Embed Size (px)
Citation preview

BIOQUÍMICA HUMANA
2009
TÉCNICAS DE SEPARACIÓN
DE PROTEÍNAS:
CROMATOGRAFÍA, ELECTROFORESIS




7
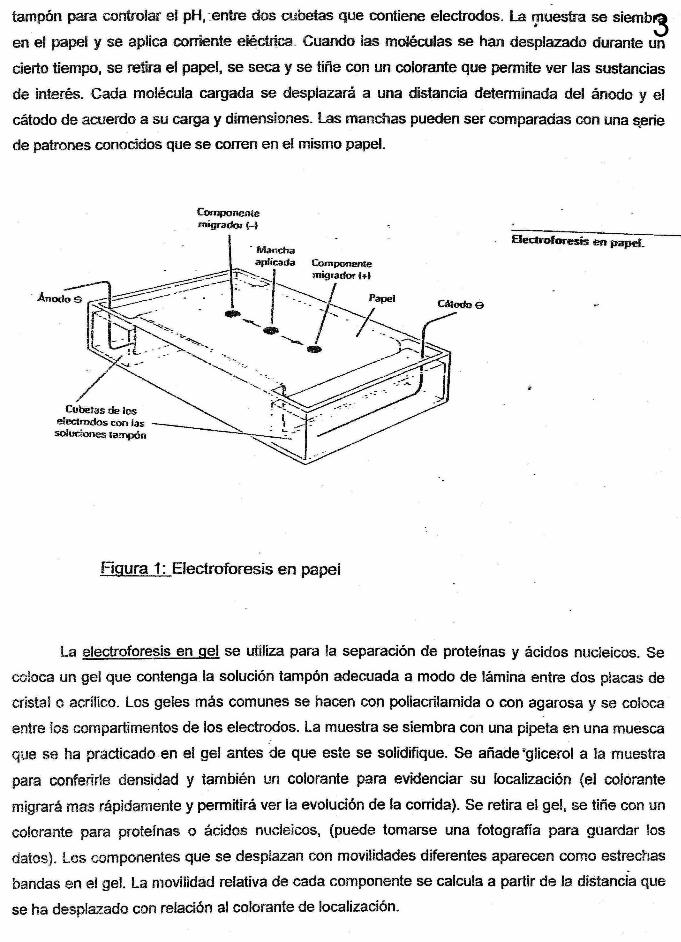
Bolitas de polimero poroso
1. Se agrega una mezcla de proteínas a la columna que contiene el polímero poroso
2. Las proteínas se separan por tamaño: las moléculas más grandes pasan libremente y salen en las primeras fracciones
1 2 3 4 5 6

8
Electroforesis de proteínas en geles de poliacrilamida
Introducción y técnica básica
La electroforesis de proteínas en geles con una matriz de poliacrilamida, comúnmente denominada electroforesis en poliacrilamida (PAGE, 'polyacrilamide gel electrophoresis') es sin duda alguna una de las técnicas más ampliamente usada para caracterizar mezclas complejas de proteínas. La electroforesis en poliacrilamida es un método conveniente, rápido y económico a nivel de muestra pues se requieren sólo cantidades del orden de microgramos de proteína.
Las proteínas presentan una carga eléctrica neta si se encuentran en un medio que tenga un pH diferente al de su punto isoeléctrico y por eso tienen la propiedad de desplazarse cuando se someten a un campo eléctrico. La matriz de poliacrilamida es un buen soporte para este método pues la migración de las proteínas en su seno no sólo es proporcional a la carga neta sino también al tamaño y forma de las proteínas.
Una ventaja importante de los geles de poliacrilamida es que son químicamente inertes, transparentes y estables en un amplio rango de pHs, temperatura y fuerza iónica (cantidad de iones presentes en la solución).
Algunas caraterísitcas destacables de la electroforesis en geles de poliacrilamida son :
? Los geles de poliacrilamida se forman por la polimerización de la acrilamida por acción de un agente entrecruzador ('cross-linking'), la bis-acrilamida
? La porosidad del gel la determina las proporciones relativas de poliacrilamida y bis-acrilamida.
? El porcentaje total de acrilamida/bisacrilamida determina el rango de separación del gel. Habitualmente los geles se denominan en función del % de acrilamida/bisacrilamida que contienen. Así, la mayoría de las proteínas se separan bien en el rango 5 a 10%. Un menor porcentaje (mayor tamaño de poro) es mejor para separar proteínas de gran tamaño.
En función del estado de las proteínas (nativo o desnaturalizado) a lo largo del proceso electroforético éstas se clasifican en electroforesis nativas o desnaturalizantes.
? una electroforesis desnaturalizante, la más común, es la que somete a las proteínas a migración asegurando la completa desnaturalización (pérdida de la estructura tridimensional). En esta situación la migración es proporcional a la carga y al tamaño de la molécula pero no a su forma. El agente desnaturalizante más empleado es el sodiododecilsulfato o SDS, un detergente.

9
? una electroforesis nativa es la que somete a las proteínas a migración sin desnaturalización. En esta situación las proteínas migran en función de su carga, de su tamaño y de su forma.
Tipos de electroforesis
SDS-PAGE
SDS-PAGE es la electroforesis de proteínas más ampliamente usada. Su nombre significa la electroforesis en geles de poliacrilamida que se realiza en presencia de SDS ('SDS-polyacrilamide gel electrophoresis'). Fue descrito por Laemmli (Lammeli (1970), Nature, 277, p. 680) Se trata de un tipo de electroforesis desnaturalizante en la que las muestras se desnaturalizan por calor en presencia de agentes desnaturalizantes (beta-mercaptoetanol, que destruye los puentes disulfuro, SDS que desnaturaliza y recubre a la proteína), y se separan como cadenas polipeptídicas aisladas.
El SDS es un detergente de acción desnaturalizante que se une a las cadenas polipeptídicas desnaturalizadas con una relación constante de SDS por g de proteína, uniéndose aproximadamente una molécula de SDS por cada dos aminoácidos de la cadena. Esta unión masiva de moléculas de SDS bloquea la carga propia de la molécula de proteína y le confiere al complejo una carga neta negativa proporcional a su masa, haciendo que todas las proteínas acomplejadas con SDS viajen hacia el ánodo. La separación de los complejos SDS-proteína es proporcional sólo a la masa de la proteína pues todas tienen la misma carga por unidad de masa. Se puede entonces determinar el peso molecular aparente de cualquier proteína por comparación con un patrón de proteínas de pesos moleculares conocidos. Las movilidades de las proteínas en los geles de SDS-PAGE son funciones lineales del logaritmo de su peso molecular.

10
Métodos de transferencia a filtros
Introducción
La transferencia de proteínas o 'blotting' supone la inmovilización de las proteínas sobre membranas sintéticas, seguido de la detección empleando sistemas especialmente diseñados para la tinción de 'blots'. El método más potente es el denominado 'Western blot' en el que las proteínas son separadas en primer lugar mediante electroforesis en geles de poliacrilamida y posteriormente se transfieren mediante la aplicación de un campo eléctrico perpendicular al gel a una membrana. El procedimiento es análogo al desarrollado por el Prof. E. Southern para la separación de fragmentos de ADN ('Southern blotting') y por ello se le denominó 'Western'.
Trabajar con las proteínas fijadas sobre una membrana tiene ventajas sobre el emplearlas dentro del propio gel:
1) Son más rápidas de teñir y desteñir; 2) se detectan cantidades menores de proteínas pues se concentran en la superficie y no se diluyen en todo el espesor del gel; 3) el 'blot' es un registo conveniente y mas cómodo de manipular que el gel; 4) las membranas son mucho más fáciles de manipular que el propio gel
Todo procedimiento de 'blotting' consta de 5 etapas:
1- Inmobilización de proteínas sobre la membrana generalmente mediante electrotransferencia.
En este método, descrito por Towbin, las proteínas son transferidas desde el gel a la membrana electroforéticamente. La ventaja de este proceso es su corta duración (de 30 min a pocas horas), lo que reduce notablemente el efecto de difusión de las bandas. El procedimiento se inicia apilando sucesivamente sobre una esponja plana papel de filtro empapado en buffer de transferencia, el
gel, la membrana en contacto directo con el gel, más papel de filtro y finalmente una esponja plana. Este conjunto se recoge entre dos capas de plástico perforado y se introduce en un tanque en el que se encuentra una solución salina (buffer de transferencia) y dos electrodos planos (diseñados para conseguir un campo uniforme en toda la superficie del gel). Se dispone de forma que el gel quede hacia el ánodo (-) y la membrana hacia el cátodo (+). La carga neta de las proteínas en el caso de los geles de SDS-PAGE es negativa debida a la carga del SDS.

11
Una vez realizada la transferencia la membrana se puede analizar inmediatamente o bien conservarla en frío (2 a 8ºC) durante meses.
Es importante incorporar en el gel a transferir un control de la transferencia que habitualmente es el propio carril donde se han cargado los marcadores de peso molecular que al transferirlos se tiñen sobre el filtro para ver si la transferencia de toda la gama de pesos moleculares ha sido correcta.
Existen otras variantes a la transferencia electroforética descrita previamente.
1.1.- Tinción de proteínas en la membrana como control del proceso de transferencia.
Habitualmente se emplean técnicas de tinción de proteínas sobre el filtro como control de la transferencia. Existen diferentes métodos para teñir todas las proteínas presentes en el filtro, pero los que más interés despiertan son los métodos de tinción específicos de proteínas totales. Para teñir las proteínas en los filtros se emplean habitualmente el Ponceau. La tinción es rápida pero no es permanente y puede desaparecer en el procesamiento posterior. Como la tinción desaparece es compatible con casi todos los procedimientos de detección con anticuerpos.
2-Saturación de todos los lugares de unión de proteínas de la membrana no ocupados para evitar la unión no específica de anticuerpos, que son proteínas.
Una etapa común a todos los procedimientos de inmunodetección es lo que comúnmente se denomina 'bloqueo de la membrana', para prevenir la unión no específica del sistema de detección a la membrana, con el riesgo asociado de tener un elevado 'background' o falsos positivos. Se han descrito numerosas soluciones bloqueantes, y todas ellas son efectivas. Las dos soluciones de bloqueo que son compatibles con casi cualquier sistema de detección son las soluciones de leche desnatada y las soluciones de albúmina sérica bovina (BSA).
3-Incubación del 'blot' con anticuerpo primarios contra la/s proteína/s de interés.
El tipo de anticuerpos empleado en la detección de las proteínas transferidas a filtro no es determinante. Existen tanto excelentes anticuerpos policlonales con alta especificidad y sensibilidad como monoclonales. Sin embargo los monoclonales se pueden obtener en mayor cantidad y mantienen siempre sus propiedades de reconocimiento independientemente del lote, pero son mucho más onerosos.
4-Incubación del 'blot' con anticuerpos secundarios, o reactivos que actúan de ligando del anticuerpo primario unidos a enzimas u otros marcadores.
Como anticuerpos secundarios se suelen emplear anticuerpos obtenidos inoculando en la especie productoras inmunoglobulinas de la especie a detectar (anticuerpos anti-especie). Estos anticuerpos tienen unidos en forma
12
Proteína a determinar
Membrana
Y
Proteína bloqueante
Primer anticuerpo
Segundo anticuerpo conjugado
Yatomo
Radioactivo Placa radiográfica
Proteína a determinar
Membrana
Y
Proteína bloqueante
Primer anticuerpo
Segundo anticuerpo conjugado
Yatomo
Radioactivo Placa radiográfica
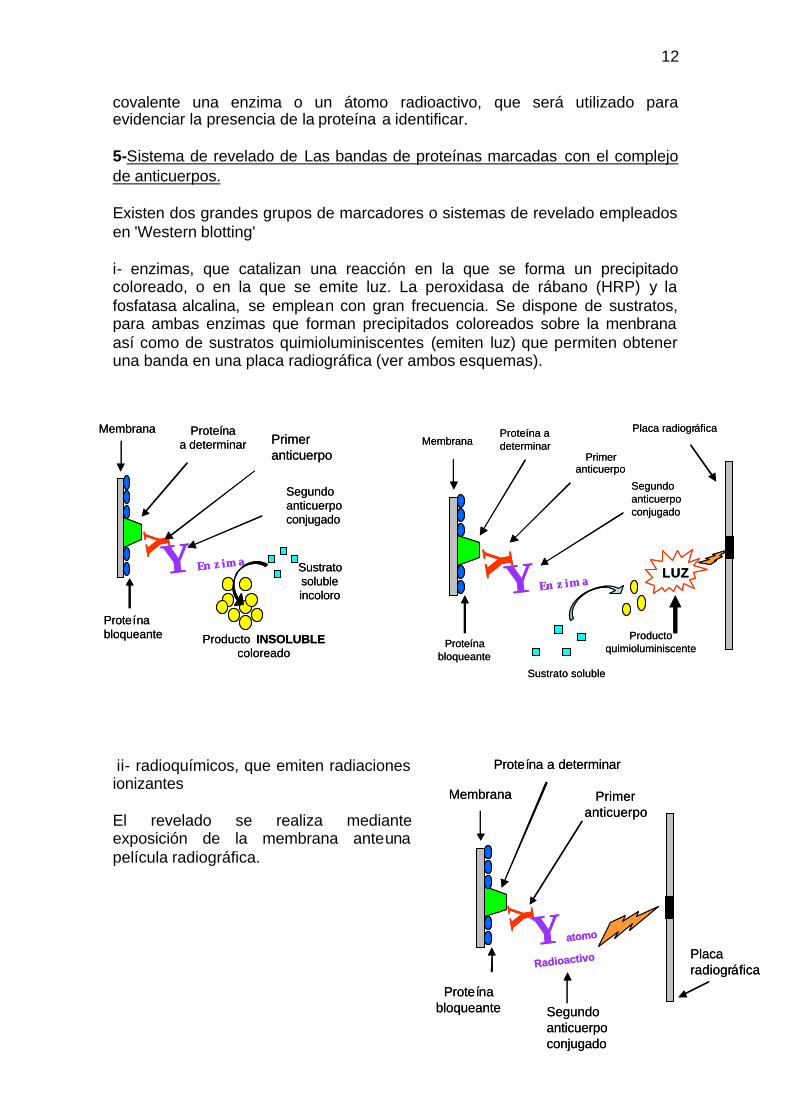
covalente una enzima o un átomo radioactivo, que será utilizado para evidenciar la presencia de la proteína a identificar.
5-Sistema de revelado de Las bandas de proteínas marcadas con el complejo de anticuerpos.
Existen dos grandes grupos de marcadores o sistemas de revelado empleados en 'Western blotting'
i- enzimas, que catalizan una reacción en la que se forma un precipitado coloreado, o en la que se emite luz. La peroxidasa de rábano (HRP) y la fosfatasa alcalina, se emplean con gran frecuencia. Se dispone de sustratos, para ambas enzimas que forman precipitados coloreados sobre la menbrana así como de sustratos quimioluminiscentes (emiten luz) que permiten obtener una banda en una placa radiográfica (ver ambos esquemas).
ii- radioquímicos, que emiten radiaciones ionizantes
El revelado se realiza mediante exposición de la membrana anteuna película radiográfica.
Primer anticuerpo
Sustrato solubleincoloro
Membrana
Y
Proteína a determinar
Proteína bloqueante
Segundo anticuerpo conjugado
YEnzima
Producto INSOLUBLEcoloreado
Primer anticuerpo
Sustrato solubleincoloro
Membrana
Y
Proteína a determinar
Proteína bloqueante
Segundo anticuerpo conjugado
YEnzima
Producto INSOLUBLEcoloreado
Placa radiográficaMembrana
Y
Proteína a determinar
Proteína bloqueante
Primer anticuerpo
Segundo anticuerpo conjugado
YEnzimaLUZ
Productoquimioluminiscente
Sustrato soluble
Placa radiográficaMembrana
Y
Proteína a determinar
Proteína bloqueante
Primer anticuerpo
Segundo anticuerpo conjugado
YEnzimaLUZ
Productoquimioluminiscente
Sustrato soluble
13
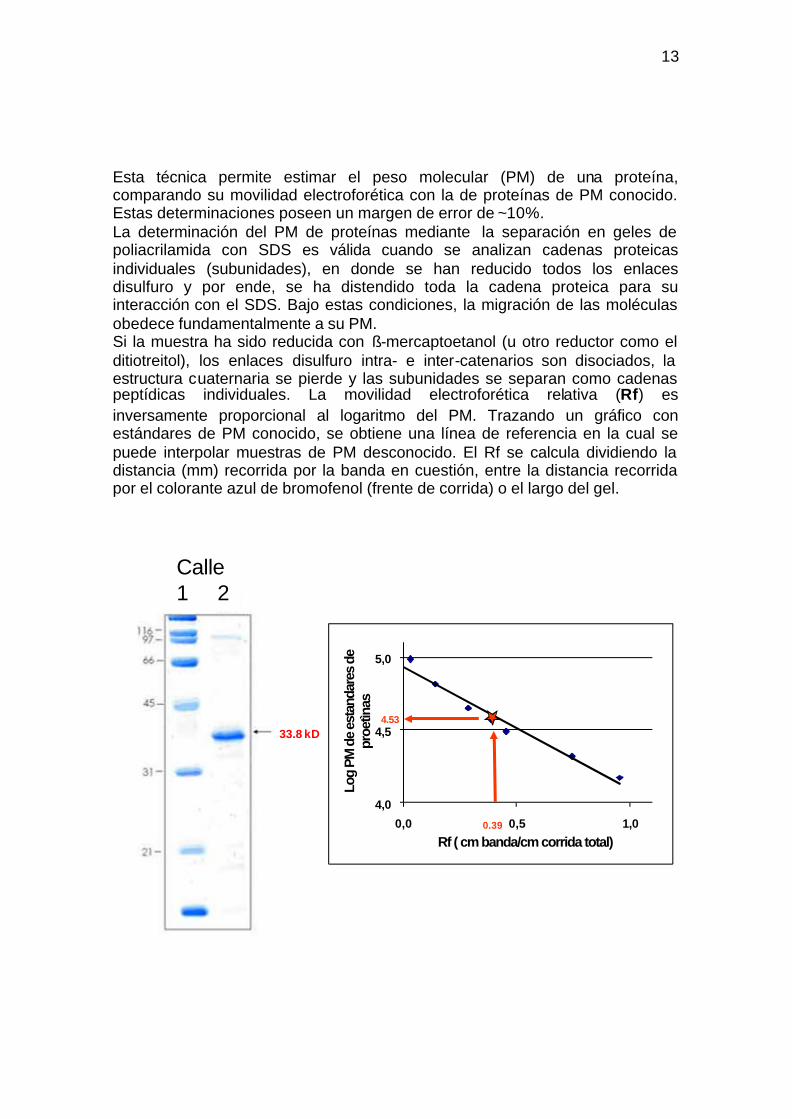
Esta técnica permite estimar el peso molecular (PM) de una proteína, comparando su movilidad electroforética con la de proteínas de PM conocido. Estas determinaciones poseen un margen de error de ~10%. La determinación del PM de proteínas mediante la separación en geles de poliacrilamida con SDS es válida cuando se analizan cadenas proteicas individuales (subunidades), en donde se han reducido todos los enlaces disulfuro y por ende, se ha distendido toda la cadena proteica para su interacción con el SDS. Bajo estas condiciones, la migración de las moléculas obedece fundamentalmente a su PM. Si la muestra ha sido reducida con ß-mercaptoetanol (u otro reductor como el ditiotreitol), los enlaces disulfuro intra- e inter-catenarios son disociados, la estructura cuaternaria se pierde y las subunidades se separan como cadenas peptídicas individuales. La movilidad electroforética relativa (Rf) es inversamente proporcional al logaritmo del PM. Trazando un gráfico con estándares de PM conocido, se obtiene una línea de referencia en la cual se puede interpolar muestras de PM desconocido. El Rf se calcula dividiendo la distancia (mm) recorrida por la banda en cuestión, entre la distancia recorrida por el colorante azul de bromofenol (frente de corrida) o el largo del gel.
Calle 1 2
33.8 kD
4,0
4,5
5,0
0,0 0,5 1,0
Log
PM d
e es
tand
ares
de
proe
tìnas
Rf ( cm banda/cm corrida total)
4.53
0.39
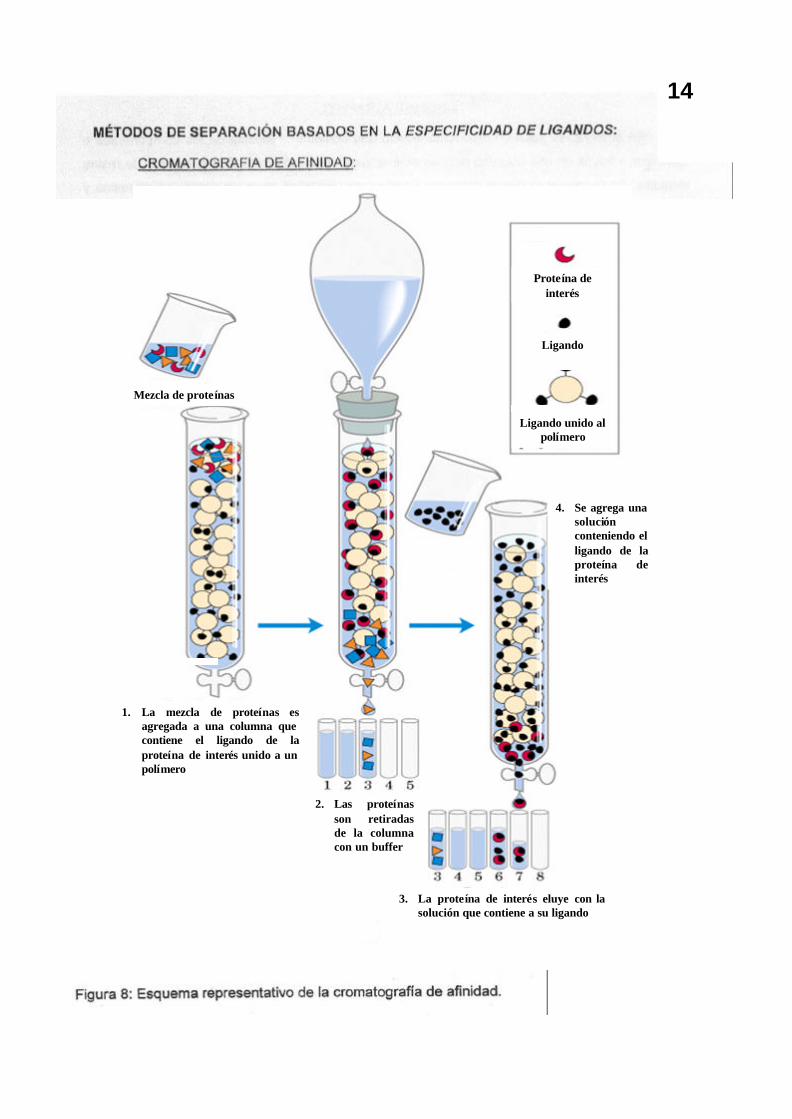
3. La proteína de interés eluye con la solución que contiene a su ligando
Mezcla de proteínas
1. La mezcla de proteínas es agregada a una columna que contiene el ligando de la proteína de interés unido a un polímero
2. Las proteínas son retiradas de la columna con un buffer
4. Se agrega una solución conteniendo el ligando de la proteína de interés
Proteína de interés
Ligando
Ligando unido al polímero
Mezcla de proteínas
1. La mezcla de proteínas es agregada a una columna que contiene el ligando de la proteína de interés unido a un polímero
2. Las proteínas son retiradas de la columna con un buffer
4. Se agrega una solución conteniendo el ligando de la proteína de interés
Proteína de interés
Ligando
Ligando unido al polímero
14

15

16





























