Embed Size (px)
Citation preview

LABORATORIO DI CHIMICA FISICALABORATORIO DI CHIMICA FISICAA.AA.A 2011/20122011/2012
I MODULO – 6 crediti(Anna Corrias)
Tecniche di Analisi Termica e Diffrazione di Raggi X
Analisi TermicaCenni teorici
Descrizione esperienze di laboratorio:(Decomposizione termica di Sali; Costruzione del Diagramma di Stato Cd-Bi;
Preparazione leghe Cd-Bi)
Testi consigliati: G. Della Gatta, A Lucci, Principi e Applicazioni di Calorimetria e Analisi Termica,
Casa Editrice PiccinA. R. West, Solid State Chemistry and Its Applications, John Wiley & Sons, Cap. 4.
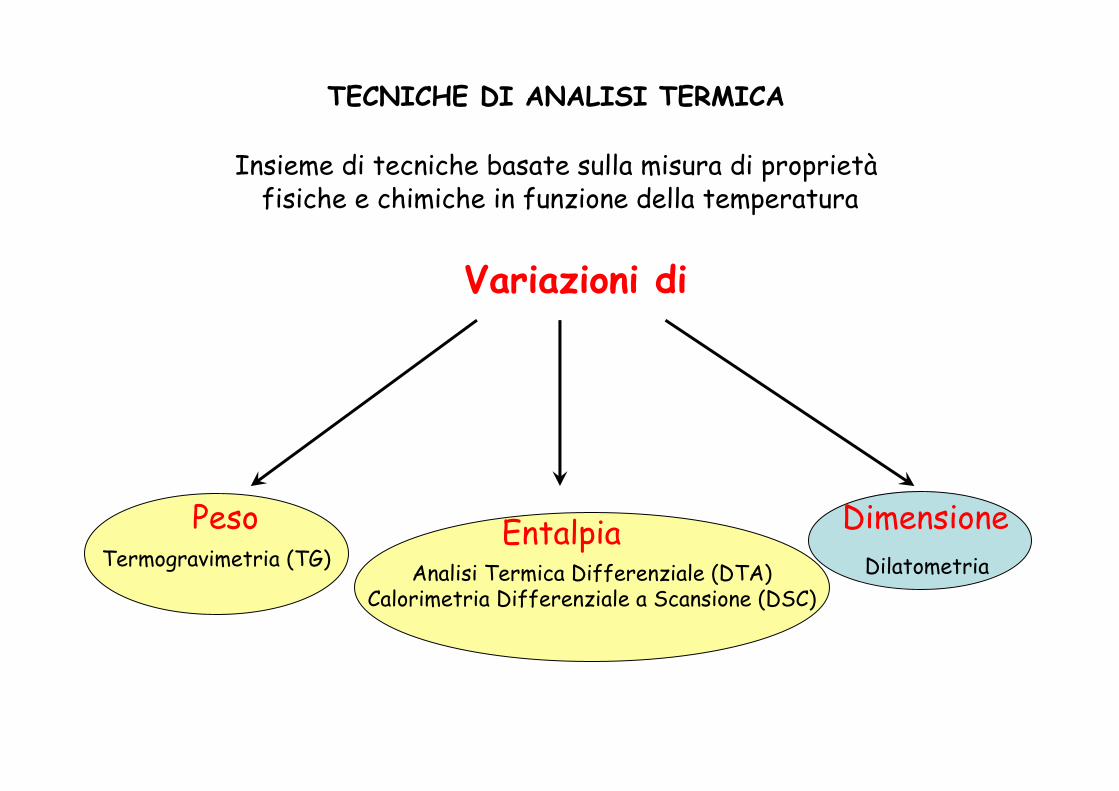
TECNICHE DI ANALISI TERMICA
Insieme di tecniche basate sulla misura di proprietàfisiche e chimiche in funzione della temperatura
Variazioni di
Peso Entalpia DimensioneTermogravimetria (TG) DilatometriaAnalisi Termica Differenziale (DTA)
Calorimetria Differenziale a Scansione (DSC)
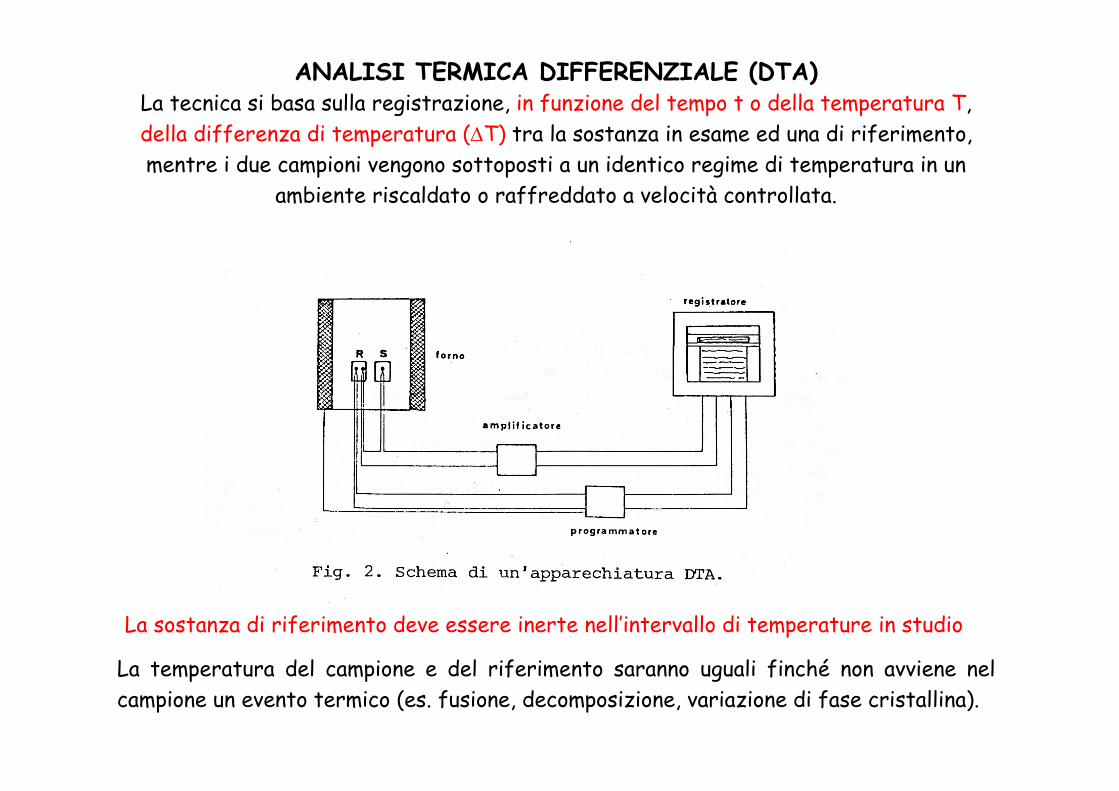
ANALISI TERMICA DIFFERENZIALE (DTA)La tecnica si basa sulla registrazione, in funzione del tempo t o della temperatura T, della differenza di temperatura (ΔT) tra la sostanza in esame ed una di riferimento, mentre i due campioni vengono sottoposti a un identico regime di temperatura in un
ambiente riscaldato o raffreddato a velocità controllata.
La temperatura del campione e del riferimento saranno uguali finché non avviene nel campione un evento termico (es. fusione, decomposizione, variazione di fase cristallina).
La sostanza di riferimento deve essere inerte nell’intervallo di temperature in studio
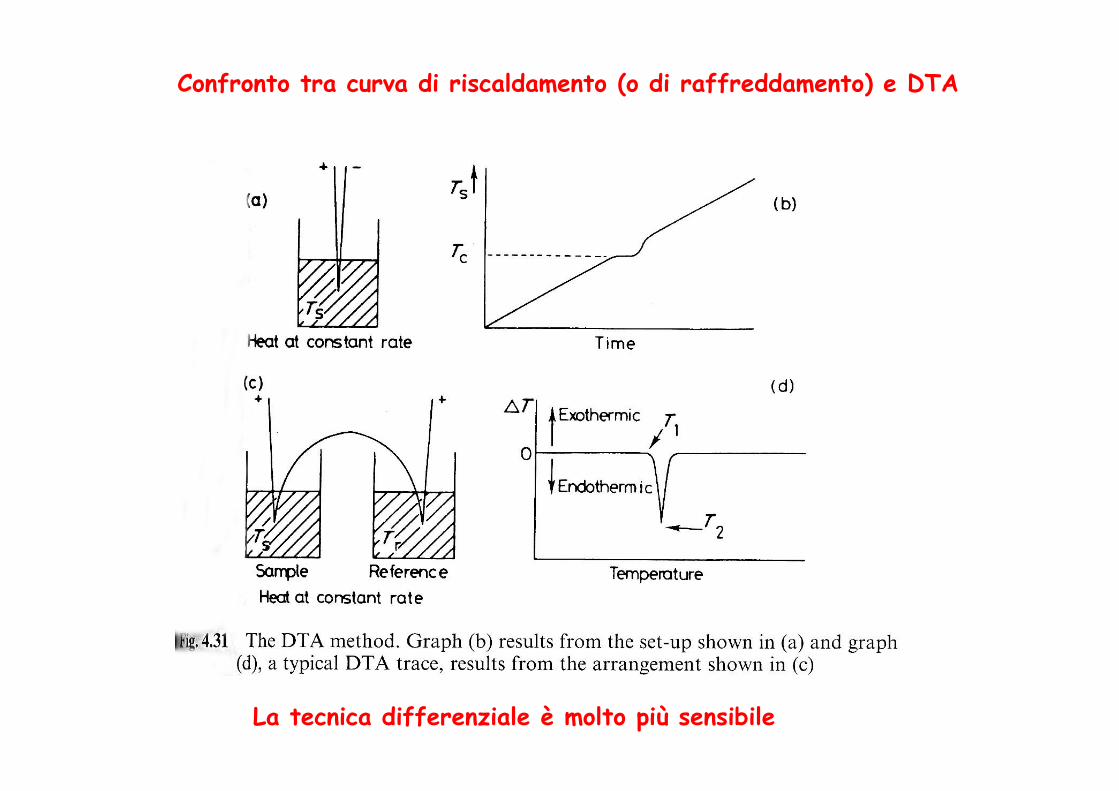
Confronto tra curva di riscaldamento (o di raffreddamento) e DTA
La tecnica differenziale è molto più sensibile

DTA richiede:
Piccole quantità di sostanza per il campione e per il riferimento
Forno capace di creare identiche condizioni termiche intorno al campione e al riferimento
Determinazione della velocità di riscaldamento e della differenza di temperatura tra la sostanza in esame e il riferimento con termocoppie a
contatto con le due sostanze
Portacampioni in:alluminio, allumina, nichel, platino
Termocoppie:cromel/alumel,platino/platino rodio

Requisiti del forno:
Zona con temperatura uniforme
Capacità di riscaldare (e raffreddare) senza inerzia termica
Posizione rispetto ai campioni costante
Requisiti programmatore:
Ampia gamma di velocità
Possibilità di programmare riscaldamenti, raffreddamenti e di mantenere condizioni isoterme
Non generare segnali elettrici che possano influenzare il segnale DTA
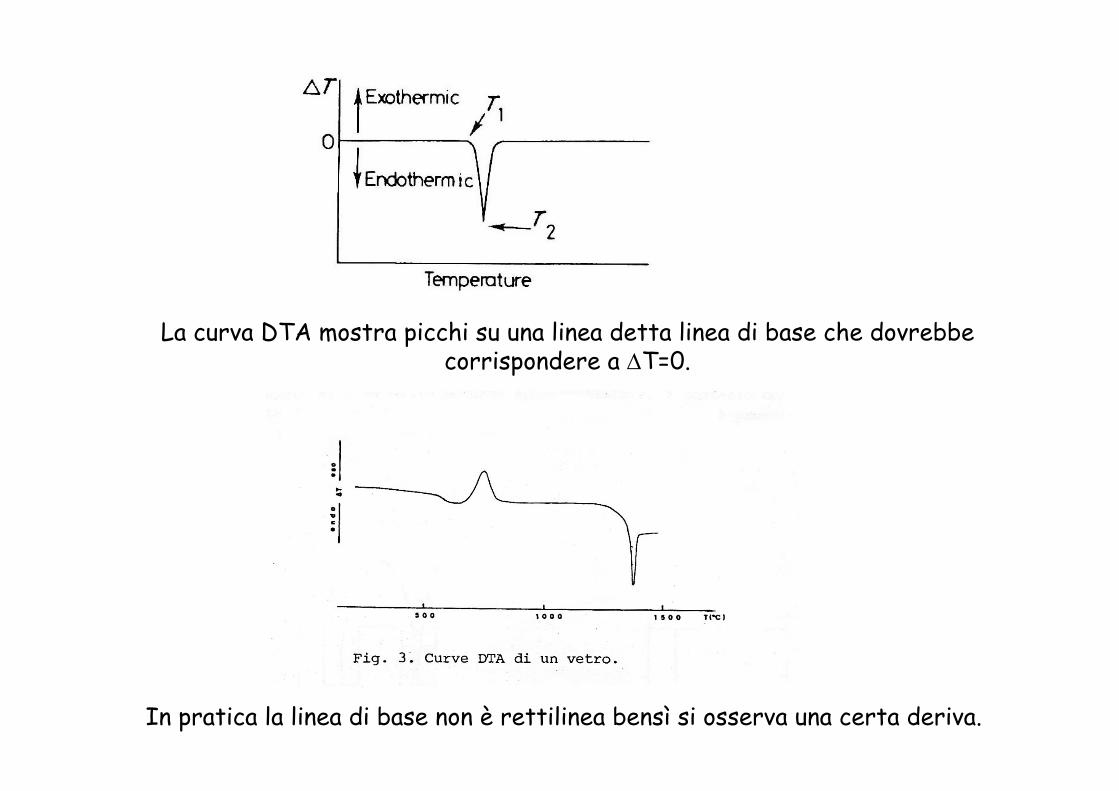
La curva DTA mostra picchi su una linea detta linea di base che dovrebbe corrispondere a ΔT=0.
In pratica la linea di base non è rettilinea bensì si osserva una certa deriva.


Il significato fisico del punto A corrispondente all’inizio del picco e del punto B corrispondente al massimo del picco dipende dalla sistemazione delle termocoppie
Se le termocoppie sono immerse nel campione la temperatura di transizione è molto prossima a quella del massimo del picco.
Se le termocoppie sono a contatto col fondo del portacampione la temperatura di transizione è vicina a quella dello scostamento della linea di base.

Area del picco
m massa di sostanza che si trasformaΔH variazione di entalpia associata alla trasformazioneλ conduttivita’ termica del campioneg fattore geometrico
gHmA
⋅Δ⋅
=λ
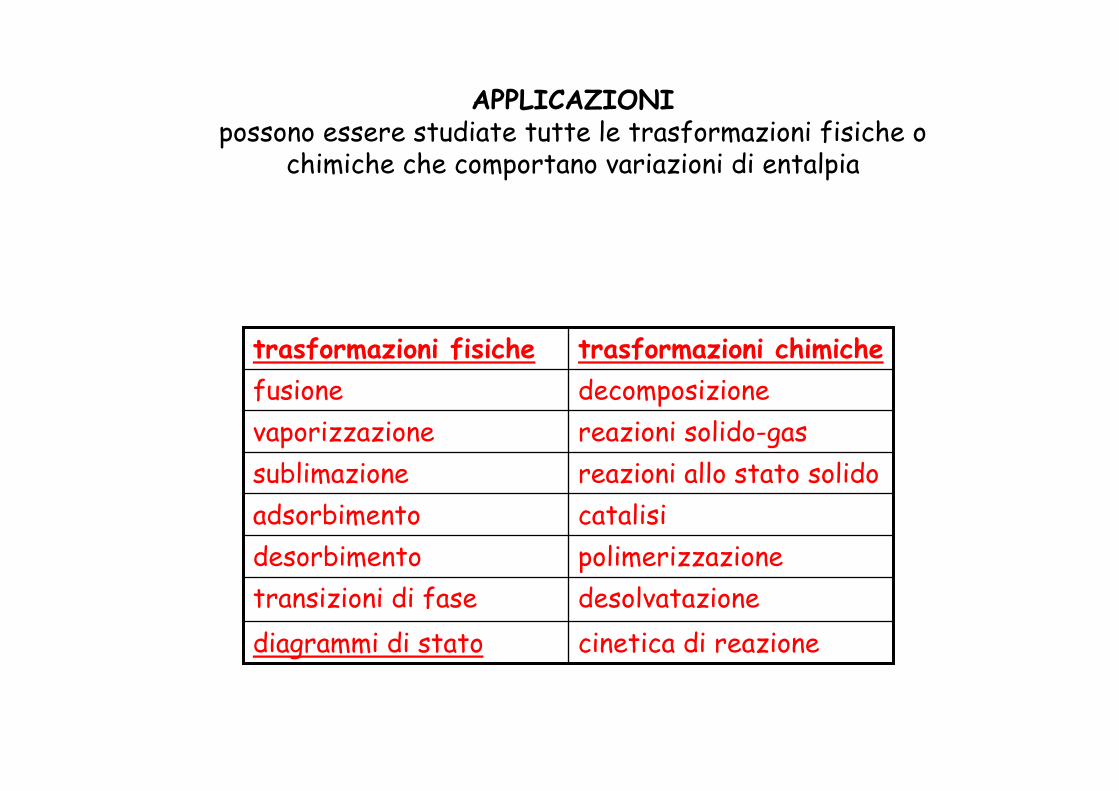
APPLICAZIONIpossono essere studiate tutte le trasformazioni fisiche o
chimiche che comportano variazioni di entalpia
cinetica di reazionediagrammi di statodesolvatazionetransizioni di fasepolimerizzazionedesorbimentocatalisiadsorbimentoreazioni allo stato solido sublimazionereazioni solido-gasvaporizzazionedecomposizionefusionetrasformazioni chimichetrasformazioni fisiche
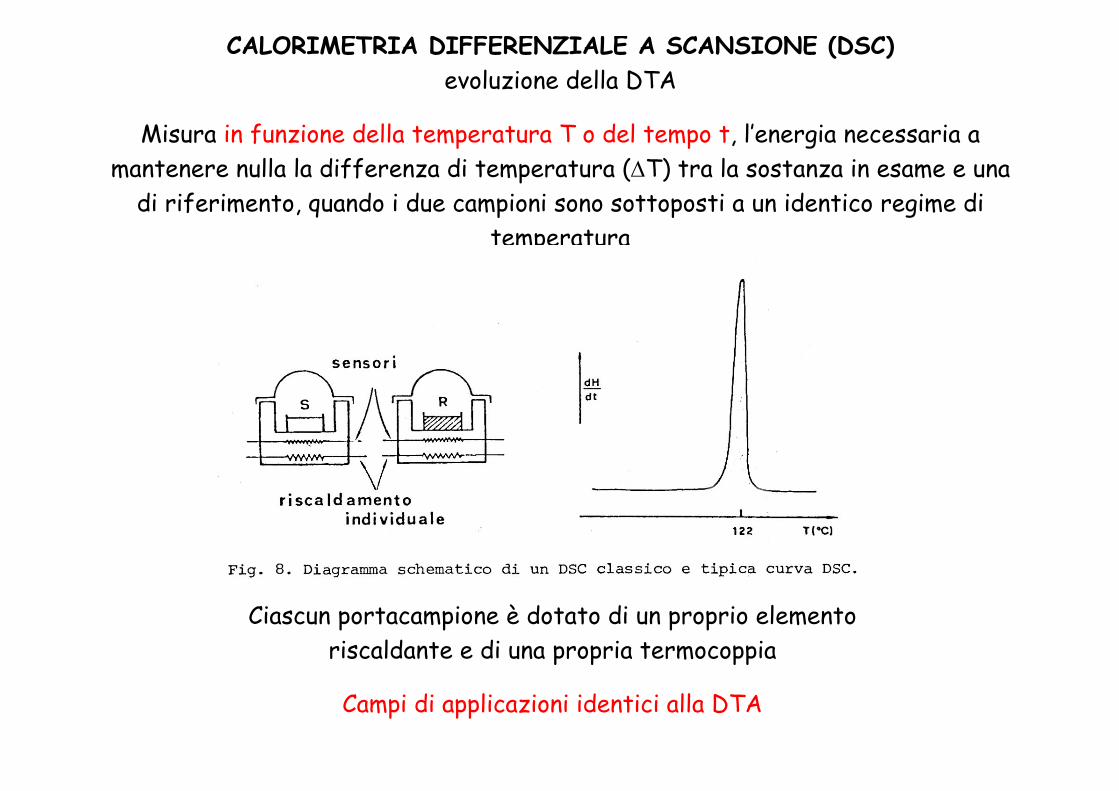
CALORIMETRIA DIFFERENZIALE A SCANSIONE (DSC)evoluzione della DTA
Misura in funzione della temperatura T o del tempo t, l’energia necessaria a mantenere nulla la differenza di temperatura (ΔT) tra la sostanza in esame e una
di riferimento, quando i due campioni sono sottoposti a un identico regime di temperatura
Ciascun portacampione è dotato di un proprio elemento riscaldante e di una propria termocoppia
Campi di applicazioni identici alla DTA
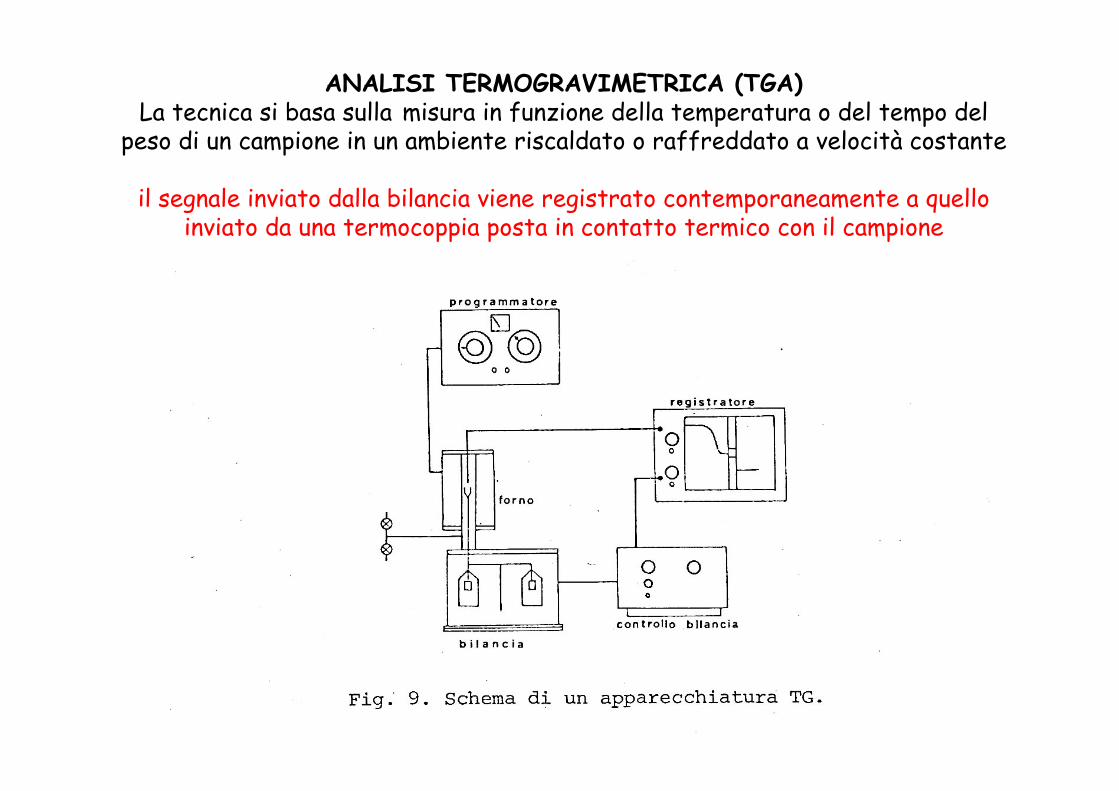
ANALISI TERMOGRAVIMETRICA (TGA)La tecnica si basa sulla misura in funzione della temperatura o del tempo del
peso di un campione in un ambiente riscaldato o raffreddato a velocità costante
il segnale inviato dalla bilancia viene registrato contemporaneamente a quello inviato da una termocoppia posta in contatto termico con il campione

Requisiti:
Peso e temperatura misurati con continuità e separatamente
Forno senza inerzia termica
Zona a temperatura uniforme intorno al campione il più ampia possibile
Velocità di riscaldamento costante nell’intervallo di temperatura in esame
Il portacampione non deve reagire con la sostanza in esame o con i prodotti delle trasformazioni
Si deve poter operare in atmosfera controllata, con diverse velocità di riscaldamento e in condizioni isoterme.

La curva TG presenta dei gradini da cui si possono valutare le variazioni di peso.
La derivata della curva (DTG) presenta dei picchi, le cui aree sono proporzionali alla variazione di peso del campione, e permette di rivelare piccole variazioni di
pendenza che possono non essere visibili nella TG
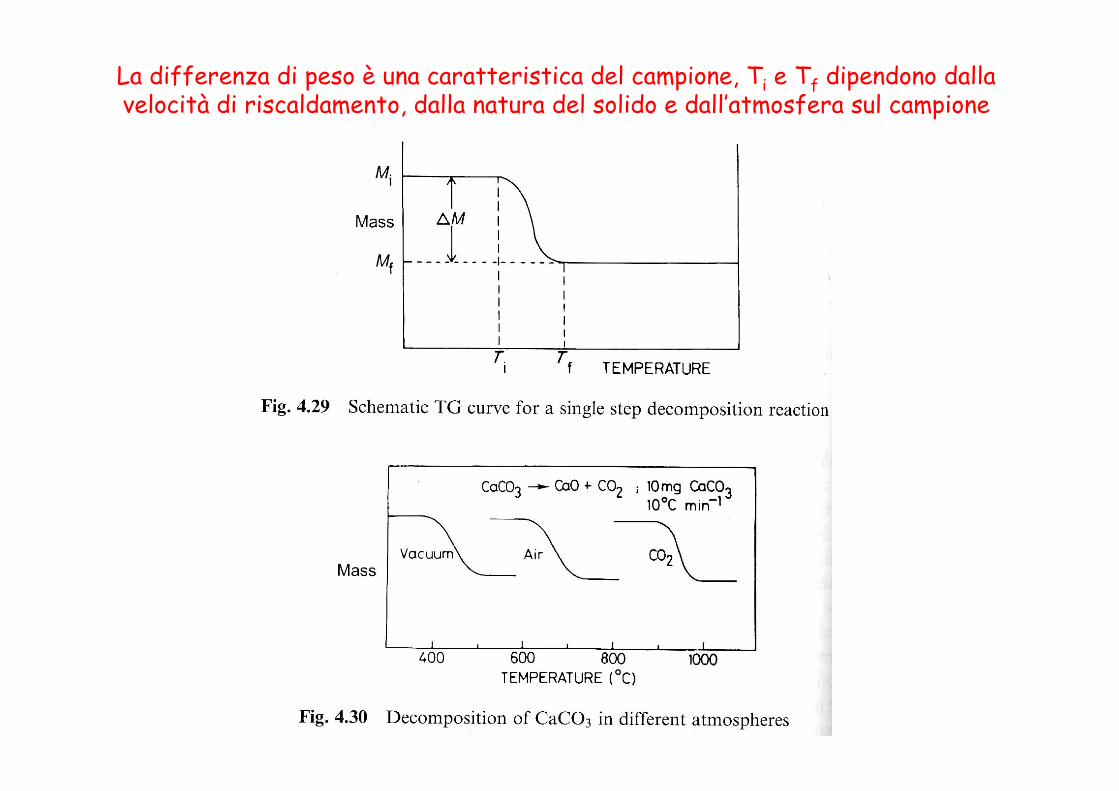
La differenza di peso è una caratteristica del campione, Ti e Tf dipendono dalla velocità di riscaldamento, dalla natura del solido e dall’atmosfera sul campione

cinetica di trasformazionedeterminazione di cenerideterminazione di composti volatilideterminazione di umidita’reazioni solido-gasreazioni allo stato solidodistillazionecorrosione dei metallidecomposizioni termiche
Applicazioni

Esempi di applicazioni di DTA e TGATGA rivela solo fenomeni accompagnati da cambiamenti di peso.
DTA rivela anche quelli (come le transizioni di fase) non accompagnati da variazioni di peso
La curva DTG assomiglia alla curva DTA e il loro confronto permette di distinguere tra le trasformazioni che avvengono con variazioni di peso e
entalpia da quelle che avvengono con sola variazione di entalpia
Al4(Si4O10) (OH)8

Seguire le trasformazioni in riscaldamento e successivamente in raffreddamento consente di distinguere le trasformazioni reversibili da quelle irreversibili
Si osserva inoltre il fenomeno dell’isteresi:il picco esotermico che si osserva in raffreddamento appare a temperature più basse di quello corrispondente, endotermico, che si osserva in riscaldamento

TGA/SDTA851 Mettler Toledo
massima temperatura del forno 1100 °C volume massimo del campione 100 μlpeso massimo del campione 1 grisoluzione 1 μg
Strumentazione usata per le esperienze di Laboratorio consente di ottenere simultaneamente TGA e DTA


Il sensore di temperatura del campione (termocoppia R-type, platino/platino-rodio) e’ posizionato al di sotto del portacampione
Misura il segnale SDTA (differenza tra temperatura del campione e valore impostato della temperatura) contemporaneamente al Peso del campione
N.B. Non esiste un campione di riferimento!!!Il sensore di temperatura del forno (termocoppia R-type) è posizionato
sulla superficie del forno stesso.

Nella cella che contiene il forno deve continuamente fluire un gas protettivo (nel nostro caso Argon), per eliminare eventuali gas dannosi per la bilancia che si possono formare come
conseguenza delle reazioni che avvengono durante il trattamento termico.
E’ inoltre possibile introdurre del gas (che chiameremo gas reattivo) che lambisce direttamente il campione (nel nostro caso possiamo usare Ar o O2 o una miscela dei due).
Capillare gas reattivo
Regolatore flusso gas protettivo
Regolatore flusso gas reattivi

Deve inoltre essere usato un criotermostato che convoglia acqua a temperatura controllata (22 °C) per assicurare la riproducibilità del segnale della bilancia
e per dissipare il calore dal forno

Lo strumento è interfacciato con un computer. Il software consente di gestire le misure e analizzare i dati

ESPERIENZE DI LABORATORIO
Applicazioni tipiche delle tecniche di analisi termica
TGA (DTA): decomposizione di sali (CuSO4·5H2O)
Identificazione delle perdite in peso; ipotesi sugli stadi del processo di decomposizione
DTA: Costruzione di un diagrammi di fase solido-liquido
Preparazione di leghe Cd-Bi
DTA di leghe a diversa composizione

Sistemi binari con immiscibilità completa allo stato solido
A e B (es. Pb e Sn, o Cd e Bi) sono completamente miscibili allo stato liquido.
A e B sono completamente insolubili allo stato solido.
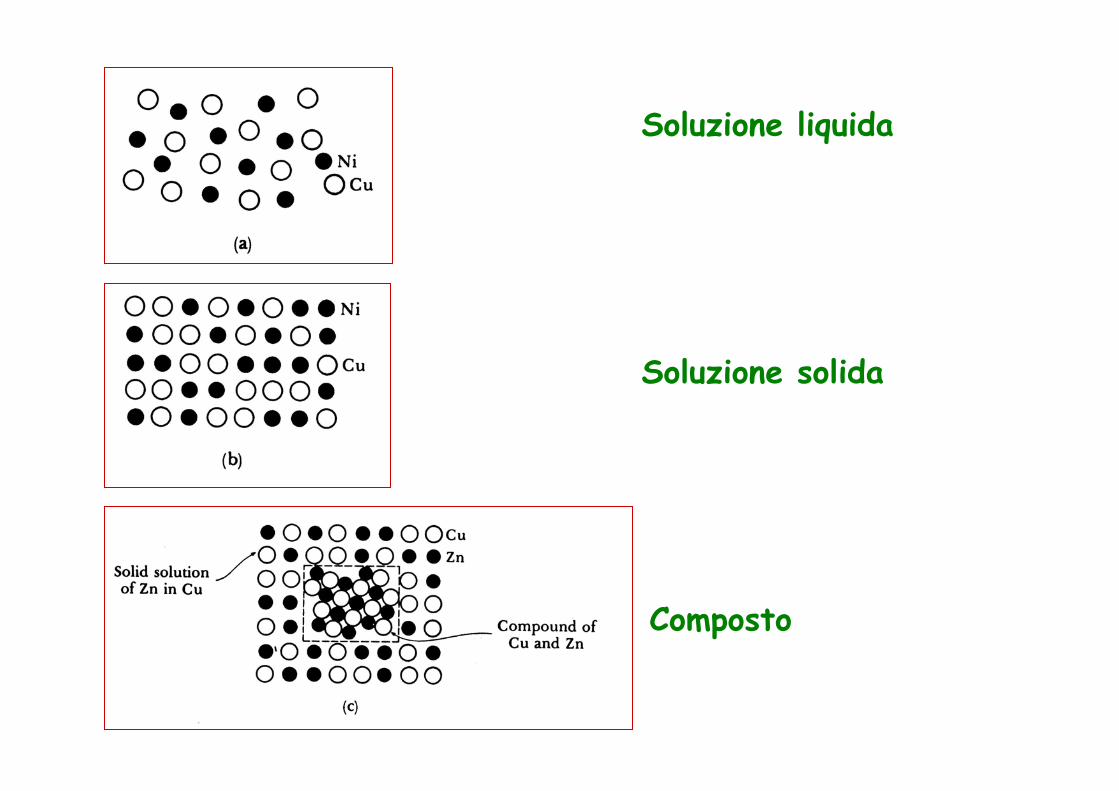
Soluzione liquida
Soluzione solida
Composto

Raffreddamento di una miscela con composizione XA=0.70, XB=0.30
T1
T2
T3
T4
A T1 abbiamo 1 sola fase liquida di composizione Pb70% Sn30%
A T2 inizia la deposizione di Pb solido. La composizione del liquido residuo è data dalla curva de liquidus.
Durante il raffreddamento il liquido si arricchische in Sn
T3 è la temperatura eutettica. Ho 3 fasi in equilibrio Pb, Sn e il liquido. Il sistema è invariante, non posso raffreddare finché tutto il liquido non si è solidificato.
A T4 il sistema è tutto solido. Si osservano i cristalli di Pb ottenuti durante la deposizione primaria immersi in una matrice microscristallina di composizione eutettica (Pb 40% Sn 60%)
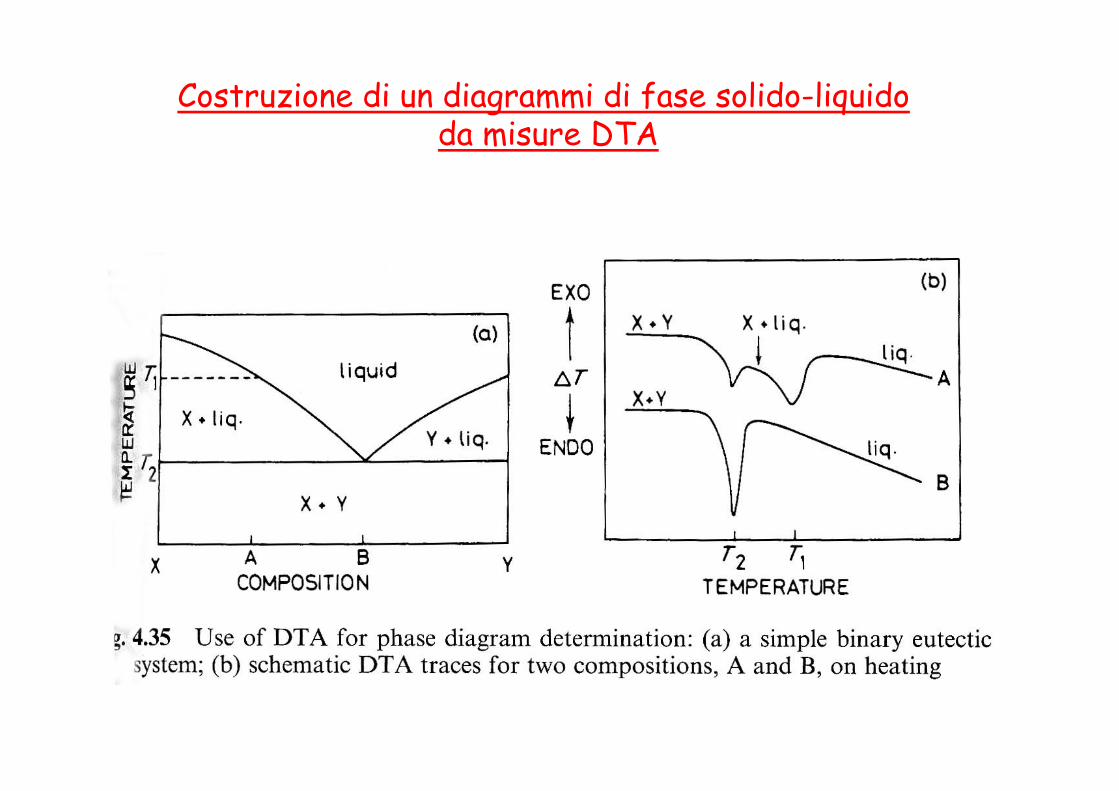
Costruzione di un diagrammi di fase solido-liquido da misure DTA

Preparazione delle leghe Cd-Bi
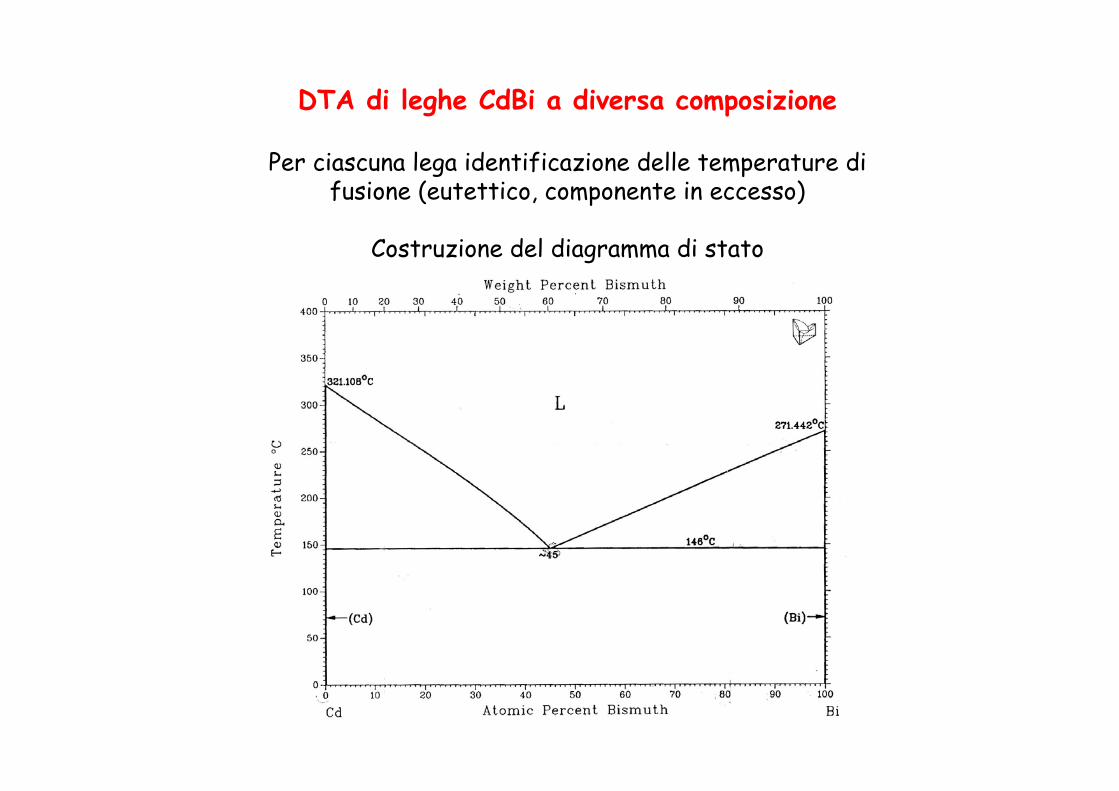
DTA di leghe CdBi a diversa composizione
Per ciascuna lega identificazione delle temperature di fusione (eutettico, componente in eccesso)
Costruzione del diagramma di stato

Diffrazione di raggi X su polveri
Cenni di cristallochimica
•Generazione dei raggi X
•Diffrazione dei raggi X da parte dei cristalli
•Equazioni di Laue e Legge di Bragg
•Metodi diffrattometrici
•Motodo delle Polveri (Solidi policristallini)
•Applicazioni e descrizione esercitazioni
Testi Consigliati:
A. R. West, Solid State Chemistry and its Application, John Wiley & Sons

Il cristallo è un corpo anisotropo omogeneo costituito da un ordine periodico tridimensionale di atomi o ioni o molecole
La distribuzione di ioni atomi o molecole è periodicamente omogenea in tre dimensioni
I solidi possono presentarsi in forma di: monocristalli (periodicità perfetta su tutto il solido),
policristalli (grani di dimensione variabile separati da bordi di grano
I solidi possono anche essereAmorfi o non-cristallini
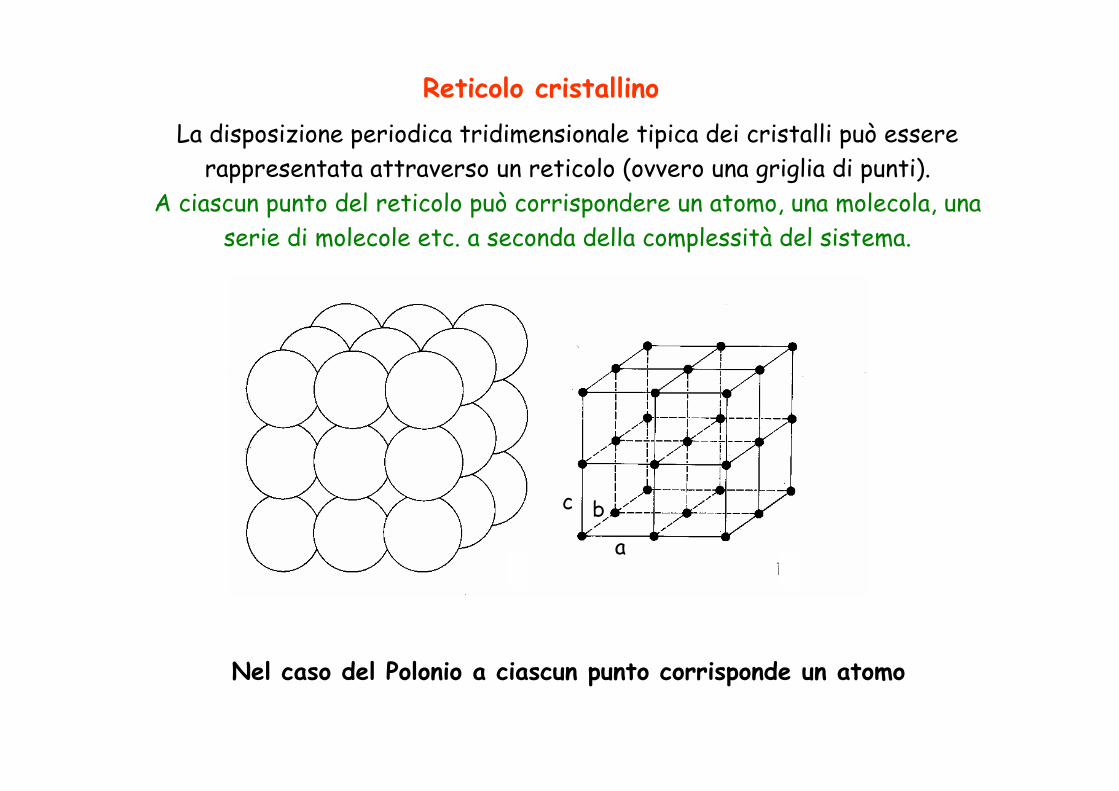
Reticolo cristallinoLa disposizione periodica tridimensionale tipica dei cristalli può essere
rappresentata attraverso un reticolo (ovvero una griglia di punti). A ciascun punto del reticolo può corrispondere un atomo, una molecola, una
serie di molecole etc. a seconda della complessità del sistema.
a
c b
Nel caso del Polonio a ciascun punto corrisponde un atomo
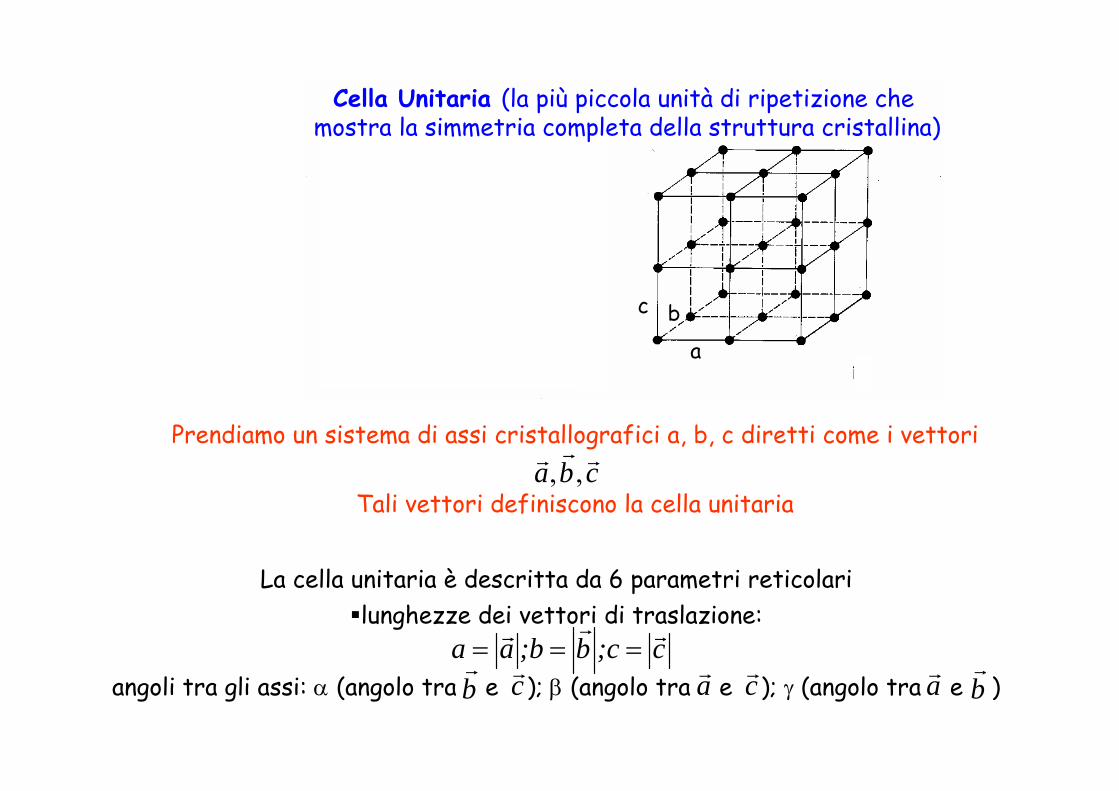
Prendiamo un sistema di assi cristallografici a, b, c diretti come i vettori
Tali vettori definiscono la cella unitaria cba rrr ,,
a
c b
La cella unitaria è descritta da 6 parametri reticolarilunghezze dei vettori di traslazione:
angoli tra gli assi: α (angolo tra e ); β (angolo tra e ); γ (angolo tra e ) c;cb;baa rrr
===br
cr cr br
arar
Cella Unitaria (la più piccola unità di ripetizione che mostra la simmetria completa della struttura cristallina)

Triclino
Monoclino
Esagonale
Romboedrico
Ortorombico
Tetragonale
Cubico
Lunghezze e angoli degli assi
Sistema
°===≠≠ 90; γβαcba
°===≠= 90; γβαcba
°===== 90; γβαcba
°≠==== 90; γβαcba
Sette forme differenti di cella unitaria - Sette Sistemi cristallini
°=°==≠= 120;90; γβαcba
°>°==≠≠ 90;90; βγαcba
°≠≠≠≠≠ 90; γβαcba
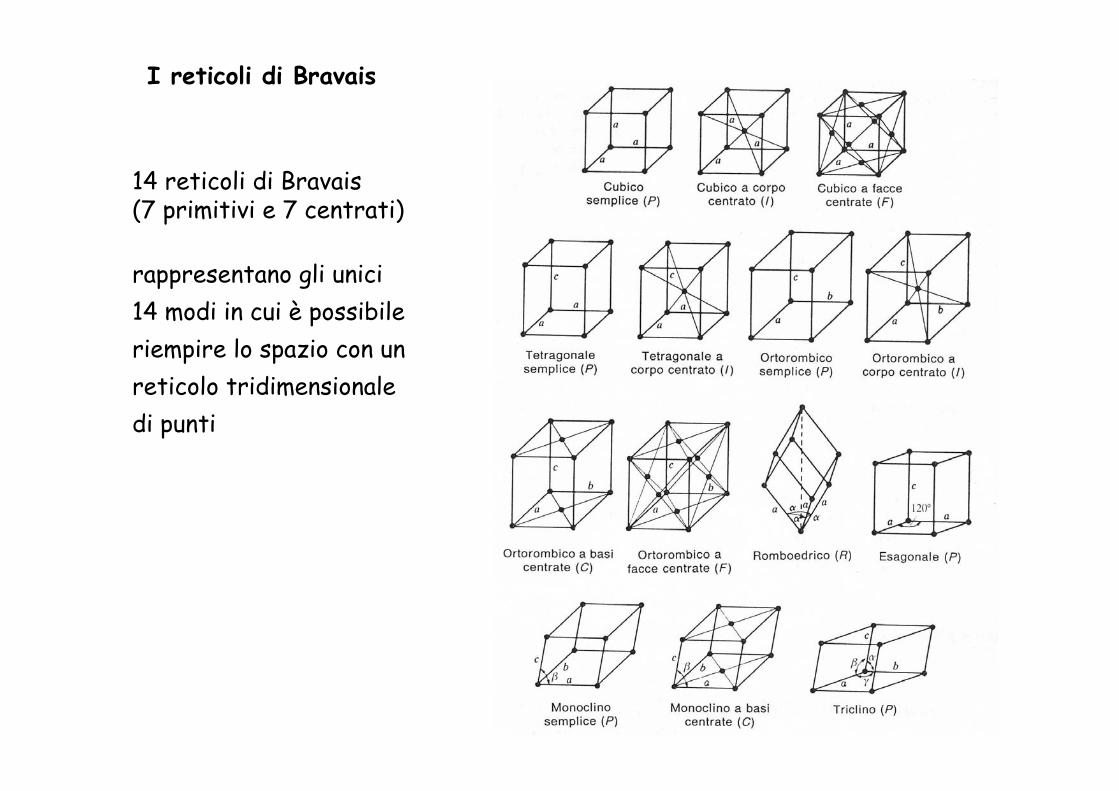
I reticoli di Bravais
14 reticoli di Bravais(7 primitivi e 7 centrati)
rappresentano gli unici 14 modi in cui è possibile riempire lo spazio con un reticolo tridimensionale di punti
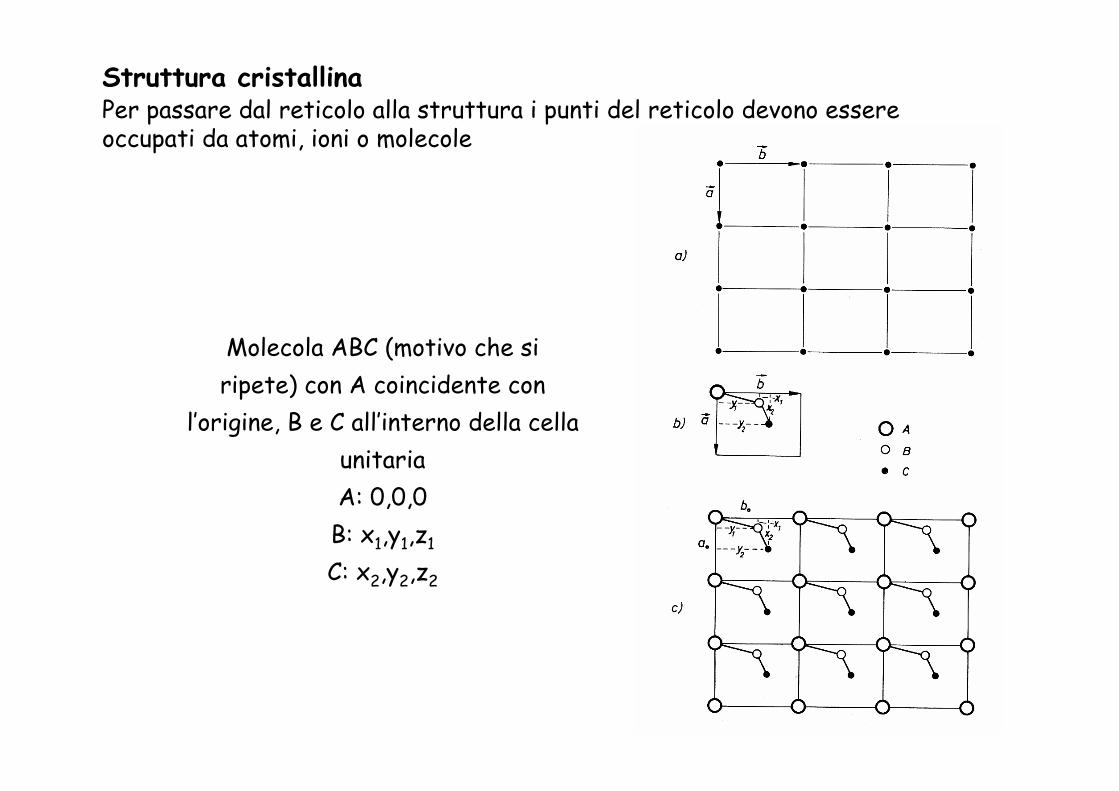
Molecola ABC (motivo che si ripete) con A coincidente con
l’origine, B e C all’interno della cella unitariaA: 0,0,0
B: x1,y1,z1
C: x2,y2,z2
Struttura cristallinaPer passare dal reticolo alla struttura i punti del reticolo devono essere occupati da atomi, ioni o molecole

Piani reticolari
Piano interseca gli assi a, b,c nei punti m00, 0n0, 00p
Le coordinate delle intercette sui tre assi (m,n,p) definiscono completamente la posizione del piano reticolare. Però una delle intercette può essere ∞
Gli esperimenti di diffrazione forniscono segnali che corrispondono a piani reticolari

Per definire univocamente il piano e evitare indici pari a ∞si usano i cosiddetti indici di Miller (hkl)
Il piano è in realtà uno dei tanti piani di una “Famiglia” tra loro paralleli e equidistanti
Il primo piano della famiglia a partire dall’origine intercetta gli assi nei punti a/h; b/k; c/l dove h,k e l sono gli indici di Miller
Gli indici di Miller (h,k,l) sono dati quindi dal rapporto tra la lunghezza di un asse e l’intercetta del piano sull’asse stesso
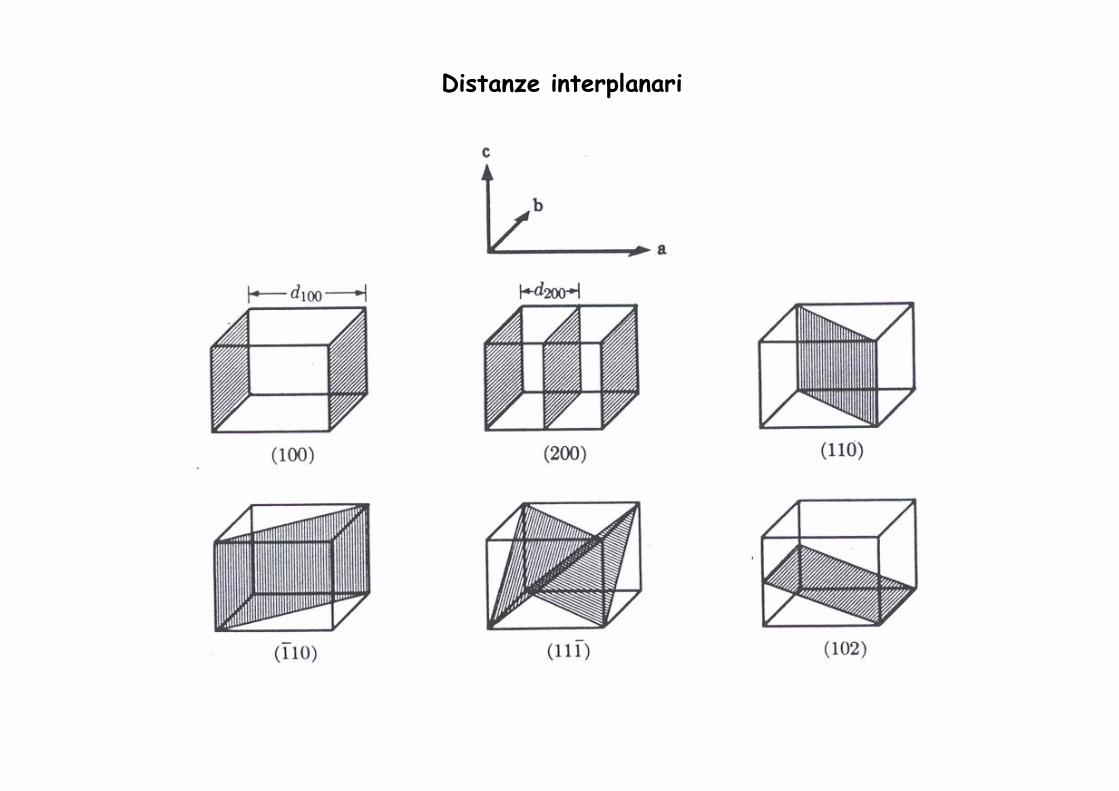
Distanze interplanari
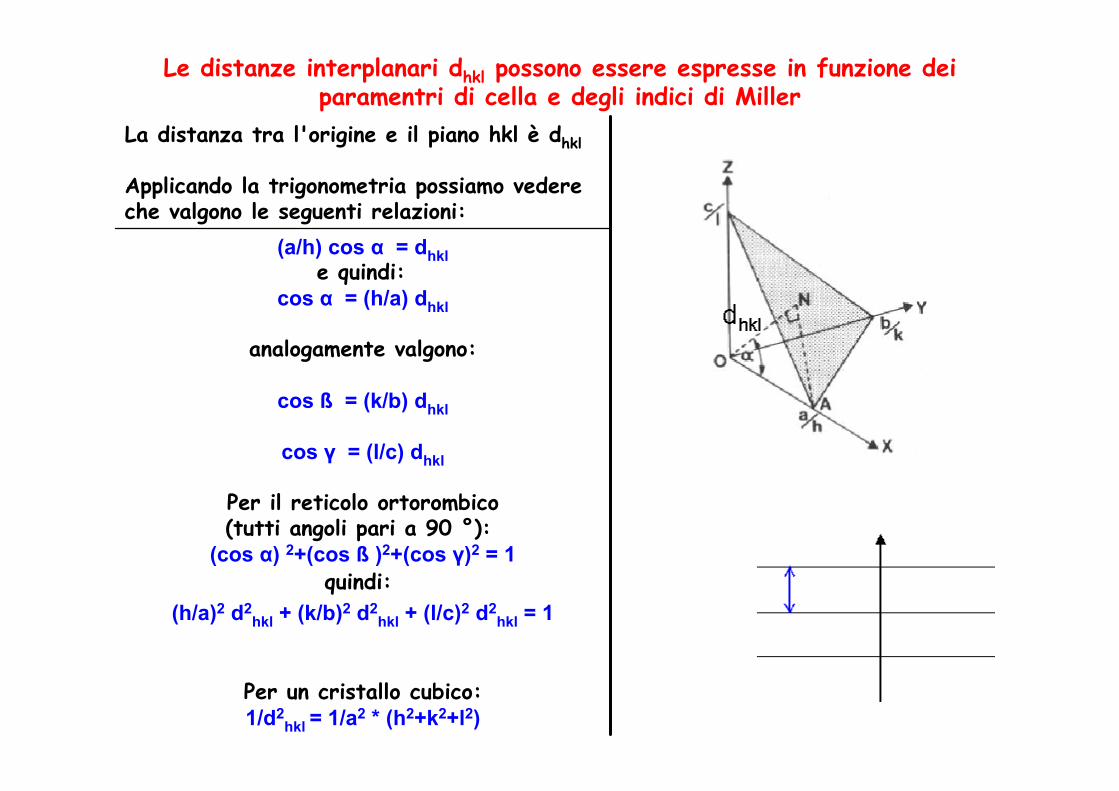
Le distanze interplanari dhkl possono essere espresse in funzione dei paramentri di cella e degli indici di Miller
(a/h) cos α = dhkle quindi:
cos α = (h/a) dhkl
analogamente valgono:
cos ß = (k/b) dhkl
cos γ = (l/c) dhkl
Per il reticolo ortorombico(tutti angoli pari a 90 °):
(cos α) 2+(cos ß )2+(cos γ)2 = 1quindi:
(h/a)2 d2hkl + (k/b)2 d2
hkl + (l/c)2 d2hkl = 1
Per un cristallo cubico: 1/d2
hkl = 1/a2 * (h2+k2+l2)
La distanza tra l'origine e il piano hkl è dhkl
Applicando la trigonometria possiamo vedere che valgono le seguenti relazioni:

Monoclino
Esagonale
Cubico
Tetragonale
Ortorombico
2
2
2
2
2
2
2
1cl
bk
ah
dhkl
++=
2
2
2
22
21
cl
akh
dhkl
++
=
2
222
21
alkh
dhkl
++=
2
2
2
22
2 341
cl
akhkh
dhkl
+++
=
ββ
ββ 42222
2
2
2
22
2
2 sincos2
sinsin1
cahl
cl
bk
ah
dhkl
+++=
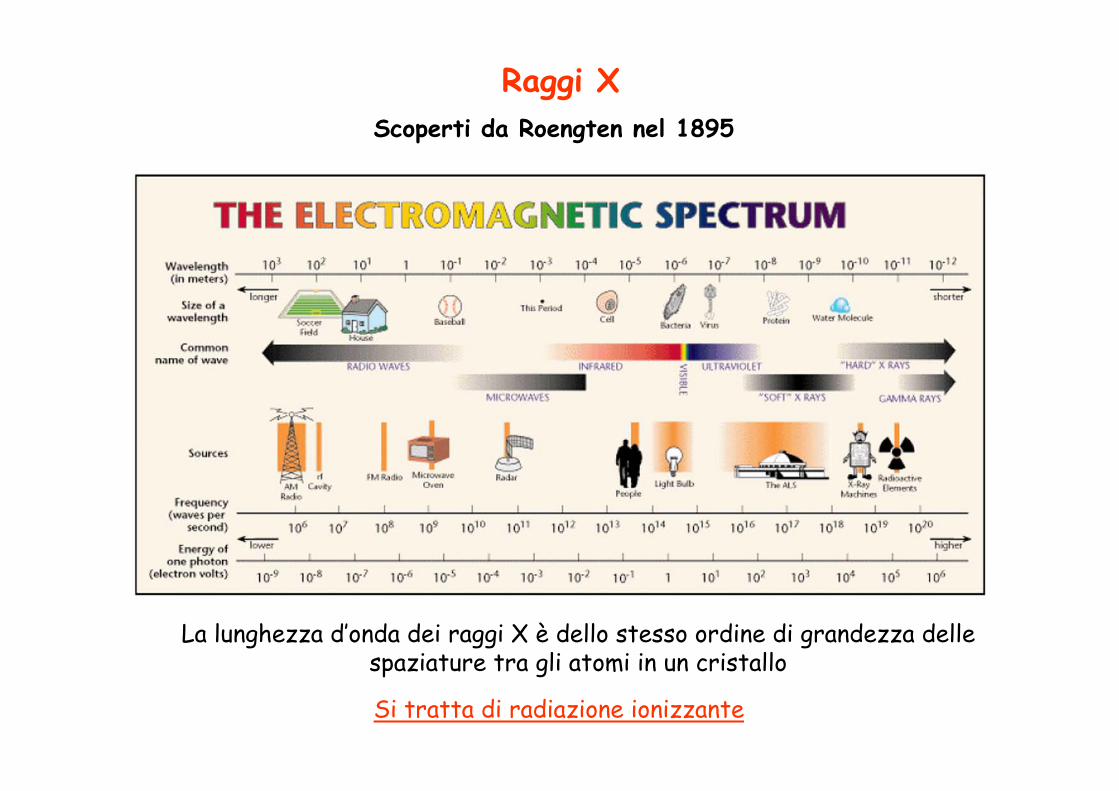
Raggi X
La lunghezza d’onda dei raggi X è dello stesso ordine di grandezza delle spaziature tra gli atomi in un cristallo
Si tratta di radiazione ionizzante
Scoperti da Roengten nel 1895

I raggi X possono essere prodotti utilizzando due modi principali :
• Eccitazione di elettroni di core negli atomi– Questo è il metodo usato nei tubi a raggi X, nei
dispositivi di laboratorio
• Accelerazione di elettroni liberi– Metodo usato nei sincrotroni

Tubo a raggi X
target X-rays
W
Vacuum
Caratteristiche:Usato in laboratorioCosto ~ migliaia di EuroRichiede acqua e alta tensione
Come funziona:Elettroni prodotti da un filamento di tungsteno riscaldato (catodo) accelerati da una elevata ddpColpiscono il bersaglio (anodo) costituito da un elemento metallicoVengono emessi raggi X

• Spettro di emissione di un tubo a raggi X
Radiazione bianca (Brehmsstralung) dovuta al frenamento e perdita di energia degli elettroni a seguito degli urti con gli atomi del bersaglio
minmaxmax
λhchveVE ===
VeVhc 12400
min ==λ
λ misurato in ÅV misurato in Volts
L’energia massima dei fotoni (e quindi la minima lunghezza d’onda) dipende SOLO dall’energia degli elettroni incidenti ed è indipendente dalla natura del materiale.
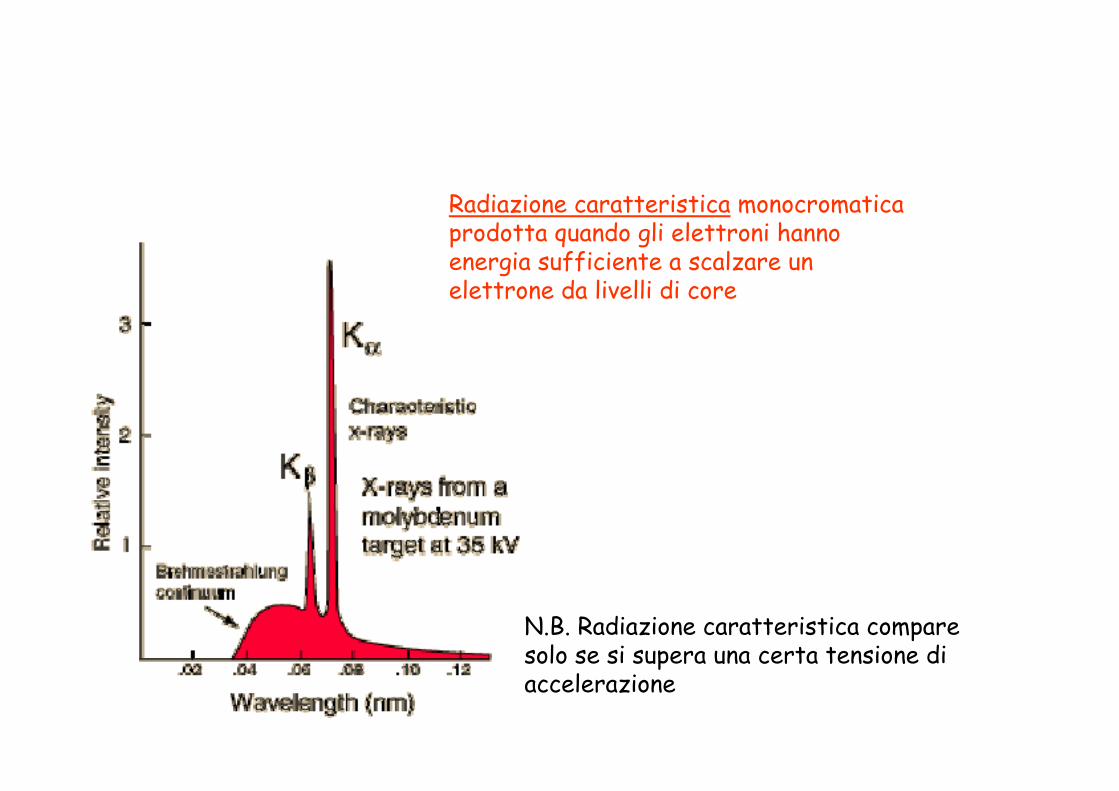
Radiazione caratteristica monocromatica prodotta quando gli elettroni hanno energia sufficiente a scalzare un elettrone da livelli di core
N.B. Radiazione caratteristica compare solo se si supera una certa tensione di accelerazione
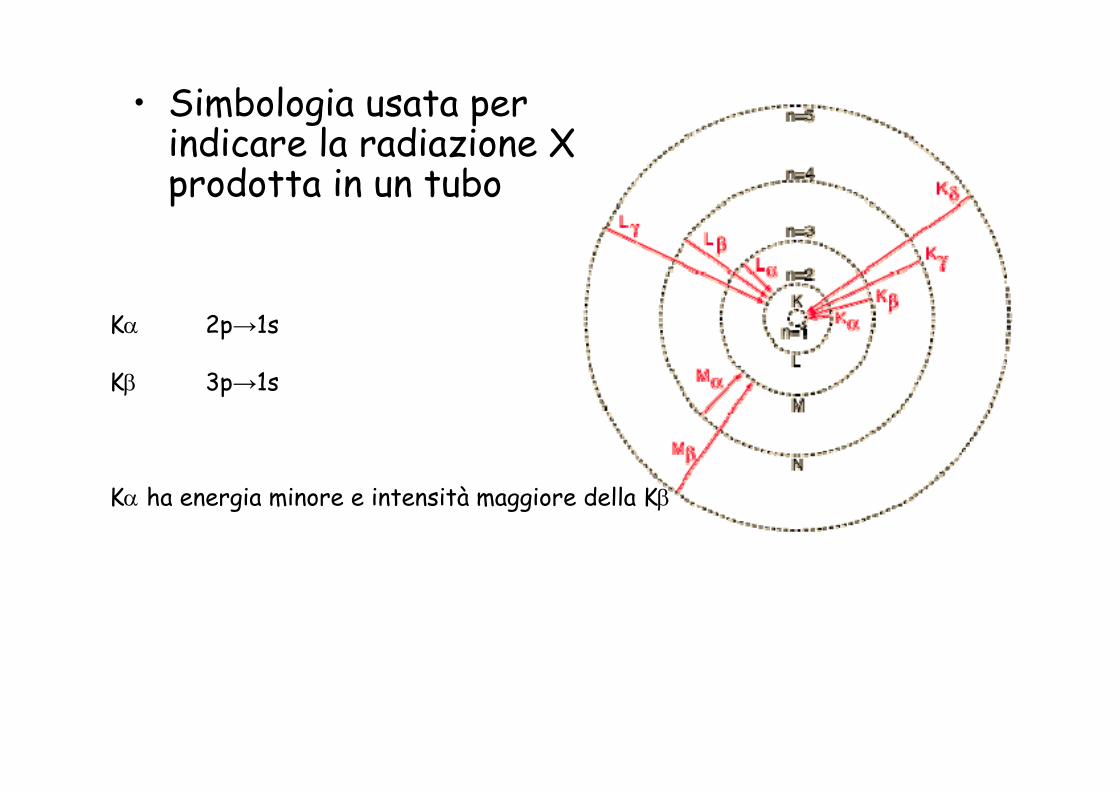
• Simbologia usata per indicare la radiazione X prodotta in un tubo
Kα 2p→1s
Kβ 3p→1s
Kα ha energia minore e intensità maggiore della Kβ

La lunghezza d’onda della radiazione prodotta dipende dal Numero Atomico del metallo usato come bersaglio
Legge di Moseley
All’aumentare di Z ν aumenta e λ diminuisce
Elementi utilizzati come bersaglio e lunghezza d’onda della radiazione X (Å)
)( σ−= Zkv
0.56080.56380.5594Ag
0.71070.71350.7093Mo
1.54181.54431.5405Cu
1.93731.93991.9360Fe
2.29092.29352.2896CrKαKα2Kα1Anodo
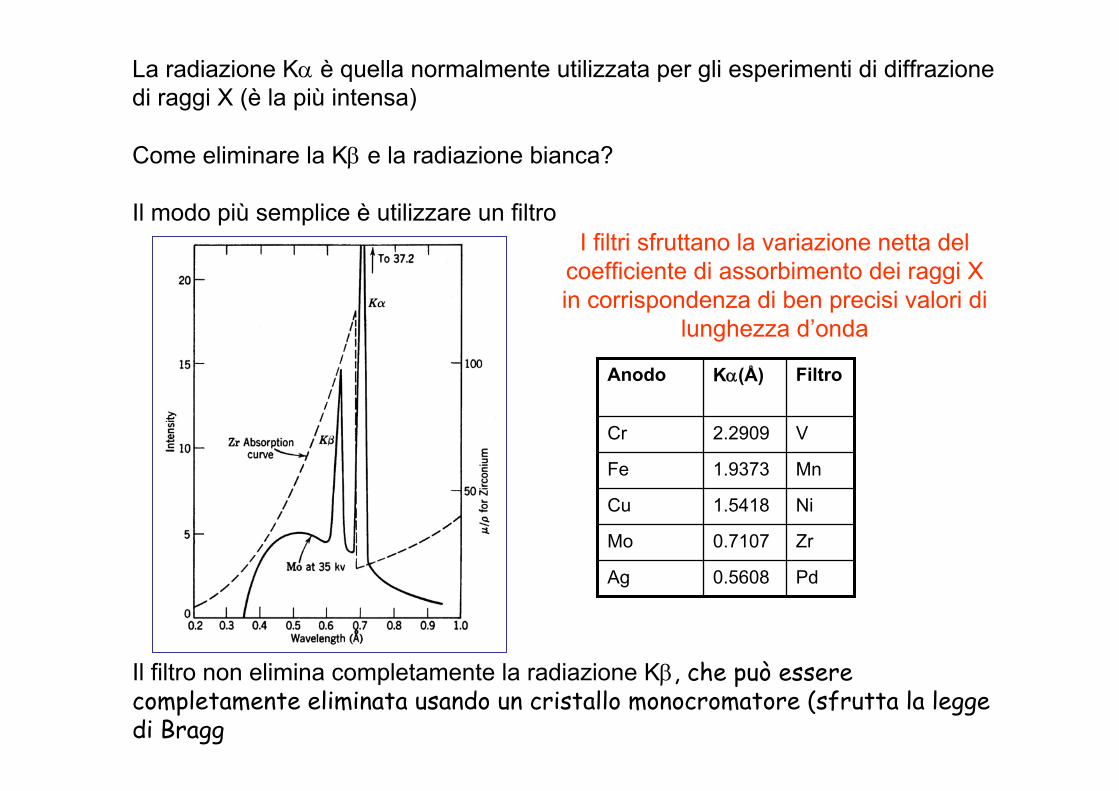
La radiazione Kα è quella normalmente utilizzata per gli esperimenti di diffrazionedi raggi X (è la più intensa)
Come eliminare la Kβ e la radiazione bianca?
Il modo più semplice è utilizzare un filtro
Il filtro non elimina completamente la radiazione Kβ, che può essere completamente eliminata usando un cristallo monocromatore (sfrutta la legge di Bragg
Pd0.5608Ag
Zr0.7107Mo
Ni1.5418Cu
Mn1.9373Fe
V2.2909Cr
FiltroKα(Å)Anodo
I filtri sfruttano la variazione netta del coefficiente di assorbimento dei raggi X in corrispondenza di ben precisi valori di
lunghezza d’onda
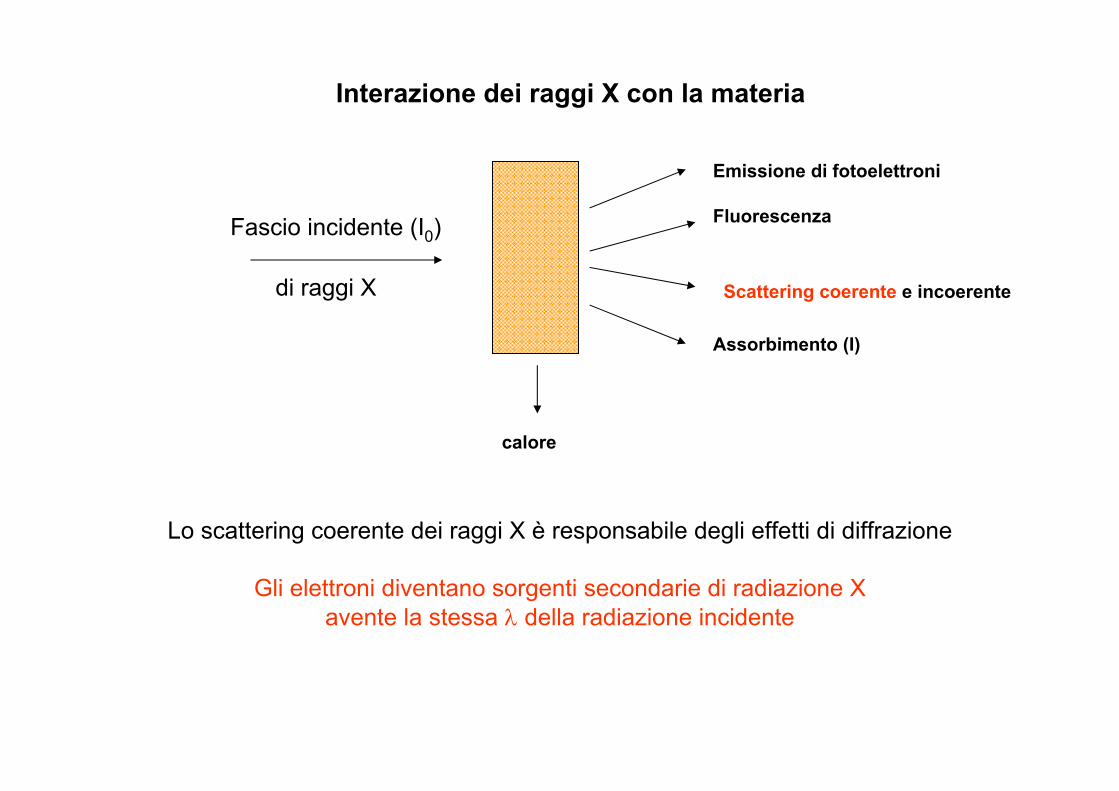
Fascio incidente (I0)
di raggi X
Emissione di fotoelettroni
Fluorescenza
Assorbimento (I)
Scattering coerente e incoerente
calore
Interazione dei raggi X con la materia
Lo scattering coerente dei raggi X è responsabile degli effetti di diffrazione
Gli elettroni diventano sorgenti secondarie di radiazione X avente la stessa λ della radiazione incidente

Il fenomeno della diffrazione
La diffrazione è un complesso fenomeno di diffusione (o scattering) e interferenza originato dall’interazione dei raggi X con un reticolo cristallino.
Il processo di diffusione (o scattering)
L’interazione di un’onda elettromagnetica con la materia avviene essenzialmente
attraverso due processi di scattering:
a) scattering elastico: i fotoni della radiazione incidente vengono deviati
in ogni direzione dello spazio senza perdita di energia.
b) scattering non-elastico: il fotone cede parte della sua energia.
Questo fenomeno non dà luogo a processi di interferenza.

Interazione raggi X con:Una singola particella
Un materiale cristallino
La particella diffonde ilfascio incidente
uniformemente in tutte le direzioni
I fasci diffusi si combinanoconstruttivamente in certe
direzioni
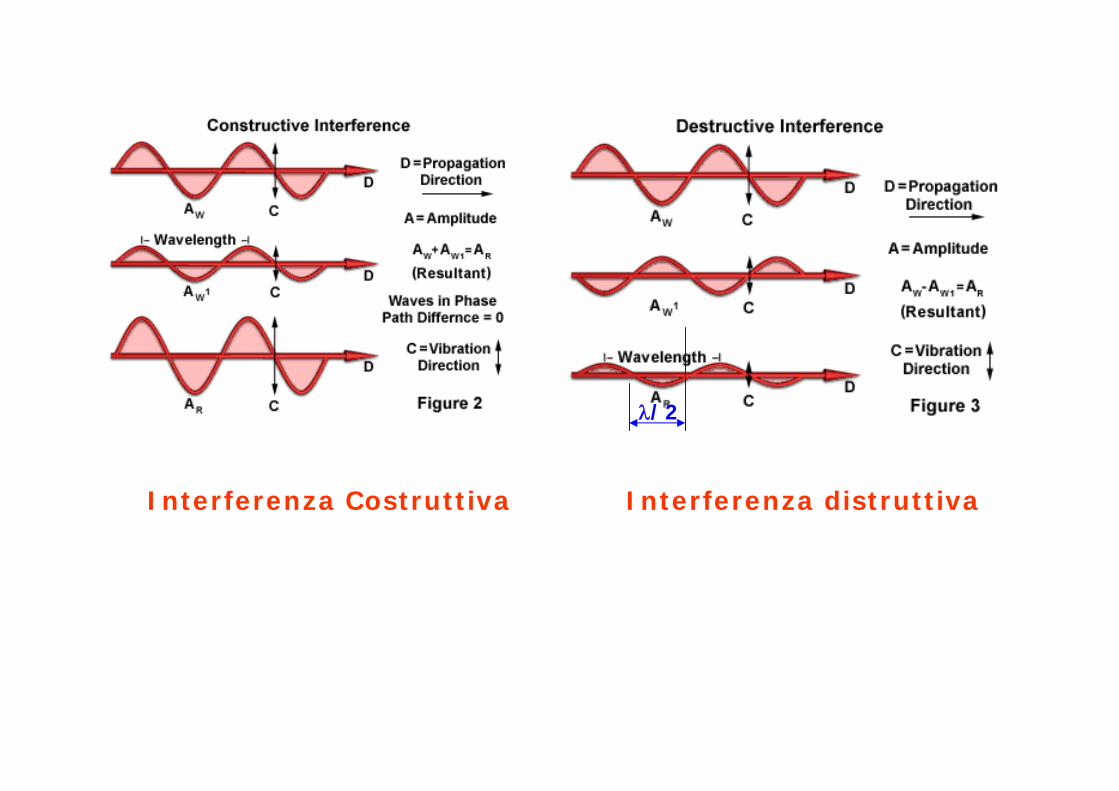
Interferenza Costruttiva Interferenza distruttiva
λ/2
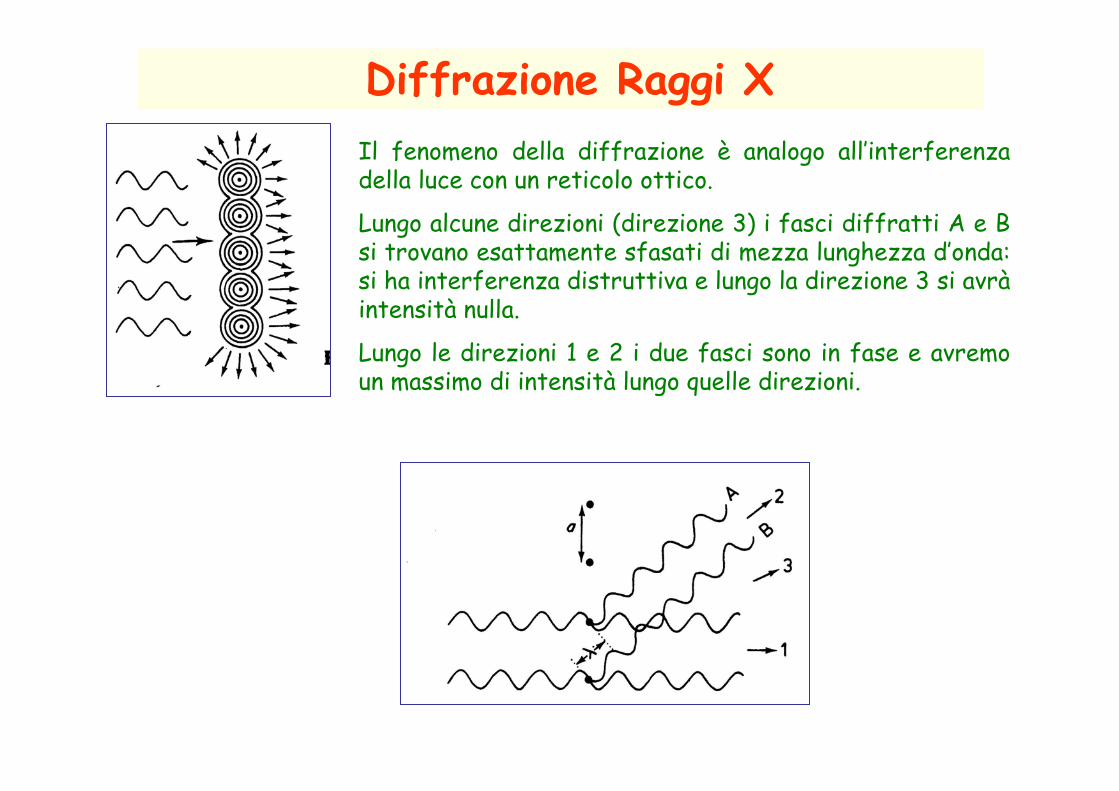
Diffrazione Raggi XIl fenomeno della diffrazione è analogo all’interferenza della luce con un reticolo ottico.
Lungo alcune direzioni (direzione 3) i fasci diffratti A e B si trovano esattamente sfasati di mezza lunghezza d’onda: si ha interferenza distruttiva e lungo la direzione 3 si avrà intensità nulla.
Lungo le direzioni 1 e 2 i due fasci sono in fase e avremo un massimo di intensità lungo quelle direzioni.

Condizioni di Laue
Max von Laue interpretò la diffrazione di raggi X da parte dei cristalli in analogia con la diffrazione della luce da parte di un reticolo ottico: la disposizione periodica
tridimensionale degli atomi corrisponde a un reticolo tridimensionale di diffrazione
Partiamo da un reticolo monodimensionale costituito da centri di scattering nei nodi reticolari
Interferenza è costruttiva solo se la differenza di cammino ottico dei raggi scatterati da due contigui è pari a un multiplo della lunghezza d’onda
Radiazione S0 incide con angolo di incidenza φsu un filare monodimensionale.
Radiazione diffratta S forma un angolo θ con il fascio incidente
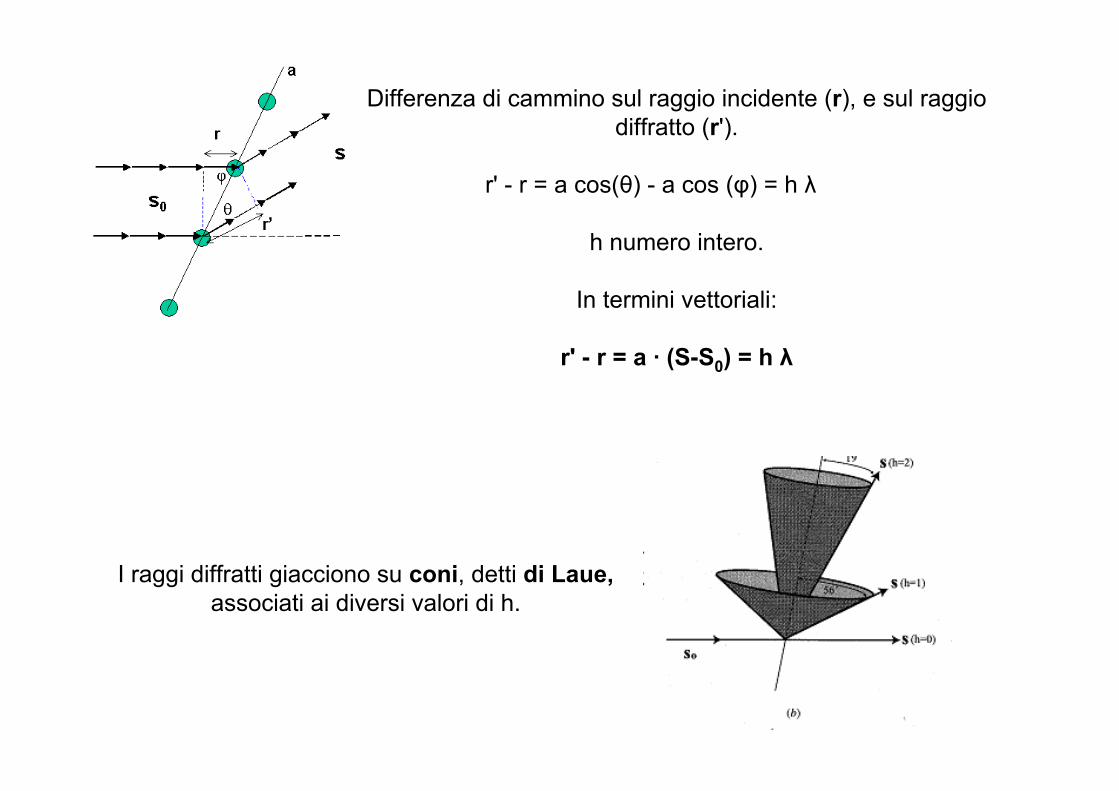
Differenza di cammino sul raggio incidente (r), e sul raggio diffratto (r').
r' - r = a cos(θ) - a cos (φ) = h λ
h numero intero.
In termini vettoriali:
r' - r = a · (S-S0) = h λ
I raggi diffratti giacciono su coni, detti di Laue,associati ai diversi valori di h.

Il reticolo è tridimensionale per cui
dobbiamo scrivere relazioni analoghe per le altre due direzioni
Condizioni di Laue per la diffrazione:
a . (S-S0) = h λb . (S-S0) = k λc . (S-S0) = l λ
Le tre equazioni di Laue devono essere contemporaneamente soddisfatte,
la diffrazione avviene solo lungo le direzioni comuni a tre superfici coniche.
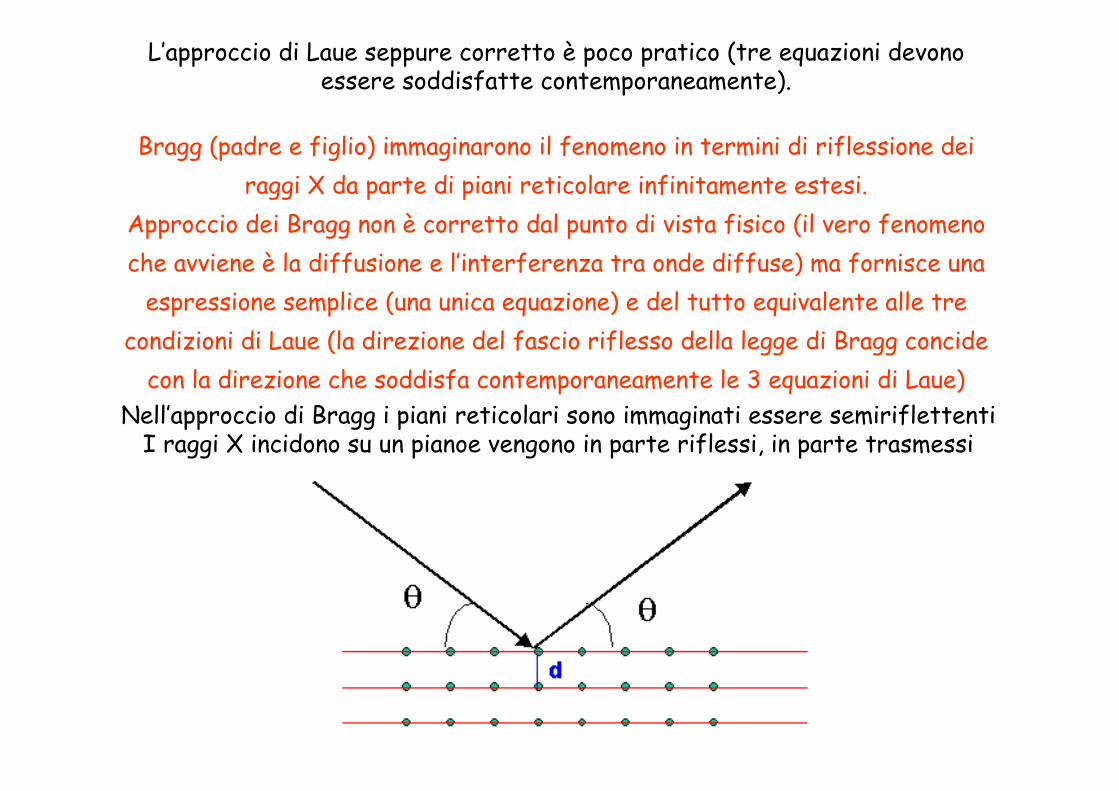
Nell’approccio di Bragg i piani reticolari sono immaginati essere semiriflettentiI raggi X incidono su un pianoe vengono in parte riflessi, in parte trasmessi
L’approccio di Laue seppure corretto è poco pratico (tre equazioni devono essere soddisfatte contemporaneamente).
Bragg (padre e figlio) immaginarono il fenomeno in termini di riflessione dei raggi X da parte di piani reticolare infinitamente estesi.
Approccio dei Bragg non è corretto dal punto di vista fisico (il vero fenomeno che avviene è la diffusione e l’interferenza tra onde diffuse) ma fornisce una
espressione semplice (una unica equazione) e del tutto equivalente alle tre condizioni di Laue (la direzione del fascio riflesso della legge di Bragg concide
con la direzione che soddisfa contemporaneamente le 3 equazioni di Laue)

r + r = dhkl sin(θ) + dhkl sin(θ) = n λ
2dhkl sin(θ) = n λ
Legge di Bragg
La riflessione avviene anche sui piani sottostanti
2dnh nk nl sin(θ) = λ
Interferenza è costruttiva solo se la differenza di cammino tra i raggi riflessi da piani contigui è pari a un multiplo della lunghezza d’onda
N.B. La direzione dei fascio diffratto prevista dalle tre condizioni di Laue coincide con quella prevista dalla legge di Bragg

2d sinθ = λd = distanza interplanare
La direzione dei raggi diffratti dipende UNICAMENTE dal reticolo di traslazione, cioè dai parametri della cella elementare, indipendentemente dagli atomi che essa contiene
PROPORZIONALITÀ INVERSA TRA sinθ e d
strutture con d grandi mostreranno pattern di diffrazione compressi, e viceversa per strutture con d piccoli
1/d = (2/λ) sinθ
1/d ∝ sinθ

Un cristallo di Fe (bcc a=2.866 Å) viene sottoposto a un esperimento di
diffrazione di Raggi X utilizzando la radiazione Cr Kα (λ=2.291 Å)
•Calcolare i valori delle distanze interplanari dhkl
•Calcolare gli angoli di Bragg
•Calcolare gli angoli di Bragg usando la radiazione Mo Kα (λ=0.7107 Å)
Esercizio
N.B. in effetti si osservano solo riflessi con h+k+l=2n

A seconda della simmetria del cristallo l’intensità dei segnali è sistematicamente uguale a zero per certi valori di hkl
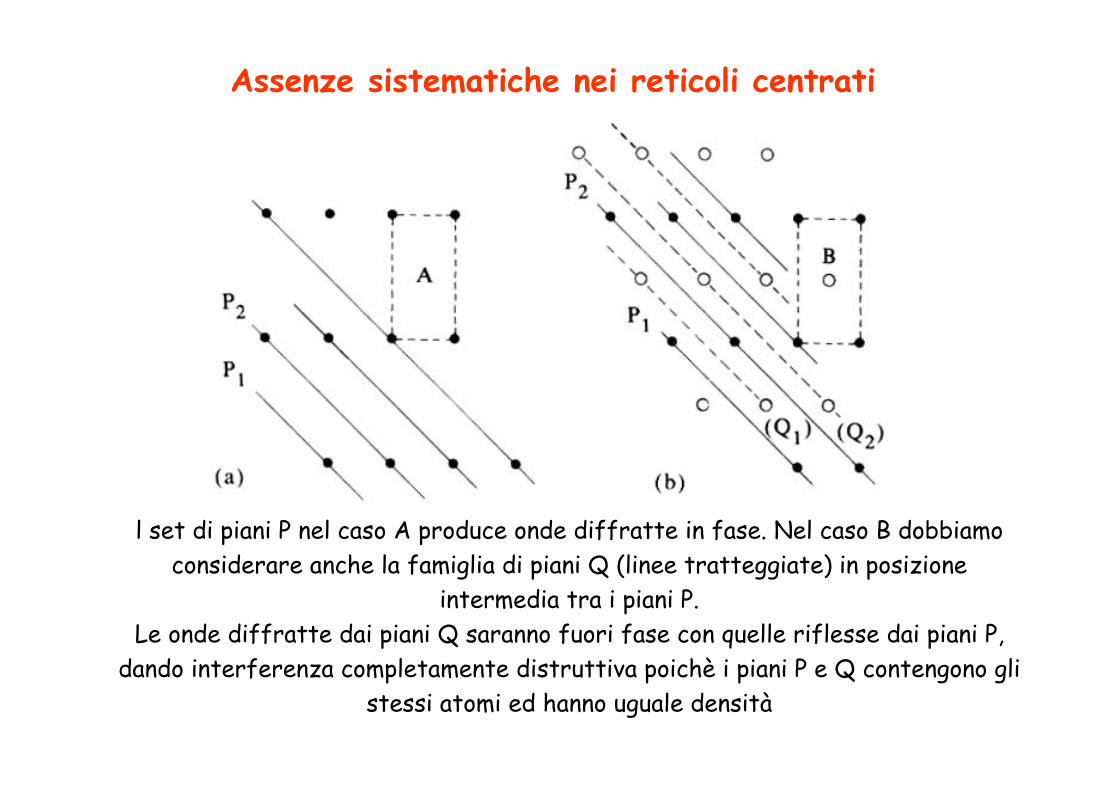
Assenze sistematiche nei reticoli centrati
l set di piani P nel caso A produce onde diffratte in fase. Nel caso B dobbiamo considerare anche la famiglia di piani Q (linee tratteggiate) in posizione
intermedia tra i piani P. Le onde diffratte dai piani Q saranno fuori fase con quelle riflesse dai piani P,
dando interferenza completamente distruttiva poichè i piani P e Q contengono gli stessi atomi ed hanno uguale densità

Tecniche sperimentali
L’esperimento di diffrazione di raggi X richiede:
Sorgente (tubo o sincrotrone)Strumenti di laboratorio usano tubo a raggi X
Campione (monocristallo o polvere)Monocristallo (o cristallo singolo) più adatto per l’analisi strutturaleCampione policristallino più semplice usato soprattutto per analisi
qualitativa e quantitativa
Rivelatore (lastra fotografica o metodi a contatore)Metodi a lastra fotografica hanno solo interesse storico, ma oggi si
usano anche contatori bidimensionali che forniscono pattern di diffrazione molto simili a quelli delle lastre fotografiche

Rivelatori per Raggi X usati in diffrazione
Film fotografico: elevata accuratezza risolutiva, ma scarsa accuratezza nella misura dell'intensità.
Scintillatore: Materiale che emette luce quando irradiato con raggi X. Un fotomoltiplicatore rivela la luce e emette un pulso. Accurata misura delle intensità ed delle posizioni, difetto di poter misurare una sola intensità diffratta alla volta
Rivelatori CCD (Charged Couple Device)Rivelatore bidimensionale a stato solido e di tipo quantico La stessa "simultaneità" di una lastra, con migliore misura delle intensità diffratte. Peccano in potere risolutivo, a causa delle dimensioni dei chip.
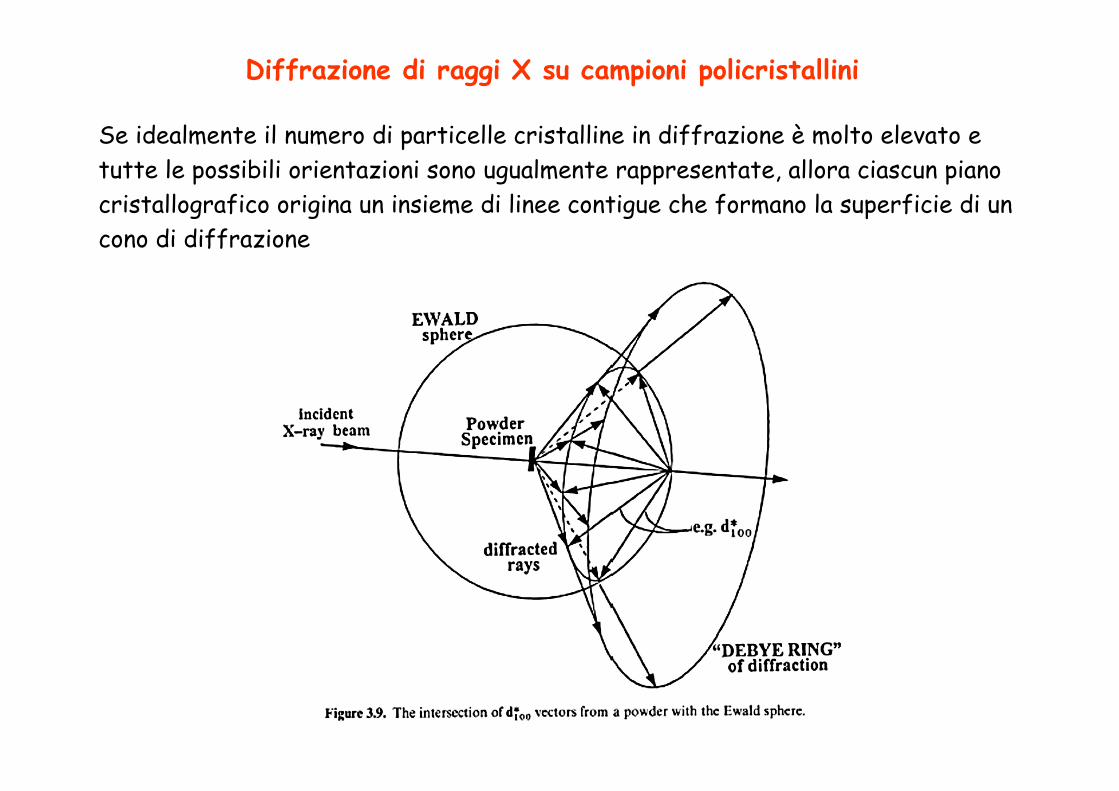
Diffrazione di raggi X su campioni policristallini
Se idealmente il numero di particelle cristalline in diffrazione è molto elevato e tutte le possibili orientazioni sono ugualmente rappresentate, allora ciascun piano cristallografico origina un insieme di linee contigue che formano la superficie di un cono di diffrazione
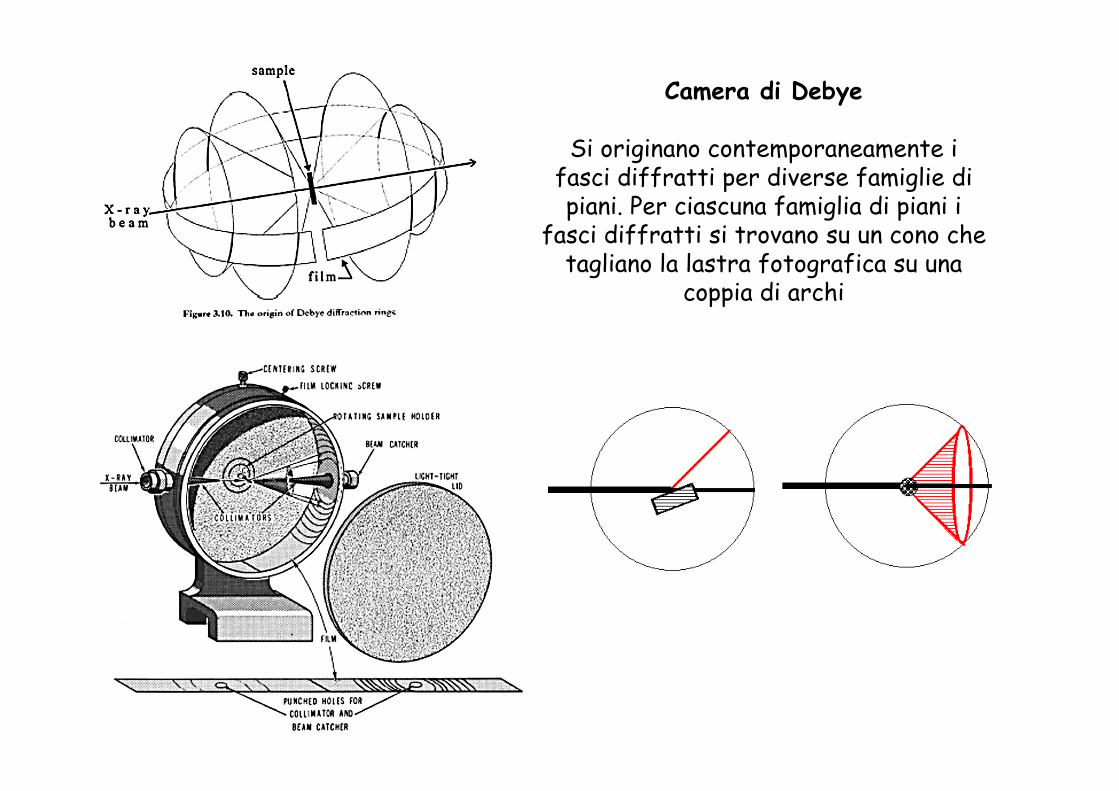
Camera di Debye
Si originano contemporaneamente i fasci diffratti per diverse famiglie di piani. Per ciascuna famiglia di piani i
fasci diffratti si trovano su un cono che tagliano la lastra fotografica su una
coppia di archi
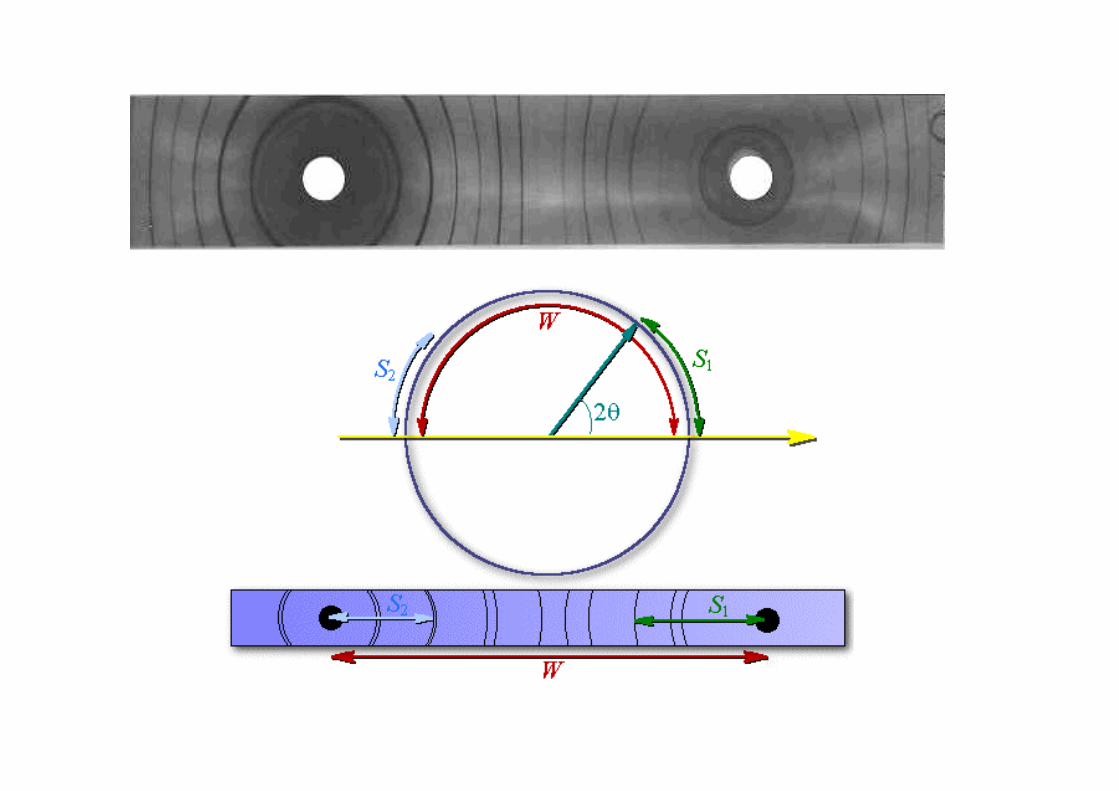

Diffrattometri per polveri (campioni policristallini)
Si varia con continuità e sincronicamente l’angolo tra fascio incidente e campione e quello tra campione e rivelatore
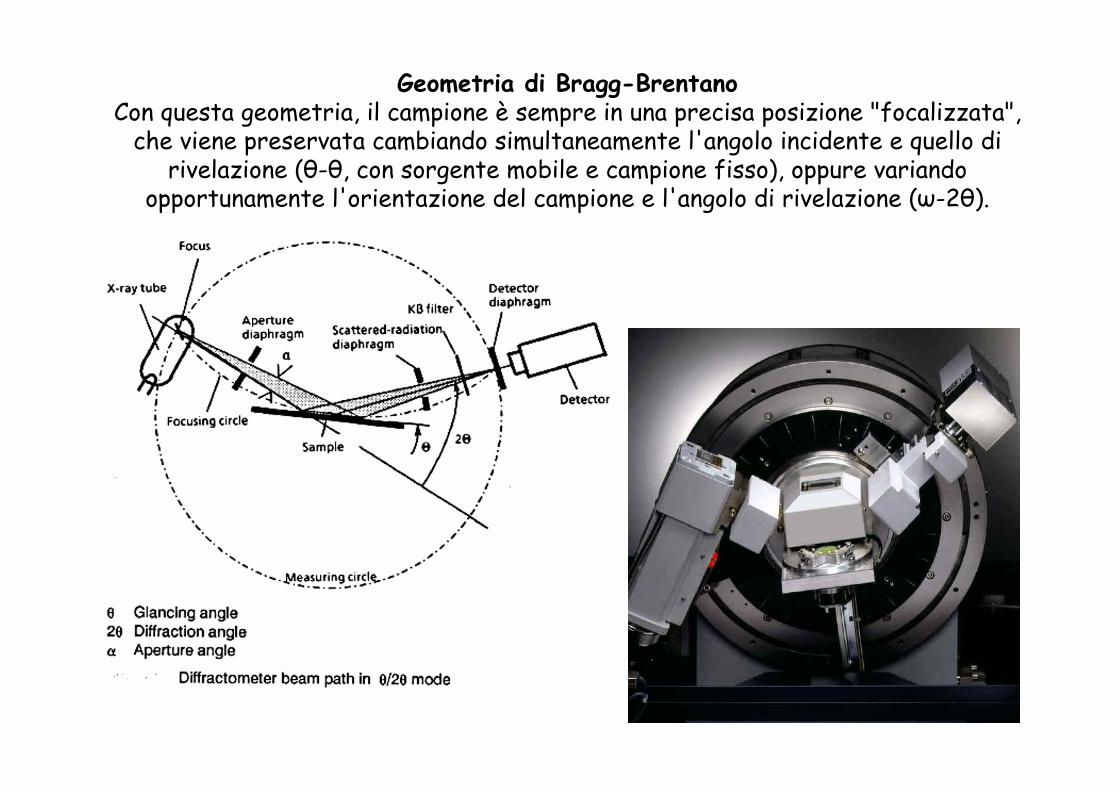
Geometria di Bragg-BrentanoCon questa geometria, il campione è sempre in una precisa posizione "focalizzata",
che viene preservata cambiando simultaneamente l'angolo incidente e quello di rivelazione (θ-θ, con sorgente mobile e campione fisso), oppure variando
opportunamente l'orientazione del campione e l'angolo di rivelazione (ω-2θ).
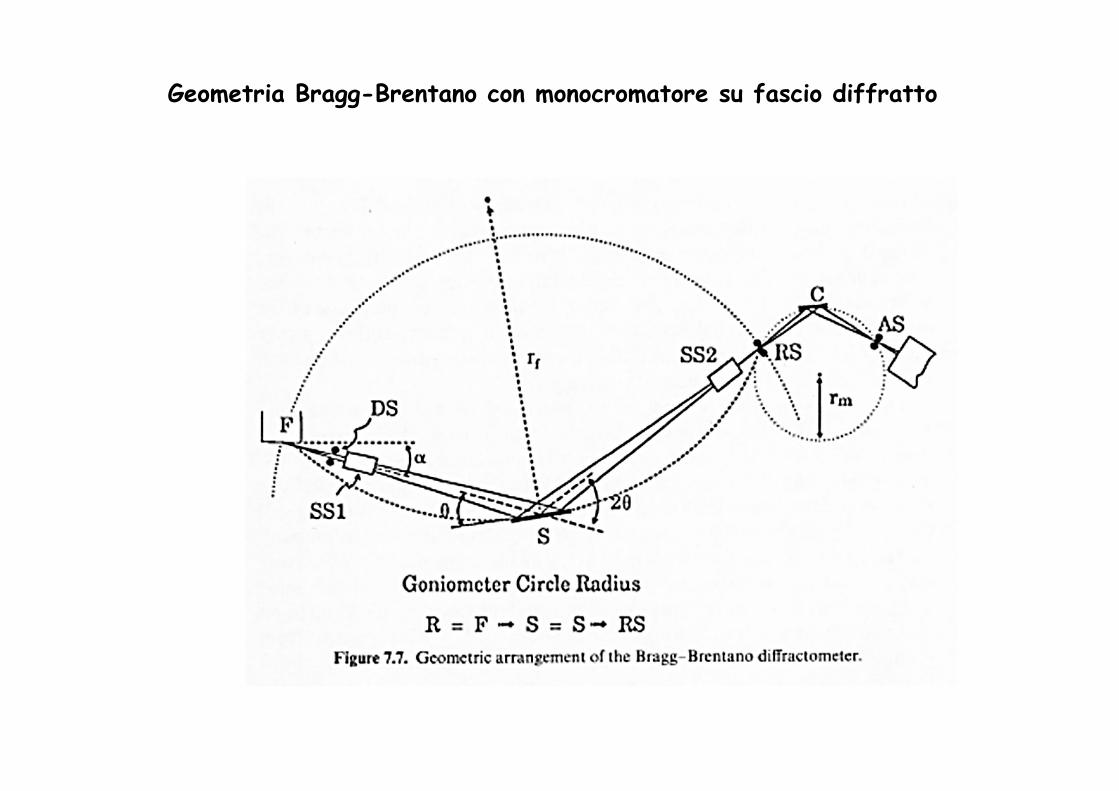
Geometria Bragg-Brentano con monocromatore su fascio diffratto
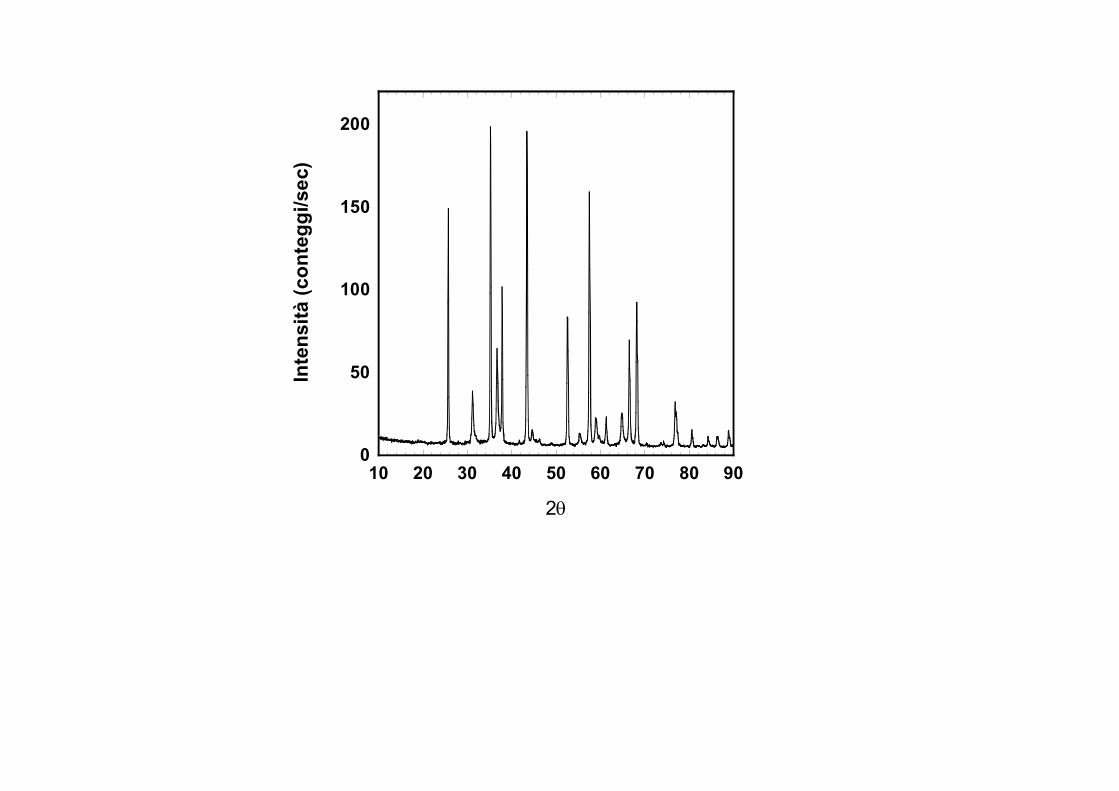
0
50
100
150
200
10 20 30 40 50 60 70 80 90
Inte
nsità
(con
tegg
i/sec
)
2θ

Quantità osservabiliPosizione dei picchiIntensità dei picchi
Forma dei picchiFondo sottostante i picchi
Posizione dei picchi: Dipende esclusivamente dalla cella elementare del materiale in esame. E possibile dai dati di polveri determinare e affinare le costanti di cella con elevata precisione. Su questo dato viene in gran parte
basata il riconoscimento di fasi incognite
Intensità dei picchi: L'intensità diffratta si ottiene integrando l'area di ciascun picco, dopo aver sottratto il contributo di fondo. Una misura
approssimata si ottiene dal massimo valore dei conteggi di ciascun picco. Le intensità diffratte da ciascuna fase presente in una miscela di un campione
polifasico sono proporzionali alla frazione di quella fase.
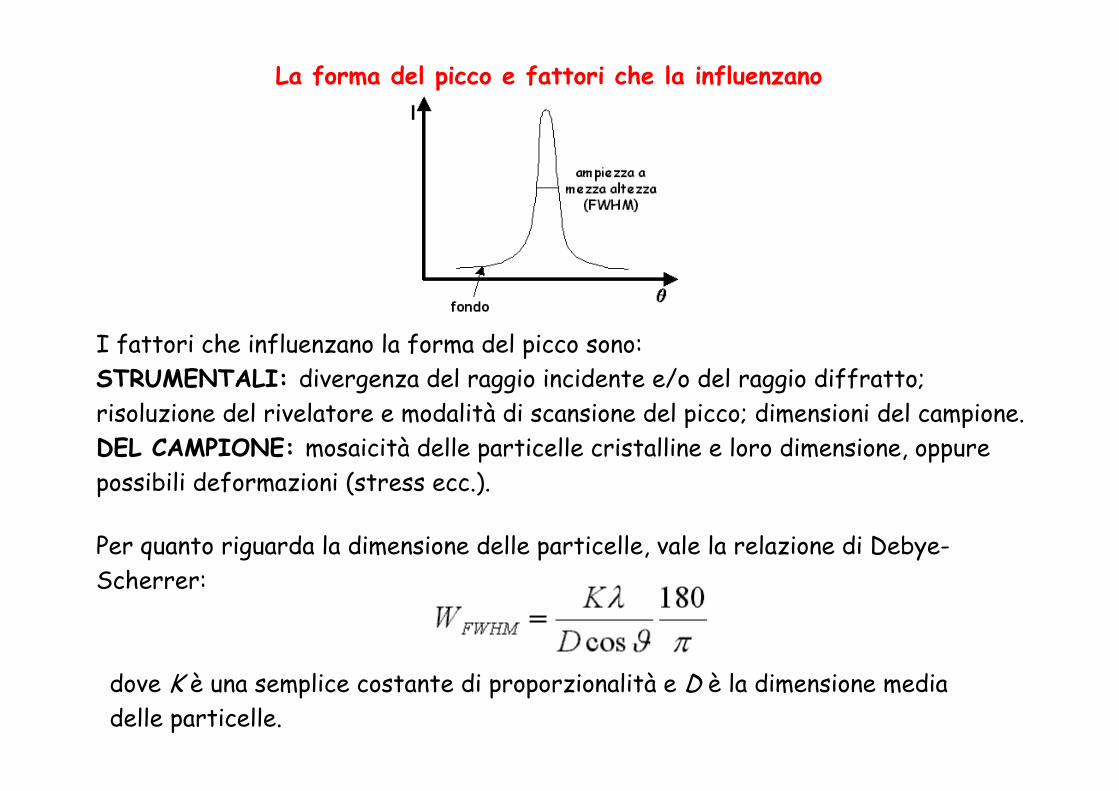
La forma del picco e fattori che la influenzano
I fattori che influenzano la forma del picco sono:STRUMENTALI: divergenza del raggio incidente e/o del raggio diffratto; risoluzione del rivelatore e modalità di scansione del picco; dimensioni del campione.DEL CAMPIONE: mosaicità delle particelle cristalline e loro dimensione, oppure possibili deformazioni (stress ecc.).
Per quanto riguarda la dimensione delle particelle, vale la relazione di Debye-Scherrer:
dove K è una semplice costante di proporzionalità e D è la dimensione media delle particelle.

•selezione del campione (microcristallinità)•macinazione per migliorare l’omogeneità riducendo le dimensioni delle particelle (ma non troppo per evitare l’allargamento dei picchi)•deposizione del campione su supporto•centratura del supporto nel goniometro•scansione (selezionando il tipo di scansione, la velocità ecc.)
Procedura sperimentale
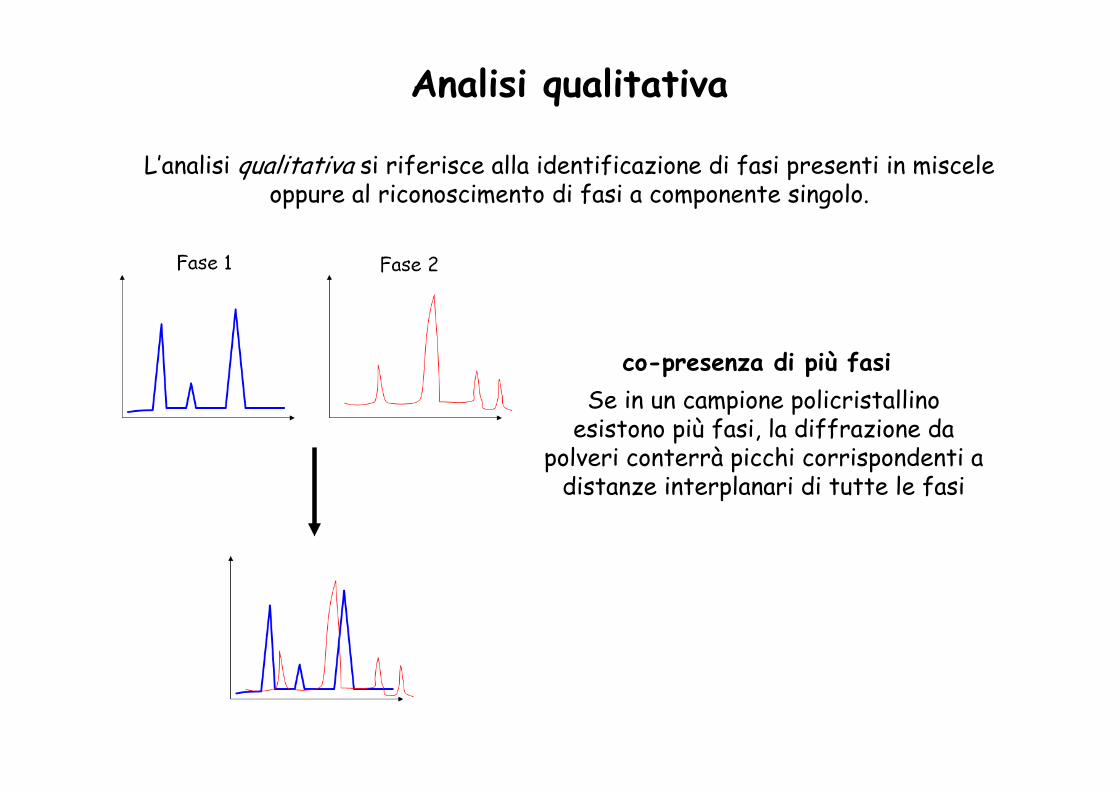
Analisi qualitativa
L’analisi qualitativa si riferisce alla identificazione di fasi presenti in miscele oppure al riconoscimento di fasi a componente singolo.
co-presenza di più fasiSe in un campione policristallino
esistono più fasi, la diffrazione da polveri conterrà picchi corrispondenti a
distanze interplanari di tutte le fasi

La struttura cristallina di molte fasi solide è nota, perché identificata con metodi diffrattometrici a partire dalla introduzione di queste tecniche, cioè a
partire dalla prima metà del XX secolo.
La principale "risorsa" di informazioni per l’identificazione di fasi ignote è il Powder Diffraction File, ossia un archivio elettronico (o cartaceo) dove sono contenute informazioni cristallografiche per più di 300000 fasi inorganiche
ed organiche.
La diffrazione è una informazione primaria, che combinata con l’analisi elementare identifica senza ambiguità una certa fase cristallina.
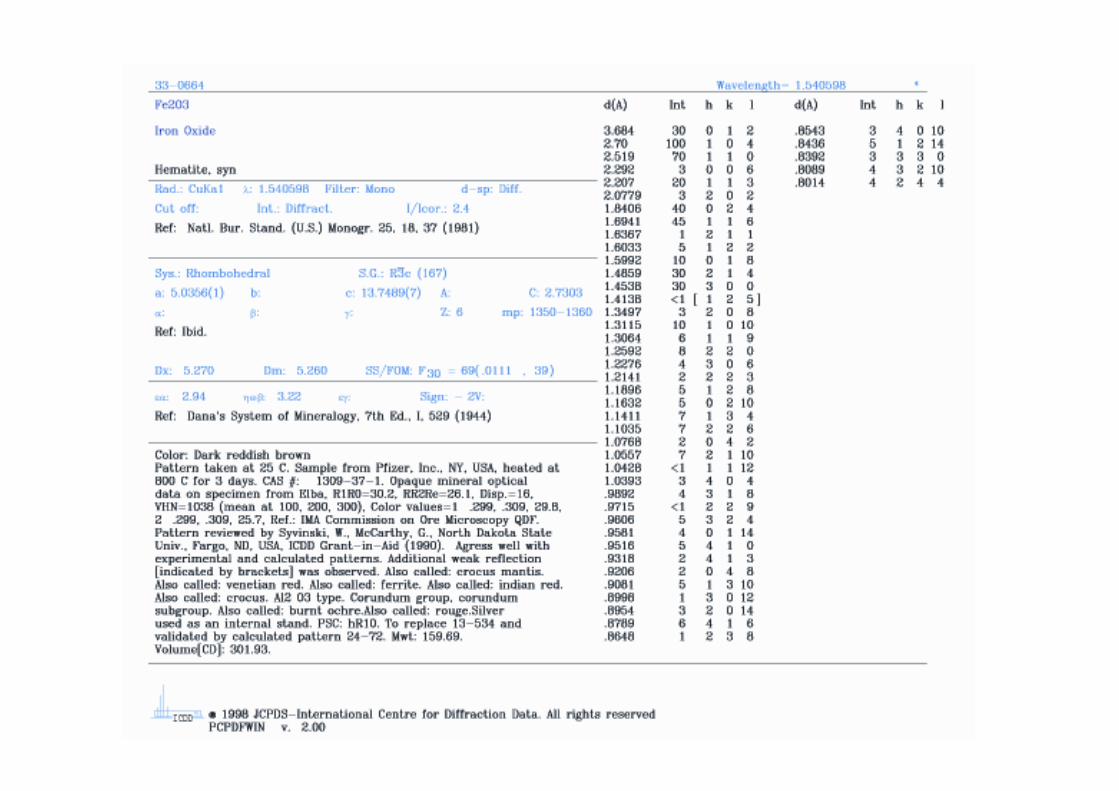
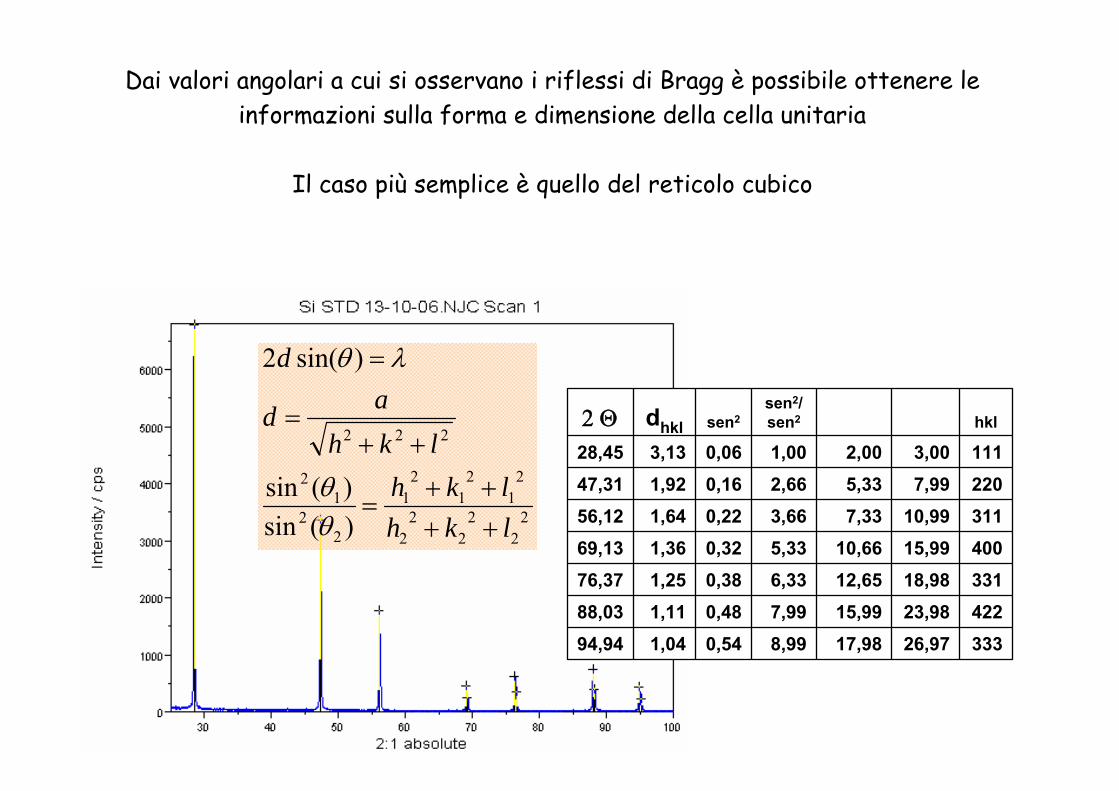
Dai valori angolari a cui si osservano i riflessi di Bragg è possibile ottenere le informazioni sulla forma e dimensione della cella unitaria
Il caso più semplice è quello del reticolo cubico
33326,9717,988,990,541,0494,9442223,9815,997,990,481,1188,0333118,9812,656,330,381,2576,3740015,9910,665,330,321,3669,1331110,997,333,660,221,6456,122207,995,332,660,161,9247,311113,002,001,000,063,1328,45hkl
sen2/sen2sen2dhkl2 Θ
22
22
22
21
21
21
22
12
222
)(sin)(sin
)sin(2
lkhlkh
lkhad
d
++++
=
++=
=
θθ
λθ





























