Embed Size (px)
Citation preview

Spektrometria mas MALDI-TOF/TOF
Warszawa, 16.04.2012
Kazimierz Dąbrowski Katedra Chemii i Technologii Polimerów Wydział Chemiczny PW

Rok Odkrycie Nazwiska badaczy
1911 Zbudowanie pierwszego spektrometru mas Joseph John Thomson
1918 Opracowanie źródła jonów EI Arthur Jeffrey Dempster
1930 Zastosowanie spektrometrii mas w chemii organicznej R. Conrad
1942 Pierwszy sprzedany spektrometr mas Consolidated Energy Corporation
1946 Analizator czasu przelotu (TOF) William E. Stephens
1953 Spektrometr o podwójnym ogniskowaniu E. G. Johnson i A. O. Nier
1953 Kwadrupolowy analizator masy W. Paul i H. Steinwedel
1958 Połączenie spektrometru mas z chromatografem gazowym (GC)
1966 Sekwencjonowanie peptydów przy pomocy spektrometru mas K. Biemann, C. Cone, B. R. Webster i B. P. Arsenault
1966 Jonizacja Chemiczna (CI) B. Munson i F. H. Field
1968 Jonizacja przez Elektrorozpylanie (ESI) Malcom Dole
1981 Metoda jonizacji przez bombardowanie szybkimi atomami (FAB) Michael Barber
1983 Opracowanie metody jonizacji przez desorpcję laserem przy udziale matrycy – MALDI (Nagroda Nobla z Chemii w 2002 roku)
Koichi Tanaka, Michael Karas i Franz Hillenkamp
1984 Pułapka jonowa G. C. Statford, P. E. Kelly, J. E. P. Syka, W. E. Reynolds i J. F. J. Todd
1984 Wykorzystanie elektrorozpylania (ESI) do analizy biopolimerów (Nagroda Nobla w 2002 roku)
Gall Lydia (ZSRR), John Fenn (USA)
Najważniejsze odkrycia w historii spektrometrii mas

Spektrometr masowy – instrument pozwalający na precyzyjny pomiar stosunku masy do ładunku (m/z) analizowanych substancji.
Rozdzielczość spektrometru – wartość liczbowa informująca o możliwości rozróżnienia na widmie masowym pików o zbliżonych masach. W przypadku pojedynczego piku wartość określająca dokładność oznaczenia masy cząsteczkowej (atomowej) substancji analizowanej. Jeśli spektrometr masowy w danym momencie analizy posiada rozdzielczość R=1000 istnieje możliwość rozróżnienia pików o m/z=1000 oraz m/z=1001. Dla izolowanego piku rozdzielczość definiuje jego szerokość połówkową, tzn. dla R=1000 i piku o m/z=1000 stosunek jego wysokości do szerokości w 0,5 wysokości wynosi co najmniej 10 (H/L0,5h>=10)
Jon molekularny – jon obdarzony ładunkiem (ładunkami) powstający w wyniku fragmentacji próbki w źródle jonów
Jon fragmentacyjny – jon powstały w wyniku spontanicznej fragmentacji substancji (np. podczas jonizacji metodą EI) lub uzyskany techniką tandemowej spektrometrii masowej. Dostarcza informacji o strukturze substancji analizowanej.
Addukt - jon powstały poprzez przyłączenie do analizowanej substancji np. jonu sodowego
Proteom - PROTEin complement of the genOME (ogół białek kodowanych przez genom)
Dalton - jednostka masy, dokładnie odpowiada 1,0000 na skali mas atomowych
Dekonwolucja - uzyskanie rzeczywistej masy substancji z widma pików wielokrotnie zjonizowanych
Matryca - niskocząsteczkowe związki organiczne absorbujące promieniowanie lasera.
Podstawowe definicje

Najczęściej stosowane skróty
EI (Electron Impact) - jonizacja elektronami MALDI (Matrix-Assisted Laser Desorption/Ionization) - jonizacja laserem wspomagana matrycą ESI (Electrospray Ionization) - jonizacja przez rozpylanie w polu elektrycznym HPLC (High Performance Liquid Chromatography) - wysokosprawna chromatografia cieczowa) MS/MS (Tandem Mass Spectrometry) - tandemowa spektrometria masowa TOF (Time of Flight Analyser) - analizator czasu przelotu PSD (Post Source Decay) - rozpad poza źródłem jonów m/z - stosunek wartości masy do liczby ładunków DIOS (Desorption/Ionization on Porous Silicon) - desorpcja/jonizacja na porowatym krzemie ICP (Inductively Coupled Plasma) - jonizacja plazmą wzbudzoną indukcyjnie

Idea działania spektrometru mas
• źródło jonów – urządzenie, w którym następuje jonizacja cząsteczek przy użyciu różnorodnych technik, z których część prowadzi do pękania wiązań chemicznych na skutek czego dochodzi do ich podziału na mniejsze fragmenty. Inne techniki powodują tylko naładowanie cząsteczek bez ich fragmentacji,
• analizator – w którym wcześniej powstałe jony ulegają rozdziałowi na podstawie stosunku ich masy do ładunku.
• detektor – urządzenie "zliczające" jony napływające z analizatora.

Jonizacja próbki
Twarda technika jonizacji Jon molekularny typu [M+.] Bogate widmo fragmentacyjne
Łagodna technika jonizacji Pozorny jon molekularny [M+H]+ lub [M-H]-
Brak fragmentacji

Jonizacja próbki cd.


Jonizacja próbki – lasery używane w technice MALDI
Nitrogen laser: pro: well structured energy profile contra: slow (maximum 50Hz)
Nd:YAG laser: pro: fast (up to 1000Hz) contra: Gaussian energy profile (non-structured)
Smartbeam/Smartbeam II (modified Nd:YAG laser): pro: fast (up to 1000Hz) pro: well structured energy profile
A. Holle, A. Haase, M. Kayser, J. Höhndorf, Journal of Mass Spectrometry, 41, 705-716 (2006)

Matryce
Procesy zachodzące pod wpływem impulsu laserowego Absorpcja promieniowania głównie przez materiał matrycy. Odparowanie próbki na głębokość 2-3 l i wyrzucenie strumienia gazów prostopadle do jej powierzchni. Dysocjacja termiczna matrycy. Tworzenie jonów (głównie H+, Na+, K+). Reakcje jonów z badaną substancją i matrycą. Możliwe drogi: - dysocjacja termiczna z utworzeniem pary kation-anion - oderwanie elektronu - oderwanie bądź przyłączenie protonu - przyłączenie kationu bądź anionu

Matryce
Pożądanymi cechami matrycy MALDI są: •dość niska masa cząsteczkowa, co sprzyja łatwemu odparowaniu, ale wystarczająco duża, by odparowanie nie nastąpiło przed pomiarem, np. w czasie przygotowywania próbki; •rozpuszczalność w rozpuszczalniku kompatybilnym z analitem; •kwasowość, by ułatwić protonowanie cząsteczek analitu; •obecność grup polarnych (hydrofilowych) w cząsteczce, co umożliwia rozpuszczanie matrycy w roztworach wodnych; •stabilność w warunkach wysokiej próżni; •wspomaganie jonizacji analitu; •zdolność intensywnej absorpcji promieniowania UV lasera; zwykle wymóg ten spełnia związek, posiadający układ sprzężonych wiązań podwójnych C=C (dlatego często matrycami są pochodne aromatycznych kwasów karboksylowych, często nienasyconych, np. kwasu cynamonowego).

Matryce
Sinapic acid Kwas 3-5-dimetoksy-4-hydroksy cynamonowy
SINA proteiny
Gentisic acid Kwas 2-5-dihroksy benzoesowy
DHB peptydy
2-(4-Hydroxyphenylazo)benzoic acid Kwas 2-(4-hydroksyfenylazo)- benzoesowy
HABA Peptydy, polimery
Dithranol 1,8-Dihydroksyantracen-9(10H)-on
DIT polimery
α-Cyano-4-hydroxycinnamic acid Kwas α-Cyano-4-hroksy cynamonowy
CHCA, α-CHCA Peptydy, lipidy
2,4,6-Trihydroxyacetophenone 2,4,6-trihroksy acetofenon
THAP oligonukleotydy

Tryb liniowy

Tryb liniowy
Jak zwiększyć rozdzielczość? • Pulsed ion extraction – polepszenie ogniskowania jonów • Optymalizacja przygotowania próbki – homogenizacja • Użycie reflektronu

Optymalne przygotowanie próbki

Najczęściej stosowane metody nanoszenia próbki (analitu i matrycy) to:
1. Metoda wysychającej kropli (dried droplet method) – jednowarstwowa: Przyrządza się osobno roztwór próbki i roztwór matrycy w tym samym rozpuszczalniku, lub – jeśli to niemożliwe – w dwóch kompatybilnych; niekiedy jeszcze używa się trzeciego roztworu - środka kationizującego, np. soli metalu (możliwe są różne rozpuszczalniki i stężenie). Wszystkie roztwory miesza się, a uzyskaną mieszaninę (0,5÷1 μl) umieszcza na płytce MALDI (MALDI target) i pozostawia do wyschnięcia na powietrzu. Wadą tej metody jest powolne wysychanie próbki, co może prowadzić do rozdzielenia kryształów matrycy próbki i soli kationizującej. Modyfikacjami tej metody są: zastosowanie odparowywania próżniowego lub odparowywanie w strumieniu ultraczystego azotu. W obu przypadkach otrzymuje się drobniejsze kryształy, lepszą rozdzielczość i powtarzalność oraz intensywność sygnałów.
2. Metoda cienkiej warstwy (thin-layer method) – dwuwarstwowa: Roztwór matrycy w odpowiednim rozpuszczalniku (np. dla CHCA – roztwór nasycony w acetonie) nanosi się na płytkę MALDI i pozwala mu się wyschnąć, otrzymując cienką warstwę matrycy. Następnie 1 μl roztworu analitu nanosi się na wierzch uzyskanej powierzchni matrycy i suszy.
3. Nanoszenie przez rozpylanie. Wariantami tej metody są: osadzanie strumieniem powietrza (air spray deposition) i osadzanie przez elektrosprej (electrospray sample depositon).
4. Metoda mieszania ciał stałych (solid/solid sample preparation): Metoda polega na bardzo dokładnym mieszaniu drobno sproszkowanych matrycy i próbki (bez rozpuszczalnika) i prasowaniu całości w pastylkę. Stosuje się ją dla niektórych poliamidów, nierozpuszczalnych w pospolitych rozpuszczalnikach organicznych

Epot = zeU Ekin = 1/2mv2
zeU = 1/2mv2 v = (2zeU/m)1/2
t = L×(1/2eU)1/2×(m/z)1/2
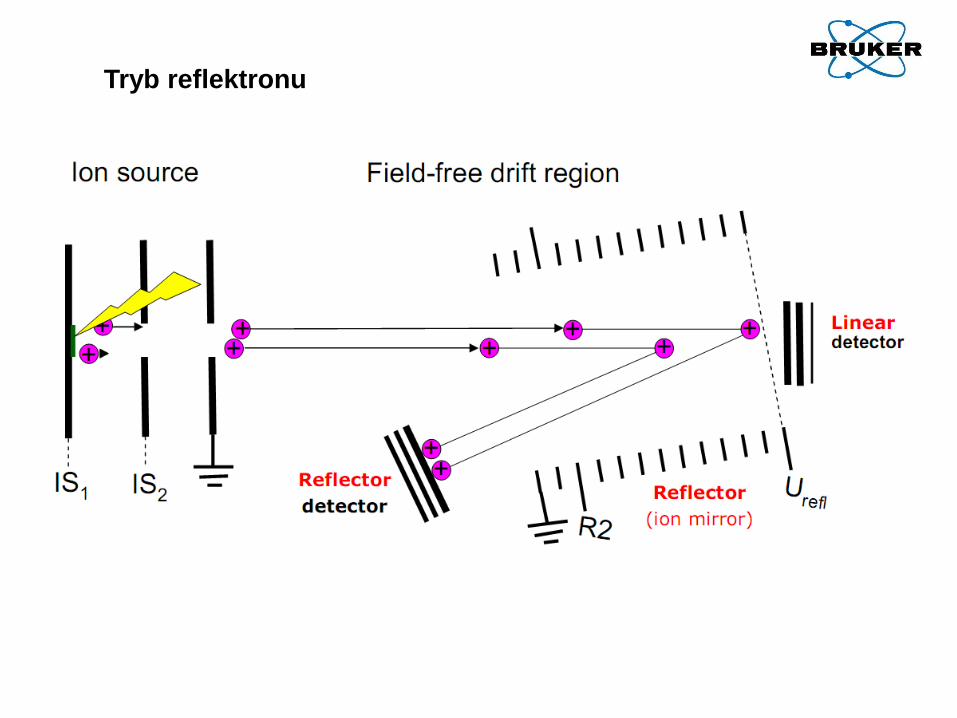
Tryb reflektronu

Tryb reflektronu


Profil izotopowy

Profil izotopowy
C41H69N13O14S [M+H]+: 1000.4880 [M+H]+: 1001.1409
C112H164N29O34S2
[M+H]+: 2524.1510 [M+H]+: 2525.8196
C253H363N55O75S [M+H]+: 5404.6075 [M+H]+: 5407.9984
Masa monoizotopowa Masa średnia

Kalibracja
tof= tdelay + tacc + tdrift
F = E q = M a
tacc = d √(2m/Uze)
d = a/2 tacc2 E = U/d q = z e
tdrift= L √(m/2zeU)
tof=tdelay + d √(2m/Uze) + L √(m/2zeU)
tof=tdelay + (d √(2/Ue)+L √(1/2Ue)) × √(m/z)
t = C0+C1√(m/z)

MALDI–ToF/ToF
Ion path in TOF1 region (linear TOF) Ion path in TOF2 region (reflectorTOF) Ion source1 = MALDI ion source Ion source2 = LIFT re-acceleration cell PCIS = Timed ion gate PLMS = Post LIFT meta stable suppressor

MALDI–ToF/ToF

Montaudo G, J. Polym. Sci. A: Polym. Chem. 34, 1996, 439-447
Za pomocą MALDI można wyznaczyć następujące parametry polimerów: • Mn, Mw, PD • grupy końcowe polimeru • budowę kopolimeru
MALDI–ToF w badaniach polimerów

A6B4C
A7B3C
A8B2C
A9BC
A10C
A7B4C
A8B3C
A9B2C
A10BC
A11C
A8B4C
A9B3C
A10B2C
A11BC
A11C
A B C

Polimery polarne: poliwęglany, politlenki olefin Polikwasy, poliestry, polimery słabo polarne (np. polistyren) Polisacharydy, teflon, poliolefiny
mass discrimination effect

Liu, J.; Loewe, R. S.; McCullough, R. D. Macromolecules1999, 32, 5777 -5785.
MALDI mass spectra of a poly(alkylthiophene) fractionated with acetone, hexanes, methylene chloride, THF, and chloroform


• Badane mogą być związki stałe, ewentualnie ciecze nielotne (ciśnienie w komorze pomiarowej < 10-7 Torr). • Do pomiaru używamy zazwyczaj 5 mg substancji. Próbki można dostarczać albo już odważone (proszę podać masę
próbki), lub w większych pojemnikach. • W razie tzw. wyższej konieczności do pomiaru wystarczy: substancji typu biologicznego (np. peptydu) 0,1 - 10 pmol,
polimeru 50 - 200 pmol. • Można również dostarczyć próbkę w postaci roztworu w dowolnym, ale koniecznie lotnym i niekorodującym
rozpuszczalniku (prosimy o podanie stężenia roztworu !) • Możliwy jest pomiar próbki nierozpuszczalnej, ale wówczas musi być ona w postaci możliwie drobnego proszku.
Należy jednak podkreślić, że wyniki takiego pomiaru będą znacznie gorszej jakości, niż próbek rozpuszczalnych. • Kategorycznie odmawiamy przyjmowania podejrzanych, dymiących cieczy zawierających stężone lotne kwasy (np.
HCI, HNO3) oraz substancji korodujących typu POCI3, wolne aminy, fenole, merkaptany itd. • Aby proces tworzenia jonów przebiegał możliwie wydajnie, próbka powinna być jak najczystsza. W szczególności
nie powinna zawierać: • nadmiaru soli nieorganicznych z grupy litowców i miedziowców, • detergentów !!!, • buforów, zwłaszcza fosforanowych, • substancji silnie alkalizujących (reagują z matrycą),
• W przypadku polimerów, co do których zachodzi podejrzenie, że mogą zawierać frakcje znacznie różniące się masą cząsteczkową ( np. Mn = 1 500 i Mn = 30 000) niezbędne jest wstępne rozdzielenie próbki na frakcje.
• Prosimy o podanie spodziewanej masy cząsteczkowej, bądź przynajmniej zakwalifikowanie próbki do jednego z następujących przedziałów: < 5000; 5000-20000; > 20000
• Do badanej próbki (lub grupy próbek) prosimy dołączyć „Zlecenie wykonania badania MALDI-ToF” (druk dostępny na stronie: www.ch.pw.edu.pl/~maldi )
Wskazówki dla PT Klientów


• M. Karas, D. Bachmann, F. Hillenkamp; Analytical Chemistry, 57, 2935-2939 (1985)
• K. Tanaka, H. Waiki, Y. Ido, S. Akita, Y. Yoshida, T. Yoshida; Rapid Communications in Mass Spectrometry, 2, 151-153 (1988)
• R. C. Beavis, B. Chait, K.G. Standing; Rapid Communications in Mass Spectrometry, 3, 233-237 (1989)
• M. Karas, M. Glückmann, J. Schäfer; Journal of Mass Spectrometry, 35, 1-12, (2000)
• R. Zenobi and R. Knochenmuss; Mass Spectrometry Reviews, 17, 337-366 (1998)
• G. Montaudo, M. S. Montaudo, and F. Samperi, “ Mass Spectrometry of Polymers ”, ed. G. Montaudo and R. P. Lattimer, 2002 , CRC Press, Boca Raton, FL, 419.
Polecana literatura

Dziękuję za uwagę…