Embed Size (px)
Citation preview
Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und WirkstoffherstellungsverordnungVom 27. Oktober 2006
(BAnz. Nr. 210 vom 09.11.2006 S. 6887; 18.07.2008 S. 2798; 21.08.2009 S. 2890; 09.06.2011 S. 2901 11; 14.09.2011 S. 3414 11a;::01.02.2012 S. 626 12)
Gemäß § 2 Nr. 3 der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV ) macht das Bundesministerium für Gesundheit die jeweils aktuelle Fassung des EG-GMP Leitfadens in deut scher Sprache im Bundesanzeiger oder im elektronischen Bundesanzeiger bekannt. Der EG-GMP Leitfaden ist der Leitfaden für die Gute Herstellungspraxis für Arzneimittel und Prüfpräparate ein schließlich seiner Anhänge, mit dem die Kommission der Europäischen Gemeinschaften die ausführlichen Leitlinien nach Artikel 4 der Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel (ABl. EG Nr. L S. 67) und nach Artikel 51 der Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel (ABl. EG Nr. L 311 S. 1) veröffentlicht hat. Er dient zur Auslegung der Grundsätze und Leitlinien der Guten Herstellungspraxis (GMP) gemäß Artikel 3 Abs. 2 der Richtlinie 2003/94/EG der Kommission vom 8. Oktober 2003 zur Festlegung der Grundsätze und Leitlinien der Guten Herstellungspraxis für Humanarzneimittel und für zur Anwendung beim Menschen bestimmte Prüfpräparate (ABl. EU Nr. L 262 S. 22) und gemäß Artikel 3 der Richtlinie 91/412/EWG der Kommission vom 23. Juli 1991 zur Festlegung der Grundsätze und Leitlinien der Guten Herstellungspraxis für Tierarzneimittel (ABl. EG Nr. L 228 S. 70).Der EG-GCP Leitfaden wurde von der Europäischen Kommission als Eudralex Band 4 "EC Leitlinien für die Gute Herstellungspraxis, Humanarzneimittel und Tierarzneimittel" auf deren Website veröffentlicht Die Übersetzung aus dem englischen Original erfolgte durch das Bundesministerium für Gesundheit.Der Leitfaden besteht aus einer Einleitung, Anlage 1 dieser Bekanntmachung und zwei Teilen
Teil I beinhaltet die GMP Grundsatze für die Herstellung von Arzneimitteln und wird hiermit gemäß § 2 Nr. 3 AMWHV in Anlage 2 dieser Bekanntmachung veröffentlicht.
•
Teil II umfasst die Gute Herstellungspraxis für Wirkstoffe, die als Ausgangsstoffe für die Arzneimittelherstellung eingesetzt werden und wird hiermit gemäß § 2 Nr. 3 AMWHV in Anlage 3 dieser Bekanntmachung veröffentlicht.
•
Die Anhange zum EG-GMP Leitfaden werden in deutscher Sprache zu einem späteren Zeitpunkt gesondert bekanntgegeben. Der Leitfaden wird künftig auch auf der Website des Bundesministeriums für Gesundheit. www.bmg.bund.de, abrufbar sein.
Eudralex Die Regelung der Arzneimittel in der Europäischen Union
Band 4 EU Leitlinien für die Gute Herstellungspraxis Humanarzneimittel und Tierarzneimitteln 1
Geschichte des Dokuments Datum
Die erste Ausgabe des Leitfadens wurde veröffentlicht, einschließlich eines Anhangs über die Herstellung steriler Arzneimittel.
1989
Die zweite Ausgabe wurde veröffentlicht, mit der die Richtlinien der Kommission 91/356/EWG vom 13. Juni 1991 und 91/412/EWG vom 23. Juli 1991 zur Festlegung der Grundsätze und Leitlinien der Guten Herstellungspraxis für Human- und Tierarzneimittel vollzogen wurden. Die zweite Ausgabe umfasst zwölf weitere Anhänge.
Januar 1992
Eine Aktualisierung der Verweise auf rechtliche Grundlagen wurde durchgeführt. In der Zwischenzeit wurde der Leitfaden soweit erforderlich auf der Website der Europäischen Kommission aktualisiert, verschiedene Anhänge wurden zugefügt.
August 2004
Umstrukturierung des GMP Leitfadens, der damit aus Teil I für Human- und Tierarzneimittel und aus Teil II für als Ausgangsstoffe eingesetzte Wirkstoffe besteht und die Richtlinien 2004/27/EG und 2004/28/EG vollzieht. Der aktuelle Leitfaden enthält 17 Anhänge, der frühere Anhang 18 wurde ersetzt.
Oktober 2005
Datum des Inkrafttretens eines neuen Anhangs 19 ist der 1. Juni 2006. Dezember 2005
Änderungen des EG-GMP Leitfadens sowie Einfügung des neuen Anhangs 20. 18. Juli 2008
EinleitungDie pharmazeutische Industrie der Europäischen Union erhält hohe Qualitätssicherungsstandards bei der Entwicklung, Herstellung und Kontrolle von Arzneimitteln aufrecht. Ein Zulassungssystem gewährleistet, dass alle Arzneimittel durch eine zuständige Behörde bewertet werden, um den derzeitigen Sicherheits-, Qualitäts- und Wirksamkeitsanforderungen zu entsprechen. Die Regelung der Herstellungserlaubnisse gewährleistet, dass alle für den europäischen Markt zugelassenen Produkte nur von autorisierten Herstellern hergestellt werden, deren Tätigkeiten regelmäßig durch die zuständigen Behörden überprüft werden. Herstellungserlaubnisse sind für alle Arzneimittelhersteller in der Europäischen Gemeinschaft vorgeschrieben, gleichgültig, ob die Produkte innerhalb oder außerhalb der Gemeinschaft verkauft werden.Die Kommission hat zwei Richtlinien verabschiedet, in denen die Grundsätze und Leitlinien der Guten Herstellungspraxis (GMP) für Arzneimittel festgelegt werden. Die Richtlinie 2003/94/EG gilt für Humanarzneimittel, die Richtlinie 91/412/EWG für Tierarzneimittel. Detaillierte Leitlinien, die deren Grundsätzen entsprechen, sind im Leitfaden für die Gute Herstellungspraxis veröffentlicht worden, der für die Bewertung von Anträgen auf eine Herstellungserlaubnis herangezogen wird und als Grundlage für Inspektionen bei den Arzneimittelherstellern dient.Die Grundsätze der Guten Herstellungspraxis und die detaillierten Leitlinien gelten für sämtliche Tätigkeiten, für die eine Erlaubnis gemäß Artikel 40 der Richtlinie 2001/83/EG und Artikel 44 der Richtlinie 2001/82/EG , geändert durch die Richtlinien 2004/27/EG bzw. 2004/28/EG, vorgeschrieben ist. Sie gelten ferner für alle anderen Verfahren der Arzneimittelherstellung im großen Maßstab, wie beispielsweise in Krankenhäusern und für die Herstellung von Produkten für klinische Prüfungen.
Seite 1 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Alle Mitgliedstaaten und auch die Industrie stimmen darin überein, dass für die Herstellung von Tierarzneimitteln dieselben Anforderungen der Guten Herstellungspraxis gelten wie für die Herstellung von Humanarzneimitteln. Bestimmte detaillierte Anpassungen an die Leitlinien der Guten Herstellungspraxis sind in zwei speziellen, die Tierarzneimittel und immunologischen Tierarzneimittel betreffenden Anhängen enthalten.Der Leitfaden ist in zwei Teile mit grundlegenden Anforderungen und speziellen Anhängen unterteilt. Teil I beinhaltet die GMP Grundsätze für die Herstellung von Arzneimitteln. Teil II umfasst die Gute Herstellungspraxis für Wirkstoffe, die als Ausgangsstoffe eingesetzt werden.Die Kapitel des Teils I zu den "Grundlegenden Anforderungen" sind mit Überschriften versehen, die den Grundsätzen in den Richtlinien 2003/94/EG und 91/412/EWG entsprechen. Kapitel 1 über das Qualitätsmanagement umreißt das grundsätzliche Konzept der für die Herstellung von Arzneimitteln geltenden Qualitätssicherung. Jedes weitere Kapitel beinhaltet einen Grundsatz, der die Zielsetzungen der Qualitätssicherung des jeweiligen Kapitels umreißt sowie einen Text mit ausreichenden Details, um den Herstellern die wesentlichen Aspekte bewusst zu machen, die bei der Anwendung des Grundsatzes zu berücksichtigen sind.Teil II wurde neu erstellt auf der Basis einer Leitlinie, die im Rahmen der ICH entwickelt und als ICH Q7a über "Wirkstoffe" veröffentlicht und im Jahre 2001 als Anhang 18 des GMP-Leitfadens für eine Verwendung auf freiwilliger Basis übernommen wurde. Gemäß den Artikeln 47 bzw. 51 der Richtlinien 2001/83/EG und 2001/82/EG in ihrer geänderten Fassung sollen von der EU-Kommission ausführliche Leitlinien über die Grundsätze der Guten Herstellungspraxis für Wirkstoffe, die als Ausgangsstoffe benutzt werden, verabschiedet und veröffentlicht werden. Der frühere Anhang 18 wurde durch den neuen Teil II des GMP-Leitfadens ersetzt, der eine erweiterte Anwendung für Human- und für Tierarzneimittel findet.Zusätzlich zu den in den Teilen I und II beschriebenen allgemeinen Aspekten der Guten Herstellungspraxis enthält dieser Leitfaden eine Reihe von Anhängen mit Details über spezielle Tätigkeitsbereiche. Für bestimmte Produktionsverfahren gelten mehrere Anhänge gleichzeitig (z.B. Anhänge über sterile Zubereitungen und über Radiopharmaka und/oder biologische Arzneimittel * .Kapitel 1 des Leitfadens Teil I über das Qualitätsmanagement ist überarbeitet worden, um die Aspekte des Qualitäts-Risikomanagements im Rahmen des Qualitätssystems aufzunehmen. In zukünftigen Revisionen des Leitfadens wird die Gelegenheit ergriffen werden, Elemente des Qualitäts-Risikomanagements aufzunehmen, wenn immer dies erforderlich ist.Der neue Anhang 20, der der ICH Q9 Guideline entspricht, bietet Anleitung für einen systematischen Zugang zum Qualitäts-Risikomanagement und führt zur Übereinstimmung mit GMP und anderen Qualitätsanforderungen. Er beinhaltet Grundsätze, die einzuhalten sind, und Optionen für Prozesse, Methoden und Werkzeuge, die genutzt werden können, wenn ein formaler Qualitäts-Risikomanagement-Maßstab angelegt wird. Obwohl der EGGMP Leitfaden primär an Hersteller gerichtet ist, hat die ICH Q9 Guideline Relevanz für andere .Qualitätsrichtlinien und beinhaltet spezifische Abschnitte für Überwachungsbehörden. Jedoch ist aus Gründen des Zusammenhangs und der Vollständigkeit die ICH Q9 Guideline vollständig in Anhang 20 zum EG-GMP Leitfaden überführt worden.Ein Glossar mit den gängigsten im Leitfaden benutzten Ausdrücken befindet sich - jeweils - am Schluss des Teils I und des Teils II.Der Leitfaden befasst sich nicht mit Arbeitssicherheitsaspekten, die im Falle der Herstellung bestimmter Arzneimittel, wie hochwirksamer, biologischer und radioaktiver Arzneimittel von besonderer Bedeutung sein können. Diese Aspekte werden jedoch in anderen gemeinschaftlichen und nationalen Rechtsvorschriften geregelt.Der Leitfaden geht von dem Grundsatz aus, dass den in der Zulassung festgelegten Anforderungen an die Sicherheit, Qualität und Wirksamkeit der Arzneimittel von den Zulassungsinhabern bei allen Tätigkeiten im Zusammenhang mit der Herstellung, der Kontrolle und dem Inverkehrbringen entsprochen wird.Seit vielen Jahren werden Arzneimittel nach den Leitlinien der Guten Herstellungspraxis hergestellt. Für die Arzneimittelherstellung gelten keine CEN/ISO-Normen. Die Pharmaunternehmen können die von den europäischen Normenorganisationen CEN/ISO festgelegten harmonisierten Normen als Instrumente zur Einführung eines Qualitätssystems im Arzneimittelbereich nach eigenem Ermessen anwenden. Die CEN/ISO-Normen wurden berücksichtigt, doch wurde ihre Terminologie nicht übernommen. Es wird anerkannt, dass andere als die in dem Leitfaden beschriebenen Methoden geeignet sein können, die Ziele der Grundsätze der Qualitätssicherung zu verwirklichen. Der Leitfaden beabsichtigt keinesfalls die Entwicklung neuer Konzepte oder Technologien einzuschränken, sofern diese validiert sind und ein Niveau der Qualitätssicherung gewährleisten, welches dem in dem Leitfaden beschriebenen mindestens gleichwertig ist. Anhang 20 stellt mit seinen Grundsätzen, Methoden und Instrumenten einen systematischen Zugang zur Verfügung, mit dem eine solche Gleichwertigkeit aufgezeigt werden kann.Der Leitfaden wird in regelmäßigen Abständen überprüft. Revisionen werden auf der Website der Europäischen Kommission unter http:/ec.europa.eu/enterprise/pharmaceuticals/eudralex/ homev4.htm veröffentlicht. * ) Die Anhänge werden in einem separaten Dokument übersetzt.
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Teil I (1/2) EG-GMP Leitfaden - Leitfaden der Guten Herstellungspraxis
Teil I
Vom 27. Oktober 2006 (BAnz. Nr. 210 vom 09.11.2006 S. 6887;::18.07.2008 S. 2798)
Geschichte des Dokuments Datum
Überarbeitung zur Einfügung eines neuen Kapitels 1 zur Produktqualitätsüberprüfung. Die überarbeitete Version soll zum 1. Januar 2006 in Kraft treten mit der Festlegung, dass eine erste Produktqualitätsüberprüfung in 2006 erwartet wird, die einen Mindestzeitraum von sechs Monaten umfasst. Nachfolgende Berichte sollen den vollen zwölfmonatigen Zeitraum umfassen.
Oktober 2005
Überarbeitung zur Einfügung eines neuen Kapitels 6 zum fortlaufenden Stabilitätsprogramm und zur Anpassung des Abschnitts 6.14 über Referenzproben. Die überarbeitete Version soll zum 1. Juni 2006 in Kraft treten.
Oktober 2005
Überarbeitung zur Einfügung eines neuen Abschnitts 8.7 über Anforderungen zu Fälschungen und Verschieben des Originalabschnitts 8.7 in den modifizierten Abschnitt 8.16. Die überarbeitete Version soll zum 1. Februar 2006 in Kraft treten.
Dezember 2005
Änderungen des EG-GMP Leitfadens sowie Einfügung des neuen Anhangs 20. 18. Juli 2008
Seite 2 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Kapitel 1 Qualitätsmanagement
GrundsätzeDer Inhaber einer Herstellungserlaubnis muss Arzneimittel so herstellen, dass ihre Eignung für den vorgesehenen Gebrauch gewährleistet ist, sie den im Rahmen der Zulassung spezifizierten Anforderungen entsprechen und die Patienten keiner Gefahr wegen unzureichender Sicherheit, Qualität oder Wirksamkeit aussetzen. Für die Erreichung dieses Qualitätszieles ist die Geschäftsleitung eines Unternehmens verantwortlich und die Beteiligung und Einsatzbereitschaft der Mitarbeiter in vielen verschiedenen Abteilungen und auf allen Ebenen eines Unternehmens sowie die der Zulieferer und Vertriebsunternehmen erforderlich. Um das Ziel zuverlässig zu erreichen, muss das Unternehmen über ein umfassend geplantes und korrekt implementiertes System der Qualitätssicherung verfügen, das die Gute Herstellungspraxis und damit die Qualitätskontrolle beinhaltet. Dieses System sollte vollständig dokumentiert sein und seine Funktionstüchtigkeit überwacht werden. Alle Bereiche des Qualitätssicherungssystems sollten angemessen mit kompetentem Personal sowie mit geeigneten und ausreichenden Räumlichkeiten und Ausrüstungen ausgestattet sein. Für den Inhaber der Herstellungserlaubnis und für die sachkundige(n) Person(en) bestehen zusätzliche rechtliche Verpflichtungen.Die Grundkonzepte der Qualitätssicherung, der Guten Herstellungspraxis, der Qualitätskontrolle und des Qualitäts-Risikomanagements sind miteinander verflochten. Sie werden im Folgenden beschrieben, um ihre Verflechtung und grundlegende Bedeutung für die Herstellung und Prüfung von Arzneimitteln zu unterstreichen.Qualitätssicherung1.1 Qualitätssicherung ist ein weit reichendes Konzept, das alle Bereiche abdeckt, die im Einzelnen oder insgesamt die Qualität eines Produktes beeinflussen. Sie stellt die Gesamtheit allervorgesehenen Maßnahmen dar, die getroffen werden, um sicherzustellen, dass Arzneimittel die für den beabsichtigten Gebrauch erforderliche Qualität aufweisen. Qualitätssicherung umfasst daher die Gute Herstellungspraxis sowie weitere Faktoren, die über den Rahmen dieses Leitfadens hinausgehen.Durch ein für die Herstellung von Arzneimitteln geeignetes Qualitätssicherungssystem sollte sichergestellt werden, dass
Arzneimittel unter Berücksichtigung der Anforderungen der Guten Herstellungspraxis und der Guten Laborpraxis konzipiert und entwickelt werden;
i.
Herstellungs- und Prüfverfahren klar spezifiziert sind und die Regeln der Guten Herstellungspraxis beinhalten; ii.Verantwortungsbereiche auf der Leitungsebene eindeutig festgelegt sind; iii.Vereinbarungen für die Herstellung, die Lieferung und den Einsatz der richtigen Ausgangsstoffe und des korrekten Verpackungsmaterials getroffen sind;
iv.
alle notwendigen Prüfungen der Zwischenprodukte sowie alle weiteren Inprozesskontrollen und Validierungen durchgeführt werden;
v.
das Fertigprodukt nach den festgelegten Verfahren ordnungsgemäß angefertigt und geprüft wird; vi.Arzneimittel nicht verkauft oder abgegeben werden, bevor eine Sachkundige Person bescheinigt hat, dass jede Herstellungscharge in Übereinstimmung mit den in der Zulassung festgelegten Anforderungen und allen anderen für die Herstellung, Prüfung und Freigabe von Arzneimitteln relevanten Vorschriften hergestellt und geprüft wurde;
vii.
ausreichende Vorkehrungen bestehen, um so weit wie möglich sicherzustellen, dass die Arzneimittel so gelagert, vertrieben und anschließend gehandhabt werden, dass die Qualität während ihrer Haltbarkeitsdauer erhalten bleibt;
viii.
ein Verfahren der Selbstinspektion und/oder Qualitätsüberprüfung zur regelmäßigen Bewertung der Wirksamkeit und Eignung des Qualitätssicherungssystems eingeführt ist.
ix.
Gute Herstellungspraxis für Arzneimittel (GMP)1.2 Gute Herstellungspraxis ist der Teil der Qualitätssicherung, der gewährleistet, dass die Produkte gleich bleibend nach den Qualitätsstandards hergestellt und geprüft werden, die der vorgesehenen Verwendung und den Zulassungsunterlagen oder der Produktspezifikation entsprechen.Gute Herstellungspraxis betrifft sowohl die Produktion als auch die Qualitätskontrolle. Die grundlegenden Anforderungen der Guten Herstellungspraxis sind folgende:
alle Herstellungsvorgänge sind klar definiert, werden unter Einbeziehung der vorliegenden Erfahrungen systematisch überprüft und sind nachweislich geeignet, gleich bleibend Arzneimittel hervorzubringen, die die erforderliche Qualität aufweisen und ihren Spezifikationen entsprechen;
i.
kritische Herstellungsschritte und wesentliche Prozessänderungen sind validiert; ii.alle für die Gute Herstellungspraxis erforderlichen Voraussetzungen sind erfüllt, insbesondere:◦
angemessen qualifiziertes und geschultes Personal; ◦
geeignete, ausreichend große Räumlichkeiten; ◦
geeignete Ausrüstung und Versorgungseinrichtungen; ◦
einwandfreie Materialien, Behältnisse und Etiketten; ◦
genehmigte Verfahrensbeschreibungen und Anweisungen; ◦
geeignete Lagerung und geeigneter Transport;◦
Anweisungen und Verfahrensbeschreibungen sind als Vorschriften in klarer und eindeutiger Sprache schriftlich abgefasst und gelten speziell für die vorhandenen Anlagen;
iii.
das ausführende Personal ist in der ordnungsgemäßen Ausführung der Verfahren geschult; iv.während der Herstellung werden manuell und/oder mit Aufzeichnungsgeräten Protokolle erstellt, aus denen hervorgeht, dass alle nach den festgelegten Verfahren und Anweisungen erforderlichen Schritte tatsächlich durchgeführt wurden und die erhaltene Menge und Qualität des Produktes den Erwartungen entsprach. Alle wesentlichen Abweichungen werden vollständig aufgezeichnet und untersucht;
v.
Herstellungsprotokolle einschließlich Aufzeichnungen über den Vertrieb, anhand derer sich die vollständige Geschichte einer Charge zurückverfolgen lässt, werden in zugänglicher und nachvollziehbarer Form aufbewahrt;
vi.
der Vertrieb der Produkte (Großhandel) erfolgt so, dass jedes Qualitätsrisiko minimiert wird; vii.es besteht ein System, mit dem jede Herstellungscharge von der Bereitstellung oder dem Verkauf zurückgerufen werden kann; viii.
Seite 3 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Beanstandungen von im Handel befindlichen Produkten werden überprüft, die Ursachen von Qualitätsmängeln untersucht, geeignete Maßnahmen bezüglich der fehlerhaften Produkte ergriffen und Vorkehrungen getroffen, um ein Wiederauftreten der Fehler zu verhindern.
ix.
Qualitätskontrolle1.3 Qualitätskontrolle ist der Teil der Guten Herstellungspraxis, der sich mit Probenahme, Spezifikationen und Prüfungen sowie Organisations-, Dokumentations- und Freigabeverfahren befasst, mit denen gewährleistet wird, dass die jeweils notwendigen und relevanten Prüfungen tatsächlich durchgeführt werden und dass sowohl die benötigten Materialien als auch die hergestellten Produkte für Verkauf oder Auslieferung erst freigegeben werden, wenn ihre Qualität als zufrieden stellend beurteilt wurde.Die grundlegenden Anforderungen an die Qualitätskontrolle sind folgende:
geeignete Einrichtungen, geschultes Personal und genehmigte Verfahrensbeschreibungen sind verfügbar für die Probenahme, nähere Kontrolle und Prüfung von Ausgangsstoffen, Verpackungsmaterial, Zwischenprodukten, Bulkware sowie Fertigprodukten und, soweit dies die Gute Herstellungspraxis erfordert, für die Überwachung der Umgebung;
i.
Proben von Ausgangsstoffen, Verpackungsmaterial, Zwischenprodukten, Bulkware und Fertigprodukten werden durch Personen und nach Methoden entnommen, die von der Qualitätskontrolle genehmigt wurden;
ii.
die Prüfmethoden sind validiert; iii.Protokolle, die zeigen, dass alle erforderlichen Probenahmen, Kontroll- und Prüfverfahren tatsächlich durchgeführt wurden, werden manuell und/oder mit Aufzeichnungsgeräten angefertigt. Jede Abweichung wird vollständig protokolliert und untersucht;
iv.
die Fertigprodukte enthalten die Wirkstoffe, die qualitativ und quantitativ der zugelassenen Zusammensetzung entsprechen, weisen die erforderliche Reinheit auf, befinden sich in den richtigen Behältnissen und sind ordnungsgemäß gekennzeichnet;
v.
Protokolle werden erstellt über die Prüfung der Materialien, Zwischenprodukte und Bulkware sowie Fertigprodukte; die Ergebnisse werden mit den Anforderungen der Spezifikation verglichen. Zur Produktbewertung gehören die Überprüfung und Beurteilung der jeweiligen Herstellungsdokumentation und eine Bewertung eventueller Abweichungen von den festgelegten Verfahren;
vi.
keine Produktcharge wird vor der Bescheinigung durch eine Sachkundige Person, dass die Charge mit den in der Zulassung festgelegten Anforderungen übereinstimmt, für den Verkauf oder die Auslieferung freigegeben;
vii.
Rückstellmuster von Ausgangsstoffen und Produkten werden in ausreichender Menge aufbewahrt, um das Produkt nötigenfalls später untersuchen zu können. Das Produkt wird in seiner endgültigen Verpackung aufbewahrt, es sei denn, es handelt sich um außergewöhnlich große Packungen.
viii.
Produktqualitätsüberprüfung1.4 Es sollten regelmäßig periodische oder wiederkehrende Qualitätsüberprüfungen aller zugelassenen Arzneimittel einschließlich der nur für den Export bestimmten Produkte mit dem Ziel durchgeführt werden, die Beständigkeit des gegenwärtigen Prozesses und die Geeignetheit der aktuellen Spezifikationen sowohl für die Ausgangsstoffe als auch für das Fertigprodukt zu verifizieren, um Trends hervorzuheben sowie Verbesserungsmöglichkeiten für Produkte und Abläufe zu identifizieren. Solche Überprüfungen sollten normalerweise unter Berücksichtigung vorhergehender Überprüfungen jährlich durchgeführt und dokumentiert werden und mindestens Folgendes beinhalten:
Eine Überprüfung der für das Produkt eingesetzten Ausgangsstoffe und Verpackungsmaterialien, besonders von solchen, die aus neuen Quellen bezogen werden.
i.
Eine Überprüfung kritischer Inprozesskontrollen und der Ergebnisse von Fertigproduktprüfungen.ii.Eine Überprüfung aller Chargen, die den festgelegten Spezifikationen nicht entsprachen und der dazugehörigen Untersuchungen.iii.Eine Überprüfung aller signifikanten Abweichungen oder Nicht-Übereinstimmungen, der dazugehörigen Untersuchungen und der Effektivität daraus resultierender Korrektiv- und Präventivmaßnahmen.
iv.
Eine Überprüfung aller durchgeführten Änderungen am Prozess oder den Analysenmethoden.v.Eine Überprüfung der eingereichten/genehmigten/abgelehnten Änderungen im Zulassungsdossier, einschließlich solcher, die sich auf Arzneimittel beziehen, die ausschließlich für den Export bestimmt sind.
vi.
Eine Überprüfung aller Ergebnisse des Stabilitätsüberwachungsprogramms und etwaiger negativer Trends.vii.Eine Überprüfung aller qualitätsbezogenen Rückgaben, Beanstandungen und Rückrufe und der zu diesem Zeitpunkt durchgeführten Untersuchungen.
viii.
Eine Überprüfung der Angemessenheit aller früheren Korrekturmaßnahmen an Herstellungsprozessen oder der Ausrüstung.ix.Eine Überprüfung der postmarketing Verpflichtungen, die nach dem Inverkehrbringen neu zugelassener Arzneimittel oder nach Durchführung einer Zulassungsänderung bestehen.
x.
Den Qualifizierungsstatus relevanter Ausrüstung und Betriebsmittel, z.B. Heizung/Be- und Entlüftung/Klimatisierung, Wasser, komprimierte Gase etc.
xi.
Eine Überprüfung aller vertraglichen Vereinbarungen wie in Kapitel 7 beschrieben, um sicherzustellen, dass sie auf dem neuesten Stand sind.
xii.
Der Hersteller und, sofern nicht identisch, der Zulassungsinhaber sollten die Ergebnisse dieser Überprüfung bewerten und es sollte eine Einschätzung erfolgen, ob korrektive oder präventive Maßnahmen oder eine Revalidierung durchzuführen sind. Die Gründe für solche Korrektivmaßnahmen sollten dokumentiert werden. Vereinbarte Korrektiv- und Präventivmaßnahmen sollten zeitnah und effizient abgeschlossen werden. Es sollten Verfahren auf Leitungsebene für die laufende Leitung und Überwachung dieser Maßnahmen vorhanden sein und die Wirksamkeit dieser Maßnahmen im Rahmen von Selbstinspektionen verifiziert werden. Qualitätsüberprüfungen können nach Produkttyp, z.B. feste Darreichungsformen, flüssige Darreichungsformen, Sterilprodukte, etc. zusammengefasst durchgeführt werden, wo dies wissenschaftlich gerechtfertigt ist.Wenn der Zulassungsinhaber nicht der Hersteller des Produktes ist, sollte eine technische Vereinbarung zwischen den verschiedenen Parteien bestehen, die ihre jeweiligen Verantwortlichkeiten bei der Durchführung der Qualitätsüberprüfung festlegt. Die Sachkundige Person, die für die abschließende Chargenfreigabe verantwortlich ist, sollte zusammen mit dem Zulassungsinhaber sicherstellen, dass die Qualitätsüberprüfung in angemessener Zeit und richtig durchgeführt wird.Qualitäts-Risikomanagement1.5 Qualitäts-Risikomanagement ist ein systematischer Prozess für die Bewertung, Kontrolle, Kommunikation und Überprüfung der Risiken eines Arzneimittels. Es kann sowohl prospektiv als auch retrospektiv angewandt werden.
Seite 4 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

1.6 Das Qualitäts-Risikomanagement-System soll sicherstellen, dass
die Bewertung des Risikos für die Qualität auf wissenschaftlichen Erkenntnissen (und) Erfahrungen mit dem Prozess basiert und letztlich in Zusammenhang gebracht wird mit dem Schutz des Patienten
•
der Grad der Bemühungen, der Förmlichkeit und Dokumentation des Qualitäts-Risikomanagements in Einklang steht mit dem Grad des Risikos.
•
Beispiele für die Prozesse und Anwendungen des Qualitäts-Risikomanagements können unter anderem in Anhang 20 gefunden werden
Kapitel 2 Personal
GrundsätzeDer Aufbau und die Erhaltung eines zufrieden stellenden Qualitätssicherungssystems und die einwandfreie Herstellung von Arzneimitteln hängen wesentlich vom Personal ab. Daher muss qualifiziertes Personal in ausreichender Zahl vorhanden sein, um alle in der Verantwortung des Herstellers liegenden Aufgaben auszuführen. Die individuellen Verantwortungsbereiche sollten von jedem einzelnen klar verstanden und aufgezeichnet sein. Jeder Mitarbeiter sollte mit den ihn angehenden Grundsätzen der Guten Herstellungspraxis vertraut sein und zu Beginn seiner Tätigkeit und fortlaufend geschult werden. Die Schulung sollte auch die jeweils notwendigen Hygieneunterweisungen umfassen.Allgemeine Anforderungen2.1 Der Hersteller sollte über Personal in ausreichender Zahl und mit der erforderlichen Qualifikation und praktischen Erfahrung verfügen. Die jedem einzelnen zugewiesenen Verantwortungsbereiche sollten nicht so umfangreich sein, dass sich daraus irgendwelche Qualitätsrisiken ergeben.2.2 Der Hersteller muss ein Organisationsschema haben. Mitarbeitern in verantwortlicher Stellung sollten spezifische Aufgaben zugewiesen sein, die in Arbeitsplatzbeschreibungen schriftlich niedergelegt sind. Diese Mitarbeiter sollten die entsprechenden Vollmachten haben, um ihrer Verantwortung gerecht werden zu können. Ihre Aufgaben können auf hierfür benannte, ausreichend qualifizierte Vertreter übertragen werden. Zwischen den Verantwortungsbereichen des mit der Anwendung der Guten Herstellungspraxis befassten Personals dürfen keine Lücken oder unbegründete Überlappungen bestehen.Personal in Schlüsselstellungen2.3 Zum Personal in Schlüsselstellungen gehören der Leiter der Herstellung, der Leiter der Qualitätskontrolle sowie, wenn nicht mindestens einer der beiden Genannten für die in Artikel 51 der Richtlinie 2001/83/EG 1 beschriebenen Aufgaben verantwortlich ist, die hierfür zuständige(n) Sachkundige(n) Person(en). Schlüsselstellungen sollten normalerweise mit Vollzeitbeschäftigten besetzt werden. Die Leiter der Herstellung und der Qualitätskontrolle müssen voneinander unabhängig sein. In großen Unternehmen kann es notwendig sein, einige der unter den Nummern 2.5, 2.6 und 2.7 genannten Funktionen zu delegieren
2.4 Die Aufgaben der Sachkundigen Person (en) sind in Artikel 51 der Richtlinie 2001/83/EG 2 vollständig beschrieben und können folgendermaßen zusammengefasst werden:
für die innerhalb der Europäischen Gemeinschaften hergestellten Arzneimittel muss eine Sachkundige Person sicherstellen, dass jede Charge in Übereinstimmung mit den Richtlinien und der Zulassung produziert und geprüft wurde 3;
a.
für außerhalb der Europäischen Gemeinschaften hergestellte Arzneimittel muss eine Sachkundige Person sicherstellen, dass jede importierte Charge im Einfuhrland den in Artikel 51 Abs. 1 (b) 4 aufgeführten Prüfungen unterzogen wurde;
b.
eine Sachkundige Person muss zu dem Zeitpunkt, zu dem die Arbeitsgänge durchgeführt werden und vor jeder Freigabe in einem Register oder einem gleichwertigen Dokument bescheinigen, dass jede Herstellungscharge den Bestimmungen des Artikels 515 genügt.
c.
Die für diese Aufgaben verantwortlichen Personen müssen die in Artikel 496 der o. a. Richtlinie niedergelegten Qualifikationsanforderungen erfüllen, sie müssen dem Inhaber der Herstellungserlaubnis zur Ausübung ihrer Funktionen ständig und fortlaufend zur Verfügung stehen, um ihren Verpflichtungen nachkommen zu können. Ihre Verpflichtungen können delegiert werden, jedoch nur an andere Sachkundige Personen.2.5 Der Leiter der Herstellung hat im Allgemeinen folgende Verantwortlichkeiten:
Sicherstellung, dass die Produkte gemäß den entsprechenden Anweisungen hergestellt und gelagert werden, um die erforderliche Qualität zu erhalten;
i.
Genehmigung der Anweisungen für die Herstellungsvorgänge und Sicherstellung, dass diese genau eingehalten werden; ii.Sicherstellung, dass die Herstellungsprotokolle von einer befugten Person überprüft und unterschrieben werden, bevor sie an die Abteilung für Qualitätskontrolle weitergegeben werden;
iii.
Kontrolle der Wartung, der Räumlichkeiten und der Ausrüstung seiner Abteilung; iv.Sicherstellung, dass die notwendigen Validierungen durchgeführt werden; v.Sicherstellung, dass die erforderliche anfängliche und fortlaufende Schulung des Personals seiner Abteilung durchgeführt und entsprechend den jeweiligen Erfordernissen angepasst wird.
vi.
2.6 Der Leiter der Qualitätskontrolle hat im Allgemeinen folgende Verantwortlichkeiten:
Billigung oder, falls er es für erforderlich hält, Zurückweisung von Ausgangsstoffen, Verpackungsmaterial, Zwischenprodukten, Bulkware und Fertigprodukten;
i.
Auswertung der Chargenprotokolle; ii.Sicherstellung, dass alle erforderlichen Prüfungen durchgeführt werden; iii.Genehmigung von Spezifikationen, Anweisungen zur Probenahme, Prüfmethoden und anderen Verfahren zur Qualitätskontrolle; iv.Zustimmung zur Beauftragung von Analysenlabors, die im Lohnauftrag arbeiten, sowie deren Überwachung; v.Kontrolle der Wartung, der Räumlichkeiten und der Ausrüstung seiner Abteilung; vi.Sicherstellung, dass die notwendigen Validierungen durchgeführt werden; vii.Sicherstellung, dass die erforderliche anfängliche und fortlaufende Schulung des Personals seiner Abteilung durchgeführt und entsprechend den jeweiligen Erfordernissen angepasst wird.
viii.
Seite 5 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Weitere Aufgaben der Abteilung für Qualitätskontrolle sind in Kapitel 6 zusammengefasst.2.7 Die Leiter der Herstellung und der Qualitätskontrolle teilen im Allgemeinen einige die Qualität betreffende Verantwortungsbereiche untereinander auf oder üben die Verantwortung gemeinsam aus. Je nach nationalen Regelungen können dies sein:
Genehmigung schriftlicher Verfahrensbeschreibungen und anderer Dokumente einschl. Ergänzungen; •Überwachung und Kontrolle der Umgebungsbedingungen bei der Herstellung; •Betriebshygiene; •Validierung von Verfahren; •Schulung; •Genehmigung und Überwachung von Materiallieferanten; •Zustimmung zur Beauftragung der Hersteller, die im Lohnauftrag arbeiten, sowie deren Überwachung; •Festlegung und Überwachung der Lagerungsbedingungen für Material und Produkte; •Aufbewahrung von Protokollen; •Überwachung der Einhaltung der Anforderungen der Guten Herstellungspraxis; •Überprüfungen, Untersuchungen und Entnahme von Proben zur Überwachung von Faktoren, die die Produktqualität beeinflussen können.
•
Schulung2.8 Der Hersteller sollte für die Schulung aller Personen sorgen, die Aufgaben in den Produktionsbereichen oder in Kontrolllaboratorien zu erfüllen haben (einschließlich des technischen, Wartungs- und Reinigungspersonals). Auch anderes Personal, dessen Tätigkeit die Produktqualität beeinflussen könnte, sollte geschult werden.2.9 Neben der theoretischen und praktischen Basisschulung in der Guten Herstellungspraxis sollten neu eingestellte Personen den ihnen jeweils zugewiesenen Aufgaben entsprechend geschult werden. Darüber hinaus sollte eine fortlaufende Schulung durchgeführt und deren effiziente Umsetzung in die Praxis periodisch bewertet werden. Es sollten Schulungsprogramme zur Verfügung stehen, die je nach Inhalt vom Leiter der Herstellung oder vom Leiter der Qualitätskontrolle genehmigt sind. Aufzeichnungen über die Schulung sollten aufbewahrt werden.2.10 Personal, das in Bereichen mit besonderen Kontaminationsrisiken arbeitet (z.B. in reinen Bereichen oder in Bereichen, in denen mit hochaktiven, toxischen, infektiösen oder sensibilisierenden Stoffen umgegangen wird), sollte eine spezielle Schulung erhalten.2.11 Besucher oder ungeschultes Personal sollten möglichst keine Herstellungs- und Qualitätskontrollbereiche betreten. Wenn dies jedoch unumgänglich ist, sollten sie vorher insbesondere über Personalhygiene und die vorgeschriebene Schutzkleidung informiert werden. Sie sollten streng beaufsichtigt werden.2.12 Im Rahmen der Schulung sollten das Konzept der Qualitätssicherung und alle Maßnahmen, die dessen Verständnis und Anwendung verbessern können, ausführlich diskutiert werden.Personalhygiene2.13 Es sollten detaillierte Hygieneprogramme erstellt und den unterschiedlichen Erfordernissen im Betrieb angepasst werden. Darin sollten Vorschriften zu Gesundheit, hygienischem Verhalten und Bekleidung des Personals enthalten sein. Diese Vorschriften sollten von jedem, der bei der Durchführung seiner Aufgaben Herstellungs- und Qualitätskontrollbereiche betritt, verstanden und sehr genau befolgt werden. Hygieneprogramme sollten von der Geschäftsleitung unterstützt und im Rahmen der Schulung eingehend diskutiert werden.2.14 Jeder Mitarbeiter sollte bei der Einstellung ärztlich untersucht werden. Der Hersteller muss dafür sorgen, dass Anweisungen vorhanden sind, mit denen sichergestellt wird, dass ihm Änderungen des Gesundheitszustandes des Personals, die von Bedeutung für die Produktqualität sein könnten, gemeldet werden. Nach der Einstellungsuntersuchung sollten, wenn aus betrieblichen oder aus Gründen der persönlichen Gesundheit nötig, Folgeuntersuchungen durchgeführt werden.2.15 Es sollten Vorkehrungen getroffen werden, die, soweit es praktisch möglich ist, sicherstellen, dass in der Arzneimittelherstellung niemand beschäftigt wird, der an einer ansteckenden Krankheit leidet oder offene Verletzungen an unbedeckten Körperstellen aufweist.2.16 Jede Person, die die Herstellungsbereiche betritt, sollte eine den jeweils auszuführenden Arbeiten angepasste Schutzkleidung tragen.2.17 Essen, Trinken, Kaugummikauen oder Rauchen sowie die Aufbewahrung von Speisen, Getränken, Tabakerzeugnissen oder Medikamenten für den persönlichen Gebrauch sollten in den Produktions- und Lagerbereichen verboten sein. Allgemein sollte jedes unhygienische Verhalten innerhalb der Herstellungsbereiche oder in jedem anderen Bereich, in dem das Produkt beeinträchtigt werden könnte, verboten sein.2.18 Der direkte Kontakt zwischen den Händen eines Beschäftigten und dem offenen Produkt sollte ebenso vermieden werden wie der direkte Kontakt mit irgendeinem Ausrüstungsteil, das mit den Produkten in Berührung kommt.2.19 Das Personal sollte angehalten werden, die Handwaschgelegenheiten zu benutzen.2.20 Alle speziellen Erfordernisse bei der Herstellung besonderer Präparategruppen, z.B. steriler Zubereitungen, werden in den Anhängen abgehandelt.
Kapitel 3 Räumlichkeiten und Ausrüstung
GrundsätzeRäumlichkeiten und Ausrüstung müssen so angeordnet, geplant, konstruiert, nachgerüstet und instand gehalten sein, dass sie für die vorgesehenen Arbeitsgänge geeignet sind. Ihre Anordnung und Ausgestaltung müssen darauf ausgerichtet sein, das Risiko von Fehlern auf ein Minimum herabzusetzen und eine gründliche Reinigung und Wartung zu erlauben, um Kreuzkontamination, Staub- oder Schmutzansammlungen und ganz allgemein jeden die Qualität des Produktes beeinträchtigenden Effekt zu vermeiden.Räumlichkeiten Allgemeine Anforderungen3.1 Die Räumlichkeiten sollten so gelegen sein, dass das umgebungsbedingte Kontaminationsrisiko für Material oder Produkte unter Berücksichtigung der Schutzmaßnahmen bei der Herstellung minimal ist.3.2 Die Räumlichkeiten sollten sorgfältig instand gehalten werden; Reparatur- und Wartungsarbeiten sollten keine Gefahr für die Qualität der Produkte darstellen. Sie sollten nach detaillierten, schriftlich festgelegten Verfahren gereinigt ,und, falls notwendig, desinfiziert werden.3.3 Beleuchtung, Temperatur, Luftfeuchtigkeit und Belüftung sollten geeignet und so beschaffen sein, dass sie weder direkt noch indirekt die Arzneimittel während der Herstellung und Lagerung oder das einwandfreie Funktionieren der Ausrüstung nachteilig beeinflussen.3.4 Die Räumlichkeiten sollten so ausgelegt und ausgestattet sein, dass der größtmögliche Schutz gegen das Eindringen von Insekten oder anderen Tieren gewährleistet ist.
Seite 6 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

3.5 Es sollten Vorkehrungen getroffen werden, um den Zutritt Unbefugter zu verhindern. Produktions-, Lagerungs- und Qualitätskontrollbereiche sollten von Personal, das dort nicht arbeitet, nicht als Durchgang benutzt werden.Produktionsbereiche3.6 Um das Risiko einer ernsten Gesundheitsschädigung durch Kreuzkontamination zu minimieren, müssen für die Produktion bestimmter Arzneimittel, wie solche aus hochsensibilisierenden Stoffen (z.B. Penicilline) oder biologische Zubereitungen (z.B. aus lebenden Mikroorganismen), fest zugeordnete und in sich geschlossene Räume/Einrichtungen zur Verfügung stehen. Die Produktion weiterer Produkte, wie bestimmter Antibiotika, Hormone, Zytostatika, hochwirksamer Arzneimittel sowie von Erzeugnissen, die keine Arzneimittel sind, sollte nicht in den gleichen Räumen/ Einrichtungen erfolgen. Für diese Produkte kann jedoch in Ausnahmefällen das Prinzip der Produktion in Kampagnen in den gleichen Räumen/Einrichtungen unter der Voraussetzung akzeptiert werden, dass spezielle Vorsichtsmaßnahmen getroffen werden und die notwendigen Validierungen durchgeführt sind. Die Herstellung technischer Gifte, wie Pestizide und Herbizide, sollte in Räumlichkeiten, die zu Herstellung von Arzneimitteln benutzt werden, nicht erlaubt sein.3.7 Die Räumlichkeiten sollten möglichst so angeordnet sein, dass die Produktion in logisch aufeinander folgenden Schritten erfolgen kann, entsprechend der Reihenfolge der Arbeitsgänge und den erforderlichen Reinheitsklassen.3.8 Ausreichende Arbeits- und Zwischenprodukt-Lagerflächen im Produktionsbereich sollten die ordnungsgemäße und folgerichtige Aufstellung der Ausrüstung und Bereitstellung der Materialien ermöglichen, um das Risiko von Verwechslungen unterschiedlicher Arzneimittel oder ihrer Bestandteile zu minimieren, Kreuzkontamination zu vermeiden und um die Gefahr, irgendeinen Produktions- oder Kontrollschritt auszulassen oder falsch anzuwenden, zu verringern.3.9 Wo Ausgangsstoffe und primäres Verpackungsmaterial, Zwischenprodukte oder Bulkware der Umgebung ausgesetzt sind, sollten die Innenflächen (Wände, Fußböden, Decken) glatt und frei von Rissen und offenen Fugen sein. Sie sollten keine Partikel abgeben und sich leicht und gründlich reinigen und, wenn nötig, desinfizieren lassen.3.10 Rohrleitungen, Beleuchtungskörper, Belüftungseinrichtungen und andere Versorgungsanlagen sollten so konstruiert und angebracht sein, dass keine schwer zu reinigenden Stellen entstehen. Für Wartungszwecke sollten sie möglichst von außerhalb der Produktionsbereiche zugänglich sein.3.11 Abflüsse sollten ausreichend groß und mit Rückstauklappe versehen sein. Offene Abflussrinnen sollten möglichst vermieden werden. Wenn sie jedoch erforderlich sind, sollten sie nicht zu tief sein, damit sie leicht gereinigt und desinfiziert werden können.3.12 Produktionsbereiche sollten wirkungsvoll belüftet sein, mit Belüftungssystemen (einschließlich Temperatur- und, falls nötig, Luftfeuchtigkeits- und Filterkontrollsystemen), die den dort gehandhabten Produkten, den durchgeführten Arbeitsgängen sowie der äußeren Umgebung angemessen sind.3.13 Das Abwiegen von Ausgangsstoffen sollte üblicherweise in einem separaten, für diesen Zweck konstruierten Wägeraum durchgeführt werden.3.14 An Stellen, an denen Staub entstehen kann (z.B. bei der Probenahme, bei Abwiegen, Mischen, Verarbeiten und Abpacken trockener Produkte), sollten besondere Maßnahmen ergriffen werden, um Kreuzkontamination zu vermeiden und die Reinigung zu erleichtern.3.15 Die Räumlichkeiten, in denen Arzneimittel verpackt werden, sollten so konstruiert und ausgestattet sein, dass Verwechslungen und Kreuzkontamination vermieden werden.3.16 Produktionsbereiche sollten gut beleuchtet sein, besonders dort, wo prozessbegleitend visuelle Kontrollen durchgeführt werden.3.17 Inprozesskontrollen dürfen innerhalb des Produktionsbereichs durchgeführt werden, vorausgesetzt, dass sie kein Risiko für die Produktion darstellen.Lagerbereiche3.18 Die Lagerräume sollten ausreichend groß sein, um eine ordnungsgemäße Lagerung der verschiedenen Kategorien von Materialien und Produkten zu erlauben: Ausgangsstoffe und Verpackungsmaterial, Zwischenprodukte, Bulkware und Fertigprodukte, in Quarantäne befindliche, freigegebene, zurückgewiesene, zurückgegebene oder zurückgerufene Produkte.3.19 Die Lagerräume sollten so konstruiert oder nachgerüstet sein, dass gute Lagerungsbedingungen gewährleistet sind. Vor allem sollten sie sauber und trocken sein sowie in einem angemessenen Temperaturbereich gehalten werden. Wenn besondere Lagerungsbedingungen (z.B. hinsichtlich Temperatur, Luftfeuchtigkeit) erforderlich sind, so sollten diese geschaffen, kontrolliert und überwacht werden.3.20 In den Annahme- und Versandbereichen sollten die Materialien und Produkte vor dem Wetter geschützt sein. Annahmebereiche sollten so konstruiert und ausgestattet sein, dass Behälter mit eingehenden Materialien erforderlichenfalls vor der Einlagerung gereinigt werden können.3.21 Wenn der Quarantänestatus durch Lagerung in abgetrennten Bereichen gewährleistet wird, müssen diese deutlich gekennzeichnet und der Zutritt für Befugte eingeschränkt werden. Jedes an die Stelle der räumlichen Quarantäne tretende System sollte die gleiche Sicherheit bieten.3.22 Normalerweise sollte die Probenahme von Ausgangsstoffen in einem abgetrennten Bereich erfolgen. Wenn die Probenahme jedoch im Lagerbereich durchgeführt wird, sollte diese in einer Weise erfolgen, die Verunreinigungen oder Kreuzkontamination verhindert.3.23 Zurückgewiesene, zurückgerufene oder zurückgegebene Materialien oder Produkte sollten in abgesonderten Bereichen gelagert werden.3.24 Hochaktive Materialien oder Produkte sollten in sicheren und geschützten Bereichen gelagert werden.3.25 Bedrucktes Verpackungsmaterial wird als kritisch für die Konformität des Arzneimittels angesehen, spezielle Aufmerksamkeit sollte daher auf die sichere und geschützte Lagerung dieser Materialien gerichtet werden.Qualitätskontrollbereiche3.26 In der Regel sollten Kontrolllaboratorien von den Produktionsbereichen abgetrennt sein. Dies gilt insbesondere für Laboratorien zur Kontrolle von biologischen und mikrobiologischen Produkten sowie von Radiopharmaka, die zudem auch voneinander getrennt sein sollten.3.27 Kontrolllaboratorien sollten so konstruiert sein, dass sie sich für die darin vorgesehenen Arbeitsgänge eignen. Sie müssen ausreichend groß sein, damit Verwechslungen und Kreuzkontamination vermieden werden. Für die Aufbewahrung von Proben und Protokollen sollte ausreichender und geeigneter Raum vorhanden sein.3.28 Separate Räume können notwendig sein, um empfindliche Instrumente vor Erschütterungen, elektrischen Störeinwirkungen, Feuchtigkeit usw. zu schützen.3.29 In Laboratorien, in denen mit besonderen Substanzen wie biologischen oder radioaktiven Proben umgegangen wird, sind spezielle Maßnahmen erforderlich.Nebenbereiche3.30 Aufenthalts- und Erfrischungsräume sollten von anderen Bereichen getrennt sein.3.31 Umkleide- und Waschräume sowie Toiletten sollten leicht erreichbar und der Benutzerzahl angemessen sein. Toiletten sollten nicht in direkter Verbindung mit Produktions- oder Lagerräumen stehen.
Seite 7 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

3.32 Werkstätten sollten, soweit möglich, von Produktionsbereichen getrennt sein. Wenn Einzelteile und Werkzeuge im Produktionsbereich gelagert werden, sollten sie in dafür vorgesehenen Räumen oder Schränken aufbewahrt werden.3.33 Räume, in denen Tiere gehalten werden, sollten von den anderen Bereichen gut isoliert sein und über einen seperaten Eingang (Tierzugang) und seperate Belüftungsanlagen verfügen.Ausrüstung3.34 Die Herstellungsausrüstung sollte so konstruiert, angeordnet und gewartet werden, dass sie für den vorgesehenen Zweck geeignet ist.3.35 Reparatur- und Wartungsarbeiten sollten die Qualität der Produkte nicht in irgendeiner Weise gefährden.3.36 Die Herstellungsausrüstung sollte so konstruiert sein, dass sie sich leicht und gründlich reinigen lässt. Sie sollte nach detaillierten, schriftlichen Verfahren gereinigt und nur sauber und trocken aufbewahrt werden.3.37 Die zum Waschen und Reinigen verwendete Ausrüstung sollte so gewählt und eingesetzt werden, dass sie selbst keine Quelle der Verunreinigung darstellt.3.38 Die Ausrüstung sollte so installiert sein, dass keine Gefahr eines Fehlers oder einer Verunreinigung besteht.3.39 Die für die Produktion verwendete Ausrüstung sollte für die Produkte kein Risiko darstellen. Kein mit dem Produkt in Berührung kommendes Ausrüstungsteil darf mit diesem so in Wechselwirkung treten, dass die Produktqualität beeinträchtigt wird und damit ein Risiko entsteht (egal ob reaktiv, additiv oder absorptiv).3.40 Für Produktions- und Kontrollzwecke sollten im geeigneten Wäge- und Messbereich und mit der erforderlichen Genauigkeit arbeitende Waagen und Messgeräte zur Verfügung stehen.3.41 Die Mess-, Wäge-, Aufzeichnungs- und Kontrollausrüstung sollte kalibriert sein und in bestimmten Abständen mit geeigneten Methoden überprüft werden. Geeignete Aufzeichnungen hierüber sollten aufbewahrt werden.3.42 Fest installierte Rohrleitungen sollten deutlich mit der Angabe des Inhalts und, wo angezeigt, mit der Fließrichtung gekennzeichnet sein.3.43 Leitungen für destilliertes und demineralisiertes Wasser und, wo angezeigt, andere Wasserleitungen sollten nach schriftlich festgelegten Verfahren desinfiziert werden, die genaue Angaben über die akzeptable mikrobiologische Verunreinigung und die bei Überschreitung der Grenzwerte zu treffenden Maßnahmen enthalten.3.44 Schadhafte Ausrüstung sollte, wenn möglich, aus Produktions- und Qualitätskontrollbereichen entfernt oder zumindest deutlich als schadhaft gekennzeichnet werden.
Kapitel 411 Dokumentation *
GrundsätzeEine gute Dokumentation ist ein wesentlicher Teil des Qualitätssicherungssystems und Schlüsselfunktion einer Herstellung in Übereinstimmung mit den GMP-Anforderungen. Die verschiedenen Arten der Dokumente und eingesetzten Medien sollten vollständig im Qualitätsmanagementsystem des Herstellers definiert sein. Die Dokumentation kann vielfältig geführt werden, einschließlich in Papierform oder mit elektronischen oder photographischen Medien. Hauptziel des genutzten Dokumentationssystems muss es sein alle Aktivitäten, die direkt oder indirekt die Qualitätsaspekte des Arzneimittels beeinflussen, zu kontrollieren, zu überwachen und aufzuzeichnen. Das Qualitätsmanagementsystem sollte ausreichende und detaillierte Instruktionen beinhalten zur Erleichterung eines allgemeinen Verständnisses über die Anforderungen, auch um eine ausreichende Dokumentation der verschiedenen Prozesse und Bewertung etwaiger Beobachtungen vorzusehen, so dass eine fortlaufende Anwendung der Anforderungen demonstriert werden kann.Es gibt zwei grundsätzliche Dokumentationstypen, um GMPÜbereinstimmung zu erreichen und zu dokumentieren: Vorschriften (Anweisungen, Anforderungen) und Protokolle/Berichte. Eine geeignete gute Dokumentationspraxis sollte entsprechend dem jeweiligen Dokumentationstyp angepasst sein.Geeignete Kontrollen sollten eingeführt werden um die Genauigkeit, Richtigkeit, Verfügbarkeit und Lesbarkeit der Dokumente sicherzustellen. Anweisungen sollten fehlerfrei und schriftlich verfügbar sein. Der Ausdruck "schriftlich" bedeutet aufgezeichnet oder dokumentiert in Medien, von denen Daten in einer für Menschen lesbaren Form wiedergegeben werden können.Erforderliche GMP-Dokumentation (nach Typus)Site Master File:Ein Dokument, das die GMP-bezogenen Aktivitäten des Herstellers beschreibt.Vorschriften (Anweisungen oder Anforderungen):Spezifikationen beschreiben im Einzelnen die Anforderungen, denen die Produkte oder Materialien, die bei der Herstellung eingesetzt oder erzielt werden, entsprechen müssen. Sie dienen als Grundlage der Qualitätsbewertung.Herstellungsvorschriften, Verarbeitungs-, Verpackungs- und Prüfanweisungen geben Details an für alle eingesetzten Ausgangsmaterialien, Ausrüstungen und Computersysteme (soweit zutreffend) und legen alle Anforderungen an die Verarbeitungs- und Verpackungsvorgänge, Probenahme und Prüfung fest. Inprozesskontrollen und zum Einsatz kommende prozessanalytische Technologien sollten, soweit relevant, mit ihren Akzeptanzkriterien festgelegt werden.Verfahrensbeschreibungen (auch Standardarbeitsanweisungen oder SOP's) geben Anweisungen für die Durchführung bestimmter Arbeitsgänge.Anweisungen geben Instruktionen für die Durchführung und die Aufzeichnung bestimmter besonderer Arbeitsgänge.Technische Vereinbarungen sind Abmachungen zwischen Vertragsgebern und Vertragsnehmern bei fremd vergebenen Aktivitäten. Protokolle/Berichtstypen:Protokolle belegen für die verschiedenen Aktionen den Nachweis der Übereinstimmung mit den Anweisungen, z.B. Aktivitäten, Ereignisse, Untersuchungen und im Falle der Chargenherstellung den Werdegang jeder Charge, einschließlich ihres Vertriebs. Die Protokolle beinhalten die Rohdaten, die für die Erzeugung anderer Protokolle eingesetzt werden. Für die elektronischen Protokolle sollten die Nutzer festlegen, welche Daten als Rohdaten genutzt werden. Mindestens solche Daten sollten als Rohdaten festgelegt werden, die für Qualitätsentscheidungen dienen.Analysenzertifikate stellen eine Zusammenfassung von Testergebnissen bereit, die von Proben des Produkts oder der Materialien * erhalten wurden, einschließlich einer Bewertung der Übereinstimmung mit einer festgelegten Spezifikation.Berichte dokumentieren die Durchführung bestimmter Aufgaben, Projekte oder Überprüfungen, zusammen mit Ergebnissen, Schlussfolgerungen und Empfehlungen.Erzeugung und Kontrolle der Dokumentation4.1 Alle Dokumentationstypen sollten festgelegt und befolgt werden. Die Anforderungen gelten für alle Dokumentarten und ihre Medien. Komplexe Systeme müssen verstanden werden, gut dokumentiert, validiert und adäquate Kontrollen vorhanden sein. Viele Dokumente (Anweisungen und/oder Protokolle) können in gemischter Form vorliegen, z.B. einige Elemente in elektronischer Form oder andere in
Seite 8 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Papierform. Die Beziehungen zwischen und Kontrollmaßnahmen für Masterdokumente, offizielle Kopien, Datenbearbeitung und Aufzeichnungen müssen festgelegt werden sowohl für die gemischte Form als auch für die einheitlichen Systeme. Für elektronische Dokumente wie Vorlagen, Formulare und Masterdokumente sollten geeignete Kontrollen eingeführt werden. Geeignete Kontrollen um die Richtigkeit und Vollständigkeit der Protokolle während der Aufbewahrungszeit sicherzustellen, sollten eingerichtet sein.4.2 Unterlagen sollten sorgfältig konzipiert, erstellt, überprüft und verteilt werden. Sie sollten den jeweiligen Vorgaben der Produktspezifikation, der Herstellungserlaubnis und der Zulassungsunterlagen, soweit zutreffend, entsprechen. Die Erstellung von Vervielfältigungen der Originalunterlagen als Arbeitsunterlagen sollte nicht zu Fehlern durch den Vervielfältigungsprozess führen.4.3 Unterlagen, die Anweisungen enthalten, sollten von geeigneten und dazu befugten Personen genehmigt, unterzeichnet und datiert werden. Der Inhalt der Unterlagen sollte eindeutig und leicht erkennbar sein. Das Datum des Inkrafttretens sollte festgelegt sein.4.4 Unterlagen, die Anweisungen enthalten, sollten übersichtlich gestaltet und leicht zu kontrollieren sein. Stil und Sprache der Dokumente sollten mit ihrem vorgesehenen Gebrauch übereinstimmen. Standardarbeitsanweisungen, Arbeitsanweisungen und Methoden sollten in verbindlicher Befehlsform geschrieben sein.4.5 Unterlagen innerhalb des Qualitätsmanagementsystems sollten regelmäßig überprüft und auf dem neuesten Stand gehalten werden.4.6 Unterlagen sollten nicht handgeschrieben sein. Wenn jedoch Daten eingetragen werden müssen, sollte dafür genügend Platz zur Verfügung stehen.Gute Dokumentationspraxis4.7 Daten, die per Hand eingetragen werden, sollten in klarer, lesbarer und nicht zu entfernender Handschrift gemacht werden.4.8 Protokolle sollten zum Zeitpunkt des jeweiligen Vorgangs so angefertigt oder vervollständigt werden, dass sich alle wichtigen, die Herstellung der Arzneimittel betreffenden Tätigkeiten rückverfolgen lassen.4.9 Jede Änderung einer Eintragung in einem Dokument sollte abgezeichnet und datiert sein. Trotz Änderung sollte die ursprüngliche Information lesbar bleiben. Sofern angezeigt, sollte der Grund für die Änderung protokolliert werden.Aufbewahrung der Dokumente4.10 Es sollte klar festgelegt werden, welches Protokoll mit welcher Herstellungsaktivität zusammenhängt und wo sich dieses Protokoll befindet. Es sollten sichere, und soweit angezeigt, validierte Kontrollmaßnahmen vorhanden sein, mit denen die Vollständigkeit und Richtigkeit der Protokolle während der Aufbewahrungszeit sichergestellt wird.4.11 Besondere Anforderungen gelten für die Chargendokumentation, die ein Jahr über das Verfalldatum der betroffenen Charge aufzubewahren ist oder mindestens fünf Jahre nach der Zertifizierung der Charge durch die sachkundige Person, wobei der längere Zeitraum gilt. Für Prüfpräparate muss die Chargendokumentation mindestens fünf Jahre nach Abschluss oder formellem Abbruch der letzten klinischen Prüfung, bei der die Charge eingesetzt wurde, aufbewahrt werden. In Verbindung mit spezifischen Produkttypen (z.B. Arzneimittel für neuartige Therapien) können durch Gesetzgebung andere Anforderungen an die Aufbewahrung der Dokumentation festgelegt und längere Aufbewahrungszeiten vorgeschrieben sein.4.12 Für andere Dokumentationstypen hängt die Aufbewahrungszeit von den Geschäftsaktivitäten ab, auf die sie sich beziehen. Entscheidende Dokumente, einschließlich Rohdaten (z.B. bezüglich der Validierung oder der Stabilität), auf die Angaben in den Zulassungsunterlagen zurückgehen, sollten so lange aufbewahrt werden, wie die Zulassung gültig ist. Es kann akzeptiert sein, bestimmte Dokumente auszusondern (z.B. Rohdaten, die die Validierungs- oder Stabilitätsberichte unterstützen), wenn diese Daten ersetzt wurden durch ein komplettes Set neuer Daten. Die Rechtfertigung dafür sollte dokumentiert werden und dabei die Anforderungen an die Aufbewahrungszeit der Chargendokumentation berücksichtigen; z.B. im Falle von Daten für eine Prozessvalidierung sollten die begleitenden Rohdaten für einen Zeitraum aufbewahrt werden, der mindestens dem der Chargenprotokolle entspricht, deren Freigabe durch diesen Validierungsvorgang unterstützt wurde.Der folgende Abschnitt zeigt einige Beispiele der erforderlichen Dokumente auf. Das Qualitätsmanagementsystem sollte alle Dokumente beschreiben, die für die Produktqualität und die Patientensicherheit erforderlich sind.Spezifikationen4.13 Für Ausgangsstoffe, Verpackungsmaterial und Fertigprodukte sollten von der hierfür verantwortlichen Person genehmigte und datierte Spezifikationen vorliegen.Spezifikationen für Ausgangsstoffe und Verpackungsmaterial4.14 Die Spezifikationen für Ausgangsstoffe und primäres oder bedrucktes Verpackungsmaterial sollten beinhalten oder, soweit zutreffend, Verweise auf entsprechende Vorschriften angeben für:
eine Beschreibung der Materialien mit a.der festgesetzten Bezeichnung und dem internen Referenzcode; ◦
Bezugnahme auf eine Arzneibuchmonographie, sofern vorhanden; ◦
der Angabe der zugelassenen Lieferanten und, wenn sinnvoll, des Originalherstellers der Produkte; ◦
einem Muster des bedruckten Verpackungsmaterials;◦
Vorschriften für die Probenahme und Prüfung; b.qualitative und quantitative Anforderungen mit den zulässigen Grenzwerten; c.Lagerungsbedingungen und Vorsichtsmaßnahmen; d.die maximale Lagerungsdauer bis zu einer Nachkontrolle. Spezifikationen für Zwischenprodukte und Bulkwaree.
4.15 Spezifikationen für Zwischenprodukte und Bulkware sollten für kritische Schritte oder dann zur Verfügung stehen, wenn diese als solche bezogen oder vertrieben werden. Die Spezifikationen sollten denen für Ausgangsstoffe oder, soweit zutreffend, denen für Fertigprodukte entsprechen.Spezifikationen für Fertigprodukte4.16 Die Spezifikationen für Fertigprodukte sollten folgende Angaben oder Verweise auf entsprechende Vorschriften beinhalten:
zu den festgesetzten Produktnamen und sofern zutreffend, zum internen Referenzcode; a.zur Zusammensetzung; b.eine Beschreibung der Darreichungsform und der Einzelheiten der Verpackung; c.Vorschriften für die Probenahme und Prüfung; d.zu den qualitativen und quantitativen Anforderungen mit den zulässigen Grenzwerten; e.zu Lagerungsbedingungen und etwaige Vorsichtsmaßnahmen; f.zur Haltbarkeitsdauer.g.
Seite 9 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffherste...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Herstellungsvorschriften und VerarbeitungsanweisungenFür alle herzustellenden Produkte und jede Chargengröße sollten ordnungsgemäß genehmigte Herstellungsvorschriften und Verarbeitungsanweisungen vorliegen.4.17 Die Herstellungsvorschriften sollten beinhalten:
den Produktnamen mit einem Produktreferenzcode, der auf die Spezifikation des Produkts hinweist; a.die Beschreibung der Darreichungsform, die Stärke des Produkts und die Chargengröße; b.eine Auflistung aller einzusetzenden Ausgangsstoffe mit den jeweiligen Mengen, bezeichnet mit den festgesetzten Namen und eindeutigen Referenzcodes; auch jede Substanz, die im Endprodukt nicht mehr enthalten ist sollte aufgeführt werden;
c.
Angaben zur erwarteten Endausbeute mit den zulässigen Grenzwerten und, soweit zutreffend, zur Ausbeute auf relevanten Zwischenstufen.
d.
4.18 Die Verarbeitungsanweisungen sollten beinhalten:
Angaben zur Verarbeitungsstätte und der wichtigsten zu verwendenden Ausrüstung; a.die Methoden oder eine Verweisung auf die Methoden, nach denen die kritischen Teile der Ausrüstung vorzubereiten sind (z.B. Reinigung, Montage, Kalibrierung, Sterilisation);
b.
Kontrollen, dass Ausrüstung und Arbeitsbereich frei von allen vorherigen, für den anlaufenden Vorgang nicht erforderlichen Produkte, Unterlagen oder Materialien sind und dass die Ausrüstung sauber und betriebsbereit ist;
c.
detaillierte schrittweise Verarbeitungsanweisungen (z.B. Materialkontrollen, Vorbehandlungen, Reihenfolge der Materialzugabe, kritische Prozessparameter [Zeiten, Temperaturen usw.]);
d.
Anweisungen für alle Inprozesskontrollen mit Grenzwerten; e.erforderlichenfalls die Anforderungen an die Lagerung der Bulkware, einschließlich der Behältnisse, der Kennzeichnung und, soweit zutreffend, spezieller Lagerungsbedingungen;
f.
alle besonderen Vorsichtsmaßnahmen, die zu beachten sind. Verpackungsanweisungeng.
4.19 Für jedes Produkt, jede Packungsgröße und jeden Packungstyp sollten ordnungsgemäß genehmigte Verpackungsanweisungen vorliegen. Diese sollten folgende Angaben oder Verweise auf entsprechende Vorschriften beinhalten:
Name des Produkts, einschließlich der Chargenbezeichnung der Bulkware und des Fertigproduktes; a.Beschreibung der Darreichungsform und, soweit zutreffend, der Stärke; b.die Packungsgröße, ausgedrückt in Zahl, Gewicht oder Volumen des Produktes im Endbehältnis; c.eine vollständige Auflistung aller für eine Standardchargengröße erforderlichen Verpackungsmaterialien nach Art, Größe und Menge, mit Angabe der Codierung oder Referenzzahl, die sich auf die Spezifikation des jeweiligen Verpackungsmaterials bezieht;
d.
soweit angezeigt, ein Muster oder eine Kopie des betreffenden bedruckten Verpackungsmaterials sowie Muster, die erkennen lassen, wo Chargenbezeichnung und Haltbarkeitsdauer des Produkts angegeben werden sollen;
e.
Kontrollen, dass Ausrüstung und Arbeitsbereich frei von allen vorherigen, für den anlaufenden Verpackungsvorgang nicht erforderlichen Produkte, Unterlagen oder Materialien sind (line clearance) und dass die Ausrüstung sauber und betriebsbereit ist;
f.
besondere Vorsichtsmaßnahmen, die zu beachten sind, einschließlich einer sorgfältigen Überprüfung des Verpackungsbereichs und der Ausrüstung, um die vollständige Räumung der Anlage vor Beginn der Verpackungsvorgänge (line clearance) sicherzustellen;
g.
eine Beschreibung der Verpackungsvorgänge mit allen wichtigen Nebenarbeiten und der einzusetzenden Ausrüstung; h.Einzelheiten zu Inprozesskontrollen mit Anweisungen für die Probenahme und den zulässigen Grenzwerten.i.
Protokolle der Chargenfertigung4.20 Für jede hergestellte Charge sollte ein Chargenverarbeitungsprotokoll angefertigt werden. Dieses sollte auf den entsprechenden Teilen der gültigen, genehmigten Herstellungsvorschrift und den zugehörigen Verarbeitungsanweisungen beruhen und folgende Informationen beinhalten:
Name und Chargenbezeichnung des Produkts; a.Daten und Zeiten des Verarbeitungsbeginns, von wichtigen Zwischenstufen und des Verarbeitungsendes; b.Identifizierung (Namenszeichen) des Bearbeiters/der Bearbeiter, der/die die verschiedenen, signifikanten Herstellungsschritte und, soweit zutreffend, Namenszeichen der Person, die diese Arbeitsgänge kontrolliert hat;
c.
die Chargennummer und/oder die Analysenkontrollnummer sowie die tatsächlich eingewogene Menge jedes Ausgangsstoffs (einschließlich der Chargenbezeichnung und der Menge jedes zugesetzten wiederverwerteten oder umgearbeiteten Materials);
d.
jeder relevante Verarbeitungsvorgang und jedes besondere Vorkommnis sowie die wichtigste eingesetzte Ausrüstung; e.Aufzeichnungen über die Inprozesskontrollen und die Namenszeichen der Person(en), die sie ausgeführt hat/haben, sowie die erhaltenen Ergebnisse;
f.
die Ausbeute des in den verschiedenen relevanten Herstellungsstufen erzielten Produkts; g.Angaben zu speziellen Problemen, einschließlich Einzelheiten zu jeder Abweichung von der Herstellungsvorschrift und den Verarbeitungsanweisungen mit Unterschrift der Person, die die Abweichung gebilligt hat;
h.
Billigung durch die für die Verarbeitungsvorgänge verantwortliche Person.i.
Hinweis:Soweit ein validierter Prozess kontinuierlich gesteuert und überwacht wird, können automatisch erzeugte Aufzeichnungen auf zusammenfassende Ergebnisberichte über die Übereinstimmung und Abweichungen/outof-specifications (OOS) Berichte beschränkt werden.Protokolle der Chargenverpackung4.21 Für jede Charge oder Teilcharge sollte ein Verpackungsprotokoll erstellt werden. Dieses sollte auf den entsprechenden Teilen der Verpackungsanweisungen beruhen.Das Protokoll über die Chargenverpackung sollte folgende Informationen beinhalten:
Seite 10 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Name und Chargenbezeichnung des Produkts; a.Datum/Daten und Zeiten der Verpackungsvorgänge; b.Identifizierung (Namenszeichen) des/der Bearbeiter der verschiedenen signifikanten Verpackungsschritte und, soweit zutreffend, Namenszeichen der Person, die diese Arbeitsgänge kontrolliert hat;
c.
Aufzeichnungen über Identitätskontrollen und Überprüfung auf Übereinstimmung mit den Verpackungsanweisungen, einschließlich der Ergebnisse von Inprozesskontrollen;
d.
Einzelheiten zu den durchgeführten Verpackungsvorgängen, einschließlich der Angaben über die verwendete Ausrüstung und die eingesetzten Verpackungslinien;
e.
wenn möglich, Proben des verwendeten bedruckten Verpackungsmaterials, einschließlich Muster mit der Chargenkennzeichnung, dem Aufdruck des Verfalldatums und anderen zusätzlichen Aufdrucken;
f.
Angaben zu speziellen Problemen oder ungewöhnlichen Vorkommnissen bei Abweichung von der Herstellungsvorschrift oder den Verarbeitungsanweisungen mit der Unterschrift der zuständigen Person, die die Abweichung gebilligt hat;
g.
Mengen und Referenznummern oder andere Angaben zur Identifizierung aller bereitgestellten, verwendeten, vernichteten oder ins Lager zurückgegebenen bedruckten Verpackungsmaterialien und der Bulkware sowie die Menge des erzielten Produkts, um eine entsprechende Bilanzierung zu ermöglichen. Auf diese Information kann verzichtet werden, soweit robuste elektronische Kontrollen während des Verpackungsvorgangs vorhanden sind;
h.
Billigung durch die für die Verarbeitungsvorgänge verantwortliche Person.i.
Verfahrensbeschreibungen und Protokolle Wareneingang4.22 Es sollten schriftliche Verfahrensbeschreibungen und Protokolle für die Annahme jeder Lieferung eines jeden Ausgangsstoffs (einschließlich Bulkware, Zwischenprodukte oder Fertigprodukte) und jedes primären, sekundären und bedruckten Verpackungsmaterials vorhanden sein.4.23 Die Protokolle des Wareneingangs sollten beinhalten:
den Namen des Materials auf dem Lieferschein und den Behältnissen; a.den firmenintern gebräuchlichen Namen und/oder Materialcode (wenn dieser sich von Buchstabe a unterscheidet); b.das Datum des Wareneingangs; c.den Namen des Lieferanten und des Herstellers; d.die Chargenbezeichnung oder Referenznummer des Herstellers; e.die Gesamtmenge und die Anzahl der erhaltenen Behältnisse; f.die der Charge nach dem Eingang zugewiesene Chargenbezeichnung; g.besondere Bemerkungenh.
4.24 Es sollten schriftliche Verfahrensbeschreibungen vorliegen für die interne Kennzeichnung, die Quarantäne und die Lagerung der Ausgangsstoffe, des Verpackungsmaterials und, soweit zutreffend, anderer Materialien.Probenahme4.25 Es sollten schriftliche Verfahrensbeschreibungen für die Probenahme vorliegen, die Angaben enthalten über die Methoden der Probenahme und die einzusetzende Ausrüstung, die zu entnehmenden Probenmengen und alle Vorsichtsmaßnahmen, die zu beachten sind, um eine Verunreinigung des Materials oder sonstige Qualitätsminderungen zu vermeiden.Prüfung4.26 Es sollten schriftliche Verfahrensbeschreibungen für die Prüfung von Materialien und Produkten auf den verschiedenen Herstellungsstufen vorliegen, in denen die Prüfmethoden und die einzusetzende Ausrüstung angegeben sind. Die ausgeführten Prüfungen sollten protokolliert werden.Sonstiges4.27 Es sollten schriftliche Verfahrensbeschreibungen für die Freigabe und Zurückweisung von Materialien und Produkten zur Verfügung stehen. Dies gilt besonders für die Freigabe des Fertigprodukts durch die Sachkundige(n) Person(en). Es sollte ein System vorhanden sein, mit dem spezielle Beobachtungen und jede Änderung kritischer Daten angezeigt werden.4.28 Es sollten Protokolle über den Vertrieb einer jeden Produktcharge angefertigt und aufbewahrt werden, um erforderlichenfalls den Rückruf der Charge zu erleichtern.4.29 Es sollten schriftliche Festlegungen über Grundsätze, Verfahren, Anweisungen, Berichte und die zugehörigen Protokolle über durchgeführte Maßnahmen oder über getroffene Schlussfolgerungen, soweit zutreffend, vorliegen für:
Validierung und Qualifizierung von Prozessen, Ausrüstungen und Systemen; •Montage und Kalibrierung der Ausrüstung; •Technologietransfer; •Wartung, Reinigung und Desinfektion; •personalbezogene Belange, einschließlich einer Unterschriftenliste, Schulung in GMP und technischen Angelegenheiten, Kleidung und Hygiene und Nachweis der Wirksamkeit der Schulung;
•
Umgebungskontrollen; •Bekämpfung von Ungeziefer; •Beanstandungen; •Rückrufe; •Rückgaben; •Change Control; •Untersuchungen von Abweichungen und Nicht-Übereinstimmungen; •interne Audits zu Qualitäts-/GMP-Übereinstimmung; •Zusammenfassung von Berichten, soweit angezeigt (z.B. Produktqualitätsüberprüfungen); •Lieferantenaudits.•
4.30 Für die wichtigsten Teile der Herstellungs- und Prüfausrüstung sollten klare Gebrauchsanweisungen zur Verfügung stehen.
Seite 11 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

4.31 Für sehr wichtige oder kritische Ausrüstungsteile für die analytische Testung und die Produktion sowie die entsprechenden Herstellungsbereiche sollten Logbücher geführt werden. In den Logbüchern sollten zeitlich geordnete Aufzeichnungen über die Benutzung der Herstellungsbereiche, der Ausrüstungsteile, der Methoden, Kalibrierungen, Wartungen, Reinigungs- oder Reparaturarbeiten vermerkt werden, mit Datum und Angabe der Personen, die diese Tätigkeiten ausgeführt haben.4.32 Ein Inventar der Dokumente sollte im Qualitätsmanagementsystem enthalten sein.*) Im anderen Falle kann die Zertifizierung ganz oder teilweise auf der Abschätzung der "real time data" (zusammenfassende Berichte und "exception reports") der chargenbezogenen prozessanalytischen Technologie (PAT) oder den Parametern oder Messwerten entsprechend den Zulassungsunterlagen basieren.
weiter
Bekanntmachung zu § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV )
Vom 8. August 2011 (BAnz. Nr. 125 vom 19.08.2011 S. 2901)
Die Europäische Kommission hat den Anhang 11 zum Leitfaden für die Gute Herstellungspraxis für Arzneimittel- und Prüfpräparate (EG-GMP Leitfaden) geändert und im Januar 2011 in englischer Sprache auf ihrer Internetseite veröffentlicht. Darüber hinaus hat die Kommission auch Kapitel 4 des EG-GMP Leitfadens in Zusammenhang mit der Änderung des Anhangs 11 geändert, um dem steigenden Einsatz elektronischer Dokumente im GMP-Bereich Rechnung zu tragen. Der neue Anhang 11 und das überarbeitete Kapitel 4 des EG-GMP Leitfadens sind ab dem 30. Juni 2011 anzuwenden.Hiermit werden der vom Bundesministerium für Gesundheit in die deutsche Sprache übersetzte Anhang 11 und das Kapitel 4 des EG-GMP Leitfadens nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung vom 3. November 2006 (BGBl. I S. 2523), die zuletzt durch Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung vom 26. März 2008 (BGBl. I S. 521) geändert worden ist, bekannt gemacht (Anlage 1 und 2).Der Leitfaden und die nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung bekannt gemachten Anhänge sind auch auf der Internetseite des Bundesministeriums für Gesundheit, www.bmg.bund.de, abrufbar.umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Teil I (2/2)
Kapitel 5 Produktion
GrundsätzeDie Produktionsvorgänge müssen nach klar definierten Verfahren erfolgen; sie müssen den Grundsätzen der Guten Herstellungspraxis entsprechen, um zu Produkten zu führen, die die erforderliche Qualität aufweisen und mit der Herstellungserlaubnis und den jeweiligen Zulassungsunterlagen übereinstimmen. Allgemeine Anforderungen5.1 Die Produktion sollte von sachkundigem Personal ausgeführt und überwacht werden. 5.2 Jeder Umgang mit Materialien und Produkten, z.B. Wareneingang und Quarantäne, Probenahme, Lagerung, Kennzeichnung, Bereitstellung, Verarbeitung, Verpackung und Vertrieb, sollte in Übereinstimmung mit schriftlich festgelegten Verfahren oder Anweisungen durchgeführt und - wenn nötig - protokolliert werden. 5.3 Alle eingehenden Materialien sollten kontrolliert werden, um sicherzustellen, dass die Lieferung der Bestellung entspricht. Behältnisse sollten erforderlichenfalls gereinigt und mit den vorgeschriebenen Angaben gekennzeichnet werden. 5.4 Schäden an Behältnissen und alle anderen Probleme, die die Materialqualität beeinträchtigen könnten, sollten untersucht, protokolliert und der Qualitätskontrollabteilung gemeldet werden. 5.5 Eingehende Materialien und Fertigprodukte sollten sofort nach Eingang oder Verarbeitung bis zu ihrer Freigabe für Verwendung oder Vertrieb durch getrennte Lagerung oder durch geeignete administrative Maßnahmen in Quarantäne gehalten werden. 5.6 Zwischenprodukte und Bulkware, die als solche gekauft werden, sollten bei der Annahme wie Ausgangsstoffe behandelt werden. 5.7 Alle Materialien und Produkte sollten unter geeigneten, vom Hersteller festgelegten Bedingungen sowie übersichtlich gelagert werden, um eine Trennung nach Chargen und die Umwälzung des Lagerbestandes zu ermöglichen. 5.8 Kontrollen der Ausbeuten und eine Bilanzierung der Mengen sollten nötigenfalls durchgeführt werden, um sicherzustellen, dass keine über die zulässigen Grenzen hinausgehenden Diskrepanzen auftreten. 5.9 Die Bearbeitung unterschiedlicher Produkte sollte nicht gleichzeitig oder nacheinander in demselben Raum durchgeführt werden, es sei denn, es besteht keine Gefahr der Verwechslung oder Kreuzkontamination. 5.10 Auf jeder Verarbeitungsstufe sollten Produkte und Materialien vor mikrobieller und anderer Verunreinigung geschützt werden. 5.11 Bei Arbeiten mit trockenen Materialien und Produkten sollten spezielle Vorkehrungen getroffen werden, um eine Staubbildung und -ausbreitung zu verhüten. Dies gilt besonders für den Umgang mit hochaktiven oder sensibilisierenden Materialien. 5.12 Während der gesamten Verarbeitungszeit sollten alle verwendeten Materialien, Behältnisse mit Bulkware, wichtigen Ausrüstungsteile und, soweit angemessen, auch Räume, beschriftet oder auf andere Weise mit einem Hinweis auf das verarbeitete Produkt oder Material, seiner Chargenbezeichnung und gegebenenfalls seiner Stärke gekennzeichnet werden. Soweit angezeigt, sollte in diesem Hinweis auch die Herstellungsstufe vermerkt sein. 5.13 Etiketten oder Hinweise an Behältnissen, Ausrüstung oder Räumen sollten klar und eindeutig sein und der firmenintern festgelegten Aufmachung entsprechen. Es ist oft hilfreich. den Status (z.B. in Quarantäne. angenommen. zurückgewiesen. sauber usw.) außer in Worten auch mit verschiedenen Farben anzuzeigen. 5.14 Durch Kontrollen sollte sichergestellt werden, dass Rohrleitungen und andere Ausrüstungsteile, die für den Transport eines Produkts von einem Bereich in einen anderen verwendet werden, vorschriftsmäßig miteinander verbunden sind. 5.15 Jede Abweichung von Anweisung und Verfahrensbeschreibungen sollte weitestgehend vermieden werden. Wenn Abweichungen vorkommen. sollten sie schriftlich von einer dafür zuständigen Person. soweit angemessen in Zusammenarbeit mit der Qualitätskontrollabteilung, gebilligt werden. 5.16 Der Zutritt zu den Produktionsbereichen sollte nur Befugten gestattet sein. 5.17 In der Regel sollten Erzeugnisse, die keine Arzneimittel sind, nicht in Bereichen und mit Ausrüstungsteilen produziert werden, die für die Produktion von Arzneimitteln bestimmt sind. Verhütung von Kreuzkontamination bei der Produktion5.18 Die Kontamination eines Ausgangsstoffs oder eines Produkts mit einem anderen Material oder Produkt muss vermieden werden. Die Gefahr einer unbeabsichtigten Kreuzkontamination resultiert aus der unkontrollierten Freisetzung von Staub, Gasen, Dämpfen, Aerosolen oder Organismen von in der Verarbeitung befindlichen Materialien und Produkten, aus Rückständen in der Ausrüstung oder aus der
Seite 12 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Arbeitskleidung. Das resultierende Risiko ist je nach Typ des verunreinigenden Stoffes und des betroffenen Produkts unterschiedlich groß. Hochsensibilisierende Stoffe, biologische Zubereitungen mit lebenden Organismen, bestimmte Hormone, Zytostatika und andere hochwirksame Stoffe zählen zu den gefährlichsten Verunreinigungen. Bei Produkten, die infundiert oder injiziert, in großen Dosen und/oder über einen langen Zeitraum verabreicht werden, ist eine Kontamination am schwerwiegendsten. 5.19 Kreuzkontamination sollte durch geeignete technische oder organisatorische Maßnahmen vermieden werden, z.B. durch:
Produktion in räumlich abgetrennten Bereichen (erforderlich für Produkte wie Penicilline, Lebendimpfstoffe, Zubereitungen, die lebende Bakterien enthalten, und einige andere biologische Präparate) oder in Kampagnen (zeitlich getrennt) mit anschließender gründlicher Reinigung;
a.
geeignete Schleusen und Abzüge;b.Minimierung des Risikos einer Kontamination, verursacht durch Rezirkulation oder Wiedereintritt von unbehandelter oder ungenügend behandelter Luft;
c.
Belassen der Schutzkleidung in Bereichen, in denen Produkte verarbeitet werden, von denen ein besonders großes Risiko einer Kreuzkontamination ausgeht;
d.
Verwendung von Reinigungs- und Dekontaminationsverfahren mit bekannter Wirksamkeit, da die ungenügende Reinigung der Ausrüstung eine häufige Ursache der Kreuzkontamination ist;
e.
Einsatz "geschlossener Systeme" bei der Produktion;f.Prüfung auf Rückstände und Verwendung von Etiketten, die den Reinigungsstatus der Ausrüstung angeben.g.
5.20 Die Maßnahmen zur Verhütung der Kreuzkontamination und ihre Wirksamkeit sollten in regelmäßigen Abständen nach festgelegten Verfahren überprüft werden. Validierung5.21 Validierungsstudien sollten die Gute Herstellungspraxis stärken und nach festgelegten Verfahren durchgeführt werden. Die Ergebnisse und Schlussfolgerungen sollten protokolliert werden. 5.22 Wenn eine neue Herstellungsvorschrift oder Verarbeitungsmethode eingeführt wird, sollte deren Eignung für den Routinebetrieb nachgewiesen werden. es sollte gezeigt werden, dass der definierte Prozess bei Einsatz der festgelegten Materialien und Ausrüstung zu einem Produkt führt, das gleiche bleibend die erforderliche Qualität aufweist. 5.23 Wesentliche Änderungen des Herstellungsprozesses, einschließlich aller Ausrüstungs- oder Materialänderungen, die die Produktqualität und/oder die Reproduzierbarkeit des Prozesses beeinflussen können, sollten validiert werden. 5.24 Arbeitsgänge und Verfahren sollten in regelmäßigen Abständen einer kritischen Revalidierung unterzogen werden, um sicherzustellen, dass sie weiterhin zu den gewünschten Ergebnissen führen. Ausgangsstoffe5.25 Der Einkauf der Ausgangsstoffe ist ein wichtiger Vorgang, an dem Personal beteiligt sein sollte, das über die Lieferanten sehr genaue und gründliche Kenntnisse hat. 5.26 Ausgangsstoffe sollten nur von zugelassenen Lieferanten bezogen werden, die in der entsprechenden Spezifikation genannt werden und, wo möglich, direkt beim Hersteller. Es wird empfohlen, dass der pharmazeutische Hersteller die von ihm festgelegten Spezifikationen für Ausgangsstoffe mit den Lieferanten diskutiert. Es ist von Vorteil, dass alle Gesichtspunkte der Produktion und Kontrolle des jeweiligen Ausgangsstoffs, einschließlich der Anforderungen an dessen Handhabung, Kennzeichnung und Verpackung sowie Beanstandungen und Zurückweisungsverfahren mit dem Hersteller und Lieferanten erörtert werden. 5.27 Bei jeder Lieferung sollte kontrolliert werden, ob Verpackung und Verschluss der Behältnisse unversehrt sind und die Angaben auf dem Lieferschein und den Etiketten des Lieferanten übereinstimmen. 5.28 Wenn eine Materiallieferung aus verschiedenen Chargen besteht, muss jede Charge hinsichtlich Probenahme, Prüfung und Freigabe einzeln betrachtet werden. 5.29 Im Lagerbereich befindliche Ausgangsstoffe sollten in geeigneter Weise gekennzeichnet sein (siehe Nummer 5.13). Die Kennzeichnung sollte mindestens folgende Informationen enthalten:
den festgesetzten Namen des Produkts und, soweit zutreffend, einen internen Referenzcode;•die beim Wareneingang zugewiesene Chargenbezeichnung;•soweit angezeigt, den Status des Inhalts (z.B. in Quarantäne, in der Prüfung, freigegeben, zurückgewiesen);•soweit angebracht, ein Verfalldatum oder ein Datum, nach dem eine Nachprüfung erforderlich ist.•
Bei vollständig computergesteuerten Lagersystemen müssen die obigen Informationen nicht unbedingt in lesbarer Form auf dem Etikett enthalten sein. 5.30 Mit geeigneten Verfahren oder Maßnahmen sollte die Identität des Inhalts eines jeden Behältnisses mit Ausgangsstoffen sichergestellt werden. Behältnisse mit Bulkware, aus denen Proben entnommen worden sind, sollten eindeutig entsprechend gekennzeichnet werden (siehe Nummer 6.13). 5.31 Es sollten nur Ausgangsstoffe verwendet werden, die von der Qualitätskontrolle freigegeben wurden und deren Haltbarkeitsdauer nicht überschritten ist. 5.32 Ausgangsstoffe sollten nur von den hierzu beauftragten Personen nach schriftlich festgelegten Verfahren zur Verarbeitung bereitgestellt werden, um sicherzustellen, dass die richtigen Stoffe in saubere und ordnungsgemäß gekennzeichnete Behältnisse genau eingewogen oder abgemessen werden. 5.33 Jedes zur Verarbeitung bereitgestellte Material und sein Gewicht oder Volumen sollten unabhängig kontrolliert werden. Die Kontrolle sollte protokolliert werden. 5.34 Die für jede einzelne Charge bereitgestellten Materialien sollten beieinander gehalten und deutlich entsprechend gekennzeichnet werden. Verarbeitungsvorgänge: Zwischenprodukte und Bulkware5.35 Vor jeden Verarbeitungsvorgang sollte sichergestellt werden, dass Arbeitsbereich und Ausrüstung sauber und frei von allen für die geplanten Arbeitsgänge nicht benötigten Ausgangsstoffen, Produkten, Produktrückständen oder Unterlagen sind. 5.36 Zwischenprodukte und Bulkware sollten unter geeigneten Bedingungen aufbewahrt werden. 5.37 Kritische Vorgänge sollten validiert sein (siehe "Validierung" in diesem Kapitel). 5.38 Alle erforderlichen Inprozess- und Umgebungskontrollen sollten durchgeführt und protokolliert werden. 5.39 Jede signifikante Abweichung von der erwarteten Ausbeute sollte protokolliert und untersucht werden. Verpackungsmaterial
Seite 13 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

5.40 Dem Einkauf, der Handhabung und der Kontrolle des primären und bedruckten Verpackungsmaterials sollte ebensoviel Aufmerksamkeit gewidmet werden wie den Ausgangsstoffen. 5.41 Besondere Vorsicht ist bei bedruckten Materialien geboten. Sie sollten unter ausreichend sicheren Bedingungen gelagert werden, um unbefugten Zugriff auszuschließen. Lose Etiketten und andere lose, bedruckte Materialien sollten in separaten, geschlossenen Behältnissen aufbewahrt und transportiert werden, um Verwechslungen zu vermeiden. Verpackungsmaterial sollte nur nach einem genehmigten und dokumentierten Verfahren von dazu befugtem Personal für den Gebrauch ausgegeben werden. 5.42 Jede Lieferung oder Charge von bedrucktem oder primärem Verpackungsmaterial sollte eine spezifische Kennzahl oder Markierung erhalten. 5.43 Überholtes oder veraltetes primäres oder bedrucktes Verpackungsmaterial sollte vernichtet werden. Die Vernichtung sollte protokolliert werden. Verpackungsvorgänge5.44 Bei der Planung der Verpackungsvorgänge muss besonders darauf geachtet werden, dass das Risiko von Kreuzkontamination, Untermischung oder Verwechslungen minimiert wird. Unterschiedliche Produkte sollten nicht in unmittelbarer Nähe zueinander verpackt werden, es sei denn, sie sind räumlich voneinander getrennt. 5.45 Vor Beginn der Verpackungsvorgänge sollte sichergestellt werden, dass der Arbeitsbereich, die Verpackungslinien, die Druckmaschinen und andere Ausrüstung sauber und frei von allen vorher verwendeten Produkten, Materialien oder Unterlagen sind, wenn diese für den anlaufenden Vorgang nicht benötigt werden. Die vollständige Räumung der Anlage sollte anhand einer geeigneten Checkliste erfolgen. 5.46 Der Name und die Chargenbezeichnung des jeweils zu verpackenden Produkts sollten an jedem Verpackungsplatz oder jeder Verpackungslinie angezeigt sein. 5.47 Alle einzusetzenden Produkte und Verpackungsmaterialien sollten bei Anlieferung an die Verpackungsabteilung hinsichtlich Menge, Identität und Übereinstimmung mit den Verpackungsanweisungen kontrolliert werden. 5.48 Zu füllende Behältnisse sollten sauber sein. Alle Verunreinigungen wie Glas- oder Metallteilchen sollten sorgfältig vermieden bzw. entfernt werden. 5.49 Normalerweise sollte das Etikettieren so schnell wie möglich auf das Abfüllen und Verschließen folgen. Wenn dies nicht der Fall ist, sollten geeignete Verfahren angewandt werden, um Verwechslungen oder Falschetikettierungen auszuschließen. 5.50 Die einwandfreie Durchführung jedes Druckvorganges (z.B. Aufdruck von Codenummern, Verfalldaten l. der getrennt oder während des Verpackens erfolgt, sollte kontrolliert und protokolliert werden. Bei nichtmaschinellem Drucken sollte besonders aufmerksam verfahren werden. Dieses sollte auch in regelmäßigen Abständen überprüft werden. 5.51 Besondere Sorgfalt sollte walten, wenn lose Etiketten verwendet und Aufdrucke nicht auf der Verpackungsanlage selbst (offline) angebracht werden. Etiketten auf Rollen sind losen Etiketten normalerweise vorzuziehen, da sich Untermischungen so besser vermeiden lassen. 5.52 Es sollte kontrolliert werden, dass elektronische Code-Lesegeräte, Etikettenzähler oder ähnliche Geräte einwandfrei arbeiten. 5.53 Gedruckte und geprägte Informationen auf Verpackungsmaterialien sollten deutlich, lichtecht und abriebfest sein. 5.54 Die laufende Kontrolle des Produkts auf der Anlage während des Verpackens (online) sollte mindestens Folgendes beinhalten:
das allgemeine Aussehen der Packungen;a.die Vollständigkeit der Packungen;b.den Einsatz der richtigen Produkte und Verpackungsmaterialien;c.die Richtigkeit der Aufdrucke;d.die einwandfreie Funktion der Überwachungsvorrichtungen der Anlage.e.
Von der Verpackungslinie entfernte Proben sollten nicht wieder in den Prozess eingeschleust werden. 5.55 Produkte, die an einem ungewöhnlichen Vorgang beteiligt waren, sollten nur nach besonderer Inspektion, Untersuchung und Genehmigung durch dazu befugtes Personal wieder in den Prozess eingeschleust werden. Darüber sollten detaillierte Aufzeichnungen aufbewahrt werden. 5.56 Jede bei der Bilanzierung festgestellte signifikante oder ungewöhnliche Diskrepanz zwischen der Menge an Bulkware und den bedruckten Verpackungsmaterialien und der Anzahl der fertig gestellten Einheiten sollte vor der Freigabe untersucht und ausreichend begründet werden. 5.57 Nach Beendigung eines Verpackungsvorgangs sollte ungebrauchtes, mit der Chargenbezeichnung versehenes Verpackungsmaterial vernichtet und dieser Vorgang protokolliert werden. Bedrucktes, nicht mit der Chargenbezeichnung versehenes Material sollte nur nach einem schriftlich festgelegten Verfahren ins Lager zurückgegeben werden. Fertigprodukte5.58 Fertigprodukte sollten bis zu ihrer endgültigen Freigabe unter vom Hersteller festgelegten Bedingungen in Quarantäne gehalten werden. 5.59 Die vor der Freigabe von Fertigprodukten zum Verkauf erforderliche Bewertung des Fertigprodukts und der Dokumentation wird in Kapitel 6 (Qualitätskontrolle) beschrieben. 5.60 Nach der Freigabe sollten Fertigprodukte als verfügbarer Bestand unter vom Hersteller festgelegten Bedingungen gelagert werden. Zurückgewiesene, wiederverwertete und zurückgegebene Materialien5.61 Zurückgewiesene Materialien und Produkte sollten klar als solche gekennzeichnet und gesondert in nicht allgemein zugänglichen Bereichen gelagert werden. Sie sollten entweder an den Lieferanten zurückgegeben oder, soweit angemessen, umgearbeitet oder vernichtet werden. Die jeweils durchgeführte Maßnahme sollte von dazu befugtem Personal genehmigt und protokolliert werden. 5.62 Die Umarbeitung von zurückgewiesenen Produkten sollte die Ausnahme sein. Die Umarbeitung ist nur zulässig, wenn die Qualität des Endprodukts nicht beeinträchtigt wird, wenn die Spezifikationen eingehalten werden und wenn die Umarbeitung in Übereinstimmung mit einem definierten und genehmigten Verfahren nach Abschätzung der dabei bestehenden Risiken durchgeführt wird. Die Umarbeitung sollte protokolliert werden. 5.63 Das vollständige oder teilweise Einbringen früherer Chargen von der erforderlichen Qualität in eine Charge desselben Produkts auf einer bestimmten Herstellungsstufe sollte vorher genehmigt werden. Die Wiederverwertung sollte in Übereinstimmung mit einem festgelegten Verfahren nach Abschätzung der dabei bestehenden Risiken, einschließlich einer möglichen Auswirkung auf die Haltbarkeitsdauer, durchgeführt werden. Die Wiederverwertung sollte protokolliert werden. 5.64 Über die Notwendigkeit zusätzlicher Prüfungen des Fertigprodukts, das umgearbeitet oder in das ein wiederverwertetes Produkt eingebracht wurde, sollte von der Qualitätskontrollabteilung entschieden werden.
Seite 14 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

5.65 Aus dem Handel zurückgegebene, der Kontrolle des Herstellers zwischenzeitlich entzogene Produkte sollten vernichtet werden, es sei denn, sie weisen zweifelsfrei die erforderliche Qualität auf. Für erneuten Verkauf, Umetikettierung oder für ein Einbringen in eine spätere Charge können sie nur in Betracht kommen, wenn sie von der Qualitätskontrollabteilung nach einem schriftlich festgelegten Verfahren kritisch beurteilt wurden. Bei dieser Beurteilung sollten die Art des Produkts, evtl. erforderliche besondere Lagerungsbedingungen, sein Zustand und seine Geschichte sowie die Zeitspanne seit seiner Auslieferung berücksichtigt werden. Wenn irgendein Zweifel über die Qualität des Produkts aufkommt, sollte eine erneute Auslieferung oder erneute Verwendung nicht in Erwägung gezogen werden. Eine grundlegende chemische Aufarbeitung zur Rückgewinnung des Wirkstoffs kann jedoch möglich sein. Jede durchgeführte Maßnahme sollte in geeigneter Weise protokolliert werden.
Kapitel 6 Qualitätskontrolle
GrundsätzeDie Qualitätskontrolle befasst sich mit Probenahme, Spezifikationen und Prüfung sowie Organisation, Dokumentation und Freigabeverfahren, die sicherstellen, dass die jeweils notwendigen und relevanten Prüfungen durchgeführt und weder Materialien für den Einsatz noch Produkte für den Verkauf oder die Auslieferung freigegeben werden, bevor ihre Qualität als zufrieden stellend beurteilt wurde. Die Qualitätskontrolle ist nicht auf Laborarbeiten beschränkt, sondern muss bei allen die Produktqualität betreffenden Entscheidungen beteiligt sein. Die Unabhängigkeit von der Produktion ist für das ordnungsgemäße Arbeiten der Qualitätskontrolle von grundlegender Bedeutung (siehe auch Kapitel 1). Allgemeine Anforderungen6.1 Jeder Inhaber einer Herstellungserlaubnis sollte über eine Qualitätskontrollabteilung verfügen. Diese Abteilung sollte von anderen Abteilungen unabhängig sein und unter der Leitung einer Person mit angemessener Qualifikation und Erfahrung stehen, die ein oder mehrere Kontrolllaboratorien zur Verfügung hat. Es müssen ausreichende Mittel verfügbar sein, damit alle Maßnahmen der Qualitätskontrolle wirksam und zuverlässig ausgeführt werden können. 6.2 Die wichtigsten Pflichten des Leiters der Qualitätskontrolle sind in Kapitel 2 zusammengefasst. Die Qualitätskontrollabteilung insgesamt hat noch weitere Aufgaben, wie die Festlegung, Validierung und Ausführung aller Qualitätskontrollverfahren, Aufbewahrung von Rückstellmustern von Materialien und Produkten, Sicherstellung der ordnungsgemäßen Kennzeichnung der Behältnisse, die die Materialien und Produkte enthalten, Sicherstellung der Überwachung der Produktstabilität, Mitwirkung an der Untersuchung von Beanstandungen hinsichtlich der Produktqualität usw. Alle diese Vorgänge sollten gemäß schriftlich festgelegten Verfahren durchgeführt und, wenn nötig, protokolliert werden. 6.3 Die Bewertung des Fertigprodukts sollte alle relevanten Faktoren umfassen, einschließlich der Produktionsbedingungen, der Ergebnisse von Inprozesskontrollen, der nochmaligen Überprüfung der Herstellungs- (einschließlich Verpackungsdokumentation, der Übereinstimmung mit den Spezifikationen des Fertigprodukts und der Überprüfung der fertigen Packung. 6.4 Das Personal der Qualitätskontrolle sollte Zugang zu den Produktionsbereichen haben, um Proben zu nehmen und Untersuchungen durchzuführen, soweit angebracht. Gute Kontrolllabor-Praxis6.5 Räumlichkeiten und Ausrüstung von Kontrolllaboratorien sollten den in Kapitel 3 beschriebenen allgemeinen und besonderen Anforderungen an Qualitätskontrollbereiche entsprechen. 6.6 Das Personal, die Räumlichkeiten und die Ausrüstung in den Laboratorien sollten den Aufgaben entsprechen, die sich aus der Art und dem Umfang der Herstellungstätigkeiten ergeben. Der Einsatz externer Laboratorien in Übereinstimmung mit den in Kapitel 7 "Prüfung im Lohnauftrag" beschriebenen Grundsätzen kann aus bestimmten Gründen akzeptiert werden. Dies sollte jedoch in den Protokollen der Qualitätskontrolle vermerkt werden. Dokumentation6.7 Laborunterlagen sollten den im Kapitel 4 genannten Anforderungen entsprechen. Ein wesentlicher Teil der dort genannten Dokumentation betrifft die Qualitätskontrolle. Die folgenden Unterlagen sollten der Qualitätskontrollabteilung ohne Weiteres zur Verfügung stehen:
Spezifikationen;•Probenahmeverfahren;•Prüfverfahren und Prüfprotokolle (einschließlich analytischer Arbeitsblätter und/oder Laborjournale);•Analysenberichte und/oder -zertifikate;•soweit erforderlich, Daten aus der Überwachung der Umgebungsbedingungen;•soweit zutreffend, Protokolle über die Validierung der Prüfmethoden;•Verfahrensbeschreibungen für und Protokolle über die Kalibrierung von Geräten und Wartung der Ausrüstung.•
6.8 Alle Unterlagen der Qualitätskontrolle, die sich auf ein Chargenprotokoll beziehen, sollten ein Jahr über das Verfalldatum der Charge und mindestens fünf Jahre über das Ausstellen der Bescheinigung gemäß Artikel 51 Abs. 3 der Richtlinie 2001/83/EG 8 hinaus aufbewahrt werden. 6.9 Es wird empfohlen, einige Daten (z.B. Ergebnisse der analytischen Prüfung, Ausbeuten, Umgebungskontrollen usw.) so aufzubewahren, dass Trends ermittelt werden können. 6.10 Zusätzlich zu den zu einem Chargenprotokoll gehörenden Informationen sollten andere Originalunterlagen wie Laborjournale und/oder -aufzeichnungen aufbewahrt werden und schnell zur Verfügung stehen. Probenahme6.11 Die Probenahme sollte nach genehmigten, schriftlich festgelegten Verfahren erfolgen, die folgende Angaben enthalten:
Methode der Probenahme;•einzusetzende Ausrüstung;•zu entnehmende Probenmenge;•Anweisungen für jede erforderliche Unterteilung der Probe;•Art und Zustand des zu verwendenden Probenbehältnisses;•identifizierende Kennzeichnung von Behältnissen, aus denen Proben gezogen werden;•alle zu beachtenden spezifischen Vorsichtsmaßnahmen, insbesondere hei der Probenahme von sterilen oder gefährlichen Materialien;
•
Seite 15 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Lagerungsbedingungen;•Anweisungen für die Reinigung und Aufbewahrung der Probenahmeausrüstung.•
6.12 Referenzproben sollten für die Material- oder Produktcharge, der sie entnommen wurden, repräsentativ sein. Es können weitere Proben entnommen werden, um sehr kritische Prozessschritte zu überwachen (z.. B. Prozessbeginn oder Prozessende). 6.13 Die Probenbehältnisse sollten Etiketten tragen mit Angabe des Inhalts, der Chargenbezeichnung, des Datums der Probenahme und der Behältnisse, aus denen die Proben entnommen wurden. 6.14 Weitere Anleitungen zu Referenz- und Rückstellmustern sind in Anhang 19 beschrieben. Prüfung6.15 Analytische Methoden sollten validiert sein. Alle in den Zulassungsunterlagen beschriebenen Prüfungen sollten in Übereinstimmung mit den genehmigten Methoden durchgeführt werden. 6.16 Die erhaltenen Ergebnisse sollten protokolliert und daraufhin kontrolliert werden, ob sie miteinander in Einklang stehen. Alle Berechnungen sollten kritisch überprüft werden. 6.17 Die durchgeführten Prüfungen sollten protokolliert werden. Die Protokolle sollten mindestens folgende Angaben enthalten:
Name des Materials oder Produkts und gegebenenfalls Darreichungsform;a.Chargenbezeichnung und, soweit angebracht, Hersteller und/ oder Lieferant;b.Bezugnahme auf die jeweiligen Spezifikationen und Prüfverfahren;c.Prüfergebnisse, einschließlich Beobachtungen und Berechnungen, sowie Referenz zu Analysenzertifikaten;d.Daten der Prüfung;e.Namenszeichen der Personen, die die Prüfungen durchgeführt haben;f.soweit angebracht, Namenszeichen der Personen, die die Prüfungen und Berechnungen verifiziert haben;g.klare Aussage zur Freigabe oder Zurückweisung (oder eine andere Entscheidung hinsichtlich des Status) mit Datum und Unterschrift der hierfür als verantwortlich bestellten Person.
h.
6.18 Alle Inprozesskontrollen, auch die im Produktionsbereich vom dortigen Personal durchgeführten, sollten nach Methoden erfolgen, die von der Qualitätskontrolle genehmigt sind. Die Ergebnisse sollten protokolliert werden. 6.19 Auf die Qualität von Laborreagenzien, Volumenmessgeräten, volumetrischen Lösungen, Referenzstandards und Kulturmedien sollte besonders geachtet werden. Ihre Zubereitung sollte nach schriftlich festgelegten Verfahren erfolgen. 6.20 Laborreagenzien, die für längeren Gebrauch vorgesehen sind, sollten mit dem Datum ihrer Zubereitung und der Unterschrift der Person versehen sein, die sie hergestellt hat. Das Verfalldatum von instabilen Reagenzien und von Kulturmedien sowie besondere Aufbewahrungsbedingungen sollten auf dem Etikett angegeben werden. Außerdem sollten bei volumetrischen Lösungen das Datum der letzten Einstellung und der jeweils gültige Faktor vermerkt sein. 6.21 Falls nötig, sollte das Eingangsdatum von jeder für die Prüfungen verwendeten Substanz (z.B. Reagenzien und Referenzstandards) auf dem Behältnis vermerkt werden. Anweisungen für Gebrauch und Aufbewahrung sollten befolgt werden. In bestimmten Fällen kann eine Identitätsprüfung und/oder eine andere Prüfung der Reagenzien nach Erhalt oder vor Gebrauch nötig sein. 6.22 Tiere, die bei der Prüfung von Bestandteilen, Materialien oder Produkten eingesetzt werden, sollten, soweit angebracht, vor ihrer Verwendung in Quarantäne gehalten werden. Sie sollten so gehalten und kontrolliert werden, dass ihre Eignung für die beabsichtigte Verwendung gesichert ist. Sie sollten identifiziert werden und ausreichende Aufzeichnungen über die Geschichte ihrer Verwendung sollten aufbewahrt werden. Fortlaufendes Stabilitätsprogramm6.23 Nach der Markteinführung sollte die Stabilität des Arzneimittels nach einem kontinuierlichen geeigneten Verfahren überwacht werden, dass das Auffinden stabilitätsbezogener Fragen oder Probleme (z.B. Änderungen des Gehaltes an Verunreinigungen oder im Dissolutionsverhalten) in Bezug auf die Arzneiform und ihre Verpackung erlaubt. 6.24 Zweck des fortlaufenden Stabilitätsprogramms ist, das Produkt während seiner Haltbarkeitsdauer zu überwachen und festzustellen, dass das Produkt unter den seiner Kennzeichnung entsprechenden Lagerungsbedingungen seine Spezifikationen erfüllt und dies auch für die gesamte Haltbarkeitsdauer erwartet werden kann. 6.25 Dies gilt hauptsächlich für das Arzneimittel in seiner Verkaufsverpackung, jedoch sollte auch die Einbeziehung von Bulkware in das Programm erwogen werden. Zum Beispiel sollte, wenn Bulkware vor ihrer Verpackung und/oder ihrem Versand von einer Herstellungsstätte zu einer Verpackungsstätte über einen langen Zeitraum gelagert wird, der Einfluss dieser Konditionen auf die Stabilität des verpackten Produkts beurteilt und unter Umgebungsbedingungen überprüft werden. Zusätzlich sollten Zwischenprodukte, die über längere Zeiträume gelagert und eingesetzt werden, Berücksichtigung finden. Stabilitätsstudien zu dem für die Verabreichung zubereiteten (rekonstituierten) Produkt werden während der Produktentwicklung durchgeführt und müssen nicht kontinuierlich überwacht werden. Jedoch kann erforderlichenfalls auch die Stabilität von zubereiteten Produkten überwacht werden. 6.26 Das fortlaufende Stabilitätsprogramm sollte in einem schriftlichen Plan nach den allgemeinen Regeln in Kapitel 4 beschrieben und die Ergebnisse sollten in einem formalisierten Bericht niedergelegt werden. Die im Rahmen des fortlaufenden Stabilitätsprogramms verwendete Ausrüstung (u. a. Klimakammern) sollte qualifiziert sein und gewartet werden, den allgemeinen Regeln des Kapitels 3 und Anhang 15 folgend. 6.27 Der Plan für ein fortlaufendes Stabilitätsprogramm sollte sich bis zum Endpunkt der jeweiligen Haltbarkeitsdauer erstrecken und mindestens folgende Angaben beinhalten:
Anzahl der Chargen pro Stärke und unterschiedlicher Chargengröße, sofern zutreffend;•relevante physikalische, chemische, mikrobiologische und biologische Prüfverfahren;•Akzeptanzkriterien;•Bezugnahme auf Prüfverfahren;•Beschreibung des Verschlusssystems der Behältnisse;•Prüfintervalle (Zeitpunkte);•Beschreibung der Lagerungsbedingungen (es sollten - im Einklang mit der Produktkennzeichnung - standardisierte ICH-Bedingungen für Langzeitstudien verwendet werden);
•
sonstige für das jeweilige Arzneimittel spezifische Parameter.•
Seite 16 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

6.28 Der Plan für das fortlaufende Stabilitätsprogramm kann sich von dem der ursprünglichen Langzeitstabilitätsstudie in den Zulassungsunterlagen unterscheiden, vorausgesetzt, dass dies begründet und im Plan dokumentiert ist (z.B. Prüffrequenz oder bei Aktualisierungen bzgl. ICH-Empfehlungen). 6.29 Die Anzahl der geprüften Chargen und die Prüffrequenz sollten eine ausreichende Datenmenge liefern, um Trendanalysen zu ermöglichen. Sofern dies nicht anderweitig gerechtfertigt werden kann, sollte von jedem hergestellten Produkt mindestens eine Charge pro Jahr für jede Stärke und, falls erforderlich, jedes Primärverpackungsmaterial in das Stabilitätsprogramm einbezogen werden (es sei denn, im entsprechenden Jahr wurde keine Charge hergestellt) . Für Produkte, für deren fortlaufende Stabilitätsüberwachung normalerweise eine Prüfung unter Verwendung von Tieren erforderlich ist und keine geeigneten alternativen, validierten Prüfverfahren zur Verfügung stehen, kann für die Entscheidung über die Prüffrequenz eine Nutzen-Risiko-Bewertung herangezogen werden. Das Prinzip der "Bracketing und Matrixing Designs" kann angewendet werden, wenn dies im Plan wissenschaftlich begründet wird. 6.30 In bestimmten Situationen sollten zusätzliche Chargen in das fortlaufende Stabilitätsprogramm einbezogen werden. Zum Beispiel sollten fortlaufende Stabilitätsstudien nach jeder signifikanten Änderung oder Abweichung vom Herstellungsprozess oder der Verpackung durchgeführt werden. Der Einschluss jedes Aufarbeitungs-, Umarbeitungs-, oder Rückgewinnungsprozesses in das Programm sollte ebenfalls in Betracht gezogen werden. 6.31 Ergebnisse von fortlaufenden Stabilitätsstudien sollten dem Schlüsselpersonal, insbesondere der/den Sachkundigen Person(en) zur Verfügung gestellt werden. Wenn die fortlaufenden Stabilitätsstudien in einer anderen Betriebsstätte als der Herstellungsstätte der Bulkware oder des Endprodukts durchgeführt werden, sollte eine schriftliche Vereinbarung zwischen den beteiligten Parteien vorliegen. Die Ergebnisse der fortlaufenden Stabilitätsstudien sollten zur Überprüfung durch die zuständige Behörde in der Herstellungsstätte verfügbar sein. 6.32 Ergebnisse außerhalb der Spezifikation oder signifikante atypische Trends sollten untersucht werden. Jedes bestätigte außerhalb der Spezifikation liegende Ergebnis oder ein signifikanter negativer Trend sollte den jeweils zuständigen Behörden gemeldet werden. Eine mögliche Auswirkung auf auf dem Markt befindliche Chargen sollte in Übereinstimmung mit Kapitel 8 des GMP Leitfadens und in Abstimmung mit der jeweiligen zuständigen Behörde sorgfältig geprüft werden. 6.33 Eine Zusammenfassung aller generierten Daten, einschließlich aller vorläufig getroffenen Schlussfolgerungen zum Programm, sollte schriftlich erstellt und aufbewahrt werden. Diese Zusammenfassung sollte einer regelmäßigen Überprüfung unterzogen werden.
Kapitel 7 Herstellung und Prüfung im Lohnauftrag
GrundsätzeHerstellung und Prüfung im Lohnauftrag müssen genau definiert, vereinbart und kontrolliert werden, um Missverständnisse zu vermeiden, aus denen sich ein Produkt oder eine Arbeit von ungenügender Qualität ergeben könnte. Zwischen Auftraggeber und Auftragnehmer muss ein schriftlicher Vertrag bestehen, der die Aufgaben jeder Seite klar festlegt. Aus dem Vertrag muss eindeutig hervorgehen, auf welche Weise die Sachkundige Person, die jede Produktcharge für den Verkauf freigibt, ihrer Verantwortung voll gerecht wird. Anmerkung: Dieses Kapitel behandelt die Verantwortlichkeiten der Hersteller gegenüber den zuständigen Behörden der Mitgliedstaaten im Hinblick auf die Erteilung von Zulassungen und Herstellungserlaubnissen. Es soll keinesfalls die jeweilige Haftung des Auftraggebers bzw. Auftragnehmers gegenüber dem Verbraucher beeinflussen. Dies wird durch andere Bestimmungen der Gemeinschaft und durch anderes nationales Recht geregelt. Allgemeine Anforderungen7.1 Es sollte ein schriftlicher Vertrag bestehen, der die Herstellung und/oder Prüfung im Lohnauftrag und alle damit in Zusammenhang stehenden technischen Vereinbarungen umfasst. 7.2 Alle Vereinbarungen über die Herstellung und Prüfung im Lohnauftrag, einschließlich aller vorgeschlagenen Änderungen an technischen oder anderen Vereinbarungen, sollten mit der Zulassung des betreffenden Produkts in Einklang stehen. Der Auftraggeber7.3 Der Auftraggeber ist verantwortlich für die Beurteilung, ob der Auftragnehmer kompetent ist, die erforderlichen Arbeiten erfolgreich auszuführen. Er hat durch den Vertrag sicherzustellen, dass die in diesem Leitfaden dargelegten Grundsätze und Leitlinien der Guten Herstellungspraxis befolgt werden. 7.4 Der Auftraggeber sollte dem Auftragnehmer alle nötigen Informationen liefern, damit dieser die in Auftrag gegebenen Arbeiten korrekt in Übereinstimmung mit der Zulassung und allen weiteren rechtlichen Vorschriften ausführen kann. Der Auftraggeber sollte sicherstellen, dass der Auftragnehmer sich über alle Probleme im Klaren ist, die mit dem Produkt oder der Arbeit in Zusammenhang stehen und die ein Risiko für seine Räumlichkeiten, die Ausrüstung, das Personal oder für andere Materialien oder Produkte darstellen könnten. 7.5 Der Auftraggeber sollte sicherstellen, dass alle vom Auftragnehmer an ihn gelieferten verarbeiteten Produkte und Materialien ihren Spezifikationen entsprechen oder die Erzeugnisse durch eine Sachkundige Person freigegeben wurden. Der Auftragnehmer7.6 Der Auftragnehmer muss über geeignete Räumlichkeiten und Ausrüstung, Sachkenntnis und Erfahrung sowie über kompetentes Personal verfügen, um die ihm vom Auftraggeber übertragenen Arbeiten zufrieden stellend ausführen zu können. Auftragsherstellung kann nur von einem Hersteller übernommen werden, der eine Herstellungserlaubnis besitzt. 7.7 Der Auftragnehmer sollte sicherstellen, dass alle an ihn gelieferten Produkte und Materialien für ihren vorgesehenen Zweck geeignet sind. 7.8 Der Auftragnehmer sollte ohne vorherige Bewertung und Genehmigung der Vereinbarungen durch den Auftraggeber keine ihm vertraglich übertragene Arbeit an eine dritte Partei weitergeben. Vereinbarungen zwischen Auftragnehmer und einer dritten Partei sollten sicherstellen, dass die Informationen über Herstellung und Prüfung in gleicher Weise zur Verfügung stehen wie zwischen dem ursprünglichen Auftraggeber und dem Auftragnehmer. 7.9 Der Auftragnehmer sollte alles unterlassen, was die Qualität des für den Auftraggeber hergestellten und/oder geprüften Produkts ungünstig beeinflussen könnte. Der Vertrag7.10 Zwischen Auftraggeber und Auftragnehmer sollte ein Vertrag geschlossen werden, der ihre jeweiligen Verantwortlichkeiten hinsichtlich Herstellung und Qualitätskontrolle des Produkts festlegt. Technische Aspekte des Vertrags sollten von kompetenten Personen abgefasst werden, die über geeignete Kenntnisse in pharmazeutischer Technologie, Analytik und der Guten Herstellungspraxis verfügen. Alle Vereinbarungen über Herstellung und Prüfung müssen mit der Zulassung übereinstimmen und von beiden Parteien anerkannt sein. 7.11 In dem Vertrag sollte festgelegt werden, auf welche Weise die Sachkundige Person, die die Chargen zum Verkauf freigibt. sicherstellt, dass jede Charge in Übereinstimmung mit den im Rahmen der Zulassung spezifizierten Anforderungen hergestellt und geprüft wurde.
Seite 17 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

7.12 Der Vertrag sollte klar beschreiben, wer für den Materialeinkauf, für die Prüfung und Freigabe von Materialien, für die Durchführung der Produktion und Qualitätskontrollen, einschließlich Inprozesskontrollen, verantwortlich ist und in wessen Verantwortungsbereich Probenahme und Prüfung fallen. Im Falle der Prüfung im Lohnauftrag sollte aus dem Vertrag hervorgehen, ob der Auftragnehmer in den Räumlichkeiten des Herstellers Proben nehmen soll. 7.13 Herstellungs-, Prüf- und Vertriebsprotokolle sowie Referenzproben sollten vom Auftraggeber aufbewahrt werden oder ihm zur Verfügung stehen. Alle für die Qualitätsbewertung eines Produkts relevanten Aufzeichnungen müssen im Falle einer Beanstandung oder eines vermuteten Mangels zugänglich und in den Verfahrensbeschreibungen des Auftraggebers betreffend Produktmängel/Rückrufe aufgeführt sein. 7.14 Der Vertrag sollte denn Auftraggeber gestatten, die Einrichtungen des Auftragnehmers zu besichtigen. 7.15 Im Falle der Prüfung im Lohnauftrag sollte sich der Auftragnehmer darüber im Klaren sein, dass er der Inspektion durch die zuständigen Behörden unterworfen ist.
Kapitel 8 Beanstandungen und Produktrückruf
GrundsätzeAlle Beanstandungen und andere Informationen über möglicherweise fehlerhafte Produkte müssen nach schriftlich festgelegten Verfahren sorgfältig überprüft werden. Um für alle Eventualitäten vorbereitet zu sein und in Übereinstimmung mit Artikel 117 der Richtlinie 2001/83/EG bzw. Artikel 84 der Richtlinie 2001/82/EG sollten systematische Vorkehrungen getroffen werden, damit erforderlichenfalls Produkte mit erwiesenen oder vermuteten Mängeln schnell und wirkungsvoll vom Markt zurückgerufen werden. Beanstandungen8.1 Es sollte eine verantwortliche Person benannt werden, die die Beanstandungen bearbeitet und die einzuleitenden Maßnahmen bestimmt. Sie sollte über ausreichendes Hilfspersonal verfügen. Wenn diese Person nicht mit der Sachkundigen Person identisch ist, sollte die letztere auf jede Beanstandung, Überprüfung und jeden Rückruf aufmerksam gemacht werden. 8.2 Es sollten schriftliche Verfahrensbeschreibungen vorliegen, die die im Falle einer Beanstandung wegen eines möglichen Produktmangels zu treffenden Maßnahmen beinhalten, einschließlich der Notwendigkeit, einen Rückruf in Betracht zu ziehen. 8.3 Jede Beanstandung wegen eines Produktmangels sollte mit allen Originalinformationen aufgezeichnet und gründlich untersucht werden. Die für die Qualitätskontrolle verantwortliche Person sollte in der Regel an der Untersuchung solcher Probleme beteiligt sein. 8.4 Wenn ein Produktmangel in einer Charge entdeckt wird oder ein entsprechender Verdacht besteht, sollte die Kontrolle anderer Chargen erwogen werden, um festzustellen, ob diese ebenfalls betroffen sind. Es sollten insbesondere die Chargen überprüft werden, die Material aus der fehlerhaften Charge enthalten können. 8.5 Alle aufgrund einer Beanstandung getroffenen Entscheidungen und Maßnahmen sollten aufgezeichnet werden. In den entsprechenden Chargenprotokollen sollte auf diese Aufzeichnungen verwiesen werden. 8.6 Die Aufzeichnungen über Beanstandungen sollten regelmäßig daraufhin überprüft werden, ob sie Hinweise auf spezifische oder sich wiederholende Probleme enthalten, die besondere Aufmerksamkeit und möglicherweise den Rückruf eines Produkts vom Markt erfordern. 8.7 Besondere Aufmerksamkeit sollte darauf gerichtet werden, ob eine Beanstandung durch eine Fälschung verursacht wurde. 8.8 Die zuständigen Behörden sollten benachrichtigt werden, wenn ein Hersteller eine Maßnahme aufgrund möglicherweise fehlerhafter Herstellung, Produktzersetzung, Nachweisen einer Fälschung oder anderer ernsthafter Qualitätsprobleme in Erwägung zieht. Rückrufe8.9 Es sollte eine Person benannt werden, die für die Durchführung und Koordination von Rückrufen verantwortlich ist. Sie sollte ausreichendes Hilfspersonal zur Verfügung haben, um alle Aspekte der Rückrufe mit der nötigen Dringlichkeit behandeln zu können. Diese verantwortliche Person sollte normalerweise unabhängig von Vertrieb und Marketing sein. Wenn diese Person nicht mit der Sachkundigen Person identisch ist, sollte die letztere auf jeden Rückruf aufmerksam gemacht werden. 8.10 Es sollten schriftliche, regelmäßig überprüfte und, wenn nötig, aktualisierte Vorschriften zur Verfügung stehen, um für die Organisierung eines eventuellen Rückrufs vorbereitet zu sein. 8.11 Die Maßnahmen für einen Rückruf sollten unverzüglich und jederzeit in Gang gesetzt werden können. 8.12 Die zuständigen Behörden aller Länder, in die die Produkte möglicherweise geliefert wurden, sollten unverzüglich benachrichtigt werden, wenn beabsichtigt wird, Produkte wegen eines erwiesenen oder vermuteten Mangels zurückzurufen. 8.13 Die Vertriebsprotokolle sollten der (den) für Rückrufe verantwortlichen Person(en) ohne Weiteres zur Verfügung stehen und ausreichende Informationen über Großhändler und direkt belieferte Kunden (einschließlich Adressen, Telefon- und/oder Telefaxnummern während und außerhalb der Arbeitszeit sowie der gelieferten Chargen und Mengen) enthalten, auch für exportierte Produkte und Ärztemuster. 8.14 Zurückgerufene Produkte sollten als solche gekennzeichnet sowie getrennt und gesichert gelagert werden, solange eine Entscheidung über ihr Schicksal aussteht. 8.15 Der Ablauf der Rückrufaktion sollte aufgezeichnet werden. Ein Abschlussbericht sollte erstellt werden, der eine Bilanzierung der ausgelieferten und zurückerhaltenen Produktmengen enthält. 8.16 Die Wirksamkeit der Rückrufverfahren sollte regelmäßig bewertet werden.
Kapitel 9 Selbstinspektion
GrundsätzeUm die Anwendung und Beachtung der Regeln der Guten Herstellungspraxis zu überwachen und um Vorschläge für notwendige Korrekturmaßnahmen zu machen, sollten Selbstinspektionen durchgeführt werden. 9.1 Personalbezogene Belange, Räumlichkeiten, Ausrüstung, Dokumentation, Produktion, Qualitätskontrolle, Vertrieb von Arzneimitteln, Vorkehrungen zur Behandlung von Beanstandungen und Abwicklung von Rückrufen sowie die Durchführung von Selbstinspektionen sollten in regelmäßigen Abständen nach einer im voraus festgelegten Programm überprüft werden, um ihre Übereinstimmung mit den Grundsätzen der Qualitätssicherung festzustellen. 9.2 Selbstinspektionen sollten unabhängig und ausführlich von einem oder mehreren beauftragten Experten des Unternehmen durchgeführt werden. Unabhängige Audits durch externe Sachverständige können ebenfalls nützlich sein. 9.3 Jede Selbstinspektion sollte protokolliert werden. Die Protokolle sollten alle während der Inspektion gemachten Beobachtungen und, soweit zutreffend, Vorschläge für Korrekturmaßnahmen enthalten. Über die anschließend ergriffenen Maßnahmen sollten ebenfalls Aufzeichnungen geführt werden. Glossar
Seite 18 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Die folgenden Definitionen beziehen sich auf die Begriffe, wie sie in diesem Leitfaden Teil 1 verwendet werden. In anderem Zusammen hang können sie davon abweichende Bedeutungen haben. ArzneimittelStoffe oder Zubereitungen aus Stoffen, die dazu bestimmt sind Krankheiten von Menschen oder Tieren zu behandeln oder zu verhüten. Stoffe oder Zubereitungen aus Stoffen, die zur Anwendung am Menschen oder Tieren bestimmt sind, um eine medizinische Diagnose zu stellen oder um physiologische Funktionen bei Mensch oder Tier wiederherzustellen, zu korrigieren oder zu verändern werden gleichermaßen als Arzneimittel angesehen. AusgangsstoffJeder bei der Herstellung eines Arzneimittels verwendete Stoff ausgenommen Verpackungsmaterial. BilanzierungEin Vergleich zwischen der theoretischen und der tatsächlich hergestellten oder verwendeten Produkt- oder Materialmenge unter angemessener Berücksichtigung der normalen Schwankungen. BulkwareJedes Produkt, das außer der Endverpackung alle Verarbeitungsstufen durchlaufen hat. ChargeEine in einem Arbeitsgang oder in einer Reihe von Arbeitsgängen gefertigte, als homogen zu erwartende definierte Menge an Ausgangsstoff, Verpackungsmaterial oder Produkt. Anmerkung: Für bestimmte Herstellungsstufen kann es notwendig sein, eine Charge in eine bestimmte Anzahl von Teilchargen aufzuteilen, die später zu einer homogenen endgültigen Charge vereinigt werden. Bei kontinuierlichem Betrieb muss die Charge einer definierten Fraktion der Produktion entsprechen, die durch ihre angestrebte Homogenität charakterisiert ist. In Anhang 1 der Richtlinie 2001/83/EG, geändert durch die Richtlinie 2003/63/EG 9, wird eine Charge für die Kontrolle des Fertigprodukts folgendermaßen definiert: "Für die Kontrolle des Fertigarzneimittels bedeutet Charge eines Arzneimittels die Gesamtheit der Einheiten einer pharmazeutischen Darreichungsform, die aus der gleichen Ursprungsmasse von Material stammen und der gleichen Abfolge von Herstellungs- und/oder Sterilisierungsabläufen unterzogen wurden, bzw. im Falle eines kontinuierlichen Herstellungsprozesses die Gesamtheit aller Einheiten, die in einem bestimmten Zeitraum hergestellt werden." Chargenbezeichnung (Chargennummer)Eine charakteristische Kombination von Zahlen und/oder Buchstaben, die eine Charge eindeutig bezeichnet. Computergestütztes SystemEin System zur Eingabe von Daten, elektronischen Verarbeitung und Ausgabe von Informationen, die entweder zur Dokumentation oder zur automatischen Steuerung verwendet werden. Fertigprodukt Ein Arzneimittel, das alle Produktionsstufen, einschließlich der Verpackung in sein endgültiges Behältnis, durchlaufen hat. HerstellerInhaber einer Herstellungserlaubnis gemäß Artikel 40 der Richtlinie 2001/83/EG bzw. Artikel 44 der Richtlinie 2001/82/EG. HerstellungAlle Arbeitsgänge wie Beschaffung von Material und Produkten, Produktion, Qualitätskontrolle, Freigabe, Lagerung und Vertrieb von Arzneimitteln und die dazugehörigen Kontrollen. Inprozesskontrolle Kontrollen im Verlauf der Produktion eines Arzneimittels zur Überwachung und gegebenenfalls Anpassung des Prozesses, um zu gewährleisten, dass das Produkt seiner Spezifikation entspricht. Die Überwachung der Umgebung oder der Ausrüstung kann auch als Teil der Inprozesskontrolle angesehen werden. KalibrierungArbeitsgänge, durch die unter genau bestimmten Bedingungen die Beziehung zwischen den durch ein Messgerät oder ein Messsystem angezeigten oder den sich aus einer Materialmessung ergebenden Werten und den entsprechenden bekannten Werten eines Referenzstandards bestimmt wird. KreuzkontaminationVerunreinigung eines Materials oder eines Produktes mit einem anderen Material oder Produkt. ProduktionAlle mit der Anfertigung eines Arzneimittels verbundenen Arbeitsgänge vom Materialeingang über die Verarbeitung und Verpackung bis zur Fertigstellung als Fertigprodukt. Protokollsiehe Kapitel 4QualifizierungBeweisführung, dass Ausrüstungsgegenstände einwandfrei arbeiten und tatsächlich zu den erwarteten Ergebnissen führen. Der Begriff Validierung wird manchmal um das Konzept der Qualifizierung erweitert. Qualitätskontrollesiehe Kapitel 1QuarantäneDer Status von Ausgangsstoffen oder Verpackungsmaterial, von Zwischen-, Bulk- oder Fertigprodukten, die getrennt gelagert oder durch andere geeignete Maßnahmen von der Verwendung oder Abgabe ausgeschlossen werden, solange die Entscheidung über ihre Freigabe oder Zurückweisung aussteht. Reiner BereichEin Bereich mit kontrollierten Umweltbedingungen hinsichtlich partikulärer und mikrobieller Kontaminationen, der so konstruiert ist und genutzt wird, dass das Eindringen, Entstehen und Verbleiben von Kontaminanten vermindert wird. Anmerkung: Die verschiedenen Reinheitsklassen werden in den ergänzenden Leitlinien für die Herstellung steriler Arzneimittel definiert. RückgabeZurücksenden eines Arzneimittels an den Hersteller oder Vertreiber, unabhängig davon, ob ein Qualitätsmangel vorliegt oder nicht. Spezifikationsiehe Kapitel 4Sterilität
Seite 19 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Sterilität ist die Abwesenheit lebender Organismen. Die Bedingungen der Sterilitätsprüfung werden im Europäischen Arzneibuch beschrieben. SystemWird im Sinne eines geregelten Mechanismus ineinander greifender Aktivitäten und Techniken verwendet, die so miteinander verbunden sind, dass ein organisiertes Ganzes entsteht. Wiederaufbereitung (Reprocessing) Die erneute Bearbeitung einer ganzen oder von Teilen einer Charge ungenügender Qualität, von einer bestimmten Produktionsstufe ausgehend, mit dem Ziel, in einem oder mehreren zusätzlichen Arbeitsgängen eine Qualität zu erreichen, die den Anforderungen genügt. ValidierungBeweisführung in Übereinstimmung mit den Grundsätzen der Guten Herstellungspraxis, dass Verfahren, Prozesse, Ausrüstungsgegenstände, Materialien, Arbeitsgänge oder Systeme tatsächlich zu den erwarteten Ergebnissen führen (siehe auch Qualifizierung). VerfahrenBeschreibung durchzuführender Arbeitsgänge, zu ergreifender Vorsichtsmaßnahmen und sonstiger Maßnahmen, die in direkter oder indirekter Beziehung zur Herstellung eines Arzneimittels stehen. VerpackungAlle Arbeitsgänge, einschließlich Abfüllen und Kennzeichnen, die eine Bulkware durchlaufen muss, um zu einem Fertigprodukt zu werden. Anmerkung: Steriles Abfüllen wird in der Regel nicht als Teil des Verpackens betrachtet. Die abgefüllten, aber nicht endgültig verpackten Primärbehälter sind als Bulkware anzusehen. VerpackungsmaterialJedes für die Verpackung eines Arzneimittels verwendete Material, ausgenommen die für Transport oder Versand verwendete äußere Umhüllung. Je nachdem, ob das Verpackungsmaterial für einen direkten Kontakt mit dem Produkt vorgesehen ist oder nicht, wird es als primär oder sekundär bezeichnet. WiederverwertungDas vollständige oder teilweise Einbringen früherer Chargen von der erforderlichen Qualität in eine andere Charge auf einer genau bestimmten Herstellungsstufe. ZwischenproduktTeilweise bearbeitetes Material, das noch weitere Produktionsstufen durchlaufen muss, bevor es zur Bulkware wird. ______________ 1) für Tierarztmittel Artikel 55 der Richtlinie 2001/82/EG 2) für Tierarzneimittel Artikel 55 der Richtlinie 2001/82/EG 3) Gemäß der Richtlinie 75/319/EWG (jetzt kodifizierte Richtlinie 2001/83/EG) und der Entscheidung des Europäischen Gerichtshofs (Fall 247/81) brauchen Arzneimittel, die in der EG durch eine sachkundige Person ordnungsgemäß kontrolliert worden sind, in keinem anderen Mitgliedstaat der Gemeinschaften nochmals kontrolliert oder nachgeprüft zu werden. 4) für Tierarzneimittel Artikel 55 Abs. 1 (b) der Richtlinie 2001/82/EG 5) für Tierarzneimittel Artikel 55 der Richtlinie 2001/82/EG 6) für Tierarzneimittel Artikel 53 der Richtlinie 2001/82/EG 7) für Tierarzneimittel Artikel 55 der Richtlinie 2001/82/EG 8) Artikel 55 Abs. 3 der Richtlinie 2001/82/EG 9) auch in Anhang I der Richtlinie 2001/82/EG
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Teil II (1/3)
Teil II - Grundlegende Anforderungen für Wirkstoffe zur Verwendung als Ausgangsstoffe
Vom 27. Oktober 2006 (BAnz. Nr. 210 vom 09.11.2006 S. 6887;::18.07.2008 S. 2798)
Geschichte des Dokuments Datum
Annahme als ICH Step 4 Dokument November 2000
Veröffentlichung als GMP Anhang 18 durch die Europäische Kommission. Juli 2001
Überarbeitung der Einleitung zur Erfüllung der Anforderungen aus den Richtlinien 2001/83/EG und 2001/82/EG, geändert durch die Richtlinien 2004/27/EG und 2004/28/EG, nach Abschluss einer öffentlichen Anhörung, der Adhoc-Arbeitsgruppe der GMP-Inspektoren, dem Pharmazeutischen Ausschuss und dem Veterinärpharmazeutischen Ausschuss. Nach einem Umstrukturierungsprozess des GMP-Leitfadens Veröffentlichung der neuen Anleitung als GMP-Leitfaden Teil II, der den früheren Anhang 18 ersetzt.
Oktober 2005
Fristablauf in den Mitgliedstaaten für die Umsetzung der neuen rechtlichen Vorgaben für Wirkstoffe, die als Ausgangsstoffe in der Herstellung von Human- oder Tierarzneimitteln verwendet werden.
30. Oktober 2005
Abschnitt 1 Einleitung
Diese Leitlinie wurde im November 2000 als Anhang 18 zum GMP-Leitfaden, der die Zustimmung der EU zu dem ICH Q7A Dokument widerspiegelt, veröffentlicht und seither von Herstellern und GMP-Inspektoraten auf freiwilliger Basis genutzt. Artikel 46 (f) der Richtlinie 2001/83/EG und Artikel 50 (f) der Richtlinie 2001/82/EG, geändert durch die Richtlinie 2004/27/EG bzw. 2004/28/EG, legt den Inhabern von Herstellungserlaubnissen als neue Verpflichtung auf, nur noch Wirkstoffe einzusetzen, die im Einklang mit den Regeln der
Seite 20 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Guten Herstellungspraxis für Ausgangsstoffe hergestellt worden sind. Die Richtlinien sagen weiterhin aus, dass die Grundsätze der Guten Herstellungspraxis für Wirkstoffe als ausführliche Leitlinien zu übernehmen sind. Die Mitgliedstaaten sind übereingekommen, dass der Text des früheren Anhangs 18 die Basis für die ausführlichen Leitlinien bilden soll, sodass er nunmehr Teil II des GMP-Leitfadens darstellt. 1.1 ZielsetzungDiese Leitlinien sollen als Anleitung bezüglich der Guten Herstellungspraxis (GMP) für die Herstellung von Wirkstoffen im Rahmen eines geeigneten Qualitätsmanagementsystems dienen. Sie sollen außerdem dabei helfen zu gewährleisten, dass alle Wirkstoffe die Anforderungen an Qualität und Reinheit erfüllen, welche sie zu besitzen vorgeben oder laut Deklaration besitzen sollen. In diesen Leitlinien umfasst der Begriff "Herstellen" alle Abläufe von Warenannahme, Produktion, Verpackungen, Umpacken, Kennzeichnen, Umetikettieren, Qualitätskontrolle, Freigabe, Lagerung und Vertrieb von Wirkstoffen sowie die damit verbundenen Kontrollen. Der Ausdruck "sollen" weist in diesem Zusammenhang auf Empfehlungen hin, von deren Anwendbarkeit auszugehen ist, es sei denn, sie sind nachweislich nicht anwendbar oder wurden in einem relevanten Anhang des GMP-Leitfadens geändert oder durch eine Alternative ersetzt, die mindestens ein gleichwertiges Maß an Qualitätssicherung gewährleistet. Der GMP-Leitfaden als Ganzes deckt weder Sicherheitsaspekte bezüglich des an der Herstellung beteiligten Personals noch Umweltschutzaspekte ab. Derartige Kontrollen fallen unter die Eigenverantwortlichkeit des Herstellers und werden durch andere Rechtsvorschriften geregelt. Diese Leitlinien beabsichtigen nicht, Zulassungsanforderungen zu definieren oder Arzneibuchanforderungen zu modifizieren und greifen nicht in die Möglichkeit der zuständigen Behörde ein, spezifische Anforderungen bezüglich der Wirkstoffe im Zusammenhang mit Zulassungen/Herstellungserlaubnissen zu stellen. Sämtliche aus Zulassungsanforderungen hervorgehende Verpflichtungen sind einzuhalten. 1.2 GeltungsbereichDiese Leitlinien gelten für die Herstellung von Wirkstoffen sowohl für Human- als auch Tierarzneimittel. Sie gelten für die Herstellung steriler Wirkstoffe nur bis zu dem Punkt unmittelbar vor der Sterilisation des Wirkstoffs. Die Sterilisation und die aseptische Aufbereitung steriler Wirkstoffe werden nicht abgedeckt, sondern sollten im Einklang mit den GMP-Grundsätzen und Leitlinien, wie in der Richtlinie 2003/94/EG festgelegt und im GMP-Leitfaden einschließlich dessen Anhang 1 beschrieben, durchgeführt werden. Im Falle von Ektoparasitika zur Anwendung bei Tieren können andere Standards als diese Leitlinien angewendet werden, sofern diese sicherstellen, dass die Wirkstoffe gleichwertige Qualität besitzen. Ausgenommen von diesen Leitlinien sind Vollblut und Plasma, weil die ausführlichen Anforderungen für die Gewinnung und Testung von Blut durch die Richtlinie 2002/98/EG und die diese Richtlinie unterstützenden technischen Anforderungen festgelegt werden; einbezogen sind jedoch Wirkstoffe, die unter Verwendung von Blut oder Plasma als Rohmaterial hergestellt werden. Schließlich gelten diese Leitlinien nicht für Arzneimittelbulkware. Sie gelten für alle anderen Wirkstoffe, die in den Anhängen des GMP-Leitfadens Einschränkungen unterliegen, insbesondere den Anhängen 2 bis 7, in denen ergänzende Erläuterungen für bestimmte Wirkstofftypen gegeben werden. Die Anhänge werden konsequent überarbeitet; aber in der Zwischenzeit, und nur, bis deren Überarbeitung abgeschlossen ist, können Hersteller wählen, ob sie weiterhin gemäß Teil I der grundlegenden Anforderungen und den für die jeweiligen Produkte relevanten Anhängen verfahren oder ob sie bereits Teil II anwenden. Abschnitt 19 enthält Anleitungen, die nur für die Herstellung von Wirkstoffen gelten, die speziell für die Herstellung von klinischen Prüfpräparaten verwendet werden, obwohl anzumerken ist, dass ihre Anwendung in diesem Fall wenn auch empfohlen, nicht durch Gemeinschaftsrecht gefordert wird. Ein "Wirkstoff-Startmaterial" ("Active Substance Starting Material") ist ein Rohmaterial, Zwischenprodukt oder Wirkstoff, der für die Produktion eines Wirkstoffes verwendet wird und der als wichtiges Strukturelement in die Struktur des Wirkstoffes eingebaut wird. Ein Wirkstoff-Startmaterial kann ein Handelsartikel, ein von einem oder mehreren Lieferanten im Rahmen eines Lohnauftrags oder Handelsübereinkommens erworbenes Material oder ein in der eigenen Anlage produziertes Material sein. Wirkstoff-Startmaterialien haben im Regelfall definierte chemische Eigenschaften und eine definierte Struktur. Der Hersteller sollte eine Begründung für den Punkt, an dem die Produktion eines Wirkstoffs beginnt, festlegen und dokumentieren. Bei synthetischen Prozessen ist dies bekanntlich der Punkt, an dem "Wirkstoff-Startmaterialen" in den Prozess eingeführt werden. Bei anderen Prozessen (z.B. Fermentation, Extraktion, Reinigung etc.) sollte die oben erwähnte Begründung von fall zu Fall genommen werden. Tabelle 1 gibt Erläuterungen zu dem Punkt, an dem das "Wirkstoff-Startmaterial" normalerweise in den Prozess eingeführt wird. Von diesem Punkt an sollten geeignete GMP-Maßnahmen gemäß diesen Leitlinien auf die folgenden Zwischenprodukt- und/oder Wirkstoffherstellungsschritte angewendet werden. Dies schließt die Validierung kritischer Prozessschritte, deren Einfluss auf die Qualität des Wirkstoffs festgestellt wurde, ein. Es sei jedoch darauf hingewiesen, dass die Tatsache, dass ein Hersteller einen Prozessschritt validiert, diesen Schritt nicht autoasiatisch zu einem kritischen Schritt macht. Die Anweisungen im vorliegenden Dokument werden üblicherweise auf die in Tabelle 1 grau unterlegten Schritte angewendet. Aus ihr ist nicht abzuleiten, dass alle dort aufgeführten Schritte vollzogen werden sollten. Das Ausmaß der GMP-Anwendung bei der Wirkstoffherstellung sollte mit dem Prozessfortschritt von frühen Herstellungsschritten bis zu den letzten Schritten, der Reinigung und Verpackung zunehmen. Die physikalische Verarbeitung von Wirkstoffen wie z.B. Granulierung, Dragierung oder physikalische Beeinflussung der Korngröße (z.B. Mahlen, Mikronisieren) sollten mindestens entsprechend der Maßstäbe dieser Leitlinien durchgeführt werden. Diese Leitlinien gelten nicht für Schritte, die vor dem ersten Einsatz des oben definierten "Wirkstoff-Startmaterials" liegen. 1 Das Glossar in Abschnitt 20 des Teils II sollte nur im Zusammenhang mit Teil II angewendet werden. Einige der dort gebrauchten Ausdrücke sind auch bereits im Teil I des GMP-Leitfadens definiert; diese sollten bei Anwendung des Teils I auch nur im Zusammenhang mit Teil I benutzt werden. Tabelle 1: Anwendung dieses Leitfadens auf die Wirkstoffherstellung
Herstellungstyp Anwendung des Leitfadens auf Schritte (grau markiert) des jeweiligen Herstellungstyps
chemische Herstellung Produktion des Ausgangsstoffs für den Wirkstoff (API starting material)
Einbringen des Ausgangsstoffs für den Wirkstoff (API starting material) in den Prozess
Produktion von Zwischenprodukt(en)
Isolierung und Reinigung
physikalische Verarbeitung und Verpackung
aus tierischen Quellen stammender Wirkstoff
Gewinnung des Organs, der Flüssigkeit oder des Gewebes
Zerlegung, Mischen und/oder initiale Verarbeitung (API starting material)
Einbringen des Ausgangsstoffs für den Wirkstoff (API starting material) in den Prozess
Isolierung und Reinigung
physikalische Verarbeitung und Verpackung
aus pflanzlichen Quellen extrahierter Wirkstoff
Gewinnung der Pflanze
Zerlegung und initiale Extraktion(en)
Einbringen des Ausgangsstoffs für den
Isolierung und Reinigung
physikalische Verarbeitung und Verpackung
Seite 21 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Wirkstoff (API starting material) in den Prozess
als Wirkstoff verwendete Kräuterextrakte
Gewinnung der Pflanzen
Zerschneiden und initiale Extraktion(en)
- weitere Extraktion
physikalische Verarbeitung und Verpackung
aus zerkleinerten oder pulverisierten Kräutern bestehender Wirkstoff
Gewinnung der Pflanzen und/oder Anbau und Ernte
Zerschneiden/ Zerkleinerung
- - physikalische Verarbeitung und Verpackung
Biotechnologie: Fermentation/Zellkultur
Anlegen einer Masterzellbank und einer Arbeitszellbank
Pflege der Arbeitszellbank
Zellkultur und/oder Fermentation
Isolierung und Reinigung
physikalische Verarbeitung und Verpackung
"Klassische" Fermentation zur Wirkstoffherstellung
Anlegen einer Zellbank
Pflege der Zellbank Zugabe der Zellen zur Fermentation
Isolierung und Reinigung
physikalische Verarbeitung und Verpackung
zunehmende GMP-Anforderungen ->
Abschnitt 2 Qualitätsmanagement
2.1 Grundsätze2.10 Alle an der Herstellung beteiligten Personen sollten für die Qualität verantwortlich sein. 2.11 Jeder Hersteller sollte ein effektives Qualitätsmanagementsystem erstellen, dokumentieren und umsetzen, an dem das Management und das entsprechende Herstellungspersonal aktiv beteiligt sind. 2.12 Das Qualitätsmanagementsystem sollte die Organisationsstruktur, Verfahren, Prozesse und Ressourcen sowie alle Aktivitäten umfassen, die notwendig sind, um sicherzustellen, dass der Wirkstoff die vorgesehenen Spezifikationen für Qualität und Reinheit erfüllt. Alle qualitätsrelevanten Aktivitäten sollten definiert und dokumentiert werden. 2.13 Es sollten eine oder mehrere Qualitäts(sicherungs)einheit(en) vorhanden sein, die von der Produktion unabhängig sind und sowohl Aufgaben der Qualitätssicherung (QS) als auch der Qualitätskontrolle (QK) übernehmen. Dies kann in Abhängigkeit von Größe und Struktur des Unternehmens in Form getrennter QS- und QK-Einheiten oder einer einzelnen Person oder Gruppe erfolgen. 2.14 Die zur Freigabe von Zwischenprodukten und Wirkstoffen berechtigten Personen sollten festgelegt werden. 2.15 Alle qualitätsrelevanten Aktivitäten sollten zum Zeitpunkt ihrer Durchführung protokolliert werden. 2.16 Jede Abweichung von festgelegten Verfahren sollte dokumentiert und begründet werden. Kritische Abweichungen sollten untersucht und die Untersuchungen sowie die daraus resultierenden Schlussfolgerungen dokumentiert werden. 2.17 Es sollten keine Materialien freigegeben oder verwendet werden, die noch nicht einer vollständigen, zufriedenstellenden Bewertung durch die Qualitäts(sicherungs)einheit(en) unterzogen wurden, es sei denn, es wurden geeignete Systeme installiert, die ein derartiges Vorgehen zulassen (z.B. Freigabe unter Quarantäne, wie in Abschnitt 10.20 beschrieben, oder die Verwendung von Rohmaterialien oder Zwischenprodukten, deren Bewertung noch nicht abgeschlossen ist). 2.18 Es sollten Verfahren für die rechtzeitige Benachrichtigung des verantwortlichen Führungspersonals über behördliche Inspektionen, schwerwiegende GMP-Mängel, Produktfehler und damit zusammenhängende Maßnahmen (z.B. qualitätsbezogene Beanstandungen, Rückrufe, behördliche Maßnahmen etc.) vorhanden sein. 2.2 Verantwortlichkeiten der Qualitäts (sicherungs) einheit(en)2.20 Die Qualitäts (sicherungs) einheit (en) sollte(n) bei allen qualitätsbezogenen Angelegenheiten hinzugezogen werden. 2.21 Die Qualitäts (sicherungs) einheit(en) sollte(n) sämtliche qualitätsrelevanten Dokumente überprüfen und genehmigen. 2.22 Die Hauptverantwortlichkeiten der unabhängigen Qualitäts (sicherungs) einheit (en) sollten nicht delegiert werden. Diese Verantwortlichkeiten sollten schriftlich niedergelegt werden und folgende Punkte, auf die sie jedoch nicht beschränkt sein müssen, umfassen:
Freigabe oder Zurückweisung sämtlicher Wirkstoffe; Freigabe oder Zurückweisung von Zwischenprodukten für den Einsatz außerhalb der Kontrolle der Herstellerfirma;
1.
Schaffung eines Systems zur Freigabe oder Zurückweisung von Rohmaterialien, Zwischenprodukten, Verpackungs- und Etikettiermaterial;
2.
Überprüfung der vollständigen Chargenherstellungs- und -prüfprotokolle auf kritische Prozessschritte vor der Freigabe des Wirkstoffs für den Vertrieb;
3.
Sicherstellung, dass kritische Abweichungen untersucht und behoben werden;4.Genehmigung aller Spezifikationen und Muster-Herstellungsanweisungen;5.Genehmigung aller Verfahren, die die Qualität der Zwischenprodukte oder Wirkstoffe beeinflussen;6.Sicherstellung, dass interne Audits (Selbstinspektionen) durchgeführt werden;7.Genehmigung von Lohnherstellern für Zwischenprodukte und Wirkstoffe;8.Genehmigung von Änderungen, die potenziell die Qualität der Zwischenprodukte oder Wirkstoffe beeinflussen;9.Überprüfung und Genehmigung von Validierungsplänen und -aufzeichnungen;10.Sicherstellung, dass qualitätsbezogene Beanstandungen untersucht und behoben werden;11.Sicherstellung, dass für die Wartung und Kalibrierung kritischer Ausrüstung wirksame Systeme verwendet werden;12.Sicherstellung, dass die Materialien in geeigneter Weise getestet und die Ergebnisse aufgezeichnet werden;13.Sicherstellung, dass gegebenenfalls Stabilitätsdaten für Zwischenprodukte und. oder Wirkstoffe vorhanden sind, die die Wiederholungsprüfungs- oder Verfallsdaten sowie Lagerbedingungen stützen;
14.
Durchführung von Produktqualitätsüberprüfungen (gemäß Nummer 2.5).15.
2.3 Verantwortung für ProduktionsaktivitätenDie Verantwortung für Produktionsaktivitäten sollte schriftlich niedergelegt werden und folgende Punkte, auf die sie jedoch nicht beschränkt sein muss, umfassen:
Seite 22 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Vorbereitung. Überprüfung, Genehmigung und Verteilung der Arbeitsanweisungen für die Produktion von Zwischenprodukten oder Wirkstoffen gemäß schriftlicher Verfahren;
1.
Produktion von Wirkstoffen und gegebenenfalls Zwischenprodukten gemäß vorher genehmigter Arbeitsanweisungen;2.Überprüfung sämtlicher Chargenherstellungsprotokolle und Sicherstellung, dass diese vollständig erstellt und unterzeichnet wurden;
3.
Sicherstellung, dass alle Produktionsabweichungen gemeldet und bewertet wurden sowie dass kritische Abweichungen untersucht und entsprechende Schlussfolgerungen festgehalten wurden;
4.
Sicherstellung, dass die Produktionsstätten sauber und erforderlichenfalls desinfiziert sind;5.Sicherstellung, dass die notwendigen Kalibrierungen vorgenommen und Aufzeichnungen aufbewahrt werden;6.Sicherstellung, dass Räumlichkeiten und Ausrüstung gewartet und hierüber Aufzeichnungen aufbewahrt werden;7.Sicherstellung, dass Validierungspläne und -berichte überprüft und genehmigt werden;8.Bewertung vorgeschlagener Änderungen an Produkt, Prozess oder Ausrüstung;9.Sicherstellung, dass neue und gegebenenfalls veränderte Einrichtungen und Ausrüstungsgegenstände qualifiziert werden.10.
2.4 Interne Audits (Selbstinspektion)2.40 Um die Einhaltung der GMP-Grundsätze für Wirkstoffe zu verifizieren, sollten regelmäßig interne Audits gemäß einem genehmigten Zeitplan durchgeführt werden. 2.41 Feststellungen aus derartigen Audits sowie Korrekturmaßnahmen sollten dokumentiert und dem verantwortlichen Führungspersonal der Firma vorgelegt werden. Vereinbarte Korrekturmaßnahmen sollten zeitnah und auf wirksame Art und Weise durchgeführt werden. 2.5 Produktqualitätsüberprüfungen2.50 Für Wirkstoffe sollten regelmäßige Produktqualitätsüberprüfungen mit dem Ziel durchgeführt werden, die Beständigkeit eines Prozesses zu verifizieren. Derartige Überprüfungen sollten im Regelfall jährlich vorgenommen und dokumentiert werden und mindestens die folgenden Punkte umfassen:
Überprüfung der Ergebnisse aus kritischen Inprozesskontrollen und kritischen Wirkstofftests;•Überprüfung aller Chargen, die die festgelegte(n) Spezifikation(en) nicht erfüllt haben;•Überprüfung aller kritischen Abweichungen oder Nichteinhaltungen und damit verbundener Untersuchungen;•Überprüfung aller an den Prozessen oder den Prüfmethoden vorgenommenen Änderungen;•Überprüfung der Ergebnisse des Stabilitätsüberwachungsprogramms;•Überprüfung aller qualitätsbezogenen Rückgaben, Beanstandungen und Rückrufe;•Überprüfung der Angemessenheit von Korrekturmaßnahmen.•
2.51 Die Ergebnisse dieser Überprüfung sollten bewertet und eine Einschätzung abgegeben werden, ob Korrekturmaßnahmen oder eine Revalidierung durchgeführt werden sollten; Gründe für solche Korrekturmaßnahmen sollten dokumentiert werden. Vereinbarte Korrekturmaßnahmen sollten zeitnah und auf wirksame Art durchgeführt werden.
Abschnitt 3 Personal
3.1 Qualifikationen des Personals3.10 Für die Durchführung und Überwachung der Zwischenprodukt- und Wirkstoffherstellung sollte eine ausreichende Anzahl von Mitarbeitern, die durch angemessene Ausbildung, Schulung und/oder Erfahrung qualifiziert sind, zur Verfügung stehen. 3.11 Die Verantwortlichkeiten aller an der Herstellung von Zwischenprodukten und Wirkstoffen beteiligten Mitarbeiter sollten schriftlich festgelegt werden. 3.12 Schulungen sollten regelmäßig von qualifizierten Personen durchgeführt werden und mindestens die Tätigkeiten, die ein Mitarbeiter ausführt, sowie GMP in Bezug auf die Funktionen des Mitarbeiters abdecken. Aufzeichnungen über Schulungen sollten aufbewahrt werden. Schulungsmaßnahmen sollten periodisch bewertet werden. 3.2 Personalhygiene3.20 Das Personal sollte gute Hygiene- und Gesundheitsverhaltensweisen praktizieren. 3.21 Die Mitarbeiter sollten saubere, für die von ihnen vorgenommenen Herstellungsaktivitäten geeignete Kleidung tragen, die im Bedarfsfall gewechselt wird. Zusätzliche Schutzkleidung, wie z.B. Kopf-, Gesichts-, Hand- und Armbedeckungen, sollten wenn nötig getragen werden, um Zwischenprodukte und Wirkstoffe vor Verunreinigungen zu schützen. 3.22 Das Personal sollte direkten Kontakt mit Zwischenprodukten oder Wirkstoffen vermeiden. 3.23 Rauchen, Essen, Trinken, Kaugummikauen und die Aufbewahrung von Lebensmitteln sollten auf bestimmte ausgewiesene, von den Herstellungsbereichen getrennte Bereiche beschränkt sein. 3.24 Mitarbeiter, die unter einer ansteckenden Krankheit leiden oder an unbedeckten Körperflächen offene Wunden haben, sollten nicht an Aktivitäten beteiligt sein, die die Qualität von Wirkstoffen beeinträchtigen könnten. Jede Person, die, ganz gleich zu welchem Zeitpunkt, an einer wahrnehmbaren Krankheit leidet oder offene Wunden hat (festgestellt entweder durch ärztliche Untersuchung oder durch Beobachtung des Vorgesetzten), sollte von Aktivitäten ausgeschlossen werden, bei denen der Gesundheitszustand die Qualität der Wirkstoffe beeinträchtigen könnte, bis der Zustand behoben ist oder qualifiziertes medizinisches Personal festgestellt hat, dass ein Einsatz der entsprechenden Person die Sicherheit oder Qualität des Wirkstoffs nicht gefährdet. 3.3 Berater3.30 Berater, die in Fragen der Herstellung und Kontrolle von Zwischenprodukten oder Wirkstoffen beraten, sollten über ausreichende Ausbildung, Schulung und Erfahrung oder eine Kombination dieser drei verfügen, um Beratungen für den Bereich, für den sie beauftragt sind, vorzunehmen. 3.31 Es sollten Aufzeichnungen über den Namen, die Adresse, die Qualifikationen und die Art der von den Beratern erbrachten Dienstleistung vorhanden sein.
Abschnitt 4 Gebäude und Anlagen
4.1 Design und Bauart
Seite 23 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

4.10 Die für die Herstellung von Zwischenprodukten und Wirkstoffen verwendeten Gebäude und Anlagen sollten so gelegen, beschaffen und konstruiert sein, dass sie die Reinigung und Wartung sowie die entsprechenden Betriebstätigkeiten nach Art und Stufe der Herstellung erleichtern. Anlagen sollten außerdem so beschaffen sein, dass das Kontaminationspotenzial minimiert wird. Falls mikrobiologische Spezifikationen für Zwischenprodukte oder Wirkstoffe festgesetzt wurden, sollten Anlagen darüber hinaus so konstruiert sein, dass sie die Exposition gegenüber unzulässigen mikrobiellen Verunreinigungen entsprechend begrenzen. 4.11 Gebäude und Anlagen sollten genügend Raum für eine ordentliche Platzierung von Ausrüstung und Material bieten, um Verwechslungen und Kontamination zu vermeiden. 4.12 Wo die Ausrüstung selbst ausreichend Schutz für das Material bietet (z.B. geschlossene Systeme), kann sie im Freien aufgestellt werden. 4.13 Der Material- und Personalfluss durch Gebäude oder Anlagen sollte so geregelt sein, dass Verwechslungen oder Kontamination vermieden werden. 4.14 Für die folgenden Aktivitäten sollten definierte Bereiche oder andere Kontrollsysteme vorhanden sein:
Annahme, Identifizierung, Probenahme und Quarantäne eingehender Materialien bis zur Freigabe oder Zurückweisung;•Quarantäne vor der Freigabe oder Zurückweisung von Zwischenprodukten und Wirkstoffen;•Probenahme von Zwischenprodukten und Wirkstoffen;•Lagerung zurückgewiesener Materialien vor der weiteren Verfügung (z.B. Rückgabe, Umarbeitung oder Vernichtung);•Lagerung freigegebener Materialien;•Produktionsvorgänge;•Verpackungs- und Etikettiervorgänge;•Labortätigkeiten.•
4.15 Dem Personal sollten geeignete und saubere Waschanlagen und Toiletten zur Verfügung stehen. Die Waschanlagen sollten je nach Bedarf mit warmem und kaltem Wasser, Seife oder einen Reinigungsmittel, Heißlufttrockner oder Einweghandtüchern ausgestattet sein. Die Waschräume und Toiletten sollten von den Herstellungsbereichen getrennt, von diesen aus jedoch leicht zugänglich sein. Falls erforderlich, sollten geeignete Duschvorrichtungen und/ oder Umkleidemöglichkeiten vorhanden sein. 4.16 Laborbereiche/-tätigkeiten sollten üblicherweise von den Produktionsbereichen getrennt gehalten werden. Einige Laborbereiche, vor allem die für Inprozesskontrollen, dürfen in Produktionsbereichen liegen, vorausgesetzt, dass die Produktionsvorgänge die Genauigkeit der Labormessungen und umgekehrt das Labor und die dort ablaufenden Vorgänge den Produktionsprozess, die Zwischenprodukte oder die Wirkstoffe nicht beeinträchtigen. 4.2 Betriebsmittel4.20 Alle Betriebsmittel, die Einfluss auf die Produktqualität haben könnten (z.B. Dampf, Gase, Druckluft sowie Heizung, Belüftung und Klimatisierung), sollten qualifiziert und ausreichend überwacht werden. Bei Überschreiten der festgelegten Grenzwerte sollten Maßnahmen ergriffen werden. Über diese Betriebssysteme sollten Aufzeichnungen verfügbar sein. 4.21 Falls erforderlich, sollten geeignete Belüftungs-, Luftfiltrations- und Abluftsysteme vorhanden sein. Diese Systeme sollten so entworfen und angelegt sein, dass das Risiko einer Kontamination und Kreuzkontamination auf ein Minimum beschränkt bleibt und über Steuerungsmöglichkeiten für Luftdruck, Mikroorganismen (falls erforderlich), Staub, Feuchtigkeit und Temperatur entsprechend der jeweiligen Herstellungsstufe verfügen. Bereiche, in denen Wirkstoffe der Umgebung ausgesetzt sind, sollten besondere Aufmerksamkeit erhalten. 4.22 Wenn die Produktionsbereiche mit Umluft versorgt werden, sollten entsprechende Maßnahmen getroffen werden, um Kontaminations- und Kreuzkontaminationsrisiken zu kontrollieren. 4.23 Fest installierte Rohrleitungen sollten ordnungsgemäß identifiziert werden. Dies kann durch die Identifizierung einzelner Leitungen, durch Dokumentation, Computerkontrollsysteme oder Alternativen zu diesen Möglichkeiten erfolgen. Rohrleitungen sollten so angebracht sein, dass ein Kontaminationsrisiko für Zwischenprodukte oder Wirkstoffe vermieden wird. 4.24 Abflüsse sollten ausreichend groß sein und gegebenenfalls mit einem Rohrtrenner oder einer geeigneten Vorrichtung ausgestattet sein, um einen Abwasserrückfluss zu verhindern. 4.3 Wasser4.30 Das für die Wirkstoffherstellung verwendete Wasser sollte für seinen vorgesehenen Einsatz nachweislich geeignet sein. 4.31 Liegt keine anderweitige Begründung vor, sollte Prozesswasser mindestens den Leitlinien der Weltgesundheitsorganisation (WHO) für Trinkwasserqualität entsprechen. 4.32 Falls Trinkwasser für eine Gewährleistung der Wirkstoffqualität nicht ausreicht und strengere chemische und/oder mikrobiologische Spezifikationen für die Wasserqualität notwendig sind, sind geeignete Spezifikationen für die physikalischen/chemischen Eigenschaften, die Gesamtkeimzahl, unzulässige Organismen und/ oder Endotoxine festzulegen. 4.33 Wird das für den Prozess verwendete Wasser vom Hersteller zwecks Erreichen einer bestimmten Qualität aufbereitet, sollte der Aufbereitungsprozess validiert und unter Festlegung geeigneter Aktionsgrenzen überwacht werden. 4.34 Beabsichtigt oder beansprucht der Hersteller eines nichtsterilen Wirkstoffs die Eignung dieses Wirkstoffs für eine Weiterverarbeitung zu sterilen Arzneimitteln, sollte das in den abschließenden Isolierungs- und Aufreinigungsschritten verwendete Wasser überwacht und auf seine Gesamtkeimzahl, unzulässige Organismen sowie Endotoxine kontrolliert werden. 4.4 Containment (separate Bereiche)4.40 Für die Produktion von stark allergisierenden Materialien (wie z.B. Penizilline oder Cephalosporine) sollten Mono-Produktionsbereiche (dedicated production areas), die Anlagen, Belüftungsvorrichtungen und/oder Prozessausrüstung einschließen können, verwendet werden. 4.41 Mono-Produktionsbereiche sollten auch erwogen werden, wenn infektiöse Materialien oder solche mit hoher pharmakologischer Aktivität oder Toxizität (z.B. bestimmte Steroide oder zytotoxische, gegen Krebs wirksame Stoffe) verarbeitet werden, es sei denn, es sind validierte Inaktivierungs- und/oder Reinigungsverfahren festgelegt und regelmäßig durchgeführt. 4.42 Es sollten geeignete Maßnahmen fest- und umgesetzt werden, die eine Kreuzkontamination von Mitarbeitern, Materialien etc., die sich von einem Mono-Produktionsbereich in den anderen bewegen, verhindern. 4.43 Alle Produktionsaktivitäten (einschließlich Verwiegen, Mahlen oder Verpacken) mit hochtoxischen, nichtpharmazeutischen Materialien wie Herbizide und Pestizide sollten nicht in den für die Wirkstoffproduktion vorgesehenen Räumlichkeiten und/oder der entsprechenden Ausrüstung durchgeführt werden. Diese hoch toxischen, nichtpharmazeutischen Materialien sollten separat von Wirkstoffen gehandhabt und gelagert werden. 4.5 Beleuchtung
Seite 24 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

4.50 In allen Bereichen sollte für angemessene Beleuchtung gesorgt werden, um Reinigung, Wartung und ordnungsgemäße Betriebsaktivitäten zu erleichtern. 4.6 Abwasser und AbfallAbwasser, Müll und andere Abfülle (z.B. Feststoffe, flüssige Stoffe oder gasförmige Nebenprodukte aus der Herstellung) in und aus Gebäuden sowie der unmittelbaren Umgebung sollten sicher, rasch und auf hygienische Art und Weise entsorgt werden. Behälter und/oder Rohrleitungen für Abfallstoffe sollten deutlich gekennzeichnet sein. 4.7 Betriebshygiene und Wartung4.70 Alle für die Herstellung von Zwischenprodukten und Wirkstoffen eingesetzten Gebäude sollten ordnungsgemäß gewartet und instand gesetzt sowie in einem sauberen Zustand gehalten werden. 4.71 Es sollten schriftliche Anweisungen erstellt werden, die die Verantwortlichkeiten, die Zeitpläne, Methoden, Ausrüstung und Materialien für die Reinigung von Gebäuden und Anlagen festlegen. 4.72 Wenn nötig, sollten schriftliche Anweisungen auch für den Einsatz geeigneter Rodentizide, Insektizide, Fungizide, Begasungsmittel sowie Reinigungs- und Desinfektionsmittel ausgearbeitet werden, um eine Kontamination von Ausrüstung, Rohmaterialien, Pack-/Etikettiermaterialien, Zwischenprodukten und Wirkstoffen zu verhindern.
Abschnitt 5 Prozessausrüstung
5.1 Design und Bauart5.10 Die für die Herstellung von Zwischenprodukten und Wirkstoffen eingesetzte Ausrüstung sollte die erforderliche Beschaffenheit und angemessene Größe aufweisen sowie sich an einem für die vorgesehene Verwendung, Reinigung, Desinfektion (wo zutreffend) und Wartung geeigneten Ort befinden. 5.11 Ausrüstungsgegenstände sollten so konstruiert sein, dass mit Rohmaterialien, Zwischenprodukten oder Wirkstoffen in Berührung kommende Oberflächen die Qualität der Zwischenprodukte und Wirkstoffe nicht über die amtlichen oder anderweitig festgelegten Spezifikationen hinaus verändern. 5.12 Produktionsausrüstung sollte nur innerhalb ihres qualifizierten Bedienungsrahmens verwendet werden. 5.13 Größere Ausrüstungsgegenstände (z.B. Rührwerke, Lagerbehälter) sowie fest installierte Herstellungslinien, die für die Produktion von Zwischenprodukten oder Wirkstoffen eingesetzt werden, sollten ordnungsgemäß identifiziert sein. 5.14 Alle mit dem Betrieb der Ausrüstung in Verbindung stehenden Substanzen, wie z.B. Schmiermittel, Heizflüssigkeiten oder Kühlmittel, sollten nicht so mit Zwischenprodukten oder Wirkstoffen in Berührung kommen, dass ihre Qualität über die amtlich oder anderweitig festgelegten Spezifikationen hinaus verändert wird. Jegliche Abweichung von diesem Prinzip sollte bewertet werden, um sicherzustellen, dass keine schädlichen Einflüsse auf die Eignung des Materials für seinen Verwendungszweck auftreten. Wann immer möglich, sollten lebensmittelechte Schmiermittel und Öle verwendet werden. 5.15 Wenn erforderlich, sollten geschlossene Systeme eingesetzt werden. Werden offene Systeme verwendet bzw. wird die Ausrüstung geöffnet, sollten geeignete Vorsichtsmaßnahmen getroffen werden, um das Risiko einer Kontamination zu minimieren. 5.16 Für die Ausrüstung und kritische Installationen (z.B. Instrumenten- und Betriebsmittelsysteme) sollten aktuelle Zeichnungen vorhanden sein. 5.2 Wartung und Reinigung der Ausrüstung5.20 Für die vorbeugende Wartung der Ausrüstung sollten Zeitpläne und Verfahren (einschließlich der Zuweisung von Verantwortlichkeiten) ausgearbeitet werden. 5.21 Es sollten schriftliche Verfahren für die Reinigung der Ausrüstung und ihre anschließende Freigabe für die Herstellung von Zwischenprodukten und Wirkstoffen erstellt werden. Die Reinigungsverfahren sollten detailliert genug sein, um es dem Bedienpersonal zu ermöglichen, jede Art von Ausrüstung auf reproduzierbare und wirksame Art zu reinigen. Derartige Verfahren sollten folgende Punkte einschließen:
Verantwortungszuweisung für die Reinigung der Ausrüstung;•Reinigungszeitpläne, die, soweit angezeigt, Desinfektionszeitpläne einschließen;•eine vollständige Beschreibung der für die Reinigung verwendeten Methoden und Materialien, einschließlich der Verdünnung von Reinigungsmitteln;
•
soweit angezeigt, Instruktionen für die Demontage sowie die anschließende Montage eines jeden Ausrüstungsgegenstands, um eine ordnungsgemäße Reinigung zu gewährleisten;
•
Anweisungen für das Entfernen oder Unkenntlichmachen vorheriger Chargenkennzeichnungen;•Arbeitsanweisungen für den Schutz gereinigter Ausrüstungsgegenstände gegen Verunreinigung vor dem Einsatz;•Inspektion der Ausrüstung auf Reinheit unmittelbar vor Gebrauch, falls praktikabel;•Festlegung eines maximalen Zeitraums zwischen dem Ende der Fertigung und der Reinigung der Ausrüstung, falls erforderlich.•
5.22 Ausrüstungsgegenstände und Utensilien sollten gereinigt, gelagert und, soweit angezeigt, desinfiziert oder sterilisiert werden, um Kontamination oder Übertragung von Material zu verhindern, das die Qualität des Zwischenprodukts oder Wirkstoffs über die amtlich oder anderweitig festgelegten Spezifikationen hinaus verändern würde. 5.23 Wird die Ausrüstung für kontinuierliche Produktion oder Kampagnenproduktion von aufeinander folgenden Chargen des gleichen Zwischenprodukts oder Wirkstoffs eingesetzt, sollte die Ausrüstung in angemessenen Abständen gereinigt werden, um Bildung und Übertragung von Verunreinigungen (z.B. Abbauprodukten oder unzulässigen Mengen von Mikroorganismen) zu verhindern. 5.24 Ausrüstungsgegenstände, die nicht ausschließlich der Produktion eines bestimmten Produkts vorbehalten sind, sollten zwischen der Produktion verschiedener Materialien gereinigt werden, um eine Kreuzkontamination zu verhindern. 5.25 Akzeptanzkriterien für Rückstände und die Wahl der Reinigungsverfahren und -mittel sollten festgelegt und begründet werden. 5.26 Die Ausrüstung sollte hinsichtlich ihres Inhalts und ihres Reinheitsstatus auf geeignete Weise gekennzeichnet werden. 5.3 Kalibrierung5.30 Kontroll-, Wäge-, Mess-, Überwachungs- und Testgeräte, die für die Gewährleistung der Qualität von Zwischenprodukten oder Wirkstoffen kritisch sind, sollten gemäß schriftlich festgehaltenen Verfahren und einem festgelegten Zeitplan kalibriert werden. 5.31 Die Ausrüstungskalibrierung sollte unter Verwendung von Standards vorgenommen werden, die auf zertifizierte Standards zurückzuführen sind, falls solche existieren. 5.32 Aufzeichnungen dieser Kalibrierungen sollten aufbewahrt werden. 5.33 Der aktuelle Kalibrierstatus kritischer Ausrüstung sollte bekannt und verifizierbar sein.
Seite 25 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

5.34 Instrumente, die die Kalibrierkriterien nicht erfüllen, sollten nicht verwendet werden. 5.35 Bei kritischen Instrumenten sollten Abweichungen von genehmigten Kalibrierstandards untersucht werden um festzustellen, ob diese die Qualität des Zwischenprodukts, der Zwischenprodukte oder des Wirkstoffs/der Wirkstoffe, die unter Verwendung dieser Ausrüstung seit der letzten erfolgreichen Kalibrierung hergestellt wurden, beeinflusst haben könnten. 5.4 Computergestützte Systeme5.40 Von GMP betroffene computergestützte Systeme sollten validiert werden. Die Tiefe und das Ausmaß der Validierung hängen davon ab, wie vielfältig, komplex und kritisch die Computeranwendung ist. 5.41 Eine geeignete Installations- und Funktionsqualifizierung sollte die Eignung der Computerhard- und -software für die Durchführung der vorgesehenen Aufgaben ausweisen. 5.42 Im Handel erhältliche Software, die qualifiziert wurde, erfordert nicht das gleiche Maß an Testung. Wenn ein bestehendes System bei der Installation nicht validiert wurde, könnte eine retrospektive Validierung durchgeführt werden, wenn hierfür geeignete Dokumentation zur Verfügung steht. 5.43 Computersysteme sollten über ausreichende Kontrollmechanismen verfügen, um unberechtigten Zugriff auf oder Veränderungen von Daten zu verhindern. Es sollte Kontrollen zur Verhinderung von Datenverlusten geben (wenn z.B. das System abgeschaltet und die Daten noch nicht erfasst wurden). Es sollten Aufzeichnungen über alle Datenänderungen, die ursprünglichen Einträge, die Personen, die die Änderung vornahmen, sowie den Zeitpunkt der Änderung vorhanden sein. 5.44 Für die Bedienung und Wartung von computergestützten Systemen sollten schriftliche Verfahren zur Verfügung stehen. 5.45 Wenn kritische Daten von Hand eingegeben werden, sollte eine zusätzliche Überprüfung der Richtigkeit des Eintrags vorgenommen werden. Dies kann durch einen zweiten Mitarbeiter oder das System selbst erfolgen. 5.46 Mit Computersystemen zusammenhängende Vorfälle, die die Qualität von Zwischenprodukten oder Wirkstoffen oder die Verlässlichkeit von Aufzeichnungen oder Testergebnissen beeinflussen könnten, sollten festgehalten und untersucht werden. 5.47 Änderungen am computergestützten System sollten anhand eines Änderungsverfahrens durchgeführt und formal genehmigt, dokumentiert und geprüft werden. Aufzeichnungen aller Änderungen, einschließlich Modifikationen und Aufrüstung der Hardware, Software und anderer kritischer Bestandteile des Systems, sollten aufbewahrt werden. Diese Aufzeichnungen sollten belegen, dass sich das System nach wie vor in einem validierten Zustand befindet. 5.48 Wenn ein Systemzusammenbruch oder -versagen zu einem dauerhaften Verlust von Aufzeichnungen führen würde, sollte ein Backup-System installiert werden. Für alle computergestützten Systeme sollte eine Form des Datenschutzes eingerichtet werden. 5.49 Daten können zusätzlich zur computergestützten Erfassung mit einer zweiten Methode aufgezeichnet werden.
Abschnitt 6 Dokumentation und Protokolle
6.1 Dokumentationssystem und Spezifikationen6.10 Alle mit der Herstellung von Zwischenprodukten oder Wirkstoffen verbundenen Dokumente sollten gemäß schriftlich festgelegten Verfahren erstellt, überprüft, genehmigt und verteilt werden. Diese Dokumente können in Papierform oder elektronisch erstellt werden. 6.11 Das Ausstellen, Überarbeiten, Ersetzen und Einziehen sämtlicher Dokumente sollte kontrolliert und zu jedem Dokument eine Überarbeitungshistorie geführt werden. 6.12 Für die Aufbewahruug aller erforderlichen Dokumente z.B. Berichte zur Entwicklungshistorie, Scale-up (Maßstabserweiterung), technischem Transfer, Prozessvalidierung, Schulungen Produktion, Kontrollen und Vertrieb) sollte ein Verfahren erstellt werden. Die Aufbewahrungszeiträume für diese Dokumente sollten festgelegt werden. 6.13 Alle Produktions-, Kontroll- und Vertriebsberichte sollten mindestens ein Jahr über das Verfallsdatum einer Charge hinaus aufbewahrt werden. Bei Wirkstoffen mit Wiederholungsprüfungsdatum sollten die Aufzeichnungen nach dem vollständigen Vertrieb einer Charge mindestens drei Jahre lang aufbewahrt werden. 6.14 Wenn Einträge in Aufzeichnungen vorgenommen werden, sollte dies unauslöschlich in den dafür vorgesehenen Feldern unmittelbar nach der Durchführung der jeweiligen Aktivitäten geschehen und die den Eintrag vornehmende Person identifizieren. Korrekturen von Einträgen sollten mit einem Datum versehen und unterschrieben werden und den Originaleintrag lesbar lassen. 6.15 Während ihres Aufbewahrungszeitraums sollten Originale oder Kopien von Aufzeichnungen an dem Ort unverzüglich zu Verfügung stehen, an dem die in diesen Dokumenten beschriebenen Aktivitäten durchgeführt wurden. Aufzeichnungen, die ohne größere Zeitverzögerung von einem anderen Standort elektronisch oder anderweitig beschafft werden können, sind zulässig. 6.16 Spezifikationen, Anweisungen, Verfahren und Aufzeichnungen können entweder im Original oder als echte Kopien, wie z.B. Fotokopien, Mikrofilm, Mikrofiche oder andere genaue Wiedergaben der Originalaufzeichnungen, aufbewahrt werden. Werden Verkleinerungstechniken wie Mikroverfilmung oder elektronische Aufzeichnungen verwendet, sollten geeignete Einrichtungen zum Abrufen der Daten und eine Möglichkeit zur Erstellung einer Printversion kurzfristig zugänglich sein. 6.17 Für Rohmaterialien, Zwischenprodukte (falls notwendig), Wirkstoffe, Etikettier- und Verpackungsmaterialien sollten Spezifikationen festgelegt und dokumentiert werden. Zusätzlich sind möglicherweise Spezifikationen für bestimmte andere Materialien erforderlich, wie z.B. Prozesshilfen, Dichtungen oder andere bei der Produktion von Zwischenprodukten oder Wirkstoffen verwendete Materialien, die die Qualität beeinträchtigen könnten. Für Inprozesskontrollen sollten Akzeptanzkriterien festgelegt und dokumentiert werden. 6.18 Falls elektronische Unterschriften auf Dokumenten verwendet werden, sollten sie authentifiziert und sicher sein. 6.2 Reinigung der Ausrüstung und Protokolle über den Gebrauch6.20 Aufzeichnungen über Gebrauch, Reinigung, Desinfektion und/oder Sterilisation und Wartung größerer Ausrüstung sollten Datum, Zeit (falls erforderlich), Produkt und Chargennummer jeder in der betreffenden Ausrüstung verarbeiteten Charge sowie die Person, die die Reinigung und Wartung durchgeführt hat, beinhalten. 6.21 Wenn die Ausrüstung nur für die Mono-Produktion eines Zwischenprodukts oder Wirkstoffs verwendet wird, sind individuelle Ausrüstungsaufzeichnungen nicht nötig, sofern die Chargen des Zwischenprodukts oder des Wirkstoffs lückenlos aufeinander folgen. In Fällen, in denen Mono-Anlagen eingesetzt werden, können die Aufzeichnungen zu Reinigung, Wartung und Verwendung Teil des Chargenprotokolls sein oder getrennt gepflegt werden. 6.3 Protokolle über Rohmaterialien, Zwischenprodukte, Kennzeichnungs- und Verpackungsmaterialien der Wirkstoffe6.30 Unter anderem sollten die Aufzeichnungen mindestens Folgendes enthalten:
den Namen des Herstellers, Identität und Menge der Lieferung jeder Charge von Rohmaterialien, Zwischenprodukten oder Etikettier- und Verpackungsmaterialien für Wirkstoffe; den Namen des Lieferanten; falls bekannt, die Kontrollnummer(n) des Lieferanten, ansonsten eine andere Identifikationsnummer; die beim Wareneingang zugewiesene Nummer und das Datum des Wareneingangs;
•
die Ergebnisse aller durchgeführten Tests oder Untersuchungen und die hieraus gezogenen Schlussfolgerungen;•
Seite 26 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Protokolle, mit Hilfe derer die Verwendung der Materialien rückverfolgt werden kann;•Dokumentation der Untersuchung und der Überprüfung des Etikettier- und Packmaterials für Wirkstoffe auf Übereinstimmung mit den festgesetzten Spezifikationen;
•
die endgültige Entscheidung über zurückgewiesene Rohmaterialien, Zwischenprodukte oder Etikettier- und Verpackungsmaterialien für Wirkstoffe.
•
6.31 Es sollten (genehmigte) Muster-Etiketten zum Vergleich mit den ausgegebenen Etiketten vorhanden sein. 6.4 Muster-Herstellungsanweisungen (Muster-Herstellungs- und -Kontrollberichte)6.40 Um von Charge zu Charge Gleichförmigkeit zu gewährleisten, sollten Muster-Herstellungsanweisungen für jedes Zwischenprodukt und jeden Wirkstoff von einer Person erstellt, datiert und unterschrieben und von einem Vertreter der Qualitätssicherungseinheit(en) unabhängig davon überprüft, datiert und unterschrieben werden. 6.41 Muster-Herstellungsanweisungen sollten folgende Punkte umfassen:
den Namen des herzustellenden Zwischenprodukts oder Wirkstoffs und gegebenenfalls einen eindeutigen Referenzcode für das Dokument;
•
eine vollständige Liste der Rohmaterialien und Zwischenprodukte mit ausreichend spezifischen Namen oder Codes, um etwaige besondere Qualitätsmerkmale identifizieren zu können;
•
eine genaue Angabe der Menge oder des Mengenverhältnisses eines jeden verwendeten Rohmaterials oder Zwischenprodukts, einschließlich seiner Maßeinheit. Ist die Menge nicht festgelegt, sollte die Berechnung für jede Chargengröße oder Produktionsrate beigefügt werden. Abweichungen der Mengen sollten angegeben werden, wo sie begründet sind;
•
den Produktionsort und die zu verwendende größere Ausrüstung;•detaillierte Herstellungsanweisungen, einschließlich: •
Reihenfolge, die zu beachten ist,◦
Grenzbereiche für die zu verwendenden Prozessparameter,◦
Anweisungen für die Probenahme und Inprozesskontrollen gegebenenfalls samt ihrer Akzeptanzkriterien,◦
Zeitgrenzen für die Durchführung einzelner Prozessschritte und/oder gegebenenfalls des gesamten Prozesses;◦
den zu erwartenden Ausbeutebereich in geeigneten Phasen der Verarbeitung oder zu bestimmten Zeitpunkten;◦
falls erforderlich besondere Anmerkungen und Vorsichtsmaßnahmen, die zu befolgen sind, oder Querverweise auf diese;◦
Anweisungen für die Lagerung von Zwischenprodukten oder Wirkstoffen einschließlich der Etikettier- und Verpackungsmaterialien, um ihre Eignung für die Verarbeitung zu gewährleisten, sowie gegebenenfalls spezielle Lagerbedingungen mit Zeitbegrenzung.
◦
6.5 Chargenprotokolle (Protokolle über die Chargenherstellung und -prüfung)6.50 Für jedes Zwischenprodukt und jeden Wirkstoff sollten Chargenprotokolle erstellt werden, die die vollständige Information zur Produktion und Kontrolle jeder Charge beinhalten. Die Chargenprotokolle sollten vor ihrer Ausgabe überprüft werden, um zu gewährleisten, dass es sich um die korrekte Version und eine lesbare, akkurate Reproduktion der entsprechenden Muster-Herstellungsvorschrift handelt. Wird das Chargenprotokoll anhand eines separaten Teils des Muster-Dokuments erstellt, sollte dieses Dokument einen Verweis auf die verwendete aktuelle Muster-Herstellungsvorschrift aufweisen. 6.51 Diese Protokolle sollten bei ihrer Ausgabe mit einer unverwechselbaren Chargen- oder Identifikationsnummer versehen, datiert und unterschrieben sein. Bei kontinuierlicher Produktion kann bis zur Zuteilung einer endgültigen Nummer der Produktcode zusammen mit dem Datum und der Zeit als unverwechselbare Kennzeichnung gelten. 6.52 Die Dokumentation der Fertigstellung jedes wichtigen Schritts in den Chargenprotokollen (Chargenherstellungs- und -prüfprotokollen) sollte Folgendes einschließen:
Datum und gegebenenfalls Zeiten;•Angabe der verwendeten größeren Ausrüstung (z.B. Reaktoren, Trockner, Mühlen etc.);•spezifische Identifikation jeder Charge, einschließlich Gewichte, Maße und Chargennummern der in der Produktion eingesetzten Rohmaterialien, Zwischenprodukte und möglicherweise aufgearbeiteten Materialien;
•
tatsächliche Messergebnisse für kritische Prozessparameter;•jede erfolgte Probenahme;•Unterschriften der Personen, die die kritischen Betriebsschritte durchgeführt und direkt überwacht oder überprüft haben;•Inprozess- und Labortestergebnisse;•Ist-Ausbeute in geeigneten Phasen oder Zeiten;•Beschreibung der Verpackung und des Etiketts des Zwischenprodukts oder Wirkstoffs;•ein repräsentatives Etikett des Wirkstoffs oder Zwischenprodukts, wenn dieser/dieses in den Handel gebracht wird;•festgestellte Abweichungen, ihre Bewertung, Untersuchung (falls zutreffend) oder•bei separater Lagerung einen Verweis auf eine derartige Untersuchung;•Ergebnisse der Freigabeprüfung.•
6.53 Für die Untersuchung kritischer Abweichungen oder den Fall, dass eine Zwischenprodukt- oder Wirkstoffcharge ihre Spezifikationen nicht erfüllt, sollten schriftliche Verfahren festgelegt und eingehalten werden. Eine Untersuchung sollte auf weitere Chargen, die von der konkreten Nichterfüllung oder Abweichung betroffen sein könnten, ausgedehnt werden. 6.6 Prüfprotokolle6.60 Prüfprotokolle der Laborkontrollen sollten die vollständigen Daten aller durchgeführten Tests einschließlich (physikalischer) Prüfungen und (chemischer) Bestimmungen beinhalten, um eine Einhaltung der festgelegten Spezifikationen und Standards sicherzustellen, und zwar:
eine Beschreibung der zur Prüfung erhaltenen Proben, einschließlich Name oder Herkunft des Materials, Chargennummer oder einer anderen unverwechselbaren Codierung, Datum der Probenahme und gegebenenfalls die Menge und das Datum, an dem die Probe zur Prüfung einging;
•
Angaben zu oder Verweise auf alle verwendeten Prüfmethoden;•
Seite 27 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Angaben zu Gewicht oder Maß der Probe, die für den jeweiligen Test gemäß den Vorgaben der Prüfmethode verwendet wurde; Daten zur oder Querverweise auf die Herstellung und Prüfung von Referenzstandards, Reagenzien und Standardlösungen;
•
eine vollständige Aufzeichnung sämtlicher während der einzelnen Tests erzeugten Rohdaten, zusätzlich zu Grafiken, Tabellen und Spektren aus Laborinstrumenten, auf denen ordnungsgemäß angegeben ist, um welches spezifische Material und welche geprüfte Charge es sich handelt;
•
eine Aufzeichnung aller im Zusammenhang mit dem Test vorgenommenen Berechnungen, einschließlich z.B. der Maßeinheiten, Umrechnungsfaktoren und Äquivalenzfaktoren;
•
eine Darlegung der Testergebnisse und ein Vergleich mit den festgelegten Akzeptanzkriterien;•die Unterschrift der Person, die den jeweiligen Test durchgeführt hat sowie das Datum/die Daten der Prüfung;•das Datum und die Unterschrift einer zweiten Person, um nachzuweisen, dass die Originalprotokolle auf Richtigkeit, Vollständigkeit und Einhaltung der festgelegten Standards hin überprüft wurden.
•
6.61 Weiterhin sollten vollständige Aufzeichnungen zu folgenden Punkten geführt werden:
jegliche Modifikationen an einer festgelegten Prüfmethode;•periodische Kalibrierungen der Laborinstrumente, Apparate, Mess- und Aufzeichnungsgeräte;•alle an Wirkstoffen vorgenommenen Stabilitätstests;•Untersuchungen zu Ergebnissen außerhalb der Spezifikation (OOS).•
6.7 Überprüfung der Chargenprotokolle6.70 Für die Überprüfung und Genehmigung von Chargenherstellungsprotokollen und Prüfprotokollen, einschließlich der Protokolle über Verpackung und Etikettierung, sollten schriftlich Verfahren niedergelegt und eingehalten werden, um vor der Freigabe oder dem Vertrieb einer Charge festzustellen, ob ein Zwischenprodukt oder Wirkstoff die festgelegten Spezifikationen einhält. 6.71 Die Chargenherstellung und -prüfprotokolle kritischer Prozessschritte sollten von der (den) Qualitätssicherungseinheit(en) vor Freigabe oder Vertrieb einer Wirkstoffcharge überprüft und genehmigt werden. Herstellungs- und Prüfprotokolle nichtkritischer Prozessschritte können unter Einhaltung von durch die Qualitätssicherungseinheit(en) genehmigten Verfahren von qualifiziertem Produktionspersonal oder anderen Einheiten überprüft werden. 6.72 Alle Abweichungen, Untersuchungen und DOS-Berichte sollten vor der Freigabe einer Charge als Teil der allgemeinen Überprüfung des Chargenprotokolls einer Prüfung unterzogen werden. 6.73 Die Qualitäts(sicherungs)einheit(en) darf (dürfen) der Produktionseinheit die Verantwortung und Vollmacht für die Freigabe von Zwischenprodukten mit Ausnahme derjenigen übertragen, die den Kontrollbereich der Herstellerfirma verlassen.
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Teil II (2/3)
Abschnitt 7 Materialmanagement
7.1 Allgemeine Kontrollen7.10 Es sollte schriftliche Verfahren für die Annahme, Identifikation, Quarantäne, Lagerung, Handhabung, Probenahme, Prüfung und Genehmigung oder Zurückweisung von Materialien geben. 7.11 Hersteller von Zwischenprodukten und/oder Wirkstoffen sollten über ein System zur Bewertung der Lieferanten von kritischen Materialien verfügen. 7.12 Materialien sollten nach einer festgesetzten Spezifikation von einem oder mehreren von der (den) Qualitätssicherungseinheit(en) genehmigten Lieferanten bezogen werden. 7.13 Wenn der Lieferant eines kritischen Materials nicht gleichzeitig Hersteller des Materials ist, sollte dem Hersteller von Zwischenprodukten und/oder Wirkstoffen der Name und die Adresse des Materialherstellers bekannt sein. 7.14 Eine Veränderung der Lieferquelle für kritische Rohmaterialien sollte gemäß Abschnitt 13 Änderungskontrolle vollzogen werden. 7.2 Wareneingang und Quarantäne7.20 Beim Wareneingang und vor der Abnahme sollte jedes Behältnis oder jede Gruppe von Behältnissen mit Material visuell auf korrekte Etikettierung (einschließlich Korrelation zwischen Lieferanten- und interner Werksbezeichnung, sofern sie sich unterscheiden), Beschädigung des Behältnisses oder des Siegels und Hinweise auf Manipulation oder Kontamination untersucht werden. Materialien sollten so lange in Quarantäne gehalten werden, bis sie bemustert, je nach Erfordernis untersucht oder getestet und anschließend für den Gebrauch freigegeben wurden. 7.21 Bevor eingehende Materialien mit vorhandenen Beständen (z.B. Lösungsmittel oder Silovorräte) gemischt werden, sollten sie als korrekt identifiziert und falls erforderlich geprüft sowie freigegeben werden. Es sollte Verfahren geben, die eine falsche Einordnung in die vorhandenen Bestände verhindern. 7.22 Wenn Bulklieferungen in Mehrzweck-Tankwagen zugestellt werden, sollte gewährleistet werden, dass durch das Transportmittel keine Kreuzkontamination entsteht. Möglichkeiten, diese Gewährleistung zu erbringen, könnten einen oder mehrere der folgenden Punkte einschließen:
Reinigungszertifikat;•Prüfen auf Spuren von Verunreinigungen;•Audit des Lieferanten.•
7.23 Große Behältnisse zur Lagerung und die zugehörigen Rohrverzweigungen, Füll- und Ablassleitungen sollten ordnungsgemäß gekennzeichnet sein. 7.24 Jedem Materialbehältnis oder jeder Gruppe von Behältnissen (Chargen) sollte ein unverwechselbarer Code oder eine Chargen- oder Eingangsnummer zugeordnet werden. Diese Nummer sollte Bestandteil der Dokumentation über die Weiterverwendung jeder Charge werden. Es sollte ein System geben, mit Hilfe dessen der Status jeder Charge bestimmt werden kann. 7.3 Probenahme und Prüfung eingehender Materialien für die Produktion7.30 Außer bei unter Nummer 7.32 beschriebenen Materialien sollte mindestens ein Identitätstest an jeder Charge eines Materials durchgeführt werden. Statt weiterer Tests kann ein Analysenzertifikat des Lieferanten verwendet werden, vorausgesetzt, dass der Hersteller über ein System zur Lieferantenbewertung verfügt.
Seite 28 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

7.31 Die Lieferantengenehmigung sollte eine Bewertung einschließen, die ausreichende Beweise (z.B. die Qualitätshistorie) dafür erbringt, dass der Hersteller kontinuierlich Material liefern kann, das die entsprechenden Spezifikationen erfüllt. Bevor die eigene Prüfung reduziert wird, sollte an mindestens drei Chargen eine Vollanalyse durchgeführt werden. Es sollte jedoch mindestens in angemessenen Zeitabständen eine Vollanalyse durchgeführt und mit dem Analysenzertifikat verglichen werden. Die Zuverlässigkeit von Analysenzertifikaten sollte in regelmäßigen Abständen geprüft werden. 7.32 Prozesshilfen, gefährliche oder hoch toxische Rohmaterialien und andere besondere Materialien oder Materialien, die in andere Betriebe innerhalb des Kontrollbereichs der Firma transferiert werden, brauchen nicht getestet zu werden, wenn das Analysenzertifikat des Herstellers vorliegt und zeigt, dass diese Rohmaterialien den festgelegten Spezifikationen entsprechen. Eine visuelle Prüfung der Behältnisse und Etiketten sowie ein Festhalten der Chargennummern sollte dabei helfen, die Identität dieser Materialien festzustellen. Das Weglassen einer Prüfung vor Ort sollte begründet und dokumentiert werden. 7.33 Proben sollten repräsentativ sein für die Materialcharge, der sie entnommen wurden. Die Methoden zur Probenahme sollten die Anzahl der zu bemusternden Behältnisse, welcher Teil des Behältnisses zu bemustern ist und die Menge an Material, die aus jedem Behältnis zu entnehmen ist, festlegen. Die Zahl der zu untersuchenden Behältnisse und die Probengröße sollten auf einem Probenahmeplan basieren, der berücksichtigt, wie kritisch und variabel ein Material ist, wie die Qualitätshistorie des Lieferanten aussieht und welche Materialmengen für die Analyse erforderlich sind. 7.34 Die Probenahme sollte an definierten Stellen und mit Hilfe von Verfahren stattfinden, die eine Kontamination des beprobten Materials sowie anderer Materialien verhindern. 7.35 Behältnisse, aus denen Proben entnommen werden, sollten vorsichtig geöffnet und im Anschluss wieder verschlossen werden. Sie sollten markiert werden, um anzuzeigen, dass eine Probe entnommen wurde. 7.4 Lagerung7.40 Materialien sollten so gehandhabt und gelagert werden, dass Abbau, Kontamination und Kreuzkontamination verhindert werden. 7.41 Materialien, die in Fibertrommeln, Beuteln oder Kisten aufbewahrt werden, sollten nicht auf dem Boden und gegebenenfalls in Abständen voneinander gelagert werden, um eine Reinigung und Inspektion zu ermöglichen. 7.42 Materialien sollten unter Bedingungen und über Zeiträume gelagert werden, die ihre Qualität nicht beeinträchtigen und sollten normalerweise so gesteuert werden, dass die ältesten Lagerbestände zuerst verbraucht werden. 7.43 Bestimmte Materialien können in geeigneten Behältnissen im Freien gelagert werden, vorausgesetzt, dass ihre Kennzeichnung lesbar bleibt und die Behältnisse vor dem Offnen und der Verwendung ordnungsgemäß gereinigt werden. 7.44 Zurückgewiesene Materialien sollten im Rahmen eines Quarantänesystems, das eine unbefugte Verwendung für die Herstellung verhindert, identifiziert und kontrolliert werden. 7.5 Nachbewertung7.50 Materialien sollten gegebenenfalls nachbewertet werden, um ihre Eignung für den Gebrauch festzustellen (z.B. nach längerer Lagerung oder Einwirkung von Hitze oder Feuchtigkeit).
Abschnitt 8 Produktion und Inprozesskontrollen
8.1 Produktionsaktivitäten8.10 Rohmaterialien für die Herstellung von Zwischenprodukten und Wirkstoffen sollten unter geeigneten Bedingungen, die ihre Eignung für den Verwendungszweck nicht beeinflussen, verwogen oder abgemessen werden. Die Wäge- und Messgeräte sollten geeignete Genauigkeit für den beabsichtigten Verwendungszweck bieten. 8.11 Wird ein Material für den späteren Gebrauch in der Produktion unterteilt, sollte das Behältnis, in das das Material gefüllt wird, geeignet sein und eine Kennzeichnung erhalten, der die folgenden Informationen zu entnehmen sind:
Materialname und/oder Code;•Eingangs- oder Kontrollnummer;•Gewicht oder Maß des Materials im neuen Behältnis;•falls erforderlich, das Datum für eine Nachbewertung oder Wiederholungsprüfung.•
8.12 Kritische Wäge-, Mess- oder Unterteilungsaktivitäten sollten von einer zweiten Person bezeugt oder einer gleichwertigen Kontrolle unterzogen werden. Vor der Verwendung sollte das Produktionspersonal verifizieren, dass es sich um die Materialien handelt, die im Chargenprotokoll für das herzustellende Zwischenprodukt oder den entsprechenden Wirkstoff angegeben sind. 8.13 Andere kritische Aktivitäten sollten bezeugt oder einer entsprechenden Kontrolle unterzogen werden. 8.14 Auf bestimmten Stufen des Produktionsprozesses sollte die Ist-Ausbeute mit der erwarteten Ausbeute verglichen werden. Die erwartete Ausbeute einschließlich geeigneter Bereiche sollte auf der Grundlage früherer Labor-, Entwicklungs- oder Herstellungsdaten festgelegt werden. Abweichungen bei der Ausbeute, die mit kritischen Prozessschritten in Verbindung stehen, sollten untersucht werden, um ihre Auswirkung oder potenzielle Auswirkung auf die Qualität der betroffenen Chargen zu bestimmen. 8.15 Alle Abweichungen sollten dokumentiert und erklärt werden. Jede kritische Abweichung sollte untersucht werden. 8.16 Der Prozessstatus größerer Ausrüstungseinheiten sollte entweder an den einzelnen Ausrüstungseinheiten oder durch geeignete Dokumentation, entsprechende Computersteuerungssysteme oder andere Mittel angegeben werden. 8.17 Auf- oder umzuarbeitende Materialien sollten angemessen kontrolliert werden, um eine unbefugte Verwendung zu verhindern. 8.2 Zeitbegrenzungen8.20 Wenn in der Muster-Herstellungsvorschrift (vgl. Nummer 6.41) Zeitgrenzen angegeben sind, sollten diese eingehalten werden, um die Qualität von Zwischenprodukten oder Wirkstoffen sicherzustellen. Abweichungen sollten dokumentiert und bewertet werden. Zeitgrenzen können unpassend sein, wenn auf einen Zielwert hingearbeitet wird (z.B. pH-Anpassung, Hydrierung, Trocknen bis zu einem festgelegten Wert), weil der Abschluss von Reaktionen oder Prozessschritten durch Inprozessprobenahme und -prüfung bestimmt wird. 8.21 Zwischenprodukte, die zur späteren Weiterverarbeitung aufbewahrt werden, sollten unter angemessenen Bedingungen gelagert werden, um ihre Eignung für den Verwendungszweck sicherzustellen. 8.3 Inprozessprobenahme und -kontrollen8.30 Zur Überwachung des Prozessfortschritts und zur Kontrolle des Ablaufs von Prozessschritten, die Schwankungen in den Qualitätseigenschaften von Zwischenprodukten und Wirkstoffen verursachen, sollten schriftliche Verfahren festgelegt werden. Inprozesskontrollen und die zugehörigen Akzeptanzkriterien sollten auf Basis der Information, die während der Entwicklungsphase gesammelt wurde, oder auf Grund historischer Daten festgelegt werden. 8.31 Die Akzeptanzkriterien sowie die Art und der Umfang der Prüfung können von der Natur des herzustellenden Zwischenprodukts oder Wirkstoffs, der durchzuführenden Reaktion oder des durchzuführenden Prozessschritts und dem Ausmaß, in dem der Prozess zu
Seite 29 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Schwankungen in der Produktqualität führt, abhängen. Während bei früheren Prozessstufen weniger strenge Inprozesskontrollen angemessen sein können, erfordern spätere Prozessstufen (z.B. Isolierungs- und Reinigungsschritte) möglicherweise striktere Kontrollen. 8.32 Kritische Inprozesskontrollen (und kritische Prozessüberwachung) einschließlich der Kontrollpunkte und -methoden sollten schriftlich festgehalten und von der (den) Qualitätssicherungseinheit (en) genehmigt werden. 8.33 Inprozesskontrollen können von qualifiziertem Produktionspersonal vorgenommen und der Prozess ohne die vorherige Genehmigung durch die Qualitätssicherungseinheit(en) angepasst werden, sofern die Anpassung innerhalb vorher festgelegter, von der (den) Qualitätssicherungseinheit(en) genehmigter Grenzen erfolgt. Alle Prüfungen und deren Ergebnisse sollten als Teil des Chargenprotokolls umfassend dokumentiert werden. 8.34 Schriftliche Verfahren sollten die Probenahmemethoden für Inprozessmaterialien, Zwischenprodukte und Wirkstoffe beschreiben. Probenahmepläne und -verfahren sollten auf wissenschaftlich fundierten Probenahmepraktiken beruhen. 8.35 Die Inprozessprobenahme sollte unter Verwendung von Verfahren durchgeführt werden, die eine Kontamination des bemusterten Materials sowie anderer Zwischenprodukte oder Wirkstoffe verhindern. Es sollten Verfahren erstellt werden, die die Unversehrtheit von Proben nach ihrer Entnahme sicherstellen. 8.36 Untersuchungen zum "Testergebnis außerhalb der Spezifikation" (OOS) sind normalerweise nicht erforderlich für Inprozessprüfungen, die zum Zwecke der Prozessüberwachung und/oder -anpassung erfolgen. 8.4 Mischen (Verschneiden) von Zwischenprodukt- oder Wirkstoffchargen8.40 Für die Zwecke des vorliegenden Dokuments ist Mischen als ein Prozess definiert, bei dem Materialien innerhalb der gleichen Spezifikation mit dem Ziel kombiniert werden, ein homogenes Zwischenprodukt oder einen homogenen Wirkstoff herzustellen. Das Mischen von Teilen einer Charge während des Prozesses (z.B. die Vereinigung mehrerer Zentrifugenladungen aus einer einzigen Kristallisierungscharge) oder die Kombination von Fraktionen aus mehreren Chargen für die weitere Verarbeitung wird als Teil des Produktionsprozesses und nicht als Mischen betrachtet. 8.41 Chargen außerhalb der Spezifikation sollten nicht zum Zweck der Spezifikationsentsprechung mit anderen Chargen gemischt werden. Jede einer Mischung hinzugefügte Charge sollte vor dem Mischen nach einem definierten Prozess hergestellt und einzeln getestet worden sein sowie nachweislich innerhalb der entsprechenden Spezifikationen liegen. 8.42 Zulässige Mischaktivitäten beinhalten, beschränken sich jedoch nicht auf:
das Mischen kleiner Chargen, um die Chargengröße zu vergrößern;•das Mischen von Nachläufen (d. h. relativ kleiner Mengen isolierten Materials) aus Chargen desselben Zwischenprodukts oder Wirkstoffs, um eine einzige Charge zu erzeugen.
•
8.43 Mischvorgänge sollten ordnungsgemäß kontrolliert und dokumentiert werden. Die gemischte Charge sollte, wenn nötig, auf eine Einhaltung der festgelegten Spezifikationen geprüft werden. 8.44 Das Chargenprotokoll des Mischprozesses sollte eine Rückverfolgung bis zu den einzelnen Chargen, aus denen die Mischung besteht, erlauben. 8.45 In Fällen, in denen die physikalischen Eigenschaften des Wirkstoffs kritisch sind (z.B. Wirkstoffe, die für feste orale Darreichungsformen oder Suspensionen vorgesehen sind), sollten die Mischvorgänge validiert werden, um die Homogenität der gemischten Charge zu belegen. Die Validierung sollte eine Prüfung der kritischen Eigenschaften (z.B. Korngrößenverteilung, Schütt- und Stampfdichte), die vom Mischprozess beeinflusst werden könnten, einschließen. 8.46 Wenn Mischen die Stabilität beeinträchtigen könnte, sollte eine Stabilitätsprüfung der endgültigen, gemischten Chargen durchgeführt werden. 8.47 Das Verfalls- oder das Wiederholungsprüfungsdatum der gemischten Charge sollte auf dem Herstellungsdatum des ältesten Nachlaufs bzw. der ältesten Charge in der Mischung basieren. 8.5 Kontaminationskontrolle8.50 Materialreste können in Folgechargen des gleichen Zwischenprodukts oder Wirkstoffes eingebracht werden, wenn sie adäquat kontrolliert werden. Dies schließt beispielsweise Produktreste an den Wänden eines Mikronisators, eine Schicht feuchter Kristalle, die nach dem Entleeren in einer Zentrifugentrommel zurückbleiben, und die unvollständige Entfernung von Flüssigkeiten oder Kristallen aus einem Verarbeitungsgefäß beim Transfer des Materials zum nächsten Prozessschritt ein. Derartige Übertragungen sollten nicht zum Einbringen von Abbauprodukten oder mikrobieller Kontamination, die das festgelegte Wirkstoff-Verunreinigungsprofil beeinträchtigen könnten, führen. 8.51 Produktionsaktivitäten sollten so vollzogen werden, dass eine Kontamination der Zwischenprodukte oder Wirkstoffe durch andere Materialien verhindert wird. 8.52 Bei der Handhabung von Wirkstoffen nach der Reinigung sollten Vorsichtsmaßnahmen zur Vermeidung von Kontamination ergriffen werden.
Abschnitt 9 Verpackung und Kennzeichnung zur Identifizierung von Wirkstoffen und Zwischenprodukten
9.1 Allgemeine Anforderungen9.10 Für die Entgegennahme, Identifikation, Quarantäne, Probenahme, Untersuchung und/oder Prüfung und Freigabe und die Handhabung von Verpackungs- und Kennzeichnungsmaterialien sollten schriftliche Arbeitsanweisungen vorliegen. 9.11 Verpackungs- und Kennzeichnungsmaterialien sollten festgelegten Spezifikationen entsprechen. Materialien, die Spezifikationen nicht erfüllen, sollten zurückgewiesen werden, um ihre Verwendung für Vorgänge, für die sie ungeeignet sind, zu verhindern. 9.12 Für jede Etiketten- und Packmittellieferung sollten Aufzeichnungen über Entgegennahme, Untersuchung oder Prüfungen sowie den Status (angenommen oder zurückgewiesen) aufbewahrt werden. 9.2 Verpackungsmaterialien9.20 Behältnisse sollten ausreichenden Schutz gegen Verderb oder Kontamination des Zwischenprodukts oder Wirkstoffs, die während des Transports und der empfohlenen Lagerung auftreten könnten, bieten. 9.21 Behältnisse sollten sauber und, sofern aufgrund der Beschaffenheit des Zwischenprodukts oder Wirkstoffs angezeigt, desinfiziert sein, um ihre Eignung für den beabsichtigten Verwendungszweck sicherzustellen. Derartige Behältnisse sollten sich nicht reaktiv, additiv oder absorptiv verhalten und dadurch die Qualität des Zwischenprodukts oder Wirkstoffs über die spezifizierten Grenzwerte hinaus verändern. 9.22 Werden Behältnisse wiederverwendet, sollten sie gemäß dokumentierter Verfahren gereinigt und alle vorherigen Etiketten beseitigt oder unlesbar gemacht werden. 9.3 Vergabe von Etiketten und Kontrolle9.30 Der Zugang zu den Lagerbereichen für Etiketten sollte auf autorisiertes Personal beschränkt sein.
Seite 30 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

9.31 Es sollten Verfahren zum mengenmäßigen Abgleichen der ausgegebenen, verwendeten und zurückgegebenen Etiketten sowie zum Bewerten gefundener Diskrepanzen zwischen der Zahl der etikettierten Behältnisse und der Zahl der ausgegebenen Etiketten verwendet werden. Derartige Diskrepanzen sollten untersucht und die Untersuchungen von der (den) Qualitätssicherungseinheit (en) genehmigt werden. 9.32 Alle überschüssigen Etiketten, die mit Chargennummern oder anderen chargenbezogenen Aufdrucken versehen sind, sollten vernichtet werden. Zurückgegebene Etiketten sollten aufbewahrt und so gelagert werden, dass Vermischungen verhindert werden und für die richtige Identifizierung gesorgt ist. 9.33 Überholte und veraltete Etiketten sollten vernichtet werden. 9.34 Druckvorrichtungen, die zum Drucken von Etiketten für Verpackungsvorgänge benutzt werden, sollten kontrolliert werden, um sicherzustellen, dass alle Aufdrucke den Druckvorgaben des Chargenherstellungsprotokolls entsprechen. 9.35 Bedruckte, für eine bestimmte Charge ausgegebene Etiketten sollten sorgfältig auf richtige Identität und Übereinstimmung mit den Spezifikationen im Muster-Herstellungsprotokoll überprüft werden. Die Ergebnisse dieser Überprüfung sollten dokumentiert werden. 9.36 Ein bedrucktes Etikett, das repräsentativ für die verwendeten Etiketten ist, sollte dem Chargenherstellungsprotokoll beigefügt werden. 9.4 Verpackungs- und Kennzeichnungsvorgänge9.40 Es sollten dokumentierte Verfahren vorhanden sein, mit Hilfe derer sichergestellt werden kann, dass die richtigen Packmittel und Etiketten verwendet werden. 9.41 Die Kennzeichnungsvorgänge sollten so angelegt sein, dass sie Vermischungen verhindern. Es sollte eine körperliche oder räumliche Trennung von Vorgängen, an denen andere Zwischenprodukte oder Wirkstoffe beteiligt sind, erfolgen. 9.42 Etiketten, die für Zwischenprodukt- oder Wirkstoffbehältnisse verwendet werden, sollten den Namen oder den Identifikationscode, die Chargennummer des Produkts und, sofern diese Information für die Sicherstellung der Qualität des Zwischenprodukts oder Wirkstoffs kritisch ist, die Lagerbedingungen anzeigen. 9.43 Soll ein Zwischenprodukt oder Wirkstoff an einen Bereich weitergegeben werden, der außerhalb der Kontrolle des Materialmanagementsystems des Herstellers liegt, sollten der Name und die Adresse des Herstellers, die Inhaltsmengen, besondere Transportbedingungen sowie gesetzliche Vorgaben ebenfalls auf dem Etikett angegeben werden. Bei Zwischenprodukten oder Wirkstoffen mit einem Verfallsdatum sollte das Verfallsdatum auf dem Etikett und dem Analysenzertifikat angegeben werden. Bei Zwischenprodukten oder Wirkstoffen mit einem Wiederholungsprüfungsdatum sollte das Wiederholungsprüfungsdatum auf dem Etikett und/oder auf dem Analysenzertifikat angegeben werden. 9.44 Verpackungs- und Etikettieranlagen sollten direkt vor Gebrauch inspiziert werden, um sicherzustellen, dass sämtliche Materialien, die für den nächsten Verpackungsvorgang nicht benötigt werden, entfernt worden sind. Diese Untersuchung sollte in den Chargenherstellungsprotokollen, dem Anlagen-Logbuch oder einem anderen Dokumentationssystem dokumentiert werden. 9.45 Verpackte und etikettierte Zwischenprodukte oder Wirkstoffe sollten überprüft werden, um sicherzustellen, dass alle Behältnisse und Verpackungen einer Charge das richtige Etikett tragen. Diese Überprüfung sollte Teil des eigentlichen Verpackungsvorgangs sein. Die Ergebnisse dieser Überprüfungen sollten im Chargenherstellungs- oder -prüfprotokoll festgehalten werden. 9.46 Zwischenprodukt- oder Wirkstoffbehältnisse, die außerhalb des Kontrollbereichs des Herstellers versandt werden, sollten so versiegelt sein, dass der Empfänger bei beschädigtem oder fehlendem Siegel vor der Möglichkeit gewarnt wird, dass am Inhalt Veränderungen vorgenommen worden sein könnten.
Abschnitt 10 Lagerung und Vertrieb
10.1 Lagerverfahren10.10 Es sollten Einrichtungen für die Lagerung aller Materialien unter geeigneten Bedingungen (z.B. kontrollierter Temperatur und Feuchtigkeit, sofern notwendig) vorhanden sein. Über diese Bedingungen sollten Aufzeichnungen geführt werden, wenn sie für den Erhalt der Materialeigenschaften kritisch sind. 10.11 Besteht kein alternatives System, mit Hilfe dessen eine unbeabsichtigte oder unbefugte Verwendung von unter Quarantäne stehenden, zurückgewiesenen, zurückgesandten oder rückgerufenen Materialien verhindert werden kann, sollten für die vorübergehende Lagerung dieser Materialien so lange separate Lagerbereiche zugewiesen werden, bis eine Entscheidung über ihre weitere Verwendung gefallen ist. 10.2 Vertriebsverfahren10.20 Wirkstoffe und Zwischenprodukte sollten nur zum Vertrieb an Dritte freigegeben werden, wenn sie zuvor von der (den) Qualitätssicherungseinheit(en) freigegeben wurden. Wirkstoffe und Zwischenprodukte können unter Quarantäne an andere, der Kontrolle der Firma unterstehende Bereiche weitergegeben werden, wenn dies von der (den) Qualitätssicherungseinheit(en) genehmigt wurde und angemessene Kontrollmechanismen sowie eine entsprechende Dokumentation vorhanden sind. 10.21 Wirkstoffe und Zwischenprodukte sollten so transportiert werden, dass ihre Qualität nicht beeinträchtigt wird. 10.22 Spezielle Transport- oder Lagerbedingungen für einen Wirkstoff oder ein Zwischenprodukt sollten auf dem Etikett angegeben werden. 10.23 Der Hersteller sollte sicherstellen, dass das mit dem Transport der Wirkstoffe oder Zwischenprodukte beauftragte Unternehmen die erforderlichen Transport- und Lagerbedingungen kennt und sie befolgt. 10.24 Es sollte ein System vorhanden sein, mit Hilfe dessen der Vertrieb jeder Zwischenprodukt- und/oder Wirkstoffcharge jederzeit nachvollzogen werden kann, um einen Rückruf zu ermöglichen.
Abschnitt 11 Laborkontrollen
11.1 Allgemeine Kontrollen11.10 Der (Den) unabhängigen Qualitäts (sicherungs) einheit (en) sollten geeignete Laboreinrichtungen zur Verfügung stehen. 11.11 Es sollten dokumentierte Verfahren für die Probenahme, Prüfung, Freigabe oder Zurückweisung von Materialien sowie die Aufzeichnung und Lagerung von Labordaten existieren. Laborprotokolle sollten gemäß Nummer 6.6 geführt werden. 11.12 Sämtliche Spezifikationen, Probenahmepläne und Prüfverfahren sollten wissenschaftlich fundiert und geeignet sein, um sicherzustellen, dass die Rohmaterialien, Zwischenprodukte, Wirkstoffe, Etiketten und Packmaterialien den festgelegten Qualitäts- und/oder Reinheitsstandards entsprechen. Spezifikationen und Prüfverfahren sollten mit den in der Anmeldung enthaltenen übereinstimmen. Zusätzlich zu denen in der Anmeldung können weitere Spezifikationen vorhanden sein. Spezifikationen, Probenahmepläne und Prüfverfahren, einschließlich deren Änderungen, sollten von der zuständigen Organisationseinheit erstellt und von der (den) Qualitätssicherungseinheit(en) überprüft und genehmigt werden.
Seite 31 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

11.13 Für Wirkstoffe sollten gemäß akzeptierter Standards und im Einklang mit dem Herstellungsprozess geeignete Spezifikationen festgelegt werden. Die Spezifikationen sollten eine Kontrolle der Verunreinigungen (z.B. organische Verunreinigungen, anorganische Verunreinigungen und Restlösemittel) enthalten. Wenn der Wirkstoff eine Spezifikation für mikrobiologische Reinheit hat, sollten geeignete Aktionsgrenzen für die Gesamtkeimzahl und unzulässige Organismen festgelegt und eingehalten werden. Wenn der Wirkstoff eine Spezifikation für Endotoxine hat, sollten geeignete Aktionsgrenzen festgelegt und eingehalten werden. 11.14 Laborkontrollen sollten zum Zeitpunkt ihrer Durchführung dokumentiert werden. Jedes Abweichen von den oben beschriebenen Verfahren sollte dokumentiert und erklärt werden. 11.15 Alle Ergebnisse außerhalb der Spezifikation (DOS) sollten gemäß einem vorgeschriebenen Verfahren untersucht und dokumentiert werden. Dieses Verfahren sollte eine Datenanalyse, eine Bewertung, ob ein wichtiges Problem besteht, eine Aufgabenverteilung für die Korrekturmaßnahmen und Schlussfolgerungen verlangen. Erneutes Probenziehen und/oder Wiederholungsprüfungen nach Erhalt von DOS-Ergebnissen sollten gemäß einem dokumentierten Verfahren durchgeführt werden. 11.16 Reagenzien und Standardlösungen sollten nach schriftlichen Verfahren hergestellt und etikettiert werden. An analytischen Reagenzien und Standardlösungen sollte gegebenenfalls ein "Verwendbar bis"-Datum angebracht werden. 11.17 Primäre Referenzstandards sollten, soweit für die Herstellung von Wirkstoffen notwendig, beschafft werden. Die Quelle jedes primären Referenzstandards sollte dokumentiert werden. Es sollten Aufzeichnungen über Lagerung und Verwendung jedes primären Referenzstandards gemäß den Empfehlungen des Lieferanten geführt werden. Primäre Referenzstandards, die aus einer amtlich anerkannten Quelle stammen, werden normalerweise ohne Prüfung verwendet, sofern sie unter Bedingungen gelagert werden, die den Empfehlungen des Lieferanten entsprechen. 11.18 Wenn ein primärer Referenzstandard aus einer amtlich anerkannten Quelle nicht erhältlich ist, sollte ein "hauseigener Primärstandard" geschaffen werden. Es sollten geeignete Tests durchgeführt werden um Identität und Reinheit des primären Referenzstandards vollständig belegen zu können. Eine angemessene Dokumentation dieser Tests sollte aufbewahrt werden. 11.19 Sekundäre Referenzstandards sollten ordnungsgemäß hergestellt, identifiziert, getestet, genehmigt und gelagert werden. Die Eignung jeder Charge eines sekundären Referenzstandards sollte vor dem ersten Gebrauch durch Vergleich mit einem primären Referenzstandard festgestellt werden. Jede Charge eines sekundären Referenzstandards sollte in periodischen Abständen nach einem schriftlichen Plan requalifiziert werden. 11.2 Prüfung von Zwischenprodukten und Wirkstoffen11.20 An jeder Charge eines Zwischenprodukts und Wirkstoffs sollten geeignete Labortests durchgeführt werden, um die Einhaltung der Spezifikationen zu überprüfen. 11.21 Für jeden Wirkstoff sollte normalerweise ein Verunreinigungsprofil erstellt werden, das die vorhandenen identifizierten und unidentifizierten Verunreinigungen in einer typischen, mit einem bestimmten kontrollierten Produktionsprozess hergestellten Charge beschreibt. Das Verunreinigungsprofil sollte die Identität oder eine qualitativanalytische Kennzeichnung (z.B. Retentionszeit), den Bereich jeder beobachteten Verunreinigung und eine Klassifizierung jeder identifizierten Verunreinigung (z.B. anorganisch, organisch, Lösungsmittel) beinhalten. Das Verunreinigungsprofil hängt normalerweise vom jeweiligen Herstellungsprozess und der Herkunft des Wirkstoffs ab. Verunreinigungsprofile sind für Wirkstoffe pflanzlichen Ursprungs oder aus tierischem Gewebe im Normalfall nicht erforderlich. Gesichtspunkte zur Biotechnologie werden in der ICH-Leitlinie Q6B behandelt. 11.22 Das Verunreinigungsprofil sollte in geeigneten Abständen mit dem Verunreinigungsprofil im Zulassungsantrag oder mit historischen Daten verglichen werden, um Veränderungen am Wirkstoff, die aus Modifikationen der Ausgangsmaterialien, der Geräteparameter oder des Produktionsprozesses resultieren, festzustellen. 11.23 Ist eine bestimmte mikrobielle Qualität spezifiziert, sollten entsprechende mikrobiologische Tests an jeder Charge eines Zwischenprodukts und Wirkstoffs durchgeführt werden. 11.3 Validierung von Prüfverfahrenvgl. Abschnitt 1211.4 Analysenzertifikate11.40 Für jede Zwischenprodukt- oder Wirkstoffcharge sollte auf Anfrage ein authentisches Analysenzertifikat ausgestellt werden. 11.41 Informationen zum Namen des Zwischenprodukts oder Wirkstoffs gegebenenfalls einschließlich seiner (Güte-)Klasse, der Chargennummer und des Freigabedatums sollten auf dem Analysenzertifikat angegeben werden. Bei Zwischenprodukten oder Wirkstoffen mit einem Verfallsdatum sollte das Verfallsdatum auf dem Etikett und dem Analysenzertifikat angegeben werden. Bei Zwischenprodukten oder Wirkstoffen mit einem Wiederholungsprüfungsdatum sollte dieses Datum auf dem Etikett und/oder dem Analysenzertifikat angegeben sein. 11.42 Das Zertifikat sollte alle gemäß Arzneibuch- oder Kundenanforderungen durchgeführten Tests einschließlich der Akzeptanzgrenzen und der erhaltenen numerischen Ergebnisse (falls die Testergebnisse numerischer Art sind) auflisten. 11.43 Zertifikate sollten datiert und von autorisiertem Personal der Qualitätssicherungseinheit(en) unterschrieben sein sowie den Namen, die Adresse und die Telefonnummer des Originalherstellers enthalten. Wird die Analyse von einem umverpackenden oder aufarbeitenden Unternehmen durchgeführt, sollte das Analysenzertifikat den Namen, die Adresse und die Telefonnummer dieses Unternehmens und einen Verweis auf den Namen des Originalherstellers enthalten. 11.44 Werden von oder im Namen eines umverpackenden, aufarbeitenden Unternehmens, Vertreters oder Händlers neue Zertifikate ausgestellt, sollten diese Zertifikate den Namen, die Adresse und die Telefonnummer des Labors, das die Analyse vornahm, anzeigen. Sie sollten außerdem einen Verweis auf Namen und Adresse des Originalherstellers und auf das Originalchargenzertifikat, dessen Kopie beigefügt sein sollte, enthalten. 11.5 Stabilitätsprüfung von Wirkstoffen11.50 Für die Überwachung der Stabilitätseigenschaften von Wirkstoffen sollte ein dokumentiertes, kontinuierliches Testprogramm entwickelt werden, dessen Ergebnisse für die Bestätigung geeigneter Lagerbedingungen und Wiederholungsprüfungs- oder Verfallsdaten verwendet werden sollten. 11.51 Die bei der Stabilitätsprüfung verwendeten Prüfverfahren sollten validiert sein und Hinweise auf die Stabilität geben. 11.52 Stabilitätsproben sollten in Behältnissen gelagert werden, die den Marktbehältnissen nachempfunden sind. Wenn ein Wirkstoff zum Beispiel in Beuteln in Fibertrommeln vermarktet wird, können die Stabilitätsproben in Beuteln aus dem gleichen Material und in kleineren Trommeln mit einer ähnlichen oder identischen Materialzusammensetzung wie die vermarkteten Trommeln verpackt werden. 11.53 Im Regelfall sollten die ersten drei Chargen aus kommerzieller Produktion dem Stabilitätsmonitoringprogramm unterzogen werden, um das Wiederholungsprüfungs- oder Verfallsdatum zu bestätigen. Wenn jedoch Daten aus vorherigen Untersuchungen zeigen, dass man erwarten kann, dass der Wirkstoff mindestens zwei Jahre stabil ist, können weniger als drei Chargen verwendet werden. 11.54 Im Anschluss daran sollte mindestens eine Charge des hergestellten Wirkstoffs pro Jahr (außer wenn in diesem Jahr keine Charge produziert wird) in das Stabilitätsmonitoringprogramm einbezogen und mindestens einmal jährlich getestet werden, um die Stabilität zu bestätigen.
Seite 32 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

11.55 Bei Wirkstoffen mit kurzer Haltbarkeitsdauer sollten häufigere Tests durchgeführt werden. Zum Beispiel sollten bei biotechnologischen/biologischen und anderen Wirkstoffen mit einer Haltbarkeitsdauer von einem Jahr oder darunter in den ersten drei Monaten monatlich und danach alle drei Monate Stabilitätsproben gezogen und getestet werden. Existieren Daten, die bestätigen, dass die Stabilität des Wirkstoffs nicht gefährdet ist, kann ein Verzicht auf bestimmte Testintervalle (z.B. 9-Monats-Tests) in Betracht gezogen werden. 11.56 Erforderlichenfalls sollten die Stabilitätslagerungsbedingungen der ICH-Stabilitätsleitlinie entsprechen. 11.6 Festlegen von Verfalls- und Wiederholungstestdaten11.60 Wenn beabsichtigt ist, ein Zwischenprodukt aus dem Kontrollbereich des Materialmanagementsystems des Herstellers abzugeben und ihm ein Verfalls- oder Retestdatum zugeordnet ist, sollten unterstützende Stabilitätsinformationen vorhanden sein (z.B. veröffentlichte Daten, Testergebnisse). 11.61 Ein Wirkstoffverfalls- oder Retestdatum sollte auf einer Bewertung von aus Stabilitätsuntersuchungen erhaltenen Daten basieren. Es ist allgemeine Praxis, ein Wiederholungsprüfungsdatum zu verwenden und kein Verfallsdatum. 11.62 Vorläufige Verfalls- oder Retestdaten für Wirkstoffe können auf Pilotchargengrößen basieren, sofern
die Pilotchargen mit Hilfe einer Herstellungsmethode und einem Verfahren produziert werden, die den endgültigen, bei der Fertigung im Produktionsmaßstab verwendeten Prozess simulieren und
1.
die Qualität des Wirkstoffs vergleichbar ist mit dem im Produktionsmaßstab herzustellenden Material.2.
11.63 Zum Zweck einer Wiederholungsprüfung sollte eine repräsentative Probe gezogen werden. 11.7 Rückhalte-/Rückstellmuster11.70 Die Verpackung und Lagerung von Rückhalte-/Rückstellmustern dient einer potenziellen zukünftigen Bewertung der Qualität von Wirkstoffchargen und nichtzukünftigen Stabilitätstests. 11.71 Von jeder Wirkstoffcharge sollten ordnungsgemäß gekennzeichnete Rückhalte-/Rückstellmuster ein Jahr über das vom Hersteller zugewiesene Verfallsdatum der Charge oder drei Jahre über den Vertrieb der Charge hinaus aufbewahrt werden, je nachdem, welcher Zeitraum länger ist. Bei Wirkstoffen mit Wiederholungsprüfungsdaten sollten ähnliche Rückstellmuster bis drei Jahre nach dem vollständigen Vertrieb der Charge durch den Hersteller aufbewahrt werden. 11.72 Das Rückhalte-/Rückstellmuster sollte im gleichen Verpackungssystem, in dem der Wirkstoff gelagert wird, oder in einem äquivalenten oder besser schützenden als dem vermarkteten Verpackungssystem gelagert werden. Es sollten ausreichende Mengen zurückgestellt werden, um mindestens zwei Vollanalysen gemäß Arzneibuch oder, wenn keine Arzneibuchmonographie existiert, zwei Vollanalysen nach der Spezifikation durchführen zu können.
Abschnitt 12 Validierung
12.1 Validierungspolitik12.10 Die Politik, die Absichten und das Vorgehen einer Firma im Bereich Validierung, einschließlich der Validierung von Produktionsprozessen, Reinigungsverfahren, Analysenmethoden, Inprozesskontroll-Testverfahren, computergestützter Systeme und der für die Entwicklung, Überprüfung, Genehmigung und Dokumentation jeder Validierungsphase verantwortlichen Personen, sollten dokumentiert werden. 12.11 Kritische Parameter/Eigenschaften sollten im Regelfall während der Entwicklungsphase oder auf der Grundlage historischer Daten festgestellt und die für einen reproduzierbaren Betrieb benötigten Bereiche definiert werden. Dies sollte folgende Punkte einschließen:
eine Definition des Wirkstoffs hinsichtlich seiner kritischen Produkteigenschaften;•eine Identifikation der Prozessparameter, die die kritischen Qualitätseigenschaften des Wirkstoffs beeinflussen könnten;•eine Bestimmung der Bandbreite für jeden kritischen Prozessparameter, der erwartungsgemäß bei routinemäßiger Herstellung und Prozesskontrolle verwendet wird.
•
12.12 Die Validierung sollte auf die Vorgänge ausgedehnt werden, die als kritisch für die Qualität und die Reinheit des Wirkstoffs eingestuft wurden. 12.2 Validierungsdokumentation12.20 Es sollte ein schriftlicher Validierungsplan entwickelt werden, aus dem hervorgeht, wie die Validierung eines bestimmten Prozesses durchzuführen ist. Der Plan sollte von der (den) Qualitäts (sicherungs) einheit (en) sowie anderen beauftragten Einheiten überprüft und genehmigt werden. 12.21 Der Validierungsplan sollte sowohl kritische Prozessschritte und Akzeptanzkriterien als auch die Art der durchzuführenden Validierung (z.B. retrospektiv, prospektiv, begleitend) und die Zahl der Prozessläufe spezifizieren. 12.22 Es sollte ein Validierungsbericht mit Querverweisen auf den Validierungsplan erstellt werden, der die Ergebnisse der Validierung zusammenfasst, Abweichungen kommentiert und die erforderlichen Schlussfolgerungen zieht, einschließlich Änderungsvorschlägen zum Beheben von Mängeln. 12.23 Änderungen am Validierungsplan sollten mit entsprechender Begründung dokumentiert «erden. 12.3 Qualifizierung12.30 Bevor mit Prozessvalidierungsaktivitäten begonnen wird, sollte eine ordnungsgemäße Qualifizierung der kritischen Ausrüstung und der Hilfseinrichtungen abgeschlossen sein. Bei einer Qualifizierung werden im Regelfall die folgenden Schritte einzeln oder gemeinsam durchlaufen:
Designqualifizierung (DQ): dokumentierte Verifizierung, dass das für Anlagen. Ausrüstung oder Systeme vorgeschlagene Design für den vorgesehenen Zweck geeignet ist.
•
Installationsqualifizierung (IQ): dokumentierte Verifizierung, dass die Ausrüstung oder Systeme, so wie sie installiert sind oder modifiziert wurden, mit dem genehmigten Design, den Empfehlungen des Herstellers und, oder den Nutzeranforderungen übereinstimmen.
•
Funktionsqualifizierung (OQ): dokumentierte Verifizierung, dass die Ausrüstung oder Systeme, so wie sie installiert sind oder modifiziert wurden, im Rahmen der vorgesehenen Operationsbereiche nach Plan funktionieren.
•
Verfahrensqualifizierung (PQ): dokumentierte Verifizierung, dass die Ausrüstung und die Hilfseinrichtungen, so wie sie miteinander verbunden sind, effektiv und reproduzierbar auf der Grundlage von genehmigten Prozessmethoden und -spezifikationen funktionieren.
•
Seite 33 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

12.4 Vorgehensweisen bei der Prozessvalidierung12.40 Prozessvalidierung (PV) ist der dokumentierte Nachweis, dass der Prozess, wenn er innerhalb etablierter Parameter abläuft, effektiv und reproduzierbar ein Zwischenprodukt oder einen Wirkstoff hervorbringen kann, das/der die im voraus festgelegten Spezifikationen und Qualitätseigenschaften erfüllt. 12.41 Es gibt drei Ansätze für die Validierung. Die prospektive Validierung ist der bevorzugte Weg, aber es gibt Ausnahmefälle, in denen die anderen beiden Ansätze gewählt werden können. Die Ansätze und ihre Anwendbarkeit sind unten aufgeführt. 12.42 Eine prospektive Validierung sollte im Regelfall für alle Wirkstoffprozesse, wie in Nummer 12.12 bestimmt, durchgeführt werden. Die an einem Wirkstoffprozess durchgeführte Validierung sollte abgeschlossen sein, bevor das aus diesem Wirkstoff hergestellte Fertigarzneimittel in den Handel gelangt. 12.43 Eine begleitende Validierung kann vorgenommen werden, wenn keine Daten aus wiederholten Produktionsansätzen zur Verfügung stehen, weil nur eine begrenzte Zahl von Wirkstoffchargen produziert wurde, Wirkstoffchargen selten hergestellt werden oder Wirkstoffchargen mit Hilfe eines validierten Prozesses, der modifiziert wurde, hergestellt werden. Auf der Grundlage sorgfältiger Überwachung und Prüfung der Wirkstoffchargen können diese vor Abschluss einer begleitenden Validierung freigegeben und für Fertigarzneimittel für den Vertrieb verwendet werden. 12.44 Eine Ausnahme kann gemacht werden für die retrospektive Validierung bewährter Prozesse, die ohne signifikante Änderung der Wirkstoffqualität aufgrund von Änderungen bei Rohmaterialien, Ausrüstung, Systemen, Anlagen oder dem Produktionsprozess abliefen. Diese Vorgehensweise kann zum Zuge kommen, wenn:
die kritischen Qualitätseigenschaften und Prozessparameter identifiziert wurden;1.geeignete Inprozess-Akzeptanzkriterien und -kontrollen festgelegt wurden;2.kein signifikantes Prozess-/Produktversagen nachzuweisen ist, ausgenommen Bedienungsfehler oder Ausrüstungsversagen, das nicht mit der Eignung der Ausrüstung zusammenhängt;
3.
für die bekannten Wirkstoffe Verunreinigungsprofile erstellt wurden.4.
12.45 Die für die retrospektive Validierung ausgewählten Chargen sollten repräsentativ für alle im Überprüfungszeitraum hergestellten Chargen sein, einschließlich derer, die die Spezifikationen nicht erfüllten. Ihre Zahl sollte ausreichen, um die Beständigkeit des Prozesses nachzuweisen. Rückstellmuster können geprüft werden, um Daten für eine retrospektive Validierung des Prozesses zu erhalten. 12.5 Prozessvalidierungsprogramm12.50 Die Anzahl der Prozessläufe für eine Validierung sollte von der Komplexität des Prozesses oder dem Umfang der geplanten Prozessänderung abhängen. Für die prospektive und die begleitende Validierung sollten als Richtwert drei aufeinanderfolgende, erfolgreiche Produktionschargen verwendet werden. Es gibt jedoch Situationen, in denen zusätzliche Prozessläufe gerechtfertigt sind, um die Beständigkeit des Prozesses zu belegen (z.B. komplexe Wirkstoffprozesse oder solche mit verzögerter Fertigstellung). Für die retrospektive Validierung sollten im Regelfall Daten von zehn bis dreißig aufeinanderfolgenden Chargen untersucht werden, um die Beständigkeit eines Prozesses festzustellen, jedoch können bei entsprechender Begründung weniger Chargen überprüft werden. 12.51 Die kritischen Prozessparameter sollten während der Prozessvalidierungsstudien kontrolliert und überwacht werden. Prozessparameter ohne Beziehung zur Qualität, wie z.B. Variablen, die kontrolliert werden, um den Energieverbrauch und den Einsatz der Ausrüstung zu minimieren, brauchen bei der Prozessvalidierung nicht berücksichtigt zu werden. 12.52 Die Prozessvalidierung sollte bestätigen, dass das Verunreinigungsprofil jedes Wirkstoffs innerhalb der spezifizierten Grenzen liegt. Das Verunreinigungsprofil sollte vergleichbar oder besser sein als historische Daten und gegebenenfalls das während der Prozessentwicklung erstellte Profil oder das Profil von Chargen, die für zentrale klinische oder toxikologische Studien verwendet wurden. 12.6 Periodische Überprüfung validierter Systeme12.60 Systeme und Prozesse sollten regelmäßig bewertet werden, um zu verifizieren, dass sie weiterhin in validem Zustand operieren. Sind am System oder Prozess keine größeren Veränderungen vorgenommen worden und eine Qualitätsüberprüfung bestätigt, dass das System oder der Prozess beständig ein innerhalb seiner Spezifikationen liegendes Material hervorbringt, besteht normalerweise keine Notwendigkeit zur Revalidierung. 12.7 Reinigungsvalidierung12.70 Reinigungsverfahren sollten im Regelfall validiert werden. Allgemein sollte die Reinigungsvalidierung sich auf Situationen oder Prozessschritte richten, bei denen Kontamination oder Materialübertragung das größte Risiko für die Wirkstoffqualität darstellen. In den frühen Phasen der Produktion mag es zum Beispiel unnötig sein, Reinigungsverfahren für die Ausrüstung zu validieren, wenn Rückstände durch die nachfolgenden Reinigungsschritte beseitigt werden. 12.71 Die Validierung von Reinigungsverfahren sollte die Art, wie die Ausrüstung tatsächlich eingesetzt wird, widerspiegeln. Wenn verschiedene Wirkstoffe oder Zwischenprodukte in derselben Ausrüstung hergestellt werden und diese Ausrüstung mit demselben Verfahren gereinigt wird, kann ein repräsentatives Zwischenprodukt oder ein derartiger Wirkstoff für die Reinigungsvalidierung ausgewählt werden. Diese Auswahl sollte auf der Löslichkeit und der Schwierigkeit der Reinigung basieren sowie auf der Berechnung von Rückstandsgrenzen auf der Grundlage von Wirksamkeit, Toxizität und Stabilität. 12.72 Der Reinigungsvalidierungsplan sollte die zu reinigende Ausrüstung, Verfahren, Materialien, zulässige Reinigungsgrade. die zu überwachenden und zu kontrollierenden Parameter und die Analysenmethoden beschreiben. Der Plan sollte weiterhin angeben, welche Art von Proben auf welche Weise zu entnehmen und wie sie zu etikettieren sind. 12.73 Die Probenahme sollte je nach Gegebenheit Wischen, Spülen oder alternative Methoden (z.B. direkte Extraktion) umfassen um sowohl unlösliche als auch lösliche Rückstände nachweisen zu können. Die verwendeten Probenahmemethoden sollten eine quantitative Bestimmung der nach der Reinigung auf den Ausrüstungsoberflächen verbleibenden Rückstandsmengen zulassen. Wischtests sind mitunter unpraktikabel, wenn die mit dem Produkt in Kontakt kommenden Oberflächen aufgrund des Ausrüstungsdesigns und/oder prozessbedingter Einschränkungen schwer zugänglich sind (z.B. Innenflächen von Schläuchen, Transportleitungen, Reaktionsgefäße mit kleinen Öffnungen oder Behälter für toxisches Material sowie kleine, komplizierte Ausrüstungsgegenstände wie Mikronisatoren und Hochdruckhomogenisatoren). 12.74 Es sollten validierte Analysenmethoden, die empfindlich genug sind, Rückstände oder Kontaminanten nachzuweisen, verwendet werden. Die Nachweisgrenze für jede Analysenmethode sollte ausreichend empfindlich sein, um das festgelegte zulässige Rückstands- oder Kontaminationsniveau zu detektieren. Der mit der Methode erreichbare Wiederfindungsgrad sollte festgelegt werden. Rückstandsgrenzen sollten praktikabel, erreichbar und verifizierbar sein und den schädlichsten Rückstand als Grundlage haben. Grenzwerte können auf der Grundlage der geringsten bekannten pharmakologischen, toxikologischen oder physiologischen Aktivität des Wirkstoffs oder seines schädlichsten Bestandteils festgelegt werden. 12.75 Bei Prozessen, bei denen die Gesamtkeimzahl oder Endotoxine im Wirkstoff vermindert werden müssen, oder bei Prozessen, bei denen eine derartige Kontamination von Bedeutung sein könnte (z.B. nichtsterile Wirkstoffe, die für die Herstellung steriler Produkte verwendet werden), sollten Untersuchungen zur Ausrüstungsreinigung/ -desinfektion eine mögliche mikrobiologische oder Endotoxinkontamination ansprechen.
Seite 34 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

12.76 Reinigungsverfahren sollten nach der Validierung in geeigneten Zeitabständen überwacht werden, um sicherzustellen, dass sie auch während der Routineproduktion wirksam sind. Die Reinheit der Ausrüstung kann durch analytische Tests und, wenn möglich, visuelle Überprüfung überwacht werden. Mittels Sichtprüfung kann es möglich sein, eine grobe Kontamination in einem kleinen Bereich, die durch Probenziehen und/oder Analyse sonst unentdeckt bleiben würde, zu entdecken. 12.8 Validierung von Prüfverfahren12.80 Analysenmethoden sollten validiert sein, es sei denn die verwendete Methode ist Teil der relevanten Pharmakopöe oder eines anderen anerkannten Standard-Referenzwerks. Die Eignung aller verwendeten Testmethoden sollte dennoch unter den tatsächlichen Einsatzbedingungen verifiziert und dokumentiert werden. 12.81 Bei der Methodenvalidierung sollten Merkmale einbezogen werden, die in den ICH-Leitlinien analytischer Validierung von Prüfmethoden beschrieben sind. Das Ausmaß der analytischen Validierung sollte den Zweck der Analyse und das Stadium des Wirkstoffherstellungsprozesses widerspiegeln. 12.82 Bevor mit der Validierung von Analysenmethoden begonnen wird, sollte die ordnungsgemäße Qualifizierung der Prüfausrüstung betrachtet werden. 12.83 Über jede Änderung einer validierten Analysenmethode sollten vollständige Aufzeichnungen aufbewahrt werden. Diese Aufzeichnungen sollten den Grund für die Änderung und geeignete Daten beinhalten, die belegen, dass mit der geänderten Methode ebenso genaue und verlässliche Ergebnisse erhalten werden wie mit der herkömmlichen Methode.
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Teil II (3/3)
Abschnitt 13 Änderungskontrolle (Change Control)
13.10 Es sollte ein formales Änderungskontrollsystem zur Bewertung aller Änderungen, die Einfluss auf die Produktion und die Kontrolle eines Zwischenprodukts oder Wirkstoffs haben könnten, geschaffen werden. 13.11 In schriftlichen Arbeitsanweisungen sollten die Identifikation, Dokumentation, die ordnungsgemäße Überprüfung und die Genehmigung von Änderungen bei Rohmaterialien, Spezifikationen, Analysenmethoden, Anlagen, unterstützenden Systemen, Ausrüstung (einschließlich Computer-Hardware), Verarbeitungsschritten, Etikettier- und Verpackungsmaterialien und Computer-Software beschrieben sein. 13.12 Alle Vorschläge für GMP-relevante Änderungen sollten von den verantwortlichen organisatorischen Einheiten schriftlich formuliert, überprüft und genehmigt und von der/den Qualitätssicherungseinheit(en) überprüft und genehmigt werden. 13.13 Die potenziellen Auswirkungen der vorgeschlagenen Änderung auf die Qualität des Zwischenprodukts oder Wirkstoffs sollte bewertet werden. Ein Klassifizierungsverfahren kann dabei helfen, den Grad des Prüfens, der Validierung und der Dokumentation festzustellen, der notwendig ist, um Änderungen an einem validierten Prozess zu rechtfertigen. Änderungen können in Abhängigkeit von Art und Ausmaß der Änderungen sowie den Auswirkungen, die sie auf den Prozess haben können, klassifiziert werden (z.B. als geringfügig oder größer). Mit Hilfe einer wissenschaftlichen Beurteilung sollte bestimmt werden, welche zusätzlichen Prüf- und Validierungsmaßnahmen geeignet sind, um eine Änderung an einem validierten Prozess zu rechtfertigen. 13.14 Bei der Umsetzung genehmigter Änderungen sollten Maßnahmen getroffen werden, die sicherstellen, dass alle von den Änderungen betroffenen Dokumente überarbeitet werden. 13.15 Nachdem eine Änderung umgesetzt wurde, sollte eine Bewertung der ersten nach der Änderung produzierten oder getesteten Chargen vorgenommen werden. 13.16 Die potenziellen Auswirkungen kritischer Änderungen auf festgelegte Wiederholungsprüfungs- oder Verfallsdaten sollten bewertet werden. Wenn nötig, können Proben des mit dem modifizierten Prozess hergestellten Zwischenprodukts oder Wirkstoffs einem beschleunigten Stabilitätsprogramm unterzogen und/oder in das Stabilitätsmonitoringprogramm aufgenommen werden. 13.17 Die aktuellen Hersteller von Darreichungsformen sollten über Änderungen von festgelegten Produktions- und Prozesskontrollverfahren, die Auswirkungen auf die Wirkstoffqualität haben können, informiert werden.
Abschnitt 14 Zurückweisung und Wiederverwendung von Materialien
14.1 Zurückweisung14.10 Zwischenprodukte und Wirkstoffe, die festgelegte Spezifikationen nicht erfüllen, sollten als solche ausgewiesen und unter Quarantäne gestellt werden. Diese Zwischenprodukte oder Wirkstoffe können wie nachstehend beschrieben aufgearbeitet oder umgearbeitet werden. Die endgültige Verfügung über zurückgewiesene Materialien sollte dokumentiert werden. 14.2 Aufarbeitung (reprocessing)14.20 Ein Zwischenprodukt oder einen Wirkstoff, einschließlich eines solchen, das/der den Standards oder Spezifikationen nicht entspricht, wieder in den Prozess zurückzuführen und eine Aufarbeitung vorzunehmen durch Wiederholung eines Kristallisationsschrittes oder anderer geeigneter chemischer oder physikalischer Manipulationsschritte (z.B. Destillation, Filtration, Chromatographie, Mahlen), die Teil des festgelegten Herstellungsprozesses sind, wird generell als zulässig angesehen. Wird eine derartige Aufarbeitung jedoch bei einer Mehrzahl der Chargen angewandt, sollte sie zum Bestandteil des standardmäßigen Herstellungsprozesses gemacht werden. 14.21 Die Fortführung eines Prozessschrittes, nachdem ein Inprozesskontrolltest gezeigt hat, dass der Schritt unfertig ist, wird als Teil des normalen Prozesses betrachtet. Dies wird nicht als Aufarbeitung bezeichnet. 14.22 Das Wiedereinführen von nicht umgesetztem Material in einen Prozess und die Wiederholung einer chemischen Reaktion werden als Aufarbeitung bezeichnet, wenn sie nicht Teil eines festgelegten Prozesses sind. Dieser Aufarbeitung sollte eine sorgfältige Bewertung vorausgehen, um sicherzustellen, dass die Qualität des Zwischenprodukts oder Wirkstoffs durch die mögliche Bildung von Nebenprodukten und überreagierten Materialien nicht beeinträchtigt wird. 14.3 Umarbeitung (reworking)14.30 Bevor die Entscheidung getroffen wird, Chargen, die den festgelegten Standards oder Spezifikationen nicht entsprechen, umzuarbeiten, sollten die Gründe für eine solche Abweichung untersucht werden. 14.31 Umgearbeitete Chargen sollten einer angemessenen Bewertung, Prüfung, wenn nötig Stabilitätsprüfung und Dokumentation unterzogen werden, um aufzuzeigen, dass das umgearbeitete Produkt mit dem im Originalprozess hergestellten Produkt qualitativ gleichwertig ist. Eine begleitende Validierung ist oftmals die angemessene Vorgehensweise für Umarbeitungsverfahren. Dies erlaubt, dass ein Protokoll (Plan) das Umarbeitungsverfahren, die Art der Ausführung und die erwarteten Ergebnisse festlegt. Wenn nur eine Charge umgearbeitet werden soll, kann ein Bericht geschrieben und die Charge freigegeben werden, sobald sie als akzeptabel eingestuft wurde.
Seite 35 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

14.32 Die Verfahren sollten einen Vergleich der Verunreinigungsprofile der umgearbeiteten Chargen mit Chargen, die mit dem herkömmlichen Prozess hergestellt wurden, vorsehen. Sind routinemäßige Analysenmethoden zur Charakterisierung der umgearbeiteten Charge ungeeignet, sollten zusätzliche Methoden eingesetzt werden. 14.4 Rückgewinnung von Materialien und Lösungsmitteln14.40 Die Rückgewinnung von Reaktanden, Zwischenprodukten oder Wirkstoffen (z.B. aus Mutterlauge oder Filtraten) wird unter der Voraussetzung als zulässig betrachtet, dass es für die Rückgewinnung genehmigte Verfahren gibt und die rückgewonnenen Materialien Spezifikationen erfüllen, die für ihren beabsichtigten Einsatz geeignet sind. 14.41 Lösungsmittel können rückgewonnen und im gleichen oder in anderen Prozessen wiederverwendet werden, wenn die Rückgewinnungsmethode kontrolliert und überwacht wird, um sicherzustellen, dass die Lösungsmittel entsprechende Standards erfüllen, bevor sie wiederverwendet oder mit anderen genehmigten Materialien gemischt werden. 14.42 Frische und rückgewonnene Lösungsmittel und Reagenzien können vereinigt werden, wenn durch adäquate Prüfung ihre Eignung für alle Herstellungsprozesse, in denen sie evtl. verwendet werden, festgestellt worden ist. 14.43 Der Einsatz rückgewonnener Lösungsmittel, Mutterlaugen und anderer rückgewonnener Materialien sollte ordnungsgemäß dokumentiert werden. 14.5 Rückgaben14.50 Zurückgesandte Zwischenprodukte oder Wirkstoffe sollten als solche ausgewiesen und unter Quarantäne gestellt werden. 14.51 Wenn die Bedingungen, unter denen zurückgesandte Zwischenprodukte oder Wirkstoffe vor oder während der Rückkehr gelagert oder transportiert wurden oder wenn der Zustand ihrer Behälter Zweifel über ihre Qualität aufkommen lassen, sollten die zurückgegebenen Zwischenprodukte oder Wirkstoffe je nach Umstand aufgearbeitet, umgearbeitet oder vernichtet werden. 14.52 Es sollten Aufzeichnungen über zurückgesandte Zwischenprodukte oder Wirkstoffe geführt werden. Die Dokumentation sollte bei jeder Rückgabe Folgendes beinhalten:
Name und Adresse des Warenempfängers;•Zwischenprodukt oder Wirkstoff, Chargennummer und zurückgesandte Menge;•Grund der Rückgabe;•Verwendung oder Entsorgung des zurückgesandten Zwischenprodukts oder Wirkstoffs.•
Abschnitt 15 Beanstandungen und Rückrufe
15.10 Alle qualitätsbezogenen Beanstandungen, ganz gleich ob sie in mündlicher oder schriftlicher Form eingehen, sollten gemäß einem schriftlichen Verfahren dokumentiert und untersucht werden. 15.11 Beanstandungsprotokolle sollten Folgendes umfassen: - Name und Adresse des Beanstandenden;
Name (und falls zutreffend Titel) und Telefonnummer der Person, die die Beanstandung einreicht;•Art der Beanstandung (einschließlich Name und Chargennummer des Wirkstoffs);•Eingangsdatum der Beanstandung;•zunächst ergriffene Maßnahmen (einschließlich Datum und Nennung der Person, die die Maßnahmen ergreift);•ergriffene Folgemaßnahmen;•an den Beanstandenden übermittelte Antwort (einschließlich des Absendedatums der Antwort);•eine abschließende Entscheidung über die Zwischenprodukt- oder Wirkstoffcharge oder -partie.•
15.12 Die Aufzeichnungen zu Beanstandungen sollten aufbewahrt werden, um Trends, produktbezogene Häufigkeit und Schwere der Beanstandung mit dem Ziel bewerten zu können, zusätzliche und, falls nötig, sofortige Korrekturmaßnahmen zu ergreifen. 15.13 Es sollte schriftliche Anweisungen geben, die die Voraussetzungen beschreiben, unter denen ein Rückruf eines Zwischenprodukts oder Wirkstoffs in Betracht zu ziehen ist. 15.14 Das Rückrufverfahren sollte darlegen, wer an der Bewertung der Informationen beteiligt sein sollte, wie ein Rückruf in die Wege geleitet werden sollte, wer über einen Rückruf informiert werden sollte und wie das zurückgerufene Material behandelt werden sollte. 15.15 Im Falle einer schwerwiegenden oder möglicherweise lebensbedrohlichen Situation sollten die lokalen, nationalen und/ oder internationalen Behörden informiert und ihr Rat gesucht werden.
Abschnitt 16 Lohnhersteller (einschließlich Labors)
16.10 Alle Lohnhersteller (einschließlich Labors) sollten die im vorliegenden Leitfaden beschriebene Gute Herstellungspraxis einhalten. Es sollte besondere Aufmerksamkeit darauf verwandt werden, eine Kreuzkontamination zu verhindern und Rückverfolgbarkeit zu gewährleisten. 16.11 Lohnhersteller (einschließlich Labors) sollten vom Auftraggeber bewertet werden, um eine GMP-Compliance der spezifischen, am Vertragsort ausgeführten Aktivitäten zu gewährleisten. 16.12 Es sollte einen schriftlichen und genehmigten Vertrag oder ein formales Abkommen zwischen dem Auftraggeber und dem Auftragnehmer geben, der/das die GMP-Verantwortlichkeiten jeder Vertragspartei einschließlich der Qualitätsmaßnahmen im Detail festlegt. 16.13 Der Vertrag sollte dem Auftraggeber erlauben, die Anlagen des Auftragnehmers auf GMP-Entsprechung zu auditieren. 16.14 Ist eine Untervergabe von Aufträgen erlaubt, sollte der Auftragnehmer ohne die vorherige Bewertung und Genehmigung der Vereinbarung durch den Auftraggeber keine ihm im Rahmen des Vertrags anvertrauten Arbeiten an einen Dritten abgeben. 16.15 Herstellungs- und Laborprotokolle sollten dort aufbewahrt werden, wo die Tätigkeiten durchgeführt werden, und leicht verfügbar sein. 16.16 Änderungen an Prozess, Ausrüstung, Prüfmethoden, Spezifikationen oder anderen Vertragsvereinbarungen sollten nur vorgenommen werden, wenn der Auftraggeber hierüber informiert ist und derartige Änderungen genehmigt.
Abschnitt 17 Vertreter, Makler, Händler, Großhändler, Umverpacker und Umetikettierer
17.1 Anwendbarkeit
Seite 36 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

17.10 Dieser Abschnitt bezieht sich auf jede nicht mit dem Originalhersteller identische Partei, die mit Wirkstoffen oder Zwischenprodukten handelt und/oder diese in Besitz nimmt, umverpackt, umetikettiert, manipuliert, vertreibt oder lagert. 17.11 Alle Vertreter, Makler, Händler, Großhändler, umverpackenden und umetikettierenden Unternehmen sollten die in diesem Leitfaden beschriebene Gute Herstellungspraxis einhalten. 17.2 Rückverfolgbarkeit vertriebener Wirkstoffe und Zwi schenprodukte17.20 Vertreter, Makler, Händler, Großhändler, umverpackende oder umetikettierende Unternehmen sollten für eine vollständige Rückverfolgbarkeit der von ihnen vertriebenen Wirkstoffe und Zwischenprodukte sorgen. Die aufzubewahrenden und jederzeit verfügbaren Dokumente sollten folgende Punkte umfassen:
Identität des Originalherstellers;•Adresse des Originalherstellers;•Bestellungsaufträge;•Frachtbriefe (Transportdokumentation);•Empfangsbescheinigungen;•Name oder Bezeichnung des Wirkstoffs oder Zwischenprodukts;•Chargennummer des Herstellers;•Transport- und Vertriebsaufzeichnungen;•Sämtliche authentische Analysenzertifikate einschließlich derer des Originalherstellers;•Wiederholungsprüfungs- oder Verfallsdatum.•
17.3 Qualitätsmanagement17.30 Vertreter, Makler, Händler, Großhändler, umverpackende und umettikettierende Unternehmen sollten ein effektives Qualitätsmanagement, wie in Abschnitt 2 beschrieben, einrichten, dokumentieren und umsetzen. 17.4 Umverpackung, Umetikettierung und Lagerung von Wirkstoffen und Zwischenprodukten17.40 Die Umverpackung, Umetikettierung und Lagerung von Wirkstoffen und Zwischenprodukten sollte unter ordnungsgemäßen GMP-Kontrollen, so wie sie im vorliegenden Leitfaden dargelegt sind, erfolgen, um Verwechslungen und Beeinträchtigungen hinsichtlich Identität oder Reinheit der Wirkstoffe und Zwischenprodukte zu vermeiden. 17.41 Umverpackungsmaßnahmen sollten unter geeigneten Umgebungsbedingungen stattfinden, um Kontamination und Kreuzkontamination zu vermeiden. 17.5 Stabilität17.50 Wird ein Wirkstoff oder Zwischenprodukt in einen anderen Typ von Behälter als den vom Wirkstoff- oder Zwischenprodukthersteller verwendeten Behälter umgepackt, sollten Stabilitätsuntersuchungen zur Rechtfertigung der zugewiesenen Verfalls- oder Wiederholungsprüfungsdaten durchgeführt werden. 17.6 Weitergabe von Informationen17.60 Vertreter, Makler, Händler, Großhändler, umverpackende oder umetikettierende Unternehmen sollten alle vom Wirkstoff- oder Zwischenprodukthersteller erhaltenen qualitäts- oder zulassungsbezogenen Informationen an den Kunden sowie Informationen vom Kunden an den Wirkstoff- oder Zwischenprodukthersteller weiterleiten. 17.61 Der Vertreter, Makler, Händler, Großhändler, umverpackende oder umetikettierende Betrieb, der den Wirkstoff oder das Zwischenprodukt an den Kunden liefert, sollte den Namen des Originalwirkstoff- oder Zwischenproduktherstellers und die erhaltene(n) Chargennummer(n) an den Kunden weiterleiten. 17.62 Der Vertreter sollte außerdem den Zulassungsbehörden auf Anfrage die Identität des Originalwirkstoff- und Zwischenproduktherstellers nennen. Der Originalhersteller kann in Abhängigkeit von der Rechtsbeziehung zwischen dem Originalwirkstoff- und Zwischenprodukthersteller und seinen ermächtigten Vertretern die Zulassungsbehörden direkt oder über seine ermächtigten Vertreter ansprechen. ("Ermächtigt" bezieht sich in diesem Kontext auf eine Ermächtigung durch den Hersteller.) 17.63 Die in Nummer 11.4 darlegten speziellen Anleitungen für Analysenzertifikate sollten befolgt werden. 17.7 Umgang mit Beanstandungen und Rückrufen17.70 Vertreter, Makler, Händler, Großhändler, umverpackende oder umetikettierende Unternehmen sollten Aufzeichnungen über Beanstandungen und Rückrufe gemäß Abschnitt 15 für alle Beanstandungen und Rückrufe führen, von denen sie Kenntnis erhalten. 17.71 Falls die Situation dies erfordert, sollten die Vertreter, Makler, Händler, Großhändler, umverpackenden oder umetikettierenden Unternehmen die Beanstandungen zusammen mit dem Originalwirkstoff- oder Zwischenprodukthersteller überprüfen, um zu bestimmen, ob weitere Maßnahmen entweder bei anderen Kunden, die möglicherweise mit dem entsprechenden Wirkstoff oder Zwischenprodukt beliefert wurden, oder bei der Zulassungsbehörde oder bei beiden veranlasst werden sollten. Die Untersuchung der Gründe für die Beanstandung oder den Rückruf sollte von der zuständigen Partei durchgeführt und dokumentiert werden. 17.72 Wird eine Beanstandung an den Originalwirkstoff- oder Zwischenprodukthersteller weitergeleitet, sollten die von den Vertretern, Maklern, Händlern, Großhändlern, umverpackenden oder umetikettierenden Unternehmen geführten Aufzeichnungen alle Stellungnahmen des Originalwirkstoff- oder Zwischenproduktherstellers beinhalten (einschließlich ihres Datums und der übermittelten Information). 17.8 Umgang mit Rückgaben17.80 Rückgaben sollten gemäß Nummer 14.52 gehandhabt werden. Vertreter, Makler, Händler, Großhändler, umverpackende oder umetikettierende Unternehmen sollten eine Dokumentation über zurückgegebene Wirkstoffe und Zwischenprodukte führen.
Abschnitt 18 Spezifische Anleitung für Wirkstoffe, die mit Hilfe von Zellkulturen/Fermentation hergestellt werden
18.1 Allgemeine Anforderungen18.10 Abschnitt 18 soll auf spezifische Kontrollen für Wirkstoffe oder Zwischenprodukte, die mit Hilfe von Zellkulturen oder durch Fermentation unter Verwendung natürlicher oder rekombinanter Organismen hergestellt werden und die in den vorheriger Abschnitten nicht ausreichend abgedeckt wurden, eingehen. Es handelt sich hierbei nicht um ein eigenständiges Kapitel. Im Allgemeinen gelten die in den übrigen Abschnitten des vorliegender Dokuments beschriebenen GMP-Prinzipien. Es sei darauf hingewiesen, dass die Grundsätze der Fermentation für "klassische" Prozesse zur Produktion von kleinen Molekülen und für Prozesse, bei denen rekombinante und nicht rekombinante Organismen zur Produktion von Proteinen und/oder Polypeptiden verwendet werden die gleichen sind, obwohl sich das Ausmaß der Kontrolle unter scheiden wird. Wo es angezeigt ist, geht dieser Abschnitt auf derartige Unterschiede ein. Im Allgemeinen ist für biotechnologisch Prozesse, die für die Produktion von Proteinen und Polypeptiden verwendet werden, ein höheres Maß an Kontrolle erforderlich als für klassische Fermentationsprozesse.
Seite 37 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

18.11 Der Begriff "biotechnologischer Prozess" bezieht sich auf den Gebrauch von Zellen oder Organismen, die durch rekombinante DNA, Hybridome oder andere Technologien für die Produktion von Wirkstoffen generiert oder modifiziert wurden. Die durch biotechnologische Prozesse hergestellten Wirkstoffe bestehen normalerweise aus hochmolekularen Substanzen wie Proteinen und Polypeptiden, für die in diesem Abschnitt speziell Anleitung gegeben wird. Gewisse niedermolekulare Wirkstoffe wie Antibiotika, Aminosäuren, Vitamine und Kohlehydrate können ebenfalls durch rekombinante DNA-Technologie produziert werden. Das Ausmaß der Kontrolle für diese Art von Wirkstoffen in ähnlich dem üblichen Ausmaß bei der klassischen Fermentation. 18.12 Der Begriff "klassische Fermentation" bezieht sich auf Prozesse, die Mikroorganismen, die in der Natur vorkommen und/oder die durch konventionelle Methoden (z.B. Bestrahlung oder chemische Mutagenese) modifiziert wurden, zur Herstellung von Wirkstoffen einsetzen. Wirkstoffe, die durch "klassische Fermentation" hergestellt werden, sind normalerweise niedermolekulare Produkte wie Antibiotika, Aminosäuren, Vitamine und Kohlenhydrate. 18.13 Die Produktion von Wirkstoffen oder Zwischenprodukten aus Zellkulturen oder Fermentation beinhaltet biologische Prozesse wie die Kultivierung von Zellen oder die Extraktion und Aufreinigung von Material aus lebenden Organismen. Es sei darauf hingewiesen, dass es zusätzliche Prozessschritte geben mag, wie z.B. eine physikochemische Modifikation, die Teil des Herstellungsprozesses sind. Die verwendeten Rohmaterialien (Medien, Pufferbestandteile) können Wachstumspotenzial für mikrobiologische Kontaminanten bieten. In Abhängigkeit von der Quelle, der Herstellungsmethode und der beabsichtigten Verwendung eines Wirkstoffs oder Zwischenprodukts ist evtl. während bestimmter Stadien der Herstellung und der Überwachung des Prozesses eine Kontrolle der mikrobiellen Belastung, der Viruskontamination und/oder der Endotoxine erforderlich. 18.14 Für alle Herstellungsstufen sollten ordnungsgemäße Kontrollen eingerichtet werden, um die Qualität des Zwischenprodukts oder Wirkstoffs zu sichern. Zwar beginnt der vorliegende Leitfaden mit dem Zellkultur-/Fermentationsschritt, dennoch sollten frühere Schritte (z.B. die Pflege von Zellbanken) unter ausreichenden Prozesskontrollen stattfinden. Der vorliegende Leitfaden deckt Zellkulturen/Fermentation ab dem Punkt ab, an dem ein Vial (Fläschchen) der Zellbank zu Herstellungszwecken entnommen wird. 18.15 Es sollten geeignete Ausrüstungs- und Umgebungskontrollen vorgenommen werden, um das Risiko einer Kontamination zu minimieren. Die Akzeptanzkriterien für die Umgebungsqualität und die Häufigkeit einer Überwachung sollten vom jeweiligen Produktionsschritt und von den Produktionsbedingungen (offene, geschlossene oder abgegrenzte Systeme oder Anlagen) abhängen. 18.16 Allgemein sollten bei Prozesskontrollen folgende Punkte berücksichtigt werden:
Wartung der Arbeitszellbank (falls zutreffend);•ordnungsgemäße Beimpfung und Vermehrung der Kultur;•Kontrolle der kritischen Betriebsparameter während der Fermentation / Zellkultur;•Überwachung des Prozesses auf Zellwachstum, der Lebensfähigkeit (für die meisten Zellkulturprozesse) und Produktivität, wo erforderlich;
•
Ernte- und Aufreinigungsverfahren die Zellen, Zellreste und Medienbestandteile beseitigen, während sie das Zwischenprodukt oder den Wirkstoff gegen Kontamination, besonders mikrobiologischer Art, und Qualitätsverlust schützen;
•
Überwachung der mikrobiellen Belastung und, wo dies nötig ist, der Endotoxinmengen in geeigneten Produktionsstadien;•Fragen der Virussicherheit wie beschrieben im ICH-Leitfaden Q5A Qualität biotechnologischer Produkte: Bewertung der Virussicherheit biotechnologischer Produkte, die aus Zelllinien menschlichen oder tierischen Ursprungs stammen.
•
18.17 Wo erforderlich, sollte die Entfernung von Medienbestandteilen, Wirtszellenproteinen, anderen prozessbezogenen Verunreinigungen, produktbezogenen Verunreinigungen und Kontaminanten nachgewiesen werden. 18.2 Zellbankwartung und -protokollierung18.20 Der Zugriff auf Zellbanken sollte auf befugtes Personal begrenzt werden. 18.21 Zellbanken sollten unter Lagerbedingungen gehalten werden, die die Lebensfähigkeit der Zellen erhalten und eine Kontamination verhindern. 18.22 Es sollten Aufzeichnungen über die Verwendung der Vials (Fläschchen) aus Zellbanken und ihre Lagerbedingungen geführt werden. 18.23 Wo erforderlich, sollten Zellbanken periodisch überwacht werden, um ihre Anwendungseignung zu überprüfen. 18.24 Siehe ICH-Leitlinie Q5D Qualität biotechnologischer Produkte: Abstammung und Charakterisierung von Zellsubstraten, die in der Produktion von biotechnologischen/biologischen Produkten verwendet werden für eine ausführlichere Diskussion der Zellbankhaltung. 18.3 Zellkultur/Fermentation18.30 Wo eine aseptische Zugabe von Zellsubstraten, Medien, Puffer und Gasen notwendig ist, sollten wenn möglich geschlossene oder abgegrenzte Systeme verwendet werden. Werden die Beimpfung des anfänglichen Behälters oder anschließende Transfers oder Zugaben (Medien, Pufferlösungen) in offenen Behältern durchgeführt, sollten Kontrollen und Verfahren zur Minimierung eines Kontaminationsrisikos vorhanden sein. 18.31 Wo die Wirkstoffqualität durch mikrobielle Kontamination beeinträchtigt werden kann, sollten Manipulationen unter Einsatz offener Behälter in einer Biosicherheitswerkbank oder einer ähnlich kontrollierten Umgehung vorgenommen werden. 18.32 Das Personal sollte geeignete Schutzkleidung tragen und besondere Vorsichtsmaßnahmen hei der Handhabung der Kulturen befolgen. 18.33 Kritische Prozessparameter, zum Beispiel Temperatur, pH-Wert, Rührgeschwindigkeit, Zugabe von Gasen, Druck, sollten überwacht werden, um ihre Übereinstimmung mit dem festgelegten Prozess sicherzustellen. Zellwachstum, Lebensfähigkeit (für die meisten Zellkulturprozesse) und falls erforderlich Produktivität sollten ebenfalls überwacht werden. Kritische Parameter unterscheiden sich in der Regel von Prozess zu Prozess und bei der klassischen Fermentation brauchen bestimmte Parameter (z.B. Viabilität der Zellen) nicht überwacht zu werden. 18.34 Zellkulturausrüstung sollte nach ihrer Verwendung gereinigt und sterilisiert werden. Falls erforderlich, sollte Ausrüstung für "klassische Fermentationsprozesse" gereinigt und desinfiziert oder sterilisiert werden. 18.35 Nährmedien sollten wenn nötig vor ihrem Einsatz sterilisiert werden, um die Qualität des Wirkstoffs zu schützen. 18.36 Für den Nachweis von Kontamination und die Festlegung der zu ergreifenden Maßnahmen sollten geeignete Verfahren vorhanden sein. Dies sollte Verfahren einschließen, mit Hilfe derer die Auswirkungen der Kontamination auf das Produkt bestimmt sowie die Ausrüstung dekontaminiert und in einen Zustand rückversetzt werden kann, in dem sie für anschließende Chargen verwendbar ist. Fremdorganismen, die während des Fermentationsprozesses festgestellt werden, sollten gegebenenfalls identifiziert und ihre Wirkung auf die Produktqualität wenn nötig bewertet werden. Die Ergebnisse einer solchen Bewertung sollten bei der Verfügung über das hergestellte Material berücksichtigt werden. 18.37 Es sollten Aufzeichnungen über Kontaminationsvorfälle geführt werden. 18.38 Für mehrere Produkte eingesetzte Ausrüstungsgegenstände müssen mitunter zwischen verschiedenen Produktkampagnen zusätzlich gereinigt oder getestet werden, um das Risiko einer Kreuzkontamination zu minimieren. 18.4 Ernte, Isolierung und Reinigung
Seite 38 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

18.40 Ernteschritte, entweder zur Entfernung von Zellen oder Zellkomponenten oder zur Gewinnung von Zellkomponenten nach dem Abbruch (der Reaktion), sollten in Ausrüstung und Bereichen durchgeführt werden, die dazu ausgelegt sind, das Kontaminationsrisiko zu minimieren. 18.41 Ernte- und Reinigungsverfahren, die produzierende Organismen, Zellreste und Medienbestandteile entfernen oder inaktivieren und dabei Abbau, Kontamination und Qualitätsverlust minimieren, sollten geeignet sein sicherzustellen, dass das Zwischenprodukt oder der Wirkstoff in gleichbleibender Qualität gewonnen wird. 18.42 Alle Ausrüstungsgegenstände sollten nach Gebrauch ordnungsgemäß gereinigt und gegebenenfalls desinfiziert werden. Es können mehrere Chargen ohne Reinigung hintereinander gefahren werden, wenn die Qualität des Zwischenprodukts oder Wirkstoffs dadurch nicht beeinträchtigt wird. 18.43 Werden offene Systeme verwendet, sollte die Aufreinigung unter Umgebungsbedingungen stattfinden, die für den Erhalt der Produktqualität geeignet sind. 18.44 Zusätzliche Kontrollen. wie z.B. die Verwendung von Chromatographieharzen für nur ein Produkt, oder zusätzliche Prüfungen können erforderlich sein, wenn die Ausrüstung für verschiedene Produkte eingesetzt werden soll. 18.5 Schritte zur Virusentfernung und -inaktivierung18.50 Siehe ICH-Leitfaden Q5A Qualität biotechnologischer Produkte: Bewertung der Virussicherheit biotechnologischer Produkte, die aus Zeltlinien menschlichen oder tierischen Ursprungs stammen, für detailliertere Information. 18.51 Virusbeseitigungs- und -inaktivierungsschritte sind bei einigen Prozessen kritische Prozessschritte und sollten innerhalb validierter Parameter vollzogen werden. 18.52 Es sollten geeignete Vorsichtsmaßnahmen getroffen werden, um eine potenzielle Viruskontamination von Schritten vor der Virusbeseitigung/-inaktivierung auf solche danach zu verhindern. Deshalb sollte die offene Verarbeitung in Bereichen vorgenommen werden, die von anderen Verarbeitungsaktivitäten getrennt sind und eigene Belüftungseinheiten haben. 18.53 Für verschiedene Aufreinigungsschritte wird normalerweise nicht die gleiche Ausrüstung verwendet. Soll jedoch die gleiche Ausrüstung benutzt werden, sollte sie vor der Wiederverwendung ordnungsgemäß gereinigt und desinfiziert werden. Es sollten geeignete Vorsichtsmaßnahmen getroffen werden, um eine potenzielle Virusübertragung (z.B. durch die Ausrüstung oder die Umgebung) aus vorherigen Schritten zu verhindern.
Abschnitt 19 Wirkstoffe zur Verwendung in klinischen Prüfungen
19.1 Allgemeine Anforderungen19.10 Nicht alle in den voranstehenden Abschnitten dieses Leitfadens behandelten Kontrollen sind für die Herstellung eines neuen Wirkstoffs für Forschungszwecke während seiner Entwicklung erforderlich. Abschnitt 19 liefert eine spezielle, nur für diese Umstände geltende Anleitung. 19.11 Die Kontrollen während der Herstellung von Wirkstoffen für klinische Prüfungen sollte mit dem Entwicklungsstadium des Arzneimittels, dessen Bestandteil der Wirkstoff ist, kongruent sein. Die Herstellungs- und Prüfverfahren sollten flexibel sein, um mit zunehmendem Wissen über den Prozess und dem Voranschreiten der klinischen Prüfung eines Arzneimittels von vorklinischen Phasen zu klinischen Phasen Veränderungen zuzulassen. Erreicht die Arzneimittelentwicklung die Stufe, auf der der Wirkstoff für die Verwendung in Arzneimitteln für klinische Prüfungen produziert wird, sollten Hersteller gewährleisten, dass der Wirkstoff in geeigneten Anlagen mittels entsprechender Herstellungs- und Prüfverfahren hergestellt wird, um die Qualität des Wirkstoffs sicherzustellen. 19.2 Qualität19.20 Bei der Produktion von Wirkstoffen für klinische Prüfzwecke sollten angemessene GMP-Konzepte einschließlich eines geeigneten Genehmigungsmechanismus für jede Charge angewandt werden. 19.21 Eine (Mehrere) von der Produktion unabhängige Qualitäts(sicherungs)einheit(en) sollte(n) für die Genehmigung oder Zurückweisung jeder Wirkstoffcharge, die in klinischen Prüfungen verwendet werden soll, eingerichtet werden. 19.22 Einige der üblicherweise von der (den) Qualitäts (sicherungs)einheit(en) ausgeübten Testfunktionen können innerhalb anderer Organisationseinheiten ausgeübt werden. 19.23 Die Qualitätsmaßnahmen sollten ein System für die Prüfung von Rohmaterialien, Packmitteln, Zwischenprodukten und Wirkstoffen einschließen. 19.24 Prozess- und Qualitätsprobleme sollten bewertet werden. 19.25 Die Etikettierung von Wirkstoffen für klinische Prüfungen sollte ordnungsgemäß kontrolliert werden und sollte das Material als für Forschungszwecke bestimmt kennzeichnen. 19.3 Ausrüstung und Anlagen19.30 In allen Phasen der klinischen Entwicklung, einschließlich der Verwendung von Einrichtungen kleineren Maßstabs oder Labors zur Herstellung von Wirkstoffchargen für klinische Prüfungen, sollten Verfahren existieren, die sicherstellen, dass die Ausrüstung kalibriert, sauber und für den vorgesehenen Einsatz geeignet ist. 19.31 Die Anweisungen für die Verwendung von Anlagen sollten sicherstellen, dass Materialien in einer Art und Weise gehandhabt werden, die das Risiko einer Kontamination oder Kreuzkontamination minimiert. 19.4 Prüfung von Rohmaterialien19.40 Die für die Herstellung von Wirkstoffen für klinische Prüfungen verwendeten Rohmaterialien sollten durch Prüfung bewertet oder mit Lieferantenanalyse entgegengenommen und einem Identitätstest unterworfen werden. Wird ein Material als gefährlich eingestuft, sollte die Analyse des Lieferanten ausreichen. 19.41 In einigen Fällen kann die Eignung eines Rohmaterials vor seiner Verwendung auf Grundlage der Brauchbarkeit bei Reaktionen im kleinen Maßstab (d. h. Verwendungstests) statt lediglich auf der Grundlage analytischer Tests bestimmt werden. 19.5 Herstellung19.50 Die Produktion von Wirkstoffen zur Verwendung in klinischen Prüfungen sollte in Laborbüchern, Chargenprotokollen oder mit anderen geeigneten Mitteln dokumentiert werden. Diese Dokumente sollten Informationen zur Verwendung der Herstellungsmaterialien, der Ausrüstung, der Verarbeitung und wissenschaftliche Beobachtungen enthalten. 19.51 Die erwartete Ausbeute kann variabler und weniger genau bestimmt sein als die für kommerzielle Prozesse. Untersuchungen von Ausbeuteschwankungen werden nicht erwartet. 19.6 Validierung19.60 Eine Prozessvalidierung für die Produktion von Wirkstoffen zur Verwendung in klinischen Prüfungen ist normalerweise unangemessen, wenn eine einzige Charge hergestellt wird oder Prozessänderungen während der Wirkstoffentwicklung eine Chargenreplikation schwierig gestalten oder ungenaue Ergebnisse liefern. In diesem Entwicklungsstadium sichert eine Kombination aus Kontrollen, Kalibrierung und gegebenenfalls Ausrüstungsqualifizierung die Wirkstoffqualität.
Seite 39 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

19.61 Eine Prozessvalidierung gemäß Abschnitt 12 sollte vorgenommen werden, wenn Chargen für Handelszwecke hergestellt werden, selbst wenn derartige Chargen in Versuchs- oder kleinem Maßstab hergestellt werden. 19.7 Änderungen19.70 Änderungen während der Entwicklung werden erwartet, da Wissen hinzugewonnen und der Produktionsmaßstab erweitert wird. Jede Änderung der Produktion, Spezifikationen oder Prüfverfahren sollte ordnungsgemäß aufgezeichnet werden. 19.8 Prüfung19.80 Obwohl die zur Bewertung einer Wirkstoffcharge für klinische Prüfungen angewandten Analysenmethoden noch nicht validiert sein mögen, sollten sie wissenschaftlich fundiert sein. 19.81 Es sollte ein System zur Aufbewahrung von Rückstellmustern von jeder Charge geben. Dieses System sollte sicherstellen, dass eine ausreichende Menge jedes Rückstellmusters über einen angemessenen Zeitraum nach Genehmigung, Beendigung oder Abbruch einer Anwendung aufbewahrt wird. 19.82 Das Festlegen von Verfalls- und Wiederholungsprüfungsdaten gemäß Nummer 11.6 ist auf bekannte, für klinische Prüfungen vorgesehene Wirkstoffe anzuwenden. Bei neuen Wirkstoffen gilt Nummer 11.6 in den frühen Stadien klinischer Prüfungen im Regelfall nicht. 19.9 Dokumentation19.90 Es sollte ein System vorhanden sein, um sicherzustellen, dass die Informationen, die während der Entwicklung und Herstellung von für klinische Prüfungen vorgesehenen Wirkstoffen gesammelt wurden, dokumentiert und verfügbar sind. 19.91 Die Entwicklung und Anwendung von Analysenmethoden, auf die sich die Freigabe einer Wirkstoffcharge für klinische Prüfungen stützt, sollten angemessen dokumentiert werden. 19.92 Es sollte ein System für die Verwahrung von Herstellungs- und Prüfprotokollen sowie Dokumenten betrieben werden. Dieses System sollte sicherstellen, dass Aufzeichnungen und Dokumente über einen angemessenen Zeitraum nach Genehmigung, Beendigung oder Abbruch einer Anwendung aufbewahrt werden.
Abschnitt 20 Glossar
AkzeptanzkriterienNumerische Grenzen, Schwankungsbereiche oder andere geeignete Maßstäbe für die Akzeptanz von Prüfergebnissen. AbweichungAbgehen von einer genehmigten Instruktion oder einem festgelegten Standard. ArzneimittelDie Darreichungsform im endgültigen Primärbehältnis für die Vermarktung. (Verweis auf Q1A) Aufarbeitung (reprocessing)Das Wiedereinführen eines Zwischenprodukts oder Wirkstoffs, einschließlich eines nicht standard- oder spezifikationskonformen Produkts, in einen Prozess und die Wiederholung eines Kristallisierungsschritts oder einer anderen geeigneten chemischen oder physikalischen Manipulation (z.B. Destillation, Filtration, Chromatographie, Mahlen), die Teil des festgelegten Herstellungsprozesses sind. Die Fortsetzung eines Prozessschrittes, nachdem eine Inprozesskontrolle gezeigt hat, dass der Schritt unfertig ist, gilt als Teil des normalen Prozesses und nicht als Aufarbeitung. Ausbeute, erwarteteDie Materialmenge oder der Prozentsatz der theoretischen Ausbeute, die/der in einer dafür bestimmten Phase der Produktion auf der Grundlage vorheriger Labor-, Versuchsmaßstabs- oder Herstellungsdaten erwartet wird. Ausbeute, theoretischeDie Menge, die in einer bestimmten Phase der Produktion, berechnet auf der Grundlage der einzusetzenden Materialien und bei Abwesenheit irgendeines Verlusts oder Fehlers, in der tatsächlichen Produktion hergestellt würde. Ausgangsstoff für einen Wirkstoff (API starting material)Ein Rohmaterial, Zwischenprodukt oder ein Wirkstoff, der für die Produktion eines Wirkstoffs verwendet wird und der als bedeutsames Strukturelement in die Struktur des Wirkstoffs eingebaut wird. Ein Ausgangsstoff für einen Wirkstoff kann ein Handelsartikel, ein Material, das von einem oder mehreren Lieferanten im Rahmen eines Lohnauftrags oder Handelsabkommens erworben oder im eigenen Haus produziert wird, sein. Ausgangsstoffe für Wirkstoffe haben in der Regel definierte chemische Eigenschaften und eine definierte Struktur. Charge (oder Ansatz)Eine bestimmte Materialmenge, die so in einem Prozess oder einer Reihe von Prozessen hergestellt wird, dass man von ihrer Homogenität innerhalb spezifizierter Grenzen ausgehen kann. Bei kontinuierlicher Produktion kann eine Charge ein definierter Bruchteil der Produktion sein. Die Chargengröße kann entweder als feste Menge oder als die in einem festgelegten Zeitraum produzierte Menge definiert werden. Chargennummer (oder Ansatznummer)Eine eindeutige Kombination von Zahlen, Buchstaben und/oder Symbolen, die eine Charge (oder Ansatz) kennzeichnet und mit Hilfe derer die Produktions- und Vertriebshistorie ermittelt werden kann. Computergestütztes SystemEin Prozess oder ein Vorgang, in den ein Computersystem integriert ist. ComputersystemEine Zusammenstellung von Hardwarekomponenten und der assoziierten Software, die entworfen und zusammengefügt wurde, um eine spezifische Funktion oder eine Gruppe von Funktionen auszuführen. HerstellungAlle Vorgänge der Warenannahme, Produktion, Verpackung, Umverpackung, Etikettierung, Umetikettierung, Qualitätskontrolle, Freigabe, Lagerung und des Vertriebs von Wirkstoffen und die damit zusammenhängenden Kontrollen. Inprozesskontrolle (oder Prozesskontrolle)Während der Produktion vorgenommene Überprüfungen zur Überwachung und gegebenenfalls Anpassung des Prozesses und/oder zur Sicherstellung, dass das Zwischenprodukt oder der Wirkstoff seinen Spezifikationen entspricht. KalibrierungDer Nachweis, dass ein bestimmtes Instrument oder Gerät im Vergleich mit Ergebnissen, die mit einem Referenzstandard oder rückverfolgbaren Standard über eine angemessene Reihe von Messungen erhalten wurde, Ergebnisse innerhalb spezifizierter Grenzen hervorbringt. Kontamination
Seite 40 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Das unerwünschte Einbringen von Verunreinigungen chemischer oder mikrobiologischer Natur oder von Fremdstoffen in oder auf Rohmaterialien, Zwischenprodukten oder Wirkstoffen während der Produktion, Probenahme, Verpackung oder Umverpackung, Lagerung oder dem Transport. KreuzkontaminationKontamination eines Materials oder Produkts mit einem anderen Material oder Produkt. KritischBeschreibt einen Prozessschritt, eine Prozessbedingung, Testanforderung oder einen anderen relevanten Parameter oder Wert, der innerhalb vorbestimmter Kriterien kontrolliert werden muss, um sicherzustellen, dass ein Wirkstoff seine Spezifikationen erfüllt. LohnherstellerEin Hersteller, der im Auftrag des Originalherstellers einige Aspekte der Herstellung durchführt. LösungsmittelEine anorganische oder organische Flüssigkeit, die bei der Herstellung eines Zwischenprodukts oder Wirkstoffs als Mittel für die Zubereitung von Lösungen oder Suspensionen dient. MaterialEin allgemeiner Begriff zur Bezeichnung von Rohmaterialien (Ausgangstoffe, Reagenzien, Lösungsmittel), Prozesshilfen, Zwischenprodukten, Wirkstoffen sowie Verpackungs- und Etikettiermaterialien. Mikrobielle BelastungDie Menge und Art (z.B. unzulässig oder nicht) von Mikroorganismen, die in Rohmaterialien, Ausgangsstoffen für Wirkstoffe, Zwischenprodukten oder Wirkstoffen vorhanden sein können. Eine mikrobielle Belastung sollte nicht als Kontamination betrachtet werden, es sei denn, es sind bestimmte Mengen überschritten oder bestimmte unzulässige Organismen nachgewiesen worden. MutterlaugeDie Restflüssigkeit, die nach Kristallisations- oder Isolationsprozessen übrig bleibt. Eine Mutterlauge kann unumgesetzte Materialien, Zwischenprodukte, bestimmte Mengen an Wirkstoff und/ oder Verunreinigungen enthalten. Sie kann für die weitere Verarbeitung verwendet werden. ProduktionAlle an der Zubereitung eines Wirkstoffs beteiligten Vorgänge von der Warenannahme bis zur Verarbeitung und Verpackung des Wirkstoffs. ProzesshilfenMaterialien mit Ausnahme von Lösungsmitteln die als Hilfsmittel bei der Herstellung eines Zwischenprodukts oder Wirkstoffs verwendet werden und selbst nicht an einer chemischen oder biologischen Reaktion beteiligt sind (z.B. Filterhilfsmittel, Aktivkohle etc.). ProzesskontrolleSiehe Inprozesskontrolle. QualifizierungMaßnahmen, mit Hilfe derer nachgewiesen und dokumentiert wird, dass Ausrüstung oder Hilfssysteme sachgemäß installiert sind, ordnungsgemäß funktionieren und tatsächlich zu den erwarteten Ergebnissen führen. Die Qualifizierung ist Teil der Validierung, aber die einzelnen Qualifizierungsschritte allein stellen keine Prozessvalidierung dar. Qualitäts(sicherungs) einheit(en)Eine von der Produktion unabhängige Organisationseinheit, die sowohl Qualitätssicherungs- als auch Qualitätskontrollpflichten erfüllt. Dies kann abhängig von der Größe und Struktur eines Unternehmens in Form von separaten QS- und QK-Einheiten oder durch eine einzelne Person (oder eine Gruppe) geschehen. Qualitätskontrolle (QK)Überprüfen oder Testen, dass die Spezifikationen eingehalten werden. Qualitätssicherung (QS)Die Summe der vereinbarten Regelungen, die mit dem Ziel getroffen wurden, sicherzustellen, dass alle Wirkstoffe die für ihre Verwendung erforderliche Qualität haben und dass Qualitätssysteme unterhalten werden. QuarantäneDer Status von physisch oder durch andere effektive Mittel isolierten Materialien, bis eine Entscheidung über ihre spätere Genehmigung oder Zurückweisung getroffen worden ist. Referenzstandard, primärerEine Substanz, bei der durch eine umfangreiche Serie analytischer Prüfungen nachgewiesen wurde, dass es sich um authentisches Material handelt, das einen hohen Reinheitsgrad aufweisen sollte. Dieser Standard kann
von einer amtlich anerkannten Quelle bezogen werden oder durch1.unabhängige Synthese präpariert oder2.aus vorhandenen Produktionsmaterialien hoher Reinheit entnommen oder3.durch weitere Aufreinigung von vorhandenem Produktionsmaterial hergestellt werden.4.
Referenzstandard, sekundärerEine Substanz von feststehender, durch einen Vergleich mit einem primären Referenzstandard nachgewiesener Qualität und Reinheit, die als Referenzstandard für routinemäßige Laboranalysen verwendet wird. RohmaterialEin allgemeiner Begriff zur Bezeichnung von Ausgangsstoffen, Reagenzien, und Lösungsmitteln, die für die Produktion von Zwischenprodukten oder Wirkstoffen vorgesehen sind. SpezifikationEine Auflistung von Tests, Verweisen auf Analysenverfahren und geeigneter Akzeptanzkriterien, bei denen es sich um numerische Grenzwerte, Bereiche oder andere Kriterien für den beschriebenen Test handelt. Die Spezifikation legt die Kriterien fest, denen das Material entsprechen sollte, um als akzeptabel für den vorgesehenen Einsatz eingestuft zu werden. "Konformität mit der Spezifikation" bedeutet, dass das Material, wenn es gemäß den aufgelisteten Analysenverfahren getestet wird, die aufgelisteten Akzeptanzkriterien erfüllt. Umarbeitung (reworking)Vorgang, in dem ein nicht standard- oder spezifikationskonformes Zwischenprodukt oder ein solcher Wirkstoff einem oder mehreren Prozessschritten unterzogen wird, die sich vom festgelegten Herstellungsprozess unterscheiden, um ein Zwischenprodukt oder einen Wirkstoff von akzeptabler Qualität zu erhalten (z.B. Umkristallisieren mit einem anderen Lösungsmittel).
Seite 41 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Unterschrieben (Unterschrift)Die Abzeichnung der Person, die eine bestimmte Maßnahme oder Überprüfung vorgenommen hat. Dieses Sich-Ausweisen kann in Form von Initialen, der vollständigen, handschriftlichen Unterschrift, einem persönlichen Siegel oder einer authentifizierten und sicheren elektronischen Unterschrift vonstatten gehen. Unterschrift (unterschrieben)Siehe Definition des Begriffs "Unterschrieben". ValidierungEin dokumentiertes Programm, das ein hohes Maß an Gewissheit bietet, dass ein bestimmter Prozess, eine spezifische Methode oder ein System beständig Ergebnisse hervorbringt, die die im voraus festgelegten Akzeptanzkriterien erfüllen. ValidierungsplanEin schriftlicher Plan, der angibt, wie die Validierung durchzuführen ist und Akzeptanzkriterien definiert. Der Plan für einen Herstellungsprozess gibt zum Beispiel die Prozessausrüstung, kritische Prozessparameter/Betriebsbereiche, Produkteigenschaften, Probenahmen, zu erfassende Testdaten, die Zahl der Validierungsläufe und die akzeptablen Testergebnisse vor. VerfahrenEine dokumentierte Beschreibung der durchzuführenden Operationen, der zu ergreifenden Vorsichtsmaßnahmen und anderer anzuwendender Maßnahmen, die direkt oder indirekt mit der Herstellung eines Zwischenprodukts oder Wirkstoffs in Verbindung stehen. VerfallsdatumDas auf dem Behälter/den Etiketten eines Wirkstoffs aufgedruckte Datum, das die Zeit angibt, während der der Wirkstoff innerhalb der festgelegten Laufzeitspezifikation bleiben sollte, wenn er unter definierten Bedingungen gelagert wird, und nach dem er nicht wehr verwendet werden sollte. VerpackungsmaterialJedes für den Schutz eines Zwischenprodukts oder Wirkstoffs während Lagerung und Transport vorgesehene Material. VerunreinigungJeder in einem Zwischenprodukt oder Wirkstoff vorhandener Bestandteil, der nicht der gewünschte selbst ist. VerunreinigungsprofilEine Beschreibung der identifizierten und nicht identifizierten Verunreinigungen in einem Wirkstoff. Wiederholungsprüfungsdatum (Retestdatum)Das Datum, an dem ein Material erneut untersucht werden sollte, um sicherzustellen, dass es sich weiterhin für eine Verwendung eignet. WirkstoffJede Substanz oder Substanzmischung, die für die Herstellung eines Arzneimittels verwendet werden soll und die bei ihrer Verwendung in der Arzneimittelproduktion ein wirksamer Bestandteil des Arzneimittels wird. Derartige Substanzen haben den Zweck, die pharmakologische Wirksamkeit oder einen anderen direkten Effekt in der Diagnose, Heilung, Linderung, Behandlung oder Vorbeugung einer Krankheit beizubringen oder die Struktur und Funktion des Körpers zu beeinflussen. ZwischenproduktEin Material, das bei Bearbeitungsschritten eines Wirkstoffs produziert und das weiterer molekularer Veränderung oder Reinigungen unterzogen werden muss, bevor es zum Wirkstoff wird. Zwischenprodukte können isoliert oder nicht isoliert werden. (Anmerkung: Dieser Leitfaden befasst sich nur mit solchen Zwischenprodukten, die nach dem Punkt produziert werden, den die Firma als den Punkt definiert hat, an dem die Produktion des Wirkstoffs beginnt.) ________ 1) Folgender Hinweis (der auf die deutsche Fassung keine Auswirkung hat) befindet sich an dieser Stelle in der englischen Version: "Im Rest dieser Leitlinien wird wiederholt der Ausdruck "Active Pharmaceutical Ingredient" (API) verwendet. Er sollte als austauschbar mit dem Ausdruck "Active Substance" betrachtet werden
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 1
Herstellung steriler Arzneimittel Anhang 1
Stand: BAnz. Nr. 52 vom 04.04.2008 S. 1217Gemäß § 2 Nr. 3 der AMWHV macht das Bundesministerium für Gesundheit die jeweils aktuelle Fassung des EG-GMP Leitfadens in deutscher Sprache im Bundesanzeiger oder im elektronischen Bundesanzeiger bekannt. Der EG-GMP Leitfaden ist der Leitfaden für die Gute Herstellungspraxis für Arzneimittel und Prüfpräparate einschließlich seiner Anhänge, mit dem die Kommission der Europäischen Gemeinschaften die ausführlichen Leitlinien nach Artikel 47 der Richtlinie 2001/83/EG 2 und nach Artikel 51 der Richtlinie 2001/82/EG 3 veröffentlicht hat.Die Teile I und II des Leitfadens wurden mit der Bekanntmachung des Bundesministeriums für Gesundheit vom 27. Oktober 2006 (BAnz. S. 6887) veröffentlicht. Der Anhang 1 zu diesem Leitfaden ist am 14. Februar 2008 in einer überarbeiteten Fassung von der europäischen Kommission veröffentlicht worden. Dieser Anhang beinhaltet Leitlinien für die Herstellung steriler Arzneimittel und wird hiermit gemäß § 2 Nr. 3 AMWHV als Anlage 1 zu dieser Bekanntmachung veröffentlicht.Der Anhang 1 in seiner überarbeiteten Form findet für Gewebeeinrichtungen mit dem Datum des Inkrafttretens der Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung (BGBl. I S. 521) Anwendung. Für das Verbördeln von lyophilisierten Vials findet er ab dem 1. März 2010 Anwendung. Im Übrigen findet er ab dem 1. März 2009 Anwendung.
GrundsatzFür die Herstellung steriler Produkte gelten besondere Anforderungen, um das Risiko einer Kontamination mit Mikroorganismen, Partikeln und Pyrogenen möglichst gering zu halten. Vieles hängt von der Fertigkeit, Schulung und dem Verhalten des betreffenden Personals ab. Die Qualitätssicherung ist hier von besonderer Bedeutung und die Herstellung muss streng nach sorgfältig festgelegten und validierten Methoden und Verfahren erfolgen. Die Sterilität oder andere Qualitätsaspekte dürfen sich nicht alleine auf den letzten Herstellungsschritt oder die Prüfung des Endproduktes stützen.Anmerkung:Dieser Leitfaden enthält keine detaillierten Methoden zur Bestimmung der mikrobiologischen und Partikelreinheit der Luft, Oberflächen usw. Es wird auf andere Kompendien wie EN/ISO Standards verwiesen.Allgemeine Anforderungen
Seite 42 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi
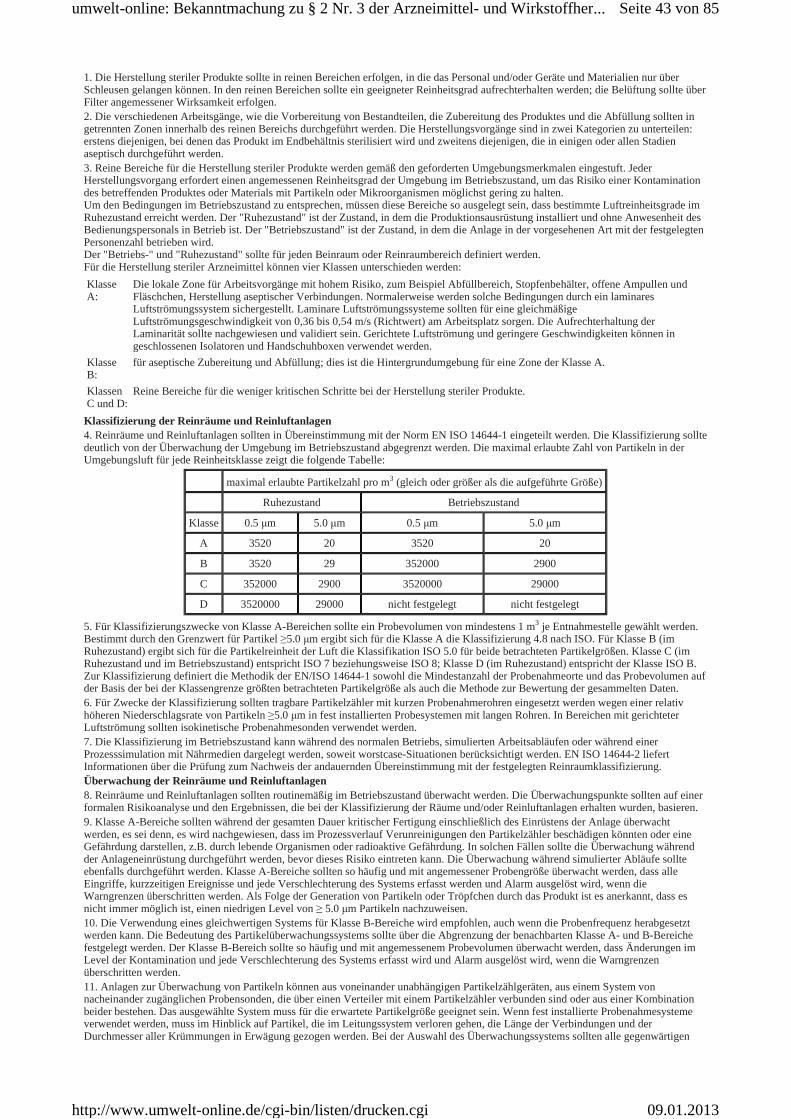
1. Die Herstellung steriler Produkte sollte in reinen Bereichen erfolgen, in die das Personal und/oder Geräte und Materialien nur über Schleusen gelangen können. In den reinen Bereichen sollte ein geeigneter Reinheitsgrad aufrechterhalten werden; die Belüftung sollte über Filter angemessener Wirksamkeit erfolgen.2. Die verschiedenen Arbeitsgänge, wie die Vorbereitung von Bestandteilen, die Zubereitung des Produktes und die Abfüllung sollten in getrennten Zonen innerhalb des reinen Bereichs durchgeführt werden. Die Herstellungsvorgänge sind in zwei Kategorien zu unterteilen: erstens diejenigen, bei denen das Produkt im Endbehältnis sterilisiert wird und zweitens diejenigen, die in einigen oder allen Stadien aseptisch durchgeführt werden.3. Reine Bereiche für die Herstellung steriler Produkte werden gemäß den geforderten Umgebungsmerkmalen eingestuft. Jeder Herstellungsvorgang erfordert einen angemessenen Reinheitsgrad der Umgebung im Betriebszustand, um das Risiko einer Kontamination des betreffenden Produktes oder Materials mit Partikeln oder Mikroorganismen möglichst gering zu halten. Um den Bedingungen im Betriebszustand zu entsprechen, müssen diese Bereiche so ausgelegt sein, dass bestimmte Luftreinheitsgrade im Ruhezustand erreicht werden. Der "Ruhezustand" ist der Zustand, in dem die Produktionsausrüstung installiert und ohne Anwesenheit des Bedienungspersonals in Betrieb ist. Der "Betriebszustand" ist der Zustand, in dem die Anlage in der vorgesehenen Art mit der festgelegten Personenzahl betrieben wird. Der "Betriebs-" und "Ruhezustand" sollte für jeden Beinraum oder Reinraumbereich definiert werden. Für die Herstellung steriler Arzneimittel können vier Klassen unterschieden werden:
Klasse A:
Die lokale Zone für Arbeitsvorgänge mit hohem Risiko, zum Beispiel Abfüllbereich, Stopfenbehälter, offene Ampullen und Fläschchen, Herstellung aseptischer Verbindungen. Normalerweise werden solche Bedingungen durch ein laminares Luftströmungssystem sichergestellt. Laminare Luftströmungssysteme sollten für eine gleichmäßige Luftströmungsgeschwindigkeit von 0,36 bis 0,54 m/s (Richtwert) am Arbeitsplatz sorgen. Die Aufrechterhaltung der Laminarität sollte nachgewiesen und validiert sein. Gerichtete Luftströmung und geringere Geschwindigkeiten können in geschlossenen Isolatoren und Handschuhboxen verwendet werden.
Klasse B:
für aseptische Zubereitung und Abfüllung; dies ist die Hintergrundumgebung für eine Zone der Klasse A.
Klassen C und D:
Reine Bereiche für die weniger kritischen Schritte bei der Herstellung steriler Produkte.
Klassifizierung der Reinräume und Reinluftanlagen4. Reinräume und Reinluftanlagen sollten in Übereinstimmung mit der Norm EN ISO 14644-1 eingeteilt werden. Die Klassifizierung sollte deutlich von der Überwachung der Umgebung im Betriebszustand abgegrenzt werden. Die maximal erlaubte Zahl von Partikeln in der Umgebungsluft für jede Reinheitsklasse zeigt die folgende Tabelle:
maximal erlaubte Partikelzahl pro m3 (gleich oder größer als die aufgeführte Größe)
Ruhezustand Betriebszustand
Klasse 0.5 µm 5.0 µm 0.5 µm 5.0 µm
A 3520 20 3520 20
B 3520 29 352000 2900
C 352000 2900 3520000 29000
D 3520000 29000 nicht festgelegt nicht festgelegt
5. Für Klassifizierungszwecke von Klasse A-Bereichen sollte ein Probevolumen von mindestens 1 m3 je Entnahmestelle gewählt werden. Bestimmt durch den Grenzwert für Partikel ≥5.0 µm ergibt sich für die Klasse A die Klassifizierung 4.8 nach ISO. Für Klasse B (im Ruhezustand) ergibt sich für die Partikelreinheit der Luft die Klassifikation ISO 5.0 für beide betrachteten Partikelgrößen. Klasse C (im Ruhezustand und im Betriebszustand) entspricht ISO 7 beziehungsweise ISO 8; Klasse D (im Ruhezustand) entspricht der Klasse ISO B. Zur Klassifizierung definiert die Methodik der EN/ISO 14644-1 sowohl die Mindestanzahl der Probenahmeorte und das Probevolumen auf der Basis der bei der Klassengrenze größten betrachteten Partikelgröße als auch die Methode zur Bewertung der gesammelten Daten.6. Für Zwecke der Klassifizierung sollten tragbare Partikelzähler mit kurzen Probenahmerohren eingesetzt werden wegen einer relativ höheren Niederschlagsrate von Partikeln ≥5.0 µm in fest installierten Probesystemen mit langen Rohren. In Bereichen mit gerichteter Luftströmung sollten isokinetische Probenahmesonden verwendet werden.7. Die Klassifizierung im Betriebszustand kann während des normalen Betriebs, simulierten Arbeitsabläufen oder während einer Prozesssimulation mit Nährmedien dargelegt werden, soweit worstcase-Situationen berücksichtigt werden. EN ISO 14644-2 liefert Informationen über die Prüfung zum Nachweis der andauernden Übereinstimmung mit der festgelegten Reinraumklassifizierung.Überwachung der Reinräume und Reinluftanlagen8. Reinräume und Reinluftanlagen sollten routinemäßig im Betriebszustand überwacht werden. Die Überwachungspunkte sollten auf einer formalen Risikoanalyse und den Ergebnissen, die bei der Klassifizierung der Räume und/oder Reinluftanlagen erhalten wurden, basieren.9. Klasse A-Bereiche sollten während der gesamten Dauer kritischer Fertigung einschließlich des Einrüstens der Anlage überwacht werden, es sei denn, es wird nachgewiesen, dass im Prozessverlauf Verunreinigungen den Partikelzähler beschädigen könnten oder eine Gefährdung darstellen, z.B. durch lebende Organismen oder radioaktive Gefährdung. In solchen Fällen sollte die Überwachung während der Anlageneinrüstung durchgeführt werden, bevor dieses Risiko eintreten kann. Die Überwachung während simulierter Abläufe sollte ebenfalls durchgeführt werden. Klasse A-Bereiche sollten so häufig und mit angemessener Probengröße überwacht werden, dass alle Eingriffe, kurzzeitigen Ereignisse und jede Verschlechterung des Systems erfasst werden und Alarm ausgelöst wird, wenn die Warngrenzen überschritten werden. Als Folge der Generation von Partikeln oder Tröpfchen durch das Produkt ist es anerkannt, dass es nicht immer möglich ist, einen niedrigen Level von ≥ 5.0 µm Partikeln nachzuweisen.10. Die Verwendung eines gleichwertigen Systems für Klasse B-Bereiche wird empfohlen, auch wenn die Probenfrequenz herabgesetzt werden kann. Die Bedeutung des Partikelüberwachungssystems sollte über die Abgrenzung der benachbarten Klasse A- und B-Bereiche festgelegt werden. Der Klasse B-Bereich sollte so häufig und mit angemessenem Probevolumen überwacht werden, dass Änderungen im Level der Kontamination und jede Verschlechterung des Systems erfasst wird und Alarm ausgelöst wird, wenn die Warngrenzen überschritten werden.11. Anlagen zur Überwachung von Partikeln können aus voneinander unabhängigen Partikelzählgeräten, aus einem System von nacheinander zugänglichen Probensonden, die über einen Verteiler mit einem Partikelzähler verbunden sind oder aus einer Kombination beider bestehen. Das ausgewählte System muss für die erwartete Partikelgröße geeignet sein. Wenn fest installierte Probenahmesysteme verwendet werden, muss im Hinblick auf Partikel, die im Leitungssystem verloren gehen, die Länge der Verbindungen und der Durchmesser aller Krümmungen in Erwägung gezogen werden. Bei der Auswahl des Überwachungssystems sollten alle gegenwärtigen
Seite 43 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Risiken der bei den Herstellungsschritten verwendeten Stoffe in Betracht gezogen werden, zum Beispiel solche mit lebenden Organismen oder Radiopharmaka.12. Das Probevolumen bei der Überwachung mittels vollautomatisierter Systeme hängt üblicherweise von der Probenahmerate des verwendeten Systems ab. Das Probevolumen bei der Überwachung muss nicht notwendigerweise das gleiche wie im Falle der formalen Klassifizierung der Reinräume und Reinluftanlagen betragen.13. In Bereichen der Klassen A und B hat die Überwachung der Zahl der Partikel ≥ 5.0 µm eine besondere Bedeutung, da diese ein wichtiges Nachweisinstrument für die frühzeitige Aufdeckung von Fehlern ist. Eine gelegentliche Anzeige von gezählten Partikeln ≥ 5.0 µm kann aufgrund elektronischer Einflüsse, Streulicht, Zufall, etc. fehlerhaft sein. In jedem Fall ist eine aufeinander folgende oder regelmäßige niedrige Zählrate ein Anzeichen einer möglichen Kontamination und sollte untersucht werden. Solche Ereignisse könnten frühzeitig Fehler im HVAC-System oder bei der Abfüllausrüstung anzeigen oder können Anzeichen eines mangelhaften Vorgehens bei der Maschineneinrüstung oder im Routinebetrieb darstellen.14. Die Partikelgrenzwerte der Tabelle für den Ruhezustand sollten nach einer kurzen "clean up"-Phase von 15 bis 20 Minuten (Richtwert) in einem unbemannten Zustand nach Beendigung der Arbeitsvorgänge erreicht werden.15. Die Überwachung der Bereiche der Klasse C und D im Betriebszustand sollte in Übereinstimmung mit den Grundsätzen des Qualitätsrisikomanagements durchgeführt werden. Die Anforderungen und die Warn-/Eingriffsgrenzen hängen von der Art der durchgeführten Tätigkeiten ab, aber die empfohlene "clean up"-Zeit sollte erreicht werden.16. Weitere Charakteristika wie Temperatur und relative Feuchte hängen vom Produkt und von der Art der durchgeführten Arbeitsvorgänge ab. Diese Parameter sollten die festgelegten Grenzwerte der Reinheitsklassen nicht störend beeinflussen.17. Die folgende Tabelle enthält Beispiele von in den verschiedenen Reinheitsklassen auszuführenden Arbeiten (siehe auch Absätze 28 bis 35):
Klasse Beispiele von Arbeitsschritten für im Endbehältnis sterilisierte Produkte (siehe Abschnitte 28 bis 30)
A Abfüllung von Produkten, wenn ein ungewöhnliches Risiko vorliegt
C Zubereitung von Lösungen, wenn ein ungewöhnliches Risiko vorliegt. Abfüllung von Produkten
D Zubereitung von Lösungen und Bestandteilen für anschließende Abfüllung
Klasse Beispiele von Arbeitsschritten für aseptische Zubereitungen (siehe Abschnitte 31 bis 35)
A Aseptische Zubereitung und Abfüllung
C Zubereitung von Lösungen, die filtriert werden
D Handhabung von Bestandteilen nach dem Waschen
18. Wo aseptische Herstellungsvorgänge durchgeführt werden, sollten häufige Kontrollen, beispielsweise unter Verwendung von Sedimentationsplatten, und Entnahmen von volumetrischen Luft- und Oberflächenproben durchgeführt werden (z.B. Tupfer und Kontaktplatten). Die Probenahmeverfahren während der Arbeit sollten den Schutz der Bereiche nicht beeinträchtigen. Die Überwachungsergebnisse sollten bei der Überprüfung der Chargenunterlagen für die Freigabe des Fertigproduktes berücksichtigt werden. Oberflächen und das Personal sollten nach kritischen Arbeitsgängen überwacht werden. Zusätzliche mikrobiologische Überwachung ist auch außerhalb der Produktionsvorgänge erforderlich, z.B. nach Validierung von Systemen, Reinigung und Desinfektion.19. Empfohlene Grenzwerte für die mikrobiologische Überwachung reiner Bereiche im Betriebszustand:
Empfohlene Grenzwerte für die mikrobiologische Kontamination (a)
Klasse Luftprobe KBE/m3
Sedimentationsplatten (Durchmesser 90 mm)
KBE/4 Stunden (b)
Kontaktplatten (Durchmesser 55 mm)
KBE/Platte
Handschuhabdruck 5 Finger KBE/Handschuh
A < 1 < 1 < 1 < 1
B 10 5 5 5
C 100 50 25 -
D 200 100 50 -
Anmerkungen:(a) Hierbei handelt es sich um Durchschnittswerte.(b) Einzelne Sedimentationsplatten können weniger als 4 Stunden exponiert werden.20. Für die Ergebnisse der Partikel- und mikrobiologischen Überwachung sind geeignete Warn- und Aktionslimits festzulegen. Für den Fall, dass diese Grenzen überschritten werden, sind Verfahren für Korrekturmaßnahmen anzugeben.Isolatortechnologie21. Die Anwendung der Isolatortechnologie, um menschliches Eingreifen in die Arbeitsbereiche möglichst gering zu halten, kann zu einer erheblichen Reduzierung des Risikos umgebungsbedingter mikrobiologischer Kontamination von aseptisch hergestellten Produkten führen. Es gibt zahlreiche Modelle von Isolatoren und Transfersystemen. Der Isolator und die Umgebung sollten so ausgelegt sein, dass die für die jeweiligen Zonen geforderte Luftqualität erreicht werden kann. Isolatoren werden aus unterschiedlichen mehr oder weniger stichfesten und leckdichten Materialien hergestellt. Transfersysteme gibt es als ein- oder doppeltürige Modelle bis hin zu völlig versiegelten Systemen mit Sterilisationsmechanismen.22. Der Transfer von Materialien in und aus dem Isolator stellt eine der größten potenziellen Kontaminierungsquellen dar. Das Innere des Isolators ist im Allgemeinen der Bereich für Arbeiten mit hohem Kontaminationsrisiko. Es wird zugestanden, dass im Arbeitsbereich innerhalb der Isolatoren nicht bei allen Modellen eine laminare Luftströmung erwartet werden kann.23. Die für die Umgebung geforderte Klassifizierung der Luft hängt von der Auslegung des Isolators und seiner Anwendung ab. Die Luftqualität sollte überwacht werden und für aseptische Verfahren zumindest der Klasse D entsprechen.24. Die Isolatoren sollten erst nach geeigneter Validierung eingeführt werden. Bei der Validierung sind alle kritischen Faktoren der Isolatortechnologie zu berücksichtigen, z.B. Luftqualität innerhalb und außerhalb (Umgebung) des Isolators, Desinfizierung, Transfervorgang und Unversehrtheit des Isolators.25. Die Überwachung sollte routinemäßig durchgeführt werden und häufige Tests auf Leckdichte des Isolators und des Handschuh/Armel-Systems einschließen.Blas-/Füll-/Verschlusstechnologie (blow/fill/seal technology)26. Blas-/Füll-/Verschlussanlagen sind Maschinen, in denen in einem kontinuierlichen Arbeitsgang Behältnisse aus einem thermoplastischen Granulat geformt, gefüllt und versiegelt werden. Blas-/Füll-/Verschluss-Ausrüstung, das zur aseptischen Herstellung
Seite 44 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

verwendet wird und mit einer effizienten Luftdusche der Luftqualität Klasse A ausgestattet ist, kann in einer Umgebung von mindestens Klasse C installiert werden, sofern Schutzkleidung der Klasse A/B getragen wird. Die Umgebung sollte im Ruhezustand den Grenzwerten für Mikroorganismen und Partikel und während der Arbeit lediglich den Grenzwerten für Mikroorganismen entsprechen. Blas-/Füll-/Verschlussanlagen, die zur Herstellung von im verschlossenen Endbehältnis sterilisierten Produkten verwendet werden, sollten in einer Umgebung von mindestens Klasse D installiert werden.27. Wegen dieser speziellen Technologie ist zumindest besonders auf folgende Einzelheiten zu achten:
Auslegung und Qualifikation der Anlagen•Validierung und Reproduzierbarkeit der Reinigung und Sterilisierung vor Ort•Umgebung des reinen Bereiches, in dem sich die Anlage befindet•Schulung und Bekleidung der Bediener•Eingreifen in die kritische Zone der Anlage, einschließlich jeder aseptischen Montage vor Beginn der Abfüllung.•
Im Endbehältnis sterilisierte Produkte28. Die Zubereitung der Bestandteile und der meisten Produkte sollte zumindest in einer Umgebung der Reinheitsklasse D erfolgen, um das Risiko der Kontamination mit Mikroorganismen und Partikeln gering zu halten und eine geeignete Umgebung für die Filtration und Sterilisation zu schaffen. Bei Produkten, die ein ungewöhnliches Risiko einer Kontamination durch Mikroorganismen aufweisen, weil sie beispielsweise das Wachstum der Mikroorganismen aktiv fördern, oder vor der Sterilisation lange Zeit aufbewahrt werden müssen oder notwendigerweise nicht vorwiegend in geschlossenen Gefäßen hergestellt werden, sollte die Zubereitung in einer Umgebung der Reinheitsklasse C erfolgen.29. Das Abfüllen von im Endbehältnis sterilisierten Produkten sollte zumindest in einer Umgebung der Reinheitsklasse C erfolgen.30. Wenn das Produkt ein ungewöhnliches Risiko für eine umgebungsbedingte Kontamination aufweist, weil beispielsweise der Füllvorgang langsam ist oder weithalsige Behältnisse verwendet werden bzw. das Produkt notwendigerweise für mehr als einige Sekunden vor dem Verschließen exponiert ist, sollte der Füllvorgang in einer Zone der Reinheitsklasse A mit einer Umgebung von mindestens Klasse C erfolgen. Die Zubereitung und Abfüllung von Salben, Cremes, Suspensionen und Emulsionen sollte vor der Sterilisation im verschlossenen Endbehältnis in der Regel unter Bedingungen der Reinheitsklasse C erfolgen.Aseptische Zubereitung31. Die Bestandteile sollten nach dem Waschen in einer Umgebung von mindestens Klasse D gehandhabt werden. Die Handhabung von sterilen Ausgangsmaterialien und Bestandteilen sollte, außer wenn diese später einer Sterilisierung oder Filtration mit einem Mikroorganismen zurückhaltenden Filter unterzogen werden, in einem Bereich der Reinheitsklasse A mit einer Umgebung der Reinheitsklasse B erfolgen.32. Die Zubereitung von Lösungen, die während des Prozesses steril filtriert werden müssen, sollte in einem Bereich der Reinheitsklasse C erfolgen. Wenn keine Filtration erfolgt, sollte die Zubereitung von Materialien und Produkten in einem Bereich der Reinheitsklasse A mit einer Umgebung der Klasse B erfolgen.33. Handhabung und Abfüllung aseptisch zubereiteter Produkte sollten in einem Bereich der Reinheitsklasse A mit einer Umgebung der Reinheitsklasse B erfolgen.34. Vor dem endgültigen Verschließen sollte der Transfer von partiell verschlossenen Behältnissen, wie sie beim Gefriertrocknen verwendet werden, entweder in einem Bereich der Reinheitsklasse A mit einem Umgebungsbereich der Klasse B oder in geschlossenen Transfersystemen in einem Bereich der Reinheitsklasse B erfolgen.35. Die Zubereitung und Abfüllung von Salben, Cremes, Suspensionen und Emulsionen sollte in einem Bereich der Reinheitsklasse A mit einer Umgebung der Reinheitsklasse B erfolgen, wenn das Produkt exponiert ist und anschließend nicht filtriert wird.Personal36. In reinen Bereichen sollte nur die unbedingt nötige Zahl von Personen anwesend sein; dies gilt besonders für aseptische Arbeitsvorgänge. Inspektionen und Kontrollen sollten so weit wie möglich von außen erfolgen.37. Das gesamte in reinen Bereichen tätige Personal (einschließlich des Reinigungs- und Wartungspersonals) sollte regelmäßig in den für die sachgemäße Herstellung steriler Produkte wichtigen Disziplinen geschult werden. Die Schulung sollte auch Hygiene und Grundlagen der Mikrobiologie umfassen. Wenn nicht entsprechend geschulte betriebsfremde Personen (z.B. solche, die mit Bau- oder Wartungsarbeiten beauftragt sind) reine Bereiche betreten müssen, sollten diese sehr sorgfältig angewiesen und beaufsichtigt werden.38. Personal, das mit anderen als den im laufenden Herstellungsprozess eingesetzten tierischen Geweben oder Kulturen von Mikroorganismen arbeitet, sollte Herstellungsbereiche für sterile Produkte nicht betreten, es sei denn, dass strenge und klar definierte Zugangsanweisungen eingehalten werden.39. Ein hoher Standard an persönlicher Hygiene und Sauberkeit ist unerlässlich. Das mit der Herstellung von sterilen Zubereitungen befasste Personal sollte angewiesen werden, alle Umstände zu melden, die zu einer Freisetzung von nach Zahl oder Art ungewöhnlichen Verunreinigungen führen können. Regelmäßige Gesundheitskontrollen in dieser Hinsicht sind wünschenswert. Eine hierfür benannte zuständige Person sollte entscheiden, welche Maßnahmen in Bezug auf Mitarbeiter ergriffen werden müssen, von denen ein nicht vertretbares mikrobiologisches Risiko ausgehen könnte.40. Armbanduhren, Makeup und Schmuck sollten in reinen Bereichen nicht getragen werden.41. Umkleiden und Waschen sollten nach schriftlich festgelegten Verfahren erfolgen, um die Kontamination der für reine Bereiche bestimmten Kleidung oder das Einschleusen von Verunreinigungen in die reinen Bereiche möglichst gering zu halten.42. Die Kleidung und deren Qualität müssen dem Arbeitsvorgang und der Reinheitsklasse des Arbeitsbereichs angepasst sein. Sie ist so zu tragen, dass das Produkt vor Kontamination geschützt ist.43. Nachfolgend wird die in den einzelnen Reinheitsklassen erforderliche Kleidung beschrieben:44. Straßenkleidung sollte nicht in Umkleideräume gebracht werden, die zu Räumen der Reinheitsklassen B und C führen. Jedem Mitarbeiter in einem Raum der Reinheitsklasse A/B sollte für jede Arbeitsperiode saubere sterile (sterilisierte oder angemessen desinfizierte) Schutzkleidung zur Verfügung gestellt werden. Handschuhe sollten während der Arbeit regelmäßig desinfiziert werden. Gesichtsmasken und Handschuhe sollten zumindest für jede Arbeitsperiode gewechselt werden.45. Reinraumkleidung sollte so gewaschen oder gereinigt und gehandhabt werden, dass sie keine zusätzlichen Verunreinigungen aufnimmt, die später wieder abgegeben werden können. Diese Verfahren sollten nach schriftlich festgelegten Anweisungen durchgeführt werden. Für diese Kleidung sind separate Wasch- und Reinigungsmöglichkeiten wünschenswert. Ungeeignete Behandlung der Kleidung kann die Fasern angreifen und die Gefahr erhöhen, dass Partikel abgegeben werden.Räumlichkeiten46. In reinen Bereichen sollten alle exponierten Oberflächen glatt, undurchlässig und ohne Risse sein, um eine Abgabe oder Ansammlung von Partikeln oder Mikroorganismen möglichst gering zu halten und die wiederholte Anwendung von Reinigungs- und gegebenenfalls Desinfektionsmitteln zu ermöglichen.
Seite 45 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

47. Um die Ansammlung von Staub zu vermindern und das Reinigen zu erleichtern, sollten keine unzugänglichen Nischen und möglichst wenig vorstehende Leisten, Regale, Schränke und Ausrüstungsgegenstände vorhanden sein. Türen sollten so konstruiert sein, dass für die Reinigung unzugängliche Stellen vermieden werden. Schiebetüren sind aus diesem Grund unerwünscht.
- Reinheitsklasse D:
Haar und gegebenenfalls Bart sollten bedeckt sein. Es sollten allgemein übliche Schutzkleidung und geeignete Schuhe oder Überschuhe getragen werden. Geeignete Maßnahmen sollten ergriffen werden, um jegliche Kontamination von außerhalb des reinen Bereichs zu vermeiden.
- Reinheitsklasse C:
Haar und gegebenenfalls Bart bzw. Schnurrbart sollten bedeckt sein. Es sollten ein ein- oder zweiteiliger Anzug mit geschlossenem Bund an den Handgelenken und hohem Kragen sowie geeignete Schuhe oder Überschuhe getragen werden. Die Kleidungsstücke sollten nahezu keine Fasern oder Partikel abgeben.
- Reinheitsklasse A/B:
Eine Kopfbedeckung sollte Haar und gegebenenfalls Bart bzw. Schnurrbart vollständig abdecken. Sie sollte in den Kragen des Anzugs gesteckt werden. Eine Gesichtsmaske sollte getragen werden, um eine Abgabe von Tröpfchen zu verhindern. Es sollten geeignete sterilisierte, nicht gepuderte Gummi- oder Plastikhandschuhe und sterilisiertes oder des- infiziertes Schuhwerk getragen werden. Die Hosenbeine sollten in das Schuhwerk und die Ärmel in die Handschuhe gesteckt werden. Die Schutzkleidung sollte nahezu keine Fasern oder Partikel abgeben und vom Körper abgegebene Partikel zurückhalten.
48. Eingezogene Decken sollten versiegelt sein, um Verunreinigungen aus dem darüberliegenden Raum zu verhindern.49. Rohre und Leitungen sollten so verlegt sein, dass keine schwer zu reinigenden Nischen, unversiegelte Öffnungen und Oberflächen entstehen.50. Ausgüsse und Abflüsse sollten in für die aseptische Herstellung benutzten Bereichen der Reinheitsklassen A/B verboten sein. In anderen Bereichen sollten Geruchsverschlüsse zwischen der Maschine oder dem Ausguss und den Abflüssen eingebaut sein. Im Fußboden befindliche Abflüsse in Räumen einer niedrigeren Reinheitsklasse sollten mit Rückstauklappen oder Verschlüssen ausgestattet sein, um eine Rückströmung zu verhindern.51. Umkleideräume sollten als Schleusen ausgelegt sein und so genutzt werden, dass die einzelnen Umkleidevorgänge voneinander getrennt erfolgen und auf diese Weise die Kontamination der Schutzkleidung mit Mikroorganismen und Partikeln möglichst gering ist. Sie sollten von gefilterter Luft wirksam durchströmt werden. Die letzte Zone des Umkleideraums sollte im Ruhezustand dieselbe Reinheitsklasse aufweisen wie der anschließende Bereich. Zuweilen sind separate Umkleideräume zum Betreten und Verlassen der reinen Bereiche wünschenswert. Handwaschbecken sollten im Allgemeinen nur im ersten Teil der Umkleideräume vorhanden sein.52. Schleusentüren sollten nicht gleichzeitig geöffnet werden. Das gleichzeitige Offnen von mehr als einer Tür sollte durch eine Sperre oder ein visuelles und/oder akustisches Warnsystem verhindert werden.53. Die Versorgung mit gefilterter Luft sollte so sein, dass unter allen Betriebsbedingungen gegenüber angrenzenden Bereichen mit niedrigerem Reinheitsgrad ein Überdruck aufrechterhalten und der Bereich wirksam durchströmt wird. Angrenzende Räume unterschiedlicher Reinheitsgrade sollten einen Druckunterschied von 10 bis 15 Pascal (Richtwert) aufweisen. Besonders zu achten ist auf den Schutz der Zone des höchsten Risikos, das heißt die unmittelbare Umgebung, der ein Produkt und gereinigte Bestandteile, die mit dem Produkt in Berührung kommen, ausgesetzt sind. Die verschiedenen Empfehlungen zu Luftzufuhr und Druckunterschieden müssen gegebenenfalls angepasst werden, wenn die Verbreitung beispielsweise pathogener, hochgiftiger, radioaktiver oder lebender viraler bzw. bakterieller Materialien oder Produkte verhindert werden muss. Bei bestimmten Arbeitsvorgängen kann eine Dekontamination der Anlagen und eine Aufbereitung der Abluft aus einem reinen Bereich erforderlich sein.54. Es sollte nachgewiesen werden, dass die Luftführung kein Kontaminationsrisiko darstellt; z.B. sollte sichergestellt sein, dass die Luftströmung Partikel, die von einer Person abgegeben werden oder bei einer Tätigkeit bzw. an einer Maschine anfallen, nicht in eine Zone mit höherem Risiko hineinträgt.55. Es sollte ein Warnsystem vorhanden sein, das Störungen in der Luftzufuhr meldet. Soweit ein Druckunterschied zwischen verschiedenen Bereichen wichtig ist, sollte zwischen diesen Bereichen ein Druckmessgerät angebracht sein. Diese Druckunterschiede sollten regelmäßig aufgezeichnet oder dokumentiert werden.Ausrüstung56. Durch die Trennwand zwischen einem Bereich der Reinheitsklasse A oder B und einem Arbeitsbereich mit niedrigerem Luftreinheitsgrad sollte kein Förderband laufen, es sei denn, das Band selbst wird kontinuierlich sterilisiert (z.B. in einem Sterilisiertunnel).57. Soweit möglich sollten Ausrüstung, Armaturen und Bedienungselemente so ausgelegt und installiert sein, dass Bedienungsvorgänge, Wartungs- und Reparaturarbeiten außerhalb des reinen Bereichs vorgenommen werden können. Wenn eine Sterilisation erforderlich ist, sollte dies möglichst erst nach vollständiger Installation erfolgen.58. Wenn eine Wartung der Ausrüstung innerhalb des reinen Bereichs durchgeführt wurde, sollte der Raum gereinigt, desinfiziert und/oder gegebenenfalls sterilisiert werden, bevor der Betrieb wieder aufgenommen wird, wenn die geforderten Standards hinsichtlich Sauberkeit und/oder Asepsis während der Wartungsarbeiten nicht aufrechterhalten wurden.59. Wasseraufbereitungs- und Verteilungsanlagen sollten so ausgelegt, konstruiert und gewartet werden, dass Wasser von geeigneter Qualität zuverlässig erzeugt wird. Sie sollten nicht über die vorgesehene Kapazität hinaus betrieben werden. Wasser für Injektionszwecke sollte so aufbereitet, gelagert und verteilt werden, dass mikrobielles Wachstum verhindert wird. Dies wird z.B. durch konstante Zirkulation bei Temperaturen über 70 °C erreicht.60. Die gesamte Ausrüstung, einschließlich der Sterilisatoren, der Systeme zur Luftaufbereitung und Filtration der Abluft und der Gasfilter sowie Systeme zur Wasserbehandlung, Wasseraufbereitung, Wasserlagerung und Verteilung sollten nach Plan gewartet und validiert werden. Ihre Wiederinbetriebnahme sollte genehmigt werden.Betriebshygiene61. Die Betriebshygiene in reinen Bereichen ist besonders wichtig. Diese sollten gründlich nach einem schriftlich festgelegten Programm gereinigt werden. Wenn Desinfektionsmittel verwendet werden, sollten mehrere typen eingesetzt werden. Es sollten regelmäßige mikrobiologische Kontrollen erfolgen, um die Entwicklung resistenter Stämme aufzudecken.62. Desinfektionsmittel und Detergenzien sollten auf mikrobiologische Verunreinigungen überprüft werden. Verdünnungen sollten in vorher gereinigten Behältnissen aufbewahrt und nur für eine festgelegte Zeit gelagert werden, wenn sie nicht sterilisiert sind. In Bereichen der Reinheitsklassen A und B verwendete Desinfektionsmittel und Detergenzien sollten vor Gebrauch steril sein.63. Begasung von reinen Bereichen kann nützlich sein, um eine mikrobiologische Kontamination an unzugänglichen Stellen zu verringern.Verarbeitungsverfahren64. Bei allen Verarbeitungsschritten, einschließlich der der Sterilisierung vorangehenden Schritte, sollten Vorkehrungen getroffen werden, um eine Kontamination möglichst gering zu halten.65. Präparate mikrobiologischen Ursprungs sollten nicht in Bereichen hergestellt oder abgefüllt werden, in denen andere Arzneimittel verarbeitet werden; allerdings können Impfstoffe aus abgetöteten Organismen oder aus Bakterienextrakten nach der Inaktivierung in denselben Räumen abgefüllt werden wie andere sterile Arzneimittel.
Seite 46 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

66. Die Validierung aseptischer Verfahren sollte die Prozesssimulation mit einem Nährmedium einschließen. Die Auswahl des Nährmediums sollte basierend auf der Darreichungsform des Produktes und Selektivität, Klarheit, Konzentration und Eignung zur Sterilisation des Nährmediums getroffen werden.67. Die Prozesssimulation sollte das routinemäßig durchgeführte aseptische Herstellungsverfahren möglichst weitgehend simulieren und alle kritischen aufeinanderfolgenden Herstellungsstufen umfassen. Es sollte ebenfalls alle Eingriffe, die bekanntermaßen während der normalen Produktion auftreten, sowie worstcase-Situationen berücksichtigen.68. Prozesssimulationen sollten zur Erstvalidierung mit drei aufeinanderfolgenden, erfolgreichen Simulationen pro Schicht durchgeführt werden und nach festgesetzten Intervallen und nach jeder signifikanten Änderung von Lüftungssystem, Ausrüstung, Verfahren oder Anzahl der Arbeitsschichten wiederholt werden. Normalerweise sollten Prozesssimulationen zweimal im Jahr pro Schicht und pro Verfahren wiederholt werden.69. Die Zahl der für ein Nährmedium verwendeten Behältnisse sollte ausreichend sein, damit eine gültige Bewertung durchgeführt werden kann. Für kleine Chargen sollte die Zahl der für das Nährmedium verwendeten Behältnisse zumindest der Größe der Produktcharge entsprechen. Das Ziel sollte ein Nullwachstum sein, die folgenden Vorgaben sollten erfüllt werden:
Bei einer Abfüllung von weniger als 5.000 Behältnissen sollten keine verunreinigten Einheiten nachgewiesen werden.•Bei einer Abfüllung zwischen 5.000 und 10.000 Behältnissen: •
Eine (1) verunreinigte Einheit sollte zu einer Ursachenforschung führen, einschließlich der Erwägung, die Simulation mit Nährmedien zu wiederholen;
a.
Zwei (2) verunreinigte Einheiten sollten Grund sein, eine Revalidierung zu erwägen, gefolgt von einer Ursachenforschung.
b.
Bei einer Abfüllung von mehr als 10.000 Behältnissen: •Eine (1) verunreinigte Einheit sollte zu einer Ursachenforschung führen; a.Zwei (2) verunreinigte Einheiten sollten Grund sein, eine Revalidierung zu erwägen, gefolgt von einer Ursachenforschung.
b.
70. Bei jeder Größe des Abfüllvorgangs können sporadisch auftretende Fälle mikrobiologischer Verunreinigung ein Hinweis auf eine Verunreinigung auf niedriger Stufe sein, die untersucht werden sollte. Die Untersuchung grober Fehler sollte den möglichen Einfluss auf die Sicherheit der Sterilität aller Chargen einschließen, die seit der letzten erfolgreichen Nährmediensimulation hergestellt wurden.71. Es sollte dafür Sorge getragen werden, dass Validierungen nicht die Verarbeitungsverfahren gefährden.72. Ausgangswasser, Wasseraufbereitungsanlagen und aufbereitetes Wasser sollten regelmäßig auf chemische und biologische Verunreinigungen sowie gegebenenfalls auf Endotoxine überprüft werden. Aufzeichnungen über die Ergebnisse dieser Kontrollen und über jede durchgeführte Maßnahme sollten aufbewahrt werden.73. Bei den Tätigkeiten in reinen Bereichen und besonders während aseptischer Arbeitsvorgänge sollte es nur ein Minimum an Aktivität geben. Das Personal sollte sich kontrolliert und methodisch bewegen, um eine übermäßige Abgabe von Partikeln und Organismen durch übertriebene Aktivität zu vermeiden. Die Raumtemperatur und -feuchtigkeit sollten wegen der Art der Schutzkleidung nicht unangenehm hoch sein.74. Die Ausgangsstoffe sollten nur minimal mikrobiell verunreinigt sein. In den Spezifikationen sollten Anforderungen an die mikrobiologische Qualität enthalten sein, wenn sich die Notwendigkeit hierfür aus den Kontrolluntersuchungen ergeben hat.75. Behältnisse und Materialien, die leicht Fasern abgeben können, sollten in reinen Bereichen möglichst nicht verwendet werden.76. Wo notwendig, sollten Maßnahmen getroffen werden, um die Verunreinigung des Endproduktes mit Partikeln möglichst gering zu halten.77. Bestandteile, Behältnisse und Ausrüstung sollten nach dem letzten Reinigungsvorgang so gehandhabt werden, dass sie nicht erneut verunreinigt werden.78. Der Zeitraum zwischen Waschen, Trocknen und Sterilisieren sowie zwischen Sterilisation und Gebrauch von Bestandteilen, Behältnissen und Ausrüstung sollte so kurz wie möglich und entsprechend den Lagerungsbedingungen befristet sein.79. Der Zeitraum zwischen dem Beginn der Herstellung einer Lösung und ihrer Sterilisation oder Filtration durch ein Bakterien zurückhaltendes Filter sollte so kurz wie möglich sein. je nach Zusammensetzung und vorgeschriebenen Lagerungsbedingungen sollte für jedes Produkt ein zulässiger Höchstwert festgelegt sein.80. Vor der Sterilisation sollte die mikrobiologische Grundbelastung (bioburden) untersucht werden. In Abhängigkeit von der Effizienz des eingesetzten Verfahrens sollten betriebliche Grenzen der Verunreinigung unmittelbar vor der Sterilisation festgelegt werden. Eine Untersuchung der mikrobiologischen Grundbelastung sollte bei jeder Charge, sowohl für aseptisch abgefüllte als auch für endsterilisierte Produkte, durchgeführt werden. Wenn für endsterilisierte Produkte Overkill-Sterilisationsbedingungen gewählt werden, kann die mikrobiologische Grundbelastung auch in angemessenen festgelegten Abständen überwacht werden. Für Systeme mit parametrischer Freigabe sollte die Prüfung auf mikrobiologische Grundbelastung für jede Charge durchgeführt werden und als Inprozesskontrolle bedacht werden. Wo es angemessen ist, sollte das Maß an Endotoxinen überwacht werden. Alle Lösungen, insbesondere großvolumige Infusionsflüssigkeiten, sollten einer Filtration unterzogen werden, bei der Mikroorganismen zurückgehalten werden, wenn möglich unmittelbar vor der Abfüllung.81. Bestandteile, Behältnisse, Ausrüstung und alle sonstigen Gegenstände, die in einem reinen Bereich benötigt werden, in dem aseptisch gearbeitet wird, sollten sterilisiert und durch zweitürige in die Wand eingelassene Sterilisatoren eingeschleust werden. Dies kann auch nach einem anderen Verfahren geschehen, wenn es gleichermaßen wirksam vor Verunreinigung schützt. Nichtbrennbare Gase sollten durch Mikroorganismen zurückhaltende Filter geleitet werden.82. Die Wirksamkeit eines jeden neuen Verfahrens sollte validiert werden. Die Validierung sollte dann in regelmäßigen Abständen unter Berücksichtigung gewonnener Erfahrungen oder bei jeder wesentlichen Änderung des Verfahrensablaufs oder der Ausrüstung verifiziert werden.Sterilisation83. Alle Sterilisationsverfahren sollten validiert werden. Besondere Vorsicht ist geboten, wenn die angewandte Sterilisationsmethode nicht in der gültigen Ausgabe des Europäischen Arzneibuchs beschrieben ist, oder wenn sie für andere Produkte als einfache wässrige oder ölige Lösungen eingesetzt wird. Wo möglich, ist die Hitzesterilisation die Methode der Wahl. In jedem Fall muss das gewählte Sterilisationsverfahren in Übereinstimmung mit der Zulassung und der Herstellungserlaubnis sein.84. Bevor ein Sterilisationsverfahren eingeführt wird, sollte nachgewiesen werden, dass es für das Produkt geeignet ist und die gewünschten Sterilisationsbedingungen in allen Teilen der Ladung jedes vorgesehenen Sterilisationsgutes erreicht werden. Dieser Nachweis sollte gegebenenfalls durch physikalische Messungen und biologische Indikatoren erfolgen. Die Gültigkeit des Verfahrens sollte in regelmäßigen Abständen, mindestens jährlich verifiziert werden und immer dann, wenn wesentliche Veränderungen an der Ausrüstung vorgenommen wurden. Die Ergebnisse sollten aufgezeichnet werden.
Seite 47 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

85. Um eine wirksame Sterilisation zu erzielen, muss die erforderliche Behandlung für das gesamte Material durchgeführt werden, und das Verfahren sollte so konzipiert sein, dass dieses Ziel mit Sicherheit erreicht wird.86. Für alle Sterilisationsverfahren sollten validierte Beladungsmuster festgelegt werden.87. Biologische Indikatoren sollten als eine zusätzliche Methode der Sterilisationskontrolle angesehen werden. Sie sollten entsprechend den Angaben des Herstellers gelagert und verwendet und ihre Qualität durch Positivkontrollen überprüft werden. Werden biologische Indikatoren eingesetzt, sollten strenge Vorkehrungen getroffen werden, um jede mikrobiologische Kontamination durch die Indikatoren zu verhindern.88. Nichtsterilisierte und sterilisierte Produkte sollten auf eindeutige Weise voneinander unterschieden werden. Jeder Korb, jedes tablett oder jedes andere Behältnis mit Produkten oder Bestandteilen sollte deutlich mit dem Namen des Materials, seiner Chargennummer sowie mit dem Hinweis auf erfolgte bzw. nicht erfolgte Sterilisation gekennzeichnet sein. Es können gegebenenfalls Indikatoren, wie Autoklavenbänder verwendet werden, um anzuzeigen, ob eine Charge (oder Teilcharge) den Sterilisationsprozess durchlaufen hat oder nicht. Diese Indikatoren geben jedoch keine zuverlässigen Hinweise darauf, ob die Charge tatsächlich steril ist.89. Sterilisationsaufzeichnungen sollten für jeden Sterilisationsvorgang vorliegen. Sie sind als Teil des Chargenfreigabeverfahrens zu genehmigen.Hitzesterilisation90. Jeder Hitzesterilisationszyklus sollte in einem Zeit/Temperatur-Diagramm in ausreichend großem Maßstab oder mittels anderer geeigneter Geräte mit ausreichender Genauigkeit und Präzision aufgezeichnet werden. Die Position der für die Kontrolle und/oder Aufzeichnung verwendeten Temperatursonden sollte während der Validierung festgelegt und gegebenenfalls auch gegen eine zweite unabhängige an der gleichen Stelle befindliche Temperatursonde kontrolliert werden.91. Chemische oder biologische Indikatoren können ebenfalls verwendet werden, aber sie sollten physikalische Messungen nicht ersetzen.92. Die Aufheizzeit muss ausreichend bemessen sein, damit die gesamte Ladung die erforderliche Temperatur erreicht, bevor mit der Messung der Sterilisationszeit begonnen wird.Diese Zeit muss für jede Art Sterilisationsgut ermittelt werden.93. Nach der Hochtemperaturphase eines Hitzesterilisationszyklus sollten Vorkehrungen getroffen werden, damit das sterilisierte Material während des Abkühlens nicht kontaminiert wird. Kühlflüssigkeit oder Kühlgase, die mit dem Produkt in Berührung kommen, sollten steril sein, es sei denn, es kann nachgewiesen werden, dass kein undichtes Behältnis zur Verwendung freigegeben würde.Feuchte Hitze94. Um den Vorgang zu überwachen, sollten sowohl die Temperatur als auch der Druck kontrolliert werden. Kontrollinstrumente sollten normalerweise unabhängig von Überwachungsgeräten und Aufzeichnungsdiagrammen sein. Wenn für diese Anwendungen automatische Kontroll- und Überwachungssysteme verwendet werden, sollten sie validiert sein, um sicherzustellen, dass kritische Verfahrensanforderungen eingehalten werden. System- und Zyklusfehler sollten vom System registriert und vom Bedienungspersonal festgestellt werden. Die Aufzeichnungen des unabhängigen Temperaturanzeigers sollten routinemäßig während der gesamten Sterilisationszeit mit dem Diagrammschreiber verglichen werden. Bei Sterilisatoren mit einem Abfluss am Kammerboden kann es auch nötig sein, die Temperatur während der Sterilisation an dieser Stelle aufzuzeichnen. Wenn der Zyklus auch eine Vakuumphase umfasst, sollte die Kammer regelmäßig auf ihre Dichtigkeit geprüft werden.95. Sterilisiergut sollte, soweit es sich nicht um Produkte in dicht verschlossenen Behältnissen handelt, in Material eingepackt sein, das die Entfernung der Luft und das Eindringen von Dampf erlaubt, aber eine Rekontamination nach der Sterilisation verhindert. Alle Teile der Ladung sollten bei der erforderlichen Temperatur für die erforderliche Zeit mit dem Sterilisiermedium in Kontakt sein.96. Es sollte sichergestellt werden, dass der für die Sterilisation verwendetet Dampf von geeigneter Qualität ist und keine Zusätze in solchen Mengen enthält, die eine Kontamination des Produktes oder der Ausrüstung verursachen könnten.Trockene Hitze97. Das angewandte Verfahren sollte die Luftzirkulation innerhalb der Kammer und die Aufrechterhaltung eines Überdrucks beinhalten, um das Eindringen von unsteriler Luft zu verhindern. Wenn Luft zugeführt wird, sollte diese durch ein HEPA-Filter geleitet werden. Wenn mit dieser Methode auch Pyrogene entfernt werden sollen, sollten auch Belastungstests mit Endotoxinen als Teil der Validierung durchgeführt werden.Strahlensterilisation98. Die Strahlensterilisation wird hauptsächlich zur Sterilisation hitzeempfindlicher Materialien und Produkte eingesetzt. Viele Arzneimittel und einige Verpackungsmaterialien sind strahlungsempfindlich. Daher ist diese Methode nur zulässig, wenn experimentell bestätigt wurde, dass das Produkt nicht nachteilig beeinflusst wird. UV-Bestrahlung ist in der Regel keine akzeptable Sterilisationsmethode.99. Während des Sterilisationsverfahrens sollte die Strahlendosis gemessen werden. Zu diesem Zweck sollten von der Dosisleistung unabhängige Dosimeter verwendet werden, die die vom Produkt selbst empfangene Dosis quantitativ erfassen. Dosimeter sollten in ausreichender Zahl nahe genug beieinander in die Ladung eingebracht werden, um zu gewährleisten, dass sich immer ein Dosimeter in der Kammer befindet. Sofern Dosimeter aus Kunststoff eingesetzt werden, sollten diese innerhalb ihrer Kalibrierungsfrist verwendet werden. Die Dosimeter sollten, nachdem sie der Strahlung ausgesetzt waren, innerhalb kurzer Zeit abgelesen werden.100. Biologische Indikatoren können als zusätzliche Kontrolle verwendet werden.101. Validierungsverfahren sollten sicherstellen, dass die Auswirkungen von Änderungen der Beladungsdichte berücksichtigt werden.102. Die Verfahren für die Handhabung der Materialien sollten Verwechslungen von bestrahlten und nicht bestrahlten Materialien verhindern. Strahlungsempfindliche Farbscheiben sollten ebenfalls auf jeder Packung verwendet werden, um zwischen bestrahlten und nicht bestrahlten Packungen zu unterscheiden.103. Die gesamte Strahlendosis sollte innerhalb einer im Voraus festgelegten Zeitspanne verabreicht werden.Ethylenoxid-Sterilisation104. Diese Methode sollte nur eingesetzt werden, wenn keine andere anwendbar ist. Während der Prozessvalidierung sollte nachgewiesen werden, dass das Produkt nicht beschädigt wird und Entgasungsbedingungen und -zeit so gewählt sind, dass Rückstände an Gas und Reaktionsprodukten auf ein festgelegtes und für das jeweilige Produkt oder Material akzeptables Niveau reduziert werden.105. Der direkte Kontakt zwischen Gas und Zellen der Mikroorganismen ist ausschlaggebend. Vorsichtsmaßnahmen sollten getroffen werden, um das Vorhandensein von Organismen zu verhindern, die in Materialien wie Kristallen oder getrocknetem Protein eingeschlossen sein könnten. Art und Menge des Verpackungsmaterials können den Vorgang wesentlich beeinflussen.106. Bevor die Materialien dem Gas ausgesetzt werden, sollten sie ins Gleichgewicht mit der für den Prozess erforderlichen Feuchtigkeit und Temperatur gebracht werden. Die hierfür erforderliche Zeit sollte unter Berücksichtigung der Notwendigkeit festgelegt werden, die Zeitspanne vor der Sterilisation möglichst kurz zu halten.107. Jeder Sterilisationszyklus sollte mit geeigneten biologischen Indikatoren überwacht werden, wobei eine angemessene Zahl von Testeinheiten über die gesamte Ladung verteilt ist. Die dabei erhaltenen Informationen sollten Bestandteil des Chargenprotokolls sein.
Seite 48 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

108. Zu jedem Sterilisationszyklus sollten Aufzeichnungen über die Dauer des gesamten Zyklus, den Druck, die Temperatur und die Feuchtigkeit innerhalb der Kammer während des Prozesses sowie über die Gaskonzentration und die insgesamt verwendete Gasmenge geführt werden. Druck und Temperatur sollten als Diagramme während des Prozesses aufgezeichnet werden. Die Aufzeichnungen sollten Bestandteil des Chargenprotokolls sein.109. Nach der Sterilisation sollte die Ladung unter kontrollierten Bedingungen gut belüftet gelagert werden, damit die Rückstände an Gas und Reaktionsprodukten auf das festgelegte Niveau reduziert werden können. Dieser Prozess sollte validiert werden.Filtration von Arzneimitteln, die nicht im Endbehäl tnis sterilisiert werden können110. Wenn eine Sterilisation im Endbehältnis durchgeführt werden kann, ist die alleinige Filtration nicht ausreichend. Im Hinblick auf derzeit verfügbare Methoden sollte der Dampfsterilisation der Vorzug gegeben werden. Wenn das Produkt nicht im Endbehältnis sterilisiert werden kann, können Lösungen oder Flüssigkeiten durch ein steriles Filter mit einer nominellen Porengröße von 0,22 Mikron (oder weniger) oder ein anderes Filter mit mindestens gleichem Rückhaltevermögen für Mikroorganismen in ein zuvor sterilisiertes Behältnis filtriert werden. Solche Filter können die meisten Bakterien und Schimmelpilze, aber nicht alle Viren oder Mykoplasmen abtrennen. Eine Ergänzung des Filtrationsprozesses durch eine Hitzebehandlung in gewissem Maße sollte erwogen werden.111. Da im Vergleich zu anderen Sterilisationsverfahren bei der Sterilfiltration zusätzliche potenzielle Risiken bestehen, kann eine zweite Filtration unmittelbar vor der Abfüllung durch ein weiteres sterilisiertes Filter, das Mikroorganismen zurückhält, ratsam sein. Die letzte Sterilfiltration sollte so nah wie möglich am Ort der Abfüllung durchgeführt werden.112. Die Abgabe von Fasern durch das Filter sollte minimal sein.113. Die Unversehrtheit des sterilisierten Filters sollte vor und unmittelbar nach jeder Verwendung durch eine geeignete Methode, wie den Blasendrucktest ("bubble point test"), den Gasdiffusionstest ("diffusive flow test") oder den Druckhaltetest ("pressure hold test") überprüft werden. Bei der Validierung sollten die benötigte Filtrationszeit für ein bekanntes Volumen an Bulklösung und der bei der Filtration anzuwendende Druckunterschied bestimmt sowie alle signifikanten Abweichungen hiervon während der routinemäßigen Herstellung aufgezeichnet und untersucht werden. Die Ergebnisse dieser Überprüfungen sollten im Chargenprotokoll festgehalten werden. Die Unversehrtheit kritischer Gas- und Luftfilter sind nach der Anwendung zu überprüfen. Die Unversehrtheit anderer Filter ist in geeigneten Abständen zu überprüfen.114. Das gleiche Filter sollte nicht länger als einen Arbeitstag verwendet werden, es sei denn, die darüber hinausgehende Verwendung ist validiert worden.115. Das Filter sollte das Produkt durch Absorption von Inhaltsstoffen oder durch Freisetzung von Substanzen in das Produkt nicht nachteilig beeinflussen.Fertigstellung steriler Produkte116. Partiell verschlossene Behältnisse, wie sie beim Gefriertrocknen verwendet werden, sollten solange unter Klasse A-Bedingungen behalten werden, bis der Stopfen vollständig eingeführt ist.117. Behältnisse sollten nach hinreichend validierten Methoden verschlossen werden. Durch Schmelzen geschlossene Behältnisse, z.B. Glas- oder Kunststoffampullen, sollten auf 100 %-ige Unversehrtheit getestet werden. Proben anderer Behältnisse sollten nach geeigneten Verfahren auf Unversehrtheit überprüft werden.118. Das Verschlusssystem für aseptisch abgefüllte Behältnisse ist nicht vollständig ausgefertigt, bis die Aluminiumkappe auf dem verschlossenen Behältnis verbördelt ist. Das Verbördeln der Kappe sollte daher so schnell wie möglich nach dem Einsetzen des Stopfens erfolgen.119. Da die zum Verbördeln der Kappen verwendeten Anlagen große Mengen von nicht vermehrungsfähigen Partikeln erzeugen können, sollte sich die Anlage separat befinden und mit einer angemessenen Luftabsaugung ausgestattet sein.120. Das Verbördeln der Kappen kann als aseptischer Prozess mit sterilisierten Kappen oder als reiner Prozess außerhalb der aseptischen Kernzone erfolgen. Wenn die letztgenannte Möglichkeit angewendet wird, sollten die Vials unter Klasse A-Bedingungen aufbewahrt werden, bis sie den aseptischen Bereich verlassen; danach sollten die verschlossenen Vials durch eine Klasse A-Luftversorgung geschützt werden, bis die Kappe verbördelt ist.121. Behältnisse mit fehlenden oder nicht richtig sitzenden Stopfen sollten vor der Ausstattung mit Kappen aussortiert werden. Wo Eingriffe durch Menschen an der Kappenstation notwendig sind, sollte eine entsprechende Technik eingesetzt werden, um den direkten Kontakt mit den Behältnissen zu verhindern und um mikrobiologische Verunreinigung zu minimieren.122. Bereiche mit Zugangsbeschränkungen und Isolatoren können dabei helfen, die benötigten Bedingungen sicherzustellen und direkte Eingriffe von Menschen in den Verschlussvorgang zu minimieren.123. Unter Vakuum verschlossene Behälter sollten nach Ablauf einer geeigneten, im Voraus festgelegten Frist auf das Fortbestehen des Vakuums geprüft werden.124. Abgefüllte Behältnisse mit Parenteralia sollten einzeln auf Fremdkontamination oder sonstige Defekte geprüft werden. Visuelle Kontrollen sollten unter geeigneten und kontrollierten Bedingungen hinsichtlich Beleuchtung und Hintergrund erfolgen. Das die Prüfung durchführende Personal sollte regelmäßigen Sehtests (gegebenenfalls mit Brille) unterzogen werden und bei der Kontrolltätigkeit häufige Pausen einlegen können. Wenn andere Prüfmethoden eingesetzt werden, sollte das Verfahren validiert sein und das ordnungsgemäße Funktionieren der Ausrüstung regelmäßig kontrolliert werden. Die Ergebnisse sollten aufgezeichnet werden.Qualitätskontrolle125. Die am Fertigprodukt durchgeführte Sterilitätsprüfung sollte nur als letzte einer Reihe von Kontrollmaßnahmen zur Gewährleistung der Sterilität betrachtet werden. Der Test sollte für das (die) betreffende(n) Produkt(e) validiert sein.126. In den Fällen, in denen eine parametrische Freigabe zugelassen wurde, sollte besonderer Wert auf die Validierung und Überwachung des gesamten Herstellungsverfahrens gelegt werden.127. Die für die Prüfung auf Sterilität entnommenen Proben sollten für die gesamte Charge repräsentativ sein, jedoch insbesondere auch Proben umfassen, die solche Teile der Charge enthalten, für die das größte Kontaminationsrisiko anzunehmen ist. Zum Beispiel sollten:
bei aseptisch abgefüllten Produkten die Proben Behältnisse einschließen, die zu Anfang und zum Ende der Charge und nach jeder wesentlichen Arbeitsunterbrechung abgefüllt wurden;
a.
bei im Endbehältnis hitzesterilisierten Produkten Proben vom potenziell kältesten Teil der Ladung entnommen werden.b.
____________ 1) Artikel 1 der Verordnung vom 3. November 2006 (BGBl. I S. 2523)2) Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel (AB1. EG Nr. L 311 S. 67), geändert durch die Richtlinie 2004/27/EG des Europäischen Parlaments und des Rates vom 31. März 2004 (ABl. EU Nr. L 136 S. 34)
Seite 49 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

3) Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel (ABl. EG Nr. L 311 S. 1), geändert durch die Richtlinie 2004/28/EG des Europäischen Parlaments und des Rates vom 31. März 2004 (ABl. EU Nr. L 136 S. 58)
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 3
Herstellung von Radiopharmaka Grundsatz Anhang 3
Stand: BAnz. Nr. 124 vom 21.08.2009 S. 2890Die Herstellung von Radiopharmaka muss in Übereinstimmung mit den Grundsätzen der Guten Herstellungspraxis für Arzneimittel Teil I und II durchgeführt werden. Dieser Anhang spricht insbesondere einige der Verfahrensweisen an, die spezifisch für Radiopharmaka sein können. Anmerkung i. Die Zubereitung von Radiopharmaka in Radiopharmazeutischen Einrichtungen (Krankenhäusern oder bestimmten Apotheken) unter Verwendung von Radionuklidgeneratoren und Kits für Radioaktive Arzneimittel mit Zulassung oder nationaler Lizenz wird von dieser Leitlinie nicht erfasst, soweit nicht von einer nationalen Regelung anderweitig festgelegt. Anmerkung ii. Gemäß der Regelungen zum Schutz vor Radioaktivität sollte sichergestellt werden, dass jede medizinische Anwendung unter der klinischen Verantwortung eines Arztes steht. In diagnostischen und therapeutischen nuklearmedizinischen Einrichtungen sollte ein Experte der medizinischen Physik vorhanden sein. Anmerkung iii. Dieser Anhang gilt auch für Radiopharmaka, die in der klinischen Prüfung eingesetzt werden. Anmerkung iv. Der Transport von Radiopharmaka ist durch die EURATOM-Richtlinien und die Anforderungen an den Strahlenschutz geregelt. Anmerkung v. Es wird anerkannt, dass andere als in diesem Anhang beschriebenen Methoden geeignet sein können, die grundsätzlichen Ziele der Qualitätssicherung zu verwirklichen. Andere Methoden sollten validiert sein und ein Niveau der Qualitätssicherung gewährleisten, welches dem in diesem Anhang mindestens gleichwertig ist. Einführung1. Herstellung und Umgang mit Radiopharmaka bergen potenzielle Risiken. Der Grad der Risiken hängt insbesondere ab von der Art der Strahlung, der Strahlungsenergie und der Halbwertzeiten der radioaktiven Isotope. Besondere Aufmerksamkeit ist der Vermeidung von Kreuzkontaminationen, dem Vermeiden von Radionuklidkontaminationen und der Abfallentsorgung beizumessen. 2. Aufgrund der kurzen physikalischen Halbwertzeit mancher Radionuklide kann es erforderlich sein, einige Radiopharmaka schon vor Beendigung aller Qualitätsprüfungen freizugeben. In diesem Falle ist die exakte und detaillierte Beschreibung des gesamten Freigabeverfahrens, einschließlich der Verantwortlichkeit des involvierten Personals und der fortdauernden Bewertung der Wirksamkeit des Qualitätssicherungssystems besonders wichtig. Die bei der Herstellung von Radiopharmaka oft kleinen Produktchargen erfordern besondere Aufmerksamkeit. 3. Diese Leitlinie ist anzuwenden auf industrielle Hersteller, Nuklearchemische/Radiopharmazeutische und klinische Einrichtungen und Institute sowie Zentren für die Positronenemissionstomographie (PET) für die Herstellung und Qualitätskontrolle folgender Produkttypen:
Radiopharmaka•Positronen aussendende Radiopharmaka für die PET•Radioaktive Vorstufen (Präkursoren bzw. Markerzubereitungen) für die Herstellung von Radiopharmaka•Radionuklid-Generatoren•
Art der Herstellung
Nicht-GMP * GMP Teil II & I (ansteigend) einschließlich relevanter Anhänge
Zadiopharmaka PET Radiopharmaka Zadioaktive Vorstufen (Präkursor)
in Reaktoren oder einem Zyklotron stattfindende Herstellungsprozesse
Chemische Synthese
Reinigungs-Schritte
Formulierung und Zubereitung
Aseptische Herstellung oder Endsterilisation
Zadionuklid-Generatoren
in Reaktoren oder einem Zyklotron stattfindende Herstellungsprozesse
Verarbeitung
*) target- und Transfersystem vom Zyklotron zur Syntheseapparatur können als erster Schritt der Wirkstoffherstellung angesehen werden.
4. Der Hersteller des radioaktiven Arzneimittels sollte die Schritte für die Herstellung des Wirkstoffs und des fertigen Arzneimittels sowie die für die spezifischen Prozess-/ Herstellungsschritte zur Anwendung kommenden GMPAnforderungen (Teil I oder Teil II) beschreiben und begründen. 5. Bei der Zubereitung von Radiopharmaka sind die Strahlenschutzvorschriften zu beachten. 6. Radiopharmaka, die parenteral angewendet werden, sollen steril sein und, soweit übertragbar, entsprechend den in Eudralex Volume 4, Anhang 1, beschriebenen Bedingungen für die Herstellung steriler Arzneimittel hergestellt werden. 7. Spezifikationen und Prüfverfahren in der Qualitätskontrolle für die am häufigsten eingesetzten Radiopharmaka sind im Europäischen Arzneibuch oder in den Zulassungsunterlagen aufgeführt. Klinische Prüfungen8. Radiopharmaka, die in der klinischen Prüfung als Prüfpräparate eingesetzt werden sollen, sollten zusätzlich in Übereinstimmung mit den Grundsätzen des Eudralex Volume 4 Anhang 13 hergestellt werden. Qualitätssicherung9. Die Qualitätssicherung ist bei der Herstellung von Radiopharmaka von noch größerer Bedeutung aufgrund deren besonderer Charakteristika, kleinen Produktionschargen und der unter bestimmten Umständen erforderlichen Anwendung des Produktes, bevor alle Freigabetests beendet sind. 10. Wie bei allen Arzneimitteln müssen die Produkte gut geschützt sein gegen Kontaminationen und Kreuzkontaminationen. Jedoch muss auch das Personal und die Umwelt vor radioaktiver Strahlung geschützt werden. Das bedeutet, dass die Rolle eines effektiven Qualitätssicherungssystems von größter Bedeutung ist.
Seite 50 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

11. Es ist wichtig, dass die beim Monitoring der Räumlichkeiten und Arbeitsabläufe erhaltenen Daten als Teil des Freigabeprozesses gründlich dokumentiert und evaluiert werden. 12. Die Grundsätze der Qualifizierung und Validierung sollten auf die Herstellung von Radiopharmaka angewendet werden. Zur Festlegung des Ausmaßes der Qualifizierung/Validierung sollte ein Verfahren des Risikomanagements eingesetzt werden, das neben den Anforderungen der Guten Herstellungspraxis auch die des Strahlenschutzes einbezieht. Personal13. Alle Herstellungstätigkeiten sollten unter der Verantwortung von Personen mit zusätzlicher Qualifikation im Strahlenschutz ausgeführt werden. Personen, die in Produktions-, analytischen Kontroll- und Freigabeaktivitäten von Radiopharmaka involviert sind, sollten angemessen in den für Radiopharmaka spezifischen Aspekten des Qualitätsmanagementsystems geschult sein. Die sachkundige Person (QP) sollte die Gesamtverantwortung für die Freigabe der Produkte haben. 14. Alle Personen (einschließlich Wartungs- und Reinigungspersonal), die in Bereichen arbeiten, in denen Radiopharmaka hergestellt werden, sollten geeignete zusätzliche Schulungen erhalten, die auf die Besonderheiten der dort eingesetzten Verfahren und der hergestellten Produkte abgestellt sind. 15. Wenn Produktionsbereiche zusammen mit Forschungseinrichtungen genutzt werden, muss das Personal der Forschungseinrichtungen entsprechend in GMP-Regelungen ausgebildet werden und die QA Funktion muss die Forschungsaktivitäten überprüfen und genehmigen, um sicherzustellen, dass sie keine Gefährdung für die Herstellung der Radiopharmaka darstellen. Räume und EinrichtungenAllgemeines16. Radioaktive Produkte sollten in kontrollierten Bereichen (bezogen auf die Umwelt und den Strahlenschutz) hergestellt werden. Alle Herstellungsschritte sollten in in sich geschlossenen Räumlichkeiten durchgeführt werden, die ausschließlich der Herstellung von Radiopharmaka dienen. 17. Es sollten Maßnahmen etabliert und eingeführt werden, um eine Kreuzkontamination von Personen, Material, Radionukliden usw. zu vermeiden. Wann immer möglich, sollte die Herstellung in geschlossener Ausrüstung erfolgen. Wenn offene Produktionsanlagen genutzt oder Produktionsanlagen geöffnet werden, sollten Vorkehrungen getroffen werden, um das Risiko einer Kontamination zu minimieren. Mittels einer Risikoeinschätzung sollte nachgewiesen werden, dass die vorgesehenen Reinraumbedingungen für die Art des herzustellenden Produkts geeignet sind. 18. Der Zutritt zu den Herstellungsbereichen sollte über eine Umkleideschleuse erfolgen und nur berechtigtem Personal möglich sein. 19. Arbeitsbereiche und ihre Umgebung sollten im Hinblick auf Radioaktivität, Partikelreinheit und mikrobiologische Qualität, wie während der Leistungsqualifizierung (PO) festgelegt, überwacht werden. 20. Wartungs-, Kalibrierungs- und Qualifizierungsprogramme sollten durchgeführt werden, um sicherzustellen, dass alle Räumlichkeiten und Einrichtungen, die bei der Herstellung von Radiopharmaka eingesetzt werden, geeignet und qualifiziert sind. Diese Aktivitäten sollten von dafür qualifiziertem Personal durchgeführt und dokumentiert werden. 21. Es sollten Vorsichtsmaßnahmen getroffen werden, um eine radioaktive Kontamination innerhalb der Einrichtung zu vermeiden. An Ort und Stelle sollten angemessene Kontrollen vorhanden sein, um jede radioaktive Kontamination festzustellen, sowohl direkt mit Hilfe eines Strahlendetektors als auch indirekt mittels Wischmethoden. 22. Die Ausrüstungen und Produktionsanlagen sollten so beschaffen sein, dass ihre Oberflächen, die mit dem Produkt in Kontakt kommen, nicht radioaktiv, additiv oder absorptiv sind und dadurch die Qualität der Radiopharmaka verändern. 23. Bei Luftströmen, die aus Bereichen abgezogen werden, in denen mit Radiopharmaka gearbeitet wird, sollte eine Rezirkulation vermieden werden, es sei denn, dass dies begründet werden kann. Die Abluftführungen sollten so konstruiert sein, dass eine Umweltkontamination durch radioaktive Partikel und Gase minimiert wird. Es sollten geeignete Maßnahmen ergriffen werden, um die kontrollierten Bereiche vor Partikeln und mikrobieller Kontamination zu schützen. 24. Um ein Entweichen von radioaktiven Partikeln zu vermeiden, kann es erforderlich sein, dass der Luftdruck in den Räumen, in denen Produkte exponiert sind, niedriger sein muss als in den angrenzenden Räumen. Jedoch ist es auch erforderlich die Produkte vor einer Kontamination aus der Umgebung zu schützen. Dies kann zum Beispiel erreicht werden durch den Gebrauch von Luftschleusen oder anderen Barriere-Technologien, die als Druckfallen wirken. Sterilherstellung25. Sterile Radiopharmaka können in solche eingeteilt werden, die aseptisch hergestellt werden, und in solche, die endsterilisiert werden. Die Einrichtung sollte den Reinheitsgrad der Umgebung aufrechterhalten, der dem jeweiligen Herstellungsvorgang angemessen ist. Bei sterilen Produkten sollte der Arbeitsbereich, in dem Produkte oder Behältnisse möglicherweise der Umwelt ausgesetzt werden, die Anforderungen an den Reinheitsgrad der Umgebung erfüllen, die in Eudralex Volume 4, Anhang 1, festgelegt sind. 26. Für die Herstellung von Radiopharmaka kann eine Risikobewertung eingesetzt werden, um die geeigneten Druckdifferenzen, Luftrichtungen und Luftqualitäten festzulegen. 27. Im Falle der Verwendung geschlossener und automatisierter Systeme (zur chemischen Synthese, Reinigung, online Sterilfiltration) sollte eine Umgebung der Reinheitsklasse C (z.B. in der Heißzelle) ausreichend sein. Geschlossene Heißzellen sollten bei gefilterter Luftzufuhr einen hohen Grad an Luftreinheit erreichen. Aseptische Herstellungsaktivitäten müssen in einer Umgebung der Reinheitsklasse A ausgeführt werden. 28. Vor dem Beginn der Herstellung ist der Zusammenbau sterilisierter Ausrüstungs- und Verbrauchsgegenstände (Schläuche, sterilisierte Filter und sterile geschlossene und verplombte Vials, die an ein Befüllungsrohr angeschlossen sind) unter aseptischen Bedingungen durchzuführen. Dokumentation29. Alle Dokumente, die in Zusammenhang mit der Herstellung der Radiopharmaka stehen, sollten nach schriftlich festlegten Verfahren erstellt, überprüft, genehmigt und verteilt werden. 30. Spezifikationen sollten für Ausgangsstoffe, Kennzeichnungs- und Verpackungsmaterial, kritische Zwischenprodukte und die Radiopharmaka-Endprodukte vorhanden sein. Spezifikationen sollten auch vorhanden sein für andere kritische Dinge, die im Herstellungsprozess eingesetzt werden, wie Prozesshilfen, Dichtungen, Kits für Sterilfilter, die kritische Auswirkungen auf die Qualität haben könnten. 31. Für die Radiopharmaka sollten Akzeptanzkriterien erstellt werden, einschließlich der Kriterien für die Freigabe und die am Ende der Haltbarkeitsdauer notwendige Qualität (z.B. chemische Identität des Isotops, radioaktiver Gehalt, Reinheit und spezifische Aktivität). 32. Protokolle sollten Aussagen über die wesentlichen verwendeten Herstellungsanlagen sowie die an diesen durchgeführte Reinigungen, Desinfektionen oder Sterilisationen und deren Wartungen ebenso enthalten wie den Produktnamen, die Chargenbezeichnung (soweit vorhanden), das Datum, die Uhrzeit und die Unterschrift der Person, die die jeweilige Tätigkeit durchgeführt hat. 33. Die Protokolle sollten mindestens 3 Jahre aufbewahrt werden, soweit dafür in nationalen Anforderungen kein anderer Zeitraum festgelegt wurde. Produktion
Seite 51 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

34. Die gleichzeitige Produktion verschiedener Radiopharmaka im gleichen Arbeitsbereich (z.B. in einer Heißzelle oder in einer LAF-Einheit) sollte vermieden werden, um das Risiko einer Kreuzkontamination oder Verwechslungen möglichst niedrig zu halten. 35. Besondere Beachtung gilt der Validierung einschließlich der Validierung von Computersystemen, die in Übereinstimmung mit Eudralex Volume 4, Anhang 11, durchgeführt werden sollte. Neue Herstellungsprozesse sollten im Voraus validiert werden. 36. Die kritischen Parameter sollten üblicherweise vor oder während der Validierung identifiziert und der notwendige Rahmen für reproduzierbare Vorgänge festgelegt werden. 37. Integritätstests der Membranfilter sollten bei aseptisch abgefüllten Produkten unter Berücksichtigung des notwendigen Strahlenschutzes und Aufrechterhaltung der Filtersterilität durchgeführt werden. 38. Wegen der Strahlengefährdung ist es akzeptabel, dass die Kennzeichnung über die wesentlichen Angaben auf dem Primärbehältnis bereits im Voraus (vor der Herstellung) durchgeführt wird. Sterile, leere, verschlossene Vials können vor dem Abfüllen mit den bereits bekannten Informationen gekennzeichnet werden, vorausgesetzt, dass damit nicht der Sterilität geschadet oder die visuelle Kontrolle der gefüllten Vials verhindert werden. Qualitätskontrolle39. Es kann erforderlich sein, dass einige Radiopharmaka auf der Basis einer Bewertung der vorliegenden Chargendokumentation und bevor alle chemischen und mikrobiologischen Tests beendet wurden, ausgeliefert und angewendet werden. Die Freigabe von Radiopharmaka kann in zwei oder mehreren Phasen erfolgen, vor und nach der vollständigen analytischen Testung:
Bewertung der Chargenprotokolle über die Herstellungsbedingungen und die bis zu diesem Zeitpunkt erfolgten analytischen Tests durch eine dafür festgelegte Person, bevor die Radiopharmaka im Quarantänestatus zur klinischen Einrichtung transportiert werden dürfen.
a.
Bewertung der endgültigen analytischen Testergebnisse unter Sicherstellung, dass alle Abweichungen von den normalen Verfahren vor der dokumentierten Zertifizierung durch die Sachkundige Person (QP) dokumentiert, begründet und entsprechend freigegeben wurden.
b.
Sofern bestimmte Freigabeparameter vor der Anwendung des Produktes noch nicht verfügbar sind, sollte die sachkundige Person (QP) das radioaktive Arzneimittel unter Vorbehalt zur Anwendung freigeben. Die endgültige Freigabe des radioaktiven Arzneimittels kann erst erfolgen, wenn der sachkundigen Person alle Freigabetestergebnisse vorliegen. 40. Die meisten Radiopharmaka sind nur für den Gebrauch innerhalb eines kurzen Zeitrahmens geeignet. Die Verwendbarkeitsfrist muss klar festgelegt sein. 41. Auf Radionukliden mit längeren physikalischen Halbwertzeiten basierende Radiopharmaka sollten vor der Freigabe durch die sachkundige Person (QP) vollständig getestet werden, um zu zeigen, dass sie alle relevanten Freigabekriterien erfüllen. 42. Vor der Testung können die Proben gelagert werden, um ein ausreichendes Abklingen der Radioaktivität zu ermöglichen. Alle Tests, einschließlich des Steriltests, sollten sobald als möglich durchgeführt werden. 43. Es sollte ein schriftliches Verfahren etabliert werden, das die Bewertung der Herstellung und der analytischen Daten, die vor dem Vertrieb der Charge zu berücksichtigen sind, detailliert festschreibt. 44. Produkte, die die Akzeptanzkriterien nicht erfüllen, sollten zurückgewiesen werden. Falls das Material umgearbeitet wird, sollte dies nach im Voraus festgelegten Verfahren erfolgen; das Endprodukt sollte zur Freigabe die Akzeptanzkriterien erfüllen. Zurückgegebene Produkte dürfen nicht umgearbeitet werden und müssen als radioaktiver Abfall gelagert werden. 45. Ein Verfahren sollte auch die Maßnahmen beschreiben, die die sachkundige Person (QP) zu ergreifen hat, falls unbefriedigende Testergebnisse (Outof-Specification) nach dem erfolgten Vertrieb aber vor dem Verfalldatum erhalten werden. Solche Ereignisse sollten untersucht werden, um durch korrektive und vorbeugende Maßnahmen zukünftige Wiederholungen zu vermeiden. Dieser Vorgang ist zu dokumentieren. 46. Falls erforderlich sollten dem verantwortlichen Klinikpersonal die notwendigen Informationen gegeben werden. Um dies zu erleichtern, sollte für Radiopharmaka ein Rückverfolgungsverfahren eingeführt werden. 47. Es sollte ein System vorhanden sein, mit dem die Qualität des Ausgangsmaterials festgestellt werden kann. Die Lieferantenzulassung sollte eine Bewertung beinhalten, dass das Material mit ausreichender Sicherheit ständig den Spezifikationen entsprechen wird. Das Ausgangs- und das Verpackungsmaterial und die kritischen Prozesshilfen sollten nur von zugelassenen Lieferanten bezogen werden. Referenzproben und Rückstellmuster48. Für Radiopharmaka sollten ausreichende Proben jedes formulierten Bulkprodukts für mindestens sechs Monate aufbewahrt werden, soweit nichts anderes durch das Risiko-Managementsystem gerechtfertigt wird. 49. Proben der Ausgangsstoffe (außer im Herstellungsprozess verwendete Lösungsmittel, Gase oder Wasser) müssen für mindestens zwei Jahre nach Freigabe des Produkts aufbewahrt werden. Diese Frist kann verkürzt werden, wenn die Haltbarkeitsdauer des Materials, wie in der relevanten Spezifikation angegeben, kürzer ist. 50. Für die Probenahme und die Aufbewahrung der Rückstellmuster von Ausgangsmaterialien und von Produkten, die für den Einzelfall oder in kleinen Mengen hergestellt werden oder deren Lagerung besondere Probleme bereiten könnten, können mit Zustimmung der zuständigen Behörde andere Festlegungen getroffen werden. Vertrieb51. Der Vertrieb des Endprodukts ist, bevor alle erforderlichen Testergebnisse vorliegen, unter kontrollierten Bedingungen für Radiopharmaka akzeptabel, vorausgesetzt das Produkt wird in der Nuklearmedizinischen Einrichtung erst angewendet, nachdem es von einer dafür vorgesehenen Person aufgrund von zufrieden stellenden Testergebnissen für die Anwendung freigegeben wurde. Glossar Zubereiten:Die Herstellung von anwendungsbereiten radioaktiven Arzneimittel in nuklearmedizinischen Einrichtungen aus einem Kit für ein radioaktives Arzneimittel, einem Radionuklidgeneratoreluat oder einer Markerzubereitung (radioaktive Vorstufe, Radionuklidprecursor). Kits für ein radioaktives Arzneimittel, Radionuklidgeneratoren und Markerzubereitungen sollten eine Zulassung oder nationale Lizenz haben. Herstellung: Produktion, Qualitätskontrolle, Freigabe und Vertrieb von Radiopharmaka aus Wirkstoffen und Ausgangsstoffen. Heißzelle:Abgeschirmte Arbeitsstationen für die Herstellung und Handhabung von radioaktivem Material. Heißzellen müssen nicht als Isolator konstruiert sein. Sachkundige Person:QP gemäß der Richtlinie 2001/83/EG und 2001/82/EG . Die Verantwortlichkeiten der QP sind näher ausgeführt in Eudralex Volume 4, Anhang 16.
Seite 52 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

weiter
Bekanntmachung zu § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung - AMWHV Vom 16. Juli 2009
(BAnz. Nr. 124 vom 21.08.2009 S. 2890) Die Europäische Kommission hat die Anhänge 3 und 7 zum Leitfaden für die Gute Herstellungspraxis für Arzneimittel und Prüfpräparate (EG-GMP Leitfaden) geändert und am 1. September 2008 in englischer Sprache auf ihrer Internetseite veröffentlicht. Anhang 3 ist seit dem 1. März 2009, Anhang 7 ab dem 1. September 2009 anzuwenden. Hiermit werden die vom Bundesministerium für Gesundheit in die deutsche Sprache übersetzten Anhänge 3 und 7 zum EG-GMP Leitfaden nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung vom 3. November 2006 (BGBl. I S. 2523), die zuletzt durch Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung vom 26. März 2008 (BGBl. I S. 521) geändert worden ist, im Anschluss an die Bekanntmachung vom 18. Juli 2008 (BAnz. S. 2798) bekannt gemacht. umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 6 .
Herstellung medizinischer Gase Anhang 6 11a zum EU-Leitfaden für die Gute Herstellungspraxis
(GMP)
Stand: (BAnz. Nr. 148 vom 29.09.2011 S. 3414)
Geschichte des Dokuments Datum
Der Anhang wurde als Folge der Restrukturierung des GMP Leitfadens und der Notwendigkeit, die Anforderungen in Teil II des Leitfadens im Hinblick auf seine Anwendbarkeit für medizinische Gase anzupassen, überarbeitet. Es besteht ein Bedarf klarer zu definieren, was als Ausgangsmaterial gegenüber einer pharmazeutischen Bulkware anzusehen ist. Die Gelegenheit wurde auch für eine grundsätzliche Überarbeitung des Anhangs genutzt.
Februar 2007
öffentliche Konsultation Juli 2007 bis Dezember 2007
Angenommen von der Europäischen Kommission 31. Januar 2010
Datum des Inkrafttretens 31. Juli 2010
GrundsätzeGase, die die Arzneimitteldefinition der Richtlinie 2001/83/EG oder der Richtlinie 2001/82/EG erfüllen (medizinische Gase), unterliegen den dort festgelegten Anforderungen, einschließlich der Anforderungen an ihre Herstellung. In diesem Zusammenhang befasst sich dieser Anhang mit der Herstellung von Gas-Wirkstoffen und mit der Herstellung medizinischer Gase.Die Abgrenzung zwischen der Herstellung des Wirkstoffs und der Herstellung des Arzneimittels sollte in den Zulassungsunterlagen klar beschrieben werden. Üblicherweise gehören die Herstellungs- und Reinigungsschritte des Gases zum Bereich der Wirkstoffherstellung. Die Gase treten in den pharmazeutischen Bereich ein ab der ersten Lagerung des Gases für diese Zweckbestimmung.Die Herstellung der Gas-Wirkstoffe sollte mit den grundlegenden Anforderungen des Leitfadens (Teil II) übereinstimmen, mit den relevanten Anforderungen dieses Anhangs und mit den anderen Anhängen des Leitfadens, soweit dies von Bedeutung ist.Die Herstellung der medizinischen Gase sollte mit den grundlegenden Anforderungen des Leitfadens (Teil I) übereinstimmen, mit den relevanten Teilen dieses Anhangs und mit den anderen Anhängen des Leitfadens, soweit dies von Bedeutung ist.Im Ausnahmefall eines kontinuierlichen Prozesses, in dem keine Zwischenlagerung des Gases von der Herstellung des Wirkstoffs bis zur Herstellung des Arzneimittels möglich ist, sollte der gesamte Prozess (vom Ausgangsmaterial des Wirkstoffs bis zum Arzneimittel) als dem pharmazeutischen Bereich zugehörig betrachtet werden. Dies sollte klar in den Zulassungsunterlagen angegeben werden.Der Anhang behandelt nicht die Herstellung und Handhabung medizinischer Gase in Krankenhäusern, es sei denn, dass dies als industrielle Zubereitung oder Herstellung angesehen wird. Relevante Abschnitte dieses Anhangs können jedoch als Basis für derartige Tätigkeiten dienen.Herstellung von Gas-WirkstoffenGas-Wirkstoffe können durch chemische Synthese oder aus natürlichen Quellen, sofern erforderlich nach Durchführung von Reinigungsschritten (z.B. in einer Lufttrennungsanlage), gewonnen werden.1. Der Prozess dieser beiden Methoden für die Herstellung der Gas-Wirkstoffe sollte dem Leitfaden Teil II entsprechen. Jedoch sind die Anforderungen
an das Ausgangsmaterial für Wirkstoffe (Teil II, Kapitel 7) nicht anwendbar auf die Herstellung des Gas-Wirkstoffs durch Lufttrennung (jedoch sollte der Hersteller sicherstellen, dass die Außenluft für den etablierten Prozess geeignet ist und jede Änderung der Qualität der Außenluft nicht die Qualität des Gas-Wirkstoffs beeinflusst);
a.
an die fortlaufenden Stabilitätsstudien (Teil II, Kapitel 11.5), die eingesetzt werden um die Lagerungsbedingungen und Verfalldaten/Wiederholungstestdaten (Teil II, Kapitel 11.6) zu bestätigen, nicht anwendbar in Fällen, in denen die Anfangsstabilitätsstudien durch bibliographische Daten (siehe Note for Guidance CPMP/QWP/1719/00) ersetzt wurden; und
b.
an die Referenzproben/Rückstellmuster (Teil II, Kapitel 11.7) nicht anwendbar auf Gas-Wirkstoffe, sofern nichts anderes festgelegt ist.
c.
2. Die Herstellung der Gas-Wirkstoffe durch einen kontinuierlichen Prozess (z.B. Lufttrennung) sollte kontinuierlich hinsichtlich der Qualität überwacht werden. Die Ergebnisse dieser Überwachung sollte im Hinblick auf eine Trendanalyse aufbewahrt werden.3. Außerdem
sollten Transfer und Auslieferungen der Gas-Wirkstoffe als Bulkware den gleichen Anforderungen genügen wie für medizinische Gase (Nummer 19 bis 21 dieses Anhangs);
a.
sollte die Abfüllung der Gas-Wirkstoffe in Gasflaschen oder mobile Flüssiggasbehälter den gleichen Anforderungen genügen wie die medizinischen Gase (Nummer 22 bis 37 dieses Anhangs) sowie Leitfaden Teil II Kapitel 9.
b.
Seite 53 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Herstellung der medizinischen GaseDie Herstellung medizinischer Gase erfolgt üblicherweise in geschlossenen Systemen. Insofern ist die Gefahr einer Kontamination des Produktes durch die Umwelt minimal. Jedoch ist die Gefahr von Kontaminationen (oder Kreuzkontaminationen mit anderen Gasen) gegeben, insbesondere bei einem Wiedergebrauch der Behälter.4. Die Anforderungen an Gasflaschen sollten auch für Flaschenbündel gelten (soweit die Lagerung und der Transport nicht unter besonderem Schutz erfolgen).Personal5. Alle Mitarbeiter, die in Herstellung und Vertrieb der medizinischen Gase involviert sind, sollten ein für diese Produkttypen spezifisches GMP-Training erhalten. Sie sollten sich der kritischen wichtigen Aspekte sowie der für Patienten potenziellen Gefahren dieser Arzneimittel bewusst sein. Die Trainingprogramme sollten auch die Tankfahrzeug-Lastwagenfahrer einschließen.6. Das Personal von Zulieferern, das die Qualität des medizinischen Gases beeinflussen könnte (wie beispielsweise das Personal, das mit der Wartung der Gasflaschen oder Ventile befasst ist), sollte in geeigneter Weise geschult werden.Räume und Ausrüstungen Räume7. Gasflaschen und mobile Flüssiggasbehälter sollten in von nichtmedizinischen Gasen getrennten Bereichen überprüft, vorbereitet, befüllt und gelagert werden. Zwischen diesen beiden Bereichen sollte kein Austausch von Gasflaschen und mobilen Flüssiggasbehältern stattfinden. Jedoch könnte die Überprüfung, Vorbereitung, Befüllung und Lagerung anderer Gase in den gleichen Bereichen akzeptiert werden, wenn diese mit den Spezifikationen der medizinischen Gase übereinstimmen und die Herstellungsvorgänge entsprechend GMPStandards erfolgen.8. Um die Gefahr einer Verwechslung zu vermeiden, sollten die Räumlichkeiten ausreichend Platz für Herstellungs-, Prüfungs- und Lagerungsvorgänge bieten. Die Räumlichkeiten sollten so ausgelegt sein, dass sie
separate, gekennzeichnete Bereiche für unterschiedliche Gase; a.die klare Identifizierung und Trennung von Gasflaschen/ mobilen Flüssiggasbehältern entsprechend ihrem Status (z.B. "Warten auf Kontrolle", "in Erwartung der Befüllung", "Quarantäne", "zertifiziert", "zurückgewiesen", "vorbereitet zur Auslieferung")
b.
vorsehen. Die einzusetzende Methode, um diese verschiedenen Stufen der Abtrennung zu erreichen, hängt ab von Art, Umfang und von der Komplexität des Gesamtprozesses. Generell können entsprechend markierte Bodenflächen, Unterteilungen, Abtrennungen und Schilder oder andere angemessene Mittel dafür verwendet werden.9. Leere aussortierte oder gewartete Gasflaschen/Heim-Flüssiggasbehälter sowie befüllte Gasflaschen/Heim-Flüssiggasbehälter sollten vor ungünstigen Wetterbedingungen geschützt gelagert werden. Befüllte Gasflaschen/mobile Flüssiggasbehälter sollten in sauberem Zustand und entsprechend der Umgebungsbedingungen, in denen sie eingesetzt werden sollen, gelagert werden.10. Spezifische Lagerungsbedingungen sollten entsprechend der Zulassungsunterlagen vorgesehen werden (z.B. für Gasmischungen, bei denen eine Phasentrennung bei Frost erfolgt).Ausrüstung11. Die Ausrüstung sollte gewährleisten, dass das richtige Gas in den richtigen Behälter gefüllt wird. Es sollten üblicherweise keine Querverbindungen zwischen Rohleitungen mit unterschiedlichen Gasen vorhanden sein. Sofern Querverbindungen erforderlich sind (z.B. Ausrüstungen zum Befüllen von Mischungen), sollte eine Qualifizierung sicherstellen, dass kein Risiko einer Kreuzkontamination zwischen den beiden Gasen besteht. Außerdem sollten die Abfüllstationen mit spezifischen Verbindungen ausgerüstet sein. Diese Verbindungen können unterschiedlichen nationalen oder internationalen Standards unterliegen. Der Gebrauch von Verbindungen, die unterschiedlichen Standards in derselben Abfüllstätte genügen, sollte sorgfältig überwacht werden, ebenso der Gebrauch von Adaptern, die in einigen Bereichen benötigt werden um spezifische Verbindungssysteme bei der Abfüllung zu überbrücken.12. Tanks und Tankfahrzeuge sollten immer nur für ein Gas sowie eine genau definierte Gasqualität bestimmt sein. Jedoch können medizinische Gase in den gleichen Tanks, anderen Behältern für die Zwischenlagerung oder Tankfahrzeugen gelagert oder transportiert werden wie nicht medizinische Gase, vorausgesetzt, dass die Qualität der letztgenannten mindestens der Qualität des medizinischen Gases vergleichbar ist und der GMP Standard eingehalten wird. In diesen Fällen sollte ein Qualitätsrisikomanagement durchgeführt und dokumentiert werden.13. Ein gemeinsames System, mit dem das Gas an Abfüllstationen für medizinische und nicht medizinische Gase geliefert wird, ist nur akzeptabel, wenn mit einer validierten Methode ein Rückfluss von der Gasleitung nicht medizinischer Gase zu der der medizinischen Gase verhindert wird.14. Abfüllstationen sollten entweder nur für ein einziges medizinisches Gas oder nur für ein bestimmtes Gemisch medizinischer Gase verwendet werden. In Ausnahmefällen kann die für medizinische Gase vorgesehene Abfüllstation auch für die Abfüllung von Gasen für andere Zwecke zulässig sein, wenn dies gerechtfertigt erscheint und kontrolliert erfolgt. In solchen Fällen sollte die Qualität des nicht medizinischen Gases dem des medizinischen Gases mindestens gleichwertig sein und die GMP Standards eingehalten werden. Die Abfüllung sollte dann in Kampagnen erfolgen.15. Reparatur- und Wartungsarbeiten (einschließlich Reinigung und Spülen) der Ausrüstung sollten die Qualität der medizinischen Gase nicht beeinträchtigen. Insbesondere sollten in Verfahrensbeschreibungen Maßnahmen festgelegt werden, die nach Reparatur- und Wartungsarbeiten unter Einbeziehung von Verletzungen der Systemintegrität zu ergreifen sind. Es sollte insbesondere gezeigt werden, dass die Ausrüstung frei von Kontaminationen ist, die die Qualität des Endproduktes vor seiner Freigabe zur Anwendung negativ beeinflussen könnte. Protokolle darüber sollten aufbewahrt werden.16. Eine Verfahrensbeschreibung sollte festlegen, welche Maßnahmen zu ergreifen sind, wenn ein Tankfahrzeug (nachdem es nicht medizinische Gase entsprechend Nummer 12 transportiert hat oder nach Wartungsmaßnahmen) in die Abfüllstation für medizinische Gase zurückkehrt. Diese sollten eine analytische Testung einschließen.Dokumentation17. Die im Chargenprotokoll von Gasflaschen/mobilen Flüssiggasbehältern enthaltenden Daten müssen die wichtigen Aspekte des Abfüllvorgangs für jeden Behälter nachvollziehbar belegen. Folgende Informationen sollten verfügbar sein.
Produktname; a.Chargenbezeichnung; b.Datum und Zeitpunkt des Abfüllprozesses; c.Identifizierung der Person(en) für jeden wichtigen Arbeitsschritt (z.B. Prüfung der vollständigen Räumung der Anlage, Entgegennahme, Vorbereitung der Abfüllung, Abfüllung etc.);
d.
Angabe der Charge/Chargen für die Befüllung, wie in Nummer 22 angegeben, einschließlich der Angabe des Status; e.eingesetzte Ausrüstung (z.B. Abfüllstation); f.
Seite 54 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Anzahl der Gasflaschen/mobilen Flüssiggasbehälter vor der Befüllung, einschließlich deren individueller Referenzcode und der Wasserkapazität(en)
g.
vor der Abfüllung durchgeführte Tätigkeiten (siehe Nummer 30)h.wichtige Parameter, die erforderlich sind, um eine korrekte Abfüllung bei Standardbedingungen zu belegen; i.die Ergebnisse von Überprüfungen, die die Füllung der Gasflaschen/mobilen Flüssiggasbehälter belegen; j.ein Muster des Chargenetiketts; k.Spezifikation des Endprodukts und Ergebnisse der Qualitätskontrollen (einschließlich der Bezugnahme auf den Kalibrierungsstatus der Testausrüstung);
l.
Anzahl der zurückgewiesenen Gasflaschen/mobilen Flüssiggasbehälter mit individuellen Angaben zur Identifizierung und den Gründen für die Zurückweisung;
m.
eine detaillierte Auflistung aller Probleme und ungewöhnlichen Vorkommnisse sowie eine Auflistung aller Abweichungen von den Füllanweisungen mit der Unterschrift der Person, die die Abweichungen genehmigt hat;
n.
Freigabebestätigung durch die Sachkundige Person mit Datum und Unterschrift.o.
18. Protokolle sollten für jede Gascharge, die zur Auslieferung in die Tanks von Krankenhäusern vorgesehen ist, aufbewahrt werden. Diese Protokolle sollten, soweit zutreffend, Folgendes enthalten (die aufzuzeichnenden Einzelheiten können je nach lokaler Gesetzgebung variieren):
Produktname; a.Chargenbezeichnung; b.Angaben zur Identifizierung des Tanks (Tankfahrzeugs), für den die Charge zertifiziert wurde; c.Datum und Zeitpunkt der Befüllung; d.Identifizierung der Person(en), die die Befüllung des Tanks (Tankfahrzeugs) vorgenommen hat; e.Referenz zu dem beliefernden Tankfahrzeug (Tank), Referenz zum Ausgangsgas, soweit angebracht; f.Relevante Einzelheiten bezüglich der Befüllungsmaßnahme; g.Spezifikation des Endprodukts und Ergebnisse der Qualitätskontrollen (einschließlich der Bezugnahme auf den Kalibrierungsstatus der Testausrüstung);
h.
eine detaillierte Auflistung aller Probleme und ungewöhnlichen Vorkommnisse sowie eine Auflistung aller Abweichungen von den Füllanweisungen mit der Unterschrift der Person, die die Abweichungen genehmigt hat;
i.
Freigabebestätigung durch die Sachkundige Person mit Datum und Unterschrift.j.
ProduktionTransfer und Auslieferungen von kryogenem und verflüssigtem Gas19. Die Überführung des kryogenen oder verflüssigten Gases aus dem Primärlager, einschließlich der vorausgehenden Kontrollen, sollte nach schriftlichen Anweisungen durchgeführt werden, damit jegliche Kontamination vermieden wird. Die Transferleitungen sollten mit einem Rückschlagventil oder einer geeigneten Alternative ausgestattet sein. Die flexiblen Verbindungen, Verbindungsschläuche und Verbindungsstücke sollten vor ihrem Einsatz mit dem betreffenden Gas durchgespült werden.20. Die 'Transferschläuche zum Befüllen von 'Tanks und 'Tankfahrzeugen sollten mit produktspezifischen Verbindungsstücken ausgerüstet sein. Die Verwendung von Verbindungsstücken, die die Verbindung von Tanks und Tankfahrzeugen ermöglichen, die nicht für das gleiche Gas vorgesehen sind, sollte ausreichend kontrolliert werden.21. Gaslieferungen können in Lagertanks eingefüllt werden, die schon das gleiche Gas enthalten, vorausgesetzt dass die Ergebnisse der Untersuchung einer Probe belegen, dass die Qualität des gelieferten Gases akzeptabel ist. Diese Probe kann entnommen werden vor der Einfüllung des angelieferten Gases aus der Lieferung oder nach der Einfüllung in den Lagertank.Anmerkung: Siehe spezifische Regelungen in Nummer 42 zur Befüllung von Tanks beim Empfänger.Füllung und Etikettierung der Gasflaschen und mobilen Flüssiggasbehälter22. Die Chargen(n) eines Gases/von Gasen sollte(n) vor der Befüllung von Gasflachen und mobilen Flüssiggasbehältern festgelegt, entsprechend ihrer Spezifikationen geprüft und zum Abfüllen freigegeben sein.23. Bei kontinuierlichem Betrieb wie unter Grundsätze beschrieben, sollten adäquate Inprozesskontrollen durchgeführt werden, um sicherzustellen, dass das Gas den Spezifikationen entspricht.24. Gasflaschen, mobile Flüssiggasbehälter und Ventile sollten den einschlägigen technischen Spezifikationen und den Zulassungen entsprechen. Sie sollten einem einzigen medizinischen Gas zugeordnet werden oder einer festgelegten Gasmischung. Gasflaschen sollten mit einer Farbkodierung entsprechend relevanter Standards versehen sein. Sie sollten vorzugsweise mit einem Minimalrestdruckventil mit Rückströmverhinderung ausgerüstet sein, um einen angemessenen Kontaminationsschutz zu gewährleisten.25. Gasflaschen, mobile Flüssiggasbehälter und Ventile sollten vor ihrem ersten Gebrauch in der Produktion überprüft und ausreichend gewartet werden. Sofern GE-gekennzeichnete Medizinprodukte verwendet werden, sollte die Wartung die Anleitungen des Medizinprodukteherstellers beachten.26. Überprüfungen und Wartungsarbeiten sollten nicht die Qualität und Sicherheit des Arzneimittels beeinträchtigen. Wasser für die Durchführung der Wasserdruckprüfung sollte zumindest Trinkwasserqualität haben.27. Als Teil der Überprüfungen und Wartungsarbeiten sollten die Gasflaschen vor dem Einbau des Ventils einer Sichtkontrolle unterzogen werden, um sicher zu stellen, dass sie nicht mit Wasser oder anderen Verunreinigungen kontaminiert sind. Dies sollte erfolgen,
wenn die Gasflaschen neu sind und erstmalig für medizinische Gase verwendet werden; •wenn eine Wasserdruck- oder ähnliche Prüfung durchgeführt wurde, bei der das Ventil entfernt wurde; •immer wenn das Ventil entfernt wurde.•
Das Ventil sollte nach dem Anbringen geschlossen bleiben, um durch die Flaschenöffnung eintretende Verunreinigungen zu vermeiden.28. Wartungs- und Reparaturarbeiten von Gasflaschen, mobilen Flüssiggasbehältern und Ventilen erfolgen unter der Verantwortung des Arzneimittelherstellers. Sofern diese durch Subunternehmen erfolgen, sollten sie nur durch genehmigte Subunternehmen durchgeführt werden; dafür sollten Verträge einschließlich der technischen Vereinbarungen erstellt werden. Die Subunternehmer sollten auditiert werden, um sicherzustellen, dass angemessene Standards aufrechterhalten werden.
Seite 55 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

29. Die Rückverfolgbarkeit von Gasflaschen, mobilen Flüssiggasbehältern und Ventilen sollte mit Hilfe eines entsprechenden Systems möglich sein.30. Vor Abfüllung sollten folgende Prüfungen durchgeführt werden:
bei Gasflaschen sollte eine nach festgelegten Verfahren durchgeführte Prüfung sicherstellen, dass ein positiver Restdruck in jeder Flasche vorhanden ist;
a.
wenn die Gasflasche mit einem Minimalrestdruckventil ausgerüstet ist und kein Anzeichen für einen Restdruck vorhanden ist, sollte das fehlerfreie Funktionieren des Ventils überprüft werden, und falls das Ventil nicht ausreichend funktioniert, sollte die Gasflasche zur Wartung gegeben werden,
◦
wenn die Gasflasche nicht mit einem Minimalrestdruckventil ausgestattet ist und kein Anzeichen für einen Restdruck vorhanden ist, sollte die Gasflasche für weitere Maßnahmen aussortiert werden, um sicherzustellen, dass sie nicht durch Wasser oder andere Verunreinigungen kontaminiert ist; die zusätzlichen Maßnahmen könnten in visuellen Überprüfungen, gefolgt von Reinigungsmaßnahmen nach validierten Verfahren, bestehen;
◦
Kontrolle, dass alle früheren Chargenetiketten entfernt wurden; b.Kontrolle, dass alle beschädigten Produktetiketten entfernt und ersetzt wurden; c.visuelle äußere Kontrolle jeder Gasflasche, jedes mobilen Flüssiggasbehälters und Ventils auf Beulen, Schweißschäden, Ablagerungen oder andere Schäden sowie auf Verunreinigungen durch Öl oder Schmiermittel; eine Reinigung sollte durchgeführt werden, sofern erforderlich;
d.
Kontrolle des Ventilanschlusses jeder Flasche oder jedes mobilen Flüssiggasbehälters darauf, ob es sich um den richtigen Anschluss für das jeweilige medizinische Gas handelt;
e.
Kontrolle des Datums des nächsten anstehenden Tests der Ventile (im Falle von Ventilen, die regelmäßig zu testen sind); f.Überprüfung der Gasflasche oder des mobilen Flüssiggasbehälters darauf, dass Tests, die durch nationale oder internationale Regelungen erforderlich sind (z.B. ob die Wasserdruck- oder eine gleichwertige Prüfung), durchgeführt wurden und noch ihre Gültigkeit besitzen; und
g.
Kontrolle, ob jeder Behälter mit der erforderlichen Farbmarkierung gemäß den Zulassungsunterlagen (Farbmarkierung nach nationalen/internationalen Standards) versehen ist.
h.
31. Eine Charge sollte für die Abfüllung definiert werden.32. Flaschen, die zum Nachfüllen zurückgegeben wurden, sollten in Einklang mit den Zulassungsunterlagen sorgfältig vorbehandelt werden, um Verunreinigungen zu vermeiden. Diese Maßnahmen, die Entleerung und/oder Leerströmen einschließen, sollten validiert sein.Anmerkung: Bei komprimierten Gasen sollte eine maximale theoretische Verunreinigung von 500 ppm v/v bei einem Fülldruck von 200 bar (und äquivalent für andere Fülldrücke) nicht überschritten werden.33. Mobile Flüssiggasbehälter, die zum Nachfüllen zurückgegeben wurden, sollten in Einklang mit den Zulassungsunterlagen sorgfältig vorbehandelt werden, um Verunreinigungen zu vermeiden. Insbesondere sollten mobile Flüssiggasbehälter ohne Restdruck nach validierten Verfahren vorbereitet werden.34. Es sollten geeignete Kontrollen durchgeführt werden, um sicherzustellen, dass jede Gasflasche/jeder mobile Flüssiggasbehälter ordnungsgemäß gefüllt wurden.35. Jede gefüllte Gasflasche sollte mit Hilfe eines geeigneten Verfahrens vor Anbringen des Originalitätsverschlusses auf Lecks kontrolliert werden (siehe Nummer 36). Durch die Testmethode sollte keine Kontamination des inneren Ventilauslasses erfolgen. Wenn Probenahmen und Untersuchungen erforderlich sind, sollten diese vor dem Lecksuchtest abgeschlossen werden.36. Nach dem Befüllen sollten Gasflaschen und mobile Flüssiggasbehälter abgedeckt werden, um die Auslassventile vor Kontaminationen zu schützen. Gasflaschen und mobile Flüssiggasbehälter sollten mit Originalverschlüssen versehen werden.37. Jede Gasflasche oder jeder mobile Flüssiggasbehälter sollte gekennzeichnet werden. Die Chargennummer und das Verfalldatum können sich auf einem gesonderten Etikett befinden.38. Bei medizinischen Gasen, die durch Mischen zweier oder mehrerer verschiedener Gase (inline vor dem Befüllen oder direkt innerhalb der Gasflasche) hergestellt werden, sollte der Mischprozess der Gase validiert sein, um sicherzustellen, dass die Gase ordnungsgemäß gemischt in jeder Gasflasche vorliegen und dass die Mischung homogen ist.Qualitätskontrolle39. Jede Charge eines medizinischen Gases (Gasflaschen, mobile Flüssiggasbehälter, Krankenhaustank) sollte in Übereinstimmung mit den Anforderungen der Zulassung getestet werden und zertifiziert sein.40. Soweit nicht anderweitig in der Zulassung festgelegt, sollten der Probenahmeplan und die durchzuführenden Analysen im Falle der Gasflaschen mit den folgenden Anforderungen übereinstimmen:
Bei Abfüllung eines einzelnen medizinischen Gases über eine Mehr-Flaschen-Abfüllstation sollte immer dann, wenn die Flaschen am Füllstand gewechselt werden, mindestens eine Flasche aus jeder Abfüllung auf Identität geprüft werden.
a.
Bei der Befüllung von einer Flasche nach der anderen mit einem einzelnen medizinischen Gas, sollte mindestens eine Flasche eines nicht unterbrochenen Abfüllvorgangs auf Identität und Gehalt geprüft werden. Ein Beispiel eines nicht unterbrochenen Abfüllvorgangs ist die Produktion innerhalb einer Arbeitsschicht mit demselben Personal, derselben Ausstattung und derselben Bulkgas-Charge.
b.
Bei medizinischen Gasen, die durch Mischen zweier oder mehrerer verschiedener Gase in einer Flasche auf derselben Abfüllstation hergestellt werden, sollte jede Gasflasche auf Identität und Gehalt aller Gaskomponenten geprüft werden. Hilfsstoffe, sofern vorhanden, können auf Identität aus einer Gasflasche per Abfüllzyklus (oder bei nicht unterbrochenen Abfüllvorgängen bei einer nacheinander erfolgenden Flaschenabfüllung) durchgeführt werden. Weniger Gasflaschen können getestet werden im Falle eines validierten automatisierten Abfüllsystems.
c.
Wenn Gase vor der Abfüllung inline gemischt werden, ist eine kontinuierliche Analyse des abzufüllenden Gemisches erforderlich. Die vorgemischten Gase sollten den gleichen Vorgaben folgen wie die medizinischen Gase, die in den Gasflaschen gemischt werden und keine kontinuierliche Analyse des abzufüllenden Gemisches erfolgt.
d.
Die Testung auf Wassergehalt sollte durchgeführt werden, soweit nicht davon abgesehen werden kann.Andere Probenahmen und Testverfahren, die mindestens einen vergleichbaren Qualitätssicherungsgrad haben, können gerechtfertigt sein.
Seite 56 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

41. Soweit in der Zulassung nichts anders bestimmt ist, sollte die Endkontrolle einen Test auf Identität und Gehalt jedes mobilen Flüssiggasbehälters vorsehen. Die chargenweise Testung sollte nur durchgeführt werden, wenn bewiesen wurde, dass die kritischen Eigenschaften des Gases, das in jedem Behältnis vor dem Wiederbefüllen zurückbleibt, gewahrt wurden.42. Aus Flüssiggasbehältern, die bei Kunden (Krankenhaustanks oder Heim-Flüssigkeitsbehälter) installiert sind und bei diesen mit Hilfe von nur für diesen Zweck genutzten mobilen Liefertanks nachgefüllt werden, brauchen nach der Füllung keine Proben entnommen zu werden, wenn ein Analysenzertifikat über den Inhalt des Tankfahrzeugs die Lieferung begleitet. Es solle jedoch gezeigt werden, dass die Spezifikation des Gases im beim Kunden installierten Flüssiggasbehälter während der aufeinander folgenden wiederholten Befüllungen aufrechterhalten bleibt.43. Referenz- und Rückstellmuster sind nicht erforderlich, soweit nicht anderweitig festgelegt.44. Kontinuierliche Stabilitätsstudien sind nicht erforderlich, wenn Anfangsstabilitätsstudien durch bibliographische Daten ersetzt wurden (siehe Note for Guidance CPMP/QWP/1719/00).Transport verpackter Gase45. Befüllte Gasflaschen und Heim-Flüssiggasbehälter sollten während des Transports geschützt werden, insbesondere sollten sie in sauberem Zustand und entsprechend der Umgebungsbedingungen, in denen sie eingesetzt werden sollen, an die Kunden ausgeliefert werden.GlossarAbfüllstationAusrüstung oder Apparatur, die so konstruiert ist, dass ein oder mehrere Gasbehälter gleichzeitig entleert und wieder gefüllt werden können.BehälterEin Behälter ist ein Flüssiggasbehälter (ein Tank, ein Tankfahrzeug oder ein anderer Typ eines mobilen Flüssiggaskessels), eine Gasflasche, ein Flaschenbündel oder jede andere Verpackung, die direkt mit dem Gas in Berührung kommt.EvakuierenEntfernung der Gasreste aus einem Behälter/System bei einem niedrigeren Druck als 1,013 bar durch Anlegen eines Vakuums.FlaschenbündelMehrere Gasflaschen, die in einem Gestell befestigt und durch Leitungen miteinander verbunden sind. Sie werden als eine Einheit behandelt und transportiert.GasJede Substanz, die bei 1,013 bar und +20 °C vollständig gasförmig ist oder bei + 50 °C einen Dampfdruck von mehr als 3 bar hat.GasflascheEin üblicherweise zylinderartiges Behältnis, das sich für komprimierte, verflüssigte oder gelöste Gase eignet und mit einer Einrichtung zur Regulierung des spontanen Gasaustritts bei atmosphärischem Druck und Raumtemperatur ausgestattet ist.Gas-WirkstoffJedes Gas, das als Wirkstoff für die Arzneimittelherstellung vorgesehen ist.Heim-FlüssiggasbehälterMobiler Flüssiggasbehälter zur Aufbewahrung von flüssigem Sauerstoff, der zuhause beim Patienten gasförmigen Sauerstoff abgibt.Komprimiertes GasEin Gas, das im Behälter unter Druck bei Temperaturen über -50 °C vollständig gasförmig ist.Kyrogenes GasEin Gas, das bei 1,013 bar und bei einer Temperatur von unter -150 °C als Flüssiggas vorliegt.LeerströmenEntfernen des Restgases aus einem Behälter/System durch Öffnen des Behälters/Systems bis zum Erreichen des atmosphärischen Drucks.LuftzerlegungTrennung atmosphärischer Luft in ihre Gasbestandteile durch fraktionierte Destillation bei Tieftemperaturen.Maximale theoretische VerunreinigungGasförmige Verunreinigung, resultierend aus dem Eindringen von Fremdbestandteilen, die trotz Vorbehandlung vor dem Abfüllvorgang bestehen bleibt. Die Schätzung der maximalen theoretischen Verunreinigung ist nur für komprimierte Gase von Bedeutung und setzt voraus, dass sich diese Gase wie ideale Gase verhalten.Medizinisches GasGas oder Gasgemisch, das als Arzneimittel eingestuft wird (entsprechend der Richtlinie 2001/83/EG und 2002/83/EG ).MinimalrestdruckventilVentil, das einen positiven Druck über dem atmosphärischen Druck in einer Gasflasche nach ihrem Gebrauch aufrechterhält, um eine Kontamination der Gasflasche zu verhindern.Mobiler FlüssiggasbehälterEin mobiler thermisch isolierter Behälter, in dem verflüssigte oder kyrogene Gase aufbewahrt werden können. Im Anhang beinhaltet dies nicht die Tankfahrzeuge.RückschlagventilVentil, das ein Strömen in nur eine Richtung erlaubt.SpülenEntfernen von Restgasen einer Gasflasche/eines Systems durch zunächst Leerströmen und Evakuieren und dann teilweises Füllen mit dem entsprechenden Gas zum Spülen bei 1,013 bar.TankEin stationärer temperaturisolierter Behälter für die Lagerung verflüssigter oder kryogener Gase. Sie werden auch als "stationäre kryogene Kessel" bezeichnet.TankfahrzeugIm Sinne dieses Anhangs ein Fahrzeug, auf dem ein temperaturisolierter Behälter befestigt ist, der für den Transport verflüssigter oder kryogener Gase verwendet wird.VentilVorrichtung zum Öffnen und Schließen von Behältern. Verflüssigtes GasEin Gas, das im Transportbehälter teilweise flüssig (oder fest) ist bei einer Temperatur oberhalb -50 °C.
Seite 57 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi
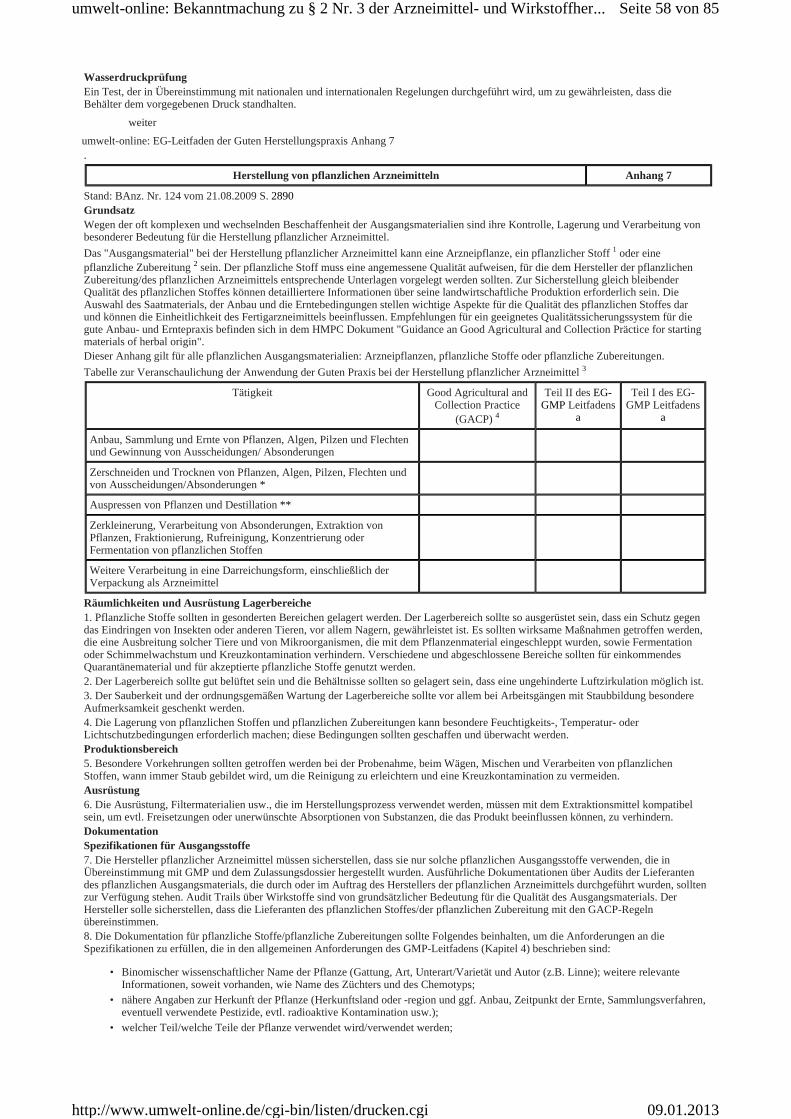
WasserdruckprüfungEin Test, der in Übereinstimmung mit nationalen und internationalen Regelungen durchgeführt wird, um zu gewährleisten, dass die Behälter dem vorgegebenen Druck standhalten.
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 7 .
Herstellung von pflanzlichen Arzneimitteln Anhang 7
Stand: BAnz. Nr. 124 vom 21.08.2009 S. 2890GrundsatzWegen der oft komplexen und wechselnden Beschaffenheit der Ausgangsmaterialien sind ihre Kontrolle, Lagerung und Verarbeitung von besonderer Bedeutung für die Herstellung pflanzlicher Arzneimittel.
Das "Ausgangsmaterial" bei der Herstellung pflanzlicher Arzneimittel kann eine Arzneipflanze, ein pflanzlicher Stoff 1 oder eine pflanzliche Zubereitung 2 sein. Der pflanzliche Stoff muss eine angemessene Qualität aufweisen, für die dem Hersteller der pflanzlichen Zubereitung/des pflanzlichen Arzneimittels entsprechende Unterlagen vorgelegt werden sollten. Zur Sicherstellung gleich bleibender Qualität des pflanzlichen Stoffes können detailliertere Informationen über seine landwirtschaftliche Produktion erforderlich sein. Die Auswahl des Saatmaterials, der Anbau und die Erntebedingungen stellen wichtige Aspekte für die Qualität des pflanzlichen Stoffes dar und können die Einheitlichkeit des Fertigarzneimittels beeinflussen. Empfehlungen für ein geeignetes Qualitätssicherungssystem für die gute Anbau- und Erntepraxis befinden sich in dem HMPC Dokument "Guidance an Good Agricultural and Collection Präctice for starting materials of herbal origin". Dieser Anhang gilt für alle pflanzlichen Ausgangsmaterialien: Arzneipflanzen, pflanzliche Stoffe oder pflanzliche Zubereitungen.
Tabelle zur Veranschaulichung der Anwendung der Guten Praxis bei der Herstellung pflanzlicher Arzneimittel 3
Tätigkeit Good Agricultural and Collection Practice
(GACP) 4
Teil II des EG-GMP Leitfadens
a
Teil I des EG-GMP Leitfadens
a
Anbau, Sammlung und Ernte von Pflanzen, Algen, Pilzen und Flechten und Gewinnung von Ausscheidungen/ Absonderungen
Zerschneiden und Trocknen von Pflanzen, Algen, Pilzen, Flechten und von Ausscheidungen/Absonderungen *
Auspressen von Pflanzen und Destillation **
Zerkleinerung, Verarbeitung von Absonderungen, Extraktion von Pflanzen, Fraktionierung, Rufreinigung, Konzentrierung oder Fermentation von pflanzlichen Stoffen
Weitere Verarbeitung in eine Darreichungsform, einschließlich der Verpackung als Arzneimittel
Räumlichkeiten und Ausrüstung Lagerbereiche1. Pflanzliche Stoffe sollten in gesonderten Bereichen gelagert werden. Der Lagerbereich sollte so ausgerüstet sein, dass ein Schutz gegen das Eindringen von Insekten oder anderen Tieren, vor allem Nagern, gewährleistet ist. Es sollten wirksame Maßnahmen getroffen werden, die eine Ausbreitung solcher Tiere und von Mikroorganismen, die mit dem Pflanzenmaterial eingeschleppt wurden, sowie Fermentation oder Schimmelwachstum und Kreuzkontamination verhindern. Verschiedene und abgeschlossene Bereiche sollten für einkommendes Quarantänematerial und für akzeptierte pflanzliche Stoffe genutzt werden. 2. Der Lagerbereich sollte gut belüftet sein und die Behältnisse sollten so gelagert sein, dass eine ungehinderte Luftzirkulation möglich ist. 3. Der Sauberkeit und der ordnungsgemäßen Wartung der Lagerbereiche sollte vor allem bei Arbeitsgängen mit Staubbildung besondere Aufmerksamkeit geschenkt werden. 4. Die Lagerung von pflanzlichen Stoffen und pflanzlichen Zubereitungen kann besondere Feuchtigkeits-, Temperatur- oder Lichtschutzbedingungen erforderlich machen; diese Bedingungen sollten geschaffen und überwacht werden. Produktionsbereich5. Besondere Vorkehrungen sollten getroffen werden bei der Probenahme, beim Wägen, Mischen und Verarbeiten von pflanzlichen Stoffen, wann immer Staub gebildet wird, um die Reinigung zu erleichtern und eine Kreuzkontamination zu vermeiden. Ausrüstung6. Die Ausrüstung, Filtermaterialien usw., die im Herstellungsprozess verwendet werden, müssen mit dem Extraktionsmittel kompatibel sein, um evtl. Freisetzungen oder unerwünschte Absorptionen von Substanzen, die das Produkt beeinflussen können, zu verhindern. DokumentationSpezifikationen für Ausgangsstoffe7. Die Hersteller pflanzlicher Arzneimittel müssen sicherstellen, dass sie nur solche pflanzlichen Ausgangsstoffe verwenden, die in Übereinstimmung mit GMP und dem Zulassungsdossier hergestellt wurden. Ausführliche Dokumentationen über Audits der Lieferanten des pflanzlichen Ausgangsmaterials, die durch oder im Auftrag des Herstellers der pflanzlichen Arzneimittels durchgeführt wurden, sollten zur Verfügung stehen. Audit Trails über Wirkstoffe sind von grundsätzlicher Bedeutung für die Qualität des Ausgangsmaterials. Der Hersteller solle sicherstellen, dass die Lieferanten des pflanzlichen Stoffes/der pflanzlichen Zubereitung mit den GACP-Regeln übereinstimmen. 8. Die Dokumentation für pflanzliche Stoffe/pflanzliche Zubereitungen sollte Folgendes beinhalten, um die Anforderungen an die Spezifikationen zu erfüllen, die in den allgemeinen Anforderungen des GMP-Leitfadens (Kapitel 4) beschrieben sind:
Binomischer wissenschaftlicher Name der Pflanze (Gattung, Art, Unterart/Varietät und Autor (z.B. Linne); weitere relevante Informationen, soweit vorhanden, wie Name des Züchters und des Chemotyps;
•
nähere Angaben zur Herkunft der Pflanze (Herkunftsland oder -region und ggf. Anbau, Zeitpunkt der Ernte, Sammlungsverfahren, eventuell verwendete Pestizide, evtl. radioaktive Kontamination usw.);
•
welcher Teil/welche Teile der Pflanze verwendet wird/verwendet werden;•
Seite 58 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

bei Verwendung von getrocknetem Pflanzenmaterial Angabe des verwendeten Trocknungsverfahrens;•Beschreibung des pflanzlichen Stoffes und seiner makround mikroskopischen Untersuchung;•geeignete Identitätsprüfungen, darunter ggf. solche auf bekannte wirksame Bestandteile oder Leitsubstanzen. Spezifische Tests zur Unterscheidung sind erforderlich, wenn damit zu rechnen ist, dass der pflanzliche Stoff gefälscht/ ersetzt wird. Für die Identifizierungszwecke sollte eine authentische Referenzprobe zur Verfügung stehen;
•
Wassergehalt der pflanzlichen Stoffe, der in Übereinstimmung mit dem Europäischen Arzneibuch bestimmt wird;•Gehaltsbestimmungen von Bestandteilen mit bekannter therapeutischer Wirksamkeit oder, sofern angezeigt, von Leitsubstanzen; geeignete Methoden zur Bestimmung einer möglichen Pestizidkontamination und dafür festgesetzte Grenzwerte in Übereinstimmung mit den Methoden des Europäischen Arzneibuchs, oder, sofern nicht vorhanden, mit einer geeigneten validierten Methode, sofern nichts anderes bestimmt wird;
•
Tests zur Bestimmung einer Pilzkontamination und/oder einer mikrobiellen Kontamination, einschließlich Aflatoxine und anderer Pilztoxine, Schädlingsbefall, und, soweit angezeigt, dafür festgesetzte Grenzwerte;
•
Prüfungen auf toxische Metalle und mögliche Verunreinigungen sowie Verfälschungen, soweit angebracht;•Prüfungen auf fremde Bestandteile, soweit angebracht;•Weitere zusätzliche Tests nach dem allgemeinen Monographien des Europäischen Arzneibuchs oder nach speziellen Monographien pflanzlicher Stoffe, soweit angebracht.
•
Jede Behandlung zur Verminderung der Pilzkontamination, der mikrobiellen Kontamination oder eines anderen Befalls sollte dokumentiert werden. Die Spezifikationen für diese Verfahren sollten verfügbar sein und die Details des Vorgehens, die Tests und die Grenzwerte für die Rückstände enthalten. Verarbeitungsanweisungen9. Die Verarbeitungsanweisungen sollten die verschiedenen an dem pflanzlichen Stoff vorgenommenen Bearbeitungsschritte beschreiben, wie das Säubern, Trocknen, Zerkleinern und Sieben, einschließlich der Trocknungsdauer und -temperaturen, sowie die zur Größenbestimmung von Fragmenten oder Partikeln verwendeten Methoden. 10. Insbesondere sollten schriftliche Anweisungen und Protokolle vorhanden sein, die sicherstellen, dass jeder Behälter mit pflanzlichen Stoffen sorgfältig untersucht wurde, um Fälschungen/Austausch oder Anwesenheit fremder Materialien, wie Metall oder Glasstücke, Tierteile oder -exkremente, Steine, Sand usw. oder Fäulnis und Anzeichen von Zersetzung festzustellen. 11. Die Verarbeitungsanweisungen sollten auch Sieb- oder andere Sicherheitsverfahren zur Entfernung von Fremdbestandteilen sowie geeignete Verfahren zur Reinigung/Selektion von Pflanzenmaterial vor der Lagerung des akzeptierten pflanzlichen Stoffes oder vor Beginn der Herstellung beschreiben. 12. Die Anweisungen zur Herstellung der pflanzlichen Zubereitung sollten Angaben zum Lösungsmittel, zur Extraktionszeit und -temperatur sowie zu allen Konzentrierungsschritten und -verfahren enthalten. Qualitätskontrolle Probenahme13. Da das unbehandelte Pflanzenmaterial/die pflanzlichen Stoffe von Natur aus eine gewisse Heterogenität aufweisen, ist die Probenahme hier mit besonderer Sorgfalt und von besonders ausgebildetem Personal vorzunehmen. jede Charge sollte durch eine eigene Dokumentation identifizierbar sein. 14. Eine Referenzprobe des Pflanzenmaterials ist erforderlich, insbesondere in solchen Fällen, in denen der pflanzliche Stoff nicht im Europäischen Arzneibuch oder in dem Arzneibuch eines anderen Mitgliedstaates beschrieben ist. Proben ungemahlenen Pflanzenmaterials sind erforderlich, wenn Pulver verwendet wird. 15. Das in der Qualitätskontrolle beschäftigte Personal sollte über besondere Sachkenntnisse und Erfahrungen auf dem Gebiet pflanzlicher Stoffe, pflanzlicher Zubereitungen und/oder pflanzlicher Arzneimittel verfügen, um Identitätsprüfungen durchführen und Fälschungen, Pilz- und Schädlingsbefall sowie mangelnde Gleichförmigkeit innerhalb einer Rohmateriallieferung usw. erkennen zu können. 16. Die Identität und Qualität von pflanzlichen Stoffen, pflanzlichen Zubereitungen und pflanzlichen Arzneimitteln sollten entsprechend der Beschreibung in den Europäischen Leitlinien zu Qualität und Spezifikationen pflanzlicher und traditionell pflanzlicher Arzneimittel und, soweit zutreffend, spezieller Monographien des Europäischen Arzneibuchs festgestellt werden. ____________ 1) Die Begriffe pflanzliche Stoffe und pflanzliche Zubereitungen der Richtlinie 2004/24/EG werden als gleichbedeutend mit den Begriffen des Europäischen Arzneibuchs pflanzliche Arzneimittel und pflanzliche Arzneizubereitungen gesehen. 2) Für diesen Anhang und soweit nicht anders bestimmt, beinhaltet der Begriff "pflanzliche Arzneimittel/Zubereitungen" die "traditionellen pflanzlichen Arzneimittel/Zubereitungen". 3) Diese Tabelle erweitert und detailliert den Abschnitt über pflanzliche Stoffe in der Tabelle 1 in Teil II des EG-GMP Leitfadens. 4) EMEA/HMPC/2468 16/2005 vom 20. Februar 2005. a) Erläuterungen Die GMP-Klassifizierung des pflanzlichen Materials hängt von dessen Verwendung durch den Inhaber der Herstellungserlaubnis ab. Das Material kann als Wirkstoff, als Zwischenprodukt oder als Arzneimittel klassifiziert werden. Es obliegt der Verantwortung des Arzneimittelherstellers, die geeignete GMP-Klassifizierung sicherzustellen. * ) Die Hersteller sollten sicherstellen, dass diese Schritte in Übereinstimmung mit der Zulassung/Registrierung durchgeführt werden. Für solche initialen Schritte, die entsprechend der Zulassung/Registrierung auf dem Feld vorgenommen werden, sind die Standards der "Good Agricultural and Collection practice for starting materials of herbal origin (GACP)" anwendbar. GMP gilt für die weiteren Zerkleinerungs- und Trocknungsschritte. ** ) Es ist akzeptabel, dass das Auspressen der Pflanzen und die Destillation auf dem Feld durchgeführt werden, soweit diese Aktivitäten zur Aufrechterhaltung der Produktqualität innerhalb der genehmigten Spezifikationen als integrierter Teil des Erntevorgangs durchgeführt werden müssen und die Anbaubedingungen in Übereinstimmung mit GACP durchgeführt werden. Diese Umstände sollten als Ausnahme gelten und von der entsprechenden Zulassung/Registrierung erfasst sein. Für die auf dem Feld durchgeführten Aktivitäten sollten eine geeignete Dokumentation, Kontrolle und Validierung gemäß der GMP-Grundsätze sichergestellt werden. Die Behörden können bezüglich dieser Aktivitäten GMP-Inspektionen zur Feststellung der Compliance durchführen.
weiter
Bekanntmachung zu § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung - AMWHV Vom 16. Juli 2009
(BAnz. Nr. 124 vom 21.08.2009 S. 2890) Die Europäische Kommission hat die Anhänge 3 und 7 zum Leitfaden für die Gute Herstellungspraxis für Arzneimittel und Prüfpräparate (EG-GMP Leitfaden) geändert und am 1. September 2008 in englischer Sprache auf ihrer Internetseite veröffentlicht. Anhang 3 ist seit dem 1. März 2009, Anhang 7 ab dem 1. September 2009 anzuwenden.
Seite 59 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Hiermit werden die vom Bundesministerium für Gesundheit in die deutsche Sprache übersetzten Anhänge 3 und 7 zum EG-GMP Leitfaden nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung vom 3. November 2006 (BGBl. I S. 2523), die zuletzt durch Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung vom 26. März 2008 (BGBl. I S. 521) geändert worden ist, im Anschluss an die Bekanntmachung vom 18. Juli 2008 (BAnz. S. 2798) bekannt gemacht. Der Leitfaden und die nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung bekannt gemachten Anhänge sind auch auf der Internetseite des Bundesministeriums für Gesundheit, www.bmg.bund.de, abrufbar. umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 11 .
Computergestützte Systeme Anhang 1111 zum EG-Leitfaden der
Guten Herstellungspraxis *
Rechtsgrundlage zur Veröffentlichung dieses Leitfadens:Artikel 47 der Richtlinie 2001/83/EG zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel und Artikel 51 der Richtlinie 2001/82/EG zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel. Dieses Dokument bietet eine Anleitung für die Auslegung der Grundsätze und Leitlinien der Guten Herstellungspraxis (GMP) für Arzneimittel entsprechend der Richtlinie 2003/94/EG für Humanarzneimittel und der Richtlinie 91/412/EWG für Tierarzneimittel.Status des Dokuments:Revision 1Grund der Änderung:Der Anhang wurde als Reaktion auf den verstärkten Einsatz computergestützter Systeme und die zunehmende Komplexität dieser Systeme überarbeitet. In der Folge wurden auch für Kapitel 4 des GMP-Leitfadens Änderungen vorgeschlagen.Termin des Inkrafttretens:30. Juni 2011GrundsätzeDer vorliegende Anhang gilt für alle Arten computergestützter Systeme, die als Bestandteil von GMP-pflichtigen Vorgängen eingesetzt werden. Ein computergestütztes System ist eine Kombination aus Software- und Hardwarekomponenten, die zusammen bestimmte Funktionen erfüllen.Die Anwendung sollte validiert, die IT-Infrastruktur sollte qualifiziert sein.Wird eine manuelle Tätigkeit durch ein computergestütztes System ersetzt, darf es in der Folge nicht zu einer Beeinträchtigung der Produktqualität, der Prozesskontrolle oder der Qualitätssicherung kommen. Dabei darf sich das Gesamtrisiko des Prozesses nicht erhöhen.Allgemeines1 RisikomanagementEin Risikomanagement sollte über den gesamten Lebenszyklus des computergestützten Systems unter Berücksichtigung von Patientensicherheit, Datenintegrität und Produktqualität betrieben werden. Als Teil eines Risikomanagementsystems sollten Entscheidungen über den Umfang der Validierung und die Sicherstellung der Datenintegrität auf einer begründeten und dokumentierten Risikobewertung des computergestützten Systems basieren.2 PersonalEs sollte eine enge Zusammenarbeit zwischen maßgeblichen Personen, wie z.B. Prozesseignern, Systemeignern und Sachkundigen Personen sowie der IT stattfinden. Alle Personen sollten über eine geeignete Ausbildung und Zugriffsrechte sowie festgelegte Verantwortlichkeiten zur Wahrnehmung der ihnen übertragenen Aufgaben verfügen.3 Lieferanten und Dienstleister3.1 Werden Dritte (z.B. Lieferanten, Dienstleister) herangezogen, um z.B. ein computergestütztes System bereitzustellen, zu installieren, zu konfigurieren, zu integrieren, zu validieren, zu warten (z.B. Fernwartung), zu modifizieren oder zu erhalten, Daten zu verarbeiten oder im Zusammenhang stehende Serviceleistungen zu erbringen, müssen formale Vereinbarungen abgeschlossen sein, in denen die Verantwortlichkeiten des Dritten eindeutig beschrieben sind. IT-Abteilungen sollten analog zu Dritten behandelt werden.3.2 Kompetenz und Zuverlässigkeit des Lieferanten sind Schlüsselfaktoren bei der Auswahl eines Produktes oder eines Dienstleisters. Die Notwendigkeit eines Audits sollte auf einer Risikobewertung basieren.3.3 Die bei kommerziell erhältlichen Standardprodukten bereitgestellte Dokumentation sollte durch Nutzer im regulierten Umfeld dahingehend überprüft werden, ob die Benutzeranforderungen erfüllt sind.3.4 Die Informationen zum Qualitätssystem und zu Audits, die Lieferanten oder Entwickler von Software und verwendeten Systemen betreffen, sollten Inspektoren auf Nachfrage zur Verfügung gestellt werden.Projektphase4 Validierung4.1 Die Validierungsdokumentation und -berichte sollten die maßgeblichen Phasen des Lebenszyklus abbilden. Hersteller sollten in der Lage sein, ihre Standards, Pläne, Akzeptanzkriterien, Vorgehensweisen und Aufzeichnungen basierend auf ihrer Risikobewertung zu begründen.4.2 Die Validierungsdokumentation sollte, sofern zutreffend, Aufzeichnungen im Rahmen der Änderungskontrolle und Berichte über alle während der Validierung beobachteten Abweichungen beinhalten.4.3 Eine aktuelle Liste aller maßgeblichen Systeme und ihrer GMP-Funktionen (Inventar) sollte zur Verfügung stehen.Für kritische Systeme sollte eine aktuelle Systembeschreibung vorliegen, welche die technische und logische Anordnung, den Datenfluss sowie Schnittstellen zu anderen Systemen oder Prozessen, sämtliche Hard- und Softwarevoraussetzungen und die Sicherheitsmaßnahmen detailliert wiedergibt.4.4 Die Benutzeranforderungen sollten die erforderlichen Funktionen des computergestützten Systems beschreiben und auf einer dokumentierten Risikobewertung sowie einer Betrachtung der möglichen Auswirkungen auf das GMP-System basieren. Die Benutzeranforderungen sollten über den Lebenszyklus des Systems verfolgbar sein.4.5 Der Nutzer im regulierten Umfeld sollte alle erforderlichen Maßnahmen ergreifen, um sicherzustellen, dass das System in Übereinstimmung mit einem geeigneten Qualitätsmanagementsystem entwickelt wurde. Der Lieferant sollte angemessen bewertet werden.4.6 Für die Validierung maßgeschneiderter Systeme oder für den Kunden spezifisch angepasster computergestützter Systeme sollte ein Verfahren vorliegen, das die formelle Bewertung und Berichterstellung zu Qualitäts- und Leistungsmerkmalen während aller Abschnitte des Lebenszyklus des Systems gewährleistet.
Seite 60 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

4.7 Die Eignung von Testmethoden und Testszenarien sollte nachgewiesen werden. Insbesondere Grenzwerte für System-/ Prozessparameter, Datengrenzen und die Fehlerbehandlung sollten betrachtet werden. Für automatisierte Testwerkzeuge und Testumgebungen sollte eine dokumentierte Bewertung ihrer Eignung vorliegen.4.8 Werden Daten in ein anderes Datenformat oder System überführt, sollte im Rahmen der Validierung geprüft werden, dass der Wert und die Bedeutung der Daten im Rahmen dieses Migrationsprozesses nicht verändert werden.Betriebsphase5 DatenUm Risiken zu minimieren sollten computergestützte Systeme, die Daten elektronisch mit anderen Systemen austauschen, geeignete Kontrollmechanismen für die korrekte und sichere Eingabe und Verarbeitung der Daten enthalten.6 Prüfung auf RichtigkeitWerden kritische Daten manuell eingegeben, sollte die Richtigkeit dieser Dateneingabe durch eine zusätzliche Prüfung abgesichert werden. Diese zusätzliche Prüfung kann durch einen zweiten Anwender oder mit Hilfe einer validierten elektronischen Methode erfolgen. Die Kritikalität und möglichen Folgen fehlerhafter oder inkorrekt eingegebener Daten für das System sollten im Risikomanagement berücksichtigt sein.7 Datenspeicherung7.1 Daten sollten durch physikalische und elektronische Maßnahmen vor Beschädigung geschützt werden. Die Verfügbarkeit, Lesbarkeit und Richtigkeit gespeicherter Daten sollten geprüft werden. Der Zugriff auf Daten sollte während des gesamten Aufbewahrungszeitraums gewährleistet sein.7.2 Es sollten regelmäßige Sicherungskopien aller maßgeblichen Daten erstellt werden. Die Integrität und Richtigkeit der gesicherten Daten sowie die Möglichkeit der Datenwiederherstellung sollten während der Validierung geprüft und regelmäßig überwacht werden.8 Ausdrucke8.1 Es sollte möglich sein, klar verständliche Kopien von elektronisch gespeicherten Daten zu erhalten.8.2 Von Protokollen, die zur Chargenfreigabe herangezogen werden, sollten Ausdrucke generiert werden können, die eine Veränderung der Daten nach ihrer Ersteingabe erkennen lassen.9 Audit TrailsBasierend auf einer Risikobewertung sollte erwogen werden, die Aufzeichnung aller GMP-relevanten Änderungen und Löschungen in das System zu integrieren (ein systemgenerierter "Audit Trail"). Bei der Änderung oder Löschung GMP-relevanter Daten sollte der Grund dokumentiert werden. Audit Trails müssen verfügbar sein, in eine allgemein lesbare Form überführt werden können und regelmäßig überprüft werden.10 Änderungs- und KonfigurationsmanagementJede Änderung an einem computergestützten System einschließlich der Systemkonfigurationen sollte kontrolliert und nach einem festgelegten Verfahren erfolgen.11 Periodische EvaluierungComputergestützte Systeme sollten periodisch evaluiert werden, um zu bestätigen, dass sie sich noch im validen Zustand befinden und die GMP-Anforderungen erfüllen. Solche Evaluierungen sollten, sofern sachgerecht, den derzeitigen Funktionsumfang, Abweichungsaufzeichnungen, Vorfälle, Probleme, Aktualisierungen, Leistung, Zuverlässigkeit, Sicherheit und Berichte zum Validierungsstatus umfassen.12 Sicherheit12.1 Es sollten physikalische und/oder logische Maßnahmen implementiert sein, um den Zugang zu computergestützten Systemen auf autorisierte Personen zu beschränken. Geeignete Maßnahmen zur Vermeidung unerlaubten Systemzugangs können die Verwendung von Schlüsseln, Kennkarten, persönlichen Codes mit Kennworten, biometrische Verfahren sowie den eingeschränkten Zugang zu Computern mit zugehöriger Ausrüstung und Datenspeicherungsbereichen einschließen.12.2 Der Umfang der Sicherheitsmaßnahmen ist von der Kritikalität des computergestützten Systems abhängig.12.3 Erteilung, Änderung und Entzug von Zugriffsberechtigungen sollten aufgezeichnet werden.12.4 Systeme zur Verwaltung von Daten und Dokumenten sollten die Identität des Anwenders, der Daten eingibt, ändert, bestätigt oder löscht, mit Datum und Uhrzeit aufzeichnen.13 VorfallmanagementAlle Vorfälle, nicht nur Systemausfälle und Datenfehler, sollten berichtet und bewertet werden. Die Ursache eines kritischen Vorfalls sollte ermittelt werden und die Basis für Korrektur- und Vorbeugemaßnahmen sein.14 Elektronische UnterschriftElektronische Aufzeichnungen können elektronisch signiert werden. Von elektronischen Unterschriften wird erwartet, dass sie
im Innenverhältnis eines Unternehmens die gleiche Bedeutung haben wie handschriftliche Signaturen,a.dauerhaft mit dem zugehörigen Dokument verbunden sind,b.die Angabe des Datums und der Uhrzeit der Signatur beinhalten.c.
15 ChargenfreigabeWird ein computergestütztes System zur Aufzeichnung der Chorgenzertifizierung und -freigabe eingesetzt, sollte durch das System sichergestellt werden, dass nur Sachkundige Personen die Chargenfreigabe zertifizieren können. Das System sollte diese Personen eindeutig identifizieren und die Identität der zertifizierenden oder freigebenden Person dokumentieren. Eine elektronische Chargenzertifizierung oder -freigabe sollte mittels elektronischer Unterschrift erfolgen.16 Kontinuität des GeschäftsbetriebesWenn computergestützte Systeme kritische Prozesse unterstützen, sollten Vorkehrungen getroffen sein, um die fortlaufende Unterstützung dieser Prozesse im Falle eines Systemausfalls sicherzustellen (z.B. durch ein manuelles oder ein alternatives System). Der erforderliche Zeitaufwand zur Inbetriebnahme dieser alternativen Verfahren sollte jeweils für ein bestimmtes System und die unterstützten Prozesse risikoabhängig festgelegt werden. Diese Verfahren sollten angemessen dokumentiert und getestet werden.17 ArchivierungDaten können archiviert werden. Diese Daten sollten auf Verfügbarkeit, Lesbarkeit und Integrität geprüft werden. Sind maßgebliche Änderungen am System erforderlich (z.B. Computer und zugehörige Ausrüstung oder Programme), sollte sichergestellt und getestet werden, ob die Daten weiterhin abrufbar sind.GlossarAnwendung:
Seite 61 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Software, die auf einer definierten Plattform/Hardware installiert ist und spezifische Funktionen bietet.Dritter:Nicht direkt vom Inhaber der Herstellungs- oder Einfuhrerlaubnis geführte Einrichtung.IT-Infrastruktur:Hardware und Software wie Netzwerksoftware und Betriebssysteme, die für die Funktionsfähigkeit der Anwendung erforderlich sind.Kommerziell erhältliche Standardsoftware:Software die kommerziell verfügbar ist und deren Eignung für den vorgesehenen Zweck durch ein breites Spektrum von Anwendern belegt ist.Kundenspezifische/für den Kunden spezifisch angepasste computergestützte Systeme:Ein computergestütztes System angepasst an einen spezifischen Geschäftsprozess.Lebenszyklus:Alle Phasen der Systemlebensdauer von den initialen Anforderungen bis zur Stillegung einschließlich Design, Spezifikation, Programmierung, Testung, Installation, Betrieb und Wartung. Prozesseigner:Die für den Geschäftsprozess verantwortliche Person. Systemeigner:Die für die Verfügbarkeit und Wartung eines computergestützten Systems und die Sicherheit der auf dem System gespeicherten Daten verantwortliche Person.
weiter
Bekanntmachung zu § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV )Vom 8. August 2011
(BAnz. Nr. 125 vom 19.08.2011 S. 2901)Die Europäische Kommission hat den Anhang 11 zum Leitfaden für die Gute Herstellungspraxis für Arzneimittel- und Prüfpräparate (EG-GMP Leitfaden) geändert und im Januar 2011 in englischer Sprache auf ihrer Internetseite veröffentlicht. Darüber hinaus hat die Kommission auch Kapitel 4 des EG-GMP Leitfadens in Zusammenhang mit der Änderung des Anhangs 11 geändert, um dem steigenden Einsatz elektronischer Dokumente im GMP-Bereich Rechnung zu tragen. Der neue Anhang 11 und das überarbeitete Kapitel 4 des EG-GMP Leitfadens sind ab dem 30. Juni 2011 anzuwenden.Hiermit werden der vom Bundesministerium für Gesundheit in die deutsche Sprache übersetzte Anhang 11 und das Kapitel 4 des EG-GMP Leitfadens nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung vom 3. November 2006 (BGBl. I S. 2523), die zuletzt durch Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung vom 26. März 2008 (BGBl. I S. 521) geändert worden ist, bekannt gemacht (Anlage 1 und 2).Der Leitfaden und die nach § 2 Nummer 3 der Arzneimittel- und Wirkstoffherstellungsverordnung bekannt gemachten Anhänge sind auch auf der Internetseite des Bundesministeriums für Gesundheit, www.bmg.bund.de, abrufbar.umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 14 .
Herstellung von Arzneimitteln aus menschlichem Blut oder Blutplasma Anhang 14 12 des EU-Leitfadens für die Gute Herstellungspraxis
Rechtsgrundlage zur Veröffentlichung der Leitfäden:Artikel 47 der Richtlinie 2001/83/EG zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel und Artikel 51 der Richtlinie 2001/82/EG zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel. Dieses Dokument gibt eine Anleitung für die Auslegung der Grundsätze und Leitlinien der Guten Herstellungspraxis (GMP) für Arzneimittel entsprechend der Richtlinie 2003/94/EG für Humanarzneimittel und der Richtlinie 91/412/EWG für Tierarzneimittel.Status des Dokuments:Revision 1Grund der Änderungen:Der Anhang wurde vor dem Hintergrund der Richtlinie 2002/98/EG und ihrer Durchführungsrichtlinien überarbeitet, die Standards zur Qualität und Sicherheit bei der Gewinnung und Testung von menschlichem Blut und Blutkomponenten für jeglichen Gebrauch festlegen, einschließlich der Herstellung von Arzneimitteln.Termin des Inkrafttretens:30. November 2011
Herstellung von Arzneimitteln aus menschlichem Blut oder PlasmaGlossarBlutBlut im Sinne der Richtlinie 2002/98/EG (Artikel 3a) ist Vollblut, das von einem Spender gewonnen und aufgearbeitet wird, entweder zur Transfusion oder zur weiteren Verarbeitung.BlutbestandteilBlutbestandteil im Sinne der Richtlinie 2002/98/EG (Artikel 3b) ist ein therapeutischer Bestandteil des Blutes (rote oder weiße Blutkörperchen, Blutplättchen und Plasma), die durch verschiedene Methoden erhalten werden können.BlutspendeeinrichtungBlutspendeeinrichtung im Sinne der Richtlinie 2002/98/EG (Artikel 3e) ist eine Struktur oder Stelle, die für einen beliebigen Teilaspekt der Gewinnung und Testung von menschlichem Blut oder Blutbestandteilen unabhängig von deren Verwendungszweck, und für deren Verarbeitung, Lagerung und Verteilung, sofern diese zur Transfusion bestimmt sind, zuständig ist. Obwohl diese Definition Krankenhausblutdepots nicht einschließt, besteht Einvernehmen darüber, dass Einrichtungen, in denen Plasmapherese stattfindet, eingeschlossen sind.BlutprodukteEin Blutprodukt im Sinne der Richtlinie 2002/98/EG (Artikel 3c) ist ein aus menschlichem Blut oder Plasma gewonnenes therapeutisches Produkt.Fraktionierung, Fraktionierungsbetrieb
Seite 62 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Fraktionierung ist der Herstellungsprozess in einem Betrieb (Fraktionierungsbetrieb), in dem die Plasmabestandteile aufgetrennt/gereinigt werden durch verschiedene physikalische und chemische Methoden, wie z.B. Präzipitation, Chromatographie.Leitlinien der guten fachlichen PraxisLeitlinien der guten fachlichen Praxis dienen zur Auslegung der Standards und Spezifikationen der Gemeinschaftsnormen für Qualitätssysteme in Blutspendeeinrichtungen, die im Anhang der Richtlinie 2005/62/EG 1 festgelegt sind.Arzneimittel aus menschlichem Blut oder menschlichem PlasmaArzneimittel aus menschlichem Blut oder Blutplasma im Sinne der Richtlinie 2001/83/EG (Artikel 1 Nummer 10) sind auf Blutbestandteile zurückgehende Arzneimittel, die industriell durch öffentliche oder private Einrichtungen hergestellt werden.Plasma zur Fraktionierung Plasma zur Fraktionierung ist der flüssige Anteil menschlichen Blutes, der nach Abtrennung der zellulären Bestandteile des in einem Behältnis mit Antikoagulantien versetzten Blutes zurückbleibt oder der durch kontinuierliche Filtration oder Zentrifugation des mit Antikoagulantien versetzten Blutes durch Apherese abgetrennt wird; es ist zur Herstellung von auf Plasma zurückgehende Arzneimittel vorgesehen, insbesondere Albumin, Gerinnungsfaktoren und Immunglobuline menschlicher Herkunft, und näher beschrieben in der Monographie des Europäischen Arzneibuchs "Menschliches Plasma zur Fraktionierung" (0853).Plasma-Stammdokumentation (Plasma Master File, PMF)Eine Plasma-Stammdokumentation, wie in Richtlinie 2001/83/EG (Anhang I Teil III Nummer 1.1.a) bezeichnet, ist ein eigenständiges Dokument, separat vom Zulassungsdossier. Es enthält alle maßgeblichen ausführlichen Angaben über die Eigenschaften des gesamten menschlichen Plasmas, das als Ausgangsmaterial und/ oder Rohstoff für die Herstellung von Sub-/Zwischenfraktionen, Hilfsstoff- und Wirkstoffbestandteilen verwendet wird, die Teil von aus Plasma hergestellten Arzneimitteln oder Medizinprodukten sind.Verarbeitung (Processing)Gemäß der Terminologie der Richtlinie 2005/62/EG ist "Verarbeitung" jeder Schritt bei der Herstellung eines Blutbestandteils, der zwischen der Gewinnung von Blut und der Bereitstellung eines Blutbestandteils durchgeführt wird, z.B. Abtrennung und Einfrieren von Blutkomponenten. In diesem Anhang bezieht sich "Verarbeitung" auch auf solche Arbeitsvorgänge, die in der Blutspendeeinrichtung durchgeführt werden und spezifisch für Plasma zur Fraktionierung sind.Sachkundige Person (QP)Die Sachkundige Person ist die Person der Richtlinie 2001/83/EG (Artikel 48).Verantwortliche Person (RP)Die Verantwortliche Person ist die Person der Richtlinie 2002/98/EG (Artikel 9).Vertragliches Drittland-FraktionierungsprogrammDies ist ein vertraglich festgelegtes Fraktionierungsprogramm, das bei einem Fraktionierer/Hersteller in der EU/dem Europäischen Wirtschaftsraum durchgeführt wird mit Plasma aus einem Drittland, wobei die daraus hergestellten Produkte nicht für den Markt der EU/des Europäischen Wirtschaftsraums vorgesehen sind.1 Anwendungsbereich1.1 Die Regelungen dieses Anhangs gelten für Arzneimittel aus menschlichem Blut oder Plasma, das in der EU/dem Europäischen Wirtschaftsraum fraktioniert oder dorthin eingeführt wird. Der Anhang gilt auch für das Ausgangsmaterial (z.B. menschliches Plasma) dieser Produkte. In Übereinstimmung mit den in der Richtlinie 2002/63/EG festgelegten Bedingungen gelten die Anforderungen auch für stabile Derivate menschlichen Blutes oder Plasmas (z.B. Albumin), das in Medizinprodukten enthalten ist.1.2 Dieser Anhang legt spezifische Anforderungen der guten Herstellungspraxis fest für die Verarbeitung, Lagerung und den Transport von menschlichem Plasma zur Fraktionierung und für die Herstellung von Arzneimitteln aus menschlichem Blut oder Plasma.1.3 Dieser Anhang behandelt spezifische Fragestellungen, wenn Ausgangsmaterial aus Drittländern eingeführt wird und für Vertragliche Drittland-Fraktionierungsprogramme.1.4 Dieser Anhang gilt nicht für Blutbestandteile, die zur Transfusion bestimmt sind.2 Grundsätze2.1 Arzneimittel aus menschlichem Blut oder Plasma (und ihre Wirkstoffe, die als Ausgangsmaterial eingesetzt werden) müssen den Grundsätzen und Leitlinien der Guten Herstellungspraxis entsprechen (wie sie in der Richtlinie 2003/94/EG der Kommission und den EG-GMP-Leitlinien festgelegt sind, die von der Europäischen Kommission veröffentlicht wurden), sowie den entsprechenden Zulassungen (Richtlinie 2001/83/EG, Artikel 46, 51). Sie werden als biologische Produkte angesehen und das Ausgangsmaterial beinhaltet biologische Substanzen wie Zellen oder Flüssigkeiten (einschließlich Blut oder Plasma) menschlichen Ursprungs (Richtlinie 2001/83/EG , Anhang I Teil I Nummer 3.2.1.1.b). Bestimmte spezielle Besonderheiten entstehen aus den biologischen Eigenschaften des Ausgangsmaterials. Z.B. krankheitsübertragende Agenzien, insbesondere Viren, können das Ausgangsmaterial kontaminieren. Die Qualität und Sicherheit dieser Produkte stützt sich daher auf die Kontrolle der Ausgangsmaterialien und deren Ursprung, ebenso wie auf die nachfolgenden Herstellungsprozesse, einschließlich der Testung auf Infektionsmarker, der Virusentfernung und der Virusinaktivierung.2.2 Grundsätzlich müssen die als Ausgangsmaterial eingesetzten Wirkstoffe den Grundsätzen und Leitlinien der Guten Herstellungspraxis entsprechen (siehe 2.1). Für das Ausgangsmaterial aus menschlichem Blut und Plasma müssen die in der Richtlinie 2002/98/EG festgelegten Anforderungen an die Gewinnung und Testung eingehalten werden. Die Gewinnung und Testung müssen in Übereinstimmung mit einem angemessenen Qualitätssystem durchgeführt werden, für das Standards und Spezifikationen im Anhang der Richtlinie 2005/62/EG festgelegt sind und ausgelegt werden in den Leitlinien zur guten fachlichen Praxis, auf die Artikel 2 (2) der Richtlinie 2005/62/EG verweist. Darüber hinaus gelten die Anforderungen der Richtlinie 2005/61/EG an die Rückverfolgbarkeit vom Spender zum Empfänger und die Meldung ernster Zwischenfälle und ernster unerwünschter Reaktionen. Zusätzlich müssen die Monographien des Europäischen Arzneibuchs beachtet werden (Richtlinie 2001/83/EG , Anhang I Teil III Nummer 1.1.b).2.3 Ausgangsmaterial zur Herstellung von Arzneimitteln aus menschlichem Blut oder Plasma, das aus Drittländern importiert wird zur Verwendung oder zum Vertrieb in der EU/dem Europäischen Wirtschaftsraum, muss Standards, die denen der Gemeinschaft vergleichbar sind, die Spezifikationen bezogen auf ein Qualitätssystem für Blutspendeeinrichtungen, wie sie in der Richtlinie 2005/62/EG (Erwägungsgrund 6, Artikel 2(3) festgelegt sind, die Anforderungen an die Rückverfolgbarkeit und die Meldung ernster Zwischenfälle und ernster unerwünschter Reaktionen der Richtlinie 2005/61/EG (Erwägungsgrund 5, Artikel 7) und die technischen Anforderungen der Richtlinie 2004/33/EG (Erwägungsgrund 4, Punkt 2.3 des Anhangs V) erfüllen.2.4 Im Falle der vertraglichen Drittland-Fraktionierungsprogramme muss das aus Drittländern importierte Plasma den Anforderungen an die Qualität und Sicherheit entsprechen, die in der Richtlinie 2002/98/EG und in Anhang V der Richtlinie 2004/33/EG festgelegt sind. Die Tätigkeiten innerhalb der EU/ dem Europäischen Wirtschaftsraum müssen vollständig mit GMP in Einklang stehen. Die von der Gemeinschaft in der Richtlinie 2005/62/EG festgelegten Standards und Spezifikationen bezüglich des Qualitätssystems für Blutspendeeinrichtungen, die Anforderungen an die Rückverfolgbarkeit und die Meldung ernster Zwischenfälle und ernster unerwünschter Reaktionen der Richtlinie 2005/61/EG und die relevanten WHO-Leitlinien und Empfehlungen sollten berücksichtigt werden.
Seite 63 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

2.5 Für alle auf die Entnahme und Testung folgenden Schritte (z.B. Verarbeitung (einschließlich der Abtrennung), Einfrieren, Lagerung und Transport zum Hersteller) gelten die Anforderungen der Richtlinie 2001/83/EG und müssen daher in Übereinstimmung mit der Guten Herstellungspraxis erfolgen. Normalerweise würden diese Tätigkeiten unter der Verantwortung der Sachkundigen Person in einer Einrichtung mit Herstellungserlaubnis ausgeführt werden. Soweit spezifische Verarbeitungsschritte bezüglich des Plasmas zur Fraktionierung in einer Blutspendeeinrichtung erfolgen, könnte jedoch eine spezielle Bestellung einer Sachkundigen Person wegen der Anwesenheit und Verantwortlichkeit einer Verantwortlichen Person nicht angemessen erscheinen. Um dieser besonderen Situation Rechnung zu tragen und um sicherzustellen, dass die gesetzliche Verantwortung der Sachkundigen Person angemessen berücksichtigt wird, sollte der Fraktionierer/Hersteller mit der Blutspendeeinrichtung einen Vertrag in Übereinstimmung mit Kapitel 7 des GMP-Leitfadens abschließen, in dem die jeweiligen Verantwortlichkeiten und die detaillierten Anforderungen festgelegt sind, um die Regelbefolgung sicherzustellen. Die Verantwortliche Person der Blutspendeeinrichtung und die Sachkundige Person des Fraktionierers/Herstellers (siehe Nummer 3.5) sollten in dieVertragsabfassung einbezogen werden. Die Sachkundige Person sollte sicherstellen, dass Audits durchgeführt werden, mit denen bestätigt wird, dass die Blutspendeeinrichtung mit den vertraglichen Festlegungen übereinstimmt.2.6 Spezielle Anforderungen an die Dokumentation und an andere Vorkehrungen bezogen auf das Ausgangsmaterial der Arzneimittel aus Plasma sind in der Plasma-Stammdokumentation festgelegt.3 Qualitätsmanagement3.1 Das Qualitätsmanagement sollte alle Schritte abdecken, von der Spenderauswahl bis zur Auslieferung des Fertigprodukts. Auf die Richtlinie 2005/61/EG zur Rückverfolgbarkeit einschließlich der Auslieferung des Plasmas an den Fraktionierer wird verwiesen und auf die Richtlinie 2006/62/EG für alle Schritte betreffend die Gewinnung und Testung des menschlichen Bluts und Plasmas für die Herstellung von Arzneimitteln.3.2 Blut oder Plasma als Ausgangsmaterial zur Herstellung von Arzneimitteln muss in Blutspendeeinrichtungen gewonnen und in Laboratorien getestet werden, die ein Qualitätssystem anwenden, das der Richtlinie 2005/62/EG entspricht und die von einer national zuständigen Behörde genehmigt wurden und regelmäßigen Inspektionen gemäß der Richtlinie 2002/98/EG unterliegen. Vertragliche Drittland-Fraktionierungsprogramme müssen vom Hersteller der zuständigen EU-Behörde entsprechend der Richtlinie 2001/83/EG angezeigt werden.3.3 Wenn Plasma aus Drittländern importiert wird, sollte es nur von genehmigten Lieferanten (z.B. Blutspendeeinrichtungen, einschließlich externer Lagerhäuser) erworben werden. Sie sollten in den vom Fraktionierer/Hersteller festgelegten Spezifikationen der Ausgangsmaterialien genannt und von der zuständigen Behörde der EU/des Europäischen Wirtschaftsraums (z.B. in Folge einer Inspektion) sowie von der Sachkundigen Person des Fraktionierers in der EU/dem Europäischen Wirtschaftsraum akzeptiert sein. Die Zertifizierung und Freigabe des Plasmas (Plasma zur Fraktionierung) als Ausgangsmaterial wird in Nummer 6.8 beschrieben.3.4 Die Qualifizierung der Lieferanten, einschließlich der Audits, sollte durch den Fraktionierer/Hersteller des Fertigprodukts nach schriftlichen Verfahren durchgeführt werden. Requalifizierungen der Lieferanten sollten in festgelegten Intervallen durchgeführt werden, unter Berücksichtigung einer risikobasierten Herangehensweise.3.5 Der Fraktionierer/Hersteller des Fertigprodukts sollte mit den Blutspendeeinrichtungen schriftliche Verträge abschließen. Mindestens die folgenden grundlegenden Aspekte sollten dabei berücksichtig werden:
Festlegung der Pflichten und entsprechenden Verantwortlichkeiten•Qualitätssystem und Dokumentationsanforderungen•Spenderauswahlkriterien und -testung•Anforderungen an die Auftrennung des Blutes in Blutkomponenten/-plasma•Einfrieren des Plasmas•Lagerung und Transport des Plasmas•Rückverfolgbarkeit und Maßnahmen nach der Entnahme (einschließlich ernster Zwischenfälle).•
Die Testergebnisse aller von der Blutspendeinrichtung gelieferten Plasmaeinheiten sollten dem Fraktionierer/Hersteller des Arzneimittels zur Verfügung stehen. Außerdem sollte jeder Fraktionierungsschritt, der durch Subunternehmen erfolgt, in einem schriftlichen Vertrag festgelegt werden.3.6 Es sollte ein formales Änderungsmanagementsystem vorhanden sein für die Planung, Auswertung und Dokumentation aller Änderungen, die die Qualität oder Sicherheit der Produkte oder deren Rückverfolgbarkeit beeinflussen könnten.3.7 Es sollte eine angemessene Strategie vorhanden sein, um das Risiko infektiöser Agenzien und entstehender infektiöser Agenzien zu minimieren. Diese Strategie sollte eine Risikoabschätzung einbeziehen, die
einen Zeitrahmen für das Vorhalten eines Lagerbestands festlegt (interner Quarantänezeitraum) vor der Verarbeitung des Plasma, z.B. um Look back-Einheiten herauszusuchen 2
•
alle Aspekte der Virusreduktion und/oder der Testung auf infektiöse Agenzien oder Surrogate beachtet•die Virusreduktionsmöglichkeiten beachtet, die Poolgröße und andere relevante Aspekte des Herstellungsprozesses.
•
4 Rückverfolgbarkeit und Maßnahmen nach der Entnahme4.1 Es muss ein System vorhanden sein, mit dem jede Spende rückverfolgt werden kann, vom Spender und der Spende über die Blutspendeeinrichtung bis zur Charge des Arzneimittels und umgekehrt.4.2 Verantwortlichkeiten für die Rückverfolgbarkeit sollten festgelegt werden (es sollten keine Lücken vorhanden sein):
vom Spender und der Spende in der Blutspendeeinrichtung zum Fraktionierungsbetrieb (dies ist die Verantwortung der Verantwortlichen Person in der Blutspendeeinrichtung)
•
vom Fraktionierungsbetrieb zum Hersteller des Arzneimittels und jeder anderen nachgeordneten Firma, ob Arzneimittelhersteller oder Hersteller eines Medizinproduktes (das ist die Verantwortung der Sachkundigen Person).
•
4.3 Daten, die zur vollständigen Rückverfolgbarkeit benötigt werden, müssen entsprechend Artikel 4 der Richtlinie 2005/61/EG und Artikel 14 der Richtlinie 2002/98/EG 3 mindestens 30 Jahre aufbewahrt werden.4.4 Die Verträge (wie in Nummer 3.5 aufgeführt) zwischen der Blutspendeeinrichtung (einschließlich der Testlaboratorien) und dem Fraktionierungsbetrieb/Hersteller sollten sicherstellen, dass die Rückverfolgbarkeit und Maßnahmen nach der Spende die vollständige Kette abdecken von der Gewinnung des Plasmas bis zu allen Herstellern, die für die Freigabe der Fertigprodukte verantwortlich sind.
Seite 64 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

4.5 Die Blutspendeeinrichtungen sollten dem Fraktionierungsbetrieb/Hersteller jeden Vorfall anzeigen, der die Qualität oder Sicherheit des Produkts beeinflussen könnte, einschließlich der in Anhang II Teil A und Anhang III Teil A der Richtlinie 2005/61/EG aufgeführten Vorfälle, sowie anderer relevanter Informationen, die im Anschluss an die Spenderakzeptanz oder die Freigabe des Plasmas gefunden wurden, z.B. Look back-Informationen 4. Wenn der Fraktionierungsbetrieb/Hersteller in einem Drittland ansässig ist, sollte die Information an die für die Freigabe in der EU/im Europäischen Wirtschaftsraum jeglicher daraus hergestellter Produkte verantwortlichen Hersteller gehen. In beiden Fällen sollte die Information, soweit diese für die Qualität und Sicherheit der Endprodukte relevant sind, auch an die für den Fraktionierungsbetrieb/Hersteller zuständige Behörde 5 gehen.4.6 Die Anzeigepflicht wie in Nummer 4.5 beschrieben gilt auch dann, wenn eine Inspektion einer Blutspendeeinrichtung durch eine zuständige Behörde zu einer Rücknahme einer bestehenden Lizenz/eines Zertifikats oder einer Genehmigung geführt hat.4.7 Die Handhabung der Information nach der Entnahme sollte in einer Verfahrensbeschreibung festgelegt sein und Verpflichtungen und Verfahren zur Information der zuständigen Behörde mit berücksichtigen. Informationen nach der Entnahme sollten verfügbar sein entsprechend der aktuellen Fassung der "Note for Guidance on Plasma Derived Medicinal Products", die vom Ausschuss für Arzneimittel (Committee for Medicinal Products for Human Use, CHMP) angenommen und von der Europäischen Arzneimittelagentur veröffentlicht wurde 6.5 Räume und Einrichtungen5.1. Um die mikrobiologische Kontamination oder das Einbringen fremder Materialien in den Plasmapool zu minimieren, sollte das Auftauen und Poolen von Plasma in einer Umgebung erfolgen, die mindestens den Grad D-Anforderungen entspricht, die in Anhang 1 des EG-GMP-Leitfadens definiert sind. Geeignete Schutzkleidung, einschließlich Gesichtsmasken und Handschuhe, sollten getragen werden. Alle anderen offenen Handhabungen während des Herstellungsprozesses sollten gemäß den entsprechenden Bedingungen des Anhangs 1 zum EG-GMP-Leitfaden durchgeführt werden.5.2 Die Überwachung der Umgebungsbedingungen sollte regelmäßig in Übereinstimmung mit Anhang 1 des EG-GMP-Leitfadens durchgeführt werden, insbesondere während des Öffnens der Plasmabehältnisse und während des anschließenden Prozesses des Auftauens und Poolens. Es sollten Akzeptanzgrenzwerte festgelegt werden.5.3 Während der Produktion von aus Plasma hergestellten Arzneimitteln werden geeignete Virusinaktivierungs- oder Virusentfernungsmaßnahmen eingesetzt und die erforderlichen Schritte sollten vorgenommen werden zur Verhinderung von Kreuzkontaminationen behandelter Produkte mit unbehandelten Produkten. Speziell dafür vorgesehene und eigene Räume und Einrichtungen sollten für Herstellungsschritte nach der Virusinaktivierungsmaßnahme eingesetzt werden.5.4 Um die Routineherstellung vor dem Risiko einer Kontamination mit Viren zu schützen, die während der Validierungsstudien eingesetzt werden, sollte die Validierung der Methoden zur Virusreduktion nicht in den Herstellungsgebäuden durchgeführt werden. Die Validierung sollte durchgeführt werden gemäß der "Note for Guidance on Virus Validation Studies: The Design, Contribution and Interpretation of Studies validating the Inactivation and Removal of Viruses" in ihrer aktuellen Version, wie sie vom Ausschuss für Arzneimittel (Committee for Medicinal Products for Human Use, CHMP) angenommen und von der Europäischen Arzneimittelagentur veröffentlicht wurde 7.6 Herstellung Ausgangsmaterial6.1 Das Ausgangsmaterial sollte allen Anforderungen aller relevanten Monographien des Europäischen Arzneibuchs und den Bedingungen, die in dem jeweiligen Dossier der Zulassung, einschließlich der Plasma-Stammdokumentation, niedergelegt sind, entsprechen. Diese Anforderungen sollten in dem schriftlichen Vertrag (siehe Nummer 3.5) zwischen der Blutspendeeinrichtung und dem Fraktionierungsbetrieb/Hersteller festgelegt sein und durch das Qualitätssystem kontrolliert werden.6.2 Das Ausgangsmaterial für ein vertragliches Drittland-Fraktionierungsprogramm sollte den in Nummer 2.4 festgelegten Anforderungen entsprechen.6.3 Abhängig von der Art der Gewinnung (z.B. entweder Vollblutentnahme oder automatische Apherese) können unterschiedliche Verarbeitungsschritte notwendig sein. Alle Verarbeitungsschritte (z.B. Zentrifugation und/oder Auftrennung, Probeentnahme, Kennzeichnung, Einfrieren) sollten in schriftlichen Verfahren festgelegt sein.6.4 Jede Verwechslung der Plasmaeinheiten und der Proben, insbesondere während der Kennzeichnung, sowie jede Kontamination, z.B. beim Abschneiden der Schlauchabschnitte/ Versiegeln der Behältnisse, muss vermieden werden.6.5 Das Einfrieren ist ein kritischer Schritt für die Gewinnung von Proteinen, die im Plasma labil sind, z.B. die Gerinnungsfaktoren. Das Einfrieren muss daher so schnell wie möglich nach der Gewinnung nach validierten Verfahren erfolgen (siehe Monographie Nummer 0853 des Europäischen Arzneibuchs "Humanplasma zur Fraktionierung" und soweit relevant, Monographie Nummer 1646 "Plasma vom Menschen [gepoolt und virusinaktiviert]").6.6 Die Lagerung und der Transport von Blut oder Plasma sollten auf jeder Stufe in der Transportkette zum Fraktionierungsbetrieb festgelegt und aufgezeichnet werden. Jede Abweichung von der festgelegten Temperatur sollte dem Fraktionierungsbetrieb angezeigt werden. Es sollten qualifizierte Ausrüstung und validierte Verfahren eingesetzt werden.Zertifizierung/Freigabe von Plasma zur Fraktionierung als Ausgangsmaterial6.7 Plasma zur Fraktionierung sollte, z.B. vom Quarantänestatus, nur durch Systeme und Verfahren freigegeben werden, die die Qualität sicherstellen, wie sie vom Hersteller des Fertigproduktes benötigt wird. Es sollte nur zum Fraktionierungsbetrieb/Hersteller ausgeliefert werden, nachdem die Verantwortliche Person (oder im Falle der Blut-/Plasmagewinnung in Drittstaaten eine Person mit vergleichbaren Verantwortlichkeiten und Qualifikationen) dokumentiert hat, dass das Plasma zur Fraktionierung den in schriftlichen Verträgen festgelegten Anforderungen und Spezifikationen entspricht und dass alle Schritte in Übereinstimmung mit der Guten fachlichen Praxis und den entsprechenden GMP-Leitlinien durchgeführt wurden.6.8 Nach Eingang im Fraktionierungsbetrieb sollten die Plasmaeinheiten unter der Verantwortung der Sachkundigen Person für die Fraktionierung freigegeben werden. Die Sachkundige Person sollte bestätigen, dass das Plasma den Anforderungen aller relevanten Monographien und den im entsprechenden Zulassungsdossier festgelegten Anforderungen, einschließlich der Plasma-Stammdokumentation, oder im Falle des Vertraglichen Drittland-Fraktionierungsprogramms den Anforderungen unter Nummer 2.4 entspricht.Verarbeitung von Plasma zur Fraktionierung6.9 Die Schritte im Fraktionierungsprozess variieren je nach Produkt und Hersteller und schließen üblicherweise verschiedene Fraktionierungs-/Reinigungsverfahren ein, von denen einige zur Inaktivierung und/oder Entfernung potentieller Verunreinigungen beitragen können.6.10 Anforderungen an die Prozesse des Poolens, der Poolprobenahme und der Fraktionierung/Reinigung und Virusinaktivierung und/oder -entfernung sollten definiert und sorgfältig eingehalten werden.6.11 Die im Virusinaktivierungsprozess verwendeten Methoden sollten unter strikter Einhaltung der validierten Verfahren und in Übereinstimmung mit den in den Validierungsstudien verwendeten Verfahren vorgenommen werden. Es sollten detaillierte Untersuchungen von Fehlern in den Virusinaktivierungsprozessen vorgenommen werden. Die Einhaltung des validierten
Seite 65 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi
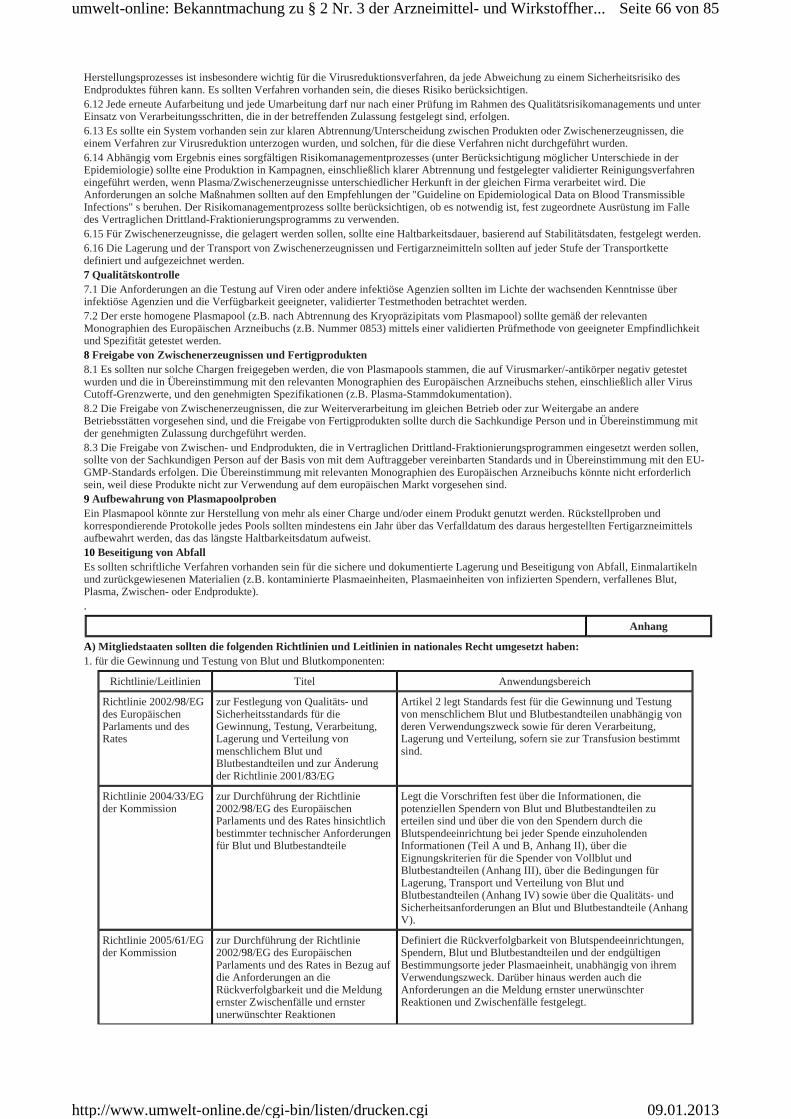
Herstellungsprozesses ist insbesondere wichtig für die Virusreduktionsverfahren, da jede Abweichung zu einem Sicherheitsrisiko des Endproduktes führen kann. Es sollten Verfahren vorhanden sein, die dieses Risiko berücksichtigen.6.12 Jede erneute Aufarbeitung und jede Umarbeitung darf nur nach einer Prüfung im Rahmen des Qualitätsrisikomanagements und unter Einsatz von Verarbeitungsschritten, die in der betreffenden Zulassung festgelegt sind, erfolgen.6.13 Es sollte ein System vorhanden sein zur klaren Abtrennung/Unterscheidung zwischen Produkten oder Zwischenerzeugnissen, die einem Verfahren zur Virusreduktion unterzogen wurden, und solchen, für die diese Verfahren nicht durchgeführt wurden.6.14 Abhängig vom Ergebnis eines sorgfältigen Risikomanagementprozesses (unter Berücksichtigung möglicher Unterschiede in der Epidemiologie) sollte eine Produktion in Kampagnen, einschließlich klarer Abtrennung und festgelegter validierter Reinigungsverfahren eingeführt werden, wenn Plasma/Zwischenerzeugnisse unterschiedlicher Herkunft in der gleichen Firma verarbeitet wird. Die Anforderungen an solche Maßnahmen sollten auf den Empfehlungen der "Guideline on Epidemiological Data on Blood Transmissible Infections" s beruhen. Der Risikomanagementprozess sollte berücksichtigen, ob es notwendig ist, fest zugeordnete Ausrüstung im Falle des Vertraglichen Drittland-Fraktionierungsprogramms zu verwenden.6.15 Für Zwischenerzeugnisse, die gelagert werden sollen, sollte eine Haltbarkeitsdauer, basierend auf Stabilitätsdaten, festgelegt werden.6.16 Die Lagerung und der Transport von Zwischenerzeugnissen und Fertigarzneimitteln sollten auf jeder Stufe der Transportkette definiert und aufgezeichnet werden.7 Qualitätskontrolle7.1 Die Anforderungen an die Testung auf Viren oder andere infektiöse Agenzien sollten im Lichte der wachsenden Kenntnisse über infektiöse Agenzien und die Verfügbarkeit geeigneter, validierter Testmethoden betrachtet werden.7.2 Der erste homogene Plasmapool (z.B. nach Abtrennung des Kryopräzipitats vom Plasmapool) sollte gemäß der relevanten Monographien des Europäischen Arzneibuchs (z.B. Nummer 0853) mittels einer validierten Prüfmethode von geeigneter Empfindlichkeit und Spezifität getestet werden.8 Freigabe von Zwischenerzeugnissen und Fertigprodukten8.1 Es sollten nur solche Chargen freigegeben werden, die von Plasmapools stammen, die auf Virusmarker/-antikörper negativ getestet wurden und die in Übereinstimmung mit den relevanten Monographien des Europäischen Arzneibuchs stehen, einschließlich aller Virus Cutoff-Grenzwerte, und den genehmigten Spezifikationen (z.B. Plasma-Stammdokumentation).8.2 Die Freigabe von Zwischenerzeugnissen, die zur Weiterverarbeitung im gleichen Betrieb oder zur Weitergabe an andere Betriebsstätten vorgesehen sind, und die Freigabe von Fertigprodukten sollte durch die Sachkundige Person und in Übereinstimmung mit der genehmigten Zulassung durchgeführt werden.8.3 Die Freigabe von Zwischen- und Endprodukten, die in Vertraglichen Drittland-Fraktionierungsprogrammen eingesetzt werden sollen, sollte von der Sachkundigen Person auf der Basis von mit dem Auftraggeber vereinbarten Standards und in Übereinstimmung mit den EU-GMP-Standards erfolgen. Die Übereinstimmung mit relevanten Monographien des Europäischen Arzneibuchs könnte nicht erforderlich sein, weil diese Produkte nicht zur Verwendung auf dem europäischen Markt vorgesehen sind.9 Aufbewahrung von PlasmapoolprobenEin Plasmapool könnte zur Herstellung von mehr als einer Charge und/oder einem Produkt genutzt werden. Rückstellproben und korrespondierende Protokolle jedes Pools sollten mindestens ein Jahr über das Verfalldatum des daraus hergestellten Fertigarzneimittels aufbewahrt werden, das das längste Haltbarkeitsdatum aufweist.10 Beseitigung von AbfallEs sollten schriftliche Verfahren vorhanden sein für die sichere und dokumentierte Lagerung und Beseitigung von Abfall, Einmalartikeln und zurückgewiesenen Materialien (z.B. kontaminierte Plasmaeinheiten, Plasmaeinheiten von infizierten Spendern, verfallenes Blut, Plasma, Zwischen- oder Endprodukte)..
Anhang
A) Mitgliedstaaten sollten die folgenden Richtlinien und Leitlinien in nationales Recht umgesetzt haben:1. für die Gewinnung und Testung von Blut und Blutkomponenten:
Richtlinie/Leitlinien Titel Anwendungsbereich
Richtlinie 2002/98/EG des Europäischen Parlaments und des Rates
zur Festlegung von Qualitäts- und Sicherheitsstandards für die Gewinnung, Testung, Verarbeitung, Lagerung und Verteilung von menschlichem Blut und Blutbestandteilen und zur Änderung der Richtlinie 2001/83/EG
Artikel 2 legt Standards fest für die Gewinnung und Testung von menschlichem Blut und Blutbestandteilen unabhängig von deren Verwendungszweck sowie für deren Verarbeitung, Lagerung und Verteilung, sofern sie zur Transfusion bestimmt sind.
Richtlinie 2004/33/EG der Kommission
zur Durchführung der Richtlinie 2002/98/EG des Europäischen Parlaments und des Rates hinsichtlich bestimmter technischer Anforderungen für Blut und Blutbestandteile
Legt die Vorschriften fest über die Informationen, die potenziellen Spendern von Blut und Blutbestandteilen zu erteilen sind und über die von den Spendern durch die Blutspendeeinrichtung bei jeder Spende einzuholenden Informationen (Teil A und B, Anhang II), über die Eignungskriterien für die Spender von Vollblut und Blutbestandteilen (Anhang III), über die Bedingungen für Lagerung, Transport und Verteilung von Blut und Blutbestandteilen (Anhang IV) sowie über die Qualitäts- und Sicherheitsanforderungen an Blut und Blutbestandteile (Anhang V).
Richtlinie 2005/61/EG der Kommission
zur Durchführung der Richtlinie 2002/98/EG des Europäischen Parlaments und des Rates in Bezug auf die Anforderungen an die Rückverfolgbarkeit und die Meldung ernster Zwischenfälle und ernster unerwünschter Reaktionen
Definiert die Rückverfolgbarkeit von Blutspendeeinrichtungen, Spendern, Blut und Blutbestandteilen und der endgültigen Bestimmungsorte jeder Plasmaeinheit, unabhängig von ihrem Verwendungszweck. Darüber hinaus werden auch die Anforderungen an die Meldung ernster unerwünschter Reaktionen und Zwischenfälle festgelegt.
Seite 66 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi
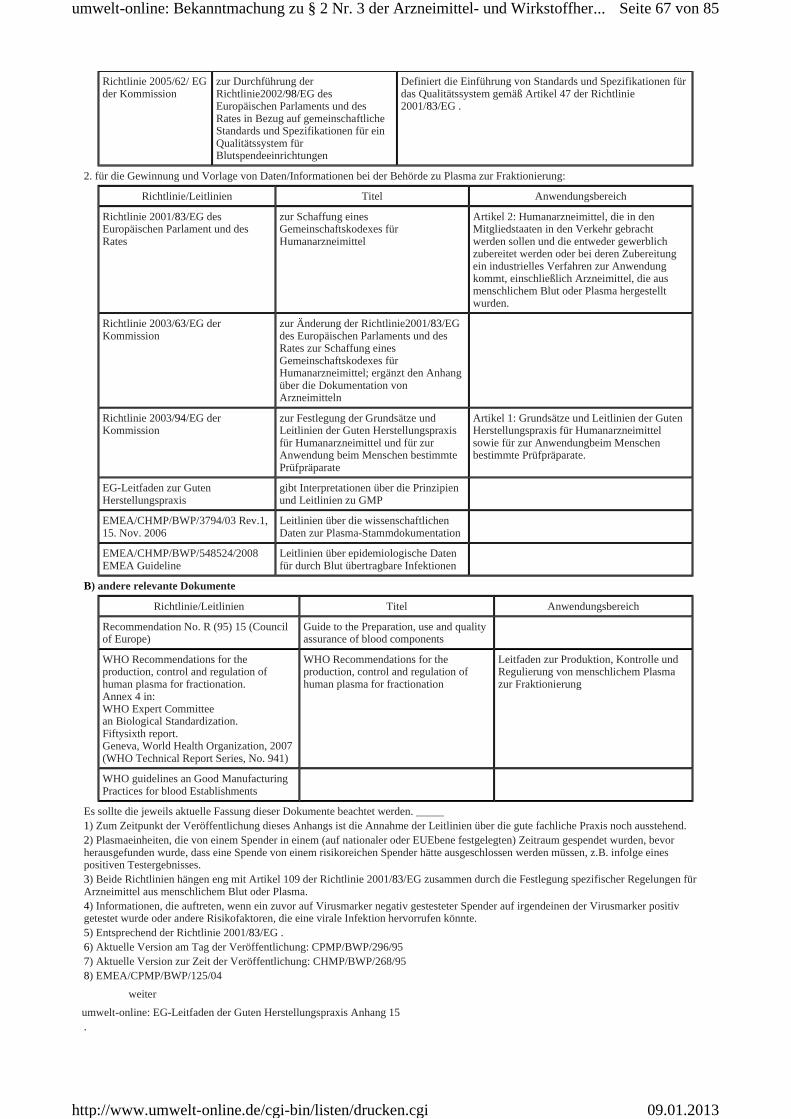
Richtlinie 2005/62/ EG der Kommission
zur Durchführung der Richtlinie2002/98/EG des Europäischen Parlaments und des Rates in Bezug auf gemeinschaftliche Standards und Spezifikationen für ein Qualitätssystem für Blutspendeeinrichtungen
Definiert die Einführung von Standards und Spezifikationen für das Qualitätssystem gemäß Artikel 47 der Richtlinie 2001/83/EG .
2. für die Gewinnung und Vorlage von Daten/Informationen bei der Behörde zu Plasma zur Fraktionierung:
Richtlinie/Leitlinien Titel Anwendungsbereich
Richtlinie 2001/83/EG des Europäischen Parlament und des Rates
zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel
Artikel 2: Humanarzneimittel, die in den Mitgliedstaaten in den Verkehr gebracht werden sollen und die entweder gewerblich zubereitet werden oder bei deren Zubereitung ein industrielles Verfahren zur Anwendung kommt, einschließlich Arzneimittel, die aus menschlichem Blut oder Plasma hergestellt wurden.
Richtlinie 2003/63/EG der Kommission
zur Änderung der Richtlinie2001/83/EG des Europäischen Parlaments und des Rates zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel; ergänzt den Anhang über die Dokumentation von Arzneimitteln
Richtlinie 2003/94/EG der Kommission
zur Festlegung der Grundsätze und Leitlinien der Guten Herstellungspraxis für Humanarzneimittel und für zur Anwendung beim Menschen bestimmte Prüfpräparate
Artikel 1: Grundsätze und Leitlinien der Guten Herstellungspraxis für Humanarzneimittel sowie für zur Anwendungbeim Menschen bestimmte Prüfpräparate.
EG-Leitfaden zur Guten Herstellungspraxis
gibt Interpretationen über die Prinzipien und Leitlinien zu GMP
EMEA/CHMP/BWP/3794/03 Rev.1, 15. Nov. 2006
Leitlinien über die wissenschaftlichen Daten zur Plasma-Stammdokumentation
EMEA/CHMP/BWP/548524/2008 EMEA Guideline
Leitlinien über epidemiologische Daten für durch Blut übertragbare Infektionen
B) andere relevante Dokumente
Richtlinie/Leitlinien Titel Anwendungsbereich
Recommendation No. R (95) 15 (Council of Europe)
Guide to the Preparation, use and quality assurance of blood components
WHO Recommendations for the production, control and regulation of human plasma for fractionation. Annex 4 in: WHO Expert Committee an Biological Standardization. Fiftysixth report. Geneva, World Health Organization, 2007 (WHO Technical Report Series, No. 941)
WHO Recommendations for the production, control and regulation of human plasma for fractionation
Leitfaden zur Produktion, Kontrolle und Regulierung von menschlichem Plasma zur Fraktionierung
WHO guidelines an Good Manufacturing Practices for blood Establishments
Es sollte die jeweils aktuelle Fassung dieser Dokumente beachtet werden. _____1) Zum Zeitpunkt der Veröffentlichung dieses Anhangs ist die Annahme der Leitlinien über die gute fachliche Praxis noch ausstehend.2) Plasmaeinheiten, die von einem Spender in einem (auf nationaler oder EUEbene festgelegten) Zeitraum gespendet wurden, bevor herausgefunden wurde, dass eine Spende von einem risikoreichen Spender hätte ausgeschlossen werden müssen, z.B. infolge eines positiven Testergebnisses.3) Beide Richtlinien hängen eng mit Artikel 109 der Richtlinie 2001/83/EG zusammen durch die Festlegung spezifischer Regelungen für Arzneimittel aus menschlichem Blut oder Plasma.4) Informationen, die auftreten, wenn ein zuvor auf Virusmarker negativ gestesteter Spender auf irgendeinen der Virusmarker positiv getestet wurde oder andere Risikofaktoren, die eine virale Infektion hervorrufen könnte.5) Entsprechend der Richtlinie 2001/83/EG .6) Aktuelle Version am Tag der Veröffentlichung: CPMP/BWP/296/957) Aktuelle Version zur Zeit der Veröffentlichung: CHMP/BWP/268/958) EMEA/CPMP/BWP/125/04
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 15 .
Seite 67 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Qualifizierung und Validierung Europäische Kommission Generaldirektion Unternehmen
(Eu Eudralex Band 4)
Anhang 15 zum EU-Leitfaden einer guten Herstellungspraxis
Erste Erörterung in Redaktionsgruppe
Erörterung in der Arbeitsgruppe "Regulierung von Arzneimitteln und Prüfungen" für die Freigabe zur Beratung
16. September 1999
Pharmazeutischer Ausschuss 28. September 1999
Zur Beratung freigegeben 30. Oktober 1999
Frist für Anmerkungen 28. Februar 2000
Abschließende Genehmigung durch Arbeitsgruppe des Prüfers Dezember 2000
Pharmazeutischer Ausschuss (zur Unterrichtung) April 2001
Datum des Anwendungsbeginns September 2001
Hinweis: Dieses Dokument basiert auf dem Empfehlungen von PICS/S PRINZIPIEN1. Der vorliegende Anhang beschreibt die auf die Herstellung von Arzneimittel anzuwendenden Prinzipien der Qualifizierung und Validierung. Es ist eine GMPAnforderung, dass der Hersteller feststellt, welche Validierungsarbeiten für den Nachweis notwendig sind, dass die kritischen Aspekte der von ihm vorgenommenen Aktivitäten unter Kontrolle stehen. Größere Änderungen an Einrichtungen, Ausrüstung und Prozessen, die die Produktqualität beeinflussen können, sollten validiert werden. Weiterhin sollte eine Risikobewertung vorgenommen werden, um Validierungsumfang und -tiefe bestimmen zu können. PLANUNG EINER VALIDIERUNG2. Alle Validierungsaktivitäten sollten geplant werden. Die Schlüsselelemente eines Validierungsprogramms sollten in einem Validierungsmasterplan (VMP) oder gleichartigen Dokumenten klar definiert und dokumentiert werden. 3. Der VMP sollte eine kurze, präzise und deutliche Zusammenfassung sein. 4. Der VMP sollte mindestens Informationen zu folgenden Punkten enthalten:
Validierungspolitik;a.organisatorische Struktur der Validierungsaktivitäten;b.zusammenfassende Darstellung der zu validierenden Einrichtungen, Anlagen, Ausrüstung und Prozesse;c.Dokumentationsformat: das für Anweisungen und Berichte zu verwendende Format;d.Planung und Zeiteinteilung;e.Änderungskontrolle;f.Verweise auf bestehende Dokumente.g.
5. Im Falle größerer Projekte kann es erforderlich sein, separate Validierungsmasterpläne zu erstellen. DOKUMENTATION6. Eine schriftliche Anweisung, aus der hervorgeht, wie die Qualifizierung und Validierung durchzuführen ist, ist zu erstellen. Die Anweisung sollte überprüft und genehmigt werden. Aus ihr sollten die kritischen Schritte und die Akzeptanzkriterien hervorgehen. 7. Ein mit Querverweisen auf die Qualifizierungs- und/oder Validierungsanweisungen versehener Bericht sollte erstellt werden, der die Ergebnisse zusammenfasst, festgestellte Abweichungen kommentiert und die notwendigen Schlussfolgerungen zieht. Letzteres schließt Empfehlungen zur Mängelbeseitigung ein. Änderungen des Plans, so wie er in der Anweisung beschrieben ist, sollten zusammen mit einer angemessenen Begründung dokumentiert werden. 8. Im Anschluss an eine zufriedenstellende Qualifizierung sollte in Form einer schriftlichen Genehmigung eine formale Freigabe für den nächsten Schritt der Qualifizierung und Validierung erfolgen. QUALIFIZIERUNG Designqualifizierung9. Der erste Schritt einer Validierung neuer Einrichtungen, Anlagen oder Ausrüstungsgegenstände sollte die Designqualifizierung (DQ) sein. 10. Die Übereinstimmung des Designs mit den GMP-Anforderungen sollte demonstriert und dokumentiert werden. Installationsqualifizierung11. Bei neuen oder veränderten Einrichtungen, Anlagen oder Ausrüstungsgegenständen ist eine Installationsqualifizierung (IQ) vorzunehmen. 12. Die IQ sollte mindestens umfassen:
Installation von Ausrüstung, Rohrleistungen, Versorgungseinrichtungen und Instrumenten nach Abgleich mit aktuellen technischen Zeichnungen und Spezifikationen;
a.
Zusammentragen und Vergleich von Betriebs- und Arbeitsanweisungen sowie Wartungsanforderungen des Lieferanten;b.Anforderungen an die Kalibrierung;c.Verifizierung der Konstruktionsmaterialien. Funktionsqualifizierungd.
13. Die Funktionsqualifizierung (OQ) sollte im Anschluss an die Installationsqualifizierung stattfinden. 14.Die OQ sollte mindestens umfassen:
Tests, die auf der Grundlage von aus den Prozessen, Anlagen und Ausrüstungsgegenständen gewonnenem Wissen entwickelt wurden;
a.
Tests, die obere und untere Betriebsgrenzen mit einschließen, zuweilen als "worst case"-Bedingungen bezeichnet.b.
Seite 68 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

15. Der Abschluß der erfolgreichen Funktionsqualifizierung sollte die Fertigstellung von Kalibrierungs-, Betriebs- und Reinigungsverfahren, Schulung des Bedienpersonals und vorbeugende Wartungsanforderungen erlauben. Er sollte die formale "Freigabe" der Einrichtungen, Anlagen und Ausrüstung ermöglichen. Leistungsqualifizierung16. Die Leistungsqualifizierung (PQ) sollte im Anschluss an die erfolgreiche Durchführung einer Installations- und Funktionsqualifizierung vorgenommen werden. 17.Die PQ sollte mindestens umfassen:
Tests mit Produktionsmaterialien, geeigneten Ersatzmaterialien oder simulierten Produkten, die auf der Grundlage von aus dem Prozess sowie den Einrichtungen, Anlagen oder Ausrüstungsgegenständen gewonnenem Wissen entwickelt wurden;
a.
Tests, die obere und untere Betriebsgrenzen mit einschließen.b.
18.Obwohl die Leistungsqualifizierung als eine eigenständige Aktivität dargestellt wird, kann es in einigen Fällen sinnvoll sein, sie zusammen mit der Funktionsqualifizierung durchzuführen. Qualifizierung bestehender (in Gebrauch befindlicher) Einrichtungen, Anlagen und Ausrüstung 19. Es sollten Daten verfügbar sein, die die Betriebsparameter und Grenzwerte für die kritischen Variablen der Betriebsausrüstung stützen und verifizieren. Weiterhin sollten die Verfahren und Aufzeichnungen für Kalibrierung, Reinigung, vorbeugende Wartung, Betriebsvorgänge und Schulungen des Bedienpersonals dokumentiert werden. PROZESSVALIDIERUNGAllgemeine Hinweise20. Die in diesem Kapitel dargelegten Anforderungen und Prinzipien gelten für die Herstellung pharmazeutischer Darreichungsformen. Abgedeckt werden die anfängliche Validierung neuer Prozesse, die anschließende Validierung veränderter Prozesse und Revalidierungen. 21. Die Prozessvalidierung sollte im Regelfall vor dem Vertrieb eines Arzneimittels abgeschlossen werden (prospektive Validierung). In Ausnahmefällen, wenn dies nicht möglich ist, kann es notwendig sein, Prozesse während der routinemäßigen Produktion zu validieren (begleitende Validierung). Prozesse, die sich bereits seit einiger Zeit im Einsatz befinden, sollten ebenfalls validiert werden (retrospektive Validierung). 22. Die zu verwendenden Einrichtungen Anlagen und Ausrüstungsgegenstände sollten qualifiziert und die analytischen Prüfmethoden validiert worden sein. Die an der Validierung beteiligten Mitarbeiter sollten angemessen geschult worden sein. 23. Einrichtungen, Anlagen, Ausrüstung und Prozesse sollten in bestimmten Zeitabständen bewertet werden, um sicherzustellen, dass sie sich weiterhin in einem validierten Zustand befinden. Prospektive Validierung24.Eine prospektive Validierung soltel mindestens umfassen:
kurze Beschreibung des Prozesses;a.zusammenfassende Darlegung der zu untersuchenden kritischen Prozessschritte;b.Auflistung der zu verwendenden Ausrüstung/Einrichtungen (einschließlich Mess-/ Überwachungs-/Protokollierausrüstung) zusammen mit ihrem jeweiligen Kalibrierstatus
c.
Spezifikationen des Fertigproduktes für die Freigabe;d.gegebenenfalls eine Auflistung der Prüfmethoden;e.die vorgeschlagenen Inprozesskontrollen einschließlich Akzeptanzkriterien;f.gegebenenfalls zusätzlich durchzuführende Prüfungen einschließlich Akzeptanzkriterien und Validierung der Methoden;g.Probenahmeplan;h.Methoden für die Aufzeichnung und Bewertung von Ergebnisseni.Funktionen und Verantwortlichkeiten;j.vorgesehener Zeitplan.k.
25. Mit Hilfe eines solchen definierten Prozesses (einschließlich spezifizierter Bestandteile) kann eine bestimmte Anzahl von Chargen des Endprodukt unter routinemäßigen Bedingungen hergestellt werden. Theoretisch sollten die Zahl der durchgeführten Prozessläufe und die entsprechenden Beobachtungen ausreichen, um das normale Ausmaß an Variationen und Trends feststellen zu können und ausreichend Daten für eine Bewertung zu erhalten. Allgemein wird es als zulässig angesehen, dass drei aufeinanderfolgende Chargen/Läufe innerhalb der endgültig festgelegten Parameter die Validierung des Prozesses darstellen. 26.Die für die Prozessvalidierung hergestellten Chargen sollten die gleiche Größe wie die für den Handel vorgesehen haben. 27. Sollten Validierungschargen verkauft oder vertrieben werden, sollten die Bedingungen, unter denen sie hergestellt werden, vollständig mit den GMPAnforderungen, inklusive zufriedenstellender Ergebnisse der Validierungsläufe, und mit der Zulassung übereinstimmen. Begleitende Validierung28. In Ausnahmefällen kann es zulässig sein, vor dem Beginn der routinemäßigen Produktion das Validierungsprogramm noch nicht abgeschlossen zu haben. 29.Die Entscheidung, eine begleitende Validierung durchzuführen, muss begründet, dokumentiert und von befugtem Personal genehmigt werden. 30. Die für eine begleitende Validierung notwendige Dokumentation entspricht der für die prospektive Validierung. Retrospektive Validierung31.Eine retrospektive Validierung ist nur bei gut etablierten Prozessen zulässig und gilt dann als ungeeignet, wenn vor Kurzem Änderungen an der Zusammensetzung des Produkts, den Betriebsverfahren oder der Ausrüstung vorgenommen wurden. 32. Die Validierung derartiger Prozesse sollte sich auf historische Daten stützen. Die hierbei zu durchlaufenden Schritte erfordern die Erstellung einer spezifischen Anweisung sowie eine Darlegung der Ergebnisse der Datenüberprüfung, die zu einer Schlussfolgerung und einer Empfehlung führen soll. 33.Die Datenquellen für eine solche Validierung sollten mindestens Chargenherstellungs- und Verpackungsprotokolle, Prozesskontrolldiagramme, Wartungslogbücher, Aufzeichnungen zu Personalwechseln, Prozessfähigkeitsuntersuchungen sowie Daten zum Fertigprodukt einschließlich Trendkarten und Stabilitätsergebnisse aus der Lagerung umfassen. 34. Die für die retrospektive Validierung gewählten Chargen sollten repräsentativ für alle während des Überprüfungszeitraums hergestellten Chargen, einschließlich derjenigen, die außerhalb der Spezifikationen lagen, sein und ausreichend in der Anzahl sein, um die
Seite 69 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Beständigkeit des Prozesses nachweisen zu können. Eine zusätzliche Überprüfung von Rückstellmustern kann erforderlich sein, um die für eine retrospektive Validierung notwendige Menge an oder Art von Daten zu erhalten. 35.Bei der retrospektiven Validierung sollten grundsätzlich Daten von zehn bis dreißig aufeinanderfolgenden Chargen untersucht werden, um die Beständigkeit des Prozesses zu bewerten. Bei entsprechender Begründung können weniger Chargen untersucht werden. REINIGUNGSVALIDIERUNG36. Eine Reinigungsvalidierung ist durchzuführen, um die Wirksamkeit eines Reinigungsverfahren zu belegen. Die Begründung für die Festlegung von Grenzwerten für die Übertragung von Produktrückständen, Reinigungsmitteln und mikrobieller Kontamination sollte logisch auf der Basis der beteiligten Materialien erfolgen. Die Grenzwerte sollten erreichbar und verifizierbar sein. 37. Es sollten validierte Prüfmethoden, die empfindlich genug sind, Rückstände oder Kontaminanten nachzuweisen, verwendet werden. Die jeweilige Prüfmethode sollte eine ausreichend empfindliche Nachweisgrenze aufweisen, um das festgelegte akzeptable Maß der Rückstände oder Kontaminenten bestimmen zu können. 38. Im Regelfall müssen nur Reinigungsverfahren für produktberührende Ausrüstungsoberflächen validiert werden. Gegebenenfalls sollten auch Teile, die keinen Kontakt mit dem Produkt haben, berücksichtigt werden. Die Zeitabstände zwischen der Verwendung und der Reinigung sowie der Reinigung und der Wiederverwendung sollten validiert werden. Reinigungsintervalle und -methoden sind festzulegen. 39. Bei Reinigungsverfahren für einander ähnliche Produkte und Prozesse gilt es als zulässig, eine repräsentative Auswahl der ähnlichen Produkte und Prozesse zu treffen. Zu diesem Zweck kann eine einzelne Validierungsuntersuchung, der ein "worst case"-Vorgehen zugrundeliegt und die alle kritischen Aspekte berücksichtigt, vorgenommen werden. 40. Normalerweise sollte ein Reinigungsverfahren dreimal hintereinander vollzogen und als erfolgreich eingestuft worden sein, um zu beweisen, dass die entsprechende Methode validiert ist. 41. Ein "Reinigen bis zur Sauberkeit" ("test until clean") gilt nicht als geeignete Alternative zur Reinigungsvalidierung. 42. Ausnahmsweise können Produkte, die die physikochemischen Eigenschaften der zu beseitigenden Substanzen simulieren, statt der Substanzen selbst verwendet werden, wenn diese entweder toxisch oder gefährlich sind. ÄNDERUNGSKONTROLLE43. Für den Fall der beabsichtigen Änderung an einem Ausgangsstoff, einem Produktbestandteil, einem Ausrüstungsgegenstand, der Prozessumgebung (oder der Anlage), der Produktions- oder Testmethode oder jeder anderen Änderung, die die Produktqualität oder Reproduzierbarkeit des Prozesses beeinflussen könnte, sollte es schriftliche Verfahren geben, die die dann zu ergreifenden Maßnahmen darlegen. Änderungskontrollverfahren sollten sicherstellen, dass ausreichend stützende Daten erzeugt werden, um beweisen zu können, dass mit Hilfe des überarbeiteten Prozesses ein der gewünschten Qualität und den genehmigten Spezifikationen entsprechendes Produkt hergestellt werden kann. 44. Alle Änderungen, die die Produktqualität oder die Reproduzierbarkeit des Prozesses beeinflussen könnten, sollten formal beantragt, dokumentiert und genehmigt werden. Die wahrscheinlichen Auswirkungen der Änderung der Einrichtungen, Anlagen und Ausrüstung auf das Produkt sollten bewertet werden. Dies schließt eine Risikoanalyse ein. Der Bedarf und das Ausmaß einer Requalifizierung oder Revalidierung sollten bestimmt werden. REVALIDIERUNG45. Einrichtungen, Anlagen, Ausrüstung und Prozesse einschließlich der Reinigung sollten in bestimmten Zeitabständen bewertet werden, um zu gewährleisten, dass sie sich weiterhin in einem validierten Zustand befinden. Wurden am Validierungsstatus keine bedeutenden Änderungen vorgenommen, erfüllt eine Überprüfung, die den Nachweis erbringt, dass die Einrichtungen, Systeme, Ausrüstung und Prozesse den vorgeschriebenen Anforderungen entsprechen, die Notwendigkeit einer Revalidierung. GLOSSAREs folgen Definitionen von mit Qualifizierung und Validierung in Zusammenhang stehenden Begriffen, die nicht im Glossar des aktuellen GMP-Leitfadens der EG enthalten sind, aber im vorliegenden Anhang verwendet werden. ÄnderungskontrolleEin formales System, durch das qualifizierte Vertreter entsprechender Fachbereiche beabsichtigte oder tatsächliche Änderungen, die den validierten Zustand von Einrichtungen, Anlagen, Ausrüstung oder Prozessen beeinflussen könnten, überprüfen. Ziel ist es, die Vorkehrungen festzulegen, die für den Nachweis und die Dokumentation der Einhaltung des validierten Zustands erforderlich sind. .
Anlage
Summe miteinander verbundener Geräte mit gemeinsamer Zweckbestimmung. Begleitende ValidierungValidierung, die während der routinemäßigen Herstellung von für den Verkauf bestimmten Produkten durchgeführt wird. Designqualifizierung (DQ)Eine dokumentierte Verifizierung, dass das für Einrichtungen, Anlagen und Ausrüstung vorgesehene Design für den entsprechenden Verwendungszweck geeignet ist. Funktionsqualifizierung (OQ)Eine dokumentierte Verifizierung, dass Einrichtungen, Anlagen und Ausrüstung, so wie sie installiert oder modifiziert wurden, im Rahmen der vorgesehenen Betriebsbereiche den Erwartungen gemäß funktionieren. Installationsqualifizierung (IQ)Eine dokumentiere Verifizierung, dass Einrichtungen, Anlagen und Ausrüstung, so wie sie installiert oder modifiziert wurden, mit dem genehmigten Design und den Empfehlungen des Herstellers übereinstimmen. Leistungsqualifizierung (PQ)Eine dokumentierte Verifizierung, dass Einrichtungen, Anlagen und Ausrüstung, so wie sie miteinander verbunden wurden, auf der Grundlage der genehmigten Prozessmethode und Produktspezifikation effektiv und reproduzierbar funktionieren. Prospektive ValidierungEine vor der routinemäßigen Produktion von für den Verkauf bestimmten Produkten durchgeführte Validierung. ProzessvalidierungDokumentierte Beweisführung, dass der Prozess innerhalb bestimmter Parameter auf effektive und reproduzierbare Art ein Arzneimittel hervorbringt, dass im voraus festgelegte Spezifikationen und Qualitätsattribute erfüllt. ReinigungsvalidierungBei der Reinigungsvalidierung handelt es sich um eine dokumentierte Beweisführung, dass ein genehmigtes Reinigungsverfahren die Ausrüstung in einen Zustand versetzt, in dem sie für die Herstellung von Arzneimitteln geeignet ist.
Seite 70 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Retrospektive Validierung Prozessvalidierung für ein Produkt, dass auf der Grundlage von zusammengetragenen Herstellungs-, Test- und Kontrollchargendaten auf den Markt gebracht wurde. RevalidierungEine Wiederholung der Prozessvalidierung, um zu gewährleisten, dass Änderungen am Prozess oder der Ausrüstung, die gemäß bestimmter Änderungskontrollverfahren vorgenommen wurden, die Prozesseigenschaften und die Produktqualität nicht beeinträchtigen. RisikoanalyseMethode, die für das Funktionieren von Ausrüstung oder Prozess kritischen Parameter zu bewerten und zu definieren. Simuliertes ProduktEin Material, das den physikalischen und, wenn möglich, den chemischen Eigenschaften (z.B. Viskosität, Korngröße, pH, etc.) des zu validierenden Produkts sehr ähnlich ist. In vielen Fällen können diese Eigenschaften mit Hilfe einer Placebocharge erreicht werden. Worst CaseEine oder mehrere Bedingungen, die obere und untere Betriebsgrenzen und die Umstände in den zugrundeliegenden Standardverfahrensanweisungen einschließen, bei denen, verglichen mit den Idealbedingungen, fehlerhafte Prozesse oder Produkte mit der größten Wahrscheinlichkeit auftreten. Diese Bedingungen führen nicht zwangsläufig zu einem Produkt- oder Prozessversagen.
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 16 .
Zertifizierung durch eine sachkundige Person und Chargenfreigabe Europäische Kommission Generaldirektion Unternehmen
(Eu Eudralex Band 4) vom 18. April 2007 (BAnz. S. 4826)
Anhang 16 zum EG-Leitfaden für die Gute Herstellungspraxis
(GMP)
1. Anwendungsbereich1.1 Dieser Anhang zum Leitfaden für die Gute Herstellungspraxis von Arzneimitteln ("der Leitfaden") enthält Leitlinien über die Zertifizierung durch eine sachkundige Person und die Chargenfreigabe von Arzneimitteln mit einer Genehmigung für das Inverkehrbringen innerhalb der Europäischen Gemeinschaft (EG) und des Europäischen Wirtschaftsraums (EWR) oder von für den Export bestimmten Arzneimitteln. Die relevanten gesetzlichen Bestimmungen sind in Artikel 22 der Richtlinie 75/319/EWG des Rates oder Artikel 30 der Richtlinie 81/851/EWG des Rates enthalten. 1.2 Dieser Anhang regelt insbesondere die Fälle, in denen eine Charge unterschiedliche Produktions- und Kontrollstufen an unterschiedlichen Orten oder bei unterschiedlichen Herstellern durchlaufen hat oder in denen ein Zwischenprodukt oder eine Bulkcharge in mehr als eine Fertigproduktcharge unterteilt wurden. Darüber hinaus berücksichtigt dieser Anhang die Freigabe von Chargen, die aus Ländern in die EG/den EWR eingeführt wurden, bei denen ein Abkommen über gegenseitige Anerkennung zwischen der Gemeinschaft und dem Drittland besteht bzw. nicht besteht. Die Leitlinie kann vorbehaltlich etwaiger Abweichungen zwischen den gesetzlichen Bestimmungen und den genaueren Leitlinien in Anhang 13 des Leitfadens auch auf Arzneimittel für Forschungszwecke angewandt werden. 1.3 Dieser Anhang beschreibt nicht alle gesetzlich möglichen Verfahren. Er bezieht sich nicht auf die Chargenfreigabe der offiziellen Überwachungsbehörde, die für spezielle Blut- und immunologische Produkte in Übereinstimmung mit Artikel 3 der Richtlinien 89/342/EWG und 89/381/EWG vorgeschrieben sein kann. 1.4 Die grundlegenden Bedingungen für die Chargenfreigabe eines Produktes werden durch die jeweilige Genehmigung für das Inverkehrbringen festgelegt. Nichts in diesem Anhang ist diesen Bestimmungen übergeordnet. 2. Grundsätze2.1 Jede Charge eines Fertigproduktes muss durch eine sachkundige Person innerhalb der EG/des EWR zertifiziert werden, bevor sie für den Verkauf oder Lieferungen innerhalb der EG/des EWR oder zum Export freigegeben wird. 2.2 Das Ziel, das mit einer derartigen Kontrolle der Chargenfreigabe verfolgt wird, ist folgendes:
Es soll sichergestellt sein, dass die Charge gemäß den Anforderungen an die Genehmigung für das Inverkehrbringen, der GMP-Leitlinien und -Richtlinien der Europäischen Gemeinschaft oder gemäß der GMP-Leitlinien eines anderen Landes, die durch ein Abkommen über die gegenseitige Anerkennung als gleichwertig angesehen wurden, hergestellt und geprüft wurde. Zudem soll die Beachtung aller anderen gesetzlichen Vorgaben vor dem Inverkehrbringen des Produktes sichergestellt sein.
•
Im Falle einer Überprüfung von Mängeln oder eines Chargenrückrufes soll zudem gewährleistet sein, dass die sachkundige Person, die die Charge und die relevanten Ergebnisse zertifiziert hat, identifiziert werden kann.
•
3. Einleitung3.1 Die Herstellung, einschließlich der Qualitätskontrolle, einer Arzneimittelcharge findet in mehreren Stufen statt, die an unterschiedlichen Orten und von unterschiedlichen Herstellern ausgeführt werden können. Jede Stufe sollte in Übereinstimmung mit der entsprechenden Genehmigung für das Inverkehrbringen, der Guten Herstellungspraxis und der Gesetze des jeweiligen Mitgliedstaates durchgeführt und von der sachkundigen Person, die die Fertigproduktcharge vor dem Inverkehrbringen zertifiziert, beachtet werden. 3.2 In industriellen Prozessen ist es jedoch nicht immer umsetzbar, dass eine einzelne sachkundige Person detailliert in alle Herstellungsverfahren involviert ist. Daher ist diese Person bei der Zertifizierung einer Fertigproduktcharge auf die Beratung und Entscheidungen anderer Personen angewiesen. Vor der Zertifizierung sollte sie entweder durch persönliche Kenntnis oder durch Bestätigung anderer sachkundiger Personen innerhalb eines von ihr anerkannten Qualitätssystems die Informationen auf ihre Zuverlässigkeit hin überprüfen. 3.3 Auch bei einer teilweisen Herstellung des Produktes in einem Drittstaat ist es erforderlich, dass Produktion und Testverfahren der Genehmigung für das Inverkehrbringen entsprechen, dass der Hersteller gemäß der jeweiligen nationalen Vorschriften eine Herstellungserlaubnis besitzt und die Herstellung gemäß einer der EG gleichwertigen Guten Herstellungspraxis durchgeführt wird. 3.4 Einige in diesem Anhang verwendeten Begriffe haben eine spezifische Bedeutung, die dem Glossar entnommen werden kann. 4. Allgemeines4.1 Vor ihrer Freigabe hat eine Fertigproduktcharge unterschiedliche Herstellungs-, Einfuhr-, Prüf- und Lagerungsverfahren in unterschiedlichen Betrieben durchlaufen. Jeder Betrieb sollte eine oder mehrere Herstellungserlaubnisse besitzen und über zumindest eine sachkundige Person verfügen. Dennoch ist die Feststellung der korrekten Herstellung einer Produktcharge, unabhängig von der Anzahl der involvierten Betriebe, die Hauptaufgabe der sachkundigen Person, die die Fertigproduktcharge vor der Freigabe zertifiziert.
Seite 71 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

4.2 Unterschiedliche Chargen eines Produktes können an unterschiedlichen Orten innerhalb der EG/des EWR hergestellt, importiert oder freigegeben werden. Beispielsweise können in einer Genehmigung für das Inverkehrbringen der Gemeinschaft mehrere Mitgliedstaaten und in einer nationalen Genehmigung für das Inverkehrbringen mehrere Einrichtungen für die Chargenfreigabe benannt werden. In diesem Fall sollte der Inhaber der Genehmigung für das Inverkehrbringen und der zur Freigabe des Produktes autorisierter Betrieb alle in eine Chargenfreigabe involvierten Einrichtungen sowie die sachkundige Person, die die Charge zertifiziert hat, exakt identifizieren können. 4.3 Die sachkundige Person kann die Fertigproduktcharge aufgrund persönlicher Kenntnis aller verwendeten Einrichtungen und Verfahren, aufgrund der Fachkenntnis involvierter Personen und des Qualitätssystems, im Rahmen dessen sie arbeiten, freigeben. Alternativ kann sie sich innerhalb eines von ihr anerkannten Qualitätssystems auf die Bestätigung einer oder mehrerer anderer sachkundiger Personen über die Einhaltung der Standards bei den Produktionszwischenstufen verlassen. Die Bestätigungen der anderen sachkundigen Personen sollten schriftlich dokumentiert und die bestätigten Sachverhalte erkennen lassen. Die diesbezüglich getroffenen systematischen Maßnahmen sollten in einer schriftlichen Vereinbarung festgehalten werden. 4.4 Die o.g. Vereinbarung ist erforderlich, wenn sich eine sachkundige Person auf die Bestätigung einer anderen sachkundigen Person verlässt. Die Vereinbarung sollte mit Kapitel 7 des Leitfadens übereinstimmen. Der Sachverständige, der die Fertigproduktcharge zertifiziert, sollte überprüfen, ob die in der Vereinbarung getroffenen Sachverhalte zutreffen. Die Form der Vereinbarung sollte der Beziehung der beiden Parteien angemessen sein, z.B. als Arbeitsanweisung innerhalb einer Firma oder als formeller Vertrag zwischen zwei Firmen, auch wenn sie der gleichen Gruppe zugehörig sind. 4.5 In dieser Vereinbarung sollte sich der Lieferant der Bulk- oder Zwischenprodukte verpflichten, den/die Empfänger von allen Abweichungen, nicht spezifizierten Ergebnissen, Abweichungen von GMP-Standards, Überprüfungen, Beanstandungen oder anderen Vorkommnissen zu unterrichten, die die sachkundige Person bei der Zertifizierung der Fertigproduktcharge zu berücksichtigen hat. 4.6 Bei Einsatz eines computergestützten Systems für die Aufzeichnung von Zertifizierungen und Chargenfreigaben sollte Anhang 11 dieses Leitfadens besondere Beachtung finden. 4.7 Die Zertifizierung der Zulassungskonformität einer Fertigproduktcharge durch eine sachkundige Person innerhalb der EG/des EWR muss für dieselbe Charge in der EG/dem EWR nicht erneut vorgenommen werden. 4.8 Unabhängig von den einzelnen Maßnahmen, die zur Zertifizierung und Freigabe von Chargen getroffen werden, sollte eine sofortige Identifizierung und ein Rückruf von Produkten, die aufgrund qualitativer Mängel der Charge ein Risiko für den Menschen darstellen, immer möglich sein. 5. Chargenüberprüfung und Freigabe innerhalb der EG/EWR hergestellter Produkte5.1 Die Herstellung an einem einzigen autorisierten ProduktionsortWurden alle Produktions- und Kontrollstufen an einem Ort durchgeführt, können gewisse Überprüfungen und Kontrollen an andere Orte abgegeben werden. Der Sachverständige des Produktionsortes, der die Zertifizierung der Fertigproduktcharge vornimmt, bleibt allerdings innerhalb eines festgelegten Qualitätssystems in der Regel persönlich für die Charge verantwortlich. Er hat die Möglichkeit, Bestätigungen anderer sachkundiger Personen des Betriebes für die Zwischenstufen heranzuziehen, für die jene verantwortlich sind. 5.2 Unterschiedliche Herstellungsstufen an unterschiedlichen Produktionsorten innerhalb desselben UnternehmensWerden unterschiedliche Herstellungsstufen einer Charge an unterschiedlichen Orten desselben Unternehmens ausgeführt (die nicht zwangsläufig die gleiche Herstellerlaubnis haben), sollte eine sachkundige Person für jede Stufe verantwortlich sein. Die Zertifizierung der Fertigproduktcharge sollte von der sachkundigen Person des Inhabers der Herstellerlaubnis vorgenommen werden, der für die Freigabe der Charge verantwortlich ist. Die sachkundige Person kann sich für alle Stufen persönlich verantwortlich zeichnen oder sich auf die Bestätigung der für die einzelnen Stufen verantwortlichen sachkundigen Personen beziehen. 5.3 Übertragung einiger Herstellungszwischenstufen an eine andere FirmaEine oder mehrere Herstellungs- oder Kontrollzwischenstufen können dem Inhaber einer Herstellungserlaubnis einer anderen Firma übertragen werden. Die sachkundige Person des Auftraggebers kann die Bestätigung der jeweiligen Stufe durch die sachkundige Person des Auftragnehmers berücksichtigen. Sie muss jedoch sicherstellen, dass diese Arbeit gemäß der in einer schriftlichen Vereinbarung fixierten Bedingungen durchgeführt wird. Die Zertifizierung der Fertigproduktcharge sollte von einer sachkundigen Person des Inhabers der Herstellerlaubnis vorgenommen werden, der für die Marktfreigabe verantwortlich ist. 5.4 Eine Bulkcharge wird an unterschiedlichen Orten zu einzelnen Fertigproduktchargen zusammengesetzt, die unter einer einzelnen Genehmigung für das Inverkehrbringen freigegeben werden. Dies ist beispielsweise unter einer nationalen Genehmigung für das Inverkehrbringen möglich, wenn alle Fertigungsstelen im gleichen Mitgliedsstaat ansässig sind oder unter einer Genehmigung für das Inverkehrbringen, der Gemeinschaft, wenn sich die Fertigungsstätten in mehreren Mitgliedsstaaten befinden. 5.4.1 Eine Alternative für die sachkundige Person des Inhabers einer Herstellerlaubnis, der die Bulkcharge herstellt, ist die Zertifizierung aller Fertigproduktchargen vor der Marktfreigabe. Sie kann hierbei entweder für alle Herstellungsstufen persönlich verantwortlich zeichnen oder auf die Herstellungsbestätigungen der sachkundigen Personen der Fertigungsstätten zurückgreifen. 5.4.2 Eine andere Alternative ist die Zertifizierung der Fertigproduktcharge vor Marktfreigabe durch die sachkundige Person des Herstellers, der die Endfertigung durchgeführt hat. Diese kann hierbei entweder für alle Herstellungsstufen persönlich verantwortlich zeichnen oder auf die Bestätigung der Bulkcharge durch die sachkundige Person des Bulkchargen-Herstellers zurückgreifen. 5.4.3 Generell sollte bei der Fertigung an unterschiedlichen Herstellungsorten eine Person, in der Regel die sachkundige Person des Herstellers der Bulkcharge, die Gesamtverantwortung für alle freigegebenen Fertigproduktchargen tragen, die von dieser Bulkcharge stammen. Diese Person muss über auftretende Qualitätsmängel von Fertigproduktchargen informiert werden, und sie muss die notwendigen Maßnahmen zur Behebung eines Mangels einer Bulkcharge einleiten. Da die Chargennummern der Bulkproduktchargen und der Fertigproduktchargen nicht notwendigerweise übereinstimmen, sollte eine Verknüpfung zwischen diesen beiden Nummern dokumentiert werden, so dass ein Auditweg gebildet werden kann. 5.5 Eine Bulkcharge wird an verschiedenen Herstellungsorten zu einzelnen Fertigproduktchargen zusammengesetzt und gemäß unterschiedlicher Genehmigung für das Inverkehrbringen freigegeben. Dies ist beispielsweise der Fall, wenn eine multinationale Organisation nationale Genehmigung für das Inverkehrbringen für ein Produkt in verschiedenen Mitgliedstaaten hält oder wenn ein Generikumhersteller Bulkware erwirbt und sie unter seiner eigenen Genehmigung für das Inverkehrbringen für den Verkauf fertigt und freigibt. 5.5.1 Eine sachkundige Person der Herstellerfirma, die die Herstellung vornimmt, zertifiziert die Fertigproduktcharge entweder, indem sie persönlich die Verantwortung für alle Herstellungsphasen übernimmt oder sich auf die Bestätigung der Bulkcharge durch eine sachkundige Person auf Seiten des Bulkherstellers bezieht. 5.5.2 Probleme bei Fertigproduktchargen, die von der Bulkcharge herrühren können, sollten der sachkundigen Person gemeldet werden, die für die Bestätigung der Bulkcharge zuständig ist. Die Person sollte in Bezug auf alle Fertigproduktchargen, die aus der fraglichen Bulkcharge entstanden sind, die notwendigen Maßnahmen ergreifen. Dies sollte in einer schriftlichen Vereinbarung festgehalten werden. 5.6 Eine Fertigproduktcharge wird von dem Inhaber einer Herstellungserlaubnis in Übereinstimmung mit seiner eigenen Genehmigung für das Inverkehrbringen erworben und für den Markt freigegeben. Dies tritt beispielsweise ein, wenn eine Lieferfirma für Generika eine Genehmigung für das Inverkehrbringen für Produkte hat, die von einer anderen Firma hergestelt werden, und Fertigprodukte einkauft, die
Seite 72 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

nicht gemäß der eigenen Genehmigung für das Inverkehrbringen zertifiziert wurden, und diese unter der eigenen Herstellerlaubnis in Übereinstimmung mit der eigenen Genehmigung für das Inverkehrbringen freigibt. In diesem Fall sollte die sachkundige Person des Einkäufers die Fertigproduktcharge vor Freigabe zertifizieren. Sie kann dabei persönlich die Verantwortung für alle Herstellungsphasen übernehmen oder auf die Bestätigung der Charge durch die sachkundige Person des Verkäufers zurückgreifen. 5.7 Die Zulassung des Kontrollabors und der Herstellerfirma basieren auf unterschiedlichen Herstellungserlaubnissen. Eine sachkundige Person kann bei der Zertifizierung einer Fertigproduktcharge entweder persönlich Verantwortung für alle Labortests übernehmen oder auf die Bestätigung der Tests und Ergebnisse der Charge durch eine andere sachkundige Person zurückgreifen. Das andere Labor und die sachkundige Person müssen nicht in demselben Mitgliedstaat ansässig sein wie der Inhaber der Herstellerlaubnis, der die Charge freigibt. Liegt eine solche Bestätigung nicht vor, sollte die zuständige sachkundige Person das für das zu zertifizierende Fertigprodukt relevante Labor und die Testverfahren persönlich kennen. 6. Chargenüberprüfung und Freigabe importierter Produkte aus Drittstaaten 6.1 Allgemeines6.1.1 Der Import von Fertigprodukten sollte von einem Einführer vorgenommen werden, wie im Glossar dieses Anhangs definiert. 6.1.2 Jede Charge importierter Fertigprodukte sollte von einer sachkundigen Person des Einführers vor der Freigabe zum Verkauf in der EG/im EWR zertifiziert werden. 6.1.3 Sofern kein Abkommen über die gegenseitige Anerkennung zwischen der Gemeinschaft und dem Drittland vorliegt (siehe Abschnitt 7), sollten vor der Zertifizierung einer Fertigproduktcharge durch die sachkundige Person jeder Charge Stichproben entnommen werden, um sie in der EG/EWR zu testen. Einfuhr und Tests müssen nicht notwendigerweise im selben Mitgliedstaat stattfinden. 6.1.4 Die Leitlinien dieses Abschnittes sollten ebenfalls auf den Import von Produkten angewandt werden, die teilweise in einem Drittstaat hergestellt werden. 6.2 Einfuhr einer gesamten Charge oder des ersten Teils einer ArzneimittelchargeDie Charge oder Teilcharge sollte von einer sachkundigen Person auf Seiten des Einführers vor der Freigabe zertifiziert werden. Die sachkundige Person kann auf die Prüf- und Testergebnisse des importierten Produktes durch eine sachkundige Person eines anderen autorisierten Herstellers zurückgreifen (d. h. innerhalb der EG/des EWR). 6.3Ein Teil einer Fertigproduktcharge wird importiert, nachdem ein anderer Teil der gleichen Charge zuvor an den gleichen oder einen anderen Ort geliefert wurde. 6.3.1 Eine sachkundige Person des Einführers, die einen Nachfolgeteil einer Charge erhält, kann auf die Testergebnisse und die Zertifizierung zurückgreifen, die von einer sachkundigen Person bei dem ersten Teil der Charge durchgeführt wurden. In diesem Fall sollte die sachkundige Person nachweisen, dass die beiden Teile tatsächlich zur gleichen Charge gehören, dass der nachfolgende Teil unter den gleichen Bedingungen wie der erste transportiert wurde und dass die entnommenen Stichproben für die gesamte Charge repräsentativ sind. 6.3.2 Die in Abschnitt 6.3.1 genannten Bedingungen werden am besten erfüllt, wenn der Hersteller des Drittstaates und der Einführer in der EG/dem EWR der gleichen Organisation angehören und unter einem gemeinsamen Qualitätssicherheitssystem arbeiten. Kann die sachkundige Person nicht mit Sicherheit feststellen, dass alle in 6.3.1 aufgelisteten Bedingungen erfüllt sind, sollten die beiden Teile als separate Chargen behandelt werden. 6.3.3 Im Falle einer Freigabe unterschiedlicher Teile einer Charge der gleichen Genehmigung für das Inverkehrbringen sollte ein Mitarbeiter, meist die sachkundige Person des Einführers des ersten Chargenteils, für die Aufbewahrung der Importaufzeichnungen aller Chargenteile sowie für die Nachvollziehbarkeit der Distribution der Chargenteile innerhalb der EG/des EWR die Verantwortung übernehmen. Jegliche Qualitätsmängel im Zusammenhang mit dieser Charge sollten ihm zugetragen werden. Er sollte die notwendigen Maßnahmen zur Behebung der Mängel koordinieren. Eine schriftliche Vereinbarung zwischen allen Einführern sollte dies sicherstellen. 6.4 Standort der Probenahme für Tests innerhalb der EG/des EWR6.4.1 Die entnommenen Proben einer Charge sollten repräsentativ sein und in der EG/dem EWR getestet werden. Um ein möglichst genaues Ergebnis zu erzielen, sollten jedoch bereits während der Herstellung im Drittstaat Proben entnommen werden. Proben für die Prüfung auf Sterilität sollten beispielsweise während der Abfüllung entnommen werden. Um jedoch ein repräsentatives Ergebnis auch nach Lagerung und Transport zu erhalten, sollten weitere Proben der Charge nach Einfuhr in die EG/den EWR entnommen werden. 6.4.2 Werden Proben in einem Drittstaat entnommen, sollten diese zusammen mit der Charge, von der sie stammen, unter den gleichen Bedingungen versandt werden. Bei separater Versendung sollte nachgewiesen werden, dass die Proben für die Charge weiterhin repräsentativ sind, z.B. indem die Lager- und Frachtbedingungen genau festgelegt und überwacht werden. Verlässt sich die sachkundige Person auf die in Drittstaaten durchgeführten Tests, sollte dies auf einer fundierten technischen Grundlage beruhen. 7. Chargenprüfung und Freigabe von Produkten aus Drittstaaten, mit denen die EG ein Abkommen über die gegenseitige Anerkennung geschlossen hat7.1 Sofern in dem Abkommen nicht anders vereinbart, entbindet dieses die sachkundige Person in der EG/dem EWR nicht von ihrer Pflicht, die Charge vor der Freigabe für Verkauf oder Auslieferung innerhalb der EG/EWR zu zertifizieren. Je nach den einzelnen vertraglichen Regelungen kann sich die sachkundige Person des Einführers jedoch auf die Bestätigung des Herstellers verlassen, dass die Charge gemäß der Genehmigung für das Inverkehrbringen und der Guten Herstellungspraxis des Drittstaates produziert und getestet wurde. Eine erneute Durchführung aller Tests ist dann nicht nötig. Die sachkundige Person kann die Charge zertifizieren, wenn sie die Bestätigung des Herstellers als ausreichend ansieht und der Transport unter den erforderlichen Bedingungen durchgeführt wurde und Erhalt und Lagerung in der EG/dem EWR ordnungsgemäß, wie in Abschnitt 8 dargestellt, erfolgt sind. 7.2 Andere Verfahren, einschließlich des Erhaltes und der Zertifizierung von Teilchargen zu unterschiedlichen Zeiten an unterschiedlichen Orten, sollten gemäß Abschnitt 6 erfolgen. 8. Routinepflichten einer sachkundigen Person8.1 Vor der Zertifizierung eines Produktes zur Freigabe sollte die sachkundige Person in Anlehnung an die o. g. Leitlinien sicherstellen, dass zumindest die nachfolgenden Anforderungen erfüllt werden:
Charge und Herstellung entsprechen der Genehmigung für das Inverkehrbringen (dies schließt, wo nötig, die Einfuhrerlaubnis ein).a.Die Herstellung erfolgte unter Einhaltung der Guten Herstellungspraxis der Europäischen Gemeinschaft, bzw. im Falle importierter Chargen aus einem Drittstaat unter als gleichwertig anerkannten Standards.
b.
Die wichtigsten Herstellungs- und Testverfahren sind validiert, die aktuellen Produktionsbedingungen und Herstellungsprotokolle wurden berücksichtigt.
c.
Alle Abweichungen oder geplanten Änderungen in Produktion oder Qualitätskontrolle wurden von der verantwortlichen Person in Übereinstimmung mit einem festgelegten System genehmigt. Alle Änderungen, die Veränderungen der Markt- und Herstellungserlaubnis erfordern, wurden der entsprechenden Behörde gemeldet und von dieser autorisiert.
d.
Seite 73 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Alle notwendigen Überprüfungen und Tests wurden durchgeführt, einschließlich aller zusätzlichen Probenahmen, Kontrollen und Überprüfungen, die aufgrund von Abweichungen oder planmäßigen Änderungen nötig sind.
e.
Die notwendige Dokumentation über Produktion und Qualitätskontrolle wurde fertiggestellt und von den Mitarbeitern genehmigt.f.Alle Audits wurden so ausgeführt, wie es das Qualitätssicherheitssystem vorsieht.g.Zudem sollte die sachkundige Person alle anderen ihr bekannten und für die Qualität der Charge relevanten Faktoren berücksichtigen.
h.
Die sachkundige Person kann je nach nationaler Gesetzgebung und Verwaltungsverfahren zu weiteren Aufgaben verpflichtet werden. 8.2 Eine sachkundige Person, die die Konformität einer Herstellungszwischenstufe gemäß Abschnitt 4.3 bestätigt, hat dieselben o.g. Verpflichtungen für diese Stufe, sofern nicht anders in einer Vereinbarung unter den sachkundigen Personen festgehalten. 8.3 Die sachkundige Person sollte ihre Kenntnisse und Erfahrung über technische und wissenschaftliche Fortschritte und Änderungen in den Verfahren der Qualitätskontrolle stets aktualisieren, soweit sie für die von ihr zu zertifizierenden Produkte relevant sind. 8.4 Muss die sachkundige Person ein Produkt zertifizieren, das sie noch nicht kennt, sei es durch Einführung neuer Produktreihen oder Arbeitsplatzwechsel, sollte sie sich mit diesem zunächst gründlich vertraut machen, um ihrer Aufgabe nachkommen zu können. Je nach den jeweiligen nationalen Bestimmungen kann die sachkundige Person dazu angehalten werden, die Behörden von den Änderungen zu unterrichten und muss eventuell eine neuerliche Zulassung beantragen. 9. GlossarEinige Begriffe werden wie nachstehend definiert in diesem Anhang verwendet. Das Glossar des Leitfadens sollte ebenfalls konsultiert werden. Abkommen über die gegenseitige Anerkennung: Eine angemessene Vereinbarung zwischen der Gemeinschaft und einem exportierenden Drittstaat, wie in Artikel 22.1 der Richtlinie 75/319/EWG und Artikel 30 der Richtlinie 81/851/EWG beschrieben. Bestätigung: Eine schriftliche Aussage, die bezeugt, dass ein Verfahren oder Test in Übereinstimmung mit der Guten Herstellungspraxis und der entsprechenden Genehmigung für das Inverkehrbringen durchgeführt wurde, wie schriftlich mit der für die Zertifizierung der Fertigproduktcharge verantwortlichen sachkundigen Person vereinbart. Bestätigung und bestätigt haben die gleiche Bedeutung. Bulkcharge: Eine Produktcharge von einer im Zulassungsantrag beschriebenen Chargengröße, bereit für die Abfüllung in ihre Endbehälter oder in Einzelbehälter zur Verpackung in Endverpackungen (eine Bulkcharge besteht beispielsweise aus einer Bulkmenge von Flüssigprodukten, festen Produkten wie tabletten oder Kapseln oder gefüllte Ampullen). Einführer: Der Inhaber einer Zulassung für den Import von Produkten aus Drittstaaten gemäß Artikel 16.3 der Richtlinie 75/319/EWG oder Artikel 24.3 der Richtlinie 81/851/EWG. Fertigproduktcharge: In Bezug auf die Kontrolle eines Fertigproduktes ist die Fertigproduktcharge in Teil 2 Abschnitt E1 in den Anhängen der Richtlinien 75/318/EWG und 81/852/EWG definiert. Im Zusammenhang mit diesem Anhang bezeichnet der Begriff vor allem die Produktcharge in ihrer Endverpackung zur Marktfreigabe. Sachkundige Person: Die in Artikel 21 der Richtlinie 75/319/EWG und Artikel 29 der Richtlinie 81/851/EWG definierte Person. Zertifizierung der Fertigproduktcharge: Die Zertifizierung in einem Register oder gleichwertigen Dokument durch eine sachkundige Person, wie in Artikel 22 der Richtlinie 75/319/EWG und Artikel 30 der Richtlinie 81/851/EWG festgelegt, vor Freigabe einer Charge zum Verkauf oder zur Distribution.
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 17 .
Parametrische Freigabe zum EU-Leitfaden für die Gute Herstellungspraxis (GMP) Europäische Kommission Generaldirektion Unternehmen
Juli 2001 (Eu Eudralex Band 4)
Anhang 17
Erste Diskussionen innerhalb des PIC/S-Rahmen Juni 1998 - August 1999
Beratung und Adhoc-Sitzung der zuständigen Stellen für die Überwachung der Guten Herstellungspraxis
September 1999 - Februar 2000
Revidierte Version März 2000
Freigabe zur Konsultierung der Industrie 6. April 2000
Vorgeschlagene Frist zur Einreichung von Kommentaren September 2000
Neuer Entwurf nach Konsultierung Januar 2001
Endgültiger Entwurf nach Beratung der zuständigen Stellen für die Überwachung der Guten Herstellungspraxis
Februar 2001
Weitergabe an den Pharmazeutischen Ausschuss Juli 2001
Vorgeschlagener Zeitpunkt der Inbetriebnahme Januar 2002
Hinweis: Dieses Dokument wurde in Zusammenarbeit mit PIC/S erstellt. Es sollte im Zusammenhang mit der Leitlinie für die Parametrische Freigabe (CPMP/QWP/3015/99) konsultiert werden, die im Februar 2001 vom Ausschuss für Arzneispezialitäten (CPMP) verabschiedet wurde. Mehr dazu unter http:/www.emea.eu.int/htms/human/qwp/qwpfin.htm 1. Grundsätze1.1 Die Definition des in diesem Anhang verwendeten Begriffes "parametrische Freigabe" basiert auf dem Vorschlag der Europäischen Organisation für Qualität: "Ein Freigabesystem, das auf Informationen, die während des Produktionsverfahrens gesammelten wurden basiert, und unter Beachtung der speziellen GMP-Anforderungen für parametrische Freigabe sicherstellt, dass ein Produkt tatsächlich die beabsichtigten Qualitätsanforderungen erfüllt". 1.2 Die parametrische Freigabe sollte mit den grundlegenden Anforderungen der Guten Herstellungspraxis, den relevanten Anhängen und den folgenden Richtlinien übereinstimmen.
Seite 74 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

2. Parametrische Freigabe2.1 Es ist allgemein anerkannt, dass umfassende Inprozesskontrollen eine größere Sicherheit für die Übereinstimmung des Produktes mit den Spezifikationen gewährleisten als die Prüfung der Fertigprodukte. 2.2 Die parametrische Freigabe kann für einige spezifische Parameter als Alternative zu Routinekontrollen von Fertigprodukten genehmigt werden. Eine Genehmigung für die parametrische Freigabe sollte gemeinsam von den Verantwortlichen für die Zulassung und den GMP-Inspektoren erteilt, abgelehnt oder zurückgezogen werden. 3. Parametrische Freigabe für sterile Produkte3.1 Dieser Abschnitt beschäftigt sich ausschließlich mit dem Teil der parametrischen Freigabe, der sich auf die Routinefreigabe von Endprodukten ohne Prüfung auf Sterilität bezieht. Ein Verzicht auf die Prüfung auf Sterilität ist nur zulässig, wenn erfolgreich nachgewiesen werden kann, dass zuvor festgelegte, validierte Sterilisationsbedingungen erfüllt sind. 3.2 Die Prüfung auf Sterilität bietet aufgrund statistischer Grenzen lediglich die Möglichkeit, grobe Fehler im Sterilitätssicherungssystem aufzudecken. 3.3 Parametrische Freigabe kann genehmigt werden, wenn die Daten, die einen korrekten Herstellungsprozess der Charge nachweisen, als Dokumentation ausreichend sicherstellen, dass das zur Gewährleistung der Sterilität der Produkte konzipierte und validierte Verfahren durchgeführt wurde. 3.4 Derzeit kann die parametrische Freigabe nur für Produkte genehmigt werden, die im verschlossenen Endbehältnis sterilisiert werden. 3.5 Sterilisationsmethoden, die den Anforderungen des Europäischen Arzneibuches entsprechen und Dampf, Trockenhitze und ionisierende Strahlung verwenden, können für die parametrische Freigabe in Betracht gezogen werden. 3.6 Ein völlig neues Produkt wird mit großer Wahrscheinlichkeit für die parametrische Freigabe als nicht geeignet betrachtet werden, da zufriedenstellende Sterilitätstestergebnisse über einen bestimmten Zeitraum hinweg einen Teil der Voraussetzungen einer Akzeptanz hierfür ausmachen. Es mag jedoch Fälle geben, bei denen sich ein neues Produkt lediglich geringfügig hinsichtlich der Sicherheit der Sterilität unterscheidet und vorhandene Sterilitätstestdaten anderer Produkte für eine Akzeptanz als aussagekräftig herangezogen werden können. 3.7 Eine Risikoanalyse des Sterilitätssicherungsystems sollte im Hinblick auf die Freigabe nichtsterilisierter Produkte durchgeführt werden. 3.8 Der Hersteller sollte in der Vergangenheit eine hohe Übereinstimmung mit den Regeln der Guten Herstellungspraxis nachgewiesen haben. 3.9 Bei der Überprüfung der Einhaltung der Guten Herstellungspraxis sollten sowohl die Historie unsteriler Produkte als auch die Ergebnisse der Sterilitätsprüfung des betroffenen Produktes und derjenigen Produkte, die das gleiche oder ein ähnliches Sterilitätssicherungsystem durchlaufen haben, in Betracht gezogen werden. 3.10 Ein erfahrener Ingenieur für Sterilisationstechnik sowie ein sachkundiger Mikrobiologe sollten normalerweise vor Ort bei der Produktion und bei der Sterilisation anwesend sein. 3.11 Das Design und die ursprüngliche Validierung des produktbezogenen Prozesses sollten sicherstellen, dass dessen Integrität unter allen relevanten Bedingungen gewährleistet werden kann. 3.12 Ein Änderungskontrollsystem sollte eine Überprüfung der Änderungen durch das Personal, welches für die Sicherstellung der Sterilität zuständig ist, vorsehen. 3.13 Ein System zur Kontrolle mikrobiologischer Verunreinigung des Produktes vor der Sterilisation sollte eingerichtet werden. 3.14 Jede Möglichkeit einer Verwechslung steriler und unsteriler Produkte sollte ausgeschlossen werden. Physische Hindernisse oder validierte elektronische Systeme können dies sicherstellen. 3.15 Die Sterilisationsaufzeichnungen sollten hinsichtlich ihrer Übereinstimmung mit den Spezifikationen von zwei unabhängigen Systemen überprüft werden. Dies können zum einen zwei Mitarbeiter oder zum anderen ein validiertes Computersystem und ein Mitarbeiter sein. 3.16 Die folgenden zusätzlichen Anforderungen sollten vor der Freigabe jeder Produktcharge überprüft werden:
Alle geplanten Wartungsarbeiten und Routineprüfungen wurden an der verwendeten Sterilisationsanlage vollständig durchgeführt.•Alle Reparaturen und Veränderungen wurden von einem Ingenieur für Sterilisationstechnik und einem Mikrobiologen geprüft und genehmigt.
•
Alle Instrumente sind kalibriert.•Der Prozess in der Sterilisationsanlage ist für die verarbeitete Produktmenge validiert.•
3.17 Nach genehmigter parametrischer Freigabe sollte die Entscheidung über die Freigabe oder Zurückweisung der Charge von den zugelassenen Spezifikationen abhängig gemacht werden. Ist eine Einhaltung der Spezifikationen der parametrischen Freigabe nicht nachgewiesen, kann auch eine erfolgreiche Sterilitätsprüfung nicht zur Freigabe der Charge führen. 4. Glossar Parametrische FreigabeEin Freigabesystem, das auf Informationen, die während des Produktionsverfahrens gesammelten wurden basiert, und unter Beachtung der speziellen GMP-Anforderungen für parametrische Freigabe sicherstellt, dass ein Produkt tatsächlich die beabsichtigten Qualitätsanforderungen erfüllt. Sterilitätssicherheitssystem Die Summe aller getroffenen Vorkehrungen, die die Sterilität eines Produktes gewährleisten sollen. Für endsterilisierte Produkte sind dies insbesondere die folgenden Stufen:
Produktdesign.a.Kenntnis und, wenn möglich, Kontrolle des mikrobiologischen Zustandes der Ausgangsstoffe und der Prozesshilfen (z.B. Gase und Schmiermittel).
b.
Überprüfung des Produktionsverfahrens auf Verunreinigung, um das Eindringen von Mikroorganismen und deren Vermehrung im Produkt zu verhindern. Dies geschieht normalerweise durch Reinigung und Desinfektion der produktberührenden Flächen, durch Arbeiten in reinen Räumen, um Verunreinigung durch die Luft zu verhindern, durch Limitierung der Prozesszeiten und, wenn notwendig, durch die Einplanung von Filtrationsschritten.
c.
Ergreifen von Präventivmaßnahmen, um eine Verwechselung steriler und unsteriler Produkte auszuschließen.d.Wahrung der Produktintegrität.e.Sterilisationsverfahren.f.
Seite 75 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Die Gesamtheit des Qualitätssicherungssystems, das das Sterilitätssicherungssystems beinhaltet, z.B. Änderungskontrolle, Ausbildung, schriftliche Verfahren, Freigabeprüfungen, vorbeugende Wartung, Analyse von Störfällen, Vorbeugung menschlichen Versagens, Validierung, Kalibrierung, usw.
g.
weiter
umwelt-online: EG-Leitfaden der Guten Herstellungspraxis Anhang 20.
Qualitäts-Risikomanagement Vorwort und Anwendungsbereich Europäische Kommission Generaldirektion Unternehmen
Vom 18. Juli 2008 (BAnz. Nr. 115 vom 01.08.2008 S. 2798)
Eudralex Band 4
Anhang 20 zum EG-Leitfaden der
Guten Herstellungspraxis
Der neue Anhang 20 zum EG-GMP Leitfaden entspricht der ICH Q9-Leitlinie zum Qualitäts-Risikomanagement. Er bietet Anleitung über einen systematischen Zugang zum Qualitäts-Risikomanagement und erleichtert so die Einhaltung von Anforderungen der Guten Herstellungspraxis und anderen Qualitätsanforderungen. Er beinhaltet Grundsätze, die anzuwenden sind, und Verfahren, Methoden und Werkzeuge, die genutzt werden können, wenn ein formaler Qualitäts-Risikomanagement-Maßstab angelegt wird. Aus Gründen der Stimmigkeit ist Kapitel 1 des Leitfadens Teil I über das Qualitätsmanagement überarbeitet worden, um die Aspekte des Qualitäts-Risikomanagements in das Qualitätssystem aufzunehmen. Eine ähnliche Überarbeitung ist auch für Teil II des Leitfadens geplant. Bei grundsätzlicher Überarbeitung anderer Abschnitte des Leitfadens werden diese gegebenenfalls ebenso an Aspekte des Qualitäts-Risikomanagements angeglichen werden. Mit der Revision der Abschnitte zum Qualitäts-Risikomanagement in Teil I und II des Leitfadens wird Qualitäts-Risikomanagement in das Qualitätssystem des Herstellers fest integriert. Der Anhang 20 beabsichtigt jedoch nicht, neue regulatorische Erwartungen zu erzeugen; er stellt vielmehr dem Hersteller eine Anzahl international anerkannter Risikomanagement-Methoden und -werkzeuge nebst einer Liste möglicher Anwendungen zur Verfügung. Nach allgemeinem Verständnis war die ICH Q9 Leitlinie in erster Linie für das Qualitäts-Risikomanagement bei Humanarzneimitteln entwickelt worden. Mit der Implementierung im Anhang 20 werden die Vorteile der Leitlinie, wie Verfahren, Methoden und Werkzeuge zum Qualitäts-Risikomanagement, auch dem Veterinärbereich zur Verfügung gestellt. Obwohl der EG-GMP Leitfaden primär an Hersteller gerichtet ist, hat die ICH Q9 Leitlinie Relevanz für andere Qualitätsleitlinien und beinhaltet spezifische Abschnitte für Überwachungsbehörden. Jedoch ist aus Gründen des Zusammenhangs und der Vollständigkeit die ICH Q9 Leitlinie vollständig in Anhang 20 zum EG-GMP Leitfaden überführt worden. Weitere Berücksichtigung in regulatorischer Hinsicht wird Schritt für Schritt erfolgen, z.B. bei der Revision der "Compilation of Community Procedures an Inspections and Exchange of Information" und einigen Leitlinien zur Qualität, die von der EMEA veröffentlicht werden. 1 EinführungDie Grundsätze des Risikomanagements werden in vielen Bereichen von Wirtschaft und Behörden einschließlich Finanzwesen, Versicherungen, Arbeitsschutz, Gesundheitswesen und Pharmakovigilanz sowie von den für diese Industriezweige zuständigen Regulierungsbehörden effektiv eingesetzt. Es gibt zwar bereits einige Beispiele für den Einsatz des Qualitäts-Risikomanagements in der pharmazeutischen Industrie; diese Beispiele sind jedoch noch eingeschränkt und nutzen bei weitem nicht das gesamte Potenzial des Risikomanagements. Darüber hinaus wurde die Bedeutung von Qualitätssystemen in der pharmazeutischen Industrie erkannt, und es wird deutlich, dass das Qualitäts-Risikomanagement eine wertvolle Komponente für ein effektives Qualitätssystem darstellt. Allgemein wird anerkannt, dass ein Risiko als die Kombination aus der Eintrittswahrscheinlichkeit eines Schadens und dem Ausmaß dieses Schadens definiert ist. Schwieriger ist es, unter allen interessierten Parteien ein gemeinsames Verständnis über die Anwendung des Risikomanagements zu erzielen, da die interessierten Parteien unterschiedliche potenzielle Gefahren sehen sowie die Wahrscheinlichkeiten des Auftretens und das Schadensausmaß jeder Gefahr unterschiedlich beurteilen. Wenn es in Verbindung mit Arzneimitteln auch viele verschiedene Interessensgruppen wie Patienten, Ärzte, Behörden und Industrie gibt, so sollte doch in Bezug auf das Management der Qualitätsrisiken immer dem Schutz des Patienten die oberste Priorität eingeräumt werden. Die Herstellung und der Einsatz von Arzneimitteln einschließlich ihrer Einzelbestandteile birgt immer ein gewisses Maß an Risiken. Das Qualitätsrisiko ist dabei nur ein Teil des Gesamtrisikos. Es muss klar sein, dass die Produktqualität über den Produkt-Lebenszyklus hinweg aufrecht erhalten werden muss, damit die für die Qualität bedeutsamen Merkmale des Arzneimittels mit den Merkmalen der in den klinischen Untersuchungen verwendeten Produkte übereinstimmen. Ein effektiver Ansatz zum Qualitäts-Risikomanagement kann außerdem die hohe Qualität von Arzneimitteln für den Patienten sicherstellen, weil er eine proaktive Methode zur Erkennung und Steuerung potenzieller Qualitätsprobleme bei der Entwicklung und Herstellung darstellt. Darüber hinaus kann beim Auftreten von Qualitätsproblemen durch Qualitäts-Risikomanagement die Entscheidungsfindung verbessert werden. Ein effektives Qualitäts-Risikomanagement kann bessere und fundiertere Entscheidungen erleichtern und bietet den Aufsichtsbehörden zuverlässigere Informationen darüber, wie gut ein Unternehmen potenzielle Risiken bewältigen kann. Es verbessert somit den Überblick über den Umfang und den Grad der direkten Überwachung. Ziel dieses Dokuments ist es, einen systematischen Ansatz für ein Qualitäts-Risikomanagement anzubieten. Es dient als Basis oder Hilfsmittel und ist zwar unabhängig, unterstützt jedoch andere ICH Qualitätsdokumente und ergänzt vorhandene Verfahren, Anforderungen, Standards und Leitlinien zur Qualitätssicherung in der pharmazeutischen Industrie und im regulatorischen Umfeld. Es stellt insbesondere Anleitungen zu den Grundsätzen und einige Instrumente des Qualitäts-Risikomanagements zur Verfügung, die effektivere und konsistente risikobasierte Entscheidungen ermöglichen. Dies gilt sowohl für die Aufsichtsbehörden als auch für die Industrie und hinsichtlich der Qualität von Wirkstoffen und Arzneimitteln über deren Produkt-Lebenszyklus hinweg. Es ist nicht beabsichtigt, neue Erwartungen über die bestehenden regulatorischen Anforderungen hinaus aufzustellen. Es ist nicht immer sinnvoll oder notwendig, anhand eines formalen Risikomanagement-Verfahrens vorzugehen (unter Nutzung anerkannter Methoden und/oder interner Vorgehensweisen, z.B. Standardarbeitsanweisungen). Auch formlose Risikomanagement-Verfahren (Nutzung empirischer Methoden und/oder interner Vorgehensweisen) können akzeptabel sein. Der angemessene Einsatz des Qualitäts-Risikomanagements erleichtert häufig die Einhaltung von behördlichen Vorschriften, er entbindet die Industrie jedoch nicht von der Verpflichtung zu deren Einhaltung und ersetzt nicht die angemessene Kommunikation zwischen Industrie und Behörde. 2 GeltungsbereichDiese Leitlinie enthält Grundsätze und Beispiele für Instrumente des Qualitäts-Risikomanagements, die auf verschiedene Aspekte pharmazeutischer Qualität angewandt werden können. Dies schließt Entwicklung, Herstellung, Vertrieb sowie Inspektion und Einreichungs-/Änderungsverfahren über den Lebenszyklus von Wirkstoffen, Arzneimitteln sowie biologischen und biotechnologischen Produkten ein (einschließlich der Verwendung von Rohmaterialien, Lösungsmitteln, Hilfsstoffen, Packmitteln und Kennzeichnungsmaterialien für Arzneimittel sowie biologische und biotechnologische Produkten).
Seite 76 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi
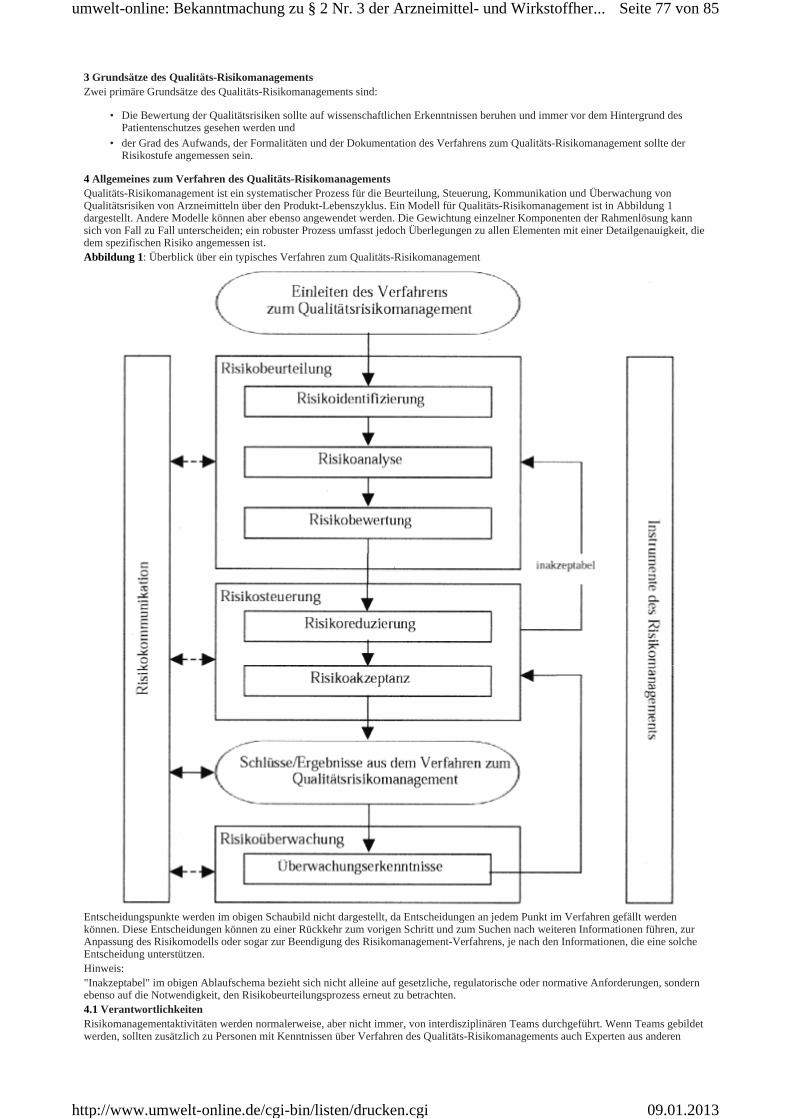
3 Grundsätze des Qualitäts-RisikomanagementsZwei primäre Grundsätze des Qualitäts-Risikomanagements sind:
Die Bewertung der Qualitätsrisiken sollte auf wissenschaftlichen Erkenntnissen beruhen und immer vor dem Hintergrund des Patientenschutzes gesehen werden und
•
der Grad des Aufwands, der Formalitäten und der Dokumentation des Verfahrens zum Qualitäts-Risikomanagement sollte der Risikostufe angemessen sein.
•
4 Allgemeines zum Verfahren des Qualitäts-RisikomanagementsQualitäts-Risikomanagement ist ein systematischer Prozess für die Beurteilung, Steuerung, Kommunikation und Überwachung von Qualitätsrisiken von Arzneimitteln über den Produkt-Lebenszyklus. Ein Modell für Qualitäts-Risikomanagement ist in Abbildung 1 dargestellt. Andere Modelle können aber ebenso angewendet werden. Die Gewichtung einzelner Komponenten der Rahmenlösung kann sich von Fall zu Fall unterscheiden; ein robuster Prozess umfasst jedoch Überlegungen zu allen Elementen mit einer Detailgenauigkeit, die dem spezifischen Risiko angemessen ist. Abbildung 1: Überblick über ein typisches Verfahren zum Qualitäts-Risikomanagement
Entscheidungspunkte werden im obigen Schaubild nicht dargestellt, da Entscheidungen an jedem Punkt im Verfahren gefällt werden können. Diese Entscheidungen können zu einer Rückkehr zum vorigen Schritt und zum Suchen nach weiteren Informationen führen, zur Anpassung des Risikomodells oder sogar zur Beendigung des Risikomanagement-Verfahrens, je nach den Informationen, die eine solche Entscheidung unterstützen. Hinweis: "Inakzeptabel" im obigen Ablaufschema bezieht sich nicht alleine auf gesetzliche, regulatorische oder normative Anforderungen, sondern ebenso auf die Notwendigkeit, den Risikobeurteilungsprozess erneut zu betrachten. 4.1 VerantwortlichkeitenRisikomanagementaktivitäten werden normalerweise, aber nicht immer, von interdisziplinären Teams durchgeführt. Wenn Teams gebildet werden, sollten zusätzlich zu Personen mit Kenntnissen über Verfahren des Qualitäts-Risikomanagements auch Experten aus anderen
Seite 77 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

geeigneten Bereichen eingebunden werden (z.B. Qualitätseinheit, Geschäftsentwicklung, Ingenieurwesen, regulatorische Angelegenheiten, Produktion, Vertrieb & Marketing, Recht, Statistik und Klinik). Die Entscheidungsträger müssen
die Verantwortung für die Koordination des Qualitäts-Risikomanagements über verschiedene Funktionen und Abteilungen hinweg in ihrer Organisation übernehmen und
•
sicherstellen, dass ein Verfahren zum Qualitäts-Risikomanagement definiert, angewendet und überprüft wird und dass angemessene Ressourcen vorhanden sind.
•
4.2 Einleiten eines Verfahrens zum Qualitäts-RisikomanagementDas Qualitäts-Risikomanagement sollte systematische Verfahren zur Koordinierung, Erleichterung und Verbesserung einer wissenschaftlich fundierten Entscheidungsfindung hinsichtlich der Risiken umfassen. Mögliche Schritte zur Einleitung und Planung eines Verfahrens zum Qualitäts-Risikomanagement können folgende Punkte umfassen:
Definition der Frage nach dem Problem bzw. Risiko einschließlich entsprechender Annahmen zum Risikopotenzial•Zusammenstellung von Hintergrundinformationen und -daten zu potenziellen Gefahren, Schäden oder Gesundheitsrisiken, die für eine Risikobeurteilung von Bedeutung sind
•
Benennung einer Führungsperson und der erforderlichen Ressourcen•Festlegung eines Zeitrahmens für die durchzuführenden Arbeiten und einer angemessenen Entscheidungsfindung für das Risikomanagement-Verfahren
•
4.3 RisikobeurteilungDie Risikobeurteilung umfasst das Erkennen von Gefahren sowie die Analyse und Bewertung von Risiken, die sich aus diesen Gefahren ergeben (siehe Definition unten). Die Beurteilung von Qualitätsrisiken beginnt mit einer klar definierten Problembeschreibung oder Fragestellung nach dem Risiko. Wenn das fragliche Risiko klar definiert ist, können geeignete Methoden zum Risikomanagement (siehe Beispiele in Abschnitt 5) und die Art der notwendigen Informationen für den Umgang mit dem jeweiligen Risiko leichter erkannt werden. Als Hilfestellung bei der klaren Definition cler Risiken für die Risikobeurteilung sind häufig drei grundsätzliche Fragen hilfreich:
Welcher Fehler kann auftreten?1.Wie hoch ist die Wahrscheinlichkeit des Auftretens?2.Was sind die Folgen (Ausmaß)?3.
Risikoidentifizierung ist die systematische Nutzung von Informationen zur Identifikation von Gefahren mit Blick auf die Frage nach dem Risiko bzw. die Problembeschreibung. Die Informationen können historische Daten, theoretische Analysen, fundierte Meinungen oder die Befürchtungen der interessierten Parteien umfassen. Die Risikoidentifizierung bezieht sich auf die Frage "Welche Fehler können auftreten?", einschließlich der Bestimmung der möglichen Konsequenzen. Dies bietet die Basis für weitere Schritte im Verfahren zum Qualitäts-Risikomanagement. Risikoanalyse ist die Einschätzung des Risikos, das mit den erkannten Gefahren verbunden ist. Es ist der qualitative oder quantitative Prozess, die Wahrscheinlichkeit des Auftretens eines Risikos mit dem Ausmaß des möglichen Schadens zu verbinden. In einigen Methoden des Risikomanagements fließt die Möglichkeit, den Fehler zu entdecken, auch in die Risikoeinschätzung ein. Risikobewertung vergleicht die erkannten und analysierten Risiken mit festgelegten Risikokriterien. Risikobewertungen berücksichtigen die Stärke der Hinweise bezogen auf die drei Ausgangsfragen. Bei der Durchführung einer effektiven Risikobeurteilung ist die Zuverlässigkeit der Datensätze von großer Bedeutung, weil sie die Qualität des Ergebnisses entscheidend beeinflusst. Die Offenlegung von Vermutungen und erkennbaren Quellen der Unsicherheit verbessern die Zuverlässigkeit dieser Ausgabe und/oder helfen, die Grenzen der Aussagefähigkeit zu erkennen. Unsicherheiten entstehen aus der Kombination von unvollständigem Prozesswissen und dessen erwarteten oder unerwarteten Variabilitäten. Typische Quellen der Unsicherheit sind Wissenslücken, Lücken im pharmazeutischen Wissen oder unzureichendes Prozessverständnis, Schadensquellen (z.B. Fehlerarten eines Prozesses, Ursachen für Variabilitäten) und die Wahrscheinlichkeit der Erkennung von Problemen. Das Ergebnis einer Risikobeurteilung ist entweder die quantitative Schätzung eines Risikos oder die qualitative Beschreibung eines Risikobereichs. Beim quantitativen Ausdrücken eines Risikos wird eine numerische Wahrscheinlichkeit verwendet. Alternativ dazu kann ein Risiko auch anhand qualitativer Merkmale beschrieben werden, z.B. "hoch", "mittel" oder "niedrig", die so detailliert wie möglich definiert sein sollten. Manchmal wird ein Risikowert" benutzt, um Merkmale in der Risikoeinstufung näher zu definieren. Bei einer quantitativen Risikobeurteilung liefert eine Risikoschätzung die Wahrscheinlichkeit einer spezifischen Konsequenz bei einer bestimmten Kombination von Risikobedingungen. Eine quantitative Risikoschätzung ist daher für jeweils eine bestimmte Konsequenz sinnvoll. Alternativ dazu verwenden manche Methoden des Risikomanagements eine relative Risikomessung zur Kombination verschiedener Stufen des Ausmaßes und der Wahrscheinlichkeit zu einer Gesamtschätzung des relativen Risikos. Die Zwischenschritte innerhalb eines Bewertungsprozesses können manchmal eine quantitative Risikoschätzung enthalten. 4.4 RisikosteuerungDie Risikosteuerung umfasst die Entscheidung zur Reduktion bzw. Akzeptanz von Risiken. Der Zweck der Risikosteuerung ist das Reduzieren von Risiken auf ein akzeptables Niveau. Der Aufwand für die Risikosteuerung sollte im angemessenen Verhältnis zur Bedeutung des Risikos stehen. Entscheidungsträger können verschiedene Methoden, einschließlich Kosten-Nutzen-Rechnung, einsetzen, um das optimale Maß der Risikosteuerung zu ermitteln. Die Risikosteuerung könnte sich auf die folgenden Fragen konzentrieren:
Liegt das Risiko oberhalb eines akzeptablen Niveaus?•Was kann getan werden, um Risiken zu reduzieren oder zu eliminieren?•Wie sieht eine angemessene Abwägung zwischen Nutzen, Risiken und Ressourcen aus?•Entstehen durch bereits identifizierte und gesteuerte Risiken neue Risiken?•
Die Risikoreduzierung konzentriert sich auf die Prozesse zur Verringerung oder Vermeidung von Qualitätsrisiken, wenn diese ein festgelegtes (akzeptables) Niveau überschreiten (siehe Abbildung 1). Die Risikoreduzierung kann Maßnahmen zur Verringerung des Ausmaßes und der Wahrscheinlichkeit eines Schadens umfassen. Prozesse, die die Erkennbarkeit von Gefahren und Qualitätsrisiken verbessern, können auch im Rahmen einer Strategie zur Risikosteuerung eingesetzt werden. Die Implementierung von Maßnahmen zur Risikoreduzierung kann neue Risiken ins System einbringen oder die Bedeutung bestehender Risiken verstärken. Es kann daher sinnvoll
Seite 78 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

sein, nach der Implementierung eines Prozesses zur Risikoreduzierung die Risikobeurteilung erneut zu überprüfen und mögliche Veränderungen bei den Risiken zu beurteilen. Risikoakzeptanz ist die Entscheidung, ein Risiko zu akzeptieren. Die Risikoakzeptanz kann eine formale Entscheidung zur Annahme des Restrisikos sein oder eine passive Entscheidung, in der die Restrisiken nicht definiert sind. Bei einigen Schadensarten können auch die besten Verfahren zum Qualitäts-Risikomanagement das Risiko nicht vollständig eliminieren. In diesen Fällen kann vereinbart werden, dass eine geeignete Strategie zum Qualitäts-Risikomanagement angewandt wurde und die Qualitätsrisiken auf ein festgelegtes (akzeptables) Niveau reduziert worden ist. Dieses (festgelegte) akzeptable Niveau hängt von einer Reihe von Parametern ab, so dass darüber von Fall zu Fall entschieden werden. 4.5 RisikokommunikationRisikokommunikation ist die gemeinsame Nutzung von Informationen zu Risiken und dem Risikomanagement zwischen den Entscheidungsträgern und anderen Beteiligten. Die Beteiligten können in jeder Phase des Verfahrens zum Risikomanagement kommunizieren (siehe Abbildung 1: gestrichelte Pfeile). Die Schlussfolgerung bzw. das Ergebnis des Verfahrens zum Qualitäts-Risikomanagement sollte angemessen kommuniziert und dokumentiert werden (siehe durchgehende Pfeile in Abbildung 1). Die Kommunikation beinhaltet diejenige zwischen interessierten Parteien, z.B. Aufsichtsbehörden und Industrie, Industrie und Patient, innerhalb eines Unternehmens, einer Branche oder einer Aufsichtsbehörde usw. Die enthaltenen Informationen können sich auf Existenz, Art, Form, Wahrscheinlichkeit, Ausmaß, Annehmbarkeit, Überwachung, Bewältigung, Erkennbarkeit und andere Aspekte zu Qualitätsrisiken beziehen. Kommunikation muss nicht für jede Risikoakzeptanz einzeln durchgeführt werden. Die Kommunikation zwischen der Industrie und den Überwachungsbehörden hinsichtlich Entscheidungen zum Qualitäts-Risikomanagement kann über bereits bestehende Kommunikationskanäle erfolgen, entsprechend der Definition in Vorschriften und Leitlinien. 4.6 RisikoüberwachungDas Risikomanagement sollte ein kontinuierlicher Teil der Verfahren zum Qualitätsmanagement sein. Ein Mechanismus zur Überprüfung und Überwachung von Ereignissen sollte implementiert werden. Die Schlussfolgerungen bzw. Ergebnisse des Verfahrens zum Risikomanagement müssen überprüft werden, um neue Erkenntnisse und Erfahrungen angemessen zu berücksichtigen. Nachdem ein Verfahren zum Qualitäts-Risikomanagement eingeleitet wurde, sollte dieser Prozess weiter bei Ereignissen angewandt werden, die sich auf eine ursprüngliche Bewertung zum Qualitäts-Risikomanagement auswirken können, unabhängig davon, ob diese Ereignisse geplant (z.B. als Ergebnisse der Produktüberwachung, Inspektionen, Audits, Änderungskontrolle) oder ungeplant sind (z.B. als Ursache aus Fehler-Untersuchungen, bei Rückrufen). Die Häufigkeit jeglicher Überprüfungen sollte entsprechend der jeweiligen Risikostufe festgelegt werden. Die Risikoüberwachung kann dazu führen, dass Entscheidungen zur Risikoakzeptanz neu überdacht werden müssen (Abschnitt 4.4). 5 Methodik des RisikomanagementsDas Qualitäts-Risikomanagement unterstützt einen wissenschaftlichen und praktischen Ansatz zur Entscheidungsfindung. Es stellt dokumentierte, transparente und reproduzierbare Verfahren zur Durchführung der einzelnen Schritte des Verfahrens zum Qualitäts-Risikomanagementprozesses bereit, aufbauend auf bestehendem Wissen über die Bewertung von Auftretenswahrscheinlichkeit, Auswirkung und manchmal Erkennbarkeit von Risiken. Qualitätsrisiken wurden bisher durch vielfältige informelle Vorgehensweisen überprüft und gelenkt (empirische und/oder interne Vorgehensweisen), die z.B. auf der Zusammenstellung von Beobachtungen, Trends und anderen Informationen beruhten. Diese Ansätze liefern weiterhin hilfreiche Informationen, um Fragestellungen zu unterstützen wie die Handhabung von Beanstandungen, Qualitätsmängel, Abweichungen und Zuordnung von Ressourcen. Zusätzlich können die pharmazeutische Industrie und Behörden die Risiken durch die Nutzung von anerkannten Risikomanagementmethoden und/oder internen Vorgehensweisen beurteilen und handhaben (z.B. Standardarbeitsanweisung). Im Folgenden findet man eine Liste solcher Instrumente, die keinen Anspruch auf Vollständigkeit erhebt (mehr Details im Anhang 1 und Abschnitt 8):
grundlegende Methoden zur Erleichterung des Risikomanagements (Fließdiagramme, Checklisten usw.)•Fehlzustandsart- und -auswirkungsanalyse (Failure Mode Effects Analysis, FMEA)•Ausfallbedeutungsanalyse (Failure Mode, Effects and Criticality Analysis, FMECA)•Fehlerbaumanalyse (Fault Tree Analysis, FTA)•Gefahrenanalyse und kritische Steuerungspunkte (HACCP)•Gefährdungs- und Betreibbarkeitsuntersuchung (HAZOP)•vorläufige Gefahrenanalyse (PHA)•Risikoeinstufung und Filterung•unterstützende statistische Methoden•
Unter Umständen empfiehlt es sich, diese Methoden für ihre Verwendung in spezifischen Bereichen mit Bezug zur Qualität von Wirkstoffen und Arzneimitteln anzupassen. Die Methoden des Qualitäts-Risikomanagements und die unterstützenden statistischen Methoden können kombiniert eingesetzt werden (z.B. Risikobeurteilung bezogen auf Auftretenswahrscheinlichkeiten). Die kombinierte Verwendung bietet eine hohe Flexibilität, die die Anwendung der Grundsätze des Qualitäts-Risikomanagements erleichtert. Das Ausmaß an Strenge und Formalität von Qualitätsriskomanagement sollte verfügbare Kenntnisse reflektieren und sollte der Komplexität und/oder Kritikalität des Sachverhalts angemessen sein. 6 Integration von Qualitäts-Risikomanagement in Abläufe von Industrie und AufsichtsbehördenQualitäts-Risikomanagement ist ein Prozess, der in Qualitätssystemen wissenschaftlich fundierte und praktische Entscheidungen unterstützt (siehe Anhang II). Wie in der Einführung beschrieben, entbindet der angemessene Einsatz des Qualitäts-Risikomanagements die Industrie nicht von der Verpflichtung zur Einhaltung der behördlichen Auflagen. Ein effektives Qualitäts-Risikomanagement erleichtert jedoch bessere und fundiertere Entscheidungen und bietet den Aufsichtsbehörden zuverlässigere Informationen darüber, wie gut ein Unternehmen potenzielle Risiken bewältigen kann; dies kann Auswirkungen auf den Umfang der behördlichen Überwachung haben. Darüber hinaus erleichtert das Qualitäts-Risikomanagement eine bessere Nutzung der Ressourcen durch alle Beteiligten. Die Schulung der Mitarbeiter in Industrie und Aufsichtsbehörden zu Verfahren des Qualitäts-Risikomanagements sorgt für ein besseres Verständnis der Entscheidungsprozesse und schafft Vertrauen in die Ergebnisse des Qualitäts-Risikomanagements. Qualitäts-Risikomanagement sollte in die bestehenden Abläufe integriert und angemessen dokumentiert werden. Anhang II enthält einige Beispiele zu Situationen, in denen ein Verfahren zum Qualitäts-Risikomanagement Informationen liefern kann, die anschließend in einer Reihe von pharmazeutischen Prozessen genutzt werden können. Diese Beispiele dienen lediglich der Illustration und sind nicht als definitiv oder als vollständig anzusehen. Diese Beispiele sollen keine neuen Erwartungen über die bestehenden behördlichen Anforderungen hinaus aufstellen. Beispiele für Abläufe in Industrie und Aufsichtsbehörden (siehe Anhang II ):
Seite 79 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Qualitätsmanagement.•
Beispiele für industrielle Abläufe und Aktivitäten (siehe Anhang II ):
Entwicklung•Einrichtungen, Ausrüstung und Infrastruktur•Materialwirtschaft•Produktion•Laborkontrolle und Stabilitätsstudien•Verpackung und Kennzeichnung•
Beispiele für behördliche Abläufe (siehe Anhang II ):
Inspektions- und Bewertungstätigkeiten•
Obwohl die Entscheidungen bei Behörden weiterhin auf regionaler Ebene erfolgen, kann doch ein allgemeines Verständnis und die Anwendung der Grundsätze des Qualitäts-Risikomanagements gegenseitiges Vertrauen schaffen und zu konsistenteren Entscheidungen bei Behörden auf Basis gemeinsamer Informationen sorgen. Diese Zusammenarbeit kann bei der Entwicklung von Standards und Leitlinien von Bedeutung sein, die Methoden des Qualitäts-Risikomanagements umfassen und unterstützen. 7 DefinitionenAnforderungen - Die expliziten oder impliziten Bedürfnisse oder Erwartungen von Patienten oder ihren Vertretern (z.B. Heilberufler, Behörden oder Gesetzgeber). "Anforderungen" in diesem Dokument verweisen nicht nur auf gesetzliche, behördliche oder normative Anforderungen, sondern auch auf einen solchen Bedarf und solche Erwartungen. Ausmaß - Ein Maß für die möglichen Konsequenzen einer Gefahr. Entscheidungsträger - Person(en) mit der Kompetenz und Autorität, um angemessen und rechtzeitig Entscheidungen zum Qualitäts-Risikomanagement zu treffen. Erkennbarkeit - Die Fähigkeit, die Existenz, das Vorhandensein oder die Tatsache einer Gefahr zu erkennen oder festzustellen. Gefahr - Die potenzielle Ursache eines Schadens (ISO/IEC Leitlinie 51). Interessierte Partei/Beteiligte - Jede Person, Gruppe oder Organisation, die Einfluss auf ein Risiko hat oder - vermeintlich oder tatsächlich - durch ein Risiko betroffen ist. Entscheidungsträger können auch interessierte Parteien sein. Im Sinne dieses Leitfadens sind die primären interessierten Parteien Patienten, Heilberufler, Aufsichtsbehörden und die Industrie. Produkt-Lebenszyklus - Alle Phasen im Lebenszyklus von der ersten Entwicklung über das Inverkehrbringen bis zur Einstellung des Produkts. Qualität - Der Grad, mit dem die Eigenschaften eines Produkts, Systems oder Verfahrens die Anforderungen erfüllen (für "Qualität" von Wirkstoffen und Arzneimitteln siehe spezifische Definition im ICH Q6A). Qualitäts-Risikomanagement - Ein systematisches Verfahren für die Beurteilung, Steuerung, Kommunikation und Überwachung von Risiken hinsichtlich der Qualität des Arzneimittels über den Produkt-Lebenszyklus hinweg. Qualitätssystem - Die Summe aller Aspekte eines Systems, das Qualitätsleitlinien implementiert und sicherstellt, dass Qualitätsziele erreicht werden. Risiko - Die Kombination aus der Eintrittswahrscheinlichkeit eines Schadens und dem Ausmaß dieses Schadens (ISO/IEC Leitlinie 51). Risikoakzeptanz - Die Entscheidung, Risiken zu akzeptieren (ISO Leitlinie 73). Risikoanalyse - Die Einschätzung des Risikos mit Rücksicht auf die damit verbundenen Gefahren. Risikobestimmung - Die systematische Anwendung von Informationen zur Erkennung potenzieller Schadensursachen (Gefahren) in Bezug auf die Frage nach dem Risiko bzw. auf die Problembeschreibung Risikobeurteilung - Ein systematischer Prozess der Organisation von Informationen zur Stützung einer zu treffenden Risikoentscheidung innerhalb eines Verfahrens zum Risikomanagement. Sie besteht aus der Erkennung von Gefahren und der Analyse und Bewertung von Risiken, die damit verbunden sind, den Gefahren ausgesetzt zu sein. Risikobewertung - Der Vergleich des geschätzten Risikos mit festgelegten Risikokriterien anhand quantitativer oder qualitativer Skalen zur Bestimmung der Bedeutung des Risikos. Risikokommunikation - Die gemeinsame Nutzung von Informationen zu Risiken und Risikomanagement zwischen dem Entscheidungsträger und anderen Beteiligten. Risikomanagement - Systematische Anwendung der Leitlinien, Verfahren und Methoden des Qualitätsmanagements bei der Bewertung, Steuerung, Kommunikation und Überwachung von Risiken. Risikoreduzierung - Aktionen zur Verringerung der Eintrittswahrscheinlichkeit eines Schadens und des Ausmaßes dieses Schadens. Risikosteuerung - Aktionen zur Implementierung von Entscheidungen aus dem Risikomanagement (ISO Leitlinie 73). Risikoüberwachung - Überprüfung oder Überwachung von Schlussfolgerungen bzw. Ergebnissen des Verfahrens zum Risikomanagement, falls notwendig unter Beachtung neuer Erkenntnisse über das oder Erfahrungen mit dem Risiko. Schaden - Gesundheitliche Schäden einschließlich des Schadens, der durch einen Verlust an Qualität oder Verfügbarkeit eines Produkts entstehen kann. Trend - Ein statistischer Begriff, der die Richtung oder den Grad einer Veränderung von (einer) Variablen darstellt. 8 LiteraturhinweiseICH Q8 Pharmaceutical Development ISO/IEC Guide 73:2002 - Risiko-Management - Wörterbuch - Leitfaden für die Berücksichtigung von Termini zum Risiko-Management in Normen. ISO/IEC Guide 51:1999 - Leitfaden für die Aufnahme von Sicherheitsaspekten in Normen. Process Mapping vom American Productivity & Quality center, 2002, ISBN 1928593739. IEC 61025 - Fehlerbaumanalyse (Fault Tree Analysis, FTA). IEC 60812 Analysetechniken für die Funktionsfähigkeit von Systemen - Verfahren für die Fehlzustandsart- und Auswirkungsanalyse (FMEA). Failure Mode and Effect Analysis, FMEA from Theory to Execution, 2. Ausgabe 2003, D. H. Stamatis, ISBN 0873895983.
Seite 80 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Guidelines for Failure Modes and Effects Analysis (FMEA) for Medical Devices, 2003 Dyadem Press, ISBN 0849319102. The Basics of FMEA, Robin McDermott, Raymond J. Mikulak, Michael R. Beauregard 1996, ISBN 0527763209. WHO Technical Report Series Nr. 908, 2003, Annex 7 Application of Hazard Analysis and Critical Control Point (HACCP) methodology to pharmaceuticals. IEC 61882 - Gefäh:rdungs- und Betreibbarkeitsuntersuchung (HAZOP). ISO 14971:2000 - Anwendung des Risikomanagements auf Medizinprodukte. ISO 7870:1993 - Control Charts (Qualitätsregelkarten). ISO 7871:1997 - Cumulative Sum Charts (Cusumkarten - Anleitung zu Qualitätslenkung und Auswertung von Daten mit Cusumtechniken). ISO 7966:1993 - Acceptance Control Charts (Annahme-Qualitätsregelkarten). ISO 8258:1991 - Shewhart Control Charts (Shewhart-Qualitätsregelkarten). What is Total Quality Control?; The Japanese Way, Kaoru Ishikawa (Translated by David J. Liu), 1985, ISBN 0139524339. .
Methoden und Instrumente des Risikomanagements Anhang I
Dieser Anhang bietet einen allgemeinen Überblick und enthält Verweise auf einige der wichtigsten Methoden, die beim Qualitäts-Risikomanagement durch Industrie und Aufsichtsbehörde eingesetzt werden können. Die Verweise sind als Hilfe gedacht, um sich weitere Kenntnisse und Details zu bestimmten Methoden aneignen zu können. Die Liste erhebt keinen Anspruch auf Vollständigkeit. Es ist wichtig anzumerken, dass es keine Instrumente oder keine Reihe von Instrumenten gibt, die auf jede Situation, in der ein Verfahren zum Qualitäts-Risikomanagement eingesetzt wird, angewendet werden können. I.1 Grundlegende Verfahren zur Erleichterung eines RisikomanagementsEinige der üblicherweise benutzten, einfachen Techniken zur Strukturierung des Risikomanagements durch das Organisieren von Daten und die Erleichterung der Entscheidungsfindung sind:
Ablaufdiagramme•Prüfblätter•Prozessübersicht•Ursache-Wirkungs-Diagramm (auch Ishikawa-Diagramm oder Fischgrät-Diagramm)•
I.2 Fehlzustandsart- und Auswirkungsanalyse (FMEA)FMEA (siehe IEC 60812) ermöglicht die Auswertung potenzieller Fehlerarten für Prozesse und ihre voraussichtliche Auswirkung auf die Ergebnisse und/oder Produktleistung. Nach der Ermittlung der Fehlerarten können potenzielle Fehler über eine Risikoreduzierung eliminiert, eingegrenzt, reduziert oder kontrolliert werden. FMEA erfordert ein gutes Verständnis von Produkten und Prozessen. FMEA bricht die Analyse komplexer Prozesse methodisch in einfacher zu verwaltende Schritte herunter. Es ist eine leistungsstarke Methode für die Zusammenfassung wichtiger Fehlerarten sowie für Faktoren, die diese Fehler verursachen, und voraussichtlicher Auswirkungen dieser Fehler. Mögliche EinsatzbereicheFMEA kann zur Priorisierung von Risiken und zur Überwachung der Effektivität von Risikosteuerungsmaßnahmen verwendet werden. FMEA lässt sich auf Ausrüstung und Einrichtungen anwenden und kann zur Analyse eines Herstellungsschritts und dessen Auswirkungen auf das Produkt oder den Prozess verwendet werden. Sie identifiziert Elemente bzw. Schritte im System, die dieses gefährden. Die Schlussfolgerungen bzw. Ergebnisse einer FMEA können als Grundlage für die Konzeption oder weitergehende Analyse dienen oder um den Ressourceneinsatz zu steuern. I.3 Ausfallbedeutungsanalyse (FMECA)FMEA kann erweitert werden, um zusätzlich das Ausmaß der Konsequenzen eines Fehlers, die jeweilige Wahrscheinlichkeit des Auftretens und seine Erkennbarkeit zu untersuchen. Durch diese Ergänzung wird die FMEA zu einer Ausfallbedeutungsanalyse (Failure Mode, Effects and Criticality Analysis, FMECA, siehe IEC 60812). Zur Durchführung einer solchen Analyse sollten Produkt- oder Prozessspezifikationen festgelegt sein. FMECA kann Punkte ermitteln, an denen zusätzliche vorbeugende Maßnahmen zur Minimierung der Risiken erforderlich sein könnten. Mögliche Einsatzbereiche FMECA sollte in der pharmazeutischen Industrie hauptsächlich bei Fehlern und Risiken in Verbindung mit Herstellungsvorgängen liegen; die Anwendung ist hierauf jedoch nicht begrenzt. Das Ergebnis einer FMECA ist eine relative Risiko-"Punktzahl" für jede Fehlerart, anhand derer die Fehlerarten nach einem relativen Risiko geordnet werden. I.4 Fehlerbaumanalyse (FTA)Das FTA-Verfahren (siehe IEC 61025) ist ein Ansatz, bei dem ein Fehler in der Funktionalität eines Produkts oder Prozesses angenommen wird. Dieses Instrument bewertet Fehler im System (oder Subsystem) einzeln nacheinander; es können jedoch auch mehrere Ursachen von Fehlern durch die Ermittlung von Kausalketten kombiniert werden. Die Ergebnisse werden grafisch als Fehlerbaum dargestellt. Auf jeder Stufe in dieser Baumstruktur sind Kombinationen von Fehlerarten mit logischen Operatoren (UND, ODER etc.) beschrieben. FTA stützt sich auf das Prozessverständnis von Experten, um Kausalzusammenhänge zu erkennen. Mögliche EinsatzbereicheFTA kann genutzt werden, um einen Fehler über seinen Weg bis zu seiner Ursache zurückzuverfolgen. FTA kann bei der Untersuchung von Reklamationen oder Abweichungen eingesetzt werden, um ihre eigentlichen Ursachen zu verstehen und sicherzustellen, dass die beabsichtigten Verbesserungen das Problem in Gänze lösen und nicht zu neuen Problemen führen. Die Fehlerbaumanalyse ist ein effektives Hilfsmittel für die Auswertung der Auswirkungen mehrerer Faktoren auf ein bestimmtes Problem. Das Ergebnis einer FTA beinhaltet eine visuelle Darstellung der Fehlerarten. Sie eignet sich gleichermaßen zur Risikobeurteilung und zur Entwicklung von Überwachungsprogrammen. I.5 Gefahrenanalyse und kritische Lenkungspunkte (HACCP)HACCP ist ein systematisches, proaktives und präventives Verfahren zur Sicherstellung der Produktqualität, -zuverlässigkeit und -sicherheit (siehe WHO Technical Report Series No 908, 2003 Annex 7). Es handelt sich dabei um einen strukturierten Ansatz, bei dem technische und wissenschaftliche Grundsätze zur Analyse, Auswertung, Vermeidung und Steuerung von Risiken oder nachteiligen Auswirkungen von Gefahren angewendet werden, die bei der Konzeption, Entwicklung, Herstellung und Verwendung von Produkten auftreten. HACCP umfasst die folgenden sieben Stufen:
Seite 81 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

1) Durchführung einer Gefahrenanalyse und Ermittlung präventiver Maßnahmen für alle Prozessschritte; 2) Festlegung der kritischen Lenkungspunkte; 3) Einrichtung kritischer Grenzwerte; 4) Einrichtung eines Systems zur Überwachung der kritischen Lenkungspunkte; 5) Einrichtung geeigneter Korrekturmaßnahmen, wenn die Überwachung zeigt, dass die kritischen Lenkungspunkte nicht unter Kontrolle sind; 6) Einrichtung eines Systems, das den effektiven Betrieb des HACCP-Systems nachweist; 7) Einrichtung eines Dokumentationssystems. Mögliche Einsatzbereiche
HACCP kann zur Ermittlung und zum Management von Risiken durch physikalische, chemische und biologische Gefahren (einschließlich mikrobiologischer Verunreinigung) verwendet werden. HACCP ist besonders nützlich, wenn das Verständnis von Produkten und Prozessen ausreicht, um die Bestimmung der kritischen Lenkungspunkte zu unterstützen. Das Ergebnis einer HACCP-Analyse sind Informationen zum Risiko-Management, die die Überwachung der kritischen Lenkungspunkte nicht nur im Herstellungsprozess, sondern auch in anderen Phasen des Lebenszyklus erleichtern. I.6 Gefährdungs- und Betreibbarkeitsuntersuchung (HAZOP)HAZOP (siehe IEC 61882) basiert auf einer Theorie, die davon ausgeht, dass Risikoereignisse durch Abweichungen von der Konzeption oder der beabsichtigten Verwendung verursacht werden. Es handelt sich dabei um ein systematisches Brainstorming-Verfahren zur Ermittlung von Gefahren anhand so genannter "Leitwörter". "Leitwörter" (z.B. Kein, Mehr, Außer, Teil von usw.) werden auf relevante Parameter (wie Verunreinigung, Temperatur) angewandt, um potenzielle Abweichungen gegenüber der üblichen oder beabsichtigten Verwendung ermitteln zu helfen. Häufig kommt hierbei ein Team von Personen mit Erfahrung zur Konzeption eines Verfahrens oder eines Produkts und seiner Anwendung zum Einsatz. Mögliche EinsatzbereicheHAZOP kann auf Herstellungsprozesse, inklusive Lohnherstellung und -formulierung ebenso wie auf vorgelagerte Lieferanten, Ausrüstung und Einrichtungen für Wirkstoffe und Arzneimittel angewandt werden. Es wurde auch bereits in der pharmazeutischen Industrie vorwiegend zur Bewertung der Betriebssicherheit eingesetzt. Wie im Falle der HACCP ist auch das Ergebnis der HAZOP-Analyse eine Liste kritischer Prozessschritte für das Risikomanagement. Dieses Ergebnis erleichtert eine regelmäßige Überwachung der kritischen Punkte im Herstellungsprozess. I.7 Vorläufige Gefahrenanalyse (PHA) PHA ist ein Analyseverfahren, bei dem frühere Erfahrungen und Kenntnisse über Gefahren oder Fehler zur Identifikation zukünftiger Gefahren, Gefahrensituationen und Ereignissen, die zu Schäden führen können, genutzt werden. Außerdem umfasst sie eine Einschätzung der Eintrittswahrscheinlichkeit bei bestimmten Arbeiten, Einrichtungen, Produkten oder Anlagen. Das Verfahren umfasst:
1) Die Ermittlung der Wahrscheinlichkeit, dass das Risikoereignis eintritt, 2) die qualitative Auswertung des Ausmaßes eines damit verbundenen möglichen Verletzungs- oder Gesundheitsschadens, 3) eine relative Einstufung der Gefahren unter Berücksichtigung der Kombination der Schwere und der Wahrscheinlichkeit des Auftretens und 4) die Ermittlung möglicher Gegenmaßnahmen. Mögliche Einsatzbereiche
PHA kann bei der Analyse vorhandener Systeme und bei der Festlegung von Prioritäten für potenzielle Gefahren hilfreich sein, wenn die Umstände die Verwendung eines weiter gehenden Verfahrens verhindern. Das Verfahren kann für die Konzeption von Produkten, Prozessen und Einrichtungen verwendet werden, wie auch für die Auswertung der Gefahrenarten für den allgemeinen Produkttyp, die Produktklasse und letztlich des spezifischen Produkts. PHA wird hauptsächlich zu einem frühen Zeitpunkt in der Projektentwicklung eingesetzt, wenn nur wenige Informationen zu den Details der Konzeption und den Betriebsabläufen vorliegen. PHA bildet daher häufig den Vorläufer für weitere Untersuchungen. Normalerweise werden die im Rahmen von PHA ermittelten Gefahren mit anderen Methoden des Risikomanagements, wie den in diesem Abschnitt beschriebenen, detaillierter bewertet. I.8 Risikoeinstufung und FilterungRisikoeinstufung und Filterung ist eine Methode zum Vergleichen und Einordnen von Risiken. Die Risikoeinstufung komplexer Systeme erfordert normalerweise eine Auswertung mehrerer verschiedener quantitativer and qualitativer Faktoren zu jedem Risiko. Die Methode umfasst das Herunterbrechen einer grundlegenden Risikofrage in so viele Komponenten wie nötig sind, um alle Faktoren, die das Risiko ausmachen, erfassen zu können. Diese Faktoren werden zu einer einzigen relativen Risiko-Punktzahl kombiniert, mit der eine Risiko-Rangfolge erstellt werden kann. "Filter" in Form von Gewichtungsfaktoren oder einer Kappung von Risiko-Punktzahlen können zur Skalierung bzw. Einpassung in die Vorgaben des Managements oder die strategischen Ziele verwendet werden. Mögliche Einsatzbereiche Mit der Risikoeinstufung und Filterung können von Aufsichtsbehörden oder Industrie Prioritäten für zu inspizierende bzw. auditierende Herstellungsstätten festgelegt werden. Die Verfahren zur Risikoeinstufung sind besonders hilfreich in den Fällen, in denen das Portfolio der Risiken und die damit verbundenen zu verwaltenden Konsequenzen sehr vielfältig und mit einer einzigen Methode nur schwer vergleichbar sind. Die Risikoeinstufung ist gut geeignet, wenn das Management quantitativ und qualitativ bewertete Risiken im gleichen organisatorischen Rahmen auswerten muss. I.9 Unterstützende statistische MethodenStatistische Methoden können das Qualitäts-Risikomanagement unterstützen und erleichtern. Sie ermöglichen eine effektive Datenbewertung, Unterstützung in der Bestimmung der Bedeutung von Datensätzen und erleichtern verlässlichere Entscheidungen. Einige der wichtigsten in der pharmazeutischen Industrie gängigen statistischen Methoden sind:
Regelkarten (Beispiele): i.Annahme-Qualitätsregelkarten (siehe ISO 7966)◦
Qualitätsregelkarten für den arithmetischen Mittelwert mit Warngrenzen (siehe ISO 7873)◦
CUSUM-Regelkarten (ISO 7871)◦
Shewhart-Qualitätsregelkarten (siehe ISO 8258)◦
gewichteter gleitender Mittelwert◦
Design of Experiments (DOE)ii.Histogrammeiii.
Seite 82 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Pareto-Diagrammeiv.Prozessfähigkeitsanalysev.
.
Mögliche Anwendungen von Qualitäts-Risikomanagement Anhang II
Dieser Anhang soll helfen, mögliche Einsatzbereiche der Prinzipien und Instrumente des Qualitäts-Risikomanagements in Industrie und Aufsichtsbehörden zu identifizieren. Die Auswahl der spezifischen Instrumente zum Risikomanagement hängt jedoch vollständig von spezifischen Faktoren und Umständen ab. Diese Beispiele wurden zur Illustration bereitgestellt; sie stellen lediglich einen Vorschlag für den potenziellen Einsatz des Qualitäts-Risikomanagements dar. Es ist nicht beabsichtigt, mit diesem Anhang neue Erwartung über die bestehenden behördlichen Anforderungen hinaus aufzustellen. II.1 Qualitäts-Risikomanagement im Rahmen des integrierten QualitätsmanagementsDokumentation
Zur Überprüfung der aktuellen Interpretation und der Anwendung behördlichen Anforderungen•Zur Bestimmung des Bedarfs für SOPs, Leitlinien etc. und/oder die Entwicklung für deren Inhalte•
Schulung
Zur Festlegung der Eignung von erstmaligen und/oder fort-. laufenden Schulungsveranstaltungen entsprechend den Ausbildungen, Erfahrungen und Arbeitsgewohnheiten der Mitarbeiter sowie der regelmäßigen Bewertung früherer Schulungen (z.B. ihrer Effektivität)
•
Zur Ermittlung des Schulungsstatus, der Erfahrung, Qualifikation und der physischen Fähigkeiten, die den Mitarbeitern eine zuverlässige Durchführung einer Tätigkeit ohne nachteilige Auswirkung auf die Qualität des Produkts ermöglichen
•
Qualitätsmängel
Zur Bereitstellung der Grundlage für die Erkennung, Bewertung und Kommunikation der potenziellen Qualitätsauswirkungen von vermuteten Qualitätsdefekten, Reklamationen, Trends, Abweichungen, Untersuchungen, Ergebnissen außerhalb der Spezifikation etc.
•
Zur Erleichterung der Risikokommunikation und Festlegung geeigneter Maßnahmen beim Umgang mit signifikanten Produktmängeln in Zusammenarbeit mit den Aufsichtsbehörden (z.B. Rückruf)
•
Audit/Inspektion
Zur Festlegung der Häufigkeit und des Umfangs von internen und externen Audits unter Berücksichtigung von Faktoren wie z.B.:•bestehende gesetzliche Vorschriften•allgemeiner Status bei der Einhaltung von Vorschriften und Historie des Unternehmens bzw. der jeweiligen Einrichtung•Ergebnisse der Aktivitäten eines Unternehmens im Bereich Qualitäts-Risikomanagement•Komplexität des Standorts•Komplexität der Herstellungsprozesse•Komplexität des Produkts und seiner therapeutischen Bedeutung•Anzahl und Bedeutung der Qualitätsdefekte (z.B. Rückruf)•Ergebnisse früherer Audits/Inspektionen•wesentliche Änderungen bei Gebäuden, Ausrüstung, Verfahren oder Personal in Schlüsselfunktionen•Erfahrung mit der Herstellung eines Produkts (z.B. Häufigkeit, Umfang, Anzahl der Chargen)•Testergebnisse von offiziellen Kontrolllaboren. Regelmäßige Überprüfung•Zur Auswahl, Bewertung und Interpretation von Datentrends im Rahmen der Überprüfung der Produktqualität•Zur Interpretation der Überwachungsdaten (z.B. Stützung einer Bewertung der Eignung der Revalidierung oder Änderungen bei der Probenahme)
•
Change-Management/Änderungskontrolle
Zum Verwalten der Änderungen entsprechend dem Wissensstand und den während der pharmazeutischen Entwicklung und der Herstellung gesammelten Erkenntnissen
•
Zur Bewertung der Bedeutung von Änderungen auf die Verfügbarkeit des Endprodukts•Zur Bewertung der Auswirkungen von Änderungen bei Einrichtungen, Ausrüstung, Material, Herstellungsverfahren oder technischen Transfers auf die Produktqualität
•
Zur Festlegung geeigneter Maßnahmen vor der Implementierung einer Änderung, d. h. zusätzliche Tests, (Re)qualifizierung, (Re)validierung oder Kommunikation mit Behörden
•
Kontinuierliche Verbesserung
Zur Erleichterung kontinuierlicher Verbesserungen in Verfahren über den Produkt-Lebenszyklus.•
II.2 Qualitäts-Risikomanagement im Rahmen behördlicher AbläufeMaßnahmen und Beurteilungen bei Inspektionen
Zur Hilfestellung bei der Zuordnung von Ressourcen, beispielsweise bei der Planung, Häufigkeit und Tiefe der Inspektion und Beurteilung (siehe Abschnitt "Audit/Inspektion" in Anhang II.1)
•
Zur Bewertung der Bedeutung von Qualitätsdefekten, potenziellen Rückrufen, Inspektionsergebnissen usw.•Zur Festlegung der Eignung und Art der Anschlussmaßnahmen im Nachgang zu Inspektionen•
Seite 83 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Zur Bewertung der von der Industrie eingereichten Informationen einschließlich Informationen zur pharmazeutischen Entwicklung•Zur Bewertung der Auswirkung vorgeschlagener Variationen oder Änderungen•Zur Benennung von Risiken, die zwischen Inspektoren und Assessoren kommuniziert werden sollten, um ein besseres Verständnis dafür zu entwickeln, wie Risiken gesteuert werden oder gesteuert werden können (z.B. parametrische Freigabe, Prozessanalytische Technologie (PAT))
•
II.3 Qualitäts-Risikomanagement im Rahmen der Entwicklung
Zur Gestaltung eines Qualitätsprodukts und dessen Herstellungsverfahrens, um die angestrebten Produktleistungsmerkmale dauerhaft zu erreichen (siehe ICH Q8)
•
Zur Erweiterung der Kenntnisse zum Produktverhalten über ein breites Spektrum von Materialeigenschaften (z.B. Partikelgrößenverteilung, Feuchtegehalt, Fließeigenschaften), Verarbeitungsoptionen und Prozessparameter
•
Zur Bewertung der kritischen Eigenschaften von Rohmaterialien, Lösungen, Ausgangsstoffen für Wirkstoffe, Wirkstoffe, Hilfsstoffe oder Packmittel
•
Zur Festlegung geeigneter Spezifikationen, Identifikation kritischer Prozessparameter und Festlegung von Prozesskontrollen (z.B. basierend auf Informationen aus pharmazeutischen Entwicklungsstudien zur klinischen Bedeutung der Qualitätsmerkmale und der Möglichkeit zur Kontrolle dieser Merkmale bei der Verarbeitung)
•
Zur Verringerung der Variabilität der Qualitätsmerkmale:•Reduzierung von Produkt- und Materialdefekten•Reduzierung von Herstellungsfehlern•Zur Bewertung des Bedarfs an zusätzlichen Untersuchungen (z.B. Bioäquivalenz, Stabilität) im Zusammenhang mit Änderungen der Chargengröße und Technologietransfer
•
Zur Nutzung des Konzepts zum "design space" (siehe ICH Q8)•
II.4 Qualitäts-Risikomanagement für Einrichtungen, Ausrüstung und VersorgungseinrichtungenKonzeption von Einrichtungen und Ausrüstung
Zur Bestimmung geeigneter Zonen bei der Konzeption von Gebäuden, Einrichtungen etc.:•Material- und Personalfluss•Minimierung von Verunreinigungen•Maßnahmen zur Schädlingsbekämpfung•Verhindern von Untermischungen•offene gegenüber geschlossenen Systemen•Reinräume versus Isolatorentechnologie•Einrichtungen bzw. Ausrüstungen, die speziellen Produkten zugeordnet oder von diesen getrennt sind•Zur Bestimmung geeigneter Produkt-Kontaktmaterialien für Ausrüstung und Behälter (z.B. Auswahl der Edelstahlsorten, Dichtungen, Schmiermittel)
•
Zur Bestimmung geeigneter Versorgungseinrichtungen (z.B. Dampf, Gase, Energiequelle, Pressluft, Heizung, Lüftung, Klima (HVAC), Wasser)
•
Zur Bestimmung geeigneter Maßnahmen zur präventiven Wartung für die zugehörige Ausrüstung (z.B. Lagerhaltung von Ersatzteilen)
•
Hygieneaspekte in den Einrichtungen•Zum Schutz des Produkts vor Umweltgefahren einschließlich chemischer, mikrobiologischer und physikalischer Gefahren (z.B. Festlegung geeigneter Bekleidung und Schutzbekleidung, Hygienebestimmungen)
•
Zum Schutz der Umwelt (z.B. Personal, Risiko der Kreuzkontamination) gegen Gefahren durch das hergestellte Produkt•
Qualifizierung der Einrichtungen/Ausrüstung/Versorgungseinrichtungen
Zur Bestimmung des Anwendungsbereichs und des Umfangs der Qualifizierung von Einrichtungen, Gebäuden und Ausrüstung für die Herstellung und/oder Laborgeräte (einschließlich geeigneter Kalibrierverfahren)
•
Reinigung von Geräten und Umweltkontrolle
Zur Differenzierung der Maßnahmen und Entscheidungen entsprechend der geplanten Nutzung (z.B. Mehrzweckanlagen vs. fest zugeordnete Anlagen, Chargenproduktion vs. kontinuierliche Herstellung)
•
Zur Festlegung akzeptabler Grenzwerte für die Reinigungsvalidierung•
Kalibrierung/vorbeugende Wartung
Zur Festlegung angemessener Kalibrierungs- und Wartungspläne•
Computersysteme und computergesteuerte Systeme
Zur Auswahl der Konzeption von Computerhardware und -software (z.B. modular, strukturiert, Fehlertoleranz)•Zur Bestimmung des Umfangs der Validierung, z.B.:•Ermittlung kritischer Leistungsparameter•Auswahl der Anforderungen und Konzeptionen•Überprüfung der Codes•Umfang der Tests und Testverfahren•Zuverlässigkeit der elektronischen Aufzeichnungen und Unterschriften•
II.5 Qualitäts-Risikomanagement im Rahmen der Materialwirtschaft
Seite 84 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi

Bewertung und Beurteilung der Lieferanten und Lohnhersteller
Zur Bereitstellung umfassender Auswertungen zu Lieferanten und Lohnherstellern (z.B. Auditing, Qualitätsvereinbarungen mit Lieferanten)
•
Ausgangsmaterial
Zur Bewertung von Unterschieden und potenziellen Qualitätsrisiken durch die Variabilität beim Ausgangsmaterial (z.B. Alter, Synthesepfad)
•
Verwendung des Materials
Zur Bestimmung, ob die Verwendung unter Quarantäne stehenden Materials angemessen ist (z.B. für die weitere interne Verarbeitung)
•
Zur Bestimmung der Zulässigkeit von Um- oder Aufarbeitungen oder der Verwendung zurückgeschickter Materialien•
Lagerungs-, Logistik- und Vertriebsbedingungen
Zur Bewertung der Eignung der Anordnungen, um die Aufrechterhaltung geeigneter Lager- und Transportbedingungen sicherzustellen (z.B. Temperatur, Feuchtigkeit, Behälterdesign)
•
Zur Festlegung der Auswirkungen unterschiedlicher Lagerungs- und Transportbedingungen (z.B. Kühlkettenmanagement) auf die Produktionsqualität, auch in Verbindung mit anderen ICH Leitlinien
•
Zur Aufrechterhaltung der Infrastruktur (z.B. Kapazität zur Sicherstellung geeigneter Versandbedingungen, Zwischenlagerung, Umgang mit gefährlichen Materialien und kontrollierten Substanzen, Zollabwicklung)
•
Zur Bereitstellung von Informationen um die Verfügbarkeit von Arzneimitteln sicherzustellen (z.B. Risikoeinstufung der Lieferkette)
•
II.6 Qualitäts-Risikomanagement als Teil der HerstellungValidierung
Zur Ermittlung des Anwendungsbereichs und des Umfangs der Aktivitäten zu Verifizierung, Qualifizierung und Validierung (z.B. Analyseverfahren, Prozesse, Ausrüstung und Reinigungsverfahren)
•
Zur Bestimmung des Umfangs der Aktivitäten zur Nachverfolgung (z.B. Probenahme, Überwachung, Revalidierung)•Zur Unterscheidung zwischen kritischen und nichtkritischen Prozessschritten zur Erleichterung der Gestaltung von Validierungsstudien
•
In-Prozess-Probenahme und -Untersuchung
Zur Abschätzung der Häufigkeit und des Umfangs von Prüfungen bei In-Prozess-Kontrollen (z.B. zur Rechtfertigung reduzierter Prüfungen unter nachgewiesen kontrollierten Bedingungen)
•
Zur Bewertung und Rechtfertigung der Verwendung von prozessanalytischen Technologien (PAT) in Verbindung mit parametrischer und Echtzeit-Freigabe
•
Produktionsplanung
Zur Festlegung einer geeigneten Produktionsplanung (z.B. Abfolge von Herstellungsverfahren, z.B. fest zugeordnet, Kampagnenherstellung und Parallelfertigung)
•
II.7 Qualitäts-Risikomanagement im Rahmen der Laborkontrolle und der StabilitätsstudienErgebnisse außerhalb der Spezifikation
Zur Identifizierung möglicher Ursachen und Korrekturmaßnahmen bei der Untersuchung von Ergebnissen außerhalb der Spezifikation
•
Zeitraum zwischen Wiederholungstests/Ablaufdatum
Zur Abschätzung der angemessenen Lagerung und Prüfung von Zwischenprodukten, Hilfs- und Ausgangsstoffen•
II.8 Qualitäts-Risikomanagement als Teil von Verpackung und KennzeichnungKonzeption der Verpackung
Zur Konzeption der Sekundärverpackung zum Schutz des primärverpackten Produkts (z.B. zur Sicherstellung der Produktauthentizität und der Lesbarkeit der Beschriftung)
•
Auswahl des Behälter-Verschlusssystems
Zur Bestimmung der kritischen Parameter des Behälter-Verschlusssystems•
Kontrolle der Kennzeichnung
Zur Konzeption der Kontrollverfahren für die Kennzeichnung unter Berücksichtigung möglicher Untermischungen verschiedener Produktetiketten, einschließlich unterschiedlicher Versionen des gleichen Etiketts
•
Seite 85 von 85umwelt-online: Bekanntmachung zu § 2 Nr. 3 der Arzneimittel- und Wirkstoffher...
09.01.2013http://www.umwelt-online.de/cgi-bin/listen/drucken.cgi





























