Embed Size (px)
Citation preview

I
USP - Universidade de São Paulo
Sergio Pereira da Costa
Aplicação de ferramentas da qualidade no desenvolvimento de métodos multiresíduos.
São Carlos
2012

II
Aplicação de ferramentas da qualidade no desenvolvimento de métodos multiresíduos.
Sergio Pereira da Costa
Dissertação apresentada ao Instituto de
Química de São Carlos, da Universidade de São
Paulo para obtenção do título de Mestre em
Ciências (Química Analítica).
Orientador: Prof. Dr. Igor Renato Bertoni Olivares
São Carlos
2012

III
AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE
TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO,
PARA FINS DE ESTUDO OU PESQUISA, DESDE QUE CITADA A FONTE.

IV
DEDICATÓRIA
À minha família que me deu suporte em todas
minhas decisões e nunca deixaram de confiar
em mim. Dedico a todos aqueles que
contribuíram de alguma forma para que este
trabalho fosse realizado da melhor forma
possível.

V
AGRADECIMENTOS
- A minha família por sempre apoiar minhas iniciativas de aprendizado e meus projetos
de vida;
- Ao Prof. Dr. Igor Renato Bertoni Olivares pelo apoio e paciência;
- A todos meus amigos que participaram diretamente ou indiretamente deste projeto,
em Especial ao Rodrigo N. Padovan e a Daniela Cordeiro;
- Um agradecimento Especial para o Prof. Dr. Vitor H. P. Pacces, pelos conselhos
concedidos e para a Prof(a) Dr(a) Ana Maria G. Peplis pelo auxílio financeiro no início
do projeto;
- Aos Funcionários que me auxiliaram e tiveram uma enorme paciência em todas
minhas dúvidas, Shirley, Andréia, Silvia, Gustavo, Marcos, Vanessa e Jeferson;
- Aos técnicos dos laboratórios do Instituto de Química pelas ajudas concedidas,
Valdecir, Renata, Cida e Guto;
- A equipe da Oficina Mecânica e Vidraria;
- A toda equipe da biblioteca, em especial para a Bernadete.
- Ao Prof. Dr. Eduardo Bessa, Prof. Dr. Marcos Lanza e a Prof(a) Dr(a) Eny Maria pelo
espaço e equipamentos concedidos para desenvolvimento do projeto, assim como para a
Equipe do Lanagro - Campinas;
- A CAPES e a FAPESP pelo apoio financeiro, ao IQSC e a USP pelo apoio institucional;
- A todos que colaboraram, direta ou indiretamente, com este trabalho.

VI
LISTA DE ILUSTRAÇÕES
Figura 1: Ciclo da garantia da qualidade analítica. (adaptado de Olivares e Antunes, 2012).
Figura 2: (A). Fluxograma da Técnica QuEChERS desenvolvida por Anastassiades e
Colaboradores em 2003. (B). Fotos ilustrativas do procedimento.
Figura 3: Gráficos da relação massa/recuperação com o volume de trabalho para analito com
KO/W = 6,0.
Figura 4: Esquema de refrigeração da fibra e sua aplicação em Cromatografia gasosa.
(GHIASVAND; HOSSEINZADEH; PAWLISZYN, 2006).
Figura 5: Ciclo PDCA.
Figura 6: Etapas para realização de um ensaio em um laboratório enfatizando a parte que será
desenvolvida no trabalho. Adaptado de Olivares, 2009.
Figura 7: Diagrama de Ishikawa para planejamento experimental da técnica QuEChERS.
Figura 8: Fluxograma do processo de trabalho para aplicação das ferramentas da qualidade.
Figura 9: (A) Extração da matriz pré-fortificada nas concentrações da faixa de trabalho; (B)
Extração da matriz limpa com posterior fortificação do extrato com as concentrações na faixa
de trabalho.
Figura 10: Diferença entre repetibilidade e reprodutibilidade intralaboratorial para análise de
precisão.
Figura 11: Gráfico (A): rampa de aquecimento da coluna inicia a 80ºC/ 1,5 min., taxa de
40ºC/min. até temperatura de 170ºC, taxa de 6,5ºC/min. até temperatura de 220ºC/7,0 min.,
taxa de 15ºC/min. até temperatura de 245ºC e por fim taxa de 50ºC/min. até temperatura de
265ºC/4 min. Gráfico (B) fluxo do gás de arraste de 0,8 mL/min.
Figura 12: Cromatograma do método cromatográfico da melhor condição para separação dos
14 compostos organoclorados; tempo de retenção: HCB, 8,017 min.; LIN, 8,410 min.; ALD,
10,817 min.; HPX, 11,722 min.; tCLD, 12,350 min.; cCLD, 12,810 min.; ppE, 13,440 min.;
DLD, 13,575 min.; PCB118, 14,723 min.; ppD, 14,990 min.; opT, 15,218 min.; ppT,16,847
min.; PCB 180, 20,383 min.; MRX, 21,153 min.
Figura 13: (A) Curva analítica para o analito Antraceno; (B) Cromatograma representativo.
Figura 14: Chromatoprobe Varian – (A) Esquema do Chromatoprobe utilizado para
dessorção térmica dos analitos da barra sortiva para o cromatógrafo; (B) Posicionamento da
barra sortiva no Chromatoprobe; (C) Acoplamento do Chromatoprobe com a barra sortiva ao
cromatógrafo.

VII
Figura 15: Projeto para confecção do Molde. (A.) Suporte de teflon com o tubo oco de aço;
(B.) suporte de teflon agregado ao molde de teflon; (C.) Esquema da barra de SBSE ou RSE
com suas dimensões para utilização em PROBE; (D.) vista do molde com seus dois lados
juntos; (E.) molde já com o sistema de travas para confecção das barras. Adaptado de Grossi,
2009.
Figura 16: Molde já finalizado: (a) Dois lados do molde e suporte de teflon; (b) Suporte com
barra ou tubo oco de aço; (c) suporte de teflon agregado ao molde; (d) vista lateral do
conjunto; (e) vista superior do conjunto; (f) Suporte com a barra de RSE já revestida com
PDMS; (g) barra de RSE.
Figura 17: Sistema RSE. (A) Tubo oco revestido com a fase PDMS no suporte de teflon; (B)
Suporte de Teflon do vial Headspace com o tubo de RSE; (C) Vial de headspace com braços
laterais, suporte e tubo de RSE; (D) Sistema RSE acoplado com sistema de refrigeração; (E)
Sistema RSE com aquecimento de 85ºC em agitador magnético; (F) Sistema RSE com vapor
na fase de headspace.
Figura 18: Sistema de refrigeração da fase sortiva - (A) Cilindro de Gás; (B) Misturador de
gases (homemade); (C) Conexão com o misturador de gases e mangueiras; (D) Esquema com
a refrigeração da barra conectado a linha de gás; (E) Vial de headspace pronto para a
extração; (F) Sistema completo.
Figura 19: Exemplo de pontos significativos e insignificativos, trabalhando com uma
distribuição normal com a tabela de probabilidade Z score para observação dos pontos a um
nível de confiança de 95%.
Figura 20: Gráfico dos efeitos de um planejamento 23 para o analito Mirex realizado em
análise multiresíduo plotado em tabela Z-Score.
Figura 21: Gráfico de Pareto mostrando os efeitos para cada fator e suas interações para o
analito Mirex.
Figura 22: Gráfico do quadrado dos fatores PSA e C18 para o Mirex, mostrando a melhor
condição de trabalho para obter maior ganho de recuperação. Valores obtidos em duplicatas
em um nível de confiança de 95%.
Figura 23: Efeitos de um planejamento 23 para o Conjunto Multiresíduo realizado pela
função de Desejabilidade plotado em tabela Z-Score.
Figura 24: Gráfico de Pareto mostrando os efeitos para cada fator e suas interações para o
Conjunto Multiresíduo.

VIII
Figura 25: Gráfico do quadrado dos fatores PSA e C18 para o Conjunto Multiresíduo,
mostrando a melhor condição de trabalho para obter maior ganho de recuperação. Valores
obtidos em duplicatas em um nível de confiança de 95%.
Figura 26: Cromatograma representativo das 10 amostras de pescado.
Figura 27: Sobreposição de amostra branca com amostra fortificada em 1 LMR: Linha
amarela representa a matriz e a linha azul representa os analitos de trabalho.
Figura 28: Cromatograma da melhor condição do planejamento experimental.
Figura 29: Proposta de fluxograma para análise da linearidade da curva.
Figura 30: Curva de calibração do método com analito Antraceno com 7 pontos.
Figura 31: Gráfico de efeito contra probabilidade Z. Considerando como resposta o fator
recuperação/massa PDMS.
Figura 32: Gráfico de pareto evidenciando os efeitos das interações de 1º, 2º e 3º ordem.

IX
LISTA DE EQUAÇÕES
Equação 1: Equação do coeficiente de partição PDMS/água.
Equação 2: Fórmula para cálculo da exatidão.
Equação 3: Fórmula para cálculo da constante de variação (CV).
Equação 4: Fórmula para cálculo da desejabilidade.
Equação 5: Fórmula para cálculo da incerteza da curva analítica.
Equação 6: Fórmula para cálculo da incerteza da recuperação.
Equação 7: Fórmula para cálculo da incerteza da precisão.
Equação 8: Fórmula para cálculo da incerteza combinada.
Equação 9: Fórmula para cálculo da incerteza expandida.
Equação 10: Fórmula para equação da curva linear.
Equação 11: Fórmula para cálculo do coeficiente de determinação.
Equação 12: Fórmula para cálculo dos resíduos da curva analítica.
Equação 13: Fórmula para cálculo de outlier – Teste de Grubbs.
Equação 14: Fórmula para cálculo da homoscedasticidade – Teste de Cochran.
Equação 15: Fórmula para cálculo a adequação do modelo linear – Teste da falta de ajuste.

X
LISTA DE TABELAS
Tabela 1: Crescimento de laboratórios acreditados no sistema ABNT NBR ISO/IEC
17025:2005.
Tabela 2: Parâmetros de validação conforme o tipo de ensaio.
Tabela 3: Propostas para análise dos parâmetros da validação.
Tabela 4: Métodos Multiresíduos.
Tabela 5: Evolução das técnicas de headspace.
Tabela 6: Limites e níveis de tolerâncias de alguns países e normas como a EPA e a FDA
para os Organoclorados de maior impacto em pescados.
Tabela 7: Avaliação da seletividade segundo a DOC-CGCRE-008 do INMETRO.
Tabela 8: Compostos organoclorados estudados.
Tabela 9: Proposta enviada a CCRC (Coordenação e Controle de Resíduos e Contaminantes).
Tabela 10: Dados de trabalho para os níveis do planejamento experimental da confecção da
fase de extração em PDMS.
Tabela 11: Aleatorização dos experimentos pelo Software Statistica 7.
Tabela 12: Rampa de aquecimento proposta para a coluna DB-5 da Agilent.
Tabela 13: Planejamento experimental com os níveis baixos e altos distribuídos
aleatoriamente para análise.
Tabela 14: área das recuperações da amostra branca fortificada sem a interferência da matriz.
Tabela 15: Recuperação dos 14 organoclorados de interesse realizado sobre a técnica
QuEChERS e injetado em GC-ECD.
Tabela 16: tabela dos fatores para o composto Mirex (MRX) em nível baixo (-1) e nível alto
(+1) com suas respectivas recuperações para cada experimento realizado aleatoriamente, foi
disposto o experimento em ordem para melhor visualização dos dados.
Tabela 17: Cálculo dos efeitos de primeira (1,2 e 3), segunda (12,13 e 23) e terceira ordem
(123).
Tabela 18: Condições de trabalho para os fatores PSA e C18.
Tabela 19: Analitos e significância de seus efeitos para um nível de confiança de 95% em
ordem decrescente em módulo (Gráficos e Figuras ver Apêndice B).
Tabela 20: Desejabilidade (di) dos 14 organoclorados.
Tabela 21: Fatores em nível baixo (-1) e nível alto (1) com suas respectivas Desejabilidade
para o experimento multiresíduo.

XI
Tabela 22: Efeitos de primeira (1,2 e 3), segunda (12,13 e 23) e terceira ordem (123).
Tabela 23: Amostras de pescado para teste de seletividade.
Tabela 24: Resultados para Curva analítica utilizando métodos dos mínimos quadrados
simples.
Tabela 25: Recuperações do dia 1, 2 (triplicata) e 3.
Tabela 26: Recuperação média e desvio padrão dos analitos de trabalho:
Tabela 27: Valores do Coeficiente de Variação (CV) para análise da precisão da técnica
QuEChERS.
Tabela 28: Incerteza padrão combinada e expandida para a técnica QuEChERS.
Tabela 29: Avaliação das curvas analíticas lineares do método QuEChERS.
Tabela 30: Recuperação de 8 barras de SBSE.
Tabela 31: Cálculo dos efeitos de primeira (1,2 e 3), segunda (12,13 e 23) e terceira ordem
(123).
Tabela 32: Resultados de MEV dos 8 experimentos realizados. Com ampliação de 150, 1000
e 5000 vezes.

XII
LISTA DE ABREVIAÇÕES
ABNT – Associação Brasileira de Normas Técnicas
ANA – Agência Nacional de Águas
ANOVA – Analyse of Variance
ANVISA – Agência Nacional de Vigilância Sanitária
AOAC – Association of Analytical Communities
BPL – Boas Prática de Laboratório
BTX – Benzeno, Tolueno e Xileno
CCRC – Coordenação e Controle de Resíduos e Contaminantes
C18 – Octadecilsilano
CONAMA – Conselho Nacional do Meio Ambiente
CV – Coeficiente de Variação
dSPE – dispersive Solid Phase Extraction
DOE – Design of Experiment – Planejamento Experimental
Embrapa – Empresa Brasileira de Pesquisa Agropecuária
EPA – Environmental Protection Agency
FDA – Food and Drugs Administration
GC-ECD – Gas Chromatography – Electron Capture Detector
GC-MS – Gas Chromatography – Mass Spectrometry
HPA – Hidrocarboneto Poliaromático
IBAMA – Instituto Brasileiro de Meio Ambiente
IEC – International Electrotechnical Commission
INMETRO – Instituo Nacional de Metrologia, Qualidade e Tecnologia
ISO – International Organization for Standardization
IUPAC – International Union of Pure and Applied Chemistry
Lanagro – Laboratórios Agropecuários
LD – Limite de Detecção
LLE – Liquid-Liquid Extraction
LMR – Limite Máximo de Resíduos
LQ – Limite de Quantificação
MAPA – Ministério da Agricultura, Pecuária e Abastecimento
MEV – Microscopia Eletronica de Varredura

XIII
MRC – Material Reference Certificate
NaCl – Cloreto de Sódio
OCPs – Organochlorines Pesticides
OECD – Organisation for Economic Co-operation and Development
PDCA – Plan – Do – Check - Act
PDMS – PoliDimetilSiloxano
PSA – Primary-Secundary Aminne
QFD – Quality Function Deployment – Desdobramento Função Qualidade
QuEChERS – Quick, Easy, Cheap, Effective, Rugged and Safe (Rápido, Fácil, Efetivo,
Robusto e Seguro)
RBC – Rede Brasileira de Calibração
RBLE – Rede Brasileira de Laboratório de Ensaios
RCRA – Resource Conservation and Recovery Act
RSD – Relative Standard Deviation
RSE – Refrigerated Sorptive Extraction
SBSE – Stir Bar Sorptive Extraction
SMA – Secretaria do Meio Ambiente do Estado de São Paulo
SPME – Solid Phase MicroExtraction
VIM – Vocabulary International Metrology
VOCs – Volatile Organic Compound
WHO – World Health Organization

XIV
LISTA DE APÊNDICES
Apêndice A: Declaração de treinamento no Laboratório Nacional Agropecuário – Lanagro –
Campinas – SP.
Apêndice B: Figuras e gráficos para os analitos de extração individuais e para o conjunto
multiresíduo no método QuEChERS.
Apêndice C: tabela de resíduos das curvas analíticas para os 14 analitos com a técnica
QuEChERS.

XV
RESUMO
O presente trabalho estuda a aplicação de ferramentas da qualidade em dois métodos
multiresíduos, sendo utilizados como instrumento para desenvolvimento de novos métodos e
otimização de métodos existentes. As ferramentas utilizadas foram o planejamento
experimental, a validação de métodos e o cálculo de incerteza de medição, as quais foram
aplicadas no método QuEChERS, já bem difundido na literatura, e no método RSE
(Refrigerated Sorptive Extraction), técnica miniaturizada de extração. Foi aplicado
planejamento experimental no método QuEChERS visando a otimização do processo de
extração de organoclorados (14 no total, sendo 12 Organoclorados e 2 PCBs) em pescado, em
que obteve-se valores de recuperação satisfatórios dentro da faixa de 70 a 120 % como é
exigido pelo protocolo de validação SANCO 12495:2011. Neste processo foi otimizado as
quantidades de sais de trabalho como NaCl, C18 e o PSA, fatores que interferem diretamente
no resultado final de recuperação. A mesma ferramenta de planejamento experimental foi
utilizada na otimização da fase de PDMS (fase extratora do sistema RSE desenvolvido)
visando obter a melhor condição de trabalho, atuando com os fatores de temperatura e tempo
de mufla, além do tempo de exposição ao meio ambiente para resfriamento entre a pré-cura e
a pós-cura. O processo de validação e cálculo de incerteza de medição foi realizado para o
método QuEChERS avaliando parâmetros primordiais para demonstrar que o método é
adequado ao uso pretendido. Foi estudado o parâmetro linearidade, pelo coeficiente de
determinação, resíduos e incerteza da curva, além da aplicação de metodologia de
investigação na adequação da curva analítica linear, com testes como outlier,
homoscedasticidade e modelo. Os dados apresentados foram satisfatórios, observando
resultados dentro do que foi planejado, inexistência de outliers pela utilização do teste de
Grubbs (foi encontrado valores perto do limite máximo do valor critico para os compostos
ppE e ppD), teste de Cochran para análise de homoscedasticidade nos resíduos em torno da
curva (todos se apresentaram homogêneos com o teste utilizado) e o teste de modelo com o
teste de falta de ajuste, em que foi averiguado se o modelo linear é o mais adequado para
metodologia. Além da linearidade, também foram avaliados os parâmetros seletividade,
precisão (com CV abaixo dos 20%) e recuperação (valores entre 70 e 120%), fatores
primordiais para o processo de validação, considerando que alguns destes também
influenciam diretamente na incerteza do resultado final. Desta maneira foi possível avaliar a
aplicabilidade do planejamento experimental no desenvolvimento de novos métodos de

XVI
extração, como no caso do RSE, bem como na otimização de métodos já bem fundamentados,
como no caso do método QuEChERS, se demonstrando como ferramenta primordial em
ambos os casos. Quanto ao método QuEChERS, também foi possível avaliar a adequação do
método quanto aos requisitos de validação e incerteza, os quais são essenciais para
confiabilidade dos resultados analíticos.

XVII
ABSTRACT
The paper studies the application of quality tools in two multiresidue methods, being
used as a tool for developing and optimization methods. The tools used were the experimental
design, method validation and measurement uncertainty, which were applied in the
QuEChERS method, already widespread in the literature, and the RSE method (Refrigerated
Sorptive Extraction), miniaturized extraction technique. Experimental design was applied in
the QuEChERS method in order to optimize the extraction process of organochlorines (14 in
total, 12 Organochlorines and 2 PCBs) in fish, which gave satisfactory recovery values within
the range of 70 to 120% as is required by the validation protocol SANCO 12495:2011. This
process was optimized the quantities of salts as NaCl, C18 and PSA, factors that directly
affect the final outcome of recovery. The same experimental design tool was used in the
optimization of extraction phase (PDMS extraction phase system developed to RSE) to obtain
the best working condition, leading with the factors as time and temperature on the muffle,
and the time of exposure to the environment to cooling between pre-cure and post-cure. The
process of validation and measurement uncertainty was conducted to evaluate the method
QuEChERS parameters to show that the method is suitable for the purpose. We studied the
linearity parameter, the determination coefficient, residues and uncertainty of the linear curve,
and the application of a methodology to observe the adequacy of the linear calibration curve,
with testing as an outlier and homoscedasticity model. The data presented were satisfactory,
no outliers was identified by the Grubbs test (values was found near the upper limit of the
critical value for compounds PPE and PPD), Cochran test for analysis of homoscedasticity in
the residue around the calibration curve (all performed with homogeneous assay) and the test
model with the lack of fit test, where it was examined whether the linear model is the most
appropriate for methodology. Besides linearity, were also evaluated parameters as selectivity,
precision (CV below 20%) and recovery (values between 70 and 120%), important factors for
the validation process, whereas some of these also has influenced of the final results of
uncertainty. In this way it was possible to evaluate the applicability of the experimental design
to develop new extraction methods, as in the case of RSE, and the optimization of the method
QuEChERS demonstrating an important tool in both cases. For the QuEChERS method was
also possible to assess the adequacy of the method for the requirements validation and
uncertainty, which are essential for reliability of analytical results.

XVIII
SUMÁRIO
1. Introdução__________________________________________________________ 01
1.1. Gestão da qualidade em laboratórios__________________________________ 01
1.2. Ferramentas da qualidade___________________________________________ 04
1.2.1. Planejamento experimental__________________________________________ 05
1.2.2. Validação de métodos______________________________________________ 06
1.2.3. Cálculo da incerteza_______________________________________________ 16
1.3. Métodos multiresíduos_____________________________________________ 17
1.3.1. Método QuEChERS (Quick, Easy, Cheap, Efficient, Rugged and Safe)______ 19
1.3.2. Método RSE (Refrigerated Sorptive Extraction)_________________________ 21
1.3.3. Aplicação de métodos multiresíduos__________________________________ 25
1.3.3.1. Análise de organoclorados em pescado________________________________ 25
2. Objetivo___________________________________________________________ 28
3. Parte experimental__________________________________________________ 29
3.1. Método QuEChERS________________________________________________ 29
3.1.1. Ferramentas da qualidade aplicadas no desenvolvimento, validação e cálculo de
incerteza para análise de organoclorados em pescado utilizando o método
QuEChERS ______________________________________________________
29
3.1.1.1. Desenvolvimento do Planejamento Experimental – Otimização QuEChERS___ 32
3.1.1.2. Parâmetros para validação__________________________________________ 34
3.1.1.2.1. Recuperação/Exatidão___________________________________________ 35
3.1.1.2.2. Linearidade/Sensibilidade______________________________ ________ 37
3.1.1.2.3. Seletividade__________________________________________________ 38
3.1.1.2.4. Precisão______________________________________________________ 39
3.1.1.3. Cálculo da Incerteza______________________________________________ 40
3.1.1.4. Método para análise de organoclorados em pescado (QuEChERS)_________ 42
3.1.1.4.1. Padrões e reagentes______________________________________________ 44
3.1.1.4.2. Materiais e equipamentos________________________________________ 44
3.1.1.4.3. Preparação das amostras e padrão__________________________________ 44
3.1.1.4.4. Método Cromatográfico__________________________________________ 46
3.2. Método RSE____________________________________________________ 49
3.2.1. Desenvolvimento do planejamento experimental – Otimização da fase extratora

XIX
RSE__________________________________________________________________ 49
3.2.1.1. Padrões e reagentes_______________________________________________ 51
3.2.1.2. Materiais e equipamentos_________________________________________ 52
3.2.1.3. Preparação das amostras e padrão___________________________________ 52
3.2.1.4. Método cromatográfico___________________________________________ 52
3.2.2. Desenvolvimento do método RSE____________________________________ 52
4. Resultados e discussão________________________________________________ 57
4.1. Aplicação das ferramentas da qualidade no desenvolvimento, validação e cálculo
da incerteza para análise de organoclorados em pescado pelo método QuEChERS ____
57
4.1.1. Planejamento experimental__________________________________________ 57
4.1.1.1.Tratamento dos dados – MIREX (MRX)_______________________________ 60
4.1.1.2. Fatores de influência no Conjunto Multiresíduo______________________ 65
4.1.1.3. Função de Desejabilidade__________________________________________ 66
4.1.2. Validação do método______________________________________________ 71
4.1.2.1. Seletividade_____________________________________________________ 71
4.1.2.2. Linearidade/Sensibilidade__________________________________________ 73
4.1.2.3. Recuperação/Exatidão____________________________________________ 75
4.1.2.4. Precisão (Repetibilidade)___________________________________________ 77
4.1.3. Cálculo de incerteza_______________________________________________ 78
4.1.4. Desenvolvimento de modelo para avaliação da curva de calibração_________ 83
4.1.4.1. Aplicação da metodologia proposta__________________________________ 88
4.2. Aplicação de ferramentas de qualidade no desenvolvimento do método RSE para
análise de organoclorados em pescado ______________________________________
93
4.2.1. Planejamento Experimental_________________________________________ 93
5. Conclusão_________________________________________________________ 98
6. Referências Bibliográficas ____________________________________________ 110

1
1. Introdução
Um dos temas em crescente expansão nos laboratórios das universidades e de empresas
privadas é a aplicação de sistemas de gestão da qualidade para garantir a confiabilidade e
rastreabilidade dos resultados gerados. Porém em laboratórios universitários, a implantação de
um sistema de gestão da qualidade, a primeira vista, não se mostra vantajoso, já que para
implantação e manutenção são necessários investimentos frequentes, os quais muitas vezes
não estão facilmente disponíveis, como em laboratórios privados (que obtêm seus recursos
pela prestação de serviços de rotina). No entanto, segundo Olivares (2009), a implantação
destes sistemas, é uma garantia de formação dos alunos de pós e de graduação, para que estes
estejam inseridos em condições reais durante sua formação, melhorando assim seu
desempenho no mercado de trabalho atual. Este conceito já é discutido na literatura
(FERNANDES et al., 2006; SANTOS et al., 2011; FERREIRA E SILVA, 2011), por ser um
sistema já bem difundido em laboratórios privados, mas ainda em processo de crescimento em
laboratórios de ensino.
O conceito de qualidade vem ganhando espaço em todos os setores de produção, com a
concretização da influência deste sistema para garantia dos processos e produtos. O sistema
ISO (International Organization for Standardization) sendo o mais conhecido mundialmente,
contêm sistemas para vários ramos de produção, como no setor empresarial com a ISO 9001,
a ISO TS 16949 para o setor automobilístico, a ISO 14001 para o meio ambiente entre outros
que vem com o objetivo de manter uma produção controlada e viavelmente econômica.
Assim os sistemas de gestão se mostram eficazes nos processos, e todos os órgãos que
necessitam de garantia de seus produtos recorrem por certificações em algum tipo de sistema
da qualidade, o que ocorre também para laboratórios privados, sejam eles de ensaio ou
calibração, e laboratórios de ensino que buscam transferir a realidade do mercado aos seus
alunos.
1.1. Gestão da Qualidade em Laboratórios
Atualmente o mercado se apresenta cada vez mais competitivo levando a necessidade
em oferecer produtos e serviços cada vez melhores, que atendam as necessidades e exigências
dos clientes, desta maneira a qualidade aparece como ferramenta primordial para as empresas

2
sobreviverem e se desenvolverem. Empresas como a Honda e a Ford, que tiveram suas vendas
em queda no ano de 2003 (WILEY, 2012) perceberam que uma forma de alavancar suas
vendas e relações de comércio, é o melhor enquadramento na gestão da qualidade visando à
satisfação do cliente. Pode-se citar também a entrada de sistemas da qualidade em
laboratórios, como o que ocorreu no Ministério da Agricultura no ano de 2003 depois de uma
missão veterinária da união européia (COSTA, 2010). Neste ano o país sofreu embargo na
exportação do mel, o que acarretou prejuízo ao mercado e uma má imagem frente aos países
importadores (PEREZ, 2007). Com isso, tem-se que os sistemas de gestão da qualidade, além
de promover estrutura de garantia para os processos, estabelecem também rastreabilidade,
evidenciando assim a competência dos analistas do laboratório com tal sistema.
Como citado anteriormente, a adesão a sistemas de gestão da qualidade é um fator de
extrema importância devido à exigência dos clientes e confiabilidade nos processos. Órgãos
do governo como MAPA (Ministério da Agricultura, Pecuária e Abastecimento), ANVISA
(Agência Nacional de Vigilância Sanitária) entre outros, exigem dos laboratórios parceiros
que estes implementem um sistema de gestão de qualidade, como a ISO/IEC 17025 e realizem
manutenção deste sistema (OLIVARES, 2009).
Este requisito é fundamental para estabelecer os controles exigidos pelos clientes. No
ano de 2010 o país foi visitado por uma missão do serviço norte-americano para verificação
do sistema de segurança alimentar do Brasil, em que os inspetores brasileiros foram avaliados
e designados aptos a realizar auditorias e por obter um sistema de inspeção organizado, além
de visita nos laboratórios nacionais agropecuários (Lanagro – Laboratórios Nacionais
Agropecuários) de campinas - SP e Porto Alegre - RS. Este fato ocorreu depois da
identificação de Ivermectina acima dos limites estabelecido nos EUA (BELTRÃO, 2010).
Assim como no MAPA e na ANVISA, esta exigência esta presente em outros órgãos
como o ANA (Agência Nacional de Águas), IBAMA (Instituto Brasileiro de Meio Ambiente
e dos Recursos Naturais Renováveis) e SMA (Secretaria do Meio Ambiente do Estado de São
Paulo). Os laboratórios atuam com sistemas da qualidade para garantir as exigências locais e
estar compatíveis com que as normas internacionais demandam. Exemplos de normas
adotadas devido a sua influência e abrangência são as normas da EPA (Environmental
Protection Agency) dos Estados Unidos que orienta, por exemplo, atividades analíticas na
área de meio ambiente e as normas da FDA (Food and Drugs Administration) relacionadas ao
monitoramento da qualidade dos alimentos. Logo, com o crescimento das exigências, o
número de laboratórios acreditados em ISO/IEC 17025 e BPL (Boas Práticas de Laboratório)

3
vêm aumentando gradualmente no decorrer dos anos, distribuídos em todo território nacional
(INMETRO, 2012).
Tabela 1: Crescimento de laboratórios acreditados no sistema ABNT NBR ISO/IEC
17025:2005.
Tipo de Acreditação
Ano ISO/IEC 17025 (RBC) ISO/IEC 17025 (RBLE) BPL
2005 264 150 15
2009 236 294 17
2011 281 424 33
Adaptado de: SCHIAVON, 2011.
Os sistemas de gestão supracitados estabelecem diferentes diretrizes, com destaque na
aplicação de ferramentas da qualidade (como a validação e cálculo de incertezas) a qual se faz
imprescindível para garantia da confiabilidade de um resultado. Desta maneira, a gestão de
qualidade em química aparece como uma nova área de pesquisa (FERNANDES et al., 2006),
visando aumentar a garantia da confiabilidade dos laboratórios, envolvendo sistema de gestão
às ferramentas da qualidade aplicadas para avaliação de novas metodologias, a qual recebe
grande destaque no monitoramento de resíduos e contaminantes em alimentos e na área
ambiental (OLIVARES, 2009).
O monitoramento de resíduos e contaminantes em alimentos destaca-se como uma
necessidade notória devido à importância do agronegócio e as exigências cada vez maiores do
mercado internacional. Esta necessidade levou a criação, através da Portaria Interministerial
No 902 de 22 de Setembro de 2008, de uma rede de laboratórios para análise de resíduos de
contaminantes em produtos de origem animal e vegetal a qual aproveita da grande experiência
de monitoramento dos Lanagro, a vivência do desenvolvimento de pesquisa da Embrapa
(Empresa Brasileira de Pesquisa Agropecuária) somadas ao conhecimento teórico, acadêmico
e de pesquisa de renomados centros de pesquisa e universidades brasileiras.
Nesta rede, destacam-se os Lanagro, Laboratórios Oficiais do MAPA, que desde 1979
são os responsáveis maiores pelo monitoramento de resíduos e contaminantes em produtos de
origem animal no país (OLIVARES, 2009). Atualmente estes laboratórios além de
apresentarem técnicos e equipamentos atualizados visando atender a demanda analítica frente
à análise de resíduos de contaminantes, também buscam a garantia da confiabilidade dos

4
resultados analíticos através da aplicação de Sistemas de Gestão da Qualidade
internacionalmente reconhecidos, principalmente em relação a ISO/IEC 17025, atendendo
além de uma pressão de mercado, também a pressão nacional e internacional quanto à
conformidade com esta norma.
Desta maneira, é inegável a importância da aplicação de Sistemas de Gestão da
Qualidade, bem como suas ferramentas, para análise de resíduos de contaminantes, análises
estas de grande destaque tanto por ser ação prioritária para a avaliação da exposição humana
através da cadeia alimentar, como para a manutenção dos mercados interno e externo (PAYÁ
et al., 2007). Devido ao interesse em se analisar um grande número de resíduos e
contaminantes nos alimentos, lista esta frequentemente atualizada devido a exigências
nacionais e internacionais, a aplicação de métodos multiresíduos adequadamente validados, e
com suas incertezas estimadas, se mostram como uma excelente ferramenta.
1.2. Ferramentas da qualidade
Dentre as várias formas de controle de processos dentro de um laboratório, algumas
ferramentas se mostram úteis para manutenção e reprodutibilidade do processo (ou análise)
estudado e desenvolvido. Ferramentas como validação e cálculo de incerteza que são
parâmetros da norma ISO/IEC 17025:2005 são requisitos obrigatórios para a manutenção do
sistema e exigidos pelos órgãos de acreditação. Estes requisitos são confirmações objetivas
para a metodologia aplicada, para observar se à aptidão do método e do resultado analítico,
com o que é exigido, seja pelo cliente, seja por uma legislação.
Há também ferramentas de qualidade para controle de processo que é mais utilizada na
área de engenharia, como método Seis Sigma, Desdobramento da Função Qualidade (QFD),
planejamento experimental ou na sigla em inglês DOE (Design Of Experiment) (JUNIOR,
2008). O planejamento experimental também se mostra uma ferramenta útil para
desenvolvimento de metodologias na área da química, já que este discute os fatores que
influenciam no processo. Essas ferramentas entre outras são fundamentais para estabelecer
um trabalho de qualidade dentro de um laboratório.
Olivares e Lopes, 2012 propõe uma forma de manter a qualidade dentro do ambiente de
um laboratório através de um ciclo, que foi denominado de Ciclo da Garantia da Qualidade
Analítica, que mantém conexões entre os requisitos da qualidade e a importância de um com o
outro (garantia da qualidade, validação e cálculo da incerteza), como peças cruciais para

5
manutenção de resultados de qualidade. Sabendo que o analista esta sempre influenciado por
fatores como validação de métodos, acreditação, rastreabilidade, teste de proficiência,
material de referência, tal procedimento é fundamental para garantia da qualidade, como
apresentado na Figura 1.
Figura 1: Ciclo da garantia da qualidade analítica. (adaptado de Olivares e Lopes, 2012).
1.2.1. Planejamento Experimental
Segundo Neto (2010) a metodologia de planejamento experimental é um termo geral
para designar o conjunto de técnicas utilizadas para experimentação sistemática além de
afirmar que o uso desta ferramenta é a aplicação de “Bom Senso”, pois se sabe que todas as
respostas são dependentes de seus fatores, e assim devem ser investigados com eficácia.
Esta metodologia pode ser utilizado por cientistas ou engenheiros para otimização de
processos, podendo assim aumentar rendimento, minimizar tempo de processos etc. Com isso,
observa-se que apesar de ser uma ferramenta aplicada na área de engenharia, adéqua-se muito
bem a metodologia laboratorial, em fase de preparação de métodos para se atingir um ponto
ótimo, independente do tipo de análise que esta se realizando. Alguns métodos de
planejamento se destacam, sendo apropriado para certos estudos, como planejamento
experimental completo para quando há poucos fatores, planejamento experimental fracionário
para quando há vários fatores (ferramentas úteis para estudos preliminares), além de outros
processos como Box-Behnken (KHAJEH, 2009) e Doehlert (SKARTLAND et al., 2011).
Um grande problema observado nesta área de planejamento experimental é quando se
observa várias respostas para uma metodologia, como por exemplo, em um método
multiresíduo em que temos várias respostas de recuperação. Desta maneira é necessário

6
avaliar estes dados da melhor forma possível, com intuito de buscar um ponto ótimo para
todos os fatores independentes e que tenham ainda assim a melhor resposta para todas as
recuperações. Harrington propôs um método para análise destes problemas e denominou de
Função de Desejabilidade (HARRINGTON apud ISLAM, 2009) que se mostrou adequado
para métodos com várias respostas. A função de Desejabilidade é uma técnica popular e bem
estabelecida para determinação simultânea do ponto ótimo de um conjunto de variáveis que
pode determinar a melhor performance de uma resposta ou mais.
A função de desejabilidade vem sendo utilizado em alguns campos da química como,
produção de biohidrogênio (WANG; WAN, 2009), otimização de múltiplas respostas na
determinação de cloroanisóis e clorofenóis (PIZARRO; GONZÁLEZ-SÁIZ; PÉREZ-DEL-
NOTARIO, 2006), determinação de hidroquinona (RUEDA et al., 2003) entre outras
aplicações.
Desta maneira, para otimização de processos analíticos, a ferramenta estatística de
planejamento experimental se mostra promissora, assim como procedimentos de validação e
cálculo de incerteza são primordiais para condução de um trabalho de qualidade na área
laboratorial.
1.2.2. Validação de Métodos
As ferramentas da qualidade visam controlar, identificar e quantificar a qualidade de um
resultado analítico através de processos estatísticos, fornecendo informações pertinentes para
que desvios ou erros possam ser corrigidos ou minimizados. Estes processos estatísticos
compreendem os estudos de Validação e Cálculos de Incerteza das metodologias analíticas.
Estas ferramentas se apresentam em Sistemas de Gestão da Qualidade para laboratórios
e cada vez mais se destacam nos laboratórios de pesquisa, uma vez que a publicação em
revistas científicas requer a apresentação de resultados consistentes, os quais podem ser
obtidos através da aplicação de ferramentas da qualidade, como por exemplo, relacionados à
validação de metodologias e cálculos de incerteza dos resultados.
A validação deve ser realizada para toda metodologia não normalizada, criada ou
desenvolvida pelo próprio laboratório, ou para metodologias normalizadas que forem
utilizadas fora de seu escopo, ampliadas ou modificadas (entende-se por metodologia
normalizada, aquelas editadas por agências reguladoras ou de normalização como ABNT,
FDA, EPA, OECD entre outras) (OLIVARES, 2006a).

7
Os protocolos de validação geralmente são desenvolvidos para determinado tipo de
análise, gerando uma diversidade de protocolos nacionais, como destacado anteriormente, e
até internacionais, como por exemplo, no desenvolvimento de metodologias analíticas
ambientais para análise de poluentes em resíduos sólidos que, nos Estados Unidos apresenta
um protocolo específico estabelecido pela EPA (Environmental Protection Agency) (EPA,
2006). Alguns protocolos internacionais muitas vezes são aplicados no Brasil devido a
exigências específicas, como no caso do monitoramento de resíduos de contaminantes em
alimentos realizado pelo Ministério da Agricultura, através dos Laboratórios Analíticos
Agropecuários (Lanagro), que devido à pressão da comunidade européia adota critérios de
validação internacionais (COMUNIDADE EUROPÉIA, 2002; SANCO, 2007).
Segundo orientação DOC-CGCRE-008 do INMETRO (INMETRO, 2011), para
confirmar que o método é apropriado, o laboratório deve validar métodos não normalizados,
métodos criados/desenvolvidos pelo próprio laboratório, métodos normalizados usados fora
dos escopos para os quais foram concebidos e ampliações e modificações de métodos
normalizados. A Tabela 2 apresenta os parâmetros de validação de acordo com o tipo de
ensaio.
Tabela 2: Parâmetros de validação conforme o tipo de ensaio.
Parâmetros
Tipo de ensaio
Qualitativo
Determinação do
componente (ou analito)
em maio teor (1).
Análise de
elementos menores
e traços (2)
Propriedades físicas
Precisão √ √ √
Seletividade √ √ √ √
Recuperação √ √ √
Robustez √ √ √ √
Sensibilidade/linearidade √ √ √
Limite de detecção √ √
Limite de quantificação √
Fonte: Documento DOC CGCRE-008 (INMETRO, 2011).
(1) Dependendo da faixa de concentração do analito pode não ser necessária a
determinação dos limites de detecção e de quantificação, como por exemplo:
determinação de sacarose em balas e determinação do teor de gordura em carnes.
por exemplo componentes maiores com concentração entre 1 a 100%,
(2) São considerados como de menor teor concentrações entre 0,01 a 1% e
elementos traços, os elementos em concentração abaixo de 0,01%.

8
A aplicação de medidas para assegurar a qualidade dos dados analíticos é cada vez mais
discutida, devido à importância das decisões que são tomadas frente aos resultados de uma
análise, como por exemplo, análises forenses; potabilidade de água; bioequivalência de
produtos farmacêuticos; diagnósticos de áreas contaminadas entre outros. Lembrando que não
há uma receita pré-estabelecida para validação de metodologias, o analista deve utilizar a
melhor forma, ou guia que melhor se adeque ao seu tipo de trabalho, A Tabela 3 abaixo
apresenta algumas abordagens de validação baseada em diferentes referências.

9
Tabela 3: Propostas para análise dos parâmetros da validação.
Referência (INMETRO, 2011) (EPA, 2006) (SANCO, 2012) (LEITE, 2008)
Metodologia Método 8081 (inseticidas
organoclorados para GC/ECD).
Seletividade
Fazer a análise com a amostra e
materiais de referência pelo
método em estudo e outros
métodos validados. Analisar
amostras contendo vários
interferentes suspeitos na
presença do analito de interesse.
Resposta no
branco e
controle de
amostra, menor
que 30% do LQ
Tem como objetivo garantir a
identidade do ativo que se deseja
determinar. De uma forma geral
podemos conceituar a seletividade
como a medida da indiferença de
método a presença, na amostra, de
espécies que poderiam interferir na
determinação do analito.
Linearidade
Verificar até que ponto a faixa
de concentração do analito
coincide com a faixa dinâmica
linear. Serão necessários vários
níveis de concentração, no
mínimo cinco, para construir a
curva analítica. Aplicação de
testes estatísticos para
Análise pela
curva de
calibração com
resíduos
menores que
20%
Com objetivo de obter proporção
direta entre a concentração de analito e
sinal obtido, confecciona uma curva
analítica de resposta. A variação dos
pontos (massas ou concentrações
distintas) numa curva, como proposta
ideal, deve ser de 20% para o menor
valor, e 15% para os demais pontos.

10
verificação dos dados obtidos. Uma curva deve conter, no mínimo, 5
pontos distintos. A curva pode ser
construída de duas formas, por
diluição de uma solução mãe ou ponto
a ponto.
Exatidão
Determinar a
recuperação
média para os
níveis
fortificados, de
70 a 120%.
Precisão
A repetibilidade de uma medida
a longo termo, por exemplo,
semanalmente, a precisão, com
instrumentos calibrados e
usando padrões comparáveis e
em dias diferentes não deve
variar mais que 15%.
Determinar a
repetibilidade
dos desvios
padrões
relativos,
determinar para
os níveis
fortificados,
menor ou igual a
O primeiro passo para a verificação da
confiabilidade de uma análise é
investigar se a mesma se repete
quando operada sequencialmente.

11
20%
Recuperação
Os processos normalmente
utilizados para avaliar a
tendência de um método são,
entre outros:
uso de materiais de referência
certificados (MRC), participação
em comparações
interlaboratoriais
e realização de ensaios de
recuperação.
A tendência pode ser expressa
como recuperação analítica,
definida como
A porcentagem do analito do
interesse para um intervalo de
recuperação na maioria dos
métodos desenvolvidos é entre
80 e 120%.
Recuperação
média para cada
analito
representativo é
na faixa de 70 a
120% com um
RSD menor que
20%.
A recuperação deveria ser executada
diretamente na matriz, mas o estado
físico da amostra pode interferir na
seletividade e no limite de
quantificação. Tendo em vista este
problema é necessário isolar os
analitos da amostra. Sobre um branco,
adicionamos a nossa espécie ou
analito, e promovemos a extração sem
interferentes.
Robustez
Pode ser obtido
no decorrer do
método/verificar
%100xadovaloresper
vadovalorobser

12
pela recuperação
média
estabelecida e o
desvio padrão
relativo.
Limite de
Quantificação
Branco da amostra.
Branco com adição de
concentrações variadas
do analito, próximas ao
LD. Medir, uma vez cada
replicata independente, a cada
nível de concentração.
O desenvolvedor deve gerar
estimativas de métodos de
quantificação e métodos de
limite de detecção dos analitos
de interesse na matriz de
interesse.
Por definição: o
nível mais baixo
no qual tenha
sido
demonstrado
que os critérios
de exatidão e
precisão foram
cumpridos,
menor ou igual
ao limites
máximos de
resíduos (LMR)
Será o valor obtido em confiabilidade
de precisão aceitável. Para o limite de
quantificação, considera-se que não se
atingiu o limite da técnica/método ou
do equipamento. Faz-se necessário ter
a condição analítica definida, em que
as espécies estejam dentro dos
parâmetros normais do equipamento
ou da técnica. Fazer um branco para
determinar interferência na região
analítica, causada pelo meio reacional,
interferência do ambiente,
manipulação, equipamento. Logo após
dilui-se a amostra com a analito em ¼
do teor do analito e se observa a
repetitividade, se não for a desejada
sxLQ 10

13
diminui a diluição. Se verificar
repetitividade, já esta estabelecido um
valor para o limite de quantificação.
Limite de
detecção
Branco da amostra.
Branco da amostra com
adição da menor concentração
aceitável do analito.
É recomendado um mínimo de 7
replicatas para a determinação
do LD. Por exemplo, no caso de
se analisar 7 alíquotas, temos 7-1
= 6 graus de liberdade de uma
matriz de branco da amostra com
adição da menor concentração
aceitável do analito. Para esses
graus de liberdade, o valor de t
unilateral, para 99% de
confiança é 3,143. O LD será
igual a 3,143 vezes o desvio
O desenvolvedor deve gerar
estimativas de métodos de
quantificação e métodos de
limite de detecção dos analitos
de interesse na matriz de
interesse.
É calculado como correspondente a
concentração que produziria um valor
do sinal medido 3 vezes maior que o
nível de ruído médio, medido com a
solução de controle ou branco,
conforme recomendado por norma
IUPAC.
stxLD n .1,1
stLD n .0 1,1

14
padrão amostral.
Efeito da matriz
Os elementos prévios no
processo de desenvolvimento do
método envolvem o uso de
padrões conhecidos ou matrizes
limpas dopadas. Uma vez que o
novo método passou por todos
os testes prévios, estar pronto
para o mais importante
demonstração de todo o processo
de desenvolvimento do método,
ou seja, como este ira atuar na
matriz real com o que é
pretendido a ser usado.
O método deve ser adequado
para a variação do tipo de
matrizes.
Comparação da
resposta do
solvente padrão
e matriz
escolhida
Sensibilidade do
instrumento
É influenciado por alguns
fatores, limite de detecção do
instrumento, limite de
Considere a espécie química ou
analito, numa solução ideal, isenta de
interferentes e na melhor condição de

15
quantificação do método e
requerimentos regulatórios da
aplicação proposta.
análise. A menor quantidade dessa
espécie, detectada pelo sistema
analítico, em confiabilidade.
Efeitos de
interferentes
Definição de falso negativo é a
resposta negativa para uma
amostra que contenha o analito
de interesse ou acima do estado
do nível de ação do método. Um
procedimento candidato deve
produzir não mais que 10% de
falsos positivos. Definição de
falso positivo é a resposta
positiva para uma amostra que
contenha o analito de Interesse
abaixo do estado do nível de
ação.
t = número de Student, n-1 = graus de liberdade, 1-α = nível de confiança. x = média, s = Desvio padrão

16
De maneira mais abrangente, uma das medidas adotadas para garantir a confiabilidade e
rastreabilidade de um resultado analítico pode ser a aplicação de um Sistema de Gestão da
Qualidade, e de forma mais pontual a validação da metodologia. Porém, apenas a validação da
metodologia não garante a concordância dos resultados analíticos entre diferentes
laboratórios, uma vez que os erros de cada laboratório podem ser diferentes, pois podem estar
relacionados aos equipamentos e instalações, assim o cálculo da incerteza também se faz
necessário, além dos procedimentos internos de controle.
1.2.3. Cálculo de Incerteza
Quando uma mesma amostra é analisada por diferentes laboratórios, com diferentes
metodologias, é esperado que os resultados analíticos apresentem concordância entre si. No
entanto, cada resultado é influenciado por diferentes fontes de incerteza durante a análise, que
variam para cada laboratório e metodologia, e alteram o resultado final. Assim, dois
laboratórios que analisem a mesma amostra por duas metodologias oficiais diferentes podem
chegar a diferentes resultados (OLIVARES, 2006a). Segundo o guia Eurachem, a incerteza é
um parâmetro associado ao resultado de uma medição, que caracteriza a dispersão de valores
que poderiam ser razoavelmente atribuídas ao mensurando (EURACHEM, 2000).
O cálculo da incerteza irá expressar o quanto se pode confiar em um resultado analítico,
considerando para isto todas as possíveis fontes de incerteza da metodologia, somando-as, e
finalmente demonstrando a faixa de variação aceitável do resultado final. Desta maneira, a
incerteza atribuída a um resultado garante a faixa de confiança do mesmo, demonstrando com
maior precisão o grau de concordância entre resultados obtidos em diferentes laboratórios.
Não existe um consenso em relação à forma de realizar o cálculo da incerteza de uma
medição, uma vez que cada metodologia apresenta diferentes fontes de incerteza, existindo
apenas alguns guias que orientam o cálculo, como no caso do Guia Eurachem/Citac adotado
pela Sociedade Brasileira de Metrologia (EURACHEM, 2000).
Ferramentas da qualidade como validação e cálculo de incerteza são obrigatórias para
laboratórios que apresentam um Sistema de Gestão da Qualidade, e atualmente cada vez mais
exigidas na pesquisa devido à necessidade em apresentar dados consistentes para publicação
em revistas científicas (GONZÁLEZ; ÁNGELES, 2007). Entre os diferentes campos da
química, a química analítica, notoriamente tem aplicado amplamente a gestão da qualidade
através de normas especificas como a ISO/IEC 17025, necessário, por exemplo, no

17
desenvolvimento e aplicação de métodos multiresíduos, muito aplicado hoje em dia devido
sua facilidade de manuseio e capacidade em avaliar simultaneamente uma grande gama de
analitos para diferentes matrizes.
1.3. Métodos Multiresíduos
Com o desenvolvimento das técnicas de análises no decorrer dos últimos anos, a busca
por um método que analise não apenas um analito, mas vários ao mesmo tempo, é uma rota
permanente, pois minimiza o trabalho do analista e consequentemente a utilização de mão de
obra e solventes orgânicos. Esta abordagem se encontra atualmente em vários campos de
pesquisa, na área ambiental, na investigação de contaminantes em alimentos e água, com os
mais diversos analitos. Assim com o aumento do rigor na fiscalização dos órgãos
responsáveis, utilizar métodos que abrangem uma maior gama de analitos se torna tendência.
Com este pensamento surgiu um novo termo, designado “Green Methods”, que é a utilização
consciente dos recursos, gerando o menor impacto possível, seja na metodologia ou no
descarte, com isso surgiu novas técnicas de extração, as técnicas miniaturizadas, que
dispensam a utilização de grandes quantidades de solventes, e são relativamente técnicas
rápidas de serem aplicadas (OLIVARES, 2006b).
Desta forma, a inovação na área de extração se torna foco com várias técnicas
miniaturizadas, ou técnicas com incidência de tecnologia que aumenta o poder extrativo, mas
que por outro lado são técnicas relativamente caras. Assim pode-se utilizar a forma mais
adequada com o respectivo analito de interesse, seja ele na área ambiental, ou na análise de
contaminantes em alimentos, como demonstrado na Tabela 4.

18
Tabela 4: Métodos Multiresíduos.
Técnica Analitos Referência
SPE (Solid Phase Extraction) Produtos farmacêuticos,
Pesticidas, Morfina.
(AZZOUZ,
BALLESTEROS, 2012;
SASANO et al., 2000;
AHADI et al., 2011)
ASE (Accelerated Sorptive
Extraction)
PHA, PCB, Hidrocarbonetos,
Organoclorados semi-
voláteis.
(RICHTER et al., 1996; HE
et al., 2009)
GPC (Gel Permeation
Chromatography) Pesticidas. (BEIZHEN et al., 2008;
MAE (Microwave Assisted-
Extraction) Produtos farmacêuticos.
(AZZOUZ,
BALLESTEROS, 2012; )
MSPD (Matrix Solid Phase
Dispersion) Multi-toxinas.
(RUBERT, SOLER,
MAÑES, 2011)
SFE (Supercritical Fluid
Extraction) Isoflavonas (KLEJDUS et al., 2010)
QuEChERS (Quick, Easy,
Cheap, Efficient, Rugged and
Safe)
Inseticidas organofosforados,
herbicidas.
(SINHA, VASUDEV, RAO,
2012)
SPME (Solid Phase
Microextraction) Contaminantes emergentes. (WANG et al., 2011)
SBSE (Stir Bar Sorptive
Extraction) Diclofenaco.
(KOLE, MILLERSHIP,
MCELNAY, 2011)
RSE (Refrigerated Sorptive
Extraction) BTEX. (CARMI et al., 2009)
Rotating Disk Extraction
Sorptive Pesticida, nonilfenol.
(GIORDANO, RICHTER,
AHUMADA, 2011)
HS-SPME (Headspace –
Solid Phase Microextraction) Organoclorados (OLIVARES, 2006b)
u-SPE (Micro Solid Phase
Extraction)
Hidrocarbonetos
poliaromáticos PAH,
(HUANG, ZHOU, XIE,
2011)

19
Entre estas técnicas de extração, algumas vêm se destacando tanto em rotina de
laboratórios como em órgãos que promovem processos de extração, como a AOAC, que vem
popularizando a técnica QuEChERS. Assim como, em técnicas miniaturizadas, como a RSE,
que se mostram promissoras para o campo de extração e que se adaptam bem a sistemas de
qualidade e tem boas respostas com as ferramentas aplicadas.
1.3.1. Método QuEChERS (Quick, Easy, Cheap, Efficient, Rugged and Safe)
Com a conscientização ambiental que todos vêm desenvolvendo em todos os campos de
trabalho, os laboratórios vêm se adaptando também com essa atual realidade, assim formas
que impactem o mínimo possível estão sendo desenvolvidas, para atender a ideais como a do
“Green Methods”. A busca por este ideal fez com que surgissem novas propostas de extração
no decorrer do tempo, como a técnica QuEChERS (Quick, Easy, Cheap, Efficient, Rugged
and Safe), apresentada na Figura 2, que foi desenvolvida pela primeira vez por Anastassiades
et al. (2003), a qual surgiu da junção da extração liquido-liquido LLE (Liquid-Liquid
Extraction) e da dSPE (Dispersive Solid Phase Extraction) com uma fase de Clean-up para
obtenção dos analitos.
A proposta inicial foi desenvolver esta técnica para extração de pesticidas em frutas e
vegetais, matriz que tem poucos interferentes. Com o decorrer do tempo a técnica se mostrou
promissora para outros tipos de matrizes e analitos, e foi modificada conforme os interesses
da análise, assim várias propostas estão presentes na literatura atual, sendo aplicada para
quase todos os tipos de analitos possíveis, e sendo já comercializada por algumas empresas,
que fornecem kits prontos para aplicação (RESTEK, 2012), além da popularização em órgãos
reguladores, tornando-se técnica oficial como na AOAC (AOAC, 2007).
A técnica é relativamente simples, pois não necessita de trabalho árduo para sua
realização, é barato, já que utilizam componentes simples e de fácil acesso, é rápido quando
comparado com outras técnicas aplicadas em rotina. Segura, pois possui boa reprodutibilidade
e atende o que é pedido hoje quando se fala em uso de componentes prejudiciais a saúde e ao
meio ambiente. A técnica se estabelece em uma fase de extração líquido-líquido com um
solvente orgânico, inicialmente realizado com acetonitrila, mas que no decorrer do tempo foi
se modificando para atender apropriadamente os trabalhos envolvidos, assim como a
aplicação dos sais. Algumas modificações já estão bem esclarecidas na literatura, como o uso
do metanol para extração de pesticidas em repolho e rabanete na fase de extração líquido-

20
líquido (NGUYEN et al., 2008), ácido acético 1% em acetonitrila para análise de produtos
veterinários em leite (COSTA, 2010), análise de clorotolanil com acidificação, pois este
depende do pH (EURL, 2011), entre várias outras propostas.
(A)
(B)
Figura 2: (A). Fluxograma da Técnica QuEChERS desenvolvida por Anastassiades e
Colaboradores em 2003. (B). Fotos ilustrativas do procedimento.

21
1.3.2. Método RSE (Refrigerated Sorptive Extraction)
As formas de preparação da amostra para análise, aplicadas em diferentes laboratórios
atualmente, aborda várias técnicas já bem difundidas. Neste preparo de amostra é de extrema
importância a obtenção de um sinal analítico adequado e é reconhecido a necessidade de
métodos analíticos rápidos e mais eficientes. Desta maneira a evolução neste campo se fez
necessário para obter limites de detecção cada vez menores, seja de amostras ambientais
(OLIVARES, 2006b), análises de drogas de controle (MELO et al., 2009), entre outros
campos. Este fator é importante, devido às exigências de órgãos nacionais e internacionais,
por um controle rígido para estabelecer limites máximos de diferentes contaminantes.
Assim como técnicas já bem difundidas como o QuEChERS, novas técnicas de extração
vem surgindo com o decorrer dos anos. Técnicas que utilizem menos solventes são
promissoras para a área analítica, assim os chamados “Green Methods” se tornam tendência, e
as formas de extração miniaturizadas acompanham este avanço.

22
Tabela 5. Evolução das técnicas de headspace.
Período
desenvolvido Técnica Observações Referências
Meados de
1965
Headspace
Estático
Piores limites de detecção
por amostrar diretamente o
gás do headspace. (WANG; MCCAFREY;
NORWOOD, 2008)
Headspace
dinâmico
“purge-and-trap”
Melhores limites de detecção
que Headspace Estático por
concentrar o gás em um trap.
(WANG; MCCAFREY;
NORWOOD, 2008)
1990 Headspace-
SPME
Melhora limites de detecção
por concentrar analitos do
headspace na fibra seguido
de injeção direta ao
cromatógrafo.
(PAWLISZYN;
ARTHUR, 1990;
BELTRAN; LÓPEZ;
HERNÁNDEZ, 2000)
1999 Headspace-SBSE
Melhora limites de detecção,
pois a SBSE apresenta mais
fase sortiva que a SPME para
concentração dos analitos em
headspace.
(BALTUSSEN et al.,
1999)
2006-2007
Headspace-
SPME com
refrigeração
Apesar de menos fase sortiva
que a SBSE, a aplicação da
SPME com refrigeração em
headspace desloca o
equilíbrio da sorção dos
analitos, concentrando estes
na fibra, atingindo assim
menores limites de detecção.
(GHIASVAND;
SETKOVA;
PAWLISZYN, 2007;
CHEN et al., 2007;
CARSEK, CUDJOE e
PAWLISZYN, 2007;
CARASEK,
PAWLISZYN, 2006;
GHIASVAND,
HOSSEINZADEH e
PAWLISZYN, 2006).

23
Como evidenciado na Tabela 5, a análise de amostras em headspace já é utilizada a um
longo período. Com os avanços no decorrer do tempo com o uso de fases extratoras iniciadas
por Pawliszyn e Arthur nos anos 90, foi observado que a técnica do uso de uma fase
compatível com o analito de interesse, ou seja, polaridade similar, poderia se obter maiores
níveis de extração, aumentando assim a eficiência dos processos. Desta forma, várias outras
técnicas surgiram como a SPME e a SBSE em headspace, assim como a SPME com
refrigeração, e como consequência o aumento dos níveis de extração. Sendo uma área
promissora, vários outros métodos multiresíduos com fase extratora surgiram como
demonstrado na Tabela 4. Estas técnicas baseiam-se em constante de partição do analito em
solução e a fase extratora, chamado de Kpdms/água que segundo a literatura tem relação com o
Koctanol/água, sendo assim passível de cálculo.
SBSE
W
W
SBSE
W
SBSE
WPDMSWOV
Vx
m
m
C
CKK //
Equação 1: Equação do coeficiente de partição PDMS/água.
Onde KO/W é a constante de equilíbrio Octanol/água, o KPDMS/W é a constante de
equilíbrio PDMS/água, mSBSE é a massa do analito na fase extratora, mW é a massa de analito
na solução, Vw é o volume da solução e VSBSE é o volume da fase extratora.
A relação dos volumes é também conhecida como fator β e quanto menor, melhor será
a relação, ou seja, quanto menor o volume de solução e maior o volume da fase, maior será a
recuperação do método, como exemplificado na Figura 3.
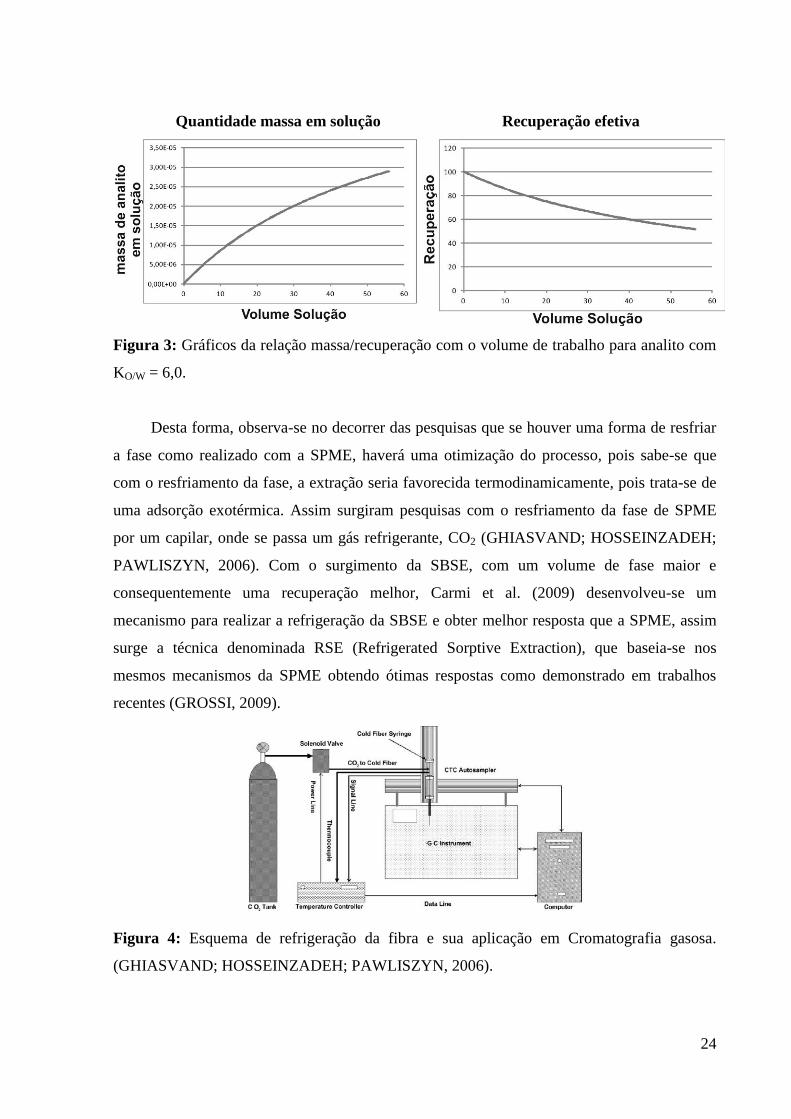
24
Quantidade massa em solução Recuperação efetiva
Figura 3: Gráficos da relação massa/recuperação com o volume de trabalho para analito com
KO/W = 6,0.
Desta forma, observa-se no decorrer das pesquisas que se houver uma forma de resfriar
a fase como realizado com a SPME, haverá uma otimização do processo, pois sabe-se que
com o resfriamento da fase, a extração seria favorecida termodinamicamente, pois trata-se de
uma adsorção exotérmica. Assim surgiram pesquisas com o resfriamento da fase de SPME
por um capilar, onde se passa um gás refrigerante, CO2 (GHIASVAND; HOSSEINZADEH;
PAWLISZYN, 2006). Com o surgimento da SBSE, com um volume de fase maior e
consequentemente uma recuperação melhor, Carmi et al. (2009) desenvolveu-se um
mecanismo para realizar a refrigeração da SBSE e obter melhor resposta que a SPME, assim
surge a técnica denominada RSE (Refrigerated Sorptive Extraction), que baseia-se nos
mesmos mecanismos da SPME obtendo ótimas respostas como demonstrado em trabalhos
recentes (GROSSI, 2009).
Figura 4: Esquema de refrigeração da fibra e sua aplicação em Cromatografia gasosa.
(GHIASVAND; HOSSEINZADEH; PAWLISZYN, 2006).

25
1.3.3. Aplicação de métodos multiresíduos
1.3.3.1. Análise de Organoclorados em Pescado
Contaminantes orgânicos presentes no meio ambiente são resultado de diferentes fontes
de poluição oriundas de atividades antrópicas como processos industriais, aplicações agrícolas
entre outros (BARREAK et al., 2009). Devido às características físico-químicas destes
contaminantes, o que oferece a eles grande persistência ao meio ambiente, e seu uso
extensivo, é possível encontrá-los na água, solo, podendo ainda promover sua acumulação em
tecidos adiposos, gerando grande preocupação por se tratar de um risco potencial para os
ecossistemas (BARREAK et al., 2009).
Entre estes, os Contaminantes Orgânicos Voláteis (VOCs) em água tem aumentado
significativamente, devido ao seu extenso uso em países industrializados, se tornando uma
preocupação ambiental e para saúde pública (LIN; LI, 2010).
Estes contaminantes são frequentemente encontrados em solos e águas subterrâneas
devido ao vazamento de tanques de combustíveis (AMARAL et al., 2010). No Brasil destaca-
se que vazamentos de tanques de combustíveis compreendem a 79% dos casos de
contaminação de solos apenas no Estado de São Paulo, correspondendo a 2.279 áreas, os
quais apresentam principalmente contaminantes como Benzeno, Tolueno e Xileno (BTX)
(CETESB, 2009).
Além de Contaminantes Voláteis, os Semi-Voláteis também são de grande preocupação
ambiental. Entre estes, os pesticidas organoclorados (OCPs), em particular, tem o potencial de
trazer sérios efeitos tóxicos ao meio ambiente devido a sua resistência biológica, química e a
fotodegradação. Os pesticidas organoclorados não apresentam efeito tóxico apenas nos
organismos aquáticos como os peixes, mas eles podem se bioacumular e biomagnificar na
cadeia alimentar exercendo efeitos carcinogênicos nos animais e seres humanos
(MMUALEFE et al., 2009). Devido à diversidade, toxicidade e mobilidade dos contaminantes
voláteis e semi-voláteis no meio ambiente, estes necessitam ser monitorados para verificar a
conformidade da concentração destes frente a diferentes limites preconizados pela legislação
visando promover segurança ao meio ambiente e a saúde publica.
No Brasil é possível destacar diferentes legislações que controlam a concentração de
contaminantes voláteis e semi-voláteis para lançamento de efluentes e enquadramento dos
corpos de água superficiais (CONAMA 357/2005) (CONAMA, 2005); contaminação de

26
águas subterrâneas (CONAMA 396/2008) (CONAMA, 2008); contaminação de solos
(CONAMA 420/2009) (CONAMA, 20009); classificação de sedimento dragado (CONAMA
344/2004) (CONAMA, 2004). Estas legislações também são um reflexo de leis
internacionais, que controlam além destas matrizes algumas outras de importância ambiental
como sedimento, estabelecido, por exemplo, pelo órgão de controle ambiental do Canadá
(CCME, 2002).
Como mencionado anteriormente, alguns contaminantes semi-voláteis, como os
pesticidas organoclorados, podem bioacumular e biomagnificar na cadeia alimentar sendo
necessário seu monitoramento nos alimentos. O monitoramento destes contaminantes em
peixes, por exemplo, é utilizado como indicador ambiental e também para fins de saúde
pública, por se tratar de um alimento, sendo definidos critérios de tolerância por diferentes
países como Suíça (SWISS CONFEDERATION, 1995), Holanda (CBI, 2010), Japão
(MINISTRY OF HEALTH, LABOUR AND WELFARE, 2006), Estados Unidos (EPA,
2000).
Estes limites são estabelecidos devido à toxicidade destas substâncias. De acordo com a
organização mundial da saúde (WHO) e a agência de proteção ambiental do meio ambiente
dos Estados Unidos (EPA), há evidências suficientes, baseados em estudos de animais e em
humanos, mostrando que alguns VOCs têm efeitos carcinogênicos e mutagênicos em
organismos vivos (LIU et al., 2009).

27
Tabela 6: Limites e níveis de tolerâncias de alguns países e normas como a EPA e a FDA para os Organoclorados de maior impacto em pescados.
Organoclorado
(EPA, 2000)
(ppm) Nível de
tolerância.
(EPA, 2000) (ppm)
CSF-diário
(SWISS
CONFEDERATION,
1995) (ppm)
(CBI, 2010) (ppm)
(MINISTRY OF
HEALTH, LABOUR,
AND WELFARE, 2006)
(ppm)
(FAO/WHO,
2012)
(ppm)
(mamíferos
marinhos)
Chlordane 0,3 0,35 0,02 0,05 0,05
Total DDT 5,0 0,34 1,0 0,5 3,0 5,0
Dieldrin 0,3 16,0 0,05 0,05 0,1 0,2
Heptaclor 0,3 9,1 0,05 0,02 0,05 0,2
Mirex 0,1
PCBs 2,0 2,0
Aldrin 0,3 0,05 0,05 0,1 0,2
Benzene
Hexacloride 0,3 1,6 0,1 0,05 0,1
2,4-D 1,0 1,0
Lindane 1,0 0,1
HCH 0,1 0,02
Endrin 0,02 0,005
Simazine 12,0 10,0

28
É importante ressaltar que no país até o momento não há padrões de controle e LMR’s
estabelecidos para estes compostos em peixe, desta maneira estes dados podem servir de
proposta aos órgãos competentes. Estes dados como valores máximos de resíduos, mostram a
importância e a urgência em estabelecer valores de controle de certos compostos nas matrizes
de importância financeira para o país.
2. Objetivo
Considerando a notória importância na aplicação das ferramentas da qualidade como
planejamento experimental, validação e cálculo de incertezas na área da química analítica, e
também considerando que nesta área os métodos multiresíduos têm sido amplamente
aplicados, este trabalho se propõe como objetivo geral a aplicação de ferramentas da
qualidade no desenvolvimento de métodos multiresíduos. Para atendimento deste objetivo,
foram traçadas as seguintes metas:
- Desenvolvimento e otimização de métodos multiresíduos (QuEChERS) para análise
de compostos organoclorados em pescado avaliando a aplicação de ferramentas de qualidade:
Planejamento experimental, Validação e cálculo de incertezas;
- Desenvolvimento de técnica analítica (RSE) para análise de compostos organoclorados
em pescado com aplicação de ferramentas da qualidade;
- Desenvolvimento de ferramentas da qualidade – método para avaliação do parâmetro
de validação linearidade.

29
3. Parte experimental
3.1. Método Quechers
3.1.1. Ferramentas da qualidade aplicadas no desenvolvimento, validação e cálculo de
incerteza para análise de organoclorados em pescado utilizando o método
QuEChERS.
O tema qualidade vem sendo aplicado em vários setores, inclusive nos laboratórios. As
ferramentas utilizadas no âmbito do sistema de gestão laboratorial constituem também
elementos úteis para controle e avaliação dos processos relativos aos métodos de ensaio.
Como mencionado nos objetivos, o presente trabalho consiste em aplicar as ferramentas da
qualidade para avaliar a metodologia estudada. É importante lembrar que a qualidade e a
gestão dos laboratórios, são baseadas no que é designado nas normas ISO/IEC 17025
(Sistema de gestão laboratorial) e as bases de controle e procedimento da ISO 9001 (Sistema
de gestão da qualidade). Assim propõe-se formular procedimentos condizentes e que
assegurem as análises apropriadas para metodologia de trabalho, identificando as melhores
situações e possíveis resoluções dos possíveis problemas encontrados.
Dos procedimentos e ferramentas já bem difundidas no âmbito da qualidade, julga-se
que um planejamento deve ser realizado com o intuito de atingir a meta proposta. Sabendo
também que o planejamento deve possuir estrutura flexível para que haja possibilidade de
adaptação às situações que eventualmente surgirem. Assim o ciclo de Deming, ou seja, a
ferramenta PDCA (Figura 5) é de fundamental importância em gestão da qualidade para
otimização do que é esperado da metodologia.

30
Figura 5: Ciclo PDCA.
Com base na ideologia do ciclo, a proposta do trabalho foi desenvolver o método
QuEChERS para análise de organoclorados em pescado. Desta forma deve-se avaliar quais
são os parâmetros que pretende-se analisar e consequentemente observar se estes atendem o
que o cliente exige ou o que a norma vigente estabelece, devendo se atentar sempre ao tipo de
analito e ao tipo de matriz de trabalho, para que não ocorra má interpretação do que é exigido.
Para análise da matriz pescado e os analitos organoclorados, observa-se que há normas bem
estabelecidas, como na União Européia através da norma SANCO 12495/2011, que estabelece
procedimentos para análise de pesticidas em alimentos com trabalhos realizados em
laboratórios acreditados. É importante ressaltar que validação é um ato documentado que
atesta que qualquer procedimento, processo, equipamento, material, operação ou sistema
realmente conduza aos resultados esperados. Este processo de validação é parte essencial da
ISO/IEC 17025.
O documento da SANCO descreve o método de validação e controle de qualidade
analítico para dar suporte à validade dos dados usados checando com os Limites Máximos de
Resíduos. Sendo assim, os objetivos específicos deste documento são: prover um sistema
harmonizado de custo-benefício do sistema da garantia da qualidade na União Européia;
assegurar a qualidade e a comparação de resultados analíticos; assegurar que a precisão
aceitável seja alcançada; assegurar que os falsos positivos ou falsos negativos não sejam
reportados e dar suporte ao que é exigido pela ISO/IEC 17025 (padrão de acreditação)
(SANCO, 2012).

31
Além desta norma da União Européia, temos também o que é vigente nos Estados
Unidos com relação às normas referentes ao meio ambiente aplicadas pela EPA.
Relacionando com o analito de trabalho através de método 8081B (EPA, 2007) e por se tratar
de uma matriz com características de bioindicador, as normas da série SW-846, referente aos
métodos de avaliação de resíduos sólidos, é um resumo oficial de métodos analíticos e
amostragem que é avaliada e aprovada pelo uso em cumprimento com as regulações do
RCRA (Resource Conservation and Recovery Act). Como o trabalho desenvolvido é focado
em um alimento, e a metodologia foi toda desenvolvida em laboratório acreditado, como é
exigido pela Resolução (EC) Nº 882/2004 do Parlamento Europeu e Conselho
(COMUNIDADE EUROPÉIA, 2004), dentro de um órgão federal (Lanagro de Campinas –
SP) pertencente ao Ministério da Agricultura, que realiza ensaios de monitoramento de
analitos orgânicos e inorgânicos em produtos cárneos para exportação, principalmente para
países europeus, optou-se pela norma SANCO 12495:2011. É importante ressaltar que
validação trata-se de um procedimento não especifico, ou seja, há vários guias a serem
seguidos, e cabe ao analista escolher qual é o mais adequado ao tipo de trabalho proposto.
Com a norma estabelecida, deve analisar o que ela exige para ser atendida, ou seja,
quais parâmetros devem ser avaliados para que esta esteja dentro dos padrões exigidos pela
Comunidade Européia. A norma é ampla, e apresenta vários controles, desde a coleta da
amostra até ao descarte dos resíduos. Mas como o trabalho proposto foi designado para
análise da aplicação das ferramentas da qualidade, este será focado na parte de
desenvolvimento e validação do método. A Figura 6 mostra as principais etapas de um ensaio
laboratorial e enfatiza os passos que serão executados no trabalho.

32
Figura 6: Etapas para realização de um ensaio em um laboratório enfatizando a parte que será
desenvolvida no trabalho. Adaptado de Olivares, 2009.
3.1.1.1. Desenvolvimento do Planejamento Experimental – Otimização QuEChERS
O planejamento experimental é uma parte fundamental para o desenvolvimento da
metodologia e uma ferramenta de extrema importância para avaliar quais são os fatores que
mais influenciam no método. Este parâmetro de qualidade pode ser visto também como um
teste de robustez do método para observar a variação dos fatores que o método suporta sem
ter seus resultados alterados.
Para o seu desenvolvimento averiguou-se qual é o fator que tem maior influência na
validação em um processo multiresíduo, neste caso observou-se que a recuperação é de maior
significância, assim foi definido os fatores que poderiam interferir nesse parâmetro. Seguindo
a rotina do QuEChERS original, observou-se que o solvente de trabalho na extração liquído-
liquído ou liquído-sólido é um importante fator, assim como a quantidade de massa de
Cloreto de Sódio (NaCl) no aumento da força iônica, quantidade de Sulfato de Magnésio (em

33
ambas as fases, extração líquido-sólido e dSPE) e a quantidade de PSA (Primary-Secundary
Amine). Todos estes fatores são essenciais na técnica original, mas deve-se atentar para a
complexidade da matriz, ou seja, o grau de interferentes existentes.
Desta forma, foi realizado um planejamento experimental para analisar os fatores que
influenciam na técnica QuEChERS, ou seja, qual o grau de influência direta na metodologia,
como seus sais e suas quantidades, e desta forma obter os melhores dados possíveis de
recuperação.
Figura 7: Diagrama de Ishikawa para planejamento experimental da técnica QuEChERS.
Com os fatores estabelecidos foi utilizado Software Statistica 7 para determinação da
aleatorização dos experimentos para evitar erros sistemáticos, foi realizado um planejamento
completo 23 com três pontos centrais para observação do desvio padrão. No início dos
estudos, se tratando de vários fatores (massa matriz, volume solvente e os sais de trabalho) foi
realizado um planejamento univariado com alguns dos fatores (massa da matriz e volume do
solvente) e depois da otimização destes fatores foi estabelecido a fixação do Sulfato de
magnésio (retirada de água residual), pois se trata de um reagente barato e por considerar que
a matriz continha pouca água residual. Estas escolhas foram satisfatórias como mostradas
posteriormente nos resultados. O planejamento foi comparado com planilhas de Excel para
confirmação dos resultados obtidos como garantia da qualidade do procedimento (Apêndice
B) dentro de um nível de confiança de 95%.

34
3.1.1.2. Parâmetros para validação
A utilização de um bom planejamento evita o retrabalho das análises laboratoriais, além
de economizar recursos que muitas vezes são limitantes das análises, como amostras
ambientais e amostras na área de pericia criminal, estruturando assim um estudo integrado
interagindo com uma multiplicidade de fatores, como a parte cultural, econômica, ambiental e
social. Após este passo foi realizado análise dos parâmetros que tem influência direta na
análise, com isso destaca-se que apesar de vários parâmetros serem exigidos dentro da
SANCO/12495 e até mesmo na DOC-CGCRE-008 do INMETRO, e considerando o guia de
validação do MAPA (MAURICIO et al., 2011) que também é baseado nestes documentos,
destaca-se que há orientação de que alterações nos parâmetros e critérios de validação podem
ser necessários devido a especificidade de cada laboratório.
Foi definido que se tratando de uma técnica de extração, um dos fatores de extrema
importância a ser avaliado, é a recuperação/exatidão e que este deve estar entre 70 e 120%, a
precisão não deve exceder 20% no coeficiente de variação (CV). Foi analisado também a
linearidade/sensibilidade do método, que não deve ter resíduo superior a 20% e coeficiente de
determinação acima de 0,98, bem como proposta a inserção de cálculo de incerteza para sua
avaliação. As extrações foram realizadas com o efeito da matriz e posteriormente fortificadas
com os 5 pontos exigidos e com as respectivas 6 replicatas para cada ponto, foi realizado
também análise da repetibilidade da metodologia e por fim o cálculo da incerteza do método.
Todos estes parâmetros foram definidos conforme exigência do guia SANCO.

35
Figura 8: Fluxograma do processo de trabalho para aplicação das ferramentas da qualidade.
3.1.1.2.1. Recuperação/Exatidão
Exatidão é a concordância entre o valor verdadeiro do analito na amostra e o extraído no
processo analítico e é um dos parâmetros chaves apara validação de metodologia. O processo
se baseia em quatro métodos principais:
- Comparação do método proposto com um método de referência;
- Uso de ensaios de recuperação na matriz;
- Estudos colaborativos;
- Uso de material de referência certificado (MRC).
Sabe-se que a melhor forma para realização deste parâmetro é a utilização de material
de referência, pois esta envolvido diretamente com padrões internacionais, garantindo assim a

36
rastreabilidade do método. A amostra é analisada e seu valor de recuperação é comparado
com o valor certificado pelo produtor do MRC. O grande problema da utilização deste
material de referência é o seu alto custo e a limitação de amostras com a matriz e analito de
interesse (BRITO et al., 2003), além da faixa de concentração desejada. Fator que já esta se
alterando no país com a acreditação de produtos de MRC pelo INMETRO. Este fator abre
portas para o surgimento de novos MRC no país conforme a demanda.
A exatidão pode também ser obtida pela comparação dos dados obtidos com um outro
método já validado. Após análise de diferentes amostras, as diferenças obtidas para cada
amostra são calculadas e comparadas com o valor desejado. Entretanto nem sempre se
encontra método de referência preexistente, impossibilitando a utilização deste procedimento.
O desenvolvimento por estudos colaborativos é conduzido com a aceitação do
desenvolvimento do método com outros laboratórios, com um mínimo absoluto de 5
participantes. O grande problema desta metodologia é manter a exatidão do método já que o
estudo envolve a estabilidade do composto, o que torna uma tarefa difícil quando se trata de
compostos com certo grau de volatilidade ou degradação. Neste processo de validação utiliza-
se análise de variância (ANOVA) para determinação de diferença significativa entre os
laboratórios envolvidos.
O ensaio de recuperação constitui o método mais utilizado em validação de processos
analíticos, e a recuperação esta relacionada com a exatidão, pois reflete a quantidade de
analito recuperado em relação à quantidade real presente na amostra. A exatidão é expressa
em porcentagem e podem possuir erros sistemáticos inerentes ao processo, como perda da
substância, medidas volumétricas imprecisas, interferentes na amostra, entre outros fatores.
(BRITO et al., 2003).
100.1 mindet
real
adoerreal
V
VVExatidão
Equação 2: Fórmula para cálculo da exatidão.

37
3.1.1.2.2. Linearidade/Sensibilidade
A linearidade de uma curva analítica é a resposta obtida (instrumentação) em função da
concentração a qual deve ser estudada em um intervalo de concentração apropriado, na prática
a linearidade é determinada através de gráficos de calibração, seguido de tratamento
estatístico. O número mínimo de pontos geralmente aceito nas curvas de calibração varia
entre 5 e 6 pontos. Seguindo a orientação da SANCO/12495:2011 foi estabelecido 5 pontos
para a curva analítica com 3 replicatas para cada ponto. As concentrações da faixa de trabalho
devem ser escolhidas entre 50% a 150% do valor esperado da amostra em estudo.
Algumas formas de realizar a curva são bem difundidas na área da química, como
fortificação da matriz em várias concentrações seguido do processo de extração. Outra forma
é a extração da matriz limpa e posteriormente fortificação do extrato (Figura 9). Também
muito aplicada é a fortificação do solvente, que não existe a interferência da matriz, esta
técnica não analisa a linearidade da metodologia e sim a do equipamento de trabalho.
(A) (B)
Figura 9: (A) Extração da matriz pré-fortificada nas concentrações da faixa de trabalho; (B)
Extração da matriz limpa com posterior fortificação do extrato com as concentrações na faixa
de trabalho.
Assim para ter-se a melhor resposta da metodologia foi realizado o procedimento de
extração do branco e posteriormente sua fortificação nas concentrações desejadas para
realização da curva na faixa de trabalho. Foi realizado fortificação nos pontos de concentração
0,5; 0,8; 1,0; 1,5 e 2,0 LMR (1 LMR é igual aos valores propostos mais a frente na tabela 9).
Após construção da curva foi desenvolvida uma sistemática de avaliação da mesma,
para se ter parâmetros que afirmam o quanto adequada é a curva em relação a metodologia

38
aplicada. Esta sistemática desenvolvida é apresentada no capítulo de resultados (4.1.4.
Desenvolvimento de modelo para avaliação da curva de calibração).
3.1.1.2.3. Seletividade
O termo especificidade, muitas vezes utilizado como sinônimo de seletividade define a
capacidade do método em detectar o analito de interesse na presença de outros componentes
da matriz. Já a seletividade refere-se à capacidade de detecção de substâncias. O processo
para demonstrar a especificidade do método depende do seu objetivo. Em diversas técnicas
analíticas (como nas análises cromatográficas, por exemplo) esse parâmetro pode ser
estabelecido pela comparação do resultado obtido com a combinação de vários fatores. Como
substâncias diferentes podem apresentar respostas similares em dadas condições deve-se
proceder à análise, seguida por outras técnicas comprobatórias (como cromatografia ou
eletroforese acoplada à espectrometria de massas). Outra maneira de avaliar a especificidade
envolve a adição de padrão analítico (muito empregada em análises por espectrometria de
absorção ou de emissão atômica) ou a comparação com padrão externo (BRITO et al., 2003).
A matriz da amostra pode conter componentes que interferem no desempenho da
medição. Os interferentes podem aumentar ou reduzir o sinal, e a magnitude do efeito também
pode depender da concentração. Experimentos para avaliação da seletividade descritos na
literatura sobre validação de métodos analíticos envolvem ensaios com padrões ou materiais
de referência, amostras com e sem o analito, além da avaliação da capacidade de identificação
do analito de interesse na presença de interferentes. Quando não há disponibilidade de
interferentes, alguns autores sugerem a avaliação da habilidade de medição do analito por
diferentes métodos, técnicas ou por meio de variações nas condições instrumentais. Se a
seletividade não for assegurada, a linearidade, a tendência e a precisão estarão seriamente
comprometidas (DOC-CGCRE-008, 2010).

39
Tabela 7: Avaliação da seletividade segundo a DOC-CGCRE-008 do INMETRO.
Procedimento Demonstrar Comentários
a) Fazer a análise com a
amostra e materiais de
referência pelo método
em estudo e outros
métodos validados.
Habilidade do método em
estudo de identificar e dosar
o analito na presença de
interferentes.
Evidências necessárias para
dar suporte e gerar
confiabilidade suficiente.
b) Analisar amostras
contendo vários
interferentes suspeitos
na presença do analito
de interesse.
Efeito de interferentes – a
presença de interferente
acentua ou inibe a detecção
ou quantificação do analito
de interesse.
Se alterar resultados,
aperfeiçoar o método ou
selecionar outro mais
adequado.
Já a SANCO/12495:2011 requer que interferentes na resposta do branco não apresentem
sinal superior a 30% em relação aquele obtido no Limite de Quantificação.
3.1.1.2.4. Precisão
É a expressão da concordância entre vários resultados analíticos obtidos para uma
mesma amostra. Pode ser determinado em condições de repetibilidade ou em condições de
reprodutibilidade intralaboratorial (OLIVARES, 2006b): intra-dias ou inter-dias, além de
precisão intermediária.
A repetibilidade pode ser obtida utilizando parâmetros iguais na análise, como amostra,
laboratório, analista e equipamento, já a reprodutibilidade intralaboratorial utiliza a mesma
amostra, mas difere em pelo menos um dos fatores, laboratório, analista ou equipamento.

40
Figura 10: Diferença entre repetibilidade e reprodutibilidade intralaboratorial para análise de
precisão.
A precisão é obtida utilizando a seguinte fórmula,
100.x
sCV
.,var médiaaéxoepadrãodesviooésiaçãodeecoeficientaéCVOnde
Equação 3: Fórmula para cálculo da constante de variação (CV).
3.1.1.3. Cálculo da incerteza
O VIM (Vocabulary International Metrology) define a incerteza como um parâmetro
“não negativo caracterizando a dispersão dos valores atribuídos ao mensurando, baseado na
informação usada”. Em outras palavras, o cálculo da incerteza demonstra o nível de confiança
de um resultado analítico. Não há um processo único para calcular a incerteza, mas a ISO/IEC
17025 (requerimento 5.4.6.2) estipula que: “Em certos casos a natureza do teste pode impedir
o cálculo da incerteza de uma forma mais rigorosa, metrológica e estatisticamente válida.
Neste caso o laboratório deverá pelo menos identificar todos os componentes da incerteza e
fazer uma estimativa razoável, e assegurar que a forma de reportar os resultados não transmita
uma impressão errada da incerteza. Estimativas razoáveis devem ser baseadas no
conhecimento da performance do método e o escopo de medidas deve fazer uso de
experiência prévia de dados de validação”.

41
Considerando as diferentes formas para calcular a incerteza, o guia
EURACHEM/CITAC aparece como referência de aplicação. De acordo com este guia, o
cálculo da incerteza envolve 4 passos básicos:
- Especificar o mensurando;
- Identificar as Fontes de incerteza; (na literatura há diferentes formas de abordagem de
escolha das fontes e cálculo da incerteza), pelas seguintes abordagens:
Bottom-up – é baseado na identificação, quantificação e combinação de todas as fontes
de incerteza. Geralmente, é a abordagem mais complexa, pois considera todas as fontes de
incerteza do método analítico. Este cálculo é aplicado usando principalmente a incerteza de
equipamentos volumétricos (por exemplo, micropipetas, balanças entre outros). Estes
geralmente tem suas incertezas expandidas declaradas em certificados de calibração.
Fitness for purpose – é baseado em uma única definição de parâmetro chamado de
função de adequação, esta abordagem é baseada principalmente em estudos de precisão e
exatidão.
Top-down – é baseado em dados obtidos por estudos interlaboratoriais (precisão). Esta
abordagem requer informações de estudos colaborativos e o cálculo é baseado em desvio
padrões de informações disponíveis.
Validation-based – é baseado em estudos de validação interlaboratoriais ou
intralaboratoriais (precisão, exatidão e robustez). Este cálculo é geralmente baseado em
desvio padrão da informação disponível.
Robustness-based – é baseado em teste de robustez de estudos interlaboratoriais. Este
cálculo também é baseado em desvio padrão das medidas.
O guia EURACHEM/CITAC cita dois métodos mais utilizados, a abordagem Bottom-
up e o Validation-based. Embora os dois métodos serem aplicáveis a metodologia, será
utilizado o Validation-based por ser considerado a forma mais fácil para prover uma boa
estimativa do cálculo da incerteza (OLIVARES E LOPES, 2012).
- Quantificar os componentes da incerteza;
- Calcular a incerteza combinada e expandida.

42
3.1.1.4. Método para análise de organoclorados em pescado (QuEChERS)
Foi desenvolvido um método multiresíduo (QuEChERS) para avaliar a extração de 14
organoclorados (Tabela 8) de matriz peixe. O objetivo deste trabalho foi aplicar diferentes
ferramentas da qualidade (Planejamento Experimental, validação e cálculo de incerteza)
segundo exigência de alguns guias de validação nacionais e internacionais. Como orientação
nacional foi utilizado o DOC-CGCRE-008 (INMETRO, 2011) e a norma ISO/IEC 17025, e
como orientação internacional a SANCO/12495/2011 (SANCO, 2012).
Tabela 8: Compostos organoclorados estudados:
Nome PM Fórmula
molecular Fórmula estrutural
Hexacicloclorobenzeno
(HCB) 288 C6H6Cl6
Heptacloro Hepóxido (HPX) 386 C10H5Cl7O
Dieldrin
(DLD) 378 C12H8Cl6O
2,2,-bis-p-clorofenil-1,1,-
dicloroetileno
(DDE)
316 C14H8Cl4
2,2,-bis-p-clorofenil-1,1,-
dicloroetano (DDD) 318 C14H8Cl4

43
Diclorodifeniltricloroetano
(DDT) 352 C14H9Cl5
Aldrin
(ALD) 365 C12H8Cl6
Chlordane
(cCLD e tCLD) 410 C10H6Cl8
Mirex
(MRX) 545 C10Cl12
Lindane
(LIN) 291 C6H6Cl6
2,3’,4,4’,5-Pentaclorobifenil
(PCB 118) 326 C12H5Cl5
2,2’,3,4,4’,5,5’-
Heptaclorobifenil
(PCB 180)
395 C12H3Cl7

44
3.1.1.4.1. Padrões e reagentes
Foram utilizado padrões de Organoclorados e PCB’s fornecidos pelo LANAGRO, todos
da Accustandard, com grau de pureza acima de 98%. Foi utilizado o n-hexano para a extração
líquido-líquido. Os sais utilizados na fase dispersiva foram o Sulfato de magnésio e Cloreto
de Sódio e na fase de clean-up foram usados o PSA e o C18. A matriz foi fornecida por
produtores credenciados ao Lanagro, com a garantia de amostras limpas, o que foi provado no
teste de seletividade realizado.
3.1.1.4.2. Materiais e equipamentos
Para realização do trabalho foi utilizado balança analítica BEL engineering Mark 160,
vortex AP56 Phoenix, centrifuga Fanem Excelsa 4 Mod. 280R, balão volumétrico,
micropipeta Ecopippette e para análise das extrações GC-ECD da marca Thermo (Thermo
Trace Gc Ultra).
3.1.1.4.3. Preparação das amostras e padrões
O método QuEChERS foi desenvolvido no ambiente de um laboratório acreditado (pré-
requisito para uma abordagem de qualidade), conforme Apêndice A, com isso tem-se que
todos equipamentos utilizados dentro do laboratório são calibrados periodicamente e
auditados pelo INMETRO.
Inicialmente foi preparada uma solução estoque com os 14 compostos organoclorados
em seus respectivos LMRs (Limite Máximos de Resíduos) e acondicionados em um
refrigerador. A matriz pescado foi fornecida por um criador local e disponibilizado ao
Lanagro-SP, esta foi limpa e triturada com o cuidado para obter apenas as partes de interesse
do peixe (filé comercialmente disponível) e posteriormente também foi acondicionado em
refrigerador para sua conservação.

45
Tabela 9: Proposta enviada a CCRC (Coordenação e Controle de Resíduos e Contaminantes).
Analito Proposta (ppb) Referência
Chlordane 300 (EPA, 2000)
Total DDT (DDD; DDT e
DDE) 5000 (EPA, 2000)
Dieldrin 300 (EPA, 2000)
Heptaclor e Heptachlor
epoxide
300 (apenas
Heptachlor Epoxide) (EPA, 2000)
Mirex 100 (EPA, 2000)
Total PCBs 2000 (EPA, 2000)
Aldrin 100 (MINISTRY OF HEALTH,
LABOUR, AND WELFARE, 2006)
Benzene hexacloride (HCB) 100 (MINISTRY OF HEALTH,
LABOUR, AND WELFARE, 2006)
Lindane 1000 (MINISTRY OF HEALTH,
LABOUR, AND WELFARE, 2006)
A solução estoque foi preparada se atentando aos LMRs de cada analito. Para isso
foram investigados quais seriam estes possíveis valores, já que o país não possui referência
para organoclorados em peixes, desta forma foram avaliados os valores internacionais destes
limites em diferentes países como destacado na Tabela 9, principalmente os valores
preconizados pelos Estados Unidos e Europa. Com base nestes valores, foi sintetizado uma
proposta de valores máximos a ser aplicado no Brasil e submetida à CCRC (Coordenação e
Controle de Resíduos e Contaminantes do MAPA).
Estes valores foram adotados como os LMRs para o trabalho, sendo assim preparada a
solução estoque. Para extração das amostras fortificadas, a matriz e o padrão foram retirados
da refrigeração e acondicionados em temperatura ambiente, seguido da pesagem de 2 g da
matriz e colocados em tubo de centrifuga de 50 mL. Nesta amostra foi adicionado 250 uL da
solução estoque atentando-se para que toda solução fosse bem dispersada na matriz e sendo
aguardado 60 minutos para interação do analito com o pescado. Após este passo foi
adicionado 8 mL de hexano para realização da primeira etapa do procedimento QuEChERS,
extração liquído-sólido, sendo agitado por 3 minutos em vortex. Em seguida foi adicionado,
0,5 g de NaCl para aumentar a força iônica do meio e 1 g de MgSO4 para remoção de água

46
residual. Esta mistura foi agitada novamente em vortex por 3 minutos e depois foi
centrifugado por 10 minutos (3500 rpm).
Na sequência, foi realizado a segunda etapa, a dSPE (dispersão da matriz em fase
sólida), onde foi coletado uma alíquota de 3 mL da primeira etapa e adicionado 500 mg de
MgSO4 para remoção da água restante, 50 mg de sorbente PSA para remoção de açucares e
ácidos graxos e 75 mg de C18 para remoção de interferentes apolares como lipídios. Esta
mistura foi agitada durante 1 minuto em vortex e centrifugada por 10 minutos (3500 rpm)
sendo o extrato final analisado em GC-ECD. É importante ressaltar que os volumes e massas
trabalhados foram pré-determinados em estudo de planejamento experimental multivariado
que será detalhado nos resultados, com exceção da massa da matriz e volume do solvente que
foram otimizados em planejamento experimental univariado, como citado anteriormente.
3.1.1.4.4. Método Cromatográfico
Para análise das extrações dos Organoclorados foi utilizado GC-ECD (Gas
Chromatograph-Electron Capture Detector) da marca Thermo (Thermo Trace Gc Ultra). O
método foi desenvolvido em uma coluna apolar OV-5MS (25m x 0,25 mm x 0,25 μm) da
Ohio Valey (95% Dimetil e 5% difenilpolisiloxano). Foi utilizado um injetor automático AS
3000 com volume de injeção de 1 μL com 10 ciclos de lavagem da seringa, pré-injeção (2
ciclos de lavagem com acetona e hexano), amostra (3 ciclos de lavagem com a amostra) e
pós-lavagem (5 ciclos de lavagem com acetona e hexano). A injeção foi no modo Splitless,
fluxo constante do gás de arraste de 0,8 mL, temperatura injetor de 250ºC, Temperatura do
detector foi fixada em 300ºC e rampa de aquecimento como descrita na Figura 11.

47
Figura 11: Gráfico (A): rampa de aquecimento da coluna inicia a 80ºC/ 1,5 min., taxa de
40ºC/min. até temperatura de 170ºC, taxa de 6,5ºC/min. até temperatura de 220ºC/7,0 min.,
taxa de 15ºC/min. até temperatura de 245ºC e por fim taxa de 50ºC/min. até temperatura de
265ºC/4 min. Gráfico (B) fluxo do gás de arraste de 0,8 mL/min.

48
Com a etapa cromatográfica otimizada, foi injetado os padrões para obter os respectivos
tempos de retenção. A Figura 12 apresenta o cromatograma para os 14 analitos.
Figura 12: Cromatograma do método cromatográfico da melhor condição para separação dos
14 compostos organoclorados; tempo de retenção: HCB, 8,017 min.; LIN, 8,410 min.; ALD,
10,817 min.; HPX, 11,722 min.; tCLD, 12,350 min.; cCLD, 12,810 min.; ppE, 13,440 min.;
DLD, 13,575 min.; PCB118, 14,723 min.; ppD, 14,990 min.; opT, 15,218 min.; ppT,16,847
min.; PCB 180, 20,383 min.; MRX, 21,153 min.

49
3.2. Método RSE
3.2.1. Desenvolvimento do planejamento experimental – Otimização da fase extratora
RSE
Antes da construção do sistema de extração, buscou-se a melhor forma de confecção da
fase de PDMS para ter-se a melhor resposta de extração no sistema. Segundo a literatura e o
fabricante, a composição adequada de polímero PDMS e seu agente de cura é a relação de
10:1(m:m). Desta forma foi realizada a mistura vigorosamente das fases e condicionada em
um sistema de vácuo para retirada de gases presente na mistura, depois deste processo foi
adicionado à mistura ao molde (destacado construção no capítulo 3.2.2. Desenvolvimento do
método RSE). Este foi fechado com o cuidado de não perder massa como demonstrado na
imagem (e) da Figura 16 e colocado em um forno à temperatura de 100ºC durante 45 minutos
para pré-cura da fase. Depois deste passo o molde foi desmontado para retirada da barra de
RSE e esta foi direcionada para mufla a alta temperatura para retirada de possíveis
interferentes e oligopolímeros que não se polimerizaram no primeiro aquecimento. No
entanto, foi observado que o tempo entre o forno e a mufla interferiam no processo. Desta
forma foi realizado um planejamento experimental com três fatores após a pré-cura: período
exposto à temperatura ambiente, tempo e temperatura de mufla para observação da robustez
na mudança destes fatores.
Tabela 10. Dados dos níveis do planejamento experimental na confecção da fase de extração
com polímero PDMS.
Fatores Nível Inferior Nível Superior
-1 +1
(1) Temperatura da mufla (ºC) 100 200
(2) Tempo de mufla (min.) 60 180
(3) Tempo a temperatura ambiente (min.) 0 240
Foi realizado um planejamento experimental completo 23
e os dados já codificados em
nível inferior (-1) e nível superior (1) foram direcionados para o Software Statistica 7 para ter-
se a aleatorização dos experimentos, e foi obtido a seguinte sequência:

50
Tabela 11: Aleatorização dos experimentos pelo Software Statistica 7.
Experimento T da mufla Tempo de mufla Tempo a temp. ambiente
2 +1 -1 -1
5 -1 -1 +1
6 +1 -1 +1
1 -1 -1 -1
7 -1 +1 +1
3 -1 +1 -1
8 +1 +1 +1
4 +1 +1 -1
Para confecção das barras de SBSE (Stir Bar Sorptive Extraction) foi utilizado metal
cilíndrico cortado na dimensão para utilização do molde, e este foi revestido com vidro para
não obter interferentes provenientes do metal e que esse não se oxide em meio aquoso. Assim,
foram confeccionadas 8 barras conforme tabela acima, e estas foram direcionada para testes
em extração de um HPA (Hidrocarboneto Poliaromático) o Antraceno em água, para
observação da recuperação. O processo de extração foi otimizado para obtenção do maior
índice de recuperação do composto analisado sendo utilizada a metodologia descrita por
Lynam, 2012.
As barras sortivas foram adicionadas em solução fortificada com Antraceno (25ug.L-1
)
volume de 10 mL e colocada sob agitação (500 rpm) durante um período de 2 horas a
temperatura ambiente. Após a extração, o Antraceno foi dessorvido da barra adicionando a
mesma em vial com 1,5mL de uma solução de Acetonitrila/Tolueno (80:20%) e colocados
durante 20 minutos em banho ultrassônico. Esta solução foi injetada em cromatógrafo gasoso
para análise do Antraceno. Algumas modificações foram realizadas no método de Lynam para
obter o analito de interesse, assim utilizou-se uma coluna DB-5 da Agilent; temperatura do
injetor de 280ºC, fluxo de gás hélio 1 mL/min e split 1:10 e a seguinte rampa de aquecimento.

51
Tabela 12: Rampa de aquecimento proposta para a coluna DB-5 da Agilent.
Temperatura (ºC) Taxa (ºC/min) Hold (min) Total (min)
80 - 1 1
200 20 1 8
280 10 2 18
Com isso foi obtido o seguinte cromatograma para o antraceno que foi quantificado em
uma curva analítica de 10 - 330 ug/L, conforme Figura 13.
(A) (B)
Figura 13: (A) Curva analítica para o analito Antraceno; (B) Cromatograma representativo.
3.2.1.1. Padrões e reagentes
Para construção do molde de confecção das barras de RSE foi utilizado um tarugo de
teflon. O polímero utilizado foi o Polidimetilsiloxano (PDMS) e seu agente de cura da Dow
Corning. Os padrões utilizados para a extração no teste de robustez foi o Antraceno e para
análise de extração foram utilizados Organoclorados e PCB’s fornecidos pelo Lanagro, todos
da Accustandard com grau de pureza acima de 98%, Cloreto de Sódio cristal PA, Quemis para
o aumento da força iônica, água destilada, Acetonitrila PANREAC 99,9%, tolueno
MALLINCKRODT para o processo de dessorção. A matriz foi fornecida por produtores
credenciados ao Lanagro, com a garantia de amostras limpas, o que foi provado no teste de
seletividade realizado.
Antraceno

52
3.2.1.2. Materiais e equipamentos
No processo de confecção das barras de RSE foi utilizado uma balança analítica Gettaka
AG 200 para pesagem do polímero e seu agente de cura, assim como para pesagem dos
analitos, bomba de vácuo para desgaseificação do polímero após mistura, forno mufla para o
processo de cura. Para o processo de extração foi utilizado vial de headspace da Agilent 20
mL modificado, barra magnética de agitação de 2 cm, agitador magnético IKA C-MAG HS7,
micropipeta 10-100 uL marca HTL Labmate+, lacre 20 mm de alumínio septo de PTFE,
lacrador e deslacrador manual ergonômico 20 mm, vial de dessorção Agilent 1,5 mL. Para o
teste de robustez foi realizado análise em GC-MS CP-3800 da VARIAN e para análise das
extrações GC-ECD.
3.2.1.3. Preparação das amostras e padrão
Os padrões utilizados no método de extração RSE foi o mesmo da técnica QuEChERS.
Para desenvolvimento da melhor condição de produção da fase de PDMS foi utilizado um
hidrocarboneto poliaromático, o Antraceno como já destacado. A matriz pescado cedida pelo
Lanagro de Campinas – SP.
3.2.1.4. Método Cromatográfico
O método utilizado foi o mesmo da técnica QuEChERS.
3.2.2. Desenvolvimento do método RSE
Para o desenvolvimento da técnica, planejou-se um molde que foi confeccionado pela
oficina mecânica do campus da USP de São Carlos. As barras de SBSE comercial,
normalmente possuem um volume maior de fase de PDMS, o que limita o trabalho apenas em
dessorção líquida, perdendo assim em sensibilidade da técnica. Devido a esse limitante, foi
construído um molde com as dimensões para ter-se 24 uL de fase sortiva para que pudesse ser
acoplado em um sistema de dessorção térmica conforme apresentado na Figura 14.

53
Figura 14: Chromatoprobe Varian – (A) Esquema do Chromatoprobe utilizado para
dessorção térmica dos analitos da barra sortiva para o cromatógrafo; (B) Posicionamento da
barra sortiva no Chromatoprobe; (C) Acoplamento do Chromatoprobe com a barra sortiva ao
cromatógrafo.
Desta maneira foi esquematizado um molde para confecção das barras de RSE (Figura
15) para que fosse possível ser acoplada ao PROBE de dessorção térmica disponível visando
a aumentar a sensibilidade do método. O molde foi adaptado de Grossi, 2009, com alterações
nas dimensões.
(A)
(C) (B)
Chromatoprobe
Suporte
Barra
sortiva
Coluna
Cromatográfica
Insert

54
Figura 15: Projeto para confecção do Molde. (A.) Suporte de teflon com o tubo oco de aço;
(B.) suporte de teflon agregado ao molde de teflon; (C.) Esquema da barra de SBSE ou RSE
com suas dimensões para utilização em PROBE; (D.) vista do molde com seus dois lados
juntos; (E.) molde já com o sistema de travas para confecção das barras. Adaptado de Grossi,
2009.
A Figura 16 apresenta o molde já finalizado, mostrando as partes de encaixe, suporte e
presilha. O molde foi confeccionado em teflon e a presilha em alumínio. A presilha foi
confeccionada para garantir que o suporte esteja bem fixo e que não haja perda do polímero
durante a confecção.
Figura 16: Molde já finalizado: (a) Dois lados do molde e suporte de teflon; (b) Suporte com
barra ou tubo oco de aço; (c) suporte de teflon agregado ao molde; (d) vista lateral do
conjunto; (e) vista superior do conjunto; (f) Suporte com a barra de RSE já revestida com
PDMS; (g) barra de RSE.

55
O sistema RSE sendo uma técnica nova de extração, é um método que necessita de
adaptações para que se atinja o ápice em eficiência, assim alguns passos foram executados
para otimização da metodologia quando comparada com o desenvolvido por Carmi, 2006. A
Figura 17 exemplifica as partes do sistema, em que é necessário uma refrigeração constante
da barra de extração para que este entre em um equilíbrio termodinâmico, entre a fase
extratora e a fase de headspace, conforme o coeficiente de equilíbrio de cada analito. Com a
refrigeração há um deslocamento deste coeficiente para a fase extratora (polímero PDMS),
aumentando assim, a eficiência do processo, com isso foi executado um procedimento de
planejamento experimental para buscar a melhor forma de confecção da fase como
demonstrado no capítulo 4.2.1. Assim, o sistema adaptado de Grossi, 2009 sofreu algumas
alterações para o trabalho proposto como, por exemplo, o sistema de vedação por anéis o-
rings, para evitar perda de analito para o meio e a dimensão da fase extratora.
Figura 17: Sistema RSE. (A) Tubo oco revestido com a fase PDMS no suporte de teflon; (B)
Suporte de Teflon do vial Headspace com o tubo de RSE; (C) Vial de headspace com braços
laterais, suporte e tubo de RSE; (D) Sistema RSE acoplado com sistema de refrigeração; (E)
Sistema RSE com aquecimento de 85ºC em agitador magnético; (F) Sistema RSE com vapor
na fase de headspace.
Com a utilização de gás para refrigeração, pode-se atingir temperaturas muito baixas o
que aumenta o leque de trabalho com o sistema, necessitando de estudos profundos
termodinâmicos para analisar viabilidade das baixas temperaturas. Com isto pode-se utilizar
CO2 como gás refrigerante e como controle desta temperatura para não se atingir temperaturas

56
mais abaixo do que se pretende pode ser injetado ar comprimido em um misturador de gases
confeccionado em vidro, como é evidenciado na imagem B da Figura 18. Todos os cabos
foram isolados para não absorver calor do ambiente e por consequência diminuir a eficiência
da refrigeração. Sendo assim, a Figura 18 mostra o sistema completo montado, pronto para o
processo de extração. Estudos futuros devem ser iniciados para estudos dos parâmetros que
influenciam diretamente no método.
Figura 18: Sistema de refrigeração da fase sortiva - (A) Cilindro de Gás; (B) Misturador de
gases (homemade); (C) Conexão com o misturador de gases e mangueiras; (D) Esquema com
a refrigeração da barra conectado a linha de gás; (E) Vial de headspace pronto para a
extração; (F) Sistema completo.
B C A
D
E F

57
4. Resultados e discussão
4.1. Aplicação das ferramentas da qualidade no desenvolvimento, validação e cálculo
da incerteza para análise de organoclorados em pescado pelo método QuEChERS
Para avaliação do método QuEChERS necessitou primeiramente definir as
concentrações de analito, ou seja, os LMRs de trabalho os quais foram apresentados na Tabela
9.
4.1.1. Planejamento experimental
Trabalhando-se com a extração de organoclorados em pescado, pelo método
QuEChERS, observou-se dificuldades na extração no intervalo que a norma SANCO 12495
exige, que são as extrações entre 70 e 120%, sendo assim uma otimização do processo de
extração foi planejada. Primeiramente fixaram-se alguns pontos que foram considerados já
bem estudados, como por exemplo, o volume de solvente orgânico (Hexano), a massa da
matriz devido a limitação de material a disposição e a quantidade de sulfato de magnésio, que
foi considerado um material de fácil acesso, não inviabilizando assim sua utilização na rotina
de trabalho, todos estes fatores foram otimizados com planejamento univariado.
A escolha das variáveis para o planejamento experimental foram, o sal da fase de
extração líquido-líquido, o NaCl (com nível baixo (-1) de 0,5 g e nível alto (+1) de 1,5 g),
para observar a influência da força iônica na extração dos analitos de interesse. Também foi
adicionado como variável no planejamento a massa de PSA com nível baixo (-1) de 50 mg e
nível alto (+1) de 150 mg) e utilização de C18 com nível baixo (-1) de 25 mg e nível alto (+1)
75 mg, na fase de Clean-up usando o método de dSPE (Dispersive Solid Phase Extraction).
Com estes dados, foi realizado um planejamento experimental 23 completo, onde foi utilizado
o Software Statistica 7 para promover a aleatoriedade para o tratamento dos dados, e como
comparação e confirmação dos resultados utilizamos a Planilha Excel, conforme Apêndice B.
Sendo assim, foi preparada a planilha de planejamento para promover a sequência de
execução dos experimentos no Software Statistica 7, a qual foi realizada com triplicata no
ponto central para observar a reprodutibilidade do experimento, desta maneira obteve-se a
seguinte sequência (Tabela 13).

58
Tabela 13: Planejamento experimental com os níveis baixos e altos distribuídos
aleatoriamente para análise.
Experimentos NaCl PSA C18
(1) -1 -1 -1
(5) -1 -1 1
(10 C) 0 0 0
(4) 1 1 -1
(7) -1 1 1
(8) 1 1 1
(2) 1 -1 -1
(6) 1 -1 1
(9 C) 0 0 0
(3) -1 1 -1
(11 C) 0 0 0
Foi realizado o método QuEChERS para extração dos organoclorados e observou-se as
recuperações das amostras fortificadas no LMR (Limite Máximo de Resíduos) dos 14 analitos
da Tabela 9. Para se obter uma referência de quanto seria a máxima extração possível, foi
utilizado uma amostra branca (sem os analitos), onde todo o processo de extração foi
realizado e ao final do mesmo, foi adicionado os analitos no solvente da fase extratora.
Observaram-se assim as seguintes áreas para a amostra branca fortificada.
Tabela 14: Área das recuperações da amostra branca fortificada sem perdas do processo de
extração.
Analito Área (Fortificado LMR 100%)
HCB 27874167
LIN 118114674
ALD 20908374
HPX 58917290
tCLD 62463266
cCLD 59969236
ppE 141377744
DLD 51645231
PCB 118 8487743
ppD 139145586
opT 124061997
ppT 159012529
PCB 180 12325377
MRX 12566941

59
Com estas áreas sendo a referência do máximo de extração dos analitos na amostra,
realizou-se a extração, obtendo as recuperações abaixo (Tabela 15).
Tabela 15: Recuperação dos 14 organoclorados de interesse realizado sobre a técnica
QuEChERS e injetado em GC-ECD.
HCB LIN ALD HPX tCLD cCLD ppE DLD PCB118 ppD opT ppT PCB180 MRX
(1) 83 79 82 81 80 83 93 72 87 83 92 91 86 89
(5) 90 82 89 83 84 88 97 71 93 84 96 94 94 94
(10C) 87 73 87 77 75 80 94 66 89 76 91 88 89 91
(4) 89 68 87 76 71 78 93 66 89 72 91 86 90 91
(7) 86 64 83 70 66 74 91 57 85 66 86 82 86 90
(8) 86 66 83 71 67 74 90 58 85 68 88 83 86 90
(2) 81 77 80 76 76 80 90 66 84 79 88 87 84 85
(6) 89 78 88 79 80 85 94 66 91 81 93 91 92 94
(9C) 92 74 91 79 76 82 94 67 90 76 91 88 91 94
(3) 91 70 90 78 72 79 94 66 88 72 90 86 89 91
(11C) 92 75 90 79 76 82 95 67 88 76 91 89 92 94
Com os resultados de recuperação obtidos com o planejamento experimental 23
completo, tratou-se os dados para observação dos efeitos dos fatores, com a intenção de
conhecer a melhor condição para o trabalho, e por conseguinte observar quais são os efeitos
significativos para um nível de confiança de 95%. Segundo Neto, 2010 quando analisado cada
efeito, este deve ser maior que o desvio de um efeito multiplicado pelo t de Student (com seus
respectivos graus de liberdade e nível de confiança) para ser considerado diferente de zero e
então significativo para o método, quando trabalhado com a tabela de probabilidade Z-score.

60
Figura 19: Exemplo de pontos significativos e insignificativos, trabalhando com uma
distribuição normal com a tabela de probabilidade Z score para observação dos pontos a um
nível de confiança de 95%.
4.1.1.1. Tratamento dos dados - MIREX (MRX)
Assim, foram tratados os dados para os compostos individualmente, para que seja
possível observar o comportamento de cada um frente à extração, porém a decisão para o
ponto ótimo para todos os analitos foi averiguada com mais cautela, por se tratar de um
método multiresíduo.
Deve-se atentar também aos efeitos gerados para cada fator individualmente, assim
como suas interações, e analisar a tendência de usar este fator em seu nível mais baixo (-1) ou
em seu nível mais alto (+1). Os efeitos são interpretados em porcentagem já que foi feito a
opção por trabalhar com porcentagem de recuperação, e sendo assim, o efeito significa o
ganho ou a perda na recuperação. Desta forma segue o exemplo de um tratamento realizado
na planilha Excel e comprovado pelo Software Statistica 7 para o composto Mirex.

61
Tabela 16: tabela dos fatores para o composto Mirex (MRX) em nível baixo (-1) e nível alto
(+1) com suas respectivas recuperações para cada experimento realizado aleatoriamente, foi
disposto os experimentos em ordem para melhor visualização dos dados.
Experimentos Fatores Recuperação (%)
Fator 1 (NaCl) Fator 2 (PSA) Fator 3 (C18) Mirex (MRX)
1 -1 -1 -1 89
2 1 -1 -1 85
3 -1 1 -1 91
4 1 1 -1 91
5 -1 -1 1 94
6 1 -1 1 94
7 -1 1 1 90
8 1 1 1 90
Assim, como procedimento para observação dos efeitos foi realizado a multiplicação
dos níveis com suas respectivas porcentagens de recuperação, como segue na tabela a seguir.
Tabela 17: Cálculo dos efeitos de primeira (1,2 e 3), segunda (12,13 e 23) e terceira ordem
(123).
Experimentos
Número de
efeitos
1 2 3 4 5 6 7
Interações 1 2 3 12 13 23 123
1
Níveis
multiplicado
pela
recuperação
-89,20 -89,20 -89,20 89,20 89,20 89,20 -89,20
2 85,50 -85,50 -85,50 -85,50 -85,50 85,50 85,50
3 -91,49 91,49 -91,49 -91,49 91,49 -91,49 91,49
4 91,34 91,34 -91,34 91,34 -91,34 -91,34 -91,34
5 -94,41 -94,41 94,41 94,41 -94,41 -94,41 94,41
6 94,08 -94,08 94,08 -94,08 94,08 -94,08 -94,08
7 -89,51 89,51 89,51 -89,51 -89,51 89,51 -89,51
8 89,64 89,64 89,64 89,64 89,64 89,64 89,64
Somatório -4,06 -1,22 10,10 4,01 3,66 -17,48 -3,09
Efeito -1,02 -0,30 2,53 1,00 0,92 -4,37 -0,77

62
Com os efeitos obtidos, estes foram colocados em uma distribuição normal com os
valores fornecidos pela tabela Z-score, assim obteve-se o seguinte gráfico.
Figura 20: Gráfico dos efeitos de um planejamento 23 para o analito Mirex realizado em
análise multiresíduo plotado em tabela Z-Score.
Com este planejamento, foi evidenciado que o efeito com maior significância foi o
proporcionado pela interação do fator 2 com o 3 (-4,37), ou seja, do PSA e o C18. Como o
fator 1 (NaCl) e suas respectivas interações tiveram efeitos muito baixo (menor que 1%), este
foi excluído do planejamento para esta resposta. Com esta exclusão, o planejamento que era
um 23, passou a ser um planejamento 2
2, com uma replicata em cada experimento, sendo
possível calcular o desvio de um fator e assim obter o valor de t crítico, que segundo a tabela
t, para 4 graus de liberdade e nível de confiança de 95%, foi de 2,59, comprovando que todos
valores de efeito acima deste valor crítico são realmente considerados significativos.
Deve-se observar que a interação dos fatores 2 e 3 (PSA e C18) foi negativo, ou seja,
quando passamos do nível -1 para o +1 existe uma diminuição na porcentagem relativa de
extração (perda de 4,37 na recuperação), sendo assim é importante utilizar os fatores 2 e 3 nas
seguintes condições.
Tabela 18: Condições de trabalho para os fatores PSA e C18.
Fator 2 (PSA) 3 (C18) Efeito
Multiplicação -1 +1 Negativo
+1 -1 Negativo

63
Observando as recuperações tem-se que a melhor condição é usar o fator 2 (PSA) no
nível mais baixo e o fator 3 (C18) no nível mais alto para este analito organoclorado (Mirex),
já que com o fator 3 no nível mais alto existe um ganho de 2,52%. Estes valores de efeitos
foram reproduzidos no programa Software Statistica 7 confirmando assim os resultados.
Figura 21: Gráfico de Pareto mostrando os efeitos para cada fator e suas interações para o
analito Mirex.
A Figura 22 demonstra graficamente o que já foi discutido anteriormente, é possível
verificar que a diagonal principal é que tem os menores valores de recuperação (87,3 e
89,6%), a diagonal principal tem os valores da interação da variáveis 2 (PSA) e 3 (C18) que
dão valores +1 e a diagonal secundária têm os maiores valores de recuperação (94,2 e 91,4%),
sendo a maior recuperação de todos os pontos em que a variável 2 esta no nível inferior (-1) e
a variável 3 esta no nível superior (+1).
Pareto Chart of Effects; Variable: Recuperação MRX
2**(3-0) design
DV: Recuperação MRX
-,30391
-,772676
,9153202
1,002565
-1,01552
2,526166
-4,37072
-0,5 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0
Effect Estimate (Absolute Value)
(2)PSA
1*2*3
1by3
1by2
(1)NaCl
(3)C18
2by3

64
Figura 22: Gráfico do quadrado dos fatores PSA e C18 para o Mirex, mostrando a melhor
condição de trabalho para obter maior ganho de recuperação. Valores obtidos em duplicatas
em um nível de confiança de 95%.
Desta forma foi realizado o planejamento e o estudo dos efeitos de todos os outros
compostos (resposta) avaliando o comportamento de cada um frente à técnica aplicada. É
importante ressaltar que o trabalho foi realizado em análise multiresíduo, e que as análises no
Apêndice B são para os analitos individuais, com exceção do conjunto multiresíduo. Assim,
são mostradas as figuras e tabelas para os outros compostos, com especial atenção para o
composto que foi o limitante na extração, ou seja, o que esteve no limiar do que é exigido pela
norma SANCO 12495 (70%), composto Dieldrin.
Predicted Means for Variable: Recuperação MRX
2**(2-0) design; MS Pure Error=1,735628
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
87,348 (84,76,89,93)
94,245 (91,66,96,83)
91,415 (88,83,94,)
89,571 (86,98,92,16)
-1 1
PSA
-1
1
C1
8

65
Tabela 19: Analitos e significância de seus efeitos para um nível de confiança de 95% em
ordem decrescente em módulo (Gráficos e Figuras ver Apêndice B).
Analitos Efeitos em ordem decrescente de
significância
Efeito significativo
para 95%
ALD 23 > 1 > 2 > 3 > 13 > 12 > 123 23
cCLD 2 > 23 > 1 > 12 > 13 > 123 > 3 2 e 23
DLD 2 > 3 > 23 > 12 > 1 > 13 > 123 2
HCB 23 > 2 > 3 > 1 > 13 > 123 > 12 23
HPX 2 > 23 > 1 > 3 > 12 > 13 > 123 2 e 23
LIN 2 > 23 > 12 > 1 > 3 > 123 > 13 2
opT 23 > 2 > 12 > 1 > 13 > 3 > 123 2 e 23
PCB 118 23 > 3 > 2 > 12 > 1 > 13 > 123 23
PCB 180 23 > 3 > 12 > 1 > 2 > 123 > 13 23
ppD 2 > 23 > 12 > 3 > 1 > 13 > 123 2
ppE 23 > 1 > 1 > 12 > 3 > 13 > 123 23
ppT 2 > 23 > 12 > 1 > 13 > 123 > 3 2
tCLD 2 > 23 > 1 > 12 > 13 > 3 > 123 2 e 23
Conjunto Multiresíduo 2 > 23 > 12 > 1 > 13 > 123 > 3 2 e 23
4.1.1.2. Fatores de influência no Conjunto Multiresíduo
Analisando o conjunto multiresíduo dos organoclorados na extração e buscando uma
forma de analisar todos juntos e assim a contribuição de cada sal presente na extração,
realizou-se uma média das recuperações de todos analitos para cada experimento realizado, e
obteve-se os seus respectivos efeitos, que é compatível com alguns dos compostos
individuais, e como foi mostrado anteriormente o analito limitante Dieldrin que esteve
próximo do que é exigido pela SANCO 12495, teve comportamento ligeiramente diferente do
conjunto, que teve como fator significativo o reagente PSA, mas que não pode-se considerar
extremamente diferente, já que tivemos um grande efeito da interação 23 mesmo não sendo
significativo para o nível de confiança trabalhado, assim pode-se seguir o que o conjunto
multiresíduo mostra, dando relevância para os fatores 2 e 3. E como mostrado, o fator 1

66
NaCl, não se mostrou significante em nenhum dos analitos, evidenciando que o efeito de
Salting-Out não se mostrou relevante quando comparado com os efeitos dos outros sais PSA
e C18.
Pode-se concluir que o planejamento foi bem executado e atendeu o que foi proposto
pelo trabalho, que foi a obtenção de extração acima do que é exigido, assim sabe-se quais são
os componentes principais, e a rota de otimização caso necessite melhorar o processo.
4.1.1.3. Função de Desejabilidade
Um grande problema visto para tomada de decisão em procedimentos que há várias
respostas é a otimização simultânea das variáveis independentes. Como forma de garantir os
resultados do conjunto multiresíduo, foi aplicada a função de Desejabilidade para obtenção
das respostas através de um tratamento estatístico conforme proposto por Harrington, 1965.
Esta proposta foi desenvolvida para determinação de respostas de análise que contêm
mais de uma variável resposta, neste caso 14 analitos organoclorados. O procedimento é
semelhante com o desenvolvido para os analitos individuais, só que neste procedimento é
encontrado um valor di (Desejabilidade) para cada recuperação, conforme proposto por
Harrington. Padroniza-se todos os pontos entre 0 e 1, e assim obtêm-se uma média
geométrica, que será a desejabilidade do procedimento, D (Desejabilidade combinada) como
demonstrado na tabela 20. Segue as fórmulas de trabalho abaixo,
cuperaçãoMenorcuperaçãoMaior
cuperaçãoMenorcuperaçãod i
ReRe
ReRe0
1
k
kxdxxddD/1
21 )...(
Onde k é o número de respostas para o experimento.
Equação 4: Fórmula para cálculo da desejabilidade.
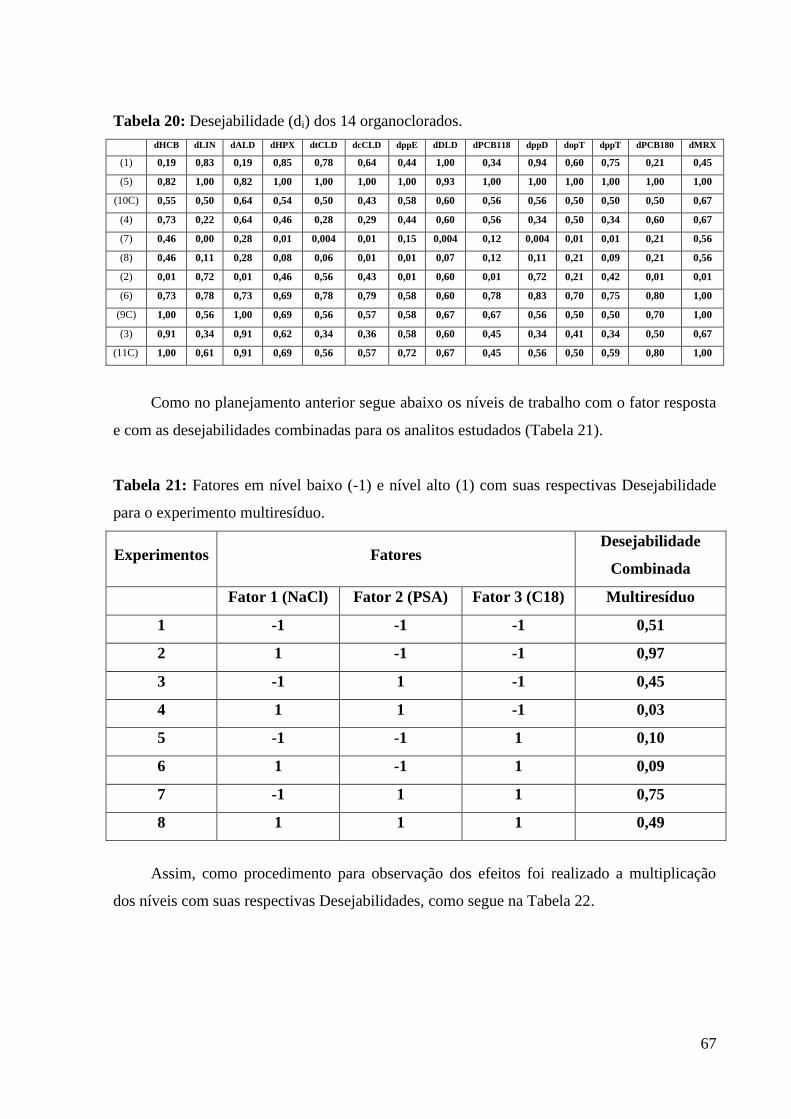
67
Tabela 20: Desejabilidade (di) dos 14 organoclorados.
dHCB dLIN dALD dHPX dtCLD dcCLD dppE dDLD dPCB118 dppD dopT dppT dPCB180 dMRX
(1) 0,19 0,83 0,19 0,85 0,78 0,64 0,44 1,00 0,34 0,94 0,60 0,75 0,21 0,45
(5) 0,82 1,00 0,82 1,00 1,00 1,00 1,00 0,93 1,00 1,00 1,00 1,00 1,00 1,00
(10C) 0,55 0,50 0,64 0,54 0,50 0,43 0,58 0,60 0,56 0,56 0,50 0,50 0,50 0,67
(4) 0,73 0,22 0,64 0,46 0,28 0,29 0,44 0,60 0,56 0,34 0,50 0,34 0,60 0,67
(7) 0,46 0,00 0,28 0,01 0,004 0,01 0,15 0,004 0,12 0,004 0,01 0,01 0,21 0,56
(8) 0,46 0,11 0,28 0,08 0,06 0,01 0,01 0,07 0,12 0,11 0,21 0,09 0,21 0,56
(2) 0,01 0,72 0,01 0,46 0,56 0,43 0,01 0,60 0,01 0,72 0,21 0,42 0,01 0,01
(6) 0,73 0,78 0,73 0,69 0,78 0,79 0,58 0,60 0,78 0,83 0,70 0,75 0,80 1,00
(9C) 1,00 0,56 1,00 0,69 0,56 0,57 0,58 0,67 0,67 0,56 0,50 0,50 0,70 1,00
(3) 0,91 0,34 0,91 0,62 0,34 0,36 0,58 0,60 0,45 0,34 0,41 0,34 0,50 0,67
(11C) 1,00 0,61 0,91 0,69 0,56 0,57 0,72 0,67 0,45 0,56 0,50 0,59 0,80 1,00
Como no planejamento anterior segue abaixo os níveis de trabalho com o fator resposta
e com as desejabilidades combinadas para os analitos estudados (Tabela 21).
Tabela 21: Fatores em nível baixo (-1) e nível alto (1) com suas respectivas Desejabilidade
para o experimento multiresíduo.
Experimentos Fatores Desejabilidade
Combinada
Fator 1 (NaCl) Fator 2 (PSA) Fator 3 (C18) Multiresíduo
1 -1 -1 -1 0,51
2 1 -1 -1 0,97
3 -1 1 -1 0,45
4 1 1 -1 0,03
5 -1 -1 1 0,10
6 1 -1 1 0,09
7 -1 1 1 0,75
8 1 1 1 0,49
Assim, como procedimento para observação dos efeitos foi realizado a multiplicação
dos níveis com suas respectivas Desejabilidades, como segue na Tabela 22.

68
Tabela 22: Efeitos de primeira (1,2 e 3), segunda (12,13 e 23) e terceira ordem (123).
Experimentos
Número de
efeitos 1 2 3 4 5 6 7
Interações 1 2 3 12 13 23 123
1
Níveis
multiplicado
pela
Desejabilidade
-0,51 -0,51 -0,51 0,51 0,51 0,51 -0,51
2 -0,97 -0,97 0,97 0,97 -0,97 -0,97 0,97
3 0,45 0,45 -0,45 0,45 -0,45 -0,45 -0,45
4 -0,03 0,03 0,03 -0,03 -0,03 0,03 -0,03
5 0,10 0,10 0,10 0,10 0,10 0,10 0,10
6 0,09 -0,09 -0,09 -0,09 -0,09 0,09 0,09
7 0,75 -0,75 0,75 -0,75 0,75 -0,75 -0,75
8 -0,49 0,49 -0,49 -0,49 0,49 -0,49 0,49
Somatório -0,61 -1,24 0,30 0,67 0,32 -1,94 -0,08
Efeito -0,15 -0,31 0,08 0,17 0,08 -0,48 -0,02
Com os efeitos obtidos, estes foram colocados em uma distribuição normal com os
valores fornecidos pela tabela Z score, assim obteve-se o seguinte gráfico.
Figura 23: Efeitos de um planejamento 23 para o Conjunto Multiresíduo realizado pela
função de Desejabilidade plotado em tabela Z-Score.

69
Estes valores de efeitos foram reproduzidos no programa Software Statistica 7 para
confirmação dos resultados.
Figura 24: Gráfico de Pareto mostrando os efeitos para cada fator e suas interações para o
Conjunto Multiresíduo.
A Figura 25 demonstra graficamente o que já foi discutido anteriormente, é possível
verificar que a diagonal principal é a que tem os menores valores de recuperação (0,267 e
0,038), a diagonal principal tem os valores da interação das variáveis 2 (PSA) e 3 (C18) que
dão valores +1 e a diagonal secundária têm os maiores valores de recuperação (0,856 e
0,468), sendo a maior recuperação de todos os pontos em que a variável 2 esta no nível
inferior (-1) e a variável 3 esta no nível superior (+1).
Pareto Chart of Effects; Variable: Desejabilidade Combinada
2**(3-0) design
DV: Desejabilidade Combinada
-,035029
,0798413
,0886807
-,168197
,1772509
-,308562
-,509205
-0,1 0,0 0,1 0,2 0,3 0,4 0,5 0,6
Effect Estimate (Absolute Value)
1*2*3
(3)C18
1by3
(1)NaCl
1by2
(2)PSA
2by3

70
Figura 25: Gráfico do quadrado dos fatores PSA e C18 para o Conjunto Multiresíduo,
mostrando a melhor condição de trabalho para obter maior ganho de recuperação. Valores
obtidos em duplicatas em um nível de confiança de 95%.
Conclui-se que a melhor condição é coerente com o que foi observado com as médias
das recuperações, assim a função de Desejabilidade é uma ferramenta útil para análise dos
fatores do experimento multiresíduo. Desta maneira fica evidente que os fatores de maior
influência são o PSA e o C18 na fase de Clean-up da técnica QuEChERS, que foi coerente
com os analitos individuais. O fator NaCl, apesar de não significante, é importante evidenciar
os valores dos efeitos de suas interações que obteve uma certa influência, só que não
significativas para o nível de confiança trabalhado, mas que somado mostrou ter relevância
sobre o método. Em relação à otimização, os níveis de NaCl não se mostraram importante,
logo seria bom fixá-lo no ponto mais baixo por economia de reagente. Já o PSA como
mostrou o estudo, é melhor utilizá-lo no nível mais baixo e o C18 no nível mais alto,
lembrando que o efeito do C18 na maioria das análises se mostrou relativamente baixo
quando não significativo, mostrando que uma otimização pode ser válido neste fator para
melhorar a recuperação do método.
Predicted Means for Variable: Desejabilidade Combinada
2**(2-0) design; MS Pure Error=,0343997
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
,267 (-,1,,63)
,856 (,49,1,22)
,468 (,1,,83)
,038 (-,33,,4)
-1 1
PSA
-1
1
C1
8

71
4.1.2. Validação do Método
4.1.2.1. Seletividade
A primeira etapa realizada para validação foi à avaliação da seletividade do método.
Este teste foi realizado analisando 10 diferentes amostras brancas de pescado verificando se
existiam interferentes nos tempos de retenção dos analitos de interesse. As amostras foram
obtidas pelas unidades COI e CTEC do Lanagro/SP, conforme destacado pela Tabela 23.
Após analisar as 10 amostras, não foram evidenciados interferentes nos tempos de retenção
dos analitos de trabalho, conforme destacado no cromatograma representativo (Figura 26) de
uma das amostras analisadas.
Tabela 23: Amostras de pescado para teste de seletividade.
Amostra Fornecedor Características
1
Unidade COI
Peixe Tambaqui – Lagoa Teresopolis
2 Peixe Tilápia (oreochromis sp)
3 Peixe Tambacu (inteiro sem vísceras e sem cabeça)
4 Peixe Tilápia (oreochromis niloticus)
5 Peixe pintado da Amazônia (pseudo latystoma ssp. X Leiarius
marmoratus)
6
Unidade
CTEC
Truta 01
7 Truta 02
8 Truta 03
9 Sardinha 01
10 Sardinha 02
A Figura 26 mostra a análise das 10 amostras dos fornecedores, mostrando que estas
não contêm nenhum interferente representativo que possa ser qualificado como resultado
positivo, mascarando assim a análise dos analitos estudados. Desta forma as amostras são
consideradas limpas para o processo e o método trabalhado seletivo.

72
Figura 26: Cromatograma representativo das 10 amostras de pescado.
A figura 27 mostra a sobreposição de uma amostra branca com uma amostra fortificada
em 1 LMR evidenciando que não existem interferentes representativos nos tempos de
retenção dos analitos de trabalho.

73
Figura 27: Sobreposição de amostra branca com amostra fortificada em 1 LMR: Linha
amarela representa a matriz e a linha azul representa os analitos de trabalho.
A Seletividade do método utilizado evidencia que a matriz nas condições de trabalhos
são apropriadas para que não haja má interpretação dos dados obtidos.
4.1.2.2. Linearidade
Para a análise da linearidade foi aplicado o coeficiente de determinação como
especificado pela DOC-CGCRE-008, análise dos resíduos conforme requisitado pela SANCO
12495:2011 e a incerteza como requisito da ISO/IEC 17025 e o guia EURACHEM/CITAC,
além do desenvolvimento de rotina para analisar se a curva linear é o mais apropriado para o
método, como será destacado em 4.1.4. “Desenvolvimento de modelo para avaliação da curva
de calibração”. As concentrações de trabalho para HCB, ALD, MRX foram de 50, 80, 100,
150 e 200 ug/Kg, para o LIN foram 500, 800, 1000, 1500 e 2000 ug/Kg, para o HPX,
tCLD,cCLD, DLD 150, 240, 300, 450 e 600 ug/Kg, para o ppE, ppD, opT, ppT 625, 1000,
1250, 1875 e 2500 ug/Kg, para o PCB 118, PCB 180 28,5, 45,6, 57,0, 85,5 e 114 ug/Kg.
Como se trata de uma análise multiresíduo, foram estabelecidas várias faixas de trabalho para

74
a curva devido ao LMR de cada analito. Os gráficos dos resíduos estão representados no
Apêndice C.
Tabela 24: Resultados para Curva analítica utilizando métodos dos mínimos quadrados
ordinários.
Analito
Faixa de
trabalho
(ug/Kg)
Equação da curva R2
Resíduos Incerteza
1
(%)
HCB 50 – 200 y = 3,34.10+5
+ 4,91.10+6
0,99
SANCO/12
495 < 20%.
O maior
valor
encontrado
foi 19,08%
do
composto
HCB
± 7,00
ALD 50 – 200 y = 3,28.10+5
– 4,65.10+5
0,99 ± 4,60
MRX 50 – 200 y = 2,67.10+5
– 2,68.10+6
0,99 ± 3,40
LIN 500 – 2000 y = 5,30.10+4
+ 9,22.10+7
0,98 ± 9,84
HPX 150 – 600 y = 1,55.10+5
+ 3,56.10+7
0,98 ± 9,33
tCLD 150 – 600 y = 1,76.10+5
+ 3,48.10+7
0,98 ± 8,33
cCLD 150 – 600 y = 1,77.10+5
+ 3,22.10+7
0,98 ± 8,80
DLD 150 – 600 y = 1,66.10+5
+ 2,23.10+7
0,98 ± 8,60
ppE 625 – 2500 y = 5,50.10+4
+ 1,17.10+8
0,98 ± 9,42
ppD 625 – 2500 y = 6,64.10+4
+ 1,09.10+8
0,98 ± 9,29
opT 625 – 2500 y = 6,17.10+4
+ 8,90.10+7
0,98 ± 8,85
ppT 625 – 2500 y = 8,15.10+4
+ 1,11.10+8
0,98 ± 8,67
PCB 118 28,5 – 114 y = 3,29.10+5
– 3,29.10+6
0,99 ± 6,67
PCB180 28,5 – 114 y = 5,02.10+5
– 4,38.10+6
0,99 ± 3,51 1 A incerteza foi calculado com o menor ponto da curva, que, segundo a teoria de Horwitz, contem o maior
desvio.
Com estes dados, pode-se observar que todos os parâmetros foram cumpridos com o
que é exigido pelas normas de trabalho, ou seja, todos os coeficientes de determinação foram
maiores que 0,98 e os resíduos não excederam os 20%, assim como exige a SANCO
12495:2011. O cálculo da incerteza não teve valor acima de 9,84% para todos os analitos de
trabalho, mostrando uma incerteza aceitável para as curvas analíticas. Considerando que a
amostra trata-se de um alimento e que os limites máximos de resíduos estão em concentrações
baixas de toxicidade, este valor de incerteza é aceitável, considerando que o guia SANCO
permite a incerteza expandida dentro da faixa de 50%, sendo assim, os valores são aceitáveis
para se chegar ao resultado pretendido.

75
4.1.2.3. Recuperação/ Exatidão
Após o planejamento experimental, foi selecionada a melhor condição e foram
realizadas extrações em 3 dias diferentes para se obter a recuperação média do método
QuEChERS. Este foi desenvolvido, sendo apresentado um cromatograma representativo na
Figura 28, que evidencia a separação dos analitos de trabalho e seus respectivos tempos de
retenção. A Tabela 25 mostra as recuperações em dias diferentes, sendo que no segundo dia
foi realizado triplicata da recuperação para observar quanto da repetibilidade dos dados.
Tabela 25: Recuperações do dia 1, 2 (triplicata) e 3.
Analitos Rec. Dia 1 (%) Rec. Dia 2 (%) Rec. dia 3 (%)
Amostra 1 Amostra 2 Amostra 3
HCB 83 112 101 107 112
LIN 79 88 84 86 94
ALD 82 109 101 107 116
HPX 81 91 87 90 100
tCLD 80 94 91 94 104
cCLD 83 97 93 96 106
ppE 93 102 101 102 109
DLD 72 74 71 74 92
PCB 118 87 100 99 99 113
ppD 83 90 88 89 98
opT 92 99 98 99 106
ppT 91 96 93 95 105
PCB 180 86 98 97 99 113
MRX 89 100 97 102 117
A exatidão expressa à concordância entre o valor real e o valor determinado pela
metodologia. Para validação da metodologia foi fortificado uma amostra conhecida e livre de
interferentes.

76
Figura 28: Cromatograma da melhor condição do planejamento experimental.
Com esta melhor condição foi analisada a recuperação/exatidão de cada analito de
trabalho, obtendo assim resultados dentro do exigido pela norma (70 a 120%), o que mostra
que a metodologia é apta a ser utilizada para o trabalho proposto, como mostrado na Tabela
26.
Tabela 26: Recuperação média (com 5 replicatas) e desvio padrão dos analitos estudados:
Analito Recuperação (%) Desvio Padrão (%)
HCB 103 12,00
LIN 86 5,33
ALD 103 12,80
HPX 90 6,86
tCLD 93 8,60
cCLD 95 8,25
ppE 101 5,53
DLD 77 8,85
PCB 118 100 9,29
ppD 90 5,55
opT 99 4,87
ppT 96 5,28
PCB 180 99 9,54
MRX 101 10,18

77
Pode se evidenciar que todos os analitos apresentaram resultados dentro do exigido, ou
seja, entre 70 e 120%.
4.1.2.4. Precisão (Reprodutibilidade Intralaboratorial)
A precisão foi realizada com 5 replicatas em 3 dias diferentes, obtendo assim valores
satisfatório para o que é exigido pela SANCO 12495:2011. A Tabela 27 mostra que os
analitos contêm um CV (Coeficiente de variação) aceitável e que os analitos com
características semelhantes possuem coeficientes de variação próximos, por exemplo, os
PCBs contêm CV próximos assim como os DDT e seus derivados.
Tabela 27: Valores do Coeficiente de Variação (CV) para análise da precisão do método
QuEChERS.
Analito Precisão – CV(%)1
Requisito SANCO 12495:2011
HCB 11,65
< 20%
LIN 6,19
ALD 12,43
HPX 7,65
tCLD 9,29
cCLD 8,69
ppE 5,46
DLD 11,54
PCB 118 9,33
ppD 6,19
opT 4,93
ppT 5,51
PCB 180 9,68
MRX 10,08 1
Os experimentos foram conduzidos em três diferentes dias, permitindo avaliar a precisão em termos de
reprotutibilidade intralaboratorial através do CV(%).

78
4.1.3. Cálculo da incerteza
Para análise do cálculo da incerteza, deve ser realizado uma estimativa razoável, e o
analista deve estabelecer quais são os parâmetros que mais interferem no processo. Segundo
estudos, é sabido que a linearidade tem grande influência na incerteza final dos métodos de
extração, sendo assim um parâmetro a ser considerado, assim como a recuperação e a
precisão, com menor impacto no resultado final, mas com influência significativa. O processo
de análise da incerteza utilizado neste trabalho é denominado método Validation based. Segue
abaixo as fórmulas de trabalho para cada parâmetro analisado.
A Linearidade foi calculada no menor ponto da curva, pois se sabe que há o maior
desvio embutido, segundo a teoria do trompete de Horwitz.
xxS
CC
npB
SIncerteza
0
1
11
Onde,
2
.1
2
10
n
cBBA
S
n
j
jj
n
j
jxx ccS1
2
Onde, B1 é a inclinação da curva, S é o desvio padrão, p é o número de medidas para
determinar C0, n é o número de medidas para a calibração, C0 é a concentração
determinada, C é o valor médio dos diferentes padrões de calibração e Cj são as
concentrações do iésimo padrão de calibração. Aj é o jésima medida dos padrões de
calibração e B0 é o intercepto.
Equação 5: Fórmula para cálculo da incerteza da curva analítica.
A Recuperação foi calculada na concentração de 1 LMR para todos analitos de trabalho,
assim como para a precisão, sendo utilizado um valor de n igual 5, ou seja, 5 análises para
obter-se a recuperação e precisão média,

79
n
SxS
Equação 6: Fórmula para cálculo da incerteza da recuperação.
A precisão foi calculada utilizando o desvio padrão das 5 análises, a sua incerteza é
exatamente o desvio padrão obtido,
Incerteza = S
Equação 7: Fórmula para cálculo da incerteza da precisão.
Com estes parâmetros principais, que contêm maior influência na incerteza final da
metodologia, calcula-se a incerteza combinada e a incerteza expandida.
Incerteza combinada
......,22 qupuqpyuc
Onde u(p)2, u(q)
2 são as incertezas dos parâmetros da validação.
Equação 8: Fórmula para cálculo da incerteza combinada.
Incerteza expandida
combinadaExpandido uKu . ,
Onde K igual a 2.
Equação 9: Fórmula para cálculo da incerteza expandida.
É importante ressaltar que os procedimentos de cálculo de incerteza adotados foram
trabalhados segundo o guia EURACHEM/CITAC (EURACHEM, 2000) e os seus dados estão
expostos na Tabela 28.

80
Tabela 28: Incerteza padrão combinada e expandida para o método QuEChERS:
Analito Fonte da
incerteza
Valor
(x)
(ug/Kg)
Incertez
a padrão
u(x)
Incerteza
padrão
(%)
Incerteza
padrão
relativa u(x)/x
Quadrado da
Incerteza
padrão relativa
(u(x)/x)2
Incerteza
combinada1
Incerteza
combinada
(%)
Incerteza
Expandida
(%) (K=2)
HCB
Curva analítica 50 3,50 7,00 0,0700 0,0049
± 14,89 ± 14,89 ± 29,79 Recuperação 100
5,36 5,36 0,0536 0,0029
Precisão 12,00 12,00 0,1200 0,0144
LIN
(Lindane)
Curva analítica 500 49,20 9,84 0,0984 0,0097
± 98,66 ± 9,86 ± 19,73 Recuperação 1000
2,38 0,24 0,0024 0,0000058
precisão 5,33 0,53 0,0053 0,000028
ALD
(Aldrin)
Curva analítica 50 2,30 4,60 0,0460 0,0021
± 14,75 ± 14,75 ± 29,51 Recuperação 100
5,72 5,72 0,0572 0,0033
precisão 12,80 12,80 0,1280 0,01638
HPX
Heptachlor
Epoxide)
Curva analítica 150 14,00 9,33 0,0933 0,0087
± 28,83 ± 9,61 ± 19,22 Recuperação 300
3,07 1,02 0,0102 0,0000104
precisão 6,86 2,29 0,0229 0,000524
tCLD
(trans-
Chlordane)
Curva analítica 150 12,50 8,33 0,0833 0,0069
± 26,64 ± 8,88 ± 17,76 Recuperação 300
3,85 1,28 0,0128 0,000164
precisão 8,60 2,87 0,0287 0,000824

81
cCLD (cis-
Chlordane)
Curva analítica 150 13,20 8,80 0,0880 0,0077
± 27,83 ± 9,28 ± 18,55 Recuperação 300
3,69 1,23 0,0123 0,000151
precisão 8,25 2,75 0,0275 0,000756
ppE
(ppDDE)
Curva analítica 625 58,90 9,42 0,0942 0,0089
± 118,08 ± 9,44 ± 18,89 Recuperação 1250
2,47 0,20 0,0020 0,0000040
precisão 5,53 0,44 0,0044 0,0000194
DLD
(Dieldrin)
Curva analítica 150 12,90 8,60 0,0860 0,0074
± 27,28 ± 9,09 ± 18,19 Recuperação 300
3,96 1,32 0,0013 0,00000169
precisão 8,85 2,95 0,0295 0,00087
PCB 118
Curva analítica 28,5 1,90 6,67 0,0667 0,0044
± 10,85 ± 19,05 ± 38,10 Recuperação 57
4,16 7,29 0,0729 0,005314
precisão 9,29 16,29 0,1630 0,02657
ppD
(ppDDD)
Curva analítica 625 58,1 9,29 0,0929 0,0086
± 116,08 ± 9,29 ± 18,57 Recuperação 1250
2,48 0,20 0,0020 0,0000040
precisão 5,55 0,44 0,0044 0,0000194
opT
(opDDT)
Curva analítica 625 55,30 8,85 0,0885 0,0078
± 110,52 ± 8,84 ± 17,68 Recuperação 1250
2,18 0,17 0,0017 0,00000289
precisão 4,87 0,39 0,0039 0,000015
ppT
(ppDDT)
Curva analítica 625 54,20 8,67 0,0867 0,0075 ± 108,41 ± 8,67 ± 17,34
Recuperação 1250 2,36 0,19 0,0019 0,00000361
82
precisão 5,28 0,42 0,0042 0,0000176
PCB 180
Curva analítica 28,50 1,00 3,51 0,0351 0,0012
± 10,64 ± 18,66 ± 37,33 Recuperação 57
4,27 7,49 0,0749 0,005610
precisão 9,54 16,74 0,1674 0,02802
MRX
Curva analítica 50 1,70 3,40 0,0340 0,0012
± 13,45 ± 13,45 ± 26,90 Recuperação 100
4,55 4,55 0,0455 0,002070
precisão 12,18 12,18 0,1218 0,01483
1 multiplica-se a o valor encontrado da incerteza combinada pela concentração de trabalho, neste caso o LMR para cada analito.
Com a utilização das ferramentas da qualidade, como o planejamento experimental, pode-se observar que foram obtidos resultados na
validação condizentes com a proposta do trabalho, sendo assim, de extrema importância a utilização de tais ferramentas para que haja a melhor
resposta. O planejamento experimental, por ser uma ferramenta que guia para a melhor forma de trabalho, proporcionou um direcionamento
correto, minimizando retrabalho e consequentemente tempo, atingindo assim resultados em conformidade com o guia de validação adotado.
Como exemplo, resíduos abaixo dos 20% para a curva analítica, com o maior valor encontrado de 19,08% para o HCB. Recuperações na faixa de
70 a 120% como requerido pela norma trabalhada, se atentando para as recuperações do Dieldrin (DLD) que ficou próximo do valor mínimo
estabelecido. Curvas analíticas com coeficientes de determinação acima do 0,98, demonstrando boa correlação entre os dados e incerteza dentro
do que foi pretendido, com incertezas inferiores a 10% para todos os analitos.
Desta forma é evidente que a utilização de tais ferramentas acrescentam valor e confiabilidade nos dados obtidos, garantindo qualidade do
método devido a aplicação de procedimentos dentro de normas e guias já bem conhecidos e difundidos na área da qualidade.

83
4.1.4. Desenvolvimento de modelo para avaliação da curva de calibração
Atualmente para a avaliação de uma curva analítica é sugerido por vários guias, alguns
parâmetros a serem analisados, como por exemplo, a DOC-CGCRE-008 que sugere análise
do coeficiente de determinação, a SANCO 12495:2011 que evidencia a análise dos resíduos e
como o guia EURACHEM que insere o cálculo de incerteza. Estes parâmetros não são todos
utilizados em rotinas, e alguns parâmetros como a coeficiente de determinação não é
considerado um parâmetro adequado para avaliação de uma curva de calibração
(BRUGGEMANA, QUAPP e WENRICH, 2006; VAN LOCO et al., 2002). Sendo assim, é de
extrema importância uma investigação para observar se a curva de calibração realmente é
adequada aos dados obtidos.
Com isso sugere-se um fluxograma como rotina de trabalho, evidenciando que uma
análise profunda dependendo do tipo de análise é necessária para assegurar os resultados
obtidos, assim propõe-se a utilização da incerteza como parâmetro primordial na análise de
uma curva analítica em rotina, além dos parâmetros já bem difundidos, como coeficiente de
determinação e resíduos. É importante ressaltar que a incerteza não é parâmetro para dizer se
uma curva analítica é aceitável ou não, mas sim evidenciar o quanto adequado esta curva é
para o trabalho utilizado. Foram utilizados também testes para análise de aptidão para a curva
linear como teste de outlier, homoscedasticidade e modelo, que é base para sustentação de
uma curva linear adequada. Assim o fluxograma da Figura 29 foi desenvolvido e aplicado ao
trabalho.
O uso da incerteza proverá um embasamento a mais para justificar um bom uso da
curva analítica, ela não mostra se a curva é linear ou não, mas evidencia o quanto apto o
modelo é para ser utilizado na representação dos dados de trabalho. Desta forma a incerteza é
uma ferramenta a mais de análise, como justificativa de adequação ao uso pretendido,
sabendo ainda que esta é exigência da qualidade, acrescentando assim maior confiabilidade no
resultado apresentado.
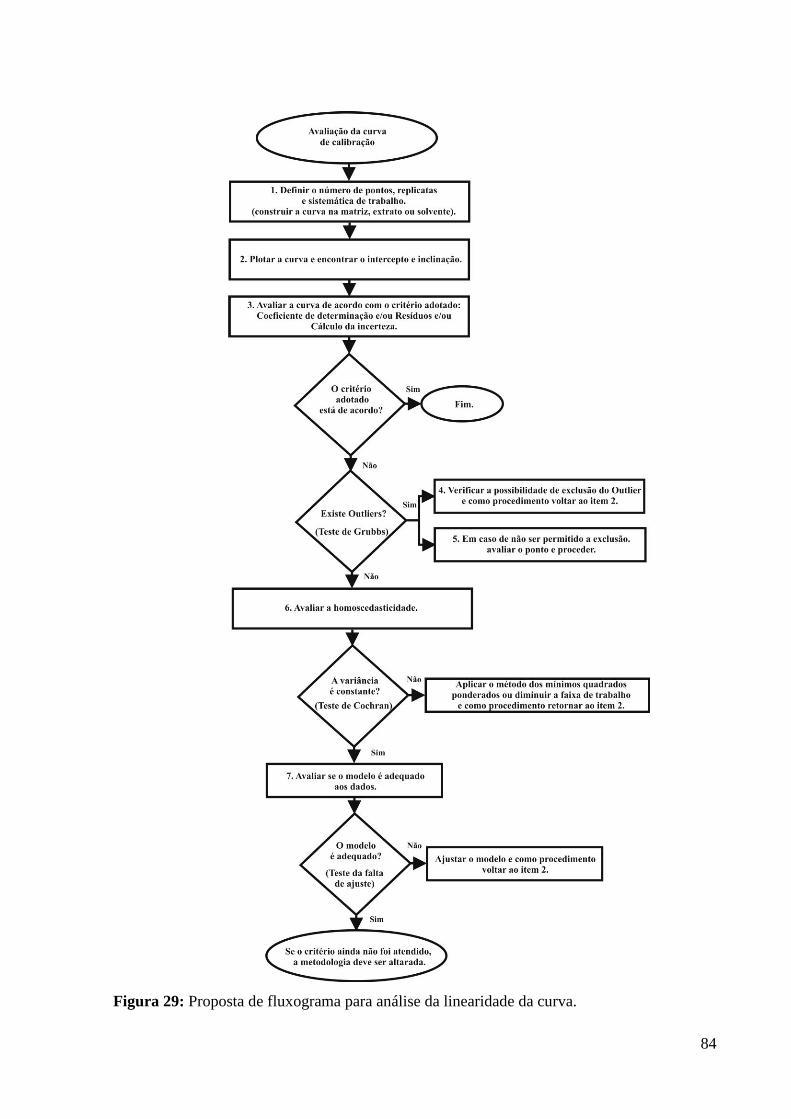
84
Figura 29: Proposta de fluxograma para análise da linearidade da curva.

85
O item 1 do fluxograma orienta o analista na definição da quantidade de pontos a serem
trabalhados, replicatas e a metodologia. Em seguida no item 2 plota-se a curva analítica com o
objetivo de obter os seus respectivos dados, como intercepto b e sua inclinação a.
y = ax + b
.int, erceptooébeanalíticacurvadainclinaçãoaéaOnde
Equação 10: Fórmula para equação da curva linear.
Após este passo, no item 3 deve-se definir qual norma, guia ou documento orientativo
será utilizado, como os citados na Tabela 3, ou outro que seja mais adequado ao método.
Muitos deles utilizam como critério de avaliação o coeficiente de determinação (Equação 11),
os resíduos (Equação 12) e como alternativa proposta pelo trabalho o uso do cálculo da
incerteza (Equação 5), sendo de responsabilidade do analista, definir qual critério é o mais
adequado. A incerteza de um resultado é um parâmetro que descreve o intervalo no qual o
valor da quantidade que esta sendo medido é esperado se encontrar, levando em conta todas
as fontes de desvios (MILLER e MILLER, 2005).
2
22
2
i
i
i
i
i
ii
yyxx
yyxx
r
.cossin
sin,,
analítiaisdosmédiaaéye
analíticoaloéyõesconcentraçdasmédiaaéxtrabalhadaãoconcentraçaéxOnde ii
Equação 11: Fórmula para cálculo do coeficiente de determinação.
100/ˆ% xyyyresíduo ii
.sinˆ analíticacurvapelapreditoaldevaloroéyOnde
Equação 12: Fórmula para cálculo dos resíduos da curva analítica.
Após definido qual parâmetro deve ser avaliado, sugere-se confrontar este parâmetro
com as normas escolhidas, por exemplo, ao se trabalhar com o coeficiente de determinação,

86
muitos guias adotam como critério valores acima de 0,98. O uso dos resíduos também é muito
utilizado, e representado na forma de porcentagem.
Utilizando a incerteza como critério, destaca-se que esta deve ser calculada por um
analista competente. O conceito de incerteza já tem uma profunda influência em muitos
aspectos da química analítica, seja ele prático ou teórico. Atualmente encontra-se difundido
em diferentes temas: validação, amostragem, apresentação de resultados, limites de
especificações, e muitos outros conceitos e atividades rotineiras em laboratórios (JIMÈNEZ-
CHACÓN e ALVAREZ-PRIETO, 2009). Após avaliar um ou todos os parâmetros
(coeficiente de determinação, resíduos e incerteza) deve ser verificado se estes são adequados
ao critério de aceitação ou exigências do cliente.
Caso não atenda algum dos critérios, deve-se inicialmente avaliar a presença de valor
discrepante que interfira na análise de alguns dos parâmetros citados anteriormente, podendo
assim aplicar aos pontos um teste de outlier, como o teste de Grubbs.
S
ySuspeitoValorG
.padrãodesviooéSOnde
Equação 13: Fórmula para cálculo de outlier – Teste de Grubbs.
Se o valor G > Gtabelado, considera-se um outlier. Conforme o item 4, deve se estudar a
possibilidade de descarte do dado considerado outlier. Caso não seja possível, deve-se refazer
o ensaio e retornar ao item 1 do fluxograma, caso seja possível, retornar ao item 2.
Se não houver nenhum ponto discrepante, sugere-se outro teste para verificar a
variância dos resíduos em torno da curva, como sugerido no item 6. Desta forma, um teste de
homoscedasticidade é aplicado para observar se os resíduos se comportam com distribuição
constante (DOQ-CGCRE-008, 2010).
N
i
iS
SC
1
2
2
max
.,,1 max desviomaioroéScalibraçãodepadrõesõesconcentraçdenúmerooéeNaiOnde
Equação 14: Fórmula para cálculo da homoscedasticidade – Teste de Cochran.

87
Se C ≤ Ctabelado então não há diferença significativa entre as variâncias das respostas no
intervalo de concentração do analito estudado e este é considerado homoscedástico. Caso
estas não sejam constante sugere-se aplicar o método dos mínimos quadrados ponderados ou
diminuir a faixa de trabalho e retornar ao ponto 2 do fluxograma 1. Caso seja constante e
ainda os critérios para os parâmetros não tenham sido atendidos, orienta-se mudar o método
de ensaio.
Após esta etapa, uma análise para verificar se o modelo linear é adequado aos dados
deve ser inserido no procedimento como descrito no item 7, como o teste da falta de ajuste
(BRUGGEMANA, QUAPP e WENRICH, 2006).
IIJ
yy
IIJ
SQ
I
i
J
J
iij
ErroErro
1 1
2
2
)(
2
ˆ
2
1
2
2
I
yy
I
SQ
I
i
iifda
fda
2
ˆ
2
1 1
2
2
IJ
yy
IJ
SQ
I
i
J
j
iij
residuoresiduo
2
2
)/()2(
Erro
fda
IIJIF
Sabendo que SQ é a soma dos quadrados, e fda é a falta de ajuste dos pontos e sinal Prev.i é
o sinal previsto na reta no iésimo ponto. Quando Ftab > F(I-2)/(IJ-I) o modelo tem característica
de linearidade, caso contrário outro modelo deve ser proposto. i=1...I, e j=1...J
Equação 15: Fórmula para cálculo a adequação do modelo linear – Teste da falta de ajuste.

88
4.1.4.1. Aplicação da metodologia proposta
Após a análise dos parâmetros é indicado observar se a curva realmente é adequada aos
dados, alguns testes são indicados para esta análise, como teste de outlier, dependência,
adequação ao modelo, tendência etc. No trabalho presente, foi julgado importante para análise
de curva linear os testes de outlier, homoscedasticidade e modelo (Tabela 29).
Foi realizado o teste de Grubbs para observar evidência de outlier nos dados obtidos.
Segundo o teste de Grubbs é calculado o valor G e se este valor for maior que o valor G
tabelado este é considerado Outlier. Considerando uma amostra de 5 replicatas para cada
ponto e utilizando um nível de confiança de 95% temos que o G crítico tabelado é de 1,715.
Foi observado que grande parte dos valores obtidos estão abaixo deste valor crítico, com
excessão do ppD e ppE que indicaram pontos com G = 1,72. Considerando que o valor
discrepante esta no limite critico de trabalho exigido, julgou-se não necessário a sua retirada
já que este ponto não interferiu na curva de forma significante, sendo assim pertinente a
utilização dos dados.
Para análise da Homoscedasticidade dos dados foi aplicado o teste de Cochran para
evidenciar se a distribuição dos resíduos é constante em torno da curva traçada. Para isto, o
teste foi aplicado e o valor encontrado foi comparado com o C crítico tabelado para amostra
de tamanho 5 e nível de confiança de 95%. O valor tabelado foi de 0,506 e todos os valores
para os analitos de trabalho foram abaixo deste valor, evidenciando homoscedasticidade dos
dados, caso estes valores fossem acima, o analista deveria diminuir a faixa de trabalho ou
aplicar o método dos mínimos quadrados ponderado para adequar os dados a curva.
Para análise de modelo, foi utilizado o teste da falta de ajuste. É sabido que há vários
outros testes, como o teste de Mandel, por exemplo, mas julgou-se o teste utilizado mais
simples e adequado ao método utilizado. Desta forma foram calculadas as somas quadradas
residuais, dos erros e da falta de ajuste e foi encontrada a razão F como demonstrado na
fórmula de falta de ajuste. O F tabelado para um nível de confiança 95% é de 3,098, o valor
de Fisher para os analitos trabalhados são todos acima deste valor, com exceção do composto
Mirex que obteve valor abaixo do F crítico, sendo assim, o modelo linear não é o mais
adequado aos dados, ou seja, outro modelo deve ser proposto para os analitos de trabalho,
como por exemplo, a função quadrática.

89
Tabela 29: Avaliação das curvas analíticas lineares do método QuEChERS.
Analito Equação Linear R2 Resíduo Incerteza
1
Outlier2
(Grubbs)
Variância3
(Cochran)
Variância4
(Levene)
Modelo5
(Mandel)
Modelo6
(Teste da Falta
de Ajuste)
HCB y=334331x +
5E+06 0,99 < 19,08% ± 7,00%
G < 1,60
(Não)
C = 0,06
(Homoscedástico)
P-valor = 0,68
L = 0,59
(Homoscedástico)
F = 1,102
(Adequado)
F = 14,83
(outro modelo
deve ser
proposto)
LIN y=53042x +
9E+07 0,98 < 7,53% ± 9,84%
G < 1,53
(Não)
C = 0,14
(Homoscedástico)
P-valor = 0,30
L = 1,31
(Heteroscedástico)
F = 9,573
(Adequado)
F = 29,32
(outro modelo
deve ser
proposto)
ALD y=327525x +
464596 0,99 < 15,56% ± 4,60%
G < 1,49
(Não)
C = 0,06
(Homoscedástico)
P-valor = 0,55
L = 0,79
(Heteroscedástico)
F= 1,314
(Adequado)
F = 14,09
(outro modelo
deve ser
proposto)
HPX y=155267x +
4E+07 0,98 < 12,58% ± 9,33%
G < 1,36
(Não)
C = 0,13
(Homoscedástico)
P-valor = 0,65
L = 0,63
Homoscedástico)
F = -0,31
(Adequado)
F = 98,80
(outro modelo
deve ser
proposto)
tCLD y=175697 +
3E+07 0,98 < 12,60% ± 8,33%
G < 1,67
(Não)
C = 0,15
(Homoscedástico)
P-valor = 0,62
L = 0,68
(Heteroscedástico)
F = -0,82
(Adequado)
F = 115,99
(outro modelo
deve ser
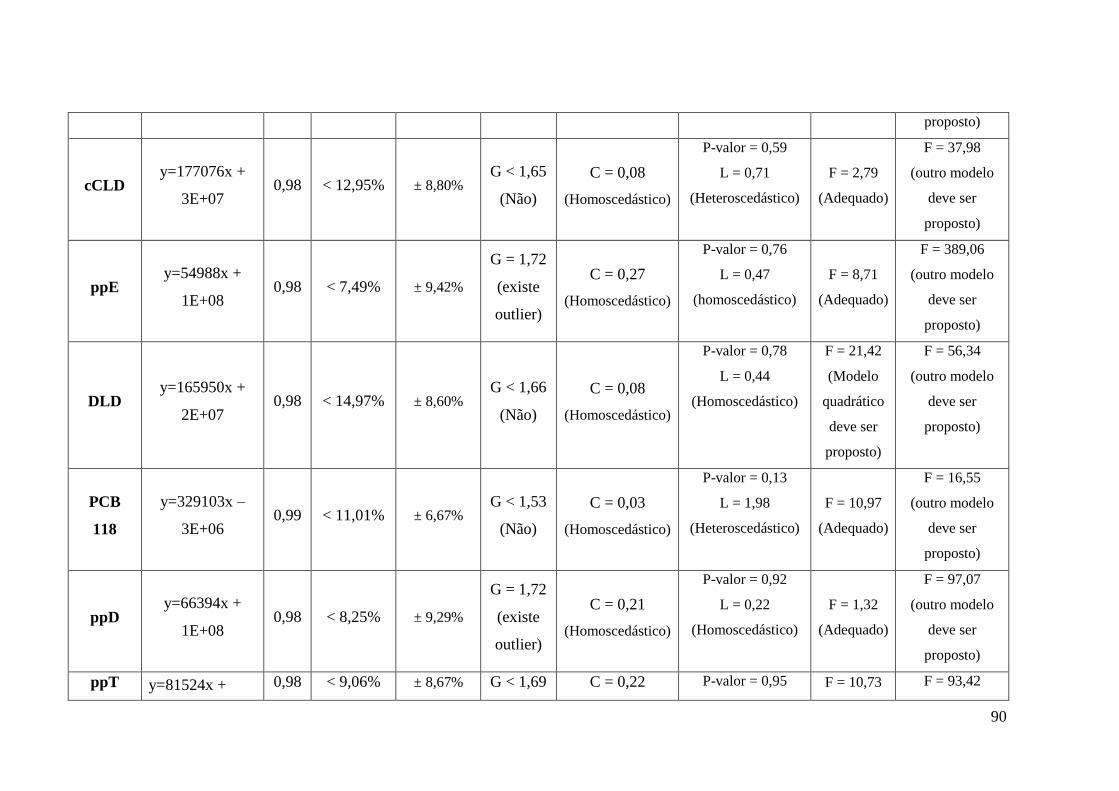
90
proposto)
cCLD y=177076x +
3E+07 0,98 < 12,95% ± 8,80%
G < 1,65
(Não)
C = 0,08
(Homoscedástico)
P-valor = 0,59
L = 0,71
(Heteroscedástico)
F = 2,79
(Adequado)
F = 37,98
(outro modelo
deve ser
proposto)
ppE y=54988x +
1E+08 0,98 < 7,49% ± 9,42%
G = 1,72
(existe
outlier)
C = 0,27
(Homoscedástico)
P-valor = 0,76
L = 0,47
(homoscedástico)
F = 8,71
(Adequado)
F = 389,06
(outro modelo
deve ser
proposto)
DLD y=165950x +
2E+07 0,98 < 14,97% ± 8,60%
G < 1,66
(Não)
C = 0,08
(Homoscedástico)
P-valor = 0,78
L = 0,44
(Homoscedástico)
F = 21,42
(Modelo
quadrático
deve ser
proposto)
F = 56,34
(outro modelo
deve ser
proposto)
PCB
118
y=329103x –
3E+06 0,99 < 11,01% ± 6,67%
G < 1,53
(Não)
C = 0,03
(Homoscedástico)
P-valor = 0,13
L = 1,98
(Heteroscedástico)
F = 10,97
(Adequado)
F = 16,55
(outro modelo
deve ser
proposto)
ppD y=66394x +
1E+08 0,98 < 8,25% ± 9,29%
G = 1,72
(existe
outlier)
C = 0,21
(Homoscedástico)
P-valor = 0,92
L = 0,22
(Homoscedástico)
F = 1,32
(Adequado)
F = 97,07
(outro modelo
deve ser
proposto)
ppT y=81524x + 0,98 < 9,06% ± 8,67% G < 1,69 C = 0,22 P-valor = 0,95 F = 10,73 F = 93,42

91
1E+08 (Não) (Homoscedástico) L = 0,18
(Homoscedástico)
(Adequado) (outro modelo
deve ser
proposto)
opT y=61726x +
9E+07 0,98 < 8,86% ± 8,85%
G < 1,70
(Não)
C = 0,17
(Homoscedástico)
P-valor = 0,97
L = 0,13
(Homoscedástico)
F = 14,15
(Adequado)
F = 36,19
(outro modelo
deve ser
proposto)
PCB
180
y=502140x –
4E+06 0,99 < 10,58% ± 3,51%
G < 1,69
(Não)
C = 0,04
(Homoscedástico)
P-valor = 0,49
L =0,90
(Heteroscedástico)
F = 3,95
(Adequado)
F = 4,03
(outro modelo
deve ser
proposto)
MRX y=267472x –
2E+06 0,99 < 6,60% ± 3,40%
G < 1,68
(Não)
C = 0,03
(Homoscedástico)
P-valor = 0,56
L = 0,76
(Heteroscedástico)
F = -1,29
(Adequado)
F = 1,05
(Adequado)
1 A incerteza dos analitos foram calculado sobre o menor ponto da curva analítica.
2 G tabelado para amostra de tamanho 5 e nível de confiança de 95% é igual a 1,715.
3 C tabelado para amostra de tamanho 5 e nível de confiança de 95% é igual a 0,506.
4 Quando o P-valor do teste é maior que o valor critico de Levene (L) para o nível de significância escolhido, não deve-se rejeitar a hipótese nula de igualdade das variâncias.
5 F tabelado para amostra de tamanho 5, graus de liberdade 1 e 2 e nível de confiança de 95% é igual a 18,513.
6 F tabelado para amostra de tamanho 5, graus de liberdade 3 e 20 e nível de confiança de 95% é igual a 3,098.
Com esta análise de curva pode-se observar os pontos e modelos que devem ser melhorados e consequentemente os caminhos mais
coerentes para se ter uma boa análise da curva analítica. Foi traçado uma curva linear como pretendido pelo fluxograma de trabalho e, por
conseguinte, a análise de resíduos, coeficiente de determinação e incerteza da curva. Os resíduos estiveram todos dentro do que é exigido pelo
guia de trabalho, a SANCO 12495:2011, os coeficientes de determinação estiveram todos acima de 0,98, mostrando boa correlação entre os

92
dados, a incerteza da curva esta dentro do pretendido, não influenciando a incerteza expandida acima do que é exigido, sendo assim satisfatória
para o método. Na aplicação dos testes para analisar se os dados estão de acordo com o modelo utilizado, trabalhou-se com o teste de outlier
(teste de Grubbs) em que foi evidenciado outlier em dois compostos (ppD e ppE) que tiveram um ponto das replicatas fora da estatística
trabalhada. No entanto, considerando que o valor discrepante esta no limite critico de trabalho exigido, julgou-se não necessário a sua retirada já
que este ponto não interferiu na curva de forma significante.
Para o teste de variância, foi utilizado o teste proposto pelo trabalho (teste de Cochran) e um teste para comparação, também muito
utilizado na literatura (teste de Levene), observando algumas disparidades entre os resultados. Com o teste de Cochran observa-se que todos os
dados são considerados homoscedásticos, ou seja, as variâncias são similares e constantes, já com o teste de Levene, observa-se que alguns dados
foram caracterizados como heteroscedásticos (LIN, ALD, cCLD, tCLD, PCB 118, PCB 180 e MRX). Com isso o analista deve analisar o rigor
que pretende para o trabalho, e se os dados forem considerados heteroscedásticos, deve-se diminuir a faixa de trabalho para atingir a
homoscedasticidade ou utilizar o método dos mínimos quadrados ponderados.
O teste do modelo foi utilizado, e assim como para o teste de variância, foi utilizado dois testes, o proposto pelo trabalho, o teste da falta
de ajuste e o teste de Mandel, como teste comparativo. Observa-se a não adequação do modelo para os compostos com o teste da falta de ajuste
com excessão do Mirex, sendo assim necessário propor um outro modelo, já o teste de Mandel, apresentou um composto em que o melhor
modelo seria a função quadrática em vez da função linear (DLD).
Desta forma, as ferramentas apresentadas evidenciam uma análise critica dos dados para a curva linear, sendo imprescindível sua
utilização para um trabalho dentro dos moldes da qualidade.

93
4.2. Aplicação das ferramentas da qualidade para análise de organoclorados em
pescado por RSE
4.2.1. Planejamento experimental
Para desenvolvimento do planejamento experimental da fase de PDMS, foi utilizado o
molde pré-estabelecido na Figura 16, e assim produzido várias barras de SBSE para obter-se
respostas de recuperações de um composto conhecido com os fatores citados na parte
experimental. Assim foram produzidas 8 barras de SBSE como destacado na Tabela 30 com
seu fator de Recuperação/massa PDMS na extração do Antraceno, que possui um Log KO/W =
4,45 e peso molecular de 178 Daltons. Foi utilizado procedimento descrito na literatura para
extração deste composto (BARCO-BONILA et al., 2011). Para quantificação da metodologia
foi construida uma curva de calibração com 7 pontos, que demonstrou um r2 de 0,994
mostrando correlação linera entre concentração e o sinal analítico.
Figura 30: Curva de calibração do método com analito Antraceno com 7 pontos.
A Tabela 30 representa as relações Recuperação/massa PDMS com cada experimento.
Foi realizada esta razão, pois na produção de cada barra de SBSE houve variação da massa de
PDMS que ficava na fase, então sabendo que a recuperação é proporcional ao volume de fase,
realizou-se este fator.
y = 49,308x + 187,42 R² = 0,994
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
0 50 100 150 200 250 300 350

94
Tabela 30: Recuperação de 8 barras de SBSE.
Experimentos Recuperação (%) Massa PDMS Recuperação/massa PDMS
1 69,40 0,02960 2344
2 88,28 0,02450 3603
3 66,63 0,02650 2514
4 56,81 0,02960 1919
5 63,34 0,02400 2639
6 78,70 0,03020 2605
7 73,50 0,02390 3075
8 71,51 0,03090 2314
Com as respostas obtidas foram multiplicados os fatores com seus respectivos
resultados para análise dos fatores significativos para a produção das fases de PDMS, como
descrito na Tabela 30. É importante ressaltar que os experimentos foram realizados de forma
aleatória para não ter-se tendências nas respostas de recuperação.
Tabela 31: Cálculo dos efeitos de primeira (1,2 e 3), segunda (12,13 e 23) e terceira ordem
(123).
Experimentos
Número de
efeitos 1 2 3 4 5 6 7
Interações 1 2 3 12 13 23 123
1
Níveis
multiplicado
pela
recuperação
-2344 -2344 -2344 2344 2344 2344 -2344
2 3603 -3603 -3603 -3603 -3603 3603 3603
3 -2514 2514 -2514 -2514 2514 -2514 2514
4 1919 1919 -1919 1919 -1919 -1919 -1919
5 -2639 -2639 2639 2639 -2639 -2639 2639
6 2605 -2605 2605 -2605 2605 -2605 -2605
7 -3075 3075 3075 -3075 -3075 3075 -3075
8 2314 2314 2314 2314 2314 2314 2314
Somatório -130 -1369 253 -2580 -1457 1658 1126
Efeito -32 -342 63 -645 -364 414 281

95
Tratando a verificação dos dados do planejamento observa-se que nem um dos fatores
são considerados signifivos para o nível de confiança de 95% e 4 graus de liberdade, que
obteve um t crítico de 864. Observa-se que todos os efeitos estão abaixo deste valor, não
sendo assim significativo para a produção da fase nos intervalos trabalhados. No entanto sabe-
se que há uma condição de trabalho que mostrou maior rendimento de recuperação nos
intervalos trabalhos: temperatura de mufla a 200ºC; sem tempo de espera em temperatura
ambiente; e tempo de mufla 60 minutos, correspondente ao experimento 2.
Figura 31: Gráfico de efeito contra probabilidade Z. Considerando como resposta o fator
recuperação/massa PDMS.
Foi construído o gráfico de pareto no Software Statistica 7 para validação da Figura 31,
evidenciando os valores dos efeitos e que nenhum deles é maior que o ponto critíco calculado,
sendo assim nenhum dos fatores teve significância sobre o processo de produção da fase,
tanto no nível baixo quanto nos níveis altos, mostrando robustez da metodologia de produção
com os fatores trabalhados.
-2
-1,5
-1
-0,5
0
0,5
1
1,5
2
-800,00 -600,00 -400,00 -200,00 0,00 200,00 400,00 600,00

96
Pareto Chart of Effects; Variable: Rec/Massa PDMS
2**(3-0) design
DV: Rec/Massa PDMS
-32,75
63,25
281,25
-342,75
-364,25
414,75
-645,25
-100 0 100 200 300 400 500 600 700 800
Effect Estimate (Absolute Value)
(1)temperatura
(3)tempo T ambiente
1*2*3
(2)tempo mufla
1by3
2by3
1by2
Figura 32: Gráfico de pareto evidenciando os efeitos das interações de 1º, 2º e 3º ordem.
Desta forma pode-se identificar robustez no método de produção das barras de SBSE
(desvio padrão na recuperação de 13%), já que nem um dos parâmetros trabalhados teve
influência significativa na extração do analito nas faixas de trabalho, esta condição foi
analisada também por Microscopia de Varredura Eletrônica (MEV) para observar se houve
alteração significativa na superfície do polímero, o que mostrou ter forma padrão.
Tabela 32: Resultados de MEV dos 8 experimentos realizados. Com ampliação de 150, 1000
e 5000 vezes.
Experimentos 150 x (100 µm) 1000x (10 µm) 5000x (1 µm)
1
2

97
A análise MEV evidenciou que o processo de cura do polímero PDMS não é perfeito, o
processo de produção deixa alguma imperfeições na superfície do polímero, como buracos e
ranhuras, evidenciado na resolução de 150x, o que foi observado em todas barras de SBSE,
isso se deve provavelmente a imperfeições do molde. Apesar disso, não foi evidenciado
nenhuma influência destas imperfeições.
3
4
5
6
7
8

98
5. Conclusão
Como pode ser observado, as ferramentas da qualidade são adequadas na otimização de
um método para ser operado em sua melhor condição (obtido com a aplicação do
planejamento experimental) e evidenciar se este método esta apto ou não para atender a
critérios estabelecidos por guias de validação. Estas ferramentas, como dito anteriormente,
são utilizados conforme decisão do analista, quando julgado ser de importância e influência
no trabalho. No método QuEChERS julgou-se que a seletividade, a recuperação, a linearidade
e a precisão foram representativos, expressando os resultados necessários para uma boa
avaliação e julgamento do método trabalhado. Como resultado, obteve-se boas respostas para
o método QuEChERS, método já bem difundida na literatura. Obteve-se seletividade ótima,
evidenciando este procedimento como trabalho prévio para todo tipo de análise. A
recuperação foi bem estabelecida, com recuperação dentro do especificado (70 e 120% de
recuperação). A precisão apresentou valores de CV abaixo dos 20%, evidenciando adequação
ao uso pretendido do método.
Com isto temos o cálculo da incerteza, como resultado final da metodologia, mostrando
a confiabilidade dos resultados, já que este fator não mostra o erro da metodologia, mas sim, o
quanto preciso a metodologia é com o resultado obtido. Desta forma o método QuEChERS se
mostrou adequado para análise de organoclorados em pescado, com vantagem de ser uma
técnica simples, barata e com vantagens visíveis, como já destacado no estudo atual.
Como proposta do trabalho, e considerando sua importância, foi selecionada a análise
da linearidade de uma forma mais profunda para evidenciar se o modelo linear é o mais
adequado, além de ser o mais utilizado no campo de química analítica, sendo a incerteza da
curva um parâmetro primordial e simples de ser aplicado, podendo ser aplicado como mais
um fator de aceitação da curva. Como foi evidenciado, a curva linear não é o mais adequado
para o método já que os resultados não se apresentaram dentro do esperado, com excessão dos
testes de Levene e Mandel aplicados, que mostraram valores condizentes para a curva linear.
Esta análise como já dito, depende do rigor que o analista pretende aplicar no trabalho, sendo
mais uma análise critica, já que os testes utilizados já são bem difundidos e aplicados.
O método RSE foi desenvolvido com o intuito de desenvolvimento da fase de PDMS,
em que foi bem sucedida, abrindo espaço para trabalhos futuros, como desenvolvimento do
sistema estabelecido, assim como a metodologia de confecção da fase mostrando que a
ferramenta da qualidade planejamento experimental pode ser utilizado em vários campos, seja

99
na otimização de um método bem estabelecido como o QuEChERS, ou no desenvolvimento
de um novo método como o RSE.
Desta forma os resultados obtidos foram muito satisfatórios, atingindo os objetivos
propostos, destacando ferramentas da qualidade as quais são bem difundidas em guias e
normas, mas pouco exploradas em pesquisas e rotinas de trabalho na química analítica,
concluindo assim quanto à importância em aplicar tais ferramentas para agregar valor ao
trabalho além de aumentar a confiabilidade nos resultados obtidos.

100
Apêndice A: Declaração de treinamento no Laboratório Nacional Agropecuário –
LANAGRO – Campinas – SP.

101
Apêndice B: Figuras e gráficos para os analitos de extração individuais e para o conjunto multiresíduo no método QuEChERS.
Analitos Figuras e Gráficos
ALD
Figura dos efeitos Gráfico de Pareto Quadrado
cCLD
Pareto Chart of Effects; Variable: Recuperação
2**(3-0) design
DV: Recuperação
,2487962
,4008573
,8851513
1,009578
1,111833
-1,5039
-6,22493
-1 0 1 2 3 4 5 6 7
Effect Estimate (Absolute Value)
1*2*3
1by2
1by3
(3)C18
(2)PSA
(1)NaCL
2by3
Predicted Means for Variable: Recuperação ALD
2**(2-0) design; MS Pure Error=1,633902
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
81,205 (78,7,83,71)
88,439 (85,93,90,95)
88,542 (86,03,91,05)
83,326 (80,82,85,84)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação cCLD
2**(3-0) design
DV: Recuperação cCLD
,3543692
,4611873
,4850045
1,705233
-1,88086
-4,72879
-7,64399
-1 0 1 2 3 4 5 6 7 8 9
Effect Estimate (Absolute Value)
(3)C18
1*2*3
1by3
1by2
(1)NaCl
2by3
(2)PSA
Predicted Means for Variable: Recuperação cCLD
2**(2-0) design; MS Pure Error=3,446684
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
81,352 (77,71,85,)
86,435 (82,79,90,08)
78,437 (74,79,82,08)
74,062 (70,42,77,71)
-1 1
PSA
-1
1
C1
8

102
DLD
HCB
Pareto Chart of Effects; Variable: Recuperação DLD
2**(3-0) design
DV: Recuperação DLD
,2715585
,6128717
-2,21048
2,782374
-4,01412
-4,62703
-6,58976
-1 0 1 2 3 4 5 6 7 8
Effect Estimate (Absolute Value)
1*2*3
1by3
(1)NaCl
1by2
2by3
(3)C18
(2)PSA
Predicted Means for Variable: Recuperação DLD
2**(2-0) design; MS Pure Error=6,538596
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
68,858 (63,84,73,88)
68,246 (63,23,73,27)
66,283 (61,26,71,3)
57,642 (52,62,62,66)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação HCB
2**(3-0) design
DV: Recuperação HCB
,2163177
,6737609
,7684077
-1,1953
1,560326
2,271846
-5,59704
-1 0 1 2 3 4 5 6 7
Effect Estimate (Absolute Value)
1by2
1*2*3
1by3
(1)NaCl
(3)C18
(2)PSA
2by3
Predicted Means for Variable: Recuperação HCB
2**(2-0) design; MS Pure Error=1,259976
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
82,019 (79,82,84,22)
89,176 (86,97,91,38)
89,888 (87,68,92,09)
85,851 (83,65,88,05)
-1 1
PSA
-1
1
C1
8

103
HPX
LIN
Pareto Chart of Effects; Variable: Recuperação HPX
2**(3-0) design
DV: Recuperação HPX
,3436652
,7388684
1,902136
-1,99002
-2,20809
-4,38223
-5,72747
-1 0 1 2 3 4 5 6 7
Effect Estimate (Absolute Value)
1*2*3
1by3
1by2
(3)C18
(1)NaCl
2by3
(2)PSA
Predicted Means for Variable: Recuperação HPX
2**(2-0) design; MS Pure Error=4,578912
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
78,305 (74,1,82,51)
80,697 (76,5,84,9)
76,96 (72,76,81,16)
70,588 (66,39,74,79)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação LIN
2**(3-0) design
DV: Recuperação LIN
,535101
,994753
-1,05004
-1,38931
1,592826
-3,20042
-12,1443
-2 0 2 4 6 8 10 12 14
Effect Estimate (Absolute Value)
1by3
1*2*3
(3)C18
(1)NaCl
1by2
2by3
(2)PSA
Predicted Means for Variable: Recuperação LIN
2**(2-0) design; MS Pure Error=2,871572
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
78,002 (74,67,81,33)
80,152 (76,83,83,48)
69,058 (65,73,72,38)
64,807 (61,48,68,13)
-1 1
PSA
-1
1
C1
8

104
opT
PCB 118
Pareto Chart of Effects; Variable: Recuperação opT
2**(3-0) design
DV: Recuperação opT
-,002536
,2834472
,4846393
-1,11435
1,995077
-3,28531
-3,89217
-0,5 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5
Effect Estimate (Absolute Value)
1*2*3
(3)C18
1by3
(1)NaCl
1by2
(2)PSA
2by3
Predicted Means for Variable: Recuperação opT
2**(2-0) design; MS Pure Error=2,728497
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
90,034 (86,79,93,28)
94,21 (90,97,97,45)
90,641 (87,4,93,88)
87,032 (83,79,90,28)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação PCB 118
2**(3-0) design
DV: Recuperação PCB 118
-,183058
-,290802
-1,03053
1,520522
-1,59178
1,640742
-4,88151
-0,5 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0 5,5
Effect Estimate (Absolute Value)
1*2*3
1by3
(1)NaCl
1by2
(2)PSA
(3)C18
2by3
Predicted Means for Variable: Recuperação PCB 118
2**(2-0) design; MS Pure Error=1,746031
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
85,325 (82,73,87,92)
91,847 (89,25,94,44)
88,614 (86,02,91,21)
85,374 (82,78,87,97)
-1 1
PSA
-1
1
C1
8

105
PCB 180
ppD
Pareto Chart of Effects; Variable: Recuperação PCB 180
2**(3-0) design
DV: Recuperação PCB 180
-,039583
-,096088
-,958967
-1,16348
1,304283
2,416382
-5,608
-1 0 1 2 3 4 5 6 7
Effect Estimate (Absolute Value)
1by3
1*2*3
(2)PSA
(1)NaCl
1by2
(3)C18
2by3
Predicted Means for Variable: Recuperação PCB 180
2**(2-0) design; MS Pure Error=1,532824
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
84,774 (82,34,87,2)
92,798 (90,37,95,23)
89,423 (86,99,91,85)
86,231 (83,8,88,66)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação ppD
2**(3-0) design
DV: Recuperação ppD
,1753415
,5156549
-1,61335
-1,63689
2,538495
-2,92867
-12,4294
-2 0 2 4 6 8 10 12 14
Effect Estimate (Absolute Value)
1*2*3
1by3
(1)NaCl
(3)C18
1by2
2by3
(2)PSA
Predicted Means for Variable: Recuperação ppD
2**(2-0) design; MS Pure Error=4,671757
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
81,213 (76,97,85,46)
82,505 (78,26,86,75)
71,712 (67,47,75,96)
67,147 (62,9,71,39)
-1 1
PSA
-1
1
C1
8

106
ppE
ppT
Pareto Chart of Effects; Variable: Recuperação ppE
2**(3-0) design
DV: Recuperação ppE
,068163
-,211156
,5013698
1,179916
-1,48561
-1,99518
-3,48608
-0,5 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0
Effect Estimate (Absolute Value)
1*2*3
1by3
(3)C18
1by2
(2)PSA
(1)NaCl
2by3
Predicted Means for Variable: Recuperação ppE
2**(2-0) design; MS Pure Error=2,711088
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
91,747 (88,51,94,98)
95,734 (92,5,98,97)
93,747 (90,51,96,98)
90,763 (87,53,94,)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação ppT
2**(3-0) design
DV: Recuperação ppT
-,096385
,1222756
,5220623
-1,57228
2,324878
-3,5353
-6,7326
-1 0 1 2 3 4 5 6 7 8
Effect Estimate (Absolute Value)
(3)C18
1*2*3
1by3
(1)NaCl
1by2
2by3
(2)PSA
Predicted Means for Variable: Recuperação ppT
2**(2-0) design; MS Pure Error=4,082315
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
89,254 (85,29,93,22)
92,693 (88,73,96,66)
86,057 (82,09,90,02)
82,425 (78,46,86,39)
-1 1
PSA
-1
1
C1
8

107
tCLD
Conjunto
Multiresíduo
Pareto Chart of Effects; Variable: Recuperação tCLD
2**(3-0) design
DV: Recuperação tCLD
,2783488
-,350243
,4230154
1,982598
-1,99025
-4,42442
-11,1707
-2 0 2 4 6 8 10 12 14
Effect Estimate (Absolute Value)
1*2*3
(3)C18
1by3
1by2
(1)NaCl
2by3
(2)PSA
Predicted Means for Variable: Recuperação tCLD
2**(2-0) design; MS Pure Error=4,074108
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
78,102 (74,14,82,06)
82,176 (78,21,86,14)
71,355 (67,39,75,32)
66,581 (62,62,70,54)
-1 1
PSA
-1
1
C1
8
Pareto Chart of Effects; Variable: Recuperação Média Conjunto Multiresíduo
2**(3-0) design
DV: Recuperação Média Conjunto Multiresíduo
,0386974
,1845351
,4531826
-1,56306
1,603434
-4,37674
-4,76287
-0,5 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0 5,5
Effect Estimate (Absolute Value)
(3)C18
1*2*3
1by3
(1)NaCl
1by2
2by3
(2)PSA
Predicted Means for Variable: Recuperação Média Conjunto Multiresíduo
2**(2-0) design; MS Pure Error=2,6268
Model includes: Main effects, 2-way inter.
(95,% confidence intervals are shown in parentheses)
82,681 (79,5,85,86)
87,097 (83,91,90,28)
82,295 (79,11,85,48)
77,957 (74,78,81,14)
-1 1
PSA
-1
1
C1
8

108
Apêndice C: tabela de resíduos das curvas analíticas para os 14 analitos com a técnica
QuEChERS.
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (HCB)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Eixo
Dispersão dos Resíduos % (ALD)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (MRX)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (LIN)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (HPX)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (tCLD)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (cCLD)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (DLD)

109
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (ppE)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (ppD)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (opT)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (ppT)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
síd
uo
s e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (PCB 118)
-20
-15
-10
-5
0
5
10
15
20
0 5 10 15 20 25
Re
s[i
du
os e
m P
orce
nta
ge
m %
Ordem
Dispersão dos Resíduos % (PCB 180)

110
6. Referências
AHADI, A.; PARTOAZAR, A.; ABEDI-KHORASGANI, M.H.; SHETAB-BOUSHEHRI,
S.V.; Comparison of liquid-liquid extraction-thin layer chromatography with solid-
phase extraction-high-performance thin layer chromatography in detection of urinary
morphine; Journal of Biomedical Research, 2011, 25(5):362-367.
AMARAL, H.I.F.; BERG, M.; BRENNWALD, M.S.; HOFER, M.; KIPFER, R.; 13
C/12
C
Analysis of Ultra-Trace Amounts of Volatile Organic Contaminants in Groundwater by
Vacuum Extraction; Environ. Sci. Technol. 2010, 44, 1023–1029.
ANASTASSIADES, M.; LEHOTAY, S. J. Fast and easy Multiresidue method
employing acetonitrile extraction/partition and “dispersive solid-phase extraction” for
the determination of pesticide residues in produce. Journal of AOAC International,
v. 86, n. 2, p. 412-431, 2003.
AOAC OFFICIAL METHOD 2007.01; Pesticide Residues in Foods by Acetonitrile
Extraction and Partitioning with Magnesium Sulfate Gas Chromatography/Mass
Spectrometry and Liquid Chromatography/Tandem Mass Spectrometry; First Action,
2007.
AZZOUZ, A.; BALLESTEROS, E.; Combined microwave-assisted extraction and
continuous solid-phase extraction prior to gás chromatography-mass spectrometry
determination of pharmaceuticals, personal care products and hormones in soils,
sediments and sludge; Science of the Total Environment, 2012.
BALTUSSEN, E.; SANDRA, P.; DAVID, F.; CRAMERS, C.A.; Stir bar sorptive
extraction (SBSE), a novel extraction technique for aqueous samples: Theory and
principles; Journal of Microcolum Separation, v. 11, p. 737, 1999.
BARCO-BONILLA, N.; ROMERO-GONZÁLEZ, R.; PLAZA-BOLAÑOS, P.;
FERNÁNDEZ-MORENO, J.L.; FRENICH, A.G.; VIDAL, J.L.M.; Comprehensive analysis
of polycyclic aromatic hydrocarbons in wastewater using stir bar sorptive extraction
and gas chromatography coupled to tandem mass spectrometry; Analytica Chimica Acta
693 (2011) 62–71.
BARREAK, S.; CREN-OLIVÉ, C.; WIEST, L.; BAUDOT, R.; ARNAUDGUILHEM, C.;
Multi-residue analysis and ultra-trace quantification of 36 priority substances from the
EuropeanWater Framework Directive by GC–MS and LC-FLD-MS/MS in surface
Waters; Talanta 79 (2009) 712–722.
BEIZHEN, H.; WEIHUA, S.; LIPING, X.; TIEFENG, S.; Determination of 33 pesticides in
tea using accelerated solvent extraction/gel permeation chromatography and solid phase
extraction/gas chromatography-mass spectrometry; Chin J Chromatogr, 2008, 26(1): 22–
28.
BELTRAN, J.; LÓPEZ, F.J.; HERNÁNDEZ, F.; Solid-Phase microextration in pesticide
residue analysis – Review; Journal of Chromatography A, v. 885, p. 389-404, 2000.
BELTRÃO, K.; Norte-americanos encerram missão ao Brasil para verificar controles
sanitários; 22 de Setembro de 2010; Disponível em: <

111
http://www.agricultura.gov.br/internacional/noticias/2010/09/norte-americanos-encerram-
missao-ao-brasil-para-verificar-controles-sanitarios > Acesso em 30 de Jan. de 2012.
BRASIL. Ministério da Agricultura, Pecuária e Abastecimento. Portaria Interministerial no
902, de 22 de Setembro de 2008. Diário Oficial da União – Institui a Rede de Laboratórios
de Resíduos e Contaminantes em produtos de origem animal e vegetal destinados ao
consumo direto e indireto. Disponível em <
http://extranet.agricultura.gov.br/sislegisconsulta/consultarlegislacao.do?operacao=visualizar
&id=19099 > Acesso em 30 de Jan. de 2012.
BRITO, N.M.; AMARANTE JR, O. P.; POLESE, L.; RIBEIRO, M. L.; Validação de
métodos analíticos: Estratégia e discussão; Pesticidas: R.Ecotoxicol. e Meio Ambiente,
Curitiba, v. 13, p. 129-146, 2003.
BRUGGEMANA L., QUAPP W., WENRICH R.; Test for non-linearity concerning linear
calibrated chemical measurements; Accreditation Quality Assurance, 2006, 625-631.
CARASEK, E.; CUDJOE, E.; PAWLISZYN, J.; Fast and sensitive method to determine
chloroanisoles in cork using an internally cooled solid-phase microextraction fiber;
Journal of Chromatography A, v. 1138, p. 10-17, 2007.
CARASEK, E.; PAWLISZYN, J.; Screening of Tropical Fruit Volatile Compounds Using
Solid-Phase Microextraction (SPME) Fibers and Internally Cooled SPME Fiber; Journal
of Agricultural and Food Chemistry, v. 54, p. 8688-8696, 2006.
CARMI, J.D.; OLIVARES, I.; GROSSI, P.; LANÇAS, F.M.; Refrigerated Sorptive
Extraction: determination of BTEX in water samples; Journal of Chromatographic
Science (2009), 47(9), 812-816.
CBI - MINISTRY OF FOREIGN AFFAIRS OF THE NETHERLANDS; The Netherlands
legislation: Maximum Residue Levels in fish and fishery products (additional
requirements), 2010.
CCME – Canadian Council of Ministers of the Environment; Canadian Sediment Quality
Guidelines for the Protection of Aquatic Life, 2002; Disponível em <
http://www.ccme.ca/assets/pdf/sedqg_summary_table.pdf > Acesso em 13 de fevereiro de
2012.
CETESB – Companhia Ambiental do Estado de São Paulo; O Gerenciamento de Áreas
Contaminadas no Estado de São Paulo - Cadastro de Áreas Contaminadas e
Reabilitadas no Estado de São Paulo, Novembro de 2009; Disponível em <
http://www.cetesb.sp.gov.br/Solo/areas_contaminadas/texto_areas_cont_nov_09_.pdf >
Acesso em 10 de fevereiro de 2012.
CHEN, Y.; BEGNAUD, F.; CHAINTREAU, A.; PAWLISZYN, J.; Analysis of flavor and
perfume using an internally cooled coated fiber device; Journal of Separation Science, v.
30, p. 1037-1043, 2007.
COMUNIDADE EUROPÉIA - Decisão da Comissão das Comunidades Européias de 12
de Agosto de 2002 (2002/657/CE). Dá execução ao disposto na Directiva 96/23/CE do

112
Conselho relativamente ao desempenho de métodos analíticos e à interpretação de resultados.
Jornal Oficial das Comunidades Européias de 17/08/2002.
COMUNIDADE EUROPÉIA (EC); Regulation (EC) nº 882/2004 of the European
Parliament and of the Council; de 29 de abril de 2004; Disponível em < http://eur-
lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:2004R0882:20060525:EN:PDF >
acessado em 09 de fevereiro de 2012.
CONAMA – Conselho Nacional do Meio Ambiente; Resolução CONAMA No 357, de 17 de
Março de 2005 - Dispõe sobre a classificação dos corpos de água e diretrizes ambientais
para o seu enquadramento, bem como estabelece as condições e padrões de lançamento
de efluentes, e dá outras providências; Disponível em <
http://www.cetesb.sp.gov.br/Agua/praias/res_conama_357_05.pdf > Acesso em 11 de
fevereiro de 2012.
CONAMA – Conselho Nacional do Meio Ambiente; Resolução CONAMA No 396, de 3 de
Abril de 2008 - Dispõe sobre a classificação e diretrizes ambientais para o
enquadramento das águas subterrâneas e dá outras providências; Disponível em <
http://www.cetesb.sp.gov.br/Solo/agua_sub/arquivos/res39608.pdf > Acesso em 11 de
fevereiro de 2012.
CONAMA – Conselho Nacional do Meio Ambiente; Resolução CONAMA No 420, de 28 de
Dezembro de 2009 - Dispõe sobre critérios e valores orientadores de qualidade do solo
quanto à presença de substâncias químicas e estabelece diretrizes para o gerenciamento
ambiental de áreas contaminadas por essas substâncias em decorrência de atividades
antrópicas; Disponível em <
http://www.fundagres.org.br/biossolido/images/downloads/resolucao_420_2009.pdf > Acesso
em 11 de fevereiro de 2012.
CONAMA – Conselho Nacional do Meio Ambiente; Resolução CONAMA Nº 344, de 25 de
Março de 2004 - Estabelece as diretrizes gerais e os procedimentos mínimos para a
avaliação do material a ser dragado em águas jurisdicionais brasileiras, e dá outras
providências.
COSTA, D.L.L.B.; Desenvolvimento e validação de um método analítico para análise
multiresíduo de produtos veterinários em leite bovino através de cromatografia líquida
acoplada à espectrometria de massas. Dissertação apresentada ao Instituto de Química de
São Carlos – USP. 2010.
DERRINGER, G.; SUICH R.; Simultaneous Optimization of Several Response Variables;
Journal of Quality Technology, Vol. 12, pp.214-219, 1980.
DOQ-CGCRE; Guidance on validation of analytical methods; Revision 3, February 2010.
EPA - ENVIRONMENTAL PROTECTION AGENCY.; Guidance for Methods
Development and Methods Validation for the RCRA Program – Phase 2: Formal
Validation. 2006. Disponível em: <
http://www.epa.gov/osw/hazard/testmethods/pdfs/methdev.pdf >. Acesso em: 30 de Jan. de
2012.

113
EPA - SW-846 METHOD 8081B; Organochlorines Pesticides By gas Chromatography;
Revision 2, February 2007; Disponível em <
http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/8081b.pdf > acessado em 09 de
feveriro de 2012.
EPA- UNITED STATES ENVIRONMENTAL PROTECTION AGENCY; Guidance for
Assessing Chemical Contaminant Data for Use in Fish Advisories – Volume 2: Risk
Assessment and Fish Consumption Limits; Third Edition; Office of Science and
Technology, Office of Water, U.S. Environmental Protection Agency, Washington, DC, 2000.
EURACHEM/CITAC; Quantifying Uncertainty in Analytical Measurement; Second
Edition; 2000.
EURACHEM - Sociedade Brasileira De Metrologia (SBM). Primeira Edição Brasileira do
Guia EURACHEM/CITAC: Determinando a incerteza de medição em química analítica.
2º ed. Rio de Janeiro: Sociedade Brasileira de Metrologia, 2000.
EURL – EUROPEAN UNION REFERENCE LABORATORY; Modified QuEChERS-
Method for the Analysis of Chlorothalonil in Fruits and Vegetables, 2011.
FAO/WHO FOOD STANDARDS; Codex Alimentarius; Disponível em <
http://www.codexalimentarius.net/pestres/data/pesticides/index.html > Acessoem 14 de
fevereiro de 2012.
FERNANDES, E.A.D.N.; BACCHI, M.A.; TAGLIAFERRO, F.S.; GONZAGA, C.L.; DE
FRANÇA, E.J.; FAVARO P.C.; FOGAÇA, A.A.; Quality system implementation in a
Brazilian university laboratory. Accred Qual Assur (2006) 10: 594–598.
FERREIRA D.C.; SILVA, C.W.M.; Implementação da ISO 17025 em laboratórios de
instituições de ensino tecnológico: Realidade ou utopia. VI Congresso Brasileiro de
Metrologia 2011. Natal – RN.
GHIASVAND, A.R.; HOSSEINZADEH, S.; PAWLISZYN, J.; New cold-fiber headspace
solid-phase microextraction device for quantitative extraction of polycyclic aromatic
hydrocarbons in sediment ; Journal of Chromatography A, 1124 (2006) 35–42.
GHIASVAND, A. R.; SETKOVA, L.; PAWLISZYN, J.; Determination of flavour profile
in Iranian fragrant rice samples using cold-fibre SPME–GC–TOF–MS; Flavour and
Fragrance Journal, v. 22, p. 377-391, 2007.
GIORDANO, A.; RICHTER, P.; AHUMADA, I.; Determination of pesticides in river
water using rotating disk sorptive extraction and gas chromatography–mass
spectrometry; Talanta 85 (2011) 2425– 2429.
GONZÁLEZ, A.G.; ÁNGELES, M.H.; A practical guide to analytical method validation,
including measurement uncertainty and accuracy profiles. Trends in Analytical
Chemistry, v. 26, n. 3, 2007.
GROSSI, P.; Desenvolvimento e aplicação de técnicas miniaturizadas de preparode
amostras para análises ambientais via GC-MS; Tese apresentado ao Instituto de Química
de São Carlos – USP; 2009.

114
HARRINGTON E.C.; The desirability function, Industrial. Quality Control 21 (1965) 494–
498 apud ISLAM, M.A.; SAKKAS, V.; ALBANIS, T.A.; Application of statistical design
of experiment with desirability function for the removal of organophosphorus pesticide
from aqueous solution by low-cost material; Journal of Hazardous Materials 170 (2009)
230–238.
HE, J.; BALASUBRAMANIAN, R.; KARTHIKEYAN, S.; JOSHI, U.M.; Determination of
semi-volatile organochlorine compounds in the atmosphere of Singapore using
accelerated solvent extraction; Chemosphere 75 (2009) 640–648.
HUANG, Y.; ZHOU, Q.; XIE, G.; Development of micro-solid phase extraction with
titanate nanotube array modified by cetyltrimethylammonium bromide for sensitive
determination of polycyclic aromatic hydrocarbons from environmental water samples;
Journal of Hazardous Materials 193 (2011) 82– 89.
INMETRO - DOC-CGCRE-008. Orientação sobre validação de métodos analíticos.
Revisão 04, Jul. 2011.
INMETRO – Instituto Nacional de Metrologia, Qualidade e Tecnologia <
http://www.inmetro.gov.br/laboratorios/rbc/lista_laboratorios.asp > Acesso em 30 de Jan. de
2012.
JIMÉNEZ-CHACÓN J., ALVAREZ-PRIETO M.; Modelling uncertainty in a
concentration range; Accreditation Quality Assurance (2009) 14: 15–27.
JUNIOR, I.M.; CIERCO, A.A.; ROCHA, A.V.; MOTA, E.B.; LEUSIN, S.; Gestão da
Qualidade; FGV Editora, 9ª edição, 2008.
KHAJEH, M.; Optimization of microwave-assisted extraction procedure for zinc and
copper determination in food samples by Box-Behnken design; Journal of Food
Composition and Analysis 22 (2009) 343–346.
KLEJDUS, B.; LOJKOVÁ, L.; PLAZA, M.; SNÒBLOVÁ, M.; STERBOVÁ, D.;
Hyphenated technique for the extraction and determination of isoflavones in algae:
Ultrasound-assisted supercritical fluid extraction followed by fast chromatography with
tandem mass spectrometry; Journal of Chromatography A, 1217 (2010) 7956–7965.
KOLE, P.L.; MILLERSHIP, J.; MCELNAY, J.C.; Determination of diclofenac from
paediatric urine samples by stir bar sorptive extraction (SBSE)–HPLC–UV technique;
Talanta 85 (2011) 1948– 1958.
LEITE, F.; Validação em análise química; Editora Átomo, 5ª edição, ampliada e atualizada,
2008.
LIN, W.; LI, Z.; Detection and Quantification of Trace Organic Contaminants in Water
Using the FT-IR-Attenuated Total Reflectance Technique; Anal. Chem. 2010, 82, 505–
515.

115
LIU, H.; LIU, Y.;WU, B.; NIAN, H.; CHEN, H.; CHIU, K.; LO, J.; Process sampling
module coupled with purge and trap–GC–FID for in situ auto-monitoring of volatile
organic compounds in wastewater; Talanta 80 (2009) 903–908.
LYNAM, K.; PAH Analyses with High Efficiency GC Columns:Column Selectionand
Best Practices; Agilent Technologies, Inc.; Dísponivel em <
http://www.chem.agilent.com/Library/applications/5990-5872EN.pdf > acessado em 24 de
maio de 2012.
MAURICIO A. Q. et al..; Manual da Garantida da Qualidade – Resíduos e
Contaminantes em alimentos; Ministério da Agricultura, Pecuária e Abastecimento; 1º
Edição; Brasília, 2011.
MELO, L.P.; NOGUEIRA, A.M.; LANÇAS, F.M.; QUEIROZ, M.E.C;
Polydimethylsiloxane/polypyrrole stir bar sorptive extraction and liquid
chromatography (SBSE/LC-UV) analysis of antidepressants in plasma samples;
Analytica Chimica Acta 633 (2009) 57–64.
MILLER J.N., MILLER J.C.; Statistics and Chemometrics for Analytical Chemistry; 5º
ed.; Person Education Limited, 2005.
MINISTRY OF HEALTH, LABOUR AND WELFARE; Positive List System for
Agricultural Chemical Residues in Foods – Japan, 2006; Disponível em <
http://www.mhlw.go.jp/english/topics/foodsafety/positivelist060228/index.html > Acesso em
13 de fevereiro de 2012.
MMUALEFE, L.C.; TORTO, N.; HUNTSMAN-MAPILA, P.; MBONGWE, B.; Headspace
solid phase microextraction in the determination of pesticides in water samples from the
Okavango Delta with gas chromatography-electron capture detection and time-of-flight
mass spectrometry; Microchemical Journal 91 (2009) 239–244.
NBR ISO/IEC 17025:2005; Requisitos gerais para a competência de laboratórios de
ensaio e calibração; 2005.
NETO, B. B.; SCARMINO, I. S.; BRUNS, R. E.; Como Fazer Experimentos; 4ª Edição,
Bookman, 2010.
NGUYEN, T.D.; YU, J.E.; LEE, D.M.; LEE, G.H.; A multiresidue method for the
determination of 107 pesticides in cabbage and radish using QuEChERS sample
preparation method and gas chromatography mass spectrometry; Food Chemistry 110
(2008) 207–213.
OLIVARES, I.R.B. Gestão de Qualidade em Laboratórios. Campinas: Editora Átomo,
2006a.
OLIVARES, I.R.B.; Desenvolvimento, otimização e validação da técnica HS-SPME-
GC/MS para análise de amostras obtidas do Rio Atibaia através da aplicação de uma
sistemática “ISO” para diagnóstico ambiental de áreas contaminadas; Tese apresentada
ao Instituto de Química de São Carlos; 2006b.

116
OLIVARES, I.R.B. Gestão de Qualidade em Laboratórios. Campinas: Editora Átomo,
2009. 146 p. 2ª edição (revisada e ampliada).
OLIVARES, I.R.B.; LOPES, F.A.: Essential steps to providing reliable results using the
analytical quality assurance cycle; Trends in analytical chemistry, Vol. 35, 109-121, 2012.
OLIVEIRA, E.C.; AGUIAR, P.F.; VALIDAÇÃO DA METODOLOGIA DA
AVALIAÇÃO DE INCERTEZA EM CURVAS DE CALIBRAÇÃO MELHOR
AJUSTADAS POR POLINÔMIOS DE SEGUNDO GRAU; Quim. Nova, Vol. 32, No. 6,
1571-1575, 2009.
PAYÁ, P.; ANASTASSIADES, M.; MACK, D.; SIGALOVA, I.; TASDELEN, B.; OLIVA,
J.; BARBA, A.; Analysis of pesticide residues using the Quick Easy Cheap Effective
Rugged and Safe (QuEChERS) pesticide multiresidue method in combination with gás
and liquid chromatography and tandem mass spectrometric detection; Anal Bioanal
Chem (2007) 389:1697–1714.
PAWLISZYN, J.; ARTHUR, C.L.; Solid-phase microextraction with thermal-desorption
using fused-silica optical fibers; Analytical Chemistry, v. 62, p. 2145-2148, 1990.
PEREZ, L. H.; Análise de Resíduos: Atendimento das exigências européias ou embargo
nas exportações de produtos brasileiros de origem animal. Análises e Indicadores do
Agronegócio, volume 2, n. 4. Disponível em: <
http://www.iea.sp.gov.br/out/verTexto.php?codTexto=8922 >. Acesso em: 30 Jan. de 2012.
PIZARRO, C.; GONZÁLEZ-SÁIZ, J.M.; PÉREZ-DEL-NOTARIO, N.; Multiple response
optimisation based on desirability functions of a microwave-assisted extraction method
for the simultaneous determination of chloroanisoles and chlorophenols in oak barrel
sawdust; Journal of Chromatography A, 1132 (2006) 8–14.
REGULATION (EC) Nº 882/2004 OF THE EUROPEAN PARLIAMENT AND OF THE
COUNCIL de 29 de abril de 2004; On official controls performed to ensure the
verification of compliance with feed and food law, animal health and animal welfare
rules. Disponível em < http://eur-
lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:2004R0882:20060525:EN:PDF >
Acesso em 27 de fevereiro de 2012.
RESTEK; Q-sep™ QuEChERS Sample Prep Packets & Tubes ;Disponível em <
http://www.restek.com/quechers > Acesso em 01 de Fevereiro de 2012.
RICHTER, B.E.; JONES, B.A.; EZZEL, J.L.; PORTER, N.L.; Accelerated Solvent
Extraction: A technique for sample preparation; Anal. Chem. 1996, 68, 1033-1039.
RUBERT, J.; SOLER, C.; MAÑES, J.; Evaluation of matrix solid-phase dispersion
(MSPD) extraction for multi-mycotoxin determination in different flours using LC–
MS/MS; Talanta 85 (2011) 206–215.
RUEDA, M.E.; SARABIA, L.A.; HERRERO, A.; ORTIZ, M.C.; Optimisation of a flow
injection system with electrochemical detection using the desirability function

117
Application to the determination of hydroquinone in cosmetics; Analytica Chimica Acta
479 (2003) 173–184.
SANCO - Method Validation and Quality Control Procedures for Pesticide Residues
Analysis in Food and Feed. Document No. SANCO/2007/3131 31/October/2007.
SANCO/12495 - Method Validation and Quality Control Procedures for Pesticide
Residues Analysis in Food and Feed. Document No. SANCO/12495/2011 - 2012.
SANTOS, M.C.H.; BERNARDES, A.T.; SANTOS, L.A.S.; ROCHA, G.M.; SILVA, A.V.F.;
A proposta do INMETRO para a disseminação da metrologia e avaliação da
conformidade na graduação em engenharia nas modalidades presencial e EAD. VI
Congresso Brasileiro de Metrologia 2011 – Natal - RN.
SASANO, R.; HAMADA, T.; KURANO, M.; FURUNO, M.; On-line coupling of solid-
phase extraction to gas chromatography with fast solvent vaporization and
concentration in an open injector liner Analysis of pesticides in aqueous samples; Journal
of Chromatography A, 896 (2000) 41–49.
SCHIAVON, B.; Pesquisa sobre dificuldades operacionais de laboratórios de ensaio após
implantação do sistema de gestão da qualidade ABNT NBR ISO/IEC 17025:2005;
Monografia apresentada ao Instituto de Química de São Carlos – USP. 2011.
SINHA, S.N.; VASUDEV, K.; RAO, M.V.V.; Quantification of organophosphate
insecticides and herbicides in vegetable samples using the ‘‘Quick Easy Cheap Effective
Rugged and Safe’’ (QuEChERS) method and a high-performance liquid
chromatography–electrospray ionisation–mass spectrometry (LC–MS/MS) technique;
Food Chemistry 132 (2012) 1574–1584.
SKARTLAND, L.K.; MJOS, S.A.; GRUNG, B.; Experimental design for modeling
retention patterns and separation efficiency in analysis of fatty acid methyl esters by gas
chromatography-mass spectrometry; Journal of Chromatography A, 1218 (2011) 6823–
6831.
THE FEDERAL AUTHORITIES OF THE SWISS CONFEDERATION; Ordonnance du
DFI sur les substances étrangères et les composants dans les denrées alimentaires –
Switzerland, 1995; Disponível em < http://www.admin.ch/ch/f/rs/8/817.021.23.fr.pdf >
Acesso em 13 de fevereiro de 2012.
SCHIAVON, B.; Pesquisa sobre dificuldades operacionais de laboratórios de ensaio após
implantação do sistema de gestão da qualidade ABNT NBR ISO/IEC 17025:2005;
Monografia apresentada ao Instituto de Química de São Carlos – USP. 2011.
VAN LOCO J., ELSKENS M., CROUX C., BEERNAERT H.; Linearity of calibration
curves: use and misuse of the correlation coefficient; Accreditation Quality Assurance,
2002, 281-285.
WANG, J.; WAN, W.; Application of desirability function based on neural network for
optimizing biohydrogen production process; International Journal of Hydrogen Energy –
34 (2009) 1253 – 1259.

118
WANG, S.; OAKES, K.D.; BRAGG, L.M.; PAWLISZYN, J.; DIXON, G.; SERVOS, M.R.;
Validation and use of in vivo solid phase micro-extraction (SPME) for the detection of
emerging contaminants in fish; Chemosphere 85 (2011) 1472–1480.
WANG, Y.; MCCAFFREY, J.; NORWOOD, D. L.; Recent Advances in Headspace Gas
Chromatography; Journal of Liquid Chromatography & Related Technologies. v. 31, p.
1823-1851, 2008.
WILEY - Total Quality Mangement. Chapter 5. Disponível em: <
http://www.wiley.com/college/sc/reid/chap5.pdf >. Acesso em: 30 de jan. 2012.





























