Embed Size (px)
Citation preview

0
UNIVERSIDAD AUTONOMA METROPOLITANAIZTAPALAPA
División de Ciencias Básicas e Ingeniería
Proyecto de LicenciaturaSíntesis, Caracterización e inserción macrociclos H2T(o-
OH)PP , H2T(3,5-OH)PP , CoT(3,5-OH)PP, Fe(OH)T(m-OH)TPP en geles de sílice.
que presenta
______________________________Q. Martha Patricia González Arévalo
____________________________M. Q. Miguel A. García Sánchez
AsesorLaboratorio de Investigación R101
Área de Química. InorgánicaDepartamento de Química
Febrero de 2005


1
Índice
Pág.Nomenclatura…………………………………………………….2.
Objetivo……………………………………………………………2.
1. Resumen………………………………………………………..2.
2. Introducción 2.
3. Experimentación
a) Prueba 1. Síntesis de H2T(o-OH)TPP………………………….5.
b) Prueba 2: Síntesis de CoT(o-OH)TPP………………………..6.
c) Síntesis del complejo CoT(3,5-OH)PP………………………..9.
d) Desmetalización………………………………………………….11.
e. Repetición de la síntesis del macrociclo…………………….12.
f) Inserción del macrociclo en sol-gel………………………….13.
4. Discusión y resultado………………………………………14.
5. Conclusiones…………………………………………………24.
6. Bibliografía…………………………………………………….25.
.

2
Síntesis, Caracterización e inserción macrociclos H2T(o-OH)PP , H2T(3,5-OH)PP , CoT(3,5-OH)PP, Fe(OH)T(m-
OH)TPP en geles de sílice.
NOMENCLATURA:H2T(o-OH)PP = Base libre de tetra-(orto-hidroxifenil)porfirinaH2T(3,5-OH)PP = Base libre de tetra-(3,5-dihidroxifenil)porfirinaCoT(3,5-OH)PP = de tetra-(3,5-dihidroxifenil)porfirina de cobalto(II ó III)Fe(OH)T(m-OH)TPP = µ-hidroxi, tetra-(3,5-dihidroxifenil)porfirina de hierro (III).
OBJETIVO.A) Sintetizar, caracterizar y purificar dos de las siguientes especie
macrocíclicas: H2T(o-OH)PP, H2(3,5-OH)PP, Co(3,5-OH)PP yFe(OH)T(3,5-OH)PP.
B) Inserción de las anteriores especies en geles de sílice por el método sol-gel.
1. Resumen.El interés principal de la presente investigación fue la síntesis y caracterización de
tetraporfirinas libres sustituidas con grupos hidroxilo (OH) en las posiciones orto
(2), H2T(o-OH)TPP y 3, 5 H2 T(3, 5-OH)TPP y sus complejos de cobalto y hierro e
insertarlas en geles de silicio por el método sol-gel. Lo anterior es motivado por las
importantes propiedades ópticas, catalíticas y fotoquímicas que estas moléculas
presentan, pero sobre todo por la posibilidad que el proceso sol-gel ofrece para
atraparlas en una matriz sólida que confiere estabilidad, dureza y las propiedades
inherentes del óxido metálico al sistema mixto.
2. Introducción.
Las porfirinas son moléculas que pertenecen a la familia de los macrociclostetrapirrólicos. Se sabe que muchas porfirinas y metaloporfirinas tienenfunciones metabólicas debido a sus sustituyentes periféricos y al metal queocupa el centro del macrociclo [1]. Por ejemplo, las porfirinas de hierro forman elgrupo prostético de varias hemoproteínas, citocromos e incluso de algunasenzimas como la peroxidasa o la catalasa. El magnesio se une a las clorinas quecolectan la energía necesaria para la fotosíntesis y el cobalto se une a la porfirinaque existe en la vitamina B12 ( fig. 1).
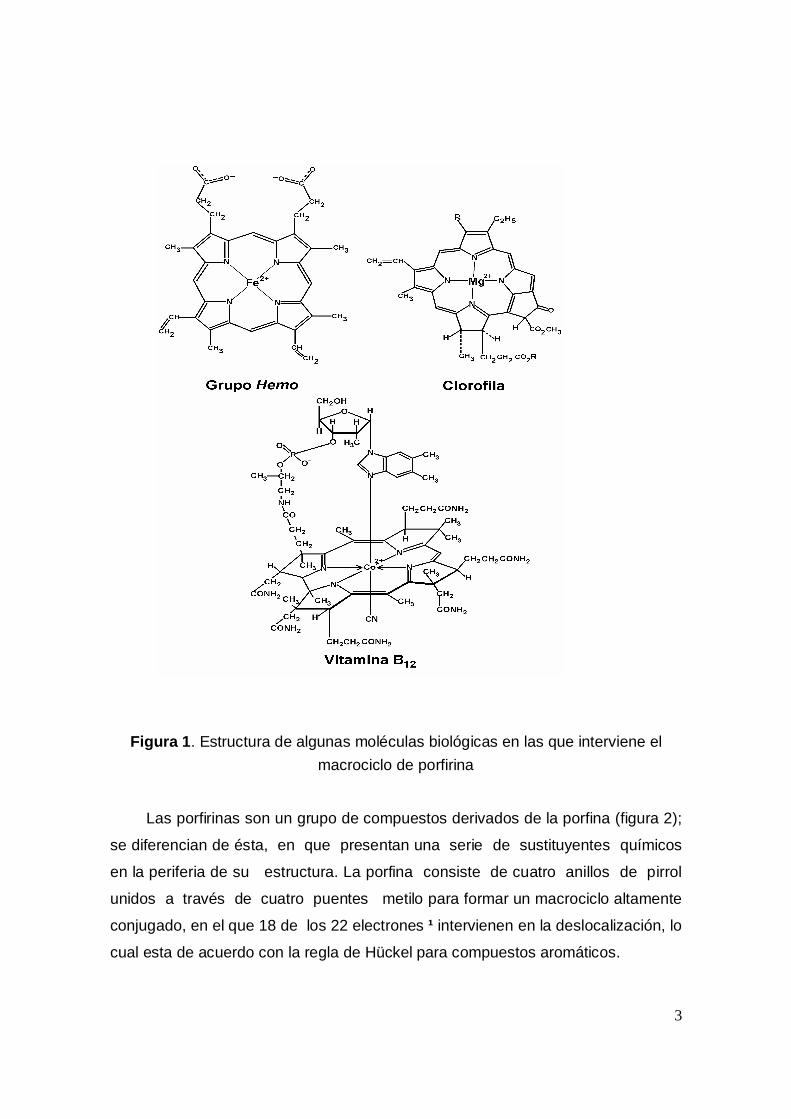
3
Figura 1. Estructura de algunas moléculas biológicas en las que interviene elmacrociclo de porfirina
Las porfirinas son un grupo de compuestos derivados de la porfina (figura 2);
se diferencian de ésta, en que presentan una serie de sustituyentes químicos
en la periferia de su estructura. La porfina consiste de cuatro anillos de pirrol
unidos a través de cuatro puentes metilo para formar un macrociclo altamente
conjugado, en el que 18 de los 22 electrones ¹ intervienen en la deslocalización, lo
cual esta de acuerdo con la regla de Hückel para compuestos aromáticos.

4
igura 2. Estructura del macrociclo de porfina.
En la estructura de las porfirinas existen 4 átomos de nitrógeno; dos
nitrógenos pirrólicos que como en el caso de las ftalocianina libre sostienen un
átomo de hidrógeno y dos nitrógenos aza que mantienen un doble enlace con un
carbono del anillo. Las metalporfirinas, son aquellas especies derivadas de las
porfirinas, que se generan cuando un catión desplaza a uno o preferentemente a
los dos hidrógenos pirrólicos e incluso puede coordinarse con los nitrógenos aza
por medio de los pares de electrones libres que estos últimos poseen. Puede
considerarse a las metaloporfirinas como la combinación de un catión cualquiera
con el anión porfinato P2-, que es un ligando tetradentado, dinegativo, rígido y
cerrado (en el que la carga negativa se deslocaliza por medio del sistema de
electrones que forman los cuatro nitrógenos). Las porfirinas son compuestos
altamente coloreados y estables, pero sus soluciones suelen ser inestables a la
luz. El macrociclo de porfirina es estable en ácido sulfúrico (que se usa como
desmetalizador), pero puede destruirse con ácido perclórico, ácido crómico, ácido
yodhídrico o permanganato.
El diámetro de la ventana central de la porfirina está restringido y por lo
tanto no puede acomodar tan fácilmente al metal cuyo radio iónico sea mayor
que 2.01A.2
Esto determina que el ión central se encuentre o no en el plano de la
molécula. Así mismo, los ligandos axiales pueden provocar el mismo efecto e
5
incluso deformar la estructura del macrociclo porfirínico. En 1883, Soret
descubrió una banda intensa de absorción (que desde entonces se nombró banda
de Soret), cerca de los 400 nm al trabajar con hemoglobina, posteriormente
Gaugee la observó en las porfirinas. Actualmente se sabe que esta banda es la
más característica e intensa ( = 400,000. ) en las porfirinas.
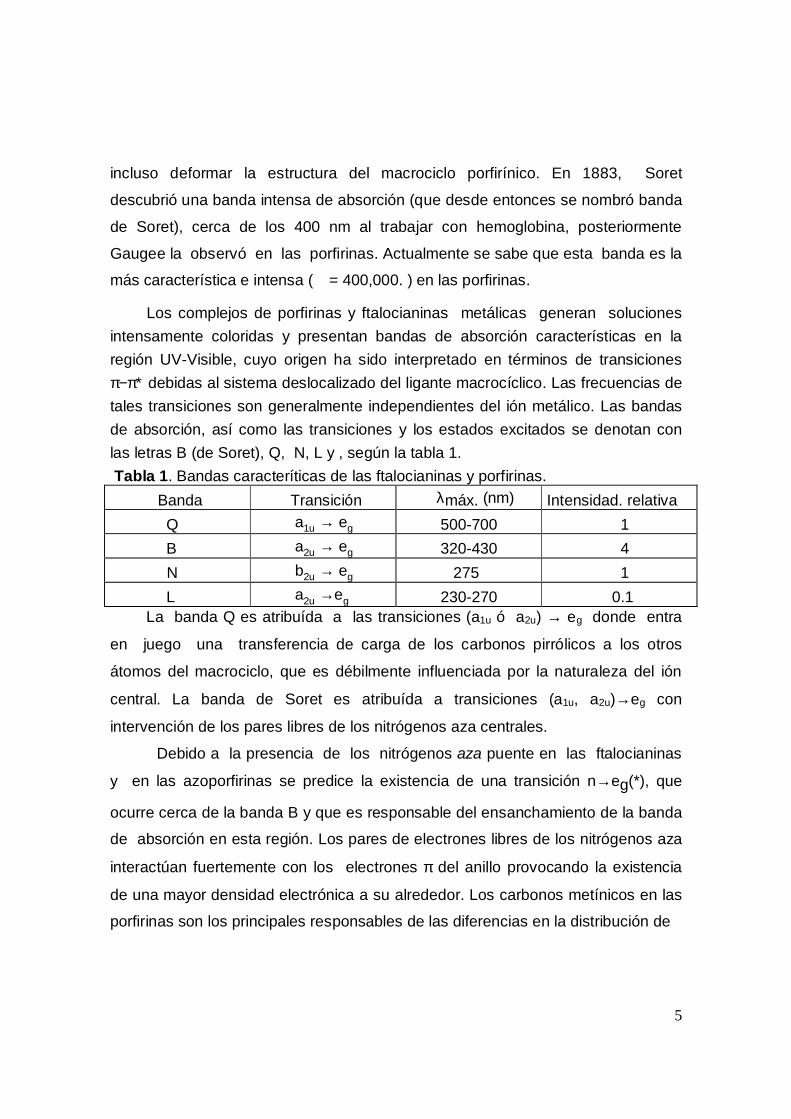
Los complejos de porfirinas y ftalocianinas metálicas generan solucionesintensamente coloridas y presentan bandas de absorción características en laregión UV-Visible, cuyo origen ha sido interpretado en términos de transicionesπ−π∗ debidas al sistema deslocalizado del ligante macrocíclico. Las frecuencias detales transiciones son generalmente independientes del ión metálico. Las bandasde absorción, así como las transiciones y los estados excitados se denotan conlas letras B (de Soret), Q, N, L y , según la tabla 1.Tabla 1. Bandas caracteríticas de las ftalocianinas y porfirinas. Banda Transición λmáx. (nm) Intensidad. relativa Q a1u → eg 500-700 1 B a2u → eg 320-430 4 N b2u → eg 275 1 L a2u →eg 230-270 0.1 La banda Q es atribuída a las transiciones (a1u ó a2u) → eg donde entra
en juego una transferencia de carga de los carbonos pirrólicos a los otros
átomos del macrociclo, que es débilmente influenciada por la naturaleza del ión
central. La banda de Soret es atribuída a transiciones (a1u, a2u)→eg con
intervención de los pares libres de los nitrógenos aza centrales.
Debido a la presencia de los nitrógenos aza puente en las ftalocianinas
y en las azoporfirinas se predice la existencia de una transición n→eg(*), que
ocurre cerca de la banda B y que es responsable del ensanchamiento de la banda
de absorción en esta región. Los pares de electrones libres de los nitrógenos aza
interactúan fuertemente con los electrones π del anillo provocando la existencia
de una mayor densidad electrónica a su alrededor. Los carbonos metínicos en las
porfirinas son los principales responsables de las diferencias en la distribución de

6
carga, ya que se ha calculado que el 85 % de la carga total se localiza en el anillo
de pirrol. Con regularidad se observa una señal próxima a la banda Q, en
algunos casos mejor resuelta que en otros y que se interpreta como una banda
vibriónica para la transición Q(0 -1). Esta señal desplazada hacia el azúl (610
nm), tiene una intensidad que en forma aproximada es 10 veces menor que la de
la transición electrónica Q(0 -0).
En las porfirinas el extenso sistema de electrones π deslocalizados confiere a
la molécula importantes propiedades espectroscópicas [2-5] y ópticas [6-8]. En el
mismo sentido, se sabe que los macrociclos libres de porfirina son excelentes
pigmentos fotoquímicos en los que se presenta el fenómeno hole-burning, por lo cual
se les ha puesto mucha atención debido a la posibilidad de usarlos en la creación de
sistema de almacenamiento de información de alta densidad [9-14]. Esta y otras
propiedades han inducido a realizar un considerable esfuerzo para atrapar este tipo
de moléculas en matrices amorfas [12, 13, 15- 21].
El proceso sol-gel es una técnica de síntesis química que permite preparar
materiales diversos como geles de óxidos, polvos, monolitos, materiales cerámicos y
películas delgadas a temperaturas inferiores a las comunmente necesarias [22-27].
La versatilidad anterior se debe a la posibilidad de controlar la fisicoquímica de las
reacciones de hidrólisis condensación. Así mismo, el hecho de que el proceso ocurra
a temperatura ambiente permite encapsular numerosos compuestos orgánicos,
organometálicos, bioquímicos e incluso biológicos en matrices inorgánicas. En
consecuencia las propiedades de estos materiales mixtos quedan mayormente
determinadas por las propiedades de la molécula alojada. En las dos últimas decadas
se ha reconocido al método sol-gel como una opción muy importante para la
preparación de nuevos y novedosos materiales, en especial en el area de óptica y
sensores químicos [24-27].
Se sabe que para insertar complejos macrocicliclos del tipo de las porfirinas y
ftalocianinas en sólidos porosos es necesario utilizar las versiones sustituidas con
grupos tales como -SO3-, -COOH, -N(R)3+, etc., que los vuelven más solubles en
agua y alcoholes y de esta forma poder ser atrapadas por el proceso sol-gel [24, 25,
27-32]. En el presente trabajo se realizó la síntesis y caracterización, asi como la

7
inserción en geles monolíticos de sílice obtenidos por el procedimiento sol-gel. La
intención de lo anterior es explorar el efecto que tiene la posición y tipo de
sustituyentes en la periféria del macrociclo de tetrafenilporfirina sobre las
propiedades y caracteristicas del sistema mixto finalmente obtenido. Estos
resultados permitiran optimizar los métodos de inserción de estas especies y las
propiedades deseadas.
3. Experimentación.
a) Prueba 1. Síntesis de H2T(o-OH)TPP.
En un matraz de bola de 500 mL se mezclaron 5 g de acetato de zinc
dihidratado (Zn(OAc)2.2H2O), 9.6 mL de salicilaldehído, después de disolver estos
fue agregado 6.3 ml de pirrol lentamente. Esta solución se mantuvo con agitación
a reflujo por un lapso de tres horas, obteniéndose una mezcla obscura. La
reacción fue seguida por espectroscopia UV-visible. Se dejo enfriar y se agrego
cloroformo, esta fue filtrada.
El producto fue seguido por UV-visible (en un espectrofotómetro UV-Visible,
Cary 5E), desafortunadamente con esta síntesis no pudimos obtener nuestro
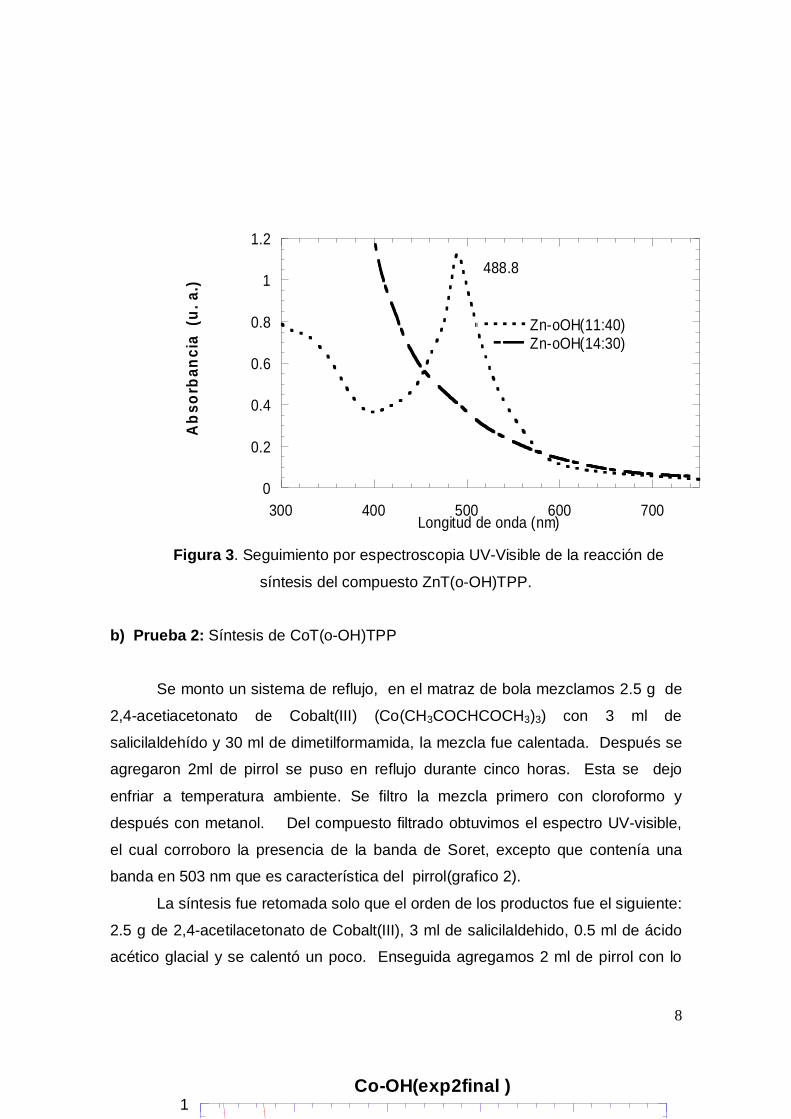
producto, este es un precipitado negro, a partir del espectro de absorción (Figura
3) pudimos corroborar que no contamos con el producto deseado, ya que no fue
posible encontrar la banda de Soret(420-430 nm) tan característica de las
porfirinas, por tal motivo desechamos el producto y descartamos la posibilidad de
utilizar acetato de zinc dihidratado(Zn(OAc)2 * 2H2O) en esta síntesis.
8
0
0.2
0.4
0.6
0.8
1
1.2
300 400 500 600 700
Zn-oOH(11:40)Zn-oOH(14:30)
Abs
orba
ncia
(u.
a.)
Longitud de onda (nm)
488.8
Figura 3. Seguimiento por espectroscopia UV-Visible de la reacción de
síntesis del compuesto ZnT(o-OH)TPP.
b) Prueba 2: Síntesis de CoT(o-OH)TPP
Se monto un sistema de reflujo, en el matraz de bola mezclamos 2.5 g de
2,4-acetiacetonato de Cobalt(III) (Co(CH3COCHCOCH3)3) con 3 ml de
salicilaldehído y 30 ml de dimetilformamida, la mezcla fue calentada. Después se
agregaron 2ml de pirrol se puso en reflujo durante cinco horas. Esta se dejo
enfriar a temperatura ambiente. Se filtro la mezcla primero con cloroformo y
después con metanol. Del compuesto filtrado obtuvimos el espectro UV-visible,
el cual corroboro la presencia de la banda de Soret, excepto que contenía una
banda en 503 nm que es característica del pirrol(grafico 2).
La síntesis fue retomada solo que el orden de los productos fue el siguiente:
2.5 g de 2,4-acetilacetonato de Cobalt(III), 3 ml de salicilaldehido, 0.5 ml de ácido
acético glacial y se calentó un poco. Enseguida agregamos 2 ml de pirrol con lo
1Co-OH(exp2final )
9
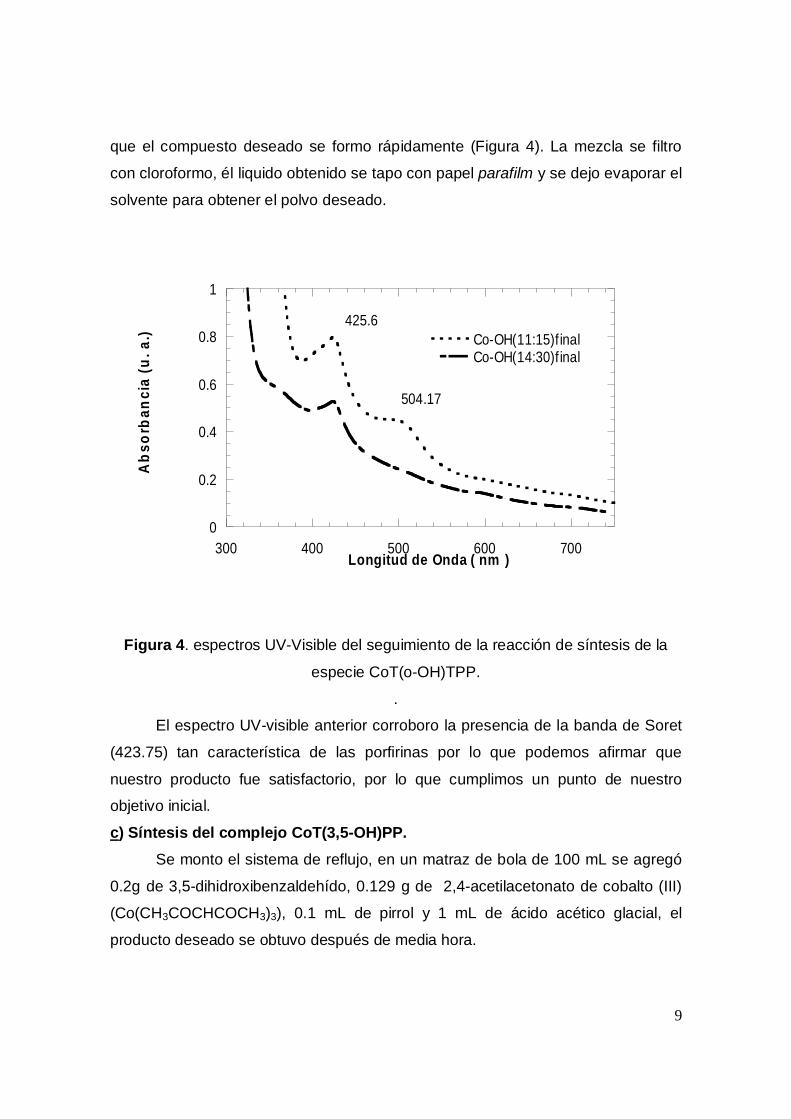
que el compuesto deseado se formo rápidamente (Figura 4). La mezcla se filtro
con cloroformo, él liquido obtenido se tapo con papel parafilm y se dejo evaporar el
solvente para obtener el polvo deseado.
0
0.2
0.4
0.6
0.8
1
300 400 500 600 700
Co-OH(11:15)f inalCo-OH(14:30)f inal
Abs
orba
ncia
(u.
a.)
Longitud de Onda ( nm )
425.6
504.17
Figura 4. espectros UV-Visible del seguimiento de la reacción de síntesis de la
especie CoT(o-OH)TPP.
.
El espectro UV-visible anterior corroboro la presencia de la banda de Soret
(423.75) tan característica de las porfirinas por lo que podemos afirmar que
nuestro producto fue satisfactorio, por lo que cumplimos un punto de nuestro
objetivo inicial.
c) Síntesis del complejo CoT(3,5-OH)PP.Se monto el sistema de reflujo, en un matraz de bola de 100 mL se agregó
0.2g de 3,5-dihidroxibenzaldehído, 0.129 g de 2,4-acetilacetonato de cobalto (III)
(Co(CH3COCHCOCH3)3), 0.1 mL de pirrol y 1 mL de ácido acético glacial, el
producto deseado se obtuvo después de media hora.
10
El polvo final fue disuelto en metanol para ser corrido en una columna
cromatográfica de alumina, el resultado de esta aplicación es que el solvente no
eluyo, se probaron otros solventes probando que la columna no servia para
nuestro fin, por lo que decidimos realizar extracciones con metanol, de estas se
obtuvo un espectro que corroboro la presencia de nuestro compuesto. El solvente
fue evaporado con un evaporador rotatorio. Despues de la realización de la
extracción con metanol pudimos obtener nuestro compuesto para realizar la
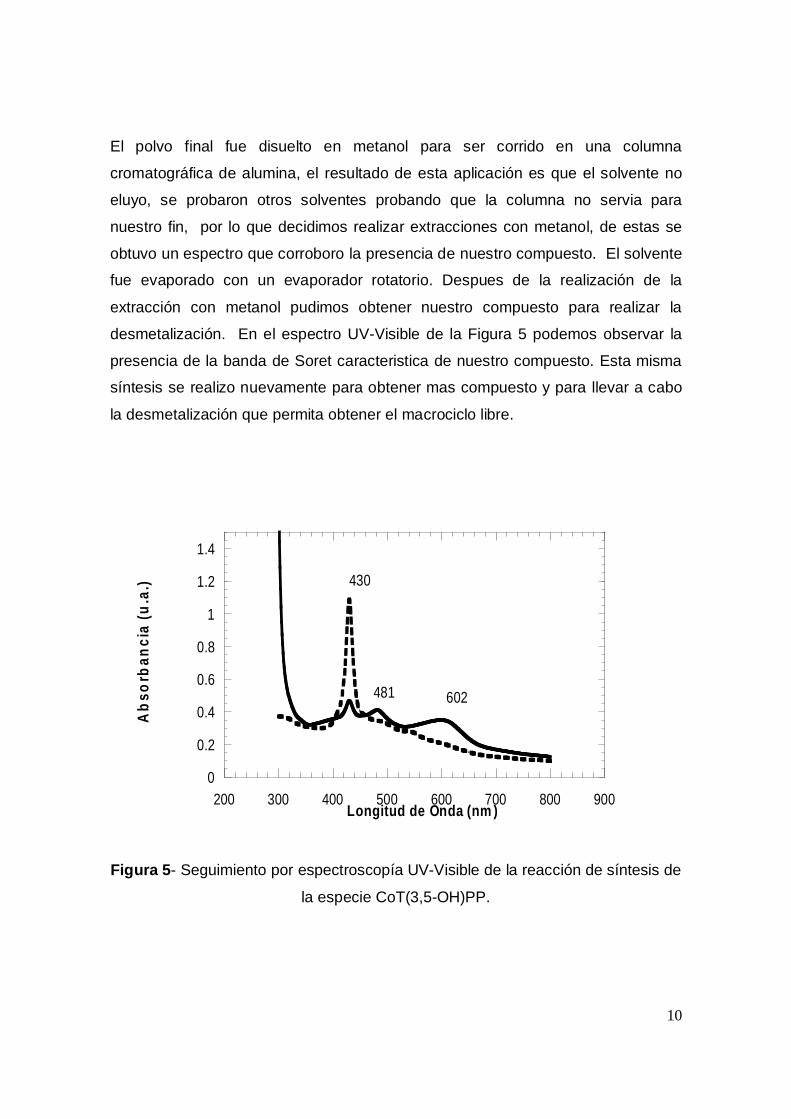
desmetalización. En el espectro UV-Visible de la Figura 5 podemos observar la
presencia de la banda de Soret caracteristica de nuestro compuesto. Esta misma
síntesis se realizo nuevamente para obtener mas compuesto y para llevar a cabo
la desmetalización que permita obtener el macrociclo libre.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
200 300 400 500 600 700 800 900
Abs
orba
ncia
(u.
a.)
Longitud de Onda (nm )
430
481 602
Figura 5- Seguimiento por espectroscopía UV-Visible de la reacción de síntesis de
la especie CoT(3,5-OH)PP.
11
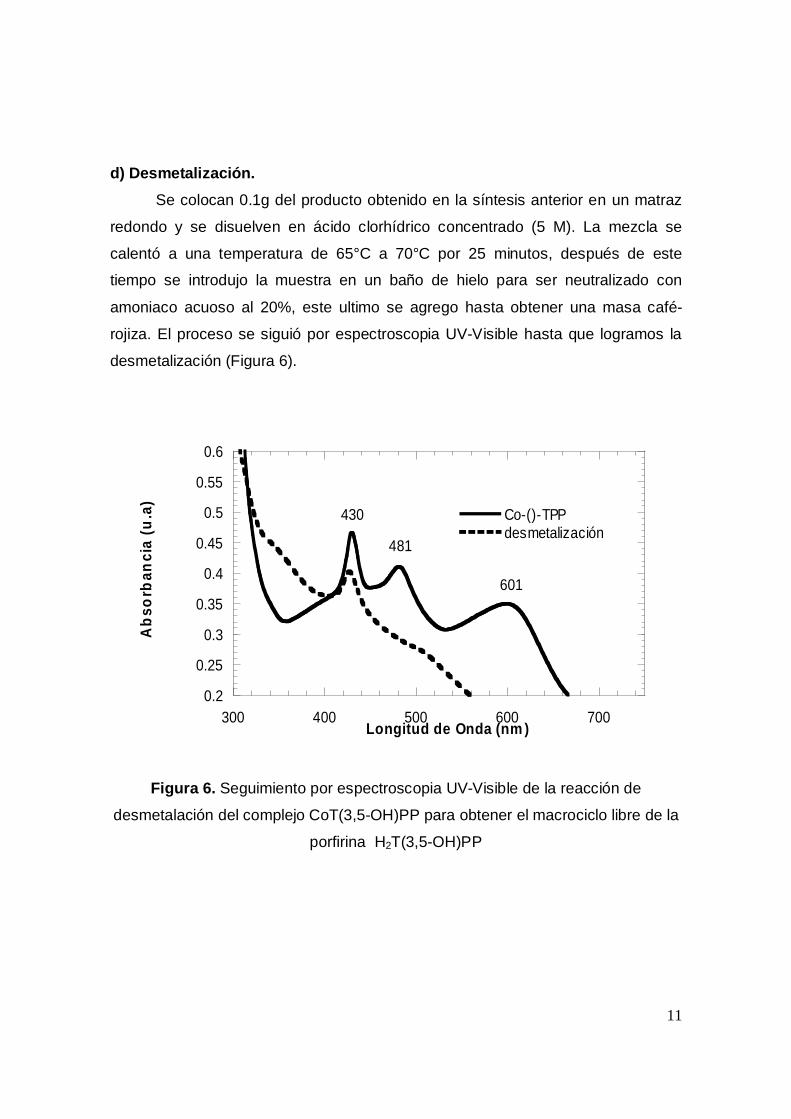
d) Desmetalización.Se colocan 0.1g del producto obtenido en la síntesis anterior en un matraz
redondo y se disuelven en ácido clorhídrico concentrado (5 M). La mezcla se
calentó a una temperatura de 65°C a 70°C por 25 minutos, después de este
tiempo se introdujo la muestra en un baño de hielo para ser neutralizado con
amoniaco acuoso al 20%, este ultimo se agrego hasta obtener una masa café-
rojiza. El proceso se siguió por espectroscopia UV-Visible hasta que logramos la
desmetalización (Figura 6).
0.2
0.25
0.3
0.35
0.4
0.45
0.5
0.55
0.6
300 400 500 600 700
Co-()-TPPdesmetalización
Abs
orba
ncia
(u.
a)
Longitud de Onda (nm )
430
481
601
Figura 6. Seguimiento por espectroscopia UV-Visible de la reacción de
desmetalación del complejo CoT(3,5-OH)PP para obtener el macrociclo libre de la
porfirina H2T(3,5-OH)PP
12
e. Repetición de la síntesis del macrociclo
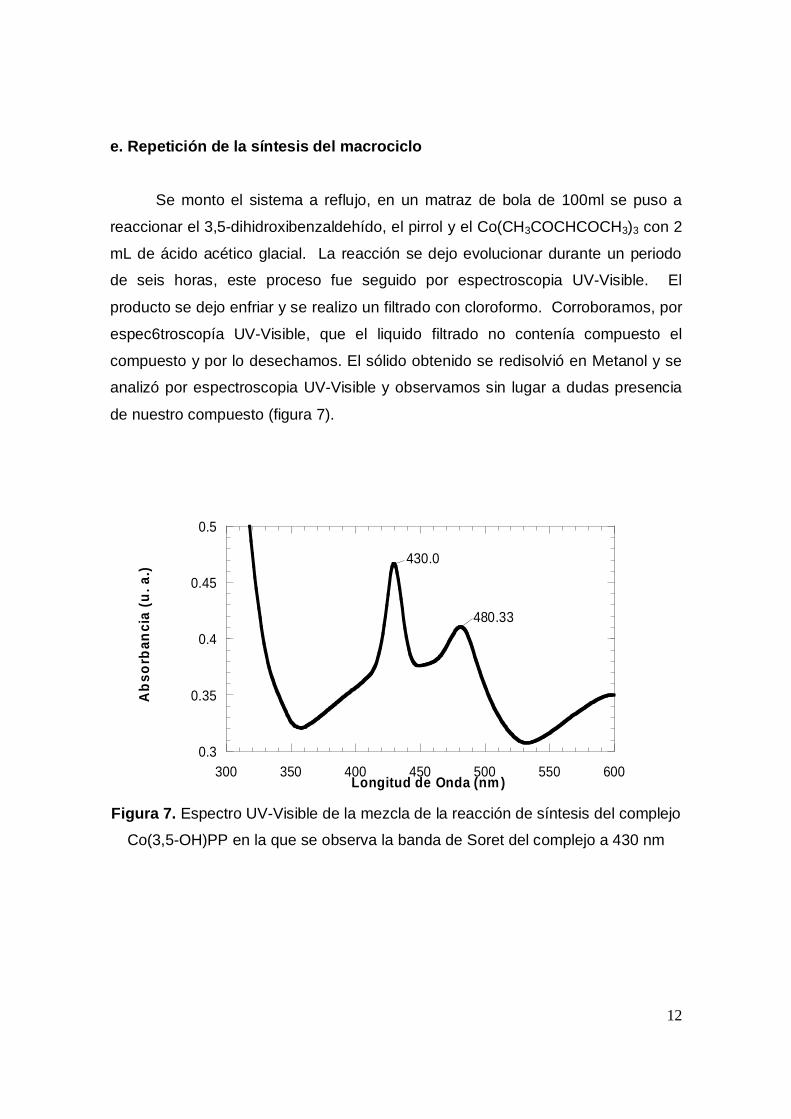
Se monto el sistema a reflujo, en un matraz de bola de 100ml se puso a
reaccionar el 3,5-dihidroxibenzaldehído, el pirrol y el Co(CH3COCHCOCH3)3 con 2
mL de ácido acético glacial. La reacción se dejo evolucionar durante un periodo
de seis horas, este proceso fue seguido por espectroscopia UV-Visible. El
producto se dejo enfriar y se realizo un filtrado con cloroformo. Corroboramos, por
espec6troscopía UV-Visible, que el liquido filtrado no contenía compuesto el
compuesto y por lo desechamos. El sólido obtenido se redisolvió en Metanol y se
analizó por espectroscopia UV-Visible y observamos sin lugar a dudas presencia
de nuestro compuesto (figura 7).
0.3
0.35
0.4
0.45
0.5
300 350 400 450 500 550 600
Abs
orba
ncia
(u.
a.)
Longitud de Onda (nm )
430.0
480.33
Figura 7. Espectro UV-Visible de la mezcla de la reacción de síntesis del complejo
Co(3,5-OH)PP en la que se observa la banda de Soret del complejo a 430 nm
13
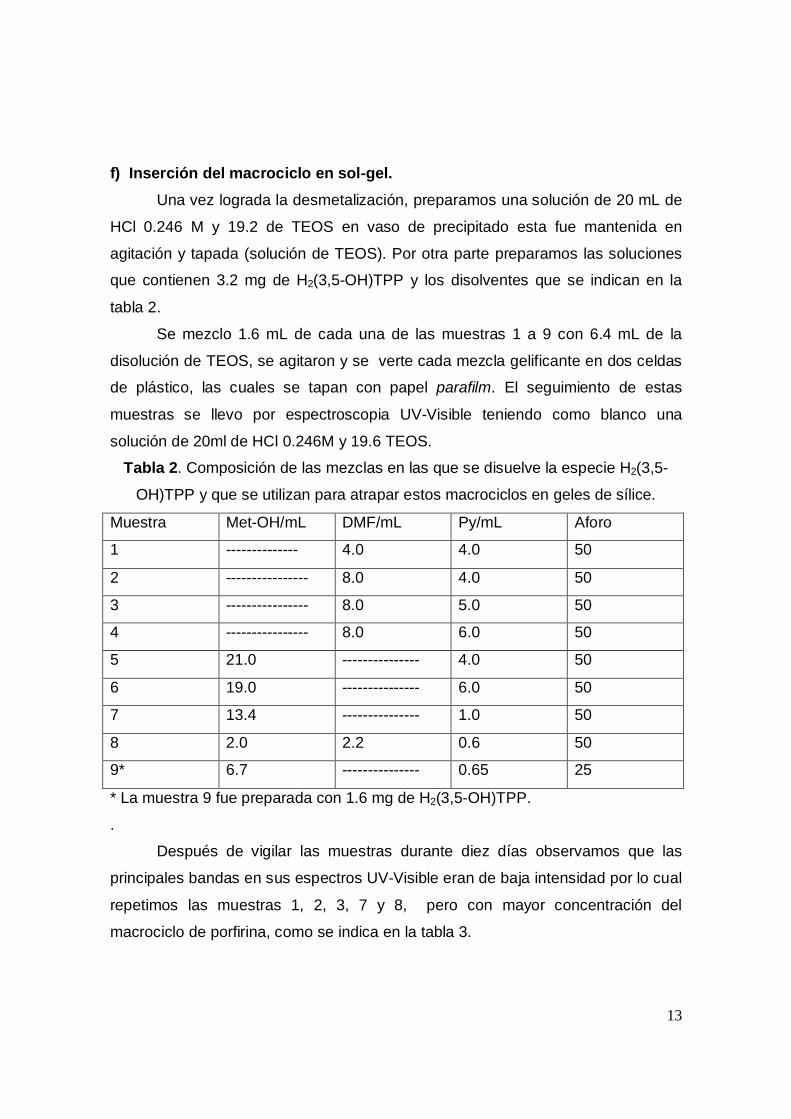
f) Inserción del macrociclo en sol-gel.Una vez lograda la desmetalización, preparamos una solución de 20 mL de
HCl 0.246 M y 19.2 de TEOS en vaso de precipitado esta fue mantenida en
agitación y tapada (solución de TEOS). Por otra parte preparamos las soluciones
que contienen 3.2 mg de H2(3,5-OH)TPP y los disolventes que se indican en la
tabla 2.
Se mezclo 1.6 mL de cada una de las muestras 1 a 9 con 6.4 mL de la
disolución de TEOS, se agitaron y se verte cada mezcla gelificante en dos celdas
de plástico, las cuales se tapan con papel parafilm. El seguimiento de estas
muestras se llevo por espectroscopia UV-Visible teniendo como blanco una
solución de 20ml de HCl 0.246M y 19.6 TEOS.
Tabla 2. Composición de las mezclas en las que se disuelve la especie H2(3,5-
OH)TPP y que se utilizan para atrapar estos macrociclos en geles de sílice.
Muestra Met-OH/mL DMF/mL Py/mL Aforo
1 -------------- 4.0 4.0 50
2 ---------------- 8.0 4.0 50
3 ---------------- 8.0 5.0 50
4 ---------------- 8.0 6.0 50
5 21.0 --------------- 4.0 50
6 19.0 --------------- 6.0 50
7 13.4 --------------- 1.0 50
8 2.0 2.2 0.6 50
9* 6.7 --------------- 0.65 25
* La muestra 9 fue preparada con 1.6 mg de H2(3,5-OH)TPP.
.
Después de vigilar las muestras durante diez días observamos que las
principales bandas en sus espectros UV-Visible eran de baja intensidad por lo cual
repetimos las muestras 1, 2, 3, 7 y 8, pero con mayor concentración del
macrociclo de porfirina, como se indica en la tabla 3.
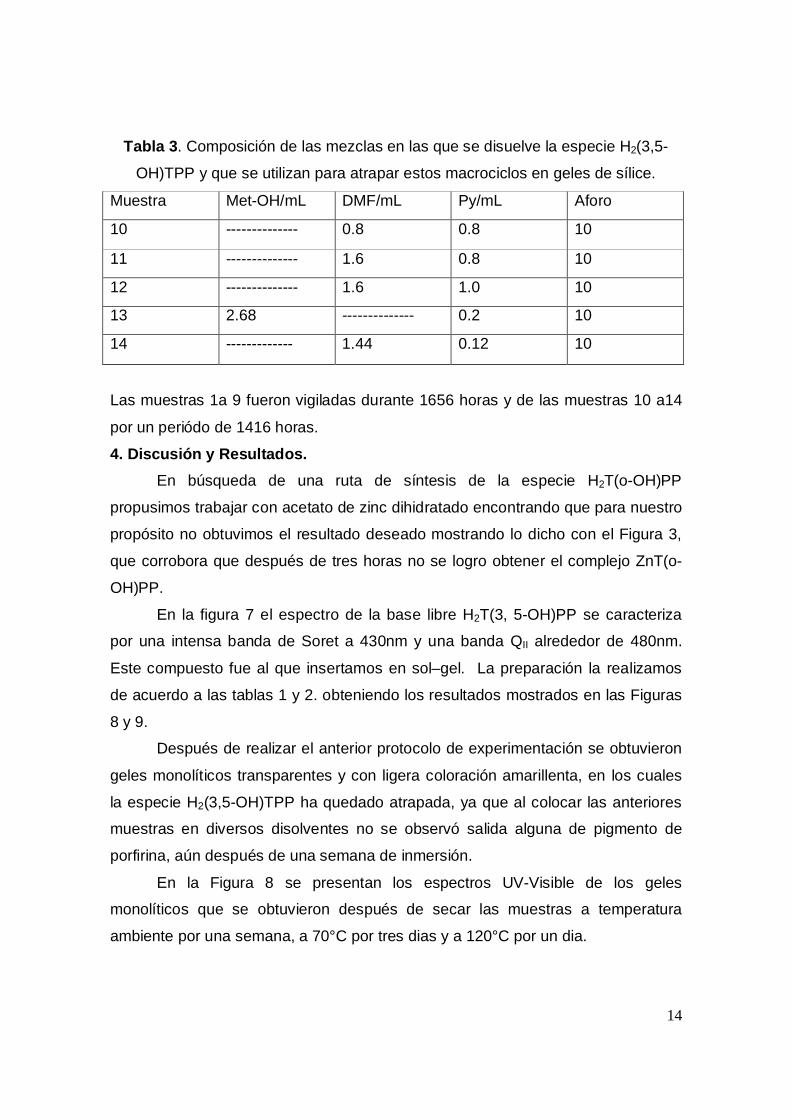
14
Tabla 3. Composición de las mezclas en las que se disuelve la especie H2(3,5-
OH)TPP y que se utilizan para atrapar estos macrociclos en geles de sílice.
Muestra Met-OH/mL DMF/mL Py/mL Aforo
10 -------------- 0.8 0.8 10
11 -------------- 1.6 0.8 10
12 -------------- 1.6 1.0 10
13 2.68 -------------- 0.2 10
14 ------------- 1.44 0.12 10
Las muestras 1a 9 fueron vigiladas durante 1656 horas y de las muestras 10 a14
por un periódo de 1416 horas.
4. Discusión y Resultados.En búsqueda de una ruta de síntesis de la especie H2T(o-OH)PP
propusimos trabajar con acetato de zinc dihidratado encontrando que para nuestro
propósito no obtuvimos el resultado deseado mostrando lo dicho con el Figura 3,
que corrobora que después de tres horas no se logro obtener el complejo ZnT(o-
OH)PP.
En la figura 7 el espectro de la base libre H2T(3, 5-OH)PP se caracteriza
por una intensa banda de Soret a 430nm y una banda QII alrededor de 480nm.
Este compuesto fue al que insertamos en sol–gel. La preparación la realizamos
de acuerdo a las tablas 1 y 2. obteniendo los resultados mostrados en las Figuras
8 y 9.
Después de realizar el anterior protocolo de experimentación se obtuvieron
geles monolíticos transparentes y con ligera coloración amarillenta, en los cuales
la especie H2(3,5-OH)TPP ha quedado atrapada, ya que al colocar las anteriores
muestras en diversos disolventes no se observó salida alguna de pigmento de
porfirina, aún después de una semana de inmersión.
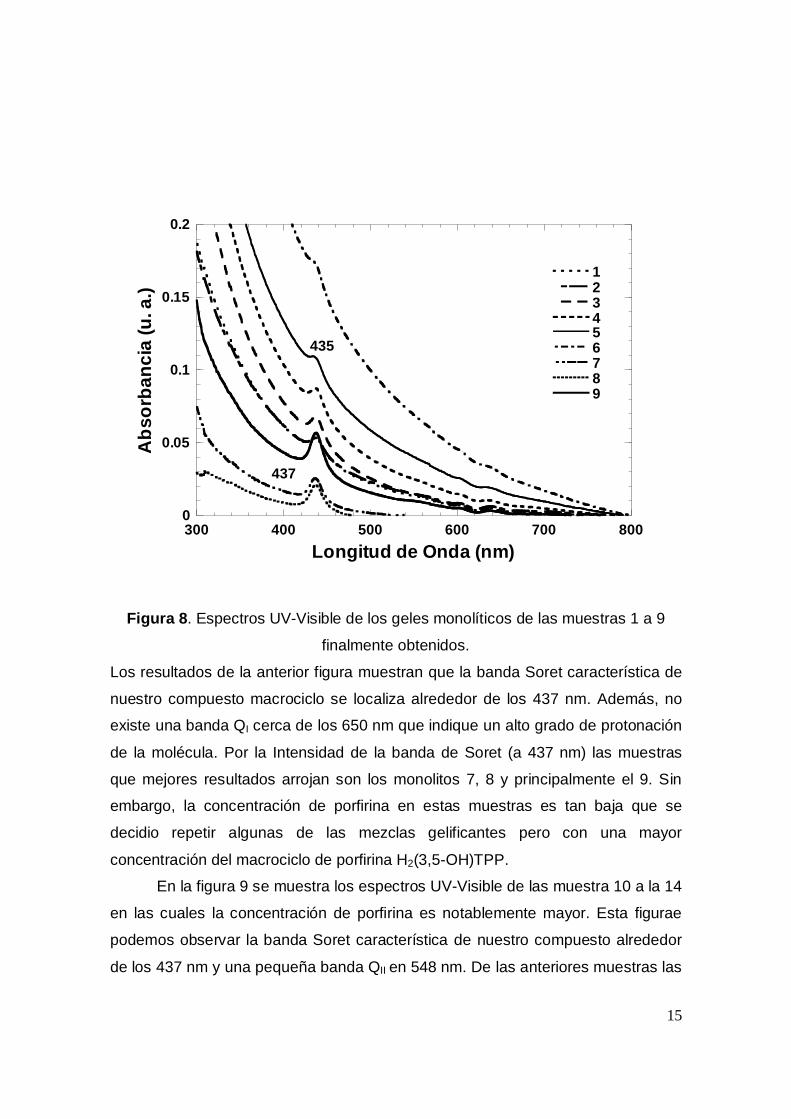
En la Figura 8 se presentan los espectros UV-Visible de los geles
monolíticos que se obtuvieron después de secar las muestras a temperatura
ambiente por una semana, a 70°C por tres dias y a 120°C por un dia.
15
0
0.05
0.1
0.15
0.2
300 400 500 600 700 800
123456789
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
435
437
Figura 8. Espectros UV-Visible de los geles monolíticos de las muestras 1 a 9
finalmente obtenidos.
Los resultados de la anterior figura muestran que la banda Soret característica de
nuestro compuesto macrociclo se localiza alrededor de los 437 nm. Además, no
existe una banda QI cerca de los 650 nm que indique un alto grado de protonación
de la molécula. Por la Intensidad de la banda de Soret (a 437 nm) las muestras
que mejores resultados arrojan son los monolitos 7, 8 y principalmente el 9. Sin
embargo, la concentración de porfirina en estas muestras es tan baja que se
decidio repetir algunas de las mezclas gelificantes pero con una mayor
concentración del macrociclo de porfirina H2(3,5-OH)TPP.
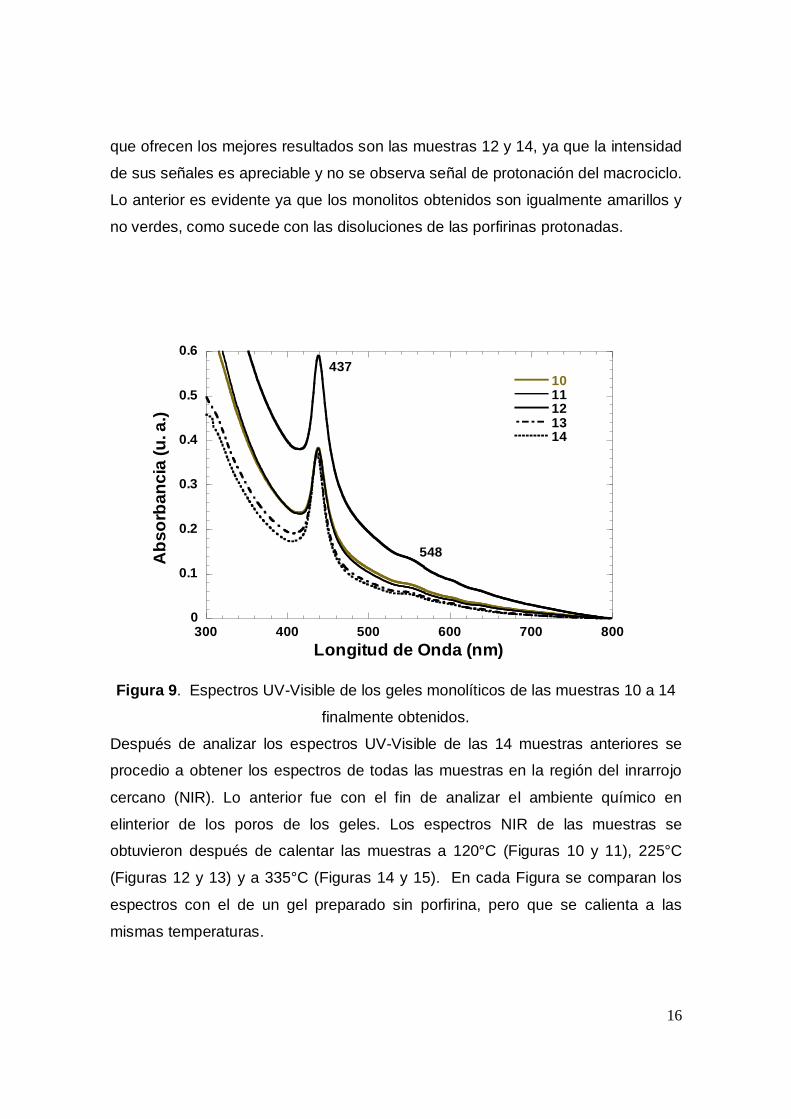
En la figura 9 se muestra los espectros UV-Visible de las muestra 10 a la 14
en las cuales la concentración de porfirina es notablemente mayor. Esta figurae
podemos observar la banda Soret característica de nuestro compuesto alrededor
de los 437 nm y una pequeña banda QII en 548 nm. De las anteriores muestras las
16
que ofrecen los mejores resultados son las muestras 12 y 14, ya que la intensidad
de sus señales es apreciable y no se observa señal de protonación del macrociclo.
Lo anterior es evidente ya que los monolitos obtenidos son igualmente amarillos y
no verdes, como sucede con las disoluciones de las porfirinas protonadas.
0
0.1
0.2
0.3
0.4
0.5
0.6
300 400 500 600 700 800
1011121314
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
437
548
Figura 9. Espectros UV-Visible de los geles monolíticos de las muestras 10 a 14
finalmente obtenidos.
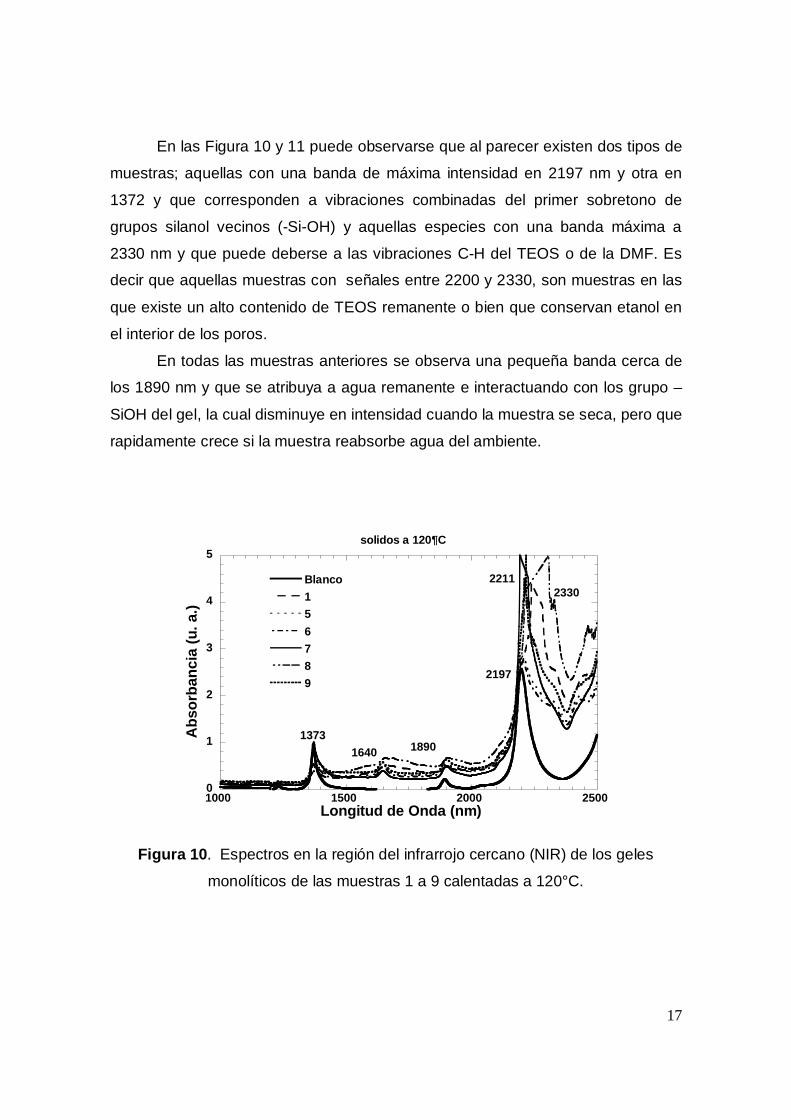
Después de analizar los espectros UV-Visible de las 14 muestras anteriores se
procedio a obtener los espectros de todas las muestras en la región del inrarrojo
cercano (NIR). Lo anterior fue con el fin de analizar el ambiente químico en
elinterior de los poros de los geles. Los espectros NIR de las muestras se
obtuvieron después de calentar las muestras a 120°C (Figuras 10 y 11), 225°C
(Figuras 12 y 13) y a 335°C (Figuras 14 y 15). En cada Figura se comparan los
espectros con el de un gel preparado sin porfirina, pero que se calienta a las
mismas temperaturas.
17
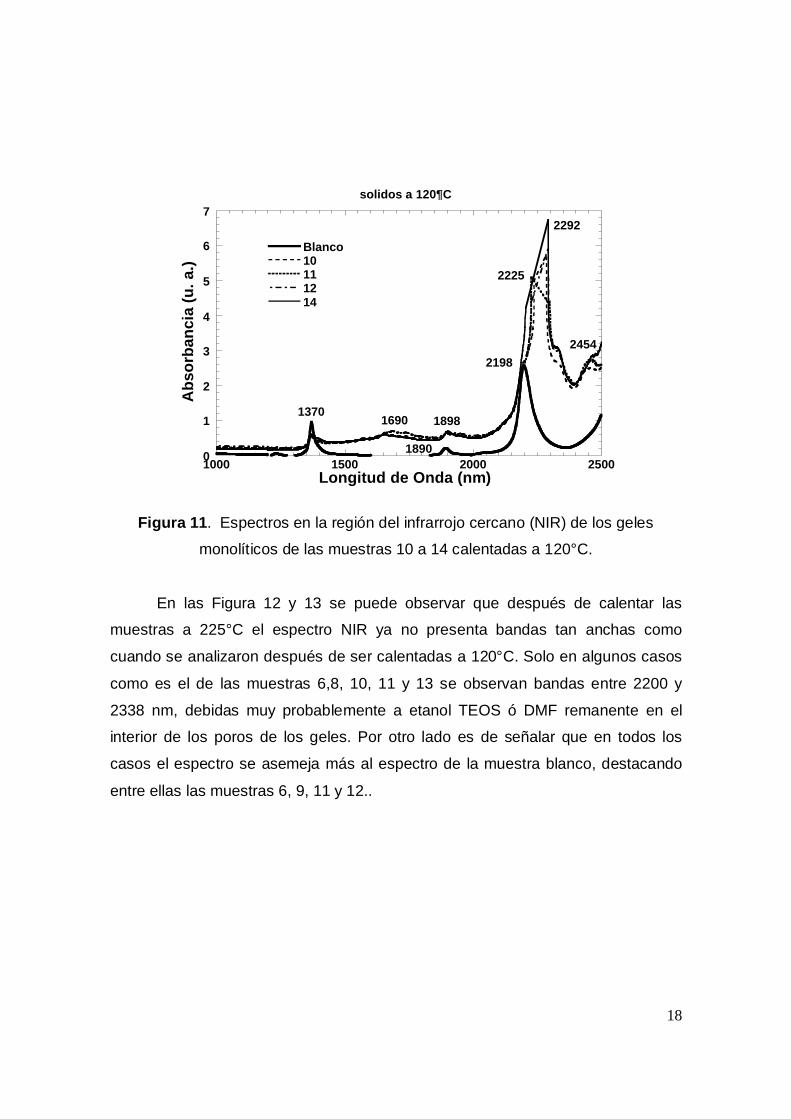
En las Figura 10 y 11 puede observarse que al parecer existen dos tipos de
muestras; aquellas con una banda de máxima intensidad en 2197 nm y otra en
1372 y que corresponden a vibraciones combinadas del primer sobretono de
grupos silanol vecinos (-Si-OH) y aquellas especies con una banda máxima a
2330 nm y que puede deberse a las vibraciones C-H del TEOS o de la DMF. Es
decir que aquellas muestras con señales entre 2200 y 2330, son muestras en las
que existe un alto contenido de TEOS remanente o bien que conservan etanol en
el interior de los poros.
En todas las muestras anteriores se observa una pequeña banda cerca de
los 1890 nm y que se atribuya a agua remanente e interactuando con los grupo –
SiOH del gel, la cual disminuye en intensidad cuando la muestra se seca, pero que
rapidamente crece si la muestra reabsorbe agua del ambiente.
0
1
2
3
4
5
1000 1500 2000 2500
solidos a 120¶C
Blanco156789
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
2197
189016401373
22112330
Figura 10. Espectros en la región del infrarrojo cercano (NIR) de los geles
monolíticos de las muestras 1 a 9 calentadas a 120°C.
18
0
1
2
3
4
5
6
7
1000 1500 2000 2500
solidos a 120¶C
Blanco10111214
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
2198
2225
2292
2454
1898
1890
16901370
Figura 11. Espectros en la región del infrarrojo cercano (NIR) de los geles
monolíticos de las muestras 10 a 14 calentadas a 120°C.
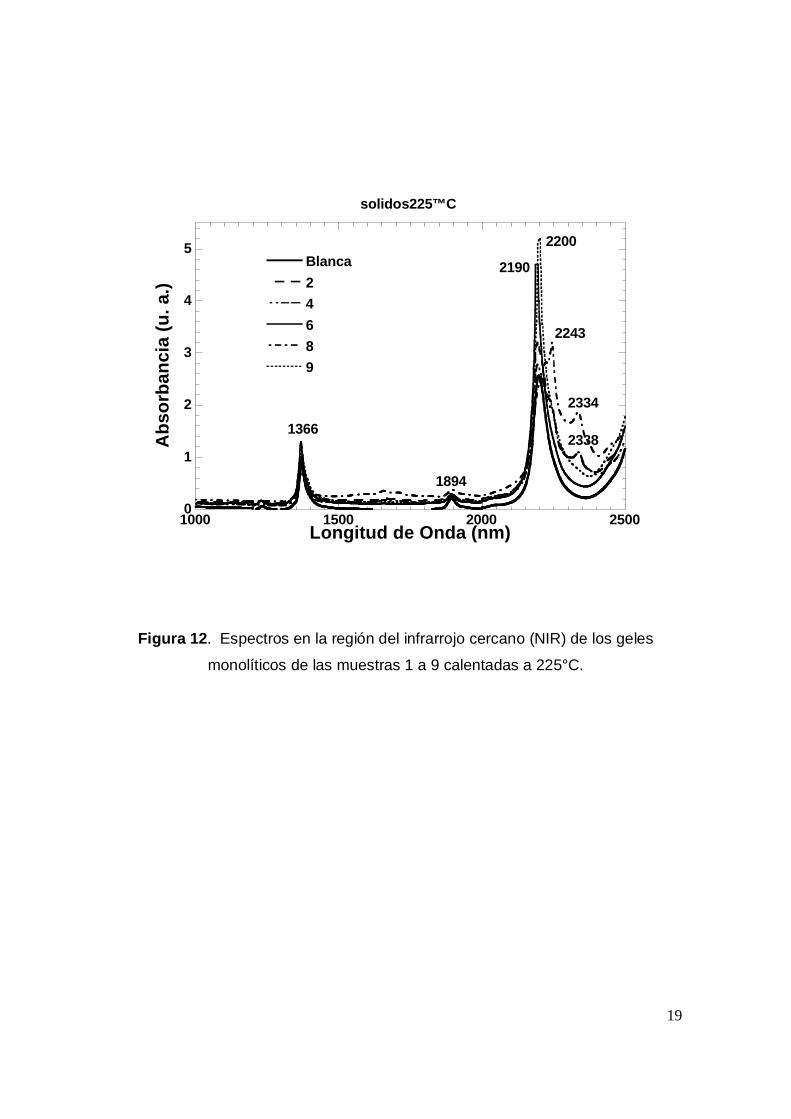
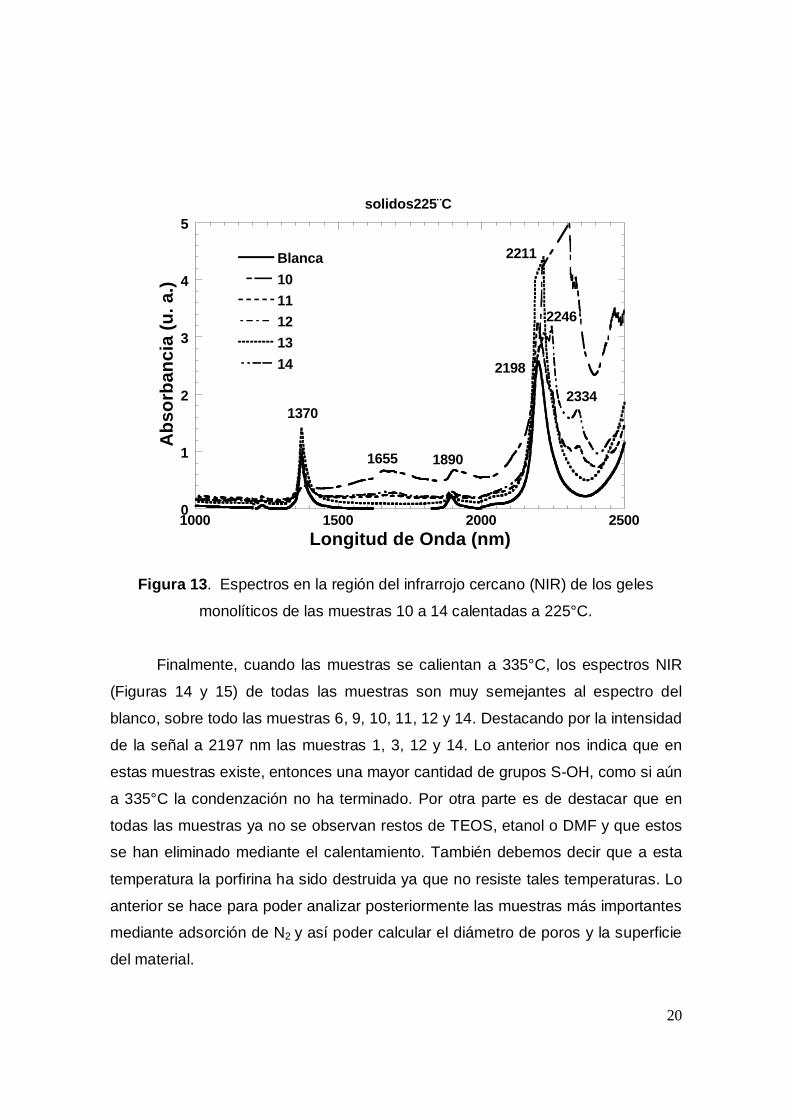
En las Figura 12 y 13 se puede observar que después de calentar las
muestras a 225°C el espectro NIR ya no presenta bandas tan anchas como
cuando se analizaron después de ser calentadas a 120°C. Solo en algunos casos
como es el de las muestras 6,8, 10, 11 y 13 se observan bandas entre 2200 y
2338 nm, debidas muy probablemente a etanol TEOS ó DMF remanente en el
interior de los poros de los geles. Por otro lado es de señalar que en todos los
casos el espectro se asemeja más al espectro de la muestra blanco, destacando
entre ellas las muestras 6, 9, 11 y 12..
19
0
1
2
3
4
5
1000 1500 2000 2500
solidos225™C
Blanca24689
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
2200
2190
2243
2334
2338
1894
1366
Figura 12. Espectros en la región del infrarrojo cercano (NIR) de los geles
monolíticos de las muestras 1 a 9 calentadas a 225°C.
20
0
1
2
3
4
5
1000 1500 2000 2500
solidos225¨C
Blanca1011121314
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
1370
18901655
2198
2211
2246
2334
Figura 13. Espectros en la región del infrarrojo cercano (NIR) de los geles
monolíticos de las muestras 10 a 14 calentadas a 225°C.
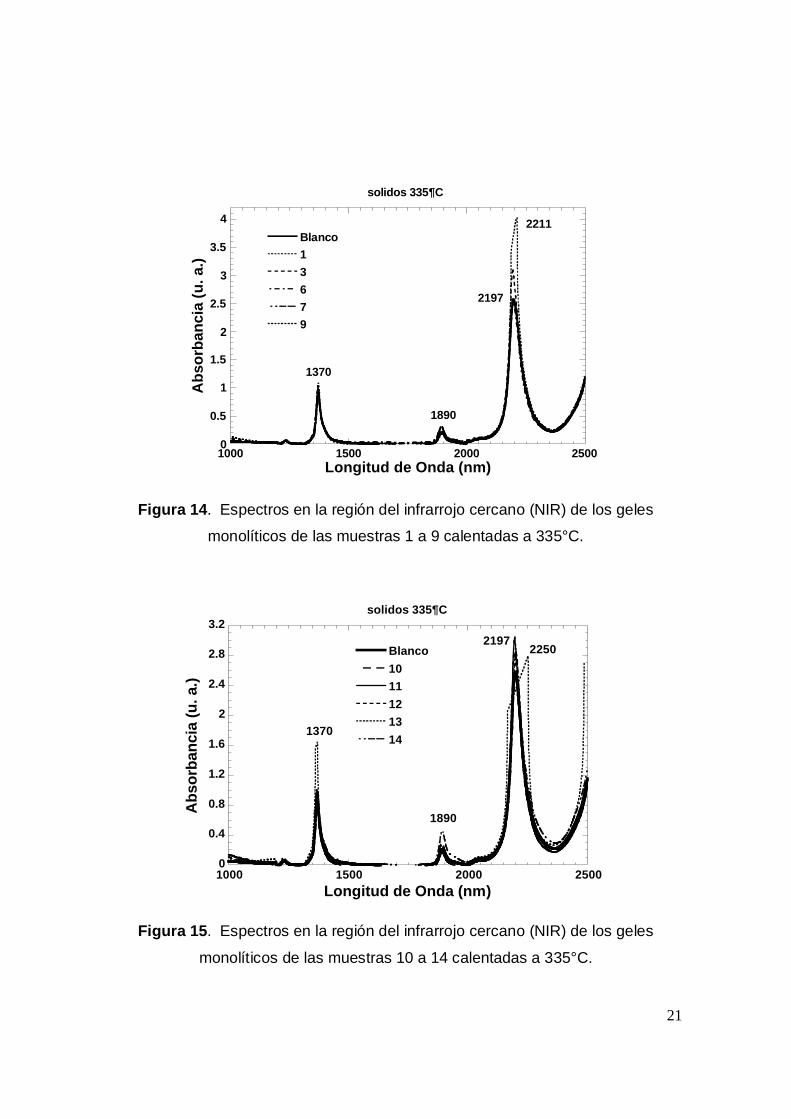
Finalmente, cuando las muestras se calientan a 335°C, los espectros NIR
(Figuras 14 y 15) de todas las muestras son muy semejantes al espectro del
blanco, sobre todo las muestras 6, 9, 10, 11, 12 y 14. Destacando por la intensidad
de la señal a 2197 nm las muestras 1, 3, 12 y 14. Lo anterior nos indica que en
estas muestras existe, entonces una mayor cantidad de grupos S-OH, como si aún
a 335°C la condenzación no ha terminado. Por otra parte es de destacar que en
todas las muestras ya no se observan restos de TEOS, etanol o DMF y que estos
se han eliminado mediante el calentamiento. También debemos decir que a esta
temperatura la porfirina ha sido destruida ya que no resiste tales temperaturas. Lo
anterior se hace para poder analizar posteriormente las muestras más importantes
mediante adsorción de N2 y así poder calcular el diámetro de poros y la superficie
del material.
21
0
0.5
1
1.5
2
2.5
3
3.5
4
1000 1500 2000 2500
solidos 335¶C
Blanco13679
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
2211
2197
1890
1370
Figura 14. Espectros en la región del infrarrojo cercano (NIR) de los geles
monolíticos de las muestras 1 a 9 calentadas a 335°C.
0
0.4
0.8
1.2
1.6
2
2.4
2.8
3.2
1000 1500 2000 2500
solidos 335¶C
Blanco1011121314
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
21972250
1890
1370
Figura 15. Espectros en la región del infrarrojo cercano (NIR) de los geles
monolíticos de las muestras 10 a 14 calentadas a 335°C.
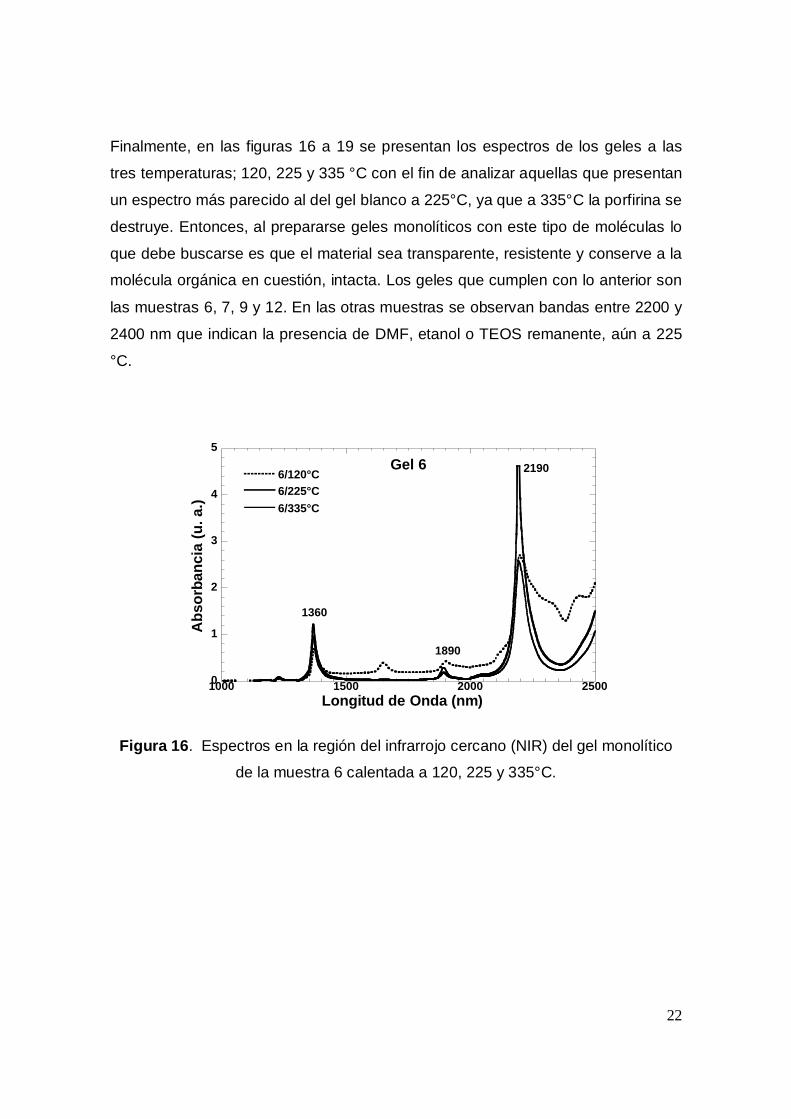
22
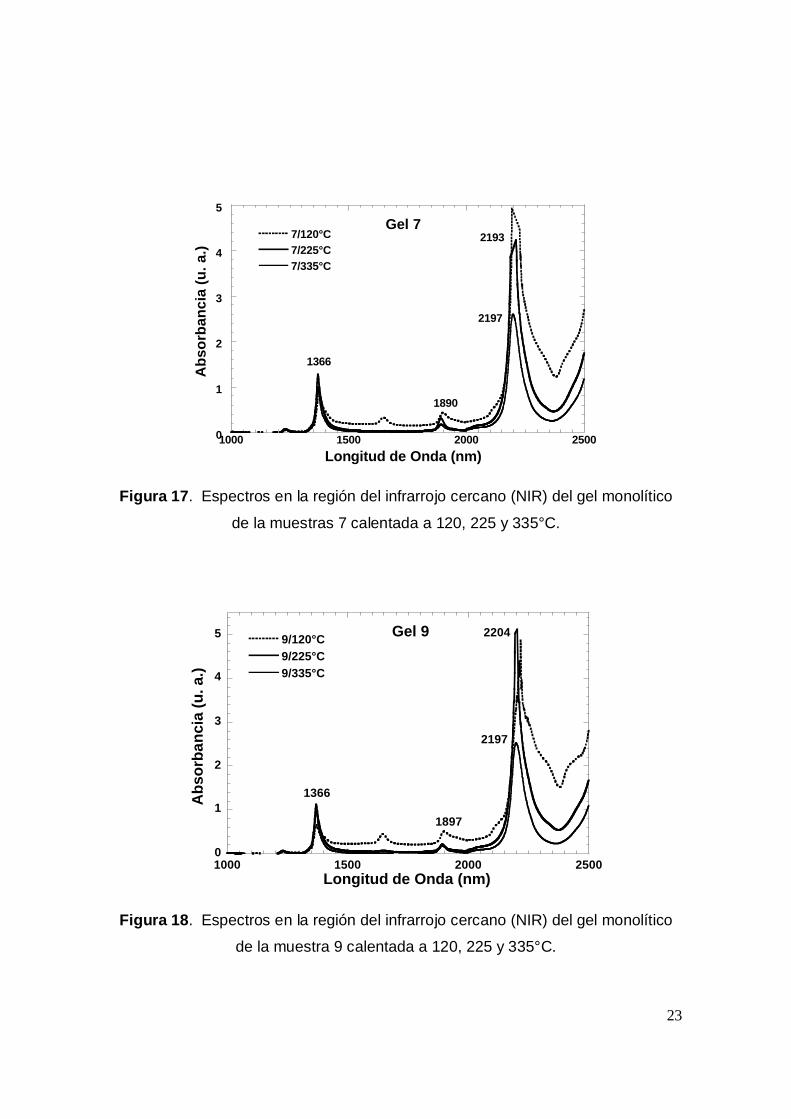
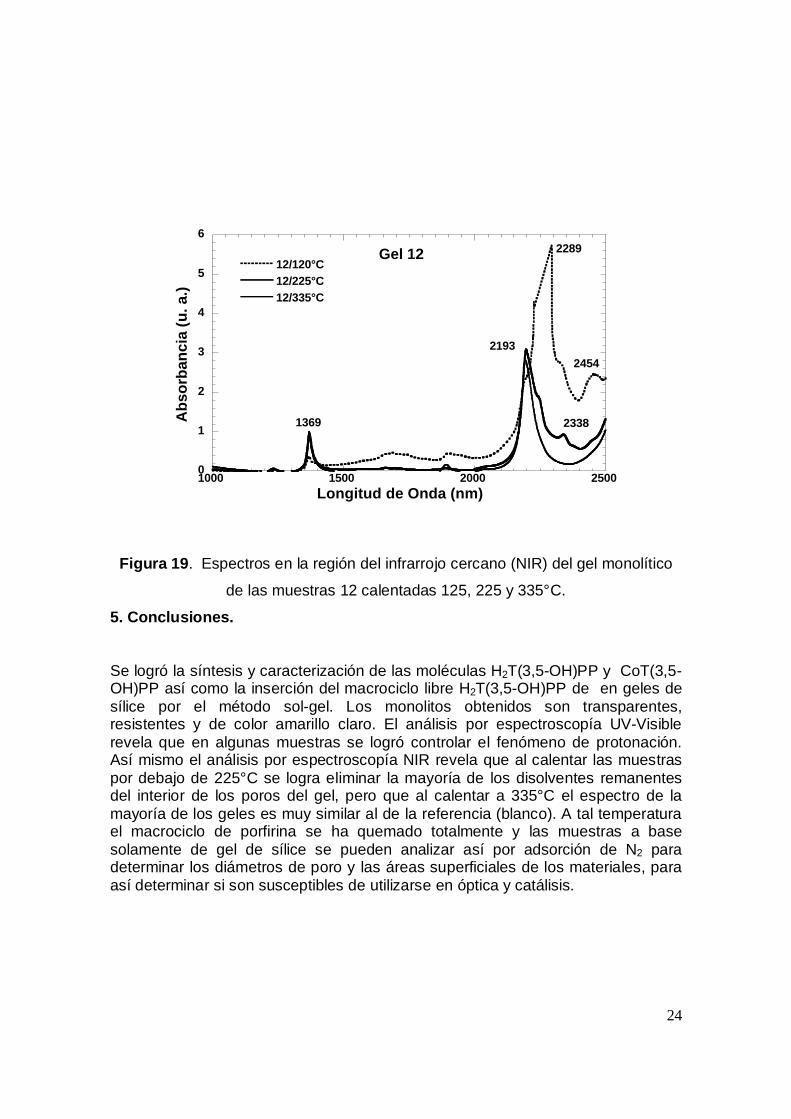
Finalmente, en las figuras 16 a 19 se presentan los espectros de los geles a las
tres temperaturas; 120, 225 y 335 °C con el fin de analizar aquellas que presentan
un espectro más parecido al del gel blanco a 225°C, ya que a 335°C la porfirina se
destruye. Entonces, al prepararse geles monolíticos con este tipo de moléculas lo
que debe buscarse es que el material sea transparente, resistente y conserve a la
molécula orgánica en cuestión, intacta. Los geles que cumplen con lo anterior son
las muestras 6, 7, 9 y 12. En las otras muestras se observan bandas entre 2200 y
2400 nm que indican la presencia de DMF, etanol o TEOS remanente, aún a 225
°C.
0
1
2
3
4
5
1000 1500 2000 2500
6/120°C6/225°C6/335°C
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
2190
1360
1890
Gel 6
Figura 16. Espectros en la región del infrarrojo cercano (NIR) del gel monolítico
de la muestra 6 calentada a 120, 225 y 335°C.
23
0
1
2
3
4
5
1000 1500 2000 2500
7/120°C7/225°C7/335°C
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
Gel 72193
2197
1890
1366
Figura 17. Espectros en la región del infrarrojo cercano (NIR) del gel monolítico
de la muestras 7 calentada a 120, 225 y 335°C.
0
1
2
3
4
5
1000 1500 2000 2500
9/120°C9/225°C9/335°C
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
Gel 9 2204
2197
1897
1366
Figura 18. Espectros en la región del infrarrojo cercano (NIR) del gel monolítico
de la muestra 9 calentada a 120, 225 y 335°C.
24
0
1
2
3
4
5
6
1000 1500 2000 2500
12/120°C12/225°C12/335°C
Abs
orba
ncia
(u. a
.)
Longitud de Onda (nm)
Gel 12 2289
24542193
23381369
Figura 19. Espectros en la región del infrarrojo cercano (NIR) del gel monolítico
de las muestras 12 calentadas 125, 225 y 335°C.
5. Conclusiones.
Se logró la síntesis y caracterización de las moléculas H2T(3,5-OH)PP y CoT(3,5-OH)PP así como la inserción del macrociclo libre H2T(3,5-OH)PP de en geles desílice por el método sol-gel. Los monolitos obtenidos son transparentes,resistentes y de color amarillo claro. El análisis por espectroscopía UV-Visiblerevela que en algunas muestras se logró controlar el fenómeno de protonación.Así mismo el análisis por espectroscopía NIR revela que al calentar las muestraspor debajo de 225°C se logra eliminar la mayoría de los disolventes remanentesdel interior de los poros del gel, pero que al calentar a 335°C el espectro de lamayoría de los geles es muy similar al de la referencia (blanco). A tal temperaturael macrociclo de porfirina se ha quemado totalmente y las muestras a basesolamente de gel de sílice se pueden analizar así por adsorción de N2 paradeterminar los diámetros de poro y las áreas superficiales de los materiales, paraasí determinar si son susceptibles de utilizarse en óptica y catálisis.

25
BIBLIOGRAFIA. [1] K. M. Smith, Porphyrins and Metalloporphyrins, Academic Press, New York,
1978.
[2] T.H. Wei, D.J. Hagan, M.J. Sence, E.W. Stryland, J. W. Perry, D.R. Coulter, Appl.
Phys. B54 (1992) 46.
[3] J.K. Duchowski, D.F. Bocian, J. Am. Chem. Soc., 112, (1990) 3312.
[4] Y. Liu, K. Shigehara, A. Yamada, Bull. Chem. Soc. Jpn. 65 (1992) 250.
[5] J.H. Weber, D.H. Buch, Inorg. Chem. 4 (1965) 469.
[6] L.A, Martarano, C. P. Wong, W. de W. Horrocks Jr., J. of Phys. Chem. 80, (1976)
2389.
[7] M. Gouterman, J. Chem. Phy. 30 (1959) 30.
[8] M. Gouterman in The Porphyrins, Physical Chemistry, Part A, Ed. D. Dolphin
Academic Press, New York 1978.
[9] N. H. Sabell, C. A. Melendres, J. Am. Chem. Soc. 86 (1982) 3926.
[10] C. Fierro, A. B. Anderson, D. A. Scherson, J. Phys. Chem. 92, (1988) 6902.
[11] T. Tani, H. Namikawa, K. Arai, A, Makishima, J. Appl. Phys. 58 (1985) 3559.
[12] H. Inoue, T. Iwamoto, A. Makishima, M. Ikemoto, K. Horie, J. Opt. Soc. Am. B
9(5) (1992) 816.
[13] H. Tanaka, J. Takahashi, J. Tsuchiya, Y. Kobayashi, Y. Kurokawa, J. Non-Cryst.
Solids 109 (1989) 164.
[14] J. Friedrich, H. Wolfrum, D. Haarer, J. Chem. Phys. 77 (1982) 2309.
[15] A. Furusawa, K. Horie, I. Mita, Chem. Phys. Lett. 161 (1989) 227.
[16] W. P. Ambrose, W. E. Moerner, Chem. Phys. 144 (1990) 71.
17.- R. Locher, A. Renn, U. P. Wild, Chem. Phys. Lett. 138 (1987) 405.
[18] L. Bartoy, J. P. Lallier, P. Battioni, D. Mansuy, New J. Chem. 16, (1972) 71.
[19] K. Kamitani, M. Uo, H. Inoue, A. Makishima, J. of Sol-Gel Science and
Technology 1, (1993) 85.
[20] X-J. Wang, L. M. Yates III, E. T. Knobbe, J. of Luminescence 60&61 (1994) 469.
[21] A. Clark, V. Terpugov, F. Medrano, M. Cervantes, D. Soto, Optical Materials
13(3) (1999) 355.
22- D. Avnir, D. Levy, R. Reisfield, J. Phys. Chem. 88(1984), 5956-5959.23- V. R. Kaufman, D. Levy, D. Avnir, J. Non-Cryst. Solids, 82, 103(1986)

26
24- A. Makishima,T. Tani, J. Am. Ceram. Soc., 69, 4(1986)25- J. Mckiernan, J. C. Pouxviel, B. Dunn, J. I. Zink, J. Phys. Chem. 93, 2129(1989).26- J. Fitremann, S. Doeuff, C. Sanchez, Ann. Chim. Fr., 15, 421(1990)27- R. Litrán, E. Blanco, M. Ramírez-Del Solar and L. Esquivias, J. of Sol-Gel
Science and Technology 8 (1997) 985.
28- M.A. García-Sánchez, C. Campero, J. of Sol-Gel Sci. Thechnol. 13(1998), 651-655.29- . M. A. García-Sánchez, A. Campero, Polyhedron, 19(2000) 2383.30- M. A. García-Sánchez, A. Campero, J. of Non-Cryst. Solids, 296 (2001) 50.
31- M. A. García-Sánchez, C. Velásquez, R. Sosa F, A. Campero, A. Muñoz F., Mat.
Chem. and Phys, 84 (2004)216.
32- M. A. García-Sánchez, A. Campero, J. of Non-Cryst. Solids, 333 (2004) 226.





























