Embed Size (px)
Citation preview

LEGISLACIÓN FARMACÉUTICA
UNIDAD DIDÁCTICA III
ATENCIÓN FARMACÉUTICA, MEDICAMENTOS Y PRODUCTOS
SANITARIOS.
TEMA 5: MEDICAMENTOS INDUSTRIALES
LEGISLACIÓN, GESTIÓN Y PLANIFICACIÓN
FARMACÉUTICA
Prof. Antonio Ramos Carrillo

Principales modificaciones en la Ley 29/2006, por la Ley 10/2013:
1.Se prohíbe la venta por correspondencia y por procedimientos telemáticos de
medicamentos y productos sanitarios sujetos a prescripción. Se prohíbe, asimismo,
la venta a domicilio y cualquier tipo de venta indirecta al público de medicamentos.
2.Asimismo el ejercicio profesional del farmacéutico en oficina de farmacia, en
establecimiento comercial detallista, en entidades o agrupaciones ganaderas o en un
servicio de farmacia hospitalaria y demás estructuras asistenciales será incompatible
con cualquier clase de intereses económicos directos de los laboratorios
farmacéuticos y/o almacenes mayoristas.
3.El precio industrial de financiación pública, establecido por el órgano competente
del Ministerio de Sanidad, Servicios Sociales e Igualdad, para los medicamentos
dispensados en oficinas de farmacia mediante receta médica oficial.
4. Cambios en infracciones y sanciones.

- R.D. 767/1993, de 21 de mayo, por el que se regula la evaluación, autorización, registro y
condiciones de dispensación de especialidades farmacéuticas y otros medicamentos de uso
humano y veterinario fabricados industrialmente (derogado).
- R.D. 2000/1995, de 7 de diciembre, por el que se modifica el R.D. 767/1993, de 21 de mayo.
- O. de 3 de marzo de 2000 que modifica el R.D. 767/1993.
- Orden SCO/3461/2003, de 26 de noviembre.
-Reglamento (CE) 726/2004/CE, del Parlamento Europeo y del Consejo de 31 de marzo, por
el que se establecen los procedimientos comunitarios para la autorización y control de los
medicamentos de uso humano y veterinario, y por el que se crea la Agencia Europea de
Medicamentos.
- Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos
sanitarios.
- R. D. 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización,
registro y condiciones de dispensación de los medicamentos de uso humano fabricados
industrialmente.
NORMAS SOBRE AUTORIZACIÓN Y REGISTRO DE MEDICAMENTOS

-R. D. 1091/2010, de 3 de septiembre, por el que se modifica el R. D. 1345/2007, de 11 de
octubre, por el que se regula el procedimiento de autorización, registro y condiciones de
dispensación de los medicamentos de uso humano fabricados industrialmente, y el R. D.
1246/2008, de 18 de julio, por el que se regula el procedimiento de autorización, registro y
farmacovigilancia de los medicamentos veterinarios fabricados industrialmente.
-Ley 10/2013, de 24 de julio, por la que se incorporan al ordenamiento jurídico español las
Directivas 2010/84/UE del Parlamento Europeo y del Consejo, de 15 de diciembre de
2010, sobre farmacovigilancia, y 2011/62/UE del Parlamento Europeo y del Consejo, de 8
de junio de 2011, sobre prevención de la entrada de medicamentos falsificados en la
cadena de suministro legal, y se modifica la Ley 29/2006.
-R. D. 686/2013, de 16 de septiembre, por el que se modifica el R. D. 1345/2007, de 11 de
octubre, por el que se regula el procedimiento de autorización, registro y condiciones de
dispensación de los medicamentos de uso humano fabricados industrialmente.
NORMAS SOBRE AUTORIZACIÓN Y REGISTRO DE MEDICAMENTOS

Esta mañana he comprado una especialidad farmacéutica que contiene este símbolo.
¿Qué significa?
Ya no existe el término de especialidad farmacéutica al ser sustituido en la
Ley 29/2006 por el de medicamento industrial.
Ejemplo de empleo de terminología obsoleta e incorrecta

“Ningún medicamento elaborado industrialmente podrá ser puesto en el
mercado sin la previa autorización de la Agencia Española de Medicamentos y
Productos Sanitarios e inscripción en el Registro de Medicamentos o sin haber
obtenido la autorización de conformidad con lo dispuesto en las normas
europeas que establecen los procedimientos comunitarios para la autorización y
control de los medicamentos de uso humano y veterinario y que regula la
Agencia Europea de Medicamentos”
Ley 29/2006, de 26 de julio, de garantías y uso
racional de los medicamentos y productos sanitarios
Capítulo II, art. 9.
Así pues, el registro de medicamentos es el último eslabón de una
cadena de actuaciones en las que se evalúa la idoneidad sanitaria
(calidad, seguridad, eficacia e información) del medicamento en cuestión
de acuerdo con unos criterios internacionalmente aceptados y sobre la
base de la información técnica y administrativa recogida en el
denominado expediente de registro.

Segura: no produciendo en condiciones normales de utilización efectos tóxicos
o indeseables desproporcionados al beneficio que procura. Los estudios toxicológicos
comprenderán ensayos de toxicidad aguda y crónica, ensayos de teratogenia,
embriotoxicidad, fertilidad, ensayos de mutagénesis y, en su caso, de carcinogénesis.
CONDICIONES PARA OTORGAR AUTORIZACIÓN SANITARIA Ley 29/2006, de 26 de julio. Art. 10: garantías exigibles para la autorización de
medicamentos
Calidad:de las materias primas,
de los productos intermedios, del
proceso de fabricación y del
producto final.
Eficaz: estudios
preclínicos y
ensayos clínicos.
En las indicaciones
terapéuticas para las
que se ofrece
Correctamente identificada y de información
precisa, de forma comprensible al paciente.
La Real Farmacopea Española es el código que establece la calidad que
deben cumplir los principios activos y excipientes que entren en la
composición de los medicamentos de uso humano y veterinario.

... Garantías exigibles a los medicamentos.
• Garantías de identificación – A cada principio activo se le atribuirá una DOE por la AEMPS, igual o lo más
aproximada a la DCI (fijada por la OMS). Será de uso obligatorio.
– Denominación del medicamento:
• Nombre de fantasía que no se confunda con la DOE.
• DOE más marca comercial o nombre del titular de la autorización de la comercialización.
• Medicamentos genéricos: DOE o DCI más nombre o marca del titular, con las siglas EFG (Equivalente Farmacéutico Genérico).
– Código Nacional de Medicamentos (establecido por el Ministerio de Sanidad, Servicios Sociales e Igualdad), que figura en el etiquetado de los medicamentos.
• Garantías de información – Ficha técnica o resumen de las características del producto aprobada por la
AEMPS.
– Prospecto de acuerdo a la ficha técnica. Deberá ser legible, claro, asegurando su comprensión por el paciente y reduciendo al mínimo los términos de naturaleza técnica.
– Etiquetado con todos los datos del medicamento: p.a., titular de la autorización, vía de administración, cantidad contenida, nº de lote de fabricación, fecha de caducidad, precauciones de conservación, condiciones de dispensación, y demás datos que reglamentariamente se determinen.

Se conoce como “procedimiento de registro” a una serie de
actuaciones que concluyen en la concesión o denegación de la
autorización de comercialización, y que son llevados a cabo en
unos plazos determinados.
Dicho procedimiento se encuentra perfectamente establecido
en la legislación vigente y es llevado a cabo con la máxima
transparencia.

II.- Procedimiento de Registro Centralizado (PRC).
TUna compañía farmacéutica realiza una única solicitud para que
se le conceda autorización en todos los países de la Unión Europea. La
autorización la concede la EMA para medicamentos derivados de la
biotecnología o considerados muy innovadores.
III.- Procedimiento de Registro de Mutuo Reconocimiento (PRMR).
TÚtil para medicamentos convencionales que una compañía
farmacéutica pretenda comercializar en varios países de la UE.
TLa autorización sanitaria la concede cada una de las agencias
nacionales de los diferentes países, que actúan reconociendo la primera
autorización concedida por una de ellas.
I-Procedimiento de registro nacional (PRN).
TLa compañía farmacéutica solicita la autorización en un único país,
donde se pretende comercializar el nuevo medicamento. La autorización es
solo válida para ese único país.
TIPOS DE PROCEDIMIENTO DE REGISTRO:

¿QUÉ ES LA EMA? Es la AGENCIA EUROPEA DE MEDICAMENTOS
• Es un organismo descentralizado de la Unión Europea que tiene su sede en Londres. Su principal responsabilidad es la protección y promoción de la salud pública y animal, mediante la evaluación y supervisión de los medicamentos de uso humano y veterinario.
• Es responsable de la evaluación científica de las solicitudes europeas de autorización de comercialización de medicamentos (procedimiento centralizado). Cuando se utiliza el procedimiento centralizado las empresas presentan a la EMA una única solicitud de autorización de comercialización.
• La seguridad de los medicamentos es controlada constantemente por la Agencia a través de una red de farmacovigilancia. La EMA adopta las medidas oportunas cuando los informes sobre efectos adversos del medicamento indican cambios en el equilibrio beneficio/riesgo de un medicamento. Con respecto a los medicamentos veterinarios, la Agencia tiene la responsabilidad de establecer límites máximos para los residuos de medicamentos en los alimentos de origen animal.
• La Agencia también participa activamente en la promoción de la innovación y la investigación en la industria farmacéutica. La EMA proporciona asesoramiento científico y asistencia a empresas farmacéuticas en la elaboración de protocolos con vistas al desarrollo de nuevos medicamentos.
= European Medicines Agency
http://www.ema.europa.eu/ema/

... EMA
• La EMA publica directrices para controlar los requisitos de comprobación de la calidad, la seguridad y la eficacia.
• La EMA contribuye a las actividades internacionales de la Unión Europea mediante su trabajo con la Farmacopea Europea o la Organización Mundial de la Salud.

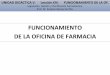
I-PROCEDIMIENTO DE REGISTRO NACIONAL. -TRAMITACIÓN DE SOLICITUDES-
ETAPAS:1.Presentación del expediente de registro
2.Estudio del expediente de registro 3.Autorización sanitaria de comercialización
4. Trámites postautorización: - Fijación de precios- Financiación por S.N.S. (En su caso)
* Para lograr la autorización sanitaria, el solicitante deberá presentarpor duplicado la correspondiente solicitud de autorización.La solicitud debe ir acompañada del comprobante del pago de tasasy del EXPEDIENTE (DOSSIER) DE REGISTRO (5 MÓDULOS).
Agencia Española de
Medicamentos y
Productos Sanitarios
Dirección General de
Cartera Básica de Servicios
del Sistema Nacional de
Salud y Farmacia

MÓDULO I: INFORMACIÓN ADMINISTRATIVA.
MÓDULO II: RESÚMENES.
MÓDULO III: INFORMACIÓN QUÍMICA ,
FARMACÉUTICA Y BIOLÓGICA.
MÓDULO IV: INFORMES NO CLÍNICOS.
MÓDULO V: INFORMES CLÍNICOS.
La Agencia Española del Medicamento y Productos
Sanitarios dispone de 210 días desde la presentación dela solicitud para resolver, concediendo o denegando,dicha autorización.
DOSSIER DE REGISTRO:

...PROCEDIMIENTO DE REGISTRO NACIONAL.
• El solicitante de registro titular en España. -Compañía Farmacéutica-
(-Avalada por el Director Técnico del Laboratorio-)
• Requisitos:
– Estar establecido en la UE.
– Contar con los medios necesarios para asumir las obligaciones que derivan de la titularidad.
– Estar inscrito en el registro de laboratorios farmacéuticos si tiene su sede en España o acreditar esta condición si tiene su sede en otro país de la UE.
• Una vez autorizada un MED debe revalidarse a los 5 años solicitándose con 6 meses de antelación.Es por tanto la Administración Sanitaria la que valora la idoneidad sanitaria de los medicamentos.
Procedimiento Nacional de registro de MED
Tramitación de solicitudes

Procedimiento Nacional de registro de MED
Tramitación de solicitudes - Información administrativa. Formulario de solicitud: identificación del medicamento, solicitante y lugares de fabricación.
Resumen de las características del producto, etiquetado y prospecto.
Informes de experto sobre calidad y estudios realizados.
Evaluación del riesgo para el medio ambiente.
- Resúmenes. Contiene información abreviada sobre:
» Resumen global de la calidad.
» Visión general de la parte no clínica.
» Visión general de la parte clínica.
» Resumen no clínico.
» Resumen clínico.
- Información química, farmacéutica y biológica. - Información general e información sobre los materiales de partida y materias primas.
- Principios activos:
- fabricación, caracterización, control, envase, estabilidad.
- Producto terminado:
- descripción y composición, desarrollo, controles, envases y estabilidad.

Procedimiento Nacional de registro de MED
- Informes no clínicos.
Pruebas:
- farmacológicas.
- farmacocinéticas.
- toxicológicas.
- Informes de estudios clínicos.
- biofarmacéuticos.
- farmacocinéticos.
- farmacodinámica humana.
- seguridad y eficacia.
- post-comercialización.
- recogida de datos y listado de pacientes.
…Tramitación de solicitudes

Procedimiento Nacional de registro de MED
Agencia Española del Medicamento y Productos Sanitarios

Procedimiento Nacional de registro de MED
Evaluación de expedientes.
La AEM debe resolver en el plazo de 210 días desde la presentación de
la solicitud.
El cómputo de días se interrumpe si son necesarias informaciones y pruebas
complementarias.
Debe abonar las tasas y subsanar información, en su caso, en 10 días.
El Comité de Medicamentos de Uso Humano es el órgano que más
directamente interviene en el procedimiento.
También interviene el Comité de Medicamentos de Veterinaria si son
veterinarios los MED.
Se estudian tres aspectos fundamentales:
- Calidad del principio activo y materia prima.
- Seguridad del medicamento.
- Eficacia del medicamento.
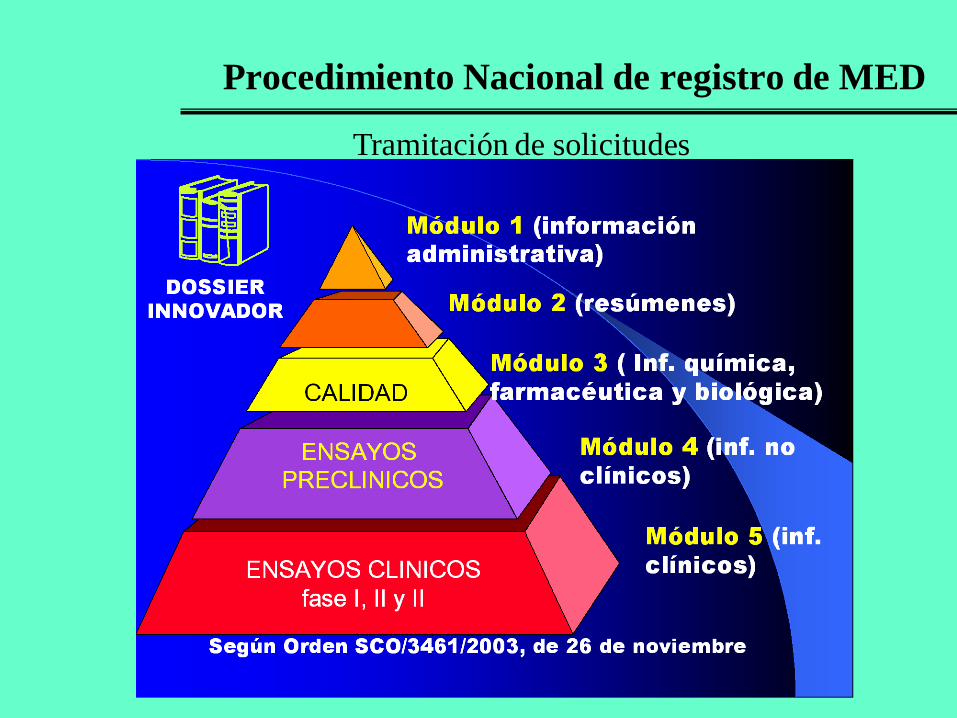
Procedimiento Nacional de registro de MED
Evaluación de expedientes.
El CODEM emite un dictamen, 30 días antes de finalizar el plazo, basado en:
el informe de calidad.
el informe de evaluación.
la ficha técnica aprobada.
AEM y PS
(Comité de Medicamentos
de Uso Humano)
Dictamen
Resolución
Positiva Autorización
y Registro
Negativa
10-15 días para alegar
Resolución definitiva Negativa
Positiva

Procedimiento Nacional de registro de MED.
Autorización e Inscripción de MED
Determina las condiciones sanitarias de comercialización de un
MED estableciendo:
Condiciones de dispensación.
Ficha técnica aprobada.
Prospecto y etiquetado.
Contenido del envase.
Condiciones de distribución.
Numero de registro (CN de 7 cifras).

Procedimiento Nacional de registro de M.
Autorización e Inscripción de EFG
Para autorizar e inscribir una EFG son necesarios como mínimos 10 años, aunque
la AEM puede aumentarlo según el tipo de medicamento.
Debe presentar los módulos:
- I (administrativo),
- II (resúmenes) y
- III (parte químico-biológica-farmacéutica)
No es necesario presentar
- parte IV (informe preclínico) y
- parte V (informe clínico).
En su lugar debe acreditarlo
mediante bibliografía y probar la
biodisponibilidad y bioequivalencia
con la EF original.

Procedimiento Nacional de registro de MED
-Modificaciones de la Autorización-
La AEM puede suspender, temporal o definitivamente una autorización de EF:
Ser nociva.
no eficaz.
riesgo previsible.
incumplimiento del laboratorio de la normativa,...
Hay un plazo de 90 días desde la instrucción del expediente.
Interviene el Comité de Seguridad de Medicamentos de Uso Humano
-Obligaciones del Titular-
Observar las condiciones de autorización.
Respetar la continuidad del servicio.
Actualizar el expediente y el procedimiento de fabricación.
Promover el uso racional del medicamento.
Realizar farmacovigilancia de ese MED (designación de responsable).
Informar de la retirada de lotes del mercado.

Procedimiento Nacional de registro de MED
Fijación de Precios y Financiación de la Seguridad Social
El precio es fijado según los establecido en el R.D. 271/1990
Los trámites de financiación por el SNS se establecen en el
R.D. 83/1993.
Interviene la Comisión Interministerial de Precios de los Medicamentos
Art. 88 Ley 29/2006. El precio industrial de financiación pública,
establecido por el órgano competente del Ministerio de Sanidad,
Servicios Sociales e Igualdad, para los medicamentos dispensados
en oficinas de farmacia mediante receta médica oficial.
No puede haber subastas solo a nivel autonómico.

En el documento de autorización deberán figurar, al menos,
los siguientes datos:
a) Nombre del medicamento.
b) Número de registro.
c) Grupo terapéutico.
d) Forma farmacéutica.
e) Vía de administración.
f) Presentaciones autorizadas con sus respectivos Códigos Nacionales.
g) Condiciones de conservación y caducidad.
h) Condiciones de prescripción y dispensación.
i) Nombre y dirección del titular de la autorización.
j) Nombre y dirección del representante del titular de la autorización de
comercialización, en su caso.
k) Nombre y dirección del fabricante, tanto del principio activo como del
medicamento en caso de que difieran.
l) Composición cualitativa y cuantitativa completa.

Procedimiento de autorización (210 días):
• Solicitud.
• Evaluación.
• Informe del Comité de Medicamentos de Uso Humano. – Alegaciones.
• Registro. – Código Nacional.
• Autorización. – Ficha Técnica//Nombre, forma y formato/Etiquetado y
Prospecto/Condiciones de comercialización.
• Fijación del precio y financiación.
Por tanto:

CÓDIGO NACIONAL (C.N.) •El C.N. es un sistema de identificación rápido, para ayudar y facilitar la gestión de las Oficinas de Farmacia y otros estamentos, haciendo posible la adquisición de medicamentos, efectos y accesorios, productos de parafarmacia que se encuentran en el mercado nacional, mediante el tratamiento informático de los indicados productos.
• El CN es adjudicado por diferentes estamentos oficiales, en función del tipo de producto:
– A los medicamentos, medicamentos a base de plantas medicinales y medicamentos homeopáticos se lo adjudica la Agencia Española de Medicamentos y Productos Sanitarios, cuando autoriza el registro.
– A los dietoterápicos y efectos y accesorios (que son respectivamente los Alimentos y Productos Sanitarios financiados por el Sistema Nacional de Salud) se lo asigna la Dirección General de Cartera Básica de Servicios del Sistema Nacional de Salud y Farmacia, según lo fija el R.D. 9/1996, que determina la necesidad de que el CN figure en el cupón precinto.
– A los productos de Parafarmacia, se lo adjudica el Consejo General de Colegios Oficiales de Farmacéuticos. Estos productos no están financiados por el S.N.S.

Medicamentos industriales

DENOMINACIÓN
DEL
MEDICAMENTO:
La denominación del
medicamento, cuando
sea una denominación
comercial o marca, no
podrá confundirse con
una DOE o una DCI, ni
inducir error sobre las
propiedades terapéuticas
o la naturaleza del
medicamento.
D.C.I.: Denominación Común Internacional
D.O.E.: Denominación Oficial Española
Nombre de fantasía

II.- Procedimiento de Registro Centralizado (PRC).
Reglamento (CE) 726/2004
• Obligatorio
– Biotecnológicos.
– Medicamentos que contienen nuevas sustancias activas contra:
• Cáncer
• SIDA
• Diabetes
• Alteraciones neurodegenerativas
– Medicamentos huérfanos.
A partir de 20 de mayo de 2008
– Extiende a medicamentos contra: • Enfermedades autoinmunes
• Enfermedades víricas
– Eventualmente, a otros medicamentos (cláusula de revisión, por mayoría cualificada).
• Optativo
– Medicamentos que contienen
nuevas sustancias activas para el resto de terapias.
– Medicamentos que usen principios activos existentes:
• Innovaciones significativas
• Interés para los pacientes

Procedimiento Centralizado o Europeo
de registro de MED
• Está regulado por el Reglamento CE 726/2004. Modificado por: • Reglamento (CE) nº 1901/2006 , Reglamento (CE) nº 1394/2007.
• Reglamento (CE) n° 219/2009, Reglamento (CE) n° 470/2009.
• Reglamento (UE) n° 1235/2010.
• Es la EMA (Agencia Europea de Medicamentos) la que dicta
autorización. Se encuentra en Londres.
• Se deben autorizar por el procedimiento centralizado los medicamentos
que figuran en el Anexo del Reglamento 726/2004.
• El expediente contiene los 5 módulos vistos anteriormente (contenidos
en la Directiva 2001/83/CE)

Procedimiento Centralizado
Día 1 Inicio del Procedimiento (CHMP-Comité de Medicamentos de Uso Humano).
Nombramiento de ponente y ponente adjunto
Día 80 Periodo mínimo para estudio de datos científicos.
Comprobación de documentos
Realización de pruebas por laboratorio oficial
Subsanación de documentación
Informe del Estado miembro sobre condiciones del fabricante
Día 210 Emisión de Dictamen por la CHMP.
NEGATIVO POSITIVO

…Procedimiento Centralizado
MEDICAMENTOS QUE DEBEN SER OBJETO DE UNA AUTORIZACIÓN COMUNITARIA
• 1. Medicamentos de uso humano desarrollados por medio de uno de los siguientes procesos biotecnológicos:
– Técnica del ADN recombinante.
– Expresión controlada de codificación de genes para las proteínas biológicamente activas en procariotas y eucariotas, incluidas las células de mamífero transformadas.
– Métodos del hibridoma y del anticuerpo monoclonal.
• 2. Medicamentos veterinarios empleados principalmente como potenciadores para fomentar el crecimiento o aumentar el rendimiento de los animales tratados.
• 3. Medicamentos de uso humano que contengan una sustancia activa nueva que, en la fecha de entrada en vigor del presente Reglamento, no estuviera autorizada en la Comunidad y cuya indicación terapéutica sea el tratamiento de alguna de las enfermedades siguientes:
- El síndrome de inmunodeficiencia adquirida.
- El cáncer.
- Los trastornos neurodegenerativos.
- La diabetes,
- Y, con efectos a partir de 20 de mayo de 2008: las enfermedades autoinmunes y otras disfunciones inmunes y las enfermedades víricas.
• 4. Los medicamentos designados como medicamentos huérfanos.

…Procedimiento Centralizado
-ORGANIGRAMA DE LA DIRECCIÓN EJECUTIVA DE LA EMA-
Dirección ejecutiva.
Evaluación de medicamentos de uso humano antes de su
autorización.
Evaluación de medicamentos de uso humano después de su
Autorización.
Medicamentos de uso veterinario e inspecciones.
Comunicaciones y redes.
Administración.

Committee for Medicinal Products for Human Use (CHMP)
Committee for Medicinal Products for Veterinary Use (CVMP)
Committee for Orphan Medicinal Products (COMP)
Committee on Herbal Medicinal Products (HMPC)
Paediatric Committee (PDCO)
Committee for Advanced Therapies (CAT)
EMA Committees
…Procedimiento Centralizado

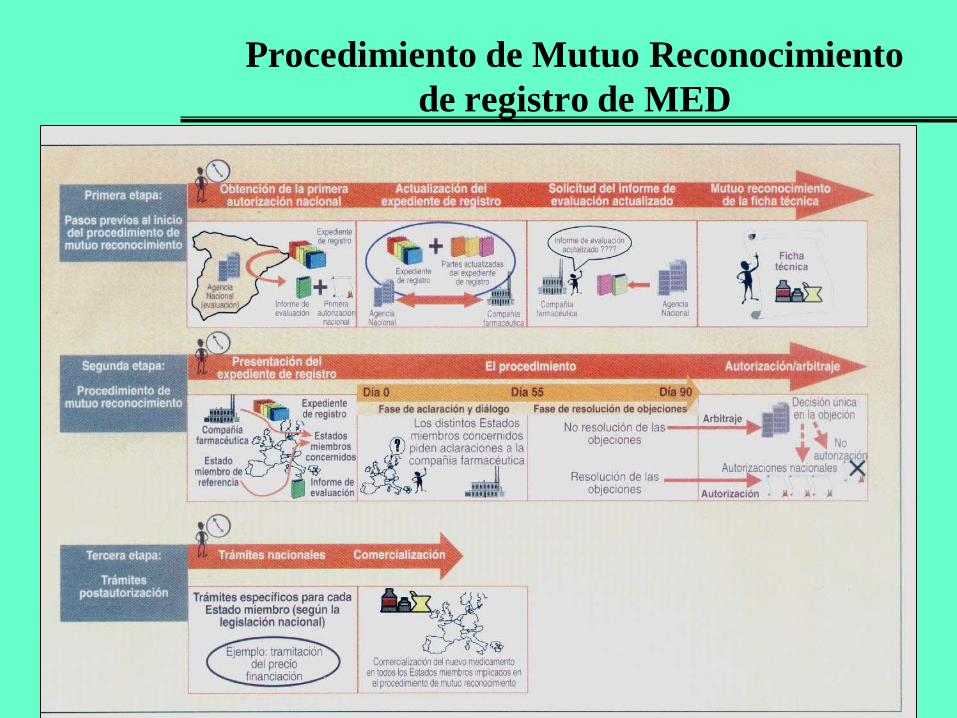
Procedimiento de Mutuo Reconocimiento
de registro de EF
Está regulado por el R.D. 2000/1995, de 7 de diciembre.
Útil para medicamentos convencionales cuando el laboratorio desea comercializarlo en varios países europeos.
No se puede usar para medicamentos del Anexo del Reglamento 726/2004.
La autorización de una agencia nacional (Estado de referencia) es reconocida por otros Estados (concernidos).
Debe actualizar el expediente de registro (5 módulos) en el estado de referencia, e indicar posteriormente los países en que se pretenda el mutuo reconocimiento.
Desde la recepción de la solicitud actualizada, la agencia nacional cuenta con 90 días para enviar informe de evaluación a un Estado concernido.
El Estado concernido cuenta también con 90 días para pronunciarse sobre su aceptación.
Si hay alegaciones u objeciones de un Estado concernido que no son subsanadas por la compañía farmacéutica, actúa la EMA como organismo de arbitraje.
Una vez aceptado el reconocimiento se realizan los trámites post-autorización.
Procedimiento de Mutuo Reconocimiento
de registro de MED

«Artículo 68. Causas de suspensión y revocación.
La Agencia Española de Medicamentos y Productos Sanitarios, podrá acordar la suspensión,
revocación o modificación de la autorización de un medicamento cuando:
1. Se considere que el medicamento es nocivo.
2. El medicamento resulte no ser terapéuticamente eficaz. La carencia de eficacia terapéutica
se valorará cuando se llegue a la conclusión de que no se pueden obtener resultados
terapéuticos del medicamento.
3. Con base a los datos de seguridad, el medicamento tenga una relación beneficio-riesgo
desfavorable.
4. El medicamento no tenga la composición cuantitativa o cualitativa autorizada, o se
incumplan las garantías de calidad, o no se ejecuten los controles de calidad exigidos.
5. Los datos e informaciones contenidos en la documentación sean erróneos o incumplan la
normativa de aplicación en la materia.
6. El modo de fabricación del medicamento o los métodos de control utilizados por el
fabricante no se ajusten a los descritos en la autorización.
7. Por cualquier otra causa, que suponga un riesgo previsible para la salud o seguridad de las
personas o animales.
8. En cualquier otro caso en el que la Comisión Europea, así lo hubiera acordado.»
Real Decreto 686/2013, de 16 de septiembre, por el que se modifica el Real
Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de
autorización, registro y condiciones de dispensación de los medicamentos de uso
humano fabricados industrialmente.

Ocho. Se añade un nuevo punto 4 al artículo 17 con la siguiente redacción:
«4. Como complemento de lo anterior, podrá concederse una autorización de
comercialización de un medicamento sujeta adicionalmente al cumplimiento de una o
varias de las condiciones siguientes:
a) Se adopten determinadas medidas para garantizar el uso seguro del medicamento
que se incluyan en el sistema de gestión de riesgos;
b) Se realicen estudios de seguridad posautorización;
c) Se cumplan las obligaciones sobre el registro o la notificación de sospechas de
reacciones adversas que sean más estrictas que las contempladas en la normativa
vigente sobre farmacovigilancia de medicamentos de uso humano.
d) Cualquier otra condición o restricción relacionada con el uso seguro y eficaz del
medicamento;
e) El sistema de farmacovigilancia sea adecuado;
f) Se realicen estudios de eficacia posautorización, cuando se planteen cuestiones
sobre la eficacia del medicamento que solo puedan resolverse después de la
comercialización de este. La obligación de realizar tales estudios se basará en los
actos delegados adoptados de conformidad con la normativa europea.»
Real Decreto 686/2013…

FORMATOS O TIPOS ESPECIALES DE
MED.
-ENVASE CLÍNICO.
- MEDICAMENTOS PUBLICITARI0S (antes E.F.P.)Medicamentos objeto de publicidad destinada alpúblico.
-MEDICAMENTOS SOMETIDOS A ESPECIALCONTROL MEDICO (E.C.M.)
-MEDICAMENTOS DE USO HOSPITALARIO YDE DIAGNÓSTICO HOSPITALARIO (H., y D.H.)

Contienen un número superior de unidades al autorizado para la
venta al público, pero igual fórmula y forma farmacéutica.
En su cartonaje debe figurar la inscripción: “Envase clínico.
Prohibida su venta al detalle”.
El número de unidades en envases clínicos, según forma
farmacéutica:
- Ampollas y viales inyectables: 50 ó 100 unidades.
- Comprimidos, grageas, cápsulas, polvos unidosis (bolsas o
papelillos): 500 ó 1000 unidades.
- Polvos: 100, 200, 500 ó 1000 gramos.
- Suspensiones o soluciones: 10 ó 20 frascos.
- Supositorios y óvulos: 100 ó 250 unidades.
Ejemplo: Augmentine® 2g/200mg I.V., 50 viales c.n.: 638676
ENVASE CLÍNICO

MEDICAMENTOS PUBLICITARIOS
Son medicamentos cuyo uso y dispensación es libre y sin receta, que se utilizan en el tratamiento de síndromes o síntomas menores que no requieran atención médica.
Ejemplo: AngileptolR sabor miel y limón. c.n.: 934091.
Están autorizadas como tales en base a los siguientes criterios:
• a) Que no se financien con fondos públicos.
• b) Que por su composición y objetivo estén concebidos y destinados para ser utilizados sin la intervención de un médico que realice el diagnóstico, la prescripción o el seguimiento del tratamiento.
• c) Que no contengan en su composición sustancias psicotrópicas ni estupefacientes.
• d) Se trate de medicamentos objeto de publicidad dirigida al público, cuya denominación no podrá ser igual o inducir a confusión con la de otro medicamento sujeto a prescripción médica o financiado con fondos públicos.
Ahora conocidos como:
Medicamentos objeto de publicidad destinada al público.

... MEDICAMENTOS PUBLICITARIOS (Medicamentos objeto de publicidad destinada al público)
• De las EFP (ya suprimido como siglas) se puede realizar publicidad dirigida al público en general, al contrario de el resto de medicamentos.
• Las características de este tipo de publicidad (art. 5. R.D. 1416/1994) son:
– Necesidad de incluir la denominación del medicamento.
– La evidencia de que lo anunciado es un medicamento.
– Invitación expresa a leer la instrucciones de uso.
– Que no se sugiera que:
• Efecto está asegurado.
• Potencia el rendimiento deportivo.
• Equipare el medicamento a un producto alimenticio, cosmético u otro producto de consumo.
• Se dirija exclusivamente a niños.
• Todos los anuncios de MP deben someterse a un control previo sanitario (CPS) por parte del Ministerio de Sanidad, Servicios Sociales e Igualdad (Dirección General de Cartera Básica de Servicios del Sistema Nacional de Salud y Farmacia).
• Pantalla azul con tres mensajes:
– El anuncio es de un medicamento.
– Leer detenidamente las instrucciones de uso.
– En caso de duda consultar al farmacéutico.
“Lea las instrucciones de este medicamento y consulte al farmacéutico”

MED. SOMETIDOS A ESPECIAL CONTROL MEDICO (E.C.M.)
En el cartonaje “Especial Control Médico”, y al lado de los
símbolos ECM.
En el prospecto “Medicamento de Especial Control Médico.
ECM. Con receta médica”.
Tienen cupón precinto diferenciado.
Se someten a este régimen:
- Derivados de la vitamina A, excepto los de aplicación tópica y la propia
vitamina A.
- Ácido acetohidroxámico.
- Talidomida.
- Clozapina.

MED. DE USO HOSPITALARIO Y DE DIAGNÓSTICO
HOSPITALARIO (H., y D.H.)
USO HOSPITALARIO: Llevan el símbolo H. Las O.F. pueden
suministrarlas a Hospitales, pero no dispensarlas al público.
Ejemplo: ZiagenR 300 mg, 60 comp. (p.a.= abacavir)
DIAGNÓSTICO HOSPITALARIO: Llevan el símbolo D.H.
Las O.F. pueden dispensarlas al público, pero el diagnóstico debe
proceder de un equipo hospitalario especializado.

Envase clínico y
Uso Hospitalario
Envase clínico

Medicamento de
Especial Control
Médico
Medicamento de
Diagnóstico
Hospitalario
Ejplo.:
Roacutan R

... D.H.

Productos de parafarmacia • En la denominación de Parafarmacia se incluyen a aquellos productos que no son
medicamentos, efectos y accesorios ni dietoterápicos o productos para la nutrición enteral y se consumen, aplican o utilizan por el ser humano y/o sobre el cuerpo y se ponen a disposición de los usuarios, de conformidad y con arreglo a lo que se establece en las Reglamentaciones Técnico–Sanitarias específicas para cada tipo de productos que existen en el mercado, así como en la normativa de carácter general aplicable a los mismos, esto es, Ley General de Consumidores y Usuarios y reguladora de la promoción y publicidad vigente.
• Los productos incluidos en esta denominación son los siguientes:
– Alimentos.
– Cosméticos y productos incluidos en el R.D. 1599/97.
– Productos Sanitarios y de diagnóstico “in vitro”.
– Productos de puericultura: Chupetes, mordedores, tetinas, cadenitas, biberones.
– Desinfectantes y biocidas para la higiene humana, plaguicidas: acaricidas, repelentes y atrayentes para la higiene humana.
– Productos de cuidado personal: cepillos dentales, instrumentos de manicura, etc.

MEDICAMENTOS VETERINARIOS Competencia:
Ministerio de Agricultura, Alimentación y Medio Ambiente.
y
Ministerio de Sanidad , Servicios Sociales e Igualdad,
sin perjuicio de las competencias de las Comunidades Autónomas.
Reglamentación: R.D. 109/1995, de 27 de enero, sobre medicamentos veterinarios.
Regula la evaluación, autorización y registro de los medicamentos veterinarios.
Orden 1 de agosto de 2000, por el que se actualiza el anexo 1 del R.D. 109/1995, de 27 de
enero.
R.D. 1470/2001, de 27 de diciembre, por el que se modifica el R.D. 109/1995,
de 27 de enero, sobre medicamentos veterinarios (B.O.E. nº 311, de 28 de diciembre).
Ley 8/2003, de 24 de abril de sanidad animal.
Ley 29/2006, de 26 de julio ----> Excluye a los medicamentos veterinarios de los
medicamentos especiales y le concede un capítulo (el III, del Título II) amplio e independiente.
R.D. 1246/2008, de 18 de julio. por el que se regula el procedimiento de autorización, registro
y farmacovigilancia de los medicamentos veterinarios fabricados industrialmente.

…MEDICAMENTOS VETERINARIOS
Son medicamentos veterinarios reconocidos:
-Los medicamentos de uso veterinario y las premezclas para piensos medicamentosos-.
Los medicamentos veterinarios
toda sustancia o combinación de sustancias que se presente como poseedora de propiedades
curativas o preventivas con respecto a las enfermedades animales, o que pueda administrarse al
animal con el fin de restablecer, corregir o modificar sus funciones fisiológicas ejerciendo una
acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico clínico
veterinario.
La premezclas para piensos medicamentosos:
todo medicamento veterinario preparado de antemano con vistas a la fabricación ulterior de
piensos medicamentosos.
Piensos medicamentosos:
toda mezcla de premezcla medicamentosa y de piensos preparada previamente a su
comercialización como tal pienso medicamentoso y destinada a ser administrada a los animales
sin transformación, en razón de las propiedades curativas, preventivas o de otras propiedades del
medicamento mencionadas en la definición de medicamento veterinario.

…MEDICAMENTOS VETERINARIOS.
Estudios y ensayos:
Se tendrán en cuenta:
- Repercusiones sobre las personas que los manejan (productos para la mezcla con piensos).
- Repercusiones sobre el medio ambiente.
- Tiempo de espera adecuado para eliminar los riesgos a las personas derivados de los residuos
metabolitos, cuando se administren a animales destinados a consumo humano.
- Tratándose de productos biológicos y de las vacunas en particular, las repercusiones epizoóticas.
Dispensación de medicamentos veterinarios: La dispensación al público de los medicamentos veterinarios se realizarán exclusivamente por:
Las Oficinas de Farmacia legalmente establecidas, que además serán las únicas autorizadas para
la elaboración y dispensación de fórmulas magistrales y preparados oficinales.
Las entidades o agrupaciones ganaderas autorizadas que cuenten con servicio farmacéutico
responsable de la custodia, conservación y dispensación de estos medicamentos para el uso
exclusivo de sus miembros.
Los establecimientos comerciales detallistas autorizados, siempre que cuenten con un servicio
farmacéutico responsable de la custodia, conservación y dispensación de estos medicamentos.

PRESCRIPCIÓN DE MEDICAMENTOS
VETERINARIOS
• La receta veterinaria será válida en todo el territorio nacional, y se editará en la
lengua oficial del Estado y en las respectivas lenguas cooficiales en las CC.AA. que
dispongan de ellas.

Ejemplos de medicamentos veterinarios.
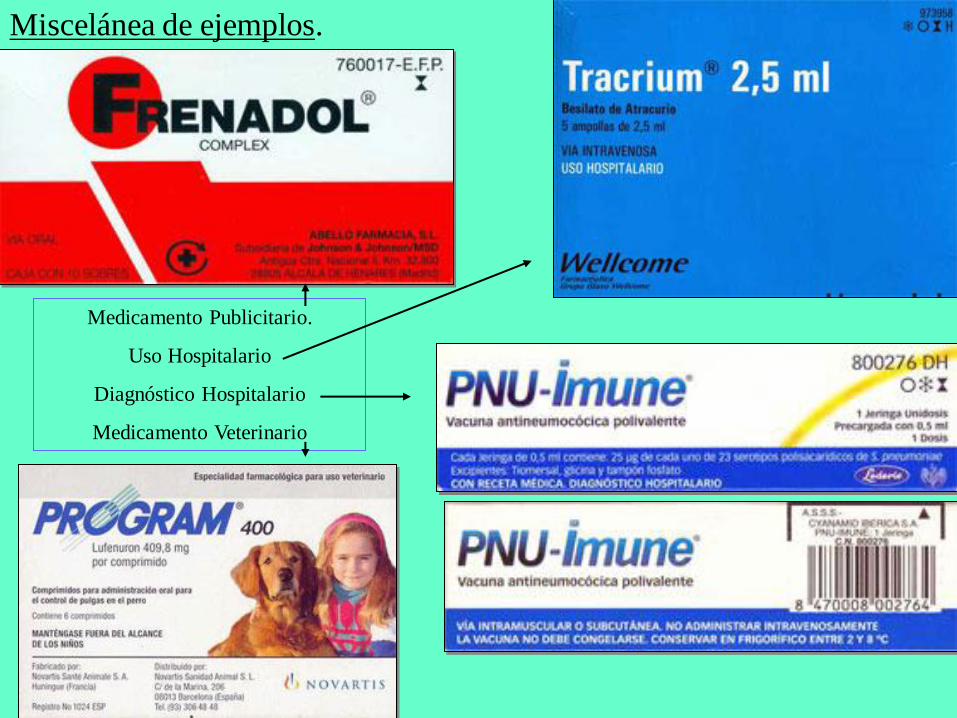
Miscelánea de ejemplos.
Medicamento Publicitario.
Uso Hospitalario
Diagnóstico Hospitalario
Medicamento Veterinario

Más ejemplos…