Embed Size (px)
Citation preview

Spin-schaltbare Eisen(II)-Komplexe für die
Verankerung auf Oberflächen
vorgelegt von
Diplom-Chemiker
Marco Haryono
aus Berlin
Von der Fakultät II – Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
—Dr. rer. nat.—
genehmigte Dissertation
Promotionsausschuss:
Vorsitzende: Prof. Dr. rer. nat. Regine von Klitzing
1. Gutachter: Prof. Dr. rer. nat. Andreas Grohmann
2. Gutachter: Prof. Dr. rer. nat. Berthold Kersting
Tag der wissenschaftlichen Aussprache: 01.12.2008
Berlin 2008
D 83

Abstract
Die Arbeit beschreibt Synthese und Charakterisierung neuer oberflächenaktiver Liganden und Eisen(II)-Spincrossover (SCO)-Verbindungen, sowie die Präparation und Charakterisierung von selbstorganisierten Monolagen (SAMs) einiger der Verbindungen auf Oberflächen. Der neue Ligand 2,6-Bis(1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin (6) wurde in einer fünfstufigen Synthese, ausgehend von Citrazinsäure, hergestellt, und zwar über folgende Zwischenstufen: 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-carbonsäure (2), Methyl-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxylat (3), Ethyl-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxylat (4) und 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methanol (5). Die Gasphasenabscheidung von 6 auf Au(111) ergab Monolagen geringer Ordnung. Der FeII-Komplex [Fe(6)2](BF4)2 (26) wurde aus 6 synthetisiert. 26 kokristallisiert unter geeigneten Bedingungen in Form zweier polymorpher Verbindungen: der High Spin (HS)-Verbindung 26a und der SCO-Verbindung 26b mit abruptem thermischen Spinübergang bei T1/2 = 272 K. Beide Verbindungen wurden charakterisiert und mittels Röntgenstrukturanalyse, SQUID-Magnetometrie und Evans-NMR-Spektroskopie auf den temperaturabhängigen Spinzustand untersucht. Ungeordnete Monolagen von 26 wurden bei Präparation aus Lösungen auf Au(111) erhalten. Der neue Ligand N-(2-Mercaptoethyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid (10) wurde aus 2 hergestellt. Im Falle von 10 ergab die Präparation aus Lösungen auf Au(111) hochkristalline Monolagen, sofern das Substrat mit PTCDA (3,4,9,10-Perylentetracarbonsäure-Dianhydrid) vorbeschichtet war. Der FeII-Komplex [Fe(10)2](BF4)2 (25) wurde mit Ligand 10 synthetisiert und mittels SQUID-Magnetometrie auf den temperaturabhängigen Spinzustand untersucht. Ungeordnete Schichten von 25 auf Au(111) resultierten bei der Präparation aus Lösung. Die neuen FeII-HS-Komplexe [Fe(3)2](BF4)2 und [Fe(4)2](BF4)2 wurden unter Verwendung der Liganden 3 bzw. 4 synthetisiert. Die neuen oberflächenaktiven Liganden N-(4-(4-Mercaptophenyl)phenyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid (12) und (2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methyl-11-mercaptoundecanoat (14) wurden ausgehend von 2 bzw. 5 synthetisiert. Zur Synthese des neuen Liganden 1-(6-(4-Carboxy-1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin-2-yl)-1H-pyrazol-4-carbonsäure (22) wurde eine achtstufige Syntheseroute ausgehend von Citrazinsäure entwickelt, wovon sieben Stufen abgeschlossen wurden. Die synthetisierten Verbindungen wurden mit Hilfe von 1H- und 13C-NMR-Spektroskopie, IR-Spektroskopie, Massenspektrometrie und/oder Elementaranalyse charakterisiert. Von 3, 23, 24, 25, 26a und 26b wurden Einkristall-Röntgenstrukturanalysen angefertigt. Temperaturabhängige Messungen der magnetischen Suszeptibilität wurden für 23, 25, 26a und 26b mittels SQUID-Magnetometrie durchgeführt. Die auf Au(111)-Oberflächen präparierten Belegungen von 6, 10, 25 und 26 wurden mittels NEXAFS-, XPS- und STM-Messungen charakterisiert.

Die vorliegende Arbeit wurde am Institut für Chemie der Technischen Universität Berlin unter Anleitung von Herrn Prof. Dr. Andreas Grohmann im Zeitraum von September 2005 bis Oktober 2008 angefertigt. An dieser Stelle möchte ich mich herzlich bedanken bei Herrn Prof. Dr. Andreas Grohmann für die interessante Aufgabenstellung, die umfangreiche Betreuung sowie die zahlreichen Anregungen und das große Interesse, mit dem er meine Arbeit begleitet hat; Herrn Prof. Dr. Berthold Kersting für die Übernahme des Zweitgutachtens, sowie Frau Prof. Dr. Regine von Klitzing für die Übernahme des Prüfungsvorsitzes; den Angestellten der TU Berlin: Herrn Dr. Heinz-Jürgen Kroth und Herrn Manfred Detlaff für die Aufnahme der NMR-Spektren, Frau Dr. Elisabeth Irran und Frau Marina Borowski für die Bestimmung von Röntgenstrukturen, Frau Alice Stöckel für die Aufnahme der Massenspektren, Frau Sigrid Imme für die Messung der IR-Spektren und die Durchführung der Elementaranalysen; Herrn Dr. Frank W. Heinemann für die Bestimmung von Röntgenstrukturen; Herrn Dr. Manfred Buck und Herrn Cai Shen für die Einführung in die Rastertunnel-mikroskopie, die Aufnahme von STM-Bildern und die in St. Andrews gewährte Gastfreundschaft; Herrn Prof. Dr. Wolfgang Kuch, Herrn Matthias Bernien, Herrn David Ball, sowie Herrn Dr. habil. Michael Zharnikov für die Durchführung von NEXAFS- und XPS-Analysen; Herrn Prof. Dr. Paul Müller, Herrn Dr. Konstantin Petukhov und Herrn Klaus Gieb für die Durchführung der SQUID-Messungen; Herrn Prof. Dr. Helmut Schwarz und Frau Dr. Maria Schlangen für die Kooperation in einer weiteren Forschungsarbeit; und nicht zuletzt danke ich allen Mitgliedern des Arbeitskreises Grohmann, darunter meinen Laborkollegen Sophie Hain, Holger Kämpf und Thomas Wagner für ihre Ratschläge und stete Hilfsbereitschaft bei chemischen Fragestellungen und die angenehme Arbeitsatmosphäre.

Inhalt:
1 Einleitung .......................................................................................................................1
1.1 Theoretischer Hintergrund......................................................................................1
1.1.1 Eisen in der Natur ...............................................................................................1
1.1.2 Spincrossover .....................................................................................................2
1.1.3 Self-Assembled Monolayers...............................................................................6
1.2 Charakterisierungsmethoden ..................................................................................7
1.2.1 SCO ....................................................................................................................7
1.2.2 SAMs..................................................................................................................8
1.3 Stand der Forschung ..............................................................................................11
1.3.1 Spincrossover und Kooperativität ....................................................................11
1.3.2 Spincrossover im Ensemble .............................................................................14
1.3.3 Spincrossover in dünnen Schichten..................................................................15
2 Eisen(II)-Komplexe mit Schwefelanker ............................................................18
2.1 Aufgabenstellung ....................................................................................................18
2.2 Synthese terdentater Liganden .............................................................................22
2.2.1 2,6-Bis(1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin (6)............................22
2.2.2 2,6-Bis(1H-pyrazol-1-yl)-4-(isothiocyanatomethyl)pyridin (8).......................29
2.2.3 N-(2-Mercaptoethyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid (10) .....32
2.2.4 N-(4-(4-Mercaptophenyl)phenyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-
carboxamid (12)................................................................................................37
2.2.5 (2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methyl-11-mercaptoundecanoat (14) .40
2.2.6 Versuchte Synthese von 1-(6-(4-Carboxy-1H-pyrazol-1-yl)
-4-(thiocyanatomethyl)pyridin-2-yl)-1H-pyrazol-4-carbonsäure (22) .............42
2.3 Synthese von Eisenkomplexen...............................................................................54
2.3.1 [Fe(3)2](BF4)2 (23)............................................................................................55
2.3.2 [Fe(4)2](BF4)2 (24)............................................................................................60
2.3.3 [Fe(10)2](BF4)2 (25)..........................................................................................63
2.4 Zusammenfassung ..................................................................................................70
2.5 Experimenteller Teil...............................................................................................71
2.5.1 Allgemeines ......................................................................................................71

2.5.2 Synthesen..........................................................................................................72
3 Polymorphie in [Fe(6)2](BF4)2 ...............................................................................94
3.1 Einleitung ................................................................................................................94
3.2 Konzept....................................................................................................................94
3.3 Ergebnisse und Diskussion ....................................................................................95
3.3.1 Synthese von [Fe(6)2](BF4)2 (26) .....................................................................95
3.3.2 Polymorph a (26a): High Spin .........................................................................96
3.3.3 Kokristallisation bei Raumtemperatur............................................................101
3.3.4 Polymorph b (26b): SCO................................................................................103
3.3.5 Vergleich der Koordinationsgeometrien der Polymorphe a und b .................109
3.3.6 Temperaturabhängige 1H-NMR-Spektroskopie .............................................111
3.3.7 Supramolekulare Strukturen der Polymorphe a und b ...................................114
3.4 Zusammenfassung ................................................................................................118
3.5 Experimentalteil ...................................................................................................119
3.5.1 Allgemeines ....................................................................................................119
3.5.2 Synthese..........................................................................................................120
4 Self-assembled Monolayers ..................................................................................124
4.1 Einleitung ..............................................................................................................124
4.2 SAM von Ligand 10 auf Au(111) ........................................................................125
4.2.1 STM................................................................................................................125
4.2.2 NEXAFS/XPS ................................................................................................130
4.2.3 Zusammenfassung ..........................................................................................135
4.3 SAM von Ligand 6 auf Au(111) ..........................................................................135
4.3.1 STM................................................................................................................135
4.3.2 NEXAFS/XPS ................................................................................................137
4.3.3 Zusammenfassung ..........................................................................................138
4.4 Volumenprobe von [Fe(5)2](BF4)2 .......................................................................139
4.5 SAM von [Fe(10)2](BF4)2 (25) auf Au(111).........................................................140
4.5.1 NEXAFS/XPS ................................................................................................140
4.5.2 STM................................................................................................................142
4.5.3 Zusammenfassung ..........................................................................................142
4.6 SAM von [Fe(6)2](BF4)2 (26) auf Au(111)...........................................................143
4.6.1 NEXAFS/XPS ................................................................................................143

4.6.2 STM................................................................................................................146
4.6.3 Zusammenfassung ..........................................................................................147
4.7 Zusammenfassung ................................................................................................148
4.8 Experimentalteil ...................................................................................................149
5 Zusammenfassung und Ausblick .......................................................................150
5.1 Zusammenfassung ................................................................................................150
5.2 Ausblick .................................................................................................................157
6 Anhang ........................................................................................................................158
6.1 Verwendete Abkürzungen ...................................................................................158
6.2 Publikationen ........................................................................................................161
7 Literaturverzeichnis ...............................................................................................163

1 Einleitung 1
1 Einleitung In der Chemie der Übergangsmetalle nimmt Eisen durch seine Relevanz für die
bioanorganische Chemie eine prominente Rolle ein. Die vorliegende Arbeit konzentriert sich
auf die Nutzbarmachung von Eisen(II)-Spincrossover(SCO)-Komplexen in funktionalisierten
Oberflächen. Ein Fernziel des Projekts ist der Einsatz derartiger Oberflächen in der
Datenspeicherung.
1.1 Theoretischer Hintergrund
1.1.1 Eisen in der Natur
Eisenkomplexe stellen die aktiven Zentren in einer Vielzahl natürlicher Verbindungen. Sie
kommen zumeist in Proteinen vor, welche für Sauerstofftransport und katalytische Prozesse
essentiell für lebende Organismen sind. Die Funktion des Metalloproteins Hämoglobin (Hb)
als Sauerstoffüberträger beruht unter anderem auf dem SCO-Effekt. Hämoglobin besteht aus
vier Einheiten des Porphyrin-Eisen(II)-Komplexes Häm (Abbildung 1.1.a), in denen das
Eisenatom über eine fünfte Koordinationsstelle an das Histidin-Stickstoffatom einer
Proteinkette geknüpft ist.
a)
N
NN
N Fe
COO–COO–
NH
N
Häm-Gruppe
proximalesHistidin
b)
Abbildung 1.1. a) Koordinationsumgebung der Eisen(II)-Zentren in Desoxy-Hb, b)
schematische Darstellung der Geometrieänderungen der Häm-Einheit in der α-Untereinheit
des Hb bei O2-Aufnahme und –Abgabe (Distanzen jeweils in Å).[1]

1 Einleitung 2
Die vier Häm-Einheiten sind durch Proteinketten miteinander verbunden. Ein Mechanismus
für die Sauerstoffaufnahme und -abgabe wurde von Perutz vorgeschlagen.[1] Desoxy-Häm
besitzt ein Fe(II)-Zentralion im High Spin-Zustand (HS). HS-Fe(II)-Komplexe weisen
typischerweise Fe–N-Bindungslängen von 2.12 - 2.18 Å auf.[2] Da die Umgebung des
Eisenions bei Anordnung in der Ringebene Fe–N-Bindungslängen zum Porphyrinliganden
von maximal 2.00 - 2.05 Å zulässt, befindet sich das Eisenion über der Ebene des
Porphyrinrings. Bei Koordination eines Sauerstoffmoleküls an die freie Koordinationsstelle
kommt es zu einem Spinübergang des Eisens zum Low Spin-Zustand (LS). Die eg-Orbitale
mit antibindendem Charakter werden beim Übergang depopuliert, wodurch eine Verkürzung
der Fe–N-Bindungslängen auf 1.98 Å resultiert und das Eisenion in die Ebene des
Porphyrinrings wandert. Der proximale Histidin-Ligand in trans-Position zum Sauerstoff
muss der Bewegung des Eisenzentrums folgen, wodurch die Konformation der zugehörigen
Proteinkette geändert wird (T-Form → R-Form). Die Änderung der Konformation überträgt
sich auf das Proteingerüst und steigert die Sauerstoff-Affinität der übrigen Häm-Einheiten. Ist
das Hämoglobin-Protein mit Sauerstoff gesättigt, tritt bei der Sauerstoffabgabe in der Zelle
der entgegengesetzte Effekt ein. Der Spinübergang stellt nach dem Perutz-Mechanismus den
Auslöser für den kooperativen Effekt dar. Weitere Beispiele für Reaktionen, die mit einem
Spinübergang einhergehen, sind katalytische Sauerstoffübertragungen auf nicht aktivierte
Substrate durch Cytochrom P450[3] und Peroxidasen.[4]
1.1.2 Spincrossover
Übergangsmetallkomplexe mit Elektronenkonfigurationen von d4 bis d7 können aufgrund der
Aufspaltung der d-Orbitale im oktaedrischen Ligandenfeld in t2g- und eg-Orbitale im Low
Spin- (LS) oder High Spin-Zustand (HS) vorliegen. Die Konfiguration des Grundzustandes ist
dabei abhängig von der Ligandenfeldstärke und von der Natur des Metallzentrums. Der
Übergang beider Grundzustände ineinander, induziert durch externe Stimuli, ist der so
genannte Spincrossover-Effekt (SCO).[5-8] Das Phänomen wurde zuerst 1937 durch Cambi
und Mitarbeiter beobachtet, die thermisch induzierte Spinübergänge in N,N’-substituierten
Tris(dithiocarbamat)Eisen(III)-Komplexen anhand der magnetischen Suszeptibilität
nachwiesen.[9] In den 1960er Jahren unternahmen König und Madeja den Versuch, die
Spincrossover-Region für Eisen(II)-Komplexe durch Substitution der anionischen Liganden
im System [Fe(phen)2(X)2] (phen = 1,10-Phenanthrolin; X = Br–, Cl–, SCN–, N3–, OCN–,
HCOO– und CN–) zu bestimmen,[10] und entdeckten im Komplex [Fe(phen)2(NCS)2] das erste
1 Einleitung 3
Beispiel für thermischen SCO in einem synthetisch hergestellten Eisen(II)-Komplex.[11]
Eisen(II)-Komplexe mit d6-Elektronenkonfiguration stellen das mit Abstand meist untersuchte
System in der Erforschung von SCO-Verbindungen dar. Zum Einen ist die d6 Low Spin-
Konfiguration durch die maximale Ligandenfeldstabilisierungsenergie energetisch günstig.
Das FeII-Ion erzeugt andererseits, z. B. verglichen mit Cobalt(III) (d6), ein schwächeres
Ligandenfeld, was sowohl HS- wie auch LS-Komplexe durch Wahl geeigneter Parameter
zugänglich macht.
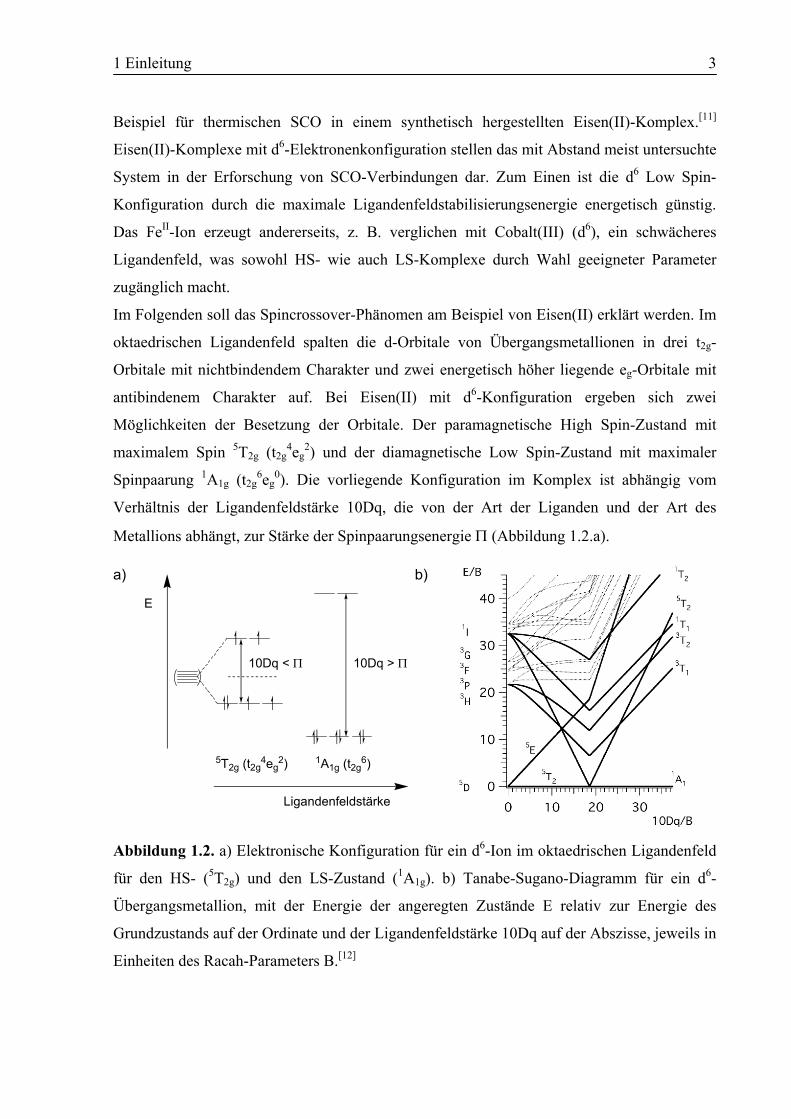
Im Folgenden soll das Spincrossover-Phänomen am Beispiel von Eisen(II) erklärt werden. Im
oktaedrischen Ligandenfeld spalten die d-Orbitale von Übergangsmetallionen in drei t2g-
Orbitale mit nichtbindendem Charakter und zwei energetisch höher liegende eg-Orbitale mit
antibindenem Charakter auf. Bei Eisen(II) mit d6-Konfiguration ergeben sich zwei
Möglichkeiten der Besetzung der Orbitale. Der paramagnetische High Spin-Zustand mit
maximalem Spin 5T2g (t2g4eg
2) und der diamagnetische Low Spin-Zustand mit maximaler
Spinpaarung 1A1g (t2g6eg
0). Die vorliegende Konfiguration im Komplex ist abhängig vom
Verhältnis der Ligandenfeldstärke 10Dq, die von der Art der Liganden und der Art des
Metallions abhängt, zur Stärke der Spinpaarungsenergie Π (Abbildung 1.2.a).
a)
10Dq < Π 10Dq > Π
5T2g (t2g4eg
2) 1A1g (t2g6)
E
Ligandenfeldstärke
b)
Abbildung 1.2. a) Elektronische Konfiguration für ein d6-Ion im oktaedrischen Ligandenfeld
für den HS- (5T2g) und den LS-Zustand (1A1g). b) Tanabe-Sugano-Diagramm für ein d6-
Übergangsmetallion, mit der Energie der angeregten Zustände E relativ zur Energie des
Grundzustands auf der Ordinate und der Ligandenfeldstärke 10Dq auf der Abszisse, jeweils in
Einheiten des Racah-Parameters B.[12]

1 Einleitung 4
Das Tanabe-Sugano-Diagramm in Abbildung 1.2.b zeigt die Aufspaltung der Terme eines d6-
Ions bei angelegtem Ligandenfeld ausgehend von den freien Ionen. Der HS-5T2g-Zustand ist
der elektronische Grundzustand bis zu einer kritischen Ligandenfeldstärke (dem Crossover-
Punkt), bei der 10Dq gleich der Spinpaarungsenergie Π ist. Ab diesem Wert wird der LS-
Zustand 1A1g als Grundzustand stabilisiert. Im Fall von Eisen(II)-Komplexen findet man meist
einen FeN6-Donorsatz mit heterocyclischen wie auch aliphatischen Stickstoffliganden.[6] Die
Ligandenfeldstärke ist zusätzlich zur Art der Liganden abhängig von der Metall-Ligand-
Bindungslänge r mit 10Dq~1/r6. Die Bindungslängen sind in HS-Komplexen größer, da die
im HS-Zustand besetzten eg-Orbitale antibindenden Charakter besitzen. Ihre Entvölkerung
beim Übergang in den LS-Zustand resultiert in einer Zunahme der Metall-Ligand-
Bindungsstärke und damit kürzeren Bindungslängen. In LS-Komplexen liegen Fe–N-
Bindungslängen von 1.95 - 2.00 Å vor, während diese in HS-Verbindungen mit 2.12 - 2.18 Å
typischerweise um ca. 0.2 Å oder 10 % länger sind.[2] Da die Ligandenfeldstärke 10Dq
abhängig von der Metall-Ligand-Bindungslänge r ist, müssen die elektronischen Zustände der
beiden Spinzustände unterschiedliche Energien aufweisen. Die Potentialtöpfe der beiden
Spinzustände sind daher in einem E/r(Fe–L)-Diagramm sowohl horizontal als auch vertikal
gegeneinander verschoben.
Abbildung 1.3. Schematische Darstellung der Potentialtöpfe des 1A1g- und des 5T2g-Zustands
für einen Eisen(II)-SCO-Komplex. Die Bedingung für einen thermischen Spinübergang ist
TkE BHL ≈Δ 0 . Die Kernkoordinate ist als r(Fe-L) bezeichnet (ΔrHL = rHS –rLS).[8]
1 Einleitung 5
Liegt die Differenz der Energien der Grundzustände 000LSHSHL EEE −=Δ in der Größenordnung
thermischer Energien, d. h. TkE BHL ≈Δ 0 , befindet sich das Metallzentrum bei tiefen
Temperaturen im LS-Zustand, während beim Übergang zu höheren Temperaturen der
Spincrossover zum HS-Zustand stattfindet. Ist die Voraussetzung des geringen
Energieunterschieds gegeben, können Übergänge auch durch Druck- oder Lichteinwirkung
(Light-induced excited spin state trapping, LIESST[13]) induziert werden.
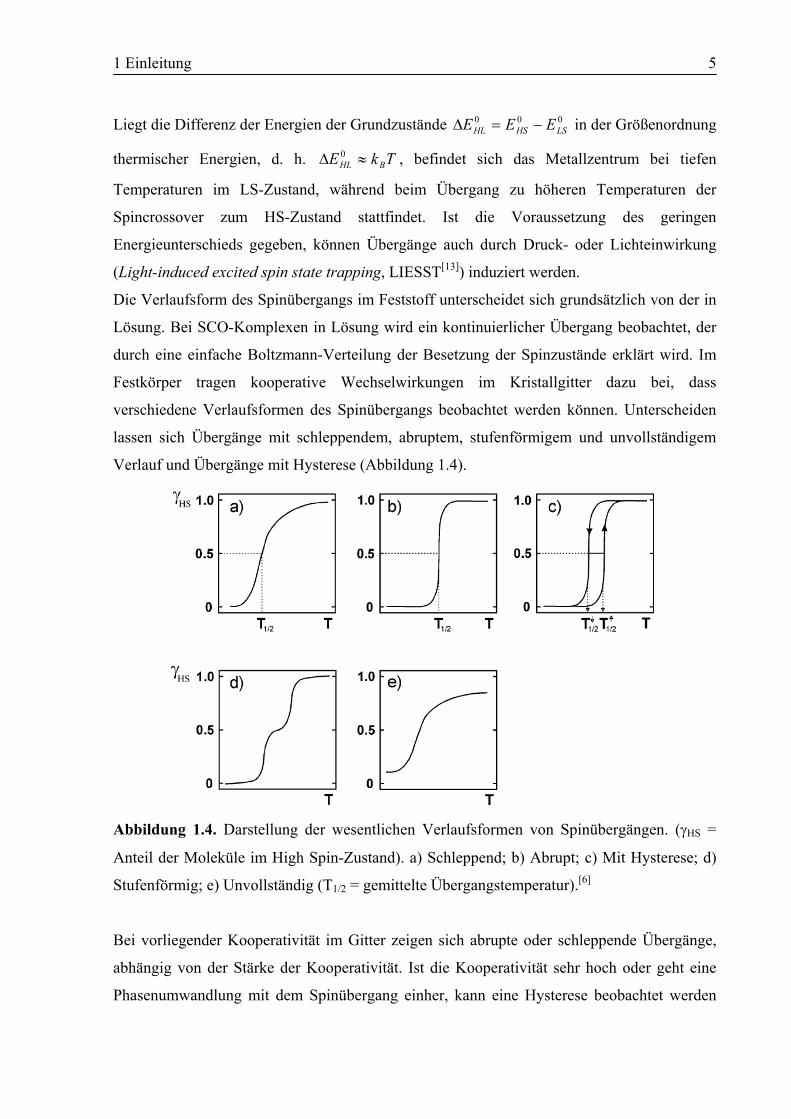
Die Verlaufsform des Spinübergangs im Feststoff unterscheidet sich grundsätzlich von der in
Lösung. Bei SCO-Komplexen in Lösung wird ein kontinuierlicher Übergang beobachtet, der
durch eine einfache Boltzmann-Verteilung der Besetzung der Spinzustände erklärt wird. Im
Festkörper tragen kooperative Wechselwirkungen im Kristallgitter dazu bei, dass
verschiedene Verlaufsformen des Spinübergangs beobachtet werden können. Unterscheiden
lassen sich Übergänge mit schleppendem, abruptem, stufenförmigem und unvollständigem
Verlauf und Übergänge mit Hysterese (Abbildung 1.4).
Abbildung 1.4. Darstellung der wesentlichen Verlaufsformen von Spinübergängen. (γHS =
Anteil der Moleküle im High Spin-Zustand). a) Schleppend; b) Abrupt; c) Mit Hysterese; d)
Stufenförmig; e) Unvollständig (T1/2 = gemittelte Übergangstemperatur).[6]
Bei vorliegender Kooperativität im Gitter zeigen sich abrupte oder schleppende Übergänge,
abhängig von der Stärke der Kooperativität. Ist die Kooperativität sehr hoch oder geht eine
Phasenumwandlung mit dem Spinübergang einher, kann eine Hysterese beobachtet werden

1 Einleitung 6
(Abbildung 1.4.c). Diese Form des Spinübergangs ist für die technische Anwendung am
interessantesten. Ein Material mit breitem Hysteresebereich um Raumtemperatur wäre zur
Anwendung in Datenspeicherung und Anzeigetechnik geeignet.[14]
1.1.3 Self-Assembled Monolayers
Selbstorganisierte Monolagen (self-assembled monolayers, SAMs) sind wohlgeordnete
Ensembles von Molekülen, die durch die Adsorption von oberflächenaktiven Substanzen
gebildet werden, welche eine Ankergruppe mit spezieller Affinität zu einem Substrat besitzen.
Endgruppe
Distanzstück(Spacer)
Ankergruppe
Substrat
Abbildung 1.5. Modellvorstellung einer SAM.[15]
Der SAM-Ansatz besitzt mehrere Vorteile gegenüber anderen Methoden der Präparation
ultradünner molekularer Lagen wie der Rotationsbeschichtung (spin-coating),[16] Langmuir-
Blodgett-Filmen,[17] organischer Molekularstrahlepitaxie (organic molecular beam epitaxy,
OMBE)[18] und chemischer Gasphasenabscheidung (chemical vapour deposition, CVD).[19]
Die Präparation von SAMs ist ein einfacher Prozess und erfordert keine kostenintensiven
Apparaturen. Die makroskopischen Eigenschaften der Oberfläche können durch synthetische
Variation der Endgruppen der Moleküle direkt beeinflusst werden. Zusätzlich ist der Aufbau
komplexerer Schichten durch die Knüpfung an bestehende SAMs durch chemische
Reaktionen auf der Oberfläche möglich.
Die meist untersuchten SAMs basieren seit den 1980er Jahren auf dem System Au/Thiol.[15,
20-22] Gold hat als Substrat den Vorteil, dass es chemisch relativ inert und elektrisch leitfähig
ist, was Untersuchungen mittels Rastertunnelmikroskopie erlaubt. Gold zeigt außerdem eine
starke spezifische Wechselwirkung mit Schwefel, auch in Anwesenheit anderer funktioneller
Gruppen. Die Stärke der Bindung von Methanthiolaten auf Au(111) (im Sinne einer
Homolyse) ist mit 45 kJ/mol beispielsweise sehr hoch.[23] Die Moleküle in Au/Thiol-SAMs
sind wie folgt aufgebaut. Die Ankergruppe besteht aus einer schwefelhaltigen funktionellen
Gruppe, bei der es sich meist um die Thiolfunktion handelt. Es sind allerdings auch SAMs mit
alternativen Ankergruppen bekannt,[20] was zusätzliche Flexibilität für die Synthese der

1 Einleitung 7
Moleküle gewährt. Das Distanzstück („spacer“) besteht in Au/Thiol-SAMs üblicherweise aus
einer Methylenkette unterschiedlicher Länge. In neueren Arbeiten werden allerdings auch
zunehmend alternative Distanzstücke gewählt, da starke intermolekulare Wechselwirkungen
wie π-π-Wechselwirkungen[24] oder H-Brückenbindungen[25] als wichtige Triebkraft zur
Ausbildung hochgeordneter Lagen erkannt wurden. Die Natur der Endgruppe ist schließlich
nur durch die Möglichkeiten der organischen Synthese beschränkt. Auf die Ordnung der SAM
hat die Endgruppe allerdings insofern einen Einfluss, als zu sperrige Endgruppen die
Kontaktflächen zwischen den Molekülen in der Schicht reduzieren können, was infolge
geringerer Wechselwirkungen zu schlechterer Ordnung führt (s.a. Kapitel 2).
1.2 Charakterisierungsmethoden
Im Folgenden werden die in dieser Arbeit verwendeten Charakterisierungsmethoden
beschrieben, die über das Maß der in der Synthese gebräuchlichen Methoden hinausgehen.
1.2.1 SCO
Die mit am meisten eingesetzte Technik zur Bestimmung des Spinzustands ist die Messung
der magnetischen Suszeptibilität χ. Eine temperaturabhängige magnetische Messung gibt den
direkten Verlauf des Spinzustandes in Abhängigkeit von der Temperatur wieder. In Lösung
erfolgt die Bestimmung von χ mit 1H-NMR-Messungen nach der Methode von Evans.[26]
Messprinzip ist die Tatsache, dass die Änderung der chemischen Verschiebung von inerten
Referenzsubstanzen durch paramagnetische Substanzen in einer Probe im NMR-Spektrometer
direkt proportional zur magnetischen Suszeptibilität der paramagnetischen Substanz ist. Für
Messungen an Feststoffen hat die Verwendung von hochempfindlichen SQUID-
Magnetometern (Superconducting Quantum Interference Device) die älteren magnetischen
Waagen nach dem Faraday- und Gouy-Prinzip weitestgehend verdrängt.[27]
Eine andere oft verwendete Methode stellt die 57Fe-Mössbauerspektroskopie dar, die neben
einer genauen Bestimmung der Konfiguration des Eisenzentrums bezüglich des Spin- und
Oxidationszustands auch weitergehende Aussagen, z. B. über die Bindungssituation in
untersuchten Komplexen liefern kann.[28] Diese Methode stand während der Anfertigung
dieser Arbeit nicht zur Verfügung und wird wegen ihrer großen Bedeutung für die
Erforschung der SCO-Verbindungen der Vollständigkeit halber an dieser Stelle genannt.
Da der Spinübergang mit einer Änderung der Metall-Ligand-Bindungslängen verbunden ist,
hat man mit der Röntgenstrukturanalyse ein weiteres wertvolles analytisches Mittel zur Hand.

1 Einleitung 8
Besonders in FeII-N6-Verbindungen ist die Änderung der Bindungslänge ΔrHL durch den
großen Spinunterschied von ΔS = 2 zwischen HS- und LS-Zustand besonders groß. Zusätzlich
lassen sich mit der Röntgenstrukturanalyse auch Verzerrungen der Koordinationsumgebung
des Zentralmetalls feststellen, die relevant für die Natur des Spinübergangs sein können.[29]
NEXAFS (Near-Edge X-Ray Absorption Fine Structure) Fe L2,3- und K-Kanten-
Spektroskopie unter Verwendung von Synchrotronstrahlung stellt eine weitere
Charakterisierungsmethode für Spinübergänge in Festkörpern dar, die anhand der
Orbitalbesetzung Informationen über Oxidationszustände und Spinzustände liefert. Ein
wichtiger Aspekt von NEXAFS-Untersuchungen ist die zusätzliche Möglichkeit der
Untersuchung von Oberflächen, was für die vorliegende Arbeit von Relevanz ist (s. u.).
Theoretische DFT-Berechnungen gewinnen verstärkt an Bedeutung für die Untersuchung von
SCO-Verbindungen. Durch Entwicklung geeigneter Hybridfunktionale (B3LYP[30],
B3LYP*[31]) können mittlerweile akkurate Vorhersagen zu potentiellen SCO-Verbindungen
getroffen werden.[32]
1.2.2 SAMs
Selbstorganisierte Monolagen lassen sich durch mikroskopische, wie auch spektroskopische
Methoden untersuchen.[33] In der vorliegenden Arbeit wurden die präparierten Monolagen
durch drei im Folgenden besprochene Methoden charakterisiert.
Die Rastertunnelmikroskopie (scanning tunneling microscopy, STM) stellt zur Untersuchung
von Oberflächen die Methode der Wahl dar.[34] Das Messprinzip besteht darin, eine Spannung
(unter 1 V) zwischen der Probenoberfläche und einer metallischen Spitze anzulegen, die sich
wenige Å voneinander entfernt befinden. Daraus resultiert ein Tunnelstrom, der zwischen
Oberfläche und STM-Spitze fließt. Die Spitze wird nun in einem x-, y-Raster über die Probe
geführt, wobei der Stromfluss durch Auslenkung der Spitze in z-Richtung konstant gehalten
wird (constant current mode).
Abbildung 1.6. STM: Messprinzip.

1 Einleitung 9
Als Ergebnis erhält man ein „topographisches“ Bild der Oberfläche. Voraussetzung ist dabei
die Leitfähigkeit sowohl des Substrats als auch der adsorbierten Moleküle. STM-Messungen
geben Informationen über die Beschaffenheit der Oberfläche bis hin zu molekularer
Auflösung. Die Untersuchungen können temperaturabhängig bei Atmosphärendruck wie auch
im Ultrahochvakuum (UHV) durchgeführt werden.
Die Röntgen-Nahkanten-Absorptions-Spektroskopie[35] (Near-Edge X-Ray Absorption Fine
Structure, NEXAFS) unter Verwendung von Synchrotronstrahlung stellt eine kombinierte
Methode dar, sowohl die Ordnung einer Monolage[36] als auch den Spinzustand von
Eisenverbindungen zu untersuchen.[37, 38] Bei der Röntgenabsorptionsspektroskopie (X-ray
absorption spectroscopy, XAS) führt die Absorption von Röntgenstrahlung dazu, dass
kernnahe Elektronen in unbesetzte Valenz-Zustände oder ins Vakuumskontinuum angeregt
werden. Das erhaltene Absorptionsspektrum setzt sich aus dem Bereich der Absorptionskante
(der bei NEXAFS untersucht wird) durch Übergänge in unbesetzte Valenzorbitale und einer
überlagerten Feinstruktur durch Übergänge ins Vakuumskontinuum zusammen.
Abbildung 1.7. Prinzip der XAS.[35]
Aus dem Intensitätsverlauf des NEXAFS-Spektrums lassen sich Rückschlüsse auf die
elektronische Struktur der unbesetzten Zustände ziehen, und durch den Vergleich mit
Referenzspektren Oxidationsstufen und Spinzustände identifizieren. Für die Untersuchung
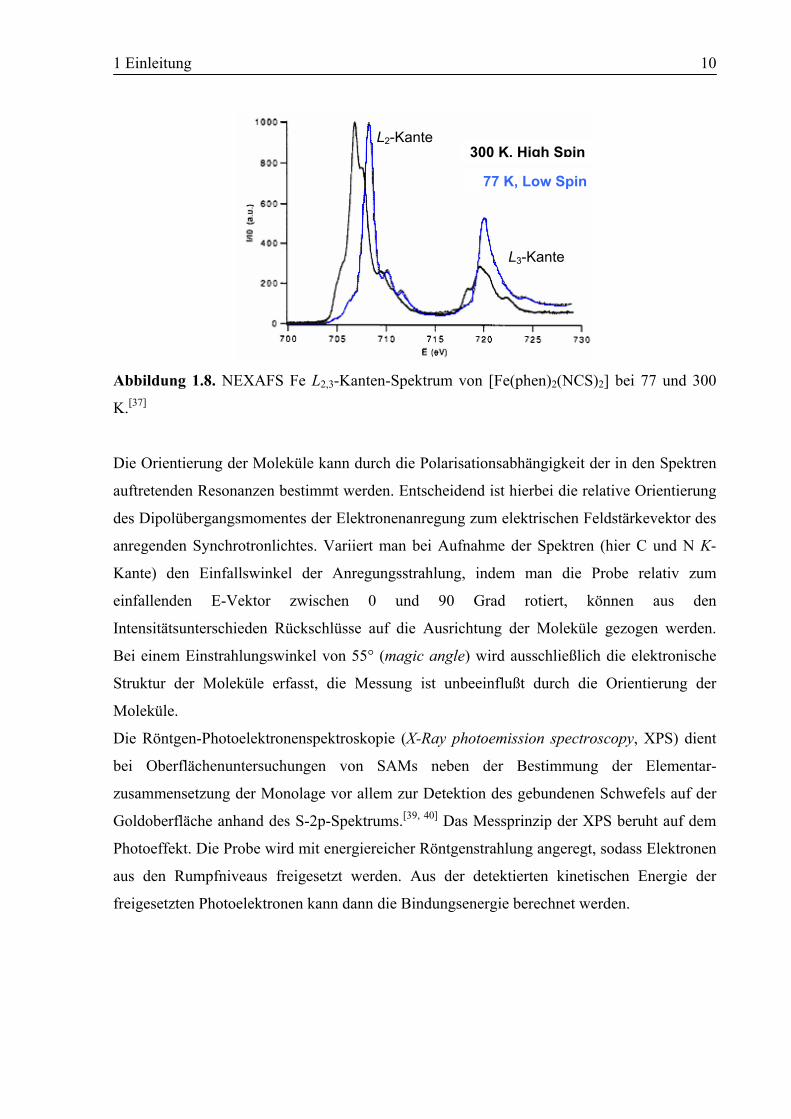
von Eisen-SCO-Verbindungen ist insbesondere der Bereich der Fe L2,3-Kanten interessant, der
durch 2p→3d-Übergänge erzeugt wird und der infolgedessen abhängig vom Spinzustand ist.
Während beim HS-Zustand des Eisen(II) (t2g4eg
2) Übergänge sowohl in die teilweise besetzten
t2g- und eg-Orbitale erfolgen können, sind im LS-Zustand durch die Vollbesetzung der t2g-
Orbitale nur Übergänge in eg-Orbitale möglich. Diese liegen energetisch höher, was das
Absorptions-Signal der Fe L2-Kante zu höherer Energie verschiebt (Abbildung 1.8).
1 Einleitung 10
Abbildung 1.8. NEXAFS Fe L2,3-Kanten-Spektrum von [Fe(phen)2(NCS)2] bei 77 und 300
K.[37]
Die Orientierung der Moleküle kann durch die Polarisationsabhängigkeit der in den Spektren
auftretenden Resonanzen bestimmt werden. Entscheidend ist hierbei die relative Orientierung
des Dipolübergangsmomentes der Elektronenanregung zum elektrischen Feldstärkevektor des
anregenden Synchrotronlichtes. Variiert man bei Aufnahme der Spektren (hier C und N K-
Kante) den Einfallswinkel der Anregungsstrahlung, indem man die Probe relativ zum
einfallenden E-Vektor zwischen 0 und 90 Grad rotiert, können aus den
Intensitätsunterschieden Rückschlüsse auf die Ausrichtung der Moleküle gezogen werden.
Bei einem Einstrahlungswinkel von 55° (magic angle) wird ausschließlich die elektronische
Struktur der Moleküle erfasst, die Messung ist unbeeinflußt durch die Orientierung der
Moleküle.
Die Röntgen-Photoelektronenspektroskopie (X-Ray photoemission spectroscopy, XPS) dient
bei Oberflächenuntersuchungen von SAMs neben der Bestimmung der Elementar-
zusammensetzung der Monolage vor allem zur Detektion des gebundenen Schwefels auf der
Goldoberfläche anhand des S-2p-Spektrums.[39, 40] Das Messprinzip der XPS beruht auf dem
Photoeffekt. Die Probe wird mit energiereicher Röntgenstrahlung angeregt, sodass Elektronen
aus den Rumpfniveaus freigesetzt werden. Aus der detektierten kinetischen Energie der
freigesetzten Photoelektronen kann dann die Bindungsenergie berechnet werden.
300 K, High Spin
77 K, Low Spin
L2-Kante
L3-Kante

1 Einleitung 11
Abbildung 1.9. Prinzip der Photoemission (Bild: W. Kuch, FU Berlin).
Das Muster der Bindungsenergien ist elementspezifisch, sodass einzelne Elemente
identifiziert und ihre Konzentration an der Oberfläche aus Intensitätsverhältnissen bestimmt
werden kann. Variationen in der Lage der Bindungsenergien (chemische Verschiebung) geben
Aufschluss über die chemische Umgebung und damit den Bindungszustand des untersuchten
Elements auf der Oberfläche.
1.3 Stand der Forschung
1.3.1 Spincrossover und Kooperativität
Der Wunsch, gezielte Synthesen von SCO-Verbindungen durchzuführen, gründet sich auf das
beträchtliche Potential derartiger Verbindungen für die technische Anwendung. In ihrer
Eigenschaft als molekulare Schalteinheiten sind SCO-Komplexe geeignet für den Einsatz in
der Datenspeicherung, Anzeige- und Sensortechnik.[14, 41, 42]
Abbildung 1.10. Schema des „Memory-Effekts“ durch Hysterese in einem SCO-Material für
die technische Anwendung. Der Spinzustand kann durch Temperatur, Druck, ein Magnetfeld
oder Lichteinstrahlung schaltbar sein.[41]

1 Einleitung 12
Obwohl der Ursprung des SCO-Effekts in den Eigenschaften des isolierten Moleküls
begründet liegt, bewirkt nur ausreichende Kooperativität im Kristallgitter die Entfaltung eines
Spinübergangs im Feststoff.[6] Weist das Kristallgitter eine ungünstige Beschaffenheit für die
Übertragung der Information über die Volumenänderung im Gitter auf, sind die Moleküle
trotz geeigneter Stärke des Ligandenfeldes nicht in der Lage, einen Spinübergang im Feststoff
zu zeigen. Beobachtet wurde dieser Effekt beispielsweise bei der Untersuchung zweier Salze
eines Eisen(II)-Komplexes des dreizähnigen Liganden 2,6-Bis(pyrazol-1-yl)pyridin (Bpp) (I),
[Fe(I)](X)2 (X = BF4, PF6).[43]
NNNN N
2+
[BF4–]2
N
NN
N
NN
N
N
N
N
Fe
I
II
Abbildung 1.11. 2,6-Bis(pyrazol-1-yl)pyridin (Bpp) (I)[44, 45] und [Fe(I)](BF4)2 (II).[46]
Die von Halcrow und Mitarbeitern synthetisierte Stammverbindung [Fe(I)](BF4)2 (II),[46]
sowie diverse andere Komplexe mit derivatisierten Bpp-Liganden, zeigen thermischen aber
auch lichtinduzierten Spincrossover.[47] Während das Tetrafluoroboratsalz des Komplexes
einen thermischen Spinübergang im Feststoff aufweist, liegt das Hexafluorophosphatsalz im
selben Temperaturbereich ausschließlich als High Spin-Verbindung vor. Im Gegensatz dazu
zeigen beide Substanzen in Lösung einen thermischen Spinübergang. Der Grund für dieses
Verhalten muss also im Einfluss zu suchen sein, den die Festkörperstruktur auf die Moleküle
ausübt. In diesem Fall unterscheiden sich die Festkörperstrukturen in der Natur der
Gegenionen im Gitter.
Auch das Vorhandensein von Lösungsmittelmolekülen in der Kristallstruktur kann den
sterischen Zwang im Gitter derart verstärken, dass ein Spinübergang unterbunden wird, wie
Bürgi et al. am Beispiel verschiedener Solvate von Tris(2-picolylamin)eisen(II)-dichlorid
(III) gezeigt haben (Tabelle 1.1).[48]

1 Einleitung 13
Tabelle 1.1. Auswirkung des Solvats auf den Spinübergang in Komplex III im Feststoff.[48]
Solvat Art des Spinübergangs
Methanol kontinuierlich
Ethanol zweistufig, ohne Hysterese
Allylalkohol zweistufig, ohne Hysterese
2-Propanol zweistufig, mit Hysterese
1-Propanol kein Übergang
Fe2+
N
N NH2
NNH2
NH2
2 Cl–
· CH3OH
· CH3CH2OH
· CH2CHCH2OH
· CH3CH2CH2OH
· (CH3)2CHOH
· (CH3)3COH
tert-Butanol kein Übergang
Auch wenn ein Spinübergang in diesem Beispiel bei mehreren Modifikationen vorliegt, hat
die Festkörperstruktur durch die Kooperativität im Gitter zusätzlich Einfluss auf den Verlauf
des Übergangs.
Bei Verbindungen mit identischer chemischer Zusammensetzung sind wenige Beispiele
bekannt, bei denen sich Strukturisomere in ihrem Spinverhalten, hauptsächlich im Verlauf des
Spinübergangs, unterscheiden.[49-51] Im Extremfall wird in einer der polymorphen
Modifikationen ein thermischer Spinübergang beobachtet, während er in einer anderen
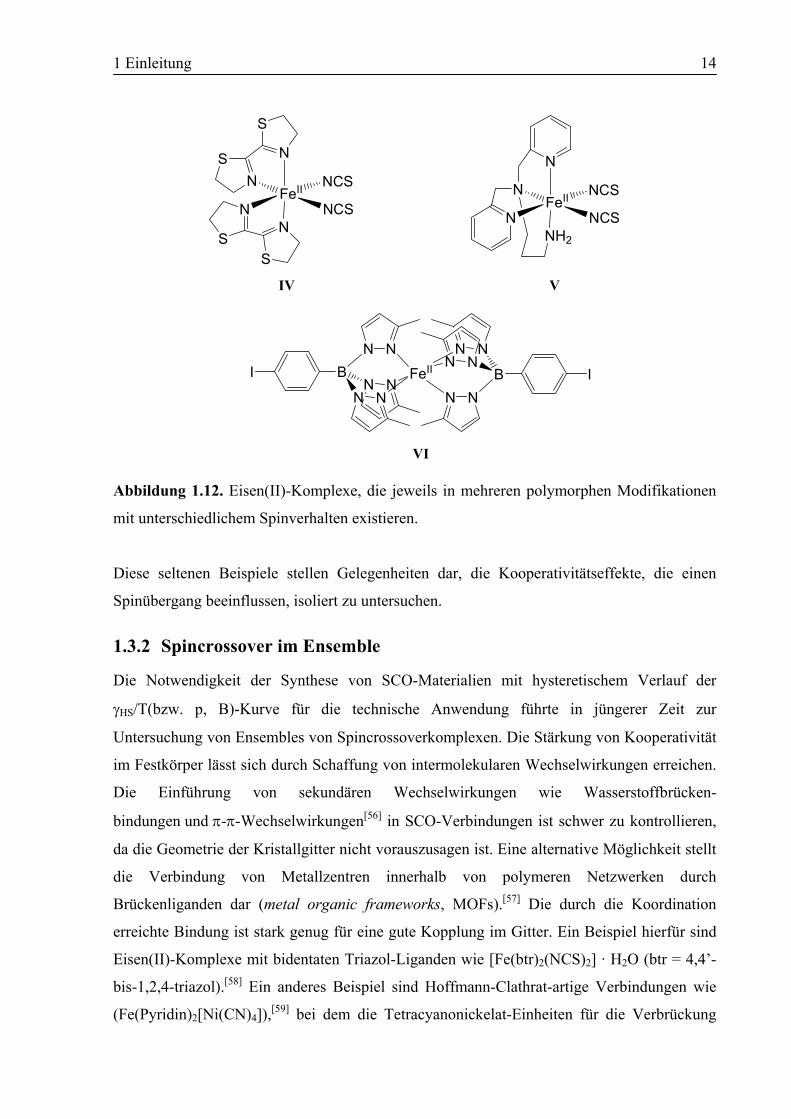
Modifikation vollständig unterdrückt ist. Ozarowski et al. konnten zwei polymorphe Formen
von Bis(2,2’-bi-2-thiazolin)bis(isothiocyanato)Eisen(II) (IV) isolieren, von denen eine einen
thermischen Spinübergang mit Hysterese zeigt, während die zweite als reine High Spin-
Verbindung vorliegt.[52] Matouzenko und Mitarbeiter beobachteten drei polymorphe Formen
in unterschiedlichen Raumgruppen von [FeII(DPPA)(NCS)2] (V) (DPPA = (3-
Aminopropyl)bis(2-pyridylmethyl)amin), wobei eine als reine High Spin-Verbindung und die
anderen beiden als SCO-Verbindungen vorliegen.[53] Bei den SCO-Verbindungen tritt beim
Übergang im einen Fall Hysterese auf, während im anderen Fall ein abrupter Übergang
stattfindet. Das jüngste Beispiel für Polymorphismus verbunden mit unterschiedlichem
Spinverhalten fanden Reger und Mitarbeiter 2005[54] in Eisen(II)-Komplexen eines
Skorpionat-Liganden[55] der dritten Generation. Es wurden zwei polymorphe Modifikationen
von Fe[(p-IC6H4)B(3-Mepz)3]2 (VI) isoliert, von denen eine im High Spin-Zustand vorliegt,
während die zweite einen abrupten thermischen Spinübergang zeigt.
III
1 Einleitung 14
N N
NCS
NCSFeII
S
NN
S
SN
N
S
NCS
NCSFeII
NH2
N
N
N
BN N
N N
I BNN
NN
INN
FeII
IV V
VI
Abbildung 1.12. Eisen(II)-Komplexe, die jeweils in mehreren polymorphen Modifikationen
mit unterschiedlichem Spinverhalten existieren.
Diese seltenen Beispiele stellen Gelegenheiten dar, die Kooperativitätseffekte, die einen
Spinübergang beeinflussen, isoliert zu untersuchen.
1.3.2 Spincrossover im Ensemble
Die Notwendigkeit der Synthese von SCO-Materialien mit hysteretischem Verlauf der
γHS/T(bzw. p, B)-Kurve für die technische Anwendung führte in jüngerer Zeit zur
Untersuchung von Ensembles von Spincrossoverkomplexen. Die Stärkung von Kooperativität
im Festkörper lässt sich durch Schaffung von intermolekularen Wechselwirkungen erreichen.
Die Einführung von sekundären Wechselwirkungen wie Wasserstoffbrücken-
bindungen und π-π-Wechselwirkungen[56] in SCO-Verbindungen ist schwer zu kontrollieren,
da die Geometrie der Kristallgitter nicht vorauszusagen ist. Eine alternative Möglichkeit stellt
die Verbindung von Metallzentren innerhalb von polymeren Netzwerken durch
Brückenliganden dar (metal organic frameworks, MOFs).[57] Die durch die Koordination
erreichte Bindung ist stark genug für eine gute Kopplung im Gitter. Ein Beispiel hierfür sind
Eisen(II)-Komplexe mit bidentaten Triazol-Liganden wie [Fe(btr)2(NCS)2] · H2O (btr = 4,4’-
bis-1,2,4-triazol).[58] Ein anderes Beispiel sind Hoffmann-Clathrat-artige Verbindungen wie
(Fe(Pyridin)2[Ni(CN)4]),[59] bei dem die Tetracyanonickelat-Einheiten für die Verbrückung

1 Einleitung 15
zwischen den Eisenzentren sorgen. Während bei den beiden genannten Beispielen
zweidimensionale Netzwerke vorliegen, lässt sich durch Austausch der jeweiligen axialen
monodentaten Liganden gegen bidentate auch die Ausbildung von dreidimensionalen
Netzwerken erreichen.[60, 61]
1.3.3 Spincrossover in dünnen Schichten
Die Funktionalisierung von Oberflächen mit SCO-Molekülen bedeutet die Verbindung der
Konzepte der Oberflächenbeschichtung und des SCO. Die Übertragung von SCO-
Eigenschaften auf Oberflächen verschiedenartiger Zusammensetzung würde einer breiteren
Anwendung von SCO-Materialien in der Technik den Weg bahnen. Bei Schaffung von
Monolagen von SCO-Molekülen wäre es, anders als im Festkörper, prinzipiell möglich,
Moleküle isoliert anzusteuern. Einzelne Moleküle könnten auf diese Weise als
Informationsträger genutzt werden, was eine sehr hohe Informationsdichte auf der so
funktionalisierten Oberfläche ermöglichen würde.
Verbindungen vom Hoffmann-Clathrat-Typ stellen das bisher einzige Beispiel für auf
Oberflächen verankerte, selbstorganisierte Lagen von SCO-Komplexen dar, allerdings handelt
es sich bei diesen Lagen nicht um Monolagen, sondern eher um SCO-Polymere.[62, 63] Die
SCO-Moleküle sind nicht direkt an die Oberfläche gebunden, vielmehr wird eine Monolage
von monodentaten Liganden in Form einer SAM auf einer Au(111)-Oberfläche verankert. Die
Anknüpfung der SCO-Zentren erfolgt über koordinative Bindungen an die freien
Donorfunktionen der SAM in einem zweiten Schritt. Die SAM bestand in einem Beispiel aus
4-Mercaptopyridin, das über die klassische Thiolat-Bindung auf der Oberfläche gebunden
wurde und im anderen Fall aus 4,4’-Azopyridin, das mit einem seiner Stickstoffatome auf
Goldoberflächen binden kann. Die erhaltenen dünnen Schichten bestanden aus ~ 25 Lagen
von SCO-Zentren. Thermischer Spincrossover mit Hysterese wurde mittels
Ramanspektroskopie aus dem Verhältnis der Intensität charakteristischer Banden der
Pyrazinschwingungen detektiert.
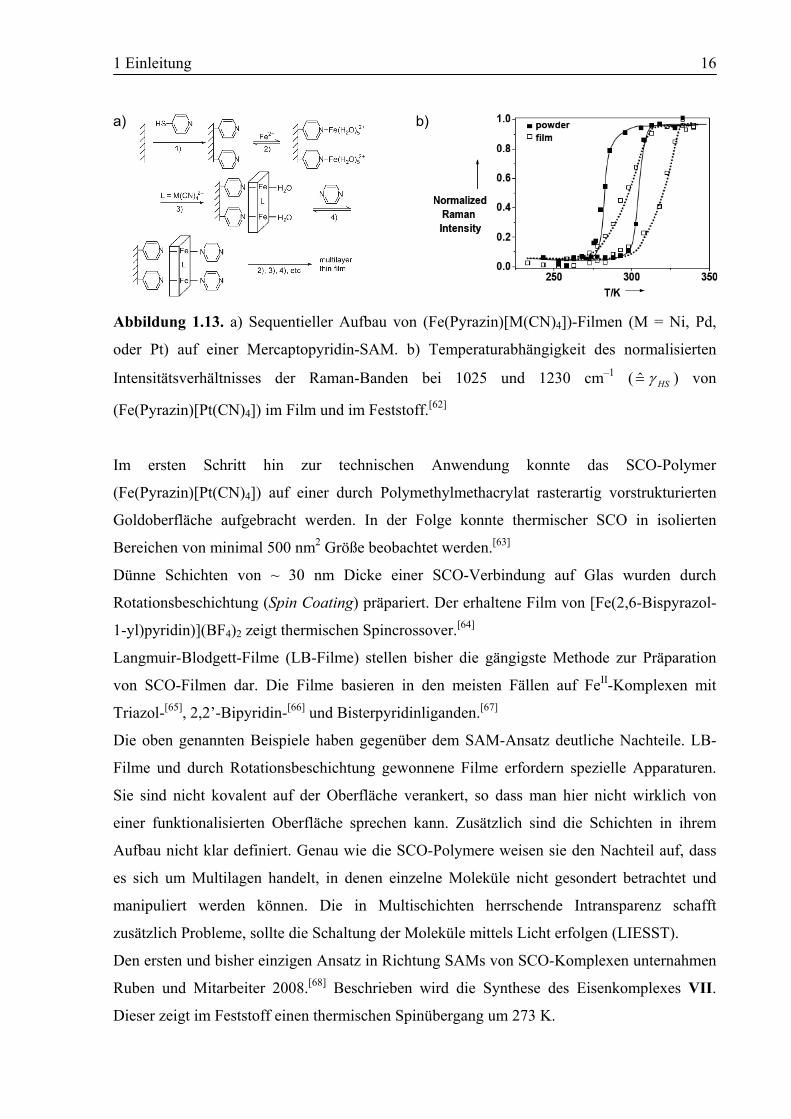
1 Einleitung 16
a) b)
Abbildung 1.13. a) Sequentieller Aufbau von (Fe(Pyrazin)[M(CN)4])-Filmen (M = Ni, Pd,
oder Pt) auf einer Mercaptopyridin-SAM. b) Temperaturabhängigkeit des normalisierten
Intensitätsverhältnisses der Raman-Banden bei 1025 und 1230 cm–1 ( HSγ=̂ ) von
(Fe(Pyrazin)[Pt(CN)4]) im Film und im Feststoff.[62]
Im ersten Schritt hin zur technischen Anwendung konnte das SCO-Polymer
(Fe(Pyrazin)[Pt(CN)4]) auf einer durch Polymethylmethacrylat rasterartig vorstrukturierten
Goldoberfläche aufgebracht werden. In der Folge konnte thermischer SCO in isolierten
Bereichen von minimal 500 nm2 Größe beobachtet werden.[63]
Dünne Schichten von ~ 30 nm Dicke einer SCO-Verbindung auf Glas wurden durch
Rotationsbeschichtung (Spin Coating) präpariert. Der erhaltene Film von [Fe(2,6-Bispyrazol-
1-yl)pyridin)](BF4)2 zeigt thermischen Spincrossover.[64]
Langmuir-Blodgett-Filme (LB-Filme) stellen bisher die gängigste Methode zur Präparation
von SCO-Filmen dar. Die Filme basieren in den meisten Fällen auf FeII-Komplexen mit
Triazol-[65], 2,2’-Bipyridin-[66] und Bisterpyridinliganden.[67]
Die oben genannten Beispiele haben gegenüber dem SAM-Ansatz deutliche Nachteile. LB-
Filme und durch Rotationsbeschichtung gewonnene Filme erfordern spezielle Apparaturen.
Sie sind nicht kovalent auf der Oberfläche verankert, so dass man hier nicht wirklich von
einer funktionalisierten Oberfläche sprechen kann. Zusätzlich sind die Schichten in ihrem
Aufbau nicht klar definiert. Genau wie die SCO-Polymere weisen sie den Nachteil auf, dass
es sich um Multilagen handelt, in denen einzelne Moleküle nicht gesondert betrachtet und
manipuliert werden können. Die in Multischichten herrschende Intransparenz schafft
zusätzlich Probleme, sollte die Schaltung der Moleküle mittels Licht erfolgen (LIESST).
Den ersten und bisher einzigen Ansatz in Richtung SAMs von SCO-Komplexen unternahmen
Ruben und Mitarbeiter 2008.[68] Beschrieben wird die Synthese des Eisenkomplexes VII.
Dieser zeigt im Feststoff einen thermischen Spinübergang um 273 K.

1 Einleitung 17
N
NN
N
NN
N
N
N
N
FeS SO
O
2+
ClO4–
2VII
Abbildung 1.14. Bis[S-(4-([2,6-(dipyrazol-1-yl)pyrid-4-yl]ethynyl)phenyl)ethanethioato]
Eisen(II)-Diperchlorat (VII).[68]
Zur Untersuchung des Ladungstransports durch Einzelmoleküle wurden in diesem Beispiel
Thioacetat-Funktionen zur Bindung an Goldoberflächen an die Liganden geknüpft. Potentiell
sind mit Komplex VII aber auch SAMs auf Au(111)-Oberflächen denkbar, die jedoch bisher
nicht präpariert wurden.
Auch in der hier vorgelegten Arbeit werden —unabhängig von Ruben— Eisen(II)komplexe
des dreizähnigen Liganden 2,6-Bis(pyrazol-1-yl)pyridin (Bpp) (I) als SCO-Einheit verwendet.
Die mit dieser Arbeit vorbereitete Synthese einer selbstorganisierten Monolage von Eisen(II)-
Spincrossoverkomplexen auf Oberflächen würde das erste Beispiel einer auf diese Art
funktionalisierten Oberfläche darstellen.

2 Eisen(II)-Komplexe mit Schwefelanker 18
2 Eisen(II)-Komplexe mit Schwefelanker
2.1 Aufgabenstellung
Die Aufgabenstellung besteht darin, Spincrossoverkomplexe kovalent an Oberflächen zu
verankern. Die Komplexe sollen durch externe Stimuli, vor allem thermisch schaltbar sein.
Eine gute zweidimensionale Ordnung der Moleküle auf der Oberfläche soll aus zwei Gründen
erreicht werden. Einerseits wird durch starke intermolekulare Kopplung innerhalb des
Ensembles ein nutzbares Schaltverhalten erzeugt. Andererseits können die funktionalisierten
Oberflächen mit den zur Verfügung stehenden analytischen Methoden besser untersucht
werden.
a) Endgruppe: Eisen(II)-SCO-Komplex
b) Distanzstück
c ) Ankergruppe: schwefelhaltige funktionelle Gruppe
Substrat: Au(111)/Glimmer
Abbildung 2.1. Abstrahiertes Modell eines SCO-Komplexes auf der Goldoberfläche.
Abbildung 2.1 verdeutlicht das Synthesekonzept. Das zu synthetisierende oberflächenaktive
Molekül soll aus folgenden Teilen bestehen:
1. Eisen(II)-SCO-Komplex (a). Der Wechsel zwischen dem diamagnetischen Low Spin-
und dem paramagnetischen High Spin-Zustand im Fall von Eisen(II) kann gut mit
physikalischen Methoden erfasst werden. Die Liganden des Komplexes müssen
synthetisch leicht zugänglich, derivatisierbar und unter den gegebenen Bedingungen gut
zu handhaben sein.

2 Eisen(II)-Komplexe mit Schwefelanker 19
2. Distanzstück (b). Das Ordnungsverhalten der Moleküle auf der Oberfläche kann durch
die Flexibilität der Moleküle und durch die Stärke der intermolekularen
Wechselwirkungen beeinflusst werden. Zwischen SCO-Einheit und Ankergruppe sollte
sich daher eine leicht austauschbare oder derivatisierbare organische Einheit befinden.
3. Ankergruppe (c). Die Bindung der SCO-Einheit an das Substrat soll kovalent erfolgen
und eine Selbstorganisation der Moleküle auf der Oberfläche erlauben. Die Präparation
der Schichten soll sowohl aus der Lösung als auch aus der Gasphase einfach möglich
sein. Die notwendigerweise reaktiven funktionellen Gruppen, die dazu in Betracht
kommen, sollen inert gegenüber einer Reaktion mit dem Eisen(II)-Zentrum der SCO-
Einheit sein. Vor allem eine Oxidation muss ausgeschlossen werden. Die Untersuchung
der beschichteten Oberfläche mit den gängigen Methoden soll gut möglich sein.
Aufgrund der genannten Gesichtspunkte wurde das System der Thiolat-Ankergruppe auf
einer Goldoberfläche gewählt. Die Knüpfung der Schwefel-Gold-Bindung kann dabei
aus diversen Schwefel-funktionellen Gruppen erfolgen.[20]
Als SCO-Einheit werden in dieser Arbeit Eisen(II)komplexe des dreizähnigen Liganden 2,6-
Bis(pyrazol-1-yl)pyridin (Bpp) (I) verwendet.[47] Komplexe dieser Art entsprechen in ihren
Eigenschaften den Vorgaben des Konzepts. Der Bpp-Ligand lässt sich durch den modularen
Charakter der Synthese sowohl am zentralen Pyridin- als auch an den seitlichen Pyrazolringen
derivatisieren. Dadurch werden die Einführung einer Ankergruppe sowie eine möglicherweise
notwendige Anpassung des Ligandenfeldes und die Schaffung intermolekularer
Wechselwirkungen ermöglicht.
Zur Verankerung auf Au(111)-Oberflächen werden üblicherweise schwefelhaltige
funktionelle Gruppen verwendet. Die Thiolgruppe für die Bildung der Schwefel-Gold-
Bindung zur Oberfläche weist trotz des sehr guten Ordnungsverhaltens in der Praxis zwei
Nachteile auf. Eine Verbindung mit SH-Gruppe „R–SH“ kann leicht zu Disulfid-Dimeren
„R–S–S–R“ oxidiert werden, beispielsweise durch Sauerstoff aus der Luft, was die Qualität
gebildeter Monolagen stark beeinflusst.[69] In homoleptischen Komplexen mit zwei Thiol-
Funktionen kann die Bildung von Disulfiden zu Polymeren führen. Darüber hinaus besitzt die
Thiolgruppe durch ihr acides Wasserstoffatom Oxidationspotential für den Eisen(II)-Kern des
Komplexes. Ein Schutz der Thiolgruppe schafft in beiden Fällen Abhilfe. Die gängigsten
schwefelhaltigen funktionellen Gruppen zur Oberflächenverankerung sind in Tabelle 2.1
aufgeführt.[20, 22, 70]

2 Eisen(II)-Komplexe mit Schwefelanker 20
Tabelle 2.1. Auswahl schwefelhaltiger funktioneller Gruppen zur Verankerung auf Gold-
Oberflächen.
Funktionelle Gruppe Strukturformel Bindungsmuster auf Au-Oberfläche
Thiol R–SH
Thioacetat R–SAc
Thiocyanat R–SCN S
R
Disulfid R–S–S–R’ S
RS
R'
Thioether R–S–R’ S
R R'
Disulfide sind in der Regel schwer löslich und bilden bei Verwendung von asymmetrischen
Disulfiden keine homogenen Schichten. Thioether tragende Moleküle bilden generell
schwächere Bindungen zur Goldoberfläche aus als Thiolate, wobei die gebildeten Monolagen
zusätzlich weniger dicht gepackt sind und geringere Ordnungen aufweisen.[71] Das
Bindungsmuster von Thioacetaten und Thiocyanaten auf Goldoberflächen entspricht dem von
Thiolen. Diese funktionellen Gruppen können also als „geschützte Thiole“ betrachtet werden,
die das Oxidationspotential der –SH-Gruppe blockieren. Thioacetate erfordern eine in situ-
Entschützung durch reaktive Hilfsreagenzien, um gute Beschichtungen zu ergeben.[72] Als
Alternative kommt somit die Thiocyanatgruppe in Betracht, die als Ankergruppe für
Oberflächenbeschichtungen erst 2004 durch Ciszek et al. näher untersucht wurde.[70] Aus
Untersuchungen zur Beschichtung von Gold-, Silber- und Platin-Oberflächen durch
Thiocyanat-tragende Moleküle mittels XPS wurde der in Abbildung 2.2 dargestellte
Mechanismus der Bindung an die Oberfläche formuliert.[73]
RS
CN
Au
S CR N
S SRRSCN R – Au(CN)2
–
Au(CN)2– S S
R R
Au Au Au
Abbildung 2.2. Mechanismus der Bindung von RSCN an ein Goldsubstrat.
Neueste Untersuchungen zeigen, dass Thiocyanate bei Beachtung geeigneter
Präparationsbedingungen Monolagen derselben hohen Qualität wie die entsprechenden Thiole

2 Eisen(II)-Komplexe mit Schwefelanker 21
ergeben. Vor allem muss auf eine hohe Reinheit der Substanzen und eine adäquate
Beschichtungsdauer geachtet werden, um der höheren Stabilität der zu spaltenden S–C-
Bindung im Thiocyanat im Vergleich zur S–H-Bindung im Thiol Rechnung zu tragen.[74]
In der Laborphase der vorliegenden Arbeit existierten keine schwefel-funktionalisierten
Derivate des Bpp. In einer 2008 erschienenen Arbeit berichten Ruben et al. von der Synthese
des SCO-Komplexes VII (s. Kapitel 1), der eine derartige Verbindung darstellt.[68] Für die
Verwendung in SAMs weist Komplex VII zwei ungünstige Merkmale auf. Die Thioacetat-
Ankergruppe hat den Nachteil, dass einer guten Beschichtung der Goldoberfläche eine
basenkatalysierte Entschützung der Acetyl-geschützten Thiolat-Funktion vorangestellt werden
muss. Darüber hinaus sind die Liganden durch das konjugierte π-System elektronisch direkt
mit der Goldoberfläche verbunden, was mit Sicherheit Einfluss auf die Stärke des
Ligandenfeldes und damit auch das SCO-Verhalten hat.
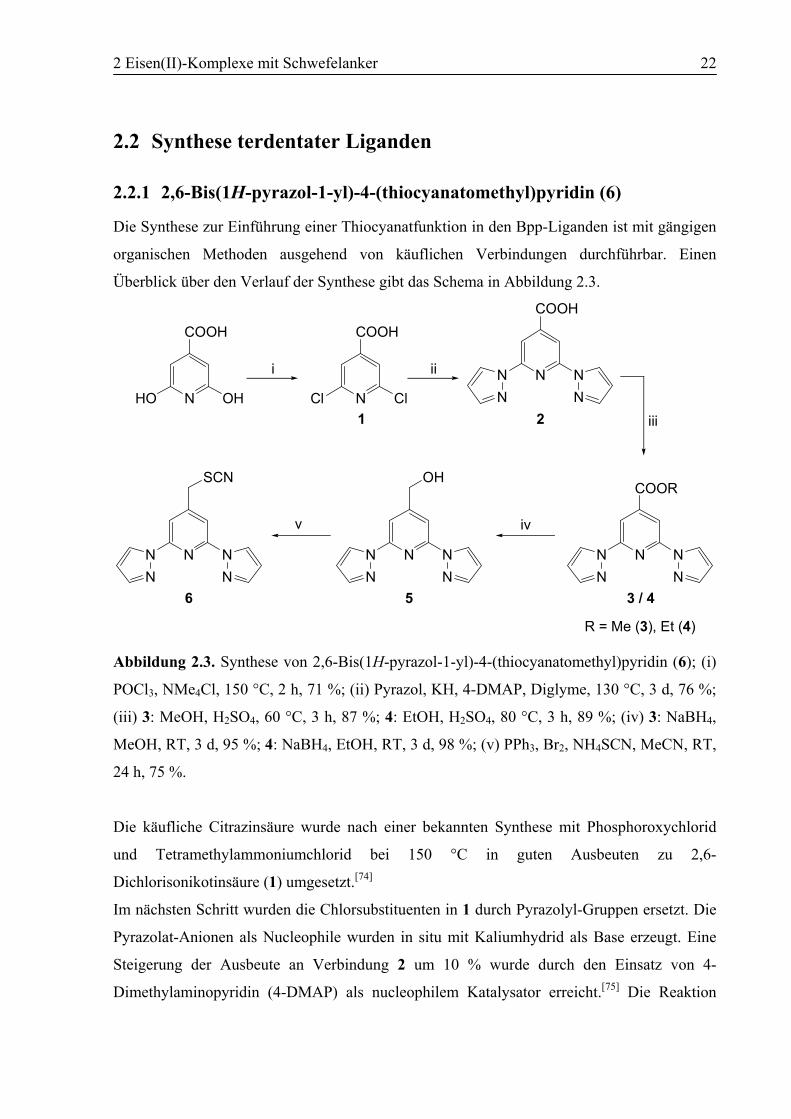
2 Eisen(II)-Komplexe mit Schwefelanker 22
2.2 Synthese terdentater Liganden
2.2.1 2,6-Bis(1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin (6)
Die Synthese zur Einführung einer Thiocyanatfunktion in den Bpp-Liganden ist mit gängigen
organischen Methoden ausgehend von käuflichen Verbindungen durchführbar. Einen
Überblick über den Verlauf der Synthese gibt das Schema in Abbildung 2.3.
NHO OH
COOH
NCl Cl
COOH
NNNN N
COOH
NNNN N
COOR
NNNN N
OH
NNNN N
SCN
i ii
iii
ivv
1 2
3 / 456
R = Me (3), Et (4)
Abbildung 2.3. Synthese von 2,6-Bis(1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin (6); (i)
POCl3, NMe4Cl, 150 °C, 2 h, 71 %; (ii) Pyrazol, KH, 4-DMAP, Diglyme, 130 °C, 3 d, 76 %;
(iii) 3: MeOH, H2SO4, 60 °C, 3 h, 87 %; 4: EtOH, H2SO4, 80 °C, 3 h, 89 %; (iv) 3: NaBH4,
MeOH, RT, 3 d, 95 %; 4: NaBH4, EtOH, RT, 3 d, 98 %; (v) PPh3, Br2, NH4SCN, MeCN, RT,
24 h, 75 %.
Die käufliche Citrazinsäure wurde nach einer bekannten Synthese mit Phosphoroxychlorid
und Tetramethylammoniumchlorid bei 150 °C in guten Ausbeuten zu 2,6-
Dichlorisonikotinsäure (1) umgesetzt.[74]
Im nächsten Schritt wurden die Chlorsubstituenten in 1 durch Pyrazolyl-Gruppen ersetzt. Die
Pyrazolat-Anionen als Nucleophile wurden in situ mit Kaliumhydrid als Base erzeugt. Eine
Steigerung der Ausbeute an Verbindung 2 um 10 % wurde durch den Einsatz von 4-
Dimethylaminopyridin (4-DMAP) als nucleophilem Katalysator erreicht.[75] Die Reaktion

2 Eisen(II)-Komplexe mit Schwefelanker 23
erfolgte in Anlehnung an eine Synthese von Verbindung 2 aus 2,6-Dibromisonikotinsäure.[76]
Die Synthese aus der Brom- anstelle der Chlorverbindung als Edukt erbringt normalerweise
deutlich höhere Ausbeuten, allerdings lässt sich 2,6-Dibromisonikotinsäure nur in schlechten
Ausbeuten und mit höherem Aufwand darstellen. Die Verwendung der gut zugänglichen
Chlorverbindung kombiniert mit dem Einsatz des nucleophilen Katalysators ergibt das beste
Verhältnis zwischen Kosten und Ausbeute für diese Synthesestufe. Ein Vergleich mit den
analytischen Daten aus der Literatur bestätigte die erfolgreiche Synthese.
Die Veresterung der Carbonsäure erfolgte sowohl zum Methyl- als auch zum Ethylester. Die
Veresterung von Verbindung 2 in MeOH, katalysiert mit Schwefelsäure, lieferte die
Methylesterverbindung 3 in guten Ausbeuten. Die Verbindung war bereits literaturbekannt
und wies identische Werte für die analytischen Daten auf.[77]
Säurekatalysierte Veresterung mit EtOH führte zur Ethylesterverbindung 4. Diese Verbindung
wurde bereits auf ähnlichem Wege synthetisiert.[76] Die Analytik zeigt die erwarteten Werte.
Farblose Einkristalle von 4 konnten aus einer Lösung der Verbindung in Chloroform durch
langsames Verdunsten bei Raumtemperatur erhalten und mittels Röntgenstrukturanalyse
untersucht werden. Verbindung 4 kristallisiert in der monoklinen Raumgruppe P21/c. Die
Einheitszelle enthält acht Moleküle von 4, mit 2 Molekülen in der asymmetrischen Einheit.
Abbildung 2.4 zeigt die Struktur eines Moleküls von 4 bei 293 K. Die aromatischen Ringe
liegen koplanar in einer Ebene mit Abweichungen zwischen den Ebenen von unter 5°. Die
Pyrazolringe sind um die Pyridin–Pyrazol-Bindungsachsen drehbar und nehmen jeweils eine
trans-Konformation bezogen auf das Pyridin-Stickstoffatom ein. Begünstigt wird diese
Konformation durch Ausbildung von intramolekularen Wasserstoffbrückenbindungen. Diese
bestehen einerseits zwischen dem Pyridin-Stickstoffatom und den pz5-Wasserstoffatomen mit
einem mittleren Abstand d(N(Pyridin)…H(pz5)) = 2.663(2) Å und andererseits zwischen den
Imin-Stickstoffatomen der Pyrazolringe und den Wasserstoffatomen in 3- und 5-Position des
Pyridinrings mit einem mittleren Abstand von d(N(pz)…H(py3/5)) = 2.518(2) Å. Im
Kristallgitter sind die Moleküle über Wasserstoffbrückenbindungen zwischen dem
Sauerstoffatom der Carbonylgruppe eines Moleküls mit einem pz5-Proton eines
Nachbarmoleküls mit einem mittleren Abstand d(OA…HB) = 2.485(2) Å zu Ketten verknüpft.
Zwischen den Ketten herrscht Zusammenhalt der Moleküle durch π-π-Stapel- und C–H…π-
Wechselwirkungen. Die trans,trans- Konfiguration der Pyrazolringe zum Pyridinring
entspricht der literaturbekannter Bpp-Liganden.[78-80]
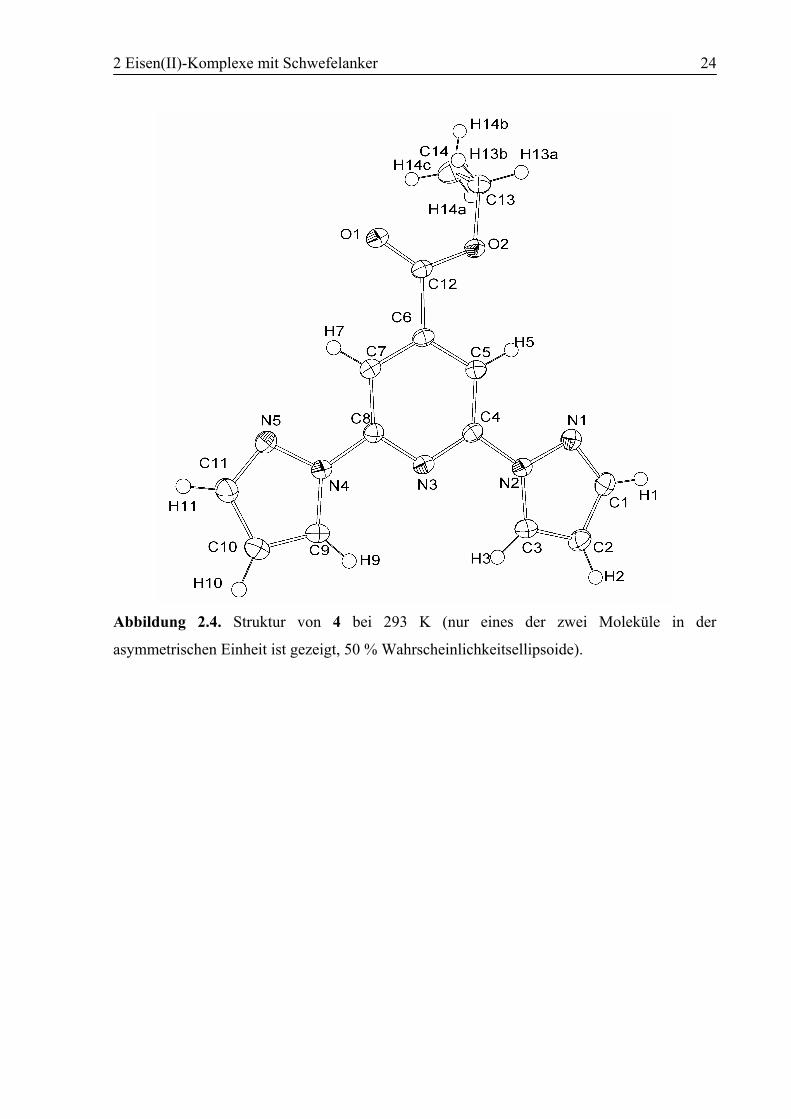
2 Eisen(II)-Komplexe mit Schwefelanker 24
Abbildung 2.4. Struktur von 4 bei 293 K (nur eines der zwei Moleküle in der
asymmetrischen Einheit ist gezeigt, 50 % Wahrscheinlichkeitsellipsoide).

2 Eisen(II)-Komplexe mit Schwefelanker 25
Tabelle 2.2. Ausgewählte Abstände [Å] und Winkel [°] für 4 bei 293 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
C4–N3 1.3320(17) N3-C4-C5 124.58(13)
C4–C5 1.3883(18) C7-C6-C5 120.29(12)
C5–C6 1.3897(19) C6-C7-C8 117.08(13)
C6–C7 1.3879(19) N3-C8-N4 114.69(12)
C7–C8 1.3928(19) C7-C8-N4 121.10(12)
C8–N3 1.3314(17) C8-N3-C4 116.92(12)
C1–C2 1.404(2) C3-C2-C1 104.91(13)
C2–C3 1.363(2) C2-C3-N2 106.52(13)
C3–N2 1.3635(18) C1-N1-N2 103.59(12)
N1–N2 1.3639(16) C3-N2-N1 112.38(12)
C4–N2 1.4127(17) C3-N2-C4 127.64(12)
C1-N1 1.3251(19) N1-N2-C4 119.92(11)
C10–C11 1.399(2) C9-C10-C11 105.39(13)
C9–C10 1.362(2) C10-C9-N4 106.22(13)
C9–N4 1.3657(18) C11-N5-N4 104.01(12)
N4–N5 1.3612(16) N5-N4-C9 112.16(12)
C8–N4 1.4061(17) C9-N4-C8 127.46(12)
C11–N5 1.3226(19) N5-N4-C8 120.37(11)
Die Reduktion der Ethylesterverbindung 4 erfolgte mit einem Überschuss (5 Äquivalente)
Natriumborhydrid in EtOH. Der Alkohol 2,6-Bis(pyrazol-1-yl)-4-(hydroxymethyl)pyridin (5)
konnte ohne weitere Aufreinigung in hoher Ausbeute gewonnen werden. Die erfolgte
Umsetzung wurde mit den zur Verfügung stehenden analytischen Methoden bestätigt. Das IR-
Spektrum zeigt den Wegfall der starken Bande der C=O-Streckschwingung der Estergruppe
bei 1729 cm–1 und eine neue breite Bande der O–H-Streckschwingung bei 3299 cm–1. Das EI-
Massenspektrum zeigt den Molekülionenpeak von 5 mit einer Intensität von 100 % bei m/z =
241. Abbildung 2.5 zeigt das 1H-NMR-Spektrum von Verbindung 5 in [D]Chloroform bei RT
mit Vergrößerungen der aufgespaltenen Signale.
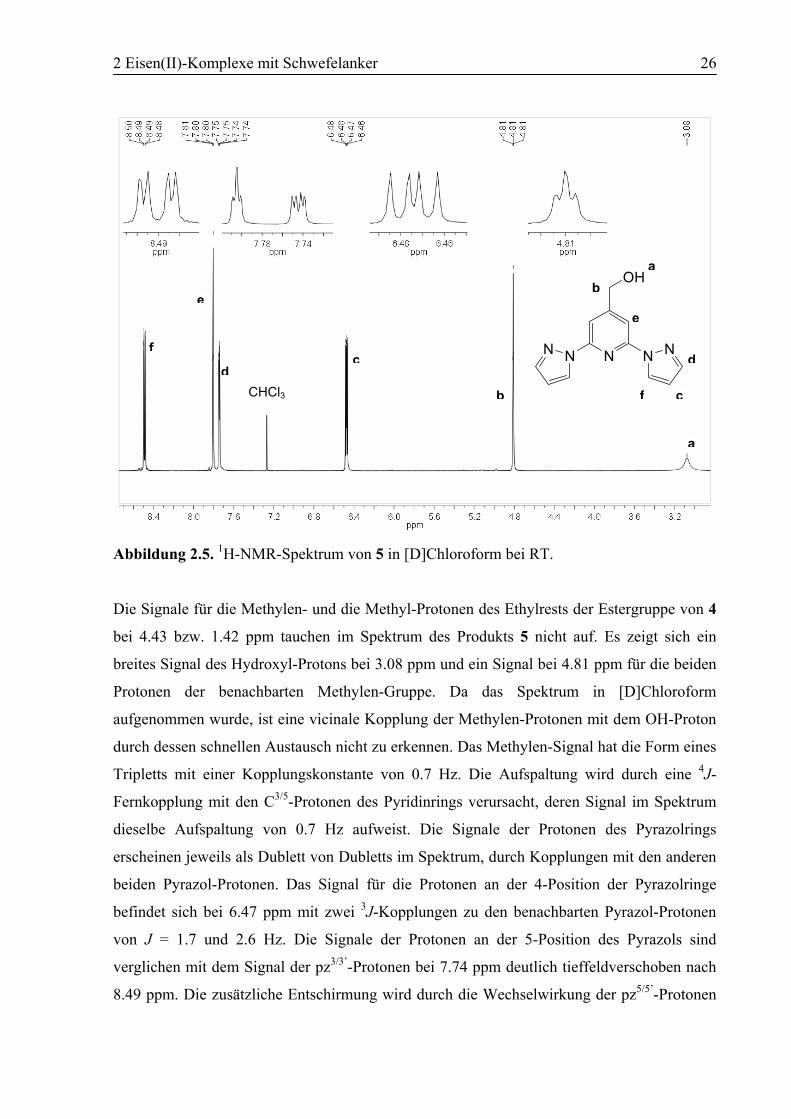
2 Eisen(II)-Komplexe mit Schwefelanker 26
Abbildung 2.5. 1H-NMR-Spektrum von 5 in [D]Chloroform bei RT.
Die Signale für die Methylen- und die Methyl-Protonen des Ethylrests der Estergruppe von 4
bei 4.43 bzw. 1.42 ppm tauchen im Spektrum des Produkts 5 nicht auf. Es zeigt sich ein
breites Signal des Hydroxyl-Protons bei 3.08 ppm und ein Signal bei 4.81 ppm für die beiden
Protonen der benachbarten Methylen-Gruppe. Da das Spektrum in [D]Chloroform
aufgenommen wurde, ist eine vicinale Kopplung der Methylen-Protonen mit dem OH-Proton
durch dessen schnellen Austausch nicht zu erkennen. Das Methylen-Signal hat die Form eines
Tripletts mit einer Kopplungskonstante von 0.7 Hz. Die Aufspaltung wird durch eine 4J-
Fernkopplung mit den C3/5-Protonen des Pyridinrings verursacht, deren Signal im Spektrum
dieselbe Aufspaltung von 0.7 Hz aufweist. Die Signale der Protonen des Pyrazolrings
erscheinen jeweils als Dublett von Dubletts im Spektrum, durch Kopplungen mit den anderen
beiden Pyrazol-Protonen. Das Signal für die Protonen an der 4-Position der Pyrazolringe
befindet sich bei 6.47 ppm mit zwei 3J-Kopplungen zu den benachbarten Pyrazol-Protonen
von J = 1.7 und 2.6 Hz. Die Signale der Protonen an der 5-Position des Pyrazols sind
verglichen mit dem Signal der pz3/3’-Protonen bei 7.74 ppm deutlich tieffeldverschoben nach
8.49 ppm. Die zusätzliche Entschirmung wird durch die Wechselwirkung der pz5/5’-Protonen
a
b
cd
e
f
CHCl3
ab
c
d
e
f
NN NN N
OH
2 Eisen(II)-Komplexe mit Schwefelanker 27
mit dem freien Elektronenpaar des Pyridin-Stickstoffatoms verursacht. Der Abstand
d(N(py)…H(pz5/5’)) beträgt in Verbindung 4 im Mittel 2.66 Å, wie aus der Röntgenstruktur
hervorgeht. Für 5 kann die gleiche Konformation mit ähnlichen Bindungsabständen erwartet
werden. Im 13C-NMR-Spektrum von Verbindung 5 sind die Signale für die Esterfunktion von
4 bei 164.48 (C=O), 62.70 (CH2) und 14.79 ppm (CH3) nicht mehr vorhanden.
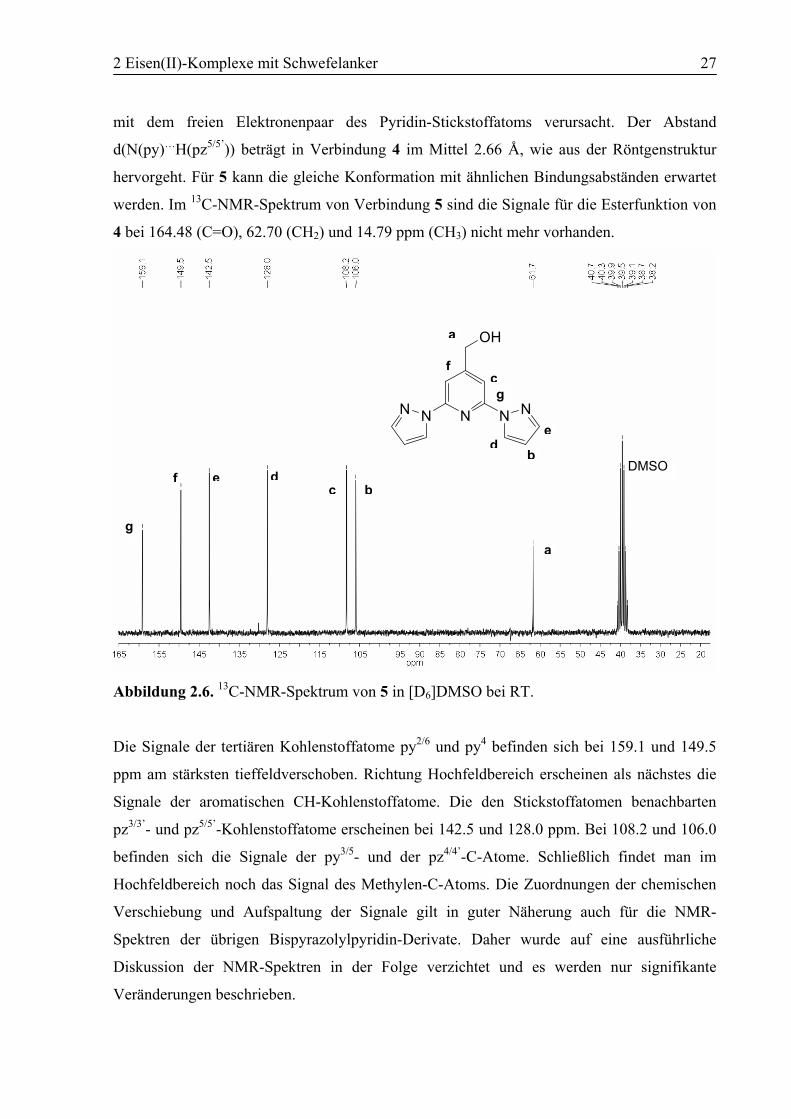
Abbildung 2.6. 13C-NMR-Spektrum von 5 in [D6]DMSO bei RT.
Die Signale der tertiären Kohlenstoffatome py2/6 und py4 befinden sich bei 159.1 und 149.5
ppm am stärksten tieffeldverschoben. Richtung Hochfeldbereich erscheinen als nächstes die
Signale der aromatischen CH-Kohlenstoffatome. Die den Stickstoffatomen benachbarten
pz3/3’- und pz5/5’-Kohlenstoffatome erscheinen bei 142.5 und 128.0 ppm. Bei 108.2 und 106.0
befinden sich die Signale der py3/5- und der pz4/4’-C-Atome. Schließlich findet man im
Hochfeldbereich noch das Signal des Methylen-C-Atoms. Die Zuordnungen der chemischen
Verschiebung und Aufspaltung der Signale gilt in guter Näherung auch für die NMR-
Spektren der übrigen Bispyrazolylpyridin-Derivate. Daher wurde auf eine ausführliche
Diskussion der NMR-Spektren in der Folge verzichtet und es werden nur signifikante
Veränderungen beschrieben.
a
bcdef
g
DMSO
a
b
c
de
f
gNN NN N
OH

2 Eisen(II)-Komplexe mit Schwefelanker 28
Die Substitution der Hydroxylgruppe durch die Thiocyanatgruppe wurde unter Nutzung eines
Protokolls von Iranpoor[81] durchgeführt. Bei dieser Methode wird aus einem Gemisch von
Triphenylphosphan, Brom und Ammoniumthiocyanat die Spezies Ph3P(SCN)2 in situ erzeugt.
Die Methode weist mehrere Vorteile auf gegenüber dem Einsatz von freien SCN–-Ionen als
Nucleophil, beispielsweise in Form von KSCN, oder der Verwendung von giftigem
Thiocyanogen ((SCN)2). Die Reaktion zeigt eine hohe Chemoselektivität. So entsteht
ausschließlich die Thiocyanat-Spezies als Produkt. Das entsprechende Isothiocyanat, welches
bei Verwendung von KSCN (s. Kap. 2.2.2) das Hauptprodukt stellt, wird nicht gebildet.
Darüber hinaus verläuft die Umsetzung schnell und bei Raumtemperatur. Bei der Reaktion
wurde zunächst das Triphenylphosphan mit Brom in einer Lösung in MeCN umgesetzt. Das
entstandene Ph3P(Br)2 wurde anschließend mit NH4SCN umgesetzt, wobei Ph3P(SCN)2
entsteht. Nach Zugabe des Alkohols 5 und anschließendem Rühren bei RT fällt als
Nebenprodukt Triphenylphosphanoxid in Form eines weißen Niederschlags aus. Dieser wurde
abgetrennt und das Filtrat säulenchromatographisch gereinigt, wobei die Thiocyanat-
Verbindung 6 als weißer Feststoff erhalten wurde. Mit der Analytik zu Verbindung 6 musste
neben der Identität der Substanz sichergestellt werden, dass es sich um das Thiocyanat- und
nicht um das Isothiocyanat-Isomer handelt (s. Kap. 2.2.2). Im Vergleich zu Verbindung 5
zeigt das 1H-NMR-Spektrum von 6 nicht länger das breite Signal des OH-Protons der
Hydroxlgruppe. Das Signal der Methylen-Protonen ist im Spektrum von 6 mit 4.21 ppm
hochfeldverschoben im Gegensatz zu 4.81 ppm für 5, entsprechend der geringeren
Elektronegativität des Schwefelatoms verglichen mit dem Sauerstoffatom der Hydroxyl-
gruppe. Aus dem gleichen Grund verschiebt sich das Signal des Kohlenstoffatoms der
Methylengruppe im 13C-NMR-Spektrum deutlich Richtung Hochfeld von 61.7 ppm in 5 nach
36.6 ppm in Verbindung 6. Das Signal des Kohlenstoffatoms der Thiocyanatgruppe erscheint
im Spektrum bei 110.6 ppm im typischen Bereich für Thiocyanate.[81] Signale für das
Kohlenstoffatom in der Isothiocyanatgruppe erscheinen im 13C-NMR-Spektrum bei tieferem
Feld um 130 ppm. Die IR-Spektroskopie stellt ein besonders geeignetes Instrument dar,
Thiocyanat-Verbindungen zu identifizieren. Die Bande der C≡N-Streckschwingung der SCN-
Gruppe erscheint im Bereich 2175–2135 cm–1 mit starker Intensität in einer
Wellenzahlenregion, in der ansonsten wenige Gruppen charakteristische Absorptionen zeigen.
Im Spektrum der Verbindung 6 befindet sich diese starke Bande bei 2160 cm–1. Zusätzlich
findet sich eine charakteristische mittelstarke Bande der ν(C–S)-Schwingung bei 618 cm–1,
welche im IR-Spektrum des Edukts nicht vorhanden ist. Die Elementaranalyse stimmt mit den

2 Eisen(II)-Komplexe mit Schwefelanker 29
berechneten Werten überein. Hinweise, dass als Nebenprodukt der Reaktion die
Isothiocyanatspezies entstanden ist, finden sich nicht. Im IR-Spektrum wäre ein Isothiocyanat
eindeutig anhand der sehr starken, breiten Doppel-Bande der asymmetrischen –N=C=S-
Streckschwingung im Bereich 2140–2080 cm–1 zu identifizieren (s. Kap. 2.2.2).
Die einzelnen Stufen der Synthese können jede für sich im Gramm-Maßstab mit guten
Ausbeuten durchgeführt werden. Hinsichtlich des Synthesekonzepts liegt mit Verbindung 6
ein dreizähniger Ligand zur Verankerung auf Oberflächen vor, der als Ankergruppe eine
Thiocyanatfunktion und als Distanzstück eine Methylengruppe aufweist.
Beschichtungsversuche von Goldoberflächen mit Ligand 6 wurden durchgeführt. Diese
Untersuchungen werden in Kapitel 4 diskutiert.
2.2.2 2,6-Bis(1H-pyrazol-1-yl)-4-(isothiocyanatomethyl)pyridin (8)
Die Thiocyanatverbindung 6 sollte ursprünglich über eine alternative Syntheseroute
hergestellt werden. Ausgehend vom Alkohol 5 sollte dabei aus der Bromverbindung 7 mittels
nucleophiler Substitution durch Thiocyanat-Ionen die Thiocyanatverbindung 6 gewonnen
werden. Infolge des ambidenten Charakters des Thiocyanat-Ions bildete sich jedoch
stattdessen die Isothiocyanatverbindung 8. Abbildung 2.7 zeigt den geplanten und den
tatsächlichen Verlauf der Synthese.
NNNN N
OH
NNNN N
Br NNNN N
SCN
NNNN N
NCS
i
ii
5 7
6
8
Abbildung 2.7. Synthese von 2,6-Bis(1H-pyrazol-1-yl)-4-(isothiocyanatomethyl)pyridin (8);
(i) PPh3, Br2, DCM, 12 h, RT, 66 %; (ii) KSCN, Diglyme, 24 h, 100 °C, 35 %.

2 Eisen(II)-Komplexe mit Schwefelanker 30
Die Alkohol-Verbindung 5 wurde mit Triphenylphosphan und elementarem Brom bei 0 °C
zusammengegeben und 12 h bei Raumtemperatur gerührt. Nach Abtrennung des entstandenen
Triphenylphosphanoxids und säulenchromatographischer Trennung konnte 2,6-Bis(1H-
pyrazol-1-yl)-4-(brommethyl)pyridin 7 in Form eines gelblichen Feststoffes gewonnen
werden. Das 1H-NMR-Spektrum von 7 zeigt im Vergleich zum Edukt 5 die erwartete
Verschiebung des Signals der Methylengruppe in Richtung Hochfeld von 4.81 ppm nach 4.48
ppm durch die schwächere Entschirmung der Methylen-Protonen durch das Bromatom. Das
breite Signal für das Hydroxyl-Proton des Alkohols ist im Spektrum der Verbindung 7 nicht
enthalten.
Die Bromverbindung 7 sollte anschließend in einer nucleophilen Substitution zum Thiocyanat
6 umgesetzt werden. Dazu wurde 7 zusammen mit Kaliumthiocyanat in Diglyme für 24 h auf
100 °C erhitzt. Nach säulenchromatographischer Trennung erhielt man Verbindung 8 als
weißen Feststoff. Die Elementaranalyse von Verbindung 8 entspricht der Summenformel
C13H10N6S, was sowohl der Thiocyanat- als auch der Isothiocyanatverbindung entspricht. Das 1H-NMR-Spektrum von 8 zeigt das Signal der Methylengruppe bei 4.80 ppm. Diese
Verschiebung entspricht der von Methylengruppen in vergleichbaren Isothiocyanat-
Verbindungen.[82] Das Signal der Methylen-Protonen in der Thiocyanatverbindung 6 erscheint
zu höherem Feld verschoben bei 4.21 ppm. Verbindungen 6 und 8 lassen sich am deutlichsten
durch IR-Spektroskopie unterscheiden. Abbildung 2.8 zeigt die IR-Spektren beider
Verbindungen als Feststoffe in KBr.
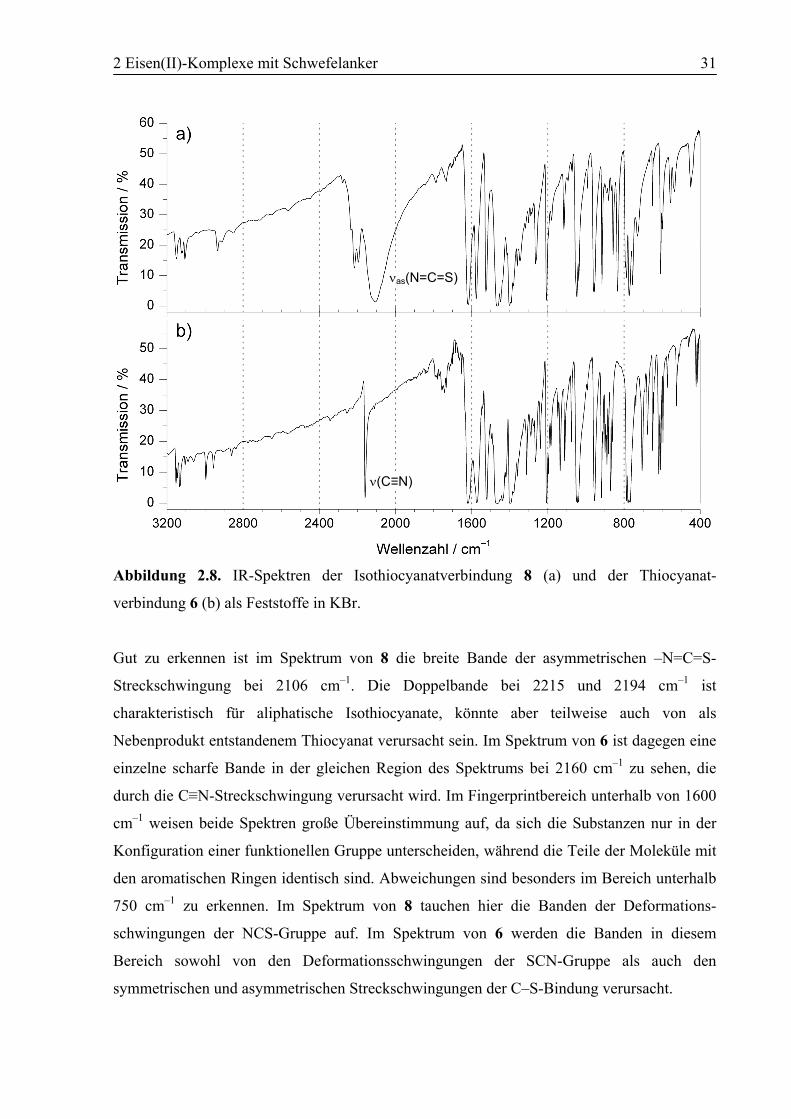
2 Eisen(II)-Komplexe mit Schwefelanker 31
Abbildung 2.8. IR-Spektren der Isothiocyanatverbindung 8 (a) und der Thiocyanat-
verbindung 6 (b) als Feststoffe in KBr.
Gut zu erkennen ist im Spektrum von 8 die breite Bande der asymmetrischen –N=C=S-
Streckschwingung bei 2106 cm–1. Die Doppelbande bei 2215 und 2194 cm–1 ist
charakteristisch für aliphatische Isothiocyanate, könnte aber teilweise auch von als
Nebenprodukt entstandenem Thiocyanat verursacht sein. Im Spektrum von 6 ist dagegen eine
einzelne scharfe Bande in der gleichen Region des Spektrums bei 2160 cm–1 zu sehen, die
durch die C≡N-Streckschwingung verursacht wird. Im Fingerprintbereich unterhalb von 1600
cm–1 weisen beide Spektren große Übereinstimmung auf, da sich die Substanzen nur in der
Konfiguration einer funktionellen Gruppe unterscheiden, während die Teile der Moleküle mit
den aromatischen Ringen identisch sind. Abweichungen sind besonders im Bereich unterhalb
750 cm–1 zu erkennen. Im Spektrum von 8 tauchen hier die Banden der Deformations-
schwingungen der NCS-Gruppe auf. Im Spektrum von 6 werden die Banden in diesem
Bereich sowohl von den Deformationsschwingungen der SCN-Gruppe als auch den
symmetrischen und asymmetrischen Streckschwingungen der C–S-Bindung verursacht.
ν(C≡N)
νas(N=C=S)

2 Eisen(II)-Komplexe mit Schwefelanker 32
Die fehlgeschlagene Synthese der Thiocyanatverbindung 6 auf dieser Route ergab die
Isothiocyanatverbindung 8. Durch das Vorliegen der analytischen Untersuchungen zur
Isothiocyanatverbindung konnte jedoch in den späteren Versuchen zur Synthese der
Thiocyanatverbindung (Kap. 2.2.1) zuverlässig auf entstandenes Isothiocyanat als
Nebenprodukt geprüft werden.
2.2.3 N-(2-Mercaptoethyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid
(10)
Nachdem Untersuchungen zur Beschichtung von Goldoberflächen mit dem
Thiocyanatliganden 6 und dessen Eisen(II)-Komplex schlechte bzw. keine Ordnung der
Moleküle auf der Oberfläche zeigten (vgl. Kapitel 4), wurde der Ligand gemäß dem flexiblen
Synthesekonzept verändert. Zwei Eigenschaften der verwendeten Moleküle sollten dabei
angepasst werden. Zum einen sind die verwendeten Moleküle in ihrer Gestalt möglicherweise
ungünstig zur Ausbildung gleichmäßiger Schichten. Der relativ schlanke Anker mit dem
Distanzstück trägt als Endgruppe den sterisch stark ausladenden Eisen(II)-Komplex.
Moleküle in geordneten Lagen auf Oberflächen sind jedoch auf signifikante intermolekulare
Wechselwirkungen angewiesen. Diese bestehen in den meisten Fällen aus van-der-Waals-
Wechselwirkungen. Durch die sterisch größere Endgruppe der hier verwendeten Moleküle
werden die Kontaktflächen zu benachbarten Molekülen kleiner und werden von den
Wechselwirkungen der Endgruppen untereinander bestimmt. Die Abstände und Winkel, die
ein solchermaßen erzeugtes Netzwerk aufweisen würde, würden nicht mit dem
darunterliegenden Au(111)-Gitter übereinstimmen. Die Moleküle können deshalb vermutlich
keine größeren geordneten Bereiche bilden.
S
R
S
R
S
R
S
R
S
R
S
R
S S S S S Sa) b)
Abbildung 2.9. Modellansicht von (a) Alkanthiolen (R = Alkylkette) und (b)
oberflächenverankerten Molekülen mit sterisch anspruchsvollen Endgruppen.
Als erstes sollte daher das Distanzstück, das in 6 aus einer Methylengruppe besteht, durch
eine längere Gruppe ersetzt werden, um den Molekülen eine höhere Flexibilität zu geben.
Durch Konformationsänderungen in der längeren Kette könnte so sterische Spannung
2 Eisen(II)-Komplexe mit Schwefelanker 33
abgebaut und die Wechselwirkungen zwischen den Molekülen erhöht werden. Eine bessere
Ordnung der Moleküle würde auch durch stärkere intermolekulare Wechselwirkungen auf der
Oberfläche erreicht werden. Daher sollte zusätzlich durch Einfügung von Amid-Funktionen in
die Distanzstücke die Ausbildung von intermolekularen Wasserstoffbrückenbindungen
erreicht werden.
S S
N N
R RO O
H
S S
N NH
R RO O
HH
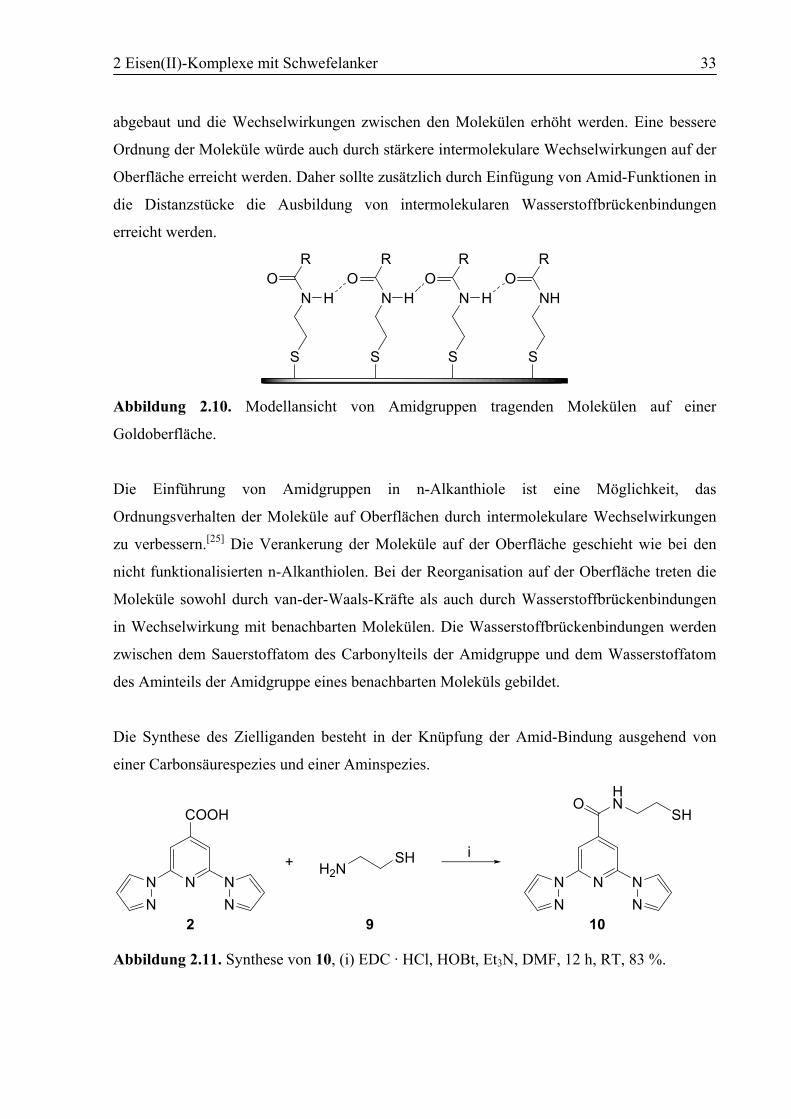
Abbildung 2.10. Modellansicht von Amidgruppen tragenden Molekülen auf einer
Goldoberfläche.
Die Einführung von Amidgruppen in n-Alkanthiole ist eine Möglichkeit, das
Ordnungsverhalten der Moleküle auf Oberflächen durch intermolekulare Wechselwirkungen
zu verbessern.[25] Die Verankerung der Moleküle auf der Oberfläche geschieht wie bei den
nicht funktionalisierten n-Alkanthiolen. Bei der Reorganisation auf der Oberfläche treten die
Moleküle sowohl durch van-der-Waals-Kräfte als auch durch Wasserstoffbrückenbindungen
in Wechselwirkung mit benachbarten Molekülen. Die Wasserstoffbrückenbindungen werden
zwischen dem Sauerstoffatom des Carbonylteils der Amidgruppe und dem Wasserstoffatom
des Aminteils der Amidgruppe eines benachbarten Moleküls gebildet.
Die Synthese des Zielliganden besteht in der Knüpfung der Amid-Bindung ausgehend von
einer Carbonsäurespezies und einer Aminspezies.
NNNN N
COOH
NNNN N
HN
SHH2N+
SHO
i
2 9 10
Abbildung 2.11. Synthese von 10, (i) EDC · HCl, HOBt, Et3N, DMF, 12 h, RT, 83 %.

2 Eisen(II)-Komplexe mit Schwefelanker 34
Zur Knüpfung der Bindung wurde die Methode von König und Geiger verwendet.[83] Statt des
Systems DCC/HOBt (DCC = Dicyclohexylcarbodiimid) wurde hier jedoch EDC · HCl/HOBt
(EDC = 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-hydrochlorid) verwendet. EDC
besitzt gegenüber DCC mehrere Vorteile.[84] Der bei der Reaktion aus dem Carbodiimid
entstehende Harnstoff ist im Fall von EDC im Gegensatz zu DCC sehr gut in polaren
Lösungsmitteln löslich und dadurch nach der Reaktion gut abzutrennen. Darüber hinaus ist
DCC auch giftiger als EDC. Die Carbonsäureverbindung 2 wurde zunächst mit EDC-
Hydrochlorid und HOBt in DMF zusammengegeben, um die Säurefunktion zu aktivieren.
Anschließend wurde Cysteamin (9) und nach 20 min Triethylamin zum Reaktionsgemisch
gegeben. Das Gemisch wurde dann 12 h bei Raumtemperatur gerührt und anschließend
wässrig aufgearbeitet. Eine säulenchromatographische Aufreinigung ist nicht notwendig, N-
(2-Mercaptoethyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid (10) entsteht in guten
Ausbeuten über 80 % bezogen auf das Edukt 2, in hoher Reinheit. Bei der analytischen
Untersuchung des Produkts konnte neben der Bestätigung einer erfolgreichen Umsetzung
auch das mögliche Vorliegen einer Disulfidverbindung anstelle der gewünschten
Thiolverbindung ausgeschlossen werden. Die Elementaranalyse von 10 stimmt gut mit den
berechneten Werten überein. Das IR-Spektrum, aufgenommen als Feststoff in KBr, bestätigt
durch den Vergleich mit dem Spektrum des Edukts 2 die erfolgreiche Umsetzung. Die
Absorptionsbanden, die den aromatischen Ringen zugeordnet werden, werden im
Produktspektrum wieder gefunden. Die Bande der C=O-Streckschwingung der
Carbonsäurefunktion in 2 bei 1725 cm–1 ist im Produktspektrum nicht mehr zu finden. Das
Vorliegen der Amid-Funktion wird im Spektrum von 10 durch drei charakteristische Banden
angezeigt, die im Spektrum von 2 nicht vorkommen. Die Bande der N–H-Streckschwingung
erscheint bei 3264 cm–1; die Amid I-Bande der C=O-Streckschwingung erscheint mit starker
Intensität bei 1643 cm–1; die Amid II-Bande der N–H-Deformations- und der C–N-
Streckschwingung findet sich bei 1543 cm–1. Bei 2556 cm–1 findet sich eine schwache Bande,
die durch die S–H-Streckschwingung der Thiolfunktion verursacht wird. In dieser Region des
Spektrums tauchen generell sehr wenige Absorptionsbanden auf, sodass die beobachtete
Bande eindeutig zugeordnet werden kann. Eine Disufidverbindung könnte möglicherweise im
IR-Spektrum anhand von zwei schwachen Banden ihrer S–S-Streckschwingung im Bereich
530-500 cm–1 identifiziert werden. Dieser Bereich ist im Spektrum von 10 frei von
Absorptionsbanden. Das 13C-NMR-Spektrum von 10 zeigt die erwarteten Signale, mit den
hinzugekommenen Signalen der beiden Methylen-C-Atome bei 43.0 und 32.1 ppm. Im 1H-

2 Eisen(II)-Komplexe mit Schwefelanker 35
NMR-Spektrum von 10 ist das breite Signal des Carboxyl-Protons des Edukts bei 14.10 ppm
nicht mehr vorhanden. Die Signale der Wasserstoffatome der Pyrazolringe und des
Pyridinringes im Aromatenbereich sind nicht oder unwesentlich verschoben. Bei 9.26 ppm
findet sich das Signal des Amid-Protons in Form eines Tripletts durch die Kopplung mit den
beiden benachbarten Methylenprotonen. Bei 2.50 ppm befindet sich ein Multiplett-Signal, das
dem Thiol-Proton zugeordnet werden kann. Die beiden letzten Signale sind im Spektrum nur
in Lösung in [D6]DMSO zu erkennen, wie es für schnell austauschende Protonen zu erwarten
ist. Sie entsprechen in ihrem Integral je einem Wasserstoffatom, wobei das Thiol-Signal vom
Resonanzsignal des Lösungsmittels bei 2.49 ppm überlagert ist. Zwei neue Signale zeigen
sich bei 3.47 und 2.69 ppm, die den Methylengruppen der Alkylkette zugeordnet sind. Beide
Signale sind jeweils in ein Dublett von Tripletts aufgespalten und entsprechen in ihren
relativen Integralen zwei Wasserstoffatomen. Die Triplettaufspaltung wird durch die vicinale
Kopplung der Methylen-Protonen untereinander verursacht. Die Dublett-Aufspaltungen
kommen durch die Kopplung der Methylengruppe bei 3.47 ppm mit dem Amid-Proton
einerseits und durch die Kopplung der Methylengruppe bei 2.69 mit dem Thiol-Proton
andererseits zustande. Wird der NMR-Probe ein Tropfen D2O zugefügt, ändert sich das
resultierende 1H-NMR-Spektrum erwartungsgemäß. Durch den Wasserstoff↔Deuterium-
Austausch verschwinden die Signale für das Amid- und das Thiol-Proton im Spektrum,
während die beiden Methylen-Signale nur noch jeweils in ein Triplett aufgespalten sind.
Abbildung 2.12 zeigt das 1H-NMR-Spektrum der Verbindung 10 mit der Zuordnung der
Signale. Ausschnitte aus dem Spektrum nach Zugabe von D2O zur Probelösung sind gezeigt.
In diesen Bereichen fallen die Signale der aciden Amid- und Thiolprotonen weg. Im Bereich
des Thiolprotons um 2.50 ppm verbleibt ein schwaches Signal, das durch das Lösungsmittel
DMSO verursacht wird.

2 Eisen(II)-Komplexe mit Schwefelanker 36
Abbildung 2.12. 1H-NMR-Spektrum von 10 in [D6]DMSO bei RT. Die Ausschnitte zeigen
die Bereiche von 9.4-8.9 und 2.8-2.4 ppm nach Zugabe von D2O zur Probelösung.
Die Untersuchungen bestätigen die Bildung der gewünschten Verbindung. Mithilfe der 1H-
sowie der IR-Spektroskopie konnte das Vorliegen der freien Thiolfunktion in der Verbindung
nachgewiesen werden. Verbindung 10 lässt sich in Ausbeuten über 80 % im Gramm-Maßstab
elementaranalysenrein reproduzieren. Die Ligandensynthese eröffnet durch den modularen
Ansatz die Möglichkeit, Derivate größerer Kettenlänge darzustellen. Dazu könnte das
eingesetzte Cysteamin durch α-Mercapto-ω-amine unterschiedlicher Methylen-Kettenlänge
ersetzt werden. Cysteamin ist allerdings das einzige günstig käuflich erhältliche α-Mercapto-
ω-amin-n-alkan. Die Synthese von Mercaptoamin-n-alkanen längerer Kette stellte sich als
schwierig heraus und wurde nach Untersuchung des Beschichtungsverhaltens des Liganden
und vor allem dessen Eisenkomplexen (Kapitel 4) zugunsten alternativer Liganden
zurückgestellt. Ein weiterer Ligand wurde noch nach dem vorgestellten Konzept synthetisiert
(s. Kapitel 2.2.4).
Mit Verbindung 10 liegt ein dreizähniger Ligand zur Verankerung auf Goldoberflächen mit
Thiolankergruppe vor. Zusätzlich ermöglicht die Struktur von 10 die Ausbildung von nicht-
+ D2O + D2Oa
b
c
d
e
fg
h
abc
de
f
g
h
NNNN N
HN
SHO

2 Eisen(II)-Komplexe mit Schwefelanker 37
bindenden intermolekularen Wechselwirkungen in Form von Wasserstoffbrückenbindungen
und Flexibilität durch eine größere Kettenlänge, womit eine höhere Ordnung der Monolagen
erreicht werden könnte. Beschichtungsversuche mit Ligand 10 auf Goldoberflächen wurden
durchgeführt und werden in Kapitel 4 behandelt.
2.2.4 N-(4-(4-Mercaptophenyl)phenyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-
carboxamid (12)
Eine in neuester Zeit untersuchte Verbindungsklasse für die Präparation von SAMs auf
Goldoberflächen stellen ω-(Biphenyl-4-yl)alkanthiole dar.[85, 86]
CH2 SHR R = H, Me
n = 0-6n
Abbildung 2.13. Allgemeine Strukturformel von ω-(Biphenyl-4-yl)alkanthiolen.
In den Monolagen dieser Verbindungen existieren intermolekulare Wechselwirkungen in
Form von π-π-Stapelwechselwirkungen zwischen den aromatischen Ringen. Moleküle mit
längeren Alkylketten bringen hierbei die besten Ergebnisse. Bei Knüpfung einer
Amidbindung der Carbonsäure 2 zu einem ω-(Biphenyl-4-yl)alkanthiol mit einem primären
Amin in der 4’-Position würde ein Ligand erhalten werden, der vergleichbar mit Ligand 10
durch Ausbildung zweier Arten sekundärer Wechselwirkungen höhere Ordnung der
Monolagen auf der Oberfläche zeigen könnte.
Die Synthese besteht in der Verknüpfung der Carbonsäure 2 mit dem Mercaptoamin 11 durch
Knüpfung einer Amidbindung nach der König-Geiger-Methode[83] unter Verwendung von
EDC statt DCC.
N
N
N
N
N
COOH
N
N
N
N
N
HN
O
SHi
2 12
H2N
SH
+
11
Abbildung 2.14. Synthese von N-(4-(4-Mercaptophenyl)phenyl)-2,6-bis(1H-pyrazol-1-
yl)pyridin-4-carboxamid (12); (i) EDC · HCl, HOBt, Et3N, DMF, 18 h, RT, 61 %.

2 Eisen(II)-Komplexe mit Schwefelanker 38
EDC · HCl, HOBt, die Carbonsäureverbindung 2 und das Mercaptoamin 11 wurden in DMF
zusammengegeben. Nach 20 min wurde Triethylamin dem Reaktionsgemisch hinzugefügt
und die resultierende Lösung 18 h bei RT gerührt. Nach Abtrennung der Nebenprodukte
durch wässrige Aufarbeitung konnte der Ligand 12 in Form eines weißen Feststoffes in 61 %
Ausbeute gewonnen werden. Das EI-Massenspektrum des Produkts zeigt den erwarteten
Molekülionenpeak mit einer Intensität von 100 % bei m/z = 438. Im Massenspektrum sind
Peaks bei m/z = 201 und 238 enthalten, die Fragmenten nach einer Spaltung der
Amidbindung im Massenspektrum entsprechen. Signale, die einem Dimer der Verbindung
(infolge Disulfidbildung) entsprechen, sind auch bei höheren Messtemperaturen nicht im
Spektrum zu finden. Das 1H-NMR-Spektrum des Produkts in [D6]DMSO zeigt die erwarteten
Signale. Das Signal des einzelnen Amid-Protons befindet sich bei 10.96 ppm. Verglichen mit
dem Signal des Amid-Protons in 10 liegt es noch weiter in Richtung Tieffeldbereich
verschoben, verursacht durch die Nachbarschaft zum aromatischen Ringsystem. Bei Zugabe
von D2O zur Probelösung ist das Signal im resultierenden Spektrum nicht zu finden. Die
Signale der Pyrazol- und der Pyridin-Protonen befinden sich kaum verschoben an den Stellen
im Spektrum wie in Verbindung 10. Das Signal des Protons am C3-Atom der Pyrazolringe bei
7.93 ppm wird dabei durch ein Multiplett überlagert, das durch zwei der Protonen des
Biphenylsystems erzeugt wird. Die restlichen sechs Protonen erscheinen in Multipletts bei
7.73 und 7.64 ppm. Das Signal des Thiol-Protons liegt bei 3.49 ppm im typischen Bereich für
Thiophenole (das S–H-Signal für Thiophenol erscheint bei 3.40 ppm).[82] Bei Zugabe von
D2O zur Probelösung ist kein Signal im resultierenden Spektrum zu erkennen. Die genaue
Identifikation wird durch die Tatsache erschwert, dass Restwasser im verwendeten D2O ein
Signal im gleichen Bereich zeigt, wie im Spektrum zu erkennen ist (Abbildung 2.15)
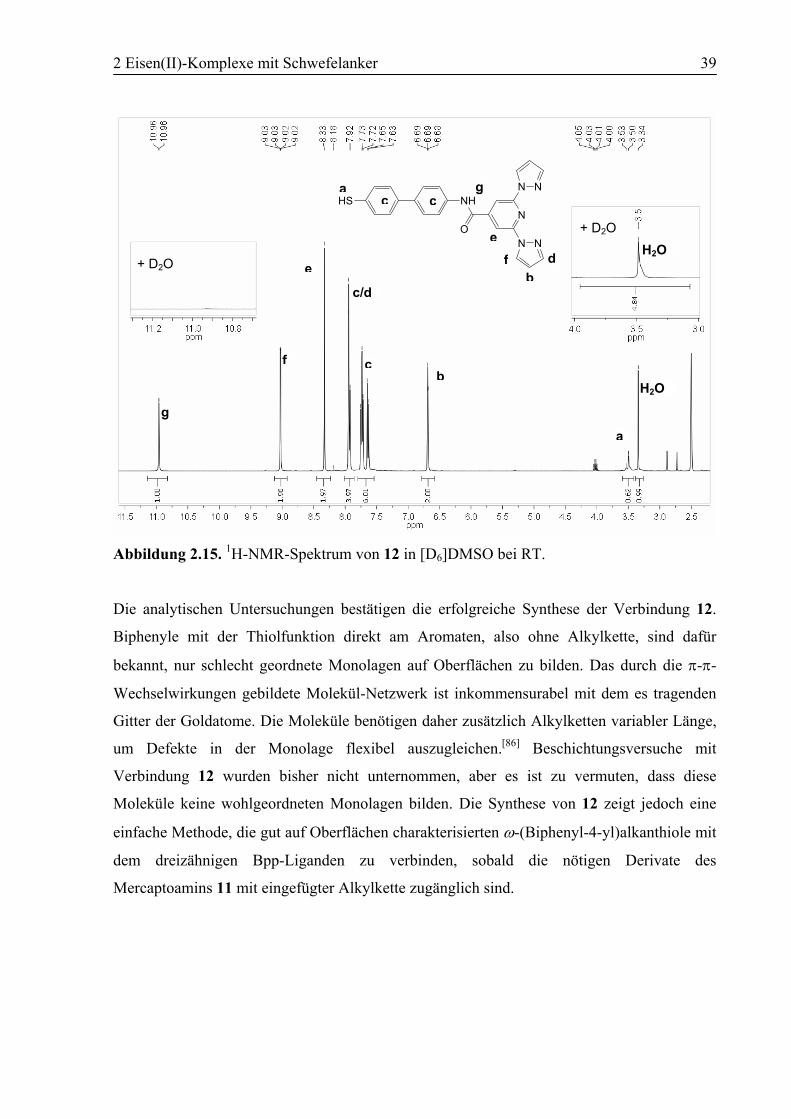
2 Eisen(II)-Komplexe mit Schwefelanker 39
Abbildung 2.15. 1H-NMR-Spektrum von 12 in [D6]DMSO bei RT.
Die analytischen Untersuchungen bestätigen die erfolgreiche Synthese der Verbindung 12.
Biphenyle mit der Thiolfunktion direkt am Aromaten, also ohne Alkylkette, sind dafür
bekannt, nur schlecht geordnete Monolagen auf Oberflächen zu bilden. Das durch die π-π-
Wechselwirkungen gebildete Molekül-Netzwerk ist inkommensurabel mit dem es tragenden
Gitter der Goldatome. Die Moleküle benötigen daher zusätzlich Alkylketten variabler Länge,
um Defekte in der Monolage flexibel auszugleichen.[86] Beschichtungsversuche mit
Verbindung 12 wurden bisher nicht unternommen, aber es ist zu vermuten, dass diese
Moleküle keine wohlgeordneten Monolagen bilden. Die Synthese von 12 zeigt jedoch eine
einfache Methode, die gut auf Oberflächen charakterisierten ω-(Biphenyl-4-yl)alkanthiole mit
dem dreizähnigen Bpp-Liganden zu verbinden, sobald die nötigen Derivate des
Mercaptoamins 11 mit eingefügter Alkylkette zugänglich sind.
a
bc
c/d
e
f
g
H2O
H2O
a
b
c
de
f
gc
+ D2O
+ D2O N
N
N
N
N
NH
O
HS

2 Eisen(II)-Komplexe mit Schwefelanker 40
2.2.5 (2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methyl-11-mercaptoundecanoat
(14)
Die Synthese von Homologen von Ligand 10 mit größerer Kettenlänge scheiterte bisher an
der schlechten Verfügbarkeit der notwendigen Mercaptoamine. Im Gegensatz dazu sind α-
Mercapto-ω-carboxyl-n-alkane mit verschiedenen Kettenlängen günstig kommerziell
erhältlich. Diese Carbonsäuren können mit einem geeigneten Alkohol eine Esterbindung
eingehen, wie z. B. mit Verbindung 5. Um die Veresterung möglichst schonend für die
Thiolfunktion durchzuführen, wurde auf säure- oder basenkatalysierte Methoden der
Veresterung verzichtet. Stattdessen wurde eine Methode von Gooßen et al. verwendet, die die
Esterbindung mittels Kupplungsreagenzien knüpft.[87] Die Methode verwendet statt der
üblichen DCC/DMAP-Kombination[88] das System Di-t-butyldicarbonat ((BOC)2O)/DMAP.
Der Vorteil der Methode ist die Art der durch das Kupplungsreagenz gebildeten
Nebenprodukte. Es handelt sich um CO2 und t-Butanol, welche infolge ihrer Flüchtigkeit
leicht abzutrennen sind.
NNNN N
OH
+ HOOC CH2 SH10
i
NNNN N
OCH2
OSH
10
5 13 14
Abbildung 2.16. Synthese von 14; (i) (BOC)2O, 4-DMAP kat., MeCN, 23 h, 60 °C, 63 %.
Zur Synthese wurde der Alkohol 5 zusammen mit 11-Mercaptoundecansäure 13, Di-t-
Butyldicarbonat und 5 Mol-% 4-DMAP in MeCN 23 h auf 60 °C erhitzt. Saure Aufarbeitung
zur Vernichtung überschüssigen Di-t-Butyldicarbonats ergab nach säulenchromatographischer
Reinigung die Thiolverbindung 14 in Form eines farblosen Öls, das sich beim Trocknen im
Hochvakuum verfestigte. Das IR-Spektrum des Produktes beinhaltet nicht mehr die
charakteristische Absorptionsbande der O–H-Streckschwingung des Edukts 5 bei 3299 cm–1.
Die durch die aromatischen Ringe verursachten Banden im Spektrum von 14 sowohl im
Bereich 3000-3150 cm–1 als auch unterhalb 1600 cm–1 stimmen gut mit denen im Spektrum
von 5 überein. Die starke Bande der C=O-Streckschwingung der Estergruppe taucht bei 1744
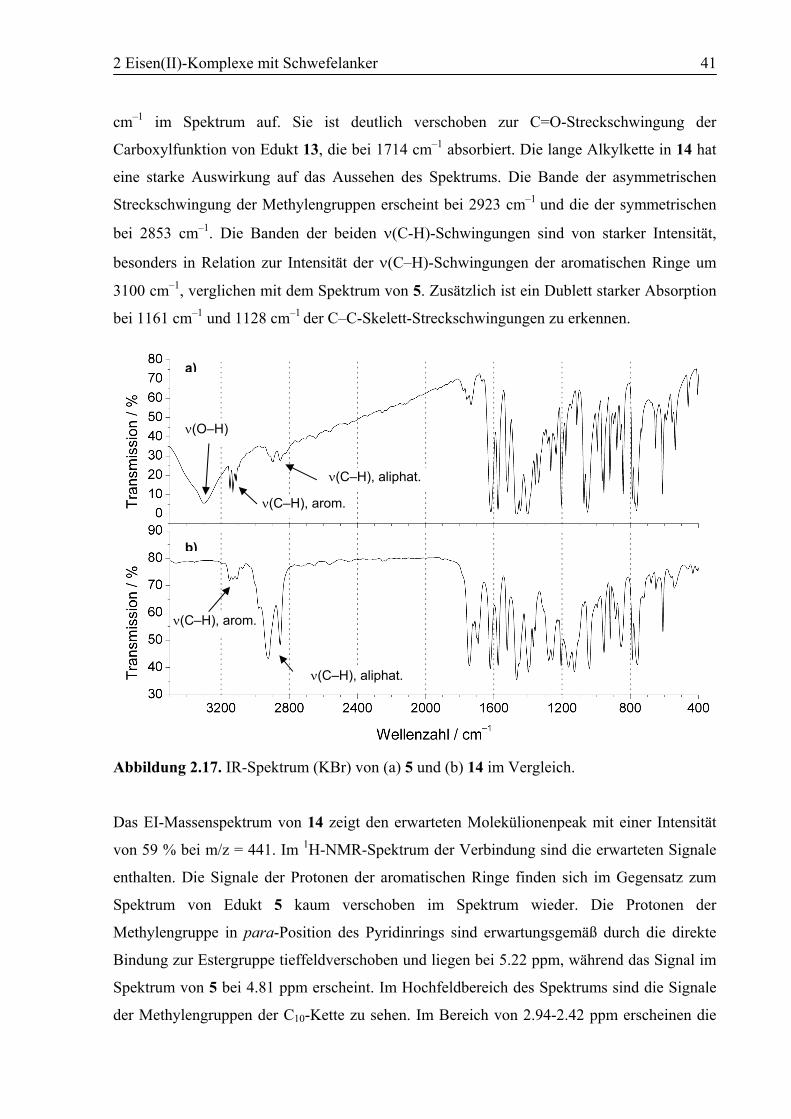
2 Eisen(II)-Komplexe mit Schwefelanker 41
cm–1 im Spektrum auf. Sie ist deutlich verschoben zur C=O-Streckschwingung der
Carboxylfunktion von Edukt 13, die bei 1714 cm–1 absorbiert. Die lange Alkylkette in 14 hat
eine starke Auswirkung auf das Aussehen des Spektrums. Die Bande der asymmetrischen
Streckschwingung der Methylengruppen erscheint bei 2923 cm–1 und die der symmetrischen
bei 2853 cm–1. Die Banden der beiden ν(C-H)-Schwingungen sind von starker Intensität,
besonders in Relation zur Intensität der ν(C–H)-Schwingungen der aromatischen Ringe um
3100 cm–1, verglichen mit dem Spektrum von 5. Zusätzlich ist ein Dublett starker Absorption
bei 1161 cm–1 und 1128 cm–1 der C–C-Skelett-Streckschwingungen zu erkennen.
Abbildung 2.17. IR-Spektrum (KBr) von (a) 5 und (b) 14 im Vergleich.
Das EI-Massenspektrum von 14 zeigt den erwarteten Molekülionenpeak mit einer Intensität
von 59 % bei m/z = 441. Im 1H-NMR-Spektrum der Verbindung sind die erwarteten Signale
enthalten. Die Signale der Protonen der aromatischen Ringe finden sich im Gegensatz zum
Spektrum von Edukt 5 kaum verschoben im Spektrum wieder. Die Protonen der
Methylengruppe in para-Position des Pyridinrings sind erwartungsgemäß durch die direkte
Bindung zur Estergruppe tieffeldverschoben und liegen bei 5.22 ppm, während das Signal im
Spektrum von 5 bei 4.81 ppm erscheint. Im Hochfeldbereich des Spektrums sind die Signale
der Methylengruppen der C10-Kette zu sehen. Im Bereich von 2.94-2.42 ppm erscheinen die
a)
b)
ν(O–H)
ν(C–H), arom.
ν(C–H), aliphat.
ν(C–H), arom.
ν(C–H), aliphat.
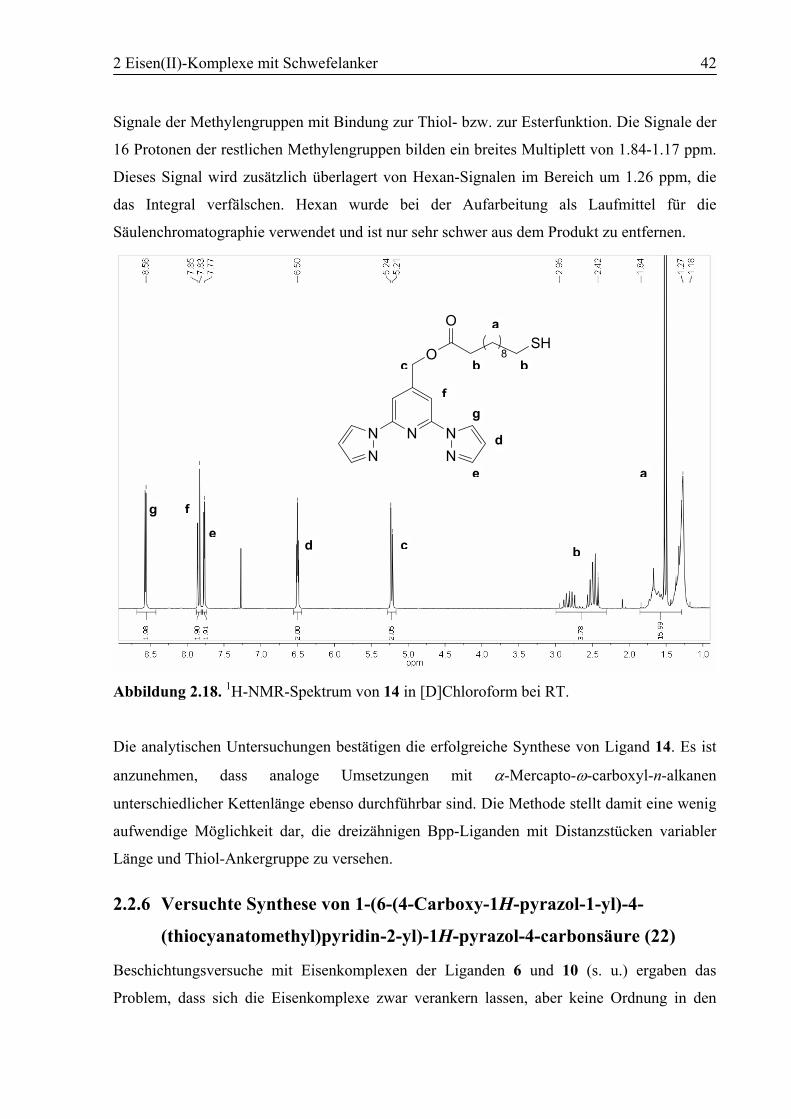
2 Eisen(II)-Komplexe mit Schwefelanker 42
Signale der Methylengruppen mit Bindung zur Thiol- bzw. zur Esterfunktion. Die Signale der
16 Protonen der restlichen Methylengruppen bilden ein breites Multiplett von 1.84-1.17 ppm.
Dieses Signal wird zusätzlich überlagert von Hexan-Signalen im Bereich um 1.26 ppm, die
das Integral verfälschen. Hexan wurde bei der Aufarbeitung als Laufmittel für die
Säulenchromatographie verwendet und ist nur sehr schwer aus dem Produkt zu entfernen.
Abbildung 2.18. 1H-NMR-Spektrum von 14 in [D]Chloroform bei RT.
Die analytischen Untersuchungen bestätigen die erfolgreiche Synthese von Ligand 14. Es ist
anzunehmen, dass analoge Umsetzungen mit α-Mercapto-ω-carboxyl-n-alkanen
unterschiedlicher Kettenlänge ebenso durchführbar sind. Die Methode stellt damit eine wenig
aufwendige Möglichkeit dar, die dreizähnigen Bpp-Liganden mit Distanzstücken variabler
Länge und Thiol-Ankergruppe zu versehen.
2.2.6 Versuchte Synthese von 1-(6-(4-Carboxy-1H-pyrazol-1-yl)-4-
(thiocyanatomethyl)pyridin-2-yl)-1H-pyrazol-4-carbonsäure (22)
Beschichtungsversuche mit Eisenkomplexen der Liganden 6 und 10 (s. u.) ergaben das
Problem, dass sich die Eisenkomplexe zwar verankern lassen, aber keine Ordnung in den
a
bcd e
fg
a
bc
d
e
fg
b
NNNN N
OSH
O
8
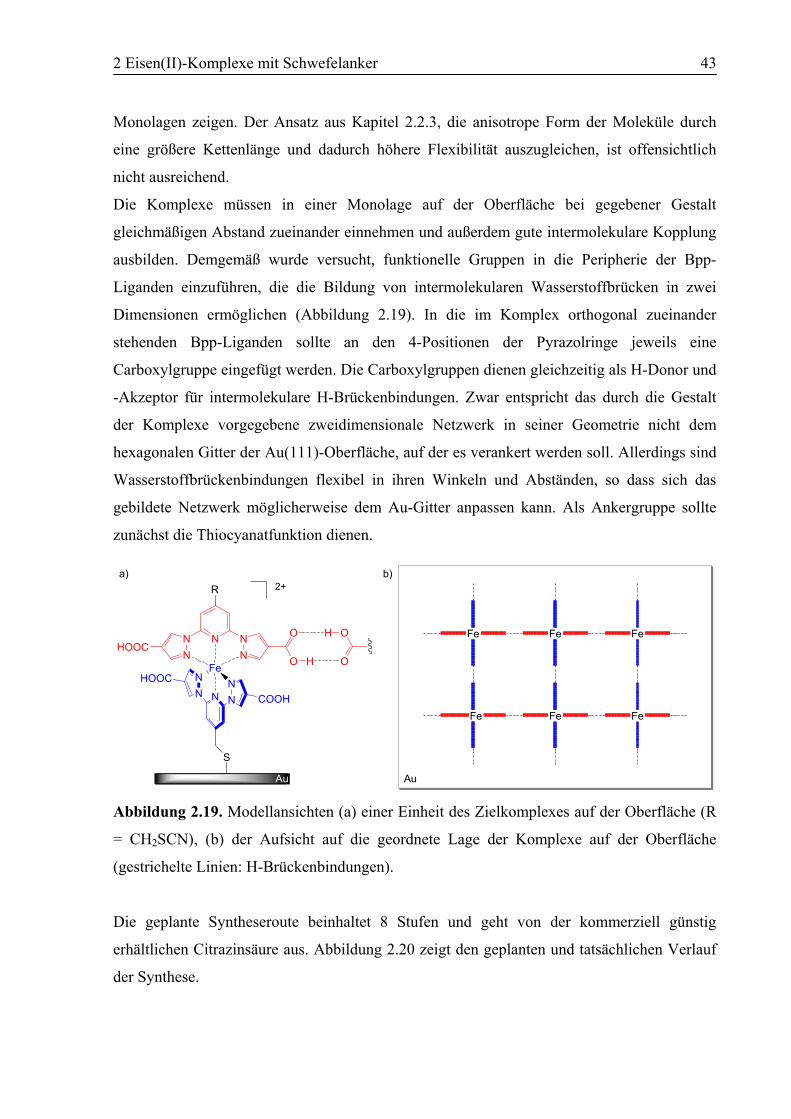
2 Eisen(II)-Komplexe mit Schwefelanker 43
Monolagen zeigen. Der Ansatz aus Kapitel 2.2.3, die anisotrope Form der Moleküle durch
eine größere Kettenlänge und dadurch höhere Flexibilität auszugleichen, ist offensichtlich
nicht ausreichend.
Die Komplexe müssen in einer Monolage auf der Oberfläche bei gegebener Gestalt
gleichmäßigen Abstand zueinander einnehmen und außerdem gute intermolekulare Kopplung
ausbilden. Demgemäß wurde versucht, funktionelle Gruppen in die Peripherie der Bpp-
Liganden einzuführen, die die Bildung von intermolekularen Wasserstoffbrücken in zwei
Dimensionen ermöglichen (Abbildung 2.19). In die im Komplex orthogonal zueinander
stehenden Bpp-Liganden sollte an den 4-Positionen der Pyrazolringe jeweils eine
Carboxylgruppe eingefügt werden. Die Carboxylgruppen dienen gleichzeitig als H-Donor und
-Akzeptor für intermolekulare H-Brückenbindungen. Zwar entspricht das durch die Gestalt
der Komplexe vorgegebene zweidimensionale Netzwerk in seiner Geometrie nicht dem
hexagonalen Gitter der Au(111)-Oberfläche, auf der es verankert werden soll. Allerdings sind
Wasserstoffbrückenbindungen flexibel in ihren Winkeln und Abständen, so dass sich das
gebildete Netzwerk möglicherweise dem Au-Gitter anpassen kann. Als Ankergruppe sollte
zunächst die Thiocyanatfunktion dienen.
N NN NN
N NN NN
Fe
S
2+R
HOOC
COOH
HOOCO
O H
H
O
O Fe
Fe
Fe
Fe
Fe
Fe
AuAu
a) b)
Abbildung 2.19. Modellansichten (a) einer Einheit des Zielkomplexes auf der Oberfläche (R
= CH2SCN), (b) der Aufsicht auf die geordnete Lage der Komplexe auf der Oberfläche
(gestrichelte Linien: H-Brückenbindungen).
Die geplante Syntheseroute beinhaltet 8 Stufen und geht von der kommerziell günstig
erhältlichen Citrazinsäure aus. Abbildung 2.20 zeigt den geplanten und tatsächlichen Verlauf
der Synthese.
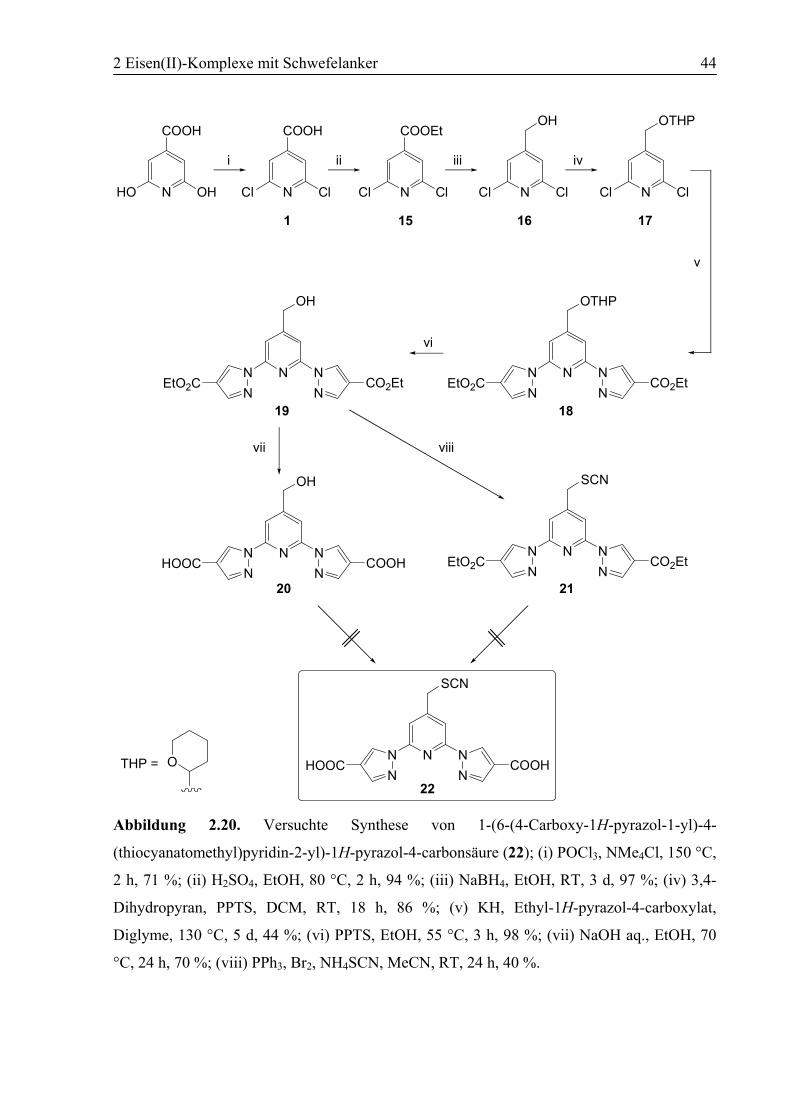
2 Eisen(II)-Komplexe mit Schwefelanker 44
N
COOH
OHHO N
COOH
ClCl N
COOEt
ClCl N ClCl
OH
N ClCl
OTHP
THP = O
NN NN N
OTHP
EtO2C CO2EtNN N
N N
OH
EtO2C CO2Et
NN NN N
OH
HOOC COOHNN N
N N
SCN
EtO2C CO2Et
NN NN N
SCN
HOOC COOH
1 15 16 17
1819
20 21
22
i ii iii iv
v
vi
vii viii
Abbildung 2.20. Versuchte Synthese von 1-(6-(4-Carboxy-1H-pyrazol-1-yl)-4-
(thiocyanatomethyl)pyridin-2-yl)-1H-pyrazol-4-carbonsäure (22); (i) POCl3, NMe4Cl, 150 °C,
2 h, 71 %; (ii) H2SO4, EtOH, 80 °C, 2 h, 94 %; (iii) NaBH4, EtOH, RT, 3 d, 97 %; (iv) 3,4-
Dihydropyran, PPTS, DCM, RT, 18 h, 86 %; (v) KH, Ethyl-1H-pyrazol-4-carboxylat,
Diglyme, 130 °C, 5 d, 44 %; (vi) PPTS, EtOH, 55 °C, 3 h, 98 %; (vii) NaOH aq., EtOH, 70
°C, 24 h, 70 %; (viii) PPh3, Br2, NH4SCN, MeCN, RT, 24 h, 40 %.

2 Eisen(II)-Komplexe mit Schwefelanker 45
Die Einführung der Carboxylfunktionen in die Pyrazolringe kann durch Verseifung der
entsprechenden Esterverbindungen geschehen. In 4-Position mit Ethylestergruppen
substituierte Pyrazole sind kommerziell erhältlich. Die Einführung der Pyrazolringe in die
2,6-Dichlorverbindung kann nicht wie in der Synthese von 5 auf der Stufe der Pyridin-4-
carboxylverbindung erfolgen, da diese Carboxylgruppe im weiteren Verlauf verestert werden
muss. Die verschiedenen Estergruppen könnten dann nicht mehr getrennt derivatisiert werden.
Stattdessen werden die Pyrazolringe in die Pyridin-4-hydroxymethylverbindung eingeführt.
Vorversuche zeigten nicht die gewünschte Reaktion der Hydroxymethylverbindung 16 mit
den Pyrazolylresten. Die Alkoholfunktion wird daher in Form eines Tetrahydropyranylethers
(THP) geschützt. Im weiteren Verlauf sollten die Alkoholgruppe durch die
Thiocyanatfunktion substituiert und die seitlichen Estergruppen verseift werden, wobei hier
die richtige Reihenfolge nicht feststand. Die Synthese von Verbindung 1 wurde bereits in
Kapitel 2.2.1 beschrieben. Die Synthesen der Verbindungen 15, 16 und 17 erfolgten in
Anlehnung an Literaturvorschriften.[79, 89]
2,6-Dichlorisonikotinsäure (1) wurde zunächst säurekatalysiert zum entsprechenden
Ethylester (15) umgesetzt. Die Synthese konnte mit relativ kurzer Reaktionszeit von 2 h bei
hohen Ausbeuten von über 90 % durchgeführt werden. Im 1H-NMR-Spektrum sind zusätzlich
zum Signal der Pyridin-Protonen die Signale der Protonen des eingefügten Ethylrests
vorhanden. Die Signale des A2B3-Spinsystems bilden ein Quartett bei 4.43 ppm und ein
Triplett bei 1.42 ppm für die Methylen- bzw. die Methylprotonen, mit einer typischen
Kopplungskonstante für die 3J-Kopplung von 7.1 Hz. Das EI-Massenspektrum enthält den
Molekülionenpeak bei m/z = 219/221 mit dem passenden Isotopenverhältnis für eine Dichlor-
Spezies.
Im nächsten Schritt wurde die Esterverbindung 15 durch Reduktion mit Natriumborhydrid
zum Alkohol 16 umgesetzt. Die Umsetzung konnte bei Raumtemperatur durchgeführt werden
und lieferte hohe Ausbeuten von über 95 % an reinem Produkt. Das 1H-NMR-Spektrum in
[D6]DMSO zeigt die erfolgreiche Abspaltung der Ethylrestes, es finden sich keine Signale für
die entsprechenden Methyl und Methylen-Protonen. Ein Signal in Form eines Tripletts bei
5.65 ppm mit einem Integral, das einem einzelnen Wasserstoffatom entspricht, wird durch das
Proton der Hydroxylgruppe verursacht. Es koppelt mit dem Signal der neuen Methylengruppe
bei 4.56 ppm (J = 5.8 Hz). Dieses Signal ist in ein Dublett von Tripletts aufgespalten. Die
Dublettaufspaltung wird durch eine 4J-Fernkopplung (J = 0.9 Hz) mit den Pyridinprotonen
verursacht, die ihrerseits eine Dublettaufspaltung erfahren.
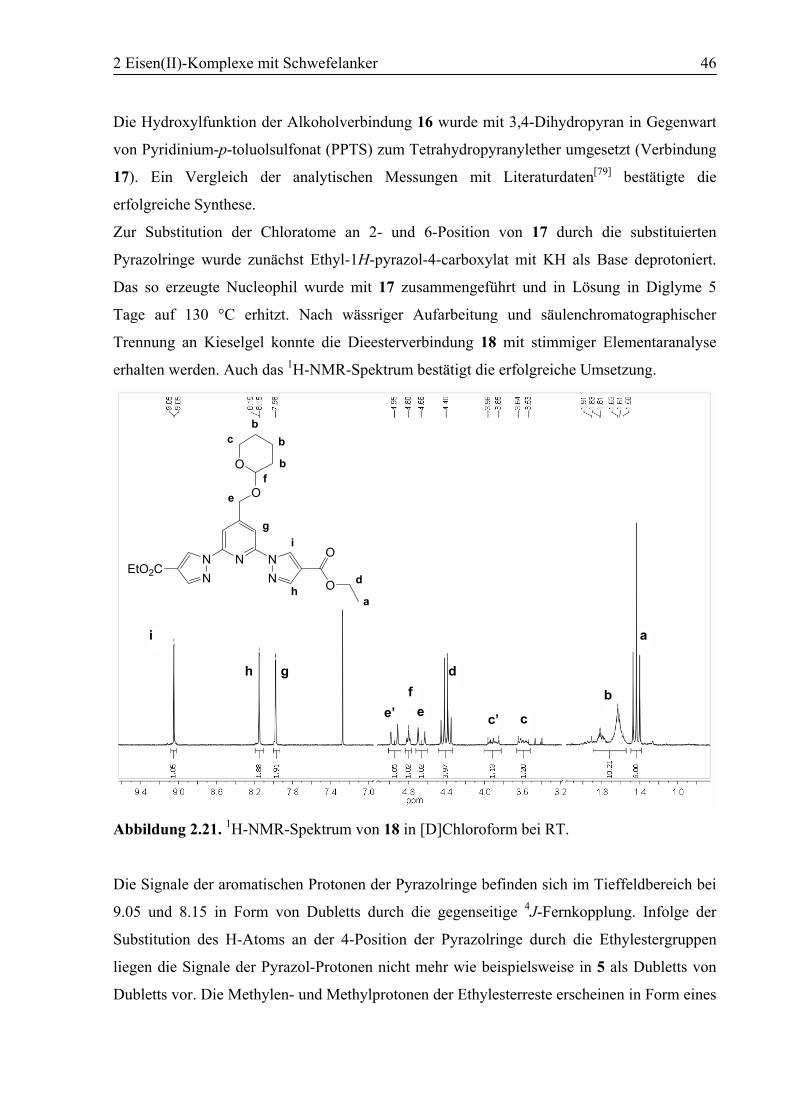
2 Eisen(II)-Komplexe mit Schwefelanker 46
Die Hydroxylfunktion der Alkoholverbindung 16 wurde mit 3,4-Dihydropyran in Gegenwart
von Pyridinium-p-toluolsulfonat (PPTS) zum Tetrahydropyranylether umgesetzt (Verbindung
17). Ein Vergleich der analytischen Messungen mit Literaturdaten[79] bestätigte die
erfolgreiche Synthese.
Zur Substitution der Chloratome an 2- und 6-Position von 17 durch die substituierten
Pyrazolringe wurde zunächst Ethyl-1H-pyrazol-4-carboxylat mit KH als Base deprotoniert.
Das so erzeugte Nucleophil wurde mit 17 zusammengeführt und in Lösung in Diglyme 5
Tage auf 130 °C erhitzt. Nach wässriger Aufarbeitung und säulenchromatographischer
Trennung an Kieselgel konnte die Dieesterverbindung 18 mit stimmiger Elementaranalyse
erhalten werden. Auch das 1H-NMR-Spektrum bestätigt die erfolgreiche Umsetzung.
Abbildung 2.21. 1H-NMR-Spektrum von 18 in [D]Chloroform bei RT.
Die Signale der aromatischen Protonen der Pyrazolringe befinden sich im Tieffeldbereich bei
9.05 und 8.15 in Form von Dubletts durch die gegenseitige 4J-Fernkopplung. Infolge der
Substitution des H-Atoms an der 4-Position der Pyrazolringe durch die Ethylestergruppen
liegen die Signale der Pyrazol-Protonen nicht mehr wie beispielsweise in 5 als Dubletts von
Dubletts vor. Die Methylen- und Methylprotonen der Ethylesterreste erscheinen in Form eines
b
c
a
c’
d
e’
h
ef
g
i
NNNN N
O
O
EtO2CO
Oa
dh
ig
ef
b
bb
c

2 Eisen(II)-Komplexe mit Schwefelanker 47
Quartetts bzw. eines Tripletts bei 4.40 und 1.43 ppm. Die Signale der Methylen-Protonen an
der 4-Position des Pyridinrings (e/e’, Abbildung 2.21) erscheinen einzeln bei 4.95 und 4.66
ppm, jeweils aufgespalten in ein Dublett von Tripletts. Durch die Nachbarschaft zum
Stereozentrum des tertiären C-Atoms des THP-Rings sind die Methylenprotonen e und e’
diastereotop. Sie zeigen untereinander eine geminale Kopplung mit J = 14.2 Hz und
zusätzlich noch eine 4J-Fernkopplung (J = 0.7 Hz) zu den Pyridin-Protonen. Deren Signal im
Spektrum ist ebenfalls in ein Triplett aufgespalten. Auch die Signale der dem Sauerstoffatom
des THP-Rings benachbarten Methylenprotonen sind diastereotop und erscheinen getrennt
voneinander im Spektrum. Das einzelne Proton am Stereozentrum erscheint als Triplett durch
die vicinale Kopplung mit der benachbarten Methylengruppe. Das Multiplett, das durch die
restlichen Methylenprotonen des THP-Rings erzeugt wird, erscheint im Bereich 1.91-1.58
ppm. Das Integral des Signals wird von in der Probe enthaltenem H2O verfälscht, welches ein
breites Resonanzsignal in derselben Region erzeugt.
Zur Abspaltung der THP-Schutzgruppe wurde Verbindung 18 mit PPTS in EtOH 3 h auf 55
°C erhitzt. Die Reaktion erfolgte mit nahezu quantitativem Umsatz von 98 %. Das 1H-NMR-
Spektrum von 19 enthält nicht mehr die Signale der THP-Gruppe aus Verbindung 18. Die
Protonen der aromatischen Ringe und die Protonen der Ethylestergruppen zeigen Signale, die
im Vergleich zu Verbindung 18 nur leicht verschoben sind. Durch den Wegfall des
Stereozentrums nach der Entschützung erscheint das Signal der Methylengruppe an der para-
Position des Pyridinrings wieder als einzelnes Signal bei 4.68 ppm. Ein neues breites Signal
eines einzelnen Protons bei 5.69 ppm wird durch die Hydroxylgruppe verursacht. Wird ein
Tropfen D2O zu der Probelösung gegeben, verschwindet das Signal im resultierenden
Spektrum.
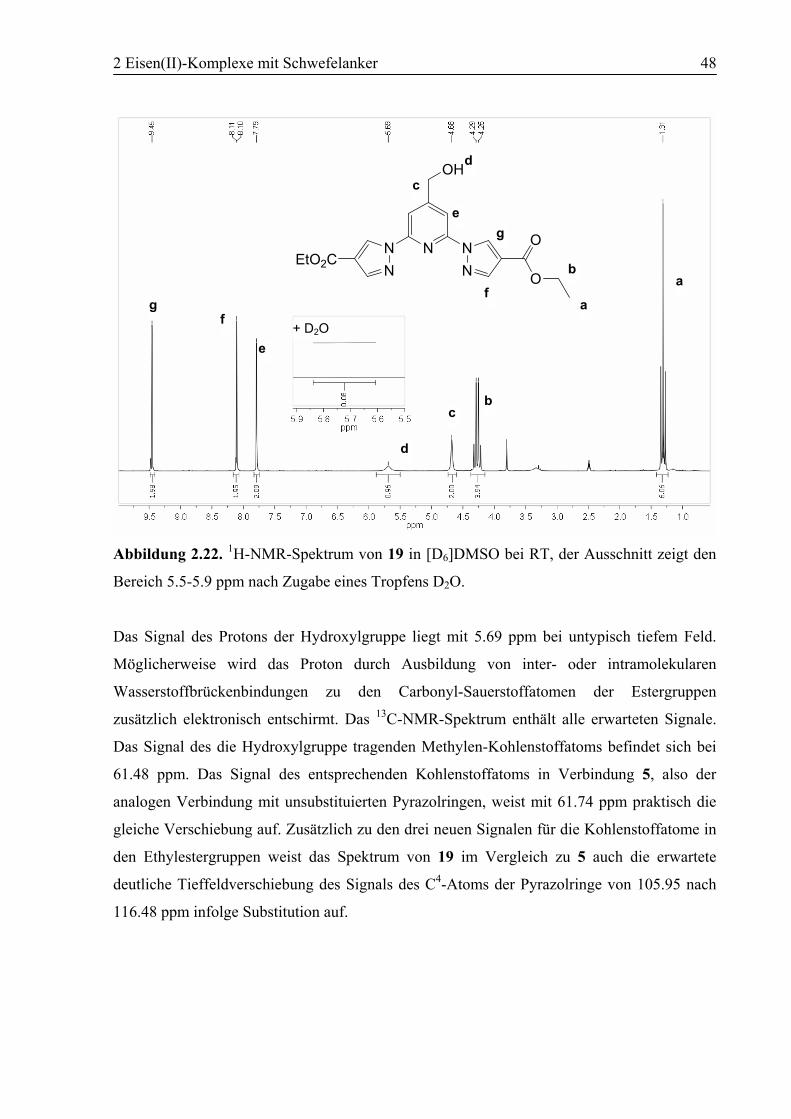
2 Eisen(II)-Komplexe mit Schwefelanker 48
Abbildung 2.22. 1H-NMR-Spektrum von 19 in [D6]DMSO bei RT, der Ausschnitt zeigt den
Bereich 5.5-5.9 ppm nach Zugabe eines Tropfens D2O.
Das Signal des Protons der Hydroxylgruppe liegt mit 5.69 ppm bei untypisch tiefem Feld.
Möglicherweise wird das Proton durch Ausbildung von inter- oder intramolekularen
Wasserstoffbrückenbindungen zu den Carbonyl-Sauerstoffatomen der Estergruppen
zusätzlich elektronisch entschirmt. Das 13C-NMR-Spektrum enthält alle erwarteten Signale.
Das Signal des die Hydroxylgruppe tragenden Methylen-Kohlenstoffatoms befindet sich bei
61.48 ppm. Das Signal des entsprechenden Kohlenstoffatoms in Verbindung 5, also der
analogen Verbindung mit unsubstituierten Pyrazolringen, weist mit 61.74 ppm praktisch die
gleiche Verschiebung auf. Zusätzlich zu den drei neuen Signalen für die Kohlenstoffatome in
den Ethylestergruppen weist das Spektrum von 19 im Vergleich zu 5 auch die erwartete
deutliche Tieffeldverschiebung des Signals des C4-Atoms der Pyrazolringe von 105.95 nach
116.48 ppm infolge Substitution auf.
+ D2O
bc
a
d
e
f g
b
c
a
d
e
f
g
NNNN N
OH
EtO2CO
O
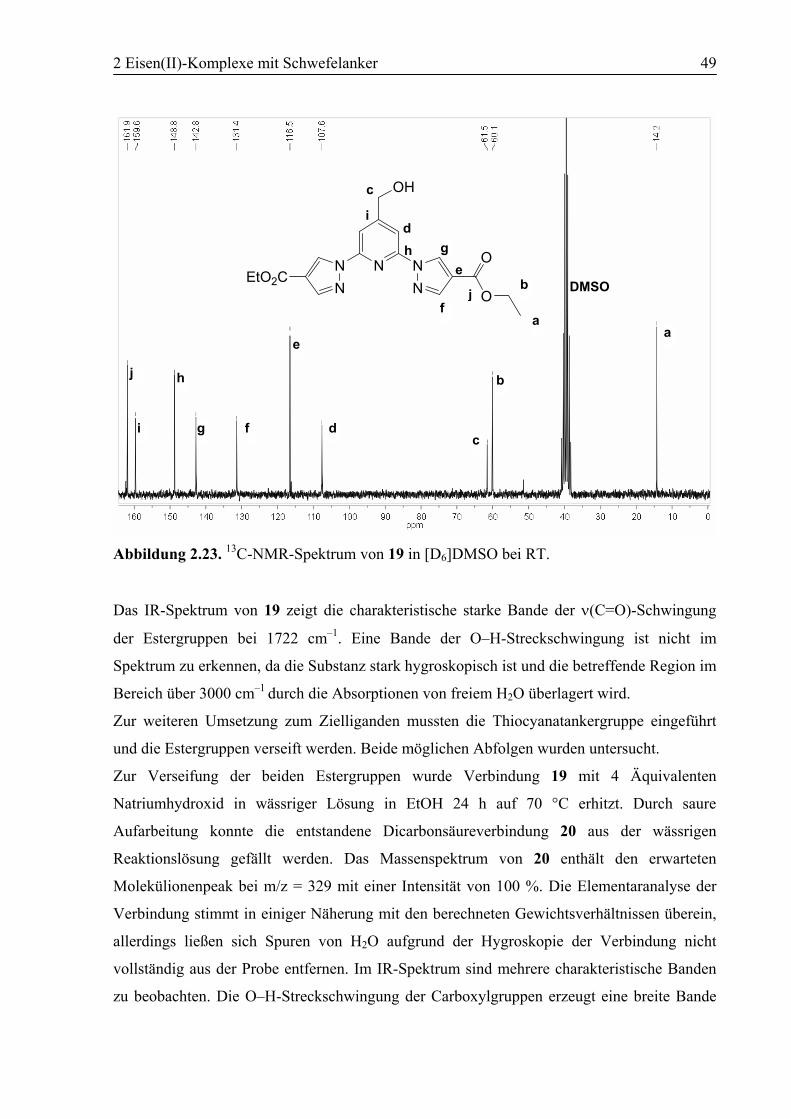
2 Eisen(II)-Komplexe mit Schwefelanker 49
Abbildung 2.23. 13C-NMR-Spektrum von 19 in [D6]DMSO bei RT.
Das IR-Spektrum von 19 zeigt die charakteristische starke Bande der ν(C=O)-Schwingung
der Estergruppen bei 1722 cm–1. Eine Bande der O–H-Streckschwingung ist nicht im
Spektrum zu erkennen, da die Substanz stark hygroskopisch ist und die betreffende Region im
Bereich über 3000 cm–1 durch die Absorptionen von freiem H2O überlagert wird.
Zur weiteren Umsetzung zum Zielliganden mussten die Thiocyanatankergruppe eingeführt
und die Estergruppen verseift werden. Beide möglichen Abfolgen wurden untersucht.
Zur Verseifung der beiden Estergruppen wurde Verbindung 19 mit 4 Äquivalenten
Natriumhydroxid in wässriger Lösung in EtOH 24 h auf 70 °C erhitzt. Durch saure
Aufarbeitung konnte die entstandene Dicarbonsäureverbindung 20 aus der wässrigen
Reaktionslösung gefällt werden. Das Massenspektrum von 20 enthält den erwarteten
Molekülionenpeak bei m/z = 329 mit einer Intensität von 100 %. Die Elementaranalyse der
Verbindung stimmt in einiger Näherung mit den berechneten Gewichtsverhältnissen überein,
allerdings ließen sich Spuren von H2O aufgrund der Hygroskopie der Verbindung nicht
vollständig aus der Probe entfernen. Im IR-Spektrum sind mehrere charakteristische Banden
zu beobachten. Die O–H-Streckschwingung der Carboxylgruppen erzeugt eine breite Bande
b
c
d
e
f
g
a
b
cd
e
f g
a
j
i
h
j
i
h
DMSO NNNN N
OH
EtO2CO
O

2 Eisen(II)-Komplexe mit Schwefelanker 50
bei 3112 cm–1, die Bande der OH-Gruppe der Alkoholfunktion ist wie bei Verbindung 19 von
Wasser in der Probe überlagert. Die Bande der C=O-Streckschwingung ist erwartungsgemäß
zu kleineren Wellenzahlen von 1722 cm–1 in 19 nach 1694 cm–1 in 20 verschoben. Im 1H-
NMR-Spektrum von 20 sind die Signale der Protonen der Ethylreste der Diesterverbindung
19 nicht mehr enthalten. Das breite Signal des einzelnen Hydroxyl-Protons ist vorhanden und
verglichen mit 19 nur leicht nach 5.73 ppm verschoben. Ein breites Signal im Tieffeldbereich
bei 12.76 ppm entspricht den zwei Protonen der Carboxylgruppen. Die Analysen bestätigen
die erfolgreiche Verseifung der beiden Esterfunktionen zur Dicarbonsäureverbindung 20.
Eine erhöhte Temperatur ist bei der Reaktion notwendig. Die analoge Reaktion durchgeführt
bei 0 °C führte zur Verseifung von nur einer der beiden Carboxylgruppen, wie dem 1H-NMR-
Spektrum des Produktes zu entnehmen war.
Die Umsetzung von Verbindung 20 zur Thiocyanatverbindung 22 gestaltete sich aufgrund der
sehr schlechten Löslichkeit der Verbindung in den verwendeten Lösungsmitteln als schwierig.
Verbindung 20 wurde mit Triphenylphosphan, Brom und Ammoniumthiocyanat in MeCN bei
Raumtemperatur umgesetzt. Das IR-Spektrum des Produkts zeigte nicht die erwartete Bande
für die C≡N-Streckschwingung der Thiocyanatgruppe im Bereich 2175-2135cm–1. Generell
war das IR-Spektrum nahezu identisch mit dem des Edukts, so dass zu vermuten ist, dass kein
Umsatz erfolgte. Ein Grund ist vermutlich die erwähnte schlechte Löslichkeit des Edukts.
Eine erhöhte Reaktionstemperatur könnte helfen, die Dicarbonsäureverbindung besser in
Lösung zu bringen. Dabei muss beachtet werden, dass unter ähnlichen Reaktionsbedingungen
(Diethylazodicarboxylat (DEAD) statt Brom) auch Carbonsäuren zu den korrespondierenden
Isothiocyanatverbindungen reagieren können.[81] Diese Nebenreaktion ist bei Raumtemperatur
gehemmt und verläuft deutlich langsamer als die Umsetzung der Alkohol-Funktionen, könnte
aber bei höherer Temperatur die Umsetzung stören.
Unter Verfolgung der alternativen Syntheseroute wurde die Diester-Alkoholverbindung 19
durch Substitution der Hydroxylgruppe zur Diester-Thiocyanatverbindung 21 umgesetzt.
Verbindung 19 wurde dazu mit dem System PPh3/Br2/NH4SCN in MeCN über 24 h bei RT
zur Reaktion gebracht. Säulenchromatographische Trennung ergab das Produkt in Form eines
hellgelben Feststoffes elementaranalysenrein in Ausbeuten um 40 %. Das IR-Spektrum der
Substanz zeigt bei 2157 cm–1 die starke, scharfe Bande der C≡N-Streckschwingung der
Thiocyanatgruppe. Zusätzlich findet sich eine schwache Bande der ν(C–S)-Schwingung bei
621 cm–1 und eine schwache Bande der asymmetrischen C–S–C-Streckschwingung bei 711
cm–1. Die drei genannten Banden sind im IR-Spektrum des Edukts 19 nicht vorhanden, das
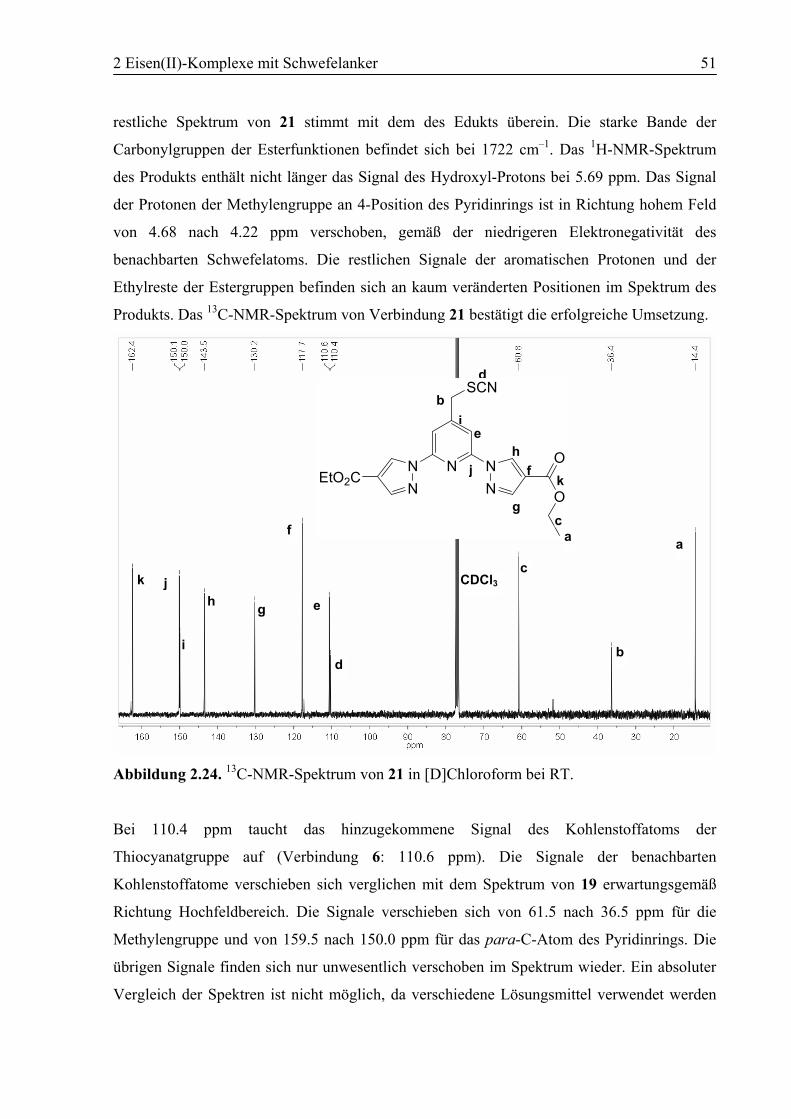
2 Eisen(II)-Komplexe mit Schwefelanker 51
restliche Spektrum von 21 stimmt mit dem des Edukts überein. Die starke Bande der
Carbonylgruppen der Esterfunktionen befindet sich bei 1722 cm–1. Das 1H-NMR-Spektrum
des Produkts enthält nicht länger das Signal des Hydroxyl-Protons bei 5.69 ppm. Das Signal
der Protonen der Methylengruppe an 4-Position des Pyridinrings ist in Richtung hohem Feld
von 4.68 nach 4.22 ppm verschoben, gemäß der niedrigeren Elektronegativität des
benachbarten Schwefelatoms. Die restlichen Signale der aromatischen Protonen und der
Ethylreste der Estergruppen befinden sich an kaum veränderten Positionen im Spektrum des
Produkts. Das 13C-NMR-Spektrum von Verbindung 21 bestätigt die erfolgreiche Umsetzung.
Abbildung 2.24. 13C-NMR-Spektrum von 21 in [D]Chloroform bei RT.
Bei 110.4 ppm taucht das hinzugekommene Signal des Kohlenstoffatoms der
Thiocyanatgruppe auf (Verbindung 6: 110.6 ppm). Die Signale der benachbarten
Kohlenstoffatome verschieben sich verglichen mit dem Spektrum von 19 erwartungsgemäß
Richtung Hochfeldbereich. Die Signale verschieben sich von 61.5 nach 36.5 ppm für die
Methylengruppe und von 159.5 nach 150.0 ppm für das para-C-Atom des Pyridinrings. Die
übrigen Signale finden sich nur unwesentlich verschoben im Spektrum wieder. Ein absoluter
Vergleich der Spektren ist nicht möglich, da verschiedene Lösungsmittel verwendet werden
NNNN N
SCN
EtO2CO
O
b
c
d
e
f
g
a
j
i
h
b
c
d
e
f
g
a
j
i
h
k
k
CDCl3
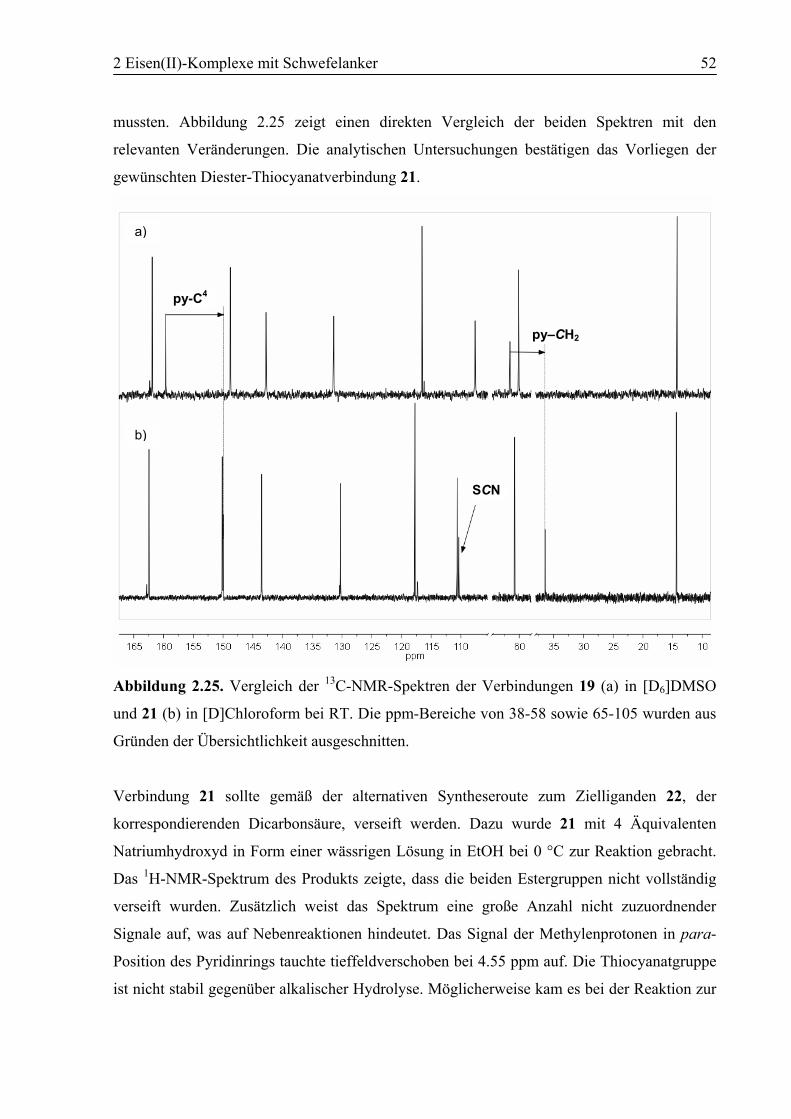
2 Eisen(II)-Komplexe mit Schwefelanker 52
mussten. Abbildung 2.25 zeigt einen direkten Vergleich der beiden Spektren mit den
relevanten Veränderungen. Die analytischen Untersuchungen bestätigen das Vorliegen der
gewünschten Diester-Thiocyanatverbindung 21.
Abbildung 2.25. Vergleich der 13C-NMR-Spektren der Verbindungen 19 (a) in [D6]DMSO
und 21 (b) in [D]Chloroform bei RT. Die ppm-Bereiche von 38-58 sowie 65-105 wurden aus
Gründen der Übersichtlichkeit ausgeschnitten.
Verbindung 21 sollte gemäß der alternativen Syntheseroute zum Zielliganden 22, der
korrespondierenden Dicarbonsäure, verseift werden. Dazu wurde 21 mit 4 Äquivalenten
Natriumhydroxyd in Form einer wässrigen Lösung in EtOH bei 0 °C zur Reaktion gebracht.
Das 1H-NMR-Spektrum des Produkts zeigte, dass die beiden Estergruppen nicht vollständig
verseift wurden. Zusätzlich weist das Spektrum eine große Anzahl nicht zuzuordnender
Signale auf, was auf Nebenreaktionen hindeutet. Das Signal der Methylenprotonen in para-
Position des Pyridinrings tauchte tieffeldverschoben bei 4.55 ppm auf. Die Thiocyanatgruppe
ist nicht stabil gegenüber alkalischer Hydrolyse. Möglicherweise kam es bei der Reaktion zur
a)
b)
py-C4
py–CH2
SCN

2 Eisen(II)-Komplexe mit Schwefelanker 53
Bildung von Disulfid-, Thiol- oder Thioetherverbindungen. Eine Erhöhung der
Reaktionstemperatur wie bei der erfolgreichen Verseifung von Verbindung 19 würde im Fall
von Verbindung 21 nur die Nebenreaktionen begünstigen. Die Verseifung müsste stattdessen
im neutralen Medium erfolgen (die Thiocyanatgruppe ist auch säurelabil). Beispielsweise ist
eine Esterverseifung mit der harten Lewissäure Aluminiumtriiodid denkbar, die unter milden
Reaktionsbedingungen verläuft.[90]
Die Umsetzungen von 20 bzw. 21 zum Zielliganden 22 konnten aus Zeitgründen nicht mehr
realisiert werden.

2 Eisen(II)-Komplexe mit Schwefelanker 54
2.3 Synthese von Eisenkomplexen
Die Bildung von Eisen(II)-Komplexen mit Bpp-Liganden folgt einem allgemeinen Muster für
die Koordination. Jeweils zwei der dreizähnigen Liganden koordinieren das Fe(II)-Ion in
meridionaler Anordnung ihrer (NNN)-Donorsätze. Das resultierende FeN6-Koordinations-
polyeder besitzt pseudooktaedrische Geometrie. Durch die Geometrie der Liganden und die
festgelegten Fe–N-Bindungslängen im Komplex weist das FeN6-Polyeder maximal D2d-
Symmetrie auf. Eine Veränderung der Anordnung der Liganden relativ zueinander bewirkt
eine Verzerrung des Koordinationspolyeders und damit eine Erniedrigung seiner Symmetrie.
Änderungen der Anordnung finden hauptsächlich durch zwei Effekte statt. Eine Drehung der
Ebenen der Liganden gegeneinander um die N(py)–Fe–N(py’)-Bindungsachse bewirkt eine
Abnahme des Winkels zwischen den Ausgleichsebenen der Liganden θ unter 90°. Eine
Kippung eines Liganden um eine gedachte Achse senkrecht zur N(py)–Fe–N(py’)-
Bindungsachse bewirkt eine Verkleinerung des N(py)-Fe-N(py’)-Winkels φ unter 180°. Bei
Abnahme der Fe–N-Bindungslängen vergrößern sich die N(pz)-Fe-N(py)-Winkel α.
Abbildung 2.26 veranschaulicht die Lage der erwähnten Winkel.
N
N
N
N
NN
N
N
N
N
Feα
θ
φ
Abbildung 2.26. Allgemeine Darstellung der Struktur des Komplexdikations in Eisen(II)-
Komplexen von Bpp-Liganden (α: Winkel N(pz)–Fe–N(py); φ: Winkel N(py)–Fe–N(py’); θ:
Winkel zwischen den Ausgleichsebenen der beiden Liganden).
Die Abweichung des Koordinationspolyeders vom idealen Oktaeder lässt sich mit den
Parametern Σ und υ[91, 92] quantifizieren, deren Definitionen in Abbildung 2.27 dargestellt
sind.

2 Eisen(II)-Komplexe mit Schwefelanker 55
∑=
−=Σ12
190
iiϕ
⎟⎟⎠
⎞⎜⎜⎝
⎛−=
S
P
VV
πυ 1100
VP = Volumen des Koordinationspolyeders
VS = Volumen der Umkugel des Koordinationspolyeders
Ideales Oktaeder: Σ = 0° Ideales Oktaeder: υ = 0 %
Abbildung 2.27. Definition der Verzerrungsparameter Σ und υ.[91, 92]
2.3.1 [Fe(3)2](BF4)2 (23)
Die Synthese erfolgte durch Reaktion von zwei Äquivalenten des Liganden 3 mit einem
Äquivalent Eisen(II)tetrafluoroborat Hexahydrat. Der Ligand wurde hierbei in leichtem
Überschuss eingesetzt, um Fe(BF4)2 möglichst quantitativ umzusetzen. Nach Zugabe der
Lösung des Eisensalzes zum Liganden in THF fiel sofort das Produkt in Form eines orangen
Niederschlags aus. Nach vier Stunden Rühren bei Raumtemperatur wurde der Niederschlag
durch Filtration abgetrennt. Nach Waschen mit THF und Trocknung im Hochvakuum konnte
der Eisenkomplex 23 als oranger Feststoff isoliert werden.
NNNN N
CO2Me
2 iN
NN
N
NN
N
N
N
N
FeMeO2C CO2Me
2+
[BF4–]2
23
3
Abbildung 2.28. Synthese von 23; (i) 0.97 Äq. Fe(BF4)2 · 6 H2O, THF, RT, 4 h, 82 %.
Das IR-Spektrum von 23 in KBr zeigt die starke Bande der B–F-Streckschwingungen bei
1052 cm–1. Zusätzlich ist die Bande der C=O-Streckschwingung der Estergruppe bei 1738
cm–1 vorhanden. Die übrigen Banden im Spektrum entsprechen denen im freien Liganden mit
nur geringen Änderungen. Das 1H-NMR-Spektrum zeigt eine paramagnetische Verbreiterung
und Verschiebung der Signale über einen Bereich von 0-70 ppm. Eine Zuordnung der Signale
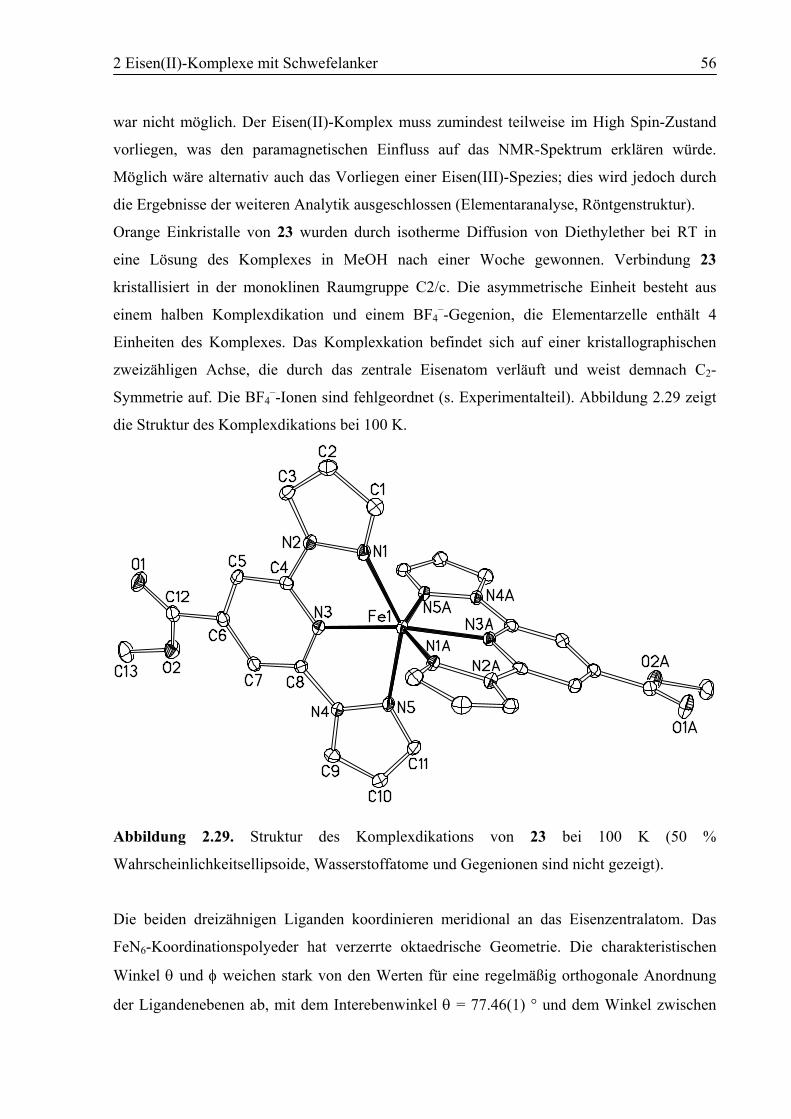
2 Eisen(II)-Komplexe mit Schwefelanker 56
war nicht möglich. Der Eisen(II)-Komplex muss zumindest teilweise im High Spin-Zustand
vorliegen, was den paramagnetischen Einfluss auf das NMR-Spektrum erklären würde.
Möglich wäre alternativ auch das Vorliegen einer Eisen(III)-Spezies; dies wird jedoch durch
die Ergebnisse der weiteren Analytik ausgeschlossen (Elementaranalyse, Röntgenstruktur).
Orange Einkristalle von 23 wurden durch isotherme Diffusion von Diethylether bei RT in
eine Lösung des Komplexes in MeOH nach einer Woche gewonnen. Verbindung 23
kristallisiert in der monoklinen Raumgruppe C2/c. Die asymmetrische Einheit besteht aus
einem halben Komplexdikation und einem BF4–-Gegenion, die Elementarzelle enthält 4
Einheiten des Komplexes. Das Komplexkation befindet sich auf einer kristallographischen
zweizähligen Achse, die durch das zentrale Eisenatom verläuft und weist demnach C2-
Symmetrie auf. Die BF4–-Ionen sind fehlgeordnet (s. Experimentalteil). Abbildung 2.29 zeigt
die Struktur des Komplexdikations bei 100 K.
Abbildung 2.29. Struktur des Komplexdikations von 23 bei 100 K (50 %
Wahrscheinlichkeitsellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Die beiden dreizähnigen Liganden koordinieren meridional an das Eisenzentralatom. Das
FeN6-Koordinationspolyeder hat verzerrte oktaedrische Geometrie. Die charakteristischen
Winkel θ und φ weichen stark von den Werten für eine regelmäßig orthogonale Anordnung
der Ligandenebenen ab, mit dem Interebenwinkel θ = 77.46(1) ° und dem Winkel zwischen
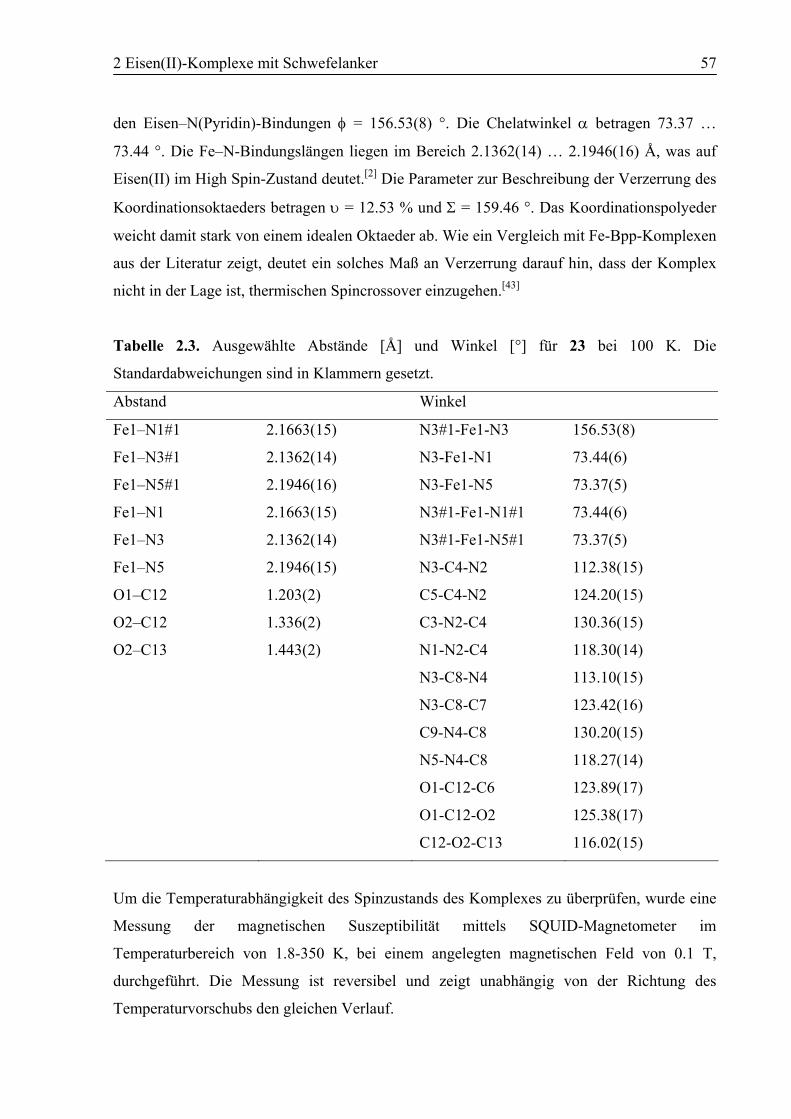
2 Eisen(II)-Komplexe mit Schwefelanker 57
den Eisen–N(Pyridin)-Bindungen φ = 156.53(8) °. Die Chelatwinkel α betragen 73.37 …
73.44 °. Die Fe–N-Bindungslängen liegen im Bereich 2.1362(14) … 2.1946(16) Å, was auf
Eisen(II) im High Spin-Zustand deutet.[2] Die Parameter zur Beschreibung der Verzerrung des
Koordinationsoktaeders betragen υ = 12.53 % und Σ = 159.46 °. Das Koordinationspolyeder
weicht damit stark von einem idealen Oktaeder ab. Wie ein Vergleich mit Fe-Bpp-Komplexen
aus der Literatur zeigt, deutet ein solches Maß an Verzerrung darauf hin, dass der Komplex
nicht in der Lage ist, thermischen Spincrossover einzugehen.[43]
Tabelle 2.3. Ausgewählte Abstände [Å] und Winkel [°] für 23 bei 100 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N1#1 2.1663(15) N3#1-Fe1-N3 156.53(8)
Fe1–N3#1 2.1362(14) N3-Fe1-N1 73.44(6)
Fe1–N5#1 2.1946(16) N3-Fe1-N5 73.37(5)
Fe1–N1 2.1663(15) N3#1-Fe1-N1#1 73.44(6)
Fe1–N3 2.1362(14) N3#1-Fe1-N5#1 73.37(5)
Fe1–N5 2.1946(15) N3-C4-N2 112.38(15)
O1–C12 1.203(2) C5-C4-N2 124.20(15)
O2–C12 1.336(2) C3-N2-C4 130.36(15)
O2–C13 1.443(2) N1-N2-C4 118.30(14)
N3-C8-N4 113.10(15)
N3-C8-C7 123.42(16)
C9-N4-C8 130.20(15)
N5-N4-C8 118.27(14)
O1-C12-C6 123.89(17)
O1-C12-O2 125.38(17)
C12-O2-C13 116.02(15)
Um die Temperaturabhängigkeit des Spinzustands des Komplexes zu überprüfen, wurde eine
Messung der magnetischen Suszeptibilität mittels SQUID-Magnetometer im
Temperaturbereich von 1.8-350 K, bei einem angelegten magnetischen Feld von 0.1 T,
durchgeführt. Die Messung ist reversibel und zeigt unabhängig von der Richtung des
Temperaturvorschubs den gleichen Verlauf.
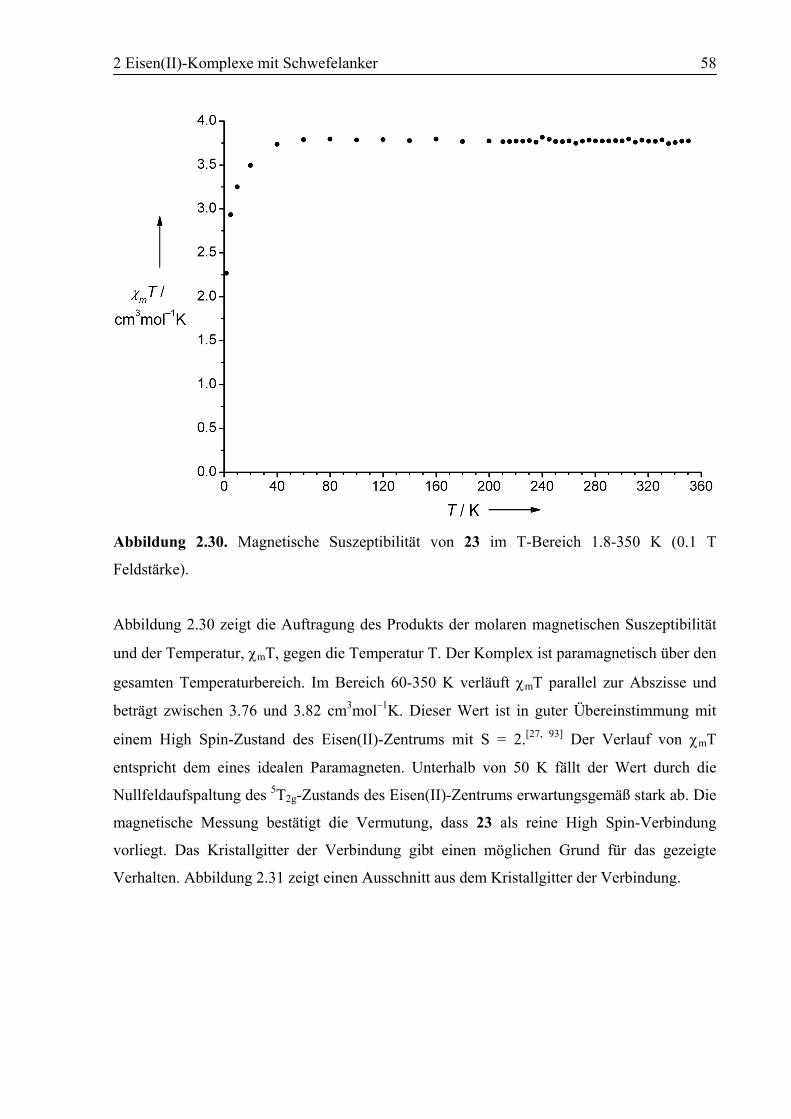
2 Eisen(II)-Komplexe mit Schwefelanker 58
Abbildung 2.30. Magnetische Suszeptibilität von 23 im T-Bereich 1.8-350 K (0.1 T
Feldstärke).
Abbildung 2.30 zeigt die Auftragung des Produkts der molaren magnetischen Suszeptibilität
und der Temperatur, χmT, gegen die Temperatur T. Der Komplex ist paramagnetisch über den
gesamten Temperaturbereich. Im Bereich 60-350 K verläuft χmT parallel zur Abszisse und
beträgt zwischen 3.76 und 3.82 cm3mol–1K. Dieser Wert ist in guter Übereinstimmung mit
einem High Spin-Zustand des Eisen(II)-Zentrums mit S = 2.[27, 93] Der Verlauf von χmT
entspricht dem eines idealen Paramagneten. Unterhalb von 50 K fällt der Wert durch die
Nullfeldaufspaltung des 5T2g-Zustands des Eisen(II)-Zentrums erwartungsgemäß stark ab. Die
magnetische Messung bestätigt die Vermutung, dass 23 als reine High Spin-Verbindung
vorliegt. Das Kristallgitter der Verbindung gibt einen möglichen Grund für das gezeigte
Verhalten. Abbildung 2.31 zeigt einen Ausschnitt aus dem Kristallgitter der Verbindung.

2 Eisen(II)-Komplexe mit Schwefelanker 59
Abbildung 2.31. Kristallpackung in 23 bei 100 K mit Blickrichtung entlang der b-Achse.
Die Komplexdikationen und die BF4–-Anionen stapeln sich in alternierenden
zweidimensionalen Schichten entlang der c-Achse. Innerhalb der Schichten der
Komplexkationen besteht ein intermolekularer Zusammenhang durch sekundäre
Wechselwirkungen in Form von Wasserstoffbrückenbindungen und π-π-Stapel-
wechselwirkungen. Zwischen den Schichten der Komplexkationen kommt es jedoch nicht
zum direkten Kontakt, aufgrund der trennenden Schicht der Anionen. Die Eisenzentren im
Kristall sind demnach nicht direkt miteinander verbunden. Eine Fortpflanzung der
Information über den Spinübergang zwischen den Molekülen ist damit erschwert.[61] Die von
Halcrow und Mitarbeitern untersuchten Fe-Bpp-Komplexe zeigen eine analoge
supramolekulare Struktur, wenn es sich um reine High Spin-Komplexe handelt.[43, 94]
Die analytischen Untersuchungen bestätigen die erfolgreiche Synthese von Verbindung 23.
Die Verbindung liegt im Feststoff im untersuchten Temperaturbereich und in Lösung bei

2 Eisen(II)-Komplexe mit Schwefelanker 60
Raumtemperatur als High Spin-Eisen(II)-Komplex vor. Weitere Untersuchungen mittels
temperaturabhängiger 1H-NMR-Spektroskopie könnten zeigen, ob das vorliegende
Ligandenfeld grundsätzlich einen thermischen Spinübergang erlaubt.
2.3.2 [Fe(4)2](BF4)2 (24)
Zur Synthese des Eisen(II)-Komplexes des Liganden 4 wurden 2 Äquivalente von 4 in
Lösung in THF vorgelegt und 0.97 Äquivalente Eisen(II)tetrafluoroborat Hexahydrat in THF
zugegeben. Direkt nach Zugabe fiel ein oranger Niederschlag aus der klaren, farblosen
Lösung aus. Es wurde noch weitere drei Stunden bei RT gerührt, um die Reaktion zu
vervollständigen und der orange Feststoff dann per Filtration isoliert, mit THF gewaschen und
im Hochvakuum getrocknet.
NNNN N
CO2Et
2 iN
NN
N
NN
N
N
N
N
FeEtO2C CO2Et
2+
[BF4–]24
24 Abbildung 2.32. Synthese von 24; (i) 0.97 Äq. Fe(BF4)2 · 6 H2O, THF, RT, 3 h, 71 %.
Das IR-Spektrum von 24 wurde in KBr aufgenommen. Eine starke Bande bei 1055 cm–1 zeigt
die B–F-Streckschwingung der Tetrafluoroborat-Gegenionen. Bei 1734 cm–1 erscheint die
starke Bande der C=O-Streckschwingung der Ethylestergruppe. Die Signale des 1H-NMR-
Spektrums in MeOH sind paramagnetisch verschoben und über einen Bereich von 0-70 ppm
verteilt. Vier breite Signale bei 69.15, 64.65, 58.29 und 49.33 ppm weisen ein Verhältnis ihrer
Integrale von 1:1:1:1 auf. Sie gehören vermutlich zu den Protonen an den aromatischen
Ringen. Die restlichen Signale im Hochfeldbereich des Spektrums von 9.48-0.96 ppm müssen
demnach von den Methyl- und Methylenprotonen der Ethylreste sowie dem Lösungsmittel
verursacht werden. Eine genaue Zuordnung ist durch den paramagnetischen Einfluss auf die
Signale nicht möglich. Das Eisenzentrum liegt in Verbindung 24 in einer paramagnetischen
Konfiguration vor.
Orange Einkristalle der Verbindung wurden durch isotherme Diffusion von Diethylether in
eine Lösung des Komplexes in MeOH über eine Woche gewonnen. Die Verbindung
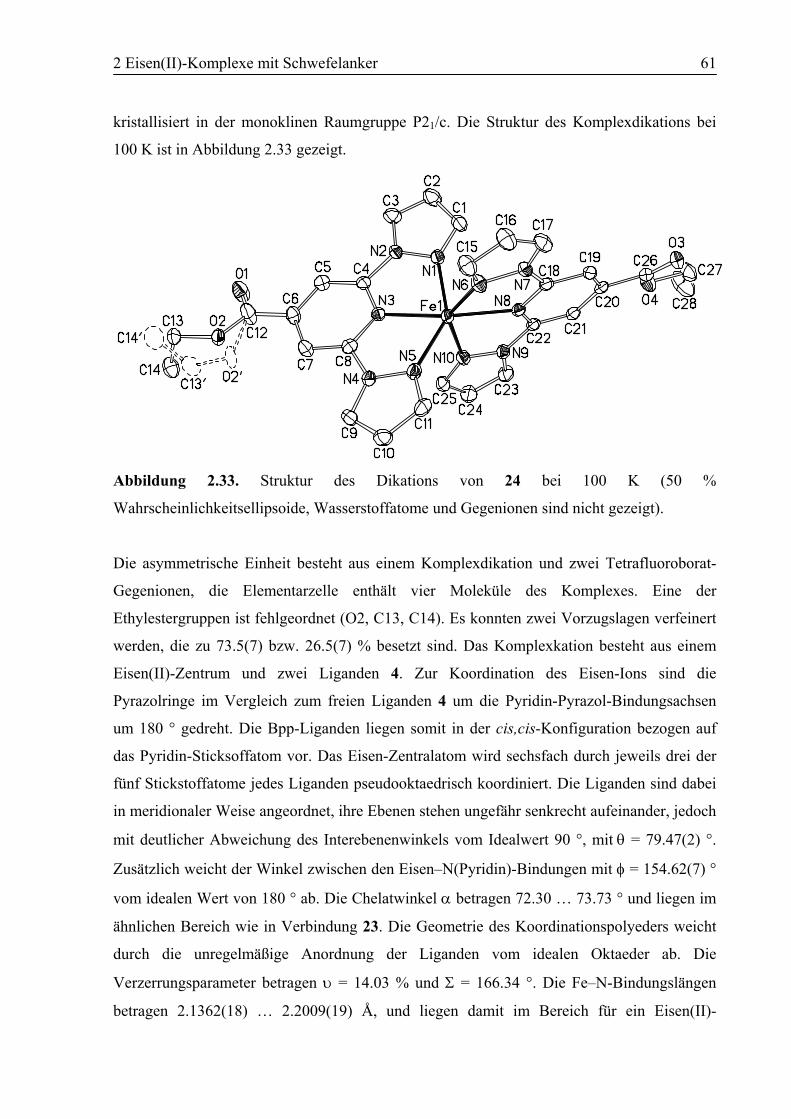
2 Eisen(II)-Komplexe mit Schwefelanker 61
kristallisiert in der monoklinen Raumgruppe P21/c. Die Struktur des Komplexdikations bei
100 K ist in Abbildung 2.33 gezeigt.
Abbildung 2.33. Struktur des Dikations von 24 bei 100 K (50 %
Wahrscheinlichkeitsellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Die asymmetrische Einheit besteht aus einem Komplexdikation und zwei Tetrafluoroborat-
Gegenionen, die Elementarzelle enthält vier Moleküle des Komplexes. Eine der
Ethylestergruppen ist fehlgeordnet (O2, C13, C14). Es konnten zwei Vorzugslagen verfeinert
werden, die zu 73.5(7) bzw. 26.5(7) % besetzt sind. Das Komplexkation besteht aus einem
Eisen(II)-Zentrum und zwei Liganden 4. Zur Koordination des Eisen-Ions sind die
Pyrazolringe im Vergleich zum freien Liganden 4 um die Pyridin-Pyrazol-Bindungsachsen
um 180 ° gedreht. Die Bpp-Liganden liegen somit in der cis,cis-Konfiguration bezogen auf
das Pyridin-Sticksoffatom vor. Das Eisen-Zentralatom wird sechsfach durch jeweils drei der
fünf Stickstoffatome jedes Liganden pseudooktaedrisch koordiniert. Die Liganden sind dabei
in meridionaler Weise angeordnet, ihre Ebenen stehen ungefähr senkrecht aufeinander, jedoch
mit deutlicher Abweichung des Interebenenwinkels vom Idealwert 90 °, mit θ = 79.47(2) °.
Zusätzlich weicht der Winkel zwischen den Eisen–N(Pyridin)-Bindungen mit φ = 154.62(7) °
vom idealen Wert von 180 ° ab. Die Chelatwinkel α betragen 72.30 … 73.73 ° und liegen im
ähnlichen Bereich wie in Verbindung 23. Die Geometrie des Koordinationspolyeders weicht
durch die unregelmäßige Anordnung der Liganden vom idealen Oktaeder ab. Die
Verzerrungsparameter betragen υ = 14.03 % und Σ = 166.34 °. Die Fe–N-Bindungslängen
betragen 2.1362(18) … 2.2009(19) Å, und liegen damit im Bereich für ein Eisen(II)-

2 Eisen(II)-Komplexe mit Schwefelanker 62
Zentralion im High Spin-Zustand.[2] Anders als in 23 und vergleichbaren reinen High Spin-
Fe-Bpp-Komplexen weist das Kristallgitter von 24 nicht die typische Stapelung
zweidimensionaler Schichten von Komplexkationen und BF4–-Anionen auf. Allerdings sind
die intermolekularen Abstände zwischen den Molekülen in Richtung der N(py)–Eisen–
N(py’)-Achse aufgrund der Größe der Ethylreste deutlich größer als in Verbindung 23.
Dadurch kommt es wiederum zur Störung der Kooperativität in dieser Richtung im Gitter,
wodurch ein Spinübergang erschwert wird.
Tabelle 2.4. Ausgewählte Abstände [Å] und Winkel [°] für 24 bei 100 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N3 2.1362(18) N3-Fe1-N8 154.62(7) O3-C26-O4 124.8(2)
Fe1–N8 2.1519(19) N8-Fe1-N10 72.30(7) O3-C26-C20 123.6(2)
Fe1–N10 2.155(2) N8-Fe1-N6 72.92(7) C26-O4-C27 115.85(19)
Fe1–N6 2.157(2) N3-Fe1-N5 73.73(7) N8-C22-N9 111.9(2)
Fe1–N5 2.177(2) N3-Fe1-N1 72.85(7) C21-C22-N9 124.6(2)
Fe1–N1 2.2009(19) C12-O2-C13 116.2(3) C23-N9-C22 131.6(2)
O1–C12 1.195(3) O1-C12-O2 124.5(3) N10-N9-C22 117.64(19)
O2–C12 1.338(3) O1-C12-C6 123.2(2) C17-N7-C18 130.9(2)
O2–C13 1.455(4) C3-N2-C4 130.1(2) N6-N7-C18 117.92(18)
O3–C26 1.202(3) N1-N2-C4 118.55(18) N8-C18-N7 112.04(19)
O4–C26 1.327(3) N3-C4-N2 112.52(19) C19-C18-N7 125.1(2)
O4–C27 1.461(3) C5-C4-N2 124.4(2)
C9-N4-C8 129.7(2)
N5-N4-C8 118.75(18)
N3-C8-N4 112.8(2)
C7-C8-N4 123.6(2)
Der Eisen(II)-Komplex 24 liegt im Feststoff vermutlich als reine High Spin-Verbindung vor.
Ein Spinübergang zum Low Spin-Zustand unterhalb 100 K ist für einen Eisen(II)-Bpp-
Komplex unwahrscheinlich. Ein Vergleich mit Eisenkomplexen von Bpp-Liganden aus der
Literatur sagt für Verbindung 24 auf Basis des Grades der Verzerrung des
Koordinationspolyeders die Abwesenheit thermischen Spincrossovers voraus.[43]

2 Eisen(II)-Komplexe mit Schwefelanker 63
Die analytischen Untersuchungen bestätigen die Synthese des Eisen(II)-Komplexes 24 als
eine High Spin-Verbindung im Feststoff bei 100 K und in Lösung bei RT.
2.3.3 [Fe(10)2](BF4)2 (25)
Die Synthese des Eisen(II)-Komplexes des Thiol-Liganden 10 erfolgte analog den
Komplexierungsreaktionen, die zu 23 und 24 führten. Zwei Äquivalente des Liganden 10
wurden mit einem Äquivalent Eisen(II)tetrafluoroborat Hexahydrat in Lösung in THF
zusammengegeben. Sofort nach Zugabe fiel ein dunkelroter Niederschlag aus, der mit THF
gewaschen und im Hochvakuum getrocknet wurde. Der Eisenkomplex 25 wurde in Form
eines roten, mikrokristallinen Feststoffes in guten Ausbeuten erhalten.
NNNN N
2 iN
NN
N
NN
N
N
N
N
Fe
2+
[BF4–]2
25
10
HN
SHO
NH
HS
OHN
O
SH
Abbildung 2.34. Synthese von 25; (i) 0.97 Äq. Fe(BF4)2 · 6 H2O, THF, RT, 18 h, 79 %.
Das IR-Spektrum als Feststoff in KBr zeigt die Anwesenheit der BF4–-Anionen durch eine
starke, breite Bande bei 1058 cm–1. Die restlichen Banden entsprechen den Banden des IR-
Spektrums des Liganden 10 mit geringer oder keiner Verschiebung. Die Elementaranalyse der
Verbindung ist durch die Anwesenheit von Restwasser, das sich durch Trocknen im
Hochvakuum nicht restlos entfernen lässt, ungenau. Das 1H-NMR-Spektrum des Komplexes
in [D4]MeOH zeigt paramagnetisch verbreiterte Signale, die über einen Bereich von 0-70 ppm
verschoben sind. Die Signale im Tieffeldbereich bei 69.58, 63.58, 59.15 und 49.01 ppm
weisen ein Integralverhältnis von 1:1:1:1 auf und werden möglicherweise durch die Protonen
der aromatischen Ringe verursacht. Die restlichen Signale im Spektrum befinden sich im
Bereich 8.23-1.09 ppm. Die Signale im 1H-NMR-Spektrum konnten aufgrund des
paramagnetischen Einflusses nicht weiter zugeordnet werden.
Orange Einkristalle von Verbindung 25 konnten durch isotherme Diffusion von Diethylether
in eine Lösung des Rohprodukts in MeOH nach einer Woche gewonnen werden. Verbindung
25 kristallisiert bei 100 K in der monoklinen Raumgruppe P21/c. Die Struktur des
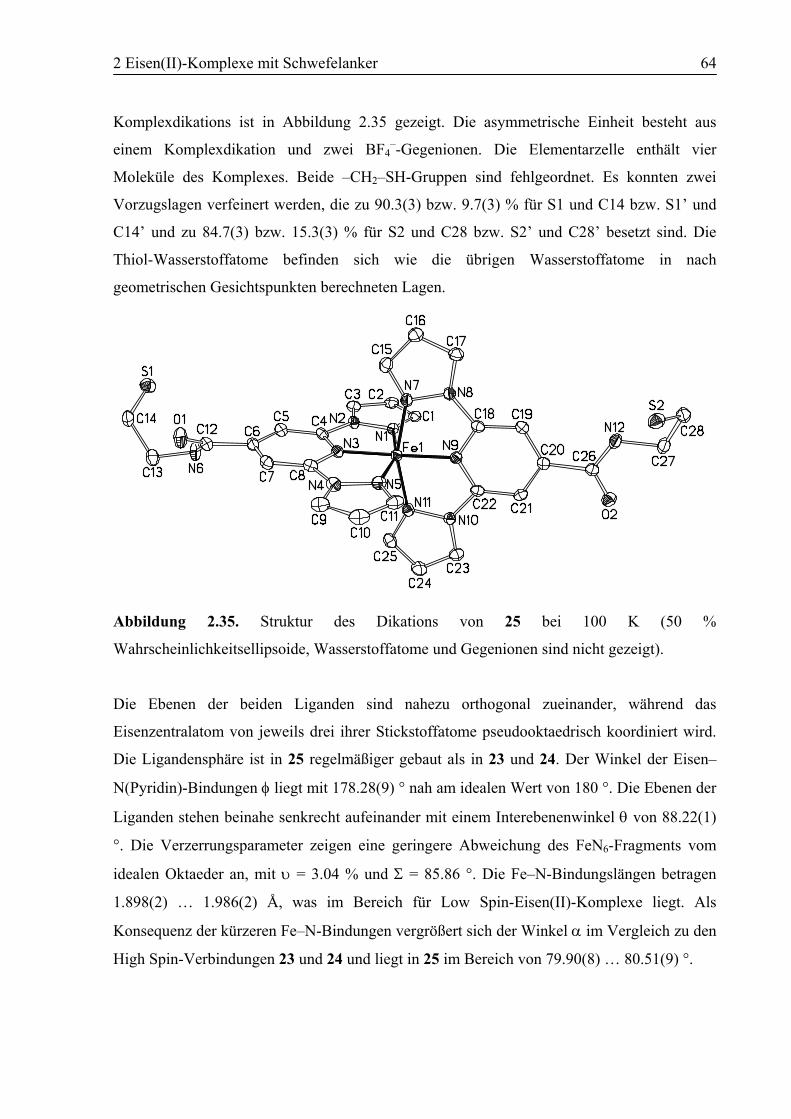
2 Eisen(II)-Komplexe mit Schwefelanker 64
Komplexdikations ist in Abbildung 2.35 gezeigt. Die asymmetrische Einheit besteht aus
einem Komplexdikation und zwei BF4–-Gegenionen. Die Elementarzelle enthält vier
Moleküle des Komplexes. Beide –CH2–SH-Gruppen sind fehlgeordnet. Es konnten zwei
Vorzugslagen verfeinert werden, die zu 90.3(3) bzw. 9.7(3) % für S1 und C14 bzw. S1’ und
C14’ und zu 84.7(3) bzw. 15.3(3) % für S2 und C28 bzw. S2’ und C28’ besetzt sind. Die
Thiol-Wasserstoffatome befinden sich wie die übrigen Wasserstoffatome in nach
geometrischen Gesichtspunkten berechneten Lagen.
Abbildung 2.35. Struktur des Dikations von 25 bei 100 K (50 %
Wahrscheinlichkeitsellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Die Ebenen der beiden Liganden sind nahezu orthogonal zueinander, während das
Eisenzentralatom von jeweils drei ihrer Stickstoffatome pseudooktaedrisch koordiniert wird.
Die Ligandensphäre ist in 25 regelmäßiger gebaut als in 23 und 24. Der Winkel der Eisen–
N(Pyridin)-Bindungen φ liegt mit 178.28(9) ° nah am idealen Wert von 180 °. Die Ebenen der
Liganden stehen beinahe senkrecht aufeinander mit einem Interebenenwinkel θ von 88.22(1)
°. Die Verzerrungsparameter zeigen eine geringere Abweichung des FeN6-Fragments vom
idealen Oktaeder an, mit υ = 3.04 % und Σ = 85.86 °. Die Fe–N-Bindungslängen betragen
1.898(2) … 1.986(2) Å, was im Bereich für Low Spin-Eisen(II)-Komplexe liegt. Als
Konsequenz der kürzeren Fe–N-Bindungen vergrößert sich der Winkel α im Vergleich zu den
High Spin-Verbindungen 23 und 24 und liegt in 25 im Bereich von 79.90(8) … 80.51(9) °.
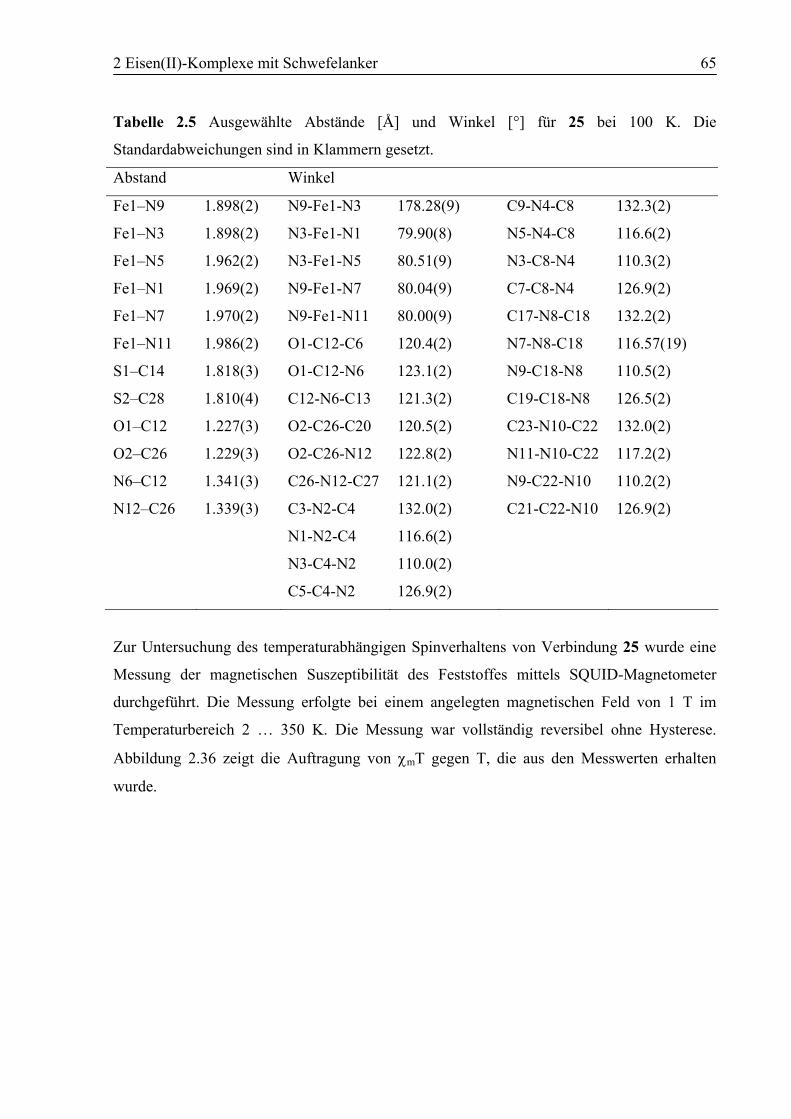
2 Eisen(II)-Komplexe mit Schwefelanker 65
Tabelle 2.5 Ausgewählte Abstände [Å] und Winkel [°] für 25 bei 100 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N9 1.898(2) N9-Fe1-N3 178.28(9) C9-N4-C8 132.3(2)
Fe1–N3 1.898(2) N3-Fe1-N1 79.90(8) N5-N4-C8 116.6(2)
Fe1–N5 1.962(2) N3-Fe1-N5 80.51(9) N3-C8-N4 110.3(2)
Fe1–N1 1.969(2) N9-Fe1-N7 80.04(9) C7-C8-N4 126.9(2)
Fe1–N7 1.970(2) N9-Fe1-N11 80.00(9) C17-N8-C18 132.2(2)
Fe1–N11 1.986(2) O1-C12-C6 120.4(2) N7-N8-C18 116.57(19)
S1–C14 1.818(3) O1-C12-N6 123.1(2) N9-C18-N8 110.5(2)
S2–C28 1.810(4) C12-N6-C13 121.3(2) C19-C18-N8 126.5(2)
O1–C12 1.227(3) O2-C26-C20 120.5(2) C23-N10-C22 132.0(2)
O2–C26 1.229(3) O2-C26-N12 122.8(2) N11-N10-C22 117.2(2)
N6–C12 1.341(3) C26-N12-C27 121.1(2) N9-C22-N10 110.2(2)
N12–C26 1.339(3) C3-N2-C4 132.0(2) C21-C22-N10 126.9(2)
N1-N2-C4 116.6(2)
N3-C4-N2 110.0(2)
C5-C4-N2 126.9(2)
Zur Untersuchung des temperaturabhängigen Spinverhaltens von Verbindung 25 wurde eine
Messung der magnetischen Suszeptibilität des Feststoffes mittels SQUID-Magnetometer
durchgeführt. Die Messung erfolgte bei einem angelegten magnetischen Feld von 1 T im
Temperaturbereich 2 … 350 K. Die Messung war vollständig reversibel ohne Hysterese.
Abbildung 2.36 zeigt die Auftragung von χmT gegen T, die aus den Messwerten erhalten
wurde.
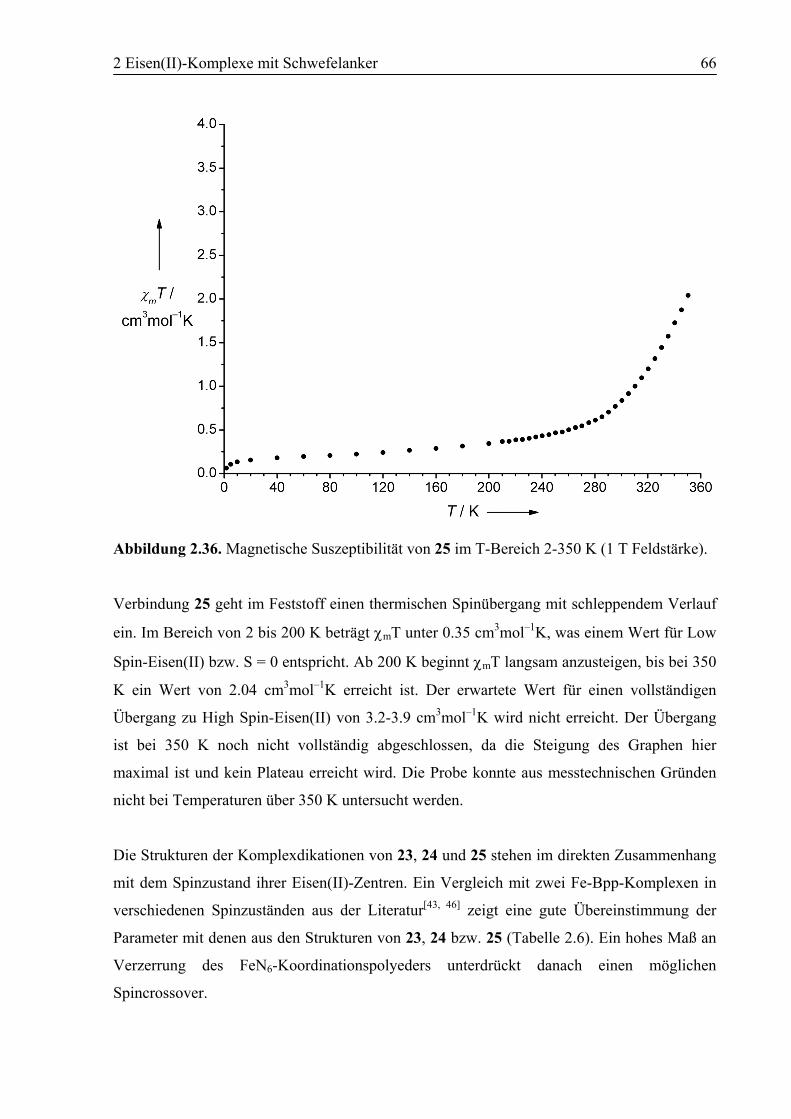
2 Eisen(II)-Komplexe mit Schwefelanker 66
Abbildung 2.36. Magnetische Suszeptibilität von 25 im T-Bereich 2-350 K (1 T Feldstärke).
Verbindung 25 geht im Feststoff einen thermischen Spinübergang mit schleppendem Verlauf
ein. Im Bereich von 2 bis 200 K beträgt χmT unter 0.35 cm3mol–1K, was einem Wert für Low
Spin-Eisen(II) bzw. S = 0 entspricht. Ab 200 K beginnt χmT langsam anzusteigen, bis bei 350
K ein Wert von 2.04 cm3mol–1K erreicht ist. Der erwartete Wert für einen vollständigen
Übergang zu High Spin-Eisen(II) von 3.2-3.9 cm3mol–1K wird nicht erreicht. Der Übergang
ist bei 350 K noch nicht vollständig abgeschlossen, da die Steigung des Graphen hier
maximal ist und kein Plateau erreicht wird. Die Probe konnte aus messtechnischen Gründen
nicht bei Temperaturen über 350 K untersucht werden.
Die Strukturen der Komplexdikationen von 23, 24 und 25 stehen im direkten Zusammenhang
mit dem Spinzustand ihrer Eisen(II)-Zentren. Ein Vergleich mit zwei Fe-Bpp-Komplexen in
verschiedenen Spinzuständen aus der Literatur[43, 46] zeigt eine gute Übereinstimmung der
Parameter mit denen aus den Strukturen von 23, 24 bzw. 25 (Tabelle 2.6). Ein hohes Maß an
Verzerrung des FeN6-Koordinationspolyeders unterdrückt danach einen möglichen
Spincrossover.
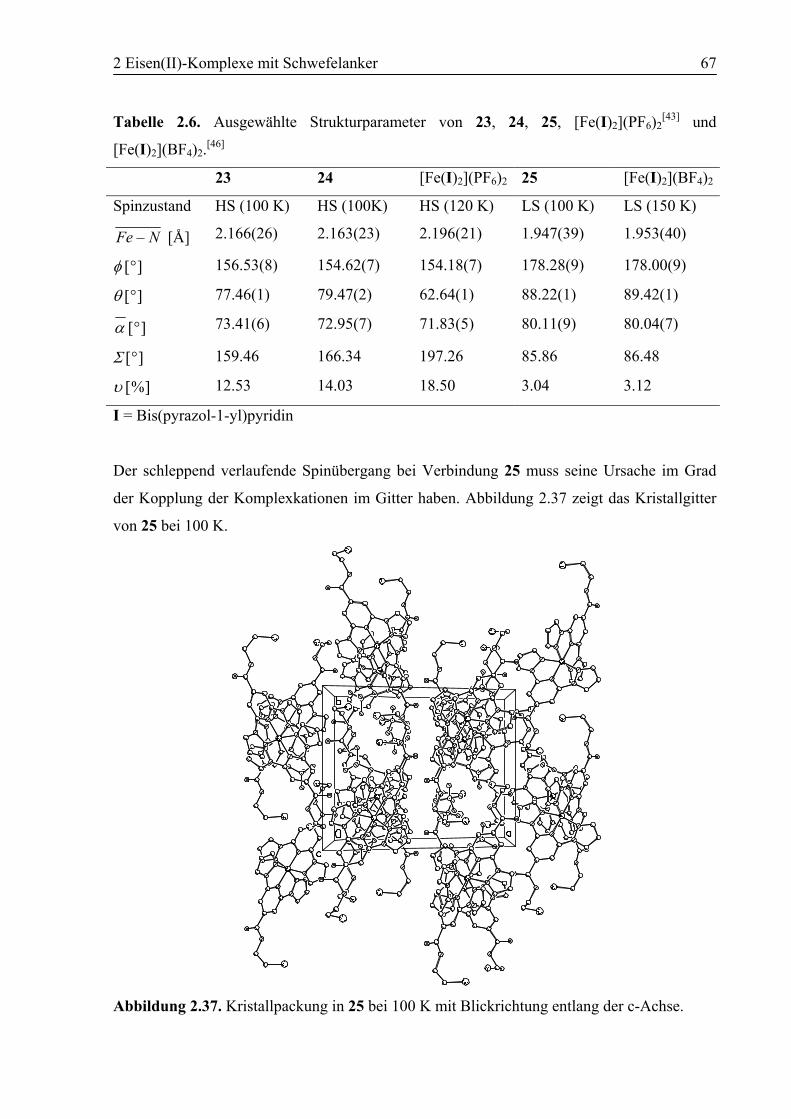
2 Eisen(II)-Komplexe mit Schwefelanker 67
Tabelle 2.6. Ausgewählte Strukturparameter von 23, 24, 25, [Fe(I)2](PF6)2[43] und
[Fe(I)2](BF4)2.[46]
23 24 [Fe(I)2](PF6)2 25 [Fe(I)2](BF4)2
Spinzustand HS (100 K) HS (100K) HS (120 K) LS (100 K) LS (150 K)
NFe – [Å] 2.166(26) 2.163(23) 2.196(21) 1.947(39) 1.953(40)
φ [°] 156.53(8) 154.62(7) 154.18(7) 178.28(9) 178.00(9)
θ [°] 77.46(1) 79.47(2) 62.64(1) 88.22(1) 89.42(1)
α [°] 73.41(6) 72.95(7) 71.83(5) 80.11(9) 80.04(7)
Σ [°] 159.46 166.34 197.26 85.86 86.48
υ [%] 12.53 14.03 18.50 3.04 3.12
I = Bis(pyrazol-1-yl)pyridin
Der schleppend verlaufende Spinübergang bei Verbindung 25 muss seine Ursache im Grad
der Kopplung der Komplexkationen im Gitter haben. Abbildung 2.37 zeigt das Kristallgitter
von 25 bei 100 K.
Abbildung 2.37. Kristallpackung in 25 bei 100 K mit Blickrichtung entlang der c-Achse.

2 Eisen(II)-Komplexe mit Schwefelanker 68
Das Kristallgitter von Verbindung 25 präsentiert einen Grenzfall in Bezug auf Fe-Bpp-
Komplexe. Bei diesen Komplexen werden vor allem zwei Typen von dreidimensionaler
Ordnung gefunden.
Reine High Spin-Verbindungen zeigen gestapelte zweidimensionale Schichten von
Komplexkationen, die durch Schichten der Anionen voneinander getrennt sind. Die N(py)–
Fe–N(py’)-Bindungsachsen der Moleküle liegen dabei parallel zu den Schichten. Die
Ausdehnung der Komplexkationen beim Spinübergang erfolgt hauptsächlich entlang dieser
Achse. Senkrecht zur Achse ist die Ausdehnung durch die Rigidität des Ligandengerüstes
deutlich schwächer. Bei Anordnung der Komplexkationen in Schichten parallel zur N(py)–
Fe–N(py’)-Bindungsachse wird die Information über den Spinübergang und die damit
einhergehende Größenveränderung zwar innerhalb der Schichten übertragen; zwischen den
Schichten findet aber durch deren „Isolation“ durch die Anionenschichten und die schwache
Ausdehnung der Moleküle senkrecht zur zentralen Bindungsachse, also auch senkrecht zu den
Schichten, keine ausreichende Kopplung statt, so dass sich die Information des Spinübergangs
nicht ohne weiteres dreidimensional im Gitter fortpflanzen kann.
N
NNN
NN
N
N
N
N
FeN
NNN
NN
N
N
N
N
FeN
NNN
NN
N
N
N
N
Fe
Anionenschicht
Richtung derstärksten räumlichen
Ausdehnung beim SCO
N
NNN
NN
N
N
N
N
FeN
NNN
NN
N
N
N
N
FeN
NNN
NN
N
N
N
N
Fe
Anionenschicht
N
NNN
NN
N
N
N
N
FeN
NNN
NN
N
N
N
N
FeN
NNN
NN
N
N
N
N
Fe
Anionenschicht Abbildung 2.38. Schema der Stapelung der Schichten im Kristallgitter von reinen High Spin-
Fe-Bpp-Komplexen.
In Fe-Bpp-Komplexen mit thermischem SCO-Verhalten liegt meist ein dreidimensionales
Netzwerk der Komplexkationen vor, in dem die Anionen gleichmäßig im Gitter verteilt sind.

2 Eisen(II)-Komplexe mit Schwefelanker 69
Kommt es trotzdem zum Aufbau einer Abfolge von alternierenden Schichten von
Komplexkationen und Anionen, liegen die N(py)–Fe–N(py’)-Bindungsachsen orthogonal zu
den Schichten. Innerhalb der Schichten wird die infolge geringer Ausdehnung der Moleküle
senkrecht zur zentralen Bindungsachse geringe Kopplung durch starke sekundäre
Wechselwirkungen zwischen den Molekülen ausgeglichen. Zwischen den Schichten sorgt die
stärkere Ausdehnung der Moleküle in diese Richtung für ausreichende Kopplung.
N NN NN
N NN NN
Fe
Anionenschicht
Richtung derstärksten räumlichen
Ausdehnung beim SCO
N NN NN
N NN NN
FeN N
N NN
N NN NN
Fe
N NN NN
N NN NN
Fe
Anionenschicht
N NN NN
N NN NN
FeN N
N NN
N NN NN
Fe
N NN NN
N NN NN
Fe
Anionenschicht
N NN NN
N NN NN
FeN N
N NN
N NN NN
Fe
Abbildung 2.39. Modell der Stapelung der Schichten im Kristallgitter von SCO-Fe-Bpp-
Komplexen. Die Anionen bilden meist keine scharf abgegrenzten Schichten, sondern befinden
sich eher zwischen den Komplexkationen verteilt.
Im Kristallgitter von 25 ordnen sich die Moleküle in Schichten in der bc-Ebene an, die in
Richtung der a-Achse gestapelt sind (Abbildung 2.37). Die N(py)–Fe–N(py’)-
Bindungsachsen der Komplexkationen sind ungefähr im 45 °-Winkel zu den Schichten
orientiert. Die Schichten der Komplexkationen sind nicht durch Schichten der BF4–-Anionen
voneinander getrennt. Die Moleküle besitzen also einerseits eingeschränkte Kopplung in
Richtung der zentralen Bindungsachsen, sind aber andererseits nicht durch Anionenschichten

2 Eisen(II)-Komplexe mit Schwefelanker 70
voneinander isoliert. Die dreidimensionale Kopplung im Gitter ist also gegeben, wodurch
Verbindung 25 einen thermischen Spinübergang zeigt. Die relativ schwache Kopplung
zwischen den Schichten bedingt allerdings einen schleppenden Verlauf des Übergangs.
Die Röntgenstrukturuntersuchungen zeigen zusammen mit den Ergebnissen der SQUID-
Untersuchungen, dass es sich bei Verbindung 25 als Feststoff um eine SCO-Verbindung
handelt. Die NMR-Untersuchung gibt einen Hinweis darauf, dass es sich beim schleppenden
Verlauf des Spinübergangs um einen reinen Festkörpereffekt handelt, da 25 in Lösung bei
Raumtemperatur im Gegensatz zum Feststoff paramagnetisch ist.
Die analytischen Untersuchungen bestätigen die erfolgreiche Synthese von Verbindung 25.
Mit dieser Verbindung liegt ein SCO-Eisen(II)-Komplex mit Schwefelankergruppen vor, wie
im Konzept geplant. Der schleppende Verlauf des Spinübergangs ist vermutlich ein Effekt der
Festkörperpackung und mag auf der Oberfläche (wie auch in Lösung) nicht mehr relevant
sein.
2.4 Zusammenfassung
Mit der Synthese der Liganden 6, 10, 12 und 14 liegen die gewünschten oberflächenaktiven
Liganden mit der Bpp-Einheit als Endgruppe vor. Die Liganden unterscheiden sich in ihren
Ankergruppen und Distanzstücken, was durch das flexible Synthesekonzept ermöglicht wird.
Die Synthese des Liganden 22 mit lateral wirksamen H-Brückenbindungsdonoren bzw. -
akzeptoren steht noch aus. Sieben Stufen der achtstufigen Syntheseroute wurden
abgeschlossen.
Verbindung 25 stellt einen oberflächenaktiven Eisen(II)-SCO-Komplex gemäß dem
zugrundegelegten Synthesekonzept dar. Die Untersuchungen zu Verbindung 25, sowie zu den
synthetisierten neuen Eisen(II)-Komplexen 23 und 24 bestätigen die von Halcrow und
Mitarbeitern entwickelte Theorie vom Zusammenhang lokaler Verzerrung und Spinverhalten
in Eisen(II)-Bpp-Komplexen.

2 Eisen(II)-Komplexe mit Schwefelanker 71
2.5 Experimenteller Teil
2.5.1 Allgemeines
Sofern nicht anders vermerkt, wurden sämtliche Reaktionen unter Stickstoffatmosphäre und
mit Hilfe von Standard-Schlenk-Technik in absoluten Lösungsmitteln durchgeführt. Die
verwendeten absoluten Lösungsmittel sind kommerziell gelagert auf Molekularsieb 3 Å
erhältlich (Restwassergehalt ≤ 50 ppm). Die eingesetzten Reagenzien wurden von den Firmen
ABCR, Acros, Alfa Aesar, Fluka, Merck, Sigma-Aldrich und VWR bezogen und ohne
weitere Reinigung verwendet. 4'-Amino-(1,1'-biphenyl)-4-thiol (11) wurde von Frau Ying
Luo, AG Haag, FU Berlin synthetisiert.
Folgende Geräte wurden zur Gewinnung der Daten verwendet:
NMR-Spektren: Die Messungen erfolgten an den Geräten Bruker ARX 200 (1H-NMR: 200
MHz; 13C-NMR: 50.32 MHz) und Bruker ARX 400 (1H-NMR: 400 MHz, 13C-NMR: 100.64
MHz). Die 13C-NMR-Spektren wurden breitbandentkoppelt aufgenommen. Sofern nicht
anders vermerkt wurden NMR-Röhrchen mit 5 mm Durchmesser verwendet und die Spektren
bei Raumtemperatur aufgenommen. Alle chemischen Verschiebungen sind in ppm relativ zur
Rest-1H- bzw. 13C-Absorption des verwendeten Lösungsmittels (interner Standard)
angegeben.
IR-Spektren: Die Proben wurden in Form von Kaliumbromid-Presslingen bzw. als Film an
einem Nicolet Magna System 750 gemessen. Die Wellenzahlen ν~ sind in cm–1 angegeben.
Die Zuordnung der Banden erfolgte anhand geeigneter Literatur.[95]
Elementaranalysen: Die quantitative Bestimmung von Kohlenstoff, Wasserstoff, Stickstoff
und Schwefel erfolgte verbrennungsanalytisch an einem Thermo Finnigan EAGER 300 (Flash
1112 Series). Die Angaben erfolgen in Gewichtsprozent.
Massenspektren: Die Aufnahme von Massenspektren erfolgte an einem Sektorfeld-
Massenspektrometer des Typs Varian 311A (EI-MS, 70 eV).

2 Eisen(II)-Komplexe mit Schwefelanker 72
2.5.2 Synthesen
2.5.2.1 2,6-Dibrom-4-carbonsäure
NBr Br
COOH
C6H3Br2NO2
M = 280.9 g/mol
weißer Feststoff
POBr3 (2.92 g, 10 mmol) und Citrazinsäure (1 g, 6.4 mmol) wurden im festen Zustand in
einem 50 ml Einhals-Kolben zusammengegeben. Auf den Kolben wurde ein Intensivkühler
aufgesetzt. Ein Schlauch am oberen Ende des Kühlers führte in einen Eimer mit Eiswasser um
entstehendes HBr-Gas abzuleiten. Es wurde für 5 h unter Rühren auf 175 °C erhitzt, wobei
sich eine braune Schmelze bildete. Über einen Zeitraum von 20 h wurde das Gemisch auf
Raumtemperatur abgekühlt und vorsichtig 15 ml dest. H2O zugefügt. Die entstandene violette
Suspension wurde abfiltriert und das Filtrat mit fünfmal 10 ml DCM extrahiert. Der
Filterrückstand wurde für 24 h mit einem Soxhlet-Extraktor extrahiert. Die vereinigten
organischen Phasen wurden über wasserfreiem Magnesiumsulfat getrocknet und das
Lösungsmittel am Rotationsverdampfer entfernt. Der erhaltene braune Feststoff wurde
zweimal hintereinander aus dest. H2O umkristallisiert und das Produkt wurde als ein weißer,
kristalliner Feststoff erhalten (2.52 g, 20 %).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 14.12 (br; 1 H; COOH), 7.98 ppm (s; 2 H;
Pyridin H).
IR (KBr): ν~ = 671 (s), 760 (m), 897(w), 924 (w), 996 (w), 1163 (m), 1221 (m), 1237 (m),
1356 (s), 1413 (m), 1536 (s), 1585 (m), 1733 (s), 2925 (s), 3076 cm–1 (s).
MS (EI, 70 eV): m/z (%) = 200 (65) / 202 (60) [M•+– Br]; 279 (51) / 281 (100) / 283 (49)
[M•+].
Elementaranalyse: Ber. (%) für C6H3Br2NO2: C 25.65, H 1.08, N 4.99; gef.: C 25.84, H
1.15, N 5.64.

2 Eisen(II)-Komplexe mit Schwefelanker 73
2.5.2.2 2,6-Dichlor-4-carbonsäure (1)
NCl Cl
COOH
C6H3Cl2NO2
M = 192.0 g/mol
hellbrauner Feststoff
Citrazinsäure (25 g, 0.161 mol, 1 Äq.) und NMe4Cl (18.37 g, 0.167 mol, 1.04 Äq) wurden in
POCl3 (74.14 g, 0.484 mol, 3 Äq.) gelöst. Unter Rühren wurde das Gemisch für 30 min auf
135 °C, und anschließend für 2 h auf 150 °C erhitzt. Die entstandene dunkelbraune Lösung
wurde nach dem Abkühlen auf Raumtemperatur in einen Kolben mit Eiswasser gegossen.
Nach weiteren 2 h Rühren wurde der ausgefallene Feststoff abfiltriert und mit dest. H2O
nachgewaschen. Das Rohprodukt wurde in 250 ml EtOAc gegeben, es wurde 20 min gerührt
und abfiltriert. Entfernung des Lösungsmittels aus dem Filtrat am Rotationsverdampfer ergab
das Produkt als hellbraunen Feststoff (21.88 g, 71 %).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 14.16 (br; 1 H; COOH), 7.86 ppm (s; 2 H;
Pyridin H).
IR (KBr): ν~ = 466 (w), 681 (s), 770 (s), 817 (w), 895 (w), 927 (w), 1002 (m), 1167 (s), 1232
(m), 1257 (s), 1368 (s), 1421 (m), 1549 (s), 1598 (w), 1724 (s), 2604 (m), 3079 cm–1 (m).
Elementaranalyse: Ber. (%) für C6H3Cl2NO2: C 37.53, H 1.57, N 7.30; gef.: C 37.87, H
1.58, N 7.26.
2.5.2.3 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-carbonsäure (2)
NNNN N
CO2H
C12H9N5O2
255.2 g/mol
weiße Nadeln
Paraffinfreies Kaliumhydrid (1.378 g, 34.4 mmol, 3.3 Äq.) wurde in einem 250 ml-Schlenk-
Kolben in 50 ml Diglyme vorgelegt. Zu der weißen Suspension wurde unter Rühren eine
Lösung von Pyrazol (2.482 g, 36.5 mmol, 3.5 Äq.) in 50 ml Diglyme über eine Spritze
gegeben. Die entstandene gelbliche Suspension wurde für 2 h bei Raumtemperatur gerührt.

2 Eisen(II)-Komplexe mit Schwefelanker 74
2,6-Dichlor-4-carbonsäure (1) (2 g, 10.4 mmol, 1 Äq.) und 4-Dimethylaminopyridin (64 mg,
0.5 mmol, 0.05 Äq.) wurden in fester Form zur Reaktionslösung gegeben. Das Gemisch
wurde für 3 d auf 130 °C erhitzt. Nach Abkühlen auf Raumtemperatur wurde das
Lösungsmittel im Hochvakuum entfernt. Es wurden 75 ml dest. H2O zugefügt und die braune
Lösung anschließend mit verd. HCl auf pH 4 gebracht. Der entstandene weiße Niederschlag
wurde abfiltriert und mit dest. H2O gewaschen. Der Feststoff wurde in einem Gemisch aus 75
ml DCM und 50 ml MeOH gelöst. Am Rotationsverdampfer wurde die Lösung bei einem
Druck von 500 mbar eingeengt, wobei ein weißer Niederschlag ausfiel. Nach Filtration und
Trocknung im Hochvakuum erhielt man das Produkt in Form von feinen weißen Nadeln
(2.038 g, 76 %).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 14.10 (br; 1 H; COOH), 8.97 (dd, J = 2.6 Hz,
0.6 Hz; 2 H; pz-H5), 8.15 (s, 2 H; Pyridin H), 7.90 (dd, J = 1.5 Hz, 0.6 Hz; 2 H; pz-H3), 6.65
ppm (dd, J = 2.6 Hz, 1.7 Hz; 2 H; pz-H4 ).
IR (KBr): ν~ = 603 (m), 680 (s), 763 (s), 783 (m), 944 (m), 974 (w), 1049 (m), 1139 (m),
1399 (s), 1466 (s), 1525 (m), 1575 (s), 1618 (m), 1725 (s), 3148 cm–1 (m).
MS (EI, 70 eV): m/z (%) = 255 (100) [M•+].
Elementaranalyse: Ber. (%) für C12H9N5O2: C 56.47, H 3.55, N 27.44; gef.: C 56.36, H 3.61,
N 28.06.
2.5.2.4 Methyl-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxylat (3)
NNNN N
CO2Me
C14H15N5O2
M = 269.3 g/mol
weiße Kristalle
In einem 50 ml-Schlenk-Kolben wurde 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-carbonsäure (2)
(0.5 g, 1.9 mmol) in MeOH (20 ml) suspendiert. Konz. H2SO4 (0.67 ml) wurde unter Rühren
zugegeben und das Reaktionsgemisch 3 h auf 60 °C erhitzt. Nach dem Abkühlen wurde das
Lösungsmittel am Rotationsverdampfer entfernt und es wurden 20 ml dest. H2O zugefügt. Der
entstandene weiße Niederschlag wurde abfiltriert, mit dest. H2O gewaschen und in einem

2 Eisen(II)-Komplexe mit Schwefelanker 75
MeOH/Wasser-Gemisch umkristallisiert. Das Produkt wurde als weißer, mikrokristalliner
Feststoff erhalten, der im Hochvakuum getrocknet wurde (0.46 g, 87 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.55 (dd, J = 2.6 Hz, 0.7 Hz; 2 H; pz-H5),
8.39 (s; 2 H; Pyridin H), 7.79 (dd, J = 1.7 Hz, 0.7 Hz; 2 H; pz-H3), 6.52 (dd, J = 2.6 Hz, 1.7
Hz; 2 H; pz-H4), 4.00 (s; 3 H; CH3).
IR (KBr): ν~ = 599 (w), 736 (m), 765 (s), 971 (s), 1052 (vs), 1167 (m), 1247 (s), 1402 (vs),
1462 (vs), 1505 (m), 1517 (s), 1579 (s), 1619 (s), 1732 (s), 2934 (w), 3131 cm–1 (m).
2.5.2.5 Ethyl-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxylat (4)
NNNN N
CO2Et
C14H13N5O2
M = 283.3 g/mol
weiße Kristalle
In einem 100 ml-Schlenk-Kolben wurde 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-carbonsäure (2)
(1.5 g, 5.8 mmol) in EtOH (60 ml) suspendiert. 2 ml konz. H2SO4 wurde unter Rühren
zugegeben und das Reaktionsgemisch 3 h auf 80 °C erhitzt. Nach dem Abkühlen wurde das
Lösungsmittel am Rotationsverdampfer entfernt und es wurden 60 ml dest. H2O zugefügt. Es
entstand ein weißer Niederschlag, der abfiltriert, mit dest. H2O gewaschen und in einem
EtOH/Wasser-Gemisch umkristallisiert wurde. Der erhaltene weiße, kristalline Feststoff
wurde im Hochvakuum getrocknet (1.48 g, 89 %). Farblose Einkristalle von 3 konnten durch
langsames Verdunsten einer Lösung der Verbindung in Chloroform erhalten werden.
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.50 (dd, J = 2.6 Hz, 0.7 Hz; 2 H; pz-H5),
8.33 (s; 2 H; Pyridin H), 7.76 (dd, J = 1.7 Hz, 0.7 Hz; 2 H; pz-H3), 6.48 (dd, J = 2.6 Hz, 1.7
Hz; 2 H; pz-H4), 4.43 (q, J = 7.1 Hz; 2 H; CH2), 1.42 ppm (t, J = 7.1 Hz; 3 H; CH3). 13C-NMR (100.6 MHz, [D1]Chloroform, 25 °C): δ = 164.48 (C=O), 151.22 (py-C2,6),
144.08 (py-C4), 143.32 (pz-C3), 127.71 (pz-C5), 109.61 (py-C3,5), 108.91 (pz-C4), 62.70
(CH2), 14.79 ppm (CH3).
IR (KBr): ν~ = 599 (m), 759 (s), 1056 (s), 1118 (s), 1236 (s), 1398 (vs), 1426 (vs), 1463 (vs),
1495 (m), 1575 (s), 1617 (s), 1729 (s), 3086 cm–1 (s).

2 Eisen(II)-Komplexe mit Schwefelanker 76
2.5.2.6 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methanol (5)
NNNN N
OH
C12H11N5O
M = 241.2 g/mol
weiße Nadeln
Methode A: Zu einer Suspension von Ethyl-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxylat (4)
(135 mg, 0.48 mmol, 1 Äq.) in EtOH wurde Natriumborhydrid (89 mg, 2.35 mmol, 5 Äq.) in
fester Form gegeben. Das Reaktionsgemisch wurde 3 d bei Raumtemperatur gerührt. Die
entstandene klare, farblose Lösung wurde am Rotationsverdampfer vom Lösungsmittel
befreit. Der zurückgebliebene Feststoff wurde mit 5 ml dest. H2O versetzt und es wurde verd.
HCl zugefügt, bis keine Gasentwicklung mehr zu bemerken war. Das Gemisch wurde nun mit
gesättigter Natriumcarbonat-Lösung versetzt, bis ein pH-Wert von 7 erreicht war. Die
wässrige Lösung wurde mit dreimal je 30 ml DCM extrahiert und die vereinigten organischen
Phasen über wasserfreiem Magnesiumsulfat getrocknet. Nach Entfernung des Lösungsmittels
am Rotationsverdampfer erhielt man das Produkt in Form von feinen weißen Nadeln (113 mg,
98 %).
Methode B: Zu einer Suspension von Methyl-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxylat
(3) (462 mg, 1.72 mmol, 1Äq.) in MeOH wurde Natriumborhydrid (324 mg, 8.58 mmol, 5
Äq.) in fester Form gegeben. Das Reaktionsgemisch wurde 3 d bei Raumtemperatur gerührt.
Die entstandene klare farblose Lösung wurde am Rotationsverdampfer vom Lösungsmittel
befreit. Der zurückgebliebene Feststoff wurde mit 30 ml dest. H2O versetzt und es wurde
verd. HCl zugefügt, bis keine Gasentwicklung mehr zu bemerken war. Das Gemisch wurde
nun mit Natriumcarbonat in fester Form versetzt, bis ein pH-Wert von 7 erreicht war. Die
wässrige Lösung wurde mit dreimal je 75 ml DCM extrahiert und die vereinigten organischen
Phasen über wasserfreiem Magnesiumsulfat getrocknet. Nach Entfernung des Lösungsmittels
am Rotationsverdampfer wurde das Produkt in Form von feinen weißen Nadeln erhalten (394
mg, 95 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.50 (dd, J = 2.6 Hz, 0.7 Hz; 2 H; pz-H5),
7.81 (t, J = 0.7 Hz; 2 H; Pyridin H), 7.74 (dd, J = 1.7 Hz, 0.7 Hz; 2 H; pz-H3), 6.47 (dd, J =
2.6 Hz, 1.7 Hz; 2 H; pz-H4), 4.81 (t, J = 0.7 Hz; 2 H; CH2), 3.08 ppm (br; 1 H; OH).

2 Eisen(II)-Komplexe mit Schwefelanker 77
13C-NMR (100.6 MHz, [D1]Chloroform, 25 °C): δ = 159.11 (py-C2,6), 149.55 (py-C4),
142.47 (pz-C3), 127.97 (pz-C5), 108.25 (py-C3,5), 105.95 (pz-C4), 61.74 ppm (CH2).
MS (EI, 70 eV): m/z (%) = 241 (100) [M•+].
IR (KBr): ν~ = 611 (m), 763 (s), 788 (s), 842 (m), 879 (w), 919 (m), 955 (m), 993 (w), 1051
(s), 1071 (s), 1178 (m), 1205 (s), 1266 (w), 1337 (m), 1357 (m), 1402 (s), 1467 (s), 1520 (m),
1576 (s), 1618 (s), 3130 (m), 3299 cm–1 (s).
Elementaranalyse: Ber. (%) für C12H11N5O: C 59.74, H 4.60, N 29.03; gef.: C 59.46, H 4.36,
N 29.22.
2.5.2.7 2,6-Bis(1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin (6)
NNNN N
SCN
C13H10N6S
M = 282.3 g/mol
weißer Feststoff
Zu einer Lösung von Ph3P (570 mg, 2.2 mmol) in MeCN (5,5 ml) in einem 50 ml-Schlenk-
Kolben wurde eine Lösung von Brom (0.11 ml, 2.2 mmol) in MeCN (1.1 ml) tropfenweise
bei Raumtemperatur zugegeben. Nach Beendigung der Zugabe wurde eine Lösung von
NH4SCN (329 mg, 4.3 mmol) in MeCN (7.4 ml) tropfenweise zugegeben. Eine Lösung von
2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methanol (5) (500 mg, 2.1 mmol) in MeCN (9 ml)
wurde zugetropft und das Reaktionsgemisch 24 h bei Raumtemperatur gerührt. Die
entstandene weiße Suspension wurde filtriert und der Feststoff mit MeCN gewaschen. Das
Filtrat wurde am Rotationsverdampfer vom Lösungsmittel befreit und das zurückgebliebene
gelbe Öl an Kieselgel säulenchromatographisch getrennt (Hexan/EtOAc = 1:1). Das Produkt
erhielt man als zweite Fraktion (Rf = 0.59). Das Thiocyanat wurde als weißer Feststoff
erhalten, der im Hochvakuum getrocknet wurde (440 mg, 75 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.57 (dd, J = 2.7 Hz, 0.6 Hz; 2 H; pz-H5),
7.90 (s, 2 H; Pyridin H), 7.79 (dd, J = 1.6 Hz, 0.6 Hz; 2 H; pz-H3), 6.53 (dd, J = 2.7 Hz, 1.7
Hz; 2 H; pz-H4), 4.21 ppm (s, 2 H; CH2).

2 Eisen(II)-Komplexe mit Schwefelanker 78
13C-NMR (100.6 MHz, [D1]Chloroform, 25 °C): δ = 150.6 (py-C2,6), 149.0 (py-C4), 142.7
(pz-C3), 127.2 (pz-C5), 110.6 (SCN), 109.0 (py-C3,5), 108.3 (pz-C4), 36.6 ppm (CH2).
IR (KBr): ν~ = 618 (m), 705 (m), 779 (s), 954 (s), 1043 (s), 1207 (s), 1396 (br), 1465 (br),
1520 (s), 1573 (s), 1620 (s), 2160 (s, –S–C≡N), 3134 cm–1 (m).
Elementaranalyse: Ber. (%) für C13H10N6S: C 55.30, H 3.57, N 29.77, S 11.36; gef.:
C 55.37, H 3.61, N 29.99, S 11.60.
2.5.2.8 2,6-Bis(1H-pyrazol-1-yl)-4-(bromomethyl)pyridin (7)
NNNN N
Br
C12H10BrN5
M = 304.1 g/mol
hellgelber Feststoff
In einem 100 ml-Schlenk-Kolben wurde eine Lösung von Triphenylphosphin (1.33 g, 5.1
mmol, 1.5 Äq.) in DCM (15 ml) bei 0 °C vorgelegt. Mit einer Fortuna-Pipette wurde eine
Lösung von Brom (0.26 ml, 5.1 mmol, 1 Äq.) in DCM (7 ml) unter Rühren langsam
zugetropft. Zu der entstandenen Suspension wurde 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-
yl)methanol (5) (0.82 g, 3.4 mmol, 1 Äq.) in 50 ml DCM gegeben. Nach 15 min bei 0 °C
wurde das Reaktionsgemisch weitere 12 h bei Raumtemperatur gerührt. Der entstandene
weiße Niederschlag wurde abfiltriert und das Filtrat am Rotationsverdampfer vom
Lösungsmittel befreit. Das entstandene gelbe Öl wurde in EtOAc gelöst und über eine kurze
Kieselgelsäule filtriert. Nach Entfernen des Lösungsmittels erhielt man das Produkt als
gelblichen Feststoff (0.68 g, 66 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.55 (dd, J = 2.6 Hz, 0.7 Hz; 2 H; pz-H5),
7.89 (s; 2 H; Pyridin H), 7.77 (dd, J = 1.7 Hz, 0.7 Hz; 2 H; pz-H3), 6.50 (dd, J = 2.6 Hz, 1.7
Hz; 2 H; pz-H4), 4.48 ppm (s; 2 H; CH2).

2 Eisen(II)-Komplexe mit Schwefelanker 79
2.5.2.9 2,6-Bis(1H-pyrazol-1-yl)-4-(isothiocyanatomethyl)pyridin (8)
NNNN N
NCS
C13H10N6S
M = 282.3 g/mol
weißer Feststoff
Zu einer Lösung von 2,6-Bis(1H-pyrazol-1-yl) 4-(bromomethyl)pyridin (7) (279 mg, 0.9
mmol, 1 Äq.) in Diglyme (11 ml) wurde Kaliumthiocyanat (1.601 g, 16.5 mmol, 18 Äq.) in
fester Form gegeben. Das Reaktionsgemisch wurde 24 h bei 100 °C unter Rühren erhitzt. Zu
der entstandenen roten Lösung wurden nach dem Abkühlen 50 ml dest. H2O gegeben. Es
wurde dreimal mit je 20 ml EtOAc extrahiert und die vereinigten organischen Phasen über
wasserfreiem Magnesiumsulfat getrocknet. Das Lösungsmittel wurde am
Rotationsverdampfer entfernt und das entstandene rote Öl säulenchromatographisch an
Kieselgel getrennt (Hexan / EtOAc = 1:1). Das Produkt bildete die erste Fraktion (Rf = 0.62).
Nach Entfernung des Lösungsmittels am Rotationsverdampfer erhielt man das Isothiocyanat
als weißen Feststoff, der im Hochvakuum getrocknet wurde (90 mg, 35 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.45 (dd, J = 2.6 Hz, 0.6 Hz; 2 H; pz-H5),
7.74 (dd, J = 1.7 Hz, 0.6 Hz; 2 H; pz-H3), 7.70 (t, J = 0.7 Hz; 2 H; Pyridin H), 6.46 (dd, J =
2.6 Hz, 1.7 Hz; 2 H; pz-H4), 4.80 ppm (t, J = 0.7 Hz; 2 H; CH2).
IR (KBr): ν~ = 608 (m), 773 (s), 834 (s), 915 (m), 958 (s), 1048 (s), 1207 (s), 1263 (m), 1401
(br), 1465 (br), 1524 (s), 1576 (s), 1620 (s), 2105 (vs, br, –N=C=S), 2194 (m), 2215 (m), 2935
(m), 3150 cm–1 (m).
MS (EI, 70 eV): m/z (%) = 282 (100) [M•+].
Elementaranalyse: Ber. (%) für C13H10N6S: C 55.30, H 3.57, N 29.77, S 11.36; gef.:
C 54.87, H 3.62, N 28.85, S 11.16.

2 Eisen(II)-Komplexe mit Schwefelanker 80
2.5.2.10 N-(2-Mercaptoethyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid (10)
NNNN N
HN
SHO
C14H14N6OS
M = 314.4 g/mol
weißer Feststoff
Zu einer Lösung von 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-carbonsäure (2) (794 mg, 3.1 mmol)
in DMF (27 ml) wurden nacheinander N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimid
Hydrochlorid (656 mg, 3.4 mmol), 1-Hydroxy-benzotriazol Hydrat (462 mg, 3.4 mmol) und
Cysteamin (240 mg, 3.1 mmol) als Feststoff gegeben. Das Gemisch wurde 20 min bei
Raumtemperatur gerührt. Triethylamin (0.87 ml, 6.2 mmol) wurde über eine Spritze
zugegeben und das Reaktionsgemisch weitere 12 h bei Raumtemperatur gerührt. Der
entstandenen Suspension wurden 50 ml dest. H2O zugefügt und die wässrige Phase mit
dreimal je 80 ml EtOAc extrahiert. Die vereinigten organischen Phasen wurden nacheinander
mit wässriger Zitronensäure-Lösung (0.05 M, 240 ml), wässriger Natriumhydrogencarbonat-
Lösung (0.5 M, 180 ml) und gesättigter Kochsalz-Lösung (120 ml) gewaschen. Die
organische Phase wurde über wasserfreiem Natriumsulfat getrocknet, das Lösungsmittel am
Rotationsverdampfer entfernt und der entstandene weiße Feststoff im Hochvakuum
getrocknet (814 mg, 83 %).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 9.26 (t, J = 5.6 Hz, 1 H; NH), 8.97 (dd, J = 2.8
Hz, 0.6 Hz; 2 H; pz-H5), 8.21 (s, 2H; Pyridin H), 7.90 (dd, J = 1.5 Hz, 0.6 Hz; 2 H; pz-H3),
6.65 (dd, J = 2.8 Hz, 1.7 Hz; 2 H; pz-H4), 3.47 (m, 2 H; N–CH2), 2.69 (m, 2 H; S–CH2); 2.50
ppm (m, 1 H; S–H). 13C-NMR (100.6 MHz, [D6]DMSO, 25 °C): δ = 163.4 (C=O), 150.1 (py-C2,6), 147.6 (py-C4),
142.9 (pz-C3), 128.3 (pz-C5), 108.7 (py-C3,5), 106.8 (pz-C4), 43.0 (NCH2), 23.1 ppm (SCH2).
IR (KBr): ν~ = 754 (m), 955 (m), 1034 (m), 1045 (m), 1209 (m), 1260 (w), 1397 (s), 1461
(vs), 1521 (m), 1543 (m), 1570 (s), 1621 (m), 1643 (m), 3264 cm–1 (br).
Elementaranalyse: Ber. (%) für C14H14N6OS: C 53.49, H 4.49, N 26.73, S 10.20; gef.:
C 53.70, H 4.48, N 26.49, S 10.32.
Dünnschichtchromatographie: Kieselgel, Hexan/ EtOAc = 1:1; Rf = 0.54.

2 Eisen(II)-Komplexe mit Schwefelanker 81
2.5.2.11 N-(4-(4-Mercaptophenyl)phenyl)-2,6-bis(1H-pyrazol-1-yl)pyridin-4-carboxamid
(12)
N
N
N
N
NHN
O
SH C24H18N6OS
M = 438.5 g/mol
weißer Feststoff
Zu einer Lösung von 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-carbonsäure (2) (50 mg, 0.20 mmol)
in DMF (6 ml) wurden nacheinander N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimid
Hydrochlorid (41 mg,0.22 mmol), 1-Hydroxy-benzotriazol Hydrat (29 mg, 0.22 mmol) und
4'-Amino-(1,1'-biphenyl)-4-thiol (39 mg, 0.20 mmol) in fester Form gegeben. Das Gemisch
wurde 20 min bei Raumtemperatur gerührt. Triethylamin (0.055 ml, 0.40 mmol) wurde über
eine Spritze zugegeben und das Reaktionsgemisch weitere 18 h bei Raumtemperatur gerührt.
Der entstandenen Suspension wurden 10 ml dest. H2O zugefügt und die wässrige Phase mit
dreimal je 30 ml EtOAc extrahiert. Die vereinigten organischen Phasen wurden nacheinander
mit wässriger Zitronensäure-Lösung (0.05 M, 40 ml), wässriger Natriumhydrogencarbonat-
Lösung (0.5 M, 30 ml) und gesättigter Kochsalz-Lösung (20 ml) gewaschen. Die organische
Phase wurde über wasserfreiem Natriumsulfat getrocknet, das Lösungsmittel am
Rotationsverdampfer entfernt und der entstandene weiße Feststoff im Hochvakuum
getrocknet (52 mg, 61 %).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 10.96 (s; 1 H; NH), 9.02 (d, J = 2.4 Hz; 2 H;
pz-H5), 8.33 (s; 2 H; Pyridin H), 7.93 (m; 4 H; bp-H/pz-H3); 7.73 (m; 4 H; bp-H), 7.64 (m; 2
H; bp-H), 6.69 (m; 2 H; pz-H4), 3.49 ppm (m; 1 H, SH).
MS (EI, 70 eV): m/z (%) = 201 (55) [M+H•+–C12H8N5O], 238 (60) [M•+–C12H10NS], 438
(100) [M•+].

2 Eisen(II)-Komplexe mit Schwefelanker 82
2.5.2.12 (2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methyl-11-mercaptoundecanoat (14)
NNNN N
OSH
O
10
C23H31N5O2S
M = 441.6 g/mol
weißer Feststoff
In einem 100 ml-Schlenkrohr wurde unter Rühren eine Lösung von 4-Dimethylaminopyridin
(2 mg, 0.02 mmol, 0.05 Äq.) in MeCN (1 ml) vorgelegt. Zu der Lösung wurden nacheinander
eine Suspension von 11-Mercaptoundecansäure (72 mg, 0.33 mmol, 1 Äq.) in MeCN (2 ml)
und 2,6-Bis(1H-pyrazol-1-yl)pyridin-4-yl)methanol (5) (80 mg, 0.33 mmol, 1 Äq.) in fester
Form gegeben. Eine Lösung von Di-tert-butyl-dicarbonat (94 mg, 0.43 mmol, 1.3 Äq.) in
MeCN (3 ml) wurde zum Reaktionsgemisch gegeben und die entstandene Suspension für 23 h
auf 60 °C erhitzt. Nach dem Abkühlen wurde der Lösung EtOAc (80 ml) zugefügt und die
organische Phase nacheinander mit verd. HCl (20 ml), NaHCO3 (2 × 40 ml), dest. H2O (2 ×
40 ml) und ges. Kochsalzlösung (40 ml) ausgeschüttelt. Die organische Phase wurde über
Magnesiumsulfat getrocknet und nach Filtration des Trockenmittels das Lösungsmittel am
Rotationsverdampfer entfernt. Das zurückgebliebene Öl wurde säulenchromatographisch an
Kieselgel gereinigt (EtOAc / Hexan = 1:3, Rf = 0.83). Das Produkt wurde in Form eines
farblosen Öls erhalten, das bei Trocknung im Hochvakuum zu einem weißen Feststoff
erstarrte (92 mg, 63 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 8.56 (dt, J = 2.6 Hz, 0.7 Hz; 2 H; pz-H5),
7.84 (dt, J = 5.5 Hz, 0.7 Hz; 2 H; Pyridin H), 7.77 (dd, J = 1.7 Hz, 0.7 Hz; 2 H; pz-H3), 6.50
(m; 2 H; pz-H4), 5.22 (dt, J = 4.8 Hz, 0.7 Hz; py–CH2), 2.94-2.42 (m; 4 H; CH2C=O/CH2–
SH), 1.84-1.17 ppm (m; 16 H; Methylen H).
IR (KBr): ν~ = 542 (w), 608 (m), 650 (w), 758 (s), 788 (s), 854 (m), 917 (m), 954 (m), 1040
(s), 1128 (s), 1161 (s), 1206 (s), 1256 (m), 1278 (m), 1398 (s), 1466 (vs), 1522 (m), 1575 (s),
1621 (s), 1692 (m), 1744 (vs), 2853 (m), 2923 (vs), 3150 cm–1 (w).
MS (EI, 70 eV): m/z (%) = 240 (100) [M•+–C11H21OS], 408 (54) [M•+–SH], 441 (59) [M•+].

2 Eisen(II)-Komplexe mit Schwefelanker 83
2.5.2.13 Ethyl 2,6-dichlorpyridin-4-carboxylat (15)
NCl Cl
CO2Et
C8H7Cl2NO2
M = 220.0 g/mol
weiße Nadeln
2,6-Dichlor-4-carbonsäure (1) (3 g, 15.6 mmol) wurde in 70 ml EtOH gelöst. Zu der klaren
Lösung wurde unter Rühren konz. H2SO4 (5 ml) gegeben und das Gemisch für 2 h unter
Rückfluss erhitzt. Nach dem Abkühlen wurde das Lösungsmittel am Rotationsverdampfer
entfernt und dest. H2O (50 ml) zugefügt. Der entstandene weiße Niederschlag wurde
abfiltriert, mit dest. H2O gewaschen und in einem EtOH/dest. H2O-Gemisch umkristallisiert.
Nach dem Trocknen im Hochvakuum erhielt man das Produkt in Form von weißen Nadeln
(3.231 g, 94 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 7.81 (s; 2 H; Pyridin H), 4.43 (q, J =
7.1 Hz; 2 H; CH2), 1.42 ppm (t, J = 7.1 Hz; 3 H; CH3). 13C-NMR (100.6 MHz, [D1]Chloroform, 25 °C): δ = 162.60 (C=O), 151.34 (py-C2,6),
142.71 (py-C4), 122.55 (py-C3,5), 62.59 (CH2), 14.04 ppm (CH3).
MS (EI, 70 eV): m/z (%) = 146 (28) / 148 (19) [M•+–C3H5O2], 174 (75) / 176 (51) [M•+–
C2H5O], 191 (100) / 193 (64) [MH•+–C2H5], 219 (59) / 221 (35) [M•+].
2.5.2.14 (2,6-Dichlorpyridin-4-yl)methanol (16)
NCl Cl
OH
C6H5Cl2NO
M = 178.0 g/mol
weißer Feststoff
Ethyl 2,6-dichlorpyridin-4-carboxylat (15) (3.23 g, 14.7 mmol, 1 Äq.) wurde in EtOH (120
ml) suspendiert. Unter Rühren wurde Natriumborhydrid (2.78 g, 73.3 mmol, 5 Äq.)
portionsweise in fester Form zugegeben. Das Reaktionsgemisch wurde 3 d bei
Raumtemperatur gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt und der
verbleibende Feststoff mit dest. H2O (50 ml) versetzt. Die Suspension wurde mit verd. HCl

2 Eisen(II)-Komplexe mit Schwefelanker 84
versetzt bis keine Gasentwicklung mehr zu beobachten war. Anschließend wurde das
Gemisch mit festem Natriumcarbonat auf pH 8 gebracht. Die wässrige Phase wurde dreimal
mit je 150 ml DCM extrahiert und die vereinigten organischen Phasen über Magnesiumsulfat
getrocknet. Nach Filtration des Trockenmittels und Entfernung des Lösungsmittels am
Rotationsverdampfer erhielt man das Produkt in Form eines weißen Feststoffes (2.54 g, 97
%).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 7.44 (t, J = 0.9 Hz; 2 H; Pyridin H), 5.65 (t, J
= 5.8 Hz; 1 H; OH), 4.56 ppm (dt, J = 5.8 Hz, 0.9 Hz; 2 H; CH2).
IR (KBr): ν~ = 607(m), 819 (m), 845 (s), 904 (m), 991 (m), 1077 (m), 1164 (s), 1234 (w),
1290 (w), 1384 (s), 1460 (m), 1544 (s), 1591 (s), 3358 cm–1 (s).
Elementaranalyse: Ber. (%) für C6H5Cl2NO: C 40.48, H 2.83, N 7.87; gef.: C 40.45, H 2.81,
N 7.68.
2.5.2.15 2,6-Dichlor-4-((tetrahydro-2H-pyran-2-yloxy)methyl)pyridin (17)
NCl Cl
O
O
C11H13Cl2NO2
M = 262.1 g/mol
weißer Feststoff
In einem 100 ml-Schlenk-Kolben wurden 3,4-Dihydropyran (0.531 ml, 5.9 mmol, 10 Äq.)
und (2,6-Dichlorpyridin-4-yl)methanol (16) (0.5 g, 2.8 mmol, 4.8 Äq.) in DCM (80 ml)
gelöst. Eine Lösung von Pyridinium-p-toluolsulfonat (0.147 g, 0.6 mmol, 1 Äq.) in DCM (5
ml) wurde langsam zur Reaktionslösung getropft. Die klare farblose Lösung wurde 18 h bei
Raumtemperatur gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt. Zum
verbliebenen weißen Feststoff wurde dest. H2O (40 ml) gegeben und mit dreimal je 40 ml
Diethylether extrahiert. Die vereinigten organischen Phasen wurden über Magnesiumsulfat
getrocknet, das Trockenmittel abfiltriert und das Lösungsmittel am Rotationsverdampfer
entfernt. Das verbliebene hellgelbe Öl wurde säulenchromatographisch an Kieselgel gereinigt

2 Eisen(II)-Komplexe mit Schwefelanker 85
(Pentan/Diethylether = 75:25, Rf = 0.68). Das Produkt wurde als farbloses Öl erhalten, das
sich beim Abkühlen verfestigt (0.635 g, 86 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 7.25 (t, J = 0.9 Hz; 2 H; Pyridin H), 4.77
(dt, J = 14.4 Hz, 0.9 Hz; 1 H; CHH’–OTHP), 4.70 (m; 1 H; THP-H2), 4.48 (dt, J = 14.4 Hz,
0.9 Hz; 1 H; CHH’–OTHP), 3.89-3.77 (m; 1 H; THP-H6 ), 3.60-3.50 (m; 1 H; THP-H6’),
1.91-1.58 ppm (m; 6 H; THP-H3,4,5).
IR (KBr): ν~ = 809 (s), 869 (m), 905 (w), 972 (w), 1035 (s), 1074 (m), 1126 (m), 1164 (vs),
1201 (w), 1250 (w), 1371 (s), 1546 (s), 1588 (m), 1732 (m), 2943 cm–1 (m).
Elementaranalyse: Ber. (%) für C11H13Cl2NO2: C 50.40, H 5.00, N 5.34; gef.: C 50.60, H
5.05, N 4.74.
2.5.2.16 Ethyl-1-(6-(4-(ethoxycarbonyl)-1H-pyrazol-1-yl)-4-((tetrahydro-2H-pyran-2-
yloxy)methyl)pyridin-2-yl)-1H-pyrazol-4-carboxylat (18)
NNNN N
O
CO2EtEtO2C
O
C23H27N5O6
M = 469.5 g/mol
weißer Feststoff
Paraffinfreies Kaliumhydrid (242 mg, 6.0 mmol, 3 Äq.) wurde in einem 250 ml-Schlenk-
Kolben in Diglyme (8 ml) vorgelegt. Zu der weißen Suspension wurde unter Rühren eine
Lösung von Ethyl-1H-pyrazol-4-carboxylat (846 mg, 6.0 mmol, 3 Äq.) in Diglyme (25 ml)
über eine Spritze gegeben. Die entstandene gelbliche Suspension wurde für 2 h bei
Raumtemperatur gerührt. 17 (527 mg, 2.0 mmol, 1 Äq.) wurde in fester Form zur
Reaktionslösung gegeben. Das Gemisch wurde für 5 d auf 130 °C erhitzt. Nach Abkühlen auf
Raumtemperatur wurde dest. H2O (150 ml) zugefügt, wobei sich zunächst eine braune, klare
Lösung bildete, aus der schließlich ein hellbrauner Niederschlag ausfiel. Der Niederschlag
wurde abfiltriert und mehrmals mit dest. H2O gewaschen. Das Rohprodukt wurde in wenig
EtOAc aufgenommen und säulenchromatographisch an Kieselgel gereinigt (Hexan/EtOAc =

2 Eisen(II)-Komplexe mit Schwefelanker 86
1:1, Rf = 0.68). Das Produkt wurde in Form eines weißen, kristallinen Feststoffes erhalten
(417 mg, 44 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 9.04 (d, J = 0.7 Hz; 2 H; pz-H5), 8.14 (d,
J = 0.7 Hz; 2 H; pz-H3), 7.97 (t, J = 0.7 Hz; 2 H; Pyridin H), 4.94 (dt, J = 14.2 Hz, 0.7 Hz; 1
H; CHH’–OTHP), 4.79 (m, 1 H; THP-H2), 4.66 (dt, J = 14.2 Hz, 0.7 Hz; 1 H; CHH’–OTHP),
4.40 (q, J = 7.1 Hz; 4 H, CH2–CH3), 3.95-3.84 (m; 1 H; THP-H6), 3.63-3.53 (m; 1 H; THP-
H6’), 1.91-1.58 (m; 6 H; THP-H3,4,5), 1.43 ppm (t, J = 7.1 Hz; 6 H; CH2–CH3).
Elementaranalyse: Ber. (%) für C23H27N5O6: C 58.84, H 5.80, N 14.92; gef.: C 58.38, H
5.60, N 14.50.
2.5.2.17 Ethyl-1-(6-(4-(ethoxycarbonyl)-1H-pyrazol-1-yl)-4-(hydroxymethyl)pyridin-2-
yl)-1H-pyrazol-4-carboxylat (19)
NNNN N
OH
CO2EtEtO2C
C18H19N5O5
M = 385.4 g/mol
weißer Feststoff
Unter Rühren wurde zu einer Lösung von 18 (405 mg, 0.86 mmol, 10 Äq.) in EtOH (22 ml),
Pyridinium-p-toluolsulfonat (22 mg, 0.09 mmol, 1 Äq.) in fester Form gegeben. Die klare
farblose Lösung wurde für 3 h auf 55 °C erhitzt. Nach dem Abkühlen wurde das
Lösungsmittel am Rotationsverdampfer entfernt. Zum verbliebenen Feststoff wurde dest. H2O
(50 ml) gegeben und dreimal mit jeweils 50 ml Diethylether extrahiert. Die vereinigten
organischen Phasen wurden noch zweimal mit jeweils 50 ml dest. H2O gewaschen und
anschließend über Magnesiumsulfat getrocknet. Das Trockenmittel wurde abfiltriert und das
Lösungsmittel am Rotationsverdampfer entfernt. Das Produkt wurde in Form eines weißen
Feststoffes erhalten (327 mg, 98 %).
1H-NMR (200 MHz, [D6]DMSO, 25 °C): δ = 9.45 (d, J = 0.7 Hz; 2 H; pz-H5), 8.10 (d, J =
0.7 Hz; 2 H; pz-H3), 7.79 (s; 2 H; Pyridin H), 5.69 (br; 1 H; OH), 4.68 (s; 2H; CH2–OH), 4.27
(q, J = 7.1 Hz; 4 H; CH2CH3), 1.31 ppm (t, J = 7.1 Hz; 6 H; CH3).

2 Eisen(II)-Komplexe mit Schwefelanker 87
13C-NMR (100.6 MHz, [D6]DMSO, 25 °C): δ = 161.83 (C=O), 159.54 (py-C4), 148.69 (py-
C2/6), 142.70 (pz-C3), 131.32 (pz-C5), 116.48 (pz-C4), 107.56 (py-C3/5), 61.48 (CH2OH), 60.04
(CH2CH3), 14.18 ppm (CH3).
IR (KBr): ν~ = 538 (w), 601 (w), 763 (m), 791 (w), 867 (w), 910 (w), 965 (m), 996 (m), 1024
(s), 1071 (m), 1131 (m), 1193 (m), 1252 (s), 1409 (m), 1471 (s), 1561 (s), 1621 (m), 1722
(vs), 2936 cm–1 (br).
2.5.2.18 1-(6-(4-Carboxy-1H-pyrazol-1-yl)-4-(hydroxymethyl)pyridin-2-yl)-1H-pyrazol-
4-carbonsäure (20)
NNNN N
OH
CO2HHO2C
C14H11N5O5
M = 329.3 g/mol
weißer Feststoff
Unter Rühren wurde die Diesterverbindung 19 (50 mg, 0.13 mmol, 1 Äq.) bei 40 °C in EtOH
(5.3 ml) gelöst. Eine wässrige 0.5 M-Natriumhydroxidlösung (2 ml, 1.00 mmol, 7.7 Äq.)
wurde zugegeben. Die entstandene klare farblose Lösung wurde für 24 h bei 70 °C gerührt.
Zur entstandenen weißen Suspension wurde dest. H2O (5 ml) gegeben, wobei sich der weiße
Niederschlag löste. Durch Zugabe einer wässrigen 1 M-Zitronensäurelösung wurde die
Reaktionslösung auf pH 4 gebracht, wobei ein weißer Niederschlag ausfiel. Der Feststoff
wurde abfiltriert und mit dest. H2O gewaschen. Nach Trocknung im Hochvakuum erhielt man
das Produkt als weißen Feststoff (29.7 mg, 70 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 12.76 (br; 2 H; COOH), 9.57 (d, J = 0.7
Hz; 2 H; pz-H5), 8.16 (d, J = 0.7 Hz; 2 H; pz-H3), 7.89 (s; 2 H; Pyridin H), 5.73 (br; 1 H; OH),
4.72 ppm (s; 2 H; CH2).
IR (KBr): ν~ = 598 (w), 771 (m), 797 (m), 848 (w), 891 (w), 998 (m), 1047 (w), 1262 (m),
1431 (s), 1479 (s), 1564 (vs), 1625 (s), 1694 (vs), 3112 cm–1 (m).
MS (EI, 70 eV): m/z (%) = 44 (55) [CO2], 284 (14) [M•+–CHO2•], 329 (100) [M•+].
Elementaranalyse: Ber. (%) für C14H11N5O5: C 51.07, H 3.37, N 21.27; gef.: C 50.76, H
3.44, N 20.40;

2 Eisen(II)-Komplexe mit Schwefelanker 88
2.5.2.19 Ethyl-1-(6-(4-(ethoxycarbonyl)-1H-pyrazol-1-yl)-4-(thiocyanatomethyl)pyridin-
2-yl)-1H-pyrazol-4-carboxylat (21)
NNNN N
SCN
CO2EtEtO2C
C19H18N6O4S
M = 426.4 g/mol
hellgelber Feststoff
In einem 25 ml-Schlenk-Kolben wurde eine Lösung von Triphenylphosphin (230 mg, 0.9
mmol, 1.05 Äq.) in MeCN (5 ml) vorgelegt. Unter Rühren wurden nacheinander eine Lösung
von Ammoniumthiocyanat (134 mg, 1.8 mmol, 2.1 Äq.) in MeCN (3 ml) und eine Lösung
von Brom (0.05 ml, 0.9 mmol, 1.05 Äq.) in MeCN (0.5 ml) zugegeben. Das Gemisch wurde
10 min bei Raumtemperatur gerührt und anschließend eine Lösung von 19 (327 mg, 0.8
mmol, 1 Äq.) in MeCN (10 ml) zugegeben. Die Reaktionslösung wurde 24 h bei
Raumtemperatur gerührt. Das Lösungsmittel wurde am Rotationsverdampfer entfernt, der
Rückstand in wenig EtOAc aufgenommen und säulenchromatographisch an Kieselgel
gereinigt (Hexan/EtOAc = 1:1, Rf = 0.70). Das Produkt wurde in Form eines hellgelben
Feststoffes erhalten (135 mg, 40 %).
1H-NMR (200 MHz, [D1]Chloroform, 25 °C): δ = 9.02 (s; 2 H; pz-H5), 8.14 (s; 2 H; pz-H3),
7.98 (s; 2 H; Pyridin H), 4.39 (q, J = 7.1 Hz; 4 H; CH2CH3), 4.22 (s; 2 H; CH2–SCN), 1.42
ppm (t, J = 7.1 Hz; 3 H; CH3). 13C-NMR (100.6 MHz, [D1]Chloroform, 25 °C): δ = 162.46 (C=O), 150.13 (py-C2/6),
149.98 (py-C4), 143.53 (pz-C3), 130.28 (pz-C5), 117.76 (pz-C4), 110.67 (py-C3/5), 110.39
(SCN), 60.82 (CH2CH3), 36.47 (CH2SCN), 14.41 ppm (CH3).
IR (KBr): ν~ = 621 (m), 711 (m), 765 (s), 791 (w), 828 (w), 873 (w), 913 (w), 966 (s), 1000
(m), 1023 (s), 1137 (m), 1171 (s), 1197 (m), 1256 (s), 1304 (m), 1407 (s), 1470 (s), 1559 (s),
1622 (s), 1718 (s), 2157 (m), 2988 (m), 3121 (m), 3416 cm–1 (br).
Elementaranalyse: Ber. (%) für C19H18N6O4S: C 53.51, H 4.25, N 19.71, S 7.52; gef.:
C 53.25, H 4.21, N 18.91, S 7.31.

2 Eisen(II)-Komplexe mit Schwefelanker 89
2.5.2.20 [Fe(C13H11N5O2)2](BF4)2 (23)
N
NN
N
NN
N
N
N
N
FeMeO2C CO2Me
2+
[BF4–]2
C26H22B2F8FeN10O4
M = 767.9 g/mol
oranger Feststoff
In einem 10 ml-Schlenkrohr wurde eine Lösung von 3 (99.6 mg, 0.4 mmol, 1 Äq.) in THF (3
ml) vorgelegt. Eine Lösung von Eisen(II)tetrafluoroborat Hexahydrat (59.3 mg, 0.2 mmol,
0.48 Äq.) in THF (2 ml) wurde zugetropft. Aus der farblosen, klaren Lösung fiel sofort ein
oranger Niederschlag aus. Das Reaktionsgemisch wurde für 4 h bei Raumtemperatur gerührt.
Der Niederschlag wurde abfiltriert und mit THF gewaschen. Nach Trocknung im
Hochvakuum wurde das Produkt in Form eines orangen Feststoffes erhalten (113 mg, 82 %).
Orange Einkristalle der Verbindung wurden durch isothermale Diffusion von Diethylether in
eine methanolische Lösung der Substanz über eine Woche bei Raumtemperatur erhalten.
1H-NMR (200 MHz, [D4]Methanol, 25 °C): δ = 68.64 (br), 64.16 (br), 57.66 (br), 49.55 (br),
8.46-2.99 ppm (m). (Alle Signale sind paramagnetisch verbreitert)
IR (KBr): ν~ = 521 (w), 599 (w), 736 (m), 765 (s), 793 (m), 912 (w), 971 (s), 1052 (vs), 1167
(m), 1265 (s), 1338 (w), 1408 (vs), 1462 (vs), 1501 (m), 1528 (s), 1582 (s), 1635 (s), 1738 (s),
2920 (w), 3131 cm–1 (m).
Elementaranalyse: Ber. (%) für C26H22B2F8FeN10O4: C 40.66, H 2.89, N 18.24; gef.:
C 40.64, H 3.06, N 18.15.

2 Eisen(II)-Komplexe mit Schwefelanker 90
2.5.2.21 [Fe(C14H13N5O2)2](BF4)2 (24)
N
NN
N
NN
N
N
N
N
FeEtO2C CO2Et
2+
[BF4–]2
C28H26B2F8FeN10O4
M = 796.0 g/mol
oranger Feststoff
In einem 10 ml-Schlenkrohr wurde eine Lösung von 4 (122.5 mg, 0.4 mmol, 1 Äq.) in THF (3
ml) vorgelegt. Unter Rühren wurde eine Lösung von Eisen(II)tetrafluoroborat Hexahydrat
(69.3 mg, 0.2 mmol, 0.48 Äq.) in THF (2 ml) zugetropft. Aus der Lösung fiel sofort ein
oranger Niederschlag aus. Die Lösung wurde für 3 h bei Raumtemperatur gerührt, der
Niederschlag abfiltriert und mit THF nachgewaschen. Nach Trocknung im Hochvakuum
wurde der Eisenkomplex in Form eines orangen Feststoffes erhalten (118 mg, 71 %). Orange
Einkristalle der Verbindung wurden durch isothermale Diffusion von Diethylether in eine
Lösung des Komplexes in MeOH über eine Woche gewonnen.
1H-NMR (200 MHz, [D4]Methanol, 25 °C): δ = 69.15 (br), 64.65 (br), 58.29 (br), 49.33 (br),
9.48-0.96 ppm (m). (Alle Signale sind paramagnetisch verbreitert)
IR (KBr): ν~ = 520 (w), 598 (w), 766 (s), 862 (w), 970 (m), 1055 (vs), 1136 (m), 1257 (s),
1286 (s), 1339 (w), 1380 (w), 1408 (s), 1474 (vs), 1499 (m), 1527 (m), 1575 (m), 1631 (m),
1734 (s), 2924 (w), 3126 cm–1 (m).
Elementaranalyse: Ber. (%) für C28H26B2F8FeN10O4: C 42.25, H 3.29, N 17.60; gef.:
C 41.51, H 3.60, N 18.18.

2 Eisen(II)-Komplexe mit Schwefelanker 91
2.5.2.22 [Fe(C14H14N6OS)2](BF4)2 (25)
N
NN
N
NN
N
N
N
N
FeHN
NH
2+
[BF4–]2
HS
O
O
SH
C28H28B2F8FeN12O2S2
M = 858.2 g/mol
orangeroter Feststoff
Eine Lösung von 10 (225 mg, 0.7 mmol, 1 Äq.) in THF (10 ml) wurde in einem 10 ml-
Schlenkrohr bei Raumtemperatur vorgelegt. Unter Rühren wurde zu der klaren, farblosen
Lösung eine Lösung von Eisen(II)tetrafluoroborat-Hexahydrat (117 mg, 0.4 mmol, 0.49 Äq.)
in THF (3 ml) gegeben. Aus der Lösung fiel sofort ein dunkelroter Niederschlag aus. Das
Reaktionsgemisch wurde für weitere 18 h bei Raumtemperatur gerührt. Der Feststoff wurde
abfiltriert, mit THF gewaschen und im Hochvakuum getrocknet. Der Eisenkomplex wurde in
Form eines orangeroten Feststoffes erhalten (235 mg, 79 %). Orange Einkristalle der
Verbindung wurden durch isothermale Diffusion von Diethylether in eine methanolische
Lösung des Rohproduktes erhalten.
1H-NMR (200 MHz, [D4]Methanol, 25 °C): δ = 69.58 (br), 63.58 (br), 59.15 (br), 49.01 (br),
8.23-1.09 ppm (m). (Alle Signale sind paramagnetisch verbreitert)
IR (KBr): ν~ = 473 (w), 520 (m), 546 (w), 600 (m), 645 (w), 767 (s), 794 (m), 869 (m), 909
(m), 972 (s), 1058 (vs), 1170 (w), 1221 (w), 1272 (m), 1291 (m), 1338 (m), 1380 (m), 1408
(s), 1460 (s), 1498 (s), 1528 (s), 1572 (s), 1632 (s), 1674 (s), 2577 (w), 2855 (w), 2930 (w),
3116 (m), 3371 cm–1 (m).
Elementaranalyse: Ber. (%) für C28H28B2F8FeN12O2S2: C 39.19, H 3.29, N 19.59, S 7.47;
gef.: C 39.89, H 3.49, N 19.37, S 7.44.

2 Eisen(II)-Komplexe mit Schwefelanker 92
2.5.2.23 Röntgenstrukturanalysen
Details zu Kristalldaten, Datensammlung und Strukturverfeinerung sind in Tabelle 2.4.1
angegeben. Die Daten wurden unter Verwendung eines Bruker-Nonius KappaCCD
Diffraktometers mit MoKα-Strahlung (λ = 0.71073 Å) und Graphit-Monochromator ermittelt
(Strukturen für 23, 24 und 25). Datensammlung und Integration erfolgte mit der COLLECT
Suite.[96] Datensammlung für Verbindung 3 erfolgte auf einem Oxford Diffraction Xcalibur S
Sapphire-Diffraktometer (Siemens) unter Verwendung von Mo-Kα-Strahlung (λ = 0.71073
Ǻ). Alle Strukturen wurden mit direkten Methoden gelöst und die Daten nach der Methode
der kleinsten Fehlerquadrate bei voller Matrix gegen F2 verfeinert, unter Benutzung des
Programms SHELXTL NT 6.12.[97] Alle Nichtwasserstoffatome wurden anisotrop verfeinert.
Alle H-Atome wurden in geometrisch optimierten Positionen berechnet und wurden mit
einem isotropen Auslenkungsparameter versehen, der dem 1.2- bzw. 1.5-fachen des
äquivalenten isotropen Auslenkungsparameters des die Wasserstoffatome tragenden C-
Atomes entspricht. Die geometrischen Parameter der Strukturen wurden mit den Programmen
IVTON[98] und Mercury v1.4.2[99] ermittelt. Fehlordnungen: 23: Ein BF4– Ion (Atome F13,
F14), zwei Vorzugslagen verfeinert (Besetzung 70(6):30(6) %); 24: Eine COOEt-Gruppe
(Atome O2, C13, C14), zwei Vorzugslagen verfeinert (Besetzung 73.5(7):26.5(7) %); 25:
Beide endständigen CH2SH-Gruppen (Atome C14, S1 bzw. C28, S2) je zwei Vorzugslagen
verfeinert (Besetzung 90.3(3):9.7(3) % bzw. 84.7(3):15.3(3) %).
2 Eisen(II)-Komplexe mit Schwefelanker 93
Tabelle 2.5.1. Kristallographische Daten für Verbindungen 3, 23, 24 und 25. 23 24 25 3
Summenformel C26H22B2F8FeN10O4 C28H26B2F8FeN10O4 C28H28 B2F8FeN12O2S2 C14H16N5O2
M [g mol–1] 767.9 796.1 858.2 286.3
Kristallsystem, Monoklin, C2/c Monoklin, P21/c Monoklin, P21/c Monoklin, P21/c
T [K] 100(2) 100(2) 100(2) 293(2)
Kristallgröße [mm] 0.10 × 0.09 × 0.07 0.16 × 0.12 × 0.09 0.28 × 0.21 × 0.10 0.14 × 0.23 × 0.26
Farbe orange orange rot farblos
a [Å] 23.212 (2) 17.5448 (10) 17.3500 (10) 13.8221 (4)
b [Å] 10.701 (1) 8.4933 (5) 13.704 (2) 19.2888 (5)
c [Å] 15.367 (2) 22.1899 (16) 15.388 (2) 10.1422 (3)
α [°] 90 90 90 90
β [°] 126.884 (4) 93.993 (6) 108.231 (9) 91.029 (2)
γ [°] 90 90 90 90
V [Å3] 3053.1 (6) 3298.6 (4) 3475.1 (7) 2703.59 (13)
Z 4 4 4 8
Dber [g cm–3] 1.671 1.603 1.640 1.407
μ [mm–1] 0.60 0.56 0.65 0.10
Abs. Korrektur SADABS[a] SADABS SADABS -
Tmin/Tmax 0.871/0.960 0.846/0.950 0.776/0.937 -
Gemessene Reflexe 28861 33867 74539 37970
Sym. unabhängig 3374 7234 7658 8952
Beobachtet[b] 2655 5017 5940 6179
Verf. Parameter 251 509 538 382
wR2 (alle Reflexe)[c] 0.071 0.092 0.108 0.143
R1 (beob. Reflexe)[d] 0.034 0.044 0.043 0.058
ρfin (max/min) [eÅ–3] 0.32/–0.28 0.48/–0.33 0.57/–0.48 0.28/–0.30
GooF = S 1.044 1.011 1.063 1.052
Wichtungsschema[e] k = 0.0228/l = 0.4610 k = 0.0335/l = 2.2752 k = 0.0404/l = 5.9029 k = 0.0667/l = 0.4059
[a] SADABS 2.06, Bruker AXS, Inc., 2002, Madison WI., U.S.A. [b] Mit Fo≥4σ(F). [c] wR2 = [Σ[w(Fo2–Fc
2)2]/Σ[w(Fo2)2]]1/2. [d] R1 = Σ||Fo|–
|Fc||/Σ|Fo| für Fo≥4σ(F). [e] w = 1/[σ2(Fo2)+(kP)2+lP] und P = (Fo
2)+2Fc2)/3.

3 Polymorphie in [Fe(6)2](BF4)2 94
3 Polymorphie in [Fe(6)2](BF4)2
3.1 Einleitung
Die gezielte Synthese von Spincrossover(SCO)-Komplexen ist von Interesse im Hinblick auf
ihr Potential in der technischen Anwendung.[14, 42] Eine „Maßschneiderung“ von SCO-
Verbindungen mit genau vorhersagbaren Eigenschaften ist bisher nicht möglich. Durch
Berücksichtigung von Merkmalen bekannter SCO-Verbindungen bei der Synthese ist
allerdings eine große Wahrscheinlichkeit gegeben, dass die Zielverbindung SCO-
Eigenschaften aufweist. Das Verständnis aller den Spinübergang ermöglichenden Einflüsse
auf einen Metallkomplex würde die Wege zu neuen SCO-Verbindungen wesentlich
verkürzen.
3.2 Konzept
Ein geeignetes System zur Untersuchung von Festkörpereffekten stellen die schon erwähnten
Eisen(II)-Komplexe von Bispyrazol-1-ylpyridin-Derivaten dar. Wie durch theoretische
Untersuchungen gezeigt wurde,[43] wird eine Jahn-Teller-Verzerrung des FeN6-
Koordinationsoktaeders im Feststoff durch die starren Bindungswinkel im Liganden
thermodynamisch stabilisiert, was mit einer Erniedrigung der Symmetrie des
Koordinationspolyeders einhergeht. Eine solche „fixierte“ Jahn-Teller-Verzerrung verhindert
Spincrossover. Bisher wurde das Zustandekommen der Jahn-Teller-Verzerrung hauptsächlich
durch den Einfluss zusätzlicher Moleküle (Lösungsmittel, Gegenionen) im Kristallgitter
erklärt. Die in diesem Kapitel geschilderten Beobachtungen lassen jedoch andere Einflüsse an
Gewicht gewinnen. Die Isolierung und Untersuchung zweier Polymorphe eines Eisen(II)-
Komplexes mit identischer Stöchiometrie, aber unterschiedlichem Spinverhalten zeigt, dass es
sich bei den bestimmenden strukturellen Einflüssen um Kooperativitätseffekte in der
Kristallpackung handelt.
Die auf diese Art mögliche isolierte Untersuchung des Festkörpereffektes kann künftig helfen,
„maßgeschneiderte“ SCO-Verbindungen zu synthetisieren.

3 Polymorphie in [Fe(6)2](BF4)2 95
3.3 Ergebnisse und Diskussion
3.3.1 Synthese von [Fe(6)2](BF4)2 (26)
Zur Synthese von Komplex 26 wurden zwei Äquivalente des Liganden 6 mit einem
Äquivalent Eisen(II)tetrafluoroborat-Hexahydrat in Tetrahydrofuran bei Raumtemperatur
umgesetzt. Der entstehende Komplex [Fe(6)2](BF4)2 ist nicht in Tetrahydrofuran löslich und
fällt sofort in Form eines gelben Niederschlags aus der Lösung aus. Der gelbe Feststoff ist in
Acetonitril, Aceton und Nitromethan gut und in Methanol ausreichend löslich.
NNNN N
SCN
2 N
NN
N
NN
N
N
N
N
FeNCS
SCN
2+
[BF4–]2
6 26
i
Abbildung 3.1. Synthese von [Fe(6)2](BF4)2 (26): i) 0.98 Äq. Fe(BF4)2 · 6 H2O, THF, RT, 18
h, 83 %.
Die Elementaranalyse von 26 stimmt gut mit den berechneten Werten überein. Die IR-
Spektren von Ligand 6 und Komplex 26 stimmen in großen Teilen überein. Das Spektrum
von 26 zeigt eine Bande bei 2156 cm–1, die der asymmetrischen Streckschwingung der
Thiocyanatgruppe zugeordnet werden kann. Eine weitere starke Bande bei 1052 cm–1 wird
durch die BF4–-Ionen verursacht.
Das 1H-NMR-Spektrum von 26 in [D3]Acetonitril bei Raumtemperatur zeigt eine starke
paramagnetische Verschiebung der Signale, die sich über einen Bereich von 0 … 70 ppm
verteilen. Eine Zuordnung der Signale war infolge der großen Halbwertsbreiten der Signale
nicht möglich. In Anbetracht des Ausmaßes der Verschiebung handelt es sich nicht um eine
Verunreinigung der Probe durch möglicherweise entstandene Eisen(III)-Spezies, sondern um
ein Vorliegen eines Großteils von 26 mit Eisen(II) im High Spin-Zustand.
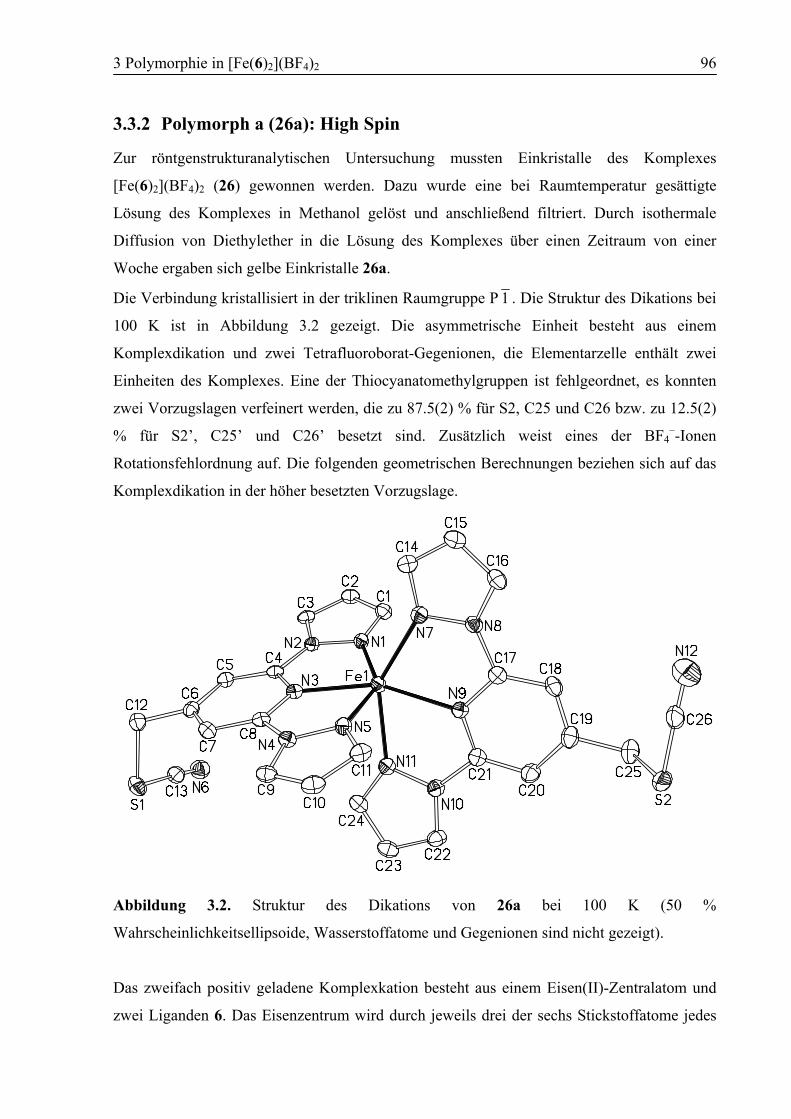
3 Polymorphie in [Fe(6)2](BF4)2 96
3.3.2 Polymorph a (26a): High Spin
Zur röntgenstrukturanalytischen Untersuchung mussten Einkristalle des Komplexes
[Fe(6)2](BF4)2 (26) gewonnen werden. Dazu wurde eine bei Raumtemperatur gesättigte
Lösung des Komplexes in Methanol gelöst und anschließend filtriert. Durch isothermale
Diffusion von Diethylether in die Lösung des Komplexes über einen Zeitraum von einer
Woche ergaben sich gelbe Einkristalle 26a.
Die Verbindung kristallisiert in der triklinen Raumgruppe P 1 . Die Struktur des Dikations bei
100 K ist in Abbildung 3.2 gezeigt. Die asymmetrische Einheit besteht aus einem
Komplexdikation und zwei Tetrafluoroborat-Gegenionen, die Elementarzelle enthält zwei
Einheiten des Komplexes. Eine der Thiocyanatomethylgruppen ist fehlgeordnet, es konnten
zwei Vorzugslagen verfeinert werden, die zu 87.5(2) % für S2, C25 und C26 bzw. zu 12.5(2)
% für S2’, C25’ und C26’ besetzt sind. Zusätzlich weist eines der BF4–-Ionen
Rotationsfehlordnung auf. Die folgenden geometrischen Berechnungen beziehen sich auf das
Komplexdikation in der höher besetzten Vorzugslage.
Abbildung 3.2. Struktur des Dikations von 26a bei 100 K (50 %
Wahrscheinlichkeitsellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Das zweifach positiv geladene Komplexkation besteht aus einem Eisen(II)-Zentralatom und
zwei Liganden 6. Das Eisenzentrum wird durch jeweils drei der sechs Stickstoffatome jedes

3 Polymorphie in [Fe(6)2](BF4)2 97
Liganden in pseudooktaedrischer Geometrie koordiniert. Die beiden Liganden ordnen sich in
meridionaler Weise in der Koordinationssphäre an, sodass ihre Ebenen senkrecht aufeinander
stehen. Das erzeugte FeN6-Koordinationspolyeder weicht durch eine unregelmäßige
Anordnung beider Liganden zueinander deutlich von der Gestalt eines idealen Oktaeders ab.
Einerseits ist der Winkel φ zwischen den Eisen–N(Pyridin)-Bindungen deutlich kleiner als der
für ein ideales Oktaeder erwartete Wert von 180° (N3–Fe1–N9 = 158.10(7)°). Andererseits
sind die Ebenen der Liganden gegeneinander um die N3–Fe1–N9-Achse verdreht, mit einem
Winkel θ zwischen den besten Ebenen der Liganden von 78.84(2)° (ideal 90°). Der Mittelwert
der Chelatwinkel α beträgt 73.2°. Abbildung 2.26 in Kapitel 2 veranschaulicht die Lage der
betrachteten Winkel.
Die Eisen–Stickstoff-Bindungslängen betragen 2.1264(18) … 2.2430(19) Å, was auf ein
Vorliegen des Eisen(II)-Zentrums im High Spin-Zustand deutet.[2] Auf die Geometrie der
Thiocyanatomethylsubstituenten und intermolekulare Wechselwirkungen wird später in
diesem Kapitel eingegangen.
Tabelle 3.1. Ausgewählte Abstände [Å] und Winkel [°] für 26a bei 100 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N1 2.153(2) N3-Fe1-N9 158.1(1)
Fe1–N3 2.126(2) N1-Fe1-N3 73.8(1)
Fe1–N5 2.180(2) N3-Fe1-N5 73.1(1)
Fe1–N7 2.168(2) N7-Fe1-N9 73.6(1)
Fe1–N9 2.138(2) N9-Fe1-N11 72.5(1)
Fe1–N11 2.243(2)
C6...N6 3.950(4) C6-C12-S1 111.1(2)
C12-S1-C13 100.3(1)
C19...N12 4.085(4) C19-C25-S2 111.2(2)
C25-S2-C26 98.7(2)
Zusätzlich wurde ein Einkristall der Verbindung 26a bei 313 K röntgenstrukturanalytisch
untersucht. Die Struktur liegt in der triklinen Raumgruppe P 1 vor. Die asymmetrische
Einheit besteht aus einem Komplexdikation und zwei Tetrafluoroborat-Gegenionen. Die
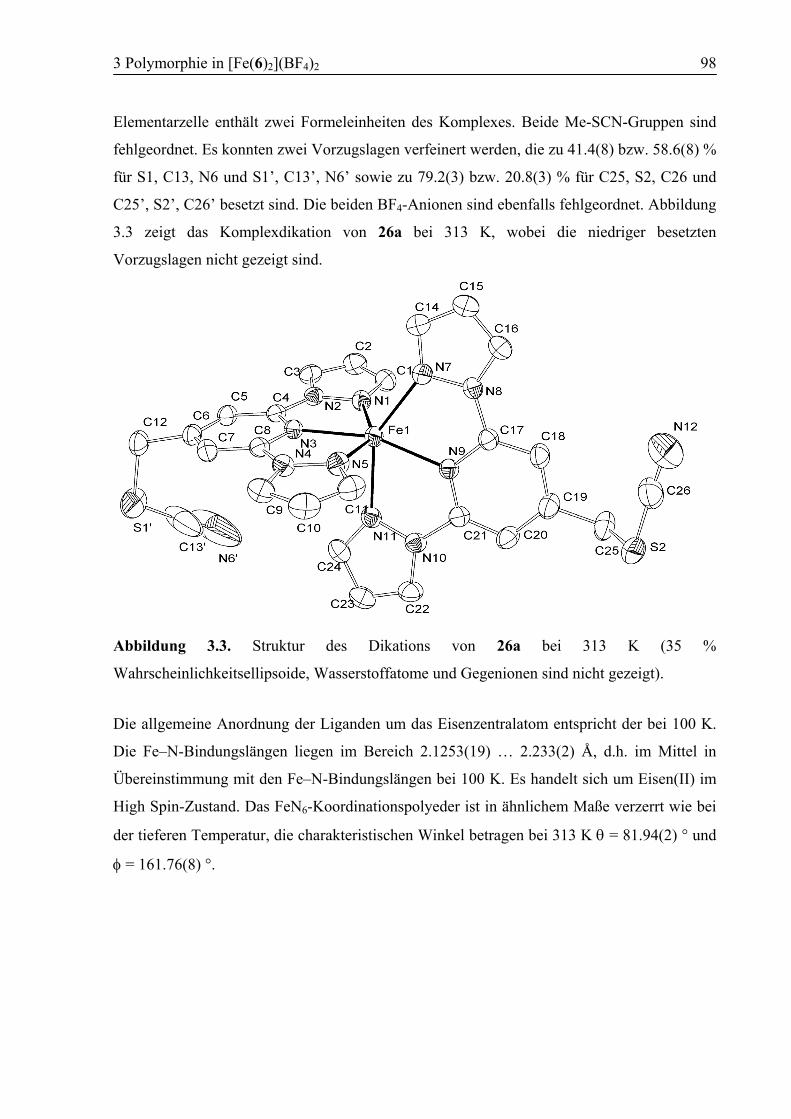
3 Polymorphie in [Fe(6)2](BF4)2 98
Elementarzelle enthält zwei Formeleinheiten des Komplexes. Beide Me-SCN-Gruppen sind
fehlgeordnet. Es konnten zwei Vorzugslagen verfeinert werden, die zu 41.4(8) bzw. 58.6(8) %
für S1, C13, N6 und S1’, C13’, N6’ sowie zu 79.2(3) bzw. 20.8(3) % für C25, S2, C26 und
C25’, S2’, C26’ besetzt sind. Die beiden BF4-Anionen sind ebenfalls fehlgeordnet. Abbildung
3.3 zeigt das Komplexdikation von 26a bei 313 K, wobei die niedriger besetzten
Vorzugslagen nicht gezeigt sind.
Abbildung 3.3. Struktur des Dikations von 26a bei 313 K (35 %
Wahrscheinlichkeitsellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Die allgemeine Anordnung der Liganden um das Eisenzentralatom entspricht der bei 100 K.
Die Fe–N-Bindungslängen liegen im Bereich 2.1253(19) … 2.233(2) Å, d.h. im Mittel in
Übereinstimmung mit den Fe–N-Bindungslängen bei 100 K. Es handelt sich um Eisen(II) im
High Spin-Zustand. Das FeN6-Koordinationspolyeder ist in ähnlichem Maße verzerrt wie bei
der tieferen Temperatur, die charakteristischen Winkel betragen bei 313 K θ = 81.94(2) ° und
φ = 161.76(8) °.
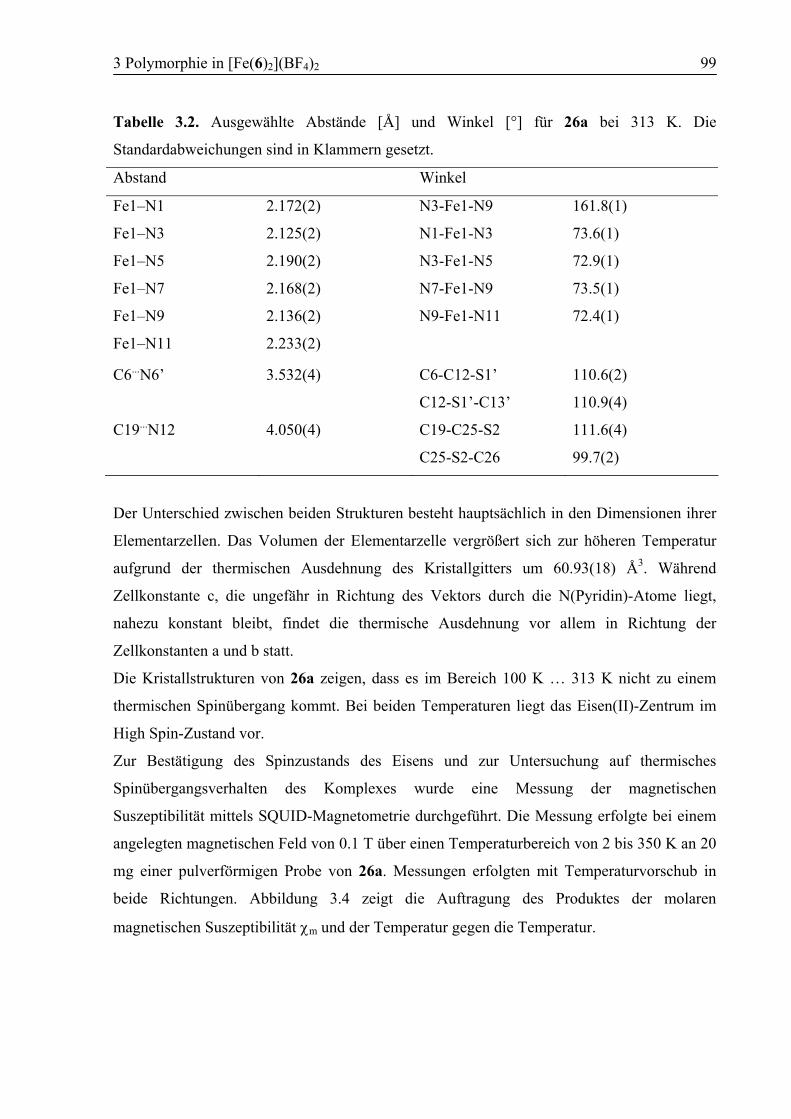
3 Polymorphie in [Fe(6)2](BF4)2 99
Tabelle 3.2. Ausgewählte Abstände [Å] und Winkel [°] für 26a bei 313 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N1 2.172(2) N3-Fe1-N9 161.8(1)
Fe1–N3 2.125(2) N1-Fe1-N3 73.6(1)
Fe1–N5 2.190(2) N3-Fe1-N5 72.9(1)
Fe1–N7 2.168(2) N7-Fe1-N9 73.5(1)
Fe1–N9 2.136(2) N9-Fe1-N11 72.4(1)
Fe1–N11 2.233(2)
C6...N6’ 3.532(4) C6-C12-S1’ 110.6(2)
C12-S1’-C13’ 110.9(4)
C19...N12 4.050(4) C19-C25-S2 111.6(4)
C25-S2-C26 99.7(2)
Der Unterschied zwischen beiden Strukturen besteht hauptsächlich in den Dimensionen ihrer
Elementarzellen. Das Volumen der Elementarzelle vergrößert sich zur höheren Temperatur
aufgrund der thermischen Ausdehnung des Kristallgitters um 60.93(18) Å3. Während
Zellkonstante c, die ungefähr in Richtung des Vektors durch die N(Pyridin)-Atome liegt,
nahezu konstant bleibt, findet die thermische Ausdehnung vor allem in Richtung der
Zellkonstanten a und b statt.
Die Kristallstrukturen von 26a zeigen, dass es im Bereich 100 K … 313 K nicht zu einem
thermischen Spinübergang kommt. Bei beiden Temperaturen liegt das Eisen(II)-Zentrum im
High Spin-Zustand vor.
Zur Bestätigung des Spinzustands des Eisens und zur Untersuchung auf thermisches
Spinübergangsverhalten des Komplexes wurde eine Messung der magnetischen
Suszeptibilität mittels SQUID-Magnetometrie durchgeführt. Die Messung erfolgte bei einem
angelegten magnetischen Feld von 0.1 T über einen Temperaturbereich von 2 bis 350 K an 20
mg einer pulverförmigen Probe von 26a. Messungen erfolgten mit Temperaturvorschub in
beide Richtungen. Abbildung 3.4 zeigt die Auftragung des Produktes der molaren
magnetischen Suszeptibilität χm und der Temperatur gegen die Temperatur.
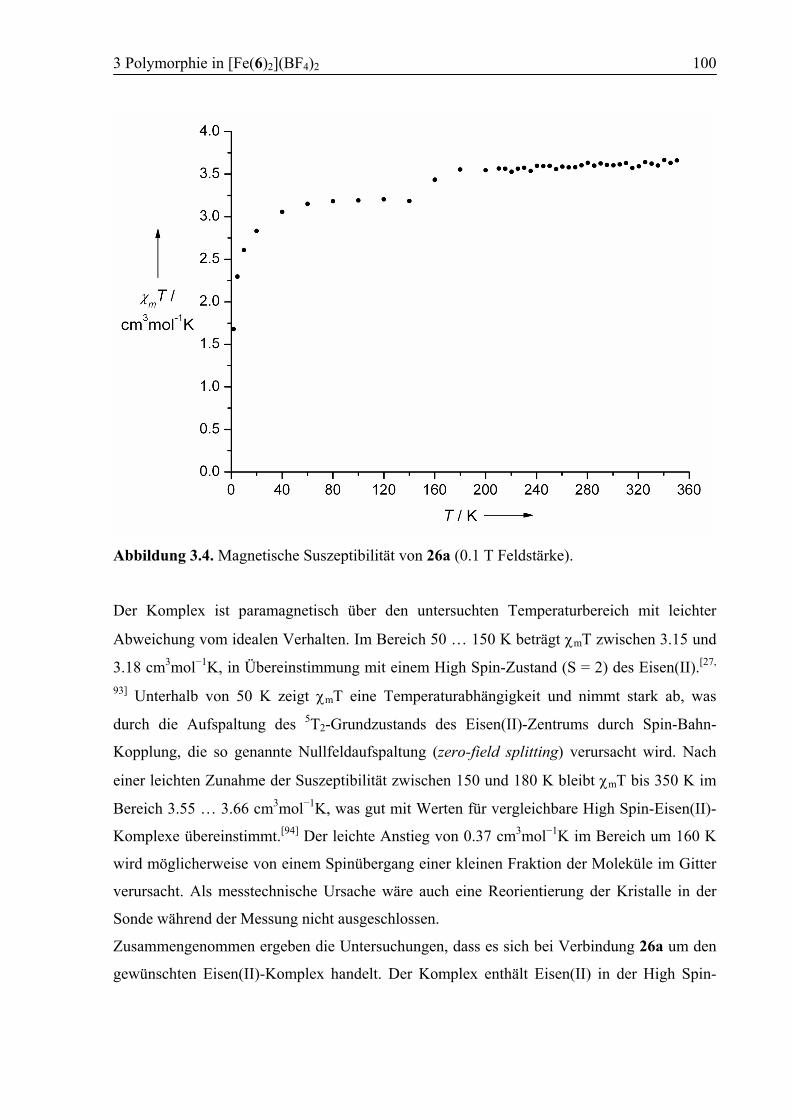
3 Polymorphie in [Fe(6)2](BF4)2 100
Abbildung 3.4. Magnetische Suszeptibilität von 26a (0.1 T Feldstärke).
Der Komplex ist paramagnetisch über den untersuchten Temperaturbereich mit leichter
Abweichung vom idealen Verhalten. Im Bereich 50 … 150 K beträgt χmT zwischen 3.15 und
3.18 cm3mol−1K, in Übereinstimmung mit einem High Spin-Zustand (S = 2) des Eisen(II).[27,
93] Unterhalb von 50 K zeigt χmT eine Temperaturabhängigkeit und nimmt stark ab, was
durch die Aufspaltung des 5T2-Grundzustands des Eisen(II)-Zentrums durch Spin-Bahn-
Kopplung, die so genannte Nullfeldaufspaltung (zero-field splitting) verursacht wird. Nach
einer leichten Zunahme der Suszeptibilität zwischen 150 und 180 K bleibt χmT bis 350 K im
Bereich 3.55 … 3.66 cm3mol−1K, was gut mit Werten für vergleichbare High Spin-Eisen(II)-
Komplexe übereinstimmt.[94] Der leichte Anstieg von 0.37 cm3mol−1K im Bereich um 160 K
wird möglicherweise von einem Spinübergang einer kleinen Fraktion der Moleküle im Gitter
verursacht. Als messtechnische Ursache wäre auch eine Reorientierung der Kristalle in der
Sonde während der Messung nicht ausgeschlossen.
Zusammengenommen ergeben die Untersuchungen, dass es sich bei Verbindung 26a um den
gewünschten Eisen(II)-Komplex handelt. Der Komplex enthält Eisen(II) in der High Spin-

3 Polymorphie in [Fe(6)2](BF4)2 101
Konfiguration im Temperaturbereich 2 … 350 K, zeigt also keinen thermischen
Spinübergang.
3.3.3 Kokristallisation bei Raumtemperatur
Da das Rohprodukt 26 in Methanol nur mäßig löslich ist, wurde der Komplex in Methanol bei
Rückflusstemperatur (60 °C) gelöst, um die Sättigung der Lösung zu erhöhen und die
Ausbeute zu steigern. Die Lösung wurde auf Raumtemperatur abgekühlt, filtriert, und es
wurde analog zum oben beschriebenen Ansatz Diethylether isotherm eindiffundieren
gelassen. Nach Ablauf von einer Woche zeigte sich, dass ein Gemisch aus zwei Kristallsorten
entstanden war (Abbildung 3.5). Zusätzlich zu einer Fraktion der bekannten gelben Kristalle
26a, die ca. 95 Gew.-% der Ausbeute ausmachten, entstand eine kleinere Fraktion von
rotbraunen Kristallen. Das Resultat ist reproduzierbar, wobei bei späteren Ansätzen die
isotherme Diffusion in druckdichten Glasgefäßen bei 0.8 bar Stickstoff-Überdruck
durchgeführt wurde, wodurch sich gleichmäßigere Ausbeuten ergaben. Erneute
Umkristallisation von Verbindung 26a unter diesen Bedingungen ergab wiederum das
Gemisch beider Kristallsorten.
Abbildung 3.5. Gemisch aus gelben (26a) und rotbraunen (26b) Kristallen.
Bei den gelben Kristallen handelte es sich um Verbindung 26a, wie die Bestimmung der
Zellkonstanten durch Einkristall-Röntgenstrukturanalyse bestätigte. Die rotbraunen Kristalle
(26b) wurden zunächst für eine oxidierte Form des Komplexes mit Eisen(III)-Zentrum
gehalten, wenngleich eine Oxidation durch den stark chelatisierenden Charakter der
Bispyrazolylpyridin-Liganden bei gleichzeitiger Redox-Stabilität der Thiocyanatgruppe
unwahrscheinlich scheint. Die Elementaranalysen beider Verbindungen stimmen gut mit der
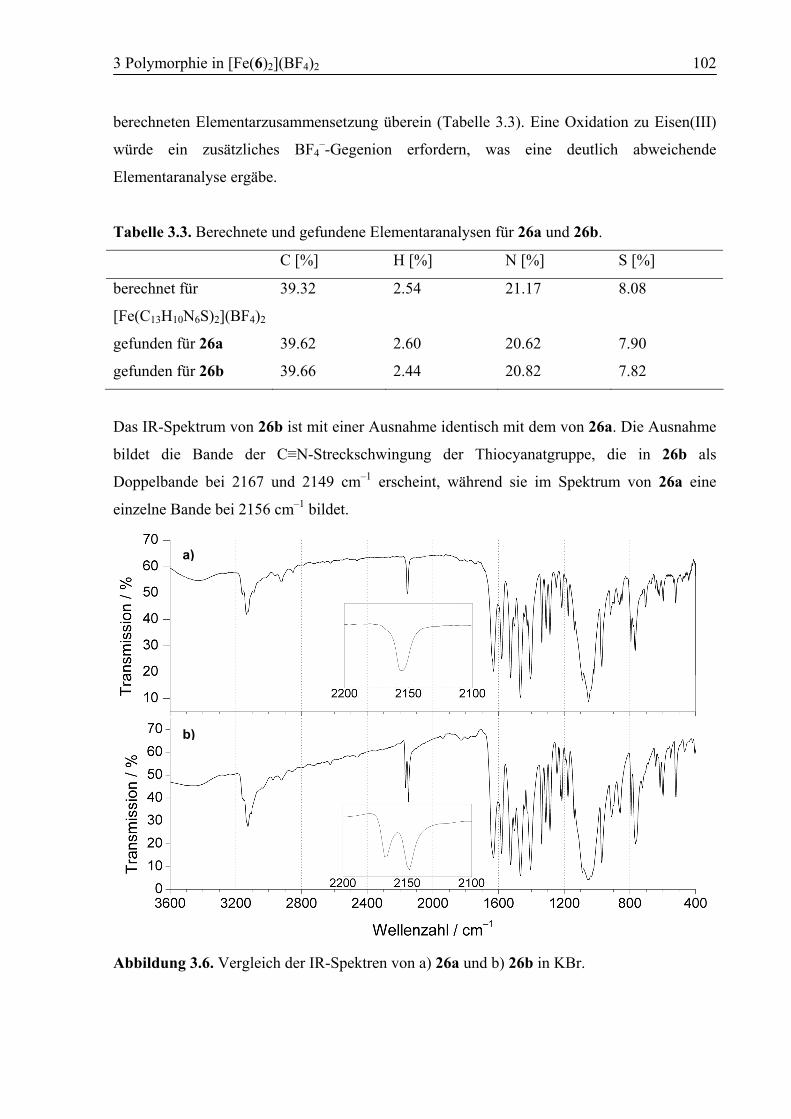
3 Polymorphie in [Fe(6)2](BF4)2 102
berechneten Elementarzusammensetzung überein (Tabelle 3.3). Eine Oxidation zu Eisen(III)
würde ein zusätzliches BF4–-Gegenion erfordern, was eine deutlich abweichende
Elementaranalyse ergäbe.
Tabelle 3.3. Berechnete und gefundene Elementaranalysen für 26a und 26b.
C [%] H [%] N [%] S [%]
berechnet für
[Fe(C13H10N6S)2](BF4)2
39.32 2.54 21.17 8.08
gefunden für 26a 39.62 2.60 20.62 7.90
gefunden für 26b 39.66 2.44 20.82 7.82
Das IR-Spektrum von 26b ist mit einer Ausnahme identisch mit dem von 26a. Die Ausnahme
bildet die Bande der C≡N-Streckschwingung der Thiocyanatgruppe, die in 26b als
Doppelbande bei 2167 und 2149 cm–1 erscheint, während sie im Spektrum von 26a eine
einzelne Bande bei 2156 cm–1 bildet.
Abbildung 3.6. Vergleich der IR-Spektren von a) 26a und b) 26b in KBr.
a)
b)
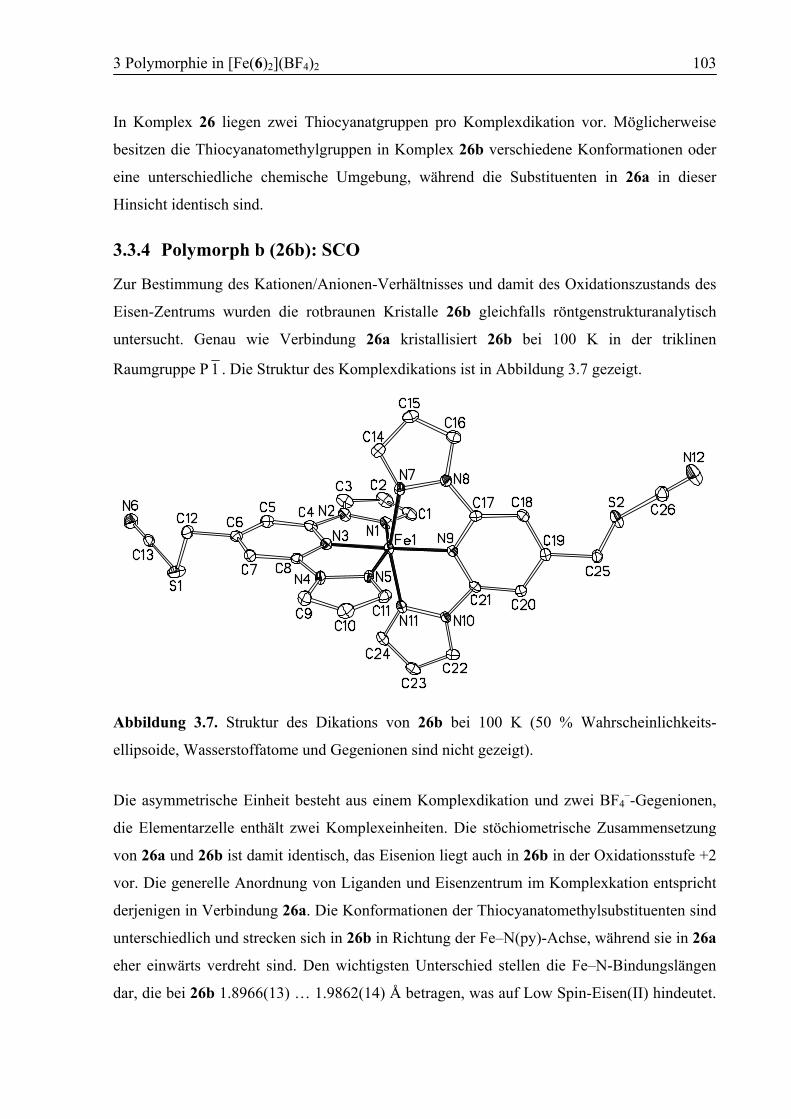
3 Polymorphie in [Fe(6)2](BF4)2 103
In Komplex 26 liegen zwei Thiocyanatgruppen pro Komplexdikation vor. Möglicherweise
besitzen die Thiocyanatomethylgruppen in Komplex 26b verschiedene Konformationen oder
eine unterschiedliche chemische Umgebung, während die Substituenten in 26a in dieser
Hinsicht identisch sind.
3.3.4 Polymorph b (26b): SCO
Zur Bestimmung des Kationen/Anionen-Verhältnisses und damit des Oxidationszustands des
Eisen-Zentrums wurden die rotbraunen Kristalle 26b gleichfalls röntgenstrukturanalytisch
untersucht. Genau wie Verbindung 26a kristallisiert 26b bei 100 K in der triklinen
Raumgruppe P 1 . Die Struktur des Komplexdikations ist in Abbildung 3.7 gezeigt.
Abbildung 3.7. Struktur des Dikations von 26b bei 100 K (50 % Wahrscheinlichkeits-
ellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Die asymmetrische Einheit besteht aus einem Komplexdikation und zwei BF4–-Gegenionen,
die Elementarzelle enthält zwei Komplexeinheiten. Die stöchiometrische Zusammensetzung
von 26a und 26b ist damit identisch, das Eisenion liegt auch in 26b in der Oxidationsstufe +2
vor. Die generelle Anordnung von Liganden und Eisenzentrum im Komplexkation entspricht
derjenigen in Verbindung 26a. Die Konformationen der Thiocyanatomethylsubstituenten sind
unterschiedlich und strecken sich in 26b in Richtung der Fe–N(py)-Achse, während sie in 26a
eher einwärts verdreht sind. Den wichtigsten Unterschied stellen die Fe–N-Bindungslängen
dar, die bei 26b 1.8966(13) … 1.9862(14) Å betragen, was auf Low Spin-Eisen(II) hindeutet.
3 Polymorphie in [Fe(6)2](BF4)2 104
Im Vergleich mit 26a bedeutet dies im Mittel eine Abnahme von 0.19 Å oder 9 % der
Bindungslängen. Durch die kürzeren Fe–N-Bindungen liegt in 26b ein größerer mittlerer
Chelatwinkel α vor (79.9°). Der Winkel N3–Fe1–N9 (φ) beträgt 178.22(6)°, während die
besten Ebenen der Liganden nahezu orthogonal aufeinander stehen (θ = 88.65(0)°). Durch
diese regelmäßigere Anordnung der Liganden erfährt das FeN6-Fragment eine Erhöhung
seiner Symmetrie auf ~D2d, verglichen mit der C2-symmetrischen Koordinationsumgebung in
26a.
Tabelle 3.4. Ausgewählte Abstände [Å] und Winkel [°] für 26b bei 100 K. Die
Standardabweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N1 1.973(1) N3-Fe1-N9 178.2(1)
Fe1–N3 1.898(1) N1-Fe1-N3 80.0(1)
Fe1–N5 1.971(1) N3-Fe1-N5 80.0(1)
Fe1–N7 1.979(1) N7-Fe1-N9 80.1(1)
Fe1–N9 1.897(1) N9-Fe1-N11 79.8(1)
Fe1–N11 1.986(1)
C6...N6 4.836(2) C6-C12-S1 108.1(1)
C12-S1-C13 99.8(1)
C19...N12 4.891(2) C19-C25-S2 108.5(1)
C25-S2-C26 100.0(1)
Eine temperaturabhängige magnetische Messung von 26b wurde an 9 mg der Verbindung
mittels SQUID-Magnetometer im Temperaturbereich 5 … 350 K durchgeführt. Die Messung
erfolgte bei einem angelegten magnetischen Feld von 0.1 T mit Temperaturvorschub in
beiden Richtungen. Die erhaltenen Messwerte waren merklich durch einen
temperaturunabhängigen Paramagnetismus[27] (TUP) beeinflusst. Dieser wird vermutlich
durch paramagnetische Reaktionsprodukte des Eisenkomplexes mit dem aus einer
Gelatinekapsel bestehenden Probenhalter verursacht (z.B. Eisenoxide). Der TUP-Anteil an
der gemessenen magnetischen Suszeptibilität konnte rechnerisch mit χm,TUP = 6.39 · 10–3
cm3mol–1 bestimmt und damit die Messwerte korrigiert werden. Abbildung 3.8 zeigt eine
Auftragung der erhaltenen, korrigierten Werte für χmT gegen T.
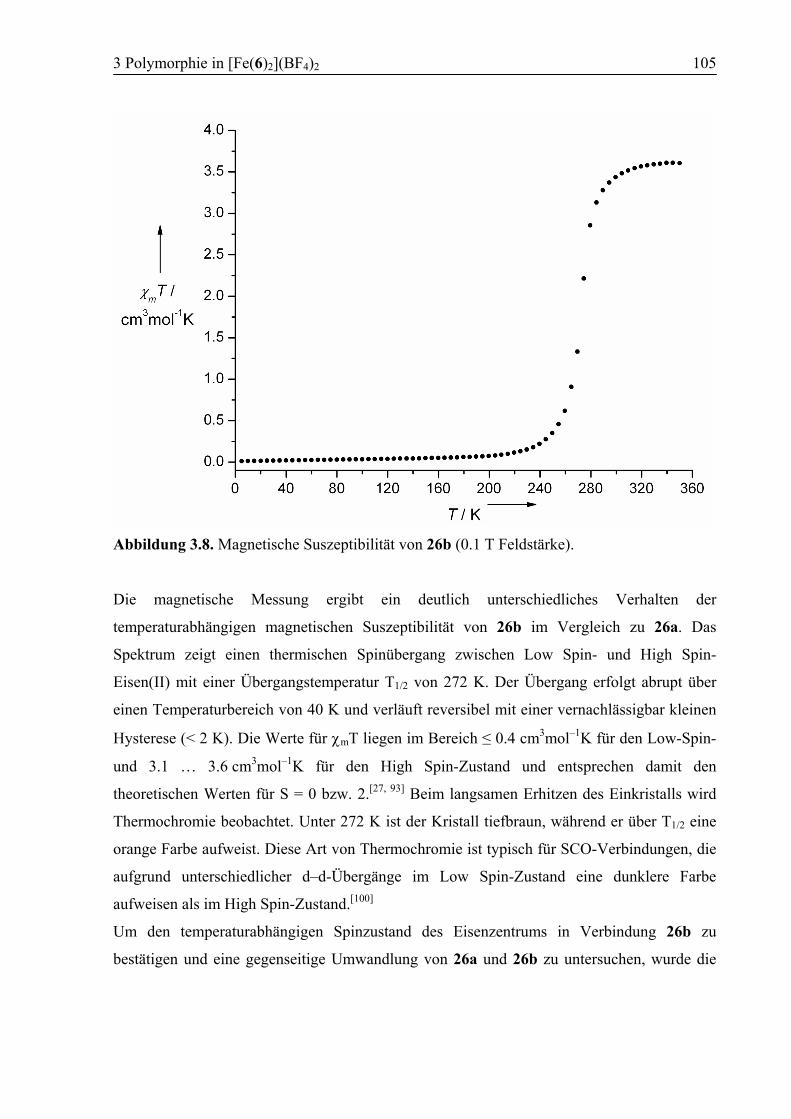
3 Polymorphie in [Fe(6)2](BF4)2 105
Abbildung 3.8. Magnetische Suszeptibilität von 26b (0.1 T Feldstärke).
Die magnetische Messung ergibt ein deutlich unterschiedliches Verhalten der
temperaturabhängigen magnetischen Suszeptibilität von 26b im Vergleich zu 26a. Das
Spektrum zeigt einen thermischen Spinübergang zwischen Low Spin- und High Spin-
Eisen(II) mit einer Übergangstemperatur T1/2 von 272 K. Der Übergang erfolgt abrupt über
einen Temperaturbereich von 40 K und verläuft reversibel mit einer vernachlässigbar kleinen
Hysterese (< 2 K). Die Werte für χmT liegen im Bereich ≤ 0.4 cm3mol–1K für den Low-Spin-
und 3.1 … 3.6 cm3mol–1K für den High Spin-Zustand und entsprechen damit den
theoretischen Werten für S = 0 bzw. 2.[27, 93] Beim langsamen Erhitzen des Einkristalls wird
Thermochromie beobachtet. Unter 272 K ist der Kristall tiefbraun, während er über T1/2 eine
orange Farbe aufweist. Diese Art von Thermochromie ist typisch für SCO-Verbindungen, die
aufgrund unterschiedlicher d–d-Übergänge im Low Spin-Zustand eine dunklere Farbe
aufweisen als im High Spin-Zustand.[100]
Um den temperaturabhängigen Spinzustand des Eisenzentrums in Verbindung 26b zu
bestätigen und eine gegenseitige Umwandlung von 26a und 26b zu untersuchen, wurde die

3 Polymorphie in [Fe(6)2](BF4)2 106
Struktur von 26b bei vier zusätzlichen Temperaturen bestimmt (150, 200, 278 und 313 K, in
dieser Reihenfolge). Alle Strukturen weisen die gleiche Raumgruppe P 1 auf.
Die Strukturen bei 150 und 200 K, also unterhalb von T1/2, entsprechen weitgehend der
Struktur von 26b, die bei 100 K bestimmt wurde. Die Zellkonstanten vergrößern sich mit
ansteigender Temperatur, was die thermische Ausdehnung des Kristallgitters widerspiegelt.
Die Fe–N-Bindungslängen unterscheiden sich nur unwesentlich voneinander und betragen im
Mittel bei allen drei Strukturen 1.95(4) Å. Erwartungsgemäß liegt das Eisen(II)-Zentrum auch
bei 150 und 200 K im Low Spin-Zustand vor.
Die Strukturen von 26b, die oberhalb T1/2, bei 278 und 313 K, bestimmt wurden, sind
einander wiederum sehr ähnlich, während sie sich von den Strukturen unterhalb 272 K
unterscheiden. In beiden Strukturen treten temperaturbedingt Fehlordnungen beider
Thiocyanatomethylgruppen auf (s. Experimentalteil). Abbildung 3.9 zeigt das
Komplexdikation von 26b bei 313 K.
Abbildung 3.9. Struktur des Dikations von 26b bei 313 K (50 %
Wahrscheinlichkeitsellipsoide, Wasserstoffatome und Gegenionen sind nicht gezeigt).
Die mittleren Fe–N-Bindungslängen betragen 2.11(4) und 2.15(4) Å, was einem typischen
Wert für High Spin-Eisen(II) entspricht. Der kleinere Wert bei 278 K wird vermutlich
dadurch verursacht, dass sich ein Teil der Moleküle im Kristallgitter noch im Low Spin-
Zustand befindet. Der Spinübergang erstreckt sich, wie in der SQUID-Messung zu sehen,
über den Bereich von 250 … 290 K und ist folglich bei 278 K noch nicht abgeschlossen. Im
3 Polymorphie in [Fe(6)2](BF4)2 107
Vergleich zu den Strukturen, die unterhalb 272 K bestimmt wurden, liegt damit eine
Erhöhung der Fe–N-Bindungslänge um 0.2(4) Å, also ~10 % vor. Dieser Wert ist
charakteristisch für einen Spincrossover in Eisen(II)-Verbindungen.
Tabelle 3.5. Ausgewählte Abstände [Å] und Winkel [°] für 26b bei 150, 200, 278 und 313 K.
Die Standardabweichungen sind in Klammern gesetzt.
Abstand 150 K 200 K Winkel 150 K 200 K
Fe1–N1 1.973(1) 1.973(2) N3-Fe1-N9 178.2(1) 178.2(1)
Fe1–N3 1.898(1) 1.898(2) N1-Fe1-N3 80.0(1) 79.9(1)
Fe1–N5 1.973(1) 1.976(2) N3-Fe1-N5 79.9(1) 79.9(1)
Fe1–N7 1.981(1) 1.981(2) N7-Fe1-N9 80.0(1) 79.9(1)
Fe1–N9 1.897(1) 1.899(1) N9-Fe1-N11 79.7(1) 79.7(1)
Fe1–N11 1.986(1) 1.985(1)
C6...N6 4.832(4) 4.834(4) C6-C12-S1 108.2(1) 108.1(1)
C12-S1-C13 99.9(1) 100.0(1)
C19...N12 4.896(4) 4.899(4) C19-C25-S2 108.3(1) 107.8(2)
C25-S2-C26 100.1(1) 100.2(4)
Abstand 278 K 313 K Winkel 278 K 313 K
Fe1–N1 2.130(2) 2.171(2) N3-Fe1-N9 174.0(1) 172.3(1)
Fe1–N3 2.075(2) 2.114(2) N1-Fe1-N3 74.5(1) 73.3(1)
Fe1–N5 2.125(2) 2.158(2) N3-Fe1-N5 74.8(1) 73.8(1)
Fe1–N7 2.133(2) 2.171(2) N7-Fe1-N9 75.5(1) 74.4(1)
Fe1–N9 2.056(2) 2.0978(2) N9-Fe1-N11 74.8(1) 73.8(1)
Fe1–N11 2.153(3) 2.182(2)
C6...N6 4.911(8) 4.936(8) C6-C12-S1 106.4(2) 106.1(2)
C12-S1-C13 99.9(2) 99.6(2)
C19...N12 4.966(8) 4.986(8) C19-C25-S2 106.8(2) 107.0(2)
C25-S2-C26 100.2(7) 99.7(7)
Aufgrund der längeren Fe–N-Bindungen bei höherer Temperatur kommt es zu einer
Verzerrung des FeN6-Koordinationspolyeders, was anhand der charakteristischen Winkel der
Struktur deutlich wird. Während der Interebenenwinkel θ nahezu unverändert bleibt, nimmt φ
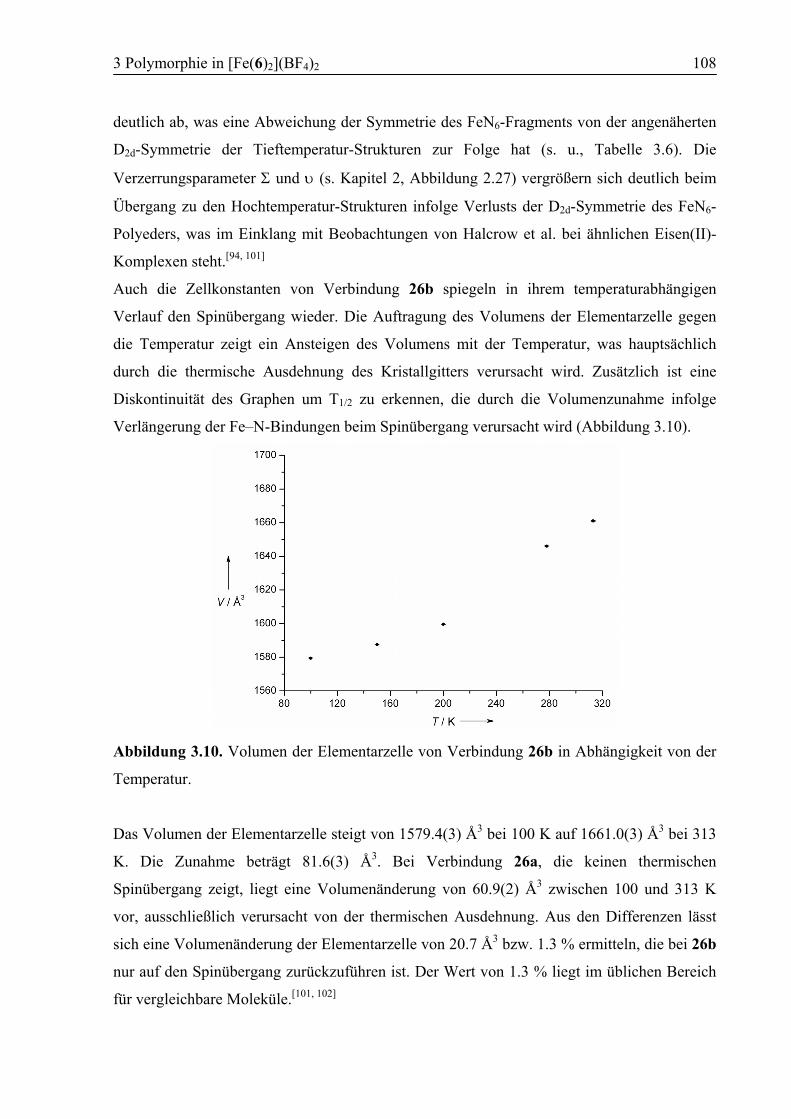
3 Polymorphie in [Fe(6)2](BF4)2 108
deutlich ab, was eine Abweichung der Symmetrie des FeN6-Fragments von der angenäherten
D2d-Symmetrie der Tieftemperatur-Strukturen zur Folge hat (s. u., Tabelle 3.6). Die
Verzerrungsparameter Σ und υ (s. Kapitel 2, Abbildung 2.27) vergrößern sich deutlich beim
Übergang zu den Hochtemperatur-Strukturen infolge Verlusts der D2d-Symmetrie des FeN6-
Polyeders, was im Einklang mit Beobachtungen von Halcrow et al. bei ähnlichen Eisen(II)-
Komplexen steht.[94, 101]
Auch die Zellkonstanten von Verbindung 26b spiegeln in ihrem temperaturabhängigen
Verlauf den Spinübergang wieder. Die Auftragung des Volumens der Elementarzelle gegen
die Temperatur zeigt ein Ansteigen des Volumens mit der Temperatur, was hauptsächlich
durch die thermische Ausdehnung des Kristallgitters verursacht wird. Zusätzlich ist eine
Diskontinuität des Graphen um T1/2 zu erkennen, die durch die Volumenzunahme infolge
Verlängerung der Fe–N-Bindungen beim Spinübergang verursacht wird (Abbildung 3.10).
Abbildung 3.10. Volumen der Elementarzelle von Verbindung 26b in Abhängigkeit von der
Temperatur.
Das Volumen der Elementarzelle steigt von 1579.4(3) Å3 bei 100 K auf 1661.0(3) Å3 bei 313
K. Die Zunahme beträgt 81.6(3) Å3. Bei Verbindung 26a, die keinen thermischen
Spinübergang zeigt, liegt eine Volumenänderung von 60.9(2) Å3 zwischen 100 und 313 K
vor, ausschließlich verursacht von der thermischen Ausdehnung. Aus den Differenzen lässt
sich eine Volumenänderung der Elementarzelle von 20.7 Å3 bzw. 1.3 % ermitteln, die bei 26b
nur auf den Spinübergang zurückzuführen ist. Der Wert von 1.3 % liegt im üblichen Bereich
für vergleichbare Moleküle.[101, 102]
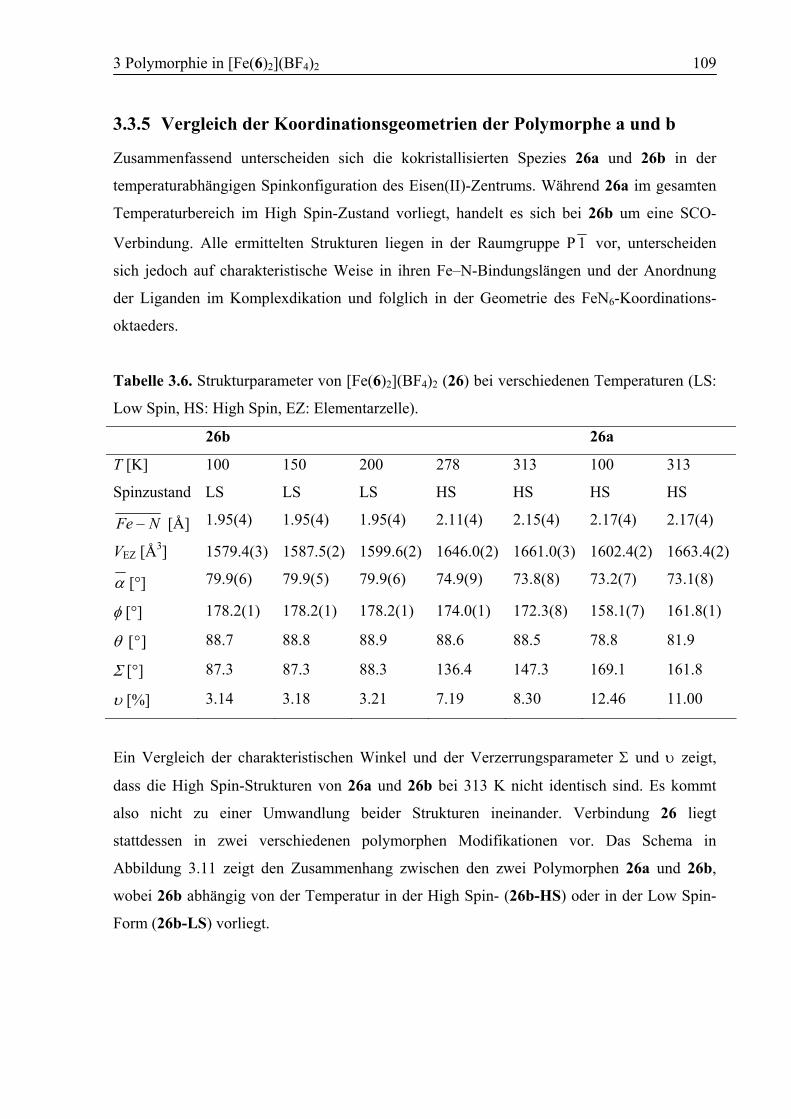
3 Polymorphie in [Fe(6)2](BF4)2 109
3.3.5 Vergleich der Koordinationsgeometrien der Polymorphe a und b
Zusammenfassend unterscheiden sich die kokristallisierten Spezies 26a und 26b in der
temperaturabhängigen Spinkonfiguration des Eisen(II)-Zentrums. Während 26a im gesamten
Temperaturbereich im High Spin-Zustand vorliegt, handelt es sich bei 26b um eine SCO-
Verbindung. Alle ermittelten Strukturen liegen in der Raumgruppe P 1 vor, unterscheiden
sich jedoch auf charakteristische Weise in ihren Fe–N-Bindungslängen und der Anordnung
der Liganden im Komplexdikation und folglich in der Geometrie des FeN6-Koordinations-
oktaeders.
Tabelle 3.6. Strukturparameter von [Fe(6)2](BF4)2 (26) bei verschiedenen Temperaturen (LS:
Low Spin, HS: High Spin, EZ: Elementarzelle).
26b 26a
T [K] 100 150 200 278 313 100 313
Spinzustand LS LS LS HS HS HS HS
NFe – [Å] 1.95(4) 1.95(4) 1.95(4) 2.11(4) 2.15(4) 2.17(4) 2.17(4)
VEZ [Å3] 1579.4(3) 1587.5(2) 1599.6(2) 1646.0(2) 1661.0(3) 1602.4(2) 1663.4(2)
α [°] 79.9(6) 79.9(5) 79.9(6) 74.9(9) 73.8(8) 73.2(7) 73.1(8)
φ [°] 178.2(1) 178.2(1) 178.2(1) 174.0(1) 172.3(8) 158.1(7) 161.8(1)
θ [°] 88.7 88.8 88.9 88.6 88.5 78.8 81.9
Σ [°] 87.3 87.3 88.3 136.4 147.3 169.1 161.8
υ [%] 3.14 3.18 3.21 7.19 8.30 12.46 11.00
Ein Vergleich der charakteristischen Winkel und der Verzerrungsparameter Σ und υ zeigt,
dass die High Spin-Strukturen von 26a und 26b bei 313 K nicht identisch sind. Es kommt
also nicht zu einer Umwandlung beider Strukturen ineinander. Verbindung 26 liegt
stattdessen in zwei verschiedenen polymorphen Modifikationen vor. Das Schema in
Abbildung 3.11 zeigt den Zusammenhang zwischen den zwei Polymorphen 26a und 26b,
wobei 26b abhängig von der Temperatur in der High Spin- (26b-HS) oder in der Low Spin-
Form (26b-LS) vorliegt.

3 Polymorphie in [Fe(6)2](BF4)2 110
[Fe(6)2](BF4)2
26
Gelbe Kristalle, 26a[Fe(6)2](BF4)2, High Spin
T = 2-350 K
Gelbe Kristalle, 26a
i
ii
Orange Kristalle, 26b-HS[Fe(6)2](BF4)2, High Spin
Braune Kristalle, 26b-LS[Fe(6)2](BF4)2, Low Spin
T > 272 K T < 272 K
und
Kristallstrukturen nicht identisch
Abbildung 3.11. Schema der Bildung der Polymorphe von 26. i) Kristallisation aus
methanolischer Lösung, gesättigt bei RT; ii) Kristallisation aus methanolischer Lösung,
gesättigt bei RF-Temperatur.
In Eisenkomplexen von Bpp-Liganden liegt stets eine charakteristische Verzerrung des FeN6-
Koordinationsoktaeders vor. Anhand des Ausmaßes dieser Verzerrung ist es möglich, aus der
Röntgenstruktur Aussagen über das thermische Spinverhalten eines Komplexes zu treffen.
Für Verbindung 26b stimmen die Werte für υ der High Spin- sowie der Low Spin-Form (313
bzw. 150 K) gut mit denen von zwei vergleichbaren Fe-Bpp-SCO-Komplexen überein
(Abbildung 3.12).

3 Polymorphie in [Fe(6)2](BF4)2 111
NNNN N
NNNN N
OH
NNNN N
SCN
L =
I 5 6
υ [%]:
High Spin
Low Spin
8.49 (290 K)
3.12 (150 K)
7.70 (300 K)
3.37 (150 K)
8.30 (313 K)
3.18 (150 K)
Abbildung 3.12. Temperaturabhängige Verzerrungsparameter υ für Komplexe
[Fe(L)2](BF4)2 (L = I,[46] 5[101] und 6 (26b)).
In der reinen High Spin-Verbindung 26a liegt υ mit 12.46 % bei 100 K deutlich höher als in
den High Spin-Strukturen dieser drei SCO-Verbindungen. In der Literatur sind diverse Fe-
Bpp-Komplexe bekannt, die ausschließlich in der High Spin-Form vorliegen. Zum Vergleich
weisen die Fe-Komplexe [Fe(I)2](A)2 (A– = ClO4–, PF6
–, SbF6–) υ-Werte zwischen 17.08 und
18.65 % bei 150 K auf.[94] Polymorph 26a verhält sich also eher wie diese reinen High Spin-
Verbindungen, die die in Kapitel 3.3 erwähnte starke Jahn-Teller-Verzerrung aufweisen,
durch die ein SCO unterbunden wird.
3.3.6 Temperaturabhängige 1H-NMR-Spektroskopie
In Anbetracht der übereinstimmenden Stöchiometrie von 26a und 26b liegt der Schluss nahe,
dass das Spinverhalten in diesem System durch die Festkörperstruktur beeinflusst wird; ein
thermischer Spinübergang wird durch die Stärke des Ligandenfeldes prinzipiell ermöglicht.
Um diese Annahme zusätzlich zu stützen, wurde die temperaturabhängige magnetische
Suszeptibilität von 26 auch in Lösung mittels 1H-NMR-Spektroskopie unter Verwendung der
Methode von Evans bestimmt.[26, 103] Auf diese Weise kann der Einfluss des Ligandenfeldes
auf das SCO-Verhalten vom Einfluss der Festkörperstruktur getrennt betrachtet werden.
Die Messung wurde an einer Probe von Verbindung 26, die nicht umkristallisiert wurde, im
Temperaturbereich 194 … 293 K durchgeführt. Als Lösungsmittel diente [D6]Aceton, dem
Tetramethylsilan als externer Standard beigefügt war ((CD3)2CO / Si(CH3)4 = 99:1 v/v). Die
magnetische Suszeptibilität wurde aus der Verschiebung des TMS-Signals in der Probe in
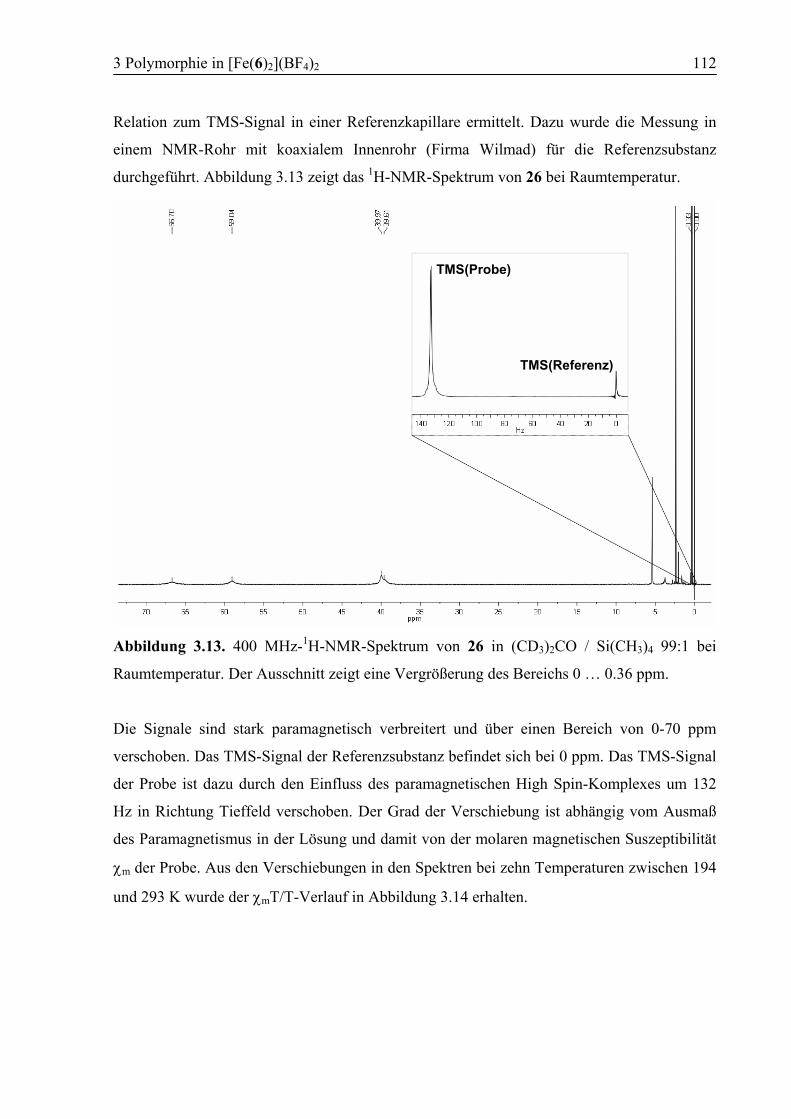
3 Polymorphie in [Fe(6)2](BF4)2 112
Relation zum TMS-Signal in einer Referenzkapillare ermittelt. Dazu wurde die Messung in
einem NMR-Rohr mit koaxialem Innenrohr (Firma Wilmad) für die Referenzsubstanz
durchgeführt. Abbildung 3.13 zeigt das 1H-NMR-Spektrum von 26 bei Raumtemperatur.
Abbildung 3.13. 400 MHz-1H-NMR-Spektrum von 26 in (CD3)2CO / Si(CH3)4 99:1 bei
Raumtemperatur. Der Ausschnitt zeigt eine Vergrößerung des Bereichs 0 … 0.36 ppm.
Die Signale sind stark paramagnetisch verbreitert und über einen Bereich von 0-70 ppm
verschoben. Das TMS-Signal der Referenzsubstanz befindet sich bei 0 ppm. Das TMS-Signal
der Probe ist dazu durch den Einfluss des paramagnetischen High Spin-Komplexes um 132
Hz in Richtung Tieffeld verschoben. Der Grad der Verschiebung ist abhängig vom Ausmaß
des Paramagnetismus in der Lösung und damit von der molaren magnetischen Suszeptibilität
χm der Probe. Aus den Verschiebungen in den Spektren bei zehn Temperaturen zwischen 194
und 293 K wurde der χmT/T-Verlauf in Abbildung 3.14 erhalten.
TMS(Referenz)
TMS(Probe)
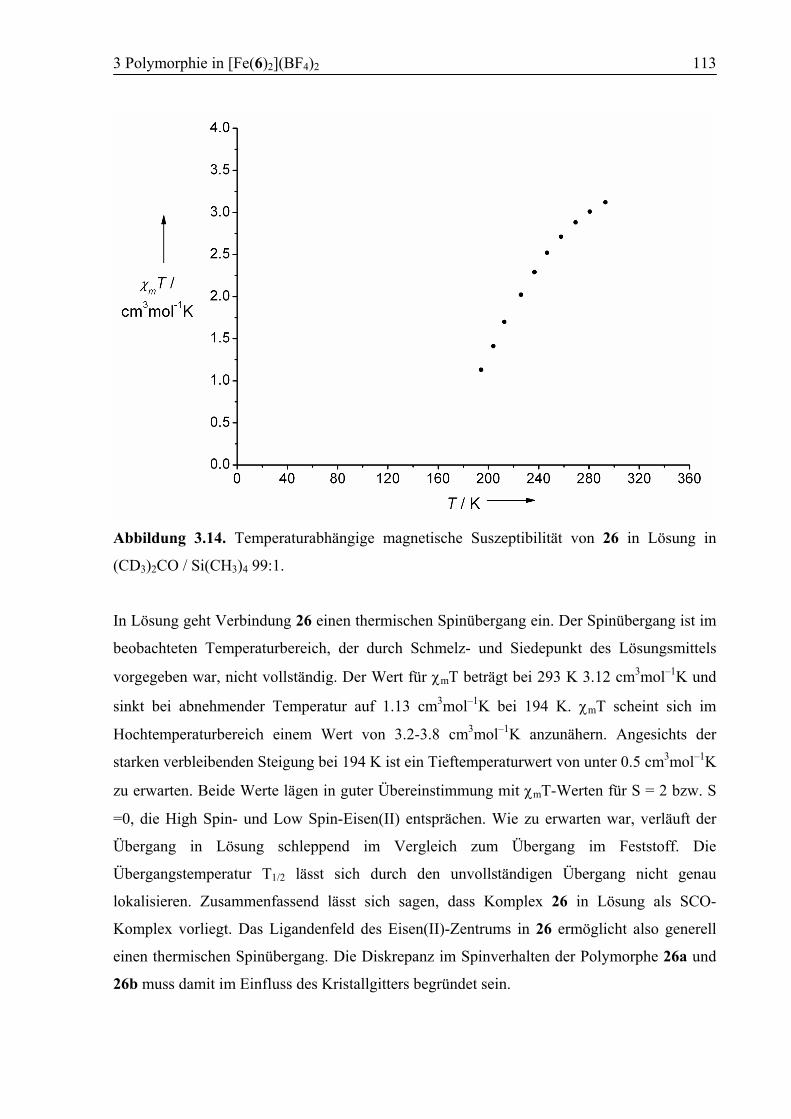
3 Polymorphie in [Fe(6)2](BF4)2 113
Abbildung 3.14. Temperaturabhängige magnetische Suszeptibilität von 26 in Lösung in
(CD3)2CO / Si(CH3)4 99:1.
In Lösung geht Verbindung 26 einen thermischen Spinübergang ein. Der Spinübergang ist im
beobachteten Temperaturbereich, der durch Schmelz- und Siedepunkt des Lösungsmittels
vorgegeben war, nicht vollständig. Der Wert für χmT beträgt bei 293 K 3.12 cm3mol–1K und
sinkt bei abnehmender Temperatur auf 1.13 cm3mol–1K bei 194 K. χmT scheint sich im
Hochtemperaturbereich einem Wert von 3.2-3.8 cm3mol–1K anzunähern. Angesichts der
starken verbleibenden Steigung bei 194 K ist ein Tieftemperaturwert von unter 0.5 cm3mol–1K
zu erwarten. Beide Werte lägen in guter Übereinstimmung mit χmT-Werten für S = 2 bzw. S
=0, die High Spin- und Low Spin-Eisen(II) entsprächen. Wie zu erwarten war, verläuft der
Übergang in Lösung schleppend im Vergleich zum Übergang im Feststoff. Die
Übergangstemperatur T1/2 lässt sich durch den unvollständigen Übergang nicht genau
lokalisieren. Zusammenfassend lässt sich sagen, dass Komplex 26 in Lösung als SCO-
Komplex vorliegt. Das Ligandenfeld des Eisen(II)-Zentrums in 26 ermöglicht also generell
einen thermischen Spinübergang. Die Diskrepanz im Spinverhalten der Polymorphe 26a und
26b muss damit im Einfluss des Kristallgitters begründet sein.

3 Polymorphie in [Fe(6)2](BF4)2 114
3.3.7 Supramolekulare Strukturen der Polymorphe a und b
Die Kristallstrukturen der beiden Polymorphe unterscheiden sich deutlich in Stärke und Art
der intermolekularen Wechselwirkungen. Für den direkten Vergleich wurden die Strukturen
von 26a und 26b bei 313 K jeweils in der höchstbesetzten Vorzugslage gewählt. Beide
Strukturen liegen im High Spin-Zustand vor.
In der Struktur von 26a bilden die Komplexdikationen zweidimensionale Schichten parallel
zur Ebene in (110)-Richtung. Innerhalb der Schichten besteht nur schwacher Zusammenhang
in Form von Van der Waals-Wechselwirkungen. Zwischen zwei Schichten befindet sich
jeweils eine Schicht BF4–-Anionen, wodurch der Zusammenhalt der Schichten durch
elektrostatische Wechselwirkungen gewährleistet wird (Abbildung 3.15).
Abbildung 3.15. Kristallpackung in 26a bei 313 K mit Blickrichtung parallel zur (110)(a,b)-
Ebene. Aus Gründen der Übersichtlichkeit sind Wasserstoffatome und Atome in niedriger
besetzten Vorzugslagen nicht eingezeichnet (schwarz: Komplexdikationen, rot: BF4–-
Anionen).
Die Struktur von Verbindung 26b zeigt eine deutlich unterschiedliche Kristallpackung. Die
BF4–-Anionen bilden hier keine zusammenhängenden Schichten, sondern sind zwischen den

3 Polymorphie in [Fe(6)2](BF4)2 115
Komplexdikationen im Gitter verteilt. Die Komplexdikationen bilden zweidimensionale
Schichten parallel zur ( 110 )-Kristallebene. Zwischen den Molekülen innerhalb dieser
Schichten bestehen nicht-kovalente intermolekulare Bindungen sowohl durch π–π-
Wechselwirkungen zwischen benachbarten Pyrazolringen als auch durch C–H…π-
Wechselwirkungen zwischen Pyrazolringen und benachbarten H-Atomen. Zwischen den
zweidimensionalen Schichten besteht zusätzlich starker Zusammenhang durch C–H…N-
Wasserstoffbrückenbindungen zwischen H-Atomen der Pyrazolringe und dem N-Atom der
Thiocyanatgruppen. Dadurch bestehen im Kristallgitter von 26b in allen drei Dimensionen
relevante Bindungskräfte.
Abbildung 3.16. Kristallpackung in 26b bei 313 K mit Blickrichtung ungefähr entlang der a-
Achse der Elementarzelle. Die grauen Ebenen zeigen die zweidimensionalen Schichten der
Komplexdikationen parallel zur ( 110 )-Kristallebene. Wasserstoffatome und Atome in
niedriger besetzten Vorzugslagen sind nicht eingezeichnet (schwarz: Komplexdikationen, rot:
BF4–-Anionen).
Die Abstände und geometrischen Parameter der nicht-kovalenten Wechselwirkungen liegen
sämtlich in charakteristischen Bereichen mit 3.8-4.0 Å (Abstand Schwerpunkt…Schwerpunkt
der Pyrazolringe) für die π–π-Wechselwirkungen,[104] 3.6 Å (Abstand H…Schwerpunkt des

3 Polymorphie in [Fe(6)2](BF4)2 116
Pyrazolrings) für die C–H…π-Wechselwirkungen[105] und 2.6-2.9 Å (Abstand H…N) für die
C–H…N-Wasserstoffbrückenbindungen.[106]
Die Richtung und Art der intermolekularen Bindungen erklärt auch die unterschiedlichen
Konformationen der Thiocyanatomethylgruppen in beiden Polymorphen. Die N-Atome der
Thiocyanatgruppen in 26b zeigen in Richtung der benachbarten Schicht, um die
Wasserstoffbrückenbindungen zu bilden. Die Thiocyanatgruppen in 26a scheinen keine
intermolekularen Wasserstoffbrückenbindungen einzugehen. Deutlich wird der Unterschied
an den Abständen C6…N6 und C19…N12, die infolge der „gestreckten“ Anordnung der
MeSCN-Gruppen in 26b um 1.4 bzw. 0.9 Å länger sind als in 26a.
Der Vergleich der beiden Strukturen zeigt, dass in Struktur 26b ein größerer
dreidimensionaler Zusammenhalt der Moleküle besteht als in 26a. Die Eisen(II)-
Komplexkationen in 26b sind durch nicht-kovalente Bindungen in allen drei Dimensionen
direkt miteinander verbunden. In 26a sind die Eisenzentren zumindest in Stapelrichtung der
Schichten nur mittelbar über die BF4–-Ionen verbunden. Ein weiterer wichtiger Aspekt bezieht
sich auf die Orientierung der Komplexdikationen relativ zur Stapelrichtung der Schichten, der
in Kapitel 2.3.3 ausgearbeitet wurde. In der Struktur von 26a sind die Komplexmoleküle mit
ihren zentralen N(py)–Fe–N(py’)-Achsen senkrecht zur Stapelrichtung der Schichten
angeordnet, während die Moleküle in 26b mit den zentralen Achsen parallel zur
Stapelrichtung stehen. Da die Längenänderung des Moleküls entlang der N(py)–Fe–N(py’)-
Achse maximal ist, kann in der Struktur von 26b die Information der Größenausdehnung
beim Spinübergang zwischen den Schichten stärker übertragen werden.
Aus der Literatur ist der Zusammenhang zwischen Packung und SCO-Verhalten bekannt.
[Fe(I)2](PF6)2 ist eine reine High Spin-Verbindung.[43] Hier zeigt die Kristallpackung wie in
26a alternierende Schichten von Komplexkationen und Anionen mit nur schwachen
Wechselwirkungen innerhalb der Kationenschichten. Die N(py)–Fe–N(py’)-Achsen sind
senkrecht zur Stapelrichtung der Schichten orientiert (Abbildung 3.17).

3 Polymorphie in [Fe(6)2](BF4)2 117
Abbildung 3.17. Kristallpackung in a) 26a und b) [Fe(I)2](PF6)2. Blick parallel zu den Fe–
N(Pyridin)-Bindungsachsen. Wasserstoffatome und Atome in niedriger besetzten
Vorzugslagen sind nicht eingezeichnet (schwarz: Komplexdikationen, rot: BF4–-(a) bzw. PF6
–-
Anionen (b)).
[Fe(I)2](BF4)2 ist im Gegensatz dazu eine SCO-Verbindung,[46] in deren Gitter die Moleküle
durch intermolekulare Wechselwirkungen einen starken dreidimensionalen Zusammenhalt
haben.
Abbildung 3.18. Kristallpackung in a) 26b und b) [Fe(I)2](BF4)2. Blick parallel zu den Fe–
N(Pyridin)-Bindungsachsen, Die 2D-Schichten befinden sich parallel zur Papierebene.
Wasserstoffatome und Atome in niedriger besetzten Vorzugslagen sind nicht eingezeichnet
(schwarz: Komplexdikationen, rot: BF4–- Anionen).
a) b)
a) b)

3 Polymorphie in [Fe(6)2](BF4)2 118
3.4 Zusammenfassung
Mit Verbindung 26 wurde ein Eisen(II)-SCO-Komplex für die Verankerung auf
Goldoberflächen gemäß dem Synthesekonzept aus Kapitel 2 synthetisiert.
Der Eisen(II)-Komplex [Fe(6)2](BF4)2 (26) kristallisiert spontan in Form zweier
unterschiedlicher Polymorphe. Bei Raumtemperatur erhält man gleichzeitig die High Spin-
(26a) und die SCO-Spezies (26b). In der Literatur sind bisher nur zwei Beispiele für die
spontane Kokristallisation einer High Spin- und einer SCO-Modifikation derselben Substanz
beschrieben. In Lösung zeigt Komplex 26 thermischen Spinübergang Der Unterschied im
Spinverhalten beider Polymorphe ist in der Festkörperstruktur begründet. Abrupter Spinübergang in Metallkomplexen erfordert effiziente Kopplung der Metallzentren,
um die Information im gesamten Gitter zu übertragen. Das Beispiel der Verbindung 26
bestätigt diese These, und der Einfluss der Festkörperstruktur lässt sich infolge identischer
Zusammensetzung der Polymorphe isoliert betrachten. Polymorph 26a zeigt wegen der in
seiner Festkörperstruktur vorhandenen schwachen intermolekularen Wechselwirkungen
schlechte dreidimensionale Kopplung der Eisenzentren und folglich keinen thermischen
Spinübergang. Im Kristallgitter von Polymorph 26b herrschen signifikante intermolekulare
Wechselwirkungen in drei Dimensionen, die hohe Kooperativität im Ensemble und damit
Spinübergang ermöglichen.
Im Hinblick auf die gezielte Synthese von SCO-Komplexen für die Verankerung auf
Oberflächen ist Verbindung 26 geeignet, grundlegende Vorgehensweisen zu entwickeln. Es
ist sinnvoll, die Eignung derartiger Verbindungen sowohl in Lösung als auch im Feststoff zu
überprüfen. Dabei gibt die Untersuchung in Lösung Aufschluss über die Eigenschaften
(Stärke) des Ligandenfeldes, und damit über die generelle Eignung als SCO-System. Das
Studium des Feststoffes hilft, kooperative intermolekulare Wechselwirkungen im
Kristallgitter zu identifizieren. Diese haben erheblichen Einfluss auf den Verlauf des
Spinübergangs und können ihn auch vollständig unterbinden. Bei einer auf einer Oberfläche
verankerten Monolage einer SCO-Verbindung spielen beide Effekte eine Rolle, wenngleich
die intermolekulare Kopplung naturgemäß nur in zwei Dimensionen erfolgen kann.

3 Polymorphie in [Fe(6)2](BF4)2 119
3.5 Experimentalteil
3.5.1 Allgemeines
Sofern nicht anders vermerkt, wurden sämtliche Reaktionen unter Stickstoffatmosphäre und
mit Hilfe von Standard-Schlenk-Technik in absoluten Lösungsmitteln durchgeführt. Die
verwendeten absoluten Lösungsmittel sind kommerziell gelagert auf Molekularsieb 3 Å
erhältlich (Restwassergehalt ≤ 50 ppm). Die eingesetzten Reagenzien wurden von den Firmen
Acros, Fluka, Sigma-Aldrich und VWR bezogen und ohne weitere Reinigung verwendet.
Folgende Geräte wurden zur Gewinnung der Daten verwendet:
NMR-Spektren: Die Messungen erfolgten an den Geräten Bruker ARX 200 (1H-NMR: 200
MHz) und Bruker ARX 400 (1H-NMR: 400 MHz). Sofern nicht anders vermerkt wurden
NMR-Röhrchen mit 5 mm Durchmesser verwendet und die Spektren bei Raumtemperatur
aufgenommen. Alle chemischen Verschiebungen sind in ppm relativ zur Rest-1H-Absorption
des verwendeten Lösungsmittels (interner Standard) angegeben. Die Bestimmung der
magnetischen Suszeptibilität in Lösung erfolgte am Bruker ARX 400 nach der Methode von
Evans[26, 103] unter Benutzung von koaxialen Innenröhrchen der Firma Wilmad mit einem
Volumen von 60 μl. Die erhaltenen Suszeptibilitätswerte wurden um den Beitrag der
diamagnetischen Suszeptibilität mit Hilfe der Pascal-Konstanten korrigiert.[107]
IR-Spektren: Die Proben wurden in Form von Kaliumbromid-Presslingen bzw. als Film an
einem Nicolet Magna System 750 gemessen. Die Wellenzahlen ν~ sind in cm–1 angegeben.
Die Zuordnung der Banden erfolgte anhand geeigneter Literatur.[95]
Elementaranalysen: Die quantitative Bestimmung von Kohlenstoff, Wasserstoff, Stickstoff
und Schwefel erfolgte verbrennungsanalytisch an einem Thermo Finnigan EAGER 300 (Flash
1112 Series). Die Angaben erfolgen in Gewichtsprozent.
Magnetische Messungen: Temperaturabhängige Messungen der magnetischen
Suszeptibilität wurden an einem SQUID-Magnetometer (Superconducting Quantum
Interference Device) des Typs Quantum Design MPMS-XL5 bei einem Feld von 0.1 T über

3 Polymorphie in [Fe(6)2](BF4)2 120
einen Temperaturbereich von 2-350 K durchgeführt. Die gemessenen Werte wurden um die
diamagnetischen Beiträge der Proben und des Probenhalters korrigiert.
3.5.2 Synthese
3.5.2.1 [Fe(C13H10N6S)2](BF4)2 (26)
N
NN
N
NN
N
N
N
N
FeNCS
SCN
2+
[BF4–]2
C26H20B2F8FeN12S2
M = 794.1 g/mol
gelber Feststoff
In einem 10 ml-Schlenkrohr wurde eine Lösung von 2,6-Bis(1H-pyrazol-1-yl)-4-
(thiocyanatomethyl)pyridin (6) (150 mg, 0.5 mmol, 1 Äq.) in Tetrahydrofuran (5 ml)
vorgelegt. Unter Rühren wurde eine Lösung von Eisen(II)tetrafluoroborat-Hexahydrat (87
mg, 0.3 mmol, 0.49 Äq.) in Tetrahydrofuran (3 ml) zum Reaktionsgemisch zugegeben. Aus
der klaren, farblosen Lösung fiel sofort ein gelber Niederschlag aus. Das Gemisch wurde 18 h
bei Raumtemperatur gerührt. Der Niederschlag wurde abfiltriert, mit Tetrahydrofuran
gewaschen und im Hochvakuum getrocknet. Das Produkt wurde in Form eines hellgelben
Feststoffes erhalten (172 mg, 83 %). Einkristalle der Verbindung wurden über zwei
verschiedene Methoden gewonnen. Methode A: Durch isothermale Diffusion von Diethylether
in eine bei Raumtemperatur gesättigte Lösung des Komplexes in Methanol ergaben sich gelbe
Einkristalle (26a). Methode B: Eine isothermale Diffusion von Diethylether in eine zuvor heiß
gesättigte Lösung des Komplexes (c = 15 mg/ml) in Methanol wurde in einem druckdichten
Glasgefäß bei 0.8 bar Stickstoff-Überdruck und Raumtemperatur durchgeführt. Es ergab sich
nach einer Woche ein Gemisch aus rotbraunen (26b) (~2 % bezogen auf das Rohprodukt) und
gelben (26a) Einkristallen.
1H-NMR (200 MHz, [D3]Acetonitril, 25 °C): δ = 66.48 (s), 58.77 (s), 39.26 (s), 37.97 (s),
4.86 ppm (s). (Alle Signale sind paramagnetisch verbreitert)

3 Polymorphie in [Fe(6)2](BF4)2 121
IR (KBr): 26a: ν~ = 521 (w), 597 (w), 704 (w), 769 (m), 793 (m), 917 (w), 971 (s), 1052 (vs),
1176 (w), 1215 (w), 1287 (m), 1311 (m), 1337 (m), 1406 (s), 1467 (vs), 1527 (s), 1581 (m),
1630 (s), 2156 (m), 3136 cm–1 (m); 26b: ν~ = 468 (w), 520 (m), 551 (w), 598 (m), 617 (m),
643 (w), 724 (w), 765 (s), 793 (m), 860 (m), 912 (m), 972 (s), 1050 (vs), 1139 (m), 1177 (m),
1214 (m), 1222 (m), 1244 (w), 1287 (m), 1312 (m), 1338 (s), 1405 (s), 1467 (s), 1503 (m),
1527 (s), 1582 (s), 1631 (s), 2149 (m), 2167 (m), 3126 cm–1 (m).
Elementaranalyse: Ber. (%) für C26H20B2F8FeN12S2: C 39.32, H 2.54, N 21.17, S 8.08; gef.
(%): 26a: C 39.62, H 2.60, N 20.62, S 7.90; 26b: C 39.66, H 2.44, N 20.82, S 7.82.
Röntgenstrukturanalysen: Details zu Kristalldaten, Datensammlung und
Strukturverfeinerung sind in Tabelle 3.7 angegeben. Die Daten wurden unter Benutzung eines
Bruker-Nonius KappaCCD Diffraktometer mit MoKα-Strahlung (λ = 0.71073 Å) und Graphit-
Monochromator ermittelt. Datensammlung und Integration erfolgte mit der COLLECT
Suite.[96] Alle Strukturen wurden mit direkten Methoden gelöst und die Daten nach der
Methode der kleinsten Fehlerquadrate bei voller Matrix gegen F2 verfeinert, unter Benutzung
des Programms SHELXTL NT 6.12.[97] Alle Nichtwasserstoffatome wurden anisotrop
verfeinert. Alle H-Atome wurden in geometrisch optimierten Positionen berechnet und
wurden mit einem isotropen Auslenkungsparameter versehen, der dem 1.2- fachen des
äquivalenten isotropen Auslenkungsparameters des die Wasserstoffatome tragenden C-
Atomes entspricht. Die Geometrischen Parameter der Strukturen wurden mit den Programmen
IVTON[98] und Mercury v1.4.2.[99] ermittelt.
Fehlordnungen („(Verbindung)_(Messtemperatur)“): 26a_100K: Eine MeSCN Gruppe
(Atome S2, C25, C26), zwei Vorzugslagen verfeinert (Besetzung 87.5(2):12.5(2) %); ein BF4–
Ion (Atome F22–F24), zwei Vorzugslagen verfeinert (Besetzung 74.9(4):25.1(4) %);
26a_313K: Beide MeSCN Gruppen (Atome S1, C13, N6 bzw. C25, S2, C26), zwei
Vorzugslagen verfeinert (Besetzung 58.6(8):41.4(8) % bzw. 79.2(3):20.8(3) %); beide BF4–
Ionen (Atome B1–F14 bzw. F22–F24), zwei Vorzugslagen verfeinert (Besetzung
52.3(8):47.7(8) % bzw. 57.6(8):42.4(8) %); 26b_200K: Eine MeSCN Gruppe (Atome S2,
C26, N12), zwei Vorzugslagen verfeinert (Besetzung 78(3):22(3) %); 26b_278K: Beide
MeSCN Gruppen (Atome S1, C13, N6 bzw. S2, C26, N12), zwei Vorzugslagen verfeinert
(Besetzung 86.4(8):13.6(8) % bzw. 64(4):36(4) %); Ein BF4– Ion (Atome F21–F24), zwei
Vorzugslagen verfeinert (Besetzung 77(2):23(2) %); 26b_313K: Beide MeSCN Gruppen
(Atome S1, C13, N6 bzw. S2, C26, N12), zwei Vorzugslagen verfeinert (Besetzung

3 Polymorphie in [Fe(6)2](BF4)2 122
80(1):20(1) % bzw. 62(4):38(4) %); beide BF4– Ionen (Atome F11–F14 bzw. F21–F24), zwei
Vorzugslagen verfeinert (Besetzung 86.4(4):13.6(4) % bzw. 74(2):26(2) %). Die Daten der
Verbindungen können kostenlos beim Cambridge Crystallographic Data Centre unter
www.ccdc.cam.ac.uk/data_request/cif anhand folgender Ablagenummern eingesehen werden:
CCDC-699523 (für 26a_100K), -699524 (für 26a_313K), -699525 (für 26b_100K), -699526
(für 26b_150K), -699527 (für 26b_200K), -699528 (für 26b_278K) and -699529 (für
26b_313K).
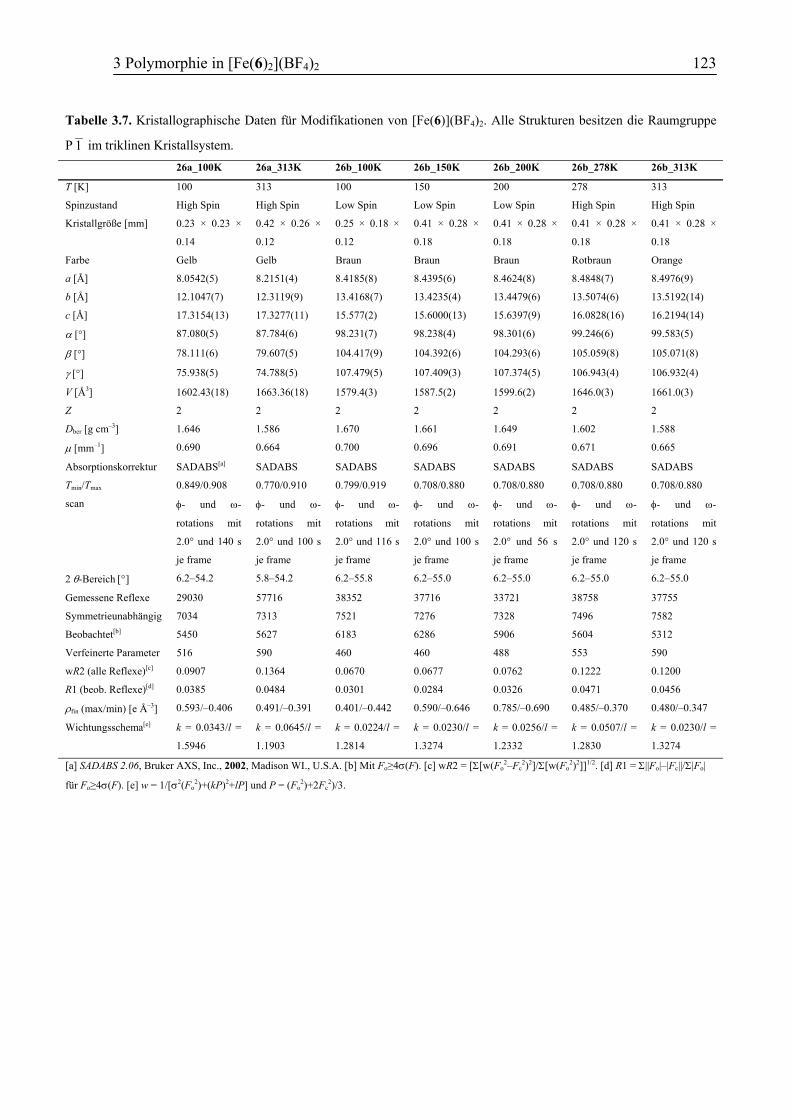
3 Polymorphie in [Fe(6)2](BF4)2 123
Tabelle 3.7. Kristallographische Daten für Modifikationen von [Fe(6)](BF4)2. Alle Strukturen besitzen die Raumgruppe
P 1 im triklinen Kristallsystem. 26a_100K 26a_313K 26b_100K 26b_150K 26b_200K 26b_278K 26b_313K
T [K] 100 313 100 150 200 278 313
Spinzustand High Spin High Spin Low Spin Low Spin Low Spin High Spin High Spin
Kristallgröße [mm] 0.23 × 0.23 ×
0.14
0.42 × 0.26 ×
0.12
0.25 × 0.18 ×
0.12
0.41 × 0.28 ×
0.18
0.41 × 0.28 ×
0.18
0.41 × 0.28 ×
0.18
0.41 × 0.28 ×
0.18
Farbe Gelb Gelb Braun Braun Braun Rotbraun Orange
a [Å] 8.0542(5) 8.2151(4) 8.4185(8) 8.4395(6) 8.4624(8) 8.4848(7) 8.4976(9)
b [Å] 12.1047(7) 12.3119(9) 13.4168(7) 13.4235(4) 13.4479(6) 13.5074(6) 13.5192(14)
c [Å] 17.3154(13) 17.3277(11) 15.577(2) 15.6000(13) 15.6397(9) 16.0828(16) 16.2194(14)
α [°] 87.080(5) 87.784(6) 98.231(7) 98.238(4) 98.301(6) 99.246(6) 99.583(5)
β [°] 78.111(6) 79.607(5) 104.417(9) 104.392(6) 104.293(6) 105.059(8) 105.071(8)
γ [°] 75.938(5) 74.788(5) 107.479(5) 107.409(3) 107.374(5) 106.943(4) 106.932(4)
V [Å3] 1602.43(18) 1663.36(18) 1579.4(3) 1587.5(2) 1599.6(2) 1646.0(3) 1661.0(3)
Z 2 2 2 2 2 2 2
Dber [g cm–3] 1.646 1.586 1.670 1.661 1.649 1.602 1.588
μ [mm–1] 0.690 0.664 0.700 0.696 0.691 0.671 0.665
Absorptionskorrektur SADABS[a] SADABS SADABS SADABS SADABS SADABS SADABS
Tmin/Tmax 0.849/0.908 0.770/0.910 0.799/0.919 0.708/0.880 0.708/0.880 0.708/0.880 0.708/0.880
scan φ- und ω-
rotations mit
2.0° und 140 s
je frame
φ- und ω-
rotations mit
2.0° und 100 s
je frame
φ- und ω-
rotations mit
2.0° und 116 s
je frame
φ- und ω-
rotations mit
2.0° und 100 s
je frame
φ- und ω-
rotations mit
2.0° und 56 s
je frame
φ- und ω-
rotations mit
2.0° und 120 s
je frame
φ- und ω-
rotations mit
2.0° und 120 s
je frame
2 θ-Bereich [°] 6.2–54.2 5.8–54.2 6.2–55.8 6.2–55.0 6.2–55.0 6.2–55.0 6.2–55.0
Gemessene Reflexe 29030 57716 38352 37716 33721 38758 37755
Symmetrieunabhängig 7034 7313 7521 7276 7328 7496 7582
Beobachtet[b] 5450 5627 6183 6286 5906 5604 5312
Verfeinerte Parameter 516 590 460 460 488 553 590
wR2 (alle Reflexe)[c] 0.0907 0.1364 0.0670 0.0677 0.0762 0.1222 0.1200
R1 (beob. Reflexe)[d] 0.0385 0.0484 0.0301 0.0284 0.0326 0.0471 0.0456
ρfin (max/min) [e Å–3] 0.593/–0.406 0.491/–0.391 0.401/–0.442 0.590/–0.646 0.785/–0.690 0.485/–0.370 0.480/–0.347
Wichtungsschema[e] k = 0.0343/l =
1.5946
k = 0.0645/l =
1.1903
k = 0.0224/l =
1.2814
k = 0.0230/l =
1.3274
k = 0.0256/l =
1.2332
k = 0.0507/l =
1.2830
k = 0.0230/l =
1.3274
[a] SADABS 2.06, Bruker AXS, Inc., 2002, Madison WI., U.S.A. [b] Mit Fo≥4σ(F). [c] wR2 = [Σ[w(Fo2–Fc
2)2]/Σ[w(Fo2)2]]1/2. [d] R1 = Σ||Fo|–|Fc||/Σ|Fo|
für Fo≥4σ(F). [e] w = 1/[σ2(Fo2)+(kP)2+lP] und P = (Fo
2)+2Fc2)/3.

4 Self-assembled Monolayers 124
4 Self-assembled Monolayers
4.1 Einleitung
Einige der in Kapitel 2 und 3 beschriebenen Liganden und Eisen(II)-Komplexe sollten in
Form selbstorganisierter Monolagen (self-assembled monolayers, SAMs) auf Au(111)-
Oberflächen aufgebracht werden. Gold hat als Substrat mehrere Vorteile.[15] Es ist chemisch
inert, vor allem gegenüber Oxidation. Vor der Beschichtung lassen sich die Goldoberflächen
durch „Ionenstrahlsputtern“,[108] d. h. Beschuss mit energiereichen Ionen, oder einfacher
durch Tempern in einer Flamme reinigen. Die im Beschichtungsprozess gebildete Au–S-
Bindung ist sehr stabil, wodurch die zu verankernden Moleküle in der Lage sind, mögliche
Verunreinigungen von der Oberfläche zu verdrängen. Als Substrat wurden bei allen Ansätzen
epitaktisch aufgewachsene Goldlagen von 300 nm Dicke auf Glimmerträgern verwendet. Die
Goldoberfläche erfährt durch das Tempern eine Rekonstruktion zu gleichmäßig strukturierten
Au(111)-Flächen. Die SAMs der Moleküle auf den Goldoberflächen wurden durch
Abscheidung aus Lösungen oder aus der Gasphase präpariert. Folgende Substanzen wurden
untersucht: die Liganden 6 und 10, und deren Eisen(II)-Komplexe [Fe(6)2](BF4)2 (26) und
[Fe(10)2](BF4)2 (25).
NNNN N
SCN
NNNN N
HN
N
NN
N
NN
N
N
N
N
FeNCS
SCN
2+
[BF4–]2
N
NN
N
NN
N
N
N
N
FeHN
NH
2+
[BF4–]2
SHO
SH
HS
O
O
6
10
26
25 Abbildung 4.1. Untersuchte Moleküle auf Oberflächen.

4 Self-assembled Monolayers 125
Die analytischen Methoden umfassten die Rastertunnelmikroskopie (STM), die Röntgen-
Nahkanten-Absorptions-Spektroskopie (NEXAFS) und die Röntgen-Photoelektronen-
Spektroskopie (XPS). Alle Messungen wurden, z. T. unter Beteiligung des Autors, in den
Arbeitskreisen Buck (St. Andrews, Großbritannien), Zharnikov (Heidelberg) bzw. Kuch
(Berlin) vorgenommen.
4.2 SAM von Ligand 10 auf Au(111)
4.2.1 STM
EtOH ist das gebräuchlichste Lösungsmittel zur Präparation von Thiol-Monolagen aus
Lösungen der zu verankernden Moleküle. SAMs, die aus Lösungen von Verbindung 10 in
EtOH bei Raumtemperatur hergestellt wurden, zeigten jedoch eine schlechte
Oberflächenstruktur mit kleinen geordneten Bereichen. Durch Erhöhung der
Präparationstemperatur auf 72 °C und Verlängerung der Beschichtungsdauer ergaben sich
besser geordnete Lagen (Abbildung 4.2.a, b).
Abbildung 4.2. STM-Bilder von 10 auf Au(111)/Glimmer. Präparation: a), b) EtOH, 72 °C, 5
d; c), d) DMF, 90 °C, 15 h.
c) d)
b) a)
50 nm 10 nm
50 nm 10 nm

4 Self-assembled Monolayers 126
Ein Wechsel zu DMF als Lösungsmittel ergab bei der gleichen Temperatur keine
Verbesserung der Schichtqualität. Durch den höheren Siedepunkt des Lösungsmittels konnten
jedoch Monolagen bei einer höheren Temperatur (90 °C) präpariert werden, die eine bessere
Ordnung zeigten (Abbildung 4.2.c, d). Die Oberfläche (Abbildung 4.2.d) zeigt streifenförmige
Strukturen in Abständen von ca. 1.5 nm. Über die gesamte Oberfläche verteilt findet man, wie
auch im Fall der bei niedrigerer Temperatur präparierten Schichten, Vertiefungen von ca. 0.25
nm Tiefe. Es handelt sich hierbei um so genannte „vacancy islands“. Diese entstehen durch
Rekonstruktion der Goldoberfläche beim Bindungsprozess in der obersten Atomlage und
stellen ein übliches Merkmal von Thiol-Monolagen auf Gold dar.[109] Ein Nachteil der so
präparierten SAMs blieb die schlechte Reproduzierbarkeit. Es zeigten sich in aufeinander
folgenden Ansätzen stark schwankende Verhältnisse von geordneten zu ungeordneten
Bereichen der Oberflächen. Grund hierfür waren möglicherweise Moleküle von 10, die durch
Wechselwirkung ihrer aromatischen Ringe mit dem Substrat flach auf der Oberfläche
adsorbiert wurden, statt über die Thiolfunktion kovalente Bindung einzugehen. Abhilfe
schaffte eine von Buck und Mitarbeitern (St. Andrews, Großbritannien) entwickelte Strategie,
und zwar die Vorbeschichtung der Goldoberfläche mit planar adsorbierten organischen
Molekülen. Das Substrat wurde mit dem Perylenderivat 3,4,9,10-Perylentetracarbonsäure-
Dianhydrid (PTCDA) vorbeschichtet, dessen planare Moleküle Monolagen hoher Ordnung
auf Goldoberflächen bilden (Abbildung 4.3).[110, 111] Die PTCDA-Moleküle adsorbieren dabei
durch Wechselwirkung des aromatischen Systems parallel zur Molekülebene flach auf der
Oberfläche und verhindern durch hohe Bedeckungsgrade die weitere Adsorption von
Fremdmolekülen. Abbildung 4.3 zeigt die aus einer DMF-Lösung von PTCDA präparierten
Monolagen auf Au(111).
OO
O
OO
O
PTCDA
Abbildung 4.3. a), b) STM-Bilder einer PTCDA-Monolage auf Au(111)/Glimmer, präpariert
aus gesättigter Lösung in DMF, RT, 5 min; c) Strukturformel von PTCDA.
a) b)
10 nm 10 Å
c)

4 Self-assembled Monolayers 127
Bei der Beschichtung der mit PTCDA vorfunktionalisierten Substrate mit 10 werden die
PTCDA-Moleküle infolge der höheren Affinität der Schwefelfunktion von 10 zur
Goldoberfläche verdrängt, und zwar offenbar unter Vermeidung einer unerwünschten
Wechselwirkung der aromatischen Ringe von 10 mit der Oberfläche. Die Beschichtung
erfolgte aus einer 1 mM Lösung von 10 in DMF bei 90 °C (Einwirkzeit 15h). Die Methode
ergab hoch geordnete Lagen von 10 auf der Au(111)-Oberfläche, bei guter
Reproduzierbarkeit (Abbildung 4.4).
c)
0 10 20 30 400.00.20.40.60.81.0
0 10 20 30 400.00.20.40.60.81.0
vacancy islands
Abbildung 4.4 STM-Bilder einer Monolage von 10 nach Vorbeschichtung der
Goldoberfläche mit PTCDA (DMF, RT, 5 min) aus Lösung (1 mM in DMF, 90 °C, 15 h). Die
durch das weiße Rechteck markierte Fläche in a) ist in Bild b) vergrößert dargestellt; Graph c)
zeigt das Höhenprofil entlang der weißen Linie in Bild b)
Die STM-Bilder zeigen in Domänen angeordnete streifenförmige Strukturen entlang der
>< 211 Richtung. Die in den ohne Vorbeschichtung gewonnen Monolagen auftretenden
Vertiefungen können auch in den Monolagen hoher Qualität (Abbildung 4.4) beobachtet
werden. Sie treten vorwiegend am Schnittpunkt von Domänen auf und weisen großenteils die
geordnete Struktur der übrigen Monolage auf, wie das Höhenprofil in Abbildung 4.4.c zeigt.
Beide Beobachtungen können auch bei SAMs von Alkanthiolen gemacht werden[109] und
a) b)
20 nm
50 nm
<110>
<112>
Höh
e [n
m]
Distanz [nm]
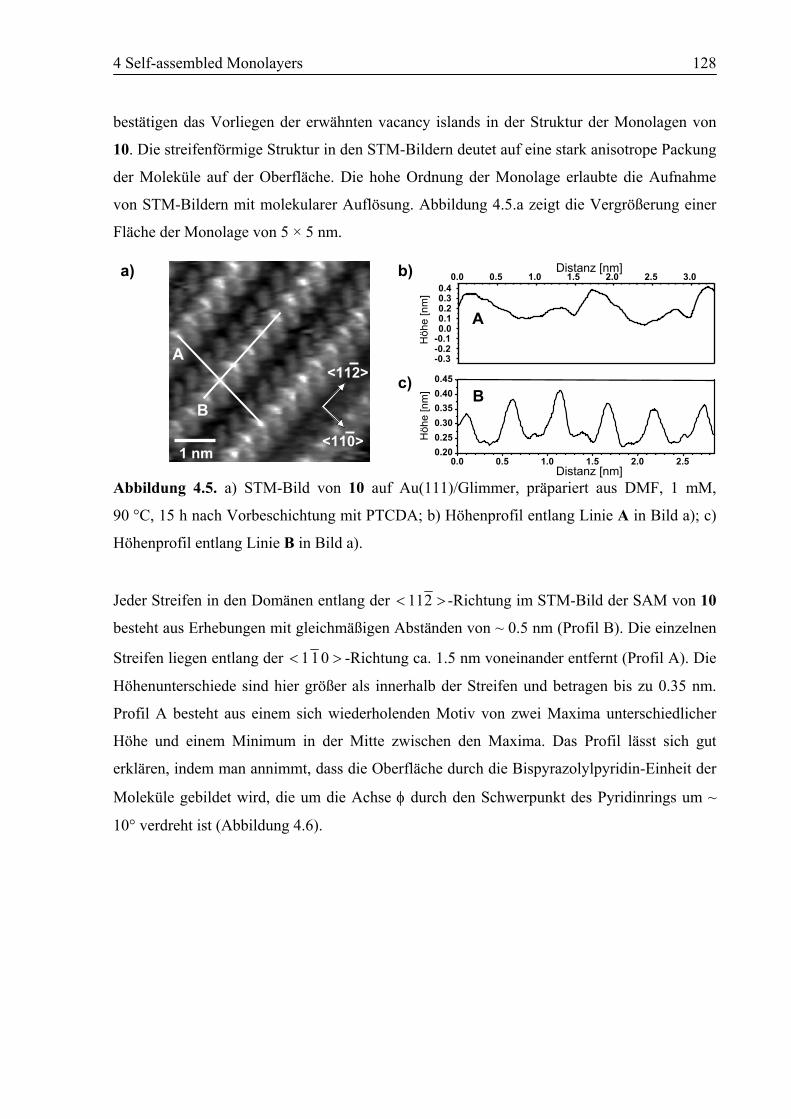
4 Self-assembled Monolayers 128
bestätigen das Vorliegen der erwähnten vacancy islands in der Struktur der Monolagen von
10. Die streifenförmige Struktur in den STM-Bildern deutet auf eine stark anisotrope Packung
der Moleküle auf der Oberfläche. Die hohe Ordnung der Monolage erlaubte die Aufnahme
von STM-Bildern mit molekularer Auflösung. Abbildung 4.5.a zeigt die Vergrößerung einer
Fläche der Monolage von 5 × 5 nm.
a) b)
c)
Abbildung 4.5. a) STM-Bild von 10 auf Au(111)/Glimmer, präpariert aus DMF, 1 mM,
90 °C, 15 h nach Vorbeschichtung mit PTCDA; b) Höhenprofil entlang Linie A in Bild a); c)
Höhenprofil entlang Linie B in Bild a).
Jeder Streifen in den Domänen entlang der >< 211 -Richtung im STM-Bild der SAM von 10
besteht aus Erhebungen mit gleichmäßigen Abständen von ~ 0.5 nm (Profil B). Die einzelnen
Streifen liegen entlang der >< 011 -Richtung ca. 1.5 nm voneinander entfernt (Profil A). Die
Höhenunterschiede sind hier größer als innerhalb der Streifen und betragen bis zu 0.35 nm.
Profil A besteht aus einem sich wiederholenden Motiv von zwei Maxima unterschiedlicher
Höhe und einem Minimum in der Mitte zwischen den Maxima. Das Profil lässt sich gut
erklären, indem man annimmt, dass die Oberfläche durch die Bispyrazolylpyridin-Einheit der
Moleküle gebildet wird, die um die Achse φ durch den Schwerpunkt des Pyridinrings um ~
10° verdreht ist (Abbildung 4.6).
0.0 0.5 1.0 1.5 2.0 2.50.200.250.300.350.400.45
0.0 0.5 1.0 1.5 2.0 2.5 3.0
-0.3-0.2-0.10.00.10.20.30.4
A
B
Distanz [nm]
Distanz [nm]
Höh
e [n
m]
Höh
e [n
m]
B
A
<110>
<112>
1 nm
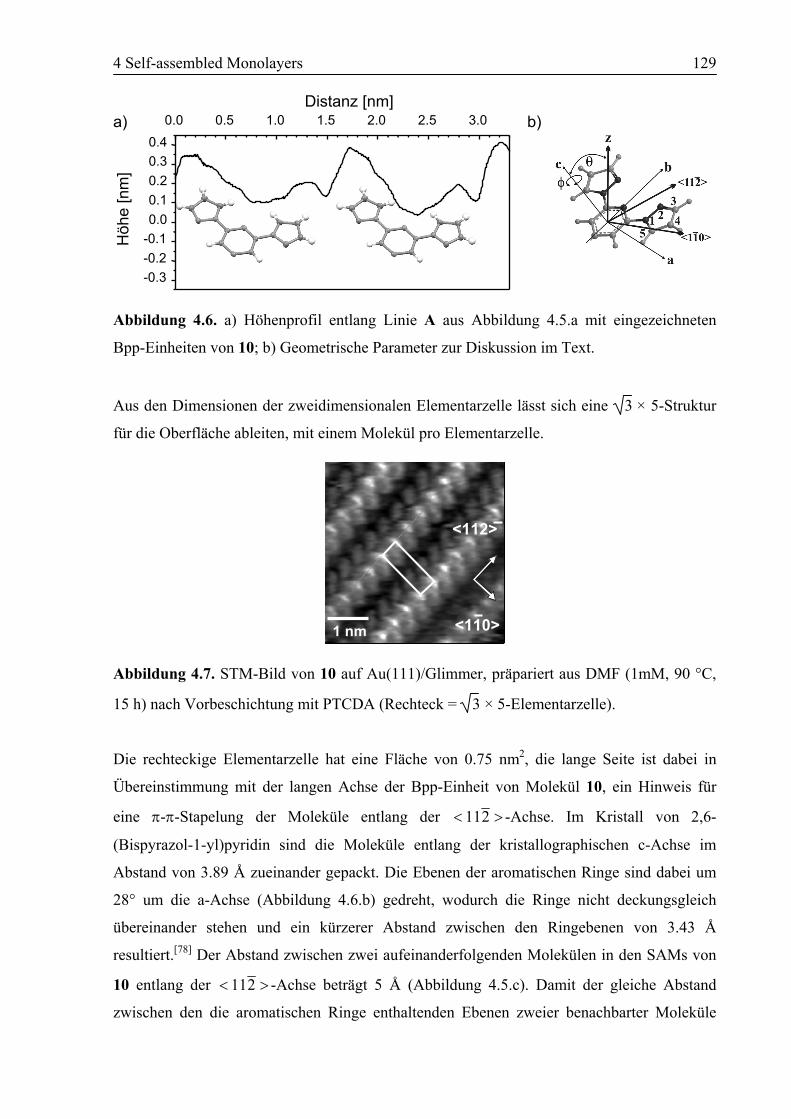
4 Self-assembled Monolayers 129
a)
b)
Abbildung 4.6. a) Höhenprofil entlang Linie A aus Abbildung 4.5.a mit eingezeichneten
Bpp-Einheiten von 10; b) Geometrische Parameter zur Diskussion im Text.
Aus den Dimensionen der zweidimensionalen Elementarzelle lässt sich eine 3 × 5-Struktur
für die Oberfläche ableiten, mit einem Molekül pro Elementarzelle.
Abbildung 4.7. STM-Bild von 10 auf Au(111)/Glimmer, präpariert aus DMF (1mM, 90 °C,
15 h) nach Vorbeschichtung mit PTCDA (Rechteck = 3 × 5-Elementarzelle).
Die rechteckige Elementarzelle hat eine Fläche von 0.75 nm2, die lange Seite ist dabei in
Übereinstimmung mit der langen Achse der Bpp-Einheit von Molekül 10, ein Hinweis für
eine π-π-Stapelung der Moleküle entlang der >< 211 -Achse. Im Kristall von 2,6-
(Bispyrazol-1-yl)pyridin sind die Moleküle entlang der kristallographischen c-Achse im
Abstand von 3.89 Å zueinander gepackt. Die Ebenen der aromatischen Ringe sind dabei um
28° um die a-Achse (Abbildung 4.6.b) gedreht, wodurch die Ringe nicht deckungsgleich
übereinander stehen und ein kürzerer Abstand zwischen den Ringebenen von 3.43 Å
resultiert.[78] Der Abstand zwischen zwei aufeinanderfolgenden Molekülen in den SAMs von
10 entlang der >< 211 -Achse beträgt 5 Å (Abbildung 4.5.c). Damit der gleiche Abstand
zwischen den die aromatischen Ringe enthaltenden Ebenen zweier benachbarter Moleküle
0.0 0.5 1.0 1.5 2.0 2.5 3.0
-0.3 -0.2 -0.1 0.0 0.1 0.2 0.3 0.4
Distanz [nm]H
öhe
[nm
]
<110>
<112>
1 nm

4 Self-assembled Monolayers 130
wie in der Kristallstruktur von Bpp resultiert, müssten die Moleküle um einen Winkel von θ =
43° um die a-Achse gekippt werden.
Die geometrischen Berechnungen und die Parameter aus den STM-Bildern sind in der
Zeichnung eines Modells der Monolage von 10 zusammengefasst, die in Abbildung 4.8
gezeigt ist.
Abbildung 4.8. Modell der SAM von 10 in der Aufsicht. Nur die Teile der Moleküle an der
Oberfläche sind gezeigt (schwarzes Rechteck = 3 × 5 Elementarzelle).
Bezüglich der Konformation der Pyrazolringe relativ zum Pyridinring können aus den STM-
Daten keine endgültigen Folgerungen getroffen werden. Die trans,trans-Orientierung in den
Festkörperstrukturen von 2,6-(Bispyrazol-1-yl)pyridin[78] und Ethyl-2,6-bis(1H-pyrazol-1-
yl)pyridin-4-carboxylat (4) (Kapitel 2) erscheint auch für die Moleküle von 10 auf der
Oberfläche wahrscheinlich. Ein Gemisch von Konformationen der einzelnen Moleküle ist
unwahrscheinlich, da eine solche statistische Verteilung in den STM-Bildern zu erkennen sein
müsste. Die trans,trans-Konformation wird durch Kontaktwinkel-Messungen der Oberflächen
gestützt, die mit einem Wert von 70° eine eher hydrophobe Oberfläche zeigen. In der cis,cis-
Konformation würden die Stickstoffatome der Pyrazolringe die Oberflächenstruktur
bestimmen und durch die Möglichkeit der Ausbildung von Wasserstoffbrückenbindungen
eine bessere Benetzbarkeit zeigen.
4.2.2 NEXAFS/XPS
Die Analyse der chemischen Zusammensetzung der Monolagen von Verbindung 10 auf
Au(111)/Glimmer wurde mittels hochauflösender Röntgen-Photoelektronenspektroskopie
(XPS) für die Bereiche der S 2p-, C 1s-, N 1s- und O 1s-Rumpfniveaus durchgeführt.
>< 011
>< 211
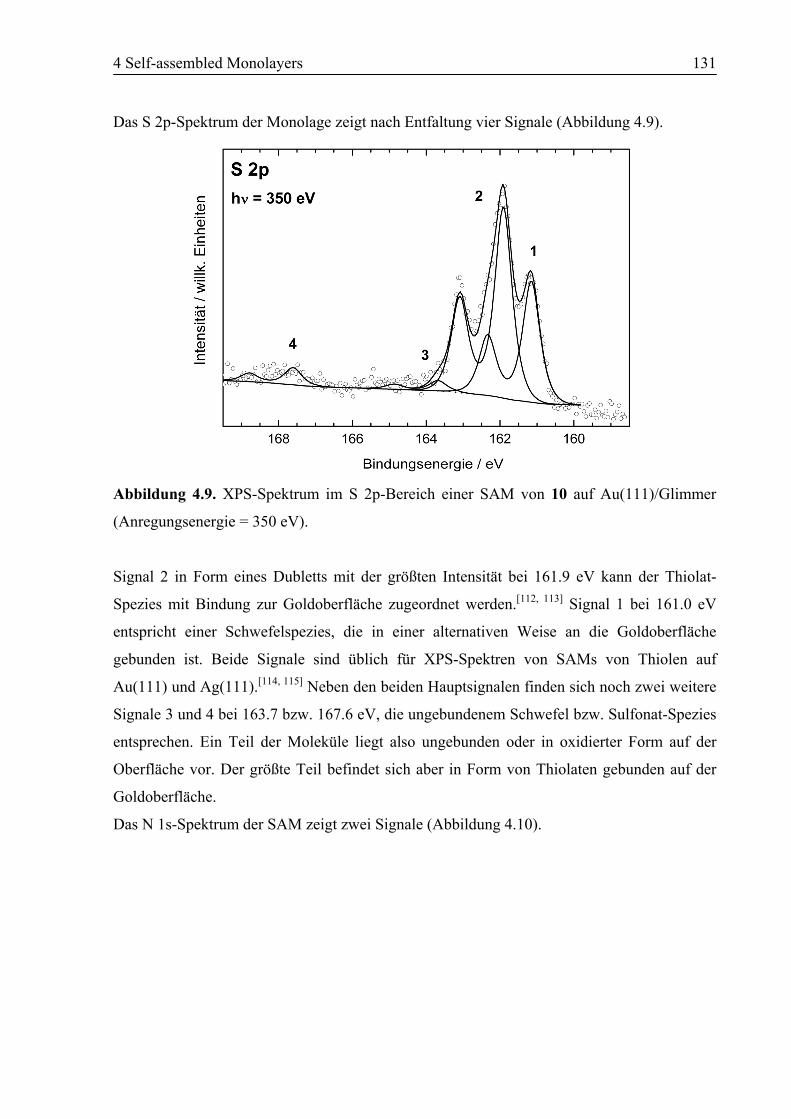
4 Self-assembled Monolayers 131
Das S 2p-Spektrum der Monolage zeigt nach Entfaltung vier Signale (Abbildung 4.9).
Abbildung 4.9. XPS-Spektrum im S 2p-Bereich einer SAM von 10 auf Au(111)/Glimmer
(Anregungsenergie = 350 eV).
Signal 2 in Form eines Dubletts mit der größten Intensität bei 161.9 eV kann der Thiolat-
Spezies mit Bindung zur Goldoberfläche zugeordnet werden.[112, 113] Signal 1 bei 161.0 eV
entspricht einer Schwefelspezies, die in einer alternativen Weise an die Goldoberfläche
gebunden ist. Beide Signale sind üblich für XPS-Spektren von SAMs von Thiolen auf
Au(111) und Ag(111).[114, 115] Neben den beiden Hauptsignalen finden sich noch zwei weitere
Signale 3 und 4 bei 163.7 bzw. 167.6 eV, die ungebundenem Schwefel bzw. Sulfonat-Spezies
entsprechen. Ein Teil der Moleküle liegt also ungebunden oder in oxidierter Form auf der
Oberfläche vor. Der größte Teil befindet sich aber in Form von Thiolaten gebunden auf der
Goldoberfläche.
Das N 1s-Spektrum der SAM zeigt zwei Signale (Abbildung 4.10).
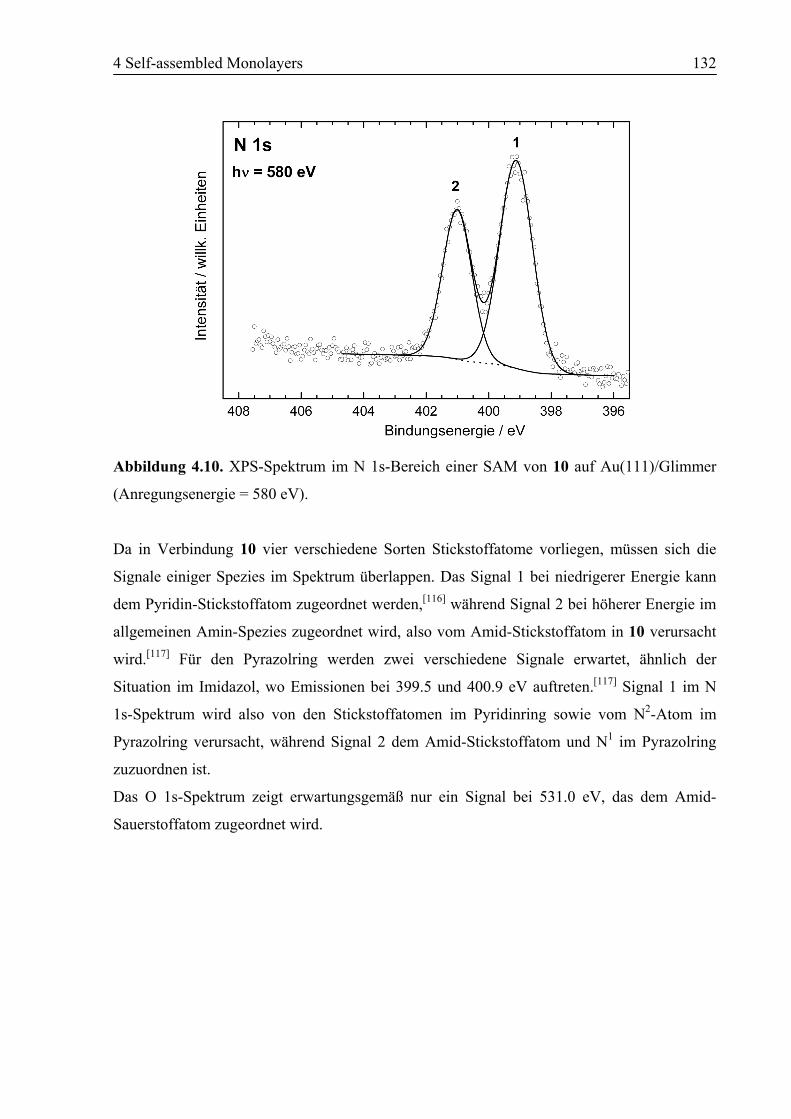
4 Self-assembled Monolayers 132
Abbildung 4.10. XPS-Spektrum im N 1s-Bereich einer SAM von 10 auf Au(111)/Glimmer
(Anregungsenergie = 580 eV).
Da in Verbindung 10 vier verschiedene Sorten Stickstoffatome vorliegen, müssen sich die
Signale einiger Spezies im Spektrum überlappen. Das Signal 1 bei niedrigerer Energie kann
dem Pyridin-Stickstoffatom zugeordnet werden,[116] während Signal 2 bei höherer Energie im
allgemeinen Amin-Spezies zugeordnet wird, also vom Amid-Stickstoffatom in 10 verursacht
wird.[117] Für den Pyrazolring werden zwei verschiedene Signale erwartet, ähnlich der
Situation im Imidazol, wo Emissionen bei 399.5 und 400.9 eV auftreten.[117] Signal 1 im N
1s-Spektrum wird also von den Stickstoffatomen im Pyridinring sowie vom N2-Atom im
Pyrazolring verursacht, während Signal 2 dem Amid-Stickstoffatom und N1 im Pyrazolring
zuzuordnen ist.
Das O 1s-Spektrum zeigt erwartungsgemäß nur ein Signal bei 531.0 eV, das dem Amid-
Sauerstoffatom zugeordnet wird.
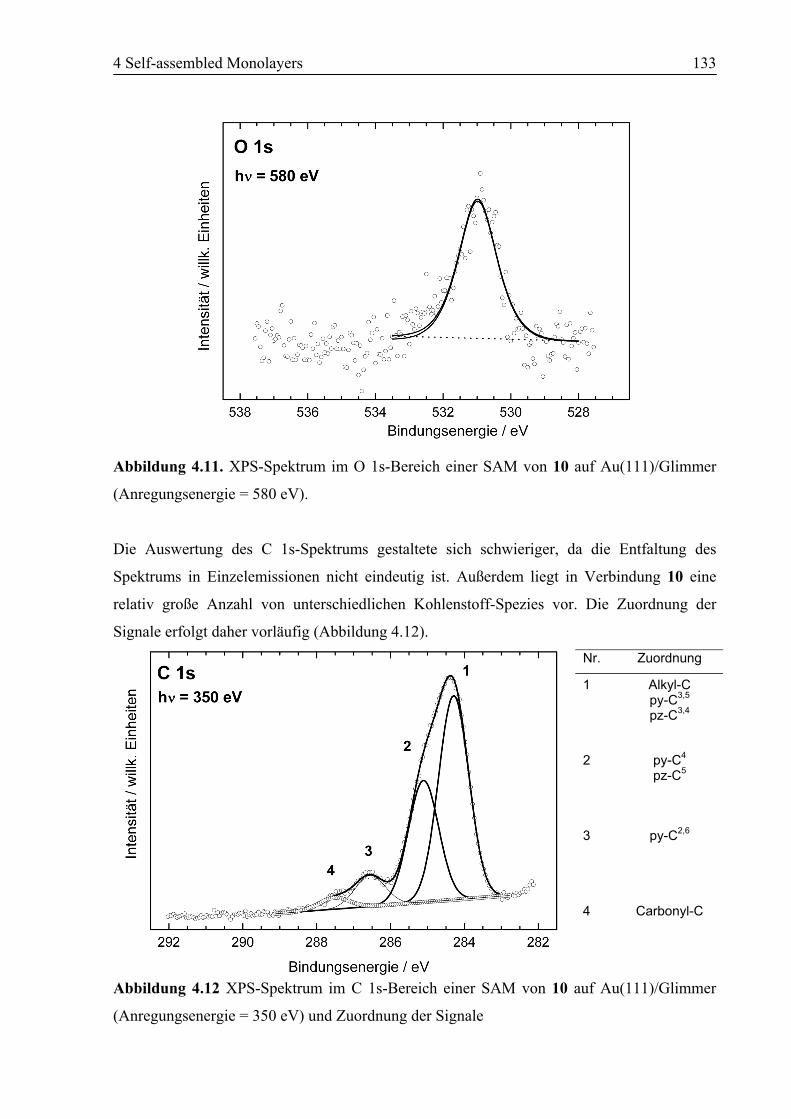
4 Self-assembled Monolayers 133
Abbildung 4.11. XPS-Spektrum im O 1s-Bereich einer SAM von 10 auf Au(111)/Glimmer
(Anregungsenergie = 580 eV).
Die Auswertung des C 1s-Spektrums gestaltete sich schwieriger, da die Entfaltung des
Spektrums in Einzelemissionen nicht eindeutig ist. Außerdem liegt in Verbindung 10 eine
relativ große Anzahl von unterschiedlichen Kohlenstoff-Spezies vor. Die Zuordnung der
Signale erfolgt daher vorläufig (Abbildung 4.12). Nr. Zuordnung
1 Alkyl-C py-C3,5 pz-C3,4
2 py-C4 pz-C5
3 py-C2,6
4 Carbonyl-C
Abbildung 4.12 XPS-Spektrum im C 1s-Bereich einer SAM von 10 auf Au(111)/Glimmer
(Anregungsenergie = 350 eV) und Zuordnung der Signale
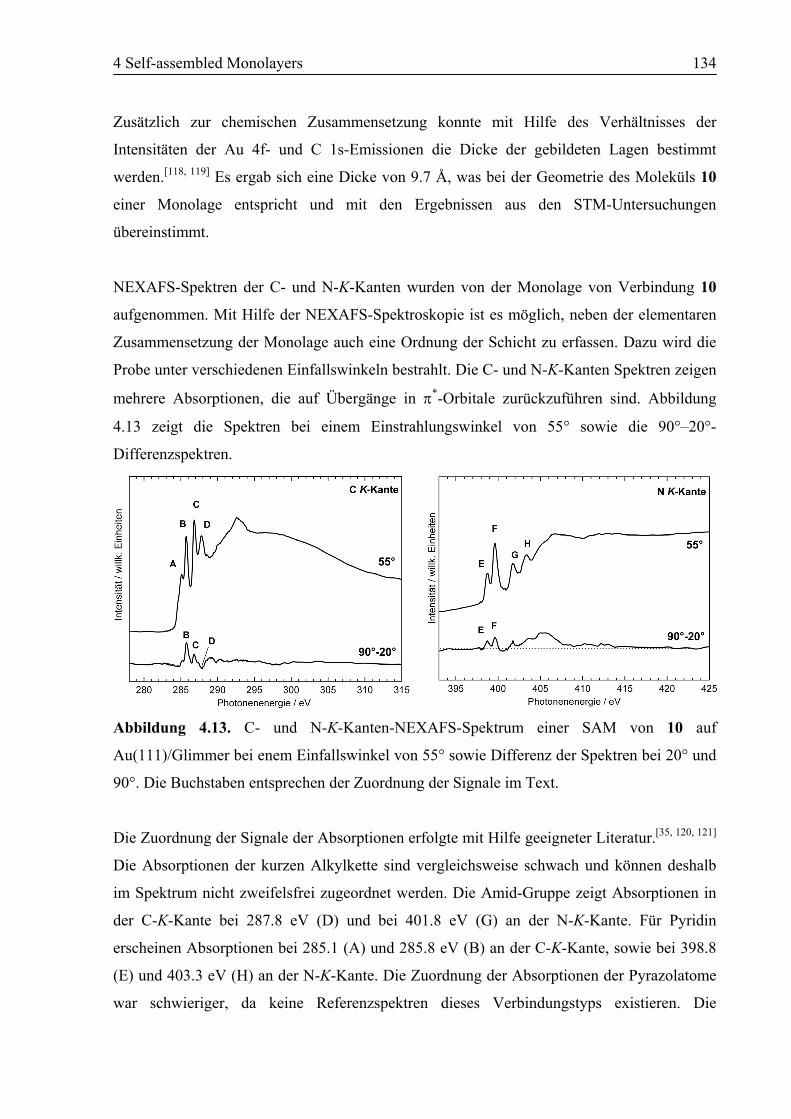
4 Self-assembled Monolayers 134
Zusätzlich zur chemischen Zusammensetzung konnte mit Hilfe des Verhältnisses der
Intensitäten der Au 4f- und C 1s-Emissionen die Dicke der gebildeten Lagen bestimmt
werden.[118, 119] Es ergab sich eine Dicke von 9.7 Å, was bei der Geometrie des Moleküls 10
einer Monolage entspricht und mit den Ergebnissen aus den STM-Untersuchungen
übereinstimmt.
NEXAFS-Spektren der C- und N-K-Kanten wurden von der Monolage von Verbindung 10
aufgenommen. Mit Hilfe der NEXAFS-Spektroskopie ist es möglich, neben der elementaren
Zusammensetzung der Monolage auch eine Ordnung der Schicht zu erfassen. Dazu wird die
Probe unter verschiedenen Einfallswinkeln bestrahlt. Die C- und N-K-Kanten Spektren zeigen
mehrere Absorptionen, die auf Übergänge in π*-Orbitale zurückzuführen sind. Abbildung
4.13 zeigt die Spektren bei einem Einstrahlungswinkel von 55° sowie die 90°–20°-
Differenzspektren.
Abbildung 4.13. C- und N-K-Kanten-NEXAFS-Spektrum einer SAM von 10 auf
Au(111)/Glimmer bei enem Einfallswinkel von 55° sowie Differenz der Spektren bei 20° und
90°. Die Buchstaben entsprechen der Zuordnung der Signale im Text.
Die Zuordnung der Signale der Absorptionen erfolgte mit Hilfe geeigneter Literatur.[35, 120, 121]
Die Absorptionen der kurzen Alkylkette sind vergleichsweise schwach und können deshalb
im Spektrum nicht zweifelsfrei zugeordnet werden. Die Amid-Gruppe zeigt Absorptionen in
der C-K-Kante bei 287.8 eV (D) und bei 401.8 eV (G) an der N-K-Kante. Für Pyridin
erscheinen Absorptionen bei 285.1 (A) und 285.8 eV (B) an der C-K-Kante, sowie bei 398.8
(E) und 403.3 eV (H) an der N-K-Kante. Die Zuordnung der Absorptionen der Pyrazolatome
war schwieriger, da keine Referenzspektren dieses Verbindungstyps existieren. Die

4 Self-assembled Monolayers 135
Zuordnung erfolgte daher durch den Vergleich mit Spektren der verwandten Verbindungen
Imidazol[122] und Pyrrol.[123] Pyrazol zeigt demnach Signale bei 286.9 eV (C) in der C-K-
Kante und 399.6 eV (F) im N-K-Kanten-Spektrum.
Die Absorptionen, die durch die Pyridin- und Pyrazol-Einheiten des Moleküls verursacht
werden, zeigen einen deutlichen Dichroismus in den Differenzspektren (Signale B, C, E und F
in Abbildung 4.13). Die Differenz-Signale sind positiv, was auf eine aufrechte Orientierung
der aromatischen Ringe und damit des gesamten Moleküls auf der Oberfläche schließen lässt.
4.2.3 Zusammenfassung
Die STM-Untersuchungen zeigen die Bildung einer hochgeordneten Monolage von 10 auf
Au(111)/Glimmer nach Vorbeschichtung der Oberfläche mit PTCDA, das eine planare
Adsorption der Moleküle verhindert. Die Monolage weist eine 3 × 5-Elementarzelle auf, in
>< 211 -Richtung findet π-π-Stapelung der aromatischen Systeme der Moleküle statt.
Zusätzlich wurde mit Hilfe der spektroskopischen Methoden das Vorhandensein der intakten
Moleküle auf der Oberfläche anhand der Elementarzusammensetzung nachgewiesen und die
Bindung in Form einer Thiolat-Bindung an die Goldoberfläche bestätigt. Verbindung 10
erfüllt damit die Anforderungen des Synthesekonzepts aus Kapitel 2.
Kontaktwinkelmessungen lassen anhand der ausgeprägten Hydrophobie der Oberfläche auf
eine trans,trans-Konfiguration der Pyrazolringe bezüglich des Pyridinrings schließen, was
einen schrittweisen Aufbau von Komplexen auf der Oberfläche potentiell erschwert..
4.3 SAM von Ligand 6 auf Au(111)
4.3.1 STM
Monolagen von Verbindung 6 wurden zunächst durch Abscheidung aus Lösungen der
Substanz in EtOH oder DMF präpariert. Die STM-Bilder der Oberflächen zeigten jedoch
keine erkennbaren geordneten Bereiche (Abbildung 4.14). Erkennbar sind „vacancy islands“
auf der Oberfläche. Es ist also davon auszugehen, dass sich geordnete Bereiche auf der
Oberfläche gebildet haben. Allerdings ist die Qualität der Filme schlecht, was sich immer
auch auf die Qualität der STM-Spitze und damit die Auflösung auswirkt.

4 Self-assembled Monolayers 136
Abbildung 4.14. STM-Bild einer Monolage von 6 auf Au(111)/Glimmer, präpariert aus
DMF, 90 °C, 48 h.
Bessere Ergebnisse erbrachte eine alternative Beschichtungsprozedur durch Abscheidung der
Moleküle aus der Gasphase. Dabei wurde eine offene Schale mit Substanz 6 in Pulverform
neben eine Au(111)/Glimmer-Oberfläche in ein verschlossenes Gefäß gestellt. Das Gefäß
wurde dann in einem Ofen 12 h auf 120 °C erhitzt.
Abbildung 4.15. STM-Bilder in unterschiedlicher Vergrößerung einer Monolage von 6 auf
Au(111)/Glimmer, präpariert durch Gasphasenabscheidung (120 °C, 12 h).
In den STM-Bildern in Abbildung 4.15 sind streifenförmige Bereiche unterschiedlicher Größe
zu erkennen, die über die gesamte Oberfläche verteilt sind. Darüberhinaus sind an den
Schnittpunkten der geordneten Domänen Bereiche vorhanden, in denen Moleküle ungeordnet
vorzuliegen scheinen, sowie vacancy islands in Form von Vertiefungen. Die Streifen bestehen
aus Reihen von Erhebungen im Abstand von ca. 0.5 nm. Der Abstand zwischen zwei Streifen
beträgt ~ 1.5 nm. Die Moleküle ordnen sich möglicherweise analog zu den SAMs von
Verbindung 10 (s. o.) aufrecht auf der Oberfläche an, wobei die Bpp-Einheit die
Oberflächenstruktur bildet. Die aromatischen Ebenen wären wie in den SAMs von 10
20 nm
50 nm 20 nm 2 nm
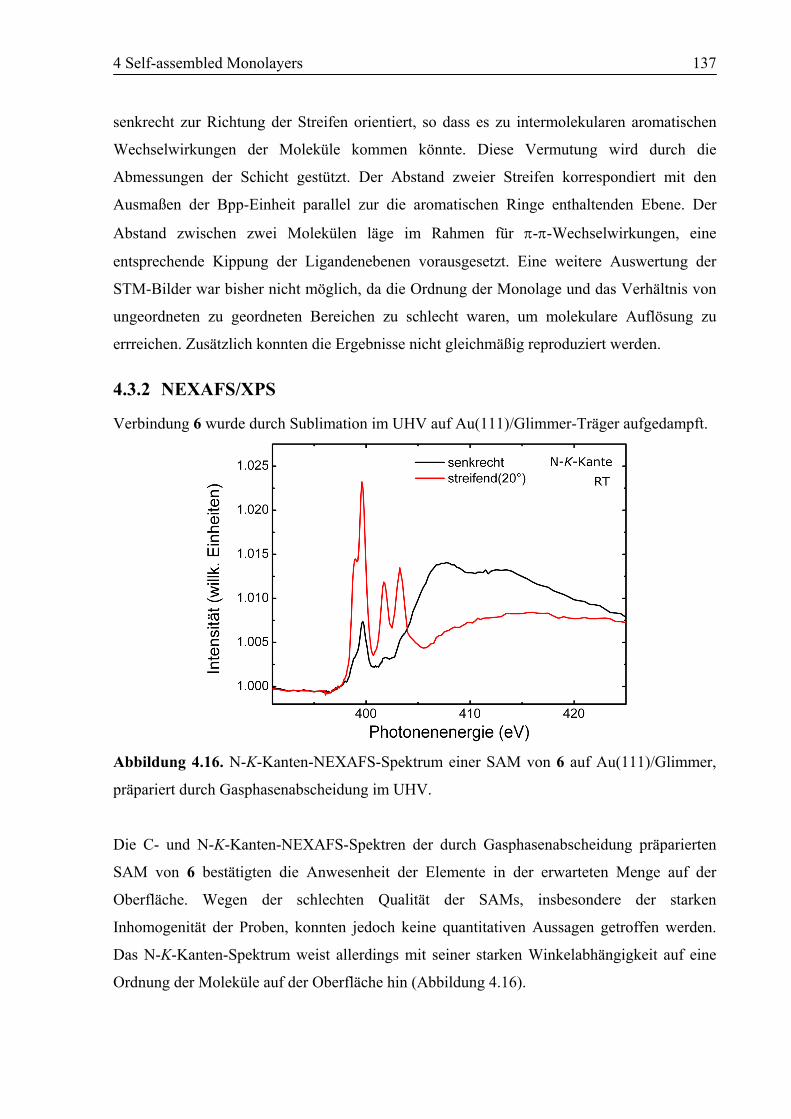
4 Self-assembled Monolayers 137
senkrecht zur Richtung der Streifen orientiert, so dass es zu intermolekularen aromatischen
Wechselwirkungen der Moleküle kommen könnte. Diese Vermutung wird durch die
Abmessungen der Schicht gestützt. Der Abstand zweier Streifen korrespondiert mit den
Ausmaßen der Bpp-Einheit parallel zur die aromatischen Ringe enthaltenden Ebene. Der
Abstand zwischen zwei Molekülen läge im Rahmen für π-π-Wechselwirkungen, eine
entsprechende Kippung der Ligandenebenen vorausgesetzt. Eine weitere Auswertung der
STM-Bilder war bisher nicht möglich, da die Ordnung der Monolage und das Verhältnis von
ungeordneten zu geordneten Bereichen zu schlecht waren, um molekulare Auflösung zu
errreichen. Zusätzlich konnten die Ergebnisse nicht gleichmäßig reproduziert werden.
4.3.2 NEXAFS/XPS
Verbindung 6 wurde durch Sublimation im UHV auf Au(111)/Glimmer-Träger aufgedampft.
Abbildung 4.16. N-K-Kanten-NEXAFS-Spektrum einer SAM von 6 auf Au(111)/Glimmer,
präpariert durch Gasphasenabscheidung im UHV.
Die C- und N-K-Kanten-NEXAFS-Spektren der durch Gasphasenabscheidung präparierten
SAM von 6 bestätigten die Anwesenheit der Elemente in der erwarteten Menge auf der
Oberfläche. Wegen der schlechten Qualität der SAMs, insbesondere der starken
Inhomogenität der Proben, konnten jedoch keine quantitativen Aussagen getroffen werden.
Das N-K-Kanten-Spektrum weist allerdings mit seiner starken Winkelabhängigkeit auf eine
Ordnung der Moleküle auf der Oberfläche hin (Abbildung 4.16).
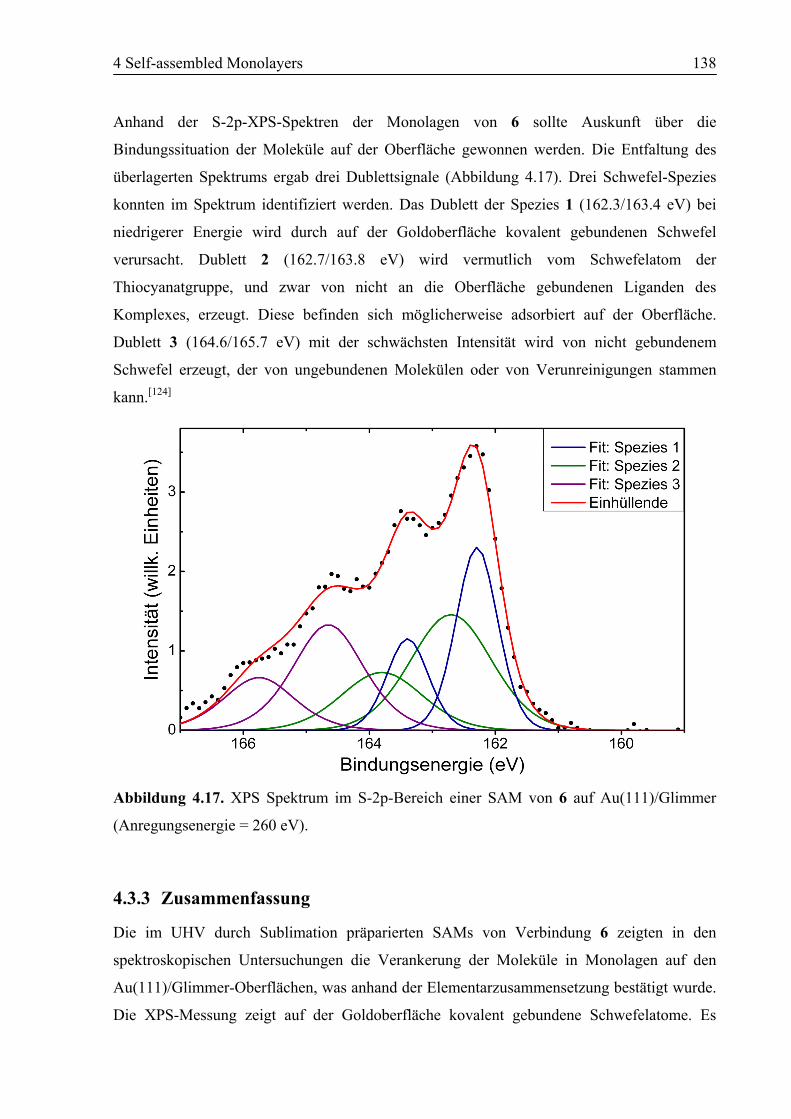
4 Self-assembled Monolayers 138
Anhand der S-2p-XPS-Spektren der Monolagen von 6 sollte Auskunft über die
Bindungssituation der Moleküle auf der Oberfläche gewonnen werden. Die Entfaltung des
überlagerten Spektrums ergab drei Dublettsignale (Abbildung 4.17). Drei Schwefel-Spezies
konnten im Spektrum identifiziert werden. Das Dublett der Spezies 1 (162.3/163.4 eV) bei
niedrigerer Energie wird durch auf der Goldoberfläche kovalent gebundenen Schwefel
verursacht. Dublett 2 (162.7/163.8 eV) wird vermutlich vom Schwefelatom der
Thiocyanatgruppe, und zwar von nicht an die Oberfläche gebundenen Liganden des
Komplexes, erzeugt. Diese befinden sich möglicherweise adsorbiert auf der Oberfläche.
Dublett 3 (164.6/165.7 eV) mit der schwächsten Intensität wird von nicht gebundenem
Schwefel erzeugt, der von ungebundenen Molekülen oder von Verunreinigungen stammen
kann.[124]
Abbildung 4.17. XPS Spektrum im S-2p-Bereich einer SAM von 6 auf Au(111)/Glimmer
(Anregungsenergie = 260 eV).
4.3.3 Zusammenfassung
Die im UHV durch Sublimation präparierten SAMs von Verbindung 6 zeigten in den
spektroskopischen Untersuchungen die Verankerung der Moleküle in Monolagen auf den
Au(111)/Glimmer-Oberflächen, was anhand der Elementarzusammensetzung bestätigt wurde.
Die XPS-Messung zeigt auf der Goldoberfläche kovalent gebundene Schwefelatome. Es
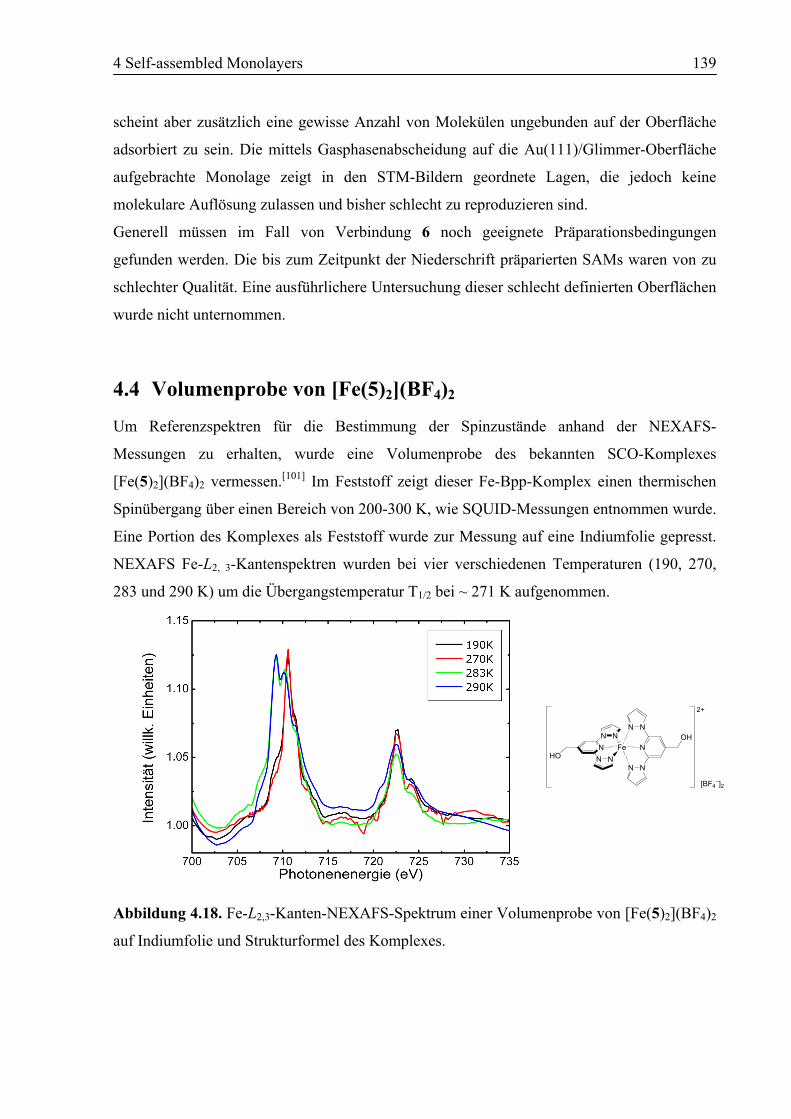
4 Self-assembled Monolayers 139
scheint aber zusätzlich eine gewisse Anzahl von Molekülen ungebunden auf der Oberfläche
adsorbiert zu sein. Die mittels Gasphasenabscheidung auf die Au(111)/Glimmer-Oberfläche
aufgebrachte Monolage zeigt in den STM-Bildern geordnete Lagen, die jedoch keine
molekulare Auflösung zulassen und bisher schlecht zu reproduzieren sind.
Generell müssen im Fall von Verbindung 6 noch geeignete Präparationsbedingungen
gefunden werden. Die bis zum Zeitpunkt der Niederschrift präparierten SAMs waren von zu
schlechter Qualität. Eine ausführlichere Untersuchung dieser schlecht definierten Oberflächen
wurde nicht unternommen.
4.4 Volumenprobe von [Fe(5)2](BF4)2
Um Referenzspektren für die Bestimmung der Spinzustände anhand der NEXAFS-
Messungen zu erhalten, wurde eine Volumenprobe des bekannten SCO-Komplexes
[Fe(5)2](BF4)2 vermessen.[101] Im Feststoff zeigt dieser Fe-Bpp-Komplex einen thermischen
Spinübergang über einen Bereich von 200-300 K, wie SQUID-Messungen entnommen wurde.
Eine Portion des Komplexes als Feststoff wurde zur Messung auf eine Indiumfolie gepresst.
NEXAFS Fe-L2, 3-Kantenspektren wurden bei vier verschiedenen Temperaturen (190, 270,
283 und 290 K) um die Übergangstemperatur T1/2 bei ~ 271 K aufgenommen.
N
NN
N
NN
N
N
N
N
FeHO
OH
2+
[BF4–]2
Abbildung 4.18. Fe-L2,3-Kanten-NEXAFS-Spektrum einer Volumenprobe von [Fe(5)2](BF4)2
auf Indiumfolie und Strukturformel des Komplexes.
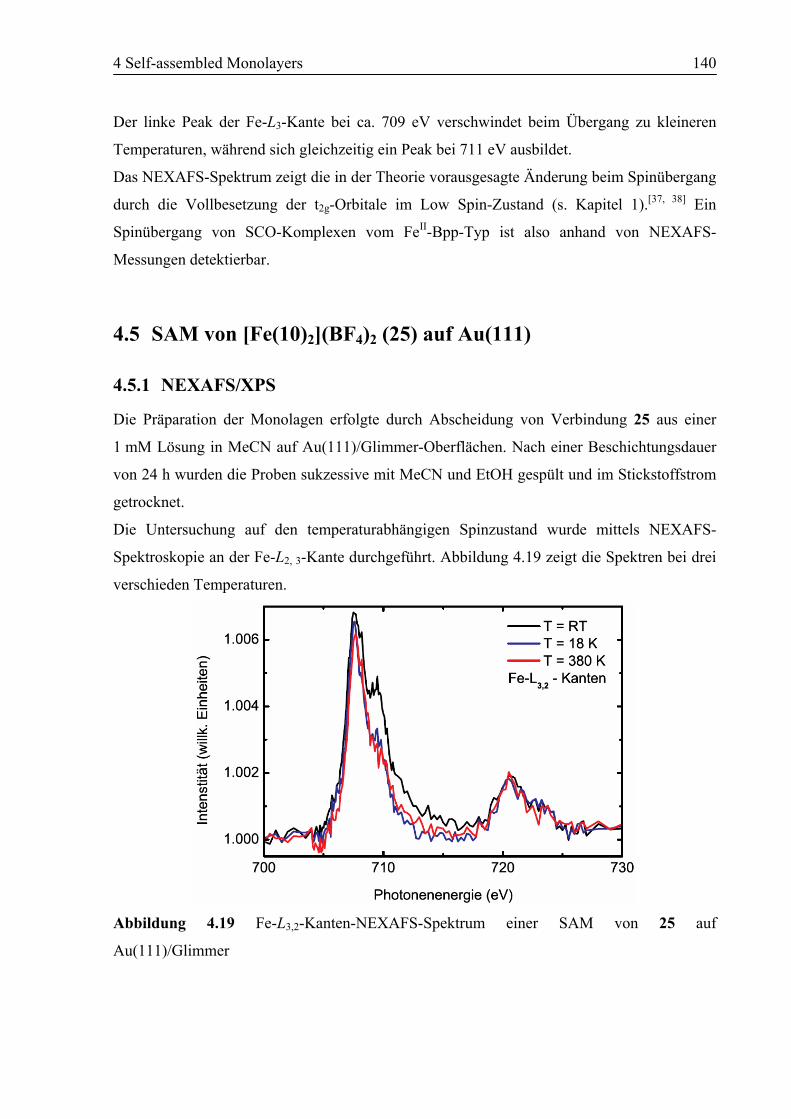
4 Self-assembled Monolayers 140
Der linke Peak der Fe-L3-Kante bei ca. 709 eV verschwindet beim Übergang zu kleineren
Temperaturen, während sich gleichzeitig ein Peak bei 711 eV ausbildet.
Das NEXAFS-Spektrum zeigt die in der Theorie vorausgesagte Änderung beim Spinübergang
durch die Vollbesetzung der t2g-Orbitale im Low Spin-Zustand (s. Kapitel 1).[37, 38] Ein
Spinübergang von SCO-Komplexen vom FeII-Bpp-Typ ist also anhand von NEXAFS-
Messungen detektierbar.
4.5 SAM von [Fe(10)2](BF4)2 (25) auf Au(111)
4.5.1 NEXAFS/XPS
Die Präparation der Monolagen erfolgte durch Abscheidung von Verbindung 25 aus einer
1 mM Lösung in MeCN auf Au(111)/Glimmer-Oberflächen. Nach einer Beschichtungsdauer
von 24 h wurden die Proben sukzessive mit MeCN und EtOH gespült und im Stickstoffstrom
getrocknet.
Die Untersuchung auf den temperaturabhängigen Spinzustand wurde mittels NEXAFS-
Spektroskopie an der Fe-L2, 3-Kante durchgeführt. Abbildung 4.19 zeigt die Spektren bei drei
verschieden Temperaturen.
Abbildung 4.19 Fe-L3,2-Kanten-NEXAFS-Spektrum einer SAM von 25 auf
Au(111)/Glimmer
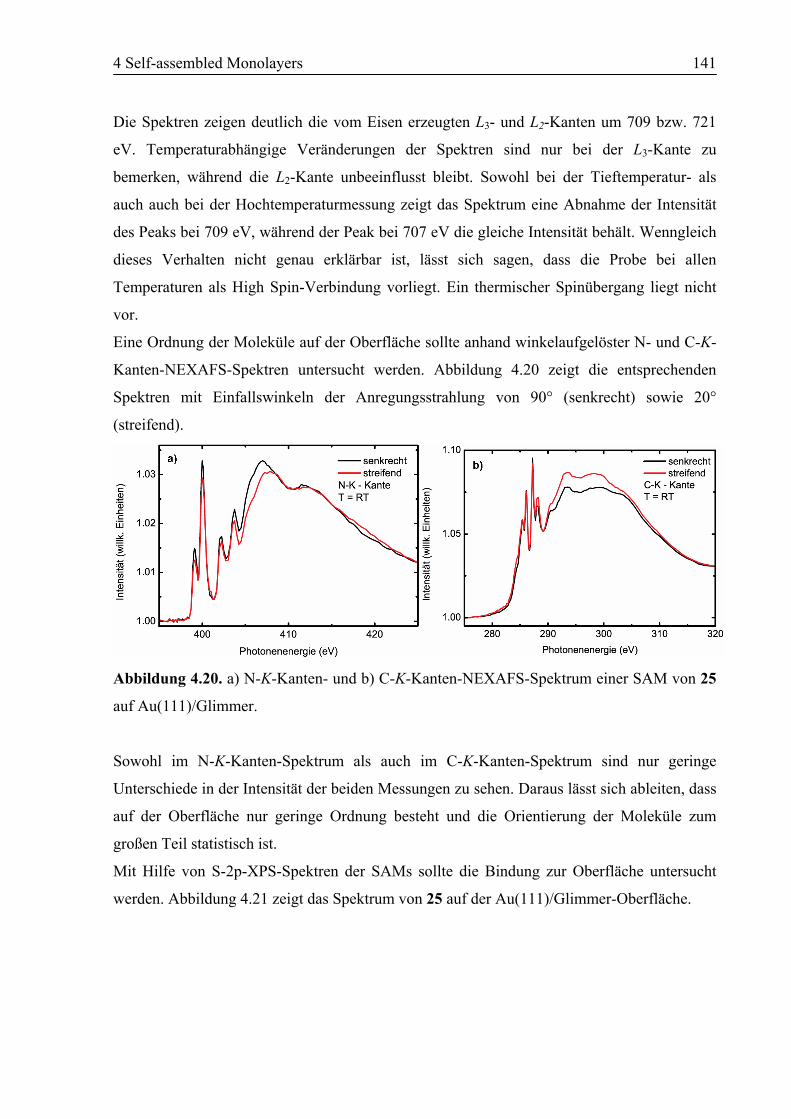
4 Self-assembled Monolayers 141
Die Spektren zeigen deutlich die vom Eisen erzeugten L3- und L2-Kanten um 709 bzw. 721
eV. Temperaturabhängige Veränderungen der Spektren sind nur bei der L3-Kante zu
bemerken, während die L2-Kante unbeeinflusst bleibt. Sowohl bei der Tieftemperatur- als
auch auch bei der Hochtemperaturmessung zeigt das Spektrum eine Abnahme der Intensität
des Peaks bei 709 eV, während der Peak bei 707 eV die gleiche Intensität behält. Wenngleich
dieses Verhalten nicht genau erklärbar ist, lässt sich sagen, dass die Probe bei allen
Temperaturen als High Spin-Verbindung vorliegt. Ein thermischer Spinübergang liegt nicht
vor.
Eine Ordnung der Moleküle auf der Oberfläche sollte anhand winkelaufgelöster N- und C-K-
Kanten-NEXAFS-Spektren untersucht werden. Abbildung 4.20 zeigt die entsprechenden
Spektren mit Einfallswinkeln der Anregungsstrahlung von 90° (senkrecht) sowie 20°
(streifend).
Abbildung 4.20. a) N-K-Kanten- und b) C-K-Kanten-NEXAFS-Spektrum einer SAM von 25
auf Au(111)/Glimmer.
Sowohl im N-K-Kanten-Spektrum als auch im C-K-Kanten-Spektrum sind nur geringe
Unterschiede in der Intensität der beiden Messungen zu sehen. Daraus lässt sich ableiten, dass
auf der Oberfläche nur geringe Ordnung besteht und die Orientierung der Moleküle zum
großen Teil statistisch ist.
Mit Hilfe von S-2p-XPS-Spektren der SAMs sollte die Bindung zur Oberfläche untersucht
werden. Abbildung 4.21 zeigt das Spektrum von 25 auf der Au(111)/Glimmer-Oberfläche.
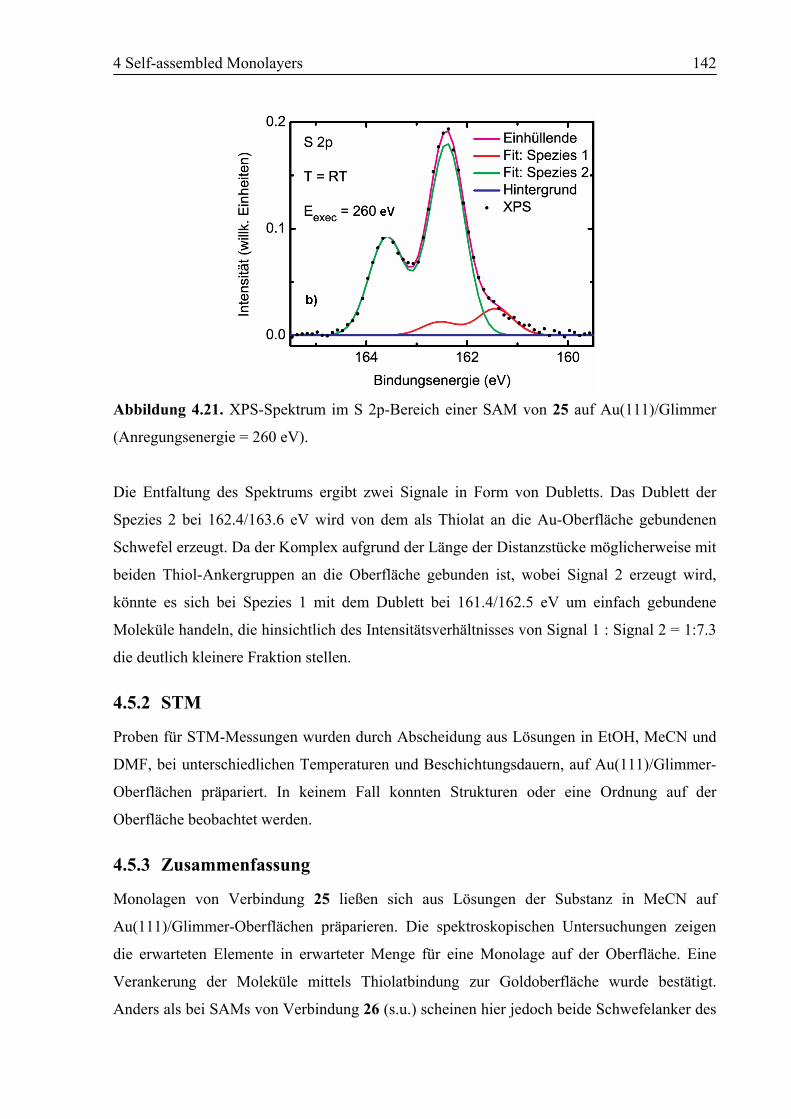
4 Self-assembled Monolayers 142
Abbildung 4.21. XPS-Spektrum im S 2p-Bereich einer SAM von 25 auf Au(111)/Glimmer
(Anregungsenergie = 260 eV).
Die Entfaltung des Spektrums ergibt zwei Signale in Form von Dubletts. Das Dublett der
Spezies 2 bei 162.4/163.6 eV wird von dem als Thiolat an die Au-Oberfläche gebundenen
Schwefel erzeugt. Da der Komplex aufgrund der Länge der Distanzstücke möglicherweise mit
beiden Thiol-Ankergruppen an die Oberfläche gebunden ist, wobei Signal 2 erzeugt wird,
könnte es sich bei Spezies 1 mit dem Dublett bei 161.4/162.5 eV um einfach gebundene
Moleküle handeln, die hinsichtlich des Intensitätsverhältnisses von Signal 1 : Signal 2 = 1:7.3
die deutlich kleinere Fraktion stellen.
4.5.2 STM
Proben für STM-Messungen wurden durch Abscheidung aus Lösungen in EtOH, MeCN und
DMF, bei unterschiedlichen Temperaturen und Beschichtungsdauern, auf Au(111)/Glimmer-
Oberflächen präpariert. In keinem Fall konnten Strukturen oder eine Ordnung auf der
Oberfläche beobachtet werden.
4.5.3 Zusammenfassung
Monolagen von Verbindung 25 ließen sich aus Lösungen der Substanz in MeCN auf
Au(111)/Glimmer-Oberflächen präparieren. Die spektroskopischen Untersuchungen zeigen
die erwarteten Elemente in erwarteter Menge für eine Monolage auf der Oberfläche. Eine
Verankerung der Moleküle mittels Thiolatbindung zur Goldoberfläche wurde bestätigt.
Anders als bei SAMs von Verbindung 26 (s.u.) scheinen hier jedoch beide Schwefelanker des
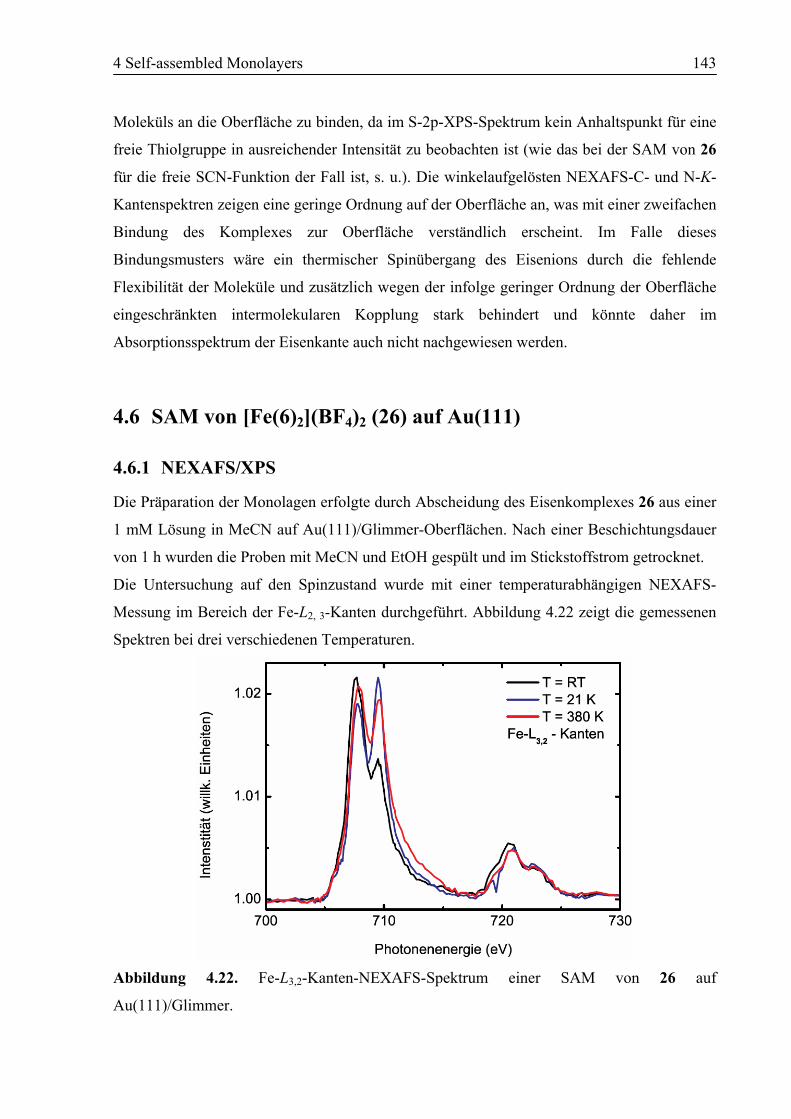
4 Self-assembled Monolayers 143
Moleküls an die Oberfläche zu binden, da im S-2p-XPS-Spektrum kein Anhaltspunkt für eine
freie Thiolgruppe in ausreichender Intensität zu beobachten ist (wie das bei der SAM von 26
für die freie SCN-Funktion der Fall ist, s. u.). Die winkelaufgelösten NEXAFS-C- und N-K-
Kantenspektren zeigen eine geringe Ordnung auf der Oberfläche an, was mit einer zweifachen
Bindung des Komplexes zur Oberfläche verständlich erscheint. Im Falle dieses
Bindungsmusters wäre ein thermischer Spinübergang des Eisenions durch die fehlende
Flexibilität der Moleküle und zusätzlich wegen der infolge geringer Ordnung der Oberfläche
eingeschränkten intermolekularen Kopplung stark behindert und könnte daher im
Absorptionsspektrum der Eisenkante auch nicht nachgewiesen werden.
4.6 SAM von [Fe(6)2](BF4)2 (26) auf Au(111)
4.6.1 NEXAFS/XPS
Die Präparation der Monolagen erfolgte durch Abscheidung des Eisenkomplexes 26 aus einer
1 mM Lösung in MeCN auf Au(111)/Glimmer-Oberflächen. Nach einer Beschichtungsdauer
von 1 h wurden die Proben mit MeCN und EtOH gespült und im Stickstoffstrom getrocknet.
Die Untersuchung auf den Spinzustand wurde mit einer temperaturabhängigen NEXAFS-
Messung im Bereich der Fe-L2, 3-Kanten durchgeführt. Abbildung 4.22 zeigt die gemessenen
Spektren bei drei verschiedenen Temperaturen.
Abbildung 4.22. Fe-L3,2-Kanten-NEXAFS-Spektrum einer SAM von 26 auf
Au(111)/Glimmer.
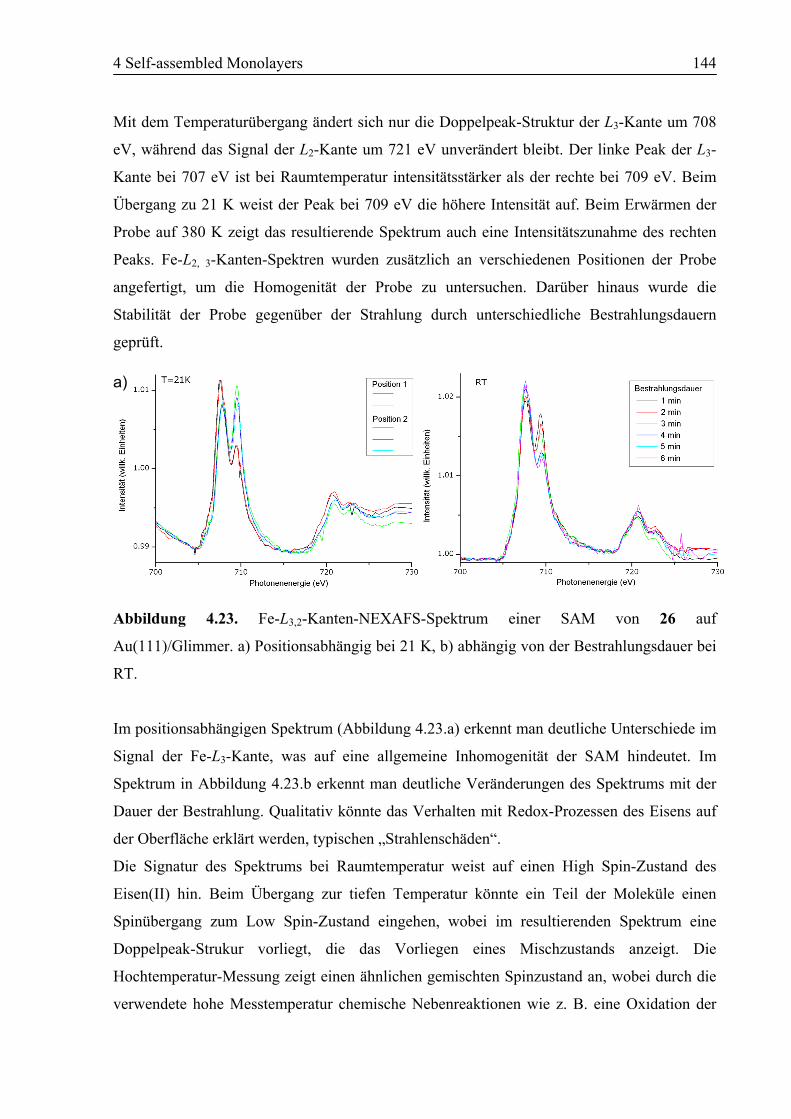
4 Self-assembled Monolayers 144
Mit dem Temperaturübergang ändert sich nur die Doppelpeak-Struktur der L3-Kante um 708
eV, während das Signal der L2-Kante um 721 eV unverändert bleibt. Der linke Peak der L3-
Kante bei 707 eV ist bei Raumtemperatur intensitätsstärker als der rechte bei 709 eV. Beim
Übergang zu 21 K weist der Peak bei 709 eV die höhere Intensität auf. Beim Erwärmen der
Probe auf 380 K zeigt das resultierende Spektrum auch eine Intensitätszunahme des rechten
Peaks. Fe-L2, 3-Kanten-Spektren wurden zusätzlich an verschiedenen Positionen der Probe
angefertigt, um die Homogenität der Probe zu untersuchen. Darüber hinaus wurde die
Stabilität der Probe gegenüber der Strahlung durch unterschiedliche Bestrahlungsdauern
geprüft.
Abbildung 4.23. Fe-L3,2-Kanten-NEXAFS-Spektrum einer SAM von 26 auf
Au(111)/Glimmer. a) Positionsabhängig bei 21 K, b) abhängig von der Bestrahlungsdauer bei
RT.
Im positionsabhängigen Spektrum (Abbildung 4.23.a) erkennt man deutliche Unterschiede im
Signal der Fe-L3-Kante, was auf eine allgemeine Inhomogenität der SAM hindeutet. Im
Spektrum in Abbildung 4.23.b erkennt man deutliche Veränderungen des Spektrums mit der
Dauer der Bestrahlung. Qualitativ könnte das Verhalten mit Redox-Prozessen des Eisens auf
der Oberfläche erklärt werden, typischen „Strahlenschäden“.
Die Signatur des Spektrums bei Raumtemperatur weist auf einen High Spin-Zustand des
Eisen(II) hin. Beim Übergang zur tiefen Temperatur könnte ein Teil der Moleküle einen
Spinübergang zum Low Spin-Zustand eingehen, wobei im resultierenden Spektrum eine
Doppelpeak-Strukur vorliegt, die das Vorliegen eines Mischzustands anzeigt. Die
Hochtemperatur-Messung zeigt einen ähnlichen gemischten Spinzustand an, wobei durch die
verwendete hohe Messtemperatur chemische Nebenreaktionen wie z. B. eine Oxidation der
a)
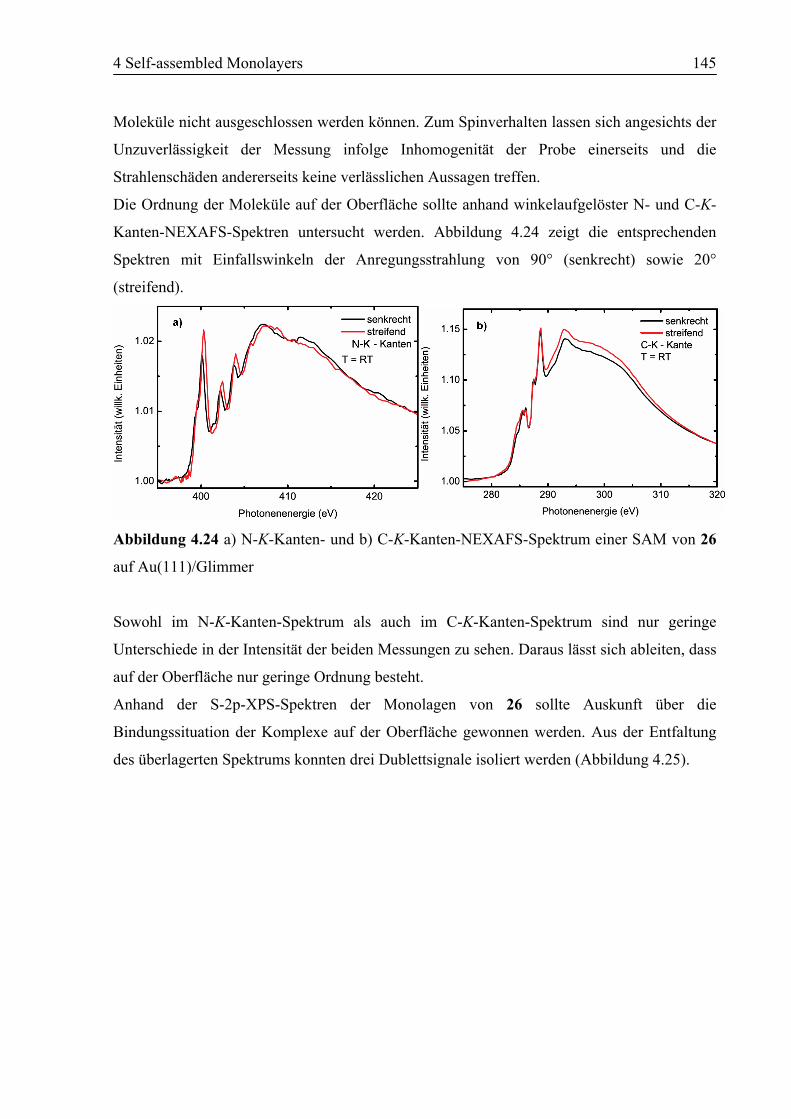
4 Self-assembled Monolayers 145
Moleküle nicht ausgeschlossen werden können. Zum Spinverhalten lassen sich angesichts der
Unzuverlässigkeit der Messung infolge Inhomogenität der Probe einerseits und die
Strahlenschäden andererseits keine verlässlichen Aussagen treffen.
Die Ordnung der Moleküle auf der Oberfläche sollte anhand winkelaufgelöster N- und C-K-
Kanten-NEXAFS-Spektren untersucht werden. Abbildung 4.24 zeigt die entsprechenden
Spektren mit Einfallswinkeln der Anregungsstrahlung von 90° (senkrecht) sowie 20°
(streifend).
Abbildung 4.24 a) N-K-Kanten- und b) C-K-Kanten-NEXAFS-Spektrum einer SAM von 26
auf Au(111)/Glimmer
Sowohl im N-K-Kanten-Spektrum als auch im C-K-Kanten-Spektrum sind nur geringe
Unterschiede in der Intensität der beiden Messungen zu sehen. Daraus lässt sich ableiten, dass
auf der Oberfläche nur geringe Ordnung besteht.
Anhand der S-2p-XPS-Spektren der Monolagen von 26 sollte Auskunft über die
Bindungssituation der Komplexe auf der Oberfläche gewonnen werden. Aus der Entfaltung
des überlagerten Spektrums konnten drei Dublettsignale isoliert werden (Abbildung 4.25).
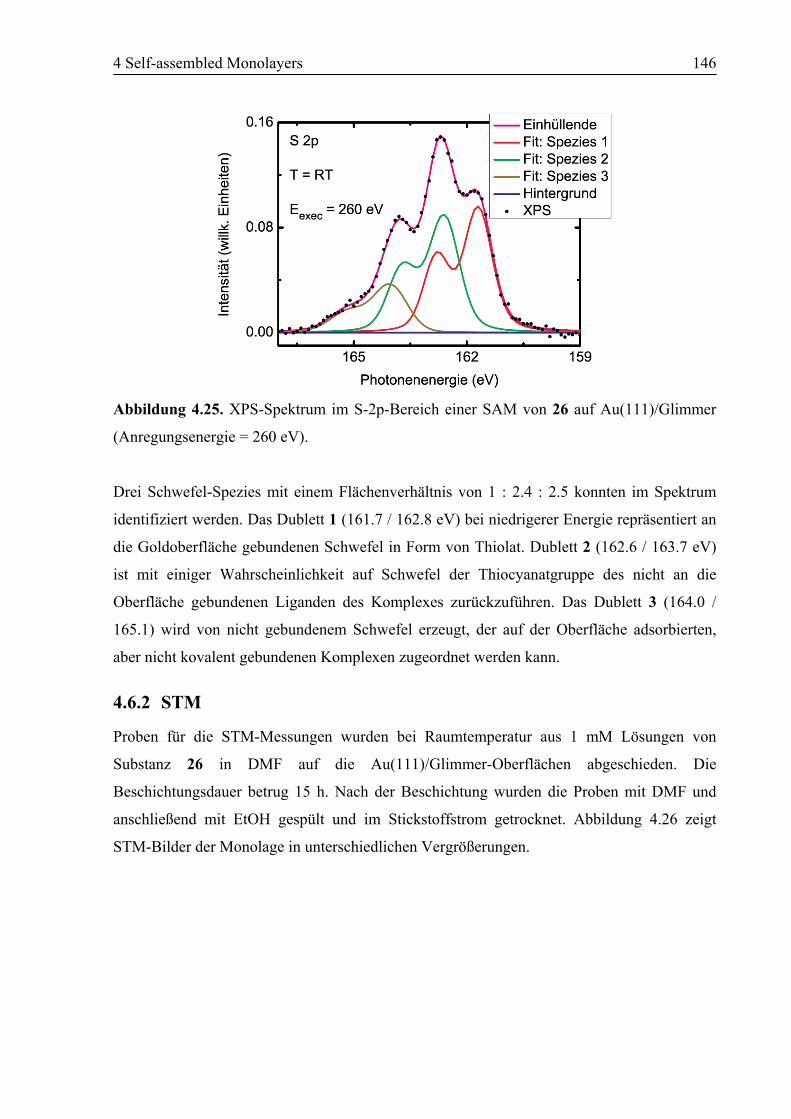
4 Self-assembled Monolayers 146
Abbildung 4.25. XPS-Spektrum im S-2p-Bereich einer SAM von 26 auf Au(111)/Glimmer
(Anregungsenergie = 260 eV).
Drei Schwefel-Spezies mit einem Flächenverhältnis von 1 : 2.4 : 2.5 konnten im Spektrum
identifiziert werden. Das Dublett 1 (161.7 / 162.8 eV) bei niedrigerer Energie repräsentiert an
die Goldoberfläche gebundenen Schwefel in Form von Thiolat. Dublett 2 (162.6 / 163.7 eV)
ist mit einiger Wahrscheinlichkeit auf Schwefel der Thiocyanatgruppe des nicht an die
Oberfläche gebundenen Liganden des Komplexes zurückzuführen. Das Dublett 3 (164.0 /
165.1) wird von nicht gebundenem Schwefel erzeugt, der auf der Oberfläche adsorbierten,
aber nicht kovalent gebundenen Komplexen zugeordnet werden kann.
4.6.2 STM
Proben für die STM-Messungen wurden bei Raumtemperatur aus 1 mM Lösungen von
Substanz 26 in DMF auf die Au(111)/Glimmer-Oberflächen abgeschieden. Die
Beschichtungsdauer betrug 15 h. Nach der Beschichtung wurden die Proben mit DMF und
anschließend mit EtOH gespült und im Stickstoffstrom getrocknet. Abbildung 4.26 zeigt
STM-Bilder der Monolage in unterschiedlichen Vergrößerungen.

4 Self-assembled Monolayers 147
Abbildung 4.26. STM-Bilder einer SAM von 26 auf Au(111)/Glimmer, präpariert aus
Lösung in DMF, RT, 15 h.
Die terassenförmige Struktur der Au(111)-Oberfläche ist gut zu erkennen. Regelmäßige
Strukturen finden sich nicht, und eine Auflösung auf molekularer Ebene ist nicht möglich.
Deutlich sind vacancy islands zu erkennen, die in großer Zahl über die Oberfläche verteilt
vorliegen. Dies ist ein Hinweis auf eine Ordnung der Moleküle, die jedoch im STM-Bild
bisher nicht aufgelöst werden konnte. Zumindest kann als sicher gelten, dass Moleküle von 26
über die gesamte Oberfläche verteilt verankert sind.
4.6.3 Zusammenfassung
SAMs von Verbindung 26 ließen sich aus einer Lösung des Komplexes in DMF auf die
Au(111)-Oberfläche aufbringen. Die spektroskopischen Untersuchungen zeigen die
erwarteten Elemente in einer Menge, die einer Monolage entspricht. Der S-2p-XPS-Messung
konnte entnommen werden, dass der Komplex mit einer Ankergruppe in Form einer
Thiolatfunktion an die Goldoberfläche bindet, während die zweite Thiocyanatfunktion
ungebunden bleibt, was im Kontrast zum doppelten Bindungsmuster in den SAMs von
Verbindung 25 steht (Abbildung 4.27). Eine Ordnung auf der Oberfläche ist den NEXAFS-C-
und N-K-Kanten-Spektren nicht zu entnehmen. Das Spektrum der Fe-L3-Kante zeigt
temperaturabhängige Veränderung, die als Low Spin- → High Spin-Übergang beim Wechsel
zu höheren Temperaturen interpretiert werden könnte. Allerdings ist die durchgeführte
Messung nicht geeignet, endgültige Aussagen zum Spinverhalten zu treffen, da wegen der
Inhomogenität der Schicht und durch die Röntgenstrahlung erzeugter Strahlenschäden keine
genaue Auswertung der relevanten Fe-L3-Kantenspektren möglich ist.
40 nm 20 nm 10 nm
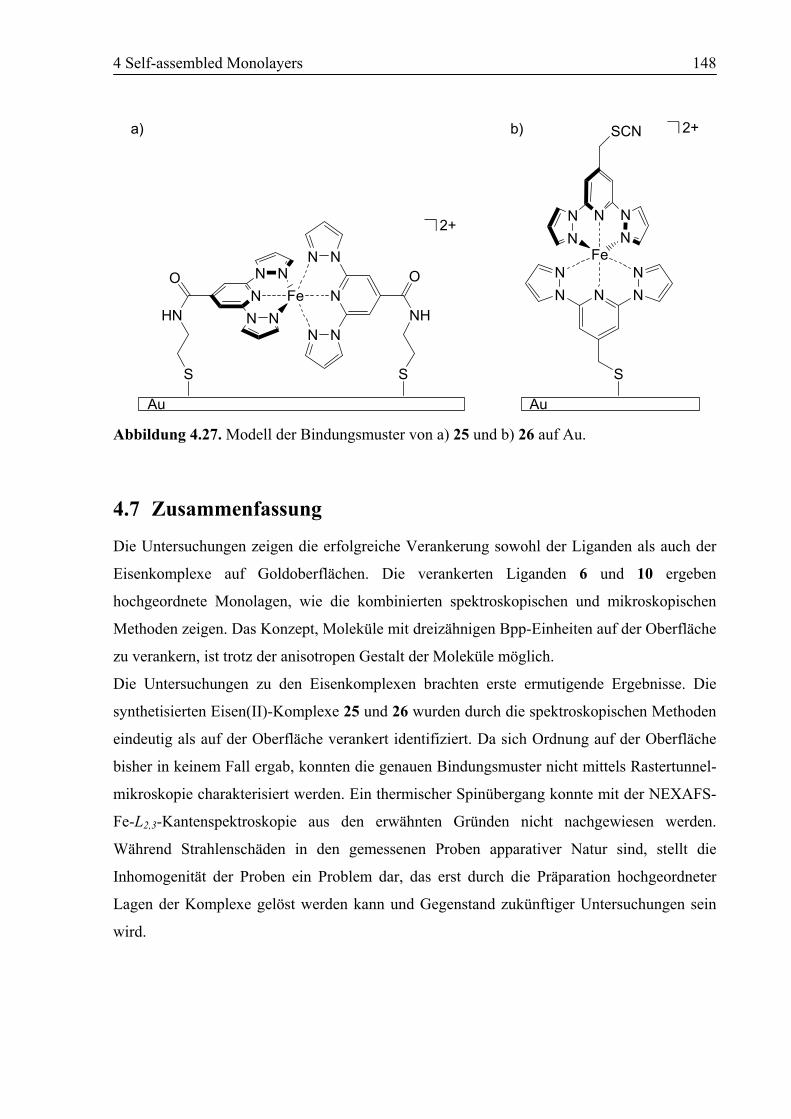
4 Self-assembled Monolayers 148
N
NN
N
NN
N
N
N
N
FeNH
O
S
HN
O
S
NNNN N
NNNN N
Fe
S
SCN
Au Au
2+
2+a) b)
Abbildung 4.27. Modell der Bindungsmuster von a) 25 und b) 26 auf Au.
4.7 Zusammenfassung
Die Untersuchungen zeigen die erfolgreiche Verankerung sowohl der Liganden als auch der
Eisenkomplexe auf Goldoberflächen. Die verankerten Liganden 6 und 10 ergeben
hochgeordnete Monolagen, wie die kombinierten spektroskopischen und mikroskopischen
Methoden zeigen. Das Konzept, Moleküle mit dreizähnigen Bpp-Einheiten auf der Oberfläche
zu verankern, ist trotz der anisotropen Gestalt der Moleküle möglich.
Die Untersuchungen zu den Eisenkomplexen brachten erste ermutigende Ergebnisse. Die
synthetisierten Eisen(II)-Komplexe 25 und 26 wurden durch die spektroskopischen Methoden
eindeutig als auf der Oberfläche verankert identifiziert. Da sich Ordnung auf der Oberfläche
bisher in keinem Fall ergab, konnten die genauen Bindungsmuster nicht mittels Rastertunnel-
mikroskopie charakterisiert werden. Ein thermischer Spinübergang konnte mit der NEXAFS-
Fe-L2,3-Kantenspektroskopie aus den erwähnten Gründen nicht nachgewiesen werden.
Während Strahlenschäden in den gemessenen Proben apparativer Natur sind, stellt die
Inhomogenität der Proben ein Problem dar, das erst durch die Präparation hochgeordneter
Lagen der Komplexe gelöst werden kann und Gegenstand zukünftiger Untersuchungen sein
wird.

4 Self-assembled Monolayers 149
4.8 Experimentalteil
Allgemeines: Folgende Chemikalien wurden kommerziell bezogen und ohne weitere
Reinigung verwendet: Ethanol, analytical reagent grade (Fisher Scientific); N,N-
Dimethylformamid (DMF), 99.9+ % HPLC grade (Aldrich); Acetonitril, 99.9%, H2O < 50
ppm, extra dry (Acros); Perylen-3,4,9,10-tetracarbonsäureanhydrid (PTCDA), >98 %
(Aldrich). Die Goldsubstrate (Georg Albert PVD, Heidelberg) bestanden aus epitaktischen
Goldlagen von 300 nm Dicke auf Glimmerträgern.
Folgende Geräte wurden zur Gewinnung der Daten verwendet:
STM-Messungen: Alle STM-Messungen wurden unter Benutzung eines PicoPlus-
Mikroskops der Firma Molecular Imaging an Luft bei Raumtemperatur ausgeführt. Die STM-
Bilder wurden im Konstant-Strom-Modus bei Tunnelströmen zwischen 10 und 100 pA und
einer Probenspannung zwischen +0.5 und +1.0 V durchgeführt.
NEXAFS/XPS-Messungen: SAMs von Verbindung 10: Die spektroskopischen Messungen
fanden am Synchrotron Speicherring MAX II am MAX-Lab in Lund, Schweden an der
„bending magnet beamline“ D1011 statt. Der Meßstand war mit einem SCIENTA SES200
Elektronenenergie-Analysator und einem „partial electron yield“ (PEY) -Detektor
ausgestattet. Die Messungen wurden unter UHV-Bedingungen bei einem Druck von < 1.5 ×
10–10 mBar durchgeführt. Für Details zur Auswertung der Messergebnisse siehe Ref[125].
SAMs von Verbindung 6, 25 und 26: Die spektroskopischen Messungen fanden am
Synchrotron-Speicherring BESSY II in Berlin an der „bending magnet beamline“ PM3 statt.
Der Messstand war mit einem SPECS PHOIBOS 100 Elektronenenergie-Analysator
ausgestattet. Die Messungen wurden unter UHV-Bedingungen bei einem Druck von < 4 × 10–
10 mbar durchgeführt.
Präparation der Oberflächen: Die Präparation der Oberflächen wird in den einzelnen
Kapiteln gesondert beschrieben.

5 Zusammenfassung und Ausblick 150
5 Zusammenfassung und Ausblick
5.1 Zusammenfassung
Die vorliegende Arbeit befasst sich mit der Nutzbarmachung von Eisen(II)-Spincrossover-
Komplexen in funktionalisierten Oberflächen. Ein Fernziel des Projekts ist der Einsatz
derartiger Oberflächen in der Datenspeicherung.
Behandelt werden die Synthese geeigneter Liganden bzw. entsprechender Eisen(II)-
Spincrossover-Komplexe und die Präparation selbstorganisierter Monolagen (self-assembled
monolayers, SAMs) sowohl der freien Liganden als auch der Komplexe auf Goldoberflächen.
In Kapitel 2 und 3 sind Synthese und Charakterisierung der neuen Liganden 6, 10, 12 und 14
bzw. der Eisen(II)-Komplexe 23, 24, 25 und 26 beschrieben. Kapitel 4 behandelt die Präpa-
ration und Charakterisierung von SAMs der Verbindungen 6, 10, 25 und 26 auf Au(111)-
Oberflächen. Kapitel 3 enthält zusätzlich Untersuchungen zum Einfluss von Festkörper-
effekten auf die Synthese von SCO-Verbindungen, und zwar am Beispiel der Polymorphie
eines Eisen(II)-Komplexes.
Für die Oberflächenverankerung geeignete Moleküle enthalten gemäß dem dieser Arbeit
zugrundegelegten Synthesekonzept folgende Strukturen:
− eine Endgruppe in Form des Eisen(II)-SCO-Komplexes,
− ein Distanzstück, über das sich das Ordnungsverhalten der Moleküle beeinflussen lässt,
− eine Ankergruppe mit spezifischer Affinität zur verwendeten Oberfläche (im konkreten
Fall schwefelhaltige funktionelle Gruppen für die Verankerung auf Goldsubstraten).
Endgruppe ([FeII-Bpp]-SCO-Komplex) Distanzstück (Spacer)
Ankergruppe
Abbildung 5.1. Modell eines oberflächenverankerten SCO-Komplexes.

5 Zusammenfassung und Ausblick 151
Die Liganden 6, 10, 12 und 14 (vgl. entsprechende Formelbilder) wurden diesem
Synthesekonzept entsprechend synthetisiert. Die Synthese von Ligand 22 wurde bis zur
vorletzten Stufe ausgearbeitet. In Anlehnung an in der Volumenphase bereits intensiv
untersuchte SCO-Komplexe wurden Komplexe von Derivaten des Bis(pyrazol-1-yl)pyridins
(Bpp) (vgl. Kap. 1, Abbildung 1.11) als SCO-Einheit gewählt.
Ligand 6 mit einer Thiocyanatfunktion als Ankergruppe wurde in einer sechsstufigen
Synthese aus käuflicher Citrazinsäure erhalten (Abbildung 5.2).
NHO OH
COOH
NCl Cl
COOH
NNNN N
COOH
NNNN N
COOR
NNNN N
OH
NNNN N
SCN
1 2
3 / 456
R = Me (3), Et (4) Abbildung 5.2. Synthese von Ligand 6.
SAMs der Verbindung wurden durch Gasphasenabscheidung auf Au(111)/Glimmer-
Oberflächen präpariert. Sowohl STM-Bilder als auch spektroskopische Untersuchungen
zeigen die erfolgreiche Verankerung der Moleküle als Monolage auf der Oberfläche. Die
Monolage enthält wohlgeordnete Bereiche; die Bereiche sind allerdings nicht gleichmäßig
über die gesamte Oberfläche verteilt, und die Ergebnisse sind schlecht reproduzierbar. Ein
möglicher Grund für die unzureichende Qualität der Monolagen ist die Adsorption einer
Mehrzahl der Moleküle über unerwünschte Wechselwirkungen ihres aromatischen Systems
mit der Oberfläche, wie aus XPS-Messungen hervorgeht.
Die Eisen(II)-Komplexe der Liganden 3, 4, 6 und 10 wurden jeweils aus zwei Äquivalenten
des Liganden und einem Äquivalent Eisen(II)-Tetrafluoroborat Hexahydrat synthetisiert
(Abbildung 5.3).
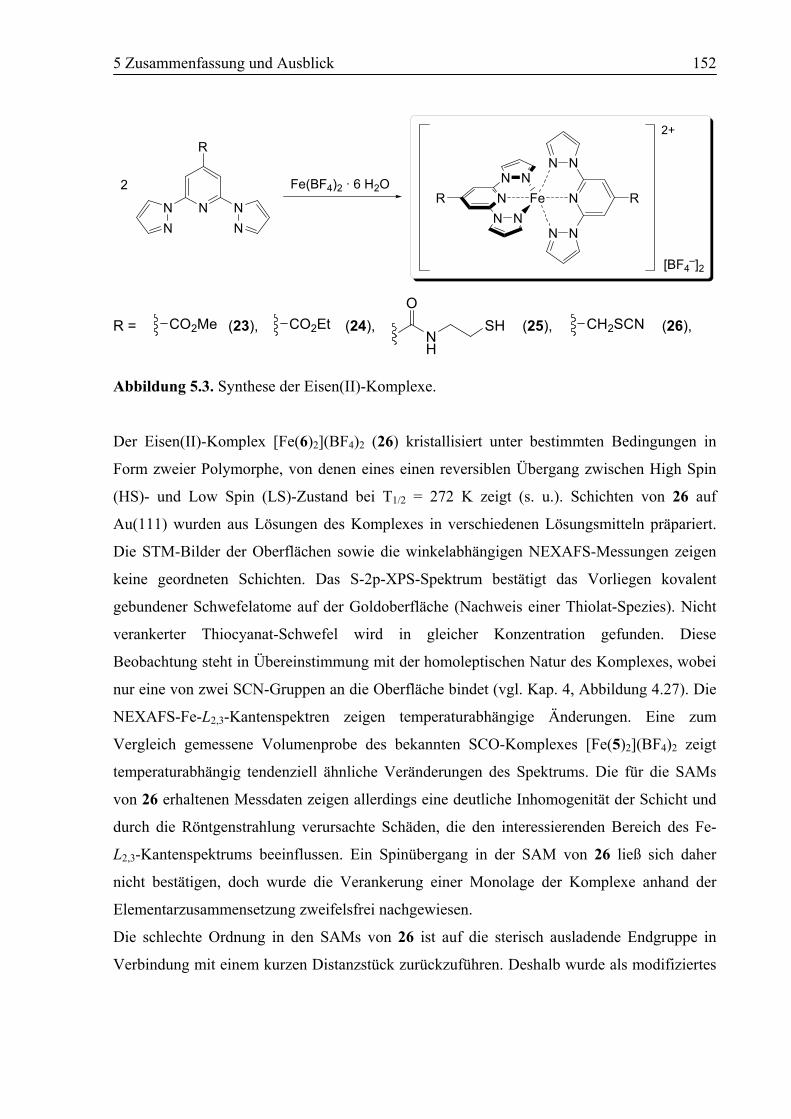
5 Zusammenfassung und Ausblick 152
NNNN N
Fe(BF4)2 · 6 H2O N
NN
N
NN
N
N
N
N
FeR R
2+
[BF4–]2
2
R
R = CO2Me (23), CO2Et (24),
NH
SHO
(25), CH2SCN (26),
Abbildung 5.3. Synthese der Eisen(II)-Komplexe.
Der Eisen(II)-Komplex [Fe(6)2](BF4)2 (26) kristallisiert unter bestimmten Bedingungen in
Form zweier Polymorphe, von denen eines einen reversiblen Übergang zwischen High Spin
(HS)- und Low Spin (LS)-Zustand bei T1/2 = 272 K zeigt (s. u.). Schichten von 26 auf
Au(111) wurden aus Lösungen des Komplexes in verschiedenen Lösungsmitteln präpariert.
Die STM-Bilder der Oberflächen sowie die winkelabhängigen NEXAFS-Messungen zeigen
keine geordneten Schichten. Das S-2p-XPS-Spektrum bestätigt das Vorliegen kovalent
gebundener Schwefelatome auf der Goldoberfläche (Nachweis einer Thiolat-Spezies). Nicht
verankerter Thiocyanat-Schwefel wird in gleicher Konzentration gefunden. Diese
Beobachtung steht in Übereinstimmung mit der homoleptischen Natur des Komplexes, wobei
nur eine von zwei SCN-Gruppen an die Oberfläche bindet (vgl. Kap. 4, Abbildung 4.27). Die
NEXAFS-Fe-L2,3-Kantenspektren zeigen temperaturabhängige Änderungen. Eine zum
Vergleich gemessene Volumenprobe des bekannten SCO-Komplexes [Fe(5)2](BF4)2 zeigt
temperaturabhängig tendenziell ähnliche Veränderungen des Spektrums. Die für die SAMs
von 26 erhaltenen Messdaten zeigen allerdings eine deutliche Inhomogenität der Schicht und
durch die Röntgenstrahlung verursachte Schäden, die den interessierenden Bereich des Fe-
L2,3-Kantenspektrums beeinflussen. Ein Spinübergang in der SAM von 26 ließ sich daher
nicht bestätigen, doch wurde die Verankerung einer Monolage der Komplexe anhand der
Elementarzusammensetzung zweifelsfrei nachgewiesen.
Die schlechte Ordnung in den SAMs von 26 ist auf die sterisch ausladende Endgruppe in
Verbindung mit einem kurzen Distanzstück zurückzuführen. Deshalb wurde als modifiziertes
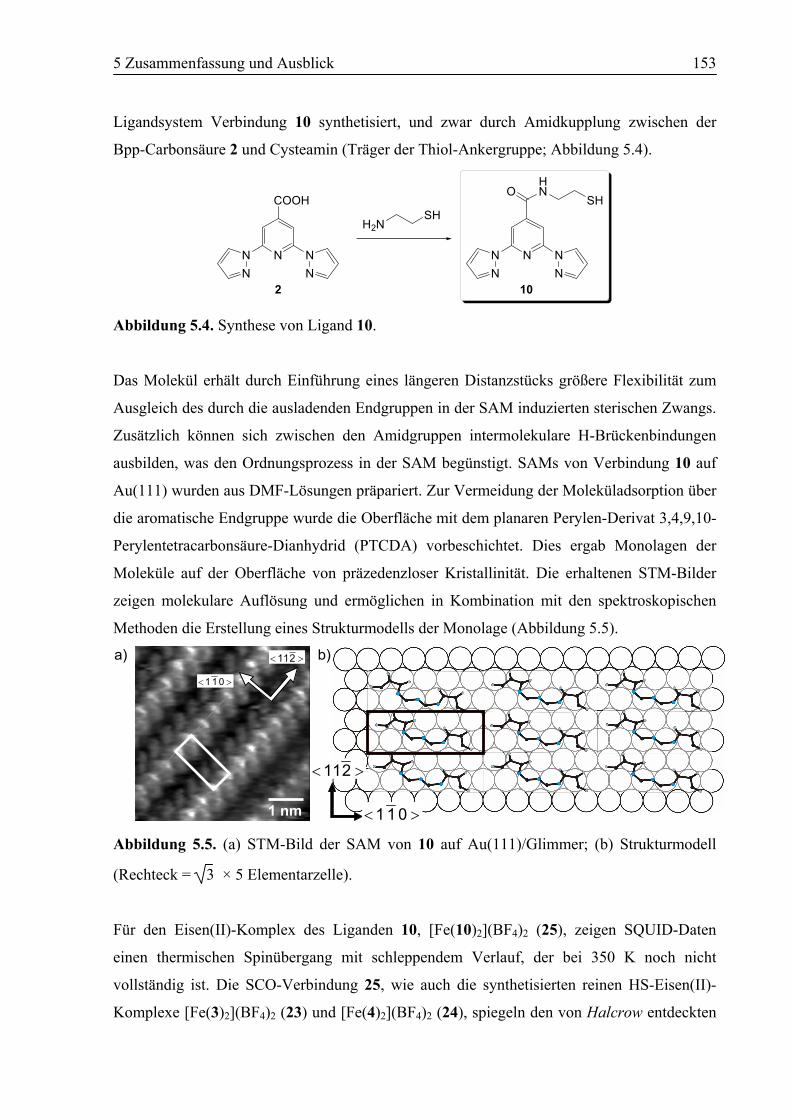
5 Zusammenfassung und Ausblick 153
Ligandsystem Verbindung 10 synthetisiert, und zwar durch Amidkupplung zwischen der
Bpp-Carbonsäure 2 und Cysteamin (Träger der Thiol-Ankergruppe; Abbildung 5.4).
NNNN N
COOH
NNNN N
HN
SHH2N
SHO
2 10
Abbildung 5.4. Synthese von Ligand 10.
Das Molekül erhält durch Einführung eines längeren Distanzstücks größere Flexibilität zum
Ausgleich des durch die ausladenden Endgruppen in der SAM induzierten sterischen Zwangs.
Zusätzlich können sich zwischen den Amidgruppen intermolekulare H-Brückenbindungen
ausbilden, was den Ordnungsprozess in der SAM begünstigt. SAMs von Verbindung 10 auf
Au(111) wurden aus DMF-Lösungen präpariert. Zur Vermeidung der Moleküladsorption über
die aromatische Endgruppe wurde die Oberfläche mit dem planaren Perylen-Derivat 3,4,9,10-
Perylentetracarbonsäure-Dianhydrid (PTCDA) vorbeschichtet. Dies ergab Monolagen der
Moleküle auf der Oberfläche von präzedenzloser Kristallinität. Die erhaltenen STM-Bilder
zeigen molekulare Auflösung und ermöglichen in Kombination mit den spektroskopischen
Methoden die Erstellung eines Strukturmodells der Monolage (Abbildung 5.5).
a)
b)
Abbildung 5.5. (a) STM-Bild der SAM von 10 auf Au(111)/Glimmer; (b) Strukturmodell
(Rechteck = 3 × 5 Elementarzelle).
Für den Eisen(II)-Komplex des Liganden 10, [Fe(10)2](BF4)2 (25), zeigen SQUID-Daten
einen thermischen Spinübergang mit schleppendem Verlauf, der bei 350 K noch nicht
vollständig ist. Die SCO-Verbindung 25, wie auch die synthetisierten reinen HS-Eisen(II)-
Komplexe [Fe(3)2](BF4)2 (23) und [Fe(4)2](BF4)2 (24), spiegeln den von Halcrow entdeckten
>< 011
>< 211
>< 211
>< 011
1 nm
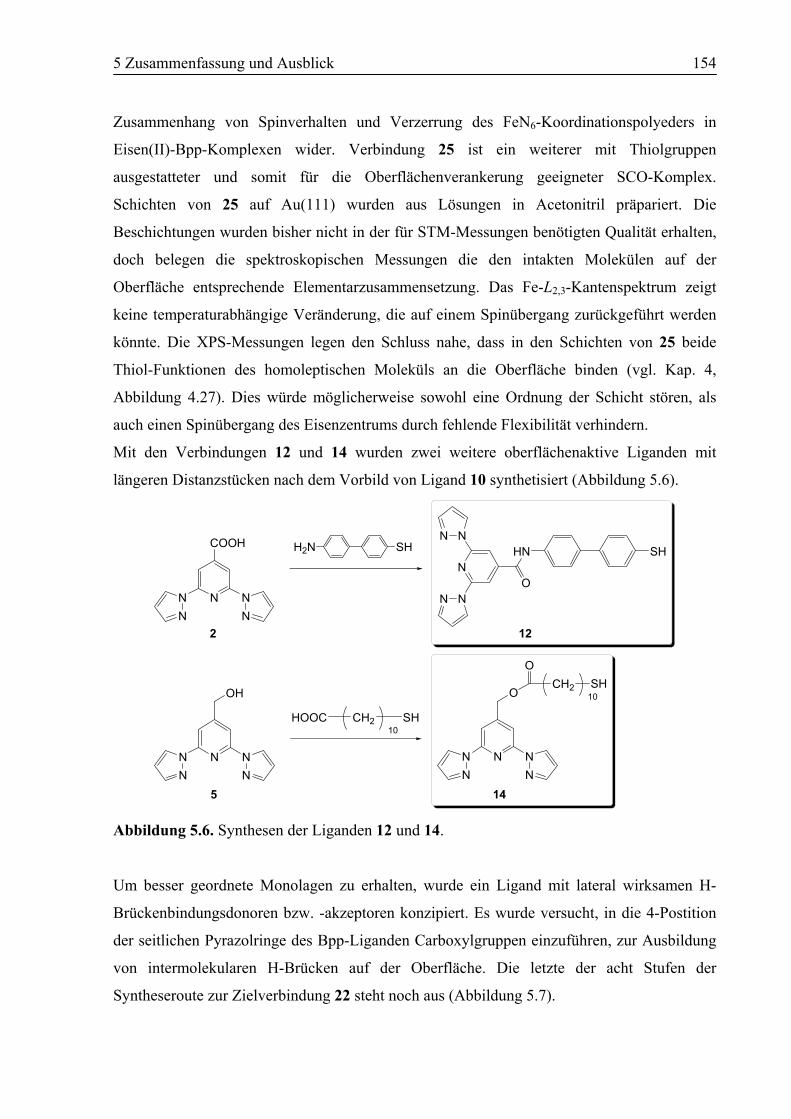
5 Zusammenfassung und Ausblick 154
Zusammenhang von Spinverhalten und Verzerrung des FeN6-Koordinationspolyeders in
Eisen(II)-Bpp-Komplexen wider. Verbindung 25 ist ein weiterer mit Thiolgruppen
ausgestatteter und somit für die Oberflächenverankerung geeigneter SCO-Komplex.
Schichten von 25 auf Au(111) wurden aus Lösungen in Acetonitril präpariert. Die
Beschichtungen wurden bisher nicht in der für STM-Messungen benötigten Qualität erhalten,
doch belegen die spektroskopischen Messungen die den intakten Molekülen auf der
Oberfläche entsprechende Elementarzusammensetzung. Das Fe-L2,3-Kantenspektrum zeigt
keine temperaturabhängige Veränderung, die auf einem Spinübergang zurückgeführt werden
könnte. Die XPS-Messungen legen den Schluss nahe, dass in den Schichten von 25 beide
Thiol-Funktionen des homoleptischen Moleküls an die Oberfläche binden (vgl. Kap. 4,
Abbildung 4.27). Dies würde möglicherweise sowohl eine Ordnung der Schicht stören, als
auch einen Spinübergang des Eisenzentrums durch fehlende Flexibilität verhindern.
Mit den Verbindungen 12 und 14 wurden zwei weitere oberflächenaktive Liganden mit
längeren Distanzstücken nach dem Vorbild von Ligand 10 synthetisiert (Abbildung 5.6).
NNNN N
OH
HOOC CH2 SH10
NNNN N
OCH2
OSH
10
5 14
NNNN N
COOH
N
N
N
N
N
HN
O
SH
2 12
H2N SH
Abbildung 5.6. Synthesen der Liganden 12 und 14.
Um besser geordnete Monolagen zu erhalten, wurde ein Ligand mit lateral wirksamen H-
Brückenbindungsdonoren bzw. -akzeptoren konzipiert. Es wurde versucht, in die 4-Postition
der seitlichen Pyrazolringe des Bpp-Liganden Carboxylgruppen einzuführen, zur Ausbildung
von intermolekularen H-Brücken auf der Oberfläche. Die letzte der acht Stufen der
Syntheseroute zur Zielverbindung 22 steht noch aus (Abbildung 5.7).
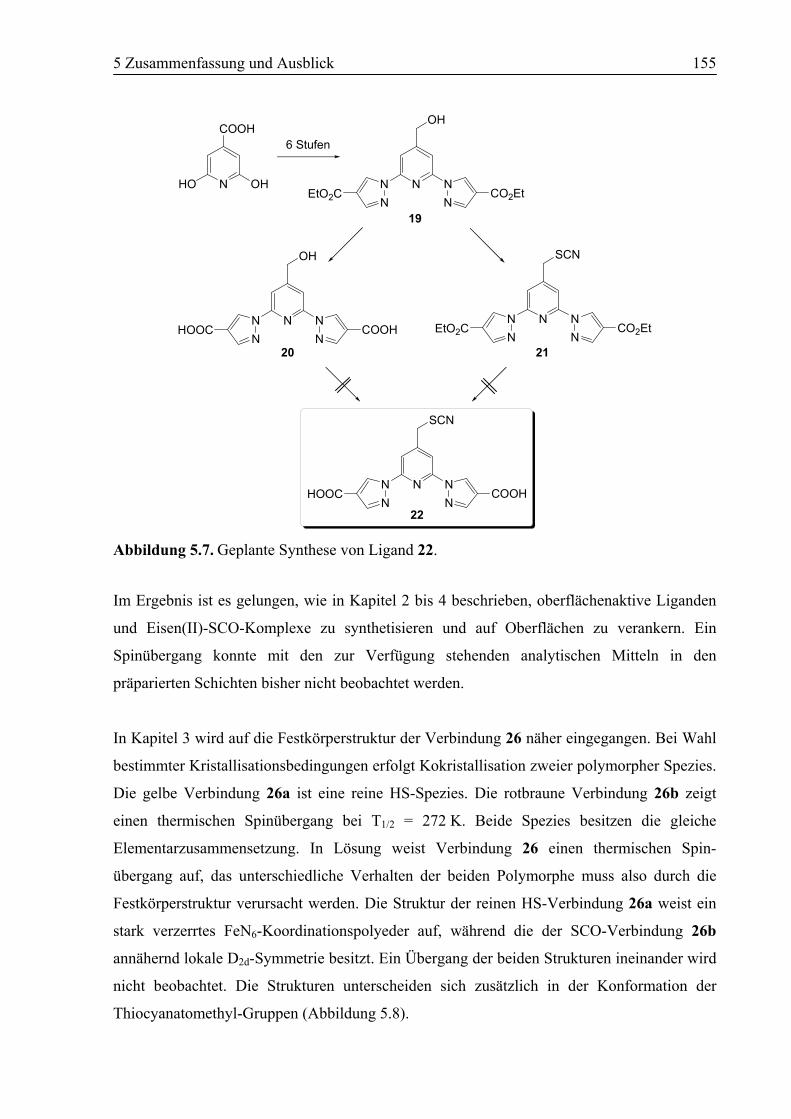
5 Zusammenfassung und Ausblick 155
N
COOH
OHHO NN NN N
OH
EtO2C CO2Et
NN NN N
OH
HOOC COOHNN N
N N
SCN
EtO2C CO2Et
NN NN N
SCN
HOOC COOH
19
20 21
22
6 Stufen
Abbildung 5.7. Geplante Synthese von Ligand 22.
Im Ergebnis ist es gelungen, wie in Kapitel 2 bis 4 beschrieben, oberflächenaktive Liganden
und Eisen(II)-SCO-Komplexe zu synthetisieren und auf Oberflächen zu verankern. Ein
Spinübergang konnte mit den zur Verfügung stehenden analytischen Mitteln in den
präparierten Schichten bisher nicht beobachtet werden.
In Kapitel 3 wird auf die Festkörperstruktur der Verbindung 26 näher eingegangen. Bei Wahl
bestimmter Kristallisationsbedingungen erfolgt Kokristallisation zweier polymorpher Spezies.
Die gelbe Verbindung 26a ist eine reine HS-Spezies. Die rotbraune Verbindung 26b zeigt
einen thermischen Spinübergang bei T1/2 = 272 K. Beide Spezies besitzen die gleiche
Elementarzusammensetzung. In Lösung weist Verbindung 26 einen thermischen Spin-
übergang auf, das unterschiedliche Verhalten der beiden Polymorphe muss also durch die
Festkörperstruktur verursacht werden. Die Struktur der reinen HS-Verbindung 26a weist ein
stark verzerrtes FeN6-Koordinationspolyeder auf, während die der SCO-Verbindung 26b
annähernd lokale D2d-Symmetrie besitzt. Ein Übergang der beiden Strukturen ineinander wird
nicht beobachtet. Die Strukturen unterscheiden sich zusätzlich in der Konformation der
Thiocyanatomethyl-Gruppen (Abbildung 5.8).
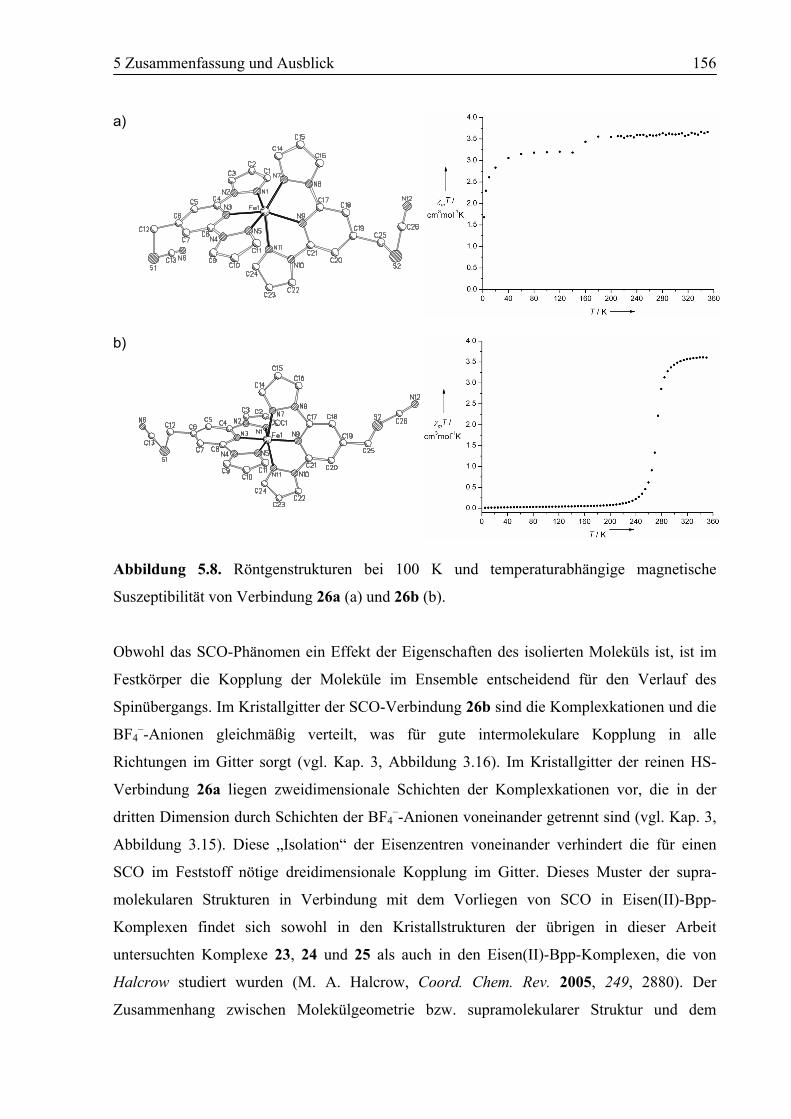
5 Zusammenfassung und Ausblick 156
a)
b)
Abbildung 5.8. Röntgenstrukturen bei 100 K und temperaturabhängige magnetische
Suszeptibilität von Verbindung 26a (a) und 26b (b).
Obwohl das SCO-Phänomen ein Effekt der Eigenschaften des isolierten Moleküls ist, ist im
Festkörper die Kopplung der Moleküle im Ensemble entscheidend für den Verlauf des
Spinübergangs. Im Kristallgitter der SCO-Verbindung 26b sind die Komplexkationen und die
BF4–-Anionen gleichmäßig verteilt, was für gute intermolekulare Kopplung in alle
Richtungen im Gitter sorgt (vgl. Kap. 3, Abbildung 3.16). Im Kristallgitter der reinen HS-
Verbindung 26a liegen zweidimensionale Schichten der Komplexkationen vor, die in der
dritten Dimension durch Schichten der BF4–-Anionen voneinander getrennt sind (vgl. Kap. 3,
Abbildung 3.15). Diese „Isolation“ der Eisenzentren voneinander verhindert die für einen
SCO im Feststoff nötige dreidimensionale Kopplung im Gitter. Dieses Muster der supra-
molekularen Strukturen in Verbindung mit dem Vorliegen von SCO in Eisen(II)-Bpp-
Komplexen findet sich sowohl in den Kristallstrukturen der übrigen in dieser Arbeit
untersuchten Komplexe 23, 24 und 25 als auch in den Eisen(II)-Bpp-Komplexen, die von
Halcrow studiert wurden (M. A. Halcrow, Coord. Chem. Rev. 2005, 249, 2880). Der
Zusammenhang zwischen Molekülgeometrie bzw. supramolekularer Struktur und dem

5 Zusammenfassung und Ausblick 157
Spinverhalten ist ein allgemeingültiges Prinzip bei SCO-Verbindungen. Bisher konnten
allerdings Einflüsse wie die Substitution der Liganden, die Natur der Anionen und das
Vorliegen von Lösungsmittelmolekülen in der Struktur nicht als Ursache für den
unterschiedlichen Verlauf des SCO ausgeschlossen werden. Die polymorphen Verbindungen
26a und 26b besitzen dagegen eine identische Zusammensetzung. Dieser Aspekt macht
Verbindung 26 vor allem für theoretische Untersuchungen interessant, mit denen Vorgaben
für die zukünftige Maßschneiderung von SCO-Materialien entwickelt werden können.
5.2 Ausblick
Für die Zukunft sind neben der Synthese der Eisen(II)-Komplexe von Verbindung 22 und
verwandten Liganden mit funktionellen Gruppen für lateral wirksame intermolekulare
Wechselwirkungen folgende Konzepte interessant.
Die Einführung einer sterisch anspruchsvollen Spacer- oder Ankergruppe mit größerem
Durchmesser als die SCO-Einheit der oberflächenaktiven Moleküle ist ein aussichtsreicher
Ansatz zur Präparation hochgeordneter Lagen. Als Distanzstück kommen hier die Bicyclo-
[2.2.2]oktan-Gruppe sowie closo-Boran-Derivate in Betracht. Auch sterisch anspruchsvolle
tripodale Ankergruppen insgesamt tetraedrischer Geometrie bzw. mit Adamantangerüst sind
bereits publiziert. Ein alternativer Ansatz ist die Vorfunktionalisierung der Oberfläche mit
einem Netzwerk aus adsorbierten organischen Molekülen, die ein Muster für die Verankerung
der SCO-Moleküle vorgeben.
Bei beiden Konzepten erlaubt die räumliche Trennung der Komplex-Fragmente allerdings
keine starke Kopplung der potentiellen SCO-Zentren. Denkbar sind dennoch graduelle
Übergänge oder die Beobachtung von Spinübergängen in Einzelmolekülen mittels STM-
Untersuchungen. Allgemein ist für die Zukunft die Synthese heteroleptischer Komplexe
geplant, bei denen nur einer der zwei dreizähnigen Liganden eine Ankergruppe trägt.
Die unterschiedlichen Konformationen der MeSCN-Gruppen in den polymorphen
Verbindungen 26a und 26b könnten das Vorliegen einer ligandvermittelten Änderung der
Ligandenfeldstärke, ähnlich dem LD-LISC-Effekt (ligand-driven light-induced spin change),
bedeuten. Aufschluss über diesen Aspekt werden theoretische Berechnungen der Strukturen
ergeben, die Gegenstand zukünftiger Arbeiten sind.

6 Anhang 158
6 Anhang
6.1 Verwendete Abkürzungen
Å Ångström (10–10m)
B Magnetische Flussdichte
BOC2O Di-tert-butyldicarbonat
Bpp 2,6-Bis(pyrazol-1-yl)pyridin
br Breit (IR und NMR)
btr 4,4’-Bis-1,2,4-triazol
CID Collision-induced dissociation
CVD Chemical vapour deposition
d Dublett (NMR); Tag
δ Chemische Verschiebung
DCC N,N'-Dicyclohexylcarbodiimid
DCM Dichlormethan
dd Dublett vom Dublett (NMR)
DEAD Azodicarbonsäurediethylester
DFT Dichtefunktionaltheorie
Diglyme Diethylenglykoldimethylether
DMAP 4-Dimethylaminopyridin
DMF N,N-Dimethylformamid
DMSO Dimethylsulfoxid
DPPA (3-Aminopropyl)bis(2-pyridylmethyl)amin)
E Energie
EDC 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid hydrochlorid
EI Elektronenstoßionisation
ESI Electrospray Ionisation
Et Ethyl
EtOH Ethanol
EZ Elementarzelle
eV Elektronenvolt

6 Anhang 159
g Gramm
h Stunde
Hb Hämoglobin
HOBt 1-Hydroxybenzotriazol
HS High Spin
IR Infrarot
l Wellenlänge
LB Langmuir-Blodgett
LD-LISC Ligand-driven light induced spin change
LIESST Light-induced excited spin state trapping
LS Low Spin
m Multiplett (NMR); mittelstark (IR)
M Molar
Me Methyl
MeCN Acetonitril
mg Milligramm
MHz Megahertz
min Minute
ml Milliliter
MOF Metal organic framework
MS Massenspektrum
m/z Masse-zu-Ladung-Verhältnis (MS)
NEXAFS Near-edge x-ray absorption fine structure spectroscopy
nm Nanometer (10–9m)
NMR Nuclear magnetic resonance
ν~ Wellenzahl (IR)
OMBE Organic molecular beam epitaxy
p Druck
phen 1,10-Phenanthrolin
pm Picometer (10–12m)
ppm Parts per million
PPTS Pyridinium-p-toluolsulfonat
py Pyridin

6 Anhang 160
pyN4 2,6-Bis(1’,3’-diamino-2’methylprop-2’yl)pyridin
pz Pyrazol
R Organischer Rest
RT Raumtemperatur
s Singulett (NMR); stark (IR)
SAM Self-assembled monolayer
SCO Spincrossover
STM Scanning tunneling microscope
T Tesla; Temperatur
t Triplett
T1/2 Gemittelte Übergangstemperatur beim Spinübergang
THF Tetrahydrofuran
THP 2-Tetrahydropyranylgruppe
UHV Ultrahochvakuum
vs Sehr stark (IR)
XAS X-ray absorption spectroscopy
XPS X-ray photoelectron spectroscopy

6 Anhang 161
6.2 Publikationen
Begutachtete Publikationen in Fachzeitschriften:
Full paper: Maria Schlangen, Johannes Neugebauer, Markus Reiher, Detlef Schröder, Jesús Pitarch López, Marco Haryono, Frank W. Heinemann, Andreas Grohmann and Helmut Schwarz „Gas-Phase C–H and N–H Bond Activation by a High Valent Nitrido-Iron Dication and
NH -Transfer to Activated Olefins”
J. Am. Chem. Soc. 2008, 130, 4285–4294.
Full paper: Cai Shen, Marco Haryono, Andreas Grohmann, Manfred Buck, Tobias Weidner, Nirmalya Ballav and Michael Zharnikov „Self-Assembled Monolayers of a Bis(pyrazol-1-yl)pyridine-Substituted Thiol on Au(111)” Langmuir 2008, DOI: 10.1021/la8019974.
Eingereichte Publikationen in Fachzeitschriften:
Full paper: Marco Haryono, Frank W. Heinemann, Konstantin Petukhov, Klaus Gieb, Paul Müller and Andreas Grohmann „Parallel crystallization of a “static” and a spin crossover polymorph of an iron(ii) complex from the same solution” Eur. J. Inorg. Chem., eingereicht.
Vorträge und Posterpräsentationen:
Vortrag: Marco Haryono „Spin-schaltbare Eisen(II)-Komplexe für die Verankerung auf Oberflächen“ „3. Koordinationschemietreffen“, Technische Universität Berlin, Februar 2007.
Poster: Marco Haryono, Sophie Hain, Andreas Grohmann, Matthias Bernien, Wolfgang Kuch, Cai Shen, Manfred Buck, Konstantin Petukhov, Paul Müller „Iron(II) spincrossover complexes for self-assembled monolayers on gold surfaces“ „GDCh Wissenschaftsforum Chemie 2007“, Universität Ulm, September 2007.

6 Anhang 162
Poster: Marco Haryono, Sophie Hain, Matthias Bernien, Wolfgang Kuch, Manfred Buck and Andreas Grohmann „Iron(II) spincrossover complexes for self-assembled monolayers on gold surfaces“ „Sfb 658, International Workshop on Molecular Switching on Surfaces“, Zeuthen b. Berlin, September 2007.
Poster: Marco Haryono, Matthias Bernien, Wolfgang Kuch, Cai Shen, Manfred Buck, Michael Zharnikov, Konstantin Petukhov, Paul Müller and Andreas Grohmann „Iron(II) Spin Crossover Complexes for Self-Assembled Monolayers on Gold Surfaces“ „4th EuCheMS Conference on Nitrogen Ligands“, Garmisch-Partenkirchen, August 2008.

7 Literaturverzeichnis 163
7 Literaturverzeichnis
[1] M. F. Perutz, G. Fermi, B. Luisi, B. Shaanan, R. C. Liddington, Acc. Chem. Res. 1987,
20, 309.
[2] E. König, Struct. Bonding 1991, 76, 51.
[3] I. G. Denisov, T. M. Makris, S. G. Sligar, I. Schlichting, Chem. Rev. 2005, 105, 2253.
[4] J. Everse, M. B. Grisham, K. E. Everse, peroxidases in chemistry and biology, Vol. 2,
crc press, boca raton, 1990.
[5] P. Gütlich, Y. Garcia, H. A. Goodwin, Chem. Soc. Rev. 2000, 29, 419.
[6] P. Gütlich, H. A. Goodwin, Top. Curr. Chem. 2004, 233, 1.
[7] P. Gütlich, A. Hauser, H. Spiering, Angew. Chem. 1994, 106, 2109.
[8] A. Hauser, Top. Curr. Chem. 2004, 233, 49.
[9] L. Cambi, L. Malatesta, Ber. Dtsch. Chem. Ges. B 1937, 70B, 2067.
[10] K. Madeja, E. Koenig, J. Inorg. Nucl. Chem. 1963, 25, 377.
[11] E. König, Chem. Commun. 1966, No. 3, 61.
[12] S. Sugano, Y. Tanabe, H. Kamimura, Multiplets of transition metal ions, Pure and
applied physics Vol. 33, Academic Press, New York, 1970.
[13] P. Gütlich, Y. Garcia, T. Woike, Coord. Chem. Rev. 2001, 219-221, 839.
[14] O. Kahn, C. J. Martinez, Science 1998, 279, 44.
[15] F. Schreiber, Prog. Surf. Sci. 2000, 65, 151.
[16] D. W. Schubert, Polym. Bull. 1997, 38, 177.
[17] A. Ulman, An Introduction to Ultrathin Organic Films: From Langmuir-Blodgett to
Self-Assembly, Academic Press, Boston, 1991.
[18] S. R. Forrest, Chem. Rev. 1997, 97, 1793.
[19] K. L. Choy, Prog. Mater. Sci. 2003, 48, 57.
[20] J. C. Love, A. Estroff Lara, K. Kriebel Jennah, G. Nuzzo Ralph, M. Whitesides
George, Chem Rev 2005, 105, 1103.
[21] R. G. Nuzzo, D. L. Allara, J. Am. Chem. Soc. 1983, 105, 4481.
[22] A. Ulman, Chem. Rev. 1996, 96, 1533.
[23] L. H. Dubois, R. G. Nuzzo, Annu. Rev. Phys. Chem. 1992, 43, 437.
[24] F. Buckel, F. Effenberger, C. Yan, A. Golzhauser, M. Grunze, Adv. Mater. 2000, 12,
901.

7 Literaturverzeichnis 164
[25] T. J. Mullen, A. A. Dameron, A. M. Andrews, P. S. Weiss, Aldrichimica Acta 2007,
40, 21.
[26] D. F. Evans, J. Chem. Soc. 1959, 2003.
[27] O. Kahn, Molecular Magnetism, VCH, New York, Heidelberg, 1993.
[28] P. Gütlich, R. Link, A. X. Trautwein, Mössbauer spectroscopy and transition metal
chemistry. Inorganic Chemistry Concepts Series No. 3, Springer, Berlin, Heidelberg,
New York, 1978.
[29] S. Alvarez, J. Am. Chem. Soc. 2003, 125, 6795.
[30] P. J. Stephens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch, J. Phys. Chem. 1994, 98,
11623.
[31] M. Reiher, O. Salomon, B. A. Hess, Theor. Chem. Acc. 2001, 107, 48.
[32] H. Paulsen, A. X. Trautwein, Top. Curr. Chem. 2004, 235, 197.
[33] C. Vericat, M. E. Vela, G. A. Benitez, J. A. M. Gago, X. Torrelles, R. C. Salvarezza,
J. Phys.: Condens. Matter 2006, 18, R867.
[34] G. E. Poirier, Chem. Rev. 1997, 97, 1117.
[35] J. Stöhr, NEXAFS Spectroscopy, Springer, New York, 1992.
[36] O. Dannenberger, K. Weiss, H. J. Himmel, B. Jager, M. Buck, C. Woll, Thin Solid
Films 1997, 307, 183.
[37] C. Cartier dit Moulin, P. Rudolf, A. M. Flank, C. T. Chen, J. Phys. Chem. 1992, 96,
6196.
[38] J. G. Chen, Surf. Sci. Rep. 1997, 30, 1.
[39] M.-C. Bourg, A. Badia, R. B. Lennox, J. Phys. Chem. B 2000, 104, 6562.
[40] D. G. Castner, K. Hinds, D. W. Grainger, Langmuir 1996, 12, 5083.
[41] A. Bousseksou, G. Molnar, G. Matouzenko, Eur. J. Inorg. Chem. 2004, 4353.
[42] J.-F. Letard, P. Guionneau, L. Goux-Capes, Top. Curr. Chem. 2004, 235, 221.
[43] J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman, M.
A. Halcrow, J. Chem. Soc., Dalton Trans. 2002, 548.
[44] D. L. Jameson, J. K. Blaho, K. T. Kruger, K. A. Goldsby, Inorg. Chem. 1989, 28,
4312.
[45] D. L. Jameson, K. A. Goldsby, J. Org. Chem. 1990, 55, 4992.
[46] J. M. Holland, C. A. Kilner, M. Thornton-Pett, M. A. Halcrow, J. A. McAllister, Z.
Lu, Chem. Commun. 2001, 577.
[47] M. A. Halcrow, Coord. Chem. Rev. 2005, 249, 2880.

7 Literaturverzeichnis 165
[48] M. Hostettler, K. W. Toernroos, D. Chernyshov, B. Vangdal, H.-B. Buergi, Angew.
Chem., Int. Ed. 2004, 43, 4589.
[49] E. König, K. Madeja, Inorg. Chem. 1967, 6, 48.
[50] E. König, K. Madeja, K. J. Watson, J. Amer. Chem. Soc. 1968, 90, 1146.
[51] M. Marchivie, P. Guionneau, J. F. Letard, D. Chasseau, Acta Crystallogr., Sect. B:
Struct. Sci. 2003, B59, 479.
[52] A. Ozarowski, B. R. McGarvey, A. B. Sarkar, J. E. Drake, Inorg. Chem. 1988, 27,
628.
[53] G. S. Matouzenko, A. Bousseksou, S. Lecocq, P. J. van Koningsbruggen, M. Perrin,
O. Kahn, A. Collet, Inorg. Chem. 1997, 36, 5869.
[54] D. L. Reger, J. R. Gardinier, M. D. Smith, A. M. Shahin, G. J. Long, L. Rebbouh, F.
Grandjean, Inorg. Chem. 2005, 44, 1852.
[55] S. Trofimenko, Polyhedron 2004, 23, 197.
[56] N. Moliner, M. C. Munoz, S. Letard, J.-F. Letard, X. Solans, R. Burriel, M. Castro, O.
Kahn, J. A. Real, Inorg. Chim. Acta 1999, 291, 279.
[57] C. Janiak, Dalton Trans. 2003, 2781.
[58] W. Vreugdenhil, J. G. Haasnoot, O. Kahn, P. Thuery, J. Reedijk, J. Am. Chem. Soc.
1987, 109, 5272.
[59] T. Kitazawa, Y. Gomi, M. Takahashi, M. Takeda, M. Enomoto, A. Miyazaki, T.
Enoki, J. Mater. Chem. 1996, 6, 119.
[60] Y. Garcia, V. Niel, M. C. Munoz, J. A. Real, Top. Curr. Chem. 2004, 233, 229.
[61] J. A. Real, A. B. Gaspar, M. C. Munoz, Dalton Trans. 2005, 2062.
[62] S. Cobo, G. Molnar, J. A. Real, A. Bousseksou, Angew. Chem., Int. Ed. 2006, 45,
5786.
[63] G. Molnar, S. Cobo, J. A. Real, F. Carcenac, E. Daran, C. Vieu, A. Bousseksou, Adv.
Mater. 2007, 19, 2163.
[64] M. Matsuda, H. Tajima, Chem. Lett. 2007, 36, 700.
[65] O. Roubeau, E. Natividad, B. Agricole, S. Ravaine, Langmuir 2007, 23, 3110.
[66] H. Soyer, E. Dupart, C. J. Gomez-Garcia, C. Mingotaud, P. Delhaes, Adv. Mater.
1999, 11, 382.
[67] Y. Bodenthin, U. Pietsch, H. Moehwald, D. G. Kurth, J. Am. Chem. Soc. 2005, 127,
3110.
[68] R. Chandrasekar, F. Schramm, O. Fuhr, M. Ruben, Eur. J. Inorg. Chem. 2008, 2649.

7 Literaturverzeichnis 166
[69] J. M. Tour, L. Jones, II, D. L. Pearson, J. J. S. Lamba, T. P. Burgin, G. M. Whitesides,
D. L. Allara, A. N. Parikh, S. Atre, J. Am. Chem. Soc. 1995, 117, 9529.
[70] J. W. Ciszek, M. P. Stewart, J. M. Tour, J. Am. Chem. Soc. 2004, 126, 13172.
[71] E. B. Troughton, C. D. Bain, G. M. Whitesides, R. G. Nuzzo, D. L. Allara, M. D.
Porter, Langmuir 1988, 4, 365.
[72] L. Cai, Y. Yao, J. Yang, D. W. Price, Jr., J. M. Tour, Chem. Mater. 2002, 14, 2905.
[73] J. W. Ciszek, J. M. Tour, Chem. Mater. 2005, 17, 5684.
[74] K. E. Henegar, S. W. Ashford, T. A. Baughman, J. C. Sih, R.-L. Gu, J. Org. Chem.
1997, 62, 6588.
[75] A. C. Spivey, S. Arseniyadis, Angew. Chem., Int. Ed. 2004, 43, 5436.
[76] T. Vermonden, D. Branowska, A. T. M. Marcelis, E. J. R. Sudholter, Tetrahedron
2003, 59, 5039.
[77] C. Rajadurai, F. Schramm, S. Brink, O. Fuhr, M. Ghafari, R. Kruk, M. Ruben, Inorg.
Chem. 2006, 45, 10019.
[78] C. A. Bessel, R. F. See, D. L. Jameson, M. R. Churchill, K. J. Takeuchi, J. Chem.
Soc., Dalton Trans. 1992, 3223.
[79] J. Elhaik, C. M. Pask, C. A. Kilner, M. A. Halcrow, Tetrahedron 2007, 63, 291.
[80] M. A. Halcrow, C. A. Kilner, M. Thornton-Pett, Acta Crystallogr., Sect. C: Cryst.
Struct. Commun. 2000, C56, 213.
[81] N. Iranpoor, H. Firouzabadi, B. Akhlaghinia, R. Azadi, Synthesis 2004, 92.
[82] SDBSWeb: http://riodb01.ibase.aist.go.jp/sdbs/ (National Institute of Advanced
Industrial Science and Technology.
[83] W. König, R. Geiger, Chem Ber 1970, 103, 788.
[84] K. Seno, K. Matumura, M. Oshima, S. Motomizu, Anal. Sci. 2008, 24, 505.
[85] P. Cyganik, M. Buck, W. Azzam, C. Woell, J. Phys. Chem. B 2004, 108, 4989.
[86] P. Cyganik, M. Buck, J. D. E. T. Wilton-Ely, C. Woell, J. Phys. Chem. B 2005, 109,
10902.
[87] L. J. Goossen, A. Doehring, Synlett 2004, 263.
[88] B. Neises, W. Steglich, Angew. Chem. 1978, 90, 556.
[89] M. Heller, U. S. Schubert, J. Org. Chem. 2002, 67, 8269.
[90] C. J. Salomon, E. G. Mata, O. A. Mascaretti, Tetrahedron 1993, 49, 3691.
[91] P. Guionneau, M. Marchivie, G. Bravic, J.-F. Letard, D. Chasseau, J. Mater. Chem.
2002, 12, 2546.

7 Literaturverzeichnis 167
[92] E. Markovicky, T. Balic-Zunic, Acta Crystallogr., Sect. B: Struct. Sci. 1998, B54, 766.
[93] C. J. O'Connor, Prog. Inorg. Chem. 1982, 29, 203.
[94] J. Elhaik, D. J. Evans, C. A. Kilner, M. A. Halcrow, Dalton Trans. 2005, 1693.
[95] G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts,
3rd ed., John Wiley & Sons Ltd, Chichester, 2001.
[96] B. V. N. COLLECT data collection software, 1998.
[97] B. A. SHELXTL NT 6.12, Inc., 2002, Madison WI., U.S.A.
[98] T. Balic Zunic, I. Vickovic, J. Appl. Crystallogr. 1996, 29, 305.
[99] C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M.
Towler, J. van de Streek, J. Appl. Crystallogr. 2006, 39, 453.
[100] U. El-Ayaan, F. Murata, Y. Fukuda, Monatsh. Chem. 2001, 132, 1279.
[101] V. A. Money, J. Elhaik, M. A. Halcrow, J. A. K. Howard, Dalton Trans. 2004, 1516.
[102] C. Carbonera, J. Sanchez Costa, V. A. Money, J. Elhaik, J. A. K. Howard, M. A.
Halcrow, J.-F. Letard, Dalton Trans. 2006, 3058.
[103] E. M. Schubert, J. Chem. Educ. 1992, 69, 62.
[104] C. Janiak, J. Chem. Soc., Dalton Trans. 2000, 3885.
[105] M. J. Calhorda, Chem. Commun. 2000, 801.
[106] R. Taylor, O. Kennard, J. Am. Chem. Soc. 1982, 104, 5063.
[107] G. A. Bain, J. F. Berry, J. Chem. Educ. 2008, 85, 532.
[108] P. Sigmund, Nucl. Instrum. Methods Phys. Res., Sect. B 1987, B27, 1.
[109] G. E. Poirier, Langmuir 1997, 13, 2019.
[110] W. Chen, H. Huang, S. Chen, L. Chen, H. L. Zhang, X. Y. Gao, A. T. S. Wee, Appl.
Phys. Lett. 2007, 91, 114102/1.
[111] N. Nicoara, E. Roman, J. M. Gomez-Rodriguez, J. A. Martin-Gago, J. Mendez, Org.
Electron. 2006, 7, 287.
[112] K. Heister, M. Zharnikov, M. Grunze, L. S. O. Johansson, J. Phys. Chem. B 2001,
105, 4058.
[113] P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh, R. G. Nuzzo,
J. Am. Chem. Soc. 1991, 113, 7152.
[114] K. Heister, H. T. Rong, M. Buck, M. Zharnikov, M. Grunze, L. S. O. Johansson, J.
Phys. Chem. B 2001, 105, 6888.
[115] A. Shaporenko, M. Brunnbauer, A. Terfort, L. S. O. Johansson, M. Grunze, M.
Zharnikov, Langmuir 2005, 21, 4370.

7 Literaturverzeichnis 168
[116] Y. Zubavichus, M. Zharnikov, Y. Yang, O. Fuchs, E. Umbach, C. Heske, A. Ulman,
M. Grunze, Langmuir 2004, 20, 11022.
[117] Y. Zubavichus, M. Zharnikov, Y. Yang, O. Fuchs, C. Heske, E. Umbach, G.
Tzvetkov, P. Netzer Falko, M. Grunze, J Phys Chem B 2005, 109, 884.
[118] C. L. A. Lamont, J. Wilkes, Langmuir 1999, 15, 2037.
[119] J. Thome, M. Himmelhaus, M. Zharnikov, M. Grunze, Langmuir 1998, 14, 7435.
[120] S. Aminpirooz, L. Becker, B. Hillert, J. Haase, Surf. Sci. 1991, 244, L152.
[121] J. A. Horsley, J. Stohr, A. P. Hitchcock, D. C. Newbury, A. L. Johnson, F. Sette, J.
Chem. Phys. 1985, 83, 6099.
[122] E. Apen, A. P. Hitchcock, J. L. Gland, J. Phys. Chem. 1993, 97, 6859.
[123] D. C. Newbury, I. Ishii, A. P. Hitchcock, Can. J. Chem. 1986, 64, 1145.
[124] K. Heister, M. Zharnikov, M. Grunze, L. S. O. Johansson, A. Ulman, Langmuir 2001,
17, 8.
[125] C. Shen, M. Haryono, A. Grohmann, M. Buck, T. Weidner, N. Ballav, M. Zharnikov,
Langmuir 2008, 24, 12883.
Berlin, 28.10.2008





























