Embed Size (px)
Citation preview

SSyynntthheessee,, CChhaarraakktteerriissiieerruunngg uunndd
UUnntteerrssuucchhuunnggeenn zzuumm RReeaakkttiioonnssvveerrhhaalltteenn vvoonn
BBoorraazziiddeenn uunndd BBoorryyllnniittrreenneenn
CC‐‐HH‐‐TTrraannssffoorrmmaattiioonn uunndd ddiirreekkttee AAmmiinniieerruunngg vvoonn AAllkkaanneenn
DDiisssseerrttaattiioonn
vvoorrggeelleeggtt vvoonn
MMaatttthhiiaass FFiilltthhaauuss
aauuss GGeellsseennkkiirrcchheenn
AApprriill 22001100

Die dieser Arbeit zugrundeliegenden Experimente sind in der Zeit von 12/2006 bis
12/2009 am Lehrstuhl für Organische Chemie II der Ruhr‐Universität Bochum
unter Anleitung von Herrn Prof. Holger F. Bettinger (seit 10/2008: Institut für
Organische Chemie der Universität Tübingen) durchgeführt worden.
Erster Referent: Prof. Holger F. Bettinger
Zweiter Referent: Prof. Martin Feigel


Für Anne, Henning & meine Eltern

Abkürzungsverzeichnis
AcCl Acetylchlorid
Ar Aryl
BDE Bindungsdissoziationsenergie
Cat Catechol (Brenzcatechin, 1,2‐Dihydroxybenzol)
DCM Dichlormethan
DFT Dichtefunktionaltheorie
Et Ethyl
Et2O Diethylether
FS Feststoff
GC Gaschromatographie
GZ Grundzustand
h Stunden
iPr Isopropyl
ISC Intersystem Crossing
LM Lösungsmittel
Me Methyl
Min. Minuten
MS Massenspektrometrie, Massenspektrum
NP Nebenprodukt
PES Potentialenergiehyperfläche
Pin Pinakol (2,3‐Dimethyl‐2,3‐butandiol)
RP Reaktionsprodukt
RS Rückstand
RSP Reaktivitäts‐Selektivitäts‐Prinzip
RT Zimmertemperatur
S Singulett
Sdp. Siedepunkt
T Triplett
THF Tetrahydrofuran
TMSCl Trimethylsilylchlorid
TMSN3 Trimethylsilylazid
Tos Tosyl
UV Ultraviolett
ÜZ Übergangszustand
WW Wechselwirkungen


Inhalt
1
Inhaltsverzeichnis 1 Ziel dieser Arbeit.......................................................................................................................... 7 2 Kurzzusammenfassung wichtiger Ergebnisse ................................................................................ 8 3 Hintergrundinformationen und Motivation ................................................................................ 10
3.1 „BN“ vs. „CC“ ........................................................................................................................... 10 3.1.1 Übersicht............................................................................................................................. 10 3.1.2 Bornitrid.............................................................................................................................. 11 3.1.3 Borazin (Borazol)................................................................................................................. 12 3.1.4 Boran‐Amin Addukte .......................................................................................................... 13 3.1.5 Aminoborane ...................................................................................................................... 15 3.1.6 Iminoborane ....................................................................................................................... 16 3.1.7 Weitere BN‐ und Organoborspezies auf Basis organischer Verbindungen. ....................... 17 3.1.8 Reaktive BN‐ und Organoborsysteme ................................................................................ 19
3.2 Nitrene ..................................................................................................................................... 20 3.2.1 Reaktivität und Reaktionsverhalten ................................................................................... 20 3.2.2 Bindungssituation ............................................................................................................... 24 3.2.3 Borylnitrene: Stand der Forschung..................................................................................... 26
4 Eigene Arbeiten ......................................................................................................................... 29 4.1 Computerchemische Untersuchungen: Borylnitrene und ihren Verwandten......................... 29
4.1.1 Zielsetzung und Motivation ................................................................................................ 29 4.1.2 Verwendete Rechenmethode............................................................................................. 30 4.1.3 Eine einfache Beschreibung der elektronischen Struktur von Borylnitrenen R2BN ........... 32
4.1.3.1 Triplett‐Borylnitren........................................................................................................ 33 4.1.3.2 Singulett‐Borylnitren ..................................................................................................... 34 4.1.3.3 Beschreibung von Borylnitrenen mit vereinfachten Konzepten der MO‐ und
Ligandenfeldtheorie......................................................................................................... 36 4.1.4 Untersuchungen zum Einfluss der Liganden R auf EST...................................................... 38
4.1.4.1 Einfache σ‐Liganden ...................................................................................................... 38 4.1.4.2 Mesomere Effekte ......................................................................................................... 39
4.1.4.2.1 ‐Donoren (σ‐Akzeptoren).................................................................................. 39 4.1.4.2.2 ‐Akzeptoren....................................................................................................... 43 4.1.4.2.3 ‐Konjugation...................................................................................................... 44
4.1.5 Rechnungen zu (H2BN)x‐Isomeren ...................................................................................... 46 4.2 Experimentelle Arbeiten.......................................................................................................... 52
4.2.1 Borazide und ihre Vorstufen............................................................................................... 52 4.2.1.1 Überblick........................................................................................................................ 52 4.2.1.2 Säure‐Base Chemie: Borazid LS‐LB Addukte.................................................................. 54 4.2.1.3 Charakterisierung und Identifizierung von Boraziden und ihren Addukten.................. 54 4.2.1.4 Synthese cyclischer und acyclischer disubstituierter Bormonochloride ....................... 56
4.2.1.4.1 Synthese disauerstoffsubstituierter Borchloride ................................................ 56 4.2.1.4.2 Synthese cyclischer diaminosubstituierter Bormonochloride ............................ 58 4.2.1.4.3 Weitere Bormonochloride .................................................................................. 59
4.2.2 Versuche zur direkten Aminierung von Kohlenwasserstoffen mithilfe von Borylnitrenen 60 4.2.2.1 Motivation und Zielsetzung........................................................................................... 60
4.2.2.1.1 Exkurs: Die Aktivierung und Funktionalisierung von Methan (und seiner höheren Homologen)......................................................................................................... 61
4.2.2.1.2 Methan: Quellen und technische Nutzung ......................................................... 61 4.2.2.1.3 Industrielle Wunschreaktionen ausgehend von Methan.................................... 63 4.2.2.1.4 Der Funktionalisierungsprozess .......................................................................... 65 4.2.2.1.5 Übergangsmetallkatalysierte C‐H‐Aktivierung .................................................... 67 4.2.2.1.6 Übergangsmetallkatalysierte Aminierung von Alkanen...................................... 68

Inhalt
2
4.2.2.1.7 Alkantransformationen mithilfe weiterer Systeme ............................................ 72 4.2.2.2 Photolyse von PinBN3 in (Cyclo)alkanen........................................................................ 73
4.2.2.2.1 Abbau‐ und Derivatisierungsexperimente an PinBNHCy: Bestimmung der Ausbeute an Aminierungsprodukten .................................................................. 78
4.2.2.2.2 Regioselektivität der C‐H‐Aminierung: 2,3‐Dimethylbutan (DMB) als Substrat.. 86 4.2.2.2.3 Mechanistische Überlegungen zur C‐H‐Aminierung: Konzertierte Insertion vs.
Abstraktion‐Rekombination................................................................................ 88 4.2.2.3 Photolyse von Dialkoxyboraziden (RO)2BN3 in Cycloalkanen ........................................ 92 4.2.2.4 Photolyse aromatischer disauerstoffsubstituierter Borazide in Cycloalkanen und
Aromaten......................................................................................................................... 97 4.2.2.4.1 Photolyse von CatBN3 und t‐BuCatBN3 in Cycloalkanen Cy‐H und Benzol .......... 97 4.2.2.4.2 Photolyse des Bisazids 171 in Cyclohexan Cy‐6‐H............................................. 104 4.2.2.4.3 Photolyse von Azidoboran 177 in den Aromaten Benzol, Toluol und Mesitylen 106 4.2.2.4.4 Photolyse von (PhO)BN3 in Cyclohexan ............................................................ 109
4.2.2.5 Photolyse stickstoff‐ und schwefelsubstituierter Borazide in Kohlenwasserstoffen .. 111 4.2.2.5.1 Photolyse von 2‐Azido‐1,3‐dimethyl‐1,3,2‐diazaborolidin in Cyclohexan ........ 111 4.2.2.5.2 Photolyse von 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin in Benzol.................... 115 4.2.2.5.3 Photolyse von 2‐Azido‐1,3,2‐dithiaborolan in Cyclohexan ............................... 117
4.2.2.6 Versuche zur präparativen Aminierung von Methan .................................................. 118 4.2.2.6.1 Versuche zur Reaktion von PinBN3 in flüssigem Methan.................................. 118 4.2.2.6.2 Reaktion von gasförmigen Methan in Lösung................................................... 120 4.2.2.6.3 Gasphasenphotoreaktion von Methan und PinBN3 .......................................... 122
4.2.2.6.3.1 Charakterisierung der Gasphasephotoprodukte ........................................ 123 4.2.2.6.3.2 Weitere Produkte und mechanistische Interpretationen........................... 124
4.2.3 Versuche zur Synthese von Borylaziridinen...................................................................... 129 4.2.3.1 Alkene als Substrate: Photolyse von PinBN3 in Tetramethylethen (TME) und Cyclohexen 130
4.2.4 Versuche zur Synthese von Boryltria‐ und tetrazolen via „Click‐Chemie“........................ 132 4.2.5 Versuche zur Synthese eines Borylnitrendimers .............................................................. 137 4.2.6 Thermolysen von Boraziden in Kohlenwasserstoffen ...................................................... 138
5 Ausblick ................................................................................................................................... 139 5.1 Substraterweiterung.............................................................................................................. 139 5.2 Erhöhung der Selektivität ...................................................................................................... 140 5.3 Die intramolekulare C‐H‐Insertion in der Synthesechemie ................................................... 142 5.4 Borylnitrene: Effiziente Stickstoffüberträger in der angewandten Forschung ?! .................. 143
6 Experimenteller Teil ................................................................................................................. 146 6.1 Allgemeines ........................................................................................................................... 146
6.1.1 Sicherheitshinweis ............................................................................................................ 146 6.1.2 Arbeitstechnik................................................................................................................... 146 6.1.3 Lösungsmittel und Chemikalien........................................................................................ 147 6.1.4 Interne Standards ............................................................................................................. 147 6.1.5 Theoretische Berechnungen............................................................................................. 148 6.1.6 Lichtquelle ........................................................................................................................ 148 6.1.7 Instrumentelle Analytik .................................................................................................... 148
6.1.7.1 NMR‐Spektroskopie..................................................................................................... 148 6.1.7.2 GC/MS‐ und GC‐Messungen........................................................................................ 149 6.1.7.3 Massenspektrometrie ................................................................................................. 149 6.1.7.4 Infrarotspektren .......................................................................................................... 149 6.1.7.5 Röntgenstrukturanalysen ............................................................................................ 150
6.2 Synthesen .............................................................................................................................. 151 6.2.1 Synthese von Pinakolborchlorid (108)[249, 323] ................................................................... 151 6.2.2 Synthese von tert‐BuCatBCl [180] ....................................................................................... 153

Inhalt
3
6.2.3 Synthese von Diisopropyloxyborchlorid (iPrO)2BCl[184, 186]................................................ 154
6.2.4 Synthese von Diethoxyborchlorid (EtO)2BCl ..................................................................... 156 6.2.5 Synthese von 2‐Chloro‐Naphto(2,3‐d)‐1,3,2‐dioxaborol .................................................. 157
6.2.5.1 Synthese von 2,3‐Bis‐(trimethylsilanyloxy)naphthalin[182, 324] ..................................... 157 6.2.5.2 Synthese von 2‐Chloro‐Naphto(2,3‐d)‐1,3,2‐dioxaborol [182, 183] ................................. 158
6.2.6 Synthese des Bischlorids 175............................................................................................ 160 6.2.6.1 Synthese von 1,2,4,5‐Tetrahydroxybenzol (173)......................................................... 160 6.2.6.2 Synthese von 1,2,4,5‐Tetrakis‐(trimethylsilanyloxy)benzol (174)[183].......................... 161 6.2.6.3 Synthese des Bisborchlorids 175[183]............................................................................ 162
6.2.7 Synthese von 2‐Chloro‐1,3‐ditosyl‐1,3,2‐diazaborolidin (202) ......................................... 164 6.2.7.1 Synthese von N,N´‐Ditosyl‐1,2‐ethandiamin (203)[270‐272]............................................ 164 6.2.7.2 Synthese von 2‐Chloro‐1,3‐ditosyl‐1,3,2‐diazaborolidin (202)[190‐192].......................... 165
6.2.8 Synthese von 2‐Chloro‐1,3‐dimethyl‐1,3,2‐diazaborolidin [189, 197] ................................... 167 6.2.9 Synthese von 2‐Chloro‐1,3,2‐dithiaborolan (117)[179, 193, 194, 325] ....................................... 169 6.2.10 Azidierung......................................................................................................................... 171 6.2.11 Allgemeine Synthesevorschrift der Azidierung[34, 172‐174]................................................... 171 6.2.12 Synthese von Pinakolborazid PinBN3 (9) ........................................................................... 172 6.2.13 Synthese von Diisopropyloxyborazid (iPrO)2BN3 (150‐(iPr)2)............................................ 173 6.2.14 Synthese von Diethoxyborazid (EtO)2BN3 (150‐(Et)2) ....................................................... 174 6.2.15 Synthese des Bisazids 171 ................................................................................................ 175 6.2.16 Synthese von 2‐Azido‐Naphto(2,3‐d)‐1,3,2‐dioxaborol (177) .......................................... 176 6.2.17 Synthese von tert‐BuCatBN3 (7‐(tBu)) .............................................................................. 177 6.2.18 Synthese von 2‐Azido‐1,3‐dimethyl‐1,3,2‐diazaborolidin (191)[108].................................. 178 6.2.19 Synthese von 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin (192)........................................... 179 6.2.20 Synthese von 2‐Azido‐1,3,2‐dithiaborolan (193) .............................................................. 180 6.2.21 Versuche zur Synthese von Boryltria‐ und tetrazolen via „Click‐Chemie“........................ 181
6.3 Photolysen ............................................................................................................................. 182 6.3.1 Allgemeine Vorschrift für Photolysen in Lösung............................................................... 182 6.3.2 Allgemeine Vorschrift für die Aufarbeitung der Photoprodukte (Abbau‐ und Derivatisierungsexperimente) .......................................................................................................... 182
6.3.2.1 Abbau durch Alkoholyse.............................................................................................. 182 6.3.2.2 Acetylierung................................................................................................................. 183
6.3.3 Repräsentative Beispiele für Photolysen in Lösung.......................................................... 183 6.3.3.1 Photolyse von PinBN3 (9) in Cycloalkanen Cy‐H. ......................................................... 184
6.3.3.1.1 Aufarbeitung von PinBNHCy (132) .................................................................... 186 6.3.3.1.2 Derivatisierung durch Acetylierung................................................................... 187
6.3.3.2 Photolyse von (iPrO)2BN3 (150‐(iPr)2) in Cyclohexan ................................................... 188 6.3.3.3 Photolyse von (EtO)2BN3 (150‐(Et)2) in Cyclohexan..................................................... 189 6.3.3.4 Photolyse von 2‐Azido‐Naphto(2,3‐d)‐1,3,2‐dioxaborol (177) in Benzol .................... 190
6.3.4 Gasphasenphotolyse: Aminierung von Methan ............................................................... 192 6.3.4.1 Vorbemerkung............................................................................................................. 192 6.3.4.2 Apparativer Aufbau ..................................................................................................... 192 6.3.4.3 Durchführung der Photolyse ....................................................................................... 194 6.3.4.4 Aufarbeitung der Photoprodukte ................................................................................ 195
6.3.5 Aminierung einer Polyethylen‐Oberfläche ....................................................................... 198 7 Anhang.....................................................................................................................................200
7.1 Kristallstrukturdaten.............................................................................................................. 200 7.2 Referenzen............................................................................................................................. 212

Inhalt
4
Abbildungsverzeichnis Abbildung 1 Borylnitrene und ihre klassischen Verwandten aus der Organischen Chemie. ......................... 7 Abbildung 2: Bindungspolarität und allgemeines Reaktivitätsmuster von BN‐Systemen............................ 11 Abbildung 3: Benzol und seine experimentell bekannten BN‐Analoga........................................................ 13 Abbildung 4: Boran‐Amin Addukte, die BN‐Analoga der Alkane.................................................................. 13 Abbildung 5: AAmmiinnoobboorraannee uunndd DDeerriivvaattee...................................................................................................... 15 Abbildung 6: Weitere repräsentative Beispiele für Borasysteme, die in enger Verwandtschaft zu reinen
organischen Verbindungen stehen. .................................................................................................... 17 Abbildung 7: Organoborverbindungen auf der Basis von Aromaten, Antiaromaten und NHCs. ................. 18 Abbildung 8: Reaktive Organoborverbindungen und ihre organischen Analoga. ........................................ 19 Abbildung 9: Wichtige Konfigurationen und elektronische Zustände des Phenylnitrens, wobei S2 und S3
Multikonfigurationscharakter besitzen. ............................................................................................. 25 Abbildung 10: Borylnitrene im einfachen Orbitalbild................................................................................... 32 Abbildung 11: Mögliche Orbitalbesetzungen des Stickstoffs in Borylnitrenen. ........................................... 33 Abbildung 12: Das Triplett‐Borylnitren im einfachen Orbitalbild................................................................. 33 Abbildung 13: Das Singulett‐Borylnitren im einfachen Orbitalbild. ............................................................. 34 Abbildung 14: Elektronenzustände bestimmter Nitrene und Carbene........................................................ 35 Abbildung 15: Stark vereinfachtes Korrelationsdiagramm für Borylnitrene. ............................................... 37 Abbildung 16 Borylnitrene R2BN mit σ‐Liganden. ........................................................................................ 38 Abbildung 17: Offene symmetrische Borylnitrene X2BN mit π‐Donorliganden X. ....................................... 39 Abbildung 18: Schematische Darstellung des Einflusses von π‐Donoren auf die elektronische Struktur in
Borylnitrenen. Oben im Lewis‐Resonanzbild, unten in der dazugehörigen Orbitaldarstellung. ........ 40 Abbildung 19: Resonanzstrukturen von CN2BN und (H2N)2BN, welche Rückschlüsse auf die π‐
Donorfähigkeit zulassen...................................................................................................................... 42 Abbildung 20: Schematische Darstellung des Einflusses von π‐Akzeptoren auf die elektronische Struktur in
Borylnitrenen. Oben im Resonanzbild, unten in der dazugehörigen Orbitaldarstellung.................... 43 Abbildung 21: Donorsubstituierte Borylnitrene und Konjugation. .............................................................. 44 Abbildung 22: Grundkörper von Borylnitrenen auf der Basis von Aromaten bzw. Antiaromaten............... 45 Abbildung 23: Berechnete (B3LYP/6‐311+G**) Geometrien für Diazadiboretidine. ................................... 48 Abbildung 24: Berechnete Strukturen für (H2BN)‐Trimere. ......................................................................... 49 Abbildung 25: Berechnete Strukturen zweier H2BN‐Tetramere. ................................................................. 50 Abbildung 26: Berechnete (B3LYP/6‐311+G**) Geometrien für CatBN3 und dessen Pyridinaddukt........... 55 Abbildung 27: Aktuelle und zukünftig denkbare Quellen von industriell nutzbarem Methan. ................... 62 Abbildung 28: Wichtige molekulare homogene Katalysatoren für die Methanaktivierung......................... 68 Abbildung 29: Kristallstruktur von PinBNHCy (R= Cy‐5, Cy‐7). ..................................................................... 75 Abbildung 30: Das PinBNHCy‐6 Dimer.......................................................................................................... 76 Abbildung 31 Berechnete (B3LYP/6‐311+G**) Geometrien und Energien für die Produkte einer
intramolekularen Insertion von (EtO)2BN. .......................................................................................... 94 Abbildung 32 Aromatische disauerstoffsubstituierte Azidoborane, die im Folgenden diskutiert werden. . 97 Abbildung 33: Denkbare isomeren Aminierungsprodukte nach Alkoholyse.............................................. 107 Abbildung 34: Isomere der Mesitylenaminierung...................................................................................... 108 Abbildung 35: Donorsubstituierte Borazide auf Stickstoff‐ und Schwefelbasis. ........................................ 111 Abbildung 36: Berechnete (B3LYP/6‐311+G**) Minimumgeometrie für Boracyclus 196.......................... 113 Abbildung 37: Kristallstruktur von Azidoborolidin 191. ............................................................................. 116 Abbildung 38: Gewünschte methylierte Azide mit Borolidingrundgerüst. ................................................ 116 Abbildung 39: Photolyseapparatur für die (beabsichtigte) Reaktion zwischen PinBN3und flüssigem Methan.
.......................................................................................................................................................... 119 Abbildung 40: Versuche zur Photolyse von Methan in einer PinBN3‐Lösung............................................. 120 Abbildung 41: Schematische Darstellung des entwickelten Gasphasenphotoreaktors. ............................ 122 Abbildung 42: Produkte aus der Reaktion von PinBN3 mit TME. ............................................................... 131 Abbildung 43: Produkte aus der Reaktion von PinBN3 mit Cyclohexen. .................................................... 131

Inhalt
5
Abbildung 44: Konzertierte 1,3‐Dipolare Cycloaddition unterschiedlicher Azide an Alkine und Nitrile. ... 134 Abbildung 45: Berechnete (B3LYP/6‐311+G**) Geometrien ausgewählter Diazoborane. ........................ 137 Abbildung 46: Farbige Borazide die interessante neue Syntheseziele bilden. ........................................... 140 Abbildung 47: Borylnitrenmetallkomplexe und ihre bekannten Verwandten. .......................................... 141 Abbildung 48: Ein Borazid mit stereochemischer Information. ................................................................. 142
Verzeichnis der Reaktionsschemata
Schema 1: Allgemeines Schema für die Aminierung von Kohlenswasserstoffen mithilfe von Borylnitrenen.9 Schema 2: Dehydrogenierung eines Boran‐Amin Addukts unter Bildung eines Aminoborans.................... 14 Schema 3: Oligomerisierung von Iminoboranen. ......................................................................................... 16 Schema 4: Synthese von Iminoboranen ausgehend von Boraziden............................................................. 17 Schema 5: 1,2‐Umlagerung von Alkylnitrenen R3CN 46. .............................................................................. 20 Schema 6: Acylnitrene und ihre Produkte.................................................................................................... 21 Schema 7: Bildung von Carbazolen über intramolekulare C‐H‐Insertion. .................................................... 21 Schema 8: Reaktionen des Phenylnitrens. ................................................................................................... 22 Schema 9: Photolyse von C6F5N3 in Gegenwart unterschiedlicher Substrate. ............................................. 23 Schema 10: Mechanismus der Iminoboranbildung...................................................................................... 26 Schema 11: Matrixexperimente mit CatBN3................................................................................................. 28 Schema 12: Die Borylnitren‐Iminoboran Umlagerung am Beispiel von Dimethylborylnitren...................... 39 Schema 13: Die Iminoboran‐Borylnitren‐Aminoborylen Umlagerung ......................................................... 46 Schema 14: Allgemeines Syntheseschema für die Herstellung der Borazide des Typs R2BN3. .................... 52 Schema 15: Hydrolyse von Boraziden unter Bildung von HN3. .................................................................... 53 Schema 16: Lewis‐Säure‐Base Reaktionen von Boraziden mit Pyridin (Py).................................................. 54 Schema 17: Sauerstoffsubstituierte Borazide, die im Rahmen dieser Arbeit abgehandelt werden. ........... 57 Schema 18: Herstellung acyclischer Borchloride des Typs (RO)2BCl durch Austauschreaktion. .................. 57 Schema 19: Synthese der in dieser Arbeit relevanten diaminosubstituierten Bormonochloride................ 58 Schema 20: Synthese von 2‐Cloro‐1,3,2‐dithiaborolan ausgehend von 1,2‐Ethan‐Dithiol. ......................... 59 Schema 21: Synthese von Diarylbormonochloriden Ar2BCl über Transmetallierung................................... 59 Schema 22: Direkte Aminierung von Alkanen mithilfe von Borylnitrenen................................................... 60 Schema 23: Aktuelle und zukünftig‐denkbare großtechnische Transformationen von Methan.. ................ 63 Schema 24: Konventionelle industrielle Synthese von Methylamin. ........................................................... 64 Schema 25: Analogie zwischen Insertion und oxidativer Addition............................................................... 66 Schema 26: Selektivitätsproblem der Methanfunktionalisierung................................................................ 66 Schema 27: Übergangsmetallkatalysierte C‐H‐Aminierungen nach dem Nitren‐Insertionsmechanismus. . 69 Schema 28: C‐H‐Aminierung nach dem C‐H‐Aktivierungsmechanismus...................................................... 70 Schema 29: Repräsentative Beispiele übergangsmetallkatalysierter C‐H‐Aminierungsreaktionen............. 71 Schema 30: Syntheseroute für die Herstellung des neuen PinBN3. ............................................................. 73 Schema 31: Photolyse von PinBN3 in Gegenwart von Cycloalkanen unterschiedlicher Ringgröße.............. 74 Schema 32: Mögliche Bildung von (HO)2BNHCy, ausgehend von PinBNHCy durch photochemische
Fragmentierung. ................................................................................................................................. 77 Schema 33: Abbau von PinBNHCy mit ROH (R = H, Alkyl). ........................................................................... 78 Schema 34: Postulierter Abbauweg von PinBNHCy mithilfe von ROH (R = H, Alkyl) ................................... 79 Schema 35: Umsetzung von PinBNHCy mit Carbonsäurechloriden RCOCl (mit R = Me, t‐Bu, Ph)............... 81 Schema 36: Umsetzung von PinBNHCy mit MeI........................................................................................... 81 Schema 37: Monoalkylierung von primären Aminen R‐NH2. A: Klassisches Beispiel über die „Tosylroute“.
B: Potentiell über „Aminoboranroute“ mit analogem Reaktivitätsmuster......................................... 82 Schema 38: Postulierter Mechanismus für die Reaktion von PinBNHCy mit Elektrophilen EX. ................... 84 Schema 39: Photochemie von PinBN3 in DMB: Untersuchungen zur Regioselektivität ............................... 86

Inhalt
6
Schema 40: Denkbare Mechanismen der Aminoborylierung....................................................................... 89 Schema 41: Photolysen von (RO)2BN3 in Gegenwart von Cy‐6‐H mit anschließender Alkoholyse. ............. 93 Schema 42: Postulierter Reaktionsweg für die intramolekulare C‐H‐Insertion unter Bildung eines 5‐
gliedrigen Boracyclus. ......................................................................................................................... 94 Schema 43: Denkbare intramolekulare C‐H‐Insertion unter Bildung eines gespannten Vierings................ 95 Schema 44: Acetylierung von 153. ............................................................................................................... 96 Schema 45: Denkbarer Reaktionspfad für die intramolekulare CH‐Insertion von (MeO)2BN...................... 96 Schema 46: Denkbarer Reaktionspfad für die Reaktion zwischen CatBN3 und verschiedenen
Kohlenwasserstoffen. ......................................................................................................................... 99 Schema 47: Abbauexperimente an CatBN3‐Photoprodukt. ....................................................................... 102 Schema 48: Experimente zur Synthese von CatBNHCy. ............................................................................. 103 Schema 49: Syntheseweg für die Herstellung des Bisazids. ....................................................................... 104 Schema 50: Denkbare isomere Photoprodukte für die vollständige Reaktion von 171 mit Cy‐6‐H........... 105 Schema 51: Photolyse von 177 in unterschiedlichen Aromaten................................................................ 106 Schema 52: Synthese von (PhO)2Cl durch Austauschreaktion. .................................................................. 109 Schema 53: Postulierter Reaktionsmechanismus für die Bildung der Hydroxyanilinderivate. .................. 110 Schema 54: Postulierter Reaktionspfad der Photolyse für 2‐Azido‐1,3‐dimethyl‐1,3,2‐diazaborolidin in
Cyclohexan........................................................................................................................................ 114 Schema 55: Syntheseroute für die Herstellung von 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin 1................ 115 Schema 56: Zielreaktion zwischen PinBN3 und Methan. ........................................................................... 118 Schema 57: Abbauwege für PinBNHMe. .................................................................................................... 124 Schema 58: Ausgeschlossene intramolekulare Me‐Umlagerung. .............................................................. 125 Schema 59: Postulierte Reaktionspfade für die Gasphasenreaktion zwischen PinBN3 und Methan......... 127 Schema 60: Borylaziridine und denkbare Folgeprodukte. ......................................................................... 129 Schema 61: Hypothetische Addition eines Borylnitrens an eine C=C‐Doppelbindung. ............................. 129 Schema 62: Allgemeines Syntheseschema für die 1,3‐dipolare Cycloaddition an Alkine bzw. Nitrile. ...... 132 Schema 63: Versuche zur Synthese von Boryltria‐ und tetrazolen. ........................................................... 133 Schema 64: Hypothetische Dimerisierung eines Triplett‐Borylnitrens. ..................................................... 137 Schema 65: Denkbare Syntheseroute für Adrenalin(derivate) über eine Borylnitren‐Insertion. .............. 143 Schema 66: Direkte Aminierung von Oberflächen. .................................................................................... 144
Tabellen
Tabelle 1: Mit unterschiedlichen Methoden berechnete EST (kcal/mol) bestimmter Borylnitrene........... 31 Tabelle 2: Berechnete ΔEST offener Borylnitrene des Typs X2BN.................................................................. 40 Tabelle 3: Cyclische Borylnitrene mit π‐Akzeptorliganden........................................................................... 43 Tabelle 4: Berechnete ΔEST bestimmter Ethen‐verbrückter Borylnitrene. ................................................... 45 Tabelle 5: Relative Energien einiger (H2BN)x‐Isomere bezogen auf HNBH................................................... 51 Tabelle 6: Ausbeuten an Aminoboranen, primären Alkylaminen und Amiden............................................ 85 Tabelle 7: Berechnete (B3LYP/6‐311+G**) Energien der Addition unterschiedlicher Azide an HCN und
HCCH. ................................................................................................................................................ 136

Ziel der Arbeit
7
1 Ziel dieser Arbeit Im Fokus dieser Arbeit liegen die aus Boraziden R2BN3 1 photochemisch
generierbaren Borylnitrene R2BN 2 und deren Folge‐ und Reaktionsprodukte.
Abbildung 1 Borylnitrene und ihre klassischen Verwandten aus der Organischen Chemie.
Diese werden ihren Verwandten aus der Kohlenstoffchemie, den Nitrenen RN 3
und Carbenen R2C 4, gegenübergestellt und hinsichtlich ihres Reaktions‐
verhaltens mit den isoelektronischen Vinylidenen R2CC 5 verglichen. Zusätzlich
soll der Einfluss der Gruppe R auf die chemischen Eigenschaften der Borazide 1
bzw. auf deren Abbauprodukte untersucht werden. Die Charakterisierung der
Borylnitrene erfolgt dabei vorrangig mithilfe klassischer Abfangexperimente in
Lösung, die indirekte Rückschlüsse zulassen. Als Grundlage der experimentellen
Untersuchungen dienen die ebenfalls in unserer Arbeitsgruppe durchgeführten
Matrixexperimente zu Systemen dieses Typs. Zusätzlich werden die in dieser
Arbeit vorgestellten experimentellen Befunde durch computerchemische
Rechnungen ergänzt. Dabei stehen fundamentale Fragestellungen zu Reaktivität,
Stabilität und elektronischen Eigenschaften von Borylnitrenen 2 im Fokus der
Untersuchungen. Darüber hinaus soll das Potential der Borylnitrene 2 in der
Synthesechemie untersucht werden.
R
B N
R
R
C C
R
R
C
R
R N
2 5 4 3

Kurzusammenfassung
8
2 Kurzzusammenfassung wichtiger Ergebnisse In unserer Arbeitsgruppe konnte erstmals das donorstabilisierte 2‐Nitreno‐1,3,2‐
benzodioxaborol 6 (Catecholborylnitren, CatBN) aus dem korrespondierenden
Azid CatBN3 7 photochemisch erzeugt und unter Bedingungen der Matrixisolation
direkt nachgewiesen und charakterisiert werden (vgl. Schema 1).[1] Bei unseren
Abfangexperimenten mit Methan als Substrat (Ar, 10 K) zeigte das
Catecholderivat 6 eine hohe Tendenz, unter Bildung von Aminoboran 8 in die
unreaktive sp3‐C‐H‐Bindung zu insertieren.[2] Da die Aktivierung und
Funktionalisierung von unreaktiven C‐H‐Alkanbindungen eine große
Herausforderung der chemischen Forschung darstellt (vgl. 4.2.2.1.1),
veranlassten uns diese Resultate, eine mögliche Methan‐Transformation auch
unter präparativen Laborbedingungen zu untersuchen. Und in der Tat konnte
eigens hierfür im Rahmen dieser Arbeit eine Apparatur entwickelt werden,
welche die photolytische Gasphasenreaktion zwischen 2‐Azido‐4,4,5,5‐
tetramethyl‐1,3‐dioxaborolan 9 (Pinakolborazid, PinBN3) bzw. Pinakolborylnitren
(PinBN) 10 und Methan unter Bildung von Aminoboran 11 erlaubt. Durch
Hydrolyse oder Alkoholyse lässt sich aus 11 die wichtige Basischemikalie
Methylamin MeNH2 freisetzen. Des Weiteren ist die Aminoborylierung auch
unter konventionellen photochemischen Bedingungen in Lösung beobachtbar[3]:
So ergibt die Photolyse (λ = 254 nm) von PinBN3 9 in (Cyclo)alkanen bei
Zimmertemperatur die erwarteten Aminoborane 11 in nahezu quantitativer
Ausbeute. Diese lassen sich anschließend unter Spaltung der BN‐Amino‐
boraneinheit leicht zu wertvollen organischen Aminoderivaten 12 abbauen.

Kurzusammenfassung
9
Schema 1: Allgemeines Schema für die Aminierung von Kohlenswasserstoffen mithilfe von Borylnitrenen.
Neben intermolekularen C‐H‐Funktionalisierungen konnten auch Produkte
intramolekularer Transformationsreaktionen nachgewiesen werden. Photolysen
an acyclischen Boraziden des Typs (RO)2BN3 (R = i‐Pr, Et) ergeben nach Alkoholyse
vicinale Aminoalkohole, deren Bildung auf cyclische intramolekulare
Aminierungsprodukte als Intermediate zurückzuführen ist. Computerchemische
und experimentelle Untersuchungen lassen vermuten, dass sich die
beobachteten Aminoborane 8 bzw. 11 über eine konzertierte C‐H‐Insertion eines
Borylnitrens in seinem Singulett‐Zustand bilden.
O
O
B NO
O
B N3
O
O
B NR
H
O
O
O
O
NH
R
R´
-N2
RH = CH4, Cycloalkane
Insertion
O
O=
"Cat" "Pin"
7 (R = Cat)
R´= H, Ac, t-BuCO, PhCO, Me, Tos
12
RH h
9 (R = Pin)
6 (R = Cat)
10 (R = Pin)
8 (R = Cat)
11 (R = Pin)

Chemie der Bor‐Stickstoff‐Verbindungen
10
3 Hintergrundinformationen und Motivation
3.1 „BN“ vs. „CC“
3.1.1 Übersicht Schon lange ist das Konzept der isoelektronischen, isolobalen, isostrukturellen
und isosteren Verwandtschaft Bestandteil chemiewissenschaftlicher
Überlegungen. Beispielsweise ist der Vergleich von Bor‐Stickstoff‐Verbindungen
mit ihren verwandten Kohlenstoffsystemen, insbesondere mit Hinblick auf die
Sonderstellung der organischen Chemie, von fundamentaler Bedeutung für das
chemische Verständnis. Obwohl bereits weitgehende Erkenntnisse über eine
Reihe von BN‐Stoffklassen vorliegen, hat das Forschungsgebiet nicht an Aktualität
verloren, wie ein kürzlich erschienender Highlight Artikel von Liu und Marder
zeigt.[3] Einige bereits gut untersuchte BN‐Verbindungen werden im Folgenden
kurz vorgestellt und ihren analogen Kohlenstoffsystemen gegenübergestellt.
Dabei besteht ein enger Zusammenhang zwischen der Wahl der hier angeführten
Beispiele und der in dieser Arbeit diskutierten Forschungsergebnisse, wodurch
die hier vorgenommene Auswahl nicht den Anspruch auf Vollständigkeit verfolgt.
Bei weiterem Interesse sei der Leser auf entsprechende Sekundärliteratur
verwiesen.[4‐6] Vorab ist festzuhalten, dass sich BN‐Verbindungen durch größere
Reaktivität gegenüber ihren C‐Analoga auszeichnen, da die BN‐Bindung durch
einen stark polaren Charakter geprägt ist (ΔEN (N/B) = 1.1) und zudem die
Bindungsstärke geringer als in den entsprechenden Kohlenstoffverbindungen
ist.[4] Den Grundlagen der Borchemie entsprechend wird das chemische
Verhalten weitgehend vom Elektronendefizit, der Lewis‐Acidität und der daraus
resultierenden Elektrophilie des Boratoms geprägt. Das Stickstoffzentrum in BN‐
Verbindungen hingegen ist von nucleophilem Charakter, obschon die Lewis‐

Chemie der Bor‐Stickstoff‐Verbindungen
11
Basizität, im Vergleich zu freien Aminoverbindungen, wegen der BN‐
Wechselwirkung (BN‐WW) herabgesetzt ist.
Abbildung 2: Bindungspolarität und allgemeines Reaktivitätsmuster von BN‐Systemen.
(Nu = Nucleophil, E= Elektrophil).
Des Weiteren bleibt anzumerken, dass grundlegend in dieser Arbeit für die
behandelten BN‐Systeme die den organischen Verbindungen analogen Lewis‐
Strukturformeln verwendet werden. Diese geben den vorliegenden
Bindungscharakter am ehesten wieder und zeigen die Verwandtschaft zu den
organischen Stoffklassen am deutlichsten auf. Auf die Angabe von
Formalladungen wird der Einfachheit halber weitgehend verzichtet.
3.1.2 Bornitrid Das einfachste BN‐System, welches in zwei unterschiedlichen Modifikationen
vorliegen kann, ist das polymer aufgebaute Bornitrid (BN)X.[5, 6] Bei der Graphit‐
analogen, formal sp2‐hybridisierten Form bildet sich eine hexagonale,
wabenartige Schichtenstapelung aus. Dabei liegen die Atome, anders als beim
Graphit, nicht versetzt übereinander sondern direkt auf Deckung. Hierbei bildet
sich eine Anordnung aus, bei der jedes Boratom von zwei Stickstoffatomen der
Nachbarschicht umgeben ist und sich analog dazu ober‐ und unterhalb eines
jeden Stickstoffatoms je ein Boratom befindet. Erklärt werden kann dies über
attraktive Wechselwirkungen zwischen B und N, die im Elektronegativitäts‐
unterschied und der daraus resultierenden Bindungspolarität begründet sind.
Prinzipiell handelt es sich hierbei um die gleichen anziehenden Kräfte, die auch
B N+ -
Nu E

Chemie der Bor‐Stickstoff‐Verbindungen
12
bei theoretischen Untersuchungen am Borazindimer, dem Grundkörper des BN‐
Graphits, ermittelt wurden.[7] Wegen der eingeschränkten Beweglichkeit der π‐
Elektronen (Lokalisation überwiegend am Stickstoff), handelt es sich beim
Bornitrid um einen elektrischen Isolator von weißer Farbe.
Zudem existiert noch das BN‐Pendant zum Diamant. Bei dieser kubisch
aufgebauten Modifikation des Bornitrids liegt sp3‐Hybridisierung mit einem BN‐
Einfachbindungsabstand von 1.56 Å (BN‐Graphit: 1.45 Å) vor. Die BN‐Analoga
bzw. BNC‐Hybride der verbleibenden Kohlenstoffallotrope (Fullerene,
Kohlenstoffnanoröhren) sind experimentell nur wenig untersucht,[8‐13]
wenngleich ausgiebige theoretische Abhandlungen zu diesen Systemen
existieren.[10, 11, 13‐19]
3.1.3 Borazin (Borazol) Das erstmals im Jahre 1926 von Stock und Pohland synthetisierte Borazin
(Borazol) 13 B3N3H3[20] verhält sich isoelektronisch und isostrukturell zu Benzol 14,
und wird deshalb auch als das „Anorganisches Benzol“ bezeichnet.[6] Diese D3d
symmetrische, sp2‐hybridisierte, planare Verbindung bildet dabei ein
regelmäßiges Sechseck mit gleichlangen BN‐Bindungsabständen von 1.44 Å aus.
Borazin besitzt ferner ähnliche physikalische Eigenschaften wie Benzol (z. B.
Dichte, Siede‐ und Schmelzpunkt, Verdampfungsenthalpie, Oberflächenspannung
usw.), unterscheidet sich jedoch stark in seinem chemischen Reaktionsverhalten.
So neigt Borazin wegen der BN‐Bindungspolarität zu Additions‐, jedoch nicht zu
Substitutionsreaktionen. Viele Forschungsarbeiten zum Borazin/Benzol‐Paar
beschäftigen sich sowohl von experimenteller als auch von theoretischer Seite
aus im Wesentlichen mit der Fragestellung, in wie weit sich das Konzept der
Aromatizität von Benzol auch auf Borazin(derivate) übertragen lässt (Grad der
Aromatizität). Kürzlich gelang es Liu et al. ein stabiles, isolierbares Benzol/Borazin
Hybridmolekül 15 herzustellen.[21] Bei der als 1,2‐Dihydro‐1,2‐azaborin

Chemie der Bor‐Stickstoff‐Verbindungen
13
bezeichneten Verbindung, ist nur eine CC‐Einheit von Benzol durch eine BN‐
Einheit gezielt ausgetauscht worden. Dabei sind ‐ wieder unter Berücksichtigung
des Aromatizitätskonzepts ‐ die experimentell erhaltenen spektroskopischen
Daten und Untersuchungen zur Reaktivität von theoretischen Betrachtungen zur
Elektronenstruktur (Resonanzenergie, elektronisches Oberflächenpotential)
ergänzt worden.
Abbildung 3: Benzol und seine experimentell bekannten BN‐Analoga.
3.1.4 Boran‐Amin Addukte
Boran‐Amin Addukte (AB) 16 können als Lewis‐Säure‐Base Addukte aufgefasst
werden, bei der das vakante p‐Orbital eines Borans mit dem doppelt besetzten
sp3‐Orbitals eines Amins in Wechselwirkung tritt. Daraus resultieren, in
Abhängigkeit der eingeführten Reste, oft stabile, feste und kristalline
Verbindungen.
Abbildung 4: Boran‐Amin Addukte, die BN‐Analoga der Alkane.
C
CC
C
CC
H
H
H
H
H
HN
BN
B
NB
H
H
H
H
H
H
CC
B
NC
CH
H
H
H
H
H
Faraday Stock Liu(1825) (1926) (2008)
1314 15
N B N..
..
B +C C B N
16

Chemie der Bor‐Stickstoff‐Verbindungen
14
Gegenwärtig erlebt diese Stoffklasse eine Renaissance, da den AB ein großes
Wasserstoffspeicherungspotential zugeschrieben wird, welches aufgrund der
prognostizierten Wasserstoff‐Energiewirtschaft von fundamentalem Interesse
ist.[22‐24] Wegen des hohen prozentualen Anteils an Wasserstoff ‐ das einfachste
AB H3BNH3 enthält allein 19.6 Gew‐% davon ‐ werden momentan massive
Bemühungen unternommen, Systeme auf AB‐Basis zu entwickeln, welche die
reversible Speicherung und Freisetzung von H2 erlauben. Insbesondere stellt
derzeit die effiziente und technische Realisierbarkeit des Dehydrogenierungs‐
schritts von AB unter Bildung eines Aminoborans R2B=NR2 17 eine große
chemische Herausforderung dar. Nichtsdestotrotz könnten AB eine Alternative zu
anderen chemischen Wasserstoffspeichern wie den viel diskutierten MOFs (metal
organic frameworks)[25‐28] werden.
Schema 2: Dehydrogenierung eines Boran‐Amin Addukts unter Bildung eines Aminoborans.
B N
H
H H
HB N
H H
HH
H H + H2
16 17

Chemie der Bor‐Stickstoff‐Verbindungen
15
3.1.5 Aminoborane
Aminoborane 17 zeigen in ihren physikalischen Eigenschaften bemerkenswerte
Analogien zur isosteren organischen Stoffklasse, den Olefinen (Alkenen).[29]
Chemisch gesehen sind sie wiederum deutlich reaktionsfreudiger als ihre
korrespondierenden CC‐Verbindungen. So attackieren Nucleophile das
elektronenarme Boratom, Elektrophile hingegen gehen mit dem Stickstoff die für
Amine typischen Reaktionen ein. Im Aminoboran 17 besteht zwischen dem B‐
und dem N‐Atom eine kovalente σ‐Bindung und eine attraktive π‐Rückbindung
zwischen dem freien Elektronenpaar des Stickstoffs und dem unbesetzten p‐
Orbital am Bor, was zur Ausbildung einer partiellen Doppelbindung mit planarer
sp2‐artiger Struktur führt (Abb. 5). Wegen des Elektronendefizits am Bor
versuchen Aminoborane 17, wie viele andere Borverbindungen auch (z. B.
Borane: 2 BH3 ↔ B2H6), eine koordinative und elektronische Sättigung durch
Assoziationsreaktionen zu erreichen. In Abhängigkeit von den elektronischen und
sterischen Eigenschaften der Reste bilden sie höhere Aggregate aus, wobei das
Borzentrum seine Koordinationszahl von 3 auf 4 erhöht. Beispielsweise führt die
Dimerisierung eines Aminoborans 17 zu einem BN‐Cyclobutan Derivat 18, dem
Produkt einer formalen [2+2]‐Cycloaddition.
Abbildung 5: AAmmiinnoobboorraannee uunndd DDeerriivvaattee..
C C
R
R R
R
B N
R
R R
R
B
N B
N
RR
RRR
R
RR
DimerMonomer
17 18
B N
..

Chemie der Bor‐Stickstoff‐Verbindungen
16
3.1.6 Iminoborane Iminoborane RBNR 19 sind sehr viel reaktiver als ihre Alkinanaloga und
gegenüber Oligomerisierung in der Regel instabil.[30‐32] Lediglich durch sterisch
anspruchsvolle Reste lassen sich einige Iminoborane in monomerer Form bei
tiefen Temperaturen isolieren. Üblicherweise führen Cyclooligomerisierungs‐
oder Polymerisierungsreaktionen zur Stabilisierung der Lewis‐sauren, polaren
Systeme. Dimerisierung führt zu BN‐Cyclobutadienen (Diazadiboretidine) 20,[33]
Trimerisierung zu substituierten Borazinen (Borazolen) 21 oder Dewar‐
Borazinderivaten 22. Ebenfalls können BN‐Verbindungen erhalten werden (23,
24), die Cyclooctatetraenen (Octahydrotetrazatetraborocine, BN‐COT) oder
Polyalkinen isoelektronisch sind.
Schema 3: Oligomerisierung von Iminoboranen.
Eine mögliche Syntheseroute, um zu symmetrischen Iminoboranen 19 bzw. ihren
Folgeprodukten zu gelangen, bietet die von Paetzold entwickelte thermische
Vakuum‐Pyrolyse von Azidoboranen R2BN3 1.[34] Hierbei findet in einer
curtiusartigen Umlagerung unter Stickstoffextrusion eine 1,2‐Verschiebung des
B NB
N B
N N
BN
B
NB
BN
N
B
BN
NB N
BN B
N
B*
BN
BN
*n
20
24
22
23
21
19

Chemie der Bor‐Stickstoff‐Verbindungen
17
Restes R statt. Auf mechanistische Aspekte dieser Synthese, insbesondere ein
mögliches Auftreten einer Borylnitren‐Zwischenstufe R2BN 2, wird an späterer
Stelle dieser Arbeit detailliert eingegangen.
Schema 4: Synthese von Iminoboranen ausgehend von Boraziden.
3.1.7 Weitere BN‐ und Organoborspezies auf Basis organischer Verbindungen.
Das Konzept des Austausches von CC‐ gegen BN‐Einheiten ist ebenfalls auf
größere Systeme übertragen worden.[35] So sind neben Polyborazinen wie dem
abgebildete BN‐Naphthalin 26[36, 37] und dem Azaboraphenalen 27,[38] bei denen
ein vollständiger Austausch der CC‐Einheiten erfolgt, auch BN‐
Hybridverbindungen bekannt. Hierzu zählt z. B. das erst kürzlich von Piers et al.
hergestellte 10a‐Aza‐10b‐borapyren 28[39] oder das bereits früher synthetisierte
BN‐Naphthalin 29.[40, 41]
Abbildung 6: Weitere repräsentative Beispiele für Borasysteme, die in enger Verwandtschaft zu reinen organischen Verbindungen stehen.
B N RRBR
RN3 -N2
1 19
B
N
B
N
B
NB
NB
NB
N
BN
H
H
H
H H
H
H
HN N
B
BN
BN
N
BN
B
NB
26 27 28 29

Chemie der Bor‐Stickstoff‐Verbindungen
18
B
R
B
R B
R
B
RR
R
BB
B B
B
OC
COOC
OC CO
B B
B
OC CO
COB
BB
B BB B
CO
COOC
CO
COOC
OC
NB
N
R
R
NC
N
R
R
D
++ +
++-
-
31 3330 3432
37
35 36
Auch Organoborane, denen aromatische oder antiaromatische Kohlenstoff‐
strukturen zugrunde liegen, sind wegen ihrer elektronischen Eigenschaften und
der besonderen vorliegenden Bindungssituation von allgemeinem chemischen
Interesse. Bei diesen ungesättigten, konjugierten, sp2‐hybridisierten Heterocyclen
macht man sich die isoelektronische Beziehung zwischen einem neutralen
Boratom und einem Carbokation zu Nutze (B ≈ C+). So konnten elektronisch[42, 43]
und/oder kinetisch[44‐46] stabilisierte Borirene 30, die den aromatischen
Cyclopropenium‐Kationen isolobal sind, erfolgreich synthetisiert und isoliert
werden. Auch findet sich in der Literatur die Beschreibung der gelungenen
Herstellung von Organoboranen auf der Basis von Borolen 31,[47‐50] Borepinen
32[51, 52] und Boratabenzolen 33. Kürzlich wurde das o. g. Konzept stark erweitert
und von aromatischen, cyclischen Boracarbonylen (34‐36) berichtet, wobei die
stoffliche Existenz dieser theoretisch postulierten Systeme noch aussteht.[53] Mit
dem 2006 entdeckten nucleophilen Borylanion 37,[54, 55] das in enger
Verwandtschaft zu N‐Heterocyclischen Carbenen (NHCs) („Arduengo‐Carbene“)
steht,[56, 57] lieferten Nozaki et al. einen weiteren wichtigen Beitrag zur
Borchemie.
Abbildung 7: Organoborverbindungen auf der Basis von Aromaten, Antiaromaten und NHCs.

Chemie der Bor‐Stickstoff‐Verbindungen
19
3.1.8 Reaktive BN‐ und Organoborsysteme Während stabile, isolierbare BN‐Systeme bereits Bestandteil eingängiger
Untersuchungen waren, sind die BN‐Analoga reaktiver Kohlenstoffintermediate
bisher nur wenig untersucht. Dazu zählen beispielsweise die in dieser Arbeit
vorgestellten Borylnitrene R2BN 2, sowie die strukturisomeren Aminoborylene
R2NB 38. Auch Organoborspezies auf der Basis von Carbokationen und Carbenen
sind nur wenig charakterisiert. So wurde das subvalente Phenylborylen PhB 39,
welches in enger chemischer Beziehung zu Phenylnitren 40 und
phenylsubstituierten Carbenen PhCR 41 steht,[58] kürzlich von H. F. Bettinger
erstmals unter Bedingungen der Matrixisolation photochemisch erzeugt und
spektroskopisch charakterisiert.[59] Auch die nach Hückel aromatischen
Benzoborirene 42 (R = H, I) konnten wegen ihrer hohen Reaktivität erst jüngst
nachgewiesen werden,[59‐62] obwohl die verwandten Cyclopropenylkationen des
Typs 43 schon lange bekannt sind.[63‐68]
Abbildung 8: Reaktive Organoborverbindungen und ihre organischen Analoga.
C C
R
R
B N
R
R
N B
R
R
NCR
B RC R
B
+
5 2 38
4243
394041

Nitrene
20
C
R
R
R
N3 C
R
R
R
N C N
RR
R
45 46 47
3.2 Nitrene
3.2.1 Reaktivität und Reaktionsverhalten
Nitrene RN 3, die sich photochemisch oder thermisch aus den entsprechenden
Aziden RN3 generieren lassen, sind hochreaktive Intermediate welche in
Abhängigkeit ihres Restes und ihrer Spinmultiplizität (Singulett bzw. Triplett)
unterschiedlichste Reaktionen eingehen können (Umlagerung, Insertion, H‐
Abstraktion, Dimerisierung, Addition, Polymerisierung usw.).[69‐71] In dieser Arbeit
werden insbesondere Nitren‐Insertionen (typisch für Singulett‐Nitrene) und
Abstraktions‐ und Dimerisierungsprozesse (typisch für Triplett‐Nitrene)
Berücksichtigung finden.
Die Spaltung von Alkylaziden R3CN3 45 setzt die entsprechenden Alkylnitrene
R3CN 46 frei, die vorrangig [1,2]‐Umlagerungen zu den korrespondierenden
Iminen 47 eingehen.[72]
Schema 5: 1,2‐Umlagerung von Alkylnitrenen R3CN 46.
Das Reaktionsspektrum der Acylnitrene RCON 48 ist breiter.[73] Neben
intramolekularen Umlagerungen unter Ausbildung von Isocyanaten 49 (Curtius‐
Reaktion),[70, 74‐77] lassen sich in Anwesenheit geeigneter Substrate auch
intermolekulare Reaktionen beobachten. So addieren Singulett‐Acylnitrene unter
Bildung von Aziridinen 50 an Alkene[78‐80] und andere ungesättigte organische
Verbindungen mit formalen C=C‐Doppelbindungen wie Aromaten, Fullerene[81]
oder Kohlenstoffnanoröhren.[82, 83] Auch C‐H‐Insertionsreaktionen sind in der
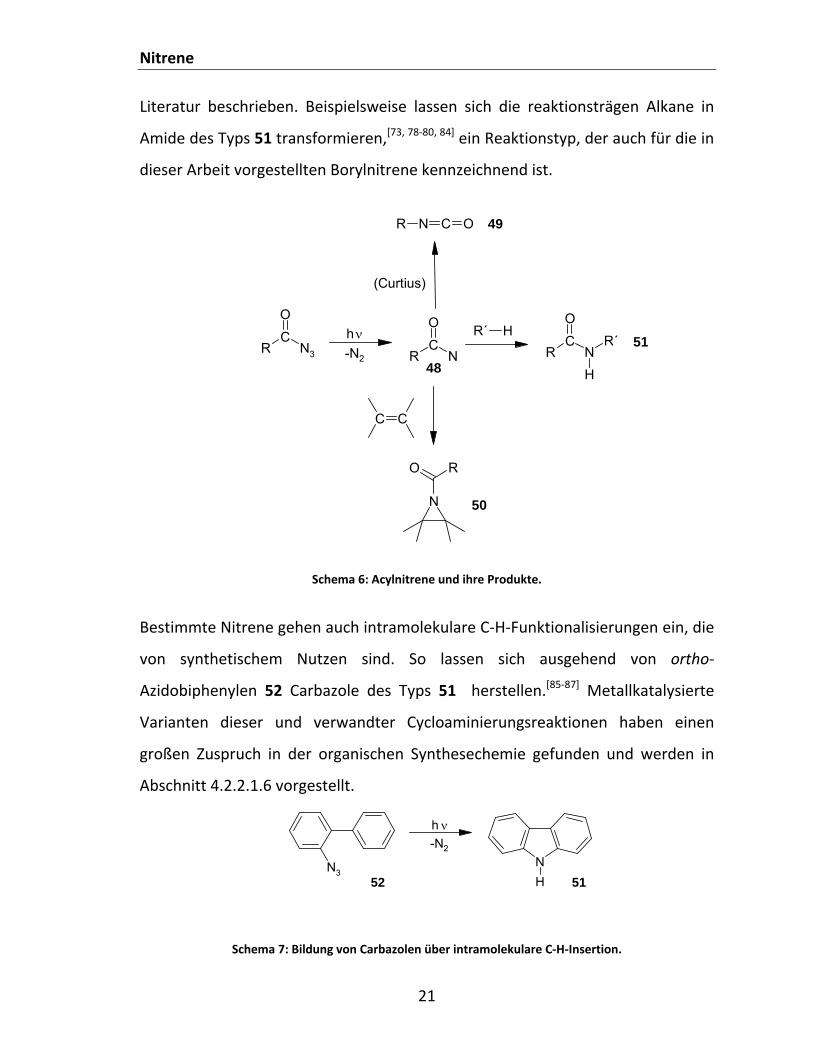
Nitrene
21
N3N
H
h -N2
52 51
Literatur beschrieben. Beispielsweise lassen sich die reaktionsträgen Alkane in
Amide des Typs 51 transformieren,[73, 78‐80, 84] ein Reaktionstyp, der auch für die in
dieser Arbeit vorgestellten Borylnitrene kennzeichnend ist.
Schema 6: Acylnitrene und ihre Produkte.
Bestimmte Nitrene gehen auch intramolekulare C‐H‐Funktionalisierungen ein, die
von synthetischem Nutzen sind. So lassen sich ausgehend von ortho‐
Azidobiphenylen 52 Carbazole des Typs 51 herstellen.[85‐87] Metallkatalysierte
Varianten dieser und verwandter Cycloaminierungsreaktionen haben einen
großen Zuspruch in der organischen Synthesechemie gefunden und werden in
Abschnitt 4.2.2.1.6 vorgestellt.
Schema 7: Bildung von Carbazolen über intramolekulare C‐H‐Insertion.
C
O
R N3C
O
R N
R N C O
C C
N
O R
C
O
R N
H
R´R´ H
(Curtius)
h-N2
48
49
51
50

Nitrene
22
N3 N
N
N
N
NEt2
N
NN
Et2NH
40-S
40-T
ISC
h-N2
53 54
55
56
57
Einfache Arylnitrene zeigen ein komplexeres Reaktionsverhalten, das stark von
den Reaktionsbedingungen, dem Substitutionsmuster und den anwesenden
Substraten abhängt.[76, 88‐90] Das aus Phenylazid 53 generierbare angeregte
Singulett‐Phenylnitren 40‐S reagiert vorrangig unter Ringerweiterungs‐reaktionen
über Cyclus 54 zu Didehydroazepin 55, welches sich mit Et2NH zu 56 abfangen
lässt. Die Interkombination ISC (engl. Inter System Crossing) zum Triplett‐GZ ist
bei RT in Lösung nicht favorisiert, so dass der Dimerisierung von 40‐T zu
Azoverbindung 57 nur eine untergeordnete Rolle zukommt. Durch die Einführung
von Schweratomen (z. B Brom) oder die Verwendung von Triplett‐Sensibilatoren
lässt sich die relative Geschwindigkeit des ISC beschleunigen.
Schema 8: Reaktionen des Phenylnitrens.
Insbesondere wurde der kinetische Einfluss von Fluorsubstituenten auf
Arylnitrene eingehend untersucht.[90‐96] So zeigt das aus dem C6F5N3 58
zugängliche Pentafluorphenylnitren 59‐S eine erhöhte Lebensdauer bei RT in
Lösung, so dass intermolekulare Insertions‐ oder Additionsreaktionen
beobachtbar sind.[95, 96]
Nitrene
23
N
FF
F
F
F
N
FF
F F
F
O
O N
H
C6F5 N
H
C6F5
NH C6F5
N
C6F5
N
C6F5
NH
C6F5
C6F5NH2
C6F5NHR
FF
F
F
F
N3
C6F5
N N
C6F5
59-T
ISC
RH
H-Abstraktion
h -N2
58
6061
59-S
65
626364
68 66
67
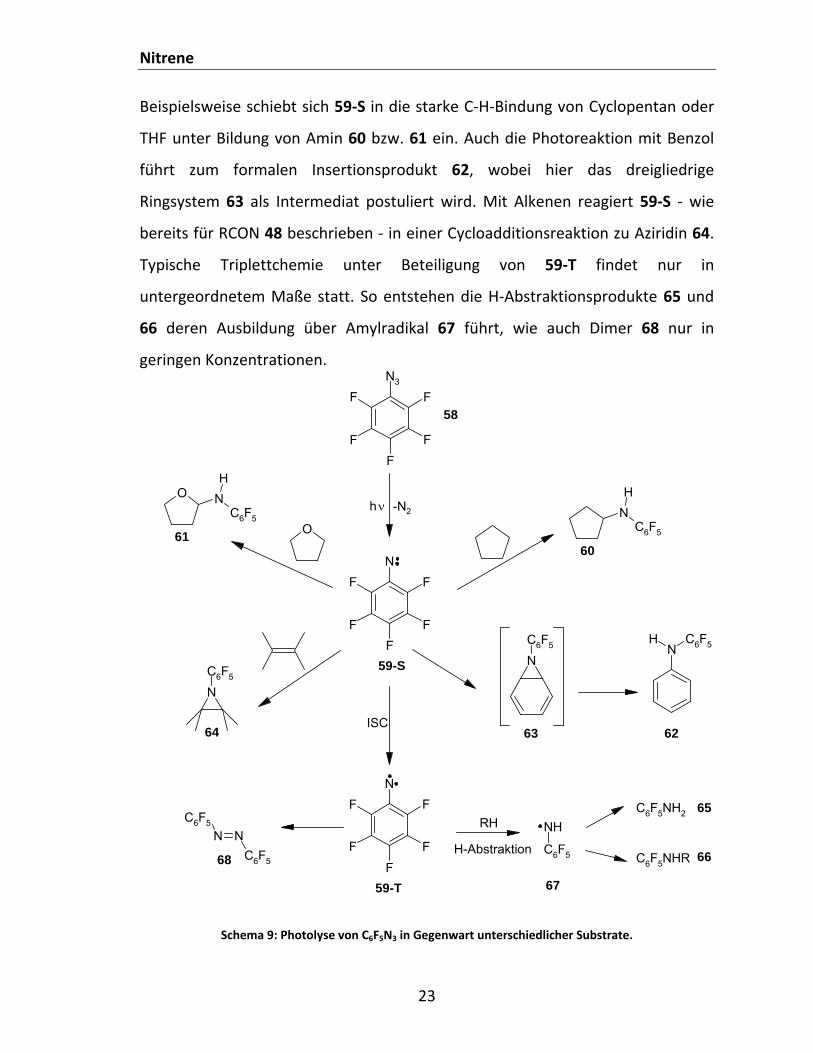
Beispielsweise schiebt sich 59‐S in die starke C‐H‐Bindung von Cyclopentan oder
THF unter Bildung von Amin 60 bzw. 61 ein. Auch die Photoreaktion mit Benzol
führt zum formalen Insertionsprodukt 62, wobei hier das dreigliedrige
Ringsystem 63 als Intermediat postuliert wird. Mit Alkenen reagiert 59‐S ‐ wie
bereits für RCON 48 beschrieben ‐ in einer Cycloadditionsreaktion zu Aziridin 64.
Typische Triplettchemie unter Beteiligung von 59‐T findet nur in
untergeordnetem Maße statt. So entstehen die H‐Abstraktionsprodukte 65 und
66 deren Ausbildung über Amylradikal 67 führt, wie auch Dimer 68 nur in
geringen Konzentrationen.
Schema 9: Photolyse von C6F5N3 in Gegenwart unterschiedlicher Substrate.

Nitrene
24
Analoge Photoprodukte lassen sich auch mit 4‐Azido‐tetrafluorobenzonitril als
Substrat gewinnen. Hier kann Cyclohexan in 75‐80 %‐iger Ausbeute in einer
intermolekularen C‐H‐Insertionsreaktion aminiert werden.[97]
Nitrene der übrigen Hauptgruppenelemente zeigen ein ähnlich vielfältiges
Reaktionsverhalten wie ihre organischen Verwandten, wobei prinzipiell die
analogen Reaktionstypen beobachtet werden können.[98‐101]
3.2.2 Bindungssituation
Wichtige elektronische Zustände von Nitrenen sollen am Beispiel des
Phenylnitrens PhN 40 vorgestellt werden (Abb. 9), da hierzu zum einen bereits
eine Fülle von experimentellen[102] und theoretischen[103‐107] Untersuchungen
vorliegen, zum anderen Parallelen zu den in dieser Arbeit vorgestellten
Borylnitrenen bestehen (vgl. 4.1).[1]
Das Singulett‐Nitren 40‐S stellt den ersten angeregten Zustand S1 dar und bildet
das photochemische Primärprodukt. Es besitzt einen iminyl‐cyclohexadienyl‐
artigen offenschaligen Diradikalcharakter (1A2‐Symmetrie),[76, 106] der durch die
Phenyleinheit stabilisiert wird. Der Triplett‐GZ T0 mit 3A2 Symmetrie liegt um etwa
17‐19 kcal/mol tiefer in der Energie, wie experimentelle[102] und theoretische[103‐
107] Untersuchungen zeigen. Die geschlossenschaligen Zustände S2 und S3 (je 1A1)
sind nahezu entartet und ähneln dem eines konventionellen Singulett‐Carbens.
Sie liegen energetisch weit über T0, wie Rechnungen zeigen ((E(T0→S2) > 30
kcal/mol).[88]

Nitrene
25
Abbildung 9: Wichtige Konfigurationen und elektronische Zustände des Phenylnitrens, wobei S2 und S3 Multikonfigurationscharakter besitzen.

Nitrene
26
3.2.3 Borylnitrene: Stand der Forschung Wie bereits erwähnt, eignen sich Azidoborane R2BN3 1 zur Herstellung
symmetrischer Iminoborane 19 bzw. ihrer Folgenprodukte. Dabei werden von
Paetzold et al. zwei mechanistische Bildungswege in Betracht gezogen.[34, 108]
Schema 10: Mechanismus der Iminoboranbildung.
Bei einer konzertierten Reaktion findet die Abspaltung des Stickstoffs synchron
mit der 1,2‐Verschiebung statt, wohingegen beim Stufenmechanismus das nach
Stickstoffextrusion intermediär auftretende Borylnitren R2BN 2 zum
entsprechenden Iminoboran 19 umlagert. Im Fall von Diarylazidoboranen Ar2BN3
wurden keine Hinweise auf Nitren‐Zwischenstufen gefunden, so dass hier von
einem Einstufenmechanismus ausgegangen wird.[34] Die Thermolysen bestimmter
Diaminoborazide R2BN3 (R = iPr2N) hingegen liefern indirekte Hinweise auf
Borylnitrene 2, wie Abfangstudien mit BEt3 unter Bildung von Aminoboran 70
zeigen.[108] Auch das Auftreten intramolekularer C‐H‐Insertionsprodukte steht mit
der Hypothese einer Nitren‐Zwischenstufe in Einklang.
B N RRBR
RN3 B
R
RN
B
R
R
N
BEt2
Et
-N2
1 192
R = Ar, iPr2N 70
-N2

Nitrene
27
Unseres Wissens nach ist der direkte Nachweis eines Borylnitrens erstmalig
unserer Gruppe gelungen.[1] Hierzu wurde Catecholborazid CatBN3 7 bei 10 K in
einer Argon‐Matrix isoliert. Die Photolyse des Azids mit kurzwelligem Licht (λ =
254 nm) liefert das erwartete Catecholborylnitren CatBN 6, das unter diesen
Bedingungen ausreichend stabil ist, um spektroskopisch (IR, UV, ESR)
charakterisiert werden zu können. Um Aussagen bezüglich der Reaktivität des
Nitrens 6 treffen zu können, wurden Abfangexperimente mit verschiedenen
Agenzien unter Matrixbedingungen durchgeführt. Es kann gezeigt werden, dass
die Stickstoffabspaltung reversibel ist, da die Azidrückbildung bei Bestrahlung mit
sichtbarem Licht beobachtet wird. Dieses Reaktionsverhalten ist sehr
ungewöhnlich und spiegelt die hohe Reaktivität des Nitrens 6 wider. Wird die
Argon‐Matrix mit CO dotiert, findet (ebenfalls bei langwelliger Bestrahlung) die
elektrophile Addition des Borylnitrens 6 an den Kohlenstoff unter Bildung des
entsprechenden Isocyanats 72 statt, welches leicht IR‐spektroskopisch
identifiziert werden kann. Besonders interessant erscheinen die Reaktionen mit
den “inerten“ Molekülen Methan und molekularen Wasserstoff zu sein. So findet
im Fall von Methan eine Insertionsreaktion des CatBN 6 in die starke sp3‐C‐H
Bindung unter Bildung von Aminoboran 8 statt.[2] Analog dazu lässt sich auch eine
Wasserstoffspaltung beobachten, wobei das symmetrische Aminoboran CatBNH2
73 entsteht.[109] CF4 hingegen ist gegenüber dem CatBN 6 stabil, und die
Reaktivität des Nitrens nicht ausreichend, um eine Bindungsspaltung unter
Bildung von 74 zu erzielen. Auch mit Triplett‐Sauerstoff kann das CatBN 6 sowohl
thermisch als auch photochemisch zur Reaktion gebracht werden, wobei je nach
Bedingungen die Oxidationsprodukte 75 und 76 IR‐spektroskopisch nachweisbar
sind. Das beobachtbare Reaktionsverhalten schreiben wir dem Borylnitren CatBN
6 in seinem geschlossenschaligen Singulett‐Zustand zu, obwohl es laut
computerchemischen Rechnungen einen Triplett‐GZ aufweist, der auch mithilfe
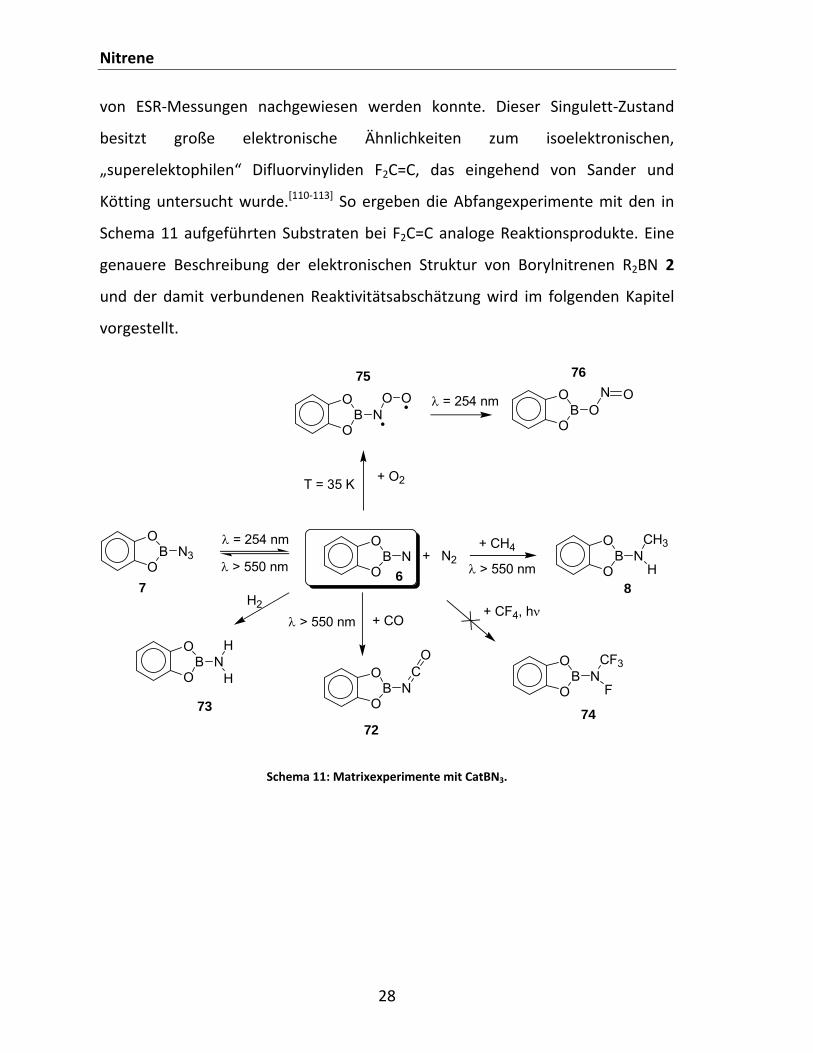
Nitrene
28
von ESR‐Messungen nachgewiesen werden konnte. Dieser Singulett‐Zustand
besitzt große elektronische Ähnlichkeiten zum isoelektronischen,
„superelektophilen“ Difluorvinyliden F2C=C, das eingehend von Sander und
Kötting untersucht wurde.[110‐113] So ergeben die Abfangexperimente mit den in
Schema 11 aufgeführten Substraten bei F2C=C analoge Reaktionsprodukte. Eine
genauere Beschreibung der elektronischen Struktur von Borylnitrenen R2BN 2
und der damit verbundenen Reaktivitätsabschätzung wird im folgenden Kapitel
vorgestellt.
Schema 11: Matrixexperimente mit CatBN3.
OB
ON
OB
ON3
= 254 nm
> 550 nm+ N2
+ CO > 550 nm
OB
ON
CO
+ O2
OB
ON
O O
T = 35 K
OB
OO
N = 254 nm O
+ CH4
OB
ON
CH3
H > 550 nm
OB
ON
CF3
F
+ CF4, h
OB
ON
H
H
H2
6
75 76
8
7472
73
7

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
29
4 Eigene Arbeiten
4.1 Computerchemische Untersuchungen: Borylnitrene und ihren Verwandten
4.1.1 Zielsetzung und Motivation
Mithilfe gängiger Bindungskonzepte der klassischen organischen Chemie[70, 71, 114‐
116] soll im Folgenden die elektronische Struktur von Borylnitrenen R2BN
beschrieben und der Einfluss der Liganden R auf die Systeme diskutiert werden.
Ziel der eigens durchgeführten systematischen computerchemischen
Berechnungen liegt dabei insbesondere in der Entwicklung eines
allgemeingültigen qualitativen Konzepts, welches anhand von Lewis‐
Resonanzformeln und einfachen Darstellungen im Orbitalmodell vorgestellt wird.
Es bleibt hierbei anzumerken, dass nur die beiden erstgenannten Spinzustände
(Singulett: 1A1 / Triplett: 3A2) bei der Diskussion der Elektronenzustände und der
Erstellung des Bindungsmodells Berücksichtigung finden. Dabei sollen u. a. die
unten aufgeführten, eng miteinander verknüpften Fragestellungen beantwortet
werden. Des Weiteren werden Untersuchungen zu H2BN‐Isomeren vorgestellt.
• Ist das Borylnitren (immer) ein lokales Minimum oder führt eine
Umlagerungen zum (cyclischen) Iminoboran?
• Welchen elektronischen Grundzustand (Singulett oder Triplett) weist ein
bestimmtes Borylnitren des Typs R2BN auf. Wie hoch ist EST?
• Welche Effekte üben die Liganden R auf die Elektronenstruktur aus?
Welchen Einfluss verursachen Akzeptor‐ bzw. Donorliganden? Lassen sich
allgemeingültige Trends feststellen?
• Welche Auswirkungen haben Konjugation, ‐ und σ‐Effekte auf EST?

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
30
4.1.2 Verwendete Rechenmethode
Es stehen unterschiedliche Rechenmethoden zur Verfügung, die sich prinzipiell in
drei Gruppen einteilen lassen.[117‐121] Die Wahl eines geeigneten Rechenniveaus
hängt dabei im Wesentlichen von der vorliegenden Problemstellung, der
erwünschten Genauigkeit und den zur Verfügung stehenden Ressourcen ab.
Bei semiempirischen Methoden fließen experimentelle Parameter einfacher
Moleküle ein, die den mathematischen Rechenaufwand gering halten und
deshalb für große Moleküle (z. B. Proteine) mit klassischen Bindungsverhältnissen
geeignet sind. Kleine Moleküle lassen sich mit ab initio Methoden genauer
untersuchen. Bei diesen quantenmechanischen Rechnungen wird die
Schrödinger‐Gleichung näherungsweise gelöst, wobei sich diese Methode in
Abhängigkeit vom verwendeten Näherungsverfahren weiter klassifizieren lässt (z.
B. Hartree‐Fock (HF), Møller‐Plesset (MP2), Coupled‐Cluster (CC)). Bei der auf
dem Hohenberg‐Kohn‐Theorem basierenden DFT‐Methode (Dichtefunktional‐
theorie) werden die Moleküleigenschaften indirekt aus der Elektronendichte‐
verteilung abgeleitet.[117, 120]
In dieser Arbeit wurde fast ausschließlich das gebräuchliche B3LYP‐
Hybridfunktional[122‐125] in Kombination mit einem 6‐311+G** Basissatz
herangezogen. Wie Berechnungen der Singulett‐Triplett Abstände an den
literaturbekannten Systemen H2BN 77,[1, 126] CatBN 6[1] und 2‐Nitreno‐1,3,2‐
dioxaborol 78[1] zeigen, sind die ermittelten Werte auf DFT‐Niveau als akzeptabel
anzusehen (vgl. Tabelle 1). Des Weiteren sind in der Literatur
computerchemische Untersuchungen zur H2BN‐Triplett‐Hyperfläche beschrieben,
bei denen ebenfalls die B3LYP Methode, hier jedoch in Verbindung mit
korrelationskonsistenten Basissätzen, Verwendung fand.[127] Da weiterhin in
dieser Dissertation nur Trends von Interesse sind, ist die erhaltene
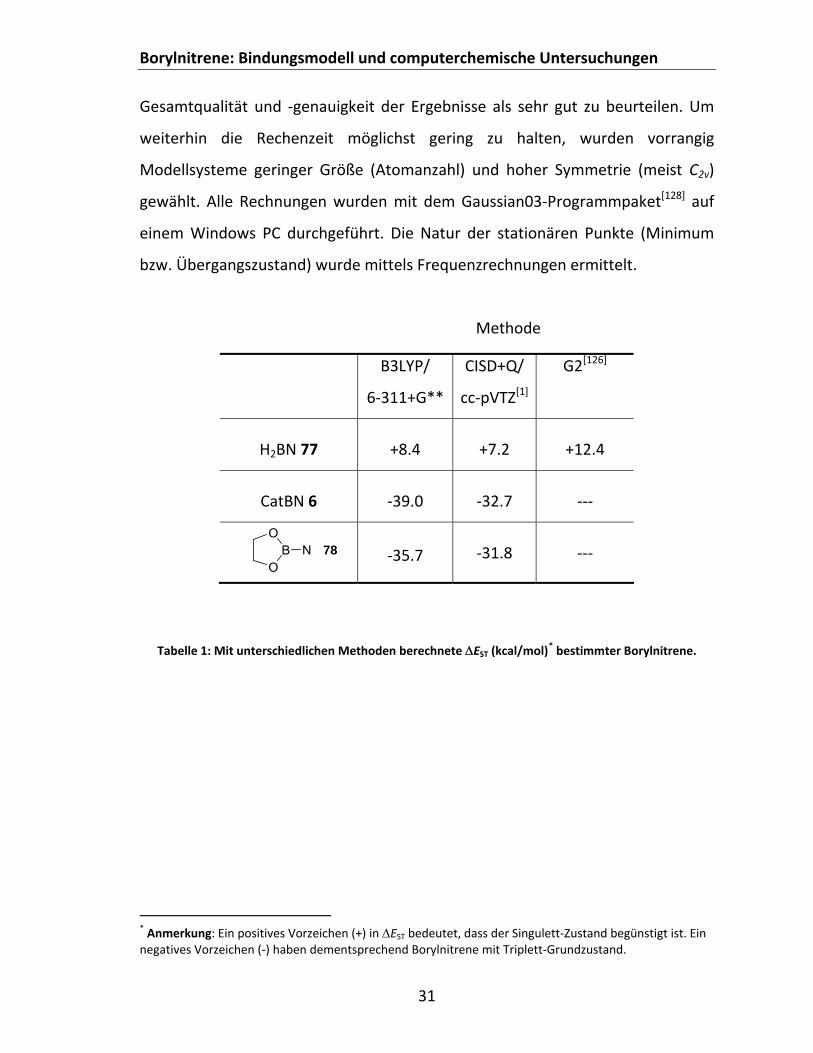
Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
31
Gesamtqualität und ‐genauigkeit der Ergebnisse als sehr gut zu beurteilen. Um
weiterhin die Rechenzeit möglichst gering zu halten, wurden vorrangig
Modellsysteme geringer Größe (Atomanzahl) und hoher Symmetrie (meist C2v)
gewählt. Alle Rechnungen wurden mit dem Gaussian03‐Programmpaket[128] auf
einem Windows PC durchgeführt. Die Natur der stationären Punkte (Minimum
bzw. Übergangszustand) wurde mittels Frequenzrechnungen ermittelt.
Methode
Tabelle 1: Mit unterschiedlichen Methoden berechnete EST (kcal/mol)* bestimmter Borylnitrene.
* Anmerkung: Ein positives Vorzeichen (+) in EST bedeutet, dass der Singulett‐Zustand begünstigt ist. Ein negatives Vorzeichen (‐) haben dementsprechend Borylnitrene mit Triplett‐Grundzustand.
B3LYP/
6‐311+G**
CISD+Q/
cc‐pVTZ[1]
G2[126]
H2BN 77
+8.4
+7.2
+12.4
CatBN 6
‐39.0
‐32.7
‐‐‐
‐35.7
‐31.8
‐‐‐ O
BO
N 78

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
32
B
R
R
px‐Orbital
pz‐Orbital
sp‐OrbitalN
4.1.3 Eine einfache Beschreibung der elektronischen Struktur von Borylnitrenen R2BN
Nach einfacher Bindungstheorie ist das Stickstoffatom im C2v symmetrischen
Borylnitren R2BN in erster Näherung sp‐hybridisiert. Eines der fünf
Valenzelektronen befindet sich in einem sp‐Hybridorbital und ist an der ‐
Bindung mit dem Bor‐Atom beteiligt, das andere sp‐artige Orbital wird durch das
„freie Elektronenpaar“ besetzt. Zudem verfügt das N‐Atom noch über zwei
orthogonal aufeinander stehende p‐Orbitale, von denen eines oberhalb und
unterhalb (pz‐Orbital), eines in der Molekülebene (px‐Orbital) liegt. Wegen der
stabilisierenden Wechselwirkung zwischen dem pz‐Orbital am Stickstoff und dem
vakanten pz‐Orbital am Bor ist das N‐pz‐Orbital gegenüber dem N‐px‐Orbital
energetisch abgesenkt, was die Aufhebung der Energieentartung zur Folge hat.
Abbildung 10: Borylnitrene im einfachen Orbitalbild.
Unter Berücksichtigung der verbliebenen vier Außenelektronen ergeben sich
formal die aufgeführten denkbaren Konfigurationen (I‐IV), welche im Folgenden
erläutert werden.
R
B N
R
R
B N
R
Singulett Triplett

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
33
sp
pz
px b2
a1
b1
B N
R
R
B NR
R
einfach besetztes px‐Orbital
doppelt‐besetztes sp‐ artiges Orbital
einfach besetztes pz‐Orbital:
von oben
Seitenansicht
pz‐Orbital
Triplett‐Borylnitren (3A2)
N I II III IV
(1A1) (3A2) (
1A2) (1A1)
Abbildung 11: Mögliche Orbitalbesetzungen des Stickstoffs in Borylnitrenen.
4.1.3.1 Triplett‐Borylnitren
Im 3A2 symmetrischen Triplett‐Nitren II‐T, der für die meisten Borylnitrene den
Grundzustand T0 darstellt, sind die beiden p‐Orbitale jeweils einfach besetzt,
wobei die Elektronen parallelen Spin aufweisen (pz(↑), px (↑)).
Abbildung 12: Das Triplett‐Borylnitren im einfachen Orbitalbild.

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
34
B N
R
R
B NR
R
leeres px‐Orbital (elektrophiles Zentrum)
doppelt‐besetztes sp‐artiges Orbital
doppelt‐besetztes pz‐Orbital
von oben
Seitenansicht
pz‐Orbital
Singulett‐Borylnitren (1A1)
4.1.3.2 Singulett‐Borylnitren
Drei unterschiedliche Singulett‐Anordnungen I‐S (1A1), III‐S (
1A2) und IV‐S (1A1)
sind denkbar (Abb. 13). Im energetisch niedrigsten Singulett‐Zustand I‐S mit 1A1‐
Symmetrie besetzen die beiden Elektronen unter Spinpaarung das N‐pz‐Orbital
(pz (↑↓), px (0)). Diese Elektronenstruktur ist energetisch günstig, da es zu einer
elektronischen Stabilisierung durch die WW mit dem vakanten pz‐Bor‐Akzeptor‐
Orbital kommt. Das in der Ebene liegende px‐Orbital ist bei dieser Konfiguration
unbesetzt (virtuelles Orbital) und verantwortlich für die extrem hohe
Elektrophilie und Reaktivität dieser Spezies. Es bestehen elektronische
Ähnlichkeiten zu Singulett‐Vinylidenen (z. B. F2CC),[110‐113, 129‐133] typischen
Singulett‐Carbenen (wie Hal2C)[57, 134‐138] sowie dem bereits diskutierten S3‐
Phenylnitren.[1, 58, 88, 105‐107]
Abbildung 13: Das Singulett‐Borylnitren im einfachen Orbitalbild.

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
35
Zusätzlich sind noch zwei weitere Singulett‐Zustände denkbar: Geht man vom
niedrigsten Triplett‐Zustand II‐T0 (3A2) aus, so erhält man durch formale
Spinumkehr eines Elektrons einen offenschaligen Singulett‐Zustand III‐S (1A2), mit
dem S1‐Zustand des Phenylnitrens vergleichbar und im Falle des CatBN 6 fast
energiegleich mit I‐S (1A1) ist (vgl. Abb. 14). Der Singulett‐Zustand IV‐S (1A1), in
dem sich beide Elektronen im px‐Orbital befinden, ist nicht favorisiert, da hier aus
Symmetriegründen keine stabilisierenden ‐Rückbindungseffekte zum Tragen
kommen.
Abbildung 14: Elektronenzustände bestimmter Nitrene und Carbene.
T0 (3A2)
S1 (1A1)
S2 (1A2)
T0 (3A2)
S1 (1A2)
S2 (1A1)
S0 (1A1)
T1 (3A2)
S2 (1A2)
S0 (1A1)
T1 (3A2)
S0 (1A1)
S3 (1A1)
H2BN CatBN PhN F2C=C F2C
E (kcal/mol)
T1 (3B1)
50
40
30
20
10
0

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
36
4.1.3.3 Beschreibung von Borylnitrenen mit vereinfachten Konzepten der MO‐ und Ligandenfeldtheorie
Wie bereits erwähnt, sind im Besonderen die Zustände I‐S und II‐T bei der
Betrachtung der Borylnitrene von Bedeutung. Welche dieser Anordnungen den
GZ darstellt, lässt sich auch mithilfe der MO‐Theorie[114, 115] und Konzepten der
Ligandenfeldtheorie[139‐141] qualitativ ableiten. Die Modellbetrachtung geht dabei
von folgender Überlegung aus: Ein Borylnitren des Typs R2BN wird formal in zwei
ungeladene Fragmente, ein N‐Atom und eine R2B‐Einheit, gespalten. Im N‐
Fragment, was formal einem sp‐hybridisierten N‐Atom entspricht, befinden sich
dann aufgrund der Hundschen Regel die beiden Elektronen einzeln mit
parallelem Spin im px‐ bzw. pz‐Orbital. Das pz‐Orbital am Bor hingegen ist
(zunächst) unbesetzt und liegt wegen der Stellung im PSE energetisch höher.
Nähern sich nun beide Fragmente aneinander an, können die p‐Orbitale gleicher
Symmetrie (z‐Richtung) in Wechselwirkung treten, was zur Aufhebung der
Entartung führt. Es gibt nun zwei unterschiedliche Möglichkeiten der
Orbitalbesetzungen, die im Wesentlichen von der relativen Lage des
Borylfragments R2B abhängen. Der linke Teil der Abbildung 15 zeigt die
Anordnung, die zu erwarten ist, wenn die Energiedifferenz zwischen den
Fragmenten relativ groß ist. Hieraus resultiert ein High‐spin System d. h. ein
Borylnitren mit Triplett‐GZ. Ist der Borylligand hingegen energetisch abgesenkt,
resultiert ein Low‐spin Borylnitren mit Singulett‐GZ. Hierbei wird die Lage des
LUMOs im Wesentlichen durch das N‐px‐Orbital bestimmt. Die High‐spin
Konfiguration tritt jedoch nur auf, wenn die Stabilisierungsenergie ΔE1 kleiner als
Der Einfachheit halber wird bei der Modellbetrachtung (siehe Korrelationsdiagramm) lediglich die
relative Lage der ‐Orbitale zueinander berücksichtigt. Sowohl die σ‐Bindung zwischen Stickstoff und Bor, als auch zwischen Rest R und Bor werden vernachlässigt. Zudem findet das doppelt besetzte sp‐artige Orbital („freies Elektronenpaar“) keine Berücksichtigung.
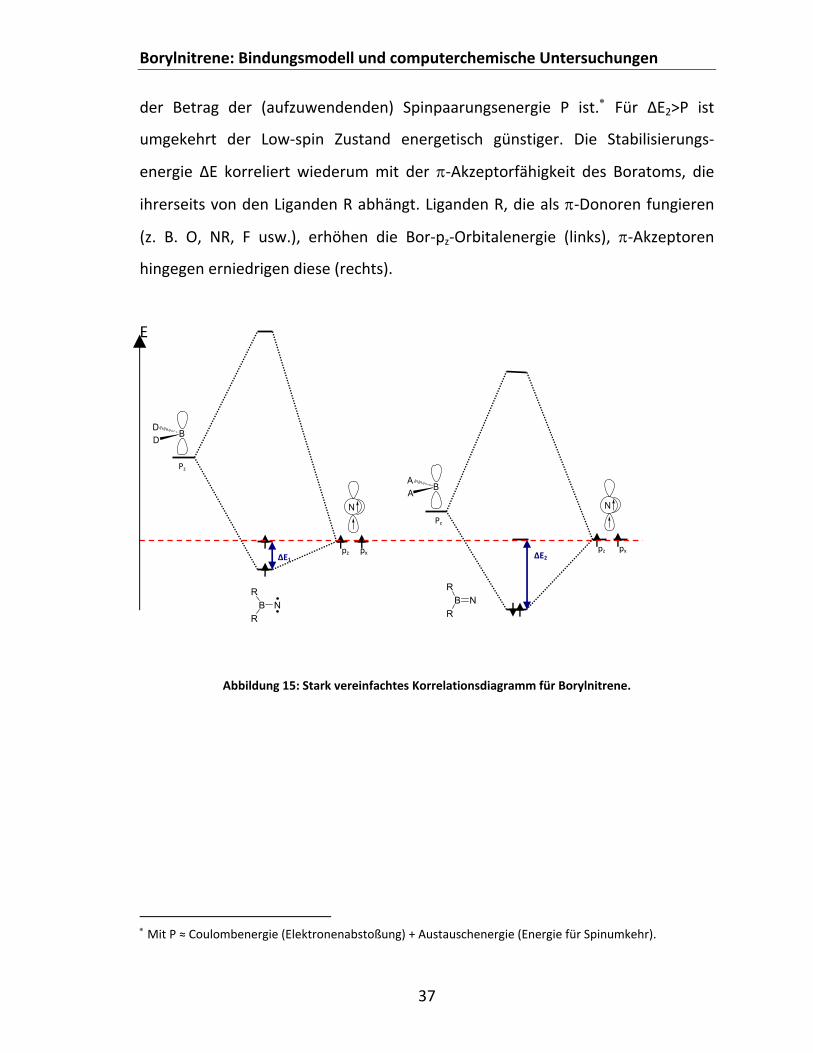
Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
37
Pz
px pz
N
ΔE1
BD
D
R
B N
R
Pz
px pz
N
ΔE2
BA
A
R
B N
R
der Betrag der (aufzuwendenden) Spinpaarungsenergie P ist. Für ΔE2>P ist
umgekehrt der Low‐spin Zustand energetisch günstiger. Die Stabilisierungs‐
energie ΔE korreliert wiederum mit der ‐Akzeptorfähigkeit des Boratoms, die
ihrerseits von den Liganden R abhängt. Liganden R, die als ‐Donoren fungieren
(z. B. O, NR, F usw.), erhöhen die Bor‐pz‐Orbitalenergie (links), ‐Akzeptoren
hingegen erniedrigen diese (rechts).
E
Abbildung 15: Stark vereinfachtes Korrelationsdiagramm für Borylnitrene.
Mit P ≈ Coulombenergie (Elektronenabstoßung) + Austauschenergie (Energie für Spinumkehr).

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
38
4.1.4 Untersuchungen zum Einfluss der Liganden R auf EST Von Carbenen R2C ist bekannt, dass die Natur der Reste R einen erheblichen
Einfluss auf die elektronischen Eigenschaften und Reaktivität hat und
verschiedensten Effekten (mesomere und induktive Effekte, Konjugation,
Hyperkonjugation, Hybridisierung, Bindungswinkel am Kohlenstoffzentrum usw.)
eine Rolle zukommt, wobei eine Quantifizierung und Abgrenzung der einzelnen
Effekte nicht immer trivial ist.[57, 69‐71, 142‐146] Im Folgenden wird der Einfluss des
Liganden R auf die elektronischen Eigenschaften von Borylnitrenen R2BN
diskutiert und hierbei insbesondere auf die Rolle von π‐Effekten eingegangen
werden. Dabei wird teilweise auf Konzepte der Carbenchemie zurückgegriffen.
Der Leser sollte berücksichtigen, dass es sich bei den vorgestellten
Bindungskonzepten lediglich um einfache Modellvorstellungen handelt.
4.1.4.1 Einfache σ‐Liganden
DFT‐Rechnungen am Stammsystem H2BN 77 zeigen, dass der Singulett‐Zustand
(77‐S) ‐ der Literatur beschrieben entsprechend [126] ‐ leicht gegenüber dem
Triplett‐Zustand (77‐T) begünstigt ist (EST = +8.4 kcal/mol). In
Übereinstimmungen mit älteren HF‐Rechnungen von Nguyen[147] und neueren ab
initio Untersuchungen in unserer Gruppe[1] wurde der Singulett‐Zustand jedoch
als Sattelpunkt erster Ordnung bestimmt.
Abbildung 16 Borylnitrene R2BN mit σ‐Liganden.
B N
H
H CH3
BCH3
N
77 79

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
39
Im Dimethylborylnitren Me2NB 79 sind beide Elektronenzustände fast
energiegleich (EST = +1.8 kcal/mol). Wiederum bildet Singulett‐Borylnitren 79‐S
einen Sattelpunkt auf der Energiehyperfläche, wobei als Minimumstruktur das
aus einer intramolekularen curtiusartigen Umlagerung resultierende Iminoboran
80 erhalten wird. Dieser theoretische Befund stimmt mit experimentellen
Ergebnissen verwandter Systeme überein. So führen Photo‐ und Thermolysen
von Diarylazidoboranen Ar2BN3 zu Iminoboranen bzw. deren Folgeprodukten,
ohne dass Hinweise für das intermediäre Auftreten von Nitren‐Zwischenstufen
vorliegen.[34]
Schema 12: Die Borylnitren‐Iminoboran Umlagerung am Beispiel von Dimethylborylnitren.
4.1.4.2 Mesomere Effekte
4.1.4.2.1 ‐Donoren (σ‐Akzeptoren)
Borylnitrene mit π‐Donorliganden weisen einen Triplett‐GZ auf, wie
Untersuchungen an den acyclischen Systemen des Typs X2BN 81 deutlich machen
(vgl. Tabelle 2).
Abbildung 17: Offene symmetrische Borylnitrene X2BN mit π‐Donorliganden X.
CH3
BCH3
N B N CH3CH3
79 80
X
B N
X
X = F, Cl, Br, OH, H2N, CN
81

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
40
S T
Tabelle 2: Berechnete ΔEST offener Borylnitrene des Typs X2BN.
Anschaulich lässt sich der Einfluss von ‐Donoren D wie folgt beschreiben:
Beginnt man mit der Betrachtung des S‐Zustands im Borylnitren entsteht durch
einen Donor D eine Konkurrenzsituation um das vakante pz‐Orbital am Bor. Der
Donor D „schiebt“ Elektronendichte in das Bor‐Akzeptororbital, wobei sich die
energetisch gewinnbringende WW zwischen dem N‐ und B‐pz‐Orbital verringert.
Ein Elektron wird deshalb unter Spinumkehr in das freie px‐Orbital am N
„gedrückt“, woraus ein Triplett‐Grundzustand resultiert.
Abbildung 18: Schematische Darstellung des Einflusses von π‐Donoren auf die elektronische Struktur in Borylnitrenen. Oben im Lewis‐Resonanzbild, unten in der dazugehörigen Orbitaldarstellung.
X = F Cl Br OH NH2 CN
X2BN
‐29.0
‐18.6
‐10.5
‐33.2
‐38.1
‐20.2
D
B N
D
D
B N
D
D
B N
D
D
B N
D
+
-
+
-
ΔEST (kcal/mol)

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
41
Am Beispiel der Singulett‐Triplett‐Abstände in den Dihalogenborylnitrenen 81‐
(Hal)2 wird zudem deutlich, dass ein komplexes Zusammenspiel zwischen Push‐
und Pull‐Effekten besteht, womit eine Analogie zu Substituenteneinflüssen bei
Carbenen auf die Spinmultiziplität besonders offensichtlich wird. Man bedenke
jedoch, dass der Trend bei Carbenen in entgegengesetzte Richtung zeigt. So
nimmt bei Dihalogencarbenen Hal2C die energetische Bevorzugung des Singulett‐
Zustands von Br zum F zu.[134‐137, 144, 145] Anhand der vorgenommen Berechnungen
können die Einflüsse der ‐Donor‐ und ‐Akzeptoreffekte jedoch nicht
unabhängig voneinander betrachtet werden, so dass sich keine einfache
Korrelation zwischen ‐Donoreigenschaft und EST ableiten lässt. Da zudem
neuere theoretische Untersuchungen an Bortrihalogenen zeigen, dass entgegen
der gängigen Lehrbuchmeinung[6]die steigende Lewis‐Acidität (LA) innerhalb der
Gruppe (LA = BF3 < BCl3 < BBr3 < BI3) nicht direkt mit der Halogen ‐
Donorfähigkeit korreliert,[148‐153] liegt in den Dihalogenborylnitrenen 81‐(Hal)2
vermutlich eine vergleichbar komplexe Bindungssituation vor wie in den
Dihalogencarbenen Hal2C vor.
Dieses komplexe Wechselspiel der konträren Bindungseffekte steht in engem Zusammenhang mit vielen experimentellen und strukturellen Befunden in der Borchemie, die auch eine Reaktivitätsabschätzung nicht immer einfach machen. So lässt sich beispielsweise die 11B‐NMR Verschiebung von Borverbindungen, die einen indirekten Hinweis auf die Elektronendichte (Abschirmung) und Bindungssituation am Bor darstellt, nicht in einem einfachen Inkrementsystem wiedergeben. Auch geometrischen Folgen können durch den Einfluss unterschiedlicher Liganden erheblich sein. Während BF3
wegen der auftretenden ‐Rückbindung des Fluors stabilisiert wird und in monomerer Form vorliegt, dimerisiert das verwandte BH3 zum B2H6. Auch die Reaktivität, die sich in der Lewis‐Acidität des
Borzentrums widerspiegelt, wird ihrerseits maßgeblich durch die am Bor befindlichen Reste, deren ‐ bzw. ‐Effekten und ihrem sterischen Anspruch bestimmt.

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
42
N
B N
C
CN
N
B N
C
NC
-
-
N
B N
NH2
H
H N
B N
NH2
H
H-
+
81-(CN)
+ +
I II I II
81-(NH2)2
Aus EST kann ferner gefolgert werden, dass nicht immer nur das direkt am Bor
befindliche Atom für die Stärke es ‐Effekts ausschlaggebend ist, sondern die
gesamte Bindungssituation Berücksichtigung finden muss. Dies zeigt der
Vergleich zwischen dem Diaminoborylnitren 81‐(H2N)2 und dem
Diisocyanoborylnitren 81‐(CN)2 deutlich. Anhand der in Grafik 19 abgebildeten
Mesomerieformeln wird unter Berücksichtigung einfacher Resonanzregeln
schnell klar, dass der Lewis‐Struktur 81‐(H2N)2 II größere Bedeutung als 81‐(CN)2
II zukommt. Damit stellt die H2N‐Gruppe einen wesentlich besseren ‐Donor als
eine CN‐Einheit dar, obwohl in beiden Fällen die Bindung über den Stickstoff
erfolgt.
Abbildung 19: Resonanzstrukturen von CN2BN und (H2N)2BN, welche Rückschlüsse auf die π‐Donorfähigkeit zulassen.

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
43
ST
XB
XN 82
4.1.4.2.2 ‐Akzeptoren
Gute ‐Donorliganden favorisieren einen Triplett‐Grundzustand und führen zu
einem großen Singulett‐Triplett Abstand. Dies lässt im Umkehrschluss erwarten,
dass gute ‐Akzeptoren den S‐Zustand in Borylnitrenen stabilisieren sollten, was
anschaulich mit einfachen qualitativen Bindungskonzepten erklärbar ist: Über die
formale BN‐Doppelbindung wird Elektronendichte in das pz‐Bor‐Akzeptororbital
„verschoben“, was wiederum zur Erhöhung der Elektronendichte am Bor und zu
attraktiven elektronischen WW zwischen den leeren Akzeptor pz‐Orbitalen der
Liganden A und dem pz‐Bororbital führt (siehe Resonanzstrukturen,
Orbitaldiagramm).
Abbildung 20: Schematische Darstellung des Einflusses von π‐Akzeptoren auf die elektronische Struktur in Borylnitrenen. Oben im Resonanzbild, unten in der dazugehörigen Orbitaldarstellung.
Die durchgeführten computerchemischen Untersuchungen an den cyclischen
Borylnitrenen 82 bestätigen diese Annnahme (vgl. Tabelle 3).
Tabelle 3: Cyclische Borylnitrene mit π‐Akzeptorliganden.
X = BH C=NH SO2
+35.6
+13.3
+10.0
A
B N
A
A
B N
A
A
B N
A

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
44
OB
ON
OB
ON
OB
ON
EST (kcal/mol) = -39.0 -40.9 -35.7
6 83-(O)2 78
4.1.4.2.3 ‐Konjugation Die vorgestellten Untersuchungen haben gezeigt, dass primäre π‐Ligandeneffekte
einen wichtigen Einfluss auf die elektronischen Eigenschaften der BN‐Einheit von
Borylnitrenen haben. Um zu überprüfen, ob sekundären π‐Konjugationseffekten
eine ähnlich zentrale Bedeutung zukommt, wurden Rechnungen an cyclischen π‐
Systemen vorgenommen.
Im Falle von sauerstoffsubstituierten Borylnitrenen 6, 83‐(O)2 und 78 zeigen
Rechnungen, dass ‐ in Übereinstimmung mit aufwendigen ab initio
Untersuchungen unserer Gruppe[1] ‐ der Einfluss des π‐Gerüstes auf EST relativ
gering und im Wesentlichen das direkt am Boratom befindliche Atom (hier
Sauerstoff) für die elektronischen Eigenschaften der BN‐Einheit ausschlaggebend
ist .
Abbildung 21: Donorsubstituierte Borylnitrene und Konjugation.
Auch die Rechnungen an den Ethen‐verbrückten Modellsystemen des Typs 83
stützen diese Annahme und zeigen, dass im Vergleich zu den offenen
Borylnitrenen X2BN 81 ähnliche Singulett‐Triplett Abstände berechnet werden
(Tabelle 4).

Borylnitrene: Bindungsmodell und computerchemische Untersuchungen
45
B
N
B
N B
N
B
NN
BMe Me
84 85 86
EST (kcal/mol) = +1.8 +2.7 -2.0 +8.1 -18.5
83-(CH2)279
XB
XN 83
ΔEST (kcal/mol)
Tabelle 4: Berechnete ΔEST bestimmter Ethen‐verbrückter Borylnitrene.
Ein merklicher Einfluss der Konjugation auf EST kann lediglich bei Borylnitrenen
festgestellt werden, deren Liganden keine primären π‐Effekte ausüben können,
wie Rechnungen an kohlenstoffsubstituierten Borylnitrenen vermuten lassen.
Abbildung 22: Grundkörper von Borylnitrenen auf der Basis von Aromaten bzw. Antiaromaten.
Die Singulett‐Borylnitrene 79, 83‐(CH2)2, 84‐86 sind Sattelpunkte erster Ordnung,
die Triplett‐Nitrene (bis auf 86‐T) bilden Minima. Die formal aromatischen
Borylnitrene (BorirenBN 84, BorepinBN 86) stabilisieren den T‐Zustand, was
durch die gewisse Delokalisation unter Beteiligung des Bor‐pz‐Orbitals erklärbar
ist. Das antiaromatische BorolBN 85 weist hingegen einen S‐GZ auf. Die π‐Einheit
im fünfgliedrigen Nitren 83‐(CH2)2 übt keinen nennenswerten Effekt auf EST aus,
da keine direkte Beteiligung mit dem Borakzeptororbital besteht.
X = O NH S PH CH2
‐40.8
‐28.6
‐29.5
+4.5
+2.7

Rechnungen zu (H2BN)x‐Isomeren
46
4.1.5 Rechnungen zu (H2BN)x‐Isomeren Es existieren bereits einige computerchemische Untersuchungen zu
unterschiedlichen (H2BN)x‐Isomeren. Da die Studien jedoch keinen vollständigen
Überblick erlauben und zudem die unterschiedlichen verwendeten
Rechenniveaus nur bedingt vergleichende Aussagen zulassen, sollen im
Folgenden einige grundlegende Ergebnisse aus den durchgeführten DFT‐
Berechnungen (B3LYP/6‐311+G**) vorgestellt und ggf. in Bezug gesetzt werden
mit literaturbekannten Daten (Ergebnisse siehe Tabelle 5).
Ausgehend vom einfachsten Iminoboran HBNH 87, das im Weiteren als
Referenzsystem dienen soll, ist durch eine typische [1,2]‐H‐Verschiebung das
bereits vorgestellte Borylnitren H2BN 77 zugänglich,[147] welches um 76.4 (77‐S)
bzw. 84.8 kcal/mol (77‐T) höher in der Energie liegt. Über eine [1,2]‐H‐
Umlagerung in entgegengesetzter Richtung gelangt man hingegen zum
strukturisomeren Aminoborylen H2NB 88. Dieses weist in guter Übereinstimmung
mit neueren Untersuchungen auf Coupled‐Cluster‐Niveau,[154] einen Singulett‐
Grundzustand auf, der lediglich um 41.2 Kcal/mol weniger stabil als HBNH 87 ist.
Das Aminoborylen H2NB 88‐T dagegen ist, analog zur Literatur, [127] fast
energiegleich mit dem Borylnitren H2BN 77‐T.
Schema 13: Die Iminoboran‐Borylnitren‐Aminoborylen Umlagerung
mit Erel (kcal/mol) = E(HBNH) – (E(H2BN)x / x) mit x = 1‐4 (Monomer‐Tetramer)
B N
H
H
N B
H
HB N HH
77 8887

Rechnungen zu (H2BN)x‐Isomeren
47
Des Weiteren wurden die Strukturen einer formalen Borylnitren‐Borylnitren‐(89),
Borylen‐Borylen‐(90), sowie Borylnitren‐Borylendimerisierung (91) bestimmt
(Strukturen und Ergebnisse siehe Tabelle 5). Bei allen Isomeren liegt hierbei eine
kumulenartige Geometrie vor. Das stabilste System ist ‐ wie zu erwarten[155] ‐ das
gemischte, BN‐konjugierte C2v‐symmetrische Dimer H2NBNBH2 91, gefolgt vom
Borylen‐ H2NBBNH2 90 und dem Nitrendimer H2BNNBH2 89, die beide jeweils die
Punktgruppe D2h aufweisen. Zudem kann als weiteres H2BN‐Dimer das BN‐
Tetrahedran 92 als stationärer Punkt bestimmt werden. Der hohe Energieinhalt
(Erel = +33.6 kcal/mol) dieser Verbindung mit C2v‐Symmetrie macht leicht
verständlich, dass ein System dieses Typs noch nicht experimentell hergestellt
wurde. Die Produkte einer formalen Borylnitren‐ bzw. Aminoborylenaddition an
Iminoboran (93 bzw. 94) bilden ‐ wie auch die entsprechenden Verbindungen
einer Insertion in die Iminoboran NH‐ bzw. BH‐Bindung (95‐97) ‐ Minima auf der
PES. Auch hier steigt wiederum die Stabilität der Systeme mit größer werdender
BN‐Konjugation. Darüber hinaus wurden typische, aus der Iminoboranchemie
bekannte, Cyclooligomerisierungsprodukte bzw. entsprechende Strukturisomere
berechnet. Für das alternierende HBNH‐Dimer 98 (1,2‐Diazadiboretidin), dessen
Bildung um 27.0 kcal/mol exotherm ist, konnte die literaturbekannte,
rautenförmige Schmetterlingsstruktur (C2v) ermittelt werden.[156‐159] Die Jahn‐
Teller‐Verzerrung sorgt dafür, dass trotz gleicher BN‐Bindungsabstände von 1.45
Å die Hückel‐Kriterien eines Antiaromats nicht erfüllt werden. Es sein daran
erinnert, dass das verwandte, planare D2h symmetrische Cyclobutadien über
Bindungsalternanz dem antiaromatischen Charakter ausweicht.[160‐163] Der ÜZ von
98 ähnelt dem des Cyclobutadien, obschon für 98‐ÜZ eine rautenförmige und
nicht quadratische Geometrie vorliegt. Der Energieunterschied zwischen dem
formal antiaromatischen 98‐ÜZ und 98 ist in Übereinstimmung mit der Literatur
nur marginal.[164] Zudem stellt das Produkt einer formalen „Syn‐Dimerisierung“

Rechnungen zu (H2BN)x‐Isomeren
48
(1,3‐Diazadiboretidin) 99 ebenfalls einen stationären Punkt dar. Auch hier ist wie
bereits in der Literatur beschrieben[164] der Übergang von 99 zu 99‐ÜZ wiederum
nahezu thermoneutral.
Abbildung 23: Berechnete (B3LYP/6‐311+G**) Geometrien für Diazadiboretidine.
Das bekannteste H2BN‐Trimer ist wohl das Borazin 13 (D3h), welches um etwa
51.1 kcal/mol stabiler ist als ein isoliertes HBNH‐Molekül 87 und damit das
thermodynamische (globale) Minimum auf der H6B3N3‐PES darstellt. Die
Geometrien der beiden anderen auf Borazin basierenden Heterocyclen 100 und
101 konnten ebenfalls ermittelt werden, wobei das partiell BN‐konjugierte 100
eine höhere thermodynamische Stabilität besitzt. Das zum Borazin 13
valenzisomere Dewar‐Borazin 102 kann überraschenderweise nicht als
stationärer Punkt identifiziert werden, obwohl Verbindungen dieses Typs
98 (C2v) 98‐ÜZ (C2h)
99 (C2) 99‐ÜZ (Cs)

Rechnungen zu (H2BN)x‐Isomeren
49
N
BN
B
NB H
H
H
H
H
H
B
NB
N
NB H
H
H
H
H
H
B
NN
N
BB H
H
H
H
H
H
B
N B
NH
H
B
N
H
H H
H N N
B
B B
N
H H
H
HH
H
13 100 101 102 103
experimentell bekannt sind. Selbst bei der Verwendung der kristallographisch
ermittelten Atomanordnung als Startgeometrie, findet die Optimierung immer
zum Borazin 13 hin statt. Für eines der beiden hypothetischen BN‐Prismane
(103) konnten indessen die strukturellen Eigenschaften berechnet werden.
Abbildung 24: Berechnete Strukturen für (H2BN)‐Trimere.
BN‐Cyclooctatetraene (BN‐COTs) sind typische experimentell zugängliche
Oligomerisierungsprodukte der Iminoborane, die eine dem 1,3,5,7‐
Cycloctatetraen (COT)[163, 165, 166] analoge Struktur aufweisen.[30, 32] Doch gelang
die Berechnung einer S4‐symmetrischen Wannenkonformation mit
alternierenden Einfach‐ und Doppelbindungen (COT = D2d) nicht. Vielmehr wird,
in Übereinstimmung mit literaturbekannten Daten,[157, 167, 168] das planare,
vollständig delokalisierte, formal antiaromatische BN‐COT 104 mit BN‐Abständen
von 1.43 Å als Minimum erhalten, welches um lediglich 4.4 kcal/mol gegenüber
Borazin 13 destabilisiert ist. Als weiteres BN‐Tetramer konnte das BN‐Cuban
105[164, 169‐171] gefunden werden, welches um 30.3 kcal/mol energetisch
gegenüber HNBH 87 abgesenkt ist.
Mit ab initio Rechnungen (MP2/6‐311+G**) gelang eine Geometrieoptimierung zum Dewar‐Borazin. Dieses ist um Erel =‐33.7 kcal/mol gegenüber dem Monomer HBNH stabilisiert und liegt damit um 18.8 kcal/mol höher in der Energie als Borazin.
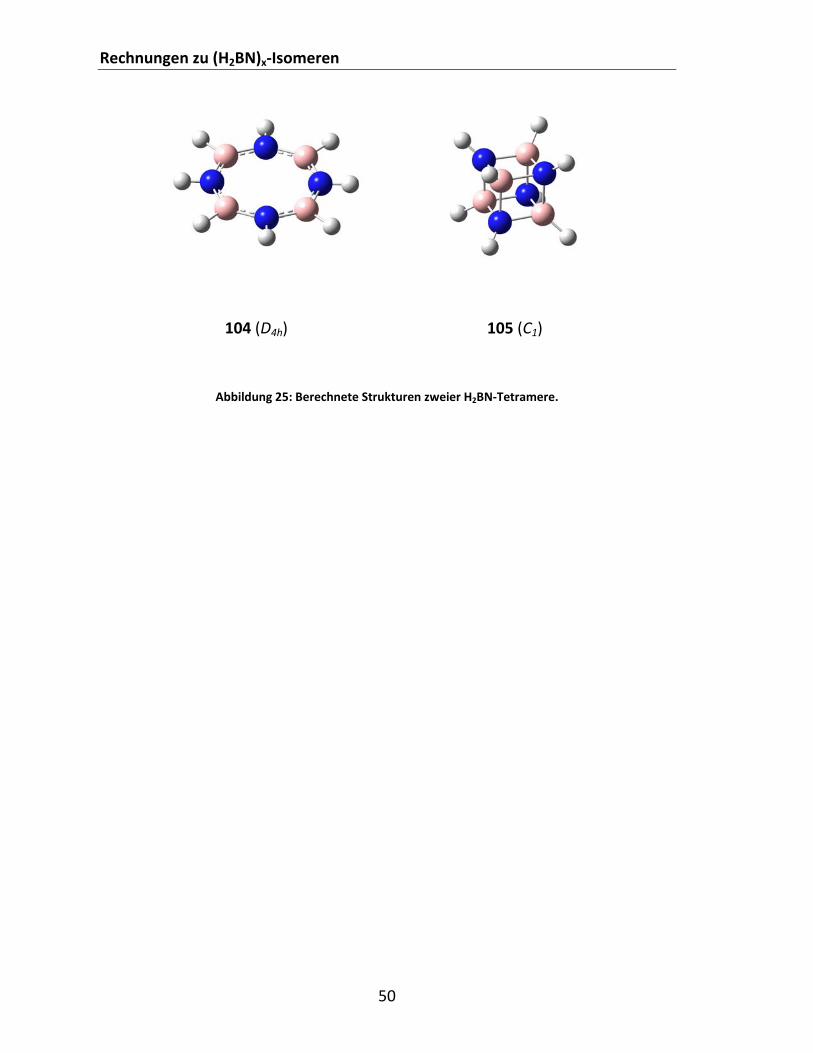
Rechnungen zu (H2BN)x‐Isomeren
50
104 (D4h) 105 (C1)
Abbildung 25: Berechnete Strukturen zweier H2BN‐Tetramere.
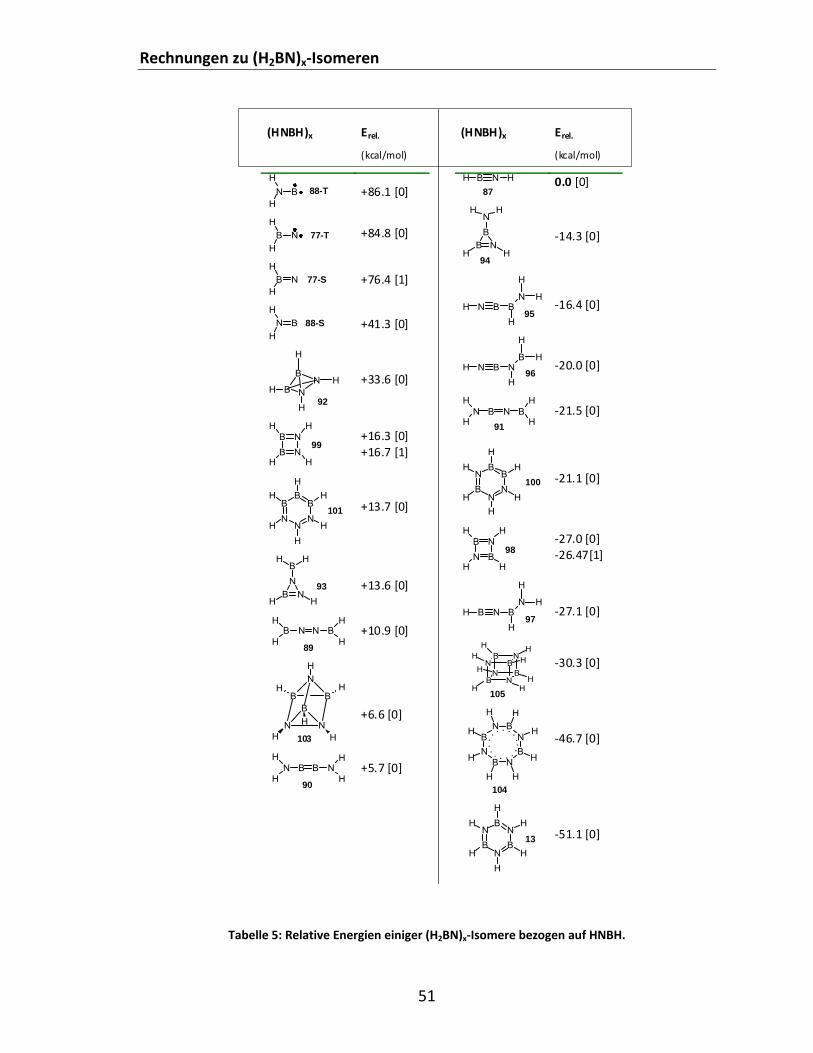
Rechnungen zu (H2BN)x‐Isomeren
51
(HNBH)x Erel.
(kcal/mol)
N B
H
H
88-T +86.1 [0]
B N
H
H
77-T +84.8 [0]
B N
H
H77-S +76.4 [1]
N B
H
H
88-S +41.3 [0]
B
B
NN H
H
H
H92
+33.6 [0]
B
B N
NHH
H H
99+16.3 [0]+16.7 [1]
B
NN
BB
N
H
H
H
H
H
H
101 +13.7 [0]
B N
N
BHH
H H
93 +13.6 [0]
N N BBH
H
H
H89
+10.9 [0]
N N
BB B
N
H H
H
HH
H
103
+6.6 [0]
B B NNH
H
H
H90
+5.7 [0]
(HNBH)x Erel.
(kcal/mol)
B N HH
870.0 [0]
B N
B
NHH
H H94
‐14.3 [0]
N BH B
H
N
H
H
95‐16.4 [0]
N BH N
H
B
H
H
96‐20.0 [0]
B N BNH
H
H
H 91
‐21.5 [0]
N
BN
BB
N
H
H
H
H
H
H
100 ‐21.1 [0]
B
N B
NHH
H H
98‐27.0 [0]‐26.47[1]
B NH B
H
N
H
H
97‐27.1 [0]
NB N
B
BN B
N
H
H
HH
H
HH H
105
‐30.3 [0]
N BN
BNB
N
B
H H
H
H
H
H
H
H
104
‐46.7 [0]
N
BN
BN
B
H
H
H
H
H
H
13 ‐51.1 [0]
Tabelle 5: Relative Energien einiger (H2BN)x‐Isomere bezogen auf HNBH.

Borazide und ihre Vorstufen
52
4.2 Experimentelle Arbeiten
4.2.1 Borazide und ihre Vorstufen
4.2.1.1 Überblick
Borazide (Azidoborane), die Vorstufen der Borylnitrene, lassen sich meist
bequem durch Substitutionsreaktionen aus den entsprechenden Halogeniden
107 (Hal = F, Cl, Br) darstellen. Dabei können sowohl ionische als auch kovalente
Azide als N3‐Quelle dienen.[172] Die in dieser Arbeit vorgestellten
Monoazidoborane R2BN3 1 wurden ausschließlich aus der Reaktion eines
Bormonochlorids R2BCl 107 mit Trimethylsilylazid (TMSN3, TMS = Trimethylsilyl) in
DCM (DCM = Dichlormethan) hergestellt.[173, 174] Dieser Syntheseweg hat sich
bewährt, da er ohne großen (apparativen) Aufwand auskommt und sich die
Aufarbeitung i. d. R. recht unproblematisch zeigt. Die umzusetzenden Borchloride
107 werden hierzu bei tiefer Temperatur (‐78 °C) mit flüssigem TMSN3
(stöchiometrisch oder im Überschuss) versetzt und für mehrere Stunden gerührt.
Langsames Aufwärmen auf RT und anschließendes Entfernen der flüchtigen
Bestandteile (LM, Kopplungsprodukt: TMSCl) liefert die gewünschten Azide 1, die
‐ je nach Reinheitsgrad‐ ggf. noch durch Umkristallisation, Umkondensation oder
Destillation weiter aufgereinigt werden müssen.
Schema 14: Allgemeines Syntheseschema für die Herstellung der Borazide des Typs R2BN3.
BR
RX B
R
RN3
1
X = Hal, R = Ar, Alky, Alkoxy, Phenoxy, Amino, Thio
Y-N3
Y = M, H, TMS
107

Borazide und ihre Vorstufen
53
Da sowohl die Borazide 1 als auch die entsprechenden –chloride 107 äußerst
wasserempfindlich sind, wurden die Synthesen unter Inertgasbedingungen
(Argon) in ausgeheizten Apparaturen und absolutierten LM durchgeführt
(Schlenk‐ und Gloveboxtechnik). Insbesondere bei der Handhabung der Azide
R2BN3 1 muss auf peniblen Feuchtigkeitsausschluss geachtet werden, da
Hydrolyse nicht nur zur Zerstörung des gewünschten Azide, sondern auch zur
Freisetzung der toxischen und hochexplosiven Stickstoffwasserstoffsäure HN3
führt.
Schema 15: Hydrolyse von Boraziden unter Bildung von HN3.
Borazide zeichnen sich im Vergleich zu den entsprechenden Borchloriden durch
größere Beständigkeit und geringere Flüchtigkeit (niedriger Dampfdruck) aus. So
zersetzt sich beispielsweise das in dieser Arbeit vorgestellte Pinakolborchlorid
PinBCl 108 bei RT innerhalb kurzer Zeit in der Glovebox, wohingegen das
korrespondierende Azid PinBN3 9 über mehrere Tage bei 20 °C stabil ist. Die
geringere Flüchtigkeit von Boraziden ist in ihrer Polarität und den daraus
resultierenden stärkeren attraktiven intermolekularen WW zu erklären.
Zusätzlich spiegelt sich das polare Verhalten dieser Substanzklasse auch im
Löslichkeitsverhalten wider. So ist die Löslichkeit von Boraziden 1 in unpolaren
LM (z. B. Hexan) geringer als die der entsprechenden ‐chloride. Durch Wahl
geeigneter Liganden R am Bor lassen sich jedoch die Löslichkeitseigenschaften
von Boraziden 1 gezielt beeinflussen und steuern.
BR
RN3 B
R
ROH
1 "
"
+ HN3 (g)H2O

Borazide und ihre Vorstufen
54
4.2.1.2 Säure‐Base Chemie: Borazid LS‐LB Addukte
Einige Borazide 1 gehen Lewis‐Säure‐Base Reaktionen ein und lassen sich
dadurch elektronisch (und kinetisch) unter Ausbildung von Azidoboraten
stabilisieren. So führt beispielsweise die Umsetzung gewisser Borazide mit Pyridin
(Py) zu den entsprechenden Addukten 109.[172‐176] Ob eine Anlagerung von
Pyridinliganden zu beobachten ist, hängt im Wesentlichen von der Lewis‐Acidität
des Boratoms im freien Borazid 1 ab. So bilden aryl‐, alkyl‐, alkoxy‐, phenoxy‐ und
thiosubstituierte Borazide die oben genannten Addukte aus, die weniger Lewis‐
sauren diaminosubstituierten Borazide hingegen nicht mehr.[172]
Schema 16: Lewis‐Säure‐Base Reaktionen von Boraziden mit Pyridin (Py).
4.2.1.3 Charakterisierung und Identifizierung von Boraziden und ihren Addukten
Die Identifizierung der (synthetisierten) Borazide ist leicht mithilfe von NMR‐ und
IR‐Messungen möglich. Das 11B‐NMR von freien Boraziden zeigt ein Singulett‐
Signal im Bereich, der typisch für ein trikoordiniertes Borzentrum ist und von den
Liganden R abhängt.[177] Empirisch konnte weiterhin festgestellt werden, dass ‐ im
Vergleich zu den eingesetzten Borchloriden ‐ für die Borazide ein Tieffeldshift von
einigen ppm (3‐5 ppm) zu beobachten ist und das Signal an Schärfe zunimmt.
Zugleich lassen sich auch mithilfe von 14N‐NMR Untersuchungen die
Stickstoffatome der Azido‐Gruppe eindeutig identifizieren.[172, 178] Teilweise sind
BR
RN3
BR
R N3
Py
Py1
R = Ar, Alky, Alkoxy, Phenoxy, Thio
109

Borazide und ihre Vorstufen
55
jedoch nicht alle drei Stickstoffatome (Nα, Nβ und Nγ) spektroskopisch
nachweisbar, da es u. U. wegen der großen Peakbreite zur Überlagerung der Nβ‐
und Nγ‐Signale kommen kann.[173] Bei Azidoboraten (Borazid‐Addukten) 109 sind
im 11B‐Spektrum, im Vergleich zu den freien Aziden 1, scharfe,
hochfeldverschobene Signale zu beobachten. Im 14N‐Spektrum lassen sich hierfür
neben den Azid‐Resonanzen Signale zuweisen, die kennzeichnend für kationische
Pyridinstickstoffe sind. IR‐spektroskopisch ist eine Identifizierung der Borazide
bzw. ihrer Addukte leicht an Hand ihrer typischen asymmetrischen
Azidvalenzschwingung (νasym‐N3 intensive Bande bei 2140‐2160 cm‐1) möglich.
Es muss lediglich sorgfältig auf eine wasserfreie Probenvorbereitung geachtet
werden. Am geeignetsten haben sich Flüssigkeits‐IR Messungen in Nyol erwiesen,
da so eine Probenvorbereitung in der Glovebox möglich ist.
CatBN3 7 CatBN3*Py 7‐Py
Abbildung 26: Berechnete (B3LYP/6‐311+G**) Geometrien für CatBN3 und dessen Pyridinaddukt.

Borazide und ihre Vorstufen
56
4.2.1.4 Synthese cyclischer und acyclischer disubstituierter Bormonochloride
Die Darstellung der benötigten Bormonochloride kann über unterschiedliche
Wege erfolgen, wobei sich drei generelle Synthesestrategien zuordnen lassen.
Anhand einiger für diese Arbeit relevanten Beispiele sollen diese kurz erläutert
werden:
• Direktsynthese
• Transmetallierungsreaktion
• Metathese/Ligandenaustauschreaktion
4.2.1.4.1 Synthese disauerstoffsubstituierter Borchloride
Für die Herstellung cyclischer fünfgliedriger disauerstoffsubstituierter Borchloride
1 eignet sich oft die Direktsynthese.[179‐181] Hierbei wird eine Lösung von
Bortrichlorid mit stöchiometrischer Menge eines vicinalen Diols 111 unter
Inertgasatmosphäre umgesetzt, wobei sich unter HCl‐Eliminierung das
gewünschte cyclische Borchlorid 112 ausbildet (vgl. Schema 17). Manchmal ist
eine Zweistufensynthese sinnvoller, insbesondere, wenn ein hygroskopisches
vicinales Diol 111 eingesetzt wird. Dazu wird das entsprechende Diol 111
zunächst durch Umsetzung mit Trimethylsilylchlorid (TMSCl) unter Bildung von
113 geschützt und durch anschließende Transmetallierungsreaktion mit
Bortrichlorid zum erwünschten Produkt 112 umgewandelt.[182, 183] Diese
Metathesereaktion kann jedoch nur (erfolgreich) bei Diolen beschritten werden,
welche über fixierte cis‐ständige Alkoholfunktionen verfügen (Präorientierung).
Dazu zählen beispielsweise die in dieser Arbeit vorgestellten 1,2‐
Dihydroxybenzole (Brenzkatechine, Catechole (engl.)) und ihre Derivate. Bei der
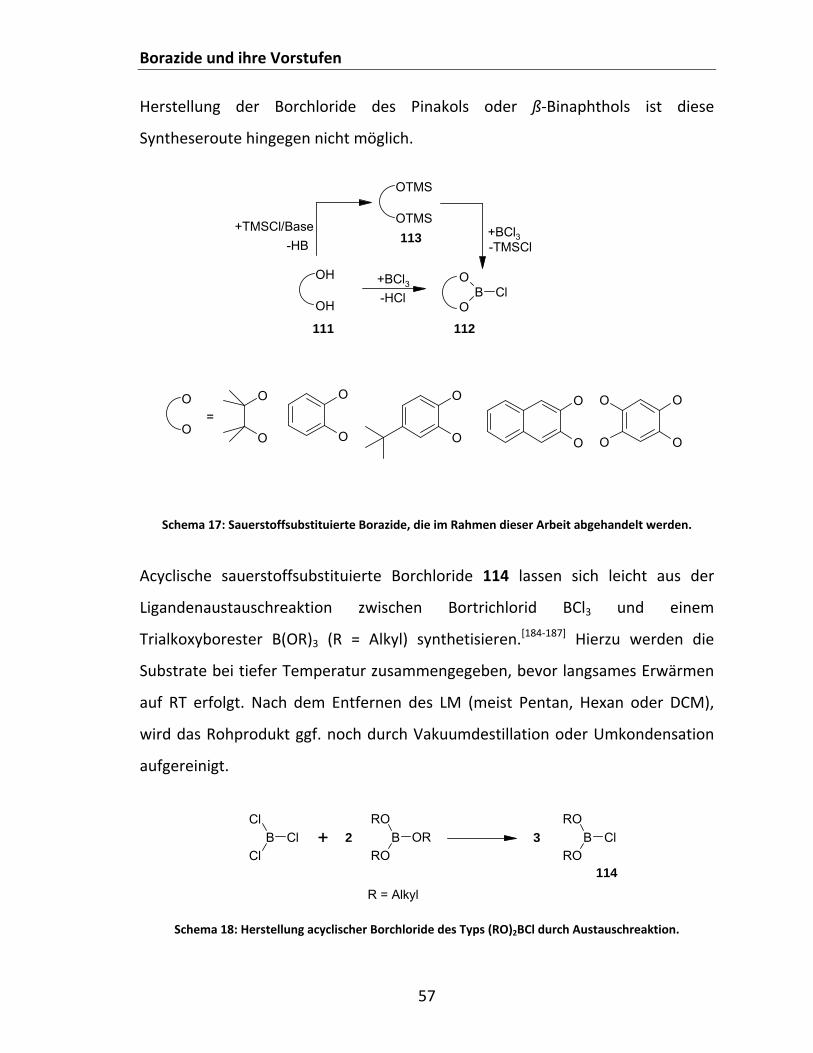
Borazide und ihre Vorstufen
57
Herstellung der Borchloride des Pinakols oder ß‐Binaphthols ist diese
Syntheseroute hingegen nicht möglich.
Schema 17: Sauerstoffsubstituierte Borazide, die im Rahmen dieser Arbeit abgehandelt werden.
Acyclische sauerstoffsubstituierte Borchloride 114 lassen sich leicht aus der
Ligandenaustauschreaktion zwischen Bortrichlorid BCl3 und einem
Trialkoxyborester B(OR)3 (R = Alkyl) synthetisieren.[184‐187] Hierzu werden die
Substrate bei tiefer Temperatur zusammengegeben, bevor langsames Erwärmen
auf RT erfolgt. Nach dem Entfernen des LM (meist Pentan, Hexan oder DCM),
wird das Rohprodukt ggf. noch durch Vakuumdestillation oder Umkondensation
aufgereinigt.
Schema 18: Herstellung acyclischer Borchloride des Typs (RO)2BCl durch Austauschreaktion.
BO
OCl
O
O
O
O
O
O
O
O
O
O
O
O
O
O=
OH
-HCl
+BCl3
OH
OTMS
OTMS
111 112
113 +BCl3-TMSCl
+TMSCl/Base
-HB
B Cl
Cl
Cl
B OR
RO
RO
B Cl
RO
RO+ 2 3
R = Alkyl
114

Borazide und ihre Vorstufen
58
4.2.1.4.2 Synthese cyclischer diaminosubstituierter Bormonochloride
Distickstoffsubstituierte cyclische Bormonochloride 115 (Diazohaloboracyclo‐
alkane) können wegen ihrer geringen NH‐Acidität nicht über Direktsynthese
hergestellt werden. In einer Eintopfsynthese wird deshalb zunächst BCl3 mit einer
nicht CH‐aciden Stickstoffbase ( i. d. R. Et3N) zum entsprechenden Boran‐Amin
Addukt BCl3*NEt3 umgesetzt. Die Austauschreaktion mit der entsprechenden
Diaminoverbindung 116 liefert dann den gewünschten Boracyclus des Typs 115,
welcher nach Abtrennung stöchiometrisch anfallender Mengen
Ammoniumchlorid (Kopplungsprodukt) ggf. noch durch Vakuumdestillation oder
Umkristallisation weiter aufgearbeitet werden muss.[188, 189] Für Stickstoffliganden
mit elektronenziehenden Gruppen (z. B. R = Tos) ist auch eine Direktsynthese
unter HCl‐Abspaltung wegen des kleineren NH‐pKS‐Wertes möglich.[190‐192]
Schema 19: Synthese der in dieser Arbeit relevanten diaminosubstituierten Bormonochloride.
BN
NCl
R
R
N
R
H
N
R
H
N
R
H
N
R
H
N
N
Me
Me
H
HN
N
Me
Me
H
HN
NH
H
Tos
Tos
-Et3NHCl
+BCl3 * Et3N
=
116 115

Borazide und ihre Vorstufen
59
4.2.1.4.3 Weitere Bormonochloride
Werden anstelle vicinaler Diole die entsprechenden Thiole eingesetzt, sind die
analogen Borathiocyclen, so wie das in der Arbeit vorgestellte 2‐Chloro‐1,3,2‐
dithiaborolan 117, durch Direktsynthese zugänglich.[179, 193, 194]
Schema 20: Synthese von 2‐Cloro‐1,3,2‐dithiaborolan ausgehend von 1,2‐Ethan‐Dithiol.
Ausgehend von Diaryldimethylzinnverbindungen Ar2Me2Sn 118, die ihrerseits aus
den Organometallsystemen 119 (Lithio‐ oder Grignardverbindungen) erhältlich
sind, können relativ durch Transmetallierung unproblematisch symmetrische
Diarylbormonochloride Ar2BCl 120 erzeugt werden.[195‐197] Die resultierenden
Borchloride bzw. ihre korrespondierenden Azide sind Vorstufen für Iminoborane
des Typs ArBNAr und wurden recht ausführlich von Paetzold et al. untersucht.[30,
34]
Schema 21: Synthese von Diarylbormonochloriden Ar2BCl über Transmetallierung.
ArB
ArCl
ArSn
Ar
Ar = z. B. Ph, Tol, Mes, C6F5
Ar-MSnCl2Me2 BCl3
M = Li, MgX
119 118 120
SB
SCl
SH
SH
117
+BCl3
-2 HCl

Borylnitrene: Aminierung von Kohlenwasserstoffen
60
4.2.2 Versuche zur direkten Aminierung von Kohlenwasserstoffen mithilfe von Borylnitrenen
4.2.2.1 Motivation und Zielsetzung
Die aus den Matrixexperimenten gewonnen Resultate veranlassten uns zur
Fragestellung, ob C‐H‐Transformationen von Kohlenwasserstoffen ‐ insbesondere
von Alkanen ‐ auch unter konventionellen Laborbedingungen mithilfe von
Borylnitrenen möglich seien (vgl. 4.2.2.1.1). Da gasförmiges Methan in
Photoexperimenten nur relativ aufwendig handhabbar ist (vgl. 4.2.2.6), wurden
zunächst photolytische Experimente zur direkten Alkanfunktionalisierung unter
klassischen Bedingungen bei RT in Lösung durchgeführt.
Schema 22: Direkte Aminierung von Alkanen mithilfe von Borylnitrenen.
Anstelle von Methan wurden zunächst die höheren homologen Cycloalkane
eingesetzt. Diese besitzen ähnliche C‐H‐Bindungsstärken wie Methan selbst (BDE
(kcal/mol) = CH4 (105.0), Cy‐5‐H (95.6), Cy‐6‐H (99.5), Cy‐7‐H (94.0), Cy‐8‐H
(95.7)),[198] bieten aber den Vorteil, dass sie bei RT als Flüssigkeiten vorliegen.
Wegen der Äquivalenz der H‐Atome bleiben weiterhin (mögliche) Probleme der
Regioselektivität (vorerst) unberücksichtigt, was zur Vereinfachung der
Dateninterpretation führen sollte.
H
R
R
R NH2
R
R
R???
"R2BN"

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
61
4.2.2.1.1 Exkurs: Die Aktivierung und Funktionalisierung von Methan (und seiner höheren Homologen)
4.2.2.1.2 Methan: Quellen und technische Nutzung
Methan CH4 ist leicht zugänglich, preiswert, derzeit noch reichlich verfügbar und
wird voraussichtlich auch in der Zukunft ein wichtiger Rohstoff bleiben.[199] Das
einfachste Alkan bildet den Hauptbestandteil von Erdgas, und auch Biogas, dem
eine immer größere Bedeutung in der nachhaltigen Energiewirtschaft zukommt,
enthält einen erheblichen Anteil an nutzbarem Methan. Gegenwärtig werden
auch Möglichkeiten in Erwägung gezogen Kohlendioxid, entweder direkt oder
gebunden in Form von Carbonat, in Methan umzuwandeln und so
klimaschädliches CO2 unter gleichzeitiger Wertschöpfung zu fixieren.[200, 201]
Zudem wird die industrielle Nutzung der großen Methanhydratreserven aktuell in
Betracht gezogen,[202] jedoch besteht (noch) die technische Herausforderung
Methanhydrat umweltfreundlich abzubauen.
Momentan wird ein Großteil des gewonnen Methans lediglich als Energieträger
verwendet und unter CO2‐Emmision verbrannt, wobei die freigesetzte
Wärmeenergie technisch genutzt wird. Obschon die Methanreserven für die
(nahe) Zukunft gesichert sind, sollte in Hinblick auf den erwarteten
Rohstoffwandel und der damit zusammenhängenden Knappheit fossiler
Energieträger, dieser C1‐Baustein sinnvoller genutzt werden.
Das größte Problem bei der Gewinnung liegt insbesondere im Risiko der Freisetzung von gasförmigem CH4 in die Atmosphäre. Da Methan selbst über ein Erwärmungspotential verfügt, welches das von CO2 um ein Vielfaches übersteigt, muss eine ökologisch verträgliche Gewinnung gewährleistet sein.

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
62
CH4
Erdgas
Biogas
CO2
Fixierung
Methanhydrat
Funktionalisierungs- "wertvollere" Produkte
Quellen
Prozess(e)
Abbildung 27: Aktuelle und zukünftig denkbare Quellen von industriell nutzbarem Methan.
Die chemische Industrie steht vor der Herausforderung Prozesse zu entwickeln,
welche die Möglichkeit bieten Methan möglichst wirtschaftlich, aber auch unter
ökologischen und nachhaltigen Gesichtspunkten, in nützlichere Grundstoffe zu
überführen („Responsible Care“). Insbesondere eine direkte Umwandlung und
Funktionalisierung wäre atomökonomisch gesehen äußerst lukrativ.
Zur Zeit sind jedoch nur wenige großtechnische Prozesse bekannt, welche die
direkte Methanderivatisierung erlauben.[203‐205] Das bekannteste Beispiel ist wohl
die thermisch initiierte radikalische Chlorierung von Methan, bei der im
Tausendjahrestonnen‐Maßstab jährlich die unterschiedlichen Chlormethane
(CHCl3, CH2Cl2, CH3Cl und CCl4) hergestellt werden. Auch die industrielle Synthese
von Blausäure (HCN) kann ausgehend von Methan erfolgen. In dem als
Andrussow‐Verfahren bezeichneten Ammonooxidations‐Prozess wird Methan
mit Ammoniak in Anwesenheit von Sauerstoff an Platin/Rhodium‐Katalysatoren
zur Reaktion gebracht.

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
63
Ansonsten wird Methan in industriellen Synthesen weitgehend nur indirekt
genutzt, wobei es vor der weiteren Umsetzung üblicherweise zunächst in
energieintensiven Prozessen in Synthesegas (CH4 + H2O CO + H2) überführt
wird. Da das Methan bei diesem Vorgang bis zu CO hochoxidiert wird, geht ein
Großteil der gespeicherten chemischen Energie ungenutzt als Wärme „verloren“.
4.2.2.1.3 Industrielle Wunschreaktionen ausgehend von Methan
Eine Wunschreaktion der chemischen Forschung, die sowohl von akademischem
als auch industriellem Interesse ist, beschäftigt sich mit der direkten
Partialoxidation von Methan zur äußerst wichtigen Grundchemikalie
Methanol.[206‐210] Aber auch die Oxidation von Methan in wichtige organische C1‐
Bausteine wie Formaldehyd oder Ameisensäure wäre wünschenswert. Zudem
werden auch Bemühungen vorgenommen, die Dehydrierung und Kupplung von
Methan zu Ethen großtechnisch zu realisieren oder eine Carboxylierung unter
Bildung von Essigsäure zu erreichen.
Schema 23: Aktuelle und zukünftig‐denkbare großtechnische Transformationen von Methan..
CH3 OH
CH3
O
OH
CH3 NH2
CH4Oxidation
CO2
CO2 + H2O + Wärme
H2O
CO + H2
CHxCly
NH3HCN
Cl2
"Wunschreaktionen""Stand der Technik"
Verb rennung
Aminierung
(Synthesegas)

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
64
CH4 CO + H2 CH3OH CH3NH2 / (CH3)2NH / (CH3)3NNH3
CH3NH2
wünschenswerte Einstufenreaktion
workup
Auch eine mögliche direkte Aminierung von Methan mit einer geeigneten
Stickstoffquelle stellt eine interessante Reaktion dar. So wäre beispielsweise die
einstufige großtechnische Darstellung von Methylamin (CH3NH2) ein überaus
erstrebenswerter Prozess. Gegenwärtig wird diese wichtige Basischemikalie
lediglich indirekt über einen kapital‐ und energieintensiven Mehrstufenprozess
aus der Reaktion von Methanol and Ammoniak (NH3) erhalten.[203] Dabei wird
Methan (oder Kohle) im ersten Syntheseschritt in einer Hochtemperatur‐
Umwandlung in Synthesegas (CO and H2) transformiert, welches dann in
Anwesenheit geeigneter Katalysatoren zu Methanol reagiert. Im nächsten
Verfahrenschritt findet bei erhöhten Temperaturen und Drücken (350‐500 °C /
15‐30 bar) die katalytische Aminierung von Methanol mithilfe von Ammoniak
statt, welches seinerseits über das Haber‐Bosch‐Verfahren (3 H2 + N2 2 NH3)
zugänglich ist. Aufgrund der geringen Selektivität des Alkylierungsschritts wegen
auftretender Mehrfachsubstitutionen, ist eine abschließende destillative
Aufreinigung der Reaktionsprodukte notwendig.
Schema 24: Konventionelle industrielle Synthese von Methylamin.

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
65
4.2.2.1.4 Der Funktionalisierungsprozess
Die größte Herausforderung einer direkten Funktionalisierung von Methan und
Alkanen im Allgemeinen liegt insbesondere in der Aktivierung der „inerten“ sp3‐C‐
H Bindung, weshalb dieser Schlüsselschritt auch mitunter als „Heiliger Gral der
Chemie“ bezeichnet wird.[211, 212] Die Reaktionsträgheit spiegelt sich dabei in
unterschiedlichen physikalischen Größen wider. So besitzt Methan eine sehr
starke C‐H‐Bindung, wie an Hand der Bindungsdissoziationsenergie von 105
kcal/mol deutlich wird.[198] Auch die geringe Acidität (pKa = 48), das hohe
Ionisierungspotential (12.5 eV), die geringe Protonenaffinität (4.4 eV), wie auch
der große HOMO‐LUMO Abstand zeigen diese Eigenschaft. Nach Schwarz
existieren mehrere Möglichkeiten einer Aktivierung von Methan, welche er am
Beispiel von Aktivierungsprozessen unter Beteiligung von reaktiven
Metallkationen erläutert hat.[213, 214] Das vorgestellte Konzept lässt sich weiter
fassen und soll wegen seiner Allgemeingültigkeit kurz vorgestellt werden: Eine
Möglichkeit der C‐H‐Aktivierung liegt in der Bindungsspaltung. Bei einem
heterolytischen Bindungsbruch bilden sich dabei die entsprechenden formalen
Methanionen. Deprotonierung führt zum Methylanion, Hydrid‐Transfer auf ein
entsprechendes Substrat zum Methylkation. Eine homolytische Bindungs‐
dissoziation hingegen, beispielsweise durch H‐Abstraktionsprozesse (vgl.
radikalische Chlorierung) führt zum ungeladenen aktivierten Methylradikal. Der
wichtigste Prozess, der auch im engen Zusammenhang mit den in dieser Arbeit
vorgestellten Ergebnissen steht, ist eine Insertion in die C‐H‐Bindung. In der
Metallorganik wird dieser Vorgang ‐ aus Sicht des betreffenden Metallszentrums
‐ auch als oxidative Addition bezeichnet.

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
66
Schema 25: Analogie zwischen Insertion und oxidativer Addition.
Eine weitere Herausforderung liegt in der Lösung des Selektivitätsproblems.
Normalerweise sind die primär gebildeten Reaktionsprodukte einer
Methanumwandlung reaktiver als Methan selbst. Dies kann dazu führen, dass
nicht das gewünschte Primärprodukt X, sondern ein nicht benötigtes
Folgeprodukt Y gebildet wird. Dieses Problem tritt z. B. bei der Partialoxidation
von Methan zu Methanol auf, da die Neigung zur Überoxidation besteht.
Schema 26: Selektivitätsproblem der Methanfunktionalisierung.
Zudem sind für die Aktivierung von Methan oft äußerst reaktive Substrate
notwendig, die nach dem Reaktivitäts‐Selektivitäts‐Prinzip (RSP)[215, 216] nur zu
geringer Produktselektivität führen. Und dennoch sind einige Systeme bekannt,
welche die Möglichkeit bieten einfache Alkane ‐ inklusive Methan ‐ zu aktivieren
und gezielt zu konvertieren.
Das RSP ist ein (immer noch) weit verbreitetes Prinzip in der Organischen Chemie welches besagt, dass eine steigende Reaktivität eines Substrates mit abnehmender Selektivität einhergeht. Meistens wird als Lehrbuchbeispiel die radikalische Halogenierung von Alkanen gewählt. Obwohl das Konzept für eine gewissen Anzahl von Reaktionstypen zutreffend ist, besitzt es eine Reihe von Schwächen (vgl. H. Mayr, A. R. Olial, Angew. Chem., 2006, 118, 1876.).
CH4 X Y
C H C X HX
X = ÜM, Reaktives Intermediat

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
67
4.2.2.1.5 Übergangsmetallkatalysierte C‐H‐Aktivierung
Der größte Erfolg wurde auf dem Gebiet der übergangsmetallvermittelten C‐H‐
Aktivierung erreicht.[217‐222] In den Pionier‐Arbeiten von Shilov und Shteinman
konnte gezeigt werden, dass bestimmte späte Übergangsmetalle in der Lage sind
die starke sp3‐C‐H‐Bindung von Methan in wässriger Lösung zu aktivieren. So
reagiert beispielsweise cis‐PtCl2(H2O)2 122 mit Methan unter Abspaltung von
einem Molekül H2O, gefolgt von anschließender C‐H‐Aktivierung. Die Addition
eines weiteren Moleküls Wasser setzt das gewünschte Transformationsprodukt
CH3OH bzw. CH3Cl, im Fall einer Chlorid‐Addition, frei. Ein verbessertes
homogenes Katalysatorsystem wurde 1998 von Periana vorgestellt.[207, 223]
Mithilfe eines Platin‐Bipyrimidin Komplexes (123) in konzentrierter Schwefel‐
säure gelang ihm die Niedertemperaturoxidation von Methan in hoher Ausbeute
und Selektivität. Zudem sind noch eine Reihe weiterer Metallspezies bekannt,
welche diesen Reaktionstyp eingehen. Hierzu zählen beispielsweise neben Pt(II)‐,
vor allem Hg(II)‐, Pd(II)‐ und Ir(II)‐Komplexe. Der Schlüsselschritt all dieser
katalytischen Aktivierungsprozesse ist in jedem Falle eine oxidative Addition der
C‐H‐Bindung von Methan an ein ungesättigtes elektrophiles Metallzentrum. Trotz
einiger Erfolge im Labormaßstab konnte dieses homogen‐katalysierte Verfahren
nicht auf den Industriemaßstab übertragen werden. Insbesondere die hohen
Katalysatorpreise, die niedrigen Reaktions‐ und Turnover‐Raten, die Instabilitäten
und das aufwendige Recycling verhinderten eine großtechnische Anwendung bis
zum jetzigen Zeitpunkt.

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
68
Abbildung 28: Wichtige molekulare homogene Katalysatoren für die Methanaktivierung.
Kürzlich erschien eine vielversprechende Arbeit von Palkovits et al. über die
Niedertemperaturoxidation von Methan zu Methanol an festen Katalysatoren,
die eine industrielle Nutzung ermöglichen könnte.[209, 224] Die Besonderheit der
vorgestellten Arbeit bildet dabei die Herstellung eines auf dem Periana‐System
basierenden, beständigen festen Polymers, welches eine heterogen‐katalysierte
Umwandlung von Methan erlaubt.
4.2.2.1.6 Übergangsmetallkatalysierte Aminierung von Alkanen
Auch die direkte Funktionalisierung höherer Alkane und Kohlenwasserstoffe ist
von großem Interesse in der Synthesechemie, da dies dem Aufbau wertvoller
organischer Verbindungen dienen kann. So stellt beispielsweise die direkte,
selektive C‐H‐Aminierung unaktivierter Substrate einen erstrebenswerten Prozess
dar. Klassisch wird eine C‐N‐Bindung meist über eine Addition eines
Stickstoffnucleophils an ein elektrophiles Kohlenstoffzentrum (z. B.
Carbonylgruppe) oder über Substitutionsreaktionen (z. B. Gabriel‐Synthese)
gebildet.
OH2PtCl
Cl OH2N N
NN
PtCl
Cl
Shilov Periana
122 123

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
69
Aufbauend auf grundlegenden Arbeiten von Breslow und Mitarbeitern sind
mittlerweile auch eine Reihe metallkatalysierter inter‐ und intramolekularer,
regio‐ und stereoselektiver C‐H‐Transformationen bekannt, die eine direkte
Aminierung erlauben und bereits in der Synthesechemie für den Aufbau
komplexer Zielstrukturen Einzug erhalten haben.[225‐231] Der Schlüsselschritt jener
Transformationen bildet meist eine C‐H‐Insertionsreaktion, bei der ein
metallgebundenes Nitren auf ein entsprechendes Substrat übertragen wird (vgl.
Schema 27). Am häufigsten werden Iminophenyliodane 125 ausgehend von
Carbamaten, Sulfonamiden oder Tosylaten in Gegenwart von Oxidationsmitteln
hergestellt, welche dann in Anwesenheit eines geeigneten Metallkomplexes in
die entsprechenden Metallnitrene des Typs 126 überführt werden. C‐H‐Insertion
bei gleichzeitiger katalytischer Rückführung des Metalls, liefert im Anschluss das
gewünschte C‐H‐Funktionalisierungsprodukt 127.
Schema 27: Übergangsmetallkatalysierte C‐H‐Aminierungen nach dem Nitren‐Insertionsmechanismus.
R NH2
R´
O
R´´O S
O
O
R N IPh
R N MC H C N
H
R
R =PhI(R´)2
+M -M
125
127126

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
70
Es sind auch Aminierungen bekannt, die über einen alternativen Mechanismus
führen (vgl. Schema 28).[226] Hierbei reagiert das eingesetzte Metall zunächst in
einer C‐H‐Aktivierungsreaktion unter Bildung von 128, bevor die Substitution
durch das nitrenoide Stickstoffsubstrat 129 (z. B. PhI=NR) und die Freisetzung des
Amins 127 sowie die Rückführung des Katalysators erfolgt.
Schema 28: C‐H‐Aminierung nach dem C‐H‐Aktivierungsmechanismus.
Neben den diskutierten Iminoiodanen sind auch Reaktionen bekannt bei denen
Azide selbst als Nitrenvorstufe Verwendung finden, was atomökonomisch und
unter Gesichtspunkten einer nachhaltigen Synthese äußerst lukrativ ist.[232‐239] So
werden Aryl‐ oder Vinylazide als Stickstoffquelle in intramolekularen
Cyclomerisierungen verwendet, wobei z. B. aromatische Indole oder Carbazole
als Produkte gebildet werden (vgl. Schema 29).[233, 236‐238] Kürzlich wurde von der
gelungenen Synthese siebengliedriger Phosphorester berichtet, die ausgehend
von Phosphorylaziden in Anwesenheit von Co(II)‐Komplexen erfolgte.[239]
Obwohl die vorgestellten Methoden ein hohes Synthesepotential mit sich
bringen, haben sie jedoch meist den Nachteil, dass es sich bei den zu
funktionalisierenden sp3‐C‐H‐Bindungen häufig um aktivierte Zentren handelt (z.
B. Allyl‐ und Benzylstellung).[225‐228, 233, 240‐242] Beispiele für die Aminierungen
unaktivierter Alkane, wie die in Schema 29 aufgezeigte Funktionalisierung von
Cyclohexan mithilfe von Azidoadamantan,[234] sind eher selten.[240, 242, 243]
R N
C H C NH
R
M HC
+M
-M127
128
129
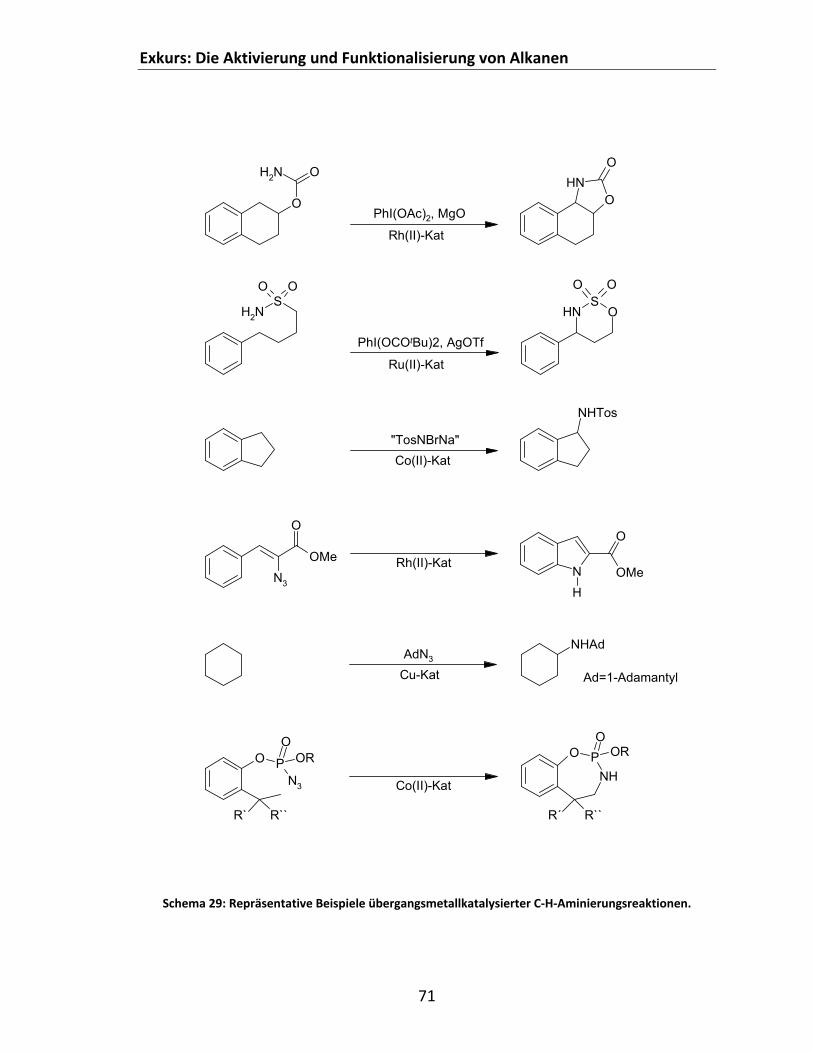
Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
71
Schema 29: Repräsentative Beispiele übergangsmetallkatalysierter C‐H‐Aminierungsreaktionen.
O
NH2 O
ONH
O
SNH2
O O
OS
NH
O O
NHTos
O
OMe
N3N
O
OMe
H
NHAd
R` R``
O P
OOR
N3
O P
NH
R´ R``
OOR
PhI(OAc)2, MgO
AdN3
Ad=1-Adamantyl
Rh(II)-Kat
PhI(OCOtBu)2, AgOTf
Ru(II)-Kat
"TosNBrNa"
Co(II)-Kat
Cu-Kat
Co(II)-Kat
Rh(II)-Kat

Exkurs: Die Aktivierung und Funktionalisierung von Alkanen
72
4.2.2.1.7 Alkantransformationen mithilfe weiterer Systeme
Neben den zuvor vorgestellten weichen elektrophilen Übergangsmetall‐
komplexen, sind auch bestimmte reaktive Spezies in der Lage, Alkane zu
funktionalisieren. Hierbei sind insbesondere die von Schwarz und Mitarbeitern
entwickelten Modellsysteme zu nennen.[212‐214, 244, 245] In seinen bahnbrechenden
Arbeiten wurden Gasphasenreaktionen sowohl von „nackten“ als auch von
„ligandgebundenen“ (Übergangs)metallkationen in Gegenwart von Methan
ausführlich untersucht. Diese massenspektroskopischen Experimente geben zwar
ein tieferes Verständnis für mechanistische Aspekte der C‐H‐Aktivierung, haben
jedoch keinen direkten synthetischen Wert. Auch gewisse enzymatische Systeme
vermögen Methan zu aktivieren.[246] Zudem haben Untersuchungen an
bestimmten Supersäuren haben gezeigt, dass diese mit Alkanen zur Reaktion
gebracht werden können.[247] Darüber hinaus auf dem Gebiet der reaktiven
Zwischenstufen bekannt, dass einige Systeme (freie Radikale R,[248] Carbene,
Nitrene, Vinylidene[110‐112] usw.) mit Alkanen ‐ teilweise auch mit Methan selbst ‐
eine C‐H‐Funktionalisierungen eingehen.

Photolyse von PinBN3 in (Cyclo)alkanen
73
4.2.2.2 Photolyse von PinBN3 in (Cyclo)alkanen
Da sich herausstellte, dass sich das in den Matrixexperimenten verwendete
Catecholborazid 7 (CatBN3) nicht zur C‐H‐Aktivierung von (Cyclo)alkanen eignet
(vgl. auch Kapitel 4.2.2.4.1), wurde das verwandte Pinakolborazid (PinBN3) 9
synthetisiert und dessen Photochemie untersucht. Das bis dato unbekannte Azid
kann ausgehend von Pinakol 131 und Bortrichlorid BCl3 und anschließender
Azidierung mit TMSN3 hergestellt werden. Im Zuge dieser Arbeiten wurde dazu
die bestehende Synthesevorschrift für die Darstellung des Pinakolborchlorids 108
(PinBCl)[249] verbessert und auf einfachen Labormaßstab angepasst.
Schema 30: Syntheseroute für die Herstellung des neuen PinBN3.
Für die Photoexperimente wurde frisch synthetisiertes, flüssiges PinBN3 9 in
einem großen Überschuss eines Cycloalkans Cy‐H (Cy5‐Cy8) gelöst und für
mehrere Stunden fortlaufend bei RT unter Argonatmosphäre mit einer
Quecksilberniederdrucklampe (λ = 254 nm) belichtet. Nach beendeter Photolyse
wurde der ggf. ausgefallene Feststoff mithilfe einer Umkehrfritte abgetrennt und
das Filtrat bis zur Trockene eingeengt. Spektroskopische und röntgenkristallo‐
graphische Untersuchungen der löslichen festen Photolyseprodukte sowie
Abbauexperimente zeigen, dass sich die erwarteten Aminoborane PinBNHCy 132
in hohen Ausbeuten bilden (vgl. Tabelle 6, S 85).
OB
ON3
OB
OCl
OH
OH
131 108 9
BCl3 TMSN3

Photolyse von PinBN3 in (Cyclo)alkanen
74
Schema 31: Photolyse von PinBN3 in Gegenwart von Cycloalkanen unterschiedlicher Ringgröße.
So weisen die löslichen Photoprodukte ein gegenüber den Edukten verbreitertes
11B‐NMR Signal bei etwa 25 ppm auf, was einer leichten Hochfeldverschiebung
von ca. 2‐3 ppm entspricht. 1H‐ und 13C‐Messungen zeigen eindeutig die zur
Pinakoleinheit zugehörigen Signale ((1H‐NMR): δ ≈ 1.2 ppm, (13C‐NMR): δ ≈ 25
ppm, δ ≈ 82 ppm) sowie die typischen Alkylresonanzen des Cycloalkangerüstes.
Besonders das neu gebildete, tieffeldverschobene im 1H‐NMR zum Multiplett
aufgespaltene PinBHNCH‐Signal bei etwa δ ≈ 3.1 ppm ((13C‐NMR): δ ≈ 51 ppm) ist
äußerst charakteristisch. IR‐Messungen geben keine Hinweise mehr auf die
Azido‐Gruppe, was für einen vollständigen Abbau des eingesetzten PinBN3 9
spricht. Für bestimmte Aminoborane PinBNHCy 132 (Cy = Cy‐5, Cy‐6, Cy‐7) gelang
es auch nach Sublimation des Rohproduktes Einkristalle zu züchten, deren
Qualität ausreichend für Röntgenuntersuchungen war (vgl. Abb. 29 und 30). Bei
allen vermessenen Strukturen liegt ein für Aminoborane typischer BN‐
Bindungsabstand von 1.385‐1.397 Å vor. Der alkenartige Doppelbindungs‐
charakter spiegelt sich zudem in der Planarität dieser Struktureinheit wider. Am
kurzen B‐O‐Abstand (B‐O‐Bindungslänge = 1.379‐1.392 Å) ist zudem indirekt der
π‐Rückbindungseffekt des Sauerstoffs mit dem vakanten pz‐Orbital am Boratom
abzulesen.[172, 174] Bei allen Insertionsprodukten liegt eine äquatoriale Anordnung
OB
ON3
Cy H
OB
ON
H
Cy
1329
CyH =
h (= 254 nm)

Photolyse von PinBN3 in (Cyclo)alkanen
75
der Pinakoleinheit in Bezug zum Cycloalkangerüst vor, was auf den sterischen
Anspruch des Boransubstituenten (PinB) zurückgeführt werden kann.
Abbildung 29: Kristallstruktur von PinBNHCy (R= Cy‐5, Cy‐7).
Innerhalb des Kristallverbands bilden die Aminoborane PinBNHCy‐5 und
PinBNHCy‐6 je zwei antiparallel zueinander stehende Dimere aus. Diese werden
über je zwei Wasserstoffbrückenbindungen zwischen dem Wasserstoffatom der
Aminoboraneinheit und einem Sauerstoff des Pinakolgerüstes stabilisiert und
bilden einen achtgliedrigen Ring aus, wie in Abb. 30 am Beispiel des Cyclohexan‐
Insertionsprodukts aufgezeigt ist. Der H••O‐Bindungsabstand beträgt im Falle
des PinBNHCy‐6 2.282 Å, und der N‐H••O‐Bindungswinkel weicht mit 176.7° nur
geringfügig von der Linearität ab. Die Wasserstoffbrücken im PinBNHCy5‐Dimer
sind etwas kürzer (2.250 Å), der N‐H••O‐Bindungswinkel mit 178.1° noch näher
an der Linearität. Die zwei O2BNH‐Einheiten spannen nahezu perfekte Ebenen in
beiden Aminoboranen auf. Bei PinBNHCy‐7 liegt hingegen kein derartiges Dimer‐
Packungsmuster vor. Vermutlich überkompensiert die Coulomb‐Repulsion, die im
größeren sterischen Anspruch des Cycloheptyl‐gerüstes begründet ist, die
Ausbildung attraktiver H‐Brückenbindungen.

Photolyse von PinBN3 in (Cyclo)alkanen
76
Abbildung 30: Das PinBNHCy‐6 Dimer.
Die Identifizierung der unlöslichen Photolyseprodukte, die in variablen Mengen
anfallen, gestaltet sich hingegen schwieriger. So bildet sich neben un‐ bzw. kaum
löslichen harzigen polymeren Produkten auch jeweils ein Feststoff definierter
Zusammensetzung bei jedem Belichtungsversuch in den unterschiedlichen
Cycloalkanen aus. Löslichkeitseigenschaften, NMR‐Messungen, sowie
Abbauexperimente lassen vermuten, dass es sich bei den unbekannten
Verbindungen um die Aminoboronsäuren (HO)2BNHCy 133 handelt. So ist eine
gute bis mäßige Löslichkeit in THF, DCM oder Et2O gegeben. In unpolaren LM, wie
den bei der Photolyse eingesetzten Cycloalkanen, ist dagegen nur eine marginale
Löslichkeit festzustellen. Im 11B‐NMR liegt ein Singulett‐Signal bei etwa δ = 22
ppm vor, was einer Hochfeldverschiebung von etwa 3 ppm gegenüber PinBNHCy
132 entspricht. Im 1H‐ bzw. 13C‐NMR gibt es keine Hinweise auf die Pinakoleinheit
mehr, die dem Cycloalkangerüst zugehörigen Resonanzen sind jedoch vorhanden.
Zudem besitzen die Verbindungen ein breites Protonsignal bei etwa δ = 8 ppm,
was für eine Boronsäureeinheit B‐OH sprechen könnte.[250] Wird das

Photolyse von PinBN3 in (Cyclo)alkanen
77
entsprechende Produkt alkoholysiert oder acetyliert (vgl. 4.2.2.2.1), ergeben sich
die gleichen Abbauprodukte, die auch für die Aminoborane 132 beobachtet
werden. Eine Bildung über hydrolytische Fragmentierung des Photoprodukts 132,
beispielsweise durch Spuren von Wasser, kann mit hoher Wahrscheinlichkeit aus
mehreren Gründen ausgeschlossen werden. So können bei der Verwendung des
perdeuterierten Cyclohexans (C6D12) als Substrat im 1H‐NMR keine Signale mehr
für B‐OH Protonen beobachtetet werden. Zudem ist aus Abbauexperimenten
bekannt, dass bei der Alkoholyse oder Hydrolyse von Aminoboranen mit
stöchiometrischen Mengen zunächst die Spaltung der BN‐ und nicht der BO‐
Bindung erfolgt (vgl. Schema 34). Da weiterhin die Menge an (HO)2BNHCy 133
gegenüber PinBNHCy 132 mit zunehmender Belichtungsdauer steigt, könnte dies
ein Hinweis darauf sein, dass das primär gebildete Aminoboran 132 eine gewisse
Photoinstabilität aufweist und photochemisch weiter zu Produkt 133 abreagiert.
Diese Hypothese steht mit Matrixexperimenten an PinBN3 9 im Einklang ,die
zeigen, dass unter bestimmten Bedingungen die Fragmentierung der
Pinakoleinheit zu beobachten ist.[251] Der genaue Bildungsmechanismus ist noch
nicht geklärt und Gegenstand aktueller Untersuchungen.
Schema 32: Mögliche Bildung von (HO)2BNHCy, ausgehend von PinBNHCy durch photochemische Fragmentierung.
OB
ON
H
Cy
OHB
OHN
H
Cy
132 133
???
h

Photolyse von PinBN3 in (Cyclo)alkanen
78
4.2.2.2.1 Abbau‐ und Derivatisierungsexperimente an PinBNHCy: Bestimmung der Ausbeute an Aminierungsprodukten
Um den Grad der C‐H‐Funktionalisierung bestimmen zu können und gleichzeitig
die gebildeten Aminoborane 132 in „wertvollere“ organische Verbindungen zu
überführen, wurden Abbau‐ und Derivatisierungsexperimente vorgenommen.
Hierzu wurde sich die bereits diskutierte Reaktivität von Aminoboranen
gegenüber elektrophilen und nucleophilen Angriffen zu Nutze gemacht. Einfache
Hydrolyse der Photolyseprodukte führt zur Freisetzung der freien
Cycloalkylamine Cy‐NH2 134, welche leicht anhand ihres charakteristischen,
fischigen Geruchs wahrnehmbar sind. Da eine Separation von den übrigen
Hydrolyseprodukten (B(OH)3 und Pinakol) nicht gelang, wurden anstelle von
Wasser Alkylalkohole ROH als Sauerstoff‐Nucleophile eingesetzt. Dabei erlaubten
GC/MS‐ und GC‐Messungen die qualitative und quantitative Analyse der
Abbauprodukte.
Schema 33: Abbau von PinBNHCy mit ROH (R = H, Alkyl).
Wird das Insertionsprodukt mit stöchiometrischer Menge Alkohol ROH
umgesetzt, lässt sich das zu erwartende Cycloalkylamin Cy‐NH2 134 eindeutig
mittels GC/MS‐Messungen anhand seines MS qualitativ nachweisen. Daneben
entsteht zudem, unabhängig vom eingesetzten Aminoboran PinBNHCy 132,
jedoch in Abhängigkeit vom verwendeten Alkohol ROH (R = Alkyl), immer die
gleiche neue Verbindung X. Das erhaltene MS von X kann dem gemischten
Borester[252] der Zusammensetzung PinBOR 135 zugeordnet werden. Wird
OB
ON
Cy
H
HN
H
Cy
132
ROH (R = H, Alkyl)
-Pin, -B(OR)3
134(Überschuss)
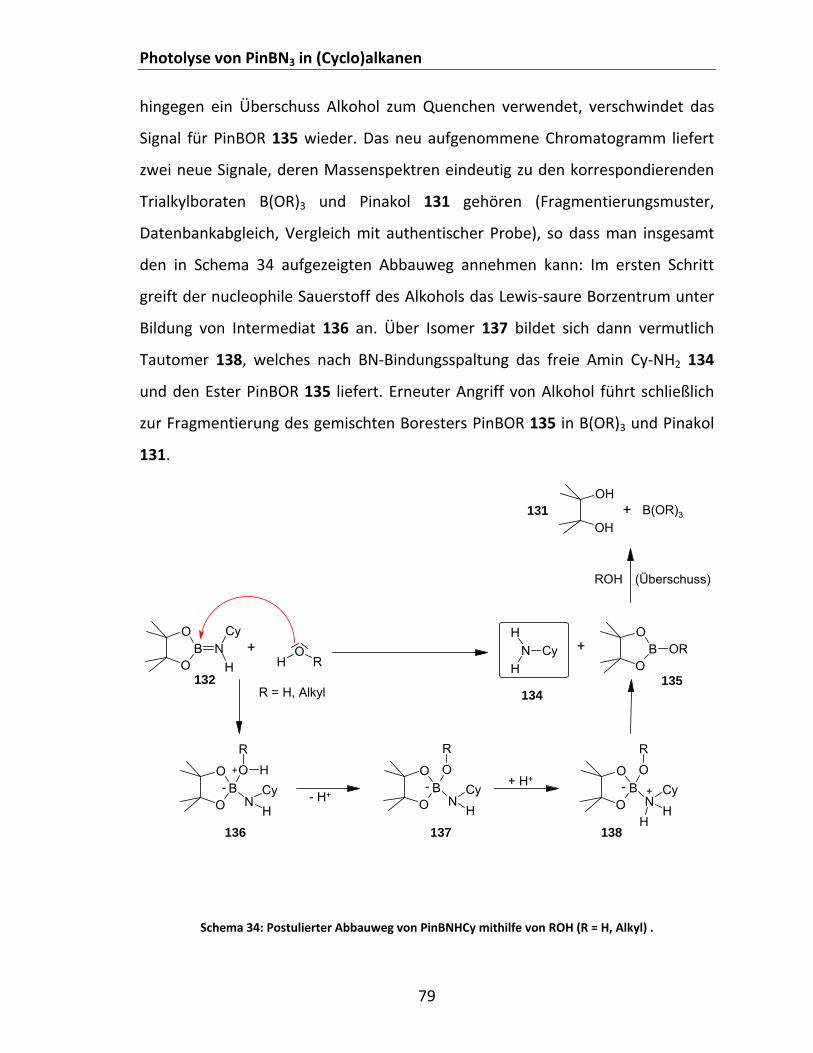
Photolyse von PinBN3 in (Cyclo)alkanen
79
OB
ON
Cy
H HO
R
OB
O
NCy
H
O
R
H
OB
O
NCy
H
O
R
OB
O
NCy
H
O
R
H
OB
OORN Cy
H
H
OH
OH
+
- H+
+ H+
R = H, Alkyl
B(OR)3
ROH (Überschuss)
+--
+ +
+
132
137
134
138136
135
-
131
hingegen ein Überschuss Alkohol zum Quenchen verwendet, verschwindet das
Signal für PinBOR 135 wieder. Das neu aufgenommene Chromatogramm liefert
zwei neue Signale, deren Massenspektren eindeutig zu den korrespondierenden
Trialkylboraten B(OR)3 und Pinakol 131 gehören (Fragmentierungsmuster,
Datenbankabgleich, Vergleich mit authentischer Probe), so dass man insgesamt
den in Schema 34 aufgezeigten Abbauweg annehmen kann: Im ersten Schritt
greift der nucleophile Sauerstoff des Alkohols das Lewis‐saure Borzentrum unter
Bildung von Intermediat 136 an. Über Isomer 137 bildet sich dann vermutlich
Tautomer 138, welches nach BN‐Bindungsspaltung das freie Amin Cy‐NH2 134
und den Ester PinBOR 135 liefert. Erneuter Angriff von Alkohol führt schließlich
zur Fragmentierung des gemischten Boresters PinBOR 135 in B(OR)3 und Pinakol
131.
Schema 34: Postulierter Abbauweg von PinBNHCy mithilfe von ROH (R = H, Alkyl) .

Photolyse von PinBN3 in (Cyclo)alkanen
80
Da eine Quantifizierung der cyclischen Amine Cy‐NH2 134 mithilfe des zur
Verfügung stehenden GC/MS‐Gerätes nicht möglich war, wurde die
Ausbeutebestimmung mittels GC‐Untersuchungen vorgenommen, da hier eine
speziell für Amine geeignete Trennsäule zur Verfügung stand. Hierzu wurden
nach beendeter Belichtung sowohl die löslichen als auch die unlöslichen
Bestandteile aus der photochemischen Reaktion von PinBN3 9 mit Cy‐H in Alkohol
aufgenommen und mit dem Ziel eines möglichst vollständigen Abbaus zu
erreichen für 30 Min in einem geschlossenen Gefäß zum Sieden erhitzt.
Anschließend erfolgte unter Zuhilfenahme eines internen Standards
(responsefaktorberichtigt) die direkte Ausbeutebestimmung am jeweiligen
Cycloalkylamin Cy‐NH2 134 (siehe Tabelle 6, S 85).
Neben Reaktionen am Lewis‐aciden Boratom in Aminoboranen können auch
typische elektrophile Angriffe am Stickstoff vorgenommen werden, wobei die
herabgesetzte Reaktivität des Stickstoffzentrums aufgrund des partiellen
Doppelbindungscharakters Berücksichtigung finden muss. Um diese Annahme zu
überprüfen, wurden Derivatisierungsexperimente mit unterschiedlichen
Elektrophilen EX bei variablen Reaktionsbedingungen vorgenommen und
ausgetestet. Und tatsächlich führt die Umsetzung der in dieser Arbeit
vorgestellten Aminoborane des Typs PinBNHCy 132 mit Elektrophilen EX nach
Aufarbeitung zu den entsprechenden substituierten sekundären Aminen ENHCy
140 (vgl. Schema 38). Als äußerst geeignet haben sich Acetylierungsreaktionen
erwiesen. Addiert man bei RT in Gegenwart katalytischer Mengen DMAP (N,N´‐
Dimethylaminopyridin)[253, 254] ein Carbonsäurechlorid RCOCl (mit R = Me, t‐Bu,
Ph) zu einer Lösung eines Aminoborans PinBNHCy 132 in Ether (Et2O) oder
Tetrahydrofuran (THF) und arbeitet die dabei gebildete Suspension mit festem
NaOH auf, werden die Amide RCONHCy 141 freigesetzt, die leicht mittels GC/MS
identifiziert werden können. Da die Amide RCONHCy 141 eine geringere Polarität

Photolyse von PinBN3 in (Cyclo)alkanen
81
aufweisen als die alkoholytisch zugänglichen primären Amine Cy‐NH2 134, ist
neben der qualitativen Analyse auch eine quantitative Bestimmung (interner
Standard, responsefaktorberichtigt) mit der GC/MS‐Standardsäule möglich. Dabei
kann empirisch festgestellt werden, dass die ermittelte Ausbeute an Amiden mit
zunehmender Größe des Restes R des Derivatisierungsreagenzes (RCOCl)
abnimmt. Möglicherweise ist dies auf sterische Effekte (Abschirmung des
Stickstoffzentrums) zurückzuführen.
Schema 35: Umsetzung von PinBNHCy mit Carbonsäurechloriden RCOCl (mit R = Me, t‐Bu, Ph).
Werden die photochemisch zugänglichen Aminoborane PinBNHCy 132 mit MeI
umgesetzt, lassen sich ebenfalls nach Aufarbeitung mit festem NaOH (oder
Alkohol) die sekundären Methylamine MeNHCy 142 gewinnen. Eine Umsetzung
kann jedoch erst bei harscheren Bedingungen beobachtet werden (höhere
Temperatur, Überschuss MeI, längere Reaktionszeit).
Schema 36: Umsetzung von PinBNHCy mit MeI.
Zusätzlich muss darauf geachtet werden, dass vor dem Aufarbeitungsschritt und
der damit verknüpften Freisetzung des freien sekundären Alkylamins MeNHCy
142, die Abtrennung des überschüssigen MeI gewährleistet wird. Ansonsten
OB
ON
Cy
H
R
O
Cl
R
O
N
H
Cy
132
1)
2) NaOH (s)
, DMAP (kat.)
- PinBOH141
OB
ON
Cy
H
MeN
H
Cy
132
1) MeI2) NaOH (s)
- PinBOH142

Photolyse von PinBN3 in (Cyclo)alkanen
82
kommt es zur typischen, aber ungewollten Mehrfachmethylierung des Amins.
Aufgrund der hohen Flüchtigkeit des MeI lässt sich dieses recht leicht nach
beendeter Reaktion im Vakuum vom Ammonium‐Intermediat entfernen, so dass
lediglich Monosubstitutionsprodukt 142 gebildet wird. Möglicherweise könnten
damit Aminoborane auch als Schutzgruppen in der klassischen organischen
Synthesechemie Zugang finden und als Hilfsreagenzien für eine Monoalkylierung
von primären Aminen eingesetzt werden (vgl. Schema 37). Um das wahre
Synthesepotential der Aminoborane abschätzen zu können, sind jedoch noch
detaillierte, systematische Untersuchungen notwendig.
Schema 37: Monoalkylierung von primären Aminen R‐NH2. A: Klassisches Beispiel über die „Tosylroute“. B: Potentiell über „Aminoboranroute“ mit analogem Reaktivitätsmuster.
NH2 R
B N
H
RX
X
Tos N
H
R Tos N
H
R
R´
B N
H
RX
X
R´
N
H
RR´
X2BCl, Base R´I
A
B
TosCl, Base R´I
H2O
H2O

Photolyse von PinBN3 in (Cyclo)alkanen
83
Bei der Verwendung der Methylierungsreagenzien Methylsulfat (Me2SO4) oder
Methyltriflat (MeOTf) anstelle von MeI wird überraschenderweise keine
Alkylierung beobachtet. Eine Erklärung zu diesem Sachverhalt steht derzeit noch
aus.
Im Einklang mit den eben diskutierten experimentell erhaltenen Resultaten sollte
der in Schema 38 abgebildete Reaktionsweg plausibel sein: Im ersten Schritt
greift der Lewis‐basische Stickstoff des Aminoborans 132 das Elektrophil EX unter
Bildung von Ammoniumsalz 143 an, womit die auftretende Trübung (Suspension)
während des Reaktionsverlaufs erklärt werden kann. Die zugesetzten
Hydroxidionen reagieren dann als Nucleophil und bewirken eine BN‐
Bindungsspaltung, die zur Freisetzung des gewünschten substituierten Amins
ENHCy 140 führt (Pfad A). Der denkbare Reaktionsweg in dem die verwendeten
OH‐‐Ionen als Base reagieren könnten (Pfad B) spielt aus zwei Gründen keine
Rolle:
• Werden anstelle von OH‐‐Ionen die schwächer basischen F‐‐Ionen
eingesetzt, die aber bekanntermaßen über eine große Affinität zum Bor
verfügen, kann ebenfalls eine Amidbildung beobachtet werden.
• Sollten die Hydroxidionen als Base fungieren, müsste die Reaktion auf der
Stufe des substituierten Aminoborans 144 stehen bleiben. Dieses sollte
aber dann durch Alkohol spaltbar sein und als ein Fragment das
gewünschte Amid 140 liefern. Da aber die Ausbeute an der
Aminoverbindung 140 durch Behandlung mit Alkohol nach erfolgter
Zugabe der Anionen nicht gesteigert werden kann, ist dieser Reaktionsweg
als unwahrscheinlich anzusehen.
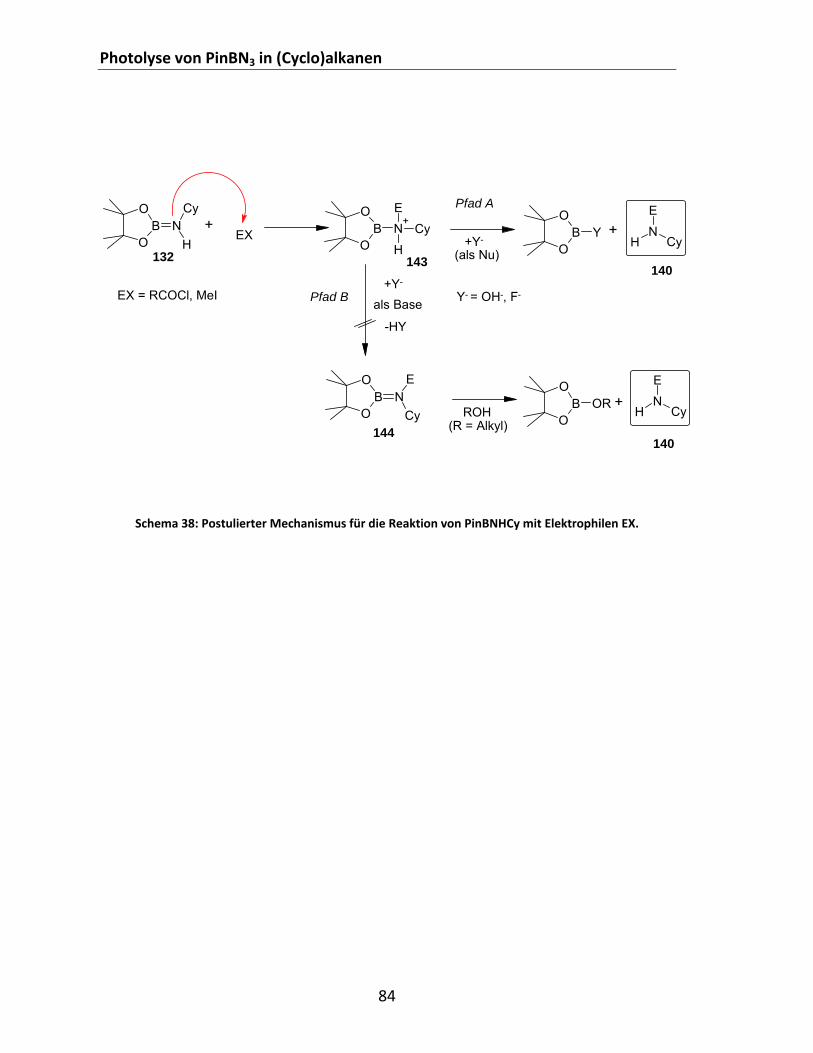
Photolyse von PinBN3 in (Cyclo)alkanen
84
Schema 38: Postulierter Mechanismus für die Reaktion von PinBNHCy mit Elektrophilen EX.
OB
ON
Cy
H OB
ON
H
Cy
E
OB
OY
HN
E
Cy
OB
ON
E
Cy OB
OOR
HN
E
Cy
+
132
EX
EX = RCOCl, MeI
++
144
+ROH
(R = Alkyl)
+Y-
+Y-
-HY
(als Nu)
als Base Y- = OH-, F-
143140
Pfad A
Pfad B
140
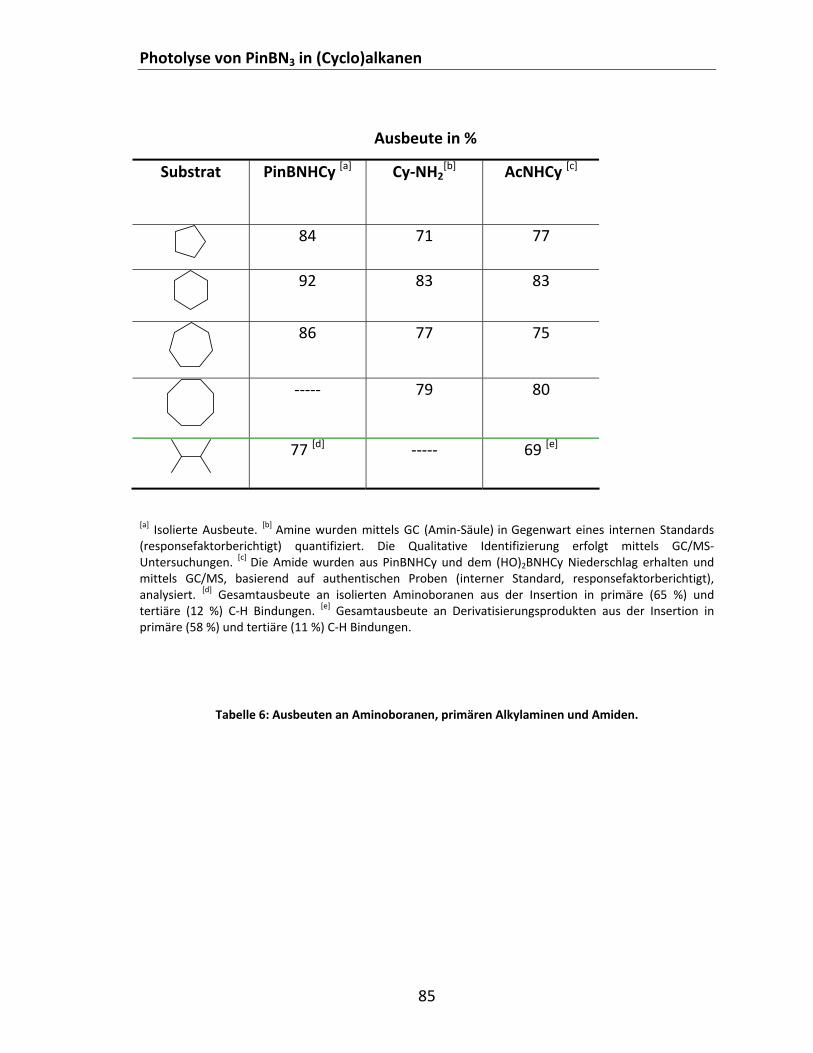
Photolyse von PinBN3 in (Cyclo)alkanen
85
Ausbeute in %
Substrat
PinBNHCy [a]
Cy‐NH2[b] AcNHCy [c]
84 71 77
92 83 83
86 77 75
‐‐‐‐‐ 79 80
77 [d] ‐‐‐‐‐ 69 [e]
[a] Isolierte Ausbeute. [b] Amine wurden mittels GC (Amin‐Säule) in Gegenwart eines internen Standards (responsefaktorberichtigt) quantifiziert. Die Qualitative Identifizierung erfolgt mittels GC/MS‐Untersuchungen. [c] Die Amide wurden aus PinBNHCy und dem (HO)2BNHCy Niederschlag erhalten und mittels GC/MS, basierend auf authentischen Proben (interner Standard, responsefaktorberichtigt), analysiert. [d] Gesamtausbeute an isolierten Aminoboranen aus der Insertion in primäre (65 %) und tertiäre (12 %) C‐H Bindungen. [e] Gesamtausbeute an Derivatisierungsprodukten aus der Insertion in primäre (58 %) und tertiäre (11 %) C‐H Bindungen.
Tabelle 6: Ausbeuten an Aminoboranen, primären Alkylaminen und Amiden.

Photolyse von PinBN3 in DMB: Untersuchungen zur Regioselektivität
86
4.2.2.2.2 Regioselektivität der C‐H‐Aminierung: 2,3‐Dimethylbutan (DMB) als Substrat
Um ein mögliches regioselektives Verhalten von Pinakolborylnitren PinBN 10 in
Bezug auf verschiedene C‐H‐Bindungen von Alkanen feststellen zu können,
wurden Abfangexperimente mit 2,3‐Dimethylbutan (DMB) als Substrat
durchgeführt. Da dieses über zwei unterschiedliche Arten von sp3‐hybridisierten
C‐H‐Bindungen (primäre und tertiäre C‐H‐Bindungen) verfügt, ist die Bildung der
zwei isomeren Produkte 146 und 147 denkbar. Wegen des Unterschiedes in den
C‐H‐Bindungsstärken (BDE: tert‐sp3‐C‐H ≈ 95.3 kcal/mol / prim‐sp3‐C‐H ≈ 100.3
kcal/mol)[198] besteht besonders unter Berücksichtigung literaturbekannter
Beispiele die Erwartung, dass bevorzugt die schwächere tertiäre Position aminiert
wird. So insertieren andere transiente Nitrene wie Acylnitrene ROCON, [84, 255]
Pentafluorphenylnitren C6F5N,[95, 96] Cyanonitren NCN[256] oder bestimmte
Phoshorylnitrene des Typs (RO)2PON[101, 257] fast ausschließlich in die schwächere
tertiäre C‐H‐Bindung von DMB.
Schema 39: Photochemie von PinBN3 in DMB: Untersuchungen zur Regioselektivität
Um den Sachverhalt für PinBN 10 zu prüfen, wurde eine Lösung von
Pinakolborazid PinBN3 9 in DMB ‐ wie bereits für Cycloalkane beschrieben ‐
photolysiert und das Rohprodukt nach dem Entfernen des überschüssigen DMB
zunächst mittels NMR‐Spektroskopie analysiert. Im 11B‐NMR Spektrum tritt ein
OB
ON3
OB
ON
H
H
H
OB
ON
H
146
+
147
-N2
h (= 254 nm)9

Photolyse von PinBN3 in DMB: Untersuchungen zur Regioselektivität
87
Signal mit der chemischen Verschiebung von δ = 24 ppm auf, das wie erwartet im
gleichen Bereich liegt, der auch für die Insertionsprodukte PinBNHCy 132
ermittelt wurde. Die Signalform lässt aber hier eine Überlagerung zweier Bande
vermuten („Schulterbildung“), was für die Anwesenheit zweier sehr ähnlicher,
aber chemisch unterschiedlicher Borspezies spricht. Auch im 1H‐ und 13C‐NMR
finden sich Hinweise auf zwei unterschiedliche Verbindungen, die sich den
erwarteten Isomeren 146 und 147 zuordnen lassen. Da die Überlagerung der
Resonanzen im 1H‐NMR eine Integration der Signale und damit eine Abschätzung
der Isomerenverteilung nur grob erlaubt, wurde das Produktverhältnis indirekt
aus den durch Acetylierung erhaltenen Amiden per GC/MS ermittelt. Bei einer
Gesamtaminierung von 69 % bezogen auf eingesetztes PinBN3 9, liegt ein
Isomerenverhältnis von etwa 6:1 (prim:tert) vor. Und obschon bevorzugt die
schwächere primäre C‐H‐Bindung aminiert wird, ist kein regioselektives Verhalten
des intermediär gebildeten PinBN 10 zu beobachten, wenn man um den
statistischen Faktor korrigiert. Dies kann wiederum nach dem Reaktivitäts‐
Selektivitäts‐Prinzip (RSP) der ungewöhnlich hohen Reaktivität des Borylnitrens
zugeschrieben werden.

Mechanismus der C‐H‐Aminierung
88
4.2.2.2.3 Mechanistische Überlegungen zur C‐H‐Aminierung: Konzertierte Insertion vs. Abstraktion‐Rekombination
Die Bildung der C‐H‐Funktionalisierungsprodukte, hier gezeigt am Beispiel der
Pinakolborylnitrenreaktion, kann prinzipiell über drei unterschiedliche
mechanistische Wege erfolgen. Der naheliegendste Reaktionspfad führt über das
aus dem Borazid 9 in situ gebildete Singulett‐Borylnitren 10‐S1, welches in einer
konzertierten Insertionsreaktion unter Bildung von Aminoboran 132 abreagiert
(Pfad A). Auch wäre die Bildung von 132 nach einem Abstraktions‐
Rekombinations‐Mechanismus durchaus denkbar. Relaxiert das zunächst
erzeugte angeregte 10‐S1 zum 10‐T0 (ISC), könnte dieses unter H‐Abstraktion
(typische Triplett‐Nitrenchemie) Radikal PinBNH• 148 bilden, welches direkt im
Lösungsmittelkäfig mit dem ebenfalls entstandenen Cycloalkylradikal Cy•
rekombiniert (Pfad B). Auch eine Aminierung nach dem Abstraktions‐
Rekombinations‐Mechanismus unter Beteiligung des offenschaligen Singulett‐
Nitrens 10‐S3 ist nicht auszuschließen (Pfad C), insbesondere unter der
Berücksichtigung des geringen energetischen Unterschieds zwischen beiden
Singulett‐Zuständen im CatBN 6.
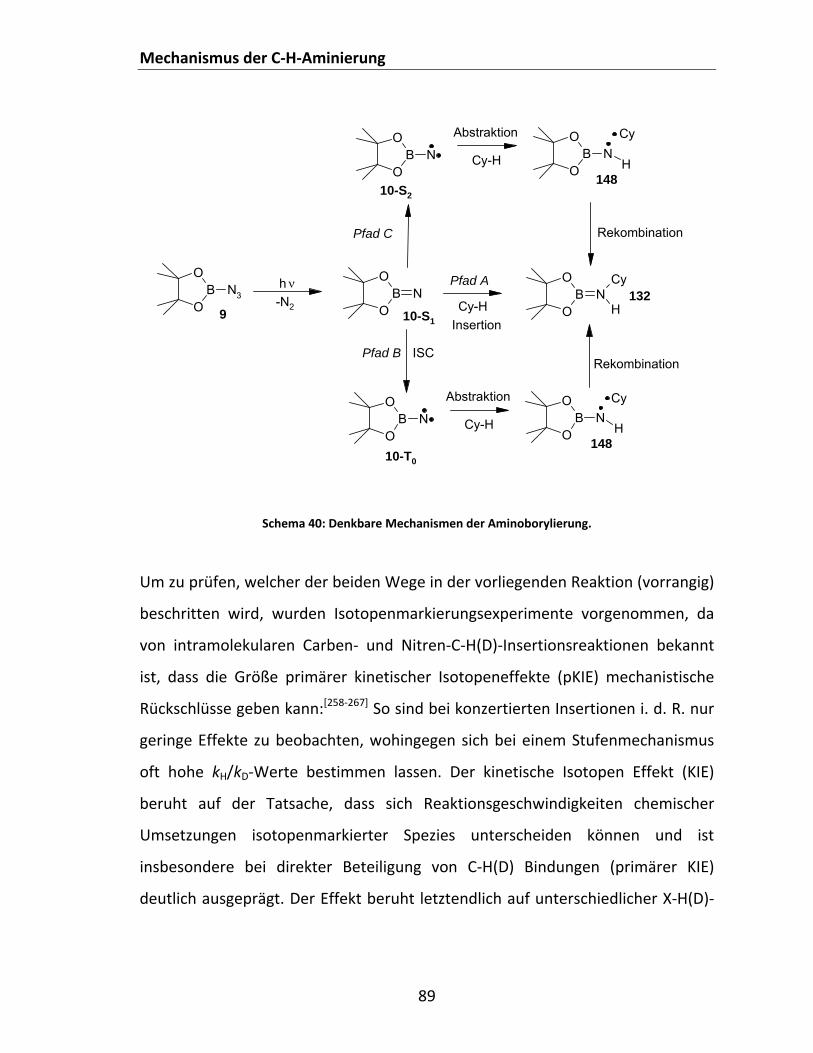
Mechanismus der C‐H‐Aminierung
89
OB
ON3
OB
ON
OB
ON
OB
ON
H
OB
ON
Cy
H
OB
ON
OB
ON
H
h-N2
Cy-H
Cy
Cy-H
Pfad A
Pfad B ISC
9 10-S1
132
14810-T0
Abstraktion
Rekombination
Insertion
Cy-H
14810-S2
Abstraktion
Pfad C Rekombination
Cy
Schema 40: Denkbare Mechanismen der Aminoborylierung.
Um zu prüfen, welcher der beiden Wege in der vorliegenden Reaktion (vorrangig)
beschritten wird, wurden Isotopenmarkierungsexperimente vorgenommen, da
von intramolekularen Carben‐ und Nitren‐C‐H(D)‐Insertionsreaktionen bekannt
ist, dass die Größe primärer kinetischer Isotopeneffekte (pKIE) mechanistische
Rückschlüsse geben kann:[258‐267] So sind bei konzertierten Insertionen i. d. R. nur
geringe Effekte zu beobachten, wohingegen sich bei einem Stufenmechanismus
oft hohe kH/kD‐Werte bestimmen lassen. Der kinetische Isotopen Effekt (KIE)
beruht auf der Tatsache, dass sich Reaktionsgeschwindigkeiten chemischer
Umsetzungen isotopenmarkierter Spezies unterscheiden können und ist
insbesondere bei direkter Beteiligung von C‐H(D) Bindungen (primärer KIE)
deutlich ausgeprägt. Der Effekt beruht letztendlich auf unterschiedlicher X‐H(D)‐

Mechanismus der C‐H‐Aminierung
90
Bindungsstärke, die wiederum in unterschiedlichen Nullpunktsschwingungs‐
energien begründet ist.
Zur Überprüfung wurde dazu PinBN3 9 zunächst in reinem D12‐Cyclohexan
photolysiert und mit den gängigen Methoden aufgearbeitet. Alkoholyse mit EtOD
führt zum erwarteten freien Amin C6D11ND2, Acetylierung entsprechend zum
Amid C6D11NHAc, wobei die ermittelten Ausbeuten im gleichen Bereich liegen wie
diejenigen die zuvor bei der Verwendung von C6H12 als Substrat ermittelt wurden.
Im Anschluss erfolgte die Belichtung von PinBN3 9 in einer 1:1‐Mischung von D12‐
/H12‐Cyclohexan unter der Fragestellung, ob das nicht‐deuterierte Cyclohexan
bevorzugt angegriffen wird. Nach der Aufarbeitung mit AcCl lassen sich mithilfe
von GC/MS‐Messungen die beiden zu erwartenden Produkte C6D11NHAc und
C6H11NHAc nachweisen. Da die Retentionszeiten der Produkte identisch sind, ist
die Quantifizierung mithilfe der Extract Ion Methode† vorgenommen und ein
Produktverhältnis von 1.0:1.1 (C6H11NHAc: C6D11NHAc) ermittelt worden.‡ Dieses
Fehlen eines ausgeprägten KIE stützt die Hypothese einer (vorrangig)
konzertierten Singulett‐Insertion.§ So wurde für die C‐H(D)‐Insertion von
Singulett‐Diethoxyphosphorylnitren (EtO)2PON in einer 1:1‐Mischung von D12‐
/H12‐Cyclohexan ein vergleichbarer kH/kD‐Wert (1.02) ermittelt.[101] Da zudem,
sofern überhaupt, nur Spuren des Cyclohexylradikal‐Rekombinationsprodukts
Cy6‐Cy6 gebildet werden, kann davon ausgegangen werden, dass die
Abstraktions‐Rekombinations‐Borylnitrenroute lediglich in untergeordnetem
† Die Extract Ion Methode, welche als Hilfsmittel in der GC/MS‐Software enthalten ist, ermöglicht eine separate Bestimmung einzelner Molekülionen. Besitzen zwei unterschiedliche Substanzen eine gleiche oder ähnliche Retentionszeit, die zu einer Signalüberlagerung führt, kann man diese nebeneinander bestimmen, vorausgesetzt sie besitzen unterschiedliche charakteristische Ionen. ‡ Das ermittelte Produktverhältnis ist das arithmetische Mittel zweier Reaktionsläufe (1.0:1.0 / 1.0:1.2). Um die absolute Güte der Daten beurteilen zu können sind noch weitere Studien notwendig. § Dieser (ausbleibende) Effekt stellt lediglich eine notwendige, aber keineswegs eine hinreichende Bedingung dar, da die Annahme besteht, dass sich die Energiebarrieren für die C‐H bzw. C‐D Abstraktion (geschwindigkeitsbestimmender Schritt) merklich voneinander unterscheiden und höher liegen als die Aktivierungsbarrieren einer konzertierten Nitren C‐H bzw. C‐D Insertion.

Mechanismus der C‐H‐Aminierung
91
Maße stattfindet. Darüber hinaus zeigen quantenmechanische Rechnungen, dass
die konzertierte Insertion des S1‐CatBN 6 in die σ‐Bindung von molekularem
Wasserstoff (BDE (kcal/mol): H2 = 104.2, CH4 = 105.0) nahezu barrierefrei
verläuft.[109]

Photolyse von (RO)2BN3: inter‐ und intramolekulare Aminierung
92
4.2.2.3 Photolyse von Dialkoxyboraziden (RO)2BN3 in Cycloalkanen
Da sich Pinakolborazid PinBN3 9 bzw. das korrespondierende Borylnitren PinBN
10 als äußerst effizientes Substrat für die C‐H‐Transformation von Alkanen
erwiesen hat, wurden systematische Experimente zur Photochemie weiterer
Azidoborane vorgenommen. Zunächst wurde das eng mit dem PinBN3 9
verwandte (iPrO)2BN3 150‐(iPr)2 untersucht. Dieses System ist chemisch sehr
ähnlich aufgebaut, verfügt aber nicht über ein rigides Ringsystem. Das ausgehend
von B(OiPr)3 und BCl3 synthetisierte neue Dialkoxyborazid 150‐(iPr)2 wurde
fortlaufend in Cyclohexan Cy‐6‐H bei RT photolysiert und das gebildete
Reaktionsprodukt anschließend mittels Isopropanol aufgearbeitet. Und wie
erhofft, bildet sich nach Alkoholyse neben dem Bortriisopropylester B(OiPr)3 das
erwartete Cyclohexylamin 134‐(Cy6) aus. Die gaschromatographisch bestimmte
Ausbeute liegt jedoch mit 46 % erheblich niedriger als im Fall der PinBN3‐
Belichtung (83 %). Zudem entsteht noch ein weiteres Alkoholyseprodukt. GC/MS‐
Messungen zeigen hierbei eindeutig (Datenbankabgleich / Vergleich mit
authentischer Probe), dass es sich bei der Verbindung um den Aminoalkohol 151‐
(Me) handelte, dessen Ausbeute bei etwa 15‐20 % liegt (GC‐Messung).
Es wurde Isopropanol als Abbaureagenz verwendet, da dies der gleiche Alkohol ist dem auch der Ligand des Borazids zu Grunde liegt. Dies bedeutet weiterhin, dass beim Quenchen mit weniger Produkten gerechnet werden kann, was eine Datenauswertung einfacher macht. Bei der Aufarbeitung des Photoprodukts aus der Reaktion von (EtO)2BN3 und Cy‐6‐H wurde dementsprechend auf Ethanol als Abbaureagenz zurückgegriffen.
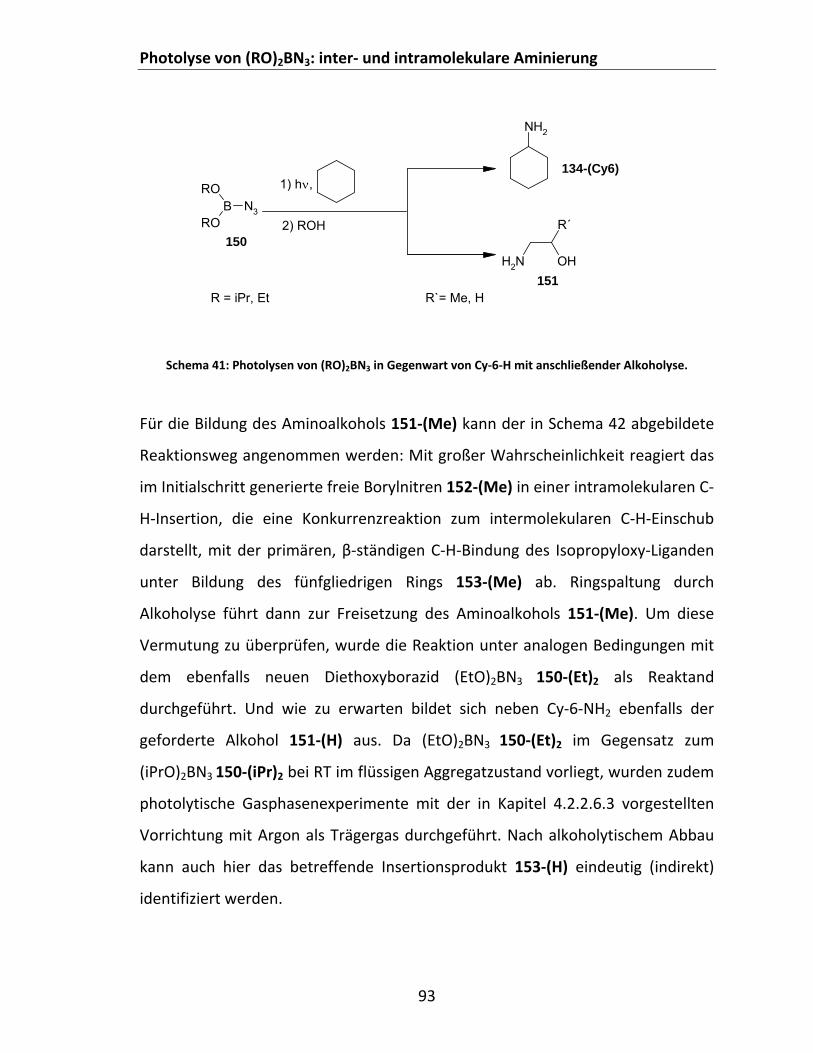
Photolyse von (RO)2BN3: inter‐ und intramolekulare Aminierung
93
Schema 41: Photolysen von (RO)2BN3 in Gegenwart von Cy‐6‐H mit anschließender Alkoholyse.
Für die Bildung des Aminoalkohols 151‐(Me) kann der in Schema 42 abgebildete
Reaktionsweg angenommen werden: Mit großer Wahrscheinlichkeit reagiert das
im Initialschritt generierte freie Borylnitren 152‐(Me) in einer intramolekularen C‐
H‐Insertion, die eine Konkurrenzreaktion zum intermolekularen C‐H‐Einschub
darstellt, mit der primären, β‐ständigen C‐H‐Bindung des Isopropyloxy‐Liganden
unter Bildung des fünfgliedrigen Rings 153‐(Me) ab. Ringspaltung durch
Alkoholyse führt dann zur Freisetzung des Aminoalkohols 151‐(Me). Um diese
Vermutung zu überprüfen, wurde die Reaktion unter analogen Bedingungen mit
dem ebenfalls neuen Diethoxyborazid (EtO)2BN3 150‐(Et)2 als Reaktand
durchgeführt. Und wie zu erwarten bildet sich neben Cy‐6‐NH2 ebenfalls der
geforderte Alkohol 151‐(H) aus. Da (EtO)2BN3 150‐(Et)2 im Gegensatz zum
(iPrO)2BN3 150‐(iPr)2 bei RT im flüssigen Aggregatzustand vorliegt, wurden zudem
photolytische Gasphasenexperimente mit der in Kapitel 4.2.2.6.3 vorgestellten
Vorrichtung mit Argon als Trägergas durchgeführt. Nach alkoholytischem Abbau
kann auch hier das betreffende Insertionsprodukt 153‐(H) eindeutig (indirekt)
identifiziert werden.
RO
ROB N3
NH2 OH
R´
NH2
1) h ,
R = iPr, Et R`= Me, H
2) ROH150
151
134-(Cy6)
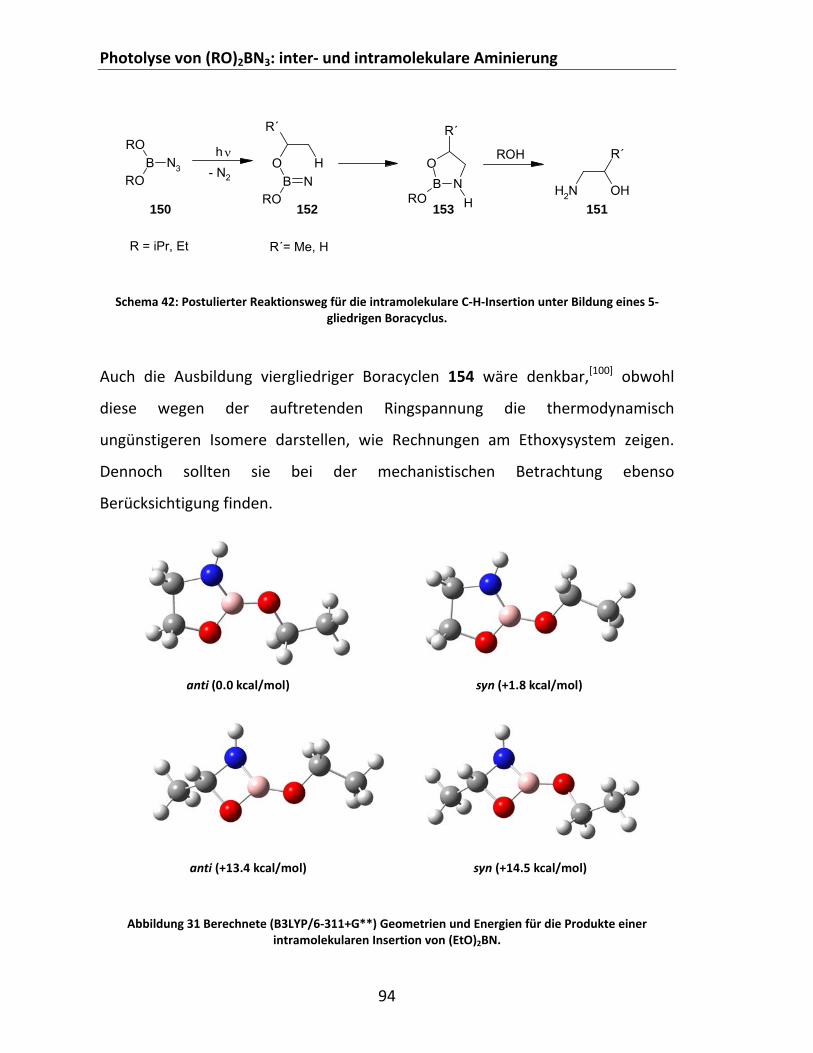
Photolyse von (RO)2BN3: inter‐ und intramolekulare Aminierung
94
anti (0.0 kcal/mol) syn (+1.8 kcal/mol)
anti (+13.4 kcal/mol) syn (+14.5 kcal/mol)
Schema 42: Postulierter Reaktionsweg für die intramolekulare C‐H‐Insertion unter Bildung eines 5‐gliedrigen Boracyclus.
Auch die Ausbildung viergliedriger Boracyclen 154 wäre denkbar,[100] obwohl
diese wegen der auftretenden Ringspannung die thermodynamisch
ungünstigeren Isomere darstellen, wie Rechnungen am Ethoxysystem zeigen.
Dennoch sollten sie bei der mechanistischen Betrachtung ebenso
Berücksichtigung finden.
Abbildung 31 Berechnete (B3LYP/6‐311+G**) Geometrien und Energien für die Produkte einer intramolekularen Insertion von (EtO)2BN.
O
BRO
R´
H
N
O
B N
R´
HRONH2 OH
R´RO
BRO
N3
h
- N2
R = iPr, Et R´= Me, H
ROH
150 152 151153

Photolyse von (RO)2BN3: inter‐ und intramolekulare Aminierung
95
Der Alkoholyseschritt von 154 sollte das Halbaminal 155 ergeben, das wegen
seiner Instabilität unter Wasserabspaltung zu Imin 156 reagiert. Je nach
Reaktionsbedingungen könnte dieses dann weiter in Ammoniak und Aceton (157‐
(Me), für R = iPr) bzw. Acetaldehyd (157‐(H), für R = Et) fragmentieren. Doch
weder die aufgeführten Verbindungen noch mögliche Umlagerungs‐ und
Abbauprodukte oder weitere denkbare Derivate können mittels GC/MS qualitativ
nachgewiesen werden.
Schema 43: Denkbare intramolekulare C‐H‐Insertion unter Bildung eines gespannten Vierings.
Um sicher zu stellen, dass der nicht geglückte Nachweis des Vierringsystem 154
nicht in der Instabilität der auftretenden Abbauprodukte begründet ist, wurden
weitere Experimente unternommen mit dem Versuch, die entstehenden
Produkte indirekt über Acetylierungsreaktionen zu identifizieren. Dazu wurde das
Photorohprodukt mit AcCl umgesetzt und nach Aufarbeitung mithilfe von GC/MS‐
Messungen analysiert. Aber auch hier können keine Produkte nachgewiesen
werden, die sich aus dem Abbau des viergliedrigen Heterocyclus 154 erklären
lassen. Es können lediglich die Acetylierungsprodukte der übrigen
Reaktionsprodukte ermittelt werden. So entstehen neben Cy‐6‐NHAc (141‐(Cy6))
auch die möglichen Acetylderivate (158‐160) des Aminoalkohols 153.
O
BRO
N
R´ Me
H O
B NRO H
R´Me
R´ Me
OH NH2
R´ Me
NH
R´ Me
O
RO
BRO
N3
NH3
-H2O
+
h
-N2
ROH
R = iPr, Et R´= Me, H
150 152 155154
156 157

Photolyse von (RO)2BN3: inter‐ und intramolekulare Aminierung
96
N O
R´
Ac
AcH N O
R´
Ac
HH N O
R´
H
AcH
O
B N
R´
HRO
1) AcCl+ +
153 158 159 160
2) NaOH (s)
O
B N
H
MeO
O
B NHMeO
B N3
MeO
MeO
h
-N2
150-(Me)2 152-(Me)2 154-(H)
Schema 44: Acetylierung von 153.
Zudem wurden Versuche unternommen, Dimethoxyborazid (MeO)2BN3 150‐
(Me)2 zu synthetisieren, da das korrespondierende Nitren 152‐(Me)2 nur über α‐
ständige C‐H‐Bindungen verfügt. Sofern eine (erzwungene) intramolekulare
Insertionsreaktion möglich sein sollte, müsste ausschließlich das Vierringsystem
154‐(H) gebildet werden.
Schema 45: Denkbarer Reaktionspfad für die intramolekulare CH‐Insertion von (MeO)2BN.
Doch trotz erheblicher Bemühungen gelang die Synthese des benötigten Azids
150‐(Me)2 nicht. So konnten das beim Azidierungsschritt zwangsläufig anfallende
Kopplungsprodukt TMSCl, sowie überschüssiges TMSN3 und das verwendete LM,
nicht abgetrennt werden wegen des geringen Siedepunktunterschiedes im
Vergleich zum Azid. Auch die Azidierung mit LiN3 bzw. NaN3 als Azidierungsmittel
misslang. Bei niedrigen Temperaturen wurde auch nach langer Reaktionszeit in
unterschiedlichen LM keine Umsetzung beobachtet (11B‐NMR Kontrolle). Die
Umsetzung bei höherer Temperatur hat sich wegen der Instabilität des
Dimethoxyborchlorids (MeO)2BCl auch als ungeeignet erwiesen.

Photolyse aromatischer Borazide in Kohlenwasserstoffen
97
4.2.2.4 Photolyse aromatischer disauerstoffsubstituierter Borazide in Cycloalkanen und Aromaten
Im Anschluss an die Arbeiten zu alkoxysubstituierten Azidoboranen wurden
Experimente zur Photochemie weiterer disauerstoffsubstituierter Borazide
vorgenommen. Um eine effizientere UV‐Absorption im längerwelligen Bereich zu
erreichen, wurden die unten aufgeführten aromatischen Borazide synthetisiert
und deren photochemisches Verhalten in Lösung untersucht.
Abbildung 32 Aromatische disauerstoffsubstituierte Azidoborane, die im Folgenden diskutiert werden.
4.2.2.4.1 Photolyse von CatBN3 und t‐BuCatBN3 in Cycloalkanen Cy‐H und Benzol
Das Reaktionsverhalten des bei den Matrixexperimenten erfolgreich eingesetzten
CatBN3 7 wurde auch unter klassischen Bedingungen in Lösung untersucht.
Zunächst erfolgte die Photolyse einer Lösung des Azids 7 ‐ wie bereits für das
PinBN3 beschrieben ‐ in Cyclohexan Cy‐6‐H. Währendessen färbte sich die zuvor
farblose, klare Lösung im Zuge der Belichtung in recht kurzer Zeit (etwa 1 h)
tiefgelb bis braun, wobei zudem ein harziges Produkt ausfiel. Anschließend wurde
das LM im Vakuum entfernt, der komplette Rückstand mit Isopropanol versetzt
OB
ON3
OB
ON3
OB
ON3
OB
ON3
OB
ON3
PhOB
PhON3
7
184177
1717-tBu

Photolyse aromatischer Borazide in Kohlenwasserstoffen
98
und die überstehende Lösung mithilfe von GC/MS‐Messungen untersucht.
Überraschenderweise lassen sich keine Hinweise auf das gewünschte
Aminierungsprodukt Cy‐6‐NH2 134‐(Cy6) finden. Auch kann kein alkoholytischer
Abbau bei erhöhter Temperatur und längerer Rührzeit beobachtet werden.
Desgleichen führt die Zugabe von Säure bzw. Base und/oder F‐‐Ionen nicht zum
freien Cyclohexylamin Cy‐6‐NH2. Des Weiteren liefern durchgeführte
Acetylierungsexperimente keine Anhaltspunkte für eine intermolekulare C‐H‐
Insertion. Auch Änderungen der Photolysebedingungen (Photolysedauer,
Konzentration des eingesetzten CatBN3, Lichtintensität usw.) blieben erfolglos. Es
kann lediglich festgestellt werden, dass mit zunehmender Belichtungsdauer die
Färbung der Reaktionsmischung immer intensiver wird und die Menge am
gebildeten Feststoff zunimmt. Da bekannt ist, dass bestimme
Catecholaminoborane zu Oligomerisierung neigen,[268] könnte dies eine mögliche
Erklärung dafür sein, dass keine Abbauprodukte beobachtet werden können. So
geht das postulierte Insertionsprodukt 8 möglicherweise aufgrund seiner
Photoinstabilität Folgereaktionen zu undefinierbaren, nicht‐ oder
schwerabbaubaren oligo‐ und polymeren Produkten ein. In der Hoffnung das
geforderte intermediär auftretende Insertionsprodukt 8 kinetisch zu stabilisieren,
wurden weitere Versuche mit dem sterisch anspruchsvolleren Cy‐8‐H als Substrat
durchgeführt. Aber auch hier misslingt die Detektion des Amins bzw. Amids nach
dem Aufarbeitungsschritt. Auch ergeben die Photolysen des verwandten tert‐
ButylCatBN3 7‐tBu in Anwesenheit von Cy‐6‐H bzw. Cy‐8‐H keine Hinweise auf
Cycloalkylaminoderivate. Ebenfalls scheiterten die Versuche, Benzol mithilfe von
7 zu aminieren. So kann kein Anilin (Alkoholyse) bzw. N‐Phenylacetamid
(Acetylierung) dokumentiert werden.

Photolyse aromatischer Borazide in Kohlenwasserstoffen
99
Schema 46: Denkbarer Reaktionspfad für die Reaktion zwischen CatBN3 und verschiedenen Kohlenwasserstoffen.
Um die Hypothese einer Oligomerisierung zu prüfen, wurden detaillierte
Analysen durchgeführt. 11B‐NMR Messungen der DCM‐löslichen Bestandteile des
kompletten Photorohprodukts zeigen ein Signal bei 24 ppm, was für ein
Aminoboran des Typs CatBNHCy 8 zu erwarten ist. Da das Signal jedoch eine
äußerst schwache Intensität aufweist, kann ‐ unter der Annahme einer guten
Löslichkeit von CatBNHCy 8 in DCM ‐ davon ausgegangen werden, dass nur
äußerst geringe Mengen des Insertionsprodukts in monomerer Form vorhanden
sind. Da weiterhin die Empfindlichkeit des GC/MS‐Gerätes gegenüber den
polaren Aminen nur gering ist, könnte dies ein denkbarer Grund für den nicht
geglückten Nachweis von Cy‐6‐NH2 134‐(Cy6) nach vorgenommener Alkoholyse
sein. Eine Resonanz im 11B‐Spektrum im Bereich von etwa 0 ppm, die man für
oligomeres (CatBNHCy)n erwarten würde (tetrakoordiniertes Borzentrum), tritt
hingegen nicht auf. Dies ist nicht überraschend, wenn man die vermutlich geringe
Löslichkeit der Oligomere in üblichen Solvenzien berücksichtigt. Eine fehlende
Umsetzung des Azids konnte mithilfe von IR‐Messungen ausgeschlossen werden,
die eindeutig den kompletten Verlust der Azid‐Bande zeigen. Werden die
löslichen und unlöslichen Bestandteile der Photoreaktion unabhängig
voneinander untersucht, ergibt sich folgendes Bild: Wird der ausgefallene
OB
ON3
R OB
ON
H
Cy
R 87
CyH =
h (= 254 nm)
R = H, t-Bu
Cy-HFolgeprodukt(e)

Photolyse aromatischer Borazide in Kohlenwasserstoffen
100
Feststoff von der Lösung separiert, zeigt dieser nur eine äußerst schlechte
Löslichkeit in üblichen Solvenzien. Auch gegenüber Säure oder Lauge verhält sich
der harzige Feststoff recht inert. Engt man hingegen die erhaltene Lösung (Cy‐6‐H
& lösliches Photoprodukt) ein, verbleibt eine kleine Menge eines gelblichen
Feststoffs. Zwar liefert auch hier die Alkoholyse kein Cy‐6‐NH2 (und auch kein
Catechol), aber es lässt sich B(OiPr)3 gaschromatographisch nachweisen. Auch die
Umsetzung der löslichen Probe mit AcCl liefert keinerlei Hinweise auf eine
intermolekulare Aminierungsreaktion. So lässt sich kein Cy‐6‐NHAc 141‐(Cy6)
identifizieren. Lediglich kleine Anteile an diacetylgeschütztem Catechol 162
können nachgewiesen werden. Da weiterhin ungeklärt blieb, ob die aus dem
postulierten Aminoboran CatBNHCy 8 zugänglichen Aminoverbindungen Cy‐6‐
NH2 134‐(Cy6) bzw. Cy‐6‐NHAc 141‐(Cy6) tatsächlich wegen ihrer geringen
Konzentration nicht bestimmbar waren, wurden weitere Messungen zu einem
alternativen Verbleib des „Borylnitren‐Stickstoffs“ vorgenommen. Dazu wurde
nach typischen Produkten gesucht, die sich aus Triplettchemie von CatBN 6
ergeben könnten (vgl. 4.2.2.6.3.2). Doch der Nachweis jener Systeme blieb aus.
Zwar ist die Konzentration des Radikaldimers Bicyclohexyl (Cy6‐Cy6) 163 etwas
höher als im Falle der Pinakolborazidbelichtung, aber mit < 5 % (unkorrigiert)
dennoch recht niedrig. Auch die Bildung von Produkten, die aus Abstraktions‐
oder Dimerisierungsprozessen entstehen könnten (CatBNH2, CatBNNBCat,
CatBNHNHCat), ist mit hoher Wahrscheinlichkeit auszuschließen. So lassen sich
nach dem Abbau keine Ammoniak‐ 164 oder Hydrazin(derivate) 165, 166
nachweisen. Auch CatBBCat 167, dessen Bildung über das Nitrendimer
CatBNNBCat durch Stickstoffextrusion möglich sein könnte, ist laut 11B‐NMR‐
Messungen nicht im Photoprodukt enthalten (Vergleich mit authentischer
Die Abtrennung des harzigen Feststoffs gestaltete sich als relativ schwierig und gelang nur zum Teil, da erhebliche Mengen des Photoprodukts an der Innenwand des Quarzrohrs verblieben.

Photolyse aromatischer Borazide in Kohlenwasserstoffen
101
Probe). Eine Umlagerung von CatBN 6 zum cyclischen Iminoboran 168 wurde
ebenfalls in Betracht gezogen. Da zum einen computerchemische
Untersuchungen zeigen, dass 168 keinen stationärer Punkt auf der PES darstellt,
zum anderen keine denkbaren Folgeprodukte von 168 im Produktgemisch
identifiziert werden können, ist auch die Beteiligung dieser Spezies im
Reaktionsverlauf als sehr unwahrscheinlich einzuschätzen.
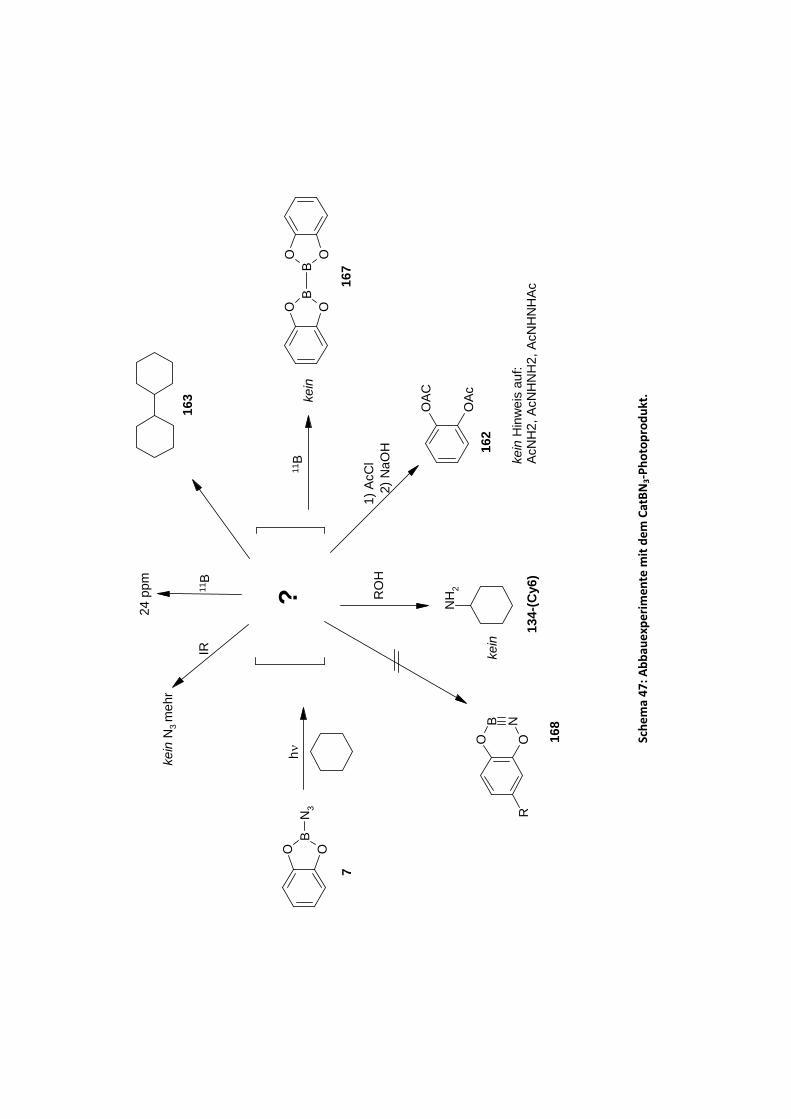
OB
ON
3
OAC
OAc
RO
NBO
NH
2
OB
OO
BO
IR
kein
N3 m
ehr
RO
H1)
AcC
l2)
NaO
H
?11B
24 p
pm
h
168
7
162
kein
Hin
wei
s au
f:Ac
NH
2, A
cNH
NH
2, A
cNH
NH
Ac
11B
163
167
134-
(Cy6
)
kein
kein
Schem
a 47: Abbau
experimen
te m
it dem
CatBN
3‐Photoprodukt.

Photolyse aromatischer Borazide in Kohlenwasserstoffen
103
Um eine mögliche (Photo)instabilität von CatBNHCy 8 direkt zu überprüfen,
wurden Versuche unternommen, das Aminoboran auf unabhängigem Weg zu
synthetisieren. Dazu wurde CatBCl 170 mit Cyclohexylamin 134‐(Cy6) in
Gegenwart einer Stickstoffbase umgesetzt. Aber das erwünschte Aminoboran 8
ließ sich selbst nach Variation der Reaktionsparameter (Verwendetes LM,
Reaktionstemperatur, eingesetzte Base, Reihenfolge der Substratzugabe usw.)
nicht rein isolieren. So bilden sich neben der Zielverbindung auch
unterschiedliche Borate (11B‐Messungen) und vermutlich das
Disubstitutionsprodukt Cat2NCy‐6.
Schema 48: Experimente zur Synthese von CatBNHCy.
Abschließend kann vorerst festgehalten werden, dass im Gegensatz zu PinBN3 9
die Photolyse von CatBN3 7 in Cycloalkanen keine oder nur geringe Anteile an
(monomeren) Aminoboranen ergibt. Ob sich dieser experimentelle Befund
ausschließlich mit der Neigung von 8 zur Oligo‐ bzw. Polymerisierung erklären
lässt oder noch andere Reaktionswege berücksichtigt werden müssen, kann
anhand der zur Verfügung stehenden Daten nicht abschließend geklärt werden.
OB
OCl
OB
ON
H
8-(Cy6)170
+Cy-6-NH2
Base
(Base = Cy-6-NH2, Et3N, Py)

Photolyse aromatischer Borazide in Kohlenwasserstoffen
104
4.2.2.4.2 Photolyse des Bisazids 171 in Cyclohexan Cy‐6‐H
Das Bisazid 171, welches ursprünglich lediglich für die Durchführung von ESR‐
Experimenten hergestellt wurde, ist auch unter photochemischen Bedingungen
in Lösung untersucht worden. Dazu wurde das unbekannte Azid in Anlehnung an
Arbeiten von Alridge et al. über eine mehrstufige Syntheseroute dargestellt.[183]
Zunächst erfolgt die Reduktion des para‐Chinons 172 mit Sn/HCl zu 1,2,4,5‐
Tetrahydroxybenzol 173 und anschließender Umwandlung in den
entsprechenden TMS‐geschützten Alkohol 174. Transmetallierung liefert das
bekannte Bischlorid 175, welches nach Azidierung mit TMSN3 das gewünschte
neue Azid 171 ergibt.
Schema 49: Syntheseweg für die Herstellung des Bisazids.
Die durchgeführten Belichtungsversuche wurden unter den bereits
beschriebenen Bedingungen durchgeführt. Da sich das Azidoboran 171 als kaum
löslich in Cy‐6‐H erwies, wurde die weiße, milchige Suspension, um eine
möglichst homogene Durchmischung zu gewährleisten, während der Photolyse
kontinuierlich gerührt. In Abhängigkeit der Photolysedauer (0.5 ‐ 3 h) und
eingesetzter Azid‐Konzentration ergibt die alkoholytische Aufarbeitung bzw.
O
O OH
OH OH
OH OH
OH
OB
OOB
OClCl
OTMS
TMSO OTMS
TMSO
OB
OOB
ON3N3
172 173
Reduktion
174
Schützen
175171
BCl3
TMSN3
Azidierung
Transmetallierung
Sn/HCl TMSCl/Et3N

Photolyse aromatischer Borazide in Kohlenwasserstoffen
105
Acetylierung eine Ausbeute an den intermolekularen Aminierungsprodukten Cy‐
6‐NH2 134‐(Cy6) bzw. Cy‐6‐NHAc 141‐(Cy6) von etwa 20‐30 %, bezogen auf ein
Mol des eingesetzten Bisazids. Warum hier im Gegensatz zu den zuvor
beschrieben Catecholboraziden 7 Produkte einer C‐H‐Insertion nachgewiesen
werden können, bleibt unklar. Möglicherweise neigt das postulierte
Photoprodukt einer zweifachen Borylnitren‐Insertion 176 weniger stark zu
Assoziations‐ und Folgereaktionen als CatBNHCy 8. Es wäre durchaus denkbar,
dass das zusätzliche Cyclohexangerüst zu einer sterischen Abschirmung und
damit zur kinetischen Stabilisierung der reaktiven Aminoboraneinheit führt.
Möglicherweise ist auch eine Oligomerisierung der wahrscheinlich entstehenden
isomeren Aminoborane 176 (syn und anti) aus Symmetriegründen ungünstiger.
Schema 50: Denkbare isomere Photoprodukte für die vollständige Reaktion von 171 mit Cy‐6‐H.
Wird die Ausbeute bezogen auf die im Molekül enthaltenen Azido‐Gruppen (2 pro Molekül) angegeben, muss sie auf 10‐15 % halbiert werden.
OB
OOB
ON3N3
OB
OOB
ONN
H
H
H OB
OOB
ONN
H
171
176-anti
176-syn
+ h (= 254 nm)

Photolyse aromatischer Borazide in Kohlenwasserstoffen
106
4.2.2.4.3 Photolyse von Azidoboran 177 in den Aromaten Benzol, Toluol und Mesitylen
Auch das auf 2,3‐Dihydroxynaphthalin basierende neue Borazid 177 wurde
mithilfe klassischer Abfangexperimente in Lösung untersucht. Da sich das Azid
ebenfalls als unlöslich in Cycloalkanen erwies, wurde es direkt mit verschiedenen
Aromaten umgesetzt, in denen es sich löslich zeigte.
Schema 51: Photolyse von 177 in unterschiedlichen Aromaten.
Erste Photoexperimente mit Benzol als Substrat ergeben nach Alkoholyse bzw.
Acetylierung neben variablen Mengen an unlöslichen polymeren Substanzen
undefinierter Zusammensetzung u. a. auch Anilin Ph‐NH2 bzw. N‐Phenylacetamid
als Produkte (Vergleich mit authentischen Proben). Obschon die erhaltene
Ausbeute mit 10‐15 % nicht von synthetischem Wert ist, spiegelt sich unter
Berücksichtigung der Aryl‐C‐H‐Bindungsstärke erneut die hohe Reaktivität des
Borylnitrens wider. Für die im Anschluss durchgeführten Untersuchungen zur
Regioselektivität, wurde zunächst Toluol als Substrat gewählt, welches nach
Zwar können einfache Aromaten (z. B. Benzol) relativ unproblematisch funktionalisiert werden, doch findet dies i. d. R. über einen anderen mechanistischen Reaktionsweg statt. So tritt normalerweise keine direkte C‐H‐Bindungsspaltung ein, sondern die C‐H‐Transformation (= Substitution des Wasserstoffs) verläuft nach einem Additions‐Eliminierungs‐Mechanismus (→ elektrophile aromatische Substitution).
OB
ON3
R1
R2 R2
R1
R2 R21) 2) ROH
R1 = R2 = H (Benzol)
R1 = Me, R2 = H (Toluol) R1 = R2 = Me (Mesitylen)
NH2
177
h

Photolyse aromatischer Borazide in Kohlenwasserstoffen
107
NH2
NH2
NH2
NH2
178 179 180 181
Aminierung und Alkoholyse die Isomere 178‐181 bilden kann. Aufgrund des
Bindungsstärkeunterschieds zwischen der Benzyl‐ und Aryl‐C‐H‐Bindung (BDE
(kcal/mol): Ph‐H (112.9), Benzyl‐H (88.5)),[198] der größer als im DMB ist, könnte
ein bevorzugter Angriff des Borylnitrens auf die Benzylstellung erwartet werden.
Abbildung 33: Denkbare isomeren Aminierungsprodukte nach Alkoholyse.
Nach beendeter Photolyse und der Zugabe von Isopropanol zur gelb‐braunen
Reaktionsmischung wurde die überstehende Lösung mittels GC/MS analysiert.
Dabei können u. a. die unterschiedlichen erwarteten Produkte identifiziert
werden. Neben Benzylamin 187 (Vergleich mit Referenzprobe), lassen sich auch
die methylsubstituierten Aniline 179‐181 anhand ihrer Massenspektren
ausmachen (keine Referenzproben vorhanden). Weil die Fragmentierungsmuster
jener Komponenten jedoch nahezu identisch sind und es zudem zu
Überlagerungen der Signale kommt, ist eine genaue Zuordnung nicht
durchführbar. Trotzdem lässt sich bereits anhand dieser Versuche qualitativ
feststellen, dass die Benzylposition vermutlich nicht bevorzugt angegriffen wird.
Um die Dateninterpretation etwas einfacher zu gestalten, wurden weitere
Photoexperimente mit Mesitylen als Substrat durchgeführt. Die GC/MS‐Analyse
der alkoholytisch abbaubaren Reaktionsprodukte liefert u. a. die in Abb. 34
dargestellten Amine 182 und 183 in geringer Ausbeute. Daneben lässt sich auch
ein nicht unerheblicher Anteil an polymeren Produkten und Dimesitylen (Mes‐

Photolyse aromatischer Borazide in Kohlenwasserstoffen
108
Mes) ausmachen, das bezogen auf eingesetztes Azid je nach
Reaktionsbedingungen bis zu 5 % (unkorrigiert) beträgt.
Abbildung 34: Isomere der Mesitylenaminierung.
Eine Umsetzung des Rohprodukts aus der Mesitylenreaktion mit AcCl erlaubt die
Ausbeuten‐ und Isomerenverhältnisbestimmung. Bei einer Gesamtausbeute von
etwa 15 % (unkorrigiert), ist die Aminierung der Benzylstellung leicht über den
statistischen Faktor (3benz. : 1ar.) hinaus bevorzugt. Möglichweise ist dies jedoch
nicht auf den Unterschied in den Bindungsstärken, sondern auf die sterische
Abschirmung der aromatischen Position durch jeweils zwei Methyl‐Gruppen
zurückzuführen.
NH2
NH2
182 183

Photolyse aromatischer Borazide in Kohlenwasserstoffen
109
4.2.2.4.4 Photolyse von (PhO)BN3 in Cyclohexan
Auch das acyclische Diphenoxyborazid (PhO)2BN3 184 sollte im Rahmen dieser
Arbeit untersucht werden. Doch konnte das benötigte Borchlorid (PhO)2BCl 185
nicht rein aus der Austauschreaktion zwischen B(OPh)3 und BCl3 gewonnen und
somit keine vollständige Umsetzung erreicht werden. Die Experimente zeigen,
dass verschiedene temperatur‐ und lösungsmittelabhängige Gleichgewichte eine
Rolle spielen, die zur Ausbildung unterschiedlicher Borspezies führen. Dennoch
wurde die Reaktionsmischung aus dem Metatheseansatz mit TMSN3 umgesetzt,
wobei sich neben dem Hauptprodukt (PhO)2BN3 184 auch noch weitere
borhaltige Systeme bilden, wie 11B‐NMR zeigten (z. B. (PhO)B(N3)2, Cl2BN3, Borate
usw.).
Schema 52: Synthese von (PhO)2Cl durch Austauschreaktion.
Die Bestrahlung (0.5‐3 h) einer Suspension des (verunreinigten) Azids 184 in Cy‐
H‐6 liefert nach dem Quenchen mit Alkohol ein bandenreiches Chromatogramm,
was u. a. Spuren des intermolekularen C‐H‐Insertionsprodukts Cy‐6‐NH2 134‐
(Cy6) aufweist. Nach Acetylierung der kompletten, inhomogenen dunklen
Reaktionsmischung lassen sich neben Cy‐6‐NHAc 141‐(Cy6) auch die drei
unterschiedlichen 2‐Aminophenolderivate 186‐188 in sehr kleinen Ausbeuten per
GC/MS finden. Die Bildung derer kann auch hier mit einer intramolekularen
Insertion als Schlüsselschritt erklärt werden. Das aus dem Azid (PhO)2BN3 184
generierte Singulett‐Borylnitren (PhO)2BN 189 insertiert in die ortho zur Oxy‐
PhO
BPhO
OPhCl
B
Cl
ClPhO
BPhO
Cl+2 1 3
185
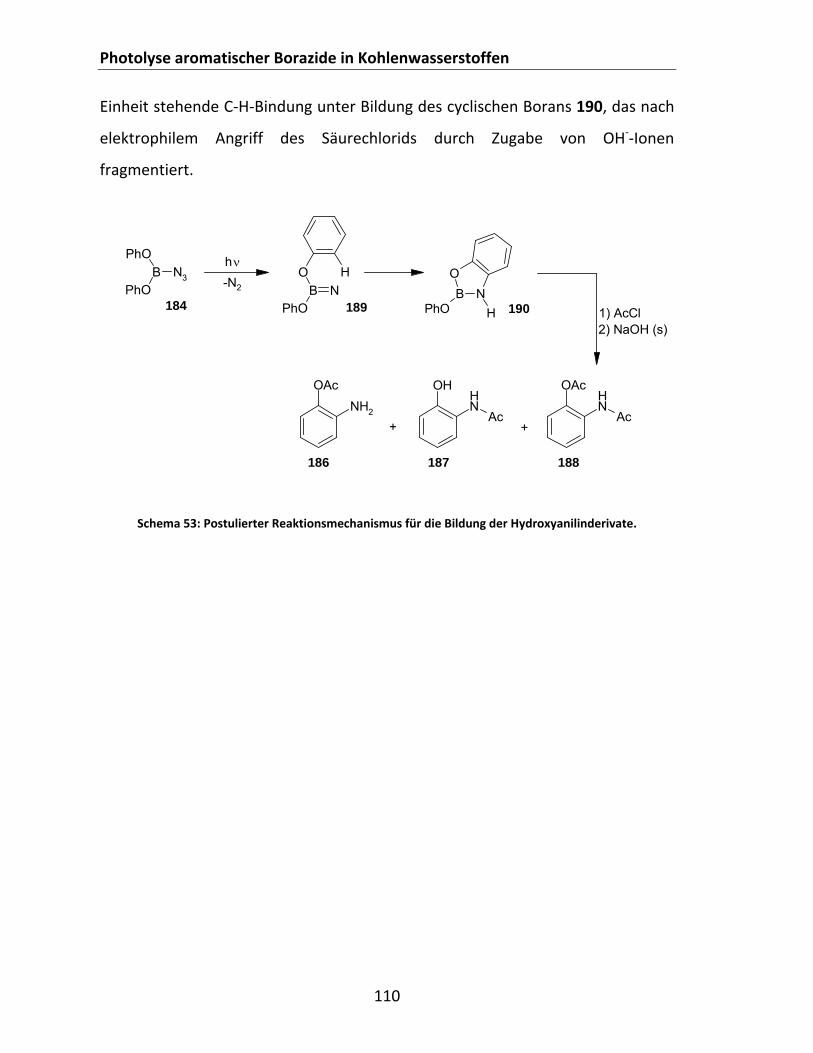
Photolyse aromatischer Borazide in Kohlenwasserstoffen
110
Einheit stehende C‐H‐Bindung unter Bildung des cyclischen Borans 190, das nach
elektrophilem Angriff des Säurechlorids durch Zugabe von OH‐‐Ionen
fragmentiert.
Schema 53: Postulierter Reaktionsmechanismus für die Bildung der Hydroxyanilinderivate.
O
BPhO
N
H
PhO
BPhO
N3 O
B NPhO H
OAc
NH2
OAc
NH
Ac
OH
NH
Ac
h
-N2
++
1) AcCl2) NaOH (s)
184 189 190
186 187 188

Photolyse stickstoff‐ und schwefelsubstituierter Borazide
111
4.2.2.5 Photolyse stickstoff‐ und schwefelsubstituierter Borazide in Kohlenwasserstoffen
Motiviert durch die geglückten grundlegenden Arbeiten zu den sauerstoff‐
substituierten Boraziden, schien uns auch die Untersuchung chemisch
verwandter Borazide interessant und aussichtsreich. Dazu wurden die in Grafik
35 abgebildeten symmetrischen amino‐ und schwefelsubstituierten Azidoborane
191‐193 synthetisiert und deren photochemisches Reaktionsverhalten in Lösung
untersucht.
Abbildung 35: Donorsubstituierte Borazide auf Stickstoff‐ und Schwefelbasis.
4.2.2.5.1 Photolyse von 2‐Azido‐1,3‐dimethyl‐1,3,2‐diazaborolidin in Cyclohexan
Obwohl sich nach der Synthese des flüssigen, literaturbekannten[108] Azidoborans
191 zeigte, dass bedauerlicherweise nur eine geringfügige Löslichkeit in
Cyclohexan besteht, wurde eine Mischung beider Komponenten für 16 h bei RT
belichtet. Um zumindest eine gewisse Durchmischung zu erreichen, wurde das
resultierende Zweiphasengemisch kontinuierlich stark gerührt und die dunkle
homogene Reaktionsmischung nach beendeter Photolyse solvolytisch mit
Isopropylalkohol aufgearbeitet. GC/MS‐Messungen der resultierenden
überstehenden Lösung liefern ein recht komplexes Chromatogramm und lediglich
Spuren des aminierten Cyclohexans Cy‐6‐NH2 134‐(Cy6). Neben einer Reihe von
unbekannten Stoffen können nur B(OiPr)3 sowie geringen Anteil an Bicyclohexyl
NB
NN3
Me
Me
SB
SN3
NB
NN3
Tos
Tos
191 192 193

Photolyse stickstoff‐ und schwefelsubstituierter Borazide
112
(Cy6‐Cy6) eindeutig identifiziert werden. Daneben bildet sich ‐ ebenfalls erst nach
der Alkoholyse ‐ eine neue Verbindung X mit einem Molekülionenpeak bei M+• =
103. Das dem Signal zugehörige Chromatogramm weist ein starkes Tailing auf,
wie es zuvor u. a. bei freien Aminen beobachtet wurde. Unter Berücksichtigung
der bereits vorgenommenen mechanistischen Untersuchungen sind zwei
Konstitutionen für Verbindung X denkbar (vgl. Schema 54). Reagiert das erzeugte
Borylnitren 195 in einer intramolekularen C‐H‐Insertionsreaktion unter
Ausbildung von Boracyclus 196, sollte nach BN‐Bindungsspaltung das Amin 197
freigesetzt werden. Lagert das Borylnitren bzw. das Azid selbst hingegen zum
Borimid 198 um, müsste sich nach Alkoholyse das Hydrazinderivat 199 bilden. Ob
eines und wenn ja, welches der beiden Isomeren gebildet wird, kann nicht
eindeutig festgestellt werden. Die Entstehung des Iminoborans 198 ist relativ
unwahrscheinlich. Zum einem geben die von Paetzold et al. durchgeführten
thermolytischen Gasphasenexperimente keine Hinweise auf das Iminoboran
dieses cyclischen Nitrens,[108] zum anderen sollte das cyclische Iminoboran 198 in
Lösung Folgereaktionen unter Bildung von Oligomeren eingehen (z. B. Bildung
des Trimers 200). Da die denkbaren Folgeprodukte der Iminoboranchemie
stabiler gegenüber Sauerstoff‐Nucleophilen sein müssten, ist eine
Fragmentierung durch eingesetztes Isopropanol bei RT nicht zu erwarten.
Darüber hinaus zeigen eigens vorgenommene computerchemische
Untersuchungen auf unterschiedlichen Niveaus (B3LYP/6‐311+G**, MP2/6‐
311+G**), dass das Iminoboran 198 keinem stationären Punkt auf der PES
entspricht. Für eine intramolekulare C‐H‐Funktionalisierung hingegen sprechen
einigen Argumente. Bei genauerer Betrachtung des MS von X kann das für
primäre Amine typische, aus einer Oniumspaltung resultierende intensive
Fragment H2C=NH2+ bei MZ 30 ausgemacht werden.[269] Des Weiteren ist das
Auftreten des Cy‐6‐NH2 134‐(Cy6) sowie des Radikalrekombinationsprodukts

Photolyse stickstoff‐ und schwefelsubstituierter Borazide
113
Bicyclohexyl (Cy6‐Cy6) ein indirekter Beweis für die Beteiligung einer Borylnitren‐
Zwischenstufe. Zudem zeigen Rechnungen, dass der viergliedrige Heterocyclus
196 eine Minimumstruktur darstellt.
Abbildung 36: Berechnete (B3LYP/6‐311+G**) Minimumgeometrie für Boracyclus 196.
Warum sich jedoch das Produkt einer intermolekularen Insertion nur in Spuren
ausgebildet hat, bleibt unklar. Möglicherweise ist dies auf die schlechte
Durchmischung zurückzuführen. Denkbar wäre auch, dass das Borylnitren 195 in
einer intermolekularen C‐H‐Insertion die N‐Methylgruppe des Liganden eines
weiteren Moleküls aminiert, was nach Abbau ebenfalls zu 197 führen würde. Um
das Löslichkeitsproblem zu umgehen, wurden Versuche unternommen, das
permethylierte Borazid 201 herzustellen (vgl. Abb. 38). Da diese erfolglos blieben,
wurde das Rohphotoprodukt auch mit AcCl umgesetzt, um ggf. zusätzliche
Hinweise auf den Reaktionsverlauf zu erhalten. Doch das entstandene
Chromatogramm lässt keine neuen mechanistischen Aussagen zu. Problematisch
bei der Umsetzung erweist sich insbesondere der Umstand, dass der N‐Ligand
ebenfalls acetyliert wird. Um die denkbare Annahme einer intramolekularen
Insertionsreaktion zu überprüfen, könnten auch Versuche in der Gasphase
sinnvoll sein. Doch zum Zeitpunkt der eben vorgestellten experimentellen
Arbeiten war die entsprechende Gasphasenphotolyseapparatur (vgl. 4.2.2.6.3)
noch nicht vorhanden.
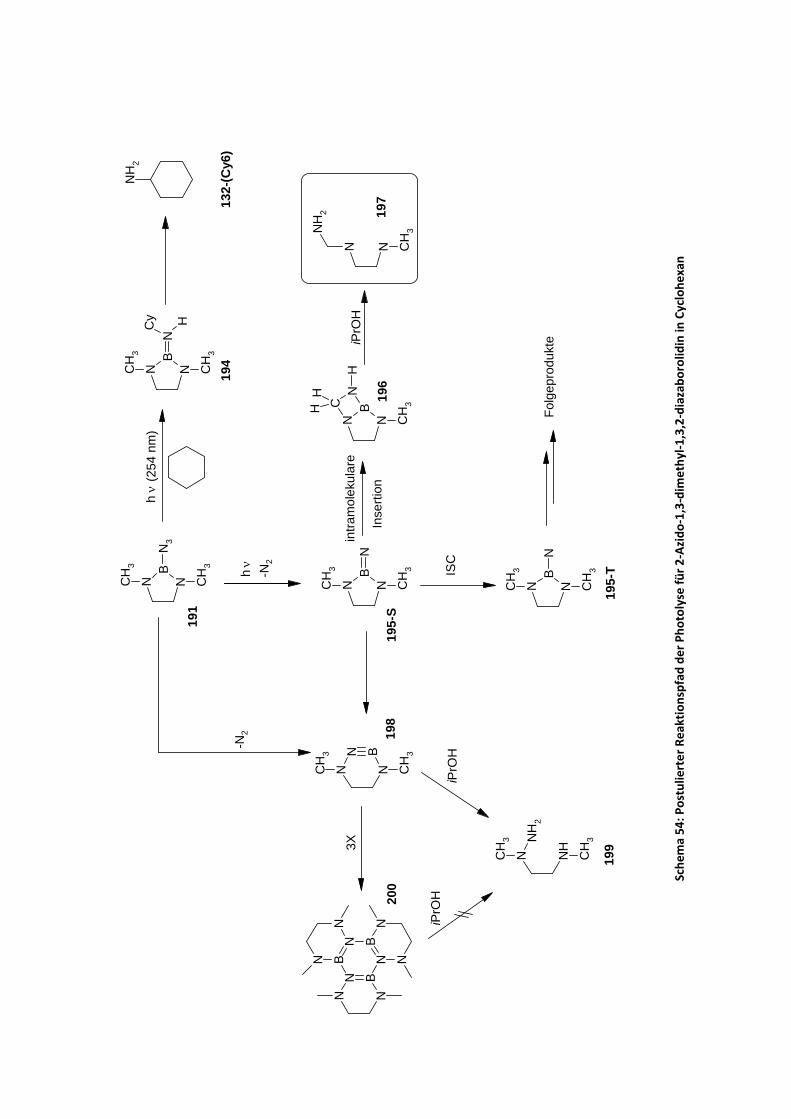
NB
NCH
3
CH
3
NN C
H3
NB
NC
H
HH
N CH
3
N
NH
2
NBN
NCH
3
CH
3
BNN N
BN N
N
BN
NN
NH
NH
2
NCH
3
CH
3
NB
NCH
3
CH
3
N3
NB
NCH
3
CH
3
N
NB
NCH
3
CH
3
NC
y H
NH
2
ISC
h
(254
nm
)
intra
mol
ekul
are
3X
h -N
2
-N2
196
198
195-
T
197
Inse
rtion
Folg
epro
dukt
e
195-
S
iPrO
H
iPrO
HiP
rOH
199
200
194
132-
(Cy6
)
191
Schem
a 54: Postulierter Reaktionspfad der Photolyse für 2‐Azido‐1,3‐dim
ethyl‐1,3,2‐diazaborolid
in in
Cyclohexan

Photolyse stickstoff‐ und schwefelsubstituierter Borazide
115
4.2.2.5.2 Photolyse von 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin in Benzol
Das dem neuen Azidoborolidin 192 zugrundeliegende Borchlorid 202 wurde
ausgehend vom ditosylierten Ethylendiamin 203[270‐272] durch Direktsynthese in
Anlehnung an Arbeiten von Corey et al. erhalten.[190‐192] Die Einführung der
Tosylgruppen beruhte auf dem Wunsch nach effizienter UV‐Absorption sowie
ausbleibender intramolekularer C‐H‐Insertion.
Schema 55: Syntheseroute für die Herstellung von 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin 1.
Durch langsames Verdunsten einer Lösung des Azids 192 in DCM konnten
Einkristalle erhalten werden, deren Qualität ausreichend hoch für eine
Röntgenstrukturbestimmung war. Im Kristallverband ist das
Diazaborolidingrundgerüst nahezu planar. Die sterisch anspruchsvollen
Tosylliganden sind verdrillt gegeneinander orientiert. Die Azido‐Gruppe ist nur
leicht gewinkelt (N(α)‐N(β)‐N(γ), 168.3°), wie es für kovalente Azide
kennzeichnend ist.[74, 178] Der N(α)‐N(β)‐Bindungsabstand (1.096 Å) ist etwas
kürzer als die N(β)‐N(γ)‐Bindungslänge (1.137 Å). Der B‐N(α)‐N(β) Winkel beträgt
144.1° und ist damit größer als derjenige im CatBN3 (119.7°)[172]
oder im 9‐Azido‐
9‐borafluoren*tert‐Pyridin (FluorenBN3tertPy).[173] Die verkürzten BN‐Bindungs‐
längen (B‐N(1), B‐N(2) = 1.426 Å, B‐N(α) = 1.417 Å) deuten auf π‐Effekte unter
Beteiligung des vakanten Bor Pz‐Orbitals hin.
NB
NN3
Tos
Tos
NH2
NH2
NH
NH
Tos
Tos
NB
NCl
Tos
Tos
192203
+TosClBase
+BCl3-HCl
+TMSN3
-TMSCl
202

Photolyse stickstoff‐ und schwefelsubstituierter Borazide
116
NB
NN3
Tos
Tos
NB
NN3
CH3
CH3204201
N1B
N2
Nβ
Nγ
Nα
Abbildung 37: Kristallstruktur von Azidoborolidin 191.
Die Photolyse einer Suspension des Azidoborans 191 in Cy‐6‐H bzw. Benzol liefert
keine Anhaltspunkte auf Cy‐6‐NH2 134(Cy6) bzw. Anilin Ph‐NH2, die Produkte
einer intermolekularen Insertionsreaktion. Nach alkoholytischer Aufarbeitung der
fast schwarzen Reaktionsmischung lassen sich nur der Bortrialkylester sowie der
tosylierte Diaminoethylenligand 203 als Fragmente gaschromatographisch
identifizieren.
Wegen der schlechten Löslichkeit der beiden Azidoborolidine 191 und 192 wurde
eine Bachelorarbeit mit dem Ziel ausgegeben, die vermutlich besser löslichen
methylierten Azidoborolidine 201 und 204 zu synthetisieren. Doch trotz
erheblicher Bemühungen gelang die Synthese der gewünschten Produkte nicht in
ausreichender Menge und Reinheit.
Abbildung 38: Gewünschte methylierte Azide mit Borolidingrundgerüst.

Photolyse stickstoff‐ und schwefelsubstituierter Borazide
117
4.2.2.5.3 Photolyse von 2‐Azido‐1,3,2‐dithiaborolan in Cyclohexan
Das aus Ethan‐1,2‐dithiol über Direktsynthese zugängliche flüssige 2‐Cloro‐1,3,2‐
dithiaborolan 117 kann leicht in das neue Azidoboran 193 überführt werden. Da
sich das feste Azidoboran ebenfalls als unlöslich in Alkanen zeigte, bestand
wiederum nur die Möglichkeit, eine Suspension unter Rühren in Cy‐6‐H zu
belichten. Nach beendeter Photolyse und alkoholytischer Umsetzung ergeben
sich wiederum keine Hinweise auf Cy‐6‐NH2 134‐(Cy6). Lediglich B(OiPr)3 und der
freie Ligand HSCH2CH2SH sind mittels GC/MS detektierbar. Da das Ethan‐1,2‐
dithiol bereits vor der Aufarbeitung in der Reaktionslösung vorhanden ist, könnte
dies ein Hinweis für die Instabilität eines primären Photoprodukts sein, was in
Anbetracht der schwachen S‐B‐Bindung nicht überraschend wäre. Wegen der
äußerst starken Geruchsbelastung, die vom Ethan‐1,2‐dithiol ausgeht, wurden
keine weiteren Untersuchungen zu diesem oder anderen schwefelhaltigen
Systemen vorgenommen.

Versuche zur präparativen Aminierung von Methan
118
4.2.2.6 Versuche zur präparativen Aminierung von Methan
Hinsichtlich der aus den Matrixexperimenten und in Lösung gewonnenen
Befunde schien uns auch eine metallfreie Methanfunktionalisierung unter
präparativen konventionellen Laborbedingungen möglich. Ziel dieses Teilprojekts
war deshalb die Aktivierung und Funktionalisierung von Methan und die
Isolierung und Charakterisierung der Reaktionsprodukte in Substanz. Dazu
wurden Experimente durchgeführt, bei denen PinBN3 9 mit Methan zur Reaktion
gebracht werden sollte.
Schema 56: Zielreaktion zwischen PinBN3 und Methan.
4.2.2.6.1 Versuche zur Reaktion von PinBN3 in flüssigem Methan
Zunächst wurden Versuche unternommen, das Insertionsprodukt PinBNHMe
132‐(Me) zu erhalten, indem eine Lösung von PinBN3 9 in flüssigem Methan
belichtet werden sollte. Dazu wurde der Versuch vorgenommen, gasförmiges
Methan mithilfe von flüssigem Stickstoff in eine mit PinBN3 9 gefüllte
Quarzglasvorlage einzukondensieren. Doch der notwendige Einkondensierungs‐
vorgang erwies sich als äußerst problematisch und nicht praktikabel. Die
Herausforderung lag dabei insbesondere im engen Temperaturbereich, in dem
Methan in der flüssigen Phase vorliegt (Sdp. ‐162 °C / Schmelzpkt. ‐182 °C). Die
Probleme, die sich bei diesem Vorgang ergaben, sind in Abb. 39 schematisch
aufgezeigt. So konnte zum einem kein Gemenge erhalten werden, in der die
Methan/Azid‐Mischung lediglich in der flüssigen Phase vorlag, zum anderen
OB
ON3
OB
ON
H
CH3CH3 H
132-(Me)9
h (= 254 nm)

Versuche zur präparativen Aminierung von Methan
119
vereiste die für die Belichtung notwenige Quarzoberfläche innerhalb kürzester
Zeit, wobei auch das Spülen mit Trockenluft nur geringen Erfolg brachte. Da es
aufgrund der Vereisung zu einer starken Absenkung der Lichtdurchlässigkeit kam,
konnte auch keine kontinuierliche Belichtung mehr vorgenommen werden.
Deshalb wurden im Anschluss Experimente einer nicht‐kontinuierlichen
Photolyse durchgeführt. Hierzu wurde zunächst das Methan/Azid‐Gemisch mit
flüssigem Stickstoff ausgefroren, das Quarzrohr von Kondenswasser befreit und
dann für kurze Zeit in die Photolysezone der UV‐Quelle positioniert. Wegen der
Umgebungstemperatur und der Wärmeabstrahlung der UV‐Lampe wurde jedoch
innerhalb kürzester Zeit der Siedepunkt des brennbaren Methans überschritten,
was zu einem starken Druckaufbau in der Apparatur führte, so dass das
Experiment aus Sicherheitsgründen abgebrochen werden musste. Da die
Photochemie des PinBN3 9 wegen seiner geringen UV‐Absorption äußert
langwierig und dadurch eine Belichtungsdauer von mehreren Stunden
erforderlich ist, wurden keine zusätzlichen Bemühungen unternommen, das
bestehende Versuchssystem weiter zu optimieren.
Abbildung 39: Photolyseapparatur für die (beabsichtigte) Reaktion zwischen PinBN3und flüssigem Methan.

Versuche zur präparativen Aminierung von Methan
120
4.2.2.6.2 Reaktion von gasförmigen Methan in Lösung
Da sich eine Photoreaktion mit flüssigem Methan technisch nicht einfach
realisieren ließ, wurde ein alternativer apparativer Aufbau entwickelt mit der
Absicht, gasförmiges Methan mit flüssigem PinBN3 9 zur Reaktion zu bringen.
Hierzu wurde das in Abb. 40 aufgezeigte Photolysesystem errichtet. Ziel der
geplanten Experimente war dabei die Herstellung einer verdünnten PinBN3‐
Lösung, durch die während der Belichtung kontinuierlich gasförmiges Methan
geleitet werden sollte, welches dann mit dem photochemisch generierten
Pinakolborylnitren 10 zum Aminoboran 132‐(Me) abreagieren könnte.
Abbildung 40: Versuche zur Photolyse von Methan in einer PinBN3‐Lösung.
Die Herausforderung liegt dabei insbesondere in der Wahl eines geeigneten
Lösungsmittels LM, welches mehreren Ansprüchen genügen muss:
• Das LM muss notwendigerweise selbst photostabil und möglichst
transparent im UV‐Abstrahlungsbereich der Lampe sein.
• Es muss sich inert gegenüber dem photochemisch erzeugten Borylnitren
PinBN 10 verhalten.

Versuche zur präparativen Aminierung von Methan
121
• Die Flüchtigkeit des LM sollte gering sein, damit es nicht durch das
strömende Methangas aus der Apparatur ausgetragen wird (Verdunstung).
• Das PinBN3 sollte sich gut im LM lösen, damit eine homogene Mischung
entsteht.
• Das LM muss frei von Wasser sein, sich ggf. trocknen lassen.
• Das LM sollte kommerziell erhältlich und möglichst preiswert sein.
Es wurde eine Reihe von LM getestet, doch keines erfüllte die oben angeführten
erforderlichen Eigenschaften gleichzeitig, so dass sich die gewünschte
Aminierungsreaktion nicht beobachten ließ. Da aus den Matrixexperimenten
bekannt ist, dass sich CF4 stabil gegenüber CatBN 6 verhält, wurden zunächst
perfluorierte Alkane (z. B. Perfluorocyclohexan C6F12) als LM eingesetzt.
Bedauerlicherweise hat sich das Edukt PinBN3 9 durchweg als unlöslich erwiesen,
was zur Ausbildung flüssiger Zwei‐Phasensystem führte. Trotz einer gewissen
Durchmischung beider Komponenten durch den Methangasstrom, können nach
beendeter Photolyse und Aufarbeitung keine intermolekularen C‐H‐
Insertionsprodukte identifiziert werden. Auch die Verwendung von
Hexafluorbenzol (C6F6) hat sich aus Löslichkeitsgründen und wegen seiner UV‐
Absorptionseigenschaften („Verschlucken“ des UV‐Lichts) als ungeeignet
herausgestellt. Die weiteren eingesetzten LM (Freon, CCl4) sind ebenfalls unter
den vorliegenden Gegebenheiten bedauerlicherweise durchweg nicht
photostabil.

Versuche zur präparativen Aminierung von Methan
122
4.2.2.6.3 Gasphasenphotoreaktion von Methan und PinBN3
Für die präparative Umwandlung von Methan in PinBNHMe 132‐(Me) bzw.
Methylamin MeNH2 134‐(Me) wurde eigens der in Abb. 41 aufgezeigte spezielle
Photoreaktor entwickelt, der die Gasphasenreaktion zwischen PinBN3 9 und
Methan erlaubt. Hierbei strömt fortlaufend Methangas durch flüssiges
Pinakolborazid 9, wobei kontinuierlich geringe Mengen Azid in die Gasphase
überführt werden. Das dabei gebildete Methan/Azid‐Gasgemisch wird durch ein
Quarzrohr geleitet und mittels einer Quecksliberniederdrucklampe (λ = 254 nm)
fortlaufend photolysiert. Dabei scheidet sich im Zuge der Belichtung innerhalb
und hinter der Belichtungszone aus den zuvor gasförmigen Edukten ein harziger,
leicht gelblicher Feststoff ab. Dieser kann größtenteils durch geschicktes
Auswaschen z. B. mit DCM unter Argongegenstrom in die nachgeschaltete, zuvor
gekühlte Vorlage gespült werden. Nach dem Entfernen des LM im Vakuum kann
das erhaltene Reaktionsprodukt direkt mittels spektroskopischer Methoden und
indirekt mithilfe von Abbauexperimenten charakterisiert werden.
Abbildung 41: Schematische Darstellung des entwickelten Gasphasenphotoreaktors.

Versuche zur präparativen Aminierung von Methan
123
4.2.2.6.3.1 Charakterisierung der Gasphasephotoprodukte
In der Tat ergeben vorgenommene NMR‐Messungen des Rohprodukts Hinweise
auf die Ausbildung des gewünschten Aminoborans PinBNHMe 132‐(Me).[273, 274]
So tritt im 11B‐Spektrum u. a. eine aminoborantypische Resonanz bei 25 ppm
auf.[2, 177] 1H‐ und 13C‐Untersuchungen lassen die zur Pinakoleinheit zugehörigen
Signale erkennen (1H = 1.22 ppm / 13C = 28.8 ppm, 80.1 ppm). Darüber hinaus
erscheint ein Signal (1H = 2.52 ppm / 13C = 23.3 ppm), welches der NMe‐Gruppe
zugeordnet werden kann. Trotzdem ist das aufgenommene Spektrum von
Verunreinigungen geprägt, welche auf weitere (Photo)produkte zurückgeführt
werden können. Da sich das gewünschte Reaktionsprodukt wegen seiner
extremen Luft‐ und Wasserempfindlichkeit nicht durch Sublimation oder
Kristallisation aufreinigen ließ, wurden wiederum Abbau‐ und
Derivatisierungsexperimente vorgenommen. Einfacher hydrolytischer Abbau
führt direkt zum freien Methylamin MeNH2 134‐(Me), welches sowohl
gaschromatographisch als auch anhand seines charakteristischen Geruchs
identifiziert werden kann. Über Variationen des pH‐Wertes und der
Wassertemperatur besteht die Möglichkeit zur Regulation der in Schema 57
aufgeführten Gleichgewichte. Wird eine alkalische Lösung (NaOH) des
hydrolysierten Photoprodukts in Ether mit Tosylchlorid umgesetzt,[270‐272] kann
das erwartete TosNMe 205 sowohl gaschromatographisch nachgewiesen als auch
in Substanz isoliert werden. Bei der Umsetzung muss jedoch beachtet werden,
dass die Zugabe bei niedriger Temperatur (0 °C) erfolgt, damit nur ein möglichst
kleiner Anteil an MeNH2 134‐(Me) in die Gasphase übergeht, ohne zu reagieren.
Die Acetylierung wird direkt ausgehend vom Photorohprodukt vorgenommen,
indem es in THF gelöst, mit AcCl versetzt wird und anschließend ‐ wie bereits für
das PinBNHCy 134‐(Cy) beschrieben ‐ mit Hydroxidionen versetzt wird. Das N‐
Methylacetamid 141‐(Me) kann dann mit GC/MS‐ und GC‐Analysen qualitativ und

Versuche zur präparativen Aminierung von Methan
124
OB
ON3
OB
ON
Me
HNH
Me
O
R NH
MeTos
-N2
HCl (aq.)MeNH3
+ Cl-
CH4
MeNH2 (aq.) MeNH2 (g)
H2O
MeNH3+ OH-
H2O
NaOH (aq.)
1) RCOCl, DMAP (kat.)
2) NaOH (s)
1) H2O, HCl
2) TosCl, NaOH
141-(Me) 132-(Me)
9
206
134-(Me)
quantitativ bestimmt werden. Bei optimierten Reaktionsbedingungen wird ‐
bezogen auf eingesetztes PinBN3 9 ‐ eine Ausbeute von 6‐8 % ermittelt.
Schema 57: Abbauwege für PinBNHMe.
4.2.2.6.3.2 Weitere Produkte und mechanistische Interpretationen
Um ausschließen zu können, dass das detektierte Methylamin 134‐(Me) nicht aus
einer intermolekularen Umlagerung einer Pinakol‐Me Gruppe auf das
Nitrenzentrum stammt, wurden Kontrollexperimente mit Argon als Trägergas
durchgeführt. Da hier kein Methylamin nachgewiesen werden konnte, ist der Pin‐
Ligand als mögliche Methyl‐Quelle eindeutig auszuschließen.

Versuche zur präparativen Aminierung von Methan
125
OB
ON
Me
OB
OH
NMe
HOB
OMe
NH-Abstraktion
Schema 58: Ausgeschlossene intramolekulare Me‐Umlagerung.
Neben den gewünschten Methylaminderivaten wurden auch Verbindungen auf
Basis von Ammoniak und Hydrazin identifiziert. So entstehen bei der Acetylierung
auch Acetamid AcNH2 sowie die mono‐ und/oder diacetylierten‐Hydrazine
AcNHNH2 bzw. AcNHNHAc. Bei der Tosylierung lassen sich analog dazu TosNH2
sowie TosNHNH2 und TosNHNHTos mithilfe von GC/MS‐Messungen eindeutig
nachweisen (Vergleich mit authentischen Proben). Trotz mangelnder Eignung der
Derivate für eine exakte Ausbeutebestimmung, lassen sich tendenzielle und
halbquantitative Aussagen treffen. Die Anteile beider Aminospezies zusammen
liegen meist unter < 5 %, wobei die Menge an Hydrazin dabei in der Regel kleiner
ist als der Anteil an Ammoniak ist. Mit den aufgezeigten Reaktionspfaden ist die
Bildung der unterschiedlichen Aminoverbindungen vereinbar: Das gewünschte
PinBNHMe 132‐(Me) bzw. MeNH2 134‐(Me) entsteht aus der postulierten
konzertierten Insertion des 10‐S in die C‐H‐Bindung von Methan. In Konkurrenz
dazu steht die Bildung der übrigen Amine, welche mit Alternativprozessen unter
Beteiligung des Triplett‐Borylnitrens 10‐T erklärt werden können. Unter
Berücksichtigung der größeren, mittleren freien Weglänge in der Gasphase und
der unbekannten Lebensdauer des 10‐S kann angenommen werden, dass ein Teil
des photochemisch intermediär erzeugten angeregten Singulett‐Borylnitrens 10‐
S in den Triplett‐Grundzustand relaxiert (ISC), bevor die Möglichkeit einer
Kollision mit einem Methanmolekül besteht. Deshalb kommt es im Anschluss zum
ISC zu den typischen H‐Abstraktions‐ und Dimerisierungsprozessen des Triplett‐

Versuche zur präparativen Aminierung von Methan
126
Nitrens. So könnte die Abstraktion eines H‐Atoms des Methans, der
Quarzoberfläche des Photolyserohrs ((Si‐OH‐Funktion) oder des bereits an der
Rohrinnenwand haftenden Photolyseprodukts (C‐H‐Bindungen) zum Radikal
PinBNH• 206 führen, welches dann, entweder unter Bildung des symmetrischen
Aminoborans PinBNH2 207 ein zweites H‐Atom abstrahiert, oder mit einem
weiteren Molekül 206 zum Dimer PinBNHNHBPin 208 rekombiniert. Auch eine
direkte Dimerisierung des freien PinBN 10‐T unter Bildung des Azoborans
PinBNNBPin 209 wäre denkbar. Hydrolyse von 208 bzw. 209 sollte dann Hydrazin
210 freisetzen, Aminoboran 207 müsste nach Spaltung Ammoniak 211 liefern.
Dass die Produkte Hydrazin und Ammoniak ebenfalls bei der Verwendung von
Argon als Trägergas gebildet werden, steht mit dieser mechanistischen
Vorstellung in Einklang. Bei diesen Untersuchungen lässt sich zusätzlich
feststellen, dass die Konzentration an Hydrazin bzw. seinen Derivaten und damit
der Dimerisierungsgrad von dem Verhältnis zwischen Trägergas und PinBN3 9
abhängt. Bei hoher Verdünnung, d. h. bei niedriger Gasströmungs‐
geschwindigkeit wird weniger Hydrazin gebildet als bei hohem Argondurchfluss.
Verständlich wird diese Beobachtung unter Berücksichtigung der sich ändernden
Stoßhäufigkeit zweier Pinakolborylspezies.
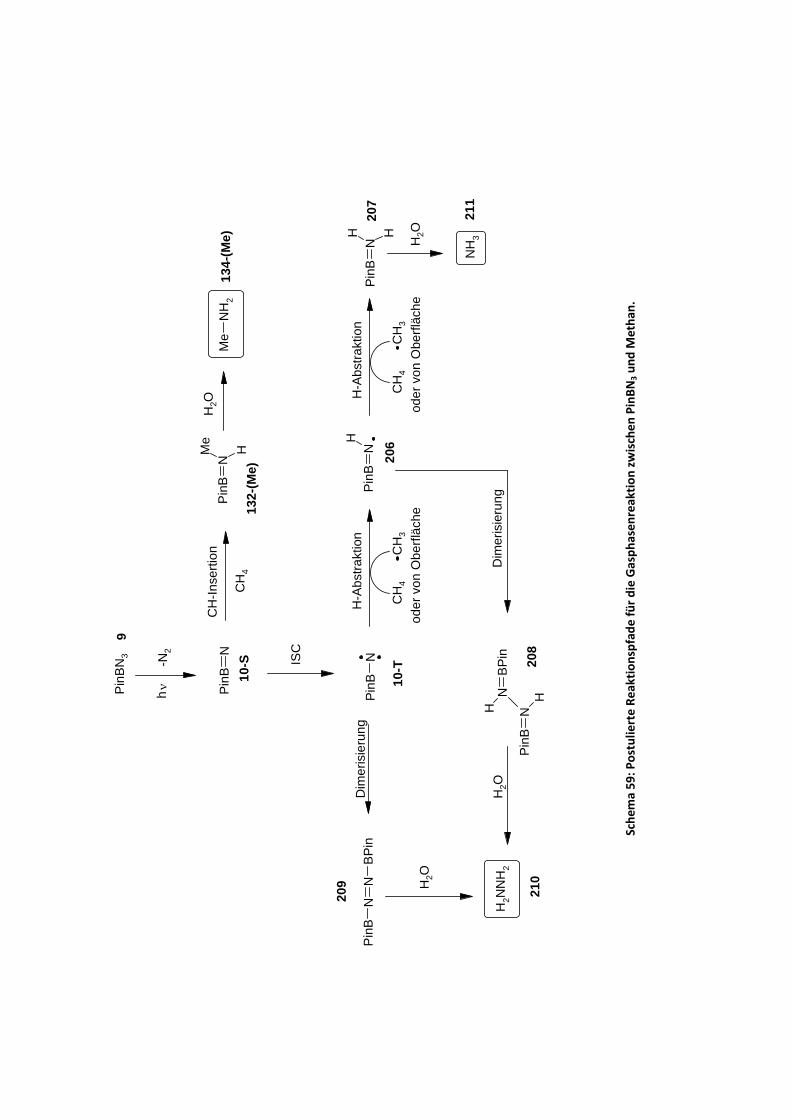
H2N
NH
2
NH
3
Pin
BN
Pin
BN
Me
H
Pin
BN
Pin
BN
NB
Pin
Pin
BN
HPi
nBN
H H
Pin
BN
H
BPi
nN
H
Me
NH
2
h
CH
4
-N2
ISC
CH
4C
H3
CH
-Inse
rtion
H-A
bstra
ktio
n
H2O
H2O
Pin
BN3
oder
von
Obe
rfläc
heH
2OC
H4
CH
3
H-A
bstra
ktio
n
oder
von
Obe
rfläc
he
H2O
208
132
-(M
e)
210
211
134
-(M
e)
10-
S
209
10-
T2
06
207
Dim
eris
ieru
ng
Dim
eris
ieru
ng
9
Schem
a 59: Postulierte Reaktionspfade für die Gasphasenreaktion zwischen PinBN3 und M
ethan
.

Versuche zur präparativen Aminierung von Methan
128
Obschon sich die Bildung der aufgeführten Aminoverbindungen MeNH2 134‐
(Me), NH3 211 und H2NNH2 210 bzw. ihre Derivate erklären lassen, muss
festgehalten werden, dass diese ‐ bezogen auf eingesetztes Pinakolborazid 9 ‐
zusammen insgesamt nur max. 15 % der theoretischen Ausbeute ergeben. Der
Verbleib des übrigen Azids könnte durch folgende Überlegungen nachvollziehbar
werden: So ist z. B. unklar, wie groß der Anteil des eingesetzten PinBN3 9 ist, der
durch die Photolyseeinheit strömt und ‐ ohne eine Photoreaktion einzugehen ‐
direkt in der gekühlten Vorlage ausgefroren wird. Weil sich jedoch in der Vorlage
auch gewisse Mengen der Photoprodukte sammeln, besteht keine (einfache)
Möglichkeit die Menge an nicht umgesetztem Azid zu ermitteln. Des Weiteren
kann empirisch festgestellt werden, dass sich der während der Photolyse
abgeschiedene Feststoff in Abhängigkeit vom Ort innerhalb des Reaktors optisch
unterscheidet. So ist der Anteil des Photoprodukts, welcher sich im gesamten
Belichtungszeitraum in der Photozone befindet, dunkler und harziger, als der Teil,
der erst hinter der UV‐Lampe bzw. erst in der Vorlage ausfällt. Dies könnte
wiederum in einer möglichen Photoinstabilität der primären Reaktionsprodukte
begründet liegen (Polymerisierung). Darüber hinaus kann eine partielle Hydrolyse
der aminierten Reaktionsprodukte meist nicht vollständig ausgeschlossen
werden.

Versuche zur Synthese von Borylaziridinen
129
4.2.3 Versuche zur Synthese von Borylaziridinen Borylaziridine des Typs 213 bilden interessante Syntheseziele, da sie
möglicherweise als nützliche Hilfsreagenzien in der organischen Synthesechemie
Verwendung finden könnten. Unter Berücksichtigung der an Aminoboranen
diskutierten Reaktivität sollten sich auch diese cyclischen Aminoborane 213 in
substituierte Aziridine 214 überführen lassen, die sich dann bei Bedarf durch
nucleophile Ringspaltung weiter zu Aminoderivaten 215 abbauen lassen.
Schema 60: Borylaziridine und denkbare Folgeprodukte.
Überraschenderweise ist unseres Wissens nach nur ein Beispiel eines
Borylaziridins in der Literatur beschrieben, wobei die Synthese über Kupplung
eines Monochloroborans mit 1‐Aziridyllithium unter Aufbau der BN‐Bindung
erfolgte.[275] Retrosynthetische Überlegungen lassen auch hoffen, dass die
Borylaziridine 213 durch die Addition eines Borylnitrens R2BN 2 an ein Alken
erhältlich seien könnten, ein Reaktionstyp, der für die verwandten Nitrene und
Carbene wohl bekannt ist und synthetischen Nutzen erfährt.[69, 70]
Schema 61: Hypothetische Addition eines Borylnitrens an eine C=C‐Doppelbindung.
N
BRR
N
EN
Nu
E
H
E Nu
213 214 215
N
BRR
NBR
R
213

Versuche zur Synthese von Borylaziridinen
130
4.2.3.1 Alkene als Substrate: Photolyse von PinBN3 in Tetramethylethen (TME) und Cyclohexen
Da sich PinBN3 9 als leistungsfähiges Substrat für die Erzeugung eines Borylnitrens
erwiesen hat, wurde dieses in Gegenwart unterschiedlicher Alkene belichtet.
Dabei sollte insbesondere die Fragestellung Berücksichtigung finden, ob die
gewünschte Borylnitrenaddition an die C=C‐Doppelbindung stattfindet oder doch
wiederum eine C‐H‐Insertionsreaktion zu beobachten ist.
Die Photolyse einer Lösung von PinBN3 9 in TME ergibt nach Alkoholyse eine
inhomogene Suspension, wobei die GC/MS‐Analyse der überstehenden Lösung
ein äußerst bandenreiches Chromatogramm liefert. Bedauerlicherweise lässt sich
das gewünschte 2,2,3,3‐Tetramethylaziridin 216, das Alkoholyseprodukt des
Borylaziridins, nur in Spuren nachweisen. Außerdem entstehen geringe Mengen
des Epoxids 217, das wahrscheinlich aufgrund von Spuren von gelöstem
Sauerstoff gebildet wird. Durch die Entgasung des TME vor dem Photolyseschritt
lässt sich die Bildung von 217 weitgehend unterdrücken. Zudem bildet sich
vermutlich das Produkt einer intermolekularen Insertionsreaktion aus. So wurde
nach Alkoholyse ein Stoff freigesetzt, dessen MS kompatibel mit dem erwarteten
Spektrum des Allylamins 218 ist. Da allerdings keine authentische Probe von 218
vorhanden war, kann der Nachweis nicht als gesichert angenommen werden.
Zudem werden erhebliche Mengen an Octamethylcyclobutan 219 erzeugt.[276]
Dieses Produkt einer [2+2]‐Cycloaddition des Substrates TME konnte durch
Kontrollversuche (Belichtung von reinem TME) bestätigt werden.

Versuche zur Synthese von Borylaziridinen
131
Abbildung 42: Produkte aus der Reaktion von PinBN3 mit TME.
Des Weiteren wurde die Photolyse von PinBN3 9 in Cyclohexen durchgeführt.
Jedoch erwies sich auch dieses Substrat unter den vorherrschenden Bedingungen
als nicht‐photostabil. Zwar lassen sich nach Aufarbeitung mit Isopropylalkohol
geringe Mengen unterschiedlicher Amine (220, 221) mittels GC/MS qualitativ
nachweisen, aber auch große Anteile an Verbindungen (222, 223), die sich aus
Dimerisierungsreaktionen von Cyclohexen ergeben.[277]
Abbildung 43: Produkte aus der Reaktion von PinBN3 mit Cyclohexen.
Da kein (geeignetes) Borazid zur Verfügung stand, welches die Bestrahlung mit
längerwelligem Licht erlaubt, bei der die betreffenden Alkene transparent und
photostabil sind, wurden die Versuche vorerst eingestellt (vgl. Ausblick).
NH2 N
HO
218 216 217 219
NH2
N H
220 221 222 223

1,3‐dipolare Cycloaddition und „Click Chemie“
132
4.2.4 Versuche zur Synthese von Boryltria‐ und tetrazolen via „Click‐Chemie“
Ein derweil hohe Aktualität aufweisendes Forschungsfeld der Chemie beschäftigt
sich mit der Cu(I)‐katalysierten 1,3‐dipolaren Cycloaddition von Aziden 224 an
Alkine 225‐(CH) und Nitrile 225‐(N) unter Bildung von Tri‐ bzw. Tetrazolen
226.[278‐290] Dieser Reaktionstyp, dessen kupferfreie Variante bereits in Arbeiten
von Huisgen in den 1960iger Jahren vorgestellt wurde,[291, 292] ist zum
Paradebeispiel einer Clickreaktion avanciert.[293]
Schema 62: Allgemeines Syntheseschema für die 1,3‐dipolare Cycloaddition an Alkine bzw. Nitrile.
Während die Verwendung organischer Azide bereits eingängig untersucht wurde,
sind in der Literatur nur wenige Beispiele beschrieben, bei denen kovalente Azide
übriger Hauptgruppenelemente als Stickstoffquelle für den Aufbau dieser
Heterocyclen dienen. Einige Arbeiten behandeln die Addition von Siliziumaziden
an ungesättigte Substrate.[294‐296] Kürzlich erschien zudem ein vielversprechender
Artikel, der die Anlagerung von Aluminiumaziden R2AlN3 an Nitrile unter Bildung
von Tetrazolen beschreibt.[297] Wegen der Verwandtschaft zwischen Aluminium,
Silizium und Bor könnte man eine analoge Reaktion auch von den in dieser Arbeit
vorgestellten Boraziden erwarten. Daher wurden die Borazide CatBN3 7 sowie
PinBN3 9 mit unterschiedlichen Alkinen und Nitrilen bei variablen
Reaktionsbedingungen umgesetzt. Aber der Nachweis der erhofften
Additionsprodukte des Typs 227 blieb sowohl in Gegenwart katalytischer Mengen
Cu(I)‐Salze als auch in Abwesenheit von Kupfer aus.
XR´R N3
X
NN
N
R´
R
X
NN
N R
R´
Cu (I)+
X = CH, N
224 225226
+
R = Organyl

1,3‐dipolare Cycloaddition und „Click Chemie“
133
Schema 63: Versuche zur Synthese von Boryltria‐ und tetrazolen.
Die nicht beobachtbare Borazidaddition lässt unterschiedliche Interpretationen
zu, wobei die kupferkatalysierte Reaktionsführung vermutlich von der Cu‐freien
Umsetzung zu unterscheiden ist.
Möglicherweise sind die eingesetzten Borazide nicht stabil gegenüber den
verwendeten Cu(I)‐Salzen. So zeigen Experimente in unserer Arbeitsgruppe, dass
sich CatBN3 bei RT instabil gegenüber Dieisennonacarbonyl Fe2(CO)9 verhält.[298]
Zudem wurde in einer kürzlich erschienen Arbeit von stabilen Cu(I)‐Phoshin‐
Boranen berichtet, bei denen CuI als Kupferquelle diente.[299] Im Falle der
Verwendung des NHC‐CuBr‐Komplexes[300] wäre auch eine Übertragung der NHC‐
Gruppe auf das Lewis‐acide Borzentrum[301] und damit eine Inaktivierung des
Katalysators durchaus denkbar. Möglicherweise reagiert das Borazid auch als
Elektrophil in einer der Hydroborierung analogen Reaktion, die für CatBH und
Alkine wohl bekannt ist.[302, 303] Damit würde sich das Azidoboran auch in
Konkurrenz zum Cu(I)‐Kat verhalten, da im Initialschritt des Ktalysekreislaufs ein
Cu‐Alkin‐π‐Komplex postuliert wird.[281, 284]
XR´B N3
O
O X
NN
N
R´
B
O
O
N
N
Mes
Mes
CuBr
+
X = CH, N R´ = Ph, C6F6,
227
(Cu)
(Cu) = CuI, CuBr,

1,3‐dipolare Cycloaddition und „Click Chemie“
134
XN N N
A B
A B
A B
NN
NX
=
XN3 +
E
X = H, Ph, Me, H2B, CatB etc.
A = B = CH
A = CH, B = N
ERef
=
ERkt
Dass eine kupferfreie 1,3‐dipolare Cycloaddition nicht beobachtet wird, ist
gleichfalls erklärbar. Da Arbeiten von Houk et al. und Anderen zu (aktivierten)
Alkinen zeigen, dass insbesondere den Übergangszuständen und
Energiebarrieren eine wichtige Bedeutung zukommt,[304‐310] wurden auch diese im
Rahmen der DFT‐Untersuchungen ermittelt (vgl. Tabelle 7). So zeigen
Rechnungen zur kupferfreien, konzertierten 1,3‐dipolaren Cycloaddition
unterschiedlicher Borazide an Alkine und Nitrile, dass die Energiebarrieren
ähnlich hoch sind wie die für die Organoazide. So ist die Addition von CatBN3 7 an
Acetylen mit 20.2 kcal/mol nahezu energiegleich mit der entsprechenden PhN3‐
Addition (19.9 kcal/mol). Dass eine kupferfreie Addition organischer Aziden erst
unter harschen Bedingungen beobachtbar ist, bei denen die Borazide ggf. nicht
mehr stabil sind, wäre ein denkbarer Grund für die nicht beobachtbare Borazid‐
Cycloaddition.
Abbildung 44: Konzertierte 1,3‐Dipolare Cycloaddition unterschiedlicher Azide an Alkine und Nitrile.
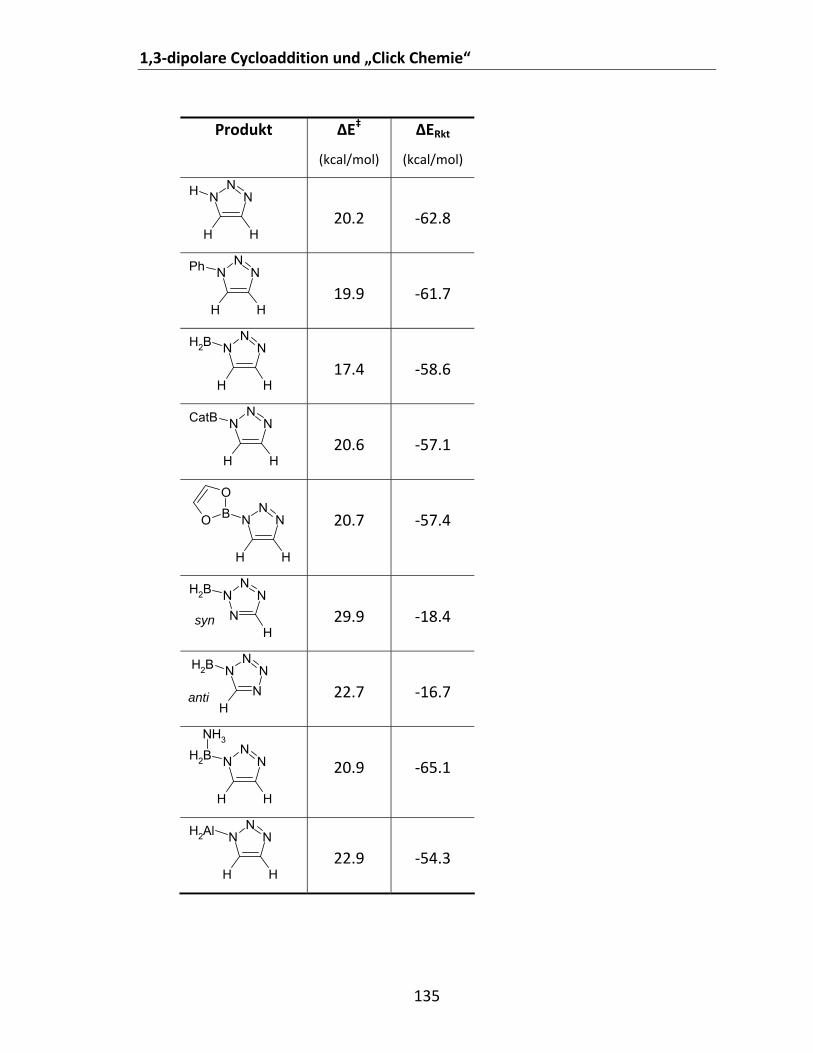
1,3‐dipolare Cycloaddition und „Click Chemie“
135
Produkt ΔE‡
(kcal/mol)
ΔERkt
(kcal/mol)
NN
NH
H H
20.2
‐62.8
NN
N
H H
Ph
19.9
‐61.7
NN
NBH2
H H
17.4
‐58.6
NN
NCatB
H H
20.6
‐57.1
B
O
O NN
N
H H
20.7
‐57.4
N
NN
NBH2
Hsyn
29.9
‐18.4
N
NN
NBH2
Hanti
22.7
‐16.7
NN
NH2B
H H
NH3
20.9
‐65.1
NN
NAlH2
H H
22.9
‐54.3

1,3‐dipolare Cycloaddition und „Click Chemie“
136
N
NN
NAlH2
Hsyn
32.9
‐16.6
N
NN
NAlH2
Hanti
28.8
‐14.3
NN
NSiH3
H H
21.5
‐57.7
Tabelle 7: Berechnete (B3LYP/6‐311+G**) Energien der Addition unterschiedlicher Azide an HCN und HCCH.

Borylnitrendimerisierung
137
R
B N
RN N
BR2
R2B2x
2-T 228
4.2.5 Versuche zur Synthese eines Borylnitrendimers
Auch Borylnitrendimere des Typs 228 bilden wegen ihrer zu erwartenden
besonderen strukturellen und elektronischen Eigenschaften interessante
Syntheseziele. So zeigen Rechnungen an den unbekannten Diazoboranen 228,
dass diese in Abhängigkeit der am Bor befindlichen Reste sowohl kumulenartige
planare als auch abgewinkelte Strukturen aufweisen können.
Abbildung 45: Berechnete (B3LYP/6‐311+G**) Geometrien ausgewählter Diazoborane.
Erste im Rahmen einer Bachelorarbeit durchgeführte Versuche, ein Diazoboran
228 über die Dimerisierung eines freien Borylnitrens 2‐T in Lösung zu erhalten,
blieben jedoch bis zum gegenwärtigen Zeitpunkt erfolglos.[311]
Schema 64: Hypothetische Dimerisierung eines Triplett‐Borylnitrens.

Thermolysen
138
4.2.6 Thermolysen von Boraziden in Kohlenwasserstoffen Neben den in der Arbeit vorgestellten Photolysen von Boraziden in Lösung und in
der Gasphase wurden auch thermolytische Experimente zur C‐H‐Aktivierung von
Kohlenwasserstoffen durchgeführt. Dazu wurden einige in der Arbeit diskutierte
Borazide (CatBN3 7, PinBN3 9, 2‐Azido‐1,3‐dimethyl‐1,3,2‐diazaborolidin 191, 2‐
Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin 192) in abgeschlossenen Glasgefäßen unter
Argonatmosphäre in hochsiedenden Substraten (Mesitylen, Cyclooctan, 1,3‐
Dimethyladamantan) für mehrere Stunden auf 200‐250 °C erhitzt und
anschließend mit den vorgestellten Methoden (Alkoholyse, Acetylierung)
aufgearbeitet. Doch trotz Azidabbaus (IR‐Kontrolle, Dunkelfärbung der
Reaktionsmischung usw.) konnten keine Aminierungsprodukte identifiziert
werden. Eine Erklärung steht noch aus und verlangt nach weiteren
experimentellen Untersuchungen.

Ausblick
139
5 Ausblick
Die vorliegenden Untersuchungen zu Borylnitrenen haben gezeigt, dass diese
sowohl das Verständnis zur BN‐Chemie erweitern und als auch zusätzlich noch
effiziente Substrate für die C‐H‐Aktivierung darstellen. Da es sich bei den
vorgestellten Ergebnissen lediglich um grundlegende Arbeiten auf einem
innovativen Forschungsfeld handelt, welche nicht im Rahmen einer Dissertation
allumfassend behandelt werden können, sollten noch weitere experimentelle
und ggf. theoretische Arbeiten folgen. Eine kleine Auswahl von Vorschlägen, sei
dazu im Folgenden kurz vorgestellt.
5.1 Substraterweiterung Es wäre wünschenswert (geeignetere) Borazide zu synthetisieren, welche
Photolysen im längerwelligen ggf. sichtbaren Bereich erlauben. So könnten z. B.
Substrate wie Alkene oder Aromaten, die selbst im kurzwelligen Bereich Licht
absorbieren und sich unter den in dieser Arbeit diskutierten Photobedingungen
als nicht photostabil erwiesen haben, effizient(er) funktionalisiert werden. Dazu
wurden bereits die in Abb. 46 aufgeführten farbigen Azide 230 und 231 als
Zielmoleküle in Betracht gezogen. Erste Herstellungsversuche zu dem auf dem
Farbstoff Alizarin basierenden Azidoboran 230 zu gelangen blieben erfolglos, da
das benötigte Bormonochlorid nicht durch Direktsynthese zugänglich ist. Durch
Versuche mit Anthrachinon als Substrat konnte gezeigt werden, dass die Lewis‐
Säure‐Base Reaktion zwischen BCl3 und einer Carbonylgruppe der chinoiden‐
Einheit eine störende Konkurrenzreaktion darstellt (11B‐NMR‐Kontrolle). Die

Ausblick
140
Synthese von 231 wurde hingegen wegen ihres hohen Syntheseaufwandes
(vorerst) zurückgestellt.
Abbildung 46: Farbige Borazide die interessante neue Syntheseziele bilden.
Um weiterhin die aufgetretenen Löslichkeitsprobleme in Zukunft zu unterbinden,
sollten diese bereits vor der Synthese der Azide ausreichende Berücksichtigung
finden. So müssten sich die Liganden durch die Einführung unpolarer Alkyl‐
Gruppen zu Aziden mit den gewünschten Löslichkeitseigenschaften modifizieren
lassen.
5.2 Erhöhung der Selektivität Auch wäre die Verfügbarkeit von Borylnitrenen wünschenswert, die eine
selektive C‐H‐Funktionalisierung erlauben. Eine Möglichkeit, ggf. die
Regioselektivität zu erhöhen, könnte nach dem RSP in der Absenkung ihrer
Reaktivität liegen. Dies könnte beispielsweise durch die Reduzierung der
Elektrophilie über die Einführung geeigneter Elektronen‐Donoren am
Borylnitrenstickstoff gelingen, wie erste theoretische Untersuchungen vermuten
lassen.[251] Auch wäre die Herstellung „gezähmter“ Borylnitrenmetallkomplexe
232, die als Borylnitrenüberträger fungieren könnten, äußerst reizvoll. Durch die
Wahl des eingesetzten Metalls sowie der am Metall als auch am Bor befindlichen
Liganden, sollten sich die chemischen Eigenschaften in weiten Bereichen gezielt
O
O
O
BO
N3
N NB
N3230 231

Ausblick
141
steuern und modifizieren lassen. Auch zeigen in unserer Arbeitsgruppe
durchgeführte computerchemischen Untersuchungen, dass Verbindungen des
Typs 232 stabil und existent sein sollten.[312] Darüber hinaus sind die BN‐analogen
(Amino)borylenmetallkomplexe (233),[313‐316] sowie die verwandten
Metallvinyliden‐ (234),[141] Metallcarben‐ (235)[141, 317, 318] und Metallnitren‐
komplexe (236)[225, 227, 228] wohl bekannt. Falls die Synthese von 232 gelänge,
könnten ergänzende Untersuchungen zum katalytischen Potential sinnvoll
werden.
Abbildung 47: Borylnitrenmetallkomplexe und ihre bekannten Verwandten.
Auch eine neue zusätzliche Methode der direkten (enantio)selektiven C‐H‐
Aminierung wäre von hohem Interesse für die Synthesechemie. Zwar existieren
bereits einige Arbeiten, welche die metallkatalysierte Aminierung von C‐H‐
Bindungen beschreiben, aber meist finden diese an aktivierten sp3‐hybridisierten
Zentren statt.[225‐228] Neben den in Abb. 47 vorgestellten Borylnitrenmetall‐
komplexen 232 mit chiralen Liganden, könnten stereospezifische Informationen
möglicherweise auch ausgehend von freien Boraziden auf ihre Substrate
übertragen werden. Hierzu wird das in der Metallorganik und Katalyse gängige
Binaphthol basierende Azid 237 als möglicher Kandidat gesehen. Erste Versuche
das benötigte Borchlorid durch Direktsynthese oder über die entsprechende
OTMS‐Verbindung zu erhalten, scheiterten jedoch.
L
M
LL
LN B
R
R
L
M
LL
LB N
R
R
L
M
LL
LC C
R
R
L
M
LL
LC
R
R
L
M
LL
LN R
232 233 234 235 236

Ausblick
142
Abbildung 48: Ein Borazid mit stereochemischer Information.
5.3 Die intramolekulare C‐H‐Insertion in der Synthesechemie
Eine intramolekulare C‐H‐Aminierungsreaktion ist ein häufig eingesetzter
Syntheseschritt für den Aufbau komplexer Zielstrukturen.[228] Da die Arbeiten an
den dialkoxysubstituierten Boraziden (RO)2BN3 zeigen, dass auch gewisse
Borylnitrene intramolekularen C‐H‐Funktionalisierungen eingehen (vgl. Kapitel
4.2.2.3), könnte man sich dieses Verhalten u. U. in der Synthesechemie nutzbar
machen. Beispielsweise sollten sich die als Hormone wirkenden, körpereigenen
Catecholamine 238[319] ausgehend von Catechol über eine mehrstufige Synthese
herstellen lassen. Den Schlüsselschritt könnte dabei eine photochemisch initiierte
intramolekulare C‐H‐Insertion eines Borylnitrens unter Aufbau von 239 bilden.
Auch die chemisch ähnlich aufgebauten pharmazeutischen Wirkstoffe Sympatol
und Ephedrin könnten über eine analoge Syntheseroute zugänglich sein.
OB
ON3
237
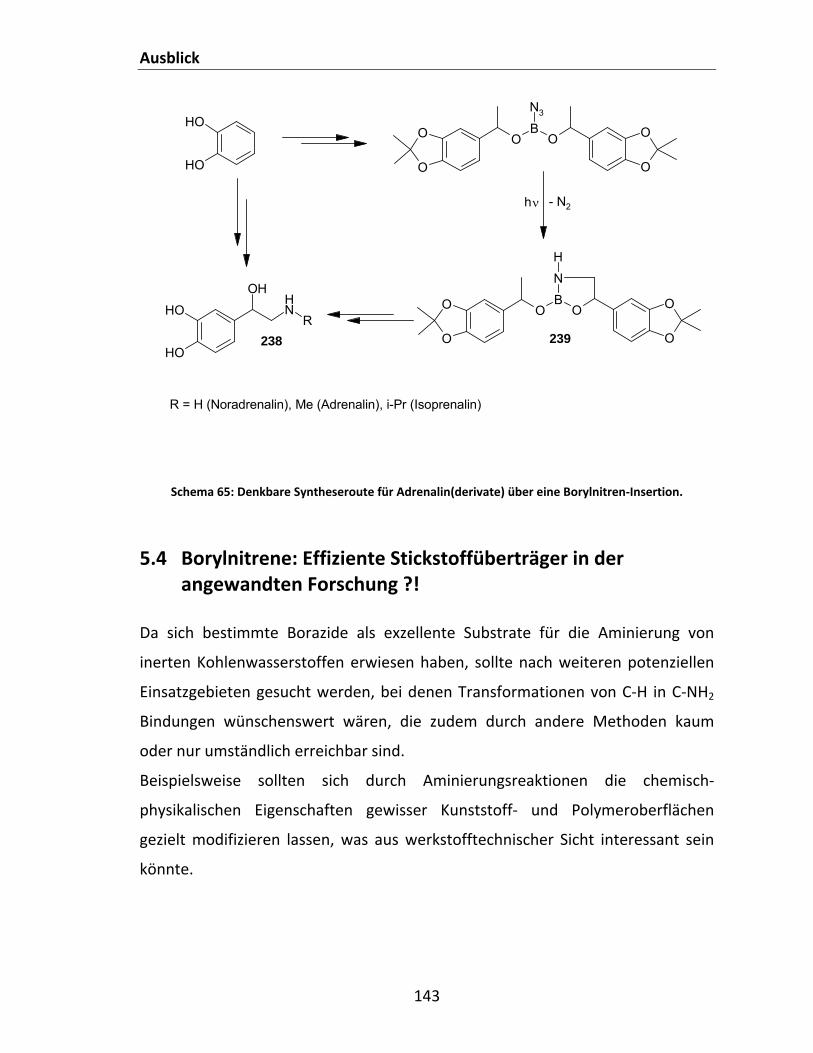
Ausblick
143
OH
OH
O
O
OB
O
N3
O
O
O
O
OB
O
N
O
O
H
OH
NH
ROH
OH
h - N2
R = H (Noradrenalin), Me (Adrenalin), i-Pr (Isoprenalin)
238 239
Schema 65: Denkbare Syntheseroute für Adrenalin(derivate) über eine Borylnitren‐Insertion.
5.4 Borylnitrene: Effiziente Stickstoffüberträger in der angewandten Forschung ?!
Da sich bestimmte Borazide als exzellente Substrate für die Aminierung von
inerten Kohlenwasserstoffen erwiesen haben, sollte nach weiteren potenziellen
Einsatzgebieten gesucht werden, bei denen Transformationen von C‐H in C‐NH2
Bindungen wünschenswert wären, die zudem durch andere Methoden kaum
oder nur umständlich erreichbar sind.
Beispielsweise sollten sich durch Aminierungsreaktionen die chemisch‐
physikalischen Eigenschaften gewisser Kunststoff‐ und Polymeroberflächen
gezielt modifizieren lassen, was aus werkstofftechnischer Sicht interessant sein
könnte.

Ausblick
144
C
H
C
H
C
H
C
NH2
C
NH2
C
NH2
1) (RO)2BN3
2) ROH oder H2O
h( = 254 nmArgon
Erste Untersuchungen an Polyethylenoberflächen lassen auf einen
Anwendungserfolg hoffen.[320] Wird Polyethylen (PET) in Gegenwart von
(EtO)2BN3 unter Argonatmosphäre belichtet und anschließend nasschemisch
(ROH, mit R = H, Alkyl) aufgearbeitet, ergeben sich Hinweise auf die Bildung von
Aminofunktionen. So zeigen IR‐Messungen des Photoprodukts, im Falle einer
PET‐Pulverbelichtung, die typischen H2N‐Valenzschwingung im Bereich >3000 cm‐
.[321, 322] Auch die Belichtung einer mit (EtO)2BN3 dünn benetzten PET‐Platte lässt
ebenfalls eine Funktionalisierung und Modifizierung der Oberfläche annehmen.
Weitere Untersuchungen zu diesem Projekt werden gegenwärtig vorgenommen.
Schema 66: Direkte Aminierung von Oberflächen.

145

Experimenteller Teil: Synthesen und Photolysen
146
6 Experimenteller Teil
6.1 Allgemeines
6.1.1 Sicherheitshinweis Da die in dieser Arbeit vorgestellten Azidoborane potentiell explosiv sind,
wurden entsprechende Schutz‐ und Vorsichtsmaßnahmen getroffen.
Die Gasphasenphotolysereaktion wurde mit äußerster Sorgfalt durchgeführt, da
das verwendete Methan(gas) hochentzündlich ist und mit Sauerstoff explosive
Gemische bildet. Insbesondere die Dichtigkeit der Apparaturen wurde eingehend
geprüft. Des Weiteren wurde die Reaktion, soweit möglich, durchgehend
beaufsichtigt.
6.1.2 Arbeitstechnik Aufgrund der Hydrolyse‐ bzw. Oxidationsempfindlichkeit fast aller in dieser Arbeit
beschriebenen Verbindungen wurden die Versuche ‐ sofern nicht anders
beschrieben ‐ unter Verwendung von Schlenk‐ und Vakuumtechnik, sowie in der
Glovebox (MBraun, Unilab) unter Argonatmosphäre (Qual. mind. 4.8)
durchgeführt. Die verwendeten Apparaturen wurden vor Versuchsbeginn im
Trockenschrank gelagert (T = 70 °C), zusätzlich im Ölpumpenvakuum ausgeheizt
und vor Benutzung mehrmals mit Argon gespült/geflutet.

Experimenteller Teil: Synthesen und Photolysen
147
6.1.3 Lösungsmittel und Chemikalien
Alle verwendeten, absolutierten LM sind entweder frisch eingesetzt (THF, Et2O)
oder vor Gebrauch über aktiviertem Molsieb (3 Å) unter Argonatmosphäre
gelagert worden. Die Trocknung von THF und Et2O erfolgte über Natrium‐
Paraffin/Benzophenon. Die verwendeten (Cyclo)alkane und Aromaten (Benzol,
Toluol, Mesitylen) wurden direkt kommerziell erworben (Acros, <50 ppm Wasser)
oder durch das Einpressen von Na‐Draht erhalten (Ausgangsqualität der LM hier:
p. A.). DCM wurde über CaH2 getrocknet und destilliert oder ebenfalls über Acros
bezogen. Alle Ausgangsverbindungen wurden käuflich erworben (SigmaAldrich,
Fluka, Acros, Baker, VWR) und ‐ sofern nicht anders beschrieben ‐ ohne weitere
Reinigung eingesetzt. Flüssige Edukte (z. B. Alkene, Alkohole) sind vor der
Benutzung über Molsieb (3 Å) aufbewahrt worden. BCl3 wurde als 1 molare
Lösung in Hexan, DCM oder Toluol verwendet und stets zügig verbraucht.
6.1.4 Interne Standards
Die Quantifizierung der Aminierungsprodukte erfolgte durch die Zugabe von
internen Standards, die i. d. R. ‐ basierend auf authentischen Proben ‐
responsefaktorberichtigt sind. Um möglichst nur kleine Integrationsfehler zu
erhalten wurde die Konzentration des betreffenden internen Standards in der
gleicher Größenordung wie die Menge des zu bestimmenden Produkts gewählt.
Des Weiteren wurde teilweise, besonders bei der Verwendung flüssiger interner
Standards (Cy‐6‐NH2, Cy‐7‐NH2), eine Lösung des betreffenden Standards
bestimmter Konzentration (meist 0.1 mmol/mL) hergestellt. Als interner Standard
fanden Hexamethylbenzol, Naphthalin, Cy‐6‐NH2, Cy‐7‐NH2 Verwendung.

Experimenteller Teil: Synthesen und Photolysen
148
6.1.5 Theoretische Berechnungen
Alle quantenmechanischen Berechnungen wurden mit dem Programmpaket
Gaussian 03 auf einem Windows‐PC durchgeführt. Es wurden fast ausschließlich
DFT‐Rechnungen auf B3LYP/6‐311+G**‐Niveau vorgenommen. Die Natur der
stationären Punkte (Minimum bzw. Übergangszustand) wurde mittels
Frequenzrechnungen ermittelt. Die Energien wurden durchweg in der
gebräuchlichen Kalorien‐Einheit angegeben. Um die Rechenzeit gering zu halten
wurden meist kleine (Modell)systeme hoher Symmetrie gewählt.
6.1.6 Lichtquelle
Photolysen wurden mithilfe einer Quecksilberniederdrucklampe der Firma
Gräntzel durchgeführt (Photoreaktor 400). Photobedingungen: Stromstärke: 200‐
220 mA, λ = 254 nm.
6.1.7 Instrumentelle Analytik
6.1.7.1 NMR‐Spektroskopie
Die NMR‐Spektren wurden mit folgenden Geräten aufgenommen: Bruker DPX‐
200 und DRX‐400 (1H und 13C‐Messungen), sowie DRX 250 (11B‐Messungen). Die
chemischen Verschiebungen δ wurden in ppm relativ zu Tetramethylsilan (1H,
13C) bzw. BF3*OEt2 (11B) angegeben. Das Strukturmuster ist als s (Singulett), d
(Dublett), t (Triplett), q (Quartett) oder m (Multiplett) angegeben. Die
Spektrenauswertung erfolgte mit dem Programm WIN‐NMR (Bruker) sowie
MestRec‐Nova.

Experimenteller Teil: Synthesen und Photolysen
149
6.1.7.2 GC/MS‐ und GC‐Messungen
GC/MS‐Messungen der Produktproben sind mit einem Gaschromatograph der
Hewlett Packard Serie ΙΙ (5890 / Detektor: Mass‐Det 5972) auf einer Zebron ZB‐
5mS (Phenomenex) Kapillar‐Säule (Länge 30 m, I.D. 0.25 mm, Film‐Dicke 0.25 μm)
mit Helium als Trägergas charakterisiert worden. GC‐Analyse wurde auf einer CP‐
Wax 51 Amin FS (Varian) Kapillar‐Säule (Länge 25 m, I.D. 0.32 mm, Film‐Dicke
1,20 μm) an einem Chromstar Sichromat (Siemens) mit Wasserstoffgas
vorgenommen. Die qualitative Analyse der Probenbestandteile erfolgte meist
mithilfe der Massenspektren (Vergleich mit authentischen Proben
/Datenbankabgleich). Zur Quantifizierung (Ausbeutbestimmung) wurden
Messungen mit internen Standards (meist responsfaktorberichtigt)
vorgenommen.
6.1.7.3 Massenspektrometrie
Die EI‐Messungen wurden mit einem Massenspektrometer Varian MAT CH5 bei
70 eV durchgeführt. Angegeben sind charakteristische Signale und ihre
Intensitäten in %.
6.1.7.4 Infrarotspektren
Die Infrarotspektren wurden an einem FT‐IR Equinox 55 Spektrometer (Bruker)
zwischen NaBr‐Platten oder als Nujol‐Verreibung zwischen NaCl‐Platten
aufgenommen.

Experimenteller Teil: Synthesen und Photolysen
150
6.1.7.5 Röntgenstrukturanalysen
Die Röntgenstrukturen wurden auf einem Oxford Xcalibur Diffraktometer unter
Verwendung von Mo‐Kα‐ bzw. Cu‐Kα‐Strahlung durchgeführt. Die Strukturlösung
und ‐verfeinerung erfolgte mit dem SHELXL‐97‐Programm. Die graphische
Darstellung wurde mit dem Programm Diamond 3.0 vorgenommen.

Experimenteller Teil: Synthesen und Photolysen
151
OH
OH
OB
OCl
108
+BCl3
Pentan, 0 °C bis RT, 3 h
-HCl
M = 162.42 g/mol
6.2 Synthesen
6.2.1 Synthese von Pinakolborchlorid (108)[249, 323]
Durchführung
Wasserfreies, pulverisiertes Pinakol wird am Platz abgewogen und zügig unter
Argongegenstrom in einen ausgeheizten Dreihalsrundkolben (1 L, 3 x NS 29
Schliffe, Aufbau: 1) Argoneinleitungshahn, 2) Tropftrichter mit Druckausgleich, 3)
Blasenzähler mit aufgesetztem Schlauch und Trichter) überführt und in Pentan
(750 mL) gelöst. Es wird auf 0 °C abgekühlt (Eisbad), wobei eine weiße Suspension
entsteht. Dann erfolgt tropfenweise über einen Zeitraum von etwa 2 h die
Zugabe von BCl3, wobei das gebildete HCl‐Gas unter leichtem Argonstrom über
einen aufgesetzten Blasenzähler in wässrige kalte NaOH eingeleitet wird.
Anschließend wird das Eisbad entfernt und für 1 h bei RT nachgerührt, bevor man
den ausgefallenen FS (Pin)B‐B(Pin) absetzen lässt. Die überstehende, klare Lösung
wird mittels Spritze (oder Kanüle) portionsweise in einen ausgeheizten
Name der Chemikalie Menge
Pinakol (Pin) m = 23.5 g (M = 118.18 g/mol, n = 0.199 mol)
BCl3 (1 M in Hexan) V = 200 mL, (c = 1 mmol/mL, n = 0.200 mol)
Pentan (abs.) V = 750 mL

Experimenteller Teil: Synthesen und Photolysen
152
Dreihalsrundkolben überführt (100 mL, Aufbau: 1) Argoneinleitungshahn, 2)
Septum, 3) kurze Brücke mit Schlenkvorlage (Vorlage vorher wiegen, Rührfisch in
Vorlage geben für anschließende Azidierung)) und die LM (Pentan, Hexan)
werden im vollen Membranpumpenvakuum (ca. 10‐20 mbar) bei RT entfernt.
Destillation des gelblichen, viskösen Rückstands ergeben 8.6 g (26 %, Methode I)
bzw. 10.3 g (32 %, Methode II) PinBCl 108 als farblose Flüssigkeit (Methode I:
Destillation im Membranvakuum (ca. 10‐20 mbar), Badtemperatur 65 °C,
Kopftemperatur ca. 40 °C, Vorlage mit Eiswasser kühlen / Methode II:
Kondensation im Ölpumpenvakuum bei RT, Vorlage mit Trockeneis/Isopropanol
oder ggf. fl. Stickstoff kühlen). Da das erhaltene PinBCl 108 nicht nur äußerst luft‐
und hydrolyseempfindlich ist, sondern sich bereits innerhalb kurzer Zeit in der
Glovebox bei RT zu zersetzen beginnt, wird es bei ‐18 °C unter Argonatmosphäre
aufbewahrt oder direkt zu PinBN3 9 weiter umgesetzt.
Charakterisierung
1H‐NMR (C6D6): δ = 0.95 ppm (s, 12 H, CH3)
13C‐NMR (C6D6): δ = 25.7 ppm (CH3), 86.8 (OCH3)
11B‐NMR (C6D6): δ = 27.5 ppm (Hydrolyseprodukt (PinBOBPin): δ = 22.4 ppm)

Experimenteller Teil: Synthesen und Photolysen
153
OH
OH OB
OCl M = 210.47 g/mol
+BCl3-HCl
-78 °C bis RT, 16 h
DCM
6.2.2 Synthese von tert‐BuCatBCl [180]
Durchführung
Zu einer Suspension von trockenem tert‐BuCatechol in DCM (60 mL) erfolgt bei ‐
78 °C (Trockeneis/Isopropanol) über einen Zeitraum von etwa 10 Min. die Zugabe
von BCl3. Man lässt die Reaktionsmischung langsam über Nacht auf RT erwärmen
und führt das gebildete HCl‐Gas über eine im Septum befindliche Nadel ab
(leichter Argonstrom). Die flüchtigen Bestandteile werden anschließend im
Ölpmpenvakuum entfernt. Zurück bleiben 2.75 g (87 %) des gewünschten tert‐
BuCatBCl als weiß‐grauer FS, der ohne weitere Aufreinigung weiter eingesetzt
wird.
Charakterisierung
1H‐NMR (CD2Cl2): δ = 1.28 ppm (s, 12 H, C(CH3)3), 6.48‐6.77 (m, 3 H, ArH)
11B‐NMR (CD2Cl2): δ = 28.4 ppm
Name der Chemikalie Menge
tert‐BuCatechol m = 2.50 g (M = 166.22 g/mol, n = 15.04 mmol)
BCl3 (1 M in Hexan) V = 16 mL (c = 1 mmol/mL, n = 16.00 mmol)
DCM (abs.) 60 mL

Experimenteller Teil: Synthesen und Photolysen
154
6.2.3 Synthese von Diisopropyloxyborchlorid (iPrO)2BCl[184, 186]
Durchführung
In einem Schlenkkolben (1000 mL) wird eine Lösung von BCl3 in Hexan mit
Pentan. (etwa 300 mL) und DCM (etwa 50 mL) verdünnt und auf ca. ‐80 °C
abgekühlt (Isopropanol/fl. Stickstoff). Dann erfolgt über einen Zeitraum von etwa
5 Min. die Zugabe von reinem B(OiPr)3 unter Bildung einer weißen, milchigen
Suspension. Das Kältebad wird gegen ein großes Eis/Wasserbad ersetzt und die
dabei gebildete klare Reaktionslösung für 16 h bei 0 °C und weitere 0.5 h bei RT
nachgerührt. Dann wird ein Teil der Reaktionslösung entnommen und mittels 11B‐
NMR überprüft, ob die Reaktion vollständig verlaufen ist. Falls noch iPrOBCl2 (δ =
31.5 ppm) vorhanden ist, werden einige Tropfen B(OiPr)3 zur Reaktionsmischung
gegeben bis das Signal verschwindet (erneute 11B‐Messung). Anschließend
werden die LM (Pentan, Hexan) im Ölpumpenvakuum bei RT entfernt. Dabei ist
darauf zu achten, dass das gewünschte Produkt nicht auch abdestilliert wird. Der
Name der Chemikalie Menge
BCl3 (1 M in Hexan) V = 100 mL (n = 0.100 mol)
B(OiPr)3
V = 45.90 mL (M = 188.08 g/mol, d = 0.818 g/mL,
c = 4.349 mmol/mL, n = 0.200 mol) 2 Äq.
Pentan (abs.) V = 300 mL
DCM (abs.) V = 50 mL
Pentan / Hexan / DCM
-80 °C bis RT, 16 h1 BCl3 + 2 B(OiPr)3 3 (iPrO)2BCl
M = 164.44 g/mol

Experimenteller Teil: Synthesen und Photolysen
155
klare, mäßig viskose Rückstand wird dann mittels Spritze in einen neuen
Schlenkkolben (100 mL, NS 14) überführt und das Produkt im Ölpumpenvakuum
bei RT über eine kurze Brücke in eine mit flüssigem Stickstoff gekühlte Vorlage
überkondensiert. Zurück bleiben 28.1 g (57 %) (iPrO)2BCl als farblose Flüssigkeit
die bei ‐18 °C unter Argonatmosphäre gelagert oder direkt zum entsprechenden
Azid (iPrO)2BN3 150‐(iPr)2 umgesetzt wird.
Charakterisierung
1H‐NMR (CDCl3): δ = 1.15 ppm (d, 12 H, OCH(CH3)2), 4.51 (sep, 2 H, OCH(CH3)2)
13C‐NMR (CDCl3): δ = 24.2 ppm (OCH(CH3)2), 69.3 (OCH(CH3)2)
11B‐NMR (CDCl3): δ = 23.2 ppm [NP: iPrOBCl2: δ = 31.5, B(OiPr)3: δ = 18.0 )]

Experimenteller Teil: Synthesen und Photolysen
156
6.2.4 Synthese von Diethoxyborchlorid (EtO)2BCl
Durchführung
Herstellung analog zur Synthese von (iPrO)2BCl. Bei der Aufarbeitung muss darauf
geachtet werden, dass beim Entfernen des LM nur ein leichtes Vakuum angelegt
wird, da das Produkt (EtO)2BCl einen niedrigen Siedepunkt besitzt und ansonsten
ebenfalls entfernt wird. Es werden 13.2 g (32 %) des gewünschten
Diethoxyborchlorids (EtO)2BCl nach Umkondensation als farbloses Öl erhalten.
Charakterisierung
1H‐NMR (CD2Cl2): δ = 1.15 ppm (t, 6 H, OCH2CH3), 3.51 (q, 4 H, OCH2CH3)
13C‐NMR (CD2Cl2): δ = 24.2 ppm (OCH2CH3), 69.3 (OCH2CH3)
11B‐NMR (CD2Cl2): δ = 23.5 ppm
Name der Chemikalie Menge
BCl3 (1 M in Hexan) V = 100 mL (n = 0.100 mol)
B(OiPr)3
V = 34.03 mL (M = 145.99 g/mol, d = 0.858 g/mL,
c = 5.877 mmol/mL, n = 0.200 mol) 2 Äq.
Pentan (abs.) V = 300 mL
DCM (abs.) V = 50 mL
Pentan / Hexan / DCM
-80 °C bis RT, 16 h1 BCl3 + 2 B(OiPr)3 3 (EtO)2BCl
M = 136.39 g/mol

Experimenteller Teil: Synthesen und Photolysen
157
OH
OH
OTMS
OTMSM = 304.53 g/mol
TMSCl/Et3N
ToluolRückfluss, 3 hRT, 16 h
6.2.5 Synthese von 2‐Chloro‐Naphto(2,3‐d)‐1,3,2‐dioxaborol
ANMERKUNG: Die Herstellung kann auch über Direktsynthese erfolgen.
6.2.5.1 Synthese von 2,3‐Bis‐(trimethylsilanyloxy)naphthalin[182, 324]
Durchführung
Zu einer Suspension von frisch sublimiertem 2,3‐Dihydroxynaphthalin in Toluol
(150 mL) erfolgt unter Argonatmosphäre sukzessiv die Zugabe von TMSCl und
Et3N. Die Reaktionsmischung wird für 3 h unter Rückfluss erhitzt und
anschließend für 16 h bei RT nachgerührt. Das gebildete Ammoniumsalz
(HNEt3Cl) wird dann mithilfe einer Umkehrfritte abgetrennt und mit Toluol (3 x 30
mL) gewaschen. Nach dem Entfernen des LM im Ölpumpenvakuum wird das
Name der Chemikalie Menge
2,3‐Dihydroxynaphthalin m = 5.00 g (M = 160.17, n = 0.031 mol)
TMSCl V = 19.0 (M = 108.64 g/mol, d = 0.856 g/mL,
c = 7.88 mmol/mL, n = 0.150 mol)
Et3N (abs.) V = 20.9 mL (M = 101.19 g/mol, d = 0.726 g/mL,
c = 7.17 mmol/mL, n = 0.150 mol)
Toluol (abs.) V = 1000 mL
Pentan nach Bedarf
Essigester nach Bedarf

Experimenteller Teil: Synthesen und Photolysen
158
OTMS
OTMS OB
OCl M = 204.42 g/mol
+BCl3
DCM, Hexan
RT bis 50 °C, 4 h
gewünschte 2,3‐Bis‐(trimethylsilanyloxy)naphthalin als grauer FS erhalten und
unter Argonatmosphäre gelagert. Falls sich durch DC‐Kontrolle noch nicht
umgesetztes Edukt bzw. monosubstituertes Diol nachweisen lässt, wird das
Rohrodukt noch mittels Flashsäulen‐Chromatographie mit einem
Pentan/Essigester (4:1)‐Gemisch zügig (!!) aufgereinigt.
Charakterisierung
1H‐NMR (CDCl3): δ = 0.35 ppm (s, 18 H, OSi(CH3)3), 7.58‐7.63 (m, 6 H, ArH)
13C‐NMR (CDCl3): δ = 0.1 ppm (OSi(CH3)3), 116.0 (ArC), 123.8 (ArC), 125.9 (ArC),
129.7 (ArC), 146.8 (ArC‐OSi(CH3)3)
6.2.5.2 Synthese von 2‐Chloro‐Naphto(2,3‐d)‐1,3,2‐dioxaborol [182, 183]
Name der Chemikalie Menge
2,3‐Bis‐(trimethylsilanyloxy)‐
naphthalin
m = 2.80 g (M = 304.53 g/mol, n = 9.19 mmol)
BCl3 (1 M in Hexan) V = 9.5 mL (c = 1 mmol/mL, n = 9.50 mmol )
DCM (abs.) V = 50 mL

Experimenteller Teil: Synthesen und Photolysen
159
Durchführung
Zu einer Lösung von 2,3‐Bis‐(trimethylsilanyloxy)naphthalin in DCM (50 mL)
erfolgt bei RT die Zugabe von BCl3. Die Reaktionsmischung wird für 1 h bei RT
nachgerührt und für weiter 3 h auf 50 °C erhitzt (verschlossene Apparatur). Man
lässt die gelbliche Lösung auf RT abkühlen und entfernt die LM im Vakuum und
wäscht den zurückbleibenden weiß‐grauen FS mit Toluol und Pentan (je 10 mL).
Trocknung im Ölpumpenvakuum liefert nach Umkristallisation aus Toluol 1.43 g
(76 %) 2‐Chloro‐Naphto(2,3‐d)‐1,3,2‐dioxaborol.
Charakterisierung
1H‐NMR (CD2Cl2): 7.64 (m, 6 H, ArH)
13C‐NMR (CD2Cl2): δ = 115.7 (ArC), 123.5 (ArC) , 125.6 (ArC), 128.3 (ArC), 139.5
(ArC‐OBClR)
11B‐NMR (CD2Cl2): δ = 27.3 ppm

Experimenteller Teil: Synthesen und Photolysen
160
O
O OH
OH OH
OHOH
OHHCl/Sn
Rückfluss, 1 h
M = 142.11 g/mol
172 173
RT, 2.5 h
6.2.6 Synthese des Bischlorids 175
6.2.6.1 Synthese von 1,2,4,5‐Tetrahydroxybenzol (173)
Durchführung
Zu einer stark gerührten Suspension von 2,5‐Dihydroxy‐1,4‐benzochinon 172 in
konzentrierter Salzsäure erfolgt über einen Zeitraum von etwa 1.5 h die Zugabe
von Sn‐Granalien. Es wird für 1 h bei RT nachgerührt und anschließend für 1 h
unter Rückfluss erhitzt. Die dunkle Reaktionsmischung wird dann heiß abfiltriert
(D‐3 Fritte) und man lässt das Filtrat langsam auf RT abkühlen. Sofern keinen FS
ausfällt wird das Filtrat bis auf 0 °C heruntergekühlt und die Glasinnenwand der
Saugflasche mit einem Glasstab angekratzt. Der abgesetzte, bräunlich‐weiße FS
wird abfiltriert (D‐3‐Fritte) und mit Wasser (100 mL) und Pentan (100 mL)
gewaschen, anschließend aus THF unkristallisiert. Zurück bleiben 17.5 g (69 %)
1,2,4,5‐Tetrahydroxybenzol 173 als nahezu weißer, kristalliner FS, das ohne
Charakterisierung weiter eingesetzt wird.
Name der Chemikalie Menge
2,5‐Dihydroxy‐1,4‐
benzochinon
m = 25.00 g (M = 140.09 g/mol, n = 0.178 mol)
Sn‐Granalien m = 25.50 g (M = 118.71 g/mol, n = 0.215 mol)
HCl (konz.) V = 540 mL

Experimenteller Teil: Synthesen und Photolysen
161
OH
OHOH
OH OTMS
OTMSTMSO
TMSOTMSCl/Et3N
Toluol173 174
M = 430.83 g/mol
RT, 16 h
Charakterisierung
MS (EI): m/z (%): 142 (100)[M +∙], 126 (10), 113 (13) 96 (27), 78 (5), 69 (12), 53
(14), 40 (15)
IR (KBr): 3400‐3200 cm‐1 (vs, OH)
6.2.6.2 Synthese von 1,2,4,5‐Tetrakis‐(trimethylsilanyloxy)benzol (174)[183]
Name der Chemikalie Menge
1,2,4,5‐Tetrahydroxy‐
benzol
m = 4.94 g (M = 142.12 g/mol, n = 0.035 mol)
TMSCl
V = 38.34 mL (M = 108.64 g/mol, d = 0.856 g/mL,
c = 7.88 mmol/mL, n = 0.303 mol)
Et3N
V = 41.82 mL (M = 101.19 g/mol, d = 0.726 g/mL,
c = 7.17 mmol/mL, n =0.300 mol)
Toluol (abs.) V = 240 mL
Hexan (abs.) nach Bedarf

Experimenteller Teil: Synthesen und Photolysen
162
OTMS
OTMSTMSO
TMSO
OB
OOB
OClCl
+BCl3
-TMSCl
Hexan
55 °C, 6 h
M = 230. 61 g/mol
174 175
Durchführung
Getrocknetes 1,2,4,5‐Tetrahydroxybenzol 173 wird unter Argonatmosphäre in
Toluol (180 mL) suspendiert, bevor die Addition von TMSCl und Et3N unter
starkem Rühren erfolgt. Es wird für 16 h bei RT nachgerührt, das gebildete
Ammoniumsalz (HNEt3Cl) mithilfe einer Umkehrfritte abgetrennt und
anschließend mit Toluol (3 x 20 mL) gewaschen. Dann wird das LM im
Ölpumpenvakuum entfernt und der Rückstand für mind. 16 h bei ‐18 °C gelagert.
Der kristalline FS (ggf. Ankratzen notwendig) wird dann in etwas Hexan
suspendiert, über eine D‐3 Fritte abfiltriert, mit wenig Hexan gewaschen und
anschließend für mehrere Stunden im Ölpumpenvakuum getrocknet. Zurück
bleiben 11.0 g (73 %) 1,2,4,5‐Tetrakis‐(trimethylsilanyloxy)benzol 174 als rosa‐
weißer FS, das unter Argonatmosphäre gelagert wird.
Charakterisierung
1H‐NMR (CDCl3): δ = 0.07 ppm (s, 36 H, (OSi(CH3)3), 6.15 (s, 2 H, ArH).
13C‐NMR (CDCl3): δ = 0.1 ppm (OSi(CH3)3), 113.4 (ArC), 139.8 (ArC‐OSi(CH3)3)
MS (EI): m/z (%): 431 (100)[M +∙], 339 (22), 286 (53), 239 (16), 213 (88), 184 (96),
155 (86), 155 (91), 91 (85), 65 (18)
6.2.6.3 Synthese des Bisborchlorids 175[183]

Experimenteller Teil: Synthesen und Photolysen
163
Durchführung
Unter Argonatmosphäre erfolgt über einen Zeitraum von etwa 1 h zu einer
Lösung von trockenem 1,2,4,5‐Tetrakis‐(trimethylsilanoxy)benzol 174 in Hexan
(150 mL) die Zugabe von BCl3. Die Reaktionsmischung wird für 6 h auf 55 °C
erhitzt, bevor die flüchtigen Bestandteile im Ölpumenvakuum bei RT entfernt
werden. Der Rückstand wird in etwas Pentan aufgenommen und über Nacht bei –
18 °C gelagert. Der ausgefallene FS wird anschließend mit wenig kaltem Pentan
gewaschen und im Ölpumpenvakuum getrocknet. Zurück bleiben 1.52 g (57 %)
des gewünschten Borchlorids 175 als weißer FS.
Charakterisierung
1H‐NMR (CDCl3): δ = 6.47 ppm (s, 2 H, ArH).
13C‐NMR (CDCl3): δ = 99.2 ppm (ArC), 144.2 (ArCOBClR)
11B‐NMR (CDCl3) δ = 28.5 ppm
Name der Chemikalie Menge
1,2,4,5‐Tetrakis‐
(trimethylsilanoxy)benzol
m = 5.00 g (M = 430.83 g/mol, n = 11.6 mmol)
BCl3 (1 M in Hexan) V = 23.5 mL (c = 1 mmol/mL, n = 23.5 mmol)
Hexan (abs.) V = 150 mL
Pentan (abs.) nach Bedarf

Experimenteller Teil: Synthesen und Photolysen
164
NH2
NH2
NH
NH
Tos
Tos 203
+TosCl
NaOH (aq.), Et2O
M = 368.47 g/mol 0 °C bis RT, 18 h
6.2.7 Synthese von 2‐Chloro‐1,3‐ditosyl‐1,3,2‐diazaborolidin (202)
6.2.7.1 Synthese von N,N´‐Ditosyl‐1,2‐ethandiamin (203)[270‐272]
Durchführung
Eine auf 0 °C gekühlte Suspension von TosCl in Et2O (200 mL) wird portionsweise
mit einer gekühlten, wässrigen NaOH/1,2‐Ethandiamin‐Lsg. versetzt und für 1 h
bei 0 °C und über Nacht bei RT gerührt. Der ausgefallene FS wird über eine große
Fritte (D‐3) abfiltriert und nacheinander mit kaltem Wasser, Et2O, MeOH (je 2 x
30 mL) gewaschen und anschließend im Ölpumpenvakuum getrocknet. Zurück
bleiben 81.0 g (88 %) N,N´‐Ditosyl‐1,2‐ethandiamin 203 als leicht grauer
amorpher FS.
Name der Chemikalie Menge
1,2‐Ethandiamin m = 15.00 g (M = 60.10 g/mol, n = 0.250 mol)
TosCl m = 95.00 g (M = 190.65 g/mol, n = 0.50 mol)
NaOH in Wasser (200 mL) m = 20.00 g (M = 40.00 g/mol, n = 0.50 mol)
Et2O V = 230 mL
MeOH V = 30 mL

Experimenteller Teil: Synthesen und Photolysen
165
NH
NH
Tos
Tos
NB
NCl
Tos
Tos
203
+BCl3
DCM, 0 °C
M = 412.72 g/mol-HCl
202
Charakterisierung
1H‐NMR (Aceton‐d6): δ = 2.38 ppm (s, 6 H, ArCH3), 2.71 (s, 4 H, CH2), 6.51 (s (br), 2
H, NH), 7.37 (d, 4 H, ArH), 7.60 (d, 4 H, ArH).
MS (EI): m/z (%): 369 (2)[M +∙], 339 (52), 303 (5), 284 (11), 261 (27), 239 (17), 213
(10), 184 (100), 155 (86), 91 (67), 65 (14)
6.2.7.2 Synthese von 2‐Chloro‐1,3‐ditosyl‐1,3,2‐diazaborolidin (202)[190‐192]
Name der Chemikalie Menge
N,N´‐Ditosyl‐1,2‐
Ethandiamin
m = 13.33 g (M = 368.47 g/mol, n = 36.18 mmol)
BCl3 (1 M in DCM) V = 36.2 mL (c = 1 mmol/mL, n = 36.20 mmol)
DCM (abs.) V = 275 mL
Toluol (abs.) V = 25 mL
Pentan (abs.) V = 25 mL

Experimenteller Teil: Synthesen und Photolysen
166
Durchführung
In einem ausgeheizten Dreihalsrundkolben (500 mL, Aufbau: 1)
Argoneinleitungshahn, 2) Septum, 3) Blasenzähler mit aufgesetztem Schlauch und
Trichter) wird eine auf 0 °C gekühlte Suspension von N,N´‐Ditosyl‐1,2‐
ethandiamin 203 in DCM (250 mL) über einen Zeitraum von etwa 30 Min.
tropfenweise mit BCl3 versetzt, wobei das gebildete HCl‐Gas unter leichtem
Argonstrom über den aufgesetzten Blasenzähler in wässrige kalte NaOH
eingeleitet wird. Nach beendeter Zugabe wird das Kältebad entfernt und die
Reaktionsmischung über Nacht bei RT nachgerührt. Anschließend wir das LM im
Vakuum entfernt und der zurückbleibende FS nacheinander mit DCM, Toluol und
Pentan (je 25 mL) gewaschen und dann im Ölpumpenvakuum getrocknet. Es
werden 10.6 g (71 %) 2‐Chloro‐1,3‐ditosyl‐1,3,2‐diazaborolidin 202 in Form eines
weiß‐grauen FS erhalten.
Charakterisierung
1H‐NMR (CDCl3): δ = 2.41 ppm (s, 6 H, ArCH3), 3.70 (s, 4 H, CH2), 7.33 (d, 4 H, ArH),
7.80 (d, 4 H, ArH)
13C‐NMR (CDCl3): δ = 21.4 ppm (ArCH3), 42.2 (CH2), 127.1 (ArC), 129.6 (ArC), 135.2
(ArC), 143.9 (ArC)
11B‐NMR (CDCl3): δ = 27.9 ppm

Experimenteller Teil: Synthesen und Photolysen
167
NH
NH
CH3
CH3
NB
NCl
CH3
CH3
+Et3N*BCl3
Hexan, 0 °C bis 50 °C
M = 132.40 g/mol- HNEt3Cl
6.2.8 Synthese von 2‐Chloro‐1,3‐dimethyl‐1,3,2‐diazaborolidin [189, 197]
Durchführung
Eine auf 0 °C gekühlte Lösung von Et3N in Hexan (500 mL), wird unter
Argonatmosphäre über einen Zeitraum von etwa 30 Min. portionsweise mit BCl3
versetzt und anschließend für weitere 15 Min. bei 0 °C gerührt. Man lässt die
Mischung auf RT erwärmen, bevor die Zugabe einer Lösung von N,N´‐Dimethyl‐
1,2‐ethandiamin in Hexan (350 mL) erfolgt. Die zähe Suspension wird für 1 h bei
RT und für weitere 3 h bei 50 °C gerührt. Dann wird das Ammoniumsalz mihilfe
einer Umkehrfritte abgetrennt und mit Hexan (3 x 100 mL) gewaschen. Das Filtrat
wird portionsweise in einen Schlenkkolben (100 mL) überführt (vgl. Synthese von
PinBCl, Destillationsapparatur) und das LM im Membranvakuum bei RT entfernt.
Anschließend wir der verbleibende flüssige RS entweder im Membranvakuum bei
Name der Chemikalie Menge
N,N´‐Dimethyl‐1,2‐
ethandiamin
V = 25 mL (M = 88.15 g/mol, d = 0.819 g/mL,
c = 9.29 mmol/mL, n = 0.232 mol)
Et3N (abs.)
V = 25 mL (M = 101.19 g/mol, d = 0.726 g/mL,
c = 7.17 mmol/mL, n = 0.465 mol)
BCl3 (1M in DCM) V = 232 mL (c = 1 mmol/mL, n = 0.232 mol )
Hexan (abs.) V = 1000 mL

Experimenteller Teil: Synthesen und Photolysen
168
etwa 55 °C Badtemperatur über eine kurze Brücken in eine gekühlte (0 °C)
Vorlage überführt (Methode I) oder im Ölpumpenvakuum in eine gekühlte
(Trockeneis oder fl. Stickstoff) Vorlage überkondensiert (Methode II). Zurück
bleiben 16.9 g (55 %, Methode I) bzw. 21.1 g (69 %, Methode II) des gewünschten
Borchlorids als farblose Flüssigkeit.
Charakterisierung
1H‐NMR (CDCl3): δ = 2.51 ppm (s, 6 H, CH3), 3.12 (s, 4 H, CH2)
13C‐NMR (CDCl3): δ = 31.7 ppm (CH3), 48.9 (CH2)
11B‐NMR (CDCl3): δ = 26.9 ppm

Experimenteller Teil: Synthesen und Photolysen
169
6.2.9 Synthese von 2‐Chloro‐1,3,2‐dithiaborolan (117)[179, 193, 194, 325]
Vorsicht: von 1,2‐Dithioethan geht eine starke Geruchsbelästigung aus ! Alle
Arbeiten werden im Abzug durchgeführt und die verwendeten Geräte nach
Gebrauch gründlich mit Natriumhypochlorit‐Lsg. gereinigt.
Durchführung
1,2‐Dithioethan wird in einen Dreihalskolben überführt (500 mL, 3 x NS 29
Schliffe, Aufbau: 1) Argoneinleitungshahn, 2) Tropftrichter mit Druckausgleich, 3)
Blasenzähler mit aufgesetztem Schlauch und Trichter), in DCM (150 mL) gelöst
und auf etwa ‐60 °C (Isopropanol/fl. Stickstoff) abgekühlt. Dann erfolgt über
einen Zeitraum von etwa 2 h tropfenweise die Zugabe von BCl3, wobei das
gebildete HCl‐Gas unter leichtem Argonstrom über den Blasenzähler in wässrige
kalte NaOH eingeleitet wird. Nach beendeter Zugabe wird das Kältebad entfernt
und die Reaktionsmischung für eine weitere Stunde bei RT nachgerührt. Die
Name der Chemikalie Menge
1,2‐Dithioethan
V = 16.77 mL (M = 94.18 g/mol, d = 1.123 g/mL,
c = 11.924 mmol/mL, n = 0.200 mol)
BCl3 (1 M in DCM) V = 200 mL, (c = 1 mmol/mL, n = 0.200 mol)
DCM (abs.) V = 150 mL
NaOCl nach Bedarf (Reinigung der Geräte)
SH
SH
SB
SCl
+BCl3
DCM
M = 138.45 g/mol
117-60 °C bis RT, 3 h
-HCl

Experimenteller Teil: Synthesen und Photolysen
170
Lösung wird anschließend mittels Spritze (oder Kanüle) portionsweise in einen
ausgeheizten Dreihalsrundkolben überführt (100 mL, Aufbau: 1)
Argoneinleitungshahn, 2) Septum, 3) kurze Brücke mit Schlenkvorlage (Vorlage
vorher wiegen, Rührfisch in Vorlage für anschließende Azidierung)) und das DCM
im Membranpumpenvakuum bei RT entfernt. Destillation des gelblichen viskösen
Rückstands („volles“ Membranvakuum (ca. 10‐20 mbar), Badtemperatur 90 °C,
Vorlage mit Eiswasser kühlen) ergibt 2‐Chloro‐1,3,2‐dithiaborolan 117 (19.9 g, 72
%) als farblose Flüssigkeit, die unter Schutzgasatmosphäre bei RT in Glovebox
aufbewahrt wird.
Charakterisierung
1H‐NMR (CDCl3): δ = 3.42 ppm (s, 4 H, CH2)
13C‐NMR (CDCl3): δ = 37.3 (CH2)
11B‐NMR (CDCl3): δ = 58.8 ppm

Experimenteller Teil: Synthesen und Photolysen
171
6.2.10 Azidierung
6.2.11 Allgemeine Synthesevorschrift der Azidierung[34, 172‐174]
Achtung: Die in dieser Arbeit vorgestellten Azidoborane sind potentiell explosiv!
Durchführung
Die gewünschten Borazide R2BN3 werden durchweg aus der Reaktion eines
Borchlorids mit TMSN3 (M = 115.21 g/mol, d = 0.876 g/mL, c = 7.604 mmol/mL)
als Azidierunsmittel herstellt. Zu einer vorgelegten DCM‐Lösung eines Borchlorids
(ca. 20 mL DCM pro g des eingesetzten Borchlorids) erfolgt unter
Argonatmosphäre bei etwa ‐80 °C (Isopropanol/fl. Stickstoff) die Zugabe von
reinem TMSN3 (1.0‐1.5 Äq.). Man lässt die Reaktionmischung langsam über Nacht
auf RT erwärmen und entfernt anschließend die flüchtigen Betandteile (DCM,
TMSCl, überschüssiges TMSN3) bei RT im Ölpumpenvakuum. Je nach
Reinheitsgrad werden die Azide noch durch Umkristallistion oder
Umkondensation (möglichst kleine Apparatur) aufgereinigt. Die Lagerung erfolgt
unter Argonatmosphäre. Die flüssigen Azide werden, sofern sie nicht zügig weiter
verwendet werden, zusätzlich noch im Kühlschrank gelagert
B Cl
R
R
B
R
R
N3
TMSN3
DCM
-80 °C bis RT, 16 h

Experimenteller Teil: Synthesen und Photolysen
172
6.2.12 Synthese von Pinakolborazid PinBN3 (9)
Besonderheiten bei der Synthese
• Aufreinigung (Umkondensation) i. d. R. nicht notwendig
Ausbeute
• m = 4.71 (90 %), farbloses Öl
Charakterisierung
1H‐NMR (C6D6): δ = 1.00 ppm (s, 12 H, CH3)
13C‐NMR (C6D6): δ = 25.6 ppm (CH3), 86.0 (OC)
11B‐NMR (C6D6): δ = 25.2 ppm
IR (Nujol): 2158 cm‐1 (νmax)
Name der Chemikalie Menge
PinBCl m = 5.00 g (M = 162.42 g/mol, n = 30.78 mmol)
TMSN3 V = 5.50 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 41.82 mmol, 1.4 Äq.)
DCM (abs.) V = 60 mL
OB
ON3 9 M = 169.99 g/mol

Experimenteller Teil: Synthesen und Photolysen
173
6.2.13 Synthese von Diisopropyloxyborazid (iPrO)2BN3 (150‐(iPr)2)
Besonderheiten bei der Synthese
• Aufreinigung i. d. R. nicht notwendig
Ausbeute
• m = 8.32 (96 %), weißer FS (ggf. auch viskoses Öl)
Charakterisierung
1H‐NMR (CDCl3): δ = 1.15 ppm (d, 12 H, OCH(CH3)2), 4.32 (sep, 2 H, OCH(CH3)2),
13C‐NMR (CDCl3): δ = 24.4 ppm (OCH(CH3)2), 67.3 (OCH(CH3)2)
11B‐NMR (CDCl3): δ = 20.5 ppm
IR (Nujol): 2151 cm‐1 (νmax)
Name der Chemikalie Menge
(iPrO)2BCl m = 8.50 g (M = 164.44 g/mol, n = 50.69 mmol)
TMSN3 V = 10.00 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 76.04 mmol, 1.5 Äq.)
DCM (abs.) V = 100 mL
B
O
O
N3
150-(iPr)2
M = 171.01 g/mol

Experimenteller Teil: Synthesen und Photolysen
174
6.2.14 Synthese von Diethoxyborazid (EtO)2BN3 (150‐(Et)2)
Besonderheiten bei der Synthese
• Nur leichtes Vakuum beim Entfernen des LM und TMSCl anlegen, da das
Produkt (EtO)2BN3 einen niedrigen Siedepunkt besitzt und ansonsten ebenfalls
entfernt wird.
• Wegen der Instabilität sollte das Produkt im Kühlschrank gelagert werden.
Ausbeute
• 4.03 g (29 %, nach Umkondensation), farbloses viskoses Öl
Charakterisierung
1H‐NMR (CD2Cl2): δ = 1.15 ppm (t, 6 H, OCH2CH3), 3.51 (q, 4 H, OCH2CH3)
13C‐NMR (CD2Cl2): δ = 24.2 ppm (OCH2CH3), 69.3 (OCH2CH3)
11B‐NMR (CD2Cl2): δ = 20.9 ppm
Name der Chemikalie Menge
(EtO)2BCl m = 13.20 g (M = 136.39 g/mol, n = 96.78 mmol)
TMSN3 V = 13.40 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 101.89 mmol, 1.05 Äq.)
DCM (abs.) V = 120 mL
B
O
O
N3
150-(Et)2
M = 142.95 g/mol

Experimenteller Teil: Synthesen und Photolysen
175
OB
OOB
ON3N3
171
M = 243.74 g/mol
6.2.15 Synthese des Bisazids 171
Besonderheiten bei der Synthese
• Produkt wird mit Benzol, Pentan (je 10 mL) gewaschen
Ausbeute
• 1.53 g (95 %), leicht gelb‐grünlicher FS
Charakterisierung
1H‐NMR (CDCl3): δ = 6.53 ppm (s, 2 H, ArH).
13C‐NMR (CDCl3): δ = 101.4 ppm (ArC), 146.3 (ArCOBN3R)
11B‐NMR (CDCl3) δ = 25.1 ppm
IR (KBr): 2149 cm‐1 (νmax)
Name der Chemikalie Menge
Bischlorid 175 m = 1.52 g ( M = 230.61 g/mol, n = 6.59 mmol)
TMSN3 V = 1.40 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 10.65 mmol, 1.6 Äq.)
DCM (abs.) V = 30 mL

Experimenteller Teil: Synthesen und Photolysen
176
OB
ON3
177
M = 210.98 g/mol
6.2.16 Synthese von 2‐Azido‐Naphto(2,3‐d)‐1,3,2‐dioxaborol (177)
Besonderheiten bei der Synthese
• Produkt wird mit Pentan (je 2 x 10 mL) gewaschen
Ausbeute
• 1,33 g (90 %), grau‐weißer FS
Charakterisierung
1H‐NMR (CD2Cl2): 7.13‐7.19 ppm (m, 4 H, ArH), 7.55‐7.59 (m, 2 H, ArH),
13C‐NMR (CD2Cl2): δ = 111.0 ppm (ArC), 122.5 (ArC) , 125.9 (ArC), 128.6 (ArC),
141.2 (ArCOBN3R)
11B‐NMR (CD2Cl2): δ = 25.1 ppm
Name der Chemikalie Menge
2‐Chloro‐Naphto(2,3‐d)‐
1,3,2‐dioxaborol
m = 1.43 g (M = 204.42 g/mol, n = 7.00 mmol)
TMSN3 V = 1.30 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 9.89 mmol, 1.4 Äq.)
DCM (abs.) V = 30 mL

Experimenteller Teil: Synthesen und Photolysen
177
OB
ON3 M = 217.03 g/mol
7-(tBu)
6.2.17 Synthese von tert‐BuCatBN3 (7‐(tBu))
Besonderheiten bei der Synthese
• Umkristallisation aus Hexan
Ausbeute
• 2.18 g (77% ), weiß‐grauer FS
Charakterisierung
1H‐NMR (CD2Cl2): δ = 1.28 ppm (s, 12 H, C(CH3)3), 6.48‐6.77 (m, 3 H, ArH)
13C‐NMR (CD2Cl2): δ = 30.0 ppm (ArC(CH3)3), 32.7 (ArC(CH3)3), 107.9 (ArC), 110.1
(ArC), 117.5 (ArC), 137.0 (ArC(CH3)3), 139.5 (ArCOBN3R)), 140.3 (ArCOBN3R)
11B‐NMR (CH2Cl2): δ = 25.8 ppm
IR (NaBr): 2160 cm‐1 (νmax)
Name der Chemikalie Menge
tert‐BuCatBCl m = 2.75 g (M = 210.47 g/mol, n = 13.07 mmol)
TMSN3 V = 2.00 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 15.2 mmol, 1.2 Äq.)
DCM (abs.) V = 40 mL

Experimenteller Teil: Synthesen und Photolysen
178
NB
N
CH3
CH3
N3 191 M = 138.97 g/mol
6.2.18 Synthese von 2‐Azido‐1,3‐dimethyl‐1,3,2‐diazaborolidin (191)[108]
Besonderheiten bei der Synthese
• Bei der Umkondensation ggf. leichtes Erwärmen des Azids notwendig
Ausbeute
• 5.12 g (65 % ) nach Umkondensation, leicht gelbliches Öl
Charakterisierung
1H‐NMR (CDCl3): δ = 2.55 ppm (s, 6 H, CH3), 3.16 (s, 4 H, CH2)
13C‐NMR (CDCl3): δ = 33.6 ppm (CH3), 51.4 (CH2)
11B‐NMR (CDCl3): δ = 23.5 ppm
MS (GC/MS): m/z (%): 138 (100)[M +∙], 123 (4), 110 (5), 95 (6), 83 (28), 69 (34), 67
(29), 55 (18), 54 (20), 42 (51), 40 (32), 28 (23)
IR (NaBr): 2153 cm‐1 (νmax)
Name der Chemikalie Menge
2‐Chloro‐1,3‐dimethyl‐
1,3,2‐diazaborolidin
m = 7.50 g ( M = 132.20 g/mol, n = 56.73 mmol)
TMSN3 V = 10.00 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 76.04 mmol, 1.3 Äq.)
DCM (abs.) V = 60 mL

Experimenteller Teil: Synthesen und Photolysen
179
NB
N
Tos
Tos
N3 192 M = 419.29 g/mol
6.2.19 Synthese von 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin (192)
Besonderheiten bei der Synthese
• Umkristallisation aus DCM, nachwaschen mit Pentan
Ausbeute
• 5.78 g (76% ), weißer, kristalliner FS
Charakterisierung
1H‐NMR (CDCl3): δ = 2.42 ppm (s, 6 H, ArCH3), 3.71 (s, 4 H, CH2), 7.33 (d, 4 H, ArH),
7.81 (d, 4 H, ArH)
13C‐NMR (CDCl3): δ = 21.5 ppm (ArCH3), 42.4 (CH2), 127.2 (ArC), 129.6 (ArC), 135.2
(ArC), 143.8 (ArC)
11B‐NMR (CDCl3): δ = 21.8 ppm
IR (Nujol): 2148 cm‐1 (νmax)
Kristallstruktur (Daten siehe Anhang)
Name der Chemikalie Menge
2‐Chloro‐1,3‐ditosyl‐
1,3,2‐diazaborolidin
m = 7.50 g ( M = 412.72 g/mol, n = 18.17 mmol)
TMSN3 V = 3.00 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 22.81 mmol, 1.3 Äq.)
DCM (abs.) V = 120 mL

Experimenteller Teil: Synthesen und Photolysen
180
SB
SN3 193 M = 145.01 g/mol
6.2.20 Synthese von 2‐Azido‐1,3,2‐dithiaborolan (193)
Besonderheiten bei der Synthese
• Äußerst sauberes Arbeiten wegen der Geruchsintensivität unerlässlich.
• Geräte nach Gebrauch mit Natriumhypochlorit‐Lsg. reinigen.
Ausbeute
• 3.55 g (94 %, ohne Aufreinigung), leicht gelblicher FS
Charakterisierung
1H‐NMR (CDCl3): δ = 3.44 ppm (s, 4 H, CH2)
13C‐NMR (CDCl3): δ = 37.6 ppm (CH2)
11B‐NMR (CDCl3): δ = 55.6 ppm
Name der Chemikalie Menge
2‐Chloro‐1,3,2‐
dithiaborolan 117
m = 3.60 g ( M = 138.45 g/mol, n = 26.00 mmol)
TMSN3 V = 5.00 mL (M = 115.21 g/mol, d = 0.876 g/mL, c =
7.604 mmol/mL, n = 38.02 mmol, 1.5 Äq.)
DCM (abs.) V = 60 mL

Experimenteller Teil: Synthesen und Photolysen
181
6.2.21 Versuche zur Synthese von Boryltria‐ und tetrazolen via „Click‐Chemie“
Vorbemerkung
Die Testreaktionen wurden in verschraubbaren kleinen Glasgefäßen (V = 25 mL)
durchgeführt, welche die zeitgleiche parallele Durchführung mehrerer Versuche
und systematische Variation der Reaktionsparameter erlauben.
Durchführung
Zu einer Lösung (verwendete LM: Cyclohexan, Toluol, Acetonitril, THF) des
betreffenden Borazids (CatBN3, PinBN3, m = 200‐300 mg) erfolgt unter
Argonatmosphäre die Zugabe stöchiometrischer Mengen eines Nitrils bzw. Alkins.
Zusätzlich wird ggf. noch ein Cu‐Kat (CuI, CuBr, Cu(NHC)Br) addiert. Die
Reaktionsmischung wird bei variablen Temperaturen (RT ‐ Siedepunkt des
betreffenden LM) gerührt (1 h ‐ 14 d). In regelmäßigen Zeitabständen werden
kleine Proben (ca. 1 mL) entnommen, alkoholysiert (iPrOH) oder acetyliert und
die Mischung mittels GC/MS analysiert.
XR´B N3
R
R X
NN
N
R´
H
X = CH, N
1)
2) iPrOH
R´= Ph, C6F6
(Cu)-Kat

Experimenteller Teil: Synthesen und Photolysen
182
6.3 Photolysen
6.3.1 Allgemeine Vorschrift für Photolysen in Lösung
Duchführung der Photolyse
Ein stark verdünnnte Lösung (ggf. Suspension) des betreffenden Azidoborans
(etwa 5‐10 mg Azid pro mL Substrat) wird bei RT unter Argonatmosphäre
fortlaufend mit einer Quecksilberniederdrucklampe (λ = 254 nm) in einem
Quarzrohr photolysiert (Dauer 0.5‐48 h, in Abhängigkeit vom eingesetzten
Azidoboran und verwendeten Substrat). Die Reaktionslösung wird anschließend
in der Glovebox filtriert (D‐3‐Fritte) und das überschüssige Substrat im Vakuum
entfernt. Der aus dem Filtrat verbleibende Rückstand wird dann spektroskopisch
analysiert (NMR‐Messungen) und ggf. durch Sublimation weiter aufgereinigt.
6.3.2 Allgemeine Vorschrift für die Aufarbeitung der Photoprodukte (Abbau‐ und Derivatisierungsexperimente)
Nach beendeter Photolyse wird das Substrat ohne vorherige Filtration im
Vakuum entfernt (Vereinigung der löslichen und unlöslichen Photoprodukte).
Anschließend wird das Rohprodukt entweder alkoholysiert oder acetyliert und
die Mischung mittels GC/MS und/oder GC analysiert.
6.3.2.1 Abbau durch Alkoholyse
Der vereinte RS wird unter Argonatmosphäre in trockenem Alkohol (i. d. R.
iPrOH) gelöst und die Reaktionsmischung für 30 Minuten bei Zimmertemperatur
gerührt. Falls sich das Rohprodukt nicht lösen sollte, wird die Reaktionsmischung
in einer verschlossenen Apparatur zusätzlich für 2 h auf 60 °C erhitzt. Die Bildung
der freien Abbauprodukte wird anschließend mittels GC/MS‐ und/oder GC‐

Experimenteller Teil: Synthesen und Photolysen
183
Messungen vorgenommen. Ggf. wird noch ein interner Standard zur
Quantifizierung zugegeben.
6.3.2.2 Acetylierung
Der gesamte vereinte RS wird in trockenem Et2O oder THF (etwa 20 mL) unter
Argonatmosphäre gelöst. Nach Zugabe katalytischer Mengen (m = 10‐20 mg)
DMAP (N,N´‐Dimethylaminopyridin) und AcCl (i. d. R. leichter Überschuss
bezogen auf eingesetztes Borazid), wird die resultierende Suspension für 18
Stunden bei RT gerührt. Nach Zugabe von festem NaOH (1.5‐3 Äq. bezogen auf
eingesetztes AcCl) wird die Reaktionsmischung für einen weiteren Tag
nachgerührt. Anschließend erfolgt ggf. die Zugabe einer Lösung des internen
Standards. Die Mischung wird filtriert und der Feststoff mit Et2O bzw. THF (3 10
mL) gewaschen. Das Filtrat wird abgetrennt und mittels GC/MS und/oder GC‐
Messungen analysiert.
6.3.3 Repräsentative Beispiele für Photolysen in Lösung Da die Durchführungen der Photoreaktionen sehr ähnlich sind, werden im
Folgenden lediglich eine Auswahl von Beispielen vorgestellt, die eindeutige
Ergebnisse lieferten.

Experimenteller Teil: Synthesen und Photolysen
184
6.3.3.1 Photolyse von PinBN3 (9) in Cycloalkanen Cy‐H.
Details zur Durchführung
• Photolysedauer 16‐18 h
• Filtration nach Photolyse
• Sublimation von PinBNHCy (Ölpumpenvakuum, Badtemperatur 60‐80 °C)
Ausbeuten
• 84‐92 % (Details siehe Tabelle 6)
Charakterisierung
PinBNH‐Cy5
1H‐NMR (CDCl3): δ 1.21 ppm (s, 12H, CH3), 1.34‐1.92 (m, 8H, CyAlkyl‐CH2), 2.18 (s,
1H, BNHCH) 3.41‐3.60 (m, 1H, BNHCH)
13C‐NMR (CDCl3): δ = 24.9 ppm (CH3), 37.6 (CyAlkyl‐CH2), 36.6 (CyAlkyl‐CH2), 53.2
(BNHCH), 82.2 (CO)
11B‐NMR (CDCl3): δ = 24.9 ppm
Name der Chemikalie Menge
PinBN3 9 m = 0.250 g (M = 169.99, n = 1.48 mmol)
Cy‐H V = 30‐35 mL (Überschuss)
OB
ON3
Cy H
OB
ON
H
Cy
1329
CyH =
h (= 254 nm)

Experimenteller Teil: Synthesen und Photolysen
185
PinBNH‐Cy6
1H‐NMR (C6D6): δ = 1.22 ppm (s, 12H, CH3), 1.30‐1.84 (m, 10H, CyAlkyl‐CH2), 2.20
(s, 1H, BNHCH), 2.97‐3.86 (m, 1H, BNHCH)
13C‐NMR (C6D6): δ = 24.3 ppm (CH3), 25.0 (CyAlkyl‐CH2), 25.5 (CyAlkyl‐CH2), 36.9
(CyAlkyl‐CH2), 49.2 (BNHCH), 81.5 (CO)
11B‐NMR (C6D6): δ = 25.1 ppm
PinBNH‐Cy7
1H‐NMR (C6D6): δ = 1.21 ppm (s, 12H, CH3), 1.38‐1.83 (m, 12H, CyAlkyl‐CH2), 2.23
(s, 1H, BNHC), 3.09‐3.18 (m, 1H, BNHCH)
13C‐NMR (C6D6): δ = 24.3 ppm (CH3), 25.5 (CyAlkyl‐CH2), 27.9 (CyAlkyl‐CH2), 38.7
(CyAlkyl‐CH2), 51.4 (BNHCH), 81.4 (CO)
11B‐NMR (C6D6): δ = 25.0 ppm
(HO)2BNH‐Cy7
1H‐NMR (CDCl3): δ = 1.36‐1.49 ppm (m, 2H), 1.51‐1.63 (m, 4H, CyAlkyl‐CH2), 1.70‐
1.80 (m, 4H, CyAlkyl‐CH2), 2.10‐2.15 (m, 4H, CyAlkyl‐CH2), 3.27‐3.34 (m, 1H,
BNHCH), 8.26 (s (breit), 2H, BOH)
13C‐NMR (CDCl3): δ = 25.3 ppm (CyAlkyl‐CH2), 27.8 (CyAlkyl‐CH2), 33.6 (CyAlkyl‐
CH2), 53.7 (BNHCH),
11B‐NMR (CDCl3): δ = 20.9 ppm

Experimenteller Teil: Synthesen und Photolysen
186
6.3.3.1.1 Aufarbeitung von PinBNHCy (132)
Abbau durch Alkoholyse
Details zur Durchführung
• RS wird in Pentan (abs.) gelöst und mit iPrOH versetzt
• Zugabe einer Pentan‐Lösung des internen Standards:
Cy‐7‐NH2 ist interner Standard im Fall von Cy‐5‐NH2, Cy‐6‐NH2, Cy‐8‐NH2
Cy‐5‐NH2 ist interner Standard für Bestimmung von Cy‐7‐NH2
Ausbeuten
• 71‐83 % (Details siehe Tabelle 6)
Name der Chemikalie Menge
iPrOH V = 10 mL (Überschuss)
Cy‐7‐NH2 in Pentan
(interner Standard)
V = 10.00 mL (c = 0.100 mmol/mL, n = 1.00 mmol )
Cy‐6‐NH2 in Pentan
(interner Standard)
V = 10.00 mL (c = 0.100 mmol/mL, n = 1.00 mmol )
OB
ON
Cy
H
HN
H
Cy
132
ROH 134
(Überschuss)

Experimenteller Teil: Synthesen und Photolysen
187
6.3.3.1.2 Derivatisierung durch Acetylierung
Details zur Durchführung
• vgl. allgemeine Vorschrift der Acetylierung
Ausbeuten
• 75‐83 % (Details siehe Tabelle 6)
Name der Chemikalie Menge
Acetylchlorid
V = 0.50 mL (M = 94.18 g/mol, d = 1.123 g/mL,
c = 11.924 mmol/mL, n = 0.200 mol)
DMAP m = 10‐20 mg (kat. Mengen)
NaOH m = 1.00 g (M = 40 g/mol, n = 25 mmol), Überschuss
Hexamethylbenzol
(interner Standard)
m = 0.240 g (M = 162.27 g/mol, n= 1.48 mmol)
Et2O (abs.) V = 70 mL
OB
ON
Cy
H
R
O
Cl
R
O
N
H
Cy
132
1)
2) NaOH (s)
, DMAP (kat.)
- PinBOH141

Experimenteller Teil: Synthesen und Photolysen
188
6.3.3.2 Photolyse von (iPrO)2BN3 (150‐(iPr)2) in Cyclohexan
Details zur Durchführung
• Photolysedauer 16 h
• Alkoholyse des kompletten RS, Addition von Pentan
• Zugabe von Cy‐7‐NH2 (interner Standard) gelöst in Pentan
Ausbeute
• Cy‐6‐NH2 (134‐(Cy6)) = 46 %
• 1‐Amino‐propan‐2‐ol (151‐(Me)) = 15‐20 %
Name der Chemikalie Menge
(iPrO)BN3 m = 0.209 g (M = 171.01 g/mol, n = 1.22 mmol)
Cy‐6‐H (abs.) V = 30 mL
iPrOH V = 5 mL (Überschuss)
Pentan (abs.) V = 15 mL
Cy‐7‐NH2 in Pentan
(interner Standard)
V = 10 mL (c = 0.100 mmol/mL, n = 1.00 mmol)
iPrO
iPrOB N3
NH2 OH
CH3
NH2
+
1) h ,16 h
2) iPrOH
Cy-6-H
134-(Cy6) 151-(Me)
150-(iPr)2

Experimenteller Teil: Synthesen und Photolysen
189
6.3.3.3 Photolyse von (EtO)2BN3 (150‐(Et)2) in Cyclohexan
Details zur Durchführung
• Photolysedauer 16 h
• Alkoholyse des kompletten RS, Addition von THF
• Zugabe von Cy‐7‐NH2 (interner Standard) gelöst in Pentan
Ausbeute
• Cy‐6‐NH2 (134‐(Cy6)) = 37 %
• 2‐Amino‐ethanol (151‐(H)) = 10‐15 %
Name der Chemikalie Menge
(EtO)BN3 m = 0.217 g (M = 142.95 g/mol, n = 1.52 mmol)
Cy‐6‐H (abs.) V = 30 mL
EtOH V = 5 mL (Überschuss)
THF (abs.) V = 15 mL
Cy‐7‐NH2 in Pentan
(interner Standard)
V = 10 mL (c = 0.100 mmol/mL, n = 1.00 mmol)
B N3
EtO
EtO NH2 OH
NH2
+
1) h ,16 h
2) EtOH
Cy-6-H
134-(Cy6) 151-(H)
150-(Et)2
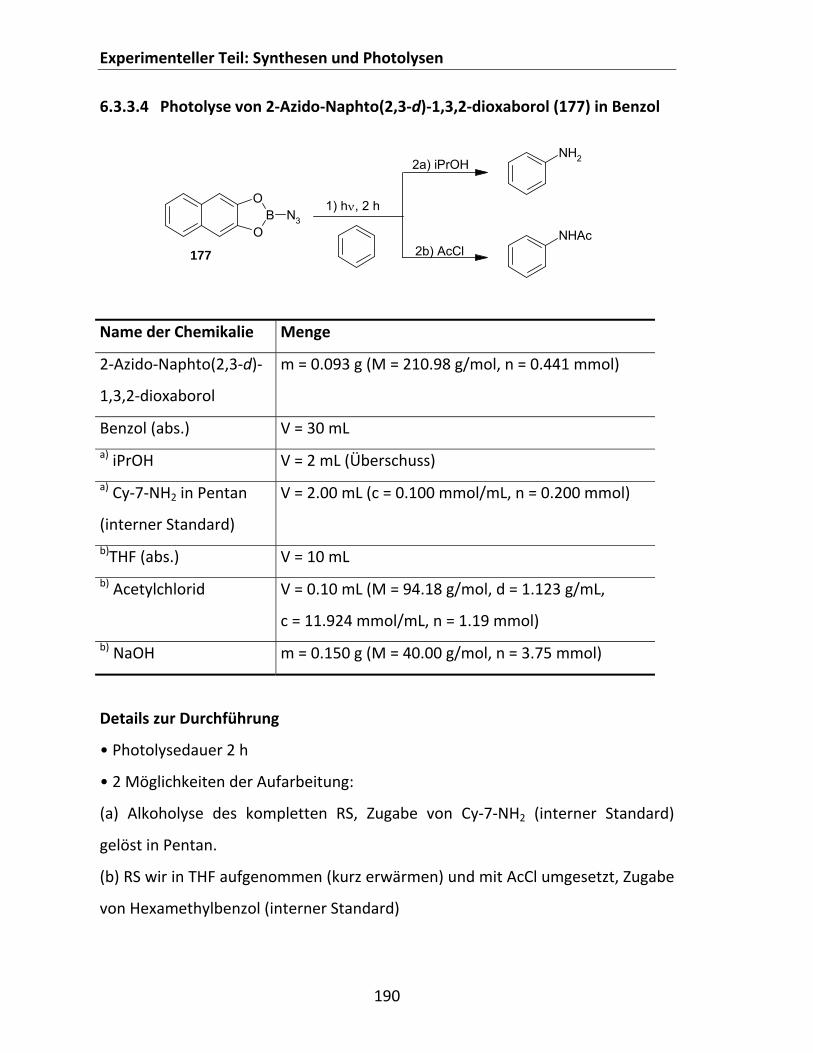
Experimenteller Teil: Synthesen und Photolysen
190
OB
ON3
NH2
NHAc
177
1) h , 2 h
2b) AcCl
2a) iPrOH
6.3.3.4 Photolyse von 2‐Azido‐Naphto(2,3‐d)‐1,3,2‐dioxaborol (177) in Benzol
Details zur Durchführung
• Photolysedauer 2 h
• 2 Möglichkeiten der Aufarbeitung:
(a) Alkoholyse des kompletten RS, Zugabe von Cy‐7‐NH2 (interner Standard)
gelöst in Pentan.
(b) RS wir in THF aufgenommen (kurz erwärmen) und mit AcCl umgesetzt, Zugabe
von Hexamethylbenzol (interner Standard)
Name der Chemikalie Menge
2‐Azido‐Naphto(2,3‐d)‐
1,3,2‐dioxaborol
m = 0.093 g (M = 210.98 g/mol, n = 0.441 mmol)
Benzol (abs.) V = 30 mL
a) iPrOH V = 2 mL (Überschuss)
a) Cy‐7‐NH2 in Pentan
(interner Standard)
V = 2.00 mL (c = 0.100 mmol/mL, n = 0.200 mmol)
b)THF (abs.) V = 10 mL
b) Acetylchlorid V = 0.10 mL (M = 94.18 g/mol, d = 1.123 g/mL,
c = 11.924 mmol/mL, n = 1.19 mmol)
b) NaOH m = 0.150 g (M = 40.00 g/mol, n = 3.75 mmol)

Experimenteller Teil: Synthesen und Photolysen
191
Ausbeute
• Ph‐NH2 (≈11 %) bei Alkoholyse
• Ph‐NHAc (≈15 %) bei Acetylierung

Experimenteller Teil: Synthesen und Photolysen
192
6.3.4 Gasphasenphotolyse: Aminierung von Methan
6.3.4.1 Vorbemerkung
Die Gasphasenphotoreaktion zwischen PinBN3 und Methan ist im Rahmen der
experimentellen Arbeiten stetig weiterentwickelt worden, um die Ausbeute an
MeNH2 bzw. PinBNHMe zu erhöhen. Die durchgeführten Modifikationen
betreffen sowohl den apparativen Aufbau selbst, als auch unterschiedliche
Strategien beim Aufarbeitungsvorgang der Reaktionsprodukte. Dennoch sind
Parametereinstellungen nicht als optimiert anzusehen.
6.3.4.2 Apparativer Aufbau
Die Gasphasenphotolyseapparatur ist eine aus mehreren Elementen bestehende
Strömungsvorrichtung (vgl. Abb.): Die wesentlichen Bestandteile bilden dabei ein
Druckminderer und „Flow‐Controler“, eine Trocknungseinheit, eine
Mischkammer, sowie ein Photolyserohr mit zugehöriger UV‐Quelle.
OB
ON3
OB
ON
H
CH3CH3 H
132-(Me)9
h (= 254 nm)

Experimenteller Teil: Synthesen und Photolysen
193
Druckminderer/Flow‐Controler“
Die Strömungsgeschwindigkeit des Methangases (Qualität mind. 4.5) wird über
einen Druckminderer sowie einen „Flow‐Controler“ reguliert. Eine Strömungs‐
und Reaktionsdauer zwischen 36‐48 h hat sich empirisch als ideal erwiesen.
Zusätzlich sind noch weitere Gaseinlassventile in die Vorrichtung vorhanden, die
beispielsweise die Zugabe von Inertgas (z. B. Argon) erlauben.v
Trocknungseinheit
Die zusammengesetzte Trocknungseinheit sorgt dafür, dass das im Methan
befindliche Restwasser entfernt wird. Das Methangas (Qualität mind. 4.5) wird
hier zunächst durch eine gekühlte (‐78 °C) Glasspirale (Durchmesser = 0.5 cm,
Länge = 6 m) geleitet, was zum Ausfrieren des mittransportierten Wassers führt.
Ggf. verbliebene Wasserspuren werden dann durch zwei nachgeschaltete gefüllte
Trockentürme (Molsieb (3 Å) / Silicagel) entfernt. Da sich das Gas während dieses
Vorgangs stark abkühlt, wird es ‐ bevor die Einleitung in das Borazid erfolgt ‐
durch eine leicht beheizte Glasspirale gleicher Bauart (30‐35 °C) geleitet, um
wieder auf Zimmertemperatur erwärmt zu werden. Zusätzlich kann bei Bedarf
auch die Methangasflasche direkt gekühlt und ein großer Teil des Restwassers
bereits in der Gasflasche einkondensiert werden.
Mischkammer
Die Mischkammer besteht aus einem zugespitzten Schlenkrohr (Länge ≈ 15 cm)
mit Gaseinleitungshahn in der sich eine flüssige Probe von PinBN3 9 befindet.
v Da die Vorrichtung es nicht erlaubt, dass das eingesetzte Methan im Kreislauf geführt wird, wird diese wegen seiner Brennbarkeit vor dem Austritt aus der Apparatur mit Inertgas vermischt. Auch die Aufarbeitung muss wegen der Hydrolyseempfindlichkeit der Photoprodukte unter Inertgasatmosphäre stattfinden.

Experimenteller Teil: Synthesen und Photolysen
194
Durch den Methangasstrom wird das Azid kontinuierlich ausgetrieben
(Verdunstung) und in das Photolyserohr überführt.
Photoreaktor
Der Photorektor besteht aus einem Quarzohr (Durchmesser = 1.7 cm, Länge = 55
cm) der von einer Quecksilberniederdrucklampe (λ = 254 nm, Belichtungszone ≈
20‐25 cm Länge) umgeben wird.
6.3.4.3 Durchführung der Photolyse
Methangas wird durch eben beschriebene Apparatur geleitet und in eine flüssige
Probe von PinBN3 9 (m = 3.00 g, M = 169.99 g/mol, 17.568 mmol) eingeströmt.
Das resultierende Methan/Azid‐Gasgemisch wird durch ein Quarzrohr geführt
und kontinuierlich mittels einer Hg‐Niederdruck Lampe (λ = 254 nm) fortlaufend
für 36‐48 h photolysiert. Dabei scheidet sich im Zuge der Belichtung ‐ innerhalb
und hinter der Belichtungszone ‐ aus den zuvor gasförmigen Edukten ein harziger,
leicht gelblicher Feststoff ab. Die gesamte Vorrichtung wird nach beendeter
Photolyse wird mit Argon gespült und der FS durch geschicktes Auswaschen mit
DCM unter Argongegenstrom in die nachgeschaltete, zuvor gekühlte Vorlage
gespült. Nach Entfernen des LM im Vakuum können die erhaltenen
Reaktionsprodukte direkt NMR‐Messungen und indirekt mithilfe von
Abbauexperimenten charakterisiert werden.

Experimenteller Teil: Synthesen und Photolysen
195
OB
ON3
OB
ON
Me
HNH
Me
O
R NH
MeTos
-N2
HCl (aq.)MeNH3
+ Cl-
CH4
MeNH2 (aq.) MeNH2 (g)
H2O
MeNH3+ OH-
H2O
NaOH (aq.)
1) RCOCl, DMAP (cat.)
2) NaOH (s)
1) H2O, HCl
2) TosCl, NaOH
6.3.4.4 Aufarbeitung der Photoprodukte
Für die Aufarbeitung des Reaktionsprodukts bestehen mehrere Möglichkeiten:
Hydrolyse liefert das freie Methylamin MeNH2. Über Variation des pH‐Wertes und
die Temperatur des Wassers kann die Gleichgewichtslage reguliert werden. Die
Acetylierung oder Tosylierung führen zu Amiden bzw. Tosylaten. Die Acetylierung
erfolgt direkt ausgehend vom Photorohprodukt, die Tosylierung kann auch vom
freien Methylamin aus vorgenommen werden. Die Derivatisierungsprodukte
können mittels GC/MS und/oder GC‐Messungen bestimmt werden, so dass
indirekte Rückschlüsse auf die Ausbeute an Methylamin MeNH2 und damit auf
den C‐H‐Aktivierungsgrad erhalten werden.

Experimenteller Teil: Synthesen und Photolysen
196
Acetylierung
Details zur Durchführung
• Photorohprodukt wird mit DCM unter Argonatmosphäre in die Vorlage gespült
• DC wird im Vakuum entfernt und RS in THF aufgenommen
• Acetylierung nach Vorschrift 6.3.3.1.2.
Ausbeute
• MeNHAc (6‐8 %)
• AcNH2, H2NNHAc, AcNHNHAc (i. d. R. zusammen < 5%)
Name der Chemikalie Menge
Acetylchlorid
V = 0.60 mL (M = 94.18 g/mol, d = 1.123 g/mL,
c = 11.924 mmol/mL, n = 8.44 mmol)
DMAP kat. Mengen
NaOH m = 0.50 g (M = 40 g/mol, n = 12.5 mmol),
Cy‐7‐NH2 in Pentan
(interner Standard)
V = 10.00 mL (c = 0.100 mmol/mL, n = 1.000 mmol)
DCM (abs.) nach Bedarf
THF (abs.) V = 20 mL

Experimenteller Teil: Synthesen und Photolysen
197
Tosylierung
Durchführung
Das Photolyseprodukt wird nach Beendigung der Belichtung zunächst mit DCM
aus dem Quarzrohr in die mit Salzsäure gefüllte Vorlage gespült und für 1 h bei RT
gerührt, anschließend für 3 h auf 50 °C erhitzt, wobei man das DCM abdampfen
lässt. Die wässrige Mischung wird dann bei 0 °C äußerst langsam (!) mit
Natronlauge versetzt, bevor TosCl gelöst in Ether (10 mL) zugegeben wird. Die
zweiphasige Mischung wird dann für 1 h bei 0 °C und für weitere 16 h bei RT
gerührt. Es erfolgt die Zugabe von Hexamethylbenzol in Et2O (interner Standard).
Die organische Phase wird abgetrennt, die Lösung mittels GC/MS bestimmt.
Ausbeute
• MeNHTos (6‐8 %)
• TosNH2, H2NNHTos, TosNHNHTos (nicht quantifizierbar)
Name der Chemikalie Menge
Tosylchlorid m = 0.80 g (M = 190.64 g/mol, n = 4.20 mmol)
HCl (1 M) V = 10 mL
NaOH (1 M) V = 20 mL
Cy‐7‐NH2 in Pentan
(interner Standard)
V = 10.00 mL (c = 0.100 mmol/mL, n = 1.000 mmol)
DCM (abs.) nach Bedarf
THF (abs.) V = 20 mL

Experimenteller Teil: Synthesen und Photolysen
198
C
H
C
H
C
H
C
NH2
C
NH2
C
NH2
1) (RO)2BN3
2) ROH oder H2O
h( = 254 nmArgon
6.3.5 Aminierung einer Polyethylen‐Oberfläche
Durchführung
Zunächst wird eine verdünnte Lösung eines flüssigen Borazids (z. B. (EtO)2BN3
150‐(Et)2, PinBN3 9) hergestellt. Das verwendete Lösungsmittel sollte dabei einen
niedrigen Siedepunkt aufweisen und sich durch Inertheit/Beständigkeit
gegenüber dem Borazid und dem PE auszeichnen (z. B. Pentan, DCM). Dann wird
eine penibel getrocknete Probe von Polyethylen (PE) in ein ausgeheiztes
Quarzrohr vorgelegt und mit der Borazidlösung versetzt. Dabei wird darauf
geachtet, dass ein möglichst gleichmäßiger Flüssigkeitsfilm an der PE‐Oberfläche
aufgetragen wird (homogene Benetzung).
Methode (I): Handelt es sich bei dem eingesetzten PE um eine Platte oder Folie,
wird diese (möglichst) waagerecht in das Photolyserohr eingeführt. Zusätzlich
besteht die Möglichkeit die Oberfläche leicht anzurauen, um eine bessere
Flüssigkeitshaftbarkeit zu erreichen.
Name der Chemikalie Menge
PE (Polyethylen) nach Bedarf
(RO)2BN3 nach Bedarf
trockenes LM: z. B.
Pentan oder DCM
nach Bedarf
Alkohol oder Wasser nach Bedarf

Experimenteller Teil: Synthesen und Photolysen
199
Methode (II): Findet PE‐Pulver Verwendung wird dieses gleichmäßig mit der
Borazidlösung vermengt, sodass eine Suspension/Paste entsteht. Durch leichtes
Schwenken des Quarzrohrs wird dann versucht die Probe gleichmäßig an der der
Innenwand haften zu lassen.
Anschließend wird das LM im Vakuum entfernt, wobei sich die Stärke und Dauer
des Vakuums nach der Flüchtigkeit des eingesetzten Borazids richtet. Dann wird
die Borazid beschichtete PE‐Probe für 1‐16 h fortlaufend unter Argonatmosphäre
mit einer Quecksilberniederdrucklampe (λ = 254 nm) belichtet. Nach Beendigung
der Photolyse wird der Feststoff mit Alkohol oder Wasser gewaschen, um die
freien NH2‐Funktion zu erhalten.
Analyse
IR: Hinweise auf NH2‐Gruppen

Anhang
200
7 Anhang
7.1 Kristallstrukturdaten General
Origin Code p-1 Database dates
Common name PinBNHCy‐6 Systematic name Structural formula
Analytical formula
Bibliographic data
Author(s) Publication title Citation Mineral name Compound source
Structure type Creation method SHELXL-97 Comments
Phase data Formula sum C12 H24 B N O2 Formula weight 225.13 g/mol Crystal system triclinic Space-group P -1 (2)
Cell parameters a=5.9731(5) Å b=10.7650(9) Å c=11.0285(8) Å α=79.999(7)° β=84.840(7)° γ=75.462(7)°
Cell ratio a/b=0.5549 b/c=0.9761 c/a=1.8464 Cell volume 675.20(144) Å3 Z 2 Calc. density 1.10727 g/cm3 Meas. density Melting point RAll 0.0792 RObs Pearson code aP80 Formula type NOP2Q12R24 Wyckoff sequence i40
Atomic parameters
Atom Ox. Wyck. Site S.O.F. x/a y/b z/c U [Å2] C11 2i 1 0.4734(5) 0.3425(2) 0.3286(3) C12 2i 1 0.6598(6) 0.3996(3) 0.3680(3) H12A 2i 1 0.61020 0.49460 0.35290 0.0460 H12B 2i 1 0.80470 0.37120 0.32030 0.0460 H12C 2i 1 0.68420 0.36950 0.45590 0.0460
Anhang
201
C13 2i 1 0.4400(6) 0.3842(3) 0.1916(3) H13A 2i 1 0.30110 0.36160 0.17010 0.0490 H13B 2i 1 0.57540 0.33970 0.14530 0.0490 H13C 2i 1 0.42200 0.47840 0.17080 0.0490 C14 2i 1 0.5136(5) 0.1932(2) 0.3735(3) C15 2i 1 0.7645(5) 0.1193(3) 0.3795(3) H15A 2i 1 0.84350 0.15560 0.43400 0.0440 H15B 2i 1 0.83950 0.12690 0.29670 0.0440 H15C 2i 1 0.77330 0.02750 0.41180 0.0440 C16 2i 1 0.3801(6) 0.1280(3) 0.3037(3) H16A 2i 1 0.38050 0.04090 0.34830 0.0470 H16B 2i 1 0.45350 0.12120 0.22120 0.0470 H16C 2i 1 0.22020 0.17970 0.29650 0.0470 O1 2i 1 0.4107(3) 0.18865(16) 0.49956(17) O2 2i 1 0.2584(4) 0.39449(17) 0.39676(18) B1 2i 1 0.2479(6) 0.3060(3) 0.5036(3) N1 2i 1 0.0950(5) 0.3323(2) 0.6032(2) H1 2i 1 -0.006(4) 0.4070(16) 0.606(3) 0.025(8) C21 2i 1 0.0658(5) 0.2428(2) 0.7151(3) H21 2i 1 0.18610 0.15990 0.71140 0.0300 C22 2i 1 0.1042(6) 0.2964(3) 0.8289(3) H22A 2i 1 -0.00020 0.38380 0.82870 0.0370 H22B 2i 1 0.26570 0.30560 0.82500 0.0370 C23 2i 1 0.0594(6) 0.2088(3) 0.9487(3) H23A 2i 1 0.17820 0.12550 0.95480 0.0440 H23B 2i 1 0.07270 0.25110 1.01970 0.0440 C24 2i 1 -0.1785(6) 0.1823(3) 0.9544(3) H24A 2i 1 -0.29770 0.26440 0.95730 0.0420 H24B 2i 1 -0.19890 0.12150 1.03060 0.0420 C25 2i 1 -0.2115(6) 0.1241(3) 0.8434(3) H25A 2i 1 -0.37090 0.11180 0.84740 0.0370 H25B 2i 1 -0.10220 0.03790 0.84480 0.0370 C26 2i 1 -0.1704(5) 0.2118(3) 0.7245(3) H26A 2i 1 -0.29200 0.29390 0.71910 0.0330 H26B 2i 1 -0.18380 0.16900 0.65380 0.0330
Anisotropic displacement parameters, in Å2
Atom U11 U22 U33 U12 U13 U23
C11 0.026(2) 0.0173(15) 0.0304(17) -0.0043(12) 0.0023(14) -0.0070(12) C12 0.031(2) 0.0185(15) 0.0445(19) -0.0108(12) 0.0023(16) -0.0065(13) C13 0.039(2) 0.0248(16) 0.0313(18) -0.0066(13) 0.0029(15) -0.0028(13) C14 0.020(2) 0.0169(14) 0.0256(16) -0.0070(11) 0.0083(13) -0.0049(11) C15 0.026(2) 0.0208(15) 0.0377(18) -0.0019(12) 0.0034(15) -0.0066(13) C16 0.034(2) 0.0226(15) 0.0409(19) -0.0118(13) 0.0017(16) -0.0109(14) O1 0.0246(15) 0.0147(10) 0.0308(12) -0.0031(8) 0.0029(9) -0.0027(8) O2 0.0223(14) 0.0152(10) 0.0303(12) -0.0021(7) 0.0047(9) -0.0038(8) B1 0.028(2) 0.0129(15) 0.0312(18) -0.0081(13) -0.0029(16) -0.0040(14) N1 0.0271(18) 0.0148(12) 0.0306(14) 0.0006(10) 0.0044(12) -0.0023(11) C21 0.028(2) 0.0144(14) 0.0315(17) -0.0043(11) 0.0019(14) -0.0013(12)

Anhang
202
C22 0.034(2) 0.0220(15) 0.0377(19) -0.0089(13) -0.0066(16) -0.0015(13) C23 0.051(3) 0.0313(17) 0.0304(17) -0.0129(15) -0.0075(17) -0.0026(14) C24 0.046(3) 0.0259(16) 0.0290(17) -0.0082(14) 0.0089(16) -0.0020(13) C25 0.030(2) 0.0248(16) 0.0366(19) -0.0090(13) 0.0052(15) -0.0022(13) C26 0.031(2) 0.0210(15) 0.0314(17) -0.0095(12) 0.0013(14) -0.0048(13)
Selected geometric informations
Atoms 1,2 d 1,2 [Å] Atoms 1,2 d 1,2 [Å] C11—O2 1.462(4) B1—N1 1.386(4) C11—C13 1.516(4) N1—C21 1.454(4) C11—C12 1.526(4) C21—C26 1.520(4) C11—C14 1.562(4) C21—C22 1.526(4) C14—O1 1.466(3) C22—C23 1.529(4) C14—C15 1.511(4) C23—C24 1.512(5) C14—C16 1.517(4) C24—C25 1.516(4) O1—B1 1.392(4) C25—C26 1.516(4) O2—B1 1.388(4) B1—N1i 6.0993(47)
Atoms 1,2,3 Angle 1,2,3 [°] Atoms 1,2,3 Angle 1,2,3 [°] O2—C11—C13 109.2(2) B1—O2—C11 106.6(2) O2—C11—C12 107.0(2) N1—B1—O2 123.8(3) C13—C11—C12 110.1(3) N1—B1—O1 124.0(3) O2—C11—C14 102.3(2) O2—B1—O1 112.2(3) C13—C11—C14 114.8(2) B1—N1—C21 126.9(2) C12—C11—C14 112.9(2) N1—C21—C26 111.4(2) O1—C14—C15 108.3(2) N1—C21—C22 110.9(2) O1—C14—C16 106.9(2) C26—C21—C22 110.9(3) C15—C14—C16 110.5(2) C21—C22—C23 112.3(2) O1—C14—C11 102.0(2) C24—C23—C22 111.3(2) C15—C14—C11 115.1(2) C23—C24—C25 111.2(3) C16—C14—C11 113.3(2) C24—C25—C26 110.9(2) B1—O1—C14 107.0(2) C25—C26—C21 112.7(2)
Atoms 1,2,3,4 Tors. an. 1,2,3,4 [°] Atoms 1,2,3,4 Tors. an. 1,2,3,4 [°]
O2—C11—C14—O1 -30.9(2) C11—O2—B1—O1 -12.5(3) C13—C11—C14—O1 -149.1(2) C14—O1—B1—N1 172.1(3) C12—C11—C14—O1 83.7(3) C14—O1—B1—O2 -8.8(3) O2—C11—C14—C15 -147.9(2) O2—B1—N1—C21 176.6(3) C13—C11—C14—C15 94.0(3) O1—B1—N1—C21 -4.4(5) C12—C11—C14—C15 -33.2(4) B1—N1—C21—C26 -113.1(3) O2—C11—C14—C16 83.6(3) B1—N1—C21—C22 122.8(3) C13—C11—C14—C16 -34.5(4) N1—C21—C22—C23 176.3(2) C12—C11—C14—C16 -161.7(3) C26—C21—C22—C23 52.0(3) C15—C14—O1—B1 146.2(2) C21—C22—C23—C24 -54.0(4) C16—C14—O1—B1 -94.7(2) C22—C23—C24—C25 55.9(3) C11—C14—O1—B1 24.5(3) C23—C24—C25—C26 -56.4(3) C13—C11—O2—B1 148.7(2) C24—C25—C26—C21 55.4(3) C12—C11—O2—B1 -92.2(2) N1—C21—C26—C25 -177.0(2) C14—C11—O2—B1 26.7(3) C22—C21—C26—C25 -53.0(3) C11—O2—B1—N1 166.6(3) (i) 1-x, 1-y, 1-z.
Selected hydrogen bonds
Extended geometric informations

Anhang
203
General Origin Code p-1 Database dates
Common name PinBNHCy‐5 Systematic name Structural formula
Analytical formula
Bibliographic data
Author(s) Publication title Citation Mineral name Compound source Structure type Creation method SHELXL-97 Comments
Phase data Formula sum C11 H22 B N O2 Formula weight 211.11 g/mol Crystal system triclinic Space-group P -1 (2)
Cell parameters a=6.0622(5) Å b=10.1366(9) Å c=10.556(1) Å α=84.346(7)° β=75.625(7)° γ=86.723(7)°
Cell ratio a/b=0.5981 b/c=0.9603 c/a=1.7413 Cell volume 624.94(119) Å3 Z 2 Calc. density 1.12182 g/cm3 Meas. density Melting point RAll 0.0559 RObs Pearson code aP74 Formula type NOP2Q11R22 Wyckoff sequence i37
Atomic parameters
Atom Ox. Wyck. Site S.O.F. x/a y/b z/c U [Å2] B1 2i 1 0.2499(3) 0.49689(16) 0.30709(15) O1 2i 1 0.41964(15) 0.49426(9) 0.19318(9) O2 2i 1 0.25091(15) 0.38267(9) 0.39105(9) C11 2i 1 0.4634(2) 0.30917(14) 0.33909(14) C12 2i 1 0.4187(3) 0.16241(14) 0.36731(15) H12A 2i 1 0.38900 0.13830 0.46210 0.0490
Anhang
204
H12B 2i 1 0.55230 0.11130 0.32210 0.0490 H12C 2i 1 0.28600 0.14260 0.33600 0.0490 C13 2i 1 0.6373(2) 0.34740(16) 0.40939(15) H13A 2i 1 0.66510 0.44240 0.38960 0.0480 H13B 2i 1 0.78010 0.29650 0.37970 0.0480 H13C 2i 1 0.57900 0.32800 0.50430 0.0480 C14 2i 1 0.5212(2) 0.36011(13) 0.19009(14) C15 2i 1 0.7732(2) 0.37019(15) 0.12533(15) H15A 2i 1 0.79320 0.40830 0.03440 0.0460 H15B 2i 1 0.84670 0.28160 0.12630 0.0460 H15C 2i 1 0.84260 0.42720 0.17340 0.0460 C16 2i 1 0.4054(3) 0.28506(15) 0.10850(15) H16A 2i 1 0.24330 0.27770 0.15220 0.0460 H16B 2i 1 0.47650 0.19610 0.09910 0.0460 H16C 2i 1 0.42170 0.33290 0.02150 0.0460 N1 2i 1 0.0930(2) 0.60231(11) 0.33496(11) H1 2i 1 -0.005(2) 0.6083(15) 0.4113(8) 0.034(4) C21 2i 1 0.0815(2) 0.72022(14) 0.24696(14) H21 2i 1 0.19540 0.70860 0.16190 0.0300 C22 2i 1 0.1291(3) 0.84939(15) 0.29760(16) H22A 2i 1 0.05390 0.85150 0.39210 0.0410 H22B 2i 1 0.29500 0.85900 0.28510 0.0410 C23 2i 1 0.0306(3) 0.95946(16) 0.21586(19) H23A 2i 1 0.15380 1.00220 0.14680 0.0540 H23B 2i 1 -0.05210 1.02790 0.27220 0.0540 C24 2i 1 -0.1333(3) 0.89225(15) 0.15378(16) H24A 2i 1 -0.28440 0.93890 0.17120 0.0450 H24B 2i 1 -0.07210 0.89260 0.05760 0.0450 C25 2i 1 -0.1520(2) 0.75092(15) 0.21802(15) H25A 2i 1 -0.18240 0.68910 0.15780 0.0370 H25B 2i 1 -0.27480 0.74510 0.29990 0.0370
Anisotropic displacement parameters, in Å2
Atom U11 U22 U33 U12 U13 U23 B1 0.0240(9) 0.0218(9) 0.0229(8) -0.0039(7) -0.0081(7) -0.0021(7) O1 0.0268(6) 0.0203(5) 0.0255(5) 0.0020(4) -0.0029(4) -0.0017(4) O2 0.0249(6) 0.0216(6) 0.0251(5) 0.0020(4) -0.0032(4) -0.0009(4) C11 0.0229(8) 0.0208(8) 0.0279(8) 0.0033(6) -0.0057(6) -0.0041(6) C12 0.0379(10) 0.0235(8) 0.0355(9) 0.0012(7) -0.0063(7) -0.0005(7) C13 0.0318(9) 0.0369(9) 0.0297(8) 0.0016(7) -0.0105(7) -0.0037(7) C14 0.0247(8) 0.0194(8) 0.0261(8) 0.0017(6) -0.0061(6) -0.0052(6) C15 0.0262(9) 0.0307(9) 0.0337(9) -0.0005(6) -0.0032(7) -0.0056(7) C16 0.0319(9) 0.0311(9) 0.0308(8) -0.0011(6) -0.0095(7) -0.0070(7) N1 0.0284(7) 0.0222(7) 0.0219(7) 0.0018(5) -0.0021(6) -0.0010(5) C21 0.0281(8) 0.0200(8) 0.0250(8) 0.0005(6) -0.0031(6) 0.0001(6) C22 0.0363(9) 0.0250(9) 0.0444(10) -0.0040(7) -0.0156(8) -0.0001(7) C23 0.0519(11) 0.0253(9) 0.0637(12) -0.0024(8) -0.0260(9) 0.0020(8) C24 0.0463(10) 0.0268(9) 0.0415(10) 0.0025(7) -0.0174(8) 0.0022(7) C25 0.0359(9) 0.0254(8) 0.0351(9) -0.0020(7) -0.0140(7) -0.0009(7)

Anhang
205
General Origin
Code PinBNHCy‐7 Database dates Common name Systematic name Structural formula
Analytical formula
Bibliographic data
Author(s) Publication title Citation Mineral name Compound source
Structure type Creation method SHELXL-97 Comments
Phase data Formula sum C13 H26 B N O2 Formula weight 239.16 g/mol Crystal system triclinic Space-group P -1 (2)
Cell parameters a=6.1606(7) Å b=10.5917(13) Å c=11.0973(16) Å α=80.183(17)° β=87.508(11)° γ=76.776(10)°
Cell ratio a/b=0.5816 b/c=0.9544 c/a=1.8013 Cell volume 694.58(261) Å3 Z 2 Calc. density 1.14345 g/cm3 Meas. density Melting point RAll 0.1098 RObs Pearson code aP92 Formula type NOP2Q13R26 Wyckoff sequence i46
Atomic parameters
Atom Ox. Wyck. Site S.O.F. x/a y/b z/c U [Å2] C1 2i 1 0.4083(5) 0.2715(3) 0.7920(3) H1 2i 1 0.31840 0.35800 0.80910 0.0290 C2 2i 1 0.2966(7) 0.2377(6) 0.6867(4) H2A 2i 1 0.34260 0.14090 0.69370 0.0790
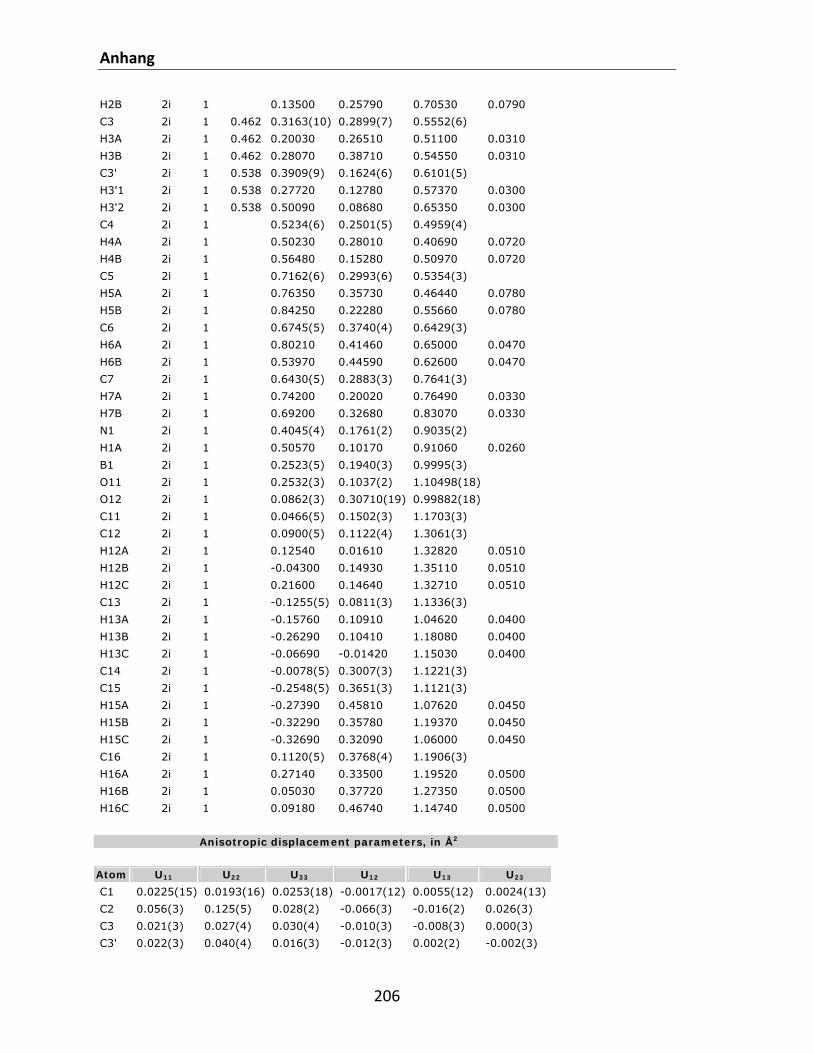
Anhang
206
H2B 2i 1 0.13500 0.25790 0.70530 0.0790 C3 2i 1 0.462 0.3163(10) 0.2899(7) 0.5552(6) H3A 2i 1 0.462 0.20030 0.26510 0.51100 0.0310 H3B 2i 1 0.462 0.28070 0.38710 0.54550 0.0310 C3' 2i 1 0.538 0.3909(9) 0.1624(6) 0.6101(5) H3'1 2i 1 0.538 0.27720 0.12780 0.57370 0.0300 H3'2 2i 1 0.538 0.50090 0.08680 0.65350 0.0300 C4 2i 1 0.5234(6) 0.2501(5) 0.4959(4) H4A 2i 1 0.50230 0.28010 0.40690 0.0720 H4B 2i 1 0.56480 0.15280 0.50970 0.0720 C5 2i 1 0.7162(6) 0.2993(6) 0.5354(3) H5A 2i 1 0.76350 0.35730 0.46440 0.0780 H5B 2i 1 0.84250 0.22280 0.55660 0.0780 C6 2i 1 0.6745(5) 0.3740(4) 0.6429(3) H6A 2i 1 0.80210 0.41460 0.65000 0.0470 H6B 2i 1 0.53970 0.44590 0.62600 0.0470 C7 2i 1 0.6430(5) 0.2883(3) 0.7641(3) H7A 2i 1 0.74200 0.20020 0.76490 0.0330 H7B 2i 1 0.69200 0.32680 0.83070 0.0330 N1 2i 1 0.4045(4) 0.1761(2) 0.9035(2) H1A 2i 1 0.50570 0.10170 0.91060 0.0260 B1 2i 1 0.2523(5) 0.1940(3) 0.9995(3) O11 2i 1 0.2532(3) 0.1037(2) 1.10498(18) O12 2i 1 0.0862(3) 0.30710(19) 0.99882(18) C11 2i 1 0.0466(5) 0.1502(3) 1.1703(3) C12 2i 1 0.0900(5) 0.1122(4) 1.3061(3) H12A 2i 1 0.12540 0.01610 1.32820 0.0510 H12B 2i 1 -0.04300 0.14930 1.35110 0.0510 H12C 2i 1 0.21600 0.14640 1.32710 0.0510 C13 2i 1 -0.1255(5) 0.0811(3) 1.1336(3) H13A 2i 1 -0.15760 0.10910 1.04620 0.0400 H13B 2i 1 -0.26290 0.10410 1.18080 0.0400 H13C 2i 1 -0.06690 -0.01420 1.15030 0.0400 C14 2i 1 -0.0078(5) 0.3007(3) 1.1221(3) C15 2i 1 -0.2548(5) 0.3651(3) 1.1121(3) H15A 2i 1 -0.27390 0.45810 1.07620 0.0450 H15B 2i 1 -0.32290 0.35780 1.19370 0.0450 H15C 2i 1 -0.32690 0.32090 1.06000 0.0450 C16 2i 1 0.1120(5) 0.3768(4) 1.1906(3) H16A 2i 1 0.27140 0.33500 1.19520 0.0500 H16B 2i 1 0.05030 0.37720 1.27350 0.0500 H16C 2i 1 0.09180 0.46740 1.14740 0.0500
Anisotropic displacement parameters, in Å2
Atom U11 U22 U33 U12 U13 U23 C1 0.0225(15) 0.0193(16) 0.0253(18) -0.0017(12) 0.0055(12) 0.0024(13) C2 0.056(3) 0.125(5) 0.028(2) -0.066(3) -0.016(2) 0.026(3) C3 0.021(3) 0.027(4) 0.030(4) -0.010(3) -0.008(3) 0.000(3) C3' 0.022(3) 0.040(4) 0.016(3) -0.012(3) 0.002(2) -0.002(3)
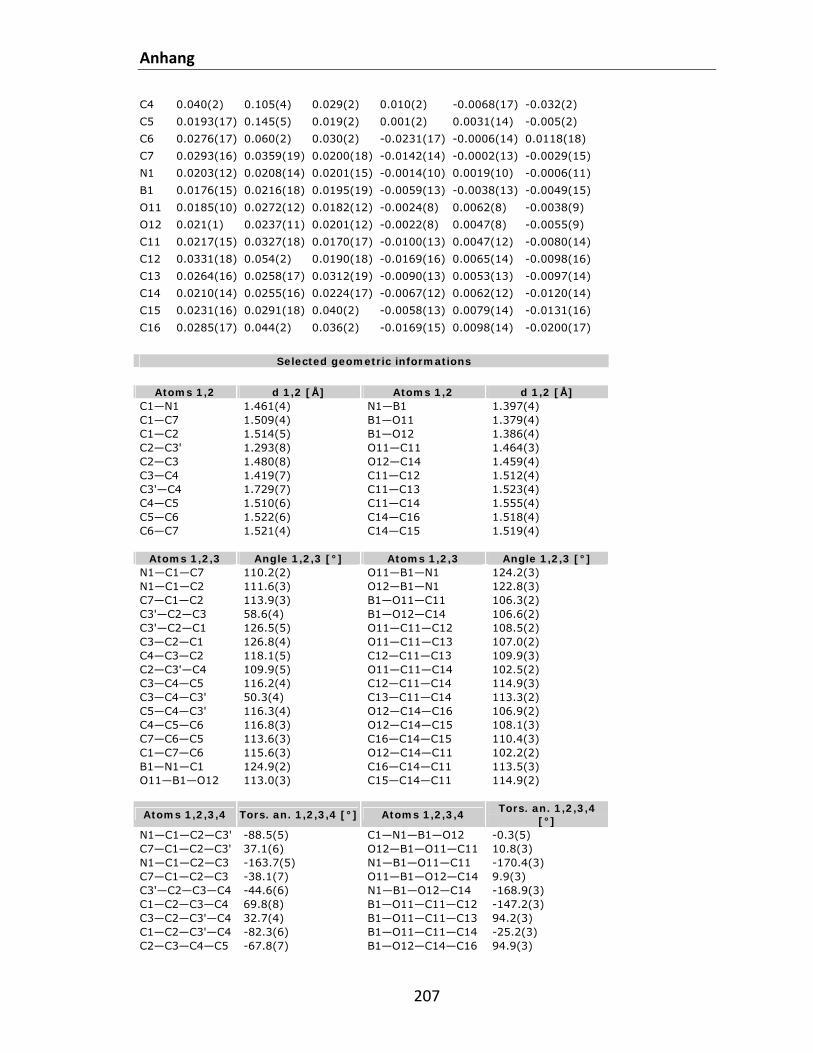
Anhang
207
C4 0.040(2) 0.105(4) 0.029(2) 0.010(2) -0.0068(17) -0.032(2)
C5 0.0193(17) 0.145(5) 0.019(2) 0.001(2) 0.0031(14) -0.005(2)
C6 0.0276(17) 0.060(2) 0.030(2) -0.0231(17) -0.0006(14) 0.0118(18) C7 0.0293(16) 0.0359(19) 0.0200(18) -0.0142(14) -0.0002(13) -0.0029(15) N1 0.0203(12) 0.0208(14) 0.0201(15) -0.0014(10) 0.0019(10) -0.0006(11) B1 0.0176(15) 0.0216(18) 0.0195(19) -0.0059(13) -0.0038(13) -0.0049(15) O11 0.0185(10) 0.0272(12) 0.0182(12) -0.0024(8) 0.0062(8) -0.0038(9) O12 0.021(1) 0.0237(11) 0.0201(12) -0.0022(8) 0.0047(8) -0.0055(9) C11 0.0217(15) 0.0327(18) 0.0170(17) -0.0100(13) 0.0047(12) -0.0080(14) C12 0.0331(18) 0.054(2) 0.0190(18) -0.0169(16) 0.0065(14) -0.0098(16) C13 0.0264(16) 0.0258(17) 0.0312(19) -0.0090(13) 0.0053(13) -0.0097(14) C14 0.0210(14) 0.0255(16) 0.0224(17) -0.0067(12) 0.0062(12) -0.0120(14) C15 0.0231(16) 0.0291(18) 0.040(2) -0.0058(13) 0.0079(14) -0.0131(16) C16 0.0285(17) 0.044(2) 0.036(2) -0.0169(15) 0.0098(14) -0.0200(17)
Selected geometric informations
Atoms 1,2 d 1,2 [Å] Atoms 1,2 d 1,2 [Å] C1—N1 1.461(4) N1—B1 1.397(4) C1—C7 1.509(4) B1—O11 1.379(4) C1—C2 1.514(5) B1—O12 1.386(4) C2—C3' 1.293(8) O11—C11 1.464(3) C2—C3 1.480(8) O12—C14 1.459(4) C3—C4 1.419(7) C11—C12 1.512(4) C3'—C4 1.729(7) C11—C13 1.523(4) C4—C5 1.510(6) C11—C14 1.555(4) C5—C6 1.522(6) C14—C16 1.518(4) C6—C7 1.521(4) C14—C15 1.519(4)
Atoms 1,2,3 Angle 1,2,3 [°] Atoms 1,2,3 Angle 1,2,3 [°] N1—C1—C7 110.2(2) O11—B1—N1 124.2(3) N1—C1—C2 111.6(3) O12—B1—N1 122.8(3) C7—C1—C2 113.9(3) B1—O11—C11 106.3(2) C3'—C2—C3 58.6(4) B1—O12—C14 106.6(2) C3'—C2—C1 126.5(5) O11—C11—C12 108.5(2) C3—C2—C1 126.8(4) O11—C11—C13 107.0(2) C4—C3—C2 118.1(5) C12—C11—C13 109.9(3) C2—C3'—C4 109.9(5) O11—C11—C14 102.5(2) C3—C4—C5 116.2(4) C12—C11—C14 114.9(3) C3—C4—C3' 50.3(4) C13—C11—C14 113.3(2) C5—C4—C3' 116.3(4) O12—C14—C16 106.9(2) C4—C5—C6 116.8(3) O12—C14—C15 108.1(3) C7—C6—C5 113.6(3) C16—C14—C15 110.4(3) C1—C7—C6 115.6(3) O12—C14—C11 102.2(2) B1—N1—C1 124.9(2) C16—C14—C11 113.5(3) O11—B1—O12 113.0(3) C15—C14—C11 114.9(2)
Atoms 1,2,3,4 Tors. an. 1,2,3,4 [°] Atoms 1,2,3,4 Tors. an. 1,2,3,4 [°]
N1—C1—C2—C3' -88.5(5) C1—N1—B1—O12 -0.3(5) C7—C1—C2—C3' 37.1(6) O12—B1—O11—C11 10.8(3) N1—C1—C2—C3 -163.7(5) N1—B1—O11—C11 -170.4(3) C7—C1—C2—C3 -38.1(7) O11—B1—O12—C14 9.9(3) C3'—C2—C3—C4 -44.6(6) N1—B1—O12—C14 -168.9(3) C1—C2—C3—C4 69.8(8) B1—O11—C11—C12 -147.2(3) C3—C2—C3'—C4 32.7(4) B1—O11—C11—C13 94.2(3) C1—C2—C3'—C4 -82.3(6) B1—O11—C11—C14 -25.2(3) C2—C3—C4—C5 -67.8(7) B1—O12—C14—C16 94.9(3)

Anhang
208
C2—C3—C4—C3' 35.6(5) B1—O12—C14—C15 -146.2(2) C2—C3'—C4—C3 -38.7(5) B1—O12—C14—C11 -24.7(3) C2—C3'—C4—C5 64.6(6) O11—C11—C14—O12 30.3(3) C3—C4—C5—C6 6.1(7) C12—C11—C14—O12 147.8(2) C3'—C4—C5—C6 -50.4(6) C13—C11—C14—O12 -84.7(3) C4—C5—C6—C7 68.8(5) O11—C11—C14—C16 -84.5(3) N1—C1—C7—C6 173.7(3) C12—C11—C14—C16 33.0(3) C2—C1—C7—C6 47.3(4) C13—C11—C14—C16 160.5(3) C5—C6—C7—C1 -86.3(4) O11—C11—C14—C15 147.1(3) C7—C1—N1—B1 133.0(3) C12—C11—C14—C15 -95.4(3) C2—C1—N1—B1 -99.4(4) C13—C11—C14—C15 32.1(4) C1—N1—B1—O11 -178.9(3)
Selected hydrogen bonds
Extended geometric informations
General Origin Code c2 Database dates
Common name 2‐Azido‐1,3‐ditosyl‐1,3,2‐diazaborolidin Systematic name Structural formula Analytical formula
Bibliographic data Author(s) Publication title Citation Mineral name Compound source Structure type Creation method SHELXL-97 Comments
Phase data Formula sum C16 H18 B N5 O4 S2 Formula weight 419.28 g/mol Crystal system monoclinic Space-group C 1 2 1 (5) Cell parameters a=20.875(5) Å b=8.8602(17) Å c=5.2408(11) Å β=102.626(18)° Cell ratio a/b=2.3560 b/c=1.6906 c/a=0.2511 Cell volume 945.88(383) Å3 Z 2 Calc. density 1.47204 g/cm3 Meas. density Melting point RAll 0.082 RObs Pearson code mC96 Formula type N2O4P5Q16R18...
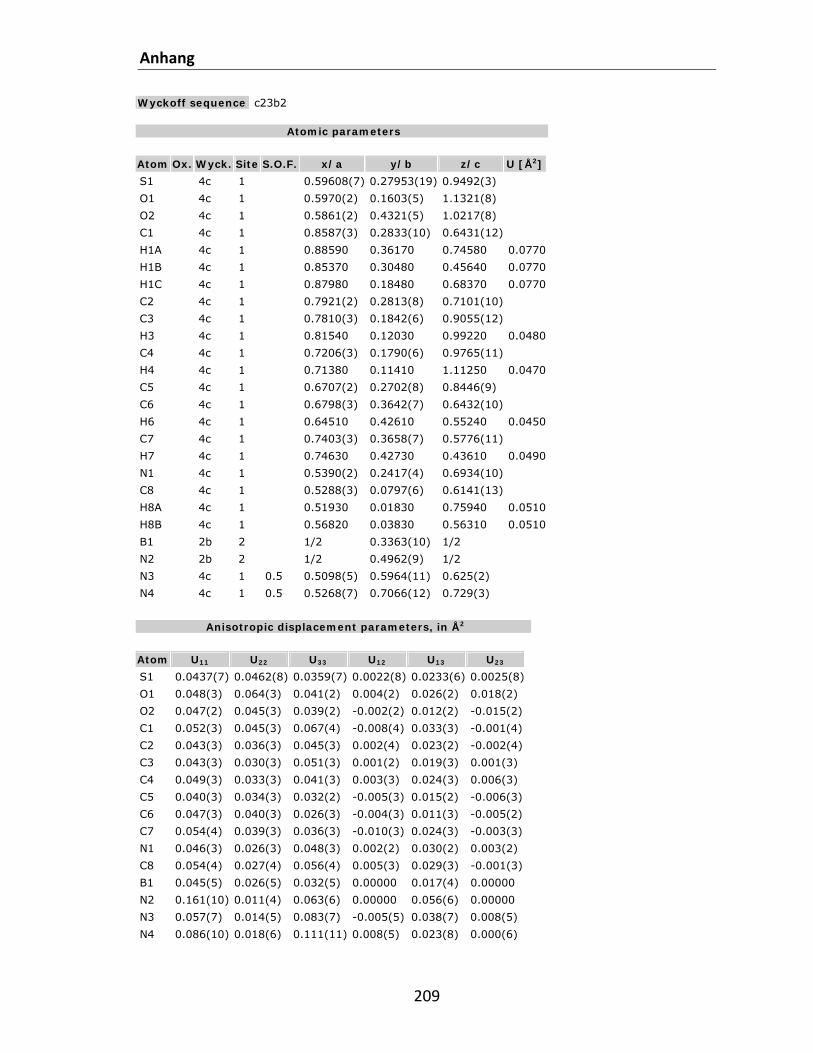
Anhang
209
Wyckoff sequence c23b2
Atomic parameters
Atom Ox. Wyck. Site S.O.F. x/a y/b z/c U [Å2] S1 4c 1 0.59608(7) 0.27953(19) 0.9492(3) O1 4c 1 0.5970(2) 0.1603(5) 1.1321(8) O2 4c 1 0.5861(2) 0.4321(5) 1.0217(8) C1 4c 1 0.8587(3) 0.2833(10) 0.6431(12) H1A 4c 1 0.88590 0.36170 0.74580 0.0770 H1B 4c 1 0.85370 0.30480 0.45640 0.0770 H1C 4c 1 0.87980 0.18480 0.68370 0.0770 C2 4c 1 0.7921(2) 0.2813(8) 0.7101(10) C3 4c 1 0.7810(3) 0.1842(6) 0.9055(12) H3 4c 1 0.81540 0.12030 0.99220 0.0480 C4 4c 1 0.7206(3) 0.1790(6) 0.9765(11) H4 4c 1 0.71380 0.11410 1.11250 0.0470 C5 4c 1 0.6707(2) 0.2702(8) 0.8446(9) C6 4c 1 0.6798(3) 0.3642(7) 0.6432(10) H6 4c 1 0.64510 0.42610 0.55240 0.0450 C7 4c 1 0.7403(3) 0.3658(7) 0.5776(11) H7 4c 1 0.74630 0.42730 0.43610 0.0490 N1 4c 1 0.5390(2) 0.2417(4) 0.6934(10) C8 4c 1 0.5288(3) 0.0797(6) 0.6141(13) H8A 4c 1 0.51930 0.01830 0.75940 0.0510 H8B 4c 1 0.56820 0.03830 0.56310 0.0510 B1 2b 2 1/2 0.3363(10) 1/2 N2 2b 2 1/2 0.4962(9) 1/2 N3 4c 1 0.5 0.5098(5) 0.5964(11) 0.625(2) N4 4c 1 0.5 0.5268(7) 0.7066(12) 0.729(3)
Anisotropic displacement parameters, in Å2
Atom U11 U22 U33 U12 U13 U23 S1 0.0437(7) 0.0462(8) 0.0359(7) 0.0022(8) 0.0233(6) 0.0025(8) O1 0.048(3) 0.064(3) 0.041(2) 0.004(2) 0.026(2) 0.018(2) O2 0.047(2) 0.045(3) 0.039(2) -0.002(2) 0.012(2) -0.015(2) C1 0.052(3) 0.045(3) 0.067(4) -0.008(4) 0.033(3) -0.001(4) C2 0.043(3) 0.036(3) 0.045(3) 0.002(4) 0.023(2) -0.002(4) C3 0.043(3) 0.030(3) 0.051(3) 0.001(2) 0.019(3) 0.001(3) C4 0.049(3) 0.033(3) 0.041(3) 0.003(3) 0.024(3) 0.006(3) C5 0.040(3) 0.034(3) 0.032(2) -0.005(3) 0.015(2) -0.006(3) C6 0.047(3) 0.040(3) 0.026(3) -0.004(3) 0.011(3) -0.005(2) C7 0.054(4) 0.039(3) 0.036(3) -0.010(3) 0.024(3) -0.003(3) N1 0.046(3) 0.026(3) 0.048(3) 0.002(2) 0.030(2) 0.003(2) C8 0.054(4) 0.027(4) 0.056(4) 0.005(3) 0.029(3) -0.001(3) B1 0.045(5) 0.026(5) 0.032(5) 0.00000 0.017(4) 0.00000 N2 0.161(10) 0.011(4) 0.063(6) 0.00000 0.056(6) 0.00000 N3 0.057(7) 0.014(5) 0.083(7) -0.005(5) 0.038(7) 0.008(5) N4 0.086(10) 0.018(6) 0.111(11) 0.008(5) 0.023(8) 0.000(6)
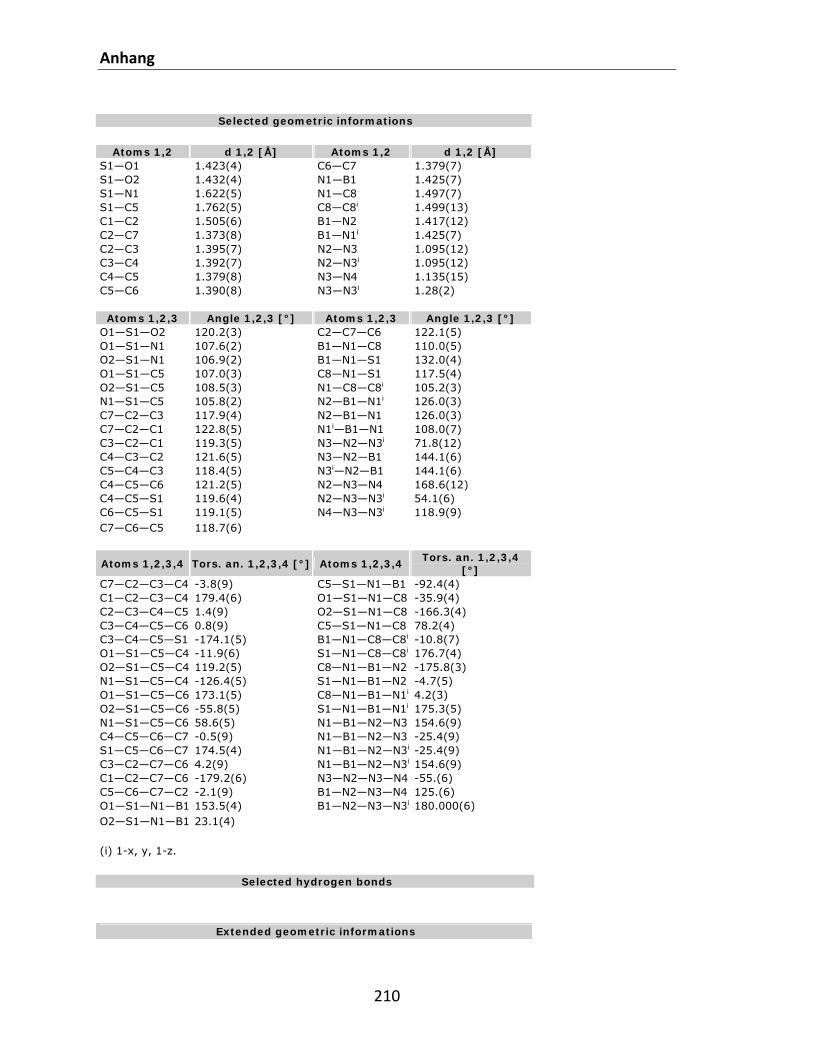
Anhang
210
Selected geometric informations
Atoms 1,2 d 1,2 [Å] Atoms 1,2 d 1,2 [Å]
S1—O1 1.423(4) C6—C7 1.379(7) S1—O2 1.432(4) N1—B1 1.425(7) S1—N1 1.622(5) N1—C8 1.497(7) S1—C5 1.762(5) C8—C8i 1.499(13) C1—C2 1.505(6) B1—N2 1.417(12) C2—C7 1.373(8) B1—N1i 1.425(7) C2—C3 1.395(7) N2—N3 1.095(12) C3—C4 1.392(7) N2—N3i 1.095(12) C4—C5 1.379(8) N3—N4 1.135(15) C5—C6 1.390(8) N3—N3i 1.28(2) Atoms 1,2,3 Angle 1,2,3 [°] Atoms 1,2,3 Angle 1,2,3 [°]
O1—S1—O2 120.2(3) C2—C7—C6 122.1(5) O1—S1—N1 107.6(2) B1—N1—C8 110.0(5) O2—S1—N1 106.9(2) B1—N1—S1 132.0(4) O1—S1—C5 107.0(3) C8—N1—S1 117.5(4) O2—S1—C5 108.5(3) N1—C8—C8i 105.2(3) N1—S1—C5 105.8(2) N2—B1—N1i 126.0(3) C7—C2—C3 117.9(4) N2—B1—N1 126.0(3) C7—C2—C1 122.8(5) N1i—B1—N1 108.0(7) C3—C2—C1 119.3(5) N3—N2—N3i 71.8(12) C4—C3—C2 121.6(5) N3—N2—B1 144.1(6) C5—C4—C3 118.4(5) N3i—N2—B1 144.1(6) C4—C5—C6 121.2(5) N2—N3—N4 168.6(12) C4—C5—S1 119.6(4) N2—N3—N3i 54.1(6) C6—C5—S1 119.1(5) N4—N3—N3i 118.9(9) C7—C6—C5 118.7(6)
Atoms 1,2,3,4 Tors. an. 1,2,3,4 [°] Atoms 1,2,3,4 Tors. an. 1,2,3,4 [°]
C7—C2—C3—C4 -3.8(9) C5—S1—N1—B1 -92.4(4) C1—C2—C3—C4 179.4(6) O1—S1—N1—C8 -35.9(4) C2—C3—C4—C5 1.4(9) O2—S1—N1—C8 -166.3(4) C3—C4—C5—C6 0.8(9) C5—S1—N1—C8 78.2(4) C3—C4—C5—S1 -174.1(5) B1—N1—C8—C8i -10.8(7) O1—S1—C5—C4 -11.9(6) S1—N1—C8—C8i 176.7(4) O2—S1—C5—C4 119.2(5) C8—N1—B1—N2 -175.8(3) N1—S1—C5—C4 -126.4(5) S1—N1—B1—N2 -4.7(5) O1—S1—C5—C6 173.1(5) C8—N1—B1—N1i 4.2(3) O2—S1—C5—C6 -55.8(5) S1—N1—B1—N1i 175.3(5) N1—S1—C5—C6 58.6(5) N1—B1—N2—N3 154.6(9) C4—C5—C6—C7 -0.5(9) N1—B1—N2—N3 -25.4(9) S1—C5—C6—C7 174.5(4) N1—B1—N2—N3i -25.4(9) C3—C2—C7—C6 4.2(9) N1—B1—N2—N3i 154.6(9) C1—C2—C7—C6 -179.2(6) N3—N2—N3—N4 -55.(6) C5—C6—C7—C2 -2.1(9) B1—N2—N3—N4 125.(6) O1—S1—N1—B1 153.5(4) B1—N2—N3—N3i 180.000(6) O2—S1—N1—B1 23.1(4) (i) 1-x, y, 1-z.
Selected hydrogen bonds
Extended geometric informations

Anhang
211

Anhang
212
7.2 Referenzen
[1] H. F. Bettinger, H. Bornemann, J. Am. Chem. Soc. 2006, 128, 11128. [2] H. F. Bettinger, M. Filthaus, H. Bornemann, I. M. Oppel, Angew. Chem. 2008, 120, 4822. [3] Z. Liu, T. B. Marder, Angew. Chem. 2008, 120, 248. [4] P. Paetzold, Pure & Appl. Chem. 1991, 63, 345. [5] A. F. Holleman, E. Wiberg, Lehrbuch der Anorganischen Chemie, 101 ed., Walter de Gruyter,
Berlin New York, 1995. [6] T. M. Klapötke, I. C. Tornieporth‐Oetting Nichtmetallchemie, Wiley‐VCH, Weinheim, 1994. [7] H. F. Bettinger, T. Kar, E. Garcia‐Sanchez, J. Phys. Chem. A 2009, 113, 353. [8] C. Y. Zhi, Y. Bando, C. C. Tang, Q. Huang, D. Golberg, J. Mater. Chem. 2008, 18, 3900. [9] M. Terrones, J. M. Romo‐Herrera, E. Cruz‐Silva, F. Lopez‐Urias, E. Munoz‐Sadoval, J. J. Velazquez‐
Salazar, H. Terrones, Y. Bando, D. Golberg, Materialstoday 2007, 10, 30. [10] O. Vostrowski, A. Hirsch, Chem. Rev. 2006, 106, 5191. [11] Z. Chen, R. B. King, Chem. Rev. 2005, 105, 3613. [12] R. Ma, D. Golberg, Y. Bando, T. Sasaki, Philos. Trans. R. Soc. London A 2004, 362, 2161. [13] V. V. Pokropivny, V. V. Skorokhod, G. S. Oleinik, A. V. Kurdyumov, T. S. Bartnitskaya, A. V.
Pokropivny, A. G. Sisonyuk, D. M. Sheichenko, J. Solid State Chem. 2000, 154, 214. [14] G. Seifert, P. W. Fowler, D. Mitchell, D. Porezag, T. Frauenheim, Chem. Phys. Lett. 1997, 268, 352. [15] J. Pattanayak, T. Kar, S. Scheiner, J. Phys. Chem. A 2002, 106, 2970. [16] T. Kar, J. Pattanayak, S. Scheiner, J. Phys. Chem. A 2003, 107, 8630. [17] X. F. Fan, Z. Zhu, Z. X. Shen, J.‐L. Kuo, J. Phys. Chem. C 2008, 12, 15691. [18] R. J. C. Batista, M. S. C. Mazzoni, H. Chacham, Chem. Phys. Lett. 2006, 421, 246. [19] S. S. Alexandre, M. S. C. Mazzoni, H. Chacham, Appl. Phys. Lett. 1999, 75, 61. [20] A. Stock, E. Pohland, Ber. Dtsch. Chem. Ges. 1926, 59, 2210. [21] A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon, S.‐Y. Liu, Angew. Chem. 2008, 120, 991. [22] T. B. Marder, Angew. Chem. 2007, 119, 8262. [23] F. H. Stephens, V. Pons, T. R. Baker, Dalton Trans. 2007, 2613. [24] C. W. Hamilton, T. R. Baker, A. Staubitz, I. Manners, Chem. Soc. Rev. 2009, 38, 279. [25] M. Dinca, J. R. Long, Angew. Chem. 2008, 120, 6870. [26] D. Zhao, D. Yuan, H.‐C. Zhou, Energy Environ. Sci. 2008, 1, 222. [27] U. Eberle, M. Felderhoff, F. Schüth, Angew. Chem. 2009, 121, 6732. [28] L. J. Murray, M. Dinca, J. R. Long, Chem. Soc. Rev. 2009, 38, 1294. [29] K. Niedenzu, Angew, Chem. 1964, 76, 168. [30] P. Paetzold, Adv. Inorg. Chem. 1987, 31, 123. [31] H. Nöth, Angew. Chem. 1988, 100, 1664. [32] J. Kiesgen, J. Münster, P. Paetzold, Chem. Ber. 1993, 126, 1559. [33] B. Thiele, P. Paetzold, U. Englert, Chem. Ber. 1992, 125, 2681. [34] P. Paetzold, R. Truppat, Chem. Ber. 1983, 116, 1531. [35] M. J. D. Bosdet, W. E. Piers, Can. J. Chem. 2008, 86, 8. [36] A. W. Laubengayer, P. C. Moews, R. F. Porter, J. Am. Chem. Soc. 1961, 83, 1337. [37] A. W. Laubengayer, O. T. Beachley, R. F. Porter, Inorg. Chem. 1965, 4, 578. [38] H. P. Ederle, H. Nöth, Eur. J. Inorg. Chem. 2009, 3398. [39] M. J. D. Bosdet, W. E. Piers, T. S. Sorensen, M. Parvez, Angew. Chem. 2007, 119, 5028. [40] X. Fang, H. Yang, J. W. Kampf, M. M. Holl, A. J. Ashe, Organometallics 2006, 25, 513. [41] M. J. S. Dewar, R. Jones, J. Am. Chem. Soc. 1968, 90, 2137. [42] C. Habben, A. Meller, Chem. Ber. 1984, 117, 2531. [43] N. Metzler, H. Nöth, Chem. Ber. 1995, 128, 711. [44] J. J. Eisch, B. Shaffi, A. L. Rheingold, J. Am. Chem. Soc. 1987, 109, 2526. [45] J. J. Eisch, B. Shafii, J. D. Odom, A. L. Rheingold, J. Am. Chem. Soc. 1990, 112, 1847. [46] C. Pues, A. Berndt, Angew. Chem. 1984, 96, 306. [47] J. J. Eisch, J. E. Galle, S. Kozima, J. Am. Chem. Soc. 1986, 108, 379.

Anhang
213
[48] H. Braunschweig, I. Fernandez, G. Frenking, T. Kupfer, Angew. Chem. 2008, 120, 1977. [49] C. Fan, W. E. Piers, M. Parvez, Angew. Chem. 2009, 121, 2999. [50] G. Varvounis, Boroles, Elsevier, 2008. [51] J. J. Eisch, J. E. Galle, B. Shafii, A. L. Rheingold, Organometallics 1990, 9, 2342. [52] L. G. Mercier, W. E. Piers, M. Parvez, Angew. Chem. 2009, 121, 6224. [53] H.‐S. Wu, H. Jiao, Z.‐X. Wang, P. v. R. Schleyer, J. Am. Chem. Soc. 2003, 125, 4428. [54] Y. Segawa, M. Yamashita, K. Nozaki, Science 2006, 314, 113. [55] H. Braunschweig, Angew. Chem. 2007, 119, 1990. [56] W. A. Herrmann, C. Köcher, Angew. Chem. 1997, 109, 2256. [57] W. Kirmse, Angew. Chem. 2004, 116, 1799. [58] M. S. Platz, Acc. Chem. Res. 1995, 28, 487. [59] H. F. Bettinger, J. Am. Chem. Soc. 2006, 128, 2534. [60] H. F. Bettinger, Chem. Commun. 2005, 2756. [61] H. F. Bettinger, R. I. Kaiser, J. Phys. Chem. A 2004, 108, 4576. [62] R. I. Kaiser, Y. T. Lee, H. F. Bettinger, Angew. Chem. 2002, 114, 2456. [63] R. Breslow, C. Yuan, J. Am. Chem. Soc. 1958, 80, 5991. [64] R. Breslow, J. Lockhart, H. Chang, J. Am. Chem. Soc. 1961, 83, 2375. [65] R. Breslow, H. Hover, H. Chang, J. Am. Chem. Soc. 1962, 84, 3168. [66] R. Breslow, J. T. Groves, J. Am. Chem. Soc. 1970, 92, 984. [67] R. Breslow, H. Chang, J. Am. Chem. Soc. 1961, 83, 2367. [68] R. Breslow, J. Am. Chem. Soc. 1957, 79, 5318. [69] T. L. Gilchrist, W. Rees, C., Carbene, Nitrene und Dehydroaromaten, UTB Hüthig, 1972. [70] C. Wentrup, Reaktive Zwischenstufen I. Radikale, Carbene, Nitrene, gespannte Ringe, Georg
Thieme Verlag, 1991. [71] R. A. Moss, M. S. Platz, M. Jones, Reactive Intermediate Chemistry, 1 ed., John Wiley & Sons,
2004. [72] H. Bock, R. Dammel, Angew. Chem. 1987, 99, 518. [73] W. Lwowski, Angew. Chem. 1967, 79, 922. [74] S. Bräse, C. Gil, K. Knepper, V. Zimmermann, Angew, Chem. 2005, 117, 5320. [75] S. Linke, G. T. Tisue, W. Lwowski, J. Am. Chem. Soc. 1967, 89, 6308. [76] G. Bucher, CRC Handbook of Organic Photochemistry and Photobiology 2004, 44/1. [77] J. Liu, S. Mandel, C. M. Hadad, M. S. Platz, J. Org. Chem. 2004, 69, 8583. [78] W. Lwowski, G. T. Tisue, J. Am. Chem. Soc. 1965, 87, 4022. [79] W. Lwowski, T. W. Mattingly, J. Am. Chem. Soc. 1965, 87, 1947. [80] G. T. Tisue, S. Linke, W. Lwowski, J. Am. Chem. Soc. 1967, 89, 6303. [81] J. Averdung, J. Mattay, D. Jacobi, W. Abraham, Tetrahedron 1995, 51, 2543. [82] M. Holzinger, O. Vostrowski, A. Hirsch, F. Hennrich, M. Kappes, R. Weiss, F. Jellen, Angew. Chem.
2001, 113, 4132. [83] M. Holzinger, J. Abraham, P. Whelan, R. Graupner, L. Ley, F. Hennrich, M. Kappes, A. Hirsch, J.
Am. Chem. Soc. 2003, 125, 8566. [84] M. Inagaki, T. Shingaki, T. Nagai, Chem. Lett. 1981, 1419. [85] P. A. S. Smith, B. B. Brown, J. Am. Chem. Soc. 1951, 73, 2435. [86] P. A. S. Smith, B. B. Bernard, J. Am. Chem. Soc. 1951, 73, 2438. [87] A. Reiser, H. Wagner, G. Bowes, Tetrahedron Lett. 1966, 23, 2635. [88] N. P. Gritsan, M. S. Platz, Chem. Rev. 2006, 106, 3844. [89] W. T. Bordon, N. P. Gritsan, C. M. Hadad, W. L. Karney, C. R. Kemnitz, M. S. Platz, Acc. Chem. Res.
2000, 33, 765. [90] N. P. Gritsan, M. S. Platz, Advances in Physical Organic Chemistry 2001, 36, 255. [91] A. Marcinek, M. S. Platz, J. Phys. Chem. 1993, 97, 12674. [92] B. A. Smith, C. J. Cramer, J. Am. Chem. Soc. 1996, 118, 5490. [93] W. L. Karney, W. T. Bordon, J. Am. Chem. Soc. 1997, 1997, 3347. [94] N. P. Gritsan, A. D. Gudmundsdottir, D. Tigelaar, Z. Zhu, W. L. Karney, C. M. Hadad, M. S. Platz, J.
Am. Chem. Soc. 2001, 123, 1951.

Anhang
214
[95] M. J. T. Young, M. S. Platz, J. Org. Chem. 1991, 56, 6403. [96] R. Poe, K. Schnapp, M. J. T. Young, J. Grayzar, M. S. Platz, J. Am. Chem. Soc. 1991, 114, 5054. [97] R. S. Pandurangi, K. Katti, V. K., C. L. Barnes, W. A. Volkert, R. R. Kuntz, J. Chem. Soc. Chem.
Commun. 1994, 1841. [98] G. Bertrand, J.‐P. Majoral, A. Baceirdo, Acc. Chem. Res. 1986, 19, 17. [99] A. Kuhn, W. Sander, Organometallics 1998, 17, 248. [100] C. G. Röser, Ph. D. thesis, Ruhr‐Universität Bochum (Bochum), 1999. [101] P. Maslak, J. Am. Chem. Soc. 1989, 111, 8201. [102] P. S. Drzaic, J. I. Brauman, J. Am. Chem. Soc. 1984, 106, 3443. [103] S.‐J. Kim, T. P. Hamilton, H. F. Schaefer III, J. Am. Chem. Soc. 1992, 114, 5359. [104] D. A. Hrovat, E. E. Waali, W. T. Borden, J. Am. Chem. Soc. 1992, 114, 8698. [105] O. Castell, V. M. Garcia, C. Bo, R. Caballol, J. Comput. Chem. 1996, 17, 42. [106] H. F. Bettinger, W. Sander, J. Am. Chem. Soc. 2003, 125, 9726. [107] M. Winkler, J. Phys. Chem. A 2008, 112, 8649. [108] W. Pieper, D. Schmitz, P. Paetzold, Chem. Ber. 1981, 114, 3801. [109] H. F. Bettinger, M. Filthaus, P. Neuhaus, Chem. Commun. 2009, 2186. [110] C. Kötting, Ph. D. thesis, Ruhr‐Universität Bochum (Bochum), 1999. [111] C. Kötting, W. Sander, J. Am. Chem. Soc. 1999, 121, 8891. [112] W. Sander, C. Kötting, Chem. Eur. J. 1999, 5, 24. [113] W. Sander, C. Kötting, R. Hübert, J. Phys. Org. Chem. 2000, 13, 561. [114] E. V. Anslyn, D. A. Dougherty, Modern Physical Organic Chemistry, Palgrave Macmillan, 2005. [115] A. Rauk, Orbital Interaction Theory of Organic Chemistry, Wiley & Sons, New York, 2001. [116] N. S. Isaacs, Reaktionszwischenstufen der Organischen Chemie, Verlag Chemie, Weinheim, 1980. [117] F. Jensen, Introduction to Computational Chemistry, Second Edition ed., Wiley, Weinheim, 2007. [118] T. M. Klapötke, A. Schulz, Quantenchemische Methoden in der Hauptgruppenchemie, Spektrum
Akademischer Verlag, Heidelberg Berlin Oxford, 1996. [119] C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, 2 ed., John Wiley &
Sons, Weinheim, 2004. [120] W. Koch, M. C. Holthausen, A Chemist´s Guide to Density Functional Theory, 2 ed., Wiley‐VCH,
Weinheim, 2001. [121] S. M. Bachrach, Computational Organic Chemistry, John Wiley & Sons
Hoboken, 2007. [122] P. J. Stevens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch, J. Phys. Chem. 1994, 98, 11623. [123] C. Lee, W. Yang, R. G. Parr, Phys. Rev. B 1988, 37, 785. [124] A. D. Becke, J. Chem. Phys. 1993, 98, 5648. [125] A. D. Becke, Phys. Rev. A 1988, 38, 3098. [126] M. L. McKee, J. Phys. Chem. 1994, 98, 13243. [127] J.‐G. Zhang, Q. S. Li, S.‐W. Zhang, Int. J. Quant. Chem. 2005, 103, 422. [128] R. C. Gaussian 03, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.;
Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al‐Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; and Pople, J. A.; Gaussian, Inc., Wallingford CT, 2004.
[129] M. M. Gallo, H. F. Schaefer III, J. Chem. Phys. 1990, 93, 865. [130] B. S. Jursic, Int. J. Quant. Chem. 1997, 62, 515.

Anhang
215
[131] M. J. Frisch, R. Krishnan, J. A. Pople, P. v. R. Schleyer, Chem. Phys. Lett. 1981, 81, 421. [132] B. J. DeLeeuw, J. T. Fermann, Y. Xie, H. F. Schaefer III, J. Am. Chem. Soc. 1993, 115, 1039. [133] J. Breidung , W. Thiel, J. Mol. Spectros. 2001, 205, 28. [134] R. L. Schwartz, G. E. Davico, T. M. Ramond, W. C. Lineberger, J. Phys. Chem. A 1999, 103, 8213. [135] C. J. Barden, H. F. Schaefer III, J. Chem. Phys. 2000, 112, 6515. [136] S. E. Worthington, C. J. Cramer, J. Phys. Org. Chem. 1997, 10, 755. [137] E. P. F. Lee, J. M. Dyke, T. G. Wright, Chem. Phys. Lett. 2000, 326, 143. [138] I. S. Ingatyev, H. F. Schaefer III, J. Am. Chem. Soc. 1997, 119, 12306. [139] F. Kober, Grundlagen der Komplexchemie, Otto Sale Verlag / Verlag Saueränder, Frankfurt a. M.,
1979. [140] L. H. Gade, Koordinationschemie, Wiley‐VCH, Weinheim, 1998. [141] C. Elschenbroich, Organometallchemie, 4 ed., Teubner, 2003. [142] W. Sander, G. Bucher, S. Wierlacher, Chem. Rev. 1993, 93, 1583. [143] J. Vignolle, X. Cattoen, D. Bourissou, Chem Rev. 2009, 109, 3333. [144] A. Nemiro, P. R. Schreiner, J. Org. Chem. 2007, 72, 9533. [145] K. Hirai, T. Itoh, H. Tomioka, Chem. Rev. 2009, 109, 3275. [146] N. G. Rondan, K. N. Houk, R. A. Moss, J. Am. Chem. Soc. 1980, 102, 1770. [147] M. T. Nguyen, J. Chem. Soc., Chem. Commun. 1987, 342. [148] B. D. Rowsell, R. J. Gillespie, G. L. Heard, Inorg. Chem. 1999, 38, 4659. [149] J. A. Plumley, J. D. Evanseck, J. Phys. Chem. A 2009, 113, 5985. [150] J. F. Liebman, Struct. Chem. 1990, 1, 395. [151] H. Hirao, K. Omoto, H. Fujimoto, J. Phys. Chem. A 1999, 103, 5807. [152] T. Brinck, J. S. Murray, P. Politzer, Inorg. Chem. 1993, 32, 2622. [153] F. Bessac, G. Frenking, Inorg. Chem. 2003, 42, 7990. [154] V. M. Rosas‐Garcia, D. T. Crawford, J. Chem. Phys. 2003, 119, 10647. [155] N. C. Baird, M. A. Whitehead, Can. J. Chem. 1967, 45, 2059. [156] A. J. Tricamo, K. J. Knaus, D. W. Ball, J. Mol. Struct. Theochem 2007, 807, 67. [157] H. Wang, H.‐S. Wu, J.‐F. Jia, Chinese J. Chem. 2006, 24, 731. [158] D. R. Armstrong, D. T. Clark, Theoret. chim. Acta (Berl.) 1972, 24, 307. [159] N. C. Baird, Inorg. Chem. 1973, 12, 473. [160] T. Bally, S. Masamune, Tetrahedron 1980, 36, 343. [161] K. B. Wiberg, Chem. Rev. 2001, 101, 1317. [162] T. Bally, Angew. Chem. 2006, 118, 6768. [163] P. Garratt, P. Vollhardt, Aromatizität, Georg Thieme Verlag, Stuttgart, 1973. [164] R. M. Minyaev, J. Struct. Chem. 2000, 41, 1. [165] F.‐G. Klärner, Angew. Chem. 2001, 113, 4099. [166] G. Schröder, Cyclooctatetraen, Verlag Chemie, Weinheim, 1965. [167] T. M. Gilbert, B. D. Gailbreath, Organometallics 2001, 20, 4727. [168] A. Y. Timoshkin, H. F. Schaefer III, J. Phys. Chem. A 2010, 114, 516. [169] A. Y. Timoshkin, Inorg. Chem. 2003, 42, 60. [170] A. Y. Timoshkin, Inorg. Chem. 2003, 48, 8145. [171] A. J. Karttunen, M. Linnolahti, T. A. Pakkanen, J. Phys. Chem. C 2008, 112, 10032. [172] W. Fraenk, Ph. D. thesis, Ludwig‐Maximilians‐Universität (München), 2001. [173] S. Biswas, I. M. Oppel, H. F. Bettinger, Inorg. Chem. 2010, in press. [174] W. Fraenk, T. Habereder, T. M. Klapötke, H. Nöth, K. Polborn, J. Chem. Soc., Dalton Trans. 1999,
4283. [175] P. Paetzold, Z. Anorg. Allg. Chem. 1995, 621, 1175. [176] P. Paetzold, Z. Anorg. Allg. Chem. 1963, 326, 58. [177] H. Nöth, B. Wrackmeyer, Nuclear Magnetic Resonance Spectroscopy of Boron Compounds,
Springer, Berlin Heidelberg New York, 1978. [178] I. C. Tornieporth‐Oetting , T. M. Klapötke, Angew. Chem. 1995, 107, 559. [179] S. G. Shore, J. L. Crist, B. Lockman, J. R. Long, A. D. Coon, J. Chem. Soc., Dalton Trans. 1972, 1123. [180] W. Gerrard, M. F. Lappert, B. A. Mountfield, J. Chem. Soc. 1959, 1529.

Anhang
216
[181] J. A. Blau, W. Gerrard, M. F. Lappert, J. Chem. Soc. 1957, 4116. [182] S. Alridge, R. J. Calder, A. Rossin, A. A. Dickinson, D. J. Willock, C. Jones, D. J. Evans, J. W. Steed,
M. E. Light, S. J. Coles, M. B. Hursthouse, J. Chem. Soc., Dalton Trans. 2002, 2020. [183] S. Alridge, R. J. Calder, M. H. Cunningham, K. M. A. Malik, J. W. Steed, J. Organomet. Chem 2000,
614‐615, 188. [184] C. D. Roy, Aust. J. Chem. 2006, 59, 834. [185] R. B. Castle, D. S. Matteson, J. Am. Chem. Soc. 1968, 90, 2194. [186] E. Wiberg, W. Sütterlin, Z. Anorg. Allg. Chem. 1931, 202, 1. [187] R. Köster, M. A. Grassberger, Liebigs Ann. Chem. 1968, 719, 169. [188] L. Weber, I. Domke, W. Greschner, K. Miqueu, A. Chrostowska, P. Baylere, Organometallics 2005,
24, 5455. [189] T.‐T. Wang, P. J. Busse, K. Niedenzu, Inorg. Chem. 1970, 9, 2150. [190] E. J. Corey, K. A. Cimprich, J. Am. Chem. Soc. 1994, 116, 3151. [191] E. J. Corey, C.‐M. Yu, S. S. Kim, J. Am. Chem. Soc. 1989, 111, 5495. [192] E. J. Corey, R. Imwinkel, S. Pikul, Y. B. Xiang, J. Am. Chem. Soc. 1989, 111, 5493. [193] J. Howarth, G. Helmchen, M. Kiefer, Tetrahedron Lett. 1993, 34, 4095. [194] A. Finch, J. Pearn, Tetrahedron Lett. 1964, 20, 173. [195] C. J. Thomas, J. C. Peters, Inorg. Chem. 2003, 42, 5055. [196] W. Fraenk, T. M. Klapötke, B. Krumm, P. Mayer, H. Nöth, H. Piotrowski, M. Suter, J. Fluor. Chem.
2001, 12, 73. [197] J. F. Janik, C. K. Narula, E. G. Gulliver, E. N. Duesler, R. T. Paine, Inorg. Chem. 1988, 27, 1222. [198] Y.‐R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, 2003. [199] J. P. Gerling, F.‐W. Wellmer, Chem. Unserer Zeit 2005, 39, 236. [200] T. Abe, M. Tanizawa, K. Watanabe, A. Taguchi, Energy Environ. Sci. 2009, 2, 315. [201] D. Jagadeesan, M. Eswaramoorthy, C. N. R. Rao, ChemSusChem 2009, 2, 878. [202] J. M. Schicks, Chem. Unserer Zeit 2008, 42, 310. [203] H.‐J. Arpe, Industrielle Organische Chemie: Bedeutende Vor‐ und Zwischenprodukte, 6 ed., Wiley‐
VCH, Weinheim, 2007. [204] R. Dittmeyer, W. Keim, G. Kreysa, A. Oberholz, Winnacker‐Küchler: Chemische Technik; Band 4:
Energieträger, Organische Grundstoffe, 5 ed., Wiley‐VCH, Weinheim, 2005. [205] R. Dittmeyer, W. Keim, G. Kreysa, A. Oberholz, Winnacker‐Küchler: Chemische Technik; Band 5:
Organische Zwischenverbindungen, Polymere, 5 ed., Wiley‐VCH, Weinheim, 2005. [206] D. E. De Vos, B. F. Sels, Angew. Chem. 2005, 117, 30. [207] D. Wolf, Angew. Chem. 1998, 110, 3545. [208] M. Muehlenhofer, T. Strassner, W. A. Herrmann, Angew. Chem. 2002, 114, 1817. [209] R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas, F. Schüth, Angew. Chem. 2009, 121, 7042. [210] G. A. Olah, G. Alain, S. G. K. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley‐VCH,
Weinheim, 2009. [211] B. A. Arndtsen, R. G. Bergman, A. T. Mobley, T. H. Peterson, Acc. Chem. Res. 1995, 28, 154. [212] H. Schwarz, Angew. Chem. 1991, 1991, 837. [213] D. Schröder, H. Schwarz, PNAS 2008, 105, 18114. [214] H. Schwarz, D. Schröder, Pure & Appl. Chem. 2000, 72, 2319. [215] P. Sykes, Reaktionsmechanismen der Organischen Chemie, 9 ed., Verlag Chemie (VCH),
Weinheim, 1988. [216] R. Brückner, Reaktionsmechanismen ‐Organische Reaktionen, Stereochemie, moderne
Synthesemethoden, 2 ed., Spektrum Akademischer Verlag, Heidelberg Berlin, 2003. [217] R. H. Crabtree, Chem. Rev. 1985, 85, 245. [218] S. S. Stahl, J. A. Labinger, J. E. Bercaw, Angew. Chem. 1998, 110, 2298. [219] G. S. Chen, J. A. Labinger, J. E. Bercaw, PNAS 2007, 104, 6915. [220] J. A. Labinger, J. E. Bercaw, Nature 2002, 417, 507. [221] Y. Boutadla, D. L. Davies, S. A. Macgregor, A. I. Poblador‐Bahamonde, Dalton Trans. 2009, 5820. [222] D. Balcells, E. Clot, O. Eisenstein, Chem. Rev. 2010, in press. [223] R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh, H. Fujii, Science 1998, 280, 560.

Anhang
217
[224] R. Palkovits, C. von Malotki, M. Baumgarten, K. Müllen, C. Baltes, M. Antonietti, P. Kuhn, J. Weber, A. Thomas, F. Schüth, ChemSusChem 2010, in press.
[225] H. M. L. Davies, J. R. Manning, Nature 2008, 451, 417. [226] F. Collet, R. H. Dodd, P. Dauban, Chem. Commun. 2009, 5061. [227] M. L. Davies, Angew. Chem. 2006, 118, 6574. [228] H. Davies, M. L., M. S. L. Long, Angew. Chem. 2005, 117, 3584. [229] M. Tobisu, N. Chatani, Angew. Chem. 2006, 118, 1713. [230] T. Graening, Nachr. Chem. 2007, 836. [231] P. Müller, C. Fruit, Chem. Rev. 2003, 103, 2905. [232] S. Cenini, E. Galo, A. Penoni, F. Ragaini, S. Tollari, Chem. Commun. 2000, 2265. [233] K. Sun, R. Sachwani, K. J. Richert, T. G. Driver, Org. Lett. 2009, 11, 3598. [234] Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari, T. H. Warren,
Angew. Chem. 2008, 120, 10109. [235] F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. L. Gotti, M. Lapadula, E. Mangioni, S. Cenini, Chem.
Eur. J. 2003, 9, 249. [236] B. J. Stokes, H. Dong, B. E. Leslie, A. L. Pumphrey, T. G. Driver, J. Am. Chem. Soc. 2007, 129, 7500. [237] M. Shen, B. E. Leslie, T. G. Driver, Angew. Chem. 2008, 120, 5134. [238] W. G. Shou, J. Li, T. Guo, Z. Lin, G. Jia, Organometallics 2009, 28, 6847. [239] H. Lu, J. Tao, J. E. Jones, L. Wojtas, X. P. Zhang, Org. Lett. 2010, 12, 1248. [240] M. Fructos, S. Trofimenko, M. M. Diaz‐Requejo, P. J. Perez, J. Am. Chem. Soc. 2006, 128, 11784. [241] J. D. Harden, J. V. Ruppel, G.‐Y. Gao, P. X. Zhang, Chem. Commun. 2007, 4644. [242] M. M. Diaz‐Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko, P. J. Perez, J. Am. Chem. Soc.
2003, 125, 12078. [243] Y. W. Chan, K. S. Chan, Organometallics 2009, 28, 6845. [244] D. Schröder, J. Roithova, Chem. Rev. 2010, in press. [245] D. Schröder, Angew. Chem. 2010, 122, 862. [246] M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müler, S. J. Lippard, Angew. Chem. 2001, 113,
2860. [247] G. A. Olah, D. G. Parker, N. Yoneda, Angew. Chem. 1978, 90, 962. [248] A. A. Fokin, P. R. Schreiner, Adv. Synth. Catal. 2003, 345, 1035. [249] G. E. Herberich, A. Fischer, Organometallics 1996, 15, 58. [250] M. Filthaus, I. M. Oppel, H. F. Bettinger, Org. Biomol. Chem. 2008, 6, 1202. [251] H. F. Bettinger, 2009, unveröffentliche Ergebnisse. [252] G. Povie, G. Villa, L. Ford, D. Pozzi, C. H. Schiesser, P. Renaud, Chem. Commun. 2010, 46, 803. [253] G. Höfle, W. Steglich, H. Vorbrüggen, Angew. Chem. 1978, 90, 602. [254] A. C. Spivey, S. Arseniyadis, Angew. Chem. 2004, 116, 5552. [255] W. Lwowski, T. J. Maricich, J. Am. Chem. Soc. 1964, 86, 3164. [256] A. G. Anastassiou, H. E. Simmons, J. Am. Chem. Soc. 1967, 89, 3177. [257] D. S. Breslow, E. I. Edward, E. C. Linsay, H. Omura, J. Am. Chem. Soc. 1976, 98, 4268. [258] S. Murata, Y. Tsubone, R. Kawai, D. Eguchi, H. Tomioka, J. Phys. Org. Chem. 2005, 18, 9. [259] A. V. Willi, Isotopeneffekte bei chemischen Reaktionen, Georg Thieme Verlag, Stuttgart New York,
1983. [260] P. Krumbiegel, Isotopeneffekte, Akademie Verlag / Pergamon Press / Vieweg & Sohn, Berlin,
1970. [261] R. A. Moss, G.‐J. Ho, W. Liu, C. Sierakowski, Tetrahedron Lett. 1993, 34, 927. [262] R. A. Moss, S. Yan, Tetrahedron Lett. 1998, 39, 9381. [263] R. A. Moss, W. Liu, Tetrahedron Lett. 1996, 37, 279. [264] P. S. Zuev, R. S. Sheridan, J. Am. Chem. Soc. 1994, 116, 4123. [265] R. A. Moss, G.‐J. Ho, J. Am. Chem. Soc. 1990, 112, 5642. [266] G. A. Sulikowski, S. Lee, Tetrahedron Lett. 1999, 1999, 8035. [267] S. Murata, Y. Tsubone, H. Tomioka, Chem. Lett. 1998, 549. [268] M. F. Lappert, M. K. Majumdar, B. P. Tilley, J. Chem. Soc (A) 1966, 1590.

Anhang
218
[269] H. Remane, R. Herzschuh, Massenspektrometrie in der organischen Chemie, Vieweg, Braunschweig, 1977.
[270] J. Romba, D. Kuppert, B. Morgenstern, C. Neis, S. Steinhauser, T. Weyhermüller, K. Hegetschweiler, Eur. J. Inorg. Chem. 2006, 314.
[271] S. Hermann, E.‐E. Roos, Chem. Ber. 1954, 87, 566. [272] J. Dale, T. Sigvartsen, Acta Chemica Scandinavica 1991, 45, 1064. [273] H. F. Bettinger, M. Filthaus, Verfahren und Vorrichtung zur Herstellung von Alkylaminen und
Alkylaminderivaten, Erfindung beim deutschen Patentamt eingereicht, 2009. [274] H. F. Bettinger, M. Filthaus, 2009, eingereicht. [275] H. D. Smith, R. J. Brotherton, Inorg. Chem. 1970, 9, 2443. [276] D. R. Arnold, V. Y. Abraitys, Chem. Commun. (London) 1967, 1053. [277] P. Kropp, J., J. J. Snyder, P. C. Rawlings, H. G. Fravel, J. Org. Chem. 1980, 45. [278] V. M. Gil, M. J. Arevalo, O. Lopez, Synthesis 2007, 11, 1589. [279] G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba, A. A. Genazzani, Med. Res. Rev.
2008, 28, 278. [280] Q. Wang, S. Chittaboina, H. N. Barnhill, Lett. Org. Chem. 2005, 2, 293. [281] V. D. Bock, H. Hiemstra, J. H. van Maarseveen, Eur. J. Org. Chem. 2006, 51. [282] J. E. Moses, A. D. Moorhouse, Chem. Soc. Rev. 2007, 36, 1249. [283] F. Amblard, J. H. Cho, R. F. Schinazi, Chem. Rev. 2009, 109, 4207. [284] M. Meldal, C. W. Tornoe, Chem. Rev. 2008, 108, 2952. [285] M. van Dijk, D. T. S. Rijkers, R. M. J. Liskamp, C. F. van Nostrum, W. E. Hennink, Bioconjugate
Chem. 2009, 20, 2001. [286] B. Engels, M. Christl, Angew. Chem. 2009, 121, 8110. [287] C. R. Becer, R. Hoogenboom, U. S. Schubert, Angew. Chem. 2009, 121, 4998. [288] J.‐F. Lutz, Angew. Chem. 2008, 120, 2212. [289] J.‐F. Lutz, Angew. Chem. 2007, 119, 1036. [290] H. C. Kolb, M. G. Finn, B. Shapless, K., Angew. Chem. 2001, 113, 2056. [291] R. Huisgen, Proc. Chem. Soc. 1961, 357. [292] R. Huisgen, Angew. Chem. 1963, 75, 604. [293] D. Kunz, Chem. Unserer Zeit 2009, 43, 22. [294] O. Tsuge, S. Urano, K. Oe, J. Org. Chem. 1980, 45, 5130. [295] A. A. Taherpour, K. Kheradmand, J. Heterocyclic Chem. 2009, 46, 131. [296] H.‐Z. Luo, H.‐Y. Ye, Acta Cryst. Sec. E 2008, E64, o136. [297] V. Aureggi, G. Sedelmeier, Angew. Chem. 2007, 119, 8592. [298] H. F. Bettinger, S. Biswas, 2008, unveröffentliche Ergebnisse. [299] M. Sircoglou, S. Bontemps, K. Miqueu, S. Ladeira, N. Saffon, L. Maron, G. Bouhadir, D. Bourissou,
Inorg. Chem. 2010, in press. [300] S. Diez‐Gonzalez, A. Correra, L. Cavallo, S. P. Nolan, Chem. Eur. J. 2006, 12, 7558. [301] J. C. Walton, Angew. Chem. 2009, 121, 1754. [302] R. Larock, S. K. Gupta, H. C. Brown, J. Am. Chem. Soc. 1972, 94, 4371. [303] H. C. Brown, S. K. Gupta, J. Am. Chem. Soc. 1972, 94, 4370. [304] D. H. Ess, G. O. Jones, K. N. Houk, Org. Lett. 2008, 10, 1633. [305] F. Schoenebeck, D. H. Ess, G. O. Jones, K. N. Houk, J. Am. Chem. Soc. 2009, 131, 8121. [306] D. H. Ess, K. N. Houk, J. Am. Chem. Soc. 2008, 130, 10187. [307] D. H. Ess, K. N. Houk, J. Am. Chem. Soc. 2007, 129, 10646. [308] D. H. Ess, K. N. Houk, J. Phys. Chem. A 2005, 109, 9542. [309] G. O. Jones, D. H. Ess, K. N. Houk, Helvetica Chimica Acta 2005, 88, 1702. [310] K. Chenoweth, D. Chenoweth, W. A. Goddard III, Org. Biomol. Chem. 2009, 7, 5255. [311] L. Schwertman, BA thesis, Ruhr‐Universität Bochum (Bochum), 2009. [312] H. F. Bettinger, Inorg. Chem. 2007, 46, 5188. [313] H. Braunschweig, C. Kollann, D. Rais, Angew. Chem. 2006, 118, 5380. [314] C. E. Anderson, H. Braunschweig, R. D. Dewhurst, Organometallics 2008, 27, 6381. [315] H. Braunschweig, D. Rais, Heteroatom Chemistry 2005, 16, 566.

Anhang
219
[316] H. Braunschweig, M. Colling, Eur. J. Inorg. Chem. 2003, 393. [317] M. Bochmann, Metallorganische Chemie der Übergangsmetalle, Wiley‐VCH, Weinheim, 1997. [318] R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons,
Hoboken, 2005. [319] G. Habermehl, P. Hammann, Naturstoffchemie, Springer‐Verlag, Berlin Heidelberg, 1992. [320] H. F. Bettinger, M. Filthaus, M. Müller, Verfahren zur N‐Funktionalisierung von Grenzflächen,
Erfindung beim deutschen Patentamt eingereicht, 2009. [321] M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme,
Stuttgart, 2005. [322] M. Reichenbächer, J. Popp, Strukturanalytik organischer und anorganischer Verbindungen,
Teubner, Wiesbaden, 2007. [323] S. Schlecht, J. F. Hartwig, J. Am. Chem. Soc. 2000, 122, 9435. [324] I. A. Akhtar, J. J. McCullough, J. Org. Chem. 1981, 46, 1447. [325] R. H. Cragg, J. P. N. Husband, A. F. Weston, J. Chem. Soc., Dalton Trans. 1973, 568.

Danksagung
Ich möchte mich bei allen Mitarbeitern des Arbeitskreises für die
hervorragende Arbeitsatmosphäre bedanken.
Matthias Böhm, Holger Bornemann, Götz Bucher, Bayram Cakir, Melanie Ertelt,
Christopher Finke, Dirk Grote, Arndt Richter, Christina Tönshoff, Elsa Sanchez,
Joel Torres, Sugumar Venkataramani, Magdalena Michalczyk, Sandra Müller,
Patrik Neuhaus, Schulz Björn, Y‐am Huynh, Konstantinos Zacharopoulos,
Tamara Munsch, Artur Mardyukov, Alexander Koch, Andre Korte, Tuhin Suvra
Khan, Saonli Roy, Arunlibertsen Lawzer, Kavitha Velappan.
Frau Barbara Schröder danke ich für die Hilfe im Laboralltag und den Versuch
mir Ordnung und Sauberkeit beizubringen.
Klaus Gomann danke ich für die Hilfe im Labor, nette Gespräche und die
manchmal notwendigen „Erziehungsmaßnahmen“.
Torsten Haenschke danke ich für die immer schnelle und umfangreiche Hilfe bei
diversen PC‐Problemen (die ich oft auch selbst verursacht habe).
Bei Heidemarie Joppich bedanke ich mich insbesondere für die tatkräftige
Unterstützung mit dem GC/MS‐Gerät.
Ulrike Steger danke ich für die Hilfe und Geduld bei Verwaltungsfragen, die
netten Gespräche und die immer gefüllte Süßigkeitendose.

Larissa Schwertmann und Sandra Müller danke ich dafür, dass sie im Rahmen
ihrer Bachelorarbeiten zu dieser Dissertation beigetragen haben.
Tammy Munsch danke ich dafür, dass sie eine so nette Labornachbarin war und
mir mit dem Englischen nach vorne geholfen hat.
Patrik Neuhaus danke ich für die Aufnahme der ESR‐Messungen und für sein
sehr umfangreiches Wissen rund um die Chemie.
Y‐am Huynh danke ich für das Korrekturlesen von diversen Postern und
Abstracts.
Christina Tönshoff danke ich dafür, dass sie mit mir zusammen die Stellung in
Bochum gehalten hat.
Ferner danke ich allen neuen und alten Mitarbeitern des AK Bettinger: Florian
Barthold, Sunanda Biswas, Rafael Bula und Matthias Müller.
Herrn Prof. Martin Feigel danke ich für die Übernahme des Koreferates.
Prof. Iris Oppel danke ich für die Aufnahme diverser Kristallstrukturen. Sowie
für ihre Geduld und ihren großen Einsatz bei „schwierigen Fällen“

Ich danke Herrn Prof. Wolfram Sander, dass er mir die Möglichkeit gegeben hat
meine Arbeit in Bchum weiterführen und beenden zu können, und ich dort die
gleiche Unterstützung bekommen habe wie jedes andere „volle“ Mitglied der
Arbeitsgruppe auch. Ich bin mir bewusst, dass dies keineswegs
selbstverständlich ist.
Meinem Doktorvater Prof. Holger F. Bettinger danke ich herzlich für das
Fördern und Fordern, für das große Interesse an meiner Arbeit, für jegliche
denkbare Unterstützung und seine unkomplizierte Art. Für die
abwechslungsreiche Arbeit, meine wissenschaftliche Freiheit und die
Möglichkeiten, an Konferenzen teilzunehmen. Ich bedanke mich hiermit auch
noch mal ausdrücklich dafür, dass ich die Arbeiten in Bochum zu Ende führen
konnte.
Meiner Frau Anne danke ich für die fortwährende Unterstützung und das
Verständnis während der ganzen Zeit. Dafür, dass sie meine meist
chemiebedingten schlechten Launen ertragen hat, sich aber genauso über jede
Kristallstruktur mit mir gefreut hat. Danke für das Korrekturlesen der Arbeit.
Meinem kleinen Henning danke ich dafür, dass er mir täglich zeigt, dass Arbeit
nicht alles ist.
Meinen Eltern danke ich für alle Unterstützung in den letzten 30 Jahren.

Publikationen
[1] H. F. Bettinger, M. Filthaus, J. Org. Chem., “Halogen‐Metal Exchange in 1,2‐
Dibromobenzene and the Possible Intermediacy of 1,2‐Dilithiobenzene”, 2007, 72,
9750.
[2] H. F. Bettinger, M. Filthaus, I. M. Oppel, Org. Biomol. Chem., “Supramolecular
structures and spontaneous resolution: the case of ortho‐substituted phenylboronic
acids” 2008, 6, 1201.
[3] H. F. Bettinger, M. Filthaus, H. Bornemann, I. M. Oppel, Angew. Chem. Int. Ed.,
“Metal‐Free Amination of Methane and Cycloalkanes Employing Borylnitrene”, 2008,
47, 4744.
[4] H. F. Bettinger, M. Filthaus, P. Neuhaus, Chem. Commun., “Insertion into dihydrogen
employing the nitrogen centre of a borylnitrene”, 2009, 2186.
[5] H. F. Bettinger, M. Filthaus, “Conversion of Methane to Methylamine at Room
Temperature”, 2009, eingereicht.
[6] H. F. Bettinger, M. Filthaus, “Inter‐ and Intra CH‐Insertion of Borylnitrenes. Influence
of Ligands and Reaction Mechanism“, 2009, in Vorbereitung.
Patente
[1] H. F. Bettinger, M. Filthaus, “Verfahren und Vorrichtung zur Herstellung von
Alkylaminen und Alkylaminderivaten“, 2009, Erfindung beim Patentamt eingereicht.
[2] H. F. Bettinger, M. Filthaus, M. Müller, “Aminierung von Oberflächen“, 2009,
Erfindung beim Patentamt eingereicht.
Tagungsbeiträge / Konferenzen
2006 M. Filthaus, H. F. Bettinger, “Beiträge zur Synthese von Bor‐Aromatischen
Verbindungen“. Vortrag: Deutsches Borchemikertreffen 2006, (September, 13‐18),
Harz.
2007 M. Filthaus, H. F. Bettinger, “Borylnitrene: Geeignet für eine direkte, effiziente
Aminierung von gesättigten Kohlenwasserstoffen?“. Vortrag: Deutsches
Borchemikertreffen 2007, (September, 3‐5), Bensheim.
M. Filthaus, H. F. Bettinger, “Halogen‐Metal Exchange in 1,2‐Dibromobenzene and
the Possible Intermediacy of 1,2‐Dilithiobenzene“. Poster Präsentation: IUPAC
Conference on Physical Organic Chemistry (July, 13‐18), Warschau, Polen.
M. Filthaus, H. F. Bettinger, “Halogen‐Metal Exchange in 1,2‐Dibromobenzene and
the Possible Intermediacy of 1,2‐Dilithiobenzene”. Poster Präsentation: Section Day
der Bochumer Research School (July), Bochum.
2008 M. Filthaus, H. F. Bettinger, “Metal‐Free Amination of Methane and (Cyclo)alkanes
Employing Borylnitrene”. Poster Präsentation: IUPAC Conference on Physical Organic
Chemistry (July, 13‐18), Santiago de Compostela, Spanien.

M. Filthaus, H. F. Bettinger, “Direct Amination of hydrocarbons via C‐H Activation
Employing Borylnitrene”. Poster Präsentation: ORCHEM 2008, 16. Lecture
Conference of the Liebig‐Vereinigung für Organische Chemie (September 1‐3),
Weimar.
M. Filthaus, H. F. Bettinger, “Präparative Methan‐Aktivierung mithilfe eines
Borylnitrens ?“. Vortrag: Deutsches Borchemikertreffen 2008, (Oktober, 10‐12),
Blaubeuren.
2009 M. Filthaus, H. F. Bettinger, “Borylnitrenes: Metal‐Free C‐H Activation of
Hydrocarbons”. Poster Präsentation: GDCh‐JCF Frühjahrssymposium (März, 11‐14,
Essen a. d. Ruhr.
M. Filthaus, H. F. Bettinger, “Borylnitrene: Direkte Metallfreie Umwandlung von
Alkanen in Amine”. Poster Präsentation: GDCh‐Wissenschaftsforum (September, 01‐
03), Frankfurt a. Main.

L eben s l au f Persönliche Daten Name, Vorname: Filthaus, Matthias Geburtsdatum: 05/10/1979 Geburtsort: Gelsenkirchen Familienstand: Verheiratet, 1 Kind Nationalität: Deutsch
Ausbildung / Studium
09/1991 – 06/2000 Schulausbildung: Besuch des Schalker Gymnasiums in Gelsenkirchen.
08/2000 – 08/2001 Zivildienst beim Diakonischen‐Werk des Kirchenkreises Gelsenkirchen
und Wattenscheid: Betreuung von psychisch Kranken.
10/2001 ‐ 11/2006 Studium der Chemie an der Ruhr‐Universität Bochum:
Bachelor of Science (03/2005). Titel der Bachelorarbeit: „Beiträge zur
chemischen Funktionalisierung von einwandigen Kohlenstoff‐
nanoröhren.
Master of Science (11/2006). Schwerpunkt: Organische Chemie. Titel
der Masterarbeit: „Beiträge zur Synthese von Bor‐Aromatischen
Verbindungen“.
seit 12/2006 Wissenschaftlicher Mitarbeiter / Doktorand am Lehrstuhl für
Organische Chemie II der Ruhr‐Universität Bochum und am Institut
für Organische Chemie der Eberhard Karls Universität Tübingen unter
Betreuung von Prof. H. F. Bettinger.









![Synthese und Charakterisierung von , Liebigit - hzdr.de · Synthese, Charakterisierung und Löslichkeit von Erdalkaliuranylcarbonaten M2[UO2(CO3)3]·xH2O; M: Mg, Ca, Sr, Ba DISSERTATION](https://img.pdfslide.tips/doc/110x75/5d5b8b6288c99342048ba22c/synthese-und-charakterisierung-von-liebigit-hzdrde-synthese-charakterisierung.jpg)









![Synthese von Phenanthridin- und Benzo[c]phenanthridin ... · Synthese von Phenanthridin- und Benzo[c]phenanthridin-Derivaten und Untersuchungen auf ihre biologische Wirkung Vom Fachbereich](https://img.pdfslide.tips/doc/110x75/5e156f76166fc450df1f62f9/synthese-von-phenanthridin-und-benzocphenanthridin-synthese-von-phenanthridin-.jpg)









