Embed Size (px)
Citation preview

Thermal Deesterification and Decarboxylation of Alternating Copolymers
of Styrene with P-Substituted t-Butyl a-Cyanoacrylates
HIROSHI ITO, IBM Research Division, Almaden Research Center, 650 Harry Road, San Jose, California 95120-6099 and
ANNE B. PADIAS and H. K. HALL, JR., Department of Chemistry, University of Arizona, Tucson, Arizona 85721
Synopsis
Alternating copolymers of styrene with 8-carboalkoxy-substituted t-butyl cr-cyanoacrylates undergo facile deesterification at 150-190°C, about 60°C below the deprotection temperature of poly( t-butyl methacrylate), and decarboxylation at 170-200°C. When the P-substituent is a methyl ester, the two events are clearly separated, with the deesterification occurring at a maximum rate a t 165°C and decarboxylation at 193°C. Anhydride formation is negligible in this case. The copolymer with t-butyl cyanofumarate exhibits simultaneous deesterification and decarboxylation events at 180°C with concomitant minor dehydration.
INTRODUCTION
Thermal behavior of poly(methacry1ic acid) (PMAA), poly(acry1ic acid) (PAA), and poly(t-butyl methacrylate) (PTBMA) has been the subject of a number of publications. Grant and Grassie' have reported that PMAA and PTBMA are converted thermally to poly(methacrylic anhydride) (PMAN) when heated to temperatures above 200°C. Matsuzaki and coworkers have found that isotactic PTBMA is converted smoothly to PMAN at 180-200"C, whereas syndiotactic PTBMA exhibits two-step weight loss; deesterification followed by dehydration.2 Geuskens et aL3 reported that the rate of water loss is about four times faster for isotactic PMAA than for atactic PMAA. The thermal behavior of random copolymers of methacrylic acid and t-butyl methacrylate has been recently studied by LaL4 Hatada and coworkers5 have shown by IR measurement that isotactic poly( apdimethylbenzyl methacrylate) (PDMBZMA) is converted to PMAN at 174°C as soon as a small amount of deprotection takes place, whereas an atactic polymer is predominantly deesterified first and then converted to PMAN. They also demonstrated that these polymers are deesterified to a certain extent upon electron beam exposure. Ito and Veda6 have recently studied thermolysis and acid-catalyzed deesterification of several polymethacrylates including PTBMA, PDMBZMA, and a cyclopropyl dimethyl carbinol ester of PMAA.
PAA dehydrates to form an intramolecular anhydride which then decom- poses to release carbon dioxide??' Maurer et al.' have recently carried out a comprehensive thermal characterization of atactic and syndiotactic PAA and found that anhydride formation commences in the same temperature region
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 27, 2871-2881 (1989) 0 1989 John Wiley & Sons, Inc. CCC 0887-624X/89/O9287 1 - 11$04.00

2872 ITO, PADIAS, AND HALL
(125-150OC) in both cases but proceeds much more gradually in the syndio- tactic polymer than in the atactic polymer. They have also found that the maximum decarboxylation due to decomposition of anhydride occurs a t about 260°C in the syndiotactic PAA, which is a substantially higher temperature than that of the atactic polymer (about 230°C). Poly(itaconic acid) (PIA) decarboxylates in aqueous solution below 100°C and conversion of P-keto acid resulting from condensation of adjacent itaconic units to cyclic ketones has been proposed as a mechanism for decarboxylation.1°
Decarboxylation has found a use in polymer synthesis and other applica- tions. For example, thermal and acid-catalyzed conversion of t-butoxylcar- bonyloxybenzene derivatives to phenol has been utilized in preparation of poly(4-hydroxystyrene)" and poly(4-hydro~y-a-methylstyrene)'~ and also in the design of dual tone resist material^'^^'^ as well as thermally developable positive resist systern~.'~ Another important application of decarboxylation to the resist technology is image reversal of positive photoresists based on diazonaphthoquinone; the dissolution-inhibiting quinonediazide is converted upon UV irradiation to polar indenecarboxylic acid, which then undergoes amine-catalyzed decarboxylation upon heating.16 Decarboxy lation of PMAA by ion beam irradiation has also been utilized in the resist design.17
In this paper we report thermal deesterification and decarboxylation of alternating copolymers of t-butyl P-carboalkoxy-a-cyanoacrylate with styrene.
EXPERIMENTAL
Monomer Synthesis
t- Butyl3-Carbomethoxy-2-Cyanoacrylate
A mixture of methyl 2-hydroxy-2-methoxyacetate (12 g, 0.1 mol) and t-butyl cyanoacetate (14 g, 0.1 mol) in 100 mL of acetonitrile containing 1 mL of acetic acid was refluxed overnight in a Soxhlet apparatus containing 3A molecular sieves. After evaporation of the solvent, the mixture was distilled in a Kugelrohr apparatus a t 8OoC/0.1 mmHg. The first fraction contained unreacted t-butyl cyanoacetate, and the second fraction was the desired compound (a viscous colorless liquid). Higher temperatures resulted in decom- position of the product. Yield: 1.8 g (10%). 'H-NMR (CDC1,): 7.35 ( s , lH), 3.9 (s,3H), 1.6 ppm (s,9H); IR (KBr): 2980, 1730, 1636, 1556, 1477, 1395, 1284, 1150, 835 cm-'. ANAL. Calcd for C,,H,,NO.,: C, 56.86; H, 6.20, N, 6.63. Found: C, 56.50; H, 6.33; N, 6.90.
t-Butyl Glyoxylate"
Isobutene (90 g) was condensed in a 500 mL pressure bottle using dry ice/methanol. Methoxyacetic acid (50 g, 0.5 mol) and 1.5 mL of sulfuric acid were slowly added. The mixture was shaken for 16 h a t room temperature. The content was cooled before opening, and the excess isobutene was evapo- rated at room temperature. Ether (250 mL) was added and the organic layer was washed twice with water and twice with 1 M aqueous sodium bicarbonate and then dried over magnesium sulfate. After evaporation of the solvent, the product was distilled at 78-8OoC/30 mmHg. Yield: 56 g (77%). 'H-NMR (CDC1,): 3.9 (s,2H), 3.4 (s,3H), 1.5 ppm (s,9H).

THERMAL DEESTERIFICATION OF STYRENE 2873
t-Butyl methoxyacetate (27 g, 0.18 mol) was mixed with 33 g of N-bromo- succinimide (NBS) in 500 mL of carbon tetrachloride in the presence of a trace of benzoyl peroxide. The mixture was refluxed for 1 h and cooled. The precipitated succinimide was filtered off and the solvent evaporated. The product was used in the next step without further purification. 'H-NMR (CDCl,): 6.0 ( s , lH), 3.5 (s,3H), 1.6 ppm (s,9H).
t-Butyl bromomethoxyacetate was added to a stirred solution of 18 g of sodium bicarbonate in 200 mL of water and stirred overnight. The aqueous solution was continuously extracted with ether for 48 h. The organic fraction was then dried over magnesium sulfate and the solvent evaporated. Reflux in a Dean-Stark trap using benzene for 4 h removed the last traces of water. After evaporation of benzene, the product was distilled a t 78-83"C/30 mmHg. Yield: 12 g (45%). 'H-NMR (CDCl,): 9.3 ( s ) , 7.3 ( s ) , 3.3 ( s ) , 1.6 ppm ( s ) . NMR shows the presence of both the hydrate and the free aldehyde.
t-Butyl3,3-Dicyanoacrylate
Distilled malononitrile (4 g, 0.06 mol) was mixed with 12 g of t-butyl glyoxylate (about 0.08 mol) in 100 mL ot acetonitrile and 1 mL of acetic acid in a Soxhlet apparatus containing 3A molecular sieves. After refluxing overnight and evaporation of the solvent, a brown solid was obtained, which was distilled in a Kugelrohr apparatus. The first fraction contained unidenti- fied materials, but the second fraction collected a t 8O0C/O.1 mmHg was crystalline. The solid was recrystallized from ether a t about 45°C. Yield: 1.7 g (14%). mp: 88°C. 'H-NMR (CDCl,): 7.1 ( s , lH), 1.6 ppm (s,9H); IR (KBr); 3071,2999, 2947, 2241,1714,1614,1375, 1353, 1258,1163, 1136,927,847 cm-'.
ANAL. Calcd for CSHl,N,O,: C, 60.66; H, 5.66; N, 15.72. Found: C, 60.68; H, 5.68; N, 15.74.
Di-t- butyl Cyanof umarate
A mixture of t-butyl glyoxylate (8 g, 0.06 mol), t-butyl cyanoacetate (7 g, 0.05 mol), 1 mL of acetic acid, and 100 mL of acetonitrile was refluxed overnight in a Soxhlet apparatus containing 3A molecular sieves. After evapo- ration of the solvent, the product was distilled in a Kugelrohr apparatus a t 8OoC/0.1 mmHg, and a colorless, viscous liquid was obtained. According to the NMR spectrum, this was a 50:50 mixture of the desired product and t-butyl cyanoacetate. Ether was added and the solution was placed at - 45°C. A solid crystallized out. Yield: 2.2 g (20%); mp: about 30°C. 'H-NMR (CDCl,): 7.2 ( s , lH), 1.6 ppm ( s , 18H). IR (KBr): 2983, 1726,1639, 1477, 1456, 1370, 1343, 1284, 1259, 1153, 1086 cm-'.
ANAL. Calcd for Cl,H19N04: C, 61.64; H, 7.56; N, 5.53. Found: C, 61.94; H, 7.61; N, 5.62.
Radical Copolymerization with Styrene or p-Methoxystyrene
All copolymerizations (1 : 1 feed ratio) were carried out with a,a-azobis(iso- butyronitrile) (AIBN) as the initiator in sulfolane or benzene under vacuum at 70°C for 16 h. Chloroform was added to dilute the polymer solution, and the polymer was precipitated in methanol and dried.
Measurements
NMR spectra were recorded in deuteriochloroform (CDCl,) on a Varian EM 360 (60 MHz) spectrometer. IR spectra were measured on an IBM IR/32

2874 ITO, PADIAS, AND HALL
FT spectrometer or on a Perkin Elmer 983 spectrometer. UV spectra were recorded on a Hewlett Packard Model 8450 UV/VIS spectrometer using thin (about 1 pm) films cast on quartz plates. Molecular weight determination was made by gel permeation chromatography (GPC) using a Waters Model 150 chromatograph equipped with 6 pStyragel@ columns a t 30°C in tetrahydrofu- ran (THF). Thus, the molecular weight reported in this paper is polystyrene- equivalent. Thermal analyses were performed on a DuPont 1090 Thermal Analyzer a t a heating rate of 5"C/min for TGA and 10"C/min for DSC under inert atmosphere. GC/MS analysis was carried out using a Hewlett Packard 5995A Gas Chromatograph/Mass Spectrometer.
RESULTS AND DISCUSSION
Heavily substituted olefins do not undergo radical homopolymerization, but electron-deficient olefins trisubstituted or tetrasubstituted with electron- withdrawing groups readily undergo 1 : 1 alternating radical copolymerization with electron-rich monomers such as styrene (St)." The alternating copoly- mers used in this study are presented in Scheme 1. The conditions and results of the copolymerizations are summarized in Table I. t-Butyl P-carboalkoxy- a-cyanoacrylates (I, 11) and t-butyl 3,3-dicyanoacrylate (111) gave high- molecular-weight, alternating copolymers with St and p-methoxystyrene, respectively, in excellent yield.
As discussed in the Introduction, tertiary esters of PMAA such as PTBMA, PDMBZMA, and cyclopropyl dimethyl carbinol ester readily deesterify upon
CO2R CN COiBu CN I I I I
I -(CH2-CH-CH-C j- -(CH2-CH-CH-C)-
CN Q I C0:Bu
OMe
I : R = M e I I : R=Bu
Scheme 1. Alternating copolymers.
I l l
TABLE I Alternating Radical Copolymerization of Trisubstituted Olefins'
Solvent Entry AIBN (monomer Analysis (%)
no.b mol% mmol/mL) Yield(%) C H N St% M,,, X M, X
I 0.3 Sulfolane (2.0) 95 67.61 6.49 4.40 49 446 153 I1 0.3 Benzene(2.0) 83 69.74 7.22 3.76 48 263 107
I11 0.26 Benzene(l.4) 90 68.53 6.20 8.74 52 329" 139'
'70"C, 16 h. bSee Scheme 1. 'Accompanied by 7.6% of a higher molecular weight fraction with M , = 9,655,000 and M,, =
5,88 5, OOO .

THERMAL DEESTERIFICATION OF STYRENE 2875
100
- 80 z E 60 m .
40-
20
- .-
CO$" I
+ C H - C H k
- LO@"
-
- . ( C )
CO$u CN - I I
+CH2-CH-CH-C+ .
- (a) 0 ' ~ ~ ' ' ' ' " " " ' " ~ " " '
40 120 200 280 360 440 Temperature ("C)
Fig. 1. TGA of (a) alternating copolymers I, 11, and 111, (b) methacrylate homopolymers, and (c) PDTBF (heating rate 5"C/min).
heating to release corresponding olefins. Thermal behavior of our copolymers is compared with that of the above-mentioned polymethacrylates as well as poly(di-t-butyl fumarate) (PDTBF)20 in Figure 1 (TGA). All the three alter- nating copolymers exhibit an onset of weight loss a t temperatures about 60" and 20°C below PTBMA and PDTBF, respectively, and in the same range as the cyclopropyl dimethyl carbinol ester. The reduced deprotection tempera- ture may be due to the electron-withdrawing effect of the CN group and the steric hindrance. The TGA weight loss a t about 170°C in I11 is about 18%, corresponding exactly to the quantitative loss of isobutene. The alternating copolymer I1 loses about 42% of its weight in the same temperature range, which is much more than the loss of 2 mol of isobutene (31.4%) can account for. The copolymer I undergoes two-step weight loss between 160 and 200"C, with the first loss (18%) corresponding to the quantitative release of isobutene (17.8%) followed by 14% weight 109.
In Scheme 2 are shown possible major thermolytic pathways in the alter- nating copolymers. Tertiary esters of PMAA readily deesterify upon heating
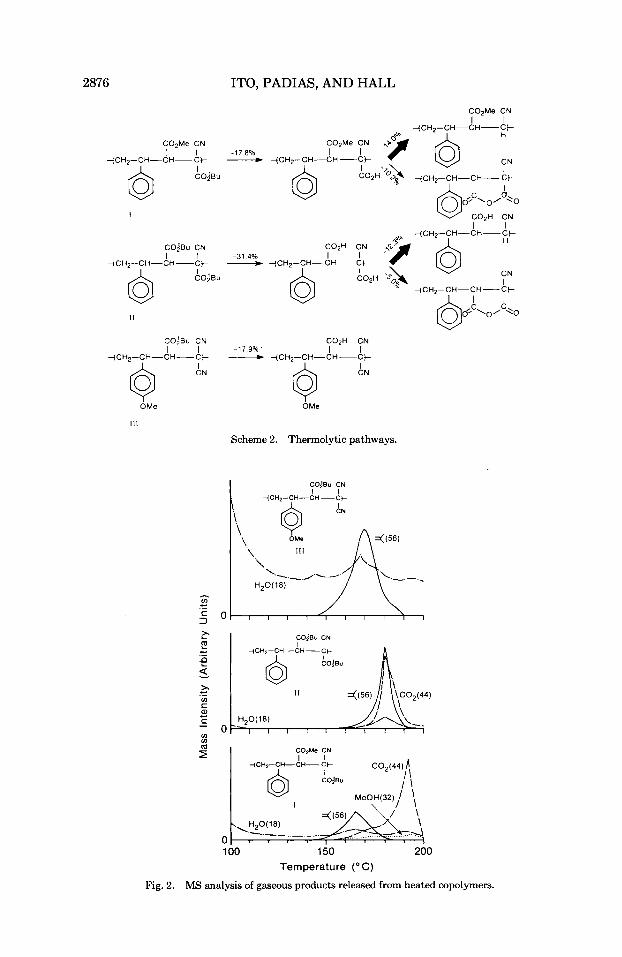
2876 ITO, PADIAS, AND HALL
P c 5f .- -P 5 2. CD C a,
+- ._
c c - 0 - . 1 u) u)
m z
C02Me CN I I
CO@u CN I I
iCHz-CH-CH-CI-
AO@U 6 HpO(18)
I , I I I
C02Me CN
+CHZ-CH-kH-CI-
C02Me CN I I -17 8% I I
+CH2-CH-CH-C+ - CN
I
6 I c0;BU
C0;Bu CN
+CHz--CH-CH-C+ - I I -31 4%
I
L0:Bu
I I
COZH CN I 1
+CHZ-CH-CH - CI- I COzH
C0;Bu CN CO2H CN
+CH2-CH-CH-C+ __t +CHp-CH-CH-Ct I I -179%' I I
I CN
I CN
111
Scheme 2. Thermolytic pathways.
I I
I H +CHz-CH-CH- CI-
CN

THERMAL DEESTERIFICATION OF STYRENE 2877
-50 1 , , . , , , , , , , , , , , , , , , , , , , 1 - 7
40 120 200 280 360 440 Temperature (" C)
Fig. 3. DSC analysis of I (heating rate lO"C/min).
with simultaneous or delayed dehydration (at about 240°C) to form an intramolecular anhydride.6 However, when the methacrylic ester units are isolated from each other by copolymerization with St or a-methylstyrene, the anhydride formation is very much suppressed. In both cases MS analysis of volatile thermal products led to better understanding of the thermal behavior of the polymers. The MS analyses of gaseous products generated upon heating the alternating copolymer powders are shown in Figure 2. The copolymer I11 having isolated t-butyl ester groups loses isobuteiie (molecular weight 56) a t a maximum rate a t 170°C, in agreement with the TGA data. An insignificant but slight increase in water generation is observed a t about 170"C, which might be due to minor intermolecular or intramolecular (discussed later) dehydration or other side reactions. Release of CO, is not observed below 200°C in 111.
The alternating copolymer I containing a carbomethoxy group on the /3-carbon exhibits two-stage gas evolution a t 165 and 193°C. The first major peak is due to evolution of isobutene (molecular weight 56) beginning at about 140°C and ceasing at about 185"C, which is followed by release of a gas with molecular weight 44 that is CO,. This indicates that the tertiary ester of I completely deesterifies and then decarboxylates upon further heating. The two-stage weight loss in TGA of I completely agrees with the quantitative loss of isobutene followed by quantitative decarboxylation (Fig. 1). DSC of I is presented as Figure 3, which shows upon the first heating an endothermic deesterification process immediately followed by an exothermic decarboxyl- ation process. The second heating shows Tg a t 160" owing to the alternating copolymer of St with P-(carbomethoxy)acrylonitrile, which was produced by thermolysis of I. In addition to a small amount of absorbed water, evolution of water seems to accompany the deesterification and decarboxylation process to a minor degree. The first dehydration, coinciding with the deesterification may be due to intramolecular anhydride formation in the acrylate diad sequences existing in a small amount. As copolymerization of t-butyl 3,3- dicyanoacrylate with St gave a copolymer containing only 30% St, existence of a diad of the acrylate units in I and I11 cannot be ighored. The second dehydration occurring along with decarboxylation might result from a minor

2878 ITO, PADIAS, AND HALL
intermolecular anhydride formation. Release of methanol from I is not signif- icant but gradually increases as the temperature reaches 150"C, with the maximum rate observed at 190-195OC, which might result from a minor concomitant intramolecular anhydride formation to form a maleic anhydride structure. It is known that copolymers of methyl methacrylate with methacrylic acid are converted to anhydride through dehydration in the methacrylic acid diad sequences, and to a lesser extent through a loss of methanol by anhydride formation involving the ester-acid diad sequences also.,' In any event, it is clear that the copolymer I undergoes deesterification and then decarboxylation almost exclusively.
Copolymer 11, bearing two adjacent t-butyl ester groups, undergoes one- stage weight loss (about 42%) a t 170-190°C. As shown in Scheme 2, if the deesterification is followed by intramolecular dehydration, the weight loss should amount to 36.6%, but if decarboxylation occurs, the total weight loss should be 43.7%. Judging from the TGA weight loss, decarboxylation appears to be a preferred pathway in 11. This is supported by the MS analysis shown in Figure 2. The deesterification is accompanied predominantly by decarbox- ylation, with a minor concomitant dehydration process with the maximum rate of all three events occurring at 180°C. This should be contrasted with the thermal behavior of PDTBF, which exhibits both deesterification and dehy- dration at 205°C with negligible or insignificant decarboxylation. In MS analysis, evolution of isobutene is always accompanied by fragments with molecular weights of 28, 39, and 41.
The decarboxylation mechanism in the copolymers I and I1 is different from that of PAA or PIA, which involves dehydration to form anhydride or j3-keto acid, which then undergoes decomposition to generate CO,. In contrast to the copolymers I and 11, tertiary esters of PMAA6 and copolymers of a,a-dimeth- ylbenzyl methacrylate with St or a-methylstyrene do not release CO, in a detectable quantity as studied by MS analysis. It has been reported that decarboxylation is insignificant in PMAA in contrast to PAA.22 It is clear that decarboxylation takes place in the ester group geminal to the CN group in the case of 11, as one compares the facile decarboxylation of I to the lack of CO, evolution from 111. It is well known in the l i t e ra t~re ,~ that geminal electron- withdrawing groups facilitate ready decarboxylation.
IR spectra of about 1.2-1.6-pm-thick films of I and I1 prebaked at 100°C (10 min) and then heated at 170°C are presented in Figure 4. The copolymer I exhibits only one ester carbonyl band a t 1740.0 cm-'. Heating a t 170°C for 30 min resulted in complete elimination of the t-butyl group as evidenced by the disappearance of absorptions a t 835.3 and 1371.6 cm-' as well as the shrink- age of the peaks a t the 1152.6 and 2950 cm-' region. The carbonyl peak became smaller without producing an OH absorption of COOH and was accompanied by a minor anhydride formation. The IR spectral changes observed upon heating clearly indicate that the acid group generated by deesterification readily undergoes decarboxylation at 170°C. Another 30 min heating at this temperature did not cause any spectral changes whatsoever. As shown in Figure 4, copolymer I1 shows only one IR carbonyl absorption
at 1730.4 cm-'. The IR spectrum measured after 90 min at 17OoC, very similar to the one obtained after 30 min, does not show absorptions a t 838.2, 1370.6, 1395.7, 2936.0, and 2980.4 cm- ', indicating that deesterification has

THERMAL DEESTERIFICATION OF STYRENE 2879
Wavenumbers (cm-')
Fig. 4. IR spectra of 1.2-1.6-pm-thick films of I and I1 heated at 100°C (10 min) and then at 170°C.
been completed; disappearance of the absorption at 1148.8 cm-' and shrink- age of the carbonyl peak a t 1730 cm- ' indicate that decarboxylation has also proceeded to completion. The new carbonyl peaks observed a t 1864.4 and 1790.2 cm-' after heating are due to formation of intramolecular five-mem- bered anhydride. In contrast to I, OH absorption of COOH remains after heating a t 170°C for 90 min, indicating that the CHCOOH is stable under these heating conditions once the neighboring COOH is destroyed by decar- boxylation. Two carbonyl absorptions observed in the heated film a t 1741.0 and 1717.8 cm-' may be due to "free" and hydrogen-bonded acids, respec- tively, since it has been reported that "free" methacrylic acid units absorb a t 1750 cm-' and its H-bonded dimers a t 1700 ~ m - ' . ~ ~ The IR study agrees with the MS analysis.
Thermolysis temperatures of tertiary esters are very much reduced in the presence of acids, and acid-catalyzed deesterification reactions have been successfully applied to the design of highly sensitive dual-tone resist system^.'^^'^ Sensitivity of these resist systems is very much reflected by the thermolysis temperature.6. l4 Since the deesterification temperature of I is as low as tnat of the cyclopropyl dimethyl carbinol ester of PMAA, the lowest deprotection temperature we have ever observed in polymers with pendant ester groups, we were tempted to formulate a resist system based on I and an onium salt cationic photoinitiator in the hope of achieving a high sensitivity. We have previously investigated %o y-radiolysis of alternating copolymers of St with olefins trisubstituted or tetrasubstituted with CN and primary ester

2880 ITO, PADIAS, AND HALL
i' C02Me CN
+CHz-CH-CH- C k I I
I CO:BU
+ 9 5 wf% Ph3SaaSbF6
- prebaked at 100°C (10 min)
-- exposed to 36 rnJIcm2 (254 nm) and postbaked at 100°C (2 min)
-.- further postbaked at 170OC (15 min)
'3 7 4000 3000 2000 1500 1000 600
Wavenumbers (cm-')
Fig. 5. IR spectra of 1.2-pm-thick film of I containing 9.5 wt% of Ph,S+-SbF, prebaked a t 100°C, exposed to 36 mJ/cm2 and postbaked a t 100°C (2 min), and further heated a t 170'C (15 min).
groups and found that these copolymers are as sensitive to ionizing radiation as PMMA but as resistant to CF, plasma as PSkZ5 The acid-catalyzed deesterification was thought to further improve the sensitivity of this type of resist materials.
IR spectra of a 1.2-pm-thick film of I containing 9.5 wt% of triphenylsulfo- nium hexafluoroantimonate are presented in Figure 5. Optical density (OD) of I is 0.068/pm a t 254 nm and the 1.2-pm-thick resist film containing 9.5 wt% of the sulfonium salt has an OD of 0.35 at the exposing wavelength of 254 nm. After exposure to 36 mJ/cm2 followed by postbaking a t 100°C for 2 min, the IR absorptions a t 835 and 1371 cm-' are very significantly reduced in intensity, indicating that the t-butyl ester group has been well converted to COOH, which is also supported by appearance of acid OH. Further postbaking a t 17OOC for 15 min resulted in the disappearance of the OH absorption and shrinkage of the carbonyl absorption, indicating that decarboxylation oc- curred during the high-temperature bake process. The carbonyl absorption of I at 1740.0 cm-' shrinks without either shifting or generating new peaks (except for a small peak a t 1790 cm-' owing to anhydride formed upon the high temperature bake), which suggests that COOCH,, COOC,H,, and COOH absorb a t the same wavenumber. Cyanoacetic acid exhibits its carbonyl absorption a t 1738.1 cm-'. The resist based on I has turned out not to be very sensitive compared with similar systems, which is presumably due to con- sumption of photochemically generated HSbF, by the CN group and is under further investigation.
SUMMARY AND CONCLUSIONS
Alternating copolymers of t-butyl P-carboalkoxy-a-cyanoacrylates with St readily lose isobutene upon heating to 170°C, which is about 60°C below the deprotection temperature of FTBMA. The subsequent major thermal event is

THERMAL DEESTERIFICATION OF STYRENE 2881
decarboxylation. Even when the P-substituent is a t-butyl ester, intramolecu- lar anhydride formation is not very significant. When the P-carbon is substi- tuted with a methyl ester, the thermal event after deesterification is almost exclusively decarboxylation. The facile decarboxylation is due to the strongly electron-withdrawing effect of the geminal CN group and is different from the mechanism in PAA or PIA which involves decomposition of cyclic anhydride or P-keto acid formed by dehydration. Although the thermal deprotection temperature of copolymers is even lower than that of PDMBZMA, the resists formulated with these copolymers in conjunction with a cationic photoinitia- tor do not exhibit expected sensitivity presumably due to consumption of photochemically generated acids by the CN group.
We thank E. Hadziioannou for the thermal analysis, R. Siemens for the GC/MS measure- ments, and C. Cole for the molecular weight determination by GPC. We are grateful to Prof. T. Otsu (Osaka City University, Japan) for providing us with PDTBF.
References 1. D. H. Grant and N. Grassie, Polymer, 1, 125 (1960). 2. K. Matsuzaki, T. Okamoto, A. Ishida, and H. Sobue, J . Polym. Sci., Part A , 2, 1105 (1964). 3. G. Geuskens, E. Hellinckx, and C. David, Eur. Polym. J., 7, 561 (1971). 4. J. H. Lai, Macromolecules, 17, 1010 (1984). 5. K. Hatada, T. Kitayama, S. Danjo, Y. Tsubokura, H. Yuki, K. Moriwaki, H. Aritome, and
6. H. Ito and M. Ueda, Macromolecules, 21, 1475 (1988). 7. A. Eisenberg, T. Yokoyama, and E. Sambalido, J . Polym. Sci., Part A-1, 7, 1717 (1969). 8. M. C. McGaugh and S. Kottle, J . Polym. Sci. Polym. Lett. Ed., 5, 817 (1967). 9. J. J. Maurer, D. J. Eustace, and C. T. Ratcliffe, Macromolecules, 20, 196 (1987).
S. Namba, Polym. Bull., 10, 45 (1983).
10. B. E. Tate, Makromol. Chem., 109, 176 (1967). 11. J. M. J. Frkhet, E. Eichler, H. Ito, and C. G. Willson, Polymer, 24, 995 (1983). 12. H. Ito, C. G. Willson, J. M. J. Frbchet, M. J. Farrall, and E. Eichler, Macromolecules, 16,
13. H. Ito and C. G. Willson, in Polymers in Electronics, T. Davidson, Ed. (American Chemical
14. H. Ito, C. G . Willson, and J. M. J. Frkhet, Proc. SPIE, 771, 24 (1987). 15. J. M. J. Frkhet, F. Bouchard, F. M. Houlihan, B. Kryczka, E. Eichler, N. Clecak, and C. G.
16. S. A. MacDonald, H. Ito, and C. G. Willson, Microelectr. Eng., 1, 269 (1983). 17. H. Hiraoka, Muter. Res. SOC. Symp. Proc., 45, 197 (1985). 18. L. A. Carpino, J . Org. Chem., 29, 2820 (1964). 19. H. K. Hall, Jr., Angew. Chem. Int. Ed. Engl., 22, 440 (1983), and references therein. 20. T. Ot-u, Polym. Prepr. Jpn., 36, 39 (1987). 21. A. Jamieson and I. C. McNeill, Eur. Polym. J., 10, 217 (1974). 22. F. X. Roux, R. Audebert, and C. Quivoron, Eur. Polym. J., 9, 815 (1973). 23. J. March, Adoanced Organic Chemistry, 3rd ed. (John Wiley & Sons, New York, 1985),
24. J. Y. Lee, P. C. Painter, and M. M. Coleman, Macromolecules, 21, 346 (1988). 25. H. Ito, C. Hrusa, H. K. Hall, Jr., and A. B Padias, J . Polym. Sci. Polym. Chem. Ed., 24,955
510 (1983).
Society, Washington, D.C., 1984), Am. Chem. SOC. Symp. Ser. 24, p. 11.
Willson, J . Zmuging Sci., 30, 59 (1986).
p. 563.
(1986).
Received June 9, 1988 Accepted October 31, 1988





























