Embed Size (px)
Citation preview

www.sciencemag.org/cgi/content/full/328/5976/363/DC1
Supporting Online Material for
Cbln1 Is a Ligand for an Orphan Glutamate Receptor δ2, a Bidirectional Synapse Organizer
Keiko Matsuda, Eriko Miura, Taisuke Miyazaki, Wataru Kakegawa, Kyoichi Emi, Sakae Narumi, Yugo Fukazawa, Aya Ito-Ishida,Tetsuro Kondo, Ryuichi Shigemoto, Masahiko
Watanabe, Michisuke Yuzaki*
*To whom correspondence should be addressed. E-mail: [email protected]
Published 16 April 2010, Science 328, 363 (2009) DOI: 10.1126/science.1185152
This PDF file includes
Materials and Methods Figs. S1 to S12 References

Supporting Online material
Materials and Methods cDNA constructs. cDNA encoding hemagglutinin (HA) was added to the 5' end of
mouse Cbln1 cDNA; two cysteine residues at amino acid positions 34 and 38 were
replaced with serine residues to create CS-Cbln1, as described previously (S1). Rat
cDNAs encoding GluD1, GluD2 and GluK2 were gifts from Dr. J. Boulter (Univ.
California at Los Angeles, Los Angeles, CA). Chimeric constructs of GluD2 and
GluK2 were created as described previously (S2). A Flag tag was added to the 3' end
of the GluK2 or GluD2 constructs. For the Fc fusion constructs, the N-terminal
domains (NTDs) of GluD2 (GluD2NTD; amino acids 1–430), GluA1 (GluA1NTD; 1–400),
GluA2 (GluA2NTD; 1–400), GluK2 (GluK2NTD; 1–424), GluD1 (GluD1NTD; 1–426) or
the extracellular domain of CD4 (a gift from Dr. Y. Oike; Sch. of Med., Keio Univ.,
Tokyo, Japan) were added immediately before the Fc domain of human IgG1.
PSD95-YFP construct was a gift from Dr. P. Sheiffele (Univ. Basel, Basel, Switzerland)
and shank2-GFP was from Dr. Okabe (Univ. Tokyo, Tokyo, Japan). The cDNA
constructs were cloned into pCAGGS vector (kindly provided by Dr. J. Miyazaki,
Osaka Univ., Osaka, Japan) or the Sindbis virus vector.
Preparation of recombinant proteins. HA-tagged Cbln1 or Fc fusion proteins were
expressed in HEK293 tSA cells (a kind gift from Dr. R. Horn, Thomas Jefferson Univ.
Med. Sch., Philadelphia, PA) as described previously (S1). The concentration of each
recombinant Cbln1 was quantified using an immunoblot analysis with purified 6 x
histidine-tagged HA–Cbln1 as the standard (S3). Fc fusion proteins were immobilized
on Protein G beads. HA-Cbln1 was incubated with biotinylated anti-HA antibody
(mouse) and then immobilized to avidin beads.
Cell cultures. Mixed cerebellar cultures were prepared from embryonic day 17 to
day-of-birth ICR, GluD2-null (ho5J) or cbln1-null mice (kindly provided by Dr. J. I.
Morgan, St. Jude Children's Research Hospital, Memphis, USA), as described
previously (S2, S4). cbln1-, GluD2-double null (cbln1/GluD2-null) mice were
generated by breeding GluD2-null and cbln1-null mice. Cells were plated at a density
1

of 2 × 105 cells on plastic coverslips (diameter, 13.5 mm) and were maintained in
DMEM⁄ F12 containing 100 μM putrescine, 30 nM sodium selenite, 0.5 ng/mL
tri-iodothyronine, 0.25 mg/ml bovine serum albumin, 3.9 mM glutamate and N3
supplement (100 μg/mL apo-transferrin, 10 μg/mL insulin and 20 nM progesterone) in
5% CO2 at 37°C. For the synapse formation assays, the cerebellar cultures were
incubated with recombinant HA-Cbln1 (3.5 μg/mL) from 7 d in vitro (DIV) through 14
DIV. For the synapse inhibition assays, wild-type cerebellar cultures were incubated
with GluD2NTD-Fc proteins (1 μg/mL) from 3 through 10 DIV. Every 3 days, half of
the culture medium was replaced with the new medium containing Fc proteins.
Heterologous synapse formation assays using HEK293 cells were performed as
described previously (S2). Briefly, HEK293 cells transfected with each cDNA
construct were added to cultured neurons at 7 DIV and cocultured for 5 d in the
presence of 5-fluoro-2-deoxyuridine (10 μM) with or without recombinant Cbln1. All
procedures related to animal care and treatment were approved by the Animal Resource
Committee of the School of Medicine, Keio University.
Cell surface biotinylation assays. For biotinylation of surface proteins, HEK293 cells
expressing GluD2 or GluD2-GluK2 chimeric proteins were treated with 0.5 mg/mL
sulfo-NHS-LC-biotin for 15 min at 4°C. After solubilization with TNE buffer (50 mM
NaF, 0.1% SDS, 1% Nonidet P-40, 20 mM EDTA, 1 μM pepstatin A, 2 μg/ml leupeptin,
10 μg/ml aprotinin, 50 mM Tris-HCl, pH 8.0), cell lysates were immunoprecipitated
using anti-GluD2 or anti-Flag antibodies, followed by detection with horse radish
peroxidase–conjugated streptavidin.
Immunohistochemistry. Cells in dissociated cultures were fixed with
phosphate-buffered saline (PBS) containing 4% paraformaldehyde for 20 min on ice,
followed by 100% methanol at -20°C for 10 min for immunostaining synaptic markers
(GluA2, synapsin I, or synaptophysin). After permeabilization with 0.4% Triton
X-100 in PBS containing 2% bovine serum albumin and 2% normal goat serum for 1 h
at room temperature, the cells were treated with primary antibodies (see below) and then
were subsequently treated with secondary antibodies that were conjugated with either
2

Alexa 405, 488, 546 (1:1000). Fluorescence images were captured using a CCD
camera attached to a fluorescence microscope or a confocal microscope.
To unmask the antigens in the slice preparations, microslicer sections were treated
with 1 mg/mL of pepsin in 0.2 N HCl at 37°C for 3 min and then were incubated
successively with 10% normal donkey serum for 20 min, mixtures of primary antibodies
overnight, and mixtures of 1:200-diluted Cy3-, Cy5-, and Alexa 488-labeled
species-specific secondary antibodies for 2 h. Photographs were taken with a confocal
laser-scanning microscope.
To quantify the accumulation of each synaptic marker in the transfected HEK293
cells or in HA-Cbln1-beads, images were randomly captured at 8 or more fields (each
filed corresponds to 450 x 600 μm) using fixed gains and exposures for each fluorescent
channel. The images were analyzed using IP-lab software. GFP-immunopositive
regions or beads regions were selected using macro "autosegmentation." The intensity
of the immunoreactivity within the segmented area was averaged, and the background
immunoreactivity within the nonsegmented area was subtracted. To quantify the
signal intensity of the presynaptic markers that were colocalized with the Purkinje cell
dendrites, an image was taken of the most distal region of the dendrites of each cell.
GluD2- or calbindin-immunopositive regions were selected using macro
"autosegmentation." The number of neurons was counted as n for the statistical
analysis.
Electron microscopy. For postembedding immunogold electron microscopy,
microslicer sections (300 μm in thickness) were cryoprotected with 30%
sucrose/phosphate buffer (PB) and frozen rapidly with liquid propane in an EM CPC
unit. Frozen sections were immersed in 0.5% uranyl acetate in methanol at -90°C in
an AFS freeze-substitution unit, infiltrated at -45°C with Lowicryl HM-20 resin, and
polymerized with UV light. After etching with saturated sodium-ethanolate solution
for 3 s, ultrathin sections on nickel grids were treated successively with 2% goat serum
albumin/0.1% Tween 20 in Tris-buffered saline (pH 7.5; GTBST) for 30 min, rabbit
Cbln1 antibody (30 μg/mL) in GTBST overnight, and colloidal gold (10
nm)-conjugated anti-rabbit IgG (1:100) in GTBST for 2 h. Finally, the grids were
3

stained with 2% uranyl acetate for 20 min and mixed lead solution for 30 s.
Photographs were taken with an H-7100 electron microscope. The density of the
immunogold particles on the electron micrographs was quantitatively analyzed using
IPLab software.
For the in vivo HA-Cbln1 administration experiments, cbln1-null, GluD2-null or
cbln1/GluD2-null mice (P50-55) were perfused transcardially with 2%
paraformaldehyde/2% glutaraldehyde in 0.1 M PB (pH 7.2) under deep pentobarbital
anesthesia. Parasagittal microslicer sections of the cerebellum (300 μm) were
postfixed for 2 h with 1% OsO4 in 0.1 M PB. After block staining in 1% aqueous
uranyl acetate solution and dehydration with graded alcohols, the sections were
embedded in Epon 812. Serial ultrathin sections (70 nm) were made using an
ultramicrotome and stained with 2% uranyl acetate for 5 min and mixed lead solution
for 2 min. Electron micrographs of the molecular layer were taken using an H-7100
electron microscope at a magnification of x4,000 and printed at a magnification of
x16,000. For the quantitative analysis of PF–Purkinje cell synapses, the asymmetrical
postsynaptic density in cerebellar cortex were randomly photographed and the
frequency of normal, free/naked and mismatched synapses were analyzed by using
MetaMorph software as described previously (S3). Three (cbln1-null, n = 1 mouse) or
six (GluD2-null, n = 2 mice; cbln1/GluD2-null, n = 2 mice) series of serial sections,
each of which consisted of ten electron micrographs, were analyzed.
For SDS-digested freeze-fracture replica labeling (SDS-FRL) of GluD2,
cbln1-null or wild-type mice at postnatal 6 to 7 weeks (n = 2 mice for each) were
perfused transcardially with 0.5% paraformaldehyde and 15% saturated picric acid in
0.1 M PB (pH 7.2) and 3 replicas of the cerebellum from individual mice were prepared
and immunolabeled with anti-GluD2 antibody (guinea pig) as described previously (S5).
Labeled replicas were investigated with a Tecnai-12 transmission electron microscope.
As GluD2 is expressed exclusively at PF–Purkinje cell synapses (S6), the exoplasmic
(E) faces of replicated plasma membranes with clusters of intramembrane particles
(IMP) labeled with immunogold particles for GluD2 were considered as PF–Purkinje
cell synapses and photomicrographed with a CCD digital camera at a magnification of
97,000x. In order to investigate GluD2 expression selectively at PF–Purkinje cell
synapses with the presynaptic structure (contacted synapse), E-face IMP clusters
4

accompanied by the protoplasmic (P) face of presynaptic structures were selected and
analyzed. The area of postsynaptic membrane specializations was measured by
demarcating outline of the IMP clusters, and the number of immunogold particles was
counted by using iTEM software. All particles within 20 nm outside from the edge of
the E-face IMP cluster were counted as synaptic labeling because they can be distant
from the epitope. The EM images were calibrated by using a calibration grid.
Specificity of the anti-GluD2 receptor antibody was confirmed previously (S5) and
background labeling assessed on the P-face of presynaptic profiles in the present study
was negligible level (average labeling density = 0.19 ± 0.016 particles / μm2, total
P-face area analyzed = 11.7 μm2).
Cbln1 binding assay using intact cells. Mixed cerebellar cultures at 14 to 16 DIV
were incubated with HA-Cbln1 or HA-CS-Cbln1 (1 μg/mL each) for 12 h, fixed with
4% paraformaldehyde, and immunostained with mouse anti-HA and rabbit
anti-calbindin. Quantification of the signal intensity was performed as described
previously (S1). HEK293 cells expressing GluD2, GluD1, GluK2, and several
chimeric mutants were incubated with HA-Cbln1 or HA-CS-Cbln1 (3.5 μg/mL each)
for 4 h and reacted with anti-HA antibody without permeabilization to selectively stain
HA-Cbln1 on the cell surface.
Cbln1 binding assay using immobilized beads in vitro. GluD2NTD-Fc or other Fc
fusion proteins immobilized on Protein G beads were incubated with various
concentrations of HA-Cbln1 in cerebellar culture medium with 1.4% bovine serum
albumin overnight. HA-Cbln1 bound to beads was recovered by magnetic separation
and was washed 4 times with PBS. The final pellet was analyzed by immunoblotting
with anti-HA antibody.
Primary antibodies. Antibodies were used at the following dilutions: anti-GFP
(chicken, 1:2,000), anti-synapsin I (rabbit, 1:500), anti-calbindin (rabbit, 1:1,000),
anti-calbindin (mouse, 1:1,000), anti-calbindin (goat, 1:200), anti-FLAG (rabbit,
1:1,000), anti-synaptophysin (mouse, 1:500), anti-HA (rabbit, 1:1000), anti-HA (mouse,
5

1:1,000), anti-EAAT4 (rabbit; 1:1,000), anti-GluD2 (rabbit; 1:2,000, guinea pig; 1:250)
(S7), anti-pan AMPA receptors (GluA; guinea pig; 1:500) (S8), and anti-Cbln1 (rabbit;
1:240), anti-homer3 (rabbit; 1:1,000) (S9), anti-shank2 (rabbit; 1:500 against 13 amino
acids at most C-terminus region), anti-gephyrin (rabbit; 1:500 against 35 amino acids at
most N-terminus region).
Functional labeling of presynaptic terminals with FM4-64. Mixed cerebellar cultures
prepared from cbln1-null mice were incubated with GluD2NTD-Fc beads with or without
Cbln1 (3.5 μg/mL) from 7 through 12 DIV. Functional presynaptic terminals were
labeled with FM4-64, as described previously (S10). Briefly, cells were incubated
with Tyrode's solution (20 mM HEPES, pH 7.2, 30 mM glucose, 129 mM NaCl, 5 mM
KCl) for 15 min at room temperature and then treated for 5 min with Tyrode's solution,
to which 5 μM of FM4-64, 80 mM of KCl, and 4 mM of CaCl2 had been added. To
quantify the presynaptic terminals on the GluD2NTD-Fc beads, the cells were fixed with
4% paraformaldehyde in PBS immediately after FM4-64 loading. After subtracting
the background level, the mean intensity of the FM4-64 fluorescence on the beads
(which was visualized by its autofluorescence) was measured using IP-lab software, as
described above.
Electrophysiology. Mice at P26-33 of age were anesthetized with the intraperitoneal
injection of ketamine (80 mg/kg body weight) and xylazine (20 mg/kg). HA-Cbln1 (1
μg/g body weight) was injected into the subarachnoidal space with a 33 gauge
microsyringe needle at a rate of 40 μL/h, as described previously (S3). Twenty-four
hours after the Cbln1 injection, a 2-μL aliquot of the solution containing the
recombinant Sindbis virus (titer, 1.0 × 108−9 TU/mL) was directly injected into the
vermis of cerebellar lobules V–VIII using a glass pipette (30 μm in diameter) and a
microinjector, as described previously (S2).
Cerebellar slices (200-μm-thick coronal sections) were prepared from the
virus-infected cerebella, and whole-cell patch-clamp recordings were performed from
Purkinje cells that emitted GFP fluorescence, as previously described (S2). The
resistances of the patch pipettes were 3 – 5 MΩ when filled with an internal solution of
6

the following composition (in mM): 65 Cs-methanesulfonate, 65 K-gluconate, 20
HEPES, 10 KCl, 1 MgCl2, 4 Na2ATP, 1 Na2GTP, 5 sucrose, and 0.4 EGTA, pH 7.25
(295 mOsm/kg). The solution used for slice storage and recording was composed as
follows (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3,
and 10 D-glucose. This solution was bubbled continuously with a mixture of 95% O2
and 5% CO2 at room temperature. Picrotoxin (100 μM) was always present in the
saline to block inhibitory synaptic transmission. To elicit the PF-evoked excitatory
postsynaptic currents (PF-EPSCs), a stimulating glass pipette was placed on the
molecular layer (square pulse, 10 μs, 0 to 200 μA), and the selective stimulation of PFs
was confirmed by the paired-pulse facilitation of PF-EPSCs with a 50-ms stimulation
interval.
Data analysis and statistics. Data are presented as the mean ± SEM, and statistical
significance was determined using a paired Student’s t test, unless otherwise noted.
7

Fig. S1. Surface transport of GluD2 and its chimeric constructs with GluK2. (A) N-terminal domains (NTD) and ligand-binding domains (LBD; consisting of S1 and S2) of GluD2 and GluK were interchanged as shown in the diagram. (B) HEK293 cells expressing each construct were surface biotinylated, solubilized, and subjected to immunoprecipitation with anti-Flag or anti-GluD2 antibody. Precipitated fractions were blotted and probed with horse radish peroxidase–conjugated avidin (upper panels) or anti-Flag or anti-GluD2 (lower panels). GluD2 with a hotfoot mutation (GluD2ho), which is retained in the endoplasmic reticulum (S11), is used as a negative control.
8

Fig. S2. Specificity of anti-Cbln1 antibody. (A) Cbln1 immunoreactivity was detected in wild-type (wt), but not cbln1-null, cerebellum. ML, molecular layer; PCL, Purkinje cell layer. Scale bar: 20 μm. (B) Postembedding immunogold EM images of endogenous Cbln1. Cbln1 was selectively immunolabeled (arrows) at parallel fiber (PF)–Purkinje cell synapses in the wild-type (wt), but not in the cbln1-null, cerebellum. Arrowheads indicate the edges of the PSD on the spines (sp) of Purkinje cells. Scale bar: 200 nm.
Fig. S3. No marked differences in the levels or profiles of Cbln1 or PSD93 were observed between wild-type and GluD2-null cerebellum. Immunoblot analyses of PSD93 (upper panel), GluD2 (middle panel) and Cbln1 (lower panel) in cbln1-null, wild-type (wt), and GluD2-null cerebella are shown. The arrowheads indicate the degradation products of Cbln1 (S12).
9

Fig. S4. Both Cbln1 and GluD2 were necessary to induce synapse formation in cultured Purkinje cells. Wild-type (wt), cbln1-null, GluD2-null and cbln1-, GluD2-double null (cbln1/GluD2-null) Purkinje cells were incubated with or without HA-Cbln1 (3.5 μg/mL) for 7 d and immunostained against calbindin (calb; green) and synaptophysin (Syn; red or white) at 14 DIV. The Purkinje cell dendrites marked by the white boxes in the top panels are enlarged in the middle and bottom panels. Scale bars: 50 μm (top panels), 20 μm (middle and bottom panels). The mean intensities of the synaptophysin immunoreactivity in the calbindin-positive areas are summarized in the lower graph. Error bars represent the SEM. At least n = 24 cells were analyzed in three independent experiments. **P = 1.04 x 10-17.
10

Fig. S5. The NTD of GluD2 (GluD2NTD) inhibits synapse formation in wild-type Purkinje cell dendrites. Wild-type mixed cerebellar cultures were incubated with GluD2NTD-Fc or HA-Fc (1 μg/mL each) from 3 through 10 DIV. Representative images of Purkinje cells immunostained against GluD2 (green) and synaptophysin (Syn; red or white) are shown. The Purkinje cell dendrites marked in the upper panels were enlarged in the lower panels. Scale bars: 50 μm (top panels), 20 μm (middle and bottom panels). The mean intensity of synaptophysin immunoreactivity in the GluD2-positive dendrites of Purkinje cells is shown in the right graph. The error bars represent the SEM. At least n = 36 cells were analyzed in three independent experiments. **P = 8.9 x 10-8.
11

Fig. S6. Ataxic phenotypes of cbln1/GluD2-null mice. (A) Representative gait patterns of wild-type (wt), cbln1-null, GluD2-null, and cbln1/GluD2-null mice. Paws of each mouse were marked with black paint. (B) Results for the rotor-rod test. Mice were placed on the rod rotating at 20 rpm. n = 3 mice for each genotype.
12
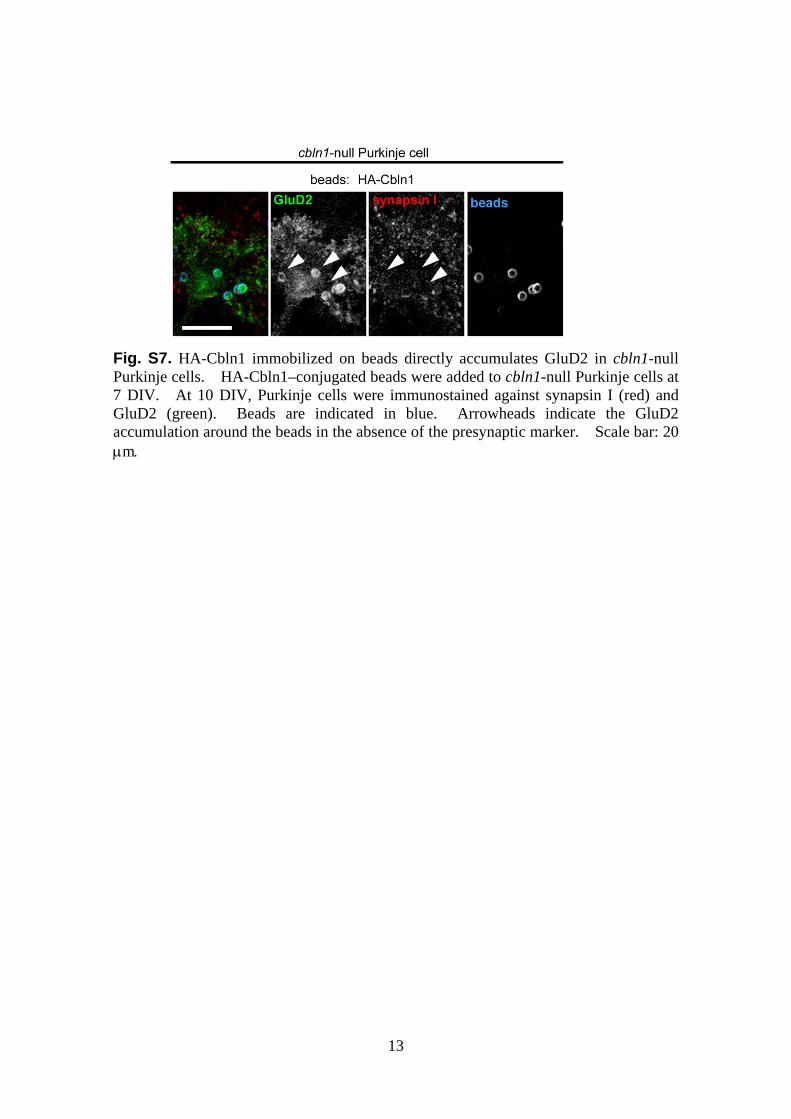
Fig. S7. HA-Cbln1 immobilized on beads directly accumulates GluD2 in cbln1-null Purkinje cells. HA-Cbln1–conjugated beads were added to cbln1-null Purkinje cells at 7 DIV. At 10 DIV, Purkinje cells were immunostained against synapsin I (red) and GluD2 (green). Beads are indicated in blue. Arrowheads indicate the GluD2 accumulation around the beads in the absence of the presynaptic marker. Scale bar: 20 μm.
13

Fig. S8. HA-Cbln1 immobilized on beads specifically accumulates GluD2 in HEK293 cells. (A, B) HEK293 cells expressing GluD2, GluK2-Flag, GluK2ext-GluD2, GluD2ext-GluK2-Flag or GluD2ΔNTD were incubated with control, HA-CS-Cbln1– or HA-Cbln1–conjugated beads for 1 d. Cells were immunostained against GluD2 or Flag (red); beads are indicated in green (A). Scale bar: 20 μm. The quantification of the intensity of GluD2 or GluK2 immunoreactivity around the beads is shown in the graph (B). The error bars represent the SEM. At least n = 20 cells for each group were analyzed in two independent experiments. **P = 6.17 x 10-6 (by Kruskal-Wallis test in a group using HEK293 cells expressing GluD2) and 4.99 x 10-9 (in the rest of the group).
14

Fig. S9. HA-Cbln1 immobilized on beads accumulates GluD2 but not EAAT4 in cbln1-null Purkinje cells. HA-Cbln1–conjugated beads were added to cbln1-null Purkinje cells at 7 DIV. At 10 DIV, Purkinje cells were immunostained against EAAT4 (red) and GluD2 (green). Beads are indicated in blue. Arrowheads indicate the GluD2 accumulation around the beads. Scale bar: 20 μm.
15

Fig. S10. HA-Cbln1–coated beads induce clustering of PSD95 or shank-2 by in a manner dependent on specific C-terminal cytoplasmic regions of GluD2. (A) Schematic drawings of the deletion mutants of GluD2. The gray boxes indicate the 4th transmembrane domain (TM4). The red and black boxes indicate the shank-binding (S13) and PDZ ligand sites, respectively. The lower numbers indicate the amino acid positions. The PDZ ligand domain was disrupted by adding the Flag tag at the end of C-terminus (GluD2-FL). Three residues essential for binding to shank (Ser 905, Thr 915, and Phe 917 (S13)) were substituted with alanines (GluD2-AAA). (B) HEK293 cells coexpressing GluD2 constructs and YFP-tagged PSD95 (or GFP-tagged shank2)
16

were incubated with HA-Cbln1–conjugated beads for 2 d. Cells were immunostained against GluD2 (red); beads are indicated in blue. Note that PSD95 and shank-2 accumulated around beads only when the responsible intracellular regions were intact. Scale bar: 20 μm.
Fig. S11. Specific immunogold labeling for GluD2 by SDS-digested freeze-fracture replica labeling in the molecular layer of mouse cerebellum. (A, B) GluD2 immunoreactivity was examined in wild-type (WT) and GluD2-null (KO) mice with anti-GluD2 antibody. Immunoparticles for GluD2 (10 nm) were accumulated over a postsynaptic membrane specialization, which was identified by clustering of intra-membrane particles on the exoplasmic-face of the plasma membrane in wild-type (A), but not in GluD2-null (B), mouse cerebellum. An arrow indicates background labeling on the protoplasmic-face of the putative presynaptic membrane (B). (C,D) Double labeling using anti-pan GluA (5 nm particles) and anti-GluD2 (15 nm particles) antibodies in a Sprague-Dawley rat (C) and a GluD2-null mouse (D). Immunoparticles for GluD2, but not those for AMPA receptors, were absent in GluD2-null mouse. (E) Quantification of immunogold particles for GluD2 in wild-type (WT) and GluD2-null mice. The number of GluD2 immunoparticles was positively correlated with the synapse size (r = 0.924, p < 0.001, Pearson). Labeling density in WT = 468 ± 125.3 particle/μm2 (Mean ± SD), n = 23; in GluD2-null = 4 ± 17.4 particle/μm2, n = 24. Scale bars: 200 nm.
17

GluD2 GluD1 GluD2 GluD1GFP
A
B
GFP
Fig. S12. Cbln1 binds to the NTD of GluD1 as well as GluD2. (A) Cbln1 binding assay using intact HEK293 cells. HEK293 cells expressing GFP and GluD2 or GluD1 were incubated with HA-Cbln1 (3.5 μg/mL) for 4 h. Bound HA-Cbln1 on transfected HEK293 cells (GFP; green) was visualized by immunostaining against HA (red). Scale bar: 50 μm. (B) Pull-down assay showing a direct interaction between HA-Cbln1 and the NTD of GluD1. The schematic diagram shows the NTD of GluD2, GluD1, or the extracellular domain of CD4 fused to Fc and coupled to magnetic protein G beads. The beads were then incubated with HA-Cbln1, and bound HA-Cbln1 was separated and quantified using an immunoblot analysis with anti-HA (upper panel) and anti-Fc (lower panel) antibodies.
HA-Cbln1
Fcbeads GluD
2-Fc
CD4-Fc
GluD1-F
c
None
und-Cbln1
Fc
+ -Cbln1
boHA
HA
HA-Cbln1
GFP
-CS-Cbln1+ -Cbln1
NTD:NTD
HA-CS-Cbln1
+HAHA
GluD2 GluD1 GluD2 GluD1GFP
bln1
Fcbeads
A
B
GFP
HA-Cbln1
HA-C
GluD2-F
c
CD4-Fc
GluD1-F
c
None
und-Cbln1
Fc
+ -Cbln1
boHA
HA
GFP
-CS-Cbln1+ -Cbln1
NTD:NTD
HA-CS-Cbln1
+HAHA
18

19
Supplemental References
S1. K. Matsuda et al., Eur J Neurosci 29, 707 (2009).
S2. W. Kakegawa et al., J Neurosci 29, 5738 (2009).
S3. A. Ito-Ishida et al., J Neurosci 28, 5920 (2008).
S4. H. Hirai et al., Nat Neurosci 8, 1534 (2005).
S5. M. Masugi-Tokita et al., J Neurosci 27, 2135 (2007).
S6. A. S. Landsend et al., J Neurosci 17, 834 (1997).
S7. T. Takeuchi et al., J Neurosci 25, 2146 (2005).
S8. M. Fukaya et al., Eur J Neurosci 24, 2177 (2006).
S9. Y. Shiraishi, A. Mizutani, S. Yuasa, K. Mikoshiba, T. Furuichi, J Comp Neurol 473,
582 (2004).
S10. T. Iijima, K. Emi, M. Yuzaki, J Neurosci 29, 5425 (2009).
S11. S. Matsuda, M. Yuzaki, Eur J Neurosci 16, 1507 (2002).
S12. D. Bao, Z. Pang, J. I. Morgan, J Neurochem 95, 618 (2005).
S13. T. Uemura, H. Mori, M. Mishina, Mol Cell Neurosci 26, 330 (2004).