaciones preliminares: acondicionamiento de la columna.
5.4.1.1. Colocar la columna en el cromatógrafo uniendo uno de los extremos
de la columna al inyector y el otro al detector. Efectuar los controles generales
del equipo para cromatografía de gases (estanquidad de los cricuitos de gases,
eficacia del detector, eficacia del sistema de división de flujo y del sistema de
registro, etc.).
5.4.1.2. Si la columna se utiliza por vez primera, es conveniente
acondicionarla previamente. Hacer pasar un ligero flujo de gas a través de la
columna, encender el equipo de cromatografía de gases e iniciar un
calentamiento gradual hasta alcanzar una temperatura al menos 20 °C superior
a la temperatura de trabajo (nota 6). Mantener dicha temperatura durante 2 horas

ELIZABETH CARACUEL PAREJA
74
como mínimo; a continuación, poner el equipo completo en condiciones de
funcionamiento (regulación del flujo de gases y de la relación de «split», ignición
de la llama, conexión con el registrador electrónico, regulación de la temperatura
del horno del detector y del inyector, etc.) y registrar la señal con una sensibilidad
al menos dos veces superior a la prevista para el análisis. El trazado de la línea
de base debe ser lineal, exento de picos de cualquier tipo y no debe presentar
deriva.
Una deriva rectilínea negativa indica que las conexiones de la columna no son
totalmente estancas; una deriva positiva indica que el acondicionamiento de la
columna es insuficiente.
Nota 6: La temperatura de acondicionamiento deberá ser siempre, como mínimo
20 °C inferior a la temperatura máxima prevista para la fase estacionaria
utilizada.
5.4.2. Elección de las condiciones de trabajo.
5.4.2.1. Las condiciones de trabajo exigidas son las siguientes:
— temperatura de la columna: 260 °C ± 5 °C,
— temperatura del inyector : 280 °C,
— temperatura del detector: 290 °C,
— velocidad lineal del gas portador: helio 20 a 35 cm/s, hidrógeno 30 a 50 cm/s,
— relación de «split» de 1/50 a 1/100,
— sensibilidad del instrumento: de 4 a 16 veces la atenuación mínima,
— sensibilidad de registro: 1 a 2 mV f.e.
— velocidad del papel: 30 a 60 cm/hora,
— cantidad de sustancia inyectada: 0,5 a 1 μl de solución de TMSE.
Estas condiciones pueden modificarse en función de las características de la
columna y del cromatógrafo, de modo que se obtengan cromatogramas que
cumplan los siguientes requisitos:
— el tiempo de retención del β-sitosterol debe ser de 20 ± 5 minutos,

ELIZABETH CARACUEL PAREJA
75
— el pico del campesterol debe ser: para el aceite de oliva (contenido medio del
3 %), 15 ± 5 % del fondo de escala; para el aceite de soja (contenido medio del
20 %), 80 ± 10 % del fondo de escala,
— se deben separar todos los esteroles presentes; es necesario que los picos
no sólo se separen, sino que se resuelvan completamente, es decir, que el trazo
del pico llegue a la línea de base antes de que se inicie el pico siguiente. No
obstante, podrá admitirse una resolución incompleta si el pico a TRR 1,02 puede
cuantificarse utilizando la perpendicular.
5.4.3. Realización del análisis.
5.4.3.1. Con la microjeringa de 10 μl tomar 1 μl de hexano, aspirar 0,5 μl de
aire y, a continuación, entre 0,5 y 1 μl de la solución problema; elevar el émbolo
de la jeringa de modo que la aguja quede vacía. Introducir la aguja a través del
septum y, después de 1 o 2 segundos, inyectar rápidamente; transcurridos unos
5 segundos, extraer la aguja lentamente.
5.4.3.2. Continuar el registro hasta la completa elución de los TMSE de los
esteroles presentes.
La línea de base debe ajustarse en todo momento a las condiciones exigidas
(5.4.1.2).
5.4.4. Identificación de los picos.
Para la identificación de los diferentes picos se utilizan los tiempos de retención
y la comparación con mezclas de TMSE de los esteroles analizadas en las
mismas condiciones.
La elución de los esteroles se efectúa en el orden siguiente: colesterol,
brasicasterol, 24-metilencolesterol, campesterol, campestanol, estigmasterol, Δ-
7-campesterol, Δ-5,23-estigmastadienol, clerosterol, β-sitosterol, sitostanol, Δ-5-
avenasterol, Δ-5,24-estigmastadienol, Δ-7-estigmastenol, Δ-7-avenasterol.
5.4.5. Determinación cuantitativa.
5.4.5.1. Calcular las áreas de los picos del α-colestanol y de los esteroles
utilizando el integrador. El factor de respuesta para el α-colestanol debe
considerarse como 1.

ELIZABETH CARACUEL PAREJA
76
5.4.5.2. Calcular del modo siguiente el contenido de cada uno de los
esteroles, expresado en mg/100 g de materia grasa:
siendo:
Ax: área del pico del esterol x
As: área del pico del α-colestanol,
ms: peso de α-colestanol añadido, en miligramos,
m: peso de la muestra tomado para la determinación, en gramos.
6. EXPRESIÓN DE LOS RESULTADOS
6.1. Se expresa el contenido de cada uno de los esteroles en mg/100 g de
materia grasa y, como «esteroles totales», su suma.
6.2. El porcentaje de cada uno de los esteroles simples es la razón entre el
área del pico correspondiente y la suma de las áreas de los picos de los
esteroles.
siendo:
Ax: área del pico de x,
ΣA: suma de las áreas de todos los picos.

ELIZABETH CARACUEL PAREJA
77
ANEXO VI
DETERMINACIÓN DEL CONTENIDO EN ERITRODIOL Y UVAOL
INTRODUCCIÓN
El eritrodiol, entendido corrientemente como conjunto de los dioles eritrodiol y
uvaol, es un componente del insaponificable, característico de algunos tipos de
materias grasas. Su concentración es mucho más elevada en los aceites de oliva
obtenidos mediante extracción que en los obtenidos mediante presión o en el
aceite de pepita de uva y, por lo tanto, su determinación puede servir para
comprobar la existencia de aceite de oliva obtenido mediante extracción.
1. OBJETO
El presente método describe un procedimiento para determinar el eritrodiol en
las materias grasas.
2. PRINCIPIO
La materia grasa se saponifica con una solución etanólica de hidróxido potásico;
a continuación, se extrae el insaponificable con éter etílico y se purifica
pasándolo por una columna de alúmina.
El fraccionamiento del insaponificable se realiza mediante cromatografía en capa
fina en placa de gel de sílice y se aíslan la banda de la fracción esterólica y la
del eritrodiol.
Los esteroles y el eritrodiol recuperados de la placa se transforman en
trimetilsililéteres y seguidamente se analiza la mezcla mediante cromatografía
de gases.
El resultado se expresa en porcentaje de eritrodiol respecto del conjunto de
eritrodiol + esteroles.
3. MATERIAL Y APARATOS
3.1. El mismo material que el indicado para el método del Anexo V
(Determinación del contenido de esteroles).
4. REACTIVOS

ELIZABETH CARACUEL PAREJA
78
4.1. Los mismos reactivos que los indicados para el método del Anexo V
(Determinación del contenido de esteroles).
4.2. Solución patrón de eritrodiol al 0,5 % en cloroformo.
5. PROCEDIMIENTO
5.1. Preparación del insaponificable
Se realiza tal como se indica en el apartado 5.1.2 del método del Anexo V
5.2. Separación del eritrodiol y los esteroles
5.2.1. Véase el apartado 5.2.1 del método del Anexo V.
5.2.2. Véase el apartado 5.2.2 del método mencionado.
5.2.3. Preparar una solución del insaponificable al 5 % en cloroformo.
Con una microjeringa de 0,1 ml depositar 0,3 ml de esta solución en una placa
cromatográfica a aproximadamente 1,5 cm del borde inferior de la placa,
formando una banda lo más fina y uniforme posible. Depositar en un extremo de
la placa, como referencia, algunos microlitros de las soluciónes de colesterol y
eritrodiol.
5.2.4. Introducir la placa en la cubeta de desarrollo, preparada como se indica
en el apartado 5.2.1. La temperatura ambiente debe ser de unos 20 °C. Tapar
inmediatamente la cubeta y dejar que se produzca la elución hasta que el frente
del disolvente se sitúe a 1 cm aproximadamente del borde superior de la placa.
Sacar ésta de la cubeta de desarrollo y evaporar el disolvente en una corriente
de aire caliente.
5.2.5. Pulverizar la placa uniformemente con la solución alcohólica de 2′, 7′-
diclorofluoresceína. Al examinar la placa a la luz ultravioleta pueden identificarse
las bandas de los esteroles y del eritrodiol mediante comparación con las
referencias; delimitar las bandas con una punta ligeramente por el exterior de los
márgenes de fluorescencia.
5.2.6. Rascar con una espátula metálica el gel de sílice contenido en las
áreas delimitadas. Reunir el material obtenido en un matraz cónico de 50 ml;
añadir 15 ml de cloroformo caliente, agitar bien y filtrar en el embudo de filtro

ELIZABETH CARACUEL PAREJA
79
poroso vertiendo el gel de sílice sobre el propio filtro. Lavar tres veces con 10 ml
de cloroformo caliente cada vez, recogiendo el filtrado en un matraz esférico de
100 ml. Evaporar hasta obtener un volumen de 4 a 5 ml, transvasar al tubo de
centrifugado de fondo cónico de 10 ml previamente tarado, evaporar hasta
sequedad mediante calentamiento suave en corriente de nitrógeno y pesar.
5.3. Preparación de los trimetilsililéteres
Se realiza tal como se indica en el apartado 5.3 del método del Anexo V.
5.4. Cromatografía de gases
Se realiza tal como se indica en el apartado 5.4 del método citado. Las
condiciones operativas de la cromatografía de gases deben cumplir los requisitos
necesarios para analizar los esteroles y, además, permitir la separación de los
TMSE del eritrodiol y del uvaol.
Una vez inyectada la muestra, dejar que se desarrolle el proceso hasta que se
produzca la elución de los esteroles presentes, el eritrodiol y el uvaol; identificar
los picos (el eritrodiol y el uvaol tienen tiempos de retención relativos, respecto
al β-sitosterol, de alrededor de 1,45 y 1,55, respectivamente) y calcular sus áreas
de la misma forma que para los esteroles.
6. EXPRESIÓN DE LOS RESULTADOS
siendo:
A1: área del pico del eritrodiol,
A2: área del pico del uvaol,
ΣAesteroles: suma de las áreas de los esteroles presentes.
Los resultados deben expresarse con una cifra decimal.

ELIZABETH CARACUEL PAREJA
80
ANEXO IX PRUEBA ESPECTROFOTOMÉTRICA EN EL ULTRAVIOLETA
INTRODUCCIÓN
La prueba espectrofotométrica en el ultravioleta puede proporcionar indicaciones
sobre la calidad de una materia grasa, su estado de conservación y las
modificaciones inducidas por los procesos tecnológicos.
Las absorciones en las longitudes de onda indicadas en el método se deben a la
presencia de sistemas diénicos y triénicos conjugados. Los valores de estas
absorciones se expresan en extinción específica E1cm 1 % (extinción de una
solución de la materia grasa al 1 % en el disolvente determinado, en un espesor
de 1 cm) que se expresará convencionalmente como K, también denominado
coeficiente de extinción.
1. OBJETO
El método describe el procedimiento de ejecución de la prueba
espectrofotométrica en el ultravioleta de las materias grasas.
2. PRINCIPIO
La materia grasa se disuelve en el disolvente requerido y se determina la
extinción de la solución a las longitudes de onda prescritas, respecto al
disolvente puro. A partir de los valores espectrofotométricos se calculan las
extinciones específicas.
3. MATERIAL Y APARATOS
3.1. Espectrofotómetro para medidas de extinción en el ultravioleta entre 220
y 360 nm, con posibilidad de lectura para cada unidad nanométrica.
3.2. Cubetas de cuarzo, con tapadera, con paso óptico de 1 cm. Las cubetas,
llenas de agua o de otro disolvente adecuado, no deben presentar entre ellas
diferencias superiores a 0,01 unidades de extinción.
3.3. Matraces aforados de 25 ml.

ELIZABETH CARACUEL PAREJA
81
3.4. Columna de cromatografía de 270 mm de longitud y 35 mm de diámetro
en la parte superior: de 270 mm de longitud y 10 mm de diámetro en la parte
inferior.
4. REACTIVOS
4.1. Isooctano (2,2,4-trimetilpentano) de calidad para espectrofotometría: debe
tener, respecto al agua destilada, una transmitancia del 60 % como mínimo a
220 nm y del 95 % como mínimo a 250 nm; o
— ciclohexano de calidad para espectrofotometría: debe tener, respecto al agua
destilada, una transmitancia del 40 % como mínimo a 220 nm y del 95 % como
mínimo a 250 nm.
4.2. Alúmina básica para cromatografía en columna, preparada y controlada
cómo se describe en el apéndice I.
4.3. n-Hexano para cromatografía.
5. PROCEDIMIENTO
5.1. La muestra debe ser perfectamente homogénea y estar exenta de
impurezas en suspensión. Los aceites líquidos a temperatura ambiente se filtran
con papel de filtro a una temperatura aproximada de 30 °C, las grasas sólidas se
homogeneizan y se filtran a una temperatura superior en 10 °C como máximo a
su temperatura de fusión.
5.2. Se pesan con precisión 0,25 g aproximadamente de la muestra preparada
y se colocan en un matraz aforado de 25 ml, se completa con el disolvente
adecuado y se homogeneiza. La solución resultante debe estar perfectamente
clara. Si presenta opalescencia o turbidez, se filtrará rápidamente con papel de
filtro.
5.3. Se llena una cubeta con la solución obtenida y se miden las extinciones,
usando como referencia el disolvente empleado, a las longitudes de onda
comprendidas entre 232 y 276 nm. Los valores de extinción obtenidos deben
estar comprendidos en el intervalo entre 0,1 y 0,8; en caso contrario es necesario
repetir la medida utilizando soluciónes más concentradas o más diluidas según
el caso.

ELIZABETH CARACUEL PAREJA
82
5.4. Cuando se quiera determinar la extinción específica después del
tratamiento con alúmina se procederá del siguiente modo: en la columna para
cromatografía se introducen 30 g de alúmina básica en suspensión en hexano;
después de asentarse el absorbente se elimina el exceso de hexano, hasta 1 cm
aproximadamente sobre el nivel superior de la alúmina.
Se disuelven 10 g de materia grasa, homogeneizada y filtrada tal como se
describe en el punto 5.1, en 100 ml de hexano y se vierte esta solución en la
columna. Se recoge el líquido eluido y se evapora totalmente el disolvente en
vacío a una temperatura inferior a 25 °C.
Con la materia grasa así obtenida se procede inmediatamente tal como se indica
en el punto 5.2.
6. EXPRESIÓN DE LOS RESULTADOS
6.1. Se expresan las extinciones específicas o coeficientes de extinción a las
diversas longitudes de onda, calculadas como sigue:
siendo:
Kλ = extinción específica a la longitud de onda lambda,
Eλ = extinción medida a la longitud de onda lambda,
c = concentración de la disolución en g por 100 ml,
e = espesor de la cubeta en cm.
Los resultados deben expresarse con dos cifras decimales.
6.2. La prueba espectrofotométrica del aceite de oliva según el método oficial
de los Reglamentos de la CEE requiere la determinación de la extinción
específica, en solución en isooctano, a las longitudes de onda de 232 y 270 nm,y
la determinación de Δ E definido como:

ELIZABETH CARACUEL PAREJA
83
donde Km es la extinción específica a la longitud de onda m, longitud de onda
de máxima absorción alrededor de 270 nm.

ELIZABETH CARACUEL PAREJA
84
ANEXO XV
1. CONTENIDO EN ACEITE DE LOS ORUJOS DE ACEITUNA
1.1. Material
— aparato de extracción apropiado tipo soxhlet, provisto de un matraz de 200 a
250 ml,
— baño por calefacción eléctrica (baño de arena, baño de agua, etc.) o placa
calefactora,
— balanza analítica,
— estufa regulada a 80 °C como máximo,
— estufa de calefacción eléctrica provista de un dispositivo de termorregulación
regulada a 103 °C ± 2 °C y que permita realizar una insuflación de aire o una
presión reducida,
— triturador mecánico fácil de limpiar y que permita el triturado del orujo sin
calentamiento y sin disminución sensible de su contenido en agua y en aceite,
— cartucho de extracción y algodón hidrófilo o papel filtro, exentos de productos
extraíbles por el hexano,
— desecador,
— criba con agujeros de 1 mm de diámetro,
— piedra pómez en pequeños granos, previamente secada.
1.2. Reactivo
n-hexano técnico cuyo residuo en evaporación completa deberá ser inferior a
0,002 g para 100 ml.
2. MODO DE OPERAR
2.1. Preparación de la muestra para ensayo
Triturar la muestra para laboratorio, si fuera necesario, en el triturador mecánico,
previamente bien limpio, para reducirla a partículas que puedan atravesar
completamente la criba.

ELIZABETH CARACUEL PAREJA
85
Utilizar aproximadamente una vigésima parte de la muestra para completar la
limpieza del triturador, tirar dicha mezcla, triturar el resto, recogerlo, mezclarlo
con cuidado y analizarlo sin demora.
2.2. Toma de muestra
Pesar inmediatamente después del triturado, alrededor de 10 g de la muestra
para ensayo con una aproximación de 0,01 g.
2.3. Preparación del cartucho de extracción
- Colocar la muestra en el cartucho y taparla con el tapón de algodón hidrófilo.
En caso de haber utilizado papel de filtro, envolver la muestra molida en dicho
papel.
2.4. Presecado
Cuando el orujo esté muy húmedo (contenido en agua y en materias volátiles
superior al 10 %), efectuar un presecado colocando durante un tiempo
conveniente el cartucho lleno (o el papel de filtro) en la estufa calentada a 80 °C
como máximo, para llevar el contenido en agua y en materias volátiles por debajo
del 10 %.
2.5. Preparación del matraz
Pesar con aproximación de 1 g el matraz que contenga 1 a 2 granos de piedra
pómez, previamente secado en la estufa a 103 °C ± 2 °C y después enfriado
durante al menos una hora en el desecador.
2.6. Primera extracción
Colocar en el aparato de extracción el cartucho (o el papel de filtro) que contenga
la muestra. Verter en el matraz la cantidad necesaria de hexano.
Adaptar el matraz al aparato de extracción y colocarlo todo sobre la placa
calefactora. Llevar la calefacción a tal estado que el caudal de reflujo sea al
menos de tres gotas por segundo (ebullición moderada, no tumultuosa).
Después de cuatro horas de extracción, dejar enfriar. Quitar el cartucho del
aparato de extracción y colocarlo en una corriente de aire para eliminar la mayor
parte del disolvente que lo impregna.

ELIZABETH CARACUEL PAREJA
86
2.7. Segunda extracción
Vaciar el cartucho en el microtriturador y triturar tan finamente como sea posible.
Colocar de nuevo cuantitativamente la mezcla en el cartucho y ésta en el aparato
de extracción.
Empezar de nuevo la extracción durante dos horas más, utilizando el mismo
matraz conteniendo la primera extracción.
La solución obtenida en el matraz de extracción deberá ser límpida. Si no fuere
así, filtrarla sobre un papel de filtro lavando varias veces el primer matraz y el
papel de filtro con hexano. Recoger el filtrado y el disolvente en un segundo
matraz previamente secado y tarado aproximadamente a 1 mg.
2.8. Eliminación del disolvente y pesada del extracto
Eliminar en el equipo de extracción la mayor parte del disolvente. Eliminar los
últimos restos de éste calentando el matraz en la estufa a 103 °C ± 2 °C durante
20 minutos. Facilitar dicha eliminación, ya sea insuflando aire de vez en cuando
o preferiblemente un gas inerte, o actuando bajo una presión reducida.
Dejar enfriar el matraz en un desecador durante al menos una hora, y pesarlo
con una precisión de 1 mg aproximadamente.
Calentar de nuevo 10 minutos en las mismas condiciones, dejar enfriar en el
desecador y pesar.
La diferencia entre los resultados de estas dos pesadas deberá ser inferior o
igual a 10 mg, si no, calentar de nuevo durante períodos de diez minutos
seguidos de enfriamento y pesada, hasta que la diferencia de peso sea, a lo
sumo, de 10 mg. Seleccionar la última pesada del matraz.
Efectuar dos determinaciones.
3. EXPRESIÓN DE LOS RESULTADOS
3.1. Modo de cálculo y fórmula
a) El extracto expresado en peso del producto tal cual será igual a:

ELIZABETH CARACUEL PAREJA
87
donde: S es el porcentaje en peso del extracto del producto tal cual,
m0 es el peso, en gramos, de la muestra,
m1 es el peso, en gramos, del extracto seco.
Tomar como resultado la media aritmética de las dos determinaciones, si las
condiciones de repetitividad se cumplen.
Expresar el resultado con un solo decimal.
b) El extracto se relacionará con la materia seca utilizando la siguiente fórmula:
donde: S es el procentaje en peso de extracto del producto tal cual,
U es su contenido en agua y en materias volátiles.
3.2. Repetitividad
La diferencia entre los resultados de las dos determinaciones, efectuadas
simultáneamente o rápidamente la una a continuación de la otra mediante el
mismo análisis, no deberá ser superior a 0,2 g de extracto de hexano por cada
100 g de muestra.
En caso contrario, repetir sobre otras dos tomas de muestra. Si esta vez la
diferencia es de nuevo superior a 0,2 g tomar como resultado la media aritmética
de las cuatro determinaciones efectuadas.

ELIZABETH CARACUEL PAREJA
88
ANEXO XVI
DETERMINACIÓN DEL ÍNDICE DE YODO
1. OBJETO
La presente norma internacional especifica un método para la determinación del
índice de yodo de las grasas y aceites animales y vegetales, en lo sucesivo
denominados grasas.
2. DEFINICIÓN
A los fines de la presente norma internacional, se aplicará la definición siguiente:
2.1. Índice de yodo: el peso de yodo absorbido por la muestra en las
condiciones de trabajo que se especifican en la presente norma internacional. El
índice de yodo se expresa en gramos de yodo por 100 gr de muestra.
3. PRINCIPIO
Disolución de la muestra problema y adición de reactivo de Wijs. Una vez
transcurrido el tiempo que se especifica, adición de solución acuosa de yoduro
potásico y valoración del yodo liberado con solución de tiosulfato sódico.
4. REACTIVOS
Todos los reactivos serán de calidad analítica reconocida.
4.1. Yoduro potásico, solución de 100 g/l, exento de yodatos o de yodo libre.
4.2. Engrudo de almidón.
Mezclar 5 g de almidón soluble con 30 ml de agua, añadir esta mezcla a 1 000
ml de agua en ebullición, hervir durante 3 minutos y dejar enfriar.
4.3. Solución volumétrica patrón de tiosulfato sódico.
c (Na2S2O3. 5H2O) = 0,1 mol/l, valorada como máximo 7 días antes de su uso.
4.4. Disolvente, preparado mezclando volúmenes iguales de ciclohexano y
ácido acético.

ELIZABETH CARACUEL PAREJA
89
4.5. Reactivo de Wijs, que contenga monocloruro de yodo en ácido acético. Se
utilizará reactivo de Wijs comercializado. Nota: El reactivo contiene 9 g de ICl3 +
9 g de I en ácido acético.
5. MATERIAL
Material ordinario de laboratorio y, en particular, lo siguiente:
5.1. Navecillas de vidrio, apropiadas para la muestra problema y que puedan
introducirse en los matraces (5.2).
5.2. Matraces erlenmeyer de 500 ml de capacidad con boca esmerilada,
provistos de sus correspondientes tapones de vidrio y perfectamente secos.
6. PREPARACIÓN DE LA MUESTRA PROBLEMA.
La muestra homogeneizada, se seca sobre sulfato sódico y se filtra.
7. PROCEDIMIENTO
7.1. Tamaño de la muestra
El peso de la muestra varía en función del índice de yodo previsto, como se
indica en el siguiente cuadro:
Índice de yodo previsto Peso de la muestra problema,gr
Menos de 5 3
5-20 1
21-50 0,40
51-100 0,20
101-150 0,13
151-200 0,10
Pesar la muestra problema con precisión de 0,1 mg en una navecilla de vidrio.
7.2. Determinación
Introducir la muestra problema en un matraz de 500 ml (5.2). Añadir 20 ml del
disolvente (4.4) para disolver la grasa. Agregar exactamente 25 ml del reactivo
de Wijs (4.5), tapar el matraz, agitar el contenido y colocar el matraz al abrigo de
la luz. No deberá utilizarse la boca para pipetear el reactivo de Wijs.

ELIZABETH CARACUEL PAREJA
90
Preparar del mismo modo un ensayo en blanco con el disolvente y el reactivo,
pero sin la muestra problema.
Para las muestras con un índice de yodo inferior a 150, mantener los matraces
en la oscuridad durante 1 hora; para las muestras con un índice de yodo superior
a 150, así como en el caso de productos polimerizados o considerablemente
oxidados, mantener en la oscuridad durante 2 horas.
Una vez transcurrido el tiempo correspondiente, agregar a cada uno de los
matraces 20 ml de solución de yoduro potásico (4.1) y 150 ml de agua.
Valorar con la disolución de tiosulfato sódico (4.3) hasta que haya desaparecido
casi totalmente el color amarillo producido por el yodo.
Añadir unas gotas de engrudo de almidón (4.2) y continuar la valoración hasta el
momento preciso en que desaparezca el color azul después de una agitación
muy intensa. Nota: Se permite la determinación potenciométrica del punto final.
7.3. Número de determinaciones
Efectuar 2 determinaciones de la muestra problema.
8. EXPRESIÓN DE LOS RESULTADOS
El índice de yodo se expresa del siguiente modo:
c: valor numérico de la concentración exacta, expresada en moles por litro, de la
solución volumétrica patrón de tiosulfato sódico (4.3) utilizada;
V1: valor numérico del volumen, expresado en mililitros, de la solución de
tiosulfato sódico (4.3) utilizada para el ensayo en blanco;
V2: valor numérico del volumen, expresado en mililitros, de la solución de
tiosulfato sódico (4.3) utilizada para la determinación;
p: valor numérico del peso, expresado en gramos, de la muestra problema (7.1).
Se tomará como resultado la media aritmética de las dos determinaciones,
siempre que se cumpla el requisito establecido con respecto a la repetibilidad.

ELIZABETH CARACUEL PAREJA
91
ANEXO XVII
MÉTODO PARA LA DETERMINACIÓN DE ESTIGMASTADIENOS EN LOS
ACEITES VEGETALES
1. OBJETIVOS
Determinación de estigmastadienos en los aceites vegetales que presentan
bajas concentraciones de dichos hidrocarburos, particularmente en el aceite de
oliva virgen y en el aceite de orujo de oliva sin refinar.
2. APLICACIÓN
Se trata de una norma aplicable a cualquier aceite de origen vegetal, teniendo
en cuenta que únicamente se obtendrán medidas fiables cuando el contenido de
estos hidrocarburos se halle incluido en un margen de 0,01 a 4,0 mg/kg. El
método resulta especialmente adecuado para detectar la presencia de aceites
vegetales refinados (aceites de oliva, orujo de oliva, girasol, palma, etc.) en el
aceite de oliva virgen, dado que los aceites refinados contienen
estigmastadienos y los aceites vírgenes no.
3. FUNDAMENTO
Aislamiento de la materia insaponificable. Separación de la fracción de
hidrocarburos esteroideos mediante cromatografía en columna de gel de sílice y
análisis mediante cromatografía de gases en columna capilar.
4. INSTRUMENTAL
4.1. Matraces de 250 ml aptos para colocarles un refrigerante de reflujo.
4.2. Embudos de decantación con capacidad para 500 ml.
4.3. Matraces de fondo redondo de 100 ml.
4.4. Rotavapor.
4.5. Columna de vidrio para cromatografía (1,5-2,0 cm de diámetro interno por
50 cm de longitud) provista de una llave de teflón y con una torunda de lana de
vidrio o un disco de vidrio unterizado en el fondo. Para preparar la columna de
gel de sílice se vierte hexano en la columna de cromatografía hasta que alcance
una altura de aproximadamente 5 cm, llenándose a continuación con una papilla

ELIZABETH CARACUEL PAREJA
92
de gel de sílice en hexano (15 g en 40 ml) mediante la ayuda de varias porciones
de hexano. Se deja sedimentar la mezcla en reposo, completándose la
sedimentación aplicando una ligera vibración. Añadir sulfato sódico anhidro
hasta una altura de aproximadamente 0,5 cm y finalmente eluir el exceso de
hexano.
4.6. Cromatógrafo de gases equipado con detector de ionización de llama,
inyector con división de flujo u «cold on -column» y horno programable con
precisión de ± 1 °C.
4.7. Columna capilar de sílice fundida (0,25 o 0,32 mm de diámetro interno ×
25 m de longitud), recubierta con una película de 0,25 μm de espesor de la fase
5 %-fenilmetilsilicona.
Nota 1.
Pueden utilizarse otras columnas con polaridades similares o inferiores.
4.8. Integrador-registrador con capacidad para modo de integración valle-valle.
4.9. Microjeringa de 5-10 μl para cromatografía de gases con aguja fija.
4.10. Calentador de tipo manta o placa eléctrica.
5. REACTIVOS
Salvo indicación en sentido contrario, únicamente se utilizarán reactivos de
calidad para análisis. Debe emplearse agua destilada o de pureza al menos
equivalente.
5.1. Hexano o una mezcla de alcanos con un intervalo de ebullición de 65-70 °
C, destilados en columna rectificadora.
Nota 2. El disolvente debe destilarse para eliminar impurezas.
5.2. Etanol de 96 v/v.
5.3. Sulfato sódico anhidro.
5.4. Solución alcohólica de hidróxido potásico al 10 %. Añadir 10 ml de agua a
50 g de hidróxido potásico, agitar y diluir seguidamente la mezcla en etanol hasta
un volumen de 500 ml.

ELIZABETH CARACUEL PAREJA
93
Nota 3. La potasa alcohólica toma color marrón con el tiempo. Debe prepararse
diariamente y se almacenará en botellas de vidrio oscuro cerradas
herméticamente.
5.5. Gel de sílice 60 para columna de cromatografia, de 70-230 mallas
(referencia 7734 de Merck o similar).
Nota 4. Por lo general, el gel de sílice puede utilizarse directamente a partir del
envase sin necesidad de tratamiento. Sin embargo, algunos lotes de sílice
pueden presentar baja actividad produciendo separaciones cromatográficas
inadecuadas. Cuando ello ocurra debe tratarse el gel de sílice de la siguiente
manera: activar el gel calentándolo a 550 °C durante un mínimo de cuatro horas.
Tras el calentamiento poner el gel de sílice a enfriar en un desecador y
trasvasarlo a continuación a un matraz con cierre hermético.
Se añade seguidamente un 2 % de agua agitando hasta que dejen de verse
grumos y el polvo fluya libremente.
Si algún lote de gel de sílice origina cromatogramas con picos que interfieran,
dicho gel será sometido al tratamiento anteriormente mencionado.
Puede considerarse como alternativa la utilización de gel de sílice 60 extrapuro
(referencia 7754 de Merck).
5.6. Solución madre (200 ppm) de colesta-3,5-dieno (Sigma, 99 % de pureza)
en hexano (10 mg en 50 ml).
5.7. Solución patrón de colesta-3,5-dieno en hexano con una concentración de
20 ppm, que se obtiene diluyendo la solución anterior.
Nota 5. Si se mantiene la temperatura por debajo de 4 °C, las soluciónes 5.6 y
5.7 permanecerán inalteradas durante un mínimo de 4 meses.
5.8. Solución de n-nonacosano en hexano con una concentración aproximada
de 100 ppm.
5.9. Gas portador para cromatografia: helio o hidrógeno de 99,9990 % de
pureza.

ELIZABETH CARACUEL PAREJA
94
5.10. Gases auxiliares para el detector de ionización de llama: aire purificado
e hidrógeno de 99,9990 % de pureza.
6. METODOLOGÍA
6.1. Preparación de la materia insaponificable:
6.1.1. Pesar 20 ± 0,1 g de aceite en un matraz de 250 ml (4.1), añadir 1 ml
de solución patrón de colesta-3,5-dieno (20μg) y 75 ml de potasa alcohólica al
10 %, colocar un refrigerante de reflujo y calentar a ebullición suave durante 30
minutos. Retirar de la fuente de calor el matraz con la muestra y dejar enfriar
ligeramente la solución (el enfriamiento no debe ser total para evitar la
decantación de la muestra). Añadir 100 ml de agua y pasar la solución a un
embudo de decantación (4.2) con ayuda de 100 ml de hexano. Agitar
enérgicamente durante 30 segundos y dejar decantar.
Nota 6. Si se produce emulsión, que no desaparece rápidamente, añadir
pequeñas cantidades de etanol.
6.1.2. Pasar la fase acuosa inferior a un segundo embudo de decantación y
someterla nuevamente a extracción con 100 ml de hexano. Separar nuevamente
la fase inferior y lavar los extractos de hexano (mezclándolos en otro embudo de
decantación) con tres porciones de 100 ml cada una de una mezcla de agua y
etanol (1: 1) hasta obtener un pH neutro.
6.1.3. Pasar la solución de hexano a través de sulfato sódico anhidro (50 g),
lavar con 20 ml de hexano y evaporar a 30 °C y presión reducida en un rotavapor
hasta sequedad.
6.2. Separación de la fracción de hidrocarburos esteroideos:
6.2.1. Introducir el residuo en la columna de fraccionamiento con ayuda de
dos porciones de 1 ml de hexano, distribuir la muestra en la columna dejando
que el nivel de solución descienda hasta la parte superior del sulfato de sodio y
comenzar la elución cromatográfica con hexano a un flujo aproximado de 1
ml/min. Desechar los primeros 25-30 ml de eluido y recoger la fracción siguiente
de 40 ml. Después, pasar esta fracción a un matraz de fondo redondo de 100 ml
(4.3).

ELIZABETH CARACUEL PAREJA
95
Nota 7. La primera fracción contiene los hidrocarburos saturados y la segunda
fracción los esteroideos. Al proseguir con la elución se obtiene escualeno y otros
compuestos relacionados. Para poder obtener una buena separación entre los
hidrocarburos saturados y los esteroideos, es necesario optimizar los volúmenes
de las fracciones. A tal efecto se debe ajustar el volumen de la primera fracción
de tal manera que cuando se analice la segunda, los picos correspondientes a
los hidrocarburos saturados sean bajos; cuando estos no aparezcan pero la
intensidad del pico correspondiente al patrón sea reducida, debe disminuirse el
volumen.
De todas formas, puesto que no se produce solapamiento de los picos durante
la cromatografía de gases, no es precisa una separación completa entre los
componentes de la primera y segunda fracciones siempre que las condiciones
de la CG estén ajustadas como se indica en 6.3.1. Por lo general no es necesario
optimizar el volumen de la segunda fracción, puesto que existe una buena
separación con los componentes que eluyen posteriormente. Sin embargo, la
presencia de un pico importante a 1,5 min, aproximadamente, por debajo del
tiempo de retención del patrón se debe al escualeno y revela una mala
separación.
6.2.2. Evaporar la segunda fracción en rotavapor a 30 °C y presión reducida
hasta sequedad. Disolver inmediatamente el residuo en 0,2 ml de hexano y
mantener la solución en el refrigerador hasta el momento de su análisis.
Nota 8. Los residuos 6.1.3 y 6.2.2 no deben mantenerse en seco a temperatura
ambiente. Una vez obtenidos, es necesario añadirles disolvente y almacenarlos
en un refrigerador.
6.3. Cromatografía de gases
6.3.1. Condiciones de trabajo para inyección con división de flujo.
— temperatura del inyector: 300 °C,
— temperatura del detector: 320 °C.
— integrador-registrador: los parámetros de integración deben ser fijados de tal
forma que la evaluación de las áreas sea correcta. La modalidad de integración
valle-valle es la más recomendable,

ELIZABETH CARACUEL PAREJA
96
— sensibilidad: aproximadamente 16 veces la atenuación mínima,
— cantidad de disolución inyectada: 1μl,
— programación de la temperatura del horno: temperatura inicial constante de
235 °C durante 6 minutos, seguida de un aumento gradual de 2 °C/min hasta
alcanzar 285 °C,
— inyección con una división de flujo de 1: 15,
— gas portador: helio o hidrógeno a 120 kPa de presión.
Dichas condiciones pueden modificarse en función de las características del
cromatógrafo y de la columna con objeto de obtener cromatogramas que
respondan a los siguientes requisitos: Pico del patrón interno situado con una
aproximación de 5 minutos en torno al tiempo citado en 6.3.2; el pico del patrón
interno alcanzará, como mínimo, un 80 % de la escala total.
El sistema de cromatografía de gases deberá comprobarse inyectando una
mezcla de la solución madre de colestadieno (5.6) y de n-nonacosano (5.8). El
pico del colesta-3,5-dieno deberá aparecer antes que el del nnonacosano; si esto
no ocurre, pueden adoptarse dos soluciónes: reducir la temperatura inicial del
horno o sustituir la columna de cromatografía de gases por otra de polaridad
inferior.
6.3.2. Identificación de picos
El pico del patrón interno aparece a un tiempo de retención de,
aproximadamente, 19 minutos, y el del estigmasta-3,5-dieno lo hace a un tiempo
de retención relativo de aproximadamente 1,29. El estigmasta- 3,5-dieno suele
ir acompañado de pequeñas cantidades de un isómero y, por lo general, ambos
originan un solo pico cromatográfico.
No obstante, si la columna es demasiado polar, o tiene un gran poder de
resolución, el isómero puede aparecer en forma de un pequeño pico justo antes
y cerca del estigmasta-3,5-dieno. En estos casos, es preciso sumar las áreas de
los dos picos. Con el fin de estar seguro de que los estigmastadienos eluyen
como un solo pico se recomienda instituir la columna por otra de menor polaridad
o de mayor diámetro interior.

ELIZABETH CARACUEL PAREJA
97
Nota 9. Para obtener una referencia de los estigmastadienos puede utilizarse el
análisis de cualquier aceite vegetal refinado empleando menor cantidad de
muestra (de 1 a 2 g). Los estigmastadienos dan lugar a un pico importante de
fácil identificación.
6.3.3. Análisis cuantitativo
El contenido de los estigmastadienos se determina aplicando la fórmula
en la cual:
Ab = área del pico de estigmastadienos (si el pico se halla dividido en dos picos,
súmese el área de los dos picos.).
Ac = área del pico del patrón interno (colestadieno).
Mc = peso de patrón añadido, en microgramos.
Mo = peso de aceite empleado, en gramos.
Límite de detección: aproximadamente 0,01 mg/kg.

4 DISEÑO DEL LABORATORIO

ELIZABETH CARACUEL PAREJA
99
A continuación, se consideran las zonas que integran el laboratorio objeto del
presente trabajo Fin de Máster.
4.1 PLANO GENERAL
En la Figura 4.1, se muestra un plano general del laboratorio.
Figura 4.1. Plano general del laboratorio

ELIZABETH CARACUEL PAREJA
100
4.2 SALA DE RECEPCIÓN
El laboratorio constará de una zona apta para la recepción y espera de los
clientes. En el plano general (Fig. 4.1), se ha nombrado como zona 2.
Las funciones de la recepción serán:
- Recepción de clientes en busca satisfacer las necesidades de comunicación
- Proporcionar al público la información que requiera
- Atender el teléfono
- Recibir y entregar la correspondencia recibida
- Actividades administrativas
- Realiza cualquier otra tarea afín que le sea asignada
4.3 DESPACHOS
Para facilitar la privacidad de los altos cargos de la empresa, habrá un despacho
privado. En el plano general (Fig. 4.1), se ha nombrado como zona 4.
Funciones del jefe de laboratorio:
- Organizar, dirigir, supervisar y coordinar el trabajo del personal a su cargo.
- Controlar, organizar y supervisar las actividades en el laboratorio.
- Repasar y validar los resultados obtenidos diariamente.
- Realizar el inventario del material que se necesite reponer.
- Comunicar a los clientes los resultados obtenidos.
- Participar en las reuniones con los demás directivos de la empresa cuando se
le requiera.
- Revisar y firmar documentos (facturas, reportes, entre otros).
4.4 SALA DE JUNTAS
En el plano general (Fig. 4.1), se ha nombrado como zona 3.
Toda empresa debe contar, en la medida de lo posible, con una sala de
reuniones. Disponer de un recinto amplio y equipado para poder llevar adelante

ELIZABETH CARACUEL PAREJA
101
presentaciones de proyectos o reuniones con clientes se demostrará necesario
conforme las actividades de la empresa progresen. El despacho de la empresa
sería insuficiente cuando se trate de reunir a todo el personal.
Las funciones principales serán:
- Reuniones de directivos para discutir nuevos proyectos.
- Reuniones de personal para informar de novedades.
- Reuniones con nuevos clientes.
- Exposición de nuevos proyectos.
4.5 PATIO DE RECEPCIÓN Y ALMACENAMIENTO DE MATERIA PRIMA
En el plano general (Fig. 4.1), se ha nombrado como zona 5.
Para una correcta recepción de la aceituna se establecerá una entrada diferente
de la entrada principal de la empresa. La descarga se realizará en el patio trasero
debido a la suciedad que traen los medios de transporte y el polvo que se
produce cuando se realiza la descarga. Una vez descargadas las cajas de
aceitunas se irán llevando a la sala de molturación según vayan a ser
procesadas.
Una vez molturadas, la mitad de cada muestra debe ser almacenada durante
unos días para posibles reclamaciones de los clientes. Por ello, las muestras
deberán ser trasladadas de nuevo al patio para dejar espacio a las muestras que
tienen que molturarse.
4.6 SALA DE MOLTURACIÓN
En el plano general (Fig. 4.1), se ha nombrado como zona 6.
Las muestras de aceituna y orujo llegan al laboratorio en bolsas de plástico
identificadas con un código de barras, y con una cantidad de 600 a 1000 gramos.
Una vez registradas, las muestras de aceituna son molidas. La primera fracción
de la muestra es desechada durante la molienda, para arrastrar los restos de
muestra anterior.
La sala de molturación contará con 3 molinos de martillos (Fig.4.2). Estos
molinos tienen un doble tambor de acero inoxidable que en su interior está

ELIZABETH CARACUEL PAREJA
102
formado por dos cribas concéntricas, dentro de las cuales se encuentran los
“martillos” dispuestos como una hélice.
La torva lleva las aceitunas hasta la parte interior del tambor donde el primer
círculo de martillos inicia la trituración. Los martillos aplastan las aceitunas, y si
este proceso se efectúa a excesiva velocidad, esta acción estresa la oliva y hace
que aumente la temperatura en este punto.
Figura 4.2. Molino de martillos y criba.
Cuando la aceituna está verde, al inicio de la cosecha, las células que contienen
el aceite son más pequeñas y es necesario que la dimensión del triturado sea
más fina. A medida que las aceitunas van madurando las células son más
grandes por lo que la dimensión del triturado puede ser más grande. Cuanto más
fina es la molturación, más “finos” (impurezas) tendrá el aceite.
Según se necesite que la pasta de molturación sea más o menos fina, la criba
exterior puede cambiarse para que el paso sea mayor o menor.
Cuando los fragmentos adquieren el tamaño determinado, la misma fuerza
centrífuga los hace pasar a la parte superior del tambor a través de la primera
criba (con pasos de un diámetro de 10 mm). Aquí el segundo círculo de martillos
sigue triturando la pasta.

ELIZABETH CARACUEL PAREJA
103
Cuando los fragmentos de pulpa y de avellano triturados alcanzan el tamaño
necesario, pasan por la criba exterior.
4.7 SALA DE ANÁLISIS
La sala de análisis estará formada por varias zonas bien diferenciadas:
1. Zona de análisis de aceituna. Método NIR
En el plano general (Fig. 4.1), se ha nombrado como zona 7A. Constará de tres
espectrofotómetros modelo Perkin Elmer (Fig.4.3).
Figura 4.3. Espectrofotómetro
Para obtener el rendimiento graso sobre húmedo de aceituna hay que seguir los
siguientes pasos:
- Homogeneizar la muestra molturada.
- Poner la pasta en la placa Petri evitando que queden espacios libres.
Figura 4.4. Pasta de aceituna en placa Petri.

ELIZABETH CARACUEL PAREJA
104
- Poner la placa en la zona de medida.
Figura 4.5. Pasta de aceituna en la zona de análisis.
- Esperar unos 50 segundos mientras se hace la lectura para obtener el
resultado.
En la pantalla del ordenador aparecerá un espectro y unos datos numéricos,
indicando el rendimiento graso, la acidez y la humedad de la muestra.
Simultáneamente se generará una base de datos con todos los datos
correspondientes a las muestras analizadas (Fig.4.6).
Figura 4.6. Resultado del análisis de aceituna.
2. Zona de análisis de aceituna. Método Soxhlet
En el plano general (Fig. 4.1), se ha nombrado como zona 7B.
El extractor Soxhlet es el método oficial para extraer el aceite. Consta de un tipo
de material de vidrio utilizado para la extracción de compuestos, contenidos en
un sólido, a través de un disolvente afín (n-hexano).
En el laboratorio habrá 3 baterías de 10 cuerpos cada una (Fig. 4.7).

ELIZABETH CARACUEL PAREJA
105
Figura 4.7. Batería de Soxhlet.
El proceso de Soxhlet sería el siguiente:
Se necesita un cuerpo extractor, un matraz y la muestra seca de aceituna
encartuchada. Se introduce la muestra en el cuerpo extractor (situado encima de
un matraz) y posteriormente se adiciona 250 ml del disolvente (2 cuerpos). Este
equipo se calienta hasta la temperatura de ebullición del disolvente para que
pueda atravesar el cuerpo una y otra vez (se evapora gracias al sistema de
calefacción y se condensa debido al sistema de refrigeración que hay encima del
cuerpo extractor), así tantas veces como sea necesario para extraer todo el
aceite que se pueda de la mezcla. Hay que tener en cuenta que, al principio, el
cuerpo extractor está de color amarillo verdoso, es decir, la muestra aún contiene
aceite, y al final el cuerpo extractor contiene el disolvente de color transparente,
y el matraz está del color del aceite. Esto quiere decir que el disolvente ya no
está extrayendo más materia grasa de la muestra.
3. Zona de análisis de aceite.
En el plano general (figura 4.1), se ha nombrado como zona 7C.
Una vez llegadas las muestras de aceite, éstas deben ponerse a filtrar antes de
ser analizadas. En ocasiones, las muestras traen demasiadas impurezas y esto
dificulta el proceso de filtración. Por ello es necesario disponer de una centrífuga
que permita separar las impurezas del aceite y facilitar el proceso de filtrado.

ELIZABETH CARACUEL PAREJA
106
En esta zona habrá varias mesas para poder hacer todas las determinaciones
pertinentes, y cada una de ellas constará de todo el material analítico
correspondiente.
4. Zona para cromatografía.
En el plano general (Fig. 4.1), se ha nombrado como zona 7C.
Otra de las zonas del laboratorio estará destinada exclusivamente a la
cromatografía. Esta zona debe estar un poco apartada del resto debido a la
sensibilidad que poseen estos equipos. Para evitar error en los resultados, habrá
una mesa que actuará como soporte del cromatógrafo.
4.8 ALMACÉN DE MUESTRAS DE ACEITE
En el plano general (Fig. 4.1), se ha nombrado como zona 8.
Una vez analizadas las muestras de aceite, hay que guardar una parte de la
muestra para posibles reclamaciones. Estas muestras deben estar bien
etiquetadas y organizadas por fechas de entrada, facilitando así su localización
ante posibles errores experimentales.

5 ÁREA COMERCIAL

ELIZABETH CARACUEL PAREJA
108
La zona de producción olivarera de la que se nutrirá el laboratorio está
perfectamente delimitada, al tratarse de una comarca natural, la de Priego de
Córdoba. El área comercial con la que se pretende trabajar abarca los municipios
de Almedinilla, Carcabuey, Fuente Tójar y Priego de Córdoba, una superficie
total de 44.818 Has., de las cuales 29.628 Has. están dedicadas al olivar
(Fig.5.1).
Figura 5.1. Zona de producción olivarera entorno al laboratorio
5.1 SERVICIOS OFERTADOS
Los servicios ofertados serán:
5.1.1 Análisis de aceituna
5.1.1.1 Índice de madurez
El índice de madurez se utiliza para determinar el momento óptimo de recogida
de la aceituna. Para su cálculo se toman 2 kg de aceitunas situadas a la altura
del operador y en las cuatro orientaciones del árbol. Se homogeniza la muestra,
se separan 100 frutos al azar y se clasifican en las siguientes categorías
(Fig.5.2):
- Clase 0: Piel verde intenso
- Clase 1: Piel verde amarillento.
- Clase 2: Piel verde con manchas rojizas en menos de la mitad del fruto. Inicio
de envero.
- Clase 3: Piel rojiza o morada en más de la mitad del fruto. Final de envero.
- Clase 4: Piel negra y pulpa blanca.

ELIZABETH CARACUEL PAREJA
109
- Clase 5: Piel negra y pulpa morada sin llegar a la mitad de la pulpa.
- Clase 6: Piel negra y pulpa morada sin llegar al hueso.
- Clase 7: Piel negra y pulpa morada totalmente hasta el hueso.
Figura 5.2. Clasificación de aceituna para el índice de madurez
Se llama Índice de Madurez (IM) al sumatorio del número de frutos de cada
categoría, por el valor numérico de su categoría, dividido por 100, es decir,
siendo A,B,C,D,E,F,G,H el número de frutos de cada clase 0,1,2,3,4,5,6,7
respectivamente, el índice de madurez es:
𝐼𝑀 =(Ax0 + Bx1 + Cx2 + Dx3 + Ex4 + Fx5 + Gx6 + Hx7)
100
Para las variedades que desarrollan normalmente el color (Picual, Hojiblanca,
Lechín, Cornicabra, etc), el momento óptimo de recolección es cuando el Índice
de Madurez alcanza valores próximos a 3,5.
Por tanto, en función del tamaño de la finca y del periodo estimado de
recolección, se debe adelantar la fecha de inicio de la recolección para que la
mayor parte de la cosecha se haga con un índice igual a 3,5.
5.1.1.2 Peso medio
Con el uso de una balanza de precisión ±0.01 g, se pesan los 100 frutos. El peso
medio se obtiene dividiendo la sumatoria del peso de los frutos entre el número
total de frutos.
PM =Ʃ peso de aceituna
nº total aceitunas

ELIZABETH CARACUEL PAREJA
110
5.1.1.3 Relación pulpa-hueso
1. Se pesan 100 frutos y se anota el peso. Después se calcula el peso medio de
una aceituna como se ha explicado en el apartado anterior.
2. Se deshuesan las aceitunas, se limpian los huesos y se dejan secar.
Posteriormente se pesan y se calcula el peso medio de un hueso.
PM Hueso =Peso Total Huesos
nº total huesos
3. La relación pulpa hueso se calcula:
P/H =Peso Medio Aceituna − Peso Medio del Hueso
Peso Medio del hueso
5.1.1.4 Humedad
Se pesan 30 ± 0.01g de masa de aceituna previamente molida y se desecan en
una estufa de aire forzado a una temperatura de 105ºC hasta pesada constante.
El cálculo de la humedad se realiza:
%Humedad =pesada inicial − pesada final
pesada inicial x 100
5.1.1.5 Contenido graso total
Fundamento
Consiste en extraer la materia grasa del fruto mediante una extracción sólido-
líquido mediante un proceso de contacto repetido de corrientes cruzadas usando
hexano.
El sistema de extracción Soxhlet consta de:
- Cuerpo extractor de vidrio donde se introduce la muestra encartuchada.

ELIZABETH CARACUEL PAREJA
111
- Matraz de fondo plano donde se recoge la masa.
- Un refrigerante de reflujo adaptado al cuerpo extractor por medio de un cierre
esmerilado.
Además, hay que tener un foco de calor (placa), que favorezca la extracción del
contenido graso.
Para obtener la pasta de aceituna seca, hay que:
1. Moler la muestra de aceituna en un molino de martillos hasta quedar una
pasta.
2. Pesar la pasta húmeda.
3. Poner la pasta en la estufa durante 24 horas.
4. Sacar la pasta de la estufa y poner 10 minutos en un desecador.
5. Pesar la pasta seca.
6. Encartuchar la pasta de aceituna en un cartucho poroso, para después
introducirlo en el cuerpo extractor.
Procedimiento de extracción
Se introduce la muestra seca de aceituna en el cuerpo extractor y posteriormente
se adiciona 250 ml del disolvente (2cuerpos). Se coge pesa el matraz que se va
a usar y se encaja con el cuerpo extractor y a su vez con el sistema de
refrigeración. Este equipo se calienta hasta la temperatura de ebullición del
disolvente para que pueda atravesar el cuerpo una y otra vez (se evapora gracias
al sistema de calefacción y se condensa debido al sistema de refrigeración que
hay encima del cuerpo extractor), así tantas veces como sea necesario para
extraer todo el aceite que se pueda de la mezcla (Fig.5.3). Hay que tener en
cuenta que, al principio, el cuerpo extractor está de color amarillo verdoso, es
decir, la muestra aún contiene aceite, y al final el cuerpo extractor contiene el
disolvente de color transparente, y el matraz está del color del aceite. Esto quiere
decir que el disolvente ya no está extrayendo más materia grasa de la muestra.
La técnica oficial indica que para el rendimiento graso de la aceituna se mantiene
el dispositivo unas 6 horas desde la primera caída del disolvente.

ELIZABETH CARACUEL PAREJA
112
Al finalizar la extracción, se destila el hexano procurando no quemar el aceite y
se deseca el matraz con el aceite en el desecador.
Figura 5.3. Esquema de extracción de materia grasa. Soxhlet
Material e instrumentación
- Muestra de aceitunas
- Balanza analítica
- Probeta
- Matraces de fondo plano
- Cuerpo extractor
- Disolvente (n-hexano)
- Placas calefactoras
Resultados
Rendimiento en base seca

ELIZABETH CARACUEL PAREJA
113
Rendimiento en Base Seca =g materia grasa extraida
g materia grasa seca en cartuchox100
Rendimiento en base húmeda
Rendimiento en Base Húmeda =materia grasa extraida
materia grasa húmeda en cartuchox100
Para hacer este proceso de extracción, se recomienda utilizar varias baterías de
Soxhlet a la vez para poder obtener el mayor número de resultados posibles en
el menor tiempo.
5.1.2 Análisis de aceite
5.1.2.1 Toma de muestra
La muestra de aceite suele recogerse en botellitas de plástico que se cierran
instantáneamente para evitar ser contaminadas. Cuando estas muestras llegan
al laboratorio, se homogenizan y se filtran a través de un papel de filtro que se
pliega manualmente adaptándose a un embudo. El modo de hacerlo se muestra
en la figura 5.4:
Figura 5.4. Modo de plegar el papel de filtro

ELIZABETH CARACUEL PAREJA
114
5.1.2.2 Grado e índice de acidez
La acidez es el % en peso de ácidos grasos libres formados por la hidrólisis de
los triglicéridos. Se expresa en función del ácido graso mayoritario: gr ácido
oleico/100 gr aceite.
Triglicérido + 3 H2O Glicerina + Ácidos grasos libres
Fundamento
Se determinan los ácidos grasos libres mediante valoración con una solución
contrastada de KOH en presencia de fenolftaleína como indicador. Se utiliza éter
etílico y alcohol etílico como disolventes del aceite.
Se neutraliza la acidez propia del disolvente agregando unas gotas de KOH
hasta ligero tinte rosa del azul alcalino; de este modo no hay interferencias en la
valoración posterior de la muestra problema.
R-COOH + KOH R-COOK + H2O
Procedimiento
Se pesa la muestra en un matraz erlenmeyer de 250 ml, según el grado de acidez
previsto, de acuerdo con la tabla siguiente (Tabla 5.1):
Tabla 5.1. Grado de acidez previsto
Grado de Acidez Previsto Peso de la muestra, g
<1 20
1-4 10
4-15 2.5
15-75 0.5
>75 0.1
Se disuelve la muestra en 50 ml de una mezcla de etanol de 95% y éter dietílico,
en proporción de volumen 1:1. Esta mezcla debe ser previamente neutralizada
exactamente en el momento de su utilización con solución de hidróxido potásico

ELIZABETH CARACUEL PAREJA
115
0.1 M en presencia de 0.3 ml de solución de fenolftaleína por cada 100 ml de
mezcla. A continuación, se valora la muestra con solución de KOH 0.1 M (ó 0.5
M si la cantidad necesaria de hidróxido potásico 0.1 M supera los 10 ml) agitando
continuamente hasta viraje del indicador azul alcalino, (azul-rosa, debe
permanecer al menos durante 10 segundos). Cada determinación debe
realizarse por duplicado.
Si la solución se enturbia durante la valoración, añadir una cantidad suficiente de
la mezcla de disolventes para que la solución se aclare.
Reactivos necesarios
- Solución al 1% de azul alcalino en etanol del 95-96% o solución al 2% de azul
alcalino (en caso de aceites de oliva muy coloreados) en etanol de 95-96%.
- Éter dietílico
- Etanol de 95%
- Solución etanólica valorada de hidróxido potásico 0.1 M ó 0.5 M
- Mezcla de éter dietílico y etanol de 95%, en proporción de volumen 1:1.
Material e instrumentación
- Matraz erlenmeyer de 250 ml (seco)
- Probeta de 50 ó 100 ml
- Bureta de 10 ml, con graduación de 0.05 ml
- Vaso de precipitado de 250 ml
- Balanza analítica
Resultados
Se calcula el grado de acidez, expresado en” porcentaje de ácido oleico”, y el
índice de acidez expresado en “mg de KOH/g de grasa”. Se tomará como
resultado la media aritmética de dos determinaciones.
El grado de acidez indica la acidez que tiene el aceite analizado. Aumenta
conforma empeora la calidad del aceite.

ELIZABETH CARACUEL PAREJA
116
Grado de acidez = V x c x M
10 x P
El índice de acidez es útil cuando se desconoce el ácido graso mayoritario en la
muestra. S
Índice de acidez = 56,1 x V x c
P
Siendo:
c: concentración exacta, en moles/litro, de la disolución de hidróxido potásico
utilizada
V: volumen en ml de la solución valorada de hidróxido potásico utilizada
M: peso molecular del ácido oleico, M= 282
P: peso en gramos de la muestra utilizada
Normalmente la acidez se expresa referida a tanto por ciento de ácido oleico.
Sólo en casos particulares, según la naturaleza de la sustancia grasa, se
expresará referida a ácido palmítico, laúrico u otros.
5.1.2.3 Índice de peróxidos
Fundamento
Se denomina "índice de peróxidos" a la cantidad (expresada en miliequivalentes
de oxígeno activo contenidos en un kilogramo de grasa) de peróxidos en la
muestra que ocasionan la oxidación del yoduro potásico en las condiciones de
trabajo descritas.
Los peróxidos son productos de oxidación de la grasa, debido a la autooxidación.
Se originan por oxidación de los ácidos grasos con dobles enlaces, que cuando
existen en una muestra, oxidan al KI, en medio ácido, según la siguiente
reacción:
O22- (peróxidos) + 2 I- (en exceso) + 4 H+ ↔ 2 H2O + I2

ELIZABETH CARACUEL PAREJA
117
El I2 liberado será proporcional a la cantidad de peróxidos existentes y se valora
con disolución de tiosulfato sódico según la reacción:
I2 + 2 S2O32- 2 I- + S4O62-
El I2 con el almidón forma un complejo azul oscuro que se destruye cuando éste
se reduce completamente a I-, visualizándose así el punto final de la valoración.
Se utiliza cloroformo como disolvente de la grasa y ácido acético para
proporcionar la acidez necesaria al medio para que se produzca la reacción.
Procedimiento
(El ensayo se realizará con luz natural difusa o con luz artificial). Un matraz
erlenmeyer de 250 ml con cuello esmerilado, provisto de tapón y seco, se llena
de gas inerte puro y seco (nitrógeno o, preferiblemente, dióxido de carbono). Con
esto se intenta evitar en la medida de lo posible el error de oxígeno u oxidación
del KI por el oxígeno del aire. Se introduce luego tan rápidamente como sea
posible la muestra de aceite que se desea ensayar, definida en función del índice
que se presupone (Tabla 5.2):
Tabla 5.2. Índice de peróxidos
Índice que se presupone (meq O2/kg) Peso Muestra (g)
0-12 5-2
12-20 2-1,2
20-30 1,2-0,8
30-50 0,8-0,5
50-90 0,5-0,3
Se agregan 10 ml de cloroformo, en el cual se disuelve rápidamente la grasa por
agitación, 15 ml de ácido acético glacial y, a continuación, 1 ml de solución
saturada de KI (que se debe preparar en el momento de usarla, disolviendo en
la mínima cantidad de agua destilada KI hasta que la solución no admita más
soluto).
Cerrar el matraz y mantenerlo en agitación durante 1 minuto, imprimiéndole un
suave movimiento de rotación, conservándolo después en la oscuridad durante

ELIZABETH CARACUEL PAREJA
118
5 minutos; transcurrido este tiempo, agregar 75 ml de agua destilada, y valorar
(agitando vigorosamente) el I2 liberado con una disolución de Na2S2O3 0.002 N
para los aceites de índices inferiores o iguales a 12 y 0.01 N para los aceites de
índices más elevados, utilizando 2 ml de solución de almidón como indicador
(agregar cerca del punto final de la valoración).
Paralelamente se efectúa un blanco, sin aceite, que debe dar índice nulo. Cada
determinación se realiza por duplicado.
Reactivos Necesarios
- Cloroformo exento de O2
- Ácido acético glacial exento de O2
- Solución acuosa saturada de KI (preparada en el momento de su uso)
- Solución indicadora de almidón al 1 % en agua destilada
- Solución acuosa de Na2S2O3 0.01 N (y 0.002 N) exactamente valorada
Material e instrumentación
- Matraz erlenmeyer de 250 ml con cierre esmerilado (seco y lleno de gas inerte)
- Probeta de 25 ó 50 ml
- Tubo graduado
- Bureta de 25 ml
Resultados
Se denomina índice de peróxidos al número de miliequivalentes de oxígeno
activo contenidos en un kilogramo de grasa, calculados a partir del yodo liberado
del yoduro potásico, en las condiciones en que se opera.
Índice de peróxidos =(V − V′) x N x 1000
P
Siendo:
c: normalidad exacta de la disolución de tiosulfato sódico (incluir factor)
V: volumen en ml de tiosulfato sódico gastado en la valoración de la muestra

ELIZABETH CARACUEL PAREJA
119
V’: volumen en ml de tiosulfato sódico gastado en la valoración del blanco
P: peso en g de la muestra utilizado en la determinación
5.1.2.4 Medida espectrofotométrica de la absorción en la región
ultravioleta de un aceite
Fundamento
Este método se funda en la medida espectrofotométrica ultravioleta del
coeficiente de extinción específica a las longitudes de onda de 232 y 270 nm.
La transparencia en la región de los 270 nm se ve afectada en el proceso de
autoxidación, y también como resultado de determinados procesos industriales.
Esto justifica que el coeficiente de extinción específica a 270 nm se tome como
criterio de calidad, que puede modificarse a través del tiempo influido por las
condiciones de conservación de la materia grasa.
K232: Es un parámetro relacionado con el contenido en hidroperóxidos. Indica la
oxidación inicial de un aceite cuantificando la absorción de luz a una longitud de
onda igual a 232 nm.
K270: El factor K270 es una prueba espectrofotométrica en el rango de longitud
de onda ultravioleta que nos puede indicar la presencia en el aceite de
compuestos de oxidación secundaria (distintos de los peróxidos) que presentan
una absorción máxima a la longitud de onda de 270 nm. Estos compuestos son
resultado del estado de conservación del aceite, de modificaciones sufridas fruto
de los procesos tecnológicos, de contaminaciones o adulteraciones.
ΔK: Se utiliza fundamentalmente como criterio de pureza, para detectar mezclas
con aceites refinados. Cuando un aceite sufre el proceso de refinación, en el
espectro aparecen tres picos (pico trieno) y la absorbancia a 270 nm es mayor.
En un aceite de oliva virgen, el ΔK=0.

ELIZABETH CARACUEL PAREJA
120
Procedimiento
La materia grasa debe estar exenta de sustancias en suspensión. Si se trata de
un aceite y se observa turbidez o sustancias en suspensión, debe ser filtrado a
la temperatura del laboratorio, a través de un papel de filtro adecuado.
Pesar, con precisión de 0.5 mg, en un matraz aforado de 10 ml la cantidad
necesaria de materia grasa, en función de su absorción en el ultravioleta (mirar
tabla). La cantidad pesada se disuelve en isoctano y se completa hasta el enrase.
La disolución debe tener una concentración tal que la absorbancia leída en el
espectrofotómetro se encuentre dentro de los límites indicados más adelante.
Como orientación y en los distintos tipos de aceites de oliva, las cantidades a
pesar serán las siguientes:
- Aceites vírgenes de oliva 100 mg
- Aceites refinados de oliva y sus mezclas con vírgenes 60 mg
- Aceites de orujo, brutos y refinados 20-40 mg
Se introduce la disolución en la cubeta de cuarzo de 1 cm de paso de luz,
llenando la cubeta de comparación con ciclohexano y se efectúan las medidas a
las longitudes de onda deseadas. En los aceites de oliva estas medidas se
realizan a 232 y 270 nm. Realizar cada determinación por duplicado.
Las lecturas de absorbancia a 270 nm deben estar comprendidas entre 0.1 y 0.8;
en el caso de situarse fuera de estos límites, debe repetirse la medida bien
diluyendo en la relación conveniente o procediendo a una nueva pesada.
Reactivos necesarios
- Isoctano, calidad UV-IR
Material e instrumentación
- Espectrofotómetro UV
- Matraz aforado de 10 ml
- Cubetas de cuarzo
- Gotero de vidrio

ELIZABETH CARACUEL PAREJA
121
- Balanza analítica
Resultados
Kλ =Aλ
𝑏 · 𝑐
siendo:
Kλ: coeficiente de extinción específica a la longitud de onda dada
Aλ: absorbancia a la longitud de onda dada
c: concentración de la disolución en g/100 ml
b: espesor de la cubeta en cm
Los resultados se expresan como coeficiente de extinción específica a la longitud
de onda en que se mide.
Niveles permitidos para el consumo alimentario
A continuación, los valores normales de los coeficientes de extinción específica
para los distintos tipos de aceites (Tabla 5.3) :
Tabla 5.3. Valores normales de los coeficientes de extinción específica
(1). Normativa de la CE, (2) Normativa del COI

ELIZABETH CARACUEL PAREJA
122
5.1.2.5 Medida de la composición de ácidos grasos
Fundamento
El método está basado en la separación y determinación por cromatografía de
gases de los ésteres metílicos de los ácidos grasos.
Es aplicable a los aceites de oliva y los aceites de orujo de oliva con un contenido
en ácidos grasos libres inferior al 3.3 %. El hidróxido potásico no produce la
esterificación de los ácidos grasos libres. Los ésteres metílicos se forman por la
transesterificación con una solución metabólica de hidróxido potásico como una
fase intermedia antes que se produzca la saponificación.
Procedimiento
Se analizan dos muestras: aceite de oliva y aceite de girasol ó aceite oliva y
aceite de oliva adulterado con un 20% de aceite de girasol.
En un tubo roscado de 5 ml pesar aproximadamente 0.1 g de la muestra de
aceite. Añadir 2 ml de heptano y agitar. Añadir 0.2 ml de la solución metabólica
2 N de hidróxido potásico, poner el tapón provisto de la junta PTFE, cerrarlo bien
y agitar enérgicamente durante 30 segundos. Dejar estar hasta que la solución
de la parte superior resulte nítida. Decantar la capa superior, que es la que
contiene los ésteres metílicos. La solución de hexano está lista para inyectarla
en el cromatógrafo. Es aconsejable mantener la solución en el frigorífico hasta el
momento de realizar el análisis cromatográfico.
a) Procedimiento cromatográfico
Se ajusta el flujo del gas portador (Helio), que debe ser adecuado para permitir
la elución del linoleato de metilo en un tiempo mínimo de 25 minutos. La presión
de entrada y el flujo necesario para conseguirlo varía según la columna y el
instrumento utilizado. Es necesario mantener el flujo constante durante todo el
análisis. El flujo gaseoso se mide con un medidor de burbuja de jabón.
Se pone en marcha la calefacción del horno, de la cámara de inyección y del
detector. Las condiciones de temperatura son las siguientes:
Temperatura del horno: 185 ºC

ELIZABETH CARACUEL PAREJA
123
Temperatura de la cámara de inyección: 220 ºC
Temperatura del detector: 220 ºC
Se inyecta aproximadamente 1.0 μl de la disolución de ésteres metílicos
utilizando una jeringa, y se retira rápidamente la aguja. Todos los picos deben
caer dentro del cromatograma por lo que se utilizará en cada caso la atenuación
de sensibilidad que sea necesaria. Una vez conseguido un cromatograma
satisfactorio se procede a la identificación de los picos.
b) Identificación de los picos
Se utilizará el criterio basado en los tiempos de retención. Si nos referimos
exclusivamente a los ácidos que entran normalmente en la composición de las
grasas naturales, sus ésteres aparecen en el cromatograma en orden creciente
de sus átomos de carbono y a su insaturación. Esto es, el palmítico (C16)
aparece delante del esteárico (C18), y los ésteres en C18 aparecen en el orden
estearato, oleato, linoleato. El éster del ácido aráquico (C20:0), usualmente,
aparece antes del linolénico (C18:3) pero, puede ocurrir lo contrario, en algunos
casos, dependiendo del tipo de columna y de las condiciones de su utilización; o
incluso superponerse el uno al otro.
Operando en condiciones constantes, los tiempos de retención son
reproducibles en cada especie química, siendo el criterio más frecuentemente
usado para su identificación.
Reactivos
- Heptano de calidad para cromatografía
- Metanol PA-ACS-ISO
- Hidróxido potásico, solución metanólica 2 N aproximadamente: disolver 11.2 g
de hidróxido potásico en 100 ml de metanol.
Material e instrumentación
- Tubos roscados provistos de tapón.
- Pipetas graduadas o automáticas de 2 ml y 0.2 ml.

ELIZABETH CARACUEL PAREJA
124
Resultados
La determinación cuantitativa se basa en el principio de que los pesos de cada
uno de los componentes separados en la mezcla son proporcionales a las áreas
comprendidas dentro de cada pico. Se utiliza el método de normalización interna.
Calcular el contenido de un componente dado (i) (expresado como porcentaje en
masa de ésteres metílicos), mediante la determinación del porcentaje que
representa el área de su pico en relación con la suma de las áreas de todos los
picos, aplicando la expresión siguiente:
%𝑐𝑜𝑚𝑝𝑜𝑛𝑒𝑛𝑡𝑒 𝑖 = 𝐴𝑖
∑𝐴 𝑥 100
siendo: Ai: área del pico correspondiente al componente i
Σ A: suma de las áreas de todos los picos integrados
En la tabla 5.4. se recogen los límites establecidos de cada ácido grado para cada
tipo de aceite.
Tabla 5.4. Reconocimiento de aceites vegetales. Fracción de ácidos grasos.
Límites

ELIZABETH CARACUEL PAREJA
125
5.1.2.6 Medida del contenido en ceras mediante cromatografía de
gases con columna capilar
Fundamento
Las ceras son aportadas al Aceite de Oliva desde distintas fuentes:
a) Fruto: el fruto está recubierto por una fina capa protectora. Estas ceras son
ésteres de ácidos grasos con alcoholes grasos de cadena larga que pueden
pasar al Aceite de Oliva durante el prensado.
b) Hojas: las hojas también contienen ceras, pero en este caso contienen mayor
cantidad de carbonos. Son aportadas cuando se recoge la aceituna en
condiciones de alta humedad o cuando el proceso de separación del fruto es
deficiente.
c) Proceso: el aumento de la temperatura durante el amasado de la pasta aporta
una mayor cantidad de ceras al Aceite de Oliva porque facilita la extracción de
las mismas. La extracción con solventes también aumenta la cantidad de ceras.
d) Almacenaje: en los aceites con alta acidez, pueden producirse esterificaciones
entre los ácidos grasos libres y alcoholes grasos. La determinación del contenido
y tipo de ceras es útil para detectar la presencia de aceite de orujo (que se
obtiene por extracción con solventes). El límite máximo de ceras en el aceite de
oliva es de 250 mg/Kg.
La determinación de ceras se realiza por cromatografía gaseosa con detector de
ionización de llama del Aceite de Oliva previamente eluído a través de una
columna de sílice para aislar las ceras de 40 a 46 átomos de carbono. La materia
grasa, a la que se habrá añadido un patrón interno adecuado, se fracciona
mediante cromatografía en columna de gel de sílice hidratado; se recupera la
fracción eluida en primer lugar en las condiciones de ensayo (cuya polaridad es
inferior a la de los triglicéridos) y, a continuación, se analiza directamente
mediante cromatografía de gases en columna capilar.
Procedimiento
1. Preparación de la columna cromatográfica

ELIZABETH CARACUEL PAREJA
126
- Suspender 15 g de gel de sílice en n-hexano e introducirlo en la columna.
Después hay que pasar 30 ml de n-hexano para eliminar las posibles impurezas
y pesar 500 mg de la muestra en el matraz Erlenmeyer y añadir el patrón interno.
- Transferir la muestra así preparada a la columna cromatográfica.
- Dejar fluir el disolvente hasta que se sitúe a 1 mm por encima del nivel superior
del absorbente, y a continuación hacer pasar 70 ml más de nhexano para
eliminar los n-alcanos presentes de forma natural. Comenzar entonces la elución
cromatográfica recogiendo 180 ml de la mezcla de nhexano/éter etílico en la
proporción 99:1.
2. Análisis por cromatografía de gases
Después de realizar las operaciones preliminares, como el acondicionamiento
de la columna y la elección de las condiciones del ensayo, se procede a la
realización del ensayo:
- Tomar 1 μl de la solución con la microjeringa de 10 μl; sacar el émbolo de la
jeringa hasta que la aguja esté vacía.
- Introducir la aguja en el sistema de inyección e inyectar rápidamente después
de 1 o 2 segundos; dejar pasar unos 5 segundos y extraer lentamente la aguja.
- Llevar a cabo el registro hasta que las ceras se hayan eluido completamente.
La línea de base debe satisfacer siempre las condiciones exigidas.
Para identificar los distintos picos del cromatograma obtenido, hay que tener en
cuenta los tiempos de retención y compararlos con mezclas de ceras analizadas en
las mismas condiciones y cuyos tiempos de retención sean conocidos.
Reactivos necesarios
- Gel de sílice de una granulometría comprendida entre 60 y 200 μm.
- N-hexano, de calidad para cromatografía.
- Éter etílico, de calidad para cromatografía.
- N-heptano, de calidad para cromatografía.
- Solución patrón de araquidato de laurilo al 0,1 % (m/v) en hexano (patrón interno).

ELIZABETH CARACUEL PAREJA
127
- Sudán 1
- Gas portador (hidrógeno o helio puro, de calidad para cromatografía de gases).
Material e intrumentación
- Matraz Erlenmeyer de 25 ml.
- Columna de vidrio para cromatografía de gases
- Cromatógrafo de gases adecuado para utilizarse con columna capilar
- Microjeringa para introducción directa en columna, de 10 μl de capacidad,
provista de una aguja cementada.
- Rotavapor.
- Horno de mufla.
- Balanza analítica
Resultados
- Calcular con ayuda del integrador las áreas de los picos correspondientes al
patrón interno y a los ésteres alifáticos de C40 a C46.
- Determinar el contenido en ceras de cada uno de los ésteres, en mg/kg de
materia grasa, aplicando la siguiente fórmula:
É𝑠𝑡𝑒𝑟 (𝑚𝑔
𝑘𝑔) =
𝐴𝑥 · 𝑚𝑠 · 1000
𝐴𝑠 · 𝑚
siendo:
Ax = el área del pico de cada éster, en milímetros cuadrados
As = el área del pico del patrón interno, en milímetros cuadrados
ms = la masa del patrón interno que se ha añadido, en miligramos
m = la masa de la muestra tomada para la determinación, en gramos.

ELIZABETH CARACUEL PAREJA
128
5.1.2.7 Medida de la concentración y composición de esteroles
Los esteroles son parámetros que caracterizan a cada tipo de grasa o aceite.
Para el Aceite de Oliva Virgen, el total de esteroles debe ser como mínimo 1000
mg/Kg. Los aceites extraídos con solvente poseen una cantidad superior, que
puede ser reducida durante el refinado.
Lo que prácticamente no varía durante el refinado son los porcentajes de la
composición que mantiene las características de la especie. De esta manera se
puede tener idea no sólo si el aceite ha sido adulterado sino además la especie
y su lugar de origen.
Se sugiere ser cuidadoso con la interpretación de este análisis en forma aislada,
ya que, en algunos aceites, esta composición sufre importantes variaciones en
su composición definida por diversas variables como: clima, suelo, edad, etc.
Los valores límites fijados internacionalmente para este perfil son: colesterol
menor o igual a 0,5 %, Brasicasterol menor o igual a 0,1%, Campesterol menor
o igual a 4 %, Estigmasterol menor que el campesterol, Delta-7-estigmasterol
menor o igual a 0,5% y mayor o igual a 93% de la suma de Betasitosterol, delta-
5-avenasterol, delta-5-23-estigmastadienol, clerosterol, sitostanol y delta 5-24-
estigmastadienol.
Fundamento
Este método consiste en la saponificación de la materia grasa, a la que se habrá
añadido α-colestanol como patrón interno, con una solución etanólica de
hidróxido potásico; a continuación, extracción del insaponificable con éter etílico.
Después se realizará la separación de la fracción de esteroles del insaponificable
extraído mediante cromatografía en placa de gel de sílice básica. Los esteroles
recuperados del gel de sílice se transforman en trimetilsililéteres y se analizan
mediante cromatografía de gases con columna capilar.
Procedimiento
a) Preparación del insaponificable.

ELIZABETH CARACUEL PAREJA
129
- Con la microjeringa de 500 μl introducir en el matraz de 250 ml un volumen de
disolución de α-colestanol al 0,2 % en cloroformo que contenga una cantidad de
α-colestanol correspondiente al 10 % aproximadamente del contenido de
esteroles en la alícuota de la muestra para la determinación.
- Evaporar en corriente de nitrógeno hasta sequedad y, a continuación, pesar
con precisión, en el mismo matraz, 5 g de muestra seca y filtrada.
- Añadir 50 ml de solución etanólica de hidróxido potásico 2N, adaptar el
refrigerante de reflujo y calentar en baño maría con ligera ebullición, agitando
enérgica e ininterrumpidamente hasta que se produzca la saponificación (la
solución se vuelve límpida). Calentar durante 20 minutos más y, a continuación,
añadir 50 ml de agua destilada por la parte superior del refrigerante; separar el
refrigerante y enfriar el matraz a 30 °C aproximadamente.
- Transvasar cuantitativamente el contenido del matraz a un embudo de
separación de 500 ml, mediante varios lavados con un total aproximado de 50
ml de agua destilada. Agregar 80 ml aproximadamente de éter etílico, agitar
enérgicamente durante unos 30 segundos y dejar reposar hasta la separación
de las fases.
- Separar la fase acuosa inferior pasándola a un segundo embudo de separación.
Efectuar otras dos extracciones de la fase acuosa por el mismo procedimiento,
utilizando cada vez de 60 a 70 ml de éter etílico.
- Reunir las fracciones etéreas en un mismo embudo de separación y lavarlas
con agua destilada (50 ml cada vez) hasta que el agua de lavado presente
reacción neutra.
- Una vez eliminada el agua de lavado, secar con sulfato sódico anhidro y filtrar
sobre sulfato sódico anhidro a un matraz de 250 ml previamente pesado, lavando
el embudo y el filtro con pequeñas cantidades de éter etílico.
- Destilar el éter hasta que queden unos pocos ml; a continuación, secar con un
vacío ligero o en una corriente de nitrógeno, completando el secado en una
estufa a 100 °C durante 15 minutos aproximadamente; dejar enfriar en un
desecador y pesar.
b) Separación de la fracción de esteroles.

ELIZABETH CARACUEL PAREJA
130
- Preparación de las placas básicas: sumergir completamente las placas con gel
de sílice en la solución etanólica 0,2N de hidróxido potásico durante 10
segundos; dejar secar las placas en campana durante dos horas y, por último,
mantenerlas en una estufa regulada a 100 °C durante una hora. Sacarlas de la
estufa y conservarlas en un desecador de cloruro de calcio hasta el momento del
uso.
- Introducir en la cubeta de desarrollo de las placas una mezcla de benceno-
acetona 95:5 (v/v) hasta una altura de 1 cm aproximadamente. Cerrar la cubeta
con su correspondiente tapa y dejar transcurrir media hora como mínimo, de
forma que se alcance el equilibrio líquido-vapor.
- Preparar una solución de insaponificable en cloroformo al 5 % y depositar 0,3
ml de dicha solución en una placa cromatográfica a unos 2 cm de uno de los
bordes, formando una línea lo más fina y uniforme posible. A la altura de la línea
de aplicación se depositan, en un extremo de la placa, de 2 a 3 μl de la solución
de referencia de para poder identificar la banda de esteroles una vez efectuado
el desarrollo.
- Introducir la placa en la cubeta de desarrollo. Se deberá mantener una
temperatura ambiente entre 15 y 20 °C. Tapar inmediatamente la cubeta y dejar
que se produzca la elución hasta que el frente del disolvente se sitúe a 1 cm
aproximadamente del borde superior de la placa. Sacar la placa de la cubeta y
evaporar el disolvente en una corriente de aire caliente o dejando la placa bajo
una campana unos minutos.
- Pulverizar la placa ligera y uniformemente con la solución de 2,7-
diclorofluoresceína. Al examinar la placa a la luz ultravioleta puede identificarse
la banda de los esteroles mediante comparación con la mancha obtenida a partir
de la solución de referencia; marcar con lápiz negro los límites de la banda a lo
largo de los márgenes de fluorescencia.
- Rascar con una espátula metálica el gel de sílice contenido en el área
delimitada. Introducir el material obtenido, finamente triturado, en el embudo
filtrante; añadir 10 ml de cloroformo caliente, mezclar cuidadosamente con la
espátula metálica y filtrar en vacío, recogiendo el filtrado en el matraz cónico
acoplado al embudo filtrante.

ELIZABETH CARACUEL PAREJA
131
- Lavar el residuo en el embudo tres veces con éter etílico (empleando cada vez
unos 10 ml), recogiendo cada vez el filtrado en el mismo matraz cónico acoplado
al embudo.
- Evaporar el filtrado hasta obtener un volumen de 4 a 5 ml, transvasar la solución
residual al tubo de ensayo de 10 ml previamente pesado, evaporar hasta
sequedad mediante calentamiento suave en corriente ligera de nitrógeno,
recoger con algunas gotas de acetona, evaporar de nuevo hasta sequedad,
introducir en una estufa a 105 °C durante unos 10 minutos, dejar enfriar en el
desecador y pesar.
El residuo que queda en el tubo de ensayo está formado por la fracción de
esteroles.
c) Preparación de los trimetilsililéteres.
- Agregar al tubo que contiene la fracción de esteroles el reactivo de silanización
formado por una mezcla de piridina-hexametildisilazano trimetilclorosilano 9:3:1
(v/v/v), a razón de 50 μl por miligramo de esteroles, evitando toda absorción de
humedad.
- Tapar el tubo y agitar cuidadosamente (sin invertir) hasta la completa disolución
de los esteroles. Dejar reposar un cuarto de hora, como mínimo, a temperatura
ambiente y centrifugar durante algunos minutos; la solución límpida queda lista
para el análisis mediante cromatografía de gases.
d) Cromatografía de gases.
Tras realizar las operaciones preliminares como el acondicionamiento de la
columna y hacer la elección de las condiciones de trabajo, se procede a la
realización del análisis.
- Con la microjeringa de 10 μl tomar 1 μl de hexano, aspirar 0,5 μl de aire y, a
continuación, entre 0,5 y 1 μl de la solución problema; elevar el émbolo de la
jeringa de modo que la aguja quede vacía. Introducir la aguja a través del septum
y, después de 1 o 2 segundos, inyectar rápidamente; transcurridos unos 5
segundos, extraer la aguja lentamente.

ELIZABETH CARACUEL PAREJA
132
- Continuar el registro hasta la completa elución de los TMSE de los esteroles
presentes.
Para la identificación de los diferentes picos se utilizan los tiempos de retención
y la comparación con mezclas de TMSE de los esteroles analizadas en las
mismas condiciones.
La elución de los esteroles se efectúa en el orden siguiente: colesterol,
brasicasterol, 24-metilencolesterol, campesterol, campestanol, estigmasterol, Δ-
7-campesterol, Δ-5,23-estigmastadienol, clerosterol, β-sitosterol, sitostanol, Δ-5-
avenasterol, Δ-5,24-estigmastadienol, Δ-7-estigmastenol, Δ-7-avenasterol.
Reactivos necesarios
- Hidróxido potásico, solución etanólica 2N aproximadamente
- Éter etílico de calidad para análisis.
- Sulfato sódico anhidro de calidad para análisis
- Placas de vidrio recubiertas con gel de sílice
- Hidróxido potásico, solución etanólica 0,2N
- Benceno para cromatografía.
- Acetona para cromatografía.
- Hexano para cromatografía.
- Éter etílico para cromatografía.
- Cloroformo de calidad para análisis.
- Solución patrón para cromatografía en capa fina
- Solución de 2,7-diclorofluoresceína al 0,2 % en etanol
- Piridina anhidra para cromatografía.
- Hexametildisilazano.
- Trimetilclorosilano.
- Solución problema de trimetilsililéteres de los esteroles

ELIZABETH CARACUEL PAREJA
133
- α-colestanol, disolución al 0,2 % (m/V) en cloroformo (patrón interno).
- Gas portador: hidrógeno o helio de calidad para cromatografía de gases.
Material e instrumentación
- Matraz de 250 ml
- Embudos de separación de 500 ml.
- Matraces de 250 ml.
- Equipo completo de cromatografía en capa fina
- Lámpara ultravioleta de una longitud de onda de 366 o 254 nm.
- Microjeringa de 100 μl y 500 μl.
- Embudo cilíndrico filtrante
- Matraz cónico para vacío de 50 ml
- Probeta de 10 ml de fondo cónico con tapón hermético.
- Equipo de cromatografía de gases que pueda funcionar con columna capilar
- Columna capilar de vidrio o sílice fundida
- Microjeringa de 10 μl para cromatografía de gases
Resultados
- Calcular las áreas de los picos del α-colestanol y de los esteroles utilizando el
integrador. El factor de respuesta para el α-colestanol debe considerarse como
1.
- Calcular del modo siguiente el contenido de cada uno de los esteroles,
expresado en mg/100 g de materia grasa:
𝑒𝑠𝑡𝑒𝑟𝑜𝑙 𝑥 = 𝐴𝑥 · 𝑚𝑠 · 100
𝐴𝑠 · 𝑚
siendo:
Ax: área del pico del esterol x
As: área del pico del α-colestanol,

ELIZABETH CARACUEL PAREJA
134
ms: peso de α-colestanol añadido, en miligramos,
m: peso de la muestra tomado para la determinación, en gramos.
El contenido de cada uno de los esteroles se expresa en mg/100 g de materia
grasa y, como «esteroles totales», su suma.
El porcentaje de cada uno de los esteroles simples es la razón entre el área del
pico correspondiente y la suma de las áreas de los picos de los esteroles.
% 𝑑𝑒𝑙 𝑒𝑠𝑡𝑒𝑟𝑜𝑙 𝑥 = 𝐴𝑥
∑𝐴· 100
siendo:
Ax: área del pico de x,
ΣA: suma de las áreas de todos los picos.
5.1.2.8 Medida de la concentración en eritrodiol y uvaol
Cuando el contenido de ceras se encuentra entre 300 y 350 mg/Kg, se
debe determinar el contenido de los alcoholes alifáticos eritrodiol y uvaol. Estos
parámetros son necesarios para distinguir si las ceras provienen un proceso de
extracción (mayor al 3,5%) o de un proceso de esterificación (menor o igual a
3,5%).
Fundamento
El eritrodiol, entendido corrientemente como conjunto de los dioles eritrodiol y
uvaol, es un componente del insaponificable, característico de algunos tipos de
materias grasas.
Su concentración es mucho más elevada en los aceites de oliva obtenidos
mediante extracción que en los obtenidos mediante presión o en el aceite de
pepita de uva y, por lo tanto, su determinación puede servir para comprobar la
existencia de aceite de oliva obtenido mediante extracción.

ELIZABETH CARACUEL PAREJA
135
La materia grasa se saponifica con una solución etanólica de hidróxido potásico;
a continuación, se extrae el insaponificable con éter etílico y se purifica
pasándolo por una columna de alúmina.
El fraccionamiento del insaponificable se realiza mediante cromatografía en capa
fina en placa de gel de sílice y se aíslan la banda de la fracción esterólica y la
del eritrodiol.
Los esteroles y el eritrodiol recuperados de la placa se transforman en
trimetilsililéteres y seguidamente se analiza la mezcla mediante cromatografía
de gases.
El resultado se expresa en porcentaje de eritrodiol respecto del conjunto de
eritrodiol + esteroles.
Procedimiento
a) Preparación del insaponificable
- Con la microjeringa de 500 μl introducir en el matraz de 250 ml un volumen de
disolución de α-colestanol al 0,2 % en cloroformo que contenga una cantidad de
α-colestanol correspondiente al 10 % aproximadamente del contenido de
esteroles en la alícuota de la muestra para la determinación.
- Evaporar en corriente de nitrógeno hasta sequedad y, a continuación, pesar
con precisión, en el mismo matraz, 5 g de muestra seca y filtrada.
- Añadir 50 ml de solución etanólica de hidróxido potásico 2N, adaptar el
refrigerante de reflujo y calentar en baño maría con ligera ebullición, agitando
enérgica e ininterrumpidamente hasta que se produzca la saponificación (la
solución se vuelve límpida). Calentar durante 20 minutos más y, a continuación,
añadir 50 ml de agua destilada por la parte superior del refrigerante; separar el
refrigerante y enfriar el matraz a 30 °C aproximadamente.
b) Separación del eritrodiol y los esteroles
- Preparación de las placas básicas: sumergir completamente las placas con gel
de sílice en la solución etanólica 0,2N de hidróxido potásico durante 10
segundos; dejar secar las placas en campana durante dos horas y, por último,
mantenerlas en una estufa regulada a 100 °C durante una hora. Sacarlas de la

ELIZABETH CARACUEL PAREJA
136
estufa y conservarlas en un desecador de cloruro de calcio hasta el momento del
uso.
- Introducir en la cubeta de desarrollo de las placas una mezcla de benceno-
acetona 95:5 (v/v) hasta una altura de 1 cm aproximadamente. Cerrar la cubeta
con su correspondiente tapa y dejar transcurrir media hora como mínimo, de
forma que se alcance el equilibrio líquido-vapor.
- Preparar una solución de insaponificable en cloroformo al 5 % y depositar 0,3
ml de dicha solución en una placa cromatográfica a unos 1,5 cm del borde inferior
de la placa, formando una banda lo más fina y uniforme posible. Depositar en un
extremo de la placa, como referencia, algunos microlitros de las soluciónes de
colesterol y eritrodiol.
- Introducir la placa en la cubeta de desarrollo. La temperatura ambiente debe
ser de unos 20 °C. Tapar inmediatamente la cubeta y dejar que se produzca la
elución hasta que el frente del disolvente se sitúe a 1 cm aproximadamente del
borde superior de la placa. Sacar ésta de la cubeta de desarrollo y evaporar el
disolvente en una corriente de aire caliente.
- Pulverizar la placa uniformemente con la solución alcohólica de 2′, 7′
diclorofluoresceína. Al examinar la placa a la luz ultravioleta pueden identificarse
las bandas de los esteroles y del eritrodiol mediante comparación con las
referencias; delimitar las bandas con una punta ligeramente por el exterior de los
márgenes de fluorescencia.
- Rascar con una espátula metálica el gel de sílice contenido en las áreas
delimitadas. Reunir el material obtenido en un matraz cónico de 50 ml; añadir 15
ml de cloroformo caliente, agitar bien y filtrar en el embudo de filtro poroso
vertiendo el gel de sílice sobre el propio filtro. Lavar tres veces con 10 ml de
cloroformo caliente cada vez, recogiendo el filtrado en un matraz esférico de 100
ml. Evaporar hasta obtener un volumen de 4 a 5 ml, transvasar al tubo de
centrifugado de fondo cónico de 10 ml previamente tarado, evaporar hasta
sequedad mediante calentamiento suave en corriente de nitrógeno y pesar.
c) Preparación de los trimetilsililéteres.

ELIZABETH CARACUEL PAREJA
137
- Agregar al tubo que contiene la fracción de esteroles el reactivo de silanización
formado por una mezcla de piridina-hexametildisilazano trimetilclorosilano 9:3:1
(v/v/v), a razón de 50 μl por miligramo de esteroles, evitando toda absorción de
humedad.
- Tapar el tubo y agitar cuidadosamente (sin invertir) hasta la completa disolución
de los esteroles. Dejar reposar un cuarto de hora, como mínimo, a temperatura
ambiente y centrifugar durante algunos minutos; la solución límpida queda lista
para el análisis mediante cromatografía de gases.
d) Cromatografía de gases.
Tras realizar las operaciones preliminares como el acondicionamiento de la
columna y hacer la elección de las condiciones de trabajo, se procede a la
realización del análisis.
- Con la microjeringa de 10 μl tomar 1 μl de hexano, aspirar 0,5 μl de aire y, a
continuación, entre 0,5 y 1 μl de la solución problema; elevar el émbolo de la
jeringa de modo que la aguja quede vacía. Introducir la aguja a través del septum
y, después de 1 o 2 segundos, inyectar rápidamente; transcurridos unos 5
segundos, extraer la aguja lentamente.
- Continuar el registro hasta la completa elución de los TMSE de los esteroles
presentes.
Las condiciones operativas de la cromatografía de gases deben cumplir los
requisitos necesarios para analizar los esteroles y, además, permitir la
separación de los TMSE del eritrodiol y del uvaol.
Una vez inyectada la muestra, dejar que se desarrolle el proceso hasta que se
produzca la elución de los esteroles presentes, el eritrodiol y el uvaol; identificar
los picos (el eritrodiol y el uvaol tienen tiempos de retención relativos, respecto
al β-sitosterol, de alrededor de 1,45 y 1,55, respectivamente) y calcular sus áreas
de la misma forma que para los esteroles.
Para la identificación de los diferentes picos se utilizan los tiempos de retención
y la comparación con mezclas de TMSE de los esteroles analizadas en las
mismas condiciones.

ELIZABETH CARACUEL PAREJA
138
Reactivos necesarios
- Hidróxido potásico, solución etanólica 2N aproximadamente
- Éter etílico de calidad para análisis.
- Sulfato sódico anhidro de calidad para análisis
- Placas de vidrio recubiertas con gel de sílice
- Hidróxido potásico, solución etanólica 0,2N
- Benceno para cromatografía.
- Acetona para cromatografía.
- Hexano para cromatografía.
- Éter etílico para cromatografía.
- Cloroformo de calidad para análisis.
- Solución patrón para cromatografía en capa fina
- Solución de 2,7-diclorofluoresceína al 0,2 % en etanol
- Piridina anhidra para cromatografía.
- Hexametildisilazano.
- Trimetilclorosilano.
- Solución problema de trimetilsililéteres de los esteroles
- α-colestanol, disolución al 0,2 % (m/V) en cloroformo (patrón interno).
- Gas portador: hidrógeno o helio de calidad para cromatografía de gases.
- Solución patrón de eritrodiol al 0,5 % en cloroformo
Material e instrumentación
- Matraz de 250 ml
- Embudos de separación de 500 ml.
- Matraces de 250 ml.

ELIZABETH CARACUEL PAREJA
139
- Equipo completo de cromatografía en capa fina
- Lámpara ultravioleta de una longitud de onda de 366 o 254 nm.
- Microjeringa de 100 μl y 500 μl.
- Embudo cilíndrico filtrante
- Matraz cónico para vacío de 50 ml
- Probeta de 10 ml de fondo cónico con tapón hermético.
- Equipo de cromatografía de gases que pueda funcionar con columna capilar
- Columna capilar de vidrio o sílice fundida
- Microjeringa de 10 μl para cromatografía de gases
Resultados
𝐸𝑟𝑖𝑡𝑟𝑜𝑑𝑖𝑜𝑙, % =𝐴1 + 𝐴2
𝐴1 + 𝐴2 + ∑𝐴𝑒𝑠𝑡𝑒𝑟𝑜𝑙𝑒𝑠· 100
siendo:
A1: área del pico del eritrodiol,
A2: área del pico del uvaol,
ΣAesteroles: suma de las áreas de los esteroles presentes.
5.1.2.9 Medida del contenido en estigmastadienos en aceites vegetales
De los hidrocarburos naturales no se determina ninguno, sin embargo, en el
proceso de refinación se puede dar lugar a la producción de estirenos debido al
efecto de la temperatura. El estigmasta-3,5-dieno se produce en la
desodorización del esterol mayoritario del aceite de oliva. Analizando el
contenido en estigmastadienos se puede saber si un aceite de oliva virgen extra
ha sido adulterado con un aceite de oliva que ha sido sometido a altas
temperaturas durante el proceso de refinación.

ELIZABETH CARACUEL PAREJA
140
Para ello hay que separar la fracción de hidrocarburos esteroideos mediante
cromatografía en columna de gel de sílice y análisis mediante cromatografía de
gases en columna capilar.
Procedimiento
a) Preparación de la materia insaponificable:
- Pesar 20 ± 0,1 g de aceite en un matraz de 250 ml, añadir 1 ml de solución
patrón de colesta-3,5-dieno (20μg) y 75 ml de potasa alcohólica al 10 %, colocar
un refrigerante de reflujo y calentar a ebullición suave durante 30 minutos. Retirar
de la fuente de calor el matraz con la muestra y dejar enfriar ligeramente la
solución. Añadir 100 ml de agua y pasar la solución a un embudo de decantación
con ayuda de 100 ml de hexano. Agitar enérgicamente durante 30 segundos y
dejar decantar.
- Pasar la fase acuosa inferior a un segundo embudo de decantación y someterla
nuevamente a extracción con 100 ml de hexano. Separar nuevamente la fase
inferior y lavar los extractos de hexano con tres porciones de 100 ml cada una
de una mezcla de agua y etanol (1: 1) hasta obtener un pH neutro.
- Pasar la solución de hexano a través de sulfato sódico anhidro (50 g), lavar con
20 ml de hexano y evaporar a 30 °C y presión reducida en un rotavapor hasta
sequedad.
b) Separación de la fracción de hidrocarburos esteroideos:
- Introducir el residuo en la columna de fraccionamiento con ayuda de dos
porciones de 1 ml de hexano, distribuir la muestra en la columna dejando que el
nivel de solución descienda hasta la parte superior del sulfato de sodio y
comenzar la elución cromatográfica con hexano a un flujo aproximado de 1
ml/min. Desechar los primeros 25-30 ml de eluido y recoger la fracción siguiente
de 40 ml. Después, pasar esta fracción a un matraz de fondo redondo de 100 ml.
La primera fracción contiene los hidrocarburos saturados y la segunda fracción
los esteroideos. Al proseguir con la elución se obtiene escualeno y otros
compuestos relacionados. Para poder obtener una buena separación entre los
hidrocarburos saturados y los esteroideos, es necesario optimizar los volúmenes
de las fracciones.

ELIZABETH CARACUEL PAREJA
141
- Evaporar la segunda fracción en rotavapor a 30 °C y presión reducida hasta
sequedad. Disolver inmediatamente el residuo en 0,2 ml de hexano y mantener
la solución en el refrigerador hasta el momento de su análisis.
c) Cromatografía de gases
El sistema de cromatografía de gases deberá comprobarse inyectando una
mezcla de la solución madre de colestadieno y de n-nonacosano. El pico del
colesta-3,5-dieno deberá aparecer antes que el del nnonacosano; si esto no
ocurre, pueden adoptarse dos soluciónes: reducir la temperatura inicial del horno
o sustituir la columna de cromatografía de gases por otra de polaridad inferior.
La identificación de picos se realiza sabiendo que el pico del patrón interno
aparece a un tiempo de retención de, aproximadamente, 19 minutos, y el del
estigmasta-3,5-dieno lo hace a un tiempo de retención relativo de
aproximadamente 1,29. El estigmasta-3,5-dieno suele ir acompañado de
pequeñas cantidades de un isómero y, por lo general, ambos originan un solo
pico cromatográfico.
No obstante, si la columna es demasiado polar, o tiene un gran poder de
resolución, el isómero puede aparecer en forma de un pequeño pico justo antes
y cerca del estigmasta-3,5-dieno. En estos casos, es preciso sumar las áreas de
los dos picos. Con el fin de estar seguro de que los estigmastadienos eluyen
como un solo pico se recomienda instituir la columna por otra de menor polaridad
o de mayor diámetro interior.
Para obtener una referencia de los estigmastadienos puede utilizarse el análisis
de cualquier aceite vegetal refinado empleando menor cantidad de muestra (de
1 a 2 g). Los estigmastadienos dan lugar a un pico importante de fácil
identificación.
Reactivos necesarios
Salvo indicación en sentido contrario, únicamente se utilizarán reactivos de
calidad para análisis. Debe emplearse agua destilada o de pureza al menos
equivalente.
- Hexano o una mezcla de alcanos con un intervalo de ebullición de 65-70 ° C,
destilados en columna rectificadora.

ELIZABETH CARACUEL PAREJA
142
- Etanol de 96 v/v.
- Sulfato sódico anhidro.
- Solución alcohólica de hidróxido potásico al 10 %. Gel de sílice 60 para columna
de cromatografiaSolución madre (200 ppm) de colesta-3,5-dieno (Sigma, 99 %
de pureza) en hexano (10 mg en 50 ml).
- Solución patrón de colesta-3,5-dieno en hexano con una concentración de 20
ppm
- Solución de n-nonacosano en hexano con una concentración aproximada de
100 ppm.
- Gas portador para cromatografia: helio o hidrógeno de 99,9990 % de pureza.
- Gases auxiliares para el detector de ionización de llama: aire purificado e
hidrógeno de 99,9990 % de pureza.
Material e instrumentación
- Matraces de 250 ml aptos para colocarles un refrigerante de reflujo.
- Embudos de decantación con capacidad para 500 ml.
- Matraces de fondo redondo de 100 ml.
- Rotavapor.
- Columna de vidrio para cromatografía (1,5-2,0 cm de diámetro interno por 50
cm de longitud) provista de una llave de teflón y con una torunda de lana de vidrio
o un disco de vidrio unterizado en el fondoCromatógrafo de gases Columna
capilar de sílice fundida.
- Integrador-registrador con capacidad para modo de integración valle-valle.
- Microjeringa de 5-10 μl para cromatografía de gases con aguja fija.
- Calentador de tipo manta o placa eléctrica.
Resultados
El contenido de los estigmastadienos se determina aplicando la fórmula:

ELIZABETH CARACUEL PAREJA
143
𝑚𝑔
𝑘𝑔𝑑𝑒 𝑒𝑠𝑡𝑖𝑔𝑚𝑎𝑠𝑡𝑎𝑑𝑖𝑒𝑛𝑜𝑠 =
𝐴𝑠 · 𝑀𝑐
𝐴𝑐 · 𝑀𝑜
Siendo:
Ab = área del pico de estigmastadienos (si el pico se halla dividido en dos picos,
súmese el área de los dos picos.).
Ac = área del pico del patrón interno (colestadieno).
Mc = peso de patrón añadido, en microgramos.
Mo = peso de aceite empleado, en gramos.
Límite de detección: aproximadamente 0,01 mg/kg.
5.1.2.10 Humedad y volátiles
Aunque en el Reglamento 2568/91 no figura como características principales de
los aceites de oliva, el Código Alimentario Español establece una tolerancia en
los aceites comestibles de un máximo de 0,1% de humedad y materias volátiles,
por lo que es fundamental este análisis.
A pesar de ser el agua inmiscible con el aceite, ésta puede existir en forma de
emulsión estabilizada por ciertos componentes. La humedad favorece la
hidrólisis, sobre todo en aquellos aceites cuya acidez inicial es elevada.
Fundamento
Se establecen las condiciones adecuadas para la determinación, en las materias
grasas, del agua y de las materias volátiles, operando en las condiciones de
ensayo.
Es aplicable a las grasas animales y vegetales, con la excepción de los aceites
secantes o semisecantes y los aceites del grupo del coco.
Procedimiento
1. Preparación de la muestra.

ELIZABETH CARACUEL PAREJA
144
- La muestra debe ser previamente homogeneizada antes de pesar la cantidad
con que se vaya a operar. Esto se logra, con las grasas fluidas, agitando
fuertemente el frasco que contiene la muestra y vertiendo rápidamente la
cantidad aproximada que se vaya a pesar en la cápsula en la que se efectúe la
desecación. Si se tratase de grasas sólidas o semisólidas a la temperatura
ambiente, calentar suavemente en baño de agua hasta conseguir el grado de
fluidez conveniente, cuidando de no llegar a fundir completamente y
homogeneizar con una espátula.
2. Técnica operatoria.
- En una cápsula, desecada previamente en estufa a 105ºC y enfriada en un
desecador, pesar, con exactitud de 1 mg, una cantidad aproximada de 5 a 10 g
de muestra, según el contenido de humedad esperado (previamente debe
pesarse la cápsula).
Colocar en la estufa, previamente regulada a 105ºC, manteniéndola allí durante
30 minutos. Sacar y pasar a un desecador donde se deja enfriar; pesar a
continuación.
Repetir este tratamiento en operaciones sucesivas hasta que la diferencia entre
dos pesadas consecutivas no exceda del 0.05%.
Material y aparatos
- Estufa de desecación, con regulación de temperatura, pudiéndose calentar
hasta 150ºC como mínimo; la regulación se efectuará entre unos límites de
oscilación de ± 2ºC.
- Cápsula de fondo plano de porcelana
- Desecador, conteniendo como agente desecante Sulfato Cálcico ó Gel de Sílice
con indicador o, en su defecto, Ácido Sulfúrico 96%.
Resultados
%Humedad y materia volátil =𝑃𝑎 − 𝑃𝑏
𝑃𝑚 𝑥 100
Siendo:

ELIZABETH CARACUEL PAREJA
145
Pa: peso, en gramos, de la cápsula con la materia grasa
Pb: peso, en gramos, de la cápsula con la grasa después de la desecación
Pm: peso, en gramos, de la muestra
5.1.2.11 Impurezas solubles en éter de petróleo
Fundamento
Permite la determinación de un conjunto de sustancias que son insolubles en un
disolvente orgánico volátil y que no hayan sido determinadas como humedad y
materia volátil.
El disolvente más utilizado para esta determinación es el éter de petróleo,
aunque también puede emplearse éter etílico.
La determinación se realiza mediante la norma UNE 55002 y se establecen los
valores máximos de impurezas, para el aceite de oliva virgen en el 0,1%.
Procedimiento
En una cápsula, previamente desecada, se pesan 10 g de aceite y se coloca en
una estufa a 105ºC hasta que se produzca la eliminación total del agua.
Una vez que la muestra está desecada, se van agregando pequeñas cantidades
de éter de petróleo a la cápsula para que se disuelva la grasa, ayudándose de
una varilla. La cantidad necesaria de disolvente es de 200 ml. A continuación, se
va filtrando a través de un filtro, previamente desecado y tarado, lavando el filtro
con pequeñas porciones de disolvente y recogiendo el filtrado en un matraz. Una
vez que el filtro está limpio de grasa se coloca en la estufa a 60ºC. El incremento
de peso del filtro dará las impurezas, que se referirán al % en la muestra.
Material e instrumentación
- Desecador
- Balanza analítica
- Estufa calefactora

ELIZABETH CARACUEL PAREJA
146
- Cápsula de porcelana
- Filtro
- Matraz erlenmeyer
Resultados
% Impurezas =Pi − Pf
P x 100
Siendo:
Pf= Peso en g del filtro seco
Pi= Peso en g del filtro seco más las impurezas
P= Peso en g de la muestra
5.1.2.12 Índice de refracción
Fundamento
Cuando una onda de cualquier tipo alcanza la frontera de dos medios distintos,
una parte de su energía se transmite al segundo medio, dando lugar en el
segundo medio a otra onda de características semejantes a las de la onda
incidente y que recibe el nombre de onda transmitida. Otra parte de la energía
se emplea en generar otra onda que se propaga hacia atrás en el primer medio
y que se llama onda reflejada.
Se llama n1 y n2 a los índices de refracción de cada medio. El índice de
refracción de un medio es el cociente entre la velocidad de la luz en el vacío y la
velocidad de la luz en ese medio. No tiene unidades y siempre es mayor o igual
que 1.
Procedimiento
Se coloca una gota de aceite en el refractómetro a 20ºC, se mira por el visor y
se anota el resultado que se observa en la escala.

ELIZABETH CARACUEL PAREJA
147
5.1.2.13 Índice de yodo. Método de Wijs
Fundamento
El índice de yodo de una materia grasa es función de su grado de insaturación.
Se determina añadiendo a la muestra un exceso de reactivo halogenante,
reduciendo el reactivo que no reacciona con yoduro potásico, y valorando el yodo
generado en la disolución con tiosulfato sódico en presencia de almidón.
Paralelamente debe prepararse un blanco, que no contenga muestra, en el que
se valorará la totalidad del reactivo añadido.
Las reacciones a considerar son:
(a) Adición del reactivo halogenante a los enlaces C-C insaturados de la muestra
R-CH=CH...-CH3 + ICl (en exceso) R-CHCl-CHI…-CH3 + exceso de ICl
(b) Reducción del exceso de reactivo halogenante con disolución de KI:
exceso de ICl + I- (K+) en exceso I2 + Cl- (K+)
(c) Valoración del yodo liberado con solución de tiosulfato sódico:
I2 + 2 S2O32- 2 I- + S4O62-
Procedimiento
(Trabajar en luz difusa a ser posible) En un matraz erlenmeyer, limpio y seco,
provisto de tapón, pesar una cantidad apropiada de muestra, en función del
índice de yodo previsto, tal y como se indica en la tabla 5.5:
Tabla 5.5. índice de Yodo previsto
Índice de Yodo previsto Peso de uestra, g
<5 3
5-20 1
21-50 0,40
51-100 0,20
101-150 0,13
151-200 0,10

ELIZABETH CARACUEL PAREJA
148
Añadir 20 ml de disolvente (una mezcla de volúmenes iguales de ciclohexano y
ácido acético) para disolver la grasa. Añadir, mediante pipeta aforada, 25 ml
exactos del reactivo de Wijs (usar una pera de goma para pipetear el reactivo y
trabajar en una campana de gases), tapar el matraz, mezclar por agitación suave
y dejar reposar al abrigo de la luz (en el interior de la taca). Para muestras con
un índice de yodo inferior a 150, mantener los matraces en la oscuridad durante
1 hora; para las muestras con un índice de yodo superior a 150, mantener en la
oscuridad durante 2 horas.
Transcurrido el tiempo correspondiente, añadir a cada uno de los matraces 20
ml de solución de KI al 10 % (p/V) y 150 ml de agua destilada.
Valorar con solución de tiosulfato sódico 0.1 M agitando constantemente hasta
que haya desaparecido casi totalmente el color amarillo producido por el yodo.
Añadir entonces unas gotas de engrudo de almidón como indicador y continuar
la valoración hasta el momento preciso en que desaparezca el color azul
después de una agitación muy intensa.
Preparar del mismo modo una prueba en blanco en idénticas condiciones, pero
sin contener materia grasa.
Se deben realizar dos determinaciones y un blanco para cada muestra.
Reactivos Necesarios
- Ciclohexano
- Ácido acético glacial
- Disolución de KI al 10 % (p/V), exenta de yodatos o de yodo libre
- Disolución de Na2S2O3. 5H2O 0.1 M (valorada como máximo 7 días antes de su
uso)
- Solución indicadora de almidón. Mezclar 5 g de almidón soluble con 30 ml de
agua, añadir la mezcla a 1000 ml de agua en ebullición, hervir durante 3 minutos
y dejar enfriar
- Reactivo de Wijs, que contenga monocloruro de yodo en ácido acético

ELIZABETH CARACUEL PAREJA
149
- Disolvente, preparado mezclando volúmenes iguales de ciclohexano y ácido
acético
Material e instrumentación
- Matraz erlenmeyer de 250 ml con boca esmerilada y provisto de tapón
- Bureta de 25 o 50 ml
- Pipeta aforada de 25 ml
- Pera
- Probeta de 50 o 100 ml
- Balanza analítica
Resultados
Convencionalmente, se calcula el índice de yodo como el peso de I2 absorbido
por la muestra en las condiciones de trabajo que se especifican en esta norma
internacional. Se expresa en gramos de yodo por 100 g de muestra.
Í𝑛𝑑𝑖𝑐𝑒 𝑑𝑒 𝑌𝑜𝑑𝑜 =12.69 𝑥 𝑐 𝑥 (𝑉1 − 𝑉2)
𝑃
Siendo:
c: concentración exacta, expresada en moles/l, de la disolución de tiosulfato
sódico utilizada
V1: volumen en ml de disolución de tiosulfato sódico utilizado para el ensayo en
blanco
V2: volumen en ml de disolución de tiosulfato sódico utilizado para la
determinación de la muestra
P: peso en g de la muestra utilizado en la determinación

ELIZABETH CARACUEL PAREJA
150
5.1.2.14 Índice de saponificación
Fundamento
Por la acción de los álcalis la grasa se saponifica. Las cantidades de álcalis
gastados en la saponificación para formar los estearatos, palmitatos, oleatos,
etc, alcalinos, dependen de la naturaleza y proporción de los ácidos que existen
en la materia grasa.
La determinación se realiza saponificando la muestra con una disolución
previamente valorada de hidróxido potásico en alcohol etílico y valorando a
continuación por retroceso el hidróxido potásico no consumido con solución
contrastada de ácido clorhídrico:
1.Saponificación de la muestra por tratamiento con una base fuerte
Triglicérido + KOH (en exceso) Jabones potásicos + Glicerina + exc KOH
2.Valoración por retroceso del exceso de base fuerte
exceso de KOH + HCl KCl + H2O
Procedimiento
Pesar, con precisión de 1 mg, en un matraz de fondo redondo de 250 ml, 2 g
aproximadamente de muestra. Agregar 25 ml exactamente medidos (con pipeta
aforada) de disolución etanólica de KOH 0.5 M. Adaptar el matraz a un
refrigerante de reflujo, llevar a ebullición y mantener durante sesenta minutos,
agitando por rotación de vez en cuando. Retirar de la fuente de calor. Una vez
que la disolución se haya enfriado en parte, sin llegar a gelatinizarse, agregar 4-
5 gotas de solución de fenolftaleína y valorar la disolución jabonosa, todavía
caliente, con disolución de ácido clorhídrico 0.5 M.
Realizar en las mismas condiciones un ensayo en blanco y hacer una sola
determinación (un blanco y una muestra).
Este método es aplicable a aceites y grasas con un contenido en ceras inferior
al 5%.

ELIZABETH CARACUEL PAREJA
151
Reactivos Necesarios
- Solución etanólica valorada de hidróxido potásico 0.5 M
- Solución acuosa valorada de HCl 0.5 M
- Solución etanólica de fenolftaleína al 1 %
Material e instrumentación
- Matraz de fondo redondo de 250 ml
- Refrigerante de reflujo
- Pipeta aforada de 25 ml
- Bureta de 25 ó 50 ml
- Balanza analítica
Resultados
El índice de saponificación expresa el peso en mg de hidróxido potásico
necesario para saponificar 1 g de materia grasa.
Í𝑛𝑑𝑖𝑐𝑒 𝑑𝑒 𝑆𝑎𝑝𝑜𝑛𝑖𝑓𝑖𝑐𝑎𝑐𝑖ó𝑛 =(𝑉 − 𝑉′)𝑥 𝑐 𝑥 56,1
𝑃
Siendo:
c: concentración exacta, en moles/l, de la disolución de HCl utilizada
V: volumen en ml de disolución de HCl 0.5 M gastado en el ensayo en blanco
V': volumen en ml de disolución de HCl 0.5 N gastado en la muestra
P: peso en g de la muestra utilizado en la determinación

ELIZABETH CARACUEL PAREJA
152
5.1.3 Análisis de subproductos
5.1.3.1 Humedad en orujos (UNE 55031)
La humedad contenida en el orujo de aceituna se determina por desecación de
la muestra, en una estufa de circulación forzada de aire.
Procedimiento
- Desmenuzar la muestra triturándola en un mortero.
- Tarar y desecar la cápsula para pesar 40 g de muestra.
- Introducir la muestra en la estufa a 105ºC durante 6-8 horas.
- Sacar la muestra de la estufa, dejarla enfriar y pesarla. Hay que repetir esta
operación hasta alcanzar una variación inferior a 0,02 g entre dos pesadas
consecutivas.
Resultados
La diferencia de peso, después de la desecación, referida a %, expresa la
humedad de la muestra.
%𝐻𝑢𝑚𝑒𝑑𝑎𝑑 = 𝑃𝑖𝑛𝑖𝑐𝑖𝑎𝑙 − 𝑃𝑓𝑖𝑛𝑎𝑙
𝑃𝑒𝑠𝑜 𝑚𝑢𝑒𝑠𝑡𝑟𝑎· 100
5.1.3.2 Materia grasa en orujos (UNE 55032)
La materia grasa total contenida en el orujo se extrae con hexano. El
procedimiento empleado es el método Soxhlet, ya explicado en el apartado
5.1.1.5. Este proceso se puede hacer sobre la pasta húmeda o seca.

ELIZABETH CARACUEL PAREJA
153
5.1.3.3 Materia grasa en alpechines
Los alpechines son efluentes líquidos compuestos por el agua de vegetación de
la aceituna y las aguas de lavado utilizadas en el proceso industrial de las
almazaras. Son considerados como subproductos y no como residuos.
Nota: en el BOJA nº41 de 1 de marzo de 2011 se publicó la Orden de la
Conserjería de Agricultura y pesca de 18 de febrero de 2011, por la que se regula
el régimen de autorización para la utilización de los efluentes líquidos resultantes
de la extracción del aceite de oliva en las almazaras, como fertilizante en suelos
agrícolas en la Comunidad Autónoma de Andalucía. En el preámbulo dice:
La extracción de aceite procedente de aceituna es una actividad económica de
gran importancia en Andalucía. Como resultado de esta actividad se obtiene un
efluente líquido, constituido por las aguas de lavado de las aceitunas y las aguas
de lavado de los aceites obtenidos mediante el sistema de extracción de dos
fases. Estos efluentes son recursos inagotables susceptibles de ser utilizados en
suelos agrícolas para restituir parte de las extracciones provocadas por el cultivo.
La determinación de la materia grasa en alpechines se puede hacer por el
método Soxhlet. Para ello es necesario eliminar previamente el agua hasta
alcanzar la desecación total de la muestra.
Procedimiento
- Se pesan 100 g de muestra en una cápsula revestida de papel de estaño.
- Poner la muestra en la estufa a 110ºC, durante 24 horas, hasta conseguir la
desecación total.
- Poner la muestra desecada junto con el papel de estaño en el cuerpo del
extractor Soxhlet.
- Proceder al proceso de extracción mediante Soxhlet, explicado en el apartado
5.1.1.5.

ELIZABETH CARACUEL PAREJA
154
5.1.4 Análisis foliar
El análisis foliar determina la cantidad de nutrientes que la planta ha absorbido y
supone la mejor manera de conocer las carencias de los cultivos. Aunque la
apariencia de un cultivo sea buena, es posible que alguno de los nutrientes no
se encuentre en cantidad suficiente, y no se lleve a cabo un desarrollo
satisfactorio. En combinación con el análisis de suelo nos permite detectar
problemas nutricionales y elaborar adecuados planes de fertilización.
Con los elementos analizados se acompaña un informe en el que se establece,
el diagnóstico nutricional de todos los nutrientes analizados, indicando en cada
caso:
• La función realizada por el nutriente en la planta
• La posible carencia o déficit del elemento en cuestión
• Interacciones entre nutrientes, teniendo en cuenta posibles antagonismos y
sinergismos entre varios elementos
• Cálculo de los equilibrios fisiológicos, índice vegetativo e índice de hierro
• Correctores de carencias a aplicar con sus dosis y épocas de aplicación, para
la solución de los problemas nutricionales detectados
Además de permitir detectar carencias nutricionales en el cultivo, el análisis foliar
es básico en los casos en que se aplica fertirrigación, una técnica cada vez más
extendida en nuestro país, ya que permite actuar de inmediato para corregir los
desequilibrios nutritivos que la planta pueda presentar incorporando a través del
agua de riego los nutrientes requeridos, que son rápidamente absorbidos por los
cultivos.
5.1.4.1 Toma de muestra
La parcela a analizar tiene que dividirse en unidades de muestreo. Cada unidad
de muestreo es un conjunto de plantas que resulten visualmente parecidas. Si
hay alguna zona claramente diferente del resto, pero muy pequeña, no se
tomarán hojas de ella. Es interesante recorrer en diagonal la unidad de muestreo.

ELIZABETH CARACUEL PAREJA
155
Las hojas a muestrear se toman de la zona media de la planta. Se deberán coger
hojas enteras y con peciolo, que no estén dañadas, atacadas por plagas ni
enfermedades y tampoco hojas que hayan tenido algún tratamiento foliar
reciente. Se cogerán las hojas de los ramos del crecimiento del año (brotes)
elegidos al azar, pero desechando los brotes “chupones” y aquellos que
presenten alguna anormalidad. Los ramos del año anterior que sustentan a los
brotes elegidos pueden “tener” o “no tener” aceitunas.
Se debe tomar la misma cantidad de hojas de las cuatro orientaciones: norte,
sur, este y oeste. Para ello es bastante práctico coger cuatro hojas por olivo
muestreado, una de cada orientación, de tal manera que por cada parcela
homogénea se muestrearán un mínimo de 50 olivos.
Se cogerá la hoja entera (limbo y peciolo) situada hacia la mitad del brote del
año, 3º o 4º par de hojas a partir del ápice, debiendo estar totalmente
desarrollada y expandida; y no presentar ningún síntoma de anormalidad (daño
por plaga o enfermedad, necrosis, etc.).
Las hojas han de estar el menor tiempo posible en contacto con las manos para
evitar problemas de contaminación. Para ello se cogerán por los bordes o
peciolos y se introducirán inmediatamente en los sobres.
Figura 5.5. Brote de olivo

ELIZABETH CARACUEL PAREJA
156
5.1.4.2 Época de muestreo
La toma de muestras se hace cuando el contenido de elementos dentro de la
hoja es prácticamente constante, y siempre en la misma época. Se han de coger
en el período de tiempo comprendido desde finales de junio hasta primeros de
agosto, siendo preferible la segunda decena del mes de julio, período en el que
mejor pueden detectarse los estados carenciales y en el que están determinados
los niveles críticos.
5.1.4.3 Preparación de la muestra
Las operaciones descritas a continuación tienen por finalidad conseguir una
muestra para el análisis lo más homogénea posible. Por ello, toda simplificación
o tratamiento insuficiente en esta operación, puede conducir a unos resultados
que no sean representativos.
Procedimiento
- Lavar la hoja con una solución de detergente no iónico, frotando ambas caras,
lavar dos veces con agua corriente y por último otras dos veces con agua
desionizada. Todo proceso de lavado no puede ser superior a 20-30 s. Este
tratamiento no debe realizarse en caso de que la hoja esté destinada a
determinaciones analíticas que permitan comprobar un tratamiento de
contaminación.
- Desecar las hojas depositadas en una bandeja de fondo de pomo a 60 ºC con
una estufa de aire forzado durante el tiempo necesario para alcanzar el peso
constante (16-24h).
- La muestra desecada, si es posible, se muele en caliente con un molinillo de
aspas metálicas comprobando previamente que no haya posible contaminación.
El tamaño de la muestra debe ser lo suficientemente grande para que sea
representativa.
- Una vez reducida la hoja a polvo, introducirla en un recipiente de cierre
hermético desecándose unas horas sin tapón y cerrándolo en caliente para su
almacenamiento.

ELIZABETH CARACUEL PAREJA
157
Reactivos necesarios
- Solución de un detergente no iónico al 1%
Materiales e instrumentación
- Estufa con aire forzado
- Bandeja de fondo de pomo
- Molinillo de aspas metálicas
- Cepillo de cerda natural suave
5.1.4.4 Mineralización de la muestra
La mineralización consiste en la destrucción de la materia orgánica por
incineración en horno de mufla a 450ºC. Es aplicable a todos los elementos
excepto aquellos en los cuales se especifique otro procedimiento.
Procedimiento
Para 2 g de muestra previamente preparada que se coloca en una cápsula de
platino o en un crisol:
- Introducir la cápsula con la muestra en el horno de mufla aun frío y elevar la
temperatura hasta 450 ºC en un tiempo de 2h. Mantener la temperatura durante
2 h y enfriar la muestra en un desecador.
- Humedecer las cenizas en 2 o 3 g de agua y 1 ml de ácido clorhídrico
concentrado que se agregará lentamente.
- Calentar sobre la placa hasta la aparición de los primeros vapores y añadir
algunos ml de agua.
- Filtrar con filtro exento de cenizas sobre un matraz de 100 ml lavando la cápsula
3 o 4 veces con agua templada.
- Pasar el filtro a la cápsula, secarlo e incinerarlo durante media hora a 550ºC
como máximo.
- Añadir 5ml de ácido fluorhídrico.

ELIZABETH CARACUEL PAREJA
158
- Llevar hasta sequedad en baño o hasta placa calefactora, filtrar sobre el mismo
matraz de 100 ml y una vez se haya enfriado el filtrado, enrasar con agua hasta
100ml.
Efectuar un ensayo en blanco a la misma vez que la muestra.
Reactivos necesarios
- Ácido clorhídrico concentrado (d=1,19 g/ml)
- Ácido fluorhídrico
Todos los reactivos deben estar exentos de los elementos que vamos a
determinar y el agua que se utilice debe ser desmineralizada.
Material e instrumentación
- Cápsula de platino o crisol
- Horno de mufla
- Probeta de polietileno de 5ml
- Material de vidrio volumétrico contrastado.
Una vez preparada la muestra para realizar los análisis convenientes se pueden
hacer las determinaciones a las hojas de olivo.
5.1.4.5 Determinación de nitrógeno
Fundamento
Atacando el material vegetal con ácido sulfúrico concentrado, a ebullición en
presencia de un catalizador, el nitrógeno se transformará en sulfato amónico. Se
destila en presencia de un exceso de hidróxido sódico y se valora el amoniaco
destilado con ácido sulfúrico 14N.
Procedimiento
- Pesar 200 mg de muestra vegetal previamente preparada e introducirla en un
matraz de 250 ml evitando que se depositen en el cuello del matraz.

ELIZABETH CARACUEL PAREJA
159
- Agregar 5 ml de ácido sulfúrico concentrado y dejar en contacto durante media
hora.
- Añadir 20 ml de catalizador y 2-3 bolas de vidrio.
- Calentar (suavemente al principio) hasta llevar a ebullición.
- Llevar a decoloración completa, que suele tener una duración de 30 min. La
duración total del calentamiento es media hora.
- Enfriar y añadir 30 ml de agua. En el momento de destilar, añadir de una sola
vez 25 ml de lejía de sosa y fijar el matraz al aparato de arrastre de vapor.
- Recoger el destilado en un vaso de 250 ml, conteniendo 0,5 ml de indicador y
10 ml de ácido bórico tocando la extremidad inferior del refrigerante en el fondo
del vaso.
- Mantener la destilación durante 2-3 min, siendo el volumen recibido de 100-120
ml.
- Añadir el indicador y valorar con ácido sulfúrico 14N. El viraje de la solución es
de verde a rojo.
Reactivos necesarios
- Sulfato potásico
- Solución de ácido bórico al 2% (20 g en 1 L agua)
- Indicador; mezclar volúmenes iguales de rojo de metilo (0.66%) y verde de
bromocresol (0.33%) en alcohol etílico de 95º.
- Lejía de sosa caústica (265g/L de NaOH)
- Ácido sulfúrico concentrado (d=1,34g/ml)
- Ácido sulfúrico 14 N
- Sulfato cúprico anhidro
- Selenio puro
- Rojo de metilo
- Verde bromocresol

ELIZABETH CARACUEL PAREJA
160
Material e instrumentación
Equipo normal de laboratorio para efectuar una digestión tipo Kjeldhal y su
siguiente valoración y destilación.
Expresión del contenido de nitrógeno
% 𝑁𝑖𝑡𝑟ó𝑔𝑒𝑛𝑜 =𝑛
10 · 𝑃
Siendo
n: volumen en ml de ácido sulfúrico 14N gastado en la valoración
P: peso en g de la muestra en hoja
5.1.4.6 Determinación de fósforo
En solución ácida, en presencia de los iones V5+ y Mo6+, el ácido fosfórico forma
un complejo amarillo, de fosfomolibdovanadatado, cuya absorbancia se mide
con el espectrofotómetro a una longitud de onda de 430 nm.
Procedimiento
Hay que realizar una curva de calibrado. Se prepararán siete disoluciones con
distintas concentraciones de vanadato amónico al 2.5%.
- Se toman siete matraces aforados de 25ml y se añade mediante pipeta los
diferentes volúmenes en cada uno de los matraces: 0,2.5,5,7.5,10,12.5 y 15 ml
de la disolución patrón.
- Añadir unos ml de agua al producto de mineralización realizado anteriormente
y añadir 5-10ml de esta disolución en cada matraz aforado de 25 ml, añadir 5 ml
de reactivo nitrovanadomolíbdico y enrasar con agua.
- Dejar reposar durante 1 hora antes de leer su absorbancia en el
espectrofotómetro (la coloración es estable durante varias horas). Transcurrido
este tiempo medimos a una longitud de onda de 430 nm.

ELIZABETH CARACUEL PAREJA
161
Hacer también una determinación del ensayo en blanco (en la curva de calibrado,
el matraz al que no se le añade analito).
- Por último, representar la curva patrón donde se expresa absorbancia frente µg
de fósforo/ml.
Reactivos necesarios
- Ácido nítrico concentrado (d=1,33g/ml)
- Molibdato de amonio al 5% en agua
- Vanadato amónico al 2,5 % (disolver 25 g de vanadato de amonio en 500 ml de
agua caliente, dejar enfriar y agregar 20 ml de ácido nítrico. Enfriar y enrasar
hasta llegar a un litro de agua)
- Reactivo nitrovanadomolíbdico (mezclar 100 ml de reactivo de molibdato de
amonio, 100 ml de vanadato de amono y 67 ml de ácido nítrico y enrasar con
agua hasta 500ml)
- Disolución patrón de 1000 µg de fósforo/ml (pesar 0,439 g de fosfato
monopotásico seco, disolver en agua y enrasar hasta 100ml)
- Disolución patrón de 100µg de fósforo/ml (llevar 10 ml de la disolución anterior
a un matraz aforado y enrasar con agua hasta 100 ml)
- Disolución patrón de 20 µg de fósforo/ml.
Todos los reactivos deben estar exentos de fósforo y el agua debe de ser
desmineralizada.
Material e instrumentación
- Espectrofotómetro
- Material de vidrio volumétrico contrastado.
Expresión de contenido de fósforo
%𝐹ó𝑠𝑓𝑜𝑟𝑜 =25 · 𝑛
𝑉 · 𝑃 · 100
Siendo

ELIZABETH CARACUEL PAREJA
162
n: concentración de fósforo en µg/ml correspondiente a la lectura
espectrofotométrica.
V: volumen en ml tomados de la disolución de las cenizas (producto de
mineralización)
P: peso en g de la muestra
5.1.4.7 Determinación de sodio
Fundamento
La emisión espectral de sodio se mide a 590 nm en el fotómetro de llama y se
comparan las lecturas obtenidas de la curva patrón. Es preciso eliminar las
interferencias debidas al calcio y a la propia ionización del sodio, mediante la
adicción de aluminio y cesio respectivamente.
Procedimiento
Hay que realizar una curva de calibrado donde preparamos seis soluciones en
matraces de 100 ml.
- Añadir respectivamente en cada matraz: 0,2,5,10,15 y 20 ml de la disolución
patrón de 10 µg de sodio/ml.
- Enrasar cada matraz con ácido clorhídrico al 2%. Las concentraciones de cada
disolución serán: 0,0.2,0.5,1,1.5 y 2 µg de sodio/ml. Efectuar medidas
fotométricas y representarlas frente a sus concentraciones correspondientes.
Determinación
Diluir la solución preparada según el método (mineralización de la muestra) de
forma que la solución a medir contenga 10 % (v/v) de la solución Cs-Al y 2% de
ácido clorhídrico.
Hacer también la determinación del ensayo en blanco.
Reactivos necesarios
- Ácido clorhídrico concentrado (d=1,19 g/ml)

ELIZABETH CARACUEL PAREJA
163
- Disolución de ácido clorhídrico al 2%
- Disolución patrón de sodio de 100 µgamos de sodio/ml (pesar 2,544 g de
cloruro sódico seco, disolver y enrasar con ácido clorhídrico al 2% y llevar a 1 L).
- Disolución patrón de sodio de 10 µgamos de sodio/ml (llevar 10 ml de la
solución patrón anterior a un matraz aforado de 100 ml y completar hasta el aforo
con ácido clorhídrico al 2%).
- Disolución patrón de 10 µg de sodio/ml de la solución patrón anterior a un
matraz aforado de 100 ml y completar con ácido clorhídrico al 2 %.
- Solución Cs-Al (disolver 12,66 g de CsCl y 134 g de AlClHO en un litro de ácido
clorhídrico al 2%).
Material e instrumentación
- Fotómetro de llama
- Material de vidrio volumétrico contrastado
Expresión del contenido de sodio
Llevar las lecturas obtenidas sobre la curva patrón y expresar el contenido de
sodio en % sobre materia seca, teniendo en cuenta las diluciones efectuadas.
5.1.4.8 Determinación de potasio
Fundamento
La emisión espectral del potasio se mide a 760 nm en un fotómetro de llama y
se comparan las lecturas obtenidas con las de la curva patrón.
Procedimiento
Hay que realizar una curva de calibrado. Hacemos ocho disoluciones donde
introducimos en cada matraz de 100 ml; 0,2,3,4,5,6,7 y 8 ml de la disolución
patrón (de concentración 1000 µg); y enrasar todos los matraces con ácido
clorhídrico al 25%. Las concentraciones de cada matraz corresponden a

ELIZABETH CARACUEL PAREJA
164
0,2,3,4,5,6,7 y 8 µg de potasio por ml. Realizar la lectura fotométrica según se
indica para la mezcla y representarla frente a las concentraciones.
Determinación
Diluir convenientemente la solución preparada según el método (mineralización
de la muestra), para que su contenido en potasio este comprendido en 0 y 8
µg/ml en medio de ácido clorhídrico 2%. Hacer la determinación del ensayo en
blanco.
Utilizar preferentemente un fotómetro equipado con llama aire-propano.
Reactivos necesarios
- Ácido clorhídrico concentrado (d=1,19 g/ml)
- Disolución de ácido clorhídrico al 2% en volumen
- Disolución patrón de 1000 µgamos de potasio/ml (pesar 1,097 g de cloruro
potásico previamente secado a 100 ºC durante 1 h; disolver y enrasar a un litro
con ácido clorhídrico al 2%)
- Disolución patrón de 100 µg de potasio/ml. Llevar 10 ml de la solución anterior
a un matraz aforado de 100 ml y enrasar con ácido clorhídrico al 2%.
Material e instrumentación
- Fotómetro de llama
- Material de vidrio volumétrico pertinente
Expresión del contenido de potasio
Llevar las lecturas obtenidas sobre la curva patrón y expresar el contenido de
potasio en porcentaje sobre materia seca, teniendo en cuenta las diluciones
efectuadas.
Para el análisis de los demás elementos (Ca, Mg, Mn y Zn) a excepción del boro,
se realiza la misma determinación que para el K; una incineración en horno Mufla
a 450 ºC y posterior extracción de las cenizas con ácido clorhídrico del 2%,
obteniéndose los resultados por espectrofotometría de absorción.

ELIZABETH CARACUEL PAREJA
165
Para la determinación del boro se realiza una calcinación a 500 ºC y el análisis
se hizo mediante el método de la azometina y posterior lectura en
espectrofotómetro.
A continuación, se presenta una tabla (tabla 5.6) donde se muestran los niveles
críticos de los nutrientes en hojas de olivo.
Tabla 5.6. Niveles críticos de los nutrientes de la hoja
Elementos Niveles
deficientes
Nivel
adecuado
Niveles
tóxicos
Unidades
Nitrógeno 1,4 1,5-2 - %BMS
Fósforo 0,05 0,1-0,3 - %BMS
Potasio 0,4 0,8-1,5 - %BMS
Calcio 0,3 1-2 - %BMS
Magnesio 0,08 0,1-0,03 - %BMS
Manganeso - 20-200 - ppm
Zinc - 10-60 - ppm
Cobre - 4-20 - ppm
Boro 14 20-150 185 ppm
Sodio - - >0,2 ppm
(BMS: Porcentaje Base Materia Seca)
(ppm: partes por millón)
5.1.4.9 Síntomas visuales de deficiencias nutritivas en el olivo
Ante posibles carencias de los elementos estudiados, el olivo presenta unos
rasgos característicos (tabla 5.7), que pueden apreciarse sin llegar a realizar un
análisis foliar. Estas deficiencias nutritivas del árbol pueden visualizarse en la
propia copa del olivo, en los brotes, hojas e incluso en el fruto.

ELIZABETH CARACUEL PAREJA
166
Tabla 5.7. Síntomas de deficiencias nutritivas
Copa del árbol Brotes
Nitrógeno
- Tamaño pequeño
- Poca densidad
- Alta defoliación
- Poco crecimiento
- Ápices necrosados
Potasio
- Tamaño normal
- Ramas péndulas
- Poco crecimiento
- Entrenudos cortos
Calcio
- Tamaño pequeño - Aceites necrosados
- Proliferación de brotes laterales
Magnesio
- Tamaño normal - Síntomas en hojas basales
- Puntas amarillas
- Aparición de bandas
Zinc - Tamaño normal Normal
Hierro - Tamaño normal Normal
Hojas Fruto
Nitrógeno
- Pequeñas
- Color amarillento
- Alta defoliación
- Poca densidad
- Apariencia normal
Potasio
- Síntomas en hojas basales
- Algo más pequeñas
- Color amarillento
Normal
Calcio
- Síntomas en hojas apicales
- Color amarillento,
abarquilladas necrosis
- Poca densidad
- Apariencia normal
Magnesio Parcialmente clorótico Maduración más temprana
Zinc
- Síntomas en hojas apicales
- Color blanquecino en zonas
internerviales
Clorótico
Hierro
- Síntomas en hojas apicales
- Color blanquecino en zonas
internerviales
Clorótico

ELIZABETH CARACUEL PAREJA
167
5.2 PLAN DE VENTAS
La implantación de un laboratorio de análisis tiene como finalidad la
amortiguación de la inversión destinada a la puesta en marcha de la empresa,
así como la rentabilidad de la misma. Para ello, se pretende ir creciendo día a
día en el sector oleícola y captando la mayor cantidad de clientes posible. Para
tal fin, se va a dar publicidad a la empresa a través de radio, prensa e internet.
Se pondrán carteles por toda la ciudad, así como en las principales cooperativas
de la zona. En el periódico se incluirá un anuncio en la parte inferior de la
contraportada durante todo el año.
El mismo anuncio será promocionado por el canal de principal de radio,
intentando que sea llamativo y capte la atención de los oyentes.
Se dedicarán dos tardes a la semana para hacer visitas a todas las cooperativas
y almazaras de la ciudad, mostrándole el catálogo de servicios que presta la
empresa, así como un listado de precios para cada determinación.
Por último, hoy en día es fundamental mantenerse activo en las redes sociales,
así como crear una página web potencial que llame la atención de los usuarios.
Para ello se pretende incluir información clara y concisa, en la que el futuro
cliente no tenga duda de los servicios que se ofertan. Además, se incluirá
información de la realización de todos los procesos y de la importancia que tienen
todos y cada uno de los servicios ofertados.

6 SEGURIDAD Y SALUD

ELIZABETH CARACUEL PAREJA
169
6.1 PREVENCIÓN DE RIESGOS LABORALES EN LABORATORIOS
6.1.1 Legislación
En España existe La Ley 31/1995, de 8 de noviembre, de Prevención de Riesgos
Laborales, que surge por la necesidad de desarrollar una política de protección
de la salud de los trabajadores mediante la prevención de los riesgos derivados
de su trabajo.
Sería de gran importancia su conocimiento por diversos motivos, ya que:
•Es un instrumento clave para minimizar los accidentes de trabajo y las
enfermedades profesionales y por tanto incrementar la calidad de vida de los
trabajadores.
•Invita a un cambio completo de conducta, criterio y mentalidad en los
trabajadores.
•La formación e información es la mejor medida de control de riesgos en el
trabajo.
6.1.2 Normas generales de seguridad
La organización de la prevención en el laboratorio debe implicar a todos los
trabajadores, independientemente del cargo que desempeñen.
En la organización de un Laboratorio se deben tener en cuenta los factores
siguientes:
•Control del cumplimiento de la normativa
•Investigación de accidentes e incidentes
•Inspecciones de seguridad
•Mecanismos administrativos de comunicación de riesgos
Por otro lado, a la hora de trabajar en Laboratorios se requieren unos hábitos
personales, tales como:
•Hay que utilizar batas o uniformes de trabajo.
•El calzado deberá ser cómodo, plano y cerrado.

ELIZABETH CARACUEL PAREJA
170
•Las personas con pelo por debajo de la nuca deben llevarlo recogido.
•El uso de lentes de contacto es altamente desaconsejado. Lo correcto es llevar
gafas graduadas.
•Los ojos no deben frotarse ni tocarse con las manos mientras se trabaja.
•No se permiten objetos personales que pudieran engancharse en los montajes.
•Está prohibido comer, beber, fumar, mascar chicle, chupar lápices o bolígrafos
i ni pipetear nunca con la boca.
•Se trabajará en grupo y no aislado o fuera de la vista de otras personas.
•Las manos deben lavarse como mínimo, al inicio y al final de la jornada laboral,
siempre que se quiten unos guantes protectores, antes y después de ir al aseo,
después de un posible contacto con sustancias irritantes, toxicas o infecciosas.
Para el secado de las manos deben utilizarse toallas de un solo uso.
•Deben emplearse guantes para impedir el contacto con la piel con los
contaminantes. Las mujeres en estado de gestación deberán recabar
información sobre los posibles efectos tóxicos para el feto provocados por
sustancias químicas o biológicas. El embarazo no es motivo para dejar de
trabajar en el laboratorio, pero si para ser mucho más cuidadosa en la labor
diaria.
Instalaciones y equipos
Las instalaciones y los equipos requieren revisiones periódicas, así como la
realización de un mantenimiento preventivo.
En ningún caso se puede dejar en marcha procesos con productos muy
inflamables, explosivos o muy tóxicos, ni aparatos en funcionamiento sin
vigilancia.
En caso de tener que dejar equipos funcionando sin ningún tipo de supervisión
estos son los riesgos asociados:
• explosión
• incendio

ELIZABETH CARACUEL PAREJA
171
• contaminación (vertidos, emisiones)
Para evitar o controlar estos riesgos habría que contar con:
• Instalaciones automáticas contraincendios (detectores de humo, alarmas…)
• Sistemas de control automático capaces de detectar cualquier variación de
temperatura, viscosidad, agitación, presión, etc., (que puedan indicar un
comportamiento anormal del proceso)
• Informar a una persona suficientemente preparada sobre las instrucciones
específicas relativas al lugar y funcionamiento de los sistemas de control.
6.2 PLAN DE EMERGENCIA
Cada laboratorio debe cumplir los siguientes requisitos:
•Disponer de un Plan de Emergencia con toda la información necesaria
(teléfonos, vías de evacuación, equipos de protección individual, etc.).
•Señalizar la ubicación de las instalaciones y aparatos o materiales existentes
para la actuación en caso de emergencia.
•Disponer de protocolos escritos de actuación para situaciones específicas,
como derrames, salpicaduras, conatos de incendio, etc.
•Adiestrar a los trabajadores sobre la actuación correcta en casos de
emergencia y accidente.
•Promover la práctica periódica de simulacros de accidentes y emergencias.
6.2.1 Actuación en caso de emergencia
Se debe tener en un lugar bien visible toda la información necesaria para la
actuación en caso de emergencia (incendio, accidente): cómo actuar, a quien
avisar, números de teléfono internos (equipos de primera y segunda
intervención, equipo de primeros auxilios…), y externos (ambulancias,
bomberos, mutua, ayuntamiento, taxis…), direcciones y otros datos que pudieran
ser útiles en caso de emergencia.

ELIZABETH CARACUEL PAREJA
172
Incendios
•Las medidas que deben adoptarse en el laboratorio para hacer frente a este
riesgo: alarmas, sistemas contraincendios automáticos, elementos de primera
intervención (extintores, mantas ignífugas, duchas de emergencia, mangueras),
procedimientos de trabajo, instalaciones adecuadas, salidas de emergencia
adecuadas, etc.
•Como norma general, en caso de evacuación, deben cerrarse las puertas.
Nunca una persona sola debe hacer frente a un incendio. La persona que
descubre el fuego debe ponerse a salvo, y lo que debe hacer en primer lugar es
avisar. Cuando se avisa se debe decir quién llama, qué ha ocurrido y dónde ha
ocurrido.
•En el laboratorio debe haber extintores portátiles adecuados a los tipos de
fuegos posibles y que resulten accesibles (deben estar cerca de los puestos de
trabajo). No deben colocarse objetos que puedan obstruir su acceso.
Accidentes
Al igual que en el caso de incendio, lo más importante es protegerse a uno
mismo, a continuación, se ha de avisar y si es posible se puede intervenir. Al
informar del accidente se ha de decir quien llama, qué ha pasado y dónde ha
pasado, explicando el tipo de accidente, número y estado actual de las víctimas.
Como norma general nunca se debe administrar líquidos, ni de provocar el
vómito a un paciente inconsciente.
Medidas a tomar ante:
Inhalación
• Respirar aire fresco.
• En caso necesario, aplicar respiración asistida (para algunos productos, como
el ácido cianhídrico, el socorrista deberá autoprotegerse).
• En caso necesario, aplicar oxígeno.
Salpicaduras en ojos o piel

ELIZABETH CARACUEL PAREJA
173
• Lavarse con agua durante 15 minutos.
• Usar ducha de seguridad/lavaojos de emergencia.
• Quitarse la ropa y objetos salpicados.
• No neutralizar.
• Acudir al médico de inmediato y mostrarle la etiqueta y/o la ficha de datos de
seguridad del producto.
Quemaduras térmicas
• Lavar abundantemente con agua fría para enfriar la zona quemada.
• No quitar la ropa pegada a la piel.
• Tapar la parte quemada con ropa limpia.
• Acudir al médico de inmediato.
• No aplicar pomadas, ni grasa, ni desinfectantes
• No dar bebidas ni alimentos.
• No dejar solo al accidentado.
Intoxicación digestiva
• Acudir al médico de inmediato y mostrarle la etiqueta/ficha de datos de
seguridad.
• No provocar el vómito ni dar de beber nada si el accidentado presenta
convulsiones o está inconsciente.
• No provocar el vómito si el producto es corrosivo o inflamable.
• En general, dar a beber abundante agua.
Indicaciones generales, en caso de ingestión de productos químicos (Fig.6.1):

ELIZABETH CARACUEL PAREJA
174
Figura 6.1. Indicaciones generales. Fuente: PANREAC
Electrocución
• Cortar la alimentación eléctrica del aparato causante del accidente.
• No suministrar alimentos, ni bebidas al accidentado.
Mareos o pérdida de conocimiento debido a una fuga tóxica que persista
Hay que protegerse del medio con un aparato respiratorio antes de aproximarse
a la víctima. Trasladar al accidentado a un lugar seguro y dejarlo recostado sobre
el lado izquierdo. Aflojarle la ropa o todo aquello que pueda oprimirlo, verificando
si ha perdido el sentido y si respira; tomarle el pulso. Activar el PAS y, practicar,
si es necesario, la reanimación cardiorrespiratoria. No suministrar alimentos,
bebidas ni productos para activar la respiración.
En caso de vertidos o derrames debe actuarse rápidamente, recogiendo
inmediatamente el producto derramado evitando su evaporación y daños sobre
las instalaciones.

ELIZABETH CARACUEL PAREJA
175
El procedimiento que debe emplearse está en función de las características del
producto: inflamable, ácido, álcali, mercurio, etc., existiendo actualmente
absorbentes y neutralizadores comercializados.
Mercurio
• Absorber con azufre, polisulfuro cálcico o amalgamantes.
• Si se ha depositado en ranuras, se puede sellar.
• Aspirar con pipeta Pasteur y guardar el metal recogido.
Líquidos inflamables
• Absorber con carbón activo o productos específicos.
Ácidos
• Neutralizar con bicarbonato o productos comerciales específicos para su
absorción y neutralización.
Bases
• Emplear productos específicos comercializados para su neutralización y
absorción.
Otros líquidos no corrosivos ni inflamables
• Absorber con vermiculita.
Medidas que tomar ante:
Fugas de gases
• Realizar mantenimiento preventivo: Inspecciones periódicas de conexiones de
botellas y de instalación de gases.
• Fugas de gases asfixiantes, corrosivos, irritantes o tóxicos: Evacuación
inmediata del laboratorio.
Fugas de oxígeno
• Es un gas comburente que puede favorecer la inflamación y corrosión de
materiales presentes.

ELIZABETH CARACUEL PAREJA
176
Fugas de gases inflamables
• Eliminar inmediatamente cualquier foco de ignición y cortar (mediante un
interruptor externo), la energía eléctrica del laboratorio.
• Ventilar bien el laboratorio
Llama en la boca de una botella
• Cerrar el grifo (si es posible) de inmediato (antes de apagar la llama).
• Si no es posible cerrar el grifo de inmediato y se prevé que se podrá cerrar una
vez apagada la llama, se utilizará extintor de polvo para extinguir el fuego y
enfriar el grifo para poder cerrarlo.
• Si no puede cerrar el grifo, habrá que evaluar, según la situación si es preferible
apagar la llama (y dejar que vaya saliendo gas) o bien dejar que el gas se vaya
quemando (alejar de la llama cualquier material que pueda incendiarse). Activar
el plan de emergencia.
6.2.2 Almacenamiento de reactivos
Tradicionalmente las existencias de reactivos se organizaban primando objetivos
como la facilidad de búsqueda y reposición de los distintos productos. En la
actualidad, la existencia de normativa respecto a la prevención de riesgos de
incendio y explosión en almacenamientos, la sensibilización respecto a
sustancias de tipo cancerígeno, la aparición de normas respecto a la seguridad
de sustancias y preparados peligrosos, entre otros, obliga a establecer unos
criterios respecto a la organización en el almacenamiento de dichas sustancias.
Existe por tanto, una necesidad de considerar aisladamente los productos desde
el punto de vista de su peligrosidad y posibles incompatibilidades entre ellos,
debido a que el almacenamiento prolongado de los productos químicos
representa en sí mismo un peligro, ya que dada la propia reactividad intrínseca
de los productos químicos pueden ocurrir distintas transformaciones:
• Formación de peróxidos inestables con el consiguiente peligro de explosión al
destilar la sustancia o por contacto.
• Polimerización de la sustancia que, aunque se trata en principio de una reacción
lenta, puede en ciertos casos llegar a ser rápida y explosiva.

ELIZABETH CARACUEL PAREJA
177
• El recipiente que contiene el producto puede atacarse y romperse por sí sólo.
• Descomposición lenta de la sustancia produciendo un gas cuya acumulación
puede hacer estallar el recipiente.
Los laboratorios, debido a que no almacenan una gran cantidad de productos
químicos, no suelen estar sujetos a la normativa que regula el almacenamiento
de productos químicos (R.D. 379/2001).
La mejor opción para evitar o controlar los riesgos es no almacenar productos
químicos durante demasiado tiempo, para ello hay que:
• Mantener el stock al mínimo,
• Utilizar un almacén externo al laboratorio,
• Almacenar en el laboratorio únicamente los productos imprescindibles que se
usan durante la jornada.
• Agrupar los productos por peligrosidades
• Separar los productos incompatibles
• Aislar los productos muy peligrosos:
- los que reaccionan con el agua (Sodio, Potasio, Litio)
- los explosivos
- los cancerígenos
- los radioactivos
• Mantener un registro actualizado de productos almacenados (indicar última
fecha de utilización y nombre del responsable).
• Emplear armarios de seguridad.
• Utilizar frigoríficos antideflagrantes para guardar productos inflamables.
Como se ha comentado anteriormente, en el laboratorio debe plantearse un
sistema ágil de control de sus existencias con objeto de evitar acumulaciones de
productos. Se hace necesaria una planificación que garantice las existencias

ELIZABETH CARACUEL PAREJA
178
durante cortos periodos de tiempo, aunque ello requiera una mayor frecuencia
de pedidos.
Este aspecto es especialmente importante cuando hablamos de acumulación de
productos inflamables que aumenta el peligro de incendio.
La experiencia demuestra que, en el caso de utilización de disolventes en un
laboratorio, si existe una buena planificación en cuanto al suministro de éstos, la
acumulación dentro del laboratorio puede reducirse hasta un 30%.
Una vez reducidas las cantidades almacenadas, hay que plantearse la
separación de productos en función de las incompatibilidades que puedan darse
entre familias de éstos. Se trata de separar ácidos de bases, oxidantes de
inflamables, venenos activos, etc.
En este sentido, se relacionan una serie de características de peligrosidad de los
productos químicos y se comentan desde el punto de vista de su manipulación y
almacenamiento. La reactividad se contempla desde las siguientes perspectivas:
- Las separaciones pueden efectuarse dedicando una serie de estanterías a una
familia determinada, de forma que a su alrededor queden pasillos. Se pueden
intercalar entre familias de reactivos incompatibles, estanterías con reactivos
inertes.
- A la hora de disponer los distintos reactivos en las baldas de sus estanterías
correspondientes, pueden colocarse los ácidos o bases fuertes en baldas
inferiores, así como los recipientes de mayor volumen. En suma, la separación y
distribución de productos con objeto de eliminar riesgos, seguirá siempre
criterios lógicos teniendo en cuenta la reactividad de las distintas sustancias.
- Ciertos productos no solo requieren la separación del resto del stock, sino
además su confinamiento por el hecho de tener ciertas propiedades físico-
químicas o biológicas. Este es el caso de los productos cancerígenos, en
particular y de las sustancias de alta toxicidad en general. En general deberán
almacenarse en recintos o armarios convenientemente rotulados y bajo llave. El
control de entradas y consumos de estos productos debe ser riguroso, prestando
especial interés al estado de los envases que los contienen, por si presentasen
defectos que puedan provocar derrames durante su manipulación.

ELIZABETH CARACUEL PAREJA
179
Estas sustancias deben contenerse en un doble recipiente que evite
dispersiones o derrames en el caso de roturas o manipulaciones incorrectas.
Este doble sistema suele ser una bolsa de plástico resistente y transparente en
el interior de un recipiente, con lo que cualquier vertido puede ser controlado con
facilidad.
En resumen, se trata de ejercer un control riguroso sobre los productos para
evitar abandonos en áreas comunes del laboratorio, obligando al personal que
los manipule a adoptar medidas de precaución.
En el caso de sustancias cuya emisión al ambiente provoque olores muy
molestos, se recomienda su confinamiento en recintos pequeños o armarios que
puedan ir equipados con un pequeño sistema de extracción, impidiendo
mediante la depresión generada, la dispersión general de los malos olores.
Cuando el almacenamiento se refiere a líquidos inflamables y combustibles,
debe planificarse bajo los criterios de la Instrucción Técnica Complementaria ITC
MIE-APQ-1 del Reglamento de Almacenamiento de Productos Químicos. En
este reglamento se establecen los requisitos de los almacenes en función de los
productos y cantidades que contienen.
6.3 GESTIÓN DE RESIDUOS
Como en toda gestión de residuos, en primer lugar, deberían no generarse
residuos o que éstos fueran mínimos. Si esto no es posible, los residuos se
deberían reutilizar. Si tampoco es posible se deberán tratar y finalmente eliminar
de forma segura. En cada laboratorio debe establecerse un procedimiento de
gestión de residuos que considere todos los tipos de residuos que se generan:
banales (no especiales o no peligrosos) o peligrosos (especiales).
Para la correcta gestión de los residuos es necesario:
• Inventariar todos los posibles residuos.
• Definir los grupos de residuos (según sus características fisicoquímicas,
peligrosidades y tratamiento/ eliminación posterior).
• Considerar las posibilidades de minimización.

ELIZABETH CARACUEL PAREJA
180
• Gestionar las compras correctamente (evitar tener stocks elevados para
disminuir la cantidad de residuos generada por reactivos caducados, o no
usados).
• Implantar sistema de recogida selectiva en función de los grupos establecidos.
• Destinar recipientes adecuados a las características de los residuos.
• Identificar y etiquetar los envases y contenedores que contienen residuos.
• Informar y formar al personal del laboratorio sobre el procedimiento de gestión
de residuos.
• Contactar con una empresa externa autorizada (gestor de residuos) para la
recogida, tratamiento y eliminación de aquellos residuos que no puedan tratarse
en el propio laboratorio.
• Cumplir con la legislación vigente. Antes de proceder al envío a gestores
autorizados, los residuos obtenidos podrían ser tratados de modo que disminuya
su peligrosidad y acondicionados en recipientes preparados al efecto.
•Los recipientes donde se deben depositar estos residuos tienen que ser de un
material y tamaño apropiados a las características del residuo a transportar.
Deben estar cerrados herméticamente y poseer una etiqueta identificativa que
informe del tipo de residuo que contienen y su peligrosidad (Fig.6.2). Puede ser
práctico clasificar los residuos según los siguientes “grupos” (en el caso de
residuos pertenecientes a un mismo grupo pueden depositarse en un mismo
recipiente):
Figura 6.2. Clasificación de residuos. Fuente: PANREAC

ELIZABETH CARACUEL PAREJA
181
No se debe desechar al vertedero habitual de basuras (residuos banales),
papeles de filtro, guantes desechados, trapos, serrín u otras materias
impregnadas de productos químicos, sin haber efectuado previamente una
eliminación, destrucción o neutralización de los mismos.
En conclusión, para una óptima gestión de residuos, el laboratorio tiene que:
• Disponer de un programa de recogida selectiva según las características de los
residuos generados.
• No acumular residuos en el laboratorio. Almacenarlos en un lugar específico y
gestionarlos según las disposiciones legales vigentes.
• Recoger el material de vidrio en contenedores especiales.
• Tratar los derrames con los productos adecuados según sus características
(ácidos, bases, disolventes, mercurio, etc.) y recogerlos como residuos.
• Minimizar los residuos, optimizando la gestión de compras, los procedimientos
de trabajo, valorando su recuperación (materiales) y su reutilización.
6.3.1 Pretratamiento de residuos
Antes de enviar al gestor, pueden ser útiles los siguientes tratamientos:
1. ÁCIDOS INORGÁNICOS, SALES Y SOLUCIÓNES ÁCIDAS
Diluir con agua aproximadamente a 1:5 y neutralizar añadiendo lentamente sodio
hidróxido en solución o en escamas (hasta pH 6-8).
Recipiente n° 3.
Productos: Ácido nítrico, ácido fosfórico, ácido sulfúrico, bifosfatos, bisulfatos,
etc.
2. ÁCIDOS ORGÁNICOS
Diluir con agua aproximadamente a 1:5 y neutralizar añadiendo lentamente sodio
hidróxido en solución o en escamas hasta pH 6-8.
Recipiente nº 3.
Productos: Ácidos acético, butírico, fenilantranílico, naftalensulfónico, succínico,
toluensulfónico, etc.

ELIZABETH CARACUEL PAREJA
182
3. BASES, AMINAS, SALES BÁSICAS Y SOLUCIÓNES BÁSICAS
Diluir con agua, aproximadamente a 1:5 y neutralizar añadiendo lentamente
ácido sulfúrico diluido (hasta pH 6-8). La solución resultante se diluye a 1:10.
Recipiente n° 3.
Productos: Dietilamina, trietanolamina, amonio hidróxido, potasio hidróxido,
sodio hidróxido, potasio carbonato, sodio carbonato.
4. CIANUROS, MERCAPTANOS
Mezclar bien en una solución de sodio hidróxido y de sodio hipoclorito, agitando
de vez en cuando. Dejar en contacto 24 horas como mínimo. Diluir con agua
abundante. Eliminar el exceso de hipoclorito con una solución de sodio tiosulfato
y neutralizar.
Recipiente n° 3.
Productos: Cianuros varios, mercaptobenzotiazol.
6.3.2 Normativa aplicable
6.3.2.1 Sustancias peligrosas
Real Decreto 363/1995, de 10 de marzo, por el que se aprueba el Reglamento
sobre notificación de sustancias nuevas y clasificación, envasado y etiquetado
de sustancias peligrosas. Transposición de la Directiva 67/548/CEE y posteriores
modificaciones.
6.3.2.2 Preparados peligrosos
Real Decreto 255/2003, de 28 de febrero, por el que se aprueba el Reglamento
sobre clasificación, envasado y etiquetado de preparados peligrosos.
Transposición de la Directiva 1999/45/CE.

7 ESTUDIO ECONÓMICO FINANCIERO

ELIZABETH CARACUEL PAREJA
184
Con objeto de conocer con precisión la rentabilidad de la nueva empresa se va
a realizar un estudio económico-financiero. Para ello se hará un inventario que
recoja todo el material necesario para la puesta en marcha del laboratorio, así
como el coste de los equipos, el alquiler de la parcela y el del personal que va a
trabajar en la empresa. Posteriormente, se aplicarán las ecuaciones
correspondientes para saber la rentabilidad que presenta la puesta en marcha
del laboratorio.
7.1 INVENTARIO
En la tabla 7.1 se recoge todo el material necesario para la sala de recepción, el
despacho y la sala de juntas.
Tabla 7.1. Material para sala de recepción, despacho y sala de juntas.
Material Cantidad Precio, € Total, €
Mesa de oficina 3 100 300
Sillón 3 60 180
Sillas plegables 20 15 300
Mesa auxiliar 1 20 20
Sofá 1 700 700
Armarios archivadores 3 450 1350
Estantería 4 70 280
Ordenador de sobremesa 3 500 1500
Teléfono inalámbrico 3 40 120
Impresora multifunción 2 150 300
Material de oficina 400
Extintor de seguridad 1 50 50
Mesa para juntas 1 200 200
Proyector 1 90 90
TOTAL 5790
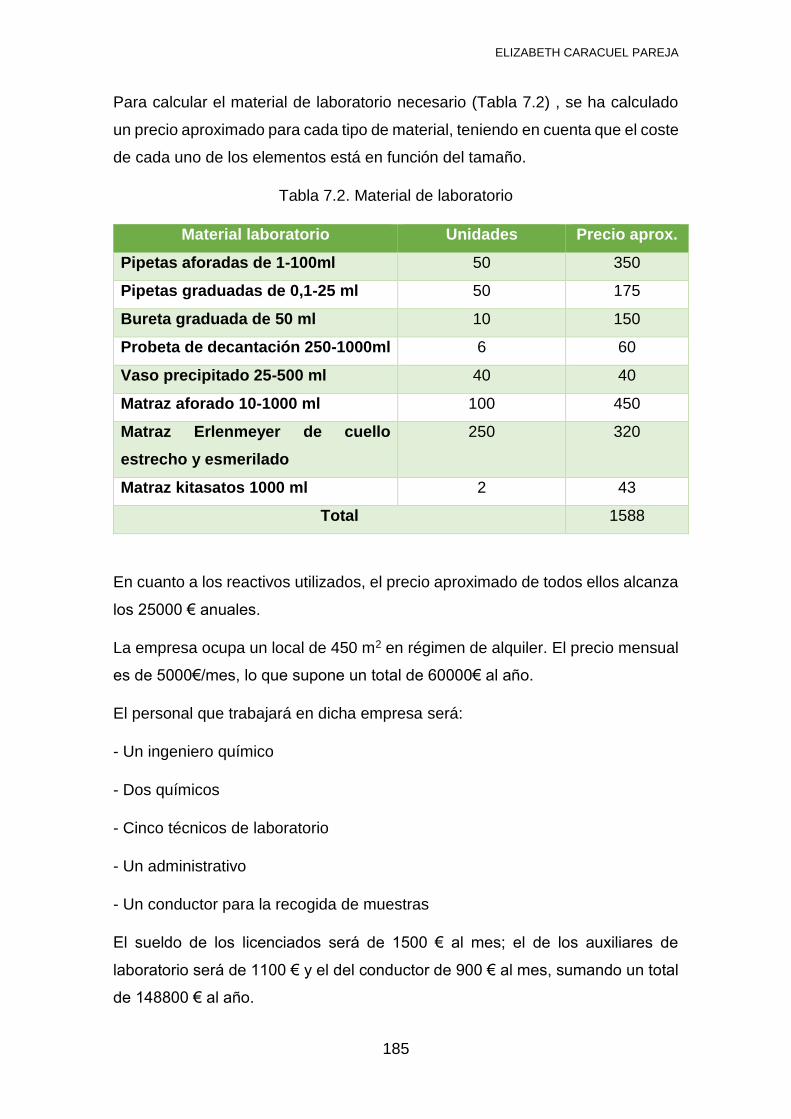
ELIZABETH CARACUEL PAREJA
185
Para calcular el material de laboratorio necesario (Tabla 7.2) , se ha calculado
un precio aproximado para cada tipo de material, teniendo en cuenta que el coste
de cada uno de los elementos está en función del tamaño.
Tabla 7.2. Material de laboratorio
Material laboratorio Unidades Precio aprox.
Pipetas aforadas de 1-100ml 50 350
Pipetas graduadas de 0,1-25 ml 50 175
Bureta graduada de 50 ml 10 150
Probeta de decantación 250-1000ml 6 60
Vaso precipitado 25-500 ml 40 40
Matraz aforado 10-1000 ml 100 450
Matraz Erlenmeyer de cuello
estrecho y esmerilado
250 320
Matraz kitasatos 1000 ml 2 43
Total 1588
En cuanto a los reactivos utilizados, el precio aproximado de todos ellos alcanza
los 25000 € anuales.
La empresa ocupa un local de 450 m2 en régimen de alquiler. El precio mensual
es de 5000€/mes, lo que supone un total de 60000€ al año.
El personal que trabajará en dicha empresa será:
- Un ingeniero químico
- Dos químicos
- Cinco técnicos de laboratorio
- Un administrativo
- Un conductor para la recogida de muestras
El sueldo de los licenciados será de 1500 € al mes; el de los auxiliares de
laboratorio será de 1100 € y el del conductor de 900 € al mes, sumando un total
de 148800 € al año.
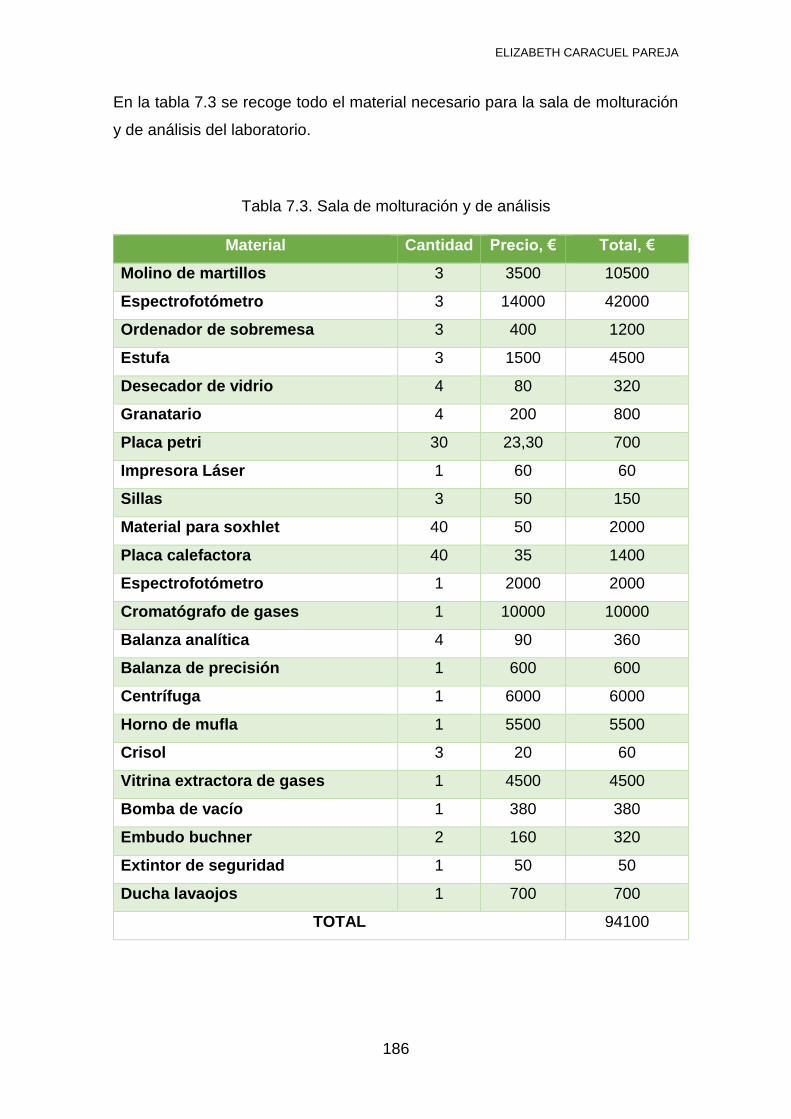
ELIZABETH CARACUEL PAREJA
186
En la tabla 7.3 se recoge todo el material necesario para la sala de molturación
y de análisis del laboratorio.
Tabla 7.3. Sala de molturación y de análisis
Material Cantidad Precio, € Total, €
Molino de martillos 3 3500 10500
Espectrofotómetro 3 14000 42000
Ordenador de sobremesa 3 400 1200
Estufa 3 1500 4500
Desecador de vidrio 4 80 320
Granatario 4 200 800
Placa petri 30 23,30 700
Impresora Láser 1 60 60
Sillas 3 50 150
Material para soxhlet 40 50 2000
Placa calefactora 40 35 1400
Espectrofotómetro 1 2000 2000
Cromatógrafo de gases 1 10000 10000
Balanza analítica 4 90 360
Balanza de precisión 1 600 600
Centrífuga 1 6000 6000
Horno de mufla 1 5500 5500
Crisol 3 20 60
Vitrina extractora de gases 1 4500 4500
Bomba de vacío 1 380 380
Embudo buchner 2 160 320
Extintor de seguridad 1 50 50
Ducha lavaojos 1 700 700
TOTAL 94100

ELIZABETH CARACUEL PAREJA
187
7.2 ESTUDIO ECONÓMICO
Se dice que una empresa es rentable cuando genera suficiente beneficio, es
decir, cuando sus ingresos son mayores que sus gastos, y la diferencia entre
ellos es considerada como aceptable.
Para calcular la rentabilidad hay que tener en cuenta los ingresos recibidos por
las ventas de la empresa, los costes generados, los impuestos, el material
inmovilizado y el capital circulante. La expresión que engloba todos estos
factores es:
𝑅 =𝑉 − 𝐶 − 𝑈
𝐼 + 𝑃𝑐· 100
Siendo:
V: ventas
C: costes
U: impuestos
I: material inmovilizado
Pc: capital circulante
Sabiendo la cantidad de determinaciones realizadas en el laboratorio y la
cantidad de cada una de ellas, se puede calcular los ingresos por ventas usando
la siguiente ecuación:
𝑉 = 𝑞 · 𝑝
Siendo
V: ingresos por ventas
q: cantidad de producto
p: precio
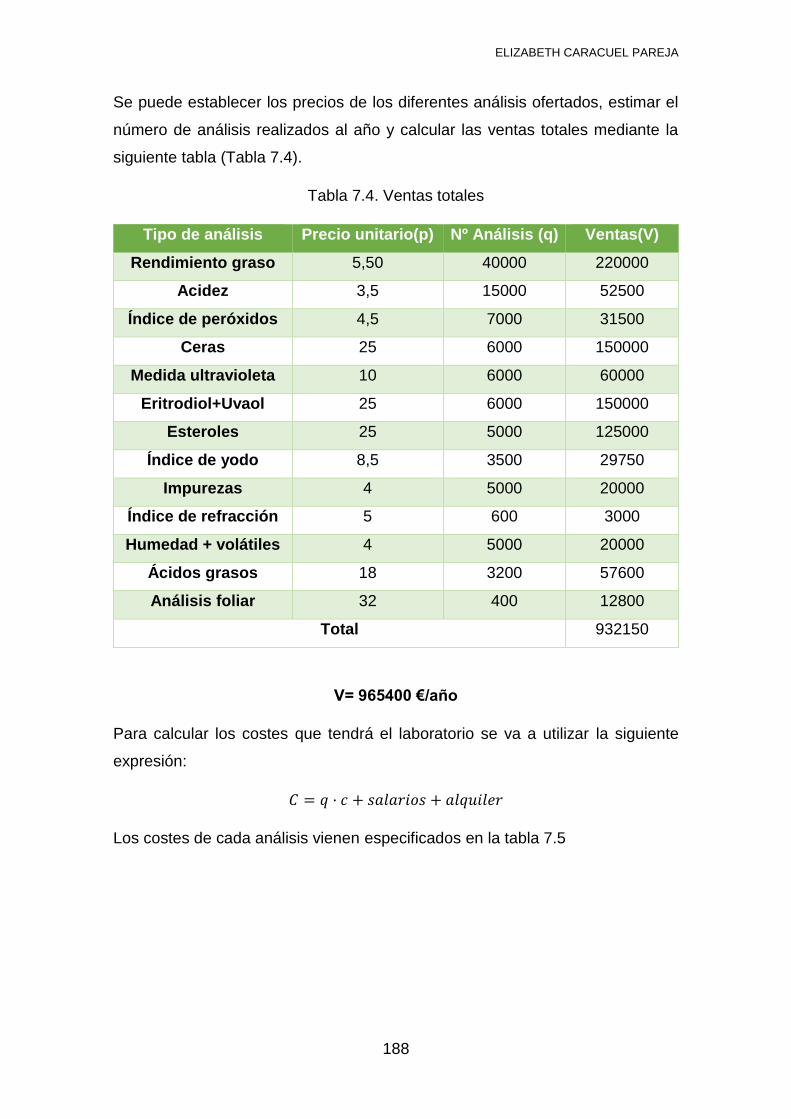
ELIZABETH CARACUEL PAREJA
188
Se puede establecer los precios de los diferentes análisis ofertados, estimar el
número de análisis realizados al año y calcular las ventas totales mediante la
siguiente tabla (Tabla 7.4).
Tabla 7.4. Ventas totales
Tipo de análisis Precio unitario(p) Nº Análisis (q) Ventas(V)
Rendimiento graso 5,50 40000 220000
Acidez 3,5 15000 52500
Índice de peróxidos 4,5 7000 31500
Ceras 25 6000 150000
Medida ultravioleta 10 6000 60000
Eritrodiol+Uvaol 25 6000 150000
Esteroles 25 5000 125000
Índice de yodo 8,5 3500 29750
Impurezas 4 5000 20000
Índice de refracción 5 600 3000
Humedad + volátiles 4 5000 20000
Ácidos grasos 18 3200 57600
Análisis foliar 32 400 12800
Total 932150
V= 965400 €/año
Para calcular los costes que tendrá el laboratorio se va a utilizar la siguiente
expresión:
𝐶 = 𝑞 · 𝑐 + 𝑠𝑎𝑙𝑎𝑟𝑖𝑜𝑠 + 𝑎𝑙𝑞𝑢𝑖𝑙𝑒𝑟
Los costes de cada análisis vienen especificados en la tabla 7.5

ELIZABETH CARACUEL PAREJA
189
Tipo de análisis Precio unitario(p) Nº Análisis (q) Costes(c)
Rendimiento graso 1,80 40000 72000
Acidez 2 15000 30000
Índice de peróxidos 4 7000 28000
Ceras 12 6000 72000
Medida ultravioleta 6 6000 36000
Eritrodiol+Uvaol 12 6000 72000
Esteroles 13 5000 65000
Índice de yodo 7 3500 24500
Impurezas 1,90 5000 9500
Índice de refracción 3 600 1800
Humedad + volátiles 3 5000 15000
Ácidos grasos 15 3200 48000
Análisis foliar 15 400 6000
Total 479800
El total de los salarios es de 148800 €/año.
El total del alquiler es de 60000 €/año.
Los costes serán:
𝐶 = 𝑞 · 𝑐 + 𝑠𝑎𝑙𝑎𝑟𝑖𝑜𝑠 + 𝑎𝑙𝑞𝑢𝑖𝑙𝑒𝑟
𝐶 = 479800 + 148800 + 60000 = 688600 €/𝑎ñ𝑜
Para calcular los impuestos se utiliza la ecuación:
𝑈 = 0,35 · (𝑉 − 𝐶) = 96880 €
Para calcular el capital circulante se utiliza la ecuación:
𝑃𝑐 = 0,2 · 𝑉 = 193080€
Para calcular el capital inmovilizado se utiliza la ecuación:
𝐼 = 𝑖𝑛𝑠𝑡𝑟𝑢𝑚𝑒𝑛𝑡𝑜𝑠 + 𝑚𝑜𝑏𝑖𝑙𝑖𝑎𝑟𝑖𝑜 + 𝑚𝑎𝑡𝑒𝑟𝑖𝑎𝑙 + 𝑟𝑒𝑎𝑐𝑡𝑖𝑣𝑜𝑠

ELIZABETH CARACUEL PAREJA
190
𝐼 = 126478€
Una vez calculado todo esto, se calcula la rentabilidad de la empresa.
𝑅 =𝑉 − 𝐶 − 𝑈
𝐼 + 𝑃𝑐· 100
Ventas=932150 €
Costes= 688600 €
Impuestos (U)= 169960 €
Capital inmovilizado (I)= 126478 €
Capitar circulante (Pc) = 193080 €
𝑹 = 𝟐𝟑, 𝟎𝟐 %

8 CONCLUSIONES

ELIZABETH CARACUEL PAREJA
192
Tras estudiar con detenimiento el mercado al que se va a destinar el laboratorio
y el proceso de análisis de cada una de las determinaciones ofertadas, se
observa que es viable la construcción de dicha empresa.
Esto queda justificado debido al cálculo de la rentabilidad de la puesta en marcha
del laboratorio, alcanzando ésta un 23,02 %. Dicho valor se considera óptimo ya
que el máximo que puede alcanzar para llegar a ser rentable sería un 30 %.
Por tanto, tras el estudio detallado del proceso de análisis junto con el estudio
económico-financiero, son razones suficientes como para justificar la inversión.

9 BIBLIOGRAFÍA

ELIZABETH CARACUEL PAREJA
194
Normas
- Norma UNE 55031 Y 55032
- Reglamento Nº 1989/2003
- Reglamento Delegado (UE) 2016/1226 de la Comisión de 4 de mayo de 2016
Libros de texto
- Benardini, E. “Tecnología de aceites y grasas”. Colección Zahira. Madrid 1986.
- López, S. “La almazara de Gérgal”. Almería 2008.
- Osuna, R. “La cooperativa agrícola Virgen del Castillo de Carcabuey,
(Córdoba)”. Carcabuey 2007
- Rosemblum, M. “La aceituna: vida y tradiciones de un noble fruto”. Barcelona
1997.
Recursos electrónicos
- www.aceitedeoliva.com
- www.agroes.es
- www.andaluciainformacion.es
- www.boe.es
- www.csrservicios.es
- www.esenciadeolivo.es
- www.juntadeandalucia.es
- www.laboratoriosoleosur.com
- www.mapama.gob.es.
- www.oleohispana.com
- www.priegodecordoba.org
- www.ruedaoliva.com




























