Embed Size (px)
Citation preview

Aust. J. Chem., 1992, 45, 71-97
The Structure and Function of Estrogens. XI* Synthesis of (&)-7(8+ 1 1a)abeu-Estradiol and its 9,ll-Didehydro Derivative*
David J. C o l l i n ~ , ~ Gary D. FallonA and Colin E, SkeneAIB
A Department of Chemistry, Monash University, Clayton, Vic. 3168. Present address: Division of Chemicals and Polymers, CSlRO,
Clayton, Vic. 3168.
Abstract
Two approaches to the synthesis of (~-7(8+1lor)abeo-estra-l,3,5(10)-triene-3,17~-diol (2a) from (rt)-l~-t-butoxy-7a~-methy1-2,3,3aor,6,7,7a-hexahydro-lH-inden-5(4H)-one (1 1) were stud- ied. A pathway involving 6-alkylation of (11) with 2-(3'-methoxypheny1)ethyl halides or sulfonate esters was unsuccessful, but conjugate addition of 3-methoxybenzyl nu- cleophiles with (~)-l~-t-butoxy-7a~-methyl-6-methylene-2,3,3a,6,7,7a-hexahydro-lH-inden- 5(4H)-one (181, prepared from ( l l ) , led to (rt)-l~-t-buto~y-60c-[2~-(3~~-methoxyphenyl)ethy1]- 7aB-methyl-2,3,3aol,6,7,7a-hexahydro-lH-inden-5(4H-one (10a). Acid-catalysed cyclization of (10a) afforded (+.)-17~-t-butoxy-3-methoxy-7(8-.ll)abeo-estra-l,3,5(10),9(ll)-tetraene (29) which upon lithium/ammonia reduction in the presence of diphenylmethane gave (*I-178- t-butoxy-3-methoxy-7(84Icr)abeoestra,1,3,5(10)-triene (31). Deprotection of (31) and (29) afforded (+)-7(8-.1la)abeo-estra-l,3,5(10)-triene-3,17~-diol (2a) and (&)-7(8-1l)abeo-estra- 1,3,5(10),9(11)-tetraene-3,178-diol (32), respectively.
Alternatively, reaction of (~)-l~-t-butoxy-7a~-methyl-6-methylene-2,3,3aor,6,7,7a-hexahy- dro-lH-inden-S(4H)-one (18) with 3-methoxybenzyl phenyl sulfoxide (23a) gave (lRS,3'SR,2RS,- 3a1SR,7a~SR)-31-t-butoxy-2-(3~~-methoxyphenyl)-3a-methyl-2,3/,3a~,4~,7/,7a~-hexahydrospiro- [~yclopropane-l,5/-[5H]inden]-6~(1~H)-one (26), reductive cleavage of which with lithium/ammo- nia afforded (10a).
The relative stereochemistries of (31) and of the spiro cyclopropyl ketone intermediate (26) were established unambiguously by X-ray crystallography.
Introduction
7(8-1la)abeo-Estradiol (2a) is of particular interest as a ring fusion analogue (annulation isomer) of estradiol (1) because, with 11/3(H) stereochemistry, the separation and relative spatial disposition of the two hydroxy groups in (2a) are virtually the same as in (1) (see Scheme 1). Compound (2a) was first synthesized by an American group who reported112 that it showed about -& of the estrogenic activity of estrone. In rationalizing this the authors commented that the edge profiles of these molecules might play a critical role in their interaction with the estrogen receptor(s). This may be so, but other factors which might be considered in explaining the lower activity of (2a)
* Part X, Aust. J. Chem., 1988, 41, 745. t Dedicated to Arthur J. Birch in recognition of his distinguished contributions to chemical science, and with thanks for stimulating years at the University of Sydney, 1953-1955 (D.J.C.).
' Pitt, C. G., and Handy, R. W., Tetrahedron, 1971, 27, 527. Wani, M. C., Rector, D. H., White, D. H., Pitt, C. G., and Kimmel, G. L., J. Med. Chem., 1977,
20, 547.
Manuscript received 24 December 1990 0004-9425/92/Ol0071$05.00

D. J. Collins, G. D. Fallon and C. E. Skene
include (i) possible consequences of in vivo quinone methide formation from estrogens, and (ii) the fact that only racemic (2a) was synthesized and tested: the lower activity of (2a) might be at least partly due to mutual antagonism of its enantiomers. Regarding the first point, although we have shown3f4 that quinone methide formation is almost certainly not an intrinsic part of estrogen action, some estrogens might nevertheless be prone to incidental in vivo oxidation to the corresponding quinone methides. This might have two important consequences. One is that if, for example, (3-7(8+lla)abeo-estradiol (2a) were converted into the corresponding quinone methide (4a) the latter might be expected to tautomerize by loss of a proton from C8 and/or C 1 1 to afford one or both of the dehydro derivatives (3). Formation of (3) (or its further metabolites) might at least partly explain the low net estrogenic activity of (2a). The other possible consequence of in vivo quinone methide formation pertains to estrogen-promoted carcinogenesis, which might result from attack of a nucleophilic group of DNA on the methide carbon of an estrogen-derived quinone methide.4 It is our eventual aim, therefore, to synthesize analogues of (2a) such as (2b) or (2c) in which methyl groups at C8 and/or C11 might inhibit quinone methide formation in the first place, or, if this still occurs, tautomerization of the quinone methides would be restricted in (4b)
Scheme 1
Collins, D. J., and Stone, G. M., Aust. J. Biol. Sci., 1983, 36, 305. Collins, D. J., Stone, G. M., and Axelson, M., Aust. J. Biol. Sci., 1983, 36, 315.

Structure and Function of Estrogens. XI
or prevented in (4c). Moreover, the presence of flanking methyl groups in the quinone methides (4b) or (4c), if they were generated, might sterically inhibit nucleophilic attack at C9. Our finding5 that the phenol (S), which has a highly hindered benzylic carbon atom and no homobenzylic protons, could not be oxidized to the corresponding quinone methide by the usual methods supports the notion that, for example, compound (2c) should be far less prone to in vivo quinone methide formation than (2a).
Pitt and Handy1 synthesized (2a) by two routes. The first, more protracted route, involved synthesis and ring contraction of a D-homo analogue of (2a). In the second, shorter route, the key step was the Michael addition of the anion of 2-methylcyclopentane-1,3-dione with the conjugated enone (6) (see Scheme 1). Our immediate objective was to devise a new synthesis of the estradiol analogue (Za), amenable both to the introduction of one, two or three of the blocking methyl groups at C 8 and/or C 11, and to the synthesis of enantiomerically pure (2a-c) and related compounds, with steroid-like absolute stereochemistry.
In this paper, we describe a new synthetic route to (2)-7(8+11a)abeo- estradiol (2a). The key intermediate in this synthesis is la-t-butoxy-7aa- methyl-6-methylene-2,3,3aa,6,7,7a-hexahydro-lH-inden-5(4H)-one (18) which unambiguously establishes the required trans-C/D stereochemistry in (2a).
(7a) R = H (7b) R = COOH (7c) R = COOMe
(9) + the 4-epimer
X
Me0
* o& Meo6 ' (10a) R = H (11) (12a) X = OH
(lob) R = COOMe (12b) X =Br (12c) X = I (12d) X = OTs (12e) X = OMS
R' R~
(13a) (13b) %Me3 H BU' BU' M e O O C , & , - MeOOClllll rw (13c) H H R'O (13d) H TiC13 i k (14)
Scheme 2
Collins, D. J., and Jacobs, H. A,, Aust. J. Chem., 1986, 39, 2095.

D. J. Collins, G. D. Fallon and C. E. Skene
Because the hydrindanone (1 1), the precursor of (l8), is available enantiomerically pure,6 this route will be adaptable to the synthesis of enantiomerically pure (2a-c).
Results and Discussion
Attempted Synthesis of (10a) by Alkylation of (Ill-Scheme 2
The original aim was to effect alkylation of the trans-hydrindanone (11) at C6 with the 2-(3'-methoxypheny1)ethyl halides (12b) or (12c) or with the corresponding sulfonate esters (12d) or (12e) to give the key intermediate (10a) (Scheme 2). Several attempts to alkylate the 1,l-ethylenedioxy analogue of (1 1) (directly, or as its trimethylsilyl en01 ether) with 2-(3'-methoxypheny1)ethyl bromide (12b) or the corresponding tosylate (12d) failed.' Attention was then turned to the preparation and alkylation of the previously unknown enolic /3-keto ester (13a). Carboxylation of the t-butoxy enone (7a)8 as described by Micheli et aL6 yielded the corresponding keto acid (7b) which upon catalytic hydrogenation over 10% palladium/barium sulfate (cf. Hajos and parrishg) and thermal decarboxylation gave the trans-fused hydrindanone (1 1). Treatment of compound (11) with sodium hydride in tetrahydrofuran, then with dimethyl carbonate at reflux afforded 59% of the pure crystalline B-keto ester (13a) which was shown by its i.r. and lH n.m.r. data to exist completely in the en01 form. Attachment of the methoxycarbonyl group at C6 rather than C4 was expected on the basis of previous experience with substitution in rrans-fused hydrindan- 5-ones.9 Unambiguous proof of this was provided by direct comparison of (13a) with the authentic 4-regioisomer (8b).9 This was prepared by hydrogenation of the unsaturated ester (7c), obtained by diazomethane esterification of (7b). Prior to attempted alkylation of (13a) to give the desired 6-substituted intermediate (lob), 6-methylation of (13a) was effected by treatment with sodium methoxide/methyl iodide in benzene. This afforded 63% yield of a single diastereomer which is tentatively assigned the 6B-methyl stereochemistry depicted in (14). This is expected to result from pseudo-axial attack of methyl iodide at C6 on the enolate anion. Also, the formation of the 6/3-methyl derivative should be favoured on steric grounds: the 1,3-diaxial interaction of a 6/3-methoxycarbonyl group with the 7aB-methyl group would be larger than the corresponding 1,3-dimethyl interaction. Base-catalysed methylation of the regioisomeric methyl 5-oxohydrindan-4-carboxylate derivative (8b) gave a mixture of the 4-methyl epimers in the ratio of about 4 : 3. The major, crystalline epimer, which was fully characterized, is tentatively assigned the stereochemistry shown in (9) on the grounds that the chemical shift (6 0.93) of its angular methyl group is slightly upfield of that (6 0.99) for its epimer in which the methoxycarbonyl group is 1,3-diaxially related to the angular methyl group. Compound (9) and its epimer may be useful in due course in the synthesis of compound (2c).
Micheli, R. A., Hajos, Z. G., Cohen, N., Parrish, D. R., Portland, L. A., Sciamanna, W., Scott, M. A., and Wehrli, P. A., J. Org. Chem., 1975, 40, 675.
Collins, D. J., and Fitzgerald, A. D., unpublished data. Hajos, Z. G., Micheli, R. A., Parrish, D. R., and Oliveto, E. P., J. Org. Chem., 1967, 32, 3008. Hajos, Z. G., and Parrish, D. R., J. Org. Chem., 1973, 38, 3239.

Structure and Function of Estrogens. XI
Although 6-methylation of (13a) was readily achieved, attempts at base- catalysed alkylation of (13a) with the 2-(3'-methoxypheny1)ethyl halides (12b) or (12c) under a variety of conditions with different bases and solvents failed, giving starting materials, and in some cases 3-methoxystyrene, formed by B-elimination from (12b) or (12c). With the intention of avoiding this base-catalysed elimination problem, the ester (13a) was converted into the corresponding trimethylsilyl en01 ether (13b) in order to attempt Lewis-acid- catalysed alkylation. Although it has been shown by Reetz and coworkers that ketene silyl acetals1° and silyl en01 ethers1l only undergo Lewis-acid-catalysed alkylation with tertiary or benzylic, SN~-reactive halides and acetates, it was considered possible that a B-phenylethyl system, although primary, might react through involvement of the corresponding phenonium ion. In the event, treatment of the trimethylsilyl ether of (13b) with 2-(3'-methoxypheny1)ethyl p-toluenesulfonate (12d) in dichloromethane in the presence of titanium tetrachloride at -80" for 1 h, then at room temperature, gave only starting materials, together with 55% of the 1B-hydroxy B-keto ester (13c) (enolic). Cleavage of the t-butyl ether group presumably proceeded by elimination of methylpropene to give the titanium complex (13d) which was hydrolysed on workup.
Synthesis of (10a) via the 6-Methylene Ketone (18)-Schemes 3-5
When attempts to effect the direct alkylation of the ketone (11) to give (loa), or of (13a) to give (lob), were unsuccessful, attention was turned to the preparation of the 6-methylene ketone (18) which was expected to yield (10a) by the 1,4-addition of appropriate nucleophiles (Scheme 5).
Reaction of the ketone (11) with dimethylamine hydrochloride and paraformaldehyde in ethanol at reflux for 6 . 5 h gave 46% of the Man- nich base (IS), which afforded the methiodide (16); 32% of the starting material (11) was recovered (Scheme 3). With prolonged reaction times the major product isolated was the Diels-Alder dimer (20) of the 6-methylene ketone (18) formed by elimination from the Mannich base (15). The 'H n.m.r. spectrum of the latter showed it to be a single diastereomer which is tentatively assigned the 6a-equatorial stereochemistry shown in (15). In an attempt to prepare the 6-methylene ketone (18), an ethanolic solution of the Mannich base methodide (16) was stirred at room temperature for 1 h with solid potassium carbonate (cf. Kulkarni and Agarwal12), but workup gave mainly the spiro dimer (20). When this reaction was repeated at a temperature between -10 and -20°, rapid and careful workup gave a product which showed 1~ n.m.r. signals at 6 5.15 and 5-95 , consistent with expectations for the exocyclic methylene protons in (18). Upon storage overnight at 0°, this product had largely undergone dimerization to give (20). To enable direct comparison of their behaviour, the known regioisomeric 4-methylene ketone (17) was prepared from the acid (8a) as described by Micheli et aL6 At room temperature the 4-methylene ketone (17) was not as prone to dimerization as its 6-regioisomer (18), but when
lo Reetz, M. T., and Schwellrus, K. , Tetrahedron Lett., 1978, 1455. Reetz, M. T., Hiittenhain, S., and Hubner, F., Synth. Commun., 1981, 1 1 , 217.
l2 Kulkarni, Y. D., and Agarwal, V. K. , J. Indian Chem. Soc., 1982, 59 , 380.

D. J. Collins, G. D. Fallon and C. E. Skene
heated in ethanol it afforded the dimer (19).6 The dimer (19) had different physical properties from the dimer (20) which was obtained from the Mannich base (15), providing unambiguous proof that (IS), and (18), are 6-substituted hydrindanones. The tentative assignment of the relative stereochemistry of the spiro carbon atom in (20) is based on considerations used for the analogous assignment in the dimer (19).6
(8a) R = COOH (11) R = H
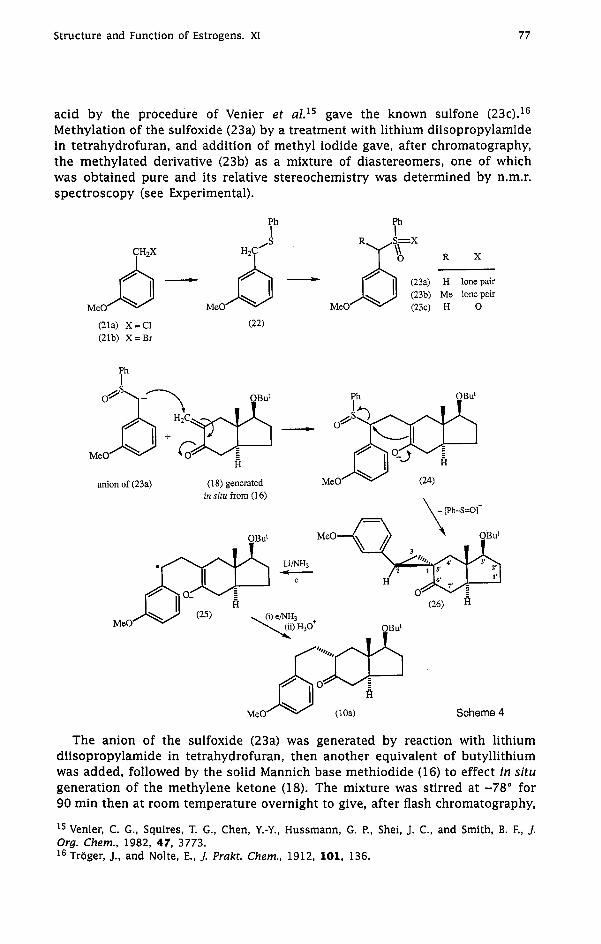
Due to the propensity of the 6-methylene ketone (18) to dimerize at room temperature, its generation and use in situ from the Mannich base methiodide (16) was planned. The first nucleophile to be used in reactions with (18) thus generated was the anion derived from 3-methoxybenzyl phenyl sulfoxide (23a), which was prepared as shown in Scheme 4. Treatment of a benzene solution of 3-methoxybenzyl bromide (21b) and benzenethiol with aqueous sodium hydroxide containing tetrabutylammonium bromide (cf. Herriott13) gave 87% of the sulfide (22). Oxidation of this with iodobenzene diacetate in acetic acid containing concentrated sulfuric acid (cf. castrillon14) afforded the sulfoxide (23a). Further oxidation of (23a) with trifluoroperoxyacetic
l 3 Herriott, A. W., Synthesis, 1975, 447. l4 Castrillon, J., Rev. Latinoam. Quim., 1982, 13, 102 (Chem. Abstr., 1983, 99, 52586j).
Structure and Function of Estrogens. XI
acid by the procedure of Venier et al.15 gave the known sulfone (23c).16 Methylation of the sulfoxide (23a) by a treatment with lithium diisopropylamide in tetrahydrofuran, and addition of methyl iodide gave, after chromatography, the methylated derivative (23b) as a mixture of diastereomers, one of which was obtained pure and its relative stereochemistry was determined by n.m.r. spectroscopy (see Experimental).
H lone pair Me lone pair H 0
Me0 R
(18) generated Me0 anion of (23a) in situ from (16)
Me0 Scheme 4
The anion of the sulfoxide (23a) was generated by reaction with lithium diisopropylamide in tetrahydrofuran, then another equivalent of butyllithium was added, followed by the solid Mannich base methiodide (16) to effect in situ generation of the methylene ketone (18). The mixture was stirred at -78" for 90 min then at room temperature overnight to give, after flash chromatography,
l5 Venier, C. G., Squires, T. G., Chen, Y.-Y., Hussmann, G. P., Shei, J. C., and Smith, B. F., J. Org. Chem., 1982, 47, 3773. l6 Trdger, J., and Nolte, E., J. Prakt. Chem., 1912, 101, 136.

D. J. Collins, G. D. Fallon and C. E. Skene
34% of the cyclopropyl ketone (26). The 'H n.m.r. signal for the angular methyl group in (26) was shifted upfield at 0 .45 ppm [cf. 1 a09 ppm for the corresponding methyl group in (IS)]. The 13C n.m.r. spectrum of (26) showed signals for two upfield methine carbon atoms at 39.9 and 42.2 ppm, and a signal for the spiro quaternary carbon atom at 34.1 ppm, consistent with structure (26). The i.r. spectrum showed carbonyl absorption at 1675 cm-l for the arylcyclopropyl carbonyl system. The relative stereochemistry depicted in structure (26) was revealed by X-ray crystallography (see below). Formation of a cyclopropane ring by further reaction of an intermediate enolate such as (24) with a leaving group on the y-carbon atom has ample precedent." In a similar base-catalysed reaction of the Mannich base methiodide (16) [= (18)] with the sulfone (23c), the yield of the cyclopropyl ketone (26) obtained after chromatography was only 21%.
Scheme 5
l7 March, J., 'Advanced Organic Chemistry' 3rd Edn (Wiley-Interscience: New York 1985).

Structure and Function of Estrogens. XI
It was expected that treatment of the arylcyclopropyl ketone (26) with lithium/ammonia should result in reductive cleavage via the anion radical (25) to give compound (10a). Indeed this was achieved, but the yield of the purified ketone (10a) was only 36%. Cyclization of (10a) to give the 7(8-1l)abeo steroid derivative (29) (Scheme 5) was effected by treatment of (10a) with methanolic hydrochloric acid at c. 0" for 16 h. The lH n.m.r. spectrum of the crude product (86% yield) showed no olefinic proton, indicating virtual absence of the regioisomer of (29) having an 8,9-double bond. The pure compound (29) showed A,,, 278 nm ( E 15300) consistent with the presence of a styrenoid chromophore, and the 13C n.m.r. spectrum showed five quaternary carbon atoms in the olefinic/aromatic region, and six methylene groups.
Because the overall yield of (10a) obtained by the sequence (23a)+[(16) - (18)l -, (26) - (10a) was low (12%), the more direct preparation of (10a) by conjugate addition of 3-methoxybenzylmagnesium chloride onto the 6-methylene ketone (18) was attempted. Although compound (18) might have again been generated in situ from the Mannich base methiodide (16), the decision was made to generate (18) by flash vacuum pyrolytic cleavage of the pure dimer (20). Indeed, flash vacuum pyrolysis of (20) at 500" cleanly gave the 6-methylene ketone (18) as a colourless oil, trapped at liquid-nitrogen temperature. Immediate measurement of the lH n.m.r. spectrum showed the substance to be essentially pure, but it rapidly dimerized at room temperature. For preparative use it was stored for the minimum time at -7B0, and introduced rapidly into a cold (0") tetrahydrofuran solution of 3-methoxybenzylmagnesium chloride, and the mixture was stirred overnight at c. 0". This afforded 46% of the crude ketone (10a) which was treated directly with methanolic hydrochloric acid to give the 7(8-1l)abeo-estradiol derivative (29) in 32% overall yield from the dimer (20) (Scheme 5). Deprotection of compound (29) by treatment with boron tribromide in dichloromethane yielded 7(8-1l)abeo-estra-1,3,5(10),9(11)- tetraene-3,17/3-diol (32), required for bioassay. Initial attempts to reduce the 9,ll-double bond in compound (29) by reaction with lithium/ammonia at -78" gave no reaction. Surprisingly, a similar experiment carried out at the temperature of refluxing ammonia (-33") gave, after flash chromatography, 31% of the phenol (30) in which the styrenoid double bond was retained. A small amount of the expected reduction product (31) was also detected. Formation of the phenol (30) could have resulted from either the homolytic or heterolytic cleavage of the methyl-oxygen bond to give the methyl radical or methyl anion respectively. In some of his earlier work, Birch found1* that anisole and some of its derivatives suffered significant cleavage to give the corresponding phenols, probably by the expulsion of methyl radicals.lg In our experience, 0-methyl cleavage in the reaction of p-methoxystyrene or stilbene derivatives with lithium/ammonia is unusual. For example, reduction of the 9(11)-double bond in 17,17-ethylenedioxy-3-methoxyestra-1,3,5(10),9(ll)-tetraene proceeded very cleanly to give the corresponding estrone derivative which was >95% pure before recrys ta l l i~at ion .~~ It appears that in the case of compound (29) protonation of the derived anion radical (28) by ammonia at C 9 or C 11 is slow compared
l8 Birch, A. J., J. Chem. Soc., 1947, 102. l9 Birch, A. J., personal communication. 20 Collins, D. J., and Sjovall, J., Aust. J. Chem., 1983, 36, 339.

D. J. Collins, G. D. Fallon and C. E. Skene
with expulsion of a methyl radical as shown in Scheme 5 to give the mesomeric anion (27a,b). It was expected that in the presence of a better proton donor than ammonia the reduction of (29) to (31) should proceed in competition with Me-0 cleavage. Indeed, reaction of compound (29) with lithium/ammonia at -78" for 5 . 5 h in the presence of 3 equiv. of diphenylmethane gave 67% of the dihydro compound (31) after flash chromatography, but there was still 30% of the 0-demethylated compound (30) produced. The 1~ and 13c n.m.r. spectra of (31) showed that it was a single stereoisomer, and X-ray crystallography established unambiguously that it had 9a(H),llB(H) stereochemistry as shown in structure (3 1). Sequential deprotection of (3 1) was accomplished by refluxing it with 4 M hydrochloric acid in dioxan to give compound (33), which was not purified, but heated with pyridine hydrochloride at 205" under nitrogen for 40 min (cf. Sheehan et to give racemic 7(8-1la)abeo-estradiol (2a) in 85% overall yield. The melting point and lH n.m.r. data for compound (2a) were in agreement with the reported data.2
Preliminary bioassay22 of racemic (2a) was consistent with previous rep0rtsl9~ on the estrogenic activity of this compound. Full details of the activity of (2a) and (32) will be published in due course when the enantiomerically pure compounds, and the corresponding methylated derivatives (2b) and (2c) are available for direct comparison.
Stereochemistry
It was shown by X-ray crystallography that the spiro cyclopropyl ketone (26) has the relative stereochemistry depicted in Fig. 1; the methylene group of the spiro cyclopropane ring attached to C(6) [C 5' on formula (26)l of the hydrindanone moiety is trans-related to the angular methyl group at C(7a) [C 3a' on formula (26)l.
From examination of molecular models it was considered that metal/ammonia reduction of the 9(11)-double bond in compound (29) should give compound (3 1) with 9a,ll/3-stereochemistry in preference to the 9/3,lloc-trans stereoisomer, or to the two diastereomers with a B/C cis ring fusion, but it was necessary to establish this unambiguously. Indeed, X-ray crystallography confirmed the stereochemistry of compound (31), as shown in Fig. 2.
Data processing was carried out in the usual ~ a y , ~ ~ - ~ ~ and full details are given in the Experimental section.
Experimental
Organic Chemistry
Melting points and boiling points are uncorrected. Bulb-to-bulb distillations were carried out with a Buchi GKR-50 tube oven, and only oven temperatures are given. Microanalyses were performed by Amdel, Australian Microanalytical Service, Melbourne, and by the Analytical
21 Sheehan, J. C., Erman, W. F., and Cruickshank, P. A., J. Am. Chem. Soc., 1957, 79, 147. 22 Stone, G. M., unpublished data. 23 Fallon, G. D., and Gatehouse, B. M., J. Solid State Chem., 1980, 34, 193. 24 Ibers, J. A., and Hamilton, W. C., (Eds) 'International Tables for X-ray Crystallography' Vol. 4 (Kynoch Press: Birmingham 1974). 25 Sheldrick, G. M., SHEW(-76 Program for Cystal Structure Determination, Cambridge, England, 1975.

Structure and Function of Estrogens. XI
Fig. 1. Molecular structure of compound (26) with the crystallographer's numbering scheme. Hydrogen atoms have been omitted. Thermal ellipsoids are scaled to 50% probability.
Fig. 2. Molecular structure of compound (31) with the crystallographer's numbering scheme. Hydrogen atoms have been included particularly so that the B/C ring junction can be clearly seen. Thermal ellipsoids are scaled to 50% probability, except for the hydrogen atoms which are of arbitrary size.

D. J. Collins, G. D. Fallon and C. E. Skene
Unit of the Research School of Chemistry, Australian National University, Canberra. Infrared spectra were measured with either a Jasco IRA-1 infrared spectrophotometer or a Jasco A-100 infrared spectrophotometer. Ultraviolet spectra were measured with a Hitachi 150-20 spectrophotometer. 'H n.m.r. spectra were recorded at 6OMHz with a Varian EM360A spectrometer, at 200 MHz with a Bruker AC200 spectrometer, or at 300 MHz with a Bruker AM300 instrument. 13c n.m.r. spectra were measured at 50.3 or 75.5 MHz, respectively, with the Bruker instruments, or at 22.6 MHz with a Bruker WH90 spectrometer. Chemical shifts (6) are measured in ppm downfield from SiMe4. 13c n.m.r. signals were determined to be due to methyl, methylene, methine or quaternary carbon atoms by using distortion enhancement by polarization transfer (DEPT), and assignments were made accordingly. Mass spectra and accurate masses were measured at 70 eV with a V.G. Micromass 7070F spectrometer. Only the molecular ion (if observed) and principal ion peaks with intensities >lo% are reported. Flash column chromatography was performed with Merck silica gel 60 (No. 9385, 230-400 mesh) or Florisil (60-100 mesh). Thin-layer chromatography was performed by using Polygram silica gel/UV254 precoated plastic sheets (0.25 mm), with fluorescent indicator UVZ54. Light petroleum refers to the fraction of b.p. 65-70'.
Flash vacuum pyrolysis.-The substrate was sublimed from the closed end of a long unpacked silica tube, and the central region (300 by 25 mm i.d.) was heated with an external electric furnace. The temperature was measured with a thermocouple placed on the external wall of the pyrolysis tube. The volatile products were collected on a surface cooled with liquid nitrogen. Pressures were measured with a Dynavac TM8 thermistor gauge attached to the exit elbow about 20 cm from the heated zone.
(a) 2-(3'-Methoxypheny1)ethyl Bromide (12b)
Reduction of 2-(3'-methoxypheny1)acetic acid with lithium aluminium hydride gave 2-(3'- methoxypheny1)ethanol (12a), b.p. 146-5"/18 mm (lit.26 14S0/13 mm). 'H n.m.r. 6 (300 MHz, CDC13) 2.24, br s , OH (exch.); 2.79, t, J 6 . 7 Hz, H 2,2; 3 - 76, apparent s, OMe, overlapping H l , l ; 6.7-6-85, m, H 2/,4',6'; 7 - 20, dd, J 7-8 , 8 - 7 Hz, H 5'. 13c n.m.r. 6 (75 - 5 MHz, CDC13) 39.2, C2; 55.1, OMe; 63.4, C1; 111.7, C4'; 114.7, C2'; 121.3, C6'; 129.4, C5'; 140.2, Cl'; 159.7, C3/.
2-(3'-Methoxypheny1)ethanol (12a) (1.0 g, 6 . 6 mmol) and triphenylphosphine (2.1 g, 8 . 0 mmol) were dissolved in dry dimethylformamide (50 ml), to which was added dropwise a solution of bromine (1.5 g) in dimethylformamide (30 ml) until a yellow colour persisted (cf. Levy and stevensonZ7). The mixture was stirred at 90" for 25 h, then cooled to room temperature, and water (120 ml) was added. Extraction with ether gave an oily solid which was triturated with light petroleum. Filtration and evaporation of the filtrate gave a yellow oil (1.4 g), bulb-to-bulb distillation of which yielded pure 2-(3'-methoxypheny1)ethyl bromide (12b) a s a colourless liquid (1 .21 g, 85%), b.p. 85-90e/3 mm (lit.28 130-142'/9 mm). 'H n.m.r. 6 (200MHz, CDC13), 3-08, t, J 7.6Hz, H2,2; 3.51, t, J 7.6Hz, H1,l ; 3-74, s, OMe; 6-75 , m, H2/,4/,6/; 7.20, apparent t, J 8 - 0 Hz, H 5'. 13c n.mr. 5 (50.3 MHz, CDCl3) 32-8, C1; 39.3, C2; 55.1, OMe; 112.1, C4'; 114.4, C2'; 120.9, C6'; 129.5, C5'; 140.3, C l l ; 159.7, C3'.
A mixture of red phosphorus (2.28 g, 0.074 moll and 2-(3'-methoxypheny1)ethanol (12a) (10.8 g, 0.071 mol) in benzene (200 ml) was gently refluxed under a Soxhlet extractor containing iodine (12.0 g, 0.094 mol). After 3 h the mixture was cooled, poured onto an ice-salt mixture, and the product was isolated with ether to give a blue-green oil which was filtered through silica gel in 4% ether/light petroleum. Distillation gave pure 2-(3'- methoxypheny1)ethyl iodide (12c) (12 - 7 g, 68%), b.p. 130°/0. 1 mm (lit.29 85-9O0/0 .01 mm).
Hunter, J. H., and Hogg, J. A., J. Am. Chem. Soc., 1949, 71, 1922. 27 Levy. D., and Stevenson, R., J. Org. Chem., 1965, 30, 3469. 28 Bachmann, W. E., and Thomas, D. G., J. Am. Chem. Soc., 1942, 64, 94. 29 Crispin, D. J., Vanstone, A. E., and Whitehurst, J. S., J. Chem. Soc. C, 1970, 10.

Structure and Function of Estrogens. XI
'H n.m.r. 6 (300MHz, CDC13) 3.14, t J 7.6Hz, H2,2; 3-33, dt, J 0.8, 7 .6Hz, H1,l; 3.79, s, OMe; 6.70-6.85, m, H 2',4',6/; 7.22, apparent t, J 7 .8 Hz, H 5'. 13c n.m.r. 6 (75.5 MHz, CDCl3) 5.2, C1; 40.4, C2; 55.1, OMe; 112.1, C4'; 114.1, C2'; 120.6, C6'; 129.5, C5'; 142.1, Cl'; 159.7, C3'.
(c) 2-(3'-Methoxypheny1)ethyl p-Toluenesulfonate (12d) (Crispin et al.29)
To a solution of 2-(3'-methoxypheny1)ethanol (12a) (3.0 g, 19.7 mmol) in pyridine (15 ml) was added p-toluenesulfonyl chloride (7.5 g, 39.3 mmol) portionwise during 15 min, then the mixture was kept at 4" for 12 h. The product was isolated with ether, and chromatographed on basic alumina. Elution with light petroleum containing dichloromethane (30%) gave 2-(3'-rnethoxyphenyl)ethyl p-toluenesulfonate (1 2d) as a colourless oil (5 - 1 g, 84%) which slowy crystallized, m.p. 34-35' (lit.29 35"). 'H n.m.r. 6 (300 MHz, CDC13) 2.42, s, Me; 2.91, t, J 7.OHz, H2,2; 3.74, s, OMe; 4.20, t, J 7-OHz, H1,l ; 6-6-6.85, m, H2/,4',6/; 7.15, apparent t, J 7.7 Hz, H 5'; 7-20-8.0, m, 4 aromatic H (tosyl). 13C n.m.r. 6 (75-5 MHz, CDC13) 21.6, Me; 35.3, C2; 55.1, OMe; 70.5, C1; 112.2, C4'; 114.4, C2'; 121.2, C6/; 127.0, 127-7, 2xCH (tosyl); 129.5, C 5'; 129-7, 130.2, 2xCH (tosyl); 132-8, C4" (tosyl); 137-7, C 1'; 144-6, C 1" (tosyl); 159.6, C 3'.
(dl 2-(3'-Methoxypheny1)ethyl Methanesulfonate (12e)
A mixture of 2-(3'-methoxypheny1)ethanol (12a) (2 - 0 g, 13 mmol) and triethylamine (2.6 ml, 1 8 - 7 mmol) in dry dichloromethane (130 ml) was stirred at -10' under nitrogen. Methanesulfonyl chloride (1.05 ml, 16 -2 mmol) was added dropwise, and stirring was continued at -10" for I h; then the mixture was poured onto ice, and the product was extracted with dichloromethane. The extract was washed with ice-cold water (x2), 4% aqueous oxalic acid (x2), 2% sodium bicarbonate solution (x2) and 10% sodium chloride solution (x2), then dried (NazS04) and evaporated at <30° to give 2-(3'-methoxypheny1)ethyl methanesulfonate (12e) as a pale yellow oil (2.95 g, 97.5%). 'H n.m.r. 6 (300 MHz, CDC13) 2.85, s, OS02Me; 3.02, t, J 6 - 9 Hz, H2,2; 3.75, s, OMe; 4.40, t, J 6 . 9 Hz, H1,l ; 6.77-6-83, m, H2',4/,6'; 7-23, apparent t, J 7.7Hz, H5'. 13c n.m.r. 6 (75.5 MHz, CDC13) 35-6, C2; 37-2, OS02Me; 55.2, OMe; 70.2, C1; 112.3, C4/; 114.7, C2'; 121-2, C6/; 129.7, C5'; 137.8, C 1'; 159-8, C3'. Mass spectrum: m/z 230 (M, 23%), 135 (20), 134 (loo), 121 (33), 91 (18), 79 (10).
(e) (d-1~-t-Butoxy-7a~-methyl-2,3,3aa,6,7,7a-hexahydro-1 H-inden-5(4H)-one (1 1)
Carboxylation of I~-t-butoxy-7a~-methyl-2,3,7,7a-tetrahydro-lH-inden-5(6H)-one (7a) in dimethylformamide with magnesium methyl carbonate as described by Micheli et and crystallization of the product from acetone yielded (+)-l~-t-butoxy-7a~-methy1-5-0~0- 2,3,5,6,7,7a-hexahydro-IH-indene-4-carboxylic acid (7b), m.p. 157" (dec.) [lit.9 159.5' (dec.)]. 'H n.m.r. 6 (200 MHz, CDC13) 1 .11, apparent s, Me and CMe3; 1 .75-2.2, m, H 2,2,7,7; 2.6-2.9, m, H3,3; 3-1-3.3, m, H6,6; 3-71, dd, J 7-2 , 10.3Hz, H1; 13.10, br s, OH. 13c n.m.r. 6 (50.3MHz, CDC13) 16.2, Me; 28.6, CMe3; 29.9, 31.4, C3,7; 31.8, C2; 33-5, C6; 48-3 , C7a; 73.4, OCMe3; 78.7, C1; 120-3, C4; 164.2, C3a; 196.3, COOH; 202.9, C5.
The above acid (6.0 g, 0.225 mol) was dissolved in methanol (150 ml), and hydrogenated over 10% palladium on barium sulfate (0-75 g) at room temperature and atmospheric pressure. After I h, removal of the catalyst and evaporation in vacuum at c. 40' gave the saturated acid (8a) as a brown oil. This was then heated under vacuum at 90' for 30 min to effect decarboxylation. Bulb-to-bulb distillation of the residue gave pure (+)-l~-t-butoxy-7a~-methyl-2,3,3aa,6,7,7a-hexahydro-lH-inden-5(4H)-one (11) as a colourless oil (4.8 g, 95%), b.p. 15O0/0.35 mm [Micheli et aL6 reported m.p. 39.5-40- 5" for the (+)-enantiornet-]. v,, (film) 1710 cm-l. 'H n.m.r. 6 (300 MHz, CDC13) 0.98, s, Me; 1.15, s, CMe3; 1.35-2.45,m,H3aand5xCHz; 3 - 4 7 , d d , J 7 - 6 , 8 - 6 H z , H l . 13~n.m.r .6 (50 .3MHz, CDC13) 10.2, Me; 25.8, C3; 28.6, CMe3; 31.8, C7; 35-2, C2; 37.3, C6; 42.0, C7a; 42.8, C4; 44-5, C3a; 72-4, OCMe3; 79-3, C l ; 211.6, C5.

D. J. Collins, G. D. Fallon and C. E. Skene
(f) Methyl (+)-l~-t-Butoxy-5-hydroxy-7a~-methyl-2,3,3aa,4,7,7a-hexahydro-l H-indene- 6-carboxylate* (13a) (Cf. Caselli et aL30)
Sodium hydride (80% in paraffin) (1 - 6 g , 0.053 moll was washed with light petroleum, then suspended in dry tetrahydrofuran (50 rnl) with stirring under nitrogen. Dimethyl carbonate (6.3 ml, 0.075 mol) was added, and the mixture was heated to reflux. (6-1P-t- Butoxy-7a~-methyl-2,3,3aa,6,7,7a-hexahydro-lH-inden-5(4H)-one (1 1) (5.5 g, 0.024 mol) in tetrahydrofuran (30 ml) was then added dropwise, and refluxing was continued overnight. The chilled mixture was acidified to pH 6 with glacial acetic acid; water (400 ml) was added, and the product was extracted with chloroform. Evaporation of the washed, dried (Na2S04) extract gave a viscous yellow oil which solidified. Recrystallization from methanol afforded methyl (~-l~-t-butoxy-5-hydroxy7a~-methyl-2,3,3aa4,7,7a-hexahydro-l H-indene-6- carboxylate (13a) (4.1 g, 59%), m.p. 85-86" (Found: C, 68-0; H, 9.6. C16H2604 requires C, 68.1; H, 9.3%). Vmax (Nujol) 1650s, 1600% 1460m, 1440% 1380m, 1360m, 1320% 1305% 1265s, 1200s, 1150m, 1110m, 1090w, 1055m, 1040w, 1030m, 1010w, 940w, 905w, 870w, 835m, 780w cm-I. 'H n.m.r. S (300 MHz, CDC13) 0.75, s, Me; 1 .17, s, CMe3; 1 - 3-2.4, m, H3a and 4xCH2; 3-53, t, J 8.6Hz, H1; 3.80, s, OMe; 12.31, s, enolic OH. 13c n.m.r. 6 (75.5 MHz, CDC13) 10.6, Me; 25-6, 31.4, 32.0, 35.0, 4xCH2; 28.8, CMe3; 39.8, C3a; 41.5, C7a; 51-4, OMe; 72.4, OCMe3; 80.0, C1; 96-8 , C6; 172.1, 173.6, C5 and COOMe. Mass spectrum: m/z 282 (M, 2%), 226 (12), 225 (20), 194 (13), 193 (32), 176 (24), 166 (lo), 150 (lo), 137 (18), 109 (lo), 81 (12), 57 (loo), 55 (15).
(g) Methyl (~-1~-t-Butoxy-6~,7a~-dimethyl-5-oxo-2,3,3aa,4,5,6,7,7a-octahydro-IH-indene- 6-carboxylate (1 4)
To a supension of sodium methoxide (0.12 g, 2 .2 mmol) in sodium-dried benzene (20 ml) under dry nitrogen was added methyl (+)-1~-t-butoxy-5-hydroxy-7a~methyl-2,3,3aa,4,7,7a- hexahydro-1H-indene-6-carboxylate (13a) (0.5 g, 1 . 7 mmol), and stirring was continued for 30 min. Methyl iodide (2.5 g, 18 mmol) in benzene (20 ml) was added dropwise during 15 min; the mixture was stirred at room temperature for 2 h, then refluxed overnight. The excess of methyl iodide was evaporated, water (30 ml) was added, and the mixture was acidified with glacial acetic acid (1 ml). The product was extracted with benzene, and evaporation of the washed (5% sodium chloride solution), dried (MgS04) extract gave a colourless oil (0.49 g) which solidified. Recrystallization from light petroleum afforded methyl (9-1B-t- butoxy-6B, 7a~-dimethyl-5-oxo-2,3,3aa,4,5,6,7,7a-octahydro-l H-indene-6-carboxylate (14) as a colourless solid (0.33 g, 63%), m.p. 108-110" (Found: M+*, 296.200-cO.003. C17H2804 requires M+', 296.199). v,,, (Nujol) 1720s, 1460m, 1435w, 1390w, 1385w, 1315w, 1295w, 1285w, 126511-1, 1195s, 1135n-1, 1115m, 1100m, 1065x11, 985w, glow, 840w cm-l. 'H n.m.r. 6 (300MHz, CDC13) 0.85, s, 7a-Me; 1-14, s , CMe3; 1.29, s , 6-Me; 1.4-2.75, m, H3a and 4xCH2; 3.41, t, J 7.9 Hz, H 1; 3-72, s, OMe. 13c n.m.r. 6 (75.5 MHz, CDC13) 11.1, 7a-Me; 23.6, 6-Me; 25-7, 31.5, 42.0, 48.4, 4xCH2; 28.7, CMe3; 42.7, C7a; 45.6, C3a; 52.5, OMe; 53-7, C6; 72.6, OCMe3; 80.0, C 1; 175.1, COOMe; 208.4, C5. Mass spectrum: m/z 297 (M+1, 0.2%), 296 (M, 0.2), 240 (12), 183 (25), 180 ( l l ) , 163 (16), 162 (lo), 140 (271, 135 (151, 130 (12), 122 (12), 107 (141, 101 (ll), 93 (13), 83 (121, 81 (12), 69 (161, 57 (loo), 55 (14).
(h) Attempted Alkylation of Methyl (+)-1~-t-Butoxy-5-hydroxy-7a~methyl-2,3,3aa,4,7,7a- hexahydro-1 H-indene-6-carboxylate (13a) with 2-(3'-Methoxypheny1)ethyl Iodide (12c)
Sodium hydride (80% in paraffin oil) (0-066 g, 2 .2 mmol) was washed with light petroleum, then suspended in dry tetrahydrofuran (50ml) under nitrogen. Methyl (d-l~-t-butoxy-5-hydroxy-7a~methyl-2,3,3aa,4,7,7a-hexahydro-lH-indene-6-carboxylate (1 3a) (0.50 g, 1 . 8 mmol) was added, and the mixture was stirred for 15 min, then heated at
* For indexing purposes, the names of the esters (13) and (14) require the 'low- est' locant for the methoxycarbonyl group, e.g. methyl 3-t-butoxy-6-hydroxy-3a-methyl- 2,3,3a,4,7,7a-hexahydro-lH-indene-5-carboxylate (13a), and methyl 3-t-butoxy-3a,5-dimethyl- 6-oxooctahydro-lH-indene-5-carboxylate (14).
30 Caselli, A. S., Collins, D. J., and Stone, G. M., Aust. J. Chem., 1982, 35, 799.

Structure and Function of Estrogens. XI
reflux for 1 h. A solution of 2-(3'-methoxypheny1)ethyl iodide (12c) (0.58 g, 2 .2 mmol) in tetrahydrofuran (50 ml) was added dropwise during 20 min, then the mixture was refluxed for 40 h. Thin-layer chromatography indicted that the 2-(3'-methoxypheny1)ethyl iodide (12c) was being consumed, but still showed the presence of (13a); additional 2-(3'-methoxypheny1)ethyl iodide (12c) (0.50 g, 1 . 9 mmol) in tetrahydrofuran (30 ml) was added, and refluxing was continued for 24 h. The cooled mixture was poured into water, then neutralized with dilute hydrochloric acid, and extracted with ether. Evaporation of the washed, dried (NazS04) extract gave a yellow oil which partly solidified. Flash chromatography (silica gel, 4% diethyl ether/light petroleum) afforded 3-methoxystyrene (0.12 g). 'H n.m.r. 6 (300 MHz, CDC13) 3.82, s, OMe; 5-25, dd, J 0 .7 , 10-9Hz, olefinic H; 5.75, dd, J 0.6, 17.6Hz, olefinic H; 6 69, dd, J 10.8, 17.6 Hz, olefinic H; 6.75-7 - 3, m, H 2,4,5,6. 13c n.m.r. 6 (75 - 5 MHz, CDC13) 55.2, OMe; 111.5, 113.4 and 118.9, C2,4,6; 114.1, =CH2; 129.4, C5; 136.7, CH=CH2; 139.0, C1; 159.8, C3.
Further elution with light petroleum containing ether (4%) gave 2-(3'-methoxypheny1)ethyl iodide (12c) (0.28 g), identified by its i.r. spectrum, and, subsequently, the unreacted enolic 8-keto ester (1 3a) (0.38 g), identified by its 'H n.m.r. spectrum. None of the desired alkylation product (lob) was isolated.
Several other experiments involving the use of either the bromide (12b) or the iodide (12c), and various base/solvent combinations including sodium methoxide/methanol, sodium methoxide/dimethyl sulfoxide, sodium methoxide/benzene, sodium hydride/benzene, thallous ethoxide/ether, sodium amide/ammonia, lithium diisopropylamide/tetrahydrofuran and potassium/xylene, were similarly unsuccessful.
(i) Attempted Alkylation of the Trimethylsilyl Ether (13b), Prepared from Methyl (+.I-1/3- t-Butoxy-5-hydroxy-7a/3-methyl-2,3,3aa,4,7,7a-hexahydro-lH-indene-6-carboxylate (13a), with 2-(3'-Methoxypheny1)ethyl p-Toluenesulfonate (12d)
To a solution of diisopropylamine (0.197 g, 1 .95 mmol) in tetrahydrofuran (10 ml), stirred at 0" under nitrogen, was added 1 . 4 M butyllithium (1.4 ml, 2 . O mmol), and the mixture was stirred at 0" for 15 min, then chilled to -80'. A solution of methyl (+)-18-t-butoxy-5-hydroxy- 7a~-methyl-2,3,3aa,4,7,7a-hexahydro-1H-indene-6-carboxylate (13a) (0.50 g, 1 - 8 mmol) in tetrahydrofuran (5 ml) was added, stirring was continued for 30 min, then chlorotrimethylsilane (2.5 ml, 1.95 mmol) was added. The mixture was stirred at -80" for a further 15 min, then allowed to warm up to room temperature, and the solvent was removed in vacuum at 30-40'. Dichloromethane (20 ml) was added to the residue, the mixture was chilled to -80°, and treated with a solution of 2-(3'-methoxypheny1)ethyl p-toluenesulfonate (12d) (1 -73 g, 5 . 7 mmol) in dichloromethane (5 ml). Titanium tetrachloride (0.21 ml. 1 - 9 mmol) was added, and the mixture was stirred at -80" for 1 h. It was allowed to warm up to room temperature, stirred for 30 min, then poured into 2% sodium bicarbonate solution (50 ml). Extraction with dichloromethane afforded a brown oil (2.06 g). Flash chromatography (silica gel, 4% diethyl ether/light petroleum) gave 3-methoxystyrene (0.20 g, 26%). Further elution with light petroleum containing ether (4%) gave some of the enolic 8-keto ester (13a) (0.20 g, 40%), followed by unreacted tosylate (12d) (1 .14 g, 66%). Subsequent elution with diethyl ether gave methyl (-r-)-l/3,5-dihydroxy-7a~-methyl-2,3,3aa,4,7,7a-hexahydro-I H-indene-6-carboxylate (13c) (0.22 g, 55%), m.p. 90-92' (Found: C, 63.8; H, 8 -4 . C12H1804 requires C, 63.7; H, 8.0%). vmax (film) 3400s(br) OH, 1710m, 1650s, 1610s, 1440s, 1380m, 1350s, 1300s, 1270s, 1210s, 1150m, 1115m, 1090m, 1050m, 1010w, 915w, 830m, 735m cm-'. 'H n.m.r. 6 (300 MHz, CDC13) 0.76, s, Me; 1.25-1.45, m, H 3a and 4xCHz; 3-75, s, OMe; 3.82, t, J 8 7 Hz, H 1; 12.26, s, enolic OH (exch.). 13c n.m.r. 6 (75 - 5 MHz, CDC13) 10.0, Me; 25 0, 30.8, 31-8, 34.7, C2,3,4,7; 39.9, C3a; 41.8, C7a; 51-4, OMe; 81.2, C1; 96.5, C6; 171.8, 173.4, C5 and COOMe. Mass spectrum: m/z 226 (M, loo%), 195 (27), 194 (631, 176 (21), 167 (241, 166 (18), 161 (141, 151 (17), 150 (171, 149 (14), 148 (20), 140 ( l l ) , 139 (13), 138 (15), 137 (281, 135 (65), 128 (14), 125 (lo), 123 (231, 122 (32), 121 (23), 120 (29), 113 (13), 111 (151, 110 (22), 109 (28), 108 (29), 107 (271, 106 (lo), 105 (14), 97 (32), 96 (29), 95 (24), 94 (331, 93 (441, 91 (18), 83 (24), 82 (17), 81 (50), 79 (43), 78 (39), 77 (231, 69 (27), 68 (18), 67 (251, 65 (121, 59 (20), 57 (161, 55 (87), 54 (12), 53 (32).
Use of the mesylate (12e) in a similar experiment also failed to give any of the desired product (lob).

D. J. Collins, G. D. Fallon and C. E. Skene
( j ) Methyl (+)-lfi-t-Butoxy-7afi-methyl-5-oxo-2,3,3aa,4,5,6,7,7a-octahydro-l H-indene-la- carboxylate (8b)
Methylation of lfi-t-butoxy-7afi-methyl-5-oxo-2,3,5,6,7,7a-hexahydro-lH-indene-4-carboxy- lic acid (7b) (5.0 g, 0.019 mol) with ethereal diazomethane, and trituration of the crude product with light petroleum gave a solid (4.7 g, 89%). Flash chromatography (silica gel, 4% ether/light petroleum), and recrystallization from methanol afforded pure methyl (+)-l~-t-butoxy-5-hydroxy-7a~methyl-2,3,7,7a-tetrahydro-lH-indene-4-carboxylate (7c) (enol) as a colourless solid, m.p. 85-90' (enol form) (lit.9 76-5-77", keto form). 'H n.m.r. 6 (200 MHz, CDC13) 0.94, s, Me; 1.18, s, CMe3; 1.3-2.75, m, 3xCH2; 3.73, t, J 8.0 Hz, H 1; 3.83, s, OMe; 5-72, s , H3; 10.16, s, 5-OH. 13c n.m.r. 6 (50.3 CDC13) 15.6, Me; 27.6, C7; 28.7, CMe3; 31-9, 39.1, C2,6; 46.1, C7a; 51 - 5, OMe; 72.5, OCMe3; 80.2, C 1; 98.5, C4; 117.3, C3; 138.4, C3a; 173.4, 175.4, C5 and COOMe.
Hydrogenation of (7c) (4 - 7 g, 0.017 moll in methanol (250 ml) over 10% palladium/barium sulfate (1.6 g) at room temperature and atmospheric pressure, and recrystallization of the product from light petroleum gave methyl (r)-lfi-t-butoxy-7afi-methyl-5-oxo-2,3,3aa,4,5,6,7,7a- octahydro-1H-indene-4a-carboxylate (8b) (4.2 g, 88%), m.p. 100-104' (lit.9 104-109'). 'H n.m.r.6(200MHz,CDC13)1.02,s,Me; l e14 ,s ,CMe3; 1-3-1*75,m,H3,3,7,7; 1.9-2-25,m, H2,2,3a; 2.35-2-60, m, H6,6; 3.36, d, J13.5Hz, H4; 3.74, s, OMe. 13Cn.m.r.6(50.3MHz, CDC13) 11.1, Me; 24.5, C3; 28.8, CMe3; 31.7, C7; 35.2, C2; 37.5, C6; 42.1, C7a; 46.8, C3a; 52.1, OMe; 59.0, C4; 72.8, 0CMe3; 79.1, C1; 170.0, COOMe; 206.1, C5.
(k) Methylation of Methyl (d-1~-t-Butoxy-7afi-methyl-5-oxo-2,3,3a(r,4,5,6,7,7a-octahydro-l H- indene-4a-carboxylate (8b)
To a suspension of sodium methoxide (0.35 g, 6.5 mmol) in benzene (50 ml) was added methyl (r)-lfi-t-butoxy-7a~methyl-5-oxo-2,3,3aa,4,5,6,7,7a-octahydro-lH-indene-4a- carboxylate (8b) (1.6 g, 5 7 mmol). The mixture was stirred under nitrogen for 30 min at room temperature, then heated to reflux, and methyl iodide (10 ml, 0.16 mol) was added. The mixture was refluxed overnight then cooled, and washed with 1 M sodium bicarbonate solution (x2) and 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a yellow oil (1.3 g). Flash chromatography (silica gel, 10% ether/light petroleum) gave material (0.5 g), trituration of which with cold methanol afforded a pure crystalline diastereomer, which is tentatively assigned the structure methyl (r)-lfi-t-butoxy-4fi,7afi- dimethyl-5-0x0-2,3,3aa,4,5,6,7,7a-octahydro-1 H-indene-40-carboxylate (9) (0.28 g, 17%), m.p. 62-63' (Found: C, 68.7; H, 9.6. C17H2804 requires C, 68.9; H, 9.5%). v,,, (Nujol) 1735, 1710 cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 0.93, s, 7a-Me; 1.13, s, CMe3; 1.28, s, 4-Me; 1.35-2-45,m,4xCH2; 2 .95 ,d t , J 7 . 9 , 18.2Hz,H3a; 3.37, t, J 8 . 2 H z , H l ; 3.70,s ,OMe. 13c n.m.r. 6 (50.3 MHz, CDCl3) 10 - 5, 7a-Me; 20.4, 4-Me; 20.9, C 3; 28.7, CMe3; 31.2, C 7; 36.78, 36-83, C2, 6; 42.2, C7a; 52.0, OMe; 54.7, C3a; 57.0, C4; 72.6, OCMe3; 79.6, C 1; 174-0, COOMe; 209.0, C 5. Mass spectrum: m/z 296 (M, 0-25%), 240 (16), 222 ( l l ) , 180 (lo), 135 (19 , 130 (201, 110 (19), 99 ( l l ) , 93 (111, 57 (loo), 55 (18).
Further elution with light petroleum containing ether (20%) afforded the minor diastereomer as an oil (0.20 g, 12%). lH n.m.r. 6 (60 MHz, CDC13) 0.99, s, 7a-Me; 1 10, s, CMe3; 1 .35, s, 4-Me; 1 .5-3.6, m, 4xCH2 and H 3a; 3.70, s, OMe. 13c n.m.r. 6 (50.3 MHz, CDC13) 13.0, 7a-Me; 16.4, 4-Me; 20.6, C3; 28.7, CMe3; 31.1, C7; 34.6, 3 4 8 , C2,6; 41-5, C7a; 48-7, C3a; 5 2 5 , OMe; 59-9, C4; 72.7, OCMe3; 79.7, C1; 173.4, COOMe; 210.8, C5. Mass spectrum: m/z 296 (M, 1.2%), 240 ( l l ) , 135 (13), 130 (19, 110 (lo), 93 (12), 88 ( l l ) , 81 (111, 67 (101, 59 ( l l ) , 57 (1001, 55 (23). This was not obtained analytically pure. Material (0.52 g) subsequently eluted from the columm was a mixture, probably including some starting material.
The 'H n.m.r. spectrum of the crude product showed that the ratio of the crystalline to non-crystalline epimers was approximately 4 : 3.
(1) ~d-lfi-t-Butoxy-7afi-methyl-4-methylene-2,3,3aa,6,7,7a-hexahydro-l H-inden-S(4H)-one (1 7) (Micheli et aL6)
A solution of lfi-t-butoxy-7afi-methyl-5-oxo-2,3,5,6,7,7a-hexahydro-lH-indene-4-carboxy1ic acid (7b) (2.0 g, 7 - 5 mmol) in methanol (20 ml) was hydrogenated over 10% palladium/barium

Structure and Function of Estrogens. XI
sulfate (0.25 g) at 0". Filtration and evaporation of the solvent at 4 5 " gave the keto acid (8a) as a brown semisolid foam. To this was added 40% aqueous formaldehyde solution (1 - 4 ml), piperidine (32 mg) and dimethyl sulfoxide (5 ml), and the mixture was stirred a t room temperature for 25 min. It was then poured onto a slurry of ice and saturated sodium chloride solution (30 ml), and extracted with ether (x2). The extract was washed with 1 M sodium bicarbonate solution (x2) and 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a pale yellow oil (0.78 g). Flash chromatography (silica gel, 20% ether/light petroleum) yielded (d-lB-t-butoxy-7a~-methyl-4-methylene-2,3,3aa,6,7,7a- hexahydro-1H-inden-5(4H)-one (17) as an oil which slowly crystallized (0.44 g, 25%) [Micheli et aL6 quote 56-60.5' for the (+)-enantiomer]. Vmax (film) 1695s, 1620m, 1460m, 1390s, 1360s, 1250m, 1195s, 1095s, 1055m, 1030m, 1005w, 930m, 910111, 875w, 825w, 755w cm-I. lH n.m.r. 6 (200 MHz, CDC13) 0.78, s, Me; 1.16, s , CMe3; 1-6-2.55, m, 4xCH2 and H3a; 3 -61 , dd, J 7.0, 8 .8 Hz, H 1; 5.00, m, olefinic H; 5.92, m, olefinic H. 13c n.m.r. 6 (50.3 MHz, CDC13) 11.2, Me; 22.7, C3; 28.7, CMe3; 31.7, C7; 33.8, 35.9, C2,6; 43-1, C7a; 48.9, C3a; 72-5, OCMe3; 80.0, C1; 118-0, C=CH2; 147.3, C4; 201.9, C5.
(m) The Dimer (19) of (k)-l~-t-Butoxy-7a~-methy1-4-methylene-2,3,3aa,6,7,7a-hexahydro-l H- inden-5(4 H)-one (1 7)
The 4-methylene ketone (17) (O.27g, 1 1 mmol) was refluxed in ethanol (50 ml) for 2 h then stirred at room temperature for 48 h. Evaporation of the solvent, and recrystallization of the residue from ethanol gave (liSR,3SR,3a1RS,6aSR,7SR,7a1SR,9aRS)-l1,7-di-t-butoxy-6a,7ai- dimethyl-1 ,2,2i,3~,3ai,6,6i,6a,7,71,7ai,8,9,9a-tetradecahydr0spir03H,5H-cyclopenta[f] [llben- zopyran -3,4i-[4H]inden-51(1'H)-one (19), m.p. 158-165" [lit.6 148-154" for the (-)-enantiomer]. vmax (Nujol) 1720m, 1680m, 1450s, 1365m, 1195m, logos, 1050m, 940w, 905w, 725w, cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 0.74, s, Me; 1.12, s, Me and 2xCMe3; 1.35-2.80, m, 22H; 3.35-3.55, m, H11,7. 13c n.m.r. 6 (50.3 MHz, CDC13) 11.4, 12.5, 6a- and 7ai-Me; 18.8, 19.7, 23.2, 24.3, 25.5, C1,2,3',5,9; 28.7, 2xCMe3; 31 .3, 32.2, 33.2, 35-1, 36.5, C 2',6,6/,7/,8; 42 - 9, 44.6, C 6a,7ai; 43.0, C 3ai; 54.3, C 9a; 72.2, 72.6, 2xOCMe3; 79.3, 79.7, C 11,7; 84.1, C3,4'; 102.4, C9b; 145.6, C4a; 210.1, CSi. Mass spectrum: m/z 472 (M 7%), 180 (281, 162 (13), 137 (151, 57 (loo), 55 (16).
(n) (~-l~-t-Butoxy-6a-dimethylaminomethyl-7a-methyl-2,3,3aa,6,7,7a-hexahydro-l H-inden- 5(4H)-one (1 5) and its Methiodide (1 6)
A mixture of (+)-l~-t-butoxy-7a~methyl-2,3,3aa,6,7,7a-hexahydro-lH-inden-5(4H)-one (1 1) (0.50 g, 2.2 mmol), dimethylamine hydrochloride (0.20 g, 2 . 5 mmol) and paraformaldehyde (0.14 g, 4 . 7 mmol) was refluxed in ethanol (20 mi) for 6 - 5 h. The solvent was removed in vacuum; the residue was suspended in 1 M hydrochloric acid, and washed with ether (x2). The chilled aqueous layer was basified and extracted with ether. The ether extract was washed with 10% sodium chloride solution, then dried (MgS04) and evaporated to give (k)-lfi-t-butoxy-6a- dimethylaminomethyl-7a~methyl-2,3,3aa,6,7,7a-hexahydro-lH-inden-5(4H)-one (1 5) (0.29 g, 46%) as a dark, viscous oil which slowly solidified, vmax (film) 1700s, 1465m, 1385m, 1365m, 1290w, 1265m, 1250m, 1195ms, 1150m, 1120m, 1055s, 1005w, 995w, 905w, 850w cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 1-09, s, 7a-Me; 1.15, s, CMe3; 1.35-2 -65, m, H3a,6, HCHNMe2 and 4xCH2; 2.19, s, NMe2: 2.76, dd, J 5.1, 12.4 Hz, HCHNMe2; 3.46, dd, J 7-2 , 8 - 4 Hz, H1. 13C n.m.r. 6 (50-3 MHz, CDC13) 11-1 , 7a-Me; 25.7, 31.8, 41-6, 43-1, C2,3,4,7; 28-7, CMe3; 42.7, C7a; 44.1, C3a; 45.8, C6; 45.9, NMe2; 59.2, CH2NMe2; 72.4, OCMe3; 79.1, C 1; 21 1 .7 , C 5. Mass spectrum: m/z 281 (M, 0.2%), 136 (17), 58 (100). The original ether washings were combined, washed with 10% sodium chloride solution, then dried (MgS04) and evaporated to give recovered ketone (11) (0- 16 g, 32%), which was distilled, then recycled.
To the above Mannich base (15) (2.58 g, 9 - 2 mmol) was added methyl iodide (20 ml), and the mixture was stirred under nitrogen at room temperature for 2 5 h. The viscous, yellow product was chilled in ice, and excess of methyl iodide was removed in vacuum. The residual yellow solid was triturated with ether to give the methiodide (16) as a yellow powder (3 82 g, 98%). 13c n.m.r. 6 [SO - 3 MHz, (Ds)pyridine] 12.3, 7a-Me; 25.6, 31.9, 43.3, 43.8, C2,3,4,7; 28-7, CMe3; 41 -7, 45.9, C3a,6; 43-5, C7a; 53-8, +NMe3; 66.2, CH2N+Me3;

D. J. Collins, G. D. Fallon and C. E. Skene
72.6, OCMe3; 78.8, C 1; 208.8, C5. This material was stored at 0°, and used without further purification.
(0) The Dimer (20) of (~)-1~-t-Butoxy-7a~methyl-6-methylene-2,3,3a,6,7,7a-hexahydro-l H- inden-5(4 H)-one (1 8)
(i) Directly from the ketone ( I 1) by prolonged heating in the Mannich reaction.-Amixture of Q- 1~-t-butoxy-7a~-methyl-2,3,3aa,6,7,7a-hexahydro-lH-inden-5(4H)-one (1 1) (2.0 g, 8 .9 mmol), dimethylamine hydrochloride (0.80 g, 9 .7 mmol) and paraformaldehyde (0.56 g, 18.7 mmol) was refluxed in ethanol (100 ml) for 70 h. The mixture was filtered while hot to remove unreacted paraformaldehyde. The cooled filtrate gave some of the crude dimer (20) which was collected. The filtrate was evaporated; the residue was suspended in 1 M hydrochloric acid, and extracted with ether. The extract was washed with 10% sodium chloride solution, then dried (MgS04) and evaporated to give a yellow solid. Flash chromatography of the combined crude dimer (silica gel, 5% diethyl ether/light petroleum), and recrystallization of the eluate from light petroleum gave (2RS,31SR,3a1SR,5aSR,6SR,7a1SR,8aSR)-31,6-di-t-butoxy-3a1,5a-dimethyl- Z1, 3, 3I,3a1,4,4l, 5a, 6,7,7l, 7a1, 8,8a,9-tetradecahydrospiro2H, 5H-cyclopentafg lfl]benzopyran- 2,5/-[5H]inden-6I(l IH)-one (20) (0.65 g, 31%), m.p. 167-168" (Found: C, 75.7; H, 10.6%; M+', 472 - 356+0.002. C30H4804 requires C, 76.2; H, 10.2%; Mf', 472 355). vm, (Nujol) 1730s, 1680m, 1465s, 1410w, 1390m, 1380m, 1365s, 1310w, 1260m, 1225m, 1200s, 1170m, 1140m, 1120s, 1105s, 1065m, 1035m, 1005m, 970m, 950w, 9OOm, 870w, 725w cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 0.72, s , 3a1-Me or 5a-Me; 0.79, s, 5a-Me or 3a1-Me; 1.12, s, 3'-OCMe3 or 6-OCMe3; 1.14, s, 6-OCMe3 or 3'-0CMe3; 1-3-2.7, m, 22H; 3.50, m, H3/,6. 13c n.m.r. 6 (50.3MHz, CDC13) 10.7, 13.5, 3a1-Me and 5a-Me; 23.2, 25.7, 26-3, 29.0, 31.0, 31-4, 31.9, 40.5, 41.1, 46.4, 10xCH2; 28.70, 28-72, 2xCMes; 39.7, 41.0, C7a1,8a; 41.7, 41.9, C3a1,5a; 72.3, 72.4, 2xCMe3; 78.5, C2 (1 C5I); 80.1, 81.1, C3/,6; 104.2, C4a; 143.3, C9a; 210.5, C6I. Mass spectrum: m/z 472 (M, 48%), 416 (14), 360 (12), 237 (15), 180 (17), 136 (231, 123 (12), 121 (13), 93 ( l l ) , 81 (lo), 57 (loo), 55 (12).
(ii) From the Mannich base methiodide (1 6).-A mixture of the Mannich base (15) (0.62 g, 2.2 mmol) and methyl iodide (1 - 4 ml, 22 mmol) was dissolved in ethanol (10 ml), and stirred at room temperature for 3 - 5 h. Powdered potassium carbonate (0.46 g, 3 - 3 mmol) was then added, and a white precipitate slowly formed. Sirring was continued for a further 40 min, then water (40 ml) was added, and the mixture was extracted with dichloromethane. The extract was washed with 10% sodium chloride solution, then dried (NazS04) and evaporated to give a yellow oil (0 ~ 4 7 g) which slowly solidified. Trituration with cold methanol, then recrystallization from methanol gave the dimer (20) (0.22 g, 42%), m.p. 165-167', which was identical with the material described above.
(iii) From the methiodide (16) a t lower temperatures.-When this reaction was carried out between -10 and -20' for 1 h, and care was taken to keep the product at or below 0" during workup, the 'H n.m.r. spectrum showed narrow triplets at 6 5 - 15 and 5-95 for the olefinic protons of the 6-methylene ketone (18). Integration showed this to constitute about 70% of the crude product, but upon storage overnight at 0' this underwent dimerization to give (20).
(p) 3-Methoxybenzyl Chloride (21a)
Reduction of 3-methoxybenzaldehyde with lithium aluminium hydride in ether gave 3-methoxybenzyl alcohol as a colourless oil, b.p. 140°/10 mm (lit.31 86-87'/0.001 mm). vmax (film) 3380 cm-I. 'H n.m.r. 6 (200 MHz, CDC13) 3.66, s, OMe; 3.95, br s, OH (exch.); 4.46, s, CH2; 6.7-6.85, m, H2,4,6; 7-17, apparent t, J8.OHz, H5. 13C n.m.r. 6(50-3MHz, CDC13) 55-1, OMe; 64.5, CH2; 111.6, 112.2, C2,4; 119.1, C6; 129.4, C5; 142.9, C1; 159.6, C3.
A mixture of 3-methoxybenzyl alcohol (21 .0 g, 0 - 15 mol), pyridine (4 - 0 ml, 0.05 mol) and freshly distilled thionyl chloride (20 ml, 0.27 moll was refluxed in benzene (170 ml) for 2 h, then cooled, and the solvent was removed in vacuum. Extraction with ether, and distillation of the product gave 3-methoxybenzyl chloride (21a) (20-2g , 85%), b.p. 117-12O0/25 mm (lit.31 58"/O. 1 mm). 'H n.m.r. 6 (200 MHz, CDC13) 3 -71, s , OMe; 4.47, s , CH2; 6.75-6 -95,
31 Grice, R., and Owen, L. N., J. Chem. Soc., 1963, 1947.

Structure and Function of Estrogens. XI
m, H2,4,6; 7.21, apparent t, J 8.1 Hz, H 5. 13c n.m.r. 6 (50.3 MHz, CDC13) 46.2, CH2; 55.1, OMe; 114.0, C2,4; 120.8, C6; 1 2 9 7 , C5; 138.9, C1; 159.8, C3.
(q) 3-Methoxybenzyl Bromide (21b) (Cf. Bachmann and horna as^^)
A solution of 3-methoxybenzyl alcohol (24 - 0 g, 0 - 17 mol) in benzene (60 ml) was stirred at 0' under nitrogen, and a solution of phosphorus tribromide (6 ml, 0 -064 mol) in benzene (60 ml) was added dropwise. Stirring was continued for 1 h at O o , then the mixture was heated in an oil bath at 60-75" for a further 3 h. The cooled mixture was poured onto ice; the organic layer was separated, was washed with 1 M sodium hydroxide, 1 M hydrochloric acid, and water (x2), then dried (MgS04) and evaporated to give a pale yellow oil (34.6 g). Distillation gave pure 3-methoxybenzyl bromide (21b) (30.8 g, 85%), b.p. 120-126'/15 mm (lit.32 116'/8 mm). 'H n.m.r. 6 (200 MHz, CDCl3) 3-74, s, OMe; 4.40, s, CH2; 6 -80, ddd, J 8, 2, l H z , H4; 6.90, m, H2,6; 7.21, apparent t, J 7.8Hz, H5. 13c n.m.r. 6 (50.3MHz, CDC13) 33.5, CH2; 55.2, OMe; 114.1, 114.4, C2,4; 121.2, C6; 129.7, C5; 139.1, C1; 159.6, C3.
(r) 3-Methoxybenzyl Phenyl Sulfide (22) (Cf. ~ e r r i o t l ~ )
To a solution of 3-methoxybenzyl bromide (21b) (0.91 g, 4 . 5 mmol) in benzene (4 ml) was added benzenethiol (0.50 ml, 4 . 9 mmol), a solution of sodium hydroxide (0.30 g, 7 5 mmol) in water (3 ml), and tetrabutylammonium bromide (20 mg, 0.06 mmol), and the mixture was stirred vigorously for 30 min. The organic layer was separated, and the aqueous layer was again extracted with benzene. Evaporation of the washed, dried (MgS04) extract gave a colourless oil (0.92 g). Bulb-to-bulb distillation afforded pure 3-methoxybenzyl phenyl sulfide (22) (0.91 g, 87%), b.p. 120-14O0/0 -02 mm (Found: C, 73.0; H, 6.4. C14H140S requires C, 73.0; H, 6.1%). v,,, (film) 1600s, 1595s, 1480s, 1465m, 1450m, 1435s, 1315m, 1290m, 1265s, 1230w, 1190w, 1150m, 1090m, 1045s, 1015m, 990w, 935w, glow, 870w, 780m, 73% 710m, 690s cm-'. 'H n.m.r. 6 [ZOO MHz, (D6)benzenel 3.30, s, OMe; 3-83, s, CH2; 6.55-7-3, m, 9 aromatic H. 13c n.m.r. 6 [50.3 MHz, (D6)benzene] 38.7, CH2; 54.6, OMe; 113.0, C4; 114.5, C2; 121.3, C6; 126.2, C4'; 129.0, 129.6, C2', 3',5,5',6/; 137.1, 139.2, C1,ll; 160.1, C3. Mass spectrum: m/z 230 (M, 23%), 121 (loo), 91 (19).
(s) 3-Methoxybenzyl Phenyl Sulfoxide (23a) (Cf. Castrillon 14)
A mixture of 3-methoxybenzyl phenyl sulfide (22) (4.0 g, 17 - 4 mmol), acetic acid (80 ml), concentrated sulfuric acid (1 . 7 ml) and iodobenzene diacetate (5.6 g, 17.4 mmol) was stirred at 0' for 10 min, then poured onto ice-water (300 ml). The mixture was extracted with ether ( ~ 2 ) ; the extract was washed with saturated sodium bicarbonate solution (x2), and 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a yellow oil (6.9 g). Flash chromatography (silica gel, 1 :4 ether/light petroleum) gave some starting material, then elution with ether afforded the crude sulfoxide (3.4 g). Recrystallization from ether/light petroleum yielded 3-methoxybenzyl phenyl sulfoxide (23a) as a voluminous white solid ( 2 -9 g, 68%), m.p. 58-59" (Found: C, 68.8; H, 5.6. C14H1402S requires C, 68.3; H, 5.7%). v,,, (film) 1600s, 1585m, 1490s, 1465m, 1445s, 1315w, 1295m, 1265s, 1220w, 1165m, 1155m, 1090m, 1070m, 1045s, 1000w, 940w, 920w, 870w, 790m, 755s, 740m, 695s cm-'. 'H n.m.r. 6 (200 MHz, CDC13) 3.67, s, OMe; 4.00, dd, J 12 -4, 27.0 Hz, CH2; 6.47, d, J 1.8Hz, H2; 6.60, d, J 7 . 4 H z , 6.81, m, H4,6; 7.16,apparent t, J 7 . 9 H z , H5; 7 .41 ,m, 5 aromatic H, P~(so). ' 3 ~ n.m.r. 6 (50.3 MHZ, CDC13) 55.1, OMe; 63.7, CH2; 114.2, 115. 3, C2,4; 122.7, C6; 124.4, C2',6'; 128.8, C3/,S1; 129.4, 130.5, C4',5; 131.1, C1; 142.8, Cl l ; 159.4, C3. Mass spectrum: m/z 246 (M, 1.0%), 121 (loo), 71 (24), 78 (lo), 77 (121, 65 (10).
(t) 1 -(3l-Methoxypheny1)ethyl Phenyl Sulfoxide (23b)
To a solution of diisopropylamine (0 - 32 ml, 2 - 26 mmol) in tetrahydrofuran (20 ml) stirred at 0" under nitrogen was added 1 . 7 M butyllithium (1 - 32 ml, 2 .24 mmol), and the mixture was chilled to -78". A solution of 3-methoxybenzyl phenyl sulfoxide (23a) (0.50 g, 2 . O mmol) in tetrahydrofuran (5 ml) was then added; after 15 min, methyl iodide (0.14 ml, 2 .2 mmol)
32 Woodward, R. B., J. Am. Chem. Soc., 1940, 62 , 1478.

D. J. Collins, G. D. Fallon and C. E. Skene
was added, and the mixture was allowed to warm up to room temperature. After a further 45 min, water (100 ml) was added, and the product was extracted with ether. The ether extract was washed with 10% sodium chloride solution, then dried (MgS04) and evaporated to give an oil (0-49g). Flash chromatography (silica gel, 60% ether/light petroleum) gave (d-1-(3'-methoxypheny1)ethyl phenyl sulfoxide (23b) as a mixture of diastereomers (68% RS,RS, 32% RS,SR) (0.23 g, 44%) which slowly solidified, m.p. 85-95'. vmax (film) 1600s, 1490s, 1460m, 1445s, 1370w, 1325w, 1290m, 1255s, 1170m, 1090m, 1075w, 1050s, 1035s, 1000w, 980w, 890w, 860w, 790m, 760m, 735w, 700s, cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 1.54, d, J 7 - 1 Hz, Me (RS,RS); 1.66, d, J 7 .2 Hz, ~ ' e (RS,SR); 3.66, s, OMe; 3.74, q, J 7 . 1 Hz, CH (RS,SR); 4.01, q, J 7 .1 Hz, CH (RS,RS); 6.45-6-9, m, H 2/,4/,6'; 7.1-7.45, m, H 5' and Ph (SO). 13c n.m.r. 6 (50.3 MHz, CDC13) 12.0, C2 (RS,RS); 13 -9, C 2 (RS,SR); 55 -0, OMe (both diastereomers); 64.2, C 1 (RS,RS); 67- 1, C 1 (RS,SR); 113.86, 113.89, 114.0, C 2/ and C4' (both diastereomers); 120.8, C 6' (RS,SR); 121 -0, C6' (RS,RS); 124.8, C 211,6" (RS,SR); 125 - 1, C2",6" (RS,RS); 128.2, C 3",5" (RS,RS); 128.4, C3",5/' (RS,SR);129.0, 129.4, 130.85, 130-90, C4//,5/ (both diastereomers); 135.3, C 1' (RS,RS); 136.9, C 1' (RS,SR); 140.3, C 1" (RS,RS); 142.0, C 1" (RS,SR);159.2, 159.4, C 3' (both diastereomers).
In another preparation, treatment of the crude product with propan-2-01 gave a solid which upon two recrystallizations from propan-2-01 gave the pure (RS,RS)-diastereomer of (23b), m.p. 108.5-109.5' (Found: C, 69.3; H, 6-3 . C15H1602S requires C, 69.2; H, 6.2%). lH n.m.r. 6 (200 MHz, CDC13) 1 - 55, d, J 7 - 1 Hz, Me; 3 - 68, s , OMe; 4 02, q, J 7 .2 Hz, CH; 6.47, m, H2'; 6-60, d, J 7.7Hz, 6.80, m, H4',6'; 7.1-7.45, m, H5'and Ph(S0). 13c n.m.r. 6 (50.3MHz, CDCI3) 12.0, Me; 55.1, OMe; 64.3, C1; 114-0, 114.1, C2/,4'; 121.1, C6'; 125.2, C2/',6"; 128.2, C3",5"; 129.1, 130.9, C4/',5'; 135.4, Cl'; 140.4, Cl"; 159.3, C3'. Mass spectrum: m/z 260 (M, 0- I%) , 136 (lo), 135 (loo), 134 (151, 105 (20), 103 (13), 91 (14), 79 (17), 78 (16), 77 (14), 65 (13), 51 (10). The stereochemistry was determined from the I H and 13C n.m.r. chemical shifts by analogy with the work of Kobayashi et
(u) 3-Methoxybenzyl Phenyl Sulfone (23c) (Cf. Venier et a1.15)
To a solution of 3-methoxybenzyl phenyl sulfoxide (23a) (1 . O g, 4 . 1 mmol) in trifluoroacetic acid (5 ml), stirred at O n , was added dropwise trifluoroperoxyacetic acid solution (1-65 ml) which was prepared by mixing 30% hydrogen peroxide solution (8.6 ml) with trifluoroacetic acid to a final volume of 25 ml. The mixture was heated at 30" for 75 min, then the solvent was removed in vacuum. The residue was treated with benzene, washed with 1 M sodium bicarbonate solution (x2) and 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a dark yellow solid. Recrystallization from ethanol with charcoal gave a pale yellow-green solid (0.41 g, 38%). A further recrystallization from ethyl acetate gave pure 3-methoxybenzyl phenyl sulfone (23c), m.p. 107-108" (lit.16 109'). vmax (Nujol) 1600s, 1490m, 1465m, 1445m, 1435m, 1410w, 1380m, 1310s, 1305s, 1270s, 1180s, 1160s, 1135s, 1090m, 1040m, 1005w, 945w, 890m, 800m, 775w, 745m, 690m cm-l. 'H n.m.r. b (200 MHz, CDC13) 3.67, s, OMe; 4-28 , s, CH2; 6.75, m, H2,4,6; 7-15, m, H5; 7.55, m, Ph(S02). 13c n.m.r. 6 (503MHz, CDC13) 55.2, OMe; 62.8, CH2; 114.7, 115.9, C2,4; 123.1, C6; 128.6, 128.9, C2/,3/,5',6/; 129.4, 129.5, 133.7, C1,4/,5; 137-8, C1; 159.5, C3. Mass spectrum: m/z 262 (M, 25%), 122 (301, 121 (loo), 91 (401, 78 (19), 77 (221, 65 (12), 51 (17).
(v) Reaction of I S - t - B u t o x y - 6 a - d i m e t h y l a m i n o m e t h y l - 7 a w 7,7a-hexahydro-1 H- inden-5(4 H)-one Methiodide (1 6) with 3-Methoxybenzyl Phenyl Sulfoxide (23a)
To a stirred solution of diisopropylamine (0 - 32 ml, 2 - 26 mmol) in tetrahydrofuran (20 ml), chilled to 0" under nitrogen, was added a solution of butyllithium in hexane (2 - 3 M,
1 .2 ml). The mixture was stirred at 0" for 15 min then chilled to -78', and a solution of 3-methoxybenzyl phenyl sulfoxide (23a) (0.64 g, 2 .6 mmol) in tetrahydrofuran (5 ml) was added by syringe. The mixture was stirred for 20 min, then a second equivalent of butyllithium solution (2.3 M, 1 . 1 ml) was added, and stirring was continued for a further 20 min. Solid methiodide (16) (1 - 0 g, 2.4 mmol) was added, together with washings of tetrahydrofuran (5 ml). Stirring was continued at -78" for 90 min, then the mixture was
33 Kobayashi, K., Kodama, Y., Nishio, M., Sugawara, T., and Swamura, H., Bull. Chem. Soc. Jpn, 1982, 55, 3560.

Structure and Function of Estrogens. XI
allowed to warm up to room temperature, and stirred overnight. The mixture was added to water (150 ml), and extracted with ether (x3); the extract was washed with 10% sodium chloride solution (~21 , then dried (MgS04) and evaporated to give a semi-solid (0.95 g). Flash chromatography (silica gel, light petroleum containing diethyl ether, 25%) afforded diphenyl disulfide (0.07g), m.p. 55-58" (lit.34 60.5*). Mass spectrum: m/z 219 (M, 98%), 185 (141, 154 (ZO), 110 (121, 109 (loo), 77 (12), 69 (13), 65 (38), 51 (13).
Further elution with the same solvent mixture, and recrystallization of the eluate from ethanol/water afforded (lRS,31SR,2RS,3a1SR,7a1SR)-3-t-butoxy-2-(3"-methoxypheny~-3a~- methyl-21,31,3a1,41,71, 7a1-hexahydro-spiro[cyclopropane-l,5/-[~]inden]-61(l /H)-one (26) (0.29 g, 34961, m.p. 119-120" (Found: C, 77.7; H, 9 -2 . C23H3203 requires C, 77.5; H, 9.1%). vmax (Nujol) 1675% 1600s, 15801-11, 1490m, 1460s, 1375s, 1360s, 1330m, 1290m, 1255s, 1230m, 1200s, 1170m, 1145m, 1125s, 1070s, 1060s, 1000w, 955w, 910m, 885w, 860w, 795w, 785w, 760m, 730m, 700m cm-l. 'H n.m.r. 6 (200 MHz, CDCl3) 0.45, s, Me; 1.00, s , CMe3; 1-05-2.65, m, H2,7a1 and SxCH2; 3.41, dd, J 7.7, 8 . 6 Hz, H3/; 3-78, s, OMe; 6-6-6.85, m, H211,411,611; 7.21, t, J 7.7 Hz, H5". 13c n.m.r. 6 (50.3 MHz, CDC13) 10.3, Me; 20.5, 25.8, 31-3 , 36 -7 ,41 .5 , 5xCH2; 28-5, CMe3; 34.1, C1 (=C6/); 39 .9 ,42 .2 , C2,7a1; 42.4, C3a1; 55.2, OMe; 72.2, OCMe3; 79-6, C3/; 112.5, C4I1; 115.0, C2I1; 121.5, C6I1; 128.7, C5I1; 137.9, C 1"; 159-3, C3I1; 210-7, C6'. Mass spectrum: m/z 356 (M, 80%), 187, ( l l ) , 179 (lo), 161 ( l l ) , 160 (lo), 159 (12), 147 (12), 135 (19), 143 (19), 122 (23), 121 (421, 91 (16), 81 (IS), 57 (loo), 55 (15).
In a similar reaction of the Mannich base methiodide (16) (1.30 g) with the sulfone (23c) (1 - 03 g) the yield of compound (26) obtained after flash chromatography and crystallization was only 8% (0 09 g).
The stereochemistry of compound (26) was determined by X-ray crystallography.
(w) Preparation of (d-1fi-t-Buto~y-6a-[2-(3~~-methoxyphenyl)ethyll-7a~ethyl-2,3,3ao1,6,7,7a- hexahydro-l H-inden-S(4H)-one (1 Oa) by Reduction of the Spiro Ketone (26) with Lithium/Ammonia
To anhydrous ammonia (50ml) were added small pieces of lithium (0.15 g, 22 mmol) at -78" under nitrogen. When all of the lithium had dissolved (approximately 45 min), a solution of the spiro cyclopropyl ketone (26) (0.30g, 0 -84 mmol) in tetrahydrofuran (5 ml) was added to the blue solution, and the mixture was allowed to warm up to -35". Stirring was continued at -35" for 7 h, then 1,2-dichloroethane was added dropwise until the blue colour was discharged. Ammonium chloride was added, and the ammonia was allowed to evaporate. Addition of water, and extraction with ether gave a colourless oil (0.29 g). This was dissolved in methanolic potassium hydroxide solution (2%, 10 ml), and stirred at room temperature for 3 h. The solvent was removed in vacuum, water was added, and the mixture was extracted with ether. Evaporation of the washed, dried (MgS04) extract yielded an oil (0.24 g). Flash chromatography and elution with light petroleum containing ether (10%) afforded (~)-lfi-t-butoxy-60(-[2~-(3~~-methoxyphenyl)ethy1]-7afi-methy1- 2,3,3aa,6,7,7a-hexahydro-1 H-inden-S(4H)-one (10a) as a colourless, viscous oil (0.11 g, 36%) (Found: MC', 358-251&0-003. C ~ 3 H 3 ~ 0 3 requires MC', 358.251). vmax (film) 1710s, 1600m, 1585m, 1490~1, 1460m, 1440w, 1395w, 1365m, 1320w, 1295w, 1260s, 1195s, 1155m, 1120m, 1065s, glow, 895w, 710w cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 0.99, s , Me; 1-15, s, CMe3; 1-2-2.45, m, H3a,6 and 5xCH2; 2.61, t, J 7.8Hz, H2I,2l; 3-44, t, J 7 . 9 Hz, H 1; 3.79, s, OMe; 6.74, m, H211,411,611; 7.19, m, H5I1. 13c n.m.r. 6 (50.3 MHz, CDC13) 11 .2, Me; 25.7, 31.2, 31.8, 33.4, 42.7, 43.1, C11,2/,2,3,4,7; 28.7, CMe3; 42.7, C7a; 44.7, 45-8, C3a,6; 55.1, OMe; 72.5, OCMe3; 79-3, C1; 111.2, C4I1; 114.0, C2I1; 120.8, C6"; 129-3, C5I1; 143-9, C1I1; 159.6, C3I1; 212.6, C5. Mass spectrum: m/z 358 (M, lo%), 224 ( l l ) , 189 (13), 181 (201, 168 (23), 167 (271, 150 (IS), 135 (21), 134 (48), 122 (31), 121 (30), 93 (141, 91 (141, 57 (loo), 55 (15).
Further elution with the same solvent mixture yielded a mixture of the ketone (10a) and starting material (26).
To a solution of (+)-lfi-t-butoxy-6a-[21-(311-methoxyphenyl)ethy1]-7a-methyl-2,3,3ao1,6,7,7a- hexahydro-lH-inden-5(4H)-one (10a) (0.11 g, 0.31 mmol) in methanol (10 ml) at room
34 Rosenmund, K. W., and Harms, H., Chem. Ber., 1920, 53, 2226.

D. J. Collins, G. D. Fallon and C. E. Skene
temperature was added concentrated hydrochloric acid (2-Oml), dropwise with stirring during 2 min. The mixture was stirred for 5.5 h at room temperature, then kept at -1' for 16 h. The precipitate was collected, washed several times with ice-water, then dried under high vacuum to give a colourless solid (90 mg, 86%). Recrystallization from methanol/water afforded (+)-I 78-t-butoxy-3-methoxy-7(84 1)abeo-estra-1,3,5(10),9(11)-tetraene (29), m.p. 109- 110" (Found: C, 81 -4; H, 9.8. C23H3202 requires C, 81 -1; H, 9.5%). Vmax (Nujol) 1605m, 1500s, 1465s, 1390w, 1365m, 1355w, 1310m, 1285111, 1255s, 1235m, 1205m, 1165m, 1115w, 107Sm, 1055w, 1045m, glow, 855m, 825w cm-l. Am, (EtOH) 278nm ( E 15300). 'H n.m.r. 6 (200 MHz, CDC13) 0.70, s, Me; 1.17, s, CMe3; 1-4-2.45, m, H14 and 5xCH2; 2.71, t, J 5 .8Hz, H6,6; 3.56, t, J8 .OHz, H17; 3.78, s, OMe; 6.70, m, H2,4; 7.11, d , J 8 . 6 H z , H l . 13c n.m.r. 6 (50.3 MHz, CDCI3) 10.8, Me; 26-2, 28-6, 28-9, 29.6, 31.2, 44.5, 6xCH2; 28.8, CMe3; 41.0, C14; 41.7, C13; 55.2, OMe; 72.2, OCMe3; 80.6, C17; 110.7, C2; 113.4, C4; 122.6, C1; 125.6, C11; 129.8, 131.3, C9,lO; 136.9, C5; 157.7, C3. Mass spectrum: m/z 340 (M, loo%), 284 (18), 283 (39), 266 (22), 265 (39), 251 ( l l ) , 239 (17), 237 (231, 225 (261, 224 (14), 223 (21), 210 (lo), 209 (151, 187 (14), 185 (12), 171 (23), 165 (131, 147 (16), 81 (17), 57 (99), 56 (12), 55 (13).
(y) Preparation of (+)-l~-t-Butoxy-7a~-methyl-6-methylene-2,3,3aa,6,7,7a-hexahydro-l H-inden- 5(4 H)-one (1 8) by Pyrolysis of its Dimer (20)
The dimer (20) (50 mg, 0.18 mmol) was subjected to flash vacuum pyrolysis by using a sublimation temperature of 165-17O0, a furnace temperature of 500" and a pressure of 0 - 02 mm Hg. The pyrolysate was collected in a liquid-nitrogen trap. When all of the sample had sublimed, the pyrolysate (24 mg) was washed into an n.m.r. tube with (D)chloroform, and the 'H n.m.r. spectrum was measured immediately. The spectrum showed it to be essentially pure (+)-l~-t-butoxy-7a~methyl-6-methylene-2,3,3aa,6,7,7a-hexahydro-lH-inden- 5(4H)-one (18), which was unstable and dimerized to give (20) on being kept at room temperature. 'H n.m.r. 6 (200 MHz, CDC13) 0.82, s , Me; 1.17, s, CMe3; 1.35-2-7, m, H3a and 4xCH2; 3.55, t, J 8 .2 Hz, H 1; 5 - 24, m, olefinic H; 5.99, t, J 2 - 4 Hz, olefinic H.
For preparative use the methylene ketone (18) was kept chilled at -80°, then used as quickly as possible at a low reaction temperature.
(z) Preparation of 17~-t-Butoxy-3-methoxy-7(8+ll)abeo-estra-1,3,5(10),9(11)-tetraene (29) by Reaction of l~-t-Butoxy-7a~-methyl-6-methylene-2,3,3aa,6,7,7a-hexahydro-l H-inden-5(4H)-one (18) with 3-Methoxybenzylmagnesium Chloride (Cf. Cohen et a ~ . ~ ~ )
(i) Generation of the methylene ketone (18) from its dimer (20).-The dimer (20) (1.2 g) was pyrolysed at 500" as described in (y); the methylene ketone (18) was trapped at liquid-nitrogen temperature and kept chilled at -80' until it was used as described below.
(ii) 1,4-Addition of 3-methoxybenzylmagnesium chloride to the methylene ketone (1 8) to give (1 Oa), and cyclization of this to (291.-To magnesium turnings (0.85 g, 0.035 mol) in refluxing tetrahydrofuran (10 ml) under nitrogen was added dropwise a solution of 3methoxybenzyl chloride (21a) (2-73 g, 0.017 mol) in tetrahydrofuran (15 ml); then refluxing was continued for 30 min. The mixture was chilled in ice, tetrahydrofuran (30 ml) was added, followed by addition of cuprous chloride (0.55 g, 5 .6 mmol); then the mixture was stirred at 0" for 80 min. A solution of (+)-l~-t-butoxy-7a~-methyl-6-methylene-2,3,3aa,6,7,7a-hexahydro- 1H-inden-5(4H)-one (18) prepared as above in tetrahydrofuran (20 ml) was then added, and the mixture was stirred overnight. The mixture was chilled (ice bath), saturated ammonium chloride and ice were added, then the mixture was stirred for 20 min and filtered. The filter cake was washed with ether, and the filtrate was also extracted with ether (x3). The combined ether extract was washed with 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a yellow oil. Bulb-to-bulb distillation (oven temperature 17O0/0. 1 mm) gave, after a forerun of 3,3/-dimethoxybibenzyl, the crude 1,4-addition product (10a) as a colourless oil (0.84 g). In an initial run this material was subjected to flash chromatography to give pure (lOa), identified by i.r. and n.m.r. spectroscopy. For preparative use it was
35 Cohen, N., Banner, B. L., Eichel, W. F., Parrish, D. R., Saucy, G., Cassal, J.-M., Meier, W., and Fiirst, A., J. Org. Chem., 1975, 40, 681.

Structure and Function of Estrogens. XI
more efficient to effect cyclization of the crude material. Thus, the crude ketone (10a) (0.84 g) obtained from the distillation was dissolved in methanol (60 ml), and concentrated hydrochloric acid (12 ml) was added dropwise. The mixture was stirred at room temperature for 5 - 5 h, then kept at -1' for 16 h. The precipitate was collected, washed with ice-water and dried under high vacuum to give 17~-t-butoxy-3-methoxy-7(8-Il)abeo-estra-l,3,5(10),9(11)- tetraene (29) (0.56 g, 32%). This was identical ('H and 13C n.m.r.) with the material described above in (x).
To a solution of boron tribromide in dichloromethane (lo%, 1 - 4 ml) stirred under nitrogen at -78" was added a solution of 17~-t-butoxy-3-methoxy-7(8-1l)abeo-estra-l,3,5(10),9(11)- tetraene (29) (0.20 g, 0 - 59 mmol) in dichloromethane (5 ml). The mixture was allowed to come to room temperature, and then stirred overnight. Water (50 ml) was added, the mixture was extracted with ether (x3), and the extract was washed with 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a pink semisolid (0.175 g). This was dissolved in 1 M sodium hydroxide solution, then washed with peroxide-free ether (x2), and acidified with 2 M sulfuric acid. The product was extracted with ether (x3), and the extract was washed with 10% sodium chloride solution (x2), then dried (Mg5o4) and evaporated to give a pale yellow semisolid (0.100 g, 63%). Recrystallization from methanol afforded pure (+)-7(~11)abeo-estra-1,3,5(10),9(11)-tetraene-3,17~-diol (32), m.p. 201-203' (Found: C, 80.1; H, 8-2 . C18H2202 requires C, 80-0; H, 8.2%). Vmax (Nujol) 3240s(br) (OH), 1615m, 1585m, 1500s, 1465s, 1375s, 1315m, 1290m, 1250s, 1185w, 1165w, 1150w, 1120m, 1050m, 1020s, 1005w, 990w, 955w, 920w, 890w, 865w, 820m, 725w cm-l. 'H n.m.r. 6 (200 MHz, CDC13/CD30D) 0.71, s, Me; 1 .5-2-45, m, H14 and 5xCH2; 2-68, t, J 5 .7 Hz, H6,6; 3 981, t, J 8 -4Hz , H17; 6-65 , m, H2,4; 7-05, d, J 8-OHz, H l . 13c n.m.r. 6 (50.3MHz, CDC13) 10.3, Me; 25.8, 28.6, 28.9, 29.9, 30-2 , 44.3, 6xCH2; 41.3, C14; 42.3, C13; 81.6, C17; 112.7, C2; 114.7, C4; 123.0, C1; 125.9, C11; 129.2, 130.6, C9,lO; 137.3, C5; 155.1, C3. Mass spectrum: m/z 270 (M, loo%), 268 (26), 211 (32), 209 (211, 195 (101, 172 (171, 160 (121, 159 (25), 158 (22), 157 (25).
(bb) Reduction of (+.)-I 7/3-t-Butoxy-3-methoxy-7(84 1)abeo-estra-1,3,5(10),9(11)-tetraene (29) with Lithium/Ammonia
To anhydrous ammonia (50 ml) (distilled from sodium) was added lithium metal (0- 10 g) at -78" under nitrogen. When all of the lithium had dissolved (approximately 20 min), a soluton of (d-178-t-butoxy-3-methoxy-7(84 1)abeo-estra-1,3,5(10),9(11)-tetraene (29) (0- 20 g, 0 .59 mmol) in tetrahydrofuran (5 ml) was added, then the mixture was allowed to warm up to -3S0, and stirred at that temperature for 5.5 h. Ammonium chloride was added, and the ammonia was allowed to evaporate under a stream of nitrogen. The residue was treated with water, and extracted with ether (x3). The extract was washed with 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give a colourless oil (0.204 g). Flash chromatography (silica gel, 25% diethyl ether/light petroleum) afforded a colourless solid (0.060 g, 31%) which upon recrystallization from methanol gave (+)-17p-t- butoxy-7kbll)abeo-estra-1,3,5(1 0),9(11)-tetran-3-0 (30), m.p. 167-168' (Found: C, 80 6; H, 9.5. C22H3002 requires C, 80.9; H, 9.3%). vmax (Nujol) 3250s(br), 1635w, 1610m, 1585m, 1540w, 1500s, 1460s, 1375s, 1365s, 1320w, 1285m, 1245s, 1195s, 1160m, 1150m, 1120m, 1070m, 1050m, 1025w, 990w, 955w, 920w, 905w, 890w, 865w, 820m, 755w, 725w cm-l. 'H n.m.r. 6 (200 MHz, CDC13) 0.70, s, Me; 1.18, s, CMe3; 1.45-2.45, m, H 14 and 5xCH2; 2.68, t, J 5-8Hz, H6,6; 3.58, t, J 8-OHz, H17; 4.96, br s, OH (exch.); 6-65, m, H2,4; 7.06, d, J 7 .9 Hz, H l . 13C n.m.r. 6 (50-3 MHz, CDCl3) 10.8, Me; 26.1, 28.6, 28.7, 29.5, 31-1, 44.5, 6xCH2; 28.8, CMe3; 41-0, C14; 41.7, C13; 72.5, OCMe3; 80.7, C17; 112.5, C2; 114.4, C4; 122.8, C1; 125.6, C11; 129.9, 131.2, C9,lO; 137.2, C5; 153.6, C3. Mass spectrum: m/z 326 (M, loo%), 270 (19 , 269 (241, 253 (12), 252 (33), 251 (531, 225 (201, 223 (23), 211 (131, 210 (141, 209 (20), 173 (16), 157 (ll), 133 (18), 97 (131, 57 (481, 55 (11). The mass spectrum of a small amount of less polar material eluted from the column showed a peak at m/z 342, consistent with the presence of compound (31).

D. J. Collins, G. D. Fallon and C. E. Skene
(CC) (*)-I 7/3-t-B~toxy-3-methoxy-7(&lla)abeo-estra-1,3,5(1O)-triene (31)
To anhydrous ammonia (100 ml) were added small pieces of lithium metal (0.20 g) at -78' under nitrogen. When all of the lithium had dissolved, a solution of (d-17~-t-butoxy-3-methoxy- 7(8-1l)abeo-estra-1,3,5(10),9(11)-tetraene (29) (0-40 g, 1 - 2 mmol) in tetrahydrofuran (10 ml) was added, and stirring was continued for 30 min. A solution of diphenylmethane (0- 56 g, 3 . 3 mmol) in tetrahydrofuran (20 ml) was added, and the mixture was stirred at -78" for a further 5 . 5 h. Ammonium chloride was added, then the ammonia was allowed to evaporate under a stream of nitrogen. The residue was treated with water, and extracted with ether (x3). The extract was washed with 10% sodium chloride solution (~21 , then dried (MgS04) and evaporated to give an oily solid. Flash chromatography (silica gel, light petroleum) afforded diphenylmethane (0- 53 g). Elution with light petroleum containing ether (5%) then afforded (d-17~-t-butoxy-3-methoxy-7(8-.11 a)abeo-estra-13,510triene (31) (0 270 g, 67%), which upon recrystallation from methanol had m.p. 112-113' (Found: C, 80-9 ; H, 10-3. C23H3402 requires C, 80.7; H, 10.0%). Vmax (Nujol) 1605m, 1575111, 1495s, 1460s, 1420m, 1375m, 1360m, 1330w, 1300m, 1280111, 1255s, 1245s, 1220~1, 1195m, 1155m, 1125m, 1100m, 1060s, 1030m, 995w, 970w, 905w, 885w, 860w, 805w, 765w, 725w cm-l. Amax (EtOH) 284 nm ( E 2300). 'H n.m.r. 6 (200 MHz, CDC13) 0.75, s, Me; 1.15, s , CMe3; 0.8-2.3, m, H9,11,14 and 5xCH2; 2-87, m, H6,6; 3.44, dd, J 6 . 7 , 8 .6Hz, H17; 3.76, s , OMe; 6-63 , narrow m, H4; 6.70, dd, J 2-6 , 8 .5Hz, H2; 7.20, d, J 8 .3Hz, H l . 13c n.m.r. 6 (50.3 MHz, CDC13) 11.8, Me; 26.0, 29.9, 30.4, 31-2, 45.1, 6xCH2; 28-8, CMe3; 35.7, C11; 42.9, C13; 44.3, 45.3, C9,14; 55.2, OMe; 72.1, OCMe3; 80.7, C17; 113.3, C2; 113.7, C4; 126.0, C1; 133.0, C10; 138.2, C5; 157.4, C3. Mass spectrum: m/z 342 (M, 28%), 287 (151, 286 (75), 285 (83), 267 (24), 227 (211, 174 (201, 173 (98), 171 (20), 160 (30), 159 (151, 147 (391, 121 (IS), 111 (12), 57 (1001, 55 (14). The stereochemistry of (31) was determined by X-ray crystallography.
Further elution with light petroleum containing diethyl ether (20%) afforded (+)-17B-t- butoxy-7(8-ll)abeo-estra-1,3,5(10),9(11)-tetraen-3-01 (30) (0 114 g, 30%).
(dd) Deprotection of (*)-I 7~-t-Butoxy-3-methoxy-7(~lla)abeo-estra-1,3,5(10)-triene (31) to Give 7(&1 1a)abeo-Estra-1,3,5(10)-triene-3,178-diol (2a)
(i) Hydrolysis of the t-butoxy group (cf. Eder et a1.36).-~o a solution of 17p-t-butoxy-3- methoxy-7(8-.lla)abeo-estra-1,3,5(10)-trine (31) (0.300 g, 0 .81 mmol) in dioxan (6- 8 ml) was added 4 M hydrochloric acid (1 - 4 ml), and the mixture was refluxed for 5 h. The mixture was cooled, water (40 ml) was added, and the mixture was extracted with ether (x4). The extract was washed with 10% sodium chloride solution (x2), then dried (MgS04) and evaporated to give compound (33) as a white solid (0.250 g) which was not purified but used directly in the next step.
(ii) Cleavage o f the methoxy group in (33) (cf. Sheehan et al.21).-~yridine hydrochloride (11-3 g, 0.098 mol) was added to the solid, and the mixture was heated at 205" under nitrogen for 40 min. The mixture was cooled, 5% hydrochloric acid (25 ml) was added, and the mixture was extracted with ether. Evaporation of the washed, dried (MgS04) extract gave a viscous oil (0.235 g). Flash chromatography (silica gel, 50% diethyl ether/light petroleum) gave a solid (0.202 g, 85%), which upon recrystallization from methanol/water afforded (~)-7(8-lla)abeo-estra-1,3,5(10)-triene-3,17-diol (2a), mp. 190-192' (lit.2 192-195") (Found: M", 272 - l77*O.OO3. Calc. for Ci~H2402: M+*, 272- 177). Vmax (Nujol) 3320s(br), 1610m, 1585m, 1500s, 1465% 1430m, 1420x11, 1380s, 1350m, 1320m, 1290s, 1275s, 1245s, 1170m, 1155m, 1125s, 1105m, 1040s, 995111, 950w, 930w, 920111, 880m, 865w, 815m, 770w, 720w cm-'. 'H n.m.r. 6 (200MH2, CD30D) 0.76, s, Me; 0.9-2.3, m, H9,11,14 and 5xCH2; 2.75, m, H6,6; 3.65, dd, J 7.3, 8 .6Hz, H17; 6-50 , m, H2,4; 7.06, d, J 8.4Hz, H1. 13c n.m.r. 6 (50.3MHz, CDC13) 11.9, Me; 26.5, 30.7, 31.1, 31.2, 32.5, 46.0, 6xCH2; 37.3, C11; 44.4, C13; 45.5, 46-6, C9,14; 82.4, C17; 113.7, C2; 116.0, C4; 126.9, C1; 132.8, C10; 139.0, CS; 156.0, C3. Mass spectrum: m/z 272 (M, 39%), 213 (IS), 160 (28), 159 (loo), 146 (37), 145 (IS), 133 (10).
36 Eder, U., Gibian, H., Haffer, G., Neef, G., Sauer, G., and Weichert, R., Chem. Ber., 1976, 109, 2948.

Structure and Function of Estrogens. XI
Table 1. Atomic parameters for C23H3203 (26) In Tables 1-6, estimated standard deviations are given in parentheses. In Tables 1 and 4, U(eq) (A2) is the equivalent isotropic displacement coefficient defined as one-third of the
trace of the orthogonalized Uii tensor Atom lo4x lo4y 1042 103u(eq) Atom lo4x lo4y lo42 103U(eq)
C(1) -1061(3) 1280(2) 2984(3) 54(2) C(l3) -292(4) 985(3) 4957(4) 108(3) C(2) -1794(3) 1 729(3) 2373(3) 77(2) C(l4) 191(4) 2282(3) 4984(4) llO(3) C(3) -1996(3) 1341(3) 1432(3) 74(2) C(1') 1666(3) 157(3) 2316(3) 73(2) C(3a) -1430(2) 654(2) 1584(3) 53(1) C(29 1946(3) 721(3) 1839(4) 80(2) C(4) -1218(3) 290(2) 752(3) 67(2) C(3') 2593(4) 1194(4) 2262(6) 109(3) C(5) -560(3) -312(2) 998(3) 60(2) C(4') 2954(4) 11 1 l (5) 3164(7) 129(4)
57(2) -283f2) 1910(2) 51(2)
A Occupancies of the disordered methyl group are: C(7/A), 0.53(2); C(7'B). 0.47(2).
Table 2. Bond lengths (A) for C23H3203 (26)
Atoms Distance Atoms Distance Atoms Distance
Table 3. Bond angles (degrees) for C23H3203 (26) - - -
Atoms Angle Atoms Angle Atoms Angle
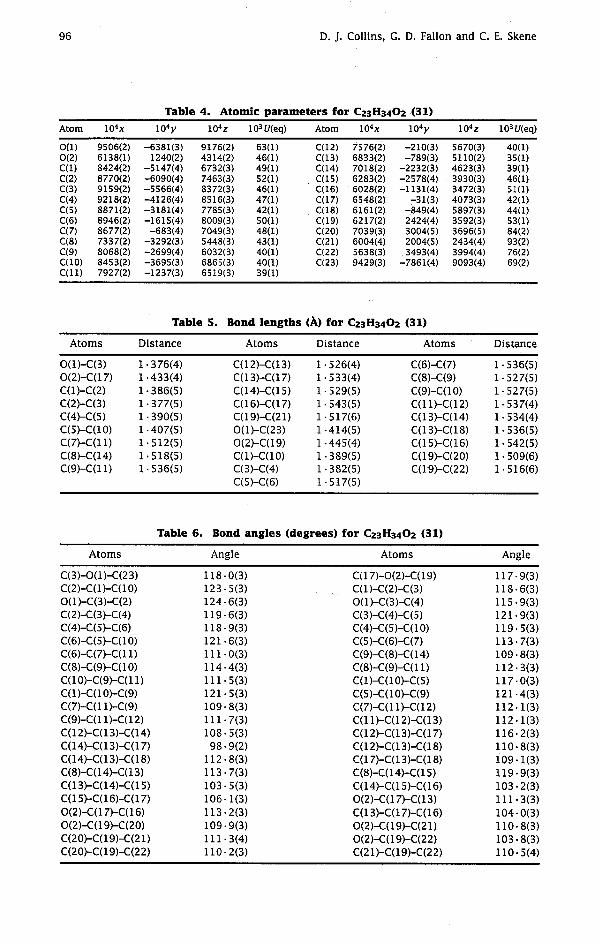
D. J. Collins, G. D. Fallon and C. E. Skene
Table 4. Atomic parameters for C23H3402 (31) Atom 104x 104y 104z 103U(eq) Atom 104x 104y 104z 103U(eq)
Table 5. Bond lengths (A) for C23H3402 (31)
Atoms Distance Atoms Distance Atoms Distance
O(1)-C(3) 1 -376(4) C(12)-C(13) 1 526(4) C(WC(7) 1 .536(5) O(2)-C(17) 1.433(4) C(13)-C(17) 1 .533(4) C(WC(9) 1 527(5) c ( l w ( 2 ) 1 .386(5) C(14)-C(15) 1 .529(5) C(9)-C(10) 1 - 527(5) C(Wc(3) 1 .377(5) C(16)-C(17) 1.543(5) C(l1)-C(12) 1 537(4) c(4)-c(5) 1 .390(5) C(19)-C(21) 1.517(6) C(13)-C(14) 1 .534(4) C(5)-C(10) 1.407(5) O(1)-C(23) 1 -414(5) C(13)-C(18) 1 .536(5) C(7)-C(l1) 1 .512(5) O(2)-C(19) 1 -445(4) C(15)-C(16) 1.542(5) C(WC(14) 1 518(5) C(1)-C(10) 1 -389(5) C(19)-C(20) 1.509(6) C(9)-C(l1) 1 .536(5) c(3)-c(4) 1 .382(5) C(19)-C(22) 1 . 5 16(6)
C(5)-C(6) 1 .517(5)
Table 6. Bond angles (degrees) for C23H3402 (31)
Atoms Angle Atoms Angle

Structure and Function of Estrogens. XI
X-Ray Crystallography
(a) The Spiro Cyclopropyl Ketone (26bFig. 1
Crystal data.-C23H3203, formula wt 356- 5, monoclinic, space group 12/a (by successful refinement). a 16.346(4), b 17.830(2), c 15.081(3) A, /3 104.01(2)', U 4265(1)A3, Dc 1 a l l , Z 8, Dm 1 - 12(l) g ~ m - ~ . F(000) 1552, p 5 3 cm-I for Cu K a radiation (A 1.5418 A). Temperature 20(1)". Cell parameters were determined from 24 accurately centred reflections, and were calculated by the standard Philips program.
Structure determination.-A representative clear acicular crystal of approximate dimensions 0 - 25 by 0 16 by 0 .11 mm was used for data collection. lntensity measurements were made on a Philips PWllOO diffractometer, with graphite-monochromatized Cu K a radiation with 6" < 2 8 I 1 30°, operating in an w scan mode with a symmetric scan range of k(O - 70+0.2 tan 8)" in w from the calculated Bragg scattering angle at a scan rate of 0.04' s-l. A total of 3627 unique data were collected (kh, +k, +I), 1905 of which were considered to be observed [ I 5 2u(1)]. Three standard reflections monitored every 4 h showed no significant variation in intensity over the data collection period. lntensity data were processed as described p r e v i o ~ s l y . ~ ~ A numerical absorption correction was applied, the maximum and minimum transmission factors being 0.932 and 0.888 respectively. The atomic scattering factors for neutral atoms were taken from ref. 24, and were corrected for anomalous dispersion by using values from ref. 24. All calculations were performed on a VAX 11/780 computer. The program used for least-squares refinement was that due to ~ h e l d r i c k . ~ ~
The structure was solved by direct methods.25 Full-matrix least-squares refinement, employing anisotropic thermal parameters for all non-hydrogen atoms and a single isotropic thermal parameter for hydrogen-which refined to 0-099(3) A2-positioned in geometrically idealized positions (C-H 0 .97 A), reduced R to 0.066 and R 1 to 0.067 at convergence, where
and w = [U~(F~)]- ' . The goodness of fit value ([I w ( I FO I - I FcI )2/(~observns - ~ ~ a r a m s ) l ~ / ~ ) was 2.32. The highest peak in the difference-Fourier synthesis was 0.25 e A-3.
Final atomic parameters are given in Table 1,* bond lengths in Table 2 and bond angles in Table 3, and Fig. 1 shows the atomic labelling scheme used.
(b) The 7(8+lla)abeo-Estradiol Derivative (31bFig. 2
Crystal data.-C23H3402, formula wt 342.5, monoclinic, space group P21/c. a 17.100(2), b 9.493(1), c 1 2 - 3 7 0 ( 1 ) ~ , /3 92.32(2)', U 2006.4(4)A3, Dc 1-13 , Z 4, Dm 1 -14(1) g ~ m - ~ . /.i
5 . 1 cm-l for Cu K a radiation (A 1.5418 A). F(000) 752. Temperature 20(1)". Cell parameters were determined as described above.
Structure determination.-As described above with the following relevant parameters. A clear tabular crystal of approximate dimensions 0.32 by 0 .17 by 0.06 mm was used for data
collection; monochromatized Cu-Ka radiation was used with 6.0" < 28 < 120 - On, symmetric w scan range of *(O. 75+0.2 tan 8)", at a scan rate of 0 - 04' s-l. 2981 unique data were collected (kh, +k, +0; 1663 were considered observed [ I 13ff(I)]. Transmission factors for absorption correction 0.967 and 0.839. Structure refinement with anisotropic thermal parameters for all non-hydrogen atoms, and a single fixed isotropic thermal parameter for hydrogen, reduced R to 0.048 and R 1 to 0.073 ( ~ = [ f f ~ ( ~ ~ ) + 0 . 0 0 6 5 ~ ~ ] - ~ ) at convergence. The goodness of fit value was 0.80. The highest peak in the final difference-Fourier synthesis was 0.21 e
Final atomic parameters are given in Table 4, bond lengths in Table 5 and bond angles in Table 6, and Fig. 2 shows the atomic labelling scheme used.
Acknowledgments
We thank the Australian Research Council for support. One of us (C.E.S.) gratefully acknowledges the receipt of a Monash Graduate Scholarship.
* Full crystallographic data for both compounds (26) and (31) have been deposited: anisotropic displacement parameters, hydrogen atom parameters, and tables of structure factors. Copies are available from the Australian Journal of Chemistry, P.O. Box 89, East Melbourne, Vic. 3002.





























