Embed Size (px)
Citation preview

©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Nature GeNetics ADVANCE ONLINE PUBLICATION �
l e t t e r s
A fundamental challenge in biology is explaining the origin of novel phenotypic characters such as new cell types�–4; the molecular mechanisms that give rise to novelties are unclear5–7. We explored the gene regulatory landscape of mammalian endometrial cells using comparative RNA-Seq and found that �,532 genes were recruited into endometrial expression in placental mammals, indicating that the evolution of pregnancy was associated with a large-scale rewiring of the gene regulatory network. About �3% of recruited genes are within 200 kb of a Eutherian-specific transposable element (MER20). These transposons have the epigenetic signatures of enhancers, insulators and repressors, directly bind transcription factors essential for pregnancy and coordinately regulate gene expression in response to progesterone and cAMP. We conclude that the transposable element, MER20, contributed to the origin of a novel gene regulatory network dedicated to pregnancy in placental mammals, particularly by recruiting the cAMP signaling pathway into endometrial stromal cells.
The defining novelties of Eutherian (placental) mammals include pro-longed internal development, maternal recognition of pregnancy, an invasive placenta and a richly vascularized uterine endometrium that can accommodate implantation8,9. An essential step in the establish-ment of pregnancy in many placental mammals is the differentiation (decidualization) of endometrial stromal cells (ESCs) in response to the hormone progesterone, the second messenger cAMP and, in some species, fetal signals10,11. Decidualization of ESCs involves extensive reprogramming of many cellular functions, including the simulta-neous silencing of cellular proliferation pathways and activation of progesterone and cAMP signaling pathways. Thus, the evolution of pregnancy was likely dependent on the evolution of ESCs and hor-mone- and cAMP-mediated cell signaling.
To better understand how the gene regulatory network in ESCs evolved in mammals, we sequenced the transcriptome from human (Homo sapiens) ESCs differentiated with progesterone and cAMP and from the endometrium of mid-pregnancy armadillo (Dasypus novemcinctus) and short-tailed opossum (Monodelphis domestica) using high-throughput Illumina sequencing (Fig. 1a). A total of 13,505,261, 13,218,476 and 14,830,816 75-bp paired-end reads were generated for human, armadillo and opossum, respectively, and
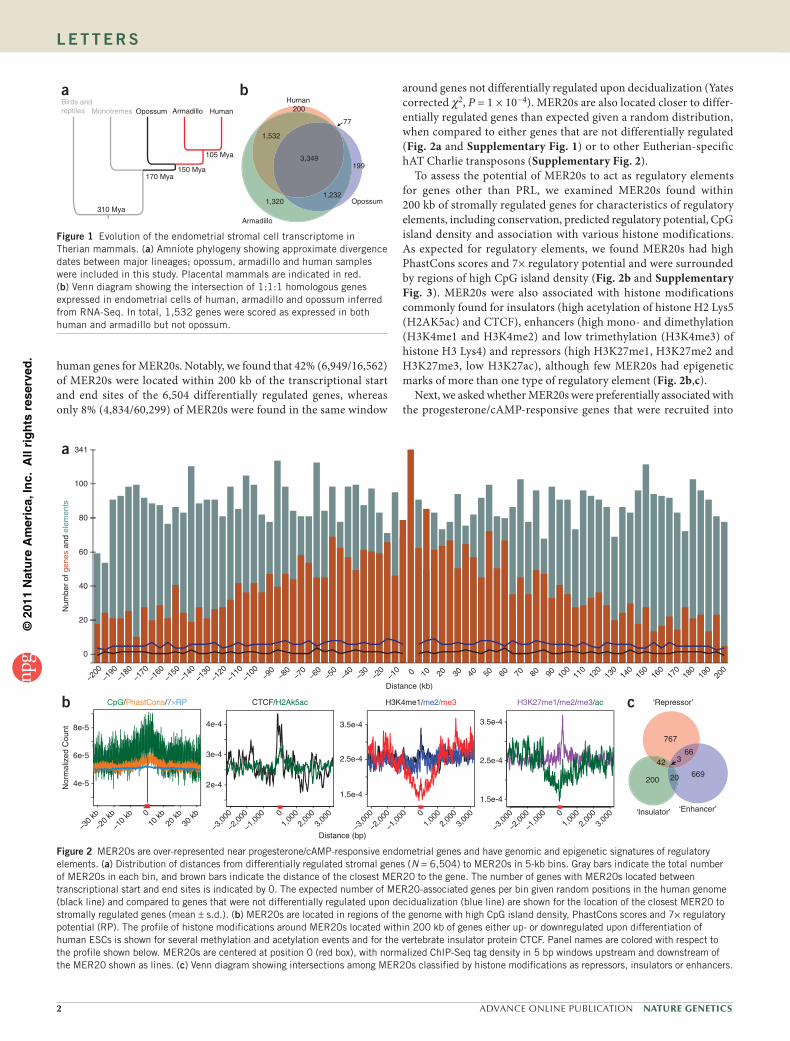
mapped to 17,550 human, 10,590 armadillo and 11,824 opossum genes. Of 9,323 1:1:1 human:armadillo:opossum orthologs, 5,158 were expressed in human ESCs, whereas 7,433 and 4,857 genes were expressed in armadillo and opossum endometrium, respectively (see Methods). We found that 1,532 genes were expressed in both human and armadillo endometrial cells but not those of opossum, whereas 199 genes were expressed in opossum but in neither human nor arma-dillo. A parsimonious interpretation of these data suggests 1,532 genes were recruited into endometrial expression during the evolution of pregnancy in placental mammals (Fig. 1b).
We annotated these 1,532 genes by their Gene Ontology (GO) terms to identify biological processes and pathways that were recruited into ESCs in placental mammals. We found that several pathways with essential roles in pregnancy and decidualization were over-represented among the recruited genes, including ‘Regulation of G-Protein Coupled Receptor Signaling’ (P = 0.006), ‘Regulation of Protein Kinase Activity’ (P = 0.002), ‘Receptor-Mediated Signaling’ (P = 4.17 × 10−5) and ‘Intracellular/Stress Activated Protein Kinase Cascade’ (P = 7.18 × 10−14), as well as more general biological processes such as ‘Signal Transduction’ (P = 2.00 × 10−8), ‘Response to Protein Stimulus’ (P = 0.008) and ‘Cell Differentiation’ (P = 7.18 × 10−14). The over-representation of genes involved in G protein–coupled receptor (GPCR) signaling is particularly interesting because GPCRs mediate the cAMP signaling pathway, which is essential for decidualization and the establishment of pregnancy10. These results suggest that recruitment of the cAMP signaling pathway into endometrial cells was likely a key innovation during the origin of pregnancy. Indeed, 54.89% (841/1,532) of recruited genes but only 37.06% (6,504/17,550) of ancestrally expressed genes were differentially regulated upon progesterone/cAMP stimulation in human ESCs (P = 5.2 × 10−50, hypergeometric test).
Although numerous progesterone/cAMP-responsive genes are expressed in human ESCs, one of the most dramatically induced is prolactin (PRL). Notably, the progesterone/cAMP-responsive enhancer of PRL in ESCs is derived from a hAT-Charlie family DNA transposon (MER20) found only in placental mammals12, suggesting MER20s have played a role in rewiring the gene regulatory landscape of ESCs. To determine if other progesterone/cAMP-responsive genes are associated with MER20s, we searched upstream, downstream and within the coding regions and introns of differentially regulated
Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammalsVincent J Lynch, Robert D Leclerc, Gemma May & Günter P Wagner
Department of Ecology and Evolutionary Biology & Yale Systems Biology Institute, Yale University, New Haven, Connecticut, USA. Correspondence should be addressed to V.J.L. ([email protected]).
Received 4 November 2010; accepted 1 August 2011; published online 25 September 2011; doi:10.1038/ng.917
©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
2 ADVANCE ONLINE PUBLICATION Nature GeNetics
l e t t e r s
human genes for MER20s. Notably, we found that 42% (6,949/16,562) of MER20s were located within 200 kb of the transcriptional start and end sites of the 6,504 differentially regulated genes, whereas only 8% (4,834/60,299) of MER20s were found in the same window
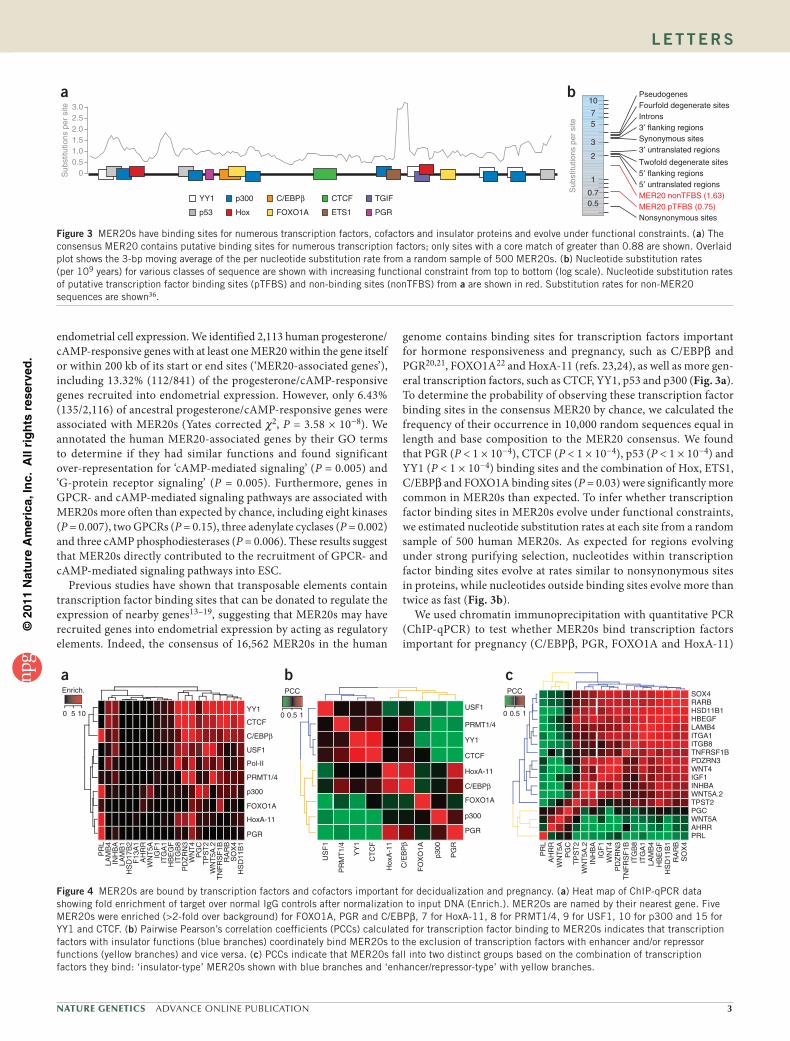
around genes not differentially regulated upon decidualization (Yates corrected χ2, P = 1 × 10−4). MER20s are also located closer to differ-entially regulated genes than expected given a random distribution, when compared to either genes that are not differentially regulated (Fig. 2a and Supplementary Fig. 1) or to other Eutherian-specific hAT Charlie transposons (Supplementary Fig. 2).
To assess the potential of MER20s to act as regulatory elements for genes other than PRL, we examined MER20s found within 200 kb of stromally regulated genes for characteristics of regulatory elements, including conservation, predicted regulatory potential, CpG island density and association with various histone modifications. As expected for regulatory elements, we found MER20s had high PhastCons scores and 7× regulatory potential and were surrounded by regions of high CpG island density (Fig. 2b and Supplementary Fig. 3). MER20s were also associated with histone modifications commonly found for insulators (high acetylation of histone H2 Lys5 (H2AK5ac) and CTCF), enhancers (high mono- and dimethylation (H3K4me1 and H3K4me2) and low trimethylation (H3K4me3) of histone H3 Lys4) and repressors (high H3K27me1, H3K27me2 and H3K27me3, low H3K27ac), although few MER20s had epigenetic marks of more than one type of regulatory element (Fig. 2b,c).
Next, we asked whether MER20s were preferentially associated with the progesterone/cAMP-responsive genes that were recruited into
Human
Armadillo
200
1,320
199
1,532
3,349
1,232
77
Opossum
HumanOpossum
a bArmadilloMonotremes
Birds andreptiles
105 Mya
150 Mya170 Mya
310 Mya
Figure 1 Evolution of the endometrial stromal cell transcriptome in Therian mammals. (a) Amniote phylogeny showing approximate divergence dates between major lineages; opossum, armadillo and human samples were included in this study. Placental mammals are indicated in red. (b) Venn diagram showing the intersection of 1:1:1 homologous genes expressed in endometrial cells of human, armadillo and opossum inferred from RNA-Seq. In total, 1,532 genes were scored as expressed in both human and armadillo but not opossum.
20010 20 30 40 50 60 70 80 90 10
011
012
013
014
015
016
017
018
019
0–1
0–2
00–1
90–1
80–1
70–1
60–1
50–1
40–1
30–1
20–1
10–1
00 –90
–80
–70
–60
–50
–40
–30
–20 0
Num
ber
of g
enes
and
ele
men
ts
0
20
40
60
80
100
341a
b cDistance (kb)
‘Repressor’
767
200
‘Enhancer’
669
‘Insulator’
6642
20
Distance (bp)
Nor
mal
ized
Cou
nt
3e-4
4e-4
2e-4
2.5e-4
3.5e-4
1.5e-4
2.5e-4
3.5e-4
1.5e-4
6e-5
8e-5
4e-5
H3K4me1/me2/me3 H3K27me1/me2/me3/acCTCF/H2Ak5acCpG/PhastCons/7×RP
3
–30
kb
–20
kb
–10
kb 030
kb20
kb10
kb
–3,0
00
–2,0
00
–1,0
00 03,
000
2,00
01,
000
–3,0
00
–2,0
00
–1,0
00 03,
000
2,00
01,
000
–3,0
00
–2,0
00
–1,0
00 03,
000
2,00
01,
000
Figure 2 MER20s are over-represented near progesterone/cAMP-responsive endometrial genes and have genomic and epigenetic signatures of regulatory elements. (a) Distribution of distances from differentially regulated stromal genes (N = 6,504) to MER20s in 5-kb bins. Gray bars indicate the total number of MER20s in each bin, and brown bars indicate the distance of the closest MER20 to the gene. The number of genes with MER20s located between transcriptional start and end sites is indicated by 0. The expected number of MER20-associated genes per bin given random positions in the human genome (black line) and compared to genes that were not differentially regulated upon decidualization (blue line) are shown for the location of the closest MER20 to stromally regulated genes (mean ± s.d.). (b) MER20s are located in regions of the genome with high CpG island density, PhastCons scores and 7× regulatory potential (RP). The profile of histone modifications around MER20s located within 200 kb of genes either up- or downregulated upon differentiation of human ESCs is shown for several methylation and acetylation events and for the vertebrate insulator protein CTCF. Panel names are colored with respect to the profile shown below. MER20s are centered at position 0 (red box), with normalized ChIP-Seq tag density in 5 bp windows upstream and downstream of the MER20 shown as lines. (c) Venn diagram showing intersections among MER20s classified by histone modifications as repressors, insulators or enhancers.
©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Nature GeNetics ADVANCE ONLINE PUBLICATION 3
l e t t e r s
endometrial cell expression. We identified 2,113 human progesterone/cAMP-responsive genes with at least one MER20 within the gene itself or within 200 kb of its start or end sites (‘MER20-associated genes’), including 13.32% (112/841) of the progesterone/cAMP-responsive genes recruited into endometrial expression. However, only 6.43% (135/2,116) of ancestral progesterone/cAMP-responsive genes were associated with MER20s (Yates corrected χ2, P = 3.58 × 10−8). We annotated the human MER20-associated genes by their GO terms to determine if they had similar functions and found significant over-representation for ‘cAMP-mediated signaling’ (P = 0.005) and ‘G-protein receptor signaling’ (P = 0.005). Furthermore, genes in GPCR- and cAMP-mediated signaling pathways are associated with MER20s more often than expected by chance, including eight kinases (P = 0.007), two GPCRs (P = 0.15), three adenylate cyclases (P = 0.002) and three cAMP phosphodiesterases (P = 0.006). These results suggest that MER20s directly contributed to the recruitment of GPCR- and cAMP-mediated signaling pathways into ESC.
Previous studies have shown that transposable elements contain transcription factor binding sites that can be donated to regulate the expression of nearby genes13–19, suggesting that MER20s may have recruited genes into endometrial expression by acting as regulatory elements. Indeed, the consensus of 16,562 MER20s in the human
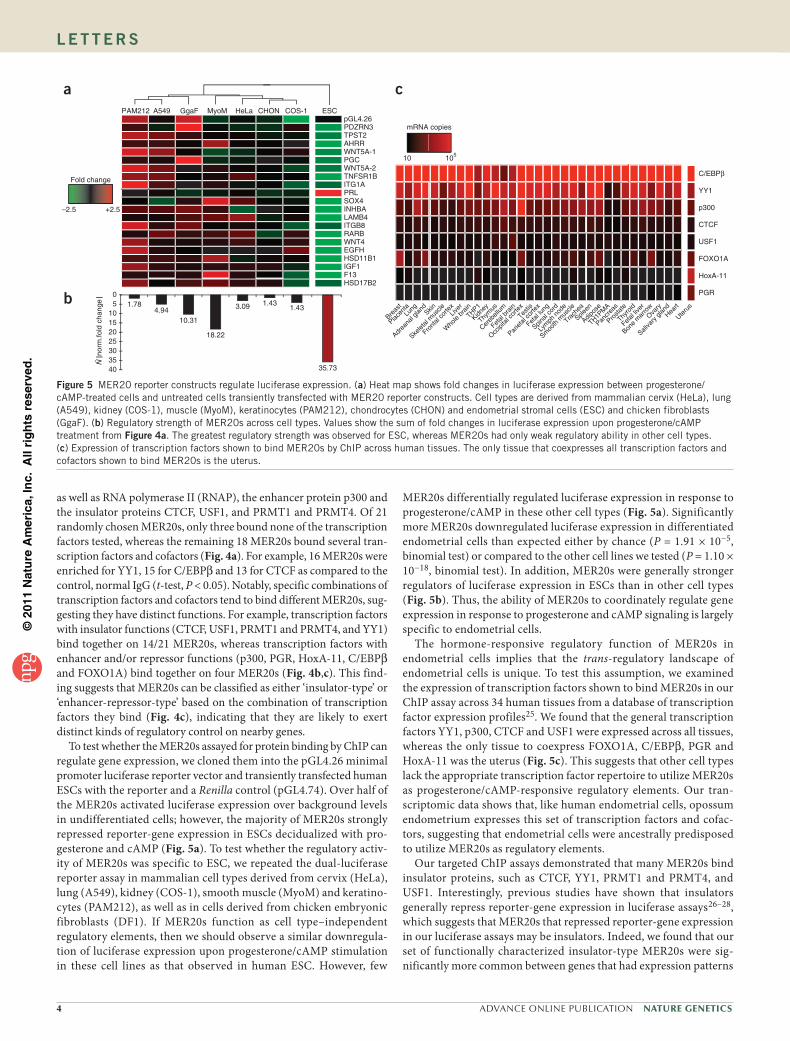
genome contains binding sites for transcription factors important for hormone responsiveness and pregnancy, such as C/EBPβ and PGR20,21, FOXO1A22 and HoxA-11 (refs. 23,24), as well as more gen-eral transcription factors, such as CTCF, YY1, p53 and p300 (Fig. 3a). To determine the probability of observing these transcription factor binding sites in the consensus MER20 by chance, we calculated the frequency of their occurrence in 10,000 random sequences equal in length and base composition to the MER20 consensus. We found that PGR (P < 1 × 10−4), CTCF (P < 1 × 10−4), p53 (P < 1 × 10−4) and YY1 (P < 1 × 10−4) binding sites and the combination of Hox, ETS1, C/EBPβ and FOXO1A binding sites (P = 0.03) were significantly more common in MER20s than expected. To infer whether transcription factor binding sites in MER20s evolve under functional constraints, we estimated nucleotide substitution rates at each site from a random sample of 500 human MER20s. As expected for regions evolving under strong purifying selection, nucleotides within transcription factor binding sites evolve at rates similar to nonsynonymous sites in proteins, while nucleotides outside binding sites evolve more than twice as fast (Fig. 3b).
We used chromatin immunoprecipitation with quantitative PCR (ChIP-qPCR) to test whether MER20s bind transcription factors important for pregnancy (C/EBPβ, PGR, FOXO1A and HoxA-11)
YY1
p53
p300
Hox
C/EBPβ
FOXO1A
CTCF
ETS1
TGIF
3.0
a b2.52.01.51.00.5
0Sub
stitu
tions
per
site
PGR
10
75
3
2
1
0.70.5
PseudogenesFourfold degenerate sitesIntrons3′ flanking regionsSynonymous sites3′ untranslated regions
Twofold degenerate sites5′ flanking regions5′ untranslated regions
Nonsynonymous sites
MER20 nonTFBS (1.63)MER20 pTFBS (0.75)
Sub
stitu
tions
per
site
Figure 3 MER20s have binding sites for numerous transcription factors, cofactors and insulator proteins and evolve under functional constraints. (a) The consensus MER20 contains putative binding sites for numerous transcription factors; only sites with a core match of greater than 0.88 are shown. Overlaid plot shows the 3-bp moving average of the per nucleotide substitution rate from a random sample of 500 MER20s. (b) Nucleotide substitution rates (per 109 years) for various classes of sequence are shown with increasing functional constraint from top to bottom (log scale). Nucleotide substitution rates of putative transcription factor binding sites (pTFBS) and non-binding sites (nonTFBS) from a are shown in red. Substitution rates for non-MER20 sequences are shown36.
a
HoxA-11
PGR
PR
LLA
MB
4
LAM
B1
HS
D17
B2
F13
A1
AH
RR
WN
T5A
IGF
1IT
GA
1
ITG
B8
PD
ZR
N3
WN
T4
PG
CT
PS
T2
WN
T5A
.2T
NF
RS
F1B
RA
RB
SO
X4
HS
D11
B1
HB
EG
F
INH
BA
PR
L
LAM
B4
AH
RR
WN
T5A
IGF
1
ITG
A1
ITG
B8
PD
ZR
N3
WN
T4
PG
CT
PS
T2
WN
T5A
.2
TN
FR
SF
1B
RA
RB
SO
X4
HS
D11
B1
HB
EG
F
INH
BA
FOXO1A
p300
PRMT1/4
Pol-II
USF1
C/EBPβ
CTCF
YY10 5 10
Enrich.
0.5 10
cPCC
USF1
PRMT1/4
YY1
CTCF
HoxA-11
C/EBPβ
FOXO1A
p300
PGR
US
F1
PR
MT
1/4
YY
1
CT
CF
Hox
A-1
1
C/E
BP
β
FO
XO
1A
p300
PG
R
0.5 10
bPCC SOX4
RARBHSD11B1HBEGFLAMB4ITGA1ITGB8TNFRSF1BPDZRN3WNT4IGF1INHBAWNT5A.2TPST2PGCWNT5AAHRRPRL
Figure 4 MER20s are bound by transcription factors and cofactors important for decidualization and pregnancy. (a) Heat map of ChIP-qPCR data showing fold enrichment of target over normal IgG controls after normalization to input DNA (Enrich.). MER20s are named by their nearest gene. Five MER20s were enriched (>2-fold over background) for FOXO1A, PGR and C/EBPβ, 7 for HoxA-11, 8 for PRMT1/4, 9 for USF1, 10 for p300 and 15 for YY1 and CTCF. (b) Pairwise Pearson’s correlation coefficients (PCCs) calculated for transcription factor binding to MER20s indicates that transcription factors with insulator functions (blue branches) coordinately bind MER20s to the exclusion of transcription factors with enhancer and/or repressor functions (yellow branches) and vice versa. (c) PCCs indicate that MER20s fall into two distinct groups based on the combination of transcription factors they bind: ‘insulator-type’ MER20s shown with blue branches and ‘enhancer/repressor-type’ with yellow branches.
©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
4 ADVANCE ONLINE PUBLICATION Nature GeNetics
l e t t e r s
as well as RNA polymerase II (RNAP), the enhancer protein p300 and the insulator proteins CTCF, USF1, and PRMT1 and PRMT4. Of 21 randomly chosen MER20s, only three bound none of the transcription factors tested, whereas the remaining 18 MER20s bound several tran-scription factors and cofactors (Fig. 4a). For example, 16 MER20s were enriched for YY1, 15 for C/EBPβ and 13 for CTCF as compared to the control, normal IgG (t-test, P < 0.05). Notably, specific combinations of transcription factors and cofactors tend to bind different MER20s, sug-gesting they have distinct functions. For example, transcription factors with insulator functions (CTCF, USF1, PRMT1 and PRMT4, and YY1) bind together on 14/21 MER20s, whereas transcription factors with enhancer and/or repressor functions (p300, PGR, HoxA-11, C/EBPβ and FOXO1A) bind together on four MER20s (Fig. 4b,c). This find-ing suggests that MER20s can be classified as either ‘insulator-type’ or ‘enhancer-repressor-type’ based on the combination of transcription factors they bind (Fig. 4c), indicating that they are likely to exert distinct kinds of regulatory control on nearby genes.
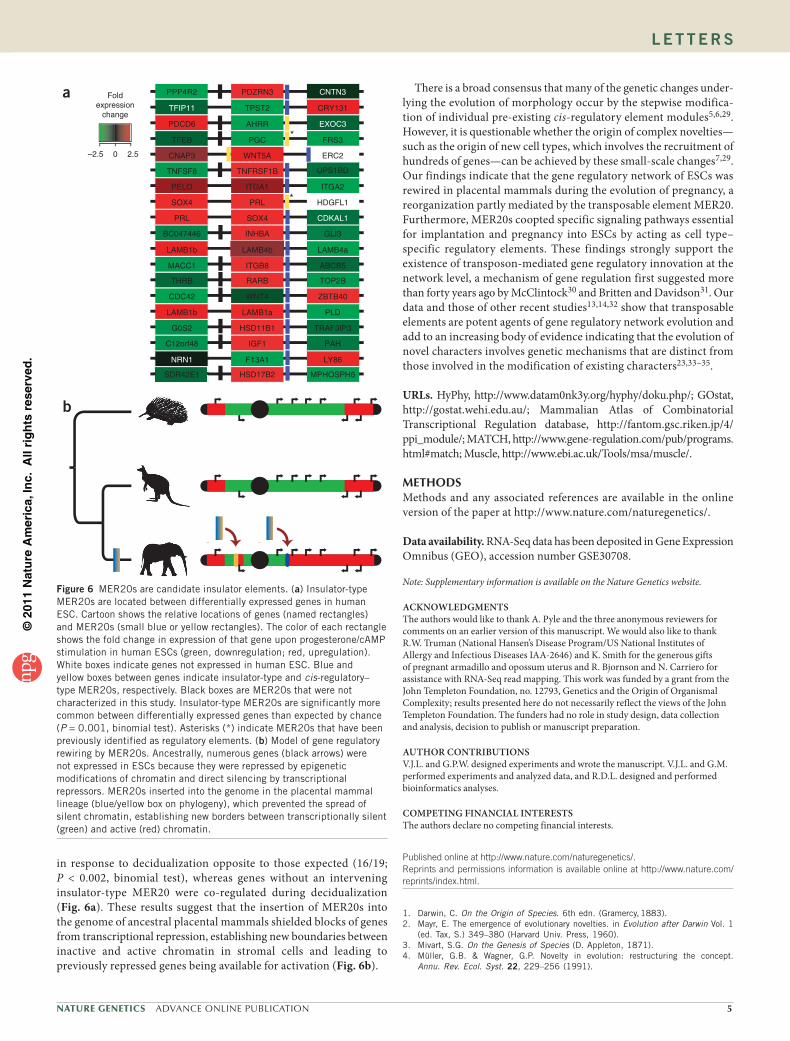
To test whether the MER20s assayed for protein binding by ChIP can regulate gene expression, we cloned them into the pGL4.26 minimal promoter luciferase reporter vector and transiently transfected human ESCs with the reporter and a Renilla control (pGL4.74). Over half of the MER20s activated luciferase expression over background levels in undifferentiated cells; however, the majority of MER20s strongly repressed reporter-gene expression in ESCs decidualized with pro-gesterone and cAMP (Fig. 5a). To test whether the regulatory activ-ity of MER20s was specific to ESC, we repeated the dual-luciferase reporter assay in mammalian cell types derived from cervix (HeLa), lung (A549), kidney (COS-1), smooth muscle (MyoM) and keratino-cytes (PAM212), as well as in cells derived from chicken embryonic fibroblasts (DF1). If MER20s function as cell type–independent regulatory elements, then we should observe a similar downregula-tion of luciferase expression upon progesterone/cAMP stimulation in these cell lines as that observed in human ESC. However, few
MER20s differentially regulated luciferase expression in response to progesterone/cAMP in these other cell types (Fig. 5a). Significantly more MER20s downregulated luciferase expression in differentiated endometrial cells than expected either by chance (P = 1.91 × 10−5, binomial test) or compared to the other cell lines we tested (P = 1.10 × 10−18, binomial test). In addition, MER20s were generally stronger regulators of luciferase expression in ESCs than in other cell types (Fig. 5b). Thus, the ability of MER20s to coordinately regulate gene expression in response to progesterone and cAMP signaling is largely specific to endometrial cells.
The hormone-responsive regulatory function of MER20s in endometrial cells implies that the trans-regulatory landscape of endometrial cells is unique. To test this assumption, we examined the expression of transcription factors shown to bind MER20s in our ChIP assay across 34 human tissues from a database of transcription factor expression profiles25. We found that the general transcription factors YY1, p300, CTCF and USF1 were expressed across all tissues, whereas the only tissue to coexpress FOXO1A, C/EBPβ, PGR and HoxA-11 was the uterus (Fig. 5c). This suggests that other cell types lack the appropriate transcription factor repertoire to utilize MER20s as progesterone/cAMP-responsive regulatory elements. Our tran-scriptomic data shows that, like human endometrial cells, opossum endometrium expresses this set of transcription factors and cofac-tors, suggesting that endometrial cells were ancestrally predisposed to utilize MER20s as regulatory elements.
Our targeted ChIP assays demonstrated that many MER20s bind insulator proteins, such as CTCF, YY1, PRMT1 and PRMT4, and USF1. Interestingly, previous studies have shown that insulators generally repress reporter-gene expression in luciferase assays26–28, which suggests that MER20s that repressed reporter-gene expression in our luciferase assays may be insulators. Indeed, we found that our set of functionally characterized insulator-type MER20s were sig-nificantly more common between genes that had expression patterns
C/EBPβ
YY1
p300
CTCF
USF1
FOXO1A
HoxA-11
PGR
10 105
mRNA copies
b
ESCCOS-1CHONHeLaMyoMGgaFA549PAM212
–2.5 +2.5
Fold change
a
4035302520151050
1.784.94
10.31
18.22
3.09 1.431.43
35.73
c
Breas
t
Placen
taLu
ng
Adrea
nal g
landSkin
Skelet
al m
uscle
Front
al co
rtexLiv
er
Who
le br
ainTHP1
Kidney
Thym
us
Cereb
ellum
Fetal
brain
Occipi
tal c
orte
x
Testis
Pariet
al co
rtex
Fetal
lung
Spinal
cord
Lym
ph n
ode
Smoo
th m
uscle
Trach
ea
Spleen
Adipos
e
TH1PM
A
Pancr
eas
Prosta
te
Thyro
id
Fetal
liver
Bone
mar
rowOva
ry
Saliva
ry g
landHea
rt
Uteru
s
pGL4.26PDZRN3TPST2AHRRWNT5A-1PGCWNT5A-2TNFSR1BITG1APRLSOX4INHBALAMB4ITGB8RARBWNT4EGFHHSD11B1IGF1F13HSD17B2
Ñ n
orm
.fold
cha
nge
Figure 5 MER20 reporter constructs regulate luciferase expression. (a) Heat map shows fold changes in luciferase expression between progesterone/cAMP-treated cells and untreated cells transiently transfected with MER20 reporter constructs. Cell types are derived from mammalian cervix (HeLa), lung (A549), kidney (COS-1), muscle (MyoM), keratinocytes (PAM212), chondrocytes (CHON) and endometrial stromal cells (ESC) and chicken fibroblasts (GgaF). (b) Regulatory strength of MER20s across cell types. Values show the sum of fold changes in luciferase expression upon progesterone/cAMP treatment from Figure 4a. The greatest regulatory strength was observed for ESC, whereas MER20s had only weak regulatory ability in other cell types. (c) Expression of transcription factors shown to bind MER20s by ChIP across human tissues. The only tissue that coexpresses all transcription factors and cofactors shown to bind MER20s is the uterus.
©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Nature GeNetics ADVANCE ONLINE PUBLICATION 5
l e t t e r s
in response to decidualization opposite to those expected (16/19; P < 0.002, binomial test), whereas genes without an intervening insulator-type MER20 were co-regulated during decidualization (Fig. 6a). These results suggest that the insertion of MER20s into the genome of ancestral placental mammals shielded blocks of genes from transcriptional repression, establishing new boundaries between inactive and active chromatin in stromal cells and leading to previously repressed genes being available for activation (Fig. 6b).
There is a broad consensus that many of the genetic changes under-lying the evolution of morphology occur by the stepwise modifica-tion of individual pre-existing cis-regulatory element modules5,6,29. However, it is questionable whether the origin of complex novelties—such as the origin of new cell types, which involves the recruitment of hundreds of genes—can be achieved by these small-scale changes7,29. Our findings indicate that the gene regulatory network of ESCs was rewired in placental mammals during the evolution of pregnancy, a reorganization partly mediated by the transposable element MER20. Furthermore, MER20s coopted specific signaling pathways essential for implantation and pregnancy into ESCs by acting as cell type–specific regulatory elements. These findings strongly support the existence of transposon-mediated gene regulatory innovation at the network level, a mechanism of gene regulation first suggested more than forty years ago by McClintock30 and Britten and Davidson31. Our data and those of other recent studies13,14,32 show that transposable elements are potent agents of gene regulatory network evolution and add to an increasing body of evidence indicating that the evolution of novel characters involves genetic mechanisms that are distinct from those involved in the modification of existing characters23,33–35.
URLs. HyPhy, http://www.datam0nk3y.org/hyphy/doku.php/; GOstat, http://gostat.wehi.edu.au/; Mammalian Atlas of Combinatorial Transcriptional Regulation database, http://fantom.gsc.riken.jp/4/ ppi_module/; MATCH, http://www.gene-regulation.com/pub/programs.html#match; Muscle, http://www.ebi.ac.uk/Tools/msa/muscle/.
METhodSMethods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Data availability. RNA-Seq data has been deposited in Gene Expression Omnibus (GEO), accession number GSE30708.
Note: Supplementary information is available on the Nature Genetics website.
AcknoWLeDGMentsThe authors would like to thank A. Pyle and the three anonymous reviewers for comments on an earlier version of this manuscript. We would also like to thank R.W. Truman (National Hansen’s Disease Program/US National Institutes of Allergy and Infectious Diseases IAA-2646) and K. Smith for the generous gifts of pregnant armadillo and opossum uterus and R. Bjornson and N. Carriero for assistance with RNA-Seq read mapping. This work was funded by a grant from the John Templeton Foundation, no. 12793, Genetics and the Origin of Organismal Complexity; results presented here do not necessarily reflect the views of the John Templeton Foundation. The funders had no role in study design, data collection and analysis, decision to publish or manuscript preparation.
AuthoR contRibutionsV.J.L. and G.P.W. designed experiments and wrote the manuscript. V.J.L. and G.M. performed experiments and analyzed data, and R.D.L. designed and performed bioinformatics analyses.
coMPetinG FinAnciAL inteRestsThe authors declare no competing financial interests.
Published online at http://www.nature.com/naturegenetics/. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
1. Darwin, C. On the Origin of Species. 6th edn. (Gramercy,1883).2. Mayr, E. The emergence of evolutionary novelties. in Evolution after Darwin Vol. 1
(ed. Tax, S.) 349–380 (Harvard Univ. Press, 1960).3. Mivart, S.G. On the Genesis of Species (D. Appleton, 1871).4. Müller, G.B. & Wagner, G.P. Novelty in evolution: restructuring the concept.
Annu. Rev. Ecol. Syst. 22, 229–256 (1991).
a
b
*
PDZRN3
TPST2
AHRR
PGC
WNT5A
TNFRSF1B
ITGA1
PRL
SOX4
INHBA
LAMB4b
ITGB8
RARB
WNT4
LAMB1a
HSD11B1
IGF1
F13A1
HSD17B2
BC047446 GLI3
CDKAL1
SOX4
PRL
HDGFL1
PPP4R2 CNTN3
TFIP11 CRY131
PDCD6 EXOC3
TFEB FRS3
CNAP3 ERC2
TNFSF8 UPS1BD
PELO ITGA2
LAMB1b
MACC1
LAMB4a
LAMB1b
ABCB5
THRB TOP2B
CDC42 ZBTB40
PLD
G0S2 TRAF3IP3
PAHC12orf48
NRN1 LY86
MPHOSPH6SDR42E1
0 2.5–2.5
Foldexpression
change
*
Figure 6 MER20s are candidate insulator elements. (a) Insulator-type MER20s are located between differentially expressed genes in human ESC. Cartoon shows the relative locations of genes (named rectangles) and MER20s (small blue or yellow rectangles). The color of each rectangle shows the fold change in expression of that gene upon progesterone/cAMP stimulation in human ESCs (green, downregulation; red, upregulation). White boxes indicate genes not expressed in human ESC. Blue and yellow boxes between genes indicate insulator-type and cis-regulatory–type MER20s, respectively. Black boxes are MER20s that were not characterized in this study. Insulator-type MER20s are significantly more common between differentially expressed genes than expected by chance (P = 0.001, binomial test). Asterisks (*) indicate MER20s that have been previously identified as regulatory elements. (b) Model of gene regulatory rewiring by MER20s. Ancestrally, numerous genes (black arrows) were not expressed in ESCs because they were repressed by epigenetic modifications of chromatin and direct silencing by transcriptional repressors. MER20s inserted into the genome in the placental mammal lineage (blue/yellow box on phylogeny), which prevented the spread of silent chromatin, establishing new borders between transcriptionally silent (green) and active (red) chromatin.

©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
� ADVANCE ONLINE PUBLICATION Nature GeNetics
l e t t e r s
5. Prud’homme, B., Gompel, N. & Carroll, S.B. Emerging principles of regulatory evolution. Proc. Natl. Acad. Sci. USA 104, 8605–8612 (2007).
6. Carroll, S.B. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134, 25–36 (2008).
7. Wagner, G.P. & Lynch, V.J. Molecular evolution of evolutionary novelties: the vagina and uterus of therian mammals. J. Exp. Zool. B Mol. Dev. Evol. 304, 580–592 (2005).
8. Mess, A. & Carter, A.M. Evolutionary transformations of fetal membrane characters in Eutheria with special reference to Afrotheria. J. Exp. Zool. B Mol. Dev. Evol. 306, 140–163 (2006).
9. Wildman, D.E. et al. Evolution of the mammalian placenta revealed by phylogenetic analysis. Proc. Natl. Acad. Sci. USA 103, 3203–3208 (2006).
10. Gellersen, B. & Brosens, J. Cyclic AMP and progesterone receptor cross-talk in endometrium: a decidualizing affair. J. Endocrinol. 178, 357–372 (2003).
11. Gellersen, B., Brosens, I.M.D. & Brosens, J.M.D. Decidualization of the human endometrium: mechanisms, functions, and clinical perspectives. Semin. Reprod. Med. 25, 445–453 (2007).
12. Gerlo, S., Davis, J.R., Mager, D.L. & Kooijman, R. Prolactin in man: a tale of two promoters. Bioessays 28, 1051–1055 (2006).
13. Bourque, G. et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 18, 1752–1762 (2008).
14. Sasaki, T. et al. Possible involvement of SINEs in mammalian-specific brain formation. Proc. Natl. Acad. Sci. USA 105, 4220–4225 (2008).
15. Kunarso, G. et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 42, 631–634 (2010).
16. Bejerano, G. et al. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 441, 87–90 (2006).
17. Jordan, I.K., Rogozin, I.B., Glazko, G.V. & Koonin, E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 19, 68–72 (2003).
18. van de Lagemaat, L.N., Landry, J.-R., Mager, D.L. & Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 19, 530–536 (2003).
19. Thornburg, B.G., Gotea, V. & Makalowski, W. Transposable elements as a significant source of transcription regulating signals. Gene 365, 104–110 (2006).
20. Christian, M. et al. Cyclic AMP-induced forkhead transcription factor, FKHR, cooperates with CCAAT/enhancer-binding protein beta in differentiating human endometrial stromal cells. J. Biol. Chem. 277, 20825–20832 (2002).
21. Mantena, S.R. et al. C/EEBP-beta is a critical mediator of steroid hormone-regulated cell proliferation and differentiation in the unterine epithelium and stroma. Proc. Natl. Acad. Sci. USA 103, 1870–1875 (2006).
22. Buzzio, O.L., Lu, Z., Miller, C.D., Unterman, T.G. & Kim, J.J. FOXO1A differentially regulates genes of decidualization. Endocrinology 147, 3870–3876 (2006).
23. Lynch, V.J. et al. Adaptive changes in the transcription factor HoxA-11 are essential for the evolution of pregnancy in mammals. Proc. Natl. Acad. Sci. USA 105, 14928–14933 (2008).
24. Hsieh-Li, H.M. et al. Hoxa 11 structure, extensive antisense transcription, and function in male and female fertility. Development 121, 1373–1385 (1995).
25. Ravasi, T. et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140, 744–752 (2010).
26. Wei, W. & Brennan, M.D. The gypsy insulator can act as a promoter-specific transcriptional stimulator. Mol. Cell. Biol. 21, 7714–7720 (2001).
27. Abhyankar, M.M., Urekar, C. & Reddi, P.P. A novel CpG-free vertebrate insulator ilences the testis-specific SP-10 gene in somatic tissues. J. Biol. Chem. 282, 36143–36154 (2007).
28. Kim, J., Kollhoff, A., Bergmann, A. & Stubbs, L. Methylation-sensitive binding of transcription factor YY1 to an insulator sequence within the paternally expressed imprinted gene, Peg3. Hum. Mol. Genet. 12, 233–245 (2003).
29. Carroll, S.B. Evolution at two levels: on genes and form. PLoS Biol. 3, e245 (2005).
30. McClintock, B. Components of action of the regulators Spm and Ac. Year B. Carnegie Inst. Wash. 64, 527–536 (1965).
31. Britten, R.J. & Davidson, E.H. Gene regulation for higher cells: a theory. Science 165, 349–357 (1969).
32. Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 9, 397–405 (2008).
33. Adamska, M. et al. The evolutionary origin of hedgehog proteins. Curr. Biol. 17, R836–R837 (2007).
34. Wagner, G.P. & Lynch, V.J. Evolutionary novelties. Curr. Biol. 20, R48–R52 (2010).
35. Oliver, K.R. & Greene, W.K. Transposable elements: powerful facilitators of evolution. Bioessays 31, 703–714 (2009).
36. Harti, D. Essential Genetics: A Genomics Perspective (Jones and Bartlett Publishers, 2010).

©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Nature GeNeticsdoi:10.1038/ng.917
oNLINE METhodSTranscriptome sequencing. Endometrial samples from mid-stage pregnant opossum and armadillo were dissected from freshly killed females to remove myometrial and placental tissue and washed in ice-cold PBS to remove blood cells; tissues were stored in RNA-Later at −80 °C until processing. Endometrial samples were isolated from whole uteri of armadillo, because they cannot be bred in captivity and tissue culture methods are not available for either armadillo or opossum stromal cells. Samples of differentiated and undiffer-entiated human endometrial stromal cells were cultured and differentiated as described below. We extracted total RNA using the Qiagen RNA-Easy Midi RNA-extraction kit followed by on-column DNase treatment (Qiagen). Total RNA quality was assayed with a Bioanalyzer 2100 (Agilent) and found to be of excellent quality. Aliquots from the total RNA samples were sequenced using the Illumina Genome Analyzer II platform by following the protocol suggested by Illumina for sequencing of cDNA samples. Two biological replicates each were sequenced for the human undifferentiated and differentiated endometrial stromal cells, and two samples dissected from different locations in the uteri of armadillo and opossum were sequenced.
Sequence analysis was performed with Bowtie, and reads were mapped to the human (GRCh37), armadillo (dasNov2) and opossum (monDom5) cDNA builds at Ensembl; two mismatches were allowed, and reads aligning to more than one cDNA were disregarded. Sequencing was performed at the W.M. Keck Microarray at the Yale University Medical School. The average read count from the two lanes of data was used for comparative transcriptome analysis.
Preliminary analysis indicated that most variability in read counts between the two replicate samples occurred for genes with under 20 reads. Therefore, subsequent analyses were based on genes with read counts greater than 20 reads. However, including all genes with reads >1 did not change our results. Differentially regulated genes were defined as those that were up- or downreg-ulated more than twofold in differentiated relative to undifferentiated human endometrial stromal cells.
We identified 1:1:1 human:armadillo:opossum orthologs from the human, armadillo and opossum cDNA builds at Ensembl using BioMart. We annotated the 1,532 derived Eutherian ESC-expressed genes by their over-represented Gene Ontology (GO) terms using GOstat with the goa_human database, a minimal path length of 3, Benjamini correction for the false discovery rate and merging GOs if their associated gene lists were inclusions or differed by less than ten genes. The background set of genes were all those found in the goa_human database.
Identification of putative transcription factor binding sites in MER20 and molecular evolution of MER20s. Potential transcription factor binding sites in the human consensus MER20 were identified using the MATCH program (see URLs) with TRANSFAC binding site matrices, with a match cut-off selected to minimize the sum of false positive and false negative results. Only binding site matches with >88% identity to the core binding site motif in the MER20 consensus are reported here.
To estimate the evolutionary rate of substitutions in MER20s, we down-loaded all MER20s from the human genome and randomly sampled 500. These 500 human MER20s were aligned with Muscle (see URLs), and alignment columns with more than 51% gapped sequences (gaps occurred outside most known or predicted binding sites and tended to occur more frequently at the 5′ and 3′ends of the sequences) were removed. The gapped trimmed sequence alignment was used to estimate site-specific substitution rates using the HyPhy batch program, siterates.bf, which implements maximum-likelihood esti-mating of substitution rates and a phylogenetic tree constructed for the 500 MER20s using PhyML under a GTR+Γ model with four gamma classes.
Cell culture. Human endometrial stromal cells immortalized with human telomerase (ATCC, cat. no. CRL-4003), HeLa, A549, COS-1, MyoM, PAM212 and chicken fibroblasts were grown in DMEM supplemented with 5% charcoal-stripped calf serum (Hyclone) and 1% antibiotic/antimycotic (ABAM). To induce decidualization, cells were treated with 0.5 mM 8-Br-cAMP (Sigma) and 1 µM of the progesterone analog,medroxyprogesterone acetate (MPA; Sigma) for 48 h. At 80% confluency, cells were collected for gene expres-sion analysis, transfected for luciferase assays using TransIT-LT1 (Mirus) according to the manufacturer′s protocol or harvested for ChIP assays.
Identification of MER20s in the human genome. We mapped the distribution of MER20s in the human genome (GRCh37) using the Repeatmasker track of the UCSC genome browser and identified 16,562 MER20s. We analyzed the distribution of distances between MER20s and differentially regulated stromal genes to determine whether MER20s were randomly distributed with respect to stromal genes or whether they were preferentially located within some distance [1,d] from the start and end sites or within (d = 0) differentially regulated genes. To generate a null distribution for the association of MER20s with stromal genes, we generated random positions in the human genome, equal in number to the set of genes scored as ‘MER20-associated’ (N = 2,113) and evaluated the distance from that position to the nearest upstream or down-stream MER20. This procedure was replicated 500 times (Fig. 2b, black line). To determine the expected random distribution and error of the background distance of MER20s to genes in the human genome, we sampled 2,113 genes that were not differentially regulated by MPA and cAMP stimulation and evaluated the distance to their nearest upstream or downstream MER20. This procedure was replicated 500 times (Fig. 2b, blue line).
Epigenetic and genomic profile of MER20s. We examined the epigenetic status of MER20s associated with stromal genes by using recent genome-wide ChIP-Seq data for 37 histone modifications, together with the histone variant H2A.Z and the insulator protein CTCF37,38. To correlate histone modifica-tions with MER20s, we counted ChIP-Seq tag density in 5-bp windows 10 kb up- and downstream of ~6,000 MER20s located within 200 kb of differentially regulated ESC genes. Note that position “0” on the x axis of Figure 2a cor-responds to the midpoint of each MER20 element.
We also annotated MER20s and the genomic region immediately around MER20s according to their CpG island density, PhastCons scores and 7× regu-latory potential by counting CpG island density, PhastCons scores and 7× regu-latory potential scores in 5-bp windows 10 kb up- and downstream of MER20s located within 200 kb of differentially regulated ESC genes; CpG island density, PhastCons scores and 7× regulatory potential data were downloaded from the UCSC genome browser and followed the definitions found there.
Chromatin immunoprecipitation and luciferase reporter assays. For chromatin immunoprecipitation (ChIP) assays, the EZ-Zyme Chromatin Prep kit (Millipore) was used following the manufacturer′s protocol. Briefly, chromatin was cross-linked with 1% formaldehyde for 10 min; this was followed by quenching with glycine and DNA fragmentation. The equivalent of 106 cells was used for each immunoprecipitation. The nuclear lysate was precleared for 1 h with protein G magnetic beads and incubated overnight at 4 °C with protein G–linked magnetic beads and 2 µg of either ChIP validated antibodies to p300, FOXO1A, PGR, YY1, HoxA-11, C/EBPβ, CTCF, USF1 or PRMT1 and PRMT4, or species-appropriate IgG as negative control (all from Santa Cruz Biotechnology). Enrichment of the MER20 targets was evaluated by qPCR using 1/50 of the immunoprecipitated chromatin as template and the Power SYBR Green PCR Master Mix (Applied Biosystems). We randomly selected 21 MER20s that span the range of distances from their associated genes (from –1 kb downstream of an end site to nearly 200 kb upstream of the start site) to test by ChIP.
The MER20s characterized by ChIP were cloned into the pGL4.26 luci-ferase reporter vector (Promega). pGL4.26 luciferase reporter constructs (100 ng) and the pGL4.74 Renilla luciferase control (20 ng) were transiently transfected into undifferentiated and differentiated ESCs, and luciferase expression was assayed using the Dual-Luciferase reporter system (Promega) 48 h after transfection. Firefly luciferase activity was normalized with respect to Renilla luciferase activity. Initially, cells for luciferase assays were grown in DMEM supplemented with 5% charcoal-stripped calf serum and 1% antibiotic/antimycotic. Cells (105) were seeded into opaque 96-well plates and either grown in the media described above or in this medium supple-mented with 0.5 mM 8-Br-cAMP (cAMP) and 1 µM medroxyprogesterone acetate (MPA).
To assess the probability of observing over-representation of downregu-lation by MER20s in luciferase assays, we used the binomial test, with the observed number of MER20s that downregulated luciferase expression in endometrial cells (19), given the sample size (21) and either an expected proportion of 0.5 (for the comparison to chance alone) or an expected

©20
11 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Nature GeNetics doi:10.1038/ng.917
proportion of 0.1 (14/140 observations from the luciferase assays in the other cell types were downregulation of luciferase expression). Raw data are provided in Supplementary Tables 1 and 2.
Gene expression profile. To identify tissues that coexpress FOXO1A, C/EBPβ, PGR, HoxA-11, YY1, p300, CTCF and USF1 (data for PRMT1 and PRMT4 are not available), we calculated the mRNA copy number across 34
tissues using the recently compiled Mammalian Atlast of Combinatorial Transcriptional Regulation database of absolutely quantified real-time PCR data (qRT-PCR). mRNA copy data were divided into ten copy bins.
37. Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
38. Wang, Z. et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 (2008).





























