Embed Size (px)
Citation preview

Microenvironment and Immunology
Systemic DC Activation Modulates the TumorMicroenvironment and Shapes the Long-LivedTumor-SpecificMemoryMediated byCD8þ TCellsKanako Shimizu1, Satoru Yamasaki1, Jun Shinga1, Yusuke Sato1, Takashi Watanabe2,Osamu Ohara2, Kiyotaka Kuzushima3, Hideo Yagita4, Yoshiko Komuro5,Miki Asakura1, and Shin-ichiro Fujii1
Abstract
Strategies to reprogram the tumormicroenvironment are beingexplored to improve cancer immunotherapy. In one approach, wehave targeted dendritic cells (DC) to improve their function withadjuvant vector cells (aAVC) that are engineered fromNKT ligand-loaded CD1dþ allogeneic cells transfected with tumor antigenmRNAs. Here, we report the finding that this approach alsoprograms local immune responses by establishing tertiary lym-phoid structures (TLS), which include expanded antigen-specificCD8þ T-cell clones, mobilized DCs, and normalized tumor
vasculature. aAVC therapy also expanded specific Vb-expressingantitumor T-cell clones, leading to the formation of long-termmemory T cells. When combined with PD-1 blockade, aAVCinfusion triggered regression of poorly immunogenic tumor cellsthat did not respond to PD-1 blockade alone, as well as expansionof antigen-specific CD8þ T-cell clones in the tumor. The findingsof this study help to inform a next-generation platform for thegeneration of efficacious cancer vaccines. Cancer Res; 76(13); 3756–66. �2016 AACR.
IntroductionCancer immunotherapy has recently enjoyed a renaissance as a
result of new and more effective approaches (1). For example,adoptive T-cell transfer with tumor antigen–specific TCR or chi-meric antigen receptor (CAR) gene-transferred T cells has shownantitumor effects on some cancers (2, 3) Also, blocking immunecheckpoints usingmAbs toCTL–associated protein-4 (CTLA-4) orprogrammed cell death protein-1 (PD-1) has demonstrated clin-ical efficacy (4). Another promising approach involves activeimmunization using a cancer vaccine, and in this context thefunction of dendritic cells (DC) appears to be very important(5, 6). The next breakthroughs for its use as an effective antitumorimmunotherapy would be the induction of memory T cells andthe modulation of tumor microenvironment in the light ofsystemic and local immune responses. Once established,memory
T cells could quickly respond to emerging cancer cells in therecurrence and metastasis. The persistence of functional memoryT cells was desired for immunotherapies. In addition, therapeuticoptions to overcome the barriers of tumormicroenvironment andto support T-cell infiltration and function in tumors are required.For this perspective, organized lymphoid aggregates found locallyat tumor sites, termed tertiary lymphoid structures (TLS), shouldbe a major focus that may lead effector T cells to recruit to tumorcells (7, 8).
For the success of immunotherapy, it has been recently arguedthat combining multiple therapies should be beneficial, becausedifferent types of immune responses may prevent tumor cellsfrom immune escape. In this point, a strategy linking innate andadaptive immune responses is attractive and promising, forexample, they can attack MHCþ or MHC� tumor cells. NKT cellshave bipotential capacity and can suppress or activate immuneresponses.Once activatedby their ligands, they can closely contactDCs andmature themsystemically in lung, liver, spleen, andbonemarrow. DCs thus licensed by activatedNKT cells via bothCD40LonNKT cells and their simultaneous production of inflammatorycytokines, for example, IFNg and TNFa (9). Then, the DCscan induce adaptive immunity in infection and tumor models(10–13). We and others have shown that coadministration of NKTcell ligand and antigen can induce adaptive immunity (10, 14, 15).
On the basis of these evidences, we previously illustrated theconcept of a unique approach of usingNKT ligand–loadedCD1dþ
allogeneic cells transfected with tumor antigen mRNA as artificialadjuvant vector cells (aAVC; refs. 16, 17). The first generation ofaAVCs demonstrated certain of its immunologic features inducingboth innate and adaptive immunity in the lymphoid organsthrough antigen-captured DCs in situ. To develop the aAVC ther-apy, we assumed that the optimal amount of target proteindelivered to in vivo DC would affect the magnitude of the ensuingimmune responses in the tumor. By the new method, aAVCs can
1Laboratory for Immunotherapy, RIKENCenter for IntegrativeMedicalScience, Yokohama, Kanagawa, Japan. 2Laboratory for IntegrativeGenomics, RIKEN Center for Integrative Medical Science, Yokohama,Kanagawa, Japan. 3Division of Immunology, Aichi Cancer CenterResearch Institute, Nagoya, Japan. 4Department of Immunology, Jun-tendo University School of Medicine,Tokyo, Japan. 5The TranslationalResearch Center, The University of Tokyo Hospital, Tokyo, Japan.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Current address for Y. Komuro: Pharmaceuticals and Medical Devices Agency,Tokyo 100-0013, Japan.
Corresponding Author: Shin-Ichiro Fujii, Laboratory for Immunotherapy, RIKENCenter for Integrative Medical Sciences (IMS), 1-7-22, Suehiro-cho, Tsurumi-ku,Yokohama, Kanagawa, 230-0045, Japan. Phone: 81-45-503-7061; Fax: 81-45-503-7061; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-15-3219
�2016 American Association for Cancer Research.
CancerResearch
Cancer Res; 76(13) July 1, 20163756
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

produce a suitable amount of protein for protein-based clinicalimmunotherapy and be developed as a current type of aAVC. Herewe assessed the antitumor effects and further analyzed the cellularmechanism against tumor in terms of local and systemic immuneresponses. This study not only demonstrates the efficacy of ourapproach, butwill alsohelp to identify thekey componentsneededfor successful future therapeutics.
Materials and MethodsReagents
Human IL2was purchased fromShionogi &Co., LTD.Humanrecombinant IL7 and IL15 were purchased from Peprotech,Inc. The clinical grade of a-GalCer was provided from Regim-mune. OVA257–264 peptide (SIINFEKL) and WT1235–243(CMTWNQMNL) were obtained from Toray Research Center,Inc. The antibodies used in this study were purchased anddescribed in Supplementary Table S1. OVA-tetramer was pur-chased from MBL. WT1-tetramer (HLA-A24) and HIV-tetramer(HLA-A24) were produced by our group. A FACSCalibur orFACSCanto II instrument and CELLQuest, Diva (BD Bios-ciences), and FlowJo (Tree Star) software were used for theanalysis. Anti-PD-1 mAb (RPMI-14) was produced as describedpreviously (18). Recombinant human WT1 was synthesized inRIKEN.
Mice and cell linesB16 and HEK293 cell lines were purchased from ATCC.
NIH3T3 and WEHI3B cell lines were obtained from the RIKENCell Bank. MO4 (19) and J558 (20) cell lines were received fromDr. R.M. Steinman (The Rockefeller University, New York, NY)and has been routinely tested for OVA expression or for IgA andH2-Kd expression by our hands, respectively. The J558-WT1 cellline was established by transfection of a human WT1 cDNAexpression vector into J558. All cell lines were tested accordingto themanufacturer's protocol and proved to bemycoplasma free(MycoplasmaDetection Kit; Minerva Biolabs). C57BL/6 or BALB/c mice were purchased from CLEA Japan. Ly5.1 congenic OT-1micewere generated by cross/backcross breeding ofOT-1with B6.Ly5.1 mice and screening for the presence of Va2 and Ly5.1 andabsence of Ly5.2 by flow cytometry. OT-1 TCR transgenic mice,CD11c-DTR/GFPmice (21), and XCR1-DTR-venusmice (22) andother all the mice were maintained under specific pathogen-freeconditions and studied in compliance with our institutionalguidelines.
Cell preparationHuman PBMCs were isolated from healthy volunteers. All
studies were approved by the RIKEN institutional reviewboard. Murine bone marrow–derived DCs were generatedin the presence of GM-CSF for 6 days as described previously(23). The preparation of aAVCs using HEK293 or NIH3T3was performed as described previously (17). Briefly, cellsresuspended in OptiMEM and RNA were transferred to acuvette and then the cell suspension was pulsed in ECM830 Square Wave Electroporation System (Harvard Appara-tus). Pulse condition was a single 500 V, 3 ms square pulse.Immediately after electroporation, the cells were transferred toculture medium and cultured in the presence of 500 ng/mL ofa-GalCer. The protein expression of transfected cells wasanalyzed by ELISA (ITEA) for OVA, flow cytometry for CD1d,
and Western blot analysis for WT1 or TRP-2 protein asdescribed previously (17).
Immunologic analysesAntigen-specific IFNg-secreting cells were performed by
ELISPOT assay as described previously (24). The cytotoxicactivity of NKT cell line was analyzed using LDH assay kitaccording to the manufacturer's instructions (Takara Bio Com-pany). Expression of immune response–related molecules inDCs and tumors was analyzed by quantitative PCR assay. In theanalysis of splenic DCs and tumor, the total RNA was isolatedusing RNeasy Mini Kit (Qiagen) and cDNA was synthesized byReverTra Ace (Toyobo) according to the manufacturer's instruc-tions. Tumor-associated DCs were sorted and directly subjectedto cDNA synthesis using a CellsDirect One-Step qRT-PCR Kit(Invitrogen) with a mixture of pooled gene-specific primers(Supplementary Table S2). Synthesized cDNA was appropri-ately diluted with water and used as template for subsequentquantitative PCR as described previously (25).
Statistical analysisThe P values were calculated with the two-side Student t test
or the Mann–Whitney U test. The log-rank test was used forsurvival calculations. P < 0.05 was considered statisticallysignificant.
ResultsGeneration of cytotoxic T cells and their infiltration into tumorsites by administration of a-GalCer-loaded, CD1d, and antigenmRNA–cotransfected cells
We previously established aAVCs, which are a-GalCer-loadedand antigen mRNA-transfected, CD1d-expressing allogeneic cells(NIH3T3 or HEK293 cells for mice and humans, respectively;ref. 17). In the current study, using a developed method byoptimizing several points of the protocol to increase proteinproduction (Supplementary Fig. S1A), we succeeded in the pro-duction of target antigen protein by aAVC at levels about 100times higher than with the previous method. As shown in Sup-plementary Fig. S1B–S1D,we verified that aAVC-OVA cells, whichwere loaded with a-GalCer and cotransfected with OVA andCD1dmRNA, highly expressed the CD1dmolecule and producedabundant OVA protein, and could directly stimulate NKT cells,but not OT-1 T cells in vitro (17). In the initial study, we assessedantitumor effects in a therapeutic model. We administered MO4cells (OVA-expressing B16 melanoma) and, after verifying thatlarge tumors (tumor size: 441.7 � 176.8 mm3) were established,mice were treated with aAVC-OVA. As shown in Fig. 1A, untreatedtumors grew larger; however, they stopped growing after theaAVC-OVA treatment and the center of tumor mass becamenecrotic. Given this striking immunotherapeutic outcome, webegan to analyze the antitumor effects in more detail. This initialstudy thus demonstrated that treatment with aAVC-OVAincreased the frequency of tumor-infiltrating T cell, especiallyCD8þ T cells in spleen and the tumor (Fig. 1B and SupplementaryFig. S2).
Prominent function of XCR1þ DCs after administration ofaAVC-OVA
We and others previously demonstrated that in vivo DCs stim-ulated by activated NKT cells produce IL12 and certain
aAVC Therapy Generates Systemic and Local Immunosurveillance
www.aacrjournals.org Cancer Res; 76(13) July 1, 2016 3757
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
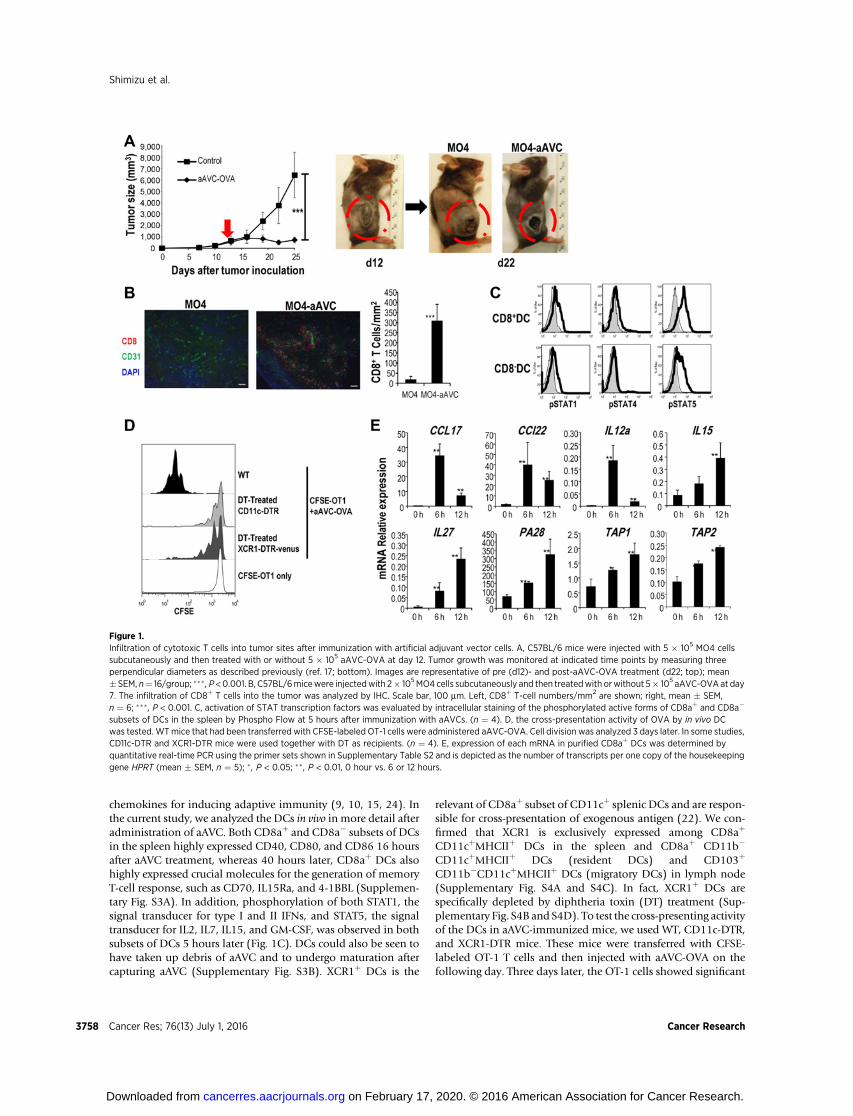
chemokines for inducing adaptive immunity (9, 10, 15, 24). Inthe current study, we analyzed the DCs in vivo in more detail afteradministration of aAVC. Both CD8aþ and CD8a� subsets of DCsin the spleen highly expressed CD40, CD80, and CD86 16 hoursafter aAVC treatment, whereas 40 hours later, CD8aþ DCs alsohighly expressed crucial molecules for the generation of memoryT-cell response, such as CD70, IL15Ra, and 4-1BBL (Supplemen-tary Fig. S3A). In addition, phosphorylation of both STAT1, thesignal transducer for type I and II IFNs, and STAT5, the signaltransducer for IL2, IL7, IL15, and GM-CSF, was observed in bothsubsets of DCs 5 hours later (Fig. 1C). DCs could also be seen tohave taken up debris of aAVC and to undergo maturation aftercapturing aAVC (Supplementary Fig. S3B). XCR1þ DCs is the
relevant of CD8aþ subset of CD11cþ splenic DCs and are respon-sible for cross-presentation of exogenous antigen (22). We con-firmed that XCR1 is exclusively expressed among CD8aþ
CD11cþMHCIIþ DCs in the spleen and CD8aþ CD11b�
CD11cþMHCIIþ DCs (resident DCs) and CD103þ
CD11b�CD11cþMHCIIþ DCs (migratory DCs) in lymph node(Supplementary Fig. S4A and S4C). In fact, XCR1þ DCs arespecifically depleted by diphtheria toxin (DT) treatment (Sup-plementary Fig. S4B and S4D). To test the cross-presenting activityof the DCs in aAVC-immunized mice, we used WT, CD11c-DTR,and XCR1-DTR mice. These mice were transferred with CFSE-labeled OT-1 T cells and then injected with aAVC-OVA on thefollowing day. Three days later, the OT-1 cells showed significant
Figure 1.Infiltration of cytotoxic T cells into tumor sites after immunization with artificial adjuvant vector cells. A, C57BL/6 mice were injected with 5 � 105 MO4 cellssubcutaneously and then treated with or without 5 � 105 aAVC-OVA at day 12. Tumor growth was monitored at indicated time points by measuring threeperpendicular diameters as described previously (ref. 17; bottom). Images are representative of pre (d12)- and post-aAVC-OVA treatment (d22; top); mean� SEM, n¼ 16/group; ��� , P <0.001. B, C57BL/6micewere injectedwith 2� 105MO4 cells subcutaneously and then treatedwith orwithout 5� 105 aAVC-OVA at day7. The infiltration of CD8þ T cells into the tumor was analyzed by IHC. Scale bar, 100 mm. Left, CD8þ T-cell numbers/mm2 are shown; right, mean � SEM,n ¼ 6; ��� , P < 0.001. C, activation of STAT transcription factors was evaluated by intracellular staining of the phosphorylated active forms of CD8aþ and CD8a�
subsets of DCs in the spleen by Phospho Flow at 5 hours after immunization with aAVCs. (n ¼ 4). D, the cross-presentation activity of OVA by in vivo DCwas tested. WTmice that had been transferred with CFSE-labeled OT-1 cells were administered aAVC-OVA. Cell division was analyzed 3 days later. In some studies,CD11c-DTR and XCR1-DTR mice were used together with DT as recipients. (n ¼ 4). E, expression of each mRNA in purified CD8aþ DCs was determined byquantitative real-time PCR using the primer sets shown in Supplementary Table S2 and is depicted as the number of transcripts per one copy of the housekeepinggene HPRT (mean � SEM, n ¼ 5); � , P < 0.05; �� , P < 0.01, 0 hour vs. 6 or 12 hours.
Shimizu et al.
Cancer Res; 76(13) July 1, 2016 Cancer Research3758
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
proliferation in WT mice, but not in CD11c-DTR or XCR-DTRmice treated with DT (Fig. 1D). These findings indicated that theCD8aþDCs that had engulfed aAVC-OVAwere activated and ableto cross-present OVA peptide to CTLs. Next, we sorted the CD8aþ
DCs and evaluated their expressionof chemokines, cytokines, andgenes involved in antigen processing,molecules that would play akey role in antigen presentation. We detected significantly elevat-ed expression of CCL17, CCL22, IL12a, IL15, IL27, TAP1, TAP2,and PA28, a component of the immunoproteasome (Fig. 1E).Taken together, aAVC therapy can deliver tumor antigens toin vivo DC.
The TLS were formed by aAVCBecause the aAVC therapy induced tumor regression, we next
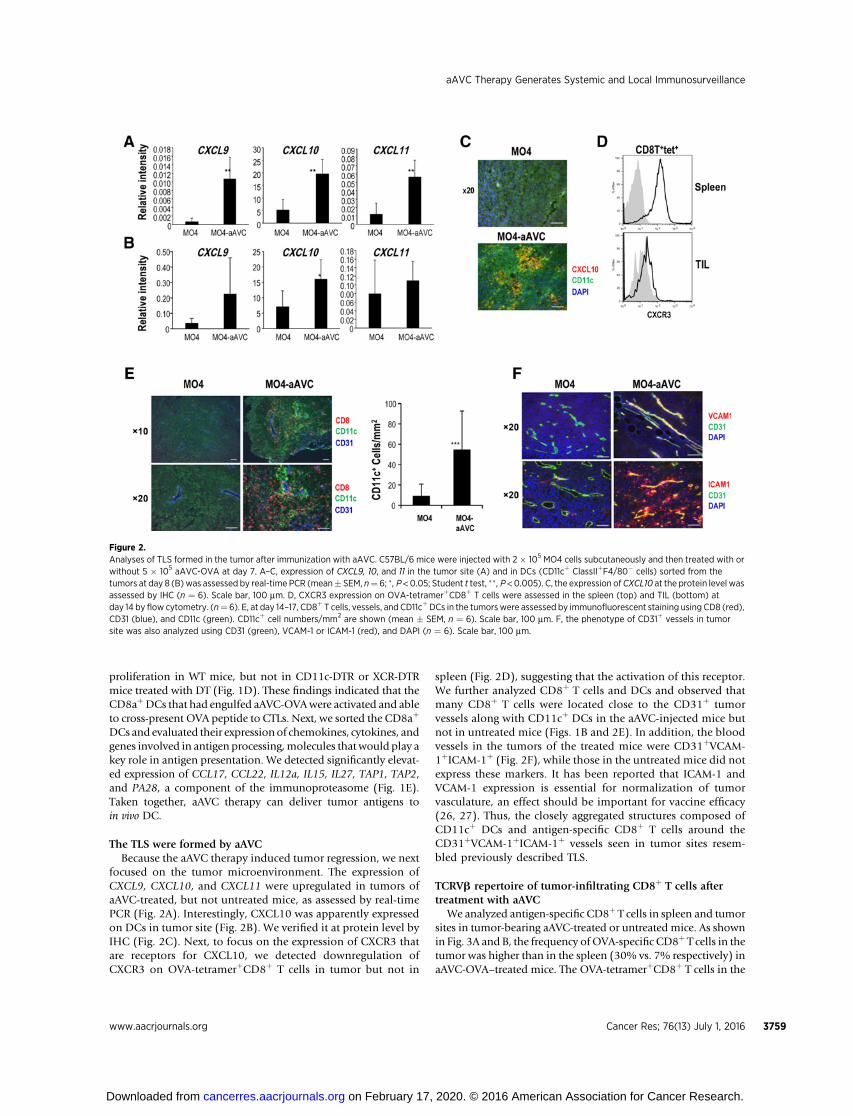
focused on the tumor microenvironment. The expression ofCXCL9, CXCL10, and CXCL11 were upregulated in tumors ofaAVC-treated, but not untreated mice, as assessed by real-timePCR (Fig. 2A). Interestingly, CXCL10 was apparently expressedon DCs in tumor site (Fig. 2B). We verified it at protein level byIHC (Fig. 2C). Next, to focus on the expression of CXCR3 thatare receptors for CXCL10, we detected downregulation ofCXCR3 on OVA-tetramerþCD8þ T cells in tumor but not in
spleen (Fig. 2D), suggesting that the activation of this receptor.We further analyzed CD8þ T cells and DCs and observed thatmany CD8þ T cells were located close to the CD31þ tumorvessels along with CD11cþ DCs in the aAVC-injected mice butnot in untreated mice (Figs. 1B and 2E). In addition, the bloodvessels in the tumors of the treated mice were CD31þVCAM-1þICAM-1þ (Fig. 2F), while those in the untreated mice did notexpress these markers. It has been reported that ICAM-1 andVCAM-1 expression is essential for normalization of tumorvasculature, an effect should be important for vaccine efficacy(26, 27). Thus, the closely aggregated structures composed ofCD11cþ DCs and antigen-specific CD8þ T cells around theCD31þVCAM-1þICAM-1þ vessels seen in tumor sites resem-bled previously described TLS.
TCRVb repertoire of tumor-infiltrating CD8þ T cells aftertreatment with aAVC
We analyzed antigen-specific CD8þ T cells in spleen and tumorsites in tumor-bearing aAVC-treated or untreated mice. As shownin Fig. 3A and B, the frequency ofOVA-specific CD8þ T cells in thetumor was higher than in the spleen (30% vs. 7% respectively) inaAVC-OVA–treated mice. The OVA-tetramerþCD8þ T cells in the
Figure 2.Analyses of TLS formed in the tumor after immunization with aAVC. C57BL/6 mice were injected with 2 � 105 MO4 cells subcutaneously and then treated with orwithout 5 � 105 aAVC-OVA at day 7. A–C, expression of CXCL9, 10, and 11 in the tumor site (A) and in DCs (CD11cþ ClassIIþF4/80� cells) sorted from thetumors at day 8 (B) was assessed by real-time PCR (mean� SEM, n¼ 6; �, P <0.05; Student t test, �� , P <0.005). C, the expression ofCXCL10 at the protein level wasassessed by IHC (n ¼ 6). Scale bar, 100 mm. D, CXCR3 expression on OVA-tetramerþCD8þ T cells were assessed in the spleen (top) and TIL (bottom) atday 14 by flow cytometry. (n¼ 6). E, at day 14–17, CD8þ T cells, vessels, and CD11cþDCs in the tumorswere assessed by immunofluorescent staining using CD8 (red),CD31 (blue), and CD11c (green). CD11cþ cell numbers/mm2 are shown (mean � SEM, n ¼ 6). Scale bar, 100 mm. F, the phenotype of CD31þ vessels in tumorsite was also analyzed using CD31 (green), VCAM-1 or ICAM-1 (red), and DAPI (n ¼ 6). Scale bar, 100 mm.
aAVC Therapy Generates Systemic and Local Immunosurveillance
www.aacrjournals.org Cancer Res; 76(13) July 1, 2016 3759
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
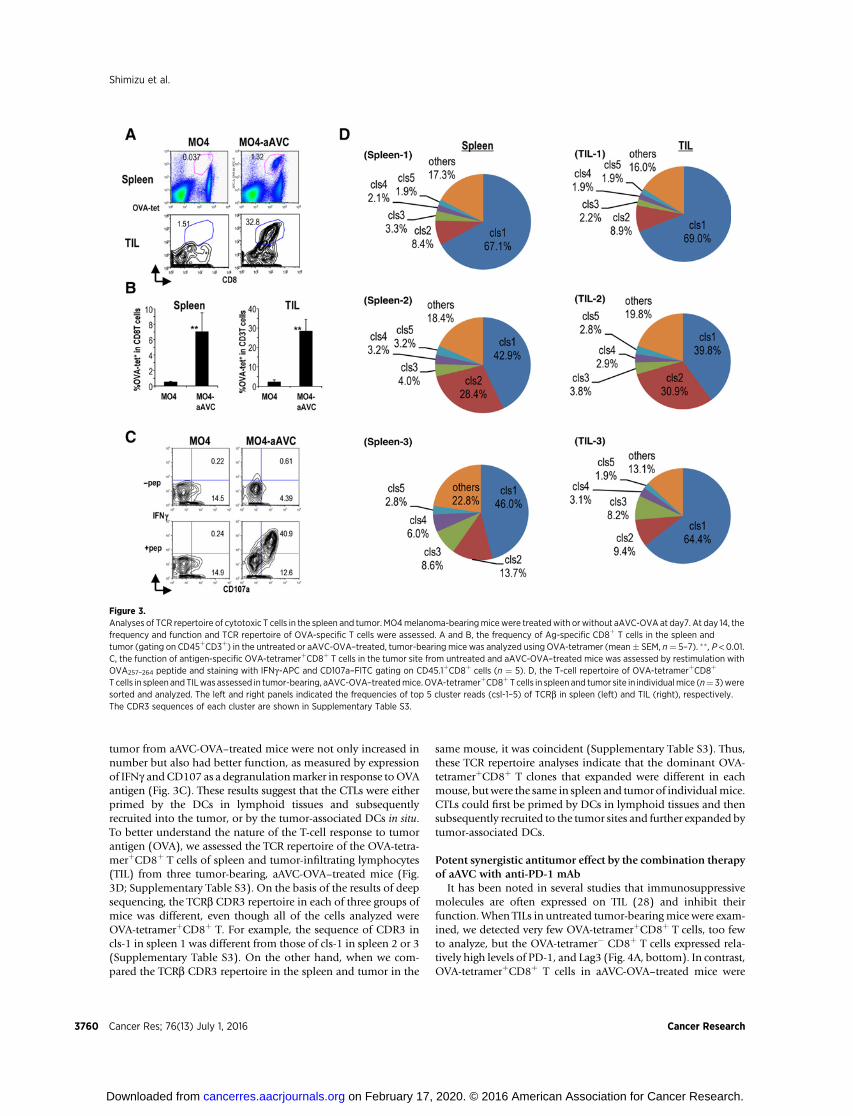
tumor from aAVC-OVA–treated mice were not only increased innumber but also had better function, as measured by expressionof IFNg andCD107 as a degranulationmarker in response toOVAantigen (Fig. 3C). These results suggest that the CTLs were eitherprimed by the DCs in lymphoid tissues and subsequentlyrecruited into the tumor, or by the tumor-associated DCs in situ.To better understand the nature of the T-cell response to tumorantigen (OVA), we assessed the TCR repertoire of the OVA-tetra-merþCD8þ T cells of spleen and tumor-infiltrating lymphocytes(TIL) from three tumor-bearing, aAVC-OVA–treated mice (Fig.3D; Supplementary Table S3). On the basis of the results of deepsequencing, the TCRb CDR3 repertoire in each of three groups ofmice was different, even though all of the cells analyzed wereOVA-tetramerþCD8þ T. For example, the sequence of CDR3 incls-1 in spleen 1 was different from those of cls-1 in spleen 2 or 3(Supplementary Table S3). On the other hand, when we com-pared the TCRb CDR3 repertoire in the spleen and tumor in the
same mouse, it was coincident (Supplementary Table S3). Thus,these TCR repertoire analyses indicate that the dominant OVA-tetramerþCD8þ T clones that expanded were different in eachmouse, butwere the same in spleen and tumor of individualmice.CTLs could first be primed by DCs in lymphoid tissues and thensubsequently recruited to the tumor sites and further expanded bytumor-associated DCs.
Potent synergistic antitumor effect by the combination therapyof aAVC with anti-PD-1 mAb
It has been noted in several studies that immunosuppressivemolecules are often expressed on TIL (28) and inhibit theirfunction.When TILs in untreated tumor-bearingmice were exam-ined, we detected very few OVA-tetramerþCD8þ T cells, too fewto analyze, but the OVA-tetramer� CD8þ T cells expressed rela-tively high levels of PD-1, and Lag3 (Fig. 4A, bottom). In contrast,OVA-tetramerþCD8þ T cells in aAVC-OVA–treated mice were
Figure 3.Analyses of TCR repertoire of cytotoxic T cells in the spleen and tumor. MO4melanoma-bearingmicewere treatedwith or without aAVC-OVA at day7. At day 14, thefrequency and function and TCR repertoire of OVA-specific T cells were assessed. A and B, the frequency of Ag-specific CD8þ T cells in the spleen andtumor (gating on CD45þCD3þ) in the untreated or aAVC-OVA–treated, tumor-bearing mice was analyzed using OVA-tetramer (mean� SEM, n¼ 5–7). �� , P < 0.01.C, the function of antigen-specific OVA-tetramerþCD8þ T cells in the tumor site from untreated and aAVC-OVA–treated mice was assessed by restimulation withOVA257–264 peptide and staining with IFNg-APC and CD107a–FITC gating on CD45.1þCD8þ cells (n ¼ 5). D, the T-cell repertoire of OVA-tetramerþCD8þ
T cells in spleen and TILwas assessed in tumor-bearing, aAVC-OVA–treatedmice. OVA-tetramerþCD8þT cells in spleen and tumor site in individualmice (n¼ 3)weresorted and analyzed. The left and right panels indicated the frequencies of top 5 cluster reads (csl-1–5) of TCRb in spleen (left) and TIL (right), respectively.The CDR3 sequences of each cluster are shown in Supplementary Table S3.
Shimizu et al.
Cancer Res; 76(13) July 1, 2016 Cancer Research3760
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
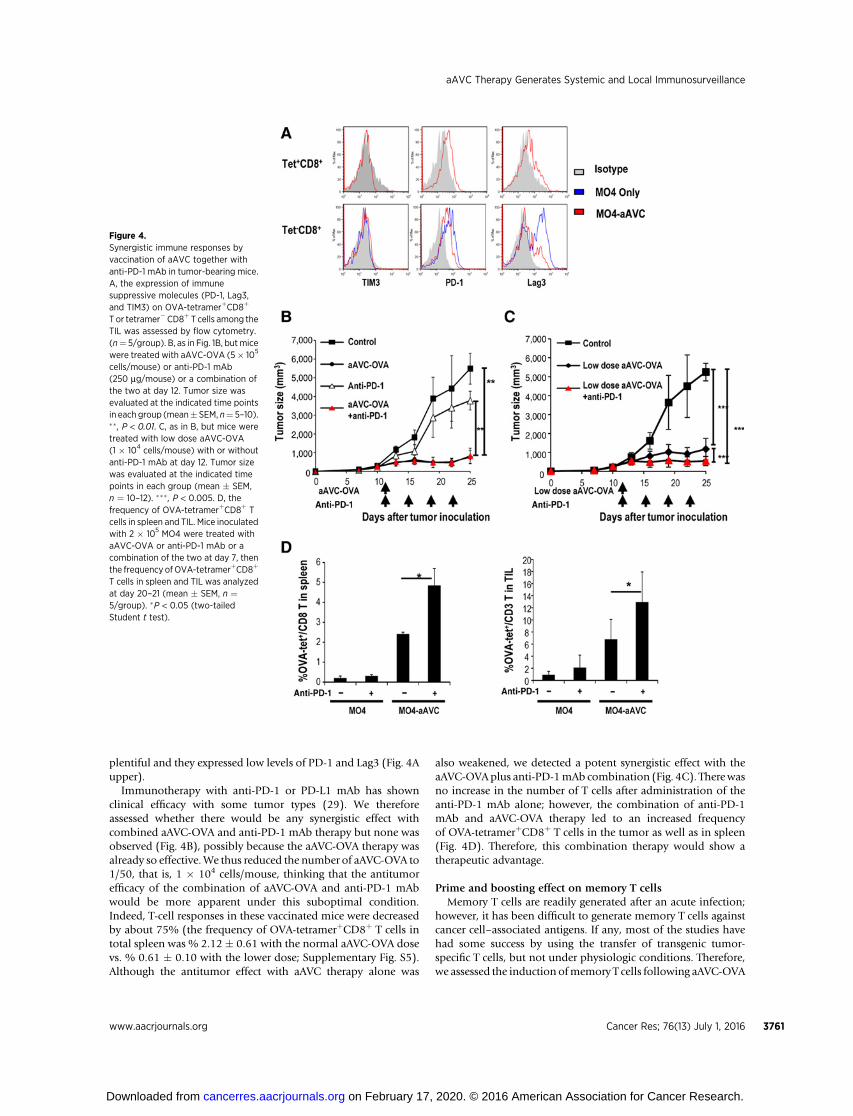
plentiful and they expressed low levels of PD-1 and Lag3 (Fig. 4Aupper).
Immunotherapy with anti-PD-1 or PD-L1 mAb has shownclinical efficacy with some tumor types (29). We thereforeassessed whether there would be any synergistic effect withcombined aAVC-OVA and anti-PD-1 mAb therapy but none wasobserved (Fig. 4B), possibly because the aAVC-OVA therapy wasalready so effective.We thus reduced the number of aAVC-OVA to1/50, that is, 1 � 104 cells/mouse, thinking that the antitumorefficacy of the combination of aAVC-OVA and anti-PD-1 mAbwould be more apparent under this suboptimal condition.Indeed, T-cell responses in these vaccinated mice were decreasedby about 75% (the frequency of OVA-tetramerþCD8þ T cells intotal spleen was % 2.12 � 0.61 with the normal aAVC-OVA dosevs. % 0.61 � 0.10 with the lower dose; Supplementary Fig. S5).Although the antitumor effect with aAVC therapy alone was
also weakened, we detected a potent synergistic effect with theaAVC-OVAplus anti-PD-1mAb combination (Fig. 4C). Therewasno increase in the number of T cells after administration of theanti-PD-1 mAb alone; however, the combination of anti-PD-1mAb and aAVC-OVA therapy led to an increased frequencyof OVA-tetramerþCD8þ T cells in the tumor as well as in spleen(Fig. 4D). Therefore, this combination therapy would show atherapeutic advantage.
Prime and boosting effect on memory T cellsMemory T cells are readily generated after an acute infection;
however, it has been difficult to generate memory T cells againstcancer cell–associated antigens. If any, most of the studies havehad some success by using the transfer of transgenic tumor-specific T cells, but not under physiologic conditions. Therefore,we assessed the induction ofmemory T cells following aAVC-OVA
Figure 4.Synergistic immune responses byvaccination of aAVC together withanti-PD-1 mAb in tumor-bearing mice.A, the expression of immunesuppressive molecules (PD-1, Lag3,and TIM3) on OVA-tetramerþCD8þ
T or tetramer�CD8þ T cells among theTIL was assessed by flow cytometry.(n¼ 5/group). B, as in Fig. 1B, butmicewere treated with aAVC-OVA (5� 105
cells/mouse) or anti-PD-1 mAb(250 mg/mouse) or a combination ofthe two at day 12. Tumor size wasevaluated at the indicated time pointsin each group (mean� SEM, n¼ 5–10).�� , P < 0.01. C, as in B, but mice weretreated with low dose aAVC-OVA(1 � 104 cells/mouse) with or withoutanti-PD-1 mAb at day 12. Tumor sizewas evaluated at the indicated timepoints in each group (mean � SEM,n ¼ 10–12). ��� , P < 0.005. D, thefrequency of OVA-tetramerþCD8þ Tcells in spleen and TIL. Mice inoculatedwith 2 � 105 MO4 were treated withaAVC-OVA or anti-PD-1 mAb or acombination of the two at day 7, thenthe frequency ofOVA-tetramerþCD8þ
T cells in spleen and TIL was analyzedat day 20–21 (mean � SEM, n ¼5/group). �P < 0.05 (two-tailedStudent t test).
aAVC Therapy Generates Systemic and Local Immunosurveillance
www.aacrjournals.org Cancer Res; 76(13) July 1, 2016 3761
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
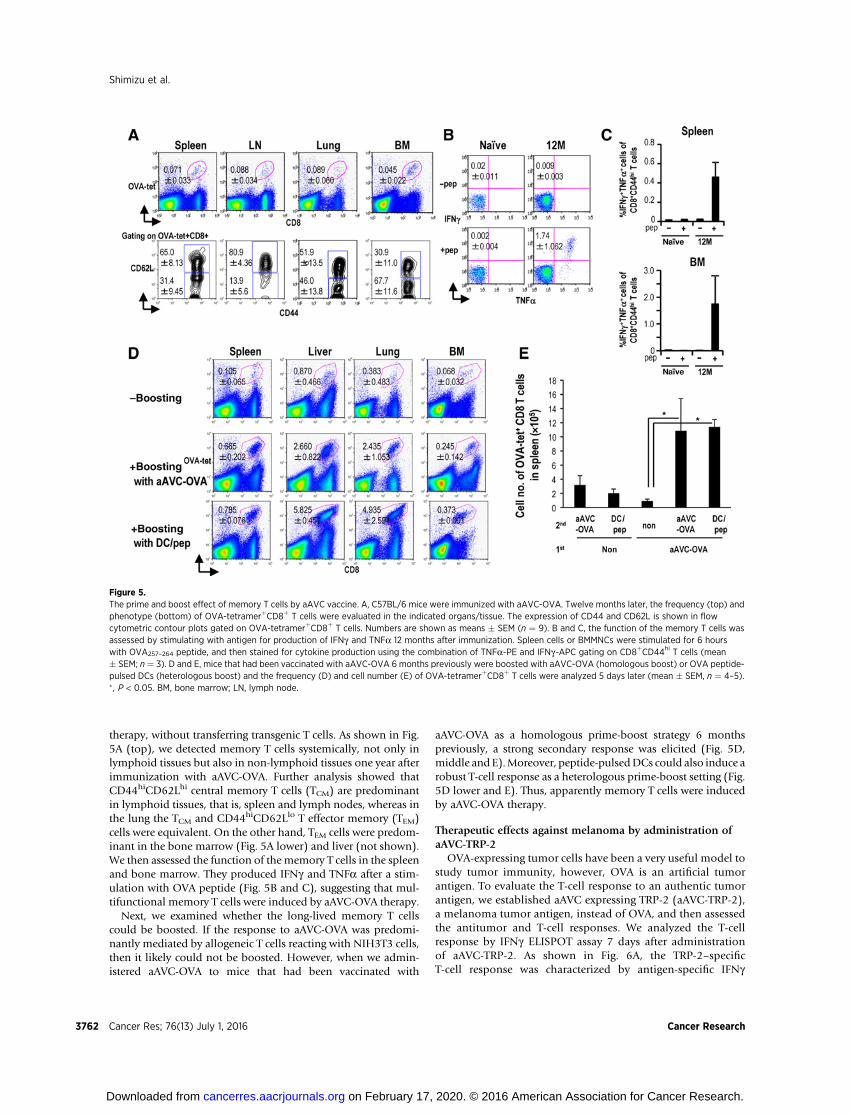
therapy, without transferring transgenic T cells. As shown in Fig.5A (top), we detected memory T cells systemically, not only inlymphoid tissues but also in non-lymphoid tissues one year afterimmunization with aAVC-OVA. Further analysis showed thatCD44hiCD62Lhi central memory T cells (TCM) are predominantin lymphoid tissues, that is, spleen and lymph nodes, whereas inthe lung the TCM and CD44hiCD62Llo T effector memory (TEM)cells were equivalent. On the other hand, TEM cells were predom-inant in the bone marrow (Fig. 5A lower) and liver (not shown).We then assessed the function of the memory T cells in the spleenand bone marrow. They produced IFNg and TNFa after a stim-ulation with OVA peptide (Fig. 5B and C), suggesting that mul-tifunctional memory T cells were induced by aAVC-OVA therapy.
Next, we examined whether the long-lived memory T cellscould be boosted. If the response to aAVC-OVA was predomi-nantly mediated by allogeneic T cells reacting with NIH3T3 cells,then it likely could not be boosted. However, when we admin-istered aAVC-OVA to mice that had been vaccinated with
aAVC-OVA as a homologous prime-boost strategy 6 monthspreviously, a strong secondary response was elicited (Fig. 5D,middle and E).Moreover, peptide-pulsedDCs could also induce arobust T-cell response as a heterologous prime-boost setting (Fig.5D lower and E). Thus, apparently memory T cells were inducedby aAVC-OVA therapy.
Therapeutic effects against melanoma by administration ofaAVC-TRP-2
OVA-expressing tumor cells have been a very useful model tostudy tumor immunity, however, OVA is an artificial tumorantigen. To evaluate the T-cell response to an authentic tumorantigen, we established aAVC expressing TRP-2 (aAVC-TRP-2),a melanoma tumor antigen, instead of OVA, and then assessedthe antitumor and T-cell responses. We analyzed the T-cellresponse by IFNg ELISPOT assay 7 days after administrationof aAVC-TRP-2. As shown in Fig. 6A, the TRP-2–specificT-cell response was characterized by antigen-specific IFNg
Figure 5.The prime and boost effect of memory T cells by aAVC vaccine. A, C57BL/6 mice were immunized with aAVC-OVA. Twelve months later, the frequency (top) andphenotype (bottom) of OVA-tetramerþCD8þ T cells were evaluated in the indicated organs/tissue. The expression of CD44 and CD62L is shown in flowcytometric contour plots gated on OVA-tetramerþCD8þ T cells. Numbers are shown as means � SEM (n ¼ 9). B and C, the function of the memory T cells wasassessed by stimulating with antigen for production of IFNg and TNFa 12 months after immunization. Spleen cells or BMMNCs were stimulated for 6 hourswith OVA257–264 peptide, and then stained for cytokine production using the combination of TNFa-PE and IFNg-APC gating on CD8þCD44hi T cells (mean� SEM; n¼ 3). D and E, mice that had been vaccinated with aAVC-OVA 6 months previously were boosted with aAVC-OVA (homologous boost) or OVA peptide-pulsed DCs (heterologous boost) and the frequency (D) and cell number (E) of OVA-tetramerþCD8þ T cells were analyzed 5 days later (mean � SEM, n ¼ 4–5).� , P < 0.05. BM, bone marrow; LN, lymph node.
Shimizu et al.
Cancer Res; 76(13) July 1, 2016 Cancer Research3762
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
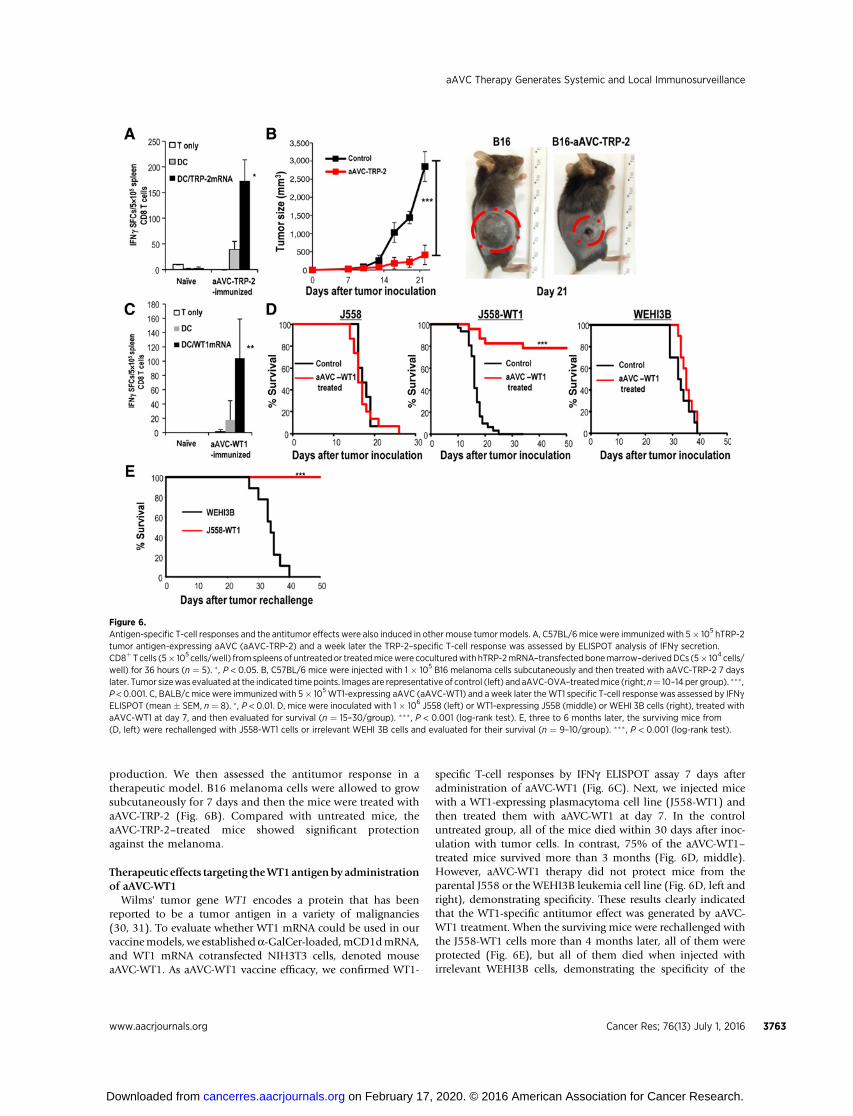
production. We then assessed the antitumor response in atherapeutic model. B16 melanoma cells were allowed to growsubcutaneously for 7 days and then the mice were treated withaAVC-TRP-2 (Fig. 6B). Compared with untreated mice, theaAVC-TRP-2–treated mice showed significant protectionagainst the melanoma.
Therapeutic effects targeting theWT1antigenby administrationof aAVC-WT1
Wilms' tumor gene WT1 encodes a protein that has beenreported to be a tumor antigen in a variety of malignancies(30, 31). To evaluate whether WT1 mRNA could be used in ourvaccinemodels, we establisheda-GalCer-loaded,mCD1dmRNA,and WT1 mRNA cotransfected NIH3T3 cells, denoted mouseaAVC-WT1. As aAVC-WT1 vaccine efficacy, we confirmed WT1-
specific T-cell responses by IFNg ELISPOT assay 7 days afteradministration of aAVC-WT1 (Fig. 6C). Next, we injected micewith a WT1-expressing plasmacytoma cell line (J558-WT1) andthen treated them with aAVC-WT1 at day 7. In the controluntreated group, all of the mice died within 30 days after inoc-ulation with tumor cells. In contrast, 75% of the aAVC-WT1–treated mice survived more than 3 months (Fig. 6D, middle).However, aAVC-WT1 therapy did not protect mice from theparental J558 or the WEHI3B leukemia cell line (Fig. 6D, left andright), demonstrating specificity. These results clearly indicatedthat the WT1-specific antitumor effect was generated by aAVC-WT1 treatment. When the surviving mice were rechallenged withthe J558-WT1 cells more than 4 months later, all of them wereprotected (Fig. 6E), but all of them died when injected withirrelevant WEHI3B cells, demonstrating the specificity of the
Figure 6.Antigen-specific T-cell responses and the antitumor effects were also induced in other mouse tumor models. A, C57BL/6mice were immunized with 5� 105 hTRP-2tumor antigen-expressing aAVC (aAVC-TRP-2) and a week later the TRP-2–specific T-cell response was assessed by ELISPOT analysis of IFNg secretion.CD8þT cells (5� 105 cells/well) from spleens of untreatedor treatedmicewere coculturedwith hTRP-2mRNA–transfected bonemarrow–derivedDCs (5� 104 cells/well) for 36 hours (n ¼ 5). � , P < 0.05. B, C57BL/6 mice were injected with 1 � 105 B16 melanoma cells subcutaneously and then treated with aAVC-TRP-2 7 dayslater. Tumor sizewas evaluated at the indicated time points. Images are representative of control (left) and aAVC-OVA–treatedmice (right; n¼ 10–14 per group). ��� ,P < 0.001. C, BALB/cmice were immunized with 5� 105WT1-expressing aAVC (aAVC-WT1) and a week later theWT1 specific T-cell response was assessed by IFNgELISPOT (mean � SEM, n ¼ 8). � , P < 0.01. D, mice were inoculated with 1 � 106 J558 (left) or WT1-expressing J558 (middle) or WEHI 3B cells (right), treated withaAVC-WT1 at day 7, and then evaluated for survival (n ¼ 15–30/group). ���, P < 0.001 (log-rank test). E, three to 6 months later, the surviving mice from(D, left) were rechallenged with J558-WT1 cells or irrelevant WEHI 3B cells and evaluated for their survival (n ¼ 9–10/group). ��� , P < 0.001 (log-rank test).
aAVC Therapy Generates Systemic and Local Immunosurveillance
www.aacrjournals.org Cancer Res; 76(13) July 1, 2016 3763
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
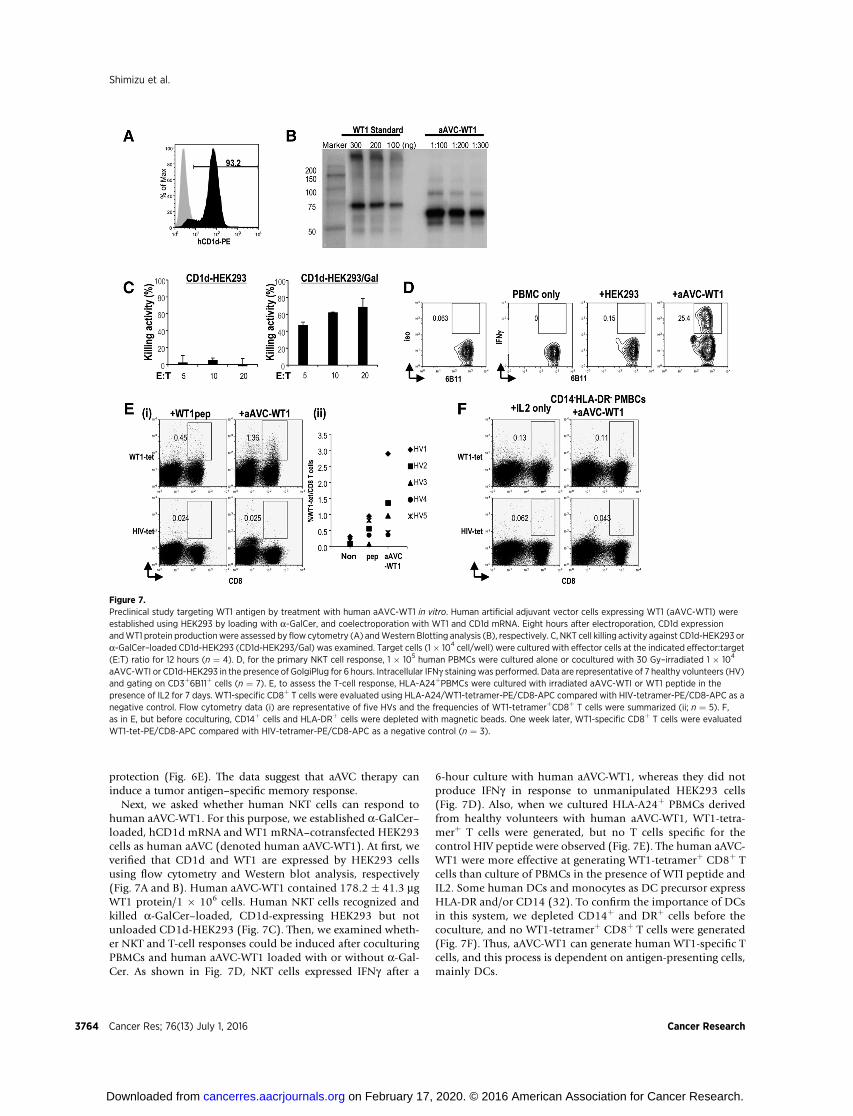
protection (Fig. 6E). The data suggest that aAVC therapy caninduce a tumor antigen–specific memory response.
Next, we asked whether human NKT cells can respond tohuman aAVC-WT1. For this purpose, we established a-GalCer–loaded, hCD1d mRNA and WT1 mRNA–cotransfected HEK293cells as human aAVC (denoted human aAVC-WT1). At first, weverified that CD1d and WT1 are expressed by HEK293 cellsusing flow cytometry and Western blot analysis, respectively(Fig. 7A and B). Human aAVC-WT1 contained 178.2 � 41.3 mgWT1 protein/1 � 106 cells. Human NKT cells recognized andkilled a-GalCer–loaded, CD1d-expressing HEK293 but notunloaded CD1d-HEK293 (Fig. 7C). Then, we examined wheth-er NKT and T-cell responses could be induced after coculturingPBMCs and human aAVC-WT1 loaded with or without a-Gal-Cer. As shown in Fig. 7D, NKT cells expressed IFNg after a
6-hour culture with human aAVC-WT1, whereas they did notproduce IFNg in response to unmanipulated HEK293 cells(Fig. 7D). Also, when we cultured HLA-A24þ PBMCs derivedfrom healthy volunteers with human aAVC-WT1, WT1-tetra-merþ T cells were generated, but no T cells specific for thecontrol HIV peptide were observed (Fig. 7E). The human aAVC-WT1 were more effective at generating WT1-tetramerþ CD8þ Tcells than culture of PBMCs in the presence of WTI peptide andIL2. Some human DCs and monocytes as DC precursor expressHLA-DR and/or CD14 (32). To confirm the importance of DCsin this system, we depleted CD14þ and DRþ cells before thecoculture, and no WT1-tetramerþ CD8þ T cells were generated(Fig. 7F). Thus, aAVC-WT1 can generate human WT1-specific Tcells, and this process is dependent on antigen-presenting cells,mainly DCs.
Figure 7.Preclinical study targeting WT1 antigen by treatment with human aAVC-WT1 in vitro. Human artificial adjuvant vector cells expressing WT1 (aAVC-WT1) wereestablished using HEK293 by loading with a-GalCer, and coelectroporation with WT1 and CD1d mRNA. Eight hours after electroporation, CD1d expressionandWT1 protein productionwere assessed by flow cytometry (A) andWestern Blotting analysis (B), respectively. C, NKT cell killing activity against CD1d-HEK293 ora-GalCer–loaded CD1d-HEK293 (CD1d-HEK293/Gal) was examined. Target cells (1� 104 cell/well) were cultured with effector cells at the indicated effector:target(E:T) ratio for 12 hours (n ¼ 4). D, for the primary NKT cell response, 1 � 105 human PBMCs were cultured alone or cocultured with 30 Gy–irradiated 1 � 104
aAVC-WTI or CD1d-HEK293 in the presence of GolgiPlug for 6 hours. Intracellular IFNg staining was performed. Data are representative of 7 healthy volunteers (HV)and gating on CD3þ6B11þ cells (n ¼ 7). E, to assess the T-cell response, HLA-A24þPBMCs were cultured with irradiated aAVC-WTI or WT1 peptide in thepresence of IL2 for 7 days. WT1-specific CD8þ T cells were evaluated using HLA-A24/WT1-tetramer-PE/CD8-APC compared with HIV-tetramer-PE/CD8-APC as anegative control. Flow cytometry data (i) are representative of five HVs and the frequencies of WT1-tetramerþCD8þ T cells were summarized (ii; n ¼ 5). F,as in E, but before coculturing, CD14þ cells and HLA-DRþ cells were depleted with magnetic beads. One week later, WT1-specific CD8þ T cells were evaluatedWT1-tet-PE/CD8-APC compared with HIV-tetramer-PE/CD8-APC as a negative control (n ¼ 3).
Shimizu et al.
Cancer Res; 76(13) July 1, 2016 Cancer Research3764
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

DiscussionIn some types of human cancer, including melanoma (33),
colorectal cancer (34), breast cancer (35) and lung cancer (36), acorrelation between a low density of CD8þ T cells in the tumorand poor prognosis has been reported. These cases where there isminimal T-cell infiltration are termed non-T-cell–inflamedtumors (37) and therefore inefficient T-cell migration into thetumor appears to be amajor limitation of cancer immunotherapy(27). We showed how aAVC can show better T-cell–mediatedantitumor efficacy at the tumor microenvironment in an aggres-sive tumor model. As a mechanistic summary, in vivo DC activa-tion can induce the formation of TLS in whichDCs recruited fromlymphoid tissues may induce the formation of ICAM-1- andVCAM-1–expressing blood vessels, resulting in normalization ofthe vasculature within the tumor. Subsequently, CXCR3-expres-sing CTLs are efficiently recruited to CXCR3 ligand-expressingDCs, resulting in the aggregation of CTLs in the tumor.Wedirectlyshowed such a mechanism for aAVC therapy in this study. Thereare some fragmentary clinical reports describing a correlationbetween the existence of tumor-associated TLS in patient samplesand clinical prognosis (8). Coexistence of effector T cells and ahigh density of mature DCs in TLS is correlated with longer termsurvival in lung cancer patients (38). The immunologic resultsobtained inmurine studies with our vaccine were similar to thosein the patients with a good clinical prognosis.
PD-1 blockade is a promising therapy because it can have abeneficial effect on approximately 30% of cancer patients (39);however, some types of cancer patients are refractory to thistreatment. There are some reports accounting for the differencesbetween the two patient groups, such as PD-L1 expression ontumors, IFNg production, and preexisting T cells in the tumor sites(37, 39). Preexisting PD-1hiCD8þ T cells in the tumor site havebeen known to be dysfunctional, but PD-1loCD8þ T cells arecapable of producing IFNg and TNFa, that is functional (39, 40).In addition, PD-1loCD8þ T cells can easily be stimulated by anti-PD-1mAb (40). Recently,many strategies have been explored thatmight be combined with the PD1 blockade therapy. In fact, weshowed here that the MO4 melanoma was resistant to anti-PD-1mAb alone, but that a combination therapy with low dose ofaAVC therapy and anti-PD-1 mAb was effective. InfiltratingPD-1loCD8þ T cells in the tumor that were generated by aAVCtreatment (Fig. 4A) is likely one reason for this successful out-come. Such an increase of antigen-specific T cells in the tumor bysynergistic combination therapy might be useful for patients whoare resistant to PD-1 blockade therapy.
Generationofmemory T cells is one hallmark of vaccine successthat may possibly control the long-term antitumor immune
response. Criteria and quality of ideal T-cell memory followingvaccination has been defined in many studies, typically in viralinfection models, and include: (i) long-term maintenance, (ii)recall response to reencounter with the same pathogen, and (iii)multifunctionality, for example, in terms of cytokine production(41–43). In this study, we verified that thememory T cells inducedby the aAVCvaccine completelymet the above criteria formemoryT cells.
Altogether, our findings with the current aAVC vaccine empha-size a novel role for DC activation in situ that shapes both local(i.e., within the tumor) and systemic (i.e., memory T cells)immune responses due to the linkage of innate and adaptiveimmunity. Further analysis of these local and systemic immuneresponses may give us important clues about how the antitumorimmunologic network operates and should be helpful for devel-oping the next generation of immunotherapy.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: K. Shimizu, S.-I. FujiiDevelopment of methodology: K. Shimizu, M. Asakura, S.-I. FujiiAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): K. Shimizu, S. Yamasaki, J. Shinga, Y. Sato, O. Ohara,H. Yagita, S.-I. FujiiAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): K. Shimizu, T. Watanabe, O. Ohara, M. Asakura,S.-I. FujiiWriting, review, and/or revision of the manuscript: K. Shimizu, H. Yagita,S.-I. FujiiAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): K. Kuzushima, Y. KomuroStudy supervision: S.-I. Fujii
AcknowledgmentsThe authors thankDrs. P.D. Burrows (University of Alabama) and I.Mellman
(Genentech) for critical reading of the manuscript and Ms. C. Oikawa, Ms. M.Sakurai, andMr.M. Kawamura (RIKEN, IMS) for providing technical assistance.
Grant SupportThiswork is supported by JSPSKAKENHI grants (K. Shimizu and S. Fujii) and
the Japan Agency for Medical Research and Development (TranslationalResearch Network Program; S. Fujii).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received December 3, 2015; revised March 22, 2016; accepted April 4, 2016;published online July 1, 2016.
References1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity
cycle. Immunity 2013;39:1–10.2. Robbins PF,Morgan RA, Feldman SA, Yang JC, Sherry RM,DudleyME, et al.
Tumor regression in patients with metastatic synovial cell sarcoma andmelanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011;29:917–24.
3. Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptiveimmunotherapy for cancer or viruses. Annu Rev Immunol 2014;32:189–225.
4. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade incancer therapy. J Clin Oncol 2015;33:1974–82.
5. Steinman RM. Decisions about dendritic cells: past, present, and future.Annu Rev Immunol 2012;30:1–22.
6. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines.Immunity 2013;39:38–48.
7. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in thetumor microenvironment. Nat Immunol 2013;14:1014–22.
8. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH.Tertiary lymphoid structures in cancer and beyond. Trends Immunol2014;35:571–80.
9. Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate toadaptive immunity via maturing dendritic cells in vivo requires CD40
www.aacrjournals.org Cancer Res; 76(13) July 1, 2016 3765
aAVC Therapy Generates Systemic and Local Immunosurveillance
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

ligation in addition to antigen presentation and CD80/86 costimulation.J Exp Med 2004;199:1607–18.
10. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of naturalkiller T cells bya-galactosylceramide rapidly induces the fullmaturation ofdendritic cells in vivo and thereby acts as an adjuvant for combined CD4and CD8 T cell immunity to a co-administered protein. J Exp Med2003;198:267–79.
11. Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Va14þ natural killer Tcellsmature dendritic cells, leading to strong adaptive immunity. ImmunolRev 2007;220:183–98.
12. Fujii S, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, et al.NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol2013;4:409.
13. Faveeuw C, Trottein F. Optimization of natural killer T cell-mediatedimmunotherapy in cancer using cell-based and nanovector vaccines.Cancer Res 2014;74:1632–8.
14. Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, SchmiegJ, Kronenberg M, et al. Natural killer T cell ligand a-galactosylceramideenhances protective immunity induced by malaria vaccines. J Exp Med2002;195:617–24.
15. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, et al. NKTcells enhance CD4þ and CD8þ T cell responses to soluble antigenin vivo through direct interaction with dendritic cells. J Immunol 2003;171:5140–47.
16. Fujii S, Goto A, Shimizu K. Antigen mRNA-transfected, allogeneic fibro-blasts loaded with NKT-cell ligand confer antitumor immunity. Blood2009;113:4262–72.
17. Shimizu K, Mizuno T, Shinga J, Asakura M, Kakimi K, Ishii Y, et al.Vaccination with antigen-transfected, NKT cell ligand-loaded, human cellselicits robust in situ immune responses by dendritic cells. Cancer Res2013;73:62–73.
18. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al.Expression of the PD-1 antigen on the surface of stimulatedmouse T and Blymphocytes. Int Immunol 1996;8:765–72.
19. Falo LD Jr, Kovacsovics-Bankowski M, Thompson K, Rock KL. Targetingantigen into the phagocytic pathway in vivo induces protective tumourimmunity. Nat Med 1995;1:649–53.
20. Guilloux Y, Bai XF, Liu X, Zheng P, Liu Y. Optimal induction ofeffector but not memory antitumor cytotoxic T lymphocytes involvesdirect antigen presentation by the tumor cells. Cancer Res 2001;61:1107–12.
21. Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T,et al. In vivo depletion of CD11cþ dendritic cells abrogates priming ofCD8þ T cells by exogenous cell-associated antigens. Immunity 2002;17:211–20.
22. Yamazaki C, Sugiyama M, Ohta T, Hemmi H, Hamada E, Sasaki I, et al.Critical roles of a dendritic cell subset expressing a chemokine receptor,XCR1. J Immunol 2013;190:6071–82.
23. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged interferon-gproducing NKT response induced with a-galactosylceramide-loaded den-dritic cells. Nat Immunol 2002;3:867–74.
24. Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S. Cross-pre-sentation of glycolipid from tumor cells loaded with a-galactosylceramideleads to potent and long-lived T cellmediated immunity via dendritic cells.J Exp Med 2007;204:2641–53.
25. Shimizu K, Sato Y, Shinga J, Watanabe T, Endo T, Asakura M, et al. KLRGþinvariant natural killer T cells are long-lived effectors. Proc Natl Acad SciU S A 2014;111:12474–9.
26. Klug F, PrakashH,Huber PE, Seibel T, BenderN,HalamaN, et al. Low-doseirradiation programs macrophage differentiation to an iNOS(þ)/M1 phe-notype that orchestrates effective T cell immunotherapy. Cancer Cell2013;24:589–602.
27. Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltrationinto tumours. Clin Exp Immunol 2014;178:1–8.
28. Gros A, Robbins PF, YaoX, Li YF, Turcotte S, Tran E, et al. PD-1 identifies thepatient-specific CD8(þ) tumor-reactive repertoire infiltrating humantumors. J Clin Invest 2014;124:2246–59.
29. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association ofPD-1, PD-1 ligands, and other features of the tumor immune microenvi-ronment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064–74.
30. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al.The prioritization of cancer antigens: a national cancer institute pilotproject for the acceleration of translational research. Clin Cancer Res2009;15:5323–37.
31. Huff V.Wilms' tumours: about tumour suppressor genes, an oncogene anda chameleon gene. Nat Rev Cancer 2011;11:111–21.
32. Schlitzer A, Ginhoux F. Organization of the mouse and human DCnetwork. Curr Opin Immunol 2014;26:90–9.
33. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW,et al. Tumor-infiltrating lymphocyte grade is an independent predictor ofsentinel lymph node status and survival in patients with cutaneousmelanoma. J Clin Oncol 2012;30:2678–83.
34. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al.Effector memory T cells, early metastasis, and survival in colorectal cancer.N Engl J Med 2005;353:2654–66.
35. Menard S, Tomasic G, Casalini P, Balsari A, Pilotti S, Cascinelli N, et al.Lymphoid infiltration as a prognostic variable for early-onset breastcarcinomas. Clin Cancer Res 1997;3:817–9.
36. Hiraoka K, Miyamoto M, Cho Y, Suzuoki M, Oshikiri T, Nakakubo Y, et al.Concurrent infiltration by CD8þ T cells and CD4þ T cells is a favourableprognostic factor in non-small-cell lung carcinoma. Br J Cancer 2006;94:275–80.
37. Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol 2015;36:250–6.
38. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al.Dendritic cells in tumor-associated tertiary lymphoid structures signal aTh1 cytotoxic immune contexture and license the positive prognostic valueof infiltrating CD8þ T cells. Cancer Res 2014;74:705–15.
39. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present,and future. J Clin Invest 2015;125:3384–91.
40. Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, et al. Athreshold level of intratumor CD8þ T-cell PD1 expression dictates ther-apeutic response to anti-PD1. Cancer Res 2015;75:3800–11.
41. Seder RA, Darrah PA, RoedererM. T-cell quality inmemory and protection:implications for vaccine design. Nat Rev Immunol 2008;8:247–58.
42. Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memoryand back. Immunity 2010;33:451–63.
43. Pulendran B, Ahmed R. Immunological mechanisms of vaccination.Nat Immunol 2011;12:509–17.
Cancer Res; 76(13) July 1, 2016 Cancer Research3766
Shimizu et al.
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

2016;76:3756-3766. Cancer Res Kanako Shimizu, Satoru Yamasaki, Jun Shinga, et al.
T Cells+CD8and Shapes the Long-Lived Tumor-Specific Memory Mediated by Systemic DC Activation Modulates the Tumor Microenvironment
Updated version
http://cancerres.aacrjournals.org/content/76/13/3756
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2016/07/08/76.13.3756.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/76/13/3756.full#ref-list-1
This article cites 43 articles, 19 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/76/13/3756.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/76/13/3756To request permission to re-use all or part of this article, use this link
on February 17, 2020. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

















![Review Role of tumor microenvironment in tumorigenesis · Review Role of tumor microenvironment in tumorigenesis Maonan Wang1,2, ... (Figure 1) [1]. Although researchers now have](https://img.pdfslide.tips/doc/110x75/5f1d575396da9a7fe415bbde/review-role-of-tumor-microenvironment-in-tumorigenesis-review-role-of-tumor-microenvironment.jpg)











