Embed Size (px)
Citation preview

1975 D. Hellwinkel, M . Haltmeier und G . Reiff 249 Liebigs Ann. Chem. 1975, 249-254
Thermische Zersetzung von 2,2‘-Biphenylylen(pheny1)jod und ahnlichen Verbindungen Dieter Hellwinkel *), Margavete Haltmeiev und Giinther R e @
Organisch-Chemisches Institut der Universitat, D-6900 Heidelberg, Im Neuenheimer Feld 7
Eingegangen am 28. Mlrz 1974
Die Thermolyse von 2,2’-Biphenylylen(pheny1)jod (4) liefert in der Hauptsache 2-Jod-m- terphenyl (5). Die Zersetzung des aus 2,2’-Biphenylylenjodoniumjodid (10) mit Methyllithium erhaltenen Reaktionsprodukts fuhrt zu 2-Jod-2’-methylbiphenyl (12) und 2-Jodbiphenyl (14). Mit Lithiunialuminiunihydrid reagiert 10 i n hohen Ausbeuten zu Biphenyl.
Thermal Decomposition of 2,2’-Biphenylylene(phenyl)iodine and Similar Compounds Thermolysis of 2,2‘-biphenylylene(phenyl)jodine (4) gives mainly 2-iodo-m-terphenyl (5). Decomposition of the reaction product from 2,2’-biphenylyleneiodonium iodide (10) and methyllithium yields 2-iodo-2’-methylbiphenyl (12) and 2-iodobiphenyl (14). Compound 10 rescts with lithium eluniinium hydride to give high yields of biphenyl.
Vor einigen Jahren 1 ) stellten wir 2-Jod-o-terphenyl (1) durch Umsetzung von Fluorbenzol niit Phenyllithium und anschlieBende Jodolyse her. Eine uber Dehydro- benzol fiihrende Reaktionssequenz entsprechend Schema 1 war hierfiir maBgeblich.
Schema 1
Da die Ausbeuten an 1 bei diesem Verfahren nicht allzu gut waren, suchten wir nach einem besseren Syntheseweg. Unter Berucksichtigung der Ergebnisse einiger typischer Thermolysereaktionen hypervalenter 2) Perarylderivate der Elemente der fiinften Hauptgruppe (z. B. 2 + 3 1 . 3 . 4 ) war zu erwarten, daB sich 2,2’-Biphenylylen(pheny1)- jod5.6) (4) beim Erwarmen glatt zu 1 unilagert. Es iiberraschte uns daher sehr, als bei der Thermolyse von 4 in Benzol, Mesitylen oder Cyclohexan als Hauptprodukt (63 bis 72% Reinausbeute) eine Substanz mit Schmp. 51 -52°C entstand, die zwar die
*) Korrespondenz bitte an diesen Autor richten. 1) G. Wittig und D. Hellwinkel, Chem. Ber. 97, 769 (1964). 2 , Vgl. J . I . Musher, Angew. Chem., 81,68 (1969); Angew. Chem., Int. Ed. Engl. 8,54 (1969). 3) G. Wittig und A . Maercker, Chem. Ber. 97, 747 (1964). 4) Vgl. auch: D. Hellwinkel in Organic Phosphorus Compounds (G. M . Kosolapoff und
5 , K . ClauJ, Chem. Ber. 88, 268 (1955). 6 ) Nach den Regeln der IUPAC-Nomenklatur auch als 5-Phenyl-SH-dibenzo[b,d]jodol ZU
L. Mcrier), 1. Aufl., Bd. 3, S. 185, John Wiley, New York 1972.
benennen.

250 D . Hellwinkel, M. Haltrneier und G . Reiff 1975
Analysendaten und die Molekulmasse eines Jodterphenyls zeigte, aber nicht rnit dem fruher auf anderem (und eindeutigen) Wege dargestellten 1 1) identisch war 7,s).
3
Dal3 es sich bei dem unbekannten Produkt um 2-Jod-m-terphenyl (5) handelte, folgte ails der Umsetzung mit Butyllithium (oder auch Lithiumaluminiumhydrid9)), die nach Hydrolyse m-Terphenyl (6) lieferte. Reaktion des aus 5 mit Butyllithium zunachst entstehenden m-Terphenyl-2-yllithiums rnit C02 fiihrte zu der bekannten m-Terphenyl-2-carbonsaure10) (7), die sich rnit Polyphosphorsaure zu 3-Phenyl- fluorenonlo) (8) und wenig 1-Phenylfluorenonlo. 11) (9) cyclisieren lieB.
h 1 -
Q OyJ \
6
Q 0 0 1 1
Weiterhin haben wir die Struktur von 5 durch Vergleich rnit einem in anderem Zusammenhange auf unabhangigem Wege synthetisierten Referenzpraparat bestatigt 12).
Auch fur Synthesezwecke hat 5 bereits Verwendung gefunden12). Fur die ungewohnliche thermische Umlagerung von 4 zu 5 fehlt bisher jede Analo-
gie13). Die bei der Thermolyse in Benzol in geringer Menge auftretenden und gas-
7) F. M. Beringer und L. L . Chang, J. Org. Chem. 36, 4055 (1971), haben die gleiche Thermo- lyse inHexan durchgefiihrt und dem dabei erhaltenenHauptprodukt ohne weitere Struktur- sicherung die Konstitution 1 zugeschrieben.
8) Vgl. auch: J . A . Cude und A . Pilbeam, J. Chem. SOC. 1964, 114. 9) Entgegen friiheren Auffassungen sind auch nicht aktivierte Halogenaromaten durch
LiAlH4 reduzierbar. Vgl. z. B.: H. C. Brown und S. Krishnamurthy, J. Org. Chem. 34, 3918 (1969).
10) D. H . Jones und W. R . Wragg, J. Chem. SOC. C: 1968,2154. 11) D . M. W. Anderson, N . Campbell, D . Leaver und W. H. Stafford, J. Chem. SOC. 1959,3992. 12) D. Hellwinkel und H. Seifert, Liebigs Ann. Chem. 762, 29 (1972).
1975 Thermolyse von 2,2’-Biphenylylen(phenyl)jod u. a. Verbindungen 25 I
chromatographisch bestimrnten Nebenprodukte Biphenyl, 2-Jodbiphenyl und 2,2‘- Dijodbiphenyl sprechen fur das Auftreten radikalischer Zwischenstufen7) ; die hohe Ausbeute an einem Jodterphenyl definierter Struktur, narnlich (5), ist hinzegen eher mit einem intramolekularen Reaktionsverlauf vereinbar. Ein solcher konnte beispiels- weise entsprechend Schema 2 aussehen 14).
Schema 2
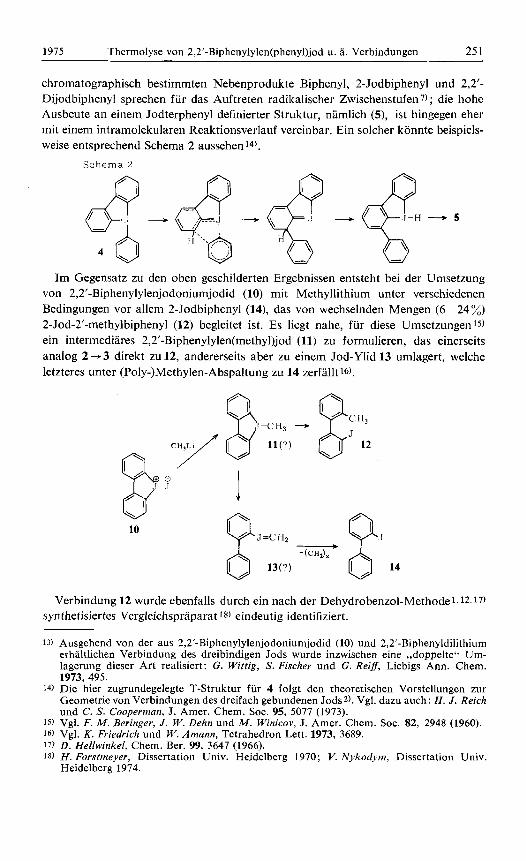
Im Gegensatz zu den oben geschilderten Ergebnissen entsteht bei der Urnsetzung von 2,2’-Biphenylylenjodoniumjodid (10) mit Methyllithium unter verschiedenen Bedingungen vor allem 2-Jodbiphenyl (14), das von wechselnden Mengen (6 -24 %) 2-Jod-2’-methylbiphenyl (12) begleitet ist. Es liegt nahe, fur diese Umsetzungen 15)
ein intermediares 2,2’-Biphenylylen(rnethyl)jod (11) zu formulieren, das einerseits analog 2 + 3 direkt zu 12, andererseits aber zu einern Jod-Ylid 13 umlagert, welche letzteres unter (Poly-)Methylen-Abspaltung zu 14 zerfallt 16).
8 /
\
10
0 . I i
0 13(?)
Verbindung 12 wurde ebenfalls durch ein nach der Dehydrobenzol-Methode 1.12.17)
syn thetisiertes Vergleichspraparat 18) eindeutig identifiziert.
13) Ausgehend von der aus 2,2’-Biphenylylenjodoniumjodid (10) und 2,2‘-Biphenyldilithium erhaltlichen Verbindung des dreibindigen Jods wurde inzwischen eine ,,doppelte“ Um- lagerung dieser Art realisiert: G. Wittig, S . Fischer und G . Rei f , Liebigs Ann. Chem. 1973, 495.
14) Die hier zugrundegelegte T-Struktur fur 4 folgt den theoretischen Vorstellungen zur Geometrie von Verbindungen des dreifach gebundenen Jods2). Vgl. dazu auch: H . J. Reich und C . S. Cooperman, J. Amer. Chem. SOC. 95, 5077 (1973).
15) Vgl. F. M. Beringer, J . W. Dehn und M. Winicov, J. Amer. Chem. SOC. 82, 2948 (1960). 16) Vgl. K . Friedrich und W. Amann, Tetrahedron Lett. 1973, 3689. 17) D. HeNwinkel, Chem. Ber. 99, 3647 (1966). 1 8 ) H . Forstmeyer, Dissertation Univ. Heidelberg 1970; V . Nykodym, Dissertation Univ.
Heidelberg 1974.

252 D . Hellwinkel, M . Hdtmeier und G. Reiff 1975
SchlieBlich setzten wir das Jodoniumsalz 10 auch mit Lithiumaluminiumhydrid in Ather urn und erhielten dabei neben 78% Biphenyl 14% 2-Jodbiphenyl. Auch hier erscheint die Formulierung des Reaktionsverlaufs uber ein Derivat 15 des dreibindigen Jods sehr attraktiv.
Weitere Untersuchungen zur Klarung der durch diese Arbeiten angeschnittenen Fragen sind ini Gange.
Diese Untersuchungen wurden mit Mitteln der NATO (Research Grant No. 520) und des Fonds der Chemisehen Indirstrie unterstutzt.
Experirnenteller Teil Alle Umsetzungen mit metallorganisclien Reagenzien wurden unter Reinststickstoff durch-
gefuhrt. Ather wurde durch Kochen rnit Natriumdraht getrocknet und anschiienend iiber Lithiumaluminiumhydrid direkt in das ReaktionsgefaR destilliert. Methyllithium und Phenyl- lithium stellte man aus Brommethan bzw. Brombenzol und Lithiumschnitzeln in Ather her's). - Fur die Dunnschichtchromatographie verwendete man Kieselgel G nach Stahl. Sichtbar wurden die Substanzen in der Jodkammer oder rnit UV-Licht gemacht. - IR-Spektren: Gerat Perkin-Elmer 221, IH-NMR-Spektren : Gerat Varian A-60, Massenspektren : Gerlt Dupont CEC 21 -1 10.
Thermolyse von 2,2'-Biphenylylen(phenyl)jod (4) in Benzol
a) 4.06 g (10 mrnol) fein gepulvertes 2,2'-BiphenyIylenjodoniuinjodid20) (10) wurden im Doppelschlenkrohr in 20 ml Ather suspendiert. Uriter Ruhren gab man bei 0°C tropfenweise 16 ml einer 0.75 M atherischen Phenyllithium-Losung zu und ruhrte weitere 5 h. Nach Ab- sitzen des zitronengelben 4 wurde das Solvens in den zweiten Schlenkrohrschenkel dekantiert. Zui Entfernung des uberschiissigen Phenyilithiums wurde rnit zuriickdestilliertem Ather gewaschen. Nach dem Trocknen fiigte man 50 ml absol. Benzol zu und erhitzte bei starkem Ruhren 1 h zurn Sieden. Die hierbei gebildete leicht getriibte Losung filtrierte man und zog das Losungsmittel i. Vak. ab. Es blieben 3.8 g braunes 01 zuriick, das durch Destillation (Sdp. 195"CjO.l Torr) gereinigt wurde. Nach Anreiben mit Athanol erhielt man 2.55 g (72%) reines 2-Jod-m-terphenyl (5) als farblose Kristalle mit Schmp. 51 --52'C.
b) Die Reaktion nach a) wurde mit dem 3fachen Ansatz wiederholt. Hierbei erhielt man 1 1 g Rohprodukt von dem 3 g saulenchromatographisch gereinigt wurden [Saule: 50 :< 4 cm; Festphase: A1203 neutral; Laufrnittel: Petrolather (40-6O0C)/Benzol = 4 : I ] . Man erhielt so 1.9g 5 rnit Schmp. 51 -52°C [nach Misch.-Schmp., DC und IR identisch mit 5 nach a)!.
19) U. Schollkopf in Methoden der organischen Chemie (Houben- Weyl-Miller), 1. Aufl..
20) J. Collette, D . MeGreer, R . Crowford, F. Chirbb und R . B. Sandin, J. Amer. Chem. SOC. Bd. 13/l , S. 86, Thieme, Stuttgart 1970.
78, 3819 (1956).

1975 Thermolyse von 2,2'-Biphenylylen(pheny1)jod u. a. Verbindungen 253
Reinausbeute 65% (bez. auf 10). - 1H-NMR: 6 = 6.80-7.60 (m; Aromaten), 7.88 ppm (d, J = 8 Hz; H o-standig zum J 21)).
C18H13J (356.2) Ber. C 60.69 H 3.68 J 35.63 Gef. C 60.57 H 3.82 J 35.64 Gef. C 60.80 H 3.88 J 35.85 MoL-Masse 356 (massenspektrom.)
Das Gaschromatogramm (SE30Glas 3.8 %, 153 cm; 149"C, nach 12 min auf 250°C mit 6"/min; 6.35 mm/nlin) des Rohprodukts weist 7 Peaks auf, von denen 4 rnit Hilfe von Vergleichs- substanzcn zugeordnet werden konnten : Biphenyl ( I -2 %), 2-Jodbiphenyl (weniger als 1 %), 2,2'-Dijodbiphenyl (weniger als 1 %), 2-Jod-m-terphenyl (5 ; ca. 85 %).
Thermolyse von 4 in Mesitylen: In einem 30-mmol-Ansatz wurde 4 wie voranstehend beschrieben hergestellt. Unter starkem Riihren wurde der Ansatz 1 h in 60 ml frisch destillier- tem Mesitylen adf 70-80°C erhitzt. Die dabei gebildete triibe Losung wurde filtriert und i. Vak. vom Solvens befreit. Man erhielt 10.7 g braunes 0 1 . Saulenchromatographische Reini- gung von 2.75 g Rohprodukt ergab 1.74 g (63 %) 5 rnit Schmp. 50-52°C (Misch.-Schmp., DC und IR bewiesen die Identitat).
Thermolyse von 4 in Cyclohexan: 17.8g (50 mol) frisch bereitetes4 wurden in 100 ml Cyclo- hexan unter intensivem Riihren 1 h zum Sieden erhitzt. Nach Filtration der triiben Losung zog man das Solvens i. Vak. ab. Ausbeute ca. 18 g gelbes 0 1 . 3 g davon wurden saulenchro- matographisch wie voranstehend gereinigt, wobei man 2 g (67%) 5 erhielt. Schmp. 50-52°C (Misch.-Schmp., DC und I R bewiesen die Identitat).
Uberfiihrung von 5 in m-Terphenyl(6) a) Mit Bufyllifhium: Zu einer Losung von 3.56 g (10 mmol) 5 in 20 ml Cyclohexarl im
Doppelschlenkrohr wurden 5.5 ml 2 N Butyllithium-Losung getropft. Nach 3 stdg. Riihren bei Raumtemp. war das gebildete m-Terphenyl-2-yllithium als farbloser, kristalliner Nieder- schlag ausgefallen. Die uberstehende Losung wurde durch die Schlenkrohrfritte dekantiert. Man loste die Lithiumverbindung in 30 ml Ather und hydrolysierte. Aus der organischen Phase erhielt man ein gelhes 01, das init Petrolather (40-60°C) iiber neutralej A1203 chro- matographiert wurde. Danach isolierte man 1.4 g (60 %) reines 6 rnit Schmp. 85 -86°C.
Cx~H14 (230.3) Ber. C 93.87 H 6.13 Gef. C 93.22 H 6.28 Gef. C 93.70 H 5.93
b) Mil Lifhiumaluminiutnhydrid: 2 g 5 wurden in 60 ml Ather rnit 0.76 g (20 mmol) LiAIH4 versetzt und 24 h geriihrt. Nach vorsichtiger Hydrolyse wurde verd. Salzslure zugefiigt, worauf man aus der athevischen Phase 1.25 g (97 %) 6 rnit Schmp. 85-87'C isolieren konnte. (Misch.-Schmp., DC und I R bewiesen die Identitat.)
Uberfuhrung von 5 in m-Terphenyl-2-carbonsaure (7): Aus 6 g (16.8 mmol) 5 stellte man wie voranstehend beschrieben m-Terphenyl-2-yllithium her und schiittete es unter Stickstoff in einen zur Halfte mit Trockeneis gefiillten 500-ml-Kolben. Die Mischung wurde wiederholt kraftig geschiittelt und 1 Tag stehengelassen. Nach Abziehen des Athers versetzte man das gelbliche Lithiumsalz von 7 mit verd. Salzsaure, schiittelte rnit Ather aus und isolierte daraus die rohe Saure 7. Mehrmaliges Umkristallisieren aus Petrolather (40-60°C) ergab farbloses 7 rnit Schmp. 134- 136°C (Lit.10) 132--133°C). Ausbeute 1.8 g (3973.
C19H1402 (274.3) Ber. C 83.19 H 5.14 Gef. C 82.73 H 5.12 Gef. C 83.16 H 5.33
21) Vgl. F. M . Beringer, P . Ganis, G. Avitabile und H. Jaffe, J. Org. Chem. 37, 879 (1972).

254 D. Hellwinkel, M. Haltmeier und G . Reiff 1975
Cyclisierung von 7 zu 3-Phenylfluorenon (8) und I-Phenylfluorenon (9): 2.1 g (7.65 mmol) 7 wurden rnit 50 ml Polyphosphorsaure so lange auf 150°C erhitzt, bis eine braune Losung entstanden war. Nach Abkuhlen goR man in 500 ml Wasser und schuttelte mehrere Male mit Chloroform aus. Nach Waschen rnit Wasser, Trocknen mit Calciumchlorid und Abziehen des Chloroforms erhielt man ein gelbes 01 , das laut DC aus 2 Substanzen bestand. Saulen- chromatographie (Saule: 70 x 4 cm) an Aluminiumoxid (neutral) mit Petrolather (4O-6O0C)/ Chloroform ( I : 1) erlaubte iiber vide Zwischenfraktionen die Reindarstellung der beiden Produkte.
Das langsamer wandernde gelbe Hauptprodukt 3-Phenylfluorenon (8) (1.2 g, 61 %) hatte den Schmp. 98-101°C (Lit.10) 101-102°C). - IR: 1710 cm-1 (CO).
C19H120 (256.3) Ber. C 89.04 H 4.72 Gef. C 88.84 H 4.97 Mol.-Masse 256 (massenspektrometr.)
Aus den Vorlaufen wurden einige Milligramm gelbes 1 -Phenylfluorenon (9) mit Schmp. 118-120°C (Lit.’[) 121 -122°C) erhalten. - IR: 1710 cm-1 (CO).
Umsetzung von 2,2’-Biphenylylenjodoniunijodid (10) mit Methyllithium und nachfolgende thermische Zersetzung
a) 4.06 g (10 mmol) 10 (fein gepulvert) wurden im Doppelschlenkrohr rnit Fritte in 20 ml Ather suspendiert. Bei -78°C wurden 8.5 ml 1.2 M Methyllithium-Losung zugetropft. Beim langsamen Erwarmen auf Raumtemp. bildete sich eine kraftig gelbe Suspension. Nach 6stdg. Ruhren bei Raumtemp. war eine gelbe Losung entstanden, die nach Abziehen des Solvens ein gelbes 0 1 lieferte. Saulenchromatographie rnit Petrolather (40-60°C) iiber neu- trales Aluminiumoxid lieferte 1.4 g (50 %) 2-Jodbiphenyl (14; identifiziert durch DC- und IR-Vergleich) und 0.18 g (6 %) 2-Jod-2’-methylbiphenyl (12) als farblose Kristalle rnit Schmp. 41-42°C (identifiziert durch 1R- und 1H-NMR-Vergleich). - IH-NMR: 8 = 2.06 (CH3), 6.80-7.45 (m; Aromaten), 7.90 ppm (d, J = 8 Hz; H o-standig zum Jod21)).
b) In einem Parallelansatz wurde die bei 0°C erhaltene gelbe Suspension iiber die Schlenk- rohrfritte filtriert, der gelbe Ruckstand mit zuruckdestilliertem Ather gewaschen, wieder filtriert und anschlieaend in 40 ml absol. Benzol zum Sieden erhitzt. Hierbei schaumte die Benzollosung (Gasentwicklung?). Aufarbeitung wie bei a) lieferte 1.2 g (43%) 14 und 0.3 g (10%) 12 rnit Schmp. 40-42°C (Identitatsnachweis durch Misch.-Schmp. und DC-Vergleich).
c) In einem dritten Parallelansatz wurde nach Abtrennen des Athers rnit 40ml Benzol versetzt und dann nur auf 30-40°C erwarmt. Hier erhielt man nach Aufarbeitung wie bei b) 0.9 g (32%) 14 und 0.7 g (24%) 12 rnit Schmp. 41 -42°C (ldentitatsnachweis durch Misch.- Schmp., IR- und DC-Vergleich).
Umsetzung von 10 mit Lithiumaluminiumhydrid: 4.06 g (10 mmol) gepulvertes 10 wurden rnit 70ml Ather und 0.8 g (21 mmol) LiAlH4 versetzt, wobei eine nur wenig getrubtelosung entstand. Nach vorsichtiger Hydrolyse versetzte man mit verd. Salzsaure und dampfte die organische Phase zur Trockne ein. Anreiben mit Methanol lieferte 1.2 g (78 %) Biphenyl (identifiziert durch DC- und IR-Vergleich). Eindunsten d.er methanolischen Mutterlauge brachte 0.4g (14%) oligen Ruckstand, der laut DC und IR zur Hauptsache aus 2-Jodbi- phenyl (14) bestand.
[95/741












![Crystallization-Induced Energy Level Change of [6,6]-Phenyl ...opac.ll.chiba-u.jp/da/curator/100085/Crystallization...1 Crystallization-Induced Energy Level Change of [6,6]-Phenyl-C61-Butyric](https://img.pdfslide.tips/doc/110x75/60dcd50502116a77a0410407/crystallization-induced-energy-level-change-of-66-phenyl-opacllchiba-ujpdacurator100085crystallization.jpg)
















